The information in this preliminary prospectus is not complete and may be changed. The securities described herein may not be sold until the registration statement filed with the U.S. Securities and Exchange Commission is declared effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
We are also registering the ordinary shares issuable from time to time upon the exercise of the pre-funded warrants, ordinary warrants and placement agent warrants offered hereby.
The public offering price per ordinary share and any pre-funded warrant will be determined at the time of pricing, and may be at a discount to the then current market price. The recent market price used throughout this prospectus may not be indicative of the final offering price. The final public offering price will be a fixed price determined through negotiation between us and investors based upon a number of factors, including our history and our prospects, the state of the biotechnology industry in which we operate, our recent operating results, including results of our pre-clinical studies and clinical trials, and the general condition of the securities markets at the time of this offering.
We have engaged H.C. Wainwright & Co., LLC, or the Placement Agent, to act as our exclusive placement agent in connection with this offering. The Placement Agent has agreed to use its reasonable best efforts to arrange for the sale of the securities offered by this prospectus. The Placement Agent is not purchasing or selling any of the securities we are offering and the Placement Agent is not required to arrange the purchase or sale of any specific number of securities or dollar amount. We have agreed to pay to the Placement Agent the placement agent fees set forth in the table, which assumes that we sell all of the securities offered by this prospectus. There is no minimum offering requirement as a condition of closing of this offering. Because there is no minimum offering amount required as a condition to closing this offering, we may sell fewer than all of the securities offered hereby, which may significantly reduce the amount of proceeds received by us. We will bear all costs associated with the offering and investors in this offering will not receive a refund in the event that we do not sell an amount of securities sufficient to pursue our business goals described in this prospectus. In addition, because there is no escrow account and no minimum offering amount, investors could be in a position where they have invested in our company, but we are unable to fulfill all of our contemplated objectives due to a lack of interest in this offering. Further, any proceeds from the sale of securities offered by us will be available for our immediate use, despite uncertainty about whether we would be able to use such funds to effectively implement our business plan. See “Plan of Distribution” on page 143 of this prospectus for more information regarding these arrangements.
We are an “emerging growth company” as defined under U.S. federal securities laws and, as such, have elected to comply with reduced public company reporting requirements. This prospectus complies with the requirements that apply to an issuer that is an emerging growth company.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities, or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
This prospectus contains summaries of certain provisions contained in some of the documents described herein, but reference is made to the actual documents for complete information. All of the summaries are qualified in their entirety by the actual documents. Copies of some of the documents referred to herein have been filed, will be filed or will be incorporated by reference as exhibits to the registration statement of which this prospectus is a part, and you may obtain copies of those documents as described below under “Where You Can Find More Information”.
The statements contained in this prospectus that are not purely historical are forward-looking statements. Certain statements in this prospectus may constitute “forward-looking statements” for purposes of the federal securities laws. These forward-looking statements include, but are not limited to, statements regarding the Company’s and the Company’s management team’s expectations, hopes, beliefs, intentions or strategies regarding the future, including statements regarding our future results of operations or financial condition, business strategy and plans, and objectives of management for future operations. In addition, any statements that refer to projections, forecasts or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking statements. The words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intends,” “may,” “might,” “plan,” “possible,” “potential,” “predict,” “project,” “should,” “will,” “would” and similar expressions may identify forward-looking statements, but the absence of these words does not mean that a statement is not forward-looking. Forward-looking statements in this prospectus may include, for example, statements about:
These forward-looking statements are based on information available as of the date of this prospectus, and current expectations, forecasts and assumptions, and involve a number of judgments, risks and uncertainties. Accordingly, forward-looking statements should not be relied upon as representing our views as of any subsequent date, and we do not undertake any obligation to update forward-looking statements to reflect events or circumstances after the date they were made, whether as a result of new information, future events or otherwise, except as may be required under applicable securities laws.
You should read this prospectus and the documents that we reference in this prospectus and have filed as exhibits to the registration statement of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. Those statements are based upon information available to us as of the date of this prospectus and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and such statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. Those statements are inherently uncertain, and investors are cautioned not to unduly rely upon those statements.
Investing in our securities involves a high degree of risk. You should carefully consider the risks and uncertainties described below together with all of the other information contained in this prospectus, including our financial statements and related notes appearing at the end of this prospectus and in the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” before deciding to invest in our securities. If any of the events or developments described below were to occur, our business, prospects, operating results and financial condition could suffer materially, the trading price of our ordinary shares could decline, and you could lose all or part of your investment. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently believe to be immaterial may also adversely affect our business.
Applications for our product candidates could fail to receive regulatory approval for many reasons, including but not limited to the following:
Clinical drug development involves a lengthy and expensive process with an uncertain outcome, and results of preclinical activity or earlier studies may not be predictive of future study results.
Events that may prevent successful or timely completion of clinical development include but are not limited to:
The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses.
Patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after it is filed. Although various extensions may be available, the life of a patent, and the protection it affords, is limited.
In addition, both the U.S. Congress and the U.S. FDA have taken steps to promote the development and approval of generic drugs and biosimilar biologics, including by providing generic and biosimilar developers a private right of action to obtain sufficient quantities of drug samples from the reference product’s manufacturer in order to conduct testing necessary to obtain approval for generic or biosimilar products.
We may be involved in lawsuits to protect or enforce our current patent applications or future patents, which could be expensive, time consuming, and unsuccessful.
Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and may also export otherwise infringing products to territories where we have patent protection but where enforcement is not as strong as that in the United States. These products may compete with Silexin’s products and our current patent application, future patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our current patent applications or future patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Conditions in the Middle East and in Israel may harm our operations.
In particular, on October 7, 2023, Hamas terrorists infiltrated Israel’s southern border from the Gaza Strip and conducted a series of attacks on civilian and military targets. Hamas also launched extensive rocket attacks on the Israeli population and industrial centers located along Israel’s border with the Gaza Strip and in other areas within the State of Israel. These attacks resulted in thousands of deaths and injuries, and Hamas additionally kidnapped many Israeli civilians and soldiers. Following the attack, Israel’s security cabinet declared war against Hamas and commenced a military campaign against Hamas and these terrorist organizations, in parallel to continued rocket and terror attacks against Israel by Hamas. Upon the start of the war, the Israeli military began to call-up reservists for active duty.
In parallel with the war against Hamas that began in October 2023, there have been continued hostilities along Israel’s northern border, as the Hezbollah terrorist organization based in Lebanon has conducted rocket, drone, and, more recently, missile attacks against Israel, and there have also been rocket, drone and missile attacks against Israel by the Houthi movement in Yemen as well continued hostilities with various rebel militia groups in Syria and Iraq. The hostilities with Hezbollah have escalated recently, prompting Israel to send its ground forces into southern Lebanon to destroy terrorist positions and infrastructure used by Hezbollah for attacks on northern and central Israel, which had caused the displacement of residents in northern Israel since early in the war. In November 2024, a ceasefire was brokered between Israel and Hezbollah.
Iran has furthermore entered the conflict against Israel, shooting ballistic missiles at Israel twice during the current war (on April 13, 2024 and October 1, 2024). Iran is furthermore widely believed to be developing nuclear weapons, with which it has threatened Israel and is also believed to have a strong influence among extremist groups in the region, such as Hamas in Gaza, Hezbollah in Lebanon, the Houthi movement in Yemen and various rebel militia groups in Syria and Iraq. There is a risk that other terrorist organizations, including Palestinian military organizations in the West Bank as well as other hostile countries will join the hostilities against Israel. Such clashes may escalate into a greater regional conflict.
Our commercial insurance does not cover losses that may occur as a result of events associated with the security situation in the Middle East. Although the Israeli government currently covers the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, we cannot assure you that this government coverage will be maintained. Any losses or damages incurred by us could have a material adverse effect on our business.
Finally, political conditions within Israel may also affect our operations. Israel held five general elections between 2019 and 2022, and prior to October 2023, the Israeli government pursued extensive changes to Israel’s judicial system, which sparked extensive political debate and unrest. To date, these initiatives have been substantially put on hold. Actual or perceived political instability in Israel or any negative changes in the political environment, may individually or in the aggregate adversely affect the Israeli economy and, in turn, our business, financial condition, results of operations and growth prospects.
If we fail to maintain compliance with Nasdaq continued listing requirements, our shares may be delisted from the Nasdaq Global Market.
If our securities are delisted from Nasdaq, we may seek to list them on other markets or exchanges or the ordinary shares may trade on the pink sheets. In the event of such delisting, our shareholders’ ability to trade, or obtain quotations of the market value of, our securities would be severely limited because of lower trading volumes and transaction delays. These factors could contribute to lower prices and larger spreads in the bid and ask prices for our securities. In addition, the substantially decreased trading in our securities and decreased market liquidity of our securties as a result of the loss of market efficiencies associated with Nasdaq and the loss of federal preemption of state securities laws, which could materially adversely affect our ability to obtain financing on acceptable terms, if at all, and may result in the potential loss of confidence by investors, suppliers, customers and employees and fewer business development opportunities. Additionally, the market price of our securities decline further and shareholders may lose some or all of their investment. There can be no assurance that our securities, if delisted from Nasdaq in the future, would be listed on another national or international securities exchange or on a national quotation service, the Over-The-Counter Markets or the pink sheets.
Broad market and industry fluctuations, as well as general economic, political, regulatory and market conditions, may also negatively impact the market price of our ordinary shares. In the past, companies that have experienced volatility in the market price of their securities have been subject to securities class action litigation. We may be the target of this type of litigation in the future, which could result in substantial expenses and divert our management’s attention.
A substantial number of our ordinary shares may be issued pursuant to the White Lion Purchase Agreement, as well as the conversion terms of our outstanding convertible notes (consisting of the A&R Sponsor Promissory Note and the EarlyBird Convertible Note), which could cause (i) the price of the ordinary shares to decline, and (ii) substantial dilution to our existing shareholders.
The requirement that we repay the EarlyBird Convertible Note could adversely affect our business plan, liquidity, financial condition, and results of operations.
If not converted, we are required to repay, in cash, principal that is outstanding under the EarlyBird Convertible Note, as well as interest thereon, in connection with equity financings by us, or upon the maturity of the note on December 31, 2025. That obligation could have important consequences on our business. In particular, it could:
No assurances can be given that we will be successful in making the required payments under the EarlyBird Convertible Note. If we are unable to make the required cash payments, there could be a default under the EarlyBird Convertible Note. In such event, or if a default otherwise occurs under the EarlyBird Convertible Note:
Because we have no current plans to pay cash dividends on our ordinary shares for the foreseeable future, you may not receive any return on investment unless you sell New Silexion ordinary shares for a price greater than that which you paid for it.
The PFIC status of New Silexion could result in adverse U.S. federal income tax consequences to U.S. Holders.
In general, a non-U.S. corporation is a PFIC for U.S. federal income tax purposes for any taxable year in which, after applying certain look-through rules, either (i) 50% or more of the average value of its assets (generally determined on the basis of a weighted quarterly average) consists of assets that produce, or are held for the production of, passive income, or (ii) 75% or more of its gross income consists of passive income. Passive income generally includes dividends, interest, rents and royalties (other than rents or royalties derived from the active conduct of a trade or business) and gains from the disposition of passive assets. Cash and cash equivalents generally are passive assets. The value of goodwill will generally be treated as an active or passive asset based on the nature of the income produced in the activity to which the goodwill is attributable. For purposes of the PFIC rules, a non-U.S. corporation that owns, directly or indirectly, at least 25% by value of the stock of another corporation is treated as if it held its proportionate share of the assets of the other corporation and received directly its proportionate share of the income of the other corporation.
If a U.S. person is treated as owning at least 10% of the shares of New Silexion, such person may be subject to adverse U.S. federal income tax consequences.
The Company may be subject to securities litigation, which is expensive and could divert management attention, including securities class action and derivative lawsuits which could result in substantial costs.
Securities class action lawsuits and derivative lawsuits are often brought against public companies that have entered into merger or business combination agreements. Even if the lawsuits are without merit, defending against these claims can result in substantial costs and divert management time and resources. An adverse judgment could result in monetary damages, which could have a negative impact on our liquidity and financial condition. We cannot predict whether any such lawsuits will be filed.
To the extent that any of our options or warrants listed above are exercised, new options are issued under our equity incentive plan and subsequently exercised or we issue additional ordinary shares in the future, there will be further dilution to new investors participating in this offering.
We have never declared or paid any dividends on ordinary shares. We anticipate that we will retain all of our future earnings, if any, for use in the operation and expansion of our business and do not anticipate paying cash dividends in the foreseeable future. Any decision to declare and pay dividends in the future will depend on, among other things, the consent of our lender(s), our results of operations, cash requirements, financial condition, contractual restrictions, funds lawfully available therefor and other factors that our board of directors may deem relevant.
The registrant, Silexion Therapeutics Corp (formerly known as Biomotion Sciences), a Cayman Islands exempted company (“New Silexion”) is providing the following unaudited pro forma condensed combined financial information to aid you in your analysis of the financial aspects of the business combination and related transactions (collectively, the “Transactions”) involving Moringa Acquisition Corp (“Moringa”) and Silexion Therapeutics Ltd. (“Silexion”). The following unaudited pro forma condensed combined financial information presents the combination of the financial information of Moringa and Silexion, as adjusted to give effect to the Transactions. Capitalized terms included in this section containing unaudited pro forma condensed combined financial information and not otherwise defined in this section have the same meaning as is provided elsewhere in this prospectus.
Moringa is a Cayman Islands exempted company that was originally a blank check company that was formed for the purpose of effecting a merger, share exchange, asset acquisition, share purchase, recapitalization, reorganization or similar business combination with one or more businesses. As of June 30, 2024 and December 31, 2023, there was approximately $5.924 million and $5.698 million, respectively, held in Moringa’s trust account, which was held for the benefit of Moringa’s public shareholders (the “Trust Account”). The Trust Account was liquidated upon the completion of the Transactions, as described below.
Silexion, an Israeli company that was known previously as Silenseed Ltd., is a pioneering biopharma developmental-stage company dedicated to the development of innovative treatments for pancreatic cancer.
New Silexion is a newly formed entity that was formed for the purpose of effecting the Transactions, and now serves as a publicly-traded holding company of its subsidiaries — including Moringa and Silexion — after the Closing of the Transactions.
On August 15, 2024, the parties completed the previously-reported Transactions pursuant to that certain amended and restated business combination agreement, dated April 3, 3024 (the “Business Combination Agreement”) by and among New Silexion (then known as Biomotion Sciences), August M.S. Ltd., an Israeli company and a wholly owned subsidiary of New Silexion (“Merger Sub 1”), Moringa Acquisition Merger Sub Corp, a Cayman Islands exempted company and a wholly owned subsidiary of New Silexion (“Merger Sub 2”), Moringa and Silexion.
Immediately prior to the Closing, seven directors were appointed to New Silexion’s board of directors, of whom five were designated by Silexion and two were designated by Moringa’s sponsor (Moringa Sponsor, LP, a Cayman Islands exempted limited partnership and/or its wholly-owned subsidiary, Moringa Sponsor US L.P., a Delaware limited partnership) (collectively, the “Moringa Sponsor”).
In connection with the Closing, the ordinary shares and warrants of New Silexion were approved for listing on the Nasdaq Global Market and began trading under the symbols “SLXN” and “SLXNW”, respectively.
The Business Combination was accounted for as a reverse recapitalization in accordance with US GAAP. Under this method of accounting, Silexion was treated as the accounting acquirer and Moringa was treated as the “acquired” company for financial reporting purposes. Silexion was determined to be the accounting acquirer based on evaluation of the following facts and circumstances:
Under the reverse recapitalization accounting method, the Business Combination is deemed to be the equivalent of a capital transaction in which Silexion has issued shares for the net assets of Moringa. The net assets of Moringa are stated at fair value, with no goodwill or other intangible assets recorded. Operations prior to the Business Combination are of Silexion.
The unaudited pro forma condensed combined financial statements were prepared in accordance with Article 11 of SEC Regulation S-X, as amended by the final rule, SEC Release No. 33-10786 “Amendments to Financial Disclosures About Acquired and Disposed Businesses”. The adjustments presented in the unaudited pro forma condensed combined financial statements (collectively, the “Pro Forma Adjustments”) have been identified and presented to provide relevant information necessary for an understanding of New Silexion upon consummation of the Business Combination and related transactions.
The unaudited pro forma condensed combined financial information is for illustrative purposes only and is not necessarily indicative of what the actual results of operations and financial position would have been had the Transactions included in the Pro Forma Adjustments taken place on the dates indicated, nor are they indicative of the future results of operations or financial position of New Silexion. The Pro Forma Adjustments are based on currently available information and certain assumptions and estimates that New Silexion believes are reasonable under the circumstances. Management performed a comprehensive review of the accounting policies between the two entities. Management is not aware of any significant accounting policy differences and has therefore not made any adjustments to the pro forma condensed combined financial information related to any potential differences.
See accompanying notes to the unaudited pro forma condensed combined financial information.
See accompanying notes to the unaudited pro forma condensed combined financial information.
1. Description of the Business Combination
On August 15, 2024, the parties completed the previously-reported transactions (the “Transactions” or “Business Combination”) pursuant to that certain amended and restated business combination agreement, dated April 3, 3024 (the “Business Combination Agreement”) by and among the registrant (“New Silexion”, which was then known as Biomotion Sciences), August M.S. Ltd., an Israeli company and a wholly owned subsidiary of New Silexion (“Merger Sub 1”), Moringa Acquisition Merger Sub Corp, a Cayman Islands exempted company and a wholly owned subsidiary of New Silexion (“Merger Sub 2”), Moringa and Silexion.
In addition, at the Closing, each of the following additional actions was taken, in accordance with the terms of the Business Combination Agreement (in certain cases, as modified by subsequent agreements):
In connection with the Moringa extraordinary general meeting at which the three proposals related to the Business Combination—the Business Combination Proposal, the Merger Proposal and the Articles Amendment Proposal— were approved by the requisite majorities of Moringa’s shareholders, holders of an aggregate of 427,297 Moringa public shares properly exercised their right to have their shares redeemed for a full pro rata portion of the Trust Account holding the proceeds from Moringa’s IPO, which was approximately $11.575 per share, or $4,946,046 in the aggregate. Also prior to the Closing, Moringa reached agreement with EarlyBird on a reduction, from $4.025 million to $1.6 million, in the aggregate, of the fee payable to EarlyBird under the Business Combination Marketing Agreement, dated February 19, 2021 (the “Marketing Agreement”), entered into by Moringa with EarlyBird at the time of Moringa’s initial public offering. Pursuant to the final invoice provided by EarlyBird under the Marketing Agreement at the Closing, Moringa paid $350,000 of cash to EarlyBird at the Closing, and New Silexion issued to EarlyBird a convertible promissory note, due December 31, 2025, in an amount of $1.25 million to be paid by New Silexion to EarlyBird in cash and/or via conversion of outstanding amounts into ordinary shares of New Silexion. After the redemption payments, the payment to EarlyBird, and the satisfaction of amounts due to parties that arranged financing and/or capital markets advisory services in connection with the Closing, the remaining balance of the Trust Account immediately prior to the Closing of $333,936 was used to partially fund the Business Combination.
Also in connection with the Closing, New Silexion entered into an ordinary share purchase agreement for an equity line of credit with White Lion, whereby New Silexion will be able to request to sell to White Lion, and White Lion will be required to purchase, via private placement transactions, up to $15.0 million of New Silexion ordinary shares from time to time after the Closing, up until December 31, 2025 (unless the agreement is terminated sooner), subject to certain limitations and conditions as described therein. The number of New Silexion ordinary shares that New Silexion may require White Lion to purchase in any single sales notice will depend on a number of factors, including the type of purchase notice that New Silexion delivers. Similarly, the purchase price to be paid by White Lion for any shares that New Silexion requires them to purchase will depend on the type of sales notice that New Silexion delivers. New Silexion has also granted registration rights to White Lion pursuant to an accompanying registration rights agreement.
Various issuances and other actions impacted the final number of New Silexion ordinary shares that were issued upon the Closing of the Business Combination, including the following:
2. Basis of Pro Forma Presentation
The unaudited pro forma condensed combined financial statements were prepared in accordance with Article 11 of SEC Regulation S-X, as amended by the final rule, SEC Release No. 33-10786 “Amendments to Financial Disclosures About Acquired and Disposed Businesses”. The adjustments presented in the unaudited pro forma condensed combined financial statements (collectively, the “Pro Forma Adjustments”) have been identified and presented to provide relevant information necessary for an understanding of New Silexion upon consummation of the Business Combination and related transactions.
The historical financial information has been adjusted to reflect the Pro Forma Adjustments giving effect to the Business Combination and related transactions as described in more detail below.
The unaudited pro forma condensed combined financial information does not give effect to any anticipated synergies, operating efficiencies, tax savings, or cost savings that may be associated with the Business Combination and related transactions as described in more detail below. Moringa and Silexion have not had any historical relationship prior to the Business Combination. Accordingly, no pro forma adjustments were required to eliminate activities between the companies. However, the unaudited pro forma condensed combined financial information includes certain share-based compensation that was granted upon the consummation of the Business Combination, as well as net exercise of all Silexion warrants outstanding for New Silexion ordinary shares, which occurred upon the consummation of the Business Combination.
Therefore, Transactions-related expenses recorded in New Silexion’s statements of operations for the period ended September 30, 2024 were moved back to 2023 as a pro forma adjustment.
E) To reflect financing loss recorded by New Silexion upon Closing, equal to the difference between Moringa’s net assets and liability-classified instruments measured at fair value issued by New Silexion as part of the Business Combination. This loss was recorded in New Silexion’s statements of operations for the period ended September 30, 2024 and was moved back to 2023 as a pro forma adjustment.
F) To reflect the elimination of unrealized interest income on investments held in the Trust Account.
G) Represents an obligation to issue ordinary shares of New Silexion with a value of $337,500 (the “ELOC Commitment Shares”) to White Lion as a fee in consideration of White Lion’s commitment to purchase New Silexion ordinary shares from New Silexion under the ordinary share purchase agreement for New Silexion's equity line of credit.
H) To reflect fair value changes of New Silexion’s A&R Sponsor Promissory Note and EarlyBird Convertible Note (the “Notes”). See also Note 3(a) and (b) of New Silexion’s interim financial statements for the period ended September 30, 2024. It was assumed that fair value changes of the Notes until August 15, 2024 are equal to the unwinding of the Notes’ discount, using the discount rates in the Notes’ valuation work.
I) To reflect the transfer by GIBF of its noncontrolling interest in Silexion’s Chinese subsidiary, Silenseed (China) Ltd., to New Silexion in exchange for 203,971 New Silexion ordinary shares upon the completion of the Business Combination.
J) As the Business Combination is being reflected as if it had occurred at the beginning of the earliest period presented, the calculation of weighted average shares outstanding for basic and diluted net loss per share assumes that the shares issuable in connection with the Pro Forma Adjustments have been outstanding for the entirety of the periods presented. Weighted average shares outstanding - basic and diluted (excluding a combined 660,001 ordinary shares underlying outstanding public warrants and private placement warrants because including them would have had an anti-dilutive effect on net loss per share) for the nine-months period ended September 30, 2024, and for the year ended December 31, 2023 are calculated based on the actual pro forma number of New Silexion ordinary shares that were issued and outstanding upon the Closing, after taking into account actual redemptions of Moringa public shares.
The following table sets forth selected historical comparative share information for each of Moringa and New Silexion and unaudited pro forma condensed combined per share information for New Silexion after giving effect to the Business Combination.
The weighted average shares outstanding and net earnings per share information reflect the Business Combination as if it had occurred on January 1, 2023.
This information is only a summary and should be read together with the historical financial statements of Moringa and New Silexion and related notes. The unaudited pro forma condensed combined per share information of Moringa and New Silexion is derived from, and should be read in conjunction with, the unaudited pro forma combined financial statements and related notes included above.
The following discussion and analysis of our financial condition and results of operations (this “MD&A”) should be read in conjunction with the financial statements and the related notes included elsewhere in this prospectus. Some of the information contained in this discussion and analysis or set forth in this prospectus, including information with respect to our plans, objectives, expectations, projections, and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set out in the “Risk Factors” sections of this prospectus, our actual results could differ materially from the results described in or implied by these forward-looking statements. See also the section entitled “Special Note Regarding Forward-Looking Statements” in this prospectus.
Unless the context otherwise requires, references to the “Company,” “we,” “us” and “our” in this MD&A generally refer to Silexion Therapeutics Ltd., an Israeli company (“Silexion”), or, from and after the Business Combination, Silexion Therapeutics Corp (formerly known as Biomotion Sciences), a Cayman Islands exempted company (also referred to herein as “New Silexion”).
On August 15, 2024, New Silexion, Silexion and Moringa completed the Business Combination pursuant to the Business Combination Agreement.
We expect to continue to invest in research and development to develop SIL-204, including hiring additional employees and continuing the research and development of that product candidate. As a result, we expect that our research and development expenses will continue to increase in the future.
General and administrative expenses consist primarily of personnel costs, including share-based compensation related to directors and employees, patent application fees, office space rental costs, and maintenance expenses, external professional service costs, including legal, accounting, audit, finance, human resource services, travel expenses and other consulting fees.
We expect that our general and administrative expenses will increase in the future to fund our continued research and development activities, primarily due to increased headcount to support anticipated growth in the business and due to incremental costs associated with operating as a public company, including costs to comply with the rules and regulations applicable to public companies such as costs related to compliance and reporting obligations pursuant to the rules and regulations of the SEC and stock exchange listing standards, public relations, insurance and professional services.
The finance expenses consisted primarily of change in fair value of warrants and financial liabilities measured at fair value, interest income and exchange rate differences expenses.
| | Three-month period ended
September 30, | |
| | 2024 | | | 2023 | |
| | | (U.S. dollars, in thousands) | |
| Payroll and related expenses | | $ | 575 | | | $ | 71 | |
| Share-based compensation expenses | | | 3,413 | | | | 13 | |
| Professional services | | | 605 | | | | 51 | |
| Depreciation | | | 22 | | | | 10 | |
| Rent and maintenance | | | 18 | | | | 25 | |
| Patent registration | | | - | | | | 2 | |
| Travel expenses | | | 56 | | | | 15 | |
| Other | | | 130 | | | | 9 | |
| Total general and administrative expenses | | $ | 4,819 | | | $ | 196 | |
General and administrative expenses increased by approximately $4.6 million, or 2,300%, to $4.8 million for the three-month period ended September 30, 2024, compared to $0.2 million for the three-month period ended September 30, 2023. The increase resulted mainly from an increase in non-cash share-based compensation expenses in an amount of $3.4 million, an increase in professional services costs primarily related to one-time legal, accounting, and other expenses associated with the costs of becoming a public company and the SPAC merger in an amount of $0.6 million, and an increase in payroll and related expenses in an amount of $0.5 million.
Financial expenses, net
Financial expenses, net increased by approximately $3.7 million, or 3,700% to expense of $3.8 million for the three-month period ended September 30, 2024 compared to financial expenses, net of $0.1 million for the three-month period ended September 30, 2023. This increase was mainly due to an increase in an amount of $4.8 million in one-time loss upon entering Transactions, offset, in part, by an increase in revaluation income of financial instruments (warrants and promissory notes) in the amount of $1.1 million.
Net loss
Net loss increased by approximately $11.1 million, or 1,387.5%, to $11.9 million for the three-month period ended September 30, 2024, compared to $0.8 million for the three-month period ended September 30, 2023. The increase was mainly due to an increase in our research and development expenses, general and administrative expenses, and financial expenses including significant non-cash items related to share-based compensation, transaction costs and costs related to becoming a public company.
Comparison of Years ended December 31, 2023 and 2022 |
The following table summarizes our results of operations for the years ended December 31, 2023 and 2022:
| | | Years ended December 31, | |
| | | 2023 | | | 2022 | |
| Operating expenses: | | | | | | |
| Research and development, net | | $ | 3,708 | | | $ | 3,226 | |
| General and administrative | | | 973 | | | | 634 | |
| Total operating expenses | | | 4,681 | | | | 3,860 | |
| Operating loss | | | 4,681 | | | | 3,860 | |
| Financial expenses (income), net | | | 395 | | | | (396 | ) |
| Loss before income tax | | | 5,076 | | | | 3,464 | |
| Income tax | | | 32 | | | | 24 | |
| Net loss for the year | | $ | 5,108 | | | $ | 3,488 | |
Research and Development Expenses |
The following table summarizes our research and development expenses for the years ended December 31, 2023 and 2022:
| | | Years ended December 31, | |
| | | 2023 | | | 2022 | |
| Payroll and related expenses | | $ | 973 | | | $ | 1,192 | |
| Subcontractors and consultants | | | 2,467 | | | | 1,595 | |
| Materials | | | 13 | | | | 191 | |
| Rent and maintenance | | | 160 | | | | 175 | |
| Travel expenses | | | 37 | | | | 42 | |
| Other | | | 58 | | | | 31 | |
| Total research and development expenses | | $ | 3,708 | | | $ | 3,226 | |
Research and development expenses increased by approximately $0.5 million, or 15.6% to $3.7 million for the year ended December 31, 2023, compared to $3.2 million for the year ended December 31, 2022. The increase resulted mainly from an increase in subcontractors in the amount of $0.9 million partially offset by a decrease in payroll and related expenses in the amount of $0.2 million and a decrease in materials costs in the amount of $0.2 million.
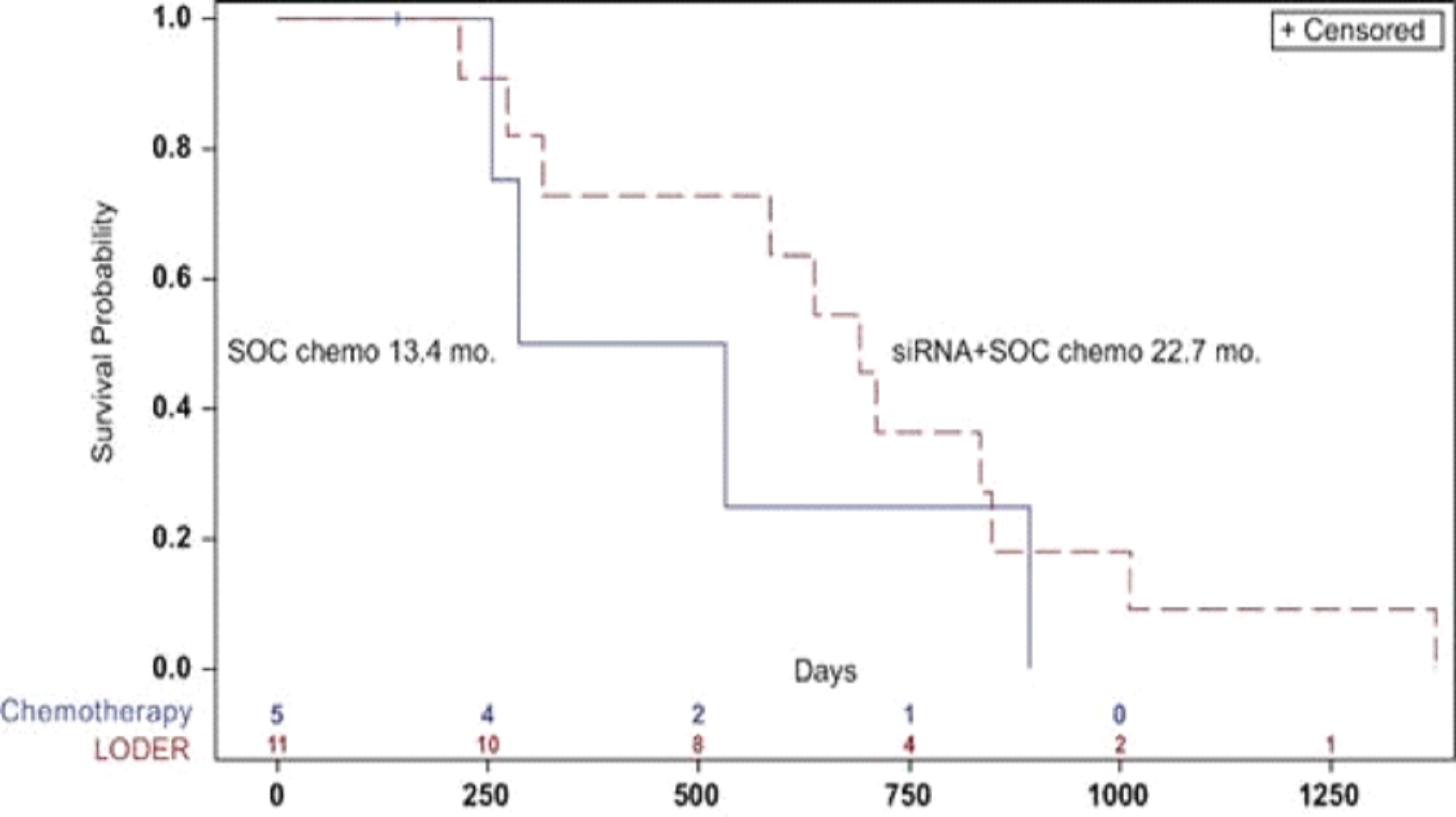
Research and development expenses for the year ended December 31, 2023 and December 31, 2022 included approximately $0.8 million and $2.6 million related to the development of Loder, respectively, and $2.9 million and $0.6 million related to the development of SIL-204B, respectively. Aggregate research and development expenses since inception for Loder program, as of December 31, 2023 and as of December 31, 2022, were approximately $18.2 million and $17.4 million, respectively. Aggregate research and development expenses since inception for SIL-204B program, as of December 31, 2023 and as of December 31, 2022, were approximately $3.6 million and $0.6 million, respectively.
