As filed with the U.S. Securities and Exchange Commission on January 3, 2025.
Registration No.
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM F-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
(Exact name of registrant as specified in its charter)
| British Virgin Islands | | 2834 | | Not Applicable |
(State or other jurisdiction of incorporation or organization) | | (Primary Standard Industrial Classification Code Number) | | (I.R.S. Employer Identification Number) |
10F-1, No.130,
Songshan Rd., Xinyi Dist.,
Taipei City 110
Taiwan
+886-2-2756-3796
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Cogency Global Inc.
122 East 42nd Street, 18th Floor
New York, NY 10168
(212) 947-7200
(Name, address, including zip code, and telephone number, including area code, of agent for service)
With a Copy to:
Keith Billotti F. Holt Goddard Seward & Kissel LLP One Battery Park Plaza New York, NY 10004 Tel: (212) 574-1200 | | Richard I. Anslow Charles Phillips Ellenoff Grossman & Schole LLP 1345 Avenue of the Americas New York, NY 10105 Tel: (212) 370-1300 |
Approximate date of commencement of proposed sale to the public: Promptly after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box: ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933.
Emerging growth company ☒
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act ☒
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the U.S. Securities and Exchange Commission, acting pursuant to such Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. We may not sell the securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and we are not soliciting any offer to buy these securities in any jurisdiction where such offer or sale is not permitted.
| Preliminary Prospectus | SUBJECT TO COMPLETION, DATED , 2025 |
Ordinary Shares

EVERFRONT BIOTECH HOLDING COMPANY LIMITED
This is an initial public offering of ordinary shares of Everfront Biotech Holding Company Limited (the “Company” or “Everfront Holding”, and when referring to the consolidated company including the Company’s subsidiaries, “we”, “us” and “our”), par value $0.01 per share (“Ordinary Shares”). We estimate that the initial public offering price will be between $ and $ per Ordinary Share.
We have reserved the symbol “EFB” for the purpose of listing our Ordinary Shares on the Nasdaq Capital Market (“Nasdaq”). This offering is contingent upon the final approval from Nasdaq for the listing of our Ordinary Shares. We will not consummate this offering if Nasdaq denies our listing application. As such, this offering may not close and our Ordinary Shares may not be approved for trading on Nasdaq.
Investing in our Ordinary Shares involves a high degree of risk, including the risk of losing your entire investment. See “Risk Factors” beginning on page 7 of this prospectus to read about factors you should consider before buying Ordinary Shares.
Upon the completion of this offering, we will have Ordinary Shares issued and outstanding. Each Ordinary Share is entitled to one vote.
As of the date hereof (after giving effect to this offering), Mr. Ho-Ching Chen holds beneficial ownership of % of our outstanding Ordinary Shares pursuant to agreements in which certain of our shareholders have given Mr. Chen voting control over their shares, including in connection with the election of our directors. This means we meet the definition of a “controlled company” under the corporate governance standards of Nasdaq and thereby qualify for exemptions from certain Nasdaq corporate governance requirements. We currently expect to rely on certain of these exemptions and may in the future elect to rely on any or all of these exemptions for so long as we remain a “controlled company.” See “Management—Corporate Governance—Controlled Company Exemptions”.
Everfront Holding is a holding company incorporated in the British Virgin Islands (“BVI”) that conducts its operations through its 99.96% owned subsidiary, Everfront Biotech Inc. (“Everfront Biotech”), in Taiwan. This is an offering of the Ordinary Shares of Everfront Holding and not equity securities of Everfront Biotech. You may never directly hold any equity interest in Everfront Holding’s operating entity.
We are an “emerging growth company” as defined under the federal securities laws and will be subject to reduced public company reporting requirements. See “Risk Factors” and “Prospectus Summary— Implications of our Being an Emerging Growth Company”.
Neither the Securities and Exchange Commission nor any state securities commission nor any other regulatory body has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| | | Per Ordinary Share | | | Total | |
| Initial public offering price(1) | | $ | | | | $ | | |
| Underwriting discounts and commissions(2) | | $ | | | | $ | | |
| Proceeds to us, before expenses(3) | | $ | | | | $ | | |
| (1) | Assumes an initial public offering price of $ per Ordinary Share, which is the midpoint of the range set forth on the cover page of this prospectus. For more information, see “Underwriting.” |
| (2) | We have agreed to reimburse the underwriters for certain expenses. For more information, see “Underwriting.” |
| (3) | We expect our cash expenses for this offering (including cash expenses payable to our underwriters for the underwriters’ out-of-pocket expenses) will not exceed $ , exclusive of the underwriters’ discounts. |
This offering is being conducted on a firm commitment basis. The underwriters are obligated to purchase all of the Ordinary Shares if they purchase any Ordinary Shares.
We have granted the underwriters an option for a period of up to 45 days to purchase up to an additional Ordinary Shares from us at the initial public offering price, less the underwriting discounts and commissions.
The underwriters expect to deliver the Ordinary Shares to purchasers in the offering on or about , 2025.
We may amend or supplement this prospectus from time to time by filing amendments or supplements as required. You should read this entire prospectus and any amendments or supplements carefully before you make your investment decision.
Joint Book-Runners
| Roth Capital Partners | | The Benchmark Company |
Prospectus dated , 2025
TABLE OF CONTENTS
We and the underwriters have not authorized any person to give you any supplemental information or to make any representations on our behalf. You should not assume that the information contained in this prospectus or any prospectus supplement are accurate as of any date other than their respective dates, regardless of the time of delivery of this prospectus or of any sale of the Ordinary Shares. This prospectus is an offer to sell only the Ordinary Shares offered hereby, but only under circumstances and in jurisdictions where it is lawful to do so. We are not making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted or where the person making the offer or sale is not qualified to do so or to any person to whom it is not permitted to make such offer or sale. The information in this registration statement is not complete and is subject to change. No person should rely on the information contained in this document for any purpose other than participating in our proposed offering, and only the prospectus dated hereof, is authorized by us to be used in connection with our proposed offering. The preliminary prospectus will only be distributed by us and no other person has been authorized by us to use this document to offer or sell any of our securities.
Until , 2025 (the 25th day after the date of this prospectus), all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
COMMONLY USED DEFINED TERMS
Unless otherwise indicated or the context requires otherwise, references in this prospectus to:
| ● | “we”, or “us” in this prospectus are to Everfront Biotech Holding Company Limited, a British Virgin Islands company and its subsidiary, Everfront Biotech Inc., a company incorporated under the laws of Taiwan, unless the context otherwise indicates; |
| ● | “BVI” are to the “British Virgin Islands”; |
| ● | “BVI Act” are to the BVI Business Companies Act (Law Revision 2020) (as amended); |
| ● | the “Company” or “Everfront Holding” are to Everfront Biotech Holding Company Limited, a BVI company; |
| ● | “Everfront Biotech” are to Everfront Biotech Inc., a company incorporated under the laws of Taiwan, with limited liability; |
| ● | “$,” “dollars,” “US$” or “U.S. dollars” are to the legal currency of the United States; |
| ● | “NTD” are to the legal currency of R.O.C.; |
| ● | “U.S. GAAP” are to generally accepted accounting principles in the United States; |
| ● | “shares” or “Ordinary Shares” are to the ordinary shares of Everfront Biotech Holding Company Limited, par value $0.01 per share; and |
| ● | “R.O.C.” or “Taiwan” refers to Taiwan, the Republic of China. |
Everfront Holding does not have any material operations of its own and Everfront Holding is a holding company with operations conducted in Taiwan through its Taiwan subsidiary, Everfront Biotech, using NTD, the currency of Taiwan. Our reporting currency is NTD. This prospectus contains translations of certain foreign currency amounts into U.S. dollars for the convenience of the reader. All translations of NTD are calculated at the rate of NTD to US$1.00, representing the exchange rate set forth in the H.10 statistical release of the Federal Reserve Board on , 2025. No representation is made that the NTD amounts could have been, or could be, converted, realized or settled into US$ at such rate, or at any other rate.
PROSPECTUS SUMMARY
The following summary is qualified in its entirety by, and should be read in conjunction with, the more detailed information and financial statements included elsewhere in this prospectus. In addition to this summary, we urge you to read the entire prospectus carefully, especially the risks of investing in Ordinary Shares, discussed under “Risk Factors” before deciding whether to buy Ordinary Shares.
Everfront Holding is a holding company incorporated in the British Virgin Islands (“BVI”) that conducts its operations through its 99.96% owned subsidiary, Everfront Biotech Inc. (“Everfront Biotech”), which is incorporated and operates in Taiwan. Everfront Holding holds no other assets and conducts no operations of its own.
Overview
We are a clinical stage biopharmaceutical company dedicated to the discovery, development and commercialization of new drugs that have potential as treatments for patients with cancers, neurodegenerative diseases and rare diseases. We are focused on new drug discovery and development, supported by our research and development (“R&D”) team established in 2010. Our team is specialized in preclinical study design, chemistry, manufacturing, and controls (“CMC”), investigational new drug (“IND”) submission, and clinical trial design and conduction. Our goal is to develop technology with the commercial potential to help people with unmet medical needs.
As of the date of this prospectus, we are conducting three clinical trials in Taiwan under the approval of the United States Food and Drug Administration (the “FDA”) and the Taiwan Food and Drug Administration (the “TFDA”) for the following product candidates. As of the date of this prospectus, we have no products approved for commercial sale and have never generated any revenue from product sales. Please also see the risk factor entitled “We are a cancer drug development company with a limited operating history.”
Cerebraca™ Wafer is a biodegradable implant produced according to Pharmaceutical Inspection Co-operation Scheme, or PIC/S, Good Manufacturing Practice, or GMP, standards. Cerebraca™ Wafer has been designed to treat glioblastoma (GBM) by directly implanting it into the GBM tumor margin after surgical resection. Cerebraca™ Wafer is characterized by its 30-day sustained drug release and 5-cm penetration depth observed in preclinical studies (IND128388, Module 2, Report 51904-12-708), with the intention that this will create a high drug concentration environment for treating the cancer. Cerebraca™ Wafer is a multi-functional drug with the potential to inhibit receptor tyrosine kinases (Epidermal Growth Factor Receptor, or EGFR, Axl-1 Receptor, and c-Mesenchymal-Epithelial Transition Factor Receptor, or cMet Receptor) to halt cancer stem cell proliferation. Additionally, it has the potential to improve patients’ quality of life by boosting Adenosine Triphosphate, or ATP, levels through Axl-mTOR (mammalian target of rapamycin) (Liu C-A, et al., Journal of Oncology. 2022; 2022.) inhibition and AMP-activated Protein Kinase, or AMPK, activation (Lee J-H, et al., International Journal of Molecular Science. 2021; 22(12): 6339.). Furthermore, Cerebraca™ Wafer has been designed to promote methylation of O6-Methylguanine-DNA Methyltransferase, or MGMT, and Programmed Cell Death Ligand 1, or PD-L1, promoters in order to help overcome chemotherapeutic resistance (Liu C-A, et al., Cancers. 2022; 14(4): 1051.) and transform a cold tumor microenvironment into an immune-active hotspot (Liu C-A, et al., Journal of Oncology. 2022; 2022.).
In the Phase I/IIa clinical trial, recurrent GBM patients with receptor tyrosine kinase markers (EGF receptor, Axl-1 receptor, and cMet receptor) who received surgical resection of GBM and implantation of the Cerebraca™ Wafer followed by temozolomide therapy showed a median overall survival of 26.2 months in the preliminary results. However, limited reliable biomarkers currently exist for GBM diagnosis and treatment selection. Identifying these biomarkers and developing accurate tests to detect them is crucial for personalized medicine but very challenging. In our next clinical study, we will recruit GBM patients with IDH wild-type (National Comprehensive Cancer Network, or NCCN, Guidelines Central Nervous System Cancers, Version 1.2024), and we will analyze biomarkers in a subgroup analysis. While the preliminary results of the Phase I/IIa clinical trial are encouraging, extending the median overall survival beyond 26.2 months will require biomarker identification before surgery, which may pose significant scientific and clinical challenges. This difficulty is multifaceted and can be attributed to several key factors.
| | ● | Challenge of Companion Diagnostics Development: There is currently no approved Companion Diagnostics for GBM treatment or diagnosis. |
| | ● | Limited Reliable Biomarkers: One of the primary challenges in GBM treatment is the scarcity of reliable biomarkers for diagnosis and treatment selection. |
| | ● | Heterogeneity of GBM: Identifying and targeting the specific molecular pathways involved in each patient’s tumor remains a significant challenge. |
| | ● | Treatment Resistance Mechanisms: The ability of GBM to adapt and resist treatment poses a significant hurdle in extending survival times further. |
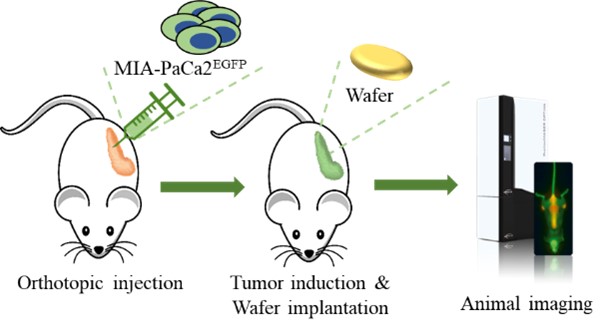
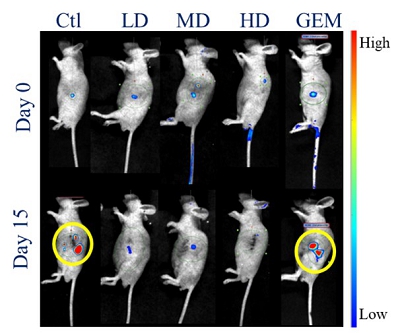
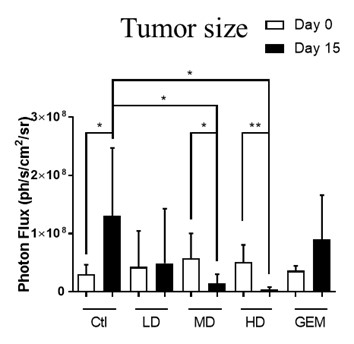
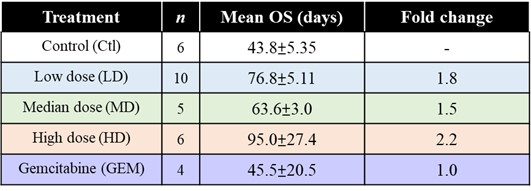
EF-009 Wafer is a biodegradable implant produced according to PIC/S GMP standards that is designed to treat pancreatic cancer by directly implanting it into the pancreatic cancer tumor margin (IND145153, Module 4, Report D06), which is intended to lead to local tumor regression. This neo-adjuvant therapy may allow patients with locally advanced pancreatic cancer to undergo tumor resection therapy. EF-009 has been designed to work by regulating DNA-methyltransferase 1, or DNMT1, and its downstream targets, Patched Domain-Containing 4, or PTCHD4, and hedgehog signaling (Huang M-H, et al., Pharmacological Research. 2019; 139: 50.), in order to help reduce pancreatic cancer growth. Additionally, our preclinical studies have demonstrated that EF-009 can have synergistic effects with existing chemotherapy drugs, including Gemcitabine, Cisplatin, 5-Fluorouracil, Irinotecan, Oxaliplatin, and Paclitaxel (Patents for cancer treatment, p.63, a combination for the treatment of cancer and its application), implying that combining EF-009 with these currently available chemotherapy drugs may help achieve improved clinical outcomes. We have received approval from both the U.S. Food and Drug Administration (“USFDA”) and Taiwan Food and Drug Administration (“TFDA”) to conduct a Phase I/IIa clinical study to evaluate the safety profile and therapeutic effects on patients with locally advanced pancreatic cancer. We have completed site selection for this study and expect to begin enrolling patients in the second quarter of 2025. Nevertheless, we will likely need to conduct a proof-of-concept study in humans to reproduce the effects observed in preclinical research. Other risks associated with using the EF-009 Wafer implant may include local inflammation caused by laparoscopic intervention and investigational product degradation, as well as surgical procedures, infection, wound bleeding, and anesthesia.
EF-031 is a soft-gel capsule formulation produced according to PIC/S GMP standards and designed to serve as a second-generation product to support the therapeutic actions of the Cerebraca™ Wafer and the EF-009 Wafer. Unlike the Cerebraca™ Wafer and the EF-009 Wafer, which require surgical implantation, EF-031 can be administered orally. In a study conducted by Pharmaron, a contract research organization (study report 51904-15-393), systemic oral administration of EF-031 revealed a 5.6 times higher concentration of EF-031 in the brain and a 64.2 times higher concentration in the pancreas compared to a plasma exposure. Nevertheless, we will likely need to conduct a proof-of-concept study in humans to replicate the effects observed in preclinical research.
HK-001 is a soft-gel capsule formulation produced according to PIC/S GMP standards and designed for the treatment of Amyotrophic Lateral Sclerosis (ALS). Animal study results indicate that administration of HK-001 may help prolong the survival of ALS animals and delay disease progression (Hsueh K-W, et al., Neuropharmacology. 2016; 108: 152.). A Phase I clinical trial on healthy subjects is ongoing to determine the maximum tolerated dose (MTD). Nevertheless, we will likely need to conduct a proof-of-concept study in humans to replicate the effects observed in preclinical research.
Corporate Information
Our principal executive offices are 10F-1, No. 130, Songshan Rd., Xinyi Dist., Taipei City 110, Taiwan, and our telephone number is +886-2-27563796. Our registered office in the British Virgin Islands is at Portcullis Chambers, 4th Floor, Ellen Skelton Building, 3076 Sir Francis Drake Highway, Road Town, Tortola, British Virgin Islands. We maintain a website at http://www.efbiotech.com. The information contained in, or accessible from, our website or any other website does not constitute a part of this prospectus.
Risk Factors Summary
An investment in our securities is subject to a number of risks, including risks related to our industry, business and corporate structure. The following summarizes some, but not all, of these risks. Please carefully consider all of the information discussed in “Risk Factors” in this prospectus beginning on page 7 for a more thorough description of these and other risks.
| | ● | We are a cancer drug development company with a limited operating history. |
| | ● | We expect our operating results to fluctuate significantly in the future as our business advances. |
| | ● | We have no products approved for commercial sale and have not generated any revenue from product sales. |
| | ● | If we fail to raise additional funds, our ability to execute our business and development strategies may be affected. |
| | ● | Raising additional capital may cause dilution to our shareholders, restrict our operations, or require us to relinquish rights to our technologies or product candidates. |
| | ● | We have never successfully completed clinical development for any of our product candidates, and we may be unable to do so. Certain of our cancer drugs are still in preclinical development and may never advance to clinical development. |
| | ● | Clinical product development involves a lengthy and expensive process, with an uncertain outcome. |
| | ● | Interim, top-line, and initial data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to confirmation, audit and verification procedures that could result in material changes in the final data. |
| | ● | We may incur additional costs or experience delays in initiating or completing, or ultimately be unable to complete, the development and commercialization of our product candidates. |
| | ● | If we experience delays or difficulties in the enrollment of patients in clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented. |
| | ● | We may not be able to replicate the results from our earlier preclinical and clinical studies in later preclinical studies and clinical trials, and this could prevent us from successfully developing, obtaining regulatory approval for and commercializing our product candidates. |
| | ● | Our growth depends on our ability to successfully develop, acquire or license new drugs. |
| | ● | Clinical trials may be subject to liability claims, which may delay or even cause our R&D plans to fail, consume our resources, cause us to incur significant liability and limit the development of our products. |
| | ● | We conduct clinical trials on some of our product candidates at locations outside the United States, approval by the FDA is critical to our development, and the FDA may not accept data from trials conducted at certain locations. |
| | ● | If clinical trials of our product candidates fail to demonstrate safety and efficacy, we may incur additional costs or delays, or ultimately fail to complete the development and commercialization of our product candidates. |
| | ● | We have no history of obtaining regulatory approval or commercialization of any new drug candidates. |
| | ● | We face substantial competition, which may result in others discovering, developing or commercializing products before, or more successfully than, we do. |
| | ● | If our current product candidates or any future product candidates do not achieve broad market acceptance, the revenue that we generate from their sales may be limited, and we may never become profitable. |
| | ● | If side effects associated with our current or future products are discovered prior to marketing and sale, we may be required to withdraw these products from the market, conduct lengthy additional clinical trials or change the labeling of our products, any of which could adversely affect our growth. |
| | ● | We may be subject to product liability claims in the future, which may consume our resources, expose us to significant liability and limit the commercialization of any products we may develop. |
| | ● | Even if our product candidates are approved for marketing, they may not be acceptable to physicians, patients, third-party payers and others in the medical community, and the market opportunity for our product candidates may be smaller than we estimate to achieve the market size required for commercial success. |
| | ● | Obtaining and enforcing pharmaceutical patents involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our business, financial position, operations and prospects, and interrupt our research activities. |
| | ● | Developments in patent law could have a negative impact on our patent positions and business. |
| | ● | If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed, respectively. |
| | ● | We and our licensors may not be able to enforce our intellectual property rights throughout the world. |
| | ● | We may seek to partner with third parties to develop and commercialize our product candidates, and if we fail to enter into such collaborations, or if such collaborations are unsuccessful, we may not be able to realize the market potential of our product candidates. |
| | ● | We are a development-stage drug discovery company and therefore face risks associated with the development of new businesses in the industry. |
| | ● | Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions. |
| | ● | We may seek orphan drug designation for certain of our product candidates, and we may be unsuccessful or may be unable to maintain the benefits associated with orphan drug designation, including the potential for market exclusivity. |
| | ● | If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business. |
| | ● | We have convertible debt that will be converted into our Ordinary Shares upon the closing of this offering, which will cause immediate and substantial dilution to our shareholders. |
| | ● | There has been no public market for our Ordinary Shares prior to this offering, and if an active trading market does not develop, you may not be able to resell our Ordinary Shares at or above the price you paid, or at all. |
| | ● | As a “controlled company” under Nasdaq Listing Rules, we plan to rely on certain exemptions from Nasdaq corporate governance rules, which means our shareholders will not have the same protections afforded to shareholders of other companies. |
| | ● | Nasdaq may apply additional and more stringent criteria for our initial and continued listing because we plan to have a small public offering and we insiders will hold a large portion of our listed securities. |
| | ● | Our Ordinary Shares may be thinly traded and you may be unable to sell at or near ask prices or at all if you need to sell your shares to raise money or otherwise desire to liquidate your shares. |
| | ● | The initial public offering price for our Ordinary Shares may not be indicative of prices that will prevail in the trading market and such market prices may be volatile. |
| | ● | Our management team has limited experience managing a public company. |
| | ● | The obligations associated with becoming a public company may strain our resources, result in more litigation and divert management’s attention from operating our business. |
| | ● | You will experience immediate and substantial dilution in the net tangible book value of Ordinary Shares purchased. |
| | ● | Substantial future sales of our Ordinary Shares or the anticipation of future sales of our Ordinary Shares in the public market could cause the price of Ordinary Shares to decline. |
| | ● | We do not intend to pay dividends for the foreseeable future. |
| | ● | If securities or industry analysts do not publish research or reports about our business, or if they publish a negative report regarding our Ordinary Shares, the price of our Ordinary Shares and trading volume could decline. |
| | ● | We may experience extreme stock price volatility unrelated to our actual or expected operating performance, financial condition or prospects, making it difficult for prospective investors to assess the rapidly changing value of our Ordinary Shares, and such volatility may subject us to securities litigation. |
| | ● | You may face difficulties in protecting your interests, and your ability to protect your rights through U.S. courts may be limited, because Everfront Holding is incorporated under British Virgin Islands law. |
| | ● | As a foreign private issuer, we are permitted to rely on certain home country rules in lieu of the corresponding corporate governance standards of the Nasdaq Listing Rules applicable to domestic U.S. issuers. This may afford less protection to holders of our shares. |
| | ● | If we cannot satisfy, or continue to satisfy, the initial listing requirements and other rules of the Nasdaq Capital Market, our securities may not be listed or may be delisted, which would negatively impact the price of our securities and your ability to sell them. |
| | ● | Because our business is conducted in New Taiwan dollars and the price of our Ordinary Shares is quoted in United States dollars, changes in currency conversion rates may affect the value of your investments. |
| | ● | We have broad discretion in the use of the net proceeds from this offering and may not use them effectively. |
| | ● | Our pre-initial public offering, or pre-IPO, shareholders will be able to sell their shares after completion of this offering subject to restrictions under Rule 144. |
| | ● | We could be deemed to be a passive foreign investment company (“PFIC”), for U.S. federal income tax purposes for any taxable year, which could result in adverse U.S. federal income tax consequences to U.S. holders of our Ordinary Shares. |
| | ● | We will be an emerging growth company within the meaning of the Securities Act upon the consummation of this offering and may take advantage of certain reduced reporting requirements. |
Implications of Being an “Emerging Growth Company”
As a company with less than $1.235 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. An “emerging growth company” may take advantage of reduced reporting requirements that are otherwise generally applicable to public companies. In particular, as an emerging growth company, we:
| | ● | may present only two years of audited financial statements and only two years of related Management’s Discussion and Analysis of Financial Condition and Results of Operations, or MD&A; |
| | | |
| | ● | are not required to provide a detailed narrative disclosure discussing our compensation principles, objectives and elements and analyzing how those elements fit with our principles and objectives, which is commonly referred to as “compensation discussion and analysis”; |
| | | |
| | ● | are not required to obtain an attestation and report from our independent registered accounting firm on its management’s assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act of 2002; |
| | | |
| | ● | are not required to obtain a non-binding advisory vote from our shareholders on executive compensation or golden parachute arrangements (commonly referred to as the “say-on-pay,” “say-on frequency” and “say-on-golden-parachute” votes); |
| | | |
| | ● | are exempt from certain executive compensation disclosure provisions requiring a pay-for-performance graph and chief executive officer, or CEO, pay ratio disclosure; |
| | | |
| | ● | are eligible to claim longer phase-in periods for the adoption of new or revised financial accounting standards under §107 of the JOBS Act; and |
| | | |
| | ● | will not be required to conduct an evaluation of our internal control over financial reporting for two years. |
We intend to take advantage of all of these reduced reporting requirements and exemptions, including the longer phase-in periods for the adoption of new or revised financial accounting standards under §107 of the JOBS Act. Our election to use the phase-in periods may make it difficult to compare our financial statements to those of non-emerging growth companies and other emerging growth companies that have opted out of the phase-in periods under §107 of the JOBS Act.
Under the JOBS Act, we may take advantage of the above-described reduced reporting requirements and exemptions for up to five years after our initial sale of common equity pursuant to a registration statement declared effective under the Securities Act of 1933, as amended, herein referred to as the Securities Act, or such earlier time that we no longer meet the definition of an emerging growth company.
We will remain an emerging growth company until the earliest of: (i) the last day of the first fiscal year in which our annual gross revenue exceeds $1.235 billion; (ii) the last day of the fiscal year during which the fifth anniversary of the date of this offering occurs; (iii) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), which would occur if the market value of Ordinary Shares that are held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter; or (iv) the date on which we have issued more than $1.00 billion in non-convertible debt securities during any three-year period.
Implications of Being a Foreign Private Issuer
Because we are incorporated in the British Virgin Islands and more than 50% of our outstanding voting securities are not directly or indirectly held by residents of the United States, we are a “foreign private issuer” under U.S. securities laws. This means that we will be subject to reporting obligations that, to some extent, are more lenient and less frequent than those of U.S. domestic reporting companies, including the following.
| | ● | We will not be required to provide as many Exchange Act reports or provide periodic and current reports as frequently, as a domestic public company. |
| | | |
| | ● | For interim reporting, we will not be required to file quarterly reports on Form 10-Q containing unaudited financial and other specific information or current reports on Form 8-K upon the occurrence of specified significant events; instead, SEC rules will only require that we comply with our home country requirements, which are less rigorous than the rules that apply to U.S. domestic public companies. |
| | | |
| | ● | We will be required to file an annual report on Form 20-F, rather than Form 10-K, and therefore will be permitted to provide less detailed disclosure on certain issues, such as executive compensation. |
| | | |
| | ● | We will be exempt from provisions of Regulation FD aimed at preventing issuers from making selective disclosures of material information. |
| | | |
| | ● | We will be exempt from compliance with the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act, including the requirement to file a proxy statement that complies with SEC rules and regulations. |
| | | |
| | ● | Section 16 of the Exchange Act, which requires insiders to file public reports of their share ownership and trading activities and establishes insider liability for profits realized from any “short-swing” trading transaction will not apply to us. |
We will cease to be a foreign private issuer, and lose the benefits of these rules, once 50% or more of our outstanding voting securities are held by U.S. residents and any of the following three conditions are true: (i) the majority of the members of our board of directors and senior management are U.S. citizens or residents; (ii) more than 50% of our assets are located in the United States; or (iii) our business is administered principally in the United States.
Both foreign private issuers and emerging growth companies are also exempt from certain more stringent executive compensation disclosure rules. Thus, even if we no longer qualify as an emerging growth company, but remain a foreign private issuer, we will continue to be exempt from the more stringent compensation disclosures required of companies that are neither an emerging growth company nor a foreign private issuer.
In addition, because we are a foreign private issuer, the Nasdaq Listing Rules permit us to follow certain home country rules rather than the corresponding corporate governance standards of Nasdaq. The standards applicable to us may be considerably different than the standards applied to domestic U.S. issuers, including requirements to:
| | ● | have a majority of our board consist of independent directors (although all of the members of the audit committee must be independent under the Exchange Act); |
| | | |
| | ● | have a compensation committee or a nominating and corporate governance committee consisting entirely of independent directors; |
| | | |
| | ● | have regularly scheduled executive sessions with only independent directors; or |
| | | |
| | ● | have executive sessions of solely independent directors each year. |
Implications of Being a Controlled Company
As of the date hereof (after giving effect to this offering), Mr. Ho-Ching Chen holds beneficial ownership of % of our outstanding Ordinary Shares pursuant to agreements in which certain of our shareholders have given Mr. Chen voting control over their shares, including in connection with the election of our directors. For so long as these agreements provide Mr. Chen with more than 50% of the voting power for the election of our directors, we will be a “controlled company” under Nasdaq rules. As a controlled company, we will be permitted not to comply with certain of Nasdaq’s corporate governance requirements, including exemptions from the following rules:
| | ● | that a majority of our board of directors must be independent directors; |
| | ● | that we have a Compensation Committee that will determine or recommend the compensation of our executive officers and that it be composed solely of independent directors; and |
| | ● | that our director nominees must be selected or recommended solely by independent directors. |
We currently intend to rely on the controlled company exemptions in respect of our Compensation Committee and Nominating and Corporate Governance Committee, each of which will include directors that are not independent under Nasdaq Listing Rules. Further, although we do not intend to rely on the exemption in respect of a majority independent board, we will have the right to use this exemption in the future. As a result, you may not have the same protections afforded to shareholders of companies that are subject to, or voluntarily follow, these Nasdaq corporate governance requirements.
Following this offering, as long as Mr. Ho-Ching Chen maintains control over a majority of the voting power of our outstanding Ordinary Shares with respect to the election of our directors, we may utilize certain of these exemptions. Accordingly, you will not have the same protections afforded to shareholders of companies that are subject to all of the Nasdaq corporate governance requirements of Nasdaq. If we cease to be a “controlled company” and our common stock continues to be listed on Nasdaq, we will be required to comply with these provisions within the applicable transition periods.
The Offering
| Ordinary Shares in this offering | | Ordinary Shares |
| | | |
| Price per Ordinary Share | | Between $ and $ per Ordinary Share |
| | | |
| Ordinary Shares outstanding prior to completion of this offering | | 52,237,745 Ordinary Shares |
| | | |
| Ordinary Shares outstanding immediately after this offering | | Ordinary Shares |
| | | |
| Transfer Agent | | |
| | | |
| Nasdaq Capital Market symbol | | We have applied to have our Ordinary Shares listed on the Nasdaq Capital Market under the symbol “EFB”. This offering is contingent upon the final approval from Nasdaq for the listing of our Ordinary Shares on the Nasdaq Capital Market, and we will not proceed to consummate this offering if Nasdaq denies our listing application. No assurance can be given that our application will be approved. |
| | | |
| Use of proceeds | | We intend to use the proceeds from this offering for (i) Glioblastoma clinical trial expenses – Cerebraca™ Wafer Phase I/IIa; (ii) Glioblastoma clinical trial expenses – Cerebraca™ Wafer Phase IIb/III; (iii) Oncology clinical trial expenses – HK-001 / EF-031; (iv) Pancreatic cancer clinical trial expenses – EF-009 Wafer; (v) general research and development expenses; (vi) patent maintenance and application expenses; and (vii) working capital and other general corporate expenses. See “Use of Proceeds.” |
| | | |
| Lock-up | | We and all of our directors and officers and shareholders that hold 5% or more of our outstanding Ordinary Shares have agreed with the representatives of the underwriters, subject to certain exceptions, not to sell, transfer, or dispose of, directly or indirectly, any Ordinary Shares or securities convertible into or exercisable or exchangeable for Ordinary Shares for a period of six months after the closing of his offering. See “Shares Eligible for Future Sale” and “Underwriting.” |
| | | |
| Risk factors | | The Ordinary Shares offered hereby involve a high degree of risk. You should read “Risk Factors” for a discussion of factors to consider before deciding to invest in our Ordinary Shares. |
Here and elsewhere in this prospectus, we have based the number of Ordinary Shares outstanding after this offering on (1) Ordinary Shares outstanding on , 2025 and (2) our issuance and sale in this offering of the number of Ordinary Shares set forth on the cover page of this prospectus. These figures do not reflect outstanding rights to purchase Ordinary Shares under our employee benefit plan or the warrants to purchase our Ordinary Shares that we will issue to the representatives of the underwriters on the closing of this offering. See “Underwriting.”
Summary Consolidated Financial Data
We have derived the following summary consolidated financial data as of June 30, 2024 and for the six months ended June 30, 2024 and 2023 from our unaudited consolidated financial statements, which you can find elsewhere in this prospectus. We have derived the following summary consolidated financial data as of and for the years ended December 31, 2023 and 2022 from our audited consolidated financial statements, which you can find elsewhere in this prospectus. We prepared these financial statements in accordance with generally accepted accounting principles of the United States of America, or U.S. GAAP. Our historical results are not necessarily indicative of the results that may occur in the future. You should read the following summary consolidated financial data in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements, including the notes thereto, which you can find elsewhere in this prospectus.
| | | For the Six Months Ended June 30, | | | For the Years Ended December 31, | |
| | | 2024 | | | 2023 | | | 2023 | | | 2022 | |
| | | US$ | | | US$ | | | US$ | | | US$ | |
| | | (Unaudited) | | | | |
| Revenue | | $ | - | | | $ | - | | | $ | - | | | $ | 79,908 | |
| Operating costs | | | - | | | | - | | | | - | | | | - | |
| Gross profit | | | - | | | | - | | | | - | | | | 79,908 | |
| Total operating expenses | | | 792,673 | | | | 984,599 | | | | 2,385,870 | | | | 1,783,902 | |
| Loss from operations | | | (792,673 | ) | | | (984,599 | ) | | | (2,385,870 | ) | | | (1,703,994 | ) |
| Total non-operating income, net | | | 4,961 | | | | 36,582 | | | | 92,673 | | | | 209,400 | |
| Loss before income tax | | | (787,712 | ) | | | (948,017 | ) | | | (2,293,197 | ) | | | (1,494,594 | ) |
| Income tax expense | | | - | | | | - | | | | - | | | | - | |
| Net loss | | $ | (787,712 | ) | | $ | (948,017 | ) | | $ | (2,293,197 | ) | | $ | (1,494,594 | ) |
| Basic and diluted earnings per share | | $ | (0.02 | ) | | $ | (0.02 | ) | | $ | (0.04 | ) | | $ | (0.03 | ) |
| | | As of June 30, | | | As of December 31, | |
| | | 2024 | | | 2023 | | | 2022 | |
| | | US$ | | | US$ | | | US$ | |
| Summary Consolidated Balance Sheet Data | | | | | | | | | | | | |
| Cash and cash equivalents | | $ | 432,932 | | | $ | 1,220,559 | | | $ | 548,138 | |
| Total current assets | | | 867,975 | | | | 1,690,317 | | | | 992,392 | |
| Total assets | | | 1,198,579 | | | | 2,065,005 | | | | 1,353,450 | |
| Total current liabilities | | | 2,645,751 | | | | 2,679,458 | | | | 174,696 | |
| Total liabilities | | | 2,652,479 | | | | 2,689,622 | | | | 175,893 | |
| Total stockholders’ (deficit) equity | | $ | (1,453,772 | ) | | $ | (624,638 | ) | | $ | 1,177,236 | |
RISK FACTORS
An investment in Ordinary Shares involves a high degree of risk. Before deciding whether to invest in Ordinary Shares, you should consider carefully the risks described below, together with all of the other information set forth in this prospectus, including the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operation” and our consolidated financial statements and related notes. If any of these risks actually occurs, our business, financial condition, results of operations or cash flow could be materially and adversely affected, which could cause the trading price of Ordinary Shares to decline, resulting in a loss of all or part of your investment. The risks described below are not the only ones that we face. Additional risks not presently known to us or that we currently deem immaterial may also affect our business. You should only consider investing in Ordinary Shares if you can bear the risk of loss of your entire investment.
Risks Related to Our Financial Position and Need for Additional Capital
We are a cancer drug development company with a limited operating history.
We are a cancer drug development company with a limited operating history. Biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. Since our inception, we have devoted substantially all of our efforts to organizing and staffing our company, acquiring and developing intellectual property, business planning, raising capital, conducting discovery, research and development and clinical trial activities, and providing general and administrative support for these operations. We have no products approved for commercial sale and therefore have never generated any revenue from product sales, and we do not expect to in the foreseeable future. We expect to continue to incur significant expenses and operating losses over the next several years and for the foreseeable future. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on cash and cash equivalent holdings, our shareholders’ equity and working capital. If we were to expend our cash resources more quickly than we anticipate as we advance into and through the drug development and approval process, our cash runway may be shorter than the target we may disclose from time to time.
We expect our operating results to fluctuate significantly in the future as our business advances.
The amount of our future losses is uncertain and our quarterly and annual operating results may fluctuate significantly or may fall below the expectations of investors or securities analysts, which may cause our share price to fluctuate or decline. Our quarterly and annual operating results may fluctuate significantly in the future due to a variety of factors, many of which are outside of our control and may be difficult to predict, including the following:
| ● | the timing and success or failure of on-going and future clinical trials for our product candidates or competing product candidates, or any other change in the competitive landscape of our industry, including consolidation among our competitors or partners; |
| ● | our ability to obtain INDs for our pipeline product candidates, successfully open clinical trial sites and recruit and retain subjects for clinical trials, and any delays caused by difficulties in such efforts; |
| ● | our ability to obtain regulatory approval for our product candidates, and the timing and scope of any such approvals we may receive; |
| ● | the timing and cost of, and level of investment in, research and development activities relating to our product candidates and any future product candidates and research-stage programs, which may change from time to time; |
| ● | the cost of manufacturing our product candidates and products, should they receive regulatory approval, which may vary depending on FDA, TFDA and other comparable foreign regulatory requirements, the quantity of production and the terms of our agreements with manufacturers; |
| ● | expenditures that we will or may incur to develop additional product candidates; |
| ● | the level of demand for our product candidates should they receive regulatory approval, which may vary significantly; |
| ● | the risk/benefit profile, cost and reimbursement policies with respect to our product candidates, if approved, and existing and potential future cancer drugs that compete with our product candidates; |
| ● | future accounting pronouncements or changes in our accounting policies. |
As a result, comparing our operating results on a period-to-period basis may not be meaningful. This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any forecasts we may provide to the market, or if the forecasts we provide to the market are below the expectations of analysts or investors, the price of our Ordinary Shares could decline substantially. Such a share price decline could occur even when we have met any previously publicly stated guidance we may provide.
We have no products approved for commercial sale and have not generated any revenue from product sales.
Our ability to become profitable depends upon our ability to generate revenue. To date, we have not generated any revenue from product sales, and we do not expect to generate any revenue from the sale of products in the near future. Our ability to generate revenue depends on a number of factors, including, but not limited to, our ability to:
| ● | successfully complete our planned preclinical studies for our EF-031 oral formulation; |
| ● | timely file INDs for our programs, and obtain clearance of these INDs to allow for commencement of such future clinical trials; |
| ● | successfully complete our Cerebraca™ Wafer and EF-009 Wafer clinical trials and any future clinical trials; |
| ● | initiate and successfully complete all safety and efficacy studies required to obtain U.S. and foreign regulatory approval for our product candidates; |
| ● | make and maintain arrangements with third-party manufacturers for clinical supply and commercial manufacturing; |
| ● | obtain and maintain patent and trade secret protection or regulatory exclusivity for our product candidates; |
| ● | launch commercial sales of our products, if and when approved, whether alone or in collaboration with others; |
| ● | obtain and maintain acceptance of our products, if and when approved, by patients, the medical community and third-party payers; |
| ● | position our products to effectively compete with other cancer drugs; |
| ● | obtain and maintain healthcare insurance coverage and adequate reimbursement for our products, if and when approved; |
| ● | enforce and defend intellectual property rights and claims; and |
| ● | maintain a continued acceptable safety profile of our products following approval. |
If we fail to raise additional funds, our ability to execute our business and development strategies may be affected.
We have limited funds, and only a small amount of service income and government subsidy income partially supports our operations. The majority of our working capital comes from capital invested by shareholders. From time to time, we may seek additional funding to provide the funds necessary to expand our R&D program and working capital. We cannot predict with 100% certainty the timing or amount of any such capital requirement.
If we do not raise sufficient funds to fund ongoing R&D activities, we may not be able to execute the intended business development plan. Even if we receive financing for near-term operations and product development, we may need additional funding in the short term. In addition, additional capital may not be available in sufficient amounts or on reasonable terms, and our ability to raise additional capital may be adversely affected by the potential deterioration in global economic conditions and subsequent disruptions. If we are unable to raise capital when required, our business, financial condition and results of operations will be materially and adversely affected and we may be forced to reduce or cease our operations.
Raising additional capital may cause dilution to our shareholders, restrict our operations, or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through one or a combination of private and public equity offerings, debt financings, collaborations, strategic alliances, and licensing arrangements. We do not have any committed external source of funds. The terms of any financing may adversely affect the holdings or the rights of our shareholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our Ordinary Shares to decline. To the extent that we raise additional capital through the sale of Ordinary Shares or securities convertible or exchangeable into our Ordinary Shares, the ownership interests of our existing shareholders will be diluted, and the terms of those securities may include liquidation or other preferences that may materially adversely affect your rights as a holder of our Ordinary Shares. Debt financing, if available, would increase our fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, acquiring, selling, or licensing intellectual property rights, and making capital expenditures, declaring dividends, repurchasing our Ordinary Shares, or other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to meet certain milestones in connection with debt financing and the failure to achieve such milestones by certain dates may force us to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us which could have a material adverse effect on our business, operating results, and prospects.
We also could be required to seek funds through arrangements with additional collaborators or otherwise at an earlier stage than otherwise would be desirable. If we raise funds through additional collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our intellectual property, future revenue streams, research programs or product candidates, grant licenses on terms that may not be favorable to us or grant rights to develop and market our product candidates that we would otherwise prefer to develop and market ourselves, any of which may have a material adverse effect on our business, operating results and prospects.
Risks Related to Clinical Development of Our Product Candidates
We have never successfully completed clinical development for any of our product candidates, and we may be unable to do so. Certain of our cancer drugs are still in preclinical development and may never advance to clinical development.
We have not yet successfully completed clinical development for any of our product candidates. Completing clinical development is a costly, complex endeavor that requires that we, among other things, enroll and complete clinical trials (including large-scale, pivotal or Phase IIb/III clinical trials), obtain regulatory approvals, manufacture a commercial scale product or arrange for a third party to do so on our behalf, and conduct sales and marketing activities necessary for successful commercialization. We received FDA and TFDA clearance of one IND application for each of Cerebraca™ Wafer and EF-009 Wafer. We may not be able to file future INDs for any of our other product candidates on the timelines we expect, if at all. Further, timelines for developing and filing INDs are subject to significant uncertainties and projected timelines can be improved upon or delayed. Moreover, we cannot be sure that submission of an IND will result in either the FDA or TFDA allowing clinical trials to begin, or that, once begun, issues will not arise that require us to suspend or terminate any clinical trials. Any guidance we receive from the FDA, TFDA or other regulatory authorities is subject to change. These regulatory authorities could change their position, including on the acceptability of our trial designs or the clinical endpoints selected, which may require us to complete additional clinical trials or result in the imposition of stricter approval conditions than we currently expect. Successful completion of our clinical trials is a prerequisite to submitting a new drug application (“NDA”) to the FDA, a marketing authorization application (“MAA”) to the European Medicines Agency (“EMA”) or other marketing applications to regulatory authorities in other jurisdictions, for each product candidate and, consequently, the regulatory approval of each product candidate. While the INDs for Cerebraca™ Wafer and EF-009 Wafer were cleared by the FDA and TFDA, it is possible that an adequate number of patients may not be enrolled on a timely basis, or at all, and the studies may not be completed (and preliminary, initial or final trial results may not be available) on time. Similarly, future clinical trials may not begin on time or be completed on schedule, if at all.
If we are required to conduct additional preclinical studies or clinical trials of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our product candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety and/or efficacy concerns, we may, among other things:
| ● | be delayed in obtaining regulatory approval for our product candidates; |
| ● | not obtain regulatory approval at all; |
| ● | obtain regulatory approval for indications or patient populations that are not as broad as intended or desired; |
| ● | be subject to post-marketing testing requirements; or |
| ● | have the product removed from the market after obtaining regulatory approval. |
Clinical product development involves a lengthy and expensive process, with an uncertain outcome.
Our preclinical studies, our Cerebraca™ Wafer and EF-009 Wafer clinical trials and future clinical trials may not be successful. It is impossible to predict when or if any of our product candidates will prove effective and safe in humans or will receive regulatory approval. Before obtaining regulatory approval from regulatory authorities for the sale of any product candidate, we must complete preclinical studies and then conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Clinical testing is expensive, difficult to design and implement, can take many years to complete and outcomes are uncertain. A failure of one or more clinical trials can occur at any stage of testing. The outcome of preclinical development testing and early clinical trials may not be predictive of the success of later clinical trials, and interim or preliminary results of a clinical trial do not necessarily predict final results (or be indicative of safety and efficacy if commercialized and used in a broader population). Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain regulatory approval of their product candidates.
Interim, top-line, and initial data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to confirmation, audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose interim, top-line or initial data from our current and future clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data, including audit and verification procedures, and as results from additional clinical trial participants become available and trial participants spend additional time on therapy. We may also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to evaluate all data fully and carefully. As a result, initial, interim and top-line data should be viewed with caution until the final data are available. In addition, we may report interim analyses of only certain endpoints rather than all endpoints. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Adverse differences between initial or interim data and final data could significantly harm our business prospects and may cause the price of our Ordinary Shares to fluctuate or decline.
Further, regulatory agencies and others may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could adversely impact the potential of a particular program, the likelihood of obtaining regulatory approval of the particular product candidate, the scope of product label, and commercialization of any approved product. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is derived from information that is typically extensive, and you or others may not agree with what we determine is material or otherwise appropriate information to include in our disclosure.
If the initial, interim or top-line data that we report differ from final or actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be significantly impaired, which could materially harm our business, operating results, prospects or financial condition.
We may incur additional costs or experience delays in initiating or completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
We may experience delays in initiating or completing our preclinical studies or clinical trials, including as a result of delays in obtaining, or failure to obtain, the FDA’s and/or TFDA’s clearance to initiate clinical trials under future INDs. Additionally, we cannot be certain that preclinical studies or clinical trials for our product candidates will not require redesign, will enroll an adequate number of subjects on time, or will be completed on schedule, if at all. We may experience numerous unforeseen events during, or as a result of, preclinical studies and clinical trials that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates, including the following:
| ● | we may receive feedback from regulatory authorities that require us to modify the design or implementation of our preclinical studies or clinical trials or to delay or terminate a clinical trial; |
| ● | regulators or institutional review boards (“IRB”), or ethics committees may delay or may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
| ● | we may experience delays in reaching, or fail to reach, agreement on acceptable terms with prospective trial sites and prospective Contract Research Organizations, or CROs, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| ● | preclinical studies or clinical trials of our product candidates may fail to show safety or efficacy or otherwise produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional preclinical studies or clinical trials, or we may decide to abandon product research or development programs; |
| ● | preclinical studies or clinical trials of our product candidates may not produce differentiated or clinically significant results across tumor types or indications; |
| ● | the number of patients required for clinical trials of our product candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials or fail to return for post-treatment follow-up at a higher rate than we anticipate; |
| ● | our third-party contractors may fail to comply with regulatory requirements, fail to maintain adequate quality controls, be unable to provide us with sufficient product supply to conduct or complete preclinical studies or clinical trials, fail to meet their contractual obligations to us in a timely manner, or at all; |
| ● | our clinical trial sites or investigators may deviate from the clinical trial protocol or drop out of the trial, which may require that we add new clinical trial sites or investigators in order to ensure our trials generate statistically significant results; |
| ● | we may elect to, or regulators or IRBs or ethics committees may require us or our investigators to, suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants in our clinical trials are being exposed to unacceptable health risks; |
| ● | the cost of clinical trials of our product candidates may be greater than we anticipate; |
| ● | the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; |
| ● | our product candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators or IRBs or ethics committees to suspend or terminate the trials, or reports may arise from preclinical or clinical testing of other cancer drugs that raise safety or efficacy concerns about our product candidates; |
| ● | regulators may revise the requirements for approving our product candidates, or such requirements may not be as we anticipate; and |
| ● | regulatory developments with respect to our competitors’ products, including any developments, litigation or public concern about the safety of such products. |
We could encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions at which such trials are being conducted or by the FDA, TFDA or other regulatory authorities. Such authorities may impose such a suspension or termination or clinical hold due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements for our clinical protocols, adverse findings upon an inspection of the clinical trial operations or trial site by the FDA, TFDA or other regulatory authorities, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates. Further, the FDA or TFDA may disagree with our clinical trial design or our interpretation of data from clinical trials or may change the requirements for approval even after each has reviewed and commented on the design for our clinical trials.
Moreover, principal investigators for our current or future clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA, TFDA or comparable foreign regulatory authorities. The FDA, TFDA or comparable foreign regulatory authority may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected the interpretation of the study. The FDA, TFDA or comparable foreign regulatory authority may therefore question the integrity of the data generated at the applicable clinical trial site, and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA, TFDA or comparable foreign regulatory authority and may ultimately lead to the denial of regulatory approval of one or more of our product candidates.
Our product development costs will also increase if we experience delays in testing or regulatory approvals. We do not know whether any of our current or future clinical trials will begin as planned, or whether any of our current or future clinical trials will need to be restructured or will be completed on schedule, if at all. Significant preclinical study or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, which would impair our ability to successfully commercialize our product candidates and may significantly harm our business, operating results, financial condition and prospects.
If we experience delays or difficulties in the enrollment of patients in clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to continue or initiate clinical trials for our product candidates if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA, TFDA or comparable foreign regulatory authorities, or as needed to provide appropriate statistical power for a given trial. In particular, because some of the indications we are pursuing are orphan indications that have small populations, our ability to enroll eligible patients may be limited or may result in slower enrollment than we anticipate.
In addition, some of our competitors have ongoing clinical trials for product candidates that treat the same indications as do our product candidates, and patients who would otherwise be eligible for our clinical trials may choose instead to enroll in clinical trials of our competitors’ product candidates.
In addition to the competitive clinical trial environment, the eligibility criteria of our clinical trials will further limit the pool of available study participants as we will require that patients have specific characteristics that we can measure to assure their cancer is either severe enough or not too advanced to include them in a study. The process of finding patients may prove costly. We also may not be able to identify, recruit or enroll a sufficient number of patients to complete our clinical studies because of the perceived risks and benefits of the product candidates under study, the availability and efficacy of competing therapies and clinical trials, the proximity and availability of clinical trial sites for prospective patients, and the patient referral practices of physicians. If patients are unwilling to participate in our studies for any reason, the timeline for recruiting patients, conducting studies, reporting initial and final trial results and obtaining regulatory approval of potential products may be delayed.
Further, if our patient recruitment for our clinical trials proceeds more slowly than we expect, this could compromise our ability to seek designations under applicable FDA expedited review and development programs, including Breakthrough Therapy Designation and Fast Track Designation (to the extent these are available to us), or otherwise seek to accelerate clinical development and regulatory timelines. Patient enrollment may be affected by other factors, including:
| ● | the severity of the disease under investigation; |
| ● | the efforts to obtain and maintain patient consents and facilitate timely enrollment in clinical trials; |
| ● | the ability to monitor patients adequately during and after treatment; |
| ● | the ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| ● | reporting of the initial results of any of our clinical trials; and |
| ● | factors we may not be able to control, including the impacts of events such as global pandemics, which may limit patients, principal investigators or staff or clinical site availability. |
We may not be able to replicate the results from our earlier preclinical and clinical studies in later preclinical studies and clinical trials, and this could prevent us from successfully developing, obtaining regulatory approval for and commercializing our product candidates.
You should not view results from our earlier preclinical studies of our programs and product candidates to be predictive of the results of subsequent preclinical studies and clinical trials. Any results from the earlier preclinical and clinical studies of our programs or our product candidates may not necessarily be predictive of the results from Later preclinical studies and clinical trials often fail to replicate or confirm the results of earlier preclinical and clinical studies for a variety of reasons, including that later studies may be more rigorous and involve a larger, more diverse patient population.
Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in early-stage development, and we cannot be certain that we will not face similar setbacks. These setbacks have been caused by, among other things, preclinical and other nonclinical findings made while clinical trials were underway, or safety, pharmacokinetic or efficacy observations made in preclinical studies and clinical trials, including previously unreported adverse events. Moreover, preclinical, nonclinical and clinical data are often susceptible to varying interpretations and analyses and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain regulatory approval.
Our growth depends on our ability to successfully develop, acquire or license new drugs.
Our current development is supported by sustained investment of time, resources and capital to identify and develop new drugs suitable for the market. If we are unable to develop new products ourselves or license new products from other parties, our ability to generate revenue and develop market share will be adversely affected. In addition, we may not recoup our investment in new drug development because our projects may be interrupted, unsuccessful, not as profitable as originally envisaged, or we may not receive the necessary capital investment. Similarly, there can be no assurance that we will be able to successfully obtain needed rights from third parties on an economically viable basis.
Clinical trials may be subject to liability claims, which may delay or even cause our R&D plans to fail, consume our resources, cause us to incur significant liability and limit the development of our products.
As we conduct clinical trials of our products, we face the inherent risk of product liability claims resulting from those trials. If any of the products we develop are suspected of causing harm or are found to be unsuitable during clinical trials, we may be liable for damages. Any product liability claims may include allegations of manufacturing defects, design defects, failure to warn of the inherent dangers of the product, negligence, strict liability, or violations of specifications. If we are unable to successfully defend a product liability claim, we may be liable for substantial damages and/or required to cease proceeding with the trial and the development of our product. Regardless of the circumstances or final outcome, liability claims can result in:
| ● | significant negative attention that damages our reputation and the reputation of our products; |
| ● | participants quitting or declining to participate in our clinical trials; |
| ● | significant costs of defending litigation; |
| ● | substantial monetary compensation to plaintiffs and other trial participants; and |
| ● | difficulty commercializing our developed products. |
We currently have insurance policies to cover liability under clinical trials, but no general liability insurance. Even if we have general liability insurance in the future, it may not fully cover any potential liability we may incur. The cost of any product liability litigation or other proceedings, even in our favor, can be substantial.
We conduct clinical trials on some of our product candidates at locations outside the United States, approval by the FDA is critical to our development, and the FDA may not accept data from trials conducted at certain locations.
We have conducted one or more clinical trials outside the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of such data is subject to certain conditions imposed by the FDA. For example, clinical trials must be carefully designed, conducted, and executed by qualified researchers in accordance with ethical principles. The trial population must also be fully representative of the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in a manner deemed clinically significant by the FDA. In addition, while these clinical trials are subject to applicable local laws, FDA’s acceptance of any trial data will depend on its determination that the trials also comply with all applicable U.S. laws and regulations. There is no guarantee that the FDA will accept data from trials conducted outside the United States. If the FDA does not accept data from a clinical trial that we decide to conduct outside the United States, it may result in the need for additional trials, which would be expensive and time-consuming, and could delay or permanently halt our development of product candidates.
In addition, the conduct of international clinical trials in the future may have a significant impact on us. The inherent risks of conducting international clinical trials include:
| ● | foreign regulatory requirements that could restrict or limit our ability to conduct our clinical trials; |
| ● | administrative burdens of conducting clinical trials under multiple foreign regulatory schema; |
| ● | foreign exchange fluctuations; and |
| ● | diminished protection of intellectual property in some countries. |
If clinical trials of our product candidates fail to demonstrate safety and efficacy, we may incur additional costs or delays, or ultimately fail to complete the development and commercialization of our product candidates.
Clinical trials are expensive, complex to design and conduct, can take years to complete, and have highly uncertain outcomes. Any failure to successfully complete preclinical and clinical development could result in additional costs for us and compromise our product commercialization milestones and ability to generate revenue from our products. In addition, if (a) we need to consider additional clinical trials or other testing of a product candidate, (b) we are unable to successfully complete clinical trials or other testing of a product candidate, (c) the results of any trials or tests are unfavorable, uncertain, or only moderately favorable, or (d) there is an unacceptable safety issue with a product candidate, in addition to incurring additional costs, we may:
| ● | suffer delays in obtaining marketing approval of candidate products; |
| ● | find there is no way to obtain marketing authorization; |
| ● | see results that do not meet the approval requirements for the broad indications or broad patient populations we had expected; |
| ● | obtain approval for labels that contain significant use or distribution restrictions or significant safety warnings, including “boxed” warnings; |
| ● | be subject to additional post-market testing or other requirements; or |
| ● | obtain marketing approval, and afterwards find that the product must be removed from the market. |
Risks Related to Obtaining Regulatory Approval for Our Product Candidates
We have no history of obtaining regulatory approval or commercialization of any new drug candidates.
Due to our limited operating history, we have never received regulatory approval or achieved commercialization for any new drug candidates. Regulatory authorities such as the FDA or the TFDA may reject New Drug Applications (or “NDAs”) for substantive review of our planned drugs or conclude after reviewing our data that our applications are insufficient to obtain regulatory approval for new drug candidates. Although our R&D units have experience participating in New Drug Application (or “NDA”) filings, the process and timeline for obtaining NDA approval can vary significantly. If the FDA does not accept or approve our planned product candidate NDA, additional clinical, preclinical or manufacturing validation studies may be required from us, which can be costly. Depending on the FDA-required research, approval of any NDA or application we submit may be significantly delayed, possibly up to several years, or may require us to spend more resources than we anticipate. Any delay in obtaining or the inability to obtain regulatory approval for any of our drug candidates will prevent us from licensing such products. If other studies are conducted and completed, the FDA may also consider them inadequate. If any of these results occur, we may be forced to abandon planned NDAs for such drug candidates, which would have a material adverse effect on our business and may result in us ceasing operations. We face similar regulatory risks in foreign jurisdictions.
Risks Related to Commercialization of Our Product Candidates
We face substantial competition, which may result in others discovering, developing or commercializing products before, or more successfully than, we do.
The development and commercialization of new products in the biopharmaceutical and related industries is highly competitive. We compete in the segments of the pharmaceutical, biotechnology, and other related markets that address structural biology-guided chemistry-based drug design to develop therapies in the fields of cancer and genetic diseases. There are other companies focusing on precision oncology to develop therapies in the fields of cancer and other diseases.
We also compete more broadly across the market for cost-effective and reimbursable cancer treatments. Some of these competitive products and therapies are based on scientific approaches that are the same as or similar to our approach, and others are based on entirely different approaches. These companies include divisions of large pharmaceutical companies and biotechnology companies of various sizes. We face competition with respect to our current product candidates, and will face competition with respect to any product candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Any product candidates that we successfully develop and commercialize will compete with currently approved therapies and new therapies that may become available in the future from segments of the pharmaceutical, biotechnology and other related markets. Key product features that would affect our ability to effectively compete with other therapeutics include the efficacy, safety and convenience of our products. We believe the principal competitive factors to our business include, among other things, our ability to identify biomarkers, the ability to successfully transition research programs into clinical development, the ability to raise capital, and the scalability of our platform, pipeline, and business.
Many of the companies that we compete against or which we may compete against in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and marketing, promoting and selling approved products than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. If these or other barriers to entry do not remain in place, other companies may be able to more directly or effectively compete with us.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we or our collaborators may develop. Our competitors also may obtain FDA, TFDA or other regulatory approval for their products sooner than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we or our collaborators are able to enter the market. The key competitive factors affecting the success of all of our product candidates, if approved, are likely to be their efficacy, safety, convenience, price, level of generic competition and novel competition that utilize the same mechanism of action and availability of reimbursement from government and other third-party payers.
If our current product candidates or any future product candidates do not achieve broad market acceptance, the revenue that we generate from their sales may be limited, and we may never become profitable.
We have never commercialized a product candidate for any indication. Even if our current product candidates and any future product candidates are approved by the appropriate regulatory authorities for marketing and sale, they may not gain acceptance among physicians, patients, third-party payers, and others in the medical community. If any product candidates for which we obtain regulatory approval do not gain an adequate level of market acceptance, we may not generate significant revenue and may not become profitable or may be significantly delayed in achieving profitability. Market acceptance of our current product candidates and any future product candidates by the medical community, patients and third-party payers will depend on a number of factors, some of which are beyond our control. For example, physicians are often reluctant to switch their patients, and patients may be reluctant to switch, from existing therapies even when new and potentially more effective or safer treatments enter the market. If public perception is influenced by claims that the use of certain precision oncology product candidates or immunotherapies and targeted therapies is unsafe, whether related to our or our competitors’ products, our potential future products may not be accepted by the general public or the medical community. Future adverse events in precision oncology, immuno-oncology or the biopharmaceutical industry could also result in greater governmental regulation, stricter labeling requirements and potential regulatory delays in the testing or approvals of our product candidates.
