QuickLinks -- Click here to rapidly navigate through this document
EXHIBIT 10.317
| |
|
|---|
| CONFIDENTIAL | | REDACTED VERSION |
[**] CERTAIN INFORMATION IN THIS EXHIBIT HAS BEEN
OMITTED AND FILED SEPARATELY WITH THE COMMISSION.
CONFIDENTIAL TREATMENT HAS BEEN REQUESTED WITH
RESPECT TO THE OMITTED PORTIONS.
Association Agreement Regarding the Sale and Servicing
of Blood Screening Products
This Association Agreement Regarding the Sale and Servicing of Blood Screening Products (this "Association Agreement") is made effective as of May 1, 2002, between America's Blood Centers ("ABC"), a charitable and non-profit corporation, having its principal office at 725 15th Street, N.W. Suite 700, Washington, D.C. 20005, and Chiron Corporation, a Delaware corporation ("Chiron") having its offices at 4560 Horton Street, Emeryville, California 94608-2916.
Background
- A.
- ABC is an association of non-profit community-based centers (each such blood center, a "Member").
- B.
- Chiron distributes, markets and sells certain Blood Screening Assays and Blood Screening Systems for use to conduct nucleic acid amplification tests to detect the presence of certain viruses in blood donation samples.
- C.
- ABC wishes to make available such amplified nucleic acid tests of blood donation samples by its Members using the Blood Screening Assays and Blood Screening Systems.
- D.
- Chiron and ABC desire to enter into an agreement on the following terms and conditions by which (i) Members may purchase Blood Screening Assays from Chiron; (ii) Members may obtain possession of and will use Blood Screening Systems that Chiron or a Third Party lessor will make available to ABC through sale or lease; and (iii) Members may obtain from Chiron installation, training, and instrument service related to the use of Blood Screening Systems.
NOW, THEREFORE, in consideration of the foregoing premises and of the mutual covenants of the parties hereinafter contained, the parties hereto hereby agree as follows:
ARTICLE 1—DEFINITIONS
ARTICLE 2—PURCHASE OF BLOOD SCREENING ASSAYS
Chiron agrees to sell, and Participating Members agree to buy, Blood Screening Assays in accordance with the terms and conditions set forth inSchedule Bhereto.
ARTICLE 3—PROVISION OF BLOOD SCREENING SYSTEMS; SOFTWARE
Chiron agrees to sell, or a Third Party designated by it and identified inSchedule Chereto (a "Third Party Lessor") will agree to lease, to Participating Members during the term hereof, Blood Screening Systems in accordance with the terms and conditions set forth inSchedule Chereto, and to license or sublicense the Software necessary for operation of the Blood Screening Assays on the
1
Blood Screening Systems also in accordance with the terms and conditions set forth inSchedule Chereto; provided that, if Blood Screening Systems are leased to ABC by a Third Party Lessor, the effectiveness of the applicable Member Supplement shall be subject to the Participating Member and the Third Party Lessor executing and delivering a separate agreement that provides for Third Party Lessor to lease Blood Screening Systems to the Participating Member pursuant to such terms and conditions (the "Third Party Systems Agreement").
ARTICLE 4—SERVICES
Installation, training and servicing of Blood Screening Systems (collectively, "Services"), shall be provided to each Participating Member by Chiron in accordance with the terms and conditions set forth inSchedule Dhereto.
ARTICLE 5—PRICES, PAYMENTS
- 5.1
- Prices.Each Participating Member agrees to pay to Chiron the prices set forth inSchedule Ehereto (as revised from time to time by mutual agreement of Chiron, ABC and the Participating Members) for the Products purchased hereunder, as determined by their respective testing volume, existing instrument base and financing selection. If a Third Party Lessor is provided for inSchedule Chereto, the prices to be paid by a Participating Member thereto will be as set forth in the Third Party Systems Agreement, as the case may be.
- 5.2
- [**]
- 5.3
- Payments.All payments shall be made on a monthly basis by each Participating Member as set forth inSchedule E. All payments due to Chiron hereunder shall be paid in full by each Participating Member in U.S. Dollars within thirty (30) days of the applicable due date. In the event of late payment, interest shall be charged at the rate of[**] from the date such payment was due until the date of actual payment, such interest to accrue daily and both before and after judgment. Invoices for amounts due from a Participating Member to a Third Party Lessor pursuant to the terms of Third Party Systems Agreement shall be payable as provided therein.
- 5.4
- Books & Records; Audit. Each Participating Member shall keep reasonably detailed and accurate records and books of account, including without limitation retaining all Data on donations tested, to enable a determination of the amounts payable to Chiron hereunder. Upon thirty (30) days written notice by Chiron, not more frequently than once per calendar year, Chiron, at its cost (except as otherwise provided below in this Section 5.4) may have the records and books of account of a Participating Member examined during reasonable business hours by an independent certified public accountant selected by Chiron for the purpose of verifying the amounts due hereunder. A copy of any final written report provided by the independent accountant to Chiron shall be given concurrently to the Participating Member. Such examination shall not be permitted unless[**] to which the books and records pertain. Where such examination results in a finding that a Participating Member underpaid Chiron[**], the Participating Member shall reimburse Chiron for its reasonable costs and expenses in conducting such examination. The Participating Member and Chiron shall promptly rectify any overpayments or underpayments by repaying such amounts together with interest thereon at the rate set forth in Section 5.3 of this Association Agreement. The parties shall endeavor to resolve any dispute between a Participating Member and Chiron as to amounts owing hereunder arising out of an audit pursuant to this Section 5.4 pursuant to Article 11 of this Association Agreement.
- 5.5
- Taxes. Each Participating Member is a non-profit, charitable corporation, exempt from the payment of sales and use taxes and shall have no tax liability on Products unless specifically legislated by a particular state. Chiron is responsible for requesting and obtaining all tax exemption numbers as required. Notwithstanding the above, if any federal, state, provincial, county or municipal sales or
2
use tax, excise or similar charge, or other tax assessment (other than that assessed against income), is assessed or charged on the sale of the Blood Screening Assays and Blood Screening Systems sold by Chiron pursuant to this Agreement, it shall be paid by the applicable Participating Member.
ARTICLE 6—CERTAIN AGREEMENTS
- 6.1
- Facilities. Each Participating Member agrees to grant Chiron reasonable access during normal business hours to inspect the facilities used for the conduct of nucleic acid amplification testing of blood donation samples. Participating Members shall perform such testing in accordance with all applicable laws and regulations and the instructions received from Chiron.
- 6.2
- Regulatory Approvals. Chiron will use commercially reasonable efforts, at Chiron's sole expense, to obtain and maintain any applicable regulatory approval for use of the Blood Screening Assays for testing of blood donations in the Territory. Chiron will be solely responsible for compliance with all regulatory requirements imposed on it in connection with the maintenance of such approvals, including all regulatory reporting requirements. ABC and the Participating Members will cooperate with Chiron to support its efforts pursuant to this Section 6.2. For clarity of understanding, Chiron has no obligation or responsibility with respect to regulatory approval or compliance relating to blood donor, blood product or blood recipient management.
- 6.3
- Regulatory Compliance.
- (a)
- The Participating Members shall be solely responsible for compliance with all reporting and other regulatory requirements imposed on them. Upon reasonable request of Chiron or any Participating Member, any Participating Member or Chiron shall provide to the requesting party copies of regulatory reports relating to the use of the Blood Screening Assays or the Blood Screening Systems.
- (b)
- Chiron shall be solely responsible for compliance with all reporting and other regulatory requirements imposed on it.
- (c)
- Any party hereunder agrees to make available to the requesting party (with authority to provide to its Affiliates or any governmental regulatory agency) such records as may be reasonably required for the requesting party to satisfy its regulatory requirements.
- (d)
- Each party agrees to provide access to their facilities and documents pertaining to this Agreement without any prior or written notice, to the FDA should it require access in accordance with any FDA policy, communication, or regulation.
- 6.4
- Data. Each Participating Member shall provide to Chiron data generated in connection with the use of the Blood Screening Assays or Blood Screening Systems sufficient to monitor the performance of the Products for quality assurance, to determine amounts due from the Participating Members hereunder, and to perform their respective regulatory obligations in the Territory (collectively the "Data"). The Data shall be deemed confidential information of the providing Participating Members, and shall be subject to Section 6.5, except that the limitations of Section 6.5 shall not apply to the uses of Data specifically authorized under Section 6.4(b) of this Association Agreement.
- (a)
- Chiron and to its Affiliates shall have the right to use the Data in connection with other regulatory applications, submissions and notifications, in any country, with respect to Blood Screening Assays, Blood Screening Systems or related products. ABC and the Participating Members agree to cooperate in providing to Chiron such information as either may reasonably believe appropriate or necessary and in applying for any such government approvals. At ABC's or any Participating Member's request, Chiron agrees to seek from such governmental
3
- 6.5
- Confidentiality. During the term of this Agreement and[**], absent the consent of the other party, (i) ABC and the Participating Members agree to keep in confidence and not to disclose to any Third Party other than their Affiliates, agents or contractors who need to know in connection with ABC and Participating Members activities under this Agreement, or use for any purpose, except pursuant to, and in order to carry out, the terms and objectives of this Agreement, any Confidential Information of Chiron, including Confidential Information of Gen-Probe which is disclosed to ABC or Participating Members by or through Chiron; and (ii) Chiron agrees to keep in confidence and not to disclose to any Third Party other than Gen-Probe and their respective Affiliates, agents or contractors who need to know in connection with Chiron activities under this Agreement, or use for any purpose, except pursuant to, and in order to carry out, the terms and objectives of this Agreement, any Confidential Information of ABC and Participating Members. Disclosures of Confidential Information to Third Parties authorized hereunder shall be permitted only if the Third Party is bound by confidentiality obligations not less restrictive than those set forth herein. Subject to this Section 6.5 and except as required by a court order issued by a court having appropriate jurisdiction, ABC and all Members agree not to disclose to any Third Party any financial terms of this Agreement, or any terms of this Agreement relating to the Blood Screening Assays and Blood Screening Systems provided hereunder, without the prior written consent of Chiron. Chiron agrees to not disclose to any Third Party any financial terms of this Agreement, or any terms of this Agreement relating to the Blood Screening Assays and Blood Screening Systems, except and only to the extent necessary to facilitate the enforcement of applicable most favored nations provisions. Notwithstanding the above, Chiron acknowledges that any disclosure by a Participating Member in violation of this Section 6.5 shall not be deemed a breach of this Agreement by ABC.
- 6.6
- Intellectual Property; Inventions. The provision by Chiron to the Participating Members of the Blood Screening Assays and Blood Screening Systems hereunder includes the implied license or sublicense under Chiron and Gen-Probe intellectual property to use the Blood Screening Assays and Blood Screening Systems in the Territory as provided herein. Except for such implied license, and except as specifically set forth herein, nothing in this Agreement conveys to any party any rights or licenses under any intellectual property of any other party. The parties do not anticipate that use of the Blood Screening Systems and Blood Screening Assays as provided herein will result in new inventions by Participating Members. However, in the event that any such new invention is made solely by a Participating Member or persons obligated to assign inventions to a Participating Member, arising from the use of the Blood Screening Systems and Blood Screening Assays, the Participating Member will own such invention. If such invention is an improvement to a Blood
4
Screening Assay or a component within a Blood Screening System, the Participating Member will provide written notice to Chiron of such invention, and the parties will commence exclusive negotiations,[**], for Chiron to obtain a worldwide license to such invention on commercially reasonable terms. If the parties fail to reach agreement on licensing terms, the Participating Member will be free to license the invention to any Third Party. If an invention is made jointly by a Participating Member or persons obligated to assign inventions to it, and Chiron or persons obligated to assign inventions to it, such invention will be owned jointly by the parties. All inventions made solely by Chiron or persons obligated to assign inventions to it will be owned by Chiron.
- 6.7
- Product Changes. In the event that any manufacturer of Blood Screening Assays or Blood Screening Systems makes a change which is required by applicable law or regulation, Chiron will provide ABC and the Participating Members with notice of such change promptly on receiving such notice from the manufacturer, and, if reasonably practicable, at least three months prior to implementation of such change. If any manufacturer makes an improvement to any Blood Screening Assays or Blood Screening Systems which is not mandated by applicable law or regulation, and if Chiron elects to make such improvement available in the Territory, Chiron will provide notice to ABC and the Participating Members of the availability of such improvement. Each Participating Member may, in its discretion, elect to implement such improvement, provided, however, that Chiron reserves the right to discontinue the sale, support and servicing of Blood Screening Assays or Blood Screening Systems that have been superceded by improved versions thereof,[**]. Each Participating Member shall be responsible, at its expense, for validation of its own procedures and satisfaction of all regulatory requirements applicable to the use of any improvements to the Blood Screening Assays or Blood Screening Systems which such Participating Member chooses to utilize. The parties acknowledge that the automated instrument system for single unit testing currently under development is not to be deemed an improvement for the purposes hereof. When commercially available, such automated system will be subject to separate terms and conditions to be negotiated at such time.
- 6.8
- Disaster Planning. Chiron shall work with ABC and the Participating Members to develop a mutually agreeable disaster preparedness and/or loss control plan to ensure continued supply of the Products in the event of a natural or man-made disaster which seriously affects or compromises production capabilities at the Product manufacturing or distribution facilities. In this connection, Chiron shall disclose to ABC and the Participating Members its backup supply capability, how it will keep the Participating Members supplied in the event of a loss or other disaster, how it segregates its own exposures, and its full fire and other loss protection measures for supply.
- 6.9
- Association Fee. Chiron agrees to pay ABC for management and service duties performed during the term of the Association Agreement in[**]. This amount shall be paid by Chiron to ABC quarterly within sixty (60) days of the close of each quarter.
ARTICLE 7—REPRESENTATIONS & WARRANTIES; WARRANTY DISCLAIMER; INDEMNIFICATION; LIMITATION OF LIABILITY; INSURANCE
- 7.1
- Chiron Representations & Warranties. Chiron hereby represents and warrants that: (a) it is duly organized, validly existing and in good standing under the laws of the jurisdiction in which it was organized; (b) this Agreement, when executed and delivered by it, will be the legal, valid and binding obligation of Chiron, enforceable against Chiron in accordance with its terms; (c) the execution, delivery and performance of this Agreement by Chiron do not and will not (i) conflict with, or constitute a breach or default under, its charter documents or any material agreement, contract, commitment, or instrument to which Chiron is a party or (ii) require the consent, approval or authorization of, or notice, declaration, filing or registration with, any Third Party or
5
any governmental or regulatory authority; (d) Chiron has not previously granted and will not grant any rights to any Third Party which are, nor contract with any Third Party in any manner which is, inconsistent with the rights granted herein.
- 7.2
- ABC Representations & Warranties. ABC hereby represents and warrants that: (a) it is duly organized, validly existing and in good standing under the laws of the jurisdiction in which it was organized; (b) this Agreement, when executed and delivered by it, will be the legal, valid and binding obligation of ABC, enforceable against ABC in accordance with its terms; (c) the execution, delivery and performance of this Agreement by ABC do not and will not (i) conflict with, or constitute a breach or default under, its charter documents or any material agreement, contract, commitment, or instrument to which ABC is a party or (ii) require the consent, approval or authorization of, or notice, declaration, filing or registration with, any Third Party or any governmental or regulatory authority; (d) ABC has not previously granted and will not grant any rights to any Third Party which are, nor contract with any Third Party in any manner which is, inconsistent with the rights granted herein.
- 7.3
- Participating Member Representations & Warranties. Each Participating Member, by executing a Member Supplement, represents and warrants that: (a) it is duly organized, validly existing and in good standing under the laws of the jurisdiction in which it was organized; (b) the Member Supplement, when executed and delivered by it, will be the legal, valid and binding obligation of it, enforceable in accordance with its terms; (c) all Products shall be used in accordance with the applicable manuals and instructions provided by Chiron and with all applicable laws and regulations; (d) it has all authority and permits required under applicable law and regulation to operate as a blood center and use the Products; (e) it is not, and it will not use in providing Chiron with any manner of service or work relating to this Agreement, a debarred person or entity under the Generic Drug Enforcement Act, or otherwise prohibited from providing such services or work, nor shall it use any investigator who has been disqualified by the FDA.
- 7.4
- Disclaimer of Warranty. EXCEPT AS SET FORTH IN SECTION 7.1 OF THIS ASSOCIATION AGREEMENT OR EXPRESSLY PROVIDED IN SCHEDULES B, C, AND D HEREOF, CHIRON MAKES NO WARRANTIES OF ANY KIND, EXPRESS OR IMPLIED, WRITTEN OR ORAL, INCLUDING WITHOUT LIMITATION ANY IMPLIED WARRANTIES OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE.
- 7.5
- Pass-Through of Warranty. Chiron agrees to pass through to each Participating Member the benefit of the manufacturer's warranties with respect to the Blood Screening Systems sold by Chiron after the effective date of the applicable Member Supplement, to the extent it is legally permitted to do so.
- 7.6
- Participating Member Responsibility for Certain Damages. In no event shall Chiron (or any Affiliate thereof) be responsible for any Damages suffered by ABC or a Participating Member arising out of a Participating Member's own negligence or willful acts or failure to act in connection with the storage, handling, or use of the Products after transfer to the Participating Member of risk of loss or damage thereto.
- 7.7
- Indemnity by Chiron. Chiron hereby indemnifies ABC and each Participating Member, their respective officers, directors, agents, and employees (the "ABC Indemnitees") and agrees to hold them harmless from and against all Direct Damages and against all Third Party Damages arising out of bodily injury claims of Third Parties, when such Damages arise from Chiron's breach of this Agreement or from the negligence of Chiron, its officers, directors, agents, employees or affiliates in the performance of Chiron's obligations under this Agreement, except to the extent arising from negligence or willful misconduct of any of the ABC Indemnitees.
6
- 7.8
- Indemnity by ABC. ABC hereby indemnifies Chiron and its respective officers, directors, agents and employees (the "Chiron Indemnitees") and agrees to hold them harmless from and against all Direct Damages and against all Third Party Damages arising out of bodily injury claims of Third Parties, when such Damages arise from ABC's breach of this Agreement or from the negligence of ABC, its officers, directors, agents or employees in the performance of its obligations under this Agreement, except to the extent arising from negligence or willful misconduct of any of the Chiron Indemnitees.
- 7.9
- Indemnity by Participating Members. Each Participating Member, by executing a Member Supplement, agrees to indemnify Chiron and its respective officers, directors, agents and employees (the "Chiron Indemnitees") and agrees to hold them harmless from and against all Direct Damages and against all Third Party Damages arising out of bodily injury claims of Third Parties, when such Damages arise from the Participating Member's breach of this Agreement or from the negligence of the Participating Member, its officers, directors, agents or employees in the performance of its obligations under this Agreement, except to the extent arising from negligence or willful misconduct of any of the Chiron Indemnitees.
- 7.10
- IP Infringement Indemnity. Chiron shall defend, at its expense, any legal action brought against ABC or a Participating Member by a Third Party to the extent that it is based on any claim that the use by a Participating Member of any Products supplied by Chiron and sold pursuant to this Agreement constitute an infringement of any patent or intellectual property rights claimed by such Third Party in the Territory. Chiron will pay all Damages finally awarded against ABC or a Participating Member in such action that are attributable to such claim. Notwithstanding the foregoing, Chiron shall have no liability hereunder to the extent that the infringement (or allegation of infringement) arises from or is attributable to (i) the use of the Products in combination with other products or materials not supplied by Chiron hereunder; (ii) part (or all) of the Products being used for a purpose other than that indicated by this Agreement; or (iii) use of the Products other than in accordance with the documentation provided by Chiron. Chiron's obligation to indemnify shall be subject to ABC or a Participating Member promptly notifying Chiron in writing of such claim and providing reasonable cooperation to Chiron in the defense of such claim or proceeding.
- 7.11
- Indemnification Procedures. Any party claiming indemnification under Section 7.7, 7.8 or 7.9 of this Association Agreement (the "Indemnitee") shall notify the party from which indemnification is claimed (the "Indemnifying Party") in writing promptly upon becoming aware of any claim to which such indemnification may apply. Failure to provide such notice shall constitute a waiver of the Indemnifying Party's indemnity obligations hereunder if, and only to the extent that, the Indemnifying Party is materially damaged thereby. The Indemnifying Party shall have the right to assume and control the defense of the claim at its own expense. If the right to assume and have sole control of the defense is exercised, the Indemnitee shall have the right to participate in, but not to control, such defense at its own expense. If the Indemnifying Party does not assume the defense of the claim, the Indemnitee may defend the claim at the Indemnifying Party's expense. The Indemnitee will not settle or compromise the claim without the prior written consent of the Indemnifying Party, and the Indemnifying Party will not settle or compromise the claim in any manner which would have an adverse effect on the Indemnitee without the consent of Indemnitee, which consent, in each case, will not be unreasonably withheld. The Indemnitee shall reasonably cooperate with the Indemnifying Party and will make available to the Indemnifying Party all pertinent information under the control of the Indemnitee.
- 7.12
- Exclusion of Consequential Damages. Excepting any obligation to indemnify against Third Party Damages as provided in this Agreement, no party to this Agreement shall be liable to any other party to this Agreement with respect to the subject matter of this Agreement under breach of contract, negligence, strict liability or any other cause of action for any Consequential Damages.
7
- 7.13
- Insurance. Chiron and the Participating Members agree to insure their potential liabilities resulting from this Agreement as follows:
- (a)
- Participating Members shall obtain and maintain, at their sole cost, a policy or policies of insurance with the coverages set forth in the applicable Member Supplement.
- (b)
- Chiron shall obtain and maintain at its sole cost, a policy or policies of insurance with the following coverages, which shall be in full force and effect for the term of this Agreement and thereafter through two years following the termination of the Agreement: a) a Commercial General Liability policy including Products and Completed Operations Liability in an amount of not less than[**] combined single limit for each occurrence; and b) Workers' Compensation coverage with statutory limits for each jurisdiction where the work required of such party under this Agreement is performed and an employers' liability policy with at least the following limits,[**] per accident,[**] per disease (policy limit), and[**] disease (each employee).
- (c)
- Each party shall provide the other with certificates of insurance evidencing the coverage required herein upon execution of this Agreement, and renewal certificates on request from the other party. Each party must notify the other within a reasonable time if there is a material change to any of the insurance policies referred to in this Section 7.13. Such certificates shall provide for thirty (30) days prior written notice to the certificate holder in the event of non-renewal of the policies, cancellation or material change in the coverage provided.
ARTICLE 8—TERM AND TERMINATION
- 8.1
- Term. This Association Agreement shall enter into force as of the Effective Date and shall continue to the[**]. Six months before the expiration of this Association Agreement, the parties shall meet to negotiate in good faith whether or not this Association Agreement shall be extended and/or revised and/or discontinued with effect from the termination date of such initial period. Each Member Supplement will become effective and terminate on its own terms; provided, however, that all Member Supplements shall terminate immediately upon the termination or expiration of this Association Agreement. Any termination of a Member Supplement shall have no impact on the continuing effectiveness of any other Member Supplement or this Association Agreement.
- 8.2
- Termination. This Association Agreement may be terminated by written notice to the other party at any time during the term of this Association Agreement, which termination shall be effective when such termination notice is received in accordance with Section 12.6 of this Association Agreement, as follows:
- (a)
- by either ABC or Chiron if the other party fails to observe, perform or otherwise breaches any of its material covenants, agreements or obligations under this Association Agreement, provided such failure continues for a period of thirty (30) days after receipt by the other party of an initial written notice thereof specifying such failure and provided further Chiron shall not have invoked its rights under Section 8.4 of this Association Agreement; or
- (b)
- by either ABC or Chiron if the other party files a petition in bankruptcy, becomes bankrupt or insolvent or subject to the reorganization of its business for the benefit of creditors under any law or regulation relating to bankruptcy, or a receiver is appointed for all or substantially all of its property or assets, or upon the making by such other party of a composition with its creditors, or upon the taking by such other party of any act for the winding up of its business, or upon any governmental authority exercising any power or authority resulting in the expropriation or confiscation of all or substantially all of its business and assets; or
- (c)
- by mutual agreement of the parties at any time without penalty.
8
- 8.3
- [**].
- 8.4
- [**].
ARTICLE 9—CONSEQUENCES OF THE TERMINATION OF THE AGREEMENT
- 9.1
- Payment Obligations Unaffected. The termination of this Association Agreement for any reason whatsoever shall not affect any party's obligations to pay any amount actually invoiced and due to Chiron prior to such termination. In the event that termination occurs at a time prior to a Participating Member having made full payment that is due and owing for any Products, the parties agree that Chiron shall have all applicable ownership rights to such Products until such payment has been made in full to Chiron.
- 9.2
- Survival of Certain Provisions. In addition, notwithstanding anything herein to the contrary, the following provisions of this Association Agreement shall survive termination of this Association Agreement: Sections 6.5, 6.6 and 7.4 through 7.12, and Articles 9, 10, 11 and 12.
- 9.3
- Return of Confidential Information. Upon termination of this Agreement for any reason whatsoever, unless required to retain such information for regulatory purposes, the parties shall immediately return to the other all confidential information in documentary or printed form and any copies or extracts thereof and shall thereafter cease to use such information but without prejudice to the then surviving confidentiality obligations provided for in Section 6.5 of this Association Agreement.
- 9.4
- No Other Payment. No indemnity or compensation in any form whatsoever shall be paid by either the party to the other in connection with a termination in accordance with the terms hereof (subject to any liability that may exist in respect of any material breach prior to such termination).
ARTICLE 10—APPLICABLE LAW
This Agreement shall be governed by and construed in accordance with the laws of the State of California, without giving effect to any conflict of law rules and regulations.
ARTICLE 11—DISPUTES
- 11.1
- To the extent that there are disputes with respect to performance under this Agreement, such disputes (other than non-payment) are not cause for Chiron to stop performance under this Agreement, but will be resolved in due course to the extent possible in accordance with this Article.
- 11.2
- The parties to this Agreement will attempt to resolve any problem or dispute arising out of, or related to, this Agreement through good faith consultation in the ordinary course of business. In the event that any problem or dispute is not so resolved, either party may upon written notice to the other request that the matter be referred to senior management officers within each respective organization with express authority to resolve the problem or issue and who are not immediately responsible for the matters contemplated by this Agreement. Such senior management officers will meet or confer at least once in good faith to negotiate a resolution.
- 11.3
- If the senior management officers are unable to resolve the problem or dispute within thirty (30) days, either party may pursue the matter as set forth in Sections 11.4 through 11.6 below. No party may institute a court proceeding until the procedure has been completed unless, and to the extent that, doing so is necessary to avoid irreparable harm.
- 11.4
- If any problem or dispute arising out of or related to this Agreement is not resolved by the parties in the manner set forth in Sections 11.2 and 11.3 above, at the request of either party, the matter will be submitted to mediation, or to such other form of dispute resolution as the parties may then agree to. A neutral person acceptable to both parties will conduct the mediation, and unless other
9
procedures are agreed to, it will be conducted in accordance with the Center for Public Resources Model Procedure for Mediation of Business Disputes.
- 11.5
- Any controversy, claim or dispute arising out of or relating to this Agreement, or the breach thereof, which is not resolved through the procedures described in Sections 11.2 through 11.4 above shall be resolved by binding arbitration. If arbitration is necessary pursuant to this paragraph, the parties shall agree upon a single arbitrator. If the parties are unable to agree on an arbitrator, then they will obtain nominations of three (3) potential arbitrators who are retired federal or state judges and each party will have the right to strike one candidate's name from the list.
- 11.6
- The prevailing party shall be entitled to recover all costs and expenses, including reasonable attorney's fees, incurred because of any legal action arising in relation to this Agreement.
