UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2018
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ___________ to ___________
Commission file number 0-19125
Ionis Pharmaceuticals, Inc.
(Exact name of Registrant as specified in its charter)
| Delaware | 33-0336973 | |
| (State or other jurisdiction of incorporation or organization) | (IRS Employer Identification No.) |
| 2855 Gazelle Court, Carlsbad, CA | 92010 | |
| (Address of Principal Executive Offices) | (Zip Code) |
760-931-9200
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
| Common Stock, $.001 Par Value | The Nasdaq Stock Market, LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ☒ | Accelerated filer ☐ | |
Non-accelerated filer ☐ | Smaller reporting company ☐ | |
Emerging growth company ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The approximate aggregate market value of the voting common stock held by non-affiliates of the Registrant, based upon the last sale price of the common stock reported on The Nasdaq Global Select Market was $4,805,287,142 as of June 30, 2018.*
The number of shares of voting common stock outstanding as of February 20, 2019 was 138,397,754.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s definitive Proxy Statement to be filed on or about April 26, 2019 with the Securities and Exchange Commission in connection with the Registrant’s annual meeting of stockholders to be held on June 6, 2019 are incorporated by reference into Part III of this Report.
| * | Excludes 21,838,695 shares of common stock held by directors and officers and by stockholders whose beneficial ownership is known by the Registrant to exceed 10 percent of the common stock outstanding at June 30, 2018. Exclusion of shares held by any person should not be construed to indicate that such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the Registrant, or that such person is controlled by or under common control with the Registrant. |
FORWARD-LOOKING STATEMENTS
This report on Form 10-K and the information incorporated herein by reference includes forward-looking statements regarding our business and the therapeutic and commercial potential of SPINRAZA (nusinersen), TEGSEDI (inotersen), WAYLIVRA (volanesorsen) and our technologies and products in development, including the business of Akcea Therapeutics, Inc., our majority-owned affiliate. Any statement describing our goals, expectations, financial or other projections, intentions or beliefs, is a forward-looking statement and should be considered an at-risk statement. Such statements are subject to certain risks and uncertainties, particularly those inherent in the process of discovering, developing and commercializing drugs that are safe and effective for use as human therapeutics, and in the endeavor of building a business around such drugs. Our forward-looking statements also involve assumptions that, if they never materialize or prove correct, could cause our results to differ materially from those expressed or implied by such forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in this report on Form 10-K, including those identified in Item 1A entitled “Risk Factors”. Although our forward-looking statements reflect the good faith judgment of our management, these statements are based only on facts and factors currently known by us. As a result, you are cautioned not to rely on these forward-looking statements.
In this report, unless the context requires otherwise, “Ionis,” “Company,” “we,” “our,” and “us” refers to Ionis Pharmaceuticals, Inc. and its subsidiaries.
TRADEMARKS
“Ionis,” the Ionis logo, and other trademarks or service marks of Ionis Pharmaceuticals, Inc. appearing in this report are the property of Ionis Pharmaceuticals, Inc. “Akcea,” the Akcea logo, and other trademarks or service marks of Akcea Therapeutics, Inc. appearing in this report are the property of Akcea Therapeutics, Inc. This report contains additional trade names, trademarks and service marks of others, which are the property of their respective owners. Solely for convenience, trademarks and trade names referred to in this report may appear without the ® or TM symbols.
CORPORATE INFORMATION
We incorporated in California in 1989 and in January 1991 we changed our state of incorporation to Delaware. In December 2015, we changed our name to Ionis Pharmaceuticals, Inc. from Isis Pharmaceuticals, Inc. Our principal offices are in Carlsbad, California. We make available, free of charge, on our website, www.ionispharma.com, our reports on Forms 10-K, 10-Q, 8-K and amendments thereto, as soon as reasonably practical after we file such materials with the Securities and Exchange Commission. Any information that we include on or link to our website is not a part of this report or any registration statement that incorporates this report by reference. You may also read and copy our filings at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-732-0330. The SEC also maintains a website that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that site is www.sec.gov.
In December 2014, we formed Akcea Therapeutics, Inc., as a Delaware corporation, with its principal office in Boston, Massachusetts. Prior to Akcea’s IPO in July 2017, we owned 100 percent of Akcea’s stock. At December 31, 2018, we owned approximately 75 percent of Akcea’s stock.
2
IONIS PHARMACEUTICALS, INC.
FORM 10-K
For the Fiscal Year Ended December 31, 2018
Table of Contents
| PART I | ||
| Page | ||
| Item 1. | Business | 4 |
| Item 1A. | Risk Factors | 39 |
| Item 1B. | Unresolved Staff Comments | 48 |
| Item 2. | Properties | 48 |
| Item 3. | Legal Proceedings | 48 |
| Item 4. | Mine Safety Disclosures | 49 |
| PART II | ||
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 49 |
| Item 6. | Selected Financial Data | 49 |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 50 |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | 75 |
| Item 8. | Financial Statements and Supplementary Data | 75 |
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure | 75 |
| Item 9A. | Controls and Procedures | 75 |
| Item 9B. | Other Information | 77 |
| PART III | ||
| Item 10. | Directors, Executive Officers and Corporate Governance | 77 |
| Item 11. | Executive Compensation | 77 |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 77 |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence | 77 |
| Item 14. | Principal Accounting Fees and Services | 77 |
| PART IV | ||
| Item 15. | Exhibits, Financial Statement Schedules | 78 |
| Signatures |
3
PART I
Item 1. Business
We are a leader in discovering and developing RNA-targeted therapeutics with sustained and growing revenues. We have created an efficient and broadly applicable drug discovery platform leveraging our expertise in antisense oligonucleotide therapeutics that we believe has fundamentally changed medicine and transformed the lives of people with devastating and often deadly diseases. Our large, diverse and advanced pipeline of over 40 first-in-class and/or best-in-class medicines addresses diseases across a broad range of therapeutic areas, targeting small, medium and large patient populations.
We have two commercial medicines approved in major markets around the world, SPINRAZA and TEGSEDI. We have at least four medicines that have entered pivotal studies or have the potential to begin pivotal studies this year, and another six medicines that could start pivotal studies in 2020. These medicines, along with the more than 30 additional medicines in our pipeline, represent multiple potential drivers of value for years to come. We believe our efficient drug discovery platform, coupled with our innovation-centric business model, provides us with the flexibility to determine the optimal development and commercialization strategy to maximize the commercial opportunity for each of our medicines and ensure that we continue to produce transformative medicines for patients who need them. We believe we are positioned to drive substantial value for patients and shareholders.
As of January 2019, SPINRAZA was approved in over 40 countries around the world, and our partner Biogen, who is responsible for global SPINRAZA commercial activities, reported that more than 6,600 patients are now on SPINRAZA therapy. In addition, Biogen plans to continue to pursue regulatory filings in additional countries. Biogen reported 2018 annual sales of SPINRAZA of more than $1.7 billion, and we earned $238 million in commercial revenues from royalties on sales of SPINRAZA. SPINRAZA is the first and only approved medicine for the treatment of spinal muscular atrophy, or SMA. SPINRAZA is the established standard-of-care for all people with this progressive, debilitating and often fatal genetic disease. In November 2018, SPINRAZA was recognized with the 2018 International Prix Galien award as Best Biotechnology Product. This prestigious honor marks the seventh Prix Galien award for SPINRAZA.
TEGSEDI, a once weekly, self-administered subcutaneous medicine, was approved in 2018 in the U.S., EU and Canada for the treatment of polyneuropathy caused by hereditary TTR amyloidosis, or hATTR, in adult patients. hATTR is a debilitating, progressive, and fatal disease. Akcea, our majority-owned affiliate focused on developing and commercializing medicines to treat patients with rare and serious diseases, launched TEGSEDI globally in late 2018. In the fourth quarter of 2018, we earned more than $2 million in TEGSEDI product sales. Akcea has an exclusive license agreement with PTC Therapeutics, or PTC, to commercialize TEGSEDI in Latin America. In January 2019, PTC filed an application for regulatory approval in Brazil with ANVISA, the Brazilian regulatory authority. ANVISA granted priority review for TEGSEDI.
We and Akcea are preparing to commercialize WAYLIVRA in the EU. The Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, adopted a positive opinion recommending conditional marketing authorization for WAYLIVRA as an adjunct to diet in adult patients with genetically confirmed familial chylomicronemia syndrome, or FCS, who are at high risk for pancreatitis, in whom response to diet and triglyceride lowering therapy has been inadequate. The positive opinion will now be referred to the European Commission, or EC, which grants marketing authorization for medicines in the EU, as well as to European Economic Area members Iceland, Liechtenstein and Norway. With this positive opinion, and, pending adoption of the positive opinion by the EC, Akcea plans to leverage its existing commercial infrastructure in Europe to market WAYLIVRA. Akcea is continuing to conduct open-label extension and early access programs. We are also focused on regulatory discussions in the U.S. We are developing WAYLIVRA to treat familial partial lipodystrophy, or FPL, a second severe and rare, genetically defined disease. FCS and FPL are orphan diseases characterized by severely high triglyceride levels that result in severe, daily symptoms and a high risk of life-threatening pancreatitis.
In addition to commercializing TEGSEDI and preparing to commercialize WAYLIVRA, Akcea is developing four other clinical-stage medicines: AKCEA-APO(a)-LRx (TQJ230), AKCEA-ANGPTL3-LRx, AKCEA-APOCIII-LRx and AKCEA-TTR-LRx, each of which could potentially treat multiple patient populations. Moving these drugs into Akcea allows us to retain substantial value from these medicines and ensures our core focus remains on innovation. As of February 2019, we owned approximately 75 percent of Akcea.
We are continuously advancing our technology and pipeline to provide the most value to patients. We have a pipeline of over 40 medicines that, like SPINRAZA and TEGSEDI, have the potential to transform the treatment of diseases with no adequate treatment today. These medicines range from treatments for rare diseases with small patient populations to more common diseases afflicting millions of patients. Our pipeline covers a broad spectrum of therapeutic areas, such as cardiometabolic diseases, neurodegenerative diseases, cancer, severe and rare diseases and others. We believe our large and diverse pipeline contains many near-, mid- and longer-term growth drivers for the company.
Our pipeline includes at least 10 potentially transformative medicines anticipated to enter pivotal clinical studies in the next two years. We anticipate at least four of these medicines will enter pivotal studies this year including: AKCEA-APO(a)-LRx, AKCEA-TTR-LRx, IONIS-HTTRx (RG6042) and IONIS-SOD1Rx. Roche recently initiated a Phase 3 study of IONIS-HTTRx for Huntington’s disease, or HD. We believe each of these medicines is a first-in-class and/or best-in-class medicine with the potential to deliver significant value to patients and shareholders. We anticipate that the data from these pivotal studies, if positive, will support global regulatory filings for each medicine.
AKCEA-APO(a)-LRx (TQJ230) – In February 2019, Novartis exercised its option to license AKCEA-APO(a)-LRx and we earned a $150 million license fee. Novartis is responsible for conducting and funding all future development and commercialization activities for AKCEA-APO(a)-LRx, including a global pivotal cardiovascular outcomes study, for which planning and initiation activities are underway. AKCEA-APO(a)-LRx targets a cardiovascular risk factor, lipoprotein(a) or Lp(a). Lp(a) is well-recognized by the medical community as a major risk factor for cardiovascular disease. Lp(a) is genetically determined at birth and there are currently no treatments available to substantially and specifically lower Lp(a). In September 2018, we reported dose-dependent and substantial reductions in Lp(a) levels in the Phase 2 clinical study in patients with established cardiovascular disease, or CVD, due to elevated levels of Lp(a), which was also the longest and largest study, regardless of phase, conducted with a LICA antisense medicine to date. Approximately 98 percent of patients who received the highest dose in the study demonstrated a reduction in Lp(a) levels to below 50 mg/dL, the recognized threshold for risk of CVD. In addition, AKCEA-APO(a)-LRx demonstrated a favorable safety and tolerability profile in the study.
4
AKCEA-TTR-LRx – We are developing AKCEA-TTR-LRx for the treatment of people with all forms of TTR amyloidosis as a once a month or even less frequent subcutaneous self-administered injection. In April 2018, we licensed to Akcea the worldwide rights to commercialize TEGSEDI and AKCEA-TTR-LRx. We plan to report data from the Phase 1/2 study this year, followed by the initiation of a pivotal program. We plan to initiate a Phase 3 study in patients with hereditary TTR amyloidosis with polyneuropathy first, followed closely by a Phase 3 study in patients with wild type and hereditary TTR cardiomyopathy, also planned for this year.
IONIS-HTTRx (RG6042) – Roche initiated the Phase 3 study of IONIS-HTTRx for HD in December 2018 and we earned a $35 million milestone payment when the first patient was dosed in the Phase 3 study in January 2019. HD is a genetic, devastating and fatal neurodegenerative disease that negatively affects psychological, cognitive and motor functions. In March 2018, we reported data from a Phase 1/2 study that demonstrated up to a 60 percent reduction in the mutant huntingtin protein, or mHTT, as observed in the cerebral spinal fluid, or CSF. It was the first study to demonstrate disease-modifying potential in HD patients. Based on preclinical data, the mHTT reductions of 40-60 percent in the CSF are predicted to result in 55-85 percent reduction in the cortex of the brain, where mHTT is highly expressed. IONIS-HTTRx demonstrated a favorable safety and tolerability profile in the study.
IONIS-SOD1Rx (BIIB067) – IONIS-SOD1Rx, for people with amyotrophic lateral sclerosis, or ALS, is the fourth medicine we anticipate we will move into pivotal studies this year. ALS is a rare, fatal neurodegenerative disease characterized by the loss of motor neurons in the brain and spinal cord resulting in an inability to control muscle movement. Scientists have identified mutations within multiple genes as causative of ALS, including mutations in the SOD1 gene. IONIS-SOD1Rx directly targets the SOD1 gene and is delivered intrathecally into the CSF. The average life expectancy for an ALS patient with the SOD1 mutation is less than five years from the time of diagnosis. Based on the positive interim analysis from the Phase 1/2 study that demonstrated proof-of-biology and proof-of-concept, in December 2018, Biogen exercised its licensing option with us to develop and commercialize IONIS-SOD1Rx. IONIS-SOD1Rx demonstrated a favorable safety and tolerability profile in the study. Biogen plans to add an additional cohort to this study to potentially support registration. We earned $40 million in payments from Biogen in the fourth quarter of 2018 when Biogen advanced and licensed IONIS-SOD1Rx.
The depth of our knowledge and expertise with antisense technology together with our strong financial position provides us the flexibility to partner our medicines at what we believe is the optimal time to maximize the near-term, mid-term and long-term value of our medicines. We have a distinct partnering strategy based on each specific medicine and the expertise and resources we and our potential partners may bring to a collaboration. We may develop and commercialize some medicines through affiliates. In general, these are medicines, like TEGSEDI, that can benefit from our internal expertise and infrastructure, have manageable development costs and have the potential for initial rare disease indications. For other medicines, we may establish collaborations to advance the medicine. We have alliances with a cadre of leading global pharmaceutical companies that are working alongside us in developing our medicines, advancing our technology, preparing to commercialize our medicines and selling our medicines. Our partners include the following companies, among others: AstraZeneca, Bayer, Biogen, GSK, Janssen, Novartis and Roche. Our partners bring resources and expertise that augment and build upon our internal capabilities. For example, we partnered AKCEA-APO(a)-LRx with Novartis because we believe Novartis brings significant resources and expertise that should accelerate our ability to deliver AKCEA-APO(a)-LRx to the large population of patients with elevated levels of Lp(a) and established CVD. As a result of Novartis exercising its option for AKCEA-APO(a)-LRx in February 2019, Novartis is responsible for conducting and funding all future development and commercialization activities, including a Phase 3 cardiovascular outcomes study Novartis is planning to conduct. We are eligible to earn additional payments from Novartis as AKCEA-APO(a)-LRx progresses.
We are now a multi-product commercial company. 2018 marks our seventh consecutive year of revenue growth. Through our partnerships, we have earned significant commercial revenue and a broad and sustaining base of research and development, or R&D, revenue in the form of license fees, upfront payments and milestone payments, while investing in advancing our pipeline and technology. Moreover, we have the potential to earn over $20 billion in future milestone payments and licensing fees from our current partnerships. We also have the potential to share in the future commercial success of our inventions and drugs resulting from our partnerships through royalty arrangements. Looking forward, we believe we have the potential to increase our commercial revenue from SPINRAZA royalties and TEGSEDI product sales from the continued growth we anticipate in the U.S., EU and other markets globally. We also have the potential to further increase our commercial revenue with the potential approval of WAYLIVRA.
We ended 2018 with a strong balance sheet with more than $2 billion in cash and short-term investments, making this the sixth year out of seven that we have been cash accretive. Our strong balance sheet provides us with the financial wherewithal to invest in expanding and advancing our pipeline, in commercializing our medicines through commercial affiliates, and advancing our technology.
5
Our Marketed Medicines – Transformational Medicines Bringing Value to Patients Today
SPINRAZA – SPINRAZA (nusinersen) injection for intrathecal use is a survival motor neuron-2, or SMN2, directed antisense oligonucleotide indicated for the treatment of SMA in pediatric and adult patients. SPINRAZA is the first and only approved medicine for the treatment of SMA and is the established standard-of-care for all people around the globe with this progressive, debilitating genetic disease. SPINRAZA is approved in over 40 countries around the world. In February 2019, SPINRAZA was approved by the China National Medical Products Association. Our partner, Biogen, who is responsible for global SPINRAZA commercial activities, reported in January 2019 that approximately 6,600 patients were on SPINRAZA therapy. Biogen reported 2018 annual sales of more than $1.7 billion, and we earned $238 million in commercial revenues from royalties on sales of SPINRAZA.
SMA is characterized by loss of motor neurons in the spinal cord and lower brain stem, resulting in severe and progressive muscular atrophy and weakness. Ultimately, individuals with the most severe type of SMA, infantile-onset, or Type 1, SMA, can become paralyzed and have difficulty performing the basic functions of life, like breathing and swallowing. Due to a loss of, or defect in, the SMN1 gene, people with SMA do not produce enough survival motor neuron, or SMN, protein, which is critical for the maintenance of motor neurons. The severity of SMA correlates with the amount of SMN protein a patient can produce on his/her own. Patients with Type 1 SMA produce very little SMN protein and do not achieve the ability to sit without support or live beyond two years without respiratory support. Patients with later-onset, or Type 2 or Type 3 SMA, produce greater amounts of SMN protein and have less severe, but still life-altering, forms of SMA.
SPINRAZA was recognized with the 2018 International Prix Galien Best Biotechnology Product award. The prestigious honor marks the seventh Prix Galien award for SPINRAZA, following country recognitions in the U.S., Germany, Italy, Belgium-Luxembourg, the Netherlands and the U.K. The International Prix Galien award is given every two years by Prix Galien International Committee members in recognition of excellence in scientific innovation to improve human health.
Biogen is conducting NURTURE, a Phase 2 open-label study of SPINRAZA in pre-symptomatic infants. Biogen presented an interim analysis of the NURTURE data at the Annual Congress of the World Muscle Society in October 2018. The interim analysis showed that SPINRAZA-treated infants achieved motor milestones in timelines more consistent with normal development than what is observed in the natural history of patients with Type 1 SMA. At the time of the interim analysis, all patients were alive and did not require respiratory intervention. All of the infants in the study were able to sit without support and 88 percent of the infants were able to walk either with assistance or independently. No new safety concerns were identified.
The safety and efficacy of SPINRAZA has been evaluated in multiple clinical studies in more than 270 patients, including two Phase 3 studies: ENDEAR, a randomized controlled study evaluating SPINRAZA in patients with infantile-onset SMA, and CHERISH, a randomized controlled study evaluating SPINRAZA in patients with later-onset SMA.
In the ENDEAR end of study analysis, or EOS, a statistically significant greater percentage of children with infant-onset SMA achieved improvement in motor milestones compared to untreated patients, with some infants in the SPINRAZA group achieving full head control, the ability to roll, sit, and stand. Additionally, infants treated with SPINRAZA demonstrated a statistically significant improvement in event-free survival compared to untreated patients.
In the CHERISH EOS there was a statistically significant and clinically meaningful improvement in motor function in children with later-onset SMA treated with SPINRAZA compared to untreated children. The majority of children treated with SPINRAZA demonstrated benefits in upper limb and general motor function, including crawling and standing with support. The overall findings from the CHERISH EOS analysis continue to support the robust efficacy and favorable safety profile of SPINRAZA across a broad patient population.
In all clinical studies, SPINRAZA demonstrated a favorable safety profile. The most common side effects of SPINRAZA included lower and upper respiratory infections, constipation, headache, back pain, and post-lumbar puncture syndrome. For additional safety information, please see www.spinraza.com (Any information that is included on or linked to this website is not part of this report or any registration statement or report that incorporates this report by reference).
TEGSEDI – TEGSEDI (inotersen) injection is a Generation 2+ antisense medicine and the world’s first and only approved subcutaneous RNA-targeting medicine designed to treat people with polyneuropathy caused by hATTR. In October 2018, the FDA approved TEGSEDI for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults. TEGSEDI is also approved in the EU and Canada for the treatment of stage 1 or stage 2 polyneuropathy in adult patients with hereditary transthyretin amyloidosis. It is administered as a once weekly, self-administered, at-home, subcutaneous injection. In March 2018, Akcea licensed TEGSEDI from us.
TTR amyloidosis that is the result of inherited mutations in the TTR gene is referred to as hATTR. There are an estimated 50,000 people worldwide with hATTR. There are two primary manifestations of hATTR: polyneuropathy and cardiomyopathy. Many people with hATTR often experience both manifestations, but often one manifestation or the other is diagnosed first and is more pronounced.
In people with hATTR, both the mutant and wild type, or wt, TTR protein builds up as fibrils in the tissues, such as peripheral nerves, heart, gastrointestinal system, eyes, kidneys, central nervous system, thyroid and bone marrow. The presence of TTR fibrils interferes with the normal function of these tissues. As the TTR protein fibrils enlarge, more tissue damage occurs and the disease worsens, resulting in poor quality of life and eventually death. We designed TEGSEDI to reduce the production of the TTR protein, the underlying cause of transthyretin amyloidosis, or ATTR.
6
Polyneuropathy due to hATTR is caused by the accumulation of misfolded mutated TTR protein in the peripheral nerves. People with polyneuropathy due to hATTR experience ongoing debilitating nerve damage throughout their body resulting in the progressive loss of sensation in the extremities that advances centrally, and loss of motor functions, such as walking. These people also accumulate TTR in other major organs, which progressively compromises their function and eventually leads to death within five to 15 years of disease onset. Cardiomyopathy caused by ATTR is the accumulation of misfolded TTR protein in the cardiac muscle.
ATTR can also result from normal, non-mutant, TTR protein forming fibrils, primarily in the heart. This form of the disease is wt-ATTR. It is estimated that more than 200,000 people worldwide have wt-ATTR. People with hATTR cardiomyopathy and wt-ATTR experience ongoing debilitating heart damage resulting in progressive heart failure, which results in death within three to five years from disease onset.
The TEGSEDI approval relied on results from the Phase 3 NEURO-TTR study in patients with hATTR amyloidosis with stage 1 and stage 2 polyneuropathy. Results from that study demonstrated that patients treated with TEGSEDI experienced significant benefit compared to patients treated with placebo across both co-primary endpoints: the Norfolk Quality of Life Questionnaire-Diabetic Neuropathy, or Norfolk QoL-DN, and modified Neuropathy Impairment Score +7, or mNIS+7, a measure of neuropathic disease progression. In July 2018, the final results from the NEURO-TTR pivotal study were published in The New England Journal of Medicine.
Thrombocytopenia and safety signals related to renal function were identified during the study. Enhanced monitoring was implemented during the study to support early detection and management of these issues. Serious platelet and renal events were infrequent and manageable with routine monitoring, which has proven effective since implementation.
Additionally, at the 60th American Society of Hematology, or ASH, Annual Meeting and Exposition held in December 2018, we presented data from the open-label extension study, or OLE, in patients with hATTR treated with TEGSEDI. The OLE is an ongoing study and is intended to evaluate the long-term efficacy and safety profile of TEGSEDI. The benefits observed from TEGSEDI in the NEURO-TTR study continued in the OLE. In addition, the OLE results demonstrated that patients who initiated TEGSEDI treatment at the start of the NEURO-TTR study, 15 months earlier, experienced greater benefit than those who received placebo treatment in the NEURO-TTR study and then initiated treatment in the OLE. Patients who began the NEURO-TTR study on placebo experienced a rapid onset of effect following TEGSEDI treatment that has been sustained for up to 2 years in the OLE. These patients further experienced improvements in quality of life and activities of daily living as measured by Norfolk QoL-DN and showed improved mNIS+7 progression compared to their rate of progression in the NEURO-TTR study. Specifically, these patients experienced a mean increase in Norfolk QoL-DN of 16.8, a 10 point improvement over projected placebo values and a mean increase in mNIS+7 of 34 points from baseline, a 24 point improvement over projected placebo values. No new safety concerns were identified in the OLE.
The product label for TEGSEDI in the U.S. has a boxed warning for thrombocytopenia and glomerulonephritis and requires periodic blood and urine monitoring. TEGSEDI has a Risk Evaluation and Mitigation Strategy, or REMS, program. For TEGSEDI’s full prescribing information, including boxed warnings, please see www.tegsedi.com (Any information that is included on or linked to this website is not part of this report or any registration statement or report that incorporates this report by reference).
We developed TEGSEDI under a collaboration agreement we had with GSK. Under the agreement, we are required to pay GSK a nominal royalty on net sales of TEGSEDI.
See our separate section below where we further discuss Akcea, our affiliate focused on developing and commercializing medicines to treat people with serious and rare diseases.
Drug Discovery and Development
Introduction to Drug Discovery
Proteins are essential working molecules in a cell. Almost all human diseases result from inappropriate protein production, improper protein activity or loss of a protein. Antisense medicines can modify the production of proteins by targeting RNAs. In this way, antisense medicines can reduce the production of a disease-causing protein, modify the protein produced or increase the production of a protein that, when absent, causes diseases. Antisense medicines also can treat diseases by targeting and reducing RNAs that may be causing diseases (so called “toxic RNAs”). RNAs are naturally occurring molecules in the body that primarily act as messengers that carry the information the cell needs to produce proteins from the DNA/genes to the protein making complex in the cell. When our antisense medicines bind to the specific RNAs of a particular gene, they will ultimately alter the production of the protein encoded in the target gene or, in the case of disease-causing RNAs, degrade the toxic RNA.
Our Development Projects
We are a leader in the discovery and development of an exciting class of RNA-targeted medicines called antisense oligonucleotide, or ASO, medicines, or just antisense medicines. With our proprietary drug discovery platform, we can rapidly identify medicines from a wealth of potential targets to treat a broad range of diseases. We focus our efforts in therapeutic areas in which our medicines will work best, efficiently screening many targets in parallel and carefully selecting the best candidates. By combining this efficiency with our rational approach to selecting disease targets, we have built a large and diverse portfolio of medicines we designed to treat a variety of health conditions, such as cardiometabolic diseases, neurodegenerative diseases, cancer, severe and rare diseases and others. We are developing antisense medicines for systemic and local delivery (e.g., intrathecal, intraocular, oral and aerosol).
7
We plan to continue to add new medicines to our pipeline, building a broad proprietary portfolio of medicines to treat many diseases and creating opportunities to generate substantial revenue. We also continue to improve our scientific understanding of our medicines, including how our medicines impact the biological processes of the diseases we target.
With our expertise in discovering and characterizing novel antisense medicines, our scientists can optimize the properties of our antisense medicines against each particular target. Our scientists have made significant advances in chemical modifications we use in our antisense medicines, such as with our Generation 2+ antisense medicines, which have increased potency and an improved side effect profile over our earlier generation medicines. Our scientists have further improved upon our second-generation chemistry with our Generation 2.5 chemistry, an advancement that further increases the potency of our medicines, which broadens the organs and tissues in which our medicines can work. We currently have 13 Generation 2.5 medicines in development, and we anticipate that more of our future medicines will incorporate our Generation 2.5 chemistry.
In addition to improving the chemical foundation of our medicines, we have also created LIgand-Conjugated Antisense, or LICA, technology, which we design to enhance the effective uptake and activity of our medicines in particular tissues. With our LICA technology we attach specific chemical structures or molecules to our antisense medicines. With our first LICA conjugate, a complex sugar-like molecule called N-acetylgalactosamine, or GalNac, we have shown an increase in medicinal potency of over 30-fold for liver targets, compared to non-conjugated antisense medicines. We currently have 13 LICA medicines in development, including two medicines that combine our Generation 2.5 chemistry and LICA technology.
We have utilized our chemistry advancements, such as Generation 2.5 and LICA, to expand the therapeutic and commercial opportunities of our pipeline. These advancements, along with the manufacturing and analytical processes that are the same for all of our medicines, shorten our timeline from initial concept to the first human dose, when compared to early development timelines for other drug modalities like small molecule and antibody drugs.

The above table lists the medicines in our pipeline that are in registration for marketing authorization or in clinical trials. The table includes the disease indication, a partner (if the medicine is partnered), and the development status of each medicine. Typically, the names of our medicines incorporate the target of the medicine. For example, with IONIS-HTTRx, the RNA produced from the huntingtin gene, represented by the acronym HTT, is the target of the medicine. Unless indicated otherwise, the majority of medicines in our pipeline are Generation 2+ antisense medicines. We differentiate medicines discovered at Ionis but being developed by Akcea by using “AKCEA”, instead of “IONIS” at the beginning of the medicine name, such as AKCEA-ANGPTL3-LRx. We differentiate our Generation 2.5 medicines by adding a “2.5” notation at the end of the medicine name, such as IONIS-JBI1-2.5Rx. We differentiate our LICA medicines by adding an “L” at the end of the medicine name, such as IONIS-PKK-LRx. As the medicines in our pipeline advance in clinical development, we will adopt nonproprietary names given to each medicine from the U.S. Adopted Names Council. For example, inotersen is a nonproprietary name that we obtained for IONIS-TTRRx. Once we or our partners establish a brand name, we will adopt the brand name. For example, TEGSEDI is the brand name for inotersen.
8
With a pipeline as large and advanced as ours, we have a number of clinical events each year as we initiate new clinical studies, complete and report data from clinical studies, and add numerous new medicines to our pipeline.
WAYLIVRA – Potential Approval in Europe Following Positive CHMP Opinion
WAYLIVRA (volanesorsen) – WAYLIVRA is a Generation 2+ antisense medicine we and Akcea are developing to treat people with FCS and FPL, which are severe, rare, genetically defined diseases characterized by extremely elevated triglyceride levels and a high risk of life-threatening pancreatitis. We are preparing to commercialize WAYLIVRA in the EU.
In February 2019, the CHMP of the EMA adopted a positive opinion recommending conditional marketing authorization for WAYLIVRA as an adjunct to diet in adult patients with genetically confirmed FCS who are at high risk for pancreatitis, in whom response to diet and triglyceride lowering therapy has been inadequate. The positive opinion will now be referred to the EC, which grants marketing authorization for medicines in the EU, as well as to European Economic Area members Iceland, Liechtenstein and Norway. With this positive opinion, and, pending adoption of the positive opinion by the EC, Akcea plans to leverage its existing commercial infrastructure in Europe to market WAYLIVRA.
In August 2018, we received a complete response letter, or CRL, from the Division of Metabolism and Endocrinology Products of the FDA regarding the NDA for WAYLIVRA. We are continuing our discussions with the FDA regarding WAYLIVRA.
Due to the high levels of triglycerides in their blood, people with FCS may suffer from many chronic health issues including severe, recurrent abdominal pain, fatigue, high risk of life-threatening pancreatitis and abnormal enlargement of the liver or spleen. It is estimated to affect 3,000 to 5,000 people in treatable markets. In addition, people with FCS must adhere to a very strict, low-fat diet. FPL is a rare, orphan disease that is estimated to affect 3,000 to 5,000 patients worldwide. Patients with FPL typically have diabetes and other metabolic abnormalities, including elevated triglycerides, which increases their risk of pancreatitis. As a result of these factors, people with FCS and FPL are often unable to work, adding to the burden of these diseases. In severe cases, patients can have bleeding into the pancreas, serious tissue damage, infection and cyst formation, as well as damage to other vital organs such as the heart, lungs and kidneys.
WAYLIVRA acts to reduce triglyceride levels by inhibiting the production of apolipoprotein C-III, or apoC-III, a protein that is a key regulator of triglyceride levels. People who have low levels of apoC-III or reduced apoC-III function have lower levels of triglycerides and a lower incidence of CVD. By inhibiting the production of apoC-III, WAYLIVRA is able to reduce their triglyceride levels.
The marketing authorization application for WAYLIVRA is based on results from the Phase 3 APPROACH study and the ongoing APPROACH Open Label Extension, or OLE, study and supported by results from the Phase 3 COMPASS study. The pivotal APPROACH study, a one-year, randomized, placebo-controlled study in 66 patients with FCS (average baseline triglycerides of 2,209 mg/dL, or 25.0 mmol/L), achieved its primary endpoint of reduction in triglycerides at three months, with a 77 percent mean reduction in triglycerides, which translated into a 1,712 mg/dL (19.3 mmol/L) mean absolute triglyceride reduction in WAYLIVRA-treated patients. We observed 50 percent of treated patients achieved triglyceride levels below 500 mg/dL, a commonly accepted threshold for pancreatitis risk. In addition, in the APPROACH study, treatment with WAYLIVRA was associated with a statistically significant reduced rate of pancreatitis attacks in the group of patients who had the highest incidence of pre-study pancreatitis and reduced abdominal pain in patients reporting pain before treatment in the study.
The most common adverse event in the studies was injection site reactions, which were mostly mild. In addition, declines in platelet counts were observed in many patients and some patients discontinued the study because of platelet declines. These platelet declines were not clinically significant in most patients and were generally well managed with monitoring and dose adjustment. Some patients discontinued participation in the APPROACH study due to other non-serious adverse events, including sweating and chills, severe fatigue, rash and injection site reaction. In the APPROACH study and the open-label extension study, the potentially treatment- related serious adverse events, or SAEs, observed were serious platelet events (grade 4 thrombocytopenia), which resolved without complication after cessation of dosing. Enhanced monitoring was implemented during the study to support early detection and management of these issues. Since implementation of the enhanced monitoring, serious platelet events have been infrequent. The COMPASS study, a six-month randomized placebo-controlled study in 113 patients with very high triglycerides (>500 mg/dL), also achieved its primary endpoint of reduction in triglycerides at three months, with a 71 percent mean reduction in triglycerides. In the COMPASS study, treatment with WAYLIVRA was associated with a statistically significant reduction in on-study pancreatitis attacks. The most common adverse event in the WAYLIVRA-treated group of patients was injection site reactions, which were mostly mild. In addition, a potentially treatment-related SAE of serum sickness reaction, from which the patient fully recovered, was reported. There have been no deaths and no treatment-related bleeding or cardiovascular events in any WAYLIVRA clinical study.
We are conducting the BROADEN study, a Phase 3 clinical trial in patients with FPL, with data anticipated this year.
An open-label extension study is ongoing for patients with FCS who have completed or meet the study criteria for the APPROACH and COMPASS studies. Additionally, we have expanded access programs, or EAPs, for WAYLIVRA. Patients in the BROADEN study are also eligible to roll over into an open-label extension study upon completing dosing in the pivotal study. We plan to commercialize WAYLIVRA through Akcea for patients with FCS and FPL, if approved in other markets.
Potential Next Wave of Pivotal Medicines
Focusing on our key fundamental strategies has created a deep and broad pipeline of over 40 first-in-class and/or best-in-class medicines that we believe have the potential to deliver significant value to patients affected by these devastating diseases, many of which have limited treatment options. We have at least four medicines that have begun pivotal studies or have the potential to begin pivotal studies this year.
9

AKCEA-APO(a)-LRx (TQJ230) – AKCEA-APO(a)-LRx is a Generation 2+ LICA medicine we designed to reduce the production of apolipoprotein(a), or Apo(a), protein in the liver to offer a direct approach for reducing lipoprotein(a), or Lp(a). Lp(a) is an independent risk factor for CVD that is composed of an apolipoprotein(a) protein bound to an LDL-cholesterol particle. Akcea initiated a collaboration with Novartis in January 2017 to advance AKCEA-APO(a)-LRx.
Akcea is developing AKCEA-APO(a)-LRx for people who are at significant risk of CVD because of their elevated levels of Lp(a). AKCEA-APO(a)-LRx inhibits the production of the Apo(a) protein, thereby reducing Lp(a). Lp(a) is a very atherogenic and thrombogenic form of LDL. Elevated Lp(a) is recognized as an independent, genetic cause of coronary artery disease, heart attack, stroke and peripheral arterial disease. Inhibiting the production of Apo(a) in the liver reduces the level of Lp(a) in blood, potentially slowing down or reversing cardiovascular disease in people with hyperlipoproteinemia(a), a condition in which individuals have levels of Lp(a) greater than 50 mg/dL.
Lp(a) is difficult to inhibit using other technologies, such as small molecules and antibodies. There are multiple genetically-determined forms of the Apo(a) molecule and creating a small molecule or antibody that can interact with multiple targets is difficult. We believe antisense technology is well suited to address hyperlipoproteinemia(a) because antisense technology specifically targets the RNA that codes for all forms of the Apo(a) molecule. As a result, it can stop the production of all the forms of the protein. Furthermore, we believe addressing elevated Lp(a) is the next important horizon in lipid-focused treatment.
We reported results of the Phase 2 study with AKCEA-APO(a)-LRx in patients with hyperlipoproteinemia(a) at the American Heart Association, or AHA, annual meeting in November 2018. In this clinical study, we observed statistically significant and dose dependent reductions from baseline in Lp(a) levels. Approximately 98 percent of patients who received the highest dose in the study demonstrated a reduction in Lp(a) levels to below 50 mg/dL, the recognized threshold for risk of CVD. This study of AKCEA-APO(a)-LRx was the longest and largest clinical study in patients with established CVD and elevated levels of Lp(a). This study was also the longest and largest clinical study of any of our LICA medicines. AKCEA-APO(a)-LRx demonstrated a favorable safety and tolerability profile in the study. Compliance in the study was almost 90 percent, which was higher than what we observed in the placebo group.
In February 2019, Novartis exercised its option to license AKCEA-APO(a)-LRx and Novartis’ preparations to initiate Phase 3 a cardiovascular outcomes study are already underway
AKCEA-TTR-LRx – We are co-developing AKCEA-TTR-LRx with Akcea to inhibit the production of transthyretin, the same protein inhibited by TEGSEDI (inotersen). There are two types of ATTR amyloidosis: hATTR amyloidosis and wt-ATTR amyloidosis.
We are developing AKCEA-TTR-LRx for the treatment of people with all forms of TTR amyloidosis as a once a month or even less frequent subcutaneous self-administered injection. We plan to report data from the Phase 1/2 study this year, followed by the initiation of a pivotal program. We plan to initiate a Phase 3 study in patients with hereditary TTR amyloidosis with polyneuropathy first, followed closely by a Phase 3 study in patients with wild type and hereditary TTR cardiomyopathy, also planned for this year.
IONIS-HTTRx – IONIS-HTTRx (RG6042) is a Generation 2+ antisense medicine we designed to target the underlying cause of HD by reducing the production of the toxic mHTT protein. Roche initiated the Phase 3 study of IONIS-HTTRx for Huntington’s disease, or HD, in December 2018 and the first patient was dosed in the Phase 3 study in January 2019. In addition to the Phase 3 study, all participants who took part in the Phase 1/2 study are eligible to continue to receive IONIS-HTTRx as part of an OLE study to assess the safety and tolerability of IONIS-HTTRx. In parallel with the Phase 3 study and the OLE, Roche initiated a natural history study in a similar patient population to the OLE. The natural history study is planned as a 15-month observational study aimed at further understanding the role of mHTT in disease progression and is anticipated to include up to 100 participants with Stage I and II HD. There is no drug treatment in the observational study, as the goal is to understand the natural progression of HD.
We completed a randomized, placebo-controlled, dose escalation, Phase 1/2 clinical study of IONIS-HTTRx in patients with early stage HD. In this study, we observed dose-dependent reductions of mHTT among patients treated with IONIS-HTTRx and IONIS-HTTRx demonstrated a favorable safety and tolerability profile. In March 2018, we reported data from the study that demonstrated up to a 60 percent reduction in the mHTT as observed in the CSF. It was the first study to demonstrate disease-modifying potential. The mHTT reductions of 40-60 percent in the CSF correspond to an estimated 55-85 percent reduction in the cortex of the brain, where mHTT is highly expressed, based on preclinical data. There were no serious adverse events reported and no participants discontinued from the study. In August 2018, the EMA granted PRIME designation to IONIS-HTTRx. EMA PRIME status is granted to medicines that may offer a major therapeutic advantage over existing treatments, or benefit patients without treatment options. The FDA and EMA granted Orphan Medicine Designation for IONIS-HTTRx to treat people with HD.
10
HD is a rare, inherited, genetic brain disorder that results in the progressive deterioration of mental abilities and physical control. In the U.S., there are approximately 30,000 individuals with symptomatic HD and more than 200,000 people at risk of inheriting HD. HD is a triplet repeat disorder and is one of a large family of genetic diseases in which the body mistakenly repeats certain gene sequences. The resulting mHTT protein is toxic and gradually damages neurons in the brain. Symptoms of HD usually appear between the ages of 30 to 50 years and continually worsen over a 10 to 25-year period. Ultimately, the weakened individual succumbs to pneumonia, heart failure or other complications. Presently, there are no disease-modifying treatments available for HD patients, with current drugs only managing some disease symptoms.
We entered into a collaboration with Roche to develop and commercialize antisense medicines to treat HD in April 2013. In December 2017, Roche exercised its licensing option to develop and commercialize IONIS-HTTRx following the completion of a Phase 1/2 randomized, placebo-controlled, dose escalation study of IONIS-HTTRx in people with HD. Roche is responsible for all IONIS-HTTRx development, regulatory and commercialization activities and costs.
IONIS-SOD1Rx (BIIB067) – IONIS-SOD1Rx is a Generation 2+ antisense medicine we designed to reduce the production of superoxide dismutase 1, or SOD1, which is a well understood genetic cause of familial amyotrophic lateral sclerosis, or ALS. We are collaborating with Biogen to develop IONIS-SOD1Rx to treat people with an inherited form of ALS, SOD1-ALS.
ALS is a rare, fatal, neurodegenerative disorder. People with ALS suffer progressive degeneration of the motor neurons, which results in a declining quality of life and ultimately death. The second most common familial form of ALS is SOD1-ALS, in which people have a mutation in the SOD1 gene that causes a progressive loss of motor neurons. As a result, people with SOD1-ALS experience muscle weakness, loss of movement, difficulty breathing and swallowing and eventually succumb to the disease. Currently, treatment options for people with ALS are extremely limited, with no medicines that significantly slow disease progression.
In December 2018, Biogen exercised its licensing option to develop and commercialize IONIS-SOD1Rx based on the positive interim analysis from the Phase 1/2 study that demonstrated proof-of-biology and proof-of-concept. Biogen is responsible for all IONIS-SOD1Rx development, regulatory and commercialization activities and costs. At the highest dose tested, treatment with IONIS-SOD1Rx over a three month period resulted in a statistically significant lowering of SOD1 protein levels in the CSF and positive numerical trends across three efficacy endpoints: slowing of clinical decline as measured by the ALS functional rating scale-revised, slowing of decline in respiratory function as measured by vital capacity and slowing of decline in muscle strength as measured by a handheld device, all compared to placebo. The safety and tolerability profile in this study supports the continued development of IONIS-SOD1Rx in ALS.
Biogen plans to add an additional cohort to this study to potentially support registration of IONIS-SOD1Rx.
Neurological Disease Franchise
We are discovering and developing antisense medicines to treat people with inadequate treatment options for both common and rare neurological diseases. According to the National Institute of Neurological Disorders and Stroke, or NINDS, at the National Institutes of Health, or NIH, a third of the 7,000 known rare diseases are neurological disorders or thought to include a neurological component.
IONIS’ Neurological Disease Clinical Pipeline

IONIS-HTTRx – See the medicine description under “Next Wave of Pivotal Medicines” section above.
AKCEA-TTR-LRx – See the medicine description under “Next Wave of Pivotal Medicines” section above.
IONIS-SOD1Rx – See the medicine description under “Next Wave of Pivotal Medicines” section above.
IONIS-MAPTRx – IONIS-MAPTRx is a Generation 2+ antisense medicine we designed to selectively reduce production of the tau protein in the brain. We are collaborating with Biogen to develop IONIS-MAPTRx to treat people with Alzheimer’s disease, or AD, and frontotemporal dementia, or FTD, common forms of dementia.
11
Microtubule-associated protein tau, or tau, is a contributor or cause of certain neurodegenerative diseases, known as tauopathies, characterized by the deposition of abnormal tau protein in neurons and non-neuronal cells in the brain. AD and FTD are characterized by predominant memory impairment and behavioral changes, resulting in a person’s inability to independently perform daily activities. AD generally occurs late in life and may progress to death in five to 20 years after the onset of the disease. FTD has a more rapid disease progression. There are approximately five million people living with AD in the U.S. and approximately 55,000 people affected by FTD in the U.S.
We and Biogen are evaluating IONIS-MAPTRx in a Phase 1/2 double-blind, randomized, placebo-controlled, dose-escalation study to evaluate the safety and activity of once-monthly intrathecal injections in patients with mild AD. We are planning to report data from this study in 2020.
IONIS-C9Rx – IONIS-C9Rx, also referred to as BIIB078, is a Generation 2+ antisense medicine we designed to selectively reduce the production of the mutated chromosome 9 open reading frame 72, or C9ORF72, gene. A mutation in this gene results in an inherited form of ALS, referred to as C9ORF72-ALS, the most prevalent genetic cause of ALS worldwide. There is substantial evidence that this mutation is responsible for a toxic gain of function repeat expansion that can lead to rapid progressive loss of motor neurons in people with C9ORF72-ALS. This is a fatal disease characterized by muscle weakness, loss of movement, and difficulty breathing and swallowing. We believe IONIS-C9Rx represents a novel approach to targeting ALS, for which there is no cure.
We and Biogen are collaborating to develop IONIS-C9Rx to treat patients with this form of ALS. In August 2018, we initiated a Phase 1/2 clinical study evaluating IONIS-C9Rx in patients with C9ORF72-ALS. The current study is a randomized, blinded, placebo-controlled study designed to assess the safety, tolerability, and pharmacokinetics of multiple ascending doses of IONIS-C9Rx administered intrathecally to adults with C9ORF72-ALS. IONIS-C9Rx is the second medicine from our Biogen collaboration targeting a familial form of ALS. The first is IONIS-SOD1Rx, designed to treat SOD1 related ALS, caused by a mutation in the SOD1 gene.
Severe and Rare Disease Franchise
Our severe and rare disease franchise is one the largest franchises in our pipeline. We are discovering and developing antisense medicines to treat people with severe and rare diseases who need new treatment options. We believe our antisense technology could offer effective therapies for these people. According to the NIH there are approximately 7,000 rare diseases, many life-threatening or fatal. Unfortunately, people with many of these severe and rare diseases have few effective therapies available. Since most of these diseases are genetic or have a genetic component, parents often pass the disease to their children, creating a legacy of the disease resulting in profound effects on the family. Due to the severe nature of these diseases and the lack of available treatments, there is an opportunity for more flexible and efficient development paths to the market. For example, SPINRAZA was approved five years after we began the Phase 1 study for it.
IONIS’ Severe and Rare Disease Clinical Pipeline

WAYLIVRA - See the medicine description under “WAYLIVRA - Under Regulatory Review for Marketing Authorization” section above.
AKCEA-TTR-LRx – See the medicine description “Next Wave of Pivotal Medicines” section above.
IONIS-GHR-LRx – IONIS-GHR-LRx is a Generation 2+ LICA medicine we designed to reduce the production of the growth hormone receptor, or GHr, to decrease the circulating level of insulin-like growth factor-1, or IGF-1. IGF-1 is a hormone primarily produced in the liver that plays an important role in childhood growth and has anabolic effects in adults. Several different diseases result from abnormally low or high levels of IGF-1, or an inappropriate response to this hormone. When produced in excess, IGF-1 results in acromegaly, a chronic, and life-threatening disease.
High levels of circulating GH and IGF-1 lead to this multisystem disease characterized by organ overgrowth and physical disfigurement, such as enlarged hands, feet, and facial features. Patients with acromegaly also experience multiple co-morbidities, such as type 2 diabetes, hypertension, and respiratory complications, as well as premature mortality. Because IGF-1 mediates the majority of the growth-promoting action of GH, reducing GHr production could in turn decrease levels of IGF-1 and provide a potential treatment to patients with acromegaly. Acromegaly is a rare disease with an estimated 25,000 patients in the U.S. Current treatments to block IGF-1 include surgical removal of the pituitary gland, which is often unsuccessful. Drug treatments to normalize IGF-1 levels are also available but are associated with potentially serious side effects.
12
We have completed a Phase 1, double-blind, placebo-controlled, dose-escalation study of IONIS-GHR-LRx in healthy volunteers. In this study, IONIS-GHR-LRx demonstrated a favorable safety and tolerability profile. There were no reports of deaths, serious adverse events or adverse events that led to study discontinuation. IONIS-GHR-LRx has the potential to bring substantial benefit to patients with acromegaly with at home, monthly subcutaneous administration.
In November 2018, we initiated the Phase 2 proof of concept clinical study of IONIS-GHR-LRx in acromegaly patients. The study is a randomized, double-blind, placebo-controlled, multi-center study in acromegaly patients uncontrolled on select long-acting somatostatin receptor ligands. Patients in the study will receive monthly subcutaneous injections for four months. We anticipate we will complete this study by the end of this year.
IONIS-TMPRSS6-LRx – IONIS-TMPRSS6-LRx is a Generation 2+ LICA medicine we designed to reduce the production of transmembrane protease, serine 6, or TMPRSS6, to treat anemia and iron toxicity in people with β-thalassemia, a disease caused by mutations in the beta globin gene. TMPRSS6 is a protein produced in the liver that is important in the regulation of the body’s iron homeostasis through the control of the iron regulatory protein hepcidin. Inhibition of TMPRSS6 leads to increased production of hepcidin, which results in more effective red blood cell production in the bone marrow and reduced iron toxicity in the liver as a result of improved control of iron availability.
Patients with β-thalassemia can experience severe anemia, marrow expansion, bone deformities, as well as iron toxicity. While the severity of anemia varies between patients, iron toxicity is a common complication leading to high rates of mortality as a result of iron accumulation in major organs, such as the heart and liver. Currently there are no effective therapies for patients with β-thalassemia. The current standard of care is managing patients’ symptoms with blood transfusions, hydroxyurea, and iron chelation.
β-thalassemia can be further subdivided into patients with transfusion-dependent thalassemia, or TDT, and non-transfusion dependent thalassemia, or NTDT, including β-thalassemia intermedia. Although transfusions are not needed to support life in patients with NTDT, the associated complications of the disease are severe and often fatal. There are approximately 20,000 people in North America and Europe who suffer from β-thalassemia intermedia.
Results from preclinical and clinical studies suggest that reducing levels of TMPRSS6 may be an effective strategy to control iron availability, improve liver iron toxicity and increase red blood cell production under conditions of β-thalassemia. In December 2018, we presented positive Phase 1 data at the ASH Annual Meeting. In a randomized, double-blind, placebo-controlled, dose-escalation Phase 1 study in healthy volunteers, we demonstrated dose-dependent reductions of serum iron and serum transferrin saturation. Additionally, we observed an increase in serum hepcidin and predicted changes in hemoglobin. IONIS-TMPRSS6-LRx demonstrated a favorable safety and tolerability profile.
We are planning to begin the Phase 2 proof of concept study of IONIS-TMPRSS6-LRx this year.
IONIS-PKKRx and IONIS-PKK-LRx – IONIS-PKKRx and IONIS-PKK-LRx are antisense medicines we designed to reduce the production of prekallikrein, or PKK, to treat people with hereditary angioedema, or HAE. It is a rare genetic disease that is characterized by rapid and painful attacks of inflammation in the hands, feet, limbs, face, abdomen, larynx and trachea and can be fatal if swelling occurs in the larynx. PKK plays an important role in the activation of inflammatory mediators associated with acute attacks of HAE. By inhibiting the production of PKK, IONIS-PKKRx and IONIS-PKK-LRx could be effective prophylactic approaches to preventing HAE attacks. In patients with frequent or severe attacks, doctors may use prophylactic treatment approaches to prevent or reduce the severity of HAE attacks.
We have completed a Phase 1 study evaluating IONIS-PKKRx in healthy volunteers and we are exploring potential development options. In this study, IONIS-PKKRx demonstrated a favorable safety and tolerability profile. We are currently evaluating IONIS-PKK-LRx in a Phase 1, randomized, double-blind, placebo-controlled, dose-escalation study in healthy volunteers. The Phase 1 study is evaluating single and multiple doses of IONIS-PKK-LRx administered subcutaneously.
IONIS-ENAC-2.5Rx – IONIS-ENAC-2.5Rx is a Generation 2.5 antisense medicine we designed to selectively reduce epithelial sodium channel, or EnaC, to treat people with cystic fibrosis, or CF. CF is an autosomal recessive disorder caused by mutations in the gene that encodes the cystic fibrosis transmembrane conductance regulator, or CFTR. CFTR is a chloride channel expressed in epithelial cells, including those in the lung. Targeting ENAC may enable treatment of all forms of CF due to various CFTR mutations, unlike existing therapeutics. CF is a multisystem disease that mostly affects the lungs, clogging airways due to mucus build-up and resulting in inflammation and infection. This disease is characterized by a progressive decline in lung function with acute periods of worsened symptoms, known as pulmonary exacerbations. CF is estimated to affect approximately 30,000 people in the U.S. and another 70,000 worldwide. Despite progress with other treatments, there remains a need for effective treatment options.
Antisense aerosol technology for lung delivery may provide a novel solution for targeting ENaC potentially enabling all patients with CF to be treated. In preclinical studies in transgenic rodents, treatment with ENaC-targeting antisense drugs specifically suppressed ENaC expression, resulting in the reduction of markers of CF mucus pathology and improved lung function. Treatment prevented manifestations of the disease from occurring and reversed existing CF.
In December 2018, we initiated a Phase 1 study of healthy volunteers in a double-blinded, placebo-controlled, dose-escalation study to evaluate the safety and efficacy of IONIS-ENAC-2.5Rx. The study will consist of four randomized single-dose cohorts and four multiple-dose cohorts.
13
Cardiometabolic and Renal Disease Franchise
Cardiovascular disease is an important area of focus for us. According to the World Health Organization, or WHO, cardiovascular disease was the number one cause of death globally. An estimated 17.9 million people died from CVD in 2016, representing 31 percent of all deaths globally. The medicines in our cardiovascular franchise target the key components of cardiovascular disease, including various atherogenic lipids, inflammation and thrombosis. Metabolic disorders are chronic diseases that affect tens of millions of people. There is a significant need for new therapies for these people. According to the Centers for Disease Control and Prevention, diabetes affects more than 30 million people in the U.S., or nine percent of the population, with type 2 diabetes constituting 90 percent of those cases.
IONIS’ Cardiometabolic and Renal Disease Clinical Pipeline

AKCEA-ANGPTL3-LRx – AKCEA-ANGPTL3-LRx is a Generation 2+ LICA medicine we designed to reduce the production of the angiopoietin-like 3, or ANGPTL3, protein. We and Akcea are developing AKCEA-ANGPTL3-LRx to treat nonalcoholic fatty liver disease, or NAFLD.
People with elevated levels of the angiopoietin-like 3, or ANGPTL3, protein have high LDL-C and triglyceride levels. Studies show this elevation is associated with an increased risk of premature heart attacks, increased arterial wall thickness, increased liver fat and multiple metabolic disorders, such as insulin resistance. In contrast, people with lower levels of ANGPTL3 have lower LDL-C and triglyceride levels, and thus lower risk of heart attacks, lower prevalence of fatty liver and lower incidence of metabolic disorders.
In preclinical studies, an analog of AKCEA-ANGPTL3-LRx inhibited the production of the ANGPTL3 protein in the liver, resulting in lower liver fat accumulation and lower blood levels of LDL-C, triglycerides and very low-density lipoprotein cholesterol, or VLDL-C. In addition, our preclinical data and initial Phase 1 data suggest that inhibiting the production of ANGPTL3 could improve other lipid parameters, including triglyceride levels and total cholesterol, as well as metabolic parameters, such as insulin sensitivity.
We have completed a Phase 1/2 program for AKCEA-ANGPTL3-LRx in healthy volunteers with elevated triglycerides. Results for the initial cohort from this study were reported at the AHA meeting in November 2016 and the data were published in The New England Journal of Medicine. We observed that the people with elevated triglycerides achieved dose-dependent, statistically significant mean reductions in ANGPTL3 of up to 83 percent. Treatment with AKCEA-ANGPTL3-LRx was also associated with statistically significant mean reductions in triglycerides of up to 66 percent, in LDL-C of up to 35 percent and in total cholesterol of up to 36 percent. In this study, AKCEA-ANGPTL3-LRx demonstrated a favorable safety and tolerability profile.
In the fourth quarter of 2017, we initiated a multicenter, randomized, double-blind, placebo-controlled dose-ranging study of AKCEA-ANGPTL3-LRx in patients with NAFLD with metabolic complications, which include hypertriglyceridemia, type 2 diabetes and nonalcoholic steatohepatitis, or NASH. We are planning to report data from this study in 2020.
Further, we have a small ongoing study of AKCEA-ANGPTL3-LRx in patients with rare hyperlipidemias.
IONIS-FXIRx and IONIS-FXI-LRx – IONIS-FXIRx and IONIS-FXI-LRx are antisense medicines we designed to reduce the production of Factor XI. Factor XI is a clotting factor produced in the liver that is important in the growth of blood clots. High levels of Factor XI increase the risk of thrombosis, which is the formation of a blood clot inside blood vessels. Thrombosis can cause heart attacks and strokes. People who are deficient in Factor XI have a lower incidence of thromboembolic events with minimal increase in bleeding risk. Although currently available anticoagulants reduce the risk of thrombosis, physicians associate these anticoagulants with increased bleeding, which can be fatal. Given the mechanism of Factor XI inhibition, we believe that our medicine can be used broadly as an anti-thrombotic in many different therapeutic settings for which additional safe and well tolerated anti-thrombotic medicines are needed.
We completed a Phase 2 open-label, comparator-controlled global study evaluating IONIS-FXIRx in people undergoing total knee replacement surgery. The study compared the safety and activity of IONIS-FXIRx to enoxaparin. In this study patients treated with 300 mg of IONIS-FXIRx experienced a seven-fold lower rate of venous thromboembolic events, such as blood clots in a deep vein or in a lung, compared to those patients treated with enoxaparin. In this study, IONIS-FXIRx demonstrated a favorable safety and tolerability profile. The data from this study were published in The New England Journal of Medicine in December 2014.
In May 2015, we exclusively licensed IONIS-FXIRx to Bayer.
14
In November 2016, we completed a Phase 2 double-blinded, randomized, placebo-controlled study of IONIS-FXIRx in people with end-stage renal disease on hemodialysis. In this Phase 2 study, patients treated with IONIS-FXIRx achieved statistically significant, dose-dependent reductions in Factor XI activity. In this study, IONIS-FXIRx demonstrated a favorable safety and tolerability profile. There were no treatment-related major or clinically relevant non-major bleeding events.
We are currently evaluating IONIS-FXIRx in a Phase 2b study in people with end-stage renal disease on hemodialysis to finalize dose selection. We are planning to report data from this study this year.
In February 2017, we amended our agreement with Bayer to advance IONIS-FXIRx and to initiate development of IONIS-FXI-LRx. We plan to develop IONIS-FXI-LRx through Phase 1. The Phase 1 study is in progress in healthy volunteers. It is a double-blind, randomized, placebo-controlled, dose-escalation study that will assess the safety and efficacy of IONIS-FXI-LRx.
AKCEA-APO(a)-LRx – See the medicine description under “Next Wave of Pivotal Medicines” section above.
AKCEA-APOCIII-LRx – AKCEA-APOCIII-LRx is a LICA medicine we designed to inhibit the production of apoC-III, the same protein inhibited by WAYLIVRA, for the broad population of people who are at risk for cardiometabolic disease due to their elevated triglyceride levels. We and Akcea are developing AKCEA-APOCIII-LRx. ApoC-III impacts triglyceride levels and may also increase inflammatory processes. This combination of effects makes apoC-III a promising target for people with LDL-C already controlled on statin therapy, but for whom triglycerides remain poorly controlled. We believe that the enhancements offered by our LICA technology can provide greater patient convenience by allowing for significantly lower doses and less frequent administration, compared to WAYLIVRA.
In October 2017, we reported positive results of a Phase 1/2 clinical study in healthy volunteers with elevated triglyceride levels. Patients in the study were treated with multiple doses at either weekly or monthly dosing intervals. Patients treated with AKCEA-APOCIII-LRx demonstrated significant dose-dependent reductions in apoC-III protein and triglycerides. In this study, AKCEA-APOCIII-LRx demonstrated a favorable safety and tolerability profile. No serious adverse events, platelet count reductions, changes in liver function or adverse events leading to treatment discontinuation were observed.
Novartis entered into a collaboration with us in January 2017 to advance AKCEA-APOCIII-LRx. In the first quarter of 2018, we initiated a Phase 2b dose-ranging study of AKCEA-APOCIII-LRx in patients with hypertriglyceridemia and established CVD. We plan to report data from this study in 2020.
