UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
(Mark One)
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended March 31, 2021
OR
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number: 001-09585

ABIOMED, Inc.
(Exact Name of Registrant as Specified in Its Charter)
Delaware |
| 04-2743260 |
(State or Other Jurisdiction of Incorporation or Organization) |
| (I.R.S. Employer Identification No.) |
|
|
|
22 Cherry Hill Drive Danvers, Massachusetts |
| 01923 |
(Address of Principal Executive Offices) |
| (Zip Code) |
(978) 646-1400
(Registrant’s Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
Title of each class | Trading symbol | Name of each exchange on which registered |
Common Stock, $0.01 par value | ABMD | The Nasdaq Stock Market LLC |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.:
Large accelerated filer | ☒ |
| Accelerated filer | ☐ |
Non-accelerated filer | ☐ |
| Smaller reporting company | ☐ |
Emerging growth company | ☐ |
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
As of September 30, 2020 the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the voting and non-voting common equity held by non-affiliates was $12,155,296,841. As of May 14, 2021, 45,295,979 shares of the registrant’s common stock, $0.01 par value, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement relating to the 2021 Annual Meeting of Stockholders are incorporated by reference into Part III (Items 10, 11, 12, 13 and 14) of this Annual Report on Form 10-K. Such Proxy Statement will be filed with the Securities and Exchange Commission within 120 days after the end of the fiscal year to which this report relates.
TABLE OF CONTENTS
|
|
| |
| Page | ||
Item 1. | 1 | ||
Item 1A. | 14 | ||
Item 1B. | 33 | ||
Item 2. | 33 | ||
Item 3. | 34 | ||
Item 4. | 34 | ||
|
|
| |
|
| ||
Item 5. | 35 | ||
Item 6. | 37 | ||
Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 38 | |
Item 7A. | 45 | ||
Item 8. | 46 | ||
Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 46 | |
Item 9A. | 47 | ||
Item 9B. | 49 | ||
|
|
| |
|
| ||
Item 10. | 50 | ||
Item 11. | 50 | ||
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 50 | |
Item 13. | Certain Relationships and Related Transactions, and Director Independence | 50 | |
Item 14. | 50 | ||
|
|
| |
|
| ||
Item 15. | 51 | ||
Item 16. | 53 | ||
EXPLANATORY NOTES
Pending Trademarks and Registered Marks
Throughout this annual report on Form 10-K (“this Report”), we refer to various trademarks, service marks and trade names that we use in our business. Abiomed, Impella, Impella 2.5, Impella 5.0, Impella LD, Impella CP, Impella RP, Impella 5.5, Impella Connect, and SmartAssist are registered trademarks of Abiomed, Inc., and are registered in the U.S. and certain foreign countries. Impella ECP, Impella XR Sheath, Impella BTR, CVAD, STEMI DTU, Automated Impella Controller and Abiomed Breethe OXY-1 System are pending trademarks of ABIOMED, Inc. Other trademarks and service marks appearing in this Report are the property of their respective holders.
Company References
Throughout this Report, “ABIOMED, Inc.,” the “Company,” “we,” “us” and “our” refer to ABIOMED, Inc. and its consolidated subsidiaries.
Industry Data and Forecasts
This Report includes data, including forecasts, obtained from industry publications and surveys and other information available to us. Data and other metrics included in this Report to describe our industry or our products are inherently uncertain and speculative in nature, and actual results for any period may materially differ. Estimates and forecasts involve uncertainties and risks and are subject to change based on various factors, including those discussed above under “Forward-Looking Statements.” While we are not aware of any misstatements regarding the third-party industry data presented in this Report, we have not independently verified any of the data from third-party sources, nor have we ascertained the underlying assumptions relied upon therein.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS AND RISK FACTOR SUMMARY
This Report, including the documents incorporated by reference in this Report, includes forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. All statements, other than statements of historical facts, may be forward-looking statements. These forward-looking statements may be accompanied by such words as “anticipate,” “believe,” “estimate,” “expect,” “forecast,” “intend,” “may,” “plan,” “potential,” “project,” “target,” “should,” “likely,” “will” and other words and terms of similar meaning. Each forward-looking statement in this Report is subject to risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statement, including, without limitation, in “Item 1. Business, “Item 1A. Risk Factors” and “Item 7A. Management’s Discussion of and Analysis of Financial Condition and Results of Operations.” Factors that could cause actual results or conditions to differ from those anticipated by these and other forward-looking statements include, among others, the items in the following list, which also summarizes some of our more principal risks:
| • | the impact of public health threats and epidemics, including the novel coronavirus (“COVID-19”) pandemic and resulting or prolonged economic downturns on our operations and financial conditions; |
| • | effects on our profitability if we are unable to manufacture our products as a result of natural or man-made disasters; |
| • | fluctuations in foreign currency exchange rates; |
| • | our dependence on Impella® products for most of our revenues; |
| • | our ability to successfully compete against our existing or potential competitors; |
| • | the acceptance of our products by cardiac surgeons and interventional cardiologists, especially those with significant influence over medical device selection and purchasing decisions; |
| • | the effect of long sales and training cycles associated with expansion into new hospital cardiac centers; |
| • | the potential for reduced market acceptance of our products and reduced revenue due to lengthy clinician training process; |
| • | our ability to effectively manage our growth; |
| • | our ability to anticipate demand for, and successfully commercialize, our products; |
| • | the impact of unsuccessful clinical trials or procedures relating to products under development; |
| • | our ability to develop additional and high-quality manufacturing capacity to support continued demand for our products; |
| • | our dependence on third-party payers to provide reimbursement to our customers of our products; |
| • | our suppliers’ failure to provide the components we require; |
| • | our reliance on distributors to sell our products in international markets; |
| • | our success in expanding our direct sales activities into international markets; |
| • | our ability to sustain profitability at levels achieved in recent years; |
| • | the unpredictability of fluctuations in our operating results; |
| • | our ability to develop and commercialize new products or acquire desirable companies, products or technologies; |
| • | inventory write-downs and other costs due to product quality issues; |
| • | risks and liabilities associated with acquisitions of other companies or businesses, including our ability to integrate acquired businesses into our operations; |
| • | the impact of consolidation in the healthcare industry on our prices; |
| • | our ability to attract and retain key personnel; |
| • | our ability to obtain governmental and other regulatory approvals and market and sell our products in certain jurisdictions; |
| • | regulatory or enforcement actions and product liability suits relating to off-label uses of our products; |
| • | the increased risk of material product liability claims and impact on our reputation and financial results; |
| • | our ability to maintain compliance with regulatory requirements and continuing regulatory review; |
| • | the impact of mandatory or voluntary product recalls; |
| • | material impairments caused by shutdowns of the U.S. federal government; |
| • | changes in healthcare reimbursement systems in the U.S. and abroad; |
| • | our ability to comply with healthcare “fraud and abuse” laws and any related penalties for non-compliance; |
| • | our failure to comply with the U.S. Foreign Corrupt Practices Act and other anti-corruption laws, export control laws, import and customs laws, trade and economic sanctions laws and other laws governing our operations; |
| • | our or our vendors’ ability to achieve and maintain high manufacturing standards; |
| • | the economic effects of “Brexit” and related impacts to relationships with our existing and future customers; |
| • | our potential “ownership change” for U.S. federal income tax purposes and our limited utilization of net operating losses from prior tax years; |
| • | our ability to maintain compliance with, and the impact on us of changes in, tax laws including U.S. Tax Reform legislation; |
| • | our ability to comply with, and the impact of any related costs or regulatory actions with respect to, environmental, health and safety requirements; |
| • | our failure to protect our intellectual property or develop or acquire additional intellectual property; |
| • | compliance with laws protecting the confidentiality of patient health information; |
| • | disruptions of critical information systems or material breaches in the security of our systems; |
| • | risks relating to our shares of common stock, including market price volatility and the potential for dilution to our stockholders’ ownership interests through the sale of additional securities; |
| • | changes in methods, estimates and judgments we use in applying our accounting policies; |
| • | changes in accounting standards, tax laws and financial reporting requirements; |
| • | the outcome of ongoing securities class action litigation relating to our public disclosures; and |
| • | other factors discussed in “Part I, Item 1A. Risk Factors” of this Report. |
The preceding list is not intended to be an exhaustive list of all of our forward-looking statements. Readers are cautioned not to place undue reliance on any forward-looking statements contained in this Report, which speak only as of the date of this Report. Any forward-looking statement made in this Report speaks only as of the date hereof. We undertake no obligation to update or revise these forward-looking statements whether as a result of new information, future events or otherwise, unless otherwise required by law. Our business is subject to risks and uncertainties, including those referenced above. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that future results, levels of activity, performance and events and circumstances reflected in the forward-looking statements will be achieved or will occur. Investors, potential investors, and others should give careful consideration to these risks and uncertainties.
PART I
ITEM 1. | BUSINESS |
Corporate Background
Our Company was founded in 1981 and is incorporated in Delaware. Our common stock is listed on the Nasdaq Global Select Market under the ticker symbol “ABMD.”
Our principal executive offices are located at 22 Cherry Hill Drive, Danvers, Massachusetts 01923. Our telephone number is (978) 646-1400. We make available, free of charge on our website located at www.abiomed.com, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and any amendments to those reports, as soon as reasonably practicable after filing such reports with or furnishing such reports to the U.S. Securities and Exchange Commission, or SEC. We also use our website for the distribution of Company information. The information we post on our website may be deemed to be material information. Accordingly, investors should monitor our website, in addition to following our press releases, SEC filings and public conference calls and webcasts. The contents of our website are not incorporated by reference into this Report.
Our Company
We are a leading provider of medical devices that provide circulatory support and oxygenation. Our products are designed to enable the heart to rest by improving blood flow and/or provide sufficient oxygenation to those in respiratory failure. We develop, manufacture and market proprietary products that are designed to enable the heart to rest, heal and recover by improving blood flow to the coronary arteries and end-organs and/or temporarily assisting the pumping function of the heart. Our products are used in the cardiac catheterization lab, or cath lab, by interventional cardiologists, the electrophysiology lab, the hybrid lab and in the heart surgery suite by cardiac surgeons. A physician may use our devices for patients who are in need of hemodynamic support prophylactically, urgently or emergently before, during or after angioplasty or heart surgery procedures. We believe that heart recovery is the optimal clinical outcome for a patient experiencing heart failure because it enhances the potential for the patient to go home with their own heart, facilitating the restoration of quality of life. In addition, we believe that, for the care of such patients, heart recovery is often the most cost-effective solution for the healthcare system.
Our strategic focus and the driver of our revenue growth is the market penetration of our family of Impella® heart pumps. The Impella device portfolio, which includes the Impella 2.5®, Impella CP®, Impella 5.0®, Impella LD®, Impella 5.5® and Impella RP® devices, has supported thousands of patients worldwide. We expect that most of our product and service revenue in the near future will be from our Impella devices. Our Impella 2.5, Impella CP, Impella 5.0, Impella LD, Impella 5.5 and Impella RP devices have U.S. Food and Drug Administration, or FDA, and CE Mark approvals which allows us to market these devices in the U.S. and European Union, respectively. We expect to continue to make additional pre-market approval, or PMA, supplement submissions for our Impella portfolio of devices for additional indications. Our Impella 2.5, Impella CP and Impella 5.0 devices have regulatory approval from the Ministry of Health, Labor and Welfare, or MHLW, in Japan. In October 2020, we also received a 510(k) clearance for the Abiomed Breethe OXY-1 System (the “OXY-1 System”) from the FDA for an all-in-one, compact cardiopulmonary bypass system.
COVID-19 Pandemic
For a discussion of the impact of the ongoing COVID-19 pandemic on our business, see “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations—COVID-19 Pandemic.”
Our Existing Products
Impella 2.5®
The Impella 2.5 device is a percutaneous micro heart pump with an integrated motor and sensors. The device is designed primarily for use by interventional cardiologists to support patients in the cath lab who may require assistance to maintain circulation. The Impella 2.5 heart pump can be quickly inserted via the femoral artery to reach the left ventricle of the heart, where it is directly deployed to draw blood out of the ventricle and deliver it to the circulatory system. This function is intended to reduce ventricular work and provide blood flow to vital organs. The Impella 2.5 heart pump is introduced with normal interventional cardiology procedures and can pump up to 2.5 liters of blood per minute.
Our Impella 2.5 device has FDA, CE Mark and MHLW approvals which allows us to market these devices in the U.S., European Union and Japan, respectively. We expect to continue to make additional PMA supplement submissions for our Impella portfolio of devices for additional indications. Our Impella 2.5, Impella CP and Impella 5.0 devices have regulatory approval from the MHLW in Japan. The Impella 2.5 device also has Health Canada approval which allows us to market the device in Canada.
1
Impella CP®
The Impella CP device provides blood flow of approximately one liter more per minute than the Impella 2.5 device and is primarily used by either interventional cardiologists to support patients in the cath lab or by cardiac surgeons in the heart surgery suite.
Our Impella CP devices has FDA and CE Mark approval which allows us to market this device in the U.S. and European Union. We expect to continue to make additional PMA supplement submissions for our Impella portfolio of devices for additional indications of Impella CP in the U.S. In March 2019, we received Japanese Pharmaceuticals and Medical Devices Agency (“PMDA”) approval from the MHLW for our Impella CP heart pump in Japan. We began selling the Impella CP heart pump in Japan in fiscal year 2020.
Impella 5.0® and Impella LD®
The Impella 5.0 and Impella LD devices are percutaneous micro heart pumps with integrated motors and sensors for use primarily in the heart surgery suite. These devices are designed to support patients who require higher levels of circulatory support as compared to the Impella 2.5 or Impella CP.
Our Impella 5.0 and Impella LD devices have FDA and CE Mark approval which allows us to market these devices in the U.S. and European Union. We expect to continue to make additional PMA supplement submissions for our Impella portfolio of devices for additional indications. Our Impella 5.0 devices have regulatory approval from the MHLW in Japan and Health Canada regulatory approval in Canada.
Impella 5.5®
The Impella 5.5 device is designed to be a percutaneous micro heart pump with integrated motors and sensors. The Impella 5.5 delivers peak flows of greater than six liters per minute. The Impella 5.5 has a motor housing that is thinner and 45% shorter than the Impella 5.0 and it improves ease of pump insertion through the vasculature.
In September 2019, the Impella 5.5 device received a PMA from the FDA for safety and efficacy in the therapy of cardiogenic shock for up to 14 days in the U.S. The Impella 5.5 pump was introduced in the U.S. through a controlled rollout at hospitals with established heart recovery protocols beginning in fiscal year 2020. The Impella 5.5 device received CE Mark approval in Europe in April 2018 is being introduced in Europe through a similar controlled rollout. We are also targeting a submission of the Impella 5.5 device to the PMDA in Japan in fiscal year 2022. The adoption of the Impella 5.5 device has decreased the utilization of Impella 5.0 and Impella LD at certain sites.
Impella RP®
The Impella RP device is a percutaneous catheter-based axial flow pump that is designed to allow greater than four liters of blood flow per minute and is intended to provide the flow and pressure needed to compensate for right side heart failure. Our Impella RP device has FDA and CE Mark approval which allows us to market these devices in the U.S. and European Union. The Impella RP is the first percutaneous single access heart pump designed for right heart support to receive FDA approval. The Impella RP device is approved to provide support of the right heart during times of acute failure for certain patients who have received a left ventricle assist device or have suffered heart failure due to AMI, a failed heart transplant, or following open heart surgery. We expect to continue to make additional PMA supplement submissions for our Impella portfolio of devices for additional indications.
Impella SmartAssist®
The Impella SmartAssist platform includes optical sensor technology for improved pump positioning and the use of algorithms that enable improved native heart assessment during the weaning process. The Impella SmartAssist platform is currently available for our Impella CP and Impella 5.5 heart pumps. The Impella SmartAssist platform is also approved under CE Mark in the European Union and other countries that require a CE Mark approval.
Impella Connect®
Impella Connect is a cloud-based technology that enables secure, remote viewing of the Automated Impella Controller, or AIC, for physicians and hospital staff from anywhere with internet connectivity. We began a controlled roll-out of Impella Connect at certain hospital sites during fiscal year 2020 and transitioned most of our higher volume customers during fiscal year 2021.
2
OXY-1 System
The OXY-1 System is a portable external respiratory assistance device that we acquired as part of our acquisition of Breethe, Inc. (“Breethe”), in April 2020 as part of our efforts to expand our product portfolio to support the needs of patients, such as those suffering from cardiogenic shock or respiratory failure, whose lungs can no longer provide sufficient oxygenation. The OXY-1 System takes venous blood from the patient, removes carbon dioxide and adds oxygen much like a human lung, and returns the oxygenated blood safely back to the patient. In October 2020, the OXY-1 System received a 510(k) clearance from the FDA for an all-in-one, compact cardiopulmonary bypass system. In the third quarter of fiscal year 2021, we initiated a controlled launch of the OXY-1 System at a limited number of hospitals in the U.S, that we expect to continue in fiscal year 2022.
Our Product Pipeline
Impella ECP™
The Impella ECP device is designed for blood flow of greater than three liters per minute. It is intended to be delivered on a standard sized catheter and will include an expandable inflow in the left ventricle. The Impella ECP device has completed the first stage in its FDA early feasibility study (“EFS”) in December 2020 by enrolling and treating five patients. The prospective, multi-center, non-randomized EFS is designed to allow us, study investigators, and the FDA to make qualitative assessments about the safety and feasibility of Impella ECP use in high-risk percutaneous coronary intervention (“PCI”) patients. In January 2021, we received approval from the FDA to expand the EFS for the Impella ECP device based on their review of results from the first five patients. The Impella ECP device is still in development and has not been approved for commercial use or sale.
Impella XR Sheath™
The Impella XR Sheath is a low-profile sheath that expands and recoils, allowing for small bore access and closure with certain Impella heart pumps. It inserts at 10 French (Fr) and the flexible, nitinol braids momentarily expand during insertion, then recoil, simplifying access for complex interventions. The Impella XR sheath is intended to produce less trauma at the arterial access site compared to large bore sheaths. In December 2020, the Impella XR Sheath for our Impella 2.5 device received a 510(k) clearance from the FDA. We are currently developing an Impella XR Sheath for our Impella CP device and anticipate making an FDA submission in fiscal year 2022.
Impella BTR™
The Impella BTR device is designed to be a percutaneous micro heart pump with integrated motors and sensors. The Impella BTR device is designed to be smaller, provide up to one year of hemodynamic support and is expected to allow for greater than five liters of blood flow per minute. The Impella BTR device also includes a wearable driver designed for hospital discharge. The Impella BTR pump is still in development and has not been approved for commercial use or sale.
Our Markets
According to the Heart Disease and Stroke Statistics 2021 Update Report from the American Heart Association, or AHA, coronary heart disease, or CHD, is the number one cause of death in the U.S. According to the report, 47% of women and 36% of men over the age of 45 will die within five years of their first heart attack, and CHD causes approximately one of every seven deaths in the U.S. CHD is a condition of the coronary arteries that causes reduced blood flow and insufficient oxygen delivery to the affected portion of the heart. CHD leads to acute myocardial infarction, or AMI, commonly known as a heart attack, which may lead to heart failure, a condition in which the heart is unable to pump enough blood to the body’s major organs.
A broad spectrum of therapies exists for the treatment of patients in early stages of CHD. Angioplasty procedures and stents are commonly used in the cath lab to restore and increase blood flow to the heart. These treatments are often successful in slowing the progression of heart disease, extending life, and/or improving the quality of life for some period of time. Patients presenting with acute cardiac injuries potentially have recoverable hearts. Treatment for these patients in pre-shock in the cath lab is primarily focused on hemodynamic stabilization. Acute heart failure patients in profound shock typically require treatment in the surgery suite. These are patients suffering from cardiogenic shock after a heart attack, post-cardiotomy cardiogenic shock or myocarditis complicated with cardiogenic shock. Chronic heart failure patients have hearts that are unlikely to be recoverable due to left and/or right-side heart failure and their conditions cause their hearts to fail over time. Limited therapies exist today for patients with severe, end-stage, or chronic heart failure.
In more severe cases of heart failure, patients are sent directly to the surgery suite for coronary bypass or valve replacement surgery. The most severe acute heart failure patients are in profound cardiogenic shock, including those suffering from myocarditis (a viral attack of the heart), or from those suffering from an impaired ability of the heart to pump blood after a heart attack or heart surgery. These patients typically require treatments involving the use of mechanical circulatory support devices that provide increased blood flow and reduce the stress on the heart. Many less severe patients in the cath lab could also benefit from circulatory support devices or other clinical treatment, which could potentially prevent them from entering into profound shock.
3
Percutaneous assist devices, like the Impella portfolio of devices, are mechanical devices that help the failing heart pump blood or take over the pumping function of the failing heart. Percutaneous assist devices allow for less invasive placement and removal and can be done through a small puncture in the leg in the cath lab, electrophysiology lab, or operating room. We believe heart recovery is a preferred clinical outcome for patients, since it generally lowers the overall relative cost to the healthcare system versus alternative therapies and treatment paths that may require multiple surgeries, lengthy or repeated hospital stays, chronic therapeutic and immunosuppressant drugs and other related healthcare costs.
Research and Product Development
Since our founding in 1981, we have gained substantial expertise in circulatory support through the development of many product platforms to support heart patients. This includes our Impella platform that we currently market and other technologies that we have supported, and sold in the past, which we do not actively market currently. Our current strategy is to develop a complete portfolio of products across the continuum of care in heart recovery, primarily focused in the area of circulatory care. We intend to continue to use this experience to develop additional circulatory support products as well as making enhancements to our existing products, with an emphasis on improving access and closure with our devices. In addition, we have a number of new products at various stages of development, some of which integrate the Impella technology platform including the Impella ECP, Impella XR Sheath and Impella BTR devices.
We expended $121.9 million, $98.8 million and $93.5 million on research and development in fiscal years 2021, 2020 and 2019, respectively. Our research and development expenditures include costs related to clinical trials and studies for our current and anticipated future products.
We are pursuing additional randomized control trials in high-risk PCI and cardiogenic shock, such as the STEMI DTU Study and PROTECT IV Study, to complement our clinical evidence and best practices to optimize patient outcomes.
STEMI DTU™ Study
In November 2018, we announced the results of our FDA-approved prospective multi-center feasibility study, “STEMI Door to Unloading with Impella CP system in acute myocardial infarction” (“STEMI DTU”). The study, which enrolled 50 patients at 10 sites, focused on the feasibility and safety of unloading the left ventricle using the Impella CP heart pump prior to primary PCI in patients presenting with ST segment elevation myocardial infarction, or STEMI, without cardiogenic shock, with the hypothesis that this will potentially reduce infarct size. The hypothesis of this novel approach to treating STEMI patients, based on extensive mechanistic research, is that unloading the left ventricle prior to PCI reduces myocardial workload and oxygen demand and also initiates a cardio-protective effect at the myocardial cell level, which may alleviate myocardial damage caused by reperfusion injury at the time of revascularization. The intent of this study was to help refine the protocol and lay the groundwork for a future pivotal study with more sites and patients and will be designed for statistical significance.
In April 2019, the FDA approved the initiation of the STEMI DTU pivotal randomized controlled trial. The prospective, multi-center, two-arm trial plans to enroll 668 patients undergoing treatment for a STEMI heart attack at up to 60 sites. Half the patients will be randomized to receive delayed reperfusion after 30 minutes of left ventricular unloading with the Impella CP. The other half will receive immediate reperfusion, the current standard of care. The STEMI DTU trial will test the hypothesis that unloading the left ventricle for 30 minutes prior to reperfusion will reduce myocardial damage from a heart attack and lead to a reduction in future heart failure related events. The trial allows for an adaptive design, which permits adjustments to the study sample size after an interim analysis. We began the trial in the third quarter of fiscal 2020 and we estimate that it will take three to four years to complete enrollment.
PROTECT IV Study
In April 2021, we announced that the first patient has been enrolled in PROTECT IV, a large, prospective, multi-center randomized controlled trial that is designed to provide the level of clinical evidence needed to achieve a Class I guideline recommendation for Impella in high-risk PCI. The two-arm trial will compare the benefits of high-risk PCI with Impella (primarily Impella CP) versus high-risk PCI without Impella support. The primary endpoint of the study is the composite of all-cause death, stroke, myocardial infarction or hospitalization for cardiovascular causes at a minimum of one year. The trial has an adaptive design. It aims to enroll 1,252 consecutive qualified patients at more than 100 hospital sites across the U.S. and Europe. The PROTECT IV trial will leverage advancements in technology and best practices learned since the completion of the PROTECT II randomized controlled trial and the FDA PMA for the Impella 2.5 heart pump for high-risk PCI. Data from PROTECT II found, when compared to intra-aortic balloon pump, Impella 2.5 led to a 29% reduction in MACCE, defined as composite of death, stroke, myocardial infarction and repeat procedures, at 90 days. PROTECT IV also builds on PROTECT III, a contemporary, prospective, single-arm FDA post-approval study of Impella 2.5 and Impella CP for high-risk PCI.
4
Sales, Clinical Support, Marketing and Field Service
Our clinical support personnel consist primarily of registered nurses and other personnel with considerable experience in either the surgery suite or the cath lab, and they play a critical role in training and educating physicians in the use of our products. In recent years, we have significantly increased the number of our direct sales and clinical support personnel in the U.S, Germany and Japan.
Manufacturing
We manufacture our products in Danvers, Massachusetts, Aachen, Germany and Halethorpe, Maryland. Our Aachen facility performs final assembly and manufactures most of our disposable Impella devices, including the Impella 2.5, Impella CP, Impella 5.0, Impella 5.5, Impella LD and Impella RP. Our Danvers facility also manufactures and performs final assembly for the Impella CP device, Impella 5.5, and certain Impella subsystems and accessories, including our Impella AIC, the console that powers our Impella devices. Our Halethorpe, Maryland facility manufactures the OXY-1 System. In addition, we rely on third-party suppliers to provide us with components used in our existing products and products under development. For example, we outsource some of the manufacturing for components and circuit cards within our consoles.
We have expanded our manufacturing capacity in all our facilities to support demand for our products. We believe our existing manufacturing facilities provide sufficient physical capacity to meet anticipated demand for at least the next twelve months. Our U.S. and German manufacturing facilities are certified as being in compliance with standards established by the International Organization for Standardization, or ISO, and operate under the FDA’s good manufacturing practice requirements for medical devices set forth in the Quality System Regulation, or QSR.
Intellectual Property
We have developed, and our business depends on, significant know-how and proprietary technology. To protect our know-how and proprietary technology, we rely on trade secret laws, trademarks, patents, copyrights, and confidentiality agreements and other contracts. However, these methods afford only limited protection. Others may independently develop equivalent proprietary information or technology, gain access to our trade secrets or disclose or use such secrets or technology without our approval.
A substantial portion of our intellectual property rights relating to the Impella devices and other products under development, such as the Impella ECP, Impella XR Sheath and Impella BTR devices, are in the form of trade secrets and patents. We protect our trade secrets and proprietary knowledge in part through confidentiality agreements with employees, consultants and other parties. We cannot assure you that our trade secrets will not become known to or be independently developed by our competitors.
We own or have rights to numerous U.S. and foreign patents. Patents filed both in the U.S. and Europe generally have a life of 20 years from the filing date. Our U.S. and foreign patents have expiration dates ranging from 2020 to 2036 and beyond as we continue to innovate and file for new patent applications. We also own or have rights to certain pending U.S. and foreign patent applications. We believe patents will issue pursuant to such applications, but cannot guarantee it. Moreover, neither the timing of any issuance, the scope of protection, nor the actual issue date of these pending applications can be forecasted with precision. Where we have licensed patent rights from third parties, we could be required to pay royalties.
Our patents may not provide us with competitive advantages. Our pending or future patent applications may not be issued. Others may hold or obtain patents that cover aspects or uses of our innovations. The patents of others may render our patents obsolete, limit our ability to patent or practice our innovations, or otherwise have an adverse effect on our ability to conduct business. Because foreign patents may afford less protection than U.S. patents, our foreign patent estate may not adequately protect our technology.
The medical device industry is characterized by a large number of patents and by frequent and consequential intellectual property litigation. Our products and technologies could infringe on the proprietary rights of third parties. If third parties successfully assert infringement or other claims against us, we may not be able to sell our products or we may have to pay significant damages and ongoing royalties. In addition, patent or intellectual property disputes or litigation may be costly, result in product development delays, or divert the efforts and attention of our management and technical personnel. If any such disputes or litigation arise, we may seek to enter into a royalty or licensing arrangement. However, such an arrangement may not be available on commercially acceptable terms, if at all. We may decide, in the alternative, to litigate the claims or seek to design around the patented or otherwise protected proprietary technology, which may also be costly and time consuming.
The U.S. government may obtain certain rights to use or disclose technical data developed under government contracts that supported the development of some of our products. We retain the right to obtain patents on any inventions developed under those contracts, provided we follow prescribed procedures and are subject to a non-exclusive, non-transferable, royalty-free license to the U.S. government.
5
Competition
Competition among providers of treatments for the failing heart is intense and subject to rapid technological change and evolving industry requirements and standards. We compete with many companies that have greater financial, product development, sales and marketing resources and experience than we do. Furthermore, new product development and technological change characterize the areas in which we compete. Our present or future products could be rendered obsolete or uneconomical as a result of technological advances by one or more of our present or future competitors or by other therapies, including drug therapies. We must continue to develop and commercialize new products and technologies to remain competitive in the cardiovascular medical technology industry. We believe that we compete primarily on the basis of clinical superiority supported by extensive data, and innovative features that enhance patient benefit, product performance, ease of use and reliability. Customer and clinical support, and data that demonstrate both improvement in a patient's quality of life and a product's cost-effectiveness are additional aspects of competition.
The cardiovascular segment of the medical technology industry is dynamic and subject to significant change due to cost-of-care considerations, regulatory reform, industry and customer consolidation and evolving patient needs. The ability to provide products and technologies that demonstrate value and improve clinical outcomes is becoming increasingly important for medical technology manufacturers.
We are aware of other cardiac assist device research efforts in the U.S., Canada, Europe and Japan. In addition, there are a number of companies, including Abbott Laboratories, Medtronic, Edwards Lifesciences, Boston Scientific, LivaNova, Terumo Heart, Teleflex, Getinge (Maquet Cardiovascular) and several early-stage companies, that are developing medical devices that provide circulatory support and oxygenation, including implantable left ventricular assist devices, miniaturized rotary ventricular assist devices and cardiopulmonary bypass devices that directly and indirectly compete with our products.
Third-Party Reimbursement
Our products and services are generally purchased by hospitals that rely on third-party payers to cover and reimburse the costs of related patient care. In the U.S., as well as in many foreign countries, government-funded or private insurance programs pay the cost of a significant portion of a patient’s medical expenses. No uniform policy of coverage or reimbursement for medical technology exists among all these payers. Therefore, coverage and reimbursement can differ significantly from payer to payer and by jurisdiction.
Third-party payers may include government healthcare programs such as Medicare or Medicaid, private insurers or managed care organizations. The Centers for Medicare & Medicaid Services (“CMS”) is responsible for administering the Medicare program in the U.S. and, along with its contractors, establishes coverage and reimbursement policies for the Medicare program. Medicare’s coverage and reimbursement policies are particularly significant to our business because a large percentage of the population for which our products are intended includes individuals who are Medicare beneficiaries. In addition, private payers often follow the coverage and reimbursement policies of Medicare. We cannot assure that government or private third-party payers will continue to cover and reimburse the procedures using our products in whole or in part in the future or that payment rates for reimbursement will be adequate. If governmental and private payers’ policies do not cover surgical procedures performed using our products, we may not be able to generate the revenues necessary to support our business.
Medicare payment may be made, in appropriate cases, for procedures performed in the in-patient hospital setting using our technology. Medicare generally reimburses hospitals in which the procedures are performed based upon prospectively determined amounts. For hospital in-patient stays, the prospective payment generally is determined by the patient’s condition and other patient data and procedures performed during the in-patient stay, using a classification system known as International Classification of Diseases, or ICD, and medical severity diagnosis-related groups, or MS DRGs. Prospective rates are adjusted for, among other things, regional differences, co-morbidity and complications. Hospitals performing in-patient procedures using our devices generally do not receive separate Medicare reimbursement for the specific costs of purchasing or implanting our products. Rather, reimbursement for these costs is bundled with the MS DRG-based payments made to hospitals for the procedures during which our devices are implanted, removed, or replaced. Because prospective payments are based on predetermined rates and may be less than a hospital’s actual costs in furnishing care, hospitals have incentives to lower their in-patient operating costs by utilizing products, devices and supplies that will reduce the length of in-patient stays, decrease labor or otherwise lower their costs. Thus, hospitals may decide not to use our products if reimbursement amounts are insufficient to cover any additional costs incurred when purchasing our products.
Coverage and reimbursement for procedures to implant, remove or replace our products are generally established in the U.S. market. For instance, Medicare covers the use of LVADs when used for support of blood circulation post-cardiotomy, as a temporary life-support system until a human heart becomes available for transplant, or as destination therapy for patients who require permanent mechanical cardiac support, when the use is consistent with FDA approval and FDA-approved labeling instructions, as applicable. Coverage and reimbursement for procedures to implant the Impella 2.5, Impella CP, Impella 5.0, Impella LD, Impella 5.5 and Impella RP devices are also established for in-hospital use by Medicare including ICD-10 for procedures and MS DRG coding. Actual coverage and payment may vary by local Medicare fiscal intermediary or third-party insurer. Our Impella devices are also covered by commercial and/or Medicare plans of many third-party insurers including Aetna, Humana, Cigna, Blue Cross Blue Shield, and United Healthcare.
6
In September 2020, CMS released final Medicare payment levels for inpatient hospital discharges for fiscal year 2021 (October 1, 2020 through September 30, 2021). The Final Rule for the IPPS update includes ICD-10 coding and confirms assignment of percutaneous Impella implantation to MS-DRG 215 for Other Heart Assist System Implant, and maintains bi-ventricular Impella (MS-DRG 1), ECpella (MS-DRG 3), and Impella hospital transfer / support (MS-DRG 268) for the receiving hospital. The Final Rule also set the relative weight for MS-DRG 215 equal to the average of the proposed FY2021 rate and FY2020 rate, an 11% reduction, instead of the proposed 24% reduction. In addition, CMS increased rates for MS-DRGs 1, 3, and 268 by 8%, 3%, and 5%, respectively. The Final Rule will be effective for the 12 months beginning on October 1, 2020 for all Medicare hospital inpatient discharges.
In April 2021, the CMS released a draft of proposed Medicare payment levels for inpatient hospital discharges after October 1, 2021. The April 2021 proposed rule, or the Proposed Rule, for the IPPS update includes reassigning select ICD-10-PCS codes from MS-DRG 215 to additional cardiovascular related MS-DRGs to align clinical populations and better reflect hospital resources. For the sickest patients who utilize extensive resources, hospitals are eligible to receive additional outlier payments, which may collectively increase reimbursement in future years. The AHA and CMS have established a system of care around the utilization of percutaneous heart pumps. The history and creation of this dedicated payment system with Impella implant/explant, bi-ventricular, ECPella, and transfer reimbursement allow some of the most critically ill patients the potential to survive and achieve native heart recovery. As highlighted by CMS, the FDA has granted Emergency Use Authorizations (“EUAs”) to us for COVID-19. For the duration of the public health emergency, hospitals treating patients diagnosed with COVID-19 receive an additional 20 percent in reimbursement. The Proposed Rule for the IPPS is open for public comment until June 28, 2021. The final rulemaking may differ substantially from this proposal and will take effect for the year beginning October 1, 2021.
In addition to payments to hospitals for procedures using our technology, Medicare makes separate payments to physicians for their professional services when they perform surgeries to implant, remove, replace or repair our devices or when they perform percutaneous insertion and removal of Impella devices. Physicians generally bill for such services using a coding system known as Current Procedural Terminology, or CPT, codes. Physician services performed in connection with the implantation, removal or repositioning of our approved products are billed using a variety of CPT codes. Generally, Medicare payment levels for physician services are based on the Medicare Physician Fee Schedule and are revised annually by CMS. Physicians may choose not to use our products if reimbursement amounts do not justify the additional costs expended when employing our products.
In general, third-party reimbursement programs in the U.S. and abroad, whether government-funded or commercially insured, are developing a variety of increasingly sophisticated methods of controlling healthcare costs, including prospective reimbursement and capitation programs, group purchasing, reducing benefit coverage, requiring second opinions prior to major surgery, negotiating reductions to charges on patient bills, promoting healthier lifestyle initiatives and exploring more cost-effective methods of delivering healthcare. These types of cost containment programs, as well as legislative or regulatory changes to reimbursement policies, could limit the amount which healthcare providers may be willing to pay for our medical devices.
Government Regulation and Other Matters
Our products and facilities are subject to regulation by numerous government agencies, including the FDA, European Union Member States competent authorities, and the Japanese Pharmaceuticals and Medical Devices Agency, to confirm compliance with the various laws and regulations governing the development, testing, manufacturing, labeling, marketing, and distribution of our products. We are also governed by federal, state, local, and international laws of general applicability, such as those regulating employee health and safety, and the protection of the environment. Overall, the amount and scope of domestic and foreign laws and regulations applicable to our business has increased over time. Compliance with these regulations has not had a material effect on our capital expenditures, earnings, or competitive position to date, but new regulations or amendments to existing regulations to make them more stringent could have such an effect in the future. We cannot estimate the expenses we may incur to comply with potential new laws or changes to existing laws, or the other potential effects these laws may have on our business.
United States Regulation
In the U.S., the FDA has responsibility for regulating medical devices under the authority of the Federal Food, Drug and Cosmetic Act, or FFDCA. The FDA regulates design, development, testing, clinical studies, manufacturing, labeling, distribution, import, export, sale promotion, and record keeping for medical devices, and reporting of adverse events, recalls, or other field actions by manufacturers and users to identify potential problems with marketed medical devices. Many of the devices that we develop, manufacture and market are in a category for which the FDA has implemented stringent clinical investigation and pre-market clearance or approval requirements. The process of obtaining FDA clearance or approval to market a product is resource intensive, lengthy, and costly. FDA review may involve delays that adversely affect the marketing and sale of our products. Some our products are pending regulatory clearance or approval to begin commercial sales in various markets. Ultimately, the FDA may not authorize the commercial release of a medical device if it determines the device is not safe and effective or does not meet other standards for clearance or approval. Additionally, even if a product is cleared or approved, the FDA may impose restrictions requiring postmarket testing and surveillance programs to monitor the effects of these products once commercialized.
7
The FDA has the authority to halt the distribution of certain medical devices, detain or seize adulterated or misbranded medical devices, order the repair, replacement, or refund of the costs of such devices, or preclude the importation of devices that are or appear to be violative. The FDA also conducts inspections to determine compliance with the QSR concerning the manufacturing and design of devices and medical device reporting regulations, recall regulations, clinical testing regulations, and other requirements. The FDA may withdraw product clearances or approvals due to failure to comply with regulatory standards, or the occurrence of unforeseen problems following initial approval, and require notification of health professionals and others with regard to medical devices that present unreasonable risks of harm to the public health. Additionally, the failure to comply with FDA or comparable regulatory standards or the discovery of previously unknown product problems could result in fines, delays, or suspensions of regulatory clearances or approvals, seizures, injunctions, recalls, refunds, civil money penalties, or criminal prosecution. Our compliance with applicable regulatory requirements is subject to continual review. Moreover, the FDA and several other U.S. agencies administer controls over the export of medical devices from the U.S. and the import of devices into the U.S., which could also subject us to sanctions for noncompliance.