ARTICLE 12—MISCELLANEOUS
- 12.1
- Force Majeure. Each party shall be excused from any delay in performance or from failure to perform in accordance with the terms of this Agreement, to the extent that such delay or failure to perform results from a Force Majeure Event. The provisions of this paragraph shall apply only if such party shall have used its reasonable efforts to avoid such Force Majeure Event. Such party shall give notice to the other promptly in writing upon learning of the Force Majeure Event. The affected party's time for performance shall be extended for the period of the delay or inability to perform due to such Force Majeure Event and notwithstanding any provision herein to the contrary, the affected party shall not be liable for any Damages arising out of such Force Majeure Event.
- 12.2
- Exclusion of Convention of Vienna for the International Sale of Goods. The parties hereby expressly exclude the application to this Agreement of the terms of the Convention of Vienna for the International Sale of Goods.
- 12.3
- Equal Employment Opportunity. None of the parties will discriminate, in terms and conditions of employment, against employees or applicants because of age, race, color, religion, sex, national origin, qualified disability or any other basis protected by applicable state or local law. The parties agree to abide by all federal, state and local employment and labor law notice posting requirements.
- 12.4
- Assignment. This Agreement shall not be directly or indirectly assigned or otherwise transferred by ABC or, as to a Member Supplement, by any Participating Member, nor, except as expressly provided hereunder, may any right or obligations of ABC or any Participating Member hereunder be assigned or transferred (whether voluntarily, by operation of law or otherwise) without the consent of Chiron. Chiron may assign and transfer to an Affiliate (provided Chiron remains a guarantor of such Affiliate's obligations hereunder), or to a Third Party possessing sufficient capitalization to satisfy its obligations under the Agreement that is acquiring all or substantially all of the business of Chiron, the rights and obligations of Chiron hereunder without further action by ABC or any Participating Member. This Agreement shall inure to the benefit of and be binding upon the parties hereto and their respective successors and permitted assigns.
- 12.5
- Subcontracting. Chiron's obligations under this contract may not be subcontracted without the prior written consent of the effected Participating Members. Any attempt to subcontract without such consent will be null and void and of no effect. Chiron must require that all subcontractors approved by the affected Participating Members be bound by the terms of this Agreement and to assume toward Chiron all obligations and responsibilities which Chiron assumes toward the affected Participating Members. Chiron must make available to each approved subcontractor, prior to the execution of any subcontract agreement, a copy of this Agreement to which the
10
subcontractor will be bound. For purposes of any subcontracts entered into pursuant to this Section 12.5, the term "Chiron" as used in this Agreement, will include any and all subcontractors.
- 12.6
- Notices. Any Notice or request required or permitted to be given in connection with this Agreement shall be deemed to have been sufficiently given if sent by pre-paid registered or certified mail, by courier or by facsimile at the address set forth below or to such other address as may have been notified in writing.
| |
| |
|
|---|
| | | If to Chiron: | | Chiron Corporation
Attention: President, Blood Testing
4560 Horton Street
Emeryville, CA 94608
Telephone: (510) 923-2416
Fax: (510) 655-8556
cc: General Counsel |
|
|
If to ABC: |
|
America's Blood Centers
Attention: Jim MacPherson, Chief Executive Officer
725 15th Street, N.W., Suite 700
Washington, DC 20005
Telephone: (202) 393-5725
Fax: (202) 393-1282 |
A notice, consent, approval or other communication takes effect from the time it is received unless a later time is specified in it, and receipt shall be deemed to occur as follows:
- (a)
- if it is sent by mail, seven (7) calendar days after posting;
- (b)
- if it is sent by courier, on the date and at the time shown on the courier's standard written confirmation of receipt;
- (c)
- if it is sent by facsimile, on the date and at the time shown on a successful transmission report by the machine from which the facsimile was sent.
- 12.7
- Entire Agreement. This Agreement constitutes the entire agreement between the parties in respect of the subject matter hereof. This Agreement cancels and supersedes any and all pre-existing agreements, either oral or in writing between the parties, including without limitation the Agreement, dated as of April 15, 2000 by and among Chiron, ABC and Gen-Probe, as amended. There are not and shall not be any oral statements, representations, warranties, undertakings or agreements between the parties other than as provided by this Agreement and any mutually accepted written amendments hereto.
- 12.8
- No Waiver. The failure on the part of either party hereto to exercise or enforce any right conferred upon it by this Agreement shall not be a waiver of any such right nor shall any single or partial exercise of any right or power hereunder or further exercise thereof operate so as to bar the later exercise or enforcement thereof.
- 12.9
- Nature of Relationship. Nothing herein contained shall be deemed to be or construed as constituting either party the agent or partner of the other party. The relationship between Chiron and each Participating Member shall be that of an independent contractor. No party shall have the right, title or authority to enter into any contract, agreement or commitment on behalf of the other or to bind the other party in any manner whatsoever. In no event shall ABC have any responsibility for the financial obligations of any Participating Member, nor shall any Participating Member have any responsibility for the financial obligations of any other Participating Member.
11
|
|
|---|
| 12.10 | Conflict Between Terms and Conditions. In the event of any conflict between the terms and conditions of this Association Agreement and any terms and conditions that may be set forth in a Member Supplement or on any invoice or purchase order or other similar document, the terms and conditions of this Association Agreement govern. |
12.11 |
Compliance with Applicable Law. In performing this Agreement, the parties shall comply with all applicable laws. Nothing in this Agreement shall be construed so as to require the violation of law, and wherever there is any conflict between any provision of this Agreement and any rule of mandatory law, the latter shall prevail, but in such event, the affected provision of this Agreement shall be ineffective only to the extent necessary to comply with the applicable law, and the parties hereto undertake to replace the invalid and/or unenforceable provision by a valid and/or enforceable provision, the nature and scope of which will come as close as possible to the contractual provision to be replaced. |
12.12 |
Counterparts. This Association Agreement and any Member Supplement may be executed in two or more counterparts, each of which shall be deemed to be an original and each of which shall constitute one and the same Agreement. |
12.13 |
Severability. In the event that any provision of this Agreement is found to be invalid or in conflict with any applicable law or regulation, the affected provision of this Agreement shall be limited or eliminated only to the extent necessary to comply with such law or regulation, and the remainder of this Agreement shall remain in full force and effect, provided that the remainder of this Agreement is consistent with the economic intentions of the parties as evidenced by this Agreement as a whole. |
IN WITNESS WHEREOF, the parties hereto, through their authorized representatives, have set their hands as of the date first above written, whereby they evidence their intent to be legally bound.
| CHIRON CORPORATION | | AMERICA'S BLOOD CENTERS |
By: |
|
/s/ WILLIAM G. GREEN
|
|
By: |
|
/s/ JIM MACPHERSON
|
Name: |
|
William G. Green |
|
Name: |
|
Jim MacPherson |
Title: |
|
President of Blood Testing |
|
Title: |
|
CEO |
12
| |
|
|---|
| CONFIDENTIAL | | REDACTED VERSION |
SCHEDULE A
Definitions
Capitalized terms used and not otherwise defined in this Agreement shall have the following meanings:
- 1.1
- "Affiliate" shall mean (i) any corporation or business entity of which securities or other ownership interests representing fifty percent (50%) or more of the equity or fifty percent (50%) or more of the ordinary voting power or fifty percent (50%) or more of the general partnership interests are, at the time such determination is being made, owned, Controlled (as hereinafter defined) or held, directly or indirectly, by such corporation or business entity, or (ii) any other corporation or business entity which, at the time such determination is being made, is Controlling, Controlled by or under common Control with, such corporation or business entity. For purposes of this definition, "Control", whether used as a noun or verb, refers to the possession, direct or indirect, of the power to direct, or cause the direction of, the management or policies of any corporation or business entity, whether through the ownership of voting securities, by contract or otherwise.
- 1.2
- "Agreement" shall mean collectively this Association Agreement, including the Schedules hereto, and all Member Supplements, including the Exhibits thereto.
- 1.3
- "Blood Screening Assays" shall mean the TMA Assays purchased by Chiron from Gen-Probe from time to time and available for sale hereunder, as more specifically described in one or more schedules, labeled B-1, B-2, etc.
- 1.4
- "Blood Screening Field" shall mean the testing of human blood, plasma, platelets or other blood products intended for transfusion or further processing for other administration to humans, including autologous donors, as limited by the Intended Use for each Blood Screening Assay, as set forth in the applicable Package Insert, as amended from time to time.
- 1.5
- "Blood Screening Systems" shall mean the instrument(s) and related Software for DNA/RNA amplified assay processing that Chiron has available for sale or lease hereunder, as more specifically described inSchedule C, as it may be amended from time to time.
- 1.6
- "Confidential Information" means any and all technical, business and other information and materials disclosed by or on behalf of such party to the other party pursuant to this Agreement or during discussions leading to this Agreement, except to the extent that the receiving party can provide evidence that such information:
- (a)
- is known to the receiving party prior to its disclosure by the disclosing party; or
- (b)
- is obtained by the receiving party from a source other than the disclosing party which source (i) did not require the receiving party to hold such information in confidence; or (ii) did not limit or restrict the receiving party's use thereof, or
- (c)
- has become public knowledge otherwise than through the fault of the receiving party; or
- (d)
- has been developed by the receiving party independently of the information received from the disclosing party as shown by the receiving party's written records; or
- (e)
- is required to be disclosed by the receiving party by law or for the purpose of complying with governmental regulations and/or the obligations of the receiving party to a licensing or regulatory authority in connection with this Agreement.
13
- 1.7
- "Consequential Damages" means consequential damages as defined by California law, including loss of profit or loss of business opportunity, and all Third Party Damages.
- 1.8
- "Damages" means Direct Damages and Third Party Damages, collectively.
- 1.9
- "Direct Damages" means costs or expenses incurred by a party that are not Consequential Damages, including without limitation, the incremental additional costs of substitute products and costs or expenses of product recall.
- 1.10
- "Documentation" shall mean text material that describes the design, functions, operation, or use of the Software and that is delivered by Chiron to Participating Members. Documentation for the Software shall be the same that is provided to licensees of such Software generally.
- 1.11
- "Effective Date" means the date set forth on the first page of this Agreement.
- 1.12
- "FDA" shall mean the United States Food and Drug Administration, or the successor thereto.
- 1.13
- "Force Majeure Event" shall mean a cause beyond the reasonable control of a party, including without limitation, fires, floods, epidemics, quarantine restrictions, strikes, war, earthquake, acts of God, labor difficulties, riot, failure of public utilities, freight embargoes, unusually severe weather conditions, delays in delivery of goods or services by suppliers or subcontractors to such party, loss of goods in transit, and governmental or court action.
- 1.14
- "Gen-Probe" shall mean Gen-Probe Incorporated, a Delaware corporation.
- 1.15
- "HCV" means the Hepatitis C virus.
- 1.16
- "HIV-1" means Human Immunodeficiency virus type 1.
- 1.17
- "Member" shall have the meaning set forth in the Recitals.
- 1.18
- "Member Supplement" shall mean the agreement executed after the Effective Date of this Association Agreement by and among Chiron and a Member and incorporating by reference the terms and conditions of this Association Agreement.
- 1.19
- "Package Insert" shall mean the package insert approved by the FDA for the applicable Blood Screening Assay, as the same may be amended from time to time.
- 1.20
- "Participating Member" shall mean a Member electing to utilize the Blood Screening Assays and Blood Screening Systems that memorializes its election by execution of a Member Supplement.
- 1.21
- "Person" shall mean an individual, corporation, partnership, limited liability company, trust, business trust, association, joint stock company, joint venture, pool, syndicate, sole proprietorship, unincorporated organization, governmental authority or any other form of entity not specifically listed herein.
- 1.22
- "Pooled Testing" means the conduct of testing on pools of samples from blood donations as follows. For the purposes of this Agreement, Pooled Testing consists of:
- 1.23
- "Primary Operators" means the employees of Participating Members trained by Chiron at Chiron's facility.
- 1.24
- [**].
- 1.25
- "Products" shall mean Blood Screening Assays, Blood Screening Systems and Software.
14
- 1.26
- "Reagent Utilization Factor" means, with respect to a specified time period, the quantity of Blood Screening Assay tests consumed by an applicable Participating Member during such period divided by the number of Reportable Results obtained by such Participating Member during such period.
- 1.27
- "Reportable Result" means a result obtained through the use of a Blood Screening Assay and Blood Screening System in Pooled Testing or Single Unit Testing from which it is determined to release for use or hold and not use (a) a blood donation intended for transfusion or for further processing for other administration to humans or (b) a product derived from such donation.
- 1.28
- "Secondary Operator" means the employees of Participating Members trained at the facility of a Participating Member by either a Primary Operator or Chiron.
- 1.29
- "Single Unit Testing" means testing of blood donations consisting of (i) testing a sample from each individual blood donation using a Blood Screening Assay, and (ii) follow up discriminatory testing of positive results for HIV-1 or HCV.
- 1.30
- "Software" means the following software programs:
CPT-16 Pooling Software, Version 2.0.0.2
Procleix™ Assay Software, Version 2.0.0.76
Procleix™ Assay Software LHP, Version 2.1
Procleix™ System Software, Version 3.0.3.4
Procleix™ Protocol, Version 2.1.0.0
Procleix™ Worklist Editor, Version 3.0.3.2
- 1.31
- "Specifications" shall mean, in respect of each Product, the specifications, tests, procedures, process description, and other information relating to such Product and packaging thereof prepared by Chiron or its Affiliates provided to Participating Members by Chiron under separate cover, which may be amended from time to time by Chiron in writing; provided, however, that the foregoing information shall be consistent with the product registration in the Territory from time to time. As to Blood Screening Assays, "Specifications" shall mean the information set forth in the current Package Insert.
- 1.32
- "Tecan Instrument" means a Tecan Genesis 150/8 Instrument, sold by Chiron or leased by a Third Party Lessor hereunder for use in the performance of the assays using the Blood Screening Assays.
- 1.33
- "Territory" means the United States of America, including Puerto Rico, Guam and all other protectorates.
- 1.34
- "Testing Centers" mean the locations identified on Exhibit 2 to the Member Supplements.
- 1.35
- "Third Party" shall mean any Person other than ABC, its Members, Gen-Probe, Chiron and their respective Affiliates.
- 1.36
- "Third Party Damages" means any liability arising out of a Third Party's claim (whether arising out of fault, strict liability or otherwise) in the form of an obligation, loss, fine, judgment for damages, arbitration award, settlement amount, penalty or claim, and all reasonable costs and expenses related thereto (including reasonable costs of investigation, fees and expenses payable to outside counsel, independent accountants and similar professional advisors or consultants, but not including any corporate allocation for use, of similar in-house services or facilities).
- 1.37
- "TMA Assay" shall mean an in vitro diagnostic assay based on or utilizing transcription-mediated amplification.
15
| |
|
|---|
| CONFIDENTIAL | | REDACTED VERSION |
SCHEDULE B-1
TERMS AND CONDITIONS FOR PURCHASE OF PROCLEIX™ HIV-1/HCV
BLOOD SCREENING ASSAY
Chiron will supply the Procleix™ HIV-1/HCV Blood Screening Assay to the Participating Members on the following terms and conditions:
- 1.
- Description of Blood Screening Assays. The Procleix™ HIV-1/HCV Blood Screening Assays that will be supplied by Chiron to the Participating Members hereunder are described in the current Package Insert, included as Attachment B-1 to this Schedule B-1 and include the following components:
Procleix™ HIV-1/HCV Assay
Internal Control Reagent
Target Capture Reagent
Amplification Reagent
Enzyme Reagent
Probe Reagent
Selection Reagent
Procleix™ Negative Calibrator
Procleix™ HIV-1 Positive Calibrator
Procleix™ HCV Positive Calibrator
- 2.
- Supply Obligation. During the term of this Agreement Chiron agrees to supply to each Participating Member, at the prices specified inSchedule E, all Blood Screening Assays necessary to permit each Participating Member to conduct screening of blood donations for the presence of HIV-1 and /or HCV in the Territory. Each Participating Member agrees to store and handle all Blood Screening Assays in accordance with the Package Insert. Any Blood Screening Assays lost or damaged due to failure to comply with such storage and handling instructions will be replaced at the affected Participating Member's sole expense, which expense reflects the Reagent Utilization Factor premium specified inSchedule E.
- 3.
- Purchase Obligation. During the term of this Agreement, each Participating Member agrees to acquire 100% of its requirements of Blood Screening Assays necessary to conduct screening by nucleic acid testing of blood donations for the presence of HIV-1 and/or HCV. However, nothing in this Agreement will restrict the right of a Participating Member to evaluate new technologies or to perform, in its discretion, any confirmatory and supplementary nucleic acid testing.
- 4.
- Other Supplies. All supplies necessary for the conduct of tests using the Blood Screening Assays, other than Blood Screening Assays, Blood Screening Systems and such disposables and calibrators included with each Blood Screening Assay Kit, shall be purchased separately by each Participating Member and the price of such supplies is not included in the fees established by this Agreement. Certain of such supplies and the associated prices thereof as of the date of execution of this Agreement are identified inSchedule E. Such prices of supplies shall increase from time to time in the event of, and in the amount of, any price increases by the manufacturers of such supplies.
- 5.
- Packing and Delivery of Blood Screening Assays. All Blood Screening Assays will be suitably packed and transported to ensure safe transport to the Participating Members in accordance with the
16
manufacturer's instructions and delivered Carriage and Insurance paid to the Participating Member's designated Testing Centers, and each delivery will be accompanied by a packing slip indicating the quantity delivered. Blood Screening Assays will be delivered during normal business hours and accompanied by storage instructions and arrive at the temperatures specified by the manufacturer and set out in the Package Insert. The Blood Screening Assays will be delivered in kit form, with appropriate distribution between screening tests, discriminatory tests and related calibrators.
- 6.
- Shipping. Except as set forth below, all shipping and handling charges for the Blood Screening Systems and the regular monthly shipments of Blood Screening Assays will be borne entirely by Chiron on shipments to the Testing Centers. All shipping and handling charges for shipping requests other than to a Testing Center, and all incremental shipping and handling charges for Modified Orders and Changed Orders, will be borne by the requesting Participating Member.
- 7.
- Forecasts and Orders.
- 7.1
- Each Participating Member will provide Chiron with a written twelve month forecast of its requirements of Blood Screening Assays (each a "Product Requirements Forecast") on an annual basis, including requested delivery dates, which forecast shall constitute such Participating Member's standing firm purchase order for each month contained therein. Not less than ninety (90) days prior to each anniversary of the effective date of the applicable Member Supplement, each Participating Member will provide such a Product Requirements Forecast. Participating Members shall deliver any modifications to the standing firm purchase order for Blood Screening Assays for delivery during the next following month on or prior to the first day of the immediately preceding calendar month (for example, a Participating Member will provide requested modifications to the February standing firm purchase order—for deliveries to be made during the month of February—prior to January 1). All purchase order modification requests must be received at least 30 days prior to a requested delivery date. Except as otherwise agreed, Chiron agrees to accept modifications to standing firm purchase orders which do not differ by more than[**] from the quantities forecast for delivery in the applicable month in the current Product Requirements Forecast. Participating Members may adjust their Product Requirements Forecast from time to time with the consent of Chiron, such consent to not be unreasonably withheld.
- 7.2
- If the variance between the modification requested and the current Product Requirements Forecast exceeds the[**] referenced in Section 7.1 of thisSchedule B-1, Chiron will notify the requesting Participating Member within five (5) business days after receipt of the modification request as to whether it accepts such modification request. If a modification request is accepted, Chiron agrees to deliver such amounts of Blood Screening Assays on the delivery date(s) stated. If a modification request is not accepted, the parties will use all reasonable endeavors to agree on further modifications to the order and/or delivery date (a "Modified Order"), subject to Chiron's manufacturer's accommodating Chiron's requests for changes in forecasted amounts.
- 8.
- Changes to Orders. If a Participating Member requests any modifications to quantities of Blood Screening Assays ordered or the delivery schedule or locations reflected in such order within 30 days of a delivery date (a "Changed Order"), Chiron will use reasonable commercial efforts to comply with such request.
- 9.
- IQA Procedures. Prior to the initial delivery of Blood Screening Assays, Chiron, ABC and the Participating Members shall mutually agree upon procedures for quality assurance testing of Blood Screening Assays received by Participating Members ("IQA Procedures"). Chiron agrees to replace Blood Screening Assays which fail to conform to the Specifications as set forth in the Package Insert. The IQA Procedures shall include procedures for notice to Chiron and for implementing the return of Blood Screening Assays for replacement. If testing by Chiron or its supplier confirms the non-conformity of the Blood Screening Assays, Chiron will bear the shipping costs associated
17
with replacement. If Chiron's testing provides reasonably acceptable evidence to the applicable Participating Member that the Blood Screening Assays do conform to the specifications in question, the shipping costs will be borne by the applicable Participating Member.
- 10.
- Technical Support. Chiron will provide Participating Members with technical support to facilitate the operation by Participating Members of the Blood Screening Assays on the Blood Screening Systems.
- 11.
- Nature of Testing. Each Participating Member agrees that Blood Screening Assays supplied by Chiron hereunder shall be used by the Participating Member solely to conduct Pooled Testing with a pool size of 16. If a Participating Member desires to change its testing from Pooled Testing using a pool size of 16 to Single Unit Testing or any other pool size, the Participating Member and Chiron shall enter into an addendum to the applicable Member Supplement reflecting any additional Blood Testing Systems, changes to the fees or prices payable underSchedule Eof this Agreement or any other amendments that may be applicable by reason of such change in the nature of the Participating Member's testing of blood donations.
- 12.
- Warranty; Limitation of Liability.
- 12.1
- All Blood Screening Assays supplied by Chiron shall be manufactured in accordance with, and shall conform with, the Specifications, provided that ABC uses the Blood Screening Assays in accordance with the instructions provided by Chiron. Any Blood Screening Assays failing to meet such Specifications shall be replaced by Chiron at Chiron's cost unless the defect was caused by an act or omission of a Participating Member.
- 12.2
- Chiron disclaims all other warranties and further limits its liability in accordance with Sections 7.4 and 7.12 of this Association Agreement, respectively.
18
ATTACHMENT B-1
Package Insert
Procleix™HIV-1/HCV Assay
ForIn Vitro Diagnostic Use
1000 Test Kit, 5000 Test Kit
| |
|
|---|
| TABLE OF CONTENTS | | |
| | INTENDED USE | | 1 |
| | SUMMARY AND EXPLANATION OF THE TEST | | 2 |
| | PRINCIPLES OF THE PROCEDURE | | 2 |
| | MATERIALS PROVIDED | | 4 |
| | MATERIALS REQUIRED, SOLD SEPARATELY | | 5 |
| | MATERIALS REQUIRED BUT NOT PROVIDED | | 6 |
| | REAGENTS | | 7 |
| | STORAGE INSTRUCTIONS | | 8 |
| | PRECAUTIONS | | 9 |
| | REAGENT PREPARATION | | 11 |
| | SPECIMEN COLLECTION, STORAGE, AND HANDLING | | 12 |
| | PROCEDURAL NOTES | | 13 |
| | INSTRUCTIONS FOR USE | | 16 |
| | | Target Capture | | 16 |
| | | Amplification | | 18 |
| | | Hybridization Protection Assay (HPA) | | 19 |
| | | Procleix HIV-1 and HCV
Discriminatory Assays | | 20 |
| | QUALITY CONTROL PROCEDURES | | 21 |
| | | Acceptance Criteria for the Procleix
HIV-1/HCV Assay and Procleix
HIV-1 and HCV Discriminatory Assays | | 21 |
| | | | Acceptance Criteria for the Calibration and Calculation of Cutoff | | 22 |
| | INTERPRETATION OF RESULTS | | 28 |
| | PERFORMANCE CHARACTERISTICS | | 30 |
| | PERFORMANCE OF POOLED SAMPLE TESTING | | 32 |
| | PERFORMANCE OF INDIVIDUAL DONATION TESTING | | 46 |
| | LIMITATIONS OF THE PROCEDURE | | 48 |
| | CONCLUSIONS | | 49 |
| | BIBLIOGRAPHY | | 49 |
INTENDED USE
The Procleix™ HIV-1/HCV Assay* is a qualitativein vitro nucleic acid assay system for the detection of human immunodeficiency virus type 1 and/or hepatitis C virus RNA in human plasma from donations of whole blood and blood components for transfusion. The assay is intended for use in screening individual donor samples or pools of human plasma comprised of equal aliquots of not more than 16 individual donations. This assay is intended to be used in conjunction with licensed tests for detecting antibodies to HIV-1 and HCV.
This assay may be used as an alternative to licensed HIV-1 p24 antigen tests for screening human plasma from donations of whole blood and blood components.
This assay is not intended for use as an aid in diagnosis.
*Developed and manufactured by Gen-Probe Incorporated; distributed by Chiron® Corporation.
IN0076 Rev. F
1
SUMMARY AND EXPLANATION OF THE TEST
Epidemiological studies identified human immunodeficiency virus type 1 (HIV-1) as the etiological agent of acquired immunodeficiency syndrome (AIDS)1-7 and hepatitis C virus (HCV)8-13 as the etiological agent for most blood-borne non-A, non-B hepatitis (NANBH). Both viruses are transmitted primarily by exposure to infected blood or blood products, certain body fluids or tissues, and from mother to fetus or child.
Current detection of HIV-1 infection in the blood bank setting is based on serologic screening for anti-viral antibodies by enzyme immunoassay (EIA) with confirmation by supplemental antibody tests such as Western blot or immunofluorescence assays. Although sensitivity of HIV-1 antibody detection has increased in the last few years and sensitive tests for p24 antigen (p24Ag) have been developed and implemented, a window period between infection and detectable serological markers still exists14,16-17. The screening of blood with current EIA tests results, on average, in a 22-day seronegative window17. Although addition of the p24Ag test allows earlier detection of HIV-1 infection, implementation of the p24Ag test in the U.S. yielded a very modest number of Ab(-)/Ag(+) donors with no significant reduction in the risk of infection. Several studies suggest that addition of nucleic acid-based amplification tests would reduce the window period of detection to 6-11 days, preventing more than half of the HIV-1 infections by blood transfusion14.
Nucleic Acid Testing (NAT) of whole blood donations has been in place in the United States since early 1999. Stramer et al., have reported the results of testing small pools of 16, 24 and 128 plasma samples15,30 under IND (Investigational New Drug Application) clearances from the FDA. As of November 2001, the major programs (pooled and individual donation testing) in the U.S. have tested a total of 24.9 million donations for HCV RNA and HIV-1 RNA. A total of 89 donations (1:280,000 screened units) were confirmed to be positive for HCV RNA and negative in serological testing. Similarly, nine HIV-1 RNA positive, serologically negative donations (1:2,767,000) were identified. During this testing period, two specimens were reactive for HIV-1 RNA and p24 Ag but nonreactive by HIV antibody testing.
Detection of HCV is based on serologic screening for anti-viral antibodies with enzyme-linked immunosorbent assays (ELISA) or enzyme immunoassays (EIA) and confirmation with a Strip Immunoblot Assay (e.g., RIBA® SIA). Even though the development of these tests has significantly reduced the incidence of post transfusion HCV infection in the U.S., risk of contracting HCV through transfusion still exists14,16-17. Recent studies indicate that nucleic acid-based amplification tests for HCV RNA will allow detection of HCV infection approximately 59 days earlier than the current antibody-based tests17.
The Procleix HIV-1/HCV Assay utilizes target amplification nucleic acid probe technology for the detection of HIV-1 and HCV RNA in voluntary blood donors18. The assay contains reagents which may be used for simultaneous detection of both viruses or individual viruses, HIV-1 and HCV. All three assays incorporate an Internal Control for monitoring assay performance in each individual specimen.
PRINCIPLES OF THE PROCEDURE
The Procleix HIV-1/HCV Assay involves three main steps which take place in a single tube: sample preparation; HIV-1 and HCV RNA target amplification by Transcription-Mediated Amplification (TMA)19; and detection of the amplification products (amplicon) by the Hybridization Protection Assay (HPA)20.