IONIS-DGAT2Rx – IONIS-DGAT2Rx is a Generation 2+ antisense medicine we designed to reduce the production of DGAT2, or diacylglycerol acyltransferase 2, to treat people with NASH. NASH is a common liver disease characterized by excessive triglycerides in the liver with concurrent inflammation and cellular damage. As NASH progresses, scarring, or fibrosis, begins to accumulate in the liver. Ultimately, cirrhosis of the liver develops. Currently, it is estimated that two to three percent of the general population have NASH. With the growing obesity epidemic, the number of people with NASH should also continue to rise. About 20 percent of people with NASH are reported to have a liver that does not function properly due to long-term damage, known as cirrhosis. Of those with NASH-related cirrhosis, 30 - 40 percent experience liver-related death. Currently, liver transplantation is the only treatment for advanced cirrhosis and liver failure. Because of the high prevalence of NASH, it has recently become the third most common indication for liver transplantation in the U.S.
DGAT2 is an enzyme that catalyzes the final step in triglyceride synthesis in the liver. Reducing the production of DGAT2 should therefore decrease triglyceride synthesis in the liver. In animal models of obesity and fatty liver disease, antisense inhibition of DGAT2 significantly improved NAFLD, lowered blood lipid levels and reversed diet-induced insulin resistance. NASH is a more severe form of NAFLD.
IONIS-DGAT2Rx was evaluated in a Phase 2 randomized, placebo-controlled, dose-escalation study in patients with type 2 diabetes and NAFLD. In December 2018, we reported that IONIS-DGAT2Rx substantially reduced liver fat after only three months of treatment. 50 percent of IONIS-DGAT2Rx treated patients had relative liver fat reductions of greater than or equal to 30 percent. IONIS-DGAT2Rx demonstrated a favorable safety profile with no safety concerns related to the liver, kidney or platelets. Additionally, there were no increased levels of triglycerides or cholesterol. We plan to develop a liver LICA version of IONIS-DGAT2Rx.
IONIS-AGT-LRx – IONIS-AGT-LRx is a Generation 2+ LICA medicine we designed to reduce the production of angiotensinogen to decrease blood pressure in people with treatment resistant hypertension, or TRH. Despite the availability of generic antihypertensive agents, TRH is a major contributor to cardiovascular and renal disease.
Approximately 75 million adults in the U.S. have hypertension, half of whom have uncontrolled hypertension. About 12-15 percent of patients with uncontrolled hypertension have resistant hypertension, defined as failure to achieve a blood pressure goal of 140/90 (systolic/diastolic) despite the use of three or more antihypertensive medications. Current estimates approximate that there are up to three million people with TRH in the U.S. People with TRH have been found to have a three-fold higher chance of having fatal and non-fatal cardiovascular events relative to those with controlled hypertension.
We are evaluating IONIS-AGT-LRx in a double-blinded, randomized, placebo-controlled, Phase 2 study in people with mild hypertension.
15
Oncology Franchise and Other Medicines in Development
Cancer is an area of significant unmet medical need. Cancer is an extremely complex disease that involves a large number of targets. Using our antisense technology, we can validate multiple potential cancer targets from a variety of different cancers, and rapidly identify anti-cancer drugs, which in many cases are the same or similar sequences to those used to validate the target. We preferentially select anti-cancer targets that can potentially provide a multi-faceted approach to treating cancer.
Our oncology franchise consists of anti-cancer antisense medicines that act upon biological targets associated with cancer progression, treatment resistance, and/or the tumor immune environment. We have a strategic alliance with AstraZeneca, which includes an anti-cancer collaboration that expands our anti-cancer efforts and supports a robust clinical development plan for danvatirsen and IONIS-AZ7-2.5Rx. AstraZeneca brings significant experience that enables the identification of novel genetic and epigenetic targets for cancer. Combining AstraZeneca’s expertise with our drug discovery technology, we plan to expand our oncology franchise with a number of promising new anti-cancer targets. We also have a collaboration agreement with University of Texas MD Anderson Cancer Center to identify cancer targets and create novel antisense medicines to treat cancer together.
Our Generation 2.5 chemistry enhances the potency and effectiveness of our antisense medicines, and potentially allows us to extend the applicability of our technology to cancers that are difficult to treat. For instance, STAT3 is a protein known to be important in carcinogenesis, however, it has been difficult to approach with traditional drug modalities. Data from a Phase 1b/2 clinical study of danvatirsen in combination with durvalumab, AstraZeneca’s programmed death ligand, or PD-L1, blocking antibody showed evidence of antitumor activity in people with advanced solid tumors and recurrent metastatic head and neck cancer.
IONIS’ Oncology/Other Clinical Pipeline
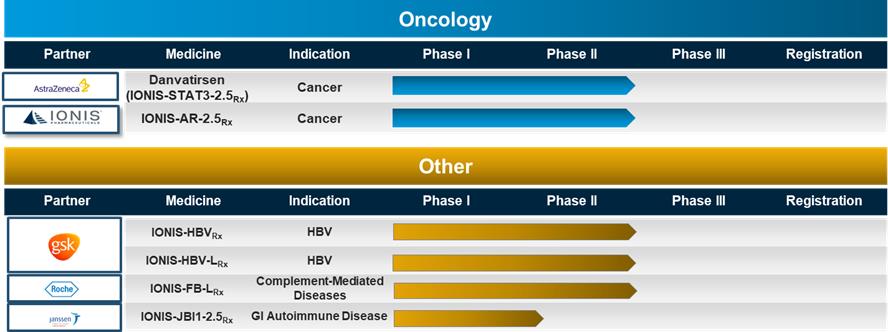
Danvatirsen (formerly IONIS-STAT3-2.5Rx) – Danvatirsen is a Generation 2.5 antisense medicine we designed to reduce the production of signal transducer and activator of transcription 3, or STAT3, to treat people with cancer. STAT3 is a protein involved in the translation of key factors critical for tumor cell growth and survival. STAT3 is over-active in a variety of cancers, including brain, lung, breast, bone, liver and multiple myeloma. Physicians believe that overactivity in STAT3 prevents cancer cell death and promotes tumor cell growth. Danvatirsen is a part of our collaboration with AstraZeneca to discover and develop anti-cancer medicines. We believe the significant potency we observed in our preclinical studies with danvatirsen broadens the therapeutic opportunities danvatirsen into many different types of cancer in which STAT3 is implicated.
In October 2018, we and AstraZeneca announced new data from a Phase 1b/2 study of danvatirsen in combination with durvalumab in recurrent metastatic head and neck cancer. The combination treatment resulted in seven percent of patients achieving a complete tumor response and 23 percent achieving either a partial or complete tumor response. This response rate is estimated to be double that with durvalumab alone, based on previous studies in this difficult to treat patient population. Results from this study demonstrated safety and tolerability profile supportive of continued development.
AstraZeneca is evaluating danvatirsen in a range of cancer types as part of a broader oncology partnership evaluating Generation 2.5 antisense therapies against undruggable targets either alone or in combination with immuno-oncology agents, including in non-small cell lung cancer, bladder cancer and head and neck cancer.
IONIS-AR-2.5Rx – IONIS-AR-2.5Rx, also known as AZD5312, is a Generation 2.5 antisense medicine we designed to treat people with prostate cancer by reducing the production of all known forms of androgen receptor, or AR, including variants of the AR gene. Prostate cancer is the second leading cause of cancer deaths in American men. Prostate cancer growth, proliferation and progression are all androgen-dependent and AR function is involved in disease progression at all stages of prostate cancer.
An open-label, dose-escalation, Phase 1/2 clinical study of IONIS-AR-2.5Rx was completed in people with advanced tumors for which the androgen receptor pathway is potentially a contributing factor. The study was primarily conducted in prostate cancer patients and it showed durable responses in a number of those patients. The medicine exhibited a favorable safety and tolerability profile supportive of continued development. In March 2017, we licensed IONIS-AR-2.5Rx to Ribo to develop and commercialize the medicine in China.
16
IONIS-HBVRx and IONIS-HBV-LRx – IONIS-HBVRx and IONIS-HBV-LRx are antisense medicines we designed to reduce the production of viral proteins associated with hepatitis B virus, or HBV. These include proteins associated with infection and replication, including the hepatitis B surface antigen, which is present in both acute and chronic infections and is associated with a poor prognosis in people with chronic HBV infection. IONIS-HBV-LRx is our first anti-infective medicine in development that incorporates our LICA technology. Together with GSK, we are evaluating IONIS-HBVRx and IONIS-HBV-LRx to treat HBV infection.
HBV infection is a serious health problem that can lead to significant and potentially fatal health conditions, including cirrhosis, liver failure and liver cancer. Chronic HBV infection is one of the most common persistent viral infections in the world. Currently available therapies, although effective in reducing circulating HBV in the blood, do not effectively inhibit HBV antigen production and secretion, which are associated with poor prognosis and increased risk of liver cancer.
We and GSK are evaluating both IONIS-HBVRx and IONIS-HBV-LRx in Phase 2 studies designed to reduce production of viral proteins associated with HBV infection.
IONIS-FB-LRx – IONIS-FB-LRx is a Generation 2+ LICA medicine we designed to reduce the production of complement factor B, or FB. FB is produced predominantly in the liver and circulates at high levels throughout the vascular system where it plays a pivotal role in an innate immunogenic cascade. Genetic association studies have shown that overactivity of this cascade has been associated with the development of several complement-mediated diseases, including dry age-related macular degeneration, or AMD.
In May 2017, we reported data from a randomized, placebo-controlled, dose-escalation Phase 1 study evaluating IONIS-FB-LRx in 54 healthy volunteers. Subjects treated with a single dose of IONIS-FB-LRx achieved dose-dependent reductions in plasma FB of up to 50 percent. Treatment with multiple doses of IONIS-FB-LRx during a six-week period resulted in greater reductions in circulating FB levels. In this study, IONIS-FB-LRx demonstrated a favorable safety and tolerability profile.
In October 2018, we entered into a new collaboration with Roche to develop IONIS-FB-LRx for the treatment of complement-mediated diseases. The first indication that we and Roche agreed to pursue is the treatment of patients with geographic atrophy, or GA, the advanced stage of dry AMD. We plan to start a Phase 2 study of IONIS-FB-LRx in people with dry AMD this year.
IONIS-JBI1-2.5Rx – IONIS-JBI1-2.5Rx is a Generation 2.5 antisense medicine we designed to treat people for an undisclosed target of gastrointestinal autoimmune disease. In December 2014, we entered into a collaboration agreement with Janssen to discover and develop antisense drugs that can be locally administered, including oral delivery, to treat autoimmune disorders of the GI tract. In July 2016, Janssen licensed IONIS-JBI1-2.5Rx from us. Janssen is currently conducting a Phase 1 study of IONIS-JBI1-2.5Rx.
Preclinical Medicines in Development
The efficiency and broad applicability of our technology enables us to develop medicines for a broad range of diseases. On average, it takes 12 to 18 months to complete the preclinical studies necessary to support clinical development. Over the last year we added eight new medicines to our preclinical pipeline.
IONIS’ Preclinical Pipeline
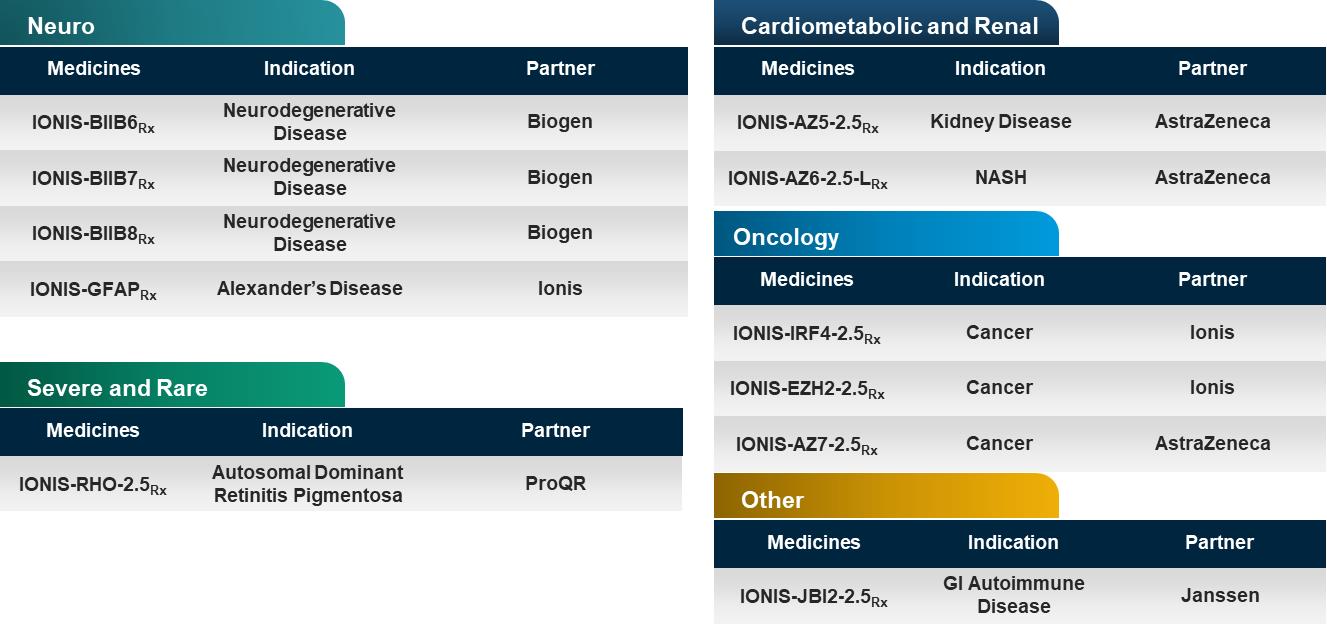
17
Akcea Therapeutics: Our Affiliate Focused on Developing and Commercializing Medicines to Treat People with Serious and Rare Diseases
We formed Akcea Therapeutics in 2015 to focus on developing and commercializing medicines to treat people with serious and rare diseases. Akcea is commercializing TEGSEDI, a medicine we discovered and developed. Additionally, Akcea is advancing a mature pipeline of five of our novel medicines, including WAYLIVRA, AKCEA-APO(a)-LRx, AKCEA-ANGPTL3-LRx, AKCEA-APOCIII-LRx, and AKCEA-TTR-LRx, all with the potential to treat multiple diseases. We discovered all of these medicines, which are based on our proprietary antisense technology. Akcea is co-developing these five drugs with us.
This report includes financial information for this separate business segment in Note 7, Segment Information and Concentration of Business Risk, in the Notes to the Consolidated Financial Statements.
TEGSEDI – See the medicine description under “Our Marketed Medicines” section above.
WAYLIVRA –See the medicine description under “WAYLIVRA – Potential Approval in Europe Following Positive CHMP
Opinion” section above.
AKCEA-APO(a)-LRx – See the medicine description under “Potential Next Wave of Pivotal Medicines” section above.
AKCEA-TTR-LRx – See the medicine description under “Potential Next Wave of Pivotal Medicines” section above.
AKCEA-ANGPTL3-LRx – See the medicine description under “Cardiometabolic and Renal Disease Pipeline” section above.
AKCEA-APOCIII-LRx – See the medicine description under “Cardiometabolic and Renal Disease Pipeline” section above.
Satellite Company Medicines in Development
We have successfully developed novel medicines we designed to treat many different diseases. In therapeutic areas that are outside of our core areas of development, we have licensed our medicines to highly focused satellite companies that have the specific expertise and resources to continue developing the medicines. For our satellite company medicines, we refer to the medicine by the partner’s name or compound number, such as ZEMDRI or ATL1102. We have listed these medicines below in our Satellite Company pipeline.
IONIS’ Satellite Company Pipeline

Antisense Technology
Our antisense technology is an innovative platform for discovering first-in-class and/or best-in-class medicines for treating disease. We believe this technology represents an important advance in the way we treat disease. Unlike most other drug technologies that work by affecting existing proteins in the body, antisense medicines target RNA, the intermediary that conveys genetic information from a gene to the protein synthesis machinery in the cell. By targeting RNA instead of proteins, we can use antisense technology to increase, decrease or alter the production of specific proteins. The unique properties of antisense technology provide several advantages over traditional drug discovery technologies.
18
These advantages include:
| ● | Direct intervention in the disease process at the genetic level by targeting RNA: antisense technology represents a direct route from gene to drug. The explosion in genomic information and RNA biology has led to the discovery of many new disease-causing proteins and RNAs and has created new opportunities that are only accessible to antisense technology. |
| ● | Precise specificity: we design antisense medicines to target a single RNA, which minimizes or eliminates the possibility our medicines will bind to unintended targets which can cause unwanted side effects. |
| ● | Good drug properties: antisense medicines distribute well throughout the body without the need for special formulations or vehicles. They also have a relatively long half-life of approximately two to four weeks in most tissues outside of the brain and spinal cord and three to four months in brain and spinal cord, which means patients and/or healthcare providers can dose our medicines weekly, monthly or even less frequently depending on the medicine and target tissue. |
| ● | Ability to combine with other drugs: because antisense medicines do not interact with the enzymes that metabolize or break down other drugs, physicians can use our medicines in combination with other drugs. |
| ● | Broad applications to multiple disease targets, multiple tissues and multiple mechanisms: there are virtually no “undruggable” targets with antisense technology. |
| ● | Utilize many different routes of administration including subcutaneous, intravenous, intrathecal, intravitreal, pulmonary and oral. |
| ● | Efficient discovery and early development: because of the efficiency of our antisense technology, our drug discovery and early development costs and success rates compare favorably to small molecule or antibody drug discovery and development. |
We develop antisense medicines to potentially treat a wide range of diseases in a number of different therapeutic areas from severe and rare diseases to diseases that affect large patient populations. We focus our efforts on diseases in which there is a large unmet medical need with limited or no current treatments or in diseases for which we believe our medicines have a competitive advantage over existing therapies.
Technology Overview
We use our core technology platform to discover and develop medicines that affect targets in the body at the genetic level. Genes contain the information necessary to produce proteins. A gene is made up of nucleotides containing the nucleoside bases: adenine, thymine, guanine, and cytosine, commonly known as A, T, G and C, which are linked together to form a two-stranded structure that resembles a twisted ladder, known as deoxyribonucleic acid, or DNA. The nucleotides on one side of the ladder bind weakly to complementary nucleotides on the other strand according to specific rules; for example, A pairs with T and G pairs with C, creating the ladder’s rungs (Figure 1). Scientists call this highly specific nucleotide pairing hybridization. The sequence or order of these nucleotides establishes the cell’s recipes for making proteins. Each protein’s instructions reside in a corresponding segment of DNA known as a gene.
19

Figure 1: Illustration of DNA.
The instructions for making a protein are transcribed from a gene, or DNA into a different genetic molecule called messenger RNA. This process starts with the partial uncoiling of the two complementary strands of the DNA. One strand acts as a template and information stored in the DNA template strand is copied into a complementary RNA (Figure 2) by an enzyme called RNA polymerase, or RNAP. Messenger RNA, or mRNA, are mature, fully processed RNA that code for proteins.
Figure 2: Transcription of information contained in a gene, or DNA, to RNA.
Ribosomes, the cell’s factories for manufacturing proteins, translate mRNA into proteins. The ribosome reads the encoded information, the mRNA’s nucleotide sequence, and in doing so, strings together amino acids to form a specific protein (Figure 3).
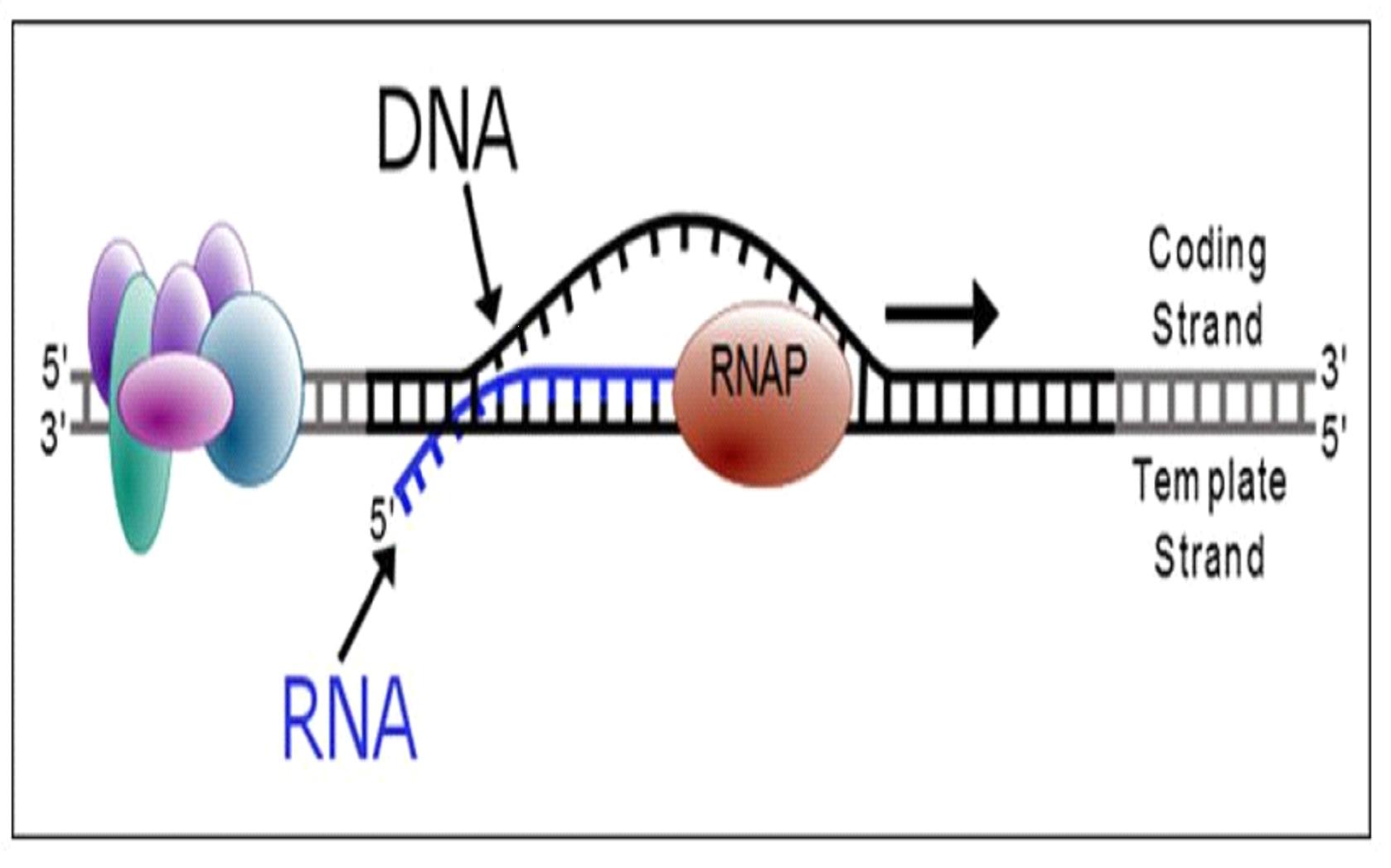
20

Figure 3: Translation of the protein-coding information contained in mRNA to protein.
We primarily use our antisense technology to interrupt the cell’s protein production process by preventing the mRNA instructions from reaching the ribosome, thus inhibiting the production of the protein. We can also design antisense medicines to increase protein production for diseases caused by the lack of a particular protein or modify the processing (or splicing) of the mRNA, which can alter the composition of the protein. The mRNA sequence of nucleotides that carries the information for protein production is called the ‘sense’ strand. Scientists call the complementary nucleotide chain that binds specifically to the sense strand the “antisense” strand. We use the information contained in mRNA to design chemical structures, that we call antisense oligonucleotides, or ASOs, or antisense medicines, which resemble DNA and RNA and are the complement of RNA. Our antisense medicines bind with high selectivity to the mRNA they were designed to target. Since each mRNA codes for a specific protein, this selectivity provides a level of specificity that is better than traditional drugs. As a result, we can design antisense medicines that selectively inhibit the disease-causing member of a protein family without interfering with other members of the protein family that might be necessary for normal cellular or bodily functions. This unique specificity means that antisense medicines may be less toxic than traditional drugs because we can design them to minimize the impact on unintended targets.
We have developed the majority of the medicines in our pipeline using our advanced screens to produce medicines with what we believe have the best possible safety and tolerability profiles. We refer to our medicines that have passed these advanced screens as Generation 2+ medicines. We continue to advance our antisense technology to create even more potent medicines that we can use in more tissues and against more targets. These advances allow us to expand the mechanisms through which we can use our medicines. These advancements provide us with greater opportunities to use our antisense medicines to treat a greater number of diseases and reach more patient populations. Today several of our early stage medicines and those entering our pipeline use our most advanced antisense technology, including our next generation chemistries, Generation 2.5, and our LICA technology.
Generation 2.5 chemistry is an advancement that we have demonstrated increases the potency of our medicines by up to 10-fold over our Generation 2+ medicines. This increase in potency enables our medicines to engage targets in a broader array of tissues. We have published data demonstrating that our Generation 2.5 medicines generally have enhanced potency over our Generation 2+ medicines and are broadly distributed throughout the body to multiple tissues including liver, kidney, lung, muscle, adipose, adrenal gland, peripheral nerves and tumor tissues. Our Generation 2.5 medicines constitute some of our recently added new medicines to our pipeline.
LICA is a chemical technology we developed that involves attaching a molecule called a ligand that binds with receptors on the surfaces of cells in a highly specific manner. Because these receptors are often found only on certain cell types, LICA allows us to increase effective delivery of our antisense medicines with higher specificity to certain cell types that express these receptors relative to non-conjugated antisense medicines. In November 2018, we published an integrated assessment of data from over 600 subjects, with more than 200 subjects on treatment for six months or longer, available from randomized placebo-controlled dose-ranging studies. The integrated assessment demonstrated with multiple Generation 2+ LICA medicines that our LICA technology for liver targets can increase potency by up to more than 30-fold over our non-LICA Generation 2+ medicines. In addition to the increase in potency, a favorable safety and tolerability profile was observed and was consistent across the entire LICA platform. There were no safety concerns related to platelets, liver or kidney function.
21
AKCEA-APO(a)-LRx further exemplifies these improvements. We designed this medicine to reduce the production of apolipoprotein(a), or Apo(a), protein in the liver to offer a direct approach for reducing lipoprotein(a), or Lp(a). The Phase 2 AKCEA-APO(a)-LRx study was the first and only medicine to selectively and robustly reduce Lp(a) levels below threshold levels associated with CVD in nearly all patients. This study included more than 280 patients, with 98 percent of patients in the high dose group achieving levels below 50 mg/dL, the recognized risk threshold for CVD. Like the integrated assessment, the safety and tolerability profile from this study was favorable and there were no safety concerns related to platelets, liver or kidney function.
We have also combined our LICA technology with our Generation 2.5 chemistry medicines to further increase potency. Although we designed our first LICA medicines to inhibit targets in the liver, we are also developing LICA conjugation technology that we can use to target other tissues, such as the pancreas, and the initial results are promising.
Antisense Targets and Mechanisms
There are more than a dozen different antisense mechanisms that we can exploit with our antisense technology. The majority of the medicines in our pipeline bind to mRNAs and inhibit the production of disease-causing proteins. However, our antisense technology is broadly applicable to many different antisense mechanisms, including modulation of RNA splicing, RNA interference, or RNAi, and enhancing protein translation to increase protein production. In May 2018, we published research showing that we can use our proprietary oligonucleotide technology with CRISPR/Cas9, a gene editing system that uses RNA to activate, target and edit specific sites on DNA. Our work in this area provides an important step toward developing potential therapeutic applications for CRISPR technology.
When using antisense technology to inhibit the production of disease-causing proteins or reduce levels of harmful RNAs, our antisense medicines bind to the target RNA via highly specific nucleotide pairing, or hybridization, and recruit a cellular enzyme called ribonuclease H1, or RNase H1, to degrade the target RNA. The antisense medicine itself remains intact during this process, so it can remain active against additional target RNA molecules and repeatedly trigger their degradation (Figure 4). Examples of our antisense medicines that use the RNase H1 mechanism to reduce disease protein production include, WAYLIVRA, TEGSEDI, IONIS-FXIRx, IONIS-FXI-LRx, AKCEA-APO(a)-LRx, IONIS-HTTRx, and others.

Figure 4: Antisense medicine using the RNase H mechanism of action.
SPINRAZA is an example of an antisense medicine that modulates RNA splicing to increase protein production of the SMN protein (Figure 5), which is critical to the health and survival of nerve cells in the spinal cord that are responsible for neuro-muscular function. The SMN protein is deficient in people with SMA. There are a number of other diseases, including cystic fibrosis and Duchenne muscular dystrophy, which may be treated by modulating splicing using antisense technology.
22

Figure 5: Antisense medicine altering splicing of the SMN2 mRNA.
Another class of RNA targets for our antisense technology are microRNAs. To date, scientists have identified more than 700 microRNAs in the human genome, and have shown that the absence or presence of specific microRNAs in various cells is associated with specific human diseases, including cancer, viral infection, metabolic disorders and inflammatory disease. To fully exploit the therapeutic opportunities of targeting microRNAs, we co-founded Regulus Therapeutics as a company focused on the discovery, development and commercialization of microRNA-based therapeutics.
We are also making progress in designing antisense medicines to target long, non-coding RNAs, or lncRNAs and RNAs that possess a toxic function in human diseases. Many of these RNAs, such as lncRNAs, do not make proteins but often cause disease by regulating the function of other genes or proteins. In 2014, we published a paper in Nature in which we were the first to show that targeted reduction of a lncRNA with an antisense compound can ameliorate certain cognitive deficits in a mouse model of Angelman syndrome, or AS. Moreover, these studies demonstrate the potential therapeutic benefits of antisense medicines for the treatment of AS.
Because the efficiency of our core technology platform can support multiple target-based antisense research programs without significantly increasing costs, we can develop antisense medicines to target a broad range of diseases, efficiently producing a large and broad proprietary portfolio of medicines. We are currently pursuing antisense drug discovery programs focused on various severe and rare diseases, cardiometabolic diseases, neurologic diseases, cancer and other diseases.
Collaborative Arrangements and Licensing Agreements
Partnering Strategy
We have established alliances with a cadre of leading global pharmaceutical companies that are working alongside us in developing our medicines, advancing our technology, preparing to commercialize our medicines and selling our medicines. Our partners include the following companies, among others: AstraZeneca, Bayer, Biogen, GSK, Janssen, Novartis and Roche. Our partners bring resources and expertise that augment and build upon our internal capabilities. The depth of our knowledge and expertise with antisense technology together with our strong financial position provides us the flexibility to partner our medicines at what we believe is the optimal time to maximize the near-term, mid-term and long-term value of our medicines. We have distinct partnering strategies that we employ based on the specific program, therapeutic area and the expertise and resources our potential partners may bring to the collaboration.
| ● | We have a strategic partnership with Biogen, which we expanded in 2018. Biogen provides expertise, tools and resources to complement our drug discovery efforts. Our broad strategic alliance with Biogen pairs Biogen’s extensive resources and expertise in neurodegenerative diseases with our antisense technology. Together we are creating a franchise of novel medicines for neurodegenerative diseases that has the potential to expand both our pipeline and Biogen’s pipeline with promising new medicines. Our development of and Biogen’s commercialization of SPINRAZA, is just one example of the power of our strategic partnership. |
| ● | We have partnerships with companies that bring significant expertise and global resources to develop and potentially commercialize medicines for a particular therapeutic area. For example, in January 2017, we initiated a collaboration with Novartis to develop and commercialize AKCEA-APO(a)-LRx and AKCEA-APOCIII-LRx. In February 2019, Novartis licensed AKCEA-APO(a)-LRx and we earned a $150 million license fee. Novartis is responsible for conducting and funding all future development and commercialization activities for AKCEA-APO(a)-LRx, including a global pivotal cardiovascular outcomes study, for which planning and initiation activities are underway. We believe Novartis brings significant resources and expertise to the collaboration that should accelerate our ability to deliver these medicines to large patient populations who have high cardiovascular risk due to inadequately treated lipid disorders. |
23
| ● | We also form early stage research and development partnerships that allow us to expand the application of our technology to new therapeutic areas. For example, we have a collaboration with Janssen that brings together our RNA-targeted technology platform and Janssen’s expertise in autoimmune disorders and therapeutic formulation to discover and develop antisense medicines to treat autoimmune disorders in the GI tract. Thus far, Janssen has licensed and is advancing two medicines under our collaboration. |
| ● | We also work with a consortium of companies that can exploit our medicines and technologies outside our primary areas of focus. We refer to these companies as satellite companies. |
Financial Benefits of Our Partnerships
Through our partnerships, we have earned both commercial revenue and a broad and sustaining base of R&D revenue in the form of license fees, upfront payments and milestone payments. Since 2007, we have received more than $4 billion in cash from upfront and licensing fees, equity purchase payments, milestone payments, research and development funding and royalties from our partnerships. We have the potential to earn more than $20 billion in future milestone payments, licensing fees and other payments from our current partnerships, not including potential royalties.
Strategic Partnership
Biogen
We have several strategic collaborations with Biogen focused on using antisense technology to advance the treatment of neurological disorders. These collaborations combine our expertise in creating antisense medicines with Biogen’s expertise in developing therapies for neurological disorders. We developed and licensed to Biogen SPINRAZA, our approved medicine to treat people with SMA. In December 2017, we entered into a collaboration with Biogen to identify new antisense medicines for the treatment of SMA. Additionally, we and Biogen are currently developing six other medicines to treat neurodegenerative diseases under our other collaborations, including IONIS-SOD1Rx for ALS, IONIS-MAPTRx for Alzheimer’s disease, IONIS-C9Rx for ALS, and IONIS-BIIB6Rx, IONIS-BIIB7Rx and IONIS-BIIB8Rx to treat undisclosed neurodegenerative diseases. In addition to these medicines, we and Biogen are evaluating numerous additional targets to develop medicines to treat neurological diseases. In April 2018, we entered into a new strategic collaboration for the treatment of neurological diseases with Biogen. From inception through February 2019, we have received over $2 billion from our Biogen collaborations, including $1 billion we received from Biogen in the second quarter of 2018 for our 2018 strategic neurology collaboration.
Spinal Muscular Atrophy Collaborations
SPINRAZA
In January 2012, we entered into a collaboration agreement with Biogen to develop and commercialize SPINRAZA, an RNA-targeted therapy for the treatment of SMA. Biogen reported in January 2019 that SPINRAZA was approved in over 40 countries around the world. In February 2019, SPINRAZA was approved in China. Biogen is responsible for global SPINRAZA commercial activities.
From inception through December 2018, we earned more than $785 million in total revenue under our SPINRAZA collaboration, including more than $350 million in revenue from SPINRAZA royalties and more than $435 million in R&D revenue. We are receiving tiered royalties ranging from 11 percent to 15 percent on any sales of SPINRAZA. We have exclusively in-licensed patents related to SPINRAZA from Cold Spring Harbor Laboratory and the University of Massachusetts. We pay Cold Spring Harbor Laboratory and the University of Massachusetts a low single digit royalty on net sales of SPINRAZA. Biogen is responsible for all further global development, regulatory and commercialization activities and costs for SPINRAZA.
New antisense medicines for the treatment of SMA
In December 2017, we entered into a collaboration agreement with Biogen to identify new antisense medicines for the treatment of SMA. Biogen will have the option to license therapies arising out of this collaboration following the completion of preclinical studies. Upon licensing, Biogen will be responsible for all further global development, regulatory and commercialization activities and costs for such therapies. Under the collaboration agreement, we received a $25 million upfront payment in December 2017. We will receive development and regulatory milestone payments from Biogen if new medicines advance towards marketing approval. In total over the term of our collaboration, we are eligible to receive up to $1.2 billion in license fees, milestone payments and other payments. In addition, we are eligible to receive tiered royalties from the mid-teens to mid-20 percent range on net sales.
Neurology Collaborations
2018 Strategic Neurology
In April 2018, we and Biogen entered into a new strategic collaboration to develop novel antisense medicines for a broad range of neurological diseases and entered into a Stock Purchase Agreement, or SPA. As part of the collaboration, Biogen gained exclusive rights to the use of our antisense technology to develop therapies for these diseases for 10 years. We are responsible for the identification of antisense drug candidates based on selected targets. Biogen is responsible for conducting IND-enabling toxicology studies for the selected target. Biogen will have the option to license the selected target after it completes the IND-enabling toxicology study. If Biogen exercises its option for a medicine, it will assume all further global development, regulatory and commercialization responsibilities and costs for that medicine. In the second quarter of 2018, we received $1 billion from Biogen, comprised of $625 million to purchase our stock at an approximately 25 percent cash premium and $375 million in an upfront payment. We are eligible to receive up to $270 million in milestone payments for each medicine that achieves marketing approval. We have generated over $1 billion in payments through February 2019, including $15 million we received in the fourth quarter of 2018 for advancing two targets under this collaboration. In addition, we are eligible to receive tiered royalties up to the 20 percent range on net sales.
24
2013 Strategic Neurology
In September 2013, we and Biogen entered into a long-term strategic relationship focused on applying antisense technology to advance the treatment of neurodegenerative diseases. As part of the collaboration, Biogen gained exclusive rights to the use of our antisense technology to develop therapies for neurological diseases and has the option to license medicines resulting from this collaboration. We will usually be responsible for drug discovery and early development of antisense medicines and Biogen will have the option to license antisense medicines after Phase 2 proof of concept. In October 2016, we expanded our collaboration to include additional research activities we will perform. If Biogen exercises its option for a medicine, it will assume all further global development, regulatory and commercialization responsibilities and costs for that medicine. We are currently advancing five medicines, IONIS-SOD1Rx, IONIS-C9Rx, IONIS-BIIB6Rx, IONIS-BIIB7Rx and IONIS-BIIB8Rx under this collaboration. In December 2018, Biogen exercised its option to license IONIS-SOD1Rx, and as a result Biogen is now responsible for all further global development, regulatory and commercialization activities and costs for IONIS-SOD1Rx.
Under the terms of the agreement, we received an upfront payment of $100 million and are eligible to receive milestone payments, license fees and royalty payments for all medicines developed through this collaboration, with the specific amounts dependent upon the modality of the molecule advanced by Biogen. For each antisense molecule that is chosen for drug discovery and development under this collaboration, we are eligible to receive up to approximately $260 million in a license fee and milestone payments per program. We have generated over $215 million through February 2019, including $40 million we earned in the fourth quarter of 2018 when Biogen advanced and licensed IONIS-SOD1Rx. In addition, we are eligible to receive tiered royalties up to the mid-teens on net sales from any antisense medicines developed under this collaboration.
Neurology
In December 2012, we and Biogen entered into a collaboration agreement to develop and commercialize up to three novel antisense medicines to treat neurodegenerative diseases. We are responsible for the development of each of the medicines through the completion of the initial Phase 2 clinical study for such medicine. Biogen has the option to license a medicine from each of the programs through the completion of the first Phase 2 study for each program. We are currently advancing IONIS-MAPTRx for Alzheimer’s disease under this collaboration. If Biogen exercises its option for a medicine, it will assume all further global development, regulatory and commercialization responsibilities and costs for that medicine. Under the terms of the agreement, we received an upfront payment of $30 million. Over the term of the collaboration, we are eligible to receive up to $210 million in a license fee and milestone payments per program, plus a mark-up on the cost estimate of the Phase 1 and 2 studies. We have generated over $55 million through February 2019. In addition, we are eligible to receive tiered royalties up to the mid-teens on net sales of any medicines resulting from each program under the agreement.
For additional details about our collaboration agreements with Biogen, including other financial information and termination provisions, see Note 6, Collaborative Arrangements and Licensing Agreements, in the Notes to the Consolidated Financial Statements.
Research, Development and Commercialization Partners
AstraZeneca
Cardiac, Renal and Metabolic Diseases Collaboration
In July 2015, we and AstraZeneca formed a collaboration to discover and develop antisense therapies for treating cardiac, renal and metabolic diseases. Under our collaboration, AstraZeneca has licensed three medicines from us: IONIS-AZ4-2.5-LRx, a medicine we designed to treat cardiovascular disease and our first medicine that combines our Generation 2.5 and LICA technology, IONIS-AZ5-2.5Rx, a medicine we designed to treat a genetically associated form of kidney disease and IONIS-AZ6-2.5-LRx, a medicine we designed to inhibit an undisclosed target to treat patients with NASH. AstraZeneca is responsible for all further global development, regulatory and commercialization activities and costs for each of the medicines it has licensed and any other future medicines AstraZeneca licenses.
Under the terms of the agreement, we received a $65 million upfront payment. We are eligible to receive license fees and milestone payments of up to more than $4 billion as medicines under this collaboration advance. We have generated over $165 million in payments through February 2019, including a $10 million milestone payment we earned in the third quarter of 2018 when AstraZeneca initiated a Phase 1 trial for IONIS-AZ4-2.5-LRx. In addition, we are eligible to receive tiered royalties up to the low teens on net sales from any product that AstraZeneca successfully commercializes under this collaboration agreement.
Oncology Collaboration
In December 2012, we entered into a collaboration agreement with AstraZeneca to discover and develop antisense medicines to treat cancer. As part of the agreement, we granted AstraZeneca an exclusive license to develop and commercialize danvatirsen for the treatment of cancer. AstraZeneca is now responsible for all global development, regulatory and commercialization activities for danvatirsen. We and AstraZeneca have evaluated danvatirsen in people with head and neck cancer, advanced lymphoma and advanced metastatic hepatocellular carcinoma. AstraZeneca is evaluating danvatirsen in combination with durvalumab, AstraZeneca’s PD-L1, blocking drug, in people with head and neck cancer, metastatic bladder cancer and metastatic non-small cell lung cancer. We and AstraZeneca also established an oncology research program. AstraZeneca has the option to license medicines resulting from the program, and if AstraZeneca exercises its option for a medicine, it will be responsible for all further global development, regulatory and commercialization activities and costs for such medicine. In the fourth quarter of 2018, we added IONIS-AZ7-2.5Rx to our preclinical pipeline, a second drug under our oncology collaboration.
25
Under the terms of this agreement, we received $31 million in upfront payments. We are eligible to receive milestone payments and license fees from AstraZeneca as programs advance in development. If AstraZeneca successfully develops danvatirsen and another medicine under the research program, we could receive license fees and milestone payments of up to more than $450 million. We have generated over $125 million in payments through February 2019, including nearly $30 million in milestone payments we achieved when AstraZeneca advanced danvatirsen and IONIS-AZ7-2.5Rx, in the fourth quarter of 2018. In addition, we are eligible to receive tiered royalties up to the mid-teens on net sales from any medicines resulting from these programs.
For additional details about our collaboration agreements with AstraZeneca, including other financial information and termination provisions, see Note 6, Collaborative Arrangements and Licensing Agreements, in the Notes to the Consolidated Financial Statements.
Bayer
In May 2015, we entered into an exclusive license agreement with Bayer to develop and commercialize IONIS-FXIRx for the prevention of thrombosis. We were responsible for completing a Phase 2 study of IONIS-FXIRx in people with end-stage renal disease on hemodialysis. Under the terms of the agreement, we received a $100 million upfront payment in the second quarter of 2015. In February 2017, we amended our agreement with Bayer to advance IONIS-FXIRx and to initiate development of IONIS-FXI-LRx, which Bayer licensed. In conjunction with the decision to advance these programs, we received a $75 million payment from Bayer. We are conducting a Phase 2b study evaluating IONIS-FXIRx in people with end-stage renal disease on hemodialysis to finalize dose selection. Additionally, we are developing IONIS-FXI-LRx through Phase 1. Following these studies and Bayer’s decision to further advance these programs, Bayer will be responsible for all global development, regulatory and commercialization activities and costs for both medicines. We are eligible to receive additional milestone payments as each medicine advances toward the market. In total over the term of this collaboration, we are eligible to receive up to $385 million in license fees, milestone payments and other payments. We have generated over $175 million through February 2019. In addition, we are eligible to receive tiered royalties in the low to high 20 percent range on gross margins of both medicines combined.
For additional details about our collaboration agreement with Bayer, including other financial information and termination provisions, see Note 6, Collaborative Arrangements and Licensing Agreements, in the Notes to the Consolidated Financial Statements.
GSK
In March 2010, we entered into an alliance with GSK using our antisense drug discovery platform to discover and develop new medicines against targets for rare and serious diseases, including infectious diseases and some conditions causing blindness. Under the terms of the agreement, we received upfront payments of $35 million. GSK is advancing two medicines targeting HBV under our collaboration: IONIS-HBVRx and IONIS-HBV-LRx. GSK is currently conducting Phase 2 studies for both of these medicines, which we designed to reduce the production of viral proteins associated with HBV infection. GSK has the exclusive option to license the medicines resulting from this alliance at Phase 2 proof-of-concept for a license fee.
Under our agreement, if GSK successfully develops these medicines and achieves pre-agreed sales targets, we could receive license fees and milestone payments of $262 million. We have generated over $162 million in payments through February 2019. In addition, we are eligible to receive tiered royalties up to the mid-teens on net sales from any product that GSK successfully commercializes under this alliance.
For additional details about our collaboration agreement with GSK, including other financial information and termination provisions, see Note 6, Collaborative Arrangements and Licensing Agreements, in the Notes to the Consolidated Financial Statements.
Janssen Biotech, Inc.
In December 2014, we entered into a collaboration agreement with Janssen Biotech, Inc. to discover and develop antisense medicines that can be locally administered, including oral delivery, to treat autoimmune disorders of the GI tract. Janssen has the option to license medicines from us through the designation of development candidates for up to three programs. Under our collaboration, Janssen licensed IONIS-JBI1-2.5Rx in July 2016 and IONIS-JBI2-2.5Rx in November 2017. Janssen is currently conducting a Phase 1 study of IONIS-JBI1-2.5Rx and IONIS-JBI2-2.5Rx is in preclinical development. Prior to option exercise we are responsible for the discovery activities to identify development candidates. If Janssen exercises an option for any of the programs, it will be responsible for the global development, regulatory and commercial activities under that program. Under the terms of the agreement, we received $35 million in upfront payments. We are eligible to receive up to more than $800 million in milestone payments and license fees for these programs. We have generated over $75 million through February 2019. In addition, we are eligible to receive tiered royalties up to the near teens on net sales from any medicines resulting from this collaboration.
For additional details about our collaboration agreement with Janssen, including other financial information and termination provisions, see Note 6, Collaborative Arrangements and Licensing Agreements, in the Notes to the Consolidated Financial Statements.
26
Roche
Huntington’s Disease
In April 2013, we formed an alliance with Hoffman-La Roche Inc. and F. Hoffmann-La Roche Ltd., collectively Roche, to develop treatments for HD based on our antisense technology. Under the agreement, we discovered and developed IONIS-HTTRx, an antisense medicine targeting HTT protein, through completion of our Phase 1/2 clinical study in people with early stage HD. In December 2017, upon completion of the Phase 1/2 study, Roche exercised its option to license IONIS-HTTRx and is now responsible for the global development, regulatory and commercialization activities for IONIS-HTTRx. Under the terms of the agreement, we received an upfront payment of $30 million in April 2013. We are eligible to receive up to $365 million in a license fee and milestone payments as IONIS-HTTRx advances. We have generated over $145 million through February 2019, including $35 million in milestone payments we generated in the first quarter of 2019 when Roche dosed the first patient in a Phase 3 study for IONIS-HTTRx. In addition, we are eligible to receive up to $136.5 million in milestone payments for each additional medicine successfully developed. We are also eligible to receive tiered royalties up to the mid-teens on net sales from any product resulting from this alliance.
IONIS-FB-LRx for Complement-Mediated Diseases
In October 2018, we entered into a collaboration agreement with Roche to develop IONIS-FB-LRx for the treatment of complement-mediated diseases. The first indication we plan to pursue is the treatment of patients with GA, the advanced stage of dry AMD. We are responsible for conducting a Phase 2 study in patients with dry AMD. In addition, we are exploring the medicine in a severe and rare renal indication. Roche has the option to license IONIS-FB-LRx at the completion of these studies. Upon licensing, Roche will be responsible for all further global development, regulatory and commercialization activities and costs. Under the terms of this agreement, we received a $75 million upfront payment in October 2018. We are eligible to receive up to $684 million in milestone payments and license fees. In addition, we are also eligible to receive tiered royalties from the high teens to twenty percent on net sales.
For additional details about our collaboration agreements with Roche, including other financial information and termination provisions, see Note 6, Collaborative Arrangements and Licensing Agreements, in the Notes to the Consolidated Financial Statements.
Akcea Collaborations
The following collaboration agreements relate to Akcea, our majority-owned affiliate. Our consolidated results include all the revenue earned and cash received under these collaboration agreements. We reflect the noncontrolling interest attributable to other owners of Akcea’s common stock in a separate line on our statement of operations and in a separate line within stockholders’ equity on our consolidated balance sheet.
Novartis
In January 2017, we and Akcea initiated a collaboration with Novartis to develop and commercialize AKCEA-APO(a)-LRx and AKCEA-APOCIII-LRx. Under the collaboration agreement, Novartis has an exclusive option to further develop and commercialize AKCEA-APO(a)-LRx and AKCEA-APOCIII-LRx. Akcea is responsible for completing a Phase 2 program, conducting an end-of-Phase 2 meeting with the FDA and providing initial quantities of API for each medicine. If Novartis exercises an option for either of these medicines, Novartis will be responsible for all further global development, regulatory and co-commercialization activities and costs for such medicine.
Akcea received a $75 million upfront payment in the first quarter of 2017. In February 2019, Novartis licensed AKCEA-APO(a)-LRx and we earned a $150 million license fee. Novartis is responsible for conducting and funding all future development, regulatory and commercialization activities for AKCEA-APO(a)-LRx, including a global pivotal cardiovascular outcomes study, for which planning and initiation activities are underway. If Novartis exercises its option for AKCEA-APOCIII-LRx, Novartis will pay Akcea a license fee equal to $150 million. In addition, Akcea is eligible to receive up to $675 million and $530 million in milestone payments related to AKCEA-APO(a)-LRx and AKCEA-APOCIII-LRx, respectively. Akcea is also eligible to receive tiered royalties in the mid-teens to low 20 percent range on net sales of AKCEA-APO(a)-LRx and AKCEA-APOCIII-LRx. Akcea will pay 50 percent of these license fees, milestone payments and royalties to us as a sublicense fee. In connection with Novartis’ license of AKCEA-APO(a)-LRx, Akcea and Novartis established a more definitive framework under which the companies would negotiate the co-commercialization of AKCEA-APO(a)-LRx in selected markets. Included in this framework is an option by which Novartis could solely commercialize AKCEA-APO(a)-LRx in exchange for Novartis paying Akcea increased commercial milestone payments based on sales of AKCEA-APO(a)-LRx. Akcea may co-commercialize IONIS-APOCIII-LRx if licensed and commercialized by Novartis in selected markets through its specialized sales force under terms and conditions to be negotiated with Novartis in the future.
In conjunction with this collaboration, we entered into a SPA with Novartis. As part of the SPA, Novartis purchased 1.6 million shares of our common stock for $100 million in the first quarter of 2017 and purchased $50 million of Akcea’s common stock at the IPO price concurrent with the IPO in July 2017.
For additional details about our collaboration agreement with Novartis, including other financial information and termination provisions, see Note 6, Collaborative Arrangements and Licensing Agreements, in the Notes to the Consolidated Financial Statements.
27
PTC Therapeutics
In August 2018, Akcea entered into an exclusive license agreement with PTC Therapeutics to commercialize TEGSEDI and WAYLIVRA in Latin America. Under the license agreement, Akcea is eligible to receive up to $26 million in payments, including $12 million which it received in the third quarter of 2018, $6 million upon the earlier of FDA or EMA approval of WAYLIVRA and up to $8 million for regulatory milestones. Akcea is eligible to receive royalties from PTC in the mid-20 percent range on net sales in Latin America for each medicine. PTC’s obligation to pay Akcea royalties begins on the earlier of 12 months after the first commercial sale of a product in Brazil or the date that PTC recognizes revenue of at least $10 million in Latin America. Consistent with the agreements between Ionis and Akcea, the companies will share all payments, including royalties.
Satellite Company Partnerships
We have a number of satellite company collaborations that expand the reach and potential of our RNA-targeting medicines into disease areas that are outside of our core focus.
For example, we have a collaboration with Alnylam Pharmaceuticals, Inc. to develop and commercialize RNAi therapeutics. Under the terms of the agreement, we exclusively licensed to Alnylam our patent estate relating to antisense motifs and mechanisms and oligonucleotide chemistry for double-stranded RNAi therapeutics in exchange for a technology access fee, participation in fees from Alnylam’s partnering programs, as well as future milestone and royalty payments from Alnylam. We also have the potential to earn a portion of payments that Alnylam receives from licenses of our technology it grants to its partners, plus royalties. We retained rights to a limited number of double-stranded RNAi therapeutic targets and all rights to single-stranded RNAi, or ssRNAi, therapeutics. In turn, Alnylam nonexclusively licensed to us its patent estate relating to antisense motifs and mechanisms and oligonucleotide chemistry to research, develop and commercialize single-stranded antisense therapeutics, ssRNAi therapeutics, and to research double-stranded RNAi compounds. We also received a license to develop and commercialize double-stranded RNAi drugs targeting a limited number of therapeutic targets on a nonexclusive basis. Additionally, In 2015, we and Alnylam entered into an alliance in which we cross-licensed intellectual property. Under this alliance, we and Alnylam each obtained exclusive license rights to four therapeutic programs. Alnylam granted us an exclusive, royalty-bearing license to its chemistry, RNA targeting mechanism and target-specific intellectual property for oligonucleotides against four targets, including FXI and Apo(a) and two other targets. In exchange, we granted Alnylam an exclusive, royalty-bearing license to our chemistry, RNA targeting mechanism and target-specific intellectual property for oligonucleotides against four other targets. Alnylam also granted us a royalty-bearing, non-exclusive license to new platform technology arising from May 2014 through April 2019 for single-stranded antisense therapeutics. In turn, we granted Alnylam a royalty-bearing, non-exclusive license to new platform technology arising from May 2014 through April 2019 for double-stranded RNAi therapeutics.
In addition to Alnylam, our satellite company collaborations include collaborations with the following companies:
| Satellite Company | Focus | |
Achaogen, Inc. | Aminoglycosides | |
| Antisense Therapeutics Limited | Inflammation, Acromegaly | |
Atlantic Pharmaceuticals Limited | Inflammation | |
| Dynacure, SAS | Muscle Disorders | |
ProQR Therapeutics N.V. | Ophthalmology | |
| Regulus Therapeutics Inc. | microRNA-targeting therapeutics | |
Suzhou Ribo Life Science Co., Ltd. | ssRNAi |
Under our satellite collaborations we are eligible to earn milestone payments, license fees and royalties. In addition, in certain cases we own equity in the company.
External Project Funding
We are pursuing discovery and development projects that provide us with new therapeutic applications for antisense medicines. These programs represent opportunities for us and our technology. In some cases, we have funded these studies through support from our partners or disease advocacy groups and foundations. Our External Project Funding partners include the following:
| ● | CHDI Foundation- Through our development collaboration, CHDI provided financial and scientific support to our Huntington’s disease drug discovery program. We have reimbursed CHDI for its support of our Huntington’s disease program out of the payments we received from Roche. |
| ● | Cystic Fibrosis Foundation- We received upfront funding from the Cystic Fibrosis Foundation to discover and advance a medicine for the treatment of cystic fibrosis. In exchange for this funding, we are obligated to pay the Cystic Fibrosis Foundation up to $18 million upon achieving specific regulatory and sales events if we advance a medicine under our collaboration. |
| ● | The Ludwig Institute; Center for Neurological Studies- We have a collaboration with the Ludwig Institute, the Center for Neurological Studies and researchers to discover and develop antisense medicines for ALS and other neurodegenerative diseases. Under this agreement, we agreed to pay the Ludwig Institute and the Center for Neurological Studies modest milestone payments and royalties on any antisense medicines resulting from the collaboration. |
28
Manufacturing
We have dedicated significant resources to develop ways to improve manufacturing efficiency and capacity. Since we can use variants of the same nucleotide building blocks and the same type of equipment to produce our oligonucleotide medicines, we found that the same techniques we used to efficiently manufacture one oligonucleotide medicine could help improve the manufacturing processes for many other oligonucleotide medicines. By developing several proprietary chemical processes to scale up our manufacturing capabilities, we have greatly reduced the cost of producing oligonucleotide medicines. For example, we have significantly reduced the cost of raw materials through improved yield efficiency, while at the same time increasing our capacity to make the medicines. Through both our internal research and development programs and collaborations with outside vendors we may achieve even greater efficiency and further cost reductions.
Our drug substance manufacturing facility is located in a 28,700 square foot building in Carlsbad, California. We purchased this building in 2017. In addition, we have a 25,800 square foot building that houses support functions for our manufacturing activities. We lease this facility under a lease that has an initial term ending in June 2021 with an option to extend the lease for up to two additional five-year periods. Our manufacturing facility is subject to periodic inspections by the FDA and foreign equivalents to ensure that it is operating in compliance with current Good Manufacturing Practices, or cGMP, requirements.
As part of our collaborations we may agree to manufacture clinical trial materials and/or commercial supply for our partners. For example, in the past we have manufactured clinical supply materials for AstraZeneca, Bayer, Biogen, GSK and Novartis.
We believe we have sufficient manufacturing capacity at our own facility or at contract manufacturing organizations, or CMOs, to meet our current internal research, development and potential commercial needs, as well as our current and future obligations under existing agreements with our partners for research, development and commercial needs. We believe our current network of CMO partners are capable of providing sufficient quantities to meet anticipated commercial demands. Additionally, we continue to evaluate relationships with additional suppliers to increase overall capacity and diversify our supply chain. While we believe that there are alternate sources of supply that can satisfy our commercial requirements, we cannot be certain that identifying and establishing relationships with such sources, if necessary, would not result in significant delay or material additional costs. We also cannot provide assurance that we will not experience a disruption in supply from our current CMO partners.
CMOs are subject to the FDA’s cGMP requirements and other rules and regulations prescribed by foreign regulatory authorities. We depend on our CMO partners for continued compliance with cGMP requirements and applicable foreign standards.
Specifically, we have the following in place for our approved medicines, SPINRAZA and TEGSEDI, our medicine currently under regulatory review, WAYLIVRA and our medicine in Phase 3 development, IONIS-HTTRx:
SPINRAZA
Pursuant to our collaboration with Biogen, Biogen is responsible for SPINRAZA drug supply. We provided Biogen with API for SPINRAZA in 2018 under our manufacturing agreement with Biogen, which ended in September 2018. Biogen has an oligonucleotide synthesis manufacturing facility that gives it the capability to manufacture SPINRAZA.
TEGSEDI
For TEGSEDI’s commercial drug supply, we are using CMOs to produce custom raw materials, API and finished goods. Our CMO partners have extensive technical expertise and cGMP experience.
WAYLIVRA
We have supplied Akcea either through our manufacturing processes or through our outside vendors, with API and finished drug product to complete Akcea’s ongoing clinical study for WAYLIVRA. We have also supplied the API and the finished drug product for WAYLIVRA’s commercial launch. We believe we have sufficient API and drug product for at least the first two years of WAYLIVRA’s commercial launch. Akcea plans to leverage our relationships with CMOs to procure its own long-term raw material and drug supplies at competitive prices in the future.
IONIS-HTTRX
Pursuant to our collaboration with Roche, Roche is responsible for IONIS-HTTRx drug supply.
LICA Medicines
We have manufactured limited supplies of our LICA medicines for our preclinical and clinical studies. We have also used CMOs to manufacture our LICA medicines. LICA enables lower doses than unconjugated oligonucleotides. With our expertise in optimizing manufacturing of oligonucleotides, we believe we can develop new processes to scale up manufacturing of our LICA medicines at commercially competitive prices.
29
Patents and Proprietary Rights
Our success depends, in part, on our ability to obtain patent protection for our products in the U.S. and other countries. We own or have exclusively licensed a substantial patent estate with numerous issued patents worldwide protecting our products and, more generally, our platform for development and commercialization of oligonucleotide therapeutics. We focus our resources on patents and new patent applications that drive value for our company.
We own or control patents that provide exclusivity for products in our pipeline and patents that provide exclusivity for our core technology in the field of antisense more generally. Our core technology patents include claims to chemically modified nucleosides and oligonucleotides as well as antisense drug designs utilizing these chemically modified nucleosides. These core claims are each independent of specific therapeutic target, nucleic acid sequence, or clinical indication. We also own a large number of patents claiming antisense compounds having nucleic acid sequences complementary to therapeutic target nucleic acids, independent of the particular chemical modifications incorporated into the antisense compound. Most importantly, we seek and obtain issued patent claims to specifically protect each of our medicines. For example, we file and seek to obtain claims covering each drug’s nucleic acid sequence and precise drug design. In sum, we maintain our competitive advantage in the field of antisense technology by protecting our core platform technology and by creating multiple layers of patent protection for each of our specific medicines in development.
Type of Patent Claim (Broadly Applicable to Specific) | ||
● Chemically Modified Nucleosides and Oligonucleotides (target and sequence independent) ● Antisense Drug Design Motifs (target and sequence independent) ● Therapeutic Methods (sequence and chemistry independent) ● Antisense Sequence (chemistry independent) ● Drug Composition |  |
Chemically Modified Nucleosides and Oligonucleotides
The most broadly applicable of our patents are those that claim modified nucleosides and oligonucleotides comprising the modified nucleosides that we incorporate into our antisense medicines to increase their therapeutic efficacy. Nucleosides and chemically modified nucleosides are the basic building blocks of our antisense medicines, therefore claims that cover any oligonucleotide incorporating one of our proprietary modified nucleosides can apply to a wide array of antisense mechanisms of action as well as several therapeutic targets. Of particular note are our patents covering our proprietary 2’-O-(2-methoxy) ethyl, or “MOE,” modified nucleosides, incorporated into many of our second generation development compounds, as well as our constrained-ethyl nucleosides, or “cEt” nucleosides incorporated into our Generation 2.5 compounds.