The following table summarizes our general and administrative expenses for the years ended December 31, 2023 and 2022:
| | | Years ended December 31, | |
| | | 2023 | | | 2022 | |
| Payroll and related expenses | | $ | 356 | | | $ | 219 | |
| Professional Services | | | 386 | | | | 197 | |
| Depreciation | | | 45 | | | | 57 | |
| Rent and maintenance | | | 86 | | | | 71 | |
| Patent registration | | | 22 | | | | 32 | |
| Travel expenses | | | 31 | | | | — | |
| Other | | | 47 | | | | 58 | |
| Total general and administrative expenses | | $ | 973 | | | $ | 634 | |
General and administrative expenses increased by approximately $0.4 million, or 66.7% to approximately $1.0 million for year ended December 31, 2023, compared to approximately $0.6 million for the year ended December 31, 2022. The increase resulted mainly from an increase in payroll and related expenses in the amount of $0.1 million and from an increase of professional services costs in the amount of $0.2 million.
Financial expenses (income), net increased by approximately $0.8 million, or 200.0% to expense of $0.4 million for the year ended December 31, 2023 compared to income of $0.4 million for the year ended December 31, 2022. This increase was mainly due to revaluation of warrants in the amount of $1.0 million for the year ended December 31, 2022 offset by a decrease of $0.2 million in foreign exchange loss.
Net loss increased by approximately $1.6 million, or 45.7% to $5.1 million for the year ended December 31, 2023, compared to $3.5 million for the year ended December 31, 2022. The increase was mainly due to increase in our research and development expenses, general and administrative expenses and financial expenses.
Liquidity and Capital Resources
Overview
Our capital requirements will depend on many factors, including the timing and extent of spending to further develop SIL-204 and initiate pre-clinical and clinical trials, support research and development efforts, investments in potential additional pipe-line products, and increased overall compensation as we continue to hire additional personnel. For the nine-month periods ended September 30, 2024 and 2023, and for the years ended December 31, 2023 and 2022, we had net losses of $14.8 million and $3.4 million, and $5.1 million and $3.5 million, respectively. As of September 30, 2024, our cash and cash equivalents totaled $2.0 million.
To date, our principal sources of liquidity have been proceeds from private offerings of our ordinary shares and convertible preferred shares, grants from the Israeli Innovation Authority, and issuance of convertible financing agreements (CFA) and Simple Agreement for Future Equity (SAFE).
Based on our current business plan, we believe our current cash and cash equivalents, and anticipated cash flow from operations, will not be sufficient to meet our anticipated cash requirements for the next 12 months from the date of this prospectus. We will need to raise additional capital to finance our operations, expand our business and pipeline, or for other reasons.
Our audited consolidated financial statements for the year ended December 31, 2023 and our unaudited condensed financial statements for the nine-month period ended September 30, 2024 included in this prospectus note that there is substantial doubt about our ability to continue as a going concern as of such date; and in its report accompanying our audited consolidated financial statements included herein, our independent registered public accounting firm included an explanatory paragraph stating that our recurring losses from operations and our cash outflows from operating activities raise substantial doubt as to our ability to continue as a going concern. This means that our management and our independent registered public accounting firm have expressed substantial doubt about our ability to continue our operations without an additional infusion of capital from external sources. Our audited consolidated financial statements have been prepared on a going concern basis and do not include any adjustments that may be necessary should we be unable to continue as a going concern. If we are unable to finance our operations, our business would be in jeopardy and we might not be able to continue operations and might have to liquidate our assets. In that case, investors might receive less than the value at which those assets are carried on our unaudited condensed financial statements for the nine-month period ended September 30, 2024 and year ended December 31, 2023, and it is likely that investors would lose all or a part of their investment.
We have lease obligations and other contractual obligations and commitments as part of our ordinary course of business. See “Note 4: Operating Leases” and “Note 6: Commitments and Contingent Liabilities” to our consolidated financial statements for the year ended December 31, 2023 for information about our lease obligations.
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements involving commitments or obligations, including contingent obligations, arising from arrangements with unconsolidated entities or persons that have or are reasonably likely to have a material current or future effect on our financial condition, results of operations, liquidity, cash requirements or capital resources.
ELOC Financing
In connection with the Closing of the Business Combination, we entered into an ordinary share purchase agreement, dated August 13, 2024, but effective as of the Closing (the “ELOC Agreement”), for an equity line of credit (the “ELOC”) with White Lion Capital, LLC (the “ELOC Investor”). Under the ELOC, we have the right to request to sell to the ELOC Investor, and the ELOC Investor is required to purchase, via private placement transactions, up to $15.0 million of our ordinary shares from time to time after the Closing, up until December 31, 2025 (unless the agreement is terminated sooner), subject to certain limitations and conditions as described therein. The number of ordinary shares that we may require the ELOC Investor to purchase in any single sales notice depends on a number of factors, including the type of purchase notice that we deliver. Similarly, the purchase price to be paid by the ELOC Investor for any shares that we require it to purchase depends on the type of sales notice that we deliver, and is derived from the market price of our ordinary shares for a certain period of time prior to our purchase request or as of the date of our purchase request. We also granted registration rights to the ELOC Investor pursuant to an accompanying registration rights agreement, also dated August 15, 2024, by and between New Silexion and the ELOC Investor (the “ELOC Registration Rights Agreement”), for which we filed a registration statement on Form S-1 (SEC file number 333-282017), which was declared effective by the SEC on September 17, 2024.
Pursuant to the ELOC Agreement, New Silexion agreed, among other things, that if our sales to the ELOC Investor under the ELOC exceed 19.99% of our total number of ordinary shares outstanding, we will seek the approval of our shareholders for the issuance of any ordinary shares under the ELOC in excess of that amount, in accordance with the Nasdaq Listing Rules, subject to certain exceptions based on the price of our ordinary shares to be sold in excess of that limit.
In consideration for the commitments of the ELOC Investor, we agreed to issue to the ELOC Investor an aggregate of $337,500 of our ordinary shares (the “ELOC Commitment Shares”) based on the closing price of the ordinary shares on the day that is the earlier of (i) the business day prior to effectiveness of the registration statement registering the resale of the shares issuable under the ELOC ordinary share purchase agreement and (ii) the business day prior to the 180th day following the date of Closing of the Business Combination.
Through December 2, 2024, we have issued and sold an aggregate of 763,713 ordinary shares (which includes the foregoing 40,602 ordinary shares issued as a commitment fee) to White Lion under the White Lion Purchase Agreement for aggregate proceeds to us of approximately $3.1 million.
Settlement of Amounts Due Under Marketing Agreement with EarlyBirdCapital
Prior to the Closing of the Business Combination, Moringa reached agreement with EarlyBirdCapital on the reduction, to $1.6 million, in the aggregate, of the fee payable to EarlyBirdCapital under the Marketing Agreement. Pursuant to the final invoice provided by EarlyBirdCapital under the Marketing Agreement at the Closing, Moringa paid $350,000 of cash to EarlyBirdCapital from the trust account at the Closing, and we issued to EarlyBirdCapital a convertible promissory note, due December 31, 2025, in an amount of $1.25 million to be paid by us to EarlyBirdCapital in cash and/or via conversion of outstanding amounts into ordinary shares (the “EarlyBird Convertible Note”).
The EarlyBird Convertible Note bears interest at a rate of 6% per annum and matures on December 31, 2025. If not repaid on or prior to that maturity date or such earlier date as to which the repayment obligation may be accelerated under the note, or not converted in accordance with the terms thereof, the rate of interest applicable to the unpaid principal amount would be adjusted to (15%) per annum. We are required to make mandatory prepayments on the note (which shall first be applied to accrued interest and then to principal) from time to time in amounts equal to ten percent (10%) of the gross proceeds received by us from any equity financings consummated by us prior to the maturity date. We are entitled to voluntarily prepay any additional part of, or all of, the principal and accrued interest, in one or more installments without penalty, prior to the maturity date.
EarlyBirdCapital, in turn, may elect, at its sole discretion, at any time on or prior to the maturity date, to elect to convert all or part of the then outstanding principal and/or accrued interest under the EarlyBird Convertible Note into ordinary shares, at a per share conversion price equal to 95% of the volume weighted average price of an ordinary share for the five trading days immediately prior to the date of our receipt of a conversion notice, provided, however, we are not required to issue, and EarlyBirdCapital may not elect to convert the principal and/or accrued interest into an aggregate number of ordinary shares that would exceed the maximum number of ordinary shares permitted by Section 5635 of the Nasdaq Listing Rules to be issued without the approval of our shareholders, unless such approval is obtained.
Through the date hereof, we have made one payment of $165,000 to EarlyBirdCapital due to amounts raised by us under the ELOC.
PIPE Financing
In connection with, and immediately prior to the Closing of, the Business Combination, Moringa raised $2.0 million via a private investment in public entity financing (the “PIPE Financing”), whereby Moringa sold to Greenstar, LP, an affiliate of the Moringa sponsor (the “PIPE Investor”), 200,000 newly issued Moringa ordinary shares (which does not reflect the reverse share split) at a price of $10.00 per share, pursuant to a subscription agreement, dated as of August 15, 2024, by and among Moringa, New Silexion and the PIPE Investor (the “PIPE Agreement”). Those 200,000 shares automatically converted upon the Closing of the Business Combination 22,223 New Silexion ordinary shares (the “PIPE Shares”).
The funds raised from the PIPE Financing, together with remaining funds in the trust account after payments to redeeming public shareholders, were used for financing support for Moringa and New Silexion, as well as for payment to service providers to whom outstanding amounts were owed by Moringa, including parties that had provided financial advisory services and capital markets advisory services to Moringa during the period leading up to the Closing.
Issuance of Amended and Restated Sponsor Promissory Note
Effective as of the Closing, we issued to the sponsor, and the sponsor accepted, in amendment and restatement, and replacement, in their entirety, of all existing promissory notes issued by Moringa to the sponsor from the IPO until the Closing (and as to which the obligations of Moringa were assigned to New Silexion upon the Closing), an amended and restated promissory note (the “A&R Sponsor Promissory Note”) in an amount of $3,433,000, which reflected the total amount owed by Moringa to the sponsor through the Closing Date. The maturity date of the A&R Sponsor Promissory Note is the 30-month anniversary of the Closing Date (i.e., February 15, 2027). Amounts outstanding under the A&R Sponsor Promissory Note may be repaid (unless otherwise decided by us) only by way of conversion into ordinary shares (“Note Shares”) in accordance with the terms set forth in the form of A&R Sponsor Promissory Note. New Silexion and the Sponsor may also convert amounts outstanding under the A&R Sponsor Promissory Note at the price per share at which we conduct an equity financing following the Closing, subject to a minimum conversion amount of $100,000, in an amount of Note Shares constituting up to thirty percent (30%) of the number of ordinary shares issued and sold by us in such equity financing. The sponsor may also elect to convert amounts of principal outstanding under the note into ordinary shares at any time following the 24-month anniversary of the date of the Closing, subject to a minimum conversion of $10,000, at a price per share equal to the volume weighted average price of the ordinary shares on the principal market on which they are traded during the 20 consecutive trading days prior to the conversion date.
Government Grants
Our research and development efforts were financed, in part, through royalty-bearing grants from the Israeli Innovation Authority, or the IIA. As of September 30, 2024, we received IIA royalty-bearing grants totaling approximately $5.8 million.
We are committed to pay royalties to the IIA at a rate of approximately 3.0% to 5.0% of the sales of all of our product candidates and other related revenues generated from such projects, that were developed, in whole or in part, using the IIA royalty-bearing grants we received under IIA programs up to the total amount of royalty-bearing grants received, linked to the U.S. dollar and bearing annual interest at rates prescribed by the IIA’s rules and guidelines.
We may in the future apply to receive additional grants from the IIA. However, we cannot predict whether we will be entitled to any future grants, or the amounts of any such grants.
Under the Israeli Innovation Law, research and development programs that meet specified criteria and are approved by a committee of the IIA are eligible for grants. A company that receives a royalty-bearing grant from the IIA is typically required to pay royalties to the IIA on income generated from products incorporating IIA-funded know-how (including income derived from services associated with such products and from IIA-funded know-how), up to 100% of the U.S. dollar-linked royalty-bearing grant amount plus interest.
The obligation to pay royalties is contingent on actual income generated from such products and services. In the absence of such income, no payment of royalties is required.
As of September 30, 2024, the total royalty amount that may be payable by the Company is approximately $5.8 million ($6.5 million including interest).
Cash Flows
Cash flows for the nine-month period ended September 30, 2024 and 2023
The following table summarizes our cash flows for the periods indicated:
| | | Nine-month period ended
September 30, | |
| | | 2024 | | | 2023 | |
| | | (U.S. dollars, in thousands) | |
Cash and cash equivalents and restricted cash at beginning of the period | | $ | 4,645 | | | | | |
Net cash used in operating activities | | | | | | | | |
Net cash provided by (used in) investing activities | | | (22 | ) | | | | |
Net cash provided by financing activities | | | 2,920 | | | | | |
Net decrease in cash and cash equivalents and restricted cash | | | | | | | | |
Translation adjustments on cash and cash equivalents and restricted cash | | | (50 | ) | | | | |
Cash and cash equivalents and restricted cash at end of the period | | | | | | | | |
Cash Flows from Operating Activities
Net cash used in operating activities increased by approximately $2 million, or 285.7%, to $2.7 million for the three-month period ended September 30, 2024, compared to $0.7 million for the three-month period ended September 30, 2023. This increase was mainly from an increase of $11.1 million in the net loss for the three-month period ended September 30, 2024, and from an increase of $0.4 million in change of net working capital, offset by (i) an increase of $5.8 million in non-cash share-based compensation and (ii) an increase of $3.6 million in non-cash financial expenses included in the net loss.
Cash Flows from Investing Activities
Net cash provided by (used in) investing activities decreased by $0.6 million, or 100%, to $0 million for the nine-month period ended September 30, 2024, compared to $0.6 million of cash provided by investing activities in the nine-month period ended September 30, 2023. This decrease was mainly due to a reduction in short-term deposits in an amount of $0.5 million, which cash was used for operating activities.
Cash Flows from Financing Activities
Net cash provided by financing activities increased by $2.4 million, or 480%, to approximately $2.9 million for the nine-month period ended September 30, 2024, compared to $0.5 million the nine-month period ended September 30, 2023. This increase was mainly due to an increase in cash received from transactions upon the closing of the Business Combination in an amount of $2.3 million and from an increase in proceeds from issuance of ordinary shares under the ELOC in an amount of $0.6 million, offset by a decrease of $0.5 million in proceeds from issuance of preferred shares, occurred in the nine-month period ended September 30, 2023.
Cash flows for the three-month periods ended September 30, 2024 and 2023
The following table summarizes our cash flows for the periods indicated:
| | | Three-month period ended
September 30, | |
| | | 2024 | | | 2023 | |
| | | (U.S. dollars, in thousands) | |
Cash and cash equivalents and restricted cash at the beginning of the period | | | | | | $ | 6,491 | |
Net cash used in operating activities | | | | | | | (685 | ) |
Net cash provided by (used in) investing activities | | | (16 | ) | | | 72 | |
Net cash provided by financing activities | | | 2,920 | | | | - | |
Net decrease in cash and cash equivalents and restricted cash | | $ | 251 | | | $ | (613 | ) |
Translation adjustments on cash and cash equivalents and restricted cash | | | 25 | | | | (66 | ) |
Cash and cash equivalents and restricted cash at the end of the period | | | | | | $ | 5,812 | |
Cash Flows from Operating Activities
Net cash used in operating activities increased by approximately $2 million, or 285.7%, to $2.7 million for the three-month period ended September 30, 2024, compared to $0.7 million for the three-month period ended September 30, 2023. This increase was mainly from an increase of $11.1 million in the net loss for the three-month period ended September 30, 2024, and from an increase of $0.4 million in change of net working capital, offset by (i) an increase of $5.8 million in share-based compensation and (ii) an increase of $3.6 million in non-cash financial expenses included in the net loss.
Cash Flows from Financing Activities
Net cash provided by financing activities increased by $2.9 million, to approximately $2.9 million for the three-month period ended September 30, 2024, compared to $0 for the three-month period ended September 30, 2023. This increase was mainly due to an increase in cash received from transactions in connection with the closing of the Business Combination in an amount of $2.3 million and from an increase in proceeds from issuance of ordinary shares under the ELOC in an amount of $0.6 million.
Cash flows for the Years ended December 31, 2023 and 2022
The following table summarizes our cash flows for the years ended December 31, 2023 and 2022:
| | | Year ended December 31, | |
| | | 2023 | | | 2022 | |
| Cash and cash equivalents and restricted cash at beginning of the period | | $ | 8,309 | | | $ | 10,083 | |
| Net cash used in operating activities | | | (4,529 | ) | | | (3,335 | ) |
| Net cash provided by (used in) investing activities | | | 573 | | | | (524 | ) |
| Net cash provided by financing activities | | | 522 | | | | 2,752 | |
| Net decrease in cash and cash equivalents and restricted cash | | $ | (3,434 | ) | | $ | (1,107 | ) |
| Translation adjustments on cash and cash equivalents and restricted cash | | | (230 | ) | | | (667 | ) |
| Cash and cash equivalents and restricted cash at the end of the period | | $ | 4,645 | | | $ | 8,309 | |
Cash Flows from Operating Activities
Net cash used in operating activities increased by approximately $1.2 million, or 36.4%, to $4.5 million for the year ended December 31, 2023, compared to $3.3 million for the year ended December 31, 2022. This increase was mainly from an increase of $1.6 million in the loss for the year, offset by a decrease of $0.6 million in non-cash financial expenses.
Cash Flows from Investing Activities
Net cash provided by (used in) investing activities increased by $1.1 million, or 220.0%, to $0.6 million for the year ended December 31, 2023, compared to investment of $0.5 million for the year ended December 31, 2022. This increase was mainly due to change short-term deposit in the amount of $1.0 million.
Cash Flows from Financing Activities
Net cash provided by financing activities decreased by $2.3 million, or 82.1%, to approximately $0.5 million for the year ended December 31, 2023, compared to $2.8 million for the year ended December 31, 2022. This decrease was mainly due to a decrease in proceeds from issuance of preferred shares which was higher in the amount of $2.2 million in 2022.
Funding Requirements
We expect to devote substantial financial resources to our ongoing and planned activities, particularly further development of SIL-204 and as we conduct our planned pre-clinical and clinical trials.
Identifying potential product candidates and conducting pre-clinical testing and clinical trials is a time-consuming, expensive, and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success. For additional information please refer to the “Risk Factors” section of this prospectus, including “Risks Related to Our Financial Condition and Capital Requirements — we have never generated any revenue from product sales and may never be profitable” and “Risks Related to the Research and Development of our Product Candidates — Silexion is heavily dependent on the success of its product candidates…”.
We expect our expenses to increase substantially in connection with our ongoing activities, particularly as we advance our pre-clinical studies and clinical trials. In addition, if we obtain marketing approval for SIL-204 in any indication or for any other product candidate we are developing or may develop in the future, we expect to incur significant commercialization expenses related to product manufacturing, sales, marketing, and distribution. Furthermore, upon the closing of the Business Combination, we expect to incur additional costs associated with operating as a public company. Accordingly, we will need to obtain substantial additional funding.
Our future capital requirements will depend on many factors, including:
| | ● | Human clinical trial costs. |
As of September 30, 2024, we had cash and cash equivalents of $2.0 million. Based on our current cash balance, as well as its history of operating losses and negative cash flows from operation, combined with its anticipated use of cash to, among other things, (i) fund the preclinical and clinical development of our products, (ii) identify and develop new product candidates, and (iii) seek approval for SIL-204 and any other product candidates we may develop, our management has concluded that we do not have sufficient cash to fund our operations for 12 months from the date of its unaudited condensed financial statements for the nine-month period ended September 30, 2024 included in this prospectus without additional financing, and as a result, there is substantial doubt about our ability to continue as a going concern.
In making this determination, applicable accounting standards prohibited us from considering the potential mitigating effect of plans that have not been fully implemented as of the date of our unaudited condensed financial statements for the nine-month period ended September 30, 2024, including, without limitation, plans to consummate the Business Combination discussed below and to raise additional capital. Our financial information throughout this prospectus, and our unaudited condensed financial statements for the nine -month period ended September 30, 2024 contained herein have been prepared on a basis that assumes that we will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. This financial information and our unaudited condensed financial statements for the nine -month period ended September 30, 2024 do not include any adjustments that may result from an unfavorable outcome of this uncertainty.
Without giving effect to the receipt of any proceeds from this offering, we currently estimate that our existing cash and cash equivalents are sufficient to fund business operations through the end of first quarter of 2025.
We have based these estimates and expectations on assumptions that may prove to be wrong, and our operating plan may change as a result of many factors currently unknown to us. We could not, as of the September 30, 2024 balance sheet date of the unaudited condensed financial statements for the three and nine-month periods ended September 30, 2024, determine the exact level of funds that will be available to us upon potential equity financings. Our expected use of funds represents our intentions based upon our current plans and business condition, which could change in the future as our plans and business condition evolve and the level of funding available to us becomes clear. In addition, changing circumstances could cause us to consume capital significantly faster than we currently anticipate, and we may need to spend more than currently expected because of circumstances beyond our control. As a result, we could deplete our capital resources sooner than we currently expect. In addition, because the successful development of SIL-204 and any studies or other product candidates that we pursue is highly uncertain, at this time we cannot reasonably estimate or know the nature, timing and costs of the efforts that will be necessary to complete the development of any product candidate.
Until such time, if ever, as we can generate substantial revenues from product sales, we expect to finance our cash needs through a combination of public and private equity offerings and debt financings, including the private placement of ordinary shares pursuant to the White Lion Purchase Agreement, strategic alliances, collaborations, and marketing, distribution, or licensing arrangements. However, adequate additional financing may not be available to us on acceptable terms, or at all, and may be impacted by the economic climate and market conditions.
Reliance on the ELOC or other similar types of equity financings as a source of ongoing funding for our operations following the Business Combination could involve significant issuances of ordinary shares by us that could cause the following impacts (among others):
| ● | significant dilution to the equity interests of our current shareholders; |
| ● | a deemed change of control of our company due to the issuance of a substantial number of ordinary shares, which may affect, among other things, our ability to use our net operating loss carry forwards, if any, and could result in a change in the officers and directors of our company relative to our current officers and directors, to the extent any shareholders build up significant beneficial ownership from ordinary shares issued pursuant to the ELOC; |
| ● | may have the effect of delaying or preventing a change of control of our company by diluting the share ownership or voting rights of a person seeking to obtain control; and |
| ● | may adversely affect prevailing market prices for our ordinary shares or warrants. |
Critical Accounting Policies and Estimates
For a description of our significant accounting policies, see Note 2 to our consolidated financial statements for the year ended December 31, 2023 and unaudited condensed consolidated financial statements for the nine-month period ended September 30, 2024 included in this prospectus.
The preparation of our consolidated financial statements for the year ended December 31, 2023 and unaudited condensed consolidated financial statements for the nine-month period ended September 30, 2024 in conformity with U.S. GAAP requires management to make estimates and assumptions in certain circumstances that affect the amounts reported in the accompanying consolidated financial statements for the year ended December 31, 2023 and unaudited condensed consolidated financial statements for the nine-month period ended September 30, 2024 and related footnotes. Actual results may differ from these estimates. We base our judgments on our experience and on various assumptions that we believe to be reasonable under the circumstances.
Of our policies, the following are considered critical to an understanding of our consolidated financial statements for the year ended December 31, 2023 and unaudited condensed consolidated financial statements for the nine-month period ended September 30, 2024 as they require the application of subjective and complex judgment, involving critical accounting estimates and assumptions impacting our consolidated financial statements for the year ended December 31, 2023 and unaudited condensed consolidated financial statements for the nine-month period ended September 30, 2024.
The critical accounting estimates relate to the following:
Share-Based Compensation
Share-based compensation expense related to share awards is recognized based on the fair value of the awards granted. The fair value of each option award is estimated on the grant date using the Black-Scholes option pricing model. The Black-Scholes option pricing model requires the input of highly subjective assumptions, including the fair value of the underlying ordinary shares (see below), the expected term of the option, and the expected volatility of the price of our ordinary shares. These estimates involve inherent uncertainties and the application of management’s judgment. The related share-based compensation expense is recognized on a straight-line basis over the requisite service period of the awards, including awards with graded vesting and no additional conditions for vesting other than service conditions. Forfeitures are accounted for as they occur instead of estimating the number of awards expected to be forfeited.
Our use of the Black-Scholes option-pricing model requires the input of highly subjective assumptions. If factors change and different assumptions are used, our share-based compensation expense could be materially different in the future.
We will continue to use judgment in evaluating the assumptions related to our share-based compensation on a prospective basis. As we continue to accumulate additional data related to our ordinary shares, we may refine our estimation process, which could materially impact our future share-based compensation expense.
Valuations of instruments convertible to our Ordinary and Preferred Shares
Our preferred shares are classified as temporary equity, as they include clauses that could constitute as in-substance redemption clauses that are outside our control. Warrants and other instruments over our Preferred Shares are classified as liabilities under ASC 480 and measured at fair value, with subsequent changes in fair value recognized in the statements of operations in each period.
The fair value of our preferred shares underlying our convertible instruments was determined by our board of directors, after considering contemporaneous third-party valuations and input from management. In the absence of a public trading market, our board of directors, with input from management, exercised significant judgment and considered various objective and subjective factors to determine the fair value of our equity including the following factors:
| | ● | History of transactions in our preferred shares; |
| | ● | Probability of an IPO scenario (including de SPAC transaction); |
| | ● | Probability of other liquidation events; |
| | | |
| | ● | Expected time to liquidation; and |
| | ● | Expected return on equity. |
The resulting equity value was then allocated to each share class based on differences in liquidation preferences of the various share classes using an Option Pricing Model (“OPM”) through the use of a series of call options and by implementing a Monte Carlo simulation. The OPM allocates the overall company value to the various share classes based on differences in liquidation preferences, using a series of call options. The OPM is appropriate to use when the range of possible future outcomes is difficult to predict. Starting from 2023, for the IPO scenario (including de SPAC transaction) we utilized a probability-weighted expected return method (“PWERM”) to allocate value among the various share classes. The PWERM involves the estimation of the value of our company under multiple future potential outcomes and estimates the probability of each potential outcome. After the value of each applicable class of shares was determined, a discount for lack of marketability (“DLOM”) was applied to arrive at the fair value of the ordinary shares on a non-marketable basis. A DLOM is applied in order to reflect the lack of a recognized market for a closely held interest and the fact that a non-controlling equity interest may not be readily transferable. A market participant purchasing this share would recognize this illiquidity associated with the shares, which would reduce the overall fair market value.
In some cases, we considered the amount of time between rounds of financing to determine whether to use a mid price calculation between two dates.
Upon completion of the Business Combination, our ordinary shares are publicly traded, and we rely on the closing price of our ordinary shares as reported on the date of the applicable valuations.
The fair value of the warrants for the purchase of preferred shares was calculated using the OPM as part of the allocation of the equity value to the various share classes (as described above). The critical accounting estimates for the valuation of those warrants include (a) the fair value of the underlying preferred shares, as mentioned above, and (b) volatility — based on peer companies’ volatility.
Recent Accounting Pronouncements
See Note 2 on page F-75 to our financial statements for the year ended December 31, 2023 included in this prospectus for a description of recent accounting pronouncements applicable to our financial statements for the year ended December 31, 2023.
Smaller Reporting Company Status
We are a “smaller reporting company,” meaning that the market value of our ordinary shares held by non-affiliates is less than $700 million and our (via its Silexion subsidiary) annual revenue was less than $100 million during the most recently completed fiscal year. We will continue to be a smaller reporting company if either (i) the market value of our shares held by non-affiliates is less than $250 million or (ii) our annual revenue was less than $100 million during the most recently completed fiscal year and the market value of our shares held by non-affiliates is less than $700 million. As a smaller reporting company, we may choose to present only the two most recent fiscal years of audited financial statements and we have reduced disclosure obligations regarding executive compensation.
Emerging Growth Company Status
Section 102(b)(1) of the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, exempts emerging growth companies from being required to comply with new or revised financial accounting standards until private companies are required to comply with the new or revised financial accounting standards. The JOBS Act provides that a company can choose not to take advantage of the extended transition period and comply with the requirements that apply to non-emerging growth companies, and any such election to not take advantage of the extended transition period is irrevocable.
We are an “emerging growth company” as defined in Section 2(a) of the Securities Act, and has elected to take advantage of the benefits of the extended transition period for new or revised financial accounting standards. We will remain an emerging growth company until the earliest of (i) the last day of the fiscal year in which the market value of ordinary shares that is held by non-affiliates exceeds $700 million as of the end of that year’s second fiscal quarter, (ii) the last day of the fiscal year in which we have total annual gross revenue of $1.235 billion or more during such fiscal year (as indexed for inflation), (iii) the date on which we have issued more than $1.0 billion in non-convertible debt in the prior three-year period, or (iv) December 31, 2025. We expect to continue to take advantage of the benefits of the extended transition period, although it may decide to early adopt such new or revised accounting standards to the extent permitted by such standards. This may make it difficult or impossible to compare our financial results with the financial results of another public company that is either not an emerging growth company or is an emerging growth company that has chosen not to take advantage of the extended transition period exemptions because of the potential differences in accounting standards used.
BUSINESS
Unless the context otherwise requires, in this “Business” section, the terms “we,” “us” and “our” generally refer to Silexion Therapeutics Ltd., an Israeli company (“Silexion”), or, from and after the Business Combination, to Silexion Therapeutics Corp (formerly known as Biomotion Sciences), a Cayman Islands exempted company (“New Silexion”).
Business Overview
We are a clinical-stage, oncology-focused biotechnology company engaged in the discovery and development of proprietary treatments for KRAS-driven cancers. The KRAS gene is an oncogene that is involved in the regulation of cell division as a result of its ability to relay external signals to the cell nucleus. Based on our research of refractory solid tumor cancers, we are actively developing a platform focused on the silencing of the KRAS oncogene using RNA-interference therapeutics. Our lead product candidate, SIL-204B, consists of locally administered small interfering RNAs, or siRNA, in an extended-release formulation, as a first-line treatment of locally advanced pancreatic cancer patients, or LAPC, in combination with standard-of-care chemotherapy.
The KRAS oncogene is considered to be the most common oncogenic gene driver in human cancers, and the most notable in pancreatic, lung, and gastrointestinal (GI) (including colorectal, esophagus, stomach, small bowel, and appendix) cancers. Considered a challenging therapeutic target due to its intrinsic characteristics, recent advances have been made at directly inhibiting the KRAS proteins produced by the mutated gene. Our platform is designed to silence the gene, and thus prevent the production of the harmful mutated KRAS proteins driving the growth of cancerous tumors.
We are currently focused on treatment for pancreatic cancer (PC) tumors bearing the KRAS G12D or KRAS G12V mutations where metastases have not been detected and are non-resectable, i.e. they are not able to be surgically removed. For our first indication, we are targeting the largest and least treatable form of localized pancreatic tumors referred to as locally advanced pancreatic cancer. LAPC represents approximately 30% of the total pancreatic cancer population. We are currently developing SIL-204B, a second-generation siRNA product candidate following a Phase 1 and Phase 2 clinical trial with our first-generation siRNA product candidate, siG12D-LODER, which we also refer to as Loder. Results from the Phase 2 clinical trial showed a trend for differences between treatment groups in patients with the KRAS G12D/V mutation, with the Loder arm suggesting an overall survival advantage of 9.3 months.
SIL-204B has been designed to optimize Loder with the aim of improving uptake into tumor cells, enhancing stability, and improving the delivery system. We plan to conduct a Phase 2/3 prospective, randomized, controlled, multinational, two-arm, open-label trial in LAPC subjects that harbor the KRAS G12D/V mutations to evaluate the efficacy, safety and tolerability of SIL-204B administered intratumorally in combination with standard of care (SoC) chemotherapy versus SoC chemotherapy only. In support of our planned Phase 2/3 trial, we held a meeting with the Federal Institute for Drugs and Medical Devices in Germany (BfArM) to discuss the planned design of the Phase 2/3 trial at which BfArM agreed, in principle, to the design. In preparation for the study, we plan to initiate toxicology studies of SIL-204B in 2025 followed by the regulatory submission in the second half of 2025 to initiate the Phase 2/3 trial and trial initiation in the first half of 2026. At this time, we are focused on the further development of the core siRNA technology underlying the Loder and SIL-204B as well as the clinical development of SIL-204B as the most optimized version of the technology.
Our Market Opportunity
Activating mutations in KRAS are among the most prevalent oncogenic driver mutations in human cancers. In a recent study of over 400,000 patients with various cancer type malignancies, 23% of adult pan-cancer samples had KRAS alterations, 88% of which were mutations, most commonly G12D, G12V, G12C, G13D and G12R, making the KRAS target a sought-out target in for many cancers (“Comprehensive pan-cancer genomic landscape of KRAS altered cancers and real-world outcomes in solid tumors”, by Jessica K. Lee, etc., NPJ Precision Oncology 2022; 6: 91). In addition, the study found that various cancers have an amplification of the non-mutated KRAS protein. Tumor types with a high prevalence of KRAS mutations included pancreatic ductal adenocarcinoma (PDAC) (92%), colorectal cancer (CRC) (49%), and non-squamous non-small cell lung cancer (NSCLC) (35%). These three cancers represent 71% of the KRAS mutant pan-tumor population studied.
The following chart shows the distribution of various alternations of the KRAS oncogene in various type of cancers:
Pancreatic Cancer
By the next decade, pancreatic cancer is expected to become the second most deadly cancer. Every year in the U.S. approximately 50,000 people die from pancreatic cancer, while approximately 61,000 new patients are diagnosed with pancreatic cancer annually (American Cancer Society Statistics 2023).


Studies have shown that pancreatic cancer patients have short survival rates compared to other cancer types (see, for example, “Prognosis and survival analysis of patients with pancreatic cancer: retrospective experience of a single institution”, by Qi Li and others, World Journal of Surgical Oncology, 2022; 20:11). There are four basic forms of pancreatic cancer: Resectable, those where a surgeon can remove a tumor; Borderline Resectable (BRPC) where the tumor is not currently permissible for surgery but prior treatment with chemotherapy or radiation could in some cases allow for surgical removal; LAPC where the tumor has surrounded more than half of a major artery or vein and is not surgically removable; and Metastatic where the cancerous tumor has spread to other organs. Of the forms of pancreatic cancer that are localized (those which have not yet spread), the largest and most life threatening is LAPC, which constitutes approximately 30% of pancreatic cancers (“Locally Advanced Pancreatic Cancer: A Review of Local Ablative Therapies”, by Alette Ruarus and others, Cancers BaseI, January 2018; 10(1):16).