During sample preparation, RNA is isolated from plasma specimens via the use of target capture. Plasma is treated with a detergent to solubilize the viral envelope, denature proteins and release viral genomic RNA. Oligonucleotides ("capture oligonucleotides") that are homologous to highly conserved
IN0076 Rev. F
2
regions of HIV-1 and HCV, are hybridized to the HIV-1 or HCV RNA target, if present, in the test specimen. The hybridized target is then captured onto magnetic microparticles that are separated from plasma in a magnetic field. Wash steps are utilized to remove extraneous plasma components from the reaction tube. Magnetic separation and wash steps are performed with the Procleix TCS.
Target amplification occurs via TMA, which is a transcription-based nucleic acid amplification method that utilizes two enzymes, MMLV reverse transcriptase and T7 RNA polymerase. The reverse transcriptase is used to generate a DNA copy (containing a promoter sequence for T7 RNA polymerase) of the target RNA sequence. T7 RNA polymerase produces multiple copies of RNA amplicon from the DNA copy template. The Procleix HIV-1/HCV Assay utilizes the TMA method to amplify regions of HIV-1 RNA and of HCV RNA.
Detection is achieved by HPA using single-stranded nucleic acid probes with chemiluminescent labels that are complementary to the amplicon. The labeled nucleic acid probes hybridize specifically to the amplicon. The Selection Reagent differentiates between hybridized and unhybridized probes by inactivating the label on unhybridized probes. During the detection step, the chemiluminescent signal produced by the hybridized probe is measured in a luminometer and is reported as Relative Light Units (RLU).
Internal Control is added to each test specimen, external quality control, or assay calibrator tube via the Target Capture Reagent that contains the Internal Control. The Internal Control in this reagent controls for specimen processing, amplification and detection steps. Internal Control signal in each tube or assay reaction is discriminated from the HIV-1/HCV signal by the differential kinetics of light emission from probes with different labels21. Internal Control specific amplicon is detected using a probe with rapid emission of light (termed flasher signal). Amplicon specific to HIV-1/HCV is detected using probes with relatively slower kinetics of light emission (termed glower signal). The Dual Kinetic Assay (DKA) is a method used to differentiate between the signals from flasher and glower labels21. When used for the simultaneous detection of HIV-1 and HCV, the Procleix HIV-1/HCV Assay differentiates between Internal Control and combined HIV-1/HCV signals but does not discriminate between individual HIV-1 and HCV signals.
Specimens found to be reactive in the Procleix HIV-1/HCV Assay must be run in individual HIV-1 and HCV Discriminatory Assays to determine if they are reactive for HIV-1, HCV or both.
The Procleix HIV-1 and HCV Discriminatory Assays utilize the same three main steps as the Procleix HIV-1/HCV Assay (target capture, TMA and HPA); the same assay procedure is followed with one difference: HIV-1-specific or HCV-specific probe reagents are used in place of the Procleix HIV-1/HCV Assay Probe Reagent.
IN0076 Rev. F
3
MATERIALS PROVIDED
| Procleix HIV-1/HCV Assay | | |
| | | 1000 Test Kit P/N 301031
5000 Test Kit P/N 301030 |
| | Internal Control Reagent | | |
| | Target Capture Reagent | | |
| | Amplification Reagent | | |
| | Enzyme Reagent | | |
| | Probe Reagent | | |
| | Selection Reagent | | |
| | Procleix Negative Calibrator | | |
| | Procleix HIV-1 Positive Calibrator | | |
| | Procleix HCV Positive Calibrator | | |
IN0076 Rev. F
4
MATERIALS REQUIRED, SOLD SEPARATELY
| |
|
|---|
Procleix HIV-1 and HCV
Discriminatory Probe Reagents | | P/N 301026 |
| |
HIV-1 Discriminatory Probe Reagent |
|
|
| | HCV Discriminatory Probe Reagent | | |
Procleix Assay Fluids |
|
P/N 301027 |
| | Wash Solution | | |
| | Oil | | |
| | Buffer for Deactivation Fluid | | |
Procleix Auto Detect Reagents |
|
P/N 301038 |
| | Auto Detect 1 | | |
| | Auto Detect 2 | | |
Disposables |
|
|
(Disposables are single use only, do not reuse.
Use of other disposables is not recommended.) | | |
| | Ten-Tube Units (TTUs) | | P/N TU0022 |
| | Ten Tip Cassettes | | P/N 104578 |
| | Sealing Cards | | P/N 102085 |
Procleix HIV-1/HCV
Proficiency Panel |
|
P/N 301034 |
| | HIV-1 Positive Panel Members | | |
| | HCV Positive Panel Members | | |
| | HIV-1 and HCV Positive Panel Member | | |
| | Negative Panel Members | | |
Procleix HIV-1 and HCV
External Quality Controls |
|
P/N 301035 |
| | HIV-1 Positive External Quality Control | | |
| | HCV Positive External Quality Control | | |
| | Negative External Quality Control | | |
Procleix HIV-1/HCV
Assay Calibrators |
|
P/N 301036 |
| | HIV-1 Positive Calibrator | | |
| | HCV Positive Calibrator | | |
| | Negative Calibrator | | |
Chiron CPT (Correlated Pipetting Transfer)
Pooling Software (Only required for pooling) |
|
P/N 10001970 |
The Chiron CPT Pooling Software, used in combination with the TECAN GENESIS RSP, performs sample scanning and pooling operations that combine aliquots from 16 individual samples into a single Master Pool Tube, which may be used for further testing.
IN0076 Rev. F
5
Equipment
All equipment is available from Chiron Corporation unless otherwise noted.
| |
|
|---|
| Procleix System | | P/N 104955 (nominal 115V)
P/N 104954 (nominal 230V) |
Dedicated fixed or adjustable repeat pipettor capable of delivering 400 µL of Target Capture Reagent with a ± 5% accuracy and a precision of< 5% CV. (Only required for manual sample pipetting method.)
Dedicated single channel pipettor capable of delivering 500 µl of specimen with a ± 5% accuracy and a precision of< 5% CV. (only required for manual sample pipetting method.)
MATERIALS REQUIRED BUT NOT PROVIDED
Eppendorf COMBITIPS repeat pipettor tips (12.5 mL, 5.0 mL, 1.25 mL) or equivalent
Disposable 1000 µL filter tips in rack
Bleach
Sterile, polypropylene conical tubes with sealing caps
Freestanding tubes are recommended in two different sizes (5 mL to 10 mL tube and> 30 mL tube). The tubes must be able to accommodate the diameter of an Eppendorf Repeat pipettor tip
TECAN GENESIS disposable 1000 µL conductive filter tips
TECAN 100 mL reagent troughs
IN0076 Rev. F
6
REAGENTS
Procleix HIV-1/HCV Assay Kit:
P/N 301031 - 1000 Test Kit
P/N 301030 - 5000 Test Kit
CONTENTS
| | Number of vials/
Volume per vial
|
|---|
Reagent Name
| | 1000
Test Kit
| | 5000
Test Kit
|
|---|
Internal Control Reagent
A HEPES buffered solution containing detergent and an RNA transcript. | | 2 X 5
mL | | 10 X 5
mL |
| Storeunopened reagent at –15° to –35°C. | | | | |
Target Capture Reagent
A HEPES buffered solution containing detergent, capture oligonucleotides and magnetic microparticles. |
|
2 X 280
mL |
|
10 X 280
mL |
Store at 2° to 8°C. (Do not freeze)
Internal Control Reagent must be added to Target Capture Reagent before use in the assay. | | | | |
Amplification Reagent
Primers, dNTPs, NTPs and co-factors in TRIS buffered solution containing PROCLIN 300 as preservative. |
|
3 X 32
mL |
|
15 X 32
mL |
| Storeunopened reagent at –15° to –35°C. | | | | |
Enzyme Reagent
MMLV Reverse Transcriptase and T7 RNA Polymerase in HEPES/TRIS buffered solution containing 0.05% sodium azide as preservative. |
|
2 X 18
mL |
|
10 X 18
mL |
| Storeunopened reagent at –15° to –35°C. | | | | |
Probe Reagent
Chemiluminescent oligonucleotide probes in succinate buffered solution containing detergent. |
|
2 X 75
mL |
|
10 X 75
mL |
| Storeunopened reagent at –15° to –35°C. | | | | |
Selection Reagent
Borate buffered solution containing surfactant. |
|
2 X 180
mL |
|
10 X 180
mL |
| Store at 15° to 30°C. | | | | |
Procleix Negative Calibrator
Defibrinated normal human plasma, nonreactive for hepatitis B surface antigen (HBsAg),
HIV-1 p24Ag, antibodies to human immunodeficiency virus type 1 (anti-HIV-1) and type 2 (anti-HIV-2), and antibodies to human hepatitis C virus (anti-HCV) when tested by FDA-licensed assays, containing gentamicin and 0.2% sodium azide as preservatives.
Store at –15° to –35°C. |
|
30 X 2
mL |
|
90 X 2
mL
 |
IN0076 Rev. F
7
Procleix HIV-1 Positive Calibrator
Inactivated HIV-1 positive plasma in defibrinated normal human plasma, nonreactive for hepatitis B surface antigen (HBsAg), and antibodies to human hepatitis C virus (anti-HCV) when tested by FDA-licensed assays, containing gentamicin and 0.2% sodium azide as preservatives.
Store at –15° to –35°C. |
|
30 X 2
mL |
|
90 X 2
mL
 |
Procleix HCV Positive Calibrator
Inactivated HCV positive plasma in defibrinated normal human plasma, nonreactive for hepatitis B surface antigen (HBsAg), and antibodies to human immunodeficiency virus type 1 (anti-HIV-1) and type 2 (anti-HIV-2), when tested by FDA-licensed assays, containing gentamicin and 0.2% sodium azide as preservatives.
Store at –15° to –35°C. |
|
30 X 2
mL |
|
90 X 2
mL
 |
STORAGE INSTRUCTIONS
- A.
- Room temperature is defined as 15° to 30°C.
- B.

The Procleix HIV-1/HCV Assay Probe Reagent and the Discriminatory Probe Reagents are light sensitive. Protect these reagents from light during storage and preparation for use.
- C.
- Target Capture Reagent (TCR) is stable when stored unopened at 2° to 8°C until the expiration date. Do not use after expiration date. If a precipitate forms in the Target Capture Reagent during storage, see instructions under REAGENT PREPARATION. DO NOT VORTEX. DO NOT FREEZE Target Capture Reagent.
NOTE: If after removing the TCR from storage at 2° to 8°C, the precipitate is allowed to settle to the bottom of the container, the likelihood of the formation of a gelatinous precipitate is increased substantially.
- D.
- Selection Reagent is stable when stored unopened at room temperature until the expiration date. Do not use after expiration date. Mix thoroughly prior to use.
- E.
- The following reagents are stable when stored unopened at room temperature until the expiration date.
Wash Solution
Oil
Auto Detect 1
Auto Detect 2
Buffer for Deactivation Fluid
|
- F.
- Once opened, Wash Solution, Oil, Selection Reagent, Buffer for Deactivation Fluid, Auto Detect 1 and Auto Detect 2 are stable for 30 days when stored at room temperature.
IN0076 Rev. F
8
- G.
- The following reagents are stable when stored unopened at -15° to -35°C until the expiration date.
Internal Control Reagent
Amplification Reagent
Enzyme Reagent
Probe Reagent
Procleix Negative Calibrator
Procleix HIV-1 Positive Calibrator
Procleix HCV Positive Calibrator
Procleix HIV-1 Discriminatory Probe Reagent
Procleix HCV Discriminatory Probe Reagent
|
Do not use after expiration date.
- H.
- After thawing, the Amplification Reagent, Enzyme Reagent, Probe Reagent, HIV-1 Discriminatory Probe Reagent and HCV Discriminatory Probe Reagent are stable when stored at 2° to 8°C for 30 days. Once completely thawed, these reagents may be kept at room temperature up to 8 hours per 24 hour period while in use, not to exceed 80 hours at room temperature. Do not refreeze Amplification, Enzyme, Probe, HIV-1 and HCV Discriminatory Probe Reagents after the initial thaw.
- I.
- After thawing, Negative, HIV-1 and HCV Positive Calibrators may be kept at room temperature up to 4 hours. These are single use vials and must be discarded after use.
- J.
- After addition of Internal Control Reagent, the working Target Capture Reagent is stable when stored at 2° to 8°C for 30 days and may be kept at room temperature up to 8 hours per 24 hour period while in use, not to exceed 80 hours at room temperature.
- K.
- If precipitate forms in the Wash Solution, Amplification Reagent, Probe Reagent, or HIV-1 and HCV Discriminatory Probe Reagents, warm to 15°to 30°C and mix thoroughly prior to use. See instructions under REAGENT PREPARATION.
- L.
- If precipitate forms in the Selection Reagent during storage, see instructions under REAGENT PREPARATION.
- M.
- Changes in the physical appearance of the reagent supplied may indicate instability or deterioration of these materials. If changes in the physical appearance of the reagents are observed (e.g., obvious changes in reagent color or cloudiness apparent with microbial contamination), they should not be used.
PRECAUTIONS
ForIn Vitro Diagnostic Use.
- A.
- Specimens may be infectious. Use Universal Precautions22,26 when performing the assay. Proper handling and disposal methods should be established according to local, state and federal regulations.23-25 Only personnel qualified as proficient in the use of the Procleix HIV-1/HCV Assay, the use of the TECAN GENESIS RSP and/or manual sample/TCR pipetting, and trained in handling infectious materials should perform this type of diagnostic procedure.
- B.
- CAUTION: Some components of this kit contain human blood products. The HIV-1 Positive Calibrator in this kit contains human plasma that is HIV-1 positive and has been heat-treated to inactivate the virus. The HCV Positive Calibrator contains human plasma that is HCV positive and has been heat-treated to inactivate the virus. The Negative Calibrator has been assayed by FDA licensed tests and found non-reactive for the presence of hepatitis B surface antigen (HBsAg), HIV-1 p24Ag and antibodies to HIV-1/2 and HCV. No known test method can offer
IN0076 Rev. F
9
complete assurance that products derived from human blood will not transmit infectious agents. All human blood sourced materials should be considered potentially infectious and should be handled with Universal Precautions.22,26 If spillage occurs, immediately disinfect, then wipe up with a 0.5% (final concentration) sodium hypochlorite solution (diluted bleach) or follow appropriate site procedures.
- C.
- Use routine laboratory precautions. Do not pipette by mouth. Do not eat, drink or smoke in designated work areas. Wear disposable gloves and laboratory coats when handling specimens and kit reagents. Wash hands thoroughly after handling specimens and kit reagents.
- D.
- This product contains sodium azide as a preservative. Do not use metal tubing for reagent transfer. If solutions containing azide compounds are disposed of in a plumbing system, they should be diluted and flushed with generous amounts of running water. These precautions are recommended to avoid accumulation of deposits in metal piping in which explosive conditions could develop.
- E.
- Avoid contact of Auto Detect Reagents 1 and 2 with skin, eyes and mucous membranes. Wash with water if contact with these reagents occurs. If spills of these reagents occur, dilute with water before wiping dry and follow appropriate site procedures.
- F.
- Dispose of all materials that have come in contact with specimens and reagents according to local, state and federal regulations.23,24 Thoroughly clean and disinfect all work surfaces.
- G.
- Use only supplied or specified required disposables.
- H.
- Do not use this kit after its expiration date. DO NOT interchange, mix, or combine reagents from kits with different master lot numbers.
- I.
- Avoid microbial and ribonuclease contamination of reagents.
- J.
- Store all assay reagents at specified temperatures. The performance of the assay may be affected by use of improperly stored assay reagents. SeeSTORAGE INSTRUCTIONS andREAGENT PREPARATION.
- K.
- Do not combine any assay reagents or fluids without specific instruction.
- L.
- Some reagents of this kit are labeled with risk and safety symbols according to the European Directive 1999/45/EC and should be handled accordingly.
IN0076 Rev. F
10
Procleix Negative Calibrator
Procleix HIV-1 Positive Calibrator
Procleix HCV Positive Calibrator
|
| Xn. Harmful | | 
R22/R32/S2
S13/S36/S46
|
| R22 | | Harmful if swallowed |
| R32 | | Contact with acid liberates very toxic gas |
| S2 | | Keep out of reach of children |
| S13 | | Keep away from food, drink, and animal feeding stuffs |
| S36 | | Wear suitable protective clothing |
| S46 | | If swallowed, seek medical advice immediately and show this container or label |
- M.
- Refer to PRECAUTIONS in other Procleix Assay package inserts and Procleix System Operator's Manuals.
REAGENT PREPARATION
This step should be performed prior to beginning Target Capture in an area that is free of template and amplicon.
- 1.
- Warm all reagents to room temperature and mix thoroughly prior to use. A dedicated waterbath at room temperature may be used to aid this process. Ensure that precipitates are dissolved. Do not use a reagent if precipitate or cloudiness is present. See step 6 for Target Capture Reagent preparation.
- 2.
- DO NOT heat Probe Reagent, HIV-1 Discriminatory Probe Reagent or HCV Discriminatory Probe Reagent above 30oC.
- 3.
- Thaw reagents upright.
- 4.
- If necessary, thaw Amplification, Probe and Enzyme Reagents at room temperature or at 2° to 8°C. Internal Control, Amplification and Probe Reagents may be mixed by vortexing. Enzyme Reagent should be mixed thoroughly by gentle inversion taking care to avoid excessive foaming. Once completely thawed, these reagents may be kept at room temperature up to 8 hours per 24 hour period while in use. These reagents are stable for 30 days when stored at 2° to 8°C. Record date of thaw (THAW DATE) for Amplification, Probe and Enzyme Reagents in the space provided on the label.
- 5.
- Precipitate will form in the Probe Reagent when stored at 2° to 8°C. Probe Reagent may be warmed in a water bath to facilitate dissolution of precipitate, but temperature in the bath should not exceed 30°C. The Probe Reagent may take up to 4 hours with periodic mixing to allow complete dissolution of precipitate if thawing is conducted on the lab bench. Ensure that precipitates in the Probe Reagent are dissolved. Do not use if precipitate or cloudiness is present.
- 6.
- Selection Reagent is stored at room temperature. If Selection Reagent has been inadvertently stored at 2° to 8°C or the temperature of the laboratory falls below 15°C, precipitate may form. If precipitate forms in the Selection Reagent during storage, heat at 60° ± 1° C for no
IN0076 Rev. F
11
more than 45 minutes, shaking the bottle frequently (every 5 to 10 minutes). Once all precipitate has gone back into solution, place the bottle in a room temperature water bath and allow the bottle to equilibrate for at least 1 hour. Do not use the Selection Reagent until it has equilibrated. The Selection Reagent must be at room temperature before use. Do not use if precipitate or cloudiness is present.
- 7.
- Prepare working Target Capture Reagent: thaw one vial of Internal Control Reagent at room temperature or 2° to 8°C. Mix the Internal Control Reagent thoroughly by inversion. Remove Target Capture Reagent (TCR) from 2° to 8°C storage. IMMEDIATELY upon removing from storage, mix vigorously (at least 10 inversions). DO NOT VORTEX. After mixing, place the TCR bottle at 22° to 30°C. Approximately every 10 minutes shake the bottle until all precipitate has disappeared. TCR precipitate should normally dissolve in about 30 minutes. If a gel is observed after performing this procedure, a new bottle must be used according to the handling recommendations above. Return the bottle with gel back to 2° to 8°C storage for subsequent use. When the Internal Control Reagent and TCR have reached room temperature, mix TCR thoroughly by inversion. Pour the entire vial of Internal Control Reagent into the TCR bottle. The total time for each of these reagents at room temperature must not exceed 8 hours, in the first 24-hour period. This is now the working Target Capture Reagent. Mix thoroughly. Use the space indicated on the TCR bottle to record the date Internal Control Reagent was added and lot number used (IC LOT). Record the expiration date of the working TCR in the space provided on the label.
- 8.
- Thaw calibrators at room temperature. These are single use vials and must be thawed prior to each run. Once thawed, use calibrators within four hours. Mix thoroughly by inversion.
- 9.
- Wash Solution is shipped at ambient temperature and stored at room temperature. Precipitates may form in the Wash Solution during shipment or during storage when temperatures fall below 15°C. Wash Solution may be incubated in a warm water bath to facilitate dissolution of precipitate. Temperature in the bath should not exceed 30°C. Ensure that precipitates in the Wash Solution are dissolved prior to use. Do not use if precipitate or cloudiness is present.
- 10.
- Once opened, Wash Solution, Oil, Selection Reagent, Buffer for Deactivation Fluid, Auto Detect 1 and Auto Detect 2 are stable for 30 days when stored at room temperature. Record the date the reagent was first opened (OPEN DATE) in the space provided on the label.
- 11.
- To prepare Deactivation Fluid, mix one part Buffer for Deactivation Fluid with one part 5% sodium hypochlorite. Deactivation Fluid is stable for 30 days when stored at room temperature.
SPECIMEN COLLECTION, STORAGE AND HANDLING
NOTE: Handle all specimens as if they are potentially infectious agents.
Take care to avoid cross-contamination during the sample handling steps. For example, discard used material without passing over open tubes.
- A.
- Plasma collected in K2EDTA, K3EDTA or in Becton Dickinson EDTA Plasma Preparation Tubes (PPT) may be used. Follow sample tube manufacturer's instructions. Specimen stability is affected by elevated temperature. Whole blood or plasma from pooled or individual donor specimens may be stored for up to 72 hours from time of draw at< 25°C; temperatures not to exceed 30°C are acceptable for no more than 24 hours. Specimens may be stored an additional five days at 2° to 8°C following centrifugation. Plasma separated from the cells may be stored for longer periods of time at<-20°C before testing.
IN0076 Rev. F
12
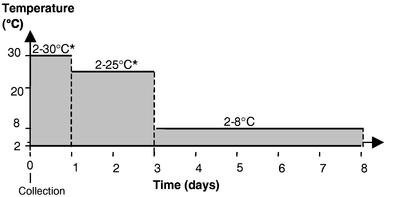 |
- B.
- Additional specimens taken from blood or plasma units collected in ACD or sodium citrate according to the collection container manufacturer's instructions may be used. ACD or sodium citrate whole blood or plasma may be stored as in A. above.
- C.
- Additional specimens may be taken from whole blood or plasma units containing CPD, CP2D, or CPDA-1 anticoagulants collected according to the collection container manufacturer's instructions. Whole blood (not plasma units) collected in these anticoagulants may be stored for up to 13 days at 2° to 8°C prior to centrifugation. At any time within this 13 day period, the whole blood unit may have been stored for up to one day at 30°C and up to two days at 25°C. Following centrifugation, the plasma may be stored for an additional five days at 2° to 8°C before testing. Plasma separated from the cells may be stored for longer periods of time at<-20°C before testing.
- D.
- No adverse effect on assay performance was observed when plasma was subjected to three freeze-thaw cycles.
- E.
- Mix thawed plasma thoroughly and centrifuge for 10 minutes at 1000 to 3000 X g before testing. Centrifugation times and speeds for thawed PPT tubes must be validated by the user.
- F.
- Other collection and storage conditions should be validated by the user. If specimens are to be shipped, they should be packaged and labeled in compliance with applicable federal and international regulations covering the transport of clinical specimens and etiologic agents.25
- G.
- False positive results may occur if cross contamination of specimens is not adequately controlled during specimen handling and processing.
- H.
- Specimen Pooling
The Chiron CPT Pooling Software, used in combination with the TECAN GENESIS RSP, performs sample scanning and pooling operations that combine aliquots from 16 individual samples into a single Master Pool Tube, which may be used for further testing.
PROCEDURAL NOTES
- A.
- RUN SIZE
IN0076 Rev. F
13
When the average run size is 55 tests or more, P/N 301030 should yield 5000 tests per kit. P/N 301031 should yield 1000 tests per kit. Smaller run sizes will result in a lower yield. Each run of up to 100 tests must contain 3 replicates each of the Negative Calibrator, the HIV-1 Positive Calibrator and the HCV Positive Calibrator.
- B.
- EQUIPMENT PREPARATION
- 1.
- Three dedicated circulating water baths must be used: one for target capture and pre-amplification (60° ± 1°C), one for amplification (41.5° ± 1°C) and one for hybridization and selection (60° ± 1°C). An additional water bath is required to be maintained at 23° ± 4°C for the step preceding detection.
- 2.
- Equilibrate circulating water baths to 60° ± 1°C for target capture and 41.5° ± 1°C for amplification incubations.
- 3.
- Prepare the TECAN GENESIS RSP for use according to instructions in the Operator's Manual.
- 4.
- Prepare the Procleix TCS for use according to instructions in the Operator's Manual.
- 5.
- Wipe work surfaces and pipettors daily with diluted bleach (0.5% sodium hypochlorite in water). Allow bleach to contact surfaces and pipettors for at least 15 minutes and then follow with a water rinse. DO NOT USE DEACTIVATION FLUID ON SURFACES.
- 6.
- Equilibrate a circulating water bath to 60° ± 1°C for hybridization and selection incubations. Prepare an additional container of water at 23° ± 4°C for cool down prior to detection.
- 7.
- Setup procedures for the Procleix HC+ Luminometer are given in the Operator's Manual.
- C.
- REAGENTS
- 1.
- Add all reagents using an Eppendorf repeat pipettor (or equivalent) capable of delivering specified volume with ± 5% accuracy and a precision of< 5% CV. Check pipettor functionality monthly and calibrate regularly.
- 2.
- To minimize waste of Amplification, Oil, Enzyme, Probe, and Selection Reagents, aliquot each reagent for a given run size. Aliquoting must be performed after reagent preparation using sterile, polypropylene conical tubes with sealing caps in an area that is template and amplicon free. The aliquoting area must be wiped down with diluted bleach (0.5% sodium hypochlorite in water) before and after the aliquoting process. The aliquoted reagents must be used the same day the aliquoting was performed. DO NOT store reagents in the aliquot conical tubes.
- D.
- WORK FLOW
- 1.
- To minimize the possibility of laboratory areas from becoming contaminated with amplicon, the laboratory area should be arranged with a uni-directional workflow. Proceed from reagent preparation to sample preparation to amplification and then to detection areas. Samples, equipment and reagents should not be returned to the area where a previous step was performed. Also, personnel may not move from the dedicated HPA area back into previous work areas without proper anti-contamination safeguards.
- 2.
- Perform reagent preparation in a template free area.
- 3.
- Perform Target Capture and Pre-Amplification steps in an amplicon-free area.
- 4.
- Perform Hybridization Protection Assay in an area separate from the reagent preparation and amplification areas.
- E.
- TEMPERATURE
IN0076 Rev. F
14
- 1.
- The Target Capture, Amplification, Hybridization and Selection reactions are temperature dependent. Therefore, it is imperative that the water baths are maintained within the specified temperature range. Use a calibrated thermometer.
- 2.
- Room temperature is defined as 15° to 30°C.
- 3.
- Detection is sensitive to temperature. The laboratory temperature in the detection area must be 21° to 27°C.
- F.
- TIME
- 1.
- The Target Capture, Amplification, and Hybridization Protection Assay steps are all time dependent. Adhere to specific times outlined in INSTRUCTIONS FOR USE. Use a calibrated timer.
- G.
- VORTEXING
- 1.
- Proper vortexing is important to the successful performance of the Procleix HIV-1/HCV Assay. Vortex equipment speed settings may vary. Start the vortexor at low speed and then adjust upward to allow reaction mixture to reach and maintain a height within the upper half of all tubes. The reaction mixture should never touch the sealing cards.It is critical to have a homogeneous mixture after the additions of the Probe Reagent and Selection Reagent.
- H.
- PIPETTING
- 1.
- All pipettors used in the Amplification and HPA steps must be dedicated. All pipettors used for manual pipetting in the Target Capture steps must be dedicated.
- 2.
- Take care to deliver reagents, excluding working TCR, to each tube without inserting pipette tip into the tube or touching the rim of the tube to minimize the chance of carryover from one tube to another.
