UNITED STATES SECURITIES AND EXCHANGE COMMISSION Washington, D.C. 20549 |
| |
| FORM 10-K |
| (Mark One) |
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | For the fiscal year ended December 31, 2011 |
| | Or |
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | For the transition period from _________________ to _______________________ |
| | |
| Commission file number: 000-50571 |
| |
| RESPONSE BIOMEDICAL CORPORATION |
| (Exact name of registrant as specified in its charter) |
| |
| Vancouver, British Columbia, Canada | 98 -1042523 |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) |
1781 - 75th Avenue W. Vancouver, British Columbia, Canada | V6P 6P2 |
| (Address of principal executive offices) | (Zip Code) |
| | |
| Registrant's telephone number, including area code: (604) 456-6010 |
Securities registered pursuant to Section 12(b) of the Act: NONE Securities registered pursuant to Section 12(g) of the Act: COMMON STOCK WITHOUT PAR VALUE |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x |
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o |
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o |
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x |
| Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. |
Large accelerated filer o | Accelerated filer o | Non-accelerated filer o (Do not check if a smaller reporting company) | Smaller reporting company x |
Indicate by check mark if the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x |
| The aggregate market value of the voting common stock held by non-affiliates of the Registrant (assuming officers, directors and 10% stockholders are affiliates), based on the last sale price for such stock on June 30, 2011: $14,136,998. The Registrant has no non-voting common stock. |
As of March 23, 2012, there were 129,078,166 shares of the Registrant's common stock outstanding. |
|
DOCUMENTS INCORPORATED BY REFERENCE |
Portions of the Registrant's Proxy Statement for the 2012 Annual Meeting of Stockholders of the Registrant to be held on June 19, 2012 are incorporated by reference into Part III of this Form 10-K. The Registrant makes available free of charge on or through its website (http://www.responsebio.com) its Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and any amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. The material is made available through the Registrant's website as soon as reasonably practicable after the material is electronically filed with or furnished to the U.S. Securities and Exchange Commission, or SEC. All of the Registrant's filings may be read or copied at the SEC's Public Reference Room at 100 F Street, N.E., Room 1580, Washington D.C. 20549. Information on the hours of operation of the SEC's Public Reference Room can be obtained by calling the SEC at 1-800-SEC-0330. The SEC maintains a website (http://www.sec.gov) that contains reports and proxy and information statements of issuers that file electronically. |
| |
RESPONSE BIOMEDICAL CORPORATION
Form 10-K – ANNUAL REPORT
For the Fiscal Year Ended December 31, 2011
Table of Contents
| | | Page |
| | | |
| PART I |
| |
| Item 1. | Business | 2 |
| Item 1A. | Risk Factors | 18 |
| Item 2. | Properties | 32 |
| Item 3. | Legal Proceedings | 32 |
| Item 4. | Mine Safety Disclosures | 32 |
| | | |
| PART II |
| | | |
| Item 5. | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 33 |
| Item 6. | Selected Financial Data | 34 |
| Item 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations | 35 |
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk | 45 |
| Item 8. | Financial Statements and Supplementary Data | 46 |
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure | 74 |
| Item 9A. | Controls and Procedures | 74 |
| Item 9B. | Other Information | 76 |
| | | |
| PART III |
| | | |
| Item 10. | Directors, Executive Officers and Corporate Governance | 76 |
| Item 11. | Executive Compensation | 76 |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 76 |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. | 76 |
| Item 14. | Principal Accountant Fees and Services | 77 |
| | | |
| PART IV |
| | | |
| Item 15. | Exhibits and Financial Statement Schedules | 78 |
PART I
SPECIAL NOTE REGARDING FORWARD LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements relating to future events and our future performance within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. In some cases, you can identify forward-looking statements by terms such as "may", "will", "should", "could", "would", "hope", "expects", "plans", "intends", "anticipates", "believes", "estimates", "projects", "predicts", "potential" and similar expressions intended to identify forward-looking statements. These forward-looking statements include, without limitation, statements relating to future events, future results, and future economic conditions in general and statements about:
| · | The development of new products, regulatory approvals of new and existing products and the expansion of the market for our current products; |
| · | Implementing aspects of our business plan and strategies; |
| · | Our financing goals and plans; |
| · | Our existing working capital and cash flows and whether and how long these funds will be sufficient to fund our operations; and |
| · | Our raising of additional capital through future equity and debt financings. |
These statements involve known and unknown risks, uncertainties and other factors, including the risks described in Part I, Item 1A. of this Annual Report on Form 10-K, which may cause our actual results, performance or achievements to be materially different from any future results, performances, time frames or achievements expressed or implied by the forward-looking statements. Given these risks, uncertainties and other factors, you should not place undue reliance on these forward-looking statements. Information regarding market and industry statistics contained in this Annual Report on Form 10-K is included based on information available to us that we believe is accurate. It is generally based on academic and other publications that are not produced for purposes of securities offerings or economic analysis. We have not reviewed or included data from all sources and cannot assure you of the accuracy of the market and industry data we have included.
Corporate Information
ITEM 1. Business
General
Response Biomedical Corporation (“Response”, “us”, or “we”) develops, manufactures and markets rapid on-site diagnostic tests for use with our RAMP® platform for clinical and environmental applications. RAMP represents a paradigm in diagnostics that provides high sensitivity and reliable information in minutes. It is ideally suited to both point of care testing and laboratory use. Response was incorporated in British Columbia in August 1980. Our principal offices are located at 1781 – 75th Avenue West, Vancouver, British Columbia, Canada. Our common stock is traded on the Toronto Stock Exchange (TSX) under the trading symbol “RBM” and quoted on the OTC markets under the symbol “RPBIF”. Our results by segment are included in our financial statements, which are included under Item 8 to this Annual Report on Form 10-K.
Our Technology – The RAMP® System
The RAMP® system is a proprietary platform technology that combines a sensitive, portable fluorescence detection system with simple lateral flow immunoassays. Although lateral flow immunoassay technology has been available for over 25 years, the market for early generation rapid immunoassays has been limited by their inability to provide the accurate, quantitative results required by the majority of test situations.
RAMP® maintains key positive attributes of lateral flow immunoassays - simplicity, specificity, reliability and rapid results – but includes a method to overcome the performance limitations of early generation immunoassays that suffer from relatively poor sensitivity and precision. By introducing a population of known antibodies that are impacted by the same conditions as the test antibodies, the ratio of a measurement of the signal from the two sets of antibodies effectively factors out uncontrolled variability, thereby providing an accurate result. Furthermore, the use of a fluorescent label in the cartridge combined with a custom optical scanner in the RAMP® Reader, or Reader, results in a reliable and sensitive detection system. The RAMP® System has demonstrated its capability to detect and quantify a wide variety of analytes with sensitivity and accuracy comparable to centralized lab systems, including multiple analytes simultaneously.
Minimal training is required to use the RAMP® System. A test is performed by adding a sample (e.g., blood, urine, saliva, water, or unknown powders) containing the analyte of interest (e.g., Myoglobin, anthrax spores) mixed with a proprietary buffer and labeled antibodies to the sample well of a cartridge. The cartridge is then inserted into the Reader, which scans the test strip and provides the result in 20 minutes or less, depending on the assay. In the absence of rapid on-site and point-of-care (POC) test results like the RAMP® test, health care providers and first responders may be forced to wait up to three days for a confirmatory result from a government- or hospital -run lab.
The RAMP® system consists of a reader and single-use disposable test cartridges, and has the potential to be adapted to more than 250 medical and non-medical tests currently performed in laboratories.
Our Markets
We develop, manufacture and sell the RAMP® system for the medical point of care market including cardiovascular tests and infectious disease tests, and the on-site environmental testing market including biodefense and vector infectious disease testing.
Medical Point-of-Care (POC) Clinical Diagnostics
Cardiovascular Testing
A major focus of our development programs in cardiovascular testing has been clinical tests for the quantification of cardiac markers. Cardiac markers are biochemical substances that are released by the heart after it has been damaged or stressed. Elevated levels of these markers can be indicative of a heart attack or congestive heart failure. There are three primary markers for the detection of a heart attack: Myoglobin, CK-MB, and Troponin I. The primary markers for congestive heart failure are B-type natriuretic peptide, or BNP, and NT-proBNP. Response has developed POC tests for each of these markers, where NT-proBNP is various countries by our international distributor network except for Japan, where BNP is sold solely.
Heart Attack/Myocardial Infarction (MI) Testing
Serial measurement of biochemical markers is now universally accepted as an important determinant in the diagnosis of a heart attack. The ideal cardiac marker is one that has high clinical sensitivity and specificity, appears soon after the onset of a heart attack, remains elevated for several days following a heart attack, and can be assayed with a rapid turnaround time.1 Today, there is no single marker that meets all of these criteria, thus necessitating testing for multiple cardiac markers. The biochemical markers that are commonly used by physicians to aid in the diagnosis of a heart attack are Myoglobin, CK-MB, Troponin I, and Troponin T. As seen in the figure below, cardiac markers follow a specific, predictable pattern of release kinetics following a coronary event. The differences in the time that it takes each marker to reach peak concentration has made it common practice for clinicians to make use of at least two different markers in tandem, an early marker such as Myoglobin and a later one such as Troponin I. More recently, international guidelines recommend the use of serial Troponin assays for definitive diagnosis of heart attacks.
Release of Cardiac Markers into the Bloodstream Following a Heart Attack2
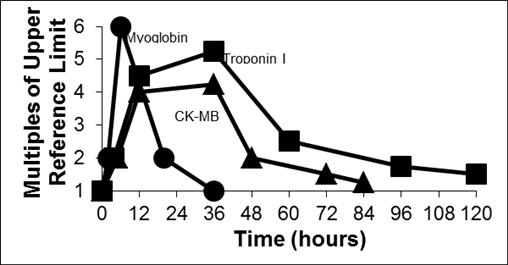
The turn-around times for results from a hospital lab can vary from as little as thirty minutes to more than two hours due to the necessity of test ordering and specimen collection, specimen transport, sample preparation, test completion and reporting. In rural settings and physicians’ offices, the turn-around time can be many hours or even days. Evidence-based clinical practice guidelines recommend that the results from cardiac marker testing be available within 60 minutes of patient presentation and ideally within thirty minutes. POC testing with products such as RAMP® could provide doctors with the information they need to diagnose and treat heart attack patients in a much shorter timeframe (e.g., less than 20 minutes from blood draw to result). In most cases, this is more likely to be within the critical window of time to minimize irreversible heart damage or death. The RAMP® System is expected to aid in the diagnosis of heart attack by enabling physicians to easily and frequently monitor changes in the levels of a patient’s cardiac markers. Early access to this information enables physicians to use accelerated care protocols, which are intended to drive earlier and better treatment decisions. According to statistics published by the U.S. Centers for Disease Control and Prevention, or CDC, approximately 6 million people visit U.S. hospital emergency departments each year with complaints of chest pain, a primary symptom of heart attack.
1 Adams JE, III, Clin Chem Acta, 1999.
2 Wu AHB, Introduction to Coronary Artery Disease (CAD) and Biochemical Markers, 1998.
Congestive Heart Failure (CHF) testing
Congestive heart failure, or CHF, is a chronic, progressive disease in which the heart muscle weakens and becomes impaired, thus impeding the heart's ability to pump enough blood to support the body's metabolic demands. CHF is the only cardiovascular disorder to show a marked increase in incidence in the past 40 years and it is expected to continue rising due in part to the aging population and better survival prospects of patients with other cardiovascular diseases.3 Many patients hospitalized with acute CHF will be re-admitted to the hospital with repeat incidence of the disease.
Previous methods for the diagnosis and assessment of CHF, which include physical examinations and chest x-rays, are not usually conclusive, making accurate diagnoses difficult. The introduction of testing for BNP and NT-proBNP dramatically changed the ability of physicians to make a qualified diagnosis and monitor the success of treatment because the level of these molecules is elevated in the blood when the heart is forced to work harder. These tests have proven to be more accurate than any other single physical or laboratory gauge of heart failure.4 Both BNP and NT-proBNP are fragments of proBNP, a neurohormone that is released by the heart in response to increased blood pressure and volume overload causing stretching of the ventricular muscle of the heart during heart failure. Both of these markers are elevated in the blood during heart failure and are sensitive and specific indicators of congestive heart failure.
Infectious Disease Testing
Influenza viruses cause seasonal epidemics associated with high morbidity and mortality, especially affecting those with underlying medical conditions and the elderly.5 Influenza is characterized by rapid start of high fever, chills, myalgia, headache, sore throat, and cough. However, even during periods of large outbreak, clinical diagnosis can be difficult due to the presence of other respiratory viruses.6 The rapid and accurate diagnosis of Influenza is important for determining appropriate treatment strategies and decreasing unnecessary use of antibiotics.7 The laboratory diagnosis of Influenza infections is based on either detection of the Influenza virus directly or isolation of the virus in cell culture, or detection of nucleic acid by polymerase chain reaction, each of which can take several hours to days before results become available.
With the recent development of different treatments for Influenza A and B and the need to begin therapy within the first 48 hours of infection8, the demand for rapid and accurate Influenza tests has grown. Being able to rapidly identify patients with Influenza at the clinic or hospital allows sites to reduce infections occurring in hospitals and reduces the amount of unnecessary or incorrect treatment and administration.
Most of the rapid Influenza tests on the market are qualitative strip tests that produce a change in color if the Influenza virus is present in sufficient quantities. Several independent evaluations demonstrate the limited clinical sensitivity or ability to detect small amounts of the Influenza virus, of the current, commercially available rapid Influenza tests.9,10,11,14 While the specificity, or ability to detect the Influenza correctly, if in sufficient quantity, of these rapid tests is generally high (median 90–95%), their limited sensitivity and false negative results remain a major concern.
3 McCullough, PA, Nowak, RM, McCord J, et al. B-type natriuretic peptide and clinical judgment in emergency diagnosis of heart failure. Clin Inv Rep. 2002;106:416-422.
4 http://www.stjohnsmercy.org/healthinfo/newsletters/heart/Aug02.asp
5 Nicholson KG, Wood JM, Zambon M (2003) Influenza. Lancet 362:1733– 1745.
6 http://www.cdc.gov/flu/about/qa/disease.htm.
7 http://www.cdc.gov/flu/professionals/treatment/0506antiviralguide.htm.
8 http://www.cdc.gov/flu/keyfacts.htm.
9 Fader RC. Comparison of the BinaxNOW Flu A Enzyme Immunochromatographic Assay and R-mix Shell Vial Culture for the 2003-2004 Influenza Season. Journal of Clinical Microbiology, December 2005; 43(12), 6133-6135.
10 Landry ML, Cohen S, Ferguson D. Comparison of BinaxNOW and Directigen for rapid detection of influenza A and B. Journal of Clinical Virology. 2004, 31, 113-115.
11 Weinberg A, Walker ML. Evaluation of Three Immunoassay Kites for Rapid Detection of Influenza Virus A and B. Clinical and Diagnostic Laboratory Immunology. March 2005, 367-370.
Respiratory Syncytial Virus (RSV)
Respiratory syncytial virus, or RSV, is a respiratory virus that infects the lungs and breathing passages. Most otherwise healthy people recover from RSV infection in 1- 2 weeks. However, infection can be severe in some people, such as certain infants, young children, and older adults. In fact, RSV is the most common cause of bronchiolitis (inflammation of the small airways in the lung) and pneumonia in children under one year of age in the United States. In addition, RSV is more often being recognized as an important cause of respiratory illness in older adults.13
On- Site Environmental Testing
Environmental tests are generally considered to be products and services used to detect and quantify substances and microbes in the environment that have potentially harmful effects to humans. We participate in two distinct areas of the environmental market. The first is biodefense, where RAMP® products are used for the detection and identification of threatening biological agents, and the second is the vector infectious diseases testing market, where a RAMP® product is used to test samples from mosquito pools for West Nile Virus to monitor the threat to humans.
We have developed and are selling RAMP® tests for the rapid detection and identification of anthrax, ricin, botulinum toxin and orthopox viruses (including smallpox). Our target market for our RAMP® Biodefense tests is primarily public safety institutions, or first responders, such as fire and police departments, military installations, emergency response teams and hazardous materials (HAZMAT) units. Government agencies and corporations that handle mail are also candidates for an on-site test for anthrax. The rapid detection and identification of biological agents is an important capability affecting the management of a bioterrorism event, forming the basis of emergency response, medical treatment and consequence management. In addition, the rapid identification of biological agents facilitates the quick dismissal of hoaxes and panic-based reports, thereby reducing the logistical burden on first responders who will be required to maintain a higher level of preparedness than in the past. In the aftermath of the terrorist attacks on September 11, 2001, there was an increased desire to be prepared for potential terrorist attacks, particularly on the part of the U.S. government, as evidenced by numerous initiatives, including the creation of the U.S. Department of Homeland Security, or DHS. The first priority of the DHS is to protect the United States against further terrorist attacks. Component agencies analyze threats and intelligence, guard borders and airports, protect critical infrastructure, and coordinate the response of the U.S. for future emergencies.
Following use of anthrax as a weapon for terrorist attacks in the United States in October 2001, we saw an opportunity to adapt the RAMP® technology for the rapid detection and identification of agents used in acts of bioterrorism and initiated development of a test for the rapid, on-site detection of Bacillus anthracis, the causative agent for anthrax, referred to as the Anthrax Test. Development of the Anthrax Test was substantially completed in April 2002 following successful initial validation by the Maryland State Department of Health where testing confirmed that the RAMP® Anthrax Test could reliably detect anthrax spores at levels lower than an infectious dose of 10,000 spores. These results were supported by further independent testing conducted by Defense Research and Development Canada in Suffield. The Anthrax Test was launched commercially in May 2002. In September 2006, our RAMP® Anthrax Test was the first and only biodetection technology approved for field use by first responders in the United States for the detection of anthrax in an independent testing program conducted by AOAC and sponsored by the U.S. Department of Homeland Security. Since the commercial launch of the Anthrax Test in May 2002, we have commercialized tests for ricin, botulinum toxin and orthopox (including smallpox), three priority bio-threat agents. Commercial sales of the ricin test and the botulinum toxin test commenced in November 2002 and the orthopox test was launched in May 2003.
12 Storch GA. Rapid diagnostic tests for influenza. Current Opinion in Pediatrics 2003, 15:77-84.
13 Centers for Disease Control & Prevention – http://www.cdc.gov/RSV/
Vector Infectious Disease Testing
West Nile Virus is an arbovirus that can cause a fatal neurological disease in humans and is commonly found in North America, Europe, Africa, the Middle East, and West Asia. Since its establishment in 1999 into the USA, the virus has spread and is now widely established from Canada to Venezuela. In the US alone, over the last five years (2007 to 2011) the CDC has reported 7417 total human WNV disease cases and a total of 300 deaths. The virus is mainly transmitted to people through the bites of infected mosquitoes, thus mosquito surveillance programs for WNV have been successfully implemented in the USA to mitigate the spread of the virus.
In North America, West Nile Virus prevalence is dependent on climate and has a specific season, beginning in May and ending in September, when the temperature drops, and the mosquito population drops.
Our Products
Cardiovascular Testing Products
Our RAMP® cardiovascular tests are intended for use primarily in hospital emergency rooms, laboratories and walk-in clinics around the world. We have obtained clearance to market these tests in the U.S., Canada, the European Union, China and other regulated jurisdictions.
In China, we sell a co-branded RAMP® Reader, RAMP®200 reader, Myoglobin, CK-MB, Troponin I and NT-proBNP products through O&D Biotech Co., Ltd. (“O&D”), who serves as our exclusive distributor and service agent, with the exception of Hong Kong Special Administrative Region and Macau Special Administrative Region. In addition, Wondfo Biotech Co. Ltd (“Wondfo”) is our exclusive distributor for the marketing and sale of the cardiac reader, assays and controls under private label OEM products bearing the Wondfo name, brand, logo and trademark in those same markets
In Japan our distributor Shionogi& Co., Ltd. (“Shionogi”) sells a rapid quantitative test for BNP under its own brand name.
In the United States, from 2009 through September 2011, Roche Diagnostics Ltd. (“Roche Diagnostics”) had served as our exclusive distributor for sale of cardiovascular products; however our agreement with Roche Diagnostics terminated effective September 30, 2011. Since that time we have resumed selling our products directly to US customers.
In addition to the above, we have agreements with regional distributors that sell our RAMP® cardiovascular products in various countries in Asia, Latin America, Europe, the Middle East and Africa.
Infectious Disease Testing Products
Influenza A/B
In 2008, our partner 3M Company (“3M”) launched their Rapid Detection Flu A+B test. This test provides hospital and physician office laboratories reliable and objective electronic results in approximately 15 minutes. For our flu tests, we were granted a Special 510(k) clearance by the U.S. Food and Drug Administration, or FDA, for an update to our RAMP® Influenza A/B Assay Package Insert to include analytical reactivity information for a strain of the 2009 H1N1 virus cultured from positive respiratory specimens. Although the RAMP® Influenza A/B Assay has been shown to detect the 2009 influenza A (H1N1) virus in cultured isolates, the performance characteristics of this device with clinical specimens that are positive for the 2009 influenza A (H1N1) virus have not been established. The RAMP® Influenza A/B Assay can distinguish between influenza A and B viruses, but it cannot differentiate influenza subtypes.
RSV
In collaboration with 3M, we began development of a RAMP® test for rapid detection of RSV, which infects virtually all children by the age of two. This product was launched in October 2009 and product sales are ongoing through our distributor, 3M.
On-Site Environmental Testing Products
Environmental tests are generally considered to be products and services used to detect and quantify substances and microbes in the environment that have potentially harmful effects to humans. We participate in two distinct areas of the environmental market. The first is biodefense, where RAMP® products are used for the detection and identification of threatening biological agents, and the second is the vector infectious diseases testing market, where a RAMP® product is used to test samples from mosquito pools for West Nile Virus to monitor the threat to humans.
Biodefense Testing Products
We market and sell our biodefense products through a network of regional distributors in the United States, and country-specific national distributors in certain other countries. These efforts are supplemented by direct sales in some geographical territories. Since October 2002, RAMP® biodefense systems have been sold in Canada, the United States, Saipan, Guam, Japan, Italy, Australia, Ireland, Israel, Korea, China, Singapore and the United Arab Emirates. Customers include UNMOVIC, the United States Air Force, the United States Army, Canadian Department of Defense, Health Canada, and the Royal Canadian Mounted Police. RAMP® Systems are being used in major U.S. markets including Chicago, Orlando, Philadelphia, Los Angeles, West Palm Beach, Atlanta, and Houston.
Vector Infectious Diseases Testing Products
The market for our West Nile Virus Test is comprised of the following end users: state public health/veterinary labs; mosquito control districts; and universities. U.S. state government testing figures for 2002 – 2003 indicated that the average state tested approximately 3,500 mosquito pools and 1,000 birds. It is estimated that approximately 279,000 tests are performed throughout North America each year to screen for West Nile Virus. We launched our WNV Test expecting that initial sales would be derived from a mixture of both direct sales and sales generated via distribution partners. On December 1, 2003, we entered into a sole distribution agreement with ADAPCO Inc., the largest distributor of mosquito control products in the United States.
Key Manufacturing, Development, Sales and Distribution Agreements
O&D Biotech, LTD. China
In February 2011, we signed an agreement with O&D that replaces an earlier agreement signed in April 2007, as the exclusive distributor of RAMP co-branded cardiovascular products in the People’s Republic of China, exclusive of Hong Kong and Macau Special Administrative Region. Under the agreement O&D is subject to certain minimum purchase levels. In the event O&D does not meet those levels, we have the option to terminate the agreement.
Wondfo Biotech Co., Ltd
In December 2009 we signed a private label OEM agreement with Wondfo which names Wondfo the exclusive distributor for the marketing and sale of private label OEM cardiac testing products in the People’s Republic of China, excluding Hong Kong and Macau. This agreement is subject to certain minimum purchase requirements by Wondfo. Should they fail to meet the minimum purchase requirements under the agreement, we may at our discretion either convert this to a non-exclusive OEM agreement or immediately terminate the agreement.
Shionogi & Co., Ltd.
In May 2006, we signed an agreement with Shionogi to market and sell our BNP tests in Japan. Under the terms of the agreement, we agreed to become the exclusive manufacturer of BNP assay kit on an OEM basis. The agreement is subject to minimum purchase levels by Shionogi. In the event Shionogi fails to meet these minimum purchase levels, it is required to pay us a percentage of the unit price for each unit that represents the shortfall. The initial term of the agreement ran for three years; however, the agreement is automatically renewed for successive periods of one year under the same terms and conditions (except for price and minimum order quantity). The agreement may be terminated by either party with twelve months notice, should either party be in breach under the terms of the agreement, or under certain other conditions.
3M Company
In September 2004 we signed a Joint Development Agreement (JDA) with 3M. This agreement was amended in November 2006 for development of tests and related reader for detection of infectious diseases.
In November 2006, we entered into a Manufacturing and Supply Agreement with 3M that was subsequently amended in November 2009 and June 2010. The agreement and related amendments set the specific products, prices and terms for the products to be provided by us to 3M. The agreement grants 3M a worldwide, royalty bearing license to all products under the agreement and requires 3M to purchase all tests, readers and related equipment under the agreement exclusively from us. Currently the supply agreement covers the Flu A/B cartridges, RSV cartridges, and the 3M branded Rapid Detection Reader.
In March 2010, we entered into a Distribution Agreement with 3M granting us a right to sell products covered under the Manufacturing and Supply Agreement in China, Hong Kong, Australia, New Zealand, Canada, Japan, Korea, Thailand and Iran. Under the terms of this agreement, we are to pay 3M a royalty on net sales of our Flu A/B cartridges, RSV cartridges and readers under ours or a third party’s brand. The initial term of this agreement is for five years, after which the parties can mutually agree to extend the agreement for another two years.
Adapco, Inc.
In April 2008 we entered into a Distribution Agreement with Adapco, which replaced an earlier agreement signed in March 2006. The initial term of the agreement was for one year, and is automatically renewed on an annual basis. This agreement appoints Adapco the exclusive distributor of tests and readers for detection of West Nile Virus in the United States; however the agreement gives us the right to sell these products directly should we choose.
Roche Diagnostics GmbH and Roche Diagnostics Ltd.
In July 2005, we secured a license from Roche Diagnostics GmbH to develop, manufacture and sell POC tests for the detection of NT pro-BNP in markets where we do not sell BNP tests.
In June 2008, we entered into a Sales and Distribution Agreement with Roche Diagnostics. That agreement granted Roche Diagnostics the rights to market our line of cardiovascular POC tests worldwide with the exception of Japan. This agreement was revised in February 2010 to limit the licensed territory to the US.
On September 2, 2011, we received notification from Roche Diagnostics that they have terminated the sales and distribution agreement between Roche and Response effective September 30, 2011. Roche Diagnostics terminated the agreement because we have not obtained the necessary approvals from the FDA to permit Roche Diagnostics to market the Response’s cardiovascular POC tests in the United States using the RAMP® 200 Reader.