Below is a curve of overall survival in LAPC patients:
Fig. Comparison of overall survival between different chemotherapy regimens in non-resected locally advanced pancreatic cancer patients. CHT indicates chemotherapy; FFX, FOLFIRINOX; Gem, gemcitabine. Reference Gemenetzis, G et al. 2019. Annals of Surgery. 270 (2):340
KRAS mutations across their various forms are responsible for approximately 92% of all pancreatic cancers, of which the KRAS G12D and KRAS12V mutations account for over 70% of the cases traced to KRAS mutations (Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531(7592), 47 – 52. https://doi.org/10.1038/nature16965 (2016) (Art. No. 7592)). Unfortunately, those with LAPC have a short survival time, about 17 months, and constitute about 30% of the total pancreatic cancer population (“Survival in Locally Advanced Pancreatic Cancer After Neoadjuvant Therapy and Surgical Resection”, by Georgio Gemenetzis, MD, and others, Annals of Surgery. 270 (2):1, March 2018). Among those patients with KRAS mutations, those with the mutation type G12D and G12V show the shortest OS among the G12x mutation type.
Distribution of KRAS Mutations in Pancreatic Cancer
Our Technology
Our research is focused on the development of a platform of therapeutics that is designed to silence the KRAS oncogene using small interfering RNA, or siRNA. This function is called interference RNA (RNAi). When the RNAi is double stranded of 19-25 nucleotides (NTs), in length, it is referred to as siRNA. This class of siRNA therapeutics exert their effect by inducing the enzymatic breakdown of the messenger (mRNA) of a targeted gene inhibiting the process called translation, which turns the message (mRNA) into a protein. The general mechanism for the silencing the oncogene is actually an evolutionary process developed by cells to protect against viruses.
We believe our approach also builds upon the validation of our target KRAS mutations as a target for cancers, as seen with the two small molecule KRAS inhibitors currently on the market for non-small cell lung cancer, and the validation of siRNA technology, as it is currently on the market for five non-oncological indications. None of these agents is appropriate for our intended primary indication, but we believe they do support our premises regarding target (KRAS) and basic technology (siRNA) for use in the oncological area. A key distinction between our technology and the existing inhibitors of KRAS is that our siRNA technology prevents the production of the protein, as indicated by a decrease in the mRNA and physiological decrease in human tumor cell line growth shown in our preclinical studies. By contrast, the existing inhibitors and to our knowledge, those in development inhibit its activity, and clinical use of the existing inhibitors has shown that they potentially leave large unmet needs.
The specific mechanism of this silencing activity is depicted in the figure below. siRNAs, usually 19-25 NTs, enter the cell as a double-standard complex. Once in the inner cell matrix, they bind to an RNA-induced silencing complex of enzymes (RISC), which splits the siRNA into two single RNA strands referred to as the passenger (sense) strand and the guide (antisense) strand. It is the antisense which is the active part. The single guide strand has complementary binding to the target mRNA and thereby acts as an inducing guide for other enzymes in the RISC complex to bind and induce specific cleavage of the now double stranded target mRNA. As the siRNA guide strand is designed to be complementary to the RNA message around the site of the mutation of the gene to be silenced, in our case the mutated KRAS oncogene, once the message (mRNA) is destroyed, the oncogene is silenced. As the sense-strand of the siRNA is designed to be specific for the KRAS, there is a specificity to the silencing of this driver of cancer.

Our first-generation siRNA product candidate, siG12D, is an extended-release formulation of siRNA. While originally designed to combat the KRAS G12D mutation in patients with LAPC, siG12D was shown to have silencing activity in other KRAS mutations including G12V as well as to a lesser degree in G12C and G12R. The product candidate is comprised of the anti-KRAS(G12D) siRNA drug substance (siG12D), formulated in a biodegradable polymeric matrix (PLGA) as solid rods in order to obtain an extended-release profile. To overcome the difficulties of a systemic drug to enter the pancreatic tumor environment and to obtain a sufficiently high level of siRNA in the pancreatic cell without inducing unnecessary side effects, the siRNA is directly delivered intratumorally using a standard ultrasound guided endoscopy (EUS). Since patients should undergo anesthesia prior to the endoscopy, the same as that required for a biopsy, the siRNA has been formulated as an extended-release formulation to increase the time between administrations. We refer to this product formulation as siG12D-LODER or Loder. Loder has undergone extensive pre-clinical testing as well as two open-label clinical trials.
Our second-generation siRNA product candidate, SIL-204B, is an optimized form of the first-generation Loder comprised of an anti-KRAS siRNA drug substance (SIL-204) encapsulated into a biodegradable polymeric matrix (PLGA). While pre-clinical testing of SIL-204B has shown silencing activity of KRAS mutations including G12D, G12V, G12C and G12R in varying degrees, it is designed to mainly combat the KRAS G12D and KRAS G12V mutations in patients with LAPC where silencing activity is more enhanced. The second-generation siRNA drug substance aims to provide improved uptake into tumor cells by introduction of a hydrophobic tail that enhances movement across the cell membrane. Additionally, second-generation siRNA drug substance includes modified nucleotides in the siRNA that enhance stability and corresponding half life. Most significantly, the delivery system has been improved in which PLGA-rods used in Loder have been replaced by PLGA-microparticles, allowing the drug substance to be injected as a suspension, facilitating administration through smaller needles. As with Loder, the administration is by ultrasound guided endoscopy (EUS), of the type typically used for pancreatic biopsies to diagnose pancreatic cancer and can be done by a typical gastrointestinal endoscopist.
As discussed further below, Silexion plans to conduct a Phase 2/3 clinical trial of SIL-204B targeting KRAS G12D and G12V mutations. The targeting of additional mutations is expected to require additional clinical trials.
We believe the optimization of Loder allows for more of a personalized medicine approach to the dosing, allowing the siRNA dose to be adjusted to the tumor size. The small volume of suspension is expected to allow for treatment of smaller tumors than those treated with Loder, and to continue treatment for a longer period of time with tumors shrinking in size. Furthermore, delivery of the extended-release PLGA formulated siRNA, SIL-204B, to be administered using a much smaller needle is expected to allow more flexibility and decrease the risk associated with administration.
��
Pre-Clinical Studies
We have conducted the evaluation of our first-generation siRNA product candidate, Loder, in pharmacology, pharmacokinetics (PK), and toxicology studies with both the siG12D and siG12D-LODER. The results of the extensive in vitro and in vivo nonclinical studies conducted with Loder have demonstrated its effects on the KRAS oncogene, tumor development, and the development of new metastases. In these studies, the mechanism of action (MOA) was elucidated and the effect of Loder on a variety of downstream processes, ultimately leading to cell apoptosis, tumor necrosis, and halting of new metastases, was investigated.
Pre-clinical data show that the siG12D silencing is very specific and effective (peak mRNA degradation effect in vivo was found to be 98%), can down-regulate signaling pathways, reduce pancreatic cancer cell viability, decelerate cancer cell migration, and suppress expression of genes involved in Epithelial-to-Mesenchymal Transition (EMT). It was demonstrated in vivo that Loder protects siG12D against degradation. The product candidate induces apoptosis and necrosis throughout the entire tumor, hinders the appearance of metastases, decelerates the development and growth of pancreatic tumors, induces an innate immune response locally within the tumor and extends survival.
siG12D was also found to be additive with FOLFIRINOX, Gemcitabine, and Gemcitabine+nab-Paclitaxel. When administered into the tumor, siG12D molecules are in part spontaneously taken up into tumor cells; the siG12D drug is distributed from the siG12D-LODER throughout the entire tumor, by diffusion and convection, and covers the entire tumor after about a week, increasing the “void volume” in the tumor microenvironment.
PK studies yielded validated methods for detecting siG12D in rat and human plasma, and confirmed distribution and absorption at the local tissue level.
The toxicological study confirmed siG12D tolerability, with minimal, not drug related, focal effect at the implantation site and No Observed Adverse Effect Level (NOAEL) 24.2 times higher than the dose in the Loder Phase 2 trial.
Taken together, these converging effects on multiple genetic, cellular, tumor tissue, tumor microenvironment, systemic levels and survival aspects act to produce a durable and prolonged anti-tumor effect both in in vitro and in vivo pancreatic cancer models.
We have also conducted pre-clinical studies of our second-generation siRNA product candidate, SIL-204B. There are three key preclinical studies which support our advancing SIL-204B into a planned Phase 2/3 clinical trial.
The first key clinical study was a dose response analysis of SIL-204 in mouse Hepa1-6 cells together with a relevant Dual Glo reporter plasmid. SIL-204 in this study showed inhibition of the drug substance across KRAS mutations G12D, G12V, G12C and G12R and the wild type to cover the KRAS amplifications. Similar to the results found with the siRNA in the Loder (siG12D) three of the mutations (G12D, G12V, and G12R) were highly inhibited. However, whereas G12C was mildly silenced (inhibited) with siG12D, it was significantly more inhibited with SIL-204 which has implications for potentially broadening the indications for SIL-204B to include all four of the major KRAS mutations and the wild-type. Additionally in this model, when a prototype of SIL-204 was tested for its silencing activity with and without the lipid lead we are conjugating to the siRNA with SIL-204, the results showed that the lipid-conjugated siRNA enhanced the silencing activity of the siRNA by two-fold. The siG12D (siRNA in the Loder) does not have the lipid conjugate.
The second key pre-clinical study examined the stability of the guide strand of SIL-204 in human serum. Whereas siG12D was completely broken down (enzymatically cleaved) in < 1 hour, SIL-204 showed complete stability at 48 hours. Consistently, when SIL-204 was administered to rats via subcutaneous injection, the plasma half-life was 5 hours. In contrast when siG12D was administered systemically to mice the half life was 1 minute.
In a third key pre-clinical study, the human pancreatic cell line CAPAN-1 harboring the KRAS G12V mutation was xenografted (transplanted) to mice and the tumor growth and necrosis (death of body tissue) in the tumor was observed following daily treatments of SIL-204 compared to the controls (untreated and vehicle treated mice). Results showed a significant growth of the tumor in the control mice while in the SIL-204-treated mice, the tumor growth was significantly curtailed, as indicated by a significant reduction in the tumor volume and weight. In addition, when analyzing tumor slices from the center of the tumor, the results showed a statistically significant increase in tumor necrosis in SIL-204 treated mice compared to untreated- and vehicle-treated mice.
Clinical Studies
Phase 1 Clinical Study
An escalating dose, multicenter Phase 1 clinical study of Loder was conducted in Israel from 2011 to 2015 at three sites enrolling a total of 15 patients with unresectable LAPC. The study was designed as an open-label, single-arm, single-administration of siG12D-LODER (concomitantly with standard of care chemotherapy), dose-escalating Phase 1 study, enrolling LAPC patients with a target tumor accessible for intra-tumor administration, with a Karnofsky performance status equal or greater than 70% and with a life expectancy of at least three months. Single escalating doses of LODER (0.025, 0.75, 1.5 or 3 mg) by insertion into the tumor were administered by standard EUS procedures. Following insertion, patients received standard of care chemotherapy as first line treatment in accordance with treating physician recommendation, Gemcitabine (nine patients) or modified FOLFIRINOX (four patients), while one patient refused to receive any chemotherapy. After study drug administration, the patients were to be monitored for signs of systemic or local treatment- related toxicity. Active follow-up comprised scheduled physical and clinical examinations, vital sign measurements, laboratory tests, abdominal CT and disease assessments.
The primary endpoint of the study was to assess the safety and tolerability of siG12D-LODER, and to define the dose-limiting toxicities (DLT) of siG12D-LODER. Secondary endpoints included the recommended Phase 2 dose and defining the maximum tolerated dose (MTD). In the event of surgery, assessment of siG12D-LODER local distribution and efficacy were to be based on histopathology measurements and RNA analysis. Progression free survival (PFS) and overall survival (OS) were to be assessed only by long term follow-up. Due to the small size of the trial, it was not powered for statistical significance. The study met its primary endpoints. The 15 patients reported a total of 125 adverse events (AEs). 111 of the reported AEs (89%) were grade 1 and 2, which were transient and resolved. The most common AE (grade 1-2) was diarrhea which was reported by seven (46.7%) patients, abdominal pain and nausea were both reported by six (40%) patients, whereas fatigue was reported by five (33.3%) patients. Thirteen severe AEs of grade 3 and one grade 4 severe were reported by 10 (66.7%) patients. The most common grade 3-4 AEs were neutropenia and cholangitis reported by three patients and two patients, respectively. Pancreatitis was reported by the investigator as possibly related to the procedure. One adverse event (cholangitis) in one patient was considered as definitely related to the EUS intervention. At the doses tested, no DLTs were observed and therefore if there was a DLT it was defined as being greater than 3mg, the highest dose administered. The study also met the secondary endpoint of establishing the Phase 2 dose of up to 3mg however the MTD was not determined.
Evidence of tumor response was observed, noting that none of the patients presented progressive disease (PD) and tumor response durability was demonstrated. The tumor marker CA19-9 decreased in 70% of relevant cases soon after siG12D-LODER administration and time to metastasis (TTM) and overall survival were comparable to (or better than) the historical data of the disease in a similar population. However, no clear dose response was observed between the dose of siG12D-LODER and OS or TTM probably due to several factors, including the small size of the study, the single dosing administration, without repeat dosing, and the level of dose at the low-dose cohort, which was selected to be ~5 times higher than then the minimal effective dose level as predicted in pre-clinical studies.
Phase 2 Clinical Study
From 2018 to 2023, we conducted a prospective, multi-center, Phase II, open label study to evaluate the efficacy, safety and tolerability of siG12D-LODER in two separate cohorts across five sites in Israel and four in the U.S.
Cohort 1 was a randomized and controlled two-arm study of 37 subjects with unresectable LAPC to assess the efficacy, safety, tolerability and pharmacokinetics of siG12D-LODER when used in combination with standard chemotherapy treatment (gemcitabine + nab-Paclitaxel) compared to gemcitabine + nab-Paclitaxel alone in subjects. Cohort 2 was a single arm study of 22 subjects with unresectable and borderline resectable LAPC to assess the efficacy, safety, and tolerability of siG12D-LODER in combination with standard of care chemotherapy treatment (gemcitabine + nab-paclitaxel or FOLFIRINOX or modified FOLFIRINOX).
For comparative purposes, we evaluated the randomized section of the trial for efficacy (Cohort 1) and both cohorts (Cohort 1 and Cohort 2) for safety. All patients who met the inclusion criteria were accepted to the trial, regardless of whether they had a KRAS mutation or which specific mutation they had.
The KRAS mutation status was however determined post-hoc in a sample of 31 patients (21 of whom were Loder treated) in the table below:
KRAS
G12x
Mutation | | Cohort 1
Arm 2
(Control) | | Cohort 1
Arm 1
(Treatment) | | Cohort 1
% Arm 1
Tx | | Cohort 2
(Treatment) | | All
Treated
% |
| R | | 5/10 | | 1/12 | | 8 | | 2/9 | | 26 (8/31) |
| D | | 2/10 | | 3/12 | | 25 | | 2/9 | | 23 (7/31) |
| V | | 3/10 | | 8/12 | | 67 | | 5/9 | | 52 (16/31) |
A total of up to eight Loders (2.8 mg) were inserted into the pancreatic tumor per single administration. Insertion was done using EUS. The trial consisted of a screening period (28 days), a treatment phase (12-week Loder treatment cycles at investigator’s discretion with concomitant chemotherapy treatment cycles) and a follow-up phase (up to six months until end of study which is defined as death, withdrawn consent or lost to follow-up). For efficacy, the primary endpoint in the randomized section of the trial (Cohort 1) was overall survival (OS), defined as the time that passed from study entry (screening visit) until death from any cause. For Cohort 2, the primary endpoint was the overall response rate (ORR) by end of treatment; ORR was defined as the proportion of subjects with best overall confirmed response (BOCR) of either a complete response (CR) or partial response (PR). Concerning the secondary endpoint of safety, the endpoints were incidence of adverse events (AEs), and serious adverse events (SAEs) overall, by severity, by relationship to each study intervention, and those that led to discontinuation of study interventions. Other secondary endpoints included ORR for Cohort 1, progression free survival, time to metastatis, time to response, duration of response and rate of disease control.
Overall, a total of 59 subjects with LAPC were enrolled in the study, 38 were treated with Loder and 48 subjects overall (81.3%) completed the study. In Cohort 1, 15/19 (78.9%) completed the Loder arm and 11/18 (61.1%) completed the standard of care arm. In Cohort 2, 21/22 (95.5%) completed the study.
In Cohort 1, OS was comparable between the two treatment arms. Median time to death was 691 days in the Loder-treated group, compared with 666 days in the standard of care group. The Hazard Ratio (Loder/SoC) was 1.47 (95% CI; 0.643, 3.365) with a non-significant p-value of 0.361 for the treatment difference. In the sub-group of subjects with D or V KRAS mutation (n=16), the median OS was 691 days for the 11 Loder treated subjects, compared to 409 days for the five SoC subjects. The Hazard Ratio was 0.59 (95% CI; 0.179, 1.963) with a non-significant p-value of 0.393. In the subjects who were not found to have D or V KRAS mutation (n=13), the median OS in the seven Loder treated subjects was 637 days compared to 937 days in the six SoC treated subjects. The hazard ratio was 2.87 (95% CI; 0.705, 11.650) with a p-value of 0.141. The ORR was 44.4% (8/18) in the Loder group compared to 27.3% in the SoC group (3/11). No subject had a CR in either group. The ORR in subjects with D or V KRAS mutation was 63.6% (7/11) in the Loder group compared to 20% in the SoC group (1/5).
In Cohort 2, the ORR was 31.6% reflecting 6/19 subjects with partial responses. No subject had CR. In subjects with D or V KRAS mutation, the ORR was 57.1% (4/7), however, these subjects also had a shorter median OS compared to subjects without these mutations (414 days vs. 671 days).
As depicted below, the survival probability in subjects of Cohort 1 bearing a KRAS G12D/V mutation compared to the control group, showed a median survival in the Loder group (n=11) of 22.8 months compared to 13.8 months in the control group (n=5).
Based on the above, although the primary endpoints of all patients were not met, the results showed a trend for differences between treatment groups in patients with the KRAS G12D/V mutation, with the Loder arm suggesting an overall survival advantage of 9.3 months. With respect to the secondary efficacy endpoints, no statistically significant differences were observed or there was insufficient data to evaluate although with respect to ORR in Cohort 1, a positive trend in the KRAS G12D/V mutation subgroup was observed. Due to the small size of the trial, it was not powered for statistical significance.
The secondary safety endpoints were met. In Cohort 1, all subjects in the Loder treatment group reported at least one moderate or severe treatment emergent adverse event (TEAE) (18 subjects). One subject was reported with a TEAE leading to death (sepsis). This event was deemed as not related to the Loder or the EUS procedure. Of the remaining 17 subjects, one subject was reported with a life-threatening TEAE, gastrointestinal disorders (colitis). The majority of moderate and severe TEAEs reported in the Loder treatment group were attributed to gastrointestinal disorders (10), blood/lymphatic disorders (14), general disorders and administration site conditions (11), metabolism and nutrition disorders (11), and investigations (10). In the SoC treatment group, all subjects reported at least one moderate or severe TEAE (11 subjects). No subjects were reported with a TEAE leading to death. Two subjects were reported with a life- threatening TEAE. The majority of moderate and severe TEAEs reported in the SoC treatment group were attributed to gastrointestinal disorders (10), blood/lymphatic disorders (9), general disorders and administration site conditions (9), metabolism and nutrition disorders (8), and investigations (9).
In Cohort 2, all subjects in the Loder treatment group reported at least one moderate or severe TEAE (20 subjects). One subject was reported with a TEAE leading to death (gastrointestinal disorder). This event was deemed as not related to the Loder or the EUS procedure. Three subjects were reported with a life-threatening TEAE. Of the three subjects, one subject was investigation (neutrophil count decreased) and two subjects were metabolism/nutrition (hyperglycaemia, hyperkalaemia). The majority of moderate and severe TEAEs reported in the Loder treatment group were attributed to gastrointestinal disorders (13), nervous system disorder (10) and investigations (11).
Manufacturing
We rely on and intend to continue to rely on third-party contract manufacturing organizations for drug substance and drug products for our clinical trials. We have agreements with contract manufacturers for the manufacturing of SIL-204B for clinical development.
Future Development Plans
We plan to conduct a Phase 2/3 prospective, randomized, controlled, multinational, two-arm, open-label trial in LAPC subjects that harbor the KRAS G12D/V mutations to evaluate the efficacy, safety and tolerability of SIL-204B administered intratumorally in combination with SoC chemotherapy versus SoC chemotherapy only. The study design, including the dose and dosing frequency, the combination with SoC chemotherapy, the proposed target population and safety monitoring is based on the experience with the preceding Phase 1 and 2 clinical studies with the first-generation siRNA product candidate, Loder.
The study is initially planned to enroll approximately 350 subjects and has an adaptive design with several segments; a screening period which includes a biopsy eligibility test for G12D/V mutations, a CT scan and physiologic profiling to ensure subjects are qualified for treatment; concurrent with this, all subjects will receive run-in SoC chemotherapy; a Phase 2 safety run-in and treatment period where initially 30 subjects will be randomized 2:1 to receive SIL-204B and SoC Chemotherapy or SoC Chemotherapy at which point a Data and Safety Monitoring Board (DSMB) will perform a safety review and recommend whether the study is safe to proceed; a Phase 2 expansion period to continue enrollment; and when approximately 60 events have occurred, an unblinded interim analysis for sample size re-estimation and futility will be conducted, to expand the trial from Phase 2 to Phase 3, which is expected to happen approximately one year from start of enrollment. Following the interim analysis, the study will continue into a Phase 3 treatment period to complete the enrollment of the remaining subjects for a total treatment period of 24 months. The study design is powered for statistical significance.
In support of our planned Phase 2/3 trial of SIL-204B, we held a meeting with the Federal Institute for Drugs and Medical Devices in Germany (BfArM) to discuss the planned design of the Phase 2/3 trial, at which BfArM agreed, in principle, to the design. In preparation for the study, we plan to initiate toxicology studies of SIL-204B in 2025 followed by the regulatory submission in late 2025 to initiate the Phase 2/3 trial. Currently, we are upscaling SIL-204 and planning for GMP production. At this time, Silexion is focused on the further development of the core siRNA technology underlying the Loder and SIL-204B as well as the clinical development of SIL-204B as the most optimized version of the technology.
We expect to apply in 2024 for Orphan Drug Designation in both the U.S. and EU. In the U.S., Orphan Drug designation by the FDA gives a company exclusive marketing rights for a seven-year period, along with other benefits to recoup the costs of researching and developing drugs to treat rare diseases. In the EU, a company receives data exclusivity for 10 years which provides protection from similar drugs being approved. We believe the size of the localized pancreatic cancer market fits the requirements for this designation however there can be no assurance that we will be granted such designation and such designation neither shortens the development time or regulatory review time of a drug nor does it increase the likelihood for any approval in the regulatory review process. Our continued development plans for SIL-204B are not dependent on whether we are granted Orphan Drug Designation.
Our Strategy
Our goal is to have a positive impact on the health and treatment of KRAS driven cancer patients, in general and initially with pancreatic cancer through the continued development and commercialization of our pipeline. Key elements of our strategy to advance toward this goal include the following:
| | ● | Advancing the clinical development of SIL-204B for the treatment of LAPC. Our Phase 2 trial with our first-generation siRNA product, Loder in LAPC patients acts as a validation of approach and foundation for our continued development efforts. As further described in “Future Development Plans”, Silexion plans to initiate toxicology studies of SIL-204B in 2025 followed by the regulatory submission in late 2025 to initiate a Phase 2/3 trial of SIL-204B powered for statistical significance. At this time, Silexion is focused on the further development of the core siRNA technology underlying the Loder and SIL-204B as well as the clinical development of SIL-204B as the most optimized version of the technology. |
| | ● | Leveraging our platform to other oncological indications harboring the KRASG12D/V mutation. |
| | ● | Advancing SIL-204B to commercialization. We have assembled a world class clinical advisory board for better understanding the market in the U.S. and EU. |
| | ● | Forming strategic alliances and collaborating with partners to augment our capabilities. We may pursue strategic alliances with other biopharmaceutical companies with well-established presences in the specialties we aim to target for our indications. This may include co-marketing, co-promotion, and co-development relationships, or a partnership with a diagnostics company to help improve availability of rapid testing. We also intend to explore options to work with partners to augment the study and treatment of patients and the impact of our product candidates, including medical professionals, healthcare professional networks, pharmacy benefit managers, insurance companies, and artificial intelligence companies. |
Silexion was established as Silenseed Ltd in 2008 as an Israeli company which underwent a name change to Silexion Therapeutics Ltd. in May 2023. On June 13, 2014, Silexion filed a registration statement on Form F-1 with the SEC, in contemplation of an initial public offering, and the listing of its shares on Nasdaq. Subsequently, the public offering process was abandoned because our board of directors was not satisfied with our valuation in the offering, and no securities were sold under the registration statement. During April 2022, our management was replaced following a health condition of the then-founder and CEO of Silexion, and the new management, after evaluating the viability of commercializing the Loder, has decided to pivot from the first-generation siRNA product candidate to the second-generation siRNA product candidate and develop SIL-204.
Competition
The biotechnology and pharmaceutical industries, and the oncology sector, are characterized by a rapid evolution of technologies, fierce competition and strong defense of intellectual property rights. While we believe that our discovery programs and technology provide us with competitive advantages, we face competition from major biotechnology and pharmaceutical companies, academic institutions, governmental agencies and public and private research institutions, among others.
Any product candidates that we successfully develop and commercialize will compete with currently approved therapies and new therapeutics that may become available in the future. Key product features that would affect our ability to effectively compete with other therapeutics include efficacy, safety and convenience of our products as compared to other available therapeutics.
There are a large number of companies developing or marketing treatments for cancer, including major biotechnology and pharmaceutical companies. These treatments consist of novel treatments based on small molecule drug products, cell-based therapies, as well as traditional chemotherapy treatments. There are also an increasing number of companies commercializing treatments and/or developing programs specifically targeting KRAS mutations, including KRAS G12D and KRAS G12V, in a variety of manners and for a variety of indications, including cancer, including Amgen Inc., Bristol-Myers Squibb Company (through the recently acquired Mirati Therapeutics, Inc.), Revolution Medicines, Inc., AstraZeneca (in collaboration with Usynova), BioNTech, Roche, Merck/Moderna, Boehringer and Gilead. Smaller and other early-stage companies may also prove to be significant competitors. In addition, academic research departments and public and private research institutions may be conducting research on compounds that could prove to be competitive.
Many of the companies against which we may compete have significantly greater financial resources and expertise in the research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Similar or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel, and establishing clinical trial sites and patient registration for clinical trials, as well as acquiring technologies complementary to, or necessary for, our programs.
Our competitors have already and/or may obtain more rapidly than we may obtain approval FDA, or other regulatory approval for commercialization of product. As a result, our competitors could establish a strong market position before we are able to enter the market with our products. The availability of coverage and reimbursement from government and other third-party payors for competing products at the time of commercialization of our products will also significantly affect the pricing and competitiveness of our products.
Intellectual Property
Our business depends, in part, on our ability to develop and maintain the proprietary aspects of our products.
We are seeking patent protection for our product (SIL-204) in International Patent Application No. PCT/IL2023/051276 filed on December 14, 2023 (the “276 Application”)
The minimum expiration date of any patent that is issued from the 276 Application is December 14, 2043. However, given typical practice with respect to regulatory-related patent term extensions, this date may be extended up to an additional five years in many countries, depending on the length of the regulatory process, to a maximum final expiration date of December 14, 2048.
The 276 Application was published on June 20, 2024 as International Patent Publication No. WO 2024/127405.
In addition to patent laws, we rely on copyright and trade secret laws to protect our proprietary rights. We also attempt to protect our trade secrets and other proprietary information through agreements with vendors, employees, and consultants.
Property and Facilities
Our principal executive officers are currently located in Modiin, Israel, where we lease a space of 450 square meters consisting of offices, under a lease agreement that will expire on July 31, 2025.
Employees
As of the date of this prospectus, we have three full time and ten part time employees. All of our employees are based in Israel. None of our employees are represented by labor unions or covered by collective bargaining agreements. We believe that we maintain good relations with all of our employees.
Grants from the Israeli Innovation Authority
During 2009 to 2020, we received several approvals from the IIA for participation in research and development activities performed by us in a total amount of $5.8 million.
The Company is obligated to pay royalties to the IIA amounting to 3%-5% of the sales of all of its product candidates and other related revenues generated from such projects, up to 100% of the grants received, linked to the U.S. dollar and bearing interest at the rate of LIBOR. The obligation to pay these royalties is contingent upon actual sales of the products and, in the absence of such sales, no payment is required. In October 2023, it was published that the interest rate of the grants will be replaced with the 12-month term SOFR published on the first trading day of each calendar year.
As of December 31, 2023, the total royalty amount that may be payable by the Company is approximately $5.8 million ($6.4 million including interest).
Legal Proceedings
From time to time, we may become involved in actions, claims, suits, and other legal proceedings arising in the ordinary course of our business, including assertions by third parties relating to intellectual property infringement, breaches of contract or warranties, or employment-related matters. We are not currently subject to any material legal proceedings.
Regulatory Environment
Government Regulation
Clinical trials, the drug approval process, and the marketing of drugs are intensively regulated in the United States and in all major foreign countries. Government authorities in the United States (including federal, state, and local authorities) and in other countries (including federal, state, and local authorities) extensively regulate, among other things, the manufacturing, research and clinical development, marketing, labeling and packaging, storage, distribution, post-approval monitoring and reporting, advertising and promotion, pricing, and export and import of pharmaceutical products, such as those we are developing. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. Government Regulation
In the United States, the Food and Drug Administration, or FDA, regulates drugs under the Federal Food, Drug, and Cosmetic Act (FDCA) and related regulations and biologics under the FDCA and the Public Health Service Act (PHSA) and its implementing regulations. FDA approval is required before any new unapproved drug or dosage form, including a new use of a previously approved drug, can be marketed in the United States. Drugs and biologics are also subject to other federal, state and local statutes and regulations. Failure to comply with the applicable United States regulatory requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include the imposition by the FDA or an Institutional Review Board, or IRB, of a clinical hold on trials, the FDA’s refusal to approve pending applications or supplements, license suspension or revocation, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, manufacture and marketing of pharmaceutical products. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, distribution, record keeping, approval, advertising and promotion of our products.
The FDA’s policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of our platforms and candidate products or any future product candidates or approval of new disease indications or label changes. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or abroad.
Marketing Approval
The process required by the FDA before product candidates may be marketed in the United States, the European Medicines Agency, or EMA, before a product can be marketed in Europe, and Medicines & Healthcare products Regulatory Agency (MHRA) for the United Kingdom. Generally involves the following:
| | ● | completion of extensive preclinical laboratory tests and preclinical animal studies, all performed in accordance with the GLP regulations; |
| | ● | submission to the FDA of an investigational new drug application, or IND, Clinical Trial Application (CTA) for Europe which must become effective or approved before human clinical studies may begin and must be updated on a regular basis; |
| | ● | approval by an independent institutional review board, or IRB, or ethics committee representing each clinical site before each clinical study may be initiated; |
| | ● | performance of adequate and well-controlled human clinical studies to establish the safety and efficacy of the product candidate for each proposed indication; |
| | ● | preparation of and submission to the FDA of a new drug application, or NDA, or biologics license application, or BLA, or for Europe a Marketing Authorization Application (MAA) after completion of all pivotal clinical studies; |
| | ● | potential review of the product application by an FDA advisory committee, where appropriate and if applicable. In the EU, the Committee for Medicinal Products for Human Use (CHMP) issues a scientific opinion to the European Commission which issues the marketing authorization; |
| | ● | a determination by the FDA within 60 days of its receipt of an NDA or BLA to file the application for review; |
| | ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities where the proposed product drug substance is produced to assess compliance with cGMP; and |
| | ● | FDA review and approval of an NDA or BLA or marketing authorization in the European Union (EU) in all European Union Member States plus Norway, Iceland and Liechtenstein, prior to any commercial marketing or sale of the drug in the United States. Note that if the centralized procedure is used, which is mandatory for all new anticancer products, a marketing authorization is issued centrally by the EU commission, which is valid immediately in all member states of the EEA (EU plus Iceland, Norway, and Liechtenstein). |
The testing and approval process requires substantial time and financial resources, and we cannot be certain that any approvals for our candidate products will be granted on a timely basis, if at all.
An IND/CTA is a request for authorization from the FDA/national regulatory authorities in Europe to administer an investigational new drug product to humans. The central focus of an IND/CTA submission is on the general investigational plan and the protocol(s) for human studies. The IND and CTA also include results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational new drug. An IND or a CTA must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to the proposed clinical studies. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before clinical studies can begin. Accordingly, submission of an IND may or may not result in the FDA allowing clinical studies to commence. In Europe, a CTA is required to be approved by regulators, which may take several months. Accordingly, submission of a CTA to European regulators may or may not result in permission to commence clinical studies in Europe.
We will need to successfully complete an extensive additional clinical trial or some clinical trials in order to be in a position to submit a new drug application to the FDA. Our planned future clinical trials for our candidate products may not begin or be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including delays in:
| | ● | obtaining regulatory approval to commence a study; |
| | ● | reaching agreement with third-party clinical trial sites and their subsequent performance in conducting accurate and reliable studies on a timely basis; |
| | ● | obtaining institutional review board approval or an Ethics Committee approval to conduct a study at a prospective site; |
| | ● | recruiting patients to participate in a study; and |
We must reach agreement with the FDA/European national authorities on the proposed protocols for our future clinical trials in the United States and EU. A separate submission apart from any IND or initial CTA application we submit must be made for each successive clinical trial to be conducted during product development. Further, an independent IRB or EC for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that site. Informed consent must also be obtained from each study subject. Regulatory authorities, an IRB or EC, a data safety monitoring board or the Sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the participants are being exposed to an unacceptable health risk.