- I.
- MANUAL SPECIMEN PIPETTING
- 1.
- When using the manual sample/TCR pipetting method, improper pipetting technique will affect the results of the assay. SeePROCEDURAL NOTES, Section H. In order to avoid the loss of Positive ID Tracking, verification of correct sample ID by a second individual is recommended.
- 2.
- Ensure that the TTU is oriented in the rack with the pointed end on the left side and the rounded end on the right side of the rack. Pipette the first calibrator into the first tube next to the pointed end of the TTU. Samples are pipetted from left to right.
- 3.
- Use a new pipette tip for each sample and dispose of the tip in a biological waste container after use. Take care to avoid cross-contamination by pipetting the specimens and discarding the used pipette tips without passing over open tubes or touching laboratory surfaces or other pieces of equipment.
- 4.
- To avoid the risk of contamination, clean and decontaminate manual sample pipettors between assay runs.
- 5.
- Ensure proper sample placement into the correct TTU position as indicated on the manual work list record.
- J.
- DECONTAMINATION
- 1.
- The extremely sensitive nature of the test makes it imperative to take all possible precautions to avoid contamination. Laboratory bench surfaces, and pipettes must be decontaminated daily with diluted bleach (0.5% sodium hypochlorite in water). Allow bleach to contact surfaces for
IN0076 Rev. F
15
- K.
- SEALING CARDS
- 1.
- When applying sealing cards, cover the TTUs with the sealing card and press gently to ensure complete contact with all of the tubes. Always use a new sealing card. DO NOT re-use sealing cards.
- 2.
- When removing sealing cards, carefully lift and peel in one continuous motion to avoid aerosols and cross contamination. Immediately dispose of card in appropriate waste container.
INSTRUCTIONS FOR USE:
PROCLEIX HIV-1/HCV ASSAY ON INDIVIDUAL DONOR PLASMA SAMPLES OR ON POOLED PLASMA SAMPLES
All specimens (individual donations or pooled samples) should be run in singlet in the initial Procleix HIV-1/HCV Assay.
Per CLIA requirements (42 CFR 493), the Procleix HIV-1 and HCV External Quality Controls or equivalent must be run every time a Procleix assay is performed. Procleix HIV-1/HCV Assay Calibrators and Discriminatory Probe Reagents are to be used with the corresponding master lot of the Procleix HIV-1/HCV Assays. The operator must check to ensure that the Procleix HIV-1/HCV Assay Calibrators and Discriminatory Probe Reagents are used with the corresponding master lot of kit reagents as indicated on the master lot sheet enclosed with each shipment of Procleix HIV-1/HCV Assay Calibrators and Discriminatory Probe Reagents.
To run the Procleix HIV-1/HCV Assay for the detection of HIV-1 and HCV RNA, follow the steps below for Target Capture, Amplification and Hybridization Protection Assay. To run the Procleix HIV-1/HCV Assay for discrimination between HIV-1 and HCV RNA, see Section D, below, prior to proceeding.
Note: Continuous Process Flow:
All process steps described below are intended to be completed in a continuous flow with a minimal, if any, delay between steps.
- A.
- TARGET CAPTURE
The Procleix HIV-1/HCV Assay has been validated using the TECAN GENESIS RSP 150/8. The use of manual pipetting requires additional operator training and demonstration of proficiency. Repeat pipettors used in this step must be dedicated for use only in the TARGET CAPTURE steps.
IF USING THE TECAN GENESIS RSP 150/8 PIPETTOR:
- 1.
- Start the Procleix Assay Software. Refer to the Procleix Assay Software Operator's Manual for software operating instructions.
- 2.
- Place TTU Carriers on the TECAN GENESIS RSP deck according to the deck layout indicated on the screen. Load sufficient Ten Tube Units (TTUs) for the run into TTU Carriers.
- 3.
- Mix working Target Capture Reagent thoroughly to resuspend microparticles. This is important before putting into the TECAN GENESIS RSP reagent trough. Put sufficient
IN0076 Rev. F
16
IN0076 Rev. F
17
IF USING THE MANUAL SAMPLE PIPETTING METHOD:
The assay results within the run report will be marked "M" indicating that the specimens were manually pipetted.
- 1.
- For sample tracking, an electronic worklist must be created using the Procleix Worklist Editor software. Refer to the Worklist Editor Operator's Manual for instructions. Verification of correct sample ID on the worklist with the specimen tubes and with the detailed assay run report by a second individual is recommended.
- 2.
- Load sufficient Ten Tube Units (TTUs) for the run into a TTU rack.
- 3.
- Thoroughly mix working TCR immediately before use to resuspend microparticles.
- 4.
- Refer to the worklist and carefully pipette 400 µL of working TCR to each reaction tube that will contain a specimen.To dispense, insert the tip approximately one quarter of the way into the tube at an angle and pipette working TCR down the side of the tube. Always pipette the working TCR first, followed by the specimen.
- 5.
- Pipette specimens.
- a.
- Refer to the worklist to identify the TTU number with the corresponding calibrator and test specimen identification numbers.
- b.
- Aspirate 500 µL of each calibrator, external quality control or test sample from its collection tube using a single channel pipettor with corresponding filtered disposable tip. Insert only the end of the pipette tip into the specimen. Do not disturb the sediment, if any.
- c.
- To dispense, insert the pipette tip halfway into the tube taking care not to touch the sides of the upper half of the tube with the pipette tip. At an angle, pipette the specimen down the side of the bottom half of the tube. Hold down the plunger of the pipettor while removing it from the tube. Take care to avoid touching the rim or the side of the tube with the pipette tip when removing it from the tube.
- 6.
- Replace pipette tip with a new tip and repeat Step 5 until all specimens have been pipetted.
- 7.
- Visually inspect tubes to ensure proper specimen volume and working TCR volume have been dispensed.
- 8.
- Cover the TTUs with sealing cards. SeePROCEDURAL NOTES. Proceed to Step 7 of section titled "If Using the TECAN GENESIS RSP 150/8 Pipettor", above.
- B.
- AMPLIFICATION
IN0076 Rev. F
18
- 5.
- Vortex the rack of TTUs a minimum of 20 seconds and until all microparticles are resuspended. Ensure that magnetic particles are no longer adhering to the walls of the tube, and are evenly dispersed in the aqueous phase.
- 6.
- Incubate the TTUs in a water bath at 60° ± 1°C for 10 minutes ± 1 minute, then at 41.5° ± 1°C for 9 to 20 minutes.
- 7.
- Leaving the rack of TTUs at 41.5° ± 1°C, carefully remove and dispose of the sealing cards. See PROCEDURAL NOTES. Proceed immediately to enzyme addition. Add 25 µL of the Enzyme Reagent into each tube using the dedicated repeat pipettor. Take care to deliver the reagent to the bottom of each tube without inserting the pipette tip into the tube or touching the rim of the tube. Place new sealing cards over the TTUs. See PROCEDURAL NOTES. Remove the rack of TTUs from the water bath and shake to mix. DO NOT VORTEX. Minimize the time the tubes are out of the water bath.
- 8.
- Incubate the rack of TTUs in the water bath at 41.5° ± 1°C for 60 minutes ± 5 minutes.
- 9.
- Remove the rack of TTUs from the water bath and transfer it to the Hybridization Protection Assay area. Rack may remain at room temperature for up to 30 minutes prior to the addition of Probe Reagent.
- C.
- HYBRIDIZATION PROTECTION ASSAY (HPA)
The repeat pipettor used in this step must be dedicated for use only in HYBRIDIZATION PROTECTION ASSAY.
A separate, dedicated location for the Hybridization Protection Assay (HPA) step is recommended to minimize amplicon contamination in the assay. This dedicated area should be on a separate bench in a separate area from the reagent and sample preparation and amplification areas.
- 1.
- Hybridization
- a.
- Carefully remove and dispose of the sealing cards. SeePROCEDURAL NOTES.
- b.
- Add 100 µL of Probe Reagent into each tube using the dedicated repeat pipettor.Take care to deliver the reagent to the bottom of each tube without inserting the pipette tip into the tube or touching the rim of the tube. Angle the pipette tip toward the sides of the tubes, not straight to the bottom, to avoid splashback.
- c.
- Cover the TTUs with sealing cards. SeePROCEDURAL NOTES.
- d.
- Vortex the rack of TTUs a minimum of 20 seconds and until a homogeneous solution is achieved. To avoid possible contamination, do not allow reaction mixture to come in contact with the sealing card. SeePROCEDURAL NOTES.
- e.
- Incubate the rack of TTUs in a dedicated water bath at 60° ± 1°C for 15 minutes ± 1 minute.
- 2.
- Selection
- a.
- Remove the rack of TTUs from the 60° ± 1°C water bath. Carefully remove and dispose of the sealing cards. SeePROCEDURAL NOTES.
- b.
- Add 250 µL of Selection Reagent to each tube using a repeat pipettor. Take care to deliver the reagent to the bottom of each tube without inserting the pipette tip into the tube or touching the rim of the tube. Angle the pipette tip toward the sides of the tubes, not straight to the bottom, to avoid splashback.
IN0076 Rev. F
19
- c.
- Cover the TTUs with sealing cards. SeePROCEDURAL NOTES. Vortex the rack of TTUs a minimum of 20 seconds and until a homogeneous solution is achieved. To avoid possible contamination, do not allow reaction mixture to come in contact with the sealing card. SeePROCEDURAL NOTES.
- d.
- Return the rack of TTUs to the 60° ± 1°C water bath for 10 minutes ± 1 minute.
- e.
- Remove the rack of TTUs from the 60° ± 1°C water bath.
- f.
- Cool the rack of TTUs in a 23° ± 4°C container of water for a minimum of 10 minutes while preparing for Detection (step 3a).
- g.
- Remove the rack of TTUs from the 23° ± 4°C container of water onto absorbent material.
- 3.
- Detection
- a.
- Prepare the Procleix HC+ Luminometer for operation as indicated in the Operator's Manual. Ensure that there are sufficient volumes of Auto Detect 1 and Auto Detect 2 to complete the tests.
- b.
- Select the "HIV-HCV" assay protocol from the Procleix System Software menu.
- c.
- Carefully remove and dispose of the sealing cards. SeePROCEDURAL NOTES.
- d.
- Before transferring TTUs to the luminometer, wipe the outside of the tubes using an absorbent tissue dampened with deionized water. This will ensure that no residue is present on the outside of the tubes and will help reduce static electricity that may affect luminometer readings.
- e.
- Transfer TTUs to the luminometer according to the software instructions. Note: Tube reading should be completed within 75 minutes after completing the selection reaction (step 2e in Selection procedure).
- f.
- When the analysis is complete, remove the TTUs from the luminometer.
- g.
- After removing the TTUs from the luminometer, add at least 1 mL Deactivation Fluid to each tube. Allow to sit at room temperature for at least 30 minutes before disposing the contents of the tubes. This will help to prevent contamination of the laboratory environment with amplicon.
- h.
- TTU racks should be decontaminated by complete immersion in diluted bleach (0.5% sodium hypochlorite in water) for a minimum of 15 minutes. The bleach should then be rinsed off with water and the rack may be allowed to air dry or may be wiped dry.
- D.
- PROCLEIX HIV-1 AND HCV DISCRIMINATORY ASSAYS
- 1.
- All reactive individual specimens should be run in singlet in the HIV-1 and/or HCV Discriminatory Assays.
- 2.
- To perform the discriminatory assays, make the following modifications to the procedure above:
- a.
- Perform all TARGET CAPTURE and AMPLIFICATION steps exactly as they are outlined above. Set up separate runs for HIV-1 and HCV Discriminatory Assays. Both Discriminatory Assays use the same Calibrators that are used in the Procleix HIV-1/HCV Assay.
IN0076 Rev. F
20
- b.
- Substitute HIV-1 or HCV Discriminatory Probe Reagent for Probe Reagent, in step C.1.b, when performing HYBRIDIZATION PROTECTION ASSAY.
- c.
- Choose the appropriate Procleix System Software protocol: "dHIV-1" for HIV-1 DISCRIMINATORY ASSAY or "dHCV" for HCV DISCRIMINATORY ASSAY, in step C.3.b, when performing the HYBRIDIZATION PROTECTION ASSAY.
QUALITY CONTROL PROCEDURES:
PROCLEIX HIV-1/HCV ASSAY ON INDIVIDUAL DONOR PLASMA SAMPLES OR ON POOLED PLASMA SAMPLES
- I.
- ACCEPTANCE CRITERIA FOR THE PROCLEIX HIV-1/HCV ASSAY AND PROCLEIX HIV-1 AND HCV DISCRIMINATORY ASSAYS
Run Validity Criteria
- A.
- A run is valid if the minimum number of calibrators is valid and calibrators meet acceptance criteria (see II.A. below). In a run, no more than 2 of the 9 calibrators may be invalid. The Procleix System Software will invalidate a run if more than 2 calibrators are invalid in a run. Cutoff values will be calculated for Internal Control (flasher) and Analyte (glower) in valid runs (see II.A. below). For Positive Calibrators or samples which are reactive for Analyte (glower signal), an Internal Control signal below the cutoff is not used to invalidate the result. All specimens in an invalid run are to be retested, except as noted in step I.B. below.
- B.
- Procleix HIV-1/HCV Assay. If more than 10% of calibrators plus specimen results (overall interpretation) are invalid in the Procleix HIV-1/HCV Assay, then the run is invalidated by the Procleix System Software and the run must be repeated. An assay run or an individual sample may also be invalidated by an operator if specific technical/ operator/ instrument difficulties were observed and documented. If individual samples are invalidated by an operator, then the 10% run validity criterion for the Procleix HIV-1/HCV Assay must be manually recalculated.
Potential reactive specimens in an invalid run due to the 10% run validity criterion must be identified by the user. Any reactive result serves as the test of record and the sample should be resolved according to the resolution algorithm for reactive specimens, as explained below in theINTERPRETATION OF RESULTSsection, step 4 or 5.
Nonreactive specimens in an invalid run due to the 10% run validity criterion are to be re-tested, as explained below in theINTERPRETATION OF RESULTSsection, step 1.
- C.
- Procleix HIV-1 and HCV Discriminatory Assays. For the Procleix HIV-1 and HCV Discriminatory Assays, the Procleix System Software will only invalidate a Procleix HIV-1 or HCV Discriminatory Assay run based on calibrator invalidity criteria described in (A) above. Specimen results are not used to invalidate the Procleix HIV-1 and HCV Discriminatory Assays. An assay run or an individual sample may be invalidated by an operator if specific technical/ operator/ instrument difficulties were observed and documented.
- D.
- When using the TECAN GENESIS RSP, specimens that are invalid due to insufficient sample or TCR are not included in the calculation of the 10% criterion.
- II.
- ACCEPTANCE CRITERIA FOR THE CALIBRATION AND CALCULATION OF CUTOFF
- A.
- Procleix HIV-1/HCV Assay
IN0076 Rev. F
21
Each individual Negative Calibrator (NC) must have an Internal Control (IC) value greater than or equal to 75,000 RLU and less than or equal to 300,000 RLU. Each individual Negative Calibrator must also have an Analyte value less than or equal to 40,000 RLU and greater than or equal to 0 RLU. If one of the Negative Calibrator values is invalid due to an IC value or an Analyte value outside of these limits, the Negative Calibrator mean (NCx) will be recalculated based upon the two acceptable values. The run is invalid and must be repeated if two or more of the three Negative Calibrator values have IC values or Analyte values that are outside of these limits.
Determination of the mean of the Negative Calibrator values (NCx) for Internal Control [NCx (Internal Control)].
Example:
Negative Calibrator
|
|
Internal Control
Relative Light Units
|
|---|
| 1 | | 124,000 |
| 2 | | 126,000 |
| 3 | | 125,000 |
| | |
|
| Total Internal Control RLU = | | 375,000 |
NCx (Internal Control) =Total Internal Control RLU = 125,000
3
Determination of the mean of the Negative Calibrator values (NCx) for Analyte [NCx (Analyte)].
Example:
Negative Calibrator
|
|
Analyte
Relative Light Units
|
|---|
| 1 | | 14,000 |
| 2 | | 16,000 |
| 3 | | 15,000 |
| | |
|
| Total Analyte RLU = | | 45,000 |
NCx (Analyte) =Total Analyte RLU = 15,000
3
HIV-1 Positive Calibrator Acceptance Criteria
Individual HIV-1 Positive Calibrator (PC) Analyte values must be less than or equal to 1,800,000 RLU and greater than or equal to 300,000 RLU. If one of the HIV-1 Positive Calibrator values is outside these limits, the HIV-1 Positive Calibrator mean (HIV-1 PCx) will be recalculated based upon the two acceptable HIV-1 Positive Calibrator values. The run is invalid and must be repeated if two or more of the three HIV-1 Positive Calibrator Analyte values are outside of these limits. IC values may not exceed 475,000 RLU.
Determination of the mean of the HIV-1 Positive Calibrator (HIV-1 PCx) values for Analyte [HIV-1 PCx(Analyte)].
IN0076 Rev. F
22
HIV-1 Positive Calibrator
|
|
Analyte
Relative Light Units
|
|---|
| 1 | | 690,000 |
| 2 | | 700,000 |
| 3 | | 710,000 |
| | |
|
| Total Analyte RLU = | | 2,100,000 |
HIV-1 PCx (Analyte) =Total Analyte RLU = 700,000
3
HCV Positive Calibrator Acceptance Criteria
Individual HCV Positive Calibrator Analyte values must be less than or equal to 900,000 RLU and greater than or equal to 200,000 RLU. If one of the HCV Positive Calibrator values is outside these limits, the HCV Positive Calibrator mean (HCV PCx) will be recalculated based upon the two acceptable HCV Positive Calibrator values. The run is invalid and must be repeated if two or more of the three HCV Positive Calibrator Analyte values are outside these limits. IC values may not exceed 475,000 RLU.
Determination of the mean of the HCV Positive Calibrator (HCV PCx) values for Analyte [HCV PCx) (Analyte)].
Example:
HCV Positive Calibrator
| | Analyte
Relative Light Units
|
|---|
| 1 | | 350,000 |
| 2 | | 360,000 |
| 3 | | 340,000 |
| | |
|
| Total Analyte RLU = | | 1,050,000 |
HCV PCx (Analyte) =Total Analyte RLU = 350,000
3
Calculation of the Internal Control Cutoff Value
Internal Control Cutoff Value = 0.5 X [NCx (Internal Control)]
Using values given in the Negative Calibrator example above:
Internal Control Cutoff Value = 0.5 X (125,000)
Internal Control Cutoff Value = 62,500 RLU
Calculation of the HIV-1/HCV Analyte Cutoff Value
Analyte Cutoff Value = NCx (Analyte) + [0.02 X HIV-1 PCx
(Analyte)]+ [0.04 X HCV PCx (Analyte)]
Using values given in the Negative Calibrator and Positive Calibrator examples above:
Analyte Cutoff Value = 15,000 + (0.02 X 700,000) + (0.04 X 350,000)
Analyte Cutoff Value = 43,000 RLU
IN0076 Rev. F
23
Summary of Acceptance Criteria for Procleix HIV-1/ HCV Assay
| Acceptance Criteria: | | |
Negative Calibrator |
|
|
| Analyte | | > 0 and< 40,000 RLU |
| Internal Control | | > 75,000 and< 300,000 RLU |
| HIV-1 Positive Calibrator | | |
| Analyte | | > 300,000 and< 1,800,000 RLU |
| Internal Control | | < 475,000 RLU |
HCV Positive Calibrator |
|
|
| Analyte | | > 200,000 and< 900,000 RLU |
| Internal Control | | < 475,000 RLU |
Summary of Cutoff Calculations for Procleix HIV-1/HCV Assay
| Analyte Cutoff = | | NC Analyte Mean RLU |
| | | + 0.02 X (HIV-1 PC Analyte Mean RLU) |
| | | + 0.04 X (HCV PC Analyte Mean RLU) |
Internal Control Cutoff = 0.5 X (Negative Calibrator IC Mean RLU)
|
- B.
- Procleix HIV-1 Discriminatory Assay
Negative Calibrator Acceptance Criteria
Each individual Negative Calibrator (NC) must have an Internal Control (IC) value greater than or equal to 75,000 RLU and less than or equal to 300,000 RLU. Each individual Negative Calibrator must also have an Analyte value less than or equal to 40,000 RLU and greater than or equal to 0 RLU. If one of the Negative Calibrator values is invalid due to an IC value or Analyte value that is outside of these limits, the Negative Calibrator mean (NCx) will be recalculated based upon the two acceptable values. The run is invalid and must be repeated if two or more of the three Negative Calibrator values have IC values or Analyte values that are outside of these limits.
Determination of the mean of the Negative Calibrator (NCx) values for Internal Control [NCx (Internal Control)].
Example:
Negative Calibrator
| | Internal Control Relative Light Units
|
|---|
| 1 | | 124,000 |
| 2 | | 126,000 |
| 3 | | 125,000 |
| | |
|
| Total Internal Control RLU = | | 375,000 |
NCx (Internal Control) =Total Internal Control RLU = 125,000
3
IN0076 Rev. F
24
Negative Calibrator
| | Analyte
Relative Light Units
|
|---|
| 1 | | 12,000 |
| 2 | | 11,000 |
| 3 | | 13,000 |
| | |
|
| Total Analyte RLU = | | 36,000 |
NCx (Analyte) =Total Analyte RLU = 12,000
3
HIV-1 Positive Calibrator Acceptance Criteria
Individual HIV-1 Positive Calibrator (PC) Analyte values must be less than or equal to 1,800,000 RLU and greater than or equal to 300,000 RLU. If one of the HIV-1 Positive Calibrator values is outside these limits, the HIV-1 Positive Calibrator mean (HIV-1 PCx) will be recalculated based upon the two acceptable HIV-1 Positive Calibrator values. The run is invalid and must be repeated if two or more of the three HIV-1 Positive Calibrator Analyte values are outside these limits. IC values may not exceed 475,000 RLU.
Determination of the mean of the HIV-1 Positive Calibrator (HIV-1 PCx) values for Analyte [HIV-1 PCx (Analyte)].
Example:
| HIV-1 Positive Calibrator | | Analyte
Relative Light Units |
| 1 | | 690,000 |
| 2 | | 700,000 |
| 3 | | 710,000 |
| | |
|
| Total Analyte RLU = | | 2,100,000 |
(HIV-1 PCx) (Analyte) =Total Analyte RLU = 700,000
3
HCV Positive Calibrator Acceptance Criteria
In the HIV-1 Discriminatory Assay, each individual HCV Positive Calibrator must have Analyte values less than or equal to 40,000 RLU and greater than or equal to 0 RLU. Each HCV Positive Calibrator must also have IC values greater than or equal to 75,000 RLU and less than or equal to 300,000 RLU. The run is invalid and must be repeated if two or more of the three calibrator values have IC values or Analyte values that are outside these limits.
Calculation of the Internal Control Cutoff Value
Internal Control Cutoff Value = 0.5 X [NCx (Internal Control)]
Using values given in the Negative Calibrator example above:
Internal Control Cutoff Value = 0.5 X (125,000)
Internal Control Cutoff Value = 62,500 RLU
Calculation of the Analyte Cutoff Value
Analyte Cutoff Value = NCx (Analyte) + [0.04 X HIV-1 PCx (Analyte)]
IN0076 Rev. F
25
Using values given in the Negative Calibrator and HIV-1 Positive Calibrator examples above:
Analyte Cutoff Value = 12,000 + (0.04 X 700,000)
Analyte Cutoff Value = 40,000 RLU
Summary of Acceptance Criteria for the Procleix HIV-1 Discriminatory Assay
| Acceptance Criteria: | | |
Negative Calibrator |
|
|
| Analyte | | > 0 and< 40,000 RLU |
| Internal Control | | > 75,000 and< 300,000 RLU |
HIV-1 Positive Calibrator |
|
|
| Analyte | | > 300,000 and< 1,800,000 RLU |
| Internal Control | | < 475,000 RLU |
HCV Positive Calibrator* |
|
|
| Analyte | | > 0 and< 40,000 RLU |
| Internal Control | | < 75,000 and< 300,000 RLU |
- *
- Note that the HCV Positive Calibrator performs similarly to the Negative Calibrator in the HIV-1 Discriminatory Assay.
Summary of Cutoff Calculations for the Procleix HIV-1 Discriminatory Assay
| Analyte Cutoff = | NC Analyte Mean RLU + 0.04 X (HIV-1 PC Analyte Mean RLU) |
Internal Control Cutoff = 0.5 X (Negative Calibrator IC Mean RLU)
|
- C.
- Procleix HCV Discriminatory Assay
Negative Calibrator Acceptance Criteria
Each individual Negative Calibrator must have an Internal Control (IC) value greater than or equal to 75,000 RLU and less than or equal to 300,000 RLU. Each individual Negative Calibrator must also have an Analyte value less than or equal to 40,000 RLU and greater than or equal to 0 RLU. If one of the Negative Calibrator values is invalid or an IC or Analyte value is outside of these limits, the Negative Calibrator mean (NCx) will be recalculated based upon the two acceptable values. The run is invalid and must be repeated if two or more of the three Negative Calibrator values have IC values or Analyte values that are outside of these limits.
Determination of the mean of the Negative Calibrator values (NCx) for Internal Control [NCx (Internal Control)].
Example:
Negative Calibrator
|
|
Internal Control
Relative Light Units
|
|---|
| 1 | | 124,000 |
| 2 | | 126,000 |
| 3 | | 125,000 |
| | |
|
| Total Analyte RLU = | | 375,000 |
NCx (Internal Control) =Total Internal Control RLU = 125,000
3
IN0076 Rev. F
26
Determination of the Analyte mean of the Negative Calibrator values (NCx) for Analyte [NCx(Analyte)].
Example:
Negative Calibrator
|
|
Analyte
Relative Light Units
|
|---|
| 1 | | 20,000 |
| 2 | | 22,000 |
| 3 | | 18,000 |
| | |
|
| Total Analyte RLU = | | 60,000 |
NCx (Analyte) =Total Analyte RLU = 20,000
3
HIV-1 Positive Calibrator Acceptance Criteria
In the HCV Discriminatory Assay, each individual HIV-1 Positive Calibrator must have Analyte values less than or equal to 40,000 RLU and greater than or equal to 0 RLU. Each HIV-1 Positive Calibrator must also have IC values greater than or equal to 75,000 RLU and less than or equal to 300,000 RLU. The run is invalid and must be repeated if two or more of the three calibrator values have IC values or Analyte values that are outside of these limits.
Individual HCV Positive Calibrator value must be less than or equal to 2,700,000 RLU and greater than or equal to 400,000 RLU. If one of the HCV Positive Calibrator values is outside these limits, the HCV Positive Calibrator mean (HCV PCx) will be recalculated based upon the two acceptable HCV Positive Calibrator values. The run is invalid and must be repeated if two or more of the three HCV Positive Calibrator values are outside these limits. IC values may not exceed 475,000 RLU.
Determination of the mean of the HCV Positive Calibrator (HCV PCx) values for Analyte [HCV PCx (Analyte)].