Premarket Regulation
The FDA classifies medical devices into one of three classes (Class I, II or III) based on the statutory framework described in the FFDCA. Our Impella products are categorized as Class III devices. Class III devices are typically life-sustaining, life-supporting or implantable devices, or new devices that have not been found to be substantially equivalent to legally marketed devices. Class III devices must generally receive PMA approval from the FDA before they can be marketed.
The PMA approval pathway requires that the applicant demonstrate to the FDA’s satisfaction, based on valid scientific evidence, that there is a reasonable assurance of the safety and effectiveness of the device for its intended use. During the PMA process, the FDA examines detailed data to assess the safety and effectiveness of the device. This information includes design, development, manufacture, labeling, advertising, preclinical testing and clinical study data. Prior to approving a PMA, the FDA may conduct an inspection of the manufacturing facilities and the clinical sites where supporting studies were conducted. The facility inspection evaluates our compliance with QSR. An inspection of clinical sites evaluates compliance with good clinical practice standards, including, for studies conducted under an investigational device exemption, or IDE, that the studies meet the requirements of FDA’s IDE regulations. Typically, the FDA will convene an advisory panel meeting to review the data presented in the PMA. The panel’s recommendation is given substantial weight but is not binding on the FDA. Under a set of performance measures that the FDA has committed to achieving in return for the receipt of user fees from manufacturers, FDA attempts to review all PMAs not requiring an advisory panel meeting within 180 “FDA days” and review of a PMA application that does require an advisory panel meeting within 320 “FDA days.” The term “FDA days” excludes the time the applicant spends responding to FDA requests for additional information. While the FDA has approved PMA applications within the allotted time period, reviews can occur over a significantly longer period.
Upon completion of its review, the FDA will either approve or deny the PMA. If the FDA’s evaluation is favorable, the PMA is approved, and the device may be marketed in the U.S. The FDA may approve a PMA with post-approval conditions such as postmarket collection of clinical data. Failure to comply with the conditions of approval can result in material adverse enforcement action, including the loss or withdrawal of the PMA approval. A PMA approval may include significant limitations on the indicated uses for which a device may be marketed. The FDA interprets the FFDCA as prohibiting the promotion of approved medical devices for unapproved uses. After approval of a PMA, a new PMA or PMA supplement is required in the event of a significant modification to the device, the device labeling, or the manufacturing process. The FDA can initiate proceedings to withdraw a PMA approval for failure to comply with regulatory requirements or the occurrence of unforeseen problems following initial marketing.
See “Item 1. Business – Our Existing Products” for additional information regarding regulatory approvals on our Impella devices. We expect to make additional PMA supplement submissions for additional indications for use for our Impella devices in the future.
When clinical trials of a device are required in order to obtain FDA approval, the sponsor of the trial is generally required to file an IDE application before commencing the trials. The FDA reviews and must approve an IDE before a clinical study may begin in the U.S. In addition, the clinical study must be approved by an Institutional Review Board, or IRB, at each clinical site. The FDA, the IRB, or we may suspend a clinical trial at any time for various reasons, including if information emerges suggesting that the subjects are being exposed to an unacceptable health risk. All clinical studies of investigational devices must be conducted in compliance with FDA requirements. Following the completion of a study, the data from the study must be collected, analyzed and presented in an appropriate submission to the FDA, either as a report submitted to the IDE file or in a marketing application such as a PMA.
8
In addition, certain medical devices can be approved by the FDA in the U.S. under a humanitarian device extension (“HDE”) rather than a PMA. In order for a device to be eligible for an HDE, there must be a qualifying target patient population of less than 8,000 patients per year for which there is no other comparable device available to treat the condition. The FDA must agree that a device meets these criteria before it can be approved under an HDE. FDA approval of an HDE also requires demonstration that the device is safe for its intended application, that it is potentially effective, and that the probable benefits outweigh the associated risks. If another device receives approval through the PMA process that addresses the same patient population as the HDE device, the HDE device may need to be withdrawn from the U.S. market. An approved HDE authorizes sales of the device to any hospital after review and approval by the hospital’s IRB. Proposed modifications to approved HDE devices, like modifications to approved PMA devices, require FDA approval through a new HDE application or an HDE supplement.
The EUA authority allows the FDA to strengthen the public health protections against biological, chemical, radiological, and nuclear agents that may be used to harm the society. Under section 564 of the Federal Food, Drug and Cosmetic Act, the FDA may allow medical countermeasures to be used in an emergency to diagnose, treat or prevent serious or life-threatening diseases or conditions caused by such agents, when there are no adequate, approved and available alternatives. Section 564 permits the FDA to authorize the introduction into interstate commerce of a drug, device or biological product intended for use in an actual or potential emergency during the effective period of a declaration. EUA candidates include products and uses that are not approved, cleared or licensed under sections 505, 510(k) and 515 of the Federal Food, Drug, and Cosmetic Act or section 351 of the Public Health Act. The FDA may issue an EUA only if, after consultation with the Director of National Institute of health (“NIH”) and the Director of Centers for Disease Control and Prevention (“CDC”), the FDA concludes that the four statutory criteria for issuance (Serious or Life-Threatening Disease or Condition, Evidence of Effectiveness, Risk-Benefit Analysis, No Alternatives) have been met. The FDA assesses the sufficiency of the effectiveness data and the risk-benefit profile of each candidate product on a case-by-case basis. The FDA recommends that requests for medical devices under consideration for EUAs include clinical evidence data to support the proposed intended use, as necessary and appropriate. The FDA provides that an EUA will be in effect for the duration of the declaration under which it was issued, unless the EUA is revoked because the criteria of issuance are no longer met or revocation is appropriate to protect public health or safety. The FDA will periodically review the circumstances and appropriateness of an EUA, including circumstances that might warrant revocation of the EUA. Such circumstances may include significant adverse inspectional findings; reports of adverse events linked to, or suspected of being caused by, the EUA product; product failure; product ineffectiveness; and availability of a preferred product.
Postmarket Regulation
The medical devices that we manufacture and distribute pursuant to regulatory clearances or approvals by the FDA and other countries’ regulatory authorities are subject to continuing regulation by those agencies. The FDA reviews design, manufacturing, and distribution practices, labeling and record keeping, and manufacturers’ required reports of adverse experience and other information to identify potential problems with marketed medical devices. Among other FDA requirements, we must comply with the FDA’s good manufacturing practice regulations for medical devices, known as the QSR. These regulations govern the methods used in, and the facilities and controls used for, the design, testing, manufacture, packaging, labeling, storage, installation, and servicing of all finished medical devices intended for human use. We must also comply with Medical Device Reporting, or MDR requirements, which require us to report to the FDA any incident in any of our products that may have caused or contributed to a death or serious injury, including medical intervention to prevent a death or serious injury, or in which any of our products malfunctioned and, if such malfunction were to recur, would be likely to cause or contribute to a death or serious injury. Labeling, advertising, and promotional activities are subject to scrutiny by the FDA and, in certain circumstances, by the Federal Trade Commission. The FDA’s enforcement policy prohibits the marketing of approved medical devices for unapproved uses. We are subject to routine inspection by the FDA for compliance with the QSR and MDR requirements, as well as other applicable regulations. If the FDA were to conclude that we are not in compliance with applicable laws or regulations, or that any of our medical devices are ineffective or pose an unreasonable health risk, the FDA could ban such medical devices, detain or seize adulterated or misbranded medical devices, order a recall, repair, replacement, or refund of such devices, and require us to notify health professionals and others that the devices present unreasonable risks of harm to the public health. The FDA may also seek a judicial injunction enjoining certain violations of the FFDCA and imposing operating restrictions and assess civil or criminal fines and penalties against our officers, employees, or us. The FDA may also recommend criminal prosecution to the U.S. Department of Justice. Conduct giving rise to civil or criminal penalties may also form the basis for private civil litigation by third-party payers or other persons allegedly harmed by our conduct. Regulatory authorities outside the U.S. enforce similar laws and regulations within their respective jurisdictions.
The FDA and other regulatory agencies actively enforce regulations prohibiting promotion of off-label uses and the promotion of products for which marketing clearance has not been obtained. If the FDA or another regulatory agency determines that our promotional materials or training constitutes promotion of an unapproved use, it could request that we modify our training or promotional materials or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and criminal penalties. Although our policy is to refrain from statements that could be considered off-label promotion of our products, the FDA or another regulatory agency could disagree and conclude that we have engaged in off-label promotion.
9
The FDA can require postmarket surveillance, or PMS, for significant risk devices, such as our medical devices, that require ongoing collection, analysis, and periodic submission to the FDA of clinical data during commercialization over a period of up to several years. The PMS data collection requirements are often burdensome and expensive. The failure to comply with the FDA’s regulations can result in enforcement action, including seizure of products, injunction, prosecution, civil fines and penalties, recall and/or suspension of FDA approval.
The FDA, in cooperation with U.S. Customs and Border Protection, or CBP, administers controls over the import and export of medical devices into and out of the U.S. International sales of our medical devices that have not received FDA approval are therefore subject to FDA export requirements. The CBP imposes its own regulatory requirements on the import of medical devices, including inspection and possible sanctions for noncompliance.
Other Regulations
We are subject to additional laws and regulations that govern our business operations, products, and technologies, including:
| • | federal, state, and foreign anti-kickback laws and regulations, which generally prohibit payments to anyone, including physicians, as an inducement to purchase or recommend a product; |
| • | the Stark law, which prohibits physicians from referring Medicare or Medicaid patients to a provider that bills these programs for the provision of certain designated health services if the physician (or a member of the physician's immediate family) has a financial relationship with that provider; |
| • | federal, state and foreign laws and regulations that protect the confidentiality of certain personal information, including patient health information, and restrict the use and disclosure of such information, in particular, the Health Insurance Portability and Accountability Act in the U.S., and the European Union’s General Data Protection Regulation (“GDPR”) (as further described in “Part I, Item 1A. Risk Factors—Risks Related to Intellectual Property, Privacy and Security—If we are found to have violated laws protecting the confidentiality of patient health information, we could be subject to civil or criminal penalties, which could increase our liabilities and harm our reputation or our business”); |
| • | the Physician Payments Sunshine Act, which requires public disclosure of the financial relationships of. healthcare professionals and teaching hospitals in the U.S. with applicable manufacturers, including medical device, pharmaceutical, and biologics companies; |
| • | the False Claims Act, which prohibits the submission of false or otherwise improper claims for payment to the United States government, including federally funded health care program, and health care fraud statutes that prohibit false statements and improper claims to any third-party payor; and |
| • | the United States Foreign Corrupt Practices Act (“FCPA”), which can be used to prosecute United States companies for arrangements with foreign government officials or other parties, or for not keeping accurate financial records or maintaining adequate internal controls to prevent and detect arrangements with foreign government officials or other parties (as further described in “Part I, Item 1A. Risk Factors—Legal, Regulatory and Compliance Risks—We are subject to the U.S. Foreign Corrupt Practices Act and other anticorruption laws, as well as export control laws, import and customs laws, trade and economic sanctions laws and other laws governing our operations”). |
Failure to comply with these laws and regulations could result in criminal liability, significant fines or penalties, negative publicity, and material costs and expenses associated with investigation enforcement activities, and individual settlement agreements that impose a government monitor for a period of several years.
We are also subject to various local, state, federal, and international laws and regulations relating regarding environmental protection, hazardous substances and health and safety. These requirements govern, among other things, matters such as safe working conditions, laboratory and manufacturing practices, the handling, storage, use and disposal of hazardous materials and wastes and, if any, the associated cleanup of properties affected by discharges, releases or exposure to pollutants, air emissions, wastewater, and occupational and human health and safety. Specifically, the use of hazardous or potentially hazardous substances in connection with our research and development and manufacturing activities, such as the manufacture of our biomaterials, is subject to compliance with federal environmental regulations and by various state and local agencies. We believe we have been, and we are in material compliance with all applicable laws and regulations (including environmental laws and regulations). We further believe that we currently have no liabilities under such requirements that could reasonably be expected to materially harm our business, results of operations or financial condition.
10
International Regulation
Internationally, the approval and regulation of medical devices is subject to a variety of laws and regulation. In Europe, our products are subject to extensive regulatory requirements. Our Impella 2.5, Impella CP, Impella 5.0, Impella LD, Impella 5.5, Impella RP, and Impella AIC are all approved under CE Mark and are available for sale in the European Union and other markets that recognize CE Mark approval. The European Union requires that medical devices may only be placed on the market if they do not compromise safety and health when properly installed, maintained, and used in accordance with their intended purpose. National laws conforming to the European Union's legislation regulate our products under the medical devices regulatory system. Although the more variable national requirements under which medical devices were formerly regulated have been substantially replaced by the European Union Medical Devices Directive, individual nations can still impose unique requirements that may require supplemental submissions. The European Union medical device laws require manufacturers to declare that their products conform to the essential regulatory requirements after which the products may be placed on the market bearing the CE Mark. Manufacturers' quality systems for products in all but the lowest risk classification are also subject to certification and audit by an independent notified body. In Europe, particular emphasis is being placed on more sophisticated and faster procedures for the reporting of adverse events to the competent authorities.
In May 2017, the European Union implemented a new regulatory requirement for medical devices under the Medical Device Regulation (“MDR”). The Medical Device Regulation became fully effective in fiscal year 2021 and introduced new requirements for many medical devices, including enhanced requirements for clinical evidence and documentation, increased focus on device identification and traceability, new definitions and registration of economic operators throughout the distribution chain, and additional postmarket surveillance and vigilance. Compliance with the Medical Device Regulation requires re-certification of many of our products to the enhanced standards.
In Japan, pre-market approval and clinical studies are required as is governmental pricing approval for medical devices. Our Impella 2.5, Impella CP, Impella 5.0 and Impella AIC have regulatory approval and are available for sale in Japan. Clinical studies are subject to a stringent Japanese “Good Clinical Practices” standard. Approval time frames from the Japanese MHLW vary from simple notifications to review periods of one or more years, depending on the complexity and risk level of the device. In addition, importation of medical devices into Japan is subject to the “Good Import Practices” regulations. As with any highly regulated market, significant changes in the regulatory environment could adversely affect future sales.
In many of the other foreign countries in which we market our products, we may be subject to regulations affecting, among other things:
| • | product standards and specifications; |
| • | packaging requirements; |
| • | labeling requirements; |
| • | marketing restrictions; |
| • | product collection and disposal requirements; |
| • | quality system requirements; |
| • | import restrictions; |
| • | tariffs; |
| • | customs and duties; and |
| • | tax requirements. |
Many of the regulations applicable to our devices and products in these countries are similar to those of the FDA. In some countries, the level of government regulation of medical devices is increasing, which can lengthen time to market and increase registration and approval costs. In many countries, the national health or social security organizations require our products to be qualified before they can be marketed and considered eligible for reimbursement.
Health Care Initiatives
Government and private sector initiatives to limit the growth of health care costs, including price regulation and competitive pricing, coverage and payment policies, comparative effectiveness reviews, technology assessments, and managed-care arrangements, are continuing in many countries where we do business, including the U.S., Canada, Europe, and Asia. As a result of these changes, the marketplace has placed increased emphasis on the delivery of more cost-effective medical therapies. For example, government programs, private health care insurance, and managed-care plans have attempted to control costs by restricting coverage and limiting the level of reimbursement for procedures or treatments, and some third-party payers require their pre-approval before new or innovative devices or therapies are utilized by patients. These various initiatives have created increased price sensitivity over medical products generally and may impact demand for our products and technologies.
11
The delivery of our products is subject to regulation by the department of Health and Human Services in the U.S. and comparable state and foreign agencies responsible for reimbursement and regulation of health care items and services. Foreign governments also impose regulations in connection with their health care reimbursement programs and the delivery of health care items and services. Reimbursement schedules regulate the amount the U.S. government will reimburse hospitals and doctors for the inpatient care of persons covered by Medicare. CMS may also review whether and/or under what circumstances a procedure or technology is reimbursable for Medicare beneficiaries. Changes in current reimbursement levels could have an adverse effect on market demand and our pricing flexibility.
Health care cost containment efforts have also prompted domestic hospitals and other customers of medical device manufacturers to consolidate into larger purchasing groups to enhance purchasing power and this trend is expected to continue. The medical device industry has also experienced some consolidation, partly in order to offer a broader range of products to large purchasers. As a result, transactions with customers are larger, more complex, and could likely involve more long-term contracts than in the past. These larger customers, due to their enhanced purchasing power, may attempt to increase pressure on product pricing.
Seasonality
Our quarterly net sales are influenced by many factors, including new product introductions, acquisitions, regulatory approvals, patient and physician holiday schedules, and other factors. Net sales in the first half of our most recent fiscal years represented 44%, 49%, and 47% of total fiscal year net sales for fiscal years 2021, 2020 and 2019, respectively. Our revenues in the first half of our fiscal years have historically generally represented a lower percentage of total annual revenues than second half revenues due to the seasonality of the U.S., European and Japanese markets, where summer vacation schedules normally result in fewer medical procedures for our products.
Human Capital
As of March 31, 2021, we had 1,725 full-time employees, including:
| • | 371 in product engineering, research and development, clinical development and regulatory; |
| • | 686 in sales, clinical support, marketing, field service and related support, 538 of whom are in the U.S. and Canada, 92 of whom are in Europe and 56 of whom are in Asia; |
| • | 482 in manufacturing; and |
| • | 186 in general and administration. |
We understand that our success depends in large part on our ability to recruit, develop and retain a productive and engaged workforce. Accordingly, investing in our employees, focusing on safety, offering competitive compensation and benefits, promoting a diverse workforce, adopting forward thinking human capital management practices and community outreach are critical elements of our corporate strategy.
| • | Health, Safety and Wellness. As a medical device manufacturer that provides on-site support to hospitals, health and safety issues are fundamental considerations in our operations. We are committed to protecting our employees by working to implement safety practices and procedures and institute training initiatives. We monitor compliance on an ongoing basis with these Company protocols as well as federal, state, local and foreign regulations to which we are subject to. In particular, this includes Occupational Safety and Health Administration, or OSHA, requirements and other similar statutory standards outside the U.S. Our training programs are focused on the particular risks that our employees deal with in our normal operations, including hazardous materials, emergency evacuation, ergonomics and biohazards. |
| • | Competitive Compensation and Benefits. Our compensation program is designed to align employee compensation with our performance and to provide the proper incentives to attract, retain and motivate employees to achieve superior results. The structure of our compensation program aims to balance incentive earnings for both short-term and long-term performance. We believe that we provide employee wages that are competitive and consistent with employee positions, skill levels, experience, knowledge and geographic location. We engage internationally recognized external compensation and benefits consulting firms to evaluate the effectiveness of our executive compensation and benefit programs and to provide benchmarking independently. We are committed to providing comprehensive benefit options that allow our employees and their families to live healthier and more secure lives. Our benefits include various health insurance and wellness programs; retirement and savings plans; an employee stock purchase plan; paid time off and family leave; and other benefits. |
| • | Diversity and Inclusion Initiatives. We value diversity as a strength because we feel a diverse workforce leads to innovative ideas and solutions that help us recover more hearts and save more lives. We are committed to building an environment where employees are encouraged to achieve their full potential. We continue to foster a vibrant and inclusive culture, where people are respected and feel a sense of belonging regardless of their race, nationality, gender, age, religion or sexual |
12
| orientation. We have targeted recruitment programs for veterans and university students, including those of diverse backgrounds. |
| • | Employee Development. We seek to offer opportunities for employee education, development and training with a goal of creating a culture of leadership and knowledge of our business and industry. We offer professional skills and leadership development programs to support the growth of our employees. Many of our training opportunities are embodied in our Code of Conduct, which emphasizes a strong commitment to educating our employees on their legal, ethical and labor-related obligations under applicable law and our own Company policies and procedures. Additionally, we offer tuition reimbursement for courses taken in pursuit of an undergraduate or graduate degree. |
| • | Community Support. We provide opportunities for and actively encourage our employees to participate in corporate giving and volunteer opportunities in the communities in which we work. We are extremely proud of the generosity and dedication of our employees who contribute to the Abiomed Citizenship and Give Back Group, which supports local and national organizations in the United States aimed supporting heart health and U.S. veterans. We also participate in the Mentoring Veterans Program, a 501(c)(3) nonprofit organization co-founded by our Chairman and Chief Executive Officer, by helping military veterans network with industry mentors in the life sciences industries. |
| • | Response to the COVID-19 Pandemic. Throughout the COVID-19 pandemic, our priorities have been to support our clinician partners, protect the well-being of our employees, and maintain continuous access to our life-saving technologies while offering front-line in-hospital support. We and our employees have worked together to institute, and maintain ongoing compliance with, stringent safety protocols to ensure continued production in our U.S. and German facilities, such as through working shift adjustments to increase social distancing, requirements for the wearing of masks and personal protective gear and for physical distancing, on-site COVID testing for our employees in both Danvers, Massachusetts and Aachen, Germany, the placement of temperature-taking stations, increased sanitizing in between shifts and readily available hand sanitizing stations in order to maintain a safe working environment for our employees. Our proactive testing program has reduced exposure with early detection, reduced employee anxiety and enabled our manufacturing facilities to operate at full capacity in line with local social distancing requirements. For a discussion of the impact of the COVID-19 pandemic on our business, including our employees, see “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations—COVID-19 Pandemic.” |
To assess and improve employee retention and engagement, we survey employees annually with the assistance of third-party consultants and take timely action to address key areas of employee concern. Our senior leadership team regularly engages with our employees to understand and identify potential opportunities for improvement.
We routinely enter into contractual agreements with our employees, which typically include confidentiality and non-competition commitments. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We have never experienced any employment-related work stoppages and we consider our employee relations to be good.
13
ITEM 1A. | RISK FACTORS |
You should carefully consider the following risks and all other information set forth in this Report, including, without limitation, our business and forward-looking information, consolidated financial statements and the related notes thereto and “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.” The risks and uncertainties we have described are not the only ones we face. If any of these risks materialize, the trading price of our common stock could fall and you could lose all or part of your investment.
Macroeconomic and External Risks
We are subject to risks associated with public health threats and epidemics, including COVID-19.
We are subject to risks associated with public health threats and epidemics, including the global health concerns relating to the ongoing COVID-19 pandemic. The global COVID-19 pandemic has adversely impacted and is likely to further adversely impact nearly all aspects of our business and markets, including our workforce and operations and the operations of our customers, suppliers and business partners. In particular, we may experience material financial or operational impacts, including:
| • | significant volatility or reductions in demand for our products; |
| • | impacts and delays to clinical trials, our pipeline milestones, or regulatory clearances and approvals; or |
| • | the inability to meet our customers’ needs or other obligations due to disruptions to our operations or the operations of our third-party partners, suppliers, contractors, logistics partners, or customers including disruptions to production, development, manufacturing, administrative and supply operations and arrangements. |
Beginning in mid-March 2020, we experienced a decline in patient utilization in our main markets in the U.S., Europe and Japan as healthcare systems diverted resources to meet the increasing demands of managing COVID-19. Our business was most impacted in the first quarter of fiscal year 2021, in terms of the decline in patients and revenue from the shelter-in-place restrictions in a majority of countries and limitations on procedures in hospitals. We experienced sequential quarterly improvement during the second quarter of fiscal year 2021 as patient procedure volume trends and availability of healthcare resources improved as certain restrictions were lifted and limitations eased during the summer of 2020. During the third quarter of fiscal year 2021, a broad resurgence of COVID-19 cases throughout the U.S. and Europe slowed our continued recovery in patient utilization. Despite these challenges, we experienced increased recovery in patient utilization and revenue during the fourth quarter of fiscal year 2021 due to the high-risk nature of the patient population that are treated with our Impella devices.
The full extent to which the pandemic will directly or indirectly impact our business, results of operations and financial condition, including but not limited to, sales, expenses, manufacturing, clinical trials, research and development costs, reserves and allowances, fair value measurements, will depend on future developments that are highly uncertain and difficult to predict. These developments include, but are not limited to, the duration and spread of the ongoing COVID-19 pandemic (including new variants of COVID-19), its severity, the actions to contain the virus or address its impact, the timing, distribution, and efficacy of vaccines and other treatments, U.S. and foreign government actions to respond to the reduction in global economic activity, and how quickly and to what extent normal economic and operating conditions can resume.
A prolonged downturn in global economic conditions may materially adversely affect our business.
Our business and results of operations were affected by worldwide economic conditions which slowed business growth in fiscal 2021. Financial markets where we operate, particularly in the U.S., Europe and Asia, have experienced disruption in recent months, including, among other things, volatility in securities prices. We are unable to predict the likely duration and severity of any disruptions in financial markets and adverse economic conditions throughout the world. These economic developments affect businesses such as ours and those of our customers in a number of ways that could result in unfavorable consequences to us. An economic downturn in the U.S. and elsewhere, including as a result of a continued or future outbreak of COVID-19 or a similar infectious disease, or reductions in the level of government funding for scientific research, may cause our current or potential customers to delay or reduce purchases, which could, in turn, result in reductions in sales of our products, materially and adversely affecting our results of operations and cash flows. Volatility and disruption of global financial markets could limit our customers’ ability to obtain adequate financing to maintain operations and proceed with planned or new capital spending initiatives, leading to a reduction in sales volume that could materially and adversely affect our results of operations and cash flow. In addition, a decline in our customers’ ability to pay as a result of the economic downturn may lead to increased difficulties in the collection of our accounts receivable, higher levels of reserves for doubtful accounts and write-offs of accounts receivable, and higher operating costs as a percentage of revenues.
14
If a natural or man-made disaster strikes our manufacturing facilities, we will be unable to manufacture our products for a substantial amount of time and our sales and profitability will decline.
Our facilities and the manufacturing equipment we use to produce our products would be costly to replace and could require substantial lead time to repair or replace. The facilities may be affected by natural or man-made disasters and in the event they were affected by a disaster, we would be forced to rely on third-party manufacturers. Although we believe we possess adequate insurance for the disruption of our business from causalities, such insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
Fluctuations in foreign currency exchange rates could result in declines in our reported sales and results of operations.
Because some of our international sales are denominated in local currencies and not in U.S. dollars, our reported sales and earnings are subject to fluctuations in foreign currency exchange rates, primarily the Euro and the Yen. At present, we do not hedge our exposure to foreign currency fluctuations from international operations. As a result, revenues and expenses occurring in the future that are denominated in foreign currencies may be translated into U.S. dollars at less favorable rates, resulting in reduced revenues and earnings.
Risks Related to Our Business, Industry and Operations
We depend on Impella® products and services for most of our revenues.
We derive, and expect to continue to derive in the near future, most of our revenues from sales of our Impella devices and related services. While we cannot fully predict what level of revenues our Impella devices will generate, we anticipate that Impella revenues will continue to account for most of our revenues in the near future. Implementation of our business strategy depends on continued revenues from of our Impella devices and services. Our ability to generate revenues from our Impella devices and services may be impaired by the factors described below:
| • | our failure to obtain approvals and maintain clearance from the FDA and foreign regulatory authorities or to comply with government regulations, or the withdrawal of market clearance or the taking of other enforcement actions that could limit or impair our ability to sell our products; |
| • | announcements by the FDA relating to our products and their impact on market perception of our product, including short-term impact; |
| • | lack of acceptance or continued acceptance by physicians, hospitals, or patients; |
| • | our reliance on specialized suppliers for certain components and materials; |
| • | manufacturing or quality control issues; |
| • | reputational risk relating to customer reviews of our products; |
| • | our inability to protect our proprietary technologies or an infringement of others’ patents; |
| • | the loss of a distributor or a distributor’s failure to perform its obligations; |
| • | our failure to compete successfully against our existing or potential competitors; |
| • | additional risks associated with selling in international markets; |
| • | long and variable sales and deployment cycles; |
| • | failure by third-party payers to provide appropriate levels of reimbursement for hospitals and physicians using our products; |
| • | our failure to comply with federal and state regulations; and |
| • | product liability claims. |
15
If we fail to compete successfully against our existing or potential competitors, our revenues or operating results may be harmed.
Competition from other companies offering circulatory care products is intense and subject to rapid technological change and evolving industry requirements and standards. We compete with companies that have greater financial, product development, sales and marketing resources and experience than we do. Our ability to compete effectively depends upon our ability to distinguish our company and our products from our competitors and their products. Factors affecting our competitive position include:
| • | the availability of other products and procedures that are technically equivalent or superior to our products, and which may be sold at lower prices; |
| • | product performance and design; |
| • | product safety; |
| • | sales, marketing and distribution capabilities; |
| • | comparable clinical outcomes; |
| • | our ability to complete clinical trials and regulatory approval processes; |
| • | success and timing of new product development and introductions; |
| • | physician and hospital acceptance of our products and the amount of time to convert physicians and hospitals into users of our products; |
| • | reimbursement approval from health care insurance providers and the cost-effectiveness of these reimbursements to our hospital customers; |
| • | our ability to integrate potential acquired businesses into our operations; |
| • | penetration into existing and new geographic markets; and |
| • | intellectual property protection. |
Our customers are primarily hospitals that have limited budgets. Physicians endorse our products to hospitals that will then choose to purchase our products and subsequently pass this cost on to patients or their insurance providers. Physicians will recommend our products based on public information regarding patient outcomes, clinical trials, and the costs and benefits of using our products when compared to other substitutes available in the market. As a result, our products compete against a broad range of medical devices and other therapies for these limited funds. Our success will depend in large part upon our ability to enhance our existing products, to develop new products to meet regulatory and customer requirements and to achieve and maintain market acceptance for our products. We believe that important competitive factors with respect to the development and commercialization of our products include the relative speed with which we can develop products, establish clinical utility, complete clinical trials and regulatory approval processes, obtain and protect reimbursement, maintain cost effectiveness for our products, and supply commercial quantities of our products to our customers.
Advances in medical technology, biotechnology and pharmaceuticals may reduce the size of the potential markets for our products or render our products obsolete. We are aware of other cardiac assist device research efforts in the U.S., Canada, Europe and Japan. In addition, there are a number of companies, including Abbott Laboratories, Medtronic, Edwards Lifesciences, CardiacAssist, Terumo Heart, Teleflex, Getinge (Maquet Cardiovascular), and several early-stage companies, that are developing heart assist products, including implantable left ventricular assist devices and miniaturized rotary ventricular assist devices that directly and indirectly compete with our products.
The commercial success of our products will require acceptance by cardiac surgeons and interventional cardiologists, a limited number of whom have significant influence over medical device selection and purchasing decisions.
We may achieve our business objectives only if our products are accepted and recommended by leading cardiac surgeons and interventional cardiologists, whose decisions are likely to be based on a determination that our products are safe and effective and represent acceptable, cost-effective methods of treatment in light of reimbursement policies with respect to our products. The commercial success of Impella devices and our other products will also require that we continue our existing relationships and develop new relationships with leading cardiac surgeons and interventional cardiologists. We cannot assure you that we can maintain our existing relationships and arrangements with leading cardiac surgeons or interventional cardiologists or that we can establish new relationships in support of our products. If cardiac surgeons and interventional cardiologists do not consider our products to be adequate for the treatment of our target cardiac patient population or if a sufficient number of these clinicians recommend and use competing products, it would seriously harm our business.
16
Expansion into hospital cardiac centers that have not historically used our products may incur long sales and training cycles that may cause our revenues and operating results to vary significantly from quarter to quarter.
Our products have lengthy sales cycles and we may incur substantial sales and marketing expenses and expend significant effort without making a sale. We sell primarily to hospitals that often have administrative requirements to introduce and expand a new technology, such as our Impella devices, at their sites. Even after making the decision to purchase our Impella devices, our customers often deploy our products slowly or infrequently. In addition, cardiac centers of hospitals that buy the majority of our products are usually led by cardiac surgeons who are heavily recruited by competing hospitals. When one of these cardiac surgeons moves to a new hospital, we sometimes experience a significant reduction in purchases by the hospital from which the physician has departed while it replaces the lead physician supporting our Impella devices. As a result, our revenues and operating results may vary significantly from quarter to quarter. In addition, product purchases often lag behind initial expressions of interest in our product by new centers due to training and education regarding the use of the products. Hospitals also need to perform internal administrative requirements prior to the initial implant procedures. These challenges in our sales initiatives may be further adversely impacted by the ongoing COVID-19 pandemic or similar infectious diseases. For more information, see “—We are subject to risks associated with public health threats and epidemics, including COVID-19.”
The training required for clinicians to use our products could reduce the market acceptance of our products and reduce our revenue.
Clinicians must be trained to use our products proficiently. It is critical to the success of our business that we ensure that there are a sufficient number of clinicians familiar with, trained on and proficient in the use of our products. Convincing clinicians to dedicate the time and energy necessary to obtain adequate training in the use of our products is challenging and we may not be successful in these efforts. Our physician education and training initiatives may also be impaired by the ongoing COVID-19 pandemic or similar infectious diseases requiring social distancing. If clinicians are not properly trained, they may misuse or ineffectively use our products. Any improper use of our products may result in unsatisfactory patient outcomes, patient injury, negative publicity or lawsuits against us, any of which could harm our reputation and affect future product sales. Furthermore, our inability to educate and train clinicians to use our products may lead to lower demand for our products.
If we do not effectively manage our growth, we may be unable to successfully develop, market and sell our products.
Our future revenue and operating results will depend on our ability to manage the anticipated growth of our business. We have experienced significant growth in recent years in which we have expanded our operations and we have increased our employee headcount. This growth has placed significant demands on our management as well as our financial and operational resources. In order to achieve our business objectives, we will need to continue to grow. However, continued growth presents numerous challenges, including:
| • | developing and retaining our global sales, marketing and administrative infrastructure and capabilities; |
| • | expanding manufacturing capacity, maintaining quality and increasing production in an efficient manner; |
| • | increasing our foreign and domestic regulatory compliance capabilities; |
| • | implementing appropriate operational, financial and IT systems and internal controls; |
| • | impact of the ongoing COVID-19 pandemic and related remote working arrangements on our ability to support our customers; |
| • | identifying, attracting and retaining qualified personnel, particularly experienced clinical staff; and |
| • | hiring, training, managing and supervising our personnel worldwide. |
Any failure to manage our growth effectively could impede our ability to successfully develop, market and sell our products, which could seriously harm our business.
17
The demand for our existing products and products under development is unproven, and we may be unable to successfully commercialize our products.
Our existing products, which have received regulatory approval for commercialization only in the last few years, and our products under development may not enjoy commercial acceptance or success, thus adversely affecting our business and operational results. We need to create new indications and geographic markets for our Impella devices and other existing products, as well as other new or future products, including achieving market acceptance among physicians, hospitals, patients and third-party payers. In particular, we must gain and maintain acceptance of our Impella devices among interventional cardiologists and cardiac surgeons. The obstacles we will face in trying to create successful commercial markets for our products include:
| • | limitations inherent in first-generation devices, and our potential inability to develop successive improvements, including increases in service life and improvements in the ease of use of our products; |
| • | introduction by other companies of new treatments, products and technologies that compete with our products; |
| • | willingness of physicians to recommend the use of our products; |
| • | timing and amount of reimbursement for these products, if any, by third-party payers, and the cost-effectiveness of using our products by our customers given these reimbursement considerations; |
| • | potential reluctance of clinicians and hospitals to obtain and support adequate training to use our products; |
| • | cost of our products; and |
| • | potential reluctance of physicians, patients, hospitals and society as a whole to accept medical devices that replace or assist the heart and risk of mechanical failure inherent in such devices. |
Several of these obstacles may be further exacerbated by the ongoing COVID-19 pandemic (including new variants of COVID-19) or similar infectious diseases. For more information, see “—We are subject to risks associated with public health threats and epidemics, including COVID-19.”
Unsuccessful clinical trials or procedures relating to products under development could have a material adverse effect on our prospects.
The regulatory approval process for new products and new indications for existing products often requires extensive clinical trials and procedures, including early clinical feasibility studies. Unfavorable or inconsistent clinical data from current or future clinical trials or procedures conducted by us, our competitors, or third parties, or perceptions regarding such clinical data, whether or not true, could adversely affect both our ability to obtain necessary approvals and the market’s view of our future prospects. Such clinical trials and procedures are inherently uncertain and there can be no assurance that these clinical trials or procedures will be completed in a timely or cost-effective manner or result in a commercially viable product or expanded indication. Failure to successfully complete these clinical trials or procedures in a timely and cost-effective manner could have a material adverse effect on our prospects. Clinical trials or procedures may experience significant setbacks even after earlier trials have shown promising results. Further, preliminary results from clinical trials or procedures may be contradicted by subsequent clinical analysis. In addition, results from our clinical trials or procedures may not be supported by actual long-term studies or clinical experience. If preliminary clinical results are later contradicted, or if initial results cannot be supported by actual long-term studies or clinical experience, our business could be adversely affected. Clinical trials or procedures may be delayed, suspended, or terminated by us, the FDA, or other regulatory authorities at any time, if it is believed that the trial participants face unacceptable health risks or for numerous other reasons. The FDA may disagree with our interpretation of the data from our clinical trials, or may find the clinical trial design, conduct or results inadequate to demonstrate safety and effectiveness of the product candidate. The FDA may also require additional pre-clinical studies or clinical trials, which could further delay approval of our products.
Our future success depends in part on the development of new circulatory assist products, and our development efforts may not be successful.
We are devoting most of our research and development and regulatory efforts, and significant financial resources, to the development of our Impella devices and product extensions of existing commercial products and new products. The development of new products and product extensions presents enormous challenges in a variety of areas, including blood compatible surfaces, blood compatible flow, manufacturing techniques, pumping mechanisms, physiological control, energy transfer, anatomical fit and surgical techniques. We may be unable to overcome all of these challenges, which could adversely affect our results of operations and prospects and limit our ability to bring new products to market or make changes to enhance existing commercial products.
18
If we are unable to develop additional, high-quality manufacturing capacity, our growth may be limited and our business could be seriously harmed.
To be successful in the long-term, we will need to increase our manufacturing capacity to support continued demand for our products. We may encounter difficulties in scaling up manufacturing of our products, including problems related to product yields, quality control and assurance, component and service availability, dependable sources of supply, adequacy of internal control policies and procedures, ability to automate certain manufacturing processes and lack of skilled personnel. For more information, see “—A pandemic, epidemic or outbreak of an infectious disease, such as COVID-19, or coronavirus, may materially and adversely impact our business, our operations and our financial results.”
If we cannot hire, train and retain enough experienced and capable scientific, technical, and manufacturing employees, we may not be able to manufacture sufficient quantities of our existing or future products on time and at an acceptable cost, which could limit market acceptance of our products or otherwise damage our business. In order to meet the expected demand for our Impella devices, we have continued to implement process improvements on the Impella production line at our manufacturing facilities in Aachen, Germany, Danvers, Massachusetts to increase the output that we can produce at these facilities. In addition to programs designed to further increase yield and capacity levels, we have expanded manufacturing employment in recent years and increased manufacturing floor space in Danvers and Aachen. We continue to work on initiatives to expand our Impella manufacturing capacity in all our manufacturing facilities. We are also working with our existing suppliers and new suppliers to ensure we are able to have sufficient inventory as we increase our manufacturing capability to support growing demand. We are and will continue outsourcing certain sub assembly production to third-party suppliers. We are also working on process improvements, such as certain automation techniques, to allow us to manufacture our products more efficiently as much of our manufacturing process is labor dependent. If we are unable to implement these process improvements on a timely basis in order to meet customer demand, it could inhibit our future revenue growth.