Clinical Studies
Clinical studies involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with current cGCP/GCP, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical studies are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety, and the efficacy criteria to be evaluated. A protocol for each clinical study and any subsequent protocol amendments must be submitted to the FDA and/or European national authorities as part of the IND or CTA. Additionally, approval must also be obtained from each clinical study site’s IRB or EC before the studies may be initiated, and the IRB/EC must monitor the study until completed. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries.
Our objective is to conduct additional clinical trials for our candidate products and, if those trials are successful, seek marketing approval from the FDA and other worldwide regulatory bodies.
For purposes of NDA approval, human clinical trials are typically conducted in phases that may overlap.
| | ● | Phase 1. The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| | ● | Phase 2. This phase involves trials in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| | ● | Phase 3. This phase involves trials undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population, often at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. |
| | ● | Phase 4. In some cases, the FDA or the EMA may condition approval of an NDA or BLA or MAA for a product candidate on the Sponsor’s agreement to conduct additional clinical studies after approval. In other cases, a sponsor may voluntarily conduct additional clinical studies after approval to gain more information about the drug. Such post-approval studies are typically referred to as Phase 4 clinical studies. |
A pivotal study is a clinical study that adequately meets regulatory agency requirements for the evaluation of a drug candidate’s efficacy and safety such that it can be used to justify the approval of the product. Generally, pivotal studies are Phase 3 studies, but the FDA or EMA may accept results from a Phase 2 study if such study’s design provides a well-controlled and reliable assessment of clinical benefit, particularly in situations where there is an unmet medical need and the results are sufficiently robust.
The FDA/EMA, the IRB/EC, or the clinical study sponsor may suspend or terminate a clinical study at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk.
Additionally, some clinical studies are overseen by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board or committee. This group provides authorization or recommendation for whether or not a study may move forward at designated check points based on access to certain data from the study. We may also suspend or terminate a clinical study based on evolving business objectives and/or competitive climate. All of these trials must be conducted in accordance with cGCP requirements in order for the data to be considered reliable for regulatory purposes.
The clinical study process can take several (2- 12 or more) years to complete, and there can be no assurance that the data collected will support FDA or EU Commission approval or licensure of the product. Government regulation may delay or prevent marketing of product candidates or new drugs for a considerable period of time and impose costly procedures upon our activities. We cannot be certain that the FDA or any other regulatory agency will grant approvals for our candidate products or any future product candidates on a timely basis, if at all. Success in early stage clinical trials does not ensure success in later stage clinical trials. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval.
The NDA Approval Process
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, detailed investigational new drug product information is submitted to the FDA in the form of an NDA or BLA requesting approval to market the product for one or more indications. Under federal law, the submission of most NDAs and BLAs is subject to an application user fee. For fiscal year 2014, the application user fee exceeds $2.1 million, and the Sponsor of an approved NDA or BLA is also subject to annual product and establishment user fees, set at $104,060 per product and $554,600 per establishment. These fees are typically increased annually. Applications for orphan drug products are exempted from the NDA and BLA user fees and may be exempted from product and establishment user fees, unless the application includes an indication for other than a rare disease or condition.
An NDA or BLA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational new drug product to the satisfaction of the FDA.
The FDA will initially review the NDA for completeness before it accepts it for filing. The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that the application is sufficiently complete to permit substantive review. After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. The FDA may refer applications for novel drug products or drug products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and, if so, under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Based on pivotal Phase 3 trial results submitted in an NDA, upon the request of an applicant, the FDA may grant a priority review designation to a product, which sets the target date for FDA action on the application at six months, rather than the standard ten months. Priority review is given where preliminary estimates indicate that a product, if approved, has the potential to provide a significant improvement compared to marketed products or offers a therapy where no satisfactory alternative therapy exists. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
After the FDA completes its initial review of an NDA, it will communicate to the sponsor that the drug will either be approved, or it will issue a complete response letter to communicate that the NDA will not be approved in its current form and inform the sponsor of changes that must be made or additional clinical, nonclinical or manufacturing data that must be received before the application can be approved, with no implication regarding the ultimate approvability of the application.
Before approving an NDA or BLA, the FDA will typically inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications.
Additionally, before approving an NDA, the FDA may inspect one or more clinical sites to assure compliance with GCPs. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable, it typically will outline the deficiencies and often will request additional testing or information. This may significantly delay further review of the application. If the FDA finds that a clinical site did not conduct the clinical trial in accordance with GCP, the FDA may determine the data generated by the clinical site should be excluded from the primary efficacy analyses provided in the NDA. Additionally, notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process for a drug requires substantial time, effort and financial resources, and this process may take several years to complete. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products.
The FDA may require, or companies may pursue, additional clinical trials after a product is approved. These so-called Phase 4 studies may be made a condition to be satisfied for continuing drug approval. The results of Phase 4 studies can confirm the effectiveness of a product candidate and can provide important safety information. In addition, the FDA now has express statutory authority to require sponsors to conduct post-market studies to specifically address safety issues identified by the agency.
Any approvals that we may ultimately receive could be withdrawn if required post-marketing trials or analyses do not meet the FDA requirements, which could materially harm the commercial prospects for our candidate products.
The FDA also has authority to require a Risk Evaluation and Mitigation Strategy, or REMS, from manufacturers to ensure that the benefits of a drug or biological product outweigh its risks. A sponsor may also voluntarily propose a REMS as part of the NDA submission. The need for a REMS is determined as part of the review of the NDA. Based on statutory standards, elements of a REMS may include “dear doctor letters,” a medication guide, more elaborate targeted educational programs, and in some cases restrictions on distribution. These elements are negotiated as part of the NDA approval, and in some cases if consensus is not obtained until after the PDUFA review cycle, the approval date may be delayed. Once adopted, REMS are subject to periodic assessment and modification.
Even if a product candidate receives regulatory approval, the approval may be limited to specific disease states, patient populations and dosages, or might contain significant limitations on use in the form of warnings, precautions or contraindications, or in the form of onerous risk management plans, restrictions on distribution, or post-marketing study requirements. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Delay in obtaining, or failure to obtain, regulatory approval for our candidate products, or obtaining approval but for significantly limited use, would harm our business. In addition, we cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
Expedited Review and Accelerated Approval Programs
A sponsor may seek approval of its product candidate under programs designed to accelerate FDA’s review and approval of NDAs and BLAs. For example, Fast Track Designation may be granted to a drug intended for treatment of a serious or life-threatening disease or condition that has potential to address unmet medical needs for the disease or condition. The key benefits of fast track designation are the eligibility for priority review, rolling review (submission of portions of an application before the complete marketing application is submitted), and accelerated approval, if relevant criteria are met. Based on results of the Phase 3 clinical study(ies) submitted in an NDA or BLA, upon the request of an applicant, the FDA may grant the NDA or BLA a priority review designation, which sets the target date for FDA action on the application at six months after the FDA accepts the application for filing. Priority review is granted where there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. If criteria are not met for priority review, the application is subject to the standard FDA review period of ten months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Under the accelerated approval program, the FDA may approve an NDA or a BLA on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Post-marketing studies or completion of ongoing studies after marketing approval are generally required to verify the drug’s clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit. In addition, the Food and Drug Administration Safety and Innovation Act (FDASIA), which was enacted and signed into law in 2012, established the new “breakthrough therapy” designation. A sponsor may seek FDA designation of its product candidate as a breakthrough therapy if the drug is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.
FDA Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
Drug manufacturers are subject to periodic unannounced inspections by the FDA and state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
We rely, and expect to continue to rely, on third parties for the production of clinical quantities of our product candidates, and expect to rely in the future on third parties for the production of commercial quantities. Future FDA and state inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA or BLA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information, imposition of post-market studies or clinical studies to assess new safety risks, or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| | ● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| | ● | fines, warning letters or holds on post-approval clinical studies; |
| | ● | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals; |
| | ● | injunctions or the imposition of civil or criminal penalties; or |
| | ● | product seizure or detention, or refusal to permit the import or export of products. |
The FDA strictly regulates marketing, labeling, advertising, and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
Orphan Designation and Exclusivity
The FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or, if it affects more than 200,000 individuals in the United States, when there is no reasonable expectation that the cost of developing and making the drug for this type of disease or condition will be recovered from sales in the United States.
Orphan drug designation entitles a party to financial incentives such as opportunities for grant funding of clinical study costs, tax advantages, and user-fee waivers. In addition, if a product receives FDA approval for the indication for which it has orphan designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity.
Patent Term Restoration
Depending upon the timing, duration, and specifics of the FDA approval of the use of our product candidates, U.S. patents that may be granted to us in the future may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA or BLA, plus the time between the submission date and the approval of that application. Only one patent applicable to an approved product is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent term for one of our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical studies and other factors involved in the filing of the relevant NDA or BLA.
Biosimilars and Exclusivity
The Patient Protection and Affordable Care Act, or Affordable Care Act, signed into law on March 23, 2010, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009 (BPCI Act), which created an abbreviated approval pathway for biological products shown to be similar to, or interchangeable with, an FDA-licensed reference biological product. This amendment to the PHSA attempts to minimize duplicative testing. Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. However, complexities associated with the larger, and often more complex, structure of biological products, as well as the process by which such products are manufactured, pose significant hurdles to implementation that are still being worked out by the FDA.
A reference biologic is granted twelve years of exclusivity from the time of first licensure of the reference product. The first biologic product submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against other biologics submitting under the abbreviated approval pathway for the lesser of (i) one year after the first commercial marketing, (ii) eighteen months after approval if there is no legal challenge, (iii) eighteen months after the resolution in the applicant’s favor of a lawsuit challenging the biologics’ patents if an application has been submitted, or (iv) 42 months after the application has been approved if a lawsuit is ongoing within the 42-month period.
Abbreviated New Drug Applications for Generic Drugs
In 1984, with passage of the Hatch-Waxman Act, Congress authorized the FDA to approve generic drugs that are the same as drugs previously approved by the FDA under the NDA provisions of the statute. To obtain approval of a generic drug, an applicant must submit an abbreviated new drug application (ANDA) to the agency. In support of such applications, a generic manufacturer may rely on the preclinical and clinical testing previously conducted for a drug product previously approved under an NDA, known as the reference listed drug (RLD).
Specifically, in order for an ANDA to be approved, the FDA must find that the generic version is identical to the RLD with respect to the active ingredients, the route of administration, the dosage form, and the strength of the drug. At the same time, the FDA must also determine that the generic drug is “bioequivalent” to the innovator drug. Under the statute, a generic drug is bioequivalent to an RLD if “the rate and extent of absorption of the [generic] drug do not show a significant difference from the rate and extent of absorption of the listed drug. . . .”
Upon approval of an ANDA, the FDA indicates that the generic product is “therapeutically equivalent” to the RLD and it assigns a therapeutic equivalence rating to the approved generic drug in its publication “Approved Drug Products with Therapeutic Equivalence Evaluations,” also referred to as the “Orange Book.” Physicians and pharmacists consider an “AB” therapeutic equivalence rating to mean that a generic drug is fully substitutable for the RLD. In addition, by operation of certain state laws and numerous health insurance programs, the FDA’s designation of an “AB” rating often results in substitution of the generic drug without the knowledge or consent of either the prescribing physician or patient.
The FDCA provides a period of five years of non-patent exclusivity for a new drug containing a new chemical entity. In cases where such exclusivity has been granted, an ANDA may not be filed with the FDA until the expiration of five years unless the submission is accompanied by a Paragraph IV certification, in which case the applicant may submit its application four years following the original product approval. The FDCA also provides for a period of three years of exclusivity if the NDA includes reports of one or more new clinical investigations, other than bioavailability or bioequivalence studies, that were conducted by or for the applicant and are essential to the approval of the application. This three-year exclusivity period often protects changes to a previously approved drug product, such as a new dosage form, route of administration, combination or indication.
Hatch-Waxman Patent Certification and the 30-Month Stay
Upon approval of an NDA or a supplement thereto, NDA sponsors are required to list with the FDA each patent with claims that cover the applicant’s product or a method of using the product. Each of the patents listed by the NDA sponsor is published in the Orange Book. When an ANDA applicant files its application with the FDA, the applicant is required to certify to the FDA concerning any patents listed for the reference product in the Orange Book, except for patents covering methods of use for which the ANDA applicant is not seeking approval.
Specifically, the applicant must certify with respect to each patent that:
| | ● | the required patent information has not been filed; |
| | ● | the listed patent has expired; |
| | ● | the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or |
| | ● | the listed patent is invalid, unenforceable or will not be infringed by the new product. |
A certification that the new product will not infringe the already approved product’s listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the applicant does not challenge the listed patents or indicates that it is not seeking approval of a patented method of use, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days after the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months after the receipt of the Paragraph IV notice, expiration of the patent, or a decision in the infringement case that is favorable to the ANDA applicant.
European Union/Rest of World Government Regulation
In addition to regulations in the United States, we will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical studies and any commercial sales and distribution of our products.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical studies or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical study application much like the IND prior to the commencement of human clinical studies. In the European Union, for example, a clinical study application, or CTA, must be submitted for each clinical protocol to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is accepted in accordance with a country’s requirements, the clinical study may proceed.
The requirements and process governing the conduct of clinical studies vary from country to country. In all cases, the clinical studies are conducted in accordance with cGCP, the applicable regulatory requirements, and the ethical principles that have their origin in the Declaration of Helsinki.
To obtain regulatory approval of an investigational medicinal product under European Union regulatory systems, we must submit a marketing authorization application. The content of the NDA or BLA filed in the United States is similar to that required in the European Union, with the exception of, among other things, country-specific document requirements.
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing product licensing, pricing, and reimbursement vary from country to country.
Countries that are part of the European Union, as well as countries outside of the European Union, have their own governing bodies, requirements, and processes with respect to the approval of pharmaceutical products. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Authorization Procedures in the European Union
Medicines can be authorized in the European Union by using either the centralized authorization procedure or national authorization procedures.
Centralized procedure
The EMA implemented the centralized procedure for the approval of human medicines to facilitate marketing authorizations that are valid throughout the European Economic Area, or EEA, which is comprised of the 27 member states of the European Union plus Norway, Iceland, and Lichtenstein. This procedure results in a single marketing authorization issued by the EMA that is valid across the EEA. The centralized procedure is compulsory for human medicines that are: derived from biotechnology processes, such as genetic engineering, contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune diseases and other immune dysfunctions, and officially designated orphan medicines.
For medicines that do not fall within these categories, an applicant has the option of submitting an application for a centralized marketing authorization to the European Commission following a favorable opinion by the EMA, as long as the medicine concerned is a significant therapeutic, scientific or technical innovation, or if its authorization would be in the interest of public health.
National authorization procedures
There are also two other possible routes to authorize medicinal products in several European Union countries, which are available for investigational medicinal products that fall outside the scope of the centralized procedure:
| | ● | Decentralized procedure. Using the decentralized procedure, an applicant may apply for simultaneous authorization in more than one European Union country of medicinal products that have not yet been authorized in any European Union country and that do not fall within the mandatory scope of the centralized procedure. |
| | ● | Mutual recognition procedure. In the mutual recognition procedure, a medicine is first authorized in one European Union Member State, in accordance with the national procedures of that country. Following this, further marketing authorizations can be sought from other European Union countries in a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization. |
In most cases, a Pediatric Investigation Plan, and/or a request for waiver or deferral, is required for submission prior to submitting a marketing authorization application. A PIP describes, among other things, proposed pediatric studies and their timing relative to clinical studies in adults.
New Chemical Entity Exclusivity
In the European Union, new chemical entities, sometimes referred to as new active substances, qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. This data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic (abbreviated) application for eight years, after which generic marketing authorization can be submitted, and the innovator’s data may be referenced, but not approved for two years. The overall ten-year period will be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
Orphan Designation and Exclusivity
In the European Union, the EMA’s Committee for Orphan Medicinal Products, or COMP, grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the European Union Community and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected). Additionally, designation is granted for products intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the medicinal product.
In the European Union, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity is granted following medicinal product approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity, and may be prolonged to a total of 12 years, if pediatric studies in accordance with a PIP are being performed.
Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Exceptional Circumstances/Conditional Approval
Orphan drugs or drugs with unmet medical needs may be eligible for EU approval under exceptional circumstances or with conditional approval. Approval under exceptional circumstances is applicable to orphan and non-orphan products and is used when an applicant is unable to provide comprehensive data on the efficacy and safety under normal conditions of use because the indication for which the product is intended is encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive evidence, when the present state of scientific knowledge does not allow comprehensive information to be provided, or when it is medically unethical to collect such information. Conditional marketing authorization is applicable to orphan medicinal products, medicinal products for seriously debilitating or life-threatening diseases, or medicinal products to be used in emergency situations in response to recognized public threats. Conditional marketing authorization can be granted on the basis of less complete data than is normally required in order to meet unmet medical needs and in the interest of public health, provided the risk-benefit balance is positive, it is likely that the applicant will be able to provide the comprehensive clinical data, and unmet medical needs will be fulfilled. Conditional marketing authorization is subject to certain specific obligations to be reviewed annually.
Accelerated Review
Under the Centralized Procedure in the European Union, the maximum timeframe for the evaluation of a marketing authorization application is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the EMA’s Committee for Medicinal Products for Human Use (CHMP)). Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, particularly from the point of view of therapeutic innovation. In this circumstance, EMA ensures that the opinion of the CHMP is given within 150 days, excluding clock stops.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we obtain regulatory approval. In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of coverage and reimbursement from third-party payors. Third-party payors include government authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the reimbursement rate that the payor will pay for the drug product. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drugs for a particular indication. Moreover, a payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. In order to obtain coverage and reimbursement for any product that might be approved for sale, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain regulatory approvals. Our product candidates may not be considered medically necessary or cost-effective. If third-party payors do not consider a product to be cost-effective compared to other available therapies, they may not cover the product after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit.
The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. By way of example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, collectively, the Healthcare Reform Law, contains provisions that may reduce the profitability of drug products, including, for example, increased rebates for drugs sold to Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. Adoption of government controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals.
In the European Community, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed to by the government. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical studies that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on cost containment measures in the United States and other countries has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Other Healthcare Laws and Compliance Requirements
If we obtain regulatory approval for any of our product candidates, we may be subject to various federal and state laws targeting fraud and abuse in the healthcare industry. These laws may impact, among other things, our proposed sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| | ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
| | ● | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent; |
| | ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
| | ● | the federal transparency laws, including the federal Physician Payment Sunshine Act, that requires drug manufacturers to disclose payments and other transfers of value provided to physicians and teaching hospitals; |
| | ● | HIPAA, as amended by HITECH and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; and |
| | ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
The Healthcare Reform Law broadened the reach of the fraud and abuse laws by, among other things, amending the intent requirement of the federal Anti-Kickback Statute and the applicable criminal healthcare fraud statutes contained within 42 U.S.C. § 1320a-7b, effective March 23, 2010. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, the Healthcare Reform Law provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act (discussed below) or the civil monetary penalties statute. Many states have adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
We are also subject to the Foreign Corrupt Practices Act, or FCPA, which prohibits improper payments or offers of payments to foreign governments and their officials for the purpose of obtaining or retaining business.
Safeguards we implement to discourage improper payments or offers of payments by our employees, consultants, and others may be ineffective, and violations of the FCPA and similar laws may result in severe criminal or civil sanctions, or other liabilities or proceedings against us, any of which would likely harm our reputation, business, financial condition and result of operations.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, exclusion from participation in government healthcare programs, such as Medicare and Medicaid and imprisonment, damages, fines and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Labeling, Marketing and Promotion
The FDA closely regulates the labeling, marketing and promotion of drugs. While doctors are free to prescribe any drug approved by the FDA for any use, a company can only make claims relating to safety and efficacy of a drug that are consistent with FDA approval, and the Company is allowed to actively market a drug only for the particular use and treatment approved by the FDA. In addition, any claims we make for our products in advertising or promotion must be appropriately balanced with important safety information and otherwise be adequately substantiated. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, injunctions and potential civil and criminal penalties. Government regulators recently have increased their scrutiny of the promotion and marketing of drugs.
Pediatric Research Equity Act
The Pediatric Research Equity Act (PREA) amended the FDCA to authorize the FDA to require certain research into drugs used in pediatric patients. The intent of PREA is to compel sponsors whose drugs have pediatric applicability to study those drugs in pediatric populations, rather than ignoring pediatric indications for adult indications that could be more economically desirable. The Secretary of Health and Human Services may defer or waive these requirements under specified circumstances.
Anti-Kickback and False Claims Laws
In the United States, the research, manufacturing, distribution, sale and promotion of drug products and medical devices are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare & Medicaid Services, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice, state Attorneys General, and other state and local government agencies. For example, sales, marketing and scientific/educational grant programs must comply with the Medicare-Medicaid Anti-Fraud and Abuse Act, as amended (the “Anti-Kickback Statute”), the False Claims Act, as amended, the privacy regulations promulgated under the Health Insurance Portability and Accountability Act, or HIPAA, and similar state laws. Pricing and rebate programs must comply with the Medicaid Drug Rebate Program requirements of the Omnibus Budget Reconciliation Act of 1990, as amended, and the Veterans Health Care Act of 1992, as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws.
In the United States, we are subject to complex laws and regulations pertaining to healthcare “fraud and abuse,” including, but not limited to, the Anti-Kickback Statute, the federal False Claims Act, and other state and federal laws and regulations. The Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid.
The federal False Claims Act prohibits anyone from knowingly presenting, or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services, including drugs, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services.
There are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. In addition, as discussed below, beginning in 2013, a similar federal requirement will require manufacturers to track and report to the federal government certain payments made to physicians and teaching hospitals made in the previous calendar year. These laws may affect our sales, marketing, and other promotional activities by imposing administrative and compliance burdens on us. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state, and soon federal, authorities.
Patient Protection and Affordable Health Care Act
In March 2010, the Patient Protection and Affordable Health Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, PPACA) was enacted, which includes measures that have or will significantly change the way health care is financed by both governmental and private insurers. The fees, discounts and other provisions of this law are expected to have a significant negative effect on the profitability of pharmaceuticals.
Many of the details regarding the implementation of PPACA are yet to be determined, and at this time, it remains unclear the full effect that PPACA would have on our business.
Other Regulations
We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future.
Israel
Clinical Testing in Israel
In order to conduct clinical testing on humans in the State of Israel, special authorization must first be obtained from the ethics committee and general manager of the institution in which the clinical studies are scheduled to be conducted, as required under the Guidelines for Clinical Trials in Human Subjects implemented pursuant to the Israeli Public Health Regulations (Clinical Trials in Human Subjects) 5741-1980, as amended from time to time, and other applicable legislation. These regulations require authorization by the institutional ethics committee and general manager as well as from the Israeli Ministry of Health, except in certain circumstances, and in the case of genetic trials, special fertility trials and complex clinical trials, an additional authorization of the Ministry of Health’s overseeing ethics committee. The institutional ethics committee must, among other things, evaluate the anticipated benefits that are likely to be derived from the project to determine if it justifies the risks and inconvenience to be inflicted on the human subjects, and the committee must ensure that adequate protection exists for the rights and safety of the participants as well as the accuracy of the information gathered in the course of the clinical testing. Since we perform a portion of the clinical studies on certain of our therapeutic candidates in Israel, we are required to obtain authorization from the ethics committee and general manager of each institution in which we intend to conduct our clinical trials, and in most cases, from the Israeli Ministry of Health.
New Silexion Corporate Information
New Silexion was formed on April 2, 2024 under the name Biomotion Sciences as a Cayman Islands exempted limited company for the purpose of effecting a business combination with Moringa and Silexion. On April 3, 2024, New Silexion entered into the Business Combination Agreement by and among New Silexion, Moringa, Silexion, and New Silexion’s two wholly-owned subsidiaries— Merger Sub 1 and Merger Sub 2. On the Closing Date of August 15, 2024, following the approval of the Business Combination (among other matters) at Moringa’s extraordinary general meeting that was held on August 6, 2024, the transactions contemplated by the Business Combination Agreement were completed. As a result, Moringa and Silexion merged with Merger Sub 2 and Merger Sub 1 pursuant to the SPAC Merger and Acquisition Merger, respectively, and became New Silexion’s wholly-owned subsidiaries, and their securityholders became securityholders of New Silexion at previously agreed-upon exchange ratios. In addition, New Silexion’s ordinary shares and warrants were listed, and began trading, on the Nasdaq Global Market on August 16, 2024, and its name was changed to Silexion Therapeutics Corp.
New Silexion’s principal executive offices are located at 12 Abba Hillel Road, Ramat Gan, Israel 5250606, and its phone number is +972-8-6286005. New Silexion’s corporate website address is www.silexion.com. Information contained on or accessible through that website is not a part of this prospectus, and the inclusion of that website address in this prospectus is an inactive textual reference only.
Properties
We currently lease 464 square meters of office space at 12 Abba Hillel Silver St, Ramat Gan, Israel. The lease term is for 24 months beginning on November 1, 2024 and ending on 31 October 2026, with an option to extend for an additional 24 months]. Monthly rent payments, including utilities, amount to approximately $12,839 per month
Access to Company Information
The Company files or furnishes periodic reports and amendments thereto, including its Annual Reports on Form 10-K, its Quarterly Reports on Form 10-Q and Current Reports on Form 8-K, proxy statements and other information with the SEC. In addition, the SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically. New Silexion’s internet address is https://www.silexion.com. The Company makes available, free of charge, its Annual Report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and all amendments to those reports as soon as reasonably practicable after such reports have been filed with or furnished to the SEC through its internet website.
MANAGEMENT
Management and Board of Directors
Our board of directors (sometimes referred to as the “New Silexion Board”) is comprised of the six directors listed below.
Each director will hold office until his or her successor is duly elected or appointed and qualified in accordance with applicable law or until his or her death, resignation or removal in accordance with law and our governing documents, including our Amended and Restated Memorandum and Articles of Association (the “Articles”), which went into effect upon the Closing of the Business Combination.
The following sets forth certain information known as of the date hereof concerning the persons who currently serve as directors and executive officers of New Silexion (who have served as such since the consummation of the Business Combination):
| Name | | Age | | Position(s) |
| Directors | | | | |
| Ilan Hadar | | 55 | | Chairman and Chief Executive Officer |
| Dror J. Abramov | | 63 | | Director |
| Ruth Alon | | 72 | | Director |
| Ilan Levin | | 58 | | Director |
| Avner Lushi | | 57 | | Director |
| Shlomo Noy | | 71 | | Director |
| Amnon Peled | | 65
| | Director |
| Executive Officers (who are not also directors) | | | | |
| Dr. Mitchell Shirvan | | 70 | | Chief Scientific and Development Officer |
| Mirit Horenshtein Hadar, CPA | | 40 | | EVP of Finance Affairs, Chief Financial Officer and Secretary |
Directors
Ilan Hadar, 55, was appointed as Chief Executive Officer of New Silexion, serving on a full-time basis, effective upon the Business Combination, and has served as our (previously, Silexion’s) Chairman of the Board since May 2024. Previously, he served as Managing Director of Silexion from April 2022 until the Business Combination. Mr. Hadar has over 20 years of multinational managerial and corporate experience with pharmaceutical and high-tech companies, as described below, over which time period he has acquired the experience and skills to serve as a valuable member of the board of directors of a company such as Biomotion Sciences. In addition to his current role at Silexion, Mr. Hadar has served as the Chief Executive Officer of Painreform Ltd (Nasdaq: PRFX) since November 2020, a position he will relinquish following the consummation of the Business Combination. Prior to joining Painreform and Silexion, Mr. Hadar served as Country Manager Israel and Chief Financial Officer at Foamix Pharmaceuticals Ltd. (currently, Nasdaq: VYNE) from 2014 until August 2020, where he was instrumental in building the organization and launching new innovative topical drugs in the U.S., and also focused on capital markets and mergers and acquisitions. Prior to his role at Foamix, Mr. Hadar was Finance Director at Pfizer PFR Pharmaceuticals Israel Ltd., where he oversaw all commercial, financial and operational activities of the local entity of the large pharmaceutical company. Before his tenure at Pfizer, Mr. Hadar served as Finance Manager at HP Indigo Ltd., a world-leading company in digital printing and, prior to that, served as Finance Director at BAE Systems, the third-largest defense company in the world, where he was responsible for all financial activities of BAE Systems Israel. From 1998 to 2006, Mr. Hadar was Chief Financial Officer at Mango DSP, a global leader of Intelligent Video Solutions. Mr. Hadar served on the board of directors of Kadimastem, a public Israeli biopharmaceutical company from 2019 to 2022. He received his MBA in Finance and Business Entrepreneurship and BA from The Hebrew University in Jerusalem, Israel. We believe Mr. Hadar is qualified to serve on our board of directors due to his extensive knowledge as Silexion’s Managing Director, and his extensive commercial, financial and managerial experience with high-tech and pharmaceutical companies, both private and public.
Dror J. Abramov, 63, was appointed as a director of New Silexion effective upon the consummation of the Business Combination. Mr. Abramov’s experience of over 15 years in multiple roles in changing and growing companies and markets within dynamic environments, and in particular his multi-disciplinary experience in R&D, consulting, taxes, sales, business development, finance and government, lend to his being an appropriate board member of Biomotion Sciences. Mr. Abramov has served as Managing Director of Hewlett Packard Inc. Israel since 2015. Mr. Abramov held several other management positions in Hewlett Packard Inc. Israel from 2006 to 2015, including finance director, general manager of imaging and printing divisions and general manager of printing and personal systems division. From 2002 through 2006, Mr. Abramov served as Chief Financial Officer of Applied Materials UK and Applied Materials Israel, both part of Applied Materials, Inc. (Nasdaq: AMAT), a nanomanufacturing company that supplies equipment, services and software for the manufacture of semiconductor chips for electronics. From 2000 through 2002, Mr. Abramov served as Vice President of business management at Avaya Communication, a cloud communications and workstream collaboration technology company. From 1997 until 2000, Mr. Abramov held positions in Mainsoft Corporation, first as director of finance and operations and later as general manager. From 1991 through 1997, Mr. Abramov served as consultant and manager at Maron, Sobel, Shor & Co., an Israeli CPA firm. Mr. Abramov is a licensed CPA and holds a Bachelor of Accounting, Master of Business Administration and Bachelor of Science in physics and computer sciences, all three degrees from Tel Aviv University. We believe Mr. Abramov is qualified to serve on our board of directors due to his extensive financial and business management experience.
Ruth Alon, 72, became a director of New Silexion effective upon the consummation of the Business Combination. Ms. Alon’s international experience of over 30 years in the high-tech medical industry and with Israeli life sciences companies, in particular, make her a prospective valuable member of the board of directors of Biomotion Sciences. Ms. Alon is the Founder and Chief Executive Officer of Medstrada, which was started in 2016. From 1997 until 2016, Ms. Alon served as a General Partner in Pitango Venture Capital, where she headed the life sciences activities and helped to facilitate the acquisition of several of the company’sportfolio companies. Currently, Ms. Alon also serves on the board of directors of a number of private and public companies as a member or chairperson, including Vascular Biogenics Ltd. (Nasdaq: VBLT), Brainsgate and KadimaStem. Ms. Alon previously worked on Wall Street where she held senior positions as a senior medical device analyst with Montgomery Securities (from 1981 to 1987) and Kidder Peabody & Co. (from 1987 to 1993). She also managed her own independent consulting business in San Francisco from 1995 to 1996, providing broad-based services to early-stage companies and venture capitalists in the medical devices industry. Ms. Alon was also instrumental in the establishment, in 2005, of Israel Life Science Industry (ILSI), a not-for-profit organization which represented, as of 2005, the mutual goals of approximately 700 Israeli life science companies. She is the Co-Founder of IATI, an umbrella organization established in 2012, representing Israel’s High Tech and Life Sciences industries. Ms. Alon holds a B.A. in Economics from the Hebrew University of Jerusalem, an M.B.A. from Boston University, and an M.Sc. from the Columbia University School of Physicians and Surgeons. We believe Ms. Alon is qualified to serve on our board of directors given her above-described extensive experience in the high-tech medical industry and with Israeli life sciences companies, in particular.
Ilan Levin, 58, who was the co-founder, Chairman and Chief Executive Officer of Moringa, was appointed as a director of New Silexion after its formation in April 2024 and has continued as a director following the consummation of the Business Combination. Mr. Levin has been involved, for approximately 25 years, as an executive and venture capital/private equity investor in high-tech, Israel-related ventures. His experience as an executive and board member in managing growth companies that develop technology, and his knowledge of that industry in Israel in particular, suit him well to serve as a director of Biomotion Sciences. From 2000 to 2018, Mr. Levin was a member of the Board and Executive Committee of Objet Ltd., which as a result of a merger with Stratasys, Inc. in 2012, formed Stratasys Ltd. (Nasdaq: SSYS), the pioneer and global leader in 3D printing. During his tenure at Objet/Stratasys, Mr. Levin held various positions including President, Vice Chairman and from 2016 to 2018, Chief Executive Officer. From 2004 to 2009, Mr. Levin was the Chief Executive Officer of CellGuide, a developer of software-based GPS for mobile devices. Since 1997, Mr. Levin has also served as a member of the board of directors and as an advisor for a wide variety of Israel-based technology-related companies, including currently serving as Chairman of Vision Sigma (TLV: VISN: IT), an Israel-based real estate and investment company. Early in his career, Mr. Levin was a practicing attorney focusing on corporate and securities related matters. In addition to his role as our Chairman and Chief Executive Officer, Mr. Levin also serves as the sole director and sole equity owner of an Israeli company that serves as the sole general partner of the Sponsor. Mr. Levin earned an LL.B. from Tel Aviv University and a B.A.Sc. in Industrial Engineering from the University of Toronto. We believe Mr. Levin is qualified to serve on our board of directors given his above-described experience in managing growth companies that develop technology, and his knowledge of that industry in Israel in particular.
