UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2006
Commission File No. 0-16614
PONIARD PHARMACEUTICALS, INC.
(Exact name of Registrant as specified in its charter)
Washington | | 91-1261311 |
(State or other jurisdiction of | | (IRS Employer Identification No.) |
incorporation or organization) | | |
7000 Shoreline Court, Suite 270, South San Francisco, CA 94080
(Address of principal executive offices)
Registrant’s telephone number, including area code: (650) 583-3774
Securities registered pursuant to Section 12(b) of the Act:
None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $.02 par value
$2.4375 Convertible Exchangeable Preferred Stock, Series 1, $.02 par value
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer o | | Accelerated filer x | | Non-accelerated filer o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
The aggregate market value of the voting and non-voting common equity held by nonaffiliates of the registrant was approximately $77.2 million as of June 30, 2006, based on a per share closing price of $0.97 on the Nasdaq Capital Market on that date.
As of March 8, 2007, 22,808,233 shares of the Registrant’s Common Stock, $.02 par value per share, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s Proxy Statement for the Registrant’s 2007 Annual Meeting of Shareholders are incorporated by reference in Part III of this Form 10-K.
PART I
IMPORTANT INFORMATION REGARDING FORWARD-LOOKING STATEMENTS
This Form 10-K contains forward-looking statements within the meaning of the Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, and in reliance upon the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. Forward-looking statements are those that predict or describe future events or trends and that do not relate solely to historical matters. In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “could,” “should,” “expect,” “plan,” “intend,” “anticipate,” “believe,” “estimate,” “predict,” “potential,” “propose,” “continue,” “assume” or other similar expressions, or the negatives of those expressions. These statements reflect our current views with respect to future events and are based on assumptions and are subject to risks and uncertainties that are difficult to predict. We have identified some of the factors that could cause future events to differ from our current expectations under the headings “Risk Factors” in Item 1A below and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Item 7 below. Given these risks and uncertainties, you should not place undue reliance on our forward-looking statements, which speak only as of the date of this report.
You should read this Form 10-K and the documents that we incorporate by reference completely and with the understanding that our actual results, performance and achievements may be materially different from any future results, performance or achievements expressed or implied by our forward-looking statements. We undertake no obligation to update publicly any forward-looking statements to reflect new information, events or circumstances after the date of this report, or to reflect the occurrence of unanticipated events.
Unless otherwise indicated, all common stock-related amounts in this report have been adjusted to reflect our one-for-six reverse stock split effective September 22, 2006.
Item 1. BUSINESS
The Company
Poniard is a biotechnology company focused on the discovery, development and commercialization of cancer therapy products. Our lead product candidate is picoplatin, a next generation platinum-based cancer therapy with an improved safety profile. An intravenous chemotherapeutic agent, picoplatin is designed to overcome platinum resistance in the treatment of solid tumors. In August 2006, we completed patient enrollment in a Phase II clinical study of picoplatin in small cell lung cancer. Based on positive interim median overall survival data from that ongoing Phase II study, we plan to initiate a pivotal Phase III SPEAR (Study of Picoplatin Efficacy After Relapse) trial of picoplatin in small cell lung cancer in the first half of 2007. We also are conducting separate Phase I/II studies of picoplatin in patients with metastatic colorectal cancer and hormone-refractory prostate cancer. Both of these Phase I/II trials are continuing to enroll patients. Additionally, we plan to initiate a Phase I study of an oral formulation of picoplatin during 2007.
Until May 2005, our lead research and development program had been skeletal targeted radiotherapy, or STR, a bone-targeting radiotherapeutic. In May 2005, we announced the immediate implementation of a strategic restructuring program to refocus our limited resources on the development of picoplatin. The restructuring plan, which was completed in June 2005, included the discontinuation of our STR development program, including halting patient enrollment in our Phase III trial of STR in multiple myeloma, ceasing operations at our Denton, Texas facility, where STR was manufactured, and reducing our workforce by approximately 50%. We recorded restructuring charges against operations totaling $1.7 million during the year ended December 31, 2005. We evaluated our STR assets in light of the restructuring and determined that a likely impairment existed on those assets. We recognized an
2
impairment loss of $3.3 million in June 2005 and an additional impairment loss of $0.4 million in December 2006. Future adjustments to the charge may be taken as assets involved are sold or otherwise disposed of. We are actively seeking a buyer for the Denton facility and our STR related assets.
We have financed our operations primarily through the sale of equity securities, technology licensing, collaborative agreements and debt instruments. We completed a $65.0 million equity financing in April 2006. As a result of that financing, entities affiliated with MPM Capital Management, or MPM, acquired beneficial ownership of an aggregate of 31.5% of our common stock outstanding immediately following the financing. Entities affiliated with Bay City Capital Management IV LLC, or BCC, acquired beneficial ownership of 19.5% of our common stock immediately outstanding following the financing. Nicholas J. Simon, a representative of MPM, and Fred B. Craves and Carl S. Goldfischer, managing directors of BCC, serve on our board of directors. We invest excess cash in investment securities that will be used to fund future operating costs. Cash used to fund operating activities for the twelve months ended December 31, 2006 totaled $17.3 million. Revenues and other income sources for 2006 were not sufficient to cover operating expenses. Cash, cash equivalents and investment securities, net of restricted cash of $0.1 million, totaled $53.7 million at December 31, 2006. We believe that our current cash, cash equivalent and investment securities balances will provide adequate resources to fund operations at least until the end of the first quarter of 2008.
Since our inception in 1984, we have dedicated substantially all of our resources to research and development. We have not generated any significant revenue from product sales to date and have operated at a loss in each year of our existence. We had a net loss of $23.3 million for the year ended December 31, 2006, a net loss of $21.0 million for the year ended December 31, 2005, and a net loss of $19.4 million for the year ended December 31, 2004. We do not anticipate that our picoplatin product candidate, or any other proposed products, will be commercially available for several years, if at all. We expect to incur additional operating losses in the future as we expand our clinical trials, increase our research and development activities and seek to commercialize picoplatin or other proposed products. Clinical studies are inherently uncertain, and our ongoing and planned trials of picoplatin or any future product candidates may not confirm the results achieved in earlier clinical and preclinical studies. If picoplatin or any future proposed products are not shown to be safe and effective, we will not receive the required regulatory approvals for commercial sale of such products. To the extent that we are successful in obtaining approvals for the commercial sale of picoplatin or any other product, we will need to secure one or more corporate partners for the manufacture, marketing and/or sale of such product. We may not be able to enter into such partnering arrangements in a timely manner or on terms acceptable to us.
Our Picoplatin Development Program
Overview of Cancer and its Treatment
Cancer is a disease characterized by the uncontrolled growth and spread of abnormal cells. Cancer cells often originate from one tissue site and invade, spread and damage other tissue beds and organs, leading to death. The National Cancer Institutes estimates that approximately 10.5 million Americans with a history of cancer in the United States were alive in January 2003 (American Cancer Society: Cancer Facts & Figures 2007).
In recent years, the diagnosis and treatment of human cancers have greatly improved. However, there is still a substantial need to improve the early diagnosis of cancer, the staging of cancer and the treatment of cancer and its metastatic spread. It is anticipated that the use of chemotherapeutics and targeted anti-cancer agents will be used both as single-agents and in combination to provide benefit to cancer patients. Often patients are treated with multiple agents in combination and with different sequence depending on the particular cancer type and severity of disease. The oncologist will often assess clinical benefit for a particular therapeutic combination by determining the impact of treatment on tumor size or spread
3
compared to tolerability features. In this regard, chemotherapeutics have continued to have significant impact on cancer treatment, especially when combined with agents that show different anti-cancer properties and different tolerability features. We believe that new treatment combinations that incorporate recently approved targeted agents with chemotherapeutics exhibiting improved safety features will be supported by physicians and their patients.
There is considerable need for new cancer treatments, as well as treatments that provide an improvement to existing therapies. In recent years, many new classes of agents that provide modest increases in patient survival have been approved for use. We anticipate that the use of multiple agents, either in combination or in sequence, will continue to provide benefit to cancer patients who have been diagnosed with disease. In addition, we believe that individualized therapies will become more prominent as tumor diagnosis and agents with different mechanisms of anti-cancer effect are approved and become available to the practicing oncologist. We also expect that early diagnosis and cancer prevention will provide for interventions that will allow patients to live longer and have a better quality of life. Current treatments for cancer include surgery, external-beam radiation and chemotherapy, including targeted pharmaceuticals, hormone therapy, cytokines, interferons, antibodies, and antibody-based radiotherapeutics. There has been substantial recent success in the combined use of both traditional chemotherapeutics, which generally destroy cells, and targeted agents, which are generally combined with more conventional chemotherapeutics for maximum effect. Occasionally, chemotherapeutics or targeted agents are used as stand-alone agents in the treatment of human cancers.
Picoplatin and Platinum-Based Chemotherapeutics
In April 2004, we acquired the rights to develop, manufacture and commercialize picoplatin, a next-generation platinum-based cancer therapeutic. In September 2006, we renegotiated the financial terms of our April 2004 license agreement and obtained exclusive worldwide rights to picoplatin. Over the past two decades, platinum-based drugs have become a critical part of modern chemotherapy treatment. Platinum-based agents such as cisplatin, carboplatin and oxaliplatin are currently used to treat a variety of tumors, including testicular, ovarian, colorectal and lung cancers. In this regard, platinum-based chemotherapeutics are administered primarily in combination with other agents, including with recently approved targeted cancer agents. The mechanism that underlies the use of platinum-based agents relies upon the targeting of tumor DNA where the platinum compound binds. Cells that undergo active cell division are prevented from completing the cell cycle by the presence of the platinum drug that is chemically bound to the DNA. The inability to proceed through normal cell division ultimately causes cell death. In some cases, treatment of cancer patients with platinum compounds leads to reduction in tumor mass due to a higher rate of tumor cell death compared with tumor cell replication.
Current platinum-based chemotherapeutics have specific limitations, including chemo-resistance and safety side effects. All platinum-based agents exhibit toxicity to the blood forming cells in the bone marrow (myelosuppression) as a dose-limiting side effect. The degree and characteristics of myelosuppression vary by platinum compound, dose and regimen. In addition, some current platinum agents show different degrees of additional safety side effects that include kidney toxicity, hearing loss, nausea, vomiting and peripheral nerve damage. As in the case of myelosuppression, these side effects vary with dose, agent, combination therapy and regimen.
For most cancers that are treated with platinum-containing regimes, patients who initially respond to platinum-containing chemotherapy but subsequently progress six months or more after chemotherapy are described as having “platinum-sensitive” disease. Patients with who initially respond to platinum-containing chemotherapy and then relapse and progress within six months after completing chemotherapy are said to have “platinum-resistant” disease. Patients with who fail to have a response or whose disease progresses during platinum-containing chemotherapy are said to have “platinum-refractory” cancer. As described below, in the case of small cell lung cancer, the distinction between platinum-sensitive and
4
platinum-resistant disease is generally drawn based on whether progression occurs before or after 90 days of completing first-line platinum-containing chemotherapy. We believe that patients would benefit from a platinum-based agent that can be used initially to prevent or delay the development of the platinum-refractory or -resistant disease and that is effective in the treatment of disease that becomes refractory or resistant to currently used platinum-based therapies.
New platinum-based chemotherapeutics that overcome both chemo-resistance and safety limitations are needed. In this regard, picoplatin has shown efficacy in preclincial and clinical studies of platinum-sensitive, -resistant and -refractory disease. Clinical evidence of activity has been observed for picoplatin in lung, ovarian and prostate cancers. In addition, evaluation of several hundred cancer patients has suggested that picoplatin treatment may result in less severe and less frequent side effects than observed with some currently marketed platinum-based agents.
Our Picoplatin Clinical Studies
We currently are evaluating picoplatin in an ongoing Phase II clinical trial in small cell lung cancer and in separate Phase I/II clinical trials in advanced colorectal and hormone-refractory prostate cancers. We plan to initiate a pivotal Phase III SPEAR (Study of Picoplatin Efficacy After Relapse) trial of picoplatin for the treatment of small cell lung cancer in the first half of 2007. In addition, we plan to initiate a Phase I clinical trial of an oral formulation of picoplatin during 2007. These programs are described below and in the section of this report entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Major Research and Development Programs.” It is important to keep in mind that clinical studies are inherently uncertain, and later trials may not confirm the results achieved in earlier clinical and preclinical studies and may not be supported by the results obtained in subsequent trials. You should refer to the section of this report entitled “Risk Factors” for a discussion of some of the factors that could materially affect our picoplatin clinical development program.
Small Cell Lung Cancer
Phase II and Planned Phase III Clinical Trials. In October 2004, we filed an investigational new drug application, or IND, with the U.S. Food and Drug Administration, or FDA, to conduct a Phase II clinical trial of intravenous picoplatin versus intravenous topotecan in patients with small cell lung cancer. Intravenous topotecan is an anti-tumor drug currently approved by the FDA as a treatment for patients who initially responded to a platinum-containing chemotherapy regime but subsequently progressed at least 60 or 90 days after completion of therapy (platinum-sensitive disease). Our Phase II trial was initiated in June 2005 in the United States and Canada, and the first patient was treated in July 2005. The objective for patient enrollment was approximately 75 patients with platinum-resistant or -refractory small cell lung cancer, defined as subjects who either (1) initially responded to first-line platinum-containing chemotherapy and then relapsed or progressed within 90 days after completion of first-line chemotherapy (resistant disease); or (2) failed to respond to or progressed during first-line platinum-containing chemotherapy (refractory disease). The clinical endpoints of the study include safety, objective tumor response rate (tumor shrinkage), time to tumor progression and overall survival.
We amended our Phase II clinical trial protocol in January 2006 from a two-arm study of picoplatin versus topotecan to a single-arm study of picoplatin. We discontinued the topotecan arm of the study because patients and investigators often were unwilling to accept this study arm. The rationale for the amendment was that the dose and schedule of topotecan approved by the FDA for use in patients with platinum-sensitive small cell lung cancer have minimal, if any, efficacy in patients with platinum-resistant or -refractory small cell lung cancer and unacceptable toxicity, thus presenting a situation in which an ineffective but toxic treatment regimen was to be used as one arm of the randomized Phase II trial. We also amended the protocol because we no longer intended to use topotecan as the comparator treatment
5
for our planned Phase III trial and wanted data in more patients treated with picoplatin to help us make a decision on whether to embark upon a large Phase III trial.
We discussed the design of a Phase III trial with the FDA in April 2006 and modified our ongoing Phase II trial to support our plans for a subsequent Phase III trial. We expanded our small cell lung cancer study to include additional clinical sites in Eastern Europe, where the greater availability of patients enabled us to more rapidly increase patient enrollment. In May 2006, we amended our Phase II protocol to provide for enrollment of a subset of patients with platinum-sensitive disease who relapsed within 91-180 days of completing first-line platinum-containing chemotherapy. We completed enrollment of our Phase II small cell lung cancer trial in August 2006. In November 2006, we announced positive interim overall survival results from the study, indicating a median overall survival of 26.7 weeks in 72 evaluable patients.
We plan to initiate a pivotal Phase III SPEAR (Study of Picoplatin Efficacy After Relapse) trial in the first half of 2007. The Phase III SPEAR trial will be undertaken pursuant to a Special Protocol Assessment, or SPA, with the FDA. The SPA is a written agreement between us and the FDA on the objectives, design and endpoints to be used as a basis of filing for accelerated approval of picoplatin and the data analysis plan necessary to support full regulatory approval of picoplatin. The Phase III trial will be an international, multi-center, open-label, controlled study to compare the efficacy and safety of picoplatin plus best supportive care with best supportive care alone as a second-line therapy. The study is expected to enroll approximately 400 patients with small cell lung cancer whose disease is refractory (non-responsive) to first-line platinum-containing (cisplatin or carboplatin) chemotherapy or whose disease responded initially to first-line platinum-containing therapy but then progressed within six months after treatment was completed. Patients will be randomized on a 2:1 ratio to receive picoplatin plus best supportive care or best supportive care alone. Best supportive care will include all medical, radiation and surgical interventions that small cell lung cancer patients should receive to palliate the symptoms and treat the complications caused by small cell lung cancer, but excludes treatment with systemic therapies intended to kill cancer cells. The primary endpoint of the planned study will be improved overall survival as measured in time from randomization to death. Secondary endpoints will include overall response rates, disease control and progression-free survival. We currently estimate that the study will take approximately 20 months to complete; however, the actual timing for completion of the study will depend on the rate of patient enrollment, survival times of all patients in the trial, as well as other factors such as patient performance status, extent of disease and the risks and uncertainties described in this report.
The FDA has designated picoplatin as an orphan drug for the treatment of small cell lung cancer under the provisions of the Orphan Drug Act, as amended. To qualify for orphan drug status, a proposed drug must be intended for use in the treatment of a condition that affects fewer than 200,000 people in the United States. Orphan drug status entitles us to exclusive marketing rights for picoplatin in the United States for seven years following market approval, if any, and qualifies us for research grants to support clinical studies, tax credits for certain research expenses and an exemption from certain application user fees. As discussed below in the section entitled “Government Regulation and Product Testing,” the manufacture and marketing of picoplatin are subject to regulation for safety, efficacy and quality by the FDA and comparable authorities in foreign countries.
Small Cell Lung Cancer and its Treatment. Approximately 15% of all lung cancer diagnoses in the United States are small cell lung cancer. In about 98% of patients, their small cell lung cancer is thought to be caused by cigarette smoking. According to a 2004 report on cancer statistics, there were approximately 34,700 new small cell lung cancer patients in the United States annually. At the time of small cell lung cancer diagnosis, approximately two-thirds of patients have metastases outside the chest and approximately one-third have limited disease confined to the chest. Small cell lung cancer consists of two stages: (1) limited, which is defined as cancer confined to the one side of the chest that can be treated with a single area of radiation therapy and (2) extensive, which is defined as disease involving both sides of the chest and/or obvious spread of the cancer beyond the chest. Surgery is only used for the very few patients with
6
limited-stage disease. Radiation therapy plus chemotherapy is the standard of care for limited-stage small cell lung cancer. Treatment with radiation therapy plus chemotherapy can cure a small percentage of limited-stage patients. Chemotherapy is the standard of care for extensive disease. Although small cell lung cancer is highly sensitive to chemotherapy and radiation therapy, most patients develop recurrent cancer.
Many chemotherapeutic agents are used to treat small cell lung cancer patients. Initial (first-line) chemotherapy regimens include cisplatin, carboplatin, etoposide, irinotecan (CPT-11), ifosfamide, cyclophosphamide, vincristine and doxorubicin. The most commonly used first-line chemotherapy regimen is combination carboplatin or cisplatin plus etoposide. Eighty percent to 100% of patients with limited disease, and 60% to 80% of patients with extensive disease, have a significant response to radiation plus chemotherapy or combination chemotherapy, respectively. The median duration of response is approximately 6 to 8 months. Median survival from the time of diagnosis for limited-stage and extensive-stage disease is approximately 14 to 20 months and 8 to 13 months, respectively.
Agents used for second-line chemotherapy include ifosfamide, paclitaxel, docetaxel, gemcitabine, topotecan, irinotecan, CAV (cyclophosphamide, doxorubicin and vincristine) and vinorelbine. Only intravenous topotecan is approved by the FDA for use as a second-line chemotherapeutic, and it is only approved for those patients with platinum-sensitive small cell lung cancer. When small cell lung cancer recurs following first-line chemotherapy, the median survival for patients treated with second-line chemotherapy is approximately 4 to 5 months.
Based on clinical and preclinical data to date, we believe that second-line picoplatin has potential activity in small cell lung cancer patients who have failed first-line platinum-containing therapy. A Phase II study was conducted by a prior licensee during 2001 and 2002 to assess the activity and tolerability of picoplatin when given intravenously as a second-line therapy to patients with small cell lung cancer. Two of 13 patients (15.4%) with platinum-resistant small cell lung cancer achieved a partial response (a decrease in the size of the tumor or in the extent of cancer in the body) with picoplatin treatment, and two additional patients (15.4%) achieved stable disease (no increase or decrease in extent or severity of the cancer). Overall, 4 of 13 patients (30.8%) with platinum-resistant small cell lung cancer achieved a partial response or stable disease with picoplatin treatment. The median survival of all 13 treated patients was approximately 6.3 months, significantly longer than that which would be expected with topotecan, the only approved chemotherapy in this population.
Metastatic Colorectal Cancer
Phase I/II Clinical Trial. In May 2006, we treated our first patient in an approximately 30-patient Phase I/II study of picoplatin in the first-line treatment of patients with metastatic colorectal cancer. The trial is being conducted in Eastern Europe and is continuing to enroll patients. The Phase I component of the trial is designed to evaluate increasing doses of picoplatin in combination with the chemotherapy agents 5-fluorouracil and leucovorin to establish an appropriate dose of picoplatin for further testing in the Phase II efficacy component of the trial. Endpoints of the study will include safety, objective tumor response rate (tumor shrinkage), time to tumor progression, progression-free survival and overall survival.
Colorectal Cancer and its Treatment. According to the American Cancer Society, colon cancer is the third most common cancer among American men and women and the second and third leading cause of cancer death in the United States for men and women, respectively. An estimated 148,000 new cases of colon cancer were diagnosed in 2006, with an estimated 55,000 deaths in 2006 (American Cancer Society, Cancer Facts and Figures 2006). Fluorouracil and leucovorin plus oxaliplatin (Eloxatin®), known as the FOLFOX regime, or FOLFOX plus bevacizumab (Avastin®), is the current standard of care for treatment of metastatic colorectal cancer in the United States. Approximately 90,000 patients received oxaliplatin-containing treatment regimes in 2005, generating approximately $1.4 billion in revenue from the treatment of early and late-stage colorectal cancers. However, approximately 82% of the patients who receive this
7
treatment develop peripheral neuropathy, and approximately 19% of all patients develop severe peripheral neuropathy. Neuropathy, a peripheral nerve function problem that can result in numbness, tingling and pricking sensations, sensitivity to touch, pain, and muscle weakness or wasting, causes an estimated 30% to 50% of patients to withdraw from oxaliplatin treatment. Picoplatin has been tested in more than 500 patients in Phase I and Phase II safety and efficacy studies. In contrast, approximately 11.5% of picoplatin treated patients in these studies developed mild or moderate peripheral neuropathy and none developed severe neuropathy.
Hormone Refractory Prostate Cancer
Phase I/II Clinical Trial. We are also conducting a Phase I/II study of picoplatin in the first-line treatment of patients with prostate cancer that is not responding to hormone treatments and has not previously been treated with chemotherapy. We treated our first patient in the approximately 12-patient trial in May 2006. The trial is being conducted in Eastern Europe and is continuing to enroll patients. The Phase I component of the trial is designed to evaluate increasing doses of picoplatin in combination with the chemotherapy agent docetaxel (Taxotere®) to establish an appropriate dose of picoplatin for further testing in the Phase II efficacy component of the trial. Endpoints of the study will include safety, reduction in prostate specific antigen (PSA), objective tumor response rate (tumor shrinkage), time to tumor progression, progression-free survival and overall survival.
Hormone-Refractory Prostate Cancer and its Treatment. Prostate cancer is the most common type of cancer among men in the United States, apart from skin cancer, and the third leading cause of death in American men. Approximately 234,000 men in the United Stated will be diagnosed with the disease in 2007, and over 27,000 will die from this disease according to the American Cancer Society. In the Europe Union, there are approximately 225,000 prostate cancer cases and 83,000 deaths annually, according to the International Agency for Research on Cancer’s GLOBOCAN 2002 database. Since the incidence of prostate cancer increases with age, the aging of the overall population is expected to further increase the number of prostate cancer patients.
Many patients diagnosed with prostate cancer initially receive surgery or radiation therapy, and some of these patients are cured. For many, however, the disease recurs. At this point the recurrent disease is treated with hormone therapy, and most patients initially respond well. The duration of response averages only 10 to 12 months, however, and the tumor cells eventually become resistant to the hormones (hormone-refractory), and the tumor again progresses. Increasingly, chemotherapy is being used as a first-line treatment for hormone-refractory prostate cancer, but few effective drugs have been identified. Docetaxel in combination with prednisone was approved by the FDA in 2004 for the treatment of patients with metastatic (stage IV) hormone-refractory prostate cancer. The majority (more than 80%) of newly-diagnosed stage IV patients who fail hormone therapy currently are treated with docetaxel either alone or in combination with other drugs. Other options include mixozantrone, estramustine or prednisone monotherapy as second-line treatment. We believe that the combination of picoplatin and docetaxel has the potential to be more effective than either docetaxel or picoplatin alone and could reduce the likelihood of chemo-resistance which occurs with platinum agents currently used to treat this patient population.
Oral Picoplatin
Planned Phase I Clinical Trial. In February 2007, we filed an IND with the FDA for an oral formulation of picoplatin. Following FDA review, we intend to initiate a Phase I clinical trial of oral picoplatin. We believe that oral picoplatin has significant potential for use in combination with other oral chemotherapies and targeted therapies, including in a refractory setting following relapse from first-line therapies. In preclinical studies, picoplatin has been shown to have up to 40% oral bioavailability and a higher therapeutic index and efficacy against platinum-sensitive and -resistant tumor variants than currently marketed platinum-based therapeutics.
8
Picoplatin Source of Supply
We have a limited supply of picoplatin drug product that was manufactured by a prior licensee and supplier. The drug product has been demonstrated to be stable for up to 30 months from the date of manufacture, which time period is not sufficient to complete our current and planned clinical trials. We have entered into separate agreements with third parties for the manufacture of picoplatin active pharmaceutical ingredient, or API, and the bulk production and distribution of finished picoplatin drug product. We currently have one supplier each of API and finished drug product. Manufacturing services under these agreements are provided on a purchase order, fixed-fee basis. Unless earlier terminated, each agreement continues for an initial term ending December 31, 2009 and may be extended beyond the initial term upon agreement of the parties. The agreements generally provide that they may be terminated by either party if there is a material breach by the other party that remains uncured or in the event of solvency or bankruptcy of the other party. We may terminate the finished drug product supply agreement at any time with one year’s advance notice. We may terminate the API manufacturing agreement if there is a change in control of the manufacturer. We have no assurance that our current suppliers will be able to manufacture sufficient picoplatin API and/or finished drug product on a timely or cost-effective basis at all times in the future. We believe that there are other contract manufacturers with the capacity to manufacture picoplatin API and finished drug product. If we are required to seek out alternative manufacturers, we may incur significant additional costs and suffer delays in, or be prevented from, completing or initiating our ongoing or planned clinical trials.
Patents and Proprietary Rights
Our policy is to aggressively protect our proprietary technologies. We have filed applications for United States and foreign patents on many aspects of our technologies.
We hold an exclusive worldwide license granted from Genzyme Corporation (successor to AnorMED, Inc.) for the development and commercial sale of picoplatin. Under the license agreement, as amended, Genzyme retains the right to prosecute its patent applications and maintain all licensed patents, with us reimbursing such expenses. We have the right to sue any third party infringers of the picoplatin patents. If we do not file suit, Genzyme, in its sole discretion, has the right to sue the infringer at its expense.
The parties executed the license agreement in April 2004, at which time we paid a one-time upfront milestone payment of $1.0 million in common stock and $1.0 million in cash. The original license agreement excluded Japan from the licensed territory and provided for $13.0 million in development and commercialization milestones, payable in cash or a combination of cash and common stock, and a royalty rate of up to 15% of product net sales after regulatory approval. The parties amended the license agreement on September 18, 2006, modifying several key financial terms and expanding the licensed territory to include Japan, thereby providing us worldwide rights. In consideration of the amendment, we paid Genzyme $5.0 million in cash on October 12, 2006 and will pay Genzyme an additional $5.0 million in cash by March 31, 2007. The amendment eliminates all development milestone payments to Genzyme. We remain obligated to pay a total of $5.0 million in commercialization milestones upon the attainment of certain levels of annual net sales of picoplatin after regulatory approval. The amendment also reduces the royalty payable to Genzyme to a maximum of 9% of annual net product sales. In addition, the amendment reduces the sharing of sublicense revenue with Genzyme for any sublicenses entered into during the first year following the amendment and eliminates the sharing of sublicense revenues on and after September 18, 2007. The license agreement may be terminated by either party for breach, or if the other party files a petition in bankruptcy or insolvency or for reorganization or is dissolved, liquidated or makes assignment for the benefit of creditors. We can terminate the license at any time upon prior written notice to Genzyme. If not earlier terminated, the license agreement will continue in effect, in each country in the territory in which the licensed product is sold or manufactured, until the earlier of (i) expiration of the last
9
valid claim of a pending or issued patent covering the licensed product in that country or (ii) a specified number of years after first commercial sale of the licensed product in that country.