The following are some of our patents in this category in key jurisdictions (U.S., Europe and Japan):
| Jurisdiction | Patent No. | Title | Expiration | Description of Claims | ||||
| United States | 7,101,993 | OLIGONUCLEOTIDES CONTAINING 2’O-MODIFIED PURINES | 2023 | Covers certain MOE nucleosides and oligonucleotides containing these nucleotides. | ||||
| United States | 7,399,845 | 6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS | 2027 | Covers our cEt nucleosides and oligonucleotides containing these nucleoside analogs. | ||||
| United States | 7,741,457 | 6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS | 2027 | Covers our cEt nucleosides and oligonucleotides containing these nucleoside analogs. | ||||
| United States | 8,022,193 | 6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS | 2027 | Covers oligonucleotides containing cEt nucleoside analogs. | ||||
| United States | 7,569,686 | COMPOUNDS AND METHODS FOR SYNTHESIS OF BICYCLIC NUCLEIC ACID ANALOGS | 2027 | Covers methods of synthesizing our cEt nucleosides. | ||||
| Europe | EP1984381 | 6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS | 2027 | Covers our cEt nucleosides and oligonucleotides containing these nucleoside analogs. | ||||
| Europe | EP2314594 | 6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS | 2027 | Covers our cEt oligonucleotides and methods of use. | ||||
| Japan | JP5342881 | 6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS | 2027 | Covers our cEt nucleosides and oligonucleotides containing these nucleoside analogs. |
30
Antisense Drug Design Motifs
We also have patents that claim oligonucleotides comprising antisense drug design motifs, or patterns of nucleoside modifications at specified positions in the oligonucleotide. Patent claims covering our antisense drug design motifs are independent of nucleic acid sequence, so they cover oligonucleotides having the recited motif, regardless of cellular target or clinical indication. The claimed motifs generally confer properties that optimize oligonucleotides for a particular antisense mechanism of action, such as ribonuclease H, or RNase H, RNAi, or splicing. We have designed oligonucleotides incorporating motifs, which we refer to as chimeric compounds or gapmers, to exploit the RNase H mechanism to achieve target RNA reduction. Almost all of our medicines, including TEGSEDI, WAYLIVRA and IONIS-HTTRx, but excluding SPINRAZA, contain this gapmer antisense drug design motif. We own a U.S. patent that covers all of our second generation MOE gapmer antisense medicines until March of 2023.
In addition, we have pursued patent claims to antisense drug design motifs incorporating bicyclic nucleoside analogs, which include both locked nucleic acids, or “LNA” and cEt. In Europe, we have been granted claims drawn to certain short gapmer oligonucleotides with bicyclic nucleosides, which include locked nucleic acids, in the wings for the treatment of cardiovascular or metabolic disorders. We have also successfully obtained issued patent claims covering our Generation 2.5 gapmer antisense drug design motifs that incorporate our cEt modified nucleosides. Santaris opposed granted European patents EP2092065 and EP2410053. In April 2015, the claims of EP2092065 were successfully upheld in amended form and in January 2017, EP2410053 was upheld with only a minor amendment. The following patents are some examples of our issued patents in this category in key jurisdictions:
| Jurisdiction | Patent/ Application No. | Title | Expiration | Description of Claims | ||||
| United States | 7,015,315 | GAPPED OLIGONUCLEOTIDES | 2023 | 2’-O-alkyl-O-alkyl gapmer oligonucleotides. | ||||
| Europe | EP2021472 | COMPOUNDS AND METHODS FOR MODULATING GENE EXPRESSION | 2027 | Short gapmer oligonucleotides, having wings of 2 bicyclic nucleosides, and a gap of 10 deoxynucleotides for the treatment of cardiovascular or metabolic disorders | ||||
| United States | 7,750,131 | 5’-MODIFIED BICYCLIC NUCLEIC ACID ANALOGS | 2027 | 5’-Methy BNA containing gapmer compounds | ||||
| Europe | EP2092065 | ANTISENSE COMPOUNDS | 2027 | Gapmer compounds having wings comprised of 2’-modifed and LNA nucleosides | ||||
| Europe | EP2410053 | ANTISENSE COMPOUNDS | 2027 | Gapmer compounds having wings comprised of 2’-MOE and bicyclic nucleosides | ||||
| Japan | JP 5665317 | ANTISENSE COMPOUNDS | 2027 | Gapmer compounds having wings comprised of 2’-MOE and bicyclic nucleosides | ||||
| Europe | EP2673361 | OLIGOMERIC COMPOUNDS COMPRISING BICYCLIC NUCLEOTIDES AND USES THEREOF | 2032 | Gapmer having at least one bicyclic nucleoside, 2’-modified nucleoside, and 2’-deoxynucleoside in either the 5’- or 3’-wing. |
LIgand-Conjugated Antisense (LICA) Technology
We have also pursued patent claims to new chemistries created to enhance targeting of antisense medicines to specific tissues and cells to improve a drug’s properties. We designed our N-acetyl-galactosamine, or GalNAc, LICA medicines to provide an increase in potency for targets in the liver. We have successfully obtained issued patent claims covering our LICA technology conjugated to any modified oligonucleotide, including gapmers, double-stranded siRNA compounds, and fully modified oligonucleotides. The following patents are some examples of our issued patents in this category:
| Jurisdiction | Patent/ Application No. | Title | Expiration | Description of Claims | ||||
| United States | 9,127,276 | CONJUGATED ANTISENSE COMPOUNDS AND THEIR USE | 2034 | Covers our primary THA LICA conjugated to any group of nucleosides, including gapmers, double-stranded siRNA compounds, and fully modified oligonucleotides | ||||
| United States | 9,181,549 | CONJUGATED ANTISENSE COMPOUNDS AND THEIR USE | 2034 | Covers our primary THA conjugate having our preferred linker and cleavable moiety conjugated to any oligomeric compound or any nucleoside having a 2’-MOE modification or a cEt modification |
Therapeutic Methods of Treatment and Antisense Drug Sequences
In addition to our broad core patents, we also own hundreds of patents, worldwide, with claims to antisense compounds having particular sequences and compounds directed to particular therapeutically important targets or methods of achieving clinical endpoints using these antisense compounds. These “Target” patents also include claims reciting the specific nucleic acid sequences utilized by our products, independent of chemical modifications and motifs. In addition, our product specific patents typically include claims combining specific nucleic acid sequences with nucleoside modifications and motifs. In this way, we seek patent claims narrowly tailored to protect our product’s specifically, in addition to the broader core antisense patents described above.
31
SPINRAZA and Survival Motor Neuron
SPINRAZA is protected from generic competition in the U.S. until at least 2030 and in Europe until 2026 by a suite of patents. These issued patents include: (i) the Bennett patent related to methods of altering mRNA processing (e.g., splicing, the mechanism of action of SPINRAZA) with a fully modified 2’MOE oligonucleotide, (ii) a patent licensed from the University of Massachusetts drawn to antisense compounds having the sequence of SPINRAZA, independent of chemical modification and uses of such compounds for treating SMA, and (iii) joint patents with Cold Spring Harbor Laboratory claiming fully modified 2’MOE compositions targeting SMN2, including the precise composition of matter of SPINRAZA and methods of using such compositions. We have filed for patent term extension, to potentially extend the term beyond 2030. With Biogen’s license of SPINRAZA, we assigned our interest in these patents to Biogen. The table below lists the key U.S. and European issued patents protecting SPINRAZA:
| Jurisdiction | Patent No. | Title | Expiration | Description of Claims | ||||
| United States | 8,361,977 | COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING | 2030 | Sequence and chemistry (full 2’-MOE) of SPINRAZA | ||||
| Europe | 1910395 | COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING | 2026 | Sequence and chemistry (full 2’-MOE) of SPINRAZA | ||||
| United States | 7,838,657 | SPINAL MUSCULAR ATROPHY (SMA) TREATMENT VIA TARGETING OF SMN2 SPLICE SITE INHIBITORY SEQUENCES | 2027 | Oligonucleotides having sequence of SPINRAZA (chemistry independent) | ||||
| United States | 8,110,560 | SPINAL MUSCULAR ATROPHY (SMA) TREATMENT VIA TARGETING OF SMN2 SPLICE SITE INHIBITORY SEQUENCES | 2025 | Methods of using antisense oligonucleotides having sequence of SPINRAZA to alter splicing of SMN2 and/or to treat SMA | ||||
| United States | 8,980,853 | COMPOSITIONS AND METHODS FOR MODULATION OF SMN2 SPLICING IN A SUBJECT | 2030 | Methods of administering SPINRAZA |
TEGSEDI and Transthyretin
We obtained issued claims covering TEGSEDI in the U.S. The issued U.S. claims protect TEGSEDI from generic competition in the U.S. until at least 2031. We are also pursuing additional patent applications designed to protect TEGSEDI in foreign jurisdictions. The table below lists the current issued patents protecting TEGSEDI in key jurisdictions:
| Jurisdiction | Patent No. | Title | Expiration | Description of Claims | ||||
| United States | 8,101,743 | MODULATION OF TRANSTHYRETIN EXPRESSION | 2025 | Antisense sequence and chemistry of TEGSEDI | ||||
| United States | 8,697,860 | DIAGNOSIS AND TREATMENT OF DISEASE | 2031 | Composition of TEGSEDI | ||||
| United States | 9,061,044 | MODULATION OF TRANSTHYRETIN EXPRESSION | 2031 | Sodium salt composition of TEGSEDI | ||||
| United States | 9,399,774 | MODULATION OF TRANSTHYRETIN EXPRESSION | 2031 | Methods of treating transthyretin amyloidosis by administering TEGSEDI | ||||
| Japan | JP5896175 | MODULATION OF TRANSTHYRETIN EXPRESSION | 2031 | Composition of TEGSEDI | ||||
| Europe | EP2563920 | MODULATION OF TRANSTHYRETIN EXPRESSION | 2031 | Composition of TEGSEDI |
WAYLIVRA and Apolipoprotein C-III
We have obtained patent claims in the U.S. drawn to the use of antisense compounds complementary to a broad active region of human Apo C-III, including the site targeted by WAYLIVRA. We have secured similar claims to compounds complementary to any site on human Apo C-III in Australia. We have also obtained issued patent claims to the specific antisense sequence and chemical composition of WAYLIVRA in the U.S., Australia, Canada, Hong Kong and Europe. The issued U.S. claims protect WAYLIVRA from generic competition in the U.S. until at least 2023. In addition, if WAYLIVRA is approved by the FDA, we will seek patent term extension to recapture a portion of the term lost during FDA regulatory review, extending the term of this patent beyond 2023. We are pursuing additional patent applications designed to protect WAYLIVRA worldwide. The table below lists the issued patents in key jurisdictions:
32
| Jurisdiction | Patent No. | Title | Expiration | Description of Claims | ||||
| United States | 9,624,496 | MODULATION OF APOLIPOPROTEIN C-III EXPRESSION | 2023 | Antisense compound specifically hybridizable within the nucleotide region of apoCIII targeted by WAYLIVRA | ||||
| United States | 7,598,227 | MODULATION OF APOLIPOPROTEIN C-III EXPRESSION | 2023 | Methods of treating hyperlipidemia, lowering cholesterol levels or lowering triglyceride levels with WAYLIVRA | ||||
| United States | 7,750,141 | MODULATION OF APOLIPOPROTEIN C-III EXPRESSION | 2023 | Antisense sequence and chemistry of WAYLIVRA | ||||
| Europe | EP1622597 | MODULATION OF APOLIPOPROTEIN C-III EXPRESSION | 2024 | Antisense sequence and chemistry of WAYLIVRA | ||||
| Europe | EP2441449 | MODULATION OF APOLIPOPROTEIN C-III EXPRESSION | 2024 | Antisense compound specifically hybridizable within the nucleotide region of apoCIII targeted by WAYLIVRA | ||||
| United States | 9,157,082 | MODULATION OF APOLIPOPROTEIN CIII (APOCIII) EXPRESSION | 2032 | Methods of using APOCIII antisense oligonucleotides for reducing pancreatitis and chylomicronemia and increasing HDL | ||||
| Japan | JP 6203707 | MODULATION OF APOLIPOPROTEIN CIII (APOCIII) EXPRESSION | 2032 | Methods of using APOCIII antisense oligonucleotides having the sequence of WAYLIVRA for treating pancreatitis | ||||
| United States | 9,593,333 | MODULATION OF APOLIPOPROTEIN C-III (APOCIII) EXPRESSION IN LIPOPROTEIN LIPASE DEFICIENT (LPLD) POPULATIONS | 2034 | Methods of using APOCIII specific inhibitors for treating lipoprotein lipase deficiency |
IONIS-HTTRx and Huntingtin
We obtained issued claims covering IONIS-HTTRx in the U.S.. The issued U.S. claims protect IONIS-HTTRx from generic competition in the U.S. until at least 2030. We are also pursuing additional patent applications designed to protect IONIS-HTTRx in foreign jurisdictions. The table below lists the current issued patents protecting IONIS-HTTRx in key jurisdictions:
| Jurisdiction | Patent No. | Title | Expiration | Description of Claims | ||||
| United States | 9,273,315 | MODULATION OF HUNTINGTIN EXPRESSION | 2030 | Composition of IONIS-HTTRx | ||||
| United States | 8,906,873 | MODULATION OF HUNTINGTIN EXPRESSION | 2030 | Methods of treating Huntington’s disease by administering IONIS-HTTRx | ||||
| Europe | EP2475675 | MODULATION OF HUNTINGTIN EXPRESSION | 2030 | Composition of IONIS-HTTRx | ||||
| Japan | JP5809146 | MODULATION OF HUNTINGTIN EXPRESSION | 2030 | Composition of IONIS-HTTRx | ||||
| United States | 7,951,934 | COMPOSITIONS AND THEIR USES DIRECTED TO HUNTINGTIN | 2027 | Antisense sequence of IONIS-HTTRx | ||||
| United States | 8,952,145 | COMPOSITIONS AND THEIR USES DIRECTED TO HUNTINGTIN | 2027 | Antisense compound specifically hybridizable within the nucleotide region of HTT targeted by IONIS-HTTRx | ||||
| Japan | 5425474 | COMPOSITIONS AND THEIR USES DIRECTED TO HUNTINGTIN | 2027 | Antisense sequence of IONIS-HTTRx | ||||
| European | EP2161038 | COMPOSITIONS AND THEIR USES DIRECTED TO HUNTINGTIN | 2027 | Antisense sequence of IONIS-HTTRx |
We seek patent protection in significant markets and/or countries for each medicine in development. We also seek to maximize patent term. In some cases, the patent term can be extended to recapture a portion of the term lost during FDA regulatory review. The patent exclusivity period for a medicine will prevent generic medicines from entering the market. Patent exclusivity depends on a number of factors including initial patent term and available patent term extensions based upon delays caused by the regulatory approval process.
33
Manufacturing Patents
We also own patents claiming methods of manufacturing and purifying oligonucleotides. These patents claim methods for improving oligonucleotide drug manufacturing, including processes for large-scale oligonucleotide synthesis and purification. These methods allow us to manufacture oligonucleotides at lower cost by, for example, eliminating expensive manufacturing steps.
We also rely on trade secrets, proprietary know-how and continuing technological innovation to develop and maintain a competitive position in antisense therapeutics.
Government Regulation
Regulation by government authorities in the U.S. and other countries is a significant component in the development, manufacture and commercialization of pharmaceutical products and services. In addition to regulations enforced by the FDA and relevant foreign regulatory authorities, we are also subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other present and potential future federal, state and local regulations.
Extensive regulation by the U.S. and foreign governmental authorities governs the development, manufacture and sale of our medicines. In particular, our medicines are subject to a number of approval requirements by the FDA in the U.S. under the Federal Food, Drug and Cosmetic Act, or FDCA, and other laws and by comparable agencies in those foreign countries in which we conduct business. The FDCA and other various federal, state and foreign statutes govern or influence the research, testing, manufacture, safety, labeling, storage, recordkeeping, approval, promotion, marketing, distribution, post-approval monitoring and reporting, sampling, quality, and import and export of our medicines. State, local, and other authorities also regulate pharmaceutical manufacturing facilities and procedures.
Our manufacturing facility and our CMOs are subject to periodic inspection by the FDA and other foreign equivalents to ensure that they are operating in compliance with cGMP requirements. In addition, marketing authorization for each new medicine may require a rigorous manufacturing pre-approval inspection by regulatory authorities. Post approval, there are strict regulations regarding changes to the manufacturing process, and, depending on the significance of the change, changes may require prior FDA approval. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use.
The FDA must approve any new medicine before a manufacturer can market it in the U.S.. In order to obtain approval, we and our partners must complete clinical studies and prepare and submit an NDA to the FDA. If the FDA approves a medicine, it will issue an approval letter authorizing commercial marketing of the medicine and may require a risk evaluation and mitigation strategy, or REMS, to help ensure the benefits of the medicine outweigh the potential risks. For example, TEGSEDI has a REMS program. The requirements for REMS can materially affect the potential market and profitability of our medicines. In foreign jurisdictions, the drug approval process is similarly demanding.
Numerous regulatory authorities in addition to the FDA, including, in the U.S., the Centers for Medicare & Medicaid Services, other divisions of the U.S. Department of Health and Human Services, the U.S. Department of Justice, and similar foreign, state and local government authorities, regulate sales, promotion and other activities following drug approval. Only those claims relating to safety and efficacy that the FDA has approved may be used in labeling. We are only allowed to use promotional communications regarding a drug that are consistent with the information in the drug’s approved labeling. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses.
For any approved medicine, domestic and foreign sales of the medicine depend, in part, on the availability and amount of reimbursement by third party payors, including governments and private health plans. Private health plans may seek to manage cost and use of our medicines by implementing coverage and reimbursement limitations. Governments may also regulate or influence coverage, reimbursement and/or pricing of our medicines to control cost or affect use. Within the EU a variety of payors pay for medicines, with governments being the primary source of payment. Negotiating pricing with governmental authorities can delay commercialization. Such pricing and reimbursement factors could impact our ability, including Akcea, and that of our commercial partners to successfully commercialize approved medicines.
In the U.S. and foreign jurisdictions, the legislative landscape continues to evolve. There have been a number of legislative and regulatory changes to the healthcare system that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels and by foreign governments that seek to reduce healthcare costs. There has also been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in efforts to bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for medicines.
Other healthcare laws that may affect our ability to operate include the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information; analogous foreign and state laws governing the privacy and security of health information, such as the General Data Protection Regulation, or GDPR, in the EU, and the California Consumer Privacy Act, or CCPA, in California, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect; and the Physician Payments Sunshine Act, which requires manufacturers of medicines, devices, biologics, and medical supplies to report annually to the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members.
34
Our operations may be directly, or indirectly through our customers, distributors, or other business partners, subject to various federal and state fraud and abuse laws, including, without limitation, anti-kickback statutes and false claims statutes. These laws may impact, among other things, our, Akcea’s, and our partners’ proposed sales, marketing and education programs.
The U.S. Foreign Corrupt Practices Act, or FCPA, prohibits certain individuals and entities, including us, from promising, paying, offering to pay, or authorizing the payment of anything of value to any foreign government official, directly or indirectly, to obtain or retain business or an improper advantage. If we violate the FCPA, it could result in large civil and criminal penalties as well as an adverse effect on our reputation, operations, and financial condition. We could also face collateral consequences such as debarment and the loss of export privileges.
Competition
Our Business in General
Some of our medicines may compete with existing therapies for market share. In addition, there are a number of companies pursuing the development of oligonucleotide-based technologies and the development of pharmaceuticals utilizing these technologies. These companies include specialized pharmaceutical firms and large pharmaceutical companies acting either independently or together with biopharmaceutical companies. Our medicines are differentiated from traditional small molecule medicines by their chemistry, how they move in the body, how they act in the body, delivery technology, and formulations.
Our approved products and our products under development address numerous markets. The diseases our medicines target for which we have or may receive marketing authorization will determine our competition. For some of our products, an important factor may be the timing of market introduction of competitive products. Accordingly, the relative speed with which we can develop products, complete the clinical trials and marketing authorization processes and supply commercial quantities of the products to the market are important competitive factors. We expect to compete among products approved for sale based on a variety of factors, including, among other things, product efficacy, safety, mechanism of action, dosing convenience, marketing and sales strategy and tactics, availability, price, and reimbursement.
Below we have included what we believe to be the competitive landscape for the following key programs:
| ● | Marketed Medicines: SPINRAZA and TEGSEDI |
| ● | Medicine under regulatory review: WAYLIVRA |
| ● | Medicine currently in pivotal trials: IONIS-HTTRx |
Marketed Medicines
SPINRAZA
We believe that the following medicines could compete with SPINRAZA:
| Medicine | Company | Medicine Description | Phase | Admin/Dosing | Efficacy(1) | Safety(1) |
Zolgensma (AVXS-101) | Novartis | Gene therapy that corrects the SMN1 gene using the AAV9 Vector | Under FDA Review | Infusion | Demonstrated an increase in survival and improvement in achievement of developmental milestones vs the natural history of SMA Type 1. Study included a small number of patients. | Generally well tolerated to date and the most commonly observed side effect was elevated liver enzymes. Study included a small number of patients. |
Risdiplam (RG7916) | PTC Therapeutics/ Roche/ SMA Foundation | A small molecule drug that modulates splicing of the SMN2 gene | 2 | Oral | Preliminary findings from Part 1 of the FIREFISH study show that infants with Type 1 SMA are meeting developmental milestones including sitting without support. Preliminary findings from Part 1 of the SUNFISH study show improvements in motor function in people with Type 2/3 SMA. Studies included a small number of patients. | Safe and well tolerated at all doses and had no drug-related or safety-related study withdrawals. Studies included a small number of patients. |
| Reldesemtiv | Cytokinetics/ Astellas | A selective, fast skeletal muscle troponin activator | 2 | Oral | The Phase 2 study demonstrated dose-dependent increases in six minute walk distance in ambulatory patients as measured at both post-baseline time points, week four and week eight | Adverse events were similar between groups receiving reldesemtiv and placebo. The most commonly observed adverse effects were headache, constipation and nausea |
| Firdapse | Catalyst/Jazz/ BioMarin | A potassium channel blocker that increases the release of acetylcholine | 2 | Oral | None reported | None reported |
| (1) | Taken from public documents including respective company press releases, company presentations, and scientific presentations. |
35
We believe that Zolgensma, if approved, may be the first medicine to compete with SPINRAZA. The FDA accepted Novartis’ biologics licensing application, or BLA, for Zolgensma, in December 2018 and regulatory action is anticipated in May 2019. The filing is supported by data from the START trial, which demonstrated an increase in survival and improvement in achievement of developmental milestones compared to the natural history of SMA Type 1. The START trial included 15 patients.
TEGSEDI
We believe that the following medicines could compete with TEGSEDI:
| Medicine | Company | Medicine Description | Phase | Admin/Dosing | Efficacy(1) | Safety(1) |
| Onpattro | Alnylam | An RNAi drug formulated with lipid nanoparticles to inhibit TTR mRNA | Approved | Infusion every 3 weeks with pre-treatment with steroids | 84.3% mean reduction in TTR at 18 months | Most common AEs more frequently observed in Onpattro arm vs. placebo were peripheral edema (29.7% vs. 22.1%) and infusion-related reactions (18.9% vs. 9.1%) |
| Tafamidis | Pfizer | A small molecule drug to stabilize TTR Protein | Commercially available in the EU for stage 1 hATTR amyloidosis with polyneuropathy. Under review in the U.S. for ATTR with cariodmyopathy with a PDUFA date in July 2019 | Daily oral capsule | In 45% of patients taking Tafamidis, nerve function either improved or stabilized, compared with 30% of patients taking placebo | Urinary tract infection, vaginal infection, upper abdominal pain and diarrhea |
| AG10 | Eidos | Small molecule that binds and stabilizes TTR in the blood | 2 | Oral | Demonstrated a statistically significant increase in serum TTR concentrations | Drug well tolerated with no safety signals |
| CRX-1008 | Corino Therapeutics | Small molecule repurposed generic drug | 2 | Daily oral dose | Shows binding and stabilization of TTR in humans | No drug related adverse events reported |
| Vutrisiran | Alnylam | An RNAi drug conjugated with GalNAC to inhibit TTR mRNA in liver cells | 3 | Monthly or quarterly | In healthy volunteers, a single dose showed mean max TTR knockdown of 97% | Injection site reactions were reported |
| (1) | Taken from public documents including respective company press releases, company presentations, and scientific presentations. Diflunisal efficacy and safety came from the published papers of two investigator sponsored studies, Berk JL, Suhr OB, Obici L, et al. Repurposing Diflunisal for Familial Amyloid Polyneuropathy: A Randomized Clinical Trial. JAMA. 2013;310(24):2658-2667 and Sekijima YS, Toja K, Morita H, et al. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015;22(2):79-83. |
TEGSEDI is a once weekly, self-administered subcutaneous medicine. TEGSEDI was approved in 2018 in the U.S., EU and Canada for the treatment of polyneuropathy caused by hATTR in adult patients. Results from our Phase 3 NEURO-TTR study demonstrated that patients treated with TEGSEDI experienced significant benefit compared to patients treated with placebo across both co-primary endpoints: the Norfolk QoL-DN and mNIS+7 a measure of neuropathic disease progression. The product label for TEGSEDI in the U.S. has a boxed warning for thrombocytopenia and glomerulonephritis and requires periodic blood and urine monitoring. TEGSEDI has a Risk Evaluation and Mitigation Strategy, or REMS, program. Our main competition in the U.S. market for TEGSEDI is ONPATTRO (patisiran), marketed by Alnylam Pharmaceuticals, Inc. Although ONPATTRO requires intravenous administration by a healthcare provider in a clinical setting every three weeks and pre-treatment with steroids, it does not have a boxed warning or REMS.
Medicine Under Regulatory Review
36
WAYLIVRA
We believe that the following medicines could compete with WAYLIVRA:
| Medicine | Company | Medicine Description | Phase | Admin/Dosing | Efficacy(1) | Safety(1) |
| Metreleptin | Novelion Therapeutics | A synthetic form of the hormone leptin | 3 | Reconstituted subcutaneous injection | 44.4% mean reduction in triglycerides at four months in patients with abnormal triglyceride levels | Anti-metreleptin antibodies, hypoglycemia, hypersensitivity, risk of T-cell lymphoma |
| Gemcabene | Gemphire Therapeutics | Monocalcium salt of a dialkyl ether dicarboxylic acid | 2 | Oral, once-daily | In a post hoc analysis (n=9) of patients with triglycerides >500 mg/dL, reductions of 59% and 60% from 150 mg and 300 mg doses, respectively, were observed | In a recent study, in the gemcabene-treatment group, the most frequently occurring adverse events were headache and infection |
| (1) | Taken from public documents including respective company press releases, company presentations, and scientific presentations. |
Metreleptin is being tested in people with FPL who also have NASH. Metreleptin is currently approved for use in the U.S. and EU in generalized lipodystrophy, or GL patients. Metreleptin does not affect apoC-III levels. ApoC-III levels have been shown to be elevated in people with FPL, and directly correlate to triglyceride levels.
Gemcabene is being studied in people with severe hypertriglyceridemia, defined as triglycerides above 500 mg/dL. Gemphire announced top-line results in June 2018 that Gemcabene met its Phase 2b primary endpoint and demonstrated statistically significant lowering of triglycerides in severe hypertriglyceridemia. The initiation of a Phase 3 study will not take place until the partial clinical hold, which was issued by the FDA in 2004, is lifted.
To date, WAYLIVRA has shown the highest percent of triglyceride reductions compared to existing treatments, such as fibrates, regardless of starting triglyceride levels prior to dosing with WAYLIVRA. Based on our broad Phase 2 data for the treatment of different patients including people with FCS, we believe that WAYLIVRA will work equally well as a single agent or in combination with other triglyceride-lowering medicines on the market. If regulatory authorities require us to implement platelet monitoring procedures in the commercial setting, which have yet to be determined, it could impact the future competitive profile of WAYLIVRA.
Medicine Currently in Pivotal Trials
IONIS-HTTRx
We believe that the following medicines could compete with IONIS-HTTRx:
| Medicine | Company | Medicine Description | Phase | Admin/Dosing | Efficacy(1) | Safety(1) |
| Laquinimod | Active Biotech | A small molecule that activates selective aryl hydrocarbon receptor | 2 | Daily oral dose | Did not meet its primary endpoint of slowing disease development, but secondary endpoint of reduction of brain atrophy was met | No drug related adverse events reported |
| OMS824 | Omeros | A small molecule that targets PDE 10 | 2 | Daily oral dose | None reported | None reported |
| Selistat | AOP Orphan | An orally active, selective SIRT1 inhibitor | 2 | Daily oral dose | None reported | Safe and tolerable in Phase 1 and Phase 2 study |
| VX15 | Vaccinex | A monoclonal antibody that blocks the activity of SEMA4D | 2 | Monthly intravenous infusions | Favored in all brain regions examined, with median increase in FDG uptake from baseline of 8.6% vs placebo control achieving significance in the majority of frontal and parietal brain regions analyzed | To date, evaluated patients showed no safety signals |
| WVE-120101/ WVE-120102 | Wave Life Sciences | Antisense drugs targeting mHTT SNP-1 and SNP-2 | 1b/2a | Intrathecal administration | None reported | None reported |
| (1) | Taken from public documents including respective company press releases, company presentations, and scientific presentations. |
37
We believe that Wave Life Sciences’ WVE-120101 and WVE-120102, being developed for Huntington’s Disease, could compete directly against IONIS-HTTRx. These medicines are antisense medicines administered intrathecally, targeting mHTT SNP-1 and SNP-2, respectively. Wave Life Sciences is currently conducting two simultaneous Phase 1b/2a clinical trials, enrolling adults with early manifest Huntington’s disease who carry a single nucleotide polymorphism, or SNP, at the SNP1 (study name: PRECISION HD1) and SNP2 (study name: PRECISION HD2) location, with their data readouts expected in the first half of 2019.
Employees
As of February 20, 2019, we employed 737 people, including 248 Akcea employees. A significant number of our management and professional employees have had prior experience with pharmaceutical, biotechnology or medical product companies. Collective bargaining agreements do not cover any of our employees, and management considers relations with our employees to be good.
Executive Officers of Ionis
The following sets forth certain information regarding our executive officers as of February 20, 2019:
| Name | Age | Position | ||
| Stanley T. Crooke, M.D., Ph.D. | 73 | Chairman, Chief Executive Officer and President | ||
| Brett P. Monia, Ph.D. | 57 | Chief Operating Officer | ||
| C. Frank Bennett, Ph.D. | 62 | Senior Vice President, Antisense Research | ||
| Damien McDevitt, Ph.D. | 52 | Chief Business Officer | ||
| Richard S. Geary, Ph.D. | 61 | Senior Vice President, Development | ||
| Elizabeth L. Hougen | 57 | Senior Vice President, Finance and Chief Financial Officer | ||
| Patrick R. O’Neil, Esq. | 45 | Senior Vice President, Legal, General Counsel, Chief Compliance Officer and Corporate Secretary |
Management Transitions
In January 2020, Dr. Crooke, our founder and Chief Executive Officer, plans to transition from Chief Executive Officer to Executive Chairman of our Board of Directors. As Executive Chairman, Dr. Crooke will continue to be responsible for the activities of the board and will remain active in the company, providing strategic advice and continuing to participate in the scientific activities. Our board has selected Dr. Monia, who has been our Chief Operating Officer for the last year and a member of our team since our founding nearly 30 years ago, to serve as our Chief Executive Officer starting in January 2020.
STANLEY T. CROOKE, M.D., Ph.D.
Chairman, Chief Executive Officer and President
Dr. Crooke is a founder of Ionis and has been Chief Executive Officer and a Director since January 1989. He was elected Chairman of the Board in February 1991. Prior to founding Ionis, from 1980 until January 1989, Dr. Crooke was employed by SmithKline Beckman Corporation, a pharmaceutical company, where his titles included President of Research and Development of SmithKline and French Laboratories.
BRETT P. MONIA, Ph.D.
Chief Operating Officer and Senior Vice President, Translational Medicine
Dr. Monia was promoted to Chief Operating Officer in January 2018 and to Senior Vice President in January 2012. From February 2009 to January 2012, Dr. Monia served as our Vice President, Drug Discovery and Corporate Development and from October 2000 to February 2009, he served as our Vice President, Preclinical Drug Discovery. From October 1989 to October 2000 he held various positions within our Molecular Pharmacology department.
C. FRANK BENNETT, Ph.D.
Senior Vice President, Antisense Research
Dr. Bennett was promoted to Senior Vice President, Antisense Research in January 2006. From June 1995 to January 2006, Dr. Bennett served as our Vice President, Research. From March 1993 to June 1995, he was Director, Molecular Pharmacology, and from May 1992 to March 1993, he was an Associate Director in our Molecular and Cellular Biology department. Prior to joining Ionis in 1989, Dr. Bennett was employed by SmithKline and French Laboratories in various research positions. He is an external member of the Scientific Advisory Board of Experimental Therapeutics Center in Singapore and the Hereditary Disease Foundation.
DAMIEN McDEVITT, Ph.D.
Chief Business Officer
Dr. McDevitt joined Ionis in June 2018 as our Chief Business Officer. In October 2018, he was appointed to the board of directors of our majority-owned affiliate, Akcea Therapeutics, Inc. Previously, Dr. McDevitt was Senior Vice President, Corporate Development at ACADIA Pharmaceuticals. Prior to ACADIA, he was at GSK for more than two decades. He served in various roles with increasing responsibility including vice president, head of business development for R&D Extended Therapy areas, head of Worldwide Business Development Asia and head of GSK’s R&D West Coast Innovation Center.
38
RICHARD S. GEARY, Ph.D.
Senior Vice President, Development
Dr. Geary was promoted to Senior Vice President, Development in August 2008. From August 2003 to August 2008, Dr. Geary served as our Vice President, Preclinical Development. From November 1995 to August 2003, he held various positions within the Preclinical Development department. Prior to joining Ionis in 1995, Dr. Geary was Senior Research Scientist and Group Leader for the bioanalytical and preclinical pharmacokinetics group in the Applied Chemistry Department at Southwest Research Institute.
ELIZABETH L. HOUGEN
Senior Vice President, Finance and Chief Financial Officer
Ms. Hougen was promoted to Senior Vice President, Finance and Chief Financial Officer in January 2013. From January 2007 to December 2012, Ms. Hougen served as our Vice President, Finance and Chief Accounting Officer and from May 2000 to January 2007, she served as our Vice President, Finance. Prior to joining Ionis in 2000, Ms. Hougen was Executive Director, Finance and Chief Financial Officer for Molecular Biosystems, Inc., a public biotechnology company.
PATRICK R. O’NEIL, Esq.
Senior Vice President, Legal, General Counsel, Chief Compliance Officer and Corporate Secretary
Mr. O’Neil was promoted to Senior Vice President, Legal and General Counsel in January 2013. Mr. O’Neil also serves as our Chief Compliance Officer and Corporate Secretary. From September 2010 to January 2013, Mr. O’Neil served as our Vice President, Legal and General Counsel and from January 2009 to September 2010, he served as our Vice President, Legal and Senior Transactions Counsel. From October 2001 to January 2009 he held various positions within our Legal department. Prior to joining Ionis, Mr. O’Neil was an associate at Cooley LLP.
Item 1A. RISK FACTORS
Investing in our securities involves a high degree of risk. You should consider carefully the following information about the risks described below, together with the other information contained in this report and in our other public filings in evaluating our business. If any of the following risks actually occur, our business could be materially harmed, and our financial condition and results of operations could be materially and adversely affected. As a result, the trading price of our securities could decline, and you might lose all or part of your investment.
Risks Associated with our Ionis Core and Akcea Therapeutics Businesses
If the market does not accept our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA, we are not likely to generate revenues or become consistently profitable.
Even if our medicines are authorized for marketing, including SPINRAZA, TEGSEDI and WAYLIVRA, our success will depend upon the medical community, patients and third-party payors accepting our medicines as medically useful, cost-effective and safe. Even when the FDA or foreign regulatory authorities authorize our or our partners’ medicines for commercialization, doctors may not prescribe our medicines to treat patients. We and our partners may not successfully commercialize additional medicines.
Additionally, in many of the markets where we may sell our medicines in the future, if we cannot agree with the government regarding the price we can charge for our medicines, then we may not be able to sell our medicines in that market. Similarly, cost control initiatives by governments or third-party payors could decrease the price received for our medicines or increase patient coinsurance to a level that makes our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA, unaffordable.
The degree of market acceptance for our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA, depends upon a number of factors, including the:
| ● | receipt and scope of marketing authorizations; |
| ● | establishment and demonstration in the medical and patient community of the efficacy and safety of our medicines and their potential advantages over competing products; |
| ● | cost and effectiveness of our medicines compared to other available therapies; |
| ● | patient convenience of the dosing regimen for our medicines; and |
| ● | reimbursement policies of government and third-party payors. |
Based on the profile of our medicines, physicians, patients, patient advocates, payors or the medical community in general may not accept and/or use any medicines that we may develop.
39
For example, the product label for TEGSEDI in the U.S. has a boxed warning for thrombocytopenia and glomerulonephritis, requires periodic blood and urine monitoring, and TEGSEDI has a Risk Evaluation and Mitigation Strategy, or REMS, program. Our main competition in the U.S. market for TEGSEDI is ONPATTRO (patisiran), marketed by Alnylam Pharmaceuticals, Inc. Although ONPATTRO requires intravenous administration and pre-treatment with steroids, it does not have a boxed warning or REMS. Additionally, in the clinical studies with WAYLIVRA, declines in platelet counts were observed in many patients and some patients discontinued the studies because of platelet declines. Therefore, we expect the product label for WAYLIVRA will require periodic blood monitoring. In each case, these label requirements could negatively affect our ability to attract and retain patients for these medicines. We believe that the enhanced monitoring we have implemented to support early detection and management of these issues can help manage these safety issues so that patients can continue treatment. Since implementation of the enhanced monitoring, serious platelet events have been infrequent. While we believe we and Akcea can better maintain patients on TEGSEDI and WAYLIVRA through patient-centric commercial approaches where we plan to have greater involvement with physicians and patients, if we cannot effectively maintain patients on TEGSEDI or WAYLIVRA, we may not be able to generate substantial revenue from TEGSEDI or WAYLIVRA sales.
If we or our partners fail to compete effectively, our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA, will not contribute significant revenues.
Our competitors engage in drug discovery throughout the world, are numerous, and include, among others, major pharmaceutical companies and specialized biopharmaceutical firms. Other companies engage in developing antisense technology. Our competitors may succeed in developing medicines that are:
| ● | priced lower than our medicines; |
| ● | reimbursed more favorably by government and other third-party payors than our medicines; |
| ● | safer than our medicines; |
| ● | more effective than our medicines; or |
| ● | more convenient to use than our medicines. |
These competitive developments could make our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA, obsolete or non-competitive.
Certain of our partners are pursuing other technologies or developing other medicines either on their own or in collaboration with others, including our competitors, to treat the same diseases our own collaborative programs target. Competition may negatively impact a partner’s focus on and commitment to our medicines and, as a result, could delay or otherwise negatively affect the commercialization of our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA.
Many of our competitors have substantially greater financial, technical and human resources than we do. In addition, many of these competitors have significantly greater experience than we do in conducting preclinical testing and human clinical studies of new pharmaceutical products, in obtaining FDA and other regulatory authorizations of such products and in commercializing such products. Accordingly, our competitors may succeed in obtaining regulatory authorization for products earlier than we do. Marketing and sales capability is another factor relevant to the competitive position of our medicines, and we will primarily rely on our partners and Akcea to provide this capability.
There are several pharmaceutical and biotechnology companies engaged in the development or commercialization of products against targets that are also targets of products in our development pipeline. For example, Zolgensma (AVXS-101), Risdiplam (RG7916), Reldesemtiv and Firdapse could compete with SPINRAZA, and ONPATTRO (approved in the U.S. and Europe for a similar indication as TEGSEDI), Tafamadis, AG10, CRX-1008 and Vutrisiran could compete with TEGSEDI. Also, Metreleptin and Gemcabene could compete with WAYLIVRA, while Laquinimod, OMS823, Selistat, VX15, WVE-120101 and WVE-120102 could compete with IONIS-HTTRx.
Following approval, our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA could be subject to regulatory limitations.
Following approval of a medicine, we and our partners must comply with comprehensive government regulations regarding the manufacture, marketing and distribution of drug products. We or our partners may not obtain the labeling claims necessary or desirable to successfully commercialize our drug products, including SPINRAZA, TEGSEDI and WAYLIVRA.
The FDA and foreign regulatory authorities have the authority to impose significant restrictions on an approved drug product through the product label and on advertising, promotional and distribution activities. For example:
| ● | In the U.S., TEGSEDI’s label contains a boxed warning for thrombocytopenia and glomerulonephritis; |
| ● | TEGSEDI requires periodic blood and urine monitoring; and |
| ● | in the U.S. TEGSEDI is available only through a Risk Evaluation and Mitigation Strategy, or REMS, program. |
In addition, when approved, the FDA or a foreign regulatory authority may condition approval on the performance of post-approval clinical studies or patient monitoring, which could be time consuming and expensive. If the results of such post-marketing studies are not satisfactory, the FDA or a foreign regulatory authority may withdraw marketing authorization or may condition continued marketing on commitments from us or our partners that may be expensive and/or time consuming to fulfill.
If we or others identify side effects after any of our drug products are on the market, or if manufacturing problems occur subsequent to regulatory approval, we or our partners may lose regulatory approval, or we or our partners may need to conduct additional clinical studies and/or change the labeling of our drug products, including SPINRAZA, TEGSEDI and WAYLIVRA.
40
We depend on our collaboration with Biogen for the development and commercialization of SPINRAZA.
We have entered into a collaborative arrangement with Biogen to develop and commercialize SPINRAZA. We entered into this collaboration primarily to:
| ● | fund our development activities for SPINRAZA; |
| ● | seek and obtain regulatory approvals for SPINRAZA; and |
| ● | successfully commercialize SPINRAZA. |
We are relying on Biogen to obtain additional regulatory approvals for SPINRAZA, and successfully commercialize SPINRAZA. In general, we cannot control the amount and timing of resources that Biogen devotes to our collaboration. If Biogen fails to further develop SPINRAZA, obtain additional regulatory approvals for SPINRAZA, or commercialize SPINRAZA, or if Biogen’s efforts are not effective, our business may be negatively affected.
Our collaboration with Biogen may not continue for various reasons. Biogen can terminate our collaboration at any time. If Biogen stops developing or commercializing SPINRAZA, we would have to seek or spend additional funding, and SPINRAZA’s commercialization may be harmed or delayed.
Our collaboration with Biogen may not result in the continued successful commercialization of SPINRAZA. If Biogen does not continue to successfully commercialize SPINRAZA, we will receive limited revenues for SPINRAZA.
If Akcea cannot optimize and maintain effective marketing and sales capabilities or enter into agreements with third parties to market and sell TEGSEDI, we may not generate significant product revenue from TEGSEDI.
To successfully commercialize TEGSEDI Akcea must successfully manage its marketing, sales and distribution capabilities or make arrangements with third parties to perform these services. Akcea may not be successful in doing so. To commercialize TEGSEDI in the initial indications Akcea is pursuing, Akcea will need to optimize and maintain a specialty sales force in each global region it expects to market TEGSEDI, supported by case managers, reimbursement specialists, partnerships with specialty pharmacies, injection training, routine blood and urine monitoring and a medical affairs team. Akcea may seek to further penetrate markets by expanding its sales force or through strategic partnerships with other pharmaceutical or biotechnology companies or third-party sales organizations.
Even though certain members of Akcea’s management team and other employees have experience commercializing drug products, Akcea has no prior experience marketing, selling or distributing drug products, and there are significant risks involved in building and managing a commercial infrastructure. It will be expensive and time consuming for Akcea to maintain its own sales force and related compliance protocols to market TEGSEDI. Akcea may never successfully optimize or manage this capability and any failure could preclude the successful commercialization of TEGSEDI. Akcea and its partners, if any, will have to compete with other companies to recruit, hire, train, manage and retain marketing and sales personnel.
Akcea incurred expenses prior to the launch of TEGSEDI to integrate and manage the associated marketing and sales infrastructure. If regulatory requirements or other factors cause the commercial launch of TEGSEDI to be less successful than expected, Akcea will have incurred expenses for having invested in these capabilities prior to realizing any significant revenue from sales of TEGSEDI. Akcea’s sales force and marketing teams may not successfully commercialize TEGSEDI.
To the extent we and Akcea decide to rely on third parties to commercialize TEGSEDI in a particular geographic market, such as the collaboration Akcea has with PTC Therapeutics to commercialize TEGSEDI in Latin America, we may receive less revenue than if Akcea commercialized TEGSEDI by itself. Further we would have less control over the sales efforts of any other third parties involved in commercializing TEGSEDI.
If Akcea cannot effectively build and manage its distribution, medical affairs, market access, marketing and sales infrastructure, or find a suitable third party to perform such functions, the commercial launch and sales of TEGSEDI may be delayed, less successful or precluded. Such events may result in decreased sales and lower revenue, which could have a material adverse effect on our business, prospects, financial condition and results of operations.
If government or other third-party payors fail to provide adequate coverage and payment rates for our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA, our revenue will be limited.
In both domestic and foreign markets, sales of our current and future products will depend in part upon the availability of coverage and reimbursement from third-party payors. The majority of people in the U.S. who would fit within our target patient populations for our medicines have their healthcare supported by a combination of Medicare coverage, other government health programs such as Medicaid, managed care providers, private health insurers and other organizations. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available. Assuming coverage is approved, the resulting reimbursement payment rates might not be enough to make our medicines affordable.
Third-party payors, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In addition, in the U.S., no uniform policy of coverage and reimbursement for drug products exists among third-party payors. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. Further, we believe that future coverage and reimbursement will likely be subject to increased restrictions both in the U.S. and in international markets. For example, in the U.S., recent health reform measures have resulted in reductions in Medicare and other healthcare funding, and there have been several U.S. Congressional inquiries and proposed federal legislation designed to, among other things, reform government program reimbursement methodologies for drug products and bring more transparency to drug pricing. Third-party coverage and reimbursement for our products or medicines may not be available or adequate in either the U.S. or international markets, which would negatively affect the potential commercial success of our products, our revenue and our profits.
41
If Biogen cannot manufacture finished drug product for SPINRAZA or the post-launch supply of the active drug substance for SPINRAZA, SPINRAZA may not maintain commercial success.
Biogen is responsible for the long-term supply of both SPINRAZA drug substance and finished drug product. Biogen may not be able to reliably manufacture SPINRAZA drug substance and drug product to support the long-term commercialization of SPINRAZA. If Biogen cannot reliably manufacture SPINRAZA drug substance and drug product, SPINRAZA may not maintain commercial success, which will harm our ability to generate revenue.
If we or our partners fail to obtain regulatory approval for our medicines, including WAYLIVRA, and additional approvals for SPINRAZA and TEGSEDI, we or our partners cannot sell them in the applicable markets.
We cannot guarantee that any of our medicines, including WAYLIVRA, will be considered safe and effective, or will be approved for commercialization. In addition, we cannot guarantee that SPINRAZA and TEGSEDI will be approved in additional markets or for additional indications. We and our partners must conduct time-consuming, extensive and costly clinical studies to show the safety and efficacy of each of our medicines before they can be approved for sale. We must conduct these studies in compliance with FDA regulations and with comparable regulations in other countries.
We and our partners may not obtain necessary regulatory approvals on a timely basis, if at all, for our medicines. It is possible that regulatory agencies will not approve our medicines, including WAYLIVRA, for marketing or additional marketing authorizations for SPINRAZA or TEGSEDI. If the FDA or another regulatory agency believes that we or our partners have not sufficiently demonstrated the safety or efficacy of any of our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA, the agency will not approve the specific medicine or will require additional studies, which can be time consuming and expensive and which will delay or harm commercialization of the medicine. For example, Akcea received a CRL from the FDA and a preliminary notice of noncompliance withdrawal letter from Health Canada for WAYLIVRA. As result, Akcea may need to submit additional data to the FDA and Health Canada or conduct additional clinical studies before obtaining marketing authorization, which would be expensive and cause delays. The CHMP of the EMA has adopted a positive opinion recommending conditional marketing authorization of WAYLIVRA as an adjunct to diet in adult patients with genetically confirmed FCS who are at high risk for pancreatitis, in whom response to diet and triglyceride lowering therapy has been inadequate. The positive opinion will now be referred to the EC, which grants marketing authorization for medicines in the European Union, as well as to European Economic Area members Iceland, Liechtenstein and Norway, however, the EC may decide not to adopt the CHMP’s positive opinion.
Failure to receive marketing authorization for WAYLIVRA or our other medicines, or failure to receive additional marketing authorizations for SPINRAZA or TEGSEDI, or delays in these authorizations could prevent or delay commercial introduction of the medicine, and, as a result, could negatively impact our ability to generate revenue from product sales.
If the results of clinical testing indicate that any of our medicines are not suitable for commercial use we may need to abandon one or more of our drug development programs.
Drug discovery and development has inherent risks and the historical failure rate for drugs is high. Antisense drugs are a relatively new approach to therapeutics. If we cannot demonstrate that our medicines are safe and effective for human use, we may need to abandon one or more of our drug development programs.
In the past, we have invested in clinical studies of medicines that have not met the primary clinical end points in their Phase 3 studies. Similar results could occur in clinical studies for our medicines, including the study of WAYLIVRA in patients with FPL and the study of IONIS-HTTRx in patients with Huntington’s disease. If any of our medicines in clinical studies, including WAYLIVRA and IONIS-HTTRx, do not show sufficient efficacy in patients with the targeted indication, it could negatively impact our development and commercialization goals for these medicines and our stock price could decline.
Even if our medicines are successful in preclinical and human clinical studies, the medicines may not be successful in late-stage clinical studies.
Successful results in preclinical or initial human clinical studies, including the Phase 2 results for some of our medicines in development, may not predict the results of subsequent clinical studies, including the Phase 3 study of WAYLIVRA in patients with FPL and the pivotal study of IONIS-HTTRx in patients with Huntington’s disease. There are a number of factors that could cause a clinical study to fail or be delayed, including:
| ● | the clinical study may produce negative or inconclusive results; |
| ● | regulators may require that we hold, suspend or terminate clinical research for noncompliance with regulatory requirements; |
| ● | we, our partners, the FDA or foreign regulatory authorities could suspend or terminate a clinical study due to adverse side effects of a medicine on subjects in the trial; |
| ● | we may decide, or regulators may require us, to conduct additional preclinical testing or clinical studies; |
| ● | enrollment in our clinical studies may be slower than we anticipate; |
| ● | people who enroll in the clinical study may later drop out due to adverse events, a perception they are not benefiting from participating in the study, fatigue with the clinical study process or personal issues; |
| ● | the cost of our clinical studies may be greater than we anticipate; and |
| ● | the supply or quality of our medicines or other materials necessary to conduct our clinical studies may be insufficient, inadequate or delayed. |
42
In addition, our current medicines, including SPINRAZA, TEGSEDI and WAYLIVRA, are chemically similar to each other. As a result, a safety observation we encounter with one of our medicines could have, or be perceived by a regulatory authority to have, an impact on a different drug we are developing. This could cause the FDA and other regulators to ask questions or take actions that could harm or delay our ability to develop and commercialize our medicines or increase our costs. For example, the FDA or other regulatory agencies could request, among other things, any of the following regarding one of our medicines: additional information or commitments before we can start or continue a clinical study, protocol amendments, increased safety monitoring, additional product labeling information, and post-approval commitments. Similarly, we have an ongoing Phase 3 study of WAYLIVRA in patients with FPL, an ongoing open-label extension study of WAYLIVRA in patients with FCS, an ongoing open-label extension study of TEGSEDI and expanded access programs for each medicine. Adverse events or results from these studies could negatively impact our current or planned marketing approval applications for WAYLIVRA in patients with FCS or the commercial opportunity for each product.
Any failure or delay in the clinical studies, including the Phase 3 study for WAYLIVRA in patients with FPL and the pivotal study of IONIS-HTTRx in patients with Huntington’s disease, could reduce the commercial potential or viability of our medicines.
If we cannot manufacture our medicines or contract with a third party to manufacture our medicines at costs that allow us to charge competitive prices to buyers, we cannot market our products profitably.
To successfully commercialize any of our medicines, we or our partner would need to establish large-scale commercial manufacturing capabilities either on our own or through a third-party manufacturer. We and Akcea will rely on third-party manufacturers to supply the drug substance and drug product for TEGSEDI and WAYLIVRA. In addition, as our drug development pipeline increases and matures, we will have a greater need for clinical trial and commercial manufacturing capacity. We have limited experience manufacturing pharmaceutical products of the chemical class represented by our medicines, called oligonucleotides, on a commercial scale for the systemic administration of a medicine. There are a small number of suppliers for certain capital equipment and raw materials that we use to manufacture our medicines, and some of these suppliers will need to increase their scale of production to meet our projected needs for commercial manufacturing. Further, we must continue to improve our manufacturing processes to allow us to reduce our drug costs. We may not be able to manufacture our medicines at a cost or in quantities necessary to make commercially successful products.
Also, manufacturers, including us, must adhere to the FDA’s current Good Manufacturing Practices regulations and similar regulations in foreign countries, which the applicable regulatory authorities enforce through facilities inspection programs. We and our contract manufacturers may not comply or maintain compliance with Good Manufacturing Practices, or similar foreign regulations. Non-compliance could significantly delay or prevent receipt of marketing authorization for our medicines, including authorizations for SPINRAZA, TEGSEDI and WAYLIVRA, or result in enforcement action after authorization that could limit the commercial success of our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA.
We depend on third parties to conduct our clinical studies for our medicines and any failure of those parties to fulfill their obligations could adversely affect our development and commercialization plans.
We depend on independent clinical investigators, contract research organizations and other third-party service providers to conduct our clinical studies for our medicines and expect to continue to do so in the future. For example, we use clinical research organizations, such as Icon Clinical Research Limited, INC Research Toronto, Inc. and Medpace for the clinical studies for our medicines, including TEGSEDI and WAYLIVRA. We rely heavily on these parties for successful execution of our clinical studies, but do not control many aspects of their activities. For example, the investigators are not our employees. However, we are responsible for ensuring that these third parties conduct each of our clinical studies in accordance with the general investigational plan and approved protocols for the study. Third parties may not complete activities on schedule or may not conduct our clinical studies in accordance with regulatory requirements or our stated protocols. The failure of these third parties to carry out their obligations or a termination of our relationship with these third parties could delay or prevent the development, marketing authorization and commercialization of our medicines, including authorizations for WAYLIVRA or additional authorizations for SPINRAZA and TEGSEDI.
Risks Associated with our Businesses as a Whole
We have incurred losses, and our business will suffer if we fail to consistently achieve profitability in the future.
Because drug discovery and development requires substantial lead-time and money prior to commercialization, our expenses have historically exceeded our revenue since we were founded in January 1989. As of December 31, 2018, we had an accumulated deficit of approximately $1.0 billion and stockholders’ equity of approximately $1,187.2 million. Most of our historical losses resulted from costs incurred in connection with our research and development programs and from selling, general and administrative costs associated with our operations. Most of our income has come from collaborative arrangements, including commercial revenue from royalties and R&D revenue, with additional income from research grants and the sale or licensing of our patents, as well as interest income. If we do not continue to earn substantial revenue, we may incur additional operating losses in the future. We may not successfully develop any additional products or achieve or sustain future profitability.
43
Our ability to use our net operating loss carryovers and certain other tax attributes may be limited.
Under the Internal Revenue Code of 1986, as amended, or the Code, a corporation is generally allowed a deduction for net operating losses, or NOLs, carried over from a prior taxable year. Under that provision, we can carryforward our NOLs to offset our future taxable income, if any, until such NOLs are used or expire. The same is true of other unused tax attributes, such as tax credits.
Under the Tax Cut and Jobs Act of 2017, or the Tax Act, federal net operating losses incurred in 2018 and in future years may be carried forward indefinitely, but the deductibility of such federal net operating losses is limited. It is uncertain if and to what extent various states will conform to the newly enacted federal tax law.
In addition, under Section 382 of the Internal Revenue Code of 1986, as amended, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50 percent change, by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income or taxes may be limited. We may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control. If an ownership change occurs and our ability to use our net operating loss carryforwards or other tax attributes is materially limited, it would harm our future operating results by effectively increasing our future tax obligations.
Since corporate partnering is a significant part of our strategy to fund the development and commercialization of our development programs, if any of our collaborative partners fail to fund our collaborative programs, or if we cannot obtain additional partners, we may have to delay or stop progress on our drug development programs.
To date, corporate partnering has played a significant role in our strategy to fund our development programs and to add key development resources. We plan to continue to rely on additional collaborative arrangements to develop and commercialize our unpartnered medicines. However, we may not be able to negotiate favorable collaborative arrangements for these drug programs. If we cannot continue to secure additional collaborative partners, our revenues could decrease and the development of our medicines could suffer.
Our corporate partners are developing and/or funding many of the medicines in our development pipeline. If any of these pharmaceutical companies stops developing and/or funding these medicines, our business could suffer and we may not have, or be willing to dedicate, the resources available to develop these medicines on our own.
Our collaborators can terminate their relationships with us under certain circumstances, many of which are outside of our control. For example, as part of a reprioritization of its pipeline and strategic review of its rare disease business, GSK declined its option on TEGSEDI and IONIS-FB-LRx.
Even with funding from corporate partners, if our partners do not effectively perform their obligations under our agreements with them, it would delay or stop the progress of our drug development and commercial programs.
In addition to receiving funding, we enter into collaborative arrangements with third parties to:
| ● | conduct clinical studies; |
| ● | seek and obtain marketing authorization; and |
| ● | manufacture, market and sell our medicines. |
Once we have secured a collaborative arrangement to further develop and commercialize one of our drug development programs, such as our collaborations with AstraZeneca, Bayer, Biogen, GSK, Janssen, Novartis and Roche, these collaborations may not continue or result in commercialized medicines, or may not progress as quickly as we first anticipated.
For example, a collaborator such as AstraZeneca, Bayer, Biogen, GSK, Janssen, Novartis or Roche, could determine that it is in its financial interest to:
| ● | pursue alternative technologies or develop alternative products that may be competitive with the drug that is part of the collaboration with us; |
| ● | pursue higher-priority programs or change the focus of its own development programs; or |
| ● | choose to devote fewer resources to our medicines than it does for its own medicines. |
If any of these occur, it could affect our partner’s commitment to the collaboration with us and could delay or otherwise negatively affect the commercialization of our medicines, including SPINRAZA.
If we do not progress in our programs as anticipated, the price of our securities could decrease.
For planning purposes, we estimate and may disclose the timing of a variety of clinical, regulatory and other milestones, such as when we anticipate a certain drug will enter the clinic, when we anticipate completing a clinical study, or when we anticipate filing an application for, or obtaining, marketing authorization. We base our estimates on present facts and a variety of assumptions. Many underlying assumptions are outside of our control. If we do not achieve milestones in accordance with our or our investors’ expectations, including milestones related to SPINRAZA, TEGSEDI and WAYLIVRA, the price of our securities could decrease.
44
If we cannot protect our patents or our other proprietary rights, others may compete more effectively against us.
Our success depends to a significant degree upon whether we can continue to develop and secure intellectual property rights to proprietary products and services. However, we may not receive issued patents on any of our pending patent applications in the U.S. or in other countries. In addition, the scope of any of our issued patents may not be sufficiently broad to provide us with a competitive advantage. Furthermore, other parties may successfully challenge, invalidate or circumvent our issued patents or patents licensed to us so that our patent rights do not create an effective competitive barrier or revenue source.
Intellectual property litigation could be expensive and prevent us from pursuing our programs.
From time to time we have to defend our intellectual property rights. If we are involved in an intellectual property dispute, we sometimes need to litigate to defend our rights or assert them against others. Disputes can involve arbitration, litigation or proceedings declared by the U.S. Patent and Trademark Office or the International Trade Commission or foreign patent authorities. Intellectual property litigation can be extremely expensive, and this expense, as well as the consequences should we not prevail, could seriously harm our business.
If a third party claims that our medicines or technology infringe its patents or other intellectual property rights, we may have to discontinue an important product or product line, alter our products and processes, pay license fees or cease certain activities. We may not be able to obtain a license to needed intellectual property on favorable terms, if at all. There are many patents issued or applied for in the biotechnology industry, and we may not be aware of patents or patent applications held by others that relate to our business. This is especially true since patent applications in the U.S. are filed confidentially for the first 18 months. Moreover, the validity and breadth of biotechnology patents involve complex legal and factual questions for which important legal issues remain.
If we fail to obtain timely funding, we may need to curtail or abandon some of our programs.
Many of our medicines are undergoing clinical studies or are in the early stages of research and development. Most of our drug programs will require significant additional research, development, preclinical and/or clinical testing, marketing authorization and/or commitment of significant additional resources prior to their successful commercialization. As of December 31, 2018, we had cash, cash equivalents and short-term investments equal to $2.1 billion. If we do not meet our goals to successfully commercialize our medicines, including SPINRAZA, TEGSEDI and WAYLIVRA, or to license our medicines and proprietary technologies, we will need additional funding in the future. Our future capital requirements will depend on many factors, such as the following:
| ● | successful commercialization for SPINRAZA and TEGSEDI; |
| ● | marketing approvals for WAYLIVRA; |
| ● | the profile and launch timing of our medicines, including TEGSEDI and WAYLIVRA; |
| ● | changes in existing collaborative relationships and our ability to establish and maintain additional collaborative arrangements; |
| ● | continued scientific progress in our research, drug discovery and development programs; |
| ● | the size of our programs and progress with preclinical and clinical studies; |
| ● | the time and costs involved in obtaining marketing authorizations; and |
| ● | competing technological and market developments, including the introduction by others of new therapies that address our markets. |
If we need additional funds, we may need to raise them through public or private financing. Additional financing may not be available at all or on acceptable terms. If we raise additional funds by issuing equity securities, the shares of existing stockholders will be diluted and the price, as well as the price of our other securities, may decline. If adequate funds are not available or not available on acceptable terms, we may have to cut back on one or more of our research, drug discovery or development programs. Alternatively, we may obtain funds through arrangements with collaborative partners or others, which could require us to give up rights to certain of our technologies or medicines.
If our planned management transition is not successful our business could suffer.
In January 2020, Dr. Crooke, our founder and Chief Executive Officer, plans to transition from Chief Executive Officer to Executive Chairman of our Board of Directors. As Executive Chairman, Dr. Crooke will continue to be responsible for the activities of the board and will remain active in the company, providing strategic advice and continuing to participate in the scientific activities. Our board has selected Dr. Monia, who has been our Chief Operating Officer for the last year and a member of our team since our founding nearly 30 years ago, to serve as our Chief Executive Officer starting in January 2020. If this transition is not successful, our business could suffer.
The loss of key personnel, or the inability to attract and retain highly skilled personnel, could make it more difficult to run our business and reduce our likelihood of success.
We are dependent on the principal members of our management and scientific staff. We do not have employment agreements with any of our executive officers that would prevent them from leaving us. The loss of our management and key scientific employees might slow the achievement of important research and development goals. It is also critical to our success that we recruit and retain qualified scientific personnel to perform research and development work. We may not be able to attract and retain skilled and experienced scientific personnel on acceptable terms because of intense competition for experienced scientists among many pharmaceutical and health care companies, universities and non-profit research institutions. In addition, failure to succeed in clinical studies may make it more challenging to recruit and retain qualified scientific personnel.
45
If the price of our securities continues to be highly volatile, this could make it harder for you to liquidate your investment and could increase your risk of suffering a loss.
The market price of our common stock, like that of the securities of many other biopharmaceutical companies, has been and is likely to continue to be highly volatile. These fluctuations in our common stock price may significantly affect the trading price of our securities. During the 12 months preceding December 31, 2018, the market price of our common stock ranged from $59.81 to $39.07 per share. Many factors can affect the market price of our securities, including, for example, fluctuations in our operating results, announcements of collaborations, clinical study results, technological innovations or new products being developed by us or our competitors, governmental regulation, marketing authorization, changes in payors’ reimbursement policies, developments in patent or other proprietary rights, public concern regarding the safety of our medicines and general market conditions.
We are exposed to potential product liability claims, and insurance against these claims may not be available to us at a reasonable rate in the future or at all.
Our business exposes us to potential product liability risks that are inherent in the testing, manufacturing, marketing and sale of therapeutic products, including potential product liability claims related to SPINRAZA, TEGSEDI and WAYLIVRA. We have clinical study insurance coverage and commercial product liability insurance coverage. However, this insurance coverage may not be adequate to cover claims against us, or be available to us at an acceptable cost, if at all. Regardless of their merit or eventual outcome, product liability claims may result in decreased demand for our drug products, injury to our reputation, withdrawal of clinical study volunteers and loss of revenues. Thus, whether or not we are insured, a product liability claim or product recall may result in losses that could be material.
Because we use biological materials, hazardous materials, chemicals and radioactive compounds, if we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
Our research, development and manufacturing activities involve the use of potentially harmful biological materials as well as materials, chemicals and various radioactive compounds that could be hazardous to human health and safety or the environment. We store most of these materials and various wastes resulting from their use at our facilities in Carlsbad, California pending ultimate use and disposal. We cannot completely eliminate the risk of contamination, which could cause:
| ● | interruption of our research, development and manufacturing efforts; |
| ● | injury to our employees and others; |
| ● | environmental damage resulting in costly clean up; and |
| ● | liabilities under federal, state and local laws and regulations governing health and human safety, as well as the use, storage, handling and disposal of these materials and resultant waste products. |
In such an event, we may be held liable for any resulting damages, and any liability could exceed our resources. Although we carry insurance in amounts and types that we consider commercially reasonable, we do not have insurance coverage for losses relating to an interruption of our research, development or manufacturing efforts caused by contamination, and the coverage or coverage limits of our insurance policies may not be adequate. If our losses exceed our insurance coverage, our financial condition would be affected.
If a natural or man-made disaster strikes our research, development or manufacturing facilities or otherwise affects our business, it could delay our progress developing and commercializing our medicines.
We manufacture our research and clinical supplies in a manufacturing facility located in Carlsbad, California. We manufacture the finished drug product for TEGSEDI and WAYLIVRA at third-party contract manufacturers. The facilities and the equipment we and our contract manufacturers use to research, develop and manufacture our medicines would be costly to replace and could require substantial lead time to repair or replace. Our facilities or our contract manufacturers may be harmed by natural or man-made disasters, including, without limitation, earthquakes, floods, fires and acts of terrorism; and if our facilities are affected by a disaster, our development and commercialization efforts would be delayed. Although we possess insurance for damage to our property and the disruption of our business from casualties, this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all. In addition, our development and commercialization activities could be harmed or delayed by a shutdown of the U.S. government, including the FDA.
Provisions in our certificate of incorporation, other agreements and Delaware law may prevent stockholders from receiving a premium for their shares.
Our certificate of incorporation provides for classified terms for the members of our board of directors. Our certificate also includes a provision that requires at least 66 2/3 percent of our voting stockholders to approve a merger or certain other business transactions with, or proposed by, any holder of 15 percent or more of our voting stock, except in cases where certain directors approve the transaction or certain minimum price criteria and other procedural requirements are met.
Our certificate of incorporation also requires that any action required or permitted to be taken by our stockholders must be taken at a duly called annual or special meeting of stockholders and may not be taken by written consent. In addition, only our board of directors, chairman of the board or chief executive officer can call special meetings of our stockholders. We have in the past, and may in the future, implement a stockholders’ rights plan, also called a poison pill, which could make it uneconomical for a third party to acquire our company on a hostile basis. In addition, our board of directors has the authority to fix the rights and preferences of, and issue shares of preferred stock, which may have the effect of delaying or preventing a change in control of our company without action by our stockholders.
46
The provisions of our convertible senior notes could make it more difficult or more expensive for a third party to acquire us. Upon the occurrence of certain transactions constituting a fundamental change, holders of the notes will have the right, at their option, to require us to repurchase all of their notes or a portion of their notes, which may discourage certain types of transactions in which our stockholders might otherwise receive a premium for their shares over the then current market prices.
These provisions, as well as Delaware law, including Section 203 of the Delaware General Corporation Law, and other of our agreements, may discourage certain types of transactions in which our stockholders might otherwise receive a premium for their shares over then current market prices, and may limit the ability of our stockholders to approve transactions that they think may be in their best interests.
Future sales of our common stock in the public market could adversely affect the trading price of our securities.
Future sales of substantial amounts of our common stock in the public market, or the perception that such sales could occur, could adversely affect trading prices of our securities. For example, we may issue approximately 10.3 million shares of our common stock upon conversion of our convertible senior notes. The addition of any of these shares into the public market may have an adverse effect on the price of our securities.
Our business is subject to changing regulations for corporate governance and public disclosure that has increased both our costs and the risk of noncompliance.
Each year we are required to evaluate our internal controls systems in order to allow management to report on and our Independent Registered Public Accounting Firm to attest to, our internal controls as required by Section 404 of the Sarbanes-Oxley Act. As a result, we continue to incur additional expenses and divert our management’s time to comply with these regulations. In addition, if we cannot continue to comply with the requirements of Section 404 in a timely manner, we might be subject to sanctions or investigation by regulatory authorities, such as the SEC, the Public Company Accounting Oversight Board, or PCAOB, or The Nasdaq Global Select Market. Any such action could adversely affect our financial results and the market price of our common stock.
The SEC and other regulators have continued to adopt new rules and regulations and make additional changes to existing regulations that require our compliance. On July 21, 2010, the Dodd-Frank Wall Street Reform and Protection Act, or the Dodd-Frank Act, was enacted. There are significant corporate governance and executive compensation-related provisions in the Dodd-Frank Act that require the SEC to adopt, or where the SEC has adopted, additional rules and regulations in these areas such as “say on pay” and proxy access. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business.
Changes in tax laws, regulations and treaties could affect our future taxable income.
A change in tax laws, treaties or regulations, or their interpretation, of any country in which we operate could materially affect us. For example, the Tax Act represented a substantial change to tax laws in the U.S. However, it did not have a material impact on our financial statements because we maintained a valuation allowance on all of our net operating losses and other deferred tax assets as of December 31, 2017. Over the next several years we expect to utilize our net operating losses and other deferred tax assets, and any future changes in tax laws could have a material effect on our business.
We could be subject to additional tax liabilities.
We are subject to U.S. federal, state, local and sales taxes in the U.S. and foreign income taxes, withholding taxes and transaction taxes in foreign jurisdictions. Significant judgment is required in evaluating our tax positions and our worldwide provision for taxes. During the ordinary course of business, there are many activities and transactions for which the ultimate tax determination is uncertain. In addition, our tax obligations and effective tax rates could be adversely affected by changes in the relevant tax, accounting and other laws, regulations, principles and interpretations, including those relating to income tax nexus, by recognizing tax losses or lower than anticipated earnings in jurisdictions where we have lower statutory rates and higher than anticipated earnings in jurisdictions where we have higher statutory rates, by changes in foreign currency exchange rates, or by changes in the valuation of our deferred tax assets and liabilities. We may be audited in various jurisdictions, and such jurisdictions may assess additional taxes, sales taxes and value-added taxes against us. Although we believe our tax estimates are reasonable, the final determination of any tax audits or litigation could be materially different from our historical tax provisions and accruals, which could have a material adverse effect on our operating results or cash flows in the period for which a determination is made.
Negative conditions in the global credit markets and financial services and other industries may adversely affect our business.
The global credit markets, the financial services industry, the U.S. capital markets, and the U.S. economy as a whole have in the past experienced periods of substantial turmoil and uncertainty characterized by unprecedented intervention by the U.S. federal government and the failure, bankruptcy, or sale of various financial and other institutions. It is possible that a crisis in the global credit markets, the U.S. capital markets, the financial services industry or the U.S. economy may adversely affect our business, vendors and prospects, as well as our liquidity and financial condition. More specifically, our insurance carriers and insurance policies covering all aspects of our business may become financially unstable or may not be sufficient to cover any or all of our losses and may not continue to be available to us on acceptable terms, or at all.
47
We are dependent on information technology systems, infrastructure and data, which exposes us to data security risks.
We are dependent upon our own or third-party information technology systems, infrastructure and data, including mobile technologies, to operate our business. The multitude and complexity of our computer systems may make them vulnerable to service interruption or destruction, disruption of data integrity, malicious intrusion, or random attacks. Likewise, data privacy or security incidents or breaches by employees or others may pose a risk that sensitive data, including our intellectual property, trade secrets or personal information of our employees, patients, customers or other business partners may be exposed to unauthorized persons or to the public. Cyber-attacks are increasing in their frequency, sophistication and intensity. Cyber-attacks could include the deployment of harmful malware, denial-of-service, social engineering and other means to affect service reliability and threaten data confidentiality, integrity and availability. Our business partners face similar risks and any security breach of their systems could adversely affect our security posture. A security breach or privacy violation that leads to disclosure or modification of or prevents access to patient information, including personally identifiable information or protected health information, could harm our reputation, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, require us to verify the correctness of database contents and otherwise subject us to litigation or other liability under laws and regulations that protect personal data, any of which could disrupt our business and/or result in increased costs or loss of revenue. Moreover, the prevalent use of mobile devices that access confidential information increases the risk of data security breaches, which could lead to the loss of confidential information, trade secrets or other intellectual property. While we have invested, and continue to invest, in the protection of our data and information technology infrastructure, there can be no assurance that our efforts will prevent service interruptions, or identify breaches in our systems, that could adversely affect our business and operations and/or result in the loss of critical or sensitive information, which could result in financial, legal, business or reputational harm to us. In addition, our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyber-attacks and other related breaches.
Item 1B. Unresolved Staff Comments
Not applicable.
Item 2. Properties
As of February 20, 2019, we occupied the following properties:
| Property Description | Location | Square Footage | Owned or Leased | Initial Lease Term End Date | Lease Extension Options | |||||
| Ionis laboratory and office space facility | Carlsbad, CA | 176,000 | Owned | |||||||
| Ionis manufacturing facility | Carlsbad, CA | 28,700 | Owned | |||||||
| Ionis manufacturing support facility | Carlsbad, CA | 25,800 | Leased | 2021 | Two, five-year options to extend | |||||
| Akcea office space facility | Boston, MA | 30,175 | Leased | 2028 | One, five-year option to extend | |||||
| Akcea office and Ionis storage space facility | Carlsbad, CA | 18,700 | Leased | 2023 | One, five-year option to extend | |||||
| 279,375 |
Item 3. Legal Proceedings
Gilead Litigation
In August 2013, Gilead Sciences Inc. filed a suit in the U.S. District Court of Northern District of California related to U.S. Patent Nos. 7,105,499 and 8,481,712, which are jointly owned by Merck Sharp & Dohme Corp. and Ionis Pharmaceuticals, Inc. In the suit Gilead asked the court to determine that Gilead’s activities do not infringe any valid claim of the named patents and that the patents are not valid. We and Merck Sharp & Dohme Corp. filed our answer denying Gilead’s noninfringement and invalidity contentions, contending that Gilead’s commercial sale and offer for sale of sofosbuvir prior to the expiration of the ‘499 and ‘712 patents infringes those patents, and requesting monetary damages to compensate for such infringement. In the trial for this case held in March 2016, the jury upheld all 10 of the asserted claims of the patents-in-suit. The jury then decided that we and Merck are entitled to four percent of $5 billion in past sales of sofosbuvir. Gilead has stated it would appeal the jury’s finding of validity. In the meantime, Gilead asserted two additional non-jury defenses: waiver and unclean hands. Although the judge rejected the waiver defense, she granted Gilead’s motion claiming that the patents are unenforceable against it under the doctrine of unclean hands. We believe this ruling is contrary to the relevant law and the facts of the case. Accordingly, in July 2016, together with Merck we appealed the decision to the Court of Appeals for the Federal Circuit. Gilead cross-appealed on the issue of validity. In April 2018, the Court of Appeals issued its ruling affirming the District Court’s finding of unenforceability based on unclean hands. Having upheld the ruling that the patents are unenforceable against Gilead, the court did not reach the question of validity. In September 2018, we filed a petition requesting a hearing before the Supreme Court, asserting that it was improper for the trial court to overturn the jury verdict on the basis of the equitable defense of unclean hands. In January 2019, the Supreme Court denied our petition. Under our agreement with Merck, Merck is responsible for the costs of this suit.
48
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock is traded publicly through The Nasdaq Global Select Market under the symbol “IONS.” As of February 20, 2019, there were approximately 541 stockholders of record of our common stock. Because many of our shares are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of stockholders represented by these record holders.
We have never paid dividends and do not anticipate paying any dividends in the foreseeable future.
Set forth below is a table and chart comparing the total return on an indexed basis of $100 invested on December 31, 2013 in our common stock, the Nasdaq Composite Index (total return) and the Nasdaq Biotechnology Index. The total return assumes reinvestment of dividends.
Performance Graph (1)

* $100 invested on December 31, 2013 in stock or index, including reinvestment of dividends. Fiscal year ending December 31.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN
Among Ionis Pharmaceuticals, Inc., the Nasdaq Composite Index,
and the Nasdaq Biotechnology Index
| Dec-13 | Dec-14 | Dec-15 | Dec-16 | Dec-17 | Dec-18 | |||||||||||||||||||
| Ionis Pharmaceuticals, Inc. | $ | 100.00 | $ | 154.97 | $ | 155.45 | $ | 120.06 | $ | 126.26 | $ | 135.69 | ||||||||||||
| Nasdaq Composite Index | $ | 100.00 | $ | 114.62 | $ | 122.81 | $ | 133.19 | $ | 172.11 | $ | 165.84 | ||||||||||||
| Nasdaq Biotechnology Index | $ | 100.00 | $ | 131.71 | $ | 140.56 | $ | 112.25 | $ | 133.67 | $ | 121.24 | ||||||||||||
| (1) | This section is not “soliciting material,” is not deemed “filed” with the SEC, is not subject to the liabilities of Section 18 of the Exchange Act and is not to be incorporated by reference in any of our filings under the Securities Act or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. |
Item 6. Selected Financial Data
This selected financial data should be read in conjunction with our audited consolidated financial statements and accompanying notes and Management’s Discussion and Analysis of Financial Condition and Results of Operations included elsewhere in this Annual Report on Form 10-K. Our historical consolidated financial information may not be indicative of our future
performance. Set forth below are our selected consolidated financial data (in millions, except per share amounts):
49
| Years Ended December 31, | ||||||||||||||||||||
| 2018 | 2017 (1) | 2016 (1) | 2015 | 2014 | ||||||||||||||||
| Consolidated Statement of Operations Data: | ||||||||||||||||||||
| Revenue | $ | 599.7 | $ | 514.2 | $ | 372.8 | $ | 283.7 | $ | 214.2 | ||||||||||
| Research, development and patent expenses | $ | 414.6 | $ | 374.6 | $ | 344.3 | $ | 322.3 | $ | 241.8 | ||||||||||
| Selling, general and administrative expenses | $ | 244.6 | $ | 108.5 | $ | 48.6 | $ | 37.2 | $ | 20.1 | ||||||||||
| Net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | 273.7 | $ | 0.3 | $ | (60.4 | ) | $ | (88.3 | ) | $ | (39.0 | ) | |||||||
| Basic net income (loss) per share attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | 2.09 | $ | 0.15 | $ | (0.50 | ) | $ | (0.74 | ) | $ | (0.33 | ) | |||||||
| Diluted net income (loss) per share attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | 2.07 | $ | 0.15 | $ | (0.50 | ) | $ | (0.74 | ) | $ | (0.33 | ) | |||||||
| Shares used in computing basic net income (loss) per share | 132.3 | 124.0 | 120.9 | 119.7 | 117.7 | |||||||||||||||
| Shares used in computing diluted net income (loss) per share | 134.1 | 126.1 | 120.9 | 119.7 | 117.7 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2018 | 2017 (1) | 2016 | 2015 | 2014 | ||||||||||||||||
| Consolidated Balance Sheet Data: | ||||||||||||||||||||
| Cash, cash equivalents and short-term investments | $ | 2,084.1 | $ | 1,022.7 | $ | 665.2 | $ | 779.2 | $ | 728.8 | ||||||||||
| Working capital | $ | 1,927.6 | $ | 925.1 | $ | 664.1 | $ | 688.1 | $ | 721.3 | ||||||||||
| Total assets | $ | 2,667.8 | $ | 1,322.8 | $ | 912.5 | $ | 947.9 | $ | 946.5 | ||||||||||
| Long-term debt and other obligations, less current portion | $ | 1,200.3 | $ | 713.9 | $ | 679.1 | $ | 598.2 | $ | 588.9 | ||||||||||
| Accumulated deficit | $ | (967.3 | ) | $ | (1,241.0 | ) | $ | (1,181.4 | ) | $ | (1,094.9 | ) | $ | (1,006.6 | ) | |||||
| Stockholders’ equity | $ | 1,187.2 | $ | 365.3 | $ | 99.6 | $ | 200.8 | $ | 257.8 | ||||||||||
| (1) | Reflects the impact of our adoption of the new revenue recognition accounting standard in 2018 (Topic 606). For additional details about our adoption of Topic 606, see Note 1, Organization and Significant Accounting Policies, in the Notes to the Consolidated Financial Statements. This change is not reflected in our consolidated statement of operations data for 2015 or 2014 or in our consolidated balance sheet data for 2016, 2015, or 2014. |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
This financial review presents our operating results for each of the three years in the period ended December 31, 2018, and our financial condition at December 31, 2018. Except for the historical information contained herein, the following discussion contains forward-looking statements that are subject to known and unknown risks, uncertainties and other factors that may cause our actual results to differ materially from those expressed or implied by such forward-looking statements. We discuss such risks, uncertainties and other factors throughout this report and specifically under Item 1A of Part I of this report, “Risk Factors.” In addition, the following review should be read in conjunction with the information presented in our consolidated financial statements and the related notes to our consolidated financial statements as indexed on page F-1.
Overview
We are a leader in discovering and developing RNA-targeted therapeutics with sustained and growing revenues. We have created an efficient and broadly applicable drug discovery platform leveraging our expertise in antisense oligonucleotide therapeutics that we believe has fundamentally changed medicine and transformed the lives of people with devastating and often deadly diseases. Our large, diverse and advanced pipeline of over 40 first-in-class and/or best-in-class medicines addresses diseases across a broad range of therapeutic areas, targeting small, medium and large patient populations.
We have two commercial medicines approved in major markets around the world, SPINRAZA and TEGSEDI. We have at least four medicines that have entered pivotal studies or have the potential to begin pivotal studies this year, and another six medicines that could start pivotal studies in 2020. These medicines, along with the more than 30 additional medicines in our pipeline, represent multiple potential drivers of value for years to come. We believe our efficient drug discovery platform, coupled with our innovation-centric business model, provides us with the flexibility to determine the optimal development and commercialization strategy to maximize the commercial opportunity for each of our medicines and ensure that we continue to produce transformative medicines for patients who need them. We believe we are positioned to drive substantial value for patients and shareholders.
As of January 2019, SPINRAZA was approved in over 40 countries around the world, and our partner Biogen, who is responsible for global SPINRAZA commercial activities, reported that more than 6,600 patients are now on SPINRAZA therapy. In addition, Biogen plans to continue to pursue regulatory filings in additional countries. Biogen reported 2018 annual sales of SPINRAZA of more than $1.7 billion, and we earned $238 million in commercial revenues from royalties on sales of SPINRAZA. SPINRAZA is the first and only approved medicine for the treatment of SMA. SPINRAZA is the established standard-of-care for all people with this progressive, debilitating and often fatal genetic disease. In November 2018, SPINRAZA was recognized with the 2018 International Prix Galien award as Best Biotechnology Product. This prestigious honor marks the seventh Prix Galien award for SPINRAZA.
50
TEGSEDI, a once weekly, self-administered subcutaneous medicine, was approved in 2018 in the U.S., EU and Canada for the treatment of polyneuropathy caused by hATTR in adult patients. hATTR is a debilitating, progressive, and fatal disease. Akcea, our majority-owned affiliate focused on developing and commercializing medicines to treat patients with rare and serious diseases, launched TEGSEDI globally in late 2018. In the fourth quarter of 2018, we earned more than $2 million in TEGSEDI product sales. Akcea has an exclusive license agreement with PTC to commercialize TEGSEDI in Latin America. In January 2019, PTC filed an application for regulatory approval in Brazil with ANVISA, the Brazilian regulatory authority. ANVISA granted priority review for TEGSEDI.
We and Akcea are preparing to commercialize WAYLIVRA in the EU. The CHMP of the EMA adopted a positive opinion recommending conditional marketing authorization for WAYLIVRA as an adjunct to diet in adult patients with genetically confirmed FCS who are at high risk for pancreatitis, in whom response to diet and triglyceride lowering therapy has been inadequate. The positive opinion will now be referred to the EC which grants marketing authorization for medicines in the EU, as well as to European Economic Area members Iceland, Liechtenstein and Norway. With this positive opinion, and, pending adoption of the positive opinion by the EC, Akcea plans to leverage its existing commercial infrastructure in Europe to market WAYLIVRA. Akcea is continuing to conduct open-label extension and early access programs. We are also focused on regulatory discussions in the U.S. We are developing WAYLIVRA to treat FPL a second severe and rare, genetically defined disease. FCS and FPL are orphan diseases characterized by severely high triglyceride levels that result in severe, daily symptoms and a high risk of life-threatening pancreatitis.
In addition to commercializing TEGSEDI and preparing to commercialize WAYLIVRA, Akcea is developing four other clinical-stage medicines: AKCEA-APO(a)-LRx (TQJ230), AKCEA-ANGPTL3-LRx, AKCEA-APOCIII-LRx and AKCEA-TTR-LRx, each of which could potentially treat multiple patient populations. Moving these drugs into Akcea allows us to retain substantial value from these medicines and ensures our core focus remains on innovation. As of February 2019, we owned approximately 75 percent of Akcea.
We are continuously advancing our technology and pipeline to provide the most value to patients. We have a pipeline of over 40 medicines that, like SPINRAZA and TEGSEDI, have the potential to transform the treatment of diseases with no adequate treatment today. These medicines range from treatments for rare diseases with small patient populations to more common diseases afflicting millions of patients. Our pipeline covers a broad spectrum of therapeutic areas, such as cardiometabolic diseases, neurodegenerative diseases, cancer, severe and rare diseases and others. We believe our large and diverse pipeline contains many near-, mid- and longer-term growth drivers for the company.
Our pipeline includes at least 10 potentially transformative medicines anticipated to enter pivotal clinical studies in the next two years. We anticipate at least four of these medicines will enter pivotal studies this year including: AKCEA-APO(a)-LRx, AKCEA-TTR-LRx, IONIS-HTTRx (RG6042) and IONIS-SOD1Rx. Roche recently initiated a Phase 3 study of IONIS-HTTRx for HD. We believe each of these medicines is a first-in-class and/or best-in-class medicine with the potential to deliver significant value to patients and shareholders. We anticipate that the data from these pivotal studies, if positive, will support global regulatory filings for each medicine.
The depth of our knowledge and expertise with antisense technology together with our strong financial position provides us the flexibility to partner our medicines at what we believe is the optimal time to maximize the near-term, mid-term and long-term value of our medicines. We have a distinct partnering strategy based on each specific medicine and the expertise and resources we and our potential partners may bring to a collaboration. We may develop and commercialize some medicines through affiliates. In general, these are medicines, like TEGSEDI, that can benefit from our internal expertise and infrastructure, have manageable development costs and have the potential for initial rare disease indications. For other medicines, we may establish collaborations to advance the medicine. We have alliances with a cadre of leading global pharmaceutical companies that are working alongside us in developing our medicines, advancing our technology, preparing to commercialize our medicines and selling our medicines. Our partners include the following companies, among others: AstraZeneca, Bayer, Biogen, GSK, Janssen, Novartis and Roche. Our partners bring resources and expertise that augment and build upon our internal capabilities. We have the potential to earn over $20 billion in future milestone payments and licensing fees from our existing partnerships.
51
Financial Highlights
The following is a summary of our financial results (in thousands):
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised) | ||||||||||||
| Total revenue | $ | 599,674 | $ | 514,179 | $ | 372,776 | ||||||
| Total operating expenses | $ | 661,046 | $ | 483,132 | $ | 392,936 | ||||||
| Income (loss) from operations | $ | (61,372 | ) | $ | 31,047 | $ | (20,160 | ) | ||||
| Net income (loss) | $ | 214,985 | $ | (10,783 | ) | $ | (60,400 | ) | ||||
| Net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | 273,741 | $ | 346 | $ | (60,400 | ) | |||||
| Cash, cash equivalents and short-term investments | $ | 2,084,072 | $ | 1,022,715 | $ | 665,223 | ||||||
Our revenue for 2018 was $599.7 million and increased significantly compared to 2017 and 2016, primarily from increased commercial revenue from SPINRAZA royalties.
Our operating expenses for 2018 were $661.0 million and continued to increase year-over-year. The increase in operating expenses was primarily due to higher SG&A expenses as we prepared to commercialize TEGSEDI and WAYLIVRA. Our SG&A expenses also increased because of fees we owed under our in-licensing agreements related to SPINRAZA. We earn tiered royalties on annual SPINRAZA sales and pay nominal fixed third-party royalties that are not tiered. R&D expenses accounted for a smaller portion of the increase in operating expenses.
52
The increase in 2018 net income attributable to Ionis’ common stockholders was primarily due to increases in revenue and the income tax benefit we recognized in the fourth quarter of 2018. Our tax benefit increased significantly in 2018 primarily due to a one-time non-cash tax benefit related to Ionis’ stand-alone deferred federal income tax assets. In the fourth quarter of 2018, we released a large portion of our valuation allowance associated with our deferred tax assets. As a result of our strong financial performance over the past few years and our outlook regarding the continued growth of our business, we determined that it was more likely than not that we would be able to realize most of our deferred income tax assets we have accumulated to offset future taxable income.
During 2018 we received more than $1.5 billion in payments from our partners, including $1 billion from Biogen for our 2018 strategic neurology collaboration. This is compared to $580 million received in 2017 and $190 million received in 2016. We believe our strong financial position should enable us to continue to execute on our corporate goals throughout 2019 and beyond.
Business Segments
We have two operating segments, our Ionis Core segment and Akcea Therapeutics, our majority-owned affiliate. Akcea is a biopharmaceutical company focused on developing and commercializing medicines to treat patients with rare and serious diseases. We provide segment financial information and results for our Ionis Core segment and our Akcea Therapeutics segment based on the segregation of revenues and expenses that our chief decision maker reviews to assess operating performance and to make operating decisions. We allocate a portion of Ionis’ development, R&D support and general and administrative expenses to Akcea for work Ionis performs on behalf of Akcea.
Critical Accounting Policies
We prepare our consolidated financial statements in conformity with accounting principles generally accepted in the United States. As such, we make certain estimates, judgments and assumptions that we believe are reasonable, based upon the information available to us. These judgments involve making estimates about the effect of matters that are inherently uncertain and may significantly impact our quarterly or annual results of operations and financial condition. Each quarter, our senior management reviews the development, selection and disclosure of such estimates with the audit committee of our board of directors. In the following paragraphs, we describe the specific risks associated with these critical accounting policies and we caution that future events rarely develop exactly as one may expect, and that best estimates may require adjustment.
The following are our significant accounting policies, which we believe are the most critical to aid in fully understanding and evaluating our reported financial results:
| ● | Assessing the propriety of revenue recognition and associated deferred revenue; |
| ● | Valuing premiums received under our collaborations; |
| ● | Determining the proper valuation of investments in marketable securities; |
| ● | Determining the appropriate cost estimates for unbilled preclinical studies and clinical development activities; and |
| ● | ncome taxes. |
Descriptions of these critical accounting policies follow.
Revenue Recognition
Adoption of New Revenue Recognition Accounting Standard (Topic 606)
In May 2014, the FASB issued accounting guidance on the recognition of revenue from customers. This guidance supersedes the revenue recognition requirements we previously followed in Accounting Standards Codification, or ASC, Topic 605, Revenue Recognition, or Topic 605, and created a new Topic 606, Revenue from Contracts with Customers, or Topic 606. Under Topic 606, an entity will recognize revenue when it transfers control of promised goods or services to customers in an amount that reflects what the entity expects to receive in exchange for the goods or services. Further, an entity will recognize revenue upon satisfying the performance obligation(s) under the related contract. We adopted Topic 606 on January 1, 2018 under the full retrospective approach, which required us to revise our prior period revenue. Under Topic 606, we were required to review all of our ongoing collaboration agreements in which we recognized revenue after January 1, 2016. We were required to assess what our revenue would have been for the period from January 1, 2016 to December 31, 2017 under Topic 606. As a result of this analysis, we determined that the cumulative revenue we would have recognized under Topic 606 decreased by $86.1 million. We recorded this amount as a cumulative adjustment to our accumulated deficit as of January 1, 2016 on our revised statement of stockholders’ equity. We have labeled our prior period financial statements “as revised” to indicate the change required under the accounting rules.
The following tables summarize the adjustments we were required to make to amounts we originally reported in 2017 and 2016 to adopt Topic 606 (in thousands, except per share amounts):
Consolidated Balance Sheet
| At December 31, 2017 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Current portion of deferred contract revenue | $ | 106,465 | $ | 18,871 | $ | 125,336 | ||||||
| Long-term portion of deferred contract revenue | $ | 72,708 | $ | 35,318 | $ | 108,026 | ||||||
| Accumulated deficit | $ | (1,187,398 | ) | $ | (53,636 | ) | $ | (1,241,034 | ) | |||
| Noncontrolling interest in Akcea Therapeutics, Inc. | $ | 87,847 | $ | (3,580 | ) | $ | 84,267 | |||||
| Total stockholders’ equity | $ | 418,719 | $ | (53,439 | ) | $ | 365,280 | |||||
53
Consolidated Statements of Operations
| Year Ended December 31, 2017 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Revenue: | ||||||||||||
| Commercial revenue: | ||||||||||||
| SPINRAZA royalties | $ | 112,540 | $ | — | $ | 112,540 | ||||||
| Licensing and other royalty revenue | 9,519 | (2,045 | ) | 7,474 | ||||||||
| Total commercial revenue | 122,059 | (2,045 | ) | 120,014 | ||||||||
| Research and development revenue under collaborative agreements | 385,607 | 8,558 | 394,165 | |||||||||
| Total revenue | $ | 507,666 | $ | 6,513 | $ | 514,179 | ||||||
| Income from operations | $ | 24,534 | $ | 6,513 | $ | 31,047 | ||||||
| Net income (loss) | $ | (17,296 | ) | $ | 6,513 | $ | (10,783 | ) | ||||
| Net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | (5,970 | ) | $ | 6,316 | $ | 346 | |||||
| Net income per share, basic and diluted | $ | 0.08 | $ | 0.07 | $ | 0.15 | ||||||
| Year Ended December 31, 2016 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Revenue: | ||||||||||||
| Commercial revenue: | ||||||||||||
| SPINRAZA royalties | $ | 883 | $ | — | $ | 883 | ||||||
| Licensing and other royalty revenue | 19,839 | 2,045 | 21,884 | |||||||||
| Total commercial revenue | 20,722 | 2,045 | 22,767 | |||||||||
| Research and development revenue under collaborative agreements | 325,898 | 24,111 | 350,009 | |||||||||
| Total revenue | $ | 346,620 | $ | 26,156 | $ | 372,776 | ||||||
| Income (loss) from operations | $ | (46,316 | ) | $ | 26,156 | $ | (20,160 | ) | ||||
| Net income (loss) | $ | (86,556 | ) | $ | 26,156 | $ | (60,400 | ) | ||||
| Net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | (86,556 | ) | $ | 26,156 | $ | (60,400 | ) | ||||
| Net income (loss) per share, basic and diluted | $ | (0.72 | ) | $ | 0.22 | $ | (0.50 | ) | ||||
Consolidated Statements of Cash Flows
| Year Ended December 31, 2017 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Net income (loss) | $ | (17,296 | ) | $ | 6,513 | $ | (10,783 | ) | ||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | ||||||||||||
| Deferred contract revenue | $ | 36,695 | $ | (6,513 | ) | $ | 30,182 | |||||
| Cash and cash equivalents at beginning of period | $ | 84,685 | $ | — | $ | 84,685 | ||||||
| Cash and cash equivalents at end of period | $ | 129,630 | $ | — | $ | 129,630 | ||||||
| Year Ended December 31, 2016 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Net income (loss) | $ | (86,556 | ) | $ | 26,156 | $ | (60,400 | ) | ||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | ||||||||||||
| Deferred contract revenue | $ | (59,150 | ) | $ | (26,156 | ) | $ | (85,306 | ) | |||
| Cash and cash equivalents at beginning of period | $ | 128,797 | $ | — | $ | 128,797 | ||||||
| Cash and cash equivalents at end of period | $ | 84,685 | $ | — | $ | 84,685 | ||||||
Under Topic 606, compared to Topic 605, our total revenue increased $6.5 million for 2017 and $26.2 million for 2016. The change in our revenue was primarily due to:
| ● | A change in how we recognize milestone payments: Topic 606 requires us to amortize more of the milestone payments we achieve, rather than recognizing the milestone payments in full in the period in which we achieved the milestone event as we did under Topic 605. This change resulted in an increase in R&D revenue recognized for 2017 and 2016 of $23.6 million and $24.1 million, respectively. |
54
| ● | A change in how we calculate revenue for payments we are recognizing into revenue over time: Under Topic 605, we amortized payments into revenue evenly over the period of our obligations. When we made a change to our estimated completion period, we recognized that change on a prospective basis. Under Topic 606, we use an input method to determine the amount we amortize each reporting period. Each period, we review our “inputs” such as our level of effort expended, including the time we estimate it will take us to complete the activities, or costs incurred relative to the total expected inputs to satisfy the performance obligation. For certain collaborations, such as Bayer, Janssen and Novartis, the input method resulted in a change to the revenue we had previously recognized using a straight-line amortization method. This change resulted in a decrease in our R&D revenue of $15.1 million for 2017. This change did not result in an impact to our 2016 R&D revenue. |
Our updated revenue recognition policy reflecting Topic 606 is as follows:
Our Revenue Sources
We generally recognize revenue when we have satisfied all contractual obligations and are reasonably assured of collecting the resulting receivable. We are often entitled to bill our customers and receive payment from our customers in advance of recognizing the revenue. In the instances in which we have received payment from our customers in advance of recognizing revenue, we include the amounts in deferred revenue on our consolidated balance sheet.
Commercial Revenue: SPINRAZA royalties and Licensing and other royalty revenue
We earn commercial revenue primarily in the form of royalty payments on net sales of SPINRAZA. We will also recognize as commercial revenue future sales milestone payments and royalties we earn under our partnerships.
Commercial Revenue: TEGSEDI product sales, net
We began adding product sales from TEGSEDI to our commercial revenue in the fourth quarter of 2018. In the U.S., TEGSEDI is distributed through an exclusive distribution agreement with a third-party logistics company, or 3PL, that takes title to TEGSEDI. The 3PL is our sole customer in the U.S. The 3PL then distributes TEGSEDI to a specialty pharmacy and a specialty distributor, which we collectively refer to as wholesalers, who then distribute TEGSEDI to health care providers and patients. In Germany, TEGSEDI is distributed through a non-exclusive distribution model with a 3PL that takes title to TEGSEDI. The 3PL is our sole customer in Germany. The 3PL in Germany then distributes TEGSEDI to hospitals and pharmacies.
Research and development revenue under collaborative agreements
We often enter into collaboration agreements to license and sell our technology on an exclusive or non-exclusive basis. Our collaboration agreements typically contain multiple elements, or performance obligations, including technology licenses or options to obtain technology licenses, research and development, or R&D, services, and manufacturing services.
Our collaboration agreements are detailed in Note 6, Collaborative Arrangements and Licensing Agreements. Under each collaboration note we discuss our specific revenue recognition conclusions, including our significant performance obligations under each collaboration.
Steps to Recognize Revenue
We use a five step process to determine the amount of revenue we should recognize and when we should recognize it. The five step process is as follows:
| 1. | Identify the contract |
Accounting rules require us to first determine if we have a contract with our partner, including confirming that we have met each of the following criteria:
| ● | We and our partner approved the contract and we are both committed to perform our obligations; |
| ● | We have identified our rights, our partner’s rights and the payment terms; |
| ● | We have concluded that the contract has commercial substance, meaning that the risk, timing, or amount of our future cash flows is expected to change as a result of the contract; and |
| ● | We believe collectability is probable. |
| 2. | Identify the performance obligations |
We next identify the distinct goods and services we are required to provide under the contract. Accounting rules refer to these as our performance obligations. We typically have only one performance obligation at the inception of a contract, which is to perform R&D services.
55
Often times we enter into a collaboration agreement in which we provide our partner with an option to license a medicine in the future. We may also provide our partner with an option to request that we provide additional goods or services in the future, such as active pharmaceutical ingredient, or API. We evaluate whether these options are material rights at the inception of the agreement. If we determine an option is a material right, we will consider the option a separate performance obligation. Historically, we have concluded that the options we grant to license a medicine in the future or to provide additional goods and services as requested by our partner are not material rights. These items are contingent upon future events that may not occur. When a partner exercises its option to license a medicine or requests additional goods or services, then we identify a new performance obligation for that item.
In some cases, we deliver a license at the start of an agreement. If we determine that our partner has full use of the license and we do not have any additional performance obligations related to the license after delivery, then we consider the license to be a separate performance obligation.
| 3. | Determine the transaction price |
We then determine the transaction price by reviewing the amount of consideration we are eligible to earn under the collaboration agreement, including any variable consideration. Under our collaboration agreements, consideration typically includes fixed consideration in the form of an upfront payment and variable consideration in the form of potential milestone payments, license fees and royalties. At the start of an agreement, our transaction price usually consists of only the upfront payment. We do not typically include any payments we may receive in the future in our initial transaction price because the payments are not probable. We reassess the total transaction price at each reporting period to determine if we should include additional payments in the transaction price.
Milestone payments are our most common type of variable consideration. We recognize milestone payments using the most likely amount method because we will either receive the milestone payment or we will not, which makes the potential milestone payment a binary event. The most likely amount method requires us to determine the likelihood of earning the milestone payment. We include a milestone payment in the transaction price once it is probable we will achieve the milestone event. Most often, we do not consider our milestone payments probable until we or our partner achieve the milestone event because the majority of our milestone payments are contingent upon events that are not within our control and are usually based on scientific progress. For example, in January 2019 we earned a $35 million milestone payment from Roche when it dosed the first patient in the Phase 3 study of IONIS-HTTRx. At December 31, 2018, we determined it was not probable that we could earn this milestone payment. As such, we did not recognize any revenue associated with it in 2018.
| 4. | Allocate the transaction price |
Next, we allocate the transaction price to each of our performance obligations. When we have to allocate the transaction price to more than one performance obligation, we make estimates of the relative stand-alone selling price of each performance obligation because we do not typically sell our goods or services on a stand-alone basis. We then allocate the transaction price to each performance obligation based on the relative stand-alone selling price.
We may engage a third party, independent valuation specialist to assist us with determining a stand-alone selling price for collaborations in which we deliver a license at the start of an agreement. We estimate the stand-alone selling price of these licenses using valuation methodologies, such as the relief from royalty method. Under this method, we estimate the amount of income, net of taxes, for the license. We then discount the projected income to present value. The significant inputs we use to determine the projected income of a license could include:
| ● | Estimated future product sales; |
| ● | Estimated royalties on future product sales; |
| ● | Contractual milestone payments; |
| ● | Expenses we expect to incur; |
| ● | Income taxes; and |
| ● | A discount rate. |
We typically estimate the selling price of R&D services by using our internal estimates of the cost to perform the specific services. The significant inputs we use to determine the selling price of our R&D services include:
| ● | The number of internal hours we estimate we will spend performing these services; |
| ● | The estimated cost of work we will perform; |
| ● | The estimated cost of work that we will contract with third parties to perform; and |
| ● | The estimated cost of API we will use. |
For purposes of determining the stand-alone selling price of the R&D services we perform and the API we will deliver, accounting guidance requires us to include a markup for a reasonable profit margin.
We do not reallocate the transaction price after the start of an agreement to reflect subsequent changes in stand-alone selling prices.
| 5. | Recognize revenue |
We recognize revenue in one of two ways, over time or at a point in time. We recognize revenue over time when we are executing on our performance obligation over time and our partner receives benefit over time. For example, we recognize revenue over time when we provide R&D services. We recognize revenue at a point in time when our partner receives full use of an item at a specific point in time. For example, we recognize revenue at a point in time when we deliver a license or API to a partner.
56
For R&D services that we recognize over time, we measure our progress using an input method. The input methods we use are based on the effort we expend or costs we incur toward the satisfaction of our performance obligation. We estimate the amount of effort we expend, including the time we estimate it will take us to complete the activities, or costs we incur in a given period, relative to the estimated total effort or costs to satisfy the performance obligation. This results in a percentage that we multiply by the transaction price to determine the amount of revenue we recognize each period. This approach requires us to make numerous estimates and use significant judgement. If our estimates or judgements change over the course of the collaboration, they may affect the timing and amount of revenue that we recognize in the current and future periods.
The following are examples of when we typically recognize revenue based on the types of payments we receive.
Commercial Revenue: SPINRAZA royalties and Licensing and other royalty revenue
We recognize royalty revenue in the period in which the counterparty sells the related product, which in certain cases may require us to estimate our royalty revenue. We recognize royalties from SPINRAZA sales in the period Biogen records the sale of SPINRAZA. Our accounting for SPINRAZA royalties did not change as a result of adopting Topic 606.
Commercial Revenue: TEGSEDI Product Sales, net
We recognize TEGSEDI product sales in the period when our customer obtains control of TEGSEDI, which occurs at a point in time upon transfer of title to the customer. We classify payments to customers or other parties in the distribution channel for services that are distinct and priced at fair value as selling, general and administrative expenses in our consolidated statements of operations. Otherwise payments to customers or other parties in the distribution channel that do not meet those criteria are classified as a reduction of revenue, as discussed further below. We exclude from revenues, taxes collected from customers relating to product sales and remitted to governmental authorities.
Reserves for TEGSEDI Product Sales
We record TEGSEDI product sales at our net sales price, or transaction price. We include in our transaction price estimated reserves for discounts, returns, chargebacks, rebates, co-pay assistance and other allowances that we offer within contracts between us and our customers, wholesalers, health care providers and other indirect customers. We estimate our reserves using the amounts we have earned or what we can claim on the associated sales. We classify our reserves as reductions of accounts receivable when the amount is payable to our customer or a current liability when the amount is payable to a party other than our customer in our consolidated balance sheet. In certain cases, our estimates include a range of possible outcomes that are probability-weighted for relevant factors such as our historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. Overall, our reserves reflect our best estimates under the terms of our respective contracts. When calculating our reserves and related product sales, we only recognize amounts to the extent that we consider it probable that we would not have to reverse in a future period a significant amount of the cumulative sales we previously recognized. The actual amounts we receive may ultimately differ from our reserve estimates. If actual amounts in the future vary from our estimates, we will adjust these estimates, which would affect our net TEGSEDI product sales in the respective period.
The following are the components of variable consideration related to TEGSEDI product sales:
Chargebacks: In the U.S., we estimate obligations resulting from contractual commitments with the government and other entities to sell products to qualified healthcare providers at prices lower than the list prices charged to our U.S. customer. Our U.S. customer charges us for the difference between what it pays for the product and the selling price to the qualified healthcare providers. We record reserves for these chargebacks related to TEGSEDI product sales to our U.S. customer during the reporting period. We also estimate the amount of product remaining in the distribution channel inventory at the end of the reporting period that we expect our customer to sell to wholesalers in future periods.
Government rebates: We are subject to discount obligations under government programs, including Medicaid programs and Medicare in the U.S. We estimate Medicaid and Medicare rebates based on a range of possible outcomes that are probability-weighted for the estimated payer mix. We record these reserves as an accrued liability on our consolidated balance sheet with a corresponding offset reducing our TEGSEDI product sales in the same period we recognize the related sale. For Medicare rebates, we also estimate the number of patients in the prescription drug coverage gap for whom we will owe an additional liability under the Medicare Part D program. On a quarterly basis, we update our estimates and record any adjustments in the period that we identify the adjustments. In Germany, pharmaceutical companies must grant a specified rebate percentage to the German government. We include this rebate in the same period we recognize the related TEGSEDI product sales, resulting in a reduction of product sales.
Trade discounts and allowances: We provide customary invoice discounts on TEGSEDI product sales to our U.S. customer for prompt payment. We record this discount as a reduction of TEGSEDI product sales in the period in which we recognize the related product revenue. In addition, we receive and pay for various distribution services from our U.S. customer and wholesalers in our U.S. distribution channel. For services we receive that are either not distinct from the sale of TEGSEDI or for which we cannot reasonably estimate the fair value, we classify such fees as a reduction of TEGSEDI product sales.
Product Returns: Our U.S. customer has return rights and the wholesalers have limited return rights primarily related to the expiration date of the TEGSEDI product. We estimate the amount of TEGSEDI product sales that our customer may return. We record our return estimate as an accrued refund liability on our consolidated balance sheet with a corresponding offset reducing our TEGSEDI product sales, in the same period we recognize the related sale. Based on our distribution model for TEGSEDI, contractual inventory limits with our customer and wholesalers and the price of TEGSEDI, we believe we will have minimal returns. Our customer in Germany only takes title to the product once it receives an order from a hospital or pharmacy and therefore does not maintain any inventory of TEGSEDI, as such we do not estimate returns in Germany.
57
Other incentives: In the U.S., we estimate reserves for other incentives including co-payment assistance we provide to patients with commercial insurance who have coverage and reside in states that allow co-payment assistance. We record a reserve for the amount we estimate we will pay for co-payment assistance. We base our reserve on the number of estimated claims and our estimate of the cost per claim related to TEGSEDI product sales that we have recognized as revenue. We record our other incentive reserve estimates as an accrued liability on our consolidated balance sheet with a corresponding offset reducing our TEGSEDI product sales, in the same period we recognize the related sale.
Research and development revenue under collaboration agreements:
Upfront Payments
When we enter into a collaboration agreement with an upfront payment, we typically record the entire upfront payment as deferred revenue if our only performance obligation is for R&D services we will provide in the future. We amortize the upfront payment into revenue as we perform the R&D services. For example, under our new collaboration agreement with Roche to develop IONIS-FB-LRx for the treatment of complement-mediated diseases, we received a $75 million upfront payment in the fourth quarter of 2018. We allocated the upfront payment to our single performance obligation, R&D services. We are amortizing the $75 million upfront payment using an input method over the estimated period of time we are providing R&D services. Refer to Note 6, Collaborative Arrangements and Licensing Agreements, for further discussion. Under Topic 605, we amortized upfront payments evenly over the period of our obligation.
Milestone Payments
We are required to include additional consideration in the transaction price when it is probable. We typically include milestone payments for R&D services in the transaction price when they are achieved. We include these milestone payments when they are achieved because there is considerable uncertainty in the research and development processes that trigger these payments under our collaboration agreements. Similarly, we include approval milestone payments in the transaction price once the medicine is approved by the applicable regulatory agency. We will recognize sales based milestone payments in the period we achieve the milestone under the sales-based royalty exception allowed under accounting rules.
We recognize milestone payments that relate to an ongoing performance obligation over our period of performance. For example, in the third quarter of 2017, we initiated a Phase 1/2a clinical study of IONIS-MAPTRx in patients with mild Alzheimer’s disease. We earned a $10 million milestone payment from Biogen related to the initiation of this study. Under Topic 606, we added this payment to the transaction price and allocated it to our R&D services performance obligation. We are recognizing revenue from this milestone payment over our estimated period of performance. Under Topic 605, this milestone payment was recognized in full in the third quarter of 2017, which was the period in which we achieved the milestone event.
Conversely, we recognize in full those milestone payments that we earn based on our partners’ activities when our partner achieves the milestone event. For example, in the third quarter of 2018, we recognized a $10 million milestone payment when AstraZeneca initiated a Phase 1 study of IONIS-AZ4-2.5-LRx. We concluded that the milestone payment was not related to our R&D services performance obligation. Therefore, we recognized this milestone payment in full in the third quarter of 2018 because we do not have any performance obligations related to this milestone payment. Our revenue recognition of milestone payments we earn based on our partners’ activities did not change as a result of adopting Topic 606.
License Fees
We generally recognize as revenue the total amount we determine to be the stand-alone selling price of a license when we deliver the license to our partner. This is because our partner has full use of the license and we do not have any additional performance obligations related to the license after delivery. For example, in the fourth quarter of 2018, we earned a $35 million license fee when Biogen licensed IONIS-SOD1Rx from us. Our recognition of license fees did not change as a result of adopting Topic 606.
Amendments to Agreements
From time to time we amend our collaboration agreements. When this occurs, we are required to assess the following items to determine the accounting for the amendment:
| 1) | If the additional goods and/or services are distinct from the other performance obligations in the original agreement; and |
| 2) | If the goods and/or services are at a stand-alone selling price. |
If we conclude the goods and/or services in the amendment are distinct from the performance obligations in the original agreement and at a stand-alone selling price, we account for the amendment as a separate agreement. If we conclude the goods and/or services are not distinct and at their stand-alone selling price, we then assess whether the remaining goods or services are distinct from those already provided. If the goods and/or services are distinct from what we have already provided, then we allocate the remaining transaction price from the original agreement and the additional transaction price from the amendment to the remaining goods and/or services. If the goods and/or services are not distinct from what we have already provided, we update the transaction price for our single performance obligation and recognize any change in our estimated revenue as a cumulative adjustment.
58
For example, in May 2015, we entered into an exclusive license agreement with Bayer to develop and commercialize IONIS-FXIRx for the prevention of thrombosis. As part of the agreement, Bayer paid us a $100 million upfront payment. At the onset of the agreement, we were responsible for completing a Phase 2 study of IONIS-FXIRx in people with end-stage renal disease on hemodialysis and for providing an initial supply of API. In February 2017, we amended our agreement with Bayer to advance IONIS-FXIRx and to initiate development of IONIS-FXI-LRx, which Bayer licensed. As part of the 2017 amendment, Bayer paid us $75 million. We are also eligible to receive milestone payments and tiered royalties on gross margins of IONIS-FXIRx and IONIS-FXI-LRx. Under the 2017 amendment, we concluded we had a new agreement with three performance obligations. These performance obligations were to deliver the license of IONIS-FXI-LRx, to provide R&D services and to deliver API. We allocated the $75 million transaction price to these performance obligations. Refer to Note 6, Collaborative Arrangements and Licensing Agreements, for further discussion of our accounting treatment for our Bayer collaboration. Our allocation of the consideration we received for the Bayer amendment did not change as a result of adopting Topic 606. However, the method in which we are recognizing revenue related to our R&D services performance obligation did change. We are amortizing revenue related to our R&D services performance obligation using the input method under Topic 606.
Multiple Agreements
From time to time, we may enter into separate agreements at or near the same time with the same partner. We evaluate such agreements to determine whether we should account for them individually as distinct arrangements or whether the separate agreements should be combined and accounted for together. We evaluate the following to determine the accounting for the agreements:
| ● | Whether the agreements were negotiated together with a single objective; |
| ● | Whether the amount of consideration in one contract depends on the price or performance of the other agreement; or |
| ● | Whether the goods and/or services promised under the agreements are a single performance obligation. |
Our evaluation involves significant judgment to determine whether a group of agreements might be so closely related that accounting guidance requires us to account for them as a combined arrangement.
For example, in the second quarter of 2018, we entered into two separate agreements with Biogen at the same time: a new strategic neurology collaboration agreement and a SPA. We evaluated the Biogen agreements to determine whether we should treat the agreements separately or combine them. We considered that the agreements were negotiated concurrently and in contemplation of one another. Based on these facts and circumstances, we concluded that we should evaluate the provisions of the agreements on a combined basis. Refer to Note 6, Collaborative Arrangements and Licensing Agreements for further discussion of the accounting treatment for the 2018 strategic neurology collaboration with Biogen.
Deferred Revenue
We are often entitled to bill our customers and receive payment from our customers in advance of our obligation to provide services or transfer goods to our partners. In these instances, we include the amounts in deferred revenue on our consolidated balance sheet.
The following table summarizes the adjustments we were required to make to our deferred revenue amounts to adopt Topic 606 (in thousands):
| At December 31, 2017 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Current portion of deferred revenue | $ | 106,465 | $ | 18,871 | $ | 125,336 | ||||||
| Long-term portion of deferred revenue | 72,708 | 35,318 | 108,026 | |||||||||
| Total deferred revenue | $ | 179,173 | $ | 54,189 | $ | 233,362 | ||||||
Our deferred revenue balance increased $54.2 million at December 31, 2017 under Topic 606, compared to Topic 605. The increase was primarily related to the change in the accounting for certain milestone payments and the way in which we amortize payments. Under Topic 605, we previously recognized the majority of the milestone payments we earned in the period we achieved the milestone event, which did not impact our deferred revenue balance. Under Topic 606 we are now amortizing more milestone payments over the period of our performance obligation, which adds to our deferred revenue balance. Additionally, under Topic 605 we amortized payments evenly over the period of our obligation. Under Topic 606, we use an input method to determine the amount we amortize each reporting period. The increase in deferred revenue relates to agreements with the following partners:
| ● | $24.2 million from Biogen; |
| ● | $15.9 million from AstraZeneca; |
| ● | $11.8 million from Novartis; and |
| ● | $ 2.3 million from other partners. |
Valuation of Premiums under our Collaborations
In conjunction with our collaboration agreements we have sold stock at a premium to our partners, including under our 2018 strategic neurology collaboration with Biogen and with Novartis in 2017. See further discussion about our valuation of the potential premium in our Fair Value Measurements policy in Note 1, Organization and Significant Accounting Policies, in the Notes to the Consolidated Financial Statements.
59
Biogen Premium
In the second quarter of 2018, we received $1 billion from Biogen, comprised of $625 million to purchase our stock at a 25 percent cash premium and $375 million in an upfront payment. At the commencement of this collaboration, we identified one performance obligation, which was to perform R&D services for Biogen. We determined the transaction price to be $552 million, comprised of $375 million from the upfront payment and $177 million for the premium paid by Biogen for its purchase of our common stock. We determined the fair value of the premium we received by using the stated premium in the SPA and applying a lack of marketability discount. We included a lack of marketability discount in our valuation of the premium because Biogen received restricted shares.
Novartis Premiums
During the first quarter of 2017, we valued the premiums under the SPA agreement with Novartis. These premiums included the premium Novartis paid us related to its $100 million purchase of our stock in the first quarter of 2017 and the premium we could have received related to Novartis’ potential purchase of our stock. These valuations required us to use level 3 inputs, which we consider to be a critical accounting policy for our results for 2017.
We determined the fair value of the premium we received and the future premium we could have received by using the stated premium in the SPA and applying a lack of marketability discount. We included a lack of marketability discount in our valuation of the premiums because Novartis received unregistered shares as part of Novartis’ $100 million equity purchase and we would have issued unregistered shares to Novartis if it had purchased our common stock. Additionally, for the future potential stock purchase, we estimated the probability of an Akcea IPO. At the inception of the agreements, we calculated the following fair values:
| ● | $28.4 million for the premium paid by Novartis for its purchase of our common stock in the first quarter of 2017; and |
| ● | $5.0 million for the potential premium Novartis would have paid if it had purchased our common stock in the future at a premium. |
Because Akcea completed its IPO before April 2018, Novartis will not purchase additional shares of Ionis stock. Therefore, this asset no longer had any value and we wrote-off the remaining potential premium Novartis would have paid to us if an Akcea IPO did not occur. We wrote off the amount to other expenses on our consolidated statement of operations during the third quarter of 2017.
Valuation of Investments in Marketable Securities
We consider all liquid investments with maturities of three months or less when we purchase them to be cash equivalents. Our short-term investments have initial maturities of greater than three months from date of purchase. We classify our short-term investments as “available-for-sale” and carry them at fair market value based upon prices for identical or similar items on the last day of the fiscal period. We record unrealized gains and losses as a separate component of comprehensive income (loss) and include net realized gains and losses in gain (loss) on investments. We use the specific identification method to determine the cost of securities sold.
We use a three-tier fair value hierarchy to prioritize the inputs used in our fair value measurements. These tiers include: Level 1, defined as observable inputs such as quoted prices in active markets for identical assets, which includes our money market funds and treasury securities classified as available-for-sale securities and our investment in equity securities in publicly held biotechnology companies; Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable, which includes our fixed income securities and commercial paper classified as available-for-sale securities; and Level 3, defined as unobservable inputs in which little or no market data exists, therefore requiring us to develop our own assumptions. We classify the majority of our securities as Level 2. We obtain the fair value of our Level 2 investments from our custodian bank or from a professional pricing service. We validate the fair value of our Level 2 investments by understanding the pricing model used by the custodian banks or professional pricing service provider and comparing that fair value to the fair value based on observable market prices.
Estimated Liability for Clinical Development Costs
We record accrued liabilities related to expenses for which service providers have not yet billed us. These liabilities are for products or services that we have received, specifically related to ongoing preclinical studies and clinical trials. These costs primarily relate to third-party clinical management costs, laboratory and analysis costs, toxicology studies and investigator grants. We have numerous medicines in concurrent preclinical studies and clinical trials at several clinical sites throughout the world. In order to ensure that we have adequately provided for ongoing preclinical and clinical development costs during the period in which we incur such costs, we maintain an accrual to cover these costs. We update our estimate for this accrual on at least a quarterly basis. The assessment of these costs is a subjective process that requires judgment. Upon settlement, these costs may differ materially from the amounts accrued in our consolidated financial statements. Our historical accrual estimates have not been materially different from our actual amounts.
60
Income Taxes
We account for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in our financial statements or tax returns. In addition, deferred tax assets are recorded for the future benefit of utilizing net operating losses and research and development credit carryforwards. Valuation allowances are provided when necessary to reduce deferred tax assets to the amount expected to be realized.
We apply the authoritative accounting guidance prescribing a threshold and measurement attribute for the financial recognition and measurement of a tax position taken or expected to be taken in a tax return. We recognize liabilities for uncertain tax positions based on a two-step process. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step requires us to estimate and measure the tax benefit as the largest amount that is more than 50 percent likely to be realized upon ultimate settlement.
We are required to use significant judgment in evaluating our uncertain tax positions and determining our provision for income taxes. Although we believe our reserves are reasonable, no assurance can be given that the final tax outcome of these matters will not be different from that which is reflected in our historical income tax provisions and accruals. We adjust these reserves for changing facts and circumstances, such as the closing of a tax audit or the refinement of an estimate. To the extent that the final tax outcome of these matters is different than the amounts recorded, such differences may impact the provision for income taxes in the period in which such determination is made.
We are also required to use significant judgment in determining any valuation allowance recorded against our deferred tax assets. In assessing the need for a valuation allowance, we consider all available evidence, including scheduled reversal of deferred tax liabilities, past operating results, the feasibility of tax planning strategies and estimates of future taxable income. Estimates of future taxable income are based on assumptions that are consistent with our plans. The assumptions we use represent our best estimates and involve inherent uncertainties and the application of our judgment. Should actual amounts differ from our estimates, the amount of our tax expense and liabilities we recognize could be materially impacted. We record a valuation allowance to reduce the balance of our net deferred tax assets to the amount we believe is more-likely-than-not to be realized.
For U.S. federal income tax purposes we are required to file separate U.S. federal income tax returns for Ionis and Akcea. We began deconsolidating Akcea for U.S. federal income tax purposes upon Akcea’s initial public offering. As a result, we are required to assess Ionis’ stand-alone and Akcea’s valuation allowances separately even though we consolidate Akcea’s financial results in our consolidated financial statements. We continue to file combined state tax returns in most jurisdictions. As a result, we continue to assess the state portion of our valuation allowance for those jurisdictions on a consolidated basis.
We have historically recorded a valuation allowance against all our net deferred tax assets due to cumulative financial statement losses. However, in the fourth quarter of 2018, we reversed the valuation allowance previously recorded against Ionis’ stand-alone U.S. federal net deferred tax assets, resulting in a one-time non-cash tax benefit of $332.1 million. Given our current stand-alone Ionis pre-tax income, and assuming we maintain this current level of Ionis stand-alone pre-tax income, we expect to generate income before taxes in the U.S. in future periods at a level that would result in us fully utilizing our U.S. federal net operating loss carryforwards and most of our existing Research and Development and Orphan Drug tax credit carryforwards over the next three years.