Example:
HCV Positive Calibrator
| | Analyte
Relative Light Units
|
|---|
| 1 | | 900,000 |
| 2 | | 1,000,000 |
| 3 | | 1,100,000 |
| | |
|
| Total Analyte RLU = | | 3,000,000 |
HCV PCx (Analyte) =Total Analyte RLU = 1,000,000
3
Calculation of the Internal Control Cutoff Value
Internal Control Cutoff Value = 0.5 X [NCx (Internal Control)]
Using values given in the Negative Calibrator example above:
Internal Control Cutoff Value = 0.5 X (125,000)
Internal Control Cutoff Value = 62,500
Calculation of the Analyte Cutoff Value
IN0076 Rev. F
27
Analyte Cutoff Value = NCx(Analyte) + [0.04 X HCV PCx(Analyte)]
Using values given in the Negative Calibrator and HCV Positive Calibrator examples above:
Analyte Cutoff Value = 20,000 + (0.04 X 1,000,000)
Analyte Cutoff Value = 60,000 RLU
Summary of Acceptance Criteria for the Procleix HCV Discriminatory Assay
| Acceptance Criteria: | | |
Negative Calibrator |
|
|
| Analyte | | > 0 and< 40,000 RLU |
| Internal Control | | > 75,000 and< 300,000 RLU |
HIV-1 Positive Calibrator* |
|
|
| Analyte | | > 0 and< 40,000 RLU |
| Internal Control | | > 75,000 and< 300,000 RLU |
HCV Positive Calibrator |
|
|
| Analyte | | > 400,000 and< 2,700,000 RLU |
| Internal Control | | < 475,000 RLU |
- *
- Note that the HIV-1 Positive Calibrator performs similarly to the Negative Calibrator in the HCV Discriminatory Assay.
Summary of Cutoff Calculations for the Procleix HCV Discriminatory Assay
| Analyte Cutoff = | | NC Analyte Mean RLU |
| | | + 0.04 X (HCV PC Analyte Mean RLU) |
| Internal Control Cutoff = | | 0.5 X (Negative Calibrator IC Mean RLU)
|
INTERPRETATION OF RESULTS
All calculations described above are performed by the Procleix System Software.Two cutoffs are determined for each assay: one for the Analyte signal (glower signal) termed the Analyte Cutoff and one for the Internal Control signal (flasher signal) termed the Internal Control Cutoff. The calculation of these cutoffs is shown above. For each sample, an Analyte signal RLU value and Internal Control signal RLU value is determined. Analyte signal RLU divided by the Analyte Cutoff is abbreviated as the Analyte Signal/Cutoff (S/CO) on the report.
For a sample with Analyte signal less than the Analyte Cutoff (i.e., Analyte S/CO < 1), the Internal Control (IC) signal must be greater than or equal to the Internal Control Cutoff (IC Cutoff) for the result to be valid. In this case the Internal Control result will be reported asValid and the sample is reported asNonReactive. For a sample with the Analyte signal less than the Analyte Cutoff (i.e., Analyte S/CO < 1) and the Internal Control signal less than the Internal Control Cutoff, the Internal Control Result will be reported asInvalidand the sample result is reported asInvalid. For all samples, the Internal Control signal may not exceed 475,000 RLU. The sample will automatically be reported asInvalid with the Procleix System Software.
IN0076 Rev. F
28
Summary of Sample Validity:
Sample
Interpretation
|
|
Internal Control
Result
|
|
|
|---|
| Nonreactive | | Valid | | Analyte S/CO < 1 and IC
> IC Cutoff and IC<
475,000 RLU |
| Reactive | | (Not used) | | Analyte S/CO> 1and IC
< 475,000 RLU
|
- 1.
- Any specimen with an invalid Procleix HIV-1/HCV Assay, Procleix HIV-1 Discriminatory Assay or Procleix HCV Discriminatory Assay result must be retested in the same assay in singlet, except as noted in step 6 below.
- 2.
- If at any point in the testing algorithm there is insufficient volume to complete the testing then an alternate specimen from the index donation (e.g., plasma unit or serology tube) may be used as long as the storage criteria in the package insert are met.
- 3.
- Specimens with a valid internal control and with an S/CO less than 1.00 in the HIV-1/HCV Assay are considered Nonreactive for HIV-1 and HCV RNA. If the Nonreactive specimen is a pool of 16, each of the 16 individual specimens comprising the pool is considered Nonreactive and no further testing is required.
- 4.
- Specimens with S/CO greater than or equal to 1.00 are considered Reactive.
- 5.
- IF THE REACTIVE SPECIMEN IS FROM AN INDIVIDUAL DONATION, then the specimen must be tested with the HIV-1 Discriminatory and HCV Discriminatory Assays.
- a.
- If an individual specimen then tests Reactive with one or both Discriminatory tests, then the specimen is considered Reactive-Discriminated and may be confirmed as described in (7) below.
- b.
- If an individual specimen then tests Nonreactive with both Discriminatory tests, then the specimen is considered Non-Discriminated. The Non-Discriminated specimen may be retested in the HIV-1/HCV Assay if sufficient sample is available.
IN0076 Rev. F
29
- (1)
- If the individual specimen tests Nonreactive in the repeated HIV-1/HCV Assay, then the specimen is considered Nonreactive for HIV-1 and HCV RNA and no further testing is required.
- (2)
- If the individual specimen tests Reactive in the repeated HIV-1/HCV Assay, then the specimen is considered Repeatedly Reactive, Non-Discriminated for HIV-1 and HCV RNA. Further clarification of these specimens may be obtained by testing an alternate specimen from the index donation with the Procleix Assays and/or by follow-up testing.
- 6.
- Potential reactive specimens in an invalid run due to the 10% run validity criterion must be identified by the user, by viewing the run report, and become the test of record. Any reactive result (analyte signal/cutoff> 1) serves as the test of record and the sample should be resolved according to the resolution algorithm for reactive specimens, as explained in theINTERPRETATION OF RESULTSsection, step 4 or 5.
- 7.
- HIV seroreactive specimens found to be Reactive-HIV-1 Discriminated in the Procleix tests may be considered positive for HIV-1 nucleic acid. HCV seroreactive specimens found to be Reactive-HCV Discriminated in the Procleix tests may be considered positive for HCV nucleic acid. The interpretation of Reactive-Discriminated specimen results on specimens that are Nonreactive by serology is unclear. Further clarification may be obtained by testing an alternate specimen from the index donation with the Procleix Assays (to eliminate false positives due to possible specimen contamination) and/or by follow-up testing to determine if the NAT reactivity of the index donation represents early pre-seroconversion infection.
PERFORMANCE CHARACTERISTICS
REPRODUCIBILITY
Procleix HIV-1/HCV Assay reproducibility was determined at three blood testing laboratories. The reproducibility study evaluated both automated pipetting using TECAN GENESIS RSP, and manual pipetting of the specimen and Target Capture Reagent (TCR) into the reaction tube. The reproducibility of the HIV-1/HCV Assay was assessed with a seven member reproducibility panel; 16 individual specimens were pipetted with the Chiron CPT Pooling Software to create each of the seven panel members (each panel member is a 16 member pool). Each pool contained from zero to three HIV-1 and/or HCV RNA positive specimens with the remaining specimens in the pool being HIV-1 and HCV RNA negative (Table I).
For determination of the reproducibility of the Procleix HIV-1 Discriminatory Assay and the Procleix HCV Discriminatory Assay, nine panel members were tested as individual specimens and not in a pool. Eight of these panel members were HIV-1 and/or HCV RNA positive, and one was HIV-1 and HCV RNA negative (Tables IIa, IIb).
The reproducibility panels were tested by a total of six operators (two at each site) with three different Clinical Lots over at least 18 nonconsecutive days. Inter- and intra-assay variability and inter-lot variability were determined. Mean S/CO, standard deviation (SD) and coefficient of variation (%CV) results are shown for panel members and for the Negative, HIV-1 Positive and HCV Positive Calibrators. Since no significant difference in assay reproducibility was observed between automated pipetting and manual pipetting, results for the two methods are combined in the Tables below (Tables I, IIa, IIb). Also, since HCV RNA positive and HIV-1 RNA positive samples containing 90 copies/mL or greater gave high (saturated) signals in all three assays, results on multiple panel members are combined.
IN0076 Rev. F
30
Table I. Reproducibility of the Procleix HIV-1/HCV Assay
| |
| |
| |
| |
| |
| | Intra-Assay
| | Inter-Assay
| | Inter-Lot
|
|---|
Specimen
| |
| | Concentration
Copies/mL
| | Number of
replicates
| | %
Agreement
| | Mean
S/CO
|
|---|
| | N
| | SD
| | %CV
| | SD
| | %CV
| | SD
| | %CV
|
|---|
| Nonreactive | | 1 | | 0 | | 320 | | 100.00 | | 0.23 | | 0.054 | | 23.5 | | 0.034 | | 14.7 | | 0.041 | | 17.7 |
| HIV-1 | | 3 | | 190, 620, 720 | | 965 | | 99.90 | | 18.75 | | 1.864 | | 9.9 | | 1.191 | | 6.4 | | 2.351 | | 12.5 |
| HIV-1/HCV | | 2 | | 620,720/90 | | 641 | | 100.00 | | 27.42 | | 2.374 | | 8.7 | | 1.840 | | 6.7 | | 2.743 | | 10.0 |
| HCV | | 1 | | 190 | | 321 | | 100.00 | | 8.70 | | 1.074 | | 12.3 | | 0.615 | | 7.1 | | 0.743 | | 8.5 |
| |
| |
| |
| | Intra-Assay
| | Inter-Assay
| | Inter-Lot
|
|---|
Specimen
| | Number of
replicates
| | %
Agreement
| | Mean
RLU
|
|---|
| | SD
| | %CV
| | SD
| | %CV
| | SD
| | %CV
|
|---|
| Negative Calibrator | | 323 | | N/A | | 10363 | | 2023 | | 19.5 | | 1740 | | 16.8 | | 1606 | | 15.5 |
| HIV-1 Positive Calibrator | | 324 | | N/A | | 858644 | | 34660 | | 4.0 | | 55020 | | 6.4 | | 141285 | | 16.5 |
| HCV Positive Calibrator | | 316 | | N/A | | 398939 | | 17511 | | 4.4 | | 15926 | | 4.0 | | 41127 | | 10.3 |
N = Number of panel members combined for this analysis.
Table IIa. Reproducibility of the Procleix HIV-1 Discriminatory Assay (excludes 10 false positive results)
| |
| |
| |
| |
| |
| | Intra-Assay
| | Inter-Assay
| | Inter-Lot
|
|---|
Specimen
| |
| | Concentration
Copies/mL
| | Number of
replicates
| | % Agreement
| | Mean
S/CO
|
|---|
| | N
| | SD
| | %CV
| | SD
| | %CV
| | SD
| | %CV
|
|---|
| Nonreactive | | 1 | | 0 | | 322 | | 100.00 | | 0.19 | | 0.050 | | 26.3 | | 0.029 | | 15.3 | | 0.024 | | 12.3 |
| HIV-1 | | 4 | | 150, 500, | | 1289 | | 100.00 | | 19.69 | | 2.391 | | 12.1 | | 1.114 | | 5.7 | | 0.883 | | 4.5 |
| | | | | 1500, 10000 | | | | | | | | | | | | | | | | | | |
| HIV-1/HCV | | 1 | | 500/500 | | 318 | | 100.00 | | 19.44 | | 1.225 | | 6.3 | | 1.373 | | 7.1 | | 1.045 | | 5.4 |
| HCV | | 3 | | 150, 500, 1500 | | 955 | | 100.00 | | 0.18 | | 0.054 | | 29.7 | | 0.038 | | 20.9 | | 0.029 | | 16.1 |
| |
| |
| |
| | Intra-Assay
| | Inter-Assay
| | Inter-Lot
|
|---|
Specimen
| | Number of
replicates
| | % Agreement
| | Mean
RLU
|
|---|
| | SD
| | %CV
| | SD
| | %CV
| | SD
| | %CV
|
|---|
| Negative Calibrator | | 323 | | N/A | | 8900 | | 2121 | | 23.8 | | 1824 | | 20.5 | | 1470 | | 16.5 |
| HIV-1 Positive Calibrator | | 320 | | N/A | | 894464 | | 57091 | | 6.4 | | 63756 | | 7.1 | | 30695 | | 3.4 |
| HCV Positive Calibrator | | 322 | | N/A | | 8686 | | 2381 | | 27.4 | | 1572 | | 18.1 | | 783 | | 9.0 |
N = Number of panel members combined for this analysis.
IN0076 Rev. F
31
Table IIb. Reproducibility of the Procleix HCV Discriminatory Assay (excludes 3 false positive results)
| |
| |
| |
| |
| |
| | Intra-Assay
| | Inter-Assay
| | Inter-Lot
|
|---|
Specimen
| |
| | Concentration
Copies/mL
| | Number of
replicates
| | % Agreement
| | Mean
S/CO
|
|---|
| | N
| | SD
| | %CV
| | SD
| | %CV
| | SD
| | %CV
|
|---|
| Nonreactive | | 1 | | 0 | | 323 | | 100.00 | | 0.13 | | 0.051 | | 39.0 | | 0.031 | | 24.1 | | 0.010 | | 8.0 |
| HIV-1 | | 4 | | 150, 500, | | 1288 | | 100.00 | | 0.13 | | 0.073 | | 55.0 | | 0.042 | | 31.5 | | 0.018 | | 13.3 |
| | | | | 1500, 10000 | | | | | | | | | | | | | | | | | | |
| HIV-1/HCV | | 1 | | 500/500 | | 323 | | 100.00 | | 21.32 | | 1.965 | | 9.2 | | 1.000 | | 4.7 | | 0.510 | | 2.4 |
| HCV | | 3 | | 150, 500, 1500 | | 966 | | 99.90 | | 21.48 | | 1.884 | | 8.8 | | 1.211 | | 5.6 | | 0.479 | | 2.2 |
| |
| |
| |
| | Intra-Assay
| | Inter-Assay
| | Inter-Lot
|
|---|
Specimen
| | Number of
replicates
| | %
Agreement
| | Mean
RLU
|
|---|
| | SD
| | %CV
| | SD
| | %CV
| | SD
| | %CV
|
|---|
| Negative Calibrator | | 320 | | N/A | | 8132 | | 2861 | | 35.2 | | 1893 | | 23.3 | | 81 | | 1.0 |
| HIV-1 Positive Calibrator | | 321 | | N/A | | 7937 | | 3089 | | 38.9 | | 2240 | | 28.2 | | 929 | | 11.7 |
| HCV Positive Calibrator | | 323 | | N/A | | 1243692 | | 57818 | | 4.6 | | 92720 | | 7.5 | | 36728 | | 3.0 |
N = Number of panel members combined for this analysis.
PERFORMANCE OF POOLED SAMPLE TESTING
Specificity of the Procleix HIV-1/HCV Assay
The HIV-1/HCV Assay was used to screen plasma pools comprised of 16 donor specimens and individual donor specimens (IDS). These specimens were tested at eight volunteer blood donor sites using three Clinical Lots of reagents. This population derived from approximately 103 geographically diverse blood donor sites in the continental US and five others which were US military blood donor sites located in Hawaii (one), Japan (two), Germany (one), and Guam (one). Pools or individual specimens with S/CO<1.0 are considered Nonreactive (NR). Individual specimens that are Reactive (R) by the HIV-1/HCV Assay and Reactive by either the HIV-1 or HCV Discriminatory Assay or both are termed Reactive-Discriminated. Individual specimens that are reactive by the HIV-1/HCV Assay but Nonreactive by both the HIV-1 or HCV Discriminatory Assays are termed Reactive-Non-Discriminated. A non-discriminated specimen which tested again as HIV-1/HCV Reactive was termed Repeatedly Reactive-Non-Discriminated. A non-discriminated specimen which tested again as HIV-1/HCV Assay Nonreactive was considered Nonreactive for HIV-1 and HCV RNA.
At the time the study was performed there was no recognized standard for establishing the presence or absence of HIV-1 RNA or HCV RNA in blood. Specificity was based on testing of blood donations from random volunteer blood donors. For the purpose of specificity calculations, specimens testing seroreactive for anti-HIV-1 and anti-HCV antibody (Ab) or HIV p24 antigen (Ag) were eliminated from the analysis.
In the pooling specificity study, 11,978 pools (191,648 donor specimens) were tested (Table III and Table IV). Specificity of pool testing relative to serology testing was 99.67% (11,625/11,663). Specificity was defined as number of pools containing all serology and Procleix HIV-1/HCV Assay nonreactive specimens (True negative, TN) divided by the sum of TN and False positive pools. False positive pools were defined as pools Reactive by the HIV-1/HCV Assay and that contained all specimens nonreactive by serology.
There were 175 (1.46%) pools initially reactive by the HIV-1/HCV Assay. All 16 individual donor specimens from each reactive pool were tested. There were 33 pools containing all Nonreactive
IN0076 Rev. F
32
individual donor specimens. One hundred and sixty-six individual donor specimens derived from the remaining 142 pools were Reactive in the HIV-1/HCV Assay. No significant differences among sites or Clinical Lots were observed.
Of the 166 HIV-1/HCV Assay reactive individual donor specimens, 138 (83.1%) were Reactive in the HCV Discriminatory Assay, 13 (7.8%) were Reactive in the HIV-1 Discriminatory Assay and 15 (9.0%) were Nonreactive in both discriminatory tests. The 15 Reactive-Non-Discriminated specimens were seronegative for HIV and HCV and were Nonreactive when retested in the HIV-1/HCV Assay. The adjusted reactive rate after removal of reactive pools containing true positive samples was 0.31% (37/11,841).
A total of 49,054 specimens (total of pooled and individual donor specimens) were run in the HIV-1/HCV Assay during these specificity studies and 185 (0.38%) specimens tested as initially invalid due to an Internal Control failure. All 185 specimens giving initially invalid results gave valid repeat testing results in the HIV-1/HCV Assay.
Table III. Procleix HIV-1/HCV Assay Reactivity in Volunteer Blood Donors
| | Procleix HIV-1/HCV Assay
Plasma Pool of 16
|
|---|
| Samples Tested* | | 11,978 |
| Initial Reactive | | 175 |
| Initial Reactive Rate | | 1.46% |
| Adjusted Reactive Rate | | 0.31% |
| Combined Mean S/CO on Negative Analytes | | 0.21 ± 0.10 |
*Combined data across all sites and Clinical Lots.
No significant differences among Clinical Lots of reagents were observed for either the Negative population Analyte mean S/CO or mean Internal Control S/CO in the negative population distribution.
Results of deconvolution of all 16 member pools are shown in Table IV. In these pivotal specificity studies (pooled and individual donation testing), no specimen was identified as a yield specimen based on the HIV-1/HCV Assay reactivity, HIV-1 and/or HCV Discriminatory Assay reactivity, reactivity by Alternative NAT and lack of HCV or HIV-1 antibody reactivity. The finding of no HIV-1 or HCV RNA yield donors in 226,205 specimens is consistent with published yields15. Rates of specimens testing reactive for both HCV RNA and HCV antibody in volunteer donors was 1 in 1,508 donors and rates of specimens testing reactive for both HIV-1 RNA and HIV-1 antibody was 1 in 14,138 donors.
IN0076 Rev. F
33
Table IV. Deconvoluted Pooling Results
| | N (%)
| | Sero-RR*
| |
| |
|
|---|
| Total pools tested | | 11,978 (100.0 | ) | N/A | | | | |
| HIV-1/HCV Assay Nonreactive pools | | 11,803 (98.54 | ) | N/A | | | | |
| HIV-1/HCV Assay Reactive pools | | 175 (1.46 | ) | N/A | | | | |
Pools with all HIV-1/HCV Assay Nonreactive IDS |
|
33 (18.9 |
) |
N/A |
|
|
|
|
| Pools with> 1 HIV-1/HCV Assay Reactive IDS | | 142 (81.1 | ) | N/A | | | | |
HIV-1/HCV Assay Reactive IDS |
|
166 (100.0 |
) |
120 |
|
|
|
|
| HIV-1 and HCV Discriminatory Reactive IDS | | 0 (0.0 | ) | 0 | | | | |
| HIV-1 Discriminatory Reactive IDS | | 13 (7.8 | ) | 13 | | | | |
| HCV Discriminatory Reactive IDS | | 138 (83.1 | ) | 131 | | | | |
| HIV-1 and HCV Discriminatory Nonreactive IDS | | 15 (9.0 | ) | 0 | | | | |
Repeat HIV-1/HCV Assay Nonreactive IDS |
|
15 (100.0 |
) |
0 |
|
|
|
|
| Repeat HIV-1/HCV Assay Reactive IDS | | 0 (0.0 | ) | 0 | | | | |
*EIA repeatedly reactive, supplemental serology positive or indeterminate.
IDS = Individual Donor Specimen.
Specificity of the Procleix HIV-1 and HCV Discriminatory Assays
In the HIV-1 and HCV Discriminatory Assays specificity study, the HIV-1 Discriminatory and HCV Discriminatory assays were run only on individual donor specimens which had previously tested as Nonreactive by the HIV-1/HCV assay. The initial reactive rates for the HIV-1 Discriminatory and HCV Discriminatory Assays were 0.24% (6/2508) and 0.29% (7/2443), respectively. In this study initially reactive specimens were not retested.
Comparison with Serology
Results generated from the pooled and individual donation testing specificity studies allow comparison of the HIV-1/HCV Assay with serology reactivity (Table V). Sixteen of 17 HIV-1 seroreactive, Western blot positive specimens were also Procleix HIV-1/HCV Assay Reactive (Table V). The one discordant specimen was Nonreactive when tested as a pool by the Procleix HIV-1/HCV Assay but was Reactive by the Procleix HIV-1/HCV Assay when tested as an individual donor specimen. All of the HIV seroreactive specimens which were Indeterminate or negative by Western Blot (91.7% of total HIV seroreactive specimens) were Nonreactive by the Procleix HIV-1/HCV Assay. Overall agreement between the Procleix HIV-1/HCV Assay and Western blot was 100% (226/226) if testing at an individual donor level is compared.
77.3% (150/194) of HCV seroreactive, Chiron RIBA HCV 3.0 (RIBA) Positive specimens were Reactive by the HIV-1/HCV Assay. 3.3% (2/60) of HCV seroreactive, RIBA Indeterminate specimens were reactive by the Procleix HIV-1/HCV Assay. All 163 RIBA Negative specimens were also Nonreactive by the Procleix HIV-1/HCV Assay. Overall agreement between the Procleix HIV-1/HCV Assay and RIBA was 89.0% (371/417). These data are consistent with previous reports that about 20% of HCV seropositives will have undetectable HCV RNA27 and estimates that approximately 20% of HCV seropositive individuals may have resolved infection28.
IN0076 Rev. F
34
Table V. Comparison of Serologic and Procleix HIV-1/HCV Assay Reactives from the Specificity Studies
| |
| | Procleix HIV-1/HCV
| |
|---|
Serology
| | Reactive
| | Nonreactive
| |
|---|
| HIV Ab RR | | POS | | 16 (7.0 | %) | 1* (0.4 | %) |
| Western Blot | | IND | | 0 (0 | %) | 74 (32.5 | %) |
| (N = 228) | | NEG | | 0 (0 | %) | 135 (59.2 | %) |
| | | N/A | | 0 (0 | %) | 2 (0.9 | %) |
| HCV Ab RR | | POS | | 150 (35.8 | %) | 44 (10.5 | %) |
| RIBA | | IND | | 2 (0.5 | %) | 58 (13.8 | %) |
| (N = 419) | | NEG | | 0 (0.0 | %) | 163 (38.9 | %) |
| | | N/A | | 1 (0.2 | %) | 1 (0.2 | %) |
*Sample contained < 50 copies/mL of HIV-1 RNA and was Procleix HIV-1/HCV negative in pool testing but reactive in individual sample testing.
Non-Specificity Studies
When tested with the Procleix HIV-1/HCV Assay, no cross-reactivity or interference was observed for naturally occurring icteric, hemolyzed or lipemic specimens or plasma containing the following substances: serum albumin (up to 225 g/L), hemoglobin (up to 5000 mg/L), billirubin (up to 200 mg/L) and lipids (up to 2,752 mg/dL).
No cross-reactivity or interference was observed in specimens from patients with autoimmune diseases or with liver diseases not caused by HCV infection. Autoimmune conditions included rheumatoid arthritis (n = 10), rheumatoid factor (n = 10), antinuclear antibody (n = 10), multiple sclerosis (n = 10), lupus (n = 10) and multiple myeloma (n = 9). Also tested were flu vaccinees (n = 10), hepatitis B vaccinees (n = 10), elevated IgM (n = 6), elevated IgG (n = 11), alcoholic liver cirrhosis (n = 10) and elevated ALT (n = 10).
No cross-reactivity or interference was observed in bacterially contaminated plasmas or in plasmas infected with other blood borne pathogens, including herpes simplex virus-1 (n = 10), herpes simplex virus-2 (n = 1), CMV (n = 10), EBV (n = 10), hepatitis A virus (n = 10), HTLV-I (n = 10), HTLV-II (n = 10), hepatitis B virus (n = 10), HIV-2 (n = 10), rubella (n = 10) and parvovirus B-19 (n = 10).
CLINICAL SENSITIVITY
Testing of Whole Blood Donor Specimens
A total of 24,764,889 donations were screened as part of pooled sample testing since early 199930. As of November 2001, eighty-eight Procleix HCV yield cases (1:281,419) and seven HIV-1 yield cases (1:3,537,841) were identified across the ten Procleix pooled testing sites. Yield cases were confirmed to be positive for either HIV-1 or HCV RNA, but negative in serology testing.
Two of the nine HIV-1 yield cases were reactive for HIV-1 RNA and p24 Ag but Nonreactive by HIV antibody testing (Table VI).
IN0076 Rev. F
35
Table VI. Summary of Procleix Assay Yield Cases
Pooled Sample Testing
|
|---|
| Number of Donations Tested | | 24,764,889 |
Procleix HCV
Yield Cases | | 88
(1:281,419) |
| Procleix HIV-1 | | 7 |
| Yield Cases | | (1:3,537,841) |
Testing of Specimens from HIV-1 and/or HCV Infected Individuals
A total of 2014 specimens positive by commercial HIV-1 RNA and HCV RNA assays (sensitivity>100 copies/mL) were obtained from four commercial vendors. Three Clinical Lots were used for all testing. These specimens were classified as HIV-1 RNA positives (n = 867), HCV RNA positives (n = 967) and both HIV-1 and HCV RNA (coinfected) positives (n = 180) based on alternate nucleic acid testing (Table VII). These specimens were also classified by disease category as described below and as shown in Table VIII. These positive samples were tested undiluted (neat) with the Procleix HIV-1/HCV Assay, HIV-1 Discriminatory Assay and the HCV Discriminatory Assay, and tested diluted 1:16 with the Procleix HIV-1/HCV Assay. All dilutions were made with processed human serum that was negative for HIV-1 RNA and antibody/antigen, and HCV RNA and antibody.
During the study, specimens known to contain <100 copies/mL of viral RNA were excluded from this analysis and therefore the sensitivity presented herein is for samples with viral RNA concentrations equal to or greater than 100 copies/mL, or of unknown viral concentration.
The sensitivity for the Procleix HIV-1/HCV and HIV-1 Discriminatory Assays for undiluted (neat) HIV-1 positive samples was 99.9% (95% CI: 99.4-100%) and 100% (95% CI: 99.6-100%), respectively. The sensitivity for the Procleix HIV-1/HCV Assay for diluted (1:16) HIV-1 positive samples was 99.0% (95% CI: 98.0-99.5%).
The sensitivity for both the Procleix HIV-1/HCV Assay and the HCV Discriminatory Assay for undiluted (neat) HCV positive samples was 99.6% (95% CI: 98.9-99.9%). The sensitivity for the Procleix HIV-1/HCV Assay for diluted (1:16) HCV positives was 99.6% (95% CI: 98.9-99.9).
The sensitivity for the Procleix HIV-1/HCV Assay, HIV-1 Discriminatory Assay and HCV Discriminatory Assay for undiluted HIV-1/HCV coinfected specimens was 100% (95% CI: 98.0-100%), 100% (95% CI: 97.9-100%) and 100% (95% CI: 92.6-100%), respectively. The sensitivity for the Procleix HIV-1/HCV Assay for HIV-1/HCV coinfected specimens was 98.9% (95% CI: 96.0-99.9) when tested at 1:16 dilution.
The overall clinical sensitivity for the Procleix HIV-1/HCV Assay, which takes into account all samples (RNA concentrations> 100 copies/mL or unknown viral concentration) tested, is 99.8% (95% CI: 99.4-99.9), that for the HIV-1 Discriminatory Assay is 100% (95% CI: 99.6-100%), and that for the HCV Discriminatory Assay is 99.6% (95% CI: 99.0-99.9%).