Our picoplatin portfolio includes United States and foreign patents and applications licensed from Genzyme, which cover the picoplatin product. With respect to picoplatin, we expect to rely primarily on US patent number 5,665,771 (expiring February 7, 2016), which is licensed to us by Genzyme, and additional licensed patents expiring in 2016 covering picoplatin in the European Union. The FDA also has designated picoplatin as an orphan drug for the treatment of small cell lung cancer under the provisions of the Orphan Drug Act, which entitles us to exclusive marketing rights for picoplatin in the United States for seven years following market approval.
A number of additional potential avenues exist which may further extend our picoplatin patent protection and exclusivity. In the United States, these include The Drug Price and Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Act, which, among other things, generally provides for patent term extension for up to five years for an issued patent covering a drug product which has undergone regulatory review before marketing. In addition, since picoplatin has not been previously approved for marketing in the United States, picoplatin may qualify for new chemical entity data exclusivity, under which the FDA bans for a period of time submissions of applications from competitors based on published data or Abbreviated New Drug Applications (ANDA) for a drug containing the same active agent. Certain patent term restoration procedures and marketing exclusivity rights also may be available for qualifying drug products in the European Union or individual foreign countries. We intend to evaluate the availability of these mechanisms for extending the patent term and marketing exclusivity for picoplatin on an individual regional or country basis. We cannot be certain that we will be successful in any efforts to extend the term of any patent relating to picoplatin or that picoplatin will be granted additional marketing exclusivity rights in the United States or abroad.
Risks associated with the protection of our patents and other proprietary technologies are described under the heading “Risk Factors” in Item 1A below. Pending or future patent applications by us or our collaborators will not necessarily result in issued patents. Moreover, the current patents that we own or license may not provide substantial protection or commercial benefit. In addition to patent protection, we rely upon trade secrets, unpatented proprietary know-how and continuing technological innovation to develop and maintain our competitive position. Third parties could acquire or independently develop the same or similar technology, or our issued patents or those licensed by us could be circumvented, invalidated or rendered obsolete by new technology. Third parties also could gain access to or disclose our proprietary technology, and we may be unable to meaningfully protect our rights in such unpatented proprietary technology.
Under United States law, although a patent has a statutory presumption of validity, the issuance of a patent is not conclusive as to its validity or as to the enforceable scope of its claims. Accordingly, the patents owned or licensed by us could be invalidated, infringed or designed around by third parties. Also, third parties could obtain patents that we would need to license or design around.
Competition
The competition for development of cancer therapies is substantial. There is intense competition from biotechnology and pharmaceutical companies, as well as academic research institutions, clinical reference laboratories and government agencies that are pursuing research and development activities similar to ours in the United States and abroad. Our initial focus for picoplatin is small cell lung cancer, the most aggressive and deadly form of lung cancer. Although platinum therapies are the preferred treatment, no FDA-approved therapies are available for patients with platinum-refractory or -resistant disease. If approved, picoplatin will be competing with existing treatment regimens, as well as emerging therapies for small cell lung cancer, and other platinum-based therapeutics. Large pharmaceutical/biotechnology
10
companies, including Bristol-Myer Squibb Company, Bayer Schering Pharma AG, Dainippon Sumitomo Pharma Co. Ltd., Eli Lilly and Company, GlaxoSmithKline PLC, Novartis AG, Pfizer Inc., Genentech, Inc., Shionogi & Co. Ltd., SK Pharma and Sanofi-Aventis Group, are marketing and/or developing therapeutics in late-stage clinical trials for the treatment of small cell lung cancer or platinum agents for the treatment of cancer. Multiple biotechnology companies are engaged in clinical trials for the treatment of small cell lung cancer and other platinum-based therapeutics, including Aeterna Zentaris Inc., Access Pharmaceuticals Inc., GPC Biotech AG, Onyx Pharmaceuticals Inc., Pharmion Corporation, Sunesis Pharmaceuticals Inc., Keryx Biopharmaceuticals Inc., Transave Inc., Vion Pharmaceuticals Inc., PharmaMar (Zeltia Group), ImmunoGen, Inc., Meabco A/S, Antigenics, Inc., Ipsen Group and Menarini Group. As we expand the utility of picoplatin into other oncology indications such as hormone-refractory prostate cancer and colorectal cancer, we will be facing additional competition from major pharmaceutical companies, biotechnology companies, research institutions and government agencies. We cannot assure you that we will be able to effectively compete with these or future third-party product development programs. Many of our existing or potential competitors have, or have access to, substantially greater financial, research and development, marketing and production resources than we do and may be better equipped than we are to develop, manufacture and market competing products. Further, our competitors may have, or may develop and introduce, new products that would render our picoplatin or any other proposed product candidates less competitive, uneconomical or obsolete.
Timing of market introduction and health care reform, both uncertainties, will affect the competitive position of our potential products. We believe that competition among products approved for sale will be based, among other things, on product safety, efficacy, reliability, availability, third-party reimbursement, price and patent protection.
Government Regulation and Product Testing
The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, manufacture, marketing and distribution of drugs. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, storage, record-keeping, approval, advertising and promotion of picoplatin and any other future drug candidates. Product development and approval within these regulatory frameworks take a number of years to accomplish, if at all, and involve the expenditure of substantial resources.
U.S. Government Regulation
In the United States, drugs and biologics are subject to rigorous regulation by the FDA under the Federal Food, Drug and Cosmetic Act of 1976, as amended, and implementing regulations. The process required by the FDA before picoplatin and any future drug candidates may be marketed in the United States generally involves the following:
· completion of extensive preclinical laboratory tests, in vivo preclinical studies and formulation studies;
· submission to the FDA of an Investigational New Drug Application (IND), which must become effective before clinical trials can commence;
· performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product candidate for each proposed indication;
· submission of a Biologic License Application (BLA) or New Drug Application (NDA) to the FDA; and
· FDA review and approval of the BLA or NDA prior to any commercial sale or shipment of the drug.
11
In addition to obtaining FDA approval for each product, each domestic drug manufacturing establishment must be registered with and inspected by the FDA. Domestic manufacturing establishments are subject to biennial inspections by the FDA and must comply with current Good Manufacturing Practice (cGMP) regulations, which are enforced by the FDA through its facilities inspection program for biologics, drugs and devices. To supply products for use in the United States, foreign manufacturing establishments must comply with cGMP regulations and are subject to periodic inspection by the FDA or by corresponding regulatory agencies in such countries under reciprocal agreements with the FDA.
Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as animal studies, to assess the potential safety and efficacy of the proposed product. Laboratories that comply with the FDA regulations regarding Good Laboratory Practice must conduct preclinical safety tests. The results of the preclinical studies are submitted to the FDA as part of an IND and are reviewed by the FDA prior to commencement of clinical trials. Unless the FDA provides comments to an IND, the IND will become effective 30 days following its receipt by the FDA. Submission of an IND does not assure FDA authorization to commence clinical trials.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or to patients under the supervision of a qualified principal investigator. Clinical trials are conducted in accordance with the FDA’s Protection of Human Subjects regulations and Good Clinical Practices under protocols that detail the objectives of the study, the parameters to be used to monitor safety, and the efficacy criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND. Further, each clinical study must be conducted under the auspices of an independent Institutional Review Board (IRB) at the institution where the study will be conducted. The IRB will consider, among other things, ethical factors, the safety of human subjects and the possible liability of the institution.
Clinical trials are typically conducted in three sequential phases, but the phases may overlap. In Phase I, the drug is tested for:
· safety (adverse effects);
· dosage tolerance;
· metabolism;
· distribution;
· excretion; and
· pharmaco-dynamics (clinical pharmacology).
In Phase II, a limited patient population is studied to:
· determine the efficacy of the drug for specific, targeted indications;
· determine dosage tolerance and optimal dosage; and
· identify possible adverse effects and safety risks.
If a compound is found to have potential activity in a disease or condition and to have an acceptable safety profile in Phase II clinical trials, Phase III clinical trials are undertaken to further evaluate clinical activity and to further test for safety within an expanded patient population at geographically dispersed clinical study sites. Often, Phase IV (post-marketing) studies are required by the FDA in order to gain more data on safety and efficacy of a drug after it has transitioned into general medical practice. With respect to picoplatin or any proposed products subject to clinical trials, there can be no assurance that Phase I, Phase II or Phase III studies will be completed successfully within any specific time period, if at all. Clinical studies are inherently uncertain, and our current picoplatin and any future clinical trials may
12
not confirm the results achieved in earlier clinical or preclinical trials. If picoplatin is not shown to be safe and effective, we will not be able to obtain the required regulatory approvals for commercial sale of that product. Furthermore, we or the FDA may suspend clinical trials at any time if it is determined that the subjects or patients are being exposed to an unacceptable health risk.
The results of the pharmaceutical development, preclinical studies and clinical trials are submitted to the FDA in the form of an NDA for approval of the marketing and commercial shipment of the drug. The testing and approval processes are likely to require substantial cost, time and effort, and there can be no assurance that any approval will be granted on a timely basis, if at all. The FDA may deny an NDA if applicable regulatory criteria are not satisfied, may require additional testing or information, or may require post-market testing and surveillance to monitor the safety of the product. If regulatory approval is granted, such approval may entail limitations on the indicated uses for which the product may be marketed. The FDA may withdraw product approvals if compliance with regulatory standards is not maintained or if problems occur following initial marketing. Among the conditions for NDA approval is the requirement that the prospective manufacturers’ quality control and manufacturing procedures conform to cGMP regulations. In complying with standards set forth in these regulations, manufacturers must continue to expend time, money and effort in the areas of production and quality control to ensure full technical compliance.
Foreign Regulation
In addition to regulation in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our proposed future products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those counties. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement also vary greatly from country to country.
Under the European Union regulatory systems, marketing authorizations may be submitted either under a centralized or mutual recognition procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The mutual recognition procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the application and assessment report, each member state must decide whether to recognize approval.
In addition to regulations in Europe and the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial distribution of our current and future product candidates.
Employees
As of March 8, 2007, we had 32 full-time employees and 6 part-time employees. Of these full-time employees, 8 hold PhD degrees, 2 hold M.D. degrees, and one holds a JD degree. Of the total full-time employees, 17 employees were engaged in research and development activities and 15 were employed in general administration. Alan Glassberg, M.D. resigned as our chief medical officer effective March 15, 2007 and will provide us consulting services as a member of our clinical advisory board. We consider our relations with employees to be good. None of our employees is covered by a collective bargaining agreement.
13
Corporate Background
We are a Washington corporation that was originally incorporated as NeoRx Corporation in 1984. We changed our name to Poniard Pharmaceuticals, Inc. and relocated our corporate headquarters from Seattle, Washington to South San Francisco in September 2006. Our principal executive office and mailing address is 7000 Shoreline Court, Suite 270, South San Francisco, California 94080, and our telephone number is (650) 583-3774.
Item 1A. RISK FACTORS
Investing in our common stock or other securities involves a high degree of risk. You should carefully read the risks and uncertainties described below and all information contained in this report before you decide to purchase our common stock. If any of the possible adverse events described below actually occurs, we may be unable to conduct our business as currently planned and our financial and operating results could be harmed. In addition the trading price of our common stock could decline due to the occurrence of any of these risks, and you may lose all or part of your investment. Please see “Special Note Regarding Forward-Looking Statements.”
Risks Related to Our Business
We have a history of operating losses, we expect to continue to incur losses, and we may never become profitable.
We have not been profitable since our formation in 1984. As of December 31, 2006, we had an accumulated deficit of $279.6 million. Our net loss for the year ended December 31, 2006 was $23.3 million. We had net losses of $21.0 million for the year ended December 31, 2005 and $19.4 million for the year ended December 31, 2004. These losses resulted principally from costs incurred in our research and development programs and from our general and administrative activities. To date, we have been engaged only in research and development activities and have not generated any significant revenue from product sales. In May 2005, we announced the discontinuation of our skeletal targeted radiotherapy (STR) development program as part of a strategic plan to refocus our limited resources on the development of picoplatin, a platinum-based cancer therapy. We do not anticipate that our picoplatin product candidate, or any other proposed products, will be commercially available for several years, if at all. We expect to incur additional operating losses in the future. These losses may increase significantly as we expand our clinical trials and increase our research and development activities and seek to commercialize picoplatin or any future product candidates.
Our ability to achieve long-term profitability is dependent upon obtaining regulatory approvals for our picoplatin product candidate and any other proposed products and successfully commercializing our products alone or with third parties.
We will need to raise additional capital to develop and commercialize our product candidates and fund operations, and our future access to capital is uncertain.
It is expensive to develop cancer therapy products and conduct clinical trials for these products. We have not generated revenue from the commercialization of any product, and we expect to continue to incur substantial net operating losses and negative cash flows from operations for the foreseeable future. On April 26, 2006, we completed a $65.0 million equity financing; however, we will require substantial additional funding to develop and commercialize picoplatin and any other proposed products and to fund our future operations.
14
Management is continuously exploring financing alternatives, including:
· raising additional capital through the public or private sale of equity or debt securities or through the establishment of credit or other funding facilities; and
· entering into strategic collaborations, which may include joint ventures or partnerships for product development and commercialization, merger, sale of assets or other similar transactions.
We may not be able to obtain the required additional capital or enter into relationships with corporate partners on a timely basis, on favorable terms, or at all. Conditions in the capital markets in general, and in the life science capital market specifically, may affect our potential financing sources and opportunities for strategic partnering. If we raise additional funds by issuing common stock or securities convertible into or exercisable for common stock, our shareholders may experience substantial dilution, and new investors could have rights superior to current security holders. If we are unable to obtain sufficient additional cash when needed, we may be forced to reduce expenses though the delay, reduction or curtailment of our picoplatin and other development and commercialization activities.
The amount of additional financing we will require in the future will depend on a number of factors, including:
· the scope and timing of our picoplatin clinical program and other research and development efforts, including the progress and costs of our ongoing Phase II and planned Phase III trials of picoplatin in small cell lung cancer;
· our ability to obtain clinical supplies of picoplatin active pharmaceutical ingredient and finished drug product in a timely and cost effective manner;
· actions taken by the FDA and other regulatory authorities;
· the timing of and amount of proceeds from any sale of the Denton facility and assets;
· the timing and amount of any milestone or other payments we might receive from or pay to potential strategic partners;
· our degree of success in commercializing picoplatin or any other product candidates;
· the emergence of competing technologies and products, and other adverse market developments;
· the acquisition or in-licensing of other products or intellectual property;
· the costs, including lease and operating costs, incurred in connection with the relocation of our corporate headquarters to South San Francisco and the planned expansion of our workforce;
· the costs of any research collaborations or strategic partnerships established;
· the costs of preparing, filing, prosecuting, maintaining and enforcing patent claims and other intellectual property rights; and
· the costs of performing our obligations under our loan with Silicon Valley Bank and Merrill Lynch Capital, including the cost of interest and other payment obligations and penalties and the cost of complying with unrestricted cash, product development and other covenants and restrictions under the loan agreement.
During 2006, we experienced significant changes to our capital structure which resulted in an ownership change, as defined under Section 382 of the Internal Revenue Code of 1986, as amended (IRC). Consequently, the amount of net operating loss carryforwards and research and experimentation credit carryforwards available to be used in future years are limited under IRC Sections 382 and 383. This limitation will result in the loss of approximately $93.3 million of our net operating loss carryforwards and
15
$9.1 million of our research and development credit carryforwards. We had net operating loss carryforwards of approximately $62.3 million available for future use as of December 31, 2006, which will expire from 2007 through 2026.
Our potential products must undergo rigorous clinical testing and regulatory approvals, which could be costly, time consuming, and subject us to unanticipated delays or prevent us from marketing any products.
The manufacture and marketing of our picoplatin product candidate and our research and development activities are subject to regulation for safety, efficacy and quality by the FDA in the United States and by comparable regulatory authorities in foreign countries.
The process of obtaining FDA and other required regulatory approvals, including foreign approvals, is expensive, often takes many years and can vary substantially depending on the type, complexity and novelty of the products involved.
We have had only limited experience in filing and pursuing applications necessary to gain regulatory approvals. This may impede our ability to obtain timely approvals from the FDA or foreign regulatory agencies. We will not be able to commercialize our product candidates until we obtain regulatory approvals, and consequently any delay in obtaining, or inability to obtain, regulatory approvals could harm our business.
If we violate regulatory requirements at any stage, whether before or after marketing approval is obtained, we may be fined, forced to remove a product from the market or experience other adverse consequences, including delay, which could materially harm our financial results. Additionally, we may not be able to obtain the labeling claims necessary or desirable for product promotion. If we or other parties identify serious side effects after any of our products are on the market, or if manufacturing problems occur, regulatory approval may be withdrawn and reformulation of our products, additional clinical trials, changes in labeling of our products, and/or additional marketing applications may be required.
The requirements governing the conduct of clinical trials and manufacturing and marketing of our proposed products outside the United States vary widely from country to country. Foreign approvals may take longer to obtain than FDA approvals and can involve additional testing. Foreign regulatory approval processes include all of the risks associated with the FDA approval processes. Also, approval of a product by the FDA does not ensure approval of the same product by the health authorities of other countries.
We may take longer to complete our clinical trials than we project, or we may be unable to complete them at all.
We completed enrollment in our Phase II clinical trial of picoplatin in small cell lung cancer in August 2006, and we continue to treat and follow patients on this study for survival. This Phase II study is an open-label, multi-center trial evaluating picoplatin in patients with small cell lung cancer that did not respond to, or which relapsed within six months after completing, prior platinum-containing chemotherapy. The endpoints of the trial include survival, response rate (tumor shrinkage), duration of response and time to progression,
Based upon positive interim overall survival data from our Phase II study, we plan to initiate an international, multi-center randomized Phase III pivotal trial of picoplatin in small cell lung cancer in the first half of 2007. The Phase III trial, which will be undertaken pursuant to an SPA with the FDA, is designed to compare the efficacy and safety of picoplatin plus best supportive care with best supportive care alone as a second-line therapy. The study is expected to enroll approximately 400 patients with small cell lung cancer whose disease did not respond to a first-line platinum-containing (cisplatin or carboplatin) chemotherapy regimen or whose disease responded initially to first-line platinum-containing therapy, but then progressed within six months after completion of treatment. Patients will be randomized on a 2:1 ratio
16
to receive picoplatin plus best supportive care or best supportive care alone. The primary endpoint of the planned study will be improved overall survival as measured in time from randomization to death. Secondary endpoints will include overall response rates, disease control and progression-free survival. We currently estimate that the study will take approximately 20 months to complete; however, the actual time to completion of the study will depend on the rate of patient enrollment, survival times of all patients in the trial, as well as other factors such as patient performance status, extent of disease and the risks and uncertainties described in this report.
In May 2006, we treated our first patient in our Phase I/II study evaluating picoplatin in the front-line treatment of patients with metastatic colorectal cancer. This study is designed to determine the safety and efficacy of picoplatin when combined with the chemotherapy agents 5-fluorouracil and leucovorin to treat patients newly diagnosed with metastatic disease. Also in May 2006, we enrolled our first patient in our Phase I/II trial of picoplatin in the first-line treatment of patients with hormone-refractory prostate cancer. This study is designed to determine the safety and efficacy of picoplatin when combined with the chemotherapy agent docetaxel. We anticipate completing enrollment of the Phase I dose-evaluation components of these trials and initiating enrollment in the Phase II efficacy components of these trials during the first half of 2007. Endpoints of these studies will include safety, disease reduction, time to progression, progression-free survival and overall survival.
The actual times for initiation and completion of our picoplatin clinical trials depend upon numerous factors, including:
· approvals and other actions by the FDA and other regulatory agencies and the timing thereof;
· our ability to open clinical sites;
· our ability to enroll qualified patients into our studies;
· our ability to obtain sufficient, reliable and affordable supplies of the picoplatin active pharmaceutical ingredient and finished drug product;
· our ability to obtain adequate additional funding or enter into strategic partnerships;
· the extent of competing trials at the clinical institutions where we conduct our trials;
· the extent of scheduling conflicts with participating clinicians and clinical institutions; and
· the identified endpoints of the studies, the extent of patient disease and patient performance status.
We may not initiate, advance or complete our picoplatin or any other proposed clinical studies as projected or achieve successful results.
We will rely on academic institutions and clinical research organizations to conduct, supervise or monitor some or all aspects of clinical trials involving picoplatin. Further, to the extent that we now or in the future participate in collaborative arrangements in connection with the development and commercialization of our proposed products, we will have less control over the timing, planning and other aspects of our clinical trials. If we fail to initiate, advance or complete, or experience delays in or are forced to curtail our current or planned clinical trials, our stock price and our ability to conduct our business could be materially negatively affected.
17
If testing of a particular product does not yield successful results, we will be unable to commercialize that product.
Our research and development programs are designed to test the safety and efficacy of our proposed products in humans through extensive preclinical and clinical testing. We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent commercialization of picoplatin or any other proposed products, including the following:
· the safety and efficacy results obtained in early human clinical trials may not be indicative of results obtained in later clinical trials;
· the results of preclinical studies may be inconclusive, or they may not be indicative of results that will be obtained in human clinical trials;
· after reviewing test results, we or any potential collaborators may abandon projects that we previously believed were promising;
· our potential collaborators or regulators may suspend or terminate clinical trials if the participating subjects or patients are being exposed to unacceptable health risks; and
· the effects of our potential products may not be the desired effects or may include undesirable side effects or other characteristics that preclude regulatory approval or limit their commercial use if approved.
Clinical testing is very expensive, can take many years, and the outcome is uncertain. The data that we may collect from our picoplatin clinical trials may not be sufficient to support regulatory approval of our proposed picoplatin product. The clinical trials of picoplatin and any other proposed products may not be initiated or completed on schedule, and the FDA or foreign regulatory agencies may not ultimately approve any of our product candidates for commercial sale. Our failure to adequately demonstrate the safety and efficacy of a cancer therapy product under development would delay or prevent regulatory approval of the product, which would prevent us from marketing the proposed product.
Success in early clinical trials may not be indicative of results obtained in later trials.
Results of early preclinical and clinical trials are based on a limited number of patients and may, upon review, be revised or negated by authorities or by later stage clinical results. Historically, the results from preclinical testing and early clinical trials often have not been predictive of results obtained in later clinical trials. A number of new drugs and therapeutics have shown promising results in initial clinical trials, but subsequently failed to establish sufficient safety and effectiveness data to obtain necessary regulatory approvals. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval.
If we cannot negotiate and maintain collaborative arrangements with third parties, our research, development, manufacturing, sales and marketing activities may not be cost-effective or successful.
Our success will depend in significant part on our ability to attract and maintain collaborative partners and strategic relationships to support the development, manufacture, sale, marketing and distribution of picoplatin and any other future product candidates in the United States and Europe.
We have entered into an exclusive worldwide license, as amended, with Genzyme Corporation (successor to AnorMED, Inc.) for the development and commercial sale of picoplatin. Under that license, we are solely responsible for the development and commercialization of picoplatin. Genzyme retains the right, at our cost, to prosecute its patent applications and maintain all licensed patents. The parties executed the license agreement in April 2004, at which time we paid a one-time upfront milestone payment of $1.0 million in common stock and $1.0 million in cash. The original agreement excluded Japan from the licensed territory and provided for $13.0 million in development and commercialization milestones,
18
payable in cash or a combination of cash and common stock, and a royalty rate of up to 15% on product net sales after regulatory approval. The parties amended the license agreement on September 18, 2006, modifying several key financial terms and expanding the licensed territory to include Japan, thereby providing us worldwide rights. In consideration of the amendment, we paid Genzyme $5.0 million in cash on October 12, 2006 and will pay Genzyme an additional $5.0 million in cash by March 31, 2007. The amendment eliminates all development milestone payments to Genzyme. Genzyme remains entitled to receive up to $5.0 million in commercialization milestones upon the attainment of certain levels of annual net sales of picoplatin after regulatory approval. The amendment also reduces the royalty payable to Genzyme to a maximum of 9% of annual net product sales. In addition, the amendment reduces the sharing of sublicense revenues for any sublicenses entered into during the first year following the amendment and eliminates the sharing of sublicense revenues with Genzyme on and after September 18, 2007. We currently plan to initiate a Phase III trial of picoplatin in small cell lung cancer in the first half of 2007, with the goal of filing an NDA with the FDA in 2009. However, because we cannot predict the length of time to regulatory approval, if any, or the extent of annual sales, if any, of picoplatin, we are unable to predict when or if the milestone and royalty payments under our license agreement with Genzyme may be triggered. The license agreement may be terminated by either party for breach, or if the other party files a petition in bankruptcy or insolvency or for reorganization or is dissolved, liquidated or makes assignment for the benefit of creditors. We can terminate the license at any time upon prior written notice to Genzyme. If not earlier terminated, the license agreement will continue in effect, in each country in the territory in which the licensed product is sold or manufactured, until the earlier of (i) expiration of the last valid claim of a pending or issued patent covering the licensed product in that country or (ii) a specified number of years after first commercial sale of the licensed product in that country. If Genzyme were to breach its obligations under the license, or if the license expires or is terminated and we cannot renew, replace, extend or preserve our rights under the license agreement, we would be unable to move forward with our current and planned picoplatin clinical studies.
On August 4, 2005, we entered into a research funding and option agreement with The Scripps Research Institute, or TSRI. Under the agreement, as amended in December 2006, we will provide TSRI an aggregate of $2.5 million over a 26-month period to fund research relating to synthesis and evaluation of novel small molecule, multi-targeted protein kinase inhibitors and focal adhesion kinase inhibitors as therapeutic agents, including for the treatment of cancer. We have the option to negotiate a worldwide exclusive license to use, enhance and develop any compounds arising from the collaboration. The research funding is payable by us to TSRI quarterly in accordance with a negotiated budget. We made an initial funding payment to TSRI of $137,500, on August 8, 2005. We paid TSRI total funding payments of $1.0 million in 2006, which amount was charged to R&D expense. The agreement provides for aggregate additional funding payments of $1.4 million in 2007. We have no assurance that the research funded under this arrangement will be successful or ultimately will give rise to any viable product candidates. Further, there can be no assurance that we will be able to negotiate, on acceptable terms, a license with respect to any compounds arising from the collaboration.
We are dependent on third-party suppliers for the timely delivery of materials and services and may experience future interruptions in supply
For our picoplatin product candidate to be successful, we need sufficient, reliable and affordable supplies of the picoplatin active pharmaceutical ingredient, or API, and finished drug product. Sources of these may be limited, and third-party suppliers may be unable to manufacture picoplatin API and finished drug product in amounts and at prices necessary to successfully commercialize our picoplatin product. Moreover, third-party manufacturers must continuously adhere to current Good Manufacturing Practice (cGMP) regulations enforced by the FDA through its facilities inspection program. If the facilities of these manufacturers cannot pass a pre-approval plant inspection, the FDA will not grant an NDA for our proposed products. In complying with cGMP and foreign regulatory requirements, any of our third-party
19
manufacturers will be obligated to expend time, money and effort in production, record-keeping and quality control to assure that our products meet applicable specifications and other requirements. If any of our third-party manufacturers or suppliers fails to comply with these requirements, we may be subject to regulatory action.