Efforts to educate the medical community and third-party payers on the benefits of our current product candidates and any future product candidates may require significant resources and may not be successful. If our current product candidates or any future product candidates are approved but do not achieve an adequate level of market acceptance, which will depend on a number of factors, we could be prevented from or significantly delayed in achieving profitability.
If side effects associated with our current or future products are discovered prior to marketing and sale, we may be required to withdraw these products from the market, conduct lengthy additional clinical trials or change the labeling of our products, any of which could adversely affect our growth.
We are currently developing the following drugs: Cerebraca™ Wafer (Glioblastoma), EF-009 Wafer (Pancreatic cancer), EF-031 (Solid tumor), HK-001 (Amyotrophic lateral sclerosis), TSCA-001 (Spinocerebellar ataxia), EF-005 (Alzheimer’s disease), EF-002 (Liver cirrhosis), EF-011 (Pulmonary fibrosis), and EF-012 (Immunotherapy). The use of certain medications can result in serious adverse events (SAEs). For instance, the administration of Cerebraca™ Wafer has been associated with recorded treatment emergent AEs such as fever, abnormal wound healing, convulsions, and headache.
After approval, if received, the occurrence of any AE would harm our future sales of these drugs and significantly increase the costs and expenses of marketing these drugs, which in turn could result in a decline in our revenue and net income. In addition, the reputation and sales of our future drugs may be adversely affected due to the serious side effects found.
We may be subject to product liability claims in the future, which may consume our resources, expose us to significant liability and limit the commercialization of any products we may develop.
If our products cause adverse side effects, we face the inherent business risk of product liability claims. Side effects or marketing or manufacturing issues related to any of our products in development may result in product liability claims or negative publicity. These risks will exist in products in clinical development and in products that receive regulatory approval for commercial sale in the future. In addition, although we have historically not encountered any issues related to product user claims because we do not yet sell our products, we do not currently have product liability insurance and cannot guarantee that we will be able to obtain commercially viable product liability insurance in the future.
As a result of selling any product we develop, there is an inherent risk of product liability claims. For example, we may be liable for damages if any of the products we develop are suspected of causing injury or are found to be unsuitable during manufacturing, marketing, or sales. Any such product liability claims may include allegations of manufacturing defects, design defects, failure to warn of the inherent dangers of the product, negligence, strict liability, or breach of regulations. Claims under consumer protection laws may also be brought. If we are unable to successfully defend against a product liability claim, we may be materially liable or required to limit the commercialization of our product candidates. Regardless of the circumstances or final outcome, liability claims can result in:
| ● | Reduced demand for our product candidates or products we may develop; |
| ● | Significant negative attention that damages our reputation; |
| ● | The resulting significant costs of litigation defense; |
| ● | Substantial monetary compensation to patients; |
| ● | Loss of income; and |
| ● | Reducing our resources and hindering the progress of our business. |
If we start selling any candidate products that are approved for marketing, we will need to increase our insurance coverage. In addition, insurance coverage is becoming more expensive. If we are unable to obtain or maintain adequate insurance coverage at an acceptable cost, or otherwise prevent potential product liability claims, this could prevent or inhibit the development and commercial production and sale of our product candidates, which could adversely affect our business, financial condition, results of operations and prospects.
We may not commercialize, market, promote or sell any product candidate in any jurisdiction without marketing approval from each country’s regulatory authorities, such as the FDA. We may never receive such approval, and we must complete extensive preclinical development and clinical trials to demonstrate the safety and efficacy of our product candidates in humans before obtaining these approvals.
Even if our product candidates are approved for marketing, they may not be acceptable to physicians, patients, third-party payers and others in the medical community, and the market opportunity for our product candidates may be smaller than we estimate to achieve the market size required for commercial success.
We have never completed the FDA approval and commercialization of new drug development processes, and even if our products receive marketing and sales approval from the relevant regulatory authorities, they may not receive adequate market acceptance from physicians, patients, third-party payers, and others in the medical community. For example, doctors are often reluctant to switch patients from existing therapies, even if new drugs may be more effective or convenient treatments enter the market. In addition, patients are often adapted to the treatment they are currently taking and do not want to switch unless their doctor recommends a replacement medication or needs to switch treatment due to lack of reimbursement for an existing treatment.
The potential market opportunity for our products is difficult to estimate accurately. Our estimates of potential market opportunity are based on a number of assumptions, including industry knowledge and publications, third-party research reports, and other surveys. While we believe that our internal assumptions are reasonable, these assumptions involve important judgments of our R&D team and are inherently uncertain, and the reasonableness of these assumptions has not been evaluated by independent sources. If any assumptions prove to be inaccurate, the actual market for our products may be smaller than our estimate of the potential market opportunity.
Risks Related to Our Intellectual Property
Obtaining and enforcing pharmaceutical patents involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our business, financial position, operations and prospects, and interrupt our research activities.
The patent positions of pharmaceutical companies and research institutions can be highly uncertain and involve complex legal and factual questions. The interpretation and breadth of claims allowed in some patents covering pharmaceutical compositions may be uncertain and difficult to determine and are often affected materially by the facts and circumstances that pertain to the patented compositions and the related patent claims. The standards of the U.S. Patent and Trademark Office, or USPTO, are sometimes uncertain and could change in the future. The standards are also, in some instances subject to interpretation by examiners at the USPTO. Consequently, the issuance and scope of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to re-examination proceedings, post-grant review and/or inter parties review in the USPTO. Foreign patents may be subject to opposition or comparable proceedings in the corresponding foreign patent office, which could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, re-examination, post-grant review, inter parties review and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
In addition, we may not be able to prevent unauthorized use of our intellectual property which could harm our business and competitive position. Changes in or different interpretations of patent laws in the U.S. and foreign countries may permit others to use our inventions and discoveries of or to develop and commercialize their new drug candidates without providing any compensation to us, or may limit the number of patents or claims that we can obtain. The laws of some countries do not protect intellectual property rights to the same extent as U.S. laws and those countries may lack adequate rules and procedures for defending and enforcing our intellectual property rights. Additionally, some of our competitors may have more resources than we do to pursue claims of infringement or misappropriation. We may conclude that even if they are infringing our patents or other intellectual property rights, the risk-adjusted costs of bringing claims against them may be too high or otherwise not in our interest. Failure to adequately protect, enforce, or be able to use our intellectual property rights could result in our competitors using our intellectual property to offer products, potentially resulting in the loss of some of our competitive advantage, a decrease in our revenue and reputational harm caused by inferior products offered by third parties, which would adversely affect our business, prospects, financial condition and operating results.
Developments in patent law could have a negative impact on our patent positions and business.
From time to time, the U.S. Supreme Court, other federal courts, the U.S. Congress or the USPTO may change the standards of patentability and any such changes may negatively affect our business. We cannot know what new developments in intellectual property law will impact future use and the value of our intellectual property rights.
In addition, the Leahy-Smith America Invents Act, or the America Invents Act, which was signed into law in 2011, includes a number of significant changes to U.S. patent law. These changes include a transition from a “first-to-invent” system to a “first-to-file” system, changes the way issued patents are challenged, and changes the way patent applications are disputed during the examination process. These changes may favor larger and more established companies that have greater resources to devote to patent application filing and prosecution. The USPTO has developed regulations and procedures to govern the full implementation of the America Invents Act, and many of the substantive changes to patent law associated with the America Invents Act, and, in particular, the first-to-file provisions, became effective on March 16, 2013.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed, respectively.
We have entered into intellectual property assignment, confidentiality and non-disclosure agreements with our employees, consultants, outside scientific and commercial collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keep confidential and not disclose to third parties any confidential information developed by the party or made known to the party by us during the course of the party’s relationship therewith. Where appropriate, they also generally require that the other party assign intellectual property developments to us. However, these agreements may not be honored and may not effectively protect our rights in our intellectual property.
Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we elect to take against such misconduct may not provide an adequate remedy to protect our interests fully. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable. For these reasons, it is possible that we would decide not to attempt to enforce our rights against a third party who we believe has engaged in unauthorized use of our trade secrets or that we would be unsuccessful in our efforts to enforce rights against such a third party.
We and our licensors may not be able to enforce our intellectual property rights throughout the world.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the U.S. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries do not favor the enforcement of patents and other intellectual property protection, especially those relating to pharmaceuticals and medical devices. This could make it difficult for us and our licensors to stop the infringement or misappropriation of our intellectual property rights. For example, some foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, some countries limit the enforceability of patents against certain types of third parties such as government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we have chosen in the past and may choose in the future not to seek patent protection in certain countries, and as a result we will not have the benefit of patent protection in those countries.
Risks Related to Our Reliance on Third Parties
We may seek to partner with third parties to develop and commercialize our product candidates, and if we fail to enter into such collaborations, or if such collaborations are unsuccessful, we may not be able to realize the market potential of our product candidates.
We may seek third-party collaborators to develop and commercialize our products, and we may partner with third parties for marketing, distribution, development, licensing or broader cooperative arrangements, including large and medium-sized pharmaceutical companies, regional and national pharmaceutical companies, non-profit organizations, government agencies, and biotechnology companies. Our ability to generate revenue from these arrangements will depend on the success of our collaborators in fulfilling the agreements they work with. Cooperation involving our products will bring us the following risks:
| ● | Collaborators may have considerable discretion to decide to invest efforts and resources in these collaborations; |
| ● | Collaborators may not pursue the development and commercialization of our product candidates, or may choose not to continue to update development or commercialization plans due to preclinical or clinical trial results, changes in a collaborator’s strategy or changes in available funding, or external factors; |
| ● | Collaborators may delay clinical trials, provide insufficient funding for clinical trial programs, stop clinical trials or abandon product candidates, repeat or conduct new clinical trials, or require new formulations of product candidates for clinical trials; |
| ● | Collaborators may develop products that compete directly or indirectly with third parties if they believe that a competing product is more likely to be successfully developed or can be commercialized on conditions that are more economically attractive than ours; |
| ● | Collaborators who have marketing and distribution rights to one or more products may not devote sufficient resources to the marketing and distribution of such products; |
| ● | Collaborators may not be able to properly defend our intellectual property rights, or may use our patents in a manner that triggers litigation that jeopardizes or invalidates our intellectual property or patents, or exposes us to potential litigation; |
| ● | Collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; |
| ● | Disputes between us and the collaborators may arise that result in delays or discontinuations in the research, development or commercialization of our product candidates, or costly litigation or arbitration; and |
| ● | The collaboration may be terminated, and if terminated, may result in the need for additional funding to pursue further development or commercialization of applicable product candidates. |
Collaboration agreements may not provide for or result in the development or commercialization of our product candidates in the most efficient manner or at all. If our collaborators participate in a business combination, the ongoing pursuit of our product development or commercialization programs may be delayed, reduced, or terminated.
Risks Related to Our Industry, Business and Operation
We are a development-stage drug discovery company and therefore face risks associated with the development of new businesses in the industry.
We are a clinical-stage new drug research and development company. Currently, there is no revenue generated by our business.
| ● | Whether we generate revenue in the future will depend on our new drug research and development results. You should consider the Company’s prospects based on the risks and uncertainties faced by a new drug discovery company with limited operating history and revenue. In particular, potential investors should consider that we may be unable to: |
| ● | determine that the processes, technologies and results of any R&D we pursue are commercially feasible; |
| ● | implement or execute our current business plans, or generate profits; |
| ● | attract and maintain qualified technical and management teams; |
| ● | raise sufficient funds in the capital market to realize our business plans; |
| ● | enter into contracts with appropriate business partners; and/or |
| ● | avoid major conflict with current political, economic and environmental changes. |
If any of the above risks occur, we may fail in our business and operations, in which case you may lose the entire amount invested in the Ordinary Shares. There can be no assurance that any efforts in our business or operations will be successful or result in the timely development of new products or ultimately generate any significant revenue and profits.
Risks Related to Government Regulation
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions.
If our clinical trials support a conclusion that one of our product candidates is safe and effective and if the FDA provides marketing approval for that product candidate in the United States, we may choose to submit marketing applications in other countries. Regulatory authorities in jurisdictions outside of the United States have requirements for approval of product candidates with which we must comply prior to marketing in those jurisdictions. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our products in certain countries. If we fail to comply with the regulatory requirements in international markets and/or fail to receive applicable regulatory approvals, our target market will be reduced, our ability to realize the full market potential of our product candidates will be harmed and future potential revenue may be less than expected by investors and analysts.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, while a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants regulatory approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including additional nonclinical studies or clinical trials as clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In short, the foreign regulatory approval process involves all of the risks associated with FDA approval. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we may charge for our products will also be subject to approval.
We may seek orphan drug designation for certain of our product candidates, and we may be unsuccessful or may be unable to maintain the benefits associated with orphan drug designation, including the potential for market exclusivity.
As part of our business strategy, we may seek orphan drug designation for certain of our product candidates, and we may be unsuccessful. Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as orphan drugs. Under the Orphan Drug Act, the FDA may designate a drug as an orphan drug if it is a product intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals annually in the United States, or a patient population of 200,000 or more in the United States where there is no reasonable expectation that the cost of developing the product will be recovered from sales in the United States. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers.
Similarly, in Europe, the European Commission, upon the recommendation of the EMA’s Committee for Orphan Medicinal Products, grants orphan drug designation to promote the development of drugs that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in Europe and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected). Additionally, designation is granted for drugs intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in Europe would be sufficient to justify the necessary investment in developing the drug. In Europe, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers.
Generally, if a product with an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to a period of marketing exclusivity, which precludes the FDA or the EMA from approving another marketing application for the same product and indication for that time period, except in limited circumstances. The applicable period is seven years in the United States and ten years in the European Union. The exclusivity period in the European Union can be reduced to six years if a product no longer meets the criteria for orphan drug designation or if the product is sufficiently profitable so that market exclusivity is no longer justified.
Even if we obtain orphan drug exclusivity for one of our product candidates, that exclusivity may not effectively protect our product candidate from competition because different products having different chemical compositions can be approved for the same condition and the same therapies can be approved for different conditions but used off-label.
Even after an orphan drug is approved, the FDA can subsequently approve the same product for the same condition if the FDA concludes that the later product is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care. In addition, a designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. Moreover, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the product to meet the needs of patients with the rare disease or condition or if another product with the same active moiety is determined to be safer, more effective, or represents a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a product nor gives the product any advantage in the regulatory review or approval process. While we may seek orphan drug designation for our product candidates, we may never receive such designations. Even if we do receive such designations, there is no guarantee that we will enjoy the benefits of those designations.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological and radioactive materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot, however, eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Risks Related to the Ordinary Shares and this Offering
We have convertible debt that will be converted into our Ordinary Shares upon the closing of this offering, which will cause immediate and substantial dilution to our shareholders.
Between June 23, 2023 and July 24, 2023, we issued and sold convertible promissory notes to certain accredited investors in the aggregate principal amount of $2,516,000. Upon the commencement of trading of our Ordinary Shares to be issued in this offering on the Nasdaq Capital Market, the outstanding principal under the notes will automatically (and without any action on the part of the note holders) be converted into our Ordinary Shares at a conversion price equal to the lesser of 30% of the initial public offering price of our Ordinary Shares or $4.00 per Ordinary Share. The issuance of our Ordinary Shares upon conversion of the notes will result in immediate dilution to the interests of our shareholders.
There has been no public market for our Ordinary Shares prior to this offering, and if an active trading market does not develop, you may not be able to resell our Ordinary Shares at or above the price you paid, or at all.
Prior to this public offering, there has been no public market for our Ordinary Shares. We expect to apply for our Ordinary Shares to be listed on the Nasdaq Capital Market. There is no guarantee that our application will be approved by the Nasdaq Capital Market. If an active trading market for our Ordinary Shares does not develop after this offering, the market price and liquidity of our Ordinary Shares will be materially adversely affected. You may not be able to sell any Ordinary Shares that you purchase in the offering at or above the public offering price. Accordingly, investors should be prepared to face a complete loss of their investment.
As a “controlled company” under Nasdaq Listing Rules, we plan to rely on certain exemptions from Nasdaq corporate governance rules, which means our shareholders will not have the same protections afforded to shareholders of other companies.
As of the date hereof (after giving effect to this offering), Mr. Ho-Ching Chen holds beneficial ownership of % of our outstanding Ordinary Shares pursuant to agreements in which certain of our shareholders have given Mr. Chen voting control over their shares, including in connection with the election of our directors. For so long as these agreements provide Mr. Chen with more than 50% of the voting power for the election of our directors, we will be a “controlled company” under Nasdaq rules. As a controlled company, we will be permitted not to comply with certain of Nasdaq’s corporate governance requirements, including exemptions from the following rules:
| | ● | that a majority of our board of directors must be independent directors; |
| | ● | that we have a Compensation Committee that will determine or recommend the compensation of our executive officers and that it be composed solely of independent directors; and |
| | ● | that our director nominees must be selected or recommended solely by independent directors. |
We currently intend to rely on the controlled company exemptions in respect of our Compensation Committee and Nominating and Corporate Governance Committee, each of which will include directors that are not independent. Further, although we do not intend to rely on the exemption in respect of a majority independent board, we will have the right to use this exemption in the future. As a result, you will not have the same protections afforded to shareholders of companies that are subject to, or voluntarily follow, these Nasdaq corporate governance requirements.
Nasdaq may apply additional and more stringent criteria for our initial and continued listing because we plan to have a small public offering and our insiders will hold a large portion of our listed securities.
Nasdaq Listing Rule 5101 provides Nasdaq with broad discretionary authority over the initial and continued listing of securities on Nasdaq and Nasdaq may use such discretion to deny initial listing, apply additional or more stringent criteria for the initial or continued listing of particular securities, or suspend or delist particular securities based on any event, condition, or circumstance that exists or occurs that makes initial or continued listing of the securities on Nasdaq inadvisable or unwarranted in the opinion of Nasdaq, even though the securities meet all enumerated criteria for initial or continued listing on Nasdaq. In addition, Nasdaq has used its discretion to deny initial or continued listing or to apply additional and more stringent criteria in the instances, including but not limited to: (i) where the company engaged an auditor that has not been subject to an inspection by a Public Accounting Oversight Board (“PCAOB”), an auditor that PCAOB cannot inspect, or an auditor that has not demonstrated sufficient resources, geographic reach, or experience to adequately perform the company’s audit; (ii) where the company planned a small public offering, which would result in insiders holding a large portion of the company’s listed securities, and Nasdaq was concerned that the offering size was insufficient to establish the company’s initial valuation, and there would not be sufficient liquidity to support a public market for the company; and (iii) where the company did not demonstrate sufficient nexus to the U.S. capital market, including having no U.S. shareholders, operations, or members of the board of directors or management. Our initial public offering will be relatively small and our insiders will hold a large portion of our listed securities following the consummation of the offering. Therefore, we may be subject to the additional and more stringent criteria of Nasdaq for our initial and continued listing, which might cause delay or even denial of our listing application.
Our Ordinary Shares may be thinly traded and you may be unable to sell at or near ask prices or at all if you need to sell your shares to raise money or otherwise desire to liquidate your shares.
When our Ordinary Shares are approved by the Nasdaq Capital Market and begin trading on the Nasdaq Capital Market, our Ordinary Shares may be “thinly-traded”, meaning that the number of persons interested in purchasing our Ordinary Shares at or near bid prices at any given time may be relatively small or non-existent. This situation may be attributable to a number of factors, including the fact that we are relatively unknown to stock analysts, stock brokers, institutional investors and others in the investment community that generate or influence sales volume, and that even if we came to the attention of such persons, they tend to be risk-averse and might be reluctant to follow an unproven company such as us or purchase or recommend the purchase of our shares until such time as we became more seasoned. As a consequence, there may be periods of several days or more when trading activity in our shares is minimal or non-existent, as compared to a seasoned issuer which has a large and steady volume of trading activity that will generally support continuous sales without an adverse effect on share price. A broad or active public trading market for our Ordinary Shares may not develop or be sustained.
The initial public offering price for our Ordinary Shares may not be indicative of prices that will prevail in the trading market and such market prices may be volatile.
The initial public offering price for our Ordinary Shares may vary from the market price of our Ordinary Shares following our initial public offering. The financial markets in the United States and other countries have experienced significant price and volume fluctuations in the last few years. If you purchase Ordinary Shares in this initial public offering, you may not be able to resell those shares at or above the initial public offering price. We cannot assure you that the initial public offering price of Ordinary Shares, or the market price following our initial public offering, will equal or exceed prices in privately negotiated transactions of our shares that have occurred from time to time prior to our initial public offering. The market price for our Ordinary Shares may be volatile and subject to wide fluctuations due to factors such as:
| | ● | the financial projections we may provide to the public, any changes in these projections or our failure to meet these projections; |
| | ● | actual or anticipated fluctuations in our quarterly operating results; |
| | ● | changes in financial estimates by securities research analysts; |
| | ● | negative publicity, studies or reports; |
| | ● | our capability to catch up with the technology innovations in the industry; |
| | ● | announcements by us or our competitors of acquisitions, strategic business relationships, joint ventures or capital commitments; |
| | ● | addition or departure of key personnel; |
| | ● | fluctuations of exchange rates between the New Taiwan dollar and the U.S. dollar; and |
| | ● | general economic or political conditions in Taiwan and the greater Asia region. |
In addition, the securities market has from time to time experienced significant price and volume fluctuations that are not related to the operating performance of particular companies. These market fluctuations may also materially and adversely affect the market price of our Ordinary Shares.
Our management team has limited experience managing a public company.
Our management has limited prior experience managing a publicly traded company. As such, our management team may encounter difficulties in managing our business as a public company and in complying with our reporting and other obligations under federal securities laws and other regulations. Their lack of prior experience in dealing with the reporting and other obligations and laws pertaining to public companies could result in our management being required to devote significant time to these activities, which may result in their devoting less time to our business and its development. It could also lead to errors in public disclosures, delays in public filings or problems with our internal controls and procedures. In addition, we will be required to hire additional personnel with the appropriate level of knowledge, experience, and training in the accounting policies, practices or internal controls over financial reporting required of public companies. Newly public companies typically have to incur significant expense in order to improve their internal controls and comply with reporting requirements, among other things, and the increase in our expenses in connection with these activities could be significant.
The obligations associated with becoming a public company may strain our resources, result in more litigation and divert management’s attention from operating our business.
Following this offering, our officers and directors will begin managing our business as a public company that is subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of the Nasdaq Capital Market, and other applicable securities rules and regulations. Complying with these rules and regulations will increase our legal, accounting and financial costs, make some activities more difficult, time-consuming or costly, and increase demand on our personnel, systems and resources. The need to establish the corporate infrastructure demanded of a public company may divert our management’s attention from implementing our business strategy. We expect to hire more employees to assist us in complying with these requirements.
In addition, changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for public companies, increasing legal and financial compliance costs, and making some activities more time-consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We intend to invest substantial resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of our management’s time and attention from business operations to compliance activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to their application and practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
Furthermore, as a result of disclosure of information in connection with this offering and in filings required of a public company, our business and financial condition has and will continue to become more visible. Such increased disclosure and visibility could result in adverse changes to our reputation and to the way our business partners and the public perceive our company, as well as shareholder activism or threatened or actual litigation, including by competitors and other third parties. If such claims are successful, our business, financial condition and results of operations could be harmed, and even if the claims do not result in litigation or are resolved in our favor, these claims, and the time and resources necessary to resolve them, could divert the resources of our management and could adversely affect our business, results of operations and financial condition.
You will experience immediate and substantial dilution in the net tangible book value of Ordinary Shares purchased.
The initial public offering price of our Ordinary Shares is substantially higher than the net tangible book value per share of our Ordinary Shares. Consequently, when you purchase our Ordinary Shares in the offering and upon completion of the offering, you will incur immediate dilution of $ per Ordinary Share, assuming an initial public offering price of $ , which is the midpoint of the price range as set forth on the cover page of this prospectus. See “Dilution”.
Substantial future sales of our Ordinary Shares or the anticipation of future sales of our Ordinary Shares in the public market could cause the price of Ordinary Shares to decline.
Sales of substantial amounts of our Ordinary Shares in the public market after this offering, or the perception that these sales could occur, could cause the market price of our Ordinary Shares to decline. An aggregate of Ordinary Shares are outstanding before the consummation of this offering and Ordinary Shares will be outstanding immediately after the consummation of this offering. Sales of these shares into the market could cause the market price of Ordinary Shares to decline.
We do not intend to pay dividends for the foreseeable future.
We currently intend to retain all available funds and future earnings, if any, for research and development and the operation and expansion of our business and do not anticipate declaring or paying any dividends in the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our board of directors after considering our financial condition, results of operations, capital requirements, contractual requirements, business prospects and other factors the board of directors deems relevant, and subject to the restrictions contained in any future financing instruments.
As of the date of this prospectus, we have not declared or paid any cash dividends on our capital shares. If we determine to pay dividends on any of our Ordinary Shares in the future, as a holding company, Everfront Holding will be dependent on receipt of funds from its Taiwan subsidiary, Everfront Biotech.
If securities or industry analysts do not publish research or reports about our business, or if they publish a negative report regarding our Ordinary Shares, the price of our Ordinary Shares and trading volume could decline.
The trading market for our Ordinary Shares may depend in part on the research and reports that industry or securities analysts publish about us or our business. We do not have any control over these analysts. If one or more of the analysts who cover us downgrade our securities, the price of our Ordinary Shares would likely decline. If one or more of these analysts cease coverage of Everfront Holding or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause the price of Ordinary Shares and the trading volume to decline.
We may experience extreme stock price volatility unrelated to our actual or expected operating performance, financial condition or prospects, making it difficult for prospective investors to assess the rapidly changing value of our Ordinary Shares, and such volatility may subject us to securities litigation.
The market for our Ordinary Shares may have, when compared to seasoned issuers, significant price volatility and we expect that our share price may continue to be more volatile than that of a seasoned issuer for the indefinite future. Recently, there have been instances of extreme stock price run-ups followed by rapid price declines and strong stock price volatility with a number of recent initial public offerings, especially among companies with relatively smaller public floats. As a relatively small-capitalization company with relatively small public float, we may experience greater stock price volatility, extreme price run-ups, lower trading volume and less liquidity than large-capitalization companies. In particular, our Ordinary Shares may be subject to rapid and substantial price volatility, low volumes of trades and large spreads in bid and ask prices. Such volatility, including any stock-run up, may be unrelated to our actual or expected operating performance, financial condition or prospects, making it difficult for prospective investors to assess the rapidly changing value of our Ordinary Shares.
In addition, if the trading volumes of our Ordinary Shares are low, persons buying or selling in relatively small quantities may easily influence prices of our Ordinary Shares. This low volume of trades could also cause the price of our Ordinary Shares to fluctuate greatly, with large percentage changes in price occurring in any trading day session. Holders of our Ordinary Shares may also not be able to readily liquidate their investment or may be forced to sell at depressed prices due to low volume trading. Broad market fluctuations and general economic and political conditions may also adversely affect the market price of our Ordinary Shares. As a result of this volatility, investors may experience losses on their investment in our Ordinary Shares. A decline in the market price of our Ordinary Shares also could adversely affect our ability to issue additional Ordinary Shares or other securities and our ability to obtain additional financing in the future. No assurance can be given that an active market in our Ordinary Shares will develop or be sustained. If an active market does not develop, holders of our Ordinary Shares may be unable to readily sell
In the past, plaintiffs have often initiated securities class action litigation against a company following periods of volatility in the market price of its securities. We may, in the future, be the target of similar litigation. Securities litigation could result in substantial costs and liabilities and could divert management’s attention and resources.
You may face difficulties in protecting your interests, and your ability to protect your rights through U.S. courts may be limited, because Everfront Holding is incorporated under British Virgin Islands law.
Everfront Holding is incorporated under the laws of the British Virgin Islands. Accordingly, our corporate affairs are governed by our memorandum and articles of association, the BVI Act and the common law of the British Virgin Islands. The rights of shareholders to take action against our directors, actions by our minority shareholders and the fiduciary duties of our directors to us under the British Virgin Islands law are to a large extent governed by the common law of the British Virgin Islands. The common law of the British Virgin Islands is derived in part from comparatively limited judicial precedent in the British Virgin Islands as well as from the common law of England, the decisions of whose courts are of persuasive authority, but are not binding, on a court in the British Virgin Islands. The rights of our shareholders and the fiduciary duties of our directors under the British Virgin Islands law are not as clearly established as they would be under statutes or judicial precedent in some jurisdictions in the United States. In particular, the British Virgin Islands has a less developed body of securities laws than the United States. Some U.S. states, such as Delaware, have more fully developed and judicially interpreted bodies of corporate law than the British Virgin Islands. In addition, British Virgin Islands companies may not have standing to initiate a shareholder derivative action in a federal court of the United States.
Certain corporate governance practices in the British Virgin Islands, where Everfront Holding is incorporated, differ significantly from requirements for companies incorporated in other jurisdictions such as the United States. We are permitted to rely on home country practice with respect to our corporate governance. Although we currently do not intend to rely on home country practice for at least one year after the initial public offering, we may elect to rely on home country practice in the future. If we choose to follow the British Virgin Islands’ practice in the future, our shareholders may be afforded less protection than they otherwise would under rules and regulations applicable to U.S. domestic issuers. See “Risk Factors – Risks Related to our Ordinary Shares and This Offering – As a foreign private issuer, we are permitted to rely on certain home country rules in lieu of the corresponding corporate governance standards corporate governance standards of the Nasdaq Listing Rules applicable to domestic U.S. issuers. This may afford less protection to holders of our shares.”
As a result of all of the above, public shareholders may have more difficulties in protecting their interests in the face of actions taken by our management, or members of our board of directors, than they would as public shareholders of a company incorporated in the United States. For a discussion of significant differences between the provisions of the BVI Act and the laws applicable to companies incorporated in the United States and their shareholders, see “Description of Share Capital—Differences in Corporate Law.”
As a foreign private issuer, we are permitted to rely on certain home country rules in lieu of the corresponding corporate governance standards of the Nasdaq Listing Rules applicable to domestic U.S. issuers. This may afford less protection to holders of our shares.
Because we are a foreign private issuer, the Nasdaq Listing Rules permit us to follow certain home country rules rather than the corresponding corporate governance standards of Nasdaq. The standards applicable to us may be considerably different than the standards applied to domestic U.S. issuers, including requirements to:
| | ● | have a majority of our board consist of independent directors (although all of the members of the audit committee must be independent under the Exchange Act); |
| | | |
| | ● | have a compensation committee or a nominating and corporate governance committee consisting entirely of independent directors; |
| | | |
| | ● | have regularly scheduled executive sessions with only independent directors; or |
| | | |
| | ● | have executive sessions of solely independent directors each year. |
We expect to rely on certain of these exemptions and may elect to rely on any or all of them in the future. As a result, you may not be provided with the benefits of certain corporate governance requirements of the Nasdaq.
If we cannot satisfy, or continue to satisfy, the initial listing requirements and other rules of the Nasdaq Capital Market, our securities may not be listed or may be delisted, which would negatively impact the price of our securities and your ability to sell them.
We will seek to have our securities approved for listing on the Nasdaq Capital Market upon consummation of this offering. We cannot assure you that we will be able to meet those initial listing requirements at that time. Even if our securities are listed on the Nasdaq Capital Market, we cannot assure you that our securities will continue to be listed on the Nasdaq Capital Market.
In addition, following this offering, in order to maintain our listing on the Nasdaq Capital Market, we will be required to comply with certain rules of the Nasdaq Capital Market, including those regarding minimum stockholders’ equity, minimum share price and certain corporate governance requirements. Even if we initially meet the listing requirements and other applicable rules of the Nasdaq Capital Market, it may not be able to continue to satisfy these requirements and applicable rules. If we are unable to satisfy the Nasdaq Capital Market criteria for maintaining our listing, our securities could be subject to delisting.
If the Nasdaq Capital Market does not list our securities, or subsequently delists our securities from trading, we could face significant consequences, including:
| | ● | a limited availability for market quotations for our securities; |
| | | |
| | ● | reduced liquidity with respect to our securities; |
| | | |
| | ● | a determination that our Ordinary Shares are a “penny stock,” which will require brokers trading in our Ordinary Share to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for our Ordinary Shares; |
| | | |
| | ● | a limited amount of news and analyst coverage; and |
| | | |
| | ● | a decreased ability to issue additional securities or obtain additional financing in the future. |
Because our business is conducted in New Taiwan dollars and the price of our Ordinary Shares is quoted in United States dollars, changes in currency conversion rates may affect the value of your investments.
Our business is conducted in Taiwan, our books and records are maintained in New Taiwan dollars, which is the currency of Taiwan, and the financial statements that we file with the SEC and provide to our shareholders are presented in United States dollars. Changes in the exchange rate between the New Taiwan dollar and the United States dollar affect the value of our assets and the results of our operations in United States dollars. The value of the New Taiwan dollar against the United States dollar and other currencies may fluctuate and is affected by, among other things, changes in Taiwan’s political and economic conditions and perceived changes in the economies of Taiwan and the United States. Any significant revaluation of the New Taiwan dollar may materially and adversely affect our cash flows, revenue and financial condition. Further, although the Ordinary Shares offered by this prospectus are denominated in United States dollars, we will need to convert the net proceeds we receive into New Taiwan dollars in order to use the funds for our business. Changes in the conversion rate between the United States dollar and New Taiwan dollar will affect that amount of proceeds we will have available for our business.
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
Our management will have broad discretion in the application of the net proceeds, including for any of the purposes described in the section entitled “Use of Proceeds,” and you will not have the opportunity as part of your investment decision to assess whether the net proceeds are being used appropriately. Because of the number and variability of factors that will determine our use of the net proceeds from this offering, their ultimate use may vary substantially from their currently intended use. The failure by our management to apply these funds effectively could harm our business.
Our pre-IPO shareholders will be able to sell their shares after completion of this offering subject to restrictions under Rule 144.
Our pre-IPO shareholders may be able to sell their Ordinary Shares under Rule 144 after completion of this offering. Because these shareholders have paid a lower price per Ordinary Share than participants in this offering, when they are able to sell their pre-IPO shares under Rule 144, they may be more willing to accept a lower sales price than the IPO price. This fact could impact the trading price of the stock following completion of the offering, to the detriment of participants in this offering. Under Rule 144, before our pre-IPO shareholders can sell their shares, in addition to meeting other requirements, they must meet the required holding period. We do not expect any of the Ordinary Shares to be sold pursuant to Rule 144 during the pendency of this offering.
We could be deemed to be a passive foreign investment company (“PFIC”), for U.S. federal income tax purposes for any taxable year, which could result in adverse U.S. federal income tax consequences to U.S. holders of our Ordinary Shares.
A non-U.S. corporation will be a PFIC for any taxable year if either (1) at least 75% of its gross income for such year consists of certain types of “passive” income; or (2) at least 50% of the value of its assets (based on an average of the quarterly values of the assets) during such year is attributable to assets that produce passive income or are held for the production of passive income, or the asset test. Based on our current and expected income and assets (taking into account the expected cash proceeds and our anticipated market capitalization following this offering), we do not presently expect to be a PFIC for the current taxable year or the foreseeable future. However, no assurance can be given in this regard because the determination of whether we are or will become a PFIC is a fact-intensive inquiry made on an annual basis that depends, in part, upon the composition of our income and assets. In addition, there can be no assurance that the Internal Revenue Service, or IRS, will agree with our conclusion or that the IRS would not successfully challenge our position. Fluctuations in the market price of our Ordinary Shares may cause us to become a PFIC for the current or subsequent taxable years because the value of our assets for the purpose of the asset test may be determined by reference to the market price of our Ordinary Shares. The composition of our income and assets may also be affected by how, and how quickly, we use our liquid assets and the cash raised in this offering. If Everfront Holding were to be or become a PFIC for any taxable year during which a U.S. Holder holds our Ordinary Shares, certain adverse U.S. federal income tax consequences could apply to such U.S. Holder and such U.S. Holder may be subject to additional reporting requirements. For a more detailed discussion of the application of the PFIC rules to us and the consequences to U.S. taxpayers if we were or are determined to be a PFIC, see “Taxation— Passive Foreign Investment Company.”
We will be an emerging growth company within the meaning of the Securities Act upon the consummation of this offering and may take advantage of certain reduced reporting requirements.
Upon the consummation of this offering, we will be an “emerging growth company,” as defined in the Jumpstart our Business Startups Act of 2012, or the JOBS Act, and we may take advantage of certain exemptions from various requirements applicable to other public companies that are not emerging growth companies including, most significantly, not being required to comply with the auditor attestation requirements of Section 404 for so long as we are an emerging growth company. As a result, if we elect not to comply with such auditor attestation requirements, our investors may not have access to certain information they may deem important.
The JOBS Act also provides that an emerging growth company does not need to comply with any new or revised financial accounting standards until such date that a private company is otherwise required to comply with such new or revised accounting standards. However, we have elected to “opt out” of this provision and, as a result, we will comply with new or revised accounting standards as required when they are adopted for public companies. This decision to opt out of the extended transition period under the JOBS Act is irrevocable.
DISCLOSURE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements, all of which are subject to risks and uncertainties. Forward-looking statements give our current expectations or forecasts of future events. You can identify these statements by the fact that they do not relate strictly to historical or current facts. You can find many (but not all) of these statements by the use of words such as “approximates,” “believes,” “hopes,” “expects,” “anticipates,” “estimates,” “projects,” “intends,” “plans,” “will,” “would,” “should,” “could,” “may” or other similar expressions in this prospectus. These statements are likely to address our growth strategy, financial results and product and development programs. You must carefully consider any such statements and should understand that many factors could cause actual results to differ from our forward-looking statements. These factors may include inaccurate assumptions and a broad variety of other risks and uncertainties, including some that are known and some that are not. No forward-looking statement can be guaranteed and actual future results may vary materially. Factors that could cause actual results to differ from those discussed in the forward-looking statements include, but are not limited to:
| | ● | our plans to develop and commercialize our product candidates; |
| | | |
| | ● | the initiation, timing, progress and results of our current and future pre-clinical studies and clinical trials and our R&D programs; |
| | | |
| | ● | our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; |
| | | |
| | ● | our ability to successfully acquire or obtain licenses for additional product candidates on reasonable terms; |
| | | |
| | ● | our ability to establish and maintain collaborations and/or obtain additional funding; |
| | | |
| | ● | our ability to obtain regulatory approval for our current and future product candidates from the FDA, TFDA and/or other relevant regulatory authorities; |
| | | |
| | ● | our expectations regarding the potential market size and the rate and degree of market acceptance of our current and future product candidates; |
| | | |
| | ● | our ability to fund our working capital requirements and expectations regarding the sufficiency of our capital resources; |
| | | |
| | ● | our intellectual property position; |
| | | |
| | ● | developments in and/or disputes concerning our intellectual property or other proprietary rights; |
| | | |
| | ● | our expectations regarding government and third-party payer coverage and reimbursement; |
| | | |
| | ● | our ability to compete in the markets we are in; |
| | | |
| | ● | the impact of government laws and regulations and the liabilities there under; |
| | | |
| | ● | our expected use of proceeds from this offering; |
| | | |
| | ● | our need to hire additional personnel and our ability to attract and retain such personnel; |
| | | |
| | ● | developments relating to our competitors and our industry; |
| | ● | we are incorporated in the British Virgin Islands, and our shareholders may have more difficulty protecting their interests than they would as shareholders of a corporation incorporated in the United States; and |
| | | |
| | ● | other risk factors as discussed under the section titled “Risk Factors”. |
We describe certain material risks, uncertainties and assumptions that could affect our business, including our financial condition and results of operations, under “Risk Factors.” We base our forward-looking statements on our management’s beliefs and assumptions based on information available to our management at the time the statements are made. We caution you that actual outcomes and results may, and are likely to, differ materially from what is expressed, implied or forecast by our forward-looking statements. Accordingly, you should be careful about relying on any forward-looking statements. Except as required under the federal securities laws, we do not have any intention or obligation to update publicly any forward-looking statements after the distribution of this prospectus, whether as a result of new information, future events, changes in assumptions, or otherwise.
Industry Data and Forecasts
This prospectus contains certain data and information that we obtained from various government and private publications. Statistical data in these publications also include projections based on a number of assumptions. You should note that the market for cancer and ALS treatments such as ours may not be as large as estimated and may not grow at the rate projected by market data, or at all. If these markets are smaller than estimated or fail to grow at the projected rate, this may have a material and adverse effect on our business and the market price of our Ordinary Shares. In addition, the new and rapidly changing nature of the industry for cancer and ALS treatments results in significant uncertainties for any projections or estimates relating to the growth prospects or future condition of our industry. Furthermore, if any one or more of the assumptions underlying the market data are later found to be incorrect, actual results may differ from the projections based on these assumptions. You should not place undue reliance on these forward-looking statements.
ENFORCEABILITY OF CIVIL LIABILITIES
Everfront Holding is incorporated in the British Virgin Islands to take advantage of certain benefits associated with being a British Virgin Islands business company, such as:
| | ● | political and economic stability; |
| | ● | an effective judicial system; |
| | ● | the absence of exchange controls or currency restrictions; and |
| | ● | the availability of professional and support services. |
However, certain disadvantages accompany incorporation in the British Virgin Islands. These disadvantages include, but are not limited to:
| | ● | the British Virgin Islands has a less developed body of securities laws as compared to the United States and these securities laws provide significantly less protection to investors as compared to the United States; and |
| | ● | British Virgin Islands companies may not have standing to sue before the federal courts of the United States. |
Everfront Holding’s memorandum and articles of association do not contain provisions requiring that disputes, including those arising under the securities laws of the United States, involving us and our officers, directors and shareholders, be arbitrated.
All of our assets are located in Taiwan. In addition, all of our directors and officers are nationals or residents of Taiwan. As a result, it may be difficult for investors to effect service of process within the United States upon us or these persons, or to enforce against us or them judgments obtained in United States courts, including judgments predicated upon the civil liability provisions of the securities laws of the United States or any state in the United States.
We have appointed Cogency Global as our agent to receive service of process upon whom process may be served in any action brought against us under the securities laws of the United States.
McW Todman & Co., our British Virgin Islands counsel, and Formosan Brothers, our Taiwan counsel, have advised us that there is uncertainty as to whether the courts of the BVI or Taiwan would (i) recognize or enforce judgments of United States courts obtained against us or our directors or officers predicated upon the civil liability provisions of the securities laws of the United States or any state in the United States or (ii) entertain original actions brought in the British Virgin Islands or Taiwan against us or our directors or officers predicated upon the securities laws of the United States or any state in the United States.
There is uncertainty with regard to British Virgin Islands law as to whether a judgment obtained from the United States courts under civil liability provisions of the securities laws will be determined by the courts of the British Virgin Islands as penal or punitive in nature. If such a determination is made, the courts of the British Virgin Islands are also unlikely to recognize or enforce the judgment against a British Virgin Islands company. Because the courts of the British Virgin Islands have yet to rule on whether such judgments are penal or punitive in nature, it is uncertain whether they would be enforceable in the British Virgin Islands. McW Todman & Co. has advised us that although there is no statutory enforcement in the British Virgin Islands of judgments obtained in the federal or state courts of the United States, in certain circumstances a judgment obtained in such jurisdiction may be recognized and enforced in the courts of the British Virgin Islands at common law, without any re-examination of the merits of the underlying dispute, by an action commenced on the foreign judgment debt in the High Court of the British Virgin Islands, provided such judgment:
| | ● | is given by a foreign court of competent jurisdiction and such foreign court had proper jurisdiction over the parties subject to such judgment; |
| | ● | imposes on the judgment debtor a liability to pay a liquidated sum for which the judgment has been given; |
| | ● | no new admissible evidence relevant to the action is submitted prior to the rendering of the judgment by the courts of the BVI; |
| | ● | is not in respect of taxes, a fine, a penalty or similar fiscal or revenue obligations of the company; and |
| | ● | was not obtained in a fraudulent manner and is not of a kind the enforcement of which is contrary to natural justice or the public policy of the British Virgin Islands. |
In appropriate circumstances, a BVI Court may give effect in the BVI to other kinds of final foreign judgments such as declaratory orders, orders for performance of contracts and injunctions.
We are also advised by our Taiwan legal counsel that a judgment obtained from the federal or state courts of the United States under civil liability provisions may be recognized and enforced in courts of Taiwan, without any re-examination of the merits of the underlying dispute, by an action initiated on the foreign judgment debt in courts of Taiwan, provided the following requirements are satisfied:
| ● | the judgment is final and conclusive; |
| ● | the United States court rendering the judgment has jurisdiction over the subject matter according to the laws of Taiwan; |
| ● | if the judgment is a default judgment against the debtor as rendered by the United States court, (a) the debtor has been duly served in the jurisdiction of the United States court within a reasonable period of time in accordance with the laws and regulations of such jurisdiction, or (b) process has been served on the debtor with judicial assistance of the government of Taiwan; |
| ● | the judgment and the United States courts procedure resulting in the judgment are not contrary to the public order or good morals of Taiwan; and |
| ● | there is mutual recognition between the United States and Taiwan, or the courts in the jurisdiction where the United States judgment is rendered do not generally refuse to recognize the validity of the judgments of Taiwan courts. |
In appropriate circumstances, a Taiwan court may give effect in Taiwan to final foreign rulings of courts, such as rulings for performance and injunctions.
USE OF PROCEEDS
We estimate that we will receive net proceeds from this offering of $ (or approximately $ if the underwriters fully exercise their option to purchase additional Ordinary Shares), after deducting the estimated underwriting discounts and the estimated offering expenses payable by us and based upon an assumed initial public offering price of $ per Ordinary Share, which is the midpoint of the price range set forth on the cover page of this prospectus.
We plan to use the net proceeds we receive from this offering for the following purposes.
| | | | | Portion of Net Proceeds | |
| Use of Proceeds | | Milestone | | Dollars | | | Percent | |
| Clinical trial expenses – Cerebraca™ Wafer Phase I/IIa | | Phase I/IIa Completion | | $ | | | | | 1.7 | % |
| Clinical trial expenses – Cerebraca™ Wafer Phase IIb/III | | Interim Analysis | | | | | | | 70.8 | |
| Clinical trial expenses – HK-001 / EF-031 | | Phase I Completion | | | | | | | 4.0 | |
| Clinical trial expenses – EF-009 Wafer | | Phase I Completion | | | | | | | 3.5 | |
| General R&D expenses | | | | | | | | | 7.0 | |
| Patent maintenance and application expenses | | | | | | | | | 3.0 | |
| Working capital and other general corporate expenses | | | | | | | | | 10.0 | |
| Total | | | | $ | | | | | 100.0 | % |
We believe the net proceeds set forth above will fund the development of our Cerebraca™ Wafer through the completion of the current Phase I/IIa clinical trial and further progress to the Phase IIb/III clinical trial. The net proceeds set forth above will support the Phase IIb/III clinical trial until the completion of the interim analysis, which will provide the primary clinical outcome of the study. The net proceeds set forth above will also fund the development of three products, including HK-001, EF-031, and EF-009 Wafer, through the completion of Phase I clinical trials. The completion of these three clinical trials will provide more applicable human data to build the foundation of the product development.
The foregoing represents our current intention based upon our present plans and business conditions to use and allocate the net proceeds of this offering. Our management, however, will have significant flexibility and discretion to apply the net proceeds of this offering. To the extent that the net proceeds we receive from this offering are not immediately used for the above purposes, we intend to invest our net proceeds in short-term, interest-bearing bank deposits or debt instruments.
DETERMINATION OF OFFERING PRICE
Since our Ordinary Shares are not listed or quoted on any exchange or quotation system, the initial public offering price of our Ordinary Shares was determined by us and the underwriters and is based on an assessment of our financial condition and prospects, comparable companies with similar sizes and businesses currently traded on U.S. capital markets, and the general condition of the securities market. It does not necessarily bear any relationship to our book value, assets, past operating results, financial condition or any other established criteria of value. Our Ordinary Shares are not listed on a public exchange, and we intend to obtain a listing on the Nasdaq Capital Market immediately prior to closing this offering.
The initial public offering price stated on the cover page of this prospectus should not be considered an indication of the actual value of the Ordinary Shares. That price is subject to change as a result of market conditions and other factors, including the depth and liquidity of the market for the Ordinary Shares, investor perception of us and general economic and market conditions, and we cannot assure you that the Ordinary Shares can be resold at or above the initial public offering price.
DIVIDEND POLICY
Subject to the BVI Act and Everfront Holding’s memorandum and articles of association, our board of directors may authorize and declare a dividend to shareholders at such time and in such an amount as they think fit if they are satisfied, on reasonable grounds, that immediately following the dividend the value of our assets will exceed our liabilities and we will be able to pay our debts as they become due. There is no further BVI statutory restriction on the amount of funds which may be distributed by us by dividend.
We have never declared or paid any cash dividends on our capital stock.
We currently intend to retain all available funds and any future earnings for research and development and to support our operations and finance the growth and development of our business. Any future determination related to our dividend policy will be made at the discretion of our board of directors and will depend upon, our results of operations, financial condition, capital requirements, contractual restrictions, business prospects and other factors our board of directors may deem relevant.
If we determine to pay dividends on any of our Ordinary Shares in the future, as a holding company, we will be dependent on receipt of funds from Everfront Biotech, our Taiwan subsidiary.
CAPITALIZATION
The following table sets forth our capitalization as of June 30, 2024:
| ● | on a pro forma basis to reflect our issuance and sale of Ordinary Shares in this offering and the use of the proceeds of this offering as set forth in “Use of Proceeds.” |
You should read this table together with our consolidated financial statements and the related notes included elsewhere in this prospectus and the information under “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
| | | June 30, 2024 | |
| | | Actual | | | Pro Forma | |
| | | US$ | | | US$ | |
| Shareholders’ equity: | | | | | | | | |
| Ordinary Shares, US$0.01 par value, 100,000,000 shares authorized actual and pro forma; 52,237,745 shares issued and outstanding, actual, shares issued and outstanding, pro forma (1) | | | | | | | | |
| Shares subscription receivable | | | | | | | | |
| Retained earnings | | | | | | | | |
| Total shareholders’ equity | | | | | | | | |
| (1) | Does not reflect outstanding rights to purchase Ordinary Shares under our employee benefit plan or the warrants to purchase our Ordinary Shares that we will issue to the representatives of the underwriters at the closing of this offering. See “Underwriting.” |
DILUTION
If you invest in our Ordinary Shares, your interest will be diluted for each Ordinary Share you purchase to the extent of the difference between the initial public offering price per Ordinary Share and our net tangible book value per Ordinary Share after this offering. Dilution results from the fact that the initial public offering price per Ordinary Share substantially exceeds the net tangible book value per Ordinary Share attributable to the existing shareholders for our presently outstanding Ordinary Shares.
Our net tangible book value as of June 30, 2024 was approximately $ , or $ per Ordinary Share. Net tangible book value represents the amount of our total consolidated tangible assets, less the amount of our total consolidated liabilities. Dilution is determined by subtracting the as adjusted net tangible book value per Ordinary Share from the initial public offering price per Ordinary Share and after deducting the estimated discounts to the underwriter and the estimated offering expenses payable by us.
After giving effect to (i) our sale of Ordinary Shares in this offering at the assumed initial public offering price of $ per Ordinary Share, which is the midpoint of the price range set forth on the cover page of this prospectus, and (ii) the deduction of our estimated offering expenses, our net tangible book value as of June 30, 2024 would have been $ , or $ per Ordinary Share. This represents an immediate increase in as adjusted net tangible book value per Ordinary Share of $ to our existing shareholders and an immediate dilution in as adjusted net tangible book value per Ordinary Share of $ to new investors purchasing Ordinary Shares in this offering. The dilution information discussed herein is illustrative only and will change based on the actual initial public offering price, number of Ordinary Shares and other terms of this offering determined at pricing.
The following table illustrates this dilution on a per Ordinary Share basis.
| Assumed initial public offering price per ordinary share | | $ | | |
| Net tangible book value per Ordinary Share as of June 30, 2024 | | $ | | |
| Increase in as adjusted net tangible book value per Ordinary Share attributable to new investors purchasing Ordinary Shares in this offering | | $ | | |
| As adjusted net tangible book value per Ordinary Share after this offering | | $ | | |
| Dilution per Ordinary Share to new investors in this offering | | $ | | |
An increase (decrease) in the assumed initial public offering price of our Ordinary Shares of US$1.00 would increase (decrease) our net tangible book value by US$ after giving effect to this offering (assuming there is no change to the number of Ordinary Shares offered by us as set forth on the cover page of this prospectus).
To the extent that we issue additional Ordinary Shares in the future, there will be further dilution to new investors participating in this offering.
The following table summarizes, on an as adjusted basis as of June 30, 2024, the differences between existing shareholders and the new investors, the total consideration paid and the average price per Ordinary Share before deducting the estimated discounts to the underwriter and the estimated offering expenses payable by us.
| | | Ordinary Shares purchased | | | Total consideration | | | Average price per ordinary | |
| | | Number | | | Percent | | | Amount | | | Percent | | | share | |
| Existing shareholders | | | | | | | | % | | $ | | | | | | % | | $ | | |
| New investors | | | | | | | | % | | $ | | | | | | % | | $ | | |
| Total | | | | | | | 100.0 | % | | $ | | | | | 100.0 | % | | $ | | |
CORPORATE HISTORY AND STRUCTURE
Our Corporate History
Everfront Biotech was incorporated under the laws of Taiwan with limited liability on December 8, 2010 by Mr. Ho-Ching Chen, our founder and Director. Everfront Holding was incorporated on September 21, 2022 under the laws of the British Virgin Islands with the sole purpose of being a holding company for Everfront Biotech. Upon incorporation, Everfront Holding issued 50,000 Ordinary Shares, par value $0.01 per share, to Mr. Ho-Ching Chen. On February 24, 2023, 87 shareholders of Everfront Biotech (including Mr. Ho-Ching Chen), who together held 99.96% of the equity interests of Everfront Biotech, sold all of their Everfront Biotech shares to Everfront Holding, and received the opportunity to reinvest this cash to purchase shares in Everfront Holdings. 272 investors, including a portion of these shareholders along with new investors, made cash investments in Everfront Holding to purchase 51,395,450 Ordinary Shares. As a result of these transactions, Everfront Holding holds 99.96% of the equity interests in Everfront Biotech, and Taiwan’s National Dong Hwa University holds the remaining 0.04%. In addition, from January 2023 to February 2023, Everfront Holding issued 792,295 Ordinary Shares to 29 new investors and one existing shareholder of Everfront Holding. As a result, Everfront Holding currently has 301 shareholders.
Corporate Structure
The following diagram illustrates our corporate structure as of the date of this prospectus:

MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with our financial statements and the related notes thereto included elsewhere in this prospectus. This discussion and analysis and other parts of this prospectus contain forward-looking statements that are based upon current beliefs, plans and expectations that involve risks, uncertainties, and assumptions. Our actual results and the timing of selected events could differ materially from those anticipated in these forward-looking statements as a result of several factors, including those set forth under “Risk Factors” and elsewhere in this prospectus. You should carefully read the “Risk Factors” section of this prospectus to gain an understanding of the important factors that could cause our actual results to differ materially from our forward-looking statements.
Overview
We are a clinical stage biopharmaceutical company dedicated to the discovery, development and commercialization of new drugs that have potential as treatments for patients with cancers, neurodegenerative diseases and rare diseases. We are focused on new drug discovery and development, supported by our research and development (“R&D”) team established in 2010. Our team is specialized in preclinical study design, chemistry, manufacturing, and controls (“CMC”), investigational new drug (“IND”) submission, and designing and conducting clinical trials. Our goal is to develop technology with the commercial potential to help people with unmet medical needs.
Recent Developments
Convertible Notes
From June 23, 2023, until July 24, 2023, we entered into securities purchase agreements with accredited investors pursuant to which we issued convertible promissory notes in the aggregate principal amount of $2,516,000. Pursuant to the terms the convertible promissory note, as amended on June 24, 2024 and December 2024, the notes mature on the earlier of (i) the date that 24 months after of the issuance date of the notes, (ii) the date when we redeem the notes, or (iii) the date of our initial public offering. The notes accrue interest at 2.40% per annum and maybe prepaid by us at any time without any penalties, provided we give prior written notice to the note holder at least ten business days before prepayment. Any outstanding and unpaid principal amount of the notes automatically converts upon the commencement of trading on the Nasdaq Stock Market or the New York Stock Exchange of our Ordinary Shares at a conversion price equal to the lesser of (i) a 30% discount to the price to the public in this offering, or (ii) $4.00 per share.
Financial Operations Overview
Revenue
We currently have no products approved for sale and have not generated any revenues from the sale of products. We have not submitted any product candidate for regulatory approval. Our revenue in 2022 is a one-time service revenue from fibroblast culture process research commissioned by a related party, Xi Yue Biomedicine Inc. (formerly Guang-Li Biomedicine Inc.).
In the future, we may generate revenue from a combination of product sales and royalties in connection with any future marketing and distribution arrangements and other collaborations, strategic alliances and license arrangements, or a combination of these approaches. However, we do not expect to receive additional revenues unless and until we receive regulatory approval for our product candidates or potentially enter into collaboration agreements. We expect that any revenue we generate will fluctuate from quarter to quarter as a result of the timing and amount of license fees, milestone and other payments, and the amount and timing of payments that we receive upon the sale of our products, to the extent any are successfully commercialized. If we fail to complete the development of our product candidates in a timely manner or obtain regulatory approval of them, our ability to generate future revenue, and our results of operations and financial position, would be materially adversely affected.
General and administrative expenses
General and administrative expenses consist primarily of salaries and related costs for personnel in executive, finance, as well as administrative functions. General and administrative expenses also include legal fees relating to patent and corporate matters; professional fees for accounting, auditing, initial public offering, tax and administrative consulting services; insurance costs; administrative travel expenses; rent expense; facility-related costs and other operating costs.
We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support our continued research and development and potential commercialization of our product candidates. We also anticipate increased expenses related to audit, legal, regulatory and tax-related services associated with maintaining compliance with exchange listing and SEC requirements, director and officer insurance premiums and investor relations costs associated with being a public company. Additionally, if we believe regulatory approval of the lead product candidate appears likely, we anticipate an increase in payroll and related expenses as a result of our preparation for commercial operations, especially as it relates to establishing a sales force and other expenses related to the sale and marketing of our product candidates.
Research and development expenses
Research and development expenses consist primarily of salaries and related expenses for personnel, preclinical costs, clinical trial costs, costs related to acquiring and manufacturing clinical trial materials, contract services, facilities costs, overhead costs and depreciation. All research and development costs are expensed as incurred. These expenses include:
| ● | expenses incurred under agreements with CROs, contract manufacturing organizations, or CMOs, as well as investigative sites and consultants that conduct our clinical trials, preclinical studies and other scientific development services; |
| ● | manufacturing scale-up expenses and the cost of acquiring and manufacturing needed for preclinical studies, including manufacturing registration and validation batches, as well as clinical trial materials; |
| ● | expenses to acquire technologies to be used in research and development; |
| ● | employee-related expenses, including salaries, related benefits expense for employees engaged in research and development functions; |
| ● | costs related to compliance with quality and regulatory requirements; and |
| ● | laboratory-related expenses, such as experimental equipment, consumables, antibodies, animals and cells. |
Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials and related product manufacturing expenses. We expect that our research and development expenses will increase substantially in connection with our planned clinical development activities in the near term and in the future. At this time, we cannot accurately estimate or know the nature, timing and costs of the efforts that will be necessary to complete the clinical development of any future product candidates, or if, when or to what extent we will generate revenues from the commercialization and sale of any of our product candidates that obtain regulatory approval. We may never succeed in achieving regulatory approval for any of our product candidates. The duration, costs and timing of clinical trials and development of our product candidates will depend on a variety of factors, including:
| ● | per patient trial costs; |
| ● | the number of patients that participate in the trials; |
| ● | the number of trials required for approval; |
| ● | the number of sites included in the trials; |
| ● | the countries in which the trials are conducted; |
| ● | the length of time required to enroll eligible patients; |
| ● | the number of doses that patients receive; |
| ● | the drop-out or discontinuation rates of patients; |
| ● | the duration of patient participation in the trials and follow-up periods; |
| ● | the potential for additional safety monitoring or other clinical trials requested by regulatory agencies; |
| ● | significant and changing government regulation; |
| ● | the timing and receipt of any regulatory approvals; |
| ● | the cost and timing of manufacturing our current or future product candidates; |
| ● | the phase of development of our current or future product candidates; |
| ● | the efficacy and safety profile of our current or future product candidates; and |
| ● | the number of product candidates we are developing. |
The successful development and commercialization of any future product candidates is highly uncertain. A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA, TFDA or another regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate will be required for the completion of clinical development of a product candidate or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development of a product candidate. In addition, the probability of success for each product candidate will depend on numerous factors, including competition, manufacturing capability and commercial viability. We will determine which programs to pursue and how much to fund each program in response to the scientific and clinical success of each product candidate, as well as an assessment of each product candidate’s commercial potential, due to the numerous risks and uncertainties associated with product development and commercialization, including, but not limited to, the following:
| ● | the timing and progress of nonclinical and clinical development activities; |
| ● | the number and scope of nonclinical and clinical trials for separate indications we decide to pursue; |
| ● | raising necessary additional funds; |
| ● | the progress of the development efforts of parties with whom we may enter into collaboration arrangements; |
| ● | our ability to maintain our current development activities and to establish new ones; |
| ● | our ability to establish new licensing or collaboration arrangements; |
| ● | the successful initiation and completion of clinical trials with safety, tolerability and efficacy profiles that are satisfactory to the U.S. Food and Drug Administration, or the FDA, the Taiwan Food and Drug Administration, or the TFDA, the European Medicines Agency, or the EMA, or any other comparable foreign regulatory authority; |
| ● | the receipt and related terms of regulatory approvals from applicable regulatory authorities; |
| ● | the availability of drug substances and drug products for use in production; |
| ● | establishing and maintaining agreements with third-party manufacturers for clinical supply for our clinical trials and commercial manufacturing, where a product candidate is approved; |
| ● | our ability to obtain and maintain patents, trade secret protection and regulatory exclusivity, both in the United States, Taiwan and internationally; |
| ● | our ability to protect our rights in our intellectual property portfolio; |
| ● | the commercialization of product candidates, if and when approved; and |
| ● | obtaining and maintaining third-party insurance coverage and adequate reimbursement; |
Interest income (expense), net
Interest income
Interest income consists of interest income earned on our cash and cash equivalents.
Foreign exchange gain (loss)
Gains or losses resulting from the application of different foreign exchange rates when cash in foreign currency is converted, or when foreign-currency receivables or payables are settled, are credited or charged to income in the year of conversion or settlement.