Competition
Medical Point-of- Care Market
The medical POC test market is comprised of five basic segments: clinical chemistry, hematology, immunoassay, blood glucose and urinalysis, plus miscellaneous other tests. Dozens of companies sell qualitative POC tests in these segments. Few companies however, participate in the quantitative POC immunoassay market. The following table summarizes our key known competitors in the POC testing market.
| | Test Market Segment |
| Company | Cardiac Markers | CHF Marker | Drugs of Abuse | Flu and Infectious Disease | Pregnancy / Ovulation | Blood Gases/ Electrolytes | Coagulation |
| Response Biomedical Corporation | Ö | Ö | | Ö | | | |
| Abbott Point of Care Inc. | Ö(1) | Ö | | | | Ö | Ö |
Becton Dickinson Corporation (4) | | | | Ö | | | |
| Dade Behring | Ö | Ö | | | | | |
| Alere Inc. | Ö | Ö | Ö | Ö | Ö | Ö | Ö |
Mitsubishi Chemical Medience Corporation (3) | Ö | Ö | | | | | |
Roche Diagnostics (2) | Ö | Ö | Ö | | | Ö | Ö |
Quidel Corporation (4) | | | | Ö | Ö | | |
| (1) | Only Troponin I, CK-MB and BNP cardiac tests at this time. |
| (2) | The Cardiac Reader measures Troponin T rather than TnI and does not measure CK-MB. This platform uses semi-quantitative technology. This limits the upper end of their NT-proBNP assay to only 20% of the entire clinical range. |
| (3) | Mitsubishi Pathfast weighs 33kg, which for some would not be considered a POC system but rather a small laboratory analyzer. |
| (4) | These companies sell rapid Influenza tests that are visually read, require precise timing and do not require an instrument. |
Certain of the competitors listed in the table above have stated their intention to broaden their category offerings. In addition to the key competitors listed above, we believe that each of the major diagnostics companies has an active interest in POC testing and, as well as being potential competitors, are also potential business partners.
Alere Inc. (formerly Inverness Medical Innovations Inc.), or Alere, has sold a three-in-one quantitative immunoassay and reader system for cardiac markers (CK-MB, Troponin I and Myoglobin) since 1999 and is currently one of the leading participants in quantitative POC cardiovascular testing on the basis of market share, revenues and technology. They also sell a “shortness of breath” panel cartridge, which includes Myoglobin, CK-MB, Troponin I, BNP and D-Dimer. While BNP is available as a stand-alone cartridge, this system can only perform Troponin I tests as part of a panel. In 2007, Biosite Incorporated, or Biosite, was acquired by Inverness Medical Innovations in a transaction valued at $1.68 billion. Based on published list prices for the Biosite products and data from the completed multi-site clinical study entitled “Evaluation of a point-of-care assay for cardiac markers for patients suspected of acute myocardial infarction”14, we believe that RAMP® has several advantages over the competing Biosite products including product performance and menu flexibility. These results were recently replicated in an unpublished trial.
14 Munjal I, Gialanella P, Goss C, McKitrick JC, Avner JR, Quiulu Pan, Litman C, Levi ML, J Clin Micro 2011Mar 49(3):1151-3.
Since 2003, Abbott Point of Care Inc., (formerly iStat Corporation) has sold a 10-minute Troponin I test for use on the i-STAT Portable Clinical Analyzer, a biosensor based technology. In 2005, Abbott Point of Care launched a CK-MB test and in 2006, launched a POC BNP test. In addition, Abbott Point of Care offers several tests for other markers in whole blood, predominantly electrolytes and blood gases. We believe the requirement for different sample types for the i-STAT markers for heart attack (TnI and CK-MB) and congestive heart failure (BNP) is a significant disadvantage as compared to the RAMP® system.
Quidel Corporation has sold rapid, qualitative tests for the detection of RSV and Influenza A and B since 2001 as the QuickVue® Influenza A+B test and the QuickVue RSV test. Based on data published in March 2011, we believe that the RAMP® RSV test branded under 3M has superior performance than the QuickVue RSV test. Binax, Inc., a division of Alere, has been selling Influenza A, Influenza B and RSV tests under the BinaxNOW® brand since 2002 and a combined Influenza A + B test since 2004. Both of these tests have received Clinical Laboratory Improvement Amendments of 1988, or CLIA, waived status, which allows for their use in physician office laboratories. Other tests available include Becton-Dickinson and Thermo Electron Corp., which produce rapid Influenza A+B tests that are not currently CLIA waived.
Other technologies that may compete against RAMP® in the future by delivering highly sensitive, quantitative results, for some POC tests include immunosensors or biosensors and nanotechnology-based approaches. Biosensor methods use specific binding molecules such as antibodies to generate a measurable signal as a direct result of binding to their target molecule (or analyte). These technologies are extremely complex and have been under development for many years with limited commercial success to date. Immunobiosensors, to date, have limited sensitivity and are not competitive with RAMP®. Although methods of testing using biosensors and nanotechnology can be fast, they generally suffer from a significant lack of accuracy, repeatability and reliability, and can be expensive to produce. Biosensors are now in limited use for selected diagnostic applications, most notably for blood glucose monitoring using non-immunoassay methods. Nanotechnology is a relatively new and growing field that deals with the use of inert micro-etched wafers, or chips, to provide templates for chemical, biochemical, and biological processes.
Much of the research effort for recent diagnostic testing has been directed toward the development of DNA hybridization probe tests. These tests identify specific gene sequences that can be associated with certain genetically based disorders, infectious diseases and the prediction of predisposition to certain medical conditions such as cancer. Several companies, such as Becton-Dickinson and Gen-Probe Inc. are now marketing specific probe tests for infectious diseases such as tuberculosis, hepatitis, Legionnaires disease and vaginitis. DNA probe technology is useful for gene markers that have been shown to be associated with specific disease states or clinical conditions. Although more useful gene sequences are being discovered all the time, we believe they will not displace the need for high-sensitivity immunoassays; there is, for instance, no genetic change when a person has a heart attack. In addition, the RAMP® format may be applicable to hybridization probe methods if a need is found for these tests to be quantitative and at the POC.
The following table summarizes our known competitors in the rapid on-site environmental biodefense testing market (note that this table may not include all biological agents for which these companies may have tests):
| Company | Biological Agent |
| Anthrax | Ricin | Botulinum Toxin | Orthopox | Brucella | Plague | Tularemia | SEB |
| Response Biomedical Corporation | Ö | Ö | Ö | Ö | | | | |
Alexeter Technologies LLC (1) | Ö | Ö | Ö | Ö | Ö | Ö | Ö | Ö |
| New Horizons Diagnostics | Ö | Ö | Ö | | | Ö | Ö | Ö |
| ADVNT Inc. | Ö | Ö | Ö | | | Ö | | Ö |
Idaho Technology Inc. (2) | Ö | Ö | Ö | Ö | Ö | Ö | Ö | |
| Tetracore, Inc. | Ö | Ö | Ö | Ö | Ö | Ö | Ö | Ö |
Smiths Detection BioSeeq Plus (3) | Ö | | | Ö | | Ö | Ö | |
| QTL/MSA | Ö | Ö | | | | | | Ö |
| (1) | Product includes a portable reader based on reflectance technology. |
| (2) | Product includes a portable reader based on polymerase chain reaction technology. |
| (3) | Product includes a portable reader based on polymerase chain reaction technology. |
A number of independent studies have been conducted on biodefense tests. The RAMP® Anthrax Test has been evaluated at four sites in the United States and Canada: DRDC Suffield,15 a division of the Canadian Department of National Defense; the Maryland State Department of Health;16 Intertox Inc.,17 a Seattle-based public and occupational health firm; and, Edgewood Chemical Biological Center, part of the U.S. Army's Aberdeen proving ground, and more recently the AOAC testing.18 Data from these four evaluations show that the RAMP® Anthrax Test meets or exceeds its product claims of reliably detecting less than 4,000 live spores, with 99 percent confidence in specificity. The CDC defines a lethal dose of anthrax as 10,000 spores.
In November 2004, the RAMP® System was the only commercially available rapid on-site anthrax detection system of those tested that met the new performance standards introduced by AOAC for rapid immunoassay-based anthrax detection systems and to receive the AOAC Official Methods Certificate 070403 stating that the RAMP® Anthrax Test performed as we claimed and that we are authorized to display the AOAC Performance Tested certification mark. All other commercially available rapid on-site anthrax detection systems tested failed to meet the AOAC’s performance standard. A further intensive, independent field testing program conducted by AOAC and sponsored by the DHS, culminated in the announcement in September 2006 that our RAMP® Anthrax Test is the first and only biodetection product approved for field use by first responders in the United States for the detection of anthrax. To date, RAMP® remains the only product with this designation by the AOAC.
Since the initial RAMP® product launch, many competitors have launched competitive products into the market place, including Alexeter Biotechnologies and ADVNT Inc., both of whom also market rapid detection tests in the US for Anthrax, Ricin and Botulinum, in addition to SEB, Y. pestis (plague) and others. Internationally, Smith Detection maintains a strong market share. We also believe that a number of diagnostics companies have an active interest in rapid on-site biodefense testing and have the potential to become either competitors or business affiliates.
We currently have approximately 346 RAMP® biodefense systems in field use by our customers (see “On-Site Environmental Testing Market, Industry Trends” and “Risk Factors”).
15 Defence Research and Development Canada, July, 2002.
16 Maryland State Department of Health, March 2002.
17Intertox Inc., July 2002.
18 The U.S. Army Aberdeen Proving Ground, November 2004.
Vector Infectious Diseases Market
Our main competitor in the rapid on-site environmental vector infectious disease testing market is VecTest, currently distributed by Thermo Scientific. VecTest is a qualitative test strip, available in multiplex format, used to detect WNV, SLE, EEE and WEE. While a study by the CDC in 2006 confirmed that RAMP® outperforms VecTest19, VecTest continues to occupy a market share due to its qualitative nature as no instrument is required to perform the testing.
Emerging competition in the detection of West Nile Virus in the US market is from RT-PCR systems, as more labs are investing in PCR technology in lieu of rapid detection.
Operations and Manufacturing
A RAMP® System consists of a Reader and kits, or Kits, of applicable RAMP® tests. Manufacturing of the Readers is currently outsourced to an electronics manufacturer that we have qualified, located in British Columbia. We manufacture all Kits in-house in order to maximize return on investment, protect proprietary technology, and ensure compliance with government and internal quality standards. Kit manufacturing includes reagent and component production, cartridge assembly and final packaging with the exception of our Chinese customers where bulk components are shipped to our distributors in China for final packaging.
In advance of expected growth of our products, we invested significantly, since 2007, to increase automation, quality and capacity of our manufacturing operations. In March 2008, we moved into our current corporate headquarters, a leased, multi-use, 46,000 square foot facility in Vancouver, British Columbia where we coordinate all support operations including customer support, technical and instrument service, production planning, shipping and receiving. The facility houses all of our operations and will allow us to achieve our projected manufacturing capacity targets during the term of the agreement. Our manufacturing scale up is ongoing, and our test production capacity has increased from approximately 500,000 tests per shift per year to approximately 2 million tests per shift per year. The initial term of the lease agreement is 15 years with two 5-year renewal options.
Where possible, we require distribution and marketing partners to provide a twelve-month rolling forecast in order to ensure timely and adequate product supply and to allow efficient production, materials, shipping and inventory planning, see “Risk Factors". We plan to meet cost and quality targets through strict scale-up validation procedures and by negotiating supplier agreements for key materials. Final packaging, inventory storage and product distribution to marketing partners will be managed in accordance with individual partner agreements.
The primary raw materials for a test cartridge consist of: antibody reagents, nitrocellulose membrane and injection molded plastic parts to act as housing for the cartridge assembly. There are several different components required to perform the test that are included with the Kit. These are a sample transfer device, a solution for diluting the test sample and reagent-containing tips (for placing the sample being tested into the cartridge). Response purchases these primary raw materials from unaffiliated domestic and international suppliers, some of which are sole suppliers. Interruptions in the delivery of these materials or services could adversely impact the Company.
Patents and Proprietary Rights
We rely on a combination of patents, trademarks, confidential procedures, contractual provisions and similar measures to protect our proprietary information. To develop and maintain our competitive position, we also rely upon continuing invention, trade secrets and technical know-how.
It has been our practice to periodically file for patent and trademark protection in the U.S. and other countries with significant markets, such as Canada, Western European countries, Japan and China. No assurance can be given that patents or trademarks will be issued to us pursuant to our applications or that our patent portfolio will provide us with a meaningful level of commercial protection. We file patent applications on our own behalf as assignee and, when appropriate, have filed and expect to continue to file, applications jointly with our collaborators. Our ability to obtain and enforce patents is uncertain and we cannot guarantee that any patents will issue from any pending or future patent applications owned by or licensed to us, see “Risk Factors”.
19 Burkhalter et al, 2006
We hold exclusive ownership to technology that originated from the University of British Columbia, which has resulted in three issued U.S. patents, one issued Canadian patent and four active foreign patents. Our key patent is also pending in China and Hong Kong. In addition Response holds an additional two patents in the U.S. These patents have expiration dates ranging from 2020 to 2028. We have pending applications for 3 additional U.S. patents and numerous foreign counterparts as well as one pending patent application filed in both the US and Europe, with joint inventorship and assignment with one of our partners. We also have two granted Design Patents related to the Ramp® 200 reader. We have four registered trademarks.
We seek to protect our trade secrets and technology by entering into confidentiality agreements with employees and third parties (such as potential licensees, customers, strategic partners and consultants). In addition, we have implemented certain security measures in our laboratories and offices. Despite such efforts, no assurance can be given that the confidentiality of our proprietary information can be maintained. Also, to the extent that consultants or contracting parties apply technical or scientific information independently developed by them to our projects, disputes may arise as to the proprietary rights to such data.
Government Regulation
Regulatory Approval
Point of Care Diagnostics
The FDA, Health Canada and comparable agencies in foreign countries impose substantial requirements upon the development, manufacturing and marketing of drugs and medical devices through the regulation of laboratory and clinical testing procedures, manufacturing, marketing and distribution by requiring labeling, registration, notification, clearance or approval, record keeping and reporting, see “Risk Factors”.
In China, clearance to market drugs and medical devices must be granted by the State Food & Drug Administration (“SFDA”). In May 2004 and November 2004, O&D received regulatory clearance from the SFDA to market the RAMP® Reader and three RAMP® cardiac marker tests in China. In November 2007, they received clearance to market the RAMP® NT-proBNP Assay and in December 2010 the clearance to market the RAMP®200 Reader was received.
As of December 7, 2003, all medical devices sold in the countries of the European Union, or the EU, are required to be compliant with the EU In-Vitro Diagnostic Directive. All new in-vitro diagnostic devices must bear a mark, called the CE Mark, to be registered and legally marketed in the EU after that date. The regulatory requirements for marketing are based on the classification of the individual products and EU member countries are not allowed to impose any additional requirements on medical device manufacturers other than the language used in product labeling. In April 2003, we fulfilled the requirements of the EU In-Vitro Diagnostic Directive Essential Requirements for the three RAMP® cardiac tests and Reader; in December 2006, we registered the NT-proBNP test as well as the RAMP® liquid cardiac marker controls used by laboratories to verify Kit performance and user technique; in May 2009, we registered the RAMP® 200 reader and in December 2009, we registered the Influenza A/B Test. Through the EC Declaration of Conformity, we are entitled to apply the CE Mark to these products. As with the FDA, future RAMP® tests may have different classifications which would require ISO 13485 registration as well as a technical file review by a registration organization, known as a Notified Body, prior to authorization to apply the CE Mark.
Prior to sale in the United States, RAMP® clinical products will typically require pre-marketing clearance through a filing with the FDA called a 510(k) submission. A 510(k) submission claims substantial equivalence to an accepted reference method or a similar, previously cleared product known as a “predicate device” and minimally takes about 100 days for approval once a submission is made. Some RAMP® tests may detect analytes or have applications, intended uses for which there are no equivalent products on the market. In such cases, the test will require pre-market approval, a process that requires clinical trials to demonstrate clinical utility, as well as safety and efficacy of the product. Including clinical trials, the pre-market approval process can take approximately two years.
Marketing clearance for the RAMP® Myoglobin Assay and RAMP® Reader was received in 2002. Marketing clearances for the RAMP® CK-MB Assay and the RAMP® Troponin I Assay on the RAMP® Reader were received in May 2004. The marketing clearance for the RAMP® Influenza A/B Assay and the RAMP® 200 Reader was received in April 2008. The marketing clearance for the RAMP® NT-proBNP Assay on the RAMP® Reader was received in July 2008. The marketing clearance for the RAMP® RSV Assay on the RAMP® 200 was received in July 2009.
As of March 23, 2012, we have received the FDA premarket clearance for Myoglobin, CK-MB, Troponin I and NT-proBNP on the RAMP® Reader, Flu and RSV on the RAMP®200 reader. We are currently developing additional tests that we will have to clear with the FDA through the 510(k) notification procedures. These new test products are crucial for our continued success in the human medical market. If we do not receive 510(k) clearance for a particular product, we will not be able to market that product in the United States until we provide additional information to the FDA and gain premarket clearance. The inability to market a new product during this time could harm our future sales in the US.
For our products to be sold in the physicians’ office lab market in the U.S., we will need to obtain waiver status under the CLIA. A CLIA-waived test is a test that employs methodologies that are so simple and accurate as to render the likelihood of erroneous results negligible and/or pose no reasonable risk of harm to the patient if the test is performed incorrectly. CLIA-waived tests are designed to be performed by less experienced and untrained personnel. The current CLIA regulations divide laboratory tests into three categories: “waived,” “moderately complex” and “highly complex.” Many of the tests performed using the RAMP system is in the “moderately complex” category. Moderately complex tests can only be performed in laboratories fulfilling certain criteria, which are fulfilled by a minority of physician office laboratories in the U.S.
In Canada, in vitro diagnostics are regulated by the Therapeutic Products Directorate of Health Canada, referred to as the TPD, and are licensed for sale through submission to the TPD. The timeline for approval is similar to that of the FDA’s 510(k) process. As of January 2003, all new and existing class II, III and IV Medical Device Licenses, or MDLs, in Canada also require a valid International Organization for Standardization, or ISO, 13485 or ISO 13488 Quality System Certificate from a registrar recognized by the Canadian Medical Devices Conformity Assessment System (“CMDCAS”). We achieved registration to the ISO 13485:2003 standard in April 2004. An MDL was issued for the Myoglobin Assay and Reader in 2002. MDLs were received for the RAMP® CK-MB Assay and RAMP® Troponin I Assay in August 2004; for the RAMP® NT-proBNP Assay in June 2007, and for RAMP® liquid cardiac marker controls used by laboratories to verify Kit performance and user technique. These controls are manufactured for Response by a U.S. company who holds the 510(k) clearance with the FDA. An MDL was received for the RAMP® Influenza A/B Assay and RAMP® 200 reader in May 2009. An MDL was received for the RAMP® RSV Assay in May 2010.
In other parts of the world, the regulatory process varies greatly and is subject to rapid change. Many developing countries only require an import permit from their own government agency or proof of approval from the regulatory agency in the manufacturer’s country of origin. We require our marketing and distribution partners to ensure that all regulatory requirements are met in order to sell RAMP® tests in their respective territories.
Clinical consultants are used to support in-house resources where necessary to develop protocols and prepare regulatory submissions for government agencies such as the FDA and the TPD. We completed multi-center clinical trials for the RAMP® Myoglobin Assay and the RAMP® Reader in 2001, for the RAMP® CK-MB Assay and the RAMP® Troponin I Assay in November 2003, for the RAMP® NT-proBNP Assay in November 2006 and for the Flu A/B Assay and the RAMP® 200 reader in May 2007, and for the RAMP® RSV Assay in November 2008.
ON-SITE ENVIRONMENTAL TESTING
Biodefense Testing
There are currently no regulatory approvals or clearances required to market on-site environmental biodefense tests in North America. There appears to be some support from the market for regulatory oversight of such testing, and regulatory agencies such as the Department of Homeland Security may in the future impose substantial requirements upon the development, manufacturing and marketing of devices through the regulation of laboratory and clinical testing procedures, manufacturing, marketing and distribution by requiring labeling, registration, pre-market notification, clearance or approval, record keeping and reporting. While additional regulatory requirements will make it more difficult for poorly performing products to participate in the market, they could also significantly increase the time and cost for companies to bring new tests to market, creating a barrier to entry.
The lack of regulatory oversight in the biodefense industry means there is virtually no independent data available for a customer to verify a manufacturer’s product claims. Since launching our Anthrax Test, we have received third party validation of the product’s performance; see “On-Site Environmental Testing Market, Competition”. Currently, however, companies do not require any form of regulatory clearance to market handheld assays for the detection of biodefense threats such as anthrax, see “Risk Factors”.
The performance and field-testing programs conducted by the AOAC in 2004 through 2006 were developed in collaboration with and funded by the DHS. One of the goals of the testing programs was to develop industry performance standards. The final form and substance of these possible standards and the impact on the industry is currently unclear.
Vector Infectious Diseases Testing
There is no regulatory clearance required for our RAMP® West Nile Virus Test because it is only used for testing mosquitoes. If we develop a Flu A or H5N1 screening test for birds, it will require regulatory clearance in some countries, such as the United States, where such tests are regulated by the Center for Veterinary Biologics division of the Department of Agriculture.
Manufacturing Regulations and Various Federal, State, Local and International Regulations
The 1976 Medical Device Amendment also requires us to manufacture our RAMP products in accordance with Good Manufacturing Practices guidelines. Current Good Manufacturing Practice requirements are set forth in the 21CFR 820 Quality System Regulation. These requirements regulate the methods used in, and the facilities and controls used for the design, manufacture, packaging, storage, installation and servicing of our medical devices intended for human use. Our manufacturing facility is subject to periodic inspections. In addition, various state regulatory agencies may regulate the manufacture of our products.
Federal, state, local and international regulations regarding the manufacture and sale of health care products and diagnostic devices may change. In addition, as we continue to sell in foreign markets, we may have to obtain additional governmental clearances in those markets.
To date, we have complied with the following federal, state, local and international regulatory requirements:
| · | United States Food and Drug Administration: In August 2007, the FDA conducted a facility inspection and verified our compliance with the 21 CFR 820 Regulation. |
| · | Health Canada Therapeutic Products Directorate: In 2004, the TPD granted our manufacturing facility Medical Device Licenses, based on the Medical Device Regulations (SOR/98-282), Section 36, for the manufacture of our medical devices . |
| · | International Organization for Standardization: In July 2004, we received our ISO 9001 certification, expanding our compliance with international quality standards. In April 2004, we received ISO 13485 Quality System certification as required by the 2003 European In Vitro Device Directive. This certified our quality system specifically to medical devices. In April 2004, we received the Canadian Medical Device Conformity Assessment System stamp on our ISO 13485 certificate to signify compliance with Health Canada regulations. In June 2010, we received our recertification to the ISO 13485:2003 Quality System Standard for medical devices. |
Research and Development Expenditure
Research and development activities relate to development of new tests and test methods, clinical trials, product improvements and optimization and enhancement of existing products. Our research and development expenses, which consist of personnel costs, facilities, materials and supplies, regulatory activities and other related expenses were $2.9 million, $4.1 million, and $5.6 million for the years ended December 31, 2011, 2010, and 2009, respectively.
Seasonal Variations in Business
Our operating results may fluctuate from quarter to quarter due to many seasonal factors. Many of our end-users are government related organizations at a federal, state/provincial or municipal level. Consequently, our sales may be tied to government budget and purchasing cycles. Sales may also be slower in the traditional vacation months, could be accelerated in the first or fourth calendar quarters by customers whose annual budgets are about to expire (especially affecting purchases of our fluorescent Readers), may be distorted by unusually large Reader shipments from time to time, or may be affected by the timing of customer cartridge ordering patterns.
Backlog
Because we ship our products shortly after we receive the orders from our customers, we generally operate with a limited order backlog. As a result, our product sales in any quarter are generally dependent on orders that we receive and ship in that quarter. As a result, any such revenue shortfall would immediately materially and adversely impact our operating results and financial condition. The sales cycle for our products can fluctuate, which may cause revenue and operating results to vary significantly from period to period. We believe this fluctuation is primarily due (i) to seasonal patterns in the decision making processes by our independent distributors and direct customers, (ii) to inventory or timing considerations by our distributors and (iii) to the purchasing requirements by various international governments to acquire our products.
Risks Attendant to Foreign Operations and Dependence
We sell in China through an exclusive distributor for RAMP branded products, O&D Biotech Co. Ltd., China (O&D), and an exclusive distributor for private labeled OEM products, Wondfo Biotech Co. Ltd. (Wondfo). Sales to O&D accounted for 44% of our total product sales in the year ended December 31, 2011. If O&D underperforms we may not be able to generate alternative distribution channels rapidly enough to prevent a significant disruption in sales generated in China, which would have an adverse impact on our business performance.
Financial Information about Industry Segments
The Company operates primarily in one business segment, the research, development, commercialization and distribution of diagnostic technologies, with primarily all of its assets and operations located in Canada. The Company’s revenues are generated from product sales primarily in China, the United States, Europe, Asia and Canada. Expenses are primarily incurred from purchases made from suppliers in Canada and the United States.
Product sales by customer location were as follows:
| | | 2011 | | | 2010 | | | 2009 | |
| | | | $ | | | $ | | | $ | |
| China | | | 5,281,063 | | | | 3,576,935 | | | | 2,273,055 | |
| United States | | | 1,551,444 | | | | 1,003,297 | | | | 3,752,552 | |
| Asia (excluding China) | | | 794,667 | | | | 844,633 | | | | 774,676 | |
| Europe | | | 654,649 | | | | 812,328 | | | | 801,629 | |
| Canada | | | 59,148 | | | | 52,872 | | | | 76,484 | |
| Other | | | 683,112 | | | | 502,065 | | | | 474,653 | |
| Total | | | 9,024,083 | | | | 6,792,130 | | | | 8,153,049 | |
Product sales by type of product were as follows:
| | | 2011 | | | 2010 | | | 2009 | |
| | | $ | | | $ | | | $ | |
| Cardiovascular | | | 7,295,501 | | | | 5,969,672 | | | | 5,984,267 | |
| Infectious Diseases | | | 587,040 | | | | 67,472 | | | | 844,384 | |
| | | 659,463 | | | | 444,895 | | | | 517,981 | |
Vector Infectious Diseases | | | 482,080 | | | | 310,090 | | | | 806,417 | |
| Total | | | 9,024,083 | | | | 6,792,130 | | | | 8,153,049 | |
For further information, please see Item 6 (“Selected Financial Data”).
Working Capital
Please see Item 6 (“Selected Financial Data”) and Item 7 (“Management's Discussion and Analysis of Financial Condition and Results of Operations").
Employees
On December 31, 2011, we had 67 full-time employees.
Available Information
Our corporate Internet address is www.responsebio.com. At the "Investors" section of this website, we make available free of charge our Annual Report on Form 10-K, our Annual Proxy statement, our quarterly reports on Form 10-Q, any Current Reports on Form 8-K, and any amendments to these reports, as soon as reasonably practicable after we electronically file them with, or furnish them to, the Securities and Exchange Commission, or the SEC, as well as our previously filed reports on Forms 20-F and 6-K. The information found on our website is not part of this Annual Report on Form 10-K. In addition to our website, the Securities and Exchange Commission, or the SEC, maintains an Internet site at www.sec.gov that contains reports, proxy and information statements, and other information regarding us and other issuers that file electronically with the SEC.
Our future performance is subject to a number of risks. If any of the following risks actually occur, our business could be harmed and the trading price of our common stock could decline. In evaluating our business, you should carefully consider the following risks in addition to the other information in this Annual Report on Form 10-K. We note these factors for investors as permitted by the Private Securities Litigation Reform Act of 1995. It is not possible to predict or identify all such factors and, therefore, you should not consider the following risks to be a complete statement of all the potential risks or uncertainties that we face.
Risks Related to Our Company
We may need to raise additional capital to fund operations. If we are unsuccessful in attracting capital to our Company, we will not be able to continue operations or will be forced to sell assets to do so. Alternatively, capital may not be available to our Company on favorable terms, or at all. If available, financing terms may lead to significant dilution to the shareholders' equity in our Company.