We have a limited supply of picoplatin drug product that was manufactured by a prior licensee and supplier. The drug product has been demonstrated to be stable for up to 30 months from the date of manufacture. Our current supply will not be sufficient for our current and planned clinical trials. We have no experience in drug formulation or manufacturing, and we lack the resources and capability to manufacture picoplatin or any other product candidate on a clinical or commercial scale. As a result, we rely on third parties to manufacture picoplatin API and finished drug product for our clinical trials. We currently have separate agreements with one supplier each of API and finished drug product. Manufacturing services under these agreements are provided on a purchase order, fixed-fee basis. Unless earlier terminated, each agreement continues for an initial term ending December 31, 2009 and may be extended beyond the initial term upon agreement of the parties. The agreements generally provide that they may be terminated by either party if there is a material breach by the other party that remains uncured or in the event of solvency or bankruptcy of the other party. We may terminate the finished drug supply agreement at any time with one year’s advance notice. We may terminate the API manufacturing agreement if there is a change in control of the manufacturer. We have no assurance that our current suppliers will be able to manufacture sufficient picoplatin API and/or finished drug product on a timely or cost-effective basis at all times in the future. If we are required to seek out alternative manufacturers, we may incur significant additional costs and suffer delays in, or be prevented from, completing or initiating our ongoing or planned clinical trials.
We also rely on third-party contractors to perform for us, or assist us with, the set-up, conduct, support and management of our clinical studies. Because these contractors provide specialized services, their activities and quality of performance may be outside our direct control. If these contractors do not perform their contractual duties or obligations, do not meet expected deadlines, or need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical trial protocols or for any other reasons, we may need to enter into new arrangements with alternative third parties. In any of these circumstances were to occur, our clinical trials may be extended, delayed or terminated or may need to be repeated, and we may not be able to obtain regulatory approval for or commercialize the product candidate being tested in such trials.
We currently have no sales and marketing staff or distribution organization. If we are unable to develop sales, marketing and distribution capabilities on our own or through collaborations with corporate partners, we may not be successful in commercializing our future products.
None of our current employees has experience selling, marketing and distributing therapeutic products. To the extent we are successful in obtaining approval for the commercial sale of picoplatin or any other product candidate, we may need to secure one or more corporate partners to conduct these activities. We may not be able to enter into partnering arrangements in a timely manner or on terms acceptable to us. To the extent that we enter into co-promotion or other licensing arrangements, our product revenues are likely to be lower than if we directly marketed and sold our products, and any revenues we receive would depend upon the efforts of third parties, which efforts may not be successful. If we are not able to secure adequate partnering arrangements, we would have to hire additional employees or consultants with expertise in sales, marketing and distribution. Employees with relevant skills may not be available to us. Additionally, any increase in the number of employees would increase our expense level and could have a material adverse effect on our financial position. If we are not successful in commercializing any future products, either on our own or through collaborations with one or more parties, our future product revenue will suffer and we may incur significant additional losses.
20
We face substantial competition in the development of cancer therapies and may not be able to compete successfully, and our potential products may be rendered obsolete by rapid technological change.
The competition for development of cancer therapies is substantial. There is intense competition from biotechnology and pharmaceutical companies, as well as academic research institutions, clinical reference laboratories and government agencies that are pursuing research and development activities similar to ours in the United States and abroad. Our initial focus for picoplatin is small cell lung cancer, the most aggressive and deadly form of lung cancer. Although platinum therapies are the preferred treatment, no FDA-approved therapies are available for patients with platinum-refractory or - -resistant disease. If approved, picoplatin will be competing with existing treatment regimens, as well as emerging therapies for small cell lung cancer and other platinum-based therapeutics. Large pharmaceutical/biotechnology companies, including Bristol-Myer Squibb Company, Bayer Schering Pharma AG, Dainippon Sumitomo Pharma Co. Ltd., Eli Lilly and Company, GlaxoSmithKline PLC, Novartis AG, Pfizer Inc., Genentech, Inc., Shionogi & Co. Ltd., SK Pharma and Sanofi-Aventis Group, are marketing and/or developing therapeutics in late-stage clinical trials for the treatment of small cell lung cancer or platinum agents for the treatment of cancer. Multiple biotechnology companies are engaged in clinical trials for the treatment of small cell lung cancer and other platinum-based therapeutics, including Aeterna Zentaris Inc., Access Pharmaceuticals Inc., GPC Biotech AG, Onyx Pharmaceuticals Inc., Pharmion Corporation, Sunesis Pharmaceuticals Inc., Keryx Biopharmaceuticals Inc., Transave Inc., Vion Pharmaceuticals Inc., PharmaMar (Zeltia Group), ImmunoGen, Inc., Meabco A/S, Antigenics, Inc., Ipsen Group and Menarini Group. As we expand the utility of picoplatin into other oncology indications such as hormone-refractory prostate cancer and colorectal cancer, we will be facing additional competition from major pharmaceutical companies, biotechnology companies, research institutions and government agencies. We cannot assure you that we will be able to effectively compete with these or future third party product development programs. Many of our existing or potential competitors have, or have access to, substantially greater financial, research and development, marketing and production resources than we do and may be better equipped than we are to develop, manufacture and market competing products. Further, our competitors may have, or may develop and introduce, new products that would render our picoplatin or any other proposed product candidates less competitive, uneconomical or obsolete.
If we are unable to protect our proprietary rights, we may not be able to compete effectively, or operate profitably.
Our success is dependent in part on obtaining, maintaining and enforcing our patents and other proprietary rights and our ability to avoid infringing the proprietary rights of others. The United States Patent and Trademark Office, or the USPTO, may not issue patents from the patent applications owned by or licensed to us. If issued, the patents may not give us an advantage over competitors with similar technologies.
The issuance of a patent is not conclusive as to its validity or enforceability and it is uncertain how much protection, if any, will be given to our patents if we attempt to enforce them and they are challenged in court or in other proceedings, such as oppositions, which may be brought in foreign jurisdictions to challenge the validity of a patent. A third party may challenge the validity or enforceability of a patent after its issuance by the USPTO. It is possible that a competitor may successfully challenge our patents or that a challenge will result in limiting their coverage. Moreover, the cost of litigation to uphold the validity of patents and to prevent infringement can be substantial. If the outcome of litigation is adverse to us, third parties may be able to use our patented invention without payment to us. Moreover, it is possible that competitors may infringe our patents or successfully avoid them through design innovation. We may need to file lawsuits to stop these activities. These lawsuits can be expensive and would consume time and other resources, even if we were successful in stopping the violation of our patent rights. In addition, there is a risk that a court would decide that our patents are not valid and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of our patents was
21
upheld, a court would refuse to stop the other party on the ground that its activities do not infringe our patents.
In addition, the protection afforded by issued patents is limited in duration. With respect to picoplatin, in the United States we expect to rely primarily on US Patent Number 5,665,771 (expiring February 7, 2016), which is licensed to us by Genzyme, and additional licensed patents expiring in 2016 covering picoplatin in the Europe Union. The FDA has also designated picoplatin as an orphan drug for the treatment of small cell lung cancer under the provisions of the Orphan Drug Act, which entitles us to exclusive marketing rights for picoplatin in the United States for seven years following market approval. We may also be able to rely on the Hatch-Waxman Act to extend the term of a U.S. patent covering picoplatin after regulatory approval, if any, of such product in the United States.
Under our license agreement with Genzyme, Genzyme retains the right to prosecute its patent applications and maintain all licensed patents, with us reimbursing such expenses. We have the right to sue any third party infringers of the picoplatin patents. If we do not file suit, Genzyme, in its sole discretion, has the right to sue the infringer at its expense.
In addition to the intellectual property rights described above, we rely on unpatented technology, trade secrets and confidential information. Therefore, others may independently develop substantially equivalent information and techniques or otherwise gain access to or disclose our technology. We may not be able to effectively protect our rights in unpatented technology, trade secrets and confidential information. We require each of our employees, consultants and advisors to execute a confidentiality agreement at the commencement of an employment or consulting relationship with us. However, these agreements may not provide effective protection of our information or, in the event of unauthorized use or disclosure, may not provide adequate remedies.
The use of our technologies could potentially conflict with the rights of others.
Our competitors or others may have or may acquire patent rights that they could enforce against us. In such case, we may be required to alter our products, pay licensing fees or cease activities. If our products conflict with patent rights of others, third parties could bring legal actions against us claiming damages and seeking to enjoin manufacturing and marketing of the affected products. If these legal actions are successful, in addition to any potential liability for damages, we could be required to obtain a license in order to continue to manufacture or market the affected products. We may not prevail in any legal action and a required license under the patent may not be available on acceptable terms.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
The cost to us of any litigation or other proceedings relating to intellectual property rights, even if resolved in our favor, could be substantial. Some of our competitors may be better able to sustain the costs of complex patent litigation because they have substantially greater resources. If there is litigation against us, we may not be able to continue our operations. If third parties file patent applications, or are issued patents claiming technology also claimed by us in pending applications, we may be required to participate in interference proceedings in the USPTO to determine priority of invention. We may be required to participate in interference proceedings involving our issued patents and pending applications. We may be required to cease using the technology or license rights from prevailing third parties as a result of an unfavorable outcome in an interference proceeding. A prevailing party in that case may not offer us a license on commercially acceptable terms.
In April 2003, we received $10.0 million from the sale to Boston Scientific Corporation, or BSC, of certain non-core patents and patent applications and the grant to BSC of exclusive license rights to certain non-core patents and patent applications. BSC originally asserted four such patents in two lawsuits against Johnson & Johnson, Inc., its subsidiary, Cordis Corporation, and Guidant Corporation, alleging
22
infringement of such patents. In both lawsuits, the defendants denied infringement and asserted invalidity and unenforceability of the patents. BSC subsequently withdrew three of the patents from the litigation, including the patents that were assigned to BSC. BSC acquired Guidant in April 2006. Although we are not currently a party to the lawsuits, our management and counsel have been deposed in connection with the lawsuits. It is possible that BSC, if it is unsuccessful or has limited success with its claims, may seek damages from us, including recovery of all or a portion of the amounts it paid to us in 2003. We cannot assess the likelihood of whether such claim will be brought against us or the extent of recovery, if any, on any such claim.
Product liability claims in excess of the amount of our insurance would adversely affect our financial condition.
The testing, manufacture, marketing and sale of picoplatin and any other proposed cancer therapy products, including past clinical and manufacturing activities in connection with our terminated STR radiotherapeutic, may subject us to product liability claims. We are insured against such risks up to a $10.0 million annual aggregate limit in connection with clinical trials of our products under development and intend to obtain product liability coverage in the future. However, insurance coverage may not be available to us at an acceptable cost. We may not be able to obtain insurance coverage that will be adequate to satisfy any liability that may arise. Regardless of merit or eventual outcome, product liability claims may result in decreased demand for a product, injury to our reputation, withdrawal of clinical trial volunteers and loss of revenues. As a result, regardless of whether we are insured, a product liability claim or product recall may result in losses that could be material.
Our past use of radioactive and other hazardous materials exposes us to the risk of material environmental liabilities, and we may incur significant additional costs to comply with environmental laws in the future.
Our past research and development and manufacturing processes, as well as the manufacturing processes that may have been used by our collaborators, involved the controlled use of hazardous and radioactive materials. As a result, we are subject to foreign, federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain materials and wastes in connection with our use of these materials. Although we believe that our safety procedures for handling and disposing of such materials complied with the standards prescribed by such laws and regulations, we may be required to incur significant costs to comply with environmental and health and safety regulations in the future. Because we have discontinued operations in facilities that have had past research and manufacturing processes where hazardous or radioactive materials have been in use, we may have significant decommissioning costs associated with the termination of operation of these facilities. These potential decommissioning costs also may reduce the market value of the facilities and may limit our ability to sell or otherwise dispose of these facilities in a timely and cost-effective manner. We have terminated our STR manufacturing operations in Denton, Texas and are actively marketing the facility for sale. In 2005, we recorded costs associated with the closure of the Denton facility of $0.9 million. We estimate costs in 2006 related to these activities at $0.2 million. These costs could increase substantially, depending on actions of regulators or if we discover previously unknown contamination in or around the facility. In addition, the risk of accidental contamination or injury from hazardous or radioactive materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any resulting damages, and any such liability could exceed our resources. Our current insurance does not cover liability for the clean-up of hazardous waste materials or other environmental risks.
23
Even if we bring products to market, changes in health care reimbursement could adversely affect our ability to effectively price our products or obtain adequate reimbursement for sales of our products.
Potential sales of our products may be affected by the availability of reimbursement from governments or other third parties, such as insurance companies. It is difficult to predict the reimbursement status of newly approved, novel medical products. In addition, third-party payors are increasingly challenging the prices charged for medical products and services. If we succeed in bringing one or more products to market, we cannot be certain that these products will be considered cost-effective and that reimbursement to the consumer will be available or will be sufficient to allow us to competitively or profitably sell our products.
The levels of revenues and profitability of biotechnology companies may be affected by the continuing efforts of government and third-party payors to contain or reduce the costs of health care through various means. For example, in certain foreign markets, pricing or profitability of prescription pharmaceuticals is subject to governmental control. In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental controls. It is uncertain what legislative proposals will be adopted or what actions federal, state or private payors for health care goods and services may take in response to any health care reform proposals or legislation. Even in the absence of statutory change, market forces are changing the health care sector. We cannot predict the effect health care reforms may have on the development, testing, commercialization and marketability of our proposed cancer therapy products. Further, to the extent that such proposals or reforms have a material adverse effect on the business, financial condition and profitability of other companies that are prospective collaborators for certain of our potential products, our ability to commercialize our products under development may be adversely affected.
The loss of key employees could adversely affect our operations.
Alan Glassberg, M.D. resigned as our chief medical officer effective March 15, 2007. Although Dr. Glassberg was an executive officer of the Company, we did not experience any material disruptions as a consequence of his resignation. Dr. Glassberg will serve on our clinical advisory board and provide us consulting services.
Susan D. Berland resigned as our chief financial officer effective July 21, 2006. Although Ms. Berland was an executive officer of the company, we did not experience any material disruptions or delays as a consequence of her resignation. Caroline M. Loewy was appointed executive vice president, strategic planning on June 23, 2006 and assumed the role of chief financial officer of the company upon Ms. Berland’s departure. Michael K. Jackson, formerly corporate controller, was appointed principal accounting officer of the company effective July 21, 2006.
As of December 31, 2006, we had a total workforce of 29 full-time employees and 6 part-time employees. In September 2006, we moved our corporate headquarters to newly leased facilities in South San Francisco. We intend to maintain clinical development and support activities and facilities in Seattle and do not have plans to relocate any of our 24 employees currently in Seattle. Our success depends, to a significant extent, on the continued contributions of our principal management and scientific personnel participating in our picoplatin development program. We have limited or no redundancy of personnel in key development areas, including finance, legal, clinical operations, regulatory affairs and quality control and assurance. The loss of the services of one or more of our employees could delay our picoplatin product development activities or any other proposed programs and research and development efforts. We do not maintain key-person life insurance on any of our officers, employees or consultants.
Competition for qualified employees among companies in the biotechnology and biopharmaceutical industry is intense. Our future success depends upon our ability to attract, retain and motivate highly skilled employees and consultants. In order to commercialize our proposed products successfully, we will in
24
the future be required to substantially expand our workforce, particularly in the areas of manufacturing, clinical trials management, regulatory affairs, business development and sales and marketing. These activities will require the addition of new personnel, including management, and the development of additional expertise by existing management personnel.
We have change of control agreements and severance agreements with all of our executive officers and consulting agreements with various of our scientific advisors. Our agreements with our executive officers provide for “at will” employment, which means that each executive may terminate his or her service with us at any time. In addition, our scientific advisors may terminate their services to us at any time.
Risks Related to Our Common Stock
Our common stock may be delisted from The Nasdaq Capital Market if we are unable to maintain compliance with Nasdaq Capital Market continued listing requirements.
Our common stock listing was transferred from The Nasdaq Global Market (formerly The Nasdaq National Market) to The Nasdaq Capital Market (formerly the Nasdaq SmallCap Market) on March 20, 2003. We elected to seek a transfer to The Nasdaq Capital Market because we had been unable to regain compliance with The Nasdaq Global Market minimum $1.00 bid price requirement for continued listing. By transferring to The Nasdaq Capital Market, we were afforded an extended grace period in which to satisfy The Nasdaq Capital Market $1.00 minimum bid price requirement. On May 6, 2003, we received notice from Nasdaq confirming that we were in compliance with the $1.00 minimum bid price requirement. We will not be eligible to relist our common stock on The Nasdaq Global Market unless and until our common stock maintains a minimum bid price of $5.00 per share for 90 consecutive trading days and we otherwise comply with the initial listing requirements for The Nasdaq Global Market. Trading on the Nasdaq Capital Market may have a negative impact on the value of our common stock, because securities trading on the Nasdaq Capital Market typically are less liquid than those traded on The Nasdaq Global Market.
On August 7, 2006, we received a notice from Nasdaq indicating that we were not in compliance with Nasdaq Marketplace Rule 4310(c)(4) (the Minimum Bid Price Requirement) because the closing bid price of our common stock had been below $1.00 per share for thirty consecutive trading days. We completed a one-for-six reverse stock split on September 22, 2006. On October 10, 2006, we received a notice from Nasdaq stating that we had regained compliance with the Minimum Bid Price Requirement because the closing bid price of our common stock had been at or above $1.00 per share for ten consecutive trading days. The closing bid price of our common stock may in the future fall below the Minimum Bid Price Requirement or we may in the future fail to meet other requirements for continued listing on the Nasdaq Capital Market. If we are unable to cure any future events of noncompliance in a timely or effective manner, our common stock could be delisted from The Nasdaq Capital Market.
If our common stock were to be delisted from The Nasdaq Capital Market, we may seek quotation on a regional stock exchange, if available. Such listing could reduce the market liquidity for our common stock. If our common stock is not eligible for quotation on another market or exchange, trading of our common stock could be conducted in the over-the-counter market on an electronic bulletin board established for unlisted securities such as the Pink Sheets or the OTC Bulletin Board. As a result, an investor would find it more difficult to dispose of, or obtain accurate quotations for the price of, our common stock.
If our common stock were to be delisted from The Nasdaq Capital Market, and our trading price remained below $5.00 per share, trading in our common stock might also become subject to the requirements of certain rules promulgated under the Securities Exchange Act of 1934, which require additional disclosure by broker-dealers in connection with any trade involving a stock defined as a “penny
25
stock” (generally, any equity security not listed on a national securities exchange or quoted on Nasdaq that has a market price of less than $5.00 per share, subject to certain exceptions). Many brokerage firms are reluctant to recommend low-priced stocks to their clients. Moreover, various regulations and policies restrict the ability of shareholders to borrow against or “margin” low-priced stocks, and declines in the stock price below certain levels may trigger unexpected margin calls. Additionally, because brokers’ commissions on low-priced stocks generally represent a higher percentage of the stock price than commissions on higher priced stocks, the current price of the common stock can result in an individual shareholder paying transaction costs that represent a higher percentage of total share value than would be the case if our share price were higher. This factor may also limit the willingness of institutions to purchase our common stock. Finally, the additional burdens imposed upon broker-dealers by these requirements could discourage broker-dealers from facilitating trades in our common stock, which could severely limit the market liquidity of the stock and the ability of investors to trade our common stock.
Our stock price is volatile and, as a result, you could lose some or all of your investment.
There has been a history of significant volatility in the market prices of securities of biotechnology companies, including our common stock. In 2005, the reported high and low closing sale prices of our common stock were $2.34 and $0.47. In 2004, the reported high and low closing sale prices were $5.78 and $1.43. The reported high and low closing sale prices during the period from January 3, 2006 through September 22, 2006 (the last trading day preceding the effectiveness of our one-for-six reverse stock split) were $1.57 and $0.50. The reported high and low closing sale prices during the period from September 25, 2006 through December 31, 2006 (after the effective date of the reverse stock split) were $7.74 and $3.00. Our stock price has been and may continue to be affected by this type of market volatility, as well as our own performance. Our business and the relative price of our common stock may be influenced by a large variety of factors, including:
· announcements by us or our competitors concerning acquisitions, strategic alliances, technological innovations, new commercial products or changes in product development strategies;
· the availability of critical materials used in developing our proposed picoplatin product;
· our ability to conduct our picoplatin clinical development program on a timely and cost-effective basis and the progress and results of our clinical trials and those of our competitors;
· developments concerning patents, proprietary rights and potential infringement;
· developments concerning potential agreements with collaborators;
· the expense and time associated with, and the extent of our ultimate success in, securing regulatory approvals;
· our available cash or other sources of funding; and
· future sales of significant amounts of our common stock by us or our shareholders.
In addition, potential public concern about the safety of our proposed picoplatin product and any other products we develop, comments by securities analysts, our ability to maintain the listing of our common stock on the Nasdaq system, and conditions in the capital markets in general and in the life science capital market specifically, may have a significant effect on the market price of our common stock. The realization of any of the risks described in this report, as well as other factors, could have a material adverse impact on the market price of our common stock and may result in a loss of some or all of your investment in our securities.
In the past, securities class action litigation often has been brought against companies following periods of volatility in their stock prices. We may in the future be the target of similar litigation. Securities
26
litigation could result in substantial costs and divert our management’s time and resources, which could cause our business to suffer.
Certain provisions in our articles of incorporation and Washington state law could discourage a change of control.
Our articles of incorporation authorize our board of directors to issue up to 200,000,000 shares of common stock and up to 2,998,425 shares of preferred stock. With respect to preferred stock, our board has the authority to determine the price, rights, preferences, privileges and restrictions, including voting rights, of those shares without any further vote or action by our shareholders. Our shareholder rights plan adopted on April 10, 1996, and the preferred stock purchase rights issued to each common shareholder thereunder, expired on April 10, 2006.
Washington law imposes restrictions on certain transactions between a corporation and significant shareholders. Chapter 23B.19 of the Washington Business Corporation Act prohibits a target corporation, with some exceptions, from engaging in particular significant business transactions with an acquiring person, which is defined as a person or group of persons that beneficially owns 10% or more of the voting securities of the target corporation, for a period of five years after the date the acquiring person first became a 10% beneficial owner of voting securities of the target corporation, unless the business transaction or the acquisition of shares is approved by a majority of the members of the target corporation’s board of directors prior to the time the acquiring person first became a 10% beneficial owner of the target corporation’s voting securities. Prohibited business transactions include, among other things:
· a merger or consolidation with, disposition of assets to, or issuance or redemption of stock to or from the acquiring person;
· termination of 5% or more of the employees of the target corporation; or
· receipt by the acquiring person of any disproportionate benefit as a shareholder.
After the five-year period, a significant business transaction may occur if it complies with “fair price” provisions specified in the statute. A corporation may not opt out of this statute. This provision may have an antitakeover effect with respect to transactions that our board does not approve in advance.
The provisions of our articles of incorporation and Washington law discussed above may have the effect of delaying, deterring or preventing a change of control of the company, even if this change would be beneficial to our shareholders. These provisions also may discourage bids for our common stock at a premium over market price and may adversely affect the market price of, and the voting and other rights of the holders of, our common stock. In addition, these provisions could make it more difficult to replace or remove our current directors and management in the event our shareholders believe this would be in the best interests of the corporation and our shareholders.
As a result of the closing of our 2006 equity financing, the number of shares of our common stock outstanding increased substantially and certain investors beneficially own significant blocks of our common stock; such common shares are generally available for resale in the public market.
On April 26, 2006, we completed a $65.0 million equity financing pursuant to a securities purchase agreement dated as of February 1, 2006. In connection with the equity financing, we issued to a small group of institutional and other accredited investors an aggregate of 15.5 million shares of common stock at a cash purchase price of $4.20 per share. Investors in the financing also received five-year warrants to purchase an aggregate of 4.6 million shares of common stock at an exercise price of $4.62 per share. Concurrent with the closing of the financing, we issued an aggregate of 1.6 million shares of common stock to the holders of our Series B preferred stock upon conversion of their outstanding Series B preferred shares. At the time of closing, the placement agent for the financing also received a five-year warrant to
27
purchase, on the same terms as the investors, 139,000 common shares. The issuance of such shares and warrants resulted in substantial dilution to shareholders who held our common stock prior to the financing.
As a result of the completion of the financing and the conversion of the Series B preferred shares, our outstanding common stock increased from approximately 5.7 million shares to approximately 22.8 million shares. Entities affiliated with MPM Capital Management (MPM) acquired beneficial ownership of 7.7 million common shares, or approximately 31.5% of our common stock outstanding immediately following the financing. Entities affiliated with Bay City Capital Management IV LLC (BCC) acquired beneficial ownership of 4.6 million common shares, or approximately 19.5% of the common shares outstanding immediately following the financing. Two of our directors, Fred B. Craves and Carl S. Goldfischer, are managing directors of BCC and possess capital and carried interests in the BCC entities that participated in the financing. We have agreed, for as long as MPM owns at least 10% of the shares of common stock and warrants purchased in the financing, to use our best efforts to cause one person designated by MPM and one person designated by mutual agreement of MPM and BCC to be nominated and elected to our board of directors. Nicholas J. Simon III, a representative of MPM, was appointed to our board of directors on April 26, 2006. Mr. Simon is a general partner of certain of the MPM entities that participated in the financing and possesses capital and carried interests in those entities.
Pursuant to the securities purchase agreement, we maintain an effective registration statement with the SEC covering the resale of the 15.5 million shares of common stock issued in the equity financing and the 4.6 million shares of common stock issuable upon exercise of the warrants. Accordingly, these shares are generally available for immediate resale in the public market. In addition, the approximately 1.6 million shares of common stock issued upon conversion of the Series B preferred stock currently are available for immediate resale pursuant to a registration statement or an exemption from registration under Rule 144(k) of the Securities Act. The market price of our common stock could fall as a result of such resales due to the increased number of shares available for sale in the market.
WHERE YOU CAN FIND MORE INFORMATION
We file annual, quarterly and current reports, as well as registration and proxy statements and other information, with the SEC. These documents may be read and copied at the SEC’s public reference rooms in Washington, DC, New York, NY and Chicago IL. Please call the SEC at 1-800-SEC-0330 for further information on the public reference rooms. Our SEC filings also are available to the public at the Internet web site maintained by the SEC at www.sec.gov. Our reports filed with the SEC after January 1, 2003, also are available on our web site, www.poniard.com. The information contained in our web site does not constitute part of, nor is it incorporated by reference into, this report. We will provide paper copies of our SEC filings free of charge upon request.
Item 1B. UNRESOLVED STAFF COMMENTS
Not Applicable.
Item 2. PROPERTIES
In September 2006, we relocated our corporate headquarters to 7000 Shoreline Court in South San Francisco, CA, where we lease 17,000 square feet of office and laboratory space under a lease that expires in July 2011.
We also currently occupy approximately 21,000 square feet of office space located at 300 Elliott Avenue West in Seattle, WA, under a lease that expires in July 2009. Through May 2006, we occupied approximately 2,900 square feet in a building and a parking area adjacent to 410 West Harrison Street, Seattle, WA. The lease on this space expired on May 31, 2006.
28
We believe that the foregoing facilities are in good condition and are adequate for their present uses.
In April 2001 we acquired a radiopharmaceutical manufacturing facility located on 12 acres in Denton, Texas. The main building is approximately 88,000 square feet and houses approximately 12,000 square feet of clean rooms. From 2001 to 2005, we used the facility to manufacture our STR radiotherapeutic compound. In May 2005, we announced the immediate implementation of a strategic restructuring program to refocus our limited resourced on the development of picoplatin. The restructuring plan, which was completed in June 2005, included cessation of manufacturing operations at the Denton facility. We are actively working to sell this facility.
Item 3. LEGAL PROCEEDINGS
Not Applicable.
Item 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
Not Applicable.
29
PART II
Item 5. MARKET FOR REGISTRANT’S COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
Our common stock is listed on the The Nasdaq Capital Market (formerly the Nasdaq SmallCap Market). The following table sets forth, for the periods indicated, the high and low sales prices for our common stock as reported on The Nasdaq Capital Market. These quotations reflect inter-dealer prices without retail mark-up, markdown or commission, and may not necessarily represent actual transactions.
| | High | | Low | |
2006 | | | | | |
First Quarter | | $ | 1.57 | | $ | 0.72 | |
Second Quarter | | 1.37 | | 0.95 | |
Third Quarter | | 5.70 | | 0.50 | (1) |
Fourth Quarter | | 7.74 | | 3.25 | |
2005 | | | | | |
First Quarter | | $ | 2.34 | | $ | 0.99 | |
Second Quarter | | 0.97 | | 0.47 | |
Third Quarter | | 1.12 | | 0.62 | |
Fourth Quarter | | 1.26 | | 0.75 | |
(1) On September 22, 2006, the Company effected a one-for-six reverse split of its outstanding common stock.