Avner Lushi, 57, was appointed as a director of New Silexion effective upon the consummation of the Business Combination. His experience of over 20 years and skills acquired in his managing roles at an Israeli life sciences venture capital fund and in life sciences investment banking make him suitable to provide similar support as a member of the board of directors of Biomotion Sciences. Mr. Lushi co-founded Guangzhou Sino-Israel Bio-industry Investment Fund (GIBF), which currently includes two approximately $100 million funds focused on introducing Israeli and other foreign companies in the field of life sciences to the Chinese market, in which he also serves as a Managing Partner & CEO of the GP since 2016. From 2004 to 2015, Mr. Lushi served as a Partner and Managing Director of Israel Healthcare Ventures (IHCV), a prominent Israeli life sciences venture capital fund. Before joining IHCV, Mr. Lushi was the Co-Founder & CEO of Life Sciences Transaction Support Ltd. (LTS), a PwC subsidiary dealing with life sciences investment banking. Since 2005, Mr. Lushi has served as an independent director on the boards of nine public companies, including, currently, Brainsway Ltd. (Nasdaq: BWAY) and Ginegar Plastic Products Ltd. In addition, he serves as a board member of several private companies as part of his role at GIBF. From 1997 to 2001, prior to turning to the private sector, he held increasingly senior roles within the Israeli Prime Minister’s Chamber and the Israeli Supreme Court. Mr. Lushi holds an LLM in Law from the Hebrew University of Jerusalem, LLB in Law and a BA in Economics from the Haifa University. We believe Mr. Lushi is qualified to serve on our board of directors due to his extensive executive and board experience with life sciences companies.
Shlomo Noy, MD PhD, 71, was appointed as a director of New Silexion effective upon the consummation of the Business Combination. Professor Noy’s international-level expertise in healthcare management, tech transfer, building ecosystems and focusing hospitals on research and clinical trials will provide important knowledge to the Biomotion Sciences board of directors in the realm of Israeli research and clinical trial activities. Professor Noy has served as Chief Medical Officer of GIBF since January 2017. Professor Noy served as the Director of the Rehabilitation Hospital at Sheba Medical Center from 1993 to 2017 and Vice President of Research and Development and Academic Affairs at Sheba Medical Center from 2000 to 2017. Prof. Noy was a Professor at Sackler School of Medicine at Tel-Aviv University from 1993 to 2016. Prof. Noy serves as a board member of several private companies as part of his role at GIBF. Prof. Noy possesses 25 years’ experience in health care management and is active in promoting research and education at an Israeli national level as well as at an international level. Prof. Noy received his MD from the Hebrew University, Hadassah Medical School, Jerusalem, Israel, and completed his MBA at the European School of Business Administration (INSEAD) Fontainebleau, France, and holds a PhD degree from Tel-Aviv University, Faculty of Medicine and Management. We believe Mr. Noy is qualified to serve on our board of directors due to his extensive medical and health care management experience.
Amnon Peled, PhD, 65, was appointed as a director in December 2024. Professor Peled has served as an associate professor at Hadassah Medical Center, Jerusalem since August 2000. Professor Peled also served as Director of the Gene Therapy Institute at Hadassah Medical Center from October 2021 to February 2024. He specializes in cytokine research, hematopoietic stem cell biology, inflammation, and cancer, leading the development of therapies now in Phase II/III trials. Professor Peled served as Founder and CEO of Biokine Therapeutics, and as its Chief Scientific Officer, from July 2000 to February 2024, where he advanced the CXCR4 antagonist BKT140/BL8040. Prof. Peled is an author of over 100 publications and holder of 200+ patents and patent applications, and holds a Bachelor’s degree in agriculture from the Hebrew University of Jerusalem, a Master’s degree in cell biology and histology from Tel Aviv University, and a Ph.D. from the Weizmann Institute. He completed postdoctoral training at Harvard Medical School and the Weizmann Institute.
Executive Officers
Mitchell Shirvan, 70, has served as the Chief Scientific and Development Officer of New Silexion since the Business Combination, and, before the Business Combination, of Silexion, since April 2022. Prior to joining Silexion, Dr. Shirvan served as the Senior Vice President of R&D and V.P. Innovation and Discovery at Foamix Pharmaceuticals Ltd. from 2014 to 2019. Dr. Shirvan has over 25 years of industry experience, previously holding positions as Chief Executive Officer at Macrocure Ltd. from 2008 to 2012. From 1992 until 2008, Dr. Shirvan held various positions of increasing responsibility at Teva Pharmaceutical Industries, including Senior Director, Strategic Business Planning and Senior Manager, Research & Development. Prior to his tenure at Teva, he was a research fellow at the U.S. National Institutes of Health. Dr. Shirvan holds a Ph.D. in microbiology from The Hebrew University of Jerusalem and an MBA from the University of Bradford.
Mirit Horenshtein Hadar, 40, has served as the Chief Financial Officer and Secretary of New Silexion since the Business Combination, and served as the Executive Vice President of Finance Affairs at Silexion before the Business Combination, beginning in January 2024. From August 2023 to January 2024, Ms. Horenshtein Hadar served as a part-time consultant in a Strategy & Corporate Finance Advisory capacity. Ms. Horenshtein Hadar has over 15 years of corporate finance experience in senior financial positions of public companies and privately held companies, in the pharmaceutical and high-tech industries, where she has been instrumental in building financial infrastructures for growth, U.S. GAAP financial reporting and FP&A functions, and has led the accounting and reporting of complex M&A transactions, integration processes and public offerings. Prior to joining Silexion, from January 2021 to December 2022, Ms. Horenshtein Hadar served as VP of Finance and then CFO Israel of Gauzy Ltd. (currently, Nasdaq: GAUZ), a nanotechnology company that develops and markets smart glass and vision control technologies. Since December 2022, Ms. Horenshtein Hadar continues to serve as an external advisor to the finance department at Gauzy. Prior to Gauzy, Ms. Horenshtein Hadar served as Senior Director of Finance and Head of FP&A, Accounting and Financial Reporting at Foamix Pharmaceuticals Ltd. (currently, Nasdaq: VYNE) from July 2016 until December 2020. Prior to Foamix, Ms. Horenshtein Hadar was a Senior Manager at PwC Israel, as an external auditor, from 2008 to 2016. Ms. Horenshtein Hadar became a Qualified CPA in 2011 and received a BA in Accounting, Economics and Business Management from Tel Aviv University.
Family Relationships
Mirit Horenshtein Hadar and Ilan Hadar are married to one another. There are no other family relationships between the individuals who serve as directors and executive officers of the Company.
Corporate Governance Practices
Overall
The Company does not qualify as a “foreign private issuer” under U.S. securities laws and is therefore not currently eligible to exempt itself from any of the Nasdaq listing rule requirements in a manner that other non-U.S. issuers often do.
Composition of the New Silexion Board After the Business Combination
New Silexion’s business and affairs are managed under the direction of the New Silexion Board. Under the terms of the Articles, the New Silexion Board may be composed of between three and nine directors, as may be amended from time to time exclusively by ordinary resolution under Cayman Islands law, being a resolution passed by a simple majority of the shareholders of New Silexion as, being entitled to do so, vote in person or by proxy at a general meeting (an “Ordinary Resolution”). The size of the New Silexion Board was set by our shareholders as seven members effective upon the Closing. Ilan Hadar serves as Chairman of the New Silexion Board. Subsequent to the Closing, Ilan Shiloah stepped down from the Board, citing competing demands on his time, thereby leaving the current size of the Board as six members.
Pursuant to the Articles, New Silexion’s directors are appointed by an Ordinary Resolution at an annual general meeting. Unless the Board resolves that the election of nominees of the Board (referred to as “Nominees”) or of nominees of any shareholders entitled to present such nomination (referred to as “Alternate Nominees”) will be determined by plurality vote, the Nominees or Alternate Nominees shall be appointed by Ordinary Resolution at the annual general meeting at which they are proposed for election.
Officers
New Silexion’s Chief Executive Officer is responsible for the company’s day-to-day management. The Chief Executive Officer is appointed by, and serves at the discretion of, the New Silexion Board, subject to his employment agreement. All other executive officers are proposed for appointment by the Chief Executive Officer, subject to approval by the New Silexion Board, and will be subject to the terms of any applicable employment or consulting agreements that Silexion may enter into with them.
Director Independence
Nasdaq listing standards require that a majority of the New Silexion Board be independent. An “independent director” is defined generally as a person who has no material relationship with the listed company (either directly or as a partner, shareholder or officer of an organization that has a relationship with the listed company) that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. In making these determinations, the New Silexion Board considers the current and prior relationships that each non-employee director has with the Company and all other facts and circumstances the New Silexion Board deems relevant in determining their independence, including the beneficial ownership of our securities by each non-employee director and the transactions described in the section titled “Certain Relationships and Related Party Transactions.”
The New Silexion Board has determined that each of Messrs. Dror J. Abramov, Ilan Levin, Avner Lushi, Amon Peled and Shlomo Noy, and Ms. Ruth Alon, meets the definition of “independent director” as defined in Nasdaq listing standards. For purposes of SEC rules applicable to members of the audit committee and compensation committee, each of Messrs Abramov and Peled and Ms. Alon are deemed independent.
Board Committees
The New Silexion Board has established an audit committee, a compensation committee and a nominating and corporate governance committee, each of which has the composition and the responsibilities described below. Each of these committees operates under a written charter, approved by the New Silexion Board and effective upon the Closing, that satisfies the applicable Nasdaq rules, copies of which are available on the investor relations portion of our website. Members will serve on these committees until their resignation or until otherwise determined by the New Silexion Board. The New Silexion Board may establish other committees as it deems necessary or appropriate from time to time.
Audit Committee
Our audit committee consists of Dror J. Abramov, Amnon Peled and Ruth Alon, with Mr. Abramov serving as chair. Ilan Shiloah had served as the third member of the audit committee from the Closing until his resignation from the New Silexion Board on September 16, 2024. Rule 10A-3 of the Exchange Act and the Nasdaq listing standards require that our audit committee be composed entirely of independent members. The Nasdaq listing standards require that the audit committee be composed of at least three members. With the resignation of Mr. Siloah from the New Silexion Board and its audit committee, we will have until the earlier of our 2025 annual general meeting or one year after Mr. Shiloah’s resignation to restore the size of the audit committee to three members. The New Silexion Board has determined that each of Messrs Abramov and Peled and Ms. Alon meets the definition of “independent director” for purposes of serving on the audit committee under Rule 10A-3 of the Exchange Act and the Nasdaq listing standards and also meets the financial literacy requirements of the Nasdaq listing standards. In addition, the New Silexion Board has determined that Mr. Abramov qualifies as an “audit committee financial expert” within the meaning of the SEC regulations.
The primary purpose of the audit committee is to discharge the responsibilities of the New Silexion Board with respect to our corporate accounting and financial reporting processes, systems of internal control and financial statement audits and to oversee our independent registered public accounting firm. The principal functions of the audit committee include, among other things:
| | ● | helping the New Silexion Board oversee our corporate accounting and financial reporting processes; |
| | ● | managing the selection, engagement, qualifications, independence, and performance of a qualified firm to serve as the independent registered public accounting firm to audit our financial statements; |
| | ● | reviewing and discussing the scope and results of the audit with the independent registered public accounting firm, and reviewing, with management and the independent accountants, our interim and year-end operating results; |
| | ● | obtaining and reviewing a report by the independent registered public accounting firm at least annually that describes our internal quality control procedures, any material issues with such procedures and any steps taken to deal with such issues when required by applicable law; |
| | ● | establishing procedures for employees to submit concerns anonymously about questionable accounting or audit matters; |
| | ● | overseeing our policies on risk assessment and risk management; |
| | ● | overseeing compliance with our code of business conduct and ethics; |
| | ● | reviewing related person transactions; and |
| | ● | approving or, as required, pre-approving audit and permissible non-audit services to be performed by the independent registered public accounting firm. |
Compensation Committee
Our compensation committee consists of Dror J. Abramov, Amon Peled and Ruth Alon. The chair of the compensation committee will be chosen from among the committee’s members. The New Silexion Board has determined that each of Messrs. Abramov and Peled and Ms. Alon meets the definition of “independent director” for purposes of serving on the compensation committee under the Nasdaq listing standards, including the heightened independence standards for members of a compensation committee.
The primary purpose of our compensation committee is to discharge the responsibilities of the New Silexion Board in overseeing our compensation policies, plans and programs and to review and determine the compensation to be paid to our executive officers, directors and other senior management, as appropriate. The principal functions of the compensation committee include, among other things:
| | ● | reviewing, approving and determining, or making recommendations to the New Silexion Board regarding the compensation of our chief executive officer, other executive officers and senior management; |
| | ● | reviewing, evaluating and recommending to the New Silexion Board succession plans for our executive officers; |
| | ● | reviewing and recommending to the New Silexion Board the compensation paid to our non-employee directors; |
| | ● | administering our equity incentive plans and other benefit programs; |
| | ● | reviewing, adopting, amending and terminating incentive compensation and equity plans, severance agreements, profit sharing plans, bonus plans, change-of-control protections and any other compensatory arrangements for our executive officers and other senior management; and |
| | ● | reviewing and establishing general policies relating to compensation and benefits of our employees, including our overall compensation philosophy. |
Corporate Governance and Nominating Committee
We have appointed a corporate governance and nominating committee that currently consists of Dror J. Abramov, Ilan Levin, Amnon Peled and Ruth Alon. The chair of the committee will be chosen from among the committee’s members. The New Silexion Board has determined that each of Messrs. Abramov, Peled and Levin, and Ms. Alon, meets the definition of “independent director” under the Nasdaq listing standards.
Our corporate governance and nominating committee is responsible for, among other things:
| | ● | identifying and evaluating candidates, including the nomination of incumbent directors for reelection and nominees recommended by shareholders, to serve on the New Silexion Board; |
| | ● | considering and making recommendations to the New Silexion Board regarding the composition and chairmanship of the committees of the New Silexion Board; |
| | ● | instituting plans or programs for the continuing education of the New Silexion Board and the orientation of new directors; |
| | ● | developing and making recommendations to the New Silexion Board regarding corporate governance guidelines and matters; |
| | ● | overseeing our corporate governance practices; |
| | ● | overseeing periodic evaluations of the New Silexion Board’s performance, including committees of the New Silexion Board; and |
| | ● | contributing to succession planning. |
Compensation Committee Interlocks and Insider Participation
None of the members of Silexion’s compensation committee is currently, or has been at any time, one of Silexion’s, Moringa’s or Silexion’s executive officers or employees. None of our executive officers serves as a member of the board of directors or compensation committee (or other committee performing equivalent functions) of any entity that had one or more executive officers serving on our board of directors or compensation committee during 2023.
Code of Business Conduct and Ethics
Prior to the completion of the Business Combination, we adopted a written code of business conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. A copy of the code is posted on the investor relations portion of our website. In addition, we intend to post on our website all disclosures that are required by law or Nasdaq listing standards concerning any amendments to, or waivers from, any provision of the code.
EXECUTIVE COMPENSATION
Silexion
Unless the context requires otherwise, references in this “Executive Compensation” section to “we,” “our,” “us” and the “Company” generally refer to our company on a consolidated basis, for all periods following the completion of the Business Combination (on August 15, 2024), and to our wholly-owned subsidiary, Silexion Therapeutics Ltd., an Israeli company (“Silexion”), and its subsidiaries, for all periods prior to the completion of the Business Combination.
Overview
The following tables and accompanying narrative set forth information about the compensation, for 2024, provided to our chief executive officer (or person serving in an equivalent position) and the two most highly compensated executive officers (other than our chief executive officer) who were serving as executive officers as of December 31, 2024, each of whom served as an executive officer of Silexion prior to the completion of the Business Combination. These executive officers consist of: Ilan Hadar, who was Silexion’s Managing Director and now serves as New Silexion’s Chairman and Chief Executive Officer; Mirit Horenshtein Hadar, who was Silexion’s EVP Finance and now serves as New Silexion’s Chief Financial Officer; and Dr. Mitchell Shirvan, who was Silexion’s— and is now New Silexion’s— Chief Scientific and Development Officer. These executive officers are referred to in this section as our “named executive officers” or “NEOs.”
This discussion may contain forward-looking statements that are based on our current plans, considerations, expectations and determinations regarding future compensation programs, in the period immediately following the Business Combination. Actual compensation programs that we adopt in the future may differ materially from the plans summarized in this discussion.
Summary Compensation Table
The following table presents summary information regarding the total compensation for services rendered in all capacities that was awarded to, earned by, or paid to our named executive officers for 2024.
| Name and Principal Position | | Year | | Base Gross
Salary
($)(1) | | | Stock
Awards
($) | | | All Other
Compensation
($)(1)(2) | | | Total
($)(1) | |
Ilan Hadar
Chief Executive Officer (formerly Managing Director of Silexion)(3) | | 2024 | | | 240,560 | | | | 1,192,785 | | | | 107,283 | | | | 1,540,628 | |
| | 2023 | | | 182,976 | | | | - | | | | 70,638 | | | | 253,614 | |
Mirit Horenshtein Hadar
Chief Financial Officer (formerly EVP Finance of Silexion)(4) | | 2024 | | | 233,532 | | | | 447,291 | | | | 89,267 | | | | 770,090 | |
| | | 2023 | | | 26,238 | | | | - | | | | - | | | | 26,238 | |
Dr. Mitchell Shirvan
Chief Scientific and Development Officer(5) | | 2024 | | | 190,286 | | | | 1,043,699 | | | | 67,505 | | | | 1,301,490 | |
| | | 2023 | | | 156,140 | | | | - | | | | 50,660 | | | | 206,800 | |
| (1) | Amounts reported for the named executive officer and paid in New Israeli Shekels are converted from New Israeli Shekels to U.S. dollars using the 2024 and 2023 (as applicable) average exchange rates as published by Bank of Israel of 3.699 and 3.689 New Israeli Shekels, respectively, to 1 U.S. Dollar. |
| (2) | The amounts in this column include payments for a leased car or car maintenance, contributions to a pension fund, compensation fund, and continuing education fund, or payments in lieu of a continuing education fund. |
| (3) | This was for a part-time (75%) position prior to, and a full-time position following, completion of the Business Combination. |
| (4) | For 2023, Ms. Horenshtein Hadar’s compensation was for a period of 4.5 months during which she served as a part-time consultant in a Strategy & Corporate Finance Advisory capacity. |
| (5) | This was for a part-time (80%) position prior to, and a full-time position following, completion of the Business Combination. |
Narrative Disclosure to Summary Compensation Table
Base Salary
The named executive officers receive base salaries to compensate them for services rendered to us. The base gross salary payable to each named executive officer is intended to provide a fixed component of compensation reflecting the executive’s skill set, experience, role, and responsibilities. The annual base salaries for Ilan Hadar, Mirit Horenshtein Hadar and Dr. Mitchell Shirvan, for 2024 and 2023 were $1,540,628 and $253,614 for Mr. Hadar, $770,090 and $26,238 for Ms. Horenshtein Hadar, and $1,301,490 and $206,800 for Dr. Shirvan, respectively.
Equity Compensation
From time to time, we have granted equity awards under the Silexion Therapeutics Ltd. 2013 Equity Incentive Plan, which was replaced by the Silexion Therapeutics Ltd. 2023 Equity Incentive Plan (the “2013 Plan” and “2023 Plan”, respectively, collectively referred to as the “Silexion Plans”), as incentives to attract, retain and motivate our named executive officers. During 2024, we granted restricted share units (RSUs) that could be settled for 16,821, 6,308 and 14,718 ordinary shares of Silexion, to Ilan Hadar, Mirit Horenshtein Hadar, and Dr. Mitchell Shirvan, respectively (each such number of Silexion ordinary shares does not reflect the exchange ratio in the Business Combination or the subsequent reverse share split of New Silexion). The vesting of those RSUs accelerated upon the Closing of the Business Combination, entitling each such officer to receive New Silexion ordinary shares in accordance with the exchange ratio upon the Closing.
During 2022, Silexion granted 32,400 and 16,200 options to purchase Silexion ordinary shares to Ilan Hadar and Dr. Mitchell Shirvan, respectively (each such number of options does not reflect the equity exchange ratio in the Business Combination or the subsequent reverse share split of New Silexion). Vesting of those options was to occur over a period of 48 months, provided that Ilan Hadar or Dr. Mitchell Shirvan, as applicable, remains engaged by Silexion (or a Silexion affiliate), and is subject to the Silexion Plans. Upon consummation of an initial public offering of Silexion’s securities or an M&A Transaction (as defined in Silexion’s articles of association), which included the Business Combination, the vesting of those options was to accelerate, such that all unvested options were to immediately vest, provided that such IPO or M&A Transaction was to be consummated after January 1, 2023.
Employment Agreements
Following the Closing, Silexion has entered or will enter into employment agreements with each of its NEOs.
Silexion 2013 Equity Incentive Plan and Silexion 2023 Equity Incentive Plan
Silexion (and, following the Business Combination, New Silexion) has maintained the Silexion Plans in order to provide additional incentives for employees, directors and consultants, and to provide incentives to attract, retain and motivate eligible persons whose present and potential contributions are important to Silexion’s (and New Silexion’s) success. The 2013 Plan was adopted on July 25, 2013 and was replaced by the 2023 Plan, which was adopted on April 4, 2023. For a complete description of the 2013 Plan and the 2023 Plan, please see “Silexion Share Incentive Plans” below.
The Silexion Plans provide for grants of options to purchase ordinary shares of Silexion (following the Business Combination, New Silexion), shares, restricted shares and RSUs. As described above, during 2022, Silexion granted option awards under the Silexion Plans to Ilan Hadar and Dr. Mitchell Shirvan and during 2024, Silexion granted RSUs to Ilan Hadar, Mirit Horenshtein Hadar, and Dr. Mitchell Shirvan, respectively.
Immediately prior to the effective time of the Acquisition Merger, all outstanding Silexion options and Silexion RSUs accelerated and became fully vested (and, in the case of RSUs, settled for underlying shares). At the effective time of the Acquisition Merger, all Silexion options outstanding immediately prior to the Acquisition Merger automatically and without any action on the part of any Silexion option holder or beneficiary thereof, were assumed by New Silexion, and each such Silexion option was converted into an option to purchase New Silexion ordinary shares (based on the equity exchange ratio for Silexion under the Business Combination Agreement).
In addition, immediately prior to the completion of the Business Combination, New Silexion adopted the 2024 Equity Incentive Plan (described below), which provides for the grant of equity-based incentive awards to its employees, directors, office holders, service providers and consultants in order to incentivize them to increase their efforts on behalf of New Silexion and to promote the success of New Silexion’s business.
Outstanding Equity Awards at Fiscal Year-End
The following table provides information regarding equity awards held by Silexion’s named executive officers that were outstanding as of December 31, 2024. The awards listed in this table were granted under the Silexion Plans, which are summarized above under “— Narrative Disclosure to Summary Compensation Table — Silexion 2013 Equity Incentive Plan and Silexion 2023 Equity Incentive Plan.”
| | | Option awards | |
| Name | | Number of securities underlying unexercised options
(#) exercisable | | | Number of securities
underlying
unexercised
options
(#) unexercisable | | | Equity
incentive
plan awards: Number of
securities
underlying
unexercised
unearned
options
(#) | | | Option
exercise price
($) | | | Option expiration date | |
Ilan Hadar
Chief Executive Officer (formerly Managing Director of Silexion | | | 14,339 | | | | - | | | -
| | | | 60.51 | | | 24/03/2032 | |
Mirit Horenshtein Hadar
Chief Financial Officer (formerly EVP Finance of Silexion | | | -- | | | | - | | | | - | | | | - | | | | - | |
Dr. Mitchell Shirvan
Chief Scientific and Development Officer( | | | 7,170 | | | | - | | | - | | | | 60.51 | | | 07/06/2032 | |
Director Compensation
| Name | | Fees earned or paid in cash
($) | | | Stock awards
($) | | | Option awards
($) | | | All other compensation
($) | | | Total
($) | |
| Ilan Hadar | | See Summary Compensation Table above
|
|
| See Summary Compensation Table above |
|
| See Summary Compensation Table above |
|
| See Summary Compensation Table above |
|
| See Summary Compensation Table above | |
| Dror Abramov | | | - | | | | 313,800 | | | | - | | | | - | | | | 313,800 | |
| Ruth Alon | | | - | | | | - | | | | - | | | | - | | | | - | |
| Ilan Levin | | | - | | | | - | | | | - | | | | 45,000 | | | | 45,000 | |
| Avner Lushi | | | - | | | | 313,800 | | | | - | | | | - | | | | 313,800 | |
| Shlomo Noy | | | - | | | | 313,800 | | | | - | | | | - | | | | 313,800 | |
| Amnon Peled | | | - | | | | - | | | | - | | | | - | | | | - | |
Ilan Shiloah (former director) | | | - | | | | 313,800 | | | | - | | | | - | | | | 313,800 | |
In the year ended December 31, 2024, we did not pay any cash fees to, but we did grant equity awards, consisting of 22,070 RSUs that could be settled for New Silexion ordinary shares (such number of RSUs reflects the one-for-nine reverse share split of New Silexion), all of which became fully vested at the Closing, to each non-employee member of our board of directors for his or her service as a director.
New Silexion Executive Compensation
Following the Closing, we are developing an executive compensation program that is designed to align compensation with our business objectives and the creation of shareholder value, while enabling us to attract, retain, incentivize and reward individuals who contribute to the long-term success of New Silexion. Decisions regarding the executive compensation program will be made by our compensation committee.
New Silexion Equity Compensation
It is anticipated that equity-based compensation will continue to be an important element of executive compensation following the consummation of the Business Combination in order to maintain a strong link between executive incentives and the creation of shareholder value. Upon the consummation of the Business Combination, we granted 78,650 RSUs, in the aggregate, to directors of New Silexion and employees of New Silexion and its subsidiaries (of which 22,070 RSUs were granted to New Silexion directors) as a means of incentivizing them and aligning their interests with those of the shareholders of the Company. We furthermore expect that the additional 63,953 New Silexion ordinary shares that will be available under the 2024 Equity Incentive Plan (as referenced below) following the consummation of the Business Combination will be an important element of the new compensation arrangements for New Silexion.
Silexion Share Incentive Plans
2013 Share Option Plan
Shares Reserved under the 2013 Share Option Plan. The total number of authorized but unissued Silexion ordinary shares available for issuance under Silexion’s 2013 Share Option Plan (the “2013 Plan”) is 33,828 (which number does not reflect the exchange ratio in the Business Combination or New Silexion’s one-for-nine reverse share split). Any option which has expired or has been cancelled, without having been exercised in full, is available again for issuance under the 2013 Plan. Upon the termination of the 2013 Plan, any unissued New Silexion ordinary shares available under the 2013 Plan shall cease to be reserved for the purpose of the 2013 Plan, other than New Silexion ordinary shares underlying then-outstanding options under the 2013 Plan.
Administration. Our board of directors, or a duly authorized compensation committee of the board of directors, has the power to administer the 2013 Plan. Subject to applicable laws and the relevant provisions of the 2013 Plan, any member of the designated committee is eligible to receive options under the 2013 Plan. The committee has the authority, subject to applicable law and our articles of association, to interpret the terms of the 2013 Plan and any option agreements granted thereunder, designate participants in the 2013 Plan, determine and amend the terms of respective option agreements, including the exercise price of an option, the fair market value of the New Silexion ordinary shares, the time and vesting schedule applicable to the option, accelerate or amend the vesting schedule applicable to an option, prescribe the forms of option agreement for use under the 2013 Plan and take all other actions and make all other determinations necessary for the administration of the 2013 Plan.
Eligibility. The 2013 Plan provides for granting options under various tax regimes, including, without limitation, in compliance with Section 102 or Section 3(i) of the Israeli Tax Ordinance (the “Ordinance”), and for awards granted to Silexion’s United States employees or service providers, including those who are deemed to be residents of the United States for tax purposes, Section 422 of the U.S. Internal Revenue Code of 1986, as amended (the “Code”) and Section 409A of the Code.
Options. All options granted pursuant to the 2013 Plan are evidenced by an option agreement, in a form approved, from time to time, by the committee in its sole discretion. The option agreement sets forth the terms and conditions of the number of shares to which the option relates and the type of option granted thereunder, the purchase price per share and the vesting schedule to which such option shall become exercisable.
Unless otherwise determined by the committee or the board of directors and as stated in the option agreement, and subject to the conditions of the 2013 Plan, options become exercisable under the following schedule: 25% of the shares covered by the option on the first anniversary of the date on which such option was granted and 12.5% of the shares covered by the option at the end of each subsequent six-month period during the second, third and fourth years from the date of grant, with the committee and/or board of directors reserving the exclusive authority to accelerate the periods for exercising an option.
Each option shall expire after ten years from the date of the grant thereof, or five years with respect to incentive stock options unless a shorter term of expiration is otherwise designated by the committee.
Grants. The 2013 Plan provides for the grant of options (including incentive stock options and nonqualified stock options).
Options granted under the 2013 Plan to Silexion employees who are U.S. residents may qualify as “incentive stock options” within the meaning of Section 422 of the Code, or may be non-qualified stock options. The exercise price of an option may not be less than the par value of the shares (if the shares bear a par value) for which such option is exercisable. The exercise price of an Incentive Stock Option may not be less than 100% of the fair market value of the underlying share on the date of grant or such other amount as may be required pursuant to the Code, and in the case of Incentive Stock Options granted to ten percent (10%) shareholders, not less than 110%.
Exercise. An option under the 2013 Plan may be exercised by providing us with a written notice of exercise and full payment of the exercise price for such shares underlying the award, if applicable, in such form and method as may be determined by the committee and the trustee and permitted by applicable law.
Transferability. No option shall be assignable, transferable or given as collateral or any right with respect to them given to any third party whatsoever, and during the lifetime of the option-holder each and all of such option-holder’s rights to purchase New Silexion ordinary shares thereunder shall be exercisable only by the option-holder.
Termination of Employment. An option may be exercised after the date of termination of option-holder’s service or employment with Silexion or any of its affiliates or termination of an affiliate’s status as such only with respect to the number of options already vested and unexpired at the time of such termination according to the vesting and expiration periods of the options set forth in the 2013 Plan, or under a different period prescribed by the committee or by the board of directors and specified in relevant option agreement, provided however, that (i) such termination is without cause, in which case the options shall be exercisable within not more than 90 days from the effective date of such termination, or (ii) such termination is the result of death or disability of the option-holder, in which case the options shall be exercisable within 12 months, and in the event of death, the option shall be exercisable by the option-holder’s estate, all in accordance the 2013 Plan. If termination of employment or service is for cause, any outstanding unexercised option (whether vested or non-vested), will immediately expire and terminate, and the option-holder shall not have any right in connection to such outstanding options.
Voting Rights. Option-holders shall not have any of the rights or privileges of shareholders of New Silexion in respect of any New Silexion ordinary shares purchasable upon the exercise of any part of an option unless and until, following exercise in accordance with the terms of the 2013 Plan and the option, registration of the option-holder as holder of such New Silexion ordinary shares in New Silexion’s register of members, but in case of options and New Silexion ordinary shares held by the trustee, subject to the provisions of the 2013 Plan.
Dividends. With respect to all New Silexion ordinary shares (in contrast to options not exercised for New Silexion ordinary shares) issued upon the exercise of options purchased by an option holder, the option holder, as a shareholder of New Silexion shall be entitled to receive dividends in accordance with the quantity of such New Silexion ordinary shares and the amended and restated memorandum and articles of association of New Silexion, and subject to any applicable taxation on distribution of dividends.
Transactions. If the outstanding shares of New Silexion shall at any time be changed or exchanged by declaration of a dividend, split, share subdivision, combination or exchange of shares, recapitalization, or any other like event of New Silexion, then in such event only and as often as the same shall occur, the number, class and kind of New Silexion ordinary shares (including New Silexion ordinary shares issuable pursuant to the 2013 Plan, in respect of which options have not yet been exercised) subject to the 2013 Plan or subject to any options granted thereunder, and the purchase prices of the options, shall be appropriately and equitably adjusted so as to maintain the proportionate number of New Silexion ordinary shares without changing the aggregate purchase price of the options.
In the event of a merger or consolidation of New Silexion or a sale of all, or substantially all, of our shares or assets or other transaction having a similar effect on New Silexion, or change in the composition of the board of directors, or such other transaction or circumstances that the New Silexion Board determines to be a relevant transaction, the merger agreement will provide for one or more of the following, without the consent of the option-holder: (i) any outstanding option will be assumed or substituted by the successor corporation; (ii) the cancellation of such options and a payment to the option-holder, as provided in the 2013 Plan; and (iii) the full exercisability of the option and full vesting of the New Silexion ordinary shares subject to the option, followed by the cancellation of the option.
2023 Equity Incentive Plan
The 2023 Equity Incentive (the “2023 Plan”) was adopted by our board of directors in 2023. The purpose of the 2023 Plan is to provide equity-based incentive awards in order to link the compensation and benefits of the individuals and entities providing services to Silexion or its affiliates with the success of Silexion and long-term shareholder value. The 2023 Plan enabled Silexion to grant options to purchase ordinary shares of Silexion (currently, following the Business Combination, New Silexion ordinary shares), restricted shares and restricted share units, all of which are referred to as “awards”.
Shares Available for Grants. The total number of shares available for issuance under the 2023 Plan was determined from time to time by our board of directors, subject to certain adjustments as described in the 2023 Plan. If any shares subject to awards under the 2023 Plan expires or otherwise terminates in accordance with the terms thereunder, such shares shall become available for future grants thereunder.
Administration. our board of directors (currently, following the Business Combination, the board of directors of New Silexion), or a duly authorized committee of its board of directors (the “Administrator”), is authorized to administer the 2023 Plan. Under the 2023 Plan, the Administrator has the authority, subject to applicable law, to interpret the terms of the 2023 Plan and any award agreements or awards granted thereunder, designate recipients of awards, determine and amend the terms of awards, including the number of awards granted, the exercise price of an award, the time and vesting schedule applicable to an award or the method of payment for an award, accelerate or amend the vesting schedule applicable to an award, prescribe the forms of agreement for use under the 2023 Plan and take all other actions and make all other determinations necessary for the administration of the 2023 Plan.