We are not profitable and have negative cash flow from operations. Based on our current cash resources, expected cash burn, and anticipated revenues, we expect that we can maintain operations through the fourth quarter of 2012. We may need to raise additional capital to fund our operations. We have relied primarily on debt and equity financings to fund our operations and commercialize our products. Additional capital may not be available, at such times or in amounts as needed by us. Even if capital is available, it might be on adverse terms. Any additional equity financing will be dilutive to our shareholders. If access to sufficient capital is not available as and when needed, our business will be materially impaired and we may be required to cease operations, curtail one or more product development programs, attempt to obtain funds through collaborative partners or others that may require us to relinquish rights to certain technologies or product candidates, or we may be required to significantly reduce expenses, sell assets, seek a merger or joint venture partner, file for protection from creditors or liquidate all our assets.
Our inability to generate sufficient cash flows may result in our Company not being able to continue as a going concern.
We have incurred significant losses to date. As at December 31, 2011, we had an accumulated deficit of $106,890,091 and had not generated positive cash flow from operations. Accordingly, there is substantial doubt about our ability to continue as a going concern. We may need to seek additional financing to support our continued operation; however, there are no assurances that any such financing can be obtained on favorable terms, if at all. In view of these conditions, our ability to continue as a going concern is dependent upon our ability to obtain such financing and, ultimately, on achieving profitable operations. The outcome of these matters cannot be predicted at this time. The consolidated financial statements for the year ended December 31, 2011 do not include any adjustments to the amounts and classification of assets and liabilities that might be necessary should we be unable to continue in business. Such adjustments could be substantial.
We have incurred substantial operating losses to date. We expect these losses to continue for the near future. If we are unable to generate sufficient revenue, positive cash flow or earnings, or raise sufficient capital to maintain operations, we may not be able to continue operating our business and be forced to sell our Company or liquidate our assets.
We have evolved from a pure development company to a commercial enterprise but to date have realized minimal operating revenues from product sales. As of December 31, 2011, we have incurred cumulative losses since inception of $106,890,091. For the fiscal years ending December 31, 2011, 2010, and 2009, we incurred losses of $5,371,312, $10,081,911, and $9,543,531, respectively. We currently are not profitable and expect operating losses to continue. Generating revenues and profits will depend significantly on our ability to successfully develop, commercialize, manufacture and market our products. The time necessary to achieve market success for any individual product is uncertain. No assurance can be given that product development efforts will be successful, that required regulatory approvals can be obtained on a timely basis, if at all, or that approved products can be successfully manufactured or marketed. Consequently, we cannot assure that we will ever generate significant revenue or achieve or sustain profitability. As well, there can be no assurance that the costs and time required to complete commercialization will not exceed current estimates. We may also encounter difficulties or problems relating to research, development, manufacturing, distribution and marketing of our products. In the event that we are unable to generate adequate revenues, cash flow or earnings, to support our operations, or we are unable to raise sufficient capital to do so, we may be forced to cease operations and either sell our business or liquidate our assets.
Current and future conditions in the global economy may have a material adverse effect on our business prospects, financial condition and results of operations.
During the second half of fiscal year 2008, the global financial crisis, particularly affecting the credit and equity markets, accelerated and the global recession deepened, with an exceptionally weak global economy in 2009 and 2010 followed by a mixed economic performance during 2011. Though we cannot predict the extent, timing or ramifications of the global financial crisis and the economic outlook in different economies, we believe that the current downturn in the world's major economies and the constraints in the credit markets have heightened or could heighten a number of material risks to our business, results of operations, cash flows and financial condition, as well as our future prospects, including the following:
| · | Credit availability and access to equity markets — Continued issues involving liquidity and capital adequacy affecting lenders could affect our ability to fully obtain credit facilities or additional debt and could affect the ability of any lenders to meet their funding requirements when we need to borrow. Further, the high level of volatility in the equity markets and the decline in our stock price may make it difficult for us to access the equity markets for additional capital at attractive prices, if at all. If we are unable to obtain credit, or access the capital markets, where required, our business could be negatively impacted. |
| · | Credit availability to our customers — We believe that many of our customers are reliant on liquidity from global credit markets and, in some cases, require external financing to fund their operations. As a consequence, if our customers lack liquidity, it would likely negatively impact their ability to pay amounts due to us. |
| · | Commitments from our customers — There is a greater risk that customers may be slower to make purchase commitments during the current economic downturn, which may negatively impact the sales of our new and existing products. |
| · | Supplier difficulties — If one or more of our suppliers experiences difficulties that result in a reduction or interruption in supplies or services to us, or they fail to meet any of our manufacturing requirements, our business could be adversely impacted until we are able to secure alternative sources, if any. |
Many of these and other factors affecting the diagnostic technology industry are inherently unpredictable and beyond our control.
We are not able to predict sales in future quarters and a number of factors affect our periodic results, which makes our quarterly operating results less predictable.
We are not able to accurately predict our sales in future quarters. A significant portion of our product sales is made through distributors, including strategic alliance partners, both domestically and internationally. As a result, our financial results, quarterly product sales, trends and comparisons are affected by seasonal factors and fluctuations in the buying patterns of end-user customers, our distributors, and by the changes in inventory levels of our products held by these distributors. For example, higher utilization rates of our BNP and NT-proBNP tests may be due to a higher number of emergency department visits by patients exhibiting shortness of breath, a symptom of heart failure and of influenza. However, higher utilization may also result from greater awareness, education and acceptance of the uses of our tests, as well as from additional users within the hospitals. Accordingly, our sales in any one quarter or period are not indicative of our sales in any future period.
We generally operate with a limited order backlog, because we ship our products shortly after we receive the orders from our customers. As a result, our product sales in any quarter are generally dependent on orders that we receive and ship in that quarter. As a result, any such revenue shortfall would immediately materially and adversely impact our operating results and financial condition. The sales cycle for our products can fluctuate, which may cause revenue and operating results to vary significantly from period to period. We believe this fluctuation is primarily due (i) to seasonal patterns in the decision making processes by our independent distributors and direct customers, (ii) to inventory or timing considerations by our distributors and (iii) to the purchasing requirements by various international governments to acquire our products.
Accordingly, we believe that period to period comparisons of our results of operations are not necessarily meaningful. In the future, our periodic operating results may vary significantly depending on, but not limited to, a number of factors, including:
| · | new product announcements made by us or our competitors; |
| · | changes in our pricing structures or the pricing structures of our competitors; |
| · | our ability to develop, introduce and market new products on a timely basis, or at all; |
| · | our manufacturing capacities and our ability to increase the scale of these capacities; |
| · | the mix of product sales between our instruments and our consumable products; |
| · | the amount we spend on research and development; and |
| · | changes in our strategy. |
We rely on a limited number of third party distributors to market and sell our products in China
We sell in China through an exclusive distributor for RAMP branded products, O&D Biotech Co. Ltd., China (O&D), and an exclusive distributor for private labeled OEM products, Wondfo Biotech Co. Ltd. (Wondfo). Sales to O&D accounted for 45% of our total product sales in the year ended December 31, 2011. If O&D underperforms we may not be able to generate alternative distribution channels rapidly enough to prevent a significant disruption in sales generated in China, which would have an adverse impact on our business performance.
Although we are a Canadian company, a small number of our products are subject to U.S. export control and economic sanctions laws. Although we did not intend to do so, we may have violated certain of these laws, and we cannot currently assess with certainty the nature and extent of fines or other penalties, if any, that U.S. governmental agencies may impose against us, our management, or other employees for these potential violations.
In the first quarter of 2012, we determined that a small number of products that we shipped to Iran may be subject to U.S. export controls and may have required a license from the U.S. Government prior to export. Although these products are manufactured in Canada, they incorporate U.S. origin components, and for that reason, they may be subject to U.S. controls. As a result, we may have violated applicable sanctions and export control laws.
Upon identifying these potential violations, we promptly began a review of our sanctions and export control compliance with our outside counsel. On March 27, 2012, we made voluntary initial filings with the Office of Foreign Assets Control of the United States Department of the Treasury, or OFAC, and with the Bureau of Industry and Security of the United States Department of Commerce, or BIS, notifying these regulatory agencies that we were conducting a review of export control matters and that we would submit any supplemental voluntary self disclosures once our internal review was complete.
Outside counsel is conducting a review of the potential export transaction at issue in preparation for filing the final voluntary disclosures to be submitted to OFAC and BIS. We cannot predict when OFAC or BIS will complete their reviews of our submissions, and resolution of these matters with the government could take substantial time and require substantial expenditure and management resources, either of which could be harmful to our business and operating results. OFAC and BIS may conclude that our actions resulted in violations that warrant the imposition of penalties that could include fines, termination of our ability to export our products, and/or referral for criminal prosecution. Penalties and potential criminal enforcement actions could be imposed against us and/or our management and certain of our employees. Penalties that the regulatory agencies may seek to impose could have a material adverse effect on our business, operating results, and financial condition. The maximum civil monetary penalty for each violation is up to $250,000 or twice the value of the transaction, whichever is greater.
A larger-than-required and high cost Facility lease and associated cash used to repay additional financial obligations associated with the Facility will negatively impact our operating results and financial position.
In May 2007, we entered into an agreement to lease a multi-use, 46,000 square foot facility in Vancouver, British Columbia, Canada. For additional information regarding the facility, see “Property, Plants and Equipment”.
This facility, which the company occupied as its main operation center in 2008, is significantly larger than required for our near term production requirements. The excess space is difficult to sublease due to the current layout of the company’s manufacturing operations and the significant availability of space in other buildings in the local real estate market. In addition to rental payments for the facility, we are obligated to repay with interest over the next 12 years the $6,784,345 balance due as of December 31, 2011 on the repayable leasehold improvement allowance.
We believe that the financial obligation associated with this facility lease and associated liabilities represent a total facilities cost significantly above the current real estate market prevailing lease rates. This factor, together with the excessive size of the facility, may adversely affect the company’s financial performance.
Should there be a downturn in our business or the markets in which we compete, we may not have a need to expand our facility as we have planned. As a result, we may then seek an alternative use for all or a portion of the property, seek to sub-lease some or all of our property and we may not exercise the option to extend the lease, any of which may have a negative impact on our operating results. We may experience unanticipated decreases in productivity and other losses due to inefficiencies relating to any such transition, or delays in obtaining any required approvals or clearances from regulatory agencies related to the validation of any new manufacturing facilities. For instance, the scale-up of manufacturing at our planned facility could result in lower than expected manufacturing output and higher than expected product costs.
Sole-source suppliers provide some of our raw materials. In the event a sole-sourced material became unavailable, there may be a delay in obtaining an alternate source, and the alternate source may require significant development to meet product specifications. It is also possible that we may not be able to locate an acceptable alternate source at all. Consequently, we may face difficulty in manufacturing, or be entirely unable to manufacture, some of our products.
Single-source suppliers provide some key components, in particular antibodies, used in the manufacture of our products. Except for one of the antibodies we use in our West Nile Virus Test and two of the antibodies we use in our B-type natriuretic peptide Test, we do not have supply agreements with any of our other antibody suppliers. We are currently negotiating supply agreements for some of the other key reagents that we use. Although we maintain inventories of some key components, including antibodies, any loss or interruption in the supply of a sole-sourced component or raw material would have a material adverse effect on our ability to manufacture these products until a new source of supply is qualified and, as a result, may temporarily or even permanently prevent us from being able to sell our products. Given the nature of variations in biological raw materials, a new supply source of antibodies may require considerable time and resources to develop manufacturing procedures to meet the required product performance levels for commercial sale. Additionally, it may require us to enter into supply agreements on commercial reasonable terms with the new suppliers to ensure supply, or at all, there could be a material adverse effect on our ability to manufacture product for commercial sale.
Interruption in the supply of any sole-sourced component or raw material would likely have a material adverse effect on our profit margins, our ability to develop and manufacture products on a timely and competitive basis, and the timing of market introductions and subsequent sales of products.
We rely significantly on third party manufacturers for some of our products and rely on third party manufacturers of certain equipment necessary for us to scale-up our internal capacity to manufacture products. If these third party manufacturers experience difficulties, our ability to serve various markets with our products may be significantly restricted.
All of our test kits, or Kits, are currently produced in-house and our portable fluorescence readers or Readers, are manufactured and supplied to us under contract. We have qualified a local contract manufacturer for the Readers, see "Operations and Manufacturing". To meet the projected demand for our products, we will require additional equipment to scale up our manufacturing processes. Some of this equipment will require customization that may increase the lead-time from the supplier. If demand for our products significantly exceeds forecast, or if the third party manufacturers of Readers or manufacturing equipment are unable to deliver to us on schedule, we may not be able to meet customer requirements.
We may not be able to adequately protect our technology and proprietary rights, and third parties may claim that we infringe on their proprietary rights. If we cannot protect our technology, companies with greater resources than us may be able to use our technology to make products that directly compete with ours. Additionally, third parties claiming that we infringe on their proprietary rights may be able to prevent us from marketing our products or force us to enter into license agreements to do so. Both situations may negatively impact our ability to generate revenues, cash flows and earnings.
The success of our technology and products is highly dependent on our intellectual property portfolio, for which we have sought protection through a variety of means, including patents (both issued and pending) and trade secrets, see "Intellectual Property". There can be no assurance that any additional patents will be issued on existing or future patent applications or on patent applications licensed from third parties. Even when such patents have been issued, there can be no assurance that the claims allowed will be sufficiently broad to protect our technologies or that the patents will provide protection against competitive products or otherwise be commercially valuable. No assurance can be given that any patents issued to or licensed to us will not be challenged, invalidated, infringed, circumvented or held unenforceable. In addition, enforcement of our patents in foreign countries will depend on the laws and procedures in those foreign jurisdictions. Monitoring and identifying unauthorized use of our technologies or licensed technologies may prove difficult, and the cost of litigation may impair the ability to guard adequately against such infringement. If we are unable to successfully defend our intellectual property, third parties may be able to use our technology to commercialize products that compete with ours. Further, defending intellectual property can be a very costly and time-consuming process. The costs and delays associated with such a defense may negatively impact our financial position.
There are many patent claims in the area of lateral flow immunoassays and some patent infringement lawsuits have occurred amongst parties, other than ourselves, with respect to patents in this area. Our commercial success may depend upon our products not infringing on any intellectual property rights of others and upon no such claims of infringement being made. In the event that a third party was able to substantiate a claim against us, it could result in us not being able to sell our products in certain markets or at all. Further, as a result we may be required to enter into license agreements with said third parties on terms that would negatively impact our ability to conduct our business. Even if such claims were found to be invalid, the dispute process would likely have a materially adverse effect on our business, results of operations and prospects. To date, to the best of our knowledge, there have been no threats of litigation, legal actions or other claims made against any of our intellectual property. Although we attempted to identify patents that pose a risk of infringement, there is no assurance that we have identified all U.S. and foreign patents that present such a risk.
In addition to patent protection, we also rely on trade secrets, proprietary know-how and technological advances which we seek to protect, in part, through confidentiality agreements with our collaborative partners, employees and consultants. There can be no assurance that these agreements will not be breached, that we will have adequate remedies for any breach, or that the trade secrets and proprietary know-how will not otherwise become known or be independently discovered by others, which could negatively impact our ability to compete in the marketplace.
To continue developing new products or enhance existing ones, we may need to obtain licenses to certain technologies and rights from third parties, and such licenses may not be available on acceptable terms, or at all. If our product development efforts are hindered, we may face considerable challenges competing in the market place with our existing products or be unable to introduce new products.
Although we believe we are able to conduct our business based on our current intellectual property portfolio, there is a risk that additional non-core technology licenses may be required in the development of new products or to enhance the performance characteristics of our existing products. We believe that such licenses would generally be available on a non-exclusive basis; however, there is no guarantee that they will be available on acceptable terms, or at all. If we are unable to license any required non-core technology, it may impede our product development capabilities, which may put us at a competitive disadvantage in the market place and negatively affect our ability to generate revenue or profits.
We must increase sales of our Cardiovascular products or we may not be able to become profitable.
Our ability to continue to be profitable and to increase profitability will depend, in part, on our ability to increase our sales volumes of our Cardiovascular line. Increasing the sales volume of our products will depend upon, among other things, our ability to:
| · | continue to improve our existing products and develop new and innovative products; |
| · | increase our sales and marketing activities; |
| · | effectively manage our manufacturing activities; and |
| · | effectively compete against current and future competitors. |
We cannot assure you that we will be able to successfully increase our sales volumes of our products to increase or sustain profitability.
Compliance with changing regulations and standards for accounting, corporate governance and public disclosure may result in additional expenses.
Changing laws, regulations and standards relating to corporate governance and public disclosure, new SEC and BC Securities Commission regulations, and Toronto Stock Exchange, or TSX, rules, are creating additional complexities and expenses for companies such as ours. In 2005, the Accounting Standards Board announced that Canadian GAAP is to be converged with International Financial Reporting Standards, or IFRS. On February 13, 2008, the Canadian Institute of Chartered Accountants confirmed that the use of IFRS is required for fiscal years beginning on or after January 1, 2011. According to Section 4.1 of National Instrument 52-107, which governs GAAP requirements in Canada, SEC issuers may use U.S. GAAP. The Company satisfies the definition of an SEC issuer and consequently converted its primary basis of accounting from Canadian GAAP to U.S. GAAP as of January 1, 2011. However, if the SEC determines at some point in 2011 to incorporate IFRS into the U.S. domestic reporting system, the first time U.S. issuers would report under that system would be approximately 2015 or 2016. Transitioning to IFRS is likely to impact how management communicates with investors, as well as how some companies conduct business with customers and vendors. The new financial reporting regime will affect internal operations and the transition will require sophisticated planning due to the many interrelated changes it will entail within the Company. The eventual conversion to IFRS will add complexity and costs to our business and require a significant investment of our time and resources to complete. Additionally, we will make every effort to ensure the effectiveness of our internal controls each year, but there is no guarantee that our efforts to do so will be successful. To maintain high standards of corporate governance and public disclosure, we intend to invest all reasonably necessary resources to comply with all other evolving standards. These investments may result in increased general and administrative expenses and a diversion of management time and attention from strategic revenue generating and cost management activities. If we fail to maintain effective internal controls and procedures for financial reporting, or the SEC requirements applicable to these, we could be unable to provide timely and accurate financial information and therefore be subject to investigation by the SEC, and civil or criminal sanctions. Additionally, ineffective internal control over financial reporting would place us at increased risk of fraud or misuse of corporate assets and could cause our stockholders, lenders, suppliers and others to lose confidence in the accuracy or completeness of our financial reports.
Management’s determination that material weaknesses existed in our internal control over financial reporting could have a material adverse impact on the Company.
We are required to maintain internal control over financial reporting to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our financial statements for external purposes. The Company determined in our amended annual report for the year ending December 31, 2010 filed under form 20-F/A that material weaknesses exist in the Company’s internal control over financial reporting. Due to these material weaknesses, management had concluded that as of December 31, 2010, the Company’s disclosure controls and procedures were not effective. If we fail to maintain effective internal controls over financial reporting and disclosure controls and procedures, our business and results of operations could be harmed, we may be unable to report properly or timely the results of our operations and investors may lose faith in the reliability of our financial statements. As a result of the material weakness identified in 2010, we or our current or former officers, directors and employees may be subject to investigation by the SEC or Canadian securities regulators, and civil or criminal sanctions, or shareholder lawsuits, any of which could result in significant expense, whether directly or indirectly through the Company’s statutory or contractual obligations to indemnify such persons, and require significant investments of management time, which may prevent management from focusing its efforts on our business operations. Ineffective internal control over financial reporting may also increase the risk of, or result in, fraud or misuse of our corporate assets. As a consequence, the market price of our securities may be harmed.
We may be subject to product liability claims, which may adversely affect our operations.
We may be held liable or incur costs to settle liability claims if any of the products we sell cause injury or are found unsuitable. Although we currently maintain product liability insurance, we cannot be assured that this insurance is adequate, and, at any time, it is possible that such insurance coverage may cease to be available on commercially reasonable terms, if at all. A product liability claim could result in liability to us greater than our total assets or insurance coverage. Moreover, product liability claims could have an adverse impact on our business even if we have adequate insurance coverage.
We rely significantly on third party distributors and alliance partners to market and sell our products. If we are unable to successfully negotiate or maintain acceptable agreements with potential distributors, our ability to access various markets with our products may be significantly restricted. Further, we may not be able to negotiate agreements that would permit us to sell our products at a profit.
Our marketing strategy in both the environmental and the medical diagnostics markets depends significantly on our ability to establish and maintain collaborative arrangements with third party distributors and alliance partners for marketing and distribution. There can be no assurance that we will be able to negotiate or maintain acceptable collaborative arrangements enabling us to sell our products in certain markets or be able to sell our products at acceptable prices or volumes. Consequently, we may not be able to generate sufficient revenue or gross margins to be profitable.
Manufacturing risks and inefficiencies may adversely affect our ability to produce products and could reduce our gross margins and increase our research and development expenses.
We are subject to manufacturing risks, including our limited manufacturing experience with newer products and processes which may hinder our ability to scale-up manufacturing. Additionally, unanticipated acceleration or deceleration of customer demand may lead to manufacturing inefficiencies. We must manufacture our products in compliance with regulatory requirements, in sufficient quantities and on a timely basis, while maintaining acceptable product quality and manufacturing costs. Significant additional resources, implementation of additional automated and semi-automated manufacturing equipment and changes in our manufacturing processes and organization have been, and are expected to continue to be, required for scale-up to meet increasing customer demand once commercialization begins, and this work may not be successfully or efficiently completed.
In addition, although we expect some of our newer products and products under development to share production attributes with our existing products, production of these products may require the development of new manufacturing technologies and expertise. It may not be possible for us, or any other party, to manufacture these products at a cost or in quantities to make these products commercially viable.
Manufacturing and quality problems have arisen and may arise in the future as we attempt to scale-up our manufacturing capacity and implement automated and semi-automated manufacturing methods. We rely on third parties for the manufacture of much of our automated and semi-automated manufacturing equipment. Consequently, implementation of automated and semi-automated manufacturing methods may not be achieved in a timely manner or at a commercially reasonable cost, or at all. In addition, we continue to make investments to improve our manufacturing processes and to design, develop and purchase manufacturing equipment that may not yield the improvements that we expect. Unanticipated acceleration and deceleration of customer demand for our products has resulted, and may continue to result, in inefficiencies or constraints related to our manufacturing, which has harmed and may in the future harm our gross margins and overall financial results. Such inefficiencies or constraints may also result in delays, lost potential product sales or loss of current or potential customers due to their dissatisfaction.
We may not be able to effectively and efficiently manage the planned growth of our operations and, as a result, we may find ourselves unable to effectively compete in the marketplace with our products resulting in lost revenue, poor operational performance, and sustained losses.
We anticipate growth in the scope of the operating and financial systems and the geographic area of operations as new products are developed and commercialized. This growth will result in increases in responsibilities for both existing and new management personnel. Managing growth effectively will require us to continue to implement and improve operational, financial and management information systems, and to successfully attract, hire on favorable terms, develop, motivate and manage employees. This growth may require additional locations and new capital equipment. If we are unable to successfully manage our expansion, we may experience an inability to take advantage of new sales opportunities, poor employee morale, an inability to attract new employees and management, an inability to generate adequate financial and other relevant reports, poor quality control and customer service and difficulty managing our operating expenses and working capital. As a consequence, we may find ourselves unable to compete effectively in the market place with our products leading to loss of revenue and poor operational performance, including sustained losses.
The research and development of our products carries substantial technical risk. We may not be able to successfully commercialize future products. As a consequence, our ability to expand our product portfolio to generate new revenue opportunities may be severely limited.
Our future growth will depend upon, among other factors, our ability to successfully develop new products and to make product improvements to meet evolving market needs. Although we believe that we have scientific and technical resources available, future products will nevertheless be subject to the risks of failure inherent in the development of products based on innovative technologies. Any specific new product in research and development may face technical challenges that may significantly increase the costs to develop that product, cause delays to commercialization or prevent us from commercializing that product at all. Although we expect to continue to expend resources on research and development efforts, to enhance existing products and develop future ones, we are unable to predict whether research and development activities will result in any commercially viable products. There can be no assurance that we will be able to successfully develop future products and tests, which would prevent us from introducing new products in the marketplace and negatively impact our ability to grow our revenues and become profitable.
We depend on our key personnel, the loss of whose services could adversely affect our business.
We are highly dependent upon the members of our management and scientific staff, who could leave Response at any time. The loss of these key individuals could impede our ability to achieve our business goals. We face competition for qualified employees from numerous industry and academic sources and there can be no assurance that we will be able to retain qualified personnel on acceptable terms. We currently do not have key man insurance in place on any of our key employees. At present, we are recruiting a Chief Executive Officer and a new executive to lead our sales, marketing, and business development team. Both positions are presently vacant to date.
In the event that we are unable to retain key personnel, and recruit qualified key personnel on favorable terms, we may not be able to successfully manage our business operations, including sales and marketing activities, product research and development and manufacturing. As a consequence, we may not be able to effectively develop and manufacture new products, negotiate strategic alliances or generate revenue from existing products.
A substantial portion of our business is in China where we have no direct presence to closely monitor and understand the rapidly expanding market.
Approximately 59% of our product revenue derives from sales of our products through our distribution channel partners in China. China is a dynamic and rapidly evolving market for medical technology including the near-patient diagnostic testing market in which the Company competes. We have neither a direct presence nor personnel in China to allow us to closely monitor and understand this market. We may not be able to anticipate changes in this market as a consequence, which could materially and adversely impact our product sales.
We may not be able to compete effectively with larger, more established entities or their products, or with future organizations or future products, which could cause our sales to decline.
In-vitro diagnostics is a well-established field in which there are a number of competitors that have substantially greater financial resources and larger, more established marketing, sales and service organizations than we do.
Our principal competitors in the human diagnostic market are Alere Inc., or Alere, Abbott Point of Care Inc., Mitsubishi Chemical Medience Corporation, Roche Diagnostics, Becton Dickinson Corporation, and Quidel Corporation. Many of our competitors have significantly larger product lines to offer and greater financial and other resources than we do. In particular, many of these competitors have large sales forces and well-established distribution channels and brand names.
Our Company is organized under the laws of British Columbia, Canada, and certain of our directors and officers and substantially all of our assets are located outside of the United States, which may make enforcement of United States judgments against us difficult.
We are organized under the laws of British Columbia, Canada, substantially all of our assets are located outside of the United States, we do not currently maintain a permanent place of business within the United States and certain of our directors and officers are resident outside the United States. As a result, it may be difficult for U.S. investors to effect service of process or enforce within the United States any judgments obtained against us or those officers or directors, including judgments predicated upon the civil liability provisions of the securities laws of the United States or any state thereof. In addition, there is uncertainty as to whether the courts of Canada would recognize or enforce judgments of United States courts obtained against us or our directors and officers predicated upon the civil liability provisions of the securities laws of the United States or any state thereof, or be competent to hear original actions brought in Canada against us or our directors and officers predicated upon the securities laws of the United States or any state thereof.
Valuation of stock-based payments, which we are required to perform for purposes of recording compensation expense under FASB – ASC 718 "Compensation - Stock Compensation", involves significant assumptions that are subject to change and difficult to predict.
On January 1, 2006, we adopted FAS 123(R), which is now codified as FASB ASC 718 Compensation – Stock Compensation, which requires that we record compensation expense in the statement of income for stock-based payments, such as stock options, using the fair value method. As long as stock-based awards are utilized as part of our compensation strategy, the requirements of ASC 718 have had, and will continue to have, a material effect on our future financial results reported under Generally Accepted Accounting Principles, and make it difficult for us to accurately predict our future financial results.
For instance, estimating the fair value of stock-based payments is highly dependent on assumptions regarding the future exercise behavior of our employees and changes in our stock price. Our stock-based payments have characteristics significantly different from those of freely traded options, and changes to the subjective input assumptions of our stock-based payment valuation models can materially change our estimates of the fair values of our stock-based payments. In addition, the actual values realized upon the exercise, expiration, early termination or forfeiture of stock-based payments might be significantly different than our estimates of the fair values of those awards as determined at the date of grant.