The closing sale price of our common stock on The Nasdaq Capital Market was $5.80 on March 8, 2007.
There were approximately 891 shareholders of record on March 8, 2007. This figure does not include the number of shareholders whose shares are held on record by a broker or clearing agency, but includes such a brokerage house or clearing agency as one holder of record.
See Part III. Item 12 for information regarding securities authorized for issuance under our incentive compensation plans.
30
Stock Price Performance Graph
The graph below compares the cumulative total shareholder return on our common stock with the cumulative shareholder return of the Nasdaq Stock Market Index (US) and the Nasdaq Pharmaceuticals Stocks Index. Sock price performance shown below is historical and not necessarily indicative of future price performance.
Comparison of Five-Year Cumulative Total Return Among Poniard Pharmaceuticals, Inc.,
Nasdaq Stock Market (US) and Nasdaq Pharmaceuticals Stocks Index (1)
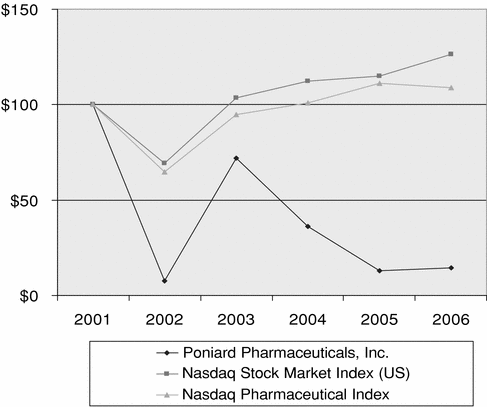
| | 2001 | | 2002 | | 2003 | | 2004 | | 2005 | | 2006 | |
Poniard Pharmaceuticals, Inc. | | $ | 100 | | | $ | 7 | | | $ | 72 | | $ | 36 | | $ | 13 | | $ | 14 | |
Nasdaq Stock Market Index (US) | | 100 | | | 69 | | | 103 | | 112 | | 115 | | 126 | |
Nasdaq Pharmaceutical Index | | 100 | | | 65 | | | 95 | | 101 | | 111 | | 109 | |
| | | | | | | | | | | | | | | | | | | | | |
(1) Assumes $100 invested on January 1, 2001, in our common stock, the Nasdaq Stock Market Index and the Nasdaq Pharmaceutical Stocks Index, an index of approximately 217 companies with common stock quoted on the Nasdaq National Market. The Primary Standard Industrial Classification Code Number (SIC) of these companies is #2835 - Pharmaceutical Companies. Total return performance for the Nasdaq Stock Market Index and the Nasdaq Pharmaceutical Stocks Index is weighted based on the market capitalization of the firms included in each index and assumes that dividends are reinvested. The Nasdaq Stock Market Index and the Nasdaq Pharmaceutical Stocks Index are produced and published by the Center for Research in Securities Pricing at the University of Chicago.
31
Item 6. SELECTED FINANCIAL DATA (IN THOUSANDS, EXCEPT PER SHARE DATA)
The following table shows selected financial data. It is important to read this selected financial data along with the “Financial Statements and Supplementary Data,” as well as the “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
| | Years Ended December 31, | |
| | 2006 | | 2005 | | 2004 | | 2003 | | 2002 | |
| | (in thousands) | |
Consolidated Statement of Operations Data: | | | | | | | | | | | |
Revenues | | $ | — | | $ | 15 | | $ | 1,015 | | $ | 10,531 | | $ | 11,054 | |
Operating expenses | | 21,234 | | 21,075 | | 20,502 | | 15,218 | | 34,949 | |
Loss from operations | | (21,234 | ) | (21,060 | ) | (19,487 | ) | (4,687 | ) | (23,895 | ) |
Net loss | | (23,294 | ) | (20,997 | ) | (19,371 | ) | (5,059 | ) | (23,093 | ) |
Net loss applicable to common shareholders | | (23,794 | ) | (21,497 | ) | (19,871 | ) | (7,535 | ) | (23,593 | ) |
Net loss per common share—basic and diluted | | $ | (1.37 | ) | $ | (3.83 | ) | $ | (3.96 | ) | $ | (1.68 | ) | $ | (5.34 | ) |
Weighted average common shares outstanding—basic and diluted | | 17,376 | | 5,611 | | 5,024 | | 4,547 | | 4,441 | |
Consolidated Balance Sheet Data: | | | | | | | | | | | |
Cash, cash equivalents and restricted cash | | $ | 44,284 | | $ | 4,523 | | $ | 16,254 | | $ | 15,166 | | $ | 6,564 | |
Investment securities | | 9,562 | | — | | 1,499 | | 12,335 | | 9,572 | |
Working capital (deficit) | | 42,299 | | (1,880 | ) | 15,689 | | 26,064 | | 14,195 | |
Total assets | | 69,067 | | 10,114 | | 27,436 | | 35,691 | | 25,993 | |
Note payable, net of current portion | | 9,975 | | — | | 3,905 | | 4,112 | | 5,182 | |
Shareholders’ equity | | $ | 46,891 | | $ | 3,173 | | $ | 20,828 | | $ | 29,490 | | $ | 17,576 | |
Item 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Introduction
The following discussion of results of operations, liquidity and capital resources contains forward-looking statements that involve risks and uncertainties. As described under the heading “Important Information Regarding Forward-Looking Statements” at the beginning of this report, our actual results may differ materially from the results discussed in these forward-looking statements. Factors that might cause or contribute to such differences include those discussed below and in the section above entitled “Risk Factors.”
Unless otherwise indicated, all common stock related amounts have been adjusted to reflect our one-for-six reverse stock split effective September 22, 2006.
Critical Accounting Policies
Basis of Revenue Recognition: To date, we do not have any significant ongoing revenue sources. On occasion, we derive significant revenue from the sale or licensing of our patented technologies and from government grants. Pursuant to the Securities and Exchange Commission Staff Accounting Bulletin No. 104 (SAB 104) and Emerging Issues Task Force Consensus No. 00-21, revenues from collaborative agreements are recognized as earned as we perform research activities under the terms of each agreement. Billings in excess of amounts earned are classified as deferred revenue. To the extent that a transaction contains multiple deliverables, we determine whether the multiple deliverables are separable, and, if separable, the revenue to be allocated to each deliverable based on fair value. If fair value is undeterminable for undelivered elements of the arrangement, revenue is deferred over the contract period or until delivery, as applicable. The revenue allocated to each deliverable is recognized following the
32
requirements of SAB 104. For a detailed description of our revenue recognition policy, refer to Note 2, Summary of Significant Accounting Policies, of the Notes to the Consolidated Financial Statements.
Impairment of Long-Lived Assets: As of December 31, 2006, we had approximately $0.5 million of property and equipment. In accounting for these long-lived assets, we make estimates about the expected useful lives of the assets, the expected residual values of the assets, and the potential for impairment based on events or circumstances. The events or circumstances could include a significant decrease in market value, a significant change in asset condition or a significant adverse change in regulatory climate. Application of the test for impairment requires judgment.
In June 2005, we recognized an asset impairment loss of $3.3 million on certain facilities and equipment resulting from our decision to terminate our STR program. The loss on the equipment at the Seattle facility was determined based on estimates of potential sales values of used equipment and other selling costs. In December 2006, we recognized an additional impairment loss of $0.4 million on the Denton manufacturing facility, which was based on our evaluation of market data for this property. We used a fair value of $2.8 million for the Denton facility in determining the total impairment loss as of December 31, 2006. Due to the inherent uncertainty of the timing of any sale of the Denton facility, we have classified this asset as a long-term asset held for sale.
Long-Term Debt: We assumed a note payable to Texas State Bank in connection with the acquisition of our radiopharmaceutical manufacturing facility in Denton, Texas. In May 2006, we paid off the $2.7 million balance outstanding on the note.
In October 2006, we entered into a loan and security agreement (the loan agreement) with Silicon Valley Bank and Merrill Lynch Capital, which is secured by a first lien on substantially all of our non-intellectual property assets. Under the loan agreement, we received capital loan proceeds of $15.0 million on October 31, 2006. The term is for 42 months with maturity on April 1, 2010. We are required to pay a 7.67% fixed interest rate on the outstanding principal balance plus a $1.35 million additional payment on the maturity date of the loan. This additional payment will be accreted to the note payable balance over the term of the loan using the effective interest rate method and reflected as additional interest expense. The loan agreement also contains covenants requiring us, not later than December 31, 2007, to provide evidence of positive Phase II data for the picoplatin drug development program and commence enrollment of patients in a Phase III trial for picoplatin. The failure to satisfy these covenants could result in acceleration of our payment obligations under the loan agreement. In connection with the loan agreement, we issued five-year warrants to purchase an aggregate of 174,418 shares of common stock at an exercise price of $4.30 per share. The portion of the loan proceeds allocable to the warrants is $540,000 based on their relative fair value, which we recorded as additional discount to notes payable. We classify the portion of the loan that is due for payment in 2008 and thereafter as a long-term payable.
Stock Compensation: Beginning January 1, 2006, we account for share-based compensation arrangements in accordance with the provisions of Statement of Financial Accounting Standards No. 123R, “Share-Based Payments,” which requires the measurement and recognition of compensation expense for all share-based payment awards to employees and directors based on estimated fair values. We use the Black-Scholes option valuation model to estimate the fair value of our stock options at the date of grant. The Black-Scholes option valuation model was developed for use in estimating the fair value of traded options which have no vesting restrictions and are fully transferable. Our employee stock options, however, have characteristics significantly different from those of traded options. For example, employee stock options are generally subject to vesting restrictions and are generally not transferable. In addition, option valuation models require the input of highly subjective assumptions, including the expected stock price volatility, the expected life of an option and the number of awards ultimately expected to vest. Changes in subjective input assumptions can materially affect the fair value estimates of an option. Furthermore, the estimated fair value of an option does not necessarily represent the value that will
33
ultimately be realized by an employee. We use historical data, and other related information as appropriate, to estimate the expected price volatility, the expected option life and the expected forfeiture rate. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of a grant. If actual results are not consistent with our assumptions and judgments used in estimating the key assumptions, we may be required to increase or decrease compensation expense, which could be material to our results of operations. See “New Accounting Pronouncements” below for additional information.
Results of Operations
Year Ended December 31, 2006 Compared with December 31, 2005
We had no revenue for 2006, while our revenues for 2005 totaled $15,000. Revenue for 2005 consisted primarily of royalty payments.
Total operating expenses increased 1% to $21.2 million for the year ended December 31, 2006, from $21.1 million for the same period in 2005. Total operating expenses for the year ended December 31, 2005 included an asset impairment charge of $3.3 million. Additionally, a restructuring charge of $1.7 million was incurred in 2005 relating to termination benefits for the reduction in staff and other costs related to the termination of our STR program.
Research and development expenses for the year ended December 31, 2006 increased 31% to $13.4 million, from $10.2 million for the same period in 2005. Among the primary components of the increase in 2006 were higher clinical costs of $4.9 million associated with our picoplatin trials and increased costs of $1.5 million for other R&D efforts, offset by decreased costs of $2.9 million related to the termination of activities related to our STR program and decreased costs of $0.4 million related to our patent portfolio maintenance.
General and administrative expenses increased 27% to $7.5 million for the year ended December 31, 2006, from $5.9 million for the same period in 2005. The increase in G&A costs was due primarily to $1.3 million of stock option expense recorded in connection with the adoption of Statement of Financial Accounting Standard 123R and $0.2 million of expense related to special shareholder meetings.
Interest expense for the year ended December 31, 2006 was $4.0 million, compared to interest expense of $0.3 million for the same period in 2005. The $3.7 million increase in interest expense for 2006 was due primarily to the amortization of debt discount in the amount of $3.5 million related to the bridge financing and $0.1 million related to the Silicon Valley Bank loan, which transactions are discussed below. The increase in interest income of $1.6 million for 2006 compared to 2005 is due to the income from the investment of excess cash from our 2006 equity financing.
We received approximately $62.0 million in net cash proceeds from the sale of common stock and warrants in April 2006 (the 2006 equity financing). In connection with the financing, we issued to a group of institutional and other accredited investors an aggregate of 15.5 million shares of common stock at a cash purchase price of $4.20 per share. Investors in the financing also received five-year warrants to purchase an aggregate of 4.6 million shares of common stock at an exercise price of $4.62 per share. As part of the 2006 equity financing, we received a $3.5 million bridge loan in February 2006 from investors in the 2006 equity financing. Pursuant the bridge loan, we issued five-year warrants to purchase an aggregate of approximately 412,000 shares of common stock at an exercise price of $4.62 per share. We used the funds received in the bridge financing for working capital pending receipt of required shareholder approvals and satisfaction of other conditions to completion of the 2006 equity financing. The outstanding principal amount of the bridge notes issued to the investors, together with $63,000 of accrued interest, automatically converted, at a conversion price of $4.20 per share, into 839,000 shares of common stock at the closing of the 2006 equity financing.
34
Cash and cash equivalents as of December 31, 2006 were $44.1 million, compared with $3.5 million at December 31, 2005.
As discussed below, we currently are conducting multiple ongoing studies of picoplatin and plan to initiate a Phase III pivotal study in the first half of 2007. In addition, we relocated our corporate headquarters to office space and laboratory facilities in South San Francisco under a five-year lease. These, as well as increases in personnel and other plans for future growth, are expected to result in significant increases in our future operating costs, including research and development and administrative expenses. We will require substantial additional funding to support our picoplatin and any other clinical development programs and to fund our operations. In the event that we do not obtain sufficient additional funds, we may be required to delay, reduce or curtail the scope of our picoplatin and other proposed development activities.
Preferred dividends on Series 1 Preferred Stock were $0.5 million in both 2006 and 2005.
Year Ended December 31, 2005 Compared with December 31, 2004
Our revenues for 2005 totaled $15,000, which primarily consisted of royalty payments. Our revenues for 2004 totaled $1.0 million, which consisted primarily of milestone payments from Boston Scientific Corporation.
Total operating expenses increased 3% to $21.1 million for the year ended December 31, 2005, from $20.5 million for the same period in 2004.
Research and development expenses for the year ended December 31, 2005 decreased 24% to $10.2 million, from $13.3 million for the same period in 2004. Among the primary components of the decrease were a $7.3 million decrease in costs related to the STR program, offset by a $3.6 million increase in picoplatin program development costs, a $0.3 million reduction in shared-cost reimbursements and a $0.3 million increase in development related overhead costs.
General and administrative expenses decreased 17% to $5.9 million for the year ended December 31, 2005, from $7.2 million for the same period in 2004. The decrease in G&A costs was due primarily to a decrease of $1.0 million for personnel related costs.
In March 2005, we raised approximately $3.8 million in net proceeds from the sale in a private placement of 3.3 million shares of common stock. In connection with this private placement, we issued five-year warrants to purchase an aggregate of 1.3 million shares of common stock at an exercise price of $2.00 per share. The warrants became exercisable beginning September 3, 2005 and are exercisable at any time during their term.
Until May 2005, our major research and development program had been STR, a bone-targeting radiotherapeutic. In May 2005, we announced the immediate implementation of a strategic restructuring program to refocus our limited resources on the development of picoplatin. The restructuring plan, which was completed in June 2005, included the discontinuation of our STR development program, including halting patient enrollment in our Phase III trial of STR in multiple myeloma, ceasing operations at our Denton, Texas facility, where STR was manufactured, and reducing our workforce by approximately 50%. We recorded restructuring charges against operations totaling $1.7 million during 2005. This charge consisted of severance costs totaling $0.9 million associated with the reduction in workforce and contract termination and decommissioning costs of $0.8 million.
We evaluated our STR assets in light of the restructuring and determined that a likely impairment existed on those assets. We recorded a charge of $3.3 million against operations in June 2005 to reflect such impairment. The asset impairment charge reflects the difference in the estimated fair value of assets as compared to the net book value of assets employed in our STR program.
35
In conjunction with our strategic restructuring, in June 2005 we negotiated the early termination of our STR-related supply agreement with the University of Missouri Research Reactor facility group (MURR). We paid MURR a fee of $368,000 in connection with such early termination. We also paid MURR $190,000 in minimum purchase requirements under the agreement during 2005.
Other income totaled $0.1 million in 2005 and 2004, respectively. These amounts consisted of interest income of $0.3 million, offset by interest expense of $0.2 million.
Preferred dividends on Series 1 Preferred Stock were $0.5 million in both 2005 and 2004.
Major Research and Development Programs
Our major research and development program during the fiscal year ended December 31, 2006 was picoplatin, a next-generation platinum-based cancer therapy. Our major research and development program during the fiscal years ended December 31, 2005 and 2004 was skeletal targeted radiotherapy (STR™), a bone-targeting radiotherapeutic. In May 2005, we discontinued our STR development program and refocused our resources on the development of picoplatin. This restructuring included terminating patient enrollment in our Phase III trial of STR in multiple myeloma, ceasing operations at our Denton facility, where STR was manufactured, and reducing our workforce by approximately 50%.
Picoplatin Program. Picoplatin is a platinum-based chemotherapeutic designed to overcome platinum resistance in the treatment of solid tumors. We completed patient enrollment in our Phase II clinical study of picoplatin in small cell lung cancer in August 2006 and, based on positive median overall survival data from that ongoing study, plan to initiate a Phase III pivotal trial of picoplatin in small cell lung cancer in the first half of 2007. In May 2006, we treated our first patients in separate Phase I/II studies evaluating picoplatin in the front-line treatment of advanced colorectal cancer and hormone-refractory prostate cancer. We expect to complete patient enrollment in these ongoing Phase I trials and initiate the Phase II components of these trials during the first half of 2007. As of December 31, 2006, we have incurred costs of approximately $14.0 million in connection with our picoplatin clinical program since the program’s inception in 2004. Total estimated costs of our picoplatin Phase II trial in small cell lung cancer are expected to be in the range of $3.0 million to $5.0 million through 2007, including the cost of drug supply. Total estimated costs of our picoplatin Phase I/II trial in colorectal cancer and our Phase I/II trial in hormone-refractory prostate cancer are expected to be in the ranges of $3.0 million to $4.0 million and $4.0 million to $5.0 million, respectively, through 2007, including the cost of drug supply. These costs could be substantially higher if we have to repeat, revise or expand the scope of any of our trials. Material cash inflows relating to our picoplatin development program will not commence unless and until we complete required clinical trials and obtain FDA marketing approvals, and then only if picoplatin finds acceptance in the marketplace. To date, we have not received any revenues from product sales of picoplatin.
The risks and uncertainties associated with completing the development of picoplatin on schedule, or at all, include the following, as well as the other risks and uncertainties described in this report:
· we may not have adequate funds to complete the development of picoplatin;
· we may be unable to secure adequate supplies of picoplatin active pharmaceutical ingredient and finished drug product in order to complete our clinical trials;
· picoplatin may not be shown to be safe and efficacious in clinical trials; and
· we may be unable to obtain regulatory approvals of the drug or may be unable to obtain such approvals on a timely basis.
36
If we fail to obtain marketing approvals for picoplatin, are unable to secure adequate clinical and commercial supplies of picoplatin active pharmaceutical ingredient and finished drug product, or do not complete development and obtain United States and foreign regulatory approvals on a timely basis, our operations, financial position and liquidity could be severely impaired, including as follows:
· we would not earn any sales revenue from picoplatin, which would increase the likelihood that we would need to obtain additional financing for our other research and development efforts; and
· our reputation among investors might be harmed, which could make it more difficult for us to obtain equity capital on attractive terms, or at all.
Because of the many risks and uncertainties relating to completion of clinical trials, receipt of marketing approvals and acceptance in the marketplace, we cannot predict the period in which material cash inflows from our picoplatin program will commence, if ever.
Discontinued STR Program. STR is a radiotherapeutic designed to deliver radiation specifically to sites of cancer in the bone and bone marrow. We have incurred costs of approximately $58.2 million in connection with the STR program since the program’s inception in 1998.
We terminated our STR development program in May 2005. Total estimated costs to complete our STR Phase III clinical trial and potentially obtain marketing approval were in the range of $35-40 million, including cost of clinical drug supply. These costs would have been substantially higher if we were required to repeat, revise or expand the scope of our trials or conduct additional clinical trials. Discontinuation of our STR development program relieved us of the annual costs associated with the program, including manufacturing, clinical trial and personnel costs. During 2004 these costs were approximately $10.2 million. During 2005 these costs were approximately $2.9 million. We are actively seeking a buyer for our Denton manufacturing facility and our other STR related assets. Given the inherent uncertainty of the timing of a sale of the Denton facility, we have classified this asset as long-term.
STR was a clinical stage product for which no marketing approvals had been obtained. We had no material cash inflows relating to our STR development and did not receive any revenues from product sales of STR. Due to our decision to curtail our STR development program, there is neither an anticipated completion date nor an expected period during which material cash inflows will commence. As a consequence of the restructuring, we are not dependent on the successful development and completion of our STR program.
Summary of Research and Development Costs. Our development administration overhead costs, consisting of rent, utilities, consulting fees, patent costs and other various overhead costs, are included in total research and development expense for each period, but are not allocated among our various projects. Our total research and development costs include the costs of various research efforts directed toward the identification and evaluation of future product candidates. These other research projects are preclinical and not considered major projects. Our total research and development costs are summarized below:
Summary of Research and Development Costs
| | 2006 | | 2005 | | 2004 | |
| | (in thousands) | |
Picoplatin | | $ | 9,058 | | $ | 4,150 | | $ | 555 | |
Discontinued programs | | 68 | | 2,864 | | 10,155 | |
Other overhead and research costs | | 4,230 | | 3,184 | | 2,621 | |
Total research and development costs | | $ | 13,356 | | $ | 10,198 | | $ | 13,331 | |
37
Liquidity and Capital Resources
We have historically suffered recurring operating losses and negative cash flows from operations. As of December 31, 2006, we had net working capital of $42.3 million, an accumulated deficit of $279.6 million and total shareholders’ equity of $46.9 million.
We have financed our operations primarily through the sale of equity securities, technology licensing, collaborative agreements and debt instruments. We invest excess cash in investment securities that will be used to fund future operating costs. Cash used for operating activities for the twelve months ended December 31, 2006 totaled $17.3 million. Revenues and other income sources for 2006 were not sufficient to cover operating expenses. Cash, cash equivalents and investment securities, net of restricted cash of $0.1 million, totaled $53.7 million at December 31, 2006 compared to $3.5 million at December 31, 2005. We believe that our cash, cash equivalent and investment securities balances will provide adequate resources to fund operations at least until the end of the first quarter of 2008.
On April 26, 2006, we completed a $65.0 million equity financing, pursuant to which we issued to a group of institutional and other accredited investors an aggregate of 15.5 million shares of common stock at a cash purchase price of $4.20 per share. Investors in the financing also received five-year warrants to purchase an aggregate of 4.6 million shares of common stock at an exercise price of $4.62 per share. We received $62.0 million in net proceeds from the financing, which we are using to fund our picoplatin clinical program and general working capital needs. Concurrent with the closing of the financing, we issued an aggregate of 1.6 million shares of common stock to the holders of our Series B preferred stock upon conversion of their outstanding Series B preferred shares. As a result of the completion of the financing and the conversion of the Series B preferred shares, our outstanding common stock increased from approximately 5.7 million shares to approximately 22.8 million shares. Entities affiliated with MPM Capital Management (MPM) acquired beneficial ownership of 7.7 million common shares, or approximately 31.5% of our common stock outstanding immediately following the financing. Entities affiliated with Bay City Capital Management IV LLC (BCC) acquired beneficial ownership of 4.6 million common shares, or approximately 19.5% of the common shares outstanding immediately following the financing. Two of our directors, Fred B. Craves and Carl S. Goldfischer, are managing directors of BCC and possess capital and carried interests in the BCC entities that participated in the financing. We have agreed, for as long as MPM owns at least 10% of the shares of common stock and warrants purchased in the financing, to use our best efforts to cause one person designated by MPM and one person designated by mutual agreement of MPM and BCC to be nominated and elected to our board of directors. Nicholas J. Simon III, a representative of MPM, was appointed to our board of directors on April 26, 2006. Mr. Simon is a general partner of certain of the MPM entities that participated in the financing and possesses capital and carried interests in those entities.
In connection with the financing, we entered into a letter agreement with Texas State Bank, pursuant to which we agreed to accelerate the maturity date of our promissory note with the Bank to June 5, 2006. The Texas State Bank note, which was secured by our radiopharmaceutical plant and other STR assets located in Denton, Texas, had an adjustable interest rate equal to the bank prime rate reported in the Wall Street Journal (8.00% at May 23, 2006). We paid off the outstanding balance of the note, $2.7 million, on May 23, 2006.
We completed the relocation of our corporate headquarters to South San Francisco in September 2006. We intend to maintain our current clinical and development and support activities in Seattle. The addition of 17,045 square feet of office and laboratory space leased in South San Francisco facility will result in a substantial increase in our future rent and operating costs. Under the lease agreement dated July 10, 2006, the annual base rent for the leased facilities is approximately $542,000 and is subject to annual adjustment based on disbursements for tenant improvements and increases in the Consumer Price Index in the San Francisco metropolitan market. Base rent is payable in monthly
38
installments of approximately $45,200. Additional rent is payable monthly based on our share of operating expenses of the project in which the leased facilities are located, as described in the lease agreement. Monthly base rent during the first seven months of the lease will average $21,000 during the construction of tenant improvements. We paid total rent (base rent and additional rent based on our share of project operating expenses) during 2006 of $143,000. We estimate total rent payable by us during 2007 will be approximately $523,000. Annual operating expenses payable under the lease may increase substantially as we move forward with our plans to establish laboratory facilities in the leased space. The initial term of the lease is 60 months. We may, upon written notice delivered at least nine months prior to expiration of the initial lease term, extend the lease for an additional three years, with rent payable at the then market rate.
In April 2004, we acquired the worldwide exclusive rights, excluding Japan, to develop, manufacture and commercialize picoplatin from AnorMED, Inc. AnorMED was acquired by Genzyme Corporation in November 2006. Under the terms of the original agreement, we paid a one-time upfront milestone payment of $1.0 million in common stock and $1.0 million in cash. The original agreement provided for development and commercialization milestone payments of up to $13.0 million, payable in cash or a combination of cash and common stock, and a royalty rate of up to 15% of net product sales after regulatory approval. The parties executed an amendment to the license agreement on September 18, 2006, modifying several key financial terms and expanding the licensed territory to include Japan, thereby providing us worldwide rights. In consideration of the amendment, we paid Genzyme $5.0 million in cash on October 12, 2006 and will pay Genzyme an additional $5.0 million in cash by March 31, 2007. The amendment eliminates $8.0 in development milestone payments to Genzyme. Genzyme remains entitled to receive up to $5.0 million in commercialization milestones upon the attainment of certain levels of annual net sales of picoplatin after regulatory approval. The amendment also reduces the royalty payable to Genzyme to a maximum of 9% of annual net product sales. In addition, the amendment reduces the sharing of sublicense revenues for any sublicenses entered into during the first year following the amendment and eliminates the sharing of sublicense revenues on and after September 18, 2007.
On October 25, 2006, we entered into a loan and security agreement with Silicon Valley Bank and Merrill Lynch Capital, pursuant to which we obtained a $15.0 million capital loan. A portion of the proceeds of the loan will be used to fund our cash payment obligations to Genzyme under the amended license agreement described above. The remaining proceeds will be used to support our strategic growth, late-stage clinical trials of picoplatin and general working capital needs. The loan is for a term of 42 months and matures on April 1, 2010. We are required to pay a 7.67% fixed interest rate on the outstanding principal balance plus a $1.35 million additional payment upon the maturity date of the loan. This additional payment will be accreted to the note payable balance over the term of the loan using the effective interest rate method and reflected as additional interest expense. All interest payable under the loan agreement and the full amount of the additional payment must be paid upon any prepayment of the loan. The loan is secured by a first lien on substantially all of our non-intellectual property assets. The loan agreement contains restrictions on our ability to, among other things, dispose of certain assets, engage in certain mergers and acquisition transactions, incur indebtedness, create liens on assets, make investments and pay dividends or repurchase stock. The loan agreement also contains covenants requiring us to maintain unrestricted cash of $7.5 million during the loan term and, not later than December 31, 2007, to provide evidence of positive Phase II data for the picoplatin drug development program and commence enrollment of patients in a Phase III trial for picoplatin. The loan contains events of default that include, among other things, nonpayment of principal and interest or fees, breaches of covenants, material adverse changes, bankruptcy and insolvency events, cross defaults to other indebtedness, material judgments, inaccuracy of representations and warranties and events constituting a change of control. The occurrence of an event of default would increase the applicable rate of interest by 5% and could result in acceleration of our payment obligations under the loan agreement.