Other income
Other income mainly includes subsidy income/government grant, clinical trial drug cost fee income and director and supervisor remuneration income.
Critical Accounting Policies and Estimates
We believe that the following accounting policies are our critical accounting policies and estimates:
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with the generally accepted accounting principles in the United States of America (the “U.S. GAAP”).
Principle of Consolidation
The consolidated financial statements include the financial statements of Everfront Biotech Holding Company Limited and its 99.96% owned subsidiary, Everfront Biotech Inc. All inter-company transactions and balances are eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the amount of revenues and expenses during the reporting periods. Actual results could differ materially from those estimates under different assumptions or circumstances.
Fair Value Measurements
FASB ASC 820, “Fair Value Measurements” defines fair value for certain financial and nonfinancial assets and liabilities that are recorded at fair value, establishes a framework for measuring fair value and expands disclosures about fair value measurements. It requires that an entity measure its financial instruments to base fair value on exit price, maximize the use of observable units and minimize the use of unobservable inputs to determine the exit price. It establishes a hierarchy which prioritizes the inputs to valuation techniques used to measure fair value. This hierarchy increases the consistency and comparability of Fair Value Measurements and related disclosures by maximizing the use of observable inputs and minimizing the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that reflect the assumptions market participants would use in pricing the assets or liabilities based on market data obtained from sources independent from us. Unobservable inputs are inputs that reflect the Company’s own assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. The hierarchy prioritizes the inputs into three broad levels based on the reliability of the inputs as follows.
| | ● | Level 1 - Inputs are quoted prices in active markets for identical assets or liabilities that we have the ability to access at the measurement date. Valuation of these instruments does not require a high degree of judgment as the valuations are based on quoted prices in active markets that are readily and regularly available. |
| | ● | Level 2 - Inputs other than quoted prices in active markets that are either directly or indirectly observable as of the measurement date, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| | ● | Level 3 - Valuations based on inputs that are unobservable and not corroborated by market data. The fair value for such assets and liabilities is generally determined using pricing models, discounted cash flow methodologies, or similar techniques that incorporate the assumptions a market participant would use in pricing the asset or liability. |
The carrying values of certain of our assets and liabilities, such as cash and cash equivalents, prepaid expenses and other current assets, accounts payable, accrued liabilities, and due to related parties approximate fair value due to their relatively short maturities.
Revenue Recognition
We adopted Accounting Standards Codification (“ASC”), Topic 606 (ASC 606), Revenue from Contracts with Customers. Pursuant to ASC 606, we recognize revenue when our customer obtains control of promised goods or services, in an amount that reflects the consideration that we expect to receive in exchange for those goods or services. To determine revenue recognition for arrangements that we determine is within the scope of ASC 606, we perform the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) we satisfy a performance obligation. We only apply the five-step model to contracts when it is probable that we will collect the consideration we are entitled to in exchange for the goods or services we transfer to the customers. At inception of the contract, once the contract is determined to be within the scope of ASC 606, we assess the goods or services promised within each contract, determines those that are performance obligations, and assesses whether each promised good or service is distinct. We then recognize as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied.
During the year ended December 31, 2022, the Company’s service revenue was derived from rendering a biological research report to a related party. Revenue was recognized when the research report was delivered to the customer.
During the year ended December 31, 2023, the Company did not generate any service revenue.
Intangible Assets
Intangible assets consist of patents and acquired software. Intangible assets are initially recognized at their respective acquisition costs. All of the Company’s intangible assets have been determined to have finite useful lives and are, therefore, amortized using the straight-line method over their estimated useful lives:
| Patents and trademarks | | 15 to 30 years |
| Acquired software | | 3 years |
Impairment of Long-Lived Assets
We have adopted Accounting Standards Codification subtopic 360-10, Property, Plant and Equipment (“ASC 360-10”). ASC 360-10 requires that long-lived assets and certain identifiable intangibles held and used by us be reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. We evaluate our long-lived assets for impairment annually or more frequently if events or changes in circumstances indicate that it might be impaired. Events relating to recoverability may include significant unfavorable changes in business conditions, recurring losses, or a forecasted inability to achieve break-even operating results over an extended period. Should impairment in value be indicated, the carrying value of intangible assets will be adjusted, based on estimates of future discounted cash flows resulting from the use and ultimate disposition of the asset. ASC 360-10 also requires assets to be disposed of be reported at the lower of the carrying amount or the fair value less costs to sell.
Long-Term Equity Investment
We account for non-marketable equity and other equity investments for which we do not have control over the investees as:
| | ● | Equity method investments when we have the ability to exercise significant influence, but not control, over the investee. Our proportionate share of the income or loss is recognized monthly and is recorded in gains (losses) on equity investments. |
| | ● | Investments in entities that are not consolidated or accounted for under the equity method are recorded as investments without readily determinable fair values. Investments without readily determinable fair values are reported on the consolidated balance sheets in investments, at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for an identical or similar investment of the same issuer. |
Prior to the adoption of the Accounting Standards Update No. 2016-01 (“ASU 2016-01”), equity investments without readily determinable fair values were recorded at cost minus other-than-temporary impairment, with other-than-temporary impairment charges included in other income (expense), net in the Consolidated Statements of Operations. Following the adoption of ASU 2016-01, these investments are measured at cost adjusted for changes in observable prices minus impairment or at net asset value, as a practical expedient, if available, with changes in measurement recognized in other income (expense), net in the Consolidated Statements of Operations. We include in Investment on the Consolidated Balance Sheets equity investments without readily determinable fair values.
Research and Development Expenses
We account for the cost of using licensing rights in research and development cost according to ASC Topic 730-10-25-1. Research and development expenses are charged to expense as incurred. Research and development expenses are comprised of costs incurred in performing research and development activities, including payroll, personnel-related costs, facilities-related overhead, and outside contracted services including clinical trial costs, manufacturing and process development costs for both clinical and preclinical materials, research costs, and other consulting services. Non-refundable advance payment for goods and services that will be used in future research and development activities are expensed when the activity has been performed or when the goods have been received rather than when the payment is made. In instances where we enter into agreements with third parties to provide research and development services, costs are expensed as services are performed.
Valuation of Deferred Tax Assets
A valuation allowance is recorded to reduce our deferred tax assets to the amount that is more likely than not to be realized. In assessing the need for the valuation allowance, management considers, among other things, projections of future taxable income and ongoing prudent and feasible tax planning strategies. If we determine that sufficient negative evidence exists, then it will consider recording a valuation allowance against a portion or all of the deferred tax assets in that jurisdiction. If, after recording a valuation allowance, our projections of future taxable income and other positive evidence considered in evaluating the need for a valuation allowance prove, with the benefit of hindsight, to be inaccurate, it could prove to be more difficult to support the realization of our deferred tax assets. As a result, an additional valuation allowance could be required, which would have an adverse impact on our effective income tax rate and results. Conversely, if, after recording a valuation allowance, we determine that sufficient positive evidence exists in the jurisdiction in which the valuation allowance was recorded, it may reverse a portion or all of the valuation allowance in that jurisdiction. In such situations, the adjustment made to the deferred tax asset would have a favorable impact on our effective income tax rate and results in the period such determination was made.
Recent Accounting Pronouncements
See the discussion of recent accounting pronouncements contained in Note 2 to the consolidated financial statements, “Summary of Significant Accounting Policies”.
Results of Operations
Comparison of the Six Months Ended June 30, 2024 and 2023
The following table summarizes our results of operations for the six months ended June 30, 2024 and 2023.
| | | For the Six Months Ended June 30, | |
| | | 2024 | | | 2023 | |
| Revenue | | $ | - | | | $ | - | |
| Operating costs | | | - | | | | - | |
| Gross profit | | | - | | | | - | |
| Operating expenses | | | | | | | | |
| General and administrative expenses | | | 478,131 | | | | 594,782 | |
| Research and development expenses | | | 314,542 | | | | 389,817 | |
| Total operating expenses | | | 792,673 | | | | 984,599 | |
| Loss from operations | | | (792,673 | ) | | | (984,599 | ) |
| Non-operating income (expense) | | | | | | | | |
| Interest income (expense), net | | | 4,961 | | | | 1,526 | |
| Other income | | | - | | | | 35,056 | |
| Total non-operating income, net | | | 4,961 | | | | 36,582 | |
| Loss before income taxes | | | (787,712 | ) | | | (948,017 | ) |
| Income tax expense | | | - | | | | - | |
| Net loss | | $ | (787,712 | ) | | $ | (948,017 | ) |
Revenues
All of our products are still in development and/or clinical trial stage. Therefore, we generated no revenue from product sales in the six months ended June 30, 2024 and 2023.
Operating costs
We did not incur any cost associated with revenues during the six months ended June 30, 2024 and 2023.
General and administrative expenses
Our general and administrative expenses were $478,131 for the six months ended June 30, 2024 as compared to $594,782 for the six months ended June 30, 2023. The decrease in general and administrative expenses in the amount of $116,651, or 19.61%, primarily resulted from most of the professional service fees and consulting fees required for our initial public offering being paid in 2023, resulting in lower expenses in 2024.
Research and development expenses
Our research and development expenses were $314,542 for the six months ended June 30, 2024, as compared to $389,817 for the six months ended June 30, 2023. The decrease in research and development expenses in the amount of $75,275, or 19.31%, was primarily driven by the decrease in Cerebraca™ Wafer Phase I/IIa clinical trial expenses. These expenses commenced in fiscal year 2021, gradually decreased in fiscal year 2022 and 2023, and further decreased in fiscal year 2024. The decrease in expenses for Cerebraca™ Wafer’s Phase I/IIa study can be attributed to completed patient recruitment, which has led to reduced activities at both hospital sites and at our CRO. The table below sets forth the approximate research and development costs for each product for each of the six months ended June 30, 2024, the years ended December 31, 2023 and 2022, as well as for periods before January 1, 2022.
| | | For the Six
Months Ended
June 30, | | | For the Year Ended
December 31, | | | Prior to
January 1, | | | | |
| Product | | 2024 | | | 2023 | | | 2022 | | | 2022 | | | Total | |
| EF-001 | | $ | 180,000 | | | $ | 100,000 | | | $ | 200,000 | | | $ | 5,700,000 | | | $ | 6,000,000 | |
| EF-009 | | | 20,000 | | | | 50,000 | | | | 20,000 | | | | 370,000 | | | | 440,000 | |
| EF-031 | | | 20,000 | | | | 50,000 | | | | 30,000 | | | | 240,000 | | | | 320,000 | |
| HK-001 | | | 5,000 | | | | 100,000 | | | | 430,000 | | | | 1,870,000 | | | | 2,400,000 | |
| TSCA-001 | | | 15,000 | | | | 20,000 | | | | 20,000 | | | | 220,000 | | | | 260,000 | |
| EF-011 | | | 5,000 | | | | 10,000 | | | | 10,000 | | | | 80,000 | | | | 100,000 | |
| EF-005 | | | 10,000 | | | | 20,000 | | | | 10,000 | | | | 90,000 | | | | 120,000 | |
| EF-002 | | | 5,000 | | | | 10,000 | | | | 10,000 | | | | 70,000 | | | | 90,000 | |
| Total | | $ | 260,000 | | | $ | 360,000 | | | $ | 730,000 | | | $ | 8,640,000 | | | $ | 9,730,000 | |
Interest income (expense), net
Our interest income (expense), net was $4,961 for the six months ended June 30, 2024, as compared to $1,526 for the six months ended June 30, 2023. The increase in net interest income (expense), net in the amount of $3,435, or 225.10%, was primarily driven by an increase in interest income on bank deposits.
Other income
Our other income was $0 for the six months ended June 30, 2024, as compared to $35,056 for the six months ended June 30, 2023. The decrease in other income in the amount of $35,056, or 100.00%, was primarily caused there being no other income in 2024.
Net loss
As a result of the above factors, our net loss was $787,712 and $948,017 for the six months ended June 30, 2024 and 2023, respectively. The decrease in net loss for the six months ended June 30, 2024 as compared to the six months ended June 30, 2023 was $160,305, or 16.91%.
Comparison of the Years Ended December 31, 2023 and 2022
The following table summarizes our results of operations for the years ended December 31, 2023 and December 31, 2022.
| | | For the Year Ended December 31, | |
| | | 2023 | | | 2022 | |
| Revenue | | $ | - | | | $ | 79,908 | |
| Operating costs | | | - | | | | - | |
| Gross profit | | | - | | | | 79,908 | |
| Operating expenses | | | | | | | | |
| General and administrative expenses | | | 1,678,178 | | | | 726,551 | |
| Research and development expenses | | | 707,692 | | | | 1,057,351 | |
| Total operating expenses | | | 2,385,870 | | | | 1,783,902 | |
| Loss from operations | | | (2,385,870 | ) | | | (1,703,994 | ) |
| Non-operating income (expense) | | | | | | | | |
| Interest income (expense), net | | | 9,969 | | | | 76 | |
| Foreign exchange gain (loss) | | | - | | | | 1,442 | |
| Other income | | | 82,704 | | | | 207,882 | |
| Total non-operating income, net | | | 92,673 | | | | 209,400 | |
| Loss before income taxes | | | (2,293,197 | ) | | | (1,494,594 | ) |
| Income tax expense | | | - | | | | - | |
| Net loss | | $ | (2,293,197 | ) | | $ | (1,494,594 | ) |
Revenues
All of our products are still in development and/or clinical trial stage. Therefore, we generated no revenue from product sales in the year ended December 31, 2023, and the revenue of $79,908 recorded in the year ended December 31, 2022 was the service revenue from the fibroblast culture process research commissioned by the related party Xi Yue Biomedicine Inc. (formerly Guang-Li Biomedicine Inc.).
Operating costs
We did not incur any costs associated with revenues during the years ended December 31, 2023 and 2022.
General and administrative expenses
General and administrative expenses were $1,678,178 for the year ended December 31, 2023 as compared to $726,551 for the year ended December 31, 2022. The increase in general and administrative expenses in the amount of $951,627 or 130.98% in the year ended December 31, 2023 was primarily caused by the increase in professional service fees and consulting fees for our initial public offering.
Research and development expenses
Research and development expenses were $707,692 for the year ended December 31, 2023 as compared to $1,057,351 for the year ended December 31, 2022. The decrease in research and development expenses in the amount of $349,659 or 33.07% in the year ended December 31, 2023, was primarily caused by the decrease in Cerebraca™ Wafer Phase I/IIa clinical trial expenses, which expenses commenced in fiscal year 2021, gradually decreased in fiscal year 2022, and further decreased in fiscal year 2023. The table below sets forth the approximate research and development costs for each product for each of the years ended December 31, 2023 and 2022, as well as for periods before January 1, 2022.
| Product | | 2023 | | | 2022 | | | Before 2022 | | | Total | |
| EF-001 | | $ | 100,000 | | | $ | 200,000 | | | $ | 5,700,000 | | | $ | 6,000,000 | |
| EF-009 | | | 50,000 | | | | 20,000 | | | | 370,000 | | | | 440,000 | |
| EF-031 | | | 50,000 | | | | 30,000 | | | | 240,000 | | | | 320,000 | |
| HK-001 | | | 100,000 | | | | 430,000 | | | | 1,870,000 | | | | 2,400,000 | |
| TSCA-001 | | | 20,000 | | | | 20,000 | | | | 220,000 | | | | 260,000 | |
| EF-011 | | | 10,000 | | | | 10,000 | | | | 80,000 | | | | 100,000 | |
| EF-005 | | | 20,000 | | | | 10,000 | | | | 90,000 | | | | 120,000 | |
| EF-002 | | | 10,000 | | | | 10,000 | | | | 70,000 | | | | 90,000 | |
| Total | | $ | 360,000 | | | $ | 730,000 | | | $ | 8,640,000 | | | $ | 9,730,000 | |
Interest income (expense), net
Interest income (expense), net was $9,969 for the year ended December 31, 2023, as compared to $76 for the year ended December 31, 2022. The increase in net interest income in the amount of $9,893 in the year ended December 31, 2023 was primarily caused by the increase in interest income on bank deposits.
Foreign exchange gain (loss)
Foreign exchange gain (loss) was $0 for the year ended December 31, 2023, as compared to $1,442 for the year ended December 31, 2022. This decrease in foreign exchange gain was mainly attributable to the absence of foreign currency accounts payable for the year ended December 31, 2023.
Other income
Other income was $82,704 for the year ended December 31, 2023, as compared to $207,882 for the year ended December 31, 2022. The decrease in other income in the amount of $125,178, or 60.22%, in the year ended December 31, 2023 was primarily caused by the decrease in research and development expenses and a decrease in related subsidy income and government grants.
Net loss
As a result of the above factors, our net loss was $2,293,197 and $1,494,594 for the years ended December 31, 2023 and 2022, respectively. The increase in net loss for the year ended December 31, 2023 as compared to the same period ended December 31, 2022 was $798,603, or 53.43%.
Liquidity and Capital Resources
Working Capital Summary
| | | As of June 30,
2024 | | | As of December 31, 2023 | |
| Current Assets | | $ | 867,975 | | | $ | 1,690,317 | |
| Current Liabilities | | | 2,645,751 | | | | 2,679,458 | |
| Working Capital | | $ | (1,777,776 | ) | | $ | (989,141 | ) |
Our working capital was a negative $1,777,776 as of June 30, 2024, which mainly reflects our convertible promissory notes. Since our inception, we have been, and currently remain, in the research and development stage, and we currently have no approved products for sale. We have incurred losses and accumulated negative cash flows from operations. As of June 30, 2024, we had an accumulated deficit of $20,789,569. We expect that our research and development expenses and general and administrative expenses will continue to increase and, as a result, we will need additional capital to fund our operations, which we may raise through equity and/or debt financings. We may also consider collaborations or selectively partnering our technology or programs.
Cash Flows
The following table summarizes our sources and uses of cash for the six months ended June 30, 2024 and 2023.
| | | For the Six Months Ended June 30, | |
| | | 2024 | | | 2023 | | | $ Change | | | % Change | |
| Net cash used in operating activities | | $ | (785,070 | ) | | $ | (1,196,133 | ) | | $ | 441,063 | | | | (34.4 | )% |
| Net cash used in investing activities | | | (320 | ) | | | (31,700 | ) | | | 31,380 | | | | (99.0 | )% |
| Net cash provided by financing activities | | | - | | | | 893,120 | | | | (893,120 | ) | | | (100.0 | )% |
| Effect of exchange rate changes on cash and cash equivalents | | | (2,237 | ) | | | (4,251 | ) | | | 2,014 | | | | (47.4 | )% |
| | | | | | | | | | | | | | | | | |
| Net decrease in cash and cash equivalents | | $ | (787,627 | ) | | $ | (338,964 | ) | | $ | (448,663 | ) | | | 132.4 | % |
Cash Flow from Operating Activities
For the six months ended June 30, 2024, operating activities used $785,070 of cash, primarily resulting from our net loss of $787,712 and net cash used by changes in our operating assets and liabilities of $22,616, partially offset by depreciation of $4,766, amortization of $17,542 and operating lease expense of $3,040. Net cash used by changes in our operating assets and liabilities primarily consisted of a $33,521 decrease in accrued expenses, mainly due to the payment of experimental consumables and patent fees for research and development payable and accrued in 2023, as well as the payment of accrued wages and related liabilities, a decrease in other receivables of $4,702, and an decrease in due from related party of $7,664.
For the six months ended June 30, 2023, operating activities used $1,196,133 of cash, primarily resulting from our net loss of $948,017 and net cash used by changes in our operating assets and liabilities of $271,829, partially offset by depreciation of $4,287, amortization of $16,504 and operating lease expense of $2,922. Net cash used by changes in our operating assets and liabilities primarily consisted of a $47,714 decrease in accrued expenses mainly due to the payment of experimental consumables and patent fees for research and development payable and accrued in 2021, as well as the payment of accrued wages and related liabilities, and a $219,640 increase in other receivables, which was primarily due to the Company entering into securities purchase agreements with accredited investors on June 23, 2023, pursuant to which the Company issued convertible promissory notes of which $200,000 of the accredited investors’ investment amount had not yet been collected.
Cash Flows from Investing Activities
Net cash used in investing activities for the six months ended June 30, 2024 was $320, primarily due to an increase in refundable deposits for renting an office.
Net cash used in investing activities for the six months ended June 30, 2023 was $31,700 and consisted primarily of purchase of equipment of $32,191 offset by a $491 decrease in other assets, which was primarily due to decreased refundable deposits for renting an office.
Cash Flows from Financing Activities
For the six months ended June 30, 2024, the net cash provided by financing activities was $0 as there were no financing activities for the six months ended June 30, 2024.
Net cash provided by financing activities for the six months ended June 30, 2023 was $893,120 and consisted primarily of (i) on June 23, 2023, the Company entered into several convertible promissory notes with accredited investors in the aggregate principal amount of $400,000, and (ii) the Company sold an aggregate of 792,295 ordinary shares of the Company, at $2.00 per share, pursuant to certain share purchase agreements. For the six months ended June 30, 2023, the total amount of subscription received was $493,120.
The following table summarizes our sources and uses of cash for the years ended December 31, 2023 and 2022.
| | | For the Year Ended December 31, | |
| | | 2023 | | | 2022 | | | $ Change | | | % Change | |
| Net cash used in operating activities | | $ | (2,279,370 | ) | | $ | (1,924,002 | ) | | $ | (355,368 | ) | | | 18.47 | % |
| Net cash used in investing activities | | | (44,930 | ) | | | (3,255 | ) | | | (41,675 | ) | | | 1,280.34 | % |
| Net cash provided by financing activities | | | 3,008,883 | | | | 2,455,395 | | | | 553,488 | | | | 22.54 | % |
| Effect of exchange rate changes on cash and cash equivalents | | | (12,162 | ) | | | (3,311 | ) | | | (8,851 | ) | | | 267.32 | % |
| | | | | | | | | | | | | | | | | |
| Net increase in cash and cash equivalents | | $ | 672,421 | | | $ | 524,827 | | | $ | 147,594 | | | | 28.12 | % |
Cash Flow from Operating Activities
For the year ended December 31, 2023, operating activities used $2,279,370 of cash, primarily resulting from our net loss of $2,293,197 and net cash used by changes in our operating assets and liabilities of $34,856, partially offset by depreciation of $9,169, amortization of $33,791 and operating lease expense of $5,723. Net cash used by changes in our operating assets and liabilities primarily consisted of an increase in due from related party of $7,848, an increase in other receivables of $4,815, and an increase in prepaid value-added tax, or VAT tax, of $11,762.
For the year ended December 31, 2022, operating activities used $1,924,002 of cash, primarily resulting from our net loss of $1,494,594 and net cash used by changes in our operating assets and liabilities of $474,339, partially offset by depreciation of $4,753, amortization of $34,207 and operating lease expense of $5,971. Net cash used by changes in our operating assets and liabilities primarily consisted of a $367,335 decrease in accrued expenses mainly due to the payment for experimental consumables and patent fees for research and development payable and accrued in 2021, as well as the payment of accrued wages and related liabilities, and a $83,903 decrease in advance from customer, which was primarily due to decreased advance of the fibroblast culture process research commissioned by the related party Xi Yue Biomedicine Inc. (formerly Guang-Li Biomedicine Inc.).
Cash Flow from Investing Activities
Net cash used in investing activities for the year ended December 31, 2023 was $44,930 and consisted primarily of purchase of equipment of $27,299, acquisition of intangible assets of $5,490 and a $12,141 increase in other assets, which was primarily due to increased leasehold improvements.
Net cash used in investing activities for the year ended December 31, 2022 was $3,255 and consisted primarily of purchase of equipment of $2,752 and a $503increase in other assets, which was primarily due to increased refundable deposits for renting an office.
Cash Flow from Financing Activities
Net cash provided by financing activities for the year ended December 31, 2023 was $3,008,883 and consisted primarily of (i) from June 23, 2023 until July 24, 2023, we entered into several convertible promissory notes with accredited investors in the aggregate principal amount of $2,516,000, and (ii) sales of 792,295 Ordinary Shares of the Company, at $2.00 per share, pursuant to certain share purchase agreements. For the year ended December 31, 2023, the total amount of capital contribution was $499,810.
Net cash provided by financing activities for the year ended December 31, 2022 was $2,455,395 and consisted primarily of (i) sales of 1,020,000 Ordinary Shares for a capital contribution of $1,067,802, (ii) receipt of $298,467 proceeds from related parties, and (iii) sales of 792,295 Ordinary Shares at $2.00 per share, pursuant to certain share purchase agreements. For the year ended December 31, 2022, the aggregate amount of subscription received in advance amounted to $1,089,126.
Funding Requirements
Our primary uses of capital are, and we expect will continue to be, compensation and related expenses, clinical research and development services, laboratory expenses, regulatory expenses, marketing, and general and administrative expenses. Based on our research and development plans and our timing expectations related to the progress of our programs, we expect that the net proceeds from this offering, together with our existing cash and cash equivalents as of December 31, 2023, will enable us to fund our operations for at least the next 18 months. We have based this estimate on assumptions that may prove to be wrong, and we could use our available capital resources sooner that we currently expect. Furthermore, our operating plan may change, and we may need additional funds sooner than planned.
The successful development of any product candidate is highly uncertain. We expect our expenses to increase substantially in connection with our ongoing activities, particularly as we advance the preclinical and clinical activities, manufacturing and clinical trials of Cerebraca™ Wafer Phase IIb/III and any future product candidates and prepare for the commercial launch of Cerebraca™ Wafer, if approved. In addition, upon the closing of this offering, we expect to incur additional costs associated with operating as a public company, including significant legal, accounting, investor relations and other expenses that we did not incur as a private company. As such, at this time, we cannot reasonably estimate or know the nature, timing and costs of the efforts that will be necessary to complete the development of our other current and future product candidates. We are also unable to predict exactly when, if ever, material net cash inflows will commence from the sale of approved product candidates. This is due to numerous risks and uncertainties in the development of our product candidates, including uncertainties in the following areas:
| | ● | the progress, timing, costs and results of our clinical trial for Cerebraca™ Wafer Phase IIb/III; |
| | ● | conducting preclinical studies and clinical trials for potential future product candidates; |
| | ● | the outcome, timing and cost of regulatory approvals by the FDA, TFDA and other foreign regulatory authorities, including the potential for the FDA, TFDA or other foreign regulatory authorities to require that we perform more studies than expected; |
| | ● | the ability of our product candidates to progress through clinical development successfully; |
| | ● | the cost of filing, prosecuting, defending and enforcing any of our patent claims and other intellectual property rights; |
| | ● | arrangements with third-party service providers and manufacturers; |
| | ● | hiring and retaining additional personnel, such as non-clinical, clinical, quality assurance, regulatory affairs, manufacturing, legal, compliance, finance, general and administrative, commercial and scientific personnel; |
| | ● | our need to implement additional infrastructure and internal systems; |
| | ● | the effect of competing technological and market developments; and |
| | ● | transitioning our organization to being a public company. |
Following this offering, we will be a publicly traded company and will incur significant legal, accounting and other expenses that we were not required to incur as a private company. In addition, the Sarbanes-Oxley Act of 2002, as well as rules adopted by the SEC and Nasdaq, require public companies to implement specified corporate governance practices that are currently not applicable to us as a private company. Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, we will first be required to furnish a report by our management on our internal control over financial reporting. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. We expect these rules and regulations will increase our legal, financial, and other compliance costs and will make some activities more time-consuming and costly.
A change in the outcome of any of these variables with respect to the development of any of our product candidates would significantly change the costs and timing associated with the development of that product candidate.
Until we can generate a sufficient amount of revenue from our products, if ever, we expect to finance future cash needs through equity and/or debt financing. We may also consider new collaborations or selectively partnering our technology or programs. Additional capital may not be available on reasonable terms, if at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our product candidates. If we raise additional funds through the issuance of additional equity or debt securities, it could result in dilution to our existing shareholders and increased fixed payment obligations, and these securities may have rights senior to those of our Ordinary Shares. If we incur indebtedness, we could become subject to covenants that would restrict our operations and potentially impair our competitiveness, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Any of these events could significantly harm our business, financial condition and prospects.
Contractual Obligations and Commitments
The following table summarizes our contractual obligations and commitments as of June 30, 2024 that will affect our future liquidity.
| | | Total | | | Less than 1 Year | | | 1-3 Years | | | 3-5 Years | | | More than 5 Years | |
| Convertible promissory notes payable(1) | | $ | 2,516,000 | | | $ | 2,516,000 | | | $ | — | | | $ | — | | | $ | — | |
| Operating lease liability | | | 13,359 | | | | 6,631 | | | | 6,728 | | | | — | | | | — | |
| Total | | $ | 2,529,359 | | | $ | 2,522,631 | | | $ | 6,728 | | | $ | — | | | $ | — | |
| (1) | From June 23, 2023 until July 24, 2023, we entered into securities purchase agreements with accredited investors pursuant to which we issued convertible promissory notes, and on June 24, 2024 and in December 2024, we entered into amendments to the convertible promissory notes to extend their maturity. As amended, each note matures on the earlier of (i) the date that is 24 months after the issuance date of the note, (ii) the date when we redeem the note at its outstanding principal amount and (iii) the date of the initial closing of our IPO. The notes accrue interest at 2.40% per annum and may be prepaid by us at any time without any penalties, provided we give prior written notice to the note holder at least ten business days before prepayment. Any outstanding and unpaid principal amount of the notes automatically convert upon the commencement of trading on the Nasdaq Stock Market or the New York Stock Exchange of our Ordinary Shares to be issued in an IPO at a conversion price equal to the lesser of (i) the price equal to a 30% discount to the actual price per ordinary share to be issued in our IPO, or (ii) $4.00 per share. |
As of June 30, 2024, we had unrecorded commitments under contract of $5,077,395. These commitments primarily consist of the transfer and licensing of intellectual property and technology of $3,830,508 and R&D outsourcing services of $1,246,887.
Off-Balance Sheet Arrangements
We did not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures, or capital resources that is material to investors.
BUSINESS
Overview
We value sustainable development and are committed to providing more promising treatment solutions for patients. Our mission is to construct effective strategies for combating hard-to-treat diseases such as glioblastoma (GBM), pancreatic cancer (PC), and amyotrophic lateral sclerosis (ALS). We designed the implantable Cerebraca™ Wafer for in-situ drug delivery. After surgical resection, implantation of the wafer at the tumor margin is designed to create a localized high drug concentration, targeting residual and or unresectable tumor cells. Additionally, the wafer’s sustained release technology is designed to facilitate continuous drug release within the brain for one month. We also developed soft-gel capsules designed to be easily swallowed as maintenance therapy following Cerebraca™ Wafer implantation. Furthermore, we are exploring the feasibility of expanding our treatment approach to pancreatic cancer. By establishing and directing the four product pipelines listed in the table below, we believe we will be able to achieve our goal.
| Product | | Formulation | | Indication | | Status | | Orphan Drug Designation | | Global Market |
| Cerebraca™ Wafer | | Implantable wafer | | GBM | | Phase I/IIa clinical trial completed (GBM patients) | | Granted | | $4 Billion/year CAGR=9%(1) |
| EF-009 Wafer | | Implantable wafer | | PC | | IND approval; Phase I/IIa clinical study patient enrollment in Q2 2025 (PC patients) | | Potential | | $8 Billion/year CAGR=9.4%(2) |
| HK-001 | | Soft-gel capsule | | ALS | | Phase I clinical trial ongoing (healthy subjects for safety profiles) | | Potential | | $1 Billion/year CAGR=13.9%(3) |
| EF-031 | | Soft-gel capsule | | GBM & PC | | IND submission in Q2 2025 (GBM and PC patients) | | Potential | | $12 Billion/year CAGR=9.3%(4) |
(1) Source: Fortune Business Insights Report, Brain Tumor Drugs, Global Market Analysis, Insights and Forecast, 2022-2029.
(2) Source: Grand View Research, Global Pancreatic Cancer Treatment Market Analysis and Segment Forecasts to 2025.
(3) Source: GlobalData, Amyotrophic Lateral Sclerosis (ALS) – Opportunity Analysis and Forecasts to 2029.
(4) Source: Fortune Business Insights Report, Brain Tumor Drugs, Global Market Analysis, Insights and Forecast, 2022-2029; and Grand View Research, Global Pancreatic Cancer Treatment Market Analysis and Segment Forecasts to 2025.
As of the date of this prospectus, we are conducting three clinical trials in Taiwan based on investigational new drug (IND) approvals from both the United States Food and Drug Administration (the “FDA”) and the Taiwan Food and Drug Administration (the “TFDA”). Additionally, we expect to submit an IND for EF-031 in the second quarter of 2025.
Cerebraca™ Wafer is a biodegradable implant produced according to Pharmaceutical Inspection Co-operation Scheme, or PIC/S, Good Manufacturing Practice, or GMP, standards. Cerebraca™ Wafer has been designed to treat glioblastoma (GBM) by directly implanting it into the GBM tumor margin after surgical resection. Cerebraca™ Wafer is characterized by its 30-day sustained drug release and 5-cm penetration depth observed in preclinical studies (IND128388, Module 2, Report 51904-12-708), with the intention that this will create a high drug concentration environment for treating the cancer. Cerebraca™ Wafer is a multi-functional drug with the potential to inhibit receptor tyrosine kinases (Epidermal Growth Factor Receptor, or EGFR, Axl-1 Receptor, and c-Mesenchymal-Epithelial Transition Factor Receptor, or cMet Receptor) to halt cancer stem cell proliferation. Additionally, it has the potential to improve patients’ quality of life by boosting Adenosine Triphosphate, or ATP, levels through Axl-mTOR (mammalian target of rapamycin) (Liu C-A, et al., Journal of Oncology. 2022; 2022.) inhibition and AMP-activated Protein Kinase, or AMPK, activation (Lee J-H, et al., International Journal of Molecular Science. 2021; 22(12): 6339.). Furthermore, Cerebraca™ Wafer has been designed to promote methylation of O6-Methylguanine-DNA Methyltransferase, or MGMT, and Programmed Cell Death Ligand 1, or PD-L1, promoters in order to help overcome chemotherapeutic resistance (Liu C-A, et al., Cancers. 2022; 14(4): 1051.) and transform a cold tumor microenvironment into an immune-active hotspot (Liu C-A, et al., Journal of Oncology. 2022; 2022.).
In the Phase I/IIa clinical trial, recurrent GBM patients with receptor tyrosine kinase markers (EGF receptor, Axl-1 receptor, and cMet receptor) who received surgical resection of GBM and implantation of the Cerebraca™ Wafer followed by temozolomide therapy showed a median overall survival of 26.2 months in the preliminary results. However, limited reliable biomarkers currently exist for GBM diagnosis and treatment selection. Identifying these biomarkers and developing accurate tests to detect them is crucial for personalized medicine but very challenging. In our next clinical study, we will recruit GBM patients with IDH wild-type (National Comprehensive Cancer Network, or NCCN, Guidelines Central Nervous System Cancers, Version 1.2024), and we will analyze biomarkers in a subgroup analysis. While the preliminary results of the Phase I/IIa clinical trial are encouraging, extending the median overall survival beyond 26.2 months will require biomarker identification before surgery, which may pose significant scientific and clinical challenges. This difficulty is multifaceted and can be attributed to several key factors.
| ● | Challenge of Companion Diagnostics Development: There is currently no approved Companion Diagnostics for GBM treatment or diagnosis. |
| ● | Limited Reliable Biomarkers: One of the primary challenges in GBM treatment is the scarcity of reliable biomarkers for diagnosis and treatment selection. |
| ● | Heterogeneity of GBM: Identifying and targeting the specific molecular pathways involved in each patient’s tumor remains a significant challenge. |
| ● | Treatment Resistance Mechanisms: The ability of GBM to adapt and resist treatment poses a significant hurdle in extending survival times further. |
EF-009 Wafer is a biodegradable implant produced according to PIC/S GMP standards that is designed to treat pancreatic cancer by directly implanting it into the pancreatic cancer tumor margin (IND145153, Module 4, Report D06), which is intended to lead to local tumor regression. This neo-adjuvant therapy may allow patients with locally advanced pancreatic cancer to undergo tumor resection therapy. EF-009 has been designed to work by regulating DNA-methyltransferase 1, or DNMT1, and its downstream targets, Patched Domain-Containing 4, or PTCHD4, and hedgehog signaling (Huang M-H, et al., Pharmacological Research. 2019; 139: 50.), in order to help reduce pancreatic cancer growth. Additionally, our preclinical studies have demonstrated that EF-009 can have synergistic effects with existing chemotherapy drugs, including Gemcitabine, Cisplatin, 5-Fluorouracil, Irinotecan, Oxaliplatin, and Paclitaxel (Patents for cancer treatment, p.63, a combination for the treatment of cancer and its application), implying that combining EF-009 with these currently available chemotherapy drugs may help achieve improved clinical outcomes. We have received approval from both the USFDA and the TFDA to conduct a Phase I/IIa clinical study to evaluate the safety profile and therapeutic effects on patients with locally advanced pancreatic cancer. We have completed site selection for this study and expect to begin enrolling patients in the second quarter of 2025. Nevertheless, we will likely need to conduct a proof-of-concept study in humans to reproduce the effects observed in preclinical research. Other risks associated with using the EF-009 Wafer implant may include local inflammation caused by laparoscopic intervention and investigational product degradation, as well as surgical procedures, infection, wound bleeding, and anesthesia.
EF-031 is a soft-gel capsule formulation produced according to PIC/S GMP standards and designed to serve as a second-generation product to support the actions of the Cerebraca™ Wafer and the EF-009 Wafer. Unlike the Cerebraca™ Wafer and the EF-009 Wafer, which require surgical implantation, EF-031 can be administered orally. In a study conducted by Pharmaron, a contract research organization (study report 51904-15-393), systemic oral administration of EF-031 revealed a 5.6 times higher concentration of EF-031 in the brain and a 64.2 times higher concentration in the pancreas compared to a plasma exposure.
HK-001 is a soft-gel capsule formulation produced according to PIC/S GMP standards and designed for the treatment of Amyotrophic Lateral Sclerosis (ALS). Animal study results indicate that administration of HK-001 may help prolong the survival of ALS animals and delay disease progression (Hsueh K-W, et al., Neuropharmacology. 2016; 108: 152.). A Phase I clinical trial on healthy subjects is ongoing to determine the maximum tolerated dose (MTD). Nevertheless, we will likely need to conduct a proof-of-concept study in humans to replicate the effects observed in preclinical research.
Our Approach
We have focused our drug development strategy on three key areas.
| ● | Systematic Integration Strategy: We operate on a semi-virtual model. Since inception we have built a core management team with in-house expertise in new drug development; worked with outsourced contract research organizations (“CROs”) and contract manufacturing organizations (“CMOs”) to accelerate product development without allocating resources to non-core activities and superfluous manpower; and used Phase I/II clinical trials to establish proof of concept and secure global authorizations. |
| ● | Drugs Candidate Proof-of-Concept Model: We perform in-vitro drug testing to accelerate the proof-of-concept process for drug candidates. The established cell models include patient-derived cancer cell lines, 3D spheroids, and induced pluripotent stem cell (iPSC)-based neurodegenerative disease models. Furthermore, we leverage in-vivo animal platforms across models for diseases such as orthotopic cancer, subcutaneous cancer, spinocerebellar ataxia, amyotrophic lateral sclerosis, and liver cirrhosis. |
| ● | Strong IP Portfolio: We aggressively pursue core patents across jurisdictions for our treatments for cancer, neurodegenerative diseases, pulmonary fibrosis, and liver cirrhosis. Our commercial success hinges on our ability to secure and sustain intellectual property and proprietary protection for our current and future product candidates, as well as for novel discoveries, product development technologies, and know-how. Equally crucial is our ability to operate without infringing on others’ proprietary rights and to prevent infringement on our own. To this end, we adopt a comprehensive IP strategy that not only secures initial protection but also manages the entire intellectual property life cycle effectively. A key aspect of our strategy includes leveraging the benefits of the Orphan Drug Act, which provides additional market exclusivity beyond standard IP protection. This designation is particularly important for rare diseases, as it incentivizes the development of treatments by offering extended periods of exclusivity, thereby enhancing our market position and commercial potential. Our policy is to develop and maintain our proprietary position by filing or in-licensing U.S. and foreign patents and applications related to our technology, inventions, and improvements crucial to our business development and implementation. We continuously seek to enhance and expand our substantive business activities and strategic collaborations through the acquisition of patents that bolster our intellectual property portfolio. Our strategies for ongoing IP life cycle management include periodic review, updating, and enforcement of IP assets to align with our evolving business objectives and market dynamics. Additionally, we rely on trademarks, trade secrets, know-how, continuing technological innovation, confidentiality agreements, and invention assignment agreements to develop and maintain our proprietary position. By integrating these measures with our comprehensive IP strategy and emphasizing the importance of orphan drug designation, we strengthen our ability to protect and commercialize our innovative treatments effectively. |
Our History and Team
Everfront Biotech was founded in 2010 with a mission: construct strategies for combating hard-to-treat diseases such as glioblastoma (GBM), pancreatic cancer, and amyotrophic lateral sclerosis. In 2023, over 14,000 Americans received a GBM diagnosis, highlighting the significance of this disease. It is now recognized that intratumor genetic heterogeneity and phenotypic diversity are key factors in GBM, influencing tumor development, progression, and treatment response. Using currently approved standard-of-care treatment methods on GBM patients, despite initial therapy success, the cancer often recurs within a short period of time, leading to short overall survival with a very poor quality of life. Throughout the past decade, no new FDA-approved drug for GBM entered the market, but Everfront Biotech remained committed to its original goal, advancing in new drug development.
We developed as our first potential product Cerebraca™ Wafer, an implantable drug with sustained-release properties, showing promising potential when combined with brain tumor surgery. Phase I/IIa clinical trials for recurrent GBM revealed impressive survival times, with no serious adverse reactions reported. These results support the potential of not only extending patient survival but also improving their quality of life post-treatment, representing a potentially revolutionary solution for neurosurgeons and GBM patients alike. Committed to sustainable development, we are also developing soft-gel capsules designed to be easily swallowed as maintenance therapy following Cerebraca™ Wafer implantation. Additionally, we are exploring the possibility of extending this treatment approach to pancreatic cancer, encompassing both implantable wafers and soft-gel capsules.
Everfront Biotech Holding Company Limited, is the holding company of our operating subsidiary, Everfront Biotech Inc., which was incorporated on September 21, 2022 in the British Virgin Islands by President Ho-Ching Chen, with Dr. Chou Pei-Wen serving as Chairman and General Manager. Dr. Shinn-Zong Lin, Dr. Horng-Jyh Harn, Dr. Tzyy-Wen Chiou and Dr. Kwei-Hang Chan currently serve on our scientific advisory board. Dr. Tza-Zen Chaung has served as CEO of Everfront Biotech Holding Company Limited since January 2023.
Our team is our most important asset. We have brought together a diverse team of experienced individuals, including experienced board members, leading scientists, and knowledgeable advisors, to effectively execute our business plan. Our management team has significant experience in cancer research and in progressing products from early-stage research to clinical trials, and ultimately to regulatory approval and commercialization. We embrace a team-work spirit, where our members work cohesively to ensure the smooth progress of every project. Their hard work is driven by a strong passion and commitment to fulfilling unmet medical needs. With expertise and experience, our expert leadership guides the way in new drug development. In Taiwan, the National Health Insurance covers all of Taiwan’s population, facilitating the efficient identification of potential subjects for clinical trials. Moreover, the adoption of good clinical practice in all medical centers ensures the presence of highly qualified biomedical researchers and doctors of medicine (“MDs”). However, Phase IIb/III clinical trials in the United States will be critical to our success.
Our Founder and Director, Mr. Ho-Ching Chen, brings extensive industry investment experience and has made significant investments across diverse sectors, including leisure, semiconductor, environmental protection, and the biotechnology and pharmaceutical industries, resulting in successful outcomes. Our Chief Executive Officer, Dr. Chaung, brings over 40 years of experience in new drug development, process development, and strategic planning. He has held key positions at renowned global pharmaceutical companies such as Merck & Company, Hoffmann-La Roche, and Bristol-Myers Squibb. Additionally, he has provided professional consulting services to the pharmaceutical and biotechnology industries in the United States and Taiwan. Our leading scientists include Dr. Jen-Wei Liu and Dr. Yuan-Sheng Li, who have extensive experience in leading teams to advance licensed patents from the research stage to the clinical stage. They oversee continuous improvements in areas such as preclinical experiments, drug formulation and manufacturing processes, drug applications, and clinical trial planning.
Our advisory team consists of highly experienced scientists with extensive academic, medical, and industry expertise including the best neurosurgeon and superintendent of Tzu-Chi medical center, Professor Shinn-Zong Lin, the pathology, toxicology and pharmacology consultant, Professor Horng-Jyh Harn, and the CMC and translational medicine consultant Professor Tzyy-Wen Chiou. Some of our company’s patents are licensed from the advisors’ groundbreaking inventions in their respective domains, which have been transferred to our company through technology transfer agreements. Throughout the research and development process, our advisors continue to provide ongoing professional advice, enabling us to advance our pipeline of new drug development and expand into niche therapeutic areas. We plan to deepen our collaboration and jointly create more innovative breakthroughs in the pharmaceutical and biotechnology sectors.
Our Pipeline
We have four products under development: the implantable Cerebraca™ Wafer for the treatment of brain tumors, the implantable EF-009 Wafer for the treatment of pancreatic cancers, a soft-gel capsule for the treatment of solid tumors (such as brain tumors and pancreatic cancer), and a soft-gel capsule for the treatment of ALS. We also continue to work to fill our pipeline and have several early stage potential product candidates with IND-enabling status.
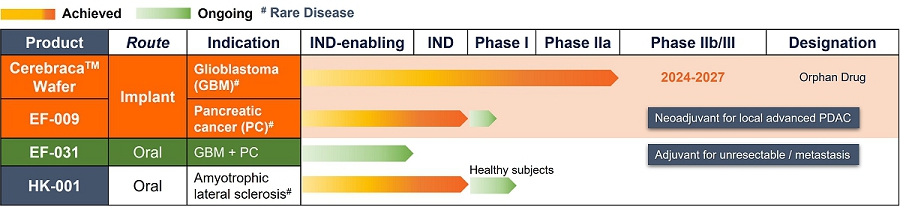
Cerebraca™ Wafer for the Treatment of Glioblastoma
Cerebraca™ Wafer is an implantable formulation carrying the active pharmaceutical ingredient (EF-API-001) using a biodegradable polymer that was designed to treat glioblastoma. It was designed to be directly implanted into the brain after surgical resection of the tumor and sustainably releases drugs over a 30-day period. The intention is to create a local high drug concentration environment for treating the cancer by leveraging the anti-tumor activity of EF-API-0015, inhibiting cancer cell immune activation in situ, and reducing chemotherapy resistance.
We have invested approximately $6 million to develop the Cerebraca™ Wafer to its current stage. A Pre-IND meeting with the FDA was conducted in 2013 to acquire critical opinions on the development strategy for Cerebraca™ Wafer from the regulatory authority. Based on the discussions, the GLP toxicology study was performed, and the PIC/s GMP production process was established. The IND submission for applying Cerebraca™ Wafer to patients with recurrent GBM was approved by the FDA and TFDA in 20166. In 2020, the Phase I dose escalation clinical trial to determine the maximum tolerated dose (“MTD”) and safety profile of Cerebraca™ Wafer on 12 patients was completed5. In 2022, the last patient of the Phase IIa clinical trial was enrolled, and subsequently a total of 10 patients completed the main study period in 2023.
We plan to conduct an End-of-Phase II meeting with the FDA at which we will consult on the Phase IIb/III clinical trial of Cerebraca™ Wafer. If we receive positive feedback from the FDA, we plan to conduct this study in the United States. Our primary plans are for a trial with 100 patients to compare the therapeutic effect of Cerebraca™ Wafer with the available treatments. We estimate that this trial will last for two years, with completion expected in 2027. However, the final clinical trial design may need to be revised based on the FDA’s comments.
5 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8870243/
6 https://clinicaltrials.gov/study/NCT03234595#study-overview
Mechanism of Action
The mechanism of action of Cerebraca™ Wafer has been studied in many peer-reviewed Science Citation Index, or SCI, journals including Oncogene, Cancers, and Journal of Oncology. It is a multiple function drug with the potential to inhibit receptor tyrosine kinases (RTK, Axl-1 and EGFR)7 to halt cancer stem cell proliferation. Additionally, it has the potential to improve patients’ quality of life by boosting ATP levels through Axl-mTOR inhibition and AMPK activation8. Furthermore, EF-API-001 promotes DNMT3B-mediated methylation of MGMT and PD-L1 promoters, which may help to overcome chemotherapeutic resistance of alkylating agent temozolomide, or TMZ, by inhibiting deoxyribonucleic acid, or DNA, repair enzyme MGMT and transforming a cold tumor microenvironment into an immune-active hotspot by inhibiting PD-L19.
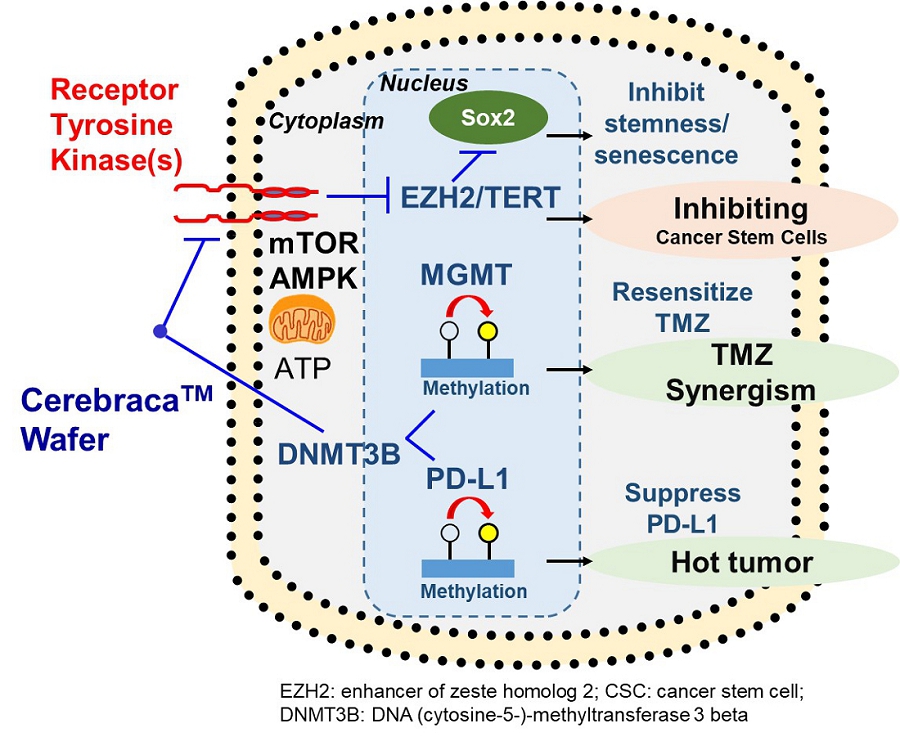
Clinical Evidence of Cerebraca™ Wafer
The Phase I clinical trial was a typical dose-escalation study utilizing a traditional 3+3 study design for determining the primary endpoint of Dose Limiting Toxicity, or DLT. DLT was to be assessed during Phase I to evaluate the safety and tolerance of Cerebraca™ Wafer and to determine the Maximum Tolerated Dose, or MTD. Data and Safety Monitoring Board (DSMB) meetings were held before dose escalation and for MTD determination. Considering the tumor size cavity limitations, the maximum feasible dose (MFD), rather than the MTD, was likely reached. The Phase I clinical trial met its primary endpoints; no increase in incidents of AEs was observed with dose escalation, and no AE met the criteria of DLT. Further, the Recommended Phase 2 Dose (RP2D) was established. The dose of six Cerebraca™ Wafer regimen was considered as the MFD and RP2D after the DSMB meeting.
The safety profile of Cerebraca™ Wafer obtained from the completed Phase I/IIa clinical trial showed no drug-related serious adverse events reported. The incidence of treatment-emergent (including tumor resection surgery and wafer implantation) serious adverse events was averaged once per patient. These serious adverse events can greatly impact the patient’s quality of life, affecting both physical and cognitive functions of patients post-surgery and throughout the course of the disease, while also increasing patients’ families’ anguish and financial load. Many patients who underwent surgical resection of GBM and implantation of Cerebraca™ Wafer left the intensive care unit (“ICU”) within two days and were discharged from the hospital within two weeks, suggesting that the treatment approach achieved a better impact on quality of life. The following table shows the incidence of treatment-emergent adverse events for Cerebraca™ Wafer.

7 https://pubmed.ncbi.nlm.nih.gov/26257061/; https://pubmed.ncbi.nlm.nih.gov/35646111/
8 https://pubmed.ncbi.nlm.nih.gov/35646111/
9 https://pubmed.ncbi.nlm.nih.gov/35205800/
In Phase IIa, ten patients were to be enrolled for assessment of the efficacy in Overall Survival (OS). The cavity surface after tumor removal was maximally covered by Cerebraca™ Wafer without exceeding the MTD defined in Phase I. Data collected from three participants in Phase I receiving treatment as designed for Phase IIa (six Cerebraca™ Wafer regimen) were incorporated into Phase IIa to reduce the patient number.
Thirteen patients with recurrent high-grade gliomas (HGG), who received six Cerebraca™ wafers (the highest dose in the clinical trial), were analyzed based on various factors including pathological tumor grade (GBM, Grade III anaplastic astrocytoma (AA), or Grade III anaplastic oligodendroglioma (AO)), cancer therapy history (Avastin-failed recurrence), biomarkers for prognosis (MGMT unmethylation (unMGMT) and isocitrate dehydrogenase 1 (IDH1) mutation (IDH1R132H)), explorative biomarkers (EGFR, Axl-1, cMet and PD-L1), and tumor resection rate (% or biopsy). Of these patients, two GBM (indicated by blue-filled columns in the table below) received Avastin, a second-line treatment. For treatment background synchronization, the efficacy analysis in overall survival (OS) focused on eleven patients with recurrent HGG (seven GBM patients with green/yellow-filled columns and four Grade III patients with red-filled column).
Seven patients with recurrent GBM isocitrate dehydrogenase wild-type (IDHwt), who received six Cerebraca™ Wafers in the Phase I/IIa trial achieved a median overall survival (OS) of 15.7 months (green/yellow-filled columns), with 57% of patients surviving for more than 12 months after treatment. In the clinical trial that combined the preliminary results from Phase I and Phase IIa for five patients receiving the highest dose of six wafers (green-filled columns), recurrent GBM patients with receptor tyrosine kinase biomarkers (EGFR, Axl-1, and cMet) showed a median OS of 26.2 months after implantation of the Cerebraca™ Wafer. Among these patients, 80% survived more than 12 months, and 40% did not experience progress or local recurrence for more than 12 months. Eleven recurrent HGG patients achieved a median OS of 13.7 months (green/yellow/red-filled columns), with 55% surviving for more than 12 months post-treatment. Recurrent HGG patients with EGFR, Axl-1 and cMet biomarkers showed a median OS of at least 22 months after Cerebraca™ Wafer treatment. In this group, 86% survived more than 12 months, and 43% had no progression or local recurrence for more than 12 months. With the main study period of the Phase IIa stage completed, we submitted to the FDA the clinical study report (“CSR”), which contained the study results and the clinical endpoints of both Phase I and Phase IIa, on September 4th, 2024. Endpoints, including the efficacy measures encompassing survival time (overall survival, OS), tumor response based on RANO criteria (progression-free survival, PFS), and local control survival (LCS, absence of new MRI T1 contrast-enhancing lesions), were achieved and are summarized in the following tables.
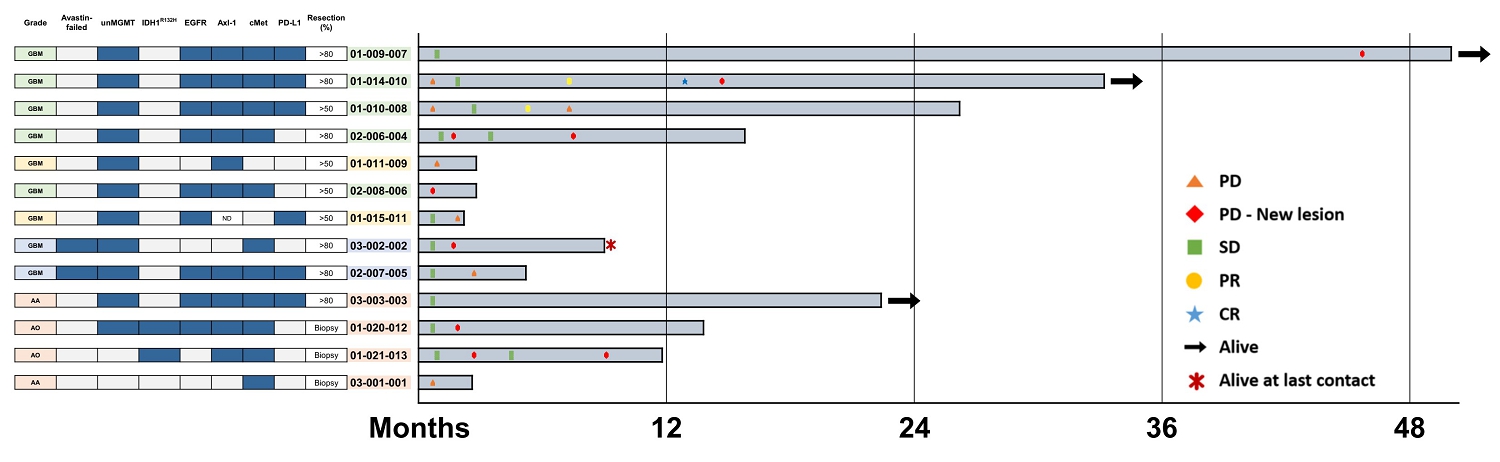
Positive marks are indicated in dark blue, negative labels are in gray, not determined (ND) is highlighted in white, Grade 3 glioma is represented in red, and GBM is depicted in green/yellow/blue.
| Efficacy | | Median OS | | OS at 6/12M | | | PFS at 6/12M | | LFS at 6/12M |
Cerebraca™ Wafer (n=5, GBM IDHwt, EGFR/Axl/cMet) | | 26.2M | | 80%/80% | | | 40%/40% | | 60%/40% |
Cerebraca™ Wafer (n=7, GBM IDHwt) | | 15.7M | | 57%/57% | | | 29%/29% | | 43%/29% |
Cerebraca™ Wafer (n=7, HGG, EGFR/Axl/cMet) | | 26.2M* | | 86%/86% | | | 43%/43% | | 57%/43% |
Cerebraca™ Wafer (n=11, HGG) | | 13.7M | | 64%/55% | | | 36%/27% | | 45%/27% |
* Patient survival data are currently being collected and updated.
EF-031 Soft-Gel Capsule for Further Improvement of Glioblastoma Treatment
The EF-031 soft-gel capsule is designed to serve as a second generation product to support the therapeutic actions of the Cerebraca™ Wafer. In pre-clinical efficacy studies using an orthotopic glioblastoma mouse model as the evaluation platform, systemic administration of EF-API-001 prolonged animal survival by 1.88 times compared to the vehicle group, demonstrating the treatment potential of systemic administration of EF-API-001. The project was then developed as EF-031 in the form of a soft-gel capsule. It was further revealed that the combination of Cerebraca™ Wafer and EF-031 resulted in a better overall survival of the orthotopic glioblastoma mouse model. The median overall survival of the Cerebraca™ Wafer + EF-031 treated orthotopic glioblastoma model was 35 days, compared to 17 days for the vehicle group, and 23 days for the Cerebraca™ Wafer alone. We believe that the development of EF-031 as our second generation of product, a next-generation GBM treatment solution (surgical resection of the GBM with the combined implantation of Cerebraca™ Wafer followed by oral administration of EF-031), can be provided. We have invested approximately $320,000 to develop the EF-031 to its current stage.
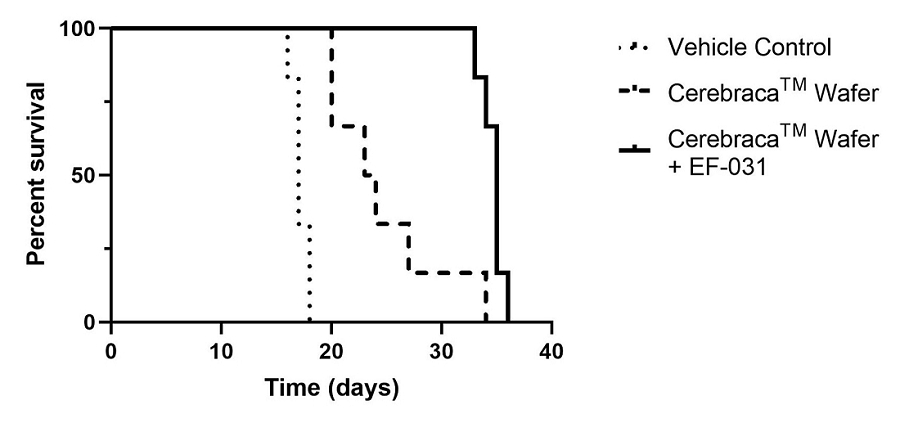
EF-009 Wafer for the treatment of Pancreatic Cancer
EF-009 Wafer is a biodegradable polymer designed to clear tumors through local release, achieving higher intra-tumoral concentrations not attainable by oral systemic administration. We have invested approximately $440,000 to develop the EF-009 Wafer to its current stage. A human pancreatic ductal adenocarcinoma orthotopic xenograft model was established to evaluate EF-009 Wafer efficacy in vivo. In the control group, the tumor continued to grow, while the chemotherapy drug gemcitabine provided limited effect on pancreatic cancer growth. Treatment with the EF-009 Wafer resulted in tumor volume reduction in a dose-dependent manner. Similar to what was observed in tumor reduction, the mean overall survival of the control and gemcitabine groups was similar (43.8 vs. 45.5 days), while EF-009 Wafer treatment resulted in a survival extension of 1.5- to 2.2-fold.