The Administrator also has the authority to determine the circumstances under which awards may be settled, cancelled, forfeited, exchanged, or surrendered under and in accordance with the 2023 Plan of any or all awards or shares, and the authority to prescribe, amend and rescind rules and regulations relating to the 2023 Plan, including the form of award agreements and rules governing the grant of awards in jurisdictions in which Silexion or any affiliate operate, or to terminate the 2023 Plan at any time before the date of expiration of its ten-year term, provided that such termination shall not materially affect the rights of grantees, to whom awards have already been granted.
Eligibility. The 2023 Plan provides for granting awards under the Israeli tax regime, including compliance with Section 102 or Section 3(i) of the Ordinance.
Grants. All awards granted pursuant to the 2023 Plan are evidenced by an award letter, in a form approved, from time to time, by the administrator in its sole discretion. The award letter sets forth the terms and conditions of the award, including the type of award, number of shares subject to such award, vesting schedule and conditions (including performance goals or measures) and the exercise price, if applicable.
Unless otherwise determined by the Administrator and stated in the award letter, and subject to the conditions of the 2023 Plan, awards vest and become exercisable under the following schedule: 25% of the shares covered by the award on the first anniversary of the vesting commencement date determined by the Administrator (and in the absence of such determination, the date on which such award was granted) and the remaining 75% of the award shall vest (equally) on a quarterly basis, over 12 quarters as of the commencement date’s first annual anniversary; provided that the grantee remains continuously as an employee or provides services to Silexion throughout such vesting dates.
Each award will expire ten years from the date of the grant thereof, unless a shorter term of expiration is otherwise designated by the Administrator.
Awards. The 2023 Plan provides for the grant of options, shares, restricted shares, RSUs, and other share-based awards.
Exercise. An award under the 2023 Plan may be exercised by providing Silexion (currently, New Silexion) a written notice of exercise and full payment of the exercise price for such shares underlying the award, if applicable, in such form and method as may be determined by the Administrator and permitted by applicable law. The 2023 Plan allows for a net exercise of awards (as may be included in the award letter or otherwise approved by the Administrator). An award may not be exercised for a fraction of a share.
Transferability. No person other than the grantee shall have any right with respect to any award granted under the 2023 Plan. No transfer of any right to any award or its underlying shares, by will or by the laws of descent, shall effectively bind Silexion (currently, New Silexion) unless and until furnished with the certain signed and notarized documents, as described in the 2023 Plan.
Termination of Engagement. In the event of termination of a grantee’s employment or service with Silexion or any of its affiliates, for any reason other than death, retirement, disability or cause, any vested and unexercised awards held by such grantee as of the date of termination may be exercised within 90-days after such date of termination, unless otherwise determined by the Administrator, but in no event later than the date of expiration of the award as set forth in the award letter. After such 90-days period, all such unexercised awards will terminate and the shares covered by such awards become available for issuance under the 2023 Plan.
In the event of termination of a grantee’s employment or service with Silexion or any of its affiliates due to such grantee’s death, retirement or permanent disability, any vested but unexercised awards shall be exercisable (a) in the case of death, by such grantee’s estate, personal representative or beneficiary, or (b) in the case of retirement or Disability, by such grantee or his/her personal representative (as the case may be), until the earlier of (i) 180 days following the date of termination for any such reason; or (ii) the date of expiration of each such as set forth in the award letter. All such grantee’s other awards shall expire upon the date of such termination.
Notwithstanding any of the foregoing, if a grantee’s employment or services with Silexion or any of its affiliates is terminated for “cause” (as defined in the 2023 Plan), all outstanding awards held by such grantee (whether vested or unvested) shall expire. With respect to any and all shares owned by such grantee pursuant to the exercise of any and all awards granted under the 2023 Plan, the board of directors of Silexion may convert such shares into deferred shares of Silexion.
Voting Rights. Until consummation of our initial public offering (or equivalent transaction, such as the Business Combination), shares underlying awards held by grantees or the trustee, as applicable, were to be voted by an irrevocable proxy assigned to a person appointed by the board of directors.
Dividends. Grantees holding shares issuable or issued upon exercise of awards granted under the 2023 Plan will, as a shareholder of New Silexion, be entitled to receive dividends and other distributions with respect to the underlying shares.
Transactions. In the event of any division or subdivision of Silexion’s (currently, New Silexion’s) issued and outstanding share capital, any distribution of bonus shares (share split or subdivision), consolidation or combination of the share capital (reverse share split), reclassification of the shares or any similar recapitalization event, as well as any reorganization, merger, consolidation, split-up, spin-off, combination, repurchase, or exchange of the shares or other securities of Silexion (currently, New Silexion), or any other change in Silexion’s (currently, New Silexion’s) corporate structure affecting the shares, the administrator may, in order to prevent diminution or enlargement of the benefits or potential benefits intended to be made available under the 2023 Plan, adjust (A) the number and class of shares that may be issued under the 2023 Plan; (B) the number, class, and exercise price of underlying shares for each outstanding award; (C) any other term of the awards that in the administrator’s opinion should be adjusted (including those concerning the vesting, exercisability and term of outstanding awards); provided, that no adjustment shall be made by reason of the distribution of subscription rights (rights offering) on outstanding shares.
Upon any such adjustment, references herein to shares and underlying shares shall be construed to mean those shares of Silexion (currently, New Silexion) subject to the 2023 Plan as determined by the Administrator following such adjustment. Any fractional shares resulting from such adjustment shall be rounded down to the nearest whole number of share and Silexion (currently, New Silexion) shall have no obligation to make any cash or other payment with respect to such fractional shares. The adjustments shall be made by the Administrator. If the applicable awards or underlying shares are deposited with a trustee, all of the shares formed by such adjustments shall also be deposited with the trustee on the same terms as such awards or underlying shares.
In the event of a merger, reorganization or consolidation of Silexion (currently, New Silexion) with or into another incorporated entity, or its acquisition by another incorporated entity by means of any transaction or series of related transactions, except any such merger, reorganization or consolidation in which the issued shares of Silexion (currently, New Silexion) as of immediately prior to such transaction continue to represent, or are converted into or exchanged for shares that represent, immediately following such merger, reorganization, or consolidation, at least a majority, by voting power, of the outstanding shares of the surviving or acquiring incorporated entity; or a sale of all, or substantially all, of our shares or assets or other transaction having a similar effect on our company, or change in the composition of the board of directors, or liquidation or dissolution, or such other transaction or circumstances that our board of directors determines to be a relevant transaction, then without the consent of the grantee, (i) unless otherwise determined by the Administrator, any outstanding award will be assumed or substituted by such successor corporation or substituted by our company or by any affiliate of the successor corporation, as determined by the Administrator, and (ii) regardless of whether or not the award is assumed or substituted, the Administrator may:
| | (a) | entitle a grantee to exercise an award, or to otherwise provide for the acceleration of such award’s vesting schedule, as to all or part of its underlying shares, including with respect to awards that would not otherwise be exercisable or vested, under such terms and conditions as the Administrator shall determine, including the cancellation of all unexercised awards upon or immediately prior to the closing of a transaction or as of such other date (the “Cut-Off Date”), and/or the termination of all awards (whether vested but un-exercised or un-vested) as of the relevant Cut-Off Date, as of which they shall no longer be exercisable by the applicable grantees; and/or |
| | (b) | provide for the cancellation of outstanding awards at or immediately prior to the closing of a transaction, and payment to the applicable grantee of a consideration determined by the Administrator to be fair in the circumstances (whether in shares, cash, other securities, property, or any combination thereof), taking into account the value of each underlying share of any such award’s vested portion as reflected by the terms of such transaction, and the exercise price of each such underlying share, and subject to such terms and conditions as determined by the Administrator. |
The Administrator shall have full authority to select the method for determining the payment to the grantees, as well as to set such payment to zero, with respect to underlying shares valued at less than their exercise price or to those of awards that would not otherwise be exercisable or vested, or to determine that such payment be made only in excess of the exercise price.
New Silexion Share Incentive Plans
2024 Equity Incentive Plan
Pursuant to written resolutions adopted by our board of directors and shareholders prior to the Closing, we have approved and adopted the 2024 Equity Incentive Plan (the “2024 Plan”), which became effective immediately upon the Closing.
Upon the Closing, a total of 63,953 New Silexion ordinary shares were reserved for issuance under the terms of the 2024 Plan, which, together with New Silexion ordinary shares allocated for issuance under the existing 2013 Plan and 2023 Plan, equaled 10% of the total number of New Silexion ordinary shares on a fully diluted basis immediately following the Closing. A summary of the other material terms of the 2024 Incentive Plan is provided below:
Administration
The compensation committee of our board of directors is the administrator of the 2024 Plan. Except as provided otherwise under the 2024 Plan, the administrator has plenary authority to grant awards pursuant to the terms of the 2024 Plan to eligible individuals, determine the types of awards and the number of shares covered by the awards, establish the terms and conditions for awards and take all other actions necessary or desirable to carry out the purpose and intent of the 2024 Plan.
Eligibility and Participation
The administrator selects the individuals who participate in the 2024 Plan. Eligibility to participate is open to officers, directors and employees of, and other individuals who provide bona fide services to or for, us or any of our subsidiaries. Our board of directors may also select as participants prospective officers, employees and individual service providers who have accepted an offer of employment or another service relationship from us or one of our subsidiaries. Any awards granted to such a prospect before the individual’s start date may not become vested or exercisable, and no shares may be issued to such individual, before the date the individual first commences performance of services with us.
Share Pool Under the 2024 Plan
The initial number of New Silexion ordinary shares allocated to the 2024 Plan is 63,953. The number of New Silexion ordinary shares available under the 2024 Plan (the “Share Pool”) will be subject to the following annual allocations and adjustments:
The Share Pool will be increased automatically on January 1 of each calendar year, beginning on January 1, 2025 and for each of the subsequent nine calendar years, through (and including) January 1, 2034, in an amount equal to the lesser of (i) 5% of the number of New Silexion ordinary shares issued and outstanding as of that January 1 date, or (ii) an amount determined by our board of directors prior to such date.
The following additional rules apply to the number of New Silexion ordinary shares available under the Share Pool on an ongoing basis:
| | ● | The Share Pool will be reduced by one share for each share made subject to an award granted under the 2024 Plan; |
| | | |
| | ● | The Share Pool will be increased by the number of unissued shares underlying or used as a reference measure for any award or portion of an award granted under the 2024 Plan that is cancelled, forfeited, expired, terminated unearned or settled in cash, in any such case without the issuance of shares; |
| | ● | The Share Pool will be increased by the number of shares that are forfeited back or surrendered for no consideration to us after issuance due to a failure to meet an award contingency or condition with respect to any award or portion of an award granted under the 2024 Plan; |
| | ● | The Share Pool shall be increased, on the exercise date, by the number of shares withheld by or surrendered (either actually or through attestation) to the Company in payment of the exercise price of any award granted under the 2024 Plan; and |
| | ● | The Share Pool shall be increased, on the relevant date, by the number of shares withheld by or surrendered (either actually or through attestation) to the Company in payment of any tax withholding obligation that arises in connection with any award granted under the 2024 Plan. |
In the event of a merger, consolidation, share rights offering, statutory share exchange or similar event affecting the Company or a share dividend, share split, reverse share split, separation, spinoff, reorganization, extraordinary dividend of cash or other property, share combination or subdivision, or recapitalization or similar event affecting the capital structure of the Company, our board of directors will make equitable and appropriate substitutions or proportionate adjustments to the Share Pool to reflect the transaction or event. Similar adjustments will be made to the award limitations described below and to the terms of outstanding awards.
ISO Award Limit
The maximum number of New Silexion ordinary shares that may be issued in connection with awards granted under the 2024 Plan that are intended to qualify as incentive stock options under Section 422 of the Code is 63,953.
Types of Awards
General. The 2024 Plan enables the grant of share awards, performance shares, restricted share units, cash-based performance units, other share-based awards, share options, share appreciation rights, and share unit awards, each of which may be granted separately or in tandem with other awards. The administrator may establish sub-plans under the 2024 Plan under which awards that qualify for preferred tax treatment for recipients in jurisdictions outside the U.S. may be granted.
We have adopted a sub-plan for Israeli participants, which provides for granting awards in compliance with Section 102 (“Section 102”) and Section 3(i) of the Israeli Income Tax Ordinance (New Version), 5721-1961, as amended (the “ITO”). Section 102 allows employees, directors and officers who are not controlling shareholders and who are considered Israeli residents for tax purposes to receive favorable tax treatment for compensation in the form of shares, options or certain other types of equity awards, subject to certain terms and conditions. Our non-employee service providers and controlling shareholders who are considered Israeli residents for tax purposes may be granted awards under Section 3(i) of the ITO, which do not provide for similar tax benefits as Section 102.
Out of the three tax tracks that are available under Section 102 ((i) the “ordinary income track” with a trustee, (ii) the “capital gains track” with a trustee and (iii) grants without a trustee and without a trust period), we have elected the “capital gain track” for grants to eligible Israeli grantees as provided above, which may allow favorable tax treatment for such grantees.
Adjustments to Awards for Corporate Transactions and Other Events
Mandatory Adjustments.
In the event of a merger, amalgamation, consolidation, share rights offering, share exchange or similar event affecting the Company (a “Corporate Event”) or a share dividend, share split, reverse share split, separation, spinoff, reorganization, extraordinary dividend of cash or other property, share combination or subdivision, or recapitalization, capital reduction distribution or similar event affecting the capital structure of the Company, the administrator will make equitable and appropriate substitutions or proportionate adjustments to:
| | ● | the aggregate number and kind of shares or other securities that may be granted to eligible individuals under the 2024 Plan; |
| | ● | the maximum number of shares or other securities that may be issued with respect to incentive share options granted under the 2024 Plan; |
| | ● | the number of shares or other securities covered by each outstanding award and the exercise price, base price or other price per share, if any, and other relevant terms of each outstanding award; and |
| | ● | all other numerical limitations relating to awards, whether contained in the 2024 Plan or in award agreements. |
Notwithstanding the foregoing, any fractional shares resulting from the above mandatory adjustments will be eliminated.
Discretionary Adjustments.
In addition to the adjustments specified above, in the case of Corporate Events, the administrator may make such other adjustments to outstanding awards as it determines to be appropriate and desirable, which adjustments may include, without limitation, (i) the cancellation of outstanding awards in exchange for payments of cash, securities or other property or a combination thereof having an aggregate value equal to the value of such awards, (ii) the substitution of securities or other property (including, without limitation, cash or other securities of the Company and securities of entities other than the Company) for the shares subject to outstanding awards, and (iii) the substitution of equivalent awards, as determined in the sole discretion of the administrator, of the surviving or successor entity or a parent thereof. The administrator may, in its discretion, adjust the performance goals applicable to any awards to reflect any unusual or non-recurring events and other extraordinary items, impact of charges for restructurings, discontinued operations and the cumulative effects of accounting or tax changes.
Repricing.
The administrator may reprice any share options or share appreciation rights without the approval of the shareholders of the Company. For this purpose, “reprice” means (i) any of the following or any other action that has the same effect: (A) lowering the exercise price or base price of an option or share appreciation right after it is granted other than an adjustment made pursuant to the provisions of the 2024 Plan, (B) any other action that is treated as a repricing under applicable accounting principles; (C) cancelling a share option or share appreciation right at a time when its exercise price or base price exceeds the fair market value of the underlying share, in exchange for another share option, share appreciation right, restricted share or other equity, unless the cancellation and exchange occurs in connection with a merger, acquisition, spin-off or other similar corporate transaction; and (ii) any other action that is considered to be a repricing under formal or informal guidance issued by the primary securities market or exchange on which the shares are listed or admitted for trading.
Treatment of Awards upon Dissolution or Liquidation or a Change in Control
Dissolution or Liquidation.
Unless the administrator determines otherwise, all awards outstanding under the 2024 Plan will terminate upon the winding up, liquidation or dissolution of the Company.
Amendment and Termination
Our board of directors or the compensation committee may terminate, amend or modify the 2024 Plan or any portion of it at any time; provided, that, (i) if required to comply with Cayman Islands law and any other applicable laws or marketplace or listing rules of a securities market or securities exchange (other than any requirement from which the Company may opt out based on any available home country exemption), the Company shall obtain shareholder approval of any 2024 Plan amendment in such a manner and to such a degree as required, and (ii) no such termination or amendment may materially impair the rights of a participant with respect to a previously granted award (other than as required to comply with applicable law or the rules of any securities exchange or market on which the shares are listed or to prevent adverse tax or accounting consequences to the Company or the participant) without such participant’s consent.
The 2024 Plan is scheduled to expire on August 14, 2034, which is ten years after the effective date of its adoption by our board of directors. After that time, no further grants may be made under the 2024 Plan, but any then-outstanding grants will remain subject to the terms of the plan.
The foregoing description of the 2024 Incentive Plan is qualified in its entirety by the full text of the 2024 Incentive Plan, which is attached to the registration statement of which this prospectus forms a part as Exhibit 10.12 and incorporated herein by reference.
Rule 10b5-1 Sales Plans
Our directors and executive officers may adopt written plans, known as Rule 10b5-1 plans, in which they will contract with a broker to buy or sell ordinary shares on a periodic basis. Under a Rule 10b5-1 plan, a broker executes trades pursuant to parameters established by the director or executive officer when entering into the plan, without further direction from them. The director or executive officer may amend a Rule 10b5-1 plan in some circumstances and may terminate a plan at any time. Our directors and executive officers also may buy or sell additional shares outside of a Rule 10b5-1 plan when they are not in possession of material nonpublic information, subject to compliance with the terms of our insider trading policy.
Emerging Growth Company Status
We are an “emerging growth company,” as defined in the JOBS Act. As an emerging growth company, it is exempt from certain requirements related to executive compensation, including the requirements to hold a nonbinding advisory vote on executive compensation and to provide information relating to the ratio of total compensation of its chief executive officer to the median of the annual total compensation of all of its employees, each as required by the Investor Protection and Securities Reform Act of 2010, which is part of the Dodd-Frank Wall Street Reform and Consumer Protection Act.
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Other than the compensation arrangements for our directors and executive officers, which are described in the section titled “Executive Compensation”, below is a description of transactions since our formation on April 2, 2024 to which we have been a party, in which:
| | ● | the amounts involved exceeded or will exceed $120,000; and |
| | ● | any of our directors, executive officers or holders of more than 5% of our share capital, or any member of the immediate family of, or person sharing the household with, the foregoing persons, had or will have a direct or indirect material interest. |
Related Party Transactions
Arrangements with Executive Officers
Ilan Hadar serves as our Chief Executive Officer and Chairman of the our Board. Mr. Hadar’s employment agreement with us, as to be amended in the period following the Closing of the Business Combination, subject to corporate approvals, will provide for him to receive an annual base salary of $357,820 (based on the average exchange rate for 2023, as published by the Bank of Israel), customary disbursements toward his providence fund, further education fund and severance pay fund, and other fringe benefits commensurate with such position. During 2022, Silexion granted Mr. Hadar 14,339 options to purchase New Silexion ordinary shares, vesting over a period of 48 months, provided that Mr. Hadar remains engaged by us (or a Silexion affiliate) at the end of each vesting period and subject to the Silexion Plan. Upon completion of the Business Combination, the vesting of all such options is expected to accelerate, such that all unvested options shall become fully vested at that time.
Mirit Horenshtein Hadar, our Executive VP of Finance, Chief Financial Officer and Secretary, is Mr. Hadar’s spouse. Ms. Horenshtein Hadar’s employment agreement with us provides for her to receive an annual base salary of $234,210 (based on the average exchange rate for 2023, as published by the Bank of Israel), as well as customary disbursements toward her providence fund, further education fund and severance pay fund, and other fringe benefits commensurate with such position.
Indemnification Agreements
On the date of, and in connection with, the Closing of the Business Combination, the Company entered into indemnification agreements with each of its directors and executive officers, which provide for indemnification and advancements by the Company of certain expenses and costs under certain circumstances. The indemnification agreements provide that we will indemnify each of its directors and executive officers against any and all expenses incurred by that director or executive officer because of his or her status as a director or officer of New Silexion, to the fullest extent permitted under Cayman law and the Articles.
The foregoing description of the indemnification agreements does not purport to be complete and is qualified in its entirety by reference to the text of the form of indemnification agreement that is filed as Exhibit 10.6 to the registration statement of which this prospectus forms a part, which is incorporated herein by reference.
Conversion Transaction with GIBF
Immediately following the Closing of the Business Combination, Guangzhou Sino-Israel Bio-Industry Investment Fund I (“GIBF”) beneficially owned 20.3% of our issued and outstanding share capital. GIBF had held the remaining 49% of the issued and outstanding share capital of our 51%-held Chinese subsidiary, Silenseed (China) Ltd. (which 49% interest had been received by it in return for its equity investments in the Chinese subsidiary, rather than equity investments directly in us). In connection with the consummation of the Business Combination, GIBF was to exchange its entire holdings in the Chinese subsidiary for preferred shares of Silexion according to the terms set out in the contract for the establishment of the Chinese subsidiary, dated August 30, 2021, as amended, which preferred shares were to be automatically converted into 203,971 New Silexion ordinary shares in accordance with the equity exchange ratio for our shareholders’ exchange of shares of Silexion for our shares under the Business Combination Agreement.
However, in order to simplify the transfer of GIBF’s 49% interest in our Chinese subsidiary to our company, in lieu of transferring that interest to Silexion, GIBF instead transferred the interest directly to New Silexion pursuant to an Agreement on Arrangements Related to Equity Interest Conversion, dated as of August 5, 2024 (the “GIBF Conversion Agreement”). As consideration for its transfer of that 49% interest to us, GIBF received 203,971 New Silexion ordinary shares at the Closing. GIBF furthermore received an additional 16,817 of our ordinary shares upon consummation of the Business Combination due to the conversion of our shares that were issued to it upon settlement of RSUs of Silexion that were subject to accelerated vesting at the Closing. Those RSUs were granted to GIBF subject to GIBF’s providing certain services to us in connection with the transfer of funds from our Chinese subsidiary to us. Avner Lushi and Shlomo Noy, both of whom serve as our directors since the Closing, share voting and investment power over the 220,788 of our ordinary shares beneficially owned by GIBF after the Closing.
PIPE Financing
In connection with, and immediately prior to the Closing of, the Business Combination, Moringa raised $2.0 million via a private investment in public entity financing (the “PIPE Financing”), whereby Moringa sold to Greenstar, LP (the “PIPE Investor”), a Cayman Islands exempted limited partnership and affiliate of the Moringa Sponsor, 200,000 newly issued Moringa ordinary shares (the “PIPE Shares”) (which does not reflect the reverse share split) at a price of $10.00 per share, pursuant to the PIPE Agreement, dated as of August 15, 2024, by and among Moringa, us and the PIPE Investor. Those 200,000 shares automatically converted upon the Closing of the Business Combination into an equivalent number of our ordinary shares (at a number prior to the effectiveness of the reverse share split), or the PIPE Shares. The PIPE Investor is entitled to customary registration rights in respect of the PIPE Shares under the PIPE Agreement, pursuant to which we have agreed that, within 60 days after the Closing Date, it will file with the SEC a registration statement registering the resale of the PIPE Shares by the PIPE Investor, and use its commercially reasonable efforts to have that registration statement be declared effective by 180 days after the Closing Date (or 90 days after the Closing Date if the SEC does not review that filing).
The funds raised from the PIPE Financing, together with remaining funds in Moringa’s trust account after payments to redeeming public shareholders of Moringa, were used for financing support for Moringa and New Silexion, as well as for payment to service providers to whom outstanding amounts were owed by Moringa, including parties that had provided financial advisory services and capital markets advisory services to Moringa during the period leading up to the Closing.
The foregoing summary provides only a brief description of the PIPE Agreement and does not purport to be complete. The summary is qualified in its entirety by the full text of the PIPE Agreement, a copy of which serves as Exhibit 10.2 to the registration statement of which this prospectus forms a part, and which is incorporated herein by reference.
Amended and Restated Sponsor Promissory Note
Effective as of the Closing, we issued to the Sponsor, and Sponsor accepted, in amendment and restatement, and replacement, in their entirety, of all existing promissory notes issued by Moringa to the Sponsor from the IPO until the Closing (and as to which the obligations of Moringa were assigned to us upon the Closing), the A&R Sponsor Promissory Note in an amount of $3,433,000, which reflected the total amount owed by Moringa to the Sponsor through the Closing Date. The maturity date of the A&R Sponsor Promissory Note is the 30-month anniversary of the Closing Date (i.e., February 15, 2027). Amounts outstanding under the A&R Sponsor Promissory Note may be repaid (unless otherwise decided by us) only by way of conversion into our ordinary shares (“Note Shares”) in accordance with the terms set forth in the form of A&R Sponsor Promissory Note. Us and the Sponsor may also convert amounts outstanding under the A&R Sponsor Promissory Note at the price per share at which we conduct an equity financing following the Closing, subject to a minimum conversion amount of $100,000, in an amount of Note Shares constituting up to thirty percent (30%) of the number of our ordinary shares issued and sold by us in such equity financing. The Sponsor may also elect to convert amounts of principal outstanding under the note into our ordinary shares at any time following the 24-month anniversary of the Closing Date, subject to a minimum conversion of $10,000, at a price per share equal to the volume weighted average price of the our ordinary shares on the principal market on which they are traded during the 20 consecutive trading days prior to the conversion date.
The foregoing summary provides only a brief description of the A&R Sponsor Promissory Note and does not purport to be complete. The summary is qualified in its entirety by the full text of the A&R Sponsor Promissory Note, a copy of which serves as Exhibit 10.5 to the registration statement of which this prospectus forms a part, and which is incorporated herein by reference.
Amended and Restated Registration Rights and Lock-Up Agreement
Prior to the Closing under the Business Combination Agreement, on August 14, 2024, us, Moringa, Moringa Sponsor, the distributees of the Sponsor Investment Shares (as defined under the Business Combination Agreement), certain of our pre-Business Combination shareholders and the PIPE Investor entered into (and EarlyBird will be bound by) an amended and restated registration rights and lock-up agreement which became effective as of the Closing of the Business Combination (the “A&R Registration Rights and Lock-Up Agreement”). Under the agreement, we have assumed Moringa’s existing obligations under Moringa’s prior registration rights agreement (entered into in connection with Moringa’s initial public offering, or IPO) and has granted registration rights to the Moringa Sponsor, the distributees of Sponsor Investment Shares, certain of our pre-Business Combination shareholders, the PIPE Investor and EarlyBird with respect to certain securities of ours.
Under the A&R Registration Rights and Lock-Up Agreement, we have agreed to provide the holders party thereto customary demand and shelf registration rights (subject to certain minimum size offerings) and piggy-back rights on primary and secondary offerings, subject to customary cut-back provisions.
Under the lock-up provisions of the agreement, lock-up periods apply following the Closing to of our securities that are held by the holders who are party to the agreement, subject to permitted transfers to certain categories of “Permitted Transferees”. Specifically, those lock-up periods apply to the following categories of securities for the following periods of time post-Closing:
| | ● | Sponsor Investment Shares and securities held by former Silexion shareholders: (A) 50% of the Sponsor Investment Shares held by the Sponsor and its distributees and 50% of the New Silexion securities held by the former Silexion shareholders who are party to the agreement, in each case, upon the Closing, are subject to a lock-up period ending on the earlier of (i) six (6) months after the completion of the Business Combination, and (ii) the date on which New Silexion will consummate a liquidation, merger, amalgamation, share exchange, reorganization, or other similar transaction after the Business Combination that results in all of our shareholders having the right to exchange their ordinary shares for cash, securities or other property, and (B) the other 50% of the Sponsor Investment Shares held by the Sponsor and its distributees and 50% of the our securities held by the former Silexion shareholders party to the agreement, in each case, upon the Closing, will be subject to a lock-up period that will end on the earliest of (x) six (6) months after the date of the consummation of the Business Combination, (y) the date on which we consummate a liquidation, merger, amalgamation, share exchange, reorganization, or other similar transaction after the Business Combination that results in all of our shareholders having the right to exchange their ordinary shares for cash, securities or other property, or (z) the date on which the closing price of our ordinary shares equals or exceeds $12.00 per share (as adjusted for share splits, share dividends, rights issuances, subdivisions, reorganizations, recapitalizations and the like) for any 20 trading days within any 30-trading day period. |
| | ● | Private Shares and Private Warrants: The lock-up period on all New Silexion ordinary shares issued pursuant to the SPAC Merger in exchange for private placement shares and private placement warrants purchased by or issued to the Sponsor and EarlyBird concurrently with Moringa’s initial public offering will remain (as provided in the documentation for Moringa’s initial public offering) 30 days after the Closing. |
| | ● | Representative Shares. The lock-up period on all Representative Shares (as defined in the Amended and Restated Registration Rights and Lock-Up Agreement) that are held by EarlyBird will remain (as provided in the documentation for Moringa’s initial public offering) three months after the Closing. |
| | ● | Note Shares and PIPE Shares. Note Shares issued to the Sponsor upon conversion of amounts due under the A&R Sponsor Promissory Note and PIPE Shares issued to the PIPE Investor will not be subject to any lock-up periods following the Closing. |
The foregoing summary provides only a brief description of the A&R Registration Rights and Lock-Up Agreement and does not purport to be complete. The summary is qualified in its entirety by the full text of the A&R Registration Rights and Lock-Up Agreement, which serves as Exhibit 10.4 to the registration statement of which this prospectus forms a part, and which is incorporated herein by reference.
BENEFICIAL OWNERSHIP OF SECURITIES
The following table sets forth information known to the Company regarding the beneficial ownership of New Silexion ordinary shares as of December 31, 2024 by:
| | ● | each person who is the beneficial owner of more than 5% of the outstanding New Silexion ordinary shares; |
| | ● | our named executive officer and directors; and |
| | ● | all of our executive officers and directors as a group. |
Beneficial ownership is determined according to the rules of the SEC, which generally provide that a person has beneficial ownership of a security if he, she or it possesses sole or shared voting or investment power over that security, including options and warrants that are currently exercisable or exercisable within 60 days. In computing the number of ordinary shares beneficially owned by a person and the percentage ownership, we include, as outstanding, those ordinary shares that are subject to warrants held by that person that are currently exercisable or exercisable within 60 days of December 31, 2024. We do not deem those shares to be outstanding, however, for the purpose of computing the percentage ownership of any other person.
Unless otherwise indicated, we believe that all persons named in the table have sole voting and investment power with respect to all New Silexion ordinary shares beneficially owned by them.
The percentage ownership of New Silexion ordinary shares is based on 1,849,132 New Silexion ordinary shares outstanding as of December 31, 2024.
Name and Address of Beneficial Owner(1) | | Number of Shares Beneficially Owned | | | Approximate
Percentage of Outstanding Ordinary Shares | |
| Directors and Executive Officers of New Silexion: | | | | | | |
| Ilan Hadar | | | 31,160 | (2) | | | 1.7 | % |
| Dror Abramov | | | 4,425 | | | | * | |
| Ruth Alon | | | 6,037 | | | | * | |
| Ilan Levin(3) | | | 229,624 | (4) | | | 12.3 | % |
| Avner Lushi(5) | | | 220,788 | (6) | | | 11.9 | % |
| Shlomo Noy(7) | | | 220,788 | (6) | | | 11.9 | % |
| Amnon Peled | | | - | | | | - | |
| Dr. Mitchell Shirvan | | | 21,888 | (8) | | | 1.2 | % |
| Mirit Horenshtein Hadar, CPA | | | 6,308 | | | | * | |
| | | | | | | | | |
| All executive officers and directors as a group (8 individuals) | | | 520,230 | | | | 27.5 | % |
| Five Percent Holders: | | | | | | | | |
| Moringa Sponsor, LP and affiliated entities (3) | | | 229,624 | (4) | | | 12.3 | % |
| Guangzhou Sino-Israel Biotech Fund(9) | | | 220,788 | (6) | | | 11.9 | % |
| Wildcat Partner Holdings LP(10) | | | 113,428 | | | | 6.1 | % |
| (1) | Unless otherwise noted, the business address of each beneficial owner listed in the above table is c/o Silexion Therapeutics Corp, 12 Abba Hillel Road, Ramat Gan, Israel 5250606. |
| (2) | Includes 14,339 New Silexion ordinary shares issuable upon exercise of options, at an exercise price of $60.48 per share, all of which are vested and currently exercisable. |
| | |
| (3) | The shares reported in this row are held of record by the Sponsor, Moringa Sponsor, LP, and/or by the PIPE Investor, Greenstar, L.P., each a Cayman Islands exempted limited partnership, as described in footnote (4) below. Moringa Partners Ltd., an Israeli company that is wholly-owned by Mr. Ilan Levin, serves as the sole general partner of each of the Sponsor and the PIPE Investor. Mr. Levin, a director of New Silexion, is the sole director of that general partner. As a result of his ownership of that general partner, Mr. Levin possesses sole voting and investment authority with respect to the shares indirectly held by the Sponsor and the PIPE Investor. The limited partnership interests of the Sponsor and the PIPE Investor are held by various individuals and entities, including Mr. Levin. Mr. Levin disclaims beneficial ownership of the securities held by the Sponsor and the PIPE Investor other than to the extent of his direct or indirect pecuniary interest in such securities. The address of each of the entities beneficially owning the shares that are reported in this row is c/o Moringa Acquisition Corp, 250 Park Avenue, 7th floor, New York, NY 10177. |
| (4) | Consists of the total of: (i) 148,592 New Silexion ordinary shares issued to the Sponsor as Sponsor Investment Shares (as defined under the Business Combination Agreement); (ii) 39,206 New Silexion ordinary shares issued to the Sponsor upon the Closing of the Business Combination due to the conversion, on a one-for-one basis, of the 352,857 Moringa private shares held by it; (iii) 19,603 New Silexion ordinary shares underlying New Silexion warrants issued to the Sponsor upon the Closing of the Business Combination due to the conversion, on a one-for-one basis, of the 176,429 Moringa private warrants held by the Sponsor (which New Silexion warrants will be exercisable beginning 30 days after the Closing Date); and (iv) 22,223 New Silexion ordinary shares issued to Greenstar, L.P., the PIPE Investor, as PIPE Shares in respect of the PIPE Financing. The foregoing beneficial ownership of New Silexion ordinary shares by the Sponsor does not include any Note Shares that may be issued to the Sponsor following the Closing upon conversion of amounts owed by New Silexion to the Sponsor under the A&R Sponsor Promissory Note, as the potential number of Note Shares, and the timing of issuance of Note Shares, cannot be determined in advance. |
| (5) | The shares reported in this row consist entirely of New Silexion ordinary shares held of record by Guangzhou Sino-Israel Biotech Fund (“GIBF”), with respect to which Mr. Lushi possesses shared voting and investment authority as a result of his serving as a Managing Partner and CEO of GIBF. |
| (6) | Includes 203,971 New Silexion ordinary shares issued to GIBF at the Closing in respect of its transfer of its noncontrolling interest in our Chinese subsidiary, Silenseed (China) Ltd., to New Silexion pursuant to the Chinese Subsidiary Transfer. |
| (7) | The shares reported in this row consist entirely of New Silexion ordinary shares held of record by GIBF, with respect to which Mr. Noy possesses shared voting and investment authority as a result of his serving as Chief Medical Officer of GIBF. |
| | |
| (8) | Includes 7,170 New Silexion ordinary shares issuable upon exercise of options, at an exercise price of $60.48 per share, all of which are vested and currently exercisable. |
| | |
| (9) | The address of this shareholder is 34 Ha’Barzel St., Tel-Aviv 6971052 Israel. Each of Avner Lushi and Shlomo Noy may be deemed to share voting and investment power over the securities beneficially owned by GIBF. |
| (10) | The address of this shareholder is 301 Commerce Street, Suite 3150, Fort Worth, Texas 76102. Len Porter may be deemed to have sole voting and investment power over the securities beneficially owned by Wildcat Partner Holdings LP. |
DESCRIPTION OF SHARE CAPITAL
In connection with the consummation of the Business Combination, New Silexion adopted the Articles. The following summary of the material terms of New Silexion’s securities is not intended to be a complete summary of the rights and preferences of such securities. You are encouraged to read the Articles and the agreements governing New Silexion’s warrants in their entirety, which serve as Exhibit 3.1 and Exhibits 4.1 and 4.2, respectively, to the registration statement of which this prospectus forms a part, along with the applicable provisions of Cayman Islands law, for a complete description of the rights and preferences of New Silexion’s securities.