39
On August 4, 2005, we entered into a research funding and option agreement with The Scripps Research Institute (TSRI). Under the agreement, as amended in December 2006, we committed to provide TSRI an aggregate of $2.5 million over a 26-month period to fund research relating to synthesis and evaluation of novel small molecule, multi-targeted protein kinase inhibitors and focal adhesion kinase inhibitors as therapeutic agents, including for the treatment of cancer. We have the option to negotiate a worldwide exclusive license to use, enhance and develop any compounds arising from the collaboration. The research funding is payable by us to TSRI quarterly in accordance with a negotiated budget. On August 8, 2005, we made an initial funding payment to TSRI of $137,500. We paid TSRI total funding payments of $1.0 million in 2006, which amount was charged to R&D expense. The agreement provides for aggregate additional funding payments of $1.4 million in 2007. We have no assurance that the research funded under this arrangement will be successful or ultimately will give rise to any viable product candidates. Further, there can be no assurance that we will be able to negotiate, on acceptable terms, a license with respect to any compounds arising from the collaboration.
In April 2004, we sold and transferred our Pretarget intellectual property to Aletheon Pharmaceuticals, Inc. Under that agreement, we could receive up to $6.6 million in milestone payments if Aletheon achieves certain development goals, plus royalties on potential future product sales. We did not receive any upfront consideration for the sale of the Pretarget property. We discontinued our clinical studies using the Pretarget technology in July 2002, and sought, both through targeted inquiries and a broad-based auction process, a buyer or licensee for the technology. The sale of the Pretarget intellectual property relieved us of the annual costs associated with maintaining the Pretarget patent estate. During 2003, we spent approximately $350,000 for the prosecution and maintenance of the Pretarget patents and trademarks. For 2004, these costs were approximately $70,000. Seattle-based Aletheon is a development stage biotherapeutics company founded by two former Poniard employees. The timing and amount of milestone payments, if any, are uncertain. The terms of the transaction were determined through arms-length negotiation.
We raised approximately $3.8 million in net proceeds from the sale of common stock and warrants in a private placement transaction in March 2005. We used the net proceeds from that financing to support our Phase II trial in picoplatin in small cell lung cancer and for general working capital, including restructuring costs associated with the termination of our STR development program. We raised approximately $9.0 million in net proceeds from the sale of common stock and warrants in a private placement transaction in February 2004. The net proceeds from that financing were used to support our STR development program, which we discontinued in May 2005, and for general working capital.
We terminated our STR manufacturing operations in Denton, Texas during the second quarter of 2005 and began actively marketing the facility for sale. In 2005, we recorded costs associated with the closure and maintenance of the Denton facility of approximately $499,000. We recorded costs of approximately $286,000 in 2006 related to these activities.
We will require substantial additional funding to develop and commercialize picoplatin and any other proposed products and to fund our operations. Management is continuously exploring financing alternatives, including:
· raising additional capital through the public or private sale of equity or debt securities or through the establishment of credit or other funding facilities; and
· entering into strategic collaborations, which may include joint ventures or partnerships for product development and commercialization, merger, sale of assets or other similar transactions.
40
Our actual capital requirements will depend upon numerous factors, including:
· the scope and timing of our picoplatin clinical program and other research and development efforts, including the progress and costs of our ongoing Phase II and planned Phase III trials of picoplatin in small cell lung cancer;
· our ability to obtain clinical supplies of picoplatin active pharmaceutical ingredient and finished drug product in a timely and cost-effective manner;
· actions taken by the FDA and other regulatory authorities;
· the timing and amounts of proceeds from any sale of the Denton facility and assets;
· the timing and amount of any milestone or other payments we might receive from potential strategic partners;
· our degree of success in commercializing picoplatin or any other cancer therapy product candidates;
· the emergence of competing technologies and products, and other adverse market developments;
· the acquisition or in-licensing of other products or intellectual property;
· the costs, including lease and operating costs, incurred in connection with the relocation of our corporate headquarters to South San Francisco and the planned expansion of our workforce;
· the costs of any research collaborations or strategic partnerships established;
· the costs of preparing, filing, prosecuting, maintaining and enforcing patent claims and other intellectual property rights; and
· the costs of performing our obligations under the loan with Silicon Valley Bank and Merrill Lynch Capital, including the cost of interest and other payment obligations and penalties and the cost of complying with unrestricted cash, product development and other covenants and restrictions under the loan agreement.
During 2006, we experienced significant changes to our capital structure which resulted in an ownership change, as defined under Section 382 of the IRC. Consequently, the amount of net operating loss carryforwards and research and experimentation credit carryforwards available to be used in future years are limited under IRC Sections 382 and 383. This limitation will result in the loss of approximately $93.3 million of our net operating loss carryforwards and $9.1 million of our research and development credit carryforwards. We had net operating loss carryforwards of approximately $62.3 million available for future use as of December 31, 2006, which will expire from 2007 through 2026.
There can be no assurance that we will be able to raise additional capital or enter into relationships with corporate partners on a timely basis, on favorable terms, or at all. Conditions in the capital markets in general, and in the life science capital market specifically, may affect our potential financing sources and opportunities for strategic partnering. Our financial statements are prepared on a going concern basis; however, our inability to obtain additional cash as needed could have a material adverse effect on our financial position, results of operations and our ability to continue in existence. Our consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
41
At December 31, 2006, we had the following contractual obligations (in thousands):
| | Total | | Less than
1 year | | 1 - 3
years | | 3 - 5
years | | More
than 5
years | |
Contractual Obligations | | | | | | | | | | | | | | | |
Long-term debt obligations: | | | | | | | | | | | | | | | |
Notes payable (2) (3) | | $ | 17,612 | | | $ | 4,879 | | | $ | 9,758 | | $ | 2,975 | | | $ | — | | |
Operating lease obligations: | | | | | | | | | | | | | | | |
Seattle office space | | 1,415 | | | 560 | | | 855 | | — | | | — | | |
South San Francisco premises (1) | | 2,484 | | | 542 | | | 1,084 | | 858 | | | — | | |
| | 3,899 | | | 1,102 | | | 1,939 | | 858 | | | — | | |
Purchase obligations: | | | | | | | | | | | | | | | |
Scripps research collaboration | | 1,350 | | | 1,350 | | | — | | — | | | — | | |
Total | | $ | 22,861 | | | $ | 7,331 | | | $ | 11,697 | | $ | 3,833 | | | $ | — | | |
(1) Lease executed in July 2006. See discussion above for details.
(2) Amounts include interest payments.
(3) Amount in “Total” column includes total principal payment of $13,881 as refected on the Consolidated Balance Sheet for the year ended December 31, 2006.
New Accounting Pronouncements
In May 2005, the Financial Accounting Standards Board (FASB) issued Statement of Financial Accounting Standards (SFAS) No. 154, “Accounting Changes and Error Corrections, a Replacement of APB Opinion No. 20 and FASB Statement No. 3.” SFAS 154 establishes, unless impracticable, retrospective application as the required method for reporting a change in accounting principle in the absence of explicit transition requirements specific to a newly adopted accounting principle. Previously, most changes in accounting principle were recognized by including the cumulative effect of the change to the new accounting principle in net income of the period of the change. Under SFAS 154, retrospective application requires the cumulative effect of the change to be reflected in the carrying amounts of assets and liabilities as of the beginning of the first period presented and financial statements for each individual prior period presented to be adjusted to reflect the effects of applying the new accounting principle. SFAS 154 carries forward the guidance in APB Opinion 20 “Accounting Changes,” requiring justification of a change in accounting principle on the basis of preferability, for reporting the correction of an error in previously issued financial statements and for a change in an accounting estimate. SFAS 154 is effective for accounting changes and corrections of errors made in fiscal years beginning after December 15, 2005. The adoption of the Statement by the Company on January 1, 2006 did not have a material impact on the Company’s consolidated financial statements.
In December 2004, the FASB issued SFAS No.123R, “Share-Based Payment.” SFAS 123R replaces SFAS 123, “Stock-Based Compensation,” issued in 1995. SFAS 123R requires that the fair value of the grant of employee stock options be reported as an expense in the results of operations. SFAS 123R eliminates the ability to account for stock-based compensation using APB 25 and requires that such transactions be recognized as compensation cost in the income statement based on their fair value on the measurement date, which is generally the date of the grant. The Statement is effective for the first annual reporting period that begins after June 15, 2005. Historically, the Company has disclosed in its footnotes the pro forma expense effect of the grants. Stock compensation expense under the prior rules would have increased reported diluted loss per share by $.03 in 2005. SFAS 123R applies to all outstanding, unvested option grants as of the effective date. The Company adopted SFAS No. 123R on January 1, 2006 using the
42
modified prospective transition method. Under the modified prospective transition method, SFAS 123R applies to new and modified option grants after January 1, 2006, and to any unvested option grants as service is rendered on or after the effective date. The attribution of compensation cost for vested option grants as of January 1, 2006 is based on the same method and on the same grant-date fair values previously determined for the pro forma disclosures required for companies that did not adopt the fair value accounting method for stock-based employee compensation. See Note 3 of the Notes to the Consolidated Financial Statements for the discussion of the effect of the adoption of this Statement on the Company’s consolidated financial statements.
In June 2006, the FASB issued Interpretation No. 48, “Accounting for Uncertainty in Income Taxes: An Interpretation of FASB Statement No. 109” (FIN 48). FIN 48 clarifies certain aspects of accounting for uncertain tax positions, including issues related to the recognition and measurement of those tax positions. FIN 48 is effective for fiscal years beginning after December 15, 2006. Accordingly, the Company will adopt FIN 48 on January 1, 2007. The Company is currently assessing the impact FIN 48 will have, if any, on its consolidated financial statements.
In September 2006, the FASB issued SFAS No. 157, “Fair Value Measurements”. SFAS 157 defines fair value, establishes a framework for measuring fair value, and expands disclosure requirements about fair value measurements. SFAS 157 applies to other accounting pronouncements that require or permit fair value measurements, but does not in itself require any new fair value measurements. The provisions of SFAS No. 157 are effective for fiscal years beginning after November 15, 2007 and interim periods within those fiscal years. The Company is currently evaluating this statement and its impact, if any, on the Company’s consolidated financial statements.
In September 2006, the SEC issued Staff Accounting Bulletin No. 108, “Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements” (SAB 108), to address diversity in practice in quantifying financial statement misstatements. SAB 108 requires the quantification of misstatements based on their impact on both the balance sheet and the income statement to determine materiality. The guidance provides for a one-time cumulative effect adjustment to correct for misstatements that were not deemed material under a company’s prior approach but are material under the SAB 108 approach. SAB 108 is effective for fiscal years ending after November 15, 2006. The adoption of this guidance did not have a material impact on the Company’s consolidated financial statements.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk
The Company’s exposure to market rate risk for changes in interest rates relates primarily to debt securities included in the Company’s investment portfolio. The Company does not have any derivative financial instruments. The Company invests in debt instruments of the U.S. Government and its agencies and in high-quality corporate issuers. Investments in both fixed rate and floating rate interest-earning instruments carry a degree of interest rate risk. Fixed rate securities may have their fair market value adversely impacted due to a rise in interest rates, while floating rate securities may produce less income than expected if interest rates decrease. Due in part to these factors, the Company’s future investment income may fall short of expectations due to changes in interest rates or the Company may experience losses in principal if forced to sell securities that have declined in market value due to changes in interest rates. At December 31, 2006, the Company owned government debt instruments totaling $2.6 million and owned corporate debt securities totaling $7.0 million. The Company’s exposure to losses as a result of interest rate changes is managed through investing primarily in securities with relatively short maturities of up to two years and securities with variable interest rates. All government debt instruments and corporate debt securities owned by the Company at December 31, 2006 had maturities of less than one year.
43
The Company’s only material outstanding debt is its loan obligation to Silicon Valley Bank and Merrill Lynch Capital. The outstanding balance of this loan was $13.9 million on December 31, 2006. The loan, which matures on April 1, 2010, bears interest at a fixed rate of 7.67%. The occurrence of an event of default under the loan, as described above, would increase the applicable rate of interest by 5% during the continuance of the event of default and could result in acceleration of the Company’s payment obligations under the loan agreement.
Investment Risk
In the past, the Company has received equity instruments under licensing agreements. These instruments are included in investment securities and are accounted for at fair value with unrealized gains or losses reported as a component of comprehensive loss and classified as accumulated other comprehensive income—unrealized gain on investment securities in shareholders’ equity. Such investments are subject to significant fluctuations in fair market value due to the volatility of the stock market. At December 31, 2005 and 2006, the Company owned no corporate equity securities.
Item 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
All financial schedules are omitted since the required information is not applicable or has been presented in the financial statements and the notes thereto.
44
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders
Poniard Pharmaceuticals, Inc.:
We have audited the accompanying consolidated balance sheets of Poniard Pharmaceuticals, Inc. and subsidiary as of December 31, 2006 and 2005, and the related consolidated statements of operations, cash flows and shareholders’ equity and comprehensive loss for each of the years in the three-year period ended December 31, 2006. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Poniard Pharmaceuticals, Inc. and subsidiary as of December 31, 2006 and 2005, and the results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2006, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 2 to the consolidated financial statements, Poniard Pharmaceuticals, Inc. and subsidiary adopted the provisions of Statement of Financial Accounting Standards No. 123 (revised 2004), Share-Based Payment, effective January 1, 2006.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the effectiveness of Poniard Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2006, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated March 15, 2007, expressed an unqualified opinion on management’s assessment of, and the effective operation of, internal control over financial reporting.
/s/ KPMG LLP
Seattle, Washington
March 15, 2007
45
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
(in thousands, except share data)
| | December 31, | |
| | 2006 | | 2005 | |
ASSETS | | | | | |
Current assets: | | | | | |
Cash and cash equivalents | | $ | 44,148 | | $ | 3,523 | |
Cash - restricted | | 136 | | 1,000 | |
Investment securities | | 9,562 | | — | |
Prepaid expenses and other current assets | | 654 | | 455 | |
Assets held for sale | | — | | 83 | |
Total current assets | | 54,500 | | 5,061 | |
Facilities and equipment, net of depreciation of $686 and $556, respectively | | 525 | | 273 | |
Other assets | | 182 | | 45 | |
Assets held for sale | | 2,624 | | 3,027 | |
Licensed products, net of accumulated amortization of $764 and $292 | | 11,236 | | 1,708 | |
Total assets | | $ | 69,067 | | $ | 10,114 | |
LIABILITIES AND SHAREHOLDERS’ EQUITY | | | | | |
Current liabilities: | | | | | |
Accounts payable | | $ | 775 | | $ | 995 | |
Accrued liabilities | | 2,520 | | 2,078 | |
Current portion of notes payable | | 3,906 | | 3,868 | |
Licensed products payable | | 5,000 | | — | |
Total current liabilities | | 12,201 | | 6,941 | |
Long-term liabilities: | | | | | |
Notes payable, net of current portion and discount of $1,753 | | 9,975 | | — | |
Total long-term liabilities | | 9,975 | | — | |
Shareholders’ equity: | | | | | |
Preferred stock, $.02 par value, 2,998,425 shares authorized: | | | | | |
Convertible preferred stock, Series 1, 205,340 shares issued and outstanding at December 31, 2006 and 2005 (entitled in liquidation to $5,175, respectively) | | 4 | | 4 | |
Convertible preferred stock, Series B, 0 and 1,575 shares issued and outstanding at December 31, 2006 and 2005 (entitled in liquidation to $15,750 at December 31, 2005) | | — | | — | |
Common stock, $.02 par value, 200,000,000 shares authorized, 22,808,233 and 5,720,382 shares issued and outstanding at December 31, 2006 and 2005, respectively | | 456 | | 114 | |
Additional paid-in capital | | 326,025 | | 258,855 | |
Accumulated deficit, including other comprehensive income of $0 at | | | | | |
December 31, 2006 and 2005 | | (279,594 | ) | (255,800 | ) |
Total shareholders’ equity | | 46,891 | | 3,173 | |
Total liabilities and shareholders’ equity | | $ | 69,067 | | $ | 10,114 | |
See accompanying notes to the consolidated financial statements.
46
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share data)
| | Years Ended December 31, | |
| | 2006 | | 2005 | | 2004 | |
Revenues | | $ | — | | $ | 15 | | $ | 1,015 | |
Operating expenses: | | | | | | | |
Research and development | | 13,356 | | 10,198 | | 13,331 | |
General and administrative | | 7,548 | | 5,948 | | 7,171 | |
Gain on sale of real estate and equipment | | (73 | ) | (158 | ) | — | |
Asset impairment loss | | 403 | | 3,346 | | — | |
Restructuring | | — | | 1,741 | | — | |
Total operating expenses | | 21,234 | | 21,075 | | 20,502 | |
Loss from operations | | (21,234 | ) | (21,060 | ) | (19,487 | ) |
Other income (expense): | | | | | | | |
Interest income | | 1,906 | | 330 | | 326 | |
Interest expense | | (3,966 | ) | (267 | ) | (210 | ) |
Total other (expense) income | | (2,060 | ) | 63 | | 116 | |
Net loss | | (23,294 | ) | (20,997 | ) | (19,371 | ) |
Preferred stock dividends | | (500 | ) | (500 | ) | (500 | ) |
Net loss applicable to common shares | | $ | (23,794 | ) | $ | (21,497 | ) | $ | (19,871 | ) |
Loss per share: | | | | | | | |
Basic and diluted loss applicable to common shares | | $ | (1.37 | ) | $ | (3.83 | ) | $ | (3.96 | ) |
Weighted average common shares outstanding—basic and diluted | | 17,376 | | 5,611 | | 5,024 | |
See accompanying notes to the consolidated financial statements.
47
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| | Years Ended December 31, | |
| | 2006 | | 2005 | | 2004 | |
Cash flows from operating activities: | | | | | | | |
Net loss | | $ | (23,294 | ) | $ | (20,997 | ) | $ | (19,371 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | |
Depreciation and amortization | | 605 | | 458 | | 800 | |
Amortization of discount on notes payable | | 3,604 | | — | | — | |
(Gain) loss on disposal of real estate and equipment | | (27 | ) | (137 | ) | 20 | |
Asset impairment loss | | 403 | | 3,346 | | — | |
Restructuring | | — | | 476 | | — | |
Increase in restricted cash to secure operating lease | | (136 | ) | — | | — | |
Stock options and warrants issued for services | | 13 | | (21 | ) | 21 | |
Stock-based employee compensation | | 1,471 | | 5 | | 340 | |
Change in operating assets and liabilities: | | | | | | | |
Prepaid expenses and other assets | | (199 | ) | 207 | | 13 | |
Accounts payable | | (220 | ) | (135 | ) | 721 | |
Accrued liabilities | | 506 | | 331 | | (24 | ) |
Net cash used in operating activities | | (17,274 | ) | (16,467 | ) | (17,480 | ) |
Cash flows from investing activities: | | | | | | | |
Proceeds from sales and maturities of investment securities | | — | | 1,500 | | 10,875 | |
Purchases of investment securities | | (9,562 | ) | — | | (33 | ) |
Facilities and equipment purchases | | (385 | ) | (84 | ) | (326 | ) |
Purchase of licensed product | | (5,000 | ) | — | | (1,000 | ) |
Proceeds from sales of equipment and facilities | | 110 | | 303 | | — | |
Net cash (used in) provided by investing activities | | (14,837 | ) | 1,719 | | 9,516 | |
Cash flows from financing activities: | | | | | | | |
Net proceeds from issuance of common stock and warrants | | 58,485 | | 3,812 | | 9,042 | |
Proceeds from bridge note payable | | 3,460 | | — | | — | |
Proceeds from bank note payable | | 15,000 | | — | | — | |
Repayment of bank notes payable principal | | (4,584 | ) | (339 | ) | (290 | ) |
Decrease (increase) in restricted cash | | 1,000 | | (1,000 | ) | — | |
Payment of notes payable issuance costs | | (144 | ) | — | | — | |
Proceeds from stock options and warrants exercised | | 19 | | 44 | | 800 | |
Preferred stock dividends | | (500 | ) | (500 | ) | (500 | ) |
Net cash provided by financing activities | | 72,736 | | 2,017 | | 9,052 | |
Net increase (decrease) in cash and cash equivalents | | 40,625 | | (12,731 | ) | 1,088 | |
Cash and cash equivalents: | | | | | | | |
Beginning of year | | 3,523 | | 16,254 | | 15,166 | |
End of year | | $ | 44,148 | | $ | 3,523 | | $ | 16,254 | |
Supplemental disclosure of non-cash financing activity: | | | | | | | |
Purchase of Licensed Products with common stock | | $ | — | | $ | — | | $ | 1,000 | |
Accrual of preferred dividend | | 500 | | 500 | | 500 | |
Increase in Licensed Products with increase in current obligations payable | | 5,000 | | — | | — | |
Warrants issued and recognition of beneficial conversion feature in connection with debt issuance | | 4,000 | | — | | — | |
Conversion of bridge loan plus interest accrued thereon into common stock | | 3,524 | | — | | — | |
Supplemental disclosure of cash paid during the period for: | | | | | | | |
Cash paid for interest | | $ | 209 | | $ | 261 | | $ | 196 | |
See accompanying notes to the consolidated financial statements.
48
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ EQUITY
AND COMPREHENSIVE LOSS
(In thousands)
| | Preferred Stock,
Series 1 | | Preferred Stock,
Series B | | Common Stock | | | | | | | |
| | Number
of
Shares | | Par
Value | | Number
of
Shares | | Par
Value | | Number
of
Shares | | Par
Value | | Additional
Paid-In
Capital | | Accumu-
lated
Deficit | | Share-
holder’s
Equity | |
Balance, December 31, 2003 | | | 205 | | | | $ | 4 | | | | 2 | | | | $ | — | | | | 4,666 | | | | $ | 93 | | | | $ | 243,832 | | | $ | (214,439 | ) | $ | 29,490 | |
Exercise of stock options and warrants | | | — | | | | — | | | | — | | | | — | | | | 136 | | | | 3 | | | | 797 | | | — | | 800 | |
Common stock issued for licensed product | | | — | | | | — | | | | — | | | | — | | | | 41 | | | | 1 | | | | 999 | | | — | | 1,000 | |
Common stock issued, net of offering costs of $763 | | | — | | | | — | | | | — | | | | — | | | | 308 | | | | 6 | | | | 9,036 | | | — | | 9,042 | |
Modification of outstanding employee options | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 340 | | | — | | 340 | |
Stock options issued for services | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 21 | | | — | | 21 | |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | (19,371 | ) | (19,371 | ) |
Unrealized gain on investment securities | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | 6 | | 6 | |
Total comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | (19,365 | ) | (19,365 | ) |
Preferred stock dividends | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | (500 | ) | (500 | ) |
Balance, December 31, 2004 | | | 205 | | | | $ | 4 | | | | 2 | | | | $ | — | | | | 5,151 | | | | $ | 103 | | | | $ | 255,025 | | | $ | (234,304 | ) | $ | 20,828 | |
Exercise of stock options and warrants | | | — | | | | — | | | | — | | | | — | | | | 16 | | | | — | | | | 44 | | | — | | 44 | |
Common stock issued, net of offering costs of $337 | | | — | | | | — | | | | — | | | | — | | | | 553 | | | | 11 | | | | 3,801 | | | — | | 3,812 | |
Modification of outstanding employee options | | | — | | | | — | | | | — | | | | — | | | | — | | | | | | | | 6 | | | — | | 6 | |
Stock options issued for services | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (21 | ) | | — | | (21 | ) |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | (20,997 | ) | (20,997 | ) |
Unrealized gain on investment securities | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | 1 | | 1 | |
Total comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | (20,996 | ) | (20,996 | ) |
Preferred stock dividends | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | (500 | ) | (500 | ) |
Balance, December 31, 2005 | | | 205 | | | | $ | 4 | | | | 2 | | | | $ | — | | | | 5,720 | | | | $ | 114 | | | | $ | 258,855 | | | $ | (255,800 | ) | $ | 3,173 | |
Exercise of stock options and warrants | | | — | | | | — | | | | — | | | | — | | | | 6 | | | | — | | | | 19 | | | — | | 19 | |
Common stock issued, net of offering costs of $3,953 | | | — | | | | — | | | | — | | | | — | | | | 14,652 | | | | 293 | | | | 58,192 | | | — | | 58,485 | |
Conversion of bridge loan and interest accrued thereon into common stock | | | — | | | | — | | | | — | | | | — | | | | 839 | | | | 17 | | | | 3,507 | | | — | | 3,524 | |
Conversion of preferred shares into common stock | | | — | | | | — | | | | (2 | ) | | | — | | | | 1,591 | | | | 32 | | | | (32 | ) | | — | | — | |
Share-based employee compensation expense | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 1,572 | | | — | | 1,572 | |
Modification of outstanding employee options | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (101 | ) | | — | | (101 | ) |
Warrants issued and recognition of beneficial conversion feature in connection with issuance of debt | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 4,000 | | | — | | 4,000 | |
Stock options and warrants issued for services | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 13 | | | — | | 13 | |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | (23,294 | ) | (23,294 | ) |
Unrealized gain on investment securities | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | — | | — | |
Total comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | (23,294 | ) | (23,294 | ) |
Preferred stock dividends | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | (500 | ) | (500 | ) |
Balance, December 31, 2006 | | | 205 | | | | $ | 4 | | | | — | | | | $ | — | | | | 22,808 | | | | $ | 456 | | | | $ | 326,025 | | | $ | (279,594 | ) | $ | 46,891 | |
See accompanying notes to the consolidated financial statements.
49
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1. Organization and Operations
Poniard is a biotechnology company focused on the discovery, development and commercialization of cancer therapy products. The consolidated financial statements include the accounts of Poniard Pharmaceuticals, Inc. and its wholly owned subsidiary, NeoRx Manufacturing Group, Inc. (the Company).
The Company has historically suffered recurring operating losses and negative cash flows from operations. As of December 31, 2006, the Company had net working capital of $42,299,000, an accumulated deficit of $279,594,000 and total shareholders’ equity of $46,891,000. The Company’s total cash, cash equivalents and investment securities, net of restricted cash of $136,000, was $53,710,000 at December 31, 2006. The Company believes that its current cash, cash equivalent and investment securities balances will provide adequate resources to fund operations at least until the end of the first quarter of 2008.
All inter-company balances and transactions have been eliminated.
Unless otherwise indicated, all common stock related amounts have been adjusted to reflect the Company’s one-for-six reverse stock split effective September 22, 2006.
NOTE 2. Summary of Significant Accounting Policies
Estimates and Uncertainties: The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Research and Development Revenues and Expenses: Revenues from collaborative agreements are recognized as earned as the Company performs research activities under the terms of each agreement. Billings in excess of amounts earned are classified as deferred revenue. Pursuant to the Securities and Exchange Commission Staff Accounting Bulletin No. 104 (SAB 104), “Revenue Recognition in Financial Statements,” non-refundable upfront technology license fees, where the Company is providing continuing services related to product development, are deferred. Such fees are recognized as revenue over the product development periods based on estimated total development costs. If the Company is not providing continuing services, revenue is recognized when the payment is due.