We depend on third-party reimbursement to our customers for market acceptance of our products. If third-party payers fail to provide coverage and appropriate levels of reimbursement for the medical procedures in which our products are used, our sales and profitability would be adversely affected.
Sales of medical devices largely depend on the reimbursement of patients’ medical expenses by government healthcare programs and private health insurers. Without the financial support of government reimbursement or third-party insurers’ payments for patient care, the market for our products will be limited. Medical products and devices incorporating new technologies are closely examined by governments and private insurers to determine whether the products and devices will be covered by reimbursement, and if so, the level of reimbursement which may apply.
In the U.S., future action by the Centers for Medicare & Medicaid Services, or CMS (which administers the Medicare program), other government agencies or private payors, may diminish payments to physicians, outpatient surgery centers and/or hospitals, which could harm our ability to market and sell our products. Private payors may adopt coverage decisions and payment amounts determined by CMS as guidelines in setting their coverage and reimbursement policies. Private payors that do not follow the Medicare guidelines may adopt different coverage and reimbursement policies for procedures performed with our products. In addition, for governmental programs, such as Medicaid, coverage and reimbursement differs from state to state. Medicaid payments to physicians and facilities are often lower than payments by other third parties, and some state Medicaid programs may not pay an adequate amount for the procedures performed with our products, if any payment is made at all. Internationally, medical reimbursement systems vary significantly from country to country, with some countries limiting medical centers spending through fixed budgets, regardless of levels of patient treatment, and other countries requiring application for, and approval of, government or third-party reimbursement. For more information, see “Part I, Item 1. Business—Third-Party Reimbursement.”
In addition, third-party payers, including private and government insurers, are increasingly requiring evidence that medical devices are cost-effective. If we are unable to demonstrate that our devices are cost-effective, the third-party payer may not reimburse the use of our products, which could reduce sales of our products to healthcare providers who depend upon reimbursement for payment. We also cannot be sure that third-party payers will continue the current levels of reimbursement to physicians and medical centers for use of our products. Any reduction in the amount of this reimbursement could harm our business. Increasing awareness of healthcare costs, public interest in healthcare reform and continuing pressure from Medicare, Medicaid, group purchasing organizations and other payers to reduce costs in the healthcare industry, as well as increasing competition from other protective products, could make it more difficult for us to sell our products at current prices.
19
If our suppliers cannot provide the components we require, our ability to manufacture our products could be harmed.
We rely on third-party suppliers to provide us with many of the components used in our existing products and products in development. For example, we outsource the manufacturing of most of our consoles and the sterilization process for our products. Relying on third-party suppliers makes us vulnerable to component part failures or obsolescence and interruptions in supply, either of which could impair our ability to conduct clinical trials or to ship our products to our customers on a timely basis. Using third-party vendors makes it difficult and sometimes impossible for us to test fully certain components, such as components on circuit boards, maintain quality control, manage inventory and production schedules and control production costs. Many of our manufacturing suppliers may be required to comply with the FDA or other regulatory manufacturing regulations and to satisfy regulatory inspections in connection with the manufacture of the components. Any failure by a supplier to comply with applicable requirements could lead to a disruption in supply. Vendor lead times to supply us with ordered components vary significantly and often can exceed six months or more. Both now, and as we expand our manufacturing capacity, we cannot be sure that our suppliers will furnish us required components when we need them or be able to provide us with sufficient inventory to support our expected growth in demand for our products. The ongoing COVID-19 pandemic(including new variants of COVID-19) or similar infectious diseases may also create disruptions in our supply chain. For more information, see “—A pandemic, epidemic or outbreak of an infectious disease, such as COVID-19, or coronavirus, may materially and adversely impact our business, our operations and our financial results.” These factors could make it more difficult for us to manufacture our products effectively and efficiently and could adversely impact our results of operations.
Some of our suppliers may be the only source for a particular component, which makes us vulnerable to significant cost increases or shortage of supply. We have many foreign suppliers for some of our parts in which we are subject to currency exchange rate volatility. Some of our vendors are small in size and may have difficulty supplying the quantity and quality of materials required for our products as our business grows. Vendors that are the sole source of certain products may decide to limit or eliminate sales of certain components due to product liability or other concerns and we might not be able to find a suitable replacement for those products. Our inventory may run out before we find alternative suppliers and we might be forced to purchase excess inventory, if available, to last until we are able to qualify an alternate supplier. If we cannot obtain a necessary component, we may need to find, test and obtain regulatory approval or clearance for a replacement component, produce the component ourselves or redesign the related product, which would cause significant delay and could increase our manufacturing costs. Any of these events could adversely impact our results of operations.
We rely on distributors to sell our products in some international markets and poor performance by a distributor could reduce our sales and harm our business.
We rely on distributors to market and sell our products in certain parts of Europe, Asia, South America and the Middle East. Many of these distributors have the exclusive right to distribute our products in their territory. We may hire distributors to market our products in additional international markets in the future. Our success in these markets will depend almost entirely upon the efforts of our distributors, over whom we have little or no control. If a distributor does not market and sell our products effectively and maintain a continued focus on the sale, distribution and support of our products up to our standards, we could lose sales and impair our ability to compete and introduce our technology in that market. From time to time, these distributors could decide to reduce their levels of inventory with regard to certain of our products due to various factors, which could have an adverse effect on our business depending on the extend of the distributor’s sales. Outbreaks of infectious diseases such as COVID-19 or similar diseases, may also create disruptions in our distribution networks, especially in foreign markets that are forced to implement extended quarantine measures due to a lack of treatment and/or testing resources. For more information, see “—A pandemic, epidemic or outbreak of an infectious disease, such as COVID-19, or coronavirus, may materially and adversely impact our business, our operations and our financial results.” We are subject to credit risk and foreign currency risk associated with shipments to our distributors and this could negatively impact our financial condition and liquidity in the future.
We may not be successful in expanding our direct sales activities into international markets.
We are seeking to expand our international sales of our products by recruiting direct sales and support teams outside the U.S. Our international operations in Germany, Japan, France, Canada, the United Kingdom, Singapore and Australia are or will be subject to a number of risks, which may vary from the risks we experience in the U.S., including:
| • | the need to obtain regulatory approvals in foreign countries before our products may be sold or used; |
| • | the need to procure reimbursement for our products in each foreign market; |
| • | the generally lower level of reimbursement available in foreign markets relative to the U.S.; |
| • | the requirement to work with distributors or other partners to sell our products in international markets; |
20
| • | longer sales cycles; |
| • | uncertainty with respect to enforcement of legal rights by local regulatory or judicial authorities; |
| • | limited protection of intellectual property rights; |
| • | difficulty and delays in collecting accounts receivable; |
| • | different income tax and sales tax environments; |
| • | difficulty in supporting patients using our products; |
| • | difficulty in attracting employees in foreign countries who want to work for a smaller U.S. based company; |
| • | different payroll, employee benefits and statutory requirements; |
| • | the adoption and expansion of trade restrictions, including the occurrence or escalation of a “trade war,” the imposition or modification of sanctions or other governmental action related to tariffs or trade agreements or policies among the governments of the United States, China and other countries; |
| • | regulatory changes and economic conditions leading up to and following “Brexit” (the United Kingdom’s recent exit from the European Union), including uncertainties as to its timing and its effect on trade laws, tariffs and taxes; |
| • | fluctuations in the values of foreign currencies; and |
| • | political and economic instability. |
If we are unable to effectively expand our sales activities in international markets, our results of operations could be negatively impacted.
The profitability we have achieved in recent years may not be indicative of our ability to sustain profitability and it is possible that we may incur losses from operations in future periods.
We recognized net income of $225.5 million, $203.0 million and $259.0 million for the fiscal years ended March 31, 2021, 2020 and 2019, respectively. The profitability we have achieved in recent years may not be indicative of our ability to sustain future profitability and it is possible that we may incur losses from operations or net losses in future periods. Any losses incurred in the future may result primarily from, among other things:
| • | integration of newly-acquired companies and products, such as Breethe and the OXY-1 System, respectively, into our operations and product lines; |
| • | changes in demand for our products; |
| • | the recent expansion of our global distribution network; |
| • | investments in new geographic markets; |
| • | ongoing product and clinical development; |
| • | costs related to new business development initiatives, such as potential acquisitions of new businesses; |
| • | legal expenses related to patent and other matters, such as the Maquet dispute and shareholder actions; |
| • | costs associated with hiring additional personnel, performing clinical trials, continuing our research and development relating to our products under development, seeking regulatory approvals and, if we receive these approvals, commencing commercial manufacturing and marketing activities; |
| • | expanded marketing initiatives, particularly with recent PMAs in the U.S.; |
| • | income and other related taxes; |
| • | increase in stock-based compensation as we hire new employees and our stock prices has continued or could expect to continue to increase in the future; |
| • | significant disruption to and volatility in national, regional and local economies and markets and our internal operations caused by pandemics; |
21
| • | significant expenditures necessary to market and manufacture in commercial quantities our approved circulatory care products; |
| • | difficulty in forecasting these expenditures; |
| • | the scope, scale and duration of the impact of the ongoing COVID-19 pandemic; and |
| • | a prolonged downturn in global economic conditions, including as a result of the ongoing COVID-19 pandemic. |
Our operating results may fluctuate unpredictably.
Historically, our annual and quarterly operating results have fluctuated widely and we expect these fluctuations to continue. Among the factors that may cause our operating results to fluctuate are:
| • | timing of customer orders and deliveries; |
| • | seasonality of sales in the U.S., European and Japanese markets, where summer vacation schedules normally result in fewer medical procedures during the first half of our fiscal year; |
| • | competitive changes, such as price changes or new product introductions that we or our competitors may make; |
| • | announcements by the FDA relating to our products and their impact on market perception of our product, including short-term impact; |
| • | reputational risk relating to customer reviews of our products; |
| • | the impact of additional investments to expand manufacturing capacity on cost of product sales; |
| • | the timing of regulatory actions, such as product approvals or recalls; |
| • | costs we incur in developing and testing our Impella heart pumps and other products; |
| • | the impact of adverse data or the perception of adverse data relating to our products and technology among the medical community; |
| • | costs we incur in anticipation of future sales, such as inventory purchases, expansion of manufacturing facilities, or establishment of international sales offices; |
| • | additional taxes; |
| • | the impact and timing of equity awards on stock-based compensation; |
| • | timing of certain marketing programs and events; |
| • | availability of physicians to use our products, as there are seasonal impacts, due to physician vacations or training events that limit their ability to be in the hospital to perform procedures that involve our products; |
| • | the impact of any businesses or technologies we may acquire in the future; |
| • | economic conditions in the healthcare industry; |
| • | gains or losses on our portfolio investments, such as Shockwave Medical, Inc. (“Shockwave Medical”); |
| • | efforts by governments, insurance companies and others to contain healthcare costs, including changes to reimbursement policies and their impact on hospitals’ ability to purchase our products; |
| • | the impact of the adoption of certain accounting standards; |
| • | clinical trial results that reveal disadvantages of our products for various markets we address, or otherwise unfavorable or inconsistent data about our products; and |
| • | the impact of a pandemic, epidemic or outbreak of an infectious disease, such as the ongoing COVID-19 pandemic, on our and our customers’ ability to operate to full capacity. |
We believe that period-to-period comparisons of our historical results are not necessarily meaningful and investors should not rely on them as an indication of our future performance. To the extent we experience the factors described above, our future operating results may not meet the expectations of securities analysts or investors, which may cause the market price of our common stock to decline.
22
We may not have sufficient funds to develop and commercialize our new products or make acquisitions of desirable companies, products or technologies.
The development, manufacture and sale of any medical device is very expensive, and we may require additional funds to make acquisitions of desirable companies, products or technologies. We cannot be sure that we will have the necessary funds to develop and commercialize our new products or acquire companies, products or technologies, or that additional funds will be available on commercially acceptable terms, if at all. We currently have no debt, and new sources of capital may not be available to us when we need it or may be available only on terms that we would find unacceptable. If we are unable to obtain the necessary funding to support these efforts, our business may be adversely affected. We believe we have sufficient liquidity to finance our operations for at least the next fiscal year based on available working capital and cash from operations. We also may evaluate other financing alternatives as necessary to fund operations, and any equity or convertible debt financing may involve dilution to our existing stockholders.
Quality issues may result in inventory write-downs and other costs.
Government regulations require us to track materials used in the manufacture of our products, so that if an issue is identified in one product it can be traced to other products that may have the same issue. An identified quality issues may require reworking or scrapping related inventory and/or recalling previous shipments. Because a malfunction in our products can possibly be life-threatening, we may be required to recall and replace, free of charge, products already in the marketplace. Any quality issue could cause us to incur significant expenses, lead to significant write-offs of inventory, injure our reputation and harm our business and financial results.
If we acquire other companies or businesses, we will be subject to risks that could hurt our business.
In April 2020, we completed the acquisition of Breethe with an aim to complement and expand our product portfolio with Breethe’s novel extracorporeal membrane oxygenation system. We may pursue acquisitions in the future to obtain complementary businesses, products or technologies. Any such acquisition may not produce the revenues, earnings or business synergies that we anticipate and could be dilutive to earnings. An acquired business, product, or technology might not perform as we expect. Our management could spend a significant amount of time, effort and money in identifying, pursuing and completing the acquisition. If we complete an acquisition, we may encounter significant difficulties and incur sizable expenses in integrating the operations and personnel of the acquired company into our operations. For more information on the acquisition of Breethe, see “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations—Acquisition of Breethe, Inc.” We may lose the services of key employees of the acquired company and we may make changes in management that impair the acquired company’s relationships with its legacy employees, vendors and customers. Furthermore, we may acquire development-stage companies that are not yet profitable, that require continued investment or become subject to events that could impair the value of our investment, which could decrease our future earnings. We may also assume significant liabilities in such a transaction.
Any of these outcomes could prevent us from realizing the anticipated benefits of an acquisition. To pay for an acquisition, we might use stock or cash. Alternatively, we might borrow money from a bank or other lender. If we use stock, our stockholders would experience dilution of their ownership interests. If we use cash or debt financing, our financial liquidity would be reduced.
If we include future milestones as part of the potential purchase price of an acquisition, then we will have to estimate the value of these milestones each reporting period and any changes underlying these estimates with respect to expected timing or valuation of these milestones could have a volatile impact on our earnings. See “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operation— Acquisition of Breethe, Inc.” We periodically make investments in medical device companies that focus on heart failure and heart pumps and other medical device technologies. The aggregate carrying amount of our portfolio of investments in medical device companies, including Shockwave Medical was $101.6 million and $94.4 million as of March 31, 2021 and 2020, respectively, and is classified within other assets in the consolidated balance sheets.
Consolidation in the healthcare industry could lead to demands for price concessions, which could have an adverse effect on our business, results of operations or financial condition.
Because healthcare costs have risen significantly over the past decade, numerous initiatives and reforms initiated by legislators, regulators and third-party payers to curb these costs have resulted in a consolidation trend in the healthcare industry to aggregate purchasing power. As the healthcare industry consolidates, competition to provide products and services to industry participants has become and will continue to become more intense. This in turn has resulted and will likely continue to result in greater pricing pressures and the exclusion of certain suppliers from important market segments as group purchasing organizations, independent delivery networks and large single accounts continue to use their market power to consolidate purchasing decisions for hospitals. We expect that market demand, government regulation, third-party coverage and reimbursement policies and societal pressures will continue to change the worldwide healthcare industry, resulting in further business consolidations and alliances among our customers, which may reduce competition, exert further downward pressure on the prices of our products and may adversely impact our business, results of operations or financial condition.
23
If we cannot attract and retain key management, scientific, sales and other personnel we need, we will not be successful.
We depend heavily on the contributions of the principal members of our business, such as financial, technical, sales and support, regulatory and clinical, operating, manufacturing and administrative management and staff, many of whom would be difficult to replace. Our key personnel include our senior officers, many of whom have very specialized scientific, medical or operational knowledge. The loss of the service of any of the key members of our senior management team, whether due to dismissal, resignation or illness, may significantly delay or prevent our achievement of our business objectives and divert remaining management’s attention to seeking qualified replacements. Our ability to attract, train and retain qualified personnel, consultants and advisors is critical to our success. For example, many members of our clinical staff are registered nurses with experience in the surgery suite or cath lab, of which only a limited number of whom seek employment with a company like ours. Competition for skilled and experienced personnel in the medical device industry is intense. We face competition for skilled and experienced management, scientific, clinical, engineering and sales personnel from numerous medical device and life sciences companies, hospitals, universities, governmental entities and other research institutions. Hiring efforts may also be compounded by intensified restrictions on travel (including during the ongoing COVID-19 pandemic). If we lose the services of any of the principal members of our management and staff and have not developed adequate succession plans, or if we are unable to attract, train and retain qualified personnel in the future, especially scientific, clinical and sales personnel, our business could be adversely affected.
Legal, Regulatory and Compliance Risks
If we fail to obtain and maintain necessary governmental approvals for our products and indications, we may be unable to market and sell our products in certain jurisdictions.
Medical devices such as ours are extensively regulated by the FDA in the U.S. and by other federal, state, local and foreign authorities. Governmental regulations relate to the testing, development, manufacturing, labeling, design, sale, promotion, distribution, importing, exporting and shipping of our products. In the U.S., before we can market a new medical device, or a new use of, or claim for, or significant modification to, an existing product, we must generally first receive Premarket Approval (“PMA”) from the FDA. This process can be expensive and lengthy, and can entail significant expenses, primarily related to clinical trials. It generally takes between one to three years to receive approval, or even longer, from the time the PMA application is submitted to the FDA. Regulatory clearances or approvals, either foreign or domestic, may not be granted on a timely basis, if at all. If we are unable to obtain regulatory approvals or clearances for use of our products under development, or if the patient populations for which they are approved are not sufficiently broad, the commercial success of these products could be limited. The FDA may also limit the claims that we can make about our products. Any significant modifications to the design, materials, or intended use of those devices require FDA approval through PMA or humanitarian device extension (“HDE”) supplemental applications.
If we do not receive FDA approval for one or more of our products, we will be unable to market and sell those products in the U.S., which would have a material adverse effect on our operations and prospects.
We also market or are beginning to market our products in international markets, including the European Union, Canada, and Japan. Regulatory approval processes differ among those jurisdictions and approval in the U.S. or any other single jurisdiction does not guarantee approval in any other jurisdiction. Obtaining foreign approvals could involve significant delays, difficulties and costs for us and could require additional clinical trials.
If the FDA or another regulatory or enforcement agency determines that we have promoted our products for one or more off-label uses, we may be subject to various penalties, including civil or criminal penalties.
The FDA, the U.S. Department of Justice, the Office of the Inspector General of Department of Health and Human Services, and other regulatory or enforcement agencies actively enforce regulations prohibiting the promotion of unapproved medical devices and the promotion of otherwise approved or cleared medical devices for unapproved uses. If any such agency determines that our promotional materials or training constitutes promotion of an unapproved use, it could request that we modify our training or promotional materials or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, recall or withdrawal, seizure, civil fine and criminal penalties. Although our policy is to refrain from statements that could be considered off-label promotion of our products, such agencies could disagree and conclude that we have engaged in off-label promotion.
To the extent a regulatory agency commences an investigation in the future, we may not be able to resolve that matter, without incurring penalties or facing significant consequences. Even if we are successful in resolving such a matter without incurring penalties, responding to a subpoena or other government inquiry could result in substantial costs and could significantly and adversely impact our reputation and divert management’s attention and resources, which could have a material adverse effect on our business, operating results, financial condition and ability to finance our operations.
24
Finally, the ability of the FDA to review and clear or approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. These factors could affect our ability to develop or commercialize new products in a timely manner, which could negatively impact our business.
Off-label use of our products may result in injuries that lead to product liability suits, which could be costly to our business.
The use of our products outside their approved indications for use, or “off-label use,” may increase the risk of injury to patients. Clinicians may use our products for off-label uses, as the FDA does not restrict or regulate a clinician’s choice of treatment within the practice of medicine. Off-label use of our products may increase the risk of product liability claims against us. Product liability claims are expensive to defend and could divert our management’s attention and result in substantial damage awards against us.
Product liability claims could damage our reputation and adversely affect our financial results.
The clinical use of medical products, even after regulatory approval, poses an inherent risk of product liability claims. We maintain limited product liability insurance coverage, subject to certain deductibles and exclusions. We cannot be sure that product liability insurance will be available in the future or will be available on acceptable terms or at reasonable costs, or that such insurance will provide us with adequate coverage against potential liabilities. We have been and anticipate that as part of our ordinary course of business we may be, subject to product liability claims alleging defects in the design, manufacture or labeling of our products. Claims against us, regardless of their merit or potential outcome, may also hurt our ability to obtain physician endorsement of our products or expand our business. As we continue to expand use or our existing products and introduce more products, we face an increased risk that a material product liability claim will be brought against us.
Some of our products are designed for patients who suffer from late-stage or end-stage heart failure, and many of these patients do not survive, even when supported by our products. There are many factors beyond our control that could result in patient death, including the condition of the patient prior to use of the product, the skill and reliability of physicians and hospital personnel using and monitoring the product and product maintenance by customers. However, the failure of our products used for clinical testing or sale could give rise to product liability claims and negative publicity.
The risk of product liability claims is heightened when we sell products that are intended to support a patient until the end of life. The finite life of our products, as well as complications associated with their use, could give rise to product liability claims whether or not the products have extended or improved the quality of a patient’s life. If we are forced to pay product liability claims in excess of our insurance coverage, our financial condition will be adversely affected.
Our products are subject to extensive regulatory requirements, including continuing regulatory review, which could affect the manufacturing and marketing of our products.
The FDA and other regulatory agencies continue to review products even after they have received initial approval. If and when the FDA or another regulatory agency clears or approves our products under development, the manufacture and marketing of these products will be subject to continuing regulation, post-approval clinical studies, including compliance with the FDA’s adverse event reporting requirements, prohibitions on promoting a product for unapproved uses, and Quality System Regulation, or QSR, requirements, which obligate manufacturers, including third-party and contract manufacturers, to adhere to stringent design, testing, control, documentation and other quality assurance procedures during the design and manufacture of a device.
Any modification to an FDA approved device that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a supplemental PMA, HDE or EUA approval. The FDA requires each manufacturer to determine in the first instance whether a modification requires approval, but the FDA may review and potentially disagree with any such decision. Modifications of this type are common with new products. We anticipate that the first generation of each of our products will undergo a number of changes, refinements, enhancements and improvements over time. If the FDA requires us to seek approval for modification of a previously approved product for which we have concluded that new clearances or approvals are unnecessary, we may be required to cease marketing or to recall the modified product until we obtain clearance or approval and we may be subject to significant regulatory fines or penalties, which could have a material adverse effect on our financial results and competitive position. We also cannot assure you that we will be successful in obtaining clearances or approvals for our modifications, if required. We and our third-party suppliers of product components are also subject to inspection and market surveillance by the FDA and other regulatory agencies for QSR and other requirements, the interpretation of which can change. Compliance with QSR and similar legal requirements can be difficult and expensive. While we continue to monitor our quality management in order to improve our overall level of compliance, our facilities are subject to periodic and unannounced inspection by U.S. and foreign regulatory agencies to audit compliance with the QSR and comparable foreign regulations. Enforcement actions resulting from failure to comply with government requirements could result in fines, suspensions of approvals or clearances, recalls or seizure of products, operating restrictions or shutdown, and criminal prosecutions that could adversely affect the manufacture and marketing of our products. The FDA or another regulatory agency could withdraw a previously approved product from the market upon receipt of newly discovered information, including a failure to comply with regulatory requirements, the occurrence of unanticipated safety problems of other defects in products following approval, or other reasons, which could adversely affect our operating results.
25
Even after receiving regulatory clearance or approval, our products may be subject to product recalls, which could harm our reputation and divert our managerial and financial resources.
The FDA and similar governmental authorities in other countries have the authority to order mandatory recall of our products or order their removal from the market if the government finds that our products might cause adverse health consequences or death. A government-mandated or voluntary recall by us could occur as a result of component failures, manufacturing errors by us or our suppliers or design defects, including labeling defects, or unanticipated safety problems. We have in the past initiated voluntary recalls for some of our products and we could do so in the future. Any recall of our products may harm our reputation with customers and divert managerial and financial resources.
Shutdowns of the U.S. federal government could materially impair our business and financial condition.
Development of our product candidates and/or regulatory approval may be delayed for reasons beyond our control. For example, over the last several years the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, CMS, IRS and the SEC, have had to furlough their government employees and stop critical activities. If a prolonged government shutdown or budget sequestration occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, in our operations as a public company, future government shutdowns could impact our ability to communicate with the SEC on various topics, such as shareholder proposals, or to have our registration statements declared effective, which could affect our ability to access the capital markets quickly.
Changes in healthcare reimbursement systems in the U.S. and abroad could reduce our revenues and profitability.
In March 2010, the U.S. federal government enacted the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (the “ACA”), which made changes to the manner in which many healthcare services are provided and paid for in the U.S. The ACA includes provisions that, among other things, reduce or limit Medicare reimbursement, require all individuals to have health insurance (with limited exceptions) and impose increased taxes on certain companies and individuals. Since its enactment, there have been numerous judicial, administrative, executive, and legislative challenges to certain aspects of the ACA, and we expect there will be additional challenges and amendments to the ACA in the future which could change the way healthcare is financed by both governmental and private insurers and which may significantly impact our industry and our business. Various portions of the ACA are currently undergoing legal and constitutional challenges in federal court, including a case challenging the constitutionality of the ACA’s individual mandate which is currently before the U.S. Supreme Court. Pending the U.S. Supreme Court’s decision, the ACA remains in effect, but it is unclear how this decision, and other efforts to repeal or further modify the ACA, particularly given the new administration, will impact the ACA and consequently our business.
Moreover, any such future actions may have significant impact on the reimbursement for healthcare services generally, including reducing significantly the number of individuals who have health insurance that can pay for our products, which could lead our health care provider customers to be more cost conscious. At the same time, certain members of the U.S. Congress have proposed measures that would expand the role of government-sponsored coverage, including single payer or so-called “Medicare-for-All” proposals, which could have far-reaching implications for the healthcare industry if enacted. Such a system could reduce our customers’ revenues, such as Medicare and other public reimbursement rates, on average could be lower than existing commercial health plan reimbursement rates. Even if legislation creating such a single-payer system is not enacted in the near term, continued introduction of legislation promoting a single-payer system by several members of the U.S. Congress could increase uncertainty for our customers and cause them to delay purchases of our products and services. Accordingly, our business and results of operations could therefore be adversely affected by any future federal or state healthcare reform legislation or regulation. In sum, even if we succeed in bringing our new products to market, uncertainties regarding future healthcare policy, legislation and regulation, as well as private market practices, could affect our ability to sell our products in commercially acceptable quantities at profitable prices in certain countries.
We must comply with healthcare “fraud and abuse” laws, and we could face penalties for non-compliance and be excluded from government healthcare programs, which would adversely affect our business, financial condition and results of operations.
Certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights may be applicable to our business. We may be subject to healthcare fraud and abuse regulation and patient privacy regulation by both the federal government and the states in which we conduct our business. The laws and regulations that govern our business operations, products, and technologies, and may affect our ability to operate, include, among others, those listed in “Part I, Item 1. Business—Government Regulation and Other Matters—Postmarket Regulation.”
26
To assist in our compliance efforts, we must adhere to many codes of ethics and conduct regarding our sales and marketing activities in the U.S. and other countries in which we operate. Failure to comply with these laws and regulations could result in criminal liability, significant fines or penalties, negative publicity, and substantial costs and expenses associated with investigation, enforcement activities, and individual settlement agreements that impose a government monitor for a period of several years.
We are subject to the U.S. Foreign Corrupt Practices Act and other anticorruption laws, as well as export control laws, import and customs laws, trade and economic sanctions laws and other laws governing our operations.
Our operations are subject to anti-corruption laws, including the FCPA, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, and other anti-corruption laws that apply in countries where we do business. The FCPA and these other laws generally prohibit us and our employees and intermediaries from authorizing, promising, offering, or providing, directly or indirectly, improper or prohibited payments, or anything else of value, to government officials or other persons to obtain or retain business or gain some other business advantage.
We and those acting on our behalf operate in a number of jurisdictions where companies in the medical device and life science industries are exposed to a high risk of potential FCPA violations associated with sales to healthcare professionals and institutions. We participate in transactions with third parties whose corrupt or illegal activities could potentially subject us to liability under the FCPA or local anti-corruption laws, even if we do not explicitly authorize or have actual knowledge of such activities. In addition, we cannot predict the nature, scope or effect of future regulatory requirements to which our international operations might be subject or the manner in which existing laws might be administered or interpreted. Compliance with the FCPA and these other laws is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, anti-corruption laws present particular challenges in the medical device industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical trials and other work have been deemed to be improper payments to government officials and have led to enforcement actions. We are also subject to other laws and regulations governing our international operations.
There is no assurance that we will be completely effective in ensuring our compliance with all applicable anti-corruption laws, including the FCPA or other legal requirements. If we are not in compliance with the FCPA and other anti-corruption laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, and legal expenses, which could have an adverse impact on our business, financial condition, results of operations and liquidity. Likewise, any investigation of any potential violations of the FCPA and other anti-corruption laws could also have an adverse impact on our reputation, our business, results of operations and financial condition. Further, the failure to comply with laws governing international business practices may result in civil and criminal penalties and suspension or debarment from government contracting.
Any failure to achieve and maintain the high manufacturing standards that our products require may seriously harm our business.
Our products require precise, high-quality manufacturing. Achieving precision and quality control requires skill and diligence by our personnel as well as our vendors. Any failure to achieve and maintain these high manufacturing standards, including the incidence of manufacturing errors, design defects or component failures could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously hurt our business. Despite our very high manufacturing standards, we cannot completely eliminate the risk of errors, defects or failures. If we or our vendors are unable to manufacture our products in accordance with necessary quality standards, or if we are unable to procure additional high-quality manufacturing facilities, our business and results of operations may be negatively affected.
The economic effects of “Brexit” may affect relationships with existing and future customers and could have an adverse impact on our business and operating results.
In June 2016, the United Kingdom held a referendum in which voters approved what is commonly referred to as Brexit, and following protracted negotiations, the United Kingdom withdrew from the European Union on January 31, 2020. A transition period ended on December 31, 2020, during which the United Kingdom and the European Union negotiated the terms of the United Kingdom’s relationship with the European Union going forward, and implemented the European Union-United Kingdom Trade and Cooperation Agreement beginning on January 1, 2021. The effects of and the perceptions as to the impact from the withdrawal of the United Kingdom from the European Union has and may continue to adversely affect business activity and economic and market conditions in the United Kingdom, the Eurozone, and globally, and could contribute to instability in global financial and foreign exchange markets, including volatility in the value of the pound sterling and the euro. In addition, Brexit and the implementation of the European Union-United Kingdom Trade and Cooperation Agreement could lead to additional . long-term political, economic, financial, trade and legal implications and have regulatory impacts applicable to our business globally and specifically in the region. If the United Kingdom were to significantly alter its regulations affecting the medical device industry, we could face significant new costs. It may also be time-consuming and expensive for us to alter our internal operations in order to comply with new regulations.
27
Additionally, as a result of Brexit, the global markets and currencies have been adversely impacted. A potential devaluation of the local currencies of our international buyers relative to the U.S. dollar may impair the purchasing power of our international buyers and could cause international buyers to decrease their participation in our marketplaces or use of our products. Finally, Brexit could lead to legal uncertainty and potentially divergent national laws and regulations as the United Kingdom determines which E.U. laws to replace or replicate, and those laws and regulations may be cumbersome, difficult or costly in terms of compliance. Any of these effects of Brexit, among others, could adversely affect our business, financial condition, operating results and cash flows.
We may undergo an “ownership change” for U.S. federal income tax purposes, which would limit our ability to utilize net operating losses from prior tax years.
If we undergo an “ownership change” for U.S. federal income tax purposes, our ability to utilize net operating loss carry-forwards from prior years to reduce taxable income in future tax years might be limited by the Internal Revenue Code, either by limiting the amount of net operating losses that can be utilized to offset taxable income in a given year, or in total over the entire carry-forward period. Certain changes in the ownership of our common stock may result in an ownership change sufficient to limit the availability of our net operating losses. Net operating losses, foreign tax credits and research and development credits have expiry dates in the U.S. and the ability to fully utilize them will be dependent upon generating taxable income in the future. The potential benefits of net operating losses and other carryforwards may be limited as a result of examinations and audits by the Internal Revenue Service (the “IRS”) and other taxing authorities. We also have net operating loss carry-forwards in other countries outside of the U.S. and our ability to use those losses in the future to offset taxable income could be limited by tax regulations in those countries.
Compliance with and changes in tax laws, including the 2017 U.S. Tax Reform legislation, could materially and adversely impact our financial condition, results of operations and cash flows.
On December 22, 2017, the Tax Cuts and Jobs Act, or Tax Reform Act, was signed into law that significantly revises the Internal Revenue Code of 1986, as amended. The Tax Reform Act contained significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a rate of 21%, effective January 1, 2018, limitation of the deduction for net operating losses to 80% of current year taxable income in respect of net operating losses generated during or after fiscal year 2018 and elimination of net operating loss carrybacks, revisions to the treatment for U.S. federal income tax purposes of foreign earnings, immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits. The U.S. Treasury Department, the IRS and other standard-setting bodies could interpret or issue guidance on how provisions of the Tax Reform Act will be applied or otherwise administered that is different from our interpretation, which may materially affect our results of operations. In addition, the current U.S. presidential administration may implement further changes to U.S. tax policy, including an increase in the U.S. corporate income tax rate. If any or all of these (or similar) proposals are ultimately enacted into law, in whole or in part, they could have a negative impact to the Company’s effective tax rate.
Additionally, the Organization for Economic Co-operation and Development (the “OECD”), the European Commission (the “EC”) and individual taxing jurisdictions where we and our affiliates do business have recently focused on issues related to the taxation of multinational corporations. The OECD has released its comprehensive plan to create an agreed set of international rules for fighting base erosion and profit shifting. In addition, the OECD, the EC and individual countries are examining changes to how taxing rights should be allocated among countries considering the digital economy. As a result, the tax laws in the U.S. and other countries in which we and our affiliates do business could change on a prospective or retroactive basis and any such changes could materially adversely affect our business
The U.S. Treasury Department, the IRS and other standard-setting bodies could interpret or issue guidance on how provisions of the Tax Reform Act will be applied or otherwise administered that is different from our interpretation. Foreign governments may enact tax laws in response to the Tax Reform Act that could result in further changes to global taxation and materially affect our financial position and results of operations. The calculation of tax exposures involves the application of complex tax laws and regulations in many jurisdictions, and there can be no assurance that we will accurately predict the ultimate outcomes of these tax estimates or that issues raised by tax authorities will be resolved at a financial cost that does not exceed our related estimates of our tax provision.
Overall, we are unable to predict what tax changes may be enacted in the future, but U.S. tax changes or changes in how international subsidiaries are taxed, including changes in how existing tax laws are interpreted or enforced, changes to U.S. corporate income tax rates or changes to accounting standards, elections or assertions could have a material adverse effect on our business, liquidity, financial condition, and/or results of operations. The uncertainty surrounding the effect of reforms on our financial results and business could also weaken confidence among investors in our financial condition. This could, in turn, have a materially adverse effect on the price of our common stock.
28
Environmental, health and safety laws, including recent environmental regulatory action regarding medical device sterilization facilities, may result in liabilities, expenses and restrictions on our operations.
Our facilities and operations, including the manufacturing of our biomaterials, are subject to various, comprehensive and frequently changing federal, state, local and foreign laws and regulations regarding environmental protection, hazardous substances and health and safety. These requirements govern, among other things, safe working conditions, laboratory and manufacturing practices, the handling, storage, use and disposal of hazardous materials and wastes and, if any, the associated cleanup of properties affected by discharges, releases or exposure to pollutants, air emissions, wastewater, and occupational and human health and safety, and may adversely affect our business. In particular, the use hazardous substances in connection with our research and development and manufacturing activities exposes us to the risk of accidental injury, contamination or other liability. If our or our suppliers’ operations result in pollution of the environment or expose individuals to hazardous substances, we could be liable for damages, expenses, and fines, and any liability could significantly exceed our insurance coverage and have a material adverse effect on our financial condition. Environmental laws also impose obligations and liability for cleanup of properties affected by hazardous materials, wastes and substance spoils or releases, which can be imposed on the parties generating or disposing of such substances or the operator of the affected property, often without regard to whether the owner or operator knew of, or was responsible for, the presence of hazardous substances. Additionally, increased costs on our suppliers stemming from compliance with new or existing environmental or health safety laws and regulations may adversely affect us, as such laws and regulations could result in operational impacts, including facility shutdowns. Suppliers may also choose to pass compliance costs to us in the form of adjusted pricing.
Recent environmental regulatory action regarding medical device sterilization may adversely impact us in the ways described above, although the outcome of this action currently remains uncertain. Many of our products require sterilization prior to sale, and we contract with third-party sterilizers to perform this service, including ethylene oxide sterilizers. In June 2020, the U.S. Environmental Protection Agency, or the EPA, finalized amendments to national emissions standards to reduce hazardous air pollutants, including emissions of ethylene oxide, by adding requirements for process vents, storage tanks, equipment in ethylene oxide service, and updating requirements for flares controlling ethylene oxide emissions, heat exchange systems, and equipment leaks. Additional regulation to address ethylene oxide emissions at sterilization facilities is expected in 2021, including revisions to the EPA’s national emissions standards for such facilities. The EPA is currently conducting a registration review of ethylene oxide to ensure that it can carry out its intended function without creating unreasonable risks to human health and the environment. EPA regulates ethylene oxide’s use as a sterilant, which is considered an antimicrobial pesticide under the Federal Insecticide, Fungicide, and Rodenticide Act.
Throughout the end of 2019 and 2020, state agencies shut down and/or temporarily suspended operations at ethylene oxide sterilization facilities in Illinois, Michigan, Georgia and Delaware. The EPA stated that it will continue to coordinate with state and local air agencies that have been working to reduce ethylene oxide emissions in their jurisdictions during the ongoing COVID-19 pandemic. Although the EPA’s Office of Inspector General released a report in March 2020 recommending that the EPA provide residents in all communities near 25 high-priority ethylene oxide-emitting facilities with a forum with the EPA or state personnel regarding exposure to ethylene oxide, the EPA Administrator immediately critiqued the report and requested that it be rescinded for appropriate revision. The FDA also voiced concerns that shutdowns may diminish the supply of available sterilization facilities and cause medical device shortages. Certain of the previously shut down facilities were permitted to resume certain operations due to increased emissions controls and/or the need to sterilize protective equipment during the pandemic.
It is currently uncertain to what extent facilities that resumed operations in response to the ongoing COVID-19 pandemic will be shut down again for environmental, health and safety concerns, or whether any additional shut-down facilities will reopen or other facilities will be required to shut down. While the sterilization facilities previously or currently shut down or suspended do not sterilize our products and are not otherwise in our supply chain, and our suppliers are not affected directly by these regulatory actions, increased scrutiny and regulation of ethylene oxide sterilization facilities in the U.S. could create additional costs for our suppliers, who may be required to take steps with respect to their sterilization processes. These costs could, in turn, be passed on to us and adversely affect our business. Also, to the extent we or our contract sterilizers are unable to sterilize our products, whether due to these regulatory or other constraints (such as capacity or availability of materials for sterilization), we may be unable to transition to other contract sterilizers, sterilizer locations or sterilization methods in a timely or cost-effective manner, or at all. Failure to transition our processes due to decreased third-party sterilization capacity could have a materially adverse impact on our results of operations and financial condition.
29
Risks Related to Intellectual Property, Privacy and Security
We own patents, trademarks, trade secrets, copyrights and other intellectual property and know-how that we believe give us a competitive advantage. If we cannot protect our intellectual property, both domestically and internationally, and develop or otherwise acquire additional intellectual property, competition could force us to lower our prices, which could hurt our profitability.
Our intellectual property rights are and will continue to be a critical component of our success. We rely and expect to continue to rely on a combination of intellectual property, including patent, trademark, copyright, trade secret and domain name protection laws, as well as confidentiality agreements with our employees and others, to protect our intellectual property and proprietary rights. If we fail to obtain and maintain adequate intellectual property protection, we may not be able to prevent third parties from using our proprietary technologies or from marketing products that are very similar or identical to ours.
A substantial portion of our intellectual property rights relating to the Impella devices and other products under development is in the form of trade secrets and patents. Unlike patents, trade secrets are only recognized under applicable law if they are kept secret by restricting their disclosure to third parties. We protect our trade secrets and proprietary knowledge in part through confidentiality agreements with employees, consultants and other parties. However, certain consultants and third parties with whom we have business relationships, and to whom in some cases we have disclosed trade secrets and other proprietary knowledge, may also provide services to other parties in the medical device industry, including companies, universities and research organizations that are developing or marketing competing products. In addition, some of our former employees who were aware of certain of our trade secrets and other proprietary knowledge in the course of their employment may seek employment with, and become employed by, our competitors. We cannot be assured that consultants, employees and other third parties with whom we have entered into confidentiality agreements will not breach the terms of such agreements by improperly using or disclosing our trade secrets or other proprietary knowledge, that we will have adequate remedies for any such breach, or that our trade secrets will not become known to or be independently developed by our competitors. The loss of trade secret protection for technologies or know-how relating to our product portfolio and products under development could adversely affect our business and our prospects.
Our business position also depends in part on our ability to maintain and defend our existing patents and obtain, maintain, and defend additional patents and other intellectual property rights. We intend to seek additional patents, but our pending and future patent applications may not result in issued patents or be granted on a timely basis. In addition, issued patents may not contain claims sufficiently broad to protect us against third parties with similar technologies or products or provide us with any competitive advantage, including exclusivity in a particular product area. The scope of our patent claims also may vary between countries, as individual countries have distinctive patent laws. We may be subject to challenges by third parties regarding our intellectual property, including, among others, claims regarding validity, enforceability, scope and effective term. Patent prosecution, related proceedings, and litigation in the U.S. and in other countries may be expensive, time consuming and ultimately unsuccessful. In addition, patents issued by foreign countries may afford less protection than is available under U.S. patent law and may not adequately protect our proprietary information. Our competitors may independently develop proprietary technologies and processes that are the same as or substantially equivalent to ours or design around our patents. Our competition may also hold or obtain intellectual property rights that would threaten our ability to develop or commercialize our product offerings. The expiration of patents on which we rely for protection of key products could diminish our competitive advantage and adversely affect our business and our prospects.
Companies in the medical device industry typically obtain patents and frequently engage in intellectual property litigation. Our products and technologies could infringe on the rights of others. If a third party successfully asserts a claim for infringement against us, we may be liable for substantial damages, be unable to sell products using that technology, or have to seek a license or redesign the related product. These alternatives may be uneconomical or impossible. Intellectual property litigation could be costly, result in product development delays and divert the efforts and attention of management from our business.
For a discussion of our material legal proceedings, including those related to patent matters, as of March 31, 2021, see “Note 16. Commitment and Contingencies – Contingencies” to our consolidated financial statements in this Report, which is incorporated by reference into this item.
If we are found to have violated laws protecting the confidentiality of patient health information, we could be subject to civil or criminal penalties, which could increase our liabilities and harm our reputation or our business.
Our business requires us to use and store personally identifiable information of our customers, vendors, employees and business partners and, in certain instances patients treated with our products in the clinical setting. We are subject to various domestic and international privacy and security regulations, including but not limited to the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) and the GDPR.
HIPAA mandates, among other things, the adoption of uniform standards for the electronic exchange of information in common healthcare transactions, as well as standards relating to the privacy and security of individually identifiable health information, which
30
require the adoption of administrative, physical and technical safeguards to protect such information. Noncompliance with HIPAA requirements can result in administrative, criminal and civil penalties. Many states have enacted comparable laws addressing the privacy and security of health information, some of which are more stringent than HIPAA. In addition, the California Consumer Privacy Act, or the CCPA, which grants expanded rights to access and delete personal information and opt out of certain personal information sharing, among other things, became effective on January 1, 2020 and will be expanded to include additional consumer rights and business obligations when the California Privacy Rights Act (the “CPRA”) goes into effect on January 1, 2023. Additional states have enacted, or are proposing similar, data privacy legislation.
The GDPR is a comprehensive update to the data protection regime in the European Economic Area which imposes requirements relating to, among other things, consent to process personal data of individuals, the information provided to individuals regarding the processing of their personal data, the security and confidentiality of personal data, and notifications in the event of data breaches and use of data by third party processors.
We are also subject to laws and regulations with respect to cross‑border transfers of such data out of certain jurisdictions in which we operate, including the EU. If we are unable to transfer data between and among countries and regions in which we operate, it could affect the manner in which we provide our services or adversely affect our financial results.
Due to the geographic scope of our operations, HIPAA, the CCPA, the GDPR and other privacy and security-related laws and regulations, which are currently in effect or may come into effect, may increase our responsibility and liability in relation to personal data that we process. We may in turn be required to put in place additional mechanisms ensuring compliance with privacy laws and regulations. If we or any of our service providers are found to be in violation of the promulgated patient privacy rules under various regimes, we could be subject to civil or criminal penalties, which could increase our liabilities, harm our reputation, and have a material adverse effect on our business, financial condition and operating results.
Disruptions of critical information systems or material breaches in the security of our systems could harm our business, customer relations and financial condition.
We rely in part on information technology, or IT, to store information, communicate with our business partners, interface with customers, maintain financial accuracy, secure our data and accurately produce our financial statements. If our IT systems do not effectively and securely collect, store, process and report relevant data for the operation of our business, whether due to equipment malfunction or constraints, software deficiencies or human error, our ability to effectively plan, forecast and execute our business plan and comply with applicable laws and regulations would be materially impaired. Any such impairment could have a material adverse effect on our results of operations, financial condition and the timeliness with which we report our operating results.
In the current environment, there are numerous and evolving risks to cybersecurity and privacy, including criminal hackers, hacktivists, state-sponsored intrusions, industrial espionage, employee malfeasance and human or technological error. High-profile security breaches at other companies and in government agencies have increased in recent years, in some cases causing significant harm, and security industry experts and government officials have warned about the risks of hackers and cyber-attacks targeting businesses. Cyber-attacks are generally becoming more sophisticated and frequent. Computer hackers and others routinely attempt to breach the security of technology products, services and systems, and to fraudulently induce employees, customers, or others to disclosure information or unwittingly provide access to systems or data. We have experienced and expect to continue to experience actual or attempted cyber-attacks of our IT systems or networks. To date, none of these actual or attempted cyber-attacks has had a material effect on our operations or financial condition. While we devote significant resources to network security, data encryption and other security measures to protect our systems and data, including our own proprietary information and the confidential and personally identifiable information of our customers, employees, business partners and patients, these measures cannot provide absolute security. The costs to eliminate or alleviate network security problems, bugs, viruses, worms, malicious software programs and security vulnerabilities could be significant, and our efforts to address these problems may not be successful, resulting potentially in the theft, loss, destruction or corruption of information we store electronically, as well as unexpected interruptions, delays or cessation of service, any of which could cause harm to our business operations. Moreover, if a computer security breach or cyber-attack affects our systems or results in the unauthorized release of proprietary or personally identifiable information, our reputation could be materially damaged, our customer confidence could be diminished, and our operations, including technical support for our devices, could be impaired. We would also be exposed to a risk of loss or litigation and potential liability, which could have a material adverse effect on our business, results of operations and financial condition. Any of these may contribute to the loss of customers and may have a material adverse effect on our business.
Risks Related to Our Common Stock
The market price of our common stock is volatile, which has in the past led to and may in the future lead to securities litigation against us. Such litigation may be costly and result in an adverse outcome.
The market price of our common stock has fluctuated widely and may continue to do so. Many factors could cause the market price of our common stock to rise and fall. Some of these factors are:
31
| • | variations in our quarterly results of operations; |
| • | status of regulatory approvals for our products; |
| • | announcements by the FDA and other regulatory authorities relating to our products and their impact on market perception of our product, including short-term impact; |
| • | reputational risk relating to customer reviews of our products; |
| • | introduction of new products by us or our competitors; |
| • | acquisitions or strategic alliances involving us or our competitors; |
| • | changes in healthcare policy or third-party reimbursement practices; |
| • | changes in estimates of our performance or recommendations by securities analysts; |
| • | the hiring or departure of key personnel; |
| • | results of clinical trials of our products; |
| • | notice of a recall or other safety issue that impacts the ability for customers to use our products; |
| • | future sales of shares of common stock in the public market; |
| • | the outcome of currently pending litigation and governmental investigations, or the initiation of additional litigation or government investigations against us; |
| • | and market conditions in the industry, particularly around reimbursement for our products and the economy as a whole. |
In addition, the stock market in general and the market for shares of medical device companies in particular have experienced extreme price and volume fluctuations in recent years. These fluctuations are often unrelated to the operating performance of individual companies. These broad market fluctuations may adversely affect the market price of our common stock. When the market price of a company’s stock drops significantly, stockholders often institute securities class action litigation against that company. For information on our ongoing securities class action, see “Note 16. Commitment and Contingencies” to our consolidated financial statements in this Report, which is incorporated by reference into this item.
We are generally obliged under our bylaws, to the extent permitted under Delaware law, to indemnify our current and former officers who are named as defendants in these types of lawsuits. While a certain amount of insurance coverage is available for expenses or losses associated with these lawsuits, this coverage may not be sufficient. Based on information currently available, we are unable to estimate reasonably a possible loss or range of possible losses, if any, with regard to the current securities class action and shareholder derivative litigation; therefore, no litigation reserve has been recorded in our consolidated balance sheet. Although we plan to defend against the securities class action and shareholder derivative litigation vigorously, there can be no assurances that a favorable final outcome will be obtained. This litigation and other future litigation against us could cause us to incur substantial costs, divert the time and attention of our management and other resources, or otherwise harm our business.
The sale of additional shares of our common stock, the issuance of restricted stock units or the exercise of outstanding options to purchase our common stock, would dilute our stockholders’ ownership interest.
We have historically issued restricted stock units and stock options to acquire our common stock and we expect to continue to issue restricted stock units and stock options to our employees and others in the future. If all outstanding stock options were exercised and all outstanding restricted stock units vested, our stockholders would suffer dilution of their ownership interest. In addition, we have issued from time to time, additional shares of our common stock in connection with acquisitions, public offerings, and other activities. Future issuances of our common stock would also result in a dilution of our stockholders’ ownership interest.
Our certificate of incorporation and Delaware law could make it more difficult for a third-party to acquire us and may prevent our stockholders from realizing a premium on our stock.
Provisions of our certificate of incorporation and Delaware General Corporation Law may make it more difficult for a third-party to acquire us, even if doing so would allow our stockholders to receive a premium over the prevailing market price of our stock. Those provisions of our certificate of incorporation and Delaware law are intended to encourage potential acquirers to negotiate with us and allow our board of directors the opportunity to consider alternative proposals in the interest of maximizing stockholder value. However, such provisions may also discourage acquisition proposals or delay or prevent a change in control, which could negatively affect our stock price.
32
The market value of our common stock could vary significantly based on market perceptions of the status of our product development efforts.
The perception of securities analysts regarding our product development efforts and controlled launches of certain products, particularly with respect to our Impella devices and the OXY-1 System, could significantly affect our stock price. As a result, the market price of our common stock has and could in the future change substantially when we or our competitors make product announcements. Many factors affecting our stock price are industry related and beyond our control.
We have not paid and do not expect to pay dividends and any return on our stockholders’ investment will likely be limited primarily to gains realized based on the value of our common stock.
We have never paid dividends on our common stock and do not anticipate paying dividends on our common stock in the foreseeable future. The payment of dividends on our common stock will depend on our earnings, financial condition and other business and economic factors affecting us at such time as our board of directors may consider relevant. If we do not pay dividends, our common stock may be less valuable because a return on our stockholders’ investment will only occur if our stock price appreciates.
General Risk Factors
We use estimates, make judgments and apply certain methods in measuring the progress of our business in determining our financial results and in applying our accounting policies. As these estimates, judgments and methods change, our assessment of the progress of our business and our results of operations could vary.
The methods, estimates and judgments we use in applying our accounting policies have a significant impact on our results of operations. Such methods, estimates and judgments are, by their nature, subject to risks, complexities, uncertainties and assumptions, and factors may arise over time that may lead us to change our methods, estimates and judgments. Changes in any of our assumptions may cause variation in our financial reporting and may adversely affect our reported financial results.
Revisions to accounting standards, tax laws and financial reporting requirements could result in changes to our standard practices and could require a significant expenditure of time, attention and resources, especially by senior management.
We must follow accounting standards, tax laws and financial reporting requirements set by the governing bodies and lawmakers in the U.S. and in other jurisdictions where we do business. From time to time, these governing bodies and lawmakers implement new and revised rules and laws, which may require changes to our accounting policies, financial reporting, and the related information we file with governing bodies. Implementing mandatory changes may require a significant expenditure of time, attention and resources. It is impossible to completely predict the impact, if any, of future changes to accounting standards and financial reporting requirements.
ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
ITEM 2. | PROPERTIES |
Our corporate offices are located in Danvers, Massachusetts. The locations and uses of our major properties as of March 31, 2021, are listed below:
Location |
| Function |
Danvers, Massachusetts (22 Cherry Hill Drive) | (1) | Corporate headquarters, research and development, regulatory and clinical affairs, manufacturing, administration, marketing, distribution |
Danvers, Massachusetts (24 - 42 Cherry Hill Drive) | (1) | Research and development, distribution, manufacturing, administration |
Halethorpe, Maryland | (2) | Research and development, manufacturing, administration |
Aachen, Germany | (1) | Research and development, regulatory and clinical affairs, manufacturing, administration, marketing, distribution |
Berlin, Germany | (2) | Research and development |
Tokyo, Japan | (2) | Administration, regulatory and clinical affairs, marketing, distribution |
| (1) | Owned properties |
| (2) | Leased property |
33
We believe our properties have been well maintained, are in good operating condition, and provide adequate capacity to support our business operations.
ITEM 3. | LEGAL PROCEEDINGS |
We are from time to time involved in various legal actions, the outcomes of which are not within our complete control and may not be known for prolonged periods of time. For a discussion of our material legal proceedings as of March 31, 2021, see “Note 16. Commitment and Contingencies – Contingencies,” which is incorporated by reference into this item.
ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
34
PART II
ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock is traded on the Nasdaq Global Market under the symbol “ABMD.”
Stockholders
As of May 14, 2021, we had approximately 357 holders of record of our common stock, including Cede & Co., the nominee of the Depository Trust Company. The number of record holders may not be representative of the number of beneficial owners of our common stock, whose shares are held in street name by banks, brokers and other nominees.
Dividends
We have never declared or paid any cash dividends on our common stock and do not anticipate paying any cash dividends on our common stock in the foreseeable future. We anticipate that we will retain all of our future earnings, if any, to support operations and to finance the growth and development of our business. Payment of any future dividends will be at the discretion of our board of directors and will depend upon our financial condition, operating results, cash needs and growth plans.
Issuer Purchases of Equity Securities
In August 2019, our Board of Directors authorized a stock repurchase program for up to $200.0 million of shares of its common stock. Under this stock repurchase program, we are authorized to repurchase shares through open market purchases, privately negotiated transactions or otherwise in accordance with applicable federal securities laws, including through Rule 10b5-1 trading plans and under Rule 10b-18 of the Securities Exchange Act of 1934 (the “Exchange Act”). The stock repurchase program has no time limit and may be suspended for periods or discontinued at any time. We are funding the stock repurchase program with our available cash and marketable securities. Through March 31, 2021, we have repurchased a total 533,336 shares at an aggregate cost of $96.2 million through this stock repurchase program. The remaining authorization under the stock repurchase program was $103.8 million as of March 31, 2021. There were no stock repurchases during our fourth quarter of fiscal year 2021.
35
Performance Graph
The following graph compares the yearly change in the cumulative total stockholder return for our last five fiscal years, based upon the market price of our common stock, with the cumulative total return on a Nasdaq Composite Index (U.S. Companies) and a peer group, the Nasdaq Medical Equipment-SIC Code 3840-3849 Index, which is comprised of medical equipment companies, for that period. The performance graph assumes the investment of $100 as of March 31, 2016 in our common stock, the Nasdaq Composite Index (U.S. Companies) and the peer group index, and the reinvestment of any and all dividends.
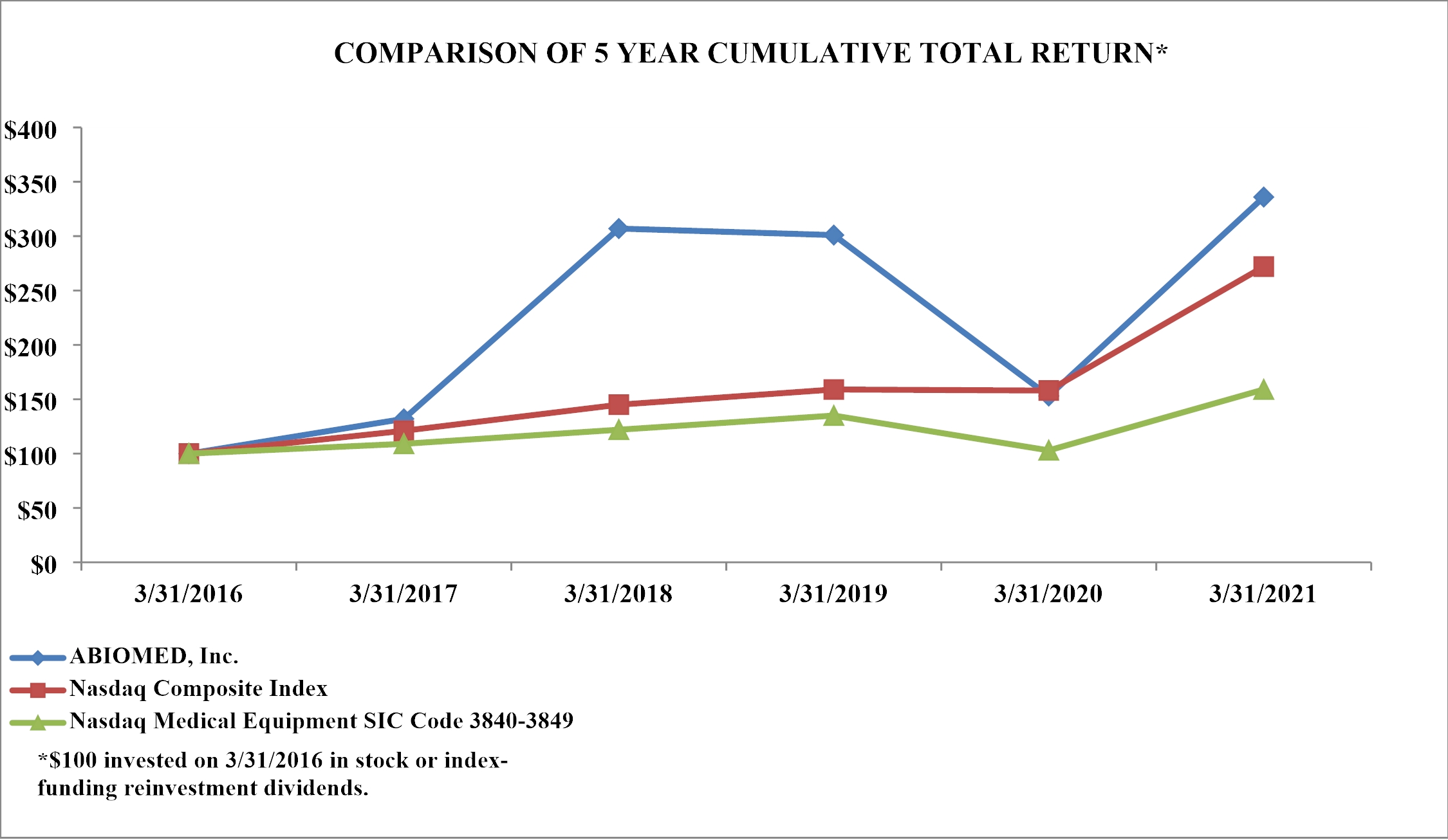
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Cumulative Total Return ($) |
| ||||||||||||||||
| 3/31/2016 |
| 3/31/2017 |
| 3/31/2018 |
| 3/31/2019 |
| 3/31/2020 |
| 3/31/2021 |
| ||||||
ABIOMED, Inc. |
| 100 |
|
| 132 |
|
| 307 |
|
| 301 |
|
| 153 |
|
| 336 |
|
Nasdaq Composite Index |
| 100 |
|
| 121 |
|
| 145 |
|
| 159 |
|
| 158 |
|
| 272 |
|
Nasdaq Medical Equipment SIC Code 3840-3849 |
| 100 |
|
| 109 |
|
| 122 |
|
| 135 |
|
| 103 |
|
| 159 |
|
This graph is not “soliciting material” under Regulation 14A or 14C of the rules promulgated under the Exchange Act, is not deemed filed with the SEC and is not to be incorporated by reference in any of our filings under the Securities Act of 1933, as amended, or the Exchange Act whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
36
ITEM 6. | SELECTED FINANCIAL DATA |
The following tables set forth, for the periods and at the dates indicated, our selected historical consolidated financial data. We have derived the selected consolidated financial data for the fiscal years ended March 31, 2021, 2020 and 2019 and as of March 31, 2021 and 2020, from our consolidated financial statements appearing elsewhere in this Report. We have derived the selected consolidated financial data for the fiscal years ended March 31, 2017 and 2016, and as of March 31, 2018, 2017 and 2016 from our consolidated financial statements not appearing elsewhere in this Report. Our historical results are not necessarily indicative of the results we may achieve in any future period. You should read the following information together with the more detailed information contained in “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the accompanying notes appearing elsewhere in this Report.
SELECTED CONSOLIDATED FINANCIAL DATA
(In thousands, except per share data)
|
| Fiscal Years Ended March 31, |
| ||||||||||||||||||||||
Statement of operations data: | 2021 |
|
| 2020 |
|
| 2019 |
|
| 2018 |
|
| 2017 |
| |||||||||||
Revenue |
| $ |
| 847,522 |
|
| $ |
| 840,883 |
|
| $ |
| 769,432 |
|
| $ |
| 593,749 |
|
| $ |
| 445,304 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of revenue |
|
|
| 161,907 |
|
|
|
| 151,305 |
|
|
|
| 129,567 |
|
|
|
| 98,581 |
|
|
|
| 70,627 |
|
Research and development |
|
|
| 121,875 |
|
|
|
| 98,759 |
|
|
|
| 93,503 |
|
|
|
| 75,297 |
|
|
|
| 66,386 |
|
Selling, general and administrative |
|
|
| 334,183 |
|
|
|
| 341,600 |
|
|
|
| 321,550 |
|
|
|
| 262,734 |
|
|
|
| 218,153 |
|
|
|
|
| 617,965 |
|
|
|
| 591,664 |
|
|
|
| 544,620 |
|
|
|
| 436,612 |
|
|
|
| 355,166 |
|
Income from operations |
|
|
| 229,557 |
|
|
|
| 249,219 |
|
|
|
| 224,812 |
|
|
|
| 157,137 |
|
|
|
| 90,138 |
|
Other income: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Investment income, net |
|
|
| 6,717 |
|
|
|
| 12,167 |
|
|
|
| 8,166 |
|
|
|
| 3,688 |
|
|
|
| 1,554 |
|
Other income (expense), net (1) |
|
|
| 51,946 |
|
|
|
| (4,561 | ) |
|
|
| 30,382 |
|
|
|
| (388 | ) |
|
|
| (349 | ) |
|
|
|
| 58,663 |
|
|
|
| 7,606 |
|
|
|
| 38,548 |
|
|
|
| 3,300 |
|
|
|
| 1,205 |
|
Income before income taxes |
|
|
| 288,220 |
|
|
|
| 256,825 |
|
|
|
| 263,360 |
|
|
|
| 160,437 |
|
|
|
| 91,343 |
|
Income tax provision (2)(3) |
|
|
| 62,695 |
|
|
|
| 53,816 |
|
|
|
| 4,344 |
|
|
|
| 48,267 |
|
|
|
| 39,227 |
|
Net income |
| $ |
| 225,525 |
|
| $ |
| 203,009 |
|
| $ |
| 259,016 |
|
| $ |
| 112,170 |
|
| $ |
| 52,116 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic net income per share |
| $ |
| 5.00 |
|
| $ |
| 4.49 |
|
| $ |
| 5.77 |
|
| $ |
| 2.54 |
|
| $ |
| 1.21 |
|
Basic weighted average shares outstanding |
|
|
| 45,140 |
|
|
|
| 45,179 |
|
|
|
| 44,911 |
|
|
|
| 44,153 |
|
|
|
| 43,238 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Diluted net income per share |
| $ |
| 4.94 |
|
| $ |
| 4.43 |
|
| $ |
| 5.61 |
|
| $ |
| 2.45 |
|
| $ |
| 1.17 |
|
Diluted weighted average shares outstanding |
|
|
| 45,674 |
|
|
|
| 45,816 |
|
|
|
| 46,151 |
|
|
|
| 45,849 |
|
|
|
| 44,658 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance sheet data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
Cash, cash equivalents and marketable securities |
| $ |
| 847,780 |
|
| $ |
| 650,911 |
|
| $ |
| 513,416 |
|
| $ |
| 399,751 |
|
| $ |
| 277,091 |
|
Working capital |
|
|
| 658,996 |
|
|
|
| 503,978 |
|
|
|
| 571,199 |
|
|
|
| 409,589 |
|
|
|
| 257,341 |
|
Total assets |
|
|
| 1,494,359 |
|
|
|
| 1,216,462 |
|
|
|
| 1,054,346 |
|
|
|
| 786,375 |
|
|
|
| 550,414 |
|
Stockholders’ equity |
|
|
| 1,329,675 |
|
|
|
| 1,065,466 |
|
|
|
| 936,890 |
|
|
|
| 689,524 |
|
|
|
| 452,071 |
|
(1) | In fiscal year 2019, we invested $25.0 million in Shockwave Medical, a medical device company. The fair value of this investment as of March 31, 2021 was $38.7 million. We recognized a pre-tax gain of $50.8 million for the year ended March 31, 2021, a pre-tax loss of $0.5 million for the year ended March 31, 2020 and pre-tax gain of $32.0 million for the year ended March 31, 2019 in other (expense) income, net. |
(2) | The income tax provision for fiscal years 2021, 2020 and 2019 included excess tax benefits of $12.1 million, $14.8 million and $69.3 million, respectively. These recognized excess tax benefits resulted from restricted stock units that vested or stock options that were exercised. |
(3) | In fiscal year 2018, we adopted ASU 2016-09 which requires that all excess tax benefits and tax deficiencies related share-based compensation arrangements be recognized as income tax benefit or expense, instead of in stockholders’ equity as previous guidance required. The income tax provision for the years ended March 31, 2021, 2020 and 2019 included excess tax benefits of $12.1 million, $14.8 million and $69.3 million, respectively. These recognized excess tax benefits resulted from restricted stock units that vested or stock options that were exercised. |
37
ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
You should read the following discussion and analysis of our financial condition and results of operations together with our “Selected Financial Data” and the consolidated financial statements and the related notes included elsewhere in “Financial Statements and Supplementary Data.” Some of the information contained in this discussion and analysis or set forth elsewhere in this Report, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties. You should read “Part I, Item 1.A Risk Factors” in this Report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a leading provider of medical devices that provide circulatory support and oxygenation. Our products are designed to enable the heart to rest by improving blood flow and/or provide sufficient oxygenation to those in respiratory failure. We develop, manufacture and market proprietary products that are designed to enable the heart to rest, heal and recover by improving blood flow to the coronary arteries and end-organs and/or temporarily assisting the pumping function of the heart. Our products are used in the cardiac catheterization lab, or cath lab, by interventional cardiologists, the electrophysiology lab, the hybrid lab and in the heart surgery suite by cardiac surgeons. A physician may use our devices for patients who are in need of hemodynamic support prophylactically, urgently or emergently before, during or after angioplasty or heart surgery procedures. We believe that heart recovery is the optimal clinical outcome for a patient experiencing heart failure because it enhances the potential for the patient to go home with their own heart, facilitating the restoration of quality of life. In addition, we believe that, for the care of such patients, heart recovery is often the most cost-effective solution for the healthcare system.
Our strategic focus and the primary driver of our revenue growth is the market penetration of our family of Impella® heart pumps. The Impella device portfolio, which includes the Impella 2.5®, Impella CP®, Impella 5.0®, Impella LD®, Impella 5.5® and Impella RP® devices, has supported thousands of patients worldwide. We expect that most of our product and service revenue in the near future will be from our Impella devices. Our Impella 2.5, Impella CP, Impella 5.0, Impella LD, Impella 5.5 and Impella RP devices have FDA and CE Mark approval which allows us to market these devices in the U.S. and European Union. We expect to continue to make additional PMA supplement submissions for our Impella portfolio of devices for additional indications. Our Impella 2.5, Impella CP and Impella 5.0 devices have regulatory approval from the MHLW in Japan.
In September 2019, the Impella 5.5 device received FDA PMA for safety and efficacy in the therapy of cardiogenic shock for up to 14 days in the U.S. The Impella 5.5 pump was introduced in the U.S. through a controlled rollout at hospitals with established heart recovery protocols beginning in the third quarter of fiscal year 2020. Impella 5.5 received CE marking approval in Europe in April 2018 and was introduced in Europe through a similar controlled rollout.
Acquisition of Breethe, Inc.
We acquired Breethe, a Maryland corporation, on April 24, 2020. Breethe is engaged in research and development of a novel extracorporeal membrane oxygenation (“ECMO”) system that we expect will complement and expand our product portfolio to more comprehensively serve the needs of patients whose lungs can no longer provide sufficient oxygenation, including some patients suffering from cardiogenic shock or respiratory failure. We acquired Breethe for $55.0 million in cash, with additional potential payouts up to a maximum of $55.0 million payable based on the achievement of certain technical, regulatory and commercial milestones. In October 2020, the OXY-1 System received a 510(k) clearance from the FDA for an all-in-one, compact cardiopulmonary bypass system. In the third quarter of fiscal year 2021, we initiated a controlled launch of the OXY-1 System at a limited number of hospitals in the U.S, with expanded U.S. commercial availability expected in fiscal year 2022.
COVID-19 Pandemic
The ongoing COVID-19 pandemic has adversely impacted and is likely to further adversely impact nearly all aspects of our business and markets, including our workforce and the operations of our customers, suppliers, and business partners.
Beginning in mid-March 2020, we experienced a decline in patient utilization in our main markets in the U.S., Europe and Japan as healthcare systems diverted resources to meet the increasing demands of managing COVID-19. Our business was most impacted in the first quarter of fiscal year 2021, in terms of the decline in patients and revenue from the shelter-in-place restrictions in a majority of countries and limitations on procedures in hospitals. We experienced sequential quarterly improvement during the second quarter of fiscal year 2021 as patient procedure volume trends and availability of healthcare resources improved as certain restrictions were lifted and limitations eased during the summer of 2020. During the third quarter of fiscal year 2021, a broad resurgence of COVID-19 cases throughout the U.S. and Europe slowed our continued recovery in patient utilization. Despite these challenges, we experienced increased recovery in patient utilization and revenue during the fourth quarter of fiscal year 2021 due to the high-risk nature of the patient population that are treated with our Impella devices.
38
While the COVID-19 pandemic remains fluid and continues to evolve differently across various geographies, we believe we are likely to continue to experience variable impacts on our business. Hospitals are generally managing the pandemic better currently than they have in the earlier part of the pandemic due to more testing, improved protocols, more experience with the effects of COVID-19 and a greater number of vaccinated caregivers. During these challenging times, our priorities have been to support our clinician partners, protect the well-being of our employees and maintain continuous access to our life-saving technologies while offering front-line in-hospital support. We have established onsite COVID-19 testing for our employees in both Danvers, Massachusetts and Aachen, Germany, set up temperature-taking stations, administered thousands of COVID-19 tests to date and provided personal protective equipment for our employees in order to maintain a safe working environment.
Our proactive testing program has reduced exposure with early detection, reduced employee anxiety and enabled our manufacturing facilities to operate at full capacity in line with local social distancing requirements. We also took proactive actions in the first half of fiscal year 2021 in order to mitigate the business impact of COVID-19 on our financial operations and we continue monitor closely the business impact of COVID-19. Despite the ongoing challenges posed by COVID-19, we continue to invest strategically in engineering, regulatory, clinical trials and manufacturing in order to support our future growth initiatives and sales and marketing activities, with a particular focus on training and education initiatives to drive utilization of our products and recovery awareness for acute heart failure patients.
We continue to closely monitor the impact of COVID-19 on all aspects of our business and geographies, including its impact on our customers, employees, suppliers, vendors, business partners and distribution channels. The full extent to which the pandemic will directly or indirectly impact our business, results of operations and financial condition, including but not limited to sales, expenses, manufacturing, clinical trials, research and development costs, reserves and allowances, fair value measurements, will depend on future developments that are highly uncertain and difficult to predict. These developments include, but are not limited to, the duration and spread of the ongoing COVID-19 pandemic (including new variants of COVID-19), its severity, the actions to contain the virus or address its impact, the timing, distribution, and efficacy of vaccines and other treatments, U.S. and foreign government actions to respond to the reduction in global economic activity, and how quickly and to what extent normal economic and operating conditions can resume.
Critical Accounting Policies and Estimates
We prepare our consolidated financial statements in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”). Preparation of the consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements, as well as the reported amounts of revenue and expenses during the reporting period. Actual results could differ from these estimates. The accounting policies we believe are critical in the preparation of our consolidated financial statements relate to revenue recognition and income taxes. Our significant accounting policies are more fully described in “Note 2. Basis of Preparation and Summary of Significant Accounting Policies” to our consolidated financial statements in this Report.
Revenue Recognition
Revenue is recognized when, or as, obligations under the terms of a contract are satisfied, which occurs when control of the promised products or services is transferred to customers. Revenue is measured as the amount of consideration we expect to receive in exchange for transferring products or services to a customer.
Product revenue is generally recognized when the customer obtains control of our product, which occurs at a point in time, and may be upon shipment or upon delivery based on the contractual shipping terms of the contract.
Service revenue is generally recognized over time as the services are rendered to the customer based on the extent of progress towards completion of the performance obligation. We recognize service revenue over the term of the service contract. Services are expected to be transferred to the customer throughout the term of the contract and we believe recognizing revenue ratably over the term of the contract best depicts the transfer of value to the customer. Revenue generated from preventative maintenance calls is recognized at a point in time when the services are provided to the customer.
Revenue from the sale of products and services are evidenced by either a contract with the customer or a valid purchase order or an invoice which includes all relevant terms of sale. We perform a review of each specific customer's credit worthiness and ability to pay prior to acceptance as a customer. Further, we perform periodic reviews of customers' creditworthiness.
39
Income Taxes
Our provision for income taxes is composed of a current and a deferred portion. The current income tax provision is calculated as the estimated taxes payable or refundable on income tax returns for the current year. The deferred income tax provision is calculated for the estimated future tax effects attributable to temporary differences and net operating loss carryforwards and tax credits using expected tax rates in effect in the years during which the differences are expected to reverse.
Deferred income taxes are recognized for the tax consequences in future years as the differences between the tax bases of assets and liabilities and their financial reporting amounts at each fiscal year end based on enacted tax laws and statutory tax rates applicable to the periods in which the differences are expected to impact taxable income.
We regularly assess our ability to realize our deferred tax assets. Assessing the realization of deferred tax assets requires significant management judgment. We consider whether a valuation allowance is needed on our deferred tax assets by evaluating all positive and negative evidence relative to our ability to recover deferred tax assets, including future reversals of existing taxable temporary differences, projected future taxable income, tax planning strategies and recent financial results.
We recognize and measure uncertain tax positions using a two-step approach. The first step is to evaluate the tax position for recognition by determining if, based on the technical merits, it is more likely than not that the position will be sustained upon audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit at the largest amount that is more likely than not of being realized upon ultimate settlement. We reevaluate these uncertain tax positions on an ongoing basis, when applicable. This evaluation is based on factors including, but not limited to, changes in facts or circumstances, new information and technical insights, and changes in tax laws. Any changes in these factors could result in the recognition of a tax benefit or an additional charge to the tax provision. When applicable, we accrue for the effects of uncertain tax positions and the related potential penalties and interest through income tax expense.
Other Investments
We periodically make investments in medical device companies that focus on heart failure and heart pumps and other medical device technologies. For investments that do not have readily determinable market values, we measure these investments at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for an identical or similar investment. We monitor any events or changes in circumstances that may have a significant effect on the fair value of investments, either due to impairment or based on observable price changes, and make any necessary adjustments.
Recent Accounting Pronouncements
Information regarding recent accounting pronouncements is included in “Note 2. Basis of Preparation and Summary of Significant Accounting Policies” to our consolidated financial statements in this Report.
Results of Operations for the Fiscal Years Ended March 31, 2021 and March 31, 2020 (“fiscal year 2021” and “fiscal year 2020”)
The following table sets forth certain consolidated statements of operations data for the periods indicated as a percentage of total revenues:
|
| Fiscal Years Ended March 31, |
|
| ||||||
|
| 2021 |
|
|
| 2020 |
|
| ||
Revenue |
|
| 100.0 |
| % |
|
| 100.0 |
| % |
Costs and expenses as a percentage of total revenue: |
|
|
|
|
|
|
|
|
|
|
Cost of revenue |
|
| 19.1 |
|
|
|
| 18.0 |
|
|
Research and development |
|
| 14.4 |
|
|
|
| 11.7 |
|
|
Selling, general and administrative |
|
| 39.4 |
|
|
|
| 40.6 |
|
|
Total costs and expenses |
|
| 72.9 |
|
|
|
| 70.4 |
|
|
Income from operations |
|
| 27.1 |
|
|
|
| 29.6 |
|
|
Other income and income tax provision, net |
|
| 0.5 |
|
|
|
| 5.5 |
|
|
Net income as a percentage of total revenue |
|
| 26.6 |
| % |
|
| 24.1 |
| % |
40
Revenue
The following table disaggregates our revenue by products and services:
|
| Fiscal Years Ended March 31, |
| |||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| (in $000's) |
| |||||
Impella product revenue |
| $ | 806,322 |
|
| $ | 806,824 |
|
Service and other revenue |
|
| 41,200 |
|
|
| 34,059 |
|
Total revenue |
| $ | 847,522 |
|
| $ | 840,883 |
|
The following table disaggregates our revenue by geographical location:
|
| Fiscal Years Ended March 31, |
| |||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| (in $000's) |
| |||||
U.S. |
| $ | 691,579 |
|
| $ | 705,409 |
|
Europe |
|
| 105,320 |
|
|
| 94,266 |
|
Japan |
|
| 42,868 |
|
|
| 35,215 |
|
Other International |
|
| 7,755 |
|
|
| 5,993 |
|
Total revenue |
| $ | 847,522 |
|
| $ | 840,883 |
|
Impella product revenue encompasses Impella 2.5, Impella CP, Impella 5.0, Impella LD, Impella 5.5, Impella RP and Impella AIC product sales and related accessories. Service and other revenue represents revenue earned on service maintenance contracts and preventative maintenance calls. The following is a discussion of our revenues for fiscal year 2021.
Total Revenue
Total revenue for fiscal year 2021 increased $6.6 million, or 1%, to $847.5 million from $840.9 million for fiscal year 2020. The main drivers of our Impella product revenue and our service and other revenue are further described below.
The growth in total revenue for the fiscal year 2021 was adversely impacted by lower Impella product revenue in the first quarter of fiscal year 2021 caused by the impact of COVID-19 pandemic in reducing hospital procedures during that time, which was offset by sequential quarterly improvement during the second, third and fourth quarters of fiscal year 2021 as patient procedure volume trends and availability of healthcare resources have improved as certain restrictions were lifted and limitations eased during those periods.
Impella Product Revenue
Impella product revenue for fiscal year 2021 decreased slightly by $0.5 million to $806.3 million from $806.8 million for fiscal year 2020. U.S. Impella product revenue for fiscal year 2021 totaled $655.2 million, down 3% compared to $674.4 million in the prior fiscal year with U.S. patient usage of Impella heart pumps down 13%. Impella product revenue outside the U.S. for fiscal year 2021 totaled $151.1 million, an increase of 14% compared to $132.4 million in the prior fiscal year. Impella product revenue outside of the U.S. increased primarily due to higher utilization in Germany and Japan.
As described above, Impella procedure volumes have varied significantly since the end of March 2020 by geography, and even by hospital, as their physicians have managed the COVID-19 pandemic. Beginning in mid-March 2020, we experienced a decline in patient utilization in the U.S., Europe and Japan as healthcare systems diverted resources to meet the increasing demands of managing COVID-19. Our business was most impacted in the first quarter of fiscal year 2021, in terms of the decline in patients and revenue from the shelter-in-place restrictions in many countries and limitations on procedures in hospitals. We experienced sequential quarterly improvement during the second, third and fourth quarters of fiscal year 2021 as patient procedure volume trends and availability of healthcare resources have improved as certain restrictions were lifted and limitations eased during those periods.
While the COVID-19 pandemic remains fluid and continues to evolve differently across various geographies, we believe we will likely continue to experience variable impacts on our business. While we cannot reliably estimate the extent to which the COVID-19 pandemic may continue to impact patient utilization and revenues of our products, our focus is on increasing patient utilization of our Impella devices in the U.S., our controlled launch of Impella 5.5, primarily in the U.S. and Europe, and our plan to continue growing our business internationally, with a continued focus on Europe and Japan.
41
Service and other revenue
Service and other revenue for fiscal year 2021 increased by $7.1 million, or 21%, to $41.2 million from $34.1 million for fiscal year 2020. The increase in service revenue was primarily due to an increase in preventative maintenance service contracts. We have expanded the number of Impella AIC consoles at many of our existing higher volume customer sites and continue to sell additional consoles to new customer sites. We expect revenue growth for service revenue to be consistent with recent history as most of these using sites in the U.S. have service contracts that normally have three-year terms.
Costs and Expenses
Cost of Revenue
Cost of revenue for fiscal year 2021 increased by $10.6 million, or 7%, to $161.9 million from $151.3 million for fiscal year 2020. Gross margin was 81% for fiscal year 2021 and 82% for fiscal year 2020.
The increase in cost of product revenue and decrease in gross margin was primarily due to increased investment in direct labor and overhead as we expanded our manufacturing capacity of our facilities in the U.S. and Germany, a higher proportion of revenue outside the U.S., sales mix and costs associated with our initial launch of Impella 5.5.
We expect that our ongoing investment in manufacturing capacity and the expansion of our Impella SmartAssist platform and Impella Connect may decrease gross margin in the near future.
Research and Development Expenses
Research and development expenses for fiscal year 2021 increased by $23.1 million, or 23%, to $121.9 million from $98.8 million for fiscal year 2020. The increase in research and development expenses was primarily due to our ongoing product development initiatives relating to our existing and pipeline products, the development of the Impella XR Sheath™, Impella ECP™ , Impella BTR devices and the OXY-1 System with the acquisition of Breethe, the expansion of our engineering organization, continued investment in our clinical trials, most notably the STEMI DTU and PROTECT IV studies, and our focus on clinical and quality initiatives for our products.
We expect research and development expenses to continue to increase with our ongoing efforts to expand our engineering, product development and clinical spending related to our initiatives to improve our existing products and develop new technologies and conduct clinical studies. Research and development expenses can fluctuate with project timing on engineering programs and clinical trials.
Selling, General and Administrative Expenses
Selling, general and administrative expenses for fiscal year 2021 decreased by $7.4 million, or 2%, to $334.2 million from $341.6 million for fiscal year 2020. The decrease in selling, general and administrative expenses was primarily due to lower stock compensation expense due to a lower number of restricted stock units granted and earned and the proactive cost actions taken early in fiscal year 2021 to mitigate the business impact of COVID-19 on our financial operations, including reducing discretionary spending primarily related to travel and marketing expenses, decreases in hiring and eliminating certain non-critical consultants and contractors, partially offset by targeted investments we made in expanding our sales distribution team and advertising. We also received an employee retention credit associated with the Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) to assist companies impacted by COVID-19.
Despite the ongoing challenges posed by COVID-19, we expect to continue to invest in sales and marketing activities, with a particular focus on training and education initiatives to drive utilization of our Impella devices and recovery awareness for acute heart failure patients. We also expect to continue to incur legal expenses for the foreseeable future related to ongoing patent litigation, securities class action litigation and other legal matters discussed in “Note 16. Commitment and Contingencies” to our consolidated financial statements. For the impact of COVID-19 on our business, see “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations—COVID-19 Pandemic.”
We continue to manage our discretionary costs closely in order to mitigate the business impact of COVID-19. Despite the ongoing challenges posed by COVID-19, including the recent global resurgence, we aim to continue to invest strategically in engineering, regulatory, clinical trials and manufacturing in order to support our future growth initiatives and sales and marketing activities, with a particular focus on training and education to drive utilization of our Impella devices and recovery awareness for acute heart failure patients.
Operating Income
Operating income decreased by $19.6 million, to $229.6 million for fiscal year 2021, compared to $249.2 million for fiscal year 2020. Operating margin was 27.1%% for fiscal year 2021 compared to 29.6%% for fiscal year 2020. The decreases in operating margins are primarily related to lower revenues as a result of the impact from the COVID-19 pandemic and increases in research and development expenses with our investments in engineering programs and clinical trials.
42
Other Income (Expense), net
Other (expense) income, net increased by $51.1 million, to $58.7 million for fiscal year 2021, compared to $7.6 million for fiscal year 2020. This increase was primarily due to the recognition of a $50.8 million pre-tax gain from our investment in Shockwave Medical in fiscal year 2021 compared to a $0.5 million loss in fiscal year 2020. We also recorded change in fair value on our investments in other private medical device companies of $1.0 million and $4.7 million for fiscal year 2021 and fiscal year 2020, respectively.
Income Tax Provision
The income tax provision increased by $8.9 million to $62.7 million for fiscal year 2021, compared to $53.8 million for fiscal year 2020. Our effective income tax rate was 21.8% in fiscal year 2021, and 21.0% in fiscal year 2020. The increase in the income tax provision and the effective tax rate for fiscal year 2021 were primarily driven by $12.1 million in excess tax benefits in fiscal year 2021, compared to $14.8 million excess tax benefits in fiscal year 2020.
Net Income
In fiscal year 2021, net income was $225.5 million, or $5.00 per basic share and $4.94 per diluted share. Net income in fiscal year 2021 included excess tax benefits related to stock-based awards of $12.1 million, or $0.27 per basic share and $0.26 per diluted share, and a $38.3 million gain, net of tax, or $0.85 per basic share and $0.84 per diluted share, related to our investment in Shockwave Medical. For more information, see “Note 10. Other Assets—Investments in Shockwave Medical.”
In fiscal year 2020, net income was $203.0 million, or $4.49 per basic share and $4.43 per diluted share. Net income in fiscal year 2020 included excess tax benefits related to stock-based awards of $14.8 million, or $0.33 per basic share and $0.32 per diluted share, and a $0.4 million loss, net of tax, related to our investment in Shockwave Medical.
Results of Operations for the Fiscal Years Ended March 31, 2020 and March 31, 2019
For a comparison of our results of operations for the fiscal years ended March 31, 2020 and March 31, 2019, see “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of our annual report on Form 10-K for the fiscal year ended March 31, 2020 filed with the SEC on May 21, 2020, which comparative information is incorporated by reference in this Report.
Liquidity and Capital Resources
As of March 31, 2021, our total cash, cash equivalents, and short and long-term marketable securities totaled $847.8 million, an increase of $196.9 million compared to $650.9 million as of March 31, 2020. The change in our cash, cash equivalents, and short and long-term marketable securities was primarily due to positive cash flows from operations, offset by cash used for investments in property, equipment and other investments, and cash used for financing activities related to equity activity.
A summary of our cash flow activities is as follows:
|
| For the Year Ended March 31, |
| |||||
|
| 2021 |
|
| 2020 |
| ||
Net cash provided by operating activities |
| $ | 274,578 |
|
| $ | 314,920 |
|
Net cash used for investing activities |
|
| (223,344 | ) |
|
| (125,455 | ) |
Net cash used for financing activities |
|
| (8,067 | ) |
|
| (117,715 | ) |
Effect of exchange rate changes on cash |
|
| (2,798 | ) |
|
| (430 | ) |
Net increase in cash and cash equivalents |
| $ | 40,369 |
|
| $ | 71,320 |
|
For a discussion of our liquidity and capital resources as of and our cash flow activities for the fiscal year ended March 31, 2020, see “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of our annual report on Form 10-K for the fiscal year ended March 31, 2020, filed with the SEC on May 21, 2020, which is incorporated by reference in this Report.
Cash Provided by Operating Activities
For fiscal year 2021, cash provided by operating activities consisted of net income of $225.5 million, plus non-cash items of $66.4 million less cash used for working capital of $17.4 million. Adjustments for non-cash items consisted primarily of $47.0 million
43
of stock-based compensation expense, a change in fair value of our investments in Shockwave Medical and other private medical technology companies of $51.0 million, a $29.4 million change in our deferred tax provision, $24.1 million of depreciation and amortization expense, $8.5 million in inventory write-downs and a change in fair value of contingent consideration of $2.4 million. The decrease in cash from changes in working capital included a $2.5 million decrease in inventory due to the mix of customer demand and production volumes related to our Impella devices and a $12.0 million decrease in accounts payable, accrued expenses and other liabilities offset by a $12.1 million increase in accounts receivable associated with timing and volume of sales and collections and a $5.2 million increase in deferred revenue.
For fiscal year 2020, cash provided by operating activities consisted of net income of $203.0 million, adjusted for non-cash items of $103.9 million plus cash from working capital of $8.0 million. Adjustments for non-cash items consisted primarily of $39.8 million of stock-based compensation expense, a change in fair value of our investments in Shockwave Medical and other private medical technology companies of $5.2 million, a $33.0 million change in our deferred tax provision, $20.4 million of depreciation and amortization expense, $4.2 million in inventory write-downs and a change in fair value of contingent consideration of $0.6 million. The increase in cash from changes in working capital included a $13.2 million increase in inventory to support demand for our Impella devices offset by a $5.6 million decrease in accounts receivable associated with collections activity and a $18.3 million increase in accounts payable and accrued expenses and a $2.8 million increase in deferred revenue.
Cash Used for Investing Activities
For fiscal year 2021, net cash used for investing activities included $159.6 million in purchases (net of maturities) of marketable securities, $52.2 million for our acquisition of Breethe and $53.4 million for the purchase of property and equipment mostly related to the $17.5 million purchase of the building adjacent to our corporate headquarters in Danvers, Massachusetts and continued expansion of manufacturing capacity, office space and research development facilities in Danvers, Massachusetts and Aachen, Germany. We also made $26.1 million of investments in medical technology companies and intangible assets during fiscal year 2021. These amounts were partially offset by $67.9 million in proceeds from the sale of Shockwave Medical securities.
For fiscal year 2020, net cash used for investing activities included $60.5 million in purchases (net of maturities) of marketable securities and other and $44.0 million for the purchase of property and equipment mostly related to the expansion of manufacturing capacity and office space and research and development facilities in Danvers and Aachen and investments in information systems. We also made $21.0 million of investments in medical technology companies and intangible assets during fiscal year 2020.
Capital expenditures for fiscal year 2022 are estimated to range from $40 million to $50 million, including, as part of long-term development of our business, additional capital expenditures for manufacturing capacity and building expansions in our Danvers and Aachen facilities and information systems development projects.
Cash Used for Financing Activities
For fiscal year 2021, net cash used for financing activities included $11.3 million for the repurchase of our common stock and $11.3 million in payments in lieu of issuance of common stock for payroll withholding taxes upon vesting of certain equity awards. These amounts were offset by $9.1 million in proceeds from the exercise of stock options and $5.5 million in proceeds from the issuance of stock under the employee stock purchase plan.
For fiscal year 2020, net cash used for financing activities included $84.9 million for the repurchase of our common stock and $41.7 million in payments in lieu of issuance of common stock for payroll withholding taxes upon vesting of certain equity awards. These amounts were offset by $3.7 million in proceeds from the exercise of stock options and $5.1 million in proceeds from the issuance of stock under the employee stock purchase plan.
Operating Capital and Liquidity Requirements
Our sources of cash liquidity are primarily from existing cash and cash equivalents, marketable securities and cash flows from operations. As of March 31, 2021, our total cash, cash equivalents, and short and long-term marketable securities totaled $847.8 million, an increase of $196.9 million compared to $650.9 million as of March 31, 2020. Marketable securities as of March 31, 2021 consisted of $615.1 million held in funds that invest in U.S. Treasury securities, government-backed securities, corporate debt securities and commercial paper. We generated operating cash flows of $274.6 million and $314.9 million for fiscal years 2021 and 2020, respectively. As of March 31, 2021, we had no debt outstanding. We believe that our sources of liquidity are sufficient to fund the current requirements of working capital, capital expenditures, and other financial commitments for at least the next twelve months.
44
Our primary liquidity requirements are to fund the following: expansion of our commercial and operational infrastructures; expansion of our manufacturing capacity and office space; the procurement and production of inventory to meet customer demand for our Impella devices; creation of new product and business development initiatives; ongoing commercial launch in Japan and expansion into potential new markets; expansion of the utilization of Impella Connect; increased clinical spending for clinical trials such as STEMI DTU and PROTECT IV; legal expenses related to ongoing patent litigation and other legal matters; and payments in lieu of issuance of common stock for payroll withholding taxes upon vesting of certain equity awards and provide for general working capital needs. To date, we have primarily funded our operations through product sales and the sale of equity securities.
Our liquidity is influenced by our ability to sell our products in a competitive industry and our customers’ ability to pay for our products. Factors that may affect liquidity primarily include our ability to penetrate the market for our products, our ability to maintain or reduce the length of the selling cycle for our products, our capital expenditures, and our ability to collect cash from customers after our products are sold. We also expect to continue to incur legal expenses for the foreseeable future related to ongoing patent litigation and other legal matters. We continue to review our short-term and long-term cash needs on a regular basis.
We also took proactive actions in the first half of fiscal year 2021 in order to mitigate the business impact of COVID-19 on our financial operations and we continue to manage discretionary costs closely in order to mitigate the business impact of COVID-19. Despite the ongoing challenges posed by COVID-19, including the recent global resurgence, we continue to invest strategically in engineering, regulatory, clinical trials and manufacturing in order to support our future growth initiatives and sales and marketing activities, with a particular focus on training and education initiatives to drive utilization of our Impella devices and recovery awareness for acute heart failure patients. We continue to closely monitor the impact of COVID-19 on all aspects of our business and geographies, including its impact on our customers, employees, suppliers, vendors, business partners and distribution channels.
Off-Balance Sheet Arrangements
We had no off-balance sheet arrangements or guarantees of third-party obligations during the periods presented. An “off-balance sheet arrangement” generally entails a transaction, agreement or other contractual arrangement to which an entity unconsolidated with us is a party under which we have any obligation arising under a guarantee contract, derivative instrument or variable interest or a retained or contingent interest in assets transferred to such entity or similar arrangement that serves as credit, liquidity or market risk support for such assets.
ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Our business and financial results are affected by fluctuations in world financial markets, including changes in currency exchange rates and interest rates. We manage these risks through a combination of normal operating and financing activities.
Investment and Interest Rate Risk
Our exposure to market risk for changes in interest rates relates primarily to our marketable securities portfolio. Our investment strategy is focused on preserving capital and supporting our liquidity requirements, while earning a reasonable market return. We invest in a variety of U.S. government and agency securities, corporate debt securities and commercial paper. The market value of our marketable securities may decline if current market interest rates rise. The fair value on marketable securities as of March 31, 2021 was $615.1 million. If market interest rates were to increase immediately and uniformly by 10% from levels as of March 31, 2021, we believe the decline in fair market value of our investment portfolio would be immaterial. Our marketable securities are recorded at fair value, and gains or losses from these securities are recognized within other comprehensive income as they occur.
Foreign Currency Exchange Rate Risk
We have foreign currency exposure to exchange rate fluctuations and particularly with respect to the Euro, British pound sterling, Japanese yen, Singapore dollar and Australian dollar. Therefore, our investment in our subsidiaries is sensitive to fluctuations in currency exchange rates. The effect of a change in currency exchange rates on our net investment in international subsidiaries is reflected in the accumulated other comprehensive income component of stockholders’ equity. If foreign exchange rates for our international subsidiaries were to have depreciated immediately and uniformly by 10% relative to the U.S. dollar from levels as of March 31, 2021, the result would have been a reduction of stockholders’ equity of approximately $21.1 million.
45
As discussed in “Note 5. Financial Instruments” to our consolidated financial statements, we have an intercompany loan agreement with our German subsidiary, Abiomed Europe GMBH. In conjunction with this intercompany loan agreement, we entered into a cross-currency swap agreement to convert the Euro denominated intercompany loan into U.S. dollars. The objective of this cross-currency swap is to hedge the variability of cash flows related to the forecasted interest and principal payments on the Euro denominated fixed rate loan against changes in the exchange rate between the U.S. dollar and the Euro. We use such a foreign-exchange-related derivative instrument to manage our exposure related to changes in the exchange rate on our intercompany loan. We do not enter into derivative instruments for any purpose other than for the cash flow hedge described above.
Credit Risk
In the normal course of business, we provide credit to our customers in the health care industry, perform credit evaluations of these customers, and maintain allowances for potential credit losses, which have historically been adequate compared to actual losses. In fiscal year 2021, we had no customers that represented 10% or more of our total revenue or accounts receivable. Credit is extended based on an evaluation of a customer’s historical financial condition and generally collateral is not required. Our history of credit losses has not been significant and we maintain an allowance for doubtful accounts based on our assessment of the collectability of accounts receivable. Accounts receivable are geographically dispersed, primarily throughout the U.S., as well as in Europe and other foreign countries where formal distributor agreements exist. Our exposure to credit losses may increase if our customers are adversely affected by changes in healthcare laws, coverage, and reimbursement, economic pressures or uncertainty associated with local or global economic recessions, disruption associated with the current COVID-19 pandemic, or other customer-specific factors.
Other Investment Risk
We are exposed to investment risks related to changes in the underlying financial condition and credit capacity of certain of our other investments. We periodically make investments in medical device companies that focus on heart failure and heart pump technologies. We monitor any events or changes in circumstances that may have a significant effect on the fair value of our other investments, either due to impairment or based on observable price changes, and make any necessary adjustments. Should these companies experience a decline in financial condition or credit capacity, or fail to meet certain development milestones, a decline in the investments’ values may occur, resulting in unrealized or realized losses. The aggregate carrying amount of our other investments was $101.6 million and $94.4 million as of March 31, 2021 and 2020, respectively, and is classified within other assets on our consolidated balance sheets. In fiscal year 2019, we invested $25.0 million in medical device company Shockwave Medical. The fair value of this investment as of March 31, 2021 was $38.7 million and we recognized a pre-tax gain of $50.8 million in other income (expense), net. Also included in other income (expense) is a change in fair value on portfolio investment of $1.0 million relating to certain of our investments in other private medical device companies.
ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The information required by this item is incorporated by reference from the discussion under the heading “Part IV, Item 15. Exhibits, Financial Statement Schedules” of this Report.
ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
46
ITEM 9A. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) and Rule 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, as of March 31, 2021. Based on this evaluation, our principal executive officer and principal financial officer concluded that, as of March 31, 2021, these disclosure controls and procedures were effective to provide reasonable assurance that information required to be disclosed by us, including our consolidated subsidiaries, in reports that we file or submit under the Exchange Act, is recorded, processed, summarized and reported, within the time periods specified in the Commission’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Act is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
Evaluation of Changes in Internal Control over Financial Reporting
During the fourth quarter of our fiscal year ended March 31, 2021, there were no changes in our internal control over financial reporting that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting. We have not experienced any material impact to our internal control over financial reporting despite the fact that certain of our employees have been working remotely due to the COVID-19 pandemic. We are continually monitoring and assessing the potential impact of COVID-19 on our internal control over financial reporting to minimize the impact on their design and operating effectiveness.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f) and Rule 15d-15(f). Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we assessed the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our assessment under the framework in Internal Control—Integrated Framework (2013), our management concluded that our internal control over financial reporting was effective as of March 31, 2021.
Important Considerations
Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Deloitte & Touche LLP, an independent registered public accounting firm that audited our financial statements for the fiscal year ended March 31, 2021, included in this Report, has issued an attestation report on the effectiveness of our internal control over financial reporting. This Report is set forth below:
47
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of ABIOMED, Inc.
Opinion on Internal Control over Financial Reporting
We have audited the internal control over financial reporting of ABIOMED, Inc. and subsidiaries (the “Company”) as of March 31, 2021, based on criteria established in Internal Control Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of March 31, 2021, based on criteria established in Internal Control — Integrated Framework (2013) issued by COSO.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the consolidated financial statements as of and for the year ended March 31, 2021, of the Company and our report dated May 21, 2021, expressed an unqualified opinion on those financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying "Management’s Report on Internal Control over Financial Reporting." Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Deloitte & Touche LLP
Boston, Massachusetts
May 21, 2021
48
ITEM 9B. | OTHER INFORMATION |
Not applicable.
49
PART III
ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by Item 10 is incorporated by reference herein from our definitive proxy statement which will be filed no later than 120 days after the close of the fiscal year covered by this Report.
ITEM 11. | EXECUTIVE COMPENSATION |
The information required by Item 11 is incorporated by reference herein from our definitive proxy statement which will be filed no later than 120 days after the close of the fiscal year covered by this Report.
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by Item 12 is incorporated by reference herein from our definitive proxy statement which will be filed no later than 120 days after the close of the fiscal year covered by this Report.
ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by Item 13 is incorporated by reference herein from our definitive proxy statement which will be filed no later than 120 days after the close of the fiscal year covered by this Report.
ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by Item 14 is incorporated by reference herein from our definitive proxy statement which will be filed no later than 120 days after the close of the fiscal year covered by this Report.
50
PART IV
ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
(a) | The following documents are filed as part of this report: |
| (1) | The following financial statements are attached hereto. |
| (2) | Consolidated financial statement schedule |
Information is contained within “Note 6. Accounts Receivable” to our consolidated financial statements in this report.
| (3) | Exhibits |
EXHIBIT INDEX
Exhibit No. |
| Description |
| Filed with this Form 10-K |
| Incorporated by Reference | ||||
|
|
| Form |
| Filing Date |
| Exhibit No. | |||
|
|
|
|
|
|
|
|
|
|
|
3.1 |
|
|
|
| S-3 |
| September 29, 1997 |
| 3.1 | |
|
|
|
|
|
|
|
|
|
|
|
3.2 |
| Amended & Restated By-Laws, as Amended and Restated February 4, 2020 |
|
|
| 10-K (File No. 001-09585) |
| May 21, 2020 |
| 3.2 |
|
|
|
|
|
|
|
|
|
|
|
3.3 |
|
|
|
| 8-K (File No. 001-09585) |
| March 21, 2007 |
| 3.4 | |
|
|
|
|
|
|
|
|
|
|
|
4.1P |
| Specimen Certificate of common stock |
|
|
| S-1 |
| June 5, 1987 |
| 4.1 |
|
|
|
|
|
|
|
|
|
|
|
4.2 |
|
|
|
| 10-K (File No. 001-09585) |
| May 21, 2020 |
| 4.2 | |
|
|
|
|
|
|
|
|
|
|
|
10.1*P |
| Form – Indemnification Agreement for Directors and Officers |
|
|
| S-1 |
| June 5, 1987 |
| 10.13 |
|
|
|
|
|
|
|
|
|
|
|
10.2* |
| Form – Employment, Nondisclosure and Non- Competition Agreement |
|
|
| 10-K (File No. 001-09585) |
| May 24, 2018 |
| 10.28 |
|
|
|
|
|
|
|
|
|
|
|
10.3* |
|
|
|
| 8-K (File No. 001-09585) |
| August 18, 2008 |
| 10.4 | |
|
|
|
|
|
|
|
|
|
|
|
10.4* |
| 1988 Employee Stock Purchase Plan, as Amended and Restated February 5, 2019 |
|
|
| 10-K (File No. 001-09585) |
| May 23, 2019 |
| 10.6 |
|
|
|
|
|
|
|
|
|
|
|
10.5* |
|
|
|
| Sch. 14A (File No. 001-09585) |
| June 22, 2018 |
| Appendix A | |
|
|
|
|
|
|
|
|
|
|
|
10.6* |
| Form –Time-Based RSU Agreement (Non-Employee Director) under the 2015 Omnibus Incentive Plan |
|
|
| 10-Q (File No. 001-09585) |
| February 5, 2016 |
| 10.4 |
|
|
|
|
|
|
|
|
|
|
|
10.7* |
| Form – Time-Based RSU Agreement (Field Employee) under the 2015 Omnibus Incentive Plan |
|
|
| 10-K (File No. 001-09585) |
| May 25, 2017 |
| 10.18 |
|
|
|
|
|
|
|
|
|
|
|
10.8* |
| Form – Performance-Based RSU Agreement (Employee) under the 2015 Omnibus Incentive Plan |
|
|
| 10-K (File No. 001-09585) |
| May 25, 2017 |
| 10.19 |
51
Exhibit No. |
| Description |
| Filed with this Form 10-K |
| Incorporated by Reference | ||||
|
|
| Form |
| Filing Date |
| Exhibit No. | |||
|
|
|
|
|
|
|
|
|
|
|
10.9* |
| Form – Time-Based Stock Option Agreement (Employee) under the 2015 Omnibus Incentive Plan |
|
|
| 10-K (File No. 001-09585) |
| May 25, 2017 |
| 10.43 |
|
|
|
|
|
|
|
|
|
|
|
10.10* |
|
|
|
| 10-Q (File No. 001-09585) |
| February 5, 2016 |
| 10.7 | |
|
|
|
|
|
|
|
|
|
|
|
10.11* |
|
|
|
| 10-Q (File No. 001-09585) |
| November 6, 2020 |
| 10.1 | |
|
|
|
|
|
|
|
|
|
|
|
10.12* |
|
|
|
| 10-Q (File No. 001-09585) |
| November 6, 2020 |
| 10.2 | |
|
|
|
|
|
|
|
|
|
|
|
10.13* |
|
|
|
| 10-Q (File No. 001-09585) |
| November 6, 2020 |
| 10.3 | |
|
|
|
|
|
|
|
|
|
|
|
10.14* |
|
|
|
| 10-Q (File No. 001-09585) |
| February 4, 2021 |
| 10.1 | |
|
|
|
|
|
|
|
|
|
|
|
10.15* |
|
|
|
| 10-Q (File No. 001-09585) |
| February 4, 2021 |
| 10.2 | |
|
|
|
|
|
|
|
|
|
|
|
10.16* |
|
|
|
| 10-Q (File No. 001-09585) |
| August 9, 2004 |
| 10.10 | |
|
|
|
|
|
|
|
|
|
|
|
10.17* |
| Amendment to Employment Agreement with Michael R. Minogue dated December 31, 2008 |
|
|
| 10-Q (File No. 001-09585) |
| February 9, 2009 |
| 10.3 |
|
|
|
|
|
|
|
|
|
|
|
10.18* |
| Amendment to Change in Control Agreement with Michael R. Minogue dated December 31, 2008 |
|
|
| 10-Q (File No. 001-09585) |
| February 9, 2009 |
| 10.4 |
|
|
|
|
|
|
|
|
|
|
|
10.19* |
|
|
|
| 10-Q (File No. 001-09585) |
| August 9, 2007 |
| 10.1 | |
|
|
|
|
|
|
|
|
|
|
|
10.20* |
|
|
|
| 10-K (File No. 001-09585) |
| May 24, 2018 |
| 10.43 | |
10.21* |
| Change of Control Severance Agreement between ABIOMED, Inc. and Todd Trapp dated April 6, 2018 |
|
|
| 10-K (File No. 001-09585) |
| May 24, 2018 |
| 10.44 |
|
|
|
|
|
|
|
|
|
|
|
10.22* |
|
| X |
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
10.23* |
| Change of Control Severance Agreement between ABIOMED, Inc and Marc A. Began dated November 1, 2018 |
| X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.24* |
|
| X |
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
21.1 |
|
| X |
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
23.1 |
| Consent of Deloitte & Touche LLP, independent registered public accounting firm |
| X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.1 |
| Rule 13a—14(a)/15d—14(a) certification of principal executive officer. |
| X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
52
Exhibit No. |
| Description |
| Filed with this Form 10-K |
| Incorporated by Reference | ||||
|
|
| Form |
| Filing Date |
| Exhibit No. | |||
31.2 |
| Rule 13a—14(a)/15d—14(a) certification of principal accounting officer |
| X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.1 |
|
| X |
|
|
|
|
|
| |
53
Exhibit No. |
| Description |
| Filed with this Form 10-K |
| Incorporated by Reference | ||||
|
|
| Form |
| Filing Date |
| Exhibit No. | |||
|
|
|
|
|
|
|
|
|
|
|
101 |
| The following financial information from the ABIOMED, Inc. Annual Report on Form 10-K for the fiscal year ended March 31, 2021, formatted in inline Extensible Business Reporting Language (XBRL): (i) Consolidated Balance Sheets as of March 31, 2021 and 2020; (ii) Consolidated Statements of Operations for the fiscal years ended March 31, 2021, 2020 and 2019; (iii) Consolidated Statements of Comprehensive Income for the fiscal years ended March 31, 2021, 2020 and 2019; (iv) Consolidated Statements of Stockholders’ Equity for the fiscal years ended March 2021, 2020 and 2019; (v) Consolidated Statements of Cash Flows for the fiscal years ended March 31, 2021, 2020 and 2019; and (vi) Notes to Consolidated Financial Statements. |
| X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
104 |
| Cover page from the ABIOMED, Inc. Annual Report on Form 10-K for the year ended March 31, 2021, formatted in inline XBRL and contained in Exhibit 101 |
| X |
|
|
|
|
|
|
* | Management contract or compensatory plan, contract or arrangement. |
P | Exhibit filed by paper |
Item 16.Form 10-K Summary.
None.
54
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
| ABIOMED, Inc. | ||
|
|
|
|
|
Dated: May 21, 2021 |
| By |
| /s/ TODD A. TRAPP |
|
|
|
| Todd A. Trapp |
|
|
|
| Vice President, Chief Financial Officer |
|
|
|
| (Principal Financial Officer) |
|
|
|
|
|
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
SIGNATURE |
| TITLE |
| DATE |
|
|
|
|
|
/s/ MICHAEL R. MINOGUE |
| Chairman, Director, President and Chief Executive Officer |
| May 21, 2021 |
Michael R. Minogue |
| (Principal Executive Officer) |
|
|
|
|
|
|
|
/s/ TODD A. TRAPP |
| Vice President, Chief Financial Officer |
| May 21, 2021 |
Todd A. Trapp |
| (Principal Financial Officer) |
|
|
|
|
|
|
|
/s/ DOROTHY E. PUHY |
| Director |
| May 21, 2021 |
Dorothy E. Puhy |
|
|
|
|
|
|
|
|
|
/s/ JEANNINE M. RIVET |
| Director |
| May 21, 2021 |
Jeannine M. Rivet |
|
|
|
|
|
|
|
|
|
/s/ ERIC A. ROSE |
| Director |
| May 21, 2021 |
Eric A. Rose |
|
|
|
|
|
|
|
|
|
/s/ MARTIN P. SUTTER |
| Director |
| May 21, 2021 |
Martin P. Sutter |
|
|
|
|
|
|
|
|
|
/s/ PAUL G. THOMAS |
| Director |
| May 21, 2021 |
Paul G. Thomas |
|
|
|
|
|
|
|
|
|
/s/ CHRIS D. VAN GORDER |
| Director |
| May 21, 2021 |
Chris D. Van Gorder |
|
|
|
|
|
|
|
|
|
/s/ MYRON L. ROLLE |
| Director |
| May 21, 2021 |
Myron L. Rolle |
|
|
|
|
|
|
|
|
|
/s/ PAULA A. JOHNSON |
| Director |
| May 21, 2021 |
Paula A. Johnson |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
55
ABIOMED, INC.
Consolidated Financial Statements
Index
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of ABIOMED, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of ABIOMED, Inc. and subsidiaries (the “Company”) as of March 31, 2021 and 2020, the related consolidated statements of operations, comprehensive income, stockholders' equity, and cash flows, for each of the three years in the period ended March 31, 2021, and the related notes (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of March 31, 2021 and 2020, and the results of its operations and its cash flows for each of the three years in the period ended March 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company's internal control over financial reporting as of March 31, 2021, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated May 21, 2021, expressed an unqualified opinion on the Company's internal control over financial reporting.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below arises from the current-period audit of the financial statements that was communicated or required to be communicated to the audit committee and that (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Valuation of investments in medical device companies – Refer to Note 10, Other Assets
Critical Audit Matter Description
At March 31, 2021, included with the Company’s other non-current assets were $63.0 million of investments in privately-held medical device companies. As these investments do not have readily determinable market values, the Company typically measures these investments at cost less any impairment, adjusted for observable price changes in orderly transactions for an identical or similar investment.
We identified the valuation of these investments as a critical audit matter because of the significant judgement management uses to estimate the investment value. Auditing the Company’s investments in privately held companies is challenging due to the subjectivity in assessing whether observable price changes have occurred for investments that are identical or similar to the investment the Company holds, and in assessing whether an investment is impaired.
F-2
How the Critical Audit Matter Was Addressed in the Audit
Our audit procedures related to the valuation of investments in privately held medical device companies included the following, among others:
| • | We tested the effectiveness of internal controls over the assessment of whether an observable price change in an orderly transaction was for an identical or similar investment, and over the evaluation of whether investments were impaired. |
| • | We considered the appropriateness of the Company’s application of accounting policy, by obtaining and reviewing the Company’s analysis, and comparing to the requirements of accounting principles generally accepted in the United States. |
| • | We tested the mathematical accuracy of the Company’s calculation of the carrying value of privately held investments, including consideration of any related impairments. |
| • | We evaluated, on a sample basis, the accounting conclusions reached by the Company as to whether any observable transactions had occurred that were identical or similar in nature through reading of the Company’s available financial and other information regarding the investee and through public searches for corroborating or contradictory information. Further, for selected investments, we evaluated the Company’s impairment conclusions considering this internal and external information. |
/s/ Deloitte & Touche LLP
Boston, Massachusetts
May 21, 2021
We have served as the Company's auditor since fiscal 2007.
F-3
ABIOMED, INC. AND SUBSIDIARIES
Consolidated Balance Sheets
(in thousands, except share data)
|
| March 31, 2021 |
|
| March 31, 2020 |
| ||
ASSETS |
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
| $ | 232,710 |
|
| $ | 192,341 |
|
Short-term marketable securities |
|
| 350,985 |
|
|
| 250,775 |
|
Accounts receivable, net |
|
| 97,179 |
|
|
| 84,650 |
|
Inventories |
|
| 81,059 |
|
|
| 90,088 |
|
Prepaid expenses and other current assets |
|
| 26,032 |
|
|
| 18,009 |
|
Total current assets |
|
| 787,965 |
|
|
| 635,863 |
|
Long-term marketable securities |
|
| 264,085 |
|
|
| 207,795 |
|
Property and equipment, net |
|
| 197,129 |
|
|
| 164,931 |
|
Goodwill |
|
| 78,568 |
|
|
| 31,969 |
|
Other intangibles, net |
|
| 42,150 |
|
|
| 14,913 |
|
Deferred tax assets |
|
| 11,380 |
|
|
| 43,336 |
|
Other assets |
|
| 113,082 |
|
|
| 117,655 |
|
Total assets |
| $ | 1,494,359 |
|
| $ | 1,216,462 |
|
LIABILITIES AND STOCKHOLDERS' EQUITY |
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Accounts payable |
| $ | 34,842 |
|
| $ | 32,774 |
|
Accrued expenses |
|
| 66,046 |
|
|
| 75,107 |
|
Deferred revenue |
|
| 24,322 |
|
|
| 19,147 |
|
Other current liabilities |
|
| 3,759 |
|
|
| 4,857 |
|
Total current liabilities |
|
| 128,969 |
|
|
| 131,885 |
|
Other long-term liabilities |
|
| 10,162 |
|
|
| 9,305 |
|
Contingent consideration |
|
| 24,706 |
|
|
| 9,000 |
|
Deferred tax liabilities |
|
| 847 |
|
|
| 806 |
|
Total liabilities |
|
| 164,684 |
|
|
| 150,996 |
|
Commitments and contingencies (Note 16) |
|
|
|
|
|
|
|
|
Stockholders' equity: |
|
|
|
|
|
|
|
|
Class B Preferred Stock, $.01 par value |
|
| — |
|
|
| — |
|
Authorized - 1,000,000 shares; Issued and outstanding - 0ne |
|
|
|
|
|
|
|
|
Common stock, $.01 par value |
|
| 453 |
|
|
| 451 |
|
Authorized - 100,000,000 shares; Issued 47,929,402 shares as of March 31, 2021 and 47,542,061 shares as of March 31, 2020 |
|
|
|
|
|
|
|
|
Outstanding 45,270,948 shares as of March 31, 2021 and 45,008,687 shares as of March 31, 2020 |
|
|
|
|
|
|
|
|
Additional paid in capital |
|
| 800,690 |
|
|
| 739,133 |
|
Retained earnings |
|
| 828,007 |
|
|
| 602,482 |
|
Treasury stock at cost 2,658,454 shares as of March 31, 2021 and 2,533,374 shares as of March 31, 2020 |
|
| (288,030 | ) |
|
| (265,411 | ) |
Accumulated other comprehensive loss |
|
| (11,445 | ) |
|
| (11,189 | ) |
Total stockholders' equity |
|
| 1,329,675 |
|
|
| 1,065,466 |
|
Total liabilities and stockholders' equity |
| $ | 1,494,359 |
|
| $ | 1,216,462 |
|
The accompanying notes are an integral part of the consolidated financial statements.
F-4
ABIOMED, INC. AND SUBSIDIARIES
Consolidated Statements of Operations
(in thousands, except per share data)
|
| Fiscal Years Ended March 31, |
| |||||||||
|
| 2021 |
|
| 2020 |
|
| 2019 |
| |||
Revenue |
| $ | 847,522 |
|
| $ | 840,883 |
|
| $ | 769,432 |
|
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
Cost of revenue |
|
| 161,907 |
|
|
| 151,305 |
|
|
| 129,567 |
|
Research and development |
|
| 121,875 |
|
|
| 98,759 |
|
|
| 93,503 |
|
Selling, general and administrative |
|
| 334,183 |
|
|
| 341,600 |
|
|
| 321,550 |
|
|
|
| 617,965 |
|
|
| 591,664 |
|
|
| 544,620 |
|
Income from operations |
|
| 229,557 |
|
|
| 249,219 |
|
|
| 224,812 |
|
Other income: |
|
|
|
|
|
|
|
|
|
|
|
|
Investment income, net |
|
| 6,717 |
|
|
| 12,167 |
|
|
| 8,166 |
|
Other income (expense), net |
|
| 51,946 |
|
|
| (4,561 | ) |
|
| 30,382 |
|
|
|
| 58,663 |
|
|
| 7,606 |
|
|
| 38,548 |
|
Income before income taxes |
|
| 288,220 |
|
|
| 256,825 |
|
|
| 263,360 |
|
Income tax provision |
|
| 62,695 |
|
|
| 53,816 |
|
|
| 4,344 |
|
Net income |
| $ | 225,525 |
|
| $ | 203,009 |
|
| $ | 259,016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic net income per share |
| $ | 5.00 |
|
| $ | 4.49 |
|
| $ | 5.77 |
|
Basic weighted average shares outstanding |
|
| 45,140 |
|
|
| 45,179 |
|
|
| 44,911 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Diluted net income per share |
| $ | 4.94 |
|
| $ | 4.43 |
|
| $ | 5.61 |
|
Diluted weighted average shares outstanding |
|
| 45,674 |
|
|
| 45,816 |
|
|
| 46,151 |
|
The accompanying notes are an integral part of the consolidated financial statements.
F-5
ABIOMED, INC. AND SUBSIDIARIES
Consolidated Statements of Comprehensive Income
(in thousands)
|
| Fiscal Years Ended March 31, |
| |||||||||
|
| 2021 |
|
| 2020 |
|
| 2019 |
| |||
Net income |
| $ | 225,525 |
|
| $ | 203,009 |
|
| $ | 259,016 |
|
Other comprehensive (loss) income: |
|
|
|
|
|
|
|
|
|
|
|
|
Foreign currency translation gain (loss) |
|
| 2,142 |
|
|
| (1,832 | ) |
|
| (11,431 | ) |
Unrealized (loss) gain on derivative instrument |
|
| (2,095 | ) |
|
| 3,999 |
|
|
| — |
|
Net unrealized (loss) gain on marketable securities |
|
| (303 | ) |
|
| 1,333 |
|
|
| 946 |
|
Other comprehensive (loss) income |
|
| (256 | ) |
|
| 3,500 |
|
|
| (10,485 | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive income |
| $ | 225,269 |
|
| $ | 206,509 |
|
| $ | 248,531 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of the consolidated financial statements.
F-6
ABIOMED, INC. AND SUBSIDIARIES
Consolidated Statements of Stockholders’ Equity
(dollars in thousands)
|
| Common Stock |
|
| Treasury Stock |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||
|
| Number of Shares |
|
| Par Value |
|
| Number of Shares |
|
| Amount |
|
| Additional Paid in Capital |
|
| Retained Earnings |
|
| Accumulated Other Comprehensive Income (Loss) |
|
| Total Stockholders' Equity |
| ||||||||
Balance, April 1, 2018 |
|
| 44,375,337 |
|
| $ | 444 |
|
|
| 1,725,312 |
|
| $ | (67,078 | ) |
| $ | 619,905 |
|
| $ | 140,457 |
|
| $ | (4,204 | ) |
|
| 689,524 |
|
Restricted stock units issued |
|
| 427,431 |
|
|
| 4 |
|
|
| - |
|
|
| - |
|
|
| (4 | ) |
|
| - |
|
|
| - |
|
|
| - |
|
Stock options exercised |
|
| 485,363 |
|
|
| 5 |
|
|
| - |
|
|
| - |
|
|
| 12,944 |
|
|
| - |
|
|
| - |
|
|
| 12,949 |
|
Stock issued under employee stock purchase plan |
|
| 12,467 |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 3,052 |
|
|
| - |
|
|
| - |
|
|
| 3,052 |
|
Stock issued to directors |
|
| 316 |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 116 |
|
|
| - |
|
|
| - |
|
|
| 116 |
|
Return of common stock to pay withholding taxes on restricted stock |
|
| (177,929 | ) |
|
| (2 | ) |
|
| 177,929 |
|
|
| (71,774 | ) |
|
| - |
|
|
| - |
|
|
| - |
|
|
| (71,776 | ) |
Stock compensation expense |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 54,494 |
|
|
| - |
|
|
| - |
|
|
| 54,494 |
|
Other comprehensive loss |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| (10,485 | ) |
|
| (10,485 | ) |
Net income |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 259,016 |
|
|
| - |
|
|
| 259,016 |
|
Balance, March 31, 2019 |
|
| 45,122,985 |
|
| $ | 451 |
|
|
| 1,903,241 |
|
| $ | (138,852 | ) |
| $ | 690,507 |
|
| $ | 399,473 |
|
| $ | (14,689 | ) |
| $ | 936,890 |
|
Restricted stock units issued |
|
| 392,872 |
|
|
| 4 |
|
|
| - |
|
|
| - |
|
|
| (4 | ) |
|
| - |
|
|
| - |
|
|
| - |
|
Stock options exercised |
|
| 85,136 |
|
|
| 2 |
|
|
| - |
|
|
| - |
|
|
| 3,747 |
|
|
| - |
|
|
| - |
|
|
| 3,748 |
|
Stock issued under employee stock purchase plan |
|
| 37,827 |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 5,103 |
|
|
| - |
|
|
| - |
|
|
| 5,103 |
|
Return of common stock to pay withholding taxes on restricted stock |
|
| (164,446 | ) |
|
| (2 | ) |
|
| 164,446 |
|
|
| (41,685 | ) |
|
| - |
|
|
| - |
|
|
| - |
|
|
| (41,687 | ) |
Stock compensation expense |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 39,781 |
|
|
| - |
|
|
| - |
|
|
| 39,781 |
|
Stock repurchase program |
|
| (465,687 | ) |
|
| (5 | ) |
|
| 465,687 |
|
|
| (84,874 | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
| (84,879 | ) |
Other comprehensive income |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 3,500 |
|
|
| 3,500 |
|
Net income |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 203,009 |
|
|
| - |
|
|
| 203,009 |
|
Balance, March 31, 2020 |
|
| 45,008,687 |
|
| $ | 451 |
|
|
| 2,533,374 |
|
| $ | (265,411 | ) |
| $ | 739,133 |
|
| $ | 602,482 |
|
| $ | (11,189 | ) |
| $ | 1,065,466 |
|
Restricted stock units issued |
|
| 140,159 |
|
|
| 2 |
|
|
| - |
|
|
| - |
|
|
| (1 | ) |
|
| - |
|
|
| - |
|
|
| 1 |
|
Stock options exercised |
|
| 215,262 |
|
|
| 2 |
|
|
| - |
|
|
| - |
|
|
| 9,073 |
|
|
| - |
|
|
| - |
|
|
| 9,075 |
|
Stock issued under employee stock purchase plan |
|
| 31,920 |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 5,479 |
|
|
| - |
|
|
| - |
|
|
| 5,479 |
|
Return of common stock to pay withholding taxes on restricted stock |
|
| (57,431 | ) |
|
| (1 | ) |
|
| 57,431 |
|
|
| (11,310 | ) |
|
| - |
|
|
| - |
|
|
| - |
|
|
| (11,311 | ) |
Stock compensation expense |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 47,006 |
|
|
| - |
|
|
| - |
|
|
| 47,006 |
|
Stock repurchase program |
|
| (67,649 | ) |
|
| (1 | ) |
|
| 67,649 |
|
|
| (11,309 | ) |
|
| - |
|
|
| - |
|
|
| - |
|
|
| (11,310 | ) |
Other comprehensive loss |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| (256 | ) |
|
| (256 | ) |
Net income |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 225,525 |
|
|
| - |
|
|
| 225,525 |
|
Balance, March 31, 2021 |
|
| 45,270,948 |
|
| $ | 453 |
|
|
| 2,658,454 |
|
| $ | (288,030 | ) |
| $ | 800,690 |
|
| $ | 828,007 |
|
| $ | (11,445 | ) |
| $ | 1,329,675 |
|
The accompanying notes are an integral part of the consolidated financial statements.
F-7
ABIOMED, INC. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
(in thousands)
|
| Fiscal Years Ended March 31, |
| |||||||||
|
| 2021 |
|
| 2020 |
|
| 2019 |
| |||
Operating activities: |
|
|
|
|
|
|
| �� |
|
|
|
|
Net income |
| $ | 225,525 |
|
| $ | 203,009 |
|
| $ | 259,016 |
|
Adjustments to reconcile net income to net cash provided by operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
| 24,097 |
|
|
| 20,430 |
|
|
| 14,121 |
|
Bad debt (recoveries) expense |
|
| (126 | ) |
|
| 487 |
|
|
| 720 |
|
Stock-based compensation |
|
| 47,006 |
|
|
| 39,781 |
|
|
| 54,494 |
|
Write-down of inventory and other |
|
| 8,518 |
|
|
| 4,249 |
|
|
| 4,252 |
|
Accretion on marketable securities |
|
| 1,977 |
|
|
| (2,731 | ) |
|
| (2,744 | ) |
Change in fair value of other investments |
|
| (50,983 | ) |
|
| 5,184 |
|
|
| (30,161 | ) |
Deferred tax provision |
|
| 29,380 |
|
|
| 32,953 |
|
|
| (7,745 | ) |
Change in fair value of contingent consideration |
|
| 2,406 |
|
|
| (575 | ) |
|
| (915 | ) |
Other non-cash operating activities |
|
| 4,164 |
|
|
| 4,108 |
|
|
| — |
|
Changes in assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
| (12,059 | ) |
|
| 5,551 |
|
|
| (22,023 | ) |
Inventories |
|
| 2,535 |
|
|
| (13,237 | ) |
|
| (37,181 | ) |
Prepaid expenses and other assets |
|
| (1,032 | ) |
|
| (5,333 | ) |
|
| (2,527 | ) |
Accounts payable |
|
| 2,629 |
|
|
| 2,581 |
|
|
| 7,976 |
|
Accrued expenses and other liabilities |
|
| (14,632 | ) |
|
| 15,676 |
|
|
| 13,406 |
|
Deferred revenue |
|
| 5,173 |
|
|
| 2,787 |
|
|
| 1,508 |
|
Net cash provided by operating activities |
|
| 274,578 |
|
|
| 314,920 |
|
|
| 252,197 |
|
Investing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of marketable securities |
|
| (556,199 | ) |
|
| (611,280 | ) |
|
| (361,602 | ) |
Proceeds from the sale and maturity of marketable securities and other |
|
| 396,643 |
|
|
| 550,788 |
|
|
| 331,886 |
|
Acquisition of Breethe Inc., net of cash acquired |
|
| (52,183 | ) |
|
| — |
|
|
| — |
|
Purchases of other investments and intangible assets |
|
| (26,104 | ) |
|
| (20,957 | ) |
|
| (42,735 | ) |
Proceeds from sale of Shockwave Medical Investment |
|
| 67,882 |
|
|
| — |
|
|
| — |
|
Purchases of property and equipment |
|
| (53,383 | ) |
|
| (44,006 | ) |
|
| (44,004 | ) |
Net cash used for investing activities |
|
| (223,344 | ) |
|
| (125,455 | ) |
|
| (116,455 | ) |
Financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from the exercise of stock options |
|
| 9,075 |
|
|
| 3,748 |
|
|
| 12,949 |
|
Taxes paid related to net share settlement upon vesting of stock awards |
|
| (11,311 | ) |
|
| (41,687 | ) |
|
| (71,776 | ) |
Repurchase of common stock |
|
| (11,310 | ) |
|
| (84,879 | ) |
|
| — |
|
Proceeds from the issuance of stock under employee stock purchase plan |
|
| 5,479 |
|
|
| 5,103 |
|
|
| 3,052 |
|
Net cash used for financing activities |
|
| (8,067 | ) |
|
| (117,715 | ) |
|
| (55,775 | ) |
Effect of exchange rate changes on cash |
|
| (2,798 | ) |
|
| (430 | ) |
|
| (1,921 | ) |
Net increase in cash and cash equivalents |
|
| 40,369 |
|
|
| 71,320 |
|
|
| 78,046 |
|
Cash and cash equivalents at beginning of year |
|
| 192,341 |
|
|
| 121,021 |
|
|
| 42,975 |
|
Cash and cash equivalents at end of year |
| $ | 232,710 |
|
| $ | 192,341 |
|
| $ | 121,021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for income taxes |
| $ | 48,693 |
|
| $ | 9,685 |
|
| $ | 5,290 |
|
Supplemental disclosure of non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment in accounts payable and accrued expenses |
|
| 1,638 |
|
|
| 2,977 |
|
|
| 4,787 |
|
Right-of-use assets obtained in exchange for lease liabilities |
|
| 2,592 |
|
|
| 15,650 |
|
|
| — |
|
The accompanying notes are an integral part of the consolidated financial statements.
F-8
ABIOMED, INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
(Dollars in thousands, except per share data)
Note 1. Nature of Operations
ABIOMED, Inc. (the “Company” or “ABIOMED”) is a provider of mechanical circulatory support devices and offers a continuum of care to heart failure patients. The Company develops, manufactures and markets proprietary products that are designed to enable the heart to rest, heal and recover by improving blood flow and/or performing the pumping function of the heart. The Company’s products are used in the cardiac catheterization lab, or cath lab, by interventional cardiologists and in the heart surgery suite by cardiac surgeons for patients who are in need of hemodynamic support prophylactically or emergently before, during or after angioplasty or heart surgery procedures.
Note 2. Basis of Preparation and Summary of Significant Accounting Policies
The accompanying consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles, or GAAP, and Regulation S-X. The information presented reflects the application of significant accounting policies described below.
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation.
COVID-19 Pandemic
The Company is subject to risks and uncertainties as a result of the ongoing COVID-19 pandemic. The ongoing COVID-19 pandemic has adversely impacted and is likely to further adversely impact the Company’s business and markets, including the Company’s workforce and the operations of its customers, suppliers, and business partners. The full extent to which the pandemic will directly or indirectly impact the Company's business, results of operations and financial condition, including sales, expenses, manufacturing, clinical trials, research and development costs, reserves and allowances, fair value measurements, asset impairment charges, contingent consideration obligations, and the effectiveness of the Company's hedging instruments, will depend on future developments that are highly uncertain and difficult to predict. These developments include, but are not limited to: the duration and spread of the ongoing COVID-19 pandemic (including new variants of COVID-19), its severity, the actions to contain the virus or address its impact, the timing, distribution, and efficacy of vaccines and other treatments, U.S. and foreign government actions to respond to the reduction in global economic activity, and how quickly and to what extent normal economic and operating conditions can resume.
Beginning in mid-March 2020, the Company experienced a decline in patient utilization in its main markets in the U.S., Europe and Japan as healthcare systems diverted resources to meet the increasing demands of managing COVID-19. The Company’s business was most impacted in the first quarter of fiscal year 2021, in terms of the decline in patients and revenue from the shelter-in-place restrictions in a majority of countries and limitations on procedures in hospitals. The Company experienced sequential quarterly improvement during the second quarter of fiscal year 2021 as patient procedure volume trends and availability of healthcare resources improved as certain restrictions were lifted and limitations eased during the summer of 2020. During the third quarter of fiscal year 2021, a broad resurgence of COVID-19 cases throughout the U.S. and Europe slowed the Company’s continued recovery in patient utilization. Despite these challenges, the Company experienced increased recovery in patient utilization and revenue during the fourth quarter of fiscal year 2021 due to the high-risk nature of the patient population that are treated with the Company’s Impella devices.
While the COVID-19 pandemic remains fluid and continues to evolve differently across various geographies, the Company believes it is likely to continue to experience variable impacts on its business. Hospitals are generally managing the pandemic better currently than they have in the earlier part of the pandemic due to more testing, improved protocols, more experience with the effects of COVID-19 and a greater number of vaccinated caregivers. During these challenging times, the Company’s priorities have been to support its clinician partners, protect the well-being of its employees and maintain continuous access to its life-saving technologies while offering front-line in-hospital support. The Company has established onsite COVID-19 testing for its employees in both Danvers, Massachusetts and Aachen, Germany, set up temperature-taking stations, administered thousands of COVID-19 tests to date and provided personal protective equipment for its employees in order to maintain a safe working environment.
F-9
The Company’s proactive testing program has reduced exposure with early detection, reduced employee anxiety and enabled its manufacturing facilities to operate at full capacity in line with local social distancing requirements. The Company also took proactive actions in the first half of fiscal year 2021 in order to mitigate the business impact of COVID-19 on its financial operations and it continues to monitor closely in order the business impact of COVID-19. Despite the ongoing challenges posed by COVID-19, including the recent global resurgence, the Company continues to invest strategically in engineering, regulatory, clinical trials and manufacturing in order to support its future growth initiatives and sales and marketing activities, with a particular focus on training and education initiatives to drive utilization of its products and recovery awareness for acute heart failure patients.
The Company continues to closely monitor the impact of COVID-19 on all aspects of its business and geographies, including its impact on its customers, employees, suppliers, vendors, business partners and distribution channels. The extent to which the COVID-19 pandemic impacts the Company’s business, results of operations, and financial condition will depend on future developments, which are highly uncertain and are difficult to predict. Even after the ongoing COVID-19 pandemic has subsided, the Company may continue to experience materially adverse impacts on its financial condition and results of operations.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. The Company bases its estimates on historical experience and various other factors believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. On an ongoing basis, the Company evaluates its estimates, including those related to revenue recognition, collectability of receivables, realizability of inventory, property and equipment, goodwill, intangible and other long-lived assets, other investments, accrued expenses, stock-based compensation, income taxes including deferred tax assets and liabilities, contingencies and litigation. Provisions for depreciation are based on their estimated useful lives using the straight-line method. Some of these estimates can be subjective and complex and, consequently, actual results may differ from these estimates under different assumptions or conditions.
Cash Equivalents and Marketable Securities
The Company classifies any marketable security with an original maturity date of 90 days or less at the time of purchase as a cash equivalent. Cash equivalents are carried on the balance sheet at fair market value. The Company classifies any marketable security with a maturity date of greater than 90 days at the time of purchase as marketable securities and classifies marketable securities with a maturity date of greater than one year from the balance sheet date as long-term marketable securities.
The Company’s marketable securities, consisting of U.S. Treasury securities, government-back securities, corporate debt securities, and commercial paper, are classified as available-for-sale securities and, accordingly, are recorded at fair value. The difference between amortized cost and fair value is included in stockholders’ equity. Marketable securities that the Company has the positive intent and ability to hold to maturity are reported at amortized cost and classified as held-to-maturity marketable securities. If the Company does not have the intent and ability to hold a marketable security to maturity, it reports the investment as available-for-sale marketable securities. The Company includes unrealized gains and, to the extent deemed temporary, unrealized losses in stockholders’ equity. If any adjustment to fair value reflects a decline in the value of the investment, the Company considers available evidence to evaluate whether the decline is “other than temporary” and, if so, marks the marketable security to market through a charge reflected on the consolidated statements of operations.
Major Customers and Concentrations of Credit Risk
The Company primarily sells its products to hospitals and distributors. NaN individual customer accounted for more than 10% of total revenues in fiscal years ended March 31, 2021, 2020 or 2019. NaN individual customer had an accounts receivable balance greater than 10% of total accounts receivable as of March 31, 2021 and 2020.
Credit is extended based on an evaluation of a customer’s historical financial condition and generally collateral is not required. The Company’s history of credit losses has not been significant and the Company maintains an allowance for doubtful accounts based on its assessment of the collectability of accounts receivable. Accounts receivables are geographically dispersed, primarily throughout the U.S., as well as in Europe and other foreign countries where formal distributor agreements exist in certain countries. The Company's exposure to credit losses may increase if its customers are adversely affected by changes in healthcare laws, coverage, and reimbursement, economic pressures or uncertainty associated with local or global economic recessions, or other customer-specific factors.
F-10
Financial instruments which potentially subject the Company to a concentration of credit risk consist of cash, cash equivalents, marketable securities, short and long-term marketable securities and accounts receivable. Management mitigates credit risk by limiting the investment type and maturity to securities that preserve capital, maintain liquidity and have a high credit quality.
Financial Instruments
The Company’s financial instruments are comprised of cash and cash equivalents, derivative instruments, marketable securities, accounts receivable, accounts payable and contingent consideration. The carrying amounts of accounts receivable and accounts payable are considered reasonable estimates of their fair value, due to the short maturity of these investments.
Derivative Instruments
The Company uses a foreign-exchange-related derivative instrument to manage its exposure related to changes in the exchange rate on its intercompany loan. The Company does not enter into derivative instruments for any purpose other than cash flow hedging. The Company does not speculate using derivative instruments. The Company recognizes all derivative instruments as either assets or liabilities in the balance sheet at their respective fair values. Changes in the fair value of the cross-currency swap designated as a hedging instrument that effectively offsets the variability of cash flows are reported in accumulated other comprehensive income (loss). These amounts subsequently are reclassified into the consolidated income statement in the same period in which the related hedged item affects earnings. For more information, see “Note 5. Financial Instruments—Derivative Instruments.”
Inventories
Inventories are stated at the lower of cost or market. Cost is based on the first in, first out method. The Company regularly reviews inventory quantities on hand and writes down to its net realizable value any inventory that it believes to be impaired. Management considers forecast demand in relation to the inventory on hand, competitiveness of product offerings, market conditions and product life cycles when determining excess and obsolescence and net realizable value adjustments. Once inventory is written down and a new cost basis is established, it is not written back up if demand increases.
Property and Equipment
Property and equipment is recorded at cost less accumulated depreciation. Land is carried at cost and is not depreciated. Depreciation is computed using the straight-line method based on estimated useful lives of three to five years for machinery and equipment, computer software, and furniture and fixtures. Building and building improvements are depreciated using the straight-line method over estimated useful lives of seven to thirty-three years. Leasehold improvements are amortized using the straight-line method over the shorter of the lease term or the estimated useful lives of the related assets. Expenditures for maintenance and repairs are expensed as incurred. Upon retirement or other disposition of assets, the costs and related accumulated depreciation are eliminated from the accounts and the resulting gain or loss is reflected in operating expenses.
Property and equipment is reviewed for impairment losses whenever events or changes in circumstances indicate the carrying amount may not be recoverable. An impairment loss would be recognized based on the amount by which the carrying value of the asset or asset group exceeds its fair value. Fair value is determined primarily using the estimated future cash flows associated with the asset or asset group under review discounted at a rate commensurate with the risk involved and other valuation techniques.
Other Investments
The Company periodically makes investments in medical device companies that focus on heart failure and heart pumps and other medical device technologies. For investments that do not have readily determinable market values, the Company measures these investments at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for an identical or similar investment. The Company monitors any events or changes in circumstances that may have a significant effect on the fair value of investments, either due to impairment or based on observable price changes, and makes any necessary adjustments.
Leases
On April 1, 2019, the Company adopted ASU 2016-02, “Leases.” This new guidance required that the Company’s lease commitments to be recognized as operating lease liabilities and right-of-use assets, which increased total assets and total liabilities that the Company reported on its consolidated balance sheet. For a discussion on the impact of this accounting policy adoption, including key accounting policies and elections, see “Note 11. Leases.”
F-11
Goodwill
Goodwill is recorded when consideration for an acquisition exceeds the fair value of the net tangible and intangible assets acquired. Goodwill is not amortized. The Company evaluates goodwill for impairment at least annually at October 31, as well as whenever events or changes in circumstances suggest that the carrying amount may not be recoverable.
In applying the goodwill impairment test, the Company assesses qualitative factors to determine whether it is more likely than not that the fair value of the reporting unit is less than its carrying value. Qualitative factors may include, but are not limited to, macroeconomic conditions, industry conditions, the competitive environment, changes in the market for the Company’s products and services, regulatory and political developments, cost factors, and entity specific factors such as strategies and overall financial performance. If, after assessing these qualitative factors, the Company determines it is more likely than not that the fair value of a reporting unit is less than its carrying value, then the Company performs a quantitative impairment test. The Company determined, after performing the qualitative review, that it is more likely than not that the fair value of each of its reporting units substantially exceeds the respective carrying amounts.
Indefinite-Lived Intangibles and In-Process Research and Development
In-process research and development, or IPR&D, assets are considered to be indefinite-lived until the completion or abandonment of the associated research and development projects. IPR&D assets represent the fair value assigned to technologies that are acquired, which at the time of acquisition have not reached technological feasibility and have no alternative future use. During the period that the IPR&D assets are considered indefinite-lived, they are tested for impairment on an annual basis on October 31, or more frequently if the Company becomes aware of any events occurring or changes in circumstances that indicate that the fair value of the IPR&D assets are less than their carrying values. If and when development is complete, which generally occurs upon regulatory approval and the Company is able to commercialize products associated with the IPR&D assets, these assets are then deemed definite-lived and are amortized based on their estimated useful lives at that point in time. If development is terminated or abandoned, the Company may have a full or partial impairment charge related to the IPR&D assets, calculated as the excess of carrying value of the IPR&D assets over fair value. The Company performed its annual impairment review for fiscal year 2021 as of October 31, 2020 and concluded that it was more likely than not that the fair value of the IPR&D assets is not less than its carrying value.
Finite-Lived Intangible Assets
The Company records finite-lived intangible assets at historical cost and amortizes over their estimated useful lives. The Company uses a straight-line method of amortization, unless a method that better reflects the pattern in which the economic benefits of the intangible asset are consumed or otherwise used up can be reliably determined. The approximate useful live for amortization of the Company’s intangible assets is 15 years for the OXY-1 System amortizable technology from the acquisition of Breethe.
The Company reviews finite-lived intangible assets subject to amortization quarterly to determine if any adverse conditions exist or a change in circumstances has occurred that would indicate impairment or a change in the remaining useful life. Conditions that may indicate impairment include, but are not limited to, a significant adverse change in legal factors or business climate that could affect the value of an asset, a product recall or an adverse action or assessment by a regulator. If the Company determines it is more likely than not that the asset is impaired based on its qualitative assessment of impairment indicators, the Company tests the intangible asset for recoverability.
Contingent Consideration
Contingent consideration represents potential milestones that the Company could pay as additional consideration for a business acquisition and is recorded as a liability and is measured at fair value using a combination of (1) an income approach, based on various revenue and cost assumptions and applying a probability to each outcome and (2) a Monte-Carlo valuation model that simulates outcomes based on management estimates. With the income approach, probabilities were applied to each potential scenario and the resulting values were discounted using a rate that considers the weighted average cost of capital, the related projections, and the overall business. The Monte-Carlo valuation model simulates estimated future revenues during the earn out-period using management's best estimates. Significant increases or decreases in any of the probabilities of success or changes in expected timelines for achievement of any of these milestones could result in a significantly higher or lower fair value of the contingent consideration liability. The fair value of the contingent consideration at each reporting date is updated by reflecting the changes in fair value reflected within research and development expenses in the Company’s consolidated statement of operations.
F-12
Accrued Expenses
As part of the process of preparing its financial statements, the Company is required to estimate accrued expenses. This process includes identifying services that third parties have performed and estimating the level of service performed and the associated cost incurred on these services as of each balance sheet date in its financial statements. Examples of estimated accrued expenses include estimates for certain payroll costs, such as bonuses and commissions; contract service fees, such as amounts due to clinical research organizations and investigators in conjunction with clinical trials; professional service fees, such as attorneys and accountants, and third-party expenses relating to marketing efforts associated with commercialization of the Company’s products. The dates in which certain services commence and end, the level of services performed on or before a given date and the cost of services is often subject to the Company’s judgment. The Company makes these judgments and estimates based upon known facts and circumstances.
Revenue Recognition
See “Note 4. Revenue Recognition” for a discussion of key accounting policies and elections related to revenue recognition.
Product Warranty
The Company generally provides a one-year warranty for certain products sold in which estimated contractual warranty obligations are recorded as an expense at the time of shipment. The Company’s products are subject to regulatory and quality standards. Future warranty costs are estimated based on historical product performance rates and related costs to repair given products. The accounting estimate related to product warranty expense involves judgment in determining future estimated warranty costs. Should actual performance rates or repair costs differ from estimates, revisions to the estimated warranty liability would be required.
Accumulated Other Comprehensive Income (Loss)
Comprehensive income (loss) is comprised of net income, plus all changes in equity of a business enterprise during a period from transactions and other events and circumstances from non-owner sources, including any foreign currency translation adjustments. These changes in equity are recorded as adjustments to accumulated other comprehensive income (loss) in the Company’s consolidated balance sheet. The components of accumulated other comprehensive income (loss) consist of foreign currency translation adjustments, unrealized gains (losses) on marketable securities, and unrealized gains (losses) on derivative instruments. There were 0 reclassifications out of accumulated other comprehensive income (loss) during the fiscal years ended March 31, 2021, 2020 and 2019.
Translation of Foreign Currencies
The functional currency of the Company’s foreign subsidiaries is their local currency. The assets and liabilities of the Company’s foreign subsidiaries are translated into U.S. dollars at exchange rates in effect at the balance sheet date. Income and expense items in the Company’s consolidated statement of operations are translated at the average exchange rates prevailing during the period. The cumulative translation effect for subsidiaries using a functional currency other than the U.S. dollar is included in accumulated other comprehensive income (loss) as a separate component of stockholders’ equity.
The Company’s intercompany accounts are denominated in the functional currency of the foreign subsidiary. Gains and losses resulting from the remeasurement of intercompany transactions that the Company considers to be of a long-term investment nature are recorded in accumulated other comprehensive income or loss as a separate component of stockholders’ equity, while gains and losses resulting from the remeasurement of intercompany transactions from those foreign subsidiaries for which the Company anticipates settlement in the foreseeable future are recorded in the consolidated statement of operations. The net foreign currency translation gains and losses recorded in the consolidated statements of operations for the fiscal years ended March 31, 2021, 2020 and 2019 were not significant.
F-13
Net Income Per Share
Basic net income per share is computed by dividing net income by the weighted average number of common shares outstanding during the fiscal year. Diluted net income per share is computed using the treasury stock method by dividing net income by the weighted average number of dilutive common shares outstanding during the fiscal year. Diluted shares outstanding is calculated by adding to the weighted average shares outstanding any potential dilutive securities outstanding for the fiscal year. Potential dilutive securities include stock options, restricted stock units, performance-based stock awards and shares to be purchased under the Company’s employee stock purchase plan. The Company’s basic and diluted net income per share were as follows (figures in tables are in thousands, except per share data):
|
| Fiscal Years Ended March 31, |
| |||||||||
Basic Net Income Per Share |
|
| 2021 |
|
|
| 2020 |
|
|
| 2019 |
|
Net income |
| $ | 225,525 |
|
| $ | 203,009 |
|
| $ | 259,016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares - basic |
|
| 45,140 |
|
|
| 45,179 |
|
|
| 44,911 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income per share - basic |
| $ | 5.00 |
|
| $ | 4.49 |
|
| $ | 5.77 |
|
|
| Fiscal Years Ended March 31, |
| |||||||||
Diluted Net Income Per Share |
|
| 2021 |
|
|
| 2020 |
|
|
| 2019 |
|
Net income |
| $ | 225,525 |
|
| $ | 203,009 |
|
| $ | 259,016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares - basic |
|
| 45,140 |
|
|
| 45,179 |
|
|
| 44,911 |
|
Effect of dilutive securities |
|
| 534 |
|
|
| 637 |
|
|
| 1,240 |
|
Weighted average shares - diluted |
|
| 45,674 |
|
|
| 45,816 |
|
|
| 46,151 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income per share - diluted |
| $ | 4.94 |
|
| $ | 4.43 |
|
| $ | 5.61 |
|
For the fiscal years ended March 31, 2021, 2020 and 2019, approximately 168,000, 232,000 and 64,000 shares of common stock underlying outstanding securities related to out-of-the-money stock options and performance-based awards where milestones were not met were not included in the computation of diluted earnings per share because their inclusion would be anti-dilutive.
Stock-Based Compensation
The Company’s stock-based compensation cost is measured at the grant date based on the fair value of the award and is recognized as an expense over the requisite service period.
The fair value of stock option grants is estimated using the Black-Scholes option pricing model. Use of the valuation model requires management to make certain assumptions with respect to selected model inputs. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant for a term consistent with the expected life of the stock options. Volatility assumptions are calculated based on historical volatility of the Company’s stock. The Company estimates the expected term of options based on historical exercise experience and estimates of future exercises of unexercised options. In addition, an expected dividend yield of 0 is used in the option valuation model because the Company does not pay cash dividends and does not expect to pay any cash dividends in the foreseeable future. Forfeitures are recorded as they occur instead of estimating forfeitures that are expected to occur.
For awards with service conditions only, the Company recognizes stock-based compensation expense on a straight-line basis over the requisite service period. For awards with service and performance-based conditions, the Company recognizes stock-based compensation expense using the graded vesting method over the requisite service period. Estimates of stock-based compensation expense for an award with performance conditions are based on the probable outcome of the performance conditions and the cumulative effect of any changes in the probability outcomes are recorded in the period in which the changes occur.
Income Taxes
The Company’s provision for income taxes is comprised of a current and deferred provision. The current income tax provision is calculated as the estimated taxes payable or refundable on income tax returns for the current fiscal year. The deferred income tax provision is calculated for the estimated future income tax effects attributable to temporary differences and carryforwards using expected tax rates in effect in the years during which the temporary differences are expected to reverse.
F-14
Deferred income taxes are recognized for the tax consequences in future years as the differences between the tax bases of assets and liabilities and their financial reporting amounts at each fiscal year end based on enacted tax laws and statutory tax rates applicable to the periods in which the differences are expected to impact taxable income.
The Company regularly assesses its ability to realize its deferred tax assets. Assessing the realization of deferred tax assets requires significant management judgment. Valuation allowances are established when necessary to reduce net deferred tax assets to the amount that is more likely than not to be realized.
The Company recognizes and measures uncertain tax positions using a two-step approach. The first step is to evaluate the tax position for recognition by determining if, based on the technical merits, it is more likely than not that the position will be sustained upon audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit at the largest amount that is more likely than not of being realized upon ultimate settlement. The Company reevaluates these uncertain tax positions on an ongoing basis, when applicable. This evaluation is based on factors including, but not limited to, changes in facts or circumstances, new information and technical insights, and changes in tax laws. Any changes in these factors could result in the recognition of a tax benefit or an additional charge to the tax provision. Refer to “Note 15. Income Taxes” for further information related to the Tax Reform Act and its impact on the Company’s financial statements. When applicable, the Company accrues for the effects of uncertain tax positions and the related potential penalties and interest through income tax expense.
Recently Adopted Accounting Pronouncements
Effective April 1, 2019, the Company adopted the Financial Accounting Standards Board (“FASB”) standard update ASU 2016-02 “Leases,” which requires lessees to identify arrangements that should be accounted for as leases. Under this guidance, for lease arrangements exceeding a one-year term, a right-of-use asset and lease obligation is recorded by the lessee for all leases on the balance sheet, whether operating or financing, while the statement of operations includes lease expense for operating leases and amortization and interest expense for financing leases. ASU 2016-02 was adopted using a modified retrospective approach for all leases existing at or entered into after the April 1, 2019. Additional information and disclosures required by this new standard are contained in “Note 11. Leases.”
In June 2016, the FASB issued ASU 2016-13, “Financial Instruments-Credit Losses (Topic 326).” This guidance will require financial instruments to be measured at amortized cost, and accounts receivables to be presented at the net amount expected to be collected. The new model requires an entity to estimate credit losses based on historical information, current information, and reasonable and supportable forecasts, including estimates of prepayments. ASU 2016-13 is effective for annual reporting periods beginning after December 31, 2019. This update did not have a material impact on the Company’s consolidated financial statements.
In January 2017, the FASB issued ASU 2017-04, “Intangibles - Goodwill and Other (Topic 350).” This new standard simplifies the accounting for goodwill impairment by removing Step 2 of the goodwill impairment test, which required companies to estimate the implied fair value of goodwill and recognize an impairment charge by the amount in which the carrying value exceeds the implied fair value. Under the new guidance, if the carrying value of a reporting unit exceeds its fair value, a goodwill impairment charge will be recorded, even if the difference is attributable to the fair value of other assets in the reporting unit. The Company adopted this standard in the first quarter of fiscal 2021, and it did not have a material impact on the Company’s consolidated financial statements.
In August 2018, the FASB issued ASU 2018-13, “Fair Value Measurement (Topic 820).” Disclosure Framework-Changes to the Disclosure Requirements for Fair Value Measurement, which modifies the disclosure requirements on fair value measurements. This ASU is effective for fiscal years beginning after December 15, 2019 and early adoption is permitted. The Company adopted this standard in the first quarter of fiscal year 2021 and did not have a material impact on its consolidated financial statements.
F-15
Recently Issued Accounting Pronouncements Not Yet Effective
In December 2019, the FASB issued ASU 2019-12, “Income Taxes (Topic 740).” This amendment to the guidance on income taxes is intended to simplify the accounting for income taxes. The amendment eliminates certain exceptions related to the approach for intraperiod tax allocation, the methodology for calculating income taxes in an interim period, and the recognition of deferred tax liabilities for outside basis differences. The amendment also clarifies existing guidance related to the evaluation of a step up in the tax basis of goodwill, and the effects of enacted changes in tax laws or rates in the effective tax rate computation, among other clarifications. ASU 2019-12 will become effective for the Company in fiscal year 2022. The Company does not expect the adoption of this standard to have a material impact on its consolidated financial statements.
In January 2020, the FASB issued ASU 2020-01, “Investments—Equity Securities (Topic 321), Investments—Equity Method and Joint Ventures (Topic 323), and Derivatives and Hedging (Topic 815),” an amendment clarifying the interaction between accounting standards related to equity securities, equity method investments, and certain derivative instruments. The guidance is effective for fiscal years beginning after December 15, 2020. ASU 2020-01 will become effective for the Company in fiscal year 2022. The Company does not expect the adoption of this standard to have a material impact on its consolidated financial statements.
No other new accounting pronouncements, issued or effective, during the period had, or are expected to have, a material impact on our consolidated financial statements.
Note 3. Acquisition
Acquisition of Breethe
The Company acquired Breethe, a Maryland corporation on April 24, 2020. Breethe is engaged in research and development of an ECMO system that we aim to complement and expand the Company’s product portfolio to more comprehensively serve the needs of patients whose lungs can no longer provide sufficient oxygenation, including patients suffering from cardiogenic shock, or respiratory failure. The Company acquired Breethe for $55.0 million in cash, with additional potential payouts up to a maximum of $55.0 million payable based on the achievement of certain technical, regulatory and commercial milestones.
Purchase Price Allocation
The acquisition of Breethe was accounted for as a business combination. The purchase price for the acquisition has been allocated to the assets acquired and liabilities assumed based on their estimated fair values.
The acquisition-date fair value of the consideration transferred is as follows:
| Total Acquisition Date Fair Value (in thousands) |
| |
Cash and other considerations | $ | 57,850 |
|
Contingent consideration |
| 13,300 |
|
Total consideration transferred | $ | 71,150 |
|
The following table summarizes the estimated fair values of the assets acquired and liabilities assumed on the date of acquisition (in thousands):
Acquired assets: |
|
|
|
Cash and cash equivalents | $ | 3,404 |
|
Property and equipment |
| 744 |
|
Goodwill |
| 44,485 |
|
In-process research and development |
| 27,000 |
|
Other assets acquired |
| 895 |
|
Total assets acquired |
| 76,528 |
|
Liabilities assumed: |
|
|
|
Accounts payable and other liabilities |
| 1,562 |
|
Deferred tax liabilities |
| 3,816 |
|
|
|
|
|
Net assets acquired | $ | 71,150 |
|
F-16
Goodwill is calculated as the difference between the acquisition-date fair value of the consideration transferred and the fair values of the assets acquired and liabilities assumed. The goodwill is not deductible for income tax purposes.
IPR&D from the acquisition of Breethe represents the estimated fair value of the Breethe ECMO technology which had not reached commercial technological feasibility nor had alternative future use at the time of the acquisition. During the third quarter of fiscal year 2021, upon receiving FDA 510(k) clearance of the OXY-1 System, in October 2020, the Company reclassified the IPR&D asset of $27 million from the acquisition of Breethe to a finite-lived developed technology intangible asset and began amortizing on a straight-line basis over an estimated useful life of 15 years (see Note 8). The Company believes the amount of purchased IPR&D assets represent fair value for these intangible assets as of the acquisition date.
Transaction costs such as legal, insurance and other costs related to the acquisition, aggregating approximately $0.9 million, have been expensed as incurred and are included in selling, general and administrative expenses in the Company’s condensed consolidated statements of operations.
The Company’s consolidated financial statements include the operating results of Breethe from the acquisition date. Separate post-acquisition operating results and pro forma results of operations for this acquisition have not been presented as the effect is not material to the Company’s financial results.
Note 4. Revenue Recognition
The Company adopted Topic 606 on April 1, 2018, using the modified retrospective method for all revenue contracts not completed as of the date of adoption. The reported results for fiscal years 2021, 2020 and 2019 reflect the application of Topic 606 guidance.
The Company has made the following accounting policy elections and elected to use certain practical expedients, as permitted by the FASB, in applying Topic 606: (1) the Company accounts for amounts collected from customers for sales and other taxes, net of related amounts remitted to tax authorities; (2) the Company does not adjust the promised amount of consideration for the effects of a significant financing component because, at contract inception, the Company expects the period between the time when the Company transfers a promised good or service to the customer and the time when the customer pays for that good or service will be one year or less; (3) the Company expenses costs to obtain a contract as they are incurred if the expected period of benefit, and therefore the amortization period, is one year or less; (4) the Company accounts for shipping and handling activities that occur after control transfers to the customer as a fulfillment cost rather than an additional promised service and these fulfillment costs are recorded as selling, general and administrative expenses; (5) the Company does not assess whether promised goods or services are performance obligations if they are immaterial in the context of the contract with the customer; and (6) the Company does not disclose the transaction price allocated to unsatisfied performance obligations when the original expected contract duration is one year or less.
The Company generates revenue primarily from the sale of Impella 2.5, Impella CP, Impella 5.0, Impella LD, Impella RP, Impella 5.5, Impella AIC and the OXY-1 System products and related accessories. The Company also earns revenue from preventative maintenance service contracts and maintenance calls.
The Company determines revenue recognition through the following steps:
| • | Identification of the contract, or contracts, with a customer |
| • | Identification of the performance obligation in the contract |
| • | Determination of the transaction price |
| • | Allocation of the transaction price to the performance obligation in the contract |
| • | Recognition of revenue when, or as, a performance obligation is satisfied |
Identification of contracts and performance obligations
The Company accounts for a contract with a customer when there is an approval and commitment from both parties, the rights of the parties are identified, payment terms are identified, the contract has commercial substance and collectability of the consideration is probable. The Company's performance obligations consist mainly of transferring control of products and services identified in the contracts, purchase orders or invoices. For each contract, the Company considers the obligation to transfer products and services to the customer, each of which are distinct, to be performance obligations.
F-17
Transaction price and allocation to performance obligations
Transaction prices of products or services are typically based on contracted rates with customers and there is only variable consideration in limited instances. To the extent that the transaction price includes variable consideration, the Company estimates the amount of variable consideration that should be included in the transaction price utilizing the expected value method or the most likely amount, depending on the circumstances, to which the Company expects to be entitled. An expected value method may be an appropriate estimate of the amount of variable consideration if an entity has a large number of contracts with similar characteristics whereas the most likely amount method may be an appropriate estimate of the amount of variable consideration if the contract has only two possible outcomes. Variable consideration is included in the transaction price if, in the Company’s judgment, it is probable that a significant future reversal of cumulative revenue under the contract will not occur. Estimates of variable consideration and determination of whether to include estimated amounts in the transaction price are based largely on an assessment of the Company’s anticipated performance and all information (historical, current and forecasted) that is reasonably available. Sales and other taxes collected on behalf of third parties are excluded from revenue.
Consistent with industry practice, the Company generally offers customers a limited right of return for its products. Product returns are typically not significant because returns are generally not allowed unless there are product order errors or the product is damaged at time of receipt. Customers typically have a limited time frame to notify the Company of any defective or non-conforming products. The Company’s limited warranty provision is accounted for using the cost accrual method and is recognized as expense when products are sold and is not considered a separate performance obligation.
If a contract contains a single performance obligation, the entire transaction price is allocated to the single performance obligation. Contracts that contain multiple performance obligations require an allocation of the transaction price based on the estimated relative standalone selling prices of the promised products or services underlying each performance obligation. The Company determines standalone selling prices based on the price at which the performance obligation is sold separately.
Revenue Recognition
Revenue is recognized when, or as, obligations under the terms of a contract are satisfied, which occurs when control of the promised products or services is transferred to customers. Revenue is measured as the amount of consideration the Company expects to receive in exchange for transferring products or services to a customer.
Product revenue is generally recognized when the customer obtains control of the Company’s product, which occurs at a point in time, and may be upon shipment or upon delivery based on the contractual shipping terms of a contract.
Service revenue is generally recognized over time as the services are rendered to the customer based on the extent of progress towards completion of the performance obligation. The Company recognizes service revenue over the term of the service contract. Services are expected to be transferred to the customer throughout the term of the contract and the Company believes recognizing revenue ratably over the term of the contract best depicts the transfer of value to the customer. Revenue generated from preventative maintenance calls is recognized at a point in time when the services are provided to the customer.
Revenue from the sale of products and services are evidenced by either a contract with the customer or a valid purchase order and an invoice which includes all relevant terms of sale and shipment of product or service provided has been incurred. The Company performs a review of each specific customer's credit worthiness and ability to pay prior to acceptance as a customer. Further, the Company performs periodic reviews of its customers' creditworthiness prospectively.
Disaggregation of Revenue
Revenue is disaggregated from contracts between product revenue and service and other revenue and by geography, which the Company believes best depicts how the nature, amount, timing, and uncertainty of revenues and cash flows are affected by economic factors. The Company generally sells its products and services through a direct sales force in the U.S. and Germany and through direct sales and distribution agreements in other international markets outside (e.g., Japan, Europe, Canada, Latin America, Asia-Pacific, Middle East).
F-18
The following table disaggregates the Company’s revenue by products and services:
|
| Fiscal Years Ended March 31, |
| |||||||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| 2019 |
|
|
| (in $000's) |
| |||||||||
Impella product revenue |
| $ | 806,322 |
|
| $ | 806,824 |
|
| $ | 741,699 |
|
Service and other revenue |
|
| 41,200 |
|
|
| 34,059 |
|
|
| 27,733 |
|
Total revenue |
| $ | 847,522 |
|
| $ | 840,883 |
|
| $ | 769,432 |
|
The following table disaggregates the Company’s revenue by geographical location:
|
| Fiscal Years Ended March 31, |
| |||||||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| 2019 |
|
|
| (in $000's) |
| |||||||||
U.S. |
| $ | 691,579 |
|
| $ | 705,409 |
|
| $ | 665,082 |
|
Europe |
|
| 105,320 |
|
|
| 94,266 |
|
|
| 57,460 |
|
Japan |
|
| 42,868 |
|
|
| 35,215 |
|
|
| 17,502 |
|
Other international |
|
| 7,755 |
|
|
| 5,993 |
|
|
| 29,388 |
|
Total revenue |
| $ | 847,522 |
|
| $ | 840,883 |
|
| $ | 769,432 |
|
Variable Consideration
Returns Reserve
The Company estimates an allowance for future sales returns based on historical return experience, which requires judgment. The Company estimates the amount of its product sales that may be returned by its customers and records this estimate as a reduction of revenue in the period the related product revenue is recognized. The Company estimates product return liabilities using the expected value method based on its historical sales information and other factors that it believes could significantly impact its expected returns, including product discontinuations, product recalls and expirations, of which it becomes aware. The Company’s cost of replacing defective products has not been material and is accounted for at the time of replacement. The Company’s returns reserve was 0t material as of March 31, 2021 and March 31, 2020.
Rebates and Discounts
The Company provides certain customers with rebates and discounts that are defined in the Company’s contract arrangements with customers and are recorded as a reduction of revenue in the period the related revenue is recognized, resulting in a reduction to revenue and the establishment of a liability, which are all included in accrued expenses in the accompanying consolidated balance sheet. Rebates normally result from performance-based offers that are primarily based on attaining contractually specified sales volumes as well as product usage. Discounts are normally from early payment incentives. The Company estimates the amount of rebates and discounts based on an estimate of the third-party’s sales and the respective rebate or discount defined in the customer contractual arrangement. Revenue adjustments that relate to performance obligations satisfied in prior periods during the twelve months ended March 31, 2021 and 2020, were not material.
Contract Balances
Deferred Revenue
The Company’s deferred revenue balance was $24.3 million and $19.1 million as of March 31, 2021 and March 31, 2020, respectively. The deferred revenue balance is comprised of product shipments in which the Company recognizes revenue when the customer obtains control of the product, and preventative maintenance service contracts in which revenue is recognized ratably over the term of the service contract. During the fiscal year ended March 31, 2021, the Company recognized $19.0 million of revenue that was included in the deferred revenue balance as of March 31, 2020. During the fiscal year ended March 31, 2020, the Company recognized $15.6 million of revenue that was included in the deferred revenue balance as of March 31, 2019.
Costs to Obtain or Fulfill a Customer Contract
The Company has certain costs to obtain and fulfill a customer contract, such as commissions and shipping costs. The Company recognizes the incremental costs of obtaining contracts as an expense when incurred if the amortization period of the assets that the Company otherwise would have recognized is one year or less. The Company accounts for shipping and handling activities related to contracts with customers as costs to fulfill the promise to transfer the associated products. These costs are included in selling, general, and administrative expenses.
F-19
Note 5. Financial Instruments
Cash Equivalents and Marketable Securities
The Company’s cash equivalents and marketable securities are invested in the following:
|
| Amortized |
|
| Gross Unrealized |
|
| Gross Unrealized |
|
| Fair Market |
| ||||
|
| Cost |
|
| Gains |
|
| Losses |
|
| Value |
| ||||
March 31, 2021: |
| (in $000's) |
| |||||||||||||
Money market funds |
| $ | 124,297 |
|
| $ | — |
|
| $ | — |
|
| $ | 124,297 |
|
Repurchase agreements |
|
| 33,000 |
|
|
| — |
|
|
| — |
|
|
| 33,000 |
|
Total cash equivalents |
|
| 157,297 |
|
|
| — |
|
|
| — |
|
|
| 157,297 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Short-term U.S. Treasury mutual fund securities |
|
| 72,221 |
|
|
| 28 |
|
|
| — |
|
|
| 72,249 |
|
Short-term government-backed securities |
|
| 128,668 |
|
|
| 13 |
|
|
| (12 | ) |
|
| 128,669 |
|
Short-term corporate debt securities |
|
| 104,253 |
|
|
| 581 |
|
|
| (2 | ) |
|
| 104,832 |
|
Short-term commercial paper |
|
| 45,237 |
|
|
| 1 |
|
|
| (3 | ) |
|
| 45,235 |
|
Total short-term marketable securities |
|
| 350,379 |
|
|
| 623 |
|
|
| (17 | ) |
|
| 350,985 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Long-term government-backed securities |
|
| 225,231 |
|
|
| 190 |
|
|
| (37 | ) |
|
| 225,384 |
|
Long-term corporate debt securities |
|
| 38,091 |
|
|
| 630 |
|
|
| (20 | ) |
|
| 38,701 |
|
Total long-term marketable securities |
|
| 263,322 |
|
|
| 820 |
|
|
| (57 | ) |
|
| 264,085 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| $ | 770,998 |
|
| $ | 1,443 |
|
| $ | (74 | ) |
| $ | 772,367 |
|
|
| Amortized |
|
| Gross Unrealized |
|
| Gross Unrealized |
|
| Fair Market |
| ||||
|
| Cost |
|
| Gains |
|
| Losses |
|
| Value |
| ||||
March 31, 2020: |
| (in $000's) |
| |||||||||||||
Money market funds |
| $ | 115,019 |
|
| $ | — |
|
| $ | — |
|
| $ | 115,019 |
|
Repurchase agreements |
|
| 20,000 |
|
|
| — |
|
|
| — |
|
|
| 20,000 |
|
Total cash equivalents |
|
| 135,019 |
|
|
| — |
|
|
| — |
|
|
| 135,019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Short-term U.S. Treasury securities |
|
| 42,236 |
|
|
| 412 |
|
|
| — |
|
|
| 42,648 |
|
Short-term government-backed securities |
|
| 67,594 |
|
|
| 401 |
|
|
| — |
|
|
| 67,995 |
|
Short-term corporate debt securities |
|
| 107,290 |
|
|
| 94 |
|
|
| (83 | ) |
|
| 107,301 |
|
Short-term commercial paper |
|
| 32,757 |
|
|
| 74 |
|
|
| — |
|
|
| 32,831 |
|
Total short-term marketable securities |
|
| 249,877 |
|
|
| 981 |
|
|
| (83 | ) |
|
| 250,775 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Long-term government-backed securities |
|
| 90,911 |
|
|
| 153 |
|
|
| — |
|
|
| 91,064 |
|
Long-term corporate debt securities |
|
| 116,110 |
|
|
| 851 |
|
|
| (230 | ) |
|
| 116,731 |
|
Total long-term marketable securities |
|
| 207,021 |
|
|
| 1,004 |
|
|
| (230 | ) |
|
| 207,795 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| $ | 591,917 |
|
| $ | 1,985 |
|
| $ | (313 | ) |
| $ | 593,589 |
|
Derivative Instruments
In October 2019, the Company entered into an intercompany agreement in which it loaned 85.0 million Euro to Abiomed Europe GMBH, its German subsidiary. In conjunction with this intercompany loan agreement, the Company entered into a cross-currency swap agreement to convert a notional amount of 85.0 million Euro equivalent to a $93.5 million denominated intercompany loan into U.S. dollars. The objective of this cross-currency swap is to hedge the variability of cash flows related to the forecasted interest and principal payments on the Euro denominated fixed rate loan against changes in the exchange rate between the U.S. dollar and the Euro. Under the terms of this cross-currency swap contract, which has been designated as a cash flow hedge, the Company will make interest payments in Euro and receive interest in U.S. dollars. Upon the maturity of this contract, the Company will pay the principal amount of the loan in Euro and receive U.S. dollars from the counterparty. The cross-currency swap is carried on the consolidated balance sheet at fair value, and changes in the fair values are recorded as unrealized gains or losses in accumulated other comprehensive income (loss). The Company does not enter into derivative instruments for any purpose other than cash flow hedging.
F-20
The following table summarizes the terms of the cross-currency swap agreement as of March 31, 2021 (dollar amounts in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
| Effective Date |
| Maturity |
| Fixed Rate |
|
| Aggregate Notional Amount (in $000's) |
| ||
Pay EUR | October 15, |
| October 15, |
| 2.75% |
|
|
| EUR 85,000 |
| |
Receive U.S.$ | 2019 |
| 2024 |
| 4.64% |
|
|
| USD 93,457 |
| |
The following table presents the fair value of the Company’s derivative instrument as follows:
|
|
|
| March 31, 2021 |
|
| March 31, 2020 |
| ||
Derivatives designated as hedging instruments under ASC 815 |
| Balance sheet classification |
| (in $000's) |
| |||||
Cross-currency swap |
| (Long-term liabilities) other assets |
| $ | (4,298 | ) |
| $ | 3,786 |
|
The Company has structured its cross-currency swap agreement to be 100% effective and, as a result, there was no net impact to earnings resulting from hedge ineffectiveness. Changes in the fair value of the cross-currency swap are designated as a hedging instrument that effectively offsets the variability of cash flows are reported in accumulated other comprehensive income (loss). These amounts subsequently are reclassified into the consolidated income statement in the same period in which the related hedged item affects earnings. The change of fair value of the cross-currency swap during the fiscal year 2021 was solely due to the appreciation of the Euro against the U.S. dollar.
For the fiscal year 2021 and 2020, the Company recorded income related to the interest rate differential of the cross-currency swaps of $1.6 million and $0.8 million, respectively, in other income (expense), net, included in the consolidated statements of operations.
Contingent Consideration
Contingent consideration represents potential milestones that the Company may pay as additional consideration related to acquired businesses. The Company has contingent consideration potentially payable related to the acquisition of ECP Entwicklungsgesellschaft mbH (“ECP”) in July 2014 and the acquisition of Breethe in April 2020. The fair value of the contingent consideration at each reporting date is updated by reflecting the changes in fair value reflected within research and development expenses in the Company’s consolidated statement of operations. Significant increases or decreases in any valuation assumptions, including probabilities of success or changes in expected timelines for achievement of any of these milestones, could result in a significantly higher or lower fair value of the contingent consideration liability. There is no assurance that any of the conditions for the milestone payments will be met.
The components of contingent consideration liability are as follows:
|
| March 31, 2021 |
|
| March 31, 2020 |
| ||
|
| (in $000's) |
| |||||
ECP |
| $ | 10,306 |
|
| $ | 9,000 |
|
Breethe |
|
| 14,400 |
|
|
| — |
|
|
| $ | 24,706 |
|
| $ | 9,000 |
|
ECP
In July 2014, the Company acquired ECP and AIS GmbH Aachen Innovative Solutions (“AIS”) for $13.0 million in cash, with additional potential payouts totaling $15.0 million based on the achievement of CE Mark approval in the European Union and a revenue-based milestone related to the development of the future Impella ECPTM expandable catheter pump technology. These potential milestone payments may be made, at the Company’s option, by a combination of cash or ABIOMED common stock.
The Company used a combination of an income approach, based on various revenue and cost assumptions and applying a probability to each outcome and a Monte-Carlo valuation model, both of which consider significant unobservable inputs. For the clinical and regulatory milestone, probabilities were applied to each potential scenario and the resulting values were discounted using a rate that considers weighted average cost of capital as well as a specific risk premium associated with the riskiness of the earn out itself, the related projections, and the overall business. The revenue-based milestone is valued using a Monte-Carlo valuation model, which simulates estimated future revenues during the earn out-period using management’s best estimates.
F-21
Key unobservable inputs include the discount rate used to present value the projected revenues and cash flows (ranging from 1.2% to 17%), the mean probability of achieving the various technical, regulatory and commercial milestones of 69% and projected revenues, which are based on the Company’s operational forecasts and long-range strategic plans.
Breethe
In April 2020, the Company acquired Breethe for $55.0 million in cash, with additional potential payouts up to a maximum of $55.0 million payable based on the achievement of certain technical, regulatory and commercial milestones.
The Company used a combination of an income approach, based on various revenue and cost assumptions and applying a probability to each outcome and a Monte-Carlo valuation model, both of which consider significant unobservable inputs. For the regulatory milestones, probabilities were applied to each potential scenario and the resulting values were discounted using a rate that considers weighted average cost of capital as well as a specific risk premium associated with the earn out itself, the related projections, and the overall business. The commercial milestones are valued using a Monte-Carlo valuation model, which simulates estimated future revenues during the earn out-period using management’s best estimates.
Key unobservable inputs include the discount rates used to present value the projected revenues and cash flows (ranging from 1.4% to 2%), the probability of achieving the various technical, regulatory and commercial milestones (estimated to range from 25% to 75%) and projected revenues, which are based on the Company’s operational forecasts and long-range strategic plans.
Fair Value Hierarchy
Fair value is defined as the price that would be received upon the sale of an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Financial assets and liabilities carried at fair value are to be classified and disclosed in one of the following three categories:
Level 1: Quoted market prices in active markets for identical assets or liabilities.
Level 2: Observable market-based inputs or unobservable inputs that are corroborated by market data.
Level 3: Unobservable inputs that are not corroborated by market data.
Level 1 primarily consists of financial instruments whose values are based on quoted market prices such as exchange-traded instruments and listed equities.
Level 2 includes financial instruments that are valued using models or other valuation methodologies. These models are primarily industry-standard models that consider various assumptions, including time value, yield curve, volatility factors, prepayment speeds, default rates, loss severity, current market and contractual prices for the underlying financial instruments, as well as other relevant economic measures. Substantially all of these assumptions are observable in the marketplace, can be derived from observable data or are supported by observable levels at which transactions are executed in the marketplace.
Level 3 is comprised of unobservable inputs that are supported by little or no market activity. Financial assets are considered Level 3 when their fair values are determined using pricing models, discounted cash flows or similar techniques and at least one significant model assumption or input is unobservable.
F-22
The following table presents the Company’s fair value hierarchy for its financial instruments measured at fair value:
|
| Level 1 |
|
| Level 2 |
|
| Level 3 |
|
| Total |
| ||||
March 31, 2021: |
| (in $000's) |
| |||||||||||||
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds |
| $ | 124,297 |
|
| $ | — |
|
| $ | — |
|
| $ | 124,297 |
|
Repurchase agreements |
|
| — |
|
|
| 33,000 |
|
|
| — |
|
|
| 33,000 |
|
Short-term U.S. Treasury securities |
|
| — |
|
|
| 72,249 |
|
|
| — |
|
|
| 72,249 |
|
Short-term government-backed securities |
|
| — |
|
|
| 128,669 |
|
|
| — |
|
|
| 128,669 |
|
Short-term corporate debt securities |
|
| — |
|
|
| 104,832 |
|
|
| — |
|
|
| 104,832 |
|
Short-term commercial paper |
|
| — |
|
|
| 45,235 |
|
|
| — |
|
|
| 45,235 |
|
Long-term government-backed securities |
|
| — |
|
|
| 225,384 |
|
|
| — |
|
|
| 225,384 |
|
Long-term corporate debt securities |
|
| — |
|
|
| 38,701 |
|
|
| — |
|
|
| 38,701 |
|
Investment in Shockwave Medical |
|
| 38,655 |
|
|
| — |
|
|
| — |
|
|
| 38,655 |
|
Liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cross currency swap agreement |
|
| — |
|
|
| 4,298 |
|
|
| — |
|
|
| 4,298 |
|
Contingent consideration |
|
| — |
|
|
| — |
|
|
| 24,706 |
|
|
| 24,706 |
|
|
| Level 1 |
|
| Level 2 |
|
| Level 3 |
|
| Total |
| ||||
March 31, 2020: |
| (in $000's) |
| |||||||||||||
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds |
| $ | 115,019 |
|
| $ | — |
|
| $ | — |
|
| $ | 115,019 |
|
Repurchase agreements |
|
| — |
|
|
| 20,000 |
|
|
| — |
|
|
| 20,000 |
|
Short-term U.S. Treasury securities |
|
| — |
|
|
| 42,648 |
|
|
| — |
|
|
| 42,648 |
|
Short-term government-backed securities |
|
| — |
|
|
| 67,995 |
|
|
| — |
|
|
| 67,995 |
|
Short-term corporate debt securities |
|
| — |
|
|
| 107,301 |
|
|
| — |
|
|
| 107,301 |
|
Short-term commercial paper |
|
| — |
|
|
| 32,831 |
|
|
| — |
|
|
| 32,831 |
|
Long-term government-backed securities |
|
| — |
|
|
| 91,064 |
|
|
| — |
|
|
| 91,064 |
|
Long-term corporate debt securities |
|
| — |
|
|
| 116,731 |
|
|
| — |
|
|
| 116,731 |
|
Cross currency swap agreement |
|
| — |
|
|
| 3,786 |
|
|
| — |
|
|
| 3,786 |
|
Investment in Shockwave Medical |
|
| 55,704 |
|
|
| — |
|
|
| — |
|
|
| 55,704 |
|
Liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contingent consideration |
|
| — |
|
|
| — |
|
|
| 9,000 |
|
|
| 9,000 |
|
The Company has determined that the estimated fair value of its money market funds and its investment in Shockwave Medical, a publicly traded medical device company, are reported as Level 1 financial assets as they are valued at quoted market prices in active markets. The investment in Shockwave Medical is classified within other assets in the consolidated balance sheet.
The Company has determined that the estimated fair value of its repurchase agreements, U.S. Treasury mutual fund securities, government-backed securities, corporate debt securities and commercial paper and cross-currency swap agreement are reported as Level 2 financial assets as they are based on model-driven valuations in which all significant inputs are observable, or can be derived from or corroborated by observable market data for substantially the full term of the asset.
Level 3 Financial Liabilities
This contingent consideration liability is reported as Level 3 as the estimated fair value of the contingent consideration related to the acquisitions of ECP and Breethe require significant management judgment or estimation. Quantitative information about the significant unobservable inputs utilized by the Company in the fair value measurements are described above.
This contingent consideration liability is reported as Level 3 as the estimated fair value of the contingent consideration related to the acquisitions of ECP and Breethe require significant management judgment or estimation and is calculated as described above.
F-23
The following table summarizes the change in fair value, as determined by Level 3 inputs, of the contingent consideration:
|
| Fiscal Years Ended March 31, |
| |||||||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| 2019 |
|
|
| (in $000's) |
| |||||||||
Level 3 liabilities, beginning balance |
| $ | 9,000 |
|
| $ | 9,575 |
|
| $ | 10,490 |
|
Additions |
|
| 13,300 |
|
|
| — |
|
|
| — |
|
Payments |
|
| — |
|
|
| — |
|
|
| — |
|
Change in fair value |
|
| 2,406 |
|
|
| (575 | ) |
|
| (915 | ) |
Level 3 liabilities, ending balance |
| $ | 24,706 |
|
| $ | 9,000 |
|
| $ | 9,575 |
|
The change in fair value of the contingent consideration was primarily due to the addition of contingent consideration related to the Breethe acquisition and the passage of time impact on fair value measurements.
Information About Uncertainty of Level 3 Fair Value Measurements
The significant unobservable inputs used in the fair value of the Company’s contingent consideration are the discount rate and forecasted financial information. Significant increases (decreases) in the discount rate would have resulted in a significantly lower (higher) fair value measurement. Significant increases (decreases) in the forecasted financial information would have resulted in a significantly higher (lower) fair value measurement. As of March 31, 2021 and 2020, the present value of expected payments related to the Company’s contingent consideration was $24.7 million and $9.0 million, respectively. The undiscounted value of the payments, assuming that all contingencies are met, would be $70.0 million and $15.0 million, respectively.
Note 6. Accounts Receivable
The components of accounts receivable are as follows:
|
| March 31, 2021 |
|
| March 31, 2020 |
| ||
|
| (in $000's) |
| |||||
Trade receivables |
| $ | 97,953 |
|
| $ | 85,852 |
|
Allowance for doubtful accounts |
|
| (774 | ) |
|
| (1,202 | ) |
|
| $ | 97,179 |
|
| $ | 84,650 |
|
The following table summarizes activity in the allowance for doubtful accounts:
|
| Fiscal Years Ended March 31, |
| |||||||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| 2019 |
|
|
| (in $000's) |
| |||||||||
Balance at beginning of year |
| $ | 1,202 |
|
| $ | 1,040 |
|
| $ | 320 |
|
Additions (recoveries) |
|
| (127 | ) |
|
| 487 |
|
|
| 720 |
|
Write-offs |
|
| (301 | ) |
|
| (325 | ) |
|
| — |
|
Balance at end of year |
| $ | 774 |
|
| $ | 1,202 |
|
| $ | 1,040 |
|
Note 7. Inventories
The components of inventories are as follows:
|
| March 31, 2021 |
|
| March 31, 2020 |
| ||
|
| (in $000's) |
| |||||
Raw materials and supplies |
| $ | 27,782 |
|
| $ | 31,411 |
|
Work-in-progress |
| $ | 35,187 |
|
|
| 35,008 |
|
Finished goods |
| $ | 18,090 |
|
|
| 23,669 |
|
|
| $ | 81,059 |
|
| $ | 90,088 |
|
The Company’s inventories relate to its Impella® and OXY-1 System product platform. Finished goods and work-in-process inventories consist of direct material, labor and overhead.
F-24
Note 8. Property and Equipment
The components of property and equipment are as follows:
|
| March 31, 2021 |
|
| March 31, 2020 |
| ||
|
| (in $000's) |
| |||||
Land |
| $ | 10,875 |
|
| $ | 7,179 |
|
Building and building improvements |
|
| 148,870 |
|
|
| 100,176 |
|
Leasehold improvements |
|
| 439 |
|
|
| 14,546 |
|
Machinery and equipment |
|
| 91,784 |
|
|
| 71,636 |
|
Furniture and fixtures |
|
| 15,608 |
|
|
| 14,600 |
|
Construction in progress |
|
| 10,906 |
|
|
| 15,075 |
|
Total cost |
|
| 278,482 |
|
|
| 223,212 |
|
Less accumulated depreciation |
|
| (81,353 | ) |
|
| (58,281 | ) |
|
| $ | 197,129 |
|
| $ | 164,931 |
|
In March 2021, the Company acquired the building adjacent to its corporate headquarters that it had been leasing in Danvers, Massachusetts. The total acquisition cost for the land and building was approximately $17.5 million, with $3.4 million being recorded to land and $13.8 million being recorded to building and building improvements. In addition, the Company reclassified $11.0 million in leasehold improvements and $4.7 million in right-of-use assets and recorded a $0.5 million adjustment to remove the prior lease liability due to the termination of the lease agreement upon the property acquisition.
Depreciation expense related to property and equipment was $23.1 million, $20.1 million, and $13.9 million for the fiscal years ending March 31, 2021, 2020 and 2019, respectively.
Note 9. Goodwill, In-Process Research & Development and Other Assets
Goodwill
The carrying amount of goodwill as of March 31, 2021 and 2020 was $78.6 million and $32.0 million, respectively, and has been recorded in connection with the Company’s acquisition of Impella Cardiosystems AG, in May 2005, ECP in July 2014 and Breethe in April 2020. The goodwill activity is as follows:
|
| (in $000's) |
| |
Balance, March 31, 2019 |
| $ | 32,601 |
|
Foreign currency translation impact |
|
| (632 | ) |
Balance, March 31, 2020 |
| $ | 31,969 |
|
Breethe acquisition |
|
| 44,485 |
|
Foreign currency translation impact |
|
| 2,114 |
|
Balance, March 31, 2021 |
| $ | 78,568 |
|
The Company evaluates goodwill at least annually on October 31, as well as whenever events or changes in circumstances suggest that the carrying amount may not be recoverable. The Company has 0 accumulated impairment losses on goodwill.
F-25
Other Intangible Assets, net
Other intangible assets consist of the following:
|
|
|
|
|
| March 31, 2021 |
|
| March 31, 2020 |
|
| ||||||||||||||||||
|
| Weighted Average Useful Life (in years) |
|
| Cost |
|
| Accumulated Amortization |
|
| Net Carrying Value |
|
| Cost |
|
| Accumulated Amortization |
|
| Net Carrying Value |
|
| |||||||
|
|
|
|
|
| (in $000's) |
|
| |||||||||||||||||||||
Finite-lived intangible assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Developed technology |
|
| 14.6 |
|
| $ | 27,000 |
|
| $ | (750 | ) |
| $ | 26,250 |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Indefinite-lived intangible assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
In-process research and development |
|
|
|
|
|
| 15,900 |
|
|
| — |
|
|
| 15,900 |
|
|
| 14,913 |
|
|
| — |
|
|
| 14,913 |
|
|
Total |
|
|
|
|
| $ | 42,900 |
|
| $ | (750 | ) |
| $ | 42,150 |
|
| $ | 14,913 |
|
|
| — |
|
| $ | 14,913 |
|
|
The Company’s finite-lived intangible asset represents developed technology associated with the estimated fair value of the OXY-1 System. Upon receiving FDA 510(k) clearance of the OXY-1 System in October 2020, the Company reclassified the IPR&D asset of $27.0 million during the quarter ended December 31, 2020 from the acquisition of Breethe (see Note 3) to finite-lived developed technology intangible assets and began amortizing the intangible asset on a straight-line basis over an estimated useful life of 15 years. The estimated fair value of developed technology was determined using a probability-weighted income approach, which discounts expected future cash flows to present value. The projected cash flow estimates for the OXY-1 System were based on certain key assumptions, including estimates of future revenue and expenses, the stage of development of the technology at the acquisition date and the time and resources needed to complete development.
The Company’s IPR&D assets represents the estimated fair value of the Impella ECPTM related to the acquisition of ECP and AIS, in July 2014. The estimated fair value of the IPR&D assets at the acquisition date was determined using a probability-weighted income approach, which discounts expected future cash flows to present value. The projected cash flow estimates for the future Impella ECPTM expandable catheter pump were based on certain key assumptions, including estimates of future revenue and expenses, taking into account the stage of development of the technology at the acquisition date and the time and resources needed to complete development.
The Company evaluates the other intangible assets at least annually on October 31, as well as whenever events or changes in circumstances suggest that the carrying amount may not be recoverable. The Company has 0 accumulated impairment losses on other intangible assets. The change in the indefinite-lived intangible assets balance for the fiscal year ended March 31, 2021 was related to foreign currency translation impact.
Note 10. Other Assets
The components of other assets are as follows:
|
| Fiscal Years Ended March 31, |
| |||||||
|
| 2021 |
|
|
|
| 2020 |
| ||
|
| (in $000's) |
| |||||||
Investment in Shockwave Medical |
| $ | 38,655 |
|
|
|
| $ | 55,704 |
|
Other investments |
|
| 62,995 |
|
|
|
|
| 38,741 |
|
Operating lease right-of-use assets (Note 11) |
|
| 6,109 |
|
|
|
|
| 11,760 |
|
Cross-currency swap (Note 5) |
|
| — |
|
|
|
|
| 3,786 |
|
Other intangible assets and other assets |
|
| 5,323 |
|
|
|
|
| 7,664 |
|
Total other assets |
| $ | 113,082 |
|
|
|
| $ | 117,655 |
|
F-26
Investment in Shockwave Medical
In fiscal year 2019, the Company invested $25.0 million in Shockwave Medical, a publicly traded medical device company. During the fiscal year ended March 31, 2021, the Company sold approximately 1.4 million of its shares for cash proceeds of $67.9 million in which it realized a gain of $47.3 million. The fair value of this investment as of March 31, 2021 and 2020 was $38.7 million and $55.7 million. Amounts recorded in other income (expense) were gains of $50.8 million and $0.5 million for the years ended March 31, 2021 and 2020, respectively. The Company held 0.3 million shares of Shockwave Medical as of March 31, 2021.
Other Investments
The Company periodically makes investments in medical device companies that focus on heart failure and heart pumps and other medical device technologies. The carrying value of the Company’s portfolio of other investments and the changes in the balance were as follows:
|
| Fiscal Years Ended March 31, |
| |||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| (in $000's) |
| |||||
Beginning balance |
| $ | 38,741 |
|
| $ | 24,584 |
|
Additions |
|
| 26,104 |
|
|
| 19,599 |
|
Reclassification |
|
| (2,000 | ) |
|
| - |
|
Impairment |
|
| (800 | ) |
|
| (750 | ) |
Change in fair value, net |
|
| 950 |
|
|
| (4,692 | ) |
Ending balance |
| $ | 62,995 |
|
| $ | 38,741 |
|
Other Intangible Assets and Other Assets
The Company’s other intangible assets and other assets is comprised primarily of license manufacturing rights to certain technology from third parties and other long-term assets such as prepaid expenses.
Note 11. Leases
Lessee
The Company has lease agreements for real estate including corporate offices and warehouse space, vehicles and certain office equipment.
At the inception of a contractual arrangement, the Company determines whether it contains a lease by assessing whether there is an identified asset and whether the contract conveys the right to control the use of the identified asset in exchange for consideration over a period of time. If both criteria are met, the Company calculates the associated lease liability and corresponding right-of-use asset upon lease commencement. Operating lease assets and liabilities are recognized based on the present value of minimum lease payments over the lease term using an appropriate discount rate. Right-of-use assets also include any lease payments made at or before lease commencement and any initial direct costs incurred and exclude any lease incentives received.
The discount rate used is the rate that the Company would have to pay to borrow on a collateralized basis over a similar term for an amount equal to the lease payments in a similar economic environment. At the lease commencement date, the discount rate implicit in the lease is used to discount the lease liability, if readily determinable. If not readily determinable or lease do not contain an implicit rate, the Company’s incremental borrowing rate is used as the discount rate. Discount rates are updated when there is a new lease or a modification to an existing lease, and the methodology is reassessed annually.
The Company records operating lease liabilities within current liabilities or long-term liabilities based upon the length of time associated with the lease payments. The Company records its operating lease right-of-use assets as long-term assets. Leases with an initial term of 12 months or less are not recognized on the consolidated balance sheet. The Company have elected the practical expedient where lease agreements with lease and non-lease components are accounted for as a single lease component for all assets. The Company’s financing leases are not material to our financial statements.
F-27
The Company adopted ASC Topic 842 on April 1, 2019 using the optional transition method. As such, the disclosures required under ASC Topic 842 are not presented for periods before the date of adoption.
The Company elected the package of transitional practical expedients for leases existing prior to the adoption date. The Company did not reassess whether existing contracts are or contain leases, leases retained their historical lease classification and initial direct costs were not reassessed for capitalization under the new standard. Operating lease assets and liabilities were recognized based on the present value of minimum lease payments over the remaining lease term as of the adoption date.
The following table presents supplemental balance sheet information related to the Company’s operating leases:
|
| March 31, 2021 |
|
| March 31, 2020 |
| ||
|
| (in $000's) |
| |||||
Assets |
|
|
|
|
|
|
|
|
Operating lease right-of-use assets in other assets |
| $ | 6,109 |
|
| $ | 11,760 |
|
Liabilities |
|
|
|
|
|
|
|
|
Operating lease liabilities in other current liabilities |
|
| 2,459 |
|
|
| 3,671 |
|
Operating lease liabilities in other long-term liabilities |
|
| 3,657 |
|
|
| 8,549 |
|
Total operating lease liabilities |
| $ | 6,116 |
|
| $ | 12,220 |
|
In March 2021, the Company acquired the building adjacent to its corporate headquarters that it had been leasing in Danvers, Massachusetts. The total acquisition cost for the land and building was approximately $17.5 million, with $3.4 million being recorded to land and $13.8 million being recorded to building and building improvements. In addition, the Company reclassified $11.0 million in leasehold improvements and $4.7 million in right-of-use assets and recorded a $0.5 million adjustment to remove the prior lease liability due to the termination of the lease agreement upon the property acquisition.
Expense charged to operations under operating leases was $4.1 million and $3.7 million during the fiscal years ended March 31, 2021 and 2020, respectively. Short-term lease expenses were not material.
Maturities of operating lease liabilities as of March 31, 2021 are as follows:
(in thousands, except lease term and discount rate) |
| |||
|
|
|
|
|
Fiscal Years Ended March 31, |
|
|
|
|
2022 |
| $ | 2,574 |
|
2023 |
|
| 1,404 |
|
2024 |
|
| 1,165 |
|
2025 |
|
| 617 |
|
2026 |
|
| 77 |
|
Thereafter |
|
| 599 |
|
Total minimum lease payments |
|
| 6,436 |
|
Less: imputed interest |
|
| (320 | ) |
Present value of operating lease liabilities |
| $ | 6,116 |
|
|
|
|
|
|
Weighted average remaining lease term |
| 4.02 |
| |
|
|
|
|
|
Weighted average discount rate |
|
| 1.96 | % |
Lessor
In March 2021, as part of the $17.5 million purchase of a building located in Danvers, Massachusetts, we assumed existing leases with third parties for a portion of the building which are classified as operating leases. The leases have annual escalating payments and the latest expires in March 2025 in accordance with the terms and conditions of the existing agreement. For the year ended March 31, 2021, operating lease income was not material.
F-28
Note 12. Accrued Expenses
Accrued expenses consisted of the following:
|
| March 31, 2021 |
|
| March 31, 2020 |
| ||
|
| (in $000's) |
| |||||
Employee compensation |
| $ | 40,954 |
|
| $ | 32,273 |
|
Research and development |
|
| 6,983 |
|
|
| 5,749 |
|
Sales and income taxes |
|
| 5,914 |
|
|
| 21,641 |
|
Marketing |
|
| 3,674 |
|
|
| 1,705 |
|
Warranty |
|
| 2,053 |
|
|
| 1,818 |
|
Professional, legal and accounting fees |
|
| 1,957 |
|
|
| 6,880 |
|
Other |
|
| 4,511 |
|
|
| 5,041 |
|
|
| $ | 66,046 |
|
| $ | 75,107 |
|
Employee compensation consists primarily of accrued bonuses, accrued commissions and accrued employee benefits as of March 31, 2021 and 2020.
Note 13. Stockholders’ Equity
Class B Preferred Stock
The Company has authorized 1,000,000 shares of Class B Preferred Stock, $.01 par value, of which the board of directors can set the designation, rights and privileges. NaN shares of Class B Preferred Stock have been issued or are outstanding.
Stock Repurchase Program
In August 2019, the Company’s Board of Directors authorized a stock repurchase program for up to $200.0 million of shares of its common stock. Under this stock repurchase program, the Company is authorized to repurchase shares through open market purchases, privately negotiated transactions or otherwise in accordance with applicable federal securities laws, including through Rule 10b5-1 trading plans and under Rule 10b-18 of the Exchange Act. The stock repurchase program has no time limit and may be suspended for periods or discontinued at any time. The Company is funding the stock repurchase program with its available cash and marketable securities. Through fiscal year 2021 and 2020, the Company has repurchased a total of 67,649 and 465,687 shares, respectively, at an aggregate cost of $11.3 million and $84.9 million, respectively. The remaining authorization under the stock repurchase program was $103.8 million as of March 31, 2021.
The following table provides stock repurchase activities:
|
| For the Year Ended March 31, |
| |||||
|
| 2021 |
|
| 2020 |
| ||
Shares repurchased |
|
| 67,649 |
|
|
| 465,687 |
|
Average price per share |
| $ | 167.19 |
|
| $ | 182.27 |
|
Value of shares repurchased (in millions) |
| $ | 11.3 |
|
| $ | 84.9 |
|
F-29
Accumulated Other Comprehensive Income (Loss)
The components of accumulated other comprehensive income (loss), are as follows (in thousands):
|
| Foreign Currency Translation Gains (Losses) |
|
| Unrealized Gains (Losses) on Investments |
|
| Unrealized Gains (Losses) on Derivative Instruments |
|
| Total |
| ||||
|
| (in $000's) |
| |||||||||||||
Balance, April 1, 2018 |
|
| (3,597 | ) |
|
| (607 | ) |
|
| — |
|
|
| (4,204 | ) |
Other comprehensive (loss) income |
|
| (11,431 | ) |
|
| 946 |
|
|
| — |
|
|
| (10,485 | ) |
Balance, March 31, 2019 |
|
| (15,028 | ) |
|
| 339 |
|
|
| — |
|
|
| (14,689 | ) |
Other comprehensive income (loss) |
|
| (1,832 | ) |
|
| 1,333 |
|
|
| 3,999 |
|
|
| 3,500 |
|
Balance, March 31, 2020 |
|
| (16,860 | ) |
|
| 1,672 |
|
|
| 3,999 |
|
|
| (11,189 | ) |
Other comprehensive (loss) income |
|
| 2,142 |
|
|
| (303 | ) |
|
| (2,095 | ) |
|
| (256 | ) |
Balance, March 31, 2021 |
| $ | (14,718 | ) |
| $ | 1,369 |
|
| $ | 1,904 |
|
|
| (11,445 | ) |
Note 14. Stock Award Plans and Stock-Based Compensation
Stock Award Plans
The Company grants stock options and restricted stock awards to employees and others. All outstanding stock options of the Company as of March 31, 2021 were granted with an exercise price equal to the fair market value on the date of grant. Outstanding stock options, if not exercised, expire 10 years from the date of grant.
2015 Stock Incentive Plan
The Company’s 2015 Amended and Restated Omnibus Incentive Plan (the “2015 Plan”) authorizes the grant of a variety of equity awards to the Company’s officers, directors, employees, consultants and advisers, including awards of unrestricted and restricted stock, restricted stock units, incentive and nonqualified stock options to purchase shares of common stock, performance share awards and stock appreciation rights. The 2015 Plan provides that options may only be granted at the current market value on the date of grant. Each share of stock issued pursuant to a stock option or stock appreciation right counts as one share against the maximum number of shares issuable under the 2015 Plan, while each share of stock issued pursuant to any other type of award counts as 1.8 shares against the maximum number of shares issuable under the 2015 Plan. The Company’s policy for issuing shares upon exercise of stock options or the vesting of its restricted stock awards and restricted stock units is to issue shares of common stock at the time of exercise or conversion. As of March 31, 2021, a total of approximately 3,334,890 shares were available for future issuance under the 2015 Plan.
Stock-Based Compensation
The following table summarizes stock-based compensation expense by financial statement line item in the Company’s consolidated statements of operations:
|
| Fiscal Years Ended March 31, |
| |||||||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| 2019 |
|
|
| (in $000's) |
| |||||||||
Cost of revenue |
| $ | 3,760 |
|
| $ | 2,641 |
|
| $ | 2,643 |
|
Research and development |
|
| 6,941 |
|
|
| 5,534 |
|
|
| 9,312 |
|
Selling, general and administrative |
|
| 36,305 |
|
|
| 31,606 |
|
|
| 42,539 |
|
|
| $ | 47,006 |
|
| $ | 39,781 |
|
| $ | 54,494 |
|
F-30
The components of stock-based compensation were as follows:
|
| Fiscal Years Ended March 31, |
| |||||||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| 2019 |
|
|
| (in $000's) |
| |||||||||
Restricted stock units |
| $ | 36,347 |
|
| $ | 28,895 |
|
| $ | 45,998 |
|
Stock options |
|
| 8,982 |
|
|
| 9,006 |
|
|
| 7,445 |
|
Employee stock purchase plan |
|
| 1,677 |
|
|
| 1,880 |
|
|
| 1,051 |
|
|
| $ | 47,006 |
|
| $ | 39,781 |
|
| $ | 54,494 |
|
Stock Options
The following table summarizes stock option activity for the fiscal year ended March 31, 2021:
|
|
|
|
|
|
|
|
|
| Weighted |
|
|
|
|
| |
|
|
|
|
|
| Weighted |
|
| Average |
|
| Aggregate |
| |||
|
|
|
|
|
| Average |
|
| Remaining |
|
| Intrinsic |
| |||
|
| Options |
|
| Exercise |
|
| Contractual |
|
| Value |
| ||||
|
| (in thousands) |
|
| Price |
|
| Term (years) |
|
| (in thousands) |
| ||||
Outstanding at beginning of period |
|
| 869 |
|
| $ | 112.03 |
|
|
| 5.30 |
|
|
|
|
|
Granted |
|
| 70 |
|
|
| 225.60 |
|
|
|
|
|
|
|
|
|
Exercised |
|
| (215 | ) |
|
| 42.16 |
|
|
|
|
|
|
|
|
|
Cancelled and expired |
|
| (13 | ) |
|
| 248.42 |
|
|
|
|
|
|
|
|
|
Outstanding at end of period |
|
| 711 |
|
| $ | 141.87 |
|
|
| 5.46 |
|
| $ | 129,912 |
|
Exercisable at end of period |
|
| 537 |
|
| $ | 104.55 |
|
|
| 4.51 |
|
| $ | 117,880 |
|
Options vested and expected to vest at end of period |
|
| 711 |
|
| $ | 141.87 |
|
|
| 5.46 |
|
| $ | 129,912 |
|
Stock options generally vest and become exercisable annually over three years. The remaining unrecognized stock-based compensation expense for unvested stock option awards as of March 31, 2021 was approximately $8.9 million and the weighted-average period over which this cost will expected to be recognized is 1.6 years.
The aggregate intrinsic value of options exercised for fiscal years 2021, 2020 and 2019 was $54.9 million, $15.0 million and $174.0 million, respectively. The total cash received as a result of employee stock option exercises during the fiscal years ended March 31, 2021, 2020 and 2019 was approximately $9.1 million, $3.7 million and $12.9 million, respectively.
The Company estimates the fair value of each stock option granted at the grant date using the Black-Scholes option valuation model. The weighted average grant-date fair values and weighted average assumptions used in the calculation of fair value of options granted were as follows:
|
| Fiscal Years Ended March 31, |
| |||||||||
|
|
| 2021 |
|
|
| 2020 |
|
|
| 2019 |
|
Valuation assumptions: |
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average grant-date fair value |
| $ | 77.82 |
|
| $ | 93.05 |
|
| $ | 141.47 |
|
Risk-free interest rate |
|
| 0.32 | % |
|
| 1.97 | % |
|
| 2.91 | % |
Expected option life (years) |
|
| 4.22 |
|
|
| 4.14 |
|
|
| 4.04 |
|
Expected volatility |
|
| 42.9 | % |
|
| 42.3 | % |
|
| 42.8 | % |
The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant for a term consistent with the expected life of the stock options. Volatility assumptions are calculated based on the historical volatility of the Company’s stock. The Company estimates the expected term of options based on historical exercise experience and estimates of future exercises of unexercised options. An expected dividend yield of 0 is used in the option valuation model because the Company does not pay cash dividends and does not expect to pay any cash dividends in the foreseeable future. Forfeitures are recorded as they occur instead of estimating forfeitures that are expected to occur.
F-31
Restricted Stock Units
The following table summarizes restricted stock unit activity for the fiscal year ended March 31, 2021:
|
| Number of Shares |
|
| Weighted Average Grant Date Fair Value |
| ||
|
| (in thousands) |
|
| (per share) |
| ||
Restricted stock units at beginning of period |
|
| 320 |
|
| $ | 263.92 |
|
Granted |
|
| 230 |
|
|
| 254.78 |
|
Vested |
|
| (139 | ) |
|
| 227.32 |
|
Forfeited |
|
| (109 | ) |
|
| 266.57 |
|
Restricted stock units at end of period |
|
| 301 |
|
| $ | 273.57 |
|
Restricted stock units generally vest annually over three years. The remaining unrecognized compensation expense for outstanding restricted stock units, including performance-based awards, as of March 31, 2021 was $51.9 million and the weighted-average period over which this cost will expected to be recognized is 1.8 years.
The weighted average grant-date fair value for restricted stock units granted during the fiscal years ended March 31, 2021, 2020 and 2019 was $254.78, $258.59 and $376.95 per share, respectively. The total fair value of restricted stock units vested in fiscal years 2021, 2020 and 2019 was $27.9 million, $99.3 million and $171.3 million, respectively.
Performance Awards
Restricted stock units include certain awards that vest subject to certain performance. The remaining unrecognized compensation expense for outstanding performance restricted stock units as of March 31, 2021 was $24.2 million and the weighted-average period over which this cost will be recognized is 1.8 years.
In November 2020, performance-based awards of restricted stock units for the potential issuance of up to 66,000 shares of common stock were issued to certain executive officers, which vest upon achievement of prescribed service milestones by the award recipients and the achievement of prescribed performance milestones by the Company.
In May 2020, performance-based awards of restricted stock units for the potential issuance of up to 62,000 shares of common stock were issued to certain executive officers and employees, which vest upon achievement of prescribed service milestones by the award recipients and the achievement of prescribed performance milestones by the Company.
In May 2019, performance-based awards of restricted stock units for the potential issuance of up to 196,580 shares of common stock were issued to certain executive officers and employees, which vest upon achievement of prescribed service milestones by the award recipients and the achievement of prescribed performance milestones by the Company. The Company did not meet the prescribed performance milestones in fiscal year 2020 and therefore 0 shares vested for these performance-based awards and the Company reversed all previously recorded stock stock-based compensation expense related to this award in the three months ended March 31, 2020.
In May 2018, performance-based awards of restricted stock units for the potential issuance of 114,000 shares of common stock were issued to certain executive officers and employees, which vest upon achievement of prescribed service milestones by the award recipients and performance milestones by the Company. The Company met the prescribed performance milestones in fiscal year 2019 such that the remaining outstanding 22,000 shares of common stock as of March 31, 2021 will vest subject to service requirements for vesting for these employees and stock-based compensation expense is being recognized accordingly over the employee’s service term.
Market-Based Awards
In May 2020, the Company awarded certain executive officers and employees a total of up to 61,762 market-based restricted stock units. These restricted stock units will vest and result in the issuance of shares of common stock based on continuing employment and the relative ranking of the total shareholder return (“TSR”) of the Company’s common stock in relation to the TSR of 20 peer companies over a two-year and three-year performance period based on a comparison of average closing stock prices during the 20 trading days prior to the first day of the performance period, reinstated dividends during each performance period and the average closing stock prices during the final 20 trading days of each performance period. The actual number of market-based restricted stock units that may be earned can range from 0% to 200% of the target number of shares. Additionally, the payout percentage is further adjusted based on the Company’s performance relative to the constituents of the S&P 500 Index on the first day
F-32
of the performance period that are still actively trading on the last day of each performance period. The restricted stock units will vest following the end of the two-year and three-year performance period, respectively.
The Company used a Monte-Carlo simulation model to estimate the grant-date fair value of the TSR restricted stock units. The fair value related to these awards are recorded as compensation expense over the period from date of grant to May 2022 and May 2023, respectively, regardless of the actual TSR outcome reached.
The table below sets forth the assumptions used to value the awards and the estimated grant-date fair value:
Risk-free interest rate |
|
| 0.2 | % |
Expected volatility |
|
| 35.5 | % |
Dividend yield |
|
| — |
|
Remaining performance period (years) |
| 1.9 - 2.9 |
| |
Estimated grant date fair value per share |
| $347.05 - $349.28 |
| |
Target performance (number of shares) |
|
| 30,881 |
|
Employee Stock Purchase Plan
The Company has an employee stock purchase plan, or ESPP. Under the ESPP, eligible employees, including officers and directors, who have completed at least three months of employment with the Company or its subsidiaries who elect to participate in the purchase plan instruct the Company to withhold a specified amount of the employee’s income each payroll period during a six-month payment period (the periods April 1—September 30 and October 1—March 31). On the last business day of each six-month payment period, the amount withheld is used to purchase shares of the Company’s common stock at an exercise price equal to 85% of the lower of its market price on the first business day or the last business day of the payment period.
Note 15. Income Taxes
The Company’s income tax provision was $62.7 million, $53.8 million and $4.3 million for fiscal years 2021, 2020 and 2019, respectively. The Company’s effective tax rate was 21.8%, 21.0% and 1.6% for fiscal years 2021, 2020 and 2019, respectively.
The components of the Company’s income tax provision are as follows:
|
| Fiscal Years Ended March 31, |
| |||||||||
|
| 2021 |
|
| 2020 |
|
| 2019 |
| |||
|
|
|
|
|
| (in $000's) |
|
|
|
|
| |
Income before provision for income taxes: |
|
|
|
|
|
|
|
|
|
|
|
|
United States |
| $ | 249,204 |
|
| $ | 214,825 |
|
| $ | 223,340 |
|
Foreign |
|
| 39,016 |
|
|
| 42,000 |
|
|
| 40,020 |
|
Income before income taxes |
|
| 288,220 |
|
|
| 256,825 |
|
|
| 263,360 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Current tax expense: |
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
| 8,624 |
|
|
| — |
|
|
| — |
|
State |
|
| 12,379 |
|
|
| 6,563 |
|
|
| 564 |
|
Foreign |
|
| 12,312 |
|
|
| 14,300 |
|
|
| 11,525 |
|
|
|
| 33,315 |
|
|
| 20,863 |
|
|
| 12,089 |
|
Deferred tax expense (benefit): |
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
| 30,413 |
|
|
| 33,239 |
|
|
| (7,153 | ) |
State |
|
| (2,382 | ) |
|
| 1,584 |
|
|
| (1,503 | ) |
Foreign |
|
| 1,349 |
|
|
| (1,870 | ) |
|
| 911 |
|
|
|
| 29,380 |
|
|
| 32,953 |
|
|
| (7,745 | ) |
Total income tax provision |
| $ | 62,695 |
|
| $ | 53,816 |
|
| $ | 4,344 |
|
F-33
The components of the Company’s net deferred taxes were as follows:
|
| March 31, 2021 |
|
| March 31, 2020 |
| ||
|
| (in $000's) |
| |||||
Deferred tax assets |
|
|
|
|
|
|
|
|
Net operating loss and tax credit carryforwards |
| $ | 27,893 |
|
| $ | 50,604 |
|
Stock-based compensation |
|
| 13,790 |
|
|
| 13,607 |
|
Nondeductible reserves and accruals |
|
| 12,097 |
|
|
| 9,570 |
|
Foreign net operating loss carryforwards |
|
| 6,856 |
|
|
| 6,778 |
|
Deferred revenue |
|
| 5,522 |
|
|
| 4,404 |
|
Depreciation and amortization |
|
| 51 |
|
|
| 114 |
|
Other, net |
|
| 312 |
|
|
| 949 |
|
|
| $ | 66,521 |
|
| $ | 86,026 |
|
Deferred tax liabilities |
|
|
|
|
|
|
|
|
Goodwill |
|
| (7,897 | ) |
|
| (7,843 | ) |
In-process research and development |
|
| (12,496 | ) |
|
| (4,564 | ) |
Depreciation |
|
| (11,747 | ) |
|
| (9,211 | ) |
Basis differences on other investments |
|
| (7,766 | ) |
|
| (6,124 | ) |
Domestic deferred tax liability on foreign net operating loss carryforwards |
|
| (415 | ) |
|
| (584 | ) |
|
|
| (40,321 | ) |
|
| (28,326 | ) |
|
|
|
|
|
|
|
|
|
Net deferred tax assets |
|
| 26,200 |
|
|
| 57,700 |
|
Valuation allowance |
|
| (15,667 | ) |
|
| (15,170 | ) |
Net deferred tax assets |
| $ | 10,533 |
|
| $ | 42,530 |
|
|
|
|
|
|
|
|
|
|
Reported as: |
|
|
|
|
|
|
|
|
Deferred tax assets |
| $ | 11,380 |
|
| $ | 43,336 |
|
Deferred tax liabilities |
|
| (847 | ) |
|
| (806 | ) |
Net deferred tax assets |
| $ | 10,533 |
|
| $ | 42,530 |
|
The significant differences between the statutory and effective income tax rate consist of the following items:
|
| Fiscal Years Ended March 31, |
|
| |||||||||
|
| 2021 |
|
| 2020 |
|
| 2019 |
|
| |||
Statutory income tax rate |
|
| 21.0 |
| % |
| 21.0 |
| % |
| 21.0 |
| % |
Increase (decrease) resulting from: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Credits |
|
| (6.0 | ) |
|
| (10.8 | ) |
|
| (1.5 | ) |
|
Rate differential on foreign operations |
|
| 4.1 |
|
|
| 3.2 |
|
|
| 0.2 |
|
|
Excess tax benefits from stock-based awards |
|
| (3.3 | ) |
|
| (5.2 | ) |
|
| (24.1 | ) |
|
State taxes, net |
|
| 3.2 |
|
|
| 3.1 |
|
|
| 0.1 |
|
|
Permanent differences |
|
| 2.4 |
|
|
| 5.0 |
|
|
| 1.8 |
|
|
Change in valuation allowance |
|
| 0.3 |
|
|
| 5.3 |
|
|
| (0.4 | ) |
|
Foreign taxes |
|
| — |
|
|
| — |
|
|
| 4.1 |
|
|
Other |
|
| 0.1 |
|
|
| (0.6 | ) |
|
| 0.4 |
|
|
Effective tax rate |
|
| 21.8 |
| % |
| 21.0 |
| % |
| 1.6 |
| % |
The Company regularly assesses its ability to realize its deferred tax assets. Assessing the realization of deferred tax assets requires significant management judgment. In determining whether its deferred tax assets are more likely than not realizable, the Company evaluates all available positive and negative evidence, and weights the evidence based on its objectivity.
F-34
As of March 31, 2021 and 2020, respectively, the Company maintained a valuation allowance of $15.7 million and $15.2 million for deferred tax assets primarily related to foreign tax credits that are expected not to be utilizable in the foreign branch basket. Based on the review of all available evidence, the Company recorded a valuation allowance to reduce these deferred tax assets to the amount that is more likely than not to be realizable as of March 31, 2021 and 2020.
Changes in the valuation allowance for deferred tax assets were as follows:
|
| Fiscal Years Ended March 31, |
| |||||||||
|
| 2021 |
|
| 2020 |
|
| 2019 |
| |||
|
| (in $000's) |
| |||||||||
Balance at beginning of year |
| $ | 15,170 |
|
| $ | 1,302 |
|
| $ | 1,652 |
|
Increase |
|
| 497 |
|
|
| 13,868 |
|
|
| — |
|
Decrease |
|
| — |
|
|
| — |
|
|
| (350 | ) |
Balance at end of year |
| $ | 15,667 |
|
| $ | 15,170 |
|
| $ | 1,302 |
|
ASU 2016-09 requires excess tax benefits and shortfalls to be recognized in the income tax provision as discrete items in the period when restricted stock units vest or stock option exercises occur, whereas previously such income tax effects were recorded as part of additional paid-in capital only when the related tax deduction resulted in a reduction of current income taxes payable. The Company recognized excess tax benefits associated with stock-based awards of $12.1 million, $14.8 million and $69.3 million as an income tax benefit for fiscal years 2021, 2020 and 2019, respectively. The amount of future excess tax benefits or shortfalls will likely fluctuate from period to period based on the price of the Company’s stock, the number of restricted stock units that vest or stock options that are exercised, and the fair value assigned to such stock-based awards under U.S. GAAP.
As of March 31, 2021, the Company had foreign net-operating losses (“NOLs”) of approximately $27.2 million. As of March 31, 2021, the Company had foreign tax credits of $13.8 million which expire in varying years from fiscal year 2029 through fiscal year 2031. In addition, as of March 31, 2021, the Company had federal and state research and development credit carryforwards of approximately $0.4 million and $12.8 million, respectively, which expire in varying years from fiscal year 2022 through fiscal year 2041.
The Company’s operating income outside the U.S. is deemed to be permanently reinvested in foreign jurisdictions. The Tax Reform Act allows for a 100% deduction for the repatriation of foreign subsidiary earnings with minimal U.S. income tax consequences other than a one-time deemed repatriation toll charge. Since most of the Company’s cash and cash equivalents are held by foreign subsidiaries which are disregarded entities for domestic tax purposes, any repatriation of such funds to the U.S. would likely have a nominal tax impact, if any.
As of March 31, 2021 and 2020, the Company has no material uncertain tax positions, and 0 interest and penalties on uncertain tax positions were recognized during fiscal years 2021, 2020 and 2019, respectively. The Company is subject to the examination of its income tax returns by the IRS and other tax authorities. The outcome of these audits cannot be predicted with certainty. The Company’s management regularly assesses the likelihood of adverse outcomes resulting from these examinations to determine the adequacy of the Company’s provision for income taxes. If any issues addressed in the Company’s tax audits are resolved in a manner not consistent with management’s expectations, the Company could be required to adjust its provision for income taxes in the period such resolution occurs. The Company’s most recent completed income tax audits were in the U.S. relating to fiscal year 2016 and in Germany, which covered fiscal years 2012 through 2015. These tax audits did not materially impact the Company’s financial statements. In October 2020, the Company was notified by the German tax authorities that they will be conducting an income tax audit on Abiomed Europe GmbH and ECP for fiscal years 2016 through 2019 beginning in January 2021. All other tax years remain subject to examination by the IRS, state and foreign tax authorities.
Note 16. Commitments and Contingencies
From time to time, the Company is involved in legal and administrative proceedings and claims of various types. In some actions, the claimants seek damages, as well as other relief, which, if granted, would require significant expenditures. The Company records a liability in its consolidated financial statements for these matters when a loss is known or considered probable and the amount can be reasonably estimated. The Company reviews these estimates each accounting period as additional information is known and adjusts the loss provision when appropriate. If a matter is both probable to result in liability and the amount of loss can be reasonably estimated, the Company estimates and discloses the possible loss or range of loss. If the loss is not probable or cannot be reasonably estimated, a liability is not recorded in its consolidated financial statements.
Thoratec Matters
F-35
Thoratec Corporation (“Thoratec”), a subsidiary of Abbott Laboratories (“Abbott”), has challenged a number of Company-owned patents in Europe in connection with the launch of Thoratec’s HeartMate PHP™ medical device (“PHP”) in Europe and the Company has counterclaimed for infringement in the District Court in Düsseldorf. The litigation was stayed pending the highest Court’s ruling on the validity and scope of the litigated patents. In September 2019, the Federal Court of Justice in Germany upheld the Company’s patents that are the subject of the patent infringement action for the sales and marketing of Thoratec’s PHP pump in Germany. Subsequently, the District Court in Düsseldorf lifted the stay, re-opened the litigation proceedings, and ruled in favor of Abiomed. The Court acknowledged that Thoratec’s PHP product infringes two of ABIOMED patents related to the key features of Impella® intravascular pump and future expandable heart pump, known as Impella ECP®. Abbott appealed and the oral hearing is set for August 26, 2021. If upheld, the verdict is provisionally enforceable, which means that ABIOMED will be able to seek a court ordered injunction preventing the sale and marketing of PHP, should Thoratec attempt to launch HeartMate PHP in Germany.
These actions relate solely to Thoratec’s ability to manufacture and sell its PHP product in Europe and have no impact on the Company's ability to manufacture or sell its Impella® line of medical devices. The actions do not expose the Company to liability risk, except under local German law that requires a losing party in a proceeding to pay a portion of the other party’s legal fees.
Maquet Matters
In December 2015, the Company received a letter from Maquet Cardiovascular LLC (“Maquet”), a subsidiary of Getinge AB, asserting that the Company’s Impella® devices infringe certain claims with guidewire, lumen, rotor, purge and sensor features, which were in two Maquet patents and one pending patent application (which has since issued as a third patent) in the U.S. and elsewhere, and attaching a draft litigation complaint. The letter encouraged the Company to take a license from Maquet. In May 2016, the Company filed suit in U.S. District Court for the District of Massachusetts (“D. Mass.” or “the Court”) against Maquet, seeking a declaratory judgment that the Company’s Impella devices do not infringe Maquet’s cited patent rights.
In August 2016, Maquet sent a letter to the Company identifying four new Maquet U.S. continuation patent filings with claims that Maquet alleges are infringed by the Company’s Impella devices. The four U.S. continuation applications have been issued as patents of Maquet but expired on September 1, 2020.
In September 2016, Maquet filed a response to the Company’s suit in D. Mass., including various counterclaims alleging that the Company’s Impella 2.5®, Impella CP®, Impella 5.0®, and Impella RP® heart pumps infringe certain claims of the three original issued U.S. patents (“2016 Action”). In July 2017, the Court granted a motion to add three of the four additional continuation patents to the 2016 Action. In April 2018, the Court conducted a Markman hearing on claim interpretation. On September 7, 2018, the judge issued a Memorandum and Order on Claim Construction, where he interpreted the disputed claim terms in the case. Maquet then filed a motion for reconsideration of the Court’s construction of one of the disputed claim terms. The motion was denied on May 22, 2019. As a result of the Court’s denial, only one of the six originally asserted patents is in dispute. The Company filed a motion for summary judgement (the “MSJ”) for the remaining patent on September 18, 2019 (non-infringement) and April 13, 2020 (invalidity). The parties argued the MSJ for non-infringement on November 19, 2019, the MSJ for invalidity on August 20, 2020 and are waiting for Court’s resolution. The Court has not set a date for trial. The only remaining patent asserted in this case expired on September 1, 2020.
In November 2017, Maquet filed a second action in D. Mass (the “2017 Action”) alleging that the Company’s Impella 2.5®, Impella CP®, and Impella 5.0® heart pumps infringe certain claims of the fourth additional U.S. continuation patent mentioned above (the seventh patent overall). Discovery in the 2017 Action is ongoing.
In a series of letters during January and February 2019, Maquet informed the Company of seven new patent applications filed from the patents in the 2016 Action and 2017 Action with claims Maquet alleges would be infringed by the Impella® products if the new applications were to issue as patents. One of the newly issued patents has been added to the 2017 Action. A Markman hearing for the newly-added patent was held on November 18, 2019. A Markman order has not been issued yet. The asserted patent in this case expired on September 1, 2020. Discovery remains ongoing.
In the 2016 Action and 2017 Action, Maquet seeks injunctive relief and monetary damages in the form of a reasonable royalty, with three times the amount for alleged willful infringement. In its responses to the Company’s counterclaims, Maquet admits that its current commercially available products do not embody the claims of the asserted patents.
The Company is unable to estimate the potential liability with respect to the legal matters noted above. There are numerous factors that make it difficult to meaningfully estimate possible loss or range of loss at this stage of the legal proceedings, including the significant number of legal and factual issues still to be resolved in the Maquet and Thoratec patent disputes.
Securities Class Action Litigation
On or about August 6, 2019, the Company received a securities class action complaint filed on behalf of a single shareholder in the U.S. District Court for the Southern District of New York (“SDNY”), on behalf of himself and persons or entities that purchased or acquired the Company’s securities between January 31, 2019 through July 31, 2019. On October 7, 2019, a similar purported class action complaint was filed by a different shareholder on behalf of himself and persons or entities that purchased or acquired the Company’s securities between November 1, 2018 and July 31, 2019. Also, on October 7, 2019, four shareholders filed applications to
F-36
be appointed lead plaintiff and for their counsel to be appointed lead counsel for the class. Two of those shareholders also filed motions to consolidate the two cases and two of the shareholders have withdrawn their applications to be lead plaintiff.
The complaints allege that the Company violated Sections 10(b) and 20(a) of and Rule 10b-5 under the Exchange Act, in connection with allegedly misleading disclosures made by the Company regarding its financial condition and results of operations. The Company believes that the allegations are without merit and plans to defend itself vigorously.
On June 29, 2020, the Court issued an order consolidating the two cases and appointed Local 705 International Brotherhood of Teamsters Pension Fund as the lead plaintiff and the Labaton Sucharow firm as lead counsel. On September 17, 2020, the lead plaintiff filed an amended complaint in which it proposed a new class period of May 3, 2018 to July 31, 2019. As prescribed by a scheduling order, the Company filed a motion to dismiss on November 16, 2020, lead plaintiff filed its opposition to that motion on January 15, 2021, and the Company filed its reply on February 24, 2021.
Shareholder Derivative Litigation
On November 6 and 7, 2019, two shareholders filed derivative actions in SDNY that were subsequently consolidated. On November 8, 2019, another shareholder filed a derivative action in Massachusetts Suffolk County Superior Court. On January 7, 2020, another shareholder derivative action was filed in the U.S. District Court for the District of Delaware. The complaints in these actions rely on many of the same allegations as in the securities class actions, and assert that, between November 1, 2018 and July 31, 2019, the directors of the Company made or allowed to be made misleading public statements regarding the Company’s growth, ultimately harming the Company.
The Company has agreed with the plaintiffs in all three actions to stay the cases pending resolution of a motion to dismiss in the securities class actions. As a result of the stay, the Delaware action has been administratively closed.
Litigation Demand
On March 3, 2020, a shareholder sent a letter to the Board of Directors asserting that the directors of the Company made or allowed to be made misleading public statements regarding the Company’s growth. The letter relies on many of the same allegations as the securities class actions and derivative actions, and demands that the Board (i) undertake an independent investigation of the directors, (ii) bring suit against the directors on behalf of the Company, and (iii) take a number of additional affirmative actions to redress the purported wrongs. On March 30, 2020, the Company, after discussions with the Board of Directors, sent a written response to the shareholder’s counsel which they responded to on June 1, 2020. The Company then sent a further response to the shareholder’s counsel on June 15, 2020, affirming the decision to defer consideration of the litigation demand pending further developments in the securities class action suit. Following the filing of the amended complaint in the securities class action, described above, the same shareholder renewed their demand on September 29, 2020. The Company responded on October 9, 2020 and once again affirmed that it will defer consideration of the demand pending further substantive developments in the securities class action suit.
On November 5, 2020, a second shareholder sent a letter to the Board of Directors that made essentially the same demands as the September 29, 2020 letter from the first shareholder. The Company responded on November 23, 2020, noting that it will defer consideration of the demand pending further substantive developments in the securities class action suit.
The Company is unable to estimate the potential liability with respect to the various legal matters noted above. There are numerous factors that make it difficult to estimate reasonably possible loss or range of loss at this stage of the legal proceedings, including the significant number of legal and factual issues still to be resolved in the securities class action litigation, as well as in the shareholder derivative litigations.
Note 17. Segment and Enterprise Wide Disclosures
The Company operates in 1 business segment—the research, development and sale of medical devices to assist or replace the pumping function of the failing heart. The Company’s chief operating decision maker (determined to be the Chief Executive Officer) does not manage any part of the Company separately, and the allocation of resources and assessment of performance are based on the Company’s consolidated operating results. International sales (sales outside the U.S. and primarily in Europe) accounted for 18%, 16% and 14% of total revenue during the fiscal years ended March 31, 2021, 2020 and 2019, respectively. As of March 31, 2021 and 2020, most of the Company’s long-lived assets are located in the U.S. except for $56.4 million and $46.7 million as of March 31, 2021 and 2020, respectively, which are located primarily in Germany.
F-37
Note 18. Quarterly Results of Operation (Unaudited)
The following is a summary of the Company’s unaudited quarterly results of operations:
|
| Fiscal Year Ended March 31, 2021 |
| ||||||||||||||||||||||
|
| 1st Quarter |
|
| 2nd Quarter |
|
| 3rd Quarter |
|
| 4th Quarter |
|
| Total Year |
| ||||||||||
|
| (in $000's) |
| ||||||||||||||||||||||
Revenue |
| $ |
| 164,850 |
|
| $ |
| 209,764 |
|
| $ |
| 231,663 |
|
| $ |
| 241,246 |
|
| $ |
| 847,522 |
|
Cost of revenue |
|
|
| 35,983 |
|
|
|
| 38,736 |
|
|
|
| 41,110 |
|
|
|
| 46,078 |
|
|
|
| 161,907 |
|
Other operating expenses |
|
|
| 94,801 |
|
|
|
| 109,692 |
|
|
|
| 119,202 |
|
|
|
| 132,364 |
|
|
|
| 456,058 |
|
Other income (1) |
|
|
| 27,010 |
|
|
|
| 11,579 |
|
|
|
| 9,384 |
|
|
|
| 10,690 |
|
|
|
| 58,663 |
|
Income before income taxes |
|
|
| 61,076 |
|
|
|
| 72,915 |
|
|
|
| 80,735 |
|
|
|
| 73,494 |
|
|
|
| 288,220 |
|
Income tax provision (2) |
|
|
| 16,488 |
|
|
|
| 10,702 |
|
|
|
| 18,867 |
|
|
|
| 16,638 |
|
|
|
| 62,695 |
|
Net income |
| $ |
| 44,588 |
|
| $ |
| 62,213 |
|
| $ |
| 61,868 |
|
| $ |
| 56,856 |
|
| $ |
| 225,525 |
|
Basic net income per share |
| $ |
| 0.99 |
|
| $ |
| 1.38 |
|
| $ |
| 1.37 |
|
| $ |
| 1.26 |
|
| $ |
| 5.00 |
|
Diluted net income per share |
| $ |
| 0.98 |
|
| $ |
| 1.36 |
|
| $ |
| 1.35 |
|
| $ |
| 1.24 |
|
| $ |
| 4.94 |
|
|
| Fiscal Year Ended March 31, 2020 |
| ||||||||||||||||||||||
|
| 1st Quarter |
|
| 2nd Quarter |
|
| 3rd Quarter |
|
| 4th Quarter |
|
| Total Year |
| ||||||||||
|
| (in $000's) |
| ||||||||||||||||||||||
Revenue |
| $ |
| 207,666 |
|
| $ |
| 204,974 |
|
| $ |
| 221,584 |
|
| $ |
| 206,658 |
|
| $ |
| 840,883 |
|
Cost of revenue |
|
|
| 37,073 |
|
|
|
| 34,867 |
|
|
|
| 39,996 |
|
|
|
| 39,369 |
|
|
|
| 151,305 |
|
Other operating expenses |
|
|
| 109,868 |
|
|
|
| 109,925 |
|
|
|
| 111,329 |
|
|
|
| 109,237 |
|
|
|
| 440,359 |
|
Other income (expense) (1) |
|
|
| 42,413 |
|
|
|
| (42,824 | ) |
|
|
| 26,757 |
|
|
|
| (18,739 | ) |
|
|
| 7,606 |
|
Income before income taxes |
|
|
| 103,138 |
|
|
|
| 17,358 |
|
|
|
| 97,016 |
|
|
|
| 39,313 |
|
|
|
| 256,825 |
|
Income tax provision (2) |
|
|
| 14,215 |
|
|
|
| 4,287 |
|
|
|
| 27,799 |
|
|
|
| 7,515 |
|
|
|
| 53,816 |
|
Net income |
| $ |
| 88,923 |
|
| $ |
| 13,071 |
|
| $ |
| 69,217 |
|
| $ |
| 31,798 |
|
| $ |
| 203,009 |
|
Basic net income per share |
| $ |
| 1.97 |
|
| $ |
| 0.29 |
|
| $ |
| 1.53 |
|
| $ |
| 0.71 |
|
| $ |
| 4.49 |
|
Diluted net income per share |
| $ |
| 1.93 |
|
| $ |
| 0.28 |
|
| $ |
| 1.51 |
|
| $ |
| 0.70 |
|
| $ |
| 4.43 |
|
| (1) | In fiscal year 2019, the Company invested $25.0 million in medical device company Shockwave Medical. The fair value of this investment as of March 31, 2021 was $38.7 million. The Company recognized a pre-tax gain of $50.8 million for the year ended March 31, 2021 and a pre-tax loss of $0.5 million for the year ended March 31, 2020 in other income (expense), net. |
| (2) | The income tax provision for the years ended March 31, 2021 and 2020 included excess tax benefits of $12.1 million and $14.8 million, respectively. These recognized excess tax benefits resulted from restricted stock units that vested or stock options that were exercised during the years ended March 31, 2021 and 2020. |
F-38