ASC 718 could also adversely impact our ability to provide accurate guidance on our future financial results as assumptions that are used to estimate the fair value of stock-based payments are based on estimates and judgments that may differ from period to period. For instance, we may be unable to accurately predict the timing, amount and form of future stock-based payments to employees. We may also be unable to accurately predict the amount and timing of the recognition of tax benefits associated with stock-based payments as they are highly dependent on the exercise behavior of our employees and the price of our stock relative to the exercise and price fair value of each outstanding stock option.
For those reasons, among others, ASC 718 may create variability and uncertainty in the compensation expense we will record in future periods, potentially negatively impacting our ability to provide accurate financial guidance. This variability and uncertainty could further adversely impact our stock price and increase our expected stock price volatility as compared to prior periods.
Risks Related to Our Industry
Products in the biomedical industry, including ours, may be subject to government regulation. Obtaining government approvals can be costly and time consuming. Any failure to obtain necessary regulatory approval will restrict our ability to sell those products and impede our ability to generate revenue.
As we operate in the biomedical industry, some of our products are subject to a wide variety of government regulation (federal, state and municipal) both within the United States and in other international jurisdictions. See "Point-of-Care (POC) Clinical Diagnostics – Regulatory Approval". For example, the FDA and comparable regulatory agencies in other countries impose substantial pre-market approval requirements on the introduction of medical products through lengthy and detailed clinical testing programs and other costly and time consuming procedures. Satisfaction of these requirements is expensive and can take a long period of time depending upon the type, complexity and novelty of the product. All medical devices manufactured for sale in the United States, regardless of country of origin, must be manufactured in accordance with Good Manufacturing Practices specified in regulations under the Federal Food, Drug, and Cosmetic Act. These practices control the product design process as well as every phase of production from incoming receipt of raw materials, components and subassemblies to product labeling, tracing of consignees after distribution and follow-up and reporting of complaint information. Both before and after a product is commercialized, we have ongoing responsibilities under the regulations of the FDA and other agencies. Noncompliance with applicable laws and the requirements of the FDA and other agencies can result in, among other things, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure of the government to grant pre-market clearance or pre-market approval for devices, withdrawal of marketing clearances or approvals, and criminal prosecution. The FDA has the authority to request recall, repair, replacement or refund of the cost of any device manufactured or distributed by us. The FDA also administers certain controls over the import and export of medical devices to and from the United States, respectively.
The U.S. Clinical Laboratory Improvement Acts of 1988 also affects our medical products. This law is intended to assure the quality and reliability of all medical testing in the United States regardless of where tests are performed. The regulations require laboratories performing clinical tests to meet specified standards in the areas of personnel qualification, administration, participation in proficiency testing, patient test management, quality control, quality assurance and inspections.
As we generate a large part of our revenues from international product sales and services for international customers, we are subject to risks inherent in international business, including currency exchange risk, difficulty in collecting accounts receivable, and possible marketing restrictions. Consequently, we may be restricted from selling our products in certain jurisdictions or our products may not be able to be sold at a profit.
There are various operational and financial risks associated with international activity. We may face difficulties and risks in our international business, including changing economic or political conditions, export restrictions, currency risks, export controls relating to technology, compliance with existing and changing regulatory requirements, tariffs and other trade barriers, longer payment cycles, problems in collecting accounts receivable, reimbursement levels, reduced protection for intellectual property, potentially adverse tax consequences, limits on repatriation of earnings, the burdens of complying with a wide variety of foreign laws, nationalization, war, insurrection, terrorism and other political risks and factors beyond our control. As a consequence, these potential international risks may prevent us from selling our products in certain jurisdictions, may make it very difficult or even impossible to collect on accounts receivable or may impose a variety of additional expenses on our business such that we cannot sell our products at a profit. For international sales, we price and invoice our products primarily in U.S. dollars and consequently incur a U.S./Canadian foreign exchange risk. We also expect that there may be a greater requirement in the future for sales to European customers to be priced and invoiced in Euros. Any significant adverse change in currency exchange rates may negatively impact our profit margins such that we may not be able to generate positive cash flow or earnings from our operations. To date, we have not made any provision for a currency-hedging program. We periodically evaluate options to mitigate our exposure to currency fluctuations, but there can be no assurance that we will be able to do so.
Sales and pricing of medical products, including ours, are affected by third-party reimbursement. Depending on our manufacturing costs, we may not be able to profitably sell our products at prices that would be acceptable to third party reimbursement programs. Consequently, we may have difficulty generating revenue, resulting in reduced profit margins and potential operating losses.
Sales of our medical products are dependent, in part, on the availability of levels of reimbursement from third-party payers, such as government agencies and private insurance companies. Reimbursement policies by such third-party payers could reduce or eliminate such reimbursements and thereby adversely affect future sales of our products. Third-party payers are increasingly challenging prices paid for medical products and the cost effectiveness of such products. Significant uncertainty exists as to the reimbursement status of newly cleared health care products. There can be no assurance that proposed products will be considered cost effective or that reimbursement from third party payers will be available or, if available, that reimbursement will not be limited, thereby adversely affecting our ability to sell products or sell our products at a profit.
Third party payers can indirectly affect the pricing or the relative attractiveness of our products by regulating the maximum amount of reimbursement provided for testing services. If the reimbursement amounts for testing services are decreased in the future, it may decrease the amount that physicians and hospitals are able to charge patients for such services and therefore the prices that we, or our distributors, can charge for our products. Consequently our ability to generate revenue and/or profits may be negatively impacted for both existing and new products.
Our business is substantially dependent on market acceptance of our products. As well, our environmental and biodefense business is affected by industry, governmental and public perceptions of these products in general. Failure to obtain or retain market acceptance for some or all of our products would have a negative impact on our revenue and ability to operate profitably.
The commercial success of our clinical tests is highly dependent upon the acceptance and adoption of the tests by the medical community. The medical community tends to be very conservative with regards to adopting new technologies and products. Often substantial data and evidence supporting product performance is required to generate market acceptance. If we are unsuccessful in generating market acceptance, our ability to generate revenue and hence profits would be severely limited.
The commercial success of our environmental and biodefense tests is dependent upon their acceptance by the public safety community and government funding agencies as being useful and cost effective. In addition, the purchase of our biodefense products in the United States (our largest potential market) by the public safety community is highly dependent on the availability of federal and state government funds dedicated to "homeland security". In the event that homeland security funds became unavailable for use (to purchase our products or otherwise) or the release of such funds was significantly delayed, it would have a negative effect on our ability to generate revenue or profits.
In addition, on July 19, 2002, the U.S. Office of Science and Technology Policy, or OSTP, now under the Office of Homeland Security, published a memorandum directed to federal mail managers and first responders to federal mail centers that raised concerns regarding the use and performance capabilities of commercially available anthrax detection equipment, and further included recommendations to the community to cease purchasing such equipment. The recommendations in this memorandum were based on an evaluation of commercially available anthrax detection equipment by the CDC, which concluded that such equipment did not pass acceptable standards for effectiveness. As both of these U.S. government agencies are considered to be influential opinion leaders, this recommendation had a negative impact on the market acceptance and adoption of biodefense products generally, including our products. Subsequently, beginning in June 2003, an 18-month study to evaluate handheld anthrax tests was performed by AOAC International and funded by DHS and the U.S. Department of Defense. In November 2004, the RAMP® System received AOAC Official Methods SM Certificate 070403 stating that the RAMP® Anthrax Test performed as we claimed. All other commercially available rapid on-site anthrax detection systems that were tested failed to meet the AOAC's performance standard. A further intensive, independent field testing program conducted by AOAC International and sponsored by the DHS, culminated in the announcement in September 2006 that our RAMP® Anthrax Test is the first and only biodetection product approved for field use by first responders in the United States for the detection of anthrax. We believe that we have adequately addressed the concerns raised by the CDC and OSTP with the public safety community, however there can be no assurance that the marketplace will continue to respond favorably. Further, it is possible that the U.S. government will enact regulations that prohibit, restrict or limit the use of equipment or funding for on-site biodefense testing, see "On-Site Environmental Testing Market, Industry Trends" and "On-Site Environmental Testing Market, Competition".
Federal, state and foreign regulations regarding the manufacture and sale of medical devices continue to evolve and are constantly subject to change. We cannot predict what regulations may come into effect in the future and what impact, if any, such regulatory changes may have on our business.
A majority of our sales are through distributors in foreign markets who sell our products or modifications of our products in their local country markets. Sales through these distributors in these markets are usually subject to the regulators in those markets. Frequently our distributors are responsible for obtaining and maintaining regulatory approval in their territories and are thus subject to all of those requirements. In the future, should we elect to build our own sales and marketing operations in certain countries outside the US, we would be subject to extensive regulations in each of those countries. We may not be successful in such new initiatives.
We operate primarily selling through distributors in highly competitive markets, with continual developments in new technologies and products. Some of our competitors have significantly greater resources than we do. Others while smaller may have a very strong market or other leadership position in a specific local market where we or our distributors compete We or our distributors may not be able to compete successfully based on many factors, including product price or performance characteristics, sales and marketing effort or customer support capabilities. An inability to successfully compete could lead to us having limited prospects for establishing market share or generating revenues.
The diagnostic industry is characterized by extensive research efforts, ongoing technological progress and intense competition. There are many public and private companies, including well-known diagnostic companies, engaged in marketing and developing products for the markets we have targeted. Many of these companies have substantially greater financial, technical and human resources than we do. Our competitors may be more successful in convincing potential customers to adopt their products over ours and hence gain greater market share. Competitors with greater financial resources may also have an advantage when dealing with suppliers, particularly sole source suppliers providing antibodies or unique reagents. Additionally, they may develop technologies and products that are more effective than any products developed by us, or that would render our technologies and products obsolete or non-competitive.
We believe our primary current competitors in the POC cardiovascular diagnostics market are: Biosite, Incorporated, or Biosite, which entered into a merger agreement with Alere in 2007; Abbott Point of Care Inc., or Abbott; and Dade Behring, Inc., or Dade. Biosite and Abbott have quantitative POC systems, and Dade produces a small quantitative bench-top system, for the detection of some cardiac markers. These three companies are currently marketing and selling their products in the cardiovascular testing market in which we compete, see "POC Clinical Diagnostics Market, Competition".
In addition, in various emerging markets such as China, there may be local competitors who sell only in that specific country. Some of these local competitors may be very strong competitors in their local markets due to factors which may include lower cost production, stronger sales, marketing and distribution capabilities, customer familiarity and preference for local suppliers and local government environments which may favor local companies and their products.
In the environmental biodefense testing market, our primary competitors are Alexeter Technologies LLC, or Alexeter, Idaho Technology Inc., and Cepheid Inc., or Cepheid. Alexeter sells rapid on-site immunoassay tests that are read by an instrument and Cepheid has a polymerase chain reaction test system being sold in this marketplace.
In the vector environmental testing market, our primary competitor is Medical Analysis Systems, Inc., which is wholly owned by Thermo Fisher Scientific, Inc. Medical Analysis Systems, Inc. markets and sells a product for the rapid detection of West Nile virus.
We believe the primary competitors in the POC Flu A/B and RSV testing market are Binax, Inc., a division of Alere, and Quidel Corporation. Both companies have qualitative POC tests for the detection of Flu A+B and RSV.
Many of our competitors have access to substantially greater technical and financial resources. In the event that we are not able to compete successfully in the marketplace, we may face limited adoption of our products by potential customers or erosion of current market share, which would seriously impede our ability to generate revenue.
In addition to the specific competitive risks from rapid diagnostic manufacturers that we face in the market for our tests, we face intense competition in the general market for diagnostic testing including companies making laboratory-based tests and analyzers, and clinical reference laboratories. Currently, the majority of diagnostic tests prescribed by physicians and other healthcare providers is performed by independent clinical reference laboratories and hospital-based laboratories using automated testing systems. Therefore, in order to achieve market acceptance for our products we will be required to demonstrate that our products provide clinical benefit and are cost-effective and time saving alternatives to automated tests traditionally used by clinical reference laboratories or hospital-based laboratories.
Companies operating in our industry may be impacted by potential healthcare reform. Such healthcare reform may include pricing restrictions on medical products, including ours, that may restrict our ability to sell our products at a profit.
Healthcare reform bills that have been before the United States Congress contemplate changes in the structure, financing and delivery of healthcare services in the United States. These and any future healthcare reforms may have a substantial impact on the operations of companies in the healthcare industry, including us. Such reforms could include product pricing restrictions or additional regulations governing the usage of medical products. No assurances can be given that any such proposals, or other current or future legislation in the United States or in other countries, will not adversely affect our product development and commercialization efforts, results of operations or financial condition. At this time, we are unaware of any recent legislation or pending legislative proposals that will negatively affect our business.
The impact of consolidation of several major competitors in the market for immunoassay testing is difficult to predict and may harm our business.
The market for immunoassay-based diagnostic testing is rapidly changing as a result of recent consolidation in the industry. Within the past five years, Siemens acquired Bayer Diagnostics, Diagnostic Products Corp. and Dade; and Biosite entered into a merger agreement with Alere. In the past three years there have been many acquisitions in the medical diagnostics market including several by Alere, helping the company expand its presence in the market for rapid diagnostic tests used in hospitals and doctors' offices. Siemens and Alere both have significant existing businesses in diagnostics and/or related markets for healthcare equipment and services. Given the period of time since the announcement of these transactions, it is unclear how these completed and proposed acquisitions will impact the competitive landscape for our products or for hospital-based diagnostic testing in general. However, because these competitors sell a broad range of product offerings to our prospective hospital customers and because of the substantially greater financial resources and more established marketing, sales and service organizations that they each have, we believe there is greater risk that these new consolidated competitors may offer discounts as a competitive tactic or may hold other competitive advantages as a result of their ability to sell a broader menu of important hospital infrastructure equipment and information systems on a combined or bundled basis.
Our business and industry is affected by seasonality, including governmental budget cycles. We may not be able to successfully scale up operations to meet demand during peak seasonal periods or scale down operations during periods of low demand, which could result in lost revenue and/or negative cash flows and losses.
Our operating results may fluctuate from quarter to quarter due to many seasonal factors. Many of our prospective customers are government related organizations at a federal, state/provincial or municipal level. Consequently, our sales may be tied to government budget and purchasing cycles. Sales may also be slower in the traditional vacation months, could be accelerated in the first or fourth calendar quarters by customers whose annual budgets are about to expire (especially affecting purchases of our fluorescent Readers), may be distorted by unusually large Reader shipments from time to time, or may be affected by the timing of customer cartridge ordering patterns. Seasonality may require us to invest significantly in additional resources, including equipment, labor and inventory to meet demand during peak seasonal periods. There can be no assurance that we will be successful in putting in place the resources to meet anticipated demand, which could lead to lost revenue opportunities. If we cannot scale down our operations and expenses sufficiently during periods of low demand for our products, we may experience significantly negative cash flow and operating losses. If we are unable to adequately forecast seasonal activity, we may experience periods of inventory shortages or excesses that would negatively impact our working capital position.
If products in the biodefense testing industry and other environmental testing segments, including ours, become subject to government legislation in the future, obtaining necessary government approvals may be very costly and time consuming. Failure to obtain government approvals will restrict our ability to sell our products and impede our ability to generate revenue.
In the biodefense and vector environmental testing markets, there is currently an absence of regulatory checks and balances and there is significant market uncertainty and misinformation. While we believe it is likely that future regulatory requirements in these markets will come into effect, the form and substance of these regulations remain highly uncertain. The effect of government regulations may be to prevent or to delay marketing and pricing of any new products for a considerable or indefinite period or to require additional studies prior to approval. Federal, state and foreign regulations, or lack thereof, regarding the sale of environmental testing devices are subject to change. We cannot predict the impact, if any, such changes may have on our business.
Risks Related to Our Common Stock
As we have a large number of warrants and stock options outstanding, our shareholders will experience dilution from these options and warrants in the event that they are exercised.
As of December 31, 2011, we had outstanding stock options to purchase an aggregate of 184,485 shares, at exercise prices between $0.34 and $10.50 and warrants to purchase an aggregate of 90,127,904 shares at a price of $0.0746, which in total represents 41% of our fully diluted outstanding share capitalization at that date. To the extent that these outstanding options and warrants are exercised, considerable dilution to the interests of our shareholders will occur.
The price of our common stock may be volatile, and a shareholder's investment in our common stock could suffer a decline in value.
There has been significant volatility in the volume and market price of our common stock, and this volatility may continue in the future. This volatility may be caused by a variety of factors, including the lack of readily available quotations, the absence of consistent administrative supervision of "bid" and "ask" quotations and generally lower trading volume. In addition, factors such as quarterly variations in our operating results, changes in financial estimates by securities analysts or our failure to meet our or their projected financial and operating results, litigation involving us, general trends relating to the medical device industry, actions by governmental agencies, national economic and stock market considerations as well as other events and circumstances beyond our control could have a significant impact on the future market price of our common stock and the relative volatility of such market price.
Because our common stock is considered a "penny stock," a shareholder may have difficulty selling shares in the secondary trading market.
Our common stock is subject to certain rules and regulations relating to "penny stock" (generally defined as any equity security that is not quoted on the Nasdaq Stock Market and that has a price less than US$5.00 per share, subject to certain exemptions). Broker-dealers who sell penny stocks are subject to certain "sales practice requirements" for sales in certain nonexempt transactions (e.g., sales to persons other than established customers and institutional "accredited investors"), including requiring delivery of a risk disclosure document relating to the penny stock market and monthly statements disclosing recent price information for the penny stock held in the account, and certain other restrictions. For as long as our common stock is subject to the rules on penny stocks, the market liquidity for such securities could be significantly limited. This lack of liquidity may also make it more difficult for us to raise capital in the future through sales of equity in the public or private markets.
Because our common stock is not traded on a national securities exchange in the U.S., a U.S. shareholder's ability to sell shares in the secondary trading market may be limited.
Our common stock is currently listed for trading in Canada on the Toronto Stock Exchange. Our common stock is also quoted in the United States on the OTC Bulletin Board. Shareholders may find it more difficult to dispose of or to obtain accurate quotations as to the price of our securities than if the securities were traded on a national securities exchange like The New York Stock Exchange, the NASDAQ Stock Market or the NYSE Amex LLC.
Our business offices are located in Vancouver, British Columbia. In May 2007, we entered into an agreement to lease a multi-use, 46,000 square foot facility. The facility has housed all of our operations since March 31, 2008 and requires us to pay approximately $160,000 per month in rent and operating expenses. Initial capital modifications to the facility were paid by the landlord and managed by us. The agreement has an initial term of 15 years with two five-year renewal options. To secure the lease, we are required to maintain a security deposit with the landlord in the form of an irrevocable letter of credit in the amount of $870,610. As part of the agreement we received a repayable leasehold improvement allowance for an amount of $7.8 million, used for additional improvements to the facility. The allowance is being repaid over the term of the operating lease at approximately $88,500 per month including interest at an annual rate of 11%, compounded monthly.
We believe that the facilities we currently lease are sufficient for our anticipated near-term needs.
ITEM 3. Legal Proceedings.
From time to time, we may be involved in litigation relating to claims arising out of our operations in the normal course of business. We are not currently a party to any legal proceedings, the adverse outcome of which, in management’s opinion, individually or in the aggregate, would have a material adverse effect on our results of operations or financial position. There are no material proceedings to which any director, officer or any of our affiliates, any owner of record or beneficially of more than five percent of any class of our voting securities is a party adverse to us or has a material interest adverse thereto.
ITEM 4. Mine Safety Disclosures
Not applicable.
PART II
ITEM 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market for Common Equity
Our common stock is traded on the Toronto Stock Exchange (“TSX”) under the trading symbol “RBM” and quoted on the OTC Bulletin Board under the symbol “RPBIF”. As of March 23, 2012, there were 129,078,166 shares of our common stock outstanding held by approximately 52 stockholders of record and 3 beneficial stockholders. The high and low sales prices of our common stock as reported on the TSX for the periods indicated are as follows:
Response Biomedical Corporation | HIGH (in CDN $) | LOW (in CDN $) |
| YEAR ENDED DECEMBER 31, 2010 | | |
| First quarter | $2.30 | $1.20 |
| Second quarter | 1.50 | 0.48 |
| Third quarter | 0.86 | 0.41 |
| Fourth quarter | 0.47 | 0.31 |
| | | |
| YEAR ENDED DECEMBER 31, 2011 | | |
| First quarter | $0.53 | $0.27 |
| Second quarter | 0.48 | 0.27 |
| Third quarter | 0.35 | 0.16 |
| Fourth quarter | 0.20 | 0.05 |
Dividend Policy
We have not declared or paid any dividends on the outstanding common shares since our inception and we do not anticipate that we will do so in the foreseeable future. The declaration of dividends on our common shares is within the discretion of the Board of Directors and will depend on the assessment of, among other factors, earnings, capital requirements and our operating and financial condition. At the present time, anticipated capital requirements are such that we intend to follow a policy of retaining earnings in order to finance the further development of the business.
Sales of Unregistered Securities
On May 21, 2009, the Company closed a public offering of gross proceeds of $12,650,000, before share issuance costs of $1,257,768, by issuing 84,333,333 units at a price of $0.15 per share. Each unit was comprised of one common share and one-half of one transferable common share purchase warrant for a total of 42,166,666 common share purchase warrants exercisable at $0.25 per share for a period of 24 months. Of the total 84,333,333 units issued, 11,000,000 were issued to Haywood Securities Inc. who was the sole underwriter of the offering. The offering was exempt under Rule 506 of Regulation D promulgated under Securities Exchange Act of 1934, as amended, or Rule 506.
On July 28, 2010, the Company closed a private placement of gross proceeds of $8,000,000, before share issuance costs of $525,080, by issuing 13,333,333 shares at a price of $0.60 per share. The shares were issued to affiliates of OrbiMed Advisors, LLC. The offering was exempt under Rule 506.
On December 29, 2011, the Company closed a rights offering of gross proceeds of $6,723,542, before share issuance costs of $685,739, by issuing 90,127,904 units at a price of $0.0746 per unit. Each unit was comprised of one common share and one common share purchase warrant for a total of 90,127,904 warrants exercisable at $0.0746 for a period of 5 years. A significant portion of the units were issued to affiliates of OrbiMed Advisors LLC. The offering was exempt under Rule 506. The Company expects to use the net proceeds for ongoing product development, market expansion, funding of future negative cash flows, capital improvements, and working capital and general corporate purposes.
Repurchases of Equity Securities
We did not repurchase any of our equity securities during the year ending December 31, 2011.
Equity Compensation Plans Information
The information required by this item regarding equity compensation plan information will be set forth in the Proxy statement as disclosed in Part III, Item 12 of this Annual Report on Form 10-K.
ITEM 6. Selected Financial Data.
The following selected financial data should be read in conjunction with our consolidated financial statements, the related notes thereto and the information contained in "Item 7 – Management's Discussion and Analysis of Financial Condition and Results of Operations".
| Year Ended December 31, | | 2011 | | | 2010 | | | 2009 | | | 2008 | | | 2007 | |
| Consolidated Statements of Income Data: | | | | | | | | | | | | | | | |
| Product sales | | $ | 9,024,083 | | | $ | 6,792,130 | | | $ | 8,153,049 | | | $ | 4,899,841 | | | $ | 3,557,244 | |
| Cost of sales | | | 6,968,832 | | | | 7,097,538 | | | | 7,933,704 | | | | 5,227,156 | | | | 3,201,626 | |
| Gross Profit | | | 2,055,251 | | | | (305,408 | ) | | | 219,345 | | | | (327,315 | ) | | | 355,618 | |
| Contract service fees and revenues | | | 449,386 | | | | 320,878 | | | | 1,793,220 | | | | 976,496 | | | | 526,872 | |
| Operating Expenses | | | 7,392,375 | | | | 9,216,256 | | | | 10,602,954 | | | | 13,653,588 | | | | 14,654,574 | |
| Income from Operations | | | (4,887,738 | ) | | | (9,200,786 | ) | | | (8,590,389 | ) | | | (13,004,407 | ) | | | (13,772,084 | ) |
| Other Expense (Income) | | | 483,574 | | | | 881,125 | | | | 953,142 | | | | 762,374 | | | | 359,037 | |
| Net Loss | | $ | (5,371,312 | ) | | $ | (10,081,911 | ) | | $ | (9,543,531 | ) | | $ | (13,766,781 | ) | | $ | (14,131,121 | ) |
| Loss per common share - basic and diluted | | $ | (0.14 | ) | | $ | (0.32 | ) | | $ | (0.43 | ) | | $ | (0.98 | ) | | $ | (1.17 | ) |
| Weighted average common shares outstanding - basic and diluted | | | 39,446,554 | | | | 31,173,823 | | | | 22,209,393 | | | | 14,067,597 | | | | 12,050,926 | |
| As of December 31, | | | 2011 | | | | 2010 | | | | 2009 | | | | 2008 | | | | 2007 | |
| Consolidated Balance Sheets Data: | | | | | | | | | | | | | | | | | | | | |
| Total assets | | $ | 20,893,266 | | | $ | 19,523,931 | | | $ | 21,464,196 | | | $ | 19,394,907 | | | $ | 17,938,351 | |
| Non-current liabilities | | | 8,235,562 | | | | 8,779,624 | | | | 9,213,763 | | | | 9,606,810 | | | | 3,021,442 | |
| Total shareholders' equity | | | 4,975,723 | | | | 8,054,075 | | | | 9,646,542 | | | | 6,760,128 | | | | 11,073,221 | |
ITEM 7. Management's Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and related notes, included in Item 8 of this Report. Unless otherwise specified, all dollar amounts are Canadian dollars.
Response Biomedical develops, manufactures and sells diagnostic tests for use with its proprietary RAMP® System, a fluorescent immunoassay-based on-site diagnostic testing platform. The RAMP® technology utilizes a unique method to account for sources of error inherent in conventional lateral flow immunoassay technologies, thereby providing the ability to quickly and accurately detect and quantify an analyte present in a liquid sample. Consequently, an end user on-site or in a point-of-care setting can rapidly obtain important diagnostic information. Response Biomedical currently has thirteen tests available for clinical and environmental testing applications and the Company has plans to commercialize additional tests.