IN0076 Rev. F
36
Table VII. The Sensitivity of the Procleix HIV-1/HCV, HIV-1 and HCV Discriminatory Assays for HIV-1 and HCV Positive Specimens with RNA Concentrations> 100 Copies/mL or Unknown
HIV-1/HCV Assay
| |
|---|
| | Sensitivity for Neat Specimens
| | Sensitivity for 1:16 Diluted Specimens
| |
|---|
Sample
| |
|---|
| | N
| | TP
| | %
| | (95% C. I.)
| | N
| | TP
| | %
| | (95% C. I.)
| |
|---|
| All | | 2014 | | 2009 | | 99.8 | | (99.4—99.9 | ) | 2012 | | 1997 | | 99.3 | | (98.8—99.6 | ) |
| HIV Only | | 867 | | 866 | | 99.9 | | (99.4—100.0 | ) | 866 | | 857 | | 99.0 | | (98.0—99.5 | ) |
| HCV Only | | 967 | | 963 | | 99.6 | | (98.9—99.9 | ) | 966 | | 962 | | 99.6 | | (98.9—99.9 | ) |
| HIV & HCV | | 180 | | 180 | | 100 | | (98.0—100.0 | ) | 180 | | 178 | | 98.9 | | (96.0—99.9 | ) |
HIV-1 Discriminatory Assay
| |
|---|
| | Sensitivity
| |
|---|
Sample
| |
|---|
| | N
| | TP
| | %
| | (95% C. I.)
| |
|---|
| All | | 1042 | | 1042 | | 100 | | (99.6—100.0 | ) |
| HIV Only | | 868 | | 868 | | 100 | | (99.6—100.0 | ) |
| HIV & HCV | | 174 | | 174 | | 100 | | (97.9—100.0 | ) |
HIV Discriminatory Assay
| |
|---|
| | Sensitivity
| |
|---|
Sample
| |
|---|
| | N
| | TP
| | %
| | (95% C. I.)
| |
|---|
| All | | 1014 | | 1010 | | 99.6 | | (99.0—99.9 | ) |
| HCV Only | | 966 | | 962 | | 99.6 | | (98.9—99.9 | ) |
| HIV & HCV | | 48 | | 48 | | 100 | | (92.6—100.0 | ) |
C. I. = Confidence interval.
The data from the above study were further analyzed according to the disease stages of the patients from whom the specimens were obtained as shown in Table VIII. A total of 296 samples were from AIDS patients (as defined by AIDS-indicative conditions and/or a CD4 count of <200/mm3), 338 from asymptomatic patients (asymptomatic, persistent generalized lymphadenopathy, or acute HIV infection), 168 from symptomatic but non-AIDS patients (not AIDS and not asymptomatic) and 240 from individuals with unknown HIV disease state29. Most of these patients were on HIV anti-viral medication. The sensitivity for HIV detection with the Procleix HIV-1/HCV Assay ranged from 99.6 to 100% for neat specimens and from 96.4 to 100% for 1:16 diluted specimens. The sensitivity for the HIV-1 Discriminatory Assay was 100%. All HIV-1 p24 Ag reactive specimens were also reactive with the Procleix HIV-1/HCV Assay when tested as undiluted (neat) or as 1:16 diluted samples, and with HIV-1 Discriminatory Assay when tested as neat samples. This was also true of all specimens excluded from the study due to low viral RNA concentrations (<100 copies/mL).
Similarly, the specimens from HCV infected patients were segregated as shown in Table IX. A total of 887 specimens were from volunteer blood donors whose donations were HCV reactive with PCR-based NAT, 53 specimens were from patients with chronic HCV infection that was first identified by blood donation screening and 75 specimens were not categorized by the vendor other than being HCV NAT or antibody positive. The sensitivity for HCV testing with the Procleix HIV-1/HCV Assay ranged from 99.5 to 100% (95% CI: 98.9-99.9%) with neat samples and 97.3 to 100% (95% CI: 93.3-100%) with 1:16 diluted samples. The sensitivity for HCV Discriminatory Assay ranged from 99.5 to 100% (95% CI: 95.2-100%) for neat samples.
During this clinical study, in which a total of 1014 HCV RNA-positive samples were tested, two of these samples were consistently reactive when tested at 1:16 dilution, but nonreactive when tested neat.
IN0076 Rev. F
37
In the same study, 13 sero-positive samples with low copy numbers of HIV or HCV (or both) were non-reactive when tested at 1:16 dilution, and reactive when tested neat.
In summary, it appears that the disease stages for HIV-1 or HCV infected individuals did not significantly affect the sensitivity for the Procleix Assays, although some low copy number HCV antibody positive specimens (2/967) were non-reactive when tested neat but reactive when tested at 1:16 dilution.
The clinical sensitivity claims of the assay are still met with the inclusion of these specimens.
IN0076 Rev. F
38
Table VIII. Sensitivity of the Procleix HIV-1/HCV and HIV-1 Discriminatory Assays for HIV-1 Positive Specimens from Individuals at Various Disease States*
| | Procleix HIV-1/HCV Assay
| | HIV-1 Discriminatory
| | HIV-1 Antibody
| | HIV-1 p24 Ag
|
|---|
| | Neat
| | 1:16 Dilution
| | Neat
| |
| |
| |
| |
| |
| |
|
|---|
Disease
| | N
Tested
| | N
R
| | %
R
| | N
Tested
| | N
R
| | %
R
| | N
Tested
| | N
R
| | %
R
| | N
Tested
| | N
R
| | %
R
| | N
Tested
| | N
R
| | %
R
|
|---|
| AIDS | | 295 | | 295 | | 100.0 | | 296 | | 295 | | 99.7 | | 296 | | 296 | | 100.0 | | 296 | | 296 | | 100.0 | | 226 | | 44 | | 19.5 |
| Symptomatic** | | 168 | | 168 | | 100.0 | | 167 | | 161 | | 96.4 | | 168 | | 168 | | 100.0 | | 168 | | 168 | | 100.0 | | 138 | | 14 | | 10.1 |
| Asymptomatic*** | | 338 | | 338 | | 100.0 | | 338 | | 338 | | 100.0 | | 338 | | 338 | | 100.0 | | 338 | | 297 | | 87.9 | | 234 | | 73 | | 31.2 |
| Unknown | | 240 | | 239 | | 99.6 | | 240 | | 238 | | 99.2 | | 240 | | 240 | | 100.0 | | 236 | | 234 | | 99.2 | | 202 | | 29 | | 14.4 |
| | |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
|
| Total | | 1041 | | 1040 | | 99.9 | | 1041 | | 1032 | | 99.1 | | 1042 | | 1042 | | 100.0 | | 1038 | | 995 | | 95.9 | | 800 | | 160 | | 20.0 |
N = Number R = Reactive
- *
- Samples with confirmed viral loads < 100 copies/mL excluded.
- **
- Symptomatic (not asymptomatic and not AIDS).
- ***
- Asymptomatic (asymptomatic, persistent generalized lymphadenopathy, or acute HIV infection).
Table IX. Sensitivity of the Procleix HIV-1/HCV and HCV Discriminatory Assays for HCV Positive Specimens From Individuals at Various Disease States*
| | Procleix HIV-1/HCV Assay
| | HIV Discriminatory Assay
| | HIV Antibody
|
|---|
| | Neat
| | 1:16 Dilution
| | Neat
| |
| |
| |
|
|---|
Disease
| | N
Tested
| | N
R
| | %
R
| | N
Tested
| | N
R
| | %
R
| | N
Tested
| | N
R
| | %
R
| | N
Tested
| | N
R
| | %
R
|
|---|
| First Time Blood | | 887 | | 883 | | 99.5 | | 886 | | 882 | | 99.5 | | 886 | | 882 | | 99.5 | | 886 | | 886 | | 100 |
| Chronic HCV | | 53 | | 53 | | 100 | | 53 | | 53 | | 100 | | 53 | | 53 | | 100 | | 53 | | 53 | | 100 |
| Unknown | | 75 | | 75 | | 100 | | 75 | | 73 | | 97.3 | | 75 | | 75 | | 100 | | 52 | | 20 | | 38.5 |
| | |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
|
| Total | | 1015 | | 1011 | | 99.6 | | 1014 | | 1008 | | 99.4 | | 1014 | | 1010 | | 99.6 | | 991 | | 959 | | 96.8 |
N = Number R = Reactive
- *
- Samples with confirmed viral loads < 100 copies/mL excluded.
Sensitivity for Pooled Samples
The clinical sensitivity of the Procleix HIV-1/HCV Assay with pooled samples was determined by testing 102 sixteen-member pools composed of one HIV-1 or HCV positive sample and 15 negative samples, and 102 sixteen-member pools composed of two HIV-1 and/or HCV positive samples and 14 negative samples. The viral load of the HIV-1 positive samples used to make the pools ranged from 1060 copies/mL to 10,018,200 copies/mL with a median of 27,490 copies/mL. The viral load of the HCV positive samples used to make the pools ranged from 1660 copies/mL to 20,200,000 copies/mL with a median of 327,000 copies/mL. All 204 (100%; 95% CI: 98.2-100%) pools containing at least one HIV-1 and/or HCV RNA positive specimen were reactive with the HIV-1/HCV Assay.
Prospective Study of Individuals at High Risk for HIV-1 and HCV Infection
Specimens from 539 individuals at high risk for infection with HIV-1 and/or HCV were tested as undiluted (neat) samples with the Procleix HIV-1/HCV, HIV-1 Discriminatory and HCV Discriminatory Assays. These samples were also tested at 1:16 dilution with the Procleix HIV-1/HCV Assay. Results
IN0076 Rev. F
39
are shown in Table X. 72.5% (391/539) had IV drug use (IVDU) as one of their risk factors. Risk factors other than IVDU included having unprotected sex, men having sex with men, occupational exposure, having sex with positive partner, and having transfusion of blood or blood products. Sensitivity was determined by comparing the Procleix Assays with an HIV-1 or HCV PCR-based assay that has a claimed analytical sensitivity of³100 copies viral RNA /mL.
Both the Procleix HIV-1/HCV Assay and HIV-1 Discriminatory Assay detected all 23 samples, either undiluted or 1:16 diluted, that were tested reactive for HIV-1 with the PCR NAT test. Of these 23 HIV-1 reactive specimens, 11 were also reactive with the HCV Discriminatory Assay and are considered samples from individuals coinfected with HIV-1 and HCV.
There was one confirmed HIV-1 yield case that appeared to be in the window period. This specimen was reactive with the Procleix HIV-1/HCV and HIV-1 Discriminatory Assays as an undiluted sample, and with the Procleix HIV-1/HCV Assay as a 1:16 diluted sample. The specimen was HIV antibody seronegative, HIV p24 Ag positive and was positive with an alternate HIV-1 nucleic acid test (NAT).
There were 268 HCV antibody and/or alternate HCV NAT positive specimens among the 520 high-risk specimens that were tested with Procleix HIV-1/HCV and HCV Discriminatory Assays. When tested as undiluted (neat) samples, 266 of the 268 positive specimens (99.3%, 95% CI: 97.3-99.9%) were reactive with both the Procleix HIV-1/HCV and HCV Discriminatory Assays. When tested as diluted samples, 254 of 259 (98.1%, 95% CI: 95.6-99.4%) were reactive with the Procleix HIV-1/HCV Assay. There were 44 HCV seropositive specimens that were nonreactive in all Procleix Assays. Of the 44, 37 were tested by an alternate HCV NAT and 34 were found to be NAT negative (considered Procleix TN for sensitivity calculations), two were equivocal, and one was positive (Procleix FN). This study resulted in three confirmed HCV yield cases that were considered true positives for the Procleix assay. These specimens were HCV antibody negative; two of which were alternate HCV NAT positive and one was QNS for alternate NAT. All three subjects later seroconverted.
Table X. Clinical Sensitivity of the Procleix HIV-1/HCV Assay in a High Risk Population
| |
| |
| |
| |
| |
| |
| | Sensitivity
|
|---|
Disease
| |
| |
| |
| |
| |
| |
|
|---|
| | Specimen
| | Number
| | TP
| | FP
| | TN
| | FN
| | %
| | (95% C. I.)
|
|---|
| HIV-1* | | Neat | | 530 | | 23 | | 0 | | 507 | | 0 | | 100 | | (85.2 - 100.0) |
| | | Diluted | | 520 | | 23 | | 0 | | 497 | | 0 | | 100 | | (85.2 - 100.0) |
HCV** |
|
Neat |
|
520 |
|
266 |
|
3 |
|
249 |
|
2 |
|
99.3 |
|
(97.3 - 99.9) |
| | | Diluted | | 508 | | 254 | | 2 | | 247 | | 5 | | 98.1 | | (95.6 - 99.4) |
- *
- Reactive in the HIV-1/HCV Assay and the HIV-1 Discriminatory Assay.
- **
- Reactive in the HIV-1/HCV Assay and the HCV Discriminatory Assay.
Analytical Sensitivity
To determine the analytical sensitivity of the Procleix HIV-1/HCV Assay and HIV-1 and HCV Discriminatory Assays for detection of HIV-1 and HCV viral RNA, HIV-1 panel members were prepared by serial dilution of negative human plasma spiked with HIV-1 (type B isolate) tissue culture supernatant. HCV panel members were made by serial dilution of a patient plasma specimen containing HCV (subtype 1a). The RNA levels in viral stocks used to make the HIV-1 panel and high titer HCV plasma used to make the HCV panel were value assigned using an in-house quantitative HIV-1 assay calibrated to the VQA standard obtained from Dr. James Bremer (Rush-Presbyterian
IN0076 Rev. F
40
Hospital, Chicago, IL) or quantitative HCV assay compared to HCV RNA WHO standard. (1 WHO IU/mL is equivalent to 2.7 copies/mL).
The panel members were tested with ten clinical lots of reagents and the test results are presented in Table XI. The Procleix HIV-1/HCV Assay and HIV-1 and HCV Discriminatory Assays achieved 100% detection for panel members containing 300 copies/mL, and > 99% detection for those members containing 100 copies/mL of HIV-1 or HCV RNA. The lower bound of 95% CI for both HIV-1 and HCV at 100 and 300 copies/mL for all assays exceeded 95%, which is consistent with a claimed analytical sensitivity of 100 copies RNA/mL. The Procleix HIV-1/HCV and Discriminatory Assays were able to detect 30 copies/mL of HIV-1 or HCV RNA at a frequency greater than 90%, with the lower bound of 95% CI ranging from 90% to 97.3%.
Table XI. Detection of HIV-1 B RNA and HCV 1a RNA in Analytical Sensitivity Panels
| | Procleix HIV-1/HCV Assay
| | HIV-1 Discriminatory Assay
|
|---|
| |
| |
| | 95%
Confidence Limits
| |
| |
| | 95%
Confidence Limits
|
|---|
HIV-1
Copies/mL
| | Number of
reactive/
tested*
| | %
Positive
| | Number of
reactive/
tested*
| | %
Positive
|
|---|
| | Lower
| | Upper
| | Lower
| | Upper
|
|---|
| 300 | | 716/716 | | 100 | | 99.5 | | 100 | | 715/715 | | 100 | | 99.5 | | 100 |
| 100 | | 719/719 | | 100 | | 99.5 | | 100 | | 718/718 | | 100 | | 99.5 | | 100 |
| 30 | | 707/720 | | 98.2 | | 96.9 | | 99.0 | | 702/713 | | 98.5 | | 97.3 | | 99.2 |
| 10 | | 573/718 | | 79.8 | | 76.7 | | 82.7 | | 592/717 | | 82.6 | | 79.6 | | 85.3 |
| 3 | | 297/718 | | 41.4 | | 37.7 | | 45.1 | | 305/717 | | 42.5 | | 38.9 | | 46.3 |
| 1 | | 112/717 | | 15.6 | | 13.0 | | 18.5 | | 139/718 | | 19.4 | | 16.5 | | 22.4 |
|
|
Procleix HIV-1/HCV Assay
|
|
HIV Discriminatory Assay
|
|---|
| |
| |
| | 95%
Confidence Limits
| |
| |
| | 95%
Confidence Limits
|
|---|
HIV 1a
Copies/mL
| | Number of
reactive/
tested*
| | %
Positive
| | Number of
reactive/
tested*
| | %
Positive
|
|---|
| | Lower
| | Upper
| | Lower
| | Upper
|
|---|
| 300 | | 718/718 | | 100 | | 99.5 | | 100 | | 720/720 | | 100 | | 99.5 | | 100 |
| 100 | | 720/720 | | 100 | | 99.5 | | 100 | | 745/746 | | 99.9 | | 99.3 | | 100 |
| 30 | | 669/718 | | 93.2 | | 91.1 | | 94.9 | | 660/716 | | 92.2 | | 90.0 | | 94.0 |
| 10 | | 470/719 | | 65.4 | | 61.8 | | 68.9 | | 458/717 | | 63.9 | | 60.2 | | 67.4 |
| 3 | | 231/718 | | 32.2 | | 28.8 | | 35.7 | | 258/717 | | 36.0 | | 32.5 | | 39.6 |
| 1 | | 70/716 | | 9.8 | | 7.7 | | 12.2 | | 104/719 | | 14.5 | | 11.0 | | 17.3 |
- *
- Invalid reactions were not re-tested.
CBER HIV-1 RNA Panel
Panel A (5 members) and Panel B (8 members) were tested in duplicate with 5 Clinical Lots using both the HIV-1/HCV Assay and the HIV-1 Discriminatory Assay. Results for both Panel A and B are shown in Table XII. For Panel A, testing with the HIV-1/HCV Assay showed reproducible detection of HIV-1 RNA at copy levels ranging from 250,000 to 100 copies/mL; the panel member at 0 copies/mL was non-reactive. Results for Panel B demonstrated reproducible detection of HIV-1 RNA at copy levels ranging from 250,000 to 50 copies/mL and non-reactive results with both negative panel members (B4 and B8). Similar results were obtained with the Discriminatory Assays.
IN0076 Rev. F
41
CBER HCV RNA Panel
This panel consisted of 10 panel members with copy levels ranging from 100,000 to 0 copies/mL. This panel was tested in duplicate with 5 Clinical Lots using both the HIV-1/HCV and HCV Discriminatory Assays. Results are shown in Table XII. Reproducible detection of HCV was obtained down to 50 copies/mL with both assays.
Table XII. Detection of HIV-1 RNA and HCV RNA in CBER panel members
| | Panel members tested and positivity rates
| |
|---|
| | A1
| | A2
| | A3
| | A4
| | A5
| | B1
| | B2
| | B3
| | B4
| | B5
| | B6
| | B7
| | B8
| |
|---|
| CBER HIV-1 RNA Panel(copies/mL) | | 250,00 | | 25,000 | | 1,000 | | 100 | | 0 | | 2,500 | | 10 | | 250,000 | | 0 | | 100 | | 50 | | 25,000 | | 0 | |
| HIV-1/HCV Assay* | | 100 | % | 100 | % | 100 | % | 100 | % | 0 | % | 100 | % | 60 | % | 100 | % | 0 | % | 100 | % | 100 | % | 100 | % | 0 | % |
| HIV-1 Discriminatory Assay** | | 100 | % | 100 | % | 100 | % | 100 | % | 0 | % | 100 | % | 100 | % | 100 | % | 0 | % | 100 | % | 100 | % | 100 | % | 0 | % |
|
|
Panel members tested and positivity rates
|
|
|---|
| | 1
| | 2
| | 3
| | 4
| | 5
| | 6
| | 7
| | 8
| | 9
| | 10
| |
|---|
| CBER HCV RNA Panel(copies/mL) | | 1,000 | | 0 | | 100,000 | | 10,000 | | 0 | | 500 | | 200 | | 50 | | 10 | | 5 | |
| HIV-1/HCV Assay* | | 100 | % | 0 | % | 100 | % | 100 | % | 0 | % | 100 | % | 100 | % | 100 | % | 90 | % | 30 | % |
| HCV Discriminatory Assay** | | 100 | % | 0 | % | 100 | % | 100 | % | 0 | % | 100 | % | 100 | % | 100 | % | 100 | % | 50 | % |
* n=10; ** n=6
WHO International Standard for HIV-1
The WHO International Standard for HIV-1 RNA (NIBSC code 97/656) with a concentration of 100,000 IU/mL was serially diluted and tested with the HIV-1/HCV Assay and the HIV-1 Discriminatory Assay. The results obtained are shown in Table XIII.
WHO International Standard for HCV
The WHO International Standard for HCV RNA (96/790) with a concentration of 100,000 international units (IU)/mL was serially diluted and tested with the HIV-1/HCV Assay and the HCV Discriminatory Assay. The results obtained are shown in Table XIII.
IN0076 Rev. F
42
Table XIII. Testing of International Standards for HIV-1 RNA (NIBSC code 97/656) and HCV RNA (NIBSC 96/790)
| | Concentrations tested and positivity rates
|
|---|
| WHO HIV-1 (97/656) | | 300
IU/mL | | 100
IU/mL | | 33.3
IU/mL | | 11.1
IU/mL | | 3.7
IU/mL | | 1.23
IU/mL | | 0
IU/mL |
| HIV-1/HCV Assay* | | 100% | | 100% | | 100% | | 77.5% | | 50% | | 32.5% | | 0% |
| HIV-1 Discriminatory Assay** | | 100% | | 100% | | 100% | | 80% | | 27.6% | | 26.6% | | 0% |
|
|
Concentrations tested and positivity rates
|
|---|
| WHO HCV (96/790) | | 110
IU/mL | | 37
IU/mL | | 11
IU/mL | | 3.7
IU/mL | | 1.1
IU/mL | | 0.37
IU/mL | | 0.11
IU/mL | | 0.04
IU/mL | | 0.00
IU/mL |
| HIV-1/HCV Assay* | | 100% | | 100% | | 100% | | 100% | | 50% | | 25% | | 0% | | 0% | | 0% |
| HCV Discriminatory Assay** | | 100% | | 100% | | 100% | | 95% | | 60% | | 25% | | 0% | | 0% | | 0% |
* n=40; ** n=30
Reactivity in Seroconverting Donors
Commercially available seroconversion panels collected from plasmapheresis donors were tested with the Procleix HIV-1/HCV Assay (neat and 1:16 diluted), HIV-1 Discriminatory (neat only) and HCV Discriminatory (neat only) Assays. Ten seroconversion panels for HIV-1 and ten panels for HCV were tested with one Clinical Lot. The test results were compared with those of the Ortho HCV 3.0 ELISA test or the Abbott Anti-HCV 2.0 test for HCV seroconversion panels, or with those of Abbott HIV-1/-2 antibody and Abbott or Coulter HIV-1 p24 antigen test for HIV-1 seroconversion panels. The Procleix HIV-1/HCV Assay was able to detect the infection with median values of 12 and 7 days earlier than the Abbott HIV-1/-2 antibody and HIV-1 p24 Ag assays, respectively, when specimens were tested neat (Table XIV). The Procleix HIV-1/HCV Assay was able to detect the infection with median values of 10 and 3 days earlier than the Abbott HIV-1/-2 antibody and HIV-1 p24 Ag tests, respectively, when specimens were tested at a 1:16 dilution. The HIV-1 Discriminatory Assay was able to detect infection with median values of 12 and 6 days earlier than the Abbott HIV-1/-2 antibody and HIV-1 p24 Ag tests, respectively, when specimens were tested neat. Reduction of the window period was observed in 9 of 10 panels when the Procleix HIV-1/HCV and HIV-1 Discriminatory Assays were used as compared to the use of the Abbott HIV Ab test alone. When compared to the HIV-1 p24 Ag assay, the Procleix HIV-1/HCV Assay detected the infection earlier in 8 of 10 panels and at the same time as HIV-1 p24 Ag in the other two panels. In all cases, HIV-1 p24 Ag reactive specimens were reactive with the Procleix HIV-1/HCV Assay and the HIV-1 Discriminatory Assay.
IN0076 Rev. F
43
Table XIV. Testing for HIV-1 RNA with the Procleix HIV-1/HCV and HIV-1 Discriminatory Assays on HIV-1 Seroconversion Panels
| | Days Earlier Detection Than HIV Antibody
| | Days Earlier Detection Than HIV-1 p24 Ag
|
|---|
| |
| |
| | HIV-1 Discriminatory
| |
| |
| | HIV-1 Discriminatory
|
|---|
| | HIV-1/HCV
| | HIV-1/HCV
|
|---|
Panel ID
|
|---|
| | Neat
| | 1/16
| | Neat
| | Neat
| | 1/16
| | Neat
|
|---|
| BCP 6240 | | 12 | | 7 | | 12 | | 7 | | 2 | | 7 |
| BCP 6248 | | 14 | | 11 | | 11 | | 7 | | 4 | | 4 |
| PRB923* | | 12 | | 17 | | 17 | | 2 | | 7 | | 7 |
| PRB926** | | 27 | | 25 | | 27 | | 7 | | 5 | | 7 |
| PRB929*** | | 11 | | 11 | | 11 | | 0 | | 0 | | 0 |
| PRB932^ | | 0 | | 0 | | 0 | | 0 | | 0 | | 0 |
| PRB943 | | 9 | | 9 | | 9 | | 2 | | 2 | | 2 |
| PRB945** | | 13 | | 10 | | 13 | | 13 | | 10 | | 13 |
| PRB946^^ | | 11 | | 7 | | 7 | | 7 | | 3 | | 3 |
| PRB950** | | 28 | | 10 | | 28 | | 18 | | 0 | | 18 |
| | |
| |
| |
| |
| |
| |
|
| Median | | 12 | | 10 | | 12 | | 7 | | 3 | | 6 |
- *
- Intermittent HIV-1/HCV reactivity at 11 and 35 days prior to ramp up is not used for this calculation.
- **
- HIV-1/HCV positive in first bleed of seroconversion panel.
- ***
- Single reactive in the HIV-1 Discriminatory Assay 14 days prior to ramp up is not used for this calculation.
- ^
- HIV antibody was negative at day 78 of the seroconversion series during HIV-1/HCV reactivity.
- ^^
- No seroconversion to HIV antibody in this panel which spans 11 days.
The Procleix HIV-1/HCV and HCV Discriminatory Assays were able to detect infection with a median value of 25 days earlier than the HCV antibody tests (Table XV) when tested diluted or undiluted. Reduction of the seroconversion window period by the Procleix HIV-1/HCV and HCV Discriminatory Assays was observed in 10 of 10 panels compared to the HCV antibody test.
IN0076 Rev. F
44
Table XV. Testing for HCV RNA with the Procleix HIV-1/HCV and HCV Discriminatory Assays on HCV Seroconversion Panels
| | Days Earlier Detection Than HCV Antibody
|
|---|
| |
| |
| | HCV Discriminatory
|
|---|
Panel ID
| | HIV-1/HCV
|
|---|
| | Neat
| | 1/16
| | Neat
|
|---|
| BCP 6213 | | 26 | | 26 | | 26 |
| BCP 6225 | | 39 | | 33 | | 39 |
| BCP 6226* | | 39 | | 39 | | 39 |
| BCP 6228* | | 31 | | 31 | | 31 |
| BCP 9045* | | 41 | | 41 | | 41 |
| PHV904* | | 14 | | 14 | | 14 |
| PHV907* | | 21 | | 21 | | 21 |
| PHV908* | | 19 | | 19 | | 19 |
| PHV914* | | 24 | | 24 | | 24 |
| PHV916* | | 23 | | 23 | | 23 |
| | |
| |
| |
|
| Median | | 25 | | 25 | | 25 |
*HIV-1/HCV Assay reactive in first bleed of seroconversion panel vs. Ortho HCV 3.0.