To date, the Company does not have any significant ongoing revenue sources. Pursuant to SAB 104 and Emerging Issues Task Force Consensus No. 00-21, “Revenue Arrangements with Multiple Deliverables,” (EITF 00-21), which became effective for revenue arrangements entered into in fiscal periods beginning after June 15, 2003, revenues from sales and licensing of intellectual property and government grants are recognized as earned. To the extent that a transaction contains multiple deliverables, the Company determines whether the multiple deliverables are separable, and, if separable, the revenue to be allocated to each deliverable based on fair value. If fair value is undeterminable for undelivered elements of the arrangement, revenue is deferred over the contract period or until delivery, as applicable. The revenue allocated to each deliverable is recognized following the requirements of SAB 104.
The Company’s revenue in the periods presented consisted primarily of proceeds from the sale and licensing of intellectual property, milestone payments received, and receipt of government grants. For the sale and licensing of intellectual property and milestone payments, revenue has been recognized as payments are due because the Company has not had continuing service or other obligations subsequent to
50
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
the sale, licensing or milestone payment. Additionally, milestone payments are based on events that represent the achievement of substantive steps in the development process and are believed to represent the fair value of achieving the milestone. Government grant revenue is recognized as earned based on completion of performance under the respective contracts whereby no ongoing obligation on the part of the Company exists. Milestone payments are recognized as revenue at the time such payments are due, based on the ratio of cumulative costs incurred to date, to total estimated development costs. Any remaining balance is deferred and recognized as revenue over the remaining development period.
Research and development costs are expensed as incurred. It is the Company’s practice to offset third-party collaborative reimbursements received as a reduction of research and development expenses. Third-party reimbursements for 2006, 2005, and 2004 were $52,000, $16,000 and $259,000, respectively.
Cash Equivalents: All highly liquid investments with an original maturity of three months or less when purchased are considered to be cash equivalents. Cash equivalents represent cash invested primarily in money market funds, federal government and agency securities and corporate debt securities.
Investment Securities: The Company considers all investment securities as available-for-sale. All securities are carried at fair value. The Company does not invest in derivative financial instruments. Unrealized gains and losses on investment securities are reported as a component of comprehensive income or loss and classified as accumulated other comprehensive income or loss - unrealized gain (loss) on investment securities in shareholders’ equity. The Company monitors investment securities for other than temporary declines in fair value and charges impairment losses to income when an other than temporary decline in estimated value occurs.
Facilities and Equipment: Facilities and equipment are stated at acquired cost, less any charges for impairment. Depreciation is provided using the straight-line method over estimated useful lives of five to seven years for equipment and furniture, three years for computer equipment and software and thirty years for buildings. Leasehold improvements are amortized using the straight-line method over the shorter of the assets’ estimated useful lives or the terms of the leases.
Impairment of Long-Lived Assets: Long-lived assets including property and equipment are reviewed for possible impairment whenever significant events or changes in circumstances, including changes in the Company’s business strategy and plans, indicate that an impairment may have occurred. An impairment is indicated when the sum of the expected future undiscounted net cash flows identifiable to that asset or asset group is less than its carrying value. Impairment losses are determined from actual or estimated fair values, which are based on market values, net realizable values or projections of discounted net cash flows, as appropriate. The Company reviews long-lived assets annually and on an as-needed basis to determine if there have been any adverse events or circumstances that would indicate that an impairment exists. As a result of these reviews, the Company recorded an impairment charge related to the restructuring activities during 2005 and an additional impairment charge in 2006. See Note 10 below for further details.
Debt Issuance Costs: Costs incurred in connection with the securing of long-term bank loans and other long-term debt are deferred and amortized as interest expense over the term of the related debt using a method that approximates the effective interest method.
Licensed Products: Licensed Products represent an exclusive license to develop, manufacture and commercialize picoplatin, a platinum-based anti-cancer agent. Licensed Products are amortized using the straight-line method over their estimated useful life of twelve years. The Company evaluates the
51
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
recoverability of Licensed Products periodically and takes into account events or circumstances that might indicate that an impairment exists. No impairment of Licensed Products was identified during 2006.
Income Taxes: The Company computes income taxes using the asset and liability method, under which deferred income taxes are provided for the temporary differences between the financial reporting basis and the tax basis of the Company’s assets and liabilities and for operating loss and tax credit carryforwards. A valuation allowance is established when necessary to reduce deferred tax assets to the amount, if any, which is expected more likely than not to be realized.
Net Loss Per Common Share: Basic and diluted loss per share are based on net loss applicable to common shares, which is comprised of net loss and preferred stock dividends in all periods presented. Shares used to calculate basic loss per share are based on the weighted average number of common shares outstanding during the period. Shares used to calculate diluted loss per share are based on the potential dilution that would occur upon the exercise or conversion of securities into common stock using the treasury stock method. The computation of diluted net loss per share excludes the following options and warrants to acquire shares of common stock for the years indicated because their effect would not be dilutive.
| | 2006 | | 2005 | | 2004 | |
Common Stock options | | 1,660,000 | | 721,000 | | 588,000 | |
Common Stock warrants | | 5,947,000 | | 538,000 | | 265,000 | |
Additionally, aggregate shares of 39,015, issuable as of December 31, 2006 upon conversion of the Company’s Series 1 convertible exchangeable preferred stock, are not included in the calculation of diluted loss per share for 2006, 2005 and 2004 because the share increments would not be dilutive. Aggregate shares of 574,398, issuable as of December 31, 2005 upon conversion of the Company’s Series B convertible preferred stock are not included in the calculation of diluted loss per share for 2005 and 2004 because the share increments would not be dilutive. All outstanding shares of the Company’s Series B convertible preferred stock were converted into the Company’s common stock and retired in April 2006.
Share-Based Compensation: Effective January 1, 2006, the Company adopted the provisions of Statement of Financial Accounting Standards (SFAS) No. 123 (revised 2004), “Share-Based Payment” (SFAS 123R), which establishes accounting for equity instruments exchanged for employee services. Under the provisions of SFAS 123R, share-based compensation cost is measured at the grant date, based on the fair value of the award, and is recognized as an expense over the employee’s requisite service period (generally the vesting period of the equity grant). Prior to January 1, 2006, the Company accounted for share-based compensation to employees in accordance with Accounting Principles Board Opinion No. 25, “Accounting for Stock Issued to Employees” (APB 25), and related interpretations. The Company also followed the disclosure requirements of SFAS No. 123, as amended by SFAS No. 148, “Accounting for Stock-Based Compensation Transition and Disclosure”. The Company elected to adopt the modified prospective transition method as provided by SFAS 123R and, accordingly, financial statement amounts for the prior years presented have not been restated to reflect the fair value method of expensing share-based compensation.
Concentration in the Available Sources of Supply of Materials: For the Company’s picoplatin product candidate to be successful, the Company needs sufficient, reliable and affordable supplies of the picoplatin active pharmaceutical ingredient (API) and finished drug product. Sources of picoplatin API and finished drug product may be limited, and third-party suppliers may be unable to manufacture API and drug
52
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
product in amounts and at prices necessary to successfully commercialize the Company’s picoplatin product. Moreover, third-party manufacturers must continuously adhere to current Good Manufacturing Practice (cGMP) regulations enforced by the FDA through its facilities inspection program. If the facilities of these manufacturers cannot pass a pre-approval plant inspection, the FDA will not grant a New Drug Application (NDA) for the Company’s proposed products. In complying with cGMP and foreign regulatory requirements, any of our third-party manufacturers will be obligated to expend time, money and effort in production, record-keeping and quality control to assure that the Company’s products meet applicable specifications and other requirements. If any of the Company’s third-party manufacturers or suppliers fails to comply with these requirements, the Company may be subject to regulatory action.
The Company relies on third parties to manufacture its picoplatin API and finished drug product for its clinical trials. The Company currently has separate agreements with one supplier each of API and finished drug product. Manufacturing services under these agreements are provided on a purchase order, fixed-fee basis. Unless earlier terminated, each agreement continues for an initial term ending December 31, 2009, and may be extended beyond the initial term upon agreement of the parties. The agreements generally provide that they may be terminated by either party if there is an uncured material breach by the other party or in the event of insolvency or bankruptcy of the other party. The Company may terminate the finished drug product supply agreement at any time with one year’s advance notice. The Company may terminate the API manufacturing agreement if there is a change in control of the manufacturer. The Company has no assurance that its current suppliers will be able to manufacture sufficient picoplatin API and/or finished drug product on a timely or cost-effective basis at all times in the future. The Company believes that there are other contract manufacturers with the capacity to manufacture picoplatin API and finished drug product. If the Company is required to seek out alternative manufacturers, it may incur significant additional costs and suffer delays in, or be prevented from, completing or initiating its ongoing or planned clinical trials.
Fair Value of Financial Instruments: The Company has financial instruments consisting of cash, cash equivalents, restricted cash, investment securities, notes receivable, accounts payable and notes payable. The fair value of all of the Company’s financial instruments, based on either the short-term nature of the instrument, current market indicators or quotes from brokers, approximates their carrying amounts.
Segment Reporting: The Company has one operating business segment, cancer therapeutics development.
New Accounting Pronouncements: In May 2005, the Financial Accounting Standards Board (FASB) issued SFAS No. 154, “Accounting Changes and Error Corrections, a Replacement of APB Opinion No. 20 and FASB Statement No. 3” (SFAS 154). SFAS 154 establishes, unless impracticable, retrospective application as the required method for reporting a change in accounting principle in the absence of explicit transition requirements specific to a newly adopted accounting principle. Previously, most changes in accounting principle were recognized by including the cumulative effect of the change to the new accounting principle in net income of the period of the change. Under SFAS 154, retrospective application requires the cumulative effect of the change to be reflected in the carrying amounts of assets and liabilities as of the beginning of the first period presented and financial statements for each individual prior period presented to be adjusted to reflect the effects of applying the new accounting principle. SFAS 154 carries forward the guidance in APB Opinion 20 “Accounting Changes,” requiring justification of a change in accounting principle on the basis of preferability, for reporting the correction of an error in previously issued financial statements and for a change in an accounting estimate. SFAS 154 is effective for
53
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
accounting changes and corrections of errors made in fiscal years beginning after December 15, 2005. The adoption of the Statement by the Company on January 1, 2006 did not have a material impact on the Company’s consolidated financial statements.
In December 2004, the FASB issued SFAS No.123R, “Share-Based Payment.” SFAS 123R replaces SFAS 123, “Stock-Based Compensation,” issued in 1995. SFAS 123R requires that the fair value of the grant of employee stock options be reported as an expense in the results of operations. SFAS 123R eliminates the ability to account for stock-based compensation using APB 25 and requires that such transactions be recognized as compensation cost in the income statement based on their fair value on the measurement date, which is generally the date of the grant. The Statement is effective for the first annual reporting period that begins after June 15, 2005. Historically, the Company has disclosed in its footnotes the pro forma expense effect of the grants (see Note 3). SFAS 123R applies to all outstanding, unvested option grants as of the effective date. The Company adopted SFAS No. 123R on January 1, 2006 using the modified prospective transition method. Under this method, SFAS 123R applies to new and modified option grants after January 1, 2006, and to any unvested option grants as service is rendered on or after the effective date. The attribution of compensation cost for vested option grants as of January 1, 2006 is based on the same method and on the same grant-date fair values previously determined for the pro forma disclosures required for companies that did not adopt the fair value accounting method for stock-based employee compensation. See Note 3 below for the discussion of the effect of the adoption of this Statement on the Company’s consolidated financial statements.
In June 2006, the FASB issued Interpretation No. 48, “Accounting for Uncertainty in Income Taxes: An Interpretation of FASB Statement No. 109” (FIN 48). FIN 48 clarifies certain aspects of accounting for uncertain tax positions, including issues related to the recognition and measurement of those tax positions. FIN 48 is effective for fiscal years beginning after December 15, 2006. Accordingly, the Company will adopt FIN 48 on January 1, 2007. The Company is currently assessing the impact FIN 48 will have, if any, on its consolidated financial statements.
In September 2006, the FASB issued SFAS No. 157, “Fair Value Measurements”. SFAS 157 defines fair value, establishes a framework for measuring fair value, and expands disclosure requirements about fair value measurements. SFAS 157 applies to other accounting pronouncements that require or permit fair value measurements, but does not in itself require any new fair value measurements. The provisions of SFAS No. 157 are effective for fiscal years beginning after November 15, 2007 and interim periods within those fiscal years. The Company is currently evaluating this statement and its impact, if any, on the Company’s consolidated financial statements.
In September 2006, the SEC issued Staff Accounting Bulletin No. 108, “Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements” (SAB 108), to address diversity in practice in quantifying financial statement misstatements. SAB 108 requires the quantification of misstatements based on their impact on both the balance sheet and the income statement to determine materiality. The guidance provides for a one-time cumulative effect adjustment to correct for misstatements that were not deemed material under a company’s prior approach but are material under the SAB 108 approach. SAB 108 is effective for fiscal years ending after November 15, 2006. The adoption of this guidance did not have a material impact on the Company’s consolidated financial statements.
54
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
NOTE 3. Stock-Based Compensation
Effective January 1, 2006, the Company adopted the provisions of SFAS 123R, using the modified prospective transition method. Under the provisions of SFAS 123R, share-based compensation cost is measured at the grant date, based on the fair value of the award, and is recognized as an expense over the employee’s requisite service period (generally the vesting period of the equity grant). Prior to January 1, 2006, the Company accounted for share-based compensation to employees in accordance with APB 25 and related interpretations.
Under SFAS 123R, the Company is required to select a valuation technique or option-pricing model that meets the criteria as stated in SFAS 123R, which includes a binomial model and the Black-Scholes-Merton (Black-Scholes) model. At the present time, the Company is continuing to use the Black-Scholes model. The adoption of SFAS 123R, applying the modified prospective transition method, as elected by the Company, requires the Company to value stock options prior to its adoption of SFAS 123R under the fair value method and expense these amounts over the stock options’ remaining vesting period. Under this transition method, compensation expense recognized during the year ended December 31, 2006 included compensation expense for all share-based awards granted prior to, but not yet vested, as of December 31, 2005, based on the grant date fair value estimated in accordance with the original provisions of SFAS 123. In accordance with the modified prospective transition method, the Company’s consolidated financial statements for years ended prior to January 1, 2006 have not been restated to reflect the impact of SFAS 123R.
As a result of adopting SFAS 123R on January 1, 2006, the Company’s loss from operations and net loss for the year ended December 31, 2006 is $1,471,000 higher than if it had continued to account for share-based compensation under the recognition and measurement provisions of APB 25, and related interpretations, as permitted by SFAS 123. Basic and diluted net loss per share for the year ended December 31, 2006 would have been $1.29 if the Company had not adopted SFAS 123R.
Had compensation cost for these stock options for employees been determined prior to January 1, 2006 using the fair value based method of accounting under SFAS No. 123, the Company’s net loss applicable to common shares and loss per share would have been the pro forma amounts indicated below (in thousands, except per share data):
| | Year ended
December 31, | |
| | 2005 | | 2004 | |
Net loss applicable to common shares: | | | | | |
As reported | | $ | (21,497 | ) | $ | (19,871 | ) |
Add: Stock-based employee compensation expense included in reported net loss | | 5 | | 340 | |
Deduct: Stock-based employee compensation determined under fair value based method for all awards | | (1,189 | ) | (1,666 | ) |
Pro forma | | $ | (22,681 | ) | $ | (21,197 | ) |
Loss per common share, basic and diluted: | | | | | |
As reported | | $ | (3.83 | ) | $ | (3.96 | ) |
Pro forma | | $ | (4.04 | ) | $ | (4.22 | ) |
55
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Disclosures for the year ended December 31, 2006 are not presented because stock-based payments were accounted for under SFAS 123R’s fair value method during this period.
The Company modified certain stock options, which had been granted to a member of the Company’s board of directors, so that such stock options would fully vest as of August 14, 2006, the date that the director retired from the board. No other modifications were made to these stock options. No other stock options held by the former director, all of which were fully vested as of August 14, 2006, were modified. The effect of this modification was a decrease in total stock compensation expense of $101,000 for the year ended December 31, 2006.
For the year ended December 31, 2006, the Company recognized stock compensation expense of $1,471,000. This amount reflects the modification of stock options described above. The remaining unrecognized compensation cost related to unvested awards at December 31, 2006, was approximately $5,038,000 and the weighted-average period of time over which this cost will be recognized is 2.2 years.
The Company records compensation expense for employee stock options based on the estimated fair value of the options on the date of grant using the Black-Scholes option-pricing model. This fair value is amortized on a straight-line basis over the requisite service periods for the grants, which is generally the vesting period. The Company uses historical data, and other related information as appropriate, to estimate the expected price volatility, the expected option life and the expected forfeiture rate. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of the grant. The weighted-average fair value per share of the Company’s stock options granted to employees was estimated to be $5.49, $4.46 and $11.22 for the years ended December 31, 2006, 2005 and 2004, respectively, using the Black-Scholes model with the following weighted-average assumptions:
| | Year ended December 31, | |
| | 2006 | | 2005 | | 2004 | |
Expected life in years | | 6.82 | | 4.00 | | 4.00 | |
Expected dividend rate | | 0.0 | % | 0.0 | % | 0.0 | % |
Risk-free interest rate | | 5.0 | % | 3.9 | % | 3.4 | % |
Expected volatility | | 105.0 | % | 122.9 | % | 120.9 | % |
56
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
The Company issues new shares of common stock upon exercise of stock options. A summary of option activity as of December 31, 2006 and changes during the three years then ended are as follows (shares and intrinsic value in thousands):
| | Number of
Shares | | Weighted
Average
Exercise
Price | | Weighted
Average
Remaining
Contractual
Term | | Aggregate
Intrinsic Value | |
Outstanding at December 31, 2003 | | | 675 | | | | $ | 22.05 | | | | | | | | | | |
Granted | | | 233 | | | | 14.25 | | | | | | | | | | |
Exercised | | | (136 | ) | | | 5.88 | | | | | | | | | | |
Forefeited/cancelled/expired | | | (182 | ) | | | 30.63 | | | | | | | | | | |
Outstanding at December 31, 2004 | | | 590 | | | | 20.06 | | | | | | | | | | |
Exercisable at December 31, 2004 | | | 309 | | | | 24.69 | | | | | | | | | | |
Granted | | | 262 | | | | 6.07 | | | | | | | | | | |
Exercised | | | (16 | ) | | | 2.83 | | | | | | | | | | |
Forefeited/cancelled/expired | | | (115 | ) | | | 15.03 | | | | | | | | | | |
Outstanding at December 31, 2005 | | | 721 | | | | 16.15 | | | | | | | | | | |
Exercisable at December 31, 2005 | | | 396 | | | | 22.42 | | | | | | | | | | |
Granted | | | 1,046 | | | | 6.44 | | | | | | | | | | |
Exercised | | | (6 | ) | | | 3.17 | | | | | | | | | | |
Forefeited/cancelled/expired | | | (101 | ) | | | 9.15 | | | | | | | | | | |
Outstanding at December 31, 2006 | | | 1,660 | | | | $ | 10.50 | | | | 8.2 | | | | $ | 366 | | |
Exercisable at December 31, 2006 | | | 587 | | | | $ | 17.55 | | | | 6.3 | | | | $ | 196 | | |
Information relating to stock options outstanding and exercisable at December 31, 2006 is as follows (in thousands, except per share data):
| | Options Outstanding | | Options Exercisable | |
Range of Exercise Prices | | | | Number
of Shares | | Weighted
Average
Remaining
Life in Years | | Weighted
Average
Exercise
Price | | Number
of Shares | | Weighted
Average
Exercise
Price | |
$2.82 - $4.62 | | | 343 | | | | 8.48 | | | | $ | 3.93 | | | | 132 | | | | $ | 3.52 | | |
$5.04 - $6.48 | | | 475 | | | | 9.47 | | | | 6.19 | | | | 20 | | | | 6.42 | | |
$6.77 - $7.44 | | | 68 | | | | 9.31 | | | | 7.00 | | | | 11 | | | | 7.44 | | |
$7.50 - $7.50 | | | 335 | | | | 9.31 | | | | 7.50 | | | | 56 | | | | 7.50 | | |
$7.68 - $109.50 | | | 439 | | | | 5.67 | | | | 23.14 | | | | 368 | | | | 25.01 | | |
| | | 1,660 | | | | 8.22 | | | | $ | 10.50 | | | | 587 | | | | $ | 17.55 | | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Cash proceeds and intrinsic value related to total stock options exercised during the years ended December 31, 2006, 2005 and 2004 are provided in the following table (dollars in thousands):
| | Year ended December 31, | |
| | 2006 | | 2005 | | 2004 | |
Proceeds from stock options exercised | | | $ | 19 | | | | $ | 44 | | | $ | 800 | |
Intrinsic value of stock options exercised | | | $ | 11 | | | | $ | 50 | | | $ | 1,328 | |
57
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
In connection with various consulting and service contracts, the Company has issued stock options to non-employees. These options are valued using a Black-Scholes option-pricing model and the total cost of the stock options are recognized over the service period. Stock options to purchase 3,333, 1,666, 8,333 and 19,166 shares of common stock were granted during 2006, 2005, 2004 and 2002, respectively. The Company recorded compensation expense (credits) of $8,000, $(21,000), and $21,000 during 2006, 2005, and 2004, respectively, due to these grants.
In April 2004, the Company extended to December 31, 2004 the time to exercise stock options, held by a former officer, to acquire approximately 26,666 shares of common stock. The Company recorded general and administrative compensation expense of $322,000 in connection with this extension. Also in April 2004, in connection with a consulting agreement with a former employee, the Company extended the vesting of the stock options to acquire approximately 10,666 shares of common stock. The Company recorded research and development compensation expense of $15,000 in connection with this extension.
NOTE 4. Liquidity and Capital Resources
The Company has financed its operations primarily through the sale of equity securities, technology licensing, collaborative agreements and debt instruments. The Company invests excess cash in investment securities that will be used to fund future operating costs. Cash used for operating activities for the twelve months ended December 31, 2006 totaled $17,274,000. Revenues and other income sources for 2006 were not sufficient to cover operating expenses.
On April 26, 2006, the Company completed a $65,000,000 equity financing, pursuant to which it issued to a group of institutional and other accredited investors an aggregate of 15,491,000 shares of common stock at a cash purchase price of $4.20 per share. Investors in the financing also received five-year warrants to purchase an aggregate of 4,643,000 shares of common stock at an exercise price of $4.62 per share. The Company received $61,945,000 in net proceeds from the financing, which it is using to fund its picoplatin clinical program and general working capital needs. Concurrent with the closing of the financing, the Company issued an aggregate of 1,591,000 shares of common stock to the holders of its Series B preferred stock upon conversion of its outstanding Series B preferred shares. As a result of the completion of the financing and the conversion of the Series B preferred shares, the Company’s outstanding common stock increased from approximately 5,722,000 shares to approximately 22,804,000 shares.
In connection with the financing, the Company entered into a letter agreement with Texas State Bank, pursuant to which the Company agreed to accelerate the maturity date of its promissory note with the Bank to June 5, 2006. The Texas State Bank note, which was secured by the Company’s radiopharmaceutical plant and other STR assets located in Denton, Texas, had an adjustable interest rate equal to the bank prime rate reported in the Wall Street Journal (8.00% at May 23, 2006). The Company paid off the outstanding balance of the note, $2,714,000, on May 23, 2006.
The Company completed the relocation of its corporate headquarters to South San Francisco in September 2006. The Company intends to maintain its current clinical and development and support activities in Seattle. The addition of 17,045 square feet of office and laboratory space leased in South San Francisco will result in a substantial increase in the Company’s future rent and operating costs. Under the lease agreement dated July 10, 2006, the annual base rent for the leased facilities is approximately $542,000 and is subject to annual adjustment based on disbursements for tenant improvements and increases in the Consumer Price Index in the San Francisco metropolitan market. Base rent is payable in monthly installments of approximately $45,200. Additional rent is payable monthly based on the Company’s share of operating expenses of the project in which the leased facilities are located, as described in the lease
58
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
agreement. Monthly base rent during the first seven months of the lease will average $21,000 during the construction of tenant improvements. The Company paid total rent (base rent and additional rent based on the Company’s share of project operating expenses) during 2006 of $143,000. The Company estimates total rent payable during 2007 will be approximately $523,000. Annual operating expenses under the lease may increase substantially as the Company moves forward with its plans to establish laboratory facilities in the leased space. The initial term of the lease is 60 months. The Company may, upon written notice delivered at least nine months prior to expiration of the initial lease term, extend the lease for an additional three years, with rent payable at the then market rate.
In April 2004, the Company acquired the worldwide exclusive rights, excluding Japan, to develop, manufacture and commercialize picoplatin from AnorMED, Inc. AnorMED was acquired by Genzyme Corporation in November 2006. Under the terms of the original agreement, the Company paid a one-time upfront milestone payment of $1,000,000 in common stock and $1,000,000 in cash. The original agreement provided for development and commercialization milestone payments of up to $13,000,000, payable in cash or a combination of cash and Company common stock, and a royalty rate of up to 15% of net product sales after regulatory approval. The parties executed an amendment to the license agreement on September 18, 2006, modifying several key financial terms and expanding the licensed territory to include Japan, thereby providing the Company worldwide rights. In consideration of the amendment, the Company paid Genzyme $5,000,000 in cash on October 12, 2006 and will pay Genzyme an additional $5,000,000 in cash by March 31, 2007. The amendment eliminates $8,000,000 in development milestone payments to Genzyme. Genzyme remains entitled to receive up to $5,000,000 in commercialization milestones upon the attainment of certain levels of annual net sales of picoplatin after regulatory approval. The amendment also reduces the royalty payable to Genzyme to a maximum of 9% of annual net product sales. In addition, the amendment reduces the sharing of sublicense revenues for any sublicenses entered into during the first year following the amendment and eliminates the sharing of sublicense revenues on and after September 18, 2007.
On October 25, 2006, the Company entered into a loan and security agreement with Silicon Valley Bank and Merrill Lynch Capital, pursuant to which it obtained a $15,000,000 capital loan. A portion of the proceeds of the loan will be used to fund the Company’s cash payment obligations to Genzyme under the amended license agreement described above. The remaining proceeds will be used to support the Company’s strategic growth, late-stage clinical trials of picoplatin and general working capital needs. The loan is for a term of 42 months and matures on April 1, 2010. The Company is required to pay a 7.67% fixed interest rate on the outstanding principal balance plus a $1,350,000 additional payment on the maturity date of the loan. This additional payment will be accreted to the note payable balance over the term of the loan using the effective interest rate method and reflected as additional interest expense. All interest payable under the loan agreement and the full amount of the additional payment must be paid upon any prepayment of the loan. The loan is secured by a first lien on substantially all of the Company’s non-intellectual property assets. The loan agreement contains restrictions on the Company’s ability to, among other things, dispose of certain assets, engage in certain mergers and acquisition transactions, incur indebtedness, create liens on assets, make investments and pay dividends or repurchase stock. The loan agreement also contains covenants requiring the Company to maintain unrestricted cash of $7,500,000 during the loan term and, not later than December 31, 2007, to provide evidence of positive Phase II data for the picoplatin drug development program and commence enrollment of patients in a Phase III trial for picoplatin. The loan contains events of default that include, among other things, nonpayment of principal and interest or fees, breaches of covenants, material adverse changes, bankruptcy and insolvency events,
59
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
cross defaults to other indebtedness, material judgments, inaccuracy of representations and warranties, and events constituting a change of control. The occurrence of an event of default would increase the applicable rate of interest by 5% and could result in acceleration of the Company’s payment obligations under the loan agreement.
On August 4, 2005, the Company entered into a research funding and option agreement with The Scripps Research Institute (TSRI). Under the agreement, as amended in December 2006, the Company committed to provide TSRI an aggregate of $2,500,000 over a 26-month period to fund research relating to synthesis and evaluation of novel small molecule, multi-targeted protein kinase inhibitors and focal adhesion kinase inhibitors as therapeutic agents, including for the treatment of cancer. The Company has the option to negotiate a worldwide exclusive license to use, enhance and develop any compounds arising from the collaboration. The research funding is payable by the Company to TSRI quarterly in accordance with a negotiated budget. On August 8, 2005, the Company made an initial funding payment to TSRI of $137,500. The Company paid TSRI total funding payments of $1,012,500 in 2006, which amount was charged to research and development expense. The agreement provides for aggregate additional funding of $1,350,000 in 2007. The Company has no assurance that the research funded under this arrangement will be successful or ultimately will give rise to any viable product candidates. Further, there can be no assurance that the Company will be able to negotiate, on acceptable terms, a license with respect to any compounds arising from the collaboration.