Our sales for any future periods are not predictable with a significant degree of certainty, and may depend on a number of factors outside of our control, including but not limited to inventory or timing considerations by our distributors. We generally operate with a limited order backlog because our products are typically shipped shortly after orders are received. Product sales in any quarter are generally dependent on orders booked and shipped in that quarter. As a result, any such revenue shortfall would negatively affect our operating results and financial condition. In addition, our sales may be adversely impacted by pricing pressure from competitors. Our ability to be consistently profitable will depend, in part, on our ability to increase the sales volumes of our products and to successfully compete with other competitors. We believe that period to period comparisons of our results of operations are not necessarily meaningful indicators of future results.
| · | On April 28, 2011, the Company announced that it has received product registration from China’s State Food & Drug Administration to sell the RAMP® 200 Reader in the Chinese market. |
| · | On May 27, 2011, the FDA informed the Company that the NTproBNP assay does not have 510 (k) clearance on the RAMP® 200 Reader. Subsequently, the Company received notification from its U.S. distributor that they have issued a stop shipment on all RAMP® 200 branded products until 510(k) clearance had been received by the FDA. |
| · | On June 13, 2011, the Company announced that it has received notification from the U.S. Food and Drug Administration, or FDA, that its NTproBNP Assay did not meet the criteria to obtain a waiver under the Clinical Laboratory Improvement Amendments of 1988, or CLIA. The Company did not forecast any sales with the assumption that its NTproBNP Assay would be CLIA waived. |
| · | On September 2, 2011, the Company announced that they have received notification from Roche Diagnostics that they have terminated, effective September 30, 2011, the sales and distribution agreement between Roche Diagnostics and the Company dated June 25, 2008. Roche Diagnostics terminated the agreement because the Company has not obtained the necessary approvals from the FDA to permit Roche Diagnostics to market the Company’s cardiovascular POC tests in the United States using the RAMP® 200 Reader. Following the termination the Company has been and is now marketing and selling the cardiovascular panel on the RAMP® Reader directly to customers in the U.S., as the Company has the necessary approvals from FDA to do so. The termination did not have a material financial effect during the year ended December 31, 2011. |
| · | On September 30, 2011, the Company announced that, as a result of an ongoing internal review of prior transactions and previous financial disclosure and following consultation with its external auditors, it has determined that it is appropriate to re-state and re-file audited financial statements for the year ended December 31,, 2010 and unaudited financial statements for the first two quarters of 2011. |
| · | On October 20, 2011, the Company announced the appointment Dr. David Wang of OrbiMed Asia to the Board of Directors of Response Biomedical, the resignation of Dr. Jonathan Wang as a Director of Response Biomedical, and the promotion of two executives to leadership positions in the Company. |
| · | On November 9, 2011, the Company announced it had restated its annual audited consolidated financial statements for the year ended December 31, 2010 and its interim unaudited consolidated financial statements for the periods ended March 31, 2011 and June 30, 2011. |
| · | On November 22, 2011, the Company announced that it entered into an agreement with affiliates of OrbiMed Advisors LLC where they have agreed to loan up to $2 million to the Company via a debt financing. In conjunction with the transaction, the affiliates of OrbiMed gained the right to appoint a total of three members of the board, an increase from the prior OrbiMed right to appoint two members of the Board. Subsequently Dr. Richard Bastiani tendered his resignation as a director of the Company. |
| · | On December 29, 2011, the Company announced that it had raised $6.7 million, before issuance costs, by issuing 90.1 million units at $0.0746 per Unit. Each unit consisted of one common share and one common share purchase warrant. Each warrant enables the holder to acquire one additional common share at an exercise price of $0.0746 per share for a period of five years after the closing of the rights offering. |
RESULTS OF OPERATIONS
Revenues, Cost of Goods Sold and Gross Margin
Fiscal 2011 versus Fiscal 2010
| | | For the Years Ended December 31, | | | Change 2010 to 2011 | | | Change 2009 to 2010 | |
| | | 2011 | | | 2010 | | | 2009 | | | Increase / (Decrease) | | | Percent Change | | | Increase / (Decrease) | | | Percent Change | |
| Product Sales | | | 9,024,083 | | | | 6,792,130 | | | | 8,153,049 | | | | 2,231,953 | | | | 33 | % | | | (1,360,919 | ) | | | (17 | %) |
| Cost of Sales | | | 6,968,832 | | | | 7,097,538 | | | | 7,933,704 | | | | (128,706 | ) | | | (2 | %) | | | (836,166 | ) | | | (11 | %) |
| Gross profit on product sales | | | 2,055,251 | | | | (305,408 | ) | | | 219,345 | | | | 2,360,659 | | | | 773 | % | | | (524,753 | ) | | | (239 | %) |
| Gross margin | | | 23 | % | | | (4 | %) | | | 3 | % | | | | | | | | | | | | | | | | |
Revenues increased 33%, or $2.23 million, in fiscal 2011 as compared to fiscal 2010. The change in total revenue is due to the following:
| · | Clinical product sales have increased 31%, or $1.85 million, primarily due to: |
| · | a $1.38 million increase in sales to our two distributors in China as a result of the expansion of the Chinese market; |
| · | a $0.52 million increase in sales of our infectious disease products in the United States due to the timing of orders by 3M; |
| · | a $0.36 million decrease in sales due to the termination of two distribution agreements in the United States and France; and |
| · | $0.31 million increase representing the sum of variances across several different markets. |
| · | Biodefense product sales have increased 48%, or $0.22 million, primarily due to a $0.29 million increase to a new distributor in China offset by insignificant variations in sales to distributors and end-users; and |
| · | Vector Infectious Disease product sales have increased 55%, or $0.17 million, due to increase in sales to our distributor as a result of the expansion of their end-user customer base. |
Gross Profit
Gross profit on product sales increased 773%, or $2.36 million, in fiscal 2011 as compared to fiscal 2010. The change in total gross profit is primarily due to the increase in gross margin to 23% from a negative gross margin of (4%) in fiscal 2010. This increase is primarily due to the following:
| · | An increase of 57%, or 0.47 million, in assays sold in fiscal 2011 compared to 2010 resulting in an increase of the absorption of fixed manufacturing overhead costs; |
| · | A decrease of $1.01 million of fixed manufacturing overhead costs in fiscal 2011 in comparison to fiscal 2010 primarily as a result of the company reorganization completed in the latter half of fiscal 2010. |
| · | A change in manufacturing processes to transition to a more automated process in addition to increases in lot sizes; and |
| · | An increase in the sales of biodefense and Vector Infectious Disease products in fiscal 2011 which earn a higher gross margin in relation to the clinical product sales as sales tend to be made to end users. |
Fiscal 2010 versus Fiscal 2009
Revenues
Revenues decreased 17%, or $1.36 million, in fiscal 2010 as compared to fiscal 2009. The change in total revenue is due to the following:
| · | Clinical product sales decreased 12%, or $0.79 million, primarily due to: |
| · | a decrease in sales made to Roche Diagnostics and 3M in fiscal 2010 in comparison to fiscal 2009 due to the reduction in demand to stock inventory levels; and |
| · | an increase in sales made to the two distributors in China. |
| · | Vector Infectious Disease product sales decreased 62%, or $0.50 million, primarily due to the loss of a key end user by our major distributor of these tests in the United States; and |
| · | Biodefense product sales decreased 14%, or $0.07 million, primarily due to a reduction in government funding due to a general decrease in biodefense attacks in the United States. |
Gross profit on product sales decreased 239%, or $0.52 million, in fiscal 2010 as compared to fiscal 2009. The change in total gross profit is primarily due to the decrease in gross margin to (4%) from a gross margin of 3% in fiscal 2009. This decrease is primarily due to the following:
| · | An increase in the provision of scrap and obsolescence provisions for inventory; and |
| · | A change in product mix as a result of an increase in sales of royalty bearing products. |
| | | For the Years Ended December 31, | | | Change 2010 to 2011 | | | Change 2009 to 2010 | |
| | | 2011 | | | 2010 | | | 2009 | | | Increase / (Decrease) | | | Percent Change | | | Increase / (Decrease) | | | Percent Change | |
| Contract service fees | | | 449,386 | | | | 320,878 | | | | 1,793,220 | | | | 128,508 | | | | 40 | % | | | (1,472,342 | ) | | | (82 | %) |
Fiscal 2011 versus Fiscal 2010
Contract service fees increased by 40%, or $0.13 million, in fiscal 2011 in comparison to fiscal 2010 due to the termination of a project agreement as further development was suspended pending changes to requirements by the FDA. Upon termination, the Company recognized the remaining revenue under the contract to offset costs incurred in accordance with the agreement.
Fiscal 2010 versus Fiscal 2009
Contract service fees decreased by 82%, or $1.47 million, in fiscal 2010 in comparison to fiscal 2009. The variability is due to the timing and performance of services required to recognize service revenue from the Company’s collaborations.
| | | For the Years Ended December 31, | | | Change 2010 to 2011 | | | Change 2009 to 2010 | |
| | | 2011 | | | 2010 | | | 2009 | | | Increase / (Decrease) | | | Percent Change | | | Increase / (Decrease) | | | Percent Change | |
| Research and development | | | 2,852,129 | | | | 4,106,266 | | | | 5,635,692 | | | | (1,254,137 | ) | | | (31 | %) | | | (1,529,426 | ) | | | (27 | %) |
| General and administrative | | | 3,625,510 | | | | 3,696,819 | | | | 3,299,042 | | | | (71,309 | ) | | | (2 | %) | | | 397,777 | | | | 12 | % |
| Marketing and business development | | | 914,736 | | | | 1,413,171 | | | | 1,668,220 | | | | (498,435 | ) | | | (35 | %) | | | (255,049 | ) | | | (15 | %) |
Fiscal 2011 versus Fiscal 2010
Research and Development Expenses
Research and Development Expenses decreased by 31%, or $1.25 million. The decrease was primarily due to a $0.75 million decrease in salary expenses and a $0.30 million decrease in professional fees as a result of the company restructuring in late fiscal 2010, and a decrease of $0.18 million in product development costs due to the decrease in the number of development projects occurring.
General and Administrative Expenses
General and Administrative Expenses decreased by $0.07 million. The variance is primarily due to a $0.52 million increase in legal expenses related to the restatement of the fiscal 2010 financial statements and financing obtained, $0.15 million increase in accounting related expenses related to the restatement of the 2010 financial statements, $0.13 million increase in board of director fees due to additional members and additional work performed in fiscal 2011, $0.09 million increase in recruiting costs, $0.07 million increase in bad debt expenses. These increases were offset by a $0.47 million decrease in salary expenses due to restructuring and a $0.59 million decrease in stock based compensation expenses as a result of the reversal of stock based compensation recognized due to the number of forfeitures of unvested options in 2011.
Marketing and Business Development Expenses
Marketing and Business Development Expenses decreased by 35%, or $0.50 million. The decrease was primarily due to a $0.36 million decrease in salary expenses due to the employee turnover in the sales department, a $0.20 million decrease in administrative costs due to the reduction of staff, and a $0.08 million decrease in samples given to customers. These decreases were somewhat offset by a $0.09 million increase in legal expenses associated with the development of sales and distribution agreements.
Fiscal 2010 versus Fiscal 2009
Research and Development Expenses
Research and Development Expenses decreased by 27%, or $1.53 million. The decrease was primarily the result of reduced expenditures for development projects totaling $0.91 million and lower payroll expenses totaling $0.53 million as a result of a restructuring of the Company that commenced in the latter half of 2010.
General and Administrative Expenses
General and Administrative Expenses increased by 12%, or $0.40 million. The increase was primarily due to one-time employee severance costs due to a corporate restructuring initiative that lead to a reduction in workforce in all departments totaling $0.80 million. These decreases were partially offset by decreases in stock based compensation expense totaling $0.35 million resulting from fully expensed stock options.
Marketing and Business Development Expenses
Marketing and Business Development Expenses decreased by 15%, or $0.26 million. The decrease was primarily due to reduced professional fees in the amount of $0.10 million and lower payroll expenditures in the amount of $0.09 million.
Other Income (Expense), Net
| | | For the Years Ended December 31, | | | Change 2010 to 2011 | | | Change 2009 to 2010 | |
| | | 2011 | | | 2010 | | | 2009 | | | Increase / (Decrease) | | | Percent Change | | | Increase / (Decrease) | | | Percent Change | |
| Interest expense | | | 864,791 | | | | 806,065 | | | | 857,731 | | | | 58,726 | | | | 7 | % | | | (51,666 | ) | | | (6 | %) |
| Interest income* | | | (16,974 | ) | | | (14,833 | ) | | | (9,016 | ) | | | (2,141 | ) | | | 14 | % | | | (5,817 | ) | | | 65 | % |
| Foreign exchange (gain)loss | | | (5,177 | ) | | | 89,893 | | | | 104,427 | | | | (95,070 | ) | | | (106 | %) | | | (14,534 | ) | | | (14 | %) |
| Warrant issue costs | | | 421,008 | | | | - | | | | - | | | | 421,008 | | | | 100 | % | | | - | | | | 100 | % |
| Unrealized gain on revaluation of warrant liability | | | (780,074 | ) | | | - | | | | - | | | | | | | | | | | | | | | | | |
*N/M – Not meaningful
Fiscal 2011 versus Fiscal 2010
Interest expenses increased by 7%, or $0.06 million. The increase was primarily due to $0.08 million in financing costs incurred as a result of the note payable incurred during the year with OrbiMed offset by decreases in the interest paid on the repayable leasehold improvement allowance.
Foreign exchange loss (gain)
Foreign exchange loss (gain) decreased by 106%, or $0.10 million. Foreign exchange gains and losses are largely due to U.S. dollar balances of cash and cash equivalents, accounts receivable and accounts payable affected by the fluctuations in the value of the U.S. dollar as compared to the Canadian dollar.
Warrant issue costs were incurred in 2011 as a result of the rights offering. These costs were allocated to the warrant liability based on the fair value of the warrant and expensed as incurred.
The change in fair value of the warrant liability decreased between the time of the closing of the rights offering and December 31, 2011 resulting in a $0.78 million gain on change in fair value. The gain was due to a $0.01 decrease in the fair market value of the shares.
Fiscal 2010 versus Fiscal 2009
Interest expense decreased by 6%, or $0.05 million. The decrease was due to the repayment of the repayable leasehold improvement allowance.
Foreign exchange loss (gain)
Foreign exchange loss (gain) decreased by 14%, or $0.01 million. Foreign exchange gains and losses are largely due to U.S. dollar balances of cash and cash equivalents, accounts receivable and accounts payable affected by the fluctuations in the value of the U.S. dollar as compared to the Canadian dollar.
Liquidity and Capital Resources
Total cash and cash equivalents and working capital at December 31, 2011, 2010 and 2009 were as follows:
| | | As at December 31, | |
| | | 2011 | | | 2010 | |
| Cash and cash equivalents | | | 7,354,802 | | | | 4,330,117 | |
| Percentage of total assets | | | 35 | % | | | 22 | % |
| Working capital (deficiency) | | | 3,815,281 | | | | 6,202,048 | |
The Company has financed its operations primarily through equity financings. As of December 31, 2011, the Company has raised approximately $103 million from the sale and issuance of equity securities and debt, net of issue costs.
The Company has sustained continuing losses since its formation and at December 31, 2011, had a deficit of approximately $106.9 million and for the year ended December 31, 2011 incurred negative cash flows from operations of $2.6 million compared to $8.2 million in 2010. Even though there is an improvement in the negative cash flow, these conditions raise substantial doubt about the Company’s ability to continue as a going concern.
Management has been able, thus far, to finance the operations through a series of equity financings. Management will continue, as appropriate, to seek other sources of financing on favorable terms; however, there are no assurances that any such financing can be obtained on favorable terms, if at all. In view of these conditions, the ability of the Company to continue as a going concern is dependent upon its ability to obtain such financing and, ultimately, on achieving profitable operations. The outcome of these matters cannot be predicted at this time.
Ongoing Sources and Uses of Cash
| | | For the Years Ended December 31, | | | Change 2010 to 2011 | | | Change 2009 to 2010 | |
| | | 2011 | | | 2010 | | | 2009 | | | Increase / (Decrease) | | | Percent Change | | | Increase / (Decrease) | | | Percent Change | |
| Cash used in operating activities | | | (2,609,682 | ) | | | (8,234,208 | ) | | | (8,202,267 | ) | | | 5,624,526 | | | | (68 | %) | | | (31,941 | ) | | | 0 | % |
| Cash used in investing activities | | | (105,985 | ) | | | (91,561 | ) | | | (127,368 | ) | | | (14,424 | ) | | | 16 | % | | | 35,807 | | | | (28 | %) |
| Cash provided by financing activities | | | 5,740,352 | | | | 7,582,415 | | | | 11,148,454 | | | | (1,842,063 | ) | | | (24 | %) | | | (3,566,039 | ) | | | (32 | %) |
| (Decrease) increase in cash during the period | | $ | 3,024,685 | | | $ | (743,354 | ) | | $ | 2,818,819 | | | $ | 3,768,039 | | | | | | | $ | (3,562,173 | ) | | | | |
As at December 31, 2011, the Company had cash and cash equivalents balance of $7.4 million as a result of a $3.0 million increase in cash during the year ended December 31, 2011. The cash increase was a result of $2.6 million cash used in operating activities, $0.1 million of cash used in investing activities, and $5.7 million of cash provided by financing activities.
The net change in non-cash working capital and non-current asset balances related to operations for the years ended December 31, 2011, 2010 and 2009 consists of the following:
Cash Used in Operating Activities
Explanations of the more significant net changes in working capital and non-current asset balances are as follows:
| | | 2011 | | | 2010 | | | 2009 | |
| Trade receivables | | | (343,635 | ) | | | 840,965 | | | | (1,068,095 | ) |
| Other receivables | | | 6,141 | | | | (54,889 | ) | | | 14,480 | |
| Inventories | | | 836,312 | | | | (855,595 | ) | | | 226,169 | |
| Prepaid expenses and other | | | (79,115 | ) | | | (23,766 | ) | | | 218,002 | |
| Accounts payable and accrued liabilities | | | 1,853,823 | | | | 99,460 | | | | (952,573 | ) |
| Deferred revenue | | | (287,564 | ) | | | (11,721 | ) | | | 535,433 | |
| · | Trade receivables increased to $1.6 million as a result of higher sales made in the last quarter of fiscal 2011 offset by improvements in the days sales outstanding. |
| · | Inventory balances decreased to $2.2 million as a result of the depletion of our reader instrument levels in finished goods as at December 31, 2011. |
| · | Accounts payable and accrued liabilities increased to $3.5 million as a result of the increase in legal and audit fees incurred with respect to the restatement of our fiscal 2010 financial statements and our rights offering in 2011 and cash management strategies developed in the last quarter of fiscal 2011. |
| · | Deferred revenue decreased to $0.4 million as a result of the decrease in collaborative agreements in fiscal 2011. |
Cash Used in Investing Activities
Net cash used in investing activities for the years ended December 31, 2011, 2010, and 2009 was $0.1 million, $0.09 million, and $0.13 million. This cash was used for the purchase of property, plant, and equipment.
Cash Provided by Financing Activities
Net cash provided by financing activities for the year ended December 31, 2011, 2010, and 2009 was $5.7 million, $7.6 million, and $11.1 million. This cash was provided by a series of equity financings during the respective years offset by cash used in the repayment of the leasehold improvement allowance.
Contractual Obligations and Contingencies
The following table summarizes our contractual commitments as of December 31, 2011 and the effect those commitments are expected to have on liquidity and cash flow in future periods.
| Payments due by Period | |
| Contractual Obligations | | Total | | | Less than 1 Year | | | 1-3 years | | | 3 - 5 years | | | More than 5 years | |
Long-term debt obligations (1) | | | 11,767,684 | | | | 1,061,746 | | | | 2,123,492 | | | | 2,123,492 | | | | 6,458,954 | |
Operating lease obligations (2) | | | 12,224,709 | | | | 990,017 | | | | 2,059,967 | | | | 2,134,165 | | | | 7,040,560 | |
Purchase obligations (3) | | | 879,524 | | | | 318,361 | | | | 364,912 | | | | 196,251 | | | | — | |
License fees (4) | | | 50,680 | | | | 26,340 | | | | 22,340 | | | | 2,000 | | | | — | |
| Total | | | 24,922,597 | | | | 2,396,464 | | | | 4,570,711 | | | | 4,455,908 | | | | 13,499,514 | |
| | (1) Long-term debt obligations consist of the repayable leasehold improvement allowance including interest and principal payments. The term of repayable leasehold improvement allowance coincides with the term of the lease mention in note (2). |
| | (2) Operating lease obligations consist of leases of the facilities and property, plant, and equipment. These lease obligations expire on various dates between 2012 and 2023. The lease for the facility, which commenced in 2008, has a term of 15 years. |
| | (3) Purchase obligations consist of obligations to purchase raw material from key suppliers and obligations to purchase property, plant, and equipment. |
| | (4) License fees consist of obligations to pay licensing fees for the use of related intellectual property to manufacture, sell, and have sold lateral flow immunoassay products. In consideration for these rights, the Company will pay non-refundable, non-creditable fees. |
Off-Balance-Sheet Arrangements
As of December 31, 2011, we had the following material off-balance-sheet arrangements as defined in Item 303(a)(4)(ii) of SEC Regulation S-K:
Under the Articles of the Company, applicable law and agreements with its directors and officers, the Company, in circumstances where the individual has acted legally, honestly and in good faith, may, or is required to indemnify its directors and officers against certain losses. The Company's liability in respect of the indemnities is not limited. The maximum potential of the future payments is unlimited. However, the Company maintains appropriate liability insurance that limits the exposure and enables the Company to recover any future amounts paid, less any deductible amounts pursuant to the terms of the respective policies, the amounts of which are not considered material.
The Company has entered into license and research agreements with third parties that include indemnification provisions that are customary in the industry. These indemnifications generally require the Company to compensate the other party for certain damages and costs incurred as a result of third party claims or damages arising from these transactions. The nature of the indemnification obligations prevents the Company from making a reasonable estimate of the maximum potential amount that it could be required to pay. To date, the Company has not made any indemnification payments under such agreements and no amount has been accrued in these financial statements with respect to these indemnification obligations.
Critical Accounting Policies and Estimates
A summary of the significant accounting policies is as follows:
Use of estimates
Our consolidated financial statements are prepared in accordance with U.S. GAAP. In the application of U.S. GAAP we are required to make estimates that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities in our consolidated financial statements. Changes in the accounting estimates from period to period are reasonably likely to occur. Accordingly, actual results could differ significantly from the estimates made by management. To the extent that there are material differences between these estimates and actual results, our future financial statement presentation of our financial condition or results of operations may be affected.
On an ongoing basis, we evaluate our estimates, including those related to revenue recognition, valuation of stock based compensation, valuation of long-lived assets, tax related contingencies, valuation of inventories, contingencies and litigation, among others. We base our estimates on historical experience and on various other assumptions, including expected trends that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources.
In addition to making critical accounting estimates, we must ensure that our financial statements are properly stated in accordance with U.S. GAAP. In many cases, the accounting treatment of a particular transaction is specifically dictated by U.S. GAAP and does not require a high degree of management judgment in its application, while in other cases, management’s judgment is required in selecting among available alternative accounting standards that allow different accounting treatment for similar transactions.
Our significant accounting policies are discussed in Note 3, “Significant Accounting Policies,” to the consolidated financial statements included in Item 8 of this Annual Report. We believe that the following are our most critical accounting policies and estimates, each of which is critical to the portrayal of our financial condition and results of operations and requires our most difficult, subjective and complex judgments. Our management has reviewed our critical accounting policies and the related disclosures with the Audit Committee of our Board of Directors.
Inventories
Raw material inventory is carried at the lower of actual cost, determined on a first-in first-out basis, and market value. Finished goods and work in process inventories are carried at the lower of weighted average cost and market value. Cost of finished goods and work in process inventories includes direct materials, direct labour and applicable overhead. The Company writes down its inventory balances for estimates of excess and obsolete amounts. These write-downs are recorded as a component of cost of sales. At the point of the write-down, a new, lower-cost basis for that inventory is established, and any subsequent improvements in facts and circumstances do not result in the restoration or increase in that newly established cost basis.
Long Lived Asset Impairment
Long-lived assets to be held and used by the Company are periodically reviewed to determine whether any events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. For long-lived assets to be held and used, the Company bases its evaluation on such impairment indicators such as the nature of the assets, the future economic benefit of the assets, any historical or future profitability measurements, as well as other external market conditions or factors that may be present. In the event that facts and circumstances indicate that the carrying amount of an asset may not be recoverable and an estimate of future undiscounted cash flows is less than the carrying amount of the asset, an impairment loss will be recognized for the difference between the carrying value and the fair value.
Deferred lease inducements
Lease inducements arising from rent-free inducements and non-repayable leasehold improvement allowances received from the landlord are being amortized over the term of the lease on a straight-line basis.
Contingent Liabilities
The Company provides for contingent liabilities when (1) it is probable that an asset has been impaired or a liability has been incurred at the date of the financial statements and (2) the amount of the loss can be reasonably estimated. Disclosure in the notes to the financial statements is required for loss contingencies that do not meet both these conditions if there is a reasonable possibility that a loss may have been incurred. The costs of defending legal claims against the Company are expensed as incurred.
Revenue recognition
Product sales are recognized when legal title passes to distributors or customers, the sales price is fixed and determinable, collection of the resulting receivables is reasonably assured and no uncertainties with regard to customer acceptance exist. Sales are recorded net of discounts and sales returns.
Contract service fees are recorded as revenue as the services are performed pursuant to the terms of the contract provided collectability is reasonably assured. Upfront fees from collaborative research arrangements which are non-refundable, require the ongoing involvement of the Company and are directly linked to specific milestones are deferred and amortized into income as services are rendered. Upfront fees from collaborative research arrangements that are non-refundable, require the ongoing involvement of the Company and are not directly linked to specific milestones are deferred and amortized into income on a straight-line basis over the term of ongoing development. Upfront fees from collaborative research arrangements that are refundable are deferred and recognized once the refundability period has lapsed. The Company earned revenue from contract service fees from collaborative research arrangements with Roche Diagnostics, 3M Company, and the Foundation for Innovative New Diagnostics (“FIND”) for the fiscal years of 2011, 2010, and 2009. The collaborative research arrangements with Roche Diagnostics were to develop a next generation Troponin assay and to develop a program, conduct clinical trials, and submit an application for the FDA waiver of the Clinical Laboratory Improvement Amendments (“CLIA”) requirements for the NT-proBNP assay. Under the agreements with Roche Diagnostics, the Company was entitled to $1,392,060 for the Troponin development project and $590,444 for the NT-proBNP assay. The collaborative research arrangement with 3M Company was to redevelop a Flu assay and under the collaborative arrangement, the Company was entitled to $113,000 U.S. Dollars. Under the agreement with FIND, the Company was entitled to $125,000 U.S. Dollars to conduct a feasibility study for the development a Tuberculosis assay.
Stock-based compensation
The Company uses the fair value method of accounting for all stock-based awards for non-employees and for all stock-based awards to employees that were granted, modified or settled since January 1, 2003. The fair value of stock options is determined using the Black-Scholes option-pricing model, which requires certain assumptions, including future stock price volatility, estimated forfeiture rates and expected time to exercise. Stock-based compensation expense is recorded net of estimated forfeitures such that expense is recorded only for those stock-based awards that are expected to vest. A forfeiture rate is estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from initial estimates. Changes to any of these assumptions could produce different fair values for stock-based compensation. The expense is amortized on a straight-line basis over the graded vesting period.
Income taxes
The Company accounts for income taxes using the liability method of tax allocation. Deferred income taxes are recognized for the future income tax consequences attributable to differences between the carrying values of assets and liabilities and their respective income tax bases. Deferred income tax assets and liabilities are measured using substantively enacted income tax rates expected to apply to taxable income in the years in which temporary differences are expected to reverse. The effect on deferred income tax assets and liabilities of a change in substantively enacted rates is included in earnings in the period that includes the substantive enactment date. Deferred income tax assets, net of a valuation allowance, are recorded in the consolidated financial statements if realization is considered more likely than not.
The Company accounts for uncertain tax positions using a “more-likely-than-not threshold for recognizing and resolving uncertain tax positions. The Company evaluates uncertain tax positions on a quarterly basis and considers various factors, including, but not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. The Company includes interest and penalties related to gross unrecognized tax benefits in the provision for income taxes.
For information on the recent accounting pronouncements impacting our business, see Note 4 of the Notes to Consolidated Financial Statements included in Item 8.
ITEM 7A. Quantitative and Qualitative Disclosures about Market Risk.
Currency Fluctuations and Exchange Risk
The Company is subject to foreign exchange risk as a significant portion of its revenues and expenditures are denominated in U.S. dollars. Significant losses may occur due to significant balances of cash held in U.S. dollars that may be affected negatively by a decline in the value of the U.S. dollar as compared to the Canadian dollar. The Company mitigates foreign exchange risk by maintaining a U.S. dollar bank account for all U.S. revenues and expenditures, thereby minimizing currency exchange. A 10% depreciation or appreciation of the Canadian dollar against the U.S. dollar would result in an increase/decrease of approximately $8,000 in the Company's loss as a result of revaluing the Company’s balance sheet items as at December 31, 2011.