Subtype Detectability
Since there are no recognized international standards for HCV or HIV-1 other than HCV subtype 1a and HIV-1 subtype B, multiple specimens and isolates (59 different HIV-1 and 53 different HCV specimens) were tested to determine detectability of these viral subtypes31. HIV-1 specimens of subtypes A, B, C, D, E, F, and G were quantified for HIV-1 RNA concentrations using commercial quantitative HIV-1 RNA assays or an in-house developed quantitative test, the latter using the same technology as the Procleix assays. HIV-1 subtypes N and O were quantified with an in-house quantitative HIV-1 RNA test. Specimens were diluted into negative human plasma to target viral concentrations of 300 or 100 copies/mL and diluted specimens were tested in the HIV-1/HCV and HIV-1 Discriminatory Assays. All HIV-1 subtypes were reactive with both the Procleix HIV-1/HCV and HIV-1 Discriminatory Assays at 300 and 100 copies/mL (Table XVI).
HCV specimens of subtypes 1, 2, 3, 4, 5 and 6 were quantified for HCV RNA using commercially available quantitative HCV RNA assays. Specimens were diluted into negative human plasma to target viral concentrations of 300 or 100 copies/mL and diluted specimens were tested with the HIV-1/HCV and HCV Discriminatory Assays. All HCV subtypes were reactive by the HIV-1/HCV and HCV Discriminatory Assays at 300 and 100 copies/mL, except one HCV subtype 2 specimen which was reactive at 300 copies/mL, but nonreactive at 100 copies/mL (Table XVI).
IN0076 Rev. F
45
Table XVI. HIV-1 and HCV Subtype Detectability
Specimen
| | Subtype
| | Copies/mL
| | HIV-1/HCV
Reactive/Total
| | HIV-1 Discriminatory
Reactive/Total
|
|---|
| HIV | | A* | | 300
100 | | 11/11
9/9 | | 11/11
9/9 |
| | | B | | 300
100 | | 10/10
10/10 | | 11/11
11/11 |
| | | C | | 300
100 | | 9/9
9/9 | | 9/9
9/9 |
| | | D | | 300
100 | | 6/6
6/6 | | 6/6
6/6 |
| | | E | | 300
100 | | 8/8
8/8 | | 8/8
8/8 |
| | | F | | 300
100 | | 5/5
5/5 | | 5/5
5/5 |
| | | G | | 300
100 | | 3/3
3/3 | | 3/3
3/3 |
| | | N | | 300
100 | | 1/1
1/1 | | 1/1
1/1 |
| | | O | | 300
100 | | 6/6
6/6 | | 6/6
6/6 |
Specimen
|
|
Subtype
|
|
Copies/mL
|
|
HIV-1/HCV
Reactive/Total
|
|
HIV Discriminatory
Reactive/Total
|
|---|
| HCV | | 1 | | 300
100 | | 10/10
10/10 | | 10/10
10/10 |
| | | 2 | | 300
100 | | 13/13
12/13 | | 13/13
13/13 |
| | | 3 | | 300
100 | | 11/11
11/11 | | 11/11
11/11 |
| | | 4 | | 300
100 | | 10/10
10/10 | | 11/11
11/11 |
| | | 5 | | 300
100 | | 4/4
4/4 | | 4/4
4/4 |
| | | 6 | | 300
100 | | 4/4
5/5 | | 4/4
5/5 |
* Two samples were quantified at < 1000 copies/mL and were reactive when tested undiluted and at 1:3 dilution.
PERFORMANCE OF INDIVIDUAL DONATION TESTING
Clinical Sensitivity
The clinical sensitivity of the HIV-1/HCV Assay, HIV-1 Discriminatory and HCV Discriminatory Assays were evaluated by testing clinical samples without dilution (neat). The HIV-1/HCV Assay was used to test a total of 867 confirmed HIV positive, 967 HCV positive and 180 HIV and HCV positive samples. As summarized in Table VII, the overall sensitivity based on this study was 99.8% (95% CI: 99.4-99.9). Specifically, the sensitivity for HIV positive samples was 99.9% (95% CI: 99.4-100.0) while
IN0076 Rev. F
46
that for HCV positive samples and HIV and HCV positive samples was 99.6% (95% CI: 98.9-99.9) and 100% (95% CI: 98.0-100.0), respectively.Three HCV positive samples tested non-reactive as undiluted samples, but reactive at 1:16 dilution. However, one of the three showed a viral load lower than the Limit of Detection for the Procleix HIV-1/HCV Assay as determined by an alternate nucleic acid test. The other two consistently tested reactive at 1:16 dilution but non-reactive with the undiluted sample and the nature of this discordance is under investigation.
The sensitivity for the discriminatory assays was evaluated as well. As shown in Table VII, the sensitivity for the HIV-1 Discriminatory Assay was 100% (95% CI: 99.6-100.0) for HIV positive samples and 100% (95% CI: 97.9-100.0) for HIV and HCV positive samples. The HCV Discriminatory Assay showed a sensitivity of 99.6% (95% CI: 98.9-99.9) for HCV positive samples and 100% (95% CI: 92.6-100.0) for HIV and HCV positive samples.
The data in the aforementioned studies were re-analyzed according to the disease stages and the results are presented in Tables VIII (for HIV/AIDS) and IX (for HCV). Overall, the assays showed similar sensitivity for samples from various disease stages.
Seroconversion Panel Testing
When a limited number of HIV-1 seroconversion panel members were tested as undiluted (neat), an average of two days earlier detection was observed as compared to 1:16 dilution (Table XIV). No difference was observed between the HIV-1/HCV Assay and HIV-1 Discriminatory Assay. Both assays were more sensitive for detecting window period samples as compared to the HIV-1 p24 Antigen assay.
For HCV seroconversion panels, no differences were observed between the Procleix HIV-1/HCV Assay and the HCV Discriminatory Assay when testing was performed on undiluted samples or 1:16 diluted samples (Table XV). Both assays were able to detect HCV infection on average 25 days earlier than the antibody test.
Clinical Specificity
The clinical specificity for individual donation testing was determined for the HIV-1/HCV assay by testing individual donor specimens that were never pooled (Table XVII). Seventy-one of 34,557 (0.21%) individual donor specimens were initially reactive in the Procleix HIV-1/HCV assay. Twenty-one of these 71 specimens were also reactive in the HCV Discriminatory Assay and were seropositive for HCV. Three of the 71 were reactive in the HIV-1 Discriminatory Assay and seropositive for HIV-1/HIV-2. Forty-five specimens were nonreactive by both the HCV and HIV-1 Discriminatory Assays and HCV and HIV-1/HIV-2 antibody assays yielding an adjusted (false) reactive rate of 0.13% (45/34,533). One specimen had incomplete assay results and could not be discriminated. One specimen was Procleix HCV Discriminated, HCV EIA repeatedly Reactive with no RIBA available. Seventeen samples with Nonreactive Procleix assay results had incomplete serologic results and were excluded from the specificity calculations, yielding a specificity in individually tested donor samples of 99.87% (34,229/34,274).
IN0076 Rev. F
47
Table XVII. Procleix HIV-1/HCV Assay Reactivity in Volunteer Blood Donors
| | Procleix HIV-1/HCV Assay
Individual Donation
|
|---|
| Samples Tested* | | 34,557 |
| Initial Reactive | | 71 |
| Initial Reactive Rate | | 0.21% |
| Adjusted Reactive Rate | | 0.13% |
| Combined Mean S/CO on Negative Analytes | | 0.17 ± 0.07 |
* Combined data across all sites and Clinical Lots.
This specificity study was conducted primarily in three military sites. The military donor population may differ from the civilian donor population. However, when sub-analyses were conducted across donor age groups, gender and race, comparable clinical specificity was observed across all categories ranging from 99.7% to 100% (all 95% confidence intervals overlapped). These sub-analyses included evaluation of 5,743 females; 1,102 donors over the age of 50; and at least 2,900 donors in each of the race categories of Black/Non-Hispanic, White/Hispanic, and White/Non-Hispanic. These results suggest that the specificity of the Procleix HIV-1/HCV Assay with individual donations is not affected by race, age or gender.
Table XVIII. Summary of Procleix Assay Yield Under IND Testing
Individual Donation Testing
|
|---|
| Number of Donations Tested | | 103,357 |
Procleix HCV
Yield Case | | 1
(1:103,357) |
Procleix HIV-1
Yield Case | | 0 |
A total of 103,357 individual donations were screened under IND from April 2000 to November 2001 (Table XVIII). One Procleix HCV yield case (1:103,357) was identified across the three Procleix individual donation test sites. The yield case was confirmed to be positive for HCV RNA, but negative in serology testing. No HIV-1 yield cases were identified with individual donation testing.
LIMITATIONS OF THE PROCEDURE
- •
- This assay has been evaluated with the Procleix instrument only.
- •
- The concentrations for HIV-1 subtype N and group O virus used for assessing analytical sensitivity were determined by an in-house quantitative test, which used the same technology as the Procleix assays. This may result in inaccurate assessment of analytical sensitivity for these viral subtypes.
- •
- The Procleix Assay may not be used to replace antibody-detection tests such as an EIA test for HIV-1 or HCV, a Western Blot for HIV-1, or a RIBA for HCV.
- •
- The clinical sensitivity for the Procleix HIV-1/HCV assay has been evaluated only for specimens with viral concentrations equal to or greater than 100 copies RNA/mL or those with unknown viral concentration.
IN0076 Rev. F
48
CONCLUSIONS
Overall specificity for the Procleix HIV-1/HCV Assay (16-member pools, single donors), HIV-1 Discriminatory Assay (single donors) and HCV Discriminatory Assay (single donors) is shown in Table XIX. Sensitivity for the Procleix HIV-1/HCV Assay (based on known HIV-1 and HCV RNA positives run neat and diluted 1:16), HIV-1 Discriminatory Assay (based on known HIV-1 RNA positives run neat), and HCV Discriminatory Assay (based on known HCV RNA positives run neat) are also shown in Table XIX.
Table XIX. Overview
| |
| | Specificity (95% C.I.)
| | Sensitivity (95% C.I.)
|
|---|
| Procleix HIV-1/HCV | | 16-member Pool | | 99.67% (99.55-99.77%) | | 99.3% (98.8-99.6%)* |
| | | Individual donation | | 99.87% (99.83-99.91%) | | 99.8% (99.4-99.9%) |
| HIV-1 Discriminatory | | Individual donation | | 99.76% (99.48-99.91%) | | 100%
(99.6-100%) |
| HCV Discriminatory | | Individual donation | | 99.71% (99.41-99.88%) | | 99.6% (99.0-99.9%) |
C.I. = Confidence intervals.
- *
- Sensitivity in 2,012 known-positive samples diluted 1:16; Sensitivity was 100% in 204 pools.
BIBLIOGRAPHY
- 1.
- Barre-Sinoussi, F., J. C. Chermann, F. Rey, M. T. Nugeyre, S. Chamaret, J. Gruest, C. Dauguet, C. Axler-Blin, F. Vezinet-Brun, C. Rouziuuz, W. Rozenbaum, and L. Montagnier. 1983. Isolation of a T-lymphotropic retrovirus from a patient at risk for Acquired Immune Deficiency Syndrome (AIDS). Science.220:868-871.
- 2.
- Popovic, M., M. G. Sarngadharan, E. Read, and R. C. Gallo. 1984. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science.224:497-500.
- 3.
- Gallo R. C., S. Z. Salahuddin, M. Popovic, G. M. Strearer, M. Kaplan, D. F. Haynas, T. J. Palker, R. Redfield, J. Oleske, B. Safai, G. White, P. Foster, and P. D. Markham. 1984. Frequent detection and isolation of cytopathic retroviruses (HTLV III) from patients with AIDS and at risk for AIDS. Science.224:500-503.
- 4.
- Piot P., F. A. Plummer, F. S. Mhalu, J-L. Lamboray, J. Chin, and J. M. Mann.1988. AIDS: An international perspective. Science.239:573-579.
- 5.
- Sarngadharan, J. G., M. Popovic, L. Broch, J. Scupbach, and R. C. Gallo. 1984. Antibodies reactive with human T-lymphotropic retroviruses (HTLV-III) in the serum of patients with AIDS. Science.224:506-508.
- 6.
- Gallo, D., J. S. Kimpton, and P. J. Dailey. 1987. Comparative studies on use of fresh and frozen peripheral blood lymphocyte specimens for isolation of human immunodeficiency virus and effects of cell lysis on isolation efficiency. J Clin Microbiol.25:1291-1294.
- 7.
- Clavel, F., D. Guetard, F. Brun-Vezinet, S. Chamaret, M. Rey, M. O. Santos-Ferraira, A. G. Laurent, C. Dauguet, C. Katlama, C. Rouzioux, D. Klatzmann, J. L. Champalimaud, and L.
IN0076 Rev. F
49
Montagnier. 1986. Isolation of a new human retrovirus from West African patients with AIDS. Science.233:343-346.
- 8.
- Alter, H. J., R. H. Purcell, J. W. Shih, J. C. Melpolder, M. Houghton, Q-L. Choo, and G. Kuo. 1989. Detection of antibody to hepatitis C virus in prospectively followed transfusion recipients with acute and chronic non-A, non-B hepatitis. N Engl J Med.321:1494-1500.
- 9.
- Esteban, J. I., A. Gonzalez, J. M. Hernandez, et al. 1990. Evaluation of antibodies to hepatitis C virus in a study of transfusion-associated hepatitis. N Engl J Med.323:1107-11200.
- 10.
- Van der Poel, C. L., H. W. Reesink, P. N. Lelie, A. Leentvaar-Kuypers, Q-L. Choo, G. Kuo, and M. Houghton. 1989.Anti-hepatitis C antibodies and non-A, non-B post-transfusion hepatitis in the Netherlands. Lancet.2:297-298.
- 11.
- Choo, Q-L., G. Kuo, A. J. Weiner, et al. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science.244:359-362.
- 12.
- Alter, H. J., P. V. Holland, Ag. Morrow, et al. 1975. Clinical and serological analysis of transfusion associated hepatitis. Lancet.2:838-841.
- 13.
- Kuo, G., Q-L. Choo, H. J. Alter, et al. 1989. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science.244:362-364.
- 14.
- Busch, M. P., S. L. Stramer, and S. H. Kleinman. 1997. Evolving applications of nucleic acid amplification assays for prevention of virus transmission by blood components and derivatives. In: Garrity G (ed): Applications of Molecular Biology to Blood Transfusion Medicine. AABB. Bethesda, MD. 123-176.
- 15.
- Stramer, S. L., S. Caglioti, and D. M. Strong. 2000. NAT of the United States and Canadian Blood Supply. Transfusion.40:1165-1168.
- 16.
- Busch, M. P., L. L. L. Lee, G. A. Satten, D. R. Henrard, H. Farzadegan, K. E. Nelson, S. Read, R. Y. Dodd, and L. R. Petersen. 1995. Time course of detection of viral and serologic markers preceding human immunodeficiency virus type 1 seroconversion: implications for screening of blood and tissue donors. Transfusion.35:91-97.
- 17.
- Schreiber, G. B., M. P. Busch, S. H. Kleinman, and J. J. Korelitz.1996. for the Retrovirus Epidemiology Study: The risk of transfusion-transmitted viral infections. The New Eng J of Med.334:1685-1690.
- 18.
- McDonough, S., C. Giachetti, Y. Yang, D. Kolk, B. Billyard, and L. Mimms.1998. High throughput assay for the simultaneous or separate detection of human immunodeficiency virus (HIV-1) and hepatitis C virus (HCV). Infusion Therapy and Transfusion Medicine.25:164-169.
- 19.
- Kacian, D. L. and T. J. Fultz. 1995. Nucleic acid sequence amplification methods. U. S. Patent 5,399,491.
- 20.
- Arnold, L. J., P. W. Hammond, W. A. Wiese, and N. C. Nelson.1989. Assay formats involving acridinium-ester-labeled DNA probes. Clin Chem.35:1588-1594.
- 21.
- Nelson, N. C., A. BenCheikh, E. Matsuda, and M. Becker.1996. Simultaneous detection of multiple nucleic acid targets in a homogeneous format. Biochem.35:8429-8438.
- 22.
- Centers for Disease Control. 1987. Recommendations for prevention of HIV transmission in health care settings. In United States Morbid. And Mortal. Weekly Rep. 36, Supplement No. 2S.
- 23.
- National Committee for Clinical Laboratory Standards.1986. Clinical laboratory hazardous waste; proposed guidelines. NCCLS Document GP5-P. Villanova, PA.
IN0076 Rev. F
50
- 24.
- U.S. Environmental Protection Agency.EPA guide for infectious waste management. Washington, DC: U.S. Environmental Protection Agency, Publication No. EPA/530-SW-86-014, 1986.
- 25.
- Title 42, Code of Federal Regulations, Part 72, 1992.
- 26.
- 29 CFR Part 1910.1030. Occupational Exposure to Bloodborne Pathogens; Final Rule, Federal Register/ Vol. 56, No. 235/ December 6, 1991.
- 27.
- Valinsky, J.E., C. Bianco. 2000. Supplemental testing for HCV: To what extent could nucleic acid amplification testing (NAT) replace HCV RIBA 3.0. Transfusion. 40, 10S, 32S.
- 28.
- Damen, M., H.L. Zaijjer, H.T.M. Cuypers, H. Vrielink, CL van der Poel, HW Reesink, PN Lelie. 1995. Reliability of the third-generation recombinant immunoblot assay for hepatitis C virus. Transfusion. 35, 745-749.
- 29.
- MMWR. 1992.41:1-9.
- 30.
- Stramer, S.L. et al. Application of Nucleic Acid Testing to Blood Borne Pathogens and Emerging Technologies OBRR/CBER/FDA Workshop—December 4, 2001.
- 31.
- Linnen, J.M., J.M. Gilker, A. Menez, A. Vaughn, A. Broulik, J. Dockter, K. Gillote-Taylor, K. Greenbaum, D.P. Kolk, L. T. Mimms, C. Giachetti. 2002. Sensitive detection of genetic variants of HIV-1 and HCV with an HIV-1/HCV assay based on transcription-mediated amplification. J. Virol. Methods. 102, 135-139.
Gen-Probe U.S. LICENSE 1592
IN0076 Rev. F
2002-02
Developed and manufactured by:
Gen-Probe Incorporated
10210 Genetic Center Drive
San Diego, CA 92121
(858) 410-8000
Distributed in U.S. by:
Chiron Corporation
4560 Horton Street
Emeryville, CA 94608-2916
Telephone (In U.S.): (800) CHIRON-8
(510) 655-8730
Distributed in rest of world by:
Chiron Ireland, Limited
United Drug House
Belgard Road, Tallaght
Dublin 24, Ireland
Chiron Blood Testing Technical Support:
(North America)
Telephone: (800) 452-6877
FAX: (800) 462-3938
(Europe, Middle East, Latin America, Africa)
Telephone: +33 (4) 78 37 99 04
FAX: +33 (4) 78 37 99 04
IN0076 Rev. F
51
(Asia/Pacific)
Telephone: +61 (4) 1044 5810
FAX: +61 (2) 9974 5411
Chiron, RIBA and Procleix are trademarks of Chiron Corporation; TECAN, GENESIS (stylized), and RSP are trademarks of Tecan AG; eppendorf (stylized) and COMBITIPS are trademarks of Eppendorf- Netheler-Hinz GmbH; PROCLIN (stylized) is a trademark of Rohm and Haas Company.
This product and its intended use are covered by one or more of the following: U.S. patent no. 5,030,557; 5,185,439; 5,283,174; 5,399,491; 5,437,990; 5,480,784; 5,585,481; 5,612,200; 5,639,604; 5,656,207; 5,656,744; 5,658,737; 5,696,251; 5,714,596; 5,750,338; 5,756,011; 5,756,709; 5,766,890; 5,827,656; 5,840,873; 5,863,719; 5,888,779; 5,948,899; 5,955,261; 6,004,745; 6,031,091; 6,074,816; 6,090,591; 6,110,678; 6,245,519; 6,252,059; 6,280,952; and international counterparts.
© 2000, 2001, 2002 Gen-Probe Incorporated
IN0076 Rev. F
52
| |
|
|---|
| CONFIDENTIAL | | REDACTED VERSION |
SCHEDULE C
TERMS AND CONDITIONS FOR USE OF BLOOD SCREENING SYSTEMS
Chiron will make available Blood Screening Systems to ABC on the following terms and conditions:
- 1.
- Sale of Blood Screening Systems. Chiron shall sell the Blood Screening Systems currently installed and in use by Participating Members as listed inExhibit 4to each Member Supplement (the "Transferred Instruments"), subject to the provisions hereof. Each Participating Member will acquire, at it's sole expense, any additional instrumentation it wishes to use in connection with the conduct of testing of blood donations, including any instruments required to perform pooling operations and sample archiving.
- 2.
- Location & Use of Blood Screening Systems. Participating Members shall use the Blood Screening Systems only at the Testing Centers identified inExhibit 2to the applicable Member Supplements. Participating Members shall not remove, customize, or alter the Blood Screening Systems, including Software, without the prior written consent of Chiron. Blood Screening Systems shall only be used to perform testing of the Participating Member's blood donations using Blood Screening Assays sold by Chiron. Chiron bears no responsibility with respect to any use of such Blood Screening Systems with Blood Screening Assays other than those supplied by Chiron hereunder. Any such alternate use shall invalidate all warranties with respect to the Blood Screening Systems.
- 3.
- Title to the Blood Screening Systems. As between Chiron and each Participating Member, upon payment of the amount specified in the applicable Member Supplement, Chiron shall transfer all right, title and interest held by it in and to the Transferred Instruments to the applicable Participating Member. Each Participating Members acquiring Transferred Instruments shall remove any markings from the Transferred Instruments which identify Chiron as the owner. If a Participating Member selects the installment payment option, title to the Transferred Instruments will transfer on execution, but the Participating Member will grant a security interest in such Transferred Instruments to Chiron as further described in the applicable Member Supplement.
- 4.
- Warranties; Limitation of Liability. Chiron represents and warrants that the Blood Screening Systems will operate in accordance with their Specifications and will be free from defects in materials and workmanship. This warranty does not apply to Blood Screening Systems (a) not installed by a Chiron representative; (b) not used or maintained in accordance with the instructions provided by Chiron or their manufacturer, including without limitation of the maintenance obligations set forth inSchedule D; (c) damaged by alteration, misuse, tampering or abuse; or (d) for which the Participating Member has failed to pay the Services Fee. Except to the extent of Chiron's obligations under Section 7.7 of this Association Agreement, Chiron's sole liability with respect to a breach of the foregoing warranty is to repair or replace the Blood Screening System. Participating Members will be responsible for all other repairs or replacements of Blood Screening Systems that fail to operate in accordance with their Specifications, including by reason of (i) any loss or damage caused by or attributable to the Participating Members or their agents (ii) a failure by the Participating Members to use the Blood Screening Systems in accordance with the manufacturer's instructions, (iii) any negligence on the part of the Participating Members or (iv) any failure by the Participating Members to carry out their obligations hereunder. Chiron disclaims all other warranties and further limits its liability in accordance with Sections 7.4 and 7.12 of this Association Agreement, respectively.
53
- 5.
- Software. Chiron shall provide to Participating Members, at no additional charge, the Software and Documentation, pursuant to the following terms and provisions:
- 5.1
- Title.
- (a)
- Chiron or the applicable licensor shall own and retain title to the Software, including all intellectual property rights embodied therein. Any copy which a Participating Member makes of the Software, in whole or in part, is and shall remain the property of Chiron or the applicable licensor.
- (b)
- If ownership of the Software or any work product does not result as provided in this Agreement or by operation of law, then the parties each assign and shall cause their respective employees, agents, and contractors to assign, without further consideration, the ownership thereof, including all associated intellectual property rights, as necessary to give effect to the ownership terms specified in this Agreement. Each party agrees to perform, at the reasonable request of the other party, such further acts as may be necessary or desirable to transfer ownership of, and to perfect and defend, the Software or work product in order to give effect to such ownership terms.
- 5.2
- Grant of Rights and Restrictions on Use
- (a)
- Grant of Rights. Chiron hereby grants, and each Participating Member hereby accepts, subject to the terms and conditions of this Agreement including thisSchedule C, a nonexclusive, nontransferable and nonassignable (except as permitted under Section 12.4 [Assignment] of this Association Agreement) object code license to use the Software at the Testing Centers solely for Participating Member's own use in connection with the operation of the Blood Screening Assays on the Blood Screening Systems during the term of the applicable Member Supplement, and to copy the Software solely for the purposes expressly authorized under this Section 5 of thisSchedule C. In addition, Chiron hereby grants to each Participating Member the right to use the Documentation in connection with its use of the Software hereunder. Documentation may not be copied. Additional copies may be obtained from Chiron at Chiron's charges then in effect. No right to use, copy, display, or print the Software or Documentation, in whole or in part, is granted, except as expressly provided in this Agreement.
- (b)
- Restrictions on Use. The grant of rights stated in Section 5.2(a) of thisSchedule Cis subject to the terms and conditions of this Agreement as well as the following restrictions:
- (i)
- Use of the Software may be subsequently transferred to other Testing Centers maintained by a Participating Member, provided (A) the total number of Testing Centers at which the Software is used by such Participating Member does not exceed the number of Testing Centers specified in the applicable Member Supplement and (B) such Participating Member provides Chiron with written notice thirty (30) days before such transfer.
- (ii)
- In the event that disaster or other circumstances prevent Participating Member from using the Software at the Testing Centers identified in the applicable Member Supplement, the effected Participating Member shall have the right to use the Software at a disaster recovery facility without prior notice to Chiron, but shall promptly notify Chiron as soon as circumstances permit.
- (iii)
- Participating Members shall not use (or cause to be used) the Software for rental, in the operation of a service bureau, or for any similar purpose; nor shall Participating Members allow access to the Software through terminals located outside Participating
54
- 5.3
- Warranties; Limitation of Liability.
- (a)
- Performance. Chiron warrants that the Software, under normal use and service, will perform all of the material functions described in the Documentation of such Software. Chiron warrants that the Documentation shall be free from material defects in materials and workmanship. If any such defect or deviation appears during the applicable periods, the Software or Documentation may be returned to Chiron for replacement by Chiron without charge.
- (b)
- Anti-Virus. Chiron warrants that to the best of its knowledge after employing reasonable technical means to detect computer viruses, the Software at the time of delivery will not contain any virus or computer software code, routines or devices (other than as set forth in the Documentation) designed to disable, damage, impair, or erase the Software or other software or data. For failure to comply with this warranty, Chiron shall, at Chiron's expense, immediately replace all copies of the affected Software in the possession of a Participating Member.
- (c)
- Right to License. Chiron warrants that it has, and on the effective date of each Member Supplement of the Software will have, full rights and authority to license the Software to Participating Members and otherwise as needed for it to perform its obligations hereunder, or has the authority to do so without infringing the rights of any Third Party.
- (d)
- Limitation of Liability. Chiron disclaims all other warranties and further limits its liability in accordance with Sections 7.4 and 7.12 of this Association Agreement.
- 5.4
- Source Code Escrow.
55
| |
|
|---|
| CONFIDENTIAL | | REDACTED VERSION |
SCHEDULE D
BLOOD SCREENING SYSTEMS AND SOFTWARE
SERVICE & SUPPORT
- 1.
- Maintenance & Troubleshooting.
- 1.1
- The Participating Member shall operate and maintain the Blood Screening Systems in accordance with Chiron and manufacturer guidelines and recommendations, which may be amended from time to time.
- 1.2
- In the event that any Testing Center resolves a problem with a Blood Screening System, such problem shall be logged in writing, including a description of the problem and the steps taken by the Participating Member to resolve it. The Participating Member shall maintain a log for each Blood Screening System and make available all such logs to Chiron Technical Support upon request.
- 1.3
- If a Testing Center is unable to resolve a problem, the Testing Center shall refer the problem to Chiron Technical Support for resolution in accordance with Section 2 of thisSchedule D.