We continue to maintain a full valuation allowance of $234.2 million against all of Akcea’s net deferred tax assets and the net state deferred tax assets of Ionis at December 31, 2018 due to uncertainties related to our ability to realize the tax benefits associated with these assets.
We evaluate our deferred tax assets regularly to determine whether adjustments to the valuation allowance are appropriate due to changes in facts or circumstances, such as changes in expected future pre-tax earnings, tax law, interactions with taxing authorities and developments in case law. In making this evaluation, we rely on our recent history of pre-tax earnings. Our material assumptions are our forecasts of future pre-tax earnings and the nature and timing of future deductions and income represented by the deferred tax assets and liabilities, all of which involve the exercise of significant judgment. Although we believe our estimates are reasonable, we are required to use significant judgment in determining the appropriate amount of valuation allowance recorded against our deferred tax assets.
We do not provide for a U.S. income tax liability and foreign withholding taxes on undistributed foreign earnings of our foreign subsidiaries.
Results of Operations
Whenever we refer to prior period results, they reflect the impact of Topic 606, which we adopted in the first quarter of 2018.
Years Ended December 31, 2018 and December 31, 2017
61
Revenue
Total revenue for 2018 was $599.7 million, compared to $514.2 million in 2017 and was comprised of the following (amounts in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Revenue: | (as revised) | |||||||
| Commercial revenue: | ||||||||
| SPINRAZA royalties | $ | 237,930 | $ | 112,540 | ||||
| TEGSEDI product sales, net | 2,237 | — | ||||||
| Licensing and other royalty revenue | 14,755 | 7,474 | ||||||
| Total commercial revenue | 254,922 | 120,014 | ||||||
| R&D revenue: | ||||||||
| Amortization from upfront payments | 124,695 | 97,646 | ||||||
| Milestone payments | 82,771 | 152,008 | ||||||
| License fees | 102,053 | 116,095 | ||||||
| Other services | 35,233 | 28,416 | ||||||
| Total R&D revenue | 344,752 | 394,165 | ||||||
| Total revenue | $ | 599,674 | $ | 514,179 | ||||
The increase in revenue in 2018 compared to 2017 was primarily due to increasing commercial revenue from SPINRAZA royalties, which more than doubled. We added TEGSEDI product sales in the fourth quarter of 2018. Additionally, we more than doubled our licensing and royalty revenue in 2018 compared to 2017, primarily from the license fee we earned from PTC Therapeutics to commercialize TEGSEDI and WAYLIVRA in Latin America.
R&D revenue from the amortization of upfront payments increased over $25 million in 2018 compared to 2017. The increase in amortization was primarily due to our 2018 strategic neurology collaboration with Biogen. Additionally, we added amortization revenue from our new collaboration with Roche to develop IONIS-FB-LRx in 2018. Our R&D revenue from milestone payments, license fees and other services continued to make a significant contribution to our financial results.
Already in the first quarter of 2019, we have earned $185 million. We earned $150 million from Novartis when it licensed AKCEA-APO(a)-LRx and $35 million from Roche when it dosed the first patient in the Phase 3 study of IONIS-HTTRx in patients with Huntington’s disease.
Operating Expenses
Operating expenses for 2018 were $661.0 million, and increased compared to $483.1 million for 2017. Our operating expenses increased year over year principally due to higher SG&A expenses as we prepared to commercialize TEGSEDI and WAYLIVRA. Our SG&A expenses also increased year over year because of fees we owed under our in-licensing agreements related to SPINRAZA. We earn tiered royalties on annual SPINRAZA sales and pay nominal fixed third-party royalties that are not tiered. R&D expenses accounted for a smaller portion of the increase in operating expenses. R&D expenses for 2018 increased compared to 2017 primarily due to increases in drug development costs related to several medicines, including AKCEA-APOCIII-LRx, as we, with Akcea, advanced these programs in development. These increases reflect the investment we are making in advancing and expanding our pipeline.
Our operating expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Ionis Core | $ | 293,175 | $ | 305,352 | ||||
| Akcea Therapeutics | 251,408 | 146,332 | ||||||
| Elimination of intercompany activity | (14,849 | ) | (54,527 | ) | ||||
| Subtotal | 529,734 | 397,157 | ||||||
| Non-cash compensation expense related to equity awards | 131,312 | 85,975 | ||||||
| Total operating expenses | $ | 661,046 | $ | 483,132 | ||||
In order to analyze and compare our results of operations to other similar companies, we believe it is important to exclude non-cash compensation expense related to equity awards from our operating expenses. We believe non-cash compensation expense is not indicative of our operating results or cash flows from our operations. Further, we internally evaluate the performance of our operations excluding it.
62
Cost of Products Sold
Our cost of products sold consisted of manufacturing costs, including certain fixed costs, transportation and freight, indirect overhead costs associated with the manufacturing and distribution of TEGSEDI, certain associated period costs. We not expect our fixed costs will increase in direct correlation to TEGSEDI product sales. We expensed a significant portion of the cost of producing TEGSEDI that Akcea is using in the commercial launch as R&D expense prior to the regulatory approval of TEGSEDI. We expect cost of products sold to increase as we deplete these inventories.
Our cost of products sold by segment were as follows (in thousands):
Year Ended December 31, | ||||
| 2018 | ||||
| Ionis Core | $ | — | ||
| Akcea Therapeutics | 11,573 | |||
| Elimination of intercompany activity | (9,913 | ) | ||
| Subtotal | 1,660 | |||
| Non-cash compensation expense related to equity awards | 160 | |||
| Total cost of products sold | $ | 1,820 | ||
For 2018, our cost of products sold was $1.7 million. We began recognizing cost of products sold in 2018 when TEGSEDI was approved. We previously recognized $0.1 million of costs to produce TEGSEDI related to the TEGSEDI commercial revenue we recognized in 2018 because we incurred these costs before we obtained regulatory approval. We did not have cost of products sold in 2017. Akcea includes the amortization for milestone payments it made to us related to the U.S. and European approvals of TEGSEDI in its cost of products sold. Akcea is recognizing this amortization over TEGSEDI’s remaining estimated patent life. This amortization is eliminated in our consolidated results. All amounts exclude non-cash compensation expense related to equity awards.
Research, Development and Patent Expenses
Our research, development and patent expenses consist of expenses for antisense drug discovery, antisense drug development, manufacturing and operations and R&D support expenses.
The following table sets forth information on research, development and patent expenses (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Research, development and patent expenses, excluding non-cash compensation expense related to equity awards | $ | 338,047 | $ | 310,123 | ||||
| Non-cash compensation expense related to equity awards | 76,557 | 64,521 | ||||||
| Total research, development and patent expenses | $ | 414,604 | $ | 374,644 | ||||
For 2018, our research, development and patent expenses were $338.0 million, compared to $310.1 million for 2017. The increase in our R&D expenses for 2018, compared to 2017 was driven primarily by increases in drug development costs related to several medicines including AKCEA-APOCIII-LRx and AKCEA-ANGPTL3-LRx, as we advanced these programs in development. All amounts exclude non-cash compensation expense related to equity awards.
Our research, development and patent expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Ionis Core | $ | 222,528 | $ | 246,390 | ||||
| Akcea Therapeutics | 120,905 | 118,260 | ||||||
| Elimination of intercompany activity | (5,386 | ) | (54,527 | ) | ||||
| Subtotal | 338,047 | 310,123 | ||||||
| Non-cash compensation expense related to equity awards | 76,557 | 64,521 | ||||||
| Total research, development and patent expenses | $ | 414,604 | $ | 374,644 | ||||
Antisense Drug Discovery
We use our proprietary antisense technology to generate information about the function of genes and to determine the value of genes as drug discovery targets. We use this information to direct our own antisense drug discovery research, and that of our partners. Antisense drug discovery is also the function that is responsible for advancing our antisense core technology.
63
As we continue to advance our antisense technology, we are investing in our drug discovery programs to expand our and our partners’ drug pipelines. Our antisense drug discovery expenses are part of our Ionis Core business segment.
Our antisense drug discovery expenses were as follows (in thousands) and are part of our Ionis Core business segment:
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Antisense drug discovery expenses, excluding non-cash compensation expense related to equity awards | $ | 61,387 | $ | 56,160 | ||||
| Non-cash compensation expense related to equity awards | 17,530 | 15,203 | ||||||
| Total antisense drug discovery expenses | $ | 78,917 | $ | 71,363 | ||||
Antisense drug discovery expenses for 2018 were $61.4 million and were slightly higher compared to $56.2 million for 2017 due to an increase in expenses we incurred in 2018 related to our expanding early stage research programs. All amounts exclude non-cash compensation expense related to equity awards.
Antisense Drug Development
The following table sets forth drug development expenses, including the breakdown for medicines in Phase 3 development and/or commercialization for which we have incurred significant costs (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| SPINRAZA | $ | — | $ | 10,996 | ||||
| WAYLIVRA | 19,397 | 22,524 | ||||||
| TEGSEDI | 19,204 | 24,880 | ||||||
| Other antisense development projects | 116,936 | 79,106 | ||||||
| Development overhead expenses | 48,754 | 43,784 | ||||||
| Total antisense drug development, excluding non-cash compensation expense related to equity awards | 204,291 | 181,290 | ||||||
| Non-cash compensation expense related to equity awards | 34,845 | 28,325 | ||||||
| Total antisense drug development expenses | $ | 239,136 | $ | 209,615 | ||||
Antisense drug development expenses were $204.3 million for 2018 and increased compared to $181.3 million for 2017. During 2018, our development expenses for AKCEA-APOCIII-LRx and AKCEA-ANGPTL3-LRx increased compared to 2017. We completed enrollment of the Phase 2 clinical study of AKCEA-APO(a)-LRx during the first quarter of 2018 and reported positive Phase 2 data in the third quarter of 2018. We also initiated a Phase 2 clinical study of AKCEA-APOCIII-LRx in patients with hypertriglyceridemia and established cardiovascular disease in the first quarter of 2018. Slightly offsetting these increases were decreased expenses for SPINRAZA, TEGSEDI and WAYLIVRA. Specifically, we have transitioned all further development of SPINRAZA to Biogen. In early 2017, we completed our Phase 3 WAYLIVRA trial in patients with FCS and our Phase 3 TEGSEDI trial in patients with hATTR with polyneuropathy. All amounts exclude non-cash compensation expense related to equity awards.
Our antisense drug development expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Ionis Core | $ | 100,090 | $ | 123,934 | ||||
| Akcea Therapeutics | 104,201 | 105,751 | ||||||
| Elimination of intercompany activity | — | (48,395 | ) | |||||
| Subtotal | 204,291 | 181,290 | ||||||
| Non-cash compensation expense related to equity awards | 34,845 | 28,325 | ||||||
| Total antisense drug development expenses | $ | 239,136 | $ | 209,615 | ||||
We may conduct multiple clinical trials on a drug candidate, including multiple clinical trials for the various indications we may be studying. Furthermore, as we obtain results from trials we may elect to discontinue clinical trials for certain drug candidates in certain indications in order to focus our resources on more promising drug candidates or indications. Our Phase 1 and Phase 2 programs are clinical research programs that fuel our Phase 3 pipeline. When our products are in Phase 1 or Phase 2 clinical trials, they are in a dynamic state in which we may adjust the development strategy for each product. Although we may characterize a product as “in Phase 1” or “in Phase 2,” it does not mean that we are conducting a single, well-defined study with dedicated resources. Instead, we allocate our internal resources on a shared basis across numerous products based on each product’s particular needs at that time. This means we are constantly shifting resources among products. Therefore, what we spend on each product during a particular period is usually a function of what is required to keep the products progressing in clinical development, not what products we think are most important. For example, the number of people required to start a new study is large, the number of people required to keep a study going is modest and the number of people required to finish a study is large. However, such fluctuations are not indicative of a shift in our emphasis from one product to another and cannot be used to accurately predict future costs for each product. And, because we always have numerous medicines in preclinical and early stage clinical research, the fluctuations in expenses from drug to drug, in large part, offset one another. If we partner a drug, it may affect the size of a trial, its timing, its total cost and the timing of the related costs.
64
Manufacturing and Operations
Expenditures in our manufacturing and operations function consist primarily of personnel costs, specialized chemicals for oligonucleotide manufacturing, laboratory supplies and outside services. Our manufacturing and operations function is responsible for providing drug supplies to antisense drug development, Akcea and our collaboration partners. Our manufacturing procedures include testing to satisfy good laboratory and good manufacturing practice requirements.
Our manufacturing and operations expenses were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Manufacturing and operations expenses, excluding non-cash compensation expense related to equity awards | $ | 39,806 | $ | 43,526 | ||||
| Non-cash compensation expense related to equity awards | 9,036 | 6,904 | ||||||
| Total manufacturing and operations expenses | $ | 48,842 | $ | 50,430 | ||||
Manufacturing and operations expenses were $39.8 million for 2018 and declined slightly compared to $43.5 million for 2017. All amounts exclude non-cash compensation expense related to equity awards.
Our manufacturing and operations expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Ionis Core | $ | 32,277 | $ | 39,098 | ||||
| Akcea Therapeutics | 12,758 | 10,440 | ||||||
| Elimination of intercompany activity | (5,229 | ) | (6,012 | ) | ||||
| Subtotal | 39,806 | 43,526 | ||||||
| Non-cash compensation expense related to equity awards | 9,036 | 6,904 | ||||||
| Total manufacturing and operations expenses | $ | 48,842 | $ | 50,430 | ||||
R&D Support
In our research, development and patent expenses, we include support costs such as rent, repair and maintenance for buildings and equipment, utilities, depreciation of laboratory equipment and facilities, amortization of our intellectual property, informatics costs, procurement costs and waste disposal costs. We call these costs R&D support expenses.
The following table sets forth information on R&D support expenses (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Personnel costs | $ | 12,968 | $ | 11,432 | ||||
| Occupancy | 8,567 | 8,236 | ||||||
| Patent expenses | 2,744 | 2,095 | ||||||
| Depreciation and amortization | 439 | 249 | ||||||
| Insurance | 1,622 | 1,735 | ||||||
| Other | 6,223 | 5,400 | ||||||
| Total R&D support expenses, excluding non-cash compensation expense related to equity awards | 32,563 | 29,147 | ||||||
| Non-cash compensation expense related to equity awards | 15,146 | 14,089 | ||||||
| Total R&D support expenses | $ | 47,709 | $ | 43,236 | ||||
R&D support expenses for 2018 were $32.6 million compared to $29.1 million for 2017. R&D support expenses increased slightly primarily related to costs associated with the expansion of our business. All amounts exclude non-cash compensation expense related to equity awards.
65
Our R&D support expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Ionis Core | $ | 28,774 | $ | 27,198 | ||||
| Akcea Therapeutics | 3,946 | 2,069 | ||||||
| Elimination of intercompany activity | (157 | ) | (120 | ) | ||||
| Subtotal | 32,563 | 29,147 | ||||||
| Non-cash compensation expense related to equity awards | 15,146 | 14,089 | ||||||
| Total R&D support expenses | $ | 47,709 | $ | 43,236 | ||||
Selling, General and Administrative Expenses
Selling, general and administrative expenses include costs associated with the pre-commercialization and commercialization activities for our medicines and costs to support our company, our employees and our stockholders. These costs include personnel and outside costs in the areas of pre-commercialization, commercialization, legal, human resources, investor relations, and finance. Additionally, we include in selling, general and administrative expenses such costs as rent, repair and maintenance of buildings and equipment, depreciation and utilities costs that we need to support the corporate functions listed above. We also include fees we owe under our in-licensing agreements related to SPINRAZA.
The following table sets forth information on selling, general and administrative expenses (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Selling, general and administrative expenses, excluding non-cash compensation expense related to equity awards | $ | 190,027 | $ | 87,034 | ||||
| Non-cash compensation expense related to equity awards | 54,595 | 21,454 | ||||||
| Total selling, general and administrative expenses | $ | 244,622 | $ | 108,488 | ||||
Selling, general and administrative expenses were $190.0 million for 2018 and significantly increased compared to $87.0 million for 2017. The increase in SG&A expenses was principally due to the cost of preparing to commercialize TEGSEDI and WAYLIVRA, and an increase in the fees we owed under our in-licensing agreements related to SPINRAZA. We project our expenses will increase as we continue to launch TEGSEDI. All amounts exclude non-cash compensation expense related to equity awards.
Our selling, general and administrative expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Ionis Core | $ | 70,647 | $ | 58,962 | ||||
| Akcea Therapeutics | 118,930 | 28,072 | ||||||
| Elimination of intercompany activity | 450 | — | ||||||
| Subtotal | 190,027 | 87,034 | ||||||
| Non-cash compensation expense related to equity awards | 54,595 | 21,454 | ||||||
| Total selling general and administrative expenses | $ | 244,622 | $ | 108,488 | ||||
Akcea Therapeutics, Inc.
The following table sets forth information on operating expenses (in thousands) for our Akcea Therapeutics business segment:
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Cost of products sold | $ | 11,573 | $ | — | ||||
| Development and patent expenses | 120,905 | 118,260 | ||||||
| Selling, general and administrative expenses | 118,930 | 28,072 | ||||||
| Total operating expenses, excluding non-cash compensation expense related to equity awards | 251,408 | 146,332 | ||||||
| Non-cash compensation expense related to equity awards | 44,275 | 17,539 | ||||||
| Total Akcea Therapeutics operating expenses | $ | 295,683 | $ | 163,871 | ||||
Operating expenses for Akcea were $251.4 million for 2018 and increased compared to $146.3 million for 2017.
66
In the third quarter of 2018, Akcea began recognizing cost of products sold expenses after the approval of TEGSEDI.
In 2017, $48.4 million of development and patent expenses was for one-time sublicensing expenses related to the Novartis collaboration recorded in the first quarter of 2017. $33.4 million of these expenses were non-cash and the remaining $15 million was paid to us. Excluding the $48.4 million of one-time expenses, Akcea’s development and patent expenses increased $51.0 million in 2018 compared to 2017 as Akcea made investments in advancing its pipeline, including AKCEA-APO(a)-LRx, and AKCEA-APOCIII-LRx. For each period presented, we allocated a portion of Ionis’ R&D support expenses, which are included in development and patent expenses in the table above, to Akcea for work we performed on behalf of Akcea.
Akcea’s SG&A expenses increased in 2018 compared to 2017, primarily because Akcea was building its commercial infrastructure and advancing the pre-commercialization activities necessary to successfully launch TEGSEDI and WAYLIVRA. For each period presented, we allocated a portion of Ionis’ G&A expenses, which were included in Akcea’s G&A expenses in the table above, to Akcea for work we performed on Akcea’s behalf.
Investment Income
Investment income for 2018 was $30.2 million compared to $8.2 million for 2017. Investment income increased primarily due to a significantly higher average cash balance and to a lesser extent an improvement in the market conditions during 2018 compared to 2017.
Interest Expense
Interest expense was $44.8 million for both 2018 and 2017. The following table sets forth information on interest expense (in thousands):
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Convertible notes: | ||||||||
| Non-cash amortization of the debt discount and debt issuance costs | $ | 35,173 | $ | 32,536 | ||||
| Interest expense payable in cash | 6,855 | 7,090 | ||||||
| Non-cash interest expense for long-term financing liability | — | 3,352 | ||||||
| Interest on mortgage for primary R&D and manufacturing facilities | 2,409 | 1,103 | ||||||
| Other | 352 | 671 | ||||||
| Total interest expense | $ | 44,789 | $ | 44,752 | ||||
In July 2017, we purchased the building that houses our primary R&D facility and the building that houses our manufacturing facility for $79.4 million and $14.0 million, respectively. As a result of the purchase of our primary R&D facility, we extinguished the financing liability we had previously recorded on our balance sheet. We financed the purchase of the buildings with mortgage debt of $51.3 million with an interest rate of 3.88 percent for our primary R&D facility and mortgage debt of $9.1 million with an interest rate of 4.2 percent for our manufacturing facility. Both mortgages mature in August 2027.
Loss on Extinguishment of Financing Liability for Leased Facility
We recognized a loss on extinguishment of the financing liability for leased facility of $7.7 million in 2017. The loss represents the difference between the amount we previously recorded as a financing liability for the leased facility and the purchase price we paid for our primary R&D facility in July 2017. This loss was non-cash and nonrecurring.
Other Expenses
Other expenses were $0.2 million in 2018 and $3.5 million for 2017. Our 2017 other expenses primarily consisted of the previously capitalized fair value of the potential premium we would have received from Novartis if Akcea had not completed its IPO. This expense was non-cash and nonrecurring.
Income Tax Benefit
We had an income tax benefit of $291.1 million for 2018, compared to $6.0 million for 2017. Our tax benefit increased significantly in 2018 primarily due to a one-time, non-cash tax benefit related to the reversal of the valuation allowance previously recorded against Ionis’ stand-alone U.S. federal net deferred tax assets of $332.1 million. Because of Ionis’ strong financial performance, on a stand-alone basis, over the past few years and our outlook regarding the continued growth of our business, we determined that it is more likely than not that we will utilize most of our deferred federal income tax assets primarily net operating loss carryforwards and research and development and orphan drug credit carryforwards. We continue to maintain a valuation allowance against certain Ionis and Akcea federal and state net deferred tax assets at December 31, 2018 due to uncertainties related to our ability to realize the tax benefits associated with these assets.
67
Net Income (Loss)
We had income of $215.0 million for 2018, compared to a net loss of $10.8 million for 2017. The increase in our net income in 2018, compared to 2017 was primarily due to our increasing revenues and income tax benefit.
Net Operating Loss and Tax Credit Carryforwards
At December 31, 2018, we had federal and California tax net operating loss carryforwards of $284.6 million and $808.7 million, respectively. Our federal tax loss carryforwards begin to expire in 2033. A portion of our California tax loss carryforwards continued to expire in 2018. At December 31, 2018, we also had federal and California research and development tax credit carryforwards of $288.9 million and $68.4 million, respectively. Our Federal research and development tax credit carryforwards began to expire in 2018. Our California research and development tax credit carryforwards are available indefinitely.
Net Loss Attributable to Noncontrolling Interest in Akcea Therapeutics, Inc.
At December 31, 2018, we owned approximately 75 percent of Akcea. The shares of Akcea third parties own represent an interest in Akcea's equity that we do not control. However, because we continue to maintain overall control of Akcea through our voting interest, we reflect the assets, liabilities and results of operations of Akcea in our consolidated financial statements. We reflect the noncontrolling interest attributable to other owners of Akcea's common stock in a separate line called “Net loss attributable to noncontrolling interest in Akcea” on our statement of operations. Our noncontrolling interest in Akcea on our statement of operations for 2018 was $58.8 million, compared to $11.1 million for 2017.
Net Income Attributable to Ionis Pharmaceuticals, Inc. Common Stockholders and Net Income per Share
We had net income attributable to our common stockholders of $273.7 million for 2018, compared $0.3 million in 2017. Basic and diluted net income per share for 2018 was $2.09 and $2.07, respectively compared to $0.15 for 2017. The increase in our net income attributable to our common stockholders in 2018, compared to 2017, was primarily due to our increasing revenues and income tax benefit, slightly offset by an increase in the net loss related to the portion of Akcea we own.
Years Ended December 31, 2017 and December 31, 2016
Revenue
Total revenue for 2017 was $514.2 million compared to $372.8 million for 2016 and was comprised of the following (amounts in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Revenue: | (as revised) | |||||||
| Commercial revenue: | ||||||||
| SPINRAZA royalties | $ | 112,540 | $ | 883 | ||||
| Licensing and other royalty revenue | 7,474 | 21,884 | ||||||
| Total commercial revenue | 120,014 | 22,767 | ||||||
| R&D revenue: | ||||||||
| Amortization from upfront payments | 97,646 | 62,415 | ||||||
| Milestone payments | 152,008 | 152,325 | ||||||
| License fees | 116,095 | 98,000 | ||||||
| Other services | 28,416 | 37,269 | ||||||
| Total R&D revenue | 394,165 | 350,009 | ||||||
| Total revenue | $ | 514,179 | $ | 372,776 | ||||
The increase in revenue in 2017 compared to 2016 was primarily due to increasing commercial revenue from SPINRAZA royalties. SPINRAZA was approved by the FDA in December 2016, making 2017 the first full year we earned commercial revenue from SPINRAZA.
Our revenue from licensing and other royalties was higher in 2016 primarily from $15 million we earned from Kastle when it acquired the global rights to develop and commercialize Kynamro.
R&D revenue from the amortization of upfront payments increased $35 million in 2017, compared to 2016, primarily due to the amortization of the upfront payment from our collaboration with Novartis which began in the first quarter of 2017.
In 2017, R&D revenue from milestone payments included over $120 million of milestone payments from Biogen, including the milestone payments for SPINRAZA approval in the EU and Japan. In 2016, R&D revenue from milestone payments included over $115 million of milestone payments from Biogen, including a $60 million milestone payment for the approval of SPINRAZA in the U.S.
68
In 2017, we earned $65 million from Bayer for the license of IONIS-FXI-LRx and $45 million from Roche for the license of IONIS-HTTRx. In 2016 we earned $91.2 million when Bayer licensed IONIS-FXIRx.
Operating Expenses
Operating expenses for 2017 were $483.1 million, and increased compared to $392.9 million for 2016. Our operating expenses increased year over year principally due to higher SG&A expenses as we prepared to commercialize TEGSEDI and WAYLIVRA. Our SG&A expenses also increased in 2017 compared to 2016 because of fees we owed under our in-licensing agreements related to SPINRAZA.
Our operating expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Ionis Core | $ | 305,352 | $ | 260,233 | ||||
| Akcea Therapeutics | 146,332 | 73,363 | ||||||
| Elimination of intercompany activity | (54,527 | ) | (12,768 | ) | ||||
| Subtotal | 397,157 | 320,828 | ||||||
| Non-cash compensation expense related to equity awards | 85,975 | 72,108 | ||||||
| Total operating expenses | $ | 483,132 | $ | 392,936 | ||||
Research, Development and Patent Expenses
The following table sets forth information on research, development and patent expenses (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Research, development and patent expenses, excluding non-cash compensation expense related to equity awards | $ | 310,123 | $ | 289,221 | ||||
| Non-cash compensation expense related to equity awards | 64,521 | 55,099 | ||||||
| Total research, development and patent expenses | $ | 374,644 | $ | 344,320 | ||||
For 2017, total research, development and patent expenses were $310.1 million, compared to $289.2 million for 2016. Our research, development and patent expenses increased slightly primarily related to expenses such as regulatory filings, manufacturing initial launch supplies and other activities in support of TEGSEDI and WAYLIVRA. If you exclude these expenses, our research, development and patent expenses decreased year-over-year. All amounts exclude non-cash compensation expense related to equity awards.
Our research, development and patent expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Ionis Core | $ | 246,390 | $ | 238,106 | ||||
| Akcea Therapeutics | 118,260 | 63,883 | ||||||
| Elimination of intercompany activity | (54,527 | ) | (12,768 | ) | ||||
| Subtotal | 310,123 | 289,221 | ||||||
| Non-cash compensation expense related to equity awards | 64,521 | 55,099 | ||||||
| Total research, development and patent expenses | $ | 374,644 | $ | 344,320 | ||||
Antisense Drug Discovery
Our antisense drug discovery expenses were as follows (in thousands) and are part of our Ionis Core business segment:
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Antisense drug discovery expenses, excluding non-cash compensation expense related to equity awards | $ | 56,160 | $ | 51,028 | ||||
| Non-cash compensation expense related to equity awards | 15,203 | 13,589 | ||||||
| Total antisense drug discovery expenses | $ | 71,363 | $ | 64,617 | ||||
Antisense drug discovery expenses for 2017 were $56.2 million and were slightly higher compared to $51.0 million for 2016, due to expenses we incurred related to advancing our early stage research programs. All amounts exclude non-cash compensation expense related to equity awards.
69
Antisense Drug Development
The following table sets forth research and development expenses for our major antisense drug development projects (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| SPINRAZA | $ | 10,996 | $ | 43,868 | ||||
| WAYLIVRA | 22,524 | 26,285 | ||||||
| TEGSEDI | 24,880 | 22,939 | ||||||
| Other antisense development products | 79,106 | 42,999 | ||||||
| Development overhead expenses | 43,784 | 42,966 | ||||||
| Total antisense drug development, excluding non-cash compensation expense related to equity awards | 181,290 | 179,057 | ||||||
| Non-cash compensation expense related to equity awards | 28,325 | 21,380 | ||||||
| Total antisense drug development expenses | $ | 209,615 | $ | 200,437 | ||||
Antisense drug development expenditures were $181.3 million for 2017 compared to $179.1 million for 2016. The expenses for SPINRAZA and WAYLIVRA declined in 2017. Specifically, we transitioned all further development of SPINRAZA to Biogen and we were completing our Phase 3 WAYLIVRA trial in patients with FCS. Additionally, we completed our Phase 3 TEGSEDI trial in patients with hATTR with polyneuropathy in 2017. Our 2017 expenses included $4.8 million of expenses related to regulatory filing activities for TEGSEDI and WAYLIVRA. Additionally, during 2017, we made investments in our other antisense development projects, including AKCEA-APO(a)-LRx and IONIS-FXIRx. All amounts exclude non-cash compensation expense related to equity awards.
Our antisense drug development expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Ionis Core | $ | 123,934 | $ | 132,418 | ||||
| Akcea Therapeutics | 105,751 | 46,639 | ||||||
| Elimination of intercompany activity | (48,395 | ) | — | |||||
| Subtotal | 181,290 | 179,057 | ||||||
| Non-cash compensation expense related to equity awards | 28,325 | 21,380 | ||||||
| Total antisense drug development expenses | $ | 209,615 | $ | 200,437 | ||||
Manufacturing and Operations
Our manufacturing and operations expenses were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Manufacturing and operations expenses, excluding non-cash compensation expense related to equity awards | $ | 43,526 | $ | 30,148 | ||||
| Non-cash compensation expense related to equity awards | 6,904 | 6,113 | ||||||
| Total manufacturing and operations expenses | $ | 50,430 | $ | 36,261 | ||||
Manufacturing and operations expenses for 2017 were $43.5 million and were higher compared to $30.1 million for 2016. All amounts exclude non-cash compensation expense related to equity awards. $11 million of the increase in manufacturing expenses was related to TEGSEDI and WAYLIVRA to prepare for the planned launches. All amounts exclude non-cash compensation expense related to equity awards.
Our manufacturing and operations expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Ionis Core | $ | 39,098 | $ | 27,341 | ||||
| Akcea Therapeutics | 10,440 | 15,455 | ||||||
| Elimination of intercompany activity | (6,012 | ) | (12,648 | ) | ||||
| Subtotal | 43,526 | 30,148 | ||||||
| Non-cash compensation expense related to equity awards | 6,904 | 6,113 | ||||||
| Total manufacturing and operations expenses | $ | 50,430 | $ | 36,261 | ||||
70
R&D Support
The following table sets forth information on R&D support expenses (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Personnel costs | $ | 11,432 | $ | 11,560 | ||||
| Occupancy | 8,236 | 7,891 | ||||||
| Patent expenses | 2,095 | 3,945 | ||||||
| Depreciation and amortization | 249 | 245 | ||||||
| Insurance | 1,735 | 1,344 | ||||||
| Other | 5,400 | 4,003 | ||||||
| Total R&D support expenses, excluding non-cash compensation expense related to equity awards | 29,147 | 28,988 | ||||||
| Non-cash compensation expense related to equity awards | 14,089 | 14,017 | ||||||
| Total R&D support expenses | $ | 43,236 | $ | 43,005 | ||||
R&D support expenses for 2017 were $29.1 million, and were essentially flat compared to $29.0 million for 2016. All amounts exclude non-cash compensation expense related to equity awards.
Our R&D support expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Ionis Core | $ | 27,198 | $ | 27,319 | ||||
| Akcea Therapeutics | 2,069 | 1,789 | ||||||
| Elimination of intercompany activity | (120 | ) | (120 | ) | ||||
| Subtotal | 29,147 | 28,988 | ||||||
| Non-cash compensation expense related to equity awards | 14,089 | 14,017 | ||||||
| Total R&D support expenses | $ | 43,236 | $ | 43,005 | ||||
Selling, General and Administrative Expenses
The following table sets forth information on selling, general and administrative expenses (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Selling, general and administrative expenses, excluding non-cash compensation expense related to equity awards | $ | 87,034 | $ | 31,607 | ||||
| Non-cash compensation expense related to equity awards | 21,454 | 17,009 | ||||||
| Total selling, general and administrative expenses | $ | 108,488 | $ | 48,616 | ||||
Selling, general and administrative expenses for 2017 were $87.0 million and increased compared to $31.6 million for 2016. The increase in SG&A expenses was principally due to the cost of preparing to commercialize TEGSEDI and WAYLIVRA and from fees we owed under our in-licensing agreements related to SPINRAZA. All amounts exclude non-cash compensation expense related to equity awards.
Our selling, general and administrative expenses by segment were as follows (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Ionis Core | $ | 58,962 | $ | 22,127 | ||||
| Akcea Therapeutics | 28,072 | 9,480 | ||||||
| Non-cash compensation expense related to equity awards | 21,454 | 17,009 | ||||||
| Total selling, general and administrative expenses | $ | 108,488 | $ | 48,616 | ||||
71
Akcea Therapeutics, Inc.
The following table sets forth information on operating expenses (in thousands) for our Akcea Therapeutics business segment:
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Development and patent expenses | $ | 118,260 | $ | 63,883 | ||||
| General and administrative expenses | 28,072 | 9,480 | ||||||
| Total operating expenses, excluding non-cash compensation expense related to equity awards | 146,332 | 73,363 | ||||||
| Non-cash compensation expense related to equity awards | 17,539 | 10,149 | ||||||
| Total Akcea Therapeutics operating expenses | $ | 163,871 | $ | 83,512 | ||||
Akcea’s operating expenses were $146.3 million for 2017 and increased compared to $73.4 million for 2016.
$48.4 million of the increase in Akcea’s development and patent expenses was for one-time sublicensing expenses related to the Novartis collaboration recorded in the first quarter of 2017. $33.4 million of these expenses were non-cash and the remaining $15 million was paid to us. For each period presented, we allocated a portion of Ionis’ R&D support expenses, which are included in development and patent expenses in the table above, to Akcea for work we performed on behalf of Akcea.
Akcea’s G&A expenses increased in 2017, compared to 2016, primarily due to Akcea continuing to build its commercial infrastructure and advance the pre-commercialization activities necessary to successfully launch WAYLIVRA. For each period presented, we allocated a portion of Ionis’ G&A expenses, which were included in Akcea’s G&A expenses in the table above, to Akcea for work we performed on Akcea’s behalf.
All amounts exclude non-cash compensation expense related to equity awards.
Investment Income
Investment income for 2017 totaled $8.2 million compared to $5.5 million for 2016. Investment income increased primarily due to a higher average cash balance and an improvement in the market conditions during 2017 compared to 2016.
Interest Expense
The following table sets forth information on interest expense (in thousands):
Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Convertible notes: | ||||||||
| Non-cash amortization of the debt discount and debt issuance costs | $ | 32,536 | $ | 25,115 | ||||
| Interest expense payable in cash | 7,090 | 6,684 | ||||||
| Non-cash interest expense for long-term financing liability | 3,352 | 6,693 | ||||||
| Interest on mortgage for primary R&D and manufacturing facilities | 1,103 | — | ||||||
| Other | 671 | 303 | ||||||
| Total interest expense | $ | 44,752 | $ | 38,795 | ||||
Loss on Extinguishment of Financing Liability for Leased Facility
We recognized a loss on extinguishment of the financing liability for leased facility of $7.7 million in 2017. The loss represents the difference between the amount we previously recorded as a financing liability for the leased facility and the purchase price we paid for our primary R&D facility in July 2017. This loss was non-cash and nonrecurring.
Early Retirement of Debt
As a result of the debt exchange we completed in December 2016, we recorded a $4.0 million non-cash loss on early retirement of debt, reflecting the early retirement of the majority of our remaining 2¾ percent convertible notes in December 2016.
Other Expenses
Other expenses were $3.5 million for 2017 and primarily consisted of the previously capitalized fair value of the potential premium we would have received from Novartis if Akcea had not completed its IPO. This expense was non-cash and nonrecurring.
72
Income Tax Expense (Benefit)
In 2017, we recorded a net income tax benefit of $6.0 million, compared to income tax expense of $2.9 million in 2016. Our tax expense flipped from an expense position in 2016 to a benefit position in 2017 primarily due to a $7.7 million reduction in our valuation allowance. As a result of the Tax Act, we reduced our valuation allowance because we are entitled to receive a tax refund for our cumulative prior year alternative minimum tax credit carryforwards.
Net Loss
Net loss for 2017 was $10.8 million compared $60.4 million for 2016. Our net loss improved for 2017 compared to 2016 primarily due to the addition of commercial revenue from SPINRAZA royalties and increased R&D revenue.
Net Loss Attributable to Noncontrolling Interest in Akcea Therapeutics, Inc.
As a result of Akcea’s IPO, beginning in July 2017, we no longer own 100 percent of Akcea. From the closing of Akcea’s IPO on July 19, 2017 through the end of 2017, we owned approximately 68 percent of Akcea. As a result, we adjusted our financial statements to reflect the portion of Akcea we no longer own, which was 32 percent at December 31, 2017. Accordingly, our consolidated statement of operations now includes a new line called “Net loss attributable to noncontrolling interests in Akcea”; our noncontrolling interest in Akcea for 2017 was $11.1 million. We also added a corresponding account to our consolidated balance sheet called “Noncontrolling interest in Akcea Therapeutics, Inc.”
Net Income (Loss) Attributable to Ionis Pharmaceuticals, Inc. Common Stockholders and Net Income (Loss) per Share
We had net income attributable to our common stockholders of $0.3 million for 2017, compared to a net loss of $60.4 million in 2016. Basic and diluted net income per share for 2017 was $0.15 compared to basic and dilutive net loss per share of $0.50 for 2016.
Liquidity and Capital Resources
We have financed our operations primarily from research and development collaborative agreements. Beginning in December 2016 we added commercial revenue from SPINRAZA royalties. From our inception through December 31, 2018, we had earned approximately $3.2 billion in revenue. We also financed our operations through the sale of our equity securities and the issuance of long-term debt. From the time we were founded through December 31, 2018, we had raised net proceeds of approximately $1.7 billion from the sale of our equity securities, not including the $182.4 million Akcea received in net proceeds from its IPO in July 2017. Additionally, we borrowed approximately $1.4 billion under long-term debt arrangements to finance a portion of our operations over the same time period.
At December 31, 2018, we had cash, cash equivalents and short-term investments of $2.1 billion and stockholders’ equity of $1,187.2 million. In comparison, we had cash, cash equivalents and short-term investments of $1.0 billion and stockholders’ equity of $365.3 million at December 31, 2017. Our cash, cash equivalents and short-term investments increased in 2018 primarily from the $1 billion payment we received from Biogen for our 2018 strategic neurology collaboration.
At December 31, 2018, we had consolidated working capital of $1.9 billion compared to $925.1 million at December 31, 2017. As of December 31, 2018, our debt and other obligations totaled $764.0 million compared to $759.8 million at December 31, 2017.
The following table summarizes our contractual obligations as of December 31, 2018. The table provides a breakdown of when obligations become due. We provide a more detailed description of the major components of our debt in the paragraphs following the table:
| Payments Due by Period (in millions) | ||||||||||||||||||||
Contractual Obligations (selected balances described below) | Total | Less than 1 year | 1-3 years | 3-5 years | After 5 years | |||||||||||||||
| Convertible senior notes (principal and interest payable) | $ | 706.1 | $ | 6.9 | $ | 699.2 | $ | — | $ | — | ||||||||||
| Building mortgage payments | $ | 80.7 | $ | 2.4 | $ | 4.8 | $ | 6.2 | $ | 67.3 | ||||||||||
| Financing arrangements (principal and interest payable) | $ | 12.7 | $ | 12.7 | $ | — | $ | — | $ | — | ||||||||||
| Other obligations (principal and interest payable) | $ | 1.0 | $ | 0.1 | $ | 0.1 | $ | 0.1 | $ | 0.7 | ||||||||||
| Operating leases | $ | 25.7 | $ | 3.1 | $ | 5.7 | $ | 5.0 | $ | 11.9 | ||||||||||
| Total | $ | 826.2 | $ | 25.2 | $ | 709.8 | $ | 11.3 | $ | 79.9 | ||||||||||
Our contractual obligations consist primarily of our convertible debt. In addition, we also have facility mortgages, facility leases, equipment financing arrangements and other obligations. Due to the uncertainty with respect to the timing of future cash flows associated with our unrecognized tax benefits, we are unable to make reasonably reliable estimates of the period of cash settlement with the respective taxing authorities. Therefore, we have excluded $68.3 million of gross unrecognized tax benefits from our contractual obligations table above.
73
1 Percent Convertible Senior Notes
In November 2014, we completed a $500 million offering of convertible senior notes, which mature in 2021 and bear interest at 1 percent. We used a substantial portion of the net proceeds from the issuance of the 1 percent convertible senior notes to repurchase $140 million in principal of our 2¾ percent convertible senior notes. As a result, the principal balance of the 2¾ percent notes following the repurchase in November 2014 was $61.2 million.
In December 2016, we issued an additional $185.5 million of 1 percent convertible senior notes in exchange for the redemption of $61.1 million of our 2¾ percent convertible senior notes. At December 31, 2018, we had a nominal amount of our 2¾ percent convertible senior notes outstanding. At December 31, 2018, we had the following 1 percent convertible senior notes outstanding (amounts in millions except price per share data):
1 Percent Convertible Senior Notes | ||||
| Outstanding principal balance | $ | 685.5 | ||
| Original issue date ($500 million of principal) | November 2014 | |||
| Additional issue date ($185.5 million of principal) | December 2016 | |||
| Maturity date | November 2021 | |||
| Interest rate | 1 percent | |||
| Conversion price per share | $ | 66.81 | ||
| Total shares of common stock subject to conversion | 10.3 | |||
Interest is payable semi-annually for the 1 percent notes. The notes are convertible under certain conditions, at the option of the note holders. We settle conversions of the notes, at our election, in cash, shares of our common stock or a combination of both. We may not redeem the 1 percent notes prior to maturity, and no sinking fund is provided for them. Holders of the 1 percent notes may require us to purchase some or all of their notes upon the occurrence of certain fundamental changes, as set forth in the indenture governing the 1 percent notes, at a purchase price equal to 100 percent of the principal amount of the notes to be purchased, plus accrued and unpaid interest.
Financing Arrangements
In June 2015, we entered into a five-year revolving line of credit agreement with Morgan Stanley Private Bank, National Association, or Morgan Stanley. We amended the credit agreement in February 2016 to increase the amount available for us to borrow. Under the amended credit agreement, Morgan Stanley will provide a maximum of $30 million of revolving credit for general working capital purposes. Any loans under the credit agreement have interest payable monthly in arrears at a borrowing rate based on our option of:
| (i) | a floating rate equal to the one-month London Interbank Offered Rate, or LIBOR, in effect plus 1.25 percent per annum; |
| (ii) | a fixed rate equal to LIBOR plus 1.25 percent for a period of one, two, three, four, six, or twelve months as elected by us; or |
| (iii) | a fixed rate equal to the LIBOR swap rate during the period of the loan. |
Additionally, we pay 0.25 percent per annum, payable quarterly in arrears, for any amount unused under the credit facility. As of December 31, 2018 we had $12.5 million in outstanding borrowings under the credit facility with a 2.31 percent fixed interest rate and a maturity date of September 2019, which we used to fund our capital equipment needs.
The credit agreement includes customary affirmative and negative covenants and restrictions. We are in compliance with all covenants of the credit agreement.
Research and Development and Manufacturing Facilities
In July 2017, we purchased the building that houses our primary R&D facility and the building that houses our manufacturing facility for $79.4 million and $14.0 million, respectively. We financed the purchase of our primary R&D facility and manufacturing facility, with mortgage debt of $51.3 million and $9.1 million, respectively. Our primary R&D facility mortgage has an interest rate of 3.88 percent. Our manufacturing facility mortgage has an interest rate of 4.20 percent. During the first five years of both mortgages we are only required to make interest payments. Both mortgages mature in August 2027.
Other Obligations
In addition to contractual obligations, we had outstanding purchase orders as of December 31, 2018 for the purchase of services, capital equipment and materials as part of our normal course of business.
We plan to continue to enter into collaborations with partners to provide for additional revenue to us and we may incur additional cash expenditures related to our obligations under any of the new agreements we may enter into. We currently intend to use our cash, cash equivalents and short-term investments to finance our activities. However, we may also pursue other financing alternatives, like issuing additional shares of our common stock, issuing debt instruments, refinancing our existing debt, or securing lines of credit. Whether we use our existing capital resources or choose to obtain financing will depend on various factors, including the future success of our business, the prevailing interest rate environment and the condition of financial markets generally.
74
Off-Balance Sheet Arrangements
We have not entered into, nor do we currently have, any off-balance sheet arrangements (as defined under SEC rules).
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
We are exposed to changes in interest rates primarily from our long-term debt arrangements and, secondarily, investments in certain short-term investments. We primarily invest our excess cash in highly liquid short-term investments of the U.S. Treasury and reputable financial institutions, corporations, and U.S. government agencies with strong credit ratings. We typically hold our investments for the duration of the term of the respective instrument. We do not utilize derivative financial instruments, derivative commodity instruments or other market risk sensitive instruments, positions or transactions to manage exposure to interest rate changes. Accordingly, we believe that, while the securities we hold are subject to changes in the financial standing of the issuer of such securities, we are not subject to any material risks arising from changes in interest rates, foreign currency exchange rates, commodity prices, equity prices or other market changes that affect market risk sensitive instruments.
Item 8. Financial Statements and Supplementary Data
We filed our consolidated financial statements and supplementary data required by this item as exhibits hereto, and listed them under Item 15(a)(1) and (2), and incorporate them herein by reference.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or Exchange Act) that are designed to ensure that information we are required to disclose in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. We designed and evaluate our disclosure controls and procedures recognizing that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance and not absolute assurance of achieving the desired control objectives.
As of the end of the period covered by this report on Form 10-K, we carried out an evaluation of our disclosure controls and procedures under the supervision of, and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer. Based on our evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of December 31, 2018.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Exchange Act Rules 13a-15(f). Our internal control over financial reporting is a process designed under the supervision of our Chief Executive Officer and Chief Financial Officer to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our financial statements for external purposes in accordance with U.S. generally accepted accounting principles.
As of December 31, 2018, we assessed the effectiveness of our internal control over financial reporting based on the criteria for effective internal control over financial reporting under the 2013 “Internal Control—Integrated Framework,” issued by the Committee of Sponsoring Organizations, or COSO, of the Treadway Commission, under the supervision of, and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer. Based on that assessment, our management concluded that we maintained effective internal control over financial reporting as of December 31, 2018.
Ernst & Young LLP, an independent registered public accounting firm, audited the effectiveness of our internal control over financial reporting as of December 31, 2018, as stated in their attestation report, which is included elsewhere herein.
Changes in Internal Control over Financial Reporting
The above assessment did not identify any change in our internal control over financial reporting that occurred during our latest fiscal quarter and that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
75
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of Ionis Pharmaceuticals, Inc.
Opinion on Internal Control over Financial Reporting We have audited Ionis Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2018, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework) (the COSO criteria). In our opinion, Ionis Pharmaceuticals, Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2018, based on the COSO criteria. We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of Ionis Pharmaceuticals, Inc. as of December 31, 2018 and 2017, and the related consolidated statements of operations, comprehensive income (loss), stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2018, and the related notes and our report dated March 1, 2019 expressed an unqualified opinion thereon. Basis for Opinion The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB. We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion. Definition and Limitations of Internal Control Over Financial Reporting A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. /s/ Ernst & Young LLP | |
| San Diego, California | |
| March 1, 2019 |
76
Item 9B. Other Information
Not applicable.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
We incorporate by reference the information required by this Item with respect to directors and the Audit Committee from the information under the caption “ELECTION OF DIRECTORS,” including in particular the information under “Nominating, Governance and Review Committee” and “Audit Committee,” contained in our definitive Proxy Statement, which we will file with the Securities and Exchange Commission within 120 days after the end of the fiscal year ended December 31, 2018 (the “Proxy Statement”).
We incorporate by reference the required information concerning our Code of Ethics from the information under the caption “Code of Ethics and Business Conduct” contained in the Proxy Statement. Our Code of Ethics and Business Conduct is posted on our website at www.ionispharma.com(1). We intend to disclose future amendments to, or waivers from, our Code of Ethics and Business Conduct on our website.
Section 16(a) Beneficial Ownership Reporting Compliance
Item 1, Part I of this Report contains information concerning our executive officers. We incorporate by reference the information required by this Item concerning compliance with Section 16(a) of the Exchange Act from the information under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” contained in the Proxy Statement.
________________
| (1) | Any information that is included on or linked to our website is not part of this Form 10-K. |
Item 11. Executive Compensation
We incorporate by reference the information required by this item to the information under the caption “EXECUTIVE COMPENSATION,” “Compensation Committee Interlocks and Insider Participation” and “COMPENSATION COMMITTEE REPORT” contained in the Proxy Statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
We incorporate by reference the information required by this item to the information under the captions “SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT” contained in the Proxy Statement.
Securities Authorized for Issuance under Equity Compensation Plans
The following table sets forth information regarding outstanding options and shares reserved for future issuance under our equity compensation plans as of December 31, 2018.
| Plan Category | Number of Shares to be Issued Upon Exercise of Outstanding Options | Weighted Average Exercise Price of Outstanding Options | Number of Shares Remaining Available for Future Issuance | ||||||||||
| Equity compensation plans approved by stockholders(a) | 11,311,944 | $ | 47.85 | 4,578,854 | (b) | ||||||||
| Total | 11,311,944 | $ | 47.85 | 4,578,854 | |||||||||
________________
| (a) | Consists of four Ionis plans: 1989 Stock Option Plan, Amended and Restated 2002 Non-Employee Directors’ Stock Option Plan, 2011 Equity Incentive Plan and Employee Stock Purchase Plan, or ESPP. |
| (b) | Of these shares, 774,816 remained available for purchase under the ESPP as of December 31, 2018. The ESPP incorporates an evergreen formula pursuant to which on January 1 of each year on the first nine anniversaries of the plan, we automatically increase the aggregate number of shares reserved for issuance under the plan by 150,000 shares. |
For additional details about our equity compensation plans, including a description of each plan, see Note 4, Stockholders’ Equity, in the Notes to the Consolidated Financial Statements.
Item 13. Certain Relationships and Related Transactions, and Director Independence
We incorporate by reference the information required by this item to the information under the captions “Independence of the Board of Directors” and “Certain Relationships and Related Transactions” contained in the Proxy Statement.
Item 14. Principal Accounting Fees and Services
We incorporate by reference the information required by this item to the information under the caption “Ratification of Selection of Independent Auditors” contained in the Proxy Statement.
77
PART IV
Item 15. Exhibits, Financial Statement Schedules
(a)(1) Index to Financial Statements
We submitted the consolidated financial statements required by this item in a separate section beginning on page F-1 of this Report.
(a)(2) Index to Financial Statement Schedules
We omitted these schedules because they are not required, or are not applicable, or the required information is shown in the consolidated financial statements or notes thereto.
(a)(3) Index to Exhibits
78
INDEX TO EXHIBITS
| Exhibit Number | Description of Document | |||
| 3.1 | Amended and Restated Certificate of Incorporation filed June 19, 1991, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2017 and incorporated herein by reference. | |||
| 3.2 | Certificate of Amendment to Restated Certificate of Incorporation, filed June 17, 2014. - Filed as an exhibit to the Registrant’s Notice of Annual Meeting and Proxy Statement, for the 2014 Annual Meeting of Stockholders, filed with the SEC on April 25, 2014, and incorporated herein by reference. | |||
| 3.3 | Certificate of Amendment to Restated Certificate of Incorporation, filed December 18, 2015. - Filed as an exhibit to the Registrant’s Current Report on Form 8-K filed December 18, 2015 and incorporated herein by reference. | |||
| 3.4 | Amended and Restated Bylaws, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed December 18, 2015 and incorporated herein by reference. | |||
| 4.1 | Certificate of Designation of the Series C Junior Participating Preferred Stock, filed as an exhibit to Registrant’s Report on Form 8-K dated filed December 13, 2000 and incorporated herein by reference. | |||
| 4.2 | Specimen Common Stock Certificate, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2017 and incorporated herein by reference. | |||
| 4.3 | Indenture, dated as of August 13, 2012, between the Registrant and Wells Fargo Bank, National Association, as trustee, including Form of 2¾ percent Convertible Senior Note due 2019, filed as an exhibit to the Registrant’s Report on Form 8-K filed August 13, 2012 and incorporated herein by reference. | |||
| 4.4 | Indenture, dated as of November 17, 2014, between the Registrant and Wells Fargo Bank, National Association, as trustee, including Form of 1.00 percent Convertible Senior Note due 2021, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed November 21, 2014 and incorporated herein by reference. | |||
| 10.1 | Form of Indemnity Agreement entered into between the Registrant and its Directors and Officers with related schedule, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2012 and incorporated herein by reference. | |||
| 10.2* | Registrant’s 1989 Stock Option Plan, as amended, filed as an exhibit to Registrant’s Notice of Annual Meeting and Proxy Statement for the 2012 Annual Meeting of Stockholders, filed with the SEC on April 16, 2012, and incorporated herein by reference. | |||
| 10.3* | Registrant’s Amended and Restated 2000 Employee Stock Purchase Plan, filed as an exhibit to Registrant’s Notice of Annual Meeting and Proxy Statement for the 2009 Annual Meeting of Stockholders, filed with the SEC on April 20, 2009, and incorporated herein by reference. | |||
| 10.4 | Form of Employee Confidential Information and Inventions Agreement, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2017 and incorporated herein by reference. | |||
| 10.5 | Collaboration and License Agreement between the Registrant and Hybridon, Inc., dated May 24, 2001, filed as an exhibit to the Registrant’s report on Form 10-Q as amended for the quarter ended June 30, 2001 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |||
| 10.6 | Amendment #1 to the Research, Development and License Agreement dated May 11, 2011 by and between the Registrant and Glaxo Group Limited, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2011 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |||
| 10.7 | Amended and Restated Collaboration and License Agreement between the Registrant and Antisense Therapeutics Ltd dated February 8, 2008, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2008 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |||
| 10.8 | Amended and Restated License Agreement between the Registrant and Atlantic Pharmaceuticals Limited dated November 30, 2009, filed as an exhibit to the Registrant’s Annual Report as Form 10-K for the year ended December 31, 2009 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |||
79
| 10.9 | Stock Purchase Agreement among the Registrant, Akcea Therapeutics, Inc. and Novartis Pharma AG dated January 5, 2017, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017 and incorporated herein by reference. | |
| 10.10 | Amendment #1 between the Registrant and Bayer AG dated February 10, 2017, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.11 | Registrant’s Amended and Restated 10b5-1 Trading Plan dated September 12, 2013, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2013 and incorporated herein by reference. | |
| 10.12* | Registrant’s Amended and Restated 2002 Non-Employee Directors’ Stock Option Plan, as amended, filed as an exhibit to the Registrant’s Notice of Annual Meeting and Proxy Statement, for the 2014 Annual Meeting of Stockholders, filed with the SEC on April 25, 2014, and incorporated herein by reference. |
| 10.13* | Form of Restricted Stock Unit Agreement for Restricted Stock Units granted under the Ionis Pharmaceuticals, Inc. Amended and Restated 2002 Non-Employee Directors’ Stock Option Plan, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2012 and incorporated herein by reference. | |
| 10.14 | Research Collaboration, Option and License Agreement between the Registrant and Biogen MA Inc. dated December 19, 2017, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2017 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.15* | Ionis Pharmaceuticals, Inc. 2011 Equity Incentive Plan, filed as an exhibit to the Registrant’s Notice of 2011 Annual Meeting of Stockholders and Proxy Statement filed with the SEC on April 28, 2011, and incorporated herein by reference. | |
| 10.16* | Form of Option Agreement under the 2011 Equity Incentive Plan, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2015 and incorporated herein by reference. | |
| 10.17* | Form of Restricted Stock Unit Agreement for Restricted Stock Units granted under the 2011 Equity Incentive Plan, filed as an exhibit to the Registrant’s Registration Statement on Form S-8 filed with the SEC on August 8, 2011, and incorporated herein by reference. | |
| 10.18 | Loan Agreement between Ionis Gazelle, LLC and UBS AG dated July 18, 2017, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed July 21, 2017 and incorporated herein by reference. | |
| 10.19* | Form of Option Agreement under the 1989 Stock Option Plan, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2015 and incorporated herein by reference. | |
| 10.20* | Form of Option Agreement for Options Granted after March 8, 2005 under the 2002 Non-Employee Director’s Stock Option Plan, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2004 and incorporated herein by reference. | |
| 10.21 | Research, Development and License Agreement between the Registrant and Glaxo Group Limited dated March 30, 2010, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.22 | Loan Agreement between Ionis Faraday, LLC and UBS AG dated July 18, 2017, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed July 21, 2017 and incorporated herein by reference. | |
| 10.23 | Research Agreement dated August 10, 2011 between the Registrant and CHDI Foundation, Inc, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2011 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.24 | Guaranty between the Registrant and UBS AG dated July 18, 2017, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed July 21, 2017 and incorporated herein by reference. | |
| 10.25 | Development, Option and License Agreement between the Registrant and Biogen Idec International Holding Ltd. dated January 3, 2012, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2012 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
80
| 10.26 | DMPK Research, Development, Option and License Agreement between the Registrant and Biogen Idec MA Inc. dated June 27, 2012, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2012 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.27 | Amendment #2 to Research, Development and License Agreement between the Registrant and Glaxo Group Limited dated October 30, 2012, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2012 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.28 | Collaboration, License and Development Agreement between the Registrant and AstraZeneca AB dated December 7, 2012, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2012 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. |
| 10.29 | Neurology Drug Discovery and Development Collaboration, Option and License Agreement between the Registrant and Biogen Idec MA Inc. dated December 10, 2012, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2012 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.30 | HTT Research, Development, Option and License Agreement among the Registrant, F. Hoffmann-La Roche Ltd and Hoffman-La Roche Inc. dated April 8, 2013, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2013 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.31 | Letter Agreement between the Registrant and CHDI Foundation, Inc. dated April 8, 2013, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2013 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.32 | Amendment #1 to Collaboration, License and Development Agreement between the Registrant and AstraZeneca AB dated August 13, 2013, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2013 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. |
| 10.33 | Letter Agreement Amendment between the Registrant and Biogen Idec International Holding Ltd dated January 27, 2014, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.34 | Amendment No. 3 to the Research, Development and License Agreement between the Registrant and Glaxo Group Limited dated July 10, 2013, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.35 | Amendment #4 to the Research, Development and License Agreement between the Registrant and Glaxo Group Limited dated April 10, 2014, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.36 | Amendment #5 to the Research, Development and License Agreement among the Registrant, Glaxo Group Limited and GlaxoSmithKline Intellectual Property Development Limited dated June 27, 2014, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.37 | Exclusive License Agreement between the Registrant and the University of Massachusetts dated January 14, 2010, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.38 | Amended and Restated Collaboration and License Agreement between the Registrant and Cold Spring Harbor Laboratory dated October 26, 2011, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.39 | Amendment to Amended and Restated Collaboration and License Agreement between the Registrant and Cold Spring Harbor Laboratory dated March 14, 2014, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. |
81
| 10.40 | Amendment #1 to the Development, Option and License Agreement between the Registrant and Biogen Idec International Holding Ltd. dated December 15, 2014, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.41 | Research Collaboration, Option and License Agreement between the Registrant and Janssen Biotech Inc. dated December 22, 2014, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.42 | Amendment No.2 to the Collaboration, License and Development Agreement between the Registrant and AstraZeneca AB dated October 15, 2014, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2014 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.43 | Strategic Collaboration Agreement between the Registrant and AstraZeneca AB dated July 31, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2015 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.44 | Amendment #6 to Research, Development and License Agreement between the Registrant, Glaxo Group Limited and GlaxoSmithKline Intellectual Property Development Limited dated September 2, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2015 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.45 | Amendment Number One to the Second Amended and Restated Strategic Collaboration and License Agreement between the Registrant and Alnylam Pharmaceuticals, Inc. dated July 13, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2015 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.46 | License Agreement between the Registrant and Bayer Pharma AG dated May 1, 2015. Portions of this exhibit have been omitted and separately filed with the SEC, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2015 and incorporated herein by reference. |
| 10.47 | Line of Credit Agreement between the Registrant and Morgan Stanley Private Bank, National Association dated June 16, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2015 and incorporated herein by reference. | |
| 10.48 | Second Amended and Restated Strategic Collaboration and License Agreement between the Registrant and Alnylam Pharmaceuticals, Inc. dated January 8, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2015 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.49 | Amendment #1 to HTT Research, Development, Option and License Agreement between the Registrant, F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. dated January 9, 2015, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2015 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.50 | Amendment No.1 to Loan Documents between the Registrant and Morgan Stanley Private Bank, National Association dated December 30, 2015, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed January 5, 2016 and incorporated herein by reference. | |
| 10.51 | Amendment No.2 to Line of Credit Agreement between the Registrant and Morgan Stanley Private Bank, National Association dated February 24, 2016, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2015 and incorporated herein by reference. | |
| 10.52 | Amendment No.3 to the Collaboration, License and Development Agreement between the Registrant and AstraZeneca AB dated January 18, 2016, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2016 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.53 | Amendment #7 to the Research, Development and License Agreement among the Registrant, Glaxo Group Limited and GlaxoSmithKline Intellectual Property Development Limited dated March 4, 2016, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2016 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. |
82
| 10.54 | First Amendment to Research Collaboration, Option and License Agreement between the Registrant and Janssen Biotech Inc. dated December 21, 2016, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2016 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.55 | Letter Agreement between the Registrant and Biogen MA Inc. dated October 28, 2016, filed as an exhibit to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2016 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.56 | Guaranty between the Registrant and UBS AG dated July 18, 2017, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed July 21, 2017 and incorporated herein by reference. | |
| 10.57 | Environmental Indemnity Agreement among the Registrant, Ionis Gazelle, LLC and UBS AG dated July 18, 2017, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed July 21, 2017 and incorporated herein by reference. | |
| 10.58 | Environmental Indemnity Agreement among the Registrant, Ionis Faraday, LLC and UBS AG dated July 18, 2017, filed as an exhibit to the Registrant’s Current Report on Form 8-K filed July 21, 2017 and incorporated herein by reference. | |
| 10.59* | Amendment to Ionis Pharmaceuticals, Inc. 2011 Equity Incentive Plan, filed as an exhibit to the Registrant’s Notice of Annual Meeting and Proxy Statement, for the 2017 Annual Meeting of Stockholders, filed with the SEC on April 10, 2017, and incorporated herein by reference. | |
| 10.60* | Registrant’s Severance Benefit Plan and Summary Plan Description dated October 18, 2018, - filed as an exhibit to the Registrant’s Current Report on form 8-K filed October 18, 2018 and incorporated herein by reference. | |
| 10.61 | Strategic Advisory Services Agreement by and between the Registrant and B. Lynne Parshall, dated January 15, 2018, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2018 and incorporated herein by reference. | |
| 10.62 | Development, Commercialization, Collaboration, and License Agreement by and between the Registrant and Akcea Therapeutics, Inc., dated March 14, 2018, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2018 and incorporated herein by reference. | |
| 10.63 | Amended and Restated Services Agreement by and between the Registrant and Akcea Therapeutics, Inc., dated March 14, 2018, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2018 and incorporated herein by reference. | |
| 10.64 | New Strategic Neurology Drug Discovery and Development Collaboration, Option and License Agreement by and between the Registrant and Biogen MA Inc., dated April 19, 2018, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2018 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| 10.65 | Stock Purchase Agreement by and between the Registrant and Biogen MA Inc., dated April 19, 2018, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2018 and incorporated herein by reference. | |
| 10.66 | Second Amendment to Research, Collaboration, Option and License Agreement by and between the Registrant and Janssen Biotech Inc., dated August 7, 2018, filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2018 and incorporated herein by reference. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | |
| Factor B Development Collaboration, Option and License Agreement by and between the Registrant, F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc., dated October 9, 2018. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | ||
| Second Amended and Restated Strategic Neurology Drug Discovery and Development Collaboration, Option and License Agreement by and between the Registrant and Biogen MA Inc, dated October 17, 2018. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | ||
| Amendment #1 to the Strategic Collaboration Agreement by and between the Registrant and AstraZeneca AB, dated October 18, 2018. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | ||
| Amendment #4 to the Collaboration, License and Development Agreement by and between the Registrant and AstraZeneca AB, dated October 18, 2018. Portions of this exhibit have been omitted and separately filed with the SEC with a request for confidential treatment. | ||
| List of Subsidiaries for the Registrant. |
83
| Consent of Independent Registered Public Accounting Firm. | ||
| 24.1 | Power of Attorney – Included on the signature page of this Annual Report on Form 10-K. | |
| Certification by Chief Executive Officer Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | ||
| Certification by Chief Financial Officer Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | ||
32.1+ | Certification Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 101 | The following financial statements from the Ionis Pharmaceuticals, Inc. Annual Report on Form 10-K for the year ended December 31, 2018, formatted in Extensive Business Reporting Language (XBRL): (i) consolidated balance sheets, (ii) consolidated statements of operations, (iii) consolidated statements of comprehensive income (loss), (iv) consolidated statements of stockholders’ equity, (v) consolidated statements of cash flows, and (vi) notes to consolidated financial statements (detail tagged). |
| * | Indicates management compensatory plans and arrangements as required to be filed as exhibits to this Report pursuant to Item 14(c). |
| + | This certification is deemed not filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or otherwise subject to the liability of that section, nor shall it be deemed incorporated by reference into any filing under the Securities Act of 133, as amended, or the Securities Exchange Act of 1934, as amended. |
84
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized on the 1st day of March, 2019.
| IONIS PHARMACEUTICALS, INC. | ||
| By: | /s/ STANLEY T. CROOKE | |
| Stanley T. Crooke, M.D., Ph.D. | ||
| Chairman of the Board, President and Chief Executive Officer (Principal executive officer) | ||
POWER OF ATTORNEY
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Stanley T. Crooke and Elizabeth L. Hougen, or any of them, his or her attorney-in-fact, each with the power of substitution, for him or her in any and all capacities, to sign any amendments to this Report, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his or her substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signatures | Title | Date | ||
| /s/ STANLEY T. CROOKE | Chairman of the Board, President, and Chief Executive Officer | March 1, 2019 | ||
| Stanley T. Crooke, M.D., Ph.D. | (Principal executive officer) | |||
| /s/ ELIZABETH L. HOUGEN | Senior Vice President, Finance and Chief Financial Officer | March 1, 2019 | ||
| Elizabeth L. Hougen | (Principal financial and accounting officer) | |||
| /s/ B. LYNNE PARSHALL | Director and Senior Strategic Advisor | March 1, 2019 | ||
| B. Lynne Parshall, J.D. | ||||
| /s/ SPENCER R. BERTHELSEN | Director | March 1, 2019 | ||
| Spencer R. Berthelsen, M.D. | ||||
| /s/ BREAUX CASTLEMAN | Director | March 1, 2019 | ||
| Breaux Castleman | ||||
| /s/ MICHAEL HAYDEN | Director | March 1, 2019 | ||
| Michael Hayden, CM OBC MB ChB PhD FRCP(C) FRSC | ||||
| /s/ JOSEPH KLEIN | Director | March 1, 2019 | ||
| Joseph Klein, III | ||||
| /s/ JOSEPH LOSCALZO | Director | March 1, 2019 | ||
| Joseph Loscalzo, M.D., Ph.D. | ||||
| /s/ FREDERICK T. MUTO | Director | March 1, 2019 | ||
| Frederick T. Muto, Esq. | ||||
| /s/ PETER N. REIKES | Director | March 1, 2019 | ||
| Peter N. Reikes | ||||
| /s/ JOSEPH H. WENDER | Director | March 1, 2019 | ||
| Joseph H. Wender |
85
IONIS PHARMACEUTICALS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page | |
| Report of Independent Registered Public Accounting Firm | F-2 |
| Consolidated Balance Sheets at December 31, 2018 and 2017 (as revised) | F-3 |
| Consolidated Statements of Operations for the years ended December 31, 2018, 2017 and 2016 (as revised) | F-4 |
| Consolidated Statements of Comprehensive Loss for the years ended December 31, 2018, 2017 and 2016 (as revised) | F-5 |
| Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2018, 2017 and 2016 (as revised) | F-6 |
| Consolidated Statements of Cash Flows for the years ended December 31, 2018, 2017 and 2016 (as revised) | F-7 |
| Notes to Consolidated Financial Statements | F-9 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of Ionis Pharmaceuticals, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Ionis Pharmaceuticals, Inc. (the Company) as of December 31, 2018 and 2017, the related consolidated statements of operations, comprehensive income (loss), stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2018, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2018 and 2017, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2018, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2018, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework), and our report dated March 1, 2019 expressed an unqualified opinion thereon.
Adoption of ASU No. 2014-09, Revenue Recognition
As discussed in Note 1 to the consolidated financial statements, the Company changed its method of accounting for revenue for all years presented, 2016 through 2018, due to the adoption of ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606).
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 1989
San Diego, California
March 1, 2019
F-2
IONIS PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share data)
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| ASSETS | (as revised*) | |||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 278,820 | $ | 129,630 | ||||
| Short-term investments | 1,805,252 | 893,085 | ||||||
| Contracts receivable | 12,759 | 62,955 | ||||||
| Inventories | 8,582 | 9,982 | ||||||
| Other current assets | 102,473 | 73,082 | ||||||
| Total current assets | 2,207,886 | 1,168,734 | ||||||
| Property, plant and equipment, net | 132,160 | 121,907 | ||||||
| Patents, net | 24,032 | 22,004 | ||||||
| Long-term deferred tax assets | 290,796 | — | ||||||
| Deposits and other assets | 12,910 | 10,129 | ||||||
| Total assets | $ | 2,667,784 | $ | 1,322,774 | ||||
LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 28,660 | $ | 24,886 | ||||
| Accrued compensation | 29,268 | 25,151 | ||||||
| Accrued liabilities | 48,361 | 66,618 | ||||||
| Current portion of long-term obligations | 13,749 | 1,621 | ||||||
| Current portion of deferred contract revenue | 160,256 | 125,336 | ||||||
| Total current liabilities | 280,294 | 243,612 | ||||||
| Long-term deferred contract revenue | 567,359 | 108,026 | ||||||
| 1 percent convertible senior notes | 568,215 | 533,111 | ||||||
| Long-term obligations, less current portion | 4,914 | 12,974 | ||||||
| Long-term mortgage debt | 59,842 | 59,771 | ||||||
| Total liabilities | 1,480,624 | 957,494 | ||||||
| Stockholders’ equity: | ||||||||
| Common stock, $0.001 par value; 300,000,000 shares authorized, 137,928,828 and 124,976,373 shares issued and outstanding at December 31, 2018 and December 31, 2017, respectively | 138 | 125 | ||||||
| Additional paid-in capital | 2,047,250 | 1,553,681 | ||||||
| Accumulated other comprehensive loss | (32,016 | ) | (31,759 | ) | ||||
| Accumulated deficit | (967,293 | ) | (1,241,034 | ) | ||||
| Total Ionis stockholders’ equity | 1,048,079 | 281,013 | ||||||
| Noncontrolling interest in Akcea Therapeutics, Inc. | 139,081 | 84,267 | ||||||
| Total stockholders’ equity | 1,187,160 | 365,280 | ||||||
| Total liabilities and stockholders’ equity | $ | 2,667,784 | $ | 1,322,774 | ||||
| * | Our 2017 amounts are revised to reflect the new revenue recognition accounting guidance, which we adopted retrospectively in the first quarter of 2018. Refer to Note 1, Organization and Significant Accounting Policies, for further information. |
See accompanying notes.
F-3
IONIS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except for per share amounts)
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Revenue: | (as revised*) | |||||||||||
| Commercial revenue: | ||||||||||||
| SPINRAZA royalties | $ | 237,930 | $ | 112,540 | $ | 883 | ||||||
| TEGSEDI product sales, net | 2,237 | — | — | |||||||||
| Licensing and other royalty revenue | 14,755 | 7,474 | 21,884 | |||||||||
| Total commercial revenue | 254,922 | 120,014 | 22,767 | |||||||||
| Research and development revenue under collaborative agreements | 344,752 | 394,165 | 350,009 | |||||||||
| Total revenue | 599,674 | 514,179 | 372,776 | |||||||||
| Expenses: | ||||||||||||
| Cost of products sold | 1,820 | — | — | |||||||||
| Research, development and patent | 414,604 | 374,644 | 344,320 | |||||||||
| Selling, general and administrative | 244,622 | 108,488 | 48,616 | |||||||||
| Total operating expenses | 661,046 | 483,132 | 392,936 | |||||||||
| Income (loss) from operations | (61,372 | ) | 31,047 | (20,160 | ) | |||||||
| Other income (expense): | ||||||||||||
| Investment income | 30,187 | 8,179 | 5,472 | |||||||||
| Interest expense | (44,789 | ) | (44,752 | ) | (38,795 | ) | ||||||
| Loss on extinguishment of financing liability for leased facility | — | (7,689 | ) | — | ||||||||
| Loss on early retirement of debt | — | — | (3,983 | ) | ||||||||
| Other expenses | (182 | ) | (3,548 | ) | — | |||||||
| Loss before income tax benefit (expense) | (76,156 | ) | (16,763 | ) | (57,466 | ) | ||||||
| Income tax benefit (expense) | 291,141 | 5,980 | (2,934 | ) | ||||||||
| Net income (loss) | 214,985 | (10,783 | ) | (60,400 | ) | |||||||
| Net loss attributable to noncontrolling interest in Akcea Therapeutics, Inc. | 58,756 | 11,129 | — | |||||||||
| Net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | 273,741 | $ | 346 | $ | (60,400 | ) | |||||
| Basic net income (loss) per share | $ | 2.09 | $ | 0.15 | $ | (0.50 | ) | |||||
| Shares used in computing basic net income (loss) per share | 132,320 | 124,016 | 120,933 | |||||||||
| Diluted net income (loss) per share | $ | 2.07 | $ | 0.15 | $ | (0.50 | ) | |||||
| Shares used in computing diluted net income (loss) per share | 134,056 | 126,098 | 120,933 | |||||||||
| * | Our 2017 and 2016 amounts are revised to reflect the new revenue recognition accounting guidance, which we adopted retrospectively in the first quarter of 2018. Refer to Note 1, Organization and Significant Accounting Policies, for further information. |
See accompanying notes.
F-4
IONIS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(In thousands)
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised*) | ||||||||||||
| Net income (loss) | $ | 214,985 | $ | (10,783 | ) | $ | (60,400 | ) | ||||
| Unrealized losses on investments, net of tax | (280 | ) | (960 | ) | (17,219 | ) | ||||||
| Reclassification adjustment for realized (gains) losses included in net loss | — | (374 | ) | 447 | ||||||||
| Currency translation adjustment | 23 | (67 | ) | (21 | ) | |||||||
| Comprehensive income (loss) | 214,728 | (12,184 | ) | (77,193 | ) | |||||||
| Comprehensive loss attributable to noncontrolling interest in Akcea Therapeutics, Inc. | (58,781 | ) | (11,224 | ) | — | |||||||
| Comprehensive income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | 273,509 | $ | (960 | ) | $ | (77,193 | ) | ||||
| * | Our 2017 and 2016 amounts are revised to reflect the new revenue recognition accounting guidance, which we adopted retrospectively in the first quarter of 2018. Refer to Note 1, Organization and Significant Accounting Policies, for further information. |
See accompanying notes.
F-5
IONIS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
Years Ended December 31, 2018, 2017 (*as revised) and 2016 (*as revised)
(In thousands)
| Common Stock | Additional Paid in | Accumulated Other Comprehensive | Accumulated | Total Ionis Stockholders’ | Noncontrolling Interest in Akcea | Total Stockholders’ | ||||||||||||||||||||||||||
| Description | Shares | Amount | Capital | Loss | Deficit | Equity | Therapeutics, Inc. | Equity | ||||||||||||||||||||||||
| Balance at December 31, 2015 | 120,351 | $ | 120 | $ | 1,309,107 | $ | (13,565 | ) | $ | (1,094,872 | ) | $ | 200,790 | $ | — | $ | 200,790 | |||||||||||||||
| Cumulative adjustment related to adopting Topic 606 revenue recognition guidance | — | — | — | — | (86,108 | ) | (86,108 | ) | — | (86,108 | ) | |||||||||||||||||||||
| Net loss | — | — | — | — | (60,400 | ) | (60,400 | ) | — | (60,400 | ) | |||||||||||||||||||||
| Change in unrealized gains (losses), net of tax | — | — | — | (16,772 | ) | — | (16,772 | ) | — | (16,772 | ) | |||||||||||||||||||||
| Foreign currency translation | — | — | — | (21 | ) | — | (21 | ) | — | (21 | ) | |||||||||||||||||||||
| Issuance of common stock in connection with employee stock plans | 1,285 | 2 | 13,706 | — | — | 13,708 | — | 13,708 | ||||||||||||||||||||||||
| 2¾ percent convertible senior notes redemption, equity portion | — | — | (128,888 | ) | — | — | (128,888 | ) | — | (128,888 | ) | |||||||||||||||||||||
| 1 percent convertible senior notes, equity portion, net of issuance costs | — | — | 43,335 | — | — | 43,335 | — | 43,335 | ||||||||||||||||||||||||
| Stock-based compensation expense | — | — | 72,108 | — | — | 72,108 | — | 72,108 | ||||||||||||||||||||||||
| Excess tax benefits from stock-based compensation awards | — | — | 1,861 | — | — | 1,861 | — | 1,861 | ||||||||||||||||||||||||
| Balance at December 31, 2016 | 121,636 | $ | 122 | $ | 1,311,229 | $ | (30,358 | ) | $ | (1,241,380 | ) | $ | 39,613 | $ | — | $ | 39,613 | |||||||||||||||
| Net income | — | — | — | — | 346 | 346 | — | 346 | ||||||||||||||||||||||||
| Change in unrealized gains (losses), net of tax | — | — | — | (1,334 | ) | — | (1,334 | ) | — | (1,334 | ) | |||||||||||||||||||||
| Foreign currency translation | — | — | — | (67 | ) | — | (67 | ) | — | (67 | ) | |||||||||||||||||||||
| Novartis stock purchase | 1,631 | 2 | 71,737 | — | — | 71,739 | — | 71,739 | ||||||||||||||||||||||||
| Issuance of common stock in connection with employee stock plans | 1,709 | 1 | 22,931 | — | — | 22,932 | — | 22,932 | ||||||||||||||||||||||||
| Stock-based compensation expense | — | — | 85,975 | — | — | 85,975 | — | 85,975 | ||||||||||||||||||||||||
| Issuance of Akcea Therapeutics, Inc. common stock in conjunction with initial public offering | — | — | 157,270 | — | — | 157,270 | — | 157,270 | ||||||||||||||||||||||||
| Noncontrolling interest in Akcea Therapeutics, Inc. in conjunction with initial public offering | — | — | (90,351 | ) | — | — | (90,351 | ) | 90,381 | 30 | ||||||||||||||||||||||
| Noncontrolling interest in Akcea Therapeutics, Inc. | — | — | (5,110 | ) | — | — | (5,110 | ) | (6,114 | ) | (11,224 | ) | ||||||||||||||||||||
| Balance at December 31, 2017 | 124,976 | $ | 125 | $ | 1,553,681 | $ | (31,759 | ) | $ | (1,241,034 | ) | $ | 281,013 | $ | 84,267 | $ | 365,280 | |||||||||||||||
| Net income | — | — | — | — | 273,741 | 273,741 | — | 273,741 | ||||||||||||||||||||||||
| Change in unrealized gains (losses), net of tax | — | — | — | (280 | ) | — | (280 | ) | — | (280 | ) | |||||||||||||||||||||
| Foreign currency translation | — | — | — | 23 | — | 23 | — | 23 | ||||||||||||||||||||||||
| Biogen stock purchase | 11,502 | 11 | 447,954 | — | — | 447,965 | — | 447,965 | ||||||||||||||||||||||||
| Issuance of common stock in connection with employee stock plans | 1,451 | 2 | 27,898 | — | — | 27,900 | — | 27,900 | ||||||||||||||||||||||||
| Share-based compensation expense | — | — | 131,312 | — | — | 131,312 | — | 131,312 | ||||||||||||||||||||||||
| Noncontrolling interest in Akcea Therapeutics, Inc. | — | — | (113,595 | ) | — | — | (113,595 | ) | 54,814 | (58,781 | ) | |||||||||||||||||||||
| Balance at December 31, 2018 | 137,929 | $ | 138 | $ | 2,047,250 | $ | (32,016 | ) | $ | (967,293 | ) | $ | 1,048,079 | $ | 139,081 | $ | 1,187,160 | |||||||||||||||
| * | Our 2017 and 2016 amounts are revised to reflect the new revenue recognition accounting guidance, which we adopted retrospectively in the first quarter of 2018. Refer to Note 1, Organization and Significant Accounting Policies, for further information. |
See accompanying notes.
F-6
IONIS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised*) | ||||||||||||
| Operating activities: | ||||||||||||
| Net income (loss) | $ | 214,985 | $ | (10,783 | ) | $ | (60,400 | ) | ||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | ||||||||||||
| Depreciation | 10,706 | 6,708 | 7,481 | |||||||||
| Amortization of patents | 1,822 | 1,641 | 1,552 | |||||||||
| Amortization of premium (discount) on investments, net | (1,013 | ) | 6,752 | 6,813 | ||||||||
| Amortization of debt issuance costs | 1,810 | 1,616 | 1,225 | |||||||||
| Amortization of convertible senior notes discount | 33,363 | 30,920 | 23,890 | |||||||||
| Amortization of long-term financing liability for leased facility | — | 3,659 | 6,693 | |||||||||
| Stock-based compensation expense | 131,312 | 85,975 | 72,108 | |||||||||
| Gain on investment in Regulus Therapeutics Inc. | — | (374 | ) | — | ||||||||
| Loss on extinguishment of financing liability for leased facility | — | 7,689 | — | |||||||||
| Loss on early retirement of debt | — | — | 3,983 | |||||||||
| Deferred income taxes (including benefit from valuation allowance release) | (290,516 | ) | — | — | ||||||||
| Non-cash losses related to patents, licensing, property, plant and equipment and strategic investments | 1,012 | 3,302 | 2,297 | |||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Contracts receivable | 47,595 | 45,088 | (96,687 | ) | ||||||||
| Inventories | 1,400 | (2,493 | ) | (590 | ) | |||||||
| Other current and long-term assets | (29,348 | ) | (58,367 | ) | 1,603 | |||||||
| Long-term income tax receivable | (223 | ) | (9,114 | ) | — | |||||||
| Accounts payable | (655 | ) | 1,784 | (10,677 | ) | |||||||
| Income taxes | (710 | ) | 435 | 1,069 | ||||||||
| Accrued compensation | 4,117 | 965 | 8,121 | |||||||||
| Accrued liabilities and deferred rent | (17,023 | ) | 28,564 | 4,720 | ||||||||
| Deferred contract revenue | 494,254 | 30,182 | (85,306 | ) | ||||||||
| Net cash provided by (used in) operating activities | 602,888 | 174,149 | (112,105 | ) | ||||||||
| Investing activities: | ||||||||||||
| Purchases of short-term investments | (1,794,735 | ) | (877,810 | ) | (300,912 | ) | ||||||
| Proceeds from the sale of short-term investments | 882,824 | 557,369 | 364,572 | |||||||||
| Purchases of property, plant and equipment | (13,608 | ) | (34,764 | ) | (7,107 | ) | ||||||
| Acquisition of licenses and other assets, net | (4,044 | ) | (3,093 | ) | (4,421 | ) | ||||||
| Purchase of strategic investments | — | (2,500 | ) | — | ||||||||
| Proceeds from the sale of Regulus Therapeutics, Inc. | — | 2,507 | 4,467 | |||||||||
| Net cash (used in) provided by investing activities | (929,563 | ) | (358,291 | ) | 56,599 | |||||||
| Financing activities: | ||||||||||||
| Proceeds from equity, net | 27,900 | 22,931 | 13,417 | |||||||||
| Proceeds from issuance of common stock in Akcea Therapeutics, Inc. from its initial public offering, net of underwriters’ discount | — | 110,438 | — | |||||||||
| Proceeds from building mortgage debt, net of issuance costs | — | 59,750 | — | |||||||||
| Proceeds from the issuance of common stock to Biogen | 447,965 | — | — | |||||||||
| Proceeds from the issuance of common stock to Novartis | — | 71,737 | — | |||||||||
| Proceeds from borrowing on line of credit facility | — | — | 4,000 | |||||||||
| Proceeds from the sale of Akcea Therapeutics, Inc. common stock to Novartis in a private placement | — | 50,000 | — | |||||||||
| Offering costs paid | — | (2,037 | ) | (818 | ) | |||||||
| Payment to settle financing liability for leased facility | — | (80,133 | ) | — | ||||||||
| Excess tax benefits from stock-based compensation awards | — | — | 1,861 | |||||||||
| Principal payments on debt and capital lease obligations | — | (3,599 | ) | (7,066 | ) | |||||||
| Net cash provided by financing activities | 475,865 | 229,087 | 11,394 | |||||||||
| Net increase (decrease) in cash and cash equivalents | 149,190 | 44,945 | (44,112 | ) | ||||||||
| Cash and cash equivalents at beginning of year | 129,630 | 84,685 | 128,797 | |||||||||
| Cash and cash equivalents at end of year | $ | 278,820 | $ | 129,630 | $ | 84,685 | ||||||
F-7
IONIS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised*) | ||||||||||||
| Supplemental disclosures of cash flow information: | ||||||||||||
| Interest paid | $ | 9,592 | $ | 8,035 | $ | 7,313 | ||||||
| Supplemental disclosures of non-cash investing and financing activities: | ||||||||||||
| Amounts accrued for capital and patent expenditures | $ | 4,428 | $ | 1,983 | $ | 3,439 | ||||||
| Purchases of property, plant and equipment included in long-term obligations | $ | 3,350 | $ | — | $ | — | ||||||
| 1 percent convertible senior notes principal issued related to our December 2016 debt exchange | $ | — | $ | — | $ | 185,450 | ||||||
2¾ percent convertible senior notes principal extinguished related to our December 2016 debt exchange | $ | — | $ | — | $ | 61,099 | ||||||
| Unpaid deferred offering costs | $ | — | $ | — | $ | 291 | ||||||
| * | Our 2017 and 2016 amounts are revised to reflect the new revenue recognition accounting guidance, which we adopted retrospectively in the first quarter of 2018. Refer to Note 1, Organization and Significant Accounting Policies, for further information. |
See accompanying notes.
F-8
IONIS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and Significant Accounting Policies
Basis of Presentation
In our consolidated financial statements we included the accounts of Ionis Pharmaceuticals, Inc. (“we”, “us” or “our”) and the consolidated results of our majority-owned affiliate, Akcea Therapeutics, Inc., which we formed in December 2014. In July 2017, Akcea completed an initial public offering, or IPO, and therefore beginning in July 2017, we no longer own 100 percent of Akcea. From the closing of Akcea’s IPO in July 2017 through mid-April 2018, we owned approximately 68 percent of Akcea. In the second, third and fourth quarters of 2018, we received additional shares of Akcea’s stock related to our license of TEGSEDI and AKCEA-TTR-LRx to Akcea, increasing our ownership percentage to approximately 75 percent. We reflected the increase in our ownership in these financial statements. In the first quarter of 2019, Akcea will pay us a $75 million sublicense fee in Akcea common stock, as a result of Novartis’ license of AKCEA-APO(a)-LRx in February 2019. We will receive 2.8 million shares of Akcea common stock for the sublicense fee. Refer to the noncontrolling interest in Akcea section in this note for further information related to our accounting for our investment in Akcea.
Organization and Business Activity
We incorporated in California on January 10, 1989. In conjunction with our IPO, we reorganized as a Delaware corporation in April 1991. We were organized principally to develop human therapeutic medicines using antisense technology. In December 2015, we changed our name from Isis Pharmaceuticals, Inc. to Ionis Pharmaceuticals, Inc.
Basic and Diluted Net Income (Loss) per Share
Basic net income (loss) per share
We compute basic net income (loss) per share by dividing the total net income (loss) attributable to our common stockholders by our weighted-average number of common shares outstanding during the period.
The calculation of total net income (loss) attributable to our common stockholders for 2018 and 2017 considered our net income for Ionis on a stand-alone basis plus our share of Akcea’s net loss for the period. During 2016, we owned 100 percent of Akcea. To calculate the portion of Akcea’s net loss attributable to our ownership for 2018 and 2017, we multiplied Akcea’s loss per share by the weighted average shares we owned in Akcea during the period. As a result of this calculation, our total net income (loss) available to Ionis common stockholders for the calculation of net income (loss) per share is different than net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders in our consolidated statements of operations for 2018 and 2017.
Our basic net income per share for 2018, was calculated as follows (in thousands, except per share amounts):
| Year Ended December 31, 2018 | Weighted Average Shares Owned in Akcea | Akcea’s Net Income (Loss) Per Share | Ionis’ Portion of Akcea’s Net Loss | |||||||||
| Common shares | 59,812 | $ | (2.74 | ) | $ | (163,938 | ) | |||||
| Akcea’s net loss attributable to our ownership | $ | (163,938 | ) | |||||||||
| Ionis’ stand-alone net income | 440,806 | |||||||||||
| Net income available to Ionis common stockholders | $ | 276,868 | ||||||||||
| Weighted average shares outstanding | 132,320 | |||||||||||
| Basic net income per share | $ | 2.09 | ||||||||||
Prior to Akcea’s IPO in July 2017, we owned Akcea series A convertible preferred stock, which included a six percent cumulative dividend. Upon completion of Akcea’s IPO in July 2017, our preferred stock was converted into common stock on a 1:1 basis. The preferred stock dividend was not paid at the IPO because the IPO was not a liquidation event or a change in control. During 2017, Akcea used a two-class method to compute its net income (loss) per share because it had both common and preferred shares outstanding during the periods. The two-class method required Akcea to calculate its net income (loss) per share for each class of stock by dividing total distributable losses applicable to preferred and common stock, including the six percent cumulative dividend contractually due to series A convertible preferred shareholders, by the weighted-average of preferred and common shares outstanding during the requisite period. Since Akcea used the two-class method, accounting rules required us to include our portion of Akcea’s net income (loss) per share for both Akcea’s common and preferred shares that we owned in our calculation of basic and diluted net income (loss) per share for year ended December 31, 2017.
F-9
We calculated our basic net income per share for 2017 as follows (in thousands, except per share amounts):
| Year Ended December 31, 2017 | Weighted Average Shares Owned in Akcea | Akcea’s Net Loss Per Share | Ionis’ Portion of Akcea’s Net Loss | |||||||||
| Common shares | 20,669 | $ | (3.08 | ) | $ | (63,638 | ) | |||||
| Preferred shares | 15,748 | (1.80 | ) | (28,346 | ) | |||||||
| Akcea’s net loss attributable to our ownership | $ | (91,984 | ) | |||||||||
| Ionis’ stand-alone net income | 110,776 | |||||||||||
| Net income available to Ionis common stockholders | $ | 18,792 | ||||||||||
| Weighted average shares outstanding | 124,016 | |||||||||||
| Basic net income per share | $ | 0.15 | ||||||||||
Dilutive net income (loss per share)
For 2018 and 2017, we had net income available to Ionis common stockholders. As a result, we computed diluted net income per share using the weighted-average number of common shares and dilutive common equivalent shares outstanding during those periods.
We calculated our diluted net income per share for 2018 as follows (in thousands except per share amounts):
| Year Ended December 31, 2018 | Income (Numerator) | Shares (Denominator) | Per-Share Amount | |||||||||
| Net income available to Ionis common stockholders | $ | 276,868 | 132,320 | $ | 2.09 | |||||||
| Effect of dilutive securities: | ||||||||||||
| Shares issuable upon exercise of stock options | — | 1,216 | ||||||||||
| Shares issuable upon restricted stock award issuance | — | 514 | ||||||||||
| Shares issuable related to our ESPP | — | 6 | ||||||||||
| Income available to Ionis common stockholders, plus assumed conversions | $ | 276,868 | 134,056 | $ | 2.07 | |||||||
We calculated our diluted net income per share for 2017 as follows (in thousands except per share amounts):
| Year Ended December 31, 2017 | Income (Numerator) | Shares (Denominator) | Per-Share Amount | |||||||||
| Net income available to Ionis common stockholders | $ | 18,792 | 124,016 | $ | 0.15 | |||||||
| Effect of dilutive securities: | ||||||||||||
| Shares issuable upon exercise of stock options | — | 1,619 | ||||||||||
| Shares issuable upon restricted stock award issuance | — | 459 | ||||||||||
| Shares issuable related to our ESPP | — | 4 | ||||||||||
| Income available to Ionis common stockholders, plus assumed conversions | $ | 18,792 | 126,098 | $ | 0.15 | |||||||
For 2018 and 2017, the calculation excluded our convertible notes because the effect on diluted earnings per share was anti-dilutive.
For 2016, we incurred a net loss; therefore, we did not include dilutive common equivalent shares in the computation of diluted net loss per share because the effect would have been anti-dilutive. Common stock from the following would have had an anti-dilutive effect on net loss per share:
| ● | 1 percent convertible senior notes; |
| ● | 2¾ percent convertible senior notes; |
| ● | Dilutive stock options; |
| ● | Unvested restricted stock units; and |
| ● | Employee Stock Purchase Plan, or ESPP. |
Revenue Recognition
Adoption of New Revenue Recognition Accounting Standard (Topic 606)
In May 2014, the FASB issued accounting guidance on the recognition of revenue from customers. This guidance supersedes the revenue recognition requirements we previously followed in Accounting Standards Codification, or ASC, Topic 605, Revenue Recognition, or Topic 605, and created a new Topic 606, Revenue from Contracts with Customers, or Topic 606. Under Topic 606, an entity will recognize revenue when it transfers control of promised goods or services to customers in an amount that reflects what the entity expects to receive in exchange for the goods or services. Further, an entity will recognize revenue upon satisfying the performance obligation(s) under the related contract. We adopted Topic 606 on January 1, 2018 under the full retrospective approach, which required us to revise our prior period revenue. Under Topic 606, we were required to review all of our ongoing collaboration agreements in which we recognized revenue after January 1, 2016. We were required to assess what our revenue would have been for the period from January 1, 2016 to December 31, 2017 under Topic 606. As a result of this analysis, we determined that the cumulative revenue we would have recognized under Topic 606 decreased by $86.1 million. We recorded this amount as a cumulative adjustment to our accumulated deficit as of January 1, 2016 on our revised statement of stockholders’ equity. We have labeled our prior period financial statements “as revised” to indicate the change required under the accounting rules.
F-10
The following tables summarize the adjustments we were required to make to amounts we originally reported in 2017 and 2016 to adopt Topic 606 (in thousands, except per share amounts):
Consolidated Balance Sheet
| At December 31, 2017 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Current portion of deferred contract revenue | $ | 106,465 | $ | 18,871 | $ | 125,336 | ||||||
| Long-term portion of deferred contract revenue | $ | 72,708 | $ | 35,318 | $ | 108,026 | ||||||
| Accumulated deficit | $ | (1,187,398 | ) | $ | (53,636 | ) | $ | (1,241,034 | ) | |||
| Noncontrolling interest in Akcea Therapeutics, Inc. | $ | 87,847 | $ | (3,580 | ) | $ | 84,267 | |||||
| Total stockholders’ equity | $ | 418,719 | $ | (53,439 | ) | $ | 365,280 | |||||
Consolidated Statements of Operations
| Year Ended December 31, 2017 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Revenue: | ||||||||||||
| Commercial revenue: | ||||||||||||
| SPINRAZA royalties | $ | 112,540 | $ | - | $ | 112,540 | ||||||
| Licensing and other royalty revenue | 9,519 | (2,045 | ) | 7,474 | ||||||||
| Total commercial revenue | 122,059 | (2,045 | ) | 120,014 | ||||||||
| Research and development revenue under collaborative agreements | 385,607 | 8,558 | 394,165 | |||||||||
| Total revenue | $ | 507,666 | $ | 6,513 | $ | 514,179 | ||||||
| Income from operations | $ | 24,534 | $ | 6,513 | $ | 31,047 | ||||||
| Net income (loss) | $ | (17,296 | ) | $ | 6,513 | $ | (10,783 | ) | ||||
| Net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | (5,970 | ) | $ | 6,316 | $ | 346 | |||||
| Net income per share, basic and diluted | $ | 0.08 | $ | 0.07 | $ | 0.15 | ||||||
| Year Ended December 31, 2016 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Revenue: | ||||||||||||
| Commercial revenue: | ||||||||||||
| SPINRAZA royalties | $ | 883 | $ | — | $ | 883 | ||||||
| Licensing and other royalty revenue | 19,839 | 2,045 | 21,884 | |||||||||
| Total commercial revenue | 20,722 | 2,045 | 22,767 | |||||||||
| Research and development revenue under collaborative agreements | 325,898 | 24,111 | 350,009 | |||||||||
| Total revenue | $ | 346,620 | $ | 26,156 | $ | 372,776 | ||||||
| Income (loss) from operations | $ | (46,316 | ) | $ | 26,156 | $ | (20,160 | ) | ||||
| Net income (loss) | $ | (86,556 | ) | $ | 26,156 | $ | (60,400 | ) | ||||
| Net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | (86,556 | ) | $ | 26,156 | $ | (60,400 | ) | ||||
| Net income (loss) per share, basic and diluted | $ | (0.72 | ) | $ | 0.22 | $ | (0.50 | ) | ||||
F-11
Consolidated Statements of Cash Flows
| Year Ended December 31, 2017 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Net income (loss) | $ | (17,296 | ) | $ | 6,513 | $ | (10,783 | ) | ||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | ||||||||||||
| Deferred contract revenue | $ | 36,695 | $ | (6,513 | ) | $ | 30,182 | |||||
| Cash and cash equivalents at beginning of year | $ | 84,685 | $ | — | $ | 84,685 | ||||||
| Cash and cash equivalents at end of year | $ | 129,630 | $ | — | $ | 129,630 | ||||||
| Year Ended December 31, 2016 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Net income (loss) | $ | (86,556 | ) | $ | 26,156 | $ | (60,400 | ) | ||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | ||||||||||||
| Deferred contract revenue | $ | (59,150 | ) | $ | (26,156 | ) | $ | (85,306 | ) | |||
| Cash and cash equivalents at beginning of year | $ | 128,797 | $ | — | $ | 128,797 | ||||||
| Cash and cash equivalents at end of year | $ | 84,685 | $ | — | $ | 84,685 | ||||||
Under Topic 606, compared to Topic 605, our total revenue increased $6.5 million for 2017 and $26.2 million for 2016. The change in our revenue was primarily due to:
| ● | A change in how we recognize milestone payments: Topic 606 requires us to amortize more of the milestone payments we achieve, rather than recognizing the milestone payments in full in the period in which we achieved the milestone event as we did under Topic 605. This change resulted in an increase in R&D revenue recognized for 2017 and 2016 of $23.7 million and $24.1 million, respectively. |
| ● | A change in how we calculate revenue for payments we are recognizing into revenue over time: Under Topic 605, we amortized payments into revenue evenly over the period of our obligations. When we made a change to our estimated completion period, we recognized that change on a prospective basis. Under Topic 606, we are required to use an input method to determine the amount we amortize each reporting period. Each period we review our “inputs” such as our level of effort expended, including the time we estimate it will take us to complete the activities or costs incurred, relative to the total expected inputs to satisfy the performance obligation. For certain collaborations, such as Bayer and Novartis, the input method resulted in a change to the revenue we had previously recognized using a straight-line amortization method. This change resulted in a decrease in our R&D revenue of $15.1 million for 2017. This change did not result in an impact to our 2016 R&D revenue. |
Our updated revenue recognition policy reflecting Topic 606 is as follows:
Our Revenue Sources
We generally recognize revenue when we have satisfied all contractual obligations and are reasonably assured of collecting the resulting receivable. We are often entitled to bill our customers and receive payment from our customers in advance of recognizing the revenue. In the instances in which we have received payment from our customers in advance of recognizing revenue, we include the amounts in deferred revenue on our consolidated balance sheet.
Commercial Revenue: SPINRAZA royalties and Licensing and other royalty revenue
We earn commercial revenue primarily in the form of royalty payments on net sales of SPINRAZA. We will also recognize as commercial revenue future sales milestone payments and royalties we earn under our partnerships.
Commercial Revenue: TEGSEDI Product Sales, net
We began adding product sales from TEGSEDI to our commercial revenue in the fourth quarter of 2018. In the U.S., TEGSEDI is distributed through an exclusive distribution agreement with a third-party logistics company, or 3PL, that takes title to TEGSEDI. The 3PL is our sole customer in the U.S. The 3PL then distributes TEGSEDI to a specialty pharmacy and a specialty distributor, which we collectively refer to as wholesalers, who then distribute TEGSEDI to health care providers and patients. In Germany, TEGSEDI is distributed through a non-exclusive distribution model with a 3PL that takes title to TEGSEDI. The 3PL is our sole customer in Germany. The 3PL in Germany then distributes TEGSEDI to hospitals and pharmacies.
F-12
Research and development revenue under collaborative agreements
We often enter into collaboration agreements to license and sell our technology on an exclusive or non-exclusive basis. Our collaboration agreements typically contain multiple elements, or performance obligations, including technology licenses or options to obtain technology licenses, research and development, or R&D, services, and manufacturing services.
Our collaboration agreements are detailed in Note 6, Collaborative Arrangements and Licensing Agreements. Under each collaboration note we discuss our specific revenue recognition conclusions, including our significant performance obligations under each collaboration.
Steps to Recognize Revenue
We use a five step process to determine the amount of revenue we should recognize and when we should recognize it. The five step process is as follows:
| 1. | Identify the contract |
Accounting rules require us to first determine if we have a contract with our partner, including confirming that we have met each of the following criteria:
| ● | We and our partner approved the contract and we are both committed to perform our obligations; |
| ● | We have identified our rights, our partner’s rights and the payment terms; |
| ● | We have concluded that the contract has commercial substance, meaning that the risk, timing, or amount of our future cash flows is expected to change as a result of the contract; and |
| ● | We believe collectability is probable. |
| 2. | Identify the performance obligations |
We next identify the distinct goods and services we are required to provide under the contract. Accounting rules refer to these as our performance obligations. We typically have only one performance obligation at the inception of a contract, which is to perform R&D services.
Often times we enter into a collaboration agreement in which we provide our partner with an option to license a medicine in the future. We may also provide our partner with an option to request that we provide additional goods or services in the future, such as active pharmaceutical ingredient, or API. We evaluate whether these options are material rights at the inception of the agreement. If we determine an option is a material right, we will consider the option a separate performance obligation. Historically, we have concluded that the options we grant to license a medicine in the future or to provide additional goods and services as requested by our partner are not material rights. These items are contingent upon future events that may not occur. When a partner exercises its option to license a medicine or requests additional goods or services, then we identify a new performance obligation for that item.
In some cases, we deliver a license at the start of an agreement. If we determine that our partner has full use of the license and we do not have any additional performance obligations related to the license after delivery, then we consider the license to be a separate performance obligation.
| 3. | Determine the transaction price |
We then determine the transaction price by reviewing the amount of consideration we are eligible to earn under the collaboration agreement, including any variable consideration. Under our collaboration agreements, consideration typically includes fixed consideration in the form of an upfront payment and variable consideration in the form of potential milestone payments, license fees and royalties. At the start of an agreement, our transaction price usually consists of only the upfront payment. We do not typically include any payments we may receive in the future in our initial transaction price because the payments are not probable. We reassess the total transaction price at each reporting period to determine if we should include additional payments in the transaction price.
Milestone payments are our most common type of variable consideration. We recognize milestone payments using the most likely amount method because we will either receive the milestone payment or we will not, which makes the potential milestone payment a binary event. The most likely amount method requires us to determine the likelihood of earning the milestone payment. We include a milestone payment in the transaction price once it is probable we will achieve the milestone event. Most often, we do not consider our milestone payments probable until we or our partner achieve the milestone event because the majority of our milestone payments are contingent upon events that are not within our control and are usually based on scientific progress. For example, in the first quarter of 2019, we earned a $35 million milestone payment from Roche when it dosed the first patient in the Phase 3 study of IONIS-HTTRx. At December 31, 2018, we determined it was not probable that we could earn this milestone payment. As such, we did not recognize any revenue associated with it in 2018.
| 4. | Allocate the transaction price |
Next, we allocate the transaction price to each of our performance obligations. When we have to allocate the transaction price to more than one performance obligation, we make estimates of the relative stand-alone selling price of each performance obligation because we do not typically sell our goods or services on a stand-alone basis. We then allocate the transaction price to each performance obligation based on the relative stand-alone selling price.
F-13
We may engage a third party, independent valuation specialist to assist us with determining a stand-alone selling price for collaborations in which we deliver a license at the start of an agreement. We estimate the stand-alone selling price of these licenses using valuation methodologies, such as the relief from royalty method. Under this method, we estimate the amount of income, net of taxes, for the license. We then discount the projected income to present value. The significant inputs we use to determine the projected income of a license could include:
| ● | Estimated future product sales; |
| ● | Estimated royalties on future product sales; |
| ● | Contractual milestone payments; |
| ● | Expenses we expect to incur; |
| ● | Income taxes; and |
| ● | A discount rate. |
We typically estimate the selling price of R&D services by using our internal estimates of the cost to perform the specific services. The significant inputs we use to determine the selling price of our R&D services include:
| ● | The number of internal hours we estimate we will spend performing these services; |
| ● | The estimated cost of work we will perform; |
| ● | The estimated cost of work that we will contract with third parties to perform; and |
| ● | The estimated cost of API we will use. |
For purposes of determining the stand-alone selling price of the R&D services we perform and the API we will deliver, accounting guidance requires us to include a markup for a reasonable profit margin.
We do not reallocate the transaction price after the start of an agreement to reflect subsequent changes in stand-alone selling prices.
| 5. | Recognize revenue |
We recognize revenue in one of two ways, over time or at a point in time. We recognize revenue over time when we are executing on our performance obligation over time and our partner receives benefit over time. For example, we recognize revenue over time when we provide R&D services. We recognize revenue at a point in time when our partner receives full use of an item at a specific point in time. For example, we recognize revenue at a point in time when we deliver a license or API to a partner.
For R&D services that we recognize over time, we measure our progress using an input method. The input methods we use are based on the effort we expend or costs we incur toward the satisfaction of our performance obligation. We estimate the amount of effort we expend, including the time we estimate it will take us to complete the activities, or costs we incur in a given period, relative to the estimated total effort or costs to satisfy the performance obligation. This results in a percentage that we multiply by the transaction price to determine the amount of revenue we recognize each period. This approach requires us to make numerous estimates and use significant judgement. If our estimates or judgements change over the course of the collaboration, they may affect the timing and amount of revenue that we recognize in the current and future periods.
The following are examples of when we typically recognize revenue based on the types of payments we receive.
Commercial Revenue: SPINRAZA royalties and Licensing and other royalty revenue
We recognize royalty revenue in the period in which the counterparty sells the related product, which in certain cases may require us to estimate our royalty revenue. We recognize royalties from SPINRAZA sales in the period Biogen records the sale of SPINRAZA. Our accounting for SPINRAZA royalties did not change as a result of adopting Topic 606.
Commercial Revenue: TEGSEDI Product Sales, net
We recognize TEGSEDI product sales in the period when our customer obtains control of TEGSEDI, which occurs at a point in time upon transfer of title to the customer. We classify payments to customers or other parties in the distribution channel for services that are distinct and priced at fair value as selling, general and administrative expenses in our consolidated statements of operations. Otherwise payments to customers or other parties in the distribution channel that do not meet those criteria are classified as a reduction of revenue, as discussed further below. We exclude from revenues, taxes collected from customers relating to product sales and remitted to governmental authorities.
Reserves for TEGSEDI Product Sales
We record TEGSEDI product sales at our net sales price, or transaction price. We include in our transaction price estimated reserves for discounts, returns, chargebacks, rebates, co-pay assistance and other allowances that we offer within contracts between us and our customers, wholesalers, health care providers and other indirect customers. We estimate our reserves using the amounts we have earned or what we can claim on the associated sales. We classify our reserves as reductions of accounts receivable when the amount is payable to our customer or a current liability when the amount is payable to a party other than our customer in our consolidated balance sheet. In certain cases, our estimates include a range of possible outcomes that are probability-weighted for relevant factors such as our historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. Overall, our reserves reflect our best estimates under the terms of our respective contracts. When calculating our reserves and related product sales, we only recognize amounts to the extent that we consider it probable that we would not have to reverse in a future period a significant amount of the cumulative sales we previously recognized. The actual amounts we receive may ultimately differ from our reserve estimates. If actual amounts in the future vary from our estimates, we will adjust these estimates, which would affect our net TEGSEDI product sales in the respective period.
F-14
The following are the components of variable consideration related to TEGSEDI product sales:
Chargebacks: In the U.S., we estimate obligations resulting from contractual commitments with the government and other entities to sell products to qualified healthcare providers at prices lower than the list prices charged to our U.S. customer. Our U.S. customer charges us for the difference between what it pays for the product and the selling price to the qualified healthcare providers. We record reserves for these chargebacks related to TEGSEDI product sales to our U.S. customer during the reporting period. We also estimate the amount of product remaining in the distribution channel inventory at the end of the reporting period that we expect our customer to sell to wholesalers in future periods.
Government rebates: We are subject to discount obligations under government programs, including Medicaid programs and Medicare in the U.S. We estimate Medicaid and Medicare rebates based on a range of possible outcomes that are probability-weighted for the estimated payer mix. We record these reserves as an accrued liability on our consolidated balance sheet with a corresponding offset reducing our TEGSEDI product sales in the same period we recognize the related sale. For Medicare rebates, we also estimate the number of patients in the prescription drug coverage gap for whom we will owe an additional liability under the Medicare Part D program. On a quarterly basis, we update our estimates and record any adjustments in the period that we identify the adjustments. In Germany, pharmaceutical companies must grant a specified rebate percentage to the German government. We include this rebate in the same period we recognize the related TEGSEDI product sales, resulting in a reduction of product sales.
Trade discounts and allowances: We provide customary invoice discounts on TEGSEDI product sales to our U.S. customer for prompt payment. We record this discount as a reduction of TEGSEDI product sales in the period in which we recognize the related product revenue. In addition, we receive and pay for various distribution services from our U.S. customer and wholesalers in our U.S. distribution channel. For services we receive that are either not distinct from the sale of TEGSEDI or for which we cannot reasonably estimate the fair value, we classify such fees as a reduction of TEGSEDI product sales.
Product Returns: Our U.S. customer has return rights and the wholesalers have limited return rights primarily related to the expiration date of the TEGSEDI product. We estimate the amount of TEGSEDI product sales that our customer may return. We record our return estimate as an accrued refund liability on our consolidated balance sheet with a corresponding offset reducing our TEGSEDI product sales, in the same period we recognize the related sale. Based on our distribution model for TEGSEDI, contractual inventory limits with our customer and wholesalers and the price of TEGSEDI, we believe we will have minimal returns. Our customer in Germany only takes title to the product once it receives an order from a hospital or pharmacy and therefore does not maintain any inventory of TEGSEDI, as such we do not estimate returns in Germany.
Other incentives: In the U.S., we estimate reserves for other incentives including co-payment assistance we provide to patients with commercial insurance who have coverage and reside in states that allow co-payment assistance. We record a reserve for the amount we estimate we will pay for co-payment assistance. We base our reserve on the number of estimated claims and our estimate of the cost per claim related to TEGSEDI product sales that we have recognized as revenue. We record our other incentive reserve estimates as an accrued liability on our consolidated balance sheet with a corresponding offset reducing our TEGSEDI product sales, in the same period we recognize the related sale.
F-15
Research and development revenue under collaboration agreements:
Upfront Payments
When we enter into a collaboration agreement with an upfront payment, we typically record the entire upfront payment as deferred revenue if our only performance obligation is for R&D services we will provide in the future. We amortize the upfront payment into revenue as we perform the R&D services. For example, under our new collaboration agreement with Roche to develop IONIS-FB-LRx for the treatment of complement-mediated diseases, we received a $75 million upfront payment in the fourth quarter of 2018. We allocated the upfront payment to our single performance obligation, R&D services. We are amortizing the $75 million upfront payment using an input method over the estimated period of time we are providing R&D services. Refer to Note 6, Collaborative Arrangements and Licensing Agreements, for further discussion. Under Topic 605, we amortized upfront payments evenly over the period of our obligation.
Milestone Payments
We are required to include additional consideration in the transaction price when it is probable. We typically include milestone payments for R&D services in the transaction price when they are achieved. We include these milestone payments when they are achieved because there is considerable uncertainty in the research and development processes that trigger these payments under our collaboration agreements. Similarly, we include approval milestone payments in the transaction price once the medicine is approved by the applicable regulatory agency. We will recognize sales based milestone payments in the period we achieve the milestone under the sales-based royalty exception allowed under accounting rules.
We recognize milestone payments that relate to an ongoing performance obligation over our period of performance. For example, in the third quarter of 2017, we initiated a Phase 1/2a clinical study of IONIS-MAPTRx in patients with mild Alzheimer’s disease. We earned a $10 million milestone payment from Biogen related to the initiation of this study. Under Topic 606, we added this payment to the transaction price and allocated it to our R&D services performance obligation. We are recognizing revenue from this milestone payment over our estimated period of performance. Under Topic 605, this milestone payment was recognized in full in the third quarter of 2017, which was the period in which we achieved the milestone event.
Conversely, we recognize in full those milestone payments that we earn based on our partners’ activities when our partner achieves the milestone event. For example, in the third quarter of 2018, we recognized a $10 million milestone payment when AstraZeneca initiated a Phase 1 study of IONIS-AZ4-2.5-LRx. We concluded that the milestone payment was not related to our R&D services performance obligation. Therefore, we recognized this milestone payment in full in the third quarter of 2018 because we do not have any performance obligations related to this milestone payment. Our revenue recognition of milestone payments we earn based on our partners’ activities did not change as a result of adopting Topic 606.
License Fees
We generally recognize as revenue the total amount we determine to be the stand-alone selling price of a license when we deliver the license to our partner. This is because our partner has full use of the license and we do not have any additional performance obligations related to the license after delivery. For example, in the fourth quarter of 2018, we earned a $35 million license fee when Biogen licensed IONIS-SOD1Rx from us. Our recognition of license fees did not change as a result of adopting Topic 606.
Amendments to Agreements
From time to time we amend our collaboration agreements. When this occurs, we are required to assess the following items to determine the accounting for the amendment:
| 1) | If the additional goods and/or services are distinct from the other performance obligations in the original agreement; and |
| 2) | If the goods and/or services are at a stand-alone selling price. |
If we conclude the goods and/or services in the amendment are distinct from the performance obligations in the original agreement and at a stand-alone selling price, we account for the amendment as a separate agreement. If we conclude the goods and/or services are not distinct and at their stand-alone selling price, we then assess whether the remaining goods or services are distinct from those already provided. If the goods and/or services are distinct from what we have already provided, then we allocate the remaining transaction price from the original agreement and the additional transaction price from the amendment to the remaining goods and/or services. If the goods and/or services are not distinct from what we have already provided, we update the transaction price for our single performance obligation and recognize any change in our estimated revenue as a cumulative adjustment.
F-16
For example, in May 2015, we entered into an exclusive license agreement with Bayer to develop and commercialize IONIS-FXIRx for the prevention of thrombosis. As part of the agreement, Bayer paid us a $100 million upfront payment. At the onset of the agreement, we were responsible for completing a Phase 2 study of IONIS-FXIRx in people with end-stage renal disease on hemodialysis and for providing an initial supply of API. In February 2017, we amended our agreement with Bayer to advance IONIS-FXIRx and to initiate development of IONIS-FXI-LRx, which Bayer licensed. As part of the 2017 amendment, Bayer paid us $75 million. We are also eligible to receive milestone payments and tiered royalties on gross margins of IONIS-FXIRx and IONIS-FXI-LRx. Under the 2017 amendment, we concluded we had a new agreement with three performance obligations. These performance obligations were to deliver the license of IONIS-FXI-LRx, to provide R&D services and to deliver API. We allocated the $75 million transaction price to these performance obligations. Refer to Note 6, Collaborative Arrangements and Licensing Agreements, for further discussion of our accounting treatment for our Bayer collaboration. Our allocation of the consideration we received for the Bayer amendment did not change as a result of adopting Topic 606. However, the method in which we are recognizing revenue related to our R&D services performance obligation did change. We are amortizing revenue related to our R&D services performance obligation using the input method under Topic 606.
Multiple Agreements
From time to time, we may enter into separate agreements at or near the same time with the same partner. We evaluate such agreements to determine whether we should account for them individually as distinct arrangements or whether the separate agreements should be combined and accounted for together. We evaluate the following to determine the accounting for the agreements:
| ● | Whether the agreements were negotiated together with a single objective; |
| ● | Whether the amount of consideration in one contract depends on the price or performance of the other agreement; or |
| ● | Whether the goods and/or services promised under the agreements are a single performance obligation. |
Our evaluation involves significant judgment to determine whether a group of agreements might be so closely related that accounting guidance requires us to account for them as a combined arrangement.
For example, in the second quarter of 2018, we entered into two separate agreements with Biogen at the same time: a new strategic neurology collaboration agreement and a stock purchase agreement, or SPA. We evaluated the Biogen agreements to determine whether we should treat the agreements separately or combine them. We considered that the agreements were negotiated concurrently and in contemplation of one another. Based on these facts and circumstances, we concluded that we should evaluate the provisions of the agreements on a combined basis. Refer to Note 6, Collaborative Arrangements and Licensing Agreements for further discussion of the accounting treatment for the 2018 strategic neurology collaboration with Biogen.
Contracts Receivable
Our contracts receivable balance represents the amounts we have billed our partners for goods we have delivered or services we have performed that are due to us unconditionally. When we bill our partners with payment terms based on the passage of time, we consider the contract receivable to be unconditional. We typically receive payment within one quarter of billing our partner. Our contracts receivable balance as of December 31, 2017 did not change when we adopted Topic 606.
Unbilled SPINRAZA Royalties
Our unbilled SPINRAZA royalties represent our right to receive consideration from Biogen in advance of when we are eligible to bill Biogen for SPINRAZA royalties. We include these unbilled amounts in other current assets on our consolidated balance sheet. Our unbilled SPINRAZA royalties as of December 31, 2017 did not change when we adopted Topic 606.
Deferred Revenue
We are often entitled to bill our customers and receive payment from our customers in advance of our obligation to provide services or transfer goods to our partners. In these instances, we include the amounts in deferred revenue on our consolidated balance sheet. During the years ended December 31, 2018 and 2017, we recognized $105.3 million and $95.1 million of revenue from amounts that were in our beginning deferred revenue balances for those periods, respectively. Refer to our revenue recognition policy above detailing how we recognize revenue for further discussion.
The following table summarizes the adjustments we were required to make to our deferred revenue amounts to adopt Topic 606 (in thousands):
| At December 31, 2017 | ||||||||||||
As Previously Reported under Topic 605 | Topic 606 Adjustment | As Revised | ||||||||||
| Current portion of deferred contract revenue | $ | 106,465 | $ | 18,871 | $ | 125,336 | ||||||
| Long-term portion of deferred contract revenue | 72,708 | 35,318 | 108,026 | |||||||||
| Total deferred revenue | $ | 179,173 | $ | 54,189 | $ | 233,362 | ||||||
F-17
Our deferred revenue balance increased $54.2 million at December 31, 2017 under Topic 606, compared to Topic 605. The increase was primarily related to the change in the accounting for certain milestone payments and the way in which we amortize payments. Under Topic 605, we previously recognized the majority of the milestone payments we earned in the period we achieved the milestone event, which did not impact our deferred revenue balance. Under Topic 606 we are now amortizing more milestone payments over the period of our performance obligation, which adds to our deferred revenue balance. Additionally, under Topic 605 we amortized payments evenly over the period of our obligation. Under Topic 606, we are required to use an input method to determine the amount we amortize each reporting period. The increase in deferred revenue relates to agreements with the following partners:
| ● | $24.2 million from Biogen; |
| ● | $15.9 million from AstraZeneca; |
| ● | $11.8 million from Novartis; and |
| ● | $ 2.3 million from other partners. |
Cost of Products Sold
We obtained the first regulatory approval for TEGSEDI in July 2018, as a result we began recognizing cost of products sold expenses related to TEGSEDI. Our cost of products sold includes manufacturing costs, transportation and freight costs and indirect overhead costs associated with the manufacturing and distribution of TEGSEDI. We also may include certain period costs related to manufacturing services and inventory adjustments in cost of products sold. Prior to obtaining regulatory approval, we expensed a significant portion of the costs we incurred to produce the TEGSEDI supply we are using in the commercial launch as research and development expense. We previously recognized $0.1 million of costs to produce TEGSEDI related to the TEGSEDI commercial revenue we recognized in 2018.
Research, Development and Patent Expenses
Our research and development expenses include wages, benefits, facilities, supplies, external services, clinical trial and manufacturing costs and other expenses that are directly related to our research and development operations. We expense research and development costs as we incur them. When we make payments for research and development services prior to the services being rendered, we record those amounts as prepaid assets on our consolidated balance sheet and we expense them as the services are provided. For the years ended December 31, 2018, 2017 and 2016, research and development expenses were $411.9 million, $372.5 million and $340.4 million, respectively. A portion of the costs included in research and development expenses are costs associated with our partner agreements. For the years ended December 31, 2018, 2017 and 2016, research and development costs of approximately $58.7 million, $59.5 million and $187.1 million, respectively, were related to our partner agreements.
We capitalize costs consisting principally of outside legal costs and filing fees related to obtaining patents. We amortize patent costs over the useful life of the patent, beginning with the date the U.S. Patent and Trademark Office, or foreign equivalent, issues the patent. The weighted average remaining amortizable life of our issued patents was 10.1 years at December 31, 2018.
The cost of our patents capitalized on our consolidated balance sheet at December 31, 2018 and 2017 was $32.7 million and $30.8 million, respectively. Accumulated amortization related to patents was $8.7 million and $8.8 million at December 31, 2018 and 2017, respectively.
Based on our existing patents, we estimate amortization expense related to patents in each of the next five years to be the following:
| Years Ending December 31, | Amortization (in millions) | |||
| 2019 | $ | 1.7 | ||
| 2020 | $ | 1.6 | ||
| 2021 | $ | 1.5 | ||
| 2022 | $ | 1.4 | ||
| 2023 | $ | 1.3 | ||
We review our capitalized patent costs regularly to ensure that they include costs for patents and patent applications that have future value. When we identify patents and patent applications that we are not actively pursuing, we write off any associated costs. In 2018, 2017 and 2016, patent expenses were $2.6 million, $2.1 million and $3.9 million, respectively, and included non-cash charges related to the write-down of our patent costs to their estimated net realizable values of $0.8 million, $0.4 million and $2.3 million, respectively.
F-18
Accrued Liabilities
Our accrued liabilities consisted of the following (in thousands):
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Clinical expenses | $ | 22,125 | $ | 16,347 | ||||
| In-licensing expenses | 12,298 | 33,790 | ||||||
| Other miscellaneous expenses | 13,938 | 16,481 | ||||||
| Total accrued liabilities | $ | 48,361 | $ | 66,618 | ||||
Noncontrolling Interest in Akcea Therapeutics, Inc.
Prior to Akcea’s IPO in July 2017, we owned 100 percent of Akcea. From the closing of Akcea’s IPO in July 2017 through mid-April 2018, we owned approximately 68 percent of Akcea. In the second, third and fourth quarters of 2018, we received additional shares of Akcea’s stock related to our license of TEGSEDI and AKCEA-TTR-LRx to Akcea, increasing our ownership percentage to approximately 75 percent. We reflected this increase in our ownership percentage in these financial statements as an adjustment to noncontrolling interest. The shares third parties own represent an interest in Akcea’s equity that is not controlled by us. However, as we continue to maintain overall control of Akcea through our voting interest, we reflect the assets, liabilities and results of operations of Akcea in our consolidated financial statements. We reflect the noncontrolling interest attributable to other owners of Akcea’s common stock in a separate line on the statement of operations and a separate line within stockholders’ equity in our consolidated balance sheet. In addition, we record a noncontrolling interest adjustment to account for the stock options Akcea grants, which if exercised, will dilute our ownership in Akcea. This adjustment is a reclassification within stockholders’ equity from additional paid-in capital to noncontrolling interest in Akcea equal to the amount of stock-based compensation expense Akcea had recognized.
Concentration of Credit Risk
Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash equivalents, short-term investments and receivables. We place our cash equivalents and short-term investments with reputable financial institutions. We primarily invest our excess cash in commercial paper and debt instruments of the U.S. Treasury, financial institutions, corporations, and U.S. government agencies with strong credit ratings and an investment grade rating at or above A-1, P-1 or F-1 by Moody’s, Standard & Poor’s, or S&P, or Fitch, respectively. We have established guidelines relative to diversification and maturities that maintain safety and liquidity. We periodically review and modify these guidelines to maximize trends in yields and interest rates without compromising safety and liquidity.
Cash, Cash Equivalents and Short-Term Investments
We consider all liquid investments with maturities of three months or less when we purchase them to be cash equivalents. Our short-term investments have initial maturities of greater than three months from date of purchase. We classify our short-term debt investments as “available-for-sale” and carry them at fair market value based upon prices on the last day of the fiscal period for identical or similar items. We record unrealized gains and losses on debt securities as a separate component of comprehensive income (loss) and include net realized gains and losses in gain (loss) on investments. We use the specific identification method to determine the cost of securities sold.
We also have equity investments of less than 20 percent ownership in publicly and privately held biotechnology companies that we received as part of a technology license or partner agreement. At December 31, 2018, we held equity investments in two publicly held companies, ProQR Therapeutics N.V., or ProQR, and Antisense Therapeutics Limited, or ATL. We also held equity investments in four privately-held companies, Atlantic Pharmaceuticals Limited, Dynacure SAS, Seventh Sense Biosystems and Suzhou Ribo Life Science Co, Ltd.
In January 2018, we adopted the amended accounting guidance related to the recognition, measurement, presentation, and disclosure of certain financial instruments. The amended guidance requires us to measure and record our equity investments at fair value. Additionally, the amended accounting guidance requires us to recognize the changes in fair value in our consolidated statement of operations, instead of through accumulated other comprehensive income. Prior to 2018, we accounted for our equity investments in privately held companies under the cost method of accounting. Under the amended guidance we account for our equity investments in privately held companies at their cost minus impairments, plus or minus changes resulting from observable price changes in orderly transactions for the identical or similar investment of the same issuer. Our adoption of this guidance did not have an impact on our results.
Inventory Valuation
We reflect our inventory on our consolidated balance sheet at the lower of cost or market value under the first-in, first-out method, or FIFO. We capitalize the costs of raw materials that we purchase for use in producing our medicines because until we use these raw materials they have alternative future uses. We include in inventory raw material costs for medicines that we manufacture for our partners under contractual terms and that we use primarily in our clinical development activities and drug products. We can use each of our raw materials in multiple products and, as a result, each raw material has future economic value independent of the development status of any single medicine. For example, if one of our medicines failed, we could use the raw materials for that medicine to manufacture our other medicines. We expense these costs as R&D expenses when we begin to manufacture API for a particular medicine if the medicine has not been approved for marketing by a regulatory agency.
F-19
We obtained the first regulatory approval for TEGSEDI in July 2018. At December 31, 2018, our physical inventory for TEGSEDI included API that we produced prior to when we obtained regulatory approval and accordingly has no cost basis as we had previously expensed the costs as R&D expenses.
We review our inventory periodically and reduce the carrying value of items we consider to be slow moving or obsolete to their estimated net realizable value based on forecasted demand compared to quantities on hand. We consider several factors in estimating the net realizable value, including shelf life of our inventory, alternative uses for our medicines in development and historical write-offs. We did not record any inventory write-offs for the years ended December 31, 2018, 2017 or 2016. Total inventory was $8.6 million and $10.0 million as of December 31, 2018 and 2017, respectively.
Property, Plant and Equipment
We carry our property, plant and equipment at cost and depreciate it on the straight-line method over its estimated useful life, which consists of the following (in thousands):
| Estimated Useful Lives | December 31, | |||||||||||
| (in years) | 2018 | 2017 | ||||||||||
| Computer software, laboratory, manufacturing and other equipment | 3 to 10 | $ | 53,496 | $ | 66,558 | |||||||
| Building, building improvements and building systems | 15 to 40 | 97,528 | 92,770 | |||||||||
| Land improvements | 20 | 2,853 | 2,853 | |||||||||
| Leasehold improvements | 5 to 15 | 18,981 | 26,748 | |||||||||
| Furniture and fixtures | 5 to 10 | 6,283 | 6,161 | |||||||||
| 179,141 | 195,090 | |||||||||||
| Less accumulated depreciation | (61,474 | ) | (87,676 | ) | ||||||||
| 117,667 | 107,414 | |||||||||||
| Land | 14,493 | 14,493 | ||||||||||
| Total | $ | 132,160 | $ | 121,907 | ||||||||
We depreciate our leasehold improvements using the shorter of the estimated useful life or remaining lease term.
Fair Value of Financial Instruments
We have estimated the fair value of our financial instruments. The amounts reported for cash, accounts receivable, accounts payable and accrued expenses approximate the fair value because of their short maturities. We report our investment securities at their estimated fair value based on quoted market prices for identical or similar instruments.
Long-Lived Assets
We evaluate long-lived assets, which include property, plant and equipment and patent costs, for impairment on at least a quarterly basis and whenever events or changes in circumstances indicate that we may not be able to recover the carrying amount of such assets. We recorded charges of $0.8 million, $0.8 million and $2.3 million for the years ended December 31, 2018, 2017 and 2016, respectively, related primarily to the write-down of intangible assets.
Use of Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the U.S. requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ from those estimates.
Stock-Based Compensation Expense
We measure stock-based compensation expense for equity-classified awards, principally related to stock options, restricted stock units, or RSUs, and stock purchase rights under our ESPP based on the estimated fair value of the award on the date of grant. We recognize the value of the portion of the award that we ultimately expect to vest as stock-based compensation expense over the requisite service period in our Consolidated Statements of Operations. We reduce stock-based compensation expense for estimated forfeitures at the time of grant and revise in subsequent periods if actual forfeitures differ from those estimates.
We use the Black-Scholes model as our method of valuing option awards and stock purchase rights under our ESPP. On the grant date, we use our stock price and assumptions regarding a number of highly complex and subjective variables to determine the estimated fair value of stock-based payment awards. These variables include, but are not limited to, our expected stock price volatility over the term of the awards, and actual and projected employee stock option exercise behaviors. Option-pricing models were developed for use in estimating the value of traded options that have no vesting or hedging restrictions and are fully transferable. Because our employee stock options have certain characteristics that are significantly different from traded options, and because changes in the subjective assumptions can materially affect the estimated value, in management’s opinion, the existing valuation models may not provide an accurate measure of the fair value of our employee stock options. Although we determine the estimated fair value of employee stock options using an option-pricing model, that value may not be indicative of the fair value observed in a willing buyer/willing seller market transaction.
F-20
We recognize compensation expense for option awards and RSUs using the accelerated multiple-option approach. Under the accelerated multiple-option approach (also known as the graded-vesting method), we recognize compensation expense over the requisite service period for each separately vesting tranche of the award as though the award were in substance multiple awards, which results in the expense being front-loaded over the vesting period.
The fair value of RSUs is based on the market price of our common stock on the date of grant. The RSUs we have granted vest annually over a four-year period.
See Note 4, Stockholders’ Equity, for additional information regarding our stock-based compensation plans.
Accumulated Other Comprehensive Loss
Accumulated other comprehensive loss is primarily comprised of unrealized gains and losses on investments, net of taxes and adjustments we made to reclassify realized gains and losses on investments from other accumulated comprehensive loss to our Consolidated Statement of Operations. The following table summarizes changes in accumulated other comprehensive loss for the years ended December 31, 2018, 2017 and 2016 (in thousands):
| Year Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Beginning balance accumulated other comprehensive loss | $ | (31,759 | ) | $ | (30,358 | ) | $ | (13,565 | ) | |||
| Unrealized losses on securities, net of tax (1) | (280 | ) | (960 | ) | (17,219 | ) | ||||||
| Amounts reclassified from accumulated other comprehensive loss | — | (374 | ) | 447 | ||||||||
| Currency translation adjustment | 23 | (67 | ) | (21 | ) | |||||||
| Net other comprehensive loss for the period | (257 | ) | (1,401 | ) | (16,793 | ) | ||||||
| Ending balance accumulated other comprehensive loss | $ | (32,016 | ) | $ | (31,759 | ) | $ | (30,358 | ) | |||
| (1) | A tax benefit of $0.3 million was included in other comprehensive loss for the year ended December 31, 2018. There was no tax benefit or expense for other comprehensive loss for the years ended December 31, 2017 or 2016. |
Convertible Debt
We account for convertible debt instruments, including our 1 percent and 2¾ percent notes that may be settled in cash upon conversion (including partial cash settlement) by separating the liability and equity components of the instruments in a manner that reflects our nonconvertible debt borrowing rate. We determine the carrying amount of the liability component by measuring the fair value of similar debt instruments that do not have the conversion feature. If no similar debt instrument exists, we estimate fair value by using assumptions that market participants would use in pricing a debt instrument, including market interest rates, credit standing, yield curves and volatilities. We use accounting estimates and assumptions when we determine the fair value of the debt component. These estimates and assumptions we use are judgmental in nature and could have a significant impact on the determination of the debt component, and the associated non-cash interest expense.
We assigned a value to the debt component of our convertible notes equal to the estimated fair value of similar debt instruments without the conversion feature, which resulted in us recording our debt at a discount. We are amortizing our debt issuance costs and debt discount over the life of the convertible notes as additional non-cash interest expense utilizing the effective interest method. For additional information, see Note 3, Long-Term Obligations and Commitments.
Segment Information
We have two operating segments, our Ionis Core segment and Akcea Therapeutics, our majority-owned affiliate. Akcea is a biopharmaceutical company focused on developing and commercializing medicines to treat patients with rare and serious diseases. We provide segment financial information and results for our Ionis Core segment and our Akcea Therapeutics segment based on the segregation of revenues and expenses that our chief decision maker reviews to assess operating performance and to make operating decisions. We allocate a portion of Ionis’ development, R&D support and general and administrative expenses to Akcea for work Ionis performs on behalf of Akcea.
Fair Value Measurements
We use a three-tier fair value hierarchy to prioritize the inputs used in our fair value measurements. These tiers include: Level 1, defined as observable inputs such as quoted prices in active markets for identical assets, which includes our money market funds and treasury securities classified as available-for-sale securities and our investment in equity securities in publicly-held biotechnology companies; Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable, which includes our fixed income securities and commercial paper classified as available-for-sale securities; and Level 3, defined as unobservable inputs in which little or no market data exists, therefore requiring us to develop our own assumptions. We classify the majority of our securities as Level 2. We obtain the fair value of our Level 2 investments from our custodian bank or from a professional pricing service. We validate the fair value of our Level 2 investments by understanding the pricing model used by the custodian banks or professional pricing service provider and comparing that fair value to the fair value based on observable market prices. During 2018 and 2017, there were no transfers between our Level 1 and Level 2 investments. When we recognize transfers between levels of the fair value hierarchy, we recognize the transfer on the date the event or change in circumstances that caused the transfer occurs.
F-21
The following tables present the major security types we held at December 31, 2018 and 2017 that are regularly measured and carried at fair value. At December 31, 2018, our ProQR investment was subject to trading restrictions through the fourth quarter of 2019, as a result we included a lack of marketability discount in valuing this investment, which is a Level 3 input. At December 31, 2017, we did not have any financial instruments that we valued using Level 3 inputs. The tables segregate each security type by the level within the fair value hierarchy of the valuation techniques we utilized to determine the respective securities’ fair value (in thousands):
At December 31, 2018 | Quoted Prices in Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | |||||||||||||
| Cash equivalents (1) | $ | 146,281 | $ | 146,281 | $ | — | $ | — | ||||||||
| Corporate debt securities (2) | 1,252,960 | — | 1,252,960 | — | ||||||||||||
| Debt securities issued by U.S. government agencies (3) | 276,612 | — | 276,612 | — | ||||||||||||
| Debt securities issued by the U.S. Treasury (4) | 260,154 | 260,154 | — | — | ||||||||||||
| Debt securities issued by states of the U.S. and political subdivisions of the states (3) | 79,942 | — | 79,942 | — | ||||||||||||
Investment in ProQR Therapeutics N.V. (5) | 1,349 | — | — | 1,349 | ||||||||||||
| Total | $ | 2,017,298 | $ | 406,435 | $ | 1,609,514 | $ | 1,349 | ||||||||
At December 31, 2017 | Quoted Prices in Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | ||||||||||
| Cash equivalents (1) | $ | 86,262 | $ | 86,262 | $ | — | ||||||
| Corporate debt securities (6) | 647,461 | — | 647,461 | |||||||||
| Debt securities issued by U.S. government agencies (3) | 136,325 | — | 136,325 | |||||||||
| Debt securities issued by the U.S. Treasury (3) | 30,818 | 30,818 | — | |||||||||
| Debt securities issued by states of the U.S. and political subdivisions of the states (7) | 93,932 | — | 93,932 | |||||||||
| Total | $ | 994,798 | $ | 117,080 | $ | 877,718 | ||||||
| (1) | Included in cash and cash equivalents on our consolidated balance sheet. |
| (2) | $50.2 million included in cash and cash equivalents on our consolidated balance sheet, with the difference included in short-term investments on our consolidated balance sheet. |
| (3) | Included in short-term investments on our consolidated balance sheet. |
| (4) | $14.2 million included in cash and cash equivalents on our consolidated balance sheet, with the difference included in short-term investments on our consolidated balance sheet. |
| (5) | Included in other current assets on our consolidated balance sheet. |
| (6) | $11.9 million included in cash and cash equivalents on our consolidated balance sheet, with the difference included in short-term investments on our consolidated balance sheet. |
| (7) | $3.5 million included in cash and cash equivalents on our consolidated balance sheet, with the difference included in short-term investments on our consolidated balance sheet. |
Novartis Future Stock Purchase
In January 2017, we and Akcea entered into a SPA with Novartis. As part of the SPA, Novartis was required to purchase $50 million of Akcea’s common stock at the IPO price or our common stock at a premium if an IPO did not occur by April 2018. Therefore, at the inception of the SPA, we recorded a $5.0 million asset representing the fair value of the potential future premium we could have received if Novartis purchased our common stock. We determined the fair value of the future premium by calculating the value based on the stated premium in the SPA and estimating the probability of an Akcea IPO. We also included a lack of marketability discount when we determined the fair value of the premium because we would have issued unregistered shares to Novartis if they had purchased our common stock. We measured this asset using Level 3 inputs and recorded it in other assets on our consolidated balance sheet. Because Akcea completed its IPO before April 2018, Novartis will not purchase additional shares of Ionis stock. Therefore, this asset no longer had any value and we wrote-off the remaining balance to other expenses on our third quarter 2017 consolidated statement of operations.
F-22
The following is a reconciliation of the potential premium we would have received if Akcea had not completed its IPO, measured at fair value on a recurring basis using significant unobservable inputs (Level 3) for 2017 (in thousands):
Year Ended December 31, 2017 | ||||
| Beginning balance of Level 3 instruments | $ | — | ||
| Value of the potential premium we will receive from Novartis at inception of the SPA (January 2017) | 5,035 | |||
| Write-off of premium to other expenses | (5,035 | ) | ||
| Ending balance of Level 3 instruments | $ | — | ||
Income Taxes
We account for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in our financial statements or tax returns. In addition, deferred tax assets are recorded for the future benefit of utilizing net operating losses and research and development credit carryforwards. We record a valuation allowance when necessary to reduce deferred tax assets to the amount we expect to realize.
We apply the authoritative accounting guidance prescribing a threshold and measurement attribute for the financial recognition and measurement of a tax position taken or expected to be taken in a tax return. We recognize liabilities for uncertain tax positions based on a two-step process. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step requires us to estimate and measure the tax benefit as the largest amount that is more than 50 percent likely to be realized upon ultimate settlement.
We are required to use significant judgment in evaluating our uncertain tax positions and determining our provision for income taxes. Although we believe our reserves are reasonable, no assurance can be given that the final tax outcome of these matters will not be different from that which is reflected in our historical income tax provisions and accruals. We adjust these reserves for changing facts and circumstances, such as the closing of a tax audit or the refinement of an estimate. To the extent that the final tax outcome of these matters is different than the amounts recorded, such differences may impact the provision for income taxes in the period in which such determination is made.
We are also required to use significant judgment in determining any valuation allowance recorded against our deferred tax assets. In assessing the need for a valuation allowance, we consider all available evidence, including scheduled reversal of deferred tax liabilities, past operating results, the feasibility of tax planning strategies and estimates of future taxable income. We base our estimates of future taxable income on assumptions that are consistent with our plans. The assumptions we use represent our best estimates and involve inherent uncertainties and the application of our judgment. Should actual amounts differ from our estimates, the amount of our tax expense and liabilities we recognize could be materially impacted. We record a valuation allowance to reduce the balance of our net deferred tax assets to the amount we believe is more-likely-than-not to be realized.
For U.S. federal income tax purposes, we are required to file separate U.S. federal income tax returns for Ionis and Akcea. We began deconsolidating Akcea for U.S. federal income tax purposes upon Akcea’s initial public offering. As a result, we are required to assess our Ionis stand-alone and Akcea’s valuation allowances separately even though we consolidate Akcea’s financial results in our consolidated financial statements. We continue to file combined state tax returns in most jurisdictions. As a result, we continue to assess the state portion of our valuation allowance for those jurisdictions on a consolidated basis.
We have historically recorded a valuation allowance against all our net deferred tax assets due to cumulative financial statement losses. However, in the fourth quarter of 2018, we reversed the valuation allowance previously recorded against our Ionis stand-alone U.S. federal net deferred tax assets, resulting in a one-time non-cash tax benefit of $332.1 million. Given our current stand-alone Ionis pre-tax income, and assuming we maintain this current level of Ionis stand-alone pre-tax income, we expect to generate income before taxes in the U.S. in future periods at a level that would result in us fully utilizing our U.S. federal net operating loss carryforwards and most of our Research and Development and Orphan Drug tax credit carryforwards over the next three years.
We continue to maintain a full valuation allowance of $234.2 million against all of Akcea’s net deferred tax assets and the net state deferred tax assets of Ionis at December 31, 2018 due to uncertainties related to our ability to realize the tax benefits associated with these assets.
We evaluate our deferred tax assets regularly to determine whether adjustments to the valuation allowance are appropriate due to changes in facts or circumstances, such as changes in expected future pre-tax earnings, tax law, interactions with taxing authorities and developments in case law. In making this evaluation, we rely on our recent history of pre-tax earnings. Our material assumptions are our forecasts of future pre-tax earnings and the nature and timing of future deductions and income represented by the deferred tax assets and liabilities, all of which involve the exercise of significant judgment. Although we believe our estimates are reasonable, we are required to use significant judgment in determining the appropriate amount of valuation allowance recorded against our deferred tax assets.
We do not provide for a U.S. income tax liability and foreign withholding taxes on undistributed foreign earnings of our foreign subsidiaries.
F-23
Impact of Recently Issued Accounting Standards
In February 2016, the FASB issued amended accounting guidance related to lease accounting, which will require us to record all leases with a term longer than one year on our balance sheet. When we record leases on our balance sheet under the new guidance, we will record a liability with a value equal to the present value of payments we will make over the life of the lease (lease liability) and an asset representing the underlying leased asset (right of use asset). The new accounting guidance requires us to determine if our leases are operating or financing leases. We will record expense for operating leases on a straight-line basis as an operating expense. If we determine a lease is a financing lease, we will record both interest and amortization expense and generally the expense will be higher in the earlier periods of the lease. We adopted this guidance on January 1, 2019 and adjusted our opening balance sheet on that date. We elected the available practical expedients. The most significant impact was the recognition of right of use assets and lease liabilities for our operating leases. We are in the process of finalizing the impact of the adoption. The adoption will not have an impact on our consolidated statement of operations or statement of cash flows.
In June 2016, the FASB issued guidance that changes the measurement of credit losses for most financial assets and certain other instruments. If we have credit losses, this updated guidance requires us to record allowances for these instruments under a new expected credit loss model. This model requires us to estimate the expected credit loss of an instrument over its lifetime, which represents the portion of the amortized cost basis we do not expect to collect. The new guidance requires us to remeasure our allowance in each reporting period we have credit losses. The new standard is effective for annual and interim periods beginning after December 15, 2019. Early adoption is permitted for periods beginning after December 15, 2018. When we adopt the new standard, we will make any adjustments to beginning balances through a cumulative-effect adjustment to accumulated deficit on that date. We plan to adopt this guidance on January 1, 2020. We are currently assessing the effects it will have on our consolidated financial statements and disclosures.
In February 2018, the FASB issued updated guidance for reclassification of tax effects from accumulated other comprehensive income (loss). The updated guidance gives entities an option to reclassify amounts included in accumulated other comprehensive income (loss) that under the Tax Act do not have a way to be relieved, and allows a one-time reclassification to retained earnings. The updated guidance is effective for all entities for fiscal years beginning after December 31, 2018, and interim periods within those fiscal years. We have decided not to record the reclassification adjustments provided by this guidance.
In June 2018, the FASB issued updated guidance to simplify the accounting for stock-based compensation expense for nonemployees. Specifically, we are now expensing grants to nonemployees in a similar manner as grants to employees. Previously, we had to re-value these grants at each reporting period to reflect the current fair value. Under the amended guidance, we value grants to nonemployees when we grant them and we will not adjust their value for future changes. We adopted this guidance in the second quarter of 2018. The updated guidance did not have a material impact to our financial results.
In November 2018, the FASB issued clarifying guidance of the interaction between the collaboration accounting guidance and the new revenue recognition guidance we adopted on January 1, 2018 (Topic 606). The clarifying guidance included the following:
| 1) | When a participant is considered a customer in a collaborative arrangement, all of the associated accounting under Topic 606 should be applied; |
| 2) | Adds “unit of account” concept to collaboration accounting guidance to align with Topic 606. This is used to determine if revenue is recognized or if a contra expense is recognized from consideration received under a collaboration; and |
| 3) | Precludes revenue from being recognized under Topic 606 when a transaction with a collaborative partner is determined not be a customer and is not directly related to the sales to third parties. |
The updated guidance is effective for public entities for fiscal years beginning after December 15, 2019, and interim periods within those fiscal years. Early adoption is permitted. We plan to adopt this guidance on January 1, 2020. We are currently assessing the effects it will have on our consolidated financial statements and disclosures.
2. Investments
As of December 31, 2018, we had primarily invested our excess cash in debt instruments of the U.S. Treasury, financial institutions, corporations, and U.S. government agencies with strong credit ratings and an investment grade rating at or above A-1, P-1 or F-1 by Moody’s, Standard & Poor’s, or S&P, or Fitch, respectively. We have established guidelines relative to diversification and maturities that maintain safety and liquidity. We periodically review and modify these guidelines to maximize trends in yields and interest rates without compromising safety and liquidity.
The following table summarizes the contract maturity of the available-for-sale securities we held as of December 31, 2018:
| One year or less | 77 | % | ||
| After one year but within two years | 20 | % | ||
| After two years but within three and one half years | 3 | % | ||
| Total | 100 | % |
As illustrated above, at December 31, 2018, 97 percent of our available-for-sale securities had a maturity of less than two years.
All of our available-for-sale securities are available to us for use in our current operations. As a result, we categorize all of these securities as current assets even though the stated maturity of some individual securities may be one year or more beyond the balance sheet date.
F-24
At December 31, 2018, we had an ownership interest of less than 20 percent in four private companies and two public companies with which we conduct business. The privately-held companies are Atlantic Pharmaceuticals Limited, Dynacure SAS, Seventh Sense Biosystems and Suzhou Ribo Life Science Co, Ltd.The publicly traded companies are ATL and ProQR.
The following is a summary of our investments (in thousands):
| December 31, 2018 | Cost (1) | Gains | Losses | Fair Value | ||||||||||||
| Available-for-sale securities: | ||||||||||||||||
| Corporate debt securities (2) | $ | 956,879 | $ | 13 | $ | (1,858 | ) | $ | 955,034 | |||||||
| Debt securities issued by U.S. government agencies | 168,839 | 3 | (104 | ) | 168,738 | |||||||||||
| Debt securities issued by the U.S. Treasury (2) | 244,640 | 15 | (77 | ) | 244,578 | |||||||||||
| Debt securities issued by states of the U.S. and political subdivisions of the states | 63,572 | — | (323 | ) | 63,249 | |||||||||||
| Total securities with a maturity of one year or less | 1,433,930 | 31 | (2,362 | ) | 1,431,599 | |||||||||||
| Corporate debt securities | 299,018 | 194 | (1,286 | ) | 297,926 | |||||||||||
| Debt securities issued by U.S. government agencies | 107,789 | 194 | (109 | ) | 107,874 | |||||||||||
| Debt securities issued by the U.S. Treasury | 15,600 | — | (24 | ) | 15,576 | |||||||||||
| Debt securities issued by states of the U.S. and political subdivisions of the states | 16,980 | — | (287 | ) | 16,693 | |||||||||||
| Total securities with a maturity of more than one year | 439,387 | 388 | (1,706 | ) | 438,069 | |||||||||||
| Total available-for-sale securities | $ | 1,873,317 | $ | 419 | $ | (4,068 | ) | $ | 1,869,668 | |||||||
| Equity securities: | ||||||||||||||||
| Total equity securities included in other current assets (3) | $ | 1,212 | 137 | — | 1,349 | |||||||||||
| Total available-for-sale and equity securities | $ | 1,874,529 | $ | 556 | $ | (4,068 | ) | $ | 1,871,017 | |||||||
| Gross Unrealized | Estimated | |||||||||||||||
| December 31, 2017 | Cost (1) | Gains | Losses | Fair Value | ||||||||||||
| Available-for-sale securities: | ||||||||||||||||
| Corporate debt securities (2) | $ | 500,599 | $ | 2 | $ | (752 | ) | $ | 499,849 | |||||||
| Debt securities issued by U.S. government agencies | 83,926 | — | (212 | ) | 83,714 | |||||||||||
| Debt securities issued by the U.S. Treasury | 29,428 | — | (17 | ) | 29,411 | |||||||||||
| Debt securities issued by states of the U.S. and political subdivisions of the states (2) | 29,240 | 4 | (122 | ) | 29,122 | |||||||||||
| Total securities with a maturity of one year or less | 643,193 | 6 | (1,103 | ) | 642,096 | |||||||||||
| Corporate debt securities | 148,663 | 8 | (1,059 | ) | 147,612 | |||||||||||
| Debt securities issued by U.S. government agencies | 52,779 | — | (168 | ) | 52,611 | |||||||||||
| Debt securities issued by the U.S. Treasury | 1,409 | — | (2 | ) | 1,407 | |||||||||||
| Debt securities issued by states of the U.S. and political subdivisions of the states | 65,550 | — | (740 | ) | 64,810 | |||||||||||
| Total securities with a maturity of more than one year | 268,401 | 8 | (1,969 | ) | 266,440 | |||||||||||
| Total available-for-sale securities | $ | 911,594 | $ | 14 | $ | (3,072 | ) | $ | 908,536 | |||||||
| (1) | We hold our available-for-sale securities at amortized cost. |
| (2) | Includes investments classified as cash equivalents on our consolidated balance sheet. |
| (3) | We recognize our equity securities at cost minus impairments, plus or minus changes resulting from observable price changes in orderly transactions for the identical or similar investment of the same issuer on our consolidated balance sheet. |
Investments we consider to be temporarily impaired at December 31, 2018 are as follows (in thousands):
Less than 12 Months of Temporary Impairment | More than 12 Months of Temporary Impairment | Total Temporary Impairment | ||||||||||||||||||||||||||
Number of Investments | Estimated Fair Value | Unrealized Losses | Estimated Fair Value | Unrealized Losses | Estimated Fair Value | Unrealized Losses | ||||||||||||||||||||||
| Corporate debt securities | 546 | $ | 1,000,461 | $ | (1,936 | ) | $ | 126,357 | $ | (1,208 | ) | $ | 1,126,818 | $ | (3,144 | ) | ||||||||||||
| Debt securities issued by U.S. government agencies | 50 | 161,312 | (109 | ) | 34,403 | (104 | ) | 195,715 | (213 | ) | ||||||||||||||||||
| Debt securities issued by the U.S. Treasury | 36 | 183,212 | (100 | ) | 1,413 | (1 | ) | 184,625 | (101 | ) | ||||||||||||||||||
| Debt securities issued by states of the U.S. and political subdivisions of the states | 49 | 13,868 | (14 | ) | 62,883 | (596 | ) | 76,751 | (610 | ) | ||||||||||||||||||
| Total temporarily impaired securities | 681 | $ | 1,358,853 | $ | (2,159 | ) | $ | 225,056 | $ | (1,909 | ) | $ | 1,583,909 | $ | (4,068 | ) | ||||||||||||
F-25
We believe that the decline in value of our debt securities is temporary and primarily related to the change in market interest rates since purchase. We believe it is more likely than not that we will be able to hold these securities to maturity. Therefore, we anticipate full recovery of our debt securities’ amortized cost basis at maturity.
3. Long-Term Obligations and Commitments
The carrying value of our long-term obligations was as follows (in thousands):
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| 1 percent convertible senior notes | $ | 568,215 | $ | 533,111 | ||||
| Long-term mortgage debt | 59,842 | 59,771 | ||||||
| Principal balance of fixed rate note with Morgan Stanley (1) | 12,500 | 12,500 | ||||||
| Leases and other obligations | 6,163 | 2,095 | ||||||
| Total | $ | 646,720 | $ | 607,477 | ||||
| Less: current portion | (13,749 | ) | (1,621 | ) | ||||
| Total Long-Term Obligations | $ | 632,971 | $ | 605,856 | ||||
| (1) | Our $12.5 million fixed rate note with Morgan Stanley is included in our current portion of long-term obligations on our consolidated balance sheet at December 31, 2018. |
Convertible Notes
In November 2014, we completed a $500 million offering of convertible senior notes, which mature in 2021 and bear interest at 1 percent. We raised $487 million of proceeds, net of issuance costs. We used a substantial portion of the net proceeds from the issuance of the 1 percent convertible senior notes to repurchase $140 million in principal of our 2¾ percent convertible senior notes at a price of $441.9 million, including accrued interest. As a result, the new principal balance of the 2¾ percent notes was $61.2 million.
In December 2016, we issued an additional $185.5 million of 1 percent convertible senior notes in exchange for the redemption of $61.1 million of our 2¾ percent convertible senior notes. As a result of the debt exchange we completed in December 2016, we recorded a $4.0 million non-cash loss on early retirement of debt, reflecting the early retirement of the majority of our remaining 2¾ percent convertible notes in December 2016.
At December 31, 2018, we had a nominal amount of our 2¾ percent convertible senior notes outstanding. At December 31, 2018, we had the following 1 percent convertible senior notes outstanding (amounts in millions except price per share data):
1 Percent Convertible Senior Notes | ||||
| Outstanding balance | $ | 685.5 | ||
| Original issue date ($500 million of principal) | November 2014 | |||
| Additional issue date ($185.5 million of principal) | December 2016 | |||
| Maturity date | November 2021 | |||
| Interest rate | 1 percent | |||
| Conversion price per share | $ | 66.81 | ||
| Total shares of common stock subject to conversion | 10.3 | |||
Interest is payable semi-annually in arrears on May 15 and November 15 of each year for the 1 percent notes. The 1 percent notes are convertible at the option of the note holders prior to July 1, 2021 only under certain conditions. On or after July 1, 2021, the notes are initially convertible into approximately 10.3 million shares of common stock at a conversion price of approximately $66.81 per share. We will settle conversions of the notes, at our election, in cash, shares of our common stock or a combination of both. We may not redeem the 1 percent notes prior to maturity, and no sinking fund is provided for them. If we undergo a fundamental change, holders may require us to purchase for cash all or any portion of their 1 percent notes at a purchase price equal to 100 percent of the principal amount of the notes to be purchased, plus accrued and unpaid interest to, but excluding, the fundamental change purchase date.
We account for our convertible notes using an accounting standard that requires us to assign a value to our convertible debt equal to the estimated fair value of similar debt instruments without the conversion feature and to record the remaining portion in equity. As a result, we recorded our convertible notes at a discount, which we are amortizing as additional non-cash interest expense over the expected life of the respective debt. We determined our nonconvertible debt borrowing rate using a combination of the present value of the debt’s cash flows and a Black-Scholes valuation model. The following table summarizes the nonconvertible borrowing rate, effective interest rate and amortization period of our debt discount for our convertible notes:
1 Percent Convertible Senior Notes Issued in November 2014 | 1 Percent Convertible Senior Notes Issued in December 2016 | ||
| Nonconvertible debt borrowing rate | 7.4 percent | 6.8 percent | |
| Effective interest rate | 7.8 percent | 7.2 percent | |
| Amortization period of debt discount | 7 years | 5 years |
F-26
Interest expense for the years ended December 31, 2018, 2017 and 2016 included $35.2 million, $32.5 million and $25.1 million, respectively, of non-cash interest expense related to the amortization of the debt discount and debt issuance costs for our convertible notes.
The following table summarizes information about the equity and liability components of our outstanding 1 percent convertible notes (in thousands). We measured the fair values of the convertible notes outstanding based on quoted market prices, which is a Level 2 measurement:
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Fair value of outstanding notes | $ | 724,966 | $ | 727,420 | ||||
| Principal amount of convertible notes outstanding | $ | 685,450 | $ | 685,450 | ||||
| Unamortized portion of debt discount | $ | 110,817 | $ | 144,112 | ||||
| Long-term debt | $ | 568,215 | $ | 533,111 | ||||
| Carrying value of equity component | $ | 219,011 | $ | 219,011 | ||||
Financing Arrangements
Line of Credit Arrangement
In June 2015, we entered into a five-year revolving line of credit agreement with Morgan Stanley Private Bank, National Association, or Morgan Stanley. We amended the credit agreement in February 2016 to increase the amount available for us to borrow. Under the amended credit agreement, we can borrow up to a maximum of $30 million of revolving credit for general working capital purposes. Under the credit agreement interest is payable monthly in arrears on the outstanding principal at a borrowing rate based on our option of:
| (i) | a floating rate equal to the one-month London Interbank Offered Rate, or LIBOR, in effect plus 1.25 percent per annum; |
| (ii) | a fixed rate equal to LIBOR plus 1.25 percent for a period of one, two, three, four, six, or twelve months as elected by us; or |
| (iii) | a fixed rate equal to the LIBOR swap rate during the period of the loan. |
Additionally, after June 1, 2016, we pay 0.25 percent per annum, payable quarterly in arrears, for any amount unused under the credit facility. As of December 31, 2018, we had $12.5 million in outstanding borrowings under the credit facility with a 2.31 percent fixed interest rate and a maturity date of September 2019, which we used to fund our capital equipment needs.
The credit agreement includes customary affirmative and negative covenants and restrictions. We are in compliance with all covenants of the credit agreement.
Research and Development and Manufacturing Facilities
In July 2017, we purchased the building that houses our primary R&D facility for $79.4 million. As a result of the purchase, we extinguished the financing liability we had previously recorded on our balance sheet. The difference between the purchase price of the facility and the carrying value of our financing liability at the time of the purchase was $7.7 million. We recognized this amount as a non-cash loss on extinguishment of financing liability for leased facility in our consolidated results of operations in the third quarter of 2017.
We also purchased our manufacturing facility in July 2017 for $14.0 million. We previously accounted for the lease on this facility as an operating lease. We capitalized the purchase price of the building as a fixed asset in the third quarter of 2017.
We financed the purchase of our primary R&D facility and our manufacturing facility, with mortgage debt of $51.3 million and $9.1 million, respectively. Our primary R&D facility mortgage has an interest rate of 3.88 percent. Our manufacturing facility mortgage has an interest rate of 4.20 percent. During the first five years of both mortgages we are only required to make interest payments. Both mortgages mature in August 2027.
F-27
Maturity Schedules
Annual debt and other obligation maturities, including fixed and determinable interest, at December 31, 2018 are as follows (in thousands):
| 2019 | $ | 22,067 | ||
| 2020 | 9,330 | |||
| 2021 | 694,774 | |||
| 2022 | 2,809 | |||
| 2023 | 3,494 | |||
| Thereafter | 68,108 | |||
| Subtotal | $ | 800,582 | ||
| Less: current portion | (12,890 | ) | ||
| Less: fixed and determinable interest | (41,837 | ) | ||
| Less: unamortized portion of debt discount | (111,426 | ) | ||
| Plus: Deferred rent | 4,960 | |||
| Total | $ | 639,389 |
Operating Leases
Ionis Leases
We lease a facility adjacent to our manufacturing facility that has laboratory and office space that we use to support our manufacturing facility. We lease this space under a non-cancelable operating lease with an initial term ending in June 2021 and an option to extend the lease for up to two five-year periods.
Additionally, we lease office space that we sublease to Akcea. We lease this space under a non-cancelable operating lease with an initial term ending in June 2023 and an option to extend the lease for one five-year period. The sublease with Akcea is eliminated in our consolidated financial statements. We also lease office equipment under non-cancelable operating leases with terms through January 2021.
Akcea Lease
On April 5, 2018, Akcea entered into an operating lease agreement for office space located in Boston, Massachusetts for its new corporate headquarters. The lease commencement date was August 15, 2018 and Akcea took occupancy in September 2018. Akcea is leasing this space under a non-cancelable operating lease with an initial term ending after 123 months and an option to extend the lease for an additional five-year term. Under the lease agreement, Akcea received a three-month free rent period, which commenced on August 15, 2018, and a tenant improvement allowance up to $3.8 million. Akcea provided the lessor with a letter of credit to secure its obligations under the lease in the initial amount of $2.4 million, to be reduced to $1.8 million on the third anniversary of the rent commencement date and to $1.2 million on the fifth anniversary of the rent commencement date if Akcea meets certain conditions set forth in the lease at each such time. The letter of credit amount is included in deposits and other assets in our consolidated balance sheet.
Annual future minimum payments under our operating leases as of December 31, 2018 are as follows (in thousands):
Operating Leases | ||||
| 2019 | $ | 3,129 | ||
| 2020 | 3,008 | |||
| 2021 | 2,725 | |||
| 2022 | 2,539 | |||
| 2023 | 2,505 | |||
| Thereafter | 11,862 | |||
| Total minimum payments | $ | 25,768 | ||
Rent expense was $2.6 million, $1.7 million and $2.0 million for the years ended December 31, 2018, 2017 and 2016. We recognized rent expense on a straight line basis over the lease term for the lease on our building adjacent to our manufacturing facility, the office building that Akcea subleases and Akcea’s office space, which resulted in a deferred rent balance of $5.0 million and $0.1 million at December 31, 2018 and 2017, respectively.
F-28
4. Stockholders’ Equity
Preferred Stock
We are authorized to issue up to 15,000,000 shares of “blank check” Preferred Stock. As of December 31, 2018, there were no shares of Preferred Stock outstanding. We have designated Series C Junior Participating Preferred Stock but have no issued or outstanding shares as of December 31, 2018.
Common Stock
At December 31, 2018 and 2017, we had 300,000,000 shares of common stock authorized, of which 137,928,828 and 124,976,373 were issued and outstanding, respectively. As of December 31, 2018, total common shares reserved for future issuance were 14,839,373.
During the years ended December 31, 2018, 2017 and 2016, we issued 1,451,000, 1,709,000 and 1,285,000 shares of common stock, respectively, for stock option exercises, vesting of restricted stock units, and ESPP purchases. We received net proceeds from these transactions of $27.9 million, $22.9 million and $13.7 million in 2018, 2017 and 2016, respectively.
Stock Plans
1989 Stock Option Plan
In June 1989, our Board of Directors adopted, and the stockholders subsequently approved, a stock option plan that, as amended, provides for the issuance of non-qualified and incentive stock options for the purchase of up to 20,000,000 shares of common stock to our employees, directors, and consultants. The plan expires in January 2024. The 1989 Plan does not allow us to grant stock bonuses or restricted stock awards and prohibits us from repricing any options outstanding under the plan unless our stockholders approve the repricing. Options vest over a four-year period, with 25 percent exercisable at the end of one year from the date of the grant and the balance vesting ratably, on a monthly basis, thereafter and have a term of seven years. At December 31, 2018, a total of 848,753 options were outstanding, of which options to purchase 831,656 shares were exercisable, and 42,925 shares were available for future grant under the 1989 Plan.
2011 Equity Incentive Plan
In March 2011, our Board of Directors adopted, and the stockholders subsequently approved, a stock option plan that provides for the issuance of stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, and performance cash awards to our employees, directors, and consultants. In June 2015 and in May 2017, after receiving approval from our stockholders, we amended our 2011 Equity Incentive Plan to increase the total number of shares reserved for issuance. We increased the shares available under our 2011 Equity Incentive Plan from 5,500,000 to 11,000,000 in June 2015 and from 11,000,000 to 16,000,000 in May 2017. The plan expires in June 2021. The 2011 Plan does not allow us to reduce the exercise price of any outstanding stock options or stock appreciation rights or cancel any outstanding stock options or stock appreciation rights that have an exercise price or strike price greater than the current fair market value of the common stock in exchange for cash or other stock awards unless our stockholders approve such action. Currently we anticipate awarding only options and restricted stock unit awards to our employees, directors and consultants. Under the 2011 Plan, stock options cannot vest in a period of less than two years and restricted stock unit awards cannot vest in a period of less than three years. We have granted restricted stock unit awards to our employees under the 2011 Plan which vest annually over a four-year period. At December 31, 2018, a total of 9,705,441 options were outstanding, of which 4,801,904 were exercisable, 1,183,154 restricted stock unit awards were outstanding, and 3,340,351 shares were available for future grant under the 2011 Plan.
Under the 2011 Plan, we may issue a stock award with additional acceleration of vesting and exercisability upon or after a change in control. In the absence of such provisions, no such acceleration will occur. The stock options and restricted stock unit awards we issue to our chief executive officer and issued to B. Lynne Parshall in her former role as chief operating officer will accelerate upon a change of control, as defined in the 2011 Plan. In addition, we implemented a change of control and severance benefit plan that provides for change of control and severance benefits to our executive officers, including our chief executive officer and chief financial officer. If we terminate one of our executive officers or if an executive officer resigns for good reason during the period that begins three months before and ends twelve months following a change in control of the company, the impacted executive officers’ stock options and RSUs vesting will accelerate for options and RSUs outstanding as of the termination date.
F-29
Corporate Transactions and Change in Control under 2011 Plan
In the event of certain significant corporate transactions, our Board of Directors has the discretion to take one or more of the following actions with respect to outstanding stock awards under the 2011 Plan:
| ● | arrange for assumption, continuation, or substitution of a stock award by a surviving or acquiring entity (or its parent company); |
| ● | arrange for the assignment of any reacquisition or repurchase rights applicable to any shares of our common stock issued pursuant to a stock award to the surviving or acquiring corporation (or its parent company); |
| ● | accelerate the vesting and exercisability of a stock award followed by the termination of the stock award; |
| ● | arrange for the lapse of any reacquisition or repurchase rights applicable to any shares of our common stock issued pursuant to a stock award; |
| ● | cancel or arrange for the cancellation of a stock award, to the extent not vested or not exercised prior to the effective date of the corporate transaction, in exchange for cash consideration, if any, as the Board, in its sole discretion, may consider appropriate; and |
| ● | arrange for the surrender of a stock award in exchange for a payment equal to the excess of (a) the value of the property the holder of the stock award would have received upon the exercise of the stock award, over (b) any exercise price payable by such holder in connection with such exercise. |
2002 Non-Employee Directors’ Stock Option Plan
In September 2001, our Board of Directors adopted, and the stockholders subsequently approved, an amendment and restatement of the 1992 Non-Employee Directors’ Stock Option Plan, which provides for the issuance of non-qualified stock options and restricted stock units to our non-employee directors. The name of the resulting plan is the 2002 Non-Employee Directors’ Stock Option Plan, or the 2002 Plan. In June 2015, after receiving approval from our stockholders, we amended our 2002 Non-Employee Directors Stock Option Plan to increase the total number of shares reserved for issuance. We increased the shares available under our 2002 Non-Employee Directors Stock Option Plan from 1,200,000 to 2,000,000. Options under this plan expire ten years from the date of grant. Options granted become exercisable in four equal annual installments beginning one year after the date of grant. At December 31, 2018, a total of 757,750 options were outstanding, of which 437,750 were exercisable, 63,099 restricted stock unit awards were outstanding, and 420,762 shares were available for future grant under the 2002 Plan.
Employee Stock Purchase Plan
In June 2009, our Board of Directors adopted, and the stockholders subsequently approved, the amendment and restatement of the ESPP and we reserved an additional 150,000 shares of common stock for issuance thereunder. In each of the subsequent years, we reserved an additional 150,000 shares of common stock for the ESPP resulting in a total of 3,674,596 shares authorized under the plan as of December 31, 2018. The ESPP permits full-time employees to purchase common stock through payroll deductions (which cannot exceed 10 percent of each employee’s compensation) at the lower of 85 percent of fair market value at the beginning of the purchase period or the end of each six-month purchase period. Under the amended and restated ESPP, employees must hold the stock they purchase for a minimum of six months from the date of purchase. During 2018, employees purchased and we issued to employees 43,416 shares under the ESPP at a weighted average price of $39.03 per share. At December 31, 2018, there were 774,816 shares available for purchase under the ESPP.
Stock Option Activity
The following table summarizes the stock option activity under our stock plans for the year ended December 31, 2018 (in thousands, except per share and contractual life data):
Number of Shares | Weighted Average Exercise Price Per Share | Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value | |||||||||||||
| Outstanding at December 31, 2017 | 9,397 | $ | 44.52 | |||||||||||||
| Granted | 3,518 | $ | 48.40 | |||||||||||||
| Exercised | (1,064 | ) | $ | 17.78 | ||||||||||||
| Cancelled/forfeited/expired | (540 | ) | $ | 52.47 | ||||||||||||
| Outstanding at December 31, 2018 | 11,311 | $ | 47.85 | 4.41 | $ | 93,663 | ||||||||||
| Exercisable at December 31, 2018 | 6,071 | $ | 46.83 | 3.22 | $ | 63,756 | ||||||||||
The weighted-average estimated fair values of options granted were $25.49, $25.42 and $26.72 for the years ended December 31, 2018, 2017 and 2016, respectively. The total intrinsic value of options exercised during the years ended December 31, 2018, 2017 and 2016 were $34.8 million, $49.5 million and $28.0 million, respectively, which we determined as of the date of exercise. The amount of cash received from the exercise of stock options was $18.9 million, $21.2 million and $12.6 million for the years ended December 31, 2018, 2017 and 2016, respectively. For the year ended December 31, 2018, the weighted-average fair value of options exercised was $50.50. As of December 31, 2018, total unrecognized compensation cost related to non-vested stock-based compensation plans was $118.3 million. We will adjust the total unrecognized compensation cost for future changes in estimated forfeitures. We expect to recognize this cost over a weighted average period of 1.2 years.
F-30
Restricted Stock Unit Activity
The following table summarizes the RSU activity for the year ended December 31, 2018 (in thousands, except per share data):
Number of Shares | Weighted Average Grant Date Fair Value Per Share | |||||||
| Non-vested at December 31, 2017 | 863 | $ | 49.55 | |||||
| Granted | 789 | $ | 51.06 | |||||
| Vested | (324 | ) | $ | 50.21 | ||||
| Cancelled/forfeited | (82 | ) | $ | 51.59 | ||||
| Non-vested at December 31, 2018 | 1,246 | $ | 50.20 | |||||
For the years ended December 31, 2018, 2017 and 2016, the weighted-average grant date fair value of RSUs granted was $51.06, $48.88 and $41.79 per RSU, respectively. As of December 31, 2018, total unrecognized compensation cost related to RSUs was $27.9 million. We will adjust the total unrecognized compensation cost for future changes in estimated forfeitures. We expect to recognize this cost over a weighted average period of 1.4 years.
Stock-based Compensation Expense and Valuation Information
The following table summarizes stock-based compensation expense for the years ended December 31, 2018, 2017 and 2016 (in thousands), which was allocated as follows and includes $44.3 million, $17.5 million and $10.1 million of stock-based compensation expense for Akcea employees in 2018, 2017 and 2016, respectively:
| Year Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Cost of products sold | $ | 160 | $ | — | $ | — | ||||||
| Research, development and patent | 76,557 | 64,521 | 55,099 | |||||||||
| Selling, general and administrative | 54,595 | 21,454 | 17,009 | |||||||||
| Total | $ | 131,312 | $ | 85,975 | $ | 72,108 | ||||||
Determining Fair Value
Valuation. We measure stock-based compensation expense for equity-classified awards, principally related to stock options, RSUs, and stock purchase rights under the ESPP at the grant date, based on the estimated fair value of the award and we recognize the expense over the employee’s requisite service period. We value RSUs based on the market price of our common stock on the date of grant.
We use the Black-Scholes model to estimate the fair value of stock options granted and stock purchase rights under our ESPP. The expected term of stock options granted represents the period of time that we expect them to be outstanding. We estimate the expected term of options granted based on actual and projected exercise patterns. We recognize compensation expense for stock options granted, RSUs, and stock purchase rights under the ESPP using the accelerated multiple-option approach. Under the accelerated multiple-option approach (also known as the graded-vesting method), we recognize compensation expense over the requisite service period for each separately vesting tranche of the award as though the award were in substance multiple awards, which results in the expense being front-loaded over the vesting period.
For the years ended December 31, 2018, 2017 and 2016, we used the following weighted-average assumptions in our Black-Scholes calculations:
Employee Stock Options:
| December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Risk-free interest rate | 2.4 | % | 1.8 | % | 1.5 | % | ||||||
| Dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Volatility | 63.0 | % | 65.9 | % | 58.7 | % | ||||||
| Expected life | 4.6 years | 4.5 years | 4.5 years | |||||||||
Board of Director Stock Options:
| December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Risk-free interest rate | 2.8 | % | 2.2 | % | 1.3 | % | ||||||
| Dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Volatility | 61.5 | % | 61.2 | % | 53.1 | % | ||||||
| Expected life | 6.6 years | 6.6 years | 6.5 years | |||||||||
F-31
ESPP:
| December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Risk-free interest rate | 1.8 | % | 0.8 | % | 0.4 | % | ||||||
| Dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Volatility | 47.3 | % | 59.9 | % | 86.4 | % | ||||||
| Expected life | 6 months | 6 months | 6 months | |||||||||
Risk-Free Interest Rate. We base the risk-free interest rate assumption on observed interest rates appropriate for the term of our stock option plans or ESPP.
Dividend Yield. We base the dividend yield assumption on our history and expectation of dividend payouts. We have not paid dividends in the past and do not expect to in the future.
Volatility. We use an average of the historical stock price volatility of our stock for the Black-Scholes model. We computed the historical stock volatility based on the expected term of the awards.
Expected Life. The expected term of stock options we have granted represents the period of time that we expect them to be outstanding. We estimated the expected term of options we have granted based on actual and projected exercise patterns.
Forfeitures. We reduce stock-based compensation expense for estimated forfeitures. We estimate forfeitures at the time of grant and revise, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We estimate forfeitures based on historical experience. Our historical forfeiture estimates have not been materially different from our actual forfeitures.
In addition to our stock plans, Akcea has its own stock plan under which it grants options and RSUs and under which it derives its stock-based compensation expense. The following are the weighted-average Black-Scholes assumptions Akcea used under its plan for the years ended December 31, 2018, 2017 and 2016:
Employee Stock Options:
| December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Risk-free interest rate | 2.8 | % | 1.9 | % | 1.6 | % | ||||||
| Dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Volatility | 77.1 | % | 79.5 | % | 71.4 | % | ||||||
| Expected life | 6.08 years | 6.06 years | 6.08 years | |||||||||
Board of Director Stock Options:
| December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Risk-free interest rate | 2.9 | % | 1.9 | % | 2.0 | % | ||||||
| Dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Volatility | 78.2 | % | 79.4 | % | 79.6 | % | ||||||
| Expected life | 6.42 years | 6.25 years | 6.08 years | |||||||||
The following summarizes the Black-Scholes input methodology for Akcea options that differs from the methodology we use for Ionis options:
Volatility. Since Akcea does not have sufficient history to estimate the volatility of its common stock, Akcea calculates its expected volatility based on a blend of its historical volatility and reported data from selected publicly traded peer companies for which historical information is available. Akcea plans to continue to use this blend to calculate its volatility until the historical volatility of its common stock is sufficient to measure expected volatility for future option grants.
Expected Life. Since Akcea does not have sufficient historical information, it uses the simplified method for estimating its expected term. Under the simplified method Akcea calculates its expected term as the average time-to-vesting and the contractual life of the options. As Akcea gains additional historical information, it will transition to calculating its expected term based on its exercise patterns.
F-32
5. Income Taxes
Loss before income tax (benefit) expense is comprised of (in thousands):
| Year Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| United States | $ | (69,576 | ) | $ | (5,289 | ) | $ | (57,466 | ) | |||
| Foreign | (6,580 | ) | (11,474 | ) | — | |||||||
| Loss before income tax (benefit) expense | $ | (76,156 | ) | $ | (16,763 | ) | $ | (57,466 | ) | |||
Our income tax (benefit) expense was as follows (in thousands):
| Year Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Current: | ||||||||||||
| Federal | $ | 438 | $ | (7,460 | ) | $ | 1,067 | |||||
| State | (1,442 | ) | 1,246 | 1,867 | ||||||||
| Foreign | 374 | 234 | — | |||||||||
| Total current income tax (benefit) expense | (630 | ) | (5,980 | ) | 2,934 | |||||||
| Deferred: | ||||||||||||
| Federal | (290,511 | ) | — | — | ||||||||
| State | — | — | — | |||||||||
| Total deferred income tax (benefit) expense | (290,511 | ) | — | — | ||||||||
| Total income tax (benefit) expense | $ | (291,141 | ) | $ | (5,980 | ) | $ | 2,934 | ||||
The reconciliation between our effective tax rate on loss from continuing operations and the statutory U.S. tax rate is as follows (in thousands):
| Year Ended December 31, | ||||||||||||||||||||||||
| 2018 | 2017 | 2016 | ||||||||||||||||||||||
| Pre-tax loss | $ | (76,156 | ) | $ | (16,763 | ) | $ | (57,466 | ) | |||||||||||||||
| Statutory rate | (15,993 | ) | 21.0 | % | (5,867 | ) | 35.0 | % | (20,113 | ) | 35.0 | % | ||||||||||||
| State income tax net of federal benefit | (2,202 | ) | 2.9 | % | 820 | (4.9 | )% | 95 | (0.2 | )% | ||||||||||||||
| Foreign | 1,735 | (2.3 | )% | 4,299 | (25.6 | )% | — | 0.0 | % | |||||||||||||||
| Net change in valuation allowance | (277,924 | ) | 364.9 | % | (86,296 | ) | 514.8 | % | 46,402 | (80.7 | )% | |||||||||||||
| Net operating loss expiration | 8,864 | (11.6 | )% | 3,987 | (23.8 | )% | — | 0.0 | % | |||||||||||||||
| TEGSEDI licensing gain | 59,583 | (78.2 | )% | — | 0.0 | % | — | 0.0 | % | |||||||||||||||
| Tax credits | (73,362 | ) | 96.3 | % | (32,769 | ) | 195.5 | % | (26,954 | ) | 46.9 | % | ||||||||||||
| Deferred tax true-up | 9,947 | (13.1 | )% | 4,848 | (28.9 | )% | 2,591 | (4.5 | )% | |||||||||||||||
| Tax rate change | (1,808 | ) | 2.4 | % | 114,832 | (685.0 | )% | — | 0.0 | % | ||||||||||||||
| Non-deductible compensation | 3,154 | (4.1 | )% | 1,575 | (9.4 | )% | 825 | (1.4 | )% | |||||||||||||||
| Other non-deductible items | (569 | ) | 0.7 | % | 2,548 | (15.2 | )% | 324 | (0.6 | )% | ||||||||||||||
| Akcea deconsolidation adjustment at IPO | — | 0.0 | % | 469 | (2.8 | )% | — | 0.0 | % | |||||||||||||||
| Stock-based compensation | (4,199 | ) | 5.5 | % | (14,337 | ) | 85.5 | % | — | 0.0 | % | |||||||||||||
| Other | 1,633 | (2.1 | )% | (89 | ) | 0.5 | % | (236 | ) | 0.4 | % | |||||||||||||
| Effective rate | $ | (291,141 | ) | 382.3 | % | $ | (5,980 | ) | 35.7 | % | $ | 2,934 | (5.1 | )% | ||||||||||
F-33
Significant components of our deferred tax assets and liabilities as of December 31, 2018 and 2017 are as follows (in thousands):
| Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Deferred Tax Assets: | ||||||||
| Net operating loss carryovers | $ | 89,717 | $ | 153,575 | ||||
| R&D credits | 313,652 | 240,290 | ||||||
| Deferred revenue | 27,381 | 54,302 | ||||||
| Stock-based compensation | 61,027 | 40,090 | ||||||
| Intangible and capital assets | 49,007 | 672 | ||||||
| Other | 8,275 | 12,164 | ||||||
| Total deferred tax assets | $ | 549,059 | $ | 501,093 | ||||
| Deferred Tax Liabilities: | ||||||||
| Convertible debt | $ | (24,018 | ) | $ | (32,391 | ) | ||
| Net deferred tax asset | $ | 525,041 | $ | 468,702 | ||||
| Valuation allowance | (234,245 | ) | (468,702 | ) | ||||
| Total net deferred tax assets and liabilities | $ | 290,796 | $ | — | ||||
We have adjusted all prior year tax amounts to reflect the tax impact of our adoption of Topic 606.
We evaluate our deferred tax assets regularly to determine whether adjustments to the valuation allowance are appropriate due to changes in facts or circumstances, such as changes in expected future pre-tax earnings, tax law, interactions with taxing authorities and developments in case law. In making this evaluation, we rely on our recent history of pre-tax earnings. Our material assumptions are our forecasts of future pre-tax earnings and the nature and timing of future deductions and income represented by the deferred tax assets and liabilities, all of which involve the exercise of significant judgment. Although we believe our estimates are reasonable, we are required to use significant judgment in determining the appropriate amount of valuation allowance recorded against our deferred tax assets.
We have historically recorded a valuation allowance against all our net deferred tax assets due to cumulative financial statement losses. However, in the fourth quarter of 2018, we reversed the valuation allowance previously recorded against Ionis’ stand-alone U.S. federal net deferred tax assets, resulting in a one-time non-cash tax benefit of $332.1 million. Given our current stand-alone Ionis pre-tax income, and assuming we maintain this current level of Ionis stand-alone pre-tax income, we expect to generate income before taxes in the U.S. in future periods at a level that would result in us fully utilizing our U.S. federal net operating loss carryforwards and most of our Research and Development and Orphan Drug tax credit carryforwards over the next three years.
We continue to maintain a full valuation allowance of $234.2 million against all of Akcea’s net deferred tax assets and the net state deferred tax assets of Ionis at December 31, 2018 due to uncertainties related to our ability to realize the tax benefits associated with these assets.
Our valuation allowance decreased by $234.5 million from December 31, 2017 to December 31, 2018. The net decrease relates primarily to the reversal of the valuation allowance previously recorded against Ionis’ stand-alone U.S. federal net deferred tax assets, offset by current year utilization of a portion of our net operating loss carry forwards.
At December 31, 2018, we had federal and state, primarily California, tax net operating loss carryforwards of $284.6 million and $808.7 million, respectively. Our federal tax loss carryforwards will begin to expire in 2033, unless we use them before then. Our California loss carryforwards continued to expire in 2018. At December 31, 2018, we also had federal and California research and development tax credit carryforwards of $288.9 million and $68.4 million, respectively. Our Federal research and development tax credit carryforwards began to expire in 2018. Our California research and development tax credit carryforwards are available indefinitely.
Utilization of the net operating loss and tax credit carryforwards may be subject to an annual limitation due to the ownership change limitations provided by the Internal Revenue Code of 1986, as amended, and similar state provisions. The annual limitation may result in the expiration of net operating losses and credits before utilization.
On December 22, 2017, the U.S. government enacted comprehensive tax legislation commonly referred to as the Tax Cuts and Jobs Act of 2017, or the Tax Act. The Tax Act made broad and complex changes to the U.S. tax code, including, but not limited to, reducing the U.S. federal corporate income tax rate to 21 percent, imposing a mandatory one-time transition tax on certain unrepatriated earnings of foreign subsidiaries and eliminating the corporate alternative minimum tax, or AMT, and changing how existing AMT credits can be realized. We were required to recognize the tax effect of the tax law changes the year of enactment. In order to calculate these effects, we were required to determine the transition tax amount, remeasure our U.S. deferred tax assets and liabilities, and consider the impact to our AMT tax credit carryforwards. For the year ended December 31, 2017, we recorded provisional amounts in accordance with that guidance where it was possible for us to make reasonable estimates of the effects of the Tax Act. We evaluated the decrease in our corporate tax rate and recorded a provisional, one-time tax expense of $107.3 million at December 31, 2017. We fully offset our tax effect by a decrease in our valuation allowance which resulted in no net tax effect in 2017. During the fourth quarter of 2018, we completed our accounting for all aspects of the Tax Act. We did not identify material changes from our 2017 provisional analysis.
F-34
We analyze filing positions in all U.S. federal, state and foreign jurisdictions where we file income tax returns, and all open tax years in these jurisdictions to determine if we have any uncertain tax positions on any of our income tax returns. We recognize the impact of an uncertain tax position on an income tax return at the largest amount that the relevant taxing authority is more-likely-than not to sustain upon audit. We do not recognize uncertain income tax positions if they have less than 50 percent likelihood of the applicable tax authority sustaining our position.
The following table summarizes our gross unrecognized tax benefits (in thousands):
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Beginning balance of unrecognized tax benefits | $ | 78,014 | $ | 66,999 | $ | 51,257 | ||||||
| Settlement of prior period tax positions | — | — | (4,033 | ) | ||||||||
| Decrease for prior period tax positions | (12,814 | ) | — | — | ||||||||
| Increase for prior period tax positions | — | 1,520 | 7,928 | |||||||||
| Increase for current period tax positions | 3,101 | 9,495 | 11,847 | |||||||||
| Ending balance of unrecognized tax benefits | $ | 68,301 | $ | 78,014 | $ | 66,999 | ||||||
Included in the balance of unrecognized tax benefits at December 31, 2018, is $55.5 million that could impact our effective tax rate, subject to our remaining valuation allowance.
We do not foresee any material changes to our gross unrecognized tax benefits within the next twelve months.
We recognize interest and/or penalties related to income tax matters in income tax expense. We did not recognize any accrued interest and penalties related to gross unrecognized tax benefits during the year ended December 31, 2018.
We are subject to taxation in the U.S. and various state and foreign jurisdictions. Our tax years for 1999 through 2018 are subject to examination by the U.S. federal, state and foreign tax authorities.
We do not provide for a U.S. income tax liability and foreign withholding taxes on undistributed foreign earnings of our foreign subsidiaries as we consider those earnings to be permanently reinvested. It is not practicable for us to calculate the amount of unrecognized deferred tax liabilities associated with these earnings.
We are subject to periodic audits by domestic and foreign tax authorities; however, we are not aware of any audits at this time. We believe that we have appropriate support for the income tax positions taken on our tax returns and our accruals for tax liabilities are adequate for all open audit years. Our conclusions are based on an assessment of many factors, including past experience and interpretations of tax law applied to the facts of each matter.
6. Collaborative Arrangements and Licensing Agreements
Strategic Partnership
Biogen
We have several strategic collaborations with Biogen focused on using antisense technology to advance the treatment of neurological disorders. These collaborations combine our expertise in creating antisense medicines with Biogen’s expertise in developing therapies for neurological disorders. We developed and licensed to Biogen SPINRAZA, our approved medicine to treat people with spinal muscular atrophy, or SMA. In December 2017, we entered into a collaboration with Biogen to identify new antisense medicines for the treatment of SMA. Additionally, we and Biogen are currently developing six other medicines to treat neurodegenerative diseases under these collaborations, including IONIS-SOD1Rx for ALS, IONIS-MAPTRx for Alzheimer’s disease, IONIS-C9Rx for ALS, and IONIS-BIIB6Rx, IONIS-BIIB7Rx and IONIS-BIIB8Rx to treat undisclosed neurodegenerative diseases. In addition to these medicines, we and Biogen are evaluating numerous additional targets to develop medicines to treat neurological diseases. In April 2018, we entered into a new strategic collaboration for the treatment of neurological diseases with Biogen. From inception through December 2018, we have received over $2.0 billion from our Biogen collaborations, including $1 billion we received from Biogen in the second quarter of 2018 for our 2018 strategic neurology collaboration.
F-35
Spinal Muscular Atrophy Collaborations
SPINRAZA
In January 2012, we entered into a collaboration agreement with Biogen to develop and commercialize SPINRAZA, an RNA-targeted therapy for the treatment of SMA. Biogen reported in January 2019 that SPINRAZA was approved in over 40 countries around the world. In February 2019, SPINRAZA was approved in China. Biogen is responsible for global SPINRAZA commercial activities.
From inception through December 2018, we earned more than $785 million in total revenue under our SPINRAZA collaboration, including more than $350 million in revenue from SPINRAZA royalties and more than $435 million in R&D revenue. We are receiving tiered royalties ranging from 11 percent to 15 percent on any net sales of SPINRAZA. We have exclusively in-licensed patents related to SPINRAZA from Cold Spring Harbor Laboratory and the University of Massachusetts. We pay Cold Spring Harbor Laboratory and the University of Massachusetts a low single digit royalty on net sales of SPINRAZA. Biogen is responsible for all further global development, regulatory and commercialization activities and costs for SPINRAZA.
Over the course of our SPINRAZA collaboration, we identified two performance obligations, which were to perform R&D services and to deliver the SPINRAZA license to Biogen. As we achieved milestone payments for our R&D services, we included these amounts in our transaction price for our R&D services performance obligation. We recognized revenue for our R&D services performance obligation over our period of performance through December 2016. We recognized the $75 million license fee for SPINRAZA as revenue when we delivered the license to Biogen in July 2016 because Biogen had full use of the license without any continuing involvement from us. Additionally, we did not have any further performance obligations related to the license after we delivered it to Biogen.
We also earned additional milestone payments subsequent to delivering the license to Biogen that we recognized in full in the period each milestone payment became probable because we did not have a performance obligation related to each milestone payment. For example, we received $90 million of milestone payments for the approval of SPINRAZA in the EU and Japan in 2017 and recognized the full amounts into revenue in the period Biogen achieved the milestone events.
New antisense medicines for the treatment of SMA
In December 2017, we entered into a collaboration agreement with Biogen to identify new antisense medicines for the treatment of SMA. Biogen will have the option to license therapies arising out of this collaboration following the completion of preclinical studies. Upon licensing, Biogen will be responsible for all further global development, regulatory and commercialization activities and costs for such therapies. Under the collaboration agreement, we received a $25 million upfront payment in December 2017. We will receive development and regulatory milestone payments from Biogen if new medicines advance towards marketing approval. In total over the term of our collaboration, we are eligible to receive up to $1.2 billion in license fees, milestone payments and other payments, including up to $80 million for the achievement of development milestones, up to $180 million for the achievement of commercialization milestones and up to $800 million for the achievement of sales milestones. In addition, we are eligible to receive tiered royalties from the mid-teens to mid-20 percent range on net sales. We will achieve the next payment of up to $60 millionfor the license of a medicine under this collaboration.
At the commencement of this collaboration, we identified one performance obligation, which was to perform R&D services for Biogen. We determined the transaction price to be the $25 million upfront payment we received when we entered into the collaboration. We allocated the transaction price to our single performance obligation. We are recognizing revenue for our R&D services performance obligation as we perform services based on our effort to satisfy our performance obligation relative to our total effort expected to satisfy our performance obligation. We currently estimate we will satisfy our performance obligation in December 2020.
Neurology Collaborations
2018 Strategic Neurology
In April 2018, we and Biogen entered into a new strategic collaboration to develop novel antisense medicines for a broad range of neurological diseases and entered into a SPA. As part of the collaboration, Biogen gained exclusive rights to the use of our antisense technology to develop therapies for these diseases for 10 years. We are responsible for the identification of antisense drug candidates based on selected targets. Biogen is responsible for conducting IND-enabling toxicology studies for the selected target. Biogen will have the option to license the selected target after it completes the IND-enabling toxicology study. If Biogen exercises its option for a medicine, it will assume all further global development, regulatory and commercialization responsibilities and costs for that medicine.
In the second quarter of 2018, we received $1 billion from Biogen, comprised of $625 million to purchase our stock at an approximately 25 percent cash premium and $375 million in an upfront payment. We are eligible to receive up to $270 million in milestone payments for each medicine that achieves marketing approval. In addition, we are eligible to receive tiered royalties up to the 20 percent range on net sales. From inception through December 2018, we have received over $1 billion in payments under this collaboration, including $15 million we received in the fourth quarter of 2018 for advancing two targets under this collaboration. We will achieve the next payment of $7.5 million if Biogen designates a target under this collaboration.
F-36
At the commencement of this collaboration, we identified one performance obligation, which was to perform R&D services for Biogen. We determined our transaction price to be $552 million, comprised of $375 million from the upfront payment and $177 million for the premium paid by Biogen for its purchase of our common stock. We determined the fair value of the premium we received by using the stated premium in the SPA and applying a lack of marketability discount. We included a lack of marketability discount in our valuation of the premium because Biogen received restricted shares. We allocated the transaction price to our single performance obligation. In the fourth quarter of 2018, we received $15 million in milestone payments when we advanced two targets under this collaboration. We added these payments to our transaction price for our R&D services performance obligation. We are recognizing revenue for our R&D services performance obligation as we perform services based on our effort to satisfy our performance obligation relative to our total effort expected to satisfy our performance obligation. We currently estimate we will satisfy our performance obligation in June 2028.
2013 Strategic Neurology
In September 2013, we and Biogen entered into a long-term strategic relationship focused on applying antisense technology to advance the treatment of neurodegenerative diseases. As part of the collaboration, Biogen gained exclusive rights to the use of our antisense technology to develop therapies for neurological diseases and has the option to license medicines resulting from this collaboration. We will usually be responsible for drug discovery and early development of antisense medicines and Biogen will have the option to license antisense medicines after Phase 2 proof of concept. In October 2016, we expanded our collaboration to include additional research activities we will perform. If Biogen exercises its option for a medicine, it will assume all further global development, regulatory and commercialization responsibilities and costs for that medicine. We are currently advancing five medicines, IONIS-SOD1Rx, IONIS-C9Rx, IONIS-BIIB6Rx, IONIS-BIIB7Rx and IONIS-BIIB8Rx under this collaboration. In the fourth quarter of 2018, Biogen licensed IONIS-SOD1Rx, and as a result Biogen now is responsible for all further global development, regulatory and commercialization activities and costs for IONIS-SOD1Rx.
Under the terms of the agreement, we received an upfront payment of $100 million and are eligible to receive milestone payments, license fees and royalty payments for all medicines developed through this collaboration, with the specific amounts dependent upon the modality of the molecule advanced by Biogen. For each antisense molecule that is chosen for drug discovery and development under this collaboration, we are eligible to receive up to approximately $260 million in a license fee and milestone payments per program. The $260 million per program consists of approximately $60 million in development milestones, including amounts related to the cost of clinical trials, and up to $130 million in milestone payments if Biogen achieves pre-specified regulatory milestones. In addition, we are eligible to receive tiered royalties up to the mid-teens on net sales from any antisense medicines developed under this collaboration. From inception through December 2018, we have received over $210 million in upfront fees, milestone payments and other payments under this collaboration, not including a $5 million milestone payment we earned in the fourth quarter of 2018 for Biogen’s initiation of a Proof of Concept study for IONIS-SOD1Rx which we received in the first quarter of 2019. We will achieve the next payment of up to $10 million if we advance a program under this collaboration.
At the commencement of our strategic neurology collaboration, we identified one performance obligation, which was to perform R&D services for Biogen. At inception, we determined the transaction price to be the $100 million upfront payment we received and allocated it to our single performance obligation. As we achieve milestone payments for our R&D services, we include these amounts in our transaction price for our R&D services performance obligation. We are recognizing revenue for our R&D services performance obligation based on our effort to satisfy our performance obligation relative to our total effort expected to satisfy our performance obligation. We currently estimate we will satisfy our performance obligation in September 2019. From inception through December 2018, we have included $145 million in total payments in the transaction price for our R&D services performance obligation. In the third quarter of 2018, we earned a $10 million milestone payment when Biogen initiated a Phase 1 study of IONIS-C9Rx. We concluded that the milestone payment was not related to our R&D services performance obligation. Therefore, we recognized this milestone payment in full in the third quarter of 2018 because we do not have any performance obligations related to this milestone payment.
We identified a second performance obligation upon Biogen’s license of IONIS-SOD1Rx in the fourth quarter of 2018 because the license we granted to Biogen is distinct from our other performance obligation. We recognized the $35 million license fee for IONIS-SOD1Rx as revenue at that time because Biogen had full use of the license without any continuing involvement from us. Additionally, we did not have any further performance obligations related to the license after we delivered it to Biogen. Additionally, in the fourth quarter of 2018 we earned a $5 million milestone when Biogen initiated a Proof-of-Concept study for IONIS-SOD1Rx. We concluded that the milestone payment was not related to our R&D services performance obligation. Therefore, we recognized this milestone payment in full in the fourth quarter of 2018 because we do not have any performance obligations related to this milestone payment.
Neurology
In December 2012, we and Biogen entered into a collaboration agreement to develop and commercialize novel antisense medicines to up to three targets to treat neurodegenerative diseases. We are responsible for the development of each of the medicines through the completion of the initial Phase 2 clinical study for such medicine. Biogen has the option to license a medicine from each of the programs through the completion of the first Phase 2 study for each program. We are currently advancing IONIS-MAPTRx for Alzheimer’s disease under this collaboration. If Biogen exercises its option for a medicine, it will assume all further global development, regulatory and commercialization responsibilities and costs for that medicine.
Under the terms of the agreement, we received an upfront payment of $30 million. Over the term of the collaboration, we are eligible to receive up to $210 million in a license fee and milestone payments per program, plus a mark-up on the cost estimate of the Phase 1 and 2 studies. The $210 million per program consists of up to $10 million in development milestone payments, plus a mark-up on the cost estimate of the Phase 1 and 2 studies and up to $130 million in milestone payments if Biogen achieves pre-specified regulatory milestones. In addition, we are eligible to receive tiered royalties up to the mid-teens on net sales of any medicines resulting from each of the three programs. From inception through December 2018, we have received $58 million in milestone payments and upfront fees under this collaboration. We will achieve the next payment of $7.5 million if we continue to advance IONIS-MAPTRx.
F-37
At the commencement of this collaboration, we identified one performance obligation, which was to perform R&D services for Biogen. At inception, we determined the transaction price to be the $30 million upfront payment we received and allocated it to our single performance obligation. As we achieve milestone payments for our R&D services, we include these amounts in our transaction price for our R&D services performance obligation. We are recognizing revenue for our R&D services performance obligation as we perform services based on our effort to satisfy our performance obligation relative to our total effort expected to satisfy our performance obligation. We currently estimate we will satisfy our performance obligation in December 2020. From inception through December 2018, we have included $40 million in total payments in the transaction price for our R&D services performance obligation.
During the years ended December 31, 2018, 2017 and 2016, we earned the following revenue from our relationship with Biogen (in millions, except percentage amounts):
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised) | ||||||||||||
| SPINRAZA royalties (commercial revenue) | $ | 237.9 | $ | 112.5 | $ | 0.9 | ||||||
| R&D revenue | 137.1 | 150.6 | 248.8 | |||||||||
| Total revenue from our relationship with Biogen | 375.0 | 263.1 | 249.7 | |||||||||
| Percentage of total revenue | 63 | % | 51 | % | 67 | % | ||||||
Our consolidated balance sheet at December 31, 2018 and 2017 included deferred revenue of $580.9 million and $93.6 million, respectively, related to our relationship with Biogen.
Research, Development and Commercialization Partners
AstraZeneca
Cardiac, Renal and Metabolic Diseases Collaboration
In July 2015, we and AstraZeneca formed a collaboration to discover and develop antisense therapies for treating cardiac, renal and metabolic diseases. Under our collaboration, AstraZeneca has licensed three medicines from us: IONIS-AZ4-2.5-LRx, a medicine we designed to treat cardiovascular disease and our first medicine that combines our Generation 2.5 and LICA technology, IONIS-AZ5-2.5Rx, a medicine we designed to treat a genetically associated form of kidney disease and IONIS-AZ6-2.5-LRx, a medicine we designed to inhibit an undisclosed target to treat patients with nonalcoholic steatohepatitis, or NASH. AstraZeneca is responsible for all further global development, regulatory and commercialization activities and costs for each of the medicines it has licensed and any other future medicines AstraZeneca licenses.
Under the terms of the agreement, we received a $65 million upfront payment. We are eligible to receive license fees and milestone payments of up to more than $4 billion as medicines under this collaboration advance, including up to $1.1 billion for the achievement of development milestones and up to $2.9 billion for regulatory milestones. In addition, we are eligible to receive tiered royalties up to the low teens on net sales from any product that AstraZeneca successfully commercializes under this collaboration agreement. From inception through December 2018, we have received over $165 million in upfront fees, license fees, milestone payments, and other payments under this collaboration, including a $10 million milestone payment we earned in the third quarter of 2018 when AstraZeneca initiated a Phase 1 trial for IONIS-AZ4-2.5-LRx. We will achieve the next payment of $10 million under this collaboration if we advance a medicine under this collaboration.
At the commencement of this collaboration, we identified one performance obligation, which was to perform R&D services for AstraZeneca. We determined the transaction price to be the $65 million upfront payment we received and we allocated it to our single performance obligation. We are recognizing revenue for our R&D services performance obligation as we perform services based on our effort to satisfy this performance obligation relative to our total effort expected to satisfy our performance obligation. We currently estimate we will satisfy this performance obligation in August 2021. As we achieve milestone payments for our R&D services, we include these amounts in our transaction price for our R&D services performance obligation. From inception through December 2018, we have included $90 million in payments in the transaction price for our R&D services performance obligation.
We identified separate performance obligations upon AstraZeneca’s license of IONIS-AZ5-2.5Rx and IONIS-AZ6-2.5-LRx in the first quarter of 2018 because the licenses are distinct from our other performance obligation and each other. We recognized each $30 million license fee in the first quarter of 2018 because AstraZeneca had full use of the licenses without any continuing involvement from us. Additionally, we did not have any further performance obligations related to the licenses after we delivered them to AstraZeneca.
In the third quarter of 2018, we earned a $10 million milestone payment when AstraZeneca initiated a Phase 1 study of IONIS-AZ4-2.5-LRx. We concluded that the milestone payment was not related to our R&D services performance obligation. Therefore, we recognized this milestone payment in full in the third quarter of 2018 because we do not have any performance obligations related to this milestone payment.
F-38
Oncology Collaboration
In December 2012, we entered into a collaboration agreement with AstraZeneca to discover and develop antisense medicines to treat cancer. As part of the agreement, we granted AstraZeneca an exclusive license to develop and commercialize danvatirsen (formerly IONIS-STAT3-2.5Rx) for the treatment of cancer. AstraZeneca is now responsible for all global development, regulatory and commercialization activities for danvatirsen. We and AstraZeneca have evaluated danvatirsen in people with head and neck cancer, advanced lymphoma and advanced metastatic hepatocellular carcinoma. AstraZeneca is evaluating danvatirsen in combination with durvalumab, AstraZeneca’s PD-L1 blocking drug, in people with head and neck cancer, metastatic bladder cancer and metastatic non-small cell lung cancer. We and AstraZeneca also established an oncology research program. AstraZeneca has the option to license medicines resulting from the program, and if AstraZeneca exercises its option for a medicine, it will be responsible for all further global development, regulatory and commercialization activities and costs for such medicine. In the fourth quarter of 2018, we added IONIS-AZ7-2.5Rx to our preclinical pipeline, a second drug under our oncology collaboration.
Under the terms of this agreement, we received $31 million in upfront payments. We are eligible to receive milestone payments and license fees from AstraZeneca as programs advance in development. If AstraZeneca successfully develops danvatirsen and another medicine under the research program, we could receive license fees and milestone payments of up to more than $450 million, including up to $152 million for the achievement of development milestones and up to $275 million for the achievement of regulatory milestones. In addition, we are eligible to receive tiered royalties up to the mid-teens on net sales from any medicines resulting from these programs. From inception through December 2018, we have received over $125 million in upfront fees, milestone payments, and other payments under this oncology collaboration, including nearly $30 million in milestone payments we achieved when AstraZeneca advanced danvatirsen and IONIS-AZ7-2.5Rx, in the fourth quarter of 2018. We will achieve the next payment of up to $25 million if we advance a medicine under our cancer research program with AstraZeneca.
At the commencement of this collaboration, we identified four performance obligations. We determined the transaction price to be the $31 million upfront payments we received. We allocated the transaction price based on the estimated stand-alone selling price of each of our performance obligations and recognized the associated revenue over the period of our performance. We recognized revenue for three of our obligations over our period of performance, which concluded in March 2014. Our remaining performance obligation was to perform R&D services. We allocated $7.6 million to this performance obligation and recognized the associated revenue over the period of our performance, which ended in February 2018. As we achieved milestone payments for our R&D services, we included these amounts in our transaction price for our R&D services performance obligation.
In the fourth quarter of 2018, we earned a $17.5 million milestone payment and a $10 million milestone payment when AstraZeneca advanced two programs under our collaboration. We recognized these milestone payments in full in the fourth quarter because we do not have any performance obligations related to these milestone payments.
During the years ended December 31, 2018, 2017 and 2016, we earned the following revenue from our relationship with AstraZeneca (in millions, except percentage amounts):
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised) | ||||||||||||
| R&D revenue | $ | 120.7 | $ | 21.6 | $ | 41.3 | ||||||
| Percentage of total revenue | 20 | % | 4 | % | 11 | % | ||||||
Our consolidated balance sheet at December 31, 2018 and 2017 included deferred revenue of $40.1 million and $57.7 million, respectively, related to our relationship with AstraZeneca.
Bayer
In May 2015, we entered into an exclusive license agreement with Bayer to develop and commercialize IONIS-FXIRx for the prevention of thrombosis. We were responsible for completing a Phase 2 study of IONIS-FXIRx in people with end-stage renal disease on hemodialysis. Under the terms of the agreement, we received a $100 million upfront payment in the second quarter of 2015. In February 2017, we amended our agreement with Bayer to advance IONIS-FXIRx and to initiate development of IONIS-FXI-LRx, which Bayer licensed. In conjunction with the decision to advance these programs, we received a $75 million payment from Bayer. We are conducting a Phase 2b study evaluating IONIS-FXIRx in people with end-stage renal disease on hemodialysis to finalize dose selection. Additionally, we are developing IONIS-FXI-LRx through Phase 1. Following these studies and Bayer’s decision to further advance these programs, Bayer will be responsible for all global development, regulatory and commercialization activities and costs for both medicines.
We are eligible to receive additional milestone payments as each medicine advances toward the market. In total over the term of this collaboration, we are eligible to receive up to $385 million in license fees, milestone payments and other payments, including up to $125 million for the achievement of development milestones and up to $110 million for the achievement of commercialization milestones. In addition, we are eligible to receive tiered royalties in the low to high 20 percent range on gross margins of both medicines combined. From inception through December 2018, we have received over $175 million from our Bayer collaboration. We will achieve the next payment of $10 million if a program advances under this collaboration.
F-39
At the commencement of this collaboration, we identified three performance obligations. We determined the transaction price to be the $100 million upfront payment we received. We allocated the transaction price based on the relative stand-alone selling prices of each of our performance obligations and recognized the associated revenue as follows:
| ● | We recognized $91.2 million for the exclusive license of IONIS-FXIRx in May 2015 because Bayer had full use of the license without any continuing involvement from us. |
| ● | We recognized $4.3 million for the R&D services for IONIS-FXIRx over the period of our performance, which ended in November 2016. |
| ● | We allocated $4.5 million for API, which we are recognizing into revenue as we deliver the API. |
In February 2017, when we amended our collaboration with Bayer, we identified two new performance obligations, one for the license of IONIS-FXI-LRx and one for R&D services. We determined the transaction price to be the $75 million payment. We allocated $64.9 million to the license of IONIS-FXI-LRx based on its estimated stand-alone selling price and recognized the associated revenue upon our delivery of the license in the first quarter of 2017. We allocated $10.1 million to our R&D services performance obligation based on an estimated stand-alone selling price. We are recognizing revenue for our R&D services performance obligation as we perform services based on our effort to satisfy our performance obligation relative to our total effort expected to satisfy our performance obligation. We currently estimate we will satisfy our R&D services performance obligation in May 2019.
During the years ended December 31, 2018, 2017 and 2016, we earned the following revenue from our relationship with Bayer (in millions, except percentage amounts):
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised) | ||||||||||||
| R&D revenue | $ | 5.0 | $ | 67.1 | $ | 5.4 | ||||||
| Percentage of total revenue | 1 | % | 13 | % | 1 | % | ||||||
Our consolidated balance sheet at December 31, 2018 and 2017 included deferred revenue of $4.3 million and $9.3 million, respectively, related to our relationship with Bayer.
GSK
In March 2010, we entered into an alliance with GSK using our antisense drug discovery platform to discover and develop new medicines against targets for rare and serious diseases, including infectious diseases and some conditions causing blindness. Under the terms of the agreement, we received upfront payments of $35 million.
GSK is advancing two medicines targeting hepatitis B virus, or HBV, under our collaboration: IONIS-HBVRx and IONIS-HBV-LRx. GSK is currently conducting Phase 2 studies for both of these medicines, which we designed to reduce the production of viral proteins associated with HBV infection. GSK has the exclusive option to license the medicines resulting from this alliance at Phase 2 proof-of-concept for a license fee.
Under our agreement, if GSK successfully develops these medicines and achieves pre-agreed sales targets, we could receive license fees and milestone payments of $262 million, including up to $47.5 million for the achievement of development milestones, up to $120 million for the achievement of regulatory milestones and up to $70 million for the achievement of commercialization milestones. In addition, we are eligible to receive tiered royalties up to the mid-teens on net sales from any product that GSK successfully commercializes under this alliance. From inception through December 2018, we have received more than $162 million in payments under this alliance with GSK. We will achieve the next payment of up to $25 million if GSK licenses a medicine under this program.
At the commencement of this collaboration, we identified one performance obligation, which was to perform R&D services for GSK. We determined the transaction price to be the $35 million upfront payments we received and allocated it to our single performance obligation. As we achieved milestone payments for our R&D services, we included these amounts in our transaction price for our R&D services performance obligation. We recognized revenue for our R&D services performance obligation over our period of performance, which ended in March 2015. We do not have any remaining performance obligations under our collaboration with GSK, however we can still earn additional payments and royalties as GSK advances these medicines.
During the years ended December 31, 2018, 2017 and 2016, we earned the following revenue from our relationship with GSK (in millions, except percentage amounts):
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised) | ||||||||||||
| R&D revenue | $ | 1.6 | $ | 14.8 | $ | 17.5 | ||||||
| Percentage of total revenue | 0 | % | 3 | % | 5 | % | ||||||
We did not have any deferred revenue from our relationship with GSK at December 31, 2018 or December 31, 2017.
F-40
Janssen Biotech, Inc.
In December 2014, we entered into a collaboration agreement with Janssen Biotech, Inc. to discover and develop antisense medicines that can be locally administered, including oral delivery, to treat autoimmune disorders of the gastrointestinal tract. Janssen has the option to license medicines from us through the designation of a development candidate for up to three programs. Under our collaboration, Janssen licensed IONIS-JBI1-2.5Rx in July 2016 and IONIS-JBI2-2.5Rx in November 2017. Janssen is currently conducting a Phase 1 study of IONIS-JBI1-2.5Rx and IONIS-JBI2-2.5Rx is in preclinical development. Prior to option exercise we are responsible for the discovery activities to identify development candidates. If Janssen exercises an option for any of the programs, it will be responsible for the global development, regulatory and commercial activities under that program. Under the terms of the agreement, we received $35 million in upfront payments. We are eligible to receive up to more than $800 million in license fees and milestone payments for these programs, including up to $175 million for the achievement of development milestones, up to $440 million for the achievement of regulatory milestones and up to $180 million for the achievement of commercialization milestones. From inception through December 2018, we have received over $75 million, including $15 million in license fees when Janssen licensed IONIS-JBI1-2.5Rx and IONIS-JBI2-2.5Rx from us. We also received $5 million in January 2018 for the initiation of a Phase 1 study of IONIS-JBI1-2.5Rx in late 2017. In addition, we are eligible to receive tiered royalties up to the near teens on net sales from any medicines resulting from this collaboration. We will achieve the next payment of $5 million if Janssen continues to advance a target under this collaboration.
At the commencement of this collaboration, we identified one performance obligation, which was to perform R&D services for Janssen. We determined the transaction price to be the $35 million upfront payments we received. We allocated the $35 million to our single performance obligation. As we achieved milestone payments for our R&D services, we included these amounts in our transaction price for our R&D services performance obligation. We recognized revenue for our R&D services performance obligation over our period of performance, which ended in November 2017.
We identified separate performance obligations each time Janssen licensed one of our medicines under our collaboration because the licenses we granted to Janssen were distinct from our other performance obligations. We recognized the $10 million license fee for IONIS-JBI1-2.5Rx in July 2016 and $5 million for the license of IONIS-JBI2-2.5Rx in November 2017, because Janssen had full use of the licenses without any continuing involvement from us. Additionally, we did not have any further performance obligations related to the licenses after we delivered them to Janssen.
During the years ended December 31, 2018, 2017 and 2016, we earned the following revenue from our relationship with Janssen (in millions, except percentage amounts):
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised) | ||||||||||||
| R&D revenue | $ | 6.6 | $ | 36.0 | $ | 24.8 | ||||||
| Percentage of total revenue | 1 | % | 7 | % | 7 | % | ||||||
We did not have any deferred revenue from our relationship with Janssen at December 31, 2018 and 2017.
Roche
Huntington’s Disease
In April 2013, we formed an alliance with Hoffman-La Roche Inc. and F. Hoffmann-La Roche Ltd., collectively Roche, to develop treatments for Huntington’s disease, or HD, based on our antisense technology. Roche had the option to license the medicines from us through the completion of the first Phase 1 trial. Under the agreement, we are responsible for the discovery and development of an antisense medicine targeting huntingtin, or HTT, protein. We evaluated a medicine targeting HTT, IONIS-HTTRx, in a Phase 1/2 clinical study in people with early stage HD.
In December 2017, upon completion of the Phase 1/2 study, Roche exercised its option to license IONIS-HTTRx and is now responsible for the global development, regulatory and commercialization activities for IONIS-HTTRx. Under the terms of the agreement, we received an upfront payment of $30 million in April 2013. In December 2016, we updated development activities for IONIS-HTTRx and as a result we were eligible for an additional $3 million payment, which we achieved in 2017. We are eligible to receive up to $365 million in a license fee and milestone payments including up to $70 million for the achievement of development milestones, up to $170 million for the achievement of regulatory milestones and up to $80 million for the achievement of commercialization milestones. In addition, we are eligible to receive up to $136.5 million in milestone payments for each additional medicine successfully developed. We are also eligible to receive tiered royalties up to the mid-teens on any net sales of any product resulting from this alliance. From inception through December 2018, we have received over $112 million in upfront fees, milestone payments and license fees for advancing IONIS-HTTRx, not including $35 million in milestone payments we earned in the first quarter of 2019 when Roche dosed the first patient in a Phase 3 study for IONIS-HTTRx. We will achieve the next payment of $15 million if Roche advances IONIS-HTTRx.
At the commencement of this collaboration, we identified one performance obligation, which was to perform R&D services for Roche. We determined the transaction price to be the $30 million upfront payment we received and allocated it to our single performance obligation. As we achieved milestone payments for our R&D services, we included these amounts in our transaction price for our R&D services performance obligation. We recognized revenue for our R&D services performance obligation over our period of performance, which ended in September 2017.
F-41
We identified a second performance obligation upon Roche’s license of IONIS-HTTRx in the fourth quarter of 2017 because the license we granted to Roche is distinct from our other performance obligation. We recognized the $45 million license fee for IONIS-HTTRx as revenue at that time because Roche had full use of the license without any continuing involvement from us. Additionally, we did not have any further performance obligations related to the license after we delivered it to Roche.
We do not have any remaining performance obligations related to IONIS-HTTRx under this collaboration with Roche, however we can still earn additional payments and royalties as Roche advances IONIS-HTTRx.
IONIS-FB-LRx for Complement-Mediated Diseases
In October 2018, we entered into a collaboration agreement with Roche to develop IONIS-FB-LRx for the treatment of complement-mediated diseases. The first indication we plan to pursue is the treatment of patients with Geographic Atrophy, or GA, the advanced stage of dry age-related macular degeneration, or AMD. We are responsible for conducting a Phase 2 study in patients with dry AMD. In addition, we are exploring the medicine in a severe and rare renal indication. Roche has the option to license IONIS-FB-LRx at the completion of these studies. Upon licensing, Roche will be responsible for all further global development, regulatory and commercialization activities and costs.
Under the terms of this agreement, we received a $75 million upfront payment in October 2018. We are eligible to receive up to $684 million in development, regulatory and sales milestone payments and license fees. In addition, we are also eligible to receive tiered royalties from the high teens to twenty percent on net sales. We will achieve the next payment of $20 million when we advance the Phase 2 study in patients with dry AMD.
At the commencement of this collaboration, we identified one performance obligation, which was to perform R&D services for Roche. We determined the transaction price to be the $75 million upfront payment we received and allocated it to our single performance obligation. We are recognizing revenue for our R&D services performance obligation as we perform services based on our effort to satisfy our performance obligation relative to our total effort expected to satisfy our performance obligation. We currently estimate we will satisfy our performance obligation in December 2022.
During the years ended December 31, 2018, 2017 and 2016, we earned the following revenue from our relationship with Roche (in millions, except percentage amounts):
| Years Ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (as revised) | ||||||||||||
| R&D revenue | $ | 8.3 | $ | 55.7 | $ | 10.7 | ||||||
| Percentage of total revenue | 1 | % | 11 | % | 3 | % | ||||||
Our consolidated balance sheet at December 31, 2018 included deferred revenue of $72.6 million related to our relationship with Roche. We did not have any deferred revenue related to our relationship with Roche at December 31, 2017.
Akcea Collaborations
The following collaboration agreements relate to Akcea, our majority-owned affiliate. Our consolidated results include all the revenue earned and cash received under these collaboration agreements. We reflect the noncontrolling interest attributable to other owners of Akcea’s common stock in a separate line on the statement of operations and in a separate line within stockholders’ equity in our consolidated balance sheet.
Novartis
In January 2017, we and Akcea initiated a collaboration with Novartis to develop and commercialize AKCEA-APO(a)-LRx and AKCEA-APOCIII-LRx. Under the collaboration agreement, Novartis has an exclusive option to further develop and commercialize AKCEA-APO(a)-LRx and AKCEA-APOCIII-LRx. Akcea is responsible for completing a Phase 2 program, conducting an end-of-Phase 2 meeting with the FDA and providing initial quantities of API for each medicine. If Novartis exercises an option for either of these medicines, Novartis will be responsible for all further global development, regulatory and co-commercialization activities and costs for such medicine.
Akcea received a $75 million upfront payment in the first quarter of 2017, of which it retained $60 million and paid us $15 million as a sublicense fee. In February 2019, Novartis licensed AKCEA-APO(a)-LRx and we earned a $150 million license fee. Akcea will pay us $75 million as a sublicense fee in 2.8 million shares of Akcea common stock. Novartis is responsible for conducting and funding all future development, regulatory and commercialization activities for AKCEA-APO(a)-LRx, including a global pivotal cardiovascular outcomes study, for which planning and initiation activities are underway. If Novartis exercises its option for AKCEA-APOCIII-LRx, Novartis will pay Akcea a license fee equal to $150 million. In addition, for AKCEA-APO(a)-LRx, Akcea is eligible to receive up to $675 million in milestone payments, including $25 million for the achievement of a development milestone, up to $290 million for the achievement of regulatory milestones and up to $360 million for the achievement of commercialization milestones. In addition, for AKCEA-APOCIII-LRx, Akcea is eligible to receive up to $530 million in milestone payments, including $25 million for the achievement of a development milestone, up to $240 million for the achievement of regulatory milestones and up to $265 million for the achievement of commercialization milestones. Akcea is also eligible to receive tiered royalties in the mid-teens to low 20 percent range on net sales of AKCEA-APO(a)-LRx and AKCEA-APOCIII-LRx. Akcea will pay 50 percent of these license fees, milestone payments and royalties to us as a sublicense fee. In connection with Novartis’ license of AKCEA-APO(a)-LRx, Akcea and Novartis established a more definitive framework under which the companies would negotiate the co-commercialization of AKCEA-APO(a)-LRx in selected markets. Included in this framework is an option by which Novartis could solely commercialize AKCEA-APO(a)-LRx in exchange for Novartis paying Akcea increased commercial milestone payments based on sales of AKCEA-APO(a)-LRx. Akcea may co-commercialize IONIS-APOCIII-LRx if licensed and commercialized by Novartis in selected markets through its specialized sales force under terms and conditions to be negotiated with Novartis in the future.
F-42
In conjunction with this collaboration, we entered into a SPA with Novartis. As part of the SPA, Novartis purchased 1.6 million shares of our common stock for $100 million in the first quarter of 2017. As part of the SPA, Novartis was required to purchase $50 million of Akcea’s common stock at the IPO price or our common stock at a premium if an IPO did not occur by April 2018. Under the SPA, in July 2017, Novartis purchased $50 million of Akcea’s common stock in a separate private placement concurrent with the completion of its IPO at a price per share equal to the IPO price.
At the commencement of this collaboration, we identified four separate performance obligations:
| ● | R&D services for AKCEA-APO(a)-LRx; |
| ● | R&D services for AKCEA-APOCIII-LRx; |
| ● | API for AKCEA-APO(a)-LRx; and |
| ● | API for AKCEA-APOCIII-LRx. |
We determined that the R&D services for each medicine and the API for each medicine were distinct from our other performance obligations.
We determined our transaction price to be $108.4 million, comprised of the following:
| ● | $75 million from the upfront payment; |
| ● | $28.4 million for the premium paid by Novartis for its purchase of our common stock at a premium in the first quarter of 2017; and |
| ● | $5.0 million for the potential premium Novartis would have paid if they purchased our common stock in the future. |
We allocated the transaction price based on the estimated stand-alone selling price of each performance obligation as follows:
| ● | $64.0 million for the R&D services for AKCEA-APO(a)-LRx; |
| ● | $40.1 million for the R&D services for AKCEA-APOCIII-LRx; |
| ● | $1.5 million for the delivery of AKCEA-APO(a)-LRx API; and |
| ● | $2.8 million for the delivery of AKCEA-APOCIII-LRx API. |
We are recognizing revenue related to the R&D services for the AKCEA-APO(a)-LRx and AKCEA-APOCIII-LRx performance obligations as we perform services based on our effort to satisfy our performance obligation relative to our total effort expected to satisfy our performance obligation. We satisfied the significant portion of our performance obligation for AKCEA-APO(a)-LRx in December 2018 and we currently estimate we will satisfy the remainder by mid-2019. We currently estimate we will satisfy the significant portion of our performance obligation for AKCEA-APOCIII-LRx by mid-2020 with the remainder by the end of 2019. We recognized the amount attributed to the API supply for AKCEA-APO(a)-LRx when we delivered it to Novartis in 2017. We recognized the amount attributed to the API supply for AKCEA-APOCIII-LRx when we delivered it to Novartis in May 2018.
Akcea is responsible for the development activities under this collaboration. As such, Akcea is recognizing the associated revenue in its statement of operations, and we reflect all of Akcea’s revenue in our consolidated results. Akcea pays us sublicense fees for payments that it receives under the collaboration and we recognize those fees as revenue in our Ionis Core operating segment results and Akcea recognizes the fees as R&D expense. In our consolidated results, we eliminate this sublicense revenue and expense. Any cash Akcea receives is included in our consolidated balance sheet.
During the years ended December 31, 2018 and 2017, we earned the following revenue from our relationship with Novartis (in millions, except percentage amounts):
| Years Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| (as revised) | ||||||||
| R&D revenue | $ | 50.6 | $ | 43.4 | ||||
| Percentage of total revenue | 8 | % | 8 | % | ||||
Our consolidated balance sheet at December 31, 2018 and 2017 included deferred revenue of $28.8 million and $70.7 million, respectively, related to our relationship with Novartis.
PTC Therapeutics
In August 2018, Akcea entered into an exclusive license agreement with PTC Therapeutics to commercialize TEGSEDI and WAYLIVRA in Latin America. Under the license agreement, Akcea is eligible to receive up to $26 million in payments, including $12 million which it received in the third quarter of 2018, $6 million upon the earlier of FDA or EMA approval of WAYLIVRA and up to $8 million for regulatory milestones. Akcea is eligible to receive royalties from PTC in the mid-20 percent range on net sales in Latin America for each medicine. PTC’s obligation to pay Akcea royalties begins on the earlier of 12 months after the first commercial sale of a product in Brazil or the date that PTC recognizes revenue of at least $10 million in Latin America. Consistent with the agreements between Ionis and Akcea, the companies will share all payments, including royalties.
At the commencement of this collaboration, we identified two performance obligations, which were the licenses Akcea granted to PTC to commercialize TEGSEDI and WAYLIVRA in Latin America in the third quarter of 2018. Akcea recognized $12 million in license fee revenue at that time because PTC had full use of both licenses without any continuing involvement from Akcea. Akcea does not have any remaining performance obligations under its collaboration with PTC. Akcea can still earn additional payments and royalties as PTC commercializes the medicines.
F-43
Akcea was responsible for the activities under this collaboration. As such, Akcea is recognizing the associated revenue in its statement of operations, and we reflect all of Akcea’s revenue in our consolidated results. Akcea pays us sublicense fees for payments that it receives under the collaboration and we recognize those fees as revenue in our Ionis Core operating segment results and Akcea recognizes the fees as SG&A expense. For example, during the third quarter of 2018, we recognized $7.2 million of sublicense revenue in our Ionis Core operating segment results related to our portion of the PTC license fee Akcea paid us. In our consolidated results, we eliminate this sublicense revenue and expense. Any cash Akcea receives is included in our consolidated balance sheet.
Our consolidated balance sheet at December 31, 2018 and 2017 did not include any deferred revenue related to our relationship with PTC.
External Project Funding
We are pursuing discovery and development projects that provide us with new therapeutic applications for antisense medicines. These programs represent opportunities for us and our technology. In some cases, we have funded these studies through support from our partners or disease advocacy groups and foundations. Our External Project Funding partners include the following:
| ● | CHDI Foundation- Through our development collaboration, CHDI provided financial and scientific support to our Huntington’s disease drug discovery program. We have reimbursed CHDI for its support of our Huntington’s disease program out of the payments we receive from Roche. |
| ● | Cystic Fibrosis Foundation- We received upfront funding from the Cystic Fibrosis Foundation to discover and advance a medicine for the treatment of cystic fibrosis. In exchange for this funding, we are obligated to pay the Cystic Fibrosis Foundation up to $18 million upon achieving specific regulatory and sales events if we advance a medicine under our collaboration. |
| ● | The Ludwig Institute; Center for Neurological Studies- We have a collaboration with the Ludwig Institute, the Center for Neurological Studies and researchers to discover and develop antisense medicines for ALS and other neurodegenerative diseases. Under this agreement, we agreed to pay the Ludwig Institute and the Center for Neurological Studies modest milestone payments and royalties on any antisense medicines resulting from the collaboration. |
In-Licensing Agreements
Our in-licensing arrangements include:
| ● | University of Massachusetts- We have a license agreement with the University of Massachusetts under which we acquired an exclusive license to the University of Massachusetts’ patent rights related to SPINRAZA. We paid the University of Massachusetts nominal amounts for license fees and milestone payments we received. We also pay a low single digit royalty on net sales of SPINRAZA. |
| ● | Cold Spring Harbor Laboratory- We have a collaboration and license agreement with the Cold Spring Harbor Laboratory under which we acquired an exclusive license to the Cold Spring Harbor Laboratory’s patent rights related to SPINRAZA. We paid Cold Spring Harbor Laboratory nominal amounts for license fees and milestone payments we received in 2017 and a low single digit royalty on net sales of SPINRAZA. Additionally, we owe a low single digit royalty on future sales of SPINRAZA. |
7. Segment Information and Concentration of Business Risk
We have two reportable segments Ionis Core and Akcea Therapeutics. At December 31, 2018, we owned approximately 75 percent of Akcea. Segment income (loss) from operations includes revenue less operating expenses attributable to each segment.
In our Ionis Core segment we are exploiting our antisense technology to generate a broad pipeline of first-in-class and/or best-in-class medicines for us and our partners. Our Ionis Core segment generates revenue from a multifaceted partnering strategy.
Akcea is a biopharmaceutical company focused on developing and commercializing medicines to treat patients with rare and serious diseases.
F-44
The following tables show our segment revenue and income (loss) from operations for 2018, 2017 and 2016 (in thousands), respectively.
| 2018 | Ionis Core | Akcea Therapeutics | Elimination of Intercompany Activity | Total | ||||||||||||
| Revenue: | ||||||||||||||||
| Commercial revenue: | ||||||||||||||||
| SPINRAZA royalties | $ | 237,930 | $ | — | $ | — | $ | 237,930 | ||||||||
| TEGSEDI product sales, net | — | 2,237 | — | 2,237 | ||||||||||||
| Licensing and other royalty revenue | 2,755 | 12,000 | — | 14,755 | ||||||||||||
| Total commercial revenue | 240,685 | 14,237 | — | 254,922 | ||||||||||||
| R&D revenue under collaborative agreements | 401,259 | 50,630 | (107,137 | ) | 344,752 | |||||||||||
| Total segment revenue | $ | 641,944 | $ | 64,867 | $ | (107,137 | ) | $ | 599,674 | |||||||
| Total operating expenses | $ | 380,212 | $ | 295,683 | $ | (14,849 | ) | $ | 661,046 | |||||||
| Income (loss) from operations | $ | 261,732 | $ | (230,816 | ) | $ | (92,288 | ) | $ | (61,372 | ) | |||||
| 2017 (as revised) | Ionis Core | Akcea Therapeutics | Elimination of Intercompany Activity | Total | ||||||||||||
| Revenue: | ||||||||||||||||
| Commercial revenue: | ||||||||||||||||
| SPINRAZA royalties | $ | 112,540 | $ | — | $ | — | $ | 112,540 | ||||||||
| Licensing and other royalty revenue | 7,474 | — | — | 7,474 | ||||||||||||
| Total commercial revenue | 120,014 | — | — | 120,014 | ||||||||||||
| R&D revenue under collaborative agreements | 405,171 | 43,401 | (54,407 | ) | 394,165 | |||||||||||
| Total segment revenue | $ | 525,185 | $ | 43,401 | $ | (54,407 | ) | $ | 514,179 | |||||||
| Total operating expenses | $ | 373,788 | $ | 163,871 | $ | (54,527 | ) | $ | 483,132 | |||||||
| Income (loss) from operations | $ | 151,397 | $ | (120,470 | ) | $ | 120 | $ | 31,047 | |||||||
| 2016 (as revised) | Ionis Core | Akcea Therapeutics | Elimination of Intercompany Activity | Total | ||||||||||||
| Revenue: | ||||||||||||||||
| Commercial revenue: | ||||||||||||||||
| SPINRAZA royalties | $ | 883 | $ | — | $ | — | $ | 883 | ||||||||
| Licensing and other royalty revenue | 21,884 | — | — | 21,884 | ||||||||||||
| Total commercial revenue | 22,767 | — | — | 22,767 | ||||||||||||
| R&D revenue under collaborative agreements | 362,657 | — | (12,648 | ) | 350,009 | |||||||||||
| Total segment revenue | $ | 385,424 | $ | — | $ | (12,648 | ) | $ | 372,776 | |||||||
| Total operating expenses | $ | 322,192 | $ | 83,512 | $ | (12,768 | ) | $ | 392,936 | |||||||
| Income (loss) from operations | $ | 63,232 | $ | (83,512 | ) | $ | 120 | $ | (20,160 | ) | ||||||
The following table shows our total assets by segment at December 31, 2018 and 2017 (in thousands), respectively.
| Total Assets | Ionis Core | Akcea Therapeutics | Elimination of Intercompany Activity | Total | ||||||||||||
| December 31, 2018 | $ | 2,975,491 | $ | 365,261 | $ | (672,968 | ) | $ | 2,667,784 | |||||||
| December 31, 2017 (as revised) | $ | 1,342,578 | $ | 268,804 | $ | (288,608 | ) | $ | 1,322,774 | |||||||
Contracts receivables at December 31, 2018 and December 31, 2017 were comprised of approximately 99 percent and 84 percent for each year from four and two significant partners, respectively.
8. Employment Benefits
We have an employee 401(k) salary deferral plan, covering all employees. Employees could make contributions by withholding a percentage of their salary up to the IRS annual limit $18,500 and $24,500 in 2018 for employees under 50 years old and employees 50 years old or over, respectively. We made approximately $5.7 million, $3.0 million and $1.7 million in matching contributions for the years ended December 31, 2018, 2017 and 2016, respectively.
F-45
9. Legal Proceedings
From time to time, we are involved in legal proceedings arising in the ordinary course of our business. Periodically, we evaluate the status of each legal matter and assess our potential financial exposure. If the potential loss from any legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Significant judgment is required to determine the probability of a loss and whether the amount of the loss is reasonably estimable. The outcome of any proceeding is not determinable in advance. As a result, the assessment of a potential liability and the amount of accruals recorded are based only on the information available to us at the time. As additional information becomes available, we reassess the potential liability related to the legal proceeding, and may revise our estimates.
Gilead Litigation
In August 2013, Gilead Sciences Inc. filed a suit in the U.S. District Court of Northern District of California related to U.S. Patent Nos. 7,105,499 and 8,481,712, which are jointly owned by Merck Sharp & Dohme Corp. and Ionis Pharmaceuticals, Inc. In the suit Gilead asked the court to determine that Gilead’s activities do not infringe any valid claim of the named patents and that the patents are not valid. We and Merck Sharp & Dohme Corp. filed our answer denying Gilead’s noninfringement and invalidity contentions, contending that Gilead’s commercial sale and offer for sale of sofosbuvir prior to the expiration of the ‘499 and ‘712 patents infringes those patents, and requesting monetary damages to compensate for such infringement. In the trial for this case held in March 2016, the jury upheld all ten of the asserted claims of the patents-in-suit. The jury then decided that we and Merck are entitled to four percent of $5 billion in past sales of sofosbuvir. Gilead has stated it would appeal the jury’s finding of validity. In the meantime, Gilead asserted two additional non-jury defenses: waiver and unclean hands. Although the judge rejected the waiver defense, she granted Gilead’s motion claiming that the patents are unenforceable against it under the doctrine of unclean hands. We believe this ruling is contrary to the relevant law and the facts of the case. Accordingly, in July 2016, together with Merck we appealed the decision to the Court of Appeals for the Federal Circuit. Gilead cross-appealed on the issue of validity. In April 2018, the Court of Appeals issued its ruling affirming the District Court’s finding of unenforceability based on unclean hands. Having upheld the ruling that the patents are unenforceable against Gilead, the court did not reach the question of validity. In September 2018, we filed a petition requesting a hearing before the Supreme Court, asserting that it was improper for the trial court to overturn the jury verdict on the basis of the equitable defense of unclean hands. In January 2019, the Supreme Court denied our petition. Under our agreement with Merck, Merck is responsible for the costs of this suit.
10. Quarterly Financial Data (Unaudited)
The following financial information reflects all normal recurring adjustments, which are, in the opinion of management, necessary for a fair statement of the results of the interim periods. Summarized quarterly data for the years ended December 31, 2018 and 2017 are as follows (in thousands, except per share data).
| 2018 Quarters | First Quarter | Second Quarter | Third Quarter | Fourth Quarter | ||||||||||||
| Revenue | $ | 144,419 | $ | 117,747 | $ | 145,395 | $ | 192,113 | ||||||||
| Operating expenses | $ | 147,720 | $ | 168,028 | $ | 163,967 | $ | 181,331 | ||||||||
| Income (loss) from operations | $ | (3,301 | ) | $ | (50,281 | ) | $ | (18,572 | ) | $ | 10,782 | |||||
| Net income (loss) | $ | (10,812 | ) | $ | (56,573 | ) | $ | (20,365 | ) | $ | 302,735 | |||||
| Net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders | $ | (1,420 | ) | $ | (40,358 | ) | $ | (4,559 | ) | $ | 320,078 | |||||
| Basic net income (loss) per share (1) (2) | $ | (0.01 | ) | $ | (0.29 | ) | $ | (0.03 | ) | $ | 2.32 | |||||
| Diluted net income (loss) per share (1) (3) | $ | (0.01 | ) | $ | (0.29 | ) | $ | (0.03 | ) | $ | 2.21 | |||||
| 2017 Quarters (as revised) | First Quarter | Second Quarter | Third Quarter | Fourth Quarter | ||||||||||||
| Revenue | $ | 115,800 | $ | 112,273 | $ | 118,314 | $ | 167,792 | ||||||||
| Operating expenses | $ | 96,315 | $ | 105,823 | $ | 107,002 | $ | 173,992 | ||||||||
| Income (loss) from operations | $ | 19,485 | $ | 6,450 | $ | 11,312 | $ | (6,200 | ) | |||||||
| Net income (loss) | $ | 8,964 | $ | (3,085 | ) | $ | (7,493 | ) | $ | (9,169 | ) | |||||
| Net income (loss) attributable to Ionis Pharmaceutical, Inc. common stockholders | 8,964 | (3,085 | ) | (2,611 | ) | (2,922 | ) | |||||||||
| Basic net income (loss) per share (1) (2) | $ | 0.07 | $ | (0.02 | ) | $ | (0.02 | ) | $ | (0.03 | ) | |||||
| Diluted net income (loss) per share (1) (3) | $ | 0.07 | $ | (0.02 | ) | $ | (0.02 | ) | $ | (0.03 | ) | |||||
| (1) | We computed net income (loss) per share independently for each of the quarters presented. Therefore, the sum of the quarterly net income (loss) per share will not necessarily equal the total for the year. |
| (2) | As discussed in Note 1, Organization and Significant Accounting Policies, we compute basic net income (loss) per share by dividing the total net income (loss) attributable to our common stockholders by our weighted-average number of common shares outstanding during the period. Our basic net income (loss) per share calculation for each of the quarters in 2018 and for the third and fourth quarters of 2017 considered our net income for Ionis on a stand-alone basis plus our share of Akcea’s net loss for the period. To calculate the portion of Akcea’s net loss attributable to our ownership, we multiplied Akcea’s loss per share by the weighted average shares we owned in Akcea during the period. As a result of this calculation, our total net income (loss) available to Ionis common stockholders for the calculation of net income (loss) per share is different than net income (loss) attributable to Ionis Pharmaceuticals, Inc. common stockholders in the consolidated statements of operations. |
F-46
Our basic net income (loss) per share for each quarter in 2018 was calculated as follows (in thousands, except per share amounts):
| Three Months Ended March 31, 2018 | Weighted Average Shares Owned in Akcea | Akcea’s Net Income (Loss) Per Share | Ionis’ Portion of Akcea’s Net Loss | |||||||||
| Common shares | 45,448 | $ | (0.44 | ) | $ | (19,997 | ) | |||||
| Akcea’s net loss attributable to our ownership | $ | (19,997 | ) | |||||||||
| Ionis’ stand-alone net income | 18,785 | |||||||||||
| Net loss available to Ionis common stockholders | $ | (1,212 | ) | |||||||||
| Weighted average shares outstanding | 125,330 | |||||||||||
| Basic net loss per share | $ | (0.01 | ) | |||||||||
| Three Months Ended June 30, 2018 | Weighted Average Shares Owned in Akcea | Akcea’s Net Income (Loss) Per Share | Ionis’ Portion of Akcea’s Net Loss | |||||||||
| Common shares | 60,832 | $ | (0.72 | ) | $ | (43,814 | ) | |||||
| Akcea’s net loss attributable to our ownership | $ | (43,814 | ) | |||||||||
| Ionis’ stand-alone net income | 5,882 | |||||||||||
| Net loss available to Ionis common stockholders | $ | (37,932 | ) | |||||||||
| Weighted average shares outstanding | 128,712 | |||||||||||
| Basic net loss per share | $ | (0.29 | ) | |||||||||
| Three Months Ended September 30, 2018 | Weighted Average Shares Owned in Akcea | Akcea’s Net Income (Loss) Per Share | Ionis’ Portion of Akcea’s Net Loss | |||||||||
| Common shares | 65,538 | $ | (0.73 | ) | $ | (47,789 | ) | |||||
| Akcea’s net loss attributable to our ownership | $ | (47,789 | ) | |||||||||
| Ionis’ stand-alone net income | 43,226 | |||||||||||
| Net loss available to Ionis common stockholders | $ | (4,563 | ) | |||||||||
| Weighted average shares outstanding | 137,346 | |||||||||||
| Basic net loss per share | $ | (0.03 | ) | |||||||||
| Three Months Ended December 31, 2018 | Weighted Average Shares Owned in Akcea | Akcea’s Net Income (Loss) Per Share | Ionis’ Portion of Akcea’s Net Loss | |||||||||
| Common shares | 67,130 | $ | (0.79 | ) | $ | (53,219 | ) | |||||
| Akcea’s net loss attributable to our ownership | $ | (53,219 | ) | |||||||||
| Ionis’ stand-alone net income | 372,913 | |||||||||||
| Net income available to Ionis common stockholders | $ | 319,694 | ||||||||||
| Weighted average shares outstanding | 137,699 | |||||||||||
| Basic net income per share | $ | 2.32 | ||||||||||
Prior to Akcea’s IPO in July 2017, we owned Akcea series A convertible preferred stock, which included a six percent cumulative dividend. Upon completion of Akcea’s IPO in July 2017, our preferred stock was converted into common stock on a 1:1 basis. The preferred stock dividend was not paid at the IPO because the IPO was not a liquidation event or a change in control. During the three months ended September 30, 2017, Akcea used a two-class method to compute its net income (loss) per share because it had both common and preferred shares outstanding during the periods. The two-class method required Akcea to calculate its net income (loss) per share for each class of stock by dividing total distributable losses applicable to preferred and common stock, including the six percent cumulative dividend contractually due to series A convertible preferred shareholders, by the weighted-average of preferred and common shares outstanding during the requisite period. Since Akcea used the two-class method, accounting rules required us to include our portion of Akcea’s net income (loss) per share for both Akcea’s common and preferred shares that we owned in our calculation of basic and diluted net income (loss) per share for the three months ended September 30, 2017.
F-47
Our basic net income (loss) per share for the three months ended September 30, 2017 was calculated as follows (in thousands, except per share amounts):
| Three Months Ended September 30, 2017 | Weighted Average Shares Owned in Akcea | Akcea’s Net Loss Per Share | Ionis’ Portion of Akcea’s Net Loss | |||||||||
| Common shares | 36,556 | $ | (0.33 | ) | $ | (12,063 | ) | |||||
| Preferred shares | 5,651 | (0.01 | ) | (57 | ) | |||||||
| Akcea’s net loss attributable to our ownership | $ | (12,120 | ) | |||||||||
| Ionis’ stand-alone net income | 10,144 | |||||||||||
| Net loss available to Ionis common stockholders | $ | (1,976 | ) | |||||||||
| Weighted average shares outstanding | 124,370 | |||||||||||
| Basic net loss per share | $ | (0.02 | ) | |||||||||
Our basic net income (loss) per share for the three months ended December 30, 2017 was calculated as follows (in thousands, except per share amounts):
| Three Months Ended December 31, 2017 | Weighted Average Shares Owned in Akcea | Akcea’s Net Loss Per Share | Ionis’ Portion of Akcea’s Net Loss | |||||||||
| Common shares | 45,448 | $ | (0.30 | ) | $ | (13,634 | ) | |||||
| Akcea’s net loss attributable to our ownership | $ | (13,634 | ) | |||||||||
| Ionis’ stand-alone net income | 10,510 | |||||||||||
| Net loss available to Ionis common stockholders | $ | (3,124 | ) | |||||||||
| Weighted average shares outstanding | 124,818 | |||||||||||
| Basic net loss per share | $ | (0.03 | ) | |||||||||
| (3) | For the three months ended December 31, 2018, we had net income available to Ionis common stockholders. As a result, we computed diluted net income per share using the weighted-average number of common shares and dilutive common equivalent shares outstanding during those periods. Diluted common equivalent shares for the three months ended December 31, 2018 consisted of the following (in thousands except per share amounts): |
| Three Months Ended December 31, 2018 | Income (Numerator) | Shares (Denominator) | Per-Share Amount | |||||||||
| Net income available to Ionis common stockholders | $ | 319,694 | 137,699 | 2.32 | ||||||||
| Effect of dilutive securities: | ||||||||||||
| Shares issuable upon exercise of stock options | — | 1,254 | ||||||||||
| Shares issuable upon restricted stock award issuance | — | 636 | ||||||||||
| Shares issuable related to our ESPP | — | 7 | ||||||||||
| Shares issuable related to our 1 percent convertible notes | 10,745 | 10,260 | ||||||||||
| Income available to Ionis common stockholders, plus assumed conversions | $ | 330,439 | 149,856 | 2.21 | ||||||||
For the three months ended March 31, 2017, we owned 100 percent of Akcea. As a result, we did not have to adjust our earnings per share calculation. For the three months ended March 31, 2017, we had net income. As a result, we computed diluted net income per share using the weighted-average number of common shares and dilutive common equivalent shares outstanding during those periods. Diluted common equivalent shares for the three months ended March 31, 2017 consisted of the following (in thousands except per share amounts):
| Three Months Ended March 31, 2017 | Income (Numerator) | Shares (Denominator) | Per-Share Amount | |||||||||
| Net income available to Ionis common stockholders | $ | 8,964 | 122,861 | $ | 0.07 | |||||||
| Effect of dilutive securities: | ||||||||||||
| Shares issuable upon exercise of stock options | — | 1,674 | ||||||||||
| Shares issuable upon restricted stock award issuance | — | 377 | ||||||||||
| Shares issuable related to our ESPP | — | 60 | ||||||||||
| Income available to Ionis common stockholders | $ | 8,964 | 124,972 | $ | 0.07 | |||||||
For the three months ended March 31, 2017, the calculation excluded the 1 percent and 2¾ percent notes because the effect on diluted earnings per share was anti-dilutive.
F-48