Interest Rate Risk
Interest rate risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market interest rates. The Company’s exposure to interest rate risk is limited as its restricted cash are long-term in nature and the interest rate related to both its repayable leasehold improvement allowance is fixed over the term of the debt.
ITEM 8. Financial Statements and Supplementary Data.
Consolidated Financial Statements
Response Biomedical Corporation
(Expressed in Canadian dollars)
(Prepared in accordance with generally accepted accounting principles used in the United States of America (U.S. GAAP))
As at December 31, 2011 and 2010 and for each of the three years in the period ended December 31, 2011
MANAGEMENT’S RESPONSIBILITY FOR FINANCIAL REPORTING
The consolidated financial statements contained in this annual report have been approved by the board of directors and were prepared by management in accordance with United States generally accepted accounting principles. Management is responsible for the preparation and integrity of the consolidated financial statements and all other information in the annual report, and for ensuring that this information is consistent, where appropriate, with the information contained in the financial statements.
Management has developed and is maintaining a system of policies and procedures and internal controls to obtain reasonable assurance that the Company’s assets are safeguarded, transactions are authorized and financial information is reliable.
The Board of Directors is responsible for ensuring that management fulfills its responsibility for financial reporting and internal control. The Board of Directors exercises this responsibility principally through the Audit Committee. The Audit Committee consists of directors not involved in the daily operations of the Company. The Audit Committee meets with management and the independent registered public accounting firm to satisfy itself that management’s responsibilities are properly discharged and to review the financial statements prior to their presentation to the Board of Directors for approval.
The independent registered public accounting firm, Ernst & Young LLP, conducted an independent audit of the consolidated financial statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Their report expresses their opinion on the consolidated financial statements of the Company. The external auditors have free and full access to the Audit Committee with respect to their findings.
| /s/ Peter A. Thompson | | /s/ Richard A. Canote | |
| | | | |
| Peter A. Thompson | | Richard A. Canote | |
| | | | |
| Interim Chief Executive Officer | | Chief Financial Officer | |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of
Response Biomedical Corporation
We have audited the accompanying consolidated balance sheets of Response Biomedical Corporation (the "Company") as at December 31, 2011 and 2010 and the related consolidated statements of loss, shareholders’ equity, and cash flows for each of the three years in the period ended December 31, 2011. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Response Biomedical Corporation at December 31, 2011 and 2010, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2011, in conformity with United States generally accepted accounting principles.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company's recurring losses from operations raise substantial doubt about its ability to continue as a going concern. Management's plans as to these matters are also described in Note 2. The 2011 consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| | | | /s/ Ernst & Young LLP |
| Vancouver, Canada | | | Chartered Accountants |
| March 29, 2012 | | | |
Response Biomedical Corporation
CONSOLIDATED BALANCE SHEETS
[See Note 2 - Basis of Presentation and Going Concern Uncertainty]
(Expressed in Canadian dollars)
| As at December 31, | | 2011 | | | 2010 | |
| | | $ | | | $ | |
| | | | | | | | | |
| ASSETS | | | | | | | | |
| Current | | | | | | | | |
| Cash and cash equivalents | | | 7,354,802 | | | | 4,330,117 | |
Trade receivables, net [note 5] | | | 1,562,305 | | | | 1,218,670 | |
| Other receivables | | | 94,744 | | | | 100,885 | |
Inventories [note 7] | | | 2,204,443 | | | | 3,040,755 | |
| Prepaid expenses and other | | | 280,968 | | | | 201,853 | |
| Total current assets | | | 11,497,262 | | | | 8,892,280 | |
| Long-term prepaid expenses | | | 61,400 | | | | 61,400 | |
Restricted deposits [note 10[iii]] | | | 900,610 | | | | 905,112 | |
Property, plant and equipment [note 8] | | | 8,433,994 | | | | 9,665,139 | |
| | | | 20,893,266 | | | | 19,523,931 | |
| | | | | | | | | |
| LIABILITIES AND SHAREHOLDERS’ EQUITY | | | | | | | | |
| Current | | | | | | | | |
Accounts payable and accrued liabilities [notes 9, 13] | | | 3,527,288 | | | | 1,673,465 | |
Lease inducements - current portion [note 10] | | | 168,939 | | | | 168,939 | |
Repayable leasehold improvement allowance -current portion [note 10] | | | 331,869 | | | | 297,449 | |
Deferred revenue - current portion [note 11] | | | 306,071 | | | | 550,379 | |
Warrant liability [note 12[b][i]] | | | 3,347,814 | | | | - | |
| Total current liabilities | | | 7,681,981 | | | | 2,690,232 | |
Lease inducements [note 10] | | | 1,703,462 | | | | 1,872,399 | |
Repayable leasehold improvement allowance [note 10] | | | 6,452,476 | | | | 6,784,345 | |
Deferred revenue [note 11] | | | 79,624 | | | | 122,880 | |
| | | | 15,917,543 | | | | 11,469,856 | |
Commitments and contingencies [notes 15 and 18] | | | | | | | | |
| Shareholders’ equity | | | | | | | | |
Common shares [note 12] | | | 99,276,253 | | | | 96,945,332 | |
Additional paid-in capital [note 12] | | | 12,589,561 | | | | 12,627,522 | |
| Deficit | | | (106,890,091 | ) | | | (101,518,779 | ) |
| Total shareholders’ equity | | | 4,975,723 | | | | 8,054,075 | |
| | | | 20,893,266 | | | | 19,523,931 | |
Response Biomedical Corporation
CONSOLIDATED STATEMENTS OF LOSS
(Expressed in Canadian dollars)
| Years ended December 31, | | 2011 | | | 2010 | | | 2009 | |
| | | $ | | | $ | | | $ | |
| | | | | | | | | | | | | |
| REVENUE | | | | | | | | | | | | |
Product sales [note 16] | | | 9,024,083 | | | | 6,792,130 | | | | 8,153,049 | |
Cost of sales [notes 7 and 15] | | | 6,968,832 | | | | 7,097,538 | | | | 7,933,704 | |
| Gross profit (loss) on product sales | | | 2,055,251 | | | | (305,408 | ) | | | 219,345 | |
| Contract service fees and revenues from collaborative research arrangements | | | 449,386 | | | | 320,878 | | | | 1,793,220 | |
| | | | 2,504,637 | | | | 15,470 | | | | 2,012,565 | |
| EXPENSES | | | | | | | | | | | | |
| Research and development | | | 2,852,129 | | | | 4,106,266 | | | | 5,635,692 | |
| General and administrative | | | 3,625,510 | | | | 3,696,819 | | | | 3,299,042 | |
| Marketing and business development | | | 914,736 | | | | 1,413,171 | | | | 1,668,220 | |
| | | | 7,392,375 | | | | 9,216,256 | | | | 10,602,954 | |
| OTHER EXPENSES (INCOME) | | | | | | | | | | | | |
Interest expense [note 10[iii]] | | | 864,791 | | | | 806,065 | | | | 857,731 | |
| Interest income | | | (16,974 | ) | | | (14,833 | ) | | | (9,016 | ) |
| Foreign exchange (gain)loss | | | (5,177 | ) | | | 89,893 | | | | 104,427 | |
Warrant issue costs [note 12[b]] | | | 421,008 | | | | - | | | | - | |
Unrealized gain on revaluation of warrant liability [note 6] | | | (780,074 | ) | | | - | | | | - | |
| | | | 483,574 | | | | 881,125 | | | | 953,142 | |
| Net loss | | | (5,371,312 | ) | | | (10,081,911 | ) | | | (9,543,531 | ) |
| Loss per common share - basic and diluted | | | (0.14 | ) | | | (0.32 | ) | | | (0.43 | ) |
| Weighted average number of common shares outstanding | | | 39,446,554 | | | | 31,173,823 | | | | 22,209,393 | |
Response Biomedical Corporation
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ EQUITY
(Expressed in Canadian dollars)
| | | Common Stock Issued and Outstanding | | | Additional paid in capital | | | Deficit | | | Total Shareholders' Equity | |
| | | # of shares | | | $ | | | $ | | | $ | | | $ | |
| Balance at December 31, 2009 | | | 25,467,152 | | | | 89,015,372 | | | | 12,068,038 | | | | (91,436,868 | ) | | | 9,646,542 | |
| Net loss | | | | | | | | | | | | | | | (10,081,911 | ) | | | (10,081,911 | ) |
| Private placement, net of issue costs | | | 13,333,333 | | | | 7,474,920 | | | | - | | | | - | | | | 7,474,920 | |
| Exercise of stock options | | | 270 | | | | 324 | | | | - | | | | - | | | | 324 | |
| Exercise of warrants | | | 149,507 | | | | 373,769 | | | | - | | | | - | | | | 373,769 | |
| Reclassification of stock based compensation on exercise of stock options | | | - | | | | 162 | | | | (162 | ) | | | - | | | | - | |
| Reclassification of value on warrants exercised | | | - | | | | 80,785 | | | | (80,785 | ) | | | - | | | | - | |
| Stock-based compensation expense | | | - | | | | - | | | | 640,431 | | | | - | | | | 640,431 | |
| Balance at December 31, 2010 | | | 38,950,262 | | | | 96,945,332 | | | | 12,627,522 | | | | (101,518,779 | ) | | | 8,054,075 | |
| Net loss | | | - | | | | - | | | | - | | | | (5,371,312 | ) | | | (5,371,312 | ) |
| Rights offering, net of issue costs | | | 90,127,904 | | | | 2,330,921 | | | | | | | | | | | | 2,330,921 | |
| Stock-based compensation expense | | | - | | | | - | | | | (37,961 | ) | | | - | | | | (37,961 | ) |
| Balance at December 31, 2011 | | | 129,078,166 | | | | 99,276,253 | | | | 12,589,561 | | | | (106,890,091 | ) | | | 4,975,723 | |
Response Biomedical Corporation
CONSOLIDATED STATEMENTS OF CASH FLOWS
(Expressed in Canadian dollars)
| Years ended December 31, | | 2011 | | | 2010 | | | 2009 | |
| | | $ | | | $ | | | $ | |
| | | | | | | | | | | | | |
| OPERATING ACTIVITIES | | | | | | | | | | | | |
| Net loss | | | (5,371,312 | ) | | | (10,081,911 | ) | | | (9,543,531 | ) |
| Add (deduct) items not involving operating cash: | | | | | | | | | | | | |
| Depreciation of plant and equipment | | | 1,337,130 | | | | 1,385,776 | | | | 1,452,126 | |
| Amortization of deferred lease inducements | | | (168,937 | ) | | | (168,939 | ) | | | (168,939 | ) |
| Restricted deposits | | | 4,502 | | | | (4,019 | ) | | | 2,155 | |
| Stock-based compensation | | | (37,961 | ) | | | 640,431 | | | | 1,037,713 | |
| Unrealized gain on revaluation of warrant liability | | | (780,074 | ) | | | - | | | | - | |
| Warrant issuance costs | | | 421,008 | | | | - | | | | - | |
| Other non-cash items | | | - | | | | - | | | | 44,793 | |
| Changes in non-cash working capital | | | | | | | | | | | | |
| Trade receivables | | | (343,635 | ) | | | 840,965 | | | | (1,068,095 | ) |
| Other receivables | | | 6,141 | | | | (54,889 | ) | | | 14,480 | |
| Inventories | | | 836,312 | | | | (855,595 | ) | | | 226,169 | |
| Prepaid expenses and other | | | (79,115 | ) | | | (23,766 | ) | | | 218,002 | |
| Accounts payable and accrued liabilities | | | 1,853,823 | | | | 99,460 | | | | (952,573 | ) |
| Deferred revenue | | | (287,564 | ) | | | (11,721 | ) | | | 535,433 | |
| Cash used in operating activities | | | (2,609,682 | ) | | | (8,234,208 | ) | | | (8,202,267 | ) |
| INVESTING ACTIVITIES | | | | | | | | | | | | |
| Purchase of property, plant and equipment | | | (105,985 | ) | | | (91,561 | ) | | | (127,368 | ) |
| Cash used in investing activities | | | (105,985 | ) | | | (91,561 | ) | | | (127,368 | ) |
| FINANCING ACTIVITIES | | | | | | | | | | | | |
| Repayment of repayable leasehold improvement allowance | | | (297,449 | ) | | | (266,598 | ) | | | (243,778 | ) |
| Proceeds from issuance of common shares, net of share issue costs | | | 2,330,921 | | | | 7,849,013 | | | | 11,392,232 | |
| Proceeds from issuance of warrants, net of warrant issue costs | | | 3,706,880 | | | | - | | | | - | |
| Cash provided by financing activities | | | 5,740,352 | | | | 7,582,415 | | | | 11,148,454 | |
| | | | | | | | | | | | | |
| Increase (decrease) in cash during the period | | | 3,024,685 | | | | (743,354 | ) | | | 2,818,819 | |
| Cash and cash equivalents, beginning of period | | | 4,330,117 | | | | 5,073,471 | | | | 2,254,652 | |
| Cash and cash equivalents, end of period | | | 7,354,802 | | | | 4,330,117 | | | | 5,073,471 | |
| | | | | | | | | | | | | |
| Supplemental Disclosure | | | | | | | | | | | | |
| Interest paid in cash | | | 769,491 | | | | 799,024 | | | | 823,002 | |
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
1. DESCRIPTION OF BUSINESS
Response Biomedical Corporation (the “Company”) was incorporated on August 20, 1980 under the predecessor to the Business Corporations Act (British Columbia). The Company is engaged in the research, development, commercialization and distribution of diagnostic technologies for the medical point of care (“POC”) and on-site environmental testing markets. POC and on-site diagnostic tests (or assays) are simple, non-laboratory based tests performed using portable hand-held devices, compact desktop analyzers, single-use test cartridges and/or dipsticks. Since 1996, the Company has developed and commercialized a proprietary diagnostic system called RAMP®.
The RAMP® System is a portable fluorescence immunoassay-based diagnostic technology that combines the performance of a clinical lab with the convenience of a dipstick test - establishing a new paradigm in diagnostic testing. Immunoassays are extremely sensitive and specific tests used to identify and measure small quantities of materials, such as proteins. A large variety of biological molecules and inorganic materials can be targeted. Accordingly, the RAMP® technology is applicable to multiple distinct market segments and many products within those segments. RAMP® tests are now commercially available for use in the early detection of heart attack, congestive heart failure, influenza A+B, the respiratory syncytial virus, environmental detection of West Nile Virus, and biodefense applications including the rapid on-site detection of anthrax, smallpox, ricin and botulinum toxin.
2. BASIS OF PRESENTATION AND GOING CONCERN UNCERTAINTY
These consolidated financial statements have been prepared by management in Canadian dollars in accordance with United States generally accepted accounting principles (“U.S. GAAP”).
Adoption of United States Generally Accepted Accounting Principles
The Company adopted U.S. GAAP as its primary basis of financial reporting commencing January 1, 2011 on a retrospective basis. All comparative financial information contained in these audited consolidated financial statements has been revised to reflect the Company’s results as if they had been historically reported in accordance with U.S. GAAP.
The Company historically prepared its consolidated financial statements in conformity with Canadian generally accepted accounting principles and provided a supplemental reconciliation to U.S. GAAP. The adoption of U.S. GAAP did not have a material change on the Company’s financial results.
Going Concern Uncertainty
The accompanying financial statements have been prepared assuming the Company will continue to operate as a going concern, which contemplates the realization of assets and liabilities and commitments in the normal course of business. However, as presented in the financial statements, as of December 31, 2011, the Company had a cash balance of $7,354,802 and an accumulated deficit of $106,890,091. The Company also incurred a net loss of $5,371,312 and negative cash flows from operations of $2,609,682 in 2011. As a result, there exists substantial doubt about the Company’s ability to continue as a going concern. The 2011 financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from uncertainty related to the Company’s ability to continue as a going concern.
Management has been able, thus far, to finance the operations through a series of equity financings. On December 29, 2011, the Company closed a rights offering for net cash proceeds of $6,037,803. Management will continue, as appropriate, to seek other sources of financing on favourable terms. However, there are no assurances that any such financing can be obtained on favourable terms, if at all. In view of these conditions, the ability of the Company to continue as a going concern is dependent upon its ability to obtain such financing and, ultimately, on achieving profitable operations. The outcome of these matters cannot be predicted at this time. The consolidated financial statements do not include any adjustments to the amounts and classification of assets and liabilities that might be necessary should the Company be unable to continue in business.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The accompanying consolidated financial statements reflect, in the opinion of management, all adjustments (which include reclassifications and normal recurring adjustments) necessary to present fairly the financial position at December 31, 2011 and 2010 and its results of operations and its cash flows for each of the three years in the period ended December 31, 2011.
3. SIGNIFICANT ACCOUNTING POLICIES
A summary of the significant accounting policies is as follows:
Principles of Consolidation
The consolidated financial statements include the accounts of the Company, and its wholly owned subsidiaries. All material intercompany accounts and transactions have been eliminated in consolidation.
Use of estimates
The preparation of these consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Areas of significant estimates include revenue recognition, stock-based compensation expense, the estimated life of property, plant and equipment, the value of the warrant liability, the resolution of uncertain tax positions, recoverability of long-lived assets and provisions for doubtful account, inventory obsolescence, and warranty accruals. Actual results could differ from those estimates.
Cash equivalents
The Company considers all highly liquid investments with an original maturity of 90 days or less, when acquired, to be cash equivalents, which are carried at fair value and are designated as held for trading.
Inventories
Raw material inventory is carried at the lower of actual cost, determined on a first-in first-out basis, and market value. Finished goods and work in process inventories are carried at the lower of weighted average cost and market value. Cost of finished goods and work in process inventories includes direct materials, direct labour and applicable overhead. The Company writes down its inventory balances for estimates of excess and obsolete amounts. These write-downs are recorded as a component of cost of sales. At the point of the write-down, a new, lower-cost basis for that inventory is established, and any subsequent improvements in facts and circumstances do not result in the restoration or increase in that newly established cost basis.
Property, plant and equipment
Property, plant and equipment is recorded at cost and depreciated over the estimated useful lives using the straight-line method as follows:
| Office and laboratory furniture and equipment | 5 years | | |
| Office and laboratory computer equipment | 3 years | | |
| Computer software | 2 years | | |
| Manufacturing molds | 5 years | | |
| Manufacturing equipment | 2 years | | |
| Leasehold improvements | Term of lease | | |
Long Lived Asset Impairment
Long-lived assets to be held and used by the Company are periodically reviewed to determine whether any events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. For long-lived assets to be held and used, the Company bases its evaluation on impairment indicators such as the nature of the assets, the future economic benefit of the assets, any historical or future profitability measurements, as well as other external market conditions or factors that may be present. In the event that facts and circumstances indicate that the carrying amount of an asset may not be recoverable and an estimate of future undiscounted cash flows is less than the carrying amount of the asset, an impairment loss will be recognized for the difference between the carrying value and the fair value.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
Leases
Leases are classified as either capital or operating leases. Leases that transfer substantially all the benefits and risks of ownership of the property to the Company are accounted for as capital leases. All other leases are accounted for as operating leases wherein rental payments are expensed in a manner that results in the total rent payments being recognized on a straight-line basis over the term of the lease.
Deferred lease inducements
Lease inducements arising from rent-free inducements and non-repayable leasehold improvement allowances received from the landlord are being amortized over the term of the lease on a straight-line basis.
Contingent Liabilities
The Company provides for contingent liabilities when (1) it is probable that an asset has been impaired or a liability has been incurred at the date of the financial statements and (2) the amount of the loss can be reasonably estimated. Disclosure in the notes to the financial statements is required for loss contingencies that do not meet both these conditions if there is a reasonable possibility that a loss may have been incurred. The costs of defending legal claims against the Company are expensed as incurred.
Revenue recognition
Product sales are recognized when legal title passes to distributors or customers, the sales price is fixed and determinable, collection of the resulting receivables is reasonably assured and no uncertainties with regard to customer acceptance exist. Sales are recorded net of discounts and sales returns.
Contract service fees are recorded as revenue as the services are performed pursuant to the terms of the contract provided collectability is reasonably assured. Upfront fees from collaborative research arrangements which are non-refundable, require the ongoing involvement of the Company and are directly linked to specific milestones are deferred and amortized into income as services are rendered. Upfront fees from collaborative research arrangements that are non-refundable, require the ongoing involvement of the Company and are not directly linked to specific milestones are deferred and amortized into income on a straight-line basis over the term of ongoing development. Upfront fees from collaborative research arrangements that are refundable are deferred and recognized once the refundability period has lapsed. The Company earned revenue from contract service fees from collaborative research arrangements with Roche Diagnostics, 3M Company, and the Foundation for Innovative New Diagnostics (“FIND”) for the fiscal years of 2011, 2010, and 2009. The collaborative research arrangements with Roche Diagnostics were to develop a next generation Troponin assay and to develop a program, conduct clinical trials, and submit an application for the FDA waiver of the Clinical Laboratory Improvement Amendments (“CLIA”) requirements for the NT-proBNP assay. Under the agreements with Roche Diagnostics, the Company was entitled to $1,392,060 for the Troponin development project and $590,444 for the NT-proBNP assay. The collaborative research arrangement with 3M Company was to redevelop a Flu assay and under the collaborative arrangement, the Company was entitled to $113,000 U.S. Dollars. Under the agreement with FIND, the Company was entitled to $125,000 U.S. Dollars to conduct a feasibility study for the development a Tuberculosis assay.
Accounts Receivable
For product sales, the Company typically invoices its customers at shipment for the sales order value of products shipped. For contract revenue, invoicing occurs based upon the terms of the specific research contract, typically one month in arrears for services rendered and any other allowable direct costs. Accounts receivable are recorded at the invoiced amount and do not bear interest. The Company does not have any off-balance sheet credit exposure related to any of its customers.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
Allowance for Doubtful Accounts
The Company evaluates the collectability of accounts receivable based on a combination of factors. In cases where the Company becomes aware of circumstances that may impair a specific customer’s ability to meet its financial obligations subsequent to the original sale, the Company will record an allowance against amounts due, and thereby reduce the net recognized receivable to the amount the Company reasonably believes will be collected. For all other customers, the Company recognizes an allowance for doubtful accounts based on the length of time the receivables are past due and consideration of other factors such as industry conditions, the current business environment and its historical experience.
Warranty accrual
The Company offers a standard limited warranty on its products. The Company estimates costs that may be incurred under its warranty program as liabilities at the time the products are sold. Factors that affect the Company’s warranty liability include the number of units sold, anticipated rate of warranty claims, and costs per claim, which require management to make estimates about future costs. The Company periodically assesses the adequacy of its recorded warranty liabilities and adjusts the amounts as necessary. The initial recognition of and subsequent adjustments to the warranty accrual are recorded to cost of sales.
Research and development costs
Research and development costs are expensed as incurred and include expenses associated with new product research and regulatory activities.
Shipping and Handling Costs
Shipping and handling costs are included in cost of revenues and are recognized as a period expense during the period in which they are incurred.
Stock-based compensation
The Company uses the fair value method of accounting for all stock-based awards for non-employees and for all stock-based awards to employees that were granted, modified or settled since January 1, 2003. The fair value of stock options is determined using the Black-Scholes option-pricing model, which requires certain assumptions, including future stock price volatility, estimated forfeiture rates and expected time to exercise. Stock-based compensation expense is recorded net of estimated forfeitures such that expense is recorded only for those stock-based awards that are expected to vest. A forfeiture rate is estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from initial estimates. Changes to any of these assumptions could produce different fair values for stock-based compensation. The expense is amortized on a straight-line basis over the graded vesting period.
Restricted deposits
Restricted deposits consists of long-term deposits pledged as security as part of certain contractual obligations. The interest earned on these deposits is recorded in interest income on the consolidated statements of loss.
Foreign currency translation
Monetary items denominated in foreign currencies are translated into Canadian dollars using exchange rates in effect at the balance sheet date. Non-monetary assets and liabilities are translated at historical exchange rates. Revenue and expense items are translated at the average exchange rate for the period. Foreign exchange gains and losses are included in the determination of loss for the year.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
Income taxes
The Company accounts for income taxes using the liability method of tax allocation. Deferred income taxes are recognized for the future income tax consequences attributable to differences between the carrying values of assets and liabilities and their respective income tax bases. Deferred income tax assets and liabilities are measured using substantively enacted income tax rates expected to apply to taxable income in the years in which temporary differences are expected to reverse. The effect on deferred income tax assets and liabilities of a change in substantively enacted rates is included in earnings in the period that includes the substantive enactment date. Deferred income tax assets, net of a valuation allowance, are recorded in the consolidated financial statements if realization is considered more likely than not.
The Company accounts for uncertain tax positions using a “more-likely-than-not threshold for recognizing and resolving uncertain tax positions. The Company evaluates uncertain tax positions on a quarterly basis and considers various factors, including, but not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. The Company includes interest and penalties related to gross unrecognized tax benefits in the provision for income taxes.
Loss per common share
Basic loss per common share is calculated using the weighted average number of common shares outstanding during the year, excluding contingently issuable shares. Diluted loss per common share is computed in accordance with the treasury stock method that uses the weighted average number of common shares outstanding during the period. The effect of potentially issuable common shares from outstanding stock options and outstanding warrants is anti-dilutive for all periods presented.
Segment Information
Operating segments are defined as components of an enterprise about which separate financial information is available that is evaluated regularly by the chief operating decision maker, or decision making group, in deciding how to allocate resources and in assessing performance. The Company’s chief operating decision maker is its Interim Chief Executive Officer. The Company has one operating segment which is dedicated to the manufacture and sale of RAMP® tests. In Note 16, the Company discloses information about Products and Services, Geographic Areas, and Major Customers.
4. RECENT ACCOUNTING PRONOUNCEMENTS
On January 1, 2011, the Company adopted Accounting Standards Update (ASU) 2010-06, Fair Value Measurements and Disclosures, which amended standards requiring additional fair value disclosures. The amended standards require disclosures of transfers in and out of Levels 1 and 2 of the fair value hierarchy, as well as requiring gross basis disclosures for purchases, sales, issuances and settlements within the Level 3 reconciliation. Additionally, the update clarifies the requirement to determine the level of disaggregation for fair value measurement disclosures and to disclose valuation techniques and inputs used for both recurring and nonrecurring fair value measurements in either Level 2 or Level 3. Because these new standards are related primarily to disclosures, the adoption did not have a significant impact on the Company’s consolidated financial statements.
On January 1, 2011, the Company adopted Accounting Standards Codification (ASC) Subtopic 605-25, Revenue Recognition - Multiple-Element Arrangements (ASC Subtopic 605-25). ASC Subtopic 605-25 provides principles for allocation of consideration among multiple-elements in an arrangement, allowing more flexibility in identifying and accounting for revenue from separate deliverables under an arrangement. ASC Subtopic 605-25 introduces an estimated selling price method for allocating revenue to the elements of a bundled arrangement if Vendor-specific objective evidence or third-party evidence of selling price is not available, and significantly expands related disclosure requirements. This standard is effective on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. The adoption of ASC Subtopic 605-25 did not have a material effect on the Company’s consolidated financial statements.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
On January 1, 2011, the Company adopted Accounting Standards Codification (ASC) Subtopic 605–28, Milestone Method of Revenue Recognition (ASC Subtopic 605-28). This standard provides guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for certain research and development transactions. Under this new standard, a company can recognize as revenue consideration that is contingent upon achievement of a milestone in the period in which it is achieved, only if the milestone meets all criteria to be considered substantive. This standard is effective for periods beginning after January 1, 2011. The adoption of this standard did not have a material effect on the Company’s consolidated financial statements.
In June, 2011, the FASB issued ASU 2011-05, Comprehensive Income (Topic 220): Presentation of Comprehensive Income. ASU 2011-05 eliminates the option to present other comprehensive income in the statement of changes in equity and provides the option to present the components of net income and comprehensive income in either one combined financial statement or two consecutive financial statements. The provisions of this ASU are applied retrospectively for interim and annual periods beginning after December 15, 2011. The adoption of this standard in fiscal 2012 is not expected to have a material effect on the Company’s consolidated financial statements.
In May, 2011, the FASB issued ASU No. 2011-04, “Fair Value Measurement.” This ASU clarifies the concepts related to highest and best use and valuation premise, blockage factors and other premiums and discounts, the fair value measurement of financial instruments held in a portfolio and of those instruments classified as a component of shareowners' equity. The guidance includes enhanced disclosure requirements about recurring Level 3 fair value measurements, the use of nonfinancial assets, and the level in the fair value hierarchy of assets and liabilities not recorded at fair value. The provisions of this ASU are effective prospectively for interim and annual periods beginning on or after December 15, 2011. The adoption of this standard in fiscal 2012 is not expected to have a material effect on the Company’s consolidated financial statements.
5. FINANCIAL INSTRUMENTS
For certain of the Company’s financial instruments, including cash and cash equivalents, trade receivables, other receivables, and accounts payable and accrued liabilities the carrying amounts approximate fair values due to their short-term nature. The fair value of the repayable leasehold improvement allowance approximates its carrying value as the fixed interest rate of 11% is considered to approximate the current market rate.
The Company has classified its cash and cash equivalents as held-for-trading. Restricted deposits are classified as held-to-maturity. Trade receivables and other receivables are classified as loans and receivables. Accounts payable, accrued and other liabilities and the repayable leasehold improvement allowance are classified as other financial liabilities.
Held-for-trading financial instruments are initially measured at fair value with subsequent changes in fair value recorded to net income. Held-to-maturity investments are measured at amortized cost using the effective interest method with changes in amortized cost recorded to net income. Loans and receivables and other financial liabilities are initially measured at amortized cost with subsequent changes in amortized cost recorded to net income. Transaction costs (except for transaction costs related to held-for-trading financial instruments which are expensed as incurred) are included in the carrying amounts of financial instruments as they are carried on the balance sheet.
Credit Risk
Credit risk is the risk of a financial loss if a customer or counterparty to a financial instrument fails to meet its obligations under a contract. The risk arises primarily from the Company’s receivables from customers.
The Company’s exposure to credit risk is dependent upon the characteristics of each customer. The Company performs ongoing credit checks on its customers and requires orders to be prepaid by certain customers.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The Company is subject to concentration risk related to its accounts receivable. The Company defines concentration risk as customers whose outstanding receivable is 10% or greater than the total receivable balance or who represent 10% or greater of total revenue. At December 31, 2011, three customers represent 82% [2010 - one customer represent 65%] of the trade receivables balance and at December 31, 2011 one customer represents 54% of the trade receivables balance. Refer to note 16 for a discussion of concentration risk on the Company’s revenues.
The Company reviews the collectability of its accounts receivable on a regular basis and establishes an allowance for doubtful accounts based on its best estimates of any potentially uncollectible accounts. As at December 31, 2011, the balance of the Company’s allowance for doubtful accounts was $Nil [2010 – $nil]. The amount written off during the year ended December 31, 2011 was $65,964 [2010 and 2009 $nil].
Liquidity Risk
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they are due. The Company continuously monitors actual and forecasted cash flows to ensure there is sufficient working capital to satisfy its operating requirements.
Pursuant to their respective terms, accounts payables, accrued liabilities, and the repayable leasehold improvement allowance are aged as follows:
| | | 2012 | | | 2013 | | | 2014 | | | 2015 | | | 2016 | | | Thereafter | |
| | | $ | | | $ | | | $ | | | $ | | | $ | | | $ | |
| Accounts payable and accrued liabilities | | | 3,527,288 | | | | - | | | | - | | | | - | | | | - | | | | - | |
| Repayable leasehold improvement allowance | | | 331,869 | | | | 370,272 | | | | 413,120 | | | | 460,926 | | | | 514,263 | | | | 4,693,894 | |
| | | | 3,859,157 | | | | 370,272 | | | | 413,120 | | | | 460,926 | | | | 514,263 | | | | 4,693,894 | |
6. FAIR VALUE MEASUREMENTS
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (“exit price”) in an orderly transaction between market participants at the measurement date. Fair value measurements of financial instruments are determined by using a fair value hierarchy that prioritizes the inputs to valuation techniques into three levels according to the relative reliability of the inputs used to estimate the fair values.
The three levels of inputs used to measure fair value are as follows:
Level 1 – Unadjusted quoted prices in active markets for identical financial instruments;
Level 2 – Inputs other than quoted prices that are observable for the financial instrument either directly or indirectly; and
Level 3 – Inputs that are not based on observable market data.
In determining fair value measurements, the Company uses the most observable inputs when available. The fair value hierarchy level at which a financial instrument is categorized is determined on the basis of the lowest level input that is significant to the fair value measurement.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| Financial Instrument carried at fair value as of December 31, 2011 |
| | | | | |
| Assets | | Level 1 | | Level 2 | | Level 3 | | | Total | |
| Cash and cash equivalents | | $ | 7,354,802 | | | | | | | $ | 7,354,802 | |
| | | | | | | | | | | | | |
| Liabilities | | | | | | | | | | | | |
| Warrant liability | | | | | | | $ | 3,347,814 | | | $ | 3,347,814 | |
As of December 31, 2011, the warrant liability is recorded at its fair value of $3,347,814. The Company reassesses the fair value of the common stock warrants at each reporting date utilizing a Black-Scholes pricing model. Inputs used in the pricing model include estimates of stock price volatility, contractual term of the warrant, and risk-free interest rate. The computation of expected volatility was based on the historical volatility of the Company’s stock.
The following table presents the changes in fair value of the Company’s total Level 3 financial liabilities for the year ended December 31, 2011:
| | | Balance at December 31, 2010 | | | Opening Balance at December 29, 2011 | | | Unrealized gain | | | Balance at December 31, 2011 | |
| Warrant Liability | | $ | - | | | $ | 4,127,888 | | | $ | (780,074 | ) | | $ | 3,347,814 | |
7. INVENTORIES
| | | 2011 | | | 2010 | |
| | | | $ | | | | $ | |
| Raw materials | | | 740,288 | | | | 876,181 | |
| Work in process | | | 524,862 | | | | 567,777 | |
| Finished goods | | | 939,293 | | | | 1,596,797 | |
| | | | 2,204,443 | | | | 3,040,755 | |
The carrying value of inventory as at December 31, 2011 includes a provision for lower of cost and market value on the Company’s RAMP® 200 Readers in the amount $102,453 [December 31, 2010 - nil] resulting from a 2012 purchase commitment of one customer. The carrying value of inventory as at December 31, 2011 also includes a provision against inventory held offsite that remain unsold in the amount of $179,176 [December 31, 2010 - $179,176]. The carrying value of inventory as at December 31, 2011 also includes a provision for obsolescence in the amount of $31,515 [December 31, 2010 - $30,658]. For the year ended December 31, 2011, inventory write-downs and obsolescence charges were $411,708 [2010 - $824,536; 2009 - $278,054].
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
8. PROPERTY, PLANT AND EQUIPMENT
| | | | | | Accumulated | | | Net book | |
| | | Cost | | | depreciation | | | Value | |
| | | $ | | | $ | | | $ | |
| | | | | | | | | | | | | |
| December 31, 2011 | | | | | | | | | | | | |
| Office furniture and equipment | | | 955,451 | | | | 715,749 | | | | 239,702 | |
| Office computer equipment | | | 282,803 | | | | 262,874 | | | | 19,929 | |
| Laboratory furniture and equipment | | | 569,901 | | | | 537,225 | | | | 32,676 | |
| Laboratory computer equipment | | | 467,907 | | | | 437,801 | | | | 30,106 | |
| Computer software | | | 409,705 | | | | 405,635 | | | | 4,070 | |
| Manufacturing equipment | | | 2,196,268 | | | | 1,372,988 | | | | 823,280 | |
| Manufacturing molds | | | 602,854 | | | | 601,944 | | | | 910 | |
| Leasehold improvements | | | 9,769,668 | | | | 2,486,347 | | | | 7,283,321 | |
| | | | 15,254,557 | | | | 6,820,563 | | | | 8,433,994 | |
| | | | | | | | | | | | | |
| December 31, 2010 | | | | | | | | | | | | |
| Office furniture and equipment | | | 955,451 | | | | 528,902 | | | | 426,549 | |
| Office computer equipment | | | 280,086 | | | | 231,932 | | | | 48,154 | |
| Laboratory furniture and equipment | | | 569,901 | | | | 515,039 | | | | 54,862 | |
| Laboratory computer equipment | | | 449,923 | | | | 420,093 | | | | 29,830 | |
| Computer software | | | 408,167 | | | | 336,342 | | | | 71,825 | |
| Manufacturing equipment | | | 2,114,201 | | | | 1,019,507 | | | | 1,094,694 | |
| Manufacturing molds | | | 601,173 | | | | 599,938 | | | | 1,235 | |
| Leasehold improvements | | | 9,769,669 | | | | 1,831,679 | | | | 7,937,990 | |
| | | | 15,148,571 | | | | 5,483,432 | | | | 9,665,139 | |
The following table shows depreciation expense allocated by type of cost:
| | | 2011 | | | 2010 | | | 2009 | |
| Cost of sales | | | 804,818 | | | | 860,858 | | | | 906,163 | |
| Research and development | | | 339,080 | | | | 318,434 | | | | 299,261 | |
| General and administrative | | | 125,147 | | | | 103,650 | | | | 146,295 | |
| Marketing and business development | | | 68,085 | | | | 102,834 | | | | 100,407 | |
| | | | 1,337,130 | | | | 1,385,776 | | | | 1,452,126 | |
As at December 31, 2011, $170,240 [2010 - $105,117] of manufacturing equipment was in the validation phase and not ready for use and therefore has not been depreciated.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
9. ACCOUNTS PAYABLE AND ACCRUED LIABILITIES
Accounts payable and accrued liabilities comprise:
| | | 2011 | | | 2010 | |
| | | $ | | | $ | |
| Trade accounts payable | | | 1,340,318 | | | | 717,648 | |
| Employee related accrued liabilities | | | 871,345 | | | | 391,760 | |
| Royalties | | | 489,593 | | | | 272,357 | |
| Other accrued liabilities | | | 826,032 | | | | 291,700 | |
| | | | 3,527,288 | | | | 1,673,465 | |
In accordance with Accounting Standards Codification Subtopic 420-10, Exit or Disposal Cost Obligation, included in employee related accruals are costs related to restructuring activities that commenced in September, 2010, focusing on a 25% reduction in the workforce. The liability was measured using fair value at the date of termination. These costs were recorded in general and administrative expenses on the consolidated statement of loss. As of December 31, 2011, the restructuring activities have been completed as follows:
| | | 2011 | | | 2010 | |
| | | $ | | | $ | |
| Balance at December 31 | | | 251,920 | | | | - | |
| Terminations under the plan | | | 125,185 | | | | 803,660 | |
| Payments made during the period | | | (377,105 | ) | | | (551,740 | ) |
| Balance as at December 31 | | | - | | | | 251,920 | |
10. LEASE INDUCEMENTS
During the year ended December 31, 2007 the Company entered into a 15 year facility lease agreement [Note 15[c][i]]. The agreement provides for lease inducements to be provided by the landlord to the Company which are summarized as follows:
| | | 2011 | | | 2010 | |
| Summarized as to: | | $ | | | $ | |
| Current Portion | | | | | | | | |
Rent-free inducement [i] | | | 54,278 | | | | 54,278 | |
Non-repayable leasehold improvement allowance [ii] | | | 114,661 | | | | 114,661 | |
| | | | 168,939 | | | | 168,939 | |
Repayable leasehold improvement allowance [iii] | | | 331,869 | | | | 297,449 | |
| Total Current Portion | | | 500,808 | | | | 466,388 | |
| | | | | | | | | |
| Long-Term Portion | | | | | | | | |
Rent-free inducement [i] | | | 547,299 | | | | 601,578 | |
Non-repayable leasehold improvement allowance [ii] | | | 1,156,163 | | | | 1,270,821 | |
| | | | 1,703,462 | | | | 1,872,399 | |
Repayable leasehold improvement allowance [iii] | | | 6,452,476 | | | | 6,784,345 | |
| Total Long-Term Portion | | | 8,155,938 | | | | 8,656,744 | |
| | | | | | | | | |
| Total | | | 8,656,746 | | | | 9,123,132 | |
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The lease inducements disclosed on the consolidated balance sheets as a result of these benefits is comprised of the following:
[i] In 2007, the Company entered into a long-term facility lease agreement that included an eight and one half month rent-free period from May 17, 2007 to February 1, 2008. The lease inducement benefit arising from the rent-free period is being amortized on a straight-line basis over the term of the operating lease as a reduction to rental expense Amortization expense for the year ended December 31, 2011 amounted to $54,278 [2010 - $54,278; 2009 - $54,278].
[ii] The Company received a non-repayable allowance for an amount of $1.7 million for expenditures related to general upgrades to the facility. The lease inducement benefit arising from the non-repayable leasehold improvement allowance is being amortized on a straight-line basis over the balance of the term of the lease beginning April 1, 2008 as a reduction to rental expense. Amortization expense for the year ended December 31, 2011 amounted to $114,661 [2010 - $114,661; 2009 - $114,661].
[iii] The Company received a repayable leasehold improvement for an amount of $7.8 million used for additional improvements to the facility. This lease inducement is being repaid over the term of the operating lease commencing February 1, 2008 at approximately $88,500 per month including interest calculated at an interest rate negotiated between the Company and the landlord. Principal repayments for the year ended December 31, 2011 amounted to $297,449 [2010 - $266,598; 2009 - $243,778], respectively. Interest payments for the year ended December 31, 2011 amounted to $764,297 [2010 - $795,148; 2009 - $822,865].
Future principal and interest repayments due to be paid are estimated as follows:
| | | Principal | | | Interest | | | Total | |
| | | $ | | | $ | | | $ | |
| 2012 | | | 331,869 | | | | 729,877 | | | | 1,061,746 | |
| 2013 | | | 370,272 | | | | 691,473 | | | | 1,061,745 | |
| 2014 | | | 413,120 | | | | 648,626 | | | | 1,061,746 | |
| 2015 | | | 460,926 | | | | 600,820 | | | | 1,061,746 | |
| 2016 | | | 514,263 | | | | 547,482 | | | | 1,061,745 | |
| Thereafter | | | 4,693,895 | | | | 1,765,060 | | | | 6,458,955 | |
| | | | 6,784,345 | | | | 4,983,338 | | | | 11,767,683 | |
To secure the lease, the Company is maintaining a security deposit with the landlord in the form of an irrevocable letter of credit in the amount of $870,610 collateralized by a term deposit with a market value of $870,610 that is presented as part of restricted deposits in the long-term asset section of the balance sheets.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
11. DEFERRED REVENUE
| | | 2011 | | | 2010 | |
| | | | $ | | | | $ | |
| Beginning balance: | | | | | | | | |
| Product sales | | | 224,708 | | | | 274,842 | |
Contract service fees and revenues from collaborative research arrangements | | | 448,551 | | | | 410,138 | |
| Additions: | | | | | | | | |
| Product sales | | | 948,532 | | | | 308,244 | |
Contract service fees and revenues from collaborative research arrangements | | | - | | | | 176,560 | |
| Recognition of revenue: | | | | | | | | |
| Product sales | | | (787,545 | ) | | | (358,378 | ) |
Contract service fees and revenues from collaborative research arrangements | | | (448,551 | ) | | | (138,147 | ) |
| Ending balance: | | | | | | | | |
| Product sales | | | 385,695 | | | | 224,708 | |
Contract service fees and revenues from collaborative research arrangements | | | - | | | | 448,551 | |
| | | | 385,695 | | | | 673,259 | |
| | | | | | | | | |
| Summarized as to: | | | | | | | | |
| Current Portion | | | | | | | | |
| Product sales | | | 306,071 | | | | 101,828 | |
Contract service fees and revenues from collaborative research arrangements | | | - | | | | 448,551 | |
| Current Portion | | | 306,071 | | | | 550,379 | |
| | | | | | | | | |
| Long-Term Portion | | | | | | | | |
| Product sales | | | 79,624 | | | | 122,880 | |
| Long-Term Portion | | | 79,624 | | | | 122,880 | |
| | | | | | | | | |
| Total | | | 385,695 | | | | 673,259 | |
12. SHARE CAPITAL AND ADDITIONAL PAID-IN CAPITAL
[a] Authorized - Unlimited common shares without par value.
[b] Issued
The Company closed a shareholder rights offering on December 29, 2011 consisting of 90,127,904 units, with each unit consisting of one common share and one common share purchase unit at a price of $0.0746 per share for total gross proceeds of $6,723,542.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
Each warrant entitles the holder thereof to purchase one common share of the Company at a price of $0.0746 per share for a period of five years after the closing date. Each warrant may only be exercised on a net cashless exercise basis, and no warrant may be exercised at a time when the exercise price equals or exceeds the current market price. Subject to certain exceptions, the holders of the warrants will be entitled to full ratchet anti-dilution price protection for a period of two years after the closing of the offering and volume weighted anti-dilution price protection thereafter. The Company accounts for warrants under the authoritative guidance on accounting for derivative financial instruments. As a result of these price protection features, the Company has classified these warrants on the accompanying balance sheet as a liability that is revalued at each balance sheet date subsequent to the initial issuance in accordance with Accounting Standards Codification (ASC) Topic 815 – Derivatives and Hedging. On the date of issuance, the Company used the Black-Scholes pricing model to value these warrants based on an assumed risk-free interest rate of 1.18%, estimated stock price volatility of 110%, and a contractual term to expiry of five years. Subsequent changes in the fair value of the warrants between the date of issuance and the balance sheet date are reflected in the consolidated statement of loss as unrealized gain (loss) on revaluation of warrant liability.
The net proceeds of the rights offering was $6,037,803 after deducting issue costs of $685,739. Of these net proceeds, $2,330,921 was allocated to common shares and $3,706,882 was allocated to the warrants. Further, of this amount allocated to the warrants, $4,127,888 was recorded as warrant liability and $421,008 of issue costs allocated to the warrants was expensed to warrant issue costs on the consolidated statement of loss.
The Company closed a private placement on July 28, 2010 consisting of 13,333,333 common shares at a price of $0.60 per share, for total gross proceeds of $8,000,000 before share issuance costs of $525,080 for net proceeds of $7,474,920.
[c] Stock option plan
At the Annual General Meeting held June 3, 2008, the Company’s shareholders’ approved a new stock option plan (“2008 Plan”). Under the plan, the Company may grant options to purchase common shares in the Company to employees, directors, officers and consultants of the Company. The exercise price of the options is determined by the Board but is equal to the fair market value of the common shares at the grant date. The Company estimates the fair value of options on the date of the grant. The options vest over the requisite service period in accordance with terms as determined by the Board, typically over four years. Stock options expire no later than five years from the date of grant.
Of the 1,700,000 stock options authorized for grant under the 2008 Plan, 1,107,570 stock options are available for grant at December 31, 2011.
The following assumptions were used to estimate the fair value of options granted during the years ended December 31, 2011, 2010, and 2009 using a Black-Scholes option-pricing model.
| | | 2011 | | | 2010 | | | 2009 | |
| | | | | | | | | | |
| | | | | | | | | | |
| Dividend yield | | | 0 | % | | | 0 | % | | | 0 | % |
| Expected volatility | | | 99 | % | | | 175 | % | | | 98 | % |
| Risk-free interest rate | | | 2.10 | % | | | 1.61 | % | | | 1.24 | % |
| Expected life in years | | | 3.45 | | | | 3.48 | | | | 3.35 | |
| Fair value per stock option | | $ | 0.25 | | | $ | 0.38 | | | $ | 0.09 | |
The expected volatility reflects the assumption that the historical volatility of common stock of the Company over a period similar to the life of the options is indicative of future trends. The Company estimates the risk-free interest rate using the Bank of Canada bond yield with a remaining term equal to the expected life of the option. The expected life of the stock options is based on historical data and current expectations.
The weighted average fair value of stock options granted during the year ended December 31, 2011 was $0.25 per share (December 31, 2010—$0.38 and December 31, 2009—$0.09).
The total fair value of options vested during the fiscal 2011, 2010, and 2009 years was $149,000, $1,150,000, and $676,000 respectively.
Total aggregate intrinsic value represents the pre-tax intrinsic value, based on the Company’s closing stock price as of December 31, 2011, that would have been received by the option holders had all option holders exercised their stock options as of that date. The intrinsic value of the options outstanding for the years was nil. Total intrinsic value of stock options exercised during fiscal 2011, 2010 and 2009 was nil, $95 and nil, respectively. Cash proceeds from stock options exercised during fiscal 2011, 2010 and 2009 were nil, $324 and nil respectively.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
At December 31, 2011, the following stock options were outstanding:
| Range of exercise price | | | Number of shares under option | | | Weighted average remaining contractual life | | | Weighted average exercise price | | | Number of options currently exercisable | | | Weighted average exercise price | |
| | | | $ | # | | | (years) | | | | | | $ | # | | | $ | | |
| | 0.34 - 1.99 | | | | 120,420 | | | | 3.14 | | | | 0.84 | | | | 31,477 | | | | 1.10 | |
| | 6.00 - 6.99 | | | | 24,440 | | | | 0.83 | | | | 6.79 | | | | 16,519 | | | | 6.74 | |
| | 7.00 - 7.99 | | | | 1,500 | | | | 1.31 | | | | 7.30 | | | | 750 | | | | 7.30 | |
| | 8.00 - 8.99 | | | | 25,490 | | | | 0.62 | | | | 8.80 | | | | 25,483 | | | | 8.80 | |
| | 10.00 - 10.50 | | | | 12,635 | | | | 0.74 | | | | 10.20 | | | | 12,633 | | | | 10.20 | |
| | 0.34– 10.50 | | | | 184,485 | | | | 2.30 | | | | 3.40 | | | | 86,862 | | | | 5.81 | |
The options expire at various dates from January 18, 2012 to June 21, 2016.
Stock option transactions and the number of stock options outstanding are summarized as follows:
| | | Number of optioned | | | Weighted average | |
| | | common shares | | | exercise price | |
| | | | # | | | | $ | |
| Balance, December 31, 2009 | | | 1,073,133 | | | | 5.93 | |
| Options granted | | | 130,238 | | | | 0.42 | |
| Options forfeited | | | (345,521 | ) | | | 5.47 | |
| Options expired | | | (3,648 | ) | | | 6.01 | |
| Options exercised | | | (270 | ) | | | 1.20 | |
| Balance, December 31, 2010 | | | 853,932 | | | | 5.28 | |
| Options granted | | | 15,600 | | | | 0.34 | |
| Options forfeited | | | (497,742 | ) | | | 5.61 | |
| Options expired | | | (187,305 | ) | | | 5.78 | |
| Balance, December 31, 2011 | | | 184,485 | | | | 3.40 | |
[d] Stock-based compensation
The following table shows stock-based compensation allocated by type of cost:
| | | 2011 | | | 2010 | | | 2009 | |
| | | $ | | | $ | | | $ | |
| | | | | | | | | | | | | |
| Cost of sales | | | 37,906 | | | | 45,636 | | | | 55,523 | |
| Research and development | | | 50,039 | | | | 109,943 | | | | 129,316 | |
| Marketing and business development | | | 21,148 | | | | 39,233 | | | | 62,428 | |
| General and administrative | | | (147,054 | ) | | | 445,619 | | | | 790,446 | |
| | | | (37,961 | ) | | | 640,431 | | | | 1,037,713 | |
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
As of December 31, 2011, the total unrecognized compensation expense related to stock options granted amounts to $41,967, which is expected to be recognized over a weighted average service period of 1.46 years.
[e] Common share purchase warrants
At December 31, 2011, there were exercisable warrants outstanding to purchase 90,127,904 shares of common stock at $0.0746 per share, expiring December, 2016.
Common share purchase warrant transactions are summarized as follows:
| | | Number of | | | Weighted average | |
| | | warrants | | | exercise price | |
| | | | # | | | | $ | |
| Balance, December 31, 2009 | | | 6,169,829 | | | | 2.3600 | |
| Warrants exercised | | | (149,507 | ) | | | 2.5000 | |
| Balance, December 31, 2010 | | | 6,020,322 | | | | 2.3600 | |
| Warrants expired | | | (6,020,322 | ) | | | (2.3600 | ) |
| Warrants issued | | | 90,127,904 | | | | 0.0746 | |
| Balance, December 31, 2011 | | | 90,127,904 | | | | 0.0746 | |
13. RELATED PARTY TRANSACTIONS
[a] The Company incurred consulting fees to a director of $127,123 for the year ended December 31, 2011 [2010 – nil; 2009 – nil]. These amounts are included in accounts payable and accrued liabilities as at December 31, 2011 [2010 – nil]. These consulting fees are included in general and administrative expenses in the consolidated statement of loss.
[b] On December 29, 2011, the Company completed a rights offering of which affiliates of OrbiMed Advisors LLC (“OrbiMed”) participated by purchasing a total of 67 million shares for $5 million. After giving effect to this transaction, the Company is now a controlled affiliate of OrbiMed.
Prior to completing the rights offering, the Company entered into a Note Purchase Agreement with affiliates of OrbiMed pursuant to which such affiliates have agreed to loan up to $2 million by way of a secured debt financing. Concurrently with the execution and delivery of the Note Purchase Agreement, the Company drew down $275,000. Subject to the satisfaction of certain conditions, the Company may draw down up to three further tranches of $575,000 at its option for a maximum potential draw of $2 million. The amounts drawn are subject to interest at 4.5% per annum. All amounts owing become due and payable on the earliest of: (i) March 31, 2012; (ii) the occurrence of an event of default followed by a declaration by the Lenders that such amounts are due and payable (or such amounts become due and payable automatically under certain circumstances); and (iii) the completion date of certain specified equity financings. In connection with the funds drawn, interest charges of $1,245 were incurred in addition to a commitment fee of $80,000.These charges are recorded as interest expenses on the consolidated statement of loss. The initial amount drawn down of $275,000 was repaid in full on December 29, 2011 with proceeds from the completion of the rights offering.
In connection with the rights offering and Note Purchase Agreement, the Company incurred and paid legal costs of $152,548 on behalf of affiliates of OrbiMed.
[c] The Company retained a law firm in which a corporate partner was a non-management member of the Board of Directors until May 3, 2010. For the year ended December 31, 2011, the Company incurred legal expenses from this law firm as a related party totaling $nil [2010 - $15,797; 2009 - $175,888] of which none remains outstanding. These legal costs are recorded in general and administrative expenses in the consolidated statement of loss.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
All related party transactions are recorded at their exchange amounts, established and agreed between the related parties.
14. INCOME TAXES
At December 31, 2011 the Company had approximately $53,761,000 [2010 - $47,143,000] of non-capital loss carry forwards, approximately $2,667,000 [2010 - $2,667,000] of federal investment tax credits and approximately $901,000 [2010 - $1,114,000] of provincial investment tax credits available to reduce taxable income and taxes payable for future years. These losses and investment tax credits expire as follows:
| Year of Expiry | | Provincial investment tax credit | | | Federal investment tax credits | | | Non-capital loss carryforwards | |
| 2012 | | | 129,000 | | | | - | | | | - | |
| 2013 | | | 93,000 | | | | - | | | | - | |
| 2014 | | | 20,000 | | | | - | | | | 4,101,000 | |
| 2015 | | | 58,000 | | | | - | | | | 6,880,000 | |
| 2016 | | | 142,000 | | | | - | | | | - | |
| 2017 | | | 205,000 | | | | - | | | | - | |
| 2018 | | | 198,000 | | | | - | | | | - | |
| 2019 | | | 56,000 | | | | 227,000 | | | | - | |
| 2020 | | | - | | | | 430,000 | | | | - | |
| 2021 | | | - | | | | 384,000 | | | | - | |
| 2022 | | | - | | | | 233,000 | | | | - | |
| 2023 | | | - | | | | 168,000 | | | | - | |
| 2024 | | | - | | | | 36,000 | | | | - | |
| 2025 | | | - | | | | 105,000 | | | | - | |
| 2026 | | | - | | | | 256,000 | | | | 7,669,000 | |
| 2027 | | | - | | | | 370,000 | | | | 8,560,000 | |
| 2028 | | | - | | | | 357,000 | | | | 4,107,000 | |
| 2029 | | | - | | | | 101,000 | | | | 7,217,000 | |
| 2030 | | | - | | | | - | | | | 9,266,000 | |
| 2031 | | | | | | | | | | | 5,961,000 | |
| | | | 901,000 | | | | 2,667,000 | | | | 53,761,000 | |
In addition, the Company has unclaimed tax deductions of approximately $11,639,035 [2010 - $11,639,035] related to scientific research and experimental development expenditures available to carry forward indefinitely to reduce taxable income of future years and other deductible temporary differences of approximately $17,894,237 [2010 - $17,894,237].
Significant components of the Company’s deferred tax assets are shown below:
| | | 2011 | | | 2010 | |
| | | $ | | | $ | |
| Deferred Tax Assets: | | | | | | | | |
| Book amortization in excess of tax capital cost allowance | | | 1,762,000 | | | | 1,428,000 | |
| Non-capital loss carry forwards | | | 13,440,000 | | | | 11,786,000 | |
| Research and development deductions and credits | | | 5,814,000 | | | | 6,027,000 | |
| Share issue costs | | | 363,000 | | | | 380,000 | |
| Unearned revenue | | | 95,000 | | | | 168,000 | |
| Unrealized foreign exchange | | | - | | | | 59,000 | |
| Free rent liability | | | 150,000 | | | | 164,000 | |
| Non-repayable lease inducements | | | 318,000 | | | | 346,000 | |
| Repayable lease inducements | | | 1,696,000 | | | | 1,770,000 | |
| Other | | | 89,000 | | | | 106,000 | |
| Total deferred tax assets | | | 23,727,000 | | | | 22,234,000 | |
| Valuation allowance | | | (23,727,000 | ) | | | (22,234,000 | ) |
| Net deferred tax assets | | | - | | | | - | |
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The potential income tax benefits relating to these deferred tax assets have not been recognized in the consolidated financial statements as their realization does not meet the requirements of “more likely than not” under the liability method of tax accounting. Accordingly, a valuation allowance has been recorded and no deferred tax assets have been recognized as at December 31, 2011 and 2010.
The reconciliation of income tax attributable to operations computed at the statutory tax rate to income tax expense (recovery), using a 26.5% [2010 – 28.5%; 2009 - 30%] statutory tax rate is as follows:
| | | 2011 | | | 2010 | | | 2009 | |
| | | $ | | | $ | | | $ | |
| | | | | | | | | | | | | |
| Income taxes (recovery) at statutory rates | | | (1,423,000 | ) | | | (2,698,000 | ) | | | (2,863,000 | ) |
| Expenses not deductible for tax purposes | | | (205,000 | ) | | | 189,000 | | | | 343,000 | |
| Non-capital losses for which no benefit has been recognized | | | 1,580,000 | | | | 2,454,000 | | | | 1,938,000 | |
| Other temporary differences for which no benefit has been recognized | | | 48,000 | | | | 55,000 | | | | 582,000 | |
| | | | - | | | | - | | | | - | |
The reconciliation of the unrecognized tax benefits of uncertain tax positions is as follows:
| | | $ | |
| | | | | |
| Balance at December 31, 2010 and 2009 | | | - | |
| Additions based on tax positions related to the current year | | | 43,500 | |
| Balance at December 31, 2011 | | | 43,500 | |
As of December 31, 2011, unrecognized benefits of approximately $43,500, if recognized, would affect the Company’s effective tax rate, and would reduce the Company’s deferred tax assets. Interest and penalties related to the unrecognized tax benefits that are accrued in the Company’s statements of loss and balance sheets as at December 31, 2011 were $23,500.
The Company is subject to taxes in Canada. The tax years which remain subject to examination as of December 31, 2011 for Canada include 2005 to 2011.
15. COMMITMENTS
[a] License agreements
[i] The Company entered into a non-exclusive license agreement, effective July 2005, as amended June 2008, to use and sublicense certain technology (“Technology”) for one of the Company’s cardiac tests. In consideration for these rights, the Company paid a non-refundable license issuance fee of $2,000,000 in the first two years after execution of the agreement and is required to pay quarterly royalties on the sale of products that incorporate the Technology. For the year ended December 31, 2011, the Company incurred an expense of $450,222 [2010 - $272,840; 2009 - $40,086] for royalties.
[ii] The company entered into a non-exclusive license and supply agreement, effective June 30, 2009 to purchase certain proprietary materials and use related intellectual property to manufacture, sell and have sold lateral flow immunoassay products. In consideration for these rights, the Company is to pay a non-refundable, non-creditable license fee, of USD$85,000 in 17 equal quarterly payments of USD$5,000 commencing December 31, 2009. For the year ended December 31, 2011, the Company incurred an expense of $19,755 [2010 - $25,344; 2009 - $Nil] for license fees.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
[iii] The Company entered into an exclusive license agreement with the University of British Columbia effective March 1996, as amended October 2003, to use and sublicense certain technology and any improvements thereon, and to manufacture, distribute and sell products in connection therewith. In consideration for these rights, the Company paid a non-refundable license fee of $10,000. Commencing in 2003 and for a period of nine years thereafter, royalties payable under this license are subject to a $2,500 quarterly minimum in addition to a $1,000 per annum maintenance fee. These payments are accrued and expensed in the year incurred. For the year ended December 31, 2011, the Company incurred a total expense of $11,000 [2010 - $11,000; 2009 - $11,000], for royalty and license fees under this agreement. The technology under this agreement is no longer used by the Company.
The minimum annual purchase commitments of material under the above licenses are as follows:
| | | | $ | |
| 2012 | | | 207,875 | |
| 2013 | | | 178,006 | |
| 2014 | | | 186,906 | |
| 2015 | | | 196,251 | |
| 2016 | | | — | |
| Thereafter | | | — | |
| | | | 769,038 | |
All royalty and license fees incurred are included in cost of sales.
[b] Supply agreement
The Company entered into a supply agreement, effective September 2003 for certain reagents for the Company’s RAMP West Nile Virus Test. In addition to paying for the reagent purchased, the Company is required to pay the supplier semi-annual royalties equal to 10% of net revenue generated from the sale of the Company’s RAMP West Nile Virus Test. The initial term of the agreement was three years from the effective date and is automatically renewed for successive periods of one year until either party terminates the agreement. For the year ended December 31, 2011, the Company incurred an expense of $45,163 [2010 - $28,640; 2009 - $75,600] for royalties to the supplier.
[c] Lease agreements
[i] The Company entered into a long-term agreement to lease a single tenant 46,000 square foot facility to house all of the Company’s operations beginning March 2008. Rent is payable from February 1, 2008 to January 31, 2023. The Company is required to pay the landlord total gross monthly payments of approximately $167,000, which is comprised of base rent, administrative and management fees, estimated property taxes and repayments of the repayable leasehold improvement allowance [Note 10[iii]].
For the year ended December 31, 2011 $1,563,507 [2010 - $1,515,196; 2009 - $1,498,776] was incurred for expenses related to base rent, administrative and management fees, estimated property taxes, rent-free inducement and interest on repayments of the repayable leasehold improvement allowance offset by amortization of both the rent-free inducement [Note 10[i]] and non-repayable leasehold improvement allowance [Note 10[ii]]. These expenses are allocated to cost of sales, research and development, general and administrative, and market and business development expenses.
[ii] The Company entered into a number of operating leases for administrative equipment.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
[iii] The minimum annual cost of lease commitments is estimated as follows:
| | | Premise | | | Equipment | | | Total | |
| | | $ | | | $ | | | $ | |
| 2012 | | | 1,998,342 | | | | 53,421 | | | | 2,051,763 | |
| 2013 | | | 2,024,728 | | | | 53,421 | | | | 2,078,149 | |
| 2014 | | | 2,051,888 | | | | 53,421 | | | | 2,105,309 | |
| 2015 | | | 2,079,843 | | | | 51,898 | | | | 2,131,741 | |
| 2016 | | | 2,108,616 | | | | 17,299 | | | | 2,125,915 | |
| Thereafter | | | 13,499,515 | | | | — | | | | 13,499,515 | |
| | | | 23,762,931 | | | | 229,460 | | | | 23,992,391 | |
[d] Purchase Commitments
As at December 31, 2011, the Company has outstanding purchase commitments of $177,204 to purchase inventory and $14,190 to purchase manufacturing equipment.
[e] Indemnification of directors and officers
Under the Articles of the Company, applicable law and agreements with its directors and officers, the Company, in circumstances where the individual has acted legally, honestly and in good faith, may, or is required to indemnify its directors and officers against certain losses. The Company's liability in respect of the indemnities is not limited. The maximum potential of the future payments is unlimited. However, the Company maintains appropriate liability insurance that limits the exposure and enables the Company to recover any future amounts paid, less any deductible amounts pursuant to the terms of the respective policies, the amounts of which are not considered material.
[f] Indemnification of third parties
The Company has entered into license and research agreements with third parties that include indemnification provisions that are customary in the industry. These indemnifications generally require the Company to compensate the other party for certain damages and costs incurred as a result of third party claims or damages arising from these transactions. The nature of the indemnification obligations prevents the Company from making a reasonable estimate of the maximum potential amount that it could be required to pay. To date, the Company has not made any indemnification payments under such agreements and no amount has been accrued in these consolidated financial statements with respect to these indemnification obligations.
16. SEGMENTED INFORMATION
The Company operates primarily in one business segment, the research, development, commercialization and distribution of diagnostic technologies, with primarily all of its assets and operations located in Canada. The Company’s revenues are generated from product sales primarily in the United States, Europe, Asia and Canada. Expenses are primarily incurred from purchases made from suppliers in Canada and the United States.
Customers that represent a concentration risk are those whose outstanding receivable is 10% or greater than the total balance or those customers who represent 10% or greater of our total revenue. Refer to note 5 for a discussion of concentration risk in relation to outstanding receivables. For the year ended December 31, 2011, $4,989,807 (55%) in product sales was generated from two customers of whom one customer represents $3,965,556 (44%) [2010 - $4,314,852 (64%); 2009 - $4,656,279 (57%) from three customers].
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
Product sales by customer location were as follows:
| | | 2011 | | | 2010 | | | 2009 | |
| | | $ | | | $ | | | $ | |
| China | | | 5,281,063 | | | | 3,576,935 | | | | 2,273,055 | |
| United States | | | 1,551,444 | | | | 1,003,297 | | | | 3,752,552 | |
| Asia (excluding China) | | | 794,667 | | | | 844,633 | | | | 774,676 | |
| Europe | | | 654,649 | | | | 812,328 | | | | 801,629 | |
| Canada | | | 59,148 | | | | 52,872 | | | | 76,484 | |
| Other | | | 683,112 | | | | 502,065 | | | | 474,653 | |
| Total | | | 9,024,083 | | | | 6,792,130 | | | | 8,153,049 | |
Product sales by type of product were as follows:
| | | 2011 | | | 2010 | | | 2009 | |
| | | $ | | | $ | | | $ | |
| Cardiovascular | | | 7,295,501 | | | | 5,969,672 | | | | 5,984,267 | |
| Infectious Diseases | | | 587,040 | | | | 67,472 | | | | 844,384 | |
| Biodefense | | | 659,463 | | | | 444,895 | | | | 517,981 | |
| Vector Infectious Diseases | | | 482,080 | | | | 310,090 | | | | 806,417 | |
| Total | | | 9,024,083 | | | | 6,792,130 | | | | 8,153,049 | |
17. COMPARATIVE FIGURES
In addition to the changes described in note 2, certain comparative figures have been reclassified from the amounts previously reported to conform to the presentation adopted in the current year. Revenues earned from one distributor previously presented in contract service fees and revenues from collaborative research arrangements have been retroactively reclassified as product sales. Also, capitalized software previously presented as intangible assets subject to amortization have been retroactively reclassified as property, plant, and equipment in addition to the amortization which has been retroactively reclassified to depreciation.
18. CONTINGENCIES
On September 2, 2011, the Company received notification from Roche Diagnostics that they have terminated, effective September 30, 2011, the sales and distribution agreement between Roche Diagnostics and the Company dated June 25, 2008. Roche Diagnostics terminated the agreement because the Company has not obtained the necessary approvals from the U.S. Food and Drug Administration (FDA) to permit Roche Diagnostics to market the Company’s cardiovascular tests for use in point-of-care settings in the United States using the RAMP® 200 Reader. This termination gives rise to loss contingencies that have a reasonable possibility of occurring but for which the potential amount of loss cannot be reasonably estimated.
In the first quarter of 2012, the Company determined that a small number of products that were shipped to Iran may be subject to U.S. export controls and may have required a license from the U.S. Government prior to export. Although these products are manufactured in Canada, they incorporate U.S. origin components, and for that reason, they may be subject to U.S. controls. As a result, applicable sanctions and export control laws may have been violated that may give rise to a maximum civil monetary penalty for each violation of up to $250,000. The Company, in conjunction with outside counsel, is conducting a review of the potential export transaction at issue in preparation for filing the final voluntary disclosures to be submitted to Office of Foreign Asset Control and Bureau of Industry and Security. The Company has not recorded any loss contingency as at December 31, 2011 as the outcome is not determinable at this point.
RESPONSE BIOMEDICAL CORPORATION
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
19. SUBSEQUENT EVENTS
The Board of Directors approved an increase to the Company’s authorized shares under its 2008 stock option plan from 1,700,000 to 24,200,000. This increase is subject to shareholder approval at the Company’s Annual General Meeting in June, 2012.
ITEM 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
Not applicable.
ITEM 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
The Company’s management, with the participation of the Company’s principal executive officer and principal financial officer, has evaluated the effectiveness of the Company’s disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, (the “Exchange Act”), as of the end of the period covered by this report.
In connection with the restatements of our financial statements for the year ended December 31, 2010 disclosed in our Form 20-F/A filed on November 9, 2011, the Company’s principal executive officer and principal financial officer had determined that due to material weaknesses in internal control over financial reporting discovered, that the Company’s disclosure controls and procedures were not effective as of December 31, 2010.
The Company implemented a remediation plan to address these material weaknesses in our Internal Control Over Financial Reporting noted in our Form 20-F/A, which is described below. Based on this remediation plan and the evaluation for the year ended December 31, 2011, the Company’s principal executive officer and principal financial officer have concluded that, as of December 31, 2011, the Company’s disclosure controls and procedures were effective.
Management’s Report on Internal Control Over Financial Reporting
We are responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Under the supervision and with the participation of the Company’s management, including its principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on criteria established in the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
As mentioned in the above section, some material weaknesses were identified in the Company’s Internal Control Over Financial Reporting in connection with the restatements that were disclosed in our Form 20-F/A filed November 9, 2011. The following material weaknesses were identified by management:
| | 1. | Control environment – The Company did not maintain an effective control environment. The control environment, which is the responsibility of senior management, sets the tone of the organization, influences the control consciousness of its people, and is the foundation for all other components of internal control over financial reporting. A tone and control consciousness that consistently emphasized adherence to accurate financial reporting and enforcement of our policies and procedures was not maintained by the prior management team. This control deficiency fostered a lack of sufficient appreciation for internal control over financial reporting, allowed for management override of internal controls in certain circumstances and resulted in an ineffective process for monitoring the adherence to our policies and procedures. |
| | 2. | Documentation, training and testing – The Company did not update its written policies and procedures with respect to internal control over financial reporting following the Company’s 2010 management reorganization and restructuring to reflect changes in the management team and reporting authorities. The Company’s written policies and procedures were not consistently followed, due to inadequate training of staff and the failures noted elsewhere. In addition, an adequate assessment of internal controls was not made by management at the time. |
| | 3. | Documentation and communication of contract terms – The Company did not maintain effective document control and records of contracts with its distributors. Certain terms of contracts were documented in oral or side agreements and were not properly disclosed to the Company’s board of directors and its external auditors or legal advisors. |
| | 4. | Revenue recognition – The Company did not maintain effective revenue recognition controls and policies. When persuasive evidence of a purchase order did not exist, when oral or side agreements existed, when contingencies existed with respect to the acceptance of the product, or when distributors did not have the ability or intent to pay independent of payment by the end-user customer, this information was not properly communicated to the Company’s board of directors, external auditors and legal advisors. Goods were shipped and revenue recognized in violation of the Company’s written policies and procedures. |
| | 5. | Anti-fraud program – The Company did not maintain an effective anti-fraud program designed to detect and prevent fraud, such as a whistle-blower program and an ongoing program to manage identified fraud risks. |
The Company’s management implemented a remediation plan, which included the following:
| | 1. | Updated the Company’s controls, policies and procedures to reflect the reorganization and restructuring that occurred in prior years. |
| | 2. | Improved the documentation of controls and the testing of controls, including the use of a qualified outside consultants, in order to ensure that the internal controls in place over financial reporting are effective or to remediate any controls that, subsequent to the appropriate testing being completed, were concluded to be ineffective. |
| | 3. | Updated the Company’s controls and procedures regarding order entry and processing. |
| | 4. | Commenced additional training of staff and management on revenue recognition. |
| | 5. | Implemented a Whistleblower Policy to facilitate reporting directly to the Company’s Audit Committee any suspected improper activity. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risks that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Accordingly, even an effective system of internal control will provide only reasonable assurance that the objectives of the internal control system are met.
At December 31, 2011, the Company recorded a material adjustment which was a result of incorrectly recognizing the effect of forfeitures on stock based compensation previously recorded. This adjustment was a result of inadequate controls related to accounting for those forfeitures, however as a result of this adjustment; we have reviewed and made improvements to our controls such that the controls currently in place are considered effective.
Based on the remediation plan of our policies and procedures and the current evaluation done for the period covered by this report, our principal executive officer and principal financial officer have concluded that our internal control over financial reporting was effective as of December 31, 2011.
Management is committed to continuing to improve the Company’s internal control processes and will continue to diligently and vigorously review the Company’s financial reporting controls and procedures.
ITEM 9B. Other Information.
PART III
ITEM 10. Directors, Executive Officers and Corporate Governance.
The information required by this item will be set forth in the Proxy Statement and is incorporated in this report by reference.
ITEM 11. Executive Compensation.
The information required by this item will be set forth in the Proxy Statement and is incorporated in this report by reference.
ITEM 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this item will be set forth in the Proxy Statement and is incorporated in this report by reference.
ITEM 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this item will be set forth in the Proxy Statement and is incorporated in this report by reference.
ITEM 14. Principal Accountant Fees and Services.
The information required by this item will be set forth in the Proxy Statement and is incorporated in this report by reference.
PART IV
ITEM 15. Exhibits and Financial Statement Schedules.
(a) (1) and (2) The financial statements and reports of independent registered public accounting firm are filed as part of this Annual Report at Item 8. The financial statement schedules are not included in this item as they are either not applicable or are included as part of the consolidated financial statements.
(b) Exhibits: The following exhibits are filed as a part of this report:
All other financial statement schedules have been omitted because they are not applicable, not required or the information required is shown in the financial statements or the notes thereto.
| | | | Incorporated by Reference |
| 3.1 | | Certificate of Incorporation dated August 20, 1980 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20-F for the year ended December 31, 2004, as filed on May 2, 2005. |
| 3.2 | | Company Act Name Change dated October 15, 1991 | | |
| 3.3 | | Articles of the Company dated April 10, 1997 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Registration Statement on Form 20-F filed on February 4, 2004. |
| 4.1 | | Escrow Agreement dated July 29, 2004 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20-F for the year ended December 31, 2004, as filed on May 2, 2005. |
| 10.1 | | Alexandria New Facility Lease Agreement dated April 24, 2007 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.2 | | Alexandria – First Amendment to Lease Agreement dated May 18, 2007 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.3 | | Creation Technologies Supply Agreement dated August 19, 2005 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Report on Form 6-K filed on April 2, 2008. |
| 10.4 | | Roche License Agreement – NT-proBNP dated July 22, 2005* | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.5 | | Roche License Agreement –Amendment 2 concluded July 26, 2005 – dated June 24, 2008* | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.6 | | Roche Sales and Supply Agreement dated June 25, 2008* | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| | | | Incorporated by Reference |
| 10.7 | | Shionogi Supply Agreement dated May 12, 2006 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.8 | | Shionogi Supply Agreement – Amendment 1 dated July 11, 2008 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.9 | | 3M Master Manufacturing and Supply Agreement dated November 30, 2006 (Redacted)* | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.10 | | 3M/Response Investment Agreement dated November 30, 2006 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Report on Form 6-K filed on December 19, 2006. |
| 10.11# | | Contract – S. Wayne Kay, Chief Executive Officer | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.12# | | Contract – D. Morris, Chief Operating Officer | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.13# | | Contract – L. Kaler, Vice President Finance and Administration | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2009 as filed on April 1, 2010. |
| 10.14# | | Contract – Paul Harris, Vice President, Research and Development | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.15# | | Short Term Incentive Plan dated March 18, 2008 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.16# | | 2008 Stock Option Plan | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.17 | | Irrevocable Commercial Letter of Credit dated May 1, 2007 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.18# | | Form of Indemnification Agreement between Response Biomedical Corp. and applicable officers | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.19 | | Demand Operating Facility Agreement dated April 26, 2007 with Schedule A | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2009 as filed on April 1, 2010. |
| | | | Incorporated by Reference |
| 10.20# | | 1996 Stock Option Plan, as amended June 21, 2004 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20-F for the year ended December 31, 2004, as filed on May 2, 2005. |
10.21# | | 2005 Stock Option Plan | | Previously filed as an exhibit to, and incorporated herein by reference from our Report on Form 6-k filed on June 23, 2005.Previously filed as an exhibit to, and incorporated herein by reference from, our Report on |
| 10.22# | | 2005 Stock Option Plan, as amended June 22, 2006 | | Form 6-K filed on April 2, 2008. |
| 10.23 | | Warrant Certificate – Othmar Iseli dated October 28, 2008 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.24 | | Warrant Certificates – various – dated October 28, 2008 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2008 as filed on March 31, 2009. |
| 10.25 | | Underwriting Agreement Public Offering dated May 7, 2009 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2009 as filed on April 1, 2010. |
| 10.26 | | Executed Warrant Indenture dated May 21, 2009 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2009 as filed on April 1, 2010. |
| 10.27 | | Form of Warrants | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2009 as filed on April 1, 2010. |
| 10.28 | | Broker’s Warrant Certificate dated May 21, 2009 | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2009 as filed on April 1, 2010. |
| 10.29 | | Form of Subscription Agreement | | Previously filed as an exhibit to, and incorporated herein by reference from, our Annual Report of Form 20F for the year ended December 31, 2009 as filed on April 1, 2010. |
| 10.30 | | Subscription Agreement dated June 27, 2010* | | Previously filed as an exhibit to, and incorporated herein by reference from, our Report on Form 6-K filed on May 5, 2011. |
| 10.31 | | Distribution Agreement with O&D Biotech Co., Ltd. China dated February 21, 2011* | | |
| 10.32 | | Note Purchase Agreement dated November 22, 2011 | | |
| 10.33 | | Standby Purchase Agreement dated November 28, 2011 | | |
| 10.34# | | Consulting Agreement | | |
| 14 | | Company's Code of Ethics | | |
| 23.1 | | Consent of Independent Registered Public Accounting Firm | | |
| 24 | | Power of Attorney (included on signature page) | | |
| | | | Incorporated by Reference |
| 31.1 | | CEO's Certification required by Rule 13A-14(a) of the Securities Exchange Act of 1934 | | |
| 31.2 | | CFO's Certification required by Rule 13A-14(a) of the Securities Exchange Act of 1934 | | |
| 32.1 | | CEO's Certification of periodic financial reports pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, U.S.C. Section 1350 | | |
| 32.2 | | CFO's Certification of periodic financial reports pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, U.S.C. Section 1350 | | |
| **101.INS | | XBRL Instance Document | | |
| **101.SCH | | XBRL Taxonomy Extension Schema Document | | |
| **101.CAL | | XBRL Taxonomy Extension Calculation Linkbase Document | | |
| **101.DEF | | XBRL Taxonomy Extension Definition Linkbase Document | | |
| **101.LAB | | XBRL Taxonomy Extension Label Linkbase Document | | |
| **101.PRE | | XBRL Taxonomy Extension Presentation Linkbase Document | | |
| # | Management compensatory plan, contract or arrangement |
| * | Confidential portion of this exhibit has been omitted and filed separately with the Commission pursuant to an application for confidential treatment under Rule 24b-2 promulgated under the Securities Exchange Act of 1934, as amended. |
| ** | XBRL information is furnished and not filed or a part of a registration statement or prospectus for purposes of sections 11 or 12 of the Securities Act of 1933, as amended, is deemed not filed for purposes of section 18 of the Securities Exchange Act of 1934, as amended, and otherwise is not subject to liability under these sections. |
Copies of the exhibits filed with this Annual Report on Form 10-K or incorporated by reference herein do not accompany copies hereof for distribution to stockholders of the Registrant. The Registrant will furnish a copy of any of such exhibits to any stockholder requesting the same for a nominal charge to cover duplicating costs.
POWER OF ATTORNEY
The registrant and each person whose signature appears below hereby appoint Dr. Peter A. Thompson and Richard A. Canote as attorney-in-fact with full power of substitution, severally, to execute in the name and on behalf of the registrant and each such person, individually and in each capacity stated below, one or more amendments to this Annual Report on Form 10-K, which amendments may make such changes in this Annual Report as the attorney-in-fact acting in the premises deems appropriate and to file any such amendments to this Annual Report on Form 10-K with the Securities and Exchange Commission.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned thereunto duly authorized.
| Dated: March 29, 2012 | Response Biomedical Corporation | |
| | | | |
| | By: | /s/ Dr. Peter A. Thompson | |
| | | Dr. Peter A. Thompson | |
| | | Acting Chief Executive Officer and | |
| | | Chairman of Board of Directors | |
| Dated: March 29, 2012 | By: | /s/ Richard A. Canote | |
| | | Richard A. Canote | |
| | | Chief Financial Officer | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | |
| By: | /s/ Dr. Peter A. Thompson | |
| | | Dr. Peter A. Thompson | |
| | | Acting Chief Executive Officer and Chairman of Board of Directors | |
| | | | |
| By: | /s/ Dr. Anthony F. Holler | |
| | | Dr. Anthony F. Holler | |
| | | Chief Financial Officer and Treasurer | |
| | | | |
| By: | /s/ Dr. Joseph D. Keegan | |
| | | Dr. Joseph D. Keegan | |
| | | Director | |
| | | | |
| By: | /s/ Clinton H. Severson | |
| | | Clinton H. Severson | |
| | | Director | |
| | | | |
| By: | /s/ Lewis J. Shuster | |
| | | Lewis J. Shuster | |
| | | Director | |
| | | | |
| By: | /s/ Dr. David Wang | |
| | | Dr. David Wang | |
| | | Director | |
82