- 1.4
- Except for routine maintenance and troubleshooting activities of Primary Operators as provided in thisSchedule D, or otherwise with the prior permission of Chiron, during the term of this Agreement only Chiron or Chiron's agents shall install, service, alter or replace the Blood Screening Systems. Chiron may inspect the Blood Screening Systems at reasonable times upon reasonable notice. Chiron will assure that activities of Primary Operators authorized under thisSchedule Dwill not invalidate any manufacturers' warranties with respect to the Blood Screening Systems.
- 2.
- Technical Support and Service.
- 2.1
- Installation. Chiron support personnel will provide installation and set up of all Blood Screening Systems, and shall ensure that the Blood Screening Systems operate in conformance with all relevant Specifications. Without limiting the foregoing, such setup and installation shall include the installation of the Software and verification of the operation of the Software in conjunction with the Blood Screening Systems. At such time as Chiron determines that such setup and installation has been completed, the Participating Member shall acknowledge the completion of such setup and installation and its acceptance of the Blood Screening Systems in writing.
- 2.2
- Project Management. Chiron will provide reasonable project management support to assist the Participating Member project manager in coordinating the activities of Chiron and the Participating Member in connection with testing blood using the Blood Screening Assays and the Blood Screening Systems.
- 2.3
- Data & Materials. The Participating Member shall make available to Chiron on a timely basis all data, information and other materials which are reasonably necessary for Chiron to perform the Services. Such data shall be treated as confidential Data in accordance with Section 6.4 of the Association Agreement. Chiron shall have no liability for any failure to perform, or for the late performance, of any Services to the extent such Services require data, information or materials possessed, prepared or generated by the Participating Member, if the Participating Member fails to provide the same in accordance with Chiron reasonable requests.
56
- 2.4
- Preventative Maintenance.
- (a)
- Chiron shall carry out scheduled Preventative Maintenance (PM) visits at six month intervals (2 per year). The PM services will be arranged at least two weeks before the due date for PM service at a time mutually agreeable between Chiron and the Participating Member. The Participating Member will be required to make the Blood Screening Systems available at a reasonable time within the working day (Monday to Friday excluding Holidays) to allow PM services to be completed within normal working hours.
- (b)
- Each PM service will cover thorough cleaning, lubrication, replacement of tubing and other parts, electronic/mechanical adjustments and safety checks, all in accordance with the Blood Screening Systems manufacturers' recommended procedures, as detailed in the applicable manufacturer's approved checklist/s. Defective components discovered during PM services will be replaced at no charge. Parts used to perform PM service will be provided by Chiron at its expense.
- (c)
- Appropriate testing will be performed in order to return the Blood Screening Systems back to the user. Tecan instruments will be "certified" as fully functional with required testing performed according to the manufacturers' specifications. Liquid precision checks will utilize the appropriate volume for testing.
- (d)
- Documentation of PM service performed will be provided to each site upon completion of all maintenance tasks. Information will be reviewed and signed by Participating Member personnel before Chiron service personnel leave the facility.
- (e)
- Additional PM service may be performed at the request of the Participating Member for an additional charge.
- 2.5
- Support and Service.
- (a)
- Chiron Technical Support is available 24 hours a day, 7 days a week including weekends and holidays via telephone. The Participating Member will refer problems to Chiron Technical Support through a central toll-free telephone number[**]. A service representative will be available at this number to receive problem referrals and provide troubleshooting assistance, from 7:30 a.m. to 5 p.m. PST, Monday through Friday, holidays excepted ("Normal Support Hours"). Messages left outside Normal Support Hours (including weekends and holidays) will automatically page the on-call Technical Support personnel. Required message information left outside Normal Support Hours should include a contact name, phone number and brief description of the problem. Telephone support will be initiated within the Priority/Response timeframes set forth below. If a problem cannot be resolved through telephone support within a reasonable amount of time, taking into account its Priority status, the problem will be escalated to Chiron's Product Support Center, for analysis by Blood Screening System specialists and if necessary, OEM support personnel. If a problem cannot be resolved following such
57
Priority
| | Response*
|
|---|
| Priority 1: | | Instrument not operating and alternate or back-up equipment not available; tests cannot be performed | | • Immediate telephone support
• Support Technician on site ASAP but no later than next morning
• Resolution within 24 hours (may be temporary workaround) |
Priority 2: |
|
Instrument operational but testing process (processing time) is affected |
|
• Telephone support within 2 hours
• Support Technician on site within 24 hours
• Resolution within 48 hours |
Priority 3: |
|
Instrument is operational; no impact on testing process |
|
• Telephone support next business day
• Work commenced within 24 hours
• Resolution within 1 week |
- *
- All timeframes are approximate; actual response times may vary due to flight availability, weather-related delays, nature of the problem, etc.
- (b)
- Chiron will maintain full records of all problem referrals and subsequent actions pursuant to FDA regulation.
- 2.6.
- Replacement Parts.
- (a)
- Chiron agrees to maintain at each Testing Center a suitable stock of replacement parts for use in Blood Screening System minor repair and maintenance at each Testing Center in the Territory. The Participating Member shall provide Chiron a secure facility at each Testing Center for storage of such replacement parts.
- (b)
- Replacement parts not available from existing on-site inventory will be delivered within the resolution timeframes in accordance with the Priority/Response time Schedule set forth above.
- (c)
- The Participating Member will document and inform Chiron if any replacement parts are used by Participating Member personnel and return to Chiron (at Chiron's expense) the part that was replaced. An inventory of replacement parts will be performed quarterly by Chiron personnel. The cost of replacement part(s) not in inventory that have not been reported used by the Participating Member will be charged to the Participating Member. Replacement parts to be placed into on site inventory will be agreed upon by Chiron and the Participating Member management. The initial inventory of replacement parts will be provided at Chiron's expense.
- 2.7.
- Replacement Instruments. If during the performance of any repair of a Blood Screening System Chiron determines in its sole discretion to substitute an entire instrument or instrument component in order to complete service in accordance with the Priority/Response Time Schedule, Chiron will remove and repair the failing instrument or instrument component. Chiron may substitute reconditioned, refurbished and/or serviceable used instrument or instrument component (tested to Chiron's quality assurance standards) for this purpose. Following completion of such repair, Chiron will replace the substituted instrument or instrument component with the repaired instrument or instrument component at the next scheduled PM service, or earlier if requested by the Participating Member.
58
- 3.
- Warranty; Limitation of Liability.
- 3.1
- Chiron warrants that the Services provided by Chiron, its agents, employees or contractors pursuant to this Agreement will be rendered in a competent workmanlike manner and in accordance with industry standards. Chiron warrants that any replacement part or substituted instrument or instrument component installed by Chiron during the performance of the Services shall function in accordance with all applicable Specifications and be free from defects. Chiron reserves the right to use reconditioned, refurbished and/or serviceable used parts (tested to Chiron's quality assurance standards) for service hereunder.
- 3.2
- The Services shall cover wear and tear and defects in design, material or workmanship, and shall include version updates to all Software for which Chiron has the right to license or sublicense, but shall exclude (i) replacement of operating supplies, necessaries, consumables or expendable parts; (ii) repairs required due to improper storage, accident, neglect, misuse, electrical stress, air conditioning, humidity control, transportation, and force majeure events, use of non-approved accessories, consumables or supplies, or causes other than intended normal use; (iii) service for Blood Screening Systems that have been tampered with, disassembled, altered, changed or modified, maintained by anyone other than Primary Operators, or repaired (or attempts have been so made) by anyone other than Chiron-authorized personnel; (iv) movement or rearrangement of the Blood Screening Systems after initial installation not authorized and supervised by Chiron personnel; and (v) service for any other equipment not manufactured or supplied by Chiron. In the case of (ii), (iii) and (iv) above, a Participating Member may request that Chiron provide service comparable to the Services described herein, and Chiron will make such services available at the then-current commercial rate for services of this type. If such service is provided, Chiron expressly disclaims any representation or warranty as to the performance of such service or the operability of the Blood Screening Systems on which such service is performed.
- 3.3
- Chiron disclaims all other warranties and further limits its liability in accordance with Sections 7.4 and 7.12 of the Association Agreement, respectively.
- 3.4
- If any applicable law implies a condition or warranty which cannot be excluded or modified in this Agreement (a "Requirement"), then such Requirement is deemed to be included in this Agreement.
59
| |
|
|---|
| CONFIDENTIAL | | REDACTED VERSION |
SCHEDULE E
PRICES
- 1.
- Fees
- 1.1
- Blood Screening Assay Fee. Each Participating Member agrees to pay to Chiron an amount equal to the number of Reportable Results achieved by such Participating Member each month, multiplied by the applicable Per Result Fee from the chart set forth in their applicable Member Supplement (the "Assay Pricing Chart"), as adjusted pursuant to Section 2 of thisSchedule E. The applicable Per Result Fee for each Participating Member is set on an annual basis using the Product Requirements Forecast provided by the Participating Member for that year.
POOLED TESTING
| |
| | Aggregate Participating Members Reportable Results
For Prior Calendar Year2
|
|---|
Three Year Term1
|
|---|
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
|
|---|
| | | | | Per Reportable Result
|
Individual Participating Member
Reportable Results
| | A
| | B
| | C
| | D
| | E
| | F
|
P1 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P2 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P3 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P4 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P5 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P6 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
- 1
- Discount applicable to Three Year Grid,[**]
- 2
- Includes all testing performed by Members; e.g., Pooled Testing and Single Unit Testing.
60
POOLED TESTING
| |
| | Aggregate Participating Members Reportable Results
For Prior Calendar Year4
|
|---|
Four Year Term3
|
|---|
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
|
|---|
| | | | | Per Reportable Result
|
Individual Participating Member
Reportable Results
| | A
| | B
| | C
| | D
| | E
| | F
|
P1 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P2 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P3 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P4 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P5 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P6 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
POOLED TESTING
| |
| | Aggregate Participating Members Reportable Results
For Prior Calendar Year4
|
|---|
Five Year Term3
|
|---|
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
|
|---|
| | | | | Per Reportable Result
|
Individual Participating Member
Reportable Results
| | A
| | B
| | C
| | D
| | E
| | F
|
P1 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P2 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P3 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P4 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P5 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
P6 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
- 3
- Discount applicable to the Four and Five Year Grid[**].
- 4
- Includes all testing performed by Members; e.g., Pooled Testing and Single Unit Testing.
61
Single Donor Testing
| |
| | Aggregate Participating Members Reportable Results
For Prior Calendar Year4
|
|---|
|
|---|
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
|
|---|
Individual Participating Member
Reportable Results
| | A
| | B
| | C
| | D
| | E
| | F
|
S0 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
S1 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
S2 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
S3 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
S4 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
S5 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
S6 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
S7 |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
On or prior to the 5th of each month, each Participating Member shall provide Chiron with a report showing the Reportable Results and Reagent Utilization Factor for the prior month for each of its Testing Centers. On or prior to the 15th of each month, Chiron shall prepare and deliver an invoice to each Participating Member based on such report, calculating the Blood Screening Assay Fee by multiplying the Reportable Results by the applicable Per Result Fee, determined in accordance with the then current Product Requirements Forecast. If a Participating Member's report is not timely received by Chiron, Chiron will prepare the invoice substituting one-twelfth of the then current Product Requirements Forecast and the parties shall "true-up" payments made to the actual Reportable Results achieved on a calendar quarter basis, rectifying any under- or over-payment on the next subsequent invoice, without application of interest. Each Participating Member shall make payment on invoices in accordance with Section 5.3 of the Association Agreement, except where modified by the applicable Member Supplement.
- 1.2
- Blood Screening Systems Fee. Each Participating Member agrees to pay to Chiron the Blood Screening Systems Fee set forth in their Member Supplement. Such amount will be due and payable in a lump sum on the effective date of the applicable Member Supplement. If a Participating Member elects to lease its Blood Screening Systems, payments will be invoiced by Chiron on a monthly basis, together with the Blood Screening Assay Fee described above. Each Participating Member shall make payment on invoices in accordance with Section 5.3 of the Association Agreement, except where modified by the applicable Member Supplement. The applicable purchase price for Blood Screening Systems will be set on a Member-by-Member basis based on the specific requirements of each Participating Member. The pricing information set forth below is provided for informational purposes only.
Blood Screening System Components
| | Unit Price
| | Monthly Lease
Payment5
|
|---|
| Pooling TECAN6 | | [**] | | $ | |
Assay TECAN7 |
|
[**] |
|
$ |
|
Procleix™ System8 |
|
[**] |
|
$ |
|
- 5
- Based on 36 month lease. Subject to credit approval.
62
- 6
- Includes CPT-16 Pooling Software, ver. 2.0.0.2
- 7
- Includes Procleix™ Assay Software, ver. 2.0.0.76 and Procleix™ Assay Software LHP, ver. 2.1
- 8
- Includes Procleix™ System Software, ver. 3.0.3.4, Procleix™ Protocol, ver. 2.1.0.0, and Procleix™ Worklist Editor, ver. 3.0.3.2
- 1.3.
- Services Fee. Each Participating Member agrees to pay to Chiron the Services Fee set forth in their Member Supplement. Such amount will be due and payable annually in a lump sum on each anniversary of the effective date of the applicable Member Supplement. If a Participating Member elects to lease its Blood Screening Systems, payments will be invoiced by Chiron on a monthly basis, together with the Blood Screening Assay Fee and Blood Screening Systems Fee described above. Each Participating Member shall make payment on invoices in accordance with Section 5.3 of the Association Agreement, except where modified by the applicable Member Supplement. The applicable purchase price for Services will be set on a Member-by-Member basis based on the specific requirements of each Participating Member. The pricing information set forth below is provided for informational purposes only.
Blood Screening System Components
| | Price/Year/Unit
| | Monthly
Installment
Payment
|
|---|
| Pooling TECAN9 | | [**] | | $ | |
Assay TECAN10 |
|
[**] |
|
$ |
|
Procleix™ System11 |
|
[**] |
|
$ |
|
- 9
- Includes all version updates to CPT-16 Pooling Software, ver. 2.0.0.2
- 10
- Includes all version updates to Procleix™ Assay Software, ver. 2.0.0.76 and Procleix™ Assay Software LHP, ver. 2.1
- 11
- Includes all version updates to Procleix™ System Software, ver. 3.0.3.4, Procleix™ Protocol, ver. 2.1.0.0, and Procleix™ Worklist Editor, ver. 3.0.3.2
- 1.4.
- NAT Tracker Fee. Each Participating Member electing to purchase the NAT Tracker software solution agrees to pay to Chiron the NAT Tracker Fee set forth in their Member Supplement. Such amount will be due and payable annually in a lump sum on the effective date of the applicable Member Supplement and on each anniversary thereafter. Participating Members electing to purchase the NAT Tracker software solution will enter into a separate software license agreement with Chiron.
- 2.
- Adjustments to Fees.
- 2.1
- Reagent Utilization Factor.
- (a)
- Chiron and each Participating Member agree to monitor the Reagent Utilization Factor at each Testing Center during the term of this Agreement. If the Reagent Utilization Factor for any Testing Center set forth on the report provided by a Participating Member pursuant to Section 1.1 of thisSchedule Eexceeds that Participating Member's target Reagent Utilization Factor, set forth in the applicable Member Supplement (the "Target RUF"),[**], Chiron and each Participating Member shall use their best endeavors to identify and correct the cause of the excess.
- (b)
- The Reagent Utilization Factor shall be calculated for each Participating Member promptly following each successive 52 week period commencing on the Effective Date of the applicable Member Supplement (each, an "Annual RUF Measuring Period"),
63
aggregating all Testing Centers. If the Reagent Utilization Factor for a Participating Member for any Annual RUF Measuring Period exceeds the applicable Target RUF, Chiron shall invoice the Participating Member on the next subsequent monthly invoice, and the Participating Member agrees to pay, the additional fee set forth in their Member Supplement.
- 2.2.
- [**].
- 3.
- [**].
- 4.
- Effect of Change in Nature of Testing. If a Participating Member elects to move from Pooled Testing to Single Unit Testing, or from Single Unit Testing to Pooled Testing, pursuant to Section 11 ofSchedule B-1, (i) the price per donation shall be adjusted pursuant to the then current Assay Pricing Chart and (ii) the Target RUF will be recalculated.
- 5.
- Additional Supplies. The following additional supplies are available from Chiron at the prices set forth below. Chiron reserves the right to increase or decrease these prices at any time, without notice to each Participating Member:
[**] [**]
[**] [**]
[**] [**]
64
EXHIBIT 10.317
| |
|
|---|
| CONFIDENTIAL | | REDACTED VERSION |
[**] CERTAIN INFORMATION IN THIS EXHIBIT HAS BEEN
OMITTED AND FILED SEPARATELY WITH THE COMMISSION.
CONFIDENTIAL TREATMENT HAS BEEN REQUESTED WITH
RESPECT TO THE OMITTED PORTIONS.
MEMBER SUPPLEMENT
This Member Supplement (this "Member Supplement"), dated as of (the "Effective Date"), is by and between Chiron Corporation, a Delaware corporation ("Chiron"), and ("ABC Member").
RECITALS
- A.
- Chiron and America's Blood Centers, an association of non-profit community-based blood centers ("ABC"), have entered into that certain Association Agreement dated as of May 1, 2002 (the "Association Agreement");
- B.
- Pursuant to and in accordance with the terms of the Association Agreement, Chiron has agreed to sell Blood Screening Assays and sell or arrange for the lease of Blood Screening Systems to ABC members, for use to conduct nucleic acid amplification tests to detect the presence of certain viruses in blood donation samples;
- C.
- , an ABC member, wishes to acquire Blood Screening Assays and Blood Screening Systems to conduct nucleic acid amplification tests to detect the presence of certain viruses in blood donation samples, in accordance with the terms of the Association Agreement, subject to the following additional terms and conditions.
AGREEMENT
NOW, THEREFORE, in consideration of the foregoing and the mutual covenants and agreements herein contained, and for other good and valuable consideration, the receipt and sufficiency of which is hereby acknowledged, the parties hereto agree as follows:
- 1.
- Definitions. All capitalized terms used but not otherwise defined in this Member Supplement shall have the meanings set forth in the Association Agreement.
- 2.
- Association Agreement. The terms and conditions of the Association Agreement, attached asExhibit 1to this Member Supplement, are adopted by the parties hereto and incorporated by reference. For the purpose of the Association Agreement and this Member Supplement, ABC MEMBER shall be deemed to be a Participating Member. In the event of any conflict between the terms and conditions of this Member Supplement and the terms and conditions of the Association Agreement, the terms and conditions of the Association Agreement govern.
- 3.
- Blood Screening Assays. In accordance withSchedule Bof the Association Agreement, Chiron shall supply ABC MEMBER with its requirements of Blood Screening Assays to permit ABC MEMBER to conduct Pooled Testing or Single Unit Testing of blood donations in the Territory at the Testing Centers listed onExhibit 2to this Member Supplement. ABC MEMBER's initial Product Requirement Forecast is attached asExhibit 3to this Member Supplement.
1
- 4.
- Blood Screening Systems. In accordance withSchedule Cof the Association Agreement, Chiron shall sell ABC MEMBER the Blood Screening Systems described onExhibit 4to this Member Supplement.
- 5.
- Fees. In accordance withSchedule Eto the Association Agreement, ABC MEMBER agrees to the following fees:
- 5.1
- Blood Screening Assay Fee. The applicable Per Result Fee is set on an annual basis from the Assay Pricing Chart below using the Product Requirements Forecast provided for that year. For the first year of the term of this Member Supplement, the applicable Per Result Fee for ABC MEMBER is .
TESTING
| |
| | Aggregate Participating Members Reportable Results
For Prior Calendar Year2
|
|---|
-Year Term1
|
|---|
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
| | [**]
|
|---|
| | | | | Per Reportable Result
|
Individual Participating Member
Reportable Results
| | A
| | B
| | C
| | D
| | E
| | F
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
|
[**] |
- 1
- Discount of[**].
- 2
- Includes all testing performed by Members; e.g., Pooled Testing and Single Unit Testing.
- 5.2
- Blood Screening Systems Fee. The Blood Screening Systems Fee for the purchase of Blood Screening Systems set forth onExhibit 4to this Member Supplement is $ , payable in a lump sum in accordance with Section 1.2 ofSchedule E. Additional components will be available to the Participating Member at the then current list price for such components.
- 5.3
- Services Fee. The Services Fee for the Blood Screening Systems set forth onExhibit 4to this Member Supplement is $ , payable annually in accordance with Section 1.3 ofSchedule E. The purchase of additional components will result in an increase in the Services Fee from and after the installation of the additional component(s). The Services Fee shall not be due or payable for Blood Screening Systems purchased new until the expiration of the applicable manufacturers' warranties.
- 5.4
- NAT Tracker Fee. The applicable license fee for the NAT Tracker software solution is based on the number of Testing Centers operating the NAT Tracker software, as set forth below, and payable annually in a lump sum in accordance with Section 1.4 ofSchedule E. Additional Testing Centers may be licensed to use the NAT Tracker software at the then current license fee.
Software
| | Price/Year
| | Number of
Testing Centers
| | Extended License
Fee
|
|---|
| NAT Tracker3 | | $ | [**] | | | | $ | |
- 3
- Includes all version updates to NAT Tracker Software, ver. 1.0E.
- 6.
- Reagent Utilization Factor. In accordance withSchedule Eto the Association Agreement, for each[**] increment by which the actual Reagent Utilization Factor during each Annual RUF
2
Measuring Period exceeds ABC MEMBER's Target RUF of , ABC MEMBER agrees to pay an additional fee[**]; provided that no additional fee shall be payable hereunder to the extent that the cause of the Reagent Utilization Factor exceeding such amount is attributable to a breach by Chiron of the limited warranty set forth in Section 12 ofSchedule B-1.
- 7.
- Training. Chiron agrees to provide the following training program for ABC MEMBER personnel.
- 7.1
- Initial Training. Chiron shall provide, at Chiron's sole expense, initial training to technical personnel from ABC MEMBER. Initial training includes assay training on the Blood Screening System and Pooled Testing training, if applicable. Training will include theory, practical training and operation/maintenance on all required equipment. Upon successful completion of training, individuals will be certified as Primary Operators. Chiron will provide an additional week of "train-the-trainer" courses for up to Primary Operators from ABC MEMBER. Upon successful completion of these courses, these Primary Operators will be issued trainer certifications. Primary Operators with trainer certifications may train Secondary Operators in accordance with Section 7.2 of this Member Supplement. Chiron also agrees to provide, at Chiron's expense, Bio Medical Instrument Technician ("BMIT") training to Primary Operators from ABC MEMBER. BMIT training includes training on the Tecan Genesis instrumentation and includes theory, diagnostic tests, troubleshooting and basic instrument repair. Upon successful completion of the course, trainees will be certified as Bio Medical Instrument Technicians. BMIT-certified Primary Operators must be re-certified annually by Chiron.
- 7.2
- On Site Training. Training of Secondary Operators will be performed on site at the Testing Centers, by Primary Operators or by Chiron support personnel. Each Secondary Operator will be required to pass proficiency certification prior to reporting results. Without limiting Section 7.1 of this Member Supplement, if the Participating Member later requires training of additional personnel due to employee turnover, such additional training will be the responsibility of the Participating Member, using Primary Operators previously trained by Chiron pursuant to this Section 7 of this Member Supplement. Chiron undertakes to co-operate with the Participating Member and if requested by the Participating Member, provide technical personnel to assist in training such additional personnel.
- 7.3
- Proficiency Panels. Chiron will provide, at no cost to the Participating Member, one proficiency panel for initial proficiency certification of each of the operators trained pursuant to Section 7.1 of this Member Supplement. If, following initial certification, the Participating Member wishes to re-certify an operator, or the Participating Member wishes to obtain proficiency panels for ongoing quality control or future training purposes, the Participating Member shall purchase such proficiency panels as may be necessary for such purposes. The Participating Member agrees to provide forecasts of its needs for proficiency panels in the same manner as is set forth in Section 7 ofSchedule B-1to this Agreement.
- 7.4
- Manuals and Documentation. The training provided under Sections 7.1 and 7.2 of this Member Supplement shall include copies of training materials, operator manuals and documentation, including maintenance and troubleshooting documentation.
- 7.5
- Changes to Blood Screening Assays, Blood Screening Systems, Software. If, in accordance with Section 6.7 of the Association Agreement, mandatory changes are made by the manufacturer to the Blood Screening Assays, Blood Screening Systems or Software during the term of this Agreement, and as a result further staff training is required, the training must be carried out by Chiron at its cost at the Testing Centers, unless the parties otherwise agree. Chiron must provide proficiency panels at its cost for any additional proficiency certification required as a result of the changes referred to in this Section 7.5 of this Member Supplement. For clarity of understanding, unless otherwise agreed the Participating Member will bear any additional
3
- 8.
- Insurance.
- 8.1
- ABC MEMBER shall obtain and maintain at its sole cost, a policy or policies of insurance with the following coverages, which shall be in full force and effect for the term of this Agreement and thereafter through two years following the termination of the Agreement: a) a Commercial General Liability policy including Products and Completed Operations Liability in an amount of not less than $ combined single limit for each occurrence; b) Workers' Compensation coverage with statutory limits for each jurisdiction where the work required of such party under this Agreement is performed and an employers' liability policy with at least the following limits, $ per accident, $ per disease (policy limit), and $ disease (each employee); and c) Professional Liability (Errors & Omissions) Insurance in an amount not less than $ each claim specifically insuring claims arising from obligations of ABC MEMBER.
- 8.2
- ABC MEMBER shall provide Chiron with certificates of insurance evidencing the coverage required herein upon execution of this Agreement, and renewal certificates on request from Chiron. ABC MEMBER must notify Chiron within a reasonable time if there is a material change to any of the insurance policies referred to in this Section. Such certificates shall provide for thirty (30) days prior written notice to the certificate holder in the event of non-renewal of the policies, cancellation or material change in the coverage provided.
- 9.
- Term. This Member Supplement shall become effective on the Effective Date of this Member Supplement, and shall terminate on the earlier to occur of (i) ( ) months from the Effective Date of this Member Supplement, or (ii) the termination or expiration of the Association Agreement. ABC MEMBER shall have the right to extend the term of this Member Supplement, subject to the continued effectiveness of the Association Agreement, by two extension periods, each one year in length. ABC MEMBER shall elect an extension by delivering written notice to Chiron not less than ninety (90) days prior to the expiration of the initial term or extension.
- 10.
- Notices. Notices, requests, waivers and other communications made pursuant to this Member Supplement or the Association Agreement shall be made in accordance with Section 12.6 of the Association Agreement and, if to ABC MEMBER, shall be addressed to ABC MEMBER as set forth below:
- 11.
- [**].
- 12.
- Survivability. Sections 2 and 9 of this Member Supplement shall survive any expiration or termination of this Member Supplement.
[SIGNATURE PAGE FOLLOWS]
4
IN WITNESS WHEREOF, the parties hereto, acting through their duly authorized officers, have executed this Agreement as of the date first set forth above.
| |
| |
|
|---|
| | | CHIRON CORPORATION |
|
|
By: |
|
|
| | | ABC MEMBER |
|
|
By: |
|
|
5
EXHIBITS
- 1
- Association Agreement
- 2
- Testing Centers
- 3
- Products Requirements Forecast
- 4
- Blood Screening Systems
6
QuickLinks
Association Agreement Regarding the Sale and Servicing of Blood Screening ProductsSCHEDULE A DefinitionsSCHEDULE B-1 TERMS AND CONDITIONS FOR PURCHASE OF PROCLEIX™ HIV-1/HCV BLOOD SCREENING ASSAYATTACHMENT B-1 Package InsertSCHEDULE C TERMS AND CONDITIONS FOR USE OF BLOOD SCREENING SYSTEMSSCHEDULE D BLOOD SCREENING SYSTEMS AND SOFTWARE SERVICE & SUPPORTSCHEDULE E PRICESMEMBER SUPPLEMENTRECITALSAGREEMENTEXHIBITS