Authorized and Outstanding Share Capital
The Articles of New Silexion authorize the issuance of 22,222,222 shares, consisting of 22,222,222 ordinary shares, $0.0009 par value per share. As of December 31, 2024, there were 1,849,132 New Silexion ordinary shares outstanding. All of such outstanding New Silexion ordinary shares were validly issued, fully paid and non-assessable.
Ordinary Shares
Voting Power
Holders of New Silexion ordinary shares are entitled to one vote in respect of each share held of record by such holder on all matters to be voted on by shareholders.
Dividends
Subject to applicable law, holders of New Silexion ordinary shares are entitled to receive dividends when, as and if declared by the New Silexion Board, payable in cash, property or shares.
Winding Up, Liquidation and Dissolution
Upon New Silexion’s winding up and liquidation and after payment in full of all amounts required to be paid to creditors, the holders of New Silexion ordinary shares are entitled to receive a pro rata share of New Silexion’s remaining assets available for distribution.
Preemptive or Other Rights
Holders of New Silexion ordinary shares will not be entitled to preemptive rights, and New Silexion ordinary shares will not be subject to conversion, redemption or sinking fund provisions.
Appointment of Directors
The New Silexion Board will consist of up to nine (9) directors and not less than three (3) directors, unless increased or decreased from time to time by Ordinary Resolution in a general meeting.
Directors will generally be appointed by an Ordinary Resolution passed at each annual general meeting.
Prior to each annual general meeting, the New Silexion Board (or a nominating committee established by the New Silexion Board for such purpose) may select, via a resolution adopted by a majority of the New Silexion Board or such committee, a number of persons to be proposed to the shareholders for appointment as directors at such annual general meeting, for service until the next annual general meeting (the “Nominees”). Any shareholder entitled under applicable law to propose one or more persons as nominees for appointment as directors at an annual general meeting (each such nominee, an “Alternate Nominee”) may make such proposal only if a written notice of such shareholder’s intent to that effect has been given to the Secretary of the Company (or, if there is no such Secretary, the chief executive officer) within the periods set out in the Articles.
Unless the New Silexion Board resolves that the appointment of Nominees or Alternate Nominees will be determined based on those Nominees or Alternate Nominees receiving the highest number of votes of Members in favor of their appointment— even if less than a majority of all Members’ votes that are cast, the Nominees or Alternate Nominees shall be appointed by Ordinary Resolution at the annual general meeting at which they are proposed for appointment. If the appointment of the Nominees or Alternate Nominees proposed to be appointed would cause the total number of directors (including those then in office) to exceed the maximum number, then any directors then in office who are not named as Nominees or Alternate Nominees will cease to hold office following the conclusion of such annual general, if any such Nominees or Alternate Nominees are appointed in such meeting, and upon such Nominees or Alternate Nominees taking office. If (a) the appointment of the Nominees or Alternate Nominees proposed to be appointed would not cause the total number of directors to exceed the maximum, or (b) no Nominees or Alternate Nominees are proposed to be appointed at an annual general meeting by either the Board or Members, or (c) no Nominees or Alternate Nominees are eventually appointed in such annual general meeting — all directors then in office shall continue to hold office until the convening of a general meeting at which Nominees or Alternate Nominees shall be proposed and appointed, and where such appointment would cause the total number of directors to exceed the maximum.
Each director will hold office until the next succeeding annual general meeting and until his or her successor is appointed and qualified, or until such director’s earlier death, resignation, disqualification or removal. The Articles will not provide for cumulative voting for the appointment of directors.
New Silexion Warrants
Following the Closing of the Business Combination and as of October 1, 2024, there were 660,001 New Silexion warrants to purchase New Silexion ordinary shares outstanding, consisting of 638,889 New Silexion public warrants issued upon conversion of an equivalent number of Moringa public warrants and 21,112 New Silexion private warrants issued upon conversion of an equivalent number of Moringa private warrants (of which 19,603 and 1,508 private warrants are held by the Sponsor and EarlyBird, respectively). In connection with the Closing, each Moringa warrant was exchanged for a New Silexion warrant on substantially similar terms, including exercise price.
Each New Silexion public warrant entitles the registered holder to purchase one New Silexion ordinary share at a price of $103.50 per share, subject to adjustment as discussed below, at any time commencing one month after the Closing. However, no New Silexion public warrants will be exercisable for cash unless there is an effective and current registration statement covering the New Silexion ordinary shares issuable upon exercise of the New Silexion public warrants and a current prospectus relating to such New Silexion ordinary shares. Notwithstanding the foregoing, if a registration statement covering the New Silexion ordinary shares issuable upon exercise of the New Silexion public warrants is not effective within 60 business days from the closing of the Business Combination, warrant holders may, until such time as there is an effective registration statement and during any period when the Company shall have failed to maintain an effective registration statement, exercise New Silexion public warrants on a cashless basis pursuant to the exemption provided by Section 3(a)(9) of the Securities Act, provided that such exemption is available. If that exemption, or another exemption, is not available, holders will not be able to exercise their warrants on a cashless basis. In the case of a cashless exercise, each holder would pay the exercise price by surrendering the warrants for that number of New Silexion ordinary shares equal to the quotient obtained by dividing (x) the product of the number of New Silexion ordinary shares underlying the warrants, multiplied by the difference between the exercise price of the warrants and the “fair market value” (defined below) by (y) the fair market value. The “fair market value” for this purpose will mean the average reported last sale price of the ordinary shares for the five trading days ending on the trading day prior to the date of exercise. The New Silexion public warrants will expire five years from the Closing of the Business Combination (i.e., on August 15, 2029) at 5:00 p.m., New York City time.
The New Silexion private warrants are identical to the New Silexion public warrants, except that each New Silexion private warrant will be exercisable for cash (even if a registration statement covering the New Silexion ordinary shares issuable upon exercise of such warrants is not effective) or on a cashless basis, at the holder’s option, and will not be redeemable by the Company, in each case so long as they are still held by the Sponsor, EarlyBird or their respective affiliates.
From and after the time the New Silexion warrants become exercisable, we may call them for redemption (excluding the New Silexion private warrants), in whole and not in part, at a price of $0.01 per warrant, upon not less than 30 days’ prior written notice of redemption to each warrant holder, if and only if
| | ● | the reported last sale price of the ordinary shares equals or exceeds $162.00 per share (as adjusted for share sub-divisions, share capitalizations, reorganizations and recapitalizations), for any 20 trading days within a 30 trading day period commencing after the warrants become exercisable and ending on the third business day prior to the notice of redemption to warrant holders; and |
| | ● | a registration statement is then in effect with respect to the New Silexion ordinary shares underlying such warrants. |
The right to exercise will be forfeited unless the New Silexion warrants are exercised prior to the date specified in the notice of redemption. On and after the redemption date, a record holder of a New Silexion warrant will have no further rights except to receive the redemption price for such holder’s warrant upon surrender of such warrant.
The redemption criteria for New Silexion warrants have been established at a price which is intended to provide warrant holders a reasonable premium to the initial exercise price and provide a sufficient differential between the then-prevailing share price and the warrant exercise price so that if the share price declines as a result of our redemption call, the redemption will not cause the share price to drop below the exercise price of the warrants.
If New Silexion calls the New Silexion warrants for redemption as described above, its management will have the option to require all holders that wish to exercise warrants prior to redemption to do so on a “cashless basis.” In such event, each holder would pay the exercise price by surrendering the warrants for that number of New Silexion ordinary shares equal to the quotient obtained by dividing (x) the product of the number of New Silexion ordinary shares underlying the warrants, multiplied by the difference between the exercise price of the warrants and the “fair market value” (defined below) by (y) the fair market value. The “fair market value” shall mean the average reported last sale price of the New Silexion ordinary shares for the 5 trading days ending on the third trading day prior to the date on which the notice of redemption is sent to the holders of warrants.
The New Silexion warrants will be issued in registered form under a warrant agreement between Continental Stock Transfer & Trust Company, as warrant agent, and New Silexion (as assignee of Moringa). The warrant agreement provides that the terms of the warrants may be amended without the consent of any holder to cure any ambiguity or correct any defective provision, but requires the approval, by written consent or vote, of the holders of at least 50% of the then outstanding public warrants in order to make any change that adversely affects the interests of the registered holders.
The exercise price and number of New Silexion ordinary shares issuable on exercise of the warrants may be adjusted in certain circumstances including in the event of a share capitalization or our recapitalization, reorganization, merger or consolidation. However, the warrants will not be adjusted for issuances of New Silexion ordinary shares at a price below their respective exercise prices.
The New Silexion warrants may be exercised upon surrender of the warrant certificate on or prior to the expiration date at the offices of the warrant agent, with the exercise form on the reverse side of the warrant certificate completed and executed as indicated, accompanied by full payment of the exercise price, by certified or official bank check payable to us, for the number of warrants being exercised. The warrant holders do not have the rights or privileges of holders of New Silexion ordinary shares and any voting rights until they exercise their warrants and receive New Silexion ordinary shares. After the issuance of New Silexion ordinary shares upon exercise of the warrants, each holder will be entitled to one vote for each share held of record on all matters to be voted on by shareholders.
Under the terms of the warrant agreement, New Silexion (as assignee of Moringa) has agreed to use its best efforts to have declared effective a registration statement relating to the New Silexion ordinary shares issuable upon exercise of the warrants and keep such registration statement and its included prospectus current until the expiration of the warrants. However, we cannot assure you that we will be able to do so and, if we do not maintain a current prospectus relating to the New Silexion ordinary shares issuable upon exercise of the warrants, holders will be unable to exercise their warrants for cash and we will not be required to net cash settle or cash settle the warrant exercise.
Warrant holders may elect to be subject to a restriction on the exercise of their warrants such that an electing warrant holder would not be able to exercise their warrants to the extent that, after giving effect to such exercise, such holder would beneficially own in excess of 9.9% of the New Silexion ordinary shares outstanding.
No fractional shares will be issued upon exercise of the New Silexion warrants. If, upon exercise of the warrants, a holder would be entitled to receive a fractional interest in a share, we will, upon exercise, round up to the nearest whole number the number of New Silexion ordinary shares to be issued to the warrant holder.
Contractual Arrangements with respect to the New Silexion private warrants
So long as the New Silexion private warrants are still held by the Sponsor, EarlyBird or their affiliates, the Company will not redeem such warrants, and the Company will allow the holders to exercise such warrants on a cashless basis (even if a registration statement covering the New Silexion ordinary shares issuable upon exercise of such warrants is not effective). However, once any of the New Silexion private warrants are transferred from the Sponsor, EarlyBird or their affiliates, these arrangements will no longer apply. Furthermore, because the New Silexion private warrants were issued in a private transaction, the holders and their transferees will be allowed to exercise the New Silexion private warrants for cash even if a registration statement covering the New Silexion ordinary shares issuable upon exercise of such warrants is not effective and receive unregistered New Silexion ordinary shares.
Listing of Securities
Our ordinary shares and New Silexion warrants are listed on the Nasdaq under the symbols “SLXN” and “SLXNW”, respectively.
Transfer Agent and Registrar
The transfer agent and registrar for the New Silexion ordinary shares and warrant agent for the New Silexion warrants is Continental Stock Transfer & Trust Company.
Anti-Takeover Provisions in Our Articles.
Some provisions of our Articles may discourage, delay or prevent a change in control of our company or management that shareholders may consider favorable.
However, under Cayman Islands law, our directors may only exercise the rights and powers granted to them under our Articles, as amended and restated from time to time, for a proper purpose and for what they believe in good faith to be in the best interests of our company.
Authorized but Unissued Shares
The authorized but unissued New Silexion ordinary shares are available for future issuance without shareholder approval, subject to any limitations imposed by the listing standards of the Nasdaq and the Articles. These additional shares may be used for a variety of corporate finance transactions, acquisitions and employee benefit plans. The existence of authorized but unissued and unreserved New Silexion ordinary shares could make more difficult or discourage an attempt to obtain control of New Silexion by means of a proxy contest, tender offer, merger or otherwise.
Shareholder Action; Extraordinary General Meetings
Our Articles provide that shareholders may take action by unanimous written resolutions or at annual or extraordinary general meetings. As a result, a holder controlling a majority of New Silexion ordinary shares would not be able to amend the Articles or remove directors without holding a meeting of shareholders called in accordance with the Articles or the shareholders of New Silexion passing unanimous written resolutions to approve the same. Further, the Articles provide that only the chairperson of the Board, or the New Silexion Board pursuant to a resolution adopted by a majority of the directors then in office — and not shareholders — may call extraordinary general meetings of shareholders, thus prohibiting a holder of New Silexion ordinary shares from calling an extraordinary general meeting. These provisions might delay the ability of shareholders to force consideration of a proposal or for shareholders controlling a majority of New Silexion to take any action, including the removal of directors.
Advance Notice Requirements for Shareholder Proposals and Director Nominations
The Articles provide that shareholders seeking to bring business before our annual general meeting, or to nominate candidates for appointment as directors at its annual general meeting, must provide timely notice. To be timely, a shareholder’s notice will need to be delivered to the Secretary of New Silexion at its principal executive offices not less than 90 days nor more than 120 calendar days prior to the one-year anniversary of the preceding year’s annual general meeting. In the event that no annual general meeting was held during the preceding year or the date of the annual general meeting is more than 30 days before or more than 60 days after such anniversary date, to be timely, a shareholder’s notice must be so delivered no earlier than the close of business on the 120th day prior to such annual general meeting and not later than the 90th day prior to such annual general meeting or, if later, the 10th day following the day on which public disclosure of the date of such annual general meeting is first made by New Silexion. The Articles also specify certain requirements as to the form and content of a shareholders’ notice. These provisions may preclude New Silexion shareholders from bringing matters before our annual general meeting or from making nominations for directors at our annual general meeting.
Supermajority Requirements for the Amendment of the Articles
Under the Companies Act (As Revised) of the Cayman Islands, the Articles may be amended by a special resolution of shareholders, being a resolution passed by not less than two-thirds (2/3) of our shareholders as being entitled to do so, vote in person or by proxy at a general meeting.
Board Vacancies
The Articles provide that any vacancy on the New Silexion Board may be filled by the affirmative vote of a majority of the directors then in office, even if less than a quorum, or by a sole remaining director, and not by our shareholders. Any director chosen to fill a vacancy will hold office until the next annual general meeting and until his or her successor is duly appointed and qualified, or until his or her earlier death, resignation, disqualification or removal. In addition, the number of directors constituting the then-authorized size of the New Silexion Board is permitted to be set only by a resolution adopted by the Board.
Exclusive Forum Selection
The Articles provide that, unless New Silexion consents in writing to the selection of an alternative forum and to the fullest extent permitted by law, the courts of the Cayman Islands have exclusive jurisdiction over any claim or dispute arising out of or in connection with the Articles or otherwise related in any way to each shareholder’s shareholding in the Company, including but not limited to (i) any derivative action or proceeding brought on behalf of the Company, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee of the Company to the Company or our shareholders, or (iii) any action asserting a claim arising pursuant to any provision of the Companies Act (As Revised) of the Cayman Islands or the Articles, and each shareholder shall be deemed to have irrevocably submitted to the exclusive jurisdiction of the courts of the Cayman Islands over all such claims or disputes.
However, such forum selection provisions will not apply to suits brought to enforce any liability or duty created by the Securities Act or the Exchange Act. The Articles also provide that, unless New Silexion consents in writing to the selection of an alternative forum, the federal district courts of the United States of America will be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act or the Exchange Act. There is uncertainty as to whether a court would enforce that exclusivity provision. Furthermore, investors cannot waive compliance with the U.S. federal securities laws and the rules and regulations thereunder.
Any person or entity purchasing or otherwise acquiring any interest in New Silexion shares shall be deemed to have notice of and consented to the forum selection provisions in the Articles.
The choice of forum provisions may limit a shareholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with New Silexion or its directors, officers, or other employees, which may discourage such lawsuits against New Silexion and its directors, officers, and other employees. Alternatively, if a court were to find the choice of forum provisions contained in the Articles to be inapplicable or unenforceable in an action, New Silexion may incur additional costs associated with resolving such action in other jurisdictions, which could harm its business, results of operations, and financial condition.
Transactions with Interested Shareholders.
Cayman Islands law does not have a business combination statute (like in Delaware) applicable to public companies prohibiting them from engaging in certain business combinations with an “interested shareholder”. As a result, we cannot avail ourselves of the types of protections afforded by a business combination statute. However, although Cayman Islands law does not regulate transactions between a company and its significant shareholders, it does provide that such transactions must be entered into bona fide in the best interests of the company and for a proper corporate purpose and not with the effect of constituting a fraud on the minority shareholders.
Limitation on Liability and Indemnification of Directors and Officers
Cayman Islands law does not limit the extent to which a company’s memorandum and articles of association may provide for indemnification of officers and directors, except to the extent any such provision may be held by the Cayman Islands courts to be contrary to public policy, such as to provide indemnification against willful default, willful neglect, actual fraud or the consequences of committing a crime. Our Articles provide that our directors and officers shall be indemnified out of the assets of New Silexion against any liability, action, proceeding, claim, demand, costs, damages or expenses, including legal expenses, whatsoever which they or any of them may incur as a result of any act or failure to act in carrying out their functions other than such liability (if any) that they may incur by reason of their own actual fraud, willful neglect or willful default. No such indemnified person shall be liable to New Silexion for any loss or damage incurred by New Silexion as a result (whether direct or indirect) of the carrying out of their functions unless that liability arises through the actual fraud, willful neglect or willful default of such indemnified person. No person shall be found to have committed actual fraud, willful neglect or willful default under the foregoing unless or until a court of competent jurisdiction shall have made a finding to that effect.. In addition, the Articles provide for the advancing of certain fees and costs to such indemnified persons and the purchase by New Silexion for insurance for the benefit of its directors and officers as further described in the Articles.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to New Silexion directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
DESCRIPTION OF SECURITIES OFFERED
We are offering up to 2,577,320 ordinary shares, together with up to 2,577,320 ordinary warrants to purchase up to 2,577,320 ordinary shares. We are also offering to those purchasers, if any, whose purchase of our ordinary shares in this offering would otherwise result in such purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding ordinary shares immediately following the consummation of this offering, the opportunity, in lieu of purchasing ordinary shares, to purchase pre-funded warrants to purchase ordinary shares. For each pre-funded warrant we sell, the number of ordinary shares we are offering will be decreased on a one-for-one basis. The pre-funded warrants will be sold together with ordinary warrants in a fixed combination, with each pre-funded warrant to purchase one ordinary share accompanied by one ordinary warrant to purchase one ordinary share. We are also registering the ordinary shares issuable from time to time upon exercise of the pre-funded warrants and ordinary warrants offered hereby.
The material terms and provisions of our ordinary shares are described under the caption “Description of Share Capital” in this prospectus.
The following summary of certain terms and provisions of the pre-funded warrants offered hereby is not complete and is subject to, and qualified in its entirety by, the provisions of pre-funded warrant the form of which has been filed as an exhibit to the registration statement of which this prospectus forms a part. Prospective investors should carefully review the terms and provisions of the form of pre-funded warrant for a complete description of the terms and conditions of the pre-funded warrants.
The term “pre-funded” refers to the fact that the purchase price of our ordinary shares in this offering includes almost the entire exercise price that will be paid under the pre-funded warrants, except for a nominal remaining exercise price of $0.0001. The purpose of the pre-funded warrants is to enable investors that may have restrictions on their ability to beneficially own more than 4.99% (or, at the election of each purchaser, 9.99%) of our outstanding ordinary shares following the consummation of this offering the opportunity to make an investment in us without triggering their ownership restrictions, by receiving pre-funded warrants in lieu of our ordinary shares which would result in such ownership of more than 4.99% (or, at the election of each purchaser, 9.99%), and receive the ability to exercise their option to purchase the shares underlying the pre-funded warrants at such nominal price at a later date.
Each pre-funded warrant is exercisable for one ordinary share, with an exercise price equal to $0.0001 per ordinary share, at any time that the pre-funded warrant is outstanding. There is no expiration date for the pre-funded warrants. The holder of a pre-funded warrant will not be deemed a holder of our underlying ordinary shares until the pre-funded warrant is exercised.
Subject to limited exceptions, a holder of pre-funded warrants will not have the right to exercise any portion of its pre-funded warrants if the holder (together with such holder’s affiliates, and any persons acting as a group together with such holder or any of such holder’s affiliates) would beneficially own a number of ordinary shares in excess of 4.99% (or, at the election of each purchaser, 9.99%) of the ordinary shares then outstanding after giving effect to such exercise.
The exercise price and the number of ordinary shares issuable upon exercise of the pre-funded warrants is subject to appropriate adjustment in the event of recapitalization events, stock dividends, share splits, share combinations, reclassifications, reorganizations or similar events affecting our ordinary shares. The pre-funded warrant holders must pay the exercise price in cash upon exercise of the pre-funded warrants, unless such pre-funded warrant holders are utilizing the cashless exercise provision of the pre-funded warrants.
Upon the holder’s exercise of a pre-funded warrant, we will issue the ordinary shares issuable upon exercise of the pre-funded warrant within two trading days following our receipt of a notice of exercise, provided that payment of the exercise price has been made (unless exercised to the extent permitted via the “cashless” exercise provision). Prior to the exercise of any pre-funded warrants to purchase ordinary shares, holders of the pre-funded warrants will not have any of the rights of holders of ordinary shares purchasable upon exercise, including the right to vote, except as set forth therein.
As an alternative to payment in immediately available funds, the holder may elect to exercise the pre-funded warrant through a cashless exercise, in which the holder would receive upon such exercise the net number of ordinary shares determined according to the formula set forth in the pre-funded warrant (in which case, the pre-funded warrants may only be exercised via a “cashless” exercise provision).
In the event of a fundamental transaction, as described in the pre-funded warrants and generally including any reorganization, recapitalization or reclassification of our ordinary shares, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding ordinary shares, or any person or group becoming the beneficial owner of more than 50% of the voting power represented by our outstanding ordinary shares, the holders of the pre-funded warrants will be entitled to receive upon exercise of the pre-funded warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the pre-funded warrants immediately prior to such fundamental transaction without regard to any limitations on exercised contained in the pre-funded warrants.
We do not intend to apply to list the pre-funded warrants on any securities exchange or other trading system. Without an active trading market, the liquidity of the pre-funded warrants will be limited.
The following summary of certain terms and provisions of the ordinary warrants offered together with the ordinary shares hereby is not complete and is subject to, and qualified in its entirety by, the form of ordinary warrant, which has been filed as an exhibit to the registration statement of which this prospectus is a part. Prospective investors should carefully review the terms and provisions set forth in the form of ordinary warrant.
Exercisability. The ordinary warrants are exercisable at any time after their original issuance and at any time up to the date that is five years after their original issuance. The ordinary warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice and, at any time a registration statement registering the issuance of the ordinary shares underlying the ordinary warrants under the Securities Act of 1933, as amended, or the Securities Act, is effective and available for the issuance of such shares, by payment in full in immediately available funds for the number of ordinary shares purchased upon such exercise. If a registration statement registering the issuance of ordinary shares underlying the ordinary warrants under the Securities Act is not effective or available the holder may, in its sole discretion, elect to exercise the ordinary warrant through a cashless exercise, in which case the holder would receive upon such exercise the net number of ordinary shares determined according to the formula set forth in the ordinary warrant. No fractional shares will be issued in connection with the exercise of an ordinary warrant. In lieu of fractional shares, we will either pay the holder an amount in cash equal to the fractional amount multiplied by the exercise price or round up to the next whole share.
Exercise Limitation. A holder will not have the right to exercise any portion of the ordinary warrant if the holder (together with its affiliates) would beneficially own in excess of 4.99% (or, at the election of each purchaser, 9.99%) of the number of ordinary shares outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the ordinary warrants. However, any holder may increase or decrease such percentage to any other percentage not in excess of 9.99%, provided that any increase in such percentage shall not be effective until 61 days following notice from the holder to us.
Exercise Price. The exercise price per whole ordinary share purchasable upon exercise of the ordinary warrants is $ per share (which will be no less than 100% of the assumed public offering price per ordinary share to be sold in this offering). The exercise price is subject to appropriate adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting our ordinary shares and also upon any distributions of assets, including cash, stock or other property to our stockholders.
Transferability. Subject to applicable laws, the ordinary warrants may be offered for sale, sold, transferred or assigned without our consent.
Exchange Listing. We do not intend to apply to list the ordinary warrants on any securities exchange or nationally recognized trading system. Without an active trading market, the liquidity of the ordinary warrants will be limited.
Fundamental Transactions. In the event of a fundamental transaction, as described in the ordinary warrants and generally including any reorganization, recapitalization or reclassification of our ordinary shares, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding ordinary shares, or any person or group becoming the beneficial owner of more than 50% of the voting power represented by our outstanding ordinary shares, the holders of the ordinary warrants will be entitled to receive upon exercise of the ordinary warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the ordinary warrants immediately prior to such fundamental transaction without regard to any limitations on exercised contained in the ordinary warrants. Additionally, as more fully described in the ordinary warrants, in the event of certain fundamental transactions, the holders of the ordinary warrants will be entitled to receive consideration in an amount equal to the Black Scholes value of the ordinary warrants on the date of consummation of such transaction.
Rights as a Stockholder. Except as otherwise provided in the ordinary warrants or by virtue of such holder’s ownership of our ordinary shares, the holder of a ordinary warrant does not have the rights or privileges of a holder of our ordinary shares, including any voting rights, until the holder exercises the ordinary warrant.
Governing Law. The ordinary warrants are governed by New York law.
The following summary of certain terms and provisions of the Placement Agent warrants is not complete and is subject to, and qualified in its entirety by, the form of Placement Agent warrant, which has been filed as an exhibit to the registration statement of which this prospectus is a part. Prospective investors should carefully review the terms and provisions set forth in the form of Placement Agent warrant.
We have agreed to issue to the Placement Agent, or its designees, up to 180,412 warrants to purchase up to 180,412 ordinary shares (which represents 7.0% of the aggregate number of ordinary shares issued in this offering and issuable upon the exercise of the warrants and pre-funded warrants issued in this offering) with an exercise price of $[•] per ordinary share (representing 125% of the assumed public offering price per ordinary share) and exercisable for five years from the date of the commencement of sales in this offering. The Placement Agent warrants issued in this offering will otherwise have substantially the same terms as the ordinary warrants. The Placement Agent warrants and underlying ordinary shares are registered on the registration statement of which this prospectus is a part.
CERTAIN MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES
The following discussion is a summary of certain material U.S. federal income tax considerations generally applicable to a “U.S. Holder” arising from the ownership and disposition of our ordinary shares and the exercise, disposition and lapse of our ordinary warrants. The ordinary shares and the ordinary warrants are referred to collectively herein as our securities. For this purpose, a “U.S. Holder” is a holder of our securities that is: (1) an individual citizen or resident of the United States, including an alien individual who is a lawful permanent resident of the United States or meets the substantial presence residency test under U.S. federal income tax laws; (2) a corporation (or entity treated as a corporation for U.S. federal income tax purposes) created or organized under the laws of the United States or the District of Columbia or any political subdivision thereof; (3) an estate, the income of which is includable in gross income for U.S. federal income tax purposes regardless of its source; (4) a trust if a court within the United States is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have authority to control all substantial decisions of the trust; or (5) a trust that has a valid election in effect to be treated as a U.S. person to the extent provided in U.S. Treasury regulations.
This discussion is not a complete analysis of all potential U.S. federal income tax consequences relating to the ownership and disposition of our securities. This summary generally considers only U.S. Holders that will own our securities as capital assets. This summary does not consider the U.S. federal tax consequences to a person that is not a U.S. Holder, nor does it describe the rules applicable to determine a taxpayer’s status as a U.S. Holder. This summary is based upon current provisions of the Code, existing U.S. Treasury Regulations promulgated thereunder, published administrative pronouncements and rulings of the U.S. Internal Revenue Service (the “IRS”), and judicial decisions, all as in effect as of the date of this prospectus. These authorities are subject to change and differing interpretation, possibly with retroactive effect. Any change or differing interpretation could alter the tax consequences to holders described in this discussion. There can be no assurance that a court or the IRS will not challenge one or more of the tax consequences described herein, and we have not obtained, nor do we intend to obtain, a ruling with respect to the U.S. federal income tax consequences to a holder of the ownership or disposition of our securities.
This discussion does not address all of the aspects of U.S. federal income taxation that may be relevant to a particular U.S. Holder based on such U.S. Holder’s particular circumstances and in particular does not discuss any estate, gift, generation-skipping transfer, state, local, excise or non-U.S. tax considerations. In addition, this discussion does not address the U.S. federal income tax treatment of a U.S. Holder who is: (1) a bank, life insurance company, regulated investment company, or other financial institution or “financial services entity;” (2) a broker or dealer in securities or foreign currency; (3) a person who acquired our ordinary shares in connection with employment or other performance of services; (4) a U.S. Holder that is subject to the U.S. alternative minimum tax; (5) a U.S. Holder that holds our securities as a hedge or as part of a hedging, straddle, conversion or constructive sale transaction or other risk-reduction transaction for U.S. federal income tax purposes; (6) a tax-exempt entity; (7) real estate investment trusts or grantor trusts; (8) a U.S. Holder that expatriates out of the United States or a former long-term resident of the United States; or (9) a person having a functional currency other than the U.S. dollar. This discussion does not address the U.S. federal income tax treatment of a U.S. Holder that owns, directly or constructively, at any time, ordinary shares representing 10% or more of the shares of our company. Additionally, the U.S. federal income tax treatment of U.S. Holders of pre-funded warrants or partnerships (or other pass-through entities) or persons who hold our securities through a partnership or other pass-through entity are not addressed.
All prospective holders of our ordinary shares, ordinary warrants or pre-funded warrants should consult their tax advisors with respect to the U.S. federal, state, local and non-U.S. tax consequences of the ownership and disposition of our ordinary shares, ordinary warrants or pre-funded warrants.
Tax Consequences of Ownership and Disposition of Our Securities
Distributions on our ordinary shares
Subject to the PFIC rules discussed below, the gross amount of any distribution on our ordinary shares that is made out of our current or accumulated earnings and profits (as determined for U.S. federal income tax purposes) generally will be taxable to a U.S. Holder as ordinary dividend income on the date such distribution is actually or constructively received. Any such dividends generally will not be eligible for the dividends received deduction allowed to corporations in respect of dividends received from U.S. corporations. To the extent that the amount of the distribution exceeds our current and accumulated earnings and profits (as determined under U.S. federal income tax principles), such excess amount will be treated first as a non-taxable return of capital to the extent of the U.S. Holder’s tax basis in our ordinary shares, and thereafter as capital gain recognized on a sale or exchange. However, it is not expected that we will maintain calculations of our earnings and profits under U.S. federal income tax principles. U.S. Holders should therefore assume that any distribution by us with respect to our ordinary shares will be reported as dividend income. U.S. Holders should consult their own tax advisors with respect to the appropriate U.S. federal income tax treatment of any distribution received from us.
Subject to the PFIC rules discussed below, dividends received by non-corporate U.S. Holders (including individuals) from a “qualified foreign corporation” may be eligible for reduced rates of taxation, provided that certain holding period requirements and other conditions are satisfied. For these purposes, a non-U.S. corporation will be treated as a qualified foreign corporation if it is eligible for the benefits of a comprehensive income tax treaty with the United States that meets certain requirements. A non-U.S. corporation is also treated as a qualified foreign corporation with respect to dividends it pays on shares that are readily tradable on an established securities market in the United States. U.S. Treasury guidance indicates that shares listed on Nasdaq (such as our ordinary shares) will be considered readily tradable on an established securities market in the United States. There can be no assurance that our ordinary shares will be considered readily tradable on an established securities market in future years. Further, we will not constitute a qualified foreign corporation for purposes of these rules if we are a PFIC for the taxable year in which we pay a dividend or for the preceding taxable year.
Subject to certain conditions and limitations, non-refundable withholding taxes (at a rate not in excess of any applicable income tax treaty rate), if any, on dividends paid by us may be treated as foreign taxes eligible for credit against a U.S. Holder’s U.S. federal income tax liability under the U.S. foreign tax credit rules. However, as a result of recent changes to the U.S. foreign tax credit rules, a withholding tax generally will need to satisfy certain additional requirements in order to be considered a creditable tax for a U.S. Holder. We have not determined whether these requirements have been met with respect to any withholding tax that may apply to dividends paid by us and, accordingly, no assurance can be given that any such withholding tax will be creditable. For purposes of calculating the U.S. foreign tax credit, dividends paid on our ordinary shares will generally be treated as income from sources outside the United States and will generally constitute passive category income or, in the case of certain U.S. Holders, general category income. The rules governing the U.S. foreign tax credit are complex. U.S. Holders should consult their tax advisors regarding the availability of the U.S. foreign tax credit under their particular circumstances.
Sale, Taxable Exchange or Other Taxable Disposition of Our Securities
Subject to the PFIC rules discussed below, upon any sale, exchange or other taxable disposition of our securities, a U.S. Holder generally will recognize gain or loss in an amount equal to the difference between (i) the sum of (x) the amount cash and (y) the fair market value of any other property, received in such sale, taxable exchange or other taxable disposition and (ii) the U.S. Holder’s adjusted tax basis in such securities. Any such gain or loss generally will be capital gain or loss and will be long-term capital gain or loss if the U.S. Holder’s holding period for such securities exceeds one year. Long-term capital gain realized by a non-corporate U.S. Holder generally will be taxable at a reduced rate. The deductibility of capital losses is subject to limitations.
This gain or loss generally will be treated as U.S. source gain or loss. Accordingly, in the event that any non-U.S. tax (including withholding tax) is imposed upon such sale, exchange or other taxable disposition, a U.S. Holder may not be able to utilize foreign tax credits in respect of such non-U.S. tax. In addition, there may be other limitations on utilizing foreign tax credits even if a U.S. Holder has foreign source income or gain in the same category from other sources. The rules governing foreign tax credits are complex and U.S. Holders are urged to consult their tax advisers regarding the creditability of foreign taxes in their particular circumstances.
Exercise, Lapse or Redemption of Our Ordinary Warrant
Subject to the PFIC rules discussed below, a U.S. Holder generally will not recognize gain or loss upon the acquisition of our ordinary share on the exercise of our ordinary warrant for cash. A U.S. Holder’s tax basis in our ordinary share received upon exercise of our ordinary warrant generally will equal the sum of the U.S. Holder’s tax basis in such ordinary warrant and the exercise price. It is unclear whether a U.S. Holder’s holding period for our ordinary share received will commence on the date of exercise of our ordinary warrant or the day following the date of exercise of such ordinary warrant; in either case, the holding period will not include the period during which the U.S. Holder held such ordinary warrant. If our ordinary warrant is allowed to lapse unexercised, a U.S. Holder generally will recognize a capital loss equal to such U.S. Holder’s tax basis in such ordinary warrant.
The tax consequences of a cashless exercise of a warrant are not clear under current law. Subject to the PFIC rules discussed below, a cashless exercise may not be taxable, either because the exercise is not a realization event or because the exercise is treated as a recapitalization for U.S. federal income tax purposes. In either situation, a U.S. Holder’s tax basis in our ordinary shares received generally should equal the U.S. Holder’s tax basis in our ordinary warrants exercised therefor. If the cashless exercise was not a realization event, it is unclear whether a U.S. Holder’s holding period for our ordinary shares received would be treated as commencing on the date of exercise of our ordinary warrants or the day following the date of exercise of such ordinary warrants; in either case, the holding period will not include the period during which the U.S. Holder held our ordinary warrants. If the cashless exercise were treated as a recapitalization, the holding period of our ordinary shares received would include the holding period of our ordinary warrants.
It is also possible that a cashless exercise could be treated in part as a taxable exchange in which gain or loss would be recognized. In such event, a U.S. Holder could be deemed to have surrendered a number of our ordinary warrants equal to the number of our ordinary shares having a value equal to the exercise price for the total number of our ordinary warrants to be exercised. In such case, subject to the PFIC rules discussed below, the U.S. Holder would recognize capital gain or loss with respect to our ordinary warrants deemed surrendered in an amount equal to the difference between the fair market value of our ordinary shares that would have been received in a regular exercise of our ordinary warrants deemed surrendered and the U.S. Holder’s tax basis in our ordinary warrants deemed surrendered. In this case, a U.S. Holder’s aggregate tax basis in our ordinary shares received would equal the sum of the U.S. Holder’s tax basis in our ordinary warrants deemed exercised and the aggregate exercise price of such ordinary warrants. It is unclear whether a U.S. Holder’s holding period for our ordinary shares would commence on the date of exercise of our ordinary warrants or the day following the date of exercise of our ordinary warrants; in either case, the holding period will not include the period during which the U.S. Holder held our ordinary warrants.
Due to the absence of authority on the U.S. federal income tax treatment of a cashless exercise, including when a U.S. Holder’s holding period would commence with respect our ordinary shares received, there can be no assurance regarding which, if any, of the alternative tax consequences and holding periods described above would be adopted by the IRS or a court of law. Accordingly, U.S. Holders should consult their tax advisors regarding the tax consequences of a cashless exercise.
Possible Constructive Distributions
The terms of our ordinary warrants provide for an adjustment to the number of our ordinary shares for which our ordinary warrants may be exercised or to the exercise price of our ordinary warrants in certain events, as discussed in the section of this prospectus entitled “Description of Securities Offered — Ordinary Warrants.” An adjustment which has the effect of preventing dilution generally is not taxable. U.S. Holders of our ordinary warrants would, however, be treated as receiving a constructive distribution from us if, for example, the adjustment increases such U.S. Holders’ proportionate interest in our assets or earnings and profits (e.g., through an increase in the number of our ordinary shares that would be obtained upon exercise or through a decrease in the exercise price of our ordinary warrants), which adjustment may be made as a result of a distribution of cash or other property to the holders of our ordinary shares. Such constructive distribution to a U.S. Holder of our ordinary warrants would be treated as if such U.S. Holder had received a cash distribution from us generally equal to the fair market value of such increased interest (taxed as described above under “— Distributions on our ordinary shares”).
PFIC Rules
In General
The treatment of U.S. Holders of our securities could be materially different from that described above if we are treated as a PFIC for U.S. federal income tax purposes.
In general, a non-U.S. corporation is a PFIC for U.S. federal income tax purposes for any taxable year in which, after applying certain look-through rules, (i) 50% or more of the value of its assets (generally determined on the basis of a weighted quarterly average) consists of assets that produce, or are held for the production of, passive income, or (ii) 75% or more of its gross income consists of passive income. Passive income generally includes dividends, interest, rents and royalties (other than rents or royalties derived from the active conduct of a trade or business) and gains from the disposition of passive assets. Cash and cash equivalents generally are passive assets. The value of goodwill will generally be treated as an active or passive asset based on the nature of the income produced in the activity to which the goodwill is attributable. For purposes of the PFIC rules, a non-U.S. corporation that owns, directly or indirectly, at least 25% by value of the stock of another corporation is treated as if it held its proportionate share of the assets of the other corporation and directly received its proportionate share of the income of the other corporation.
The application of the PFIC rules to warrants is uncertain. The Code provides that, to the extent provided in Treasury Regulations, if any person has an option to acquire shares of a PFIC, the shares will be considered as owned by that person for purposes of the PFIC rules. Under proposed Treasury Regulations that have a retroactive effective date, an option to acquire shares of a PFIC is generally treated as ownership of those PFIC shares. The remainder of this discussion assumes that the PFIC rules will apply to our ordinary warrants if we were a PFIC. However, U.S. Holders should consult their tax advisers regarding the application of the PFIC rules to our ordinary warrants prior to the finalization of the proposed Treasury Regulations.
If non-U.S. corporation is treated as a PFIC during a U.S. Holder’s holding period, it will, with respect to such U.S. Holder, always be treated as a PFIC, regardless of whether it satisfied either of the qualification tests in subsequent years, subject to certain exceptions (such as upon making a “deemed sale” election).
The adverse impact of the PFIC rules on a U.S. Holder that holds shares in a PFIC may generally be mitigated if the U.S. Holder makes a timely qualified electing fund (“QEF”) election or mark-to-market election for the PFIC’s first taxable year as a PFIC in which the U.S. Holder held (or was deemed to hold) shares, or a QEF election along with an applicable purging election (collectively, “PFIC Elections”). Further detail about the PFIC Elections is provided below under “— Our Securities”.
Based on the projected composition of our income and assets, it cannot be determined whether we will be classified as a PFIC for our current taxable year or in any future taxable year. Further, changes in the composition of our income or composition of our assets may cause us to be or become a PFIC for the current or subsequent taxable years. Whether we are treated as a PFIC for U.S. federal income tax purposes is a factual determination that must be made annually at the close of each taxable year and, thus, is subject to significant uncertainty.
If we are or become a PFIC during any year in which a U.S. Holder holds our securities, there are three separate taxation regimes that could apply to such U.S. Holder under the PFIC rules: (i) the excess distribution regime (which is the default regime), (ii) the QEF regime, or (iii) the mark-to-market regime. A U.S. Holder who holds (actually or constructively) stock in a non-U.S. corporation during any year in which such corporation qualifies as a PFIC is subject to U.S. federal income taxation under one of these three regimes. The effect of the PFIC rules on a U.S. Holder will depend upon which of these regimes applies to such U.S. Holder. However, dividends paid by a PFIC are not eligible for the lower rates of taxation applicable to qualified dividend income under any of the foregoing regimes.
If New Silexion is or becomes a PFIC during any year in which a U.S. Holder holds New Silexion securities, there are three separate taxation regimes that could apply to such U.S. Holder under the PFIC rules: (i) the excess distribution regime (which is the default regime), (ii) the QEF regime, or (iii) the mark-to-market regime. A U.S. Holder who holds (actually or constructively) stock in a non-U.S. corporation during any year in which such corporation qualifies as a PFIC is subject to U.S. federal income taxation under one of these three regimes. The effect of the PFIC rules on a U.S. Holder will depend upon which of these regimes applies to such U.S. Holder. However, dividends paid by a PFIC are not eligible for the lower rates of taxation applicable to qualified dividend income under any of the foregoing regimes.
Excess Distribution Regime. If a U.S. Holder does not make or is not eligible to make a QEF election or a mark-to-market election, as described below, the U.S. Holder will be subject to the default “excess distribution regime” under the PFIC rules with respect to (i) any gain realized on a sale or other disposition (including a pledge) of our securities, and (ii) any “excess distribution” on our securities (generally, any distributions in excess of 125% of the average of the annual distributions on our securities during the preceding three taxable years or the U.S. Holder’s holding period, for the our securities that preceded the taxable year of the distribution whichever is shorter). Generally, under this excess distribution regime:
| | ● | the U.S. Holder’s gain or excess distribution will be allocated ratably over the U.S. Holder’s holding period for our securities; |
| | ● | the amount allocated to the U.S. Holder’s taxable year in which the U.S. Holder recognized the gain or received the excess distribution, or to the period in the U.S. Holder’s holding period before the first day of our first taxable year in which we are a PFIC, will be taxed as ordinary income; |
| | ● | the amount allocated to other taxable years (or portions thereof) of the U.S. Holder and included in its holding period will be taxed at the highest tax rate in effect for that year and applicable to the U.S. Holder without regard to the U.S. Holder’s other items of income and loss for such year; and |
| | ● | an additional amount equal to the interest charge generally applicable to underpayments of tax will be imposed on the U.S. Holder with respect to the tax attributable to each such other taxable year of the U.S. Holder. |
The tax liability for amounts allocated to years prior to the year of disposition or excess distribution will be payable generally without regard to offsets from deductions, losses and expenses. In addition, gains (but not losses) realized on the sale of our securities by a U.S. Holder cannot be treated as capital gains, even if such securities are held as capital assets. Further, no portion of any distribution will be treated as qualified dividend income.
If we are, or we are treated as, a PFIC for any taxable year during which a U.S. Holder owns our securities and any entity in which we own equity interests is also a PFIC (a “lower-tier PFIC”), the U.S. Holder will be deemed to own their proportionate amount (by value) of the shares of each lower-tier PFIC and will be subject to U.S. federal income tax according to the rules described above on (i) certain distributions by a lower-tier PFIC and (ii) dispositions of shares of lower-tier PFICs, in each case, as if the U.S. Holder held such shares directly, even though the U.S. Holder will not receive any proceeds of those distributions or dispositions.
QEF Regime. A valid QEF election is effective for the taxable year for which the election is made and all subsequent taxable years and may not be revoked without the consent of the IRS. If a U.S. Holder makes a timely QEF election with respect to its direct or indirect interest in a PFIC, the U.S. Holder will be required to include in income each year its allocable portion of the ordinary earnings and net capital gains of the PFIC as QEF income inclusions, even if such portion is not distributed to the U.S. Holder. Thus, the U.S. Holder may be required to report taxable income as a result of QEF income inclusions without corresponding receipts of cash. U.S. Holders of our securities should not expect that they will receive cash distributions from us sufficient to cover their respective U.S. tax liability with respect to such QEF income inclusions. In addition, U.S. Holders of our ordinary warrants will not be able to make a QEF election with respect to such ordinary warrants.
The timely QEF election also allows the electing U.S. Holder to: (i) generally treat any gain recognized on the disposition of its shares of the PFIC as capital gain; (ii) treat its share of the PFIC’s net capital gain, if any, as long-term capital gain instead of ordinary income; and (iii) either avoid interest charges resulting from PFIC status, or make an annual election, subject to certain limitations, to defer payment of current taxes on its undistributed QEF income inclusions, subject to an interest charge on the deferred tax computed by using the statutory rate of interest applicable to an extension of time for payment of tax. In addition, net losses (if any) of a PFIC will not pass through to its shareholders and may not be carried back or forward in computing such PFIC’s ordinary earnings and net capital gain in other taxable years.
A U.S. Holder’s tax basis in our ordinary shares will be increased to reflect QEF income inclusions and will be decreased to reflect distributions of amounts previously included in income as QEF income inclusions. No portion of the QEF income inclusions attributable to ordinary income will be treated as qualified dividend income. Amounts included as QEF income inclusions with respect to direct and indirect PFICs generally will not be taxed again when distributed by such PFICs.
A U.S. Holder may make a QEF election with respect to its our ordinary shares only if we provide U.S. Holders on an annual basis with certain information, including a “PFIC annual information statement” as described in the Treasury Regulations. [If we determine that we are a PFIC for any taxable year, we will endeavor to provide to a U.S. Holder such information as the IRS may require, including a PFIC annual information statement, in order to enable a U.S. Holder to make and maintain a QEF election.]1 However, there can be no assurance that we will have timely knowledge of our status as a PFIC in the future or that we will timely provide U.S. Holders with the required information on an annual basis to allow U.S. Holders to make and maintain a QEF election with respect to our ordinary shares in the event we are treated as a PFIC for any taxable year. The failure to provide such information on an annual basis could prevent a U.S. Holder from making a QEF election or result in the invalidation or termination of a U.S. Holder’s prior QEF election. In addition, as mentioned above, U.S. Holders of our ordinary warrants will not be able to make a QEF election with respect to their warrants.
If a U.S. Holder makes a QEF election with respect to our ordinary shares in a year after our first taxable year as a PFIC in which the U.S. Holder held (or was deemed to hold) our ordinary shares, then notwithstanding such QEF election, the excess distribution regime discussed above, adjusted to take into account the QEF income inclusions resulting from the QEF election, will continue to apply with respect to our ordinary shares held by such U.S. Holder, unless the U.S. Holder makes a purging election under the PFIC rules. Under one type of purging election, the U.S. Holder will be deemed to have sold such ordinary shares at their fair market value and any gain recognized on such deemed sale will be treated as an excess distribution, as described above. As a result of such purging election, the U.S. Holder will have additional basis (to the extent of any gain recognized on the deemed sale) and, solely for purposes of the PFIC rules, a new holding period in our ordinary shares.
In addition, as mentioned above, U.S. Holders of our ordinary warrants will not be able to make a QEF election with respect to their ordinary warrants. As a result, if a U.S. Holder sells or otherwise disposes of such ordinary warrants (other than upon exercise of such ordinary warrants) and we were a PFIC at any time during the U.S. Holder’s holding period of such ordinary warrants, any gain recognized generally will be treated as an excess distribution, taxed as described above. If a U.S. Holder that exercises such ordinary warrants properly makes and maintains a QEF election with respect to the newly acquired ordinary shares (or has previously made a QEF election with respect to our ordinary shares), the QEF election will apply to the newly acquired ordinary shares. Notwithstanding such QEF election, the excess distribution rules discussed above, adjusted to take into account the current income inclusions resulting from the QEF election, will continue to apply with respect to such newly acquired ordinary shares (which, while not entirely clear, generally will be deemed to have a holding period for purposes of the PFIC rules that includes the period the U.S. Holder held the ordinary warrants), unless the U.S. Holder makes a purging election under the PFIC rules. U.S. Holders are urged to consult their tax advisors as to the application of the rules governing purging elections to their particular circumstances.
Mark-to-Market Regime. Alternatively, a U.S. Holder may make an election to mark marketable shares in a PFIC to market on an annual basis. PFIC shares generally are marketable if they are “regularly traded” on a national securities exchange that is registered with the SEC, such as Nasdaq, as we intend our ordinary shares will be, but there can be no assurance that our ordinary shares will continue to be so listed or will be “regularly traded” for purposes of these rules. Pursuant to such an election, a U.S. Holder of our ordinary shares would include in each year as ordinary income the excess, if any, of the fair market value of such stock or shares over its adjusted basis at the end of the taxable year. A U.S. Holder may treat as ordinary loss any excess of the adjusted basis of our ordinary shares over its fair market value at the end of the year, but only to the extent of the net amount previously included in income as a result of the election in prior years. A U.S. Holder’s adjusted tax basis in our ordinary shares will be increased to reflect any amounts included in income, and decreased to reflect any amounts deducted, as a result of a mark-to-market election. Any gain recognized on a disposition of our ordinary shares in a taxable year in which we are a PFIC will be treated as ordinary income and any loss will be treated as ordinary loss (but only to the extent of the net amount of income previously included as a result of a mark-to-market election, and any loss in excess of such prior inclusions generally would be treated as a capital loss). A mark-to-market election applies for the taxable year in which the election was made, and for each subsequent taxable year, unless the PFIC shares cease to be marketable or the IRS consents to the revocation of the election. U.S. Holders should also be aware that the Code and the Treasury Regulations do not allow a mark-to-market election with respect to stock of lower-tier PFICs that is non-marketable. There is also no provision in the Code, Treasury Regulations or other published authority that specifically provides that a mark-to-market election with respect to the stock of a publicly traded holding company (such as us) effectively exempts stock of any lower-tier PFICs from the negative tax consequences arising from the general PFIC rules. U.S. Holders are advised to consult their own tax advisor to determine whether the mark-to-market tax election is available to them and the consequences resulting from such election. In addition, U.S. Holders of our ordinary warrants will not be able to make a mark-to-market election with respect to such ordinary warrants.
PFIC Reporting Requirements
A U.S. Holder that owns (or is deemed to own) shares in a PFIC during any taxable year of the U.S. Holder generally is required to file an IRS Form 8621 with such U.S. Holder’s U.S. federal income tax return and provide such other information as the IRS may require. Failure to file IRS Form 8621 for each applicable taxable year may result in substantial penalties and result in the U.S. Holder’s taxable years being open to audit by the IRS until such forms are properly filed.
THE RULES DEALING WITH PFICS AND PFIC ELECTIONS ARE VERY COMPLEX AND ARE AFFECTED BY VARIOUS FACTORS IN ADDITION TO THOSE DESCRIBED ABOVE. ACCORDINGLY, U.S. HOLDERS OF OUR SECURITIES ARE URGED TO CONSULT THEIR OWN TAX ADVISORS CONCERNING THE APPLICATION OF THE PFIC RULES TO OUR SECURITIES UNDER THEIR PARTICULAR CIRCUMSTANCES.
Additional Reporting Requirements
Certain U.S. Holders may be required to file an IRS Form 926 (Return by a U.S. Transferor of Property to a Foreign Corporation) to report a transfer of property (including cash) to us. Substantial penalties may be imposed on a U.S. Holder that fails to comply with this reporting requirement, and the period of limitations on assessment and collection of U.S. federal income taxes will be extended in the event of a failure to comply. Furthermore, certain U.S. Holders who are individuals and certain entities will be required to report information with respect to such U.S. Holder’s investment in “specified foreign financial assets” on IRS Form 8938 (Statement of Specified Foreign Financial Assets), subject to certain exceptions. Specified foreign financial assets generally include any financial account maintained with a non-U.S. financial institution and should also include our ordinary shares and our ordinary warrants if they are not held in an account maintained with a U.S. financial institution. Persons who are required to report specified foreign financial assets and fail to do so may be subject to substantial penalties, and the period of limitations on assessment and collection of U.S. federal income taxes may be extended in the event of a failure to comply. U.S. Holders are urged to consult their tax advisors regarding the foreign financial asset and other reporting obligations and their application to an investment in our ordinary shares and our ordinary warrants.
Tax on Net Investment Income
U.S. Holders who are individuals, estates or trusts will generally be required to pay a 3.8% Medicare tax on their net investment income (including dividends on and gains from the sale or other disposition of our ordinary securities), or in the case of estates and trusts on their net investment income that is not distributed to beneficiaries of the estate or trust. In each case, the 3.8% Medicare tax applies only to the extent the U.S. Holder’s total adjusted income exceeds applicable thresholds.
Information Reporting and Backup Withholding
Dividend payments with respect to our ordinary shares and proceeds from the sale, exchange or redemption of our securities may be subject to information reporting to the IRS and possible backup withholding. Backup withholding will not apply, however, to a U.S. Holder who furnishes a correct taxpayer identification number and makes other required certifications, or who is otherwise exempt from backup withholding and establishes such exempt status.
Backup withholding is not an additional tax. Amounts withheld as backup withholding may be credited against a U.S. Holder’s U.S. federal income tax liability, and a U.S. Holder generally may obtain a refund of any excess amounts withheld under the backup withholding rules by timely filing the appropriate claim for refund with the IRS and furnishing any required information.
THE DISCUSSION ABOVE IS A GENERAL SUMMARY. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A U.S. HOLDER. EACH U.S. HOLDER IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES RELATING TO THE PURCHASE, OWNERSHIP AND DISPOSITION OF OUR ORDINARY SHARES, ORDINARY WARRANTS OR PRE-FUNDED WARRANTS IN LIGHT OF THE U.S. HOLDER’S OWN CIRCUMSTANCES, INCLUDING THE CONSEQUENCES OF ANY PROPOSED CHANGE IN APPLICABLE LAWS.
PLAN OF DISTRIBUTION
We have engaged H.C. Wainwright & Co., LLC, or the Placement Agent, to act as our exclusive placement agent to solicit offers to purchase the securities offered pursuant to this prospectus on a reasonable best efforts basis. The engagement agreement does not give rise to any commitment by the Placement Agent to purchase any of our securities, and the Placement Agent will have no authority to bind us by virtue of the engagement agreement. The Placement Agent is not purchasing or selling any of the securities offered by us under this prospectus, nor is it required to arrange for the purchase or sale of any specific number or dollar amount of securities, other than to use its “reasonable best efforts” to arrange for the sale of such securities by us. Therefore, we may not sell all of the securities being offered. The terms of this offering were subject to market conditions and negotiations between us, the Placement Agent and prospective investors. This is a best efforts offering and there is no minimum offering amount required as a condition to the closing of this offering. Because there is no minimum offering amount required as a condition to closing this offering, we may sell fewer than all of the securities offered hereby, which may significantly reduce the amount of proceeds received by us. The Placement Agent does not guarantee that it will be able to raise new capital in any prospective offering. The Placement Agent may engage sub-agents or selected dealers to assist with the offering.
Investors purchasing securities offered hereby will have the option to execute a securities purchase agreement with us. In addition to rights and remedies available to all purchasers in this offering under federal securities and state law, the purchasers which enter into a securities purchase agreement will also be able to bring claims of breach of contract against us. The ability to pursue a claim for breach of contract is material to larger purchasers in this offering as a means to enforce (i) a covenant to not enter into variable rate financings for a period of one year following the closing of the offering, subject to certain conditions and exceptions, and (ii) a covenant to not enter into any equity financings for 60 days from closing of the offering, subject to certain exceptions. The nature of the representations, warranties and covenants in the securities purchase agreements shall include:
| • | standard issuer representations and warranties on matters such as organization, qualification, authorization, no conflict, no governmental filings required, current in SEC filings, no litigation, labor or other compliance issues, environmental, intellectual property and title matters and compliance with various laws such as the Foreign Corrupt Practices Act; and |
| • | covenants regarding matters such as registration of warrant shares, no integration with other offerings, no stockholder rights plans, no material nonpublic information, use of proceeds, indemnification of purchasers, reservation and listing of shares of common stock, no subsequent equity sales for 60 days, subject to certain exceptions, and an agreement to not enter into variable rate financings for one (1) year from closing, subject to certain exceptions. |
The securities will be offered at a fixed price and are expected to be issued in a single closing. We expect this offering to be completed not later than two business days following the commencement of this offering, which will be the date that we enter into a securities purchase agreement to sell the securities offered hereby. We expect to close the offering on or before 2025. We will deliver all securities to be issued in connection with this offering delivery versus payment/receipt versus payment upon receipt of investor funds received by us. Accordingly, neither we nor the Placement Agent have made any arrangements to place investor funds in an escrow account or trust account since the Placement Agent will not receive investor funds in connection with the sale of the securities offered hereunder.
We expect to deliver the securities being offered pursuant to this prospectus on or about , 2025.
Fees and Expenses
The following table shows the per ordinary share and accompanying ordinary warrant and per pre-funded warrant and accompanying ordinary warrant and total placement agent fees we will pay in connection with the sale of the securities in this offering.
| | | Per Ordinary Share and
Accompanying Ordinary
Warrant | | | Per Pre-Funded
Warrant and Accompanying Ordinary Warrant | | | Total | |
| Public offering price | | |
|
|
|
|
|
|
|
|
| |
| Placement agent fees | | |
|
|
|
|
|
|
|
|
| |
| Proceeds to us, before expenses | | |
|
|
|
|
|
|
|
|
| |
We have agreed to pay the Placement Agent a cash fee equal to 7.0% of the aggregate gross proceeds raised in this offering and a management fee equal to 1.0% of the gross proceeds raised in this offering. In addition, we have agreed to pay the Placement Agent $25,000 for its non-accountable expenses and up to $100,000 for legal fees and out-of-pocket expenses and for its clearing expenses in the amount of up to $15,950. We estimate the total offering expenses of this offering that will be payable by us, excluding the Placement Agent fees and expenses, will be approximately $200,000.
Placement Agent Warrants
In addition, we have agreed to issue to the Placement Agent, or its designees, up to 180,412 warrants to purchase up to 180,412 ordinary shares (which represents 7.0% of the aggregate number of ordinary shares issued in this offering and issuable upon the exercise of the pre-funded warrants issued in this offering) with an exercise price of $2.425 per ordinary share (representing 125% of the assumed public offering price per ordinary share and accompanying common warrant) and exercisable for five years from the date of the commencement of sales in this offering. Such warrants will be subject to FINRA Rule 5110(e)(1) in that, except as otherwise permitted by FINRA rules, for a period of 180 days from the commencement of sales of this offering, the warrant shall not be sold, transferred, assigned, pledged, or hypothecated, or be the subject of any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of the securities by any person except as permitted by FINRA Rule 5110(e)(2). The Placement Agent warrants and underlying ordinary shares are registered on the registration statement of which this prospectus is a part. The form of the Placement Agent warrant will be included as an exhibit to this registration statement of which this prospectus forms a part.
We have granted the Placement Agent a right of first refusal for a period of 12 months following the closing of this offering to act as exclusive financial advisor, sole book-running manager, sole underwriter, sole placement agent or sole agent for each and every future debt financing or refinancing and public or private equity or debt offering or acquisition or disposition by us or any of our successors or subsidiaries.
Tail
We have also agreed to pay the Placement Agent a tail fee equal to the cash and warrant compensation in this offering, if any investor, who was contacted or introduced to us by the Placement Agent during the term of its engagement, provides us with capital in any public or private offering or other financing or capital raising transaction during the 12-month period following expiration or termination of our engagement of the Placement Agent.
Lock-up Agreements
We and our subsidiaries have agreed with the Placement Agent to be subject to a lock-up period of 60 days following the date of closing of the offering pursuant to this prospectus. Each of our officers and directors have agreed with the Placement Agent to be subject to a lock-up period of 90 days following the date of closing of the offering pursuant to this prospectus. This means that, during the applicable lock-up period, we and such persons may not offer for sale, contract to sell, sell, distribute, grant any option, right or warrant to purchase, pledge, hypothecate or otherwise dispose of, directly or indirectly, any of our shares of ordinary shares or any securities convertible into, or exercisable or exchangeable for, shares of ordinary shares, subject to customary exceptions. The Placement Agent may waive the terms of these lock-up agreements in its sole discretion and without notice. In addition, we have agreed to not issue any securities that are subject to a price reset based on the trading prices of our ordinary shares or upon a specified or contingent event in the future, or enter into any agreement to issue securities at a future determined price for a period of two years following the closing date of this offering, subject to an exception. The Placement Agent may waive this prohibition in its sole discretion and without notice.
Regulation M
The Placement Agent may be deemed to be an underwriter within the meaning of Section 2(a)(11) of the Securities Act, and any commissions received by it and any profit realized on the resale of the securities sold by it while acting as principal might be deemed to be underwriting discounts or commissions under the Securities Act. As an underwriter, the Placement Agent would be required to comply with the requirements of the Securities Act and the Exchange Act, including, without limitation, Rule 10b-5 and Regulation M under the Exchange Act. These rules and regulations may limit the timing of purchases and sales of our securities by the Placement Agent acting as principal. Under these rules and regulations, the Placement Agent (i) may not engage in any stabilization activity in connection with our securities and (ii) may not bid for or purchase any of our securities or attempt to induce any person to purchase any of our securities, other than as permitted under the Exchange Act, until it has completed its participation in the distribution.
Indemnification
We have agreed to indemnify the Placement Agent against certain liabilities, including certain liabilities arising under the Securities Act, or to contribute to payments that the Placement Agent may be required to make for these liabilities.
Determination of Offering Price and Ordinary Warrants Exercise Price
The actual offering price of the securities we are offering has been negotiated between us and the investors in the offering based on the trading of our ordinary shares and warrants prior to the offering, among other things. Other factors considered in determining the public offering price of the securities we are offering include our history and our prospects, the state of the biotechnology industry in which we operate, our recent operating results, including results of our pre-clinical studies and clinical trials, the general condition of the securities markets at the time of this offering, and such other factors as were deemed relevant.
Electronic Offer, Sale and Distribution of Securities
A prospectus in electronic format may be made available on the websites maintained by the Placement Agent, if any, participating in this offering and the Placement Agent may distribute prospectuses electronically. Other than the prospectus in electronic format, the information on these websites is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved or endorsed by us or the Placement Agent, and should not be relied upon by investors.
Other Relationships
From time to time, the Placement Agent or its affiliates have in the past or may in the future provide in the future, various advisory, investment and commercial banking and other services to us in the ordinary course of business, for which they have received and may continue to receive customary fees and commissions. However, except as disclosed in this prospectus, we have no present arrangements with the Placement Agent for any further services.
Listing
Our ordinary shares and warrants are currently listed on the Nasdaq Global Market under the symbols “SLXN” and “SLXNW”, respectively.
LEGAL MATTERS
The legality of the ordinary shares offered by this prospectus and certain other Cayman Islands legal matters will be passed upon for us by Conyers Dill & Pearman LLP. The legality of the ordinary warrants, pre-funded warrants and Placement Agent warrants offered by this prospectus and certain legal matters relating to U.S. law will be passed upon for us by Greenberg Traurig LLP. The Placement Agent is being represented Ellenoff Grossman & Schole LLP.
EXPERTS
The financial statements of Moringa Acquisition Corp as of December 31, 2023 and 2022, and for the two years ended December 31, 2023, included in this prospectus, have been so included in reliance on the report (which contains an explanatory paragraph relating to the Company’s ability to continue as a going concern as described in Note 1E to the financial statements) of Kesselman & Kesselman, a member firm of PricewaterhouseCoopers International Limited, an independent registered public accounting firm, given on the authority of said firm as experts in auditing and accounting.
The financial statements of Silexion Therapeutics Corp (formerly known as Silexion Therapeutics Ltd.) as of December 31, 2023 and 2022, and for the two years ended December 31, 2023, included in this prospectus have been so included in reliance on the report (which contains an explanatory paragraph relating to the Company’s ability to continue as a going concern as described in Note 1e to the financial statements) of Kesselman & Kesselman, a member firm of PricewaterhouseCoopers International Limited, an independent registered public accounting firm, given on the authority of said firm as experts in auditing and accounting.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act, with respect to the securities being offered by this prospectus. This prospectus, which constitutes part of the registration statement, does not contain all of the information in the registration statement and its exhibits. For further information with respect to New Silexion and the securities offered by this prospectus, we refer you to the registration statement and its exhibits. Statements contained in this prospectus as to the contents of any contract or any other document referred to are not necessarily complete, and in each instance, we refer you to the copy of the contract or other document filed as an exhibit to the registration statement. Each of these statements is qualified in all respects by this reference. You can read our SEC filings, including the registration statement, over the internet at the SEC’s website at www.sec.gov.
We are subject to the information reporting requirements of the Exchange Act, and we file reports, proxy statements and other information with the SEC. These reports, proxy statements and other information will be available for review at the SEC’s website at www.sec.gov. We also maintain a website at https://www.completesolaria.com/, at which you may access these materials free of charge as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC. The information contained in, or that can be accessed through, our website is not part of this prospectus.