Mechanism of Action
Aberrant DNA methylation has been shown to play an important role in carcinogenesis in PC, with approximately 80% of tumors overexpressing the DNMT1 protein. EF-009 Wafer has been designed to inhibit DNMT1 expression in both a dose-dependent and time-dependent manner11. Furthermore, microarray chip analysis of pancreatic cancer cells treated with EF-009 Wafer confirmed its ability to inhibit Hedgehog expression through the regulation of PTCHD4 gene expression11. It also suppresses the growth of PC cells and induces cell cycle arrest at the G0/G1 phase, leading to apoptosis. Such growth suppression can be restored by the overexpression of DNMT1 in PC cells11. Our study also revealed that EF-009 Wafer could reduce CD44 intracellular domain or CD44-ICD, expression, targeting pancreatic cancer stem cells.
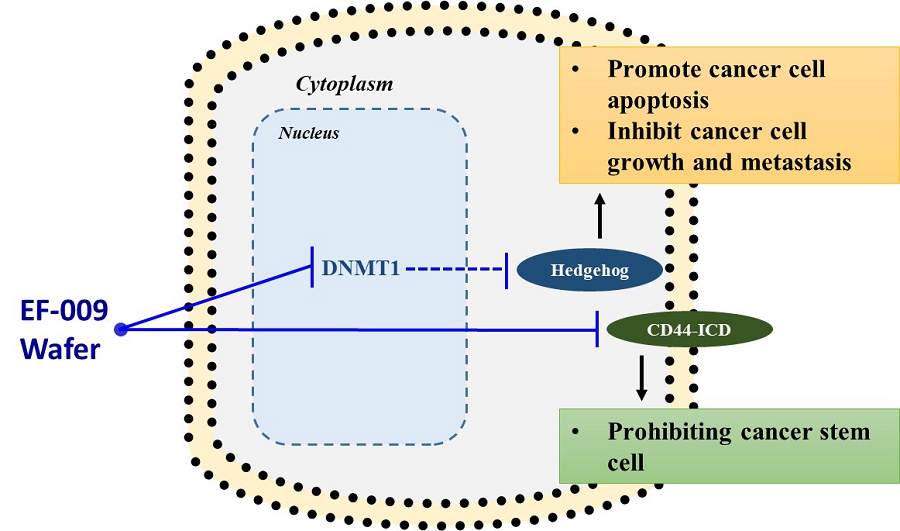
11 https://pubmed.ncbi.nlm.nih.gov/30385365/
Clinical Trials
EFB received IND Clearance from both USFDA and TFDA to initiate a Phase I/IIa clinical trial for EF-009 to treat PC. We will conduct our Phase I/IIa clinical trial of EF-009 for the treatment of pancreatic cancer at two sites in Taiwan, Tzu-Chi Medical Center and Tri-Service General Hospital, and expect the first patient to be recruited in the second quarter of 2025. In Phase I, the study will follow a “3+3” design to determine the maximum tolerated dose (MTD) of EF-009 implanted surgically in patients with pancreatic cancer. The MTD is defined as one dose level (cohort) below the dose at which dose-limiting toxicities (DLTs) were observed in 33% or more of the participants. Phase IIa is a single-arm study involving up to 12 subjects conducted to test the hypothesis that the MTD of EF-009 will increase overall survival (OS) and progression-free survival (PFS) in patients with borderline resectable and unresectable pancreatic cancer. The number of EF-009 implants will be based on the MTD dose determined in the Phase I portion of the study.
In both the Phase I and Phase IIa portions of the study, subjects will be evaluated for response every 8 weeks after EF-009 Wafer implantation for up to two years by computed tomography scan (CT), positron emission tomography-computed tomography scan (PET/CT), or magnetic resonance imaging (MRI), (at the treating investigator’s discretion) using the same method as at baseline. Tumor measurements will be assessed based on the Response Evaluation Criteria in Solid Tumors guidelines version 1.1 (RECIST v1.1). The total study duration for each subject will consist of screening, treatment, and the extended follow-up and survival follow-up periods.
The primary objective of Phase I is to determine the MTD of EF-009 by evaluating DLTs in patients with borderline resectable and unresectable pancreatic cancer. The primary objective of Phase IIa is to evaluate the effect of EF-009 on overall survival in patients with borderline resectable and unresectable pancreatic cancer. The secondary objectives of the clinical trials are to evaluate the anti-tumor efficacy, pharmacokinetics, and safety of EF-009 in patients with borderline resectable and unresectable pancreatic cancer.
HK-001 for the treatment of Amyotrophic Lateral Sclerosis (ALS)
The HK-001 capsule is in oral dosage form. We have spent approximately $2.4 million on the development of HK-001. Two improvements were observed in animal testing results (ALS mice, superoxide dismutase 1, SOD1G93A mice).
| | (1) | Delayed onset of ALS progression: Using a Basso, Beattie, Bresnahan, BBB scale score of less than 15 as the disease onset threshold, it was observed that the onset time of ALS in mice orally administered with HK-001 was delayed compared to the control drug Riluzole. |
| | (2) | Prolonged survival in ALS: The survival of ALS mice orally administered with HK-001 was superior to the untreated group and also superior to those given the control drug Riluzole. |
Mechanism of Action
The neuroprotective effect of HK-001 targets calpain and autophagy, both of which are associated with the protein degradation pathway. Inhibition of calpain results in a reduction of abnormal protein degradation. Regulation of autophagy accelerates the clearance of toxic proteins, which is associated with mutant SOD1, accumulation and related neuron degeneration.
Clinical Trials
EFB received IND Clearance from both USFDA and TFDA to initiate the Phase I clinical trial of HK-001. Eligible subjects will receive either (i) a different dosage of HK-001 or (ii) a placebo in a 3:1 ratio in one of the seven dose cohorts. After single-dose administration, followed by an independent Data and Safety Monitoring Board (DSMB) meeting for safety assessments (including the available plasma pharmacokinetic profile), the subjects will be allowed to receive either HK-001 or a placebo orally, twice a day at the study site for 14 consecutive days. A follow-up will be performed on the 28th day following the last dose administration during a site visit. The study drugs (including placebo) will be administered at the study site subject to the investigator’s instructions to either (i) perform blood sampling for pharmacokinetic evaluation or (ii) maximize treatment compliance.
IND-Enabling Stage Pipeline
In addition to our clinical product candidates, we have four potential product candidates with IND-Enabling status. IND-Enabling status implies that the preclinical studies, including therapeutic potential, pharmacology studies, pharmacokinetics studies, genotoxicity studies, safety pharmacology studies, and general toxicology studies, have been completed. These products have the potential to proceed with an IND application to the FDA for further development. Although the funds raised in this offering will not be allocated to the development of these pre-IND products, they may have the potential to be out-licensed in the future. We have invested approximately $557,000 on the development of these projects.
| Product | | Indication | | MOA | | Pre-clinical Findings | | Status |
| TSCA-001 | | Spinocerebellar ataxia (SCA) | | ● | Autophagy modulation | | ● | Motor function improvement | | IND-Enabling |
| | | | | ● | Protein aggregation reduction | | ● | Disease progression deceleration | | |
| EF-005 | | Alzheimer’s disease | | ● | β-amyloid plaque reduction | | ● | Short-term memory improvement | | IND-Enabling |
| EF-002 | | Liver cirrhosis | | ● | BMP-7 induction | | ● | Liver function recovery | | IND-Enabling |
| | | | | | | | ● | Fibrotic scar reduction | | |
| EF-011 | | Pulmonary fibrosis | | ● | Type I collagen reduction | | ● | Lung collagen deposition reduction | | IND-Enabling |
| | | | | ● | SOX2 Inhibition | | ● | Pulmonary ventilation function improvement | | |
Abbreviations: AMPK, adenosine monophosphate-activated protein kinase; BMP-7, bone morphogenetic protein 7; DNMT3B, DNA methyltransferase 3 beta; IND, investigational new drug; MOA, mechanism of action; mTOR, mammalian target of rapamycin; RTK, receptor tyrosine kinase; SOX2, sex determining region Y-box 2.
The following summarizes the preclinical studies for these product candidates.
| | ● | TSCA-001: The study was conducted on a iPSC model12 and a transgenic spinocerebellar ataxia mouse model. Our results showed that TSCA-001-promoted autophagy protected against the loss of Purkinje cells in the cerebellum, which regulates networks associated with motor functions13. |
| | | |
| | ● | EF-005: The study was conducted on a transgenic Alzheimer’s disease mouse model. Our results showed that EF-005 improved short-term memory and significantly reduced β-amyloid accumulation in the hippocampus and cortex of the transgenic animals14. |
| | | |
| | ● | EF-002: The study was conducted on a chemically induced liver fibrosis rat model. Our results showed that EF-002 activated BMP-7, further reducing liver cirrhosis and the inflammatory response15. |
| | | |
| | ● | EF-011: The study was conducted on a chemically induced pulmonary fibrosis mouse model. Our results showed that treatment of EF-011 reduced the expression of transcription factor SOX2 in the chemically induced pulmonary fibrosis mice model. Mice treated with EF-011 displayed reduced lung fibrosis and type I collagen deposition, recovering in their pulmonary ventilation function16. |
12 HH Yang et al., Int. J. Mol. Sci. 2022 Jan 26;23(3):1391. https://doi.org/10.3390/ijms23031391
13 JH Lee et al., Int. J. Mol. Sci. 2021, 22(12), 6339; https://doi.org/10.3390/ijms22126339
14 W Wuli et al., Int J Mol Sci. 2022 Sep 11;23(18):10554. https://doi.org/10.3390/ijms231810554
15 HM Chuang et al., Front Pharmacol. 2016 Apr 27:7:112. https://doi.org/10.3389/fphar.2016.00112
16 HM Chaung et al., Int J Mol Sci. 2018 Oct 4;19(10):3024. https://doi.org/10.3390/ijms19103024
Our Strategy
Our goal is to fulfill unmet medical needs by developing orphan drugs to treat rare disease. To that end, we primarily focus on the research and development of new drugs; the discovery of the application of various potential small molecules; and progressing from front-end potential product development to preclinical study, CMC, IND submission, clinical trial planning and execution, and commercialization. We are working to help patients affected by orphan diseases, enabling them to achieve faster access to better quality of life and a real chance for a cure. We plan to achieve our goal through the following.
| ● | Leveraging the advantages in application of potential candidates. Leveraging our in-house R&D platforms, we are able to advance potential drug candidates to clinical stages targeting different diseases and mechanisms of action. We have established collaborations with leading academic and industry partners to expand our pipeline and increase our chances of success. Through these collaborations, we have access to cutting-edge technologies and expertise in various therapeutic areas and various types of formulations, enabling us to accelerate the discovery and development of novel drugs. We aggressively apply for core patents across jurisdictions for our treatments for cancer, neuro-degenerative diseases, pulmonary fibrosis and liver cirrhosis. Furthermore, our patent strategy encompasses drug chemical synthesis process technology, drug formulation, and combination efficacy with other drugs or immune cell therapy. Furthermore, leveraging in-vivo animal platforms, we are able to study the efficacy of potential drugs in a more efficient way. |
| ● | Drug Delivery Platform. Leveraging our company’s expertise, we have proposed a unique drug development strategy that sets our company apart. Our focus is on developing a delicate invasive treatment strategy, underpinned by two key aspects of our company. Firstly, our local delivery platform is designed to penetrate hard-to-treat solid tumors that present biological barriers. Secondly, we maintain strong communication with clinical experts throughout the treatment strategy process. |
| ● | Developing orphan drugs by leveraging regulatory advantages and targeting unmet needs. We target rare disease patients with unmet medical needs as our primary product development strategy. We also seek to leverage regulatory advantages offered by governments to expedite drug development timelines (such as orphan drug designation, fast track, breakthrough therapy, and priority review designations), reduce costs associated with drug development (such as decreasing the number of patients or conducting single-arm clinical trials), and potentially gaining market exclusivity after orphan drug approval. |
| ● | Growing our existing business and deepening our pipeline. Leveraging the multi-function properties of our developed drug candidates, we can combine them with existing medical approaches or future new therapeutic technologies (such as immune cell therapy and cytokine-induced killer cell therapy) to expand the therapeutic applications for treating more diseases and to maximize their wider medical benefits. Once our developed drugs are approved and are successfully launched, we can address unmet medical needs in various disease areas by collaborating with other pharmaceutical companies or healthcare organizations for combination therapies. This collaboration aims to drive innovation and advance patient care. |
| ● | Benefiting from the collective experience of a deep, seasoned management team coupled with a scientific advisory board. Led by our experienced management team, our business includes, but is not limited to: (i) clinical stage project management (such as outsourcing projects, commissioned research and development projects, and investigational product production, storage and distribution projects), (ii) analytical testing, and (iii) clinical trial contracts entered into with certain professional institutions. In addition, we have established a scientific advisory board which assists us by providing professional scientific perspectives and guidance for the development of new drug projects and in evaluating potential opportunities for acquiring or licensing biotechnology innovations. |
Relevant Markets
Brain Tumors
The information presented in this sub-section has been derived from an industry report commissioned by us and prepared by Fortune Business Insights, an independent research firm, in 2021 regarding the global market for the treatment of brain tumors and forecast for 2022-2029.
Brain cancer is one of the most fatal cancers accounting for approximately 1.0%-2.0% of the total cancer types in the world. The prevalence of brain tumors is increasing rapidly owing to factors such as higher exposure to radiation, improved diagnosis, and the increasing aging population. According to the Global Cancer Observatory (“GLOBOCAN”), around 308,000 new cases of brain & central nervous system (CNS) cancer were recorded globally in 2020. Brain and CNS cancer were ranked 20th among the most common oncology diseases in 2020. The five-year prevalence of brain and CNS cancer was 10.74 per 100,000 population worldwide. Geographically, the incidence and prevalence of brain tumor is high in North America and the Asia Pacific region. Thus, the growing prevalence along with the increasing diagnosis and treatment rate in these regions is supporting the growth of brain tumor drugs in the region. The global brain tumor drug market was approximately $2.31 billion in 2021 and is expected to increase to $4.43 billion by 2029, reflecting a compound annual growth rate, or CAGR, of 9.0% over that period.
Chemotherapy dominated the brain tumor drug market, capturing a 39.2% share in 2021. Chemotherapy drugs are among the first line of therapies for a recurrent patient with aggressive brain tumors and include drugs such as temozolomide, which has been shown to have a median overall survival of 5.8 months17. On the other hand, the Gliadel® wafer, an implant carrying the chemotherapy drug Carmustine, has been shown to result in a median overall survival of 6.5 months and a 6-month survival rate of 56% in recurrent glioblastoma patients (compared to the blank wafer, which has a median overall survival of 4.6 months and a 6-month survival rate of 36%)18. In addition, these drugs are highly adopted in hospitals and cancer treatment centers making them a significant revenue generating segment. Furthermore, development of chemotherapy treatments combined with other novel therapies also contribute to the market growth. Targeted therapy is estimated to grow at the highest rate through 2029 with a CAGR of 9.7% and in 2021, it held 33.5% market share. The higher share of the segment is attributed to the growing adoption and preference for targeted drugs in emerging regions for the treatment of brain tumors owing to their ability to target only the cancerous cells and treat certain advanced cancers. The immunotherapy segment accounted for a 17.8% share of the market in 2021. Continuous research for developing immunotherapy drugs for brain tumor treatment is likely to contribute to the segment growth. In July 2020, Japan Aichi Cancer Center and NEC Corporation announced the launch of fundamental research aimed at advanced cancer immunotherapy through the fusion of AI and immunology for various cancers such as brain metastasis. Other approaches, including steroids and hormones, captured 9.5% market share in 2021, and are anticipated to grow at a rate of 6.9% through 2029.
Glioblastoma accounted for the highest share at 40.2% in the global brain tumor drugs market. According to the American Association of Neurological Surgeons (2022), glioblastoma was one of the most prevalent adult brain tumors, comprising 47.7% of all brain tumor cases in the U.S. Moreover, according to the same source, glioblastoma has an incidence of 3.21 per 100,000 population. Also, key pharmaceutical companies in collaboration with various institutes are developing drugs for the treatment of glioblastoma and this is likely to support segment growth. In October 2021, GlaxoSmithKline in collaboration with Ivy Brain Tumor Center and others initiated a clinical trial to study the efficacy of Niraparib in patients with newly diagnosed glioblastoma and recurrent glioma.
Pancreatic Cancer
The information presented in this sub-section has been derived from an industry report commissioned by us and prepared by Grand View Research, an independent research firm, in 2018 regarding the global market for the treatment of pancreatic cancer.
There are two pancreatic cancer types: endocrine and exocrine. Among them, endocrine pancreatic cancer held the major market at more than 95%. The global endocrine pancreatic cancer drug/therapy market was valued at $3,437 million in 2016 and is expected to grow at a CAGR of 9.4% through 2025. The global market size is estimated to be $7,882 million in 2025. The rise in the prevalence of obesity and the increase in alcohol consumption & tobacco use is expected to propel market growth. In addition, the increasing geriatric population prone to cancer is expected to drive the pancreatic cancer treatment market through 2025. The chemotherapy segment is expected to dominate the market with the largest revenue share through 2025. Availability of a number of drugs, use of drug combination therapy, and the combination of drug-radiation therapy is providing effective treatment options. As a result, chemotherapy is expected to remain the highest revenue generating sector of the market.
Amyotrophic Lateral Sclerosis
The information presented in this sub-section has been derived from an industry report prepared by GlobalData, an independent research firm, in 2020 Amyotrophic Lateral Sclerosis (ALS) – Opportunity Analysis and Forecasts to 2029.
Amyotrophic Lateral Sclerosis (ALS) is a fatal motor neurodegenerative disease affecting both upper and lower motor neurons, with hereditary (FALS) and idiopathic (SALS) origins. Current treatments include Sanofi’s Rilutek (riluzole) and Mitsubishi Tanabe’s Radicava (edaravone), with recent reformulations like ITF Pharma’s Tiglutik (oral suspension) and Aquestive Therapeutics’ Exservan (oral film) aiding advanced-stage patients with swallowing difficulties. In 2019, global ALS drug sales reached $282 million, with the United States accounting for $197 million. Radicava was the top-selling drug at $162 million. The ALS market is projected to grow to $1.04 billion by 2029, with a compound annual growth rate of 13.9%.
17 Bower M, et al. Cancer Chemotherapy and Pharmacology. 1997;40484.
https://pubmed.ncbi.nlm.nih.gov/9332462/
18 Gliadel® FDA label
https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/020637s029lbl.pdf
Our Collaboration Partners
In order to develop and commercialize our product candidates, we seek to collaborate and cooperate with third parties, including the following current partners and collaborative projects.
Collaboration with Buddhist Tzu Chi Hospital, Professor Horng-Jyh Harn and China Medical University (CT100004)
On June 11, 2011, we entered into a license agreement with Buddhist Tzu Chi Hospital, Professor Horng-Jyh Harn, and China Medical University (the “Buddhist Tzu Chi Hospital Agreement) for the transfer of “Angelicae Sinensis Useful for treatment of cancers” seven patents in Taiwan, Hong Kong, the United States, the European Union, People’s Republic of China (PRC), Japan and Singapore for Company’s utilization globally. The license has a term of 15 years since the date of the agreement, after which we may extend for an additional two-year term, subject to the parties’ further negotiation of contract terms. We have made total royalty payments of $81,464 as of the date of this prospectus. If any drug candidate is commercialized, we shall provide 1.5% of relevant revenue to the Buddhist Tzu Chi Hospital and Professor Horng-Jyh Harn as a royalty bonus. If payments due under the agreement remain unpaid for more than one month, Buddhist Tzu Chi Medical Foundation may terminate the agreement.
Collaboration with A+ Inc. (CT106021、CT107017、CT107020、CT108021、CT109002、CT109074、CT111071 、CT112023)
On August 25, 2017 we entered into the Contract Research Organization Service Agreement with A+ Inc. (the “A+ Inc. Agreement”), In May 2018, A+ Inc. merged with Protech Pharmaservices Corporation, we agreed to transfer the original A+ INC. Agreement to Protech Pharmaservices Corporation, and amended part of the contract content in December 2018, June 2019, January 2020, October 2020 and October 2022. The A+ Inc. Agreement term is from August 25, 2017, until completion of a clinical trial of a Phase I/IIa study to determine the maximum tolerated dose in respect of and to evaluate the safety and efficacy profile of Cerebraca™ Wafer plus adjuvant temozolomide in patients with recurrent high-grade glioma. The A+ Inc. Agreement requires strict control of the quality of clinical trials and ensuring that clinical trials are carried out according to the established plan. Under the A+ Inc. Agreement, the total clinical trial fee is $1,174,016 (excluding VAT). For the twelve months ended December 31, 2023 and 2022 we incurred total clinical trial fees of $101,387 and $161,220, respectively. As of the date of this prospectus, we have paid an aggregate amount of $890,270 under the A+ Inc. Agreement. Either party may terminate the agreement by providing notice to the other party if the inability to perform this contract is not attributable to A+ Inc. or its agents. Termination of the agreement shall be notified to the other party in writing at least thirty days in advance for termination.
Collaboration with Hualien Tzuchi Hospital, The Buddhist Tzuchi Medical Foundation and Professor Shinn-Zong Lin (CT106023)
On September 5, 2017, we entered into the “Engagement with Hualien Tzuchi Hospital, The Buddhist Tzuchi Medical Foundation to Implement Clinical Trial Agreement” with Hualien Tzuchi Hospital, The Buddhist Tzuchi Medical Foundation and Professor Shinn-Zong Lin (the “Hualien Tzuchi Hospital Agreement”), which was further amended in February 2018, July 2018, and September 2020. The Hualien Tzuchi Hospital Agreement was initially scheduled to expire on December 31, 2023 but has been extended to December 31, 2025. The Hualien Tzuchi Hospital Agreement provides for implementation of the clinical trial of a Phase I/IIa study to determine the maximum tolerated dose (“MTD”) and to evaluate the safety and efficacy profile of Cerebraca™ Wafer plus adjuvant temozolomide (“TMZ”) in patients with recurrent high-grade glioma. The total clinical trial fee is $594,721 (including VAT), which is to be paid according to the progress of the clinical trial. For the twelve months ended December 31, 2023 and 2022, we incurred clinical trial fees of $30,996 and $29,717, respectively. As of the date of this prospectus, we have paid an aggregate amount of $351,996 under the Hualien Tzuchi Hospital Agreement.
Hualien Tzuchi Hospital, The Buddhist Tzuchi Medical Foundation may terminate the agreement in writing if the Ethics Committee terminates or temporarily suspends the trial according to the applicable laws or regulations. We may terminate this Agreement by giving a 10-day prior written notice to Hualien Tzuchi Hospital, The Buddhist Tzuchi Medical Foundation without specifying reason.
Collaboration with Tri-Service General Hospital, National Defense Medical Center (CT107029)
On April 27, 2018, we entered into the Clinical Trial Agreement with Tri-Service General Hospital (TSGH), National Defense Medical Center and Protech Pharmaservices Corporation (the “Tri-Service Agreement”) for a Phase I/IIa study to determine the MTD and to evaluate the safety and efficacy profile of Cerebraca™ Wafer plus adjuvant temozolomide (TMZ) in patients with recurrent high-grade glioma. The Tri-Service Agreement term is from April 27, 2018 until completion of the Study (as defined in the Tri-Service Agreement) or until termination. The total clinical trial fee is $766,684, which is to be paid according to the progress of the clinical trial.
For the twelve months ended December 31, 2023 and 2022, we incurred no clinical trial fees. As of the date of this prospectus, we have paid an aggregate amount of $269,758 under the Tri-Service Agreement.
In the event that TSGH or Principal investigator violates any provision of this Agreement or requirements of the Protocol, Everfront may notify TSGH or Principal investigator in writing and request correction by a specified date. If TSGH or Principal investigator fails to resolve the breach before the deadline after receipt of the written notice and said violation is a serious breach, Everfront may terminate this Agreement, and Everfront shall request for indemnification, including but not limited to reasonable attorney fees.
Should Everfront fail to make payment in accordance with the payment schedule specified in the Agreement and by the deadline specified by TSGH in writing after receipt of the notice from TSGH, TSGH may terminate this Agreement with a 30-day prior written notice.
For situations other than the aforementioned provisions, Everfront may terminate this Agreement before the termination date if a written notice is given to TSGH at least 30 days before the desired rescission date.
Should the Principal Investigator resign from TSGH during the effective period of this Agreement, TSGH may reappoint a replacement principal investigator acceptable to Everfront in its sole discretion, or Everfront may terminate the Agreement. Everfront will be no longer to pay the cost apart from the payment of the termination of the contract.
Collaboration with Taichung Veterans General Hospital and Protech Pharmaservices Corporation (CT110036):
On July 6, 2021, we entered into a Clinical Trial Agreement with Taichung Veterans General Hospital and Protech Pharmaservices Corporation (the “Taichung Veterans Agreement”) for a Phase I/IIa study to determine the MTD and to evaluate the safety and efficacy profile of Cerebraca™ Wafer plus TMZ in patients with recurrent high-grade glioma. The Taichung Veterans Agreement term is July 6, 2021 to January 31, 2024. For the twelve months ended December 31, 2023 and 2022, we incurred no clinical trial fees. As of the date of this prospectus, we had paid an aggregate amount of $69,203 under the Taichung Veterans Agreement. If Taichung Veterans General Hospital discovers any event or possible event of serious adverse reaction during the course of the study, Taichung Veterans General Hospital is required to stop the study immediately and inform us of the event in order to protect the rights of the study subjects.
Taiwan Government supported projects:
In November 2017, we entered into an agreement with the Institute for Information Industry for the A+ Industrial Innovation R&D Program–Fast Track Clinical Trial Program for “Cerebraca™ Wafer Phase I/IIa Clinical Trial Plan for Recurrent Highly Malignant Glioma”, pursuant to which the Ministry of Economic Affairs shall subsidize our Phase I/IIa clinical trial of Cerebraca™ Wafer with governmental funds of approximately $1,688,341, which will be appropriated through the Institute for Information Industry. For the twelve months ended December 31, 2023 and 2022, performance guarantee notes were issued in the amounts of approximately $1,688,341 and $1,682,297, respectively. As of December 31, 2022, we had commitments under contract of approximately $5,219,278. These commitments primarily consist of the transfer and licensing of intellectual property and technology of $4,059,438 and R&D outsourcing services of $1,159,840.
In August 2017, we applied for the A+ Industrial Innovation R&D Program - Fast Track Clinical Trial Program for “Cerebraca™ Wafer Phase I/IIa Clinical Trial Plan for Recurrent Highly Malignant Glioma” administered by the Ministry of Economic Affairs (R.O.C) to the Institute for Information Industry. During the year ended December 31, 2023 and 2022, subsidy income received under the Cerebraca™ Wafer plan amounted to approximately $29,979 and $176,761, respectively.
In May 2022, we applied for the “Development Project for Production of LV-001 Functional Dropping pills by Supercritical Extraction Method” under the Small Business Innovation Research (SBIR) program of the Small and Medium Enterprise and Startup Administration, Ministry of Economic Affairs, Taiwan. During the year ended December 31, 2022, subsidy income collected under the SBIR program amounted to approximately $7,853.
Research Awards
In May 2023, Cerebraca™ Wafer secured the prestigious 2023 NBRP DEMODAY – Investor’s Choice Award, recognized for its groundbreaking technology and strategic vision in a competitive pitch setting judged by venture capitalists and industry luminaries. Additionally, Cerebraca™ Wafer, EF-009, and HK-001 were honored with the National Innovation Award on multiple occasions, spanning December 2014 to December 2021, underscoring their pivotal contributions to advancing research and development within Taiwan’s innovative landscape. Further distinction came in December 2019 and 2021, when Cerebraca™ Wafer and HK-001 were recipients of the National Innovation Award – Excelsior Award, celebrating their sustained innovation and notable achievements across various facets of research and development. Lastly, in July 2019, Everfront Biotech received the Taipei Biotech Award based on introduction of Cerebraca™ Wafer, highlighting its excellence among domestic biotechnology entities and its impactful contributions to the field, as determined by a panel of esteemed experts and scholars.
Intellectual Property
Our commercial success depends in part on our ability to obtain and maintain intellectual property and proprietary protection for our current and future product candidates, as well as novel discoveries, product development technologies, and know-how. Our commercial success also depends in part on our ability to operate without infringing on the proprietary rights of others and to prevent others from infringing our proprietary rights. Our policy is to develop and maintain protection of our proprietary position by, among other methods, filing or in-licensing U.S. and foreign patents and applications related to our technology, inventions, and improvements that are important to the development and implementation of our business.
We also rely on trademarks, trade secrets, know-how, continuing technological innovation, confidentiality agreements, and invention assignment agreements to develop and maintain our proprietary position. The confidentiality agreements are designed to protect our proprietary information and the invention assignment agreements are designed to grant us ownership of inventions and technologies that are developed for us by our employees, consultants, or other third parties. We seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in our agreements and security measures, either may be breached, and we may not have adequate remedies in the event of such occurrences. In addition, our trade secrets may otherwise become known or independently discovered by competitors.
With respect to both licensed and Company-owned intellectual property, we cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed or licensed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to or licensed by us in the future will be commercially useful in protecting commercial products we develop and methods of using and manufacturing the same.
We are actively building our intellectual property portfolio around our product candidates and our discovery programs, based on our own intellectual property as well as licensed intellectual property.
As of the date hereof, we have more than 30 granted or licensed patents in the U.S., the European Union, Taiwan, Japan, India and China; and approximately 5 pending patent applications. Our main patent portfolio includes cancer therapy, neuron diseases, and fibrosis treatment. The table below sets forth our key patent portfolio.
| Type | | Title | | JURISD. | | Status | | Applicant | | Expiration Date | | Company Rights | | PROD. Candidate Covered |
Patents for cancer treatment | | Angelicae sinensis extracts useful for the treatment of cancers | | US, TW, EP, CN, JP, | | Granted | | Buddhist Tzu Chi General Hospital | | 2024 | | Exclusive License | | Cerebraca™ Wafer, EF-009 |
| | A method for preparing isobenzofuran-1(3H)-one compound | | US, TW, EP, CN | | Granted | | Everfront Biotech Inc. | | 2035 | | Company owned | | Cerebraca™ Wafer, HK-001, EF-009 |
| | An anticancer formulation | | US, EP, JP | | Granted | | National Dong Hwa University | | 2030 | | Exclusive License | | Cerebraca™ Wafer |
| | A pharmaceutical formulation comprising EF-API-001 | | IN | | Granted | | National Dong Hwa University | | 2031 | | Exclusive License | | Cerebraca™ Wafer |
| | Method for treating brain cancer or reducing temozolomide-resistance of brain cancer cells | | US, TW, EP, CN, JP | | Granted | | China Medical University | | 2031 | | Exclusive License | | Cerebraca™ Wafer |
| | A method for treating pancreatic cancer | | US, TW, EP, CN, JP | | Granted | | China Medical University | | 2034 | | Exclusive License | | EF-009 |
| | Uses of EF-API-001 | | US, TW, EP, CN, | | Granted | | Everfront Biotech Inc. | | 2036 | | Company owned | | EF-009, Cerebraca™ Wafer |
| | A combination for the treatment of cancer and its application | | PCT, US, JP, CN, TW | | Pending | | Everfront Biotech Inc. | | 2041 | | Company owned | | EF-009, Cerebraca™ Wafer |
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the date of filing of the first non-provisional application to which priority is claimed. In the United States, the patent term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the United States Patent and Trademark Office in granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier-filed patent. In the United States, the term of a patent that covers an FDA-approved drug may also be eligible for a patent term extension of up to five years under the Hatch-Waxman Act, which is designed to compensate for the patent term lost during the FDA regulatory review process. The length of the patent term extension is calculated based on the length of time it takes for regulatory review. A patent term extension under the Hatch-Waxman Act cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be restored. Moreover, a patent can only be restored once, and thus, if a single patent is applicable to multiple products, it can only be extended based on one product. Similar provisions are available in Europe and certain other foreign jurisdictions to extend the term of a patent that covers an approved drug.
Our commercial success will also depend in part on not infringing upon the proprietary rights of third parties. It is uncertain whether the issuance of any third-party patent would require us to alter our development or commercial strategies, or our product, if approved, or product candidates or processes, obtain licenses or cease certain activities. Our breach of any license agreements or failure to obtain a license to proprietary rights that we may require to develop or commercialize our future product candidates may have an adverse impact on us. Since patent applications in the United States and certain other jurisdictions are maintained in secrecy for 18 months or potentially longer, and since publication of discoveries in the scientific or patent literature often lags actual discoveries, we cannot be certain of the priority of inventions covered by pending patent applications. Moreover, we may have to participate in interference or opposition proceedings brought by third parties or declared by the USPTO. For more information, see “Risk Factors—Risks Related to Our Intellectual Property.”
Competition
The biotechnology sector operates within a landscape characterized by rapid technological advancements, fierce competition, and a strong focus on unique proprietary products and potential offerings. If approved, Cerebraca™ Wafer holds promise across various markets. However, its competitive standing will ultimately be determined by the specific disease(s) it targets and the corresponding marketing approvals it secures. Notably, there are ongoing development efforts by other companies targeting Glioblastoma, the primary indication initially pursued by Cerebraca™ Wafer. In the event of approval, Cerebraca™ Wafer will contend with both existing and future therapies. Our competition spans a wide spectrum, encompassing larger, well-funded entities in the pharmaceutical, biopharmaceutical, and biotechnological sectors. These competitors often possess greater financial, technical, and operational resources, as well as more extensive experience and expertise in their respective programs. Additionally, we may encounter competition from universities and research institutions active in our target areas. Moreover, smaller or emerging firms could pose significant challenges, particularly through collaborative ventures with established players.
We believe our current and future competition can be grouped into three broad categories.
| | ● | Companies working to develop Janus kinase (JAK), mTOR, or Phosphoinositide 3-kinase (PI3K), inhibitors, including Roche, Pfizer, and Kazia Therapeutics. |
| | | |
| | ● | Companies with similar product candidates in Phase III clinical trial stage for GBM, including Amgen, Bayer, and Biohaven. |
| | | |
| | ● | Companies commercializing presently available treatment agents, such as Gliadel® wafer, Temodar®, and Avastin®, both as branded medicines or in generic or biosimilar form including Eisai, Merck & Co., Roche, and Amgen. |
Should we secure approval for Cerebraca™ Wafer or any forthcoming product candidates, we anticipate that their success will hinge on several pivotal competitive factors. These include efficacy, safety, tolerability, convenience, pricing, and the amount and accessibility of reimbursement from governmental and third-party payers compared to similar products in the market. Our commercial prospects may diminish or vanish altogether if rival products excel in one or more of these aspects.
Government Regulation and Product Approval
As a biopharmaceutical company that operates in the Taiwan and intends to market products in Taiwan, the United States and other jurisdictions, we are subject to extensive regulation. Government authorities in Taiwan, the United States (at the federal, state and local level) and in other countries extensively regulate, among other things, the research, development, testing, manufacturing, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of biopharmaceutical products such as those we are developing. Our product candidates must be approved by the FDA before they may be legally marketed in the United States and by the appropriate foreign regulatory agency before they may be legally marketed in foreign countries. Generally, our activities in other countries will be subject to regulation that is similar in nature and scope as that imposed in the United States, although there can be important differences. Additionally, some significant aspects of regulation in Europe are addressed in a centralized way, but country-specific regulation remains essential in many respects. The process for obtaining regulatory marketing approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
FDA Drug Development Regulations
The process of obtaining regulatory approvals and maintaining compliance with appropriate federal, state and local statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative or judicial sanctions or lead to voluntary product recalls. Administrative or judicial sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. The process required by the FDA before a drug may be marketed in the United States generally involves the following.
| ● | completion of non-clinical laboratory tests, preclinical studies according to current good laboratory practice (as adopted by the applicable regulatory authority) (“GLP”) and manufacturing of clinical supplies according to current good manufacturing practice (as adopted by the applicable regulatory authority) (“cGMP”); |
| ● | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| ● | approval by an independent institutional review board (“IRB”), at each clinical site before each trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials according to current good clinical practice (as adopted by the applicable regulatory authority) (“GCP”), to establish the safety and efficacy of the proposed product for its intended use; |
| ● | preparation and submission to the FDA of an NDA, for a drug; |
| ● | satisfactory completion of an FDA advisory committee review, if applicable; |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with cGMP; and |
| ● | payment of user fees and the FDA review and approval of the NDA. |
The testing and approval process requires substantial time, effort and financial resources and we cannot be certain that any approvals for our drug candidates, or any future drug candidates we may develop, will be granted on a timely basis, if at all.
Once a drug candidate is identified for development, it enters the non-clinical testing stage. Non-clinical tests include laboratory evaluations of product chemistry, toxicity, formulation and stability, as well as preclinical studies. An IND sponsor must submit the results of the non-clinical tests, together with manufacturing information, analytical data and any available clinical data or literature, to the FDA as part of the IND prior to commencing any testing in humans. An IND sponsor must also include a protocol detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated if the initial clinical trial lends itself to an efficacy evaluation. Some non-clinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions related to a proposed clinical trial and places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during clinical trials due to safety concerns or non-compliance and may be imposed on all products within a certain class of products. The FDA also can impose partial clinical holds, for example, prohibiting the initiation of clinical trials for certain duration or for certain doses.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCP regulations. These regulations include the requirement that all research subjects provide informed consent in writing before their participation in any clinical trial. Further, an IRB representing each institution participating in a clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review and reapprove the study at least annually. An IRB is responsible for protecting the rights of clinical trial subjects and considers, among other things, whether the risks to individuals participating in the clinical trial are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Each new clinical protocol and any amendments to the protocol must be submitted to the FDA for review, and to the IRBs for approval. Protocol detail, among other things, includes the objectives of the clinical trial, testing procedures, sublease selection and exclusion criteria, and the parameters to be used to monitor subject safety.
Human clinical trials are typically conducted in the following three sequential phases that may overlap or be combined.
| ● | Phase I. Phase I includes the initial introduction of an investigational new drug into humans. These studies are closely monitored and may be conducted in patients, but are usually conducted in healthy volunteer subjects. These studies are designed to determine the metabolic and pharmacologic actions of the drug in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. During Phase I, sufficient information about the drug’s pharmacokinetics and pharmacological effects should be obtained to permit the design of well-controlled, scientifically valid, Phase II studies. Phase I studies also evaluate drug metabolism, structure-activity relationships, and the mechanism of action in humans. These studies also determine which investigational drugs are used as research tools to explore biological phenomena or disease processes. The total number of subjects included in Phase I studies varies with the drug, but is generally in the range of 20 to 80. |
| ● | Phase II. Phase II includes the early controlled clinical studies conducted to obtain some preliminary data on the effectiveness of the drug for a particular indication or indications in patients with the disease or condition. This phase of testing also helps determine the common short-term side effects and risks associated with the drug. Phase II studies are typically well-controlled, closely monitored, and conducted in a relatively small number of patients, usually involving several hundred people. |
| ● | Phase III. Phase III studies are expanded controlled and uncontrolled trials. They are performed after preliminary evidence suggesting effectiveness of the drug has been obtained in Phase II, and are intended to gather the additional information about effectiveness and safety that is needed to evaluate the overall benefit-risk relationship of the drug. Phase III studies are designed to provide an adequate basis for extrapolating the results to the general population and transmitting that information in the physician labeling. Phase III studies usually include several hundred to several thousand people. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and clinical investigators within 15 calendar days for serious and unexpected suspected adverse events, any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator’s brochure, or any findings from other studies or animal or in vitro testing that suggest a significant risk in humans exposed to the drug candidate. Additionally, a sponsor must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction no later than 7 calendar days after the sponsor’s receipt of the information. There is no assurance that Phase I, Phase II and Phase III testing can be completed successfully within any specified period, or at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product has been associated with unexpected serious harm to subjects.
Concurrent with clinical trials, companies usually complete additional preclinical studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product drug and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product drug does not undergo unacceptable deterioration over its shelf life.
The results of product development, non-clinical studies and clinical trials, together with other detailed information regarding the manufacturing process, analytical tests conducted on the product, proposed labeling and other relevant information, are submitted to the FDA as part of an NDA requesting approval to market the new drug. The FDA reviews all NDAs submitted within 60 days of submission to ensure that they are sufficiently complete for substantive review before it accepts them for filing. If the submission is accepted for filing, the FDA begins an in-depth substantive review.
The approval process is lengthy and difficult and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive, and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA in its present form. The complete response letter usually describes all of the specific deficiencies that the FDA identified in the NDA that must be satisfactorily addressed before it can be approved. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application or request an opportunity for a hearing.
If after such review a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. Any products for which we receive FDA approval would be subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with the FDA promotion and advertising requirements. In addition, the FDA may require post-approval studies, including Phase IV clinical trials, to further assess a product’s safety and effectiveness after NDA approval and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. The FDA also may conclude that an NDA may only be approved with a Risk Evaluation and Mitigation Strategy designed to mitigate risks through, for example, a medication guide, physician communication plan, or other elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools.
In addition, the FDA has the authority to designate a drug product as an orphan drug if it is intended to treat a rare disease or condition. Typically, a rare disease or condition affects fewer than 200,000 individuals in the United States. Alternatively, it may affect more than 200,000 individuals, but there is no reasonable expectation that the costs of developing and making the product available in the United States for such diseases or conditions will be recouped from sales.
Requests for orphan drug designation must be made before submitting a New Drug Application (“NDA”). Once the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are publicly disclosed by the FDA. If a product with orphan drug designation is subsequently granted the first FDA approval for the disease or condition for which it was designated, it becomes eligible for orphan drug exclusivity. This means that the FDA cannot approve any other applications to market the same drug for the same indication for seven years from the date of such approval. There are limited exceptions to this exclusivity, such as demonstrating clinical superiority or in cases of drug supply issues. Nevertheless, competitors may still gain approval for different products for the same indication or the same product for a different indication that could be used off-label for the orphan indication.
Orphan drug exclusivity may also impede the approval of our products for seven years if a competitor gains approval first for the same product and indication, or if a product candidate falls within the scope of a competitor’s product for the same indication or disease. If one of our products receives marketing approval for a broader indication than designated as an orphan, it may not qualify for orphan drug exclusivity. It’s important to note that orphan drug status in the European Union has similar, but not identical, requirements and benefits.
TFDA Drug Development Regulations
The drug development process in Taiwan involves preclinical tests, clinical trials, manufacturing and post-market monitoring. Each stage is subject to scrutiny by the TFDA. Conducting clinical trials of drugs should strictly adhere to relevant regulations, such as medical laws, pharmaceutical laws, and GCP guidelines. The regulatory processes in Taiwan are generally similar with those in the United States, and include:
| ● | Completion of preclinical laboratory tests, preclinical animal studies and manufacturing of clinical supplies in accordance with applicable regulations. |
| ● | Preclinical Research: Before advancing to human trials, an extensive preclinical research phase is conducted to gather essential data on the drug’s safety, efficacy, and potential adverse effects. This stage involves a series of comprehensive studies conducted in laboratory settings and animal models. |
| ● | Animal testing is a vital component of preclinical research. It allows researchers to investigate the drug’s efficacy, toxicity, and pharmacokinetics in living organisms. Various animal models, such as rodents or non-human primates, are utilized to simulate human physiological responses and determine the drug’s potential effectiveness in treating specific diseases. |
| ● | Toxicity evaluations are conducted to assess the drug’s safety profile. This includes examining its potential adverse effects, identifying the maximum tolerated dose, and evaluating any potential risks associated with long-term use or specific patient populations. |
| ● | During preclinical research, meticulous data collection and analysis are performed to determine the drug’s optimal dosage, formulation, and potential routes of administration. The findings from preclinical studies provide crucial insights that guide the design and implementation of subsequent clinical trials in human subjects. |
Submission to the TFDA of an IND application, which must be approved by the TFDA before human clinical trials may begin. This application includes preclinical data, proposed clinical trial protocols, and information about the drug’s manufacturing, safety, and potential risks.
| ● | IND Application: Following successful preclinical studies, the drug developer submits an IND application to the TFDA for review and approval. The IND application represents a crucial step in the drug development process and includes a comprehensive compilation of data and information gathered during the preclinical research phase. |
| ● | The IND application provides detailed documentation of the drug’s preclinical studies, including the methodology employed, experimental results, and analysis. This data demonstrates the drug’s safety, efficacy, and potential therapeutic benefits. Additionally, the application includes information on the drug’s chemical composition, formulation, and stability, ensuring the TFDA has a comprehensive understanding of the product’s characteristics. |
| ● | The manufacturing process of the drug is outlined in the IND application, detailing the steps involved in its production, quality control measures, and adherence to Good Manufacturing Practices (GMP) standards. This information ensures that the drug is manufactured consistently and meets the required quality standards throughout the clinical trial process. |
| ● | Furthermore, the IND application provides an overview of the proposed clinical trial design. It includes details on the target patient population, the number of participants, trial duration, dosing regimens, and endpoints to be evaluated. The application also outlines the ethical considerations and safeguards in place to protect the rights and welfare of the participants involved in the clinical trials. |
| ● | The TFDA thoroughly reviews the IND application, assessing the scientific validity of the preclinical data, the drug’s safety profile, and the appropriateness of the proposed clinical trial design. This rigorous evaluation ensures that the drug’s benefits outweigh the potential risks and that the proposed clinical trials are conducted in an ethical and scientifically sound manner. |
| ● | Upon successful review and approval of the IND application by the TFDA, the drug developer can proceed with conducting clinical trials in human subjects. The IND application serves as the foundation for subsequent phases of clinical development, where the drug’s safety, efficacy, and optimal dosage are further evaluated in controlled human trials. |
Human clinical trials in Taiwan typically include the following phases and submissions:
| ● | Phase I. Phase I includes the initial introduction of an investigational new drug into humans and tests for safety, dosage, tolerance, pharmacokinetic and side effects. |
| ● | Phase II. Phase II includes the early controlled clinical studies conducted to obtain some preliminary data on the effectiveness and adverse effects of the drug for a particular indication or indications in patients with the disease or condition. |
| ● | Phase III. Phase III studies include expanded patient population to evaluate dosage tolerance, efficacy and safety. |
| ● | Submission to the TFDA of an NDA, which must be approved by the TFDA in accordance with the Taiwan Regulations for Registration of Medicinal Products (“RRMP”). Pursuant to the RRMP, applicants are required to provide enough information to support a drug’s safety and effectiveness. The NDA includes comprehensive data from preclinical and clinical studies, information on manufacturing processes, proposed labeling, and packaging details. The TFDA reviews the NDA and conducts a thorough evaluation of the drug’s safety, efficacy, and quality. This includes assessment by medical and scientific experts, examination of clinical trial data, and evaluation of manufacturing practices. If the TFDA determines that the drug’s benefits outweigh its risks and that it meets the necessary regulatory requirements, approval is granted. The approval may be subject to certain conditions, such as post-marketing surveillance or additional studies. |
| ● | Post-Marketing Surveillance: Once the drug is approved, post-marketing surveillance is conducted to monitor the drugs’ safety and effectiveness in a larger patient population. Adverse events and any emerging safety concerns are closely monitored and reported to TFDA. |
Employees
As of December 31, 2024, we had 14 full-time employees. Of these 14 full-time employees, 6 hold Ph.D. degrees, and 8 are engaged in research, development and technical operations. Substantially all of our employees are located in Taiwan. Our employees are not represented by labor unions or covered by collective bargaining agreements. We consider our relationship with our employees to be good.
Research and Development Expenses
For the year ended December 31, 2023, we had research and development expenses of $707,692, consisting of personnel-related costs, facilities-related overhead, and outside contracted services including clinical trial costs, manufacturing and process development costs for both clinical and preclinical materials, and other consulting services.
Facilities
We occupy approximately 2,409 square feet of office and laboratory space in Taiwan at 10F-1, No. 130, Songshan Rd., Xinyi Dist., Taipei City 110, Taiwan and NDHU IIIC R1-10, No.1, Sec.2, Da Hsueh Rd., Shoufeng, Hualien, Taiwan 974301. The Taipei City office is our operational headquarters, serving as the primary location for company strategy and operations, and planning functions including: finance, drug development, production, clinical trials planning, handling external business, and technology licensing contracts and negotiations. The Hualien facility functions as the Research, Development and Clinical Medical Research Center, serving as the primary location for preclinical drug development, toxicology, pharmacology, pharmacokinetics, process development, clinical project management, therapeutic application development, and research affairs related to patient pathology and pharmacology.
Legal Proceedings
From time to time, we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. We are not presently a party to any legal proceedings that, if determined adversely to us, would individually or taken together have a material adverse effect on our business, results of operations, financial condition or cash flows. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
REGULATIONS
Regulations Related to our Business Operations in Taiwan
We have substantially conducted our business through the Taiwan subsidiary, Everfront Biotech, in Taiwan. Therefore, we are principally subject to the relevant laws and regulations in Taiwan. The following sets out a summary of certain aspects of major laws and regulations which are relevant to our business operations in Taiwan.
Company Registration Regime
Under the Company Act of Taiwan, a company may not conduct its business operations or commit any juristic act in the name of a company unless it has completed the procedure for company incorporation registration. A company incorporated in Taiwan may apply for company registration, except special licenses for certain company businesses shall be obtained before the application for registration.
As our Taiwan subsidiary conducts business operations in Taiwan, it is required to obtain the company registration with the competent authority and retain good standing onward when the subsidiary is still operating. Since that our business involves “wholesale of western pharmaceutical and retail sale of western pharmaceutical”, which requires special licenses from the government, our Taiwan subsidiary is required to obtain special licenses thereof prior to applying for company registration. As such, to conduct business in Taiwan, our Taiwan subsidiary has obtained such required special licenses for the preceding businesses, completed the company registration with the Minister of Economic Affairs on December 8, 2010, and has retained good corporate standing till now.
Laws and Regulations Governing Inbound Investment
We own 99.96%of the total shares in the Taiwan subsidiary, which is a company limited by shares. The investment and holding shares are subject to laws and regulations with respect to inbound investment, including the Statute for Investment by Foreign Nationals (“Foreign Investment Regulation”), the Act Governing Relations between the People of the Taiwan Area and the Mainland Area (“Cross-strait People Relations Act”), and the Regulation Governing Permission for People of the Mainland Area to Invest in Taiwan (“PRC Investment Regulation”). These rules apply to both public and non-public company.
Investment by Foreign Investors
Under the Statute for Investment by Foreign Nationals (“Foreign Investment Statute”), any foreign investor who makes an investment in accordance with this Statute is required to submit an investment application to the Investment Commission of the Minister of Economic Affairs (“Investment Commission”) for approval. The foreign investor includes foreign legal persons when making investments within the territory of Taiwan. The investments may include:
| 1. | holding shares issued by a Taiwan company or contributing to the capital of a Taiwan company; |
| 2. | establishing a branch office, a proprietary business, or a partnership in Taiwan; and |
| 3. | providing loan(s) to the invested business referred to in the preceding two paragraphs for a period exceeding one year. |
Industries which concern national security, public order, good customs, practices, or citizen health are prohibited from accepting investment by foreign investors; investments in such industries which are restricted by laws or orders require approval of the competent authorities prior to investment. The negative list promulgated by the competent authority prescribes the prohibited and restricted industries.
For foreign-invested enterprises in which (i) the foreign investor holds, in an aggregate, more than one-third of the total shares issued by an enterprise or if a foreign investor contributes, and (ii) in an aggregate, more than one-third of the total capital amount of an enterprise, their reinvestment shall be subject to the Investment Commission’s approval.
Unless otherwise provided in the Foreign Investment Statute, if any foreign investor violates the provisions thereof or fails to perform in any manner as required by the competent authority, the competent authority may deal with the situation in the following manner:
| | 1. | revoke the investor’s right of exchange settlement against their income of the profit from their investment and the interest accrued thereon in a prescribed period of time; and |
| | 2. | revoke the approval for the investor’s investment and their rights under this Statute. |
Investment from PRC Investors
Under the Regulation Governing Permission for People of the Mainland Area to Invest in Taiwan (“PRC Investment Regulation”), PRC Investors shall apply for permission for their investment activities in Taiwan, including holding shares or capital contributions of Taiwan companies, gaining control over non-public Taiwan companies by contract or other means, or acquisition of business or property of non-public companies in Taiwan by third area companies. The business categories allowed to be invested by the PRC Investors are limited to specific categories prescribed by the competent authorities of Taiwan.
The “PRC Investors” as defined in PRC Investment Regulation refers to investors who are PRC citizens, legal persons/organizations incorporated in PRC, or Third Area Companies. The “Third Area Companies” includes any company of a third area in which more than 30% of the total shares or capital contributions are directly or indirectly held by investors from PRC or which of a third area is controlled by PRC individuals or entities. In case we are considered as a “Third Area Companies” and then as a “PRC Investor” due to 30% of the total shares in Everfront Holding being held by investors from PRC, we will be required to seek permission for the investor status switching. If the PRC Investor is a legal person, organization, or other institution invested by a political party or military, administrative, or political organization from PRC or is a Third Area Company invested thereby, the competent authorities shall restrict its investment in Taiwan.
Any violation of the preceding regulations may subject us to administrative fines, and competent authorities may order to cease or withdraw such investment, suspension of the violator’s shareholder’s rights, and revocation of company registration, according to the Cross-Strait People Relations Act.
While there is uncertainty as to whether the business operations of Everfront Biotech would be defined as businesses restricted or prohibited from PRC Investment, we currently do not fall into the definition of PRC Investor since none of our shares are held directly or indirectly by PRC Investors. Hence, our operation is not prohibited or restricted under the PRC Investment Regulation and the Cross-strait People Relations Act. However, if Everfront Holding is considered as a PRC Investor due to accepting investment from PRC in the future, we may need to apply for permission for the investment activities, or Everfront Biotech may be prohibited from receiving such investment.
Transfer in/out and Dividends between Everfront Holding and Everfront Subsidiary.
Under the Foreign Investment Statute, when we transfer or reduce our investment in our Taiwan Subsidiary, we are also required to apply for approval of the transfer or reduction with the Investment Commission. In the case of transferring shareholding, the application shall be filed jointly by the transferee and us.
For the distribution of dividends, we are not required to apply for approval with the Investment Commission to distribute dividends from our Taiwan Subsidiary. However, if we intend to exchange currencies for transferring the distributed dividends out of Taiwan, we are required to apply for exchange settlement against the distributed dividends with the Central Bank of Taiwan.
Laws and Regulations Governing Pharmaceutical Affairs
The Pharmaceutical Affairs Act regulates and controls, inter alia, medicaments, pharmaceutical firms, and other relevant matters. As our business operations in Taiwan include development, wholesale, and retail of pharmaceuticals, we are required to comply with the Pharmaceutical Affairs Act.
Pharmaceutical Firm Administration
Under the Pharmaceutical Affairs Act, pharmaceutical firms are business undertakings categorized and announced by the competent authority, such as wholesaling, retailing, importing, and exporting pharmaceuticals. Pharmaceutical firms shall file an application to the competent local authority for approval and registration, pay the license fee and obtain the business license before starting the permit operation, establish their own traceability system for tracing the source and tracking the flow of the drugs according to their industry modes, and use electronic methods to declare the information of the traceability system. We have not commenced selling our product and thus are not required to file such application to competent local authority.
Any violators of the preceding requirements may be imposed a fine of not less than NT$30,000 but not more than NT$2,000,000.
Drug Administration
Any pharmaceutical firm who intends to manufacture and import drugs in Taiwan, including pharmaceutical raw materials, shall apply with the Taiwan Food and Drug Administration for registration and market approval to obtain the drug permit license before manufacturing and importing such drugs, according to the Pharmaceutical Affairs Act. The preceding application shall be affixed with information concerning the ingredients, source of active pharmaceutical ingredients, specifications, functions, a summary of the manufacturing process, and the specification and method of testing, as well as other related information and certificates, accompanied by labels and use instructions in the original and Chinese languages, and samples. Where the pharmaceutical firm imports the raw materials for trial production, the materials for the application for the central competent authority for approval shall, instead of the preceding requirements, include (1) the pharmaceutical firm’s permit license; (2) the written trial production plan; (3) the factory registration documents issued by the Ministry of Economic Affairs, but research and development organizations without any factory are exempt from this requirement; (4) when another pharmaceutical firm is contracted to handle the importation of pharmaceutical ingredients for the trial production, the letter of instruction and the pharmaceutical firm permit licenses of the contracting party and the contractor are required.
For supplying “drugs for dined trials” of which the therapeutic efficacy and safety are not yet verified and which are provided for exclusive use in the pharmacological assessment of toxicity on animals or in clinical trials to approved teaching hospitals for use in clinical trials to confirm the safety and therapeutic efficacy, the pharmaceutical firm is required to obtain approval from the Taiwan Food and Drug Administration before supplying.
For pharmaceutical firms engaged in drug research and development, their products shall be manufactured by factories or establishments which comply with regulations promulgated by the Taiwan Food and Drug Administration.
Any pharmaceutical firms who violate the preceding requirements may be imposed a fine of not less than NT$30,000 but not more than NT$2,000,000.
New Drug Permit and Patent Linkage
The patent linkage refers to the mechanism that the holder of a new drug permit may submit the patent information regarding such drug to the designated website set up by the Ministry of Health and Welfare (“MOHW”) within 45 days after the next day of the receipt of the drug permit if the new drug permit holder deems a need to submit the patent information. In the event that the holder of a new drug permit obtains the approval of an application for an invention patent after the approval of said new drug permit from MOHW, the patent information thereof maybe submitted within 45 days after the next day of the patent issuance. If choosing not to or failing to make the preceding submissions within the 45-day period, the holder is no longer able to submit such patent information to the designated website to link the information to the new drug. The drug patent for the purpose of patent linkage shall be limited to the substance, composition or formulation, and medical use.
Once a new drug is linked with patent information, a generic drug company shall, during the application process for a generic drug permit, notify such new drug permit holder and MOHW if the generic drug is involved in a controversy against the patent corresponding to the said new drug that the generic drug company asserts the said patent shall be revoked or will not be infringed by the generic drug. When receiving the preceding notification, the new drug permit holder may choose to initiate litigation against the said generic drug company within 45 days after the next day of its receipt of said notification and notify MOHW. If the litigation is initiated and notified to MOHW, MOHW shall stay the issuance of the drug permit for the said generic drug company for 12 months as of the next day of the preceding notification from the generic drug company to the new drug permit holder.
For granting a new drug permit, the MOHW is required to publish the patent numbers or file numbers that have already been disclosed to the public as submitted by the applicant for such new drug permit. Given that we currently do not have any pharmaceutical product, we have not filed any patent information relevant to a new drug.
Medicament Advertisements
Under the Pharmaceutical Affairs Act, medicament advertisements can only be made by pharmaceutical firms that are duly registered with the authority. For publishing or disseminating medicament advertisements, pharmaceutical firms shall, before publishing or disseminating, submit all texts, drawings, or pictures constituting an advertisement to the central or municipal competent authority for approval and shall forward the approval to media enterprises carrying out the advertisement for verification. Where medicaments are required to have the prescriptions of physicians or to have been specifically designated by public notice(s) made by the central competent authority, the advertisements thereof shall be published only in academic medical journals. Interviews, news reports, or propaganda containing information implying or suggesting medical efficacy shall be regarded as advertisements of drugs.
Any pharmaceutical firm who violates the preceding requirements may be imposed a fine of not less than NT$ 200,000 but not more than NT$ 5,000,000, and the competent authority is required to announce in the newspaper the name of the person(s) responsible, the name of the drug, and the act of violation. In case of serious violation, the permit license previously granted to the said drug may also be revoked, and no application for use of the original name of the said drug shall be made within a period of two years thereafter.
All of our drug candidates are in development stage, accordingly we do not currently publish or disseminate medicament advertisements that are subject to the Pharmaceutical Affairs.
Clinical Practice for Medicaments
In Taiwan, the regulations for clinical trials are primarily governed by the Ministry of Health and Welfare (“MOHW”). The main regulatory documents that outline the requirements and guidelines for conducting clinical trials in Taiwan are the “Regulations for Good Clinical Practice (藥品優良臨床試驗作業準則, “GCP Regulation”)” and its guideline, the “Guidance for Good Clinical Practice (藥品優良臨床試驗作業指引),” as a supplementary explanation. When clinical trials are involved in human subjects, the “Medical Care Act,” “Human Subjects Research Act (人體研究法),” “Regulations on Human Trials (人體試驗管理辦法)” Pharmaceutical Affairs Act (藥事法), and Personal Data Protection Act (個人資料保護法) also apply. These regulations provide the legal framework of Good Clinical Practice (“GCP”) for the ethical, scientific, and quality standards for practical operations in the design, execution, recording, and reporting of pharmaceutical clinical trials.
Basic Principles
Under the GCP Regulation, clinical trials should be conducted in accordance with the ethical principles of the Declaration of Helsinki. Before initiated, a trial should be conducted by weighing foreseeable risks and inconveniences against the anticipated benefit for the trial subject and society and continued only if the anticipated benefits justify the risks and inconveniences. When weighing the benefits, the rights, safety, and well-being of the trial subjects are the most important considerations and should prevail over interests of science and society.
Independent Ethics Review
For the purpose of clinical trial review, the institution carrying out the trials is required to establish an ethics committee to review and evaluate the science, medical aspects, and ethics of the proposed trial. The aspects involved in the ethics review for the clinical trial include the following.
| | 1. | Clinical trial protocols: Clinical trial protocols must be submitted for review and approval by an ethics committee. |
| | | |
| | 2. | Composition of the ethics committee: The ethics committee consists of experts from various disciplines, including medical and nonscientific, and a member who is independent of the trial institution/site to ensure a multidisciplinary approach. |
| | | |
| | 3. | Competence: Ethics committee members must possess the necessary qualifications, expertise, and experience to carry out thorough and fair reviews. |
| | | |
| | 4. | Review process: The ethics committee evaluates the ethical aspects, participant protection measures, and scientific validity of the trial. |
| | | |
| | 5. | Informed consent: The ethics committee reviews the informed consent process to ensure that participants are provided with adequate information, have the capacity to give consent, and consent voluntarily. |
| | | |
| | 6. | Participant protection: The ethics committee assesses measures taken to protect the rights, safety, and welfare of trial participants, particularly vulnerable populations. |
| | | |
| | 7. | Conflict of interest: If the ethics committee members find that conflict of interest occurs, the concerned members shall recuse themselves from the review. |
Investigator
Investigators are responsible for the conduct of the clinical trial and ensuring compliance with GCP. The requirements for the investigators are as follows.
| | 1. | Qualifications: Investigators must possess appropriate qualifications, training, and experience relevant to the conduct of clinical trials. They shall have the necessary knowledge and expertise in the therapeutic area under investigation. |
| | | |
| | 2. | Responsibilities: Investigators are responsible for the overall conduct of the clinical trial and ensuring compliance with GCP and relevant regulations. They are required to strictly adhere to the approved protocol, ensuring that all trial procedures, interventions, assessments, and data collection are performed as specified. |
| | | |
| | 3. | Informed consent: Investigators must obtain informed consent from trial participants or their legally authorized representatives in accordance with applicable laws and regulations. They shall ensure that participants have been provided with sufficient information and have understood the nature, purpose, risks, and potential benefits of participating in the trial. |
| | | |
| | 4. | Participant safety: Investigators are responsible for monitoring participant safety throughout the trial. They are required to promptly report and appropriately manage any adverse events or unexpected problems encountered during the study. |
| | | |
| | 5. | Study conduct: Investigators must adhere to the approved protocol and conduct the trial in accordance with GCP and relevant regulations. They shall ensure the accurate and timely collection of trial data and maintain proper documentation of all trial-related activities. |
| | | |
| | 6. | Record-keeping: Investigators must maintain accurate and complete records of the trial, including essential documents such as the protocol, informed consent forms, adverse event reports, and any protocol deviations or amendments. |
| | | |
| | 7. | Ethics committee communication: Investigators shall promptly inform the ethics committee of any significant changes or issues related to the conduct of the trial, such as protocol deviations, serious adverse events, or participant complaints. |
| | | |
| | 8. | Supervision: Investigators shall ensure that all trial staff members receive appropriate supervision to perform their duties effectively and in compliance with GCP Regulation. |
Sponsor
Sponsors have overall responsibility for the initiation and management of the clinical trial. They must ensure compliance with GCP and have a qualified team to oversee the trial. The requirements for the sponsors are as follows.
| | 1. | Quality management, assurance, and control: The sponsor is responsible for implementing a system to manage quality throughout all stages of the trial process and maintaining systems to assure and control quality to ensure that trials are conducted, and data are generated, documented, and reported in compliance with the protocol, GCP, and applicable regulations. |
| | | |
| | 2. | Qualified personnel: The sponsor must have a qualified team to oversee the trial, including individuals with appropriate expertise in clinical research, management and verification of data, statistical analysis, and safety monitoring. |
| | | |
| | 3. | Trial design and conduct: The sponsor shall utilize qualified individuals (e.g., biostatisticians, clinical pharmacologists, and physicians) as appropriate throughout all stages of the trial process to design the protocol, conduct the analyses, and prepare interim and final clinical trial reports. |
| | | |
| | 4. | Data management: The sponsor is responsible for establishing data management procedures and systems to ensure the quality, accuracy, and integrity of the trial data. This includes implementing appropriate measures for data collection, entry, verification, and archiving. |
| | | |
| | 5. | Investigator selection and oversight: The sponsor is responsible for selecting qualified investigators and ensuring that they receive appropriate support. |
| | | |
| | 6. | Regulatory submissions: The sponsor is responsible for preparing and submitting regulatory documents and applications required for the approval and progress of the clinical trial. This includes submitting the clinical trial protocol, informed consent forms, and any amendments to the relevant regulatory authorities. |
| | | |
| | 7. | Supplying and handling investigational products: The sponsor shall supply the investigator/ trial institution with the investigational products only after the approval of the trial. |
| | 8. | Safety information: The sponsor is responsible for the ongoing safety evaluation of the investigational products throughout the study. This includes establishing procedures for the timely detection, reporting, and evaluation of adverse events and serious adverse events. |
| | | |
| | 9. | Adverse event reporting: The sponsor is responsible for ensuring that adverse events and other safety-related information are reported in accordance with regulatory requirements and within specified timelines. The sponsor shall promptly notify the relevant authorities, ethics committee, investigators, and other involved parties as required. |
| | | |
| | 10. | Record-keeping: The sponsor should maintain complete and accurate records of the trial, including all essential documents such as the protocol, informed consent forms, investigator agreements, and correspondence with regulatory authorities and ethics committees. |
| | | |
| | 11. | Monitoring: To assure the rights of trial subjects, the accuracy, completeness, and verifiability of trial data, and the compliance of the trial with applicable requirements, the sponsor shall appoint monitors to carry out activities, including (a) acting as the communication line between the sponsor and investigator; (b) verifying that the investigator has and remain adequate qualifications and resources; (c) verifying matters regarding the investigational products; and (d) verifying the compliance of other required documents and process with applicable regulations. |
Clinical Trial Protocol
The clinical trial protocol is a document that describes the objective(s), design, methodology, statistical considerations, and institution of a trial. The protocol or its referenced documents usually also gives the background and rationale for the trial. The requirements for the protocol are as follows:
| | 1. | Trial design: The protocol shall specify the trial’s primary and secondary endpoints (if any), treatment interventions, participant eligibility criteria, and sample size determination. |
| | | |
| | 2. | Study procedures: The protocol shall outline the specific procedures to be followed during the trial, including subject screening, randomization, blinding, treatment administration, assessments, and follow-up visits. It shall also specify the frequency and timing of data collection. |
| | | |
| | 3. | Subject considerations: The protocol must set out the criteria of subject inclusion and exclusion. |
| | | |
| | 4. | Treatment of subjects: The protocol must provide detailed information about the administered treatments, including the names of all products, doses, dosing schedules, and treatment periods. |
| | | |
| | 5. | Assessment of efficacy and safety: The protocol must provide the specifications of the efficacy parameters and safety parameters and the methods for assessing, recording, and analyzing thereof. |
| | | |
| | 6. | Statistical considerations: The protocol shall specify the statistical methods to be used for data analysis, including any planned interim analyses, subgroup analyses, or sensitivity analyses. It shall also address how missing data or protocol deviations will be handled in the analysis. |
Investigator’s Brochure
The investigator’s brochure (IB) is a compilation of the clinical and non-clinical data on the investigational product. Its purpose is to provide the investigators and others involved in the trial with the information to facilitate their understanding of the rationale for, and their compliance with, many key features of the protocol. The material contents of the IB shall be as follows.
| | 1. | Content: The IB should provide comprehensive and up-to-date information about the investigational product. It should include details such as the drug’s composition, physical and chemical properties, pharmacological and toxicological characteristics, and known or potential risks and adverse reactions. |
| | | |
| | 2. | Non-clinical data: The IB should summarize the non-clinical data, including non-clinical pharmacology, toxicology, pharmacokinetics, and product metabolism in animal studies. |
| | | |
| | 3. | Effects on Humans: The IB should include a thorough discussion of the known effects of the investigational product on humans. This may include information on the drug’s efficacy, safety profile, dosage regimen, and any known interactions with other drugs. |
| | | |
| | 4. | Dosage and administration: The IB should provide guidance on the recommended dosage, route of administration, and duration of treatment. It should also include information on dose adjustments for special populations, such as elderly patients or those with impaired organ function. |
As Everfront Biotech conducts clinical trials to develop drugs in Taiwan, we have adhered to all the required procedures and guidance for clinical practice when conducting clinical trials.
Personal Data Protection Laws
As we engage in the business of new drug development, including non-clinical and clinical trials on human subjects, we may collect, process, and use the personal data of these subjects. Aside from stipulations set forth in clinical practice regulations and guidelines, the main legislation governing personal data protection for our business is the Personal Data Protection Act of Taiwan.
Grounds for Collecting Personal Data
Under Article 19 of the Personal Data Protection Act, the collection or processing of personal data by a private agency shall be for specific purposes and on one of the following bases:
| | 1. | where it is expressly required by law; |
| | | |
| | 2. | where there is a contractual or quasi-contractual relationship between the private agency and the data subject, and proper security measures have been adopted to ensure the security of the personal data; |
| | | |
| | 3. | where the personal data has been disclosed to the public by the data subject or has been made public lawfully; |
| | | |
| | 4. | where it is necessary for statistics gathering or academic research by an academic institution in pursuit of public interests, provided that such data, as processed by the data provider or as disclosed by the data collector, may not lead to the identification of a specific data subject; |
| | | |
| | 5. | where consent has been given by the data subject; |
| | | |
| | 6. | where it is necessary for furthering public interest; |
| | | |
| | 7. | where the personal data is obtained from publicly available sources unless the data subject has an overriding interest in prohibiting the processing or use of such personal data; or |
| | | |
| | 8. | where the rights and interests of the data subject will not be infringed upon. |
Personal Data Notice
Under Article 8 of the Personal Data Protection Act, a private agency shall give a clear notice to the data subject of the following information when colleting their personal data in accordance with Article 19 thereof:
| | 1. | the name of the collecting agency; |
| | | |
| | 2. | the purpose of the collection; |
| | | |
| | 3. | the categories of the personal data to be collected; |
| | | |
| | 4. | the time period, territory, recipients, and methods by which the personal data is used; |
| | | |
| | 5. | the data subject’s rights under Article 3 and the methods for exercising such rights; and |
| | | |
| | 6. | the data subject’s rights and interests will be affected if he/she elects not to provide his/her personal data. |
Proper Security Measures
Under Article 27 of the Personal Data Protection Act, a private agency in possession of personal data files shall implement proper security measures to prevent personal data from being stolen, altered, damaged, destroyed, or disclosed.
Article 12 of the Enforcement Rules of the Personal Data Protection Act provides that the proper security measures shall refer to the technical or organizational measures taken by a private agency for the purpose of preventing personal data from being stolen, altered, damaged, destroyed, or disclosed. These measures may include the following and shall be proportionate to the intended purposes of personal data protection:
| | 1. | allocating management personnel and reasonable resources; |
| | | |
| | 2. | defining the scope of personal data; |
| | | |
| | 3. | establishing a mechanism of risk assessment and management of personal data; |
| | | |
| | 4. | establishing a mechanism of preventing, giving notice of, and responding to a data breach; |
| | | |
| | 5. | establishing an internal control procedure for the collection, processing, and use of personal data; |
| | | |
| | 6. | managing data security and personnel; |
| | | |
| | 7. | promoting awareness, education, and training; |
| | | |
| | 8. | managing facility security; |
| | | |
| | 9. | establishing an audit mechanism of data security; |
| | | |
| | 10. | keeping records, log files, and relevant evidence; and |
| | | |
| | 11. | implementing integrated and persistent improvements on the security and maintenance of personal data. |
In view that the trial subjects provide their personal data for clinical trials on the developing drug, our Taiwan subsidiary obtains the bases to collect their personal data by either establishing a contractual relationship with the subjects or obtaining subjects’ consent. In addition, we have developed a “Personal Data Protection Management Procedure” to ensure compliance with this legislation, under which we ensure appropriate technical and organizational measures to protect personal data from damage, theft, unauthorized alteration, or disclosure. To ensure the effectiveness of our data protection measures, professionals have been employed to monitor and manage the processing of personal data and conduct regular security reviews and assessments. Furthermore, in the ongoing clinical trials, we also adhere to relevant regulations and clearly state the purposes of data usage and the methods of data de-identification in the informed consent form.
Laws and Regulations Governing Employment and Labor Relationship
As an employer in Taiwan, Everfront Biotech is required to comply with various laws and regulations in relation to employment and labor relationship.
Work Hours and Overtime
The regular working time of employees may not exceed 8 hours a day or 40 hours a week. To have an employee perform the work overtime, the employer is required to obtain the consent of a labor union or. If there is no labor union, an approval of a labor-management conference is required.
Under Article 24 of the Labor Standards Act, an employer shall pay overtime wages to an employee using the following basis:
For the first 2 hours of overtime on the regular working day, the employee shall be paid at least one and one-third of the regular hourly rate.
For hours beyond the first 2 hours of overtime on the regular working day, the employee shall be paid at least one and two-thirds of the regular hourly rate.
When the employee is required to work on the rest days. For the first 2 hours, the employee shall be paid at least one and one-third of the regular hourly rate. As to the overtime work is over two hours, the employee shall be paid at least one and two-thirds of the regular hourly rate.
The overtime hours combined with the regular working hours shall not exceed 12 hours a day, and the total number of overtime shall not exceed 46 hours a month. However, the 46-hour-a-month limit of overtime hours may be extended to 54 hours a month and 138 hours every three months if the employer obtains the consent of a labor union or, if there is no labor union in a business entity, the approval of a labor-management conference.
An employee shall be permitted to have a break for at least 30 minutes after having worked for 4 continuous hours; provided, however, that such break may be rescheduled by the employer to be taken within other working hours if a rotation system is adopted or work of a continuous or urgent nature is involved.
Termination of Employment (Labor) Contract
Termination by the Employee
An employee may terminate a labor contract by giving prior notice of a given period based on the service period. However, an employee may terminate a labor contract without giving advance notice to the employer where one of the following situations arises:
| | 1. | an employer misrepresents any fact at the time of signing a labor contract in a manner which might mislead his/her employee and thus caused him/her to sustain damage there from; |
| | | |
| | 2. | an employer, his/her family member, or his/ her agent commits violence or grossly insults the employee; |
| | | |
| | 3. | the work specified in a labor contract is likely to be injurious to the employee’s health and the employee has requested his/her employer to improve working conditions but all in vain; |
| | | |
| | 4. | the employer, the agent of the employer, or co-worker suffers from a noted contagious disease that may infect employees working with the infected person and seriously endanger their health; |
| | | |
| | 5. | an employer fails to pay for work in accordance with the labor contract or to give sufficient work to a worker who is paid on a piecework basis; or |
| | | |
| | 6. | an employer breaches a labor contract or violates any labor statute or administrative regulation in a manner likely to adversely affect the rights and interests of the particular employee. |
Termination for Cause
Under Articles 11 and 13 of the Labor Standards Act, any employer shall not terminate a labor contract with an advance notice unless one of the following situations arises:
| 1. | The employers’ businesses are suspended or have been transferred. |
| 2. | The employers’ businesses suffer operating losses or business contractions. |
| 3. | Force majeure necessitates the suspension of business for more than one month. |
| 4. | A change in of the nature of business necessitates the reduction of the workforce, and the terminated employees cannot be reassigned to other suitable positions. |
| 5. | A particular employee is clearly not able to perform satisfactorily the duties required of the position held. |
| 6. | The employer cannot continue operating the business due to an act of God, catastrophe, or other force majeure and prior approval has been obtained from the competent authorities under the circumstances that the particular employee is on maternity leave or receiving medical treatment due to occupational accidents. |
Under Article 16 of the Labor Standards Act, advance notice shall be given when any employer terminates a labor contract pursuant to preceding provisions, and the notice period is prescribed as follows:
| 1. | Where an employee has worked continuously for more than three months, but less than one year, the notice shall be given ten days in advance. |
| 2. | Where an employee has worked continuously for more than one year but less than three years, the notice shall be given twenty days in advance. |
| 3. | Where an employee has worked continuously for more than three years, the notice shall be given thirty days in advance. |
Article 17 of the Labor Standards Act provides that any employer who terminates a labor contract in accordance with the preceding Article shall pay severance pay.
Disciplinary Discharge
Under Article 12 of the Labor Standards Act, if the employee commits the following illegal or improper acts, an employer may terminate the labor contract without advance notice, and no severance pay is required where an employee:
| 1. | misrepresents any fact at the time of signing of a labor contract in a manner which might mislead his/ her employer who sustains damage therefrom; |
| 2. | commits a violent act against or grossly insults the employer, his /her family member or agent of the employer, or a fellow worker; |
| 3. | has been sentenced to temporary imprisonment in a final and conclusive judgment and is not granted a suspended sentence or permitted to commute the sentence to payment of a fine; |
| 4. | is in serious breach of the labor contract or in serious violation of work rules; |
| 5. | deliberately damages or abuses any machinery, tool, raw materials, product, or other property of the employer or deliberately discloses any technical or confidential information of the employer, thereby causing damage to the employer; and |
| 6. | is, without good cause, absent from work for three consecutive days or for a total of six days in any month. |
Forced Discharge Due to Causes Attributable to the Employer
Under Article 14 of the Labor Standards Act, any employee may terminate a labor contract without giving advance notice to the employer in any of the following situations.
| 1. | Where an employer misrepresents any fact at the time of signing a labor contract in a manner which might mislead his/her employee who sustains damage there from. |
| 2. | Where an employer, his/her family member, or his/ her agent commits violence or grossly insults the employee. |
| 3. | Where the work specified in a labor contract is likely to be injurious to the employee’s health, and the employee has requested his/her employer to improve working conditions but all in vain. |
| 4. | The employer, the agent of the employer, or co-worker suffers from a noted contagious disease that may infect employees working with the infected person and seriously endanger their health. |
| 5. | Where an employer fails to pay for work in accordance with the labor contract or to give sufficient work to an employee who is paid on a piecework basis. |
| 6. | Where an employer breaches a labor contract or violates any labor statute or administrative regulation in a manner likely to adversely affect the rights and interests of the particular employee. |
Leave and Holidays
An employee shall have two regular days off every seven days. One day is a regular leave, and the other one is a rest day. Leave shall be granted for national holidays, holidays, and Labor Day, which are designated as holidays by the Ministry of the Interior and holidays designated by other Central Competent Authority. Under Article 38 of the Labor Standards Act, an employee who has worked continually for the same employer for a certain period of time shall be granted annual paid leave on an annual basis based on the following conditions.
| 1. | 3 days for service of 6 months or more but less than 1 year; |
| 2. | 7 days for service of 1 year or more but less than 2 years; |
| 3. | 10 days for service of 2 years or more but less than 3 years; |
| 4. | 14 days for service of 3 years or more but less than 5 years; |
| 5. | 15 days for service of 5 years or more but less than 10 years; and |
| 6. | one additional day for each year of service over 10 years up to a maximum of 30 days. |
Annual leave is to be arranged by employees. The employer, however, in the light of urgent needs of the business operation or personal factors of employees, may consult and make adjustments with employees. The employer shall inform the employee to arrange the annual leave when the employee meets the conditions for the annual leave. Wages for annual leave must be paid when the leave is not used by employees at the time of the end of such year or termination of contracts. However, the employer may negotiate with the employee for unused annual leave to extend to the following year, and wages of the extended annual leave shall be paid for those not used by employees at the end of the following year or upon the termination of contracts.
Transfer of Position
Under Article 10-1 of the Labor Standards Act, when transferring an employee to another position or location, the employer is obliged to not violate the provisions of the labor/employment agreement and also satisfy the following five principles:
| 1. | The employee shall be transferred based on the needs of business operation and without improper motives or purposes. Matters not provided for herein shall be governed by other applicable statutes; |
| 2. | The wages and other working conditions shall not be changed to be unfavorable to the employee concerned; |
| 3. | The employee shall still be able to satisfactorily perform the duties required in terms of physical ability and skills after the transfer; |
| 4. | The employer shall provide necessary assistance if the relocated workplace where is too far away for the employee concerned; and |
| 5. | The livelihood interests of the employee and his or her family shall be considered. |
After-termination-non-compete Clause
Under Article 9-1 of the Labor Standards Act, the after-termination-non-compete clause in a labor/employment contract may be enforceable provided that the following requirements have been met:
| 1. | The employer has proper business interests that require being protected; |
| 2. | The position or job of the employee entitles him or her to have access to or be able to use the employer’s trade secrets; |
| 3. | The period, area, scope of occupational activities, and prospective employers with respect to the after-termination-non-compete clause shall not exceed a reasonable range; and |
| 4. | The employer shall reasonably compensate the employee concerned who does not engage in competition activities for the losses incurred by him or her. |
The reasonable compensation shall not include the remuneration received by the employee during employment and shall be in a lump sum upon resignation or on a monthly basis to the employee after resignation. The amount of compensation per month shall be no less than 50% of one month’s average wage of the employee upon resignation.
The period of non-compete shall not exceed a maximum of two years. If such a period is more than two years, then it shall be shortened to two years.
The enforceability of the provision is subject to review by the court on a case-by-case basis over whether there is any business secret of the employer accessible by the employee concerned and whether the limitation on period, area, scope of occupational activities, and prospective employers are reasonable.
Mandatory Insurance (Social Security)
Labor Insurance
Under Article 6 of the Labor Insurance Act, employees employed by a company or firm with more than five employees above 15 full years and below 65 years of age shall all be enrolled under this Labor Insurance as insured persons through their employers or the organizations or institutes to which the employees belong, which are reckoned as the insured units.
Employers who employ four employees or fewer do not have an obligation to insure their employees under the Labor Insurance, but the employer can still opt to do so, pursuant to Article 8 thereof.
Employment Insurance
Under Article 5 of the Employment Insurance Act, an employed employee who is a Taiwan (R.O.C.) national or a foreign national, Mainland Chinese citizen, Hong Kong citizen, or Macao citizen married to a R.O.C. citizen and having acquired legal residency in R.O.C. with age over 15 and under 65 years is required to join this Employment Insurance as an insured person through his employer or the organization to which he or she belongs.
Labor Occupational Accident Insurance
Under Articles 6 to 10 of the Labor Occupational Accident Insurance and Protection Act, employees or persons employed by employers or performing work may enroll in this insurance through their employers or by themselves. An employer reckoned as an insured unit shall carry out insurance enrollment, insurance withdrawal procedure and other relevant insurance matters for its employees.
National Health Insurance
Under Articles 10 and 15 of the National Health Insurance Act, for employees of private enterprises or institutions, the enterprises or institutions for which they work for, shall be reckoned as insured units and shall set up special departments or agents to administer relevant matters of this Insurance.
Labor Pension
Under Article 6 of the Labor Pension Act, employers shall on a monthly basis contribute labor pension funds to individual labor pension accounts at the Bureau of Labor Insurance for employees covered by the Labor Pension Act.
The amount of the labor pension contribution borne by the employer for the employees designated under the Labor Standards Act shall not be less than 6% of the employee’s monthly wage, pursuant to Article 14 of the Labor Pension Act. Under Article 2 of the Labor Standards Act, an employee denotes a person who is hired by an employer to work for wages.
Gender Equality
Under the Act of Gender Equality in Employment, our Taiwan subsidiary may be subject to rules requiring us to eliminate sex discrimination and promote gender equality in Taiwan business operations from three perspectives.
| 1. | Our Taiwan subsidiary is prohibited from discriminating against employees on the basis of sex in the recruitment, hiring, evaluation, promotion, training, wage, benefits, retirement and severance, and other related systems; |
| 2. | Our Taiwan subsidiary is obliged to prevent and correct sexual harassment and establish measures for preventing sexual harassment, complaint procedures, and disciplinary measures, when hiring over thirty employees; and |
| 3. | Our Taiwan subsidiary is required to provide employees with menstrual leave, maternity leave, parental leave, time for feeding or breast milk collection, breastfeeding (breast milk collection) rooms or suitable childcare measures when having one hundred employees or more, and other measures for promoting equality in employment. |
MANAGEMENT
Executive Officers and Directors
The table below sets forth information concerning our directors, executive officers, and other key employees.
| Name | | Age | | Position |
| Dr. TZA-ZEN CHAUNG | | 72 | | Chief Executive Officer |
| PI-CHUAN CHIEN | | 56 | | Chief Financial Officer |
| Dr. YU-FANG HU | | 62 | | Chief Operating Officer |
| Dr. JEN-WEI LIU | | 41 | | Chief Scientific Officer |
| Dr. YUAN-SHENG LI | | 40 | | Chief Technology Officer |
| HO-CHING CHEN | | 67 | | Director |
| Dr. PEI-WEN CHOU | | 56 | | Director |
| HANN-CHYI LIN | | 72 | | Independent Director |
| Dr. TAI-NANG HUANG | | 74 | | Independent Director |
| YIN-HO HUANG | | 65 | | Independent Director |
DR. TZA-ZEN CHAUNG, Chief Executive Officer
Dr. Tza-Zen Chaung currently serves as our Chief Executive Officer and has served in this role since January 2023. Dr. Dr. Chaung has 40 years of experience in new drug development, pharmaceutical good manufacturing practice (“GMP”), manufacturing process development, and R&D management and strategy. Prior to serving as our Chief Executive Officer, Dr. Chaung was Senior Engineer at Merck & Company in Rahway, New Jersey from August 1982 to August 1989, where he worked on new drug development. He worked as Research Investigator with Hoffmann-La Roche in Nutley, New Jersey from June 1991 to August 1992. He worked as Group Leader with Bristol-Myers Squibb in New Brunswick, New Jersey from August 1992 to January 1995. From January 1995 to January 2023, Dr. Chaung has been working as a consultant for pharmaceutical and biotech industries in the United States and in Taiwan. Dr. Chaung obtained a Bachelor of Science degree in chemical engineering from the National Taiwan University in Taiwan, in 1974. Dr. Chaung obtained a Master of Science degree in chemical engineering from West Virginia University in West Virginia, in 1978. Dr. Chaung obtained a Doctor of Science degree from the Massachusetts Institute of Technology in Cambridge, Massachusetts in 1982.
MR. PI-CHUAN CHIEN, Chief Financial Officer
Mr. Pi-Chuan Chien currently serves as our Chief Financial Officer and has served as Chief Financial Officer since February 2023. Mr. Chien also currently serves as General Manager of Excellence Management Consulting Co., Ltd. in Taiwan, serving in this role since July 2007, where he works on introducing sophisticated cost management systems to businesses and provides financial accounting consulting services associated with U.S. initial public offering processes. Mr. Chien has accumulated considerable experience in financial statement audits and complex technical accounting topics. To this end, Mr. Chien previously served as Senior Manager in the audit practice of Ernst & Young Global Limited in Taiwan from July 2004 to July 2007. Mr. Chien previously served as Manager at BDO Taiwan from August 1995 to July 2004. Mr. Chien graduated from the National Chung Hsing University in Taiwan in 1995, with a Bachelor’s degree in accounting.
DR. YU-FANG HU, Chief Operating Officer
Dr. Yu-Fang Hu currently serves as our Chief Operating Officer and has served in this role since March 2024. Dr. Hu possesses skills and knowledge in the industrial pharmaceutical field with over 25 years of experience in organizational development and management. Dr. Hu has also successfully led and collaborated on multiple international business operation projects with relevant entities. Prior to serving as our Chief Operating Officer, Dr. Hu served in the roles of R&D Senior Manager, Vice President, and Senior Vice President of TTY Biopharm Co. Ltd., a biotech company in Taiwan, from April 1999 to September 2023, where one of his key responsibilities was development of TTY Biopharm Co. Ltd.’s product line. Since September 2023, Dr. Hu served as a Consultant with TTY Biopharm Co. Ltd. During his TTY Biopharm Co. Ltd. employment, Dr. Yu contributed to the development of liposome technology and long-acting microsphere injection platforms, launched the AmBisome generic drug (Liposomal Amphotericin B Injection) in the United States, and obtained the global contract manufacturing project for Doxil product (Liposomal Doxorubicin Injection) from Johnson & Johnson. From May 2019 to February 2021, Dr. Yu served as President & CEO of PharmaEngine Inc. (a listed company in Taiwan). Dr. Hu founded EnhanX Biopharm Inc., a clinical stage therapy development company in Taiwan, in 2017 and currently serves as Chairman of EnhanX Biopharm Inc. Dr. Hu obtained a Bachelor of Science degree in pharmacy from Kaohsiung Medical University in Taiwan in 1986. Dr. Hu obtained a Master of Science degree in pharmacy from St. John’s University in 1992. Dr Hu. obtained a Ph.D. degree in pharmacy from St. John’s University in 1999.
DR. JEN-WEI LIU, Chief Scientific Officer
Dr. Jen-Wei Liu currently serves as the Chief Scientific Officer of the Company and has served in this role since March 2024. Dr. Liu also currently serves as Director of the Clinical Research Director of the Subsidiary and has served in this role since September 2020. From December 2019 to August 2020, Dr. Liu served as Research Fellow at the Subsidiary. Dr. Liu has ten years of experience in new drug development, clinical management, and strategic planning. Dr. Liu has been involved in three (3) clinical trials regulated by the FDA and TFDA and is experienced in toxicology and pharmacokinetic studies. Dr. Liu has served as the principal investigator of national-level projects including A+ Industrial Innovation R&D Program (project number: 103-EC-17-A-20-I1-0004) from2014 to 2018, with a total budget of over $4 million. Dr. Liu has a strong scientific background and has published more than ten articles. Dr. Liu obtained a Bachelor’s degree in biological science from the department of biological science at National Sun Yat-Sen University in Taiwan in 2005. Dr. Liu obtained a Master’s degree in biotechnology from the Institute of Biotechnology at National Dong Hwa University in 2008. Dr. Liu obtained a Ph.D. degree in life sciences from the National Chung Hsing University in Taiwan, in 2013.
DR. YUAN-SHENG LI, Chief Technology Officer
Dr. Yuan-Sheng Li currently serves as the Chief Technology Officer of the Company and has served in this role since March 2024. Dr. Li also currently serves as Director of the Research, Development and Manufacturing Department for the Subsidiary and has served in this role since August 2018. From October 2017 to July 2018, Dr. Li served as Research Fellow at the Subsidiary. From July 2013 to September 2017, Dr. Li served as Associate Research Fellow at the Subsidiary. Dr. Li has ten years of experience in new drug development, pharmaceutical chemistry, manufacturing and controls, R&D management and strategy and his experience was acquired during his period of employment with the Subsidiary since July 2013.Dr. Li has co-written and published ten papers for stem cell therapy, drug formulation, and neuron science. Dr. Li graduated with a Bachelor’s degree in chemical engineering from the chemical engineering department at Chinese Culture University in Taiwan in 2006. Dr. Li graduated from the National Dong Hwa University in Taiwan, with a Ph.D. degree in biotechnology, in 2016.
MR. HO-CHING CHEN, Director
Mr. Ho-Ching Chen is our founder, and he currently serves as a Director of the Company. Mr. Chen also currently serves as a Consultant and Director of the Subsidiary and has served in both roles since December 2010.Mr. Chen is an investor and has invested in companies in the leisure industry, semiconductor industry, environmental protection industry, and the biotechnology and pharmaceutical industry. Mr. Chen is also the founder of: Sunflower Holiday Co., Ltd., a leisure industry company in Taiwan; Chiplus Semiconductor Corp., a semiconductor industry company in Taiwan; Tmg Group Wanji Technology Co., Ltd., an environmental protection industry company in Taiwan; Singapore Enviro-Hub holding Ltd., an environmental protection industry company in Singapore; Baining Biotech Co., Ltd., a biotechnology and pharmaceutical industry company in Taiwan; and Guang-Li Biomedicine Inc.(which effected a name change to “Xi Yue Biomedicine Inc.” on January 13, 2023), a biotechnology and pharmaceutical industry company in Taiwan. Mr. Chen was a major investor and served as chairman of Alishan Hotel Co., Ltd., a leisure industry company in Taiwan, from January 2015 to January 2021. Because of the pain of his family, he aspired to give back to the society, focusing on the development of new drugs such as cancer, neurodegenerative and rare diseases, hoping to benefit the community affected by diseases. Mr. Chen obtained a degree in international business administration from Ching Yun University in Taiwan, in 2005. Mr. Chen obtained a Master of Business Administration degree from Jinan University in China, in 2009.
DR. PEI-WEN CHOU, Director
Dr. Pei-Wen Chou has served as a Director of the Company since April 13, 2023. Dr. Chou also currently serves as Chief Executive Officer and President of the Subsidiary and has served in these roles since December 2010. Dr. Chou also currently serves as President and Director of Hawking Biological Technology Co. Ltd, a biotechnology company in Taiwan, and has served in these roles since January 2013 and December 2010, respectively. Dr. Chou previously served as chairman of Guang-Li Biomedicine Inc. from February 2011 to July 2014. Dr. Chou has 20 years of experience in clinical practice, establishment and management of biotech companies, and as a nurse practitioner. To this end, Dr. Chou previously served as Specialist/Leader of the cardiac surgery team and intensive care units (ICUs) in a medical center from 1993 to 2002. Dr. Chou obtained a Bachelor’s degree in nursing and health from Deh Yu College in Taiwan, in 1987. Dr. Chou obtained a Master’s degree from the graduate institute of medicine of Nobel College in 2001. Dr. Chou obtained a Ph.D. degree in homeopathy medicine from Taman University in Malaysia, in 2005.
MR. HANN-CHYI LIN, Independent Director
Mr. Hann-Chyi Lin was appointed as Independent Director of the Company on April 13, 2023. Mr. Lin also currently serves as Independent Director of Taiwan Cooperative Financial Holding Co., Ltd. in Taiwan, and has served in this role since June 2020. Mr. Lin served as Manager of First Bank’s Hong Kong branch from February 2005 to April 2006 and again from February 2007 to March 2009. Mr. Lin served as Manager of the international department of First Bank in Taiwan from February 2002 to February 2005. Mr. Lin served as Manager of First Bank in its Singapore branch from July 1997 to August 2001. Mr. Lin was employed by First Commercial Bank Co., Ltd. (“First Bank”) in its Wanhua branch in Taiwan from March 1976 to September 1979. He served as Chairman of First Securities Incorporation in Taiwan from October 2014 to November 2016. Mr. Lin is proficient in the operation of foreign exchange funds of banks, the operation and management of foreign branches, and has extensive experience in internal controls, audit system operations and overall operation and management of financial holding companies, banks and securities companies. Mr. Lin obtained a Bachelor’s degree in statistics from Feng Chia University in Taiwan in 1973.
DR. TAI-NANG HUANG, Independent Director
Dr. Tai-Nang Huang was appointed as Independent Director of the Company on April 13, 2023. Dr. Huang currently serves as General Manager of Linden Technologies, Inc. and has served in this role since April 2000. Dr. Huang has served as a member of the board of directors of Pyrexar Medical Inc. Dr. Huang has extensive experience in gene chip innovation for cancer biomarker discovery and new drug research, and drug metabolism investigations. Dr. Huang obtained a Bachelor of Science degree in chemistry from National Taiwan University, in 1972. Dr. Huang obtained a Master of Science degree in chemistry from the University of Goettingen in Germany, in 1977. Dr. Huang obtained a Ph.D. degree in chemistry from the Australian National University, in Australia, in 1980. Dr. Huang was a postdoctoral research associate at Princeton University from 1980 to 1982.
MR. YIN-HO HUANG, Independent Director
Mr. Yin-Ho Huang was appointed as Independent Director of the Company on April 13, 2023. Mr. Huang currently serves as the Director of Tianxia Law Firm and has served in this role since January 1993.Prior to joining Tianxia Law Firm, Mr. Huang was a lawyer with Jiying Law Firm in Taiwan from March 1992 to February1993. Mr. Huang is currently a practicing lawyer and patent attorney in Taiwan. Mr. Huang obtained his Bachelor’s degree from the Law Department of National Taiwan University in Taiwan in 1988.
Advisory Board
PROFESSOR SHINN-ZONG LIN, Everfront Biotech Holding Company Limited Advisor
Professor Shinn-Zong Lin currently serves as Superintendent of Hualien Tzu Chi Hospital, the President of the Buddhist Tzu Chi Bioinnovation Center in Tzu Chi Foundation, and as Professor of Neurosurgery at Tzu Chi University. Professor Lin is an authority in brain science with 40 years of clinical medicine and clinical trial experience.
Professor Lin’s qualifications: Charter Fellow of the National Academy of Inventors (USA); Fellow of the American Association for the Advancement of Science (USA); International Fellow of the American Association of Neurological Surgery (USA); Fellow of the American Institute of Medical and Biological Engineering (USA); Ph.D. degree in Physiology and Biophysics and Neurological Surgery from the State University of New York at Stony Book, USA.
PROFESSOR HORNG-JYH HARN, Everfront Biotech Holding Company Limited Advisor
Professor Horng-Jyh Harn currently serves as Associate Vice President of the Bioinnovation Center in Tzu Chi Foundation; and is also a professor of pathology at Tzu Chi University. Professor Harn has over 40 years of experience in clinical medicine and clinical trials. Additionally, Professor Harn is an expert in drug toxicity and pharmacokinetics.
Professor Harn’s qualifications: Charter Fellow of the National Academy of Inventors (USA), Ph.D. degree in Pathology from Duke University in Durham, USA.
PROFESSOR TZYY-WEN CHIOU, Everfront Biotech Holding Company Limited Advisor
Professor Tzyy-Wen Chiou is a Professor at the Department of Life Science and Graduate Institute of Biotechnology in National Dong Hwa University. Professor Chiou has conducted numerous industry-academia collaborative projects, and has thirty years’ experience in CMC, biochemical engineering, translational medicine and chemistry, manufacturing, and controls, or CMC.
Professor Chiou graduated from the Massachusetts Institute of Technology, USA, with a Ph.D. degree in Biochemical Engineering in 1992.
PROFESSOR KWEI-HANG CHAN, Everfront Biotech Holding Company Limited Advisor
Professor Kwei-Hang Chan previously served as a senior manager of the FDA drug review division. Professor Chan has 30 years of experience in new drug development and FDA regulatory review experience.
Professor Chan graduated from the University of Minnesota, USA, with a Ph.D. degree in Pharmacy in 1980.
Family Relationships
None of the directors or executive officers have a family relationship as defined in Item 401 of Regulation S-K.
Involvement in Certain Legal Proceedings
To the best of our knowledge, none of our directors or executive officers has, during the past ten years, been involved in any legal proceedings described in subparagraph (f) of Item 401 of Regulation S-K.
Board of Directors
Our board of directors will consist of five directors upon the effectiveness of the registration statement of which this prospectus forms a part. Our board of directors have determined that each of our three independent directors, Mr. Hann-Chyi Lin, Dr. Tai-Nang Huang and Mr. Yin-Ho Huang, satisfies the “independence” requirements of Rule 5605(a) (2) of the Listing Rules of Nasdaq Stock Market and Rule 10A-3 under the Exchange Act.
Duties of Directors
Under Cayman Islands law, all of our directors owe fiduciary duties to the Company, including a duty of loyalty, a duty to act honestly and a duty to act in what they consider in good faith to be in our best interests. Our directors must also exercise their powers only for a proper purpose. Our directors also have a duty to exercise the skill they actually possess and such care and diligence that a reasonably prudent person would exercise in comparable circumstances. In fulfilling their duty of care to us, our directors must ensure compliance with our memorandum and articles of association, as amended from time to time. We have the right to seek damages if a duty owed by any of our directors is breached. In limited exceptional circumstances, a shareholder may have the right to seek damages in our name if a duty owed by our directors is breached.
Terms of Directors and Executive Officers
Each of our directors holds office until a successor has been duly elected and qualified unless the director was appointed by the board of directors, in which case such director holds office until the next following annual meeting of shareholders at which time such director is eligible for re-election. All of our executive officers are appointed by and serve at the discretion of our board of directors.
Qualification
There is currently no shareholding qualification for directors, although a shareholding qualification for directors may be fixed by our shareholders by ordinary resolution.
Insider Participation Concerning Executive Compensation
Our board of directors, which currently consists of Mr. Ho-Ching Chen, Dr. Pei-Wen Chou, Mr. Hann-Chyi Lin, Mr. Yin-Ho Huang and Dr. Tai-Nang Huang and will also consist of three independent directors whose appointments will be effective as of the effectiveness of the registration statement of which this prospectus forms a part, is making all determinations regarding executive officer compensation from the time the Company first entered into employment agreements with executive officers up until the time where the three independent directors will be installed.
Committees of the Board of Directors
We will establish three committees under the board of directors immediately upon the effectiveness of the registration statement of which this prospectus forms a part: an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee. Even though we are exempted from certain corporate governance standards because we are a foreign private issuer and a controlled company, we have voluntarily adopted a charter for each of the three committees. Each committee’s members and functions are described below.
Audit Committee. Our Audit Committee will consist of Mr. Hann-Chyi Lin, Mr. Yin-Ho Huang and Dr. Tai-Nang Huang. We have determined that each of these directors will satisfy the “independence” requirements of Section 5605(a) (2) of the Nasdaq Listing Rules and Rule 10A-3 under the Exchange Act. Our board also has determined that Mr. Hann-Chyi Lin qualifies as an Audit Committee financial expert within the meaning of the SEC rules or possesses financial sophistication within the meaning of the Nasdaq Listing Rules. The Audit Committee will oversee our accounting and financial reporting processes and the audits of the financial statements of our company. The Audit Committee will be responsible for, among other things:
| ● | appointing the independent auditors and pre-approving all auditing and non-auditing services permitted to be performed by the independent auditors; |
| ● | reviewing with the independent auditors any audit problems or difficulties and management’s response; |
| ● | discussing the annual audited financial statements with management and the independent auditors; |
| ● | reviewing the adequacy and effectiveness of our accounting and internal control policies and procedures and any steps taken to monitor and control major financial risk exposures; |
| ● | reviewing and approving all proposed related party transactions; |
| ● | meeting separately and periodically with management and the independent auditors; and |
| ● | monitoring compliance with our code of business conduct and ethics, including reviewing the adequacy and effectiveness of our procedures to ensure proper compliance. |
Compensation Committee. Our Compensation Committee will consist of Mr. Yin-Ho Huang, Mr. Hann-Chyi Lin and Dr. Pei-Wen Chou. Mr. Huang and Mr. Lin are independent directors under Rule 5065(a)(2) of the Nasdaq Listing Rules, and Dr. Chou is not independent under these rules. We are relying upon our status as a “controlled company” to be exempt from Nasdaq rules that require that our Compensation Committee be composed entirely of independent directors.
The Compensation Committee will assist the board in reviewing and approving the compensation structure, including all forms of compensation, relating to our directors and executive officers. Our chief executive officer may not be present at any committee meeting during which his compensation is deliberated. The Compensation Committee will be responsible for, among other things:
| ● | reviewing and approving to the board with respect to the total compensation package for our most senior executive officers; |
| ● | approving reviewing and recommending to the board with respect to the compensation of our directors; and overseeing the total compensation package for our executives other than the most senior executive officers; |
| ● | selecting compensation consultants, legal counsel or other advisors after taking into consideration all factors relevant to that person’s independence from management; and |
| ● | programs or similar arrangements, annual bonuses, employee pension and welfare benefit plans. |
Nominating and Corporate Governance Committee. Our Nominating and Corporate Governance Committee will consist of Dr. Pei-Wen Chou, Mr. Ho-Ching Chen and Mr. Hann-Chyi Lin. Mr. Lin is an independent director under Rule 5065(a)(2) of the Nasdaq Listing Rules, and Dr. Chou and Mr. Chen are not independent under these rules. We are relying upon our status as a “controlled company” to be exempt from Nasdaq rules that require that our Nominating and Corporate Governance Committee be composed entirely of independent directors.
The Nominating and Corporate Governance Committee will assist the board of directors in selecting individuals qualified to become our directors and in determining the composition of the board and its committees. The Nominating and Corporate Governance Committee will be responsible for, among other things:
| ● | identifying and recommending nominees for election or re-election to our board of directors or for appointment to fill any vacancy; |
| ● | reviewing annually with our board of directors its current composition in light of the characteristics of independence, age, skills, experience and availability of service to us; |
| ● | identifying and recommending to our board the directors to serve as members of committees; |
| ● | advising the board periodically with respect to significant developments in the law and practice of corporate governance as well as our compliance with applicable laws and regulations, and making recommendations to our board of directors on all matters of corporate governance and on any corrective action to be taken; and |
| ● | monitoring compliance with our code of business conduct and ethics, including reviewing the adequacy and effectiveness of our procedures to ensure proper compliance. |
Code of Business Conduct and Ethics
Our board will adopt a code of business conduct and ethics that applies to our directors, officers and employees, prior to the completion of the Offering.
Corporate Governance Requirements
Foreign Private Issuer
We are a “foreign private issuer,” as defined by the SEC. As a result, in accordance with the rules and regulations of Nasdaq, we may at our option comply with certain home country governance requirements rather than complying with the corresponding Nasdaq corporate governance standard. While we currently intend to voluntarily follow all Nasdaq corporate governance rules, including rules regarding committee structure and director independence, as described above, we may in the future choose to take advantage of the following exemptions afforded to foreign private issuers:
| | ● | that a majority of our board of directors consists of independent directors; |
| | ● | that our Audit Committee have a written charter addressing the Audit Committee’s responsibilities and authority as set forth in Nasdaq Rule 5605(c)(1); |
| | ● | that our Compensation Committee have a written charter addressing the remuneration committee’s responsibilities and authority as set forth in Nasdaq Rule 5605(d); |
| | ● | that we have independent director oversight of director nominations and a formal written charter or board resolution addressing the nominations process as set forth in Nasdaq Rule 5605(e); |
| | ● | that we have a code of conduct applicable to all directors, officers and employees and from any requirement that we have a code of conduct in compliance with Section 406 of the Sarbanes-Oxley Act of 2002; |
| | ● | that we provide disclosure within four business days of any determination to grant a waiver of the code of business conduct and ethics to directors and officers; |
| | ● | that we obtain shareholder approval for certain issuances of securities, including shareholder approval of stock option plans; |
| | ● | that we adhere to certain requirements governing the review and oversight of all “related party transactions,” as defined in Item 7.B of Form 20-F; and |
| | | |
| | ● | that our board of directors shall have regularly scheduled meetings at which only independent directors are present as set forth in Nasdaq Rule 5605(b)(2). |
Although we may rely on home country corporate governance practices in lieu of certain of the rules in the Nasdaq Rule 5600 Series and Rule 5250(d), we must comply with Nasdaq’s Notification of Noncompliance requirement (Rule 5625), the Voting Rights requirement (Rule 5640) and have an audit committee that satisfies Rule 5605(c)(3), consisting of committee members that meet the independence requirements of Rule 5605(c)(2)(A)(ii).
In addition, as a foreign private issuer, we will not have to file certain public reports required of domestic issuers, including:
| | ● | annual reports on Form 10-K (instead, we will file annual reports on Form 20-F, which requires less information in certain respects); |
| | | |
| | ● | quarterly reports on Form 10-Q; |
| | | |
| | ● | current reports on Form 8-K, disclosing significant events within four days of their occurrence; and |
| | ● | Section 16 reports regarding sales of common shares by insiders. |
Accordingly, our shareholders will not have the same protections afforded to shareholders of companies that are mandatorily subject to all of the corporate governance requirements of Nasdaq and the domestic reporting requirements of the SEC. We may utilize these exemptions for as long as we continue to qualify as a foreign private issuer.
Controlled Company
As of the date hereof (after giving effect to this offering), Mr. Ho-Ching Chen holds beneficial ownership of % of our outstanding Ordinary Shares pursuant to agreements in which certain of our shareholders have given Mr. Chen voting control over their shares, including in connection with the election of our directors. For so long as these agreements provide Mr. Chen with more than 50% of the voting power for the election of our directors, we will be a “controlled company” under Nasdaq rules. As a controlled company, we will be permitted not to comply with certain of Nasdaq’s corporate governance requirements, including exemptions from the following rules:
| | ● | that a majority of our board of directors must be independent directors; |
| | ● | that we have a Compensation Committee that will determine or recommend the compensation of our executive officers and that it be composed solely of independent directors; and |
| | ● | that our director nominees must be selected or recommended solely by independent directors. |
We currently intend to rely on the controlled company exemptions in respect of our Compensation Committee and Nominating and Corporate Governance Committee, each of which will include directors that are not independent under Nasdaq Listing Rules. Further, although we do not intend to rely on the exemption in respect of a majority independent board, we will have the right to use this exemption in the future. As a result, you may not have the same protections afforded to shareholders of companies that are subject to, or voluntarily follow, these Nasdaq corporate governance requirements.
Following this offering, as long as Mr. Ho-Ching Chen maintains control over a majority of the voting power of our outstanding Ordinary Shares with respect to the election of our directors, we may utilize certain of these exemptions. Accordingly, you will not have the same protections afforded to shareholders of companies that are subject to all of the Nasdaq corporate governance requirements of Nasdaq. If we cease to be a “controlled company” and our common stock continues to be listed on Nasdaq, we will be required to comply with these provisions within the applicable transition periods.
Interested Party Transactions
A director may vote, attend a board meeting or sign a document on our behalf with respect to any contract or transaction in which he or she is interested. A director must disclose the nature of his interest to all other directors at a meeting of the board after becoming aware of the fact that he or she is interested in a transaction we have entered into or are to enter into. A general notice given to the board by any director to the effect that he is a member of any specified company or firm and is to be regarded as interested in any contract which may thereafter be made with that company or firm shall be deemed a sufficient declaration of interest in regard to any contract so made.
Remuneration and Borrowing
The directors may receive such remuneration as our board of directors may determine from time to time. Each director is entitled to be repaid or prepaid for all traveling, hotel and incidental expenses reasonably incurred or expected to be incurred in attending meetings of our board of directors or committees of our board of directors or shareholder meetings or otherwise in connection with the discharge of his or her duties as a director. The Compensation Committee will assist the directors in reviewing and approving the compensation structure for the directors. Our board of directors may exercise all the powers of the company to borrow money and to mortgage or charge our undertakings, property, assets (present and future) and uncalled capital or any part thereof, to issue debentures, debenture stock and other securities whenever money is borrowed or as security for any debt, liability or obligation of the company or of any third party.
2025 Share Compensation Plan
Prior to the consummation of this offering, we will adopt a 2025 Share Compensation Plan (the “Plan”). The Plan will provide for discretionary grants of Awards (as defined in the Plan) to our key employees, directors and consultants. The purpose of the Plan is to recognize contributions made to our company and its subsidiaries by such individuals and to provide them with additional incentive to achieve the objectives of our Company. No grants have been made under the plan as of the date hereof. Further, we do not intend to grant any equity Awards under the Plan in conjunction with the closing of this offering.
The following is a summary of the Plan and is qualified by the full text of the Plan.
Administration
The Plan will be administered by our board of directors, or, once constituted, the Compensation Committee of the board of directors (we refer to the body administering the Plan as the “Committee”).
Number of Ordinary Shares
The number of Ordinary Shares that may be issued under the Plan is the maximum aggregate number of Ordinary Shares reserved and available pursuant to the Plan and shall be the aggregate of (i) Ordinary Shares (or up to Ordinary Shares if the underwriters fully exercise the over-allotment option), % of the total issued and outstanding Ordinary Shares immediately after the consummation of this offering, and (ii) on each January 1, starting with January 1, 2025 until December 31, 2029, an additional number of Ordinary Shares equal to the lesser of (A) % of the outstanding number of Ordinary Shares (on a fully-diluted basis) on the immediately preceding December 31, and (B) such lower number of Ordinary Shares as may be determined by the Committee, subject in all cases to adjustment as provided therein. If an Award (or any portion thereof) (as described in the Plan) terminates, expires or lapses or is cancelled for any reason, any Ordinary Shares subject to the Award (or such portion thereof) shall again be available for the grant of an Award pursuant to the Plan (unless the Plan has terminated). If any Award (in whole or in part) is settled in cash or other property in lieu of Ordinary Shares, then the number of Ordinary Shares subject to such Award (or such part) shall again be available for grant pursuant to the Plan. Ordinary Shares that have actually been issued under the Plan, pursuant to Awards under the Plan shall not be returned to the Plan and shall not cause the number of Ordinary Shares available to be subject to Awards under the Plan to be increased. Subject to any required action by our shareholders, the number of Ordinary Shares covered by each outstanding Award, the number of Ordinary Shares which have been authorized for issuance under the Plan but as to which no Awards have yet been granted or which have been returned to the Plan upon cancellation or expiration of an Award, and the number of Ordinary Shares subject to grant as Incentive Stock Options (as described in the Plan), as well as the price per Ordinary Shares covered by each such outstanding Award and any other affected terms of such Awards, shall be proportionally and equitably adjusted for any increase or decrease in the number of issued Ordinary Shares resulting from a subdivision or consolidation, share dividend, amalgamation, spin-off, arrangement or consolidation, combination or reclassification of Ordinary Shares. Except as the board of directors or the Committee determines, no issuance by us of shares of any class, or securities convertible into shares of any class, shall affect, and no adjustment by reason hereof shall be made with respect to, the number or price of Ordinary Shares subject to an Award.
Types of Awards
The Plan permits the granting of any or all of the following types of awards to all grantees:
| | ● | share options, including incentive share options, or ISOs; |
| | ● | share appreciation rights, or SARs; |
| | ● | restricted shares; |
| | ● | restricted share units; and |
| | ● | share payments |
Awards granted under the Plan may, in the discretion of the Committee, be granted alone or in addition to, in tandem with or in substitution for, any other award under the Plan. The material terms of each Award will be set forth in a written award agreement between the grantee and us.
Share Options and SARs
The Committee is authorized to grant SARs and share options (including ISOs except that an ISO may only be granted to an employee of ours or our subsidiary corporation). A share option allows a grantee to purchase a specified number of our Ordinary Shares at a predetermined price per share (the “exercise price”) during a fixed period measured from the date of grant. An SAR entitles the grantee to receive the excess of the fair market value of a specified number of Ordinary Shares on the date of exercise over a predetermined exercise price per share. The exercise price of an option or an SAR will be determined by the Committee and set forth in the award agreement but the exercise price may not be less than the fair market value of a share on the grant date. The term of each option or SAR is determined by the Committee and set forth in the award agreement, except that the term may not exceed 10 years. Options may be exercised by payment of the purchase price through one or more of the following means: payment in cash, payment in check, payment in promissory note, with the approval of the Committee, by delivery of our Ordinary Shares acquired upon the exercise of such option; consideration received by us under a broker-assisted or similar cashless exercise program implemented by us in connection with the Plan; payment by such other consideration as may be approved by the Committee from time to time to the extent permitted by applicable laws; or any combination of the foregoing methods of payment.
Restricted Shares
The Committee may award restricted shares consisting of our Ordinary Shares which remain subject to a risk of forfeiture and may not be disposed of by grantees until certain restrictions established by the Committee lapse. A grantee receiving restricted shares will have all of the rights of a shareholder, including the right to vote the shares and the right to receive any dividends, except as otherwise provided in the award agreement. If the price for the restricted shares was paid in services, then upon termination as a service provider, the grantee shall no longer have any right in the unvested restricted shares and such restricted shares shall be and thereupon either cancelled or surrendered to us without consideration. If a purchase price was paid by the grantee for the restricted shares (other than in services), then upon the grantee’s termination as a service provider, we shall have the right to repurchase from the grantee the unvested restricted shares then subject to restrictions at a cash price per share equal to the price paid by the grantee for such restricted shares or such other amount as may be specified in the award agreement.
Restricted Share Units
The Committee may also grant restricted share unit awards. A restricted share unit award is the grant of a right to receive a specified number of our Ordinary Shares upon lapse of a specified forfeiture condition. If the condition is not satisfied during the restriction period, the award will lapse without the issuance of the Ordinary Shares underlying such award.
Restricted share units carry no voting or other rights associated with share ownership until the Ordinary Shares underlying the award are delivered in settlement of the award. We shall cause such Ordinary Shares to be evidenced as issued by entry in our register of shareholders promptly after the restricted share unit vests.
Share Payments
The Committee may grant share payments to any service provider in the manner determined from time to time by the Committee; provided, that unless otherwise determined by the Committee such share payments shall be made in lieu of base salary, bonus, or other cash compensation otherwise payable to such grantee, including any such compensation that has been deferred at the election of the grantee; provided, further, that not less than the par value of any Ordinary Share shall be received by us in connection with its issue pursuant to any such share payment. In accordance with applicable law, such par value may be paid through the provision of services. The number of Ordinary Shares issuable as a share payment shall be determined by the Committee and may be based upon satisfaction of such specific criteria as determined appropriate by the Committee, including specified dates for electing to receive such share payment at a later date and the date on which such share payment is to be made.
Change in Control
If there is a merger or consolidation of us with or into another corporation or a sale of substantially all of our Ordinary Shares, or, collectively, a Change in Control, we, as determined in the sole discretion of the Committee and without the consent of the grantee, may take any of the following actions:
| | (i) | accelerate or not accelerate the vesting, in whole or in part, of any award, or some or all awards, of any grantee, some grantees or all grantees; |
| | | |
| | (ii) | purchase any award for an amount of cash or Ordinary Shares equal to the value that could have been attained upon the exercise of such award or realization of the grantee’s rights had such award been currently exercisable or payable or fully vested (and, for the avoidance of doubt, if as of such date the Committee determines in good faith that no amount would have been attained upon the exercise of such award or realization of the grantee’s rights, then such award may be terminated by us without payment); or |
| | | |
| | (iii) | provide for the assumption, conversion or replacement of any award by the successor or surviving company or a parent or subsidiary of the successor or surviving company with other rights (including cash) or property selected by the Committee in its sole discretion or the assumption or substitution of such award by the successor or surviving company, or a parent or subsidiary thereof, with such appropriate adjustments as to the number and kind of Ordinary Shares and prices as the Committee deems, in its sole discretion, reasonable, equitable and appropriate. In the event the successor or surviving company refuses to assume, convert or replace outstanding awards, the awards shall fully vest and the grantee shall have the right to exercise or receive payment as to all of the Ordinary Shares subject to the award, including Ordinary Shares as to which it would not otherwise be vested, exercisable or otherwise issuable (including at the time of the Change in Control). |
Amendment to and Termination of the Plan
The Board of Directors in its sole discretion may terminate this Plan at any time. The Board of Directors may amend this Plan at any time in such respects as the Board of Directors may deem advisable; provided, that, if required to comply with applicable laws or stock exchange rules or the rules of any automated quotation systems (other than any requirement which may be disapplied by us following any available home country exemption), we shall obtain shareholder approval of any Plan amendment in such a manner and to such a degree as required. In addition, subject to the terms of the Plan, no amendment or termination of the Plan may materially and adversely affect the right of a grantee under any award granted under the Plan.
COMPENSATION
Compensation of Executive Officers
For the fiscal year ended December 31, 2023, we paid an aggregate of $135,917 in cash to our executive officers. We have not set aside or accrued any amount to provide pension, retirement or other similar benefits to our executive officers.
The following table presents information regarding the total compensation awarded to, earned by, or paid to our executive officers for the year ended December 31, 2023:
| Name | | Principal Position | | Year | | | Salary (USD) | | | Bonus (USD) | | | Option Awards (USD) | | | Other Compensation (USD) | | | Total (USD) | |
| Dr. Tza-Zen Chaung | | Chief Executive Officer | | | 2023 | | | $ | 42,900 | | | | — | | | | — | | | | — | | | $ | 42,900 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Pi-Chuan Chien | | Chief Financial Officer | | | 2023 | | | $ | 31,373 | | | | — | | | | — | | | | — | | | $ | 31,373 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Dr. Jen-Wei Liu | | Chief Scientific Officer | | | 2023 | | | $ | 34,144 | | | | — | | | | — | | | | — | | | $ | 34,144 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Dr. Yuan-Sheng Li | | Chief Technology Officer | | | 2023 | | | $ | 27,499 | | | | — | | | | — | | | | — | | | $ | 27,499 | |
We have not granted any options or issued any shares of restricted stock to our executive officers.
Employment Agreements with Executive Officers
On January 1, 2023, we and Dr. Tza-Zen Chaung entered into an employment agreement pursuant to which Dr. Chaung agreed to be employed as our Chief Executive Officer. Under the employment agreement, Dr. Chaung will receive a salary of $3,300 per month prior to completion of this offering. After completion of this offering, the salary will be adjusted to $5,000 per month. We have already awarded Dr. Chaung 10,000 Ordinary Shares, and upon the completion of this offering, Dr. Chaung will receive an additional 10,000 Ordinary Shares. In addition, if we license any technology upon Dr. Chaung’s recommendation, then we will pay to Dr. Chaung 5% of the royalty paid under the license as bonus.
On February 1, 2023, we and Mr. Pi-Chuan Chien entered into an employment agreement pursuant to which Mr. Chien agreed to be employed as our Chief Financial Officer. Under the employment agreement, Mr. Chien will receive a salary of $2,670 per month prior to completion of this offering. Upon completion of this offering, we also will award to Mr. Chien 10,000 Ordinary Shares.
On August 1, 2023, we and Dr. Jen-Wei Liu entered into an employment agreement pursuant to which Dr. Liu agreed to be employed as our Director of the Clinical Research Department. Under the employment agreement, Dr. Liu will receive an annual salary of $34,144.
On August 1, 2023, we and Dr. Yuan-Sheng Li entered into an employment agreement pursuant to which Dr. Li agreed to be employed as the Director of our Research Development and Manufacturing Department. Under the employment agreement, Dr. Li will have an annual salary of $27,499.
Incentive Compensation
We do not maintain any incentive cash or bonus programs.
Compensation of Directors
We provide only cash compensation to our non-executive directors for the time and effort necessary to serve as a member of our board of directors. For the fiscal year ended December 31, 2023, we paid an aggregate of $41,523 in cash to our directors.
The following table presents information regarding the total compensation awarded to, earned by, or paid to our non-executive directors for the year ended December 31, 2023:
| Name | | Year | | | Salary (USD) | | | Bonus (USD) | | | Option Awards (USD) | | | Other Compensation (USD) | | | Total (USD) | |
| Ho-Ching Chen | | | 2023 | | | $ | 15,409 | | | | — | | | | — | | | | — | | | $ | 15,409 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Dr. Pei-Wen Chou | | | 2023 | | | $ | 26,114 | | | | — | | | | — | | | | — | | | $ | 26,114 | |
We have not granted any options or issued any shares of restricted stock to our non-executive directors.
PRINCIPAL SHAREHOLDERS
The following table sets forth information with respect to the beneficial ownership of our Ordinary Shares as of the date of this prospectus and as adjusted to reflect our issuance and sale of Ordinary Shares offered in this offering for:
| | ● | each of our directors and executive officers; and |
| | ● | each person known to us to own beneficially more than 5.0% of our Ordinary Shares. |
The table presents beneficial ownership as determined pursuant to Rule 13d-3 under the Exchange Act. This rule attributes beneficial ownership to anyone having voting or investment power with respect to Ordinary Shares as well as to anyone having a right to purchase Ordinary Shares within 60 days, including through the exercise of an option or warrant or conversion of a security. Except as indicated below, and subject to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all Ordinary Shares shown as beneficially owned by them. Percentages are based on Ordinary Shares outstanding prior to this offering and Ordinary Shares outstanding after this offering.
As of the date of the prospectus, we have 11 shareholders of record, none of which are located in the United States.
| | | Ordinary Shares Beneficially Owned Prior to this Offering | | | Ordinary Shares Beneficially Owned After this Offering | | | Percentage of Votes Held After this Offering | |
| | | Number | | | Percent | | | Number | | | Percent | | | Percent | |
| Directors and Executive Officers(1): | | | | | | | | | | | | | | | | | | | | |
| Dr. Tza-Zen Chaung | | | 10,000 | | | | * | | | | | | | | | | | | | |
| Pi-Chuan Chien | | | — | | | | — | | | | | | | | | | | | | |
| Dr. Jen-Wei Liu | | | 125,000 | | | | * | | | | | | | | | | | | | |
| Dr. Yuan-Sheng Li | | | 127,000 | | | | * | | | | | | | | | | | | | |
| Ho-Ching Chen(2) | | | | | | | | % | | | | | | | | | | | | |
| Dr. Pei-Wen Chou | | | 840,000 | | | | 1.6 | % | | | | | | | | | | | | |
| Hann-Chyi Lin | | | 50,000 | | | | * | | | | | | | | | | | | | |
| Dr. Tai-Nang Huang | | | — | | | | — | | | | | | | | | | | | | |
| Yin-Ho Huang | | | 1,000 | | | | * | | | | | | | | | | | | | |
| All directors and executive officers as a group | | | | | | | | % | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| 5% Principal Shareholders: | | | | | | | | | | | | | | | | | | | | |
| None. | | | | | | | | | | | | | | | | | | | | |
| * | Less than 1%. |
| (1) | Unless otherwise indicated, the business address of each of the individuals is . |
| (2) | Consists of 1,088,150 Ordinary Shares that Mr. Chen owns directly and Ordinary Shares that Mr. Chen beneficially owns pursuant to certain voting proxy agreements that grant him the right to vote the Ordinary Shares on all matters, including the election of directors. Mr. Chen disclaims beneficial ownership of these Ordinary Shares except to the extent of his pecuniary interest therein. |
We are not aware of any arrangement that may, at a subsequent date, result in a change of control of our company.
RELATED PARTY TRANSACTIONS
Contractual Arrangements with Our Variable Interest Entity and Its Shareholders
See “Corporate History and Structure.”
Employment Agreements and Indemnification Agreements
See “Management—Employment Agreements and Indemnification Agreements.”
Share Compensation Plan
See “Management—2025 Share Compensation Plan.”
Other Related Party Transactions
Since January 2022, we have entered into transactions with the following related parties.
| Name of Entity or Individual | | Relationship with the Company and its subsidiary |
| Ho-Ching Chen | | Director of the Company |
| Sunflower Holiday Co., Ltd. | | Entity controlled by controlling beneficiary shareholder of Company |
| Xi Yue Biomedicine Inc. (formerly Guang-Li Biomedicine Inc.) | | Entity controlled by controlling beneficiary shareholder of Company |
| Foo Kin Construction Co., Ltd. | | Entity controlled by controlling beneficiary shareholder of Company |
| Depei Biotechnology Co., Ltd. | | Entity controlled by controlling beneficiary shareholder of Company |
Service revenue
Service revenue from related parties consisted of the following as of the periods indicated.
| | | | December 31, | | | | December 31, | |
| | | | 2023 | | | | 2022 | |
| Xi Yue Biomedicine Inc. | | $ | — | | | $ | 79,908 | |
Rent expense
Rent expense from related parties consisted of the following as of the periods indicated:
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Xi Yue Biomedicine Inc. | | $ | 1,834 | | | $ | 1,918 | |
| Foo Kin Construction Co., Ltd. | | | — | | | | 5,753 | |
| Total | | $ | 1,834 | | | $ | 7,671 | |
During the years ended December 31, 2023 and 2022, we had an office lease with Guang-Li Biomedicine Inc. on a twelve-month basis. The lease term expires on December 31, 2023 with a monthly rent of approximately $155.
On January 1, 2022, the Company had another office lease with Foo Kin Construction Co., Ltd. with the expiration date on December 31, 2022. The monthly rent was approximately $467.
Due from related party
Due from related party consisted of the following.
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Depei Biotechnology Co., Ltd. | | $ | 7,984 | | | $ | — | |
Amounts due from related party were due to the sales of supplies
Other assets
Refundable deposits from related parties consisted of the following as of the periods indicated.
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Xi Yue Biomedicine Inc. | | $ | 327 | | | $ | 326 | |
| Foo Kin Construction Co., Ltd. | | | — | | | | 488 | |
| Total | | $ | 327 | | | $ | 814 | |
Due to related parties
Amount due to related parties consisted of the following as of the periods indicated.
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Ho-Ching Chen | | $ | — | | | $ | 4,769 | |
Due to related parties were unsecured, had no written agreement, due on demand, and interest-free.
DESCRIPTION OF SHARE CAPITAL
The Company was incorporated as a BVI business company under the laws of the British Virgin Islands on September 21, 2022. As of the date of this prospectus, the Company’s authorized shares consist of 100,000,000 Ordinary Shares with a par value of $0.01 per share.
As of the date of this prospectus, there were 52,237,745 Ordinary Shares issued and outstanding.
On , 2025, the Company effectuated a forward split of its Ordinary Shares at a ratio of -for- to increase its authorized capital shares from Ordinary Shares with a par value of $ per share to Ordinary Shares with a par value of $ per share (the “Forward Split”). As a result of the Forward Split, the Company now has Ordinary Shares issued and outstanding as of the date hereof.
Ordinary Shares
General
All of our issued shares are fully paid and non-assessable. Certificates evidencing the shares are issued in registered form. There are no limitations imposed by our memorandum and articles of association on the rights of non-resident or foreign shareholders to hold or exercise voting rights on our shares. In addition, there are no provisions in our memorandum and articles of association governing the ownership threshold above which shareholder ownership must be disclosed.
Under the BVI Act, the Ordinary Shares are deemed to be issued when the name of the shareholder is entered in our register of members. If (a) information that is required to be entered in the register of members is omitted from the register or is inaccurately entered in the register, or (b) there is unreasonable delay in entering information in the register, a shareholder of the company, or any person who is aggrieved by the omission, inaccuracy or delay, may apply to the British Virgin Islands Courts for an order that the register be rectified, and the court may either refuse the application or order the rectification of the register, and may direct the company to pay all costs of the application and any damages the applicant may have sustained.
Dividends
The holders of our Ordinary Shares are entitled to such dividends as may be declared by our board of directors subject to the BVI Act.
Voting Rights
Any action required or permitted to be taken by the shareholders must be effected at a duly called meeting of the shareholders entitled to vote on such action or may be effected by a resolution of members in writing, each in accordance with the memorandum and articles of association. At each meeting of shareholders, each shareholder who is present in person or by proxy (or, in the case of a shareholder being a corporation, by its duly authorized representative) will have one vote for each share that such shareholder holds.
Transfer of Ordinary Shares
Subject to the restrictions contained in our articles of association, any of our shareholders may transfer all or any of his or her Ordinary Shares by an instrument of transfer in the usual or common form or any other form approved by our board of directors.
Liquidation
As permitted by the BVI Act and our memorandum and articles of association, we may be voluntarily liquidated under Part XII of the BVI Act by resolution of directors and resolution of shareholders if our assets exceed our liabilities and we are able to pay our debts as they fall due. We may also be wound up in circumstances where we are insolvent in accordance with the terms of the BVI Insolvency Act (Law Revision 2020).
If we are wound up and our assets available for distribution among our shareholders are more than sufficient to repay all amounts paid to us on account of the issue of shares immediately prior to the winding up, the excess shall be distributable pari passu among those shareholders in proportion to the amount paid up immediately prior to the winding up on the shares held by them, respectively. If we are wound up and our assets available for distribution among the shareholders as such are insufficient to repay the whole of the amounts paid to us on account of the issue of shares, those assets shall be distributed so that, to the greatest extent possible, the losses shall be borne by the shareholders in proportion to the amounts paid up immediately prior to the winding up on the shares held by them, respectively. If we are wound up, the liquidator appointed by us may, in accordance with the BVI Act, divide among our shareholders in specie or kind the whole or any part of our assets (whether they shall consist of property of the same kind or not) and may, for such purpose, set such value as the liquidator deems fair upon any property to be divided and may determine how such division shall be carried out as between the shareholders or different classes of shareholders.
Calls on Ordinary Shares and Forfeiture of Ordinary Shares
Our board of directors may from time to time make calls upon shareholders for any amounts unpaid on their Ordinary Shares in a notice served to such shareholders at least 14 clear days prior to the specified time of payment. The Ordinary Shares that have been called upon and remain unpaid are subject to forfeiture.
Redemption of Ordinary Shares
Subject to the provisions of the BVI Act, we may issue shares on terms that are subject to redemption, at our option or at the option of the holders, on such terms and in such manner as may be determined by our memorandum and articles of association and subject to any applicable requirements imposed from time to time by the BVI Act, the SEC, the Nasdaq Capital Market, or by any recognized stock exchange on which our securities are listed.
Variations of Rights of Shares
If at any time we are authorized to issue more than one class of shares, all or any of the rights attached to any class of shares may be amended only with the consent in writing of or by a resolution passed at a meeting of not less than 50% of the shares of the class to be affected.
General Meetings of Shareholders
Under our memorandum and articles of association, a copy of the notice of any meeting of shareholders shall be given not less than seven days before the date of the proposed meeting to those persons whose names appear as shareholders in the register of members on the date of the notice and are entitled to vote at the meeting. Our board of directors shall call a meeting of shareholders upon the written request of shareholders holding at least 30% of our outstanding voting shares. In addition, our board of directors may call a meeting of shareholders on its own motion. A meeting of shareholders may be called on short notice if at least 90% of the shares entitled to vote on the matters to be considered at the meeting have agreed to short notice of the meeting, or if all members holding shares entitled to vote on all or any matters to be considered at the meeting have waived notice and presence at the meeting shall be deemed to constitute waiver for this purpose.
At any meeting of shareholders, a quorum will be present if there are shareholders present in person or by proxy representing not less than 50% of the shares entitled to vote on the resolutions to be considered at the meeting. Such quorum may be represented by only a single shareholder or proxy. If no quorum is present within two hours of the start time of the meeting, the meeting shall be dissolved if it was requested by shareholders. In any other case, the meeting shall be adjourned to the next business day, and if shareholders representing not less than one-third of the votes of the Ordinary Shares or each class of shares entitled to vote on the matters to be considered at the meeting are present within one hour of the start time of the adjourned meeting, a quorum will be present. If not, the meeting will be dissolved. No business may be transacted at any meeting of shareholders unless a quorum is present at the commencement of business. If present, the chair of our board of directors shall be the chair presiding at any meeting of the shareholders. If the chair of our board is not present, then the members present shall choose a shareholder to act to chair the meeting of the shareholders. If the shareholders are unable to choose a chairman for any reason, then the person representing the greatest number of voting shares present in person or by proxy shall preside as chairman, failing which the oldest individual member or member representative shall take the chair.
A corporation that is a shareholder shall be deemed for the purpose of our memorandum and articles of association to be present in person if represented by its duly authorized representative. This duly authorized representative shall be entitled to exercise the same powers on behalf of the corporation which he represents as that corporation could exercise if it were our individual shareholder.
Inspection of Books and Records
Under the BVI Act, members of the general public, on payment of a nominal fee, can obtain copies of the public records of a company available at the office of the Registrar of Corporate Affairs, which will include the company’s certificate of incorporation, its memorandum and articles of association (with any amendments) and records of license fees paid to date and will also disclose any articles of dissolution, articles of merger and a register of charges if the company has elected to file such a register.
A member of the Company is also entitled, upon giving written notice to the Company, to inspect (i) our memorandum and articles of association, (ii) the register of members, (iii) the register of directors and (iv) minutes of meetings and resolutions of members and of those classes of members of which that member is a member, and to make copies and take extracts from the documents and records referred to in (i) to (iv) above. However, our directors may, if they are satisfied that it would be contrary to the company’s interests to allow a member to inspect any document, or part of a document specified in (ii) to (iv) above, refuse to permit the member to inspect the document or limit the inspection of the document, including limiting the making of copies or the taking of extracts or records. See “Where You Can Find Additional Information.” on page 121 of this prospectus. Where a company fails or refuses to permit a member to inspect a document or permits a member to inspect a document subject to limitations, that member may apply to the BVI Court for an order that he should be permitted to inspect the document or to inspect the document without limitation.
Changes in Capital
We may from time to time by resolution of shareholders or resolution of our board of directors, subject to our memorandum and articles of association:
| | ● | amend our memorandum and articles of association to increase or decrease the maximum number of shares the Company is authorized to issue; |
| | ● | split our authorized and issued shares into a larger number of shares; |
| | ● | combine our authorized and issued shares into a smaller number of shares; and |
| | ● | create new classes of shares with preferences to be determined by resolution of the board of directors to amend the memorandum and articles of association to create new classes of shares with such preferences at the time of authorization. |
Representative Warrants
We will issue to the representatives, or their respective designees, warrants (the “Representative Warrants”) to purchase an amount of our Ordinary Shares equal to up to 5% of the Ordinary Shares sold in this offering (including any Ordinary Shares sold pursuant to the underwriters’ option to purchase additional shares). The Representative Warrants will have an exercise price equal to 120% of the price to the public of the Ordinary Shares in this offering, subject to standard anti-dilution adjustments for share splits and similar transactions, and will also contain a cashless exercise provision. The Representative Warrants will be immediately exercisable upon issuance, in whole or in part, and will expire on the fifth anniversary of the commencement of sales in this offering in accordance with Financial Industry Regulatory Authority (FINRA) Rule 5110(g)(8)(A).
Pursuant to FINRA Rule 5110(e), the Representative Warrants and any Ordinary Shares issued upon exercise of the Representative Warrants shall not be sold, transferred, assigned, pledged, or hypothecated, or be the subject of any hedging, short sale, derivative, put or call transaction that would result in the effective economic disposition of the securities by any person for a period of 180 days immediately following the date of commencement of sales of this offering, except: (i) the transfer of any security by operation of law or by reason of reorganization of the issuer; (ii) the transfer of any security to any FINRA member firm participating in the offering and the officers, partners, registered persons or affiliates thereof, if all securities so transferred remain subject to these lock-up restrictions for the remainder of the time period; (iii) if the aggregate amount of our securities held by the representatives or related persons does not exceed 1% of the securities being offered; (iv) the transfer of any security that is beneficially owned on a pro-rata basis by all equity owners of an investment fund, provided that no participating member manages or otherwise directs investments by the fund and the participating members in the aggregate do not own more than 10% of the equity in the fund; (v) the exercise or conversion of any security, if all securities remain subject to the lock-up restriction set forth above for the remainder of the time period; (vi) if we meet the registration requirements of Forms S-3, F-3 or F-10; or (vii) the transfer of any security back to us in a transaction exempt from registration with the SEC.
The Representative Warrants and the Ordinary Shares underlying the Representative Warrants have been registered in the registration statement of which this prospectus is a part. See the Form of Representative Warrant that will be filed as an exhibit as part of this registration statement for more information.
Differences in Corporate Law
The BVI Act and the laws of the British Virgin Islands affecting British Virgin Islands companies like us and our shareholders differ from laws applicable to U.S. corporations and their shareholders. Set forth below is a summary of the significant differences between the provisions of the laws of the British Virgin Islands applicable to us and the laws applicable to companies incorporated in the United States and their shareholders.
Mergers and Similar Arrangements
Under the laws of the British Virgin Islands, two or more companies may merge or consolidate in accordance with Section 170 of the BVI Act. A merger means the merging of two or more constituent companies into one of the constituent companies (the “surviving company”) and a consolidation means the uniting of two or more constituent companies into a new company (the “consolidated company”). The procedure for a merger or consolidation between the company and another company (which need not be a BVI company, and which may be the company’s parent or subsidiary, but need not be) is set out in the BVI Act. In order to merge or consolidate, the directors of each constituent company must approve a written plan of merger or consolidation, which with the exception of a merger between a parent company and its subsidiary, must also be approved by a resolution of a majority of the shareholders voting at a quorate meeting of shareholders or by written resolution of the shareholders of the BVI company or BVI companies which are to merge. While a director may vote on the plan of merger or consolidation, or any other matter, even if he has a financial interest in the plan, the interested director must disclose the interest to all other directors of the company promptly upon becoming aware of the fact that he is interested in a transaction entered into or to be entered into by the company. A transaction entered into by us in respect of which a director is interested (including a merger or consolidation) is voidable by us unless the director’s interest was (a) disclosed to the board prior to the transaction or (b) the transaction is (i) between the director and the company and (ii) the transaction is in the ordinary course of the company’s business and on usual terms and conditions. Notwithstanding the above, a transaction entered into by the company is not voidable if the material facts of the interest are known to the shareholders and they approve or ratify it or the company received fair value for the transaction. In any event, all shareholders must be given a copy of the plan of merger or consolidation irrespective of whether they are entitled to vote at the meeting to approve the plan of merger or consolidation. A foreign company which is able under the laws of its foreign jurisdiction to participate in the merger or consolidation is required by the BVI Act to comply with the laws of that foreign jurisdiction in relation to the merger or consolidation. The shareholders of the constituent companies are not required to receive shares of the surviving or consolidated company but may receive debt obligations or other securities of the surviving or consolidated company, other assets, or a combination thereof. Further, some or all of the shares of a class or series may be converted into a kind of asset while the other shares of the same class or series may receive a different kind of asset. As such, not all the shares of a class or series must receive the same kind of consideration. After the plan of merger or consolidation has been approved by the directors and authorized, if required, by a resolution of the shareholders, articles of merger or consolidation are executed by each company and filed with the Registrar of Corporate Affairs in the British Virgin Islands. The merger is effective on the date that the articles of merger are registered with the Registrar or on such subsequent date, not exceeding thirty days, as is stated in the articles of merger or consolidation.
As soon as a merger becomes effective: (a) the surviving company or consolidated company (so far as is consistent with its memorandum and articles of association, as amended or established by the articles of merger or consolidation) has all rights, privileges, immunities, powers, objects and purposes of each of the constituent companies; (b) in the case of a merger, the memorandum and articles of association of any surviving company are automatically amended to the extent, if any, that changes to its memorandum and articles of association are contained in the articles of merger or, in the case of a consolidation, the memorandum and articles of association filed with the articles of consolidation are the memorandum and articles of the consolidated company; (c) assets of every description, including choses-in-action and the business of each of the constituent companies, immediately vest in the surviving company or consolidated company; (d) the surviving company or consolidated company is liable for all claims, debts, liabilities and obligations of each of the constituent companies; (e) no conviction, judgment, ruling, order, claim, debt, liability or obligation due or to become due, and no cause existing, against a constituent company or against any member, director, officer or agent thereof, is released or impaired by the merger or consolidation; and (f) no proceedings, whether civil or criminal, pending at the time of a merger by or against a constituent company, or against any member, director, officer or agent thereof, are abated or discontinued by the merger or consolidation; but: (i) the proceedings may be enforced, prosecuted, settled or compromised by or against the surviving company or consolidated company or against the member, director, officer or agent thereof, as the case may be; or (ii) the surviving company or consolidated company may be substituted in the proceedings for a constituent company. The Registrar of Corporate Affairs shall strike off the register of companies each constituent company that is not the surviving company in the case of a merger and all constituent companies in the case of a consolidation. If the directors determine it to be in the best interests of the company, it is also possible for a merger to be approved as a Court approved plan of arrangement or scheme of arrangement in accordance with the BVI Act.
A shareholder may dissent from (a) a merger if the company is a constituent company, unless the company is the surviving company and the member continues to hold the same or similar shares; (b) a consolidation if the company is a constituent company; (c) any sale, transfer, lease, exchange or other disposition of more than 50 per cent in value of the assets or business of the company if not made in the usual or regular course of the business carried on by the company but not including: (i) a disposition pursuant to an order of the court having jurisdiction in the matter, (ii) a disposition for money on terms requiring all or substantially all net proceeds to be distributed to the members in accordance with their respective interest within one year after the date of disposition, or (iii) a transfer pursuant to the power of the directors to transfer assets for the protection thereof; (d) a compulsory redemption of 10 per cent or fewer of the issued shares of the company required by the holders of 90% or more of the shares of the company pursuant to the terms of the BVI Act; and (e) a plan of arrangement, if permitted by the British Virgin Islands Court (each, an Action). A shareholder properly exercising his dissent rights is entitled to a cash payment equal to the fair value of his shares.
A shareholder dissenting from an Action must object in writing to the Action before the vote by the shareholders on the merger or consolidation, unless notice of the meeting was not given to the shareholder. If the merger or consolidation is approved by the shareholders, the company must give notice of this fact to each shareholder within 20 days who gave written objection. Such objection shall include a statement that the member proposes to demand payment for his or her shares if the Action is taken. These shareholders then have 20 days to give to the company their written election in the form specified by the BVI Act to dissent from the Action, provided that in the case of a merger, the 20 days starts when the plan of merger is delivered to the shareholder. Upon giving notice of his election to dissent, a shareholder ceases to have any shareholder rights except the right to be paid the fair value of his shares. As such, the merger or consolidation may proceed in the ordinary course notwithstanding his dissent. Within seven days of the later of the delivery of the notice of election to dissent and the effective date of the merger or consolidation, the company shall make a written offer to each dissenting shareholder to purchase his shares at a specified price per share that the company determines to be the fair value of the shares. The company and the shareholder then have 30 days to agree upon the price. If the company and a shareholder fail to agree on the price within the 30 days, then the company and the shareholder shall, within 20 days immediately following the expiration of the 30-day period, each designate an appraiser and these two appraisers shall designate a third appraiser. These three appraisers shall fix the fair value of the shares as of the close of business on the day prior to the shareholders’ approval of the transaction without taking into account any change in value as a result of the transaction.
Shareholders’ Suits
There are both statutory and common law remedies available to our shareholders as a matter of British Virgin Islands law. These are summarized below.
Prejudiced members
A shareholder who considers that the affairs of the company have been, are being, or are likely to be, conducted in a manner that is, or any act or acts of the company have been, or are, likely to be oppressive, unfairly discriminatory or unfairly prejudicial to him in that capacity, can apply to the court under Section 184I of the BVI Act, inter alia, for an order that his shares be acquired, that he be provided compensation, that the Court regulate the future conduct of the company, or that any decision of the company which contravenes the BVI Act or our memorandum and articles of association be set aside.
Derivative actions
Section 184C of the BVI Act provides that a shareholder of a company may, with the leave of the Court, bring an action in the name of the company in certain circumstances to redress any wrong done to it. Such actions are known as derivative actions. The BVI Court may only grant permission to bring a derivative action where the following circumstances apply:
| | ● | the company does not intend to bring, diligently continue or defend or discontinue proceedings; and |
| | ● | it is in the interests of the company that the conduct of the proceedings not be left to the directors or to the determination of the shareholders as a whole. |
When considering whether to grant leave, the British Virgin Islands Court is also required to have regard to the following matters:
| | ● | whether the shareholder is acting in good faith; |
| | ● | whether a derivative action is in the company’s best interests, taking into account the directors’ views on commercial matters; |
| | ● | whether the action is likely to proceed; |
| | ● | the costs of the proceedings in relation to the relief likely to be obtained; and |
| | ● | whether an alternative remedy is available. |
Just and equitable winding up
In addition to the statutory remedies outlined above, shareholders can also petition the BVI Court for the winding up of a company under the BVI Insolvency Act (Law Revision 2020) for the appointment of a liquidator to liquidate the company and the court may appoint a liquidator for the company if it is of the opinion that it is just and equitable for the court to so order. Save in exceptional circumstances, this remedy is generally only available where the company has been operated as a quasi-partnership and trust and confidence between the partners has broken down.
Indemnification of directors and executive officers and limitation of liability
Our memorandum and articles of association provides that, subject to certain limitations, we indemnify against all expenses, including legal fees, and against all judgments, fines and amounts paid in settlement and reasonably incurred in connection with legal, administrative or investigative proceedings for any person who:
| | ● | is or was a party or is threatened to be made a party to any threatened, pending or completed proceedings, whether civil, criminal, administrative or investigative, by reason of the fact that the person is or was our director; or |
| | ● | is or was, at our request, serving as a director or officer of, or in any other capacity is or was acting for, another body corporate or a partnership, joint venture, trust or other enterprise. |
These indemnities only apply if the person acted honestly and in good faith with a view to be in our best interests and, in the case of criminal proceedings, the person had no reasonable cause to believe that his conduct was unlawful. The decision of the directors as to whether the person acted honestly and in good faith and with a view to be in the best interests of the company and as to whether the person had no reasonable cause to believe that his conduct was unlawful and is, in the absence of fraud, sufficient for the purposes of the memorandum and articles of association, unless a question of law is involved. The termination of any proceedings by any judgment, order, settlement, conviction or the entering of a nolle prosequi does not, by itself, create a presumption that the person did not act honestly and in good faith and with a view to be in the best interests of the company or that the person had reasonable cause to believe that his conduct was unlawful.
This standard of conduct is generally the same as permitted under the Delaware General Corporation Law for a Delaware corporation. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers or persons controlling us under the foregoing provisions, we have been advised that in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Anti-takeover provisions in our memorandum and articles of association
Some provisions of our articles of association may discourage, delay or prevent a change in control of our company or management that shareholders may consider favorable. Under the BVI Act there are no provisions which specifically prevent the issuance of preferred shares or any such other ‘poison pill’ measures. Our memorandum and articles of association also do not contain any express prohibitions on the issuance of any preferred shares. Therefore, the directors without the approval of the holders of Ordinary Shares may issue preferred shares that have characteristics that may be deemed to be anti-takeover. Additionally, such a designation of shares may be used in connection with plans that are poison pill plans. However, under British Virgin Islands law, our directors in the exercise of their powers granted to them under our memorandum and articles of association and performance of their duties, are required to act honestly and in good faith in what the director believes to be in the best interests of our company.
Directors’ fiduciary duties
Under Delaware corporate law, a director of a Delaware corporation has a fiduciary duty to the corporation and its shareholders. This duty has two components: the duty of care and the duty of loyalty. The duty of care requires that a director act in good faith, with the care that an ordinarily prudent person would exercise under similar circumstances. Under this duty, a director must inform himself of, and disclose to shareholders, all material information reasonably available regarding a significant transaction.
The duty of loyalty requires that a director act in a manner he reasonably believes to be in the best interests of the corporation. He must not use his corporate position for personal gain or advantage. This duty prohibits self-dealing by a director and mandates that the best interest of the corporation and its shareholders take precedence over any interest possessed by a director, officer or controlling shareholder and not shared by the shareholders generally. In general, actions of a director are presumed to have been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties. Should such evidence be presented concerning a transaction by a director, a director must prove the procedural fairness of the transaction and that the transaction was of fair value to the corporation.
Under British Virgin Islands law, our directors owe fiduciary duties both at common law and under statute including, among others, a statutory duty to act honestly, in good faith, for a proper purpose and with a view to what the directors believe to be in the best interests of the company. Our directors are also required, when exercising powers or performing duties as a director, to exercise the care, diligence and skill that a reasonable director would exercise in comparable circumstances, taking into account without limitation, the nature of the company, the nature of the decision and the position of the director and the nature of the responsibilities undertaken. In the exercise of their powers, our directors must ensure neither they nor the company acts in a manner which contravenes the BVI Act or our memorandum and articles of association. A shareholder has the right to seek damages for breaches of duties owed to us by our directors.
Pursuant to the BVI Act and our memorandum and articles of association, a director of a company who has an interest in a transaction and who has declared such interest to the other directors, may:
| | (a) | vote on a matter relating to the transaction; |
| | (b) | attend a meeting of directors at which a matter relating to the transaction arises and be included among the directors present at the meeting for the purposes of a quorum; and |
| | (c) | sign a document on our behalf, or do any other thing in his or her capacity as a director, that relates to the transaction. |
In certain limited circumstances, a shareholder has the right to seek various remedies against the company in the event the directors are in breach of their duties under the BVI Act. Pursuant to Section 184B of the BVI Act, if a company or director of a company engages in, or proposes to engage in or has engaged in, conduct that contravenes the provisions of the BVI Act or the memorandum or articles of association of the company, the British Virgin Islands Court may, on application of a shareholder or director of the company, make an order directing the company or director to comply with, or restraining the company or director from engaging in conduct that contravenes the BVI Act or the memorandum or articles. Furthermore, pursuant to section 184I(1) of the BVI Act, a shareholder of a company who considers that the affairs of the company have been, are being or are likely to be, conducted in a manner that is, or any acts of the company have been, or are likely to be oppressive, unfairly discriminatory, or unfairly prejudicial to him in that capacity, may apply to the British Virgin Islands Court for an order which, inter alia, can require the company or any other person to pay compensation to the shareholders.
Shareholder action by written consent
Under the Delaware General Corporation Law, a corporation may eliminate the right of shareholders to act by written consent by amendment to its certificate of incorporation. British Virgin Islands law provides that, subject to the memorandum and articles of association of a company, an action that may be taken by members of the company at a meeting may also be taken by a resolution of members consented to in writing.
Shareholder proposals
A special meeting may be called by the board of directors or any other person authorized to do so in the governing documents, but shareholders may be precluded from calling special meetings. British Virgin Islands law and our memorandum and articles of association allow our shareholders holding 30% or more of the votes of the outstanding voting shares to requisition a shareholders’ meeting. There is no requirement under BVI law to hold shareholders’ annual general meetings, but our memorandum and articles of association do permit the directors to call such a meeting. The location of any shareholders’ meeting can be determined by the board of directors and can be held anywhere in the world.
Cumulative voting
Under the Delaware General Corporation Law, cumulative voting for elections of directors is not permitted unless the corporation’s certificate of incorporation specifically provides for it. Cumulative voting potentially facilitates the representation of minority shareholders on a board of directors since it permits the minority shareholder to cast all the votes to which the shareholder is entitled on a single director, which increases the shareholder’s voting power with respect to electing such director. As permitted under British Virgin Islands law, our memorandum and articles of association do not provide for cumulative voting. As a result, our shareholders are not afforded any less protections or rights on this issue than shareholders of a Delaware corporation.
Removal of directors
Under the Delaware General Corporation Law, a director of a corporation with a classified board may be removed only for cause with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. Under our memorandum and articles of association, directors can be removed from office, with or without cause, by a resolution of shareholders passed at a meeting of the shareholders called for the purposes of removing the director or for purposes including the removal of the director or by a written resolution passed by at least 75% of the votes of the shareholders entitled to vote. Directors can also be removed by a resolution of directors passed at a meeting of directors called for the purpose of removing the director or for purposes including the removal of the director.
Transactions with interested shareholders
The Delaware General Corporation Law contains a business combination statute applicable to Delaware public corporations whereby, unless the corporation has specifically elected not to be governed by such statute by amendment to its certificate of incorporation, it is prohibited from engaging in certain business combinations with an “interested shareholder” for three years following the date that such person becomes an interested shareholder. An interested shareholder generally is a person or group who or which owns or owned 15% or more of the target’s outstanding voting shares within the past three years. This has the effect of limiting the ability of a potential acquirer to make a two-tiered bid for the target in which all shareholders would not be treated equally. The statute does not apply if, among other things, prior to the date on which such shareholder becomes an interested shareholder, the board of directors approves either the business combination or the transaction which resulted in the person becoming an interested shareholder. This encourages any potential acquirer of a Delaware public corporation to negotiate the terms of any acquisition transaction with the target’s board of directors. British Virgin Islands law has no comparable statute and our memorandum and articles of association does not provide for the same protection afforded by the Delaware business combination statute.
Dissolution; Winding Up
Under the Delaware General Corporation Law, unless the board of directors approves the proposal to dissolve, dissolution must be approved by shareholders holding 100% of the total voting power of the corporation. Only if the dissolution is initiated by the board of directors may it be approved by a simple majority of the corporation’s outstanding shares. Delaware law allows a Delaware corporation to include in its certificate of incorporation a supermajority voting requirement in connection with dissolutions initiated by the board. Under the BVI Act and our memorandum and articles of association, we may appoint a voluntary liquidator by a resolution of the shareholders, provided that the directors have made a declaration of solvency that the company is able to discharge its debts as they fall due and that the value of the company’s assets exceed its liabilities.
Variation of rights of shares
Under the Delaware General Corporation Law, a corporation may vary the rights of a class of shares with the approval of a majority of the outstanding shares of such class, unless the certificate of incorporation provides otherwise. Under our memorandum and articles of association, if at any time our shares are divided into different classes of shares, the rights attached to any class may only be varied, whether or not we are in liquidation, with the consent in writing of or by a resolution passed at a meeting by a majority of the votes cast by those entitled to vote at a meeting of the holders of the issued shares in that class. For these purposes the creation, designation or issue of shares with rights and privileges ranking pari passu to an existing class of shares is deemed not to be a variation of the rights of such existing class and may in accordance with our memorandum and articles of association be effected by resolution of directors without shareholder approval.
Amendment of governing documents
Under the Delaware General Corporation Law, a corporation’s governing documents may be amended with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. As permitted by British Virgin Islands law, our memorandum and articles of association may be amended by a resolution of shareholders and, subject to certain exceptions, by a resolution of directors. An amendment is effective from the date it is registered at the Registry of Corporate Affairs in the British Virgin Islands.
Anti-Money Laundering Laws
In order to comply with legislation or regulations aimed at the prevention of money laundering we are required to adopt and maintain anti-money laundering procedures, and may require subscribers to provide evidence to verify their identity. Where permitted, and subject to certain conditions, we also may delegate the maintenance of our anti-money laundering procedures (including the acquisition of due diligence information) to a suitable person.
We reserve the right to request such information as is necessary to verify the identity of a subscriber. In the event of delay or failure on the part of the subscriber in producing any information required for verification purposes, we may refuse to accept the application, in which case any funds received will be returned without interest to the account from which they were originally debited.
If any person resident in the British Virgin Islands knows or suspects that another person is engaged in money laundering or terrorist financing and the information for that knowledge or suspicion came to their attention in the course of their business the person will be required to report his belief or suspicion to the Financial Investigation Agency of the British Virgin Islands, pursuant to the Proceeds of Criminal Conduct Act (Law Revision 2020). Such a report shall not be treated as a breach of confidence or of any restriction upon the disclosure of information imposed by any enactment or otherwise.
SHARES ELIGIBLE FOR FUTURE SALE
Before this initial public offering, there has not been a public market for our Ordinary Shares, and although we intend to apply for listing on the Nasdaq Capital Market, a regular trading market for our Ordinary Shares may not develop. Future sales of substantial amounts of our Ordinary Shares in the public market after our initial public offering, or the possibility of these sales occurring, could cause the prevailing market price for our Ordinary Shares to fall or impair our ability to raise equity capital in the future. Upon completion of this offering, we will have Ordinary Shares issued and outstanding. All of the Ordinary Shares sold in this offering will be freely transferable by persons other than our “affiliates” without restriction or further registration under the Securities Act.
Lock-Up Agreements
We have agreed not to, for a period of six months from the closing of this offering, offer, issue, sell, contract to sell, encumber, grant any option for the sale of, or otherwise dispose of, except in this offering, any of our Ordinary Shares or securities that are substantially similar to our Ordinary Shares, including but not limited to any options or warrants to purchase our Ordinary Shares, or any securities that are convertible into or exchangeable for, or that represent the right to receive, our Ordinary Shares or any such substantially similar securities (other than pursuant to employee stock option plans existing on, or upon the conversion or exchange of convertible or exchangeable securities outstanding as of, the date such lock-up agreement was executed), without the prior written consent of the representatives.
Furthermore, each of our directors, executive officers, and principal shareholders (holders of 5% or more of our Ordinary Shares) has also entered into a similar lock-up agreement for a period of six months from the closing of this offering, subject to certain exceptions, with respect to our Ordinary Shares and securities that are substantially similar to our Ordinary Shares. See “Underwriting”.
We are not aware of any plans by any significant shareholders to dispose of significant numbers of our Ordinary Shares. However, one or more existing shareholders or owners of securities convertible or exchangeable into or exercisable for our Ordinary Shares may dispose of significant numbers of our Ordinary Shares in the future. We cannot predict what effect, if any, future sales of our Ordinary Shares, or the availability of Ordinary Shares for future sale, will have on the trading price of our Ordinary Shares from time to time. Sales of substantial amounts of our Ordinary Shares in the public market, or the perception that these sales could occur, could adversely affect the trading price of our Ordinary Shares.
Rule 144
All of our Ordinary Shares outstanding prior to this offering are “restricted securities” as that term is defined in Rule 144 under the Securities Act and may be sold publicly in the United States only if they are subject to an effective registration statement under the Securities Act or pursuant to an exemption from the registration requirement such as those provided by Rule 144 promulgated under the Securities Act.
In general, under Rule 144 as currently in effect, beginning 90 days after the date of this prospectus, a person who is not deemed to have been our affiliate at any time during the three months preceding a sale and who has beneficially owned restricted securities within the meaning of Rule 144 for more than six months would be entitled to sell an unlimited number of those shares, subject only to the availability of current public information about us. A non-affiliate who has beneficially owned restricted securities for at least one year from the later of the date these shares were acquired from us or from our affiliate would be entitled to freely sell those shares.
A person who is deemed to be our affiliate and who has beneficially owned “restricted securities” for at least six months would be entitled to sell, within any three-month period, a number of shares that is not more than the greater of:
| | ● | 1% of the number of Ordinary Shares then outstanding, in the form of Ordinary Shares or otherwise; or |
| | ● | the average weekly trading volume of the Ordinary Shares on the Nasdaq Capital Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to such sale. |
Sales under Rule 144 by our affiliates or persons selling shares on behalf of our affiliates are also subject to certain manner of sale provisions and notice requirements and to the availability of current public information about us.
Rule 701
In general, under Rule 701 of the Securities Act as currently in effect, each of our employees, consultants, or advisors who purchases our Ordinary Shares from us in connection with a compensatory stock plan or other written agreement executed prior to the completion of this offering is eligible to resell those Ordinary Shares in reliance on Rule 144, but without compliance with some of the restrictions, including the holding period, contained in Rule 144. However, the Rule 701 shares would remain subject to lock-up arrangements and would only become eligible for sale when the lock-up period expires.
Regulation S
Regulation S provides generally that sales made in offshore transactions are not subject to the registration or prospectus-delivery requirements of the Securities Act.
TAXATION
Material U.S. Federal Income Tax Consequences Applicable to U.S. Holders of Ordinary Shares
The following sets forth the material U.S. federal income tax consequences related to an investment in Ordinary Shares. It is directed to U.S. Holders (as defined below) of Ordinary Shares and is based upon laws and relevant interpretations thereof in effect as of the date of this prospectus, all of which are subject to change. This description does not deal with all possible tax consequences relating to an investment in Ordinary Shares or U.S. tax laws, other than the U.S. federal income tax laws, such as the tax consequences under non-U.S. tax laws, state, local and other tax laws.
The following brief description applies only to U.S. Holders (defined below) that hold Ordinary Shares as capital assets and that have the U.S. dollar as their functional currency. This brief description is based on the federal income tax laws of the United States in effect as of the date of this prospectus and on U.S. Treasury regulations in effect or, in some cases, proposed, as of the date of this prospectus, as well as judicial and administrative interpretations thereof available on or before such date. All of the foregoing authorities are subject to change, which change could apply retroactively and could affect the tax consequences described below.
The brief description below of the U.S. federal income tax consequences to “U.S. Holders” will apply to you if you are a beneficial owner of Ordinary Shares and you are, for U.S. federal income tax purposes:
| | ● | an individual who is a citizen or resident of the United States; |
| | ● | a corporation (or other entity taxable as a corporation for U.S. federal income tax purposes) organized under the laws of the United States, any state thereof or the District of Columbia; |
| | ● | an estate whose income is subject to U.S. federal income taxation regardless of its source; or |
| | ● | a trust that (1) is subject to the primary supervision of a court within the United States and the control of one or more U.S. persons for all substantial decisions or (2) has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person. |
If a partnership (or other entity treated as a partnership for United States federal income tax purposes) is a beneficial owner of Ordinary Shares, the tax treatment of a partner in the partnership will depend upon the status of the partner and the activities of the partnership. Partnerships and partners of a partnership holding Ordinary Shares are urged to consult their tax advisors regarding an investment in Ordinary Shares.
EVERFRONT HOLDING URGES POTENTIAL PURCHASERS OF EVERFRONT HOLDING’S ORDINARY SHARES TO CONSULT THEIR OWN TAX ADVISORS CONCERNING THE U.S. FEDERAL, STATE, LOCAL AND NON-U.S. TAX CONSEQUENCES OF PURCHASING, OWNING AND DISPOSING OF EVERFRONT HOLDING’S ORDINARY SHARES.
The following does not address the tax consequences to any particular investor or to persons in special tax situations such as:
| | ● | regulated investment companies; |
| | ● | real estate investment trusts; |
| | ● | traders that elect to use a mark-to-market method of accounting; |
| | ● | Certain former U.S. citizens or long-term residents; |
| | ● | tax-exempt entities (including private foundations); |
| | ● | persons liable for alternative minimum tax, liable for the “base erosion and anti-avoidance tax” or required to recognize income for U.S. federal income tax purposes no later than when such income is included on an applicable financial statement; |
| | ● | persons holding Ordinary Shares as part of a straddle, hedging, conversion or integrated transaction; |
| | ● | persons that actually or constructively own 10% (by vote or value) or more of our voting shares (including by reason of owning Ordinary Shares), and, therefore, might be treated as owning shares in a “controlled foreign corporation” or “CFC” with respect to their ownership of Ordinary Shares; |
| | ● | persons who acquired Ordinary Shares pursuant to the exercise of any employee share option or otherwise as compensation; |
| | ● | persons holding Ordinary Shares through partnerships or other pass-through entities; |
| | ● | real estate investment trusts; |
| | ● | governments or agencies or instrumentalities thereof; |
| | ● | beneficiaries of a trust holding Ordinary Shares; or |
| | ● | persons holding Ordinary Shares through a trust. |
All of whom may be subject to tax rules that differ significantly from those discussed below.
The discussion set forth below is addressed only to U.S. Holders that purchase Ordinary Shares in this offering. Prospective purchasers are urged to consult their own tax advisors about the application of the U.S. federal income tax rules to their particular circumstances as well as the state, local, foreign and other tax consequences to them of the purchase, ownership and disposition of Ordinary Shares. Prospective U.S. Holders that may actually or constructively own 10% (by vote or value) or more of Everfront Holding’s voting shares (including by reason of owning Ordinary Shares) are urged to consult their tax advisors regarding the application of the “controlled foreign corporation” or “CFC” rules to their investment in Ordinary Shares.
Taxation of Dividends and Other Distributions on Ordinary Shares
Subject to the PFIC rules discussed below, the gross amount of distributions made by us to you with respect to the Ordinary Shares (including the amount of any taxes withheld there from) will generally be includable in your gross income as dividend income on the date of actual or constructive receipt by you, but only to the extent that the distribution is paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). With respect to corporate U.S. Holders, the dividends will generally not be eligible for the dividends-received deduction allowed to corporations in respect of dividends received from U.S. corporations.
With respect to non-corporate U.S. Holders, including individual U.S. Holders, dividends will be taxed at the lower capital gains rate applicable to qualified dividend income, provided that (1) the Ordinary Shares are readily tradable on an established securities market in the United States, or Everfront Holding is eligible for the benefits of an approved qualifying income tax treaty with the United States that includes an exchange of information program, (2) Everfront Holding is not a PFIC for either its taxable year in which the dividend is paid or the preceding taxable year, and (3) certain holding period requirements are met. Because there is no income tax treaty between the United States and the BVI, clause (1) above can be satisfied only if the Ordinary Shares are readily tradable on an established securities market in the United States. Ordinary Shares are considered for purpose of clause (1) above to be readily tradable on an established securities market in the United States if they are listed on the Nasdaq Capital Market. You are urged to consult your tax advisors regarding the availability of the lower rate for dividends paid with respect to Ordinary Shares, including the effects of any change in law after the date of this prospectus.
Dividends will constitute foreign source income for foreign tax credit limitation purposes. If the dividends are taxed as qualified dividend income (as discussed above), the amount of the dividend taken into account for purposes of calculating the foreign tax credit limitation will be limited to the gross amount of the dividend, multiplied by the reduced rate divided by the highest rate of tax normally applicable to dividends. The limitation on foreign taxes eligible for credit is calculated separately with respect to specific classes of income. For this purpose, dividends distributed by us with respect to Ordinary Shares will constitute “passive category income” but could, in the case of certain U.S. Holders, constitute “general category income.”
To the extent that the amount of the distribution exceeds our current and accumulated earnings and profits (as determined under U.S. federal income tax principles), it will be treated first as a tax-free return of your tax basis in your Ordinary Shares, and to the extent the amount of the distribution exceeds your tax basis, the excess will be taxed as capital gain. We do not intend to calculate our earnings and profits under U.S. federal income tax principles. Therefore, a U.S. Holder should expect that a distribution will be treated as a dividend even if a particular that distribution would otherwise be treated as a non-taxable return of capital or as capital gain under the rules described above.
If Everfront Holdings pays an “extraordinary dividend” on the Ordinary Shares (generally, a dividend in an amount which is equal to or in excess of 10% of a shareholder’s adjusted tax basis (or fair market value in certain circumstances) in the Ordinary Shares or dividends received within a one-year period that, in the aggregate, equal or exceed 20% of a shareholder’s adjusted tax basis (or fair market value upon the shareholder’s election)) that is treated as “qualified dividend income,” then any loss derived by a U.S. Holder from the sale or exchange of such Ordinary Shares will be treated as long-term capital loss to the extent of such dividend.
Taxation of Dispositions of Ordinary Shares
Subject to the PFIC rules discussed below, you will recognize taxable gain or loss on any sale, exchange or other taxable disposition of an Ordinary Share equal to the difference between the amount realized (in U.S. dollars) for the Ordinary Share and your tax basis (in U.S. dollars) in the Ordinary Shares. The gain or loss will be capital gain or loss. If you are a non-corporate U.S. Holder, including an individual U.S. Holder, who has held the Ordinary Shares for more than one year, you will be eligible for reduced tax rates. The deductibility of capital losses is subject to limitations. Any such gain or loss that you recognize will generally be treated as United States source income or loss for foreign tax credit limitation purposes.
Information Reporting and Backup Withholding
Dividend payments with respect to Ordinary Shares and proceeds from the sale, exchange or redemption of Ordinary Shares may be subject to information reporting to the U.S. Internal Revenue Service and possible U.S. backup withholding at a current rate of 24%. Backup withholding will not apply, however, to a U.S. Holder who furnishes a correct taxpayer identification number and makes any other required certification on U.S. Internal Revenue Service Form W-9 or who is otherwise exempt from backup withholding. U.S. Holders who are required to establish their exempt status generally must provide such certification on IRS Form W-9. U.S. Holders are urged to consult their tax advisors regarding the application of the U.S. information reporting and backup withholding rules.
Backup withholding is not an additional tax. Amounts withheld as backup withholding may be credited against your U.S. federal income tax liability, and you may obtain a refund of any excess amounts withheld under the backup withholding rules by filing the appropriate claim for refund with the IRS and furnishing any required information. We do not intend to withhold taxes for individual shareholders. However, transactions effected through certain brokers or other intermediaries may be subject to withholding taxes (including backup withholding), and such brokers or intermediaries may be required by law to withhold such taxes.
Under the Foreign Account Tax Compliance Act, certain U.S. Holders are required to report information relating to Ordinary Shares, subject to certain exceptions (including an exception for Ordinary Shares held in accounts maintained by certain financial institutions), by attaching a complete IRS Form 8938, Statement of Specified Foreign Financial Assets, with their tax return for each year in which they hold Ordinary Shares. Failure to report the information could result in substantial penalties. You should consult your own tax advisor regarding your obligation to file Form 8938.
Passive Foreign Investment Company (“PFIC”)
Based on our current and anticipated operations and the composition of our assets, we were not a PFIC for U.S. federal income tax purposes for the taxable year ended December 31, 2023 and the taxable year ended December 31, 2022. Depending on the amount of cash we raise in this offering, together with any other assets held for the production of passive income, it is possible that, for our taxable year ending December 31, 2024 or for any subsequent year, more than 50% of our assets may be assets which produce passive income, in which case we would be deemed a PFIC, which could have adverse U.S. federal income tax consequences for U.S. taxpayers who are shareholders. We will make this determination following the end of any particular tax year. PFIC status is a factual determination for each taxable year which cannot be made until the close of the taxable year. A non-U.S. corporation is considered a PFIC, as defined in Section 1297(a) of the U.S. Internal Revenue Code, for any taxable year if either:
| | ● | at least 75% of its gross income is passive income; or |
| | | |
| | ● | at least 50% of the value of its assets (based on an average of the quarterly values of the assets during a taxable year) is attributable to assets that produce or are held for the production of passive income (the “asset test”). |
We will be treated as owning our proportionate share of the assets and earning our proportionate share of income of any other corporation in which we own, directly or indirectly, at least 25% (by value) of the stock.
We must make a separate determination each year as to whether we are a PFIC, however, and there can be no assurance with respect to our status as a PFIC for our current taxable year or any future taxable year. Depending on the amount of cash Everfront Holding raises in this offering, together with any other assets held for the production of passive income, it is possible that, for our current taxable year or for any subsequent taxable year, more than 50% of our assets may be assets held for the production of passive income. We will make this determination following the end of any particular tax year. In addition, because the value of our assets for purposes of the asset test will generally be determined based on the market price of Ordinary Shares and because cash is generally considered to be an asset held for the production of passive income, our PFIC status will depend in large part on the market price of Ordinary Shares and the amount of cash we raise in this offering. Accordingly, fluctuations in the market price of the Ordinary Shares may cause us to become a PFIC. In addition, the application of the PFIC rules is subject to uncertainty in several respects and the composition of our income and assets will be affected by how, and how quickly, we spend the cash we raise in this offering. we are under no obligation to take steps to reduce the risk of our being classified as a PFIC, and as stated above, the determination of the value of our assets will depend upon material facts (including the market price of Ordinary Shares from time to time and the amount of cash we raise in this offering) that may not be within our control. If we are a PFIC for any year during which you hold Ordinary Shares, we will continue to be treated as a PFIC for all succeeding years during which you hold Ordinary Shares. If we cease to be a PFIC and you did not previously make a timely “mark-to-market” election as described below, however, you may avoid some of the adverse effects of the PFIC regime by making a “purging election” (as described below) with respect to your Ordinary Shares.
If we are a PFIC for any taxable year during which you hold Ordinary Shares, you will be subject to special tax rules with respect to any “excess distribution” that you receive and any gain you realize from a sale or other disposition (including a pledge) of the Ordinary Shares, unless you make a “mark-to-market” election as discussed below. Distributions you receive in a taxable year that are greater than 125% of the average annual distributions you received during the shorter of the three preceding taxable years or your holding period for the Ordinary Shares will be treated as an excess distribution. Under these special tax rules:
| | ● | the excess distribution or gain will be allocated ratably over your holding period for the Ordinary Shares (in the case of Ordinary Shares obtained through the exercise of warrants, the holding period will include the holding period of the underlying warrants); |
| | ● | the amount allocated to the current taxable year, and the amount allocated to any taxable year prior to the first taxable year in which was we were a PFIC, will be treated as ordinary income earned in the current taxable year; |
| | | |
| | ● | the amount allocated to each other year will be subject to the highest tax rate in effect for that year and the interest charge generally applicable to underpayments of tax will be imposed on the resulting tax attributable to each such year; and |
| | | |
| | ● | An additional tax equal to the interest charge generally applicable to underpayments of tax will be imposed on the tax attributable to each prior taxable year, other than a pre-PFIC year. |
The tax liability for amounts allocated to years prior to the year of disposition or “excess distribution” cannot be offset by any net operating losses for such years, and gains (but not losses) realized on the sale of the Ordinary Shares cannot be treated as capital, even if you hold the Ordinary Shares as capital assets.
A U.S. Holder of “marketable stock” (as defined below) in a PFIC may make a mark-to-market election for such stock (but not warrants) to elect out of the tax treatment discussed above. If you make a mark-to-market election for the Ordinary Shares, you will include in income each year an amount equal to the excess, if any, of the fair market value of the Ordinary Shares as of the close of your taxable year over your adjusted basis in such Ordinary Shares. You are allowed a deduction for the excess, if any, of the adjusted basis of the Ordinary Shares over their fair market value as of the close of the taxable year. However, deductions are allowable only to the extent of any net mark-to-market gains on the Ordinary Shares included in your income for prior taxable years. Amounts included in your income under a mark-to-market election, as well as gain on the actual sale or other disposition of the Ordinary Shares, are treated as ordinary income. Ordinary loss treatment also applies to the deductible portion of any mark-to-market loss on the Ordinary Shares, as well as to any loss realized on the actual sale or disposition of the Ordinary Shares, to the extent that the amount of such loss does not exceed the net mark-to-market gains previously included for such Ordinary Shares. Your basis in the Ordinary Shares will be adjusted to reflect any such income or loss amounts. If you make a valid mark-to-market election, the tax rules that apply to distributions by corporations which are not PFICs would apply to distributions by us, except that the lower applicable capital gains rate for qualified dividend income discussed above under “— Taxation of Dividends and Other Distributions on Ordinary Shares” generally would not apply.
The mark-to-market election is available only for “marketable stock”, which is stock that is traded in other than de minimis quantities on at least 15 days during each calendar quarter (“regularly traded”) on a qualified exchange or other market (as defined in applicable U.S. Treasury regulations), including the Nasdaq Capital Market. If the Ordinary Shares are regularly traded on the Nasdaq Capital Market and if you are a holder of Ordinary Shares, the mark-to-market election would be available to you were we to be or become a PFIC.
Alternatively, a U.S. Holder of stock (but not warrants) in a PFIC may make a “qualified electing fund” election under Section 1295(b) of the U.S. Internal Revenue Code (“IRC”) with respect to such PFIC to elect out of the tax treatment discussed above. A U.S. Holder who makes a valid qualified electing fund election with respect to a PFIC will generally include in gross income for a taxable year such holder’s pro rata share of the corporation’s earnings and profits for the taxable year. However, the qualified electing fund election is available only if such PFIC provides such U.S. Holder with certain information regarding its earnings and profits as required under applicable U.S. Treasury regulations. We do not currently intend to prepare or provide the information that would enable you to make a qualified electing fund election. Therefore, prospective investors should assume that a qualified electing fund election will not be available. If you hold Ordinary Shares in any year in which we are a PFIC, you will be required to file IRS Form 8621 regarding distributions received on the Ordinary Shares and any gain realized on the disposition of the Ordinary Shares. The failure to file IRS Form 8621 could result in the imposition of penalties and the extension of the statute of limitations with respect to U.S. federal income tax.
If you do not make a timely “mark-to-market” election (as described above), and if we were a PFIC at any time during the period you hold Ordinary Shares, then such Ordinary Shares will continue to be treated as stock of a PFIC with respect to you even if we cease to be a PFIC in a future year, unless you make a “purging election” for the year we cease to be a PFIC (no such election is available to warrants). A “purging election” creates a deemed sale of such Ordinary Shares at their fair market value on the last day of the last year in which we are treated as a PFIC. The gain recognized by the purging election will be subject to the special tax and interest charge rules treating the gain as an excess distribution, as described above. As a result of the purging election, you will have a new basis (equal to the fair market value of the Ordinary Shares on the last day of the last year in which we are treated as a PFIC) and holding period (which new holding period will begin the day after such last day) in your Ordinary Shares for tax purposes.
IRC Section 1014(a) provides for a step-up in basis to the fair market value for Ordinary Shares when inherited from a decedent that was previously a holder of Ordinary Shares. However, if we are determined to be a PFIC and a decedent that was a U.S. Holder did not make either a timely qualified electing fund election for our first taxable year as a PFIC in which the U.S. Holder held (or was deemed to hold) Ordinary Shares, or a mark-to-market election and ownership of those Ordinary Shares are inherited, a special provision in IRC Section 1291(e) provides that the new U.S. Holder’s basis should be reduced by an amount equal to the Section 1014 basis minus the decedent’s adjusted basis just before death. As such if we are determined to be a PFIC at any time prior to a decedent’s passing, the PFIC rules will cause any new U.S. Holder that inherits Ordinary Shares from a U.S. Holder to not get a step-up in basis under Section 1014 and instead will receive a carryover basis in those Ordinary Shares.
You are urged to consult your tax advisors regarding the application of the PFIC rules to your investment in Ordinary Shares and the elections discussed above.
Taiwan Taxation
Our business operations in Taiwan are carried out through our Taiwan subsidiary, Everfront Biotech. The following brief description of Taiwan’s laws is designed to highlight the enterprise-level taxation on our earnings, which will affect the amount of dividends, if any, that we are ultimately able to pay to our shareholders.
Enterprise Income Tax
The accounting system of a company is conducted on an accrual basis. For any enterprise having its head office within the territory of Taiwan (including any subsidiary of a foreign enterprise in Taiwan), enterprise income tax shall be levied on its total enterprise income derived within or outside the territory of Taiwan. However, if income tax has been paid on the income derived outside of the territory of Taiwan in accordance with the tax law of the source country of that income, such tax paid may, upon presentation by the taxpayer of evidence of tax payment issued by the tax office of said source country for the same business year, be deducted from the amount of tax payable by the taxpayer at the time of filing final returns on the total enterprise income, to the extent that such deduction shall not exceed the amount of tax which, computed at the applicable domestic tax rate, is increased in consequence of the inclusion of income derived from abroad.
Expenses and losses related to the operation of the business may be recognized as expenses and losses of the business only if appropriate and legitimate evidence is provided. For example, the excessive amount of the limit under the tax law or the difference between the evidence of expense and the tax law should not be recognized as current expenses or losses.
Losses incurred in business operations in previous years are generally not included in the computation for the current year. However, in the case of an enterprise organized as a company that keeps a complete set of account books, uses the Blue Returns as provided in Article 77 of the Income Tax Act in the years such losses occurred and in the year of declaring such losses, or such losses have been duly certified by a certified public accountant and declared within the prescribed period, taxation may be made on net income after deduction of losses incurred in the preceding ten years as verified and determined by the local tax authority.
In the event that the total taxable income of an enterprise is NT$120,000 or less, the enterprise is exempt from tax. If the total taxable income of an enterprise is more than NT$120,000, the income tax rate shall be 20%. However, the income tax payable shall not exceed one-half of the portion of taxable income greater than NT$120,000.
Value-added Tax
Enterprises conducting the sale of goods or services within the territory of Taiwan are subject to value-added or non-value-added business tax provided under the Value-added and Non-value-added Business Tax Act (“Business Tax Act”). For the purpose of the Business Tax Act, the sale of goods means the transfer of ownership of goods to others for consideration, and the sale of services means the supply of services to others or the provision of goods for the use of others for consideration, with the exception of professional services offered by practitioners and services rendered by individuals in employment.
Non-value-added business tax is applicable only to special food and beverage service enterprises, small business entities, and financial businesses such as banks, insurance companies, investment trusts, securities, futures, commercial paper, and pawnshops. Small business entities are enterprises whose average monthly sales do not exceed NT$200,000. As our Taiwan subsidiary does not fall into the business categories to which the non-value-added business tax applies, our business operations within Taiwan are subject to value-added business tax corresponding to the sale of goods or services. The value-added business tax rate is 5%, and the amount of business tax payable is the difference between the output tax in a tax period and the input tax in the same period.
Stamp Tax
Under the Stamp Tax Act of Taiwan, types of documents subject to the levy of stamp tax include the following.
| | ● | Receipts for monetary payments. This category includes documents, books, or records drawn upon the receipt of money, such as receipts, slips, releases, bank books, payment records, and the like issued to identify monetary payments, excluding monetary receipts for commercial invoices. The tax is levied at 0.4% of the amount received, with the exception of receipts for deposit of bid bonds, for which the tax is levied at 0.1% of the money deposited by the bidder. |
| | | |
| | ● | Deeds for sale of movables. This category includes deeds issued for the sale of movables. The tax is levied at NT$12 per piece of the deed. |
| | | |
| | ● | Contracting agreements. This category includes agreements executed to complete a specifically ordered task, such as construction contracts, printing contracts, OEM contracts, and other similar contracts. The tax is levied at 0.1% of the contract price. |
| | | |
| | ● | Contracts for the pledge, sale, transfer, and partition of real estate. This category includes pledges of lien on real estate, or deeds or contracts for sale, exchange, gratuitous transfer, or partition of real estate to be submitted to government agencies for registration. The tax is levied at 0.1% of the contract price. |
Since our Taiwan subsidiary may be contracted to provide products or services or dispose of its properties, it may be subject to the obligation to pay stamp tax for relevant business operations in Taiwan.
British Virgin Islands Taxation
The disclosure relating to tax consequences under British Virgin Islands law is the opinion of McW Todman & Co., our counsel as to BVI law.
The Government of the British Virgin Islands does not, under existing legislation, impose any income, corporate or capital gains tax, estate duty, inheritance tax, gift tax or withholding tax upon us or our shareholders who are not tax resident in the British Virgin Islands.
We and all distributions, interest and other amounts paid by us to persons who are not tax resident in the British Virgin Islands will not be subject to any income, withholding or capital gains taxes in the British Virgin Islands, with respect to the Ordinary Shares in us owned by them and dividends received on such shares, nor will they be subject to any estate or inheritance taxes in the British Virgin Islands.
No estate, inheritance, succession or gift tax, rate, duty, levy or other charge is payable by persons who are not tax resident in the British Virgin Islands with respect to any shares, debt obligations or other of our securities.
Except to the extent that we have any interest in real property in the British Virgin Islands, all instruments relating to transactions in respect of the shares, debt obligations or other of our securities, and all instruments relating to other transactions relating to our business are exempt from the payment of stamp duty in the British Virgin Islands.
There are currently no withholding taxes or exchange control regulations in the British Virgin Islands applicable to us or our shareholders.
There is no income tax treaty or convention currently in effect between the United States and the British Virgin Islands or between Taiwan and the British Virgin Islands.
British Virgin Islands Economic Substance Legislation
The British Virgin Islands, together with several other non-European Union jurisdictions, has introduced legislation aimed at addressing concerns raised by the Council of the European Union (the “EU”) as to offshore structures engaged in certain activities which attract profits without real economic activity. With effect from January 1, 2019, the Economic Substance (Companies and Limited Partnerships) Act, 2018 (the “ES Act”) came into force in the British Virgin Islands introducing certain economic substance requirements for in-scope British Virgin Islands entities which are engaged in certain “relevant activities”.
Although it is presently anticipated that the ES Act will have little material impact on us or our operations, as the legislation is relatively new and remains subject to further clarification and interpretation, it is not currently possible to ascertain the precise impact of these legislative changes on us.
British Virgin Islands Data Protection Act
The Data Protection Act, 2021 (the “BVI DPA”) came into force in the British Virgin Islands on 9 July 2021. The BVI DPA establishes a framework of rights and duties designed to safeguard individuals’ personal data, balanced against the need of public authorities, businesses and organizations to collect and use personal data for lawful purposes. The BVI DPA is centered around seven data protection principles (the General Principle, the Notice and Choice Principle, the Disclosure Principle, the Security Principle, the Retention Principle, the Data Protection Principle and the Access Principle) which require that:
| | ● | personal data must not be processed without consent unless specific conditions are met and must not be transferred outside the British Virgin Islands, unless there is proof of adequate data protection safeguards or consent from the data subject; |
| | | |
| | ● | Where consent has been given to processing of personal data, the data subject may at any time withdraw his or her consent; |
| | | |
| | ● | a data controller must inform a data subject of specific matters, for instance the purposes for which it is being collected and further processed; |
| | | |
| | ● | personal data must not be disclosed for any purpose other than the purpose for which it was to be disclosed at the time of collection or a purpose directly related thereto or to any party other than a third party of a class previously notified to the data subject; |
| | | |
| | ● | a data controller shall, when processing personal data, take practical steps to protect personal data from loss, misuse, modification, unauthorized or accidental access or disclosure, alteration or destruction; |
| | | |
| | ● | personal data must not be kept for longer than is necessary for the purpose; |
| | | |
| | ● | personal data must be accurate, complete, not misleading and kept up to date; and |
| | | |
| | ● | a data subject must be given access to his or her own personal data and be able to correct that data where it is inaccurate, incomplete, misleading or not up to date, except where a request for such access or correction is refused under the BVI DPA. |
The BVI DPA imposes specific obligations on data controllers, including the duty to (i) apply the data protection principles; and (ii) respond in a timely fashion to requests from data subjects in relation to their personal data.
The Information Commissioner is the regulator responsible for the proper functioning and enforcement of the BVI DPA. Offences under the BVI DPA include
| | ● | processing sensitive personal data in contravention of the BVI DPA; |
| | | |
| | ● | willfully obstructing the Information Commissioner or an authorized officer in the conduct of his or her duties and functions; |
| | | |
| | ● | willfully disclosing personal information in contravention of the BVI DPA; and |
| | | |
| | ● | collecting, storing or disposing of personal information in a manner that contravenes the BVI DPA. |
Offences committed under the BVI DPA may result in fines (up to US$500,000 in certain cases) or imprisonment. Further, a data subject who suffers damage or distress as a result of their data being processed in contravention of the Data Protection Act (DPA) may institute civil proceedings in the British Virgin Islands courts.
UNDERWRITING
We have entered into an underwriting agreement, dated as of , 2025, with Roth Capital Partners, LLC and the Benchmark Company, LLC, acting as the co-representatives (the “representatives”) of the underwriters named below. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to the underwriters, and the underwriters have agreed to purchase from us, Ordinary Shares. We have been approved to list our Ordinary Shares on the Nasdaq Capital Market under the symbol “EFB” upon our satisfaction of the exchange’s initial listing criteria, including the completion of this offering.
Pursuant to the terms and subject to the conditions contained in the underwriting agreement, we have agreed to sell to the underwriters named below, and the underwriters have agreed to purchase from us, the respective number of Ordinary Shares set forth opposite its name below.
| Underwriter | | Number of
Ordinary Shares | |
| Roth Capital Partners, LLC | | | | |
| The Benchmark Company, LLC | | | | |
| Total | | | | |
The underwriting agreement provides that the obligation of the underwriters to purchase the Ordinary Shares offered by this prospectus is subject to certain conditions. The underwriters are obligated to purchase all of the Ordinary Shares offered hereby if they purchase any of the Ordinary Shares.
Over-Allotment Option
We have granted the underwriters an option, exercisable for 45 days from the date of this prospectus, to purchase up to additional Ordinary Shares at the public offering price per share, less underwriting discounts and commissions, to cover over-allotments, if any, in connection with the Ordinary Shares offered by this prospectus.
Discounts, Commissions and Expenses
The underwriters propose to offer the Ordinary Shares purchased pursuant to the underwriting agreement to the public at the initial public offering price set forth on the cover page of this prospectus and to certain dealers at that price less a concession not in excess of $ per Ordinary Share. After this offering, the initial public offering price and concession may be changed by the underwriters. No such change shall change the amount of proceeds to be received by us as set forth on the cover page of this prospectus.
In connection with the sale of the Ordinary Shares to be purchased by the underwriters, the underwriters will be deemed to have received compensation in the form of underwriting commissions and discounts. The underwriters’ commissions and discounts will be 7.0% of the gross proceeds of this offering.
We have also agreed to reimburse the representatives at closing for their legal and other out of pocket expenses incurred by them in connection with the offering up to a maximum of $225,000. We will also pay to the representatives a non-accountable expense allowance equal to 1% of the total gross proceeds received from the offering, including any proceeds received from the exercise of the underwriters’ option to purchase additional Ordinary Shares.
The following table shows the underwriting discounts and commissions payable to the underwriters by us in connection with this offering (assuming both that the underwriters do not exercise and fully exercise their option to purchase additional Ordinary Shares).
| | | Per Ordinary Share | | | Total Without Over- allotment | | | Total With Over- allotment | |
| Initial public offering price | | $ | | | | $ | | | | $ | | |
| Underwriting discounts and commissions paid by us | | $ | | | | $ | | | | $ | | |
| Proceeds to us, before expenses | | $ | | | | $ | | | | $ | | |
The estimated offering expenses payable by us, exclusive of the underwriting discounts and commissions, are approximately $ . This figure includes the representatives’ non-accountable expense allowance equal to 1% of the aggregate gross proceeds received from the offering and the legal and other out of pocket expense reimbursement up to $225,000.
Representative Warrants
We will issue to the representatives, or their respective designees, warrants (the “Representative Warrants”) to purchase an amount of our Ordinary Shares equal to up to 5% of the Ordinary Shares sold in this offering (including any Ordinary Shares sold pursuant to the underwriters’ option to purchase additional shares). The Representative Warrants will have an exercise price equal to 120% of the price to the public of the Ordinary Shares in this offering, subject to standard anti-dilution adjustments for share splits and similar transactions, and will also contain a cashless exercise provision. The Representative Warrants will be immediately exercisable upon issuance, in whole or in part, and will expire on the fifth anniversary of the commencement of sales in this offering in accordance with FINRA Rule 5110(g)(8)(A).
Pursuant to FINRA Rule 5110(e), the Representative Warrants and any Ordinary Shares issued upon exercise of the Representative Warrants shall not be sold, transferred, assigned, pledged, or hypothecated, or be the subject of any hedging, short sale, derivative, put or call transaction that would result in the effective economic disposition of the securities by any person for a period of 180 days immediately following the date of commencement of sales of this offering, except: (i) the transfer of any security by operation of law or by reason of reorganization of the issuer; (ii) the transfer of any security to any FINRA member firm participating in the offering and the officers, partners, registered persons or affiliates thereof, if all securities so transferred remain subject to these lock-up restrictions for the remainder of the time period; (iii) if the aggregate amount of our securities held by the representatives or related persons does not exceed 1% of the securities being offered; (iv) the transfer of any security that is beneficially owned on a pro-rata basis by all equity owners of an investment fund, provided that no participating member manages or otherwise directs investments by the fund and the participating members in the aggregate do not own more than 10% of the equity in the fund; (v) the exercise or conversion of any security, if all securities remain subject to the lock-up restriction set forth above for the remainder of the time period; (vi) if we meet the registration requirements of Forms S-3, F-3 or F-10; or (vii) the transfer of any security back to us in a transaction exempt from registration with the SEC.
The Representative Warrants and the Ordinary Shares underlying the Representative Warrants have been registered in the registration statement of which this prospectus is a part. See the Form of Representative Warrant that will be filed as an exhibit as part of this registration statement for more information.
Tail Fee
We have also agreed to pay the representatives a tail fee equal to the cash and warrant compensation in this offering, subject to certain exceptions, with respect to any offering of our securities to the extent such financing or capital is provided to us or our affiliates by investors whom were introduced to us by the representatives during their engagement period for a period of twelve months following the earlier of the expiration or termination of our engagement with the representatives, provided this provision shall not have a duration of more than three years from the commencement of sales of this offering.
Financial Advisory Fee
On May 17, 2024, the Company and Enclave Capital LLC (“Enclave”) entered into a letter agreement (the “Letter Agreement”) pursuant to which Enclave was engaged as the Company’s financial advisor to provide financial advisory services to the Company, including, but not limited to, acting as a finder and/or arranging meetings with various prospective underwriters in connection with the offering.
Pursuant to the Letter Agreement, at the closing of the offering, the Company is required to pay Enclave a cash fee equal to $500,000.
Enclave is registered as a broker-dealer with the SEC. Enclave may be deemed to be an underwriter (as that term is defined in the Securities Act) in connection with the offering.
Indemnification
Pursuant to the underwriting agreement, we have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, or to contribute to payments that the underwriters or such other indemnified parties may be required to make in respect of those liabilities.
Discretionary Accounts
The underwriters do not intend to confirm sales of the securities offered hereby to any accounts over which they have discretionary authority.
Lock-Up Agreements
We and each of our directors, officers and shareholders that hold 5% or more of our outstanding Ordinary Shares have agreed not to offer, sell, agree to sell, directly or indirectly, or otherwise dispose of any Ordinary Shares or any securities convertible into or exchangeable for Ordinary Shares for a period of 180 days after the closing date of the offering pursuant to the underwriting agreement without the prior written consent of the representatives. These lock-up agreements provide for limited exceptions and their restrictions may be waived at any time by the representatives.
Nasdaq Listing
We have applied to have our Ordinary Shares listed on Nasdaq under the symbol “EFB”. We will not consummate this offering unless our Ordinary Shares are approved for listing on Nasdaq. There is no established public trading market for the Ordinary Shares and there is no assurance that a market will develop.
Electronic Distribution
This prospectus may be made available in electronic format on websites or through other online services maintained by the underwriters or by their affiliates. In those cases, prospective investors may view offering terms online and prospective investors may be allowed to place orders online. Other than this prospectus and the accompanying prospectus in electronic format, the information on the underwriters’ websites or our website and any information contained in any other websites maintained by the underwriters or by us is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or the underwriter in its capacity as underwriter, and should not be relied upon by investors.
Price Stabilization, Short Positions and Penalty Bids
In connection with the offering the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate covering transactions and penalty bids in accordance with Regulation M under the Exchange Act:
| | ● | Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum. |
| | | |
| | ● | Over-allotment involves sales by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase, which creates a syndicate short position. The short position may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriters is not greater than the number of shares that they may purchase in the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriters may close out any covered short position by either exercising their over-allotment option and/or purchasing shares in the open market. |
| | | |
| | ● | Syndicate covering transactions involve purchases of the Ordinary Shares in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the over-allotment option. A naked short position occurs if the underwriters sell more shares than could be covered by the over-allotment option. This position can only be closed out by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there could be downward pressure on the price of the shares in the open market after pricing that could adversely affect investors who purchase in the offering. |
| | ● | Penalty bids permit the underwriters to reclaim a selling concession from a syndicate member when the Ordinary Shares originally sold by the syndicate member is purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions. |
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of our Ordinary Shares or preventing or retarding a decline in the market price of the Ordinary Shares. As a result, the price of our Ordinary Shares may be higher than the price that might otherwise exist in the open market. These transactions may be discontinued at any time.
Neither we nor the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our Ordinary Shares. In addition, neither we nor the underwriters make any representation that the underwriter will engage in these transactions or that any transaction, if commenced, will not be discontinued without notice.
Determination of Offering Price
The initial public offering price was determined by negotiations between us and the representatives. Among the factors considered in determining the initial public offering price were our future prospects and those of our industry in general, our sales, earnings and certain other financial and operating information in recent periods, and the price-earnings ratios, price-sales ratios, market prices of securities and certain financial and operating information of companies engaged in activities similar to ours. We offer no assurances that the initial public offering price will correspond to the price at which the Ordinary Shares will trade in the public market subsequent to the offering or that an active trading market for the Ordinary Shares will develop and continue after the offering.
Other Relationships
The underwriters and their respective affiliates may, in the future provide various investment banking, commercial banking and other financial services for the Company and its affiliates for which they may in the future receive customary fees. However, except as disclosed in this prospectus, the Company has no present arrangements with the underwriters for any further services.
Offer Restrictions Outside the United States
Other than in the United States, no action has been taken by us or the underwriter that would permit a public offering of the securities offered by this prospectus in any jurisdiction where action for that purpose is required. The securities offered by this prospectus may not be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sale of any such securities be distributed or published in any jurisdiction, except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons into whose possession this prospectus comes are advised to inform themselves about and to observe any restrictions relating to this offering and the distribution of this prospectus. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities offered by this prospectus in any jurisdiction in which such an offer or a solicitation is unlawful.
EXPENSES RELATING TO THIS OFFERING
Set forth below is an itemization of the total expenses, excluding underwriting discounts, that we expect to incur in connection with this offering. With the exception of the SEC registration fee and the Nasdaq Capital Market listing fee, all amounts are estimates.
| | | USD | |
| SEC Registration Fee | | $ | | |
| FINRA Filing Fee | | $ | | |
| Nasdaq Capital Market Listing Fee | | $ | | |
| Independent Financial Advisor Fees and Expenses | | $ | | |
| Legal Fees and Expenses | | $ | | |
| Accounting Fees and Expenses | | $ | | |
| Printing and Engraving Expenses | | $ | | |
| Transfer Agent Fees and Expenses | | $ | | |
| Miscellaneous Expenses | | $ | | |
| Total Expenses | | $ | | |
We will bear all these expenses.
LEGAL MATTERS
The validity of the Ordinary Shares offered in this offering and certain other legal matters as to BVI law will be passed upon for us by McW Todman & Co., our counsel as to BVI law. The legal matters as to United States Federal and New York State law will be passed upon for us by Seward & Kissel LLP, New York, New York. The underwriters are being represented by Ellenoff Grossman & Schole LLP, New York, New York, with respect to legal matters of United States federal and New York State law. Legal matters as to Taiwan laws will be passed upon for us by Formosan Brothers.
EXPERTS
The consolidated financial statements for the years ended December 31, 2022 and 2023 included in this prospectus have been included in reliance on the report of KCCW Accountancy Corp, an independent registered public accounting firm, given on the authority of said firm as experts in auditing and accounting. The office of KCCW Accountancy Corp. is located at 3333 S. Brea Canyon Road, Suite 206, Diamond Bar, CA 91765.
INTERESTS OF NAMED EXPERTS AND COUNSEL
No expert or counsel named in this prospectus as having prepared or certified any part of this prospectus or having given an opinion upon the validity of the securities being registered or upon other legal matters in connection with the registration or offering of the Ordinary Shares was employed on a contingency basis, or had, or is to receive, in connection with the offering, a substantial interest, direct or indirect, in the registrant. Nor was any such person connected with the registrant as a promoter, managing or principal Underwriter, voting trustee, director, officer, or employee.
DISCLOSURE OF COMMISSION POSITION ON INDEMNIFICATION
Insofar as indemnification for liabilities arising under the Securities Act, may be permitted to our directors, officers or persons controlling us, we have been advised that it is the SEC’s opinion that such indemnification is against public policy as expressed in such act and is, therefore, unenforceable.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the SEC a registration statement on Form F-1, including relevant exhibits and schedules under the Securities Act, covering the Ordinary Shares offered by this prospectus. You should refer to our registration statement and its exhibits and schedules if you would like to find out more about us and the Ordinary Shares. This prospectus summarizes material provisions of contracts and other documents, and we refer you to the exhibits to our registration statement where these documents are available in full. Since the prospectus may not contain all the information that you may find important, you should review the full text of these documents.
Immediately upon the completion of this offering, we will be subject to periodic reporting and other informational requirements of the Exchange Act, as applicable to foreign private issuers. Accordingly, we will be required to file reports, including annual reports on Form 20-F, and other information with the SEC. As a foreign private issuer, we are exempt from the rules of the Exchange Act prescribing the furnishing and content of proxy statements to shareholders under the federal proxy rules contained in Sections 14(a), (b) and (c) of the Exchange Act, and our executive officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act.
The registration statements, reports and other information so filed can be inspected and copied at the public reference facilities maintained by the SEC at 100 F Street, N.E., Washington, D.C. 20549. You can request copies of these documents upon payment of a duplicating fee, by writing to the SEC. Please call the SEC at 1-800-SEC-0330 for further information on the operation of the public reference rooms. The SEC also maintains a website that contains reports, proxy statements and other information about issuers, such as us, who file electronically with the SEC. The address of that website is http://www.sec.gov. The information on that website is not a part of this prospectus.
No dealers, salesperson or other person is authorized to give any information or to represent anything not contained in this prospectus. You must not rely on any unauthorized information or representations. This prospectus is an offer to sell only the securities offered hereby, but only under circumstances and in jurisdictions where it is lawful to do so. The information contained in this prospectus is current only as of its date.
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
CONSOLIDATED BALANCE SHEETS
(UNAUDITED)
| | | June 30, 2024 | | | December 31, 2023 | |
| Assets | | | | | | | | |
| Current assets | | | | | | | | |
| Cash and cash equivalents | | $ | 432,932 | | | $ | 1,220,559 | |
| Other receivables | | | - | | | | 4,898 | |
| Due from related party | | | - | | | | 7,984 | |
| Prepaid taxes | | | 433,819 | | | | 456,005 | |
| Prepaid expenses and other current assets | | | 1,224 | | | | 871 | |
| Total current assets | | | 867,975 | | | | 1,690,317 | |
| Long-term investment, net | | | - | | | | - | |
| Property and equipment, net | | | 23,902 | | | | 32,149 | |
| Intangible assets, net | | | 292,615 | | | | 324,997 | |
| Operating lease right-of-use assets | | | 12,411 | | | | 16,098 | |
| Other assets | | | 1,676 | | | | 1,444 | |
| Total assets | | $ | 1,198,579 | | | $ | 2,065,005 | |
| | | | | | | | | |
| Liabilities and deficit | | | | | | | | |
| Current liabilities | | | | | | | | |
| Accrued expenses | | | 119,517 | | | | 157,980 | |
| Convertible notes payable | | | 2,516,000 | | | | 2,516,000 | |
| Operating lease liabilities – current portion | | | 6,631 | | | | 4,805 | |
| Other current liabilities | | | 3,603 | | | | 673 | |
| Total current liabilities | | | 2,645,751 | | | | 2,679,458 | |
| Operating lease liabilities – noncurrent portion | | | 6,728 | | | | 10,164 | |
| Total Liabilities | | | 2,652,479 | | | | 2,689,622 | |
| | | | | | | | | |
| Commitments and Contingencies | | | | | | | | |
| | | | | | | | | |
| Deficit | | | | | | | | |
| Ordinary shares, $0.01 par value, 100,000,000 authorized, 52,237,745 shares issued and outstanding as of both June 30, 2024 and December 31, 2023 | | | 522,377 | | | | 522,377 | |
| Additional paid-in capital | | | 19,093,280 | | | | 19,093,280 | |
| Accumulated deficit | | | (20,789,569 | ) | | | (20,002,006 | ) |
| Accumulated other comprehensive loss | | | (279,860 | ) | | | (238,289 | ) |
| Total Stockholders’ deficit | | | (1,453,772 | ) | | | (624,638 | ) |
| Noncontrolling interest | | | (128 | ) | | | 21 | |
| Total deficit | | | (1,453,900 | ) | | | (624,617 | ) |
| | | | | | | | | |
| Total liabilities and deficit | | $ | 1,198,579 | | | $ | 2,065,005 | |
The accompanying notes are an integral part of these consolidated financial statements.
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(UNAUDITED)
| | | Six Months Ended June 30, | |
| | | 2024 | | | 2023 | |
| Revenue | | $ | - | | | $ | - | |
| Operating costs | | | - | | | | - | |
| Gross profit | | | - | | | | - | |
| Operating expenses | | | | | | | | |
| General and administrative expenses | | | 478,131 | | | | 594,782 | |
| Research and development expenses | | | 314,542 | | | | 389,817 | |
| Total operating expenses | | | 792,673 | | | | 984,599 | |
| Loss from operations | | | (792,673 | ) | | | (984,599 | ) |
| Non-operating income (expense) | | | | | | | | |
| Interest income (expense), net | | | 4,961 | | | | 1,526 | |
| Other income | | | - | | | | 35,056 | |
| Total non-operating income, net | | | 4,961 | | | | 36,582 | |
| Loss before income taxes | | | (787,712 | ) | | | (948,017 | ) |
| Income tax expense | | | - | | | | - | |
| Net loss | | | (787,712 | ) | | | (948,017 | ) |
| Net loss attributable to noncontrolling interests | | | (149 | ) | | | (166 | ) |
| Net loss attributed to Everfront Biotech Holding Company Limited | | | (787,563 | ) | | | (947,851 | ) |
| Foreign currency translation loss | | | (41,571 | ) | | | (18,140 | ) |
| Comprehensive loss | | $ | (829,134 | ) | | $ | (965,991 | ) |
| | | | | | | | | |
| Net loss per share: | | | | | | | | |
| Basic and diluted | | $ | (0.02 | ) | | $ | (0.02 | ) |
| | | | | | | | | |
| Weighted average number of common shares outstanding: | | | | | | | | |
| Basic and diluted | | | 52,237,745 | | | | 51,795,624 | |
The accompanying notes are an integral part of these consolidated financial statements.
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
CONSOLIDATED STATEMENT OF CASH FLOWS
(UNAUDITED)
| | | Six Months Ended June 30, | |
| | | 2024 | | | 2023 | |
| Cash Flows from Operating Activities | | | | | | | | |
| Net loss | | $ | (787,712 | ) | | $ | (948,017 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | |
| Depreciation | | | 4,766 | | | | 4,287 | |
| Amortization | | | 17,452 | | | | 16,504 | |
| Operating lease expense | | | 3,040 | | | | 2,922 | |
| Net changes in operating assets and liabilities: | | | | | | | | |
| Decrease (increase) in other receivables | | | 4,702 | | | | (219,640 | ) |
| Decrease in due from related party | | | 7,664 | | | | - | |
| Increase in prepaid expenses | | | (348 | ) | | | (668 | ) |
| Increase in prepaid taxes | | | (3,668 | ) | | | (7,622 | ) |
| Decrease (increase) in other current assets | | | 17 | | | | (5,905 | ) |
| Decrease in accrued expenses | | | (33,521 | ) | | | (47,714 | ) |
| Increase in other payable | | | 511 | | | | 11,770 | |
| Increase in other current liabilities | | | 3,019 | | | | 872 | |
| Payment for operating lease liabilities | | | (992 | ) | | | (2,922 | ) |
| Net cash used in operating activities | | | (785,070 | ) | | | (1,196,133 | ) |
| | | | | | | | | |
| Cash Flows from Investing Activities | | | | | | | | |
| Purchase of property and equipment | | | - | | | | (32,191 | ) |
| (Increase) decrease in other assets | | | (320 | ) | | | 491 | |
| Net cash used in investing activities | | | (320 | ) | | | (31,700 | ) |
| | | | | | | | | |
| Cash Flows from Financing Activities | | | | | | | | |
| Proceeds from convertible notes payable | | | - | | | | 400,000 | |
| Capital contribution | | | - | | | | 493,120 | |
| Net cash provided by financing activities | | | - | | | | 893,120 | |
| | | | | | | | | |
| Effect of exchange rate changes on cash and cash equivalents | | | (2,237 | ) | | | (4,251 | ) |
| | | | | | | | | |
| Net decrease in cash and cash equivalents | | | (787,627 | ) | | | (338,964 | ) |
| | | | | | | | | |
| Cash and Cash Equivalents | | | | | | | | |
| Beginning of period | | | 1,220,559 | | | | 548,138 | |
| Ending of period | | $ | 432,932 | | | $ | 209,174 | |
| | | | | | | | | |
| Supplemental Disclosure of Cash Flows | | | | | | | | |
| Cash paid during the period for: | | | | | | | | |
| Interest | | $ | - | | | $ | - | |
| Income taxes | | $ | - | | | $ | - | |
The accompanying notes are an integral part of these consolidated financial statements.
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
CONSOLIDATED STATEMENTS OF DEFICIT
(UNAUDITED)
| | | Ordinary shares* | | | Subscription | | | Additional | | | | | | Accumulated other | | | | | | | |
| | | Number of
shares | | | Amount | | | received in
advance | | | paid-in
capital | | | Accumulated
Deficit | | | comprehensive loss | | | Noncontrolling interest | | | Total | |
| Balance at December 31, 2022 | | | 51,445,450 | | | $ | 514,455 | | | $ | 1,089,126 | | | $ | 17,512,266 | | | $ | (17,709,109 | ) | | $ | (229,502 | ) | | $ | 321 | | | $ | 1,177,557 | |
| Issuance of ordinary shares for cash | | | 792,295 | | | | 7,922 | | | | (1,089,126 | ) | | | 1,581,348 | | | | - | | | | - | | | | - | | | | 500,144 | |
| Net loss | | | - | | | | - | | | | - | | | | - | | | | (947,851 | ) | | | - | | | | (166 | ) | | | (948,017 | ) |
| Foreign currency translation | | | - | | | | - | | | | - | | | | - | | | | - | | | | (18,140 | ) | | | - | | | | (18,140 | ) |
| Balance at June 30, 2023 | | | 52,237,745 | | | $ | 522,377 | | | $ | - | | | $ | 19,093,614 | | | $ | (18,656,960 | ) | | $ | (247,642 | ) | | $ | 155 | | | $ | 711,544 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2023 | | | 52,237,745 | | | $ | 522,377 | | | $ | - | | | $ | 19,093,280 | | | $ | (20,002,006 | ) | | $ | (238,289 | ) | | $ | 21 | | | $ | (624,617 | ) |
| Net loss | | | - | | | | - | | | | - | | | | - | | | | (787,563 | ) | | | - | | | | (149 | ) | | | (787,712 | ) |
| Foreign currency translation | | | - | | | | - | | | | - | | | | - | | | | - | | | | (41,571 | ) | | | - | | | | (41,571 | ) |
| Balance at June 30, 2024 | | | 52,237,745 | | | $ | 522,377 | | | $ | - | | | $ | 19,093,280 | | | $ | (20,789,569 | ) | | $ | (279,860 | ) | | $ | (128 | ) | | $ | (1,453,900 | ) |
* The shares amounts are presented on a retroactive basis.
The accompanying notes are an integral part of these consolidated financial statements.
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
1. ORGANIZATION AND DESCRIPTION OF BUSINESS
Everfront Biotech Holding Company Limited (“Everfront BVI”, or the “Company”), which was incorporated on September 21, 2022 in the British Virgin Islands, is a holding company and has not carried out substantive business operations of its own.
Everfront Biotech Inc. (“Everfront Biotech”, or the “Subsidiary”) was incorporated under the laws of Taiwan on December 8, 2010. Everfront Biotech has been primarily engaged in new drug discovery and development since 2010.
Everfront Biotech has established its research and development (“R&D”) team since 2010, and is currently developing small molecules that are intended to treat patients with cancers, neurodegenerative diseases and rare diseases. The R&D team is specialized in preclinical study design; chemistry, manufacturing, and control (“CMC”); investigational new drug (“IND”) submission; and clinical trial design and conduction.
As of the date hereof, three clinical trials are being performed in Taiwan under the approval of the United States Food and Drug Administration (“USFDA”) and Taiwan Food and Drug Administration (“TFDA”). Additionally, the Company has four product candidates currently under IND-Enabling status.
Reorganization
From January 2023 to February 2023, Everfront BVI completed its restructuring (“Restructuring Transaction”) with the Subsidiary; pursuant to which Everfront BVI acquired 51,395,450 shares of common stock of Everfront Biotech from eighty-seven shareholders of Everfront Biotech (the “Subsidiary Shareholders”), representing 99.96% of the issued and outstanding common stock of the Subsidiary, and issued 51,395,450 ordinary shares of Everfront BVI to the Subsidiary Shareholders (or their designees).
As a result of the Restructuring Transaction, Everfront BVI directly owns 99.96% of the ownership of Everfront Biotech, and the Subsidiary Shareholders (or their designees) collectively received an aggregate of 51,395,450 ordinary shares of the Company, representing approximately 98.39% of the then issued and outstanding shares of the Company’s ordinary shares.
Reverse acquisition
For accounting purposes, the acquisition was accounted for as a reverse acquisition and treated as a recapitalization of the Company effected by a share exchange, with Everfront Biotech as the accounting acquirer. Consequently, the historical financial statements of Everfront Biotech are now the historical consolidated financial statements of the Company. The assets and liabilities of Everfront Biotech were brought forward at their book value and no goodwill was recognized.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with the generally accepted accounting principles in the United States of America (the “U.S. GAAP”).
Principle of consolidation
The consolidated financial statements include the financial statements of Everfront Biotech Holding Company Limited and its 99.96% owned subsidiary, Everfront Biotech Inc. All inter-company transactions and balances are eliminated in consolidation.
Going Concern
The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The realization of assets and the satisfaction of liabilities in the normal course of business are dependent on, among other things, the Company’s ability to operate profitably, to generate cash flows from operations, and to pursue financing arrangements to support its working capital requirements.
The Company has suffered net losses of $787,712 and $948,017 for the six months ended June 30, 2024 and 2023, respectively, and used net cash in operating activities of $785,070 and $1,196,133 for the six months ended June 30, 2024 and 2023, respectively, and has an accumulated deficit of $20,789,569 as of June 30, 2024. These facts and conditions raise substantial doubt about the Company’s ability to continue as a going concern.
Management believes that it has developed a liquidity plan, summarized below, that, if executed successfully, should provide sufficient liquidity to meet the Company’s obligations as they become due for a reasonable period of time, and allow the development of its core business. The plan includes:
● Raising cash through potential equity offerings.
While the Company’s management believes that the measures in its liquidity plan including those described above will be adequate to satisfy its liquidity requirements for the twelve months after the date that these financial statements are issued, there is no assurance that the liquidity plan will be successfully implemented. Failure to successfully implement the liquidity plan may have a material adverse effect on its business, results of operations and financial position, and may adversely affect its ability to continue as a going concern. These consolidated financial statements do not include any adjustments related to the recoverability and classification of recorded assets or the amounts and classification of liabilities or any other adjustments that might be necessary should the Company be unable to continue as a going concern.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the amount of revenues and expenses during the reporting periods. Actual results could differ materially from those results.
Fair Value Measurements
FASB ASC 820, “Fair Value Measurements” defines fair value for certain financial and nonfinancial assets and liabilities that are recorded at fair value, establishes a framework for measuring fair value and expands disclosures about fair value measurements. It requires that an entity measure its financial instruments to base fair value on exit price, maximize the use of observable units and minimize the use of unobservable inputs to determine the exit price. It establishes a hierarchy which prioritizes the inputs to valuation techniques used to measure fair value. This hierarchy increases the consistency and comparability of fair value measurements and related disclosures by maximizing the use of observable inputs and minimizing the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that reflect the assumptions market participants would use in pricing the assets or liabilities based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s own assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. The hierarchy prioritizes the inputs into three broad levels based on the reliability of the inputs as follows:
| | ● | Level 1 - Inputs are quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date. Valuation of these instruments does not require a high degree of judgment as the valuations are based on quoted prices in active markets that are readily and regularly available. |
| | ● | Level 2 - Inputs other than quoted prices in active markets that are either directly or indirectly observable as of the measurement date, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| | ● | Level 3 - Valuations based on inputs that are unobservable and not corroborated by market data. The fair value for such assets and liabilities is generally determined using pricing models, discounted cash flow methodologies, or similar techniques that incorporate the assumptions a market participant would use in pricing the asset or liability. |
The carrying values of certain assets and liabilities of the Company, such as cash and cash equivalents, prepaid expenses and other current assets, accounts payable, accrued liabilities, and due to related parties approximate fair value due to their relatively short maturities.
Cash and Cash Equivalents
The Company considers highly liquid investments with maturities of three months or less, when purchased, to be cash equivalents. As of June 30, 2024 and December 31, 2023, the Company’s cash and cash equivalents amounted to $432,932 and $1,220,559, respectively. Some of the Company’s cash deposits are held in financial institutions located in Taiwan where there is currently regulation mandated on obligatory insurance of bank accounts. The Company believes these financial institutions are of high credit quality.
Concentration of Credit Risk
The Company’s financial instruments that are exposed to concentrations of credit risk consist primarily of cash and cash equivalents. The Company places its cash and temporary cash investments in high quality credit institutions, but these investments may be in excess of Taiwan Central Deposit Insurance Corporation and the U.S. Federal Deposit Insurance Corporation’s insurance limits. The Company does not enter into financial instruments for hedging, trading or speculative purposes. Accounts are guaranteed by the Federal Deposit Insurance Corporation (FDIC) up to $250,000. As of June 30, 2024 and December 31, 2023, the Company had approximately nil and $966,305 in excess of FDIC insured limits, respectively. The Company maintains cash in state-owned banks in Taiwan. In Taiwan, the insurance coverage of each bank is NTD$3,000,000 (approximately USD$92,450) by the Taiwan Central Deposit Insurance Corporation (TCDIC). As of June 30, 2024 and December 31, 2023, the Company had approximately $325,229 and nil in excess of TCDIC insured limits, respectively. The Company has not experienced any losses in such accounts.
Revenue Recognition
The Company adopted Accounting Standards Codification (“ASC”), Topic 606 (ASC 606), Revenue from Contracts with Customers. Pursuant to ASC 606, the Company recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the consideration that the Company expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that the Company determines is within the scope of ASC 606, the Company performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the Company satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the Company will collect the consideration the Company is entitled to in exchange for the goods or services the Company transfers to the customers. At inception of the contract, once the contract is determined to be within the scope of ASC 606, the Company assesses the goods or services promised within each contract, determines those that are performance obligations, and assesses whether each promised good or service is distinct. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied.
During the six months ended June 30, 2024 and 2023, the Company had no revenue.
Property and Equipment
Property and equipment are carried at cost net of accumulated depreciation. Repairs and maintenance are expensed as incurred. Expenditures that improve the functionality of the related asset or extend the useful life are capitalized. When property and equipment is retired or otherwise disposed of, the cost and associated accumulated depreciation are removed from the consolidated balance sheet, the related gain or loss is reflected on the consolidated statement of operations. Leasehold improvements are depreciated on the straight-line method over the shorter of the remaining lease term or estimated useful life of the asset. Depreciation is calculated on the straight-line method, including property and equipment under capital leases, generally based on the following useful lives:
| | | Estimated Life
in Years |
| Machinery and equipment | | 3 ~ 6 |
| Office equipment | | 3 ~ 4 |
| Leasehold improvements | | 2 ~ 3 |
Intangible Assets
Intangible assets consist of patents and acquired software. Intangible assets are initially recognized at their respective acquisition costs. All of the Company’s intangible assets have been determined to have finite useful lives and are, therefore, amortized using the straight-line method over their estimated useful lives:
| Patents and trademarks | | 15 to 30 | | | years |
| Acquired software | | | 3 | | | years |
Impairment of Long-Lived Assets
The Company has adopted Accounting Standards Codification subtopic 360-10, Property, Plant and Equipment (“ASC 360-10”). ASC 360-10 requires that long-lived assets and certain identifiable intangibles held and used by the Company be reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The Company evaluates its long-lived assets for impairment annually or more frequently if events or changes in circumstances indicate that it might be impaired. Events relating to recoverability may include significant unfavorable changes in business conditions, recurring losses, or a forecasted inability to achieve break-even operating results over an extended period. Should impairment in value be indicated, the carrying value of intangible assets will be adjusted, based on estimates of future discounted cash flows resulting from the use and ultimate disposition of the asset. ASC 360-10 also requires assets to be disposed of be reported at the lower of the carrying amount or the fair value less costs to sell.
Lease
The Company adopted FASB Accounting Standards Codification, Topic 842, Leases (“ASC 842”). The Company applied the following practical expedients allowed under ASC 842:
| | ● | Reassessment of expired or existing contracts: The Company elected not to reassess, at the application date, whether any expired or existing contracts contained leases, the lease classification for any expired or existing leases, and the accounting for initial direct costs for any existing leases. |
| | ● | Use of hindsight: The Company elected to use hindsight in determining the lease term (that is, when considering options to extend or terminate the lease and to purchase the underlying asset) and in assessing impairment of right-to-use assets. |
| | ● | Reassessment of existing or expired land easements: The Company elected not to evaluate existing or expired land easements that were not previously accounted for as leases under ASC 840, as allowed under the transition practical expedient. Going forward, new or modified land easements will be evaluated under ASU No. 2016-02. |
| | ● | Separation of lease and non- lease components: Lease agreements that contain both lease and non-lease components are generally accounted for separately. |
| | ● | Short-term lease recognition exemption: The Company also elected the short-term lease recognition exemption and will not recognize ROU assets or lease liabilities for leases with a term less than 12 months. |
The new leasing standard requires recognition of leases on the consolidated balance sheets as right-of-use (“ROU”) assets and lease liabilities. ROU assets represent the Company’s right to use underlying assets for the lease terms and lease liabilities represent the Company’s obligation to make lease payments arising from the leases. Operating lease ROU assets and operating lease liabilities are recognized based on the present value and future minimum lease payments over the lease term at commencement date. The Company’s future minimum based payments used to determine the Company’s lease liabilities mainly include minimum based rent payments. As most of Company’s leases do not provide an implicit rate, the Company uses its estimated incremental borrowing rate based on the information available at commencement date in determining the present value of lease payments.
The Company recognized lease liabilities, with corresponding ROU assets, based on the present value of unpaid lease payments for existing operating leases longer than twelve months. The ROU assets were adjusted per ASC 842 transition guidance for existing lease-related balances of accrued and prepaid rent, unamortized lease incentives provided by lessors, and restructuring liabilities. Operating lease cost is recognized as a single lease cost on a straight-line basis over the lease term and is recorded in selling, general and administrative expenses. Variable lease payments for common area maintenance, property taxes and other operating expenses are recognized as expense in the period when the changes in facts and circumstances on which the variable lease payments are based occur.
Long-term Equity Investment
The Company accounts for non-marketable equity and other equity investments for which the Company does not have control over the investees as:
| | ● | Equity method investments when the Company has the ability to exercise significant influence, but not control, over the investee. Its proportionate share of the income or loss is recognized monthly and is recorded in gains (losses) on equity investments. |
| | ● | Investments in entities that are not consolidated or accounted for under the equity method are recorded as investments without readily determinable fair values. Investments without readily determinable fair values are reported on the consolidated balance sheets in investments, at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for an identical or similar investment of the same issuer. |
Prior to the adoption of ASU 2016-01, equity investments without readily determinable fair values were recorded at cost minus other-than-temporary impairment, with other-than-temporary impairment charges included in other income (expense), net in the Consolidated Statements of Operations. Following the adoption of ASU 2016-01, these investments are measured at cost adjusted for changes in observable prices minus impairment or at net asset value, as a practical expedient, if available, with changes in measurement recognized in other income (expense), net in the Consolidated Statements of Operations. The Company’s equity investments without readily determinable fair values are included in Investment on the Consolidated Balance Sheets.
Research and Development Expenses
The Company accounts for the cost of using licensing rights in research and development cost according to ASC Topic 730-10-25-1. Research and development expenses are charged to expense as incurred. Research and development expenses are comprised of costs incurred in performing research and development activities, including payroll, personnel-related costs, facilities-related overhead, and outside contracted services including clinical trial costs, manufacturing and process development costs for both clinical and preclinical materials, research costs, and other consulting services. Non-refundable advance payment for goods and services that will be used in future research and development activities are expensed when the activity has been performed or when the goods have been received rather than when the payment is made. In instances where the Company enters into agreements with third parties to provide research and development services, costs are expensed as services are performed.
Post-retirement and post-employment benefits
The Company’s subsidiary in Taiwan adopted the government mandated defined contribution plan pursuant to the Labor Pension Act (the “Act”) in Taiwan. Such labor regulations require that the rate of contribution made by an employer to the Labor Pension Fund per month shall not be less than 6% of the worker’s monthly salaries. Pursuant to the Act, the Company makes monthly contribution equal to 6% of employees’ salaries to the employees’ pension fund. The Company has no legal obligation for the benefits beyond the contributions made. The total amounts for such employee benefits, which were expensed as incurred, were $8,438 and $8,255 for the six months ended June 30, 2024 and 2023, respectively. Other than the above, the Company does not provide any other post-retirement or post-employment benefits.
Income Taxes
The Company accounts for income taxes using the asset and liability approach which allows the recognition and measurement of deferred tax assets to be based upon the likelihood of realization of tax benefits in future years. Under the asset and liability approach, deferred taxes are provided for the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. A valuation allowance is provided for deferred tax assets if it is more likely than not these items will expire before the Company is able to realize their benefits, or future deductibility is uncertain.
Under ASC 740, a tax position is recognized as a benefit only if it is “more likely than not” that the tax position would be sustained in a tax examination, with a tax examination being presumed to occur. The evaluation of a tax position is a two-step process. The first step is to determine whether it is more-likely-than-not that a tax position will be sustained upon examination, including the resolution of any related appeals or litigations based on the technical merits of that position. The second step is to measure a tax position that meets the more-likely-than-not threshold to determine the amount of benefits recognized in the financial statements. A tax position is measured at the largest amount of benefit that is greater than 50 percent likely of being realized upon ultimate settlement. Tax positions that previously failed to meet the more-likely-than-not recognition threshold should be recognized in the first subsequent period in which the threshold is met. Previously recognized tax positions that no longer meet the more-likely-than-not criteria should be de-recognized in the first subsequent financial reporting period in which the threshold is no longer satisfied. Penalties and interest incurred related to underpayment of income tax are classified as income tax expense in the year incurred. No significant penalty or interest relating to income taxes has been incurred for the six months ended June 30, 2024 and 2023.
Valuation of Deferred Tax Assets
A valuation allowance is recorded to reduce the Company’s deferred tax assets to the amount that is more likely than not to be realized. In assessing the need for the valuation allowance, management considers, among other things, projections of future taxable income and ongoing prudent and feasible tax planning strategies. If the Company determines that sufficient negative evidence exists, then it will consider recording a valuation allowance against a portion or all of the deferred tax assets in that jurisdiction. If, after recording a valuation allowance, the Company’s projections of future taxable income and other positive evidence considered in evaluating the need for a valuation allowance prove, with the benefit of hindsight, to be inaccurate, it could prove to be more difficult to support the realization of its deferred tax assets. As a result, an additional valuation allowance could be required, which would have an adverse impact on its effective income tax rate and results. Conversely, if, after recording a valuation allowance, the Company determines that sufficient positive evidence exists in the jurisdiction in which the valuation allowance was recorded, it may reverse a portion or all of the valuation allowance in that jurisdiction. In such situations, the adjustment made to the deferred tax asset would have a favorable impact on its effective income tax rate and results in the period such determination was made.
Loss Per Share of Common Stock
The Company calculates net loss per share in accordance with ASC Topic 260, “Earnings per Share”. Basic loss per share is computed by dividing the net loss by the weighted average number of common shares outstanding during the period. Diluted loss per share is computed similar to basic loss per share except that the denominator is increased to include the number of additional common shares that would have been outstanding if the potential common stock equivalents had been issued and if the additional common shares were dilutive. Diluted earnings per share excludes all dilutive potential shares if their effect is anti-dilutive.
Commitments and Contingencies
The Company has adopted ASC Topic 450 “Contingencies” subtopic 20, in determining its accruals and disclosures with respect to loss contingencies. Accordingly, estimated losses from loss contingencies are accrued by a charge to income when information available before financial statements are issued or are available to be issued indicates that it is probable that an asset had been impaired or a liability had been incurred at the date of the financial statements and the amount of the loss can be reasonably estimated. Legal expenses associated with the contingency are expensed as incurred. If a loss contingency is not probable or reasonably estimable, disclosure of the loss contingency is made in the financial statements when it is at least reasonably possible that a material loss could be incurred.
Foreign-currency Transactions
For the Company’s subsidiary in Taiwan, the foreign-currency transactions are recorded in New Taiwan dollars (“NTD”) at the rates of exchange in effect when the transactions occur. Gains or losses resulting from the application of different foreign exchange rates when cash in foreign currency is converted into New Taiwan dollars, or when foreign-currency receivables or payables are settled, are credited or charged to income in the year of conversion or settlement. On the balance sheet dates, the balances of foreign-currency assets and liabilities are restated at the prevailing exchange rates and the resulting differences are charged to current income except for those foreign currencies denominated investments in shares of stock where such differences are accounted for as translation adjustments under the Statements of Stockholders’ Equity (Deficit).
Translation Adjustment
The accounts of the Company’s subsidiary in Taiwan were maintained, and their financial statements were expressed, in New Taiwan Dollar (“NT$”). Such financial statements were translated into U.S. Dollars (“$” or “USD”) in accordance ASC 830, “Foreign Currency Matters”, with the NT$ as the functional currency. According to the Statement, all assets and liabilities are translated at the current exchange rate, stockholder’s deficit are translated at the historical rates and income statement items are translated at an average exchange rate for the period. The resulting translation adjustments are reported under other comprehensive income (loss) as a component of stockholders’ equity (deficit).
Recent Accounting Pronouncements
In June 2016, the Financial Accounting Standards Board (the “FASB”) issued Accounting Standards Update (the “ASU”) 2016-13 (the “ASU 2016-13”), Financial Instruments—Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. The new standard changes the accounting for credit losses for financial assets and certain other instruments, including trade receivables and contract assets that are not measured at fair value through net income. Under legacy standards, the Company recognizes an impairment of receivables when it was probable that a loss had been incurred. Under the new standard pursuant to ASU 2016-13, the Company is required to recognize estimated credit losses expected to occur over the estimated life or remaining contractual life of an asset (which includes losses that may be incurred in future periods) using a broader range of information including reasonable and supportable forecasts about future economic conditions. The guidance is effective for smaller reporting companies (the “SRC”) as defined by the SEC for fiscal years beginning after December 15, 2022, including interim periods within those fiscal years with early adoption permitted. On January 1, 2023, the Company adopted this new guidance, and the adoption did not have a material impact on the Company’s financial statements and related disclosure.
In August 2020, the FASB issued Accounting Standards Update 2020-06 (the “ASU 2020-06”), Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity, which simplifies accounting for convertible instruments by removing major separation models required under current GAAP. ASU 2020-06 removes certain settlement conditions that are required for equity contracts to qualify for the derivative scope exception, and it also simplifies the diluted earnings per share calculation in certain areas. ASU 2020-06 is effective for SRC’s fiscal years beginning after December 15, 2023, including interim periods within those fiscal years, with early adoption permitted. The Company early adopted ASU 2020-06 on January 1, 2023.
In November 2023, the FASB issued ASU No. 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures (“ASU 2023-07”). The amendments in ASU 2023-07 improve reportable segment disclosure requirements through enhanced disclosures about significant segment expenses that are regularly provided to the chief operating decision maker (CODM). In addition, the amendments enhance interim disclosure requirements, clarify circumstances in which an entity can disclose multiple segment measures of profit or loss, provide new segment disclosure requirements for entities with a single reportable segment, and contain other disclosure requirements. ASU 2023-07 will be effective for annual reporting periods beginning after December 15, 2023, and interim periods within annual reporting periods beginning after December 15, 2024. Early adoption is permitted. The Company is currently evaluating the impact of this standard on its consolidated financial statements.
In December 2023, the FASB issued ASU No. 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures (“ASU 2023-09”), which requires disclosures of incremental income tax information within the rate reconciliation and expanded disclosures of income taxes paid, among other disclosure requirements. This ASU will be effective for annual reporting periods beginning after December 15, 2024. Early adoption is permitted. ASU 2023-09 will be applied on a prospective basis with the option to apply the standard retrospectively. The Company’s management does not believe that the adoption of ASU 2023-09 will have a material impact on the Company’s consolidated financial statement presentation or disclosures.
3. PROPERTY AND EQUIPMENT
Property and equipment as of June 30, 2024 and December 31, 2023 are summarized as follows:
| | | June 30, | | | December 31, | |
| | | 2024 | | | 2023 | |
| Machinery and equipment | | $ | 135,259 | | | $ | 146,080 | |
| Office equipment | | | 27,621 | | | | 27,841 | |
| Leasehold improvements | | | 30,828 | | | | 31,916 | |
| Total | | | 193,708 | | | | 205,837 | |
| Less: accumulated depreciation | | | (169,806 | ) | | | (173,688 | ) |
| Property and equipment, net | | $ | 23,902 | | | $ | 32,149 | |
Depreciation expenses were $4,766 and $4,287 for the six months ended June 30, 2024 and 2023, respectively.
4. LEASE
The Company has no finance leases. The Company’s leases primarily include various office and laboratory spaces, copy machine, and vehicles under various operating lease arrangements. The Company’s operating leases have remaining lease terms of up to approximately five years.
| | | June 30,
2024 | | | December 31, 2023 | |
| ASSETS | | | | | | |
| Operating lease right-of-use assets | | $ | 12,411 | | | $ | 16,098 | |
| | | | | | | | | |
| LIABILITIES | | | | | | | | |
| Operating lease liabilities - current | | | 6,631 | | | | 4,805 | |
| Operating lease liabilities - noncurrent | | | 6,728 | | | | 10,164 | |
Supplemental Information
The following provides details of the Company’s lease expenses:
| | | Six Months Ended
June 30, | |
| | | 2024 | | | 2023 | |
| Operating lease expenses | | $ | 3,040 | | | $ | 2,922 | |
Other information related to leases is presented below:
| | | Six Months Ended
June30, | |
| | | 2024 | | | 2023 | |
| Cash paid for amounts included in the measurement of operating lease liabilities | | | | | | |
| Operating cash flows from operating leases | | $ | 992 | | | $ | 2,922 | |
| Weighted Average Remaining Lease Term: | | | | | | | | |
| Operating leases | | | 2.68 years | | | | 4.44 years | |
| | | | | | | | | |
| Weighted Average Discount Rate: | | | | | | | | |
| Operating leases | | | 2.98 | % | | | 2.37 | % |
The minimum future annual payments under non-cancellable leases during the next five years and thereafter, at rates now in force, are as follows:
| For the years ended June 30, | | Operating leases | |
| 2025 | | $ | 6,925 | |
| 2026 | | | 5,686 | |
| 2027 | | | 1,173 | |
| Total future minimum lease payments, undiscounted | | | 13,784 | |
| Less: Imputed interest | | | (425 | ) |
| Present value of future minimum lease payments | | $ | 13,359 | |
5. LONG-TERM INVESTMENTS
| (1) | The Company’s ownership interest of investments in unconsolidated entities as of June 30, 2024 and December 31, 2023 consists of the following: |
| | | | Ownership percentage | |
| | | | June 30, | | | | December 31, | |
| Name of related party | | | 2024 | | | | 2023 | |
| QBAS TECH. CO., LTD. | | | 3.03 | % | | | 3.03 | % |
| (2) | The extent the investee relies on the Company for its business are summarized as follows: |
| Name of related party | | The extent the investee relies on the Company for its business |
| QBAS TECH. CO., LTD. | | No specific business relationship |
| (3) | Long-term investment mainly consists of the following: |
| | | June 30,
2024 | | | December 31, 2023 | |
| QBAS TECH. CO., LTD. | | $ | 308,166 | | | $ | 326,584 | |
| Less: Other-than-temporary impairment | | | (308,166 | ) | | | (326,584 | ) |
| Investments, net | | $ | - | | | $ | - | |
Equity Investment without Readily Determinable Fair Value
Equity investments (except those accounted for under the equity method of accounting or those that result in consolidation of the Company) which do not have readily determinable fair values are recorded as equity investments without readily determinable fair value. All equity investments without readily determinable fair value are assessed for impairment when events or changes in circumstances indicate that the carrying amounts may not be recoverable, and measured at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for an identical or similar investment of the same issuer. The recoverable value of the investment was determined based on the Company’s best estimate of the amount that could be realized from the investment, which considered the latest financial information. As of June 30, 2024 and December 31, 2023, the Company recorded a full impairment equal to the cost of the equity investments without readily determinable fair values.
6. PREPAID TAXES
Prepaid taxes as of June 30, 2024 and December 31, 2023, consist of prepaid value added tax (“VAT”) in the amount of $433,819 and $456,005, respectively, which can be used to offset VAT payable when the Company incurs sales.
7. INTANGIBLE ASSETS
Intangible assets as of June 30, 2024 and December 31, 2023 are summarized as follows:
| | | June 30, 2024 | |
| | | Weighted | | | | | | | | | |
| | | Average | | Gross | | | | | | Net | |
| | | Amortization | | Carrying | | | Accumulated | | | Carrying | |
| | | Period (Years) | | Amount | | | Amortization | | | Amount | |
| Patents | | 20 | | $ | 547,055 | | | $ | 254,440 | | | $ | 292,615 | |
| Computer software cost | | 3 | | | 17,518 | | | | 17,518 | | | | - | |
| Total | | | | $ | 564,573 | | | $ | 271,958 | | | $ | 292,615 | |
| | | December 31, 2023 | |
| | | Weighted | | | | | | | | | |
| | | Average | | Gross | | | | | | Net | |
| | | Amortization | | Carrying | | | Accumulated | | | Carrying | |
| | | Period (Years) | | Amount | | | Amortization | | | Amount | |
| Patents | | 20 | | $ | 579,750 | | | $ | 254,753 | | | $ | 324,997 | |
| Computer software cost | | 3 | | | 18,564 | | | | 18,564 | | | | - | |
| Total | | | | $ | 598,314 | | | $ | 273,317 | | | $ | 324,997 | |
No impairments of intangible assets were identified for the six months ended June 30, 2024 and the year ended December 31, 2023 based on management’s assessment.
Amortization expenses were $17,452 and $16,504 for the six months ended June 30, 2024 and 2023, respectively.
Estimated amortization for the next five years and thereafter is as follows:
| For the year ended June 30, | | | |
| 2025 | | $ | 28,108 | |
| 2026 | | | 28,108 | |
| 2027 | | | 23,216 | |
| 2028 | | | 23,216 | |
| 2029 | | | 23,216 | |
| Thereafter | | | 166,751 | |
| | | $ | 292,615 | |
8. ACCRUED EXPENSES
| | | June 30, | | | December 31, | |
| | | 2024 | | | 2023 | |
| Accrued research and development | | $ | 14,960 | | | $ | 30,756 | |
| Accrued payroll and bonus | | | 25,461 | | | | 47,175 | |
| Accrued employee benefits | | | 11,030 | | | | 11,924 | |
| Accrued professional service fees | | | 65,492 | | | | 62,812 | |
| Other | | | 2,574 | | | | 5,313 | |
| Total | | $ | 119,517 | | | $ | 157,980 | |
9. CONVERTIBLE NOTES PAYABLE
| | | June 30, 2024 | | | December31, 2023 | |
| Convertible notes payable | | $ | 2,516,000 | | | $ | 2,516,000 | |
From June 23, 2023 until July 24, 2023, the Company entered into securities purchase agreements with accredited investors pursuant to which the Company issued convertible promissory notes in the aggregate principal amount of $2,516,000, and on June 24, 2024, the Company entered into amendments to the convertible promissory notes to extend their maturity. As amended, each note matures on the earlier of (i) the date that is 18 months after the issuance date of the note, (ii) the date when the Company redeems the notes, provided the Company has not completed its IPO within 18 months of the issuance date of the notes; otherwise, the notes mature on the date of initial closing of its IPO. The notes accrue interest at 2.40% per annum and maybe prepaid by the Company. Any outstanding and unpaid principal amount of the notes automatically convert upon the commencement of trading on the Nasdaq Stock Market or the New York Stock Exchange of the Company’s ordinary shares to be issued in an IPO at a conversion price equal to the lesser of (i) the price equal to a 30% discount to the actual price per ordinary share to be issued in the Company’s IPO, or (ii) $4.00 per share.
10. OTHER INCOME
Other income consists of the following for the six months ended June 30, 2024 and 2023:
| | | Six Months Ended June 30, | |
| | | 2024 | | | 2023 | |
| Subsidy income / government grant | | $ | - | | | $ | 6,999 | |
| Other | | | - | | | | 28,057 | |
| Total | | $ | - | | | $ | 35,056 | |
Subsidy income / government grant
CerebracaTM wafer: In August 2017, the Company applied for the A+ Enterprise Innovation and Research Development Program - Rapid Review Clinical Trial Plan “CerebracaTM wafer Phase I/IIa Clinical Trial Plan for Recurrent Highly Malignant Glioma” entrusted by the Ministry of Economic Affairs (R.O.C) to the Institute for Information Industry. During the six months ended June 30, 2024 and 2023, the Company did not receive the subsidy income under the CerebracaTM wafer plan.
SBIR: In May 2022, the Company applied for the “Development Project for Production of LV-001 Functional Dropping pills by Supercritical Extraction Method” under the Small Business Innovation Research (SBIR) program of the Small and Medium Enterprise Administration, Ministry of Economic Affairs. During the six months ended June 30, 2024 and 2023, subsidy income collected under the SBIR program amounted to $0 and $6,999, respectively.
11. INCOME TAXES
British Virgin Islands
Everfront Biotech Holding Company Limited was incorporated in British Virgin Islands, which does not tax income.
Taiwan
Everfront Biotech Inc. was incorporated in Taiwan. The Taiwan Income Tax Law imposes a unified enterprise income tax rate of 20% on all enterprises with taxable income greater than approximately $3,698 (NT$120,000).
The Company had net operating loss carryforwards and investment credit that may be available to reduce future years’ taxable income. Future tax benefits which may arise as a result of these loss carryforwards have not been recognized in these financial statements, as their realization is determined not likely to occur and accordingly, the Company has recorded a valuation allowance for the deferred tax asset relating to these tax loss carry-forwards.
As of June 30, 2024, the Company had foreign net operating loss carryforwards of $14,304,621 in Taiwan, which will begin to expire in 2024, if not utilized. As of June 30, 2024, the Company had tax credit of $989,878 in Taiwan arising from the Company’s research and development. The tax credit will be available to reduce taxable income and begin to expire in five years after the Company begins to generate taxable income.
The components of deferred tax assets, liabilities and valuation allowance are as follows:
| | | June 30, | | | December31, | |
| | | 2024 | | | 2023 | |
| Deferred tax asset attributable to: | | | | | | | | |
| Net operating loss carryforwards | | $ | 2,860,924 | | | $ | 2,952,108 | |
| Tax credit carryforwards | | | 989,878 | | | | 1,049,038 | |
| Unrealized impairment loss in investment | | | 61,633 | | | | 65,317 | |
| Total deferred tax asset | | | 3,912,435 | | | | 4,066,463 | |
| Less: valuation allowance | | | (3,912,435 | ) | | | (4,066,463 | ) |
| Deferred tax asset, net of valuation allowance | | $ | - | | | $ | - | |
A reconciliation of the statutory tax rate to the effective tax rate is as follows:
| | | June 30, | | | June 30, | |
| | | 2024 | | | 2023 | |
| Taiwan statutory income tax rate | | | 20 | % | | | 20 | % |
| Change in deferred tax asset valuation allowance | | | (20 | )% | | | (20 | )% |
| Effective income tax rate | | | - | % | | | - | % |
12. RELATED PARTIES TRANSACTIONS
The related parties of the company with whom transactions are reported in these financial statements are as follows:
| Name of Entity or Individual | | Relationship with the Company and its subsidiary |
| Ho-Ching Chen | | Director of the Company |
| Sunflower Holiday Co., Ltd. | | Entity controlled by controlling beneficiary shareholder of Company |
| Xi Yue Biomedicine Inc. (formerly Guang-Li Biomedicine Inc.) | | Entity controlled by controlling beneficiary shareholder of Company |
| Foo Kin Construction Co., Ltd. | | Entity controlled by controlling beneficiary shareholder of Company |
| Depei Biotechnology Co., Ltd. | | Entity controlled by controlling beneficiary shareholder of Company |
Rent expense
Rent expense from related parties consisted of the following as of the periods indicated:
| | | Six Months Ended June 30, | |
| | | 2024 | | | 2023 | |
| Xi Yue Biomedicine Inc. | | $ | 896 | | | $ | 982 | |
| | | | | | | | | |
During the six months ended June 30, 2024 and 2023, the Company has an office lease with Xi Yue Biomedicine Inc. on a twelve-month basis. The lease term expires on December 31, 2024 with a monthly rent of approximately $146.
Due from related party
Due from related party consisted of the following as of the periods indicated:
| | | June 30, | | | December 31, | |
| | | 2024 | | | 2023 | |
| Depei Biotechnology Co., Ltd. | | $ | - | | | $ | 7,984 | |
Amounts due from related party were due to the sales of supplies.
Other assets
Refundable deposits from related parties consisted of the following as of the periods indicated:
| | | June 30, | | | December 31, | |
| | | 2024 | | | 2023 | |
| Xi Yue Biomedicine Inc. | | $ | 308 | | | $ | 327 | |
Accrued expenses
Accrued expenses from related parties consisted of the following as of the periods indicated:
| | | June 30, | | | December 31, | |
| | | 2024 | | | 2023 | |
| Xi Yue Biomedicine Inc. | | $ | 154 | | | $ | 1,960 | |
13. EQUITY
Ordinary Shares
The Company was incorporated in the British Virgin Islands on September 21, 2022. The Company’s authorized share capital consists of 100,000,000 ordinary shares, with a par value of $0.01 per share. On September 21, 2022, the Company issued 50,000 ordinary shares at par value, to Mr. Ho-Ching Chen, founder and a director of the Company. From January2023 to February 2023, the Company issued 51,395,450 ordinary shares to the Subsidiary Shareholders (or their designees) during the Restructuring Transaction (see Note 1).
The equity structures as of June 30, 2024 and 2023 was presented after giving retroactive effect to the reorganization of the Company that was completed in the fiscal year 2023.
Capital Contribution
In April 2022 and June 2022, four shareholders of the Subsidiary converted their outstanding loans with the Subsidiary, in the aggregate amount of $1,572,840, in exchange for 4,190,250 common shares of the Subsidiary. In April 2022, June 2022, and September 2022, the Subsidiary sold 1,020,000 shares of the Subsidiary’s common stock to twenty-seven individuals for an aggregate amount of $1,032,528. As a result of the consummation of the Restructuring Transaction (see Note 1), the aforementioned shares of the Subsidiary were converted into the ordinary shares of the Company in the fiscal year 2023.
In January and February 2023, the Company sold an aggregate of 792,295 ordinary shares of the Company to thirty investors at a price of $2.00 per share pursuant to the stock purchase agreements. As of December 31, 2022, the aggregate amount of Subscription received in advance amounted to $1,089,126, and 792,295 ordinary shares were issued during the year ended December 31, 2023.
14. LOSS PER SHARE
Basic loss per share is computed by dividing net loss by the weighted-average number of common shares outstanding during the year. Diluted loss per share is computed by dividing net loss by the weighted-average number of common shares and dilutive potential common shares outstanding during the six months ended June 30, 2024 and 2023.
| | | Six Months Ended June 30, | |
| | | 2024 | | | 2023 | |
| Numerator: | | | | | | |
| Net loss attributable to the Company’s common stockholders | | $ | (787,563 | ) | | $ | (948,183 | ) |
| | | | | | | | | |
| Denominator: | | | | | | | | |
| Weighted-average shares outstanding: | | | | | | | | |
| Weighted-average shares outstanding - Basic | | | 52,237,745 | | | | 51,795,624 | |
| Weighted-average shares outstanding - Diluted | | | 52,237,745 | | | | 51,795,624 | |
| | | | | | | | | |
| Loss per share | | | | | | | | |
| -Basic | | $ | (0.02 | ) | | $ | (0.02 | ) |
| -Diluted | | $ | (0.02 | ) | | $ | (0.02 | ) |
Diluted loss per share takes into account the potential dilution that could occur if securities or other contracts to issue Common Stock were exercised and converted into Common Stock.
15. COMMITMENTS AND CONTINGENCIES
In November 2017, the Company entered into an agreement with the Ministry of Economic Affairs(R.O.C) to the Institute for Information Industry, for the A+ Enterprise Innovation and Research Development Program - Rapid Review Clinical Trial Plan for “CerebracaTM wafer Phase I/IIa Clinical Trial Plan for Recurrent Highly Malignant Glioma”, pursuant to which the Ministry of Economic Affairs shall subsidize our Phase I/IIa clinical trial of CerebracaTM wafer with governmental fund of $1,593,128 (or NT$51,697,000). As of June 30, 2024 and December 31, 2023, performance guarantee notes were issued of $1,593,128 (or NT$51,697,000) and $1,688,341 (or NT$51,697,000), respectively.
As of June 30, 2024, the Company had unrecorded commitments under contract of $5,077,395. These commitments primarily consist of the transfer and licensing of intellectual property and technology of $3,830,508 and R&D outsourcing services of $1,246,887.
16. SUBSEQUENT EVENTS
The Company follows the guidance in FASB ASC 855-10 for the disclosure of subsequent events. The Company evaluated subsequent events through the date the financial statements were issued and determined the Company has no material subsequent events need to be disclosed.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Everfront Biotech Holding Company Limited
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Everfront Biotech Holding Company Limited (the “Company”) as of December 31, 2023 and 2022, the related statements of operations and comprehensive loss, changes in stockholders’ equity (deficit), and cash flows for the years then ended, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2023 and 2022, and the results of its operations and its cash flows for the years ended December 31, 2023 and 2022, in conformity with the U.S. generally accepted accounting principles in the United States of America.
Consideration of the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As described in Note 2 to the financial statements, the Company incurred recurring losses from operations and has an accumulated deficit, which raises substantial doubt about its ability to continue as a going concern. Management’s plans with regard to these matters are described in Note 2. The accompanying consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
/s/ KCCW Accountancy Corp.
We have served as the Company’s auditor since 2023.
Diamond Bar, California
June 14, 2024
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
CONSOLIDATED BALANCE SHEETS
| | | December 31, 2023 | | | December 31, 2022 | |
| Assets | | | | | | | | |
| Current assets | | | | | | | | |
| Cash and cash equivalents | | $ | 1,220,559 | | | $ | 548,138 | |
| Other receivables | | | 4,898 | | | | - | |
| Due from related party | | | 7,984 | | | | - | |
| Prepaid taxes | | | 456,005 | | | | 442,370 | |
| Prepaid expenses and other current assets | | | 871 | | | | 1,884 | |
| Total current assets | | | 1,690,317 | | | | 992,392 | |
| Long-term investment, net | | | - | | | | - | |
| Property and equipment, net | | | 32,149 | | | | 6,093 | |
| Intangible assets, net | | | 324,997 | | | | 347,741 | |
| Operating lease right-of-use assets | | | 16,098 | | | | 5,298 | |
| Other assets | | | 1,444 | | | | 1,926 | |
| Total assets | | $ | 2,065,005 | | | $ | 1,353,450 | |
| | | | | | | | | |
| Liabilities and equity (deficit) | | | | | | | | |
| Current liabilities | | | | | | | | |
| Accrued expenses | | $ | 157,980 | | | $ | 162,492 | |
| Convertible notes payable | | | 2,516,000 | | | | - | |
| Due to related parties | | | - | | | | 4,769 | |
| Operating lease liabilities – current portion | | | 4,805 | | | | 5,347 | |
| Other current liabilities | | | 673 | | | | 2,088 | |
| Total current liabilities | | | 2,679,458 | | | | 174,696 | |
| Operating lease liabilities – noncurrent portion | | | 10,164 | | | | 1,197 | |
| Total Liabilities | | | 2,689,622 | | | | 175,893 | |
| | | | | | | | | |
| Commitments and contingencies | | | | | | | | |
| | | | | | | | | |
| (Deficit) equity | | | | | | | | |
| Ordinary shares, $0.01 par value, 100,000,000 authorized, 52,237,745 and 51,445,450 shares issued and outstanding * | | | 522,377 | | | | 514,455 | |
| Subscription received in advance | | | - | | | | 1,089,126 | |
| Additional paid-in capital | | | 19,093,280 | | | | 17,512,266 | |
| Accumulated deficit | | | (20,002,006 | ) | | | (17,709,109 | ) |
| Accumulated other comprehensive loss | | | (238,289 | ) | | | (229,502 | ) |
| Total Stockholders’ (deficit) equity | | | (624,638 | ) | | | 1,177,236 | |
| Noncontrolling interest | | | 21 | | | | 321 | |
| Total (deficit) equity | | | (624,617 | ) | | | 1,177,557 | |
| | | | | | | | | |
| Total liabilities and (deficit) equity | | $ | 2,065,005 | | | $ | 1,353,450 | |
* The shares amounts are presented on a retroactive basis.
The accompanying notes are an integral part of these consolidated financial statements.
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
| | | Year Ended December 31, | |
| | | 2023 | | | 2022 | |
| Revenue | | | | | | | | |
| Service revenue | | $ | - | | | $ | 79,908 | |
| Total revenue | | | - | | | | 79,908 | |
| Operating costs | | | - | | | | - | |
| Gross profit | | | - | | | | 79,908 | |
| Operating expenses | | | | | | | | |
| General and administrative expenses | | | 1,678,178 | | | | 726,551 | |
| Research and development expenses | | | 707,692 | | | | 1,057,351 | |
| Total operating expenses | | | 2,385,870 | | | | 1,783,902 | |
| Loss from operations | | | (2,385,870 | ) | | | (1,703,994 | ) |
| Non-operating income (expense) | | | | | | | | |
| Interest income (expense), net | | | 9,969 | | | | 76 | |
| Foreign exchange gain | | | - | | | | 1,442 | |
| Other income | | | 82,704 | | | | 207,882 | |
| Total non-operating income, net | | | 92,673 | | | | 209,400 | |
| Loss before income taxes | | | (2,293,197 | ) | | | (1,494,594 | ) |
| Income tax expense | | | - | | | | - | |
| Net loss | | | (2,293,197 | ) | | | (1,494,594 | ) |
| Net loss attributable to noncontrolling interests | | | (300 | ) | | | (77 | ) |
| Net loss attributed to Everfront Biotech Holding Company Limited | | | (2,292,897 | ) | | | (1,494,517 | ) |
| Foreign currency translation (loss) gain | | | (8,787 | ) | | | 49,495 | |
| Comprehensive loss | | $ | (2,301,684 | ) | | $ | (1,445,022 | ) |
| | | | | | | | | |
| Net loss per share: | | | | | | | | |
| Basic and diluted | | $ | (0.04 | ) | | $ | (0.03 | ) |
| | | | | | | | | |
| Weighted average number of Ordinary Shares outstanding: | | | | | | | | |
| Basic and diluted * | | | 52,195,027 | | | | 51,409,423 | |
* The shares amounts are presented on a retroactive basis.
The accompanying notes are an integral part of these consolidated financial statements.
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
CONSOLIDATED STATEMENT OF CASH FLOWS
| | | Year Ended December 31, | |
| | | 2023 | | | 2022 | |
| Cash Flows from Operating Activities | | | | | | | | |
| Net loss | | $ | (2,293,197 | ) | | $ | (1,494,594 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | |
| Depreciation | | | 9,169 | | | | 4,753 | |
| Amortization | | | 33,791 | | | | 34,207 | |
| Operating lease expense | | | 5,723 | | | | 5,971 | |
| Net changes in operating assets and liabilities: | | | | | | | | |
| (Increase) decrease in other receivables | | | (4,815 | ) | | | 10,068 | |
| Increase in due from related party | | | (7,848 | ) | | | - | |
| Decrease (increase) in prepaid expenses | | | 1,446 | | | | (367 | ) |
| Increase in prepaid taxes | | | (11,762 | ) | | | (27,757 | ) |
| Decrease in other current assets | | | 1,606 | | | | 1,578 | |
| Decrease in accrued expenses | | | (4,020 | ) | | | (367,335 | ) |
| Decrease in advance from customer | | | - | | | | (83,903 | ) |
| Decrease in other current liabilities | | | (1,400 | ) | | | (632 | ) |
| Payment for operating lease liabilities | | | (8,063 | ) | | | (5,991 | ) |
| Net cash used in operating activities | | | (2,279,370 | ) | | | (1,924,002 | ) |
| | | | | | | | | |
| Cash Flows from Investing Activities | | | | | | | | |
| Acquisition of long-term investments | | | - | | | | (2,752 | ) |
| Purchase of property and equipment | | | (27,299 | ) | | | - | |
| Purchase of intangible assets | | | (5,490 | ) | | | - | |
| Increase in other assets | | | (12,141 | ) | | | (503 | ) |
| Net cash used in investing activities | | | (44,930 | ) | | | (3,255 | ) |
| | | | | | | | | |
| Cash Flows from Financing Activities | | | | | | | | |
| Proceeds from convertible notes payable | | | 2,516,000 | | | | - | |
| Capital contribution | | | 499,810 | | | | 1,067,802 | |
| Subscription received in advance | | | - | | | | 1,089,126 | |
| (Repayment of) proceeds from related parties | | | (6,927 | ) | | | 298,467 | |
| Net cash provided by financing activities | | | 3,008,883 | | | | 2,455,395 | |
| | | | | | | | | |
| Effect of exchange rate changes on cash and cash equivalents | | | (12,162 | ) | | | (3,311 | ) |
| | | | | | | | | |
| Net increase in cash and cash equivalents | | | 672,421 | | | | 524,827 | |
| | | | | | | | | |
| Cash and Cash Equivalents | | | | | | | | |
| Beginning of period | | | 548,138 | | | | 23,311 | |
| Ending of period | | $ | 1,220,559 | | | $ | 548,138 | |
| | | | | | | | | |
| Supplemental Disclosure of Cash Flows | | | | | | | | |
| Cash paid during the period for: | | | | | | | | |
| Interest | | $ | - | | | $ | - | |
| Income taxes | | $ | - | | | $ | - | |
| | | | | | | | | |
| Non-cash investing and financing activities: | | | | | | | | |
| Capital contribution through debt conversion | | $ | - | | | $ | 1,532,152 | |
The accompanying notes are an integral part of these consolidated financial statements.
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
CONSOLIDATED STATEMENTS OF EQUITY (DEFICIT)
| | | Ordinary shares* | | | Subscription | | | Additional | | | | | | Accumulated other | | | Non | | | | |
| | | Number of
shares | | | Amount | | | received in advance | | | paid-in
capital | | | Accumulated Deficit | | | comprehensive loss | | | controlling interest | | | Total | |
| Balance at December 31, 2021 | | | 51,395,450 | | | $ | 513,955 | | | $ | - | | | $ | 14,913,913 | | | $ | (16,221,209 | ) | | $ | (278,997 | ) | | | - | | | $ | (1,072,338 | ) |
| Capital contribution | | | - | | | | - | | | | - | | | | 1,032,528 | | | | - | | | | - | | | | - | | | | 1,032,528 | |
| Capital contribution through debt conversion | | | - | | | | - | | | | - | | | | 1,572,840 | | | | - | | | | - | | | | - | | | | 1,572,840 | |
| Reverse acquisition adjustment | | | - | | | | - | | | | - | | | | (7,015 | ) | | | 6,617 | | | | - | | | | 398 | | | | - | |
| Issuance of Ordinary Shares for cash | | | 50,000 | | | | 500 | | | | - | | | | - | | | | - | | | | - | | | | - | | | | 500 | |
| Subscription received in advance | | | - | | | | - | | | | 1,089,126 | | | | - | | | | - | | | | - | | | | - | | | | 1,089,126 | |
| Net loss | | | - | | | | - | | | | - | | | | - | | | | (1,494,517 | ) | | | - | | | | (77 | ) | | | (1,494,594 | ) |
| Foreign currency translation | | | - | | | | - | | | | - | | | | - | | | | - | | | | 49,495 | | | | - | | | | 49,495 | |
| Balance at December 31, 2022 | | | 51,445,450 | | | | 514,455 | | | | 1,089,126 | | | | 17,512,266 | | | | (17,709,109 | ) | | | (229,502 | ) | | | 321 | | | | 1,177,557 | |
| Issuance of Ordinary Shares for cash | | | 792,295 | | | | 7,922 | | | | (1,089,126 | ) | | | 1,581,014 | | | | - | | | | - | | | | - | | | | 499,810 | |
| Net loss | | | - | | | | - | | | | - | | | | - | | | | (2,292,897 | ) | | | - | | | | (300 | ) | | | (2,293,197 | ) |
| Foreign currency translation | | | - | | | | - | | | | - | | | | - | | | | - | | | | (8,787 | ) | | | - | | | | (8,787 | ) |
| Balance at December 31, 2023 | | | 52,237,745 | | | $ | 522,377 | | | $ | - | | | $ | 19,093,280 | | | $ | (20,002,006 | ) | | $ | (238,289 | ) | | $ | 21 | | | $ | (624,617 | ) |
* The shares amounts are presented on a retroactive basis.
The accompanying notes are an integral part of these consolidated financial statements.
EVERFRONT BIOTECH HOLDING COMPANY LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. ORGANIZATION AND DESCRIPTION OF BUSINESS
Everfront Biotech Holding Company Limited (“Everfront BVI”, or the “Company”), which was incorporated on September 21, 2022 in the British Virgin Islands, is a holding company and has not carried out substantive business operations of its own.
Everfront Biotech Inc. (“Everfront Biotech”, or the “Subsidiary”) was incorporated under the laws of Taiwan on December 8, 2010. Everfront Biotech has been primarily engaged in new drug discovery and development since 2010.
Everfront Biotech has established its research and development (“R&D”) team since 2010, and is currently developing small molecules that are intended to treat patients with cancers, neurodegenerative diseases and rare diseases. The R&D team is specialized in preclinical study design; chemistry, manufacturing, and control (“CMC”); investigational new drug (“IND”) submission; and clinical trial design and conduction.
As of the date hereof, three clinical trials are being performed in Taiwan under the approval of the United States Food and Drug Administration (“USFDA”) and Taiwan Food and Drug Administration (“TFDA”). Additionally, the Company has five product candidates currently under IND-Enabling status.
Reorganization
From January 2023 to February 2023, Everfront BVI completed its restructuring (“Restructuring Transaction”) with the Subsidiary; pursuant to which Everfront BVI acquired 51,395,450 shares of common stock of Everfront Biotech from eighty-seven shareholders of Everfront Biotech (the “Subsidiary Shareholders”), representing 99.96% of the issued and outstanding common stock of the Subsidiary, and issued 51,395,450 Ordinary Shares of Everfront BVI to the Subsidiary Shareholders (or their designees).
As a result of the Restructuring Transaction, Everfront BVI directly owns 99.96% of the ownership of Everfront Biotech, and the Subsidiary Shareholders (or their designees) collectively received an aggregate of 51,395,450 Ordinary Shares of the Company, representing approximately 98.39% of the then issued and outstanding shares of the Company’s Ordinary Shares.
Reverse acquisition
For accounting purposes, the acquisition was accounted for as a reverse acquisition and treated as a recapitalization of the Company effected by a share exchange, with Everfront Biotech as the accounting acquirer. Consequently, the historical financial statements of Everfront Biotech are now the historical consolidated financial statements of the Company. The assets and liabilities of Everfront Biotech were brought forward at their book value and no goodwill was recognized.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with the generally accepted accounting principles in the United States of America (the “U.S. GAAP”).
Principle of consolidation
The consolidated financial statements include the financial statements of Everfront Biotech Holding Company Limited and its 99.96% owned subsidiary, Everfront Biotech Inc. All inter-company transactions and balances are eliminated in consolidation.
Going Concern
The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The realization of assets and the satisfaction of liabilities in the normal course of business are dependent on, among other things, the Company’s ability to operate profitably, to generate cash flows from operations, and to pursue financing arrangements to support its working capital requirements.
The Company has suffered net losses of $2,293,197 and $1,494,594 for the years ended December 31, 2023 and 2022, respectively, and used net cash in operating activities of $2,279,370 and $1,924,002 for the years ended December 31, 2023 and 2022, respectively, and has an accumulated deficit of $20,002,006 as of December 31, 2023. These facts and conditions raise substantial doubt about the Company’s ability to continue as a going concern.
Management believes that it has developed a liquidity plan, summarized below, that, if executed successfully, should provide sufficient liquidity to meet the Company’s obligations as they become due for a reasonable period of time, and allow the development of its core business. The plan includes:
● Raising cash through potential equity offerings.
While the Company’s management believes that the measures in its liquidity plan including those described above will be adequate to satisfy its liquidity requirements for the twelve months after the date that these financial statements are issued, there is no assurance that the liquidity plan will be successfully implemented. Failure to successfully implement the liquidity plan may have a material adverse effect on its business, results of operations and financial position, and may adversely affect its ability to continue as a going concern. These consolidated financial statements do not include any adjustments related to the recoverability and classification of recorded assets or the amounts and classification of liabilities or any other adjustments that might be necessary should the Company be unable to continue as a going concern.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the amount of revenues and expenses during the reporting periods. Actual results could differ materially from those results.
Fair Value Measurements
FASB ASC 820, “Fair Value Measurements” defines fair value for certain financial and nonfinancial assets and liabilities that are recorded at fair value, establishes a framework for measuring fair value and expands disclosures about fair value measurements. It requires that an entity measure its financial instruments to base fair value on exit price, maximize the use of observable units and minimize the use of unobservable inputs to determine the exit price. It establishes a hierarchy which prioritizes the inputs to valuation techniques used to measure fair value. This hierarchy increases the consistency and comparability of fair value measurements and related disclosures by maximizing the use of observable inputs and minimizing the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that reflect the assumptions market participants would use in pricing the assets or liabilities based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s own assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. The hierarchy prioritizes the inputs into three broad levels based on the reliability of the inputs as follows:
| | ● | Level 1 - Inputs are quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date. Valuation of these instruments does not require a high degree of judgment as the valuations are based on quoted prices in active markets that are readily and regularly available. |
| | ● | Level 2 - Inputs other than quoted prices in active markets that are either directly or indirectly observable as of the measurement date, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| | ● | Level 3 - Valuations based on inputs that are unobservable and not corroborated by market data. The fair value for such assets and liabilities is generally determined using pricing models, discounted cash flow methodologies, or similar techniques that incorporate the assumptions a market participant would use in pricing the asset or liability. |
The carrying values of certain assets and liabilities of the Company, such as cash and cash equivalents, prepaid expenses and other current assets, accounts payable, accrued liabilities, and due to related parties approximate fair value due to their relatively short maturities.
Cash and Cash Equivalents
The Company considers highly liquid investments with maturities of three months or less, when purchased, to be cash equivalents. As of December 31, 2023 and 2022, the Company’s cash and cash equivalents amounted to $1,220,559 and $548,138, respectively. Some of the Company’s cash deposits are held in financial institutions located in Taiwan where there is currently regulation mandated on obligatory insurance of bank accounts. The Company believes these financial institutions are of high credit quality.
Concentration of Credit Risk
The Company’s financial instruments that are exposed to concentrations of credit risk consist primarily of cash and cash equivalents. The Company places its cash and temporary cash investments in high quality credit institutions, but these investments may be in excess of Taiwan Central Deposit Insurance Corporation and the U.S. Federal Deposit Insurance Corporation’s insurance limits. The Company does not enter into financial instruments for hedging, trading or speculative purposes. Accounts are guaranteed by the Federal Deposit Insurance Corporation (FDIC) up to $250,000. As of December 31, 2023 and 2022, the Company had approximately $966,305 and $243,801 in excess of FDIC insured limits, respectively. The Company maintains cash in state-owned banks in Taiwan. In Taiwan, the insurance coverage of each bank is NTD$3,000,000 (approximately USD$97,975). As of December 31, 2023 and 2022, the Company had no cash in excess of the insured amount. The Company has not experienced any losses in such accounts.
Revenue Recognition
The Company adopted Accounting Standards Codification (“ASC”), Topic 606 (ASC 606), Revenue from Contracts with Customers. Pursuant to ASC 606, the Company recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the consideration that the Company expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that the Company determines is within the scope of ASC 606, the Company performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the Company satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the Company will collect the consideration the Company is entitled to in exchange for the goods or services the Company transfers to the customers. At inception of the contract, once the contract is determined to be within the scope of ASC 606, the Company assesses the goods or services promised within each contract, determines those that are performance obligations, and assesses whether each promised good or service is distinct. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied.
During the year ended December 31, 2022, the Company’s service revenue was derived from rendering a biological research report to a related party (see Note 12). Revenue is recognized at a point in time when the research report was delivered to the customer.
During the year ended December 31, 2023, the Company did not generate any service revenue.
Property and Equipment
Property and equipment are carried at cost net of accumulated depreciation. Repairs and maintenance are expensed as incurred. Expenditures that improve the functionality of the related asset or extend the useful life are capitalized. When property and equipment is retired or otherwise disposed of, the cost and associated accumulated depreciation are removed from the consolidated balance sheet, the related gain or loss is reflected on the consolidated statement of operations. Leasehold improvements are depreciated on the straight-line method over the shorter of the remaining lease term or estimated useful life of the asset. Depreciation is calculated on the straight-line method, including property and equipment under capital leases, generally based on the following useful lives:
| | | Estimated Life
in Years |
| Machinery and equipment | | 2 ~ 6 |
| Office equipment | | 3 ~ 4 |
| Leasehold improvements | | 2 ~ 3 |
Intangible Assets
Intangible assets consist of patents and acquired software. Intangible assets are initially recognized at their respective acquisition costs. All of the Company’s intangible assets have been determined to have finite useful lives and are, therefore, amortized using the straight-line method over their estimated useful lives:
| Patents and trademarks | | 15 to 30 years |
| Acquired software | | 3 years |
Impairment of Long-Lived Assets
The Company has adopted Accounting Standards Codification subtopic 360-10, Property, Plant and Equipment (“ASC 360-10”). ASC 360-10 requires that long-lived assets and certain identifiable intangibles held and used by the Company be reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The Company evaluates its long-lived assets for impairment annually or more frequently if events or changes in circumstances indicate that it might be impaired. Events relating to recoverability may include significant unfavorable changes in business conditions, recurring losses, or a forecasted inability to achieve break-even operating results over an extended period. Should impairment in value be indicated, the carrying value of intangible assets will be adjusted, based on estimates of future discounted cash flows resulting from the use and ultimate disposition of the asset. ASC 360-10 also requires assets to be disposed of be reported at the lower of the carrying amount or the fair value less costs to sell.
Lease
The Company adopted FASB Accounting Standards Codification, Topic 842, Leases (“ASC 842”). The Company applied the following practical expedients allowed under ASC 842:
| | ● | Reassessment of expired or existing contracts: The Company elected not to reassess, at the application date, whether any expired or existing contracts contained leases, the lease classification for any expired or existing leases, and the accounting for initial direct costs for any existing leases. |
| | ● | Use of hindsight: The Company elected to use hindsight in determining the lease term (that is, when considering options to extend or terminate the lease and to purchase the underlying asset) and in assessing impairment of right-to-use assets. |
| | ● | Reassessment of existing or expired land easements: The Company elected not to evaluate existing or expired land easements that were not previously accounted for as leases under ASC 840, as allowed under the transition practical expedient. Going forward, new or modified land easements will be evaluated under ASU No. 2016-02. |
| | ● | Separation of lease and non- lease components: Lease agreements that contain both lease and non-lease components are generally accounted for separately. |
| | ● | Short-term lease recognition exemption: The Company also elected the short-term lease recognition exemption and will not recognize ROU assets or lease liabilities for leases with a term less than 12 months. |
The new leasing standard requires recognition of leases on the consolidated balance sheets as right-of-use (“ROU”) assets and lease liabilities. ROU assets represent the Company’s right to use underlying assets for the lease terms and lease liabilities represent the Company’s obligation to make lease payments arising from the leases. Operating lease ROU assets and operating lease liabilities are recognized based on the present value and future minimum lease payments over the lease term at commencement date. The Company’s future minimum based payments used to determine the Company’s lease liabilities mainly include minimum based rent payments. As most of Company’s leases do not provide an implicit rate, the Company uses its estimated incremental borrowing rate based on the information available at commencement date in determining the present value of lease payments.
The Company recognized lease liabilities, with corresponding ROU assets, based on the present value of unpaid lease payments for existing operating leases longer than twelve months. The ROU assets were adjusted per ASC 842 transition guidance for existing lease-related balances of accrued and prepaid rent, unamortized lease incentives provided by lessors, and restructuring liabilities. Operating lease cost is recognized as a single lease cost on a straight-line basis over the lease term and is recorded in selling, general and administrative expenses. Variable lease payments for common area maintenance, property taxes and other operating expenses are recognized as expense in the period when the changes in facts and circumstances on which the variable lease payments are based occur.
Long-term Equity Investment
The Company accounts for non-marketable equity and other equity investments for which the Company does not have control over the investees as:
| | ● | Equity method investments when the Company has the ability to exercise significant influence, but not control, over the investee. Its proportionate share of the income or loss is recognized monthly and is recorded in gains (losses) on equity investments. |
| | | |
| | ● | Investments in entities that are not consolidated or accounted for under the equity method are recorded as investments without readily determinable fair values. Investments without readily determinable fair values are reported on the consolidated balance sheets in investments, at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for an identical or similar investment of the same issuer. |
Prior to the adoption of ASU 2016-01, equity investments without readily determinable fair values were recorded at cost minus other-than-temporary impairment, with other-than-temporary impairment charges included in other income (expense), net in the Consolidated Statements of Operations. Following the adoption of ASU 2016-01, these investments are measured at cost adjusted for changes in observable prices minus impairment or at net asset value, as a practical expedient, if available, with changes in measurement recognized in other income (expense), net in the Consolidated Statements of Operations. The Company’s equity investments without readily determinable fair values are included in Investment on the Consolidated Balance Sheets.
Research and Development Expenses
The Company accounts for the cost of using licensing rights in research and development cost according to ASC Topic 730-10-25-1. Research and development expenses are charged to expense as incurred. Research and development expenses are comprised of costs incurred in performing research and development activities, including payroll, personnel-related costs, facilities-related overhead, and outside contracted services including clinical trial costs, manufacturing and process development costs for both clinical and preclinical materials, research costs, and other consulting services. Non-refundable advance payment for goods and services that will be used in future research and development activities are expensed when the activity has been performed or when the goods have been received rather than when the payment is made. In instances where the Company enters into agreements with third parties to provide research and development services, costs are expensed as services are performed.
Post-retirement and post-employment benefits
The Company’s subsidiary in Taiwan adopted the government mandated defined contribution plan pursuant to the Labor Pension Act (the “Act”) in Taiwan. Such labor regulations require that the rate of contribution made by an employer to the Labor Pension Fund per month shall not be less than 6% of the worker’s monthly salaries. Pursuant to the Act, the Company makes monthly contribution equal to 6% of employees’ salaries to the employees’ pension fund. The Company has no legal obligation for the benefits beyond the contributions made. The total amounts for such employee benefits, which were expensed as incurred, were $16,379 and $17,987 for the year ended December 31, 2023 and 2022, respectively. Other than the above, the Company does not provide any other post-retirement or post-employment benefits.
Income Taxes
The Company accounts for income taxes using the asset and liability approach which allows the recognition and measurement of deferred tax assets to be based upon the likelihood of realization of tax benefits in future years. Under the asset and liability approach, deferred taxes are provided for the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. A valuation allowance is provided for deferred tax assets if it is more likely than not these items will expire before the Company is able to realize their benefits, or future deductibility is uncertain.
Under ASC 740, a tax position is recognized as a benefit only if it is “more likely than not” that the tax position would be sustained in a tax examination, with a tax examination being presumed to occur. The evaluation of a tax position is a two-step process. The first step is to determine whether it is more-likely-than-not that a tax position will be sustained upon examination, including the resolution of any related appeals or litigations based on the technical merits of that position. The second step is to measure a tax position that meets the more-likely-than-not threshold to determine the amount of benefits recognized in the financial statements. A tax position is measured at the largest amount of benefit that is greater than 50% likely of being realized upon ultimate settlement. Tax positions that previously failed to meet the more-likely-than-not recognition threshold should be recognized in the first subsequent period in which the threshold is met. Previously recognized tax positions that no longer meet the more-likely-than-not criteria should be de-recognized in the first subsequent financial reporting period in which the threshold is no longer satisfied. Penalties and interest incurred related to underpayment of income tax are classified as income tax expense in the year incurred. No significant penalty or interest relating to income taxes has been incurred for the year ended December 31, 2023 and 2022.
Valuation of Deferred Tax Assets
A valuation allowance is recorded to reduce the Company’s deferred tax assets to the amount that is more likely than not to be realized. In assessing the need for the valuation allowance, management considers, among other things, projections of future taxable income and ongoing prudent and feasible tax planning strategies. If the Company determines that sufficient negative evidence exists, then it will consider recording a valuation allowance against a portion or all of the deferred tax assets in that jurisdiction. If, after recording a valuation allowance, the Company’s projections of future taxable income and other positive evidence considered in evaluating the need for a valuation allowance prove, with the benefit of hindsight, to be inaccurate, it could prove to be more difficult to support the realization of its deferred tax assets. As a result, an additional valuation allowance could be required, which would have an adverse impact on its effective income tax rate and results. Conversely, if, after recording a valuation allowance, the Company determines that sufficient positive evidence exists in the jurisdiction in which the valuation allowance was recorded, it may reverse a portion or all of the valuation allowance in that jurisdiction. In such situations, the adjustment made to the deferred tax asset would have a favorable impact on its effective income tax rate and results in the period such determination was made.
Loss Per Share of Common Stock
The Company calculates net loss per share in accordance with ASC Topic 260, “Earnings per Share”. Basic loss per share is computed by dividing the net loss by the weighted average number of common shares outstanding during the period. Diluted loss per share is computed similar to basic loss per share except that the denominator is increased to include the number of additional common shares that would have been outstanding if the potential common stock equivalents had been issued and if the additional common shares were dilutive. Diluted earnings per share excludes all dilutive potential shares if their effect is anti-dilutive.
Commitments and Contingencies
The Company has adopted ASC Topic 450 “Contingencies” subtopic 20, in determining its accruals and disclosures with respect to loss contingencies. Accordingly, estimated losses from loss contingencies are accrued by a charge to income when information available before financial statements are issued or are available to be issued indicates that it is probable that an asset had been impaired or a liability had been incurred at the date of the financial statements and the amount of the loss can be reasonably estimated. Legal expenses associated with the contingency are expensed as incurred. If a loss contingency is not probable or reasonably estimable, disclosure of the loss contingency is made in the financial statements when it is at least reasonably possible that a material loss could be incurred.
Foreign-currency Transactions
For the Company’s subsidiary in Taiwan, the foreign-currency transactions are recorded in New Taiwan dollars (“NTD”) at the rates of exchange in effect when the transactions occur. Gains or losses resulting from the application of different foreign exchange rates when cash in foreign currency is converted into New Taiwan dollars, or when foreign-currency receivables or payables are settled, are credited or charged to income in the year of conversion or settlement. On the balance sheet dates, the balances of foreign-currency assets and liabilities are restated at the prevailing exchange rates and the resulting differences are charged to current income except for those foreign currencies denominated investments in shares of stock where such differences are accounted for as translation adjustments under the Statements of Stockholders’ Equity (Deficit).
Translation Adjustment
The accounts of the Company’s subsidiary in Taiwan were maintained, and their financial statements were expressed, in New Taiwan Dollar (“NT$”). Such financial statements were translated into U.S. Dollars (“$” or “USD”) in accordance ASC 830, “Foreign Currency Matters”, with the NT$ as the functional currency. According to the Statement, all assets and liabilities are translated at the current exchange rate, stockholder’s deficit are translated at the historical rates and income statement items are translated at an average exchange rate for the period. The resulting translation adjustments are reported under other comprehensive income (loss) as a component of stockholders’ equity (deficit).
Recent Accounting Pronouncements
In June 2016, the Financial Accounting Standards Board (the “FASB”) issued Accounting Standards Update (the “ASU”) 2016-13 (the “ASU 2016-13”), Financial Instruments—Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. The new standard changes the accounting for credit losses for financial assets and certain other instruments, including trade receivables and contract assets that are not measured at fair value through net income. Under legacy standards, the Company recognizes an impairment of receivables when it was probable that a loss had been incurred. Under the new standard pursuant to ASU 2016-13, the Company is required to recognize estimated credit losses expected to occur over the estimated life or remaining contractual life of an asset (which includes losses that may be incurred in future periods) using a broader range of information including reasonable and supportable forecasts about future economic conditions. The guidance is effective for smaller reporting companies (the “SRC”) as defined by the SEC for fiscal years beginning after December 15, 2022, including interim periods within those fiscal years with early adoption permitted. On January 1, 2023, the Company adopted this new guidance, and the adoption did not have a material impact on the Company’s financial statements and related disclosure.
In August 2020, the FASB issued Accounting Standards Update 2020-06 (the “ASU 2020-06”), Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity, which simplifies accounting for convertible instruments by removing major separation models required under current GAAP. ASU 2020-06 removes certain settlement conditions that are required for equity contracts to qualify for the derivative scope exception, and it also simplifies the diluted earnings per share calculation in certain areas. ASU 2020-06 is effective for SRC’s fiscal years beginning after December 15, 2023, including interim periods within those fiscal years, with early adoption permitted. The Company early adopted ASU 2020-06 on January 1, 2023.
In November 2023, the FASB issued ASU No. 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures (“ASU 2023-07”). The amendments in ASU 2023-07 improve reportable segment disclosure requirements through enhanced disclosures about significant segment expenses that are regularly provided to the chief operating decision maker (CODM). In addition, the amendments enhance interim disclosure requirements, clarify circumstances in which an entity can disclose multiple segment measures of profit or loss, provide new segment disclosure requirements for entities with a single reportable segment, and contain other disclosure requirements. ASU 2023-07 will be effective for annual reporting periods beginning after December 15, 2023, and interim periods within annual reporting periods beginning after December 15, 2024. Early adoption is permitted. The Company is currently evaluating the impact of this standard on its consolidated financial statements.
In December 2023, the FASB issued ASU No. 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures (“ASU 2023-09”), which requires disclosures of incremental income tax information within the rate reconciliation and expanded disclosures of income taxes paid, among other disclosure requirements. This ASU will be effective for annual reporting periods beginning after December 15, 2024. Early adoption is permitted. ASU 2023-09 will be applied on a prospective basis with the option to apply the standard retrospectively. The Company’s management does not believe that the adoption of ASU 2023-09 will have a material impact on the Company’s consolidated financial statement presentation or disclosures.
3. PROPERTY AND EQUIPMENT
Property and equipment as of December 31, 2023 and 2022 are summarized as follows:
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| | | | | | | |
| Machinery and equipment | | $ | 146,080 | | | $ | 142,148 | |
| Office equipment | | | 27,841 | | | | 3,892 | |
| Leasehold improvements | | | 31,916 | | | | 19,224 | |
| Total | | | 205,837 | | | | 165,264 | |
| Less: accumulated depreciation | | | (173,688 | ) | | | (159,171 | ) |
| Property and equipment, net | | $ | 32,149 | | | $ | 6,093 | |
Depreciation expenses were $9,169 and $4,753 for the years ended December 31, 2023 and 2022, respectively.
4. LEASE
The Company has no finance leases. The Company’s leases primarily include various office and laboratory spaces, copy machine, and vehicles under various operating lease arrangements. The Company’s operating leases have remaining lease terms of up to approximately 2.75 years.
| | | December 31,
2023 | | | December 31,
2022 | |
| ASSETS | | | | | | | | |
| Operating lease right-of-use assets | | $ | 16,098 | | | $ | 5,298 | |
| LIABILITIES | | | | | | | | |
| Operating lease liabilities - current | | | 4,805 | | | | 5,347 | |
| Operating lease liabilities - noncurrent | | | 10,164 | | | | 1,197 | |
Supplemental Information
The following provides details of the Company’s lease expenses:
| | | Year Ended
December 31, | |
| | | 2023 | | | 2022 | |
| Operating lease expenses | | $ | 5,723 | | | $ | 5,971 | |
Other information related to leases is presented below:
| | | Year Ended December 31,, | |
| | | 2023 | | | 2022 | |
| Cash paid for amounts included in the measurement of operating lease liabilities | | | | | | |
| Operating cash flows from operating leases | | $ | 8,063 | | | $ | 5,991 | |
| Weighted Average Remaining Lease Term: | | | | | | | | |
| Operating leases | | | 2.66 years | | | | 1.35 years | |
| | | | | | | | | |
| Weighted Average Discount Rate: | | | | | | | | |
| Operating leases | | | 2.97 | % | | | 2.37 | % |
The minimum future annual payments under non-cancellable leases during the next five years and thereafter, at rates now in force, are as follows:
| | | Operating leases | |
| 2024 | | $ | 5,205 | |
| 2025 | | | 6,334 | |
| 2026 | | | 4,102 | |
| Total future minimum lease payments, undiscounted | | | 15,641 | |
| Less: Imputed interest | | | (672 | ) |
| Present value of future minimum lease payments | | $ | 14,969 | |
5. LONG-TERM INVESTMENTS
| (1) | The Company’s ownership interest of investments in unconsolidated entities as of December 31, 2023 and 2022 consists of the following: |
| | | Ownership percentage | |
| | | December 31, | | | December 31, | |
| Name of related party | | 2023 | | | 2022 | |
| QBAS TECH. CO., LTD. | | | 3.03 | % | | | 3.03 | % |
| (2) | The extent the investee relies on the Company for its business are summarized as follows: |
| Name of related party | | The extent the investee relies on the Company for its business |
| QBAS TECH. CO., LTD. | | No specific business relationship |
| (3) | Long-term investment mainly consists of the following: |
| | | December 31,
2023 | | | December 31,
2022 | |
| QBAS TECH. CO., LTD. | | $ | 326,584 | | | $ | 352,415 | |
| Less: Other-than-temporary impairment | | | (326,584 | ) | | | (352,415 | ) |
| Investments, net | | $ | - | | | $ | - | |
Equity Investment without Readily Determinable Fair Value
Equity investments (except those accounted for under the equity method of accounting or those that result in consolidation of the Company) which do not have readily determinable fair values are recorded as equity investments without readily determinable fair value. All equity investments without readily determinable fair value are assessed for impairment when events or changes in circumstances indicate that the carrying amounts may not be recoverable, and measured at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for an identical or similar investment of the same issuer. The recoverable value of the investment was determined based on the Company’s best estimate of the amount that could be realized from the investment, which considered the latest financial information. As of December 31, 2023 and 2022, the Company recorded a full impairment equal to the cost of the equity investments without readily determinable fair values.
6. PREPAID TAXES
Prepaid taxes as of December 31, 2023 and 2022, consist of prepaid value added tax (“VAT”) in the amount of $456,005 and $442,370, respectively, which can be used to offset VAT payable when the Company incurs sales.
7. INTANGIBLE ASSETS
Intangible assets as of December 31, 2023 and 2022 are summarized as follows:
| | | December 31, 2023 | |
| | | Weighted
Average
Amortization | | | Gross
Carrying | | | Accumulated | | | Net
Carrying | |
| | | Period (Years) | | | Amount | | | Amortization | | | Amount | |
| Patents | | | 20 | | | $ | 579,750 | | | $ | 254,753 | | | $ | 324,997 | |
| Computer software cost | | | 3 | | | | 18,564 | | | | 18,564 | | | | - | |
| Total | | | | | | $ | 598,314 | | | $ | 273,317 | | | $ | 324,997 | |
| | | December 31, 2022 | |
| | | Weighted
Average
Amortization | | | Gross
Carrying | | | Accumulated | | | Net
Carrying | |
| | | Period (Years) | | | Amount | | | Amortization | | | Amount | |
| Patents | | | 20 | | | $ | 572,110 | | | $ | 224,369 | | | $ | 347,741 | |
| Computer software cost | | | 3 | | | | 18,498 | | | | 18,498 | | | | - | |
| Total | | | | | | $ | 590,608 | | | $ | 242,867 | | | $ | 347,741 | |
No impairments of intangible assets were identified for the years ended December 31, 2023 and 2022 based on management’s assessment.
Amortization expenses were $33,791 and $34,207 for the years ended December 31, 2023 and 2022, respectively.
Estimated amortization for the next five years and thereafter is as follows:
| As of December 31, | | | |
| 2024 | | $ | 29,788 | |
| 2025 | | | 29,788 | |
| 2026 | | | 27,195 | |
| 2027 | | | 24,604 | |
| 2028 | | | 24,604 | |
| Thereafter | | | 189,018 | |
| | | $ | 324,997 | |
8. ACCRUED EXPENSES
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Accrued research and development | | $ | 30,756 | | | $ | 75,510 | |
| Accrued payroll and bonus | | | 47,175 | | | | 34,615 | |
| Accrued employee benefits | | | 11,924 | | | | 11,100 | |
| Accrued professional service fees | | | 62,812 | | | | 27,691 | |
| Other | | | 5,313 | | | | 13,576 | |
| Total | | $ | 157,980 | | | $ | 162,492 | |
9.CONVERTIBLENOTESPAYABLE
| | | December 31,
2023 | | | December 31,
2022 | |
| Convertible notes payable | | $ | 2,516,000 | | | $ | - | |
From June 23, 2023 until July 24, 2023, the Company entered into securities purchase agreements with accredited investors pursuant to which the Company issued convertible promissory notes in the aggregate principal amount of $2,516,000. The notes mature on the earlier of (i) the 12-month anniversary of the issuance date of the notes, or (ii) the date when the Company redeems the notes, provided the Company has not completed its IPO within 12 months of the issuance date of the notes; otherwise, the notes mature on the date of initial closing of its IPO. The notes accrue interest at 2.40% per annum and maybe prepaid by the Company. Any outstanding and unpaid principal amount of the notes automatically convert upon the commencement of trading on the Nasdaq Stock Market or the New York Stock Exchange of the Company’s Ordinary Shares to be issued in an IPO at a conversion price equal to the lesser of (i) the price equal to a 30% discount to the actual price per ordinary share to be issued in the Company’s IPO, or (ii) $4.00 per share.
10. OTHER INCOME
Other income consists of the following for the years ended December 31, 2023 and 2022:
| | | December 31,
2023 | | | December 31,
2022 | |
| Subsidy income / government grant | | $ | 36,843 | | | $ | 184,614 | |
| Other | | | 45,861 | | | | 23,268 | |
| Total | | $ | 82,704 | | | $ | 207,882 | |
Subsidy income / government grant
Cerebraca™ Wafer: In August 2017, the Company applied for the A+ Enterprise Innovation and Research Development Program - Rapid Review Clinical Trial Plan “Cerebraca™ Wafer Phase I/IIa Clinical Trial Plan for Recurrent Highly Malignant Glioma” entrusted by the Ministry of Economic Affairs (R.O.C) to the Institute for Information Industry. During the years ended December 31, 2023 and 2022, subsidy income received under the Cerebraca™ Wafer plan amounted to $29,979 and $176,761, respectively.
SBIR: In May 2022, the Company applied for the “Development Project for Production of LV-001 Functional Dropping pills by Supercritical Extraction Method” under the Small Business Innovation Research (SBIR) program of the Small and Medium Enterprise Administration, Ministry of Economic Affairs. During the years ended December 31, 2023 and 2022, subsidy income collected under the SBIR program amounted to o $6,864 and $7,853, respectively.
11. INCOME TAXES
British Virgin Islands
Everfront Biotech Holding Company Limited was incorporated in British Virgin Islands, which does not tax income.
Taiwan
Everfront Biotech Inc. was incorporated in Taiwan. The Taiwan Income Tax Law imposes a unified enterprise income tax rate of 20% on all enterprises with taxable income greater than approximately $3,919 (NT$120,000).
The Company had net operating loss carryforwards and investment credit that may be available to reduce future years’ taxable income. Future tax benefits which may arise as a result of these loss carryforwards have not been recognized in these financial statements, as their realization is determined not likely to occur and accordingly, the Company has recorded a valuation allowance for the deferred tax asset relating to these tax loss carry-forwards.
As of December 31, 2023, the Company had foreign net operating loss carryforwards of $14,760,541 in Taiwan, which will begin to expire in 2024, if not utilized. As of December 31, 2023, the Company had tax credit of $1,049,038 in Taiwan arising from the Company’s research and development. The tax credit will be available to reduce taxable income and begin to expire in five years after the Company begins to generate taxable income.
The components of deferred tax assets, liabilities and valuation allowance are as follows:
| | | December 31,
2023 | | | December 31,
2022 | |
| Deferred tax asset attributable to: | | | | | | | | |
| Net operating loss carryforwards | | $ | 2,952,108 | | | $ | 2,910,958 | |
| Tax credit carryforwards | | | 1,049,038 | | | | 1,045,283 | |
| Unrealized impairment loss in investment | | | 65,317 | | | | 65,083 | |
| Total deferred tax asset | | | 4,066,463 | | | | 4,021,324 | |
| Less: valuation allowance | | | (4,066,463 | ) | | | (4,021,324 | ) |
| Deferred tax asset, net of valuation allowance | | $ | - | | | $ | - | |
A reconciliation of the statutory tax rate to the effective tax rate is as follows:
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Taiwan statutory income tax rate | | | 20 | % | | $ | 20 | % |
| Change in deferred tax asset valuation allowance | | | (20 | )% | | | (20 | )% |
| Effective income tax rate | | | -% | | | $ | - | % |
12. RELATED PARTIES TRANSACTIONS
The related parties of the Company with whom transactions are reported in these financial statements are as follows:
| Name of Entity or Individual | | Relationship with the Company and its subsidiary |
| Ho-Ching Chen | | Director of the Company |
| Sunflower Holiday Co., Ltd. | | Entity controlled by controlling beneficiary shareholder of Company |
| Xi Yue Biomedicine Inc. (formerly Guang-Li Biomedicine Inc.) | | Entity controlled by controlling beneficiary shareholder of Company |
| Foo Kin Construction Co., Ltd. | | Entity controlled by controlling beneficiary shareholder of Company |
| Depei Biotechnology Co., Ltd. | | Entity controlled by controlling beneficiary shareholder of Company |
Service revenue
Service revenue from related parties consisted of the following as of the periods indicated:
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Xi Yue Biomedicine Inc. | | $ | - | | | $ | 79,908 | |
Rent expense
Rent expense from related parties consisted of the following as of the periods indicated:
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Xi Yue Biomedicine Inc. | | $ | 1,834 | | | $ | 1,918 | |
| Foo Kin Construction Co., Ltd. | | | - | | | | 5,753 | |
| Total | | $ | 1,834 | | | $ | 7,671 | |
During the years ended December 31, 2023 and 2022, the Company has an office lease with Xi Yue Biomedicine Inc. on a twelve-month basis. The lease term expires on December 31, 2023 with a monthly rent of approximately $155.
On January 1, 2022, the Company entered into an office lease agreement with Foo Kin Construction Co., Ltd. with the expiration date on December 31, 2022. The monthly rent was approximately $467.
Due from related party
Due from related party consisted of the following as of the periods indicated:
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Depei Biotechnology Co., Ltd. | | $ | 7,984 | | | $ | - | |
Amounts due from related party were due to the sales of supplies.
Other assets
Refundable deposits from related parties consisted of the following as of the periods indicated:
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Xi Yue Biomedicine Inc. | | $ | 327 | | | $ | 326 | |
| Foo Kin Construction Co., Ltd. | | | - | | | | 488 | |
| Total | | $ | 327 | | | $ | 814 | |
Accrued expenses
Accrued expenses from related parties consisted of the following as of the periods indicated:
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Xi Yue Biomedicine Inc. | | $ | 1,960 | | | $ | 1,952 | |
| Foo Kin Construction Co., Ltd. | | | - | | | | 2,929 | |
| Total | | $ | 1,960 | | | $ | 4,881 | |
Due to related parties
Amount due to related parties consisted of the following as of the periods indicated:
| | | December 31, | | | December 31, | |
| | | 2023 | | | 2022 | |
| Ho-Ching Chen | | $ | - | | | $ | 4,769 | |
Amounts due to related parties were unsecured, had no written agreement, due on demand, and interest-free.
13. EQUITY
Ordinary Shares
The Company was incorporated in the British Virgin Islands on September 21, 2022. The Company’s authorized share capital consists of 100,000,000 Ordinary Shares, with a par value of $0.01 per share. On September 21, 2022, the Company issued 50,000 Ordinary Shares at par value, to Mr. Ho-Ching Chen, founder and a director of the Company. From January 2023 to February 2023, the Company issued 51,395,450 Ordinary Shares to the Subsidiary Shareholders (or their designees) during the Restructuring Transaction (see Note 1).
The equity structures as of December 31, 2023 and 2022 was presented after giving retroactive effect to the reorganization of the Company that was completed in the fiscal year 2023.
Capital Contribution
In April 2022 and June 2022, four shareholders of the Subsidiary converted their outstanding loans with the Subsidiary, in the aggregate amount of $1,572,840, in exchange for 4,190,250 common shares of the Subsidiary. In April 2022, June 2022, and September 2022, the Subsidiary sold 1,020,000 shares of the Subsidiary’s common stock to twenty-seven individuals for an aggregate amount of $1,032,528. As a result of the consummation of the Restructuring Transaction (see Note 1), the aforementioned shares of the Subsidiary were converted into the Ordinary Shares of the Company in the fiscal year 2023.
In January and February 2023, the Company sold an aggregate of 792,295 Ordinary Shares of the Company to thirty investors at a price of $2.00 per share pursuant to the stock purchase agreements. As of December 31, 2022, the aggregate amount of Subscription received in advance amounted to $1,089,126, and 792,295 Ordinary Shares were issued during the year ended December 31, 2023.
14. LOSS PER SHARE
Basic loss per share is computed by dividing net loss by the weighted-average number of Ordinary Shares outstanding during the year. Diluted loss per share is computed by dividing net loss by the weighted-average number of Ordinary Shares and dilutive potential Ordinary Shares outstanding during the years ended December 31, 2023 and 2022.
| | | For the Year Ended | |
| | | December 31,
2023 | | | December 31,
2022 | |
| Numerator: | | | | | | | | |
| Net loss attributable to the Company’s stockholders | | $ | (2,292,897 | ) | | $ | (1,494,517 | ) |
| | | | | | | | | |
| Denominator: | | | | | | | | |
| Weighted-average shares outstanding: | | | | | | | | |
| Weighted-average shares outstanding - Basic | | | 52,195,027 | | | | 51,409,423 | |
| Weighted-average shares outstanding - Diluted | | | 52,195,027 | | | | 51,409,423 | |
| | | | | | | | | |
| Loss per share | | | | | | | | |
| -Basic and diluted | | $ | (0.04 | ) | | $ | (0.03 | ) |
Diluted loss per share takes into account the potential dilution that could occur if securities or other contracts to issue Ordinary Shares were exercised and converted into Ordinary Shares.
15. COMMITMENTS AND CONTINGENCIES
In November 2017, the Company entered into an agreement with the Ministry of Economic Affairs(R.O.C) to the Institute for Information Industry, for the A+ Enterprise Innovation and Research Development Program - Rapid Review Clinical Trial Plan for “Cerebraca™ Wafer Phase I/IIa Clinical Trial Plan for Recurrent Highly Malignant Glioma”, pursuant to which the Ministry of Economic Affairs shall subsidize our Phase I/IIa clinical trial of Cerebraca™ Wafer with governmental fund of $1,688,341 (or NT$51,697,000). As of December 31, 2023 and 2022, performance guarantee notes were issued of $1,688,341 (orNT$51,697,000) and $1,682,297(or NT$51,697,000), respectively.
As of December 31, 2023, the Company had unrecorded commitments under contract of $5,219,278. These commitments primarily consist of the transfer and licensing of intellectual property and technology of $4,059,438 and R&D outsourcing services of $1,159,840.
16. SUBSEQUENT EVENTS
The Company follows the guidance in FASB ASC 855-10 for the disclosure of subsequent events. The Company evaluated subsequent events through the date the financial statements were issued and determined the Company has no material subsequent events need to be disclosed.
Everfront Biotech Holding Company Limited
$
Ordinary Shares
PRELIMINARY PROSPECTUS
Joint Book-Runners
| Roth Capital Partners | | The Benchmark Company |
, 2025
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 6. INDEMNIFICATION OF DIRECTORS AND OFFICERS.
Everfront Holding’s amended and restated memorandum and articles of association, which became effective on , 2025, empowers Everfront Holding to indemnify Everfront Holding’s directors and officers against certain liabilities they incur by reason of their being a director or officer of Everfront Holding.
Everfront Holding has also entered into indemnification agreements with each of Everfront Holding’s directors and executive officers in connection with this offering. Under these agreements, Everfront Holding has agreed to indemnify Everfront Holding’s directors and executive officers against certain liabilities and expenses incurred by such persons in connection with claims made by reason of their being a director or officer of Everfront Holding.
The underwriting agreement in connection with this offering also provides for indemnification of Everfront Holding and Everfront Holding’s officers, directors or persons controlling Everfront Holding for certain liabilities.
Everfront Holding intends to obtain directors’ and officer’s liability insurance coverage that will cover certain liabilities of directors and officers of Everfront Holding arising out of claims based on acts or omissions in their capacities as directors or officers.
ITEM 7. RECENT SALES OF UNREGISTERED SECURITIES.
2023 Private Offering
In January 2023 and February 2023, we sold an aggregate of 792,295 of our Ordinary Shares to thirty investors at a price of $2.00 per share pursuant to certain letters of intent we entered into with the investors. As of December 31, 2023, the aggregate amount of subscription received in advance amounted to$1,089,126, and these Ordinary Shares have not been issued.
2023 Convertible Notes
From June 23, 2023 until July 24, 2023, we issued accredited investors convertible promissory notes in the aggregate principal amount of $2,516,000, and on June 24, 2024 and in December 2024, we entered into amendments to the convertible promissory notes to extend their maturity. As amended, each note matures on the earlier of (i) the date that is 24 months after the issuance date of the note, (ii) the date when we redeem the note at its outstanding principal amount and (iii) the date of the initial closing of our IPO. The notes accrue interest at 2.40% per annum and maybe prepaid by us at any time without any penalties, provided we give prior written notice to the note holder at least ten business days before prepayment. Any outstanding and unpaid principal amount of the notes automatically convert upon the commencement of trading on the Nasdaq Stock Market or the New York Stock Exchange of our Ordinary Shares to be issued in an IPO at a conversion price equal to the lesser of (i) the price equal to a 30% discount to the actual price per ordinary share to be issued in our IPO, or (ii) $4.00 per share.
ITEM 8. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES.
(a) Exhibits
See the Exhibit Index attached to this registration statement, which is incorporated by reference herein.
(b) Financial Statement Schedules
Schedules have been omitted because the information required to be set forth therein is not applicable or is shown in the Consolidated Financial Statements or the Notes thereto.
ITEM 9. UNDERTAKINGS.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the provisions described in Item 6, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant under Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) For the purpose of determining liability under the Securities Act to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(4) For the purpose of determining any liability of the registrant under the Securities Act to any purchaser in the initial distribution of the securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
EXHIBIT INDEX
| Exhibit No. | | Description |
| 1.1+ | | Underwriting Agreement |
| 3.1 | | Memorandum and Articles of Association. |
| 3.2+ | | Form of Amended and Restated Memorandum and Articles of Association |
| 4.1+ | | Specimen Certificate for Ordinary Shares |
| 4.2+ | | Form of Representative Warrant |
| 5.1+ | | Opinion of McW Todman & Co. regarding the validity of the Ordinary Shares being registered |
| 8.1+ | | Opinion of McW Todman & Co. as to BVI tax matters (included in Exhibit 5.1) |
| 10.1 | | Technology Transfer License Agreement, dated June 11, 2011, by and between Everfront Biotech Inc. and Buddhist Tzu Chi Medical Foundation |
| 10.2 | | Contract Research Organization Service Agreement, dated August 25, 2017, by and between Everfront Biotech Inc. and A+ Inc. |
| 10.3 | | Engagement Agreement, dated September 5, 2017, by and between Everfront Biotech Inc. and Hualien Tzuchi Hospital, The Buddhist Tzuchi Medical Foundation |
| 10.4 | | Agreement, dated December 19, 2017, by and between Everfront Biotech Inc. and TTY Biopharm Company Limited, as amended by that certain Supplementary Agreement, November 29, 2018 |
| 10.5 | | Agreement, dated December 17, 2018, by and between Everfront Biotech Inc. and Protech Pharmaservices Corporation, as amended by five certain Change Orders, dated June 1, 2018, June 19, 2019, June 16, 2020, October 27, 2020 and October 3, 2022, respectively |
| 10.6 | | Clinical Trial Agreement, dated April 27, 2018, by and between Everfront Biotech Inc., Tri-Service General Hospital, National Defense Medical Center and Protech Pharmaservices Corporation |
| 10.7 | | Clinical Trial Agreement, dated June 23, 2021, by and between Everfront Biotech Inc., Taichung Veterans General Hospital and Protech Pharmaservices Corporation |
| 10.8+ | | Engagement Letter Agreement, by and between Everfront Biotech Holding Company Limited and Enclave Capital LLC |
| 10.9+ | | Form of Employment Agreement |
| 10.10+ | | Form of Indemnification Agreement with the Registrant’s directors and officers |
| 10.11 | | Form of Securities Purchase Agreement for 2023 Convertible Promissory Note Offering |
| 10.12 | | Form of Convertible Promissory Note for 2023 Convertible Promissory Note Offering |
| 10.13+ | | 2025 Share Compensation Plan |
| 10.14 | | Agreement with the Institute for Information Industry for the A+ Industrial Innovation R&D Program-Fast Track Clinical Trial Program |
| 10.15 | | Form of Amendment to Convertible Promissory Note from June 2024 |
| 10.16 | | Form of Amendment to Convertible Promissory Note from December 2024 |
| 21.1 | | List of Subsidiaries |
| 23.1 | | Consent of KCCW Accountancy Corp., Independent Registered Public Accounting Firm |
| 23.2+ | | Consent of McW Todman & Co. (included in Exhibit 5.1) |
| 23.3+ | | Consent of Formosan Brothers (included in Exhibit 99.5) |
| 23.4+ | | Consent of Fortune Business Insights |
| 23.4+ | | Consent of Grand View Research |
| 24.1 | | Power of Attorney |
| 99.1+ | | Code of Business Conduct and Ethics of the Registrant |
| 99.2 | | Audit Committee Charter |
| 99.3 | | Nominating and Corporate Governance Committee Charter |
| 99.4 | | Compensation Committee Charter |
| 99.5+ | | Opinion of Formosan Brothers regarding certain Taiwan Legal Matters |
| 107 | | Filing Fee Table |
| + | To be filed by amendment |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on the Form F-1 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in Taipei, Taiwan on January 3, 2025.
| | Everfront Biotech Holding Company Limited |
| | | |
| | By: | /s/ Tza-Zen Chaung |
| | | Tza-Zen Chaung |
| | | Chief Executive Officer |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints each of Tza-Zen Chaung, Keith Billotti and F. Holt Goddard as his or her true and lawful attorney-in-fact and agent, with full power of substitution and re-substitution, for him or her and in his name or her name, place and stead, in any and all capacities, in connection with this registration statement, including to sign and file in the name and on behalf of the undersigned as director or officer of the registrant, any and all amendments or supplements (including any and all prospectus supplements, stickers and post-effective amendments) to this registration statement with all exhibits thereto, and sign any registration statement for the same offering covered by this registration statement that is to be effective upon filing pursuant to Rule 462(b) under the Securities Act of 1933, as amended, and all post-effective amendments thereto and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, and any applicable securities exchange, securities self-regulatory body or other regulatory authority, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite or necessary to be done in connection therewith and in and about the premises, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming that said attorney-in-fact and agent, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
| /s/ Tza-Zen Chaung | | Chief Executive Officer | | January 3, 2025 |
| Tza-Zen Chaung | | (Principal Executive Officer) | | |
| | | | | |
| /s/ Pi-Chuan Chien | | Chief Financial Officer | | January 3, 2025 |
| Pi-Chuan Chien | | (Principal Accounting and Financial Officer) | | |
| | | | | |
| /s/ Mr. Ho-Ching Chen | | Director | | January 3, 2025 |
| Mr. Ho-Ching Chen | | | | |
| | | | | |
| /s/ Dr. Pei-Wen Chou | | Director | | January 3, 2025 |
| Dr. Pei-Wen Chou | | | | |
| | | | | |
| /s/ Mr. Hann-Chyi Lin | | Independent Director | | January 3, 2025 |
| Mr. Hann-Chyi Lin | | | | |
| | | | | |
| /s/ Dr. Tai-Nang Huang | | Independent Director | | January 2, 2025 |
| Dr. Tai-Nang Huang | | | | |
| | | | | |
| /s/ Mr. Yin-Ho Huang | | Independent Director | | January 3, 2025 |
| Mr. Yin-Ho Huang | | | | |
SIGNATURE OF AUTHORIZED REPRESENTATIVE IN THE UNITED STATES
Pursuant to the Securities Act of 1933 as amended, the undersigned, the duly authorized representative in the United States of America, has signed this registration statement thereto on January 2, 2025.
| | U.S. Authorized Representative |
| | | |
| | By: | /s/ Colleen A. De Vries |
| | Name: | Colleen A. De Vries |
| | Title: | Senior Vice President on behalf of Cogency Global Inc. |