The Company terminated its STR manufacturing operations in Denton, Texas during the second quarter of 2005 and began actively marketing the facility for sale. In 2005, the Company recorded costs associated with the closure and maintenance of the Denton facility totaling $499,000. The Company recorded costs totaling $286,000 in 2006 related to these activities. See Note 9 below for additional information regarding the Company’s restructuring.
The Company received approximately $3,812,000 in net proceeds from the sale of common stock and warrants in a private placement transaction in March 2005. The Company has applied the net proceeds from this financing to support its Phase II trial in picoplatin in small cell lung cancer and for general working capital, including restructuring costs associated with the termination of its STR development program. The Company raised approximately $9,042,000 in net proceeds from the sale of common stock and warrants in a private placement transaction in February 2004. The net proceeds from this financing were used to support the Company’s STR development program and for general working capital.
In April 2004, the Company sold and transferred its Pretarget intellectual property to Aletheon Pharmaceuticals, Inc. Under the agreement, the Company could receive up to $6,600,000 in milestone payments if Aletheon achieves certain development goals, plus royalties on potential future product sales. The Company did not receive any upfront consideration for the sale of the Pretarget property. The Company discontinued its clinical studies using the Pretarget technology in July 2002, and sought, both through targeted inquiries and a broad-based auction process, a buyer or licensee for the technology. The sale of the Pretarget intellectual property relieved the Company of the annual costs associated with maintaining the Pretarget patent estate. During 2003, the Company spent approximately $350,000 for the prosecution and maintenance of the Pretarget patents and trademarks. For 2004, these costs were approximately $70,000. Seattle-based Aletheon is a development stage biotherapeutics company founded by two former Poniard employees. The timing and amount of milestone payments, if any, are uncertain. The terms of the transaction were determined through arms-length negotiation.
60
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
The Company had cash, cash equivalents and investment securities totaling $53,710,000 at December 31, 2006. Company management believes that current cash, cash equivalent and investment securities balances will provide adequate resources to fund operations at least until the end of the first quarter of 2008. The Company’s actual capital requirements will depend upon numerous factors, including:
· the scope and timing of the Company’s picoplatin clinical program and other research and development efforts, including the progress and costs of the Company’s on-going Phase II and planned Phase III trials of picoplatin in small cell lung cancer;
· the Company’s ability to obtain clinical supplies of picoplatin active pharmaceutical ingredient and drug product in a timely and cost-effective manner;
· actions taken by the FDA and other regulatory authorities;
· the timing and amounts of proceeds from any sale of the Denton facility and assets;
· the timing and amount of any milestone or other payments the Company might receive from potential strategic partners;
· the Company’s degree of success in commercializing picoplatin or any other cancer therapy product candidates;
· the emergence of competing technologies and products, and other adverse market developments;
· the acquisition or in-licensing of other products or intellectual property;
· the costs, including lease and operating costs, incurred in connection with the Company’s relocation of its corporate headquarters to South San Francisco and the planned expansion of its workforce;
· the costs of any research collaborations or strategic partnerships established;
· the costs of preparing, filing, prosecuting, maintaining and enforcing patent claims and other intellectual property rights; and
· the costs of performing the Company’s obligations under the loan with Silicon Valley Bank and Merrill Lynch Capital, including the cost of interest and other payment obligations and penalties and the cost of complying with unrestricted cash, product development and other covenants and restrictions under the loan agreement.
The Company had net operating loss carryforwards of approximately $62,300,000 available for future use as of December 31, 2006, which will expire from 2007 through 2026. During 2006, the Company experienced significant changes to its capital structure which resulted in an ownership change, as defined under Section 382 of the Internal Revenue Code of 1986, as amended (IRC). Consequently, the amount of net operating loss carryforwards and research and experimentation credit carryforwards available to be used in future years are limited under IRC Sections 382 and 383. This limitation will result in the loss of approximately $93,300,000 of the Company’s net operating loss carryforwards and $9,100,000 of the Company’s research and development credit carryforwards.
There can be no assurance that the Company will be able to raise additional capital or enter into relationships with corporate partners on a timely basis, on favorable terms, or at all. Conditions in the capital markets in general, and in the life science capital market specifically, may affect the Company’s potential financing sources and opportunities for strategic partnering.
61
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
NOTE 5. Restricted Cash
At December 31, 2006, the Company had restricted cash of $136,000, in the form of a certificate of deposit. The certificate of deposit serves as collateral for a standby letter of credit issued by Silicon Valley Bank on behalf of the Company. At December 31, 2005 $1,000,000, in the form of a certificate of deposit, was restricted. Pursuant to the terms of a letter agreement with Texas State Bank, the Bank, on January 31, 2005, applied the $1,000,000 cash collateral and all interest accrued thereon to the outstanding balance of the loan.
NOTE 6. Investment Securities
Investment securities consisted of the following (in thousands):
| | December 31, | |
| | 2006 | | 2005 | |
Corporate debt securities | | 6,964 | | | — | | |
Federal government and agency securities | | 2,598 | | | — | | |
| | 9,562 | | | — | | |
Unrealized gains and losses at December 31, 2006 are as follows (in thousands):
| | Amortized | | Gross Unrealized | | Fair Market | |
| | Cost | | Gains | | (Losses) | | Value | |
Corporate debt securities | | | $ | 6,964 | | | | $ | — | | | | $ | — | | | | $ | 6,964 | | |
Federal government and agency securities | | | 2,598 | | | | — | | | | — | | | | 2,598 | | |
| | | $ | 9,562 | | | | $ | — | | | | $ | — | | | | $ | 9,562 | | |
Net unrealized loss | | | | | | | | | | | $ | — | | | | | | |
All of the debt securities owned by the Company at December 31, 2006 had maturities of less than one year.
NOTE 7. Accrued Liabilities
Accrued liabilities consist of the following (in thousands):
| | December 31, | |
| | 2006 | | 2005 | |
Clinical trials | | $ | 1,444 | | $ | 447 | |
Accrued expenses | | 312 | | 289 | |
Compensation | | 618 | | 743 | |
Restructuring | | — | | 217 | |
Decommissioning costs | | — | | 73 | |
Severance | | 10 | | 250 | |
Other | | 136 | | 59 | |
| | $ | 2,520 | | $ | 2,078 | |
62
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
NOTE 8. Notes Payable
On October 25, 2006, the Company entered into a loan and security agreement (the loan agreement) with Silicon Valley Bank and Merrill Lynch Capital. Under the loan agreement, the Company received capital loan proceeds of $15,000,000 on October 31, 2006. The Company plans to use the proceeds of the loan to fund its cash payment obligations to Genzyme Corporation under the amended license amendment described in Note 13 below and to support the Company’s strategic growth, late-stage clinical trials of picoplatin and general working capital needs. The term of the loan is 42 months, maturing on April 1, 2010, with the first monthly principal payment on November 1, 2006. The Company is required to pay a 7.67% fixed interest rate on the outstanding principal balance plus a $1,350,000 additional payment on the maturity date of the loan. This additional payment will be accreted to the note payable balance over the term of the loan using the effective interest rate method and reflected as additional interest expense. Principal and interest paid on the note during the year ended December 31, 2006 totaled $813,000. In connection with the loan agreement, the Company issued five-year warrants to purchase an aggregate of 174,418 shares of common stock at an exercise price of $4.30 per share. The fair value of the warrants using the Black-Scholes option-pricing model was approximately $611,000 based upon assumptions of expected volatility of 112%, a contractual term of five years, an expected dividend rate of zero and a risk-free rate of interest of 4.75%. The portion of the loan proceeds allocable to the warrants is $540,000 based on their relative fair value, which the Company recorded as additional discount to notes payable. The total discount of $1,890,000 is amortized to interest expense using an effective interest rate of 13.7%. All interest payable under the loan agreement and the full amount of the additional payment must be paid upon any prepayment of the loan. The loan is secured by a first lien on substantially all of the non-intellectual property assets of the Company.
The loan agreement contains restrictions on the Company’s ability to, among other things, dispose of certain assets, engage in certain mergers and acquisition transactions, incur indebtedness, create liens on assets, make investments and pay dividends or repurchase stock. The loan agreement also contains covenants requiring the Company to maintain unrestricted cash of $7,500,000 during the loan term and, not later than December 31, 2007, to provide evidence of positive Phase II data for the picoplatin drug development program and commence enrollment of patients in a Phase III trial for picoplatin. The loan contains events of default that include, among other things, nonpayment of principal and interest or fees, breaches of covenants, material adverse changes, bankruptcy and insolvency events, cross defaults to other indebtedness, material judgments, inaccuracy of representations and warranties and events constituting a change of control. The occurrence of an event of default would increase the applicable rate of interest by 5% and could result in acceleration of the Company’s payment obligations under the loan agreement. The Company is not aware of any circumstances that indicate that an event of default is likely and, therefore, has not classified any portion of the non-current loan balance as current.
In connection with the Company’s 2001 purchase of the radiopharmaceutical manufacturing plant and other assets located in Denton, Texas, the Company assumed $6,000,000 principal amount of restructured debt held by Texas State Bank, McAllen, Texas. The loan, which matured in June 2006, was secured by the assets acquired in the transaction. Principal and interest paid on the note during the year ended December 31, 2006 totaled $3,980,000.
63
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Notes payable maturities as of December 31, 2006 are as follows (in thousands):
Year | | | | | |
2007 | | $ | 3,906 | |
2008 | | 4,218 | |
2009 | | 4,560 | |
2010 | | 2,950 | |
| | 15,634 | |
Less: discount | | (1,753 | ) |
| | $ | 13,881 | |
NOTE 9. Restructuring
In May and June 2005, the Company restructured its operations and reduced its workforce by approximately 50% in connection with the implementation of its restructuring plan to refocus its resources on the development of picoplatin and discontinue its STR development program. The employees terminated as part of the reduction of staff were no longer with the Company at December 31, 2005 and did not provide future services to the Company. The Company incurred termination benefits charges of totaling $892,000 related to the reduction in staff in May and June 2005. Of this amount, $250,000 remained unpaid as of December 31, 2005 and is included in accrued expenses in the consolidated balance sheet as of December 31, 2005. This amount was paid during 2006. The Company incurred additional non-employee charges totaling $612,000 related to the discontinuation of its STR clinical trials and the closure of its radiopharmaceutical manufacturing plant and STR research facilities, primarily consisting of contract termination and decommissioning costs. The Company recorded additional charges of $237,000 for decommissioning costs during the third and fourth quarters of 2005 due to anticipated increased waste disposal costs at its radiopharmaceutical manufacturing plant in Denton, Texas and anticipated increased STR study finalization costs. Total non-employee charges totaled $849,000. Of this amount, $217,000 remained unpaid as of December 31, 2005 and is included in accrued expenses in the consolidated balance sheet as of December 31, 2005. This amount was paid during 2006.
In conjunction with the Company’s strategic restructuring, in June 2005, the Company negotiated the early termination of its STR-related supply agreement with the University of Missouri Research Reactor facility group (MURR). The Company paid MURR a fee of $368,000 in connection with such early termination. The Company also paid MURR $190,000 in minimum purchase requirements under the agreement in 2005. These two amounts are included in the non-employee charges of $612,000 discussed above.
64
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
The following table summarizes the change in the restructuring accrual from initial recognition through December 31, 2006:
Description | | | | Initial
Restructuring
Charge | | Adjustment of
Restructuing
Charge | | Adjusted
Restructuring
Charge | | Payment of
Restructuring
Obligations | | Accrued
Restructuring
Charge as of
December 31,
2005 | | Payment of
Restructuring
Obligations | | Accrued
Restructuring
Charge as of
December 31,
2006 | |
Employee termination benefits | | | $ | 892,000 | | | | $ | — | | | | $ | 892,000 | | | | $ | (642,000 | ) | | | $ | 250,000 | | | | $ | (250,000 | ) | | | $ | — | | |
Contract termination costs | | | 378,000 | | | | (10,000 | ) | | | 368,000 | | | | (366,000 | ) | | | 2,000 | | | | (2,000 | ) | | | — | | |
Other termination costs | | | 234,000 | | | | 247,000 | | | | 481,000 | | | | (266,000 | ) | | | 215,000 | | | | (215,000 | ) | | | — | | |
Sub-total | | | 612,000 | | | | 237,000 | | | | 849,000 | | | | (632,000 | ) | | | 217,000 | | | | (217,000 | ) | | | — | | |
Total | | | $ | 1,504,000 | | | | $ | 237,000 | | | | $ | 1,741,000 | | | | $ | (1,274,000 | ) | | | $ | 467,000 | | | | $ | (467,000 | ) | | | $ | — | | |
NOTE 10. Asset Impairment Loss
In June 2005, the Company recognized an asset impairment loss of $3,346,000 on certain facilities and equipment resulting from the Company’s decisions to terminate its STR program. The loss on the Denton manufacturing facility and related equipment was determined based on an appraisal study commissioned by the Company, as well as management reviews with the assistance of outside commercial real estate brokers. The Company used a fair value of $3,300,000 for the Denton facility in determining the impairment loss. This valuation was the result of weighting the range of values in the appraisal study, which varied from $3,100,000 to $5,000,000. The loss on the equipment at the Seattle facility was determined based on estimates of potential sales values of used equipment. These impairment charges established new cost bases for the impaired assets, which are reported in Assets Held for Sale in current assets and other non-current assets on the consolidated balance sheets as of December 31, 2005 and 2006, respectively. All equipment in current Assets Held for Sale was disposed of as of December 31, 2006. Given the inherent uncertainty of the timing of a sale of the Denton facility, the Company has classified this asset as non-current.
As of December 31, 2006, the Company reduced the carrying value of Denton facility in non-current Assets Held for Sale based on a fair value of $2,800,000 and recognized additional impairment loss of $403,000. This valuation adjustment is based on the Company’s review of listing prices and completed sales of comparable properties in the region and the interest of prospective buyers. The Company continues to actively seek a buyer for the Denton facility.
The following table summarizes information related to the impairment charges:
Description | | | | Impairment
Loss | | Impaired
Carrying
Value as of
June 30,
2005 | | Disposals of
Assets | | Post
Impairment
Carrying Value
as of
December 31,
2005 | | Disposals
of Assets | | Post
Impairment
Loss | | Post
Impairment
Carrying Value
as of
December 31,
2006 | |
Equipment—Seattle, WA | | | $ | 155,000 | | | $ | 45,000 | | | $ | (44,000 | ) | | | $ | 1,000 | | | | $ | (1,000 | ) | | | $ | — | | | | $ | — | | |
Equipment—Manufacturing Facility, Denton, TX | | | 589,000 | | | 183,000 | | | (101,000 | ) | | | 82,000 | | | | (82,000 | ) | | | — | | | | — | | |
Manufacturing Facility—Denton, TX | | | 2,602,000 | | | 3,027,000 | | | — | | | | 3,027,000 | | | | — | | | | (403,000 | ) | | | 2,624,000 | | |
Total | | | $ | 3,346,000 | | | $ | 3,255,000 | | | $ | (145,000 | ) | | | $ | 3,110,000 | | | | $ | (83,000 | ) | | | $ | (403,000 | ) | | | $ | 2,624,000 | | |
65
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
NOTE 11. Leases
The Company leases the office and laboratory space for its principal locations under various leasing arrangements. In July 2006, Company entered into a five-year lease for office space and laboratory space in South San Francisco. The Company relocated its corporate headquarters to these facilities in September 2006. Base rental payments under this lease are subject to annual adjustment based on disbursements for tenant improvements and increases in the Consumer Price Index in the San Francisco metropolitan market (CPISF). The first adjustment in annual rent will occur in July 2007. Additional rental payments under this lease are paid based on the Company’s share of operating expenses of the project in which the leased facilities are located.
Total rent expense under non-cancelable operating leases was approximately $958,000, $744,000 and $673,000 for 2006, 2005 and 2004, respectively. The Company recognizes rent expense on a straight-line basis over the term of each lease, including any periods of free rent.
Minimum lease payments under non-cancelable operating leases as of December 31, 2006 are as follows (in thousands):
Year | | | | | |
2007 | | $ | 1,102 | |
2008 | | 1,082 | |
2009 | | 857 | |
2010 | | 542 | |
2011 | | 316 | |
Thereafter | | — | |
Total minimum lease payments | | $ | 3,899 | |
NOTE 12. Shareholders’ Equity
Common Stock Transactions: In connection with the 2006 equity financing described in Note 4 above, the Company issued to a group of institutional and other accredited investors an aggregate of 15,491,000 shares of common stock at a cash purchase price of $4.20 per share. Investors in the financing also received five-year warrants to purchase an aggregate of 4,643,000 shares of common stock at an exercise price of $4.62 per share. Concurrent with the closing of the financing, the Company issued an aggregate of 1,591,000 shares of common stock to the holders of its Series B preferred stock upon conversion of the outstanding Series B preferred shares (the Series B shares).
As part of the 2006 equity financing, on February 1, 2006, the Company received a $3,460,000 bridge loan from investors in the 2006 equity financing. Pursuant to the bridge loan, the Company issued convertible promissory notes in the principal amount of the loan and five-year warrants to purchase an aggregate of 411,906 shares of common stock at an exercise price of $4.62 per share. The fair value attributable to the warrants using the Black-Scholes option-pricing model was approximately $1,647,000 based upon assumptions of expected volatility of 114%, a contractual term of five years, an expected dividend rate of zero and a risk-free rate of interest of 4.5%. The Company recorded the warrants’ fair value as a discount to the promissory notes payable. The convertibility of the promissory notes gave rise to a beneficial conversion feature, which the Company recorded as additional discount on the promissory notes of approximately $1,813,000. The proceeds of the bridge loan were used for working capital pending closing of the 2006 equity financing on April 26, 2006. The convertible promissory notes provided for an
66
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
interest rate of 8% per annum and, at the closing of the 2006 equity financing, the principal amount of the notes, together with approximately $63,000 of accrued interest thereon, automatically converted, at a conversion rate of $4.20 per share, into an aggregate of 838,976 shares of common stock.
On September 22, 2006, the Company’s shareholders approved a one-for-six reverse split of the Company’s outstanding common stock, which became effective at the close of business that day. As a result of the reverse split, every six shares of Company common stock outstanding at the effective time automatically were combined into one outstanding share of Company common stock. The reverse stock split did not change the number of authorized shares of Company common stock designated in the Company’s articles of incorporation, nor did it change the par value of the Company’s common stock. In lieu of fractional shares, shareholders are entitled to receive an amount in cash equal to the value of their fractional shares based on $0.57, the closing price per share of the Company’s common stock on September 22, 2006.
In March 2005, the Company raised approximately $3,812,000 in net proceeds through the sale in a private placement (the 2005 financing) of 553,333 shares of common stock. In connection with the 2005 financing, the Company issued five-year warrants to purchase an aggregate of 221,333 shares of common stock at an exercise price of $12.00 per share. In addition, the placement agent in the 2005 financing was granted a warrant, on the same terms as those received by the purchasers in that transaction, for 33,200 shares of common stock. The Company has registered the shares of common stock issued in the 2005 financing, including the shares of common stock issuable upon exercise of the related warrants, with the SEC.
In April 2004, the Company issued 40,618 shares of common stock valued at $1,000,000 as a partial payment to purchase an exclusive license to develop, manufacture and commercialize picoplatin, a platinum-based anti-cancer agent. The 40,618 shares of common stock issued in this licensing arrangement have been registered with the SEC.
In February 2004, the Company raised approximately $9,000,000 in net proceeds through the sale in a private placement (the 2004 financing) of 307,500 shares of common stock. In connection with the 2004 financing, the Company issued five-year warrants to purchase an aggregate of 153,750 shares of common stock at $42.00 per share. The 307,500 shares of common stock issued in the 2004 financing, and the shares of common stock issuable upon exercise of the warrants related thereto, have been registered with the SEC.
During 2006, the Company received approximately $19,000 in net proceeds from the issuance of approximately 6,100 shares of common stock related to the exercises of employee stock options.
During 2005, the Company received approximately $44,000 in net proceeds from the issuance of approximately 15,600 shares of common stock related to the exercises of employee stock options.
During 2004, the Company generated approximately $800,000 in net proceeds from the issuance of approximately 136,200 shares of common stock related to the exercises of employee stock options.
Preferred Stock Transactions. During 2003, the Company issued 1,575 shares of a newly created class of Series B Convertible Preferred Stock with attached warrants to buy 105,000 shares of common stock. As described above, in connection with the 2006 equity financing, the 1,575 shares of Series B shares were converted into 1,591,000 shares of common stock in April 2006. The Series B shares received by the Company were retired and cancelled and are not reissuable. The Company had 205,340 shares of Series 1
67
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Convertible Exchangeable Preferred Stock (Series 1 preferred stock) outstanding at December 31, 2006. Holders of the Series 1 preferred stock are entitled to receive an annual cash dividend of $2.4375 per share if declared by the Board, payable semi-annually on June 1 and December 1. Dividends are cumulative. Each share of Series 1 preferred stock is convertible into 0.19 shares of common stock, subject to adjustment in certain events. The Series 1 preferred stock is redeemable at the option of the Company at $25.00 per share. Holders of Series 1 preferred stock have no voting rights, except in limited circumstances. Dividends of $500,000 were paid in each of the years 2006, 2005 and 2004, respectively.
The Company’s board of directors may, without further action by the shareholders, issue preferred stock in one or more series and fix the rights and preferences thereof, including dividend rights, dividend rates, conversion rates, voting rights, terms of redemption, redemption price or prices, liquidation preferences and the number of shares constituting any series or the designations of such series.
Shareholder Rights Plan. The Company’s Shareholder Rights Plan, and all preferred share purchase rights issued thereunder, expired on April 10, 2006.
Stock Options: At December 31, 2006, the Company’s 2004 Incentive Compensation Plan (the 2004 Plan) was the only compensation plan under which options were available for grant. The Company’s 1991 Stock Option Plan for Non-Employee Directors (the Directors Plan) was terminated on March 31, 2005, and no further options can be granted under that plan. The Company’s 1994 Stock Option Plan (the 1994 Plan) was terminated on February 17, 2004 and no further options can be granted under that plan. The 2004 Plan, as amended and restated on September 13, 2006, authorizes the Company’s board or a committee appointed by the board to grant options to purchase a maximum aggregate of 4,166,666 shares of common stock, of which 2,500,000 shares are subject to shareholder approval at the 2007 annual meeting of shareholders. The 2004 Plan allows for the issuance of incentive stock options and nonqualified stock options to employees, officers, directors, agents, consultants, advisors and independent contractors of the Company, subject to certain restrictions. All option grants expire ten years from the date of grant, except in the event of earlier termination of employment or service. Option grants for employees with less than one year of service generally become exercisable at a rate of 25% after one year from the grant date with the balance vesting at a rate of 1/36th per month over the following three years. Option grants for employees with more than one year of service or for employees receiving promotions become exercisable at a rate of 1/48th per month over the following four years. As of December 31, 2006, there were 1,956,115 shares of common stock available for issuance as new option awards under the 2004 Plan, subject to shareholder approval at the 2007 annual meeting of shareholders as described below. Although no Company securities are available for issuance under the Directors Plan or the 1994 Plan, options granted prior to termination of those plans continue in effect in accordance with their terms.
On September 13, 2006, the Company issued stock option grants to employees and consultants that are subject to shareholder approval at the Company’s 2007 annual meeting of an increase in the number of shares authorized for issuance under the 2004 Plan. If the shareholders do not approve an increase in the number of shares authorized for issuance under the 2004 Plan, these grants will immediately be cancelled after the 2007 annual meeting of shareholders. No portion of any options granted subject to shareholder approval is exercisable until the date on which the shareholders approval is received. If shareholder approval is obtained, these grants would be exercisable to the extent vested. The Company has not recognized compensation expense for these grants, because a grant date as defined in SFAS 123R has not occurred. This is because they are contingent upon shareholder approval, which cannot be assured.
68
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
In May 2000, the Company amended the 1994 Plan to provide that an employee will have two years to exercise the vested portion of an option upon retirement from the Company, whereas an employee previously had three months to exercise such option. Compensation expense equal to the intrinsic value of an employee’s option at the modification date will be recorded for any employees that receive an extension of their options upon retirement. The intrinsic value at the modification date for the options subject to the modifications that were outstanding at December 31, 2006 totaled approximately $119,000.
Restricted Stock. The Company adopted a Restricted Stock Plan (the Restricted Stock Plan) in 1991, under which 400,000 shares were authorized for issuance. Under the Restricted Stock Plan, restricted stock could be granted or sold to selected employees, officers, agents, consultants, advisors and independent contractors of the Company. The Restricted Stock Plan was terminated by the Board of Directors in June 2006.
Warrants. In connection with the 2006 equity financing, the Company issued five-year warrants to purchase an aggregate of 4,230,951 shares of common stock at an exercise price of $4.62 per share. The warrants became exercisable on April 26, 2006 and, thereafter, are exercisable at any time during their term. In payment of placement agent fees for the 2006 equity financing, the Company issued five-year warrants to purchase 139,286 shares of common stock at an exercise price of $4.62 per share. In connection with the bridge notes that were issued as part of the 2006 equity financing, the Company issued five-year warrants to purchase an aggregate of 411,906 shares of common stock at an exercise price of $4.62 per share. The warrants became exercisable on February 1, 2006 and, thereafter, are exercisable at any time during their term. The shares of common stock issuable upon exercise of the 2006 financing warrants have been registered with the SEC.
In connection with an agreement in 2006 for corporate communications services, the Company issued a two-year warrant to purchase an aggregate of 1,667 shares of common stock at an exercise price of $3.66 per share. The Company recorded an expense in the amount of approximately $3,400 for the fair value of the warrant on the date the services were completed. Based upon the Black-Scholes option pricing model, the grant date fair value of the warrant was $2.06 per share using assumptions of expected volatility of 105%, contractual term of two years, expected dividend rate of zero and a risk-free interest rate of 4.8%. The warrant became exercisable upon issuance and is exercisable at any time during its term.
In connection with the loan and security agreement with Silicon Valley Bank and Merrill Lynch Capital executed on October 25, 2006, the Company issued five-year warrants to purchase an aggregate of 174,418 shares of common stock at an exercise price of $4.30 per share. The fair value of the warrants was determined to be approximately $611,000 using the Black-Scholes option pricing model with assumptions of expected volatility of 112%, contractual term of five years, expected dividend rate of zero and a risk-free interest rate of 4.8%. Based on this fair value, approximately $540,000 was ascribed to the warrants and treated as a discount against the $15,000,000 loan obtained from Silicon Valley Bank and Merrill Lynch Capital. The warrants became exercisable upon issuance and are exercisable at any time during their term.
In connection with the 2005 financing, the Company issued five-year warrants to purchase an aggregate of 221,333 shares of common stock at an exercise price of $12.00 per share. The warrants became exercisable on September 3, 2005 and, thereafter, are exercisable at any time during their term. In payment of placement agent fees for the 2005 financing, the Company issued a five-year warrant to purchase 33,200 shares of common stock at an exercise price of $12.00 per share. The warrant contains provisions requiring the adjustment of the exercise price and number of shares issuable if the Company sells (other than in connection with certain permitted transactions, such as strategic collaborations and
69
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
acquisitions approved by the board) shares of common stock at a price lower than the then-current exercise price of the warrants. The shares of common stock issuable upon exercise of the 2005 financing warrants have been registered with the SEC.
In connection with the 2004 financing, the Company issued five-year warrants to purchase an aggregate of 153,750 shares of common stock, at an exercise price of $42.00 per share. The warrants became exercisable on February 23, 2004 and, thereafter, are exercisable at any time during their term. The warrants contain provisions requiring the adjustment of the exercise price and number of shares issuable if the Company sells (other than in connection with certain permitted transactions, such as strategic collaborations and acquisitions approved by the board) shares of common stock at a price lower than the then-current exercise price of the warrants. The warrants are redeemable at the election of the Company at any time after March 24, 2006, if the volume-weighted average price of the underlying common stock for each trading day over a period of 20 consecutive trading days is equal to or greater than $63.00 per share, subject to adjustment. The shares of common stock issuable upon exercise of the 2004 financing warrants have been registered with the SEC. In payment of placement agent fees for the 2004 financing, the Company issued three-year warrants to purchase an aggregate of 5,833 shares of common stock at an exercise price of $33.24 per share. The Company recorded a charge to general and administrative expense of $118,000 for the fair value of the warrants on February 23, 2004. Based upon the Black-Scholes option-pricing model, the fair value of the warrants was $20.28 per share using assumptions of expected volatility of 124%, contractual terms of three years, expected dividend rate of zero and a risk-free rate of interest of 2.2%.
In connection with its agreement to purchase the manufacturing facility in Denton, Texas, the Company on April 19, 2001, issued to International Isotopes Inc. a three-year warrant to purchase up to 133,333 shares of common stock at a purchase price of $60.00 per share. The warrant was valued at $9.66 per share using an option pricing model with assumptions of expected volatility of 125%, contractual term of three years, expected dividend rate of zero and a risk-free rate of interest of 4.6%. This warrant expired April 19, 2004.
NOTE 13. Acquisition of Picoplatin
In April 2004, the Company acquired from AnorMED, Inc. the worldwide exclusive rights, excluding Japan, to develop, manufacture and commercialize picoplatin, a platinum-based anti-cancer agent. AnorMED, Inc. was acquired by Genzyme Corporation in November 2006. Under the terms of the agreement, the Company paid a one-time upfront milestone payment of $1,000,000 in its common stock and $1,000,000 in cash. The agreement also initially provided for $13,000,000 in development and commercialization milestones, payable in cash or a combination of cash and common stock, and a royalty rate of up to 15% on net product sales after regulatory approval. The license agreement was amended on September 18, 2006 to modify several key financial terms and expand the licensed territory to include Japan, thereby providing the Company worldwide rights. In consideration of the amendment, the Company paid Genzyme $5,000,000 in cash on October 12, 2006 and will pay Genzyme an additional $5,000,000 in cash by March 31, 2007. The amendment eliminates all development milestone payments to Genzyme. Genzyme remains entitled to receive up to $5,000,000 in commercialization milestones upon the attainment of certain levels of annual net sales of picoplatin after regulatory approval. The amendment also reduces the royalty payable to Genzyme to a maximum of 9% of annual net product sales. In addition, the amendment reduces the sharing of sublicense revenues for any sublicenses entered into during the first
70
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
year following the amendment and eliminates the sharing of sublicense revenues with Genzyme on and after September 18, 2007.
The Company accounted for this transaction by recording a $10,000,000 increase in Licensed Products and the recognition of a current liability of $10,000,000. The Company concluded that no change in the expected life of the intangible asset occurred and will, therefore, amortize the increase in Licensed Products over the same amortization period used for the original Licensed Products amount. The Company recognized $306,000 of additional amortization expense during the year ended December 31, 2006 as a result of this increase in Licensed Products.
Licensed Products consists of the picoplatin amortizable intangible with a gross amount of $12,000,000 and accumulated amortization of $764,000 and $292,000 at December 31, 2006 and 2005, respectively. Licensed Products is amortized on a straight-line basis over 12 years. The estimated annual amortization expense for Licensed Products is approximately $1,215,000 for each of the years 2007 through 2011.
NOTE 14. Revenues
The Company did not record any revenues during 2006.
Revenue in 2005 was $15,000, which consisted primarily of royalty payments received in connection with licensed intellectual property.
Revenue in 2004 was $1,015,000 and consisted primarily of $1,000,000 from milestone payments received from Boston Scientific Corporation in connection with certain intellectual property licensed to Boston Scientific Corporation in 2003.
NOTE 15. Federal Income Taxes
Temporary differences and carryforwards giving rise to deferred tax assets (liabilities) were as follows (in thousands):
| | December 31, | |
Deferred Tax Assets (Liabilities): | | | | 2006 | | 2005 | |
Net operating loss carryforwards | | $ | 21,183 | | $ | 47,008 | |
Research and experimentation credit carryforwards | | 445 | | 8,883 | |
Capitalized research and development | | 11,591 | | 11,709 | |
Property and equipment | | 1,757 | | 1,694 | |
Other | | 711 | | 718 | |
Net deferred tax assets | | 35,687 | | 70,012 | |
Deferred tax assets valuation allowance | | (35,687 | ) | (70,012 | ) |
Net deferred income taxes | | $ | — | | $ | — | |
| | | | | | | | | |
The Company has established a valuation allowance equal to the amount of its net deferred tax assets because the Company has not had taxable income since its inception and significant uncertainty exists regarding the ultimate realization of its deferred tax assets. Accordingly, no tax benefits have been recorded in the accompanying statements of operations. The valuation allowance decreased by $34,325,000 in 2006, and increased by $5,251,000 in 2005.
71
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
In April 2006, the Company experienced a significant change to its capital structure which resulted in an ownership change, as defined under Section 382 of the IRC. Consequently, the amount of net operating loss carryforwards and research and experimentation credit carryforwards available to be used in future years will be limited under IRC Sections 382 and 383. This limitation will result in the loss of approximately $93,300,000 (approximately $31,700,000 in tax benefits) of the Company’s net operating loss carryforwards and $9,100,000 of the research and development credit carryforwards. Accordingly, the deferred tax asset and related valuation allowance associated to these carryforwards were reduced in 2006 by approximately $40,800,000.
The Company has net operating loss carryforwards of approximately $62,300,000 (net of the impact of the above referenced change in ownership under IRC Section 382), which expire from 2007 through 2026. Research and experimentation credits expire from 2007 to 2026. Future changes in the Company’s ownership could result in additional limitations on the Company’s ability to utilize its remaining net operating loss carryforwards and research and experimentation credit carryforwards.
Approximately $21,159,000 of the Company’s net operating loss carryforwards at December 31, 2006, result from deductions associated with the exercise of non-qualified employee stock options, the realization of which would result in a credit to shareholders’ equity.
NOTE 16. Related Party Transactions
As a consequence of the Company’s 2006 equity financing, entities affiliated with MPM Capital Management (MPM) acquired beneficial ownership of 7,744,000 common shares, or approximately 31.5% of the Company’s common stock outstanding immediately following the financing. Entities affiliated with Bay City Capital Management IV LLC (BCC) acquired beneficial ownership of 4,643,000 common shares, or approximately 19.5% of the common shares outstanding immediately following the 2006 equity financing. Two Company directors, Fred B. Craves and Carl S. Goldfischer, are managing directors of BCC and possess capital and carried interests in the BCC entities that participated in the 2006 equity financing. The Company has agreed, for as long as MPM owns at least 10% of the shares of common stock and warrants purchased in the financing, to use its best efforts to cause one person designated by MPM and one person designated by mutual agreement of MPM and BCC to be nominated and elected to the Company’s board of directors. Nicholas J. Simon III, a representative of MPM, was appointed to the board of directors on April 26, 2006. Mr. Simon is a general partner of certain of the MPM entities that participated in the financing and possesses capital and carried interests in those entities.
NOTE 17. 401(K) Plan
The Company sponsors a 401(K) plan that covers substantially all employees. At its own discretion, the Company may make contributions to the plan on a percentage of participants’ contributions. The Company made contributions of approximately $9,000, $12,000 and $11,000 for the years ended December 31, 2006, 2005, and 2004, respectively. The Company has no other post-employment or post-retirement benefit plans.
72
PONIARD PHARMACEUTICALS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
NOTE 18. Unaudited Quarterly Data
The following table presents summarized unaudited quarterly financial data (in thousands, except per share data):
| | First
Quarter | | Second
Quarter | | Third
Quarter | | Fourth
Quarter | |
2006 | | | | | | | | | |
Revenues | | $ | — | | $ | — | | $ | — | | $ | — | |
Operating expenses | | 3,999 | | 5,211 | | 5,555 | | 6,469 | |
Net loss | | (5,799 | ) | (6,488 | ) | (4,886 | ) | (6,121 | ) |
Net loss applicable to common shares | | (5,924 | ) | (6,613 | ) | (5,011 | ) | (6,246 | ) |
Net loss per common share: | | | | | | | | | |
Basic | | (0.51 | ) | (0.37 | ) | (0.22 | ) | (0.27 | ) |
Diluted | | (0.51 | ) | (0.37 | ) | (0.22 | ) | (0.27 | ) |
2005 | | | | | | | | | |
Revenues | | $ | — | | $ | 2 | | $ | 2 | | $ | 11 | |
Operating expenses | | 5,106 | | 8,887 | | 3,094 | | 3,988 | |
Net loss | | (5,076 | ) | (8,853 | ) | (3,078 | ) | (3,990 | ) |
Net loss applicable to common shares | | (5,201 | ) | (8,978 | ) | (3,203 | ) | (4,115 | ) |
Net loss per common share: | | | | | | | | | |
Basic | | (0.98 | ) | (1.57 | ) | (0.56 | ) | (0.72 | ) |
Diluted | | (0.98 | ) | (1.57 | ) | (0.56 | ) | (0.72 | ) |
Note: Net loss per common share, basic and diluted, may not add to net loss per common share for the year due to rounding.
73
Item 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not Applicable.
Item 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Under the supervision and with the participation of the Company’s management, including the Company’s Chairman and Chief Executive Officer and the Chief Financial Officer, the Company has evaluated the effectiveness and design of its disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e)) as of the end of the period covered by this report, and, based on their evaluations, the Chairman and Chief Executive Officer and the Chief Financial Officer have concluded that these disclosure controls and procedures were effective as of December 31, 2006, in ensuring that all material information required to be disclosed in the reports that the Company files or submits under the Securities Exchange Act of 1934, as amended, have been made known to them in a timely fashion.
Management’s Annual Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting for the Company and for the assessment of the effectiveness of internal control over financial reporting. The Company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United Stated of America. The Company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the Company’s assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America, and that the Company’s receipts and expenditures are being made only in accordance with authorizations of the Company’s management and directors; and (iii) provide reasonable assurance regarding the prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies and procedures may deteriorate.
Management conducted an evaluation of the effectiveness of the system of internal control over financial reporting based on the framework in Internal Control Integrated Framework issued by the Committee Sponsoring Organizations of the Treadway Commission. Based on this evaluation, management concluded that the Company’s internal control over financial reporting was effective as of December 31, 2006. Management’s assessment of the effectiveness of the Company’s internal control over financial reporting as of December 31, 2006 has been audited by KPMG LLP, a registered independent public accounting firm, as stated in their report below.
74
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders
Poniard Pharmaceuticals, Inc.:
We have audited management’s assessment, included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting, that Poniard Pharmaceuticals, Inc. maintained effective internal control over financial reporting as of December 31, 2006, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Poniard Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting. Our responsibility is to express an opinion on management’s assessment and an opinion on the effectiveness of the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, evaluating management’s assessment, testing and evaluating the design and operating effectiveness of internal control, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, management’s assessment that Poniard Pharmaceuticals, Inc. maintained effective internal control over financial reporting as of December 31, 2006, is fairly stated, in all material respects, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Also, in our opinion, Poniard Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2006, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
75
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheet of Poniard Pharmaceuticals, Inc. and subsidiary as of December 31, 2006 and 2005, and the related consolidated statements of operations, cash flows and shareholders’ equity and comprehensive income for each of the years in the three-year period ended December 31, 2006, and our report dated March 15, 2007, expressed an unqualified opinion on those consolidated financial statements.
/s/ KPMG LLP
Seattle, Washington
March 15, 2007
Changes in internal control over financial reporting
There have been no changes in the Company’s internal control over financial reporting that occurred during the Company’s fourth fiscal quarter that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
Item 9B. OTHER INFORMATION
Not Applicable.
76
PART III
Item 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE OF THE REGISTRANT
(a) Directors and Audit Committee. The information required by this item is incorporated herein by reference to the sections captioned “Election of Directors” and “Board of Directors and Corporate Governance” in the Company’s definitive Proxy Statement for the 2007 Annual Meeting of Shareholders to be held on June 14, 2007, to be filed with the Securities and Exchange Commission, or the Commission, pursuant to Regulation 14A not later than 120 days after December 31, 2006.
(b) Executive Officers. Information with respect to the Company’s executive officers is set forth below.
Name | | | | Age | | Position with the Company | |
Gerald McMahon, PhD | | | 52 | | | Chairman, President and Chief Executive Officer | |
Caroline M. Loewy | | | 40 | | | Chief Financial Officer | |
David A. Karlin, M.D. | | | 63 | | | Senior Vice President, Clinical Development & Regulatory Affairs | |
Anna L. Wight, JD | | | 52 | | | Vice President, Legal and Secretary | |
Cheni Kwok, PhD | | | 38 | | | Vice President, Business Development | |
Business Experience
Gerald McMahon, PhD, was appointed Chief Executive Officer of the Company in May 2004 and Chairman of the Board of Directors in June 2004. Dr. McMahon was appointed President of the Company on June 15, 2005. Previously, he was President of SUGEN, Inc., a biopharmaceutical company focused on the discovery and development of novel targeted small-molecule drugs. At SUGEN, Dr. McMahon played a key role in the discovery and development of several innovative cancer products, including Sutent®, a multi-targeted protein kinase inhibitor for the treatment of advanced cancers, now in Phase III trials with Pfizer Inc. SUGEN was acquired by Pharmacia Corp. in 1999, which subsequently was acquired by Pfizer in 2003. Prior to his role at SUGEN, which he joined in 1993, Dr. McMahon held several research and development management positions at Sandoz Pharmaceuticals (now Novartis), where his responsibilities included the establishment of external collaborations and the development of corporate alliances within the United States and Europe. Dr. McMahon has contributed to more than 100 scientific publications and was a Staff Scientist and Principal Investigator at the Massachusetts Institute of Technology and Tufts University School of Medicine early in his career. He holds a B.S. in Biology and a PhD in Biochemistry from Rensselaer Polytechnic Institute.
Caroline M. Loewy was appointed Chief Financial Officer in July 2006. She initially joined the Company in June 2006 as Executive Vice President of Strategic Planning. Ms. Loewy has served in a business and financial consulting capacity to biotechnology companies since 2004. Prior thereto, she was Executive Director, Equity Research at Morgan Stanley, Inc. from March 2000 to June 2004, where she covered large cap biotechnology stocks. Previously, she was with Prudential Securities, first as an associate capital goods analyst in San Francisco from 1993 to 1996 and then as a senior biotechnology analyst in New York from 1996 to 2000. Ms. Loewy holds an M.B.A. from Carnegie Mellon, Graduate School of Business and a B.A. in economics from the University of California, Berkeley.
David A. Karlin, M.D., joined the Company as Senior Vice President of Clinical Development and Regulatory Affairs in July of 2005. Prior thereto, Dr. Karlin served as Vice President of Clinical Research at Cellegy Pharmaceuticals, Inc. from 2002 to 2005. Dr. Karlin’s experience in the biotech and pharmaceutical industry also includes positions as Vice President of Clinical Development for Genteric, Inc., a privately held company specializing in gene therapy during 2002, and Senior Medical
77
Director at Matric Pharmaceuticals, Inc., an oncology therapeutics development company (1991 to 2001). Dr. Karlin has also served as Vice President for Clinical Research and Medical Director at SciClone Pharmaceuticals, Inc. from 1995 to 1999 and held various positions at Syntex Corporation, including Director of Medical Research, from 1986 to 1995. Before joining the pharmaceutical industry, Dr. Karlin was an associate professor at Temple University School of Medicine and an assistant professor at the University of Texas M.D. Anderson Hospital and Tumor Institute. He received his M.D. from the University of Chicago and completed his residency in Internal Medicine at the University of Michigan and a fellowship in Gastroenterology and Gastrointestinal Oncology at the University of Chicago. He holds a B.S. in Biology from the University of Illinois.
Anna Lewak Wight, JD, was appointed Vice President, Legal and Secretary in September 2001. Prior thereto, she served as Director of Intellectual Property since joining the Company in 1994. Ms. Wight previously was a partner in the law firm of Morrison & Foerster, where she managed their Seattle intellectual property practice. Ms. Wight also was a partner in the intellectual property law firm of Harness, Dickey and Pierce in Michigan, where she established and chaired the Biotechnology and Medical Arts Group. Ms. Wight received a JD from Wayne State University Law School and an MS from the Genetics Program at Michigan State University.
Cheni Kwok, PhD, joined the Company as Vice President, Business Development in July 2006. Prior thereto, she was Director, Business Development at Celera Genomics, a division of Applera Corporation engaged in the discovery and development of targeted therapeutics for cancer, autoimmune and inflammatory disease, from 2004 through June 2006. From 2000 to 2004, Dr. Kwok served in various business development positions, including as Associate Director, Business Development at Exelixis, Inc., a publicly held drug discovery company. Dr. Kwok received a bachelor’s degree in biotechnology from Imperial College of Science, Technology and Medicine, University of London, U.K. and a PhD in human molecular genetics from the University of Cambridge, U.K.
(c) Compliance with Section 16(a) of the Exchange Act. The information required by this item is incorporated herein by reference to the section captioned “Section 16(a) Beneficial Ownership Reporting Compliance” in the Company’s definitive Proxy Statement for the 2007 Annual Meeting of Shareholders to be held June 14, 2007, to be filed with the Commission pursuant to Regulation 14A not later than 120 days after December 31, 2006.
(d) Code of Ethics. The information required by this item is incorporated herein by reference to the section captioned “Board of Directors and Corporate Governance” in the Company’s definitive Proxy Statement for the 2007 Annual Meeting of Shareholders to be held June14, 2006, to be filed with the Commission pursuant to Regulation 14A not later than 120 days after December 31, 2006.
Item 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated herein by reference to the sections captioned “Executive Compensation” and “Board of Directors and Corporate Governance—Compensation Committee Interlocks and Insider Participation” in the Company’s definitive Proxy Statement for the 2007 Annual Meeting of Shareholders to be held June 14, 2007, to be filed with the Commission pursuant to Regulation 14A not later than 120 days after December 31, 2006.
Item 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated herein by reference to the sections captioned “Security Ownership of Certain Beneficial Owners and Management” and “Executive Compensation-Equity Compensation Plan Information” in the Company’s definitive Proxy Statement for the 2007 Annual
78
Meeting of Shareholders to be held June 14, 2007, to be filed with the Commission pursuant to Regulation 14A not later than 120 days after December 31, 2006.
Item 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
The information required by this item is incorporated herein by reference to the sections captioned “Certain Relationships and Related Transactions with Management” and “Board of Directors and Corporate Governance” in the Company’s definitive Proxy Statement for the 2007 Annual Meeting of Shareholders to be held June 14, 2007, to be filed with the Commission pursuant to Regulation 14A not later than 120 days after December 31, 2006.
Item 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this item is incorporated herein by reference to the sections captioned “Independent Registered Public Accounting Firm” and “Ratification of Appointment of Independent Registered Public Accounting Firm” in the Company’s definitive Proxy Statement for the 2007 Annual Meeting of Shareholders to be held June 14, 2007, to be filed with the Commission pursuant to Regulation 14A not later than 120 days after December 31, 2006.
79
PART IV
Item 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
(a) (1) Financial Statements—See Index to Financial Statements.
(2) Financial Statement Schedules—Not applicable.
(3) Exhibits—See Exhibit Index filed herewith.
(b) Exhibits—See Exhibit Index filed herewith.
80
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| PONIARD PHARMACEUTICALS, INC. |
| (Registrant) |
| /s/ CAROLINE M. LOEWY |
| Caroline M. Loewy |
| Chief Financial Officer |
Date: March 15, 2007
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and as of the dates indicated:
/s/ GERALD MCMAHON | | Chairman, President and | | March 15, 2007 |
Gerald McMahon | | Chief Executive Officer | | |
/s/ FRED B. CRAVES | | Director | | March 15, 2007 |
Fred B. Craves | | | | |
/s/ E. ROLLAND DICKSON | | Director | | March 15, 2007 |
E. Rolland Dickson | | | | |
/s/ CARL S. GOLDFISCHER | | Director | | March 15, 2007 |
Carl S. Goldfischer | | | | |
/s/ ROBERT M. LITTAUER | | Director | | March 15, 2007 |
Robert M. Littauer | | | | |
/s/ DAVID R. STEVENS | | Director | | March 15, 2007 |
David R. Stevens | | | | |
/s/ NICHOLAS J. SIMON III | | Director | | March 15, 2007 |
Nicholas J. Simon III | | | | |
/s/ RONALD A. MARTELL | | Director | | March 15, 2007 |
Ronald A. Martell | | | | |
/s/ MICHAEL K. JACKSON | | Principal Accounting Officer | | March 15, 2007 |
Michael K. Jackson | | | | |
81
EXHIBIT INDEX
Exhibit | | Description | | |
3.1 | | Amended and Restated Articles of Incorporation, as amended February 7, 2007 | | (N) |
3.2 | | Restated Bylaws, as amended March 28, 2006 | | (V) |
10.1 | | Restated 1994 Stock Option Plan(‡) | | (F) |
10.2 | | 1991 Stock Option Plan for Non-Employee Directors, as amended(‡) | | (E) |
10.3 | | Reserved. | | |
10.4 | | Indemnification Agreement(‡) | | (H) |
10.5 | | Stock Option Grant Program for Nonemployee Directors under the NeoRx 2004 Incentive Compensation Plan, as amended(‡) | | (B) |
10.6 | | Stock Option Agreement, dated December 19, 2000, between NeoRx Corporation and Carl S. Goldfischer(‡) | | (I) |
10.7 | | Stock Option Agreement, dated January 17, 2001, between NeoRx Corporation and Carl S. Goldfischer(‡) | | (I) |
10.8 | | License Agreement dated as of April 2, 2004, between the Company and AnorMED, Inc. Certain portions of the agreement have been omitted pursuant to a request for confidential treatment | | (Q) |
10.9 | | Amendment No. 1 to License Agreement effective as of September 18, 2006, between the Company and AnorMED, Inc. Certain portions of the agreement have been omitted pursuant to a request for confidential treatment. | | (Y) |
10.10 | | Stock Option Grant Program for Nonemployee Directors under the NeoRx Corporation 1994 Restated Stock Option Plan(‡) | | (M) |
10.11 | | Facilities Lease dated February 15, 2002, between NeoRx Corporation and Selig Real Estate Holdings Six | | (A) |
10.12 | | Amended and Restated 2004 Incentive Compensation Plan as amended and restated June 16, 2006(‡) | | (G) |
10.13 | | Reserved. | | |
10.14 | | Key Executive Severance Agreement dated as of February 28, 2003, between the Company and Anna Wight(‡) | | (C) |
10.15 | | Amendment No. 1 dated as of March 30, 2005 to Key Executive Severance Agreement dated as of February 28, 2003, between the Company and Anna Wight(‡) | | (L) |
10.16 | | Change of Control Agreement dated as of February 28, 2003, between the Company and Anna Wight(‡) | | (C) |
10.17 | | Key Executive Severance Agreement dated as of June 23, 2005, between the Company and David A. Karlin(‡) | | (P) |
10.18 | | Change of Control Agreement dated as of June 23, 2005, between the Company and David A. Karlin(‡) | | (P) |
10.19 | | Employment Letter dated as of April 26, 2004, between the Company and Gerald McMahon(‡) | | (L) |
82
10.20 | | Key Executive Severance Agreement dated as of May 11, 2004, between the Company and Gerald McMahon(‡) | | (R) |
10.21 | | Change of Control Agreement dated as of May 11, 2004, between the Company and Gerald McMahon(‡) | | (R) |
10.22 | | Reserved. | | |
10.23 | | Reserved. | | |
10.24 | | Key Employee Severance Agreement dated as of July 11, 2006, between the Company and Michael K. Jackson(‡) | | (X) |
10.25 | | Form of Non-Qualified Stock Option Agreement under 2004 Incentive Compensation Plan(‡) | | (O) |
10.26 | | Form of Incentive Stock Option Agreement under 2004 Incentive Compensation Plan(‡) | | (O) |
10.27 | | Key Executive Severance Agreement dated as of June 23, 2006, between the Company and Caroline M. Loewy(‡) | | (S) |
10.28 | | Change of Control Agreement dated as of June 23, 2006, between the Company and Caroline M. Loewy(‡) | | (S) |
10.29 | | Executive Severance Agreement dated as of June 23, 2006, between the Company and Cheni Kwok(‡) | | (S) |
10.30 | | Change of Control Agreement dated as of July 1, 2006, between the Company and Cheni Kwok(‡) | | (S) |
10.31 | | Research Funding and Option Agreement dated August 4, 2005, between the Company and The Scripps Research Institute. Certain portions of the agreement have been omitted pursuant to a request for confidential treatment | | (U) |
10.32 | | Form of Directors’ Indemnification Agreements(‡) | | (K) |
10.33 | | Lease Agreement dated as of July 10, 2006, between the Company and ARE San Francisco No. 17 LLC | | (W) |
10.34 | | Loan and Security Agreement dated as of October 25, 2006, among the Company, Silicon Valley Bank and Merrill Lynch Capital | | (J) |
10.35 | | Secured Promissory Notes to Silicon Valley Bank and Merrill Lynch Capital | | (J) |
23.1 | | Consent of KPMG | | (Z) |
31.1 | | Rule 13a-14(a)/15d-14(a) Certification of President and Chief Executive Officer | | (Z) |
31.2 | | Rule 13a-14(a)/15d-14(a) Certification of Chief Financial Officer | | (Z) |
32.1 | | Section 1350 Certification of President and Chief Executive Officer | | (Z) |
32.2 | | Section 1350 Certification of Chief Financial Officer | | (Z) |
(‡) Management contract or compensatory plan.
(A) Filed as an exhibit to the Company’s Form 10-K for the fiscal year ended December 31, 2001, and incorporated herein by reference.
83
(B) Filed as an exhibit to the Company’s Current Report on Form 8-K filed June 16, 2005, and incorporated herein by reference.
(C) Filed as an exhibit to the Company’s Registration Statement on Form S-3/A (Registration No. 333-111344) filed on February 23, 2004, and incorporated herein by reference.
(D) Reserved.
(E) Incorporated by reference to Exhibit A to the Company’s definitive proxy statement on Schedule 14A filed April 10, 1996.
(F) Filed as an exhibit to the Company’s Form 10-K for the fiscal year ended December 31, 1995, and incorporated herein by reference.
(G) Filed as an exhibit to the Company’s Current Report on Form 8-K filed on June 21, 2006 and incorporated herein by reference.
(H) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended March 31, 1996, and incorporated herein by reference.
(I) Filed as an exhibit to the Company’s Form 10-K for the fiscal year ended December 31, 2000, and incorporated herein by reference.
(J) Filed as an exhibit to the Company’s Current Report on Form 8-K filed on October 31, 2006, and incorporated herein by reference.
(K) Filed as an exhibit to the Company’s Current Reports on Form 8-K filed on April 28, 2006 and June 27, 2006, and incorporated herein by reference.
(L) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended March 31, 2005, and incorporated herein by reference.
(M) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended June 30, 2002, and incorporated herein by reference.
(N) Filed as an exhibit to the Company’s Current Report on Form 8-K filed on September 26, 2006, and incorporated herein by reference.
(O) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended September 30, 2004, and incorporated herein by reference.
(P) Filed as an exhibit to the Company’s Current Report on Form 8-K filed on June 29, 2005, and incorporated herein by reference.
(Q) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended March 31, 2004, and incorporated herein by reference.
(R) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended June 30, 2004, and incorporated herein by reference.
(S) Filed as an exhibit to the Company’s Current Report on Form 8-K filed June 23, 2006, and incorporated herein by reference.
(T) Reserved.
(U) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended September 30, 2005, and incorporated herein by reference.
(V) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended March 31, 2006 and incorporated herein by reference.
84
(W) Filed as an exhibit to the Company’s Current Report Form 8-K filed on July 13, 2006 and incorporated herein by reference.
(X) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended June 30, 2006 and incorporated herein by reference.
(Y) Filed as an exhibit to the Company’s Form 10-Q for the quarterly period ended September 30, 2006 and incorporated herein by reference.
(Z) Filed herewith.
85