UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For The Fiscal Year Ended December 31, 2011
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File No. 0-19700
AMYLIN PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in its Charter)
| | |
Delaware
| | 33-0266089 |
(State or other jurisdiction of
incorporation or organization) | | (I.R.S. Employer
Identification No.) |
| |
9360 Towne Centre Drive San Diego, California | | 92121 |
| (Address of principal executive offices) | | (Zip Code) |
Registrant’s telephone number, including area code: (858) 552-2200
Securities registered pursuant to Section 12(b) of the Act:
| | |
Title of Each Class | | Name of each Exchange on Which Registered |
| Common Stock, $.001 par value | | The NASDAQ Stock Market, LLC |
Securities registered pursuant to Section 12(g) of the Act:
NONE
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes x No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (Section 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | | |
| Large accelerated filer | | x | | Accelerated filer | | ¨ |
| | | |
Non-accelerated filer
| | ¨ (Do not check if a smaller reporting company) | | Smaller reporting company | | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2). Yes ¨ No x
The aggregate market value of the common stock of the registrant as of June 30, 2011 held by non-affiliates was $1,225,574,756.
The number of shares outstanding of the registrant’s common stock was 147,304,316 as of February 16, 2012.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Definitive Proxy Statement to be filed with the Securities and Exchange Commission (the “Commission”) pursuant to Regulation 14A in connection with the 2012 Annual Meeting of Stockholders are incorporated herein by reference into Part III of this Annual Report. Such Definitive Proxy Statement will be filed with the Commission not later than 120 days after December 31, 2011.
You should read the following together with the more detailed information regarding our company, our common stock and our financial statements and notes to those statements appearing elsewhere in this document or incorporated by reference. The Securities and Exchange Commission, or SEC, allows us to “incorporate by reference” information that we file with the SEC, which means that we can disclose important information to you by referring you to those documents. The information incorporated by reference is considered to be part of this annual report on Form 10-K.
Except for the historical information contained herein, this annual report on Form 10-K and the information incorporated by reference contains forward-looking statements that involve risks and uncertainties. These statements include projections about our accounting and finances, plans and objectives for the future, future operating and economic performance and other statements regarding future performance. These statements are not guarantees of future performance or events. Our actual results may differ materially from those discussed here. Factors that could cause or contribute to such differences are described in Part I, Item 1A, entitled “Risk Factors,” as well as those discussed in Part II, Item 7, entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and elsewhere throughout this annual report on Form 10-K and in any other documents incorporated by reference into this annual report on Form 10-K. We disclaim any obligation to update any forward-looking statement.
PART I
Business Overview
We are a biopharmaceutical company committed to improving the lives of people with diabetes and other metabolic diseases through the discovery, development and commercialization of innovative medicines. We are marketing two first-in-class medicines to treat diabetes, BYETTA® (exenatide) injection and SYMLIN® (pramlintide acetate) injection. We are also marketing the first and only once-weekly diabetes treatment, BYDUREON™ (exenatide extended-release for injectable suspension). BYDUREON is an extended-release medication for type 2 diabetes that provides continuous glycemic control in a once-weekly dose.
In 2011 and early 2012, we executed on key initiatives for the company, including:
| | • | | Obtaining approval for BYDUREON in the United States in January 2012 and the European Union in June 2011. BYDUREON is currently approved by regulatory authorities in 33 countries and is being marketed in 12 countries worldwide in which we have also received pricing and reimbursement decisions; |
| | • | | Regaining 100% of the global rights to develop and commercialize our exenatide franchise, including BYDUREON and BYETTA, from Eli Lilly and Company and entering into discussions with potential new partners with global capabilities to develop and commercialize exenatide outside the United States; |
| | • | | Obtaining approval in the United States of BYETTA for use with insulin glargine. BYETTA is currently the only short-acting glucagon-like peptide-1, or GLP-1, receptor agonist approved in the United States for use as an add-on therapy to insulin glargine in patients with type 2 diabetes; and |
| | • | | Continuing to operate our business on an operating cash flow positive basis since the beginning of 2010. |
In 2012, we will continue our BYDUREON launch efforts and will work toward securing a development and commercialization partner with global capabilities for our exenatide franchise. We also plan to advance our metreleptin program for the treatment of lipodystophy, a very rare metabolic condition, and to advance our once-weekly and once-monthly exenatide suspension programs. We will continue our efforts to develop and obtain approval for the BYDUREON pen with the goal of making the BYDUREON pen delivery system available to patients in late 2012 or early 2013. In the near term, we will also focus on maximizing the financial contributions from BYETTA and SYMLIN and continue to operate our business with financial discipline.
We maintain a research and early development program focused on novel peptide and protein therapeutics. We have also entered into strategic alliances and business initiatives, including our strategic relationship with Biocon, Limited, or Biocon, to develop pharmaceutical products, including AC165198, a peptide hybrid drug candidate for diabetes, which was developed from our phybrid technology platform. In collaboration with Biocon, we submitted an investigational new drug application, or IND, at the end of 2011 and commenced a phase 1 study for AC165198 in early 2012.
1
Our principal executive offices are located at 9360 Towne Centre Drive, San Diego, CA 92121, and our telephone number is (858) 552-2200. We were incorporated in Delaware in September 1987. We maintain a corporate website atwww.amylin.com. The reference to our worldwide web address does not constitute incorporation by reference into this report of any of the information contained on our website.
Our periodic and current reports that we file with the SEC are available free of charge on our website atwww.amylin.com as soon as reasonably practicable after we have electronically filed them with, or furnished them to, the SEC.
Diabetes
Diabetes is a major worldwide health problem and is the sixth leading cause of death by disease in the United States. Diabetes is a complex, metabolic disorder of carbohydrate, fat and protein metabolism, primarily resulting from the failure of pancreatic beta cells to produce sufficient insulin to match the demands for insulin in the body. Insulin is a hormone that plays a central role in helping the body process, convert and store energy from glucose. Another important hormone in glucose regulation is glucagon which is released from the alpha-cells of the pancreas. Its action opposes insulin by increasing glucose appearance in the bloodstream. With the discovery of incretin hormones, GLP-1, gastric inhibitory peptide and the pancreatic hormone amylin, it is now understood that several organs and hormones play a role in maintaining glucose balance in the body. In individuals with diabetes, the relative shortage of insulin impairs the ability of glucose to enter and fuel the body’s cells and, as a result, glucose builds up in the bloodstream causing hyperglycemia (high blood sugar). Prolonged elevation of blood glucose may result in damage to the kidney, retina and nerves—and may lead to kidney failure, permanent nerve damage, blindness and amputation. High glucose also increases the risk of cardiovascular disease. Conversely, too much insulin in the bloodstream can cause hypoglycemia (low blood sugar). Individuals who manage their diabetes with insulin or other oral antidiabetic medication are especially vulnerable to swings of high to low blood sugar level and the risk of very low blood sugar which, if left untreated, can be fatal.
It is estimated that over 345 million people worldwide have diabetes. Of that population, it is estimated that approximately 90-95% have type 2 diabetes, previously known as adult-onset diabetes, and the remainder have type 1 diabetes, previously known as juvenile-onset diabetes. In the United States alone, there are approximately 26 million people, or 8.3% of the population, with diabetes. Nearly 19 million of these people have been diagnosed, while more than 7 million people with diabetes have not been diagnosed. From 1980 through 2010, newly diagnosed cases of diabetes among Americans aged 18-79 more than tripled. In addition, there are currently approximately 79 million adults aged 20 years or older in the United States with pre-diabetes, a condition that raises the risk of developing type 2 diabetes, heart disease and stroke. People with pre-diabetes have blood glucose levels higher than normal but not high enough to establish a diagnosis of diabetes.
Long-term control of blood glucose is known to limit the risk of developing diabetes-related retinal, renal and neurologic complications. A1C is the most widely used measure of long-term blood glucose control. A1C value is a recognized indicator of an individual’s average blood glucose concentrations over the preceding three- to four-month period. Lower A1C values indicate better average blood glucose control, with values in people without diabetes usually being less than 6%. The ADA suggests that people with diabetes should aim for an A1C value that is lower than 7%. It is estimated that more than half of Americans being treated for diabetes are failing to achieve recommended A1C targets and, according to research studies conducted in the United States and abroad, these patients would significantly benefit from improved glycemic control. Additionally, aggressive use of insulin and some oral medications to reduce glucose levels can be associated with an increased risk of hypoglycemia and weight gain. Consequently, there has long been a need to develop new treatment strategies that safely improve glucose control, improve the overall health profile of patients with diabetes and reduce the risk of complications.
In 2008, findings from various long-term clinical trials, including the 10-year follow up of the UK Prospective Diabetes Study and the “Action to Control Cardiovascular Risk in Diabetes,” or ACCORD, trial suggested that it is important to treat patients with less advanced diabetes earlier. These studies also suggest that it is important to lower blood glucose without weight gain and hypoglycemia which are often associated with older diabetes therapies. The cardiovascular outcomes data of these studies suggest that blood glucose control strategies employing therapies that do not promote weight gain or hypoglycemia may become increasingly valued.
For people suffering from diabetes, poor control of blood glucose levels has been shown to result in severe long-term complications. For instance, the United States Centers for Disease Control, or CDC, has stated that complications associated with diabetes include:
| | • | | heart disease and stroke; |
2
| | • | | blindness due to retinopathy, a condition manifested by damage to the retina; |
| | • | | nephropathy, or kidney disease; |
| | • | | neuropathy, a condition where there is damage to the nervous system; |
| | • | | amputations due to peripheral vascular disease; and |
Obesity is common in patients with type 2 diabetes and weight control is a major problem for many patients with both type 1 and type 2 diabetes. In fact, 85% of people with type 2 diabetes are overweight and 55% are considered obese. Weight gain is particularly common in those using insulin and certain oral medications as part of their treatment regimen. In addition, patients with diabetes frequently have wide fluctuations in blood sugar following meals. These fluctuations in blood sugar can significantly affect a patient’s quality of life. Blood glucose fluctuations, weight gain and diabetes complications may each contribute to substantial disability, reduced quality of life, reduced productivity in the workplace, increased pain and suffering and premature death. It is estimated that obesity increases the risk of cardiovascular disease in people with type 2 diabetes and cardiovascular death accounts for at least 65% of all deaths among people with diabetes. In fact, the risk of cardiovascular disease, including coronary artery disease and other atherosclerotic disorders, and death are significantly increased in the overweight population and to an even greater extent in obese patients with type 2 diabetes.
We have introduced three new treatment options for the management of diabetes, BYDUREON, BYETTA and SYMLIN. BYDUREON and BYETTA offer type 2 diabetes patients with inadequate glycemic control the opportunity to better control their blood glucose levels, with the potential for weight loss rather than weight gain. SYMLIN offers type 1 or type 2 diabetes patients with inadequate glycemic control using mealtime insulin a treatment option that can both improve glucose control and result in weight loss. These novel medicines provide new options in disease management and glucose control to millions of people suffering from diabetes.
Marketed Products
BYDUREON™ (exenatide extended-release for injectable suspension)
BYDUREON is the first and only once-weekly diabetes treatment approved for use in the United States, the European Union and other parts of the world. It is in a class of compounds called GLP-1 receptor agonists and is indicated for use in the United States as an adjunct to diet and exercise to improve glycemic control in adult patients with type 2 diabetes. BYDUREON combines exenatide, the active ingredient in BYETTA, with proprietary technology developed by us and our partner, Alkermes, Inc., or Alkermes, to provide a sustained release delivery of exenatide. The combination of potency and the glucose-dependent mechanism of action inherent in exenatide makes it well suited for use as a once weekly formulation. Common side effects with BYDUREON include nausea, which most commonly happens when first starting BYDUREON but may become less prevalent over time, headache, itching at the injection site and indigestion. BYDUREON is currently approved in 33 countries and has been launched in 12 countries worldwide, including the United States.
In October 2007, we announced positive results from our DURATION-1 pivotal comparator study comparing treatment with BYDUREON to treatment with BYETTA over a 30-week period. BYDUREON showed a statistically significant improvement in A1C of approximately 1.9% from baseline, compared to an improvement of approximately 1.5% for BYETTA. In June 2008, we announced results from the extension phase of DURATION-1 showing durable efficacy of BYDUREON over 52-weeks. In this extension phase, patients either remained on BYDUREON or switched from BYETTA to BYDUREON for an additional 22 weeks. Patients taking BYDUREON over the course of one year sustained a similar improvement in glucose control of 2.0% lower A1C and lower fasting plasma glucose from baseline.
In March 2009 and July 2009, we announced positive results from our DURATION-2 and DURATION-3 studies, respectively, the second and third in a series of six such studies designed to test the superiority of BYDUREON compared to other diabetes therapies. In DURATION-2, BYDUREON demonstrated superior glucose control with weight loss and no increase in hypoglycemia compared to maximum doses of Januvia® or Actos®. In DURATION-3, BYDUREON demonstrated superior glucose control with weight loss and with less hypoglycemia compared to insulin glargine (Lantus®). In December 2009, we announced results from DURATION-5, a head-to-head study comparing BYDUREON to BYETTA. In DURATION-5, patients taking BYDUREON experienced a statistically superior reduction in A1C compared to BYETTA.
3
In June 2010, we announced results from our DURATION-4 study, a head-to-head study that compared BYDUREON monotherapy to Januvia®, Actos® or metformin in a monotherapy setting in which patients were not achieving adequate glucose control with diet and exercise alone. Results from DURATION-4 demonstrated that BYDUREON efficacy and tolerability profile extended to monotherapy treatment. In March 2011, we announced top-line results from our DURATION-6 study that showed that once weekly BYDUREON significantly improved glucose control from baseline. Although BYDUREON did not meet the pre-specified primary endpoint of non-inferiority to once-daily Victoza® (liraglutide (rDNA origin) injection) in this study, DURATION-6 reinforced the important role of GLP-1 receptor agonists in the treatment of type 2 diabetes and demonstrated improved tolerability versus Victoza.
At the end of 2010, we published data from a retrospective, real-world analysis of over 400,000 type 2 diabetes patient outcomes which showed that BYETTA was associated with a 14-20% reduction in the risk of cardiovascular events. Given the positive effects on cardiovascular outcomes observed with exenatide in this analysis, the beneficial effects observed in clinical trials of exenatide on markers of cardiovascular risk, and the current regulatory interest in cardiovascular outcomes, in 2010 we initiated our prospective EXSCEL cardiovascular outcomes trial with a superiority design that will evaluate the effects of BYDUREON on major cardiovascular events, compared to standard of care with other antidiabetes medications. This study will give us the opportunity to demonstrate the effect of BYDUREON on cardiovascular outcomes and other end points of interest to our stakeholders. We expect results from this study will not be available for several years.
BYETTA® (exenatide) injection
BYETTA is the first approved medicine in the GLP-1 receptor agonist class of compounds. It is approved as a first-line, stand-alone medication (monotherapy) along with diet and exercise to improve glycemic control in adults with type 2 diabetes. BYETTA is also approved as an adjunctive therapy to improve glycemic control in patients with type 2 diabetes who have not achieved adequate glycemic control by using metformin, a sulfonylurea and/or a thiazolidinedione (TZD), three common oral therapies for type 2 diabetes. Further, the FDA recently approved an expanded use of BYETTA as an add-on for patients who have not achieved adequate glycemic control with insulin glargine. The type 2 diabetes treatment guidelines of the American Diabetes Association, or ADA, the European Association for the Study of Diabetes, or EASD, and the American Association of Clinical Endocrinologists, or AACE, include the GLP-1 receptor agonist class, which includes BYETTA, as a secondary treatment option for type 2 diabetes patients. Net product sales of BYETTA were $517.7 million in 2011, $559.3 million in 2010 and $667.6 million in 2009.
We estimate the number of people in the United States currently using metformin, sulfonylurea and/or a TZD to be approximately 10 million and the number of people managing their diabetes with diet and exercise alone to be approximately 3 million. More than half of all diabetes patients using oral medications are believed to have an A1C higher than the ADA’s recommendation of less than 7% and the vast majority of these patients could be candidates for BYETTA or BYDUREON.
BYETTA provides glucose control by augmenting the body’s natural physiologic processes, allowing the body to respond to blood glucose changes as they occur. BYETTA directly affects the beta cells’ responses to elevated glucose by enhancing insulin secretion; this effect dissipates as glucose levels approach the normal range. BYETTA also restores first-phase insulin response, an effect which is evident following the initial dose. BYETTA is administered twice a day by using a fixed dose injection, and requires no dose adjustments due to changes in meal size or composition, exercise or other variables. No additional glucose monitoring is required with BYETTA therapy.
The most common adverse effect of BYETTA is mild to moderate nausea, which tends to dissipate with time. Mild to moderate hypoglycemia has also been observed, but this was primarily when used in conjunction with a sulfonylurea, agents that are known to cause hypoglycemia.
In August 2008, the FDA updated a prior alert for BYETTA referencing pancreatitis. Prescriptions for BYETTA have declined since the first half of 2008. In October 2009, the FDA approved changes to the BYETTA label to incorporate updated safety information, including pancreatitis-related safety language and an expansion of existing language regarding use of BYETTA in patients with renal impairment. We continue to work to better understand the relationship between BYETTA use and pancreatitis described in some spontaneously reported cases. In keeping with our focus on patient safety, we continue to pursue our drug safety program that includes thorough investigation of individual spontaneous case reports along with clinical and epidemiologic studies. Within the detection limits of an initial epidemiology study which we provided to the FDA, we have not observed an increased incidence of pancreatitis associated with BYETTA use compared to other treatments for diabetes and thus believe a definite causal relationship between BYETTA and pancreatitis has not been proven.
4
At the end of 2011, we had a field sales force of approximately 360 individuals who target those doctors that write the majority of BYETTA and SYMLIN prescriptions. In early 2012, we entered into an agreement with a contract sales organization and to add approximately 390 sales representatives and 36 sales managers to our sales force. Our goal is to provide education, through both one-on-one interactions and educational programs, to ensure that physicians understand BYETTA, including its mechanisms of action, potential benefits, identify appropriate patients and provide important use considerations. We have refined our marketing efforts to remind both endocrinologists and primary care physicians of BYETTA’s unique benefits of glucose control with weight loss. Primary care physicians write approximately 70% of diabetes prescriptions in the United States. Additionally, more than 80% of insured lives have access to BYETTA at the lowest branded co-pays.
We continue to support initiatives to facilitate the successful initiation of therapy by primary care physicians. This effort includes: increased patient educational material for health care providers to distribute in their offices; a network of approximately 200 diabetes educators to work with physicians and their patients within their local communities; direct support to patients through the BYETTA By Your Side Program, which provides a toll-free number that allows patients to contact trained medical professionals to better understand the benefits of BYETTA therapy and to get assistance starting and using the BYETTA pen; a pharmacy support component partnering with managed care plans designed specifically to assist with patient refills; and a BYETTA website. We believe this support is helpful to patients who may be on their first injectable therapy and to primary care providers who may be less accustomed to treating patients with an injectable product earlier in the disease cycle and who have fewer resources in their offices.
We have an agreement with Eli Lilly and Company, or Lilly, that provides for the transition of development and commercialization activities for exenatide outside the United States no later than December 31, 2013 or at such later date if (i) mutually agreed by us and Lilly in the event such transition has not occurred by December 31, 2013 or (ii) in certain circumstances as set forth in our transition agreement with Lilly. We are currently in discussions with various companies with the goal of transitioning these exenatide responsibilities to one or more partners prior to such date. By the end of 2011, BYETTA was approved in 87 countries and launched in approximately 80 countries worldwide.
SYMLIN® (pramlintide acetate) injection
SYMLIN is the first and only approved medicine in a class of compounds called amylinomimetics. We began selling SYMLIN in the United States in April 2005 as adjunctive therapy to mealtime insulin to treat diabetes. Other than insulin and insulin analogues, SYMLIN is the first FDA-approved medication addressing glucose control for patients with type 1 diabetes since the discovery of insulin approximately 90 years ago. We own 100% of the global rights to SYMLIN which had net product sales of $103.9 million in 2011, $91.8 million in 2010 and $86.4 million in 2009.
SYMLIN is indicated for use in people treated with mealtime insulin alone or, in the case of patients with type 2 diabetes, treated with mealtime insulin with or without one or more oral medications to help improve blood glucose control. SYMLIN works with insulin to smooth out the peaks in blood glucose levels after meals and to give patients more stable blood glucose levels after meals and throughout the day. SYMLIN also lowers the A1C values of most patients beyond what insulin alone can achieve. SYMLIN reduces food intake and leads to weight loss in many patients. In addition, because SYMLIN works with insulin to control blood sugar, patients often need less insulin to achieve desired blood sugar levels after meals.
SYMLIN is used with insulin and has been associated with an increased risk of insulin-induced severe hypoglycemia. The risk can be reduced by appropriate patient selection, careful patient instruction and insulin dose adjustments. Other adverse effects commonly observed are primarily gastrointestinal, including nausea, which decrease over time in most patients.
Our SYMLIN marketing is focused on a physician population of approximately 30,000 who prescribe rapid acting insulin, with a goal of educating these physicians on SYMLIN, including its mechanisms of action, potential benefits, use considerations and appropriate patient selection for initiating SYMLIN therapy. These physicians write approximately 61% of all mealtime insulin prescriptions in the United States. In 2008, we launched the SymlinPen® 120 and the SymlinPen® 60 pen-injector devices for administering SYMLIN. These pre-filled, pen-injector devices feature fixed dosing to improve mealtime glucose control and can be stored at room temperature not to exceed 86 degrees F (30 degrees C) after first use. We have ceased manufacturing and distributing the Symlin vial. Our near-term goal for SYMLIN is to grow SYMLIN prescriptions by highlighting how the product addresses the key unmet needs of patients using mealtime insulin and increase awareness of SYMLIN’s potential benefits to these patients.
5
Research and Development
Product Pipeline Programs
We have late-stage and early-stage development programs in the metabolic diseases therapeutic area, with a primary focus on diabetes and diabetes-related disorders. We have developed capabilities in the discovery, biology, chemistry, and drug delivery of peptide hormones, and expertise in the clinical development and manufacturing of peptide-based therapeutics. We are leveraging this expertise to develop potential treatments for diabetes, and diabetes-related disorders and lipodystrophy.
Diabetes
We are developing a dual chamber cartridge pen configuration of BYDUREON. We believe this design will further simplify dosing for patients, allowing them to mix and administer BYDUREON from a pre-filled pen device. We currently plan to seek FDA approval for the BYDUREON pen during 2012 with a projected launch by the end of 2012 or the first quarter of 2013.
As part of the exenatide lifecycle, we are evaluating the efficacy, pharmacokinetics, tolerability and safety of a suspension formulation that uses the same exenatide-containing microsphere technology used in BYDUREON, but would eliminate the need to reconstitute the product prior to use which we believe has the potential to improve convenience for patients. In 2011, we announced results from a clinical proof-of-concept study that assessed exenatide suspension given once-monthly. The study showed once-monthly exenatide suspension improved glucose control in patients with type 2 diabetes, including reductions in A1C and fasting plasma glucose with modest weight loss and generally demonstrated comparable efficacy, safety and tolerability with BYDUREON. In late 2011, we completed an End-of-Phase 2 meeting with the FDA, and plan to commence the phase 3 programs for a weekly suspension program in 2012 and a monthly suspension program in 2013.
Rare Forms of Lipodystrophy
Lipodystrophy is a group of very rare disorders that is characterized by generalized or partial loss of adipose tissue (that can be inherited or acquired) and leptin deficiency. Lipodystrophy is often associated with metabolic abnormalities (e.g. hypertriglyceridemia, insulin resistance, and/or diabetes) that can result in life-threatening co-morbidities such as acute pancreatitis, steatohepatitis, and/or accelerated atherosclerosis. These metabolic abnormalities are often difficult to control even with high doses of currently available diabetes and lipid-lowering therapies. Not only are these treatments rendered less effective by the profound, refractory nature of the metabolic abnormalities, they also do little to correct the pathophysiological mechanisms underlying the development of such abnormalities. Thus, there is a significant unmet medical need for a therapy that effectively improves the metabolic disorders in these patients.
Because of the loss of adipose tissue in lipodystrophy, levels of the adipocyte-secreted hormone leptin are typically very low in patients suffering from this disease. Clinical studies have shown that metreleptin is a unique potential therapy for patients with lipodystrophy because it addresses key pathophysiological defects underlying the condition. In clinical studies, metreleptin treatment in patients with inherited or acquired lipodystrophy resulted in substantial and clinically meaningful improvements in fasting plasma glucose and A1C due to improved insulin sensitivity as well as marked improvements in hypertriglyceridemia.
In December 2010, we submitted to the FDA the clinical and non-clinical sections of a rolling Biologics License Application, or BLA, for the use of metreleptin to treat diabetes and/or hypertriglyceridemia in pediatric and adult patients with inherited or acquired lipodystrophy. We plan to submit the chemistry, manufacturing and controls section of the BLA to the FDA in the first half of 2012. Based on published data regarding the prevalence of this disease, we have estimated that lipodystrophy affects approximately 1,000 individuals in the United States and 3,000 individuals in other major markets for orphan drugs. Because lipodystrophy affects a very small number of patients, metreleptin for the treatment for lipodystrophy has received orphan drug designation by the FDA. Upon completion of the BLA submission, we plan to apply for fast-track and priority-review designation, which, if granted, could translate to a PDUFA action date by the end of 2012. We also plan to continue working with European regulatory agencies to gain orphan drug designation for metreleptin for the treatment for lipodystrophy in Europe.
Obesity
Obesity is a chronic condition that affects millions of people and is linked to increased health risk of several medical conditions including type 2 diabetes, high blood pressure, heart disease, stroke, osteoarthritis, sleep disorders and several types of cancers. Obesity is also rapidly becoming a major health problem in all industrialized nations and many developing
6
countries. According to NAASO (The Obesity Society), obesity is the second leading cause of preventable death in the United States. It is estimated that 66% of the adult population in the United States is overweight and nearly 72 million adult Americans are considered obese. This epidemic is not limited to the United States, but represents a major global health concern. According to the World Health Organization, or WHO, there are more than 1 billion overweight adults, of which 300 million are considered obese. Current treatments for obesity include diet, exercise, drug therapy and surgery.
Since 2006, we have been conducting non-clinical and clinical studies to assess the safety and efficacy of multiple neurohormones used in combination to treat obesity. In October 2009, we entered into a worldwide exclusive license, development and commercialization agreement with Takeda to co-develop and commercialize pharmaceutical products for the treatment of obesity and related indications. The agreement includes products to be developed from our pipeline and additional compounds from our and Takeda’s obesity research programs. In August 2011, we and Takeda announced that we discontinued the development of pramlintide/metreleptin for the treatment of obesity and will continue to discuss the potential of other assets as candidates for the treatment of obesity and related indications under the terms of our collaboration agreement.
Research Activities
A key objective of our research strategy is to develop well-differentiated, innovative compounds with the potential to advance the treatment of metabolic diseases. To achieve this goal, we are taking a highly integrated, systematic approach that combines peptide chemistry, in-vitro and in-vivo biology, and drug delivery research, in order to generate drug candidates that have promising profiles with respect to, among other things, efficacy, safety and dosing frequency.
This approach to research and development recently led to the development of AC165198, a peptide hybrid (“phybrid”) drug candidate for diabetes that exhibits dual pharmacological activity and elicits enhanced glucose-lowering and weight loss in nonclinical models. In collaboration with our alliance partner for this program, Biocon, we have filed an IND application for this compound with the FDA in late 2011 and initiated phase 1 clinical testing in January 2012.
We are also developing capabilities in scaffolding and delivery system research and development, focused on product presentations that have the potential to enhance clinical outcomes and patient convenience. Delivery systems are selected on the basis of technical feasibility, regulatory and manufacturing considerations, and market preference. They include injectable sustained-release formulations such as salt complexes, lipids, biodegradable polymer and gel systems, as well as non-invasive drug delivery systems. We are also using our resources to optimize pharmaceutical properties of peptide drugs to develop new peptide hormone analogs that may be more amenable to alternative forms of delivery.
As of December 31, 2011, we had approximately 275 full-time employees dedicated to our research and development activities. In the years ended December 31, 2011, 2010 and 2009, we incurred research and development expense of $161.2 million, $183.1 million and $198.4 million, respectively.
Strategic Relationships
Lilly Exenatide Collaboration
On November 7, 2011, we and Lilly entered into a Settlement and Termination Agreement, or the Termination Agreement, which provides for, among other things, the termination of the parties’ Collaboration Agreement and the full and final settlement and resolution of certain outstanding claims asserted by us against Lilly in the lawsuit we filed in May 2011. Under the terms of the Termination Agreement, we obtained the exclusive right to develop and commercialize exenatide products in the United States. We also obtained exclusive rights to develop and commercialize exenatide globally outside the United States subject to a transition period in which Lilly will continue to have exclusive rights to commercialize exenatide products outside the United States until no later than December 31, 2013 or at such later date if (i) mutually agreed by us and Lilly in the event such transition has not occurred by December 31, 2013 or (ii) in certain circumstances as set forth in our transition agreement with Lilly.
The Termination Agreement provides that either party may deliver a notice to the other party with respect to a particular country or countries, or an OUS Country, specifying an earlier transfer date for that country. Under the terms of the Termination Agreement, we are permitted to deliver a notice to transition an OUS Country at any time after (i) June 30, 2012 with respect to an OUS Country where Lilly has launched BYDUREON prior to March 31, 2012, or (ii) March 31, 2012 with respect to any other OUS Countries, provided that with respect to Europe, we may deliver a transition notice on or after April 1, 2012. Lilly is permitted to deliver a notice to transition an OUS Country any time on or after September 30, 2012. The transition of any OUS Country(ies) set forth in a notice will be effective 180 days after the notice date. After the transition of an OUS Country, we, or our designee, will have exclusive rights to commercialize exenatide products in that country.
7
Under the terms of the Termination Agreement, we paid Lilly an upfront payment of $250 million and have agreed to pay to Lilly a milestone payment of $150 million, payable upon approval by the FDA of a monthly exenatide suspension product. In addition, we will make quarterly payments to Lilly pursuant to a revenue sharing obligation (as described below), or the Revenue Sharing Obligation, based on sales of exenatide products and certain payments received by us from third parties.
We are required to make quarterly payments to Lilly equal to (i) 15% of net sales of exenatide products by us or any sales partners, subject to minimum guarantees in each of 2012 and 2013, and (ii) 20% of any consideration, including upfront or milestone payments, received by us from our sales partners for the grant to such sales partners of certain rights relating to exenatide products up to $1.2 billion, in the aggregate plus any interest that is accrued and compounded as follows. The Revenue Sharing Obligation will continue until the earliest to occur of (x) the date that we have paid to Lilly an amount equal to $1.2 billion, plus any interest that is accrued and compounded, (y) December 31, 2036, and (z) termination of the Revenue Sharing Obligation in accordance with the terms of the Termination Agreement. Interest will accrue on the outstanding balance of the Revenue Sharing Obligation at a rate of 2.295% quarterly. Interest will not be payable in cash, but will be added on the last day of each calendar quarter to the outstanding balance of the Revenue Sharing Obligation. We may delay payments on the Revenue Sharing Obligation for the first two quarters of 2012, however, interest will accrue and compound on any payments so delayed. Simultaneously with the execution of the Termination Agreement, we entered into a promissory note with Lilly in the initial principal amount of $1.2 billion, secured by certain of our assets and those of our subsidiaries and guaranteed by certain of our subsidiaries.
On November 7, 2011, in connection with executing the Termination Agreement, we and Lilly amended and restated our loan agreement pursuant to which Lilly previously made a $165 million unsecured loan to us. The amended and restated loan agreement extends the maturity date of our outstanding obligations made under the original loan agreement to June 30, 2016. We and Lilly also amended and restated our Exenatide Once Weekly Supply Agreement, pursuant to which we will supply commercial quantities of fixed-dose injection of exenatide administered once weekly (including related components) in accordance with a fixed pricing schedule to Lilly for sale in jurisdictions outside the United States until the transition of operations outside the United States to us as contemplated by the Termination Agreement. We and Lilly also amended our Device Development and Manufacturing Agreement pursuant to which Lilly will manufacture and supply to us a mechanical injection pen for daily use for sale in and outside the United States and a once weekly exenatide fixed-dose injectable in finished product form and all components and associated packaging for sale outside the United States in accordance with a fixed pricing schedule. These amended and restated supply agreements will expire no later than December 31, 2013. Amylin and Lilly are parties to a number of other agreements that have been entered into in connection with the Termination Agreement.
Exenatide Partner(s) Outside the United States
We are actively engaged in discussions with various companies to secure one or more partners to assume the development and commercialization responsibilities for our exenatide franchise outside the United States and will work toward transitioning these responsibilities from Lilly to our new partner(s) once an agreement is reached. We are committed to ensuring a transition of these development and commercialization responsibilities to maintain continuity of patient care. Our goal is to secure one or more partners for exenatide activities outside the United States in 2012; however, we cannot be certain that we will be able to secure any such partners on terms acceptable to us within this time frame, or at all.
Takeda Obesity Collaboration
In October 2009, we entered into a worldwide exclusive license, development and commercialization agreement with Takeda to co-develop and commercialize pharmaceutical products for the treatment of obesity and related indications. The agreement includes products to be developed from our pipeline and additional compounds from our and Takeda’s obesity research programs. Takeda will be responsible for commercializing the products in and outside the United States and will be responsible for all commercialization costs associated with the products.
We received a one-time up-front payment of $75 million from Takeda upon entering into the agreement and we are eligible to receive additional payments upon achieving certain development, commercialization and sales-based milestones. The agreement also provides for future tiered, double-digit royalty payments to us based on global product sales. We will be responsible for executing development activities for potential products through phase 2 for regulatory approval in the United States. Takeda will lead development activities beyond phase 2 in the United States and all development activities outside the United States. Generally, we will be responsible for 20% of the development costs associated with obtaining approval for products in the United States and Takeda will be responsible for 80% of such United States development costs. Takeda will also be responsible for 100% of development costs associated with obtaining approval for products outside the United States.
8
In August 2011, we and Takeda announced that we discontinued the development of pramlintide/metreleptin for the treatment of obesity and will continue to discuss the potential of other assets as candidates for the treatment of obesity and related indications under the terms of our collaboration agreement.
Early Stage Strategic Collaborations
We have established strategic relationships with other companies and we continue to assess additional opportunities for strategic relationships or in-licensing opportunities. For example, in September 2009, we entered into an exclusive agreement with Biocon to jointly develop, manufacture and commercialize pharmaceutical products, including AC165198, a novel peptide therapeutic for the potential treatment of diabetes. We will share development costs with Biocon of this program which was developed from our “Phybrid” technology platform. A Phybrid is a peptide hybrid molecule that combines the pharmacological effects of two peptide hormones into a single molecular entity. Under the terms of the agreement, we will provide expertise in peptide hormone development, particularly in the area of phybrid technology, as well as metabolic disease therapeutics. Biocon will use its expertise in recombinant microbial expression to manufacture the compound and also leverage its experience in preclinical and clinical development of diabetes products. We and Biocon submitted an IND for AC165198 at the end of 2011 and commenced a phase 1 study in January 2012.
Sales, Marketing and Distribution
We have built a sales and marketing organization that focuses on healthcare providers, managed healthcare organizations, hospitals, wholesalers and pharmacies, government purchasers and other third-party payers. Our field organization also includes our managed care organization.
In January 2012, we created two separate commercial teams focused on two distinct aspects of our commercial portfolio. One commercial team is focused on the commercialization of our exenatide franchise while the other is focused on the commercialization of our products that target specialty and orphan diseases. The exenatide commercial team will consists of 650 diabetes sales specialists who will target those physicians that account for more than 60% of all branded diabetes prescriptions, including approximately 90% of all GLP-1 prescriptions.
The specialty and orphan disease commercial team consists of 65 diabetes sales specialists who will initially focus on the commercialization of SYMLIN by promoting SYMYLIN to those physicians who write mealtime insulin prescriptions. In the near term, the specialty and orphan commercial team will also be dedicated to establishing the short-acting GLP-1 market for BYETTA for use with insulin glargine and will provide the infrastructure for the launch of metreleptin if we are successful in our efforts to receive approval for metreleptin as a treatment for rare forms of lipodystrophy.
Our field force brings a specialty approach to endocrinologists and diabetes-focused primary care physicians and is focused on targeting those doctors that write the majority of prescriptions for branded diabetes therapies. Our field force calls on endocrinologists and other physicians who have large diabetes care practices and other healthcare professionals who support their practices. Members of our sales and marketing team have extensive industry experience from a wide range of large and small companies and have substantial experience in the field of diabetes, as well as in launching and marketing pharmaceutical products.
We utilize common pharmaceutical company practices to market our products. We call on individual physicians and other healthcare professionals and other organizations and individuals involved in the prescribing, purchasing and/or distributing of human medicines. We also provide professional symposia through our extensive medical education programs. Our medical education events are conducted live, via satellite or telephone and through web-based, interactive programs. We will continue to focus on medical education efforts for BYDUREON, BYETTA and SYMLIN through thousands of programs across the United States organized by our medical affairs and external professional education organizations. We train physicians and other healthcare professionals as speakers, so that they can in turn teach their peers about how best to incorporate BYDUREON, BYETTA or SYMLIN into their patients’ diabetes treatment regimens.
We provide customer service and other related programs for our products, such as disease and product-specific websites, insurance research services, a customer service call center and order, delivery and fulfillment services. We have programs in the United States that provide qualified uninsured and underinsured patients with our products at no charge.
We sell BYDUREON, BYETTA and SYMLIN to wholesale distributors who in turn sell to retail pharmacies and government entities. Decisions made by these wholesalers and their customers regarding the levels of inventory they hold, and thus the amount of BYDUREON, BYETTA and SYMLIN they purchase, may affect the level of our product sales in any particular period.
9
Manufacturing
We have selected manufacturers that we believe comply with current Good Manufacturing Practices, or cGMP, and other applicable regulatory standards. Manufactured product is used commercially following established registration procedures and after applicable regulatory approvals have been granted by various international regulatory entities. We have established a quality control and quality assurance program, including a set of standard operating procedures, analytical methods and specifications, designed to ensure that our products and product candidates are manufactured in accordance with the applicable regulations. We require that our contract manufacturers adhere to cGMPs appropriate to the clinical or pre-clinical phase of manufacturing.
Although some materials for our drug products are currently available from a single-source or a limited number of qualified sources, we attempt to acquire an adequate inventory of such materials, establish alternative sources and/or negotiate long-term supply arrangements. We believe we do not have any significant issues obtaining suppliers; however, we cannot be certain that we will continue to be able to obtain long-term supplies of our manufacturing materials.
BYDUREON Manufacturing
We have built and are operating a facility in West Chester, Ohio to manufacture BYDUREON. Our Ohio facility obtains bulk exenatide, the active ingredient in BYDUREON, from Lonza Ltd., or Lonza, and Mallinckrodt, Inc., or Mallinckrodt, and we obtain pre-filled diluent syringes for BYDUREON from Vetter Pharma-Fertigung GmbH & Co. KG pursuant to long-term agreements with each company.
Under the terms of our development and license agreement with Alkermes, we are responsible for manufacturing the dosing formulation of BYDUREON for commercial sale and will pay Alkermes milestone payments upon achievement of development milestones and royalties ranging in the mid single digits on sales of BYDUREON. To date, we have paid an aggregate of $13 million as development milestone payments to Alkermes under the agreement. If all future milestones are achieved, we may be obligated to pay Alkermes an aggregate of $7 million in additional milestone payments. Alkermes has transferred to us its technology for manufacturing BYDUREON and is supplying us with the polymer materials required for the commercial manufacture of BYDUREON. The development and license agreement terminates on the later of 10 years from first commercial sale of product under the agreement or the expiration or invalidation of certain Alkermes patents covering such products. In addition, we can terminate the agreement at will upon 180 days written notice to Alkermes, and Alkermes can terminate the agreement pursuant to standard bankruptcy and liquidation provisions. Both parties can terminate the agreement pursuant to uncured material breach of contract terms.
BYETTA Manufacturing
We obtain exenatide, the active ingredient contained in BYETTA, from Bachem, Inc., or Bachem, and Mallinckrodt pursuant to agreements with each company. We have agreements with Wockhardt UK (Holdings) Ltd., or Wockhardt, and Baxter Pharmaceutical Solutions LLC, a subsidiary of Baxter, Inc., or Baxter, to supply us the dosage form of exenatide in cartridges. We have an agreement with Lilly to supply pens for delivery of BYETTA in cartridges which will eventually be transferred to another manufacturer.
SYMLIN Manufacturing
We obtain pramlintide acetate, the active ingredient contained in SYMLIN, from Bachem and Lonza pursuant to agreements with each company. We have an agreement with Wockhardt to supply the dosage form of SYMLIN in cartridges and an agreement with Ypsomed AG to supply SYMLIN pen components. We also have an agreement with Sharp Corporation for the assembly of the SYMLIN pen.
Competition
The biotechnology and pharmaceutical industries are highly competitive. There are many pharmaceutical companies, biotechnology companies, public and private universities and research organizations actively engaged in the research and development of products that may be similar to the products in our portfolio. A number of our largest competitors, including AstraZeneca, Bristol-Myers Squibb Company, Boehringer Ingelheim, GlaxoSmithKline, J&J (Janssen),
10
Lilly, Merck & Co., Novartis AG, Novo-Nordisk, Pfizer, Sanofi-Aventis, Roche and Takeda are pursuing the development of or are marketing pharmaceuticals that target the same diseases that we are targeting, and it is probable that the number of companies seeking to develop products and therapies for the treatment of diabetes, obesity and other metabolic disorders will increase. For example, in 2010, Novo Nordisk obtained approval of and commercially launched a GLP-1 receptor agonist to treat type 2 diabetes. Many of these companies and other existing or potential competitors have substantially greater financial, technical and human resources than we do and may be better equipped to develop, manufacture and market products. These companies may develop and introduce products and processes competitive with or superior to ours. In addition, other technologies or products may be developed that have an entirely different approach or means of accomplishing the intended purposes of our products, which might render our technology and products noncompetitive or obsolete. For example, all of our current drug products are injectable, and compete, in part, with therapies that do not require injection. We cannot be certain that we will be able to compete successfully.
SYMLIN is the only non-insulin-based drug product approved for improving blood glucose control in people with type 1 diabetes. Further, insulin and oral medications are often insufficient for many people with type 2 diabetes to achieve satisfactory glucose and weight control. BYDUREON, BYETTA or SYMLIN may be complementary to, or competitive with, these other medications.
BYDUREON, BYETTA and SYMLIN must compete with established therapies for market share. In addition, many companies are pursuing the development of novel pharmaceuticals that target diabetes. These companies may develop and introduce products competitive with or superior to BYDUREON, BYETTA or SYMLIN. Such competitive products and potential products include:
| | • | | insulins (injectable and inhaled versions); |
| | • | | TZDs and other PPAR or non-PPAR insulin sensitizers; |
| | • | | incretin mimetics/GLP-1 receptor agonists; |
| | • | | alpha-glucosidase inhibitors; and |
| | • | | sodium-glucose transporter-2 (SGLT-2) inhibitors. |
Patents, Proprietary Rights, and Licenses
We believe that patents and other proprietary rights are important to our business. Our policy is to file patent applications to protect technology, inventions and improvements that may be important to the development of our business. We also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position. We plan to enforce our issued patents and our rights to proprietary information and technology. We review third-party patents and patent applications, both to refine our own patent strategy and to identify useful licensing opportunities.
We have a number of patents, patent applications and rights to patents related to our compounds, products and technology, but we cannot be certain that issued patents will be enforceable or provide adequate protection or that pending patent applications will result in issued patents. We have also filed foreign counterparts to many of these issued patents and applications.
We may obtain patents for our compounds many years before we obtain marketing approval for them. Because patents have a limited life, which may begin to run prior to the commercial sale of the related product, the commercial value of the patent may be limited. However, we may be able to apply for patent term extensions to compensate in part for delays in obtaining marketing approval. For example, in the United States a patent term extension of 1,586 days has been granted for
11
SYMLIN, resulting in a patent expiration date of March 16, 2019, and a patent term extension of 1,287 days has been granted for BYETTA, resulting in a patent expiration date of December 1, 2016. Similar patent term extensions may be available for other products that we are developing, but we cannot be certain we will obtain them.
Included within our exenatide patent portfolio are issued patents for:
| | • | | pharmaceutical compositions containing exenatide; |
| | • | | modulating gastric emptying; |
| | • | | inhibiting glucagon secretion; |
| | • | | stimulating insulin release to treat diabetes; and |
These patents expire between 2016 and 2020. We do not have a composition of matter patent for the exenatide molecule.
Included within our pramlintide patent portfolio are issued patents for:
| | • | | pramlintide and other amylin agonist analogues; |
| | • | | pharmaceutical compositions containing amylin agonists, including pharmaceutical compositions containing pramlintide; and |
| | • | | methods for treating diabetes and related conditions using amylin agonists. |
These patents expire between 2013 and 2019.
Our SYMLIN, BYETTA and BYDUREON products are subject to the provisions of the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, which provides data exclusivity for a certain period of time. Under the Hatch-Waxman Act, the data exclusivity period for SYMLIN and BYETTA expired in 2009, such that generic manufacturers can now file Abbreviated New Drug Applications, or ANDAs, requesting the FDA’s approval of generic versions of these approved products. If an ANDA is filed for one of our approved products prior to expiration of the patents covering those products, it could result in our initiating patent infringement litigation to enforce our rights.
With respect to our drug candidates, we have patents and patent applications pending, or have licensed patents and patent applications, relevant to the development and commercialization of such drug candidates. Generally, our policy is to file foreign counterpart applications in countries with significant pharmaceutical markets.
It is important that we do not infringe patents or proprietary rights of others and that we do not violate the agreements that grant proprietary rights to us. If we do infringe patents or violate these agreements, we could be prevented from developing or selling products or from using the processes covered by those patents or agreements, or we could be required to obtain a license from the third party allowing us to use their technology. We cannot be certain that, if required, we could obtain a license to any third-party technology or that we could obtain one at a reasonable cost. If we were not able to obtain a required license, we could be adversely affected. Because patent applications are confidential for at least some period of time, there may be pending patent applications from which patents will eventually issue and prevent us from developing or selling certain products unless we can obtain a license to use the patented technology.
Patents relating to pharmaceutical, biopharmaceutical and biotechnology products, compounds and processes such as those that cover our existing products, compounds and processes and those that we will likely file in the future do not always provide complete or adequate protection. Future litigation or proceedings initiated by the United States Patent and Trademark Office regarding the enforcement or validity of our existing patents or any future patents could invalidate our patents or substantially reduce their protection. In addition, statutory or regulatory changes may adversely affect our ability to obtain protection or enforce our patents. Furthermore, our pending patent applications and patent applications filed by our collaborative partners may not result in the issuance of any patents or may result in patents that do not provide adequate protection. As a result, we may not be able to prevent third parties from developing the same compounds and products that we have developed or are developing. In addition, we do not have patent protection or we may not be able to enforce our patents in certain countries. As a result, manufacturers may be able to sell generic versions of our products in those countries.
12
We also rely on unpatented trade secrets and improvements, unpatented internal know-how and technological innovation. We protect these rights mainly through confidentiality agreements with our corporate partners, employees, consultants and vendors. These agreements provide that all confidential information developed or made known to an individual during the course of their relationship with us will be kept confidential and will not be used or disclosed to third parties except in specified circumstances. In the case of employees, the agreements provide that all inventions made by the individual while employed by us will be our exclusive property. We cannot be certain that these parties will comply with these confidentiality agreements, that we have adequate remedies for any breach, or that our trade secrets will not otherwise become known or be independently discovered by our competitors. Under some of our research and development agreements, inventions discovered in certain cases become jointly owned by us and our corporate partner and in other cases become the exclusive property of one of us. It can be difficult to determine who owns a particular invention and disputes could arise regarding those inventions.
Government Regulation
Regulation by governmental authorities in the United States and foreign countries is a significant factor in the development, manufacture and marketing of pharmaceutical products. All of our potential products will require regulatory approval by governmental agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical testing and clinical trials and other pre-market approval requirements by the FDA and regulatory authorities in foreign countries. Various federal, state and foreign statutes and regulations also govern or influence the manufacturing, safety, labeling, storage, record keeping and marketing of such products.
A number of steps must be taken before a pharmaceutical agent may be marketed in the United States. First, the pharmaceutical agent must undergo preclinical testing. Preclinical tests include laboratory evaluation of product chemistry and animal studies to assess the potential safety and activity of the product candidate and its formulations. The results of these studies must be submitted to the FDA as part of an investigational new drug application, or IND, which must be reviewed by the FDA before a proposed clinical trial can begin. Typically, clinical trials involve a three-phase process. In Phase 1, clinical trials are conducted with a small number of healthy volunteers to determine the early safety and tolerability profile and the pattern of drug distribution and metabolism. In Phase 2, clinical trials are conducted with groups of patients afflicted with a specified disease in order to determine preliminary efficacy, dosing regimens and expanded evidence of safety and tolerability. In Phase 3, large-scale, multi-center, adequate and well-controlled comparative clinical trials are typically conducted with patients afflicted with a target disease in order to provide enough data for the statistical proof of efficacy and safety required by the FDA and others. The results of the preclinical testing and clinical trials for a pharmaceutical product are then submitted to the FDA in the form of an NDA or BLA for approval to commence commercial sales. In responding to an NDA or BLA, the FDA may grant marketing approval or issue a “complete response letter,” which may include requests for additional information, indicating why the application is not ready for approval. Once a drug is approved for marketing in the United States, the FDA requires ongoing safety monitoring to ascertain any undiscovered issues related to “real-world” use of the drug. The expanded patient exposure once a drug is introduced to the marketplace can reveal new risks (as well as new benefits) that were not detectable during clinical testing.
Among the conditions for NDA approval is the requirement that the prospective manufacturer’s quality control and manufacturing procedures conform to cGMP. In complying with cGMP, manufacturers must continue to expend time, money and effort in the area of production, quality control, and quality assurance to ensure full technical compliance. Manufacturing facilities are subject to periodic inspections by the FDA to ensure compliance.
We are also subject to various federal, state, and local laws, regulations and recommendations relating to safe working conditions; laboratory and manufacturing practices; the experimental use of animals; and the use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents, used in connection with our research, development and manufacturing.
The activities required before a pharmaceutical agent may be marketed in the EU are dictated by the International Conference on Harmonization and are generally similar to those established in the United States. Approval of new drugs across the EU relies on either the centralized authorization procedure of the European Medicines Agency or national authorization procedures that allow simultaneous approval in several countries via mutual recognition or decentralization. Under the centralized procedure, the marketing application is referred for review to two review teams, each representing one of the member countries. Each reviewer then forwards an early assessment to the Committee for Medicinal Products for Human Use, or CHMP, for discussion and preparation of an initial consolidated assessment report, including a list of
13
questions requesting clarification as well as additional information. This step initiates a series of dialogues, meetings and other communications among the CHMP, the two review teams and the applicant, leading in turn to clarification, education and refinement of the original assessment reports. Ultimately, a decision is reached to either grant marketing authorization or deny the application if it is determined that the application does not satisfy the regulatory approval criteria. The clinical testing, manufacture and sale of pharmaceutical products outside of the United States and the EU are subject to regulatory approvals by other jurisdictions which may be more or less rigorous than those required by the United States or the EU.
Employees
As of December 31, 2011, we had approximately 1,300 full-time employees. A significant number of our management and professional employees have had experience with pharmaceutical, biotechnology or medical product companies. We believe that we have been highly successful in attracting skilled and experienced personnel. None of our employees are covered by collective bargaining agreements and we consider relations with our employees to be good.
Executive Officers
The names of our executive officers and certain information about them as of February 15, 2012 are set forth below:
| | | | |
Name | | Age | | Position |
| Daniel M. Bradbury | | 50 | | President, Chief Executive Officer and Director |
| Mark G. Foletta | | 51 | | Senior Vice President, Finance and Chief Financial Officer |
| Mark J. Gergen | | 49 | | Senior Vice President, Corporate Development |
| Orville G. Kolterman, M.D. | | 64 | | Senior Vice President, Chief Medical Officer |
| Harry J. Leonhardt | | 55 | | Senior Vice President, Legal and Compliance, and Corporate Secretary |
| Marcea Bland Lloyd | | 63 | | Senior Vice President, Chief Administrative Officer and General Counsel |
| Paul G. Marshall | | 52 | | Senior Vice President, Operations |
| Vincent P. Mihalik | | 61 | | Senior Vice President, Sales and Marketing and Chief Commercial Officer |
| Lloyd A. Rowland | | 55 | | Vice President, Chief Compliance Officer |
| Christian Weyer, M.D. | | 42 | | Senior Vice President, Research and Development |
Mr. Bradbury has been our Chief Executive Officer since March 2007, serving as President since June 2006 and as Chief Operating Officer since June 2003. He has served as a director since June 2006 and serves on the Finance Committee. He previously served as Executive Vice President from June 2000 until June 2003. He joined Amylin in 1994 and has held officer-level positions in Corporate Development and Marketing during that time. Prior to joining Amylin, Mr. Bradbury spent ten years at SmithKline Beecham Pharmaceuticals, where he held a number of sales and marketing positions. He is a member of the board of directors of Illumina, Inc. He also serves on the RAND Health Board of Advisors and as a board member for PhRMA, BIOCOM, the Keck Graduate Institute’s Board of Trustees and the San Diego Regional Economic Development Corporation. Mr. Bradbury serves on the UCSD Rady School of Management’s Advisory Council and the University of Miami’s Innovation Corporate Advisory Council and the University of Miami’s Diabetes Research Institute Corporate Advisory Council. He received a Bachelor of Pharmacy from Nottingham University and a Diploma in Management Studies from Harrow and Ealing Colleges of Higher Education.
Mr. Foletta has served as Senior Vice President, Finance and Chief Financial Officer since March 2006 and he previously served as Vice President, Finance and Chief Financial Officer from March 2000 to March 2006. Mr. Foletta previously served as a Principal of Triton Group Management, Inc. from 1997 to 2000. From 1986 to 1997, Mr. Foletta held a number of management positions with Intermark, Inc. and Triton Group Ltd., the most recent of which was Senior Vice President, Chief Financial Officer and Corporate Secretary. From 1982 to 1986, Mr. Foletta was with Ernst & Young, most recently serving as an Audit Manager. Mr. Foletta received a B.A. in Business Economics from the University of California, Santa Barbara. He is a Certified Public Accountant and a member of the Financial Executives Institute.
Mr. Gergen has served as Senior Vice President, Corporate Development since August 2006 and previously served as Vice President of Business Development from May 2005 to August 2006. Prior to joining us, Mr. Gergen was an independent consultant to biotech and medical technology companies for strategy, financing and corporate development. From 2003 to 2005, Mr. Gergen was Executive Vice President at CardioNet, Inc. He held various positions at Advanced Tissue Sciences, Inc. from 2000 to 2003 most recently as Chief Restructuring Officer and Acting CEO. He also served as Senior Vice President, Chief Financial and Development Officer, and Vice President, Development, General Counsel and Secretary. From 1999 to 2000, Mr. Gergen was employed at Premier, Inc. and from 1994 to 1999 he held various positions with Medtronic, Inc. From 1990 to 1994 he held various legal and corporate development positions at Jostens, Inc. and from 1986 to 1990, he practiced law at various law firms. Mr. Gergen serves on the Board of Directors of a privately held company. Mr. Gergen received a B.A. in Administration from Minot State University and a J.D. from the University of Minnesota Law School.
14
Dr. Kolterman has served as Senior Vice President, Chief Medical Officer since June 2010 and previously served as Senior Vice President, Research and Development from June 2008 to June 2010. He served as Senior Vice President, Development from March 2008 to May 2008. He also served as Senior Vice President, Clinical and Regulatory Affairs from August 2005 to March 2008, Senior Vice President, Clinical Affairs from February 1997 to August 2005, Vice President, Medical Affairs from 1993 to 1997, and Director, Medical Affairs from 1992 to 1993. From 1983 to 1992, he was Program Director of the General Clinical Research Center and Medical Director of the Diabetes Center, at the University of California, San Diego Medical Center. Since 1989, he has been Adjunct Professor of Medicine at the University of California, San Diego. From 1978 to 1983, he was Assistant Professor of Medicine in the Endocrinology and Metabolism Division at the University of Colorado School of Medicine, Denver. He was a member of the Diabetes Control and Complications Trial Study Group and presently serves as a member of the Epidemiology of Diabetes Intervention and Complications Study. He is also a past-president of the California Affiliate of the American Diabetes Association. Dr. Kolterman received his M.D. from Stanford University School of Medicine.
Mr. Leonhardt has served as our Senior Vice President, Legal and Compliance, and Corporate Secretary since September, 2011. He previously served as Vice President, Legal, Corporate Governance and Secretary from June 2010 to September, 2011 and served as Vice President Legal, Deputy General Counsel from October 2008 to June 2010. He previously served as our Vice President, Chief Intellectual Property Counsel since September 2007. Prior to joining us, Mr. Leonhardt served as Senior Vice President, General Counsel and Corporate Secretary of Senomyx, Inc. from September 2003 to September 2007. From February 2001 to September 2003 Mr. Leonhardt was Executive Vice President, General Counsel and Corporate Secretary of Genoptix, Inc. and from July 1996 to November 2000 he served as Vice President and then Senior Vice President, General Counsel and Corporate Secretary of Nanogen, Inc. From January 1990 through June 1996 Mr. Leonhardt served in various legal and management capacities at Allergan, Inc. Prior to that Mr. Leonhardt was an attorney with Lyon & Lyon LLP in Los Angeles where he represented a number of pharmaceutical, biotechnology and consumer products companies. He also serves as a board member for BIOCOM and a Special Master through the California State Bar. Mr. Leonhardt received a B.S. in Pharmacy from the University of the Sciences and a J.D. from the University of Southern California School of Law.
Ms. Lloyd has served as our Senior Vice President, Chief Administrative Officer and General Counsel since July, 2011. She previously served as Senior Vice President, Government & Corporate Affairs and General Counsel from June 2008 to July, 2011 and Senior Vice President, Legal and Corporate Affairs, and General Counsel from February 2007 to June 2008. Prior to joining us, Ms. Lloyd served as Group Senior Vice President, Chief Administrative Officer, General Counsel and Secretary of VHA Inc. from November 2004 to February 2007. Previously, she served as VHA’s General Counsel and Secretary from May 1999 to November 2004. From 1993 to April 1999, Ms. Lloyd was Vice President and Assistant General Counsel of Medtronic Inc. and served as Medtronic’s Assistant General Counsel from 1991 to 1993. From 1978 to 1991, Ms. Lloyd held various legal positions with Medtronic. Prior to joining Medtronic, Ms. Lloyd served as counsel to Pillsbury Company and Montgomery Ward & Co. and she taught Business Law at the University of Minnesota Business School. Ms. Lloyd is past Chairperson of the Executive Leadership Foundation, a member of the board of directors for California Healthcare Institute and is an associate of the Women Business Leaders of the United States Health Care Industry Foundation. She received a B.S./B.A. from Knox College and a J.D. from Northwestern University.
Mr. Marshall has served as Senior Vice President, Operations since December 2008. He previously served as Vice President Operations from December 2006 to December 2008. Prior to joining us, he was Vice President of Corporate Manufacturing at Amgen, Inc. From 2002 to 2005, Mr. Marshall served as Vice President of Recombinant Protein Manufacturing at the Bioscience Division of Baxter International. From 1999 to 2002, he was Site Head of the Baxter International Thousand Oaks facility. He joined Creative BioMolecules in 1992, first as Head of Process Development and Clinical Manufacturing and then as Head of Operations. From 1988 to 1992, Mr. Marshall held various management positions with Welgen Manufacturing Partnership (now Amgen, Rhode Island), Repligen Corporation and Damon Biotech. Mr. Marshall received a B.S. and an M.S. in Biology from the University of Massachusetts at Dartmouth and completed three years of post-graduate work concentrating in hematology and coagulation research at Brown University.
Mr. Mihalik has served as Senior Vice President, Sales and Marketing and Chief Commercial Officer since January 2009. Mr. Mihalik has over 35 years of experience across multiple commercial roles. Before joining us, Mr. Mihalik served as Vice President of Global Brand Development Diabetes and Endocrine Platform Team Leader for Lilly since 2004. Previously, he was Business Unit Head of Diabetes Care for Lilly U.S. from 2001 to 2004. From 1990 to 2001 he served in various senior management positions at other healthcare companies including Senior Vice President and General Manager for Lab Systems and Molecular Biochemical at Roche Diagnostics Corporation, President, Diabetes Care North America at
15
Boehringer Mannheim Group and President, Scientific Products Biomedical and General Manager, Pandex Diagnostic Research and Development Center for Baxter Healthcare Inc. He has a B.S. degree in Biology from The Pennsylvania State University and completed the Northwestern University Masters in Management—Executive Program.
Mr. Rowland has served as our Vice President, Chief Compliance Officer since June 2010. He previously served as Vice President, Governance and Compliance, Secretary, and Chief Compliance Officer from February 2007 to June 2010 and as Vice President, Legal, Secretary and General Counsel from September 2001 to February 2007. Prior to joining us, Mr. Rowland served in various positions at Alliance Pharmaceutical Corp., including as Vice President, General Counsel and Secretary, beginning in 1993. Earlier, Mr. Rowland served as Vice President and Senior Counsel, Finance and Securities, at Imperial Savings Association for four years. For the previous eight years, he was engaged in the private practice of corporate law with the San Diego, California law firm of Gray, Cary, Ames & Fry, and the Houston, Texas law firm of Bracewell & Patterson. He received a J.D. from Emory University.
Dr. Weyer has served as Senior Vice President, Research and Development since June 2010, and previously served as Vice President, Medical Development from September 2009 to June 2010. He previously served as Vice President of Corporate Development for Diabetes and Obesity from August 2008 to September 2009. Dr. Weyer has held leadership positions in Research, Clinical Development, Corporate Development, and Medical Affairs since joining Amylin in January 2001. Prior to joining us, Dr. Weyer was a Visiting Fellow with the National Institutes of Health, NIDDK, in Phoenix, AZ, from 1997-2000, where he conducted clinical research on the pathophysiology of obesity and type 2 diabetes in Pima Indians. He received his MD and clinical training at the Department of Metabolic Disorders, WHO Collaborating Center for Diabetes Treatment and Prevention, at the University of Düsseldorf, Germany. Dr. Weyer also holds a postdoctoral master’s degree in advanced clinical research from the University of California, San Diego, and currently serves on the program’s advisory board.
CAUTIONARY FACTORS THAT MAY AFFECT FUTURE RESULTS
Except for the historical information contained herein or incorporated by reference, this annual report on Form 10-K and the information incorporated by reference contains forward-looking statements that involve risks and uncertainties. These statements include projections about our accounting and finances, plans and objectives for the future, future operating and economic performance and other statements regarding future performance. These statements are not guarantees of future performance or events. Our actual results may differ materially from those discussed here. Factors that could cause or contribute to differences in our actual results include those discussed in the following section, as well as those discussed in Part II, Item 7 entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere throughout this annual report on Form 10-K and in any other documents incorporated by reference into this report. You should consider carefully the following risk factors, together with all of the other information included or incorporated in this annual report on Form 10-K. Each of these risk factors, either alone or taken together, could adversely affect our business, operating results and financial condition, as well as adversely affect the value of an investment in our common stock. There may be additional risks that we do not presently know of or that we currently believe are immaterial which could also impair our business and financial position.
We have a history of operating losses, anticipate future losses and may never become profitable.
We have experienced significant operating losses since our inception in 1987, including losses of $543.4 million in 2011, $152.3 million in 2010 and $186.3 million in 2009. As of December 31, 2011, we had an accumulated deficit of approximately $2.6 billion. The extent of our future losses and the timing of potential profitability are uncertain, and we may never achieve profitable operations. We have been engaged in discovering and developing drugs since inception, which has required, and will continue to require, significant research and development expenditures. Our three commercial products, BYDUREON, BYETTA and SYMLIN may not be as commercially successful as we expect and we may not succeed in commercializing any of our other drug candidates, including metreleptin to treat lipodystrophy, if approved. We may incur substantial operating losses for at least the next few years. These losses, among other things, have had and will have an adverse effect on our stockholders’ equity and working capital. Even if we become profitable, we may not remain profitable.
We began selling, marketing and distributing our first two products, BYETTA and SYMLIN, in 2005, and our third product, BYDUREON, in 2011, and we will depend heavily on the success of those products in the marketplace.
Prior to the launch of BYETTA and SYMLIN in 2005, we had never sold or marketed our own products. Our ability to generate product revenue in the near term depends solely on the success of these products and BYDUREON, which received
16
marketing authorization from the European Commission in June 2011 and FDA approval in January 2012. The ability of BYDUREON, BYETTA and SYMLIN to generate revenue at the levels we expect will depend on many factors, including the following:
| | • | | our ability to successfully launch BYDUREON in the United States; |
| | • | | our ability to secure a new partner to assist in the development and commercialization of our exenatide franchise outside the United States; |
| | • | | the ability of patients in the current uncertain economic climate to be able to afford our medications or obtain health care coverage that covers our products; |
| | • | | acceptance of and ongoing satisfaction with these novel medicines in the United States and foreign markets by the medical community, patients receiving therapy and third party payers; |
| | • | | a satisfactory efficacy and safety profile as demonstrated in a broad patient population; |
| | • | | successfully expanding and sustaining manufacturing capacity to meet demand; |
| | • | | safety concerns in the marketplace for diabetes therapies; |
| | • | | the competitive landscape for approved and developing therapies that will compete with the products; and |
| | • | | our ability to expand the indications for which we can market the products. |
If we encounter safety issues with BYDUREON, BYETTA or SYMLIN or any other drugs we market or fail to comply with extensive continuing regulations enforced by domestic and foreign regulatory authorities, it could cause us to discontinue marketing those drugs, reduce our revenues and harm our ability to generate future revenues, which would negatively impact our financial position.
BYDUREON, BYETTA and SYMLIN, in addition to any other of our drug candidates that may be approved by the FDA, will be subject to continual review by the FDA, and we cannot assure you that newly discovered or developed safety issues will not arise. With the use of any of our marketed drugs by a wide patient population, serious adverse events may occur from time to time that initially do not appear to relate to the drug itself, and only if the specific event occurs with some regularity over a period of time does the drug become suspect as having a causal relationship to the adverse event. Some patients taking BYETTA have reported developing pancreatitis. We are working to better understand the relationship between BYETTA and pancreatitis described in some spontaneously reported cases. In keeping with our focus on patient safety, we continue to pursue our drug safety program that includes thorough investigation of individual spontaneous case reports along with clinical and epidemiologic studies. Within the detection limits of an initial epidemiology study which we provided to the FDA, we have not observed an increased incidence of pancreatitis associated with BYETTA use compared to other treatments for diabetes and thus believe a definite causal relationship between BYETTA and pancreatitis has not been proved. In addition, since BYETTA was introduced, we have received other reports of adverse events, including rare reports of acute renal failure in patients using BYETTA, and in pre-clinical studies of BYDUREON, observations were made of C-cell tumors in animals. Although direct relationships have not been established, it may be difficult to rule out any particular direct relationship at any point in time for these or other reports of adverse events or observations that may be made. Any safety issues could cause us to suspend or cease marketing of our approved products, cause us to modify how we market our approved products, subject us to substantial liabilities, and adversely affect our revenues and financial condition.
Moreover, the marketing of our approved products will be subject to extensive regulatory requirements administered by the FDA and other regulatory bodies, including adverse event reporting requirements and the FDA’s general prohibition against promoting products for unapproved uses. The manufacturing facilities for our approved products are also subject to continual review and periodic inspection and approval of manufacturing modifications. Manufacturing facilities that manufacture drug products for the United States market, whether they are located inside or outside the United States, are subject to biennial inspections by the FDA and must comply with the FDA’s current good manufacturing practice, or cGMP, regulations. The FDA stringently applies regulatory standards for manufacturing. Failure to comply with any of these post-approval requirements can, among other things, result in warning letters, product seizures, recalls, fines, injunctions, suspensions or revocations of marketing licenses, operating restrictions and criminal prosecutions. Any of these enforcement actions, any unanticipated changes in existing regulatory requirements or the adoption of new requirements, or any safety issues that arise with any approved products, could adversely affect our ability to market products and generate revenues and thus adversely affect our ability to continue our business.
The manufacturers of our products and drug candidates also are subject to numerous federal, state, local and foreign laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control and hazardous substance disposal. In the future, our manufacturers may incur significant costs to comply with those laws and regulations, which could increase our manufacturing costs and reduce our ability to operate profitably.
17
We currently do not manufacture BYETTA and SYMLIN product, our bulk exenatide or pramlintide or our drug candidates and may not be able to obtain adequate supplies. This could cause delays, subject us to product shortages, or reduce product sales. If our BYDUREON manufacturing facility is damaged, rendered inoperable or does not comply with regulatory requirements, we may not be able to obtain an adequate supply of BYDUREON.
The manufacturing of sufficient quantities of newly-approved drug products and drug candidates is a time-consuming and complex process. We currently have no manufacturing capabilities for two of our three marketed drug products, BYETTA and SYMLIN. In order to successfully supply our products and continue to develop our drug candidates we rely on various third parties to provide the necessary manufacturing.
There are a limited number of manufacturers that operate under the FDA’s cGMP regulations capable of manufacturing for us. In addition, there are a limited number of bulk drug substance suppliers, cartridge manufacturers and disposable pen manufacturers. If we are not able to arrange for and maintain third-party manufacturing on commercially reasonable terms, or we lose one of our sole source suppliers used for our existing products or for some components of our manufacturing processes for our products or drug candidates, we may not be able to market our products or complete development of our drug candidates on a timely basis, if at all.
Reliance on third-party suppliers limits our ability to control certain aspects of the manufacturing process and therefore exposes us to a variety of significant risks, including, but not limited to, risks to our ability to supply or commercialize our products or conduct clinical trials, risks of reliance on the third-party for regulatory compliance and quality assurance, third-party refusal to supply on a long-term basis, or at all, the possibility of breach of the manufacturing agreement by the third-party and the possibility of termination or non-renewal of the agreement by the third-party, based on its business priorities, at a time that is costly or inconvenient for us. In addition, reliance on single-source suppliers subjects us to the risk of price increases by these suppliers which could negatively impact our operating margins. If any of these risks occur, our product supply will be interrupted resulting in lost or delayed revenues and delayed clinical trials. Our reliance on third-party manufacturers for the production of our commercial products is described in more detail below.
Our Ohio manufacturing facility has been approved by the FDA and the EMA for manufacturing BYDUREON for commercial distribution in the United States and the European Union, respectively. In order to continue manufacturing BYDUREON for these two geographic locations, we must maintain these regulatory approvals. Further, in order to manufacture BYDUREON for commercialization in additional jurisdictions, we will need to gain regulatory approvals from such jurisdictions. We cannot assure you that we will be able to continue to successfully operate our manufacturing facility in accordance with FDA or EMA regulations or the regulations of any other geographic location. In addition, we are dependent on Alkermes to supply us with commercial quantities of the polymer required to manufacture BYDUREON. We also will need to obtain sufficient supplies of diluent, solvents, devices, packaging and other components necessary for commercial manufacture of BYDUREON. We are dependent upon Mallinckrodt and Lonza to manufacture our long-term commercial supply of bulk exenatide, the active ingredient in BYDUREON, and upon single suppliers to produce components for packaging BYDUREON.
Our Ohio facility is currently the only manufacturing capacity available to us for the production of BYDUREON. Equipment failures, natural disasters, power outages or other catastrophic events, such as a severe storm or fire, could cause interruptions or delays in our ability to manufacture BYDUREON. If we experience equipment failures, or our manufacturing facility is damaged by a natural disaster or other catastrophic event, or if severe weather conditions prevent us from delivering BYDUREON to meet market needs in a timely manner, our business, financial condition and operating results could be adversely impacted.
We rely on Bachem and Mallinckrodt to manufacture our long-term commercial supply of bulk exenatide, the active ingredient in BYETTA. In addition, we rely on single-source manufacturers for some of our raw materials used by Bachem and Mallinckrodt to produce bulk exenatide. We also rely on Wockhardt and Baxter to manufacture the dosage form of BYETTA in cartridges. We are further dependent upon Lilly to supply pens for delivery of BYETTA in cartridges.
We rely on Bachem and Lonza to manufacture our commercial supply of bulk pramlintide acetate, the active ingredient contained in SYMLIN. We rely on Wockhardt for the dosage form of SYMLIN in cartridges and Ypsomed AG to manufacture the components for the SYMLIN disposable pen. We also rely on Sharp Corporation for the assembly of the SYMLIN pen.
If any of our existing or future manufacturers cease to manufacture or are otherwise unable to timely deliver sufficient quantities of BYETTA or SYMLIN, in either bulk or dosage form, or other product components, including pens for the delivery of these products, it could disrupt our ability to market our products, subject us to product shortages, reduce product sales and/or reduce our profit margins. Any delay or disruption in the manufacturing of bulk product, the dosage form of our products or other product components, including pens for delivery of our products, could also harm our reputation in the
18
medical and patient communities. In addition, we may need to engage additional manufacturers so that we will be able to continue our commercialization and development efforts for these products or drug candidates. The cost and time to establish these new manufacturing facilities would be substantial.
Our manufacturers have produced BYETTA and SYMLIN for commercial use for approximately seven years, however, unforeseeable risks related to environmental, economic, technical or other issues may be encountered as we, together with our manufacturers, continue to develop familiarity and experience with regard to manufacturing our products. Furthermore, we and the other manufacturers used for our drug candidates may not be able to produce supplies in commercial quantities if our drug candidates are approved. While we believe that business relations between us and our manufacturers are generally good, we cannot predict whether any of the manufacturers that we may use will meet our requirements for quality, quantity or timeliness for the manufacture of bulk exenatide or pramlintide acetate, dosage form of BYETTA or SYMLIN, or pens. Therefore, we may not be able to obtain necessary supplies of products with acceptable quality, on acceptable terms or in sufficient quantities, if at all. Our dependence on third parties for the manufacture of products may also reduce our gross profit margins and our ability to develop and deliver products in a timely manner.
We have a significant amount of indebtedness. We may not be able to make payments on our indebtedness, and we may incur additional indebtedness in the future, which could adversely affect our operations.
In June 2007, we issued $575 million of the 2007 Notes and in May 2011 we borrowed $165 million from Lilly under the Lilly Loan. Our ability to make payments on our debt, including the 2007 Notes and the Lilly Loan, will depend on our future operating performance and ability to generate cash and may also depend on our ability to obtain additional debt or equity financing. During three of the last five years, our operating cash flows were negative and insufficient to cover our fixed costs. We will need to use cash to pay principal and interest on our debt, thereby reducing the funds available to fund our research and development programs, strategic initiatives and working capital requirements. Our ability to generate sufficient operating cash flow and revenues to service our indebtedness and fund our operating requirements will depend on our ability, alone or with others, to successfully develop, manufacture, obtain required regulatory approvals for and market our drug products and candidates, as well as other factors, including general economic, financial, competitive, legislative and regulatory conditions, some of which are beyond our control. Our debt service increases our vulnerabilities to competitive pressures because many of our competitors are less leveraged than we are. If we are unable to generate sufficient operating cash flow and revenues to service our indebtedness and fund our operating requirements, we may be forced to reduce or defer our development programs, sell assets or seek additional debt or equity financing, which may not be available to us on satisfactory terms or at all. Our level of indebtedness may make us more vulnerable to economic or industry downturns. If we incur new indebtedness, the risks relating to our business and our ability to service our indebtedness and other financial obligations will intensify.
We have a substantial revenue sharing obligation payable to Lilly that is secured by certain of our assets. If we are unable to make payments on our revenue sharing obligation, our operations and financial position could be harmed.
In connection with the termination of our collaboration with Lilly in November 2011, we incurred a $1.2 billion revenue sharing obligation payable to Lilly, or the Revenue Sharing Obligation, and entered into a promissory note in favor of Lilly in the initial principal amount of $1.2 billion, secured by certain of our assets and certain assets of our subsidiaries. We also entered into a security agreement with Lilly pursuant to which we and our Ohio subsidiary granted to Lilly, as collateral to secure payment of principal, interest and certain expenses under the promissory note, a security interest in intellectual property relating to our exenatide products, United States regulatory approvals relating to our exenatide products, certain third party license agreements, certain deposit accounts into which counterparties of such license agreements are required to make payments, certain third party supply agreements, inventory and a supply agreement for BYDUREON between us and our Ohio subsidiary. Under the promissory note an event of default would occur if we fail to make payments under the Revenue Sharing Obligation, or upon certain other events as set forth in the promissory note. Upon the occurrence and during the continuance of an event of default, all outstanding amounts under the promissory note may be declared due and payable (and in the case of a bankruptcy event of default, such obligations will automatically become due and payable), and Lilly may exercise its rights with respect to the collateral under the security agreement. If Lilly successfully exercises its rights with respect to the collateral, Lilly will be able to, among other things, sell, lease, license or otherwise dispose of the collateral, enforce our rights in the intellectual property, license agreements and supply agreements relating to BYETTA and BYDUREON (including bringing intellectual property infringement actions against third parties and acquiring the rights to receive BYDUREON supply under an intercompany supply agreement between us and our Ohio subsidiary) and directly collect payments from counterparties of the license agreements that such counterparties would otherwise be required to pay to us. Any such successful exercise by Lilly of its rights with respect to the collateral could have a negative impact on our day-to-day operations, financial condition and operating results.
19
Our ability to generate revenues will be diminished if we fail to obtain acceptable prices or an adequate level of reimbursement for our products from third-party payers.
The continuing efforts of government, private health insurers and other third-party payers to contain or reduce the costs of health care through various means, including efforts to increase the amount of patient co-pay obligations, may limit our commercial opportunity. In the United States, the Federal government recently passed health care reform legislation. Many of the details regarding the implementation of this legislation have yet to be determined and implementation may ultimately adversely affect our business. Further, we expect that there will continue to be a number of federal and state proposals to implement government control over the pricing of prescription pharmaceuticals. In addition, increasing emphasis on managed care in the United States will continue to put pressure on the rate of adoption and pricing of pharmaceutical products.
Significant uncertainty exists as to the reimbursement status of health care products. Third-party payers, including Medicare, are challenging the prices charged for medical products and services. Government and other third-party payers increasingly are attempting to contain health care costs by limiting both coverage and the level of reimbursement for new drugs and by refusing to provide coverage for uses of approved products for disease indications for which the FDA has not granted labeling approval. Third-party insurance coverage may not be available to patients for BYDUREON, BYETTA and/or SYMLIN or any other products we discover and develop. If government and other third-party payers do not provide adequate coverage and reimbursement levels for our products, the market acceptance of these products may be reduced.
Competition in the biotechnology and pharmaceutical industries may result in competing products, superior marketing of other products and lower revenues or profits for us.
There are many companies that are seeking to develop products and therapies for the treatment of diabetes and other metabolic disorders. Our competitors include multinational pharmaceutical and chemical companies, specialized biotechnology firms and universities and other research institutions. A number of our largest competitors, including AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Lilly, Merck & Co., Novartis, Novo Nordisk, Pfizer, Sanofi-Aventis, Roche and Takeda, are pursuing the development or marketing of pharmaceuticals that target the same diseases that we are targeting. For example, in 2010, Novo Nordisk obtained approval of and commercially launched a GLP-1 receptor agonist to treat type 2 diabetes. In addition, Lilly is developing a GLP-1 receptor agonist to treat type 2 diabetes and has announced a global alliance with Boehringer Ingelheim to jointly develop and commercialize a portfolio of diabetes compounds which Lilly has announced includes a SGLT-2 inhibitor and a DPP-IV inhibitor that has been approved by the FDA and the European Commission. These products all compete for patients in the diabetes space. It is possible that the number of companies seeking to develop products and therapies for the treatment of diabetes, obesity and other metabolic disorders will increase.
Many of our competitors have substantially greater financial, technical, sales force, human and other resources than we do and may be better equipped to develop, manufacture and market technologically superior products. In addition, many of these competitors have significantly greater experience than we do in undertaking preclinical testing and human clinical studies of new pharmaceutical products and in obtaining regulatory approvals of human therapeutic products. Accordingly, our competitors may succeed in obtaining FDA approval for competing and possibly superior products. Furthermore, now that we have received FDA approval for BYDUREON, BYETTA and SYMLIN, we may also be competing against other companies with respect to our manufacturing and product distribution efficiency and sales and marketing capabilities, areas in which we have limited experience as an organization.
Our target patient population for BYETTA includes people with type 2 diabetes who have not achieved adequate glycemic control with diet and exercise or by using metformin, sulfonylurea and/or a TZD, three common oral therapies for type 2 diabetes, and insulin glargine. Our target population for BYDUREON includes people with type 2 diabetes who have not achieved adequate glycemic control with diet and exercise and our target population for SYMLIN includes people with either type 2 or type 1 diabetes whose therapy includes multiple mealtime insulin injections per day. Other products are currently in development or exist in the market that may compete directly with the products that we are developing or marketing. Various other products are available or in development to treat type 2 diabetes, including, for example:
| | • | | insulins, including injectable and inhaled versions; |
| | • | | TZDs and other PPAR or non-PPAR insulin sensitizers; |
| | • | | incretin/GLP-1 receptor agonists; |
20
| | • | | alpha-glucosidase inhibitors; and |
| | • | | sodium-glucose transporter-2 (SGLT-2) inhibitors. |
In addition, several companies are developing various approaches, including alternative delivery methods, to improve treatments for type 1 and type 2 diabetes. We cannot predict whether our products will have sufficient advantages to cause health care professionals to adopt them over other products or that our products will offer an economically feasible alternative to other products. Our products could become obsolete before we recover expenses incurred in developing these products.
Our business has a substantial risk of product liability claims, and insurance may not be adequate to cover these claims.
Our business exposes us to potential product liability risks that are inherent in the testing, manufacturing and marketing of human therapeutic products. As of December 31, 2011, we were involved in approximately 114 separate product liability cases, certain of which cases have been brought by individuals who allege they have used BYETTA and generally seek compensatory and punitive damages for alleged injuries, consisting primarily of pancreatitis and, in some cases, alleged wrongful death. We have also been notified of other claims of individuals who have not filed suit. We currently have limited product liability insurance coverage for existing claims and any future related claims and we expect to be largely self-insured for any future product liability risks that are not covered by existing insurance. Product liability claims could result in the imposition of substantial defense costs and liability on us, a recall of products, or a change in the indications for which they may be used. We cannot assure you that our insurance will provide adequate coverage against potential liabilities.
Delays in the conduct or completion of our clinical trials, the analysis of the data from our clinical trials or our manufacturing scale-up activities may result in delays in our planned filings for regulatory approvals of our products or delays in completion of post-marketing studies and requirements, and may adversely affect our ability to enter into new collaborative arrangements.
We cannot predict whether we will encounter problems with any of our completed, ongoing or planned clinical studies that will cause us to delay or suspend our ongoing and planned clinical studies, delay the analysis of data from our completed or ongoing clinical studies or perform additional clinical studies prior to receiving necessary regulatory approvals. We also cannot predict whether we will encounter delays or an inability to create manufacturing processes for drug candidates that allow us to produce drug product in sufficient quantities to be economical, otherwise known as manufacturing scale-up.
If the results of our ongoing or planned clinical studies for our drug candidates are not available when we expect or if we encounter any delay in the analysis of data from our clinical studies or if we encounter delays in our ability to scale-up our manufacturing processes:
| | • | | we may be unable to complete our development programs; |
| | • | | we may have to delay or terminate our planned filings for regulatory approval; |
| | • | | we may encounter delays in the completion of post-marketing requirements for our products; |
| | • | | we may not have the financial resources to continue research and development of any of our drug candidates; and |
| | • | | we may not be able to enter into, if we chose to do so, any additional collaborative arrangements. |
Any of the following could delay the completion of our ongoing and planned clinical studies:
| | • | | ongoing discussions with the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; |
| | • | | delays in enrolling volunteers; |
| | • | | lower than anticipated retention rate of volunteers in a clinical trial; |
| | • | | negative results of clinical studies; |
| | • | | insufficient supply or deficient quality of drug candidate materials or other materials necessary for the performance of clinical trials; |
| | • | | our inability to reach agreement with any future collaboration partner(s) regarding the scope, design, conduct or costs of clinical trials outside the United States with respect to BYETTA, BYDUREON or an exenatide suspension formulation; or |
| | • | | serious side effects experienced by study participants relating to a drug candidate, or other clinical trial observations that may pose a potential safety concern. |
21
We and Lilly terminated our exenatide development and commercialization collaboration in late 2011 whereupon we became solely responsible for the development and commercialization of our exenatide products within the United States. Accordingly, Lilly no longer shares exenatide development or commercialization costs and Lilly’s sales force is no longer available for commercializing our exenatide products within the United States. We cannot assure you that our exenatide development and commercial efforts will produce the results we expect.
In November 2011, we and Lilly terminated our 10-year collaboration under which Lilly assisted us in the development, production and commercialization of our exenatide products and shared certain exenatide development, production and commercialization costs. Further, a significant number of Lilly’s sales representatives were available for commercializing our exenatide products within the United States. As a result of the termination of our collaboration, we are now solely responsible for developing and commercializing exenatide within the United States. In addition, we will be responsible for many of the functions previously performed by Lilly during the course of our collaboration which we are in the process of transitioning from Lilly to us. While we believe we have an effective transition plan in place for assuming these responsibilities, our exenatide development, production and commercialization efforts may be impaired without the assistance or financial support of a collaboration partner. We cannot assure you that our efforts to transition Lilly’s collaboration responsibilities will proceed according to plan. In addition, we have retained a contract sales force to extend the reach of our sales and marketing organization in order to maintain a comparable level of coverage previously provided by the Lilly field force. Although the new sales organization consists of sales professionals with experience in the pharmaceutical industry, including many with experience selling diabetes products, we cannot assure you that their sales efforts will be effective or produce the results we expect.
We are substantially dependent on our arrangement with Lilly and will be substantially dependent on any future exenatide collaboration partner(s) for the development and commercialization of our exenatide products outside the United States. We are also dependent and Alkermes’ technology for the production of BYDUREON.
Upon termination of our collaboration with Lilly, we entered into an arrangement with Lilly, who currently markets its own diabetes therapies and is developing additional diabetes drug candidates, to commercialize BYDUREON and BYETTA outside the United States through a transition period until December 31, 2013 or at such later date if (i) mutually agreed by us and Lilly in the event such transition has not occurred by December 31, 2013 or (ii) in certain circumstances as set forth in our transition agreement with Lilly. We entered into this short-term arrangement with Lilly and currently seek to secure a new exenatide partner outside the United States in order to:
| | • | | fund some of our research and development activities; |
| | • | | assist us in seeking and obtaining regulatory approvals; and |
| | • | | assist us in the successful commercialization of BYDUREON, BYETTA and any other future exenatide products. |
In general, we cannot control the amount and timing of resources that any collaboration partner(s) may devote to our collaboration. If our partners fail to assist in the further development of our exenatide products or the commercialization of BYDUREON or BYETTA, or if our partner’s efforts are not effective, our business may be negatively affected. In the near-term, we are relying on Lilly, and will continue to rely on any future exenatide collaboration partner, to obtain regulatory approvals for and successfully commercialize BYETTA and BYDUREON outside the United States. Although we are currently in discussions with a number of potential partners, we cannot be certain that we will be able to secure a partner to develop and commercialize our exenatide products outside the United States on terms acceptable to us, or at all.
Our collaborations may not continue or result in additional successfully commercialized drugs. If any of our partners ceased funding and/or developing and commercializing BYETTA or BYDUREON, we would have to seek additional sources for funding and may have to delay, reduce or eliminate one or more of our commercialization and development programs for these products. If any of our partners do not successfully commercialize BYETTA or BYDUREON outside the United States, we may receive limited or no revenues from them. In addition, we are dependent on Alkermes’ technology for manufacturing BYDUREON. If Alkermes’ technology does not continue to effectively deliver exenatide in a sustained release formulation, or Alkermes does not devote sufficient resources to the collaboration, our efforts to produce sustained release formulations of exenatide could be curtailed.
If our patents are determined to be unenforceable or if we are unable to obtain new patents based on current patent applications or for future inventions, we may not be able to prevent others from using our intellectual property. If we are unable to obtain licenses to third party patent rights for required technologies, we could be adversely affected.
We own or hold exclusive rights to many issued United States patents and pending United States patent applications related to the development and commercialization of exenatide, including BYETTA and BYDUREON, SYMLIN and our other drug candidates. These patents and applications cover composition-of-matter, medical indications, methods of use,
22
formulations and other inventive results. We have issued and pending applications for formulations of BYETTA and BYDUREON, but we do not have a composition-of-matter patent covering exenatide. We also own or hold exclusive rights to various foreign patent applications that correspond to issued United States patents or pending United States patent applications.
Our success will depend in part on our ability to obtain patent protection for our products and drug candidates and technologies both in the United States and other countries. We cannot guarantee that any patents will issue from any pending or future patent applications owned by or licensed to us. Alternatively, a third party may successfully challenge or circumvent our patents. Our rights under any issued patents may not provide us with sufficient protection against competitive products or otherwise cover commercially valuable products or processes. For example, our SYMLIN, BYETTA and BYDUREON products are subject to the provisions of the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the “Hatch-Waxman Act,” which provides data exclusivity for a certain period of time. Under the Hatch-Waxman Act, the data exclusivity period for SYMLIN and BYETTA expired in 2009 such that generic manufacturers can now file Abbreviated New Drug Applications, or ANDAs, requesting the FDA’s approval of generic versions of previously-approved products. If an ANDA is filed for one of our approved products prior to expiration of the patents covering those products, it could result in our initiating patent infringement litigation to enforce our rights. We can provide no assurances that we would prevail in such an action or in any challenge related to our patent rights.
In addition, because patent applications in the United States are maintained, in general, in secrecy for 18 months after the filing of the applications, and publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be sure that the inventors of subject matter covered by our patents and patent applications were the first to invent or the first to file patent applications for these inventions. Third parties have filed, and in the future are likely to file, patent applications on inventions similar to ours. From time-to-time we have participated in, and in the future are likely to participate in, interference proceedings declared by the United States Patent and Trademark Office to determine priority of invention, which could result in a loss of our patent position. We have also participated in, and in the future are likely to participate in, opposition proceedings against our patents in other jurisdictions, such as Europe and Australia. Furthermore, we may not have identified all United States and foreign patents that pose a risk of infringement.
We also rely upon licensing opportunities for some of our technologies. We cannot be certain that we will not lose our rights to certain patented technologies under existing licenses or that we will be able to obtain a license to any required third-party technology. If we lose our licensed technology rights or if we are not able to obtain a required license, we could be adversely affected.
We may be unable to obtain regulatory clearance and pricing approval to market our drug candidates in the United States or foreign countries on a timely basis, or at all.
Our drug candidates are subject to extensive government regulations related to development, clinical trials, manufacturing and commercialization. The process of obtaining FDA and other regulatory approvals is costly, time-consuming, uncertain and subject to unanticipated delays. Regulatory authorities may refuse to approve an application for approval of a drug candidate, such as metreleptin for the treatment of lipodystrophy, if they believe that applicable regulatory criteria are not satisfied. Regulatory authorities may also require additional testing for safety and efficacy. Moreover, if the FDA grants regulatory approval of a product, the approval may be limited to specific indications or limited with respect to its distribution, and expanded or additional indications for approved drugs may not be approved, which could limit our revenues. Foreign regulatory authorities may apply similar limitations or may refuse to grant any approval. Further, even if approval is granted, there can be no assurance that the regulatory authority will grant pricing approval for the drug. Unexpected changes to the FDA or foreign regulatory approval process could also delay or prevent the approval of our drug candidates.
The data collected from clinical trials may not be sufficient to support approval of our drug candidates or additional or expanded indications by the FDA or any foreign regulatory authorities. Biotechnology stock prices have declined significantly in certain instances where companies have failed to meet expectations with respect to FDA approval or the timing for FDA approval. If the FDA’s or any foreign regulatory authority’s response is delayed or not favorable for any of our drug candidates our stock price could decline significantly.
Moreover, manufacturing facilities operated by us or by the third-party manufacturers with whom we may contract to manufacture our unapproved drug candidates may not pass an FDA or other regulatory authority inspections. Any corrective actions we may need to take as a result of regulatory inspections could cause us or any of our business partners to delay marketing these drug candidates.
Consequently, even if we believe that preclinical and clinical data are sufficient to support regulatory approval for our drug candidates, the FDA and foreign regulatory authorities may not ultimately approve our drug candidates for commercial sale in any jurisdiction. If our drug candidates are not approved, our ability to generate revenues may be limited, our manufacturing facility could become impaired, and our business will be adversely affected.
23
Litigation regarding patents and other proprietary rights may be expensive, cause delays in bringing products to market and harm our ability to operate.
Our success will depend in part on our ability to operate without infringing the proprietary rights of third parties and preventing others from infringing our patents. Challenges by pharmaceutical companies against the patents of competitors are common. Legal standards relating to the validity of patents covering pharmaceutical and biotechnological inventions and the scope of claims made under these patents are still developing. As a result, our ability to obtain and enforce patents is uncertain and involves complex legal and factual questions. Third parties may challenge, in courts or through patent office proceedings, or infringe upon, existing or future patents. In the event that a third party challenges a patent, a court or patent office may invalidate the patent or determine that the patent is not enforceable. Proceedings involving our patents or patent applications or those of others could result in adverse decisions about:
| | • | | the patentability of our inventions, products and drug candidates; and/or |
| | • | | the enforceability, validity or scope of protection offered by our patents. |
The manufacture, use or sale of any of our products or drug candidates may infringe on the patent rights of others. If we are unable to avoid infringement of the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, if we do not obtain a license, develop or obtain non-infringing technology, fail to successfully defend an infringement action or have infringing patents declared invalid, we may:
| | • | | incur substantial monetary damages; |
| | • | | encounter significant delays in bringing our drug candidates to market; and/or |
| | • | | be precluded from participating in the manufacture, use or sale of our products or drug candidates or methods of treatment requiring licenses. |
We are subject to “fraud and abuse��� and similar laws and regulations, and a failure to comply with such regulations or prevail in any litigation related to noncompliance could harm our business.
We are subject to various health care “fraud and abuse” laws, such as the Federal False Claims Act, the federal anti-kickback statute and other state and federal laws and regulations. Pharmaceutical companies have faced lawsuits and investigations pertaining to violations of these laws and regulations. We cannot guarantee that measures that we have taken to prevent such violations, including our corporate compliance program, will protect us from future violations, lawsuits or investigations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Our financial results will fluctuate, and these fluctuations may cause our stock price to fall.
Forecasting our future revenues is difficult, especially since we launched our first products, BYETTA and SYMLIN, in 2005 and recently launched a third product, BYDUREON, for commercial sale in the United States in early 2012. The level of market acceptance of these novel products may change rapidly. In addition, our customer base is highly concentrated with four customers accounting for most of our net product sales. Fluctuations in the buying patterns of these customers, which may result from seasonality, wholesaler buying decisions or other factors outside of our control, could significantly affect the level of our net sales on a period to period basis. As a result, it is reasonably likely that our financial results will fluctuate to an extent that may not meet with market expectations and that also may adversely affect our stock price. There are a number of other factors that could cause our financial results to fluctuate unexpectedly, including:
| | • | | achievement and timing of research and development milestones; |
| | • | | collaboration revenues; |
| | • | | cost and timing of clinical trials, regulatory approvals and product launches; |
| | • | | marketing and other expenses; |
| | • | | manufacturing or supply issues; and |
| | • | | potential acquisitions of businesses and technologies and our ability to successfully integrate any such acquisitions into our existing business. |
24
We may require additional financing in the future, which may not be available to us on favorable terms, or at all.
We intend to use our available cash for:
| | • | | Commercialization of BYDUREON, BYETTA and SYMLIN, including activities associated with the commercial launch of BYDUREON in 2012; |
| | • | | Satisfaction of our financial obligations to Lilly, including a $1.2 billion revenue sharing obligation; |
| | • | | Establishment of additional manufacturing sources and maintenance of our Ohio manufacturing facility; |
| | • | | Development of our pipeline candidates; |
| | • | | Our other research and development activities; |
| | • | | Other operating expenses; |
| | • | | Potential acquisitions or investments in complementary technologies or businesses; and |
| | • | | Other general corporate purposes. |
We may also be required to use our cash to pay principal and interest on outstanding debt, including $575 million in outstanding principal amount of convertible senior notes due in 2014, referred to as the 2007 Notes, and $165 million of an unsecured loan from Lilly that is due in 2016, referred to as the Lilly Loan.
If we require additional financing in the future, we cannot assure you that it will be available to us on favorable terms, or at all. Although we have previously been successful in obtaining financing through our debt and equity securities offerings, there can be no assurance that we will be able to so in the future, especially given the current adverse economic and credit conditions.
Our investments in marketable debt securities are subject to credit and market risks that may adversely affect their fair value.
We maintain a portfolio of investments in marketable debt securities which are recorded at fair value. Although we have established investment guidelines relative to diversification and maturity with the objective of maintaining safety of principal and liquidity, credit rating agencies may reduce the credit rating of our individual holdings which could adversely affect their value. Lower credit quality and other market events, such as increases in interest rates, and further deterioration in the credit markets may have an adverse effect on the fair value of our investment holdings and cash position.
Our ability to enter into and maintain third-party relationships is important to our successful development and commercialization of BYDUREON, BYETTA, SYMLIN and our other drug candidates and to our potential profitability.
We have entered into an agreement with a contract sales force to expand the reach of our sales organization for marketing BYDUREON, BYETTA and SYMLIN within the United States. If we are successful in our efforts to secure one or more collaboration partners to develop and commercialize our exenatide products outside the United States, we will be substantially dependent on these partners for such activities relating to BYDUREON and BYETTA, and any other sustained-release formulations of exenatide, if approved. We believe that we will likely need to enter into marketing and distribution arrangements with third parties for, or find a corporate partner who can provide support for, the development and commercialization of SYMLIN or our other drug candidates outside the United States. We may also enter into arrangements with third parties for the commercialization of our other drug candidates within the United States.
We may not be able to enter into marketing and distribution arrangements or find a corporate partner for SYMLIN or our other drug candidates as we deem necessary. If we are not able to enter into a marketing or distribution arrangement or find a corporate partner who can provide support for commercialization of our drug candidates as we deem necessary, we may not be able to successfully perform these marketing or distribution activities. Moreover, any new marketer or distributor or corporate partner for our drug candidates with whom we choose to contract may not establish adequate sales and distribution capabilities or gain market acceptance for our products, if any.
We may be required to redeem our convertible senior notes upon a designated event.
Holders of the 2007 Notes may require us to redeem all or any portion of their notes upon the occurrence of certain designated events which generally involve a change in control of our company. We may not have sufficient cash funds to redeem the 2007 Notes upon a designated event. If we are prohibited from redeeming the 2007 Notes, we could seek consent
25
from our lenders to redeem the 2007 Notes. If we are unable to obtain their consent, we could attempt to refinance the 2007 Notes. If we were unable to obtain a consent or refinance, we would be prohibited from redeeming the 2007 Notes. If we were unable to redeem the 2007 Notes upon a designated event, it would result in an event of default under the indentures governing the 2007 Notes. An event of default under the indentures could result in a further event of default under our other then-existing debt. In addition, the occurrence of a designated event may be an event of default under our other debt.
If our research and development programs fail to result in additional drug candidates, the growth of our business could be impaired.
Certain of our research and development programs for drug candidates are at an early stage and will require significant research, development, preclinical and clinical testing, manufacturing scale-up activities, regulatory approval and/or commitments of resources before commercialization. We cannot predict whether our research will lead to the discovery of any additional drug candidates that could generate additional revenues for us.
Our future success depends on our chief executive officer, and other key executives and our ability to attract, retain and motivate qualified personnel.
We are highly dependent on our chief executive officer, and the other principal members of our executive and scientific teams. The unexpected loss of the services of any of these persons might impede the achievement of our research, development and commercialization objectives. Recruiting and retaining qualified sales, marketing, regulatory, manufacturing, scientific and other personnel and consultants will also be critical to our success. We may not be able to attract and retain these personnel and consultants on acceptable terms given the competition between numerous pharmaceutical and biotechnology companies. We do not maintain “key person” insurance on any of our employees.
We may be unable to adequately prevent disclosure of trade secrets and other proprietary information.
In order to protect our proprietary technology and processes, we rely in part on confidentiality agreements with our corporate partners, employees, consultants, manufacturers, outside scientific collaborators and sponsored researchers and other advisors. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information.
Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
Our research and development activities and planned manufacturing activities involve the use of hazardous materials, which subject us to regulation, related costs and delays and potential liabilities.
Our research and development and our planned manufacturing activities involve the controlled use of hazardous materials, chemicals and various radioactive compounds. Although we believe that our research and development safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. In addition, as part of the development of our planned manufacturing activities, we will need to develop additional safety procedures for the handling and disposing of hazardous materials. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate any of these laws or regulations.
We are exposed to potential risks from legislation requiring companies to evaluate internal control over financial reporting.
The Sarbanes-Oxley Act requires that we report annually on the effectiveness of our internal control over financial reporting. Among other things, we must perform systems and processes evaluation and testing. We must also conduct an assessment of our internal control to allow management to report on, and our independent registered public accounting firm to attest to, our internal control over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. In connection with our Section 404 compliance efforts, we have incurred or expended, and expect to continue to incur or expend, substantial accounting and other expenses and significant management time and resources. We have implemented certain remediation activities resulting from our ongoing assessment of internal control over financial reporting. Our future assessment, or the future assessments by our independent registered public accounting firm, may reveal material weaknesses in our internal control. If material weaknesses are identified in the future we would be required to conclude that our internal
26
control over financial reporting is ineffective and we could be subject to sanctions or investigations by the Securities and Exchange Commission, the NASDAQ Stock Market or other regulatory authorities, which would require additional financial and management resources and could adversely affect the market price of our common stock.
We have implemented anti-takeover provisions that could discourage or prevent an acquisition of our company, even if the acquisition would be beneficial to our stockholders, and as a result our management may become entrenched and hard to replace.
Provisions in our certificate of incorporation and bylaws could make it more difficult for a third party to acquire us, even if doing so would benefit our stockholders. These provisions include:
| | • | | allowing our board of directors to elect a director to fill a vacancy created by the expansion of the board of directors; |
| | • | | allowing our board of directors to issue, without stockholder approval, up to 5.5 million shares of preferred stock with terms set by the board of directors; |
| | • | | limiting the ability of holders of our outstanding common stock to call a special meeting of our stockholders; and |
| | • | | preventing stockholders from taking actions by written consent and requiring all stockholder actions to be taken at a meeting of our stockholders. |
Each of these provisions, as well as selected provisions of Delaware law, could discourage potential takeover attempts, could adversely affect the trading price of our securities and could cause our management to become entrenched and hard to replace. In addition to provisions in our charter documents and under Delaware law, an acquisition of our company could be made more difficult by our employee benefits plans and our employee change in control severance plan, under which, in connection with a change in control and termination of employment, stock options and other equity grants held by our employees may become vested and our officers may receive severance benefits. We also have implemented a stockholder rights plan, also called a poison pill, which could make it uneconomical for a third party to acquire us on a hostile basis.
Our executive officers, directors and major stockholders control approximately 39% of our common stock.
As of December 31, 2011, executive officers, directors and holders of approximately 5% or more of our outstanding common stock, in the aggregate, owned or controlled approximately 39% of our outstanding common stock. As a result, these stockholders are able to influence all matters requiring approval by our stockholders, including the election of directors and the approval of corporate transactions. This concentration of ownership may also delay, deter or prevent a change in control of our company and may make some transactions more difficult or impossible to complete without the support of these stockholders.
Substantial future sales of our common stock by us or our existing stockholders or the conversion of our convertible senior notes to common stock could cause the trading price of our common stock to fall.
Sales by us or our existing stockholders of a large number of shares of our common stock in the public market or the perception that additional sales could occur could cause the trading price of our common stock to drop. Likewise, the issuance of shares of common stock upon conversion of our convertible notes or redemption of our convertible notes upon a designated event, or upon additional convertible debt or equity financings or other share issuances by us, including shares issued in connection with potential future strategic alliances, could adversely affect the trading price of our common stock. Our convertible notes are currently convertible into a total of up to 9.4 million shares. In addition, the existence of these notes may encourage short selling of our common stock by market participants.
Significant volatility in the market price for our common stock could expose us to litigation risk.
The market prices for securities of biopharmaceutical and biotechnology companies, including our common stock, have historically been highly volatile, and the market from time to time has experienced significant price and volume fluctuations that are unrelated to the quarterly operating performance of these biopharmaceutical and biotechnology companies. Since January 1, 2010, the high and low sales price of our common stock varied significantly, as shown in the following table:
| | | | | | | | |
| | | High | | | Low | |
Year ending December 31, 2012 | | | | | | | | |
First Quarter (through February 16, 2012) | | $ | 18.35 | | | $ | 10.68 | |
Year ending December 31, 2011 | | | | | | | | |
Fourth Quarter | | $ | 12.23 | | | $ | 8.03 | |
Third Quarter | | $ | 14.60 | | | $ | 9.12 | |
Second Quarter | | $ | 14.28 | | | $ | 11.00 | |
First Quarter | | $ | 16.65 | | | $ | 10.25 | |
Year ending December 31, 2010 | | | | | | | | |
Fourth Quarter | | $ | 21.95 | | | $ | 9.51 | |
Third Quarter | | $ | 22.09 | | | $ | 17.81 | |
Second Quarter | | $ | 24.21 | | | $ | 14.85 | |
First Quarter | | $ | 23.93 | | | $ | 14.13 | |
27
Given the uncertainty of our future funding, whether BYDUREON, BYETTA and SYMLIN will meet our expectations, and the regulatory approval of our other drug candidates, we may continue to experience volatility in our stock price for the foreseeable future. In addition, the following factors may significantly affect the market price of our common stock:
| | • | | our financial results and/or fluctuations in our financial results; |
| | • | | our ability to satisfy our significant financial obligations to Lilly; |
| | • | | safety issues with BYDUREON, BYETTA, SYMLIN or our product candidates; |
| | • | | any requirement to restate financial results due to changing interpretation of the application of accounting principles that would have a significant effect on Amylin’s reported results of operations and financial condition; |
| | • | | progress or set-backs in our development programs, including clinical study results; |
| | • | | determinations by regulatory authorities with respect to our drug candidates; |
| | • | | developments in our relationships with current or future collaborative partners, including any partner for the development and commercialization of exenatide outside the United States; |
| | • | | our ability to successfully execute our commercialization strategies; |
| | • | | developments in our relationships with third-party manufacturers of our products and other parties who provide services to us; |
| | • | | technological innovations or new commercial therapeutic products by us or our competitors; |
| | • | | developments in patent or other proprietary rights; and |
| | • | | governmental policy or regulation, including with respect to pricing and reimbursement. |
Broad market and industry factors also may materially adversely affect the market price of our common stock, regardless of our actual operating performance. Periods of volatility in the market price of our common stock expose us to securities class-action litigation, and we may be the target of such litigation as a result of market price volatility in the future.
| Item 1B. | Unresolved Staff Comments |
None.
Our primary administrative offices and research laboratories are located in San Diego, California. As of December 31, 2011, we had leases for approximately 576,000 square feet of office and laboratory space. Our leases on a majority of these properties expire between 2015 and 2019. We have also entered into an agreement for office space in Washington, D.C.
Our wholly-owned subsidiary, Amylin Ohio LLC, owns two buildings and 44.4 acres of land in West Chester, Ohio. The buildings, once built out for the manufacture of BYDUREON, will have approximately 565,000 square feet of manufacturing and office space.
From time to time in the ordinary course of business, we become involved in various lawsuits, claims and proceedings relating to the conduct of our business, including those pertaining to product liability, patent infringement and employment claims. As of December 31, 2011, we and Lilly were involved in approximately 114 separate product liability cases involving approximately 572 plaintiffs in various courts in the United States. Approximately 70 plaintiffs who previously filed cases have subsequently dismissed their cases or claims without prejudice. Certain of these cases have been brought by individuals who allege they have used BYETTA. They generally seek compensatory and punitive damages for alleged injuries, consisting primarily of pancreatitis and, in some cases, alleged wrongful death. Most of the cases are pending in California
28
state court, where the Judicial Council has granted our petition for a “coordinated proceeding” for all California state court cases alleging harm allegedly as a result of BYETTA use. We also have received notice from plaintiff’s counsel of additional claims by individuals who have not filed suit. While we cannot reasonably predict the outcome of any lawsuit, claim or proceeding, we and Lilly intend to vigorously defend these matters. However, if we are unsuccessful in our defense, these matters could result in a material adverse impact to our financial position and results of operations.
Lilly Lawsuit
On May 13, 2011, we filed a lawsuit against Lilly in the United States District Court for the Southern District of California titledAmylin Pharmaceuticals, Inc. v. Eli Lilly & Company (Case No. 11CV1061 JLS (NLS)) alleging, among other things, that Lilly was engaging in anticompetitive activity and breaching its strategic alliance agreements with us to maximize commercialization of exenatide. In our complaint we alleged that Lilly was engaging in improper, unlawful and anticompetitive conduct in the implementation of its global alliance agreement with Boehringer Ingelheim GmbH, or BI, to jointly develop and commercialize BI’s linagliptin product. In the lawsuit, we sought permanent injunctive relief to prevent Lilly from continuing to use the same sales force to sell both exenatide and BI’s competitive linagliptin. We also sought, among other things, compensatory, punitive and exemplary damages and that Lilly’s actions be adjudged to be (i) in violation of federal and state antitrust and unfair competition laws, (ii) in breach of our strategic alliance agreements with Lilly and (iii) in breach of the covenant of good faith and fair dealing under such strategic alliance agreements.
On November 7, 2011, we entered into a Settlement and Termination Agreement with Lilly which provided for the termination of our collaboration agreement with Lilly and the full and final settlement and resolution of certain outstanding claims by us against Lilly. Under the agreement, we and Lilly have each agreed to dismiss or withdraw all claims in the litigation, with prejudice, with each party bearing its own attorneys’ fees and costs. Each party granted the other party and its agents, employees, collaborators and other similar related parties a release with respect to all claims arising out of, or substantially similar to, the claims asserted in the litigation, the conduct complained of in legal papers leading up to the litigation, the party’s participation in the collaboration or any contractual duty under the collaboration agreement, the use of confidential information, and the research, manufacture, development, marketing and sale of the releasing party’s other products. The parties have also entered into covenants not to sue each other based on the released claims, subject to the right to defend against any action by released parties on such claims.
PART II
| Item 4. | Mine Safety Disclosures. |
Not applicable.
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock is traded on The NASDAQ Global Market under the symbol “AMLN.” The following table sets forth, for the periods indicated, the reported high and low sales price per share of our common stock on The NASDAQ Global Market:
| | | | | | | | |
| | | High | | | Low | |
Year Ended December 31, 2011 | | | | | | | | |
Fourth Quarter | | $ | 12.23 | | | $ | 8.03 | |
Third Quarter | | $ | 14.60 | | | $ | 9.12 | |
Second Quarter | | $ | 14.28 | | | $ | 11.00 | |
First Quarter | | $ | 16.65 | | | $ | 10.25 | |
Year ending December 31, 2010 | | | | | | | | |
Fourth Quarter | | $ | 21.95 | | | $ | 9.51 | |
Third Quarter | | $ | 22.09 | | | $ | 17.81 | |
Second Quarter | | $ | 24.21 | | | $ | 14.85 | |
First Quarter | | $ | 23.93 | | | $ | 14.13 | |
The last reported sale price of our common stock on The NASDAQ Global Market on February 16, 2012 was $18.07. As of February 16, 2012, there were approximately 549 shareholders of record of our common stock.
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain any future earnings for funding operations and, therefore, do not anticipate paying any cash dividends in the foreseeable future.
For information concerning prior stockholder approval of and other matters relating to our equity incentive plans, see “Equity Compensation Plan Information” under Item 12 in this annual report on Form 10-K.
29
PERFORMANCE MEASUREMENT COMPARISON
The material in this section is not “soliciting material,” is not deemed “filed” with the Securities and Exchange Commission, and is not to be incorporated by reference into any filing of Amylin under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended.
The following graph compares total stockholder returns of Amylin for the past five years to two indices: the NASDAQ Composite Total Return Index, or the NASDAQ-Composite, and the NASDAQ Pharmaceutical Index, or the NASDAQ- Pharmaceutical. The total return for our common stock and for each index assumes the reinvestment of dividends, although dividends have never been declared on our common stock, and is based on the returns of the component companies weighted according to their capitalizations as of the end of each monthly period. The NASDAQ-Composite tracks the aggregate price performance of equity securities of companies traded on the NASDAQ National Market. The NASDAQ- Pharmaceutical tracks the aggregate price performance of equity securities of pharmaceutical companies traded on the NASDAQ National Market.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among Amylin Pharmaceuticals, Inc., The NASDAQ Composite Index
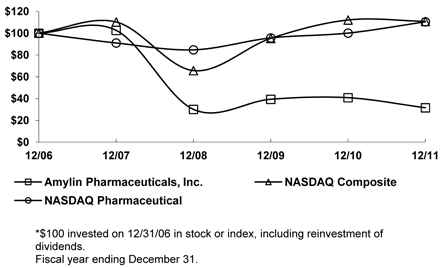
| * | $100 invested on 12/31/06 in stock or index, including reinvestment of dividends. |
Fiscal year ending December 31.
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | 12/06 | | | 12/07 | | | 12/08 | | | 12/09 | | | 12/10 | | | 12/11 | |
Amylin Pharmaceuticals, Inc. | | | 100.00 | | | | 102.58 | | | | 30.08 | | | | 39.34 | | | | 40.78 | | | | 31.55 | |
NASDAQ Composite | | | 100.00 | | | | 110.26 | | | | 65.65 | | | | 95.19 | | | | 112.10 | | | | 110.81 | |
NASDAQ Pharmaceutical | | | 100.00 | | | | 90.99 | | | | 84.71 | | | | 95.64 | | | | 100.10 | | | | 110.44 | |
30
| Item 6. | Selected Financial Data |
Please read the following selected financial data in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the Consolidated Financial Statements and related notes included elsewhere in this annual report on Form 10-K.
| | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31 | |
| | | 2011 | | | 2010 | | | 2009 | | | 2008 | | | 2007 | |
| | | (in thousands, except for per share amounts) | |
Consolidated Statements of Operations Data: | | | | | | | | | | | | | | | | | | | | |
Net product sales | | $ | 621,570 | | | $ | 651,113 | | | $ | 753,993 | | | $ | 765,342 | | | $ | 701,450 | |
Revenues under collaborative agreements | | | 29,108 | | | | 17,700 | | | | 4,426 | | | | 4,286 | | | | 19,286 | |
| | | | | | | | | | | | | | | | | | | | |
Total revenues | | | 650,678 | | | | 668,813 | | | | 758,419 | | | | 769,628 | | | | 720,736 | |
Costs and expenses: | | | | | | | | | | | | | | | | | | | | |
Cost of goods sold | | | 48,376 | | | | 61,687 | | | | 82,999 | | | | 91,596 | | | | 65,457 | |
Selling, general and administrative(1) | | | 271,224 | | | | 276,879 | | | | 330,486 | | | | 380,294 | | | | 376,064 | |
Research and development(1) | | | 161,215 | | | | 183,083 | | | | 198,440 | | | | 237,432 | | | | 231,257 | |
Collaborative profit-sharing | | | 222,545 | | | | 257,127 | | | | 302,861 | | | | 302,600 | | | | 290,934 | |
Net costs associated with reacquisition of economic interest in exenatide products(2) | | | 431,587 | | | | — | | | | — | | | | — | | | | — | |
Restructuring | | | 7,190 | | | | 16,780 | | | | 16,980 | | | | 54,926 | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Total costs and expenses | | | 1,142,137 | | | | 795,556 | | | | 931,766 | | | | 1,066,848 | | | | 963,712 | |
Interest and other (expense) income, net | | | (51,940 | ) | | | (25,372 | ) | | | (11,532 | ) | | | (9,778 | ) | | | 26,490 | |
Loss on impairment of investments | | | — | | | | (198 | ) | | | (1,377 | ) | | | (14,943 | ) | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Net loss | | | (543,399 | ) | | | (152,313 | ) | | | (186,256 | ) | | | (321,941 | ) | | | (216,486 | ) |
| | | | | | | | | | | | | | | | | | | | |
Net loss per share—basic and diluted | | $ | (3.73 | ) | | $ | (1.06 | ) | | $ | (1.32 | ) | | $ | (2.35 | ) | | $ | (1.63 | ) |
| | | | | | | | | | | | | | | | | | | | |
Shares used in calculating net loss per share—basic and diluted | | | 145,730 | | | | 143,525 | | | | 140,702 | | | | 137,006 | | | | 132,621 | |
Consolidated Balance Sheets Data: | | | | | | | | | | | | | | | | | | | | |
Cash, cash equivalents and short-term investments | | $ | 204,065 | | | $ | 442,663 | | | $ | 667,769 | | | $ | 816,838 | | | $ | 1,130,415 | |
Working capital | | $ | 125,173 | | | $ | 274,800 | | | $ | 541,313 | | | $ | 722,290 | | | $ | 1,049,024 | |
Total assets | | $ | 1,870,199 | | | $ | 1,531,429 | | | $ | 1,726,419 | | | $ | 1,727,053 | | | $ | 1,774,430 | |
Long-term obligations, excluding current portion | | $ | 1,712,927 | | | $ | 786,351 | | | $ | 938,516 | | | $ | 893,998 | | | $ | 759,388 | |
Accumulated deficit | | $ | (2,643,579 | ) | | $ | (2,100,180 | ) | | $ | (1,947,867 | ) | | $ | (1,761,611 | ) | | $ | (1,439,670 | ) |
Total stockholders’ equity (deficit) | | $ | (138,745 | ) | | $ | 343,483 | | | $ | 422,534 | | | $ | 519,277 | | | $ | 727,757 | |
| (1) | The selected financial data presented herein have been revised to conform to the current presentation. Specifically, certain administrative costs are now being allocated to research and development expense. |
| (2) | The net costs associated with the reacquisition of the economic interest in exenatide products relates to the November 7, 2011 Termination Agreement with Lilly. See Note 2 in the accompanying Consolidated Financial Statements. |
31
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
Overview
We are a biopharmaceutical company committed to improving the lives of people with diabetes and other metabolic diseases through the discovery, development and commercialization of innovative medicines. We are marketing two first-in-class medicines to treat diabetes, BYETTA® (exenatide) injection and SYMLIN® (pramlintide acetate) injection. We are also marketing the first and only once-weekly diabetes treatment, BYDUREON™ (exenatide extended-release for injectable suspension). BYDUREON is an extended-release medication for type 2 diabetes that provides continuous glycemic control in a once-weekly dose.
In 2011 and early 2012, we executed on key initiatives for the company, including:
| | • | | Obtaining approval for BYDUREON in the United States in January 2012 and the European Union in June 2011. BYDUREON is currently approved by regulatory authorities in 33 countries and is being marketed in 12 countries worldwide in which we have also received pricing and reimbursement decisions; |
| | • | | Regaining 100% of the global rights to develop and commercialize our exenatide franchise, including BYDUREON and BYETTA, from Eli Lilly and Company and entering into discussions with potential new partners with global capabilities to develop and commercialize exenatide outside the United States; |
| | • | | Obtaining approval in the United States of BYETTA for use with insulin glargine. BYETTA is currently the only short-acting glucagon-like peptide-1, or GLP-1, receptor agonist approved in the United States for use as an add-on therapy to insulin glargine in patients with type 2 diabetes; and |
| | • | | Continuing to operate our business on an operating cash flow positive basis since the beginning of 2010. |
In 2012, we will continue our BYDUREON launch efforts and will work toward securing a development and commercialization partner with global capabilities for our exenatide franchise. We also plan to advance our metreleptin program for the treatment of lipodystophy, a very rare metabolic condition, and to advance our once-weekly and once-monthly exenatide suspension programs. We will continue our efforts to develop and obtain approval for the BYDUREON pen with the goal of making the BYDUREON pen delivery system available to patients in late 2012 or early 2013. In the near term, we will also focus on maximizing the financial contributions from BYETTA and SYMLIN and continue to operate our business with financial discipline.
BYDUREON is the first and only once-weekly diabetes treatment approved for use in the United States and European Union and other parts of the world. It is in a class of compounds called GLP-1 receptor agonists and is indicated for use in the United States as an adjunct to diet and exercise to improve glycemic control in adult patients with type 2 diabetes. BYDUREON combines exenatide, the active ingredient in BYETTA, with proprietary technology developed by us and our partner, Alkermes, Inc., or Alkermes, to provide a sustained release delivery of exenatide. The combination of potency and the glucose-dependent mechanism of action inherent in exenatide makes it well suited for use as a once weekly formulation. Common side effects with BYDUREON include nausea, which most commonly happens when first starting BYDUREON but may become less prevalent over time, headache, itching at the injection site and indigestion. BYDUREON is currently approved in 33 countries and has been launched in 12 countries worldwide, including the United States.
BYETTA is the first approved medicine in the GLP-1 receptor agonist class of compounds. It is approved as a first-line, stand-alone medication (monotherapy) along with diet and exercise to improve glycemic control in adults with type 2 diabetes. BYETTA is also approved as an adjunctive therapy to improve glycemic control in patients with type 2 diabetes who have not achieved adequate glycemic control by using metformin, a sulfonylurea and/or a thiazolidinedione, orTZD, three common oral therapies for type 2 diabetes. Further, the FDA recently approved an expanded use of BYETTA as an add-on for patients who have not achieved adequate glycemic control with insulin glargine. The type 2 diabetes treatment guidelines of the American Diabetes Association, or ADA, the European Association for the Study of Diabetes, or EASD, and the American Association of Clinical Endocrinologists, or AACE, include the GLP-1 receptor agonist class, which includes BYETTA, as a secondary treatment option for type 2 diabetes patients. By the end of 2011, BYETTA was approved in 87 countries and launched in approximately 80 countries worldwide. Net product sales of BYETTA were $517.7 million in 2011, $559.3 million in 2010 and $667.6 million in 2009.
We have an agreement with Eli Lilly and Company, or Lilly, that provides for the transition of development and commercialization of activities for exenatide outside the United States no later than December 31, 2013. We are currently in discussions with various companies with the goal of transitioning these exenatide responsibilities to one or more partners prior to such date.
32
SYMLIN is the first and only approved medicine in a class of compounds called amylinomimetics. We began selling SYMLIN in the United States in April 2005 as adjunctive therapy to mealtime insulin to treat diabetes. Other than insulin and insulin analogues, SYMLIN is the first FDA-approved medication addressing glucose control for patients with type 1 diabetes since the discovery of insulin approximately 90 years ago. We own 100% of the global rights to SYMLIN which had net product sales of $103.9 million in 2011, $91.8 million in 2010 and $86.4 million in 2009.
In January 2012, we created two separate commercial teams focused on two distinct aspects of our commercial portfolio. One commercial team is focused on the commercialization of our exenatide franchise while the other is focused on the commercialization of our products that target specialty and orphan diseases. The exenatide commercial team will consist of [650] diabetes sales specialists who will target those physicians that account for more than 60% of all branded diabetes prescriptions, including approximately 90% of all GLP-1 prescriptions.
The specialty and orphan disease commercial team consists of 65 diabetes sales specialists who will initially focus on the commercialization of SYMLIN by promoting SYMYLIN to those physicians who write mealtime insulin prescriptions. In the near term, the specialty and orphan commercial team will also be dedicated to establishing the short-acting GLP-1 market for BYETTA for use with insulin glargine and will provide the infrastructure for the launch of metreleptin if we are successful in our efforts to receive approval for metreleptin as a treatment for rare forms of lipodystrophy.
Settlement and Termination of Lilly Exenatide Collaboration
On November 7, 2011, we and Lilly entered into a Settlement and Termination Agreement, or the Termination Agreement, which provides for, among other things, the termination of the parties’ Collaboration Agreement and the full and final settlement and resolution of certain outstanding claims asserted by us against Lilly in the lawsuit we filed in May 2011. Under the terms of the Termination Agreement, we obtained the exclusive right to develop and commercialize exenatide products in the United States. We also obtained exclusive rights to develop and commercialize exenatide globally outside the United States subject to a transition period in which Lilly will continue to have exclusive rights to commercialize exenatide products outside the United States until no later than December 31, 2013.
The Termination Agreement provides that either party may deliver a notice to the other party with respect to a particular country or countries, or an OUS Country, specifying an earlier transfer date for that country. Under the terms of the Termination Agreement, we are permitted to deliver a notice to transition an OUS Country at any time after (i) June 30, 2012 with respect to an OUS Country where Lilly has launched BYDUREON prior to March 31, 2012, or (ii) March 31, 2012 with respect to any other OUS Countries, provided that with respect to Europe, we may deliver a transition notice on or after April 1, 2012. Lilly is permitted to deliver a notice to transition an OUS Country any time on or after September 30, 2012. The transition of any OUS Country(ies) set forth in a notice will be effective 180 days after the notice date. After the transition of an OUS Country, we, or our designee, will have exclusive rights to commercialize exenatide products in that country. In the event the OUS transition does not occur by December 31, 2013, the agreement contains various provisions with respect to future rights and obligations of each party that extend beyond December 31, 2013.
Under the terms of the Termination Agreement, we paid Lilly an upfront payment of $250 million and have agreed to pay to Lilly a milestone payment of $150 million, payable upon approval by the FDA of a monthly exenatide suspension product. In addition, we will make quarterly payments to Lilly pursuant to a revenue sharing obligation (as described below), or the Revenue Sharing Obligation, based on sales of exenatide products and certain payments received by us from third parties.
We are required to make quarterly payments to Lilly equal to (i) 15% of net sales of exenatide products by us or any sales partners, subject to minimum guarantees in each of 2012 and 2013, and (ii) 20% of any consideration, including upfront or milestone payments, received by us from our sales partners for the grant to such sales partners of certain rights relating to exenatide products up to $1.2 billion, in the aggregate plus any interest that is accrued and compounded as follows. The Revenue Sharing Obligation will continue until the earliest to occur of (x) the date that we have paid to Lilly an amount equal to $1.2 billion, plus any interest that is accrued and compounded, (y) December 31, 2036, and (z) termination of the Revenue Sharing Obligation in accordance with the terms of the Termination Agreement. Interest will accrue on the outstanding balance of the Revenue Sharing Obligation at a rate of 2.295% quarterly. Interest will not be payable in cash, but will be added on the last day of each calendar quarter to the outstanding balance of the Revenue Sharing Obligation. We may delay payments on the Revenue Sharing Obligation for the first two quarters of 2012, however, interest will accrue and compound on any payments so delayed. Simultaneously with the execution of the Termination Agreement, we entered into a promissory note with Lilly in the initial principal amount of $1.2 billion, secured by certain of our assets and those of our subsidiaries and guaranteed by certain of our subsidiaries.
33
On November 7, 2011, in connection with executing the Termination Agreement, we and Lilly amended and restated our loan agreement pursuant to which Lilly previously made a $165 million unsecured loan to us. The amended and restated loan agreement extends the maturity date of our outstanding obligations made under the original loan agreement to June 30, 2016. We and Lilly also amended and restated our Exenatide Once Weekly Supply Agreement, pursuant to which we will supply commercial quantities of fixed-dose injection of exenatide administered once weekly (including related components) in accordance with a fixed pricing schedule to Lilly for sale in jurisdictions outside the United States until the transition of operations outside the United States to us as contemplated by the Termination Agreement. We and Lilly also amended our Device Development and Manufacturing Agreement pursuant to which Lilly will manufacture and supply to us a mechanical injection pen for daily use for sale in and outside the United States and a once weekly exenatide fixed-dose injectable in finished product form and all components and associated packaging for sale outside the United States in accordance with a fixed pricing schedule. These amended and restated supply agreements will expire no later than December 31, 2013. Amylin and Lilly are parties to a number of other agreements that have been entered into in connection with the Termination Agreement.
In October 2009, we entered into a worldwide exclusive license, development and commercialization agreement with Takeda to co-develop and commercialize pharmaceutical products for the treatment of obesity and related indications. The agreement includes products to be developed from our pipeline and additional compounds from our and Takeda’s obesity research programs. In August 2011, we and Takeda announced that we discontinued the development of pramlintide/metreleptin for the treatment of obesity and will continue to discuss the potential of other assets as candidates for the treatment of obesity and related indications under the terms of our collaboration agreement.
In December 2010, we submitted to the FDA the clinical and non-clinical sections of a rolling BLA for the use of metreleptin to treat diabetes and/or hypertriglyceridemia in pediatric and adult patients with inherited or acquired lipodystrophy. We plan to submit the chemistry, manufacturing and controls section of the BLA to the FDA in the first half of 2012. Because lipodystrophy affects a very small number of patients, estimated to be 1,000 patients in the United States and 3,000 patients world-wide, metreleptin for the treatment for lipodystrophy has received orphan drug designation by the FDA. Upon completion of the BLA submission, we plan to apply for fast-track and priority-review designation, which, if granted, could translate to a PDUFA action date by the end of 2012. We also plan to continue working with European regulatory agencies to gain orphan drug designation for metreleptin for the treatment for lipodystrophy in Europe.
We maintain a research and early development program focused on novel peptide and protein therapeutics. We have also entered into strategic alliances and business initiatives, including our strategic relationship with Biocon, Limited, or Biocon, to develop pharmaceutical products, including AC165198, which was developed from our phybrid technology platform. In collaboration with Biocon, we submitted an investigational new drug application, or IND, at the end of 2011 and commenced a phase 1 study for AC165198 in early 2012.
Since our inception in September 1987, we have devoted substantially all of our resources to our research and development programs and, more recently, to the commercialization of our products. All of our revenues prior to 2005 were derived from milestones and amortization of up-front payments under our exenatide collaboration agreement with Lilly, previous SYMLIN collaborative agreements, and previous co-promotion agreements. During 2005, we began to derive revenues from product sales of BYETTA and SYMLIN. At December 31, 2011, our accumulated deficit was approximately $2.6 billion.
At December 31, 2011, we had $204.1 million in cash, cash equivalents and short-term investments and $10.5 million of restricted cash. Although we have yet to consistently generate positive operating cash flows, we intend to continue to tightly manage our cash in 2012 while investing in working capital to support the launch of BYDUREON. Refer to the discussions under the headings “Liquidity and Capital Resources” below and “Cautionary Factors That May Affect Future Results” in Part I, Item 1A for further discussion regarding our anticipated future capital requirements.
34
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or US GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues, expenses and related disclosures of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates, including those related to revenue recognition, stock-based compensation, inventory costs, research and development expenses and income taxes. We base our estimates on historical experience and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe the following critical accounting policies affect the significant judgments and estimates used in the preparation of our consolidated financial statements (see Note 1 to our consolidated financial statements on page F-6).
Revenue Recognition
We recognize revenue from the sale of our products, license fees and milestones as earned.
Net Product Sales
We sell BYETTA and SYMLIN primarily to wholesale distributors, who in turn, sell to retail pharmacies and government entities. Decisions made by these wholesalers and their customers regarding the levels of inventory they hold, and thus the amount of BYETTA and SYMLIN they purchase, may materially affect the level of our product sales in any particular period. We recognize revenue from the sale of our products when delivery has occurred, title has transferred to the customer, the selling price is fixed or determinable, collectability is reasonably assured and we have no further obligations. We record product sales net of allowances for product returns, rebates and wholesaler chargebacks, wholesaler discounts and prescription vouchers. We are required to make significant judgments and estimates in determining some of these allowances. If actual results differ from our estimates, we will be required to make adjustments to these allowances in the future.
Product Returns
We do not offer our wholesale customers a general right of return. However, we will accept returns of products that are damaged or defective when received by the wholesale customer or for any unopened product during the period beginning six months prior to and up to 12 months subsequent to its expiration date. Product returned is generally not resalable as our products require refrigeration. We refund the sales price for product returns in cash or credit to our customers. We estimate product returns based on our historical returns experience and record an allowance for estimated returns at the time of sale. Additionally, we consider several other factors in our estimation process including our internal sales forecasts, the expiration dates of product shipped and third party data to assist us in monitoring estimated channel inventory levels and prescription trends. Actual returns could exceed our historical experience and our estimates of expected future returns due to factors such as wholesaler and retailer stocking patterns and inventory levels and/or competitive changes. To date actual returns have not differed materially from our estimates.
Rebates and Wholesaler Chargebacks
Allowances for rebates include mandated discounts under the Medicaid Drug Rebate Program and contracted discounts with commercial payors. Rebates are amounts owed after the final dispensing of the product by a pharmacy to a benefit plan participant and are based upon contractual agreements or legal requirements with private sector and public sector (e.g. Medicaid) benefit providers. The allowance for rebates is based on contractual discount rates, expected utilization under each contract and our estimate of the amount of inventory in the distribution channel that will become subject to such rebates. Our estimates for expected utilization for rebates are based on historical rebate claims and to a lesser extent third party market research data. Rebates are generally invoiced and paid quarterly in arrears so that our accrual consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual for prior quarters’ unpaid rebates and an accrual for inventory in the distribution channel.
Wholesaler chargebacks are discounts that occur when contracted customers purchase directly from an intermediary wholesale purchaser. Contracted customers, which currently consist primarily of Federal government entities purchasing off the Federal Supply Schedule, generally purchase the product at its contracted price, plus a mark-up from the wholesaler. The wholesaler, in-turn, charges back to us the difference between the price initially paid by the wholesaler and the contracted
35
price paid to the wholesaler by the customer. The allowance for wholesaler chargebacks is based on expected utilization of these programs and reported wholesaler inventory levels. Actual rebates and wholesaler chargebacks could exceed historical experience and our estimates of future participation in these programs. To date, actual rebate claims and wholesaler chargebacks have not differed materially from our estimates.
Wholesaler Discounts
Wholesaler discounts consist of prompt payment discounts and distribution service fees. We offer all of our wholesale customers a 2% prompt-pay discount within the first 30 days after the date of the invoice. Distribution service fees arise from contractual agreements with certain of our wholesale customers for distribution services they provide to us and are generally a fixed percentage of their purchases of our products in a given period. Prompt payment discounts and distribution service fees are recorded as a reduction to gross sales in the period the sales occur. The allowance for wholesaler discounts is based upon actual data of product sales to wholesale customers and not on estimates.
Prescription Vouchers
Prescription vouchers result in amounts owed to pharmacies that have redeemed vouchers for a free prescription. We provide prescription vouchers to physicians, who in turn distribute them to patients. Patients may redeem a voucher at a pharmacy for a free prescription. We reimburse the pharmacy for the price it paid the wholesaler for the medicine and record this reimbursement as a reduction to gross sales. The allowance for prescription vouchers is based on the number of unredeemed vouchers in circulation, and the estimated utilization rate. The estimated utilization rate is based on our historical utilization rates experience with prescription vouchers. The allowance for prescription vouchers could exceed historical experience and our estimates of future utilization rates. To date, actual prescription voucher utilization has not differed materially from our estimates.
Revenues Under Collaborative Agreements
Revenues under collaborative agreements consist of the amortization of product and technology license fees and milestone payments earned. Upfront product and technology license fees under multiple-element arrangements are deferred and recognized over the period of such services or performance if such arrangements require on-going services or performance. Non-refundable amounts received for substantive milestones are recognized upon achievement of the milestone. Any amounts received prior to satisfying our revenue recognition criteria are recorded as deferred revenue in the accompanying consolidated balance sheets.
Valuation of Stock-Based Compensation
We account for stock-based compensation to employees in accordance with the fair value method of accounting for stock-based compensation arrangements which requires us to expense the estimated fair value of non-cash, stock-based payments to employees.
We estimate the fair value of stock-based payments to employees using the Black-Scholes-Merton model. This estimate is affected by our stock price as well as assumptions regarding a number of inputs that require us to make significant estimates and judgments. These inputs include the expected volatility of our stock price, the expected term of employee stock options, the risk-free interest rate, expected dividends and expected forfeiture rate.
We estimate volatility based upon the historical volatility of our common stock for a period corresponding to the expected term of our employee stock options and the implied volatility of market-traded options on our common stock with various maturities between six months and two years. The determination to use implied volatility in addition to historical volatility was based upon the availability of data related to actively traded options on our common stock and our assessment that the addition of implied volatility is more representative of future stock price trends than historical volatility alone.
The expected life of our employee stock options represents the weighted-average period of time that options granted are expected to be outstanding in consideration of historical exercise patterns and the assumption that all outstanding options will be exercised at the mid-point of the then current date and their maximum contractual term.
The risk-free interest rates are based on the yield curve of United States Treasury strip securities in effect at the time of grant for periods corresponding with the expected life of our employee stock options. We have never paid dividends and do not anticipate doing so for the foreseeable future. Accordingly, we have assumed no dividend yield for purposes of estimating the fair value of our stock-based payments to employees.
36
Stock-based compensation expense recognized is based on awards ultimately expected to vest, and therefore is reduced by expected forfeitures. We estimate forfeitures based upon historical forfeiture rates, and will adjust our estimate of forfeitures if actual forfeitures differ, or are expected to differ, from such estimates. Changes in estimated forfeitures will be recognized through a cumulative adjustment in the period of the change and will also impact the amount of stock-based compensation expense in future periods.
If factors underlying the above assumptions change in future periods, the associated estimated non-cash, stock-based compensation expense that we record may differ significantly from what we have recorded in the current period.
Research and Development Expenses
Research and development costs are expensed as incurred and include: salaries, benefits, bonus, stock-based compensation, license fees, milestone payments due under license agreements, costs paid to third-party contractors to perform research, conduct clinical trials, and develop drug materials and delivery devices; and associated overhead and facilities costs. Reimbursed research and development costs under collaborative arrangements are recorded as a reduction to research and development expenses and are recognized in the period in which the related costs are incurred. Clinical trial costs are a significant component of research and development expenses and include costs associated with third-party contractors. Invoicing from third-party contractors for services performed can lag several months. We accrue the costs of services rendered in connection with third-party contractor activities based on our estimate of management fees, site management and monitoring costs and data management costs. Differences between actual clinical trial costs from estimated clinical trial costs have historically not been material and are adjusted for in the period in which they become known.
Income Taxes
We have net deferred tax assets relating primarily to net operating loss carryforwards and research and development tax credit carryforwards. Subject to certain limitations, these deferred tax assets may be used to offset taxable income in future periods. Since we have been unprofitable since inception and the likelihood of future profitability is not assured, we have established a valuation allowance for most of these net deferred tax assets in our consolidated balance sheets at December 31, 2011 and 2010. If we determine that we are able to realize a portion or all of these deferred tax assets in the future, we will record an adjustment to increase their recorded value and a corresponding adjustment to increase income or additional paid in capital, as appropriate, in that same period.
We recognize the impact of a tax position in our financial statements only if it is more likely than not that the tax position will be sustained upon examination by taxing authorities, based on the technical merits of the position. We provide estimates for unrecognized tax benefits which relate primarily to issues common among corporations in our industry. We apply a variety of methodologies in making these estimates which include advice from industry and subject experts, evaluation of public actions taken by the Internal Revenue Service and other taxing authorities, as well as our own industry experience. If our estimates are not representative of actual outcomes, our results could be materially impacted.
Inventories and Related Reserves
Inventories consist of raw materials, work-in-process and finished goods for SYMLIN and BYETTA and pre-approval inventories for BYDUREON. We maintain inventory reserves primarily for production failures and potential product expiration. The manufacturing processes for our products are complex. Deviations in the manufacturing process may result in production failures and additional inventory reserves. Obsolete inventory due to expiration may also result in additional inventory reserves. In estimating inventory obsolescence reserves, we analyze the shelf life, expiration dates and internal sales forecasts, each on a product-by-product basis.
We expense costs relating to the purchase and production of pre-approval inventories for which the sole use is pre-approval products as research and development expense in the period incurred until such time as we believe future commercialization is probable and future economic benefit is expected to be recognized. With respect to capitalization of unapproved product candidates, we produce inventory in preparation for the launch of the product and in amounts sufficient to support forecasted initial market demand. Typically, capitalization of such inventory does not begin until the product candidate is considered to have a high probability of regulatory approval. This generally will occur only after we have submitted an NDA to the FDA, and only if our assessment of the status of the regulatory review has led us to conclude there is a high probability of receiving regulatory approval. If we are aware of any specific risks or contingencies that are likely to impact the regulatory approval process or if there are any specific issues identified during our research and development process relating to safety, efficacy or manufacturing of the product candidate, we would not capitalize the related inventory.
37
We manage the levels of inventory at each stage of the manufacturing process to optimize the shelf life of the inventory and avoid product expiration issues relative to anticipated market demand following launch. On a quarterly basis, we evaluate all inventory, including inventory capitalized for which regulatory approval has not yet been obtained, to determine if any lower of cost or market adjustment is required. As our evaluation relates to pre-approval inventory, we consider several factors including expected timing of FDA approval, projected sales volume, expiration dates of the inventory and estimated selling price. Projected sales volume is based on several factors including market research, sales of similar products and competition in the market. Estimated sales price is based on the price of existing products sold for the same indications, market research and expected market demand.
Once we have capitalized inventory for a product candidate that is not yet approved, we will monitor, on a quarterly basis, the status of such candidate within the regulatory approval process. We could be required to expense previously capitalized costs related to pre-approval inventory upon a change in our judgment of future commercialization and future economic benefit expected to be recognized, due to a denial or delay of approval by the FDA, a delay in the timeline for commercialization or other potential factors.
Fair Value Measurements
US GAAP requires us to estimate the fair value of certain assets and liabilities as of the date of their acquisition or incurrence, on an ongoing basis, or both. Determining the fair value of an asset or liability requires the use of accounting estimates and assumptions which are judgmental in nature and could have a significant impact on the determination of the amount of the fair value ascribed to the asset or liability.
Valuation of Economic Interest in Exenatide Products Reacquired
We evaluated whether the Termination Agreement should be accounted for as a single transaction or as a transaction that consists of separate elements relating to the OUS operations and US operations. Because the OUS operations and US operations (1) have independent economic value and substance, (2) could be purchased or sold on an individual basis and (3) qualify as a business combination and the reacquisition of previously shared economic interests through the termination of a contract, respectively, we determined that the OUS operations and US operations should be accounted for as separate elements. The OUS operation constitutes a business, as defined by ASC 805, “Business Combinations”, and the transition of the OUS operations will be accounted for as a business combination when control transfers from Lilly to Amylin. With respect to the contract termination, certain aspects of the US operations represent the reacquisition of a previously shared economic interest that qualify as an asset for developed products and the amounts relating to the unapproved products are expensed. In allocating the consideration to the acquired elements the relative fair value of the rights acquired was used for the allocation for the US reacquired right asset for developed products, the US reacquired rights expensed for unapproved products and also for the OUS business to be acquired.
In connection with the Termination Agreement, we had a termination of a contract which resulted in us obtaining reacquired rights in the economic interest in exenatide products in the US. This economic interest represents (i) Lilly’s former share of the US gross margin for exenatide products, and (ii) our expected increase in the US estimated cash flows for the unapproved products, net of the increased cost of research and marketing. The fair value of each of these items was derived using the multi-period excess earnings discounted cash flow method to value the technology. We used a discount rate of 21% for the economic interest related to BYETTA since it was an approved product with historic revenue streams and 25% for unapproved products reflecting in part the increased uncertainties. In selecting the discount rates, we considered a number of factors, including the industry composite weighted average cost of capital, the internal rate of return of the purchase consideration allocated to the US operations, and the weighted average return on assets, which is a measure of the after-tax returns that would be required by an investor in a particular class of assets, weighted by the value of each asset relative to the total value of all assets purchased. Because the value ascribed to this asset was completed on a relative fair value basis, an increase or decrease of the interest rate by 1% would not have a significant impact on the allocation of the consideration.
We evaluate the recoverability of our long-lived assets including amortizable intangible and tangible assets in accordance with authoritative guidance. When events or changes in circumstances indicate that the carrying amount of long-lived assets may not be recoverable, we recognize such impairment in the event the net book value of such assets exceeds the future undiscounted cash flows attributable to such assets. Our acquired intangible assets with definite useful lives are amortized over the expected economic use of the asset. Our amortization policy reflects the pattern by which the economic benefits of the intangible assets are consumed, and that pattern is reliably determinable, and will be periodically evaluated for impairment.
Valuation of Economic Interest in Exenatide Products to be Reacquired as a Business
In connection with the Termination Agreement, upon completion of the OUS Transition, we will reacquire the economic interest in exenatide products for the OUS markets. This economic interest represents the OUS estimated cash flows for exenatide products, and our expected increase in the OUS estimated cash flows for the unapproved products. The fair value primarily related to the BYETTA, BYDUREON and exenatide products currently under development and were estimated using the multi-period excess earnings discounted cash flow method. We used a discount rate ranging between 15-23% depending on the state of the product or unapproved product to correspond to the related uncertainty with higher discount rates used where the uncertainty is the highest as would be the case with unapproved products. In selecting the discount rate, we considered a number of factors, including the industry composite weighted average cost of capital, the internal rate of return of the purchase consideration allocated to the OUS operations, and the weighted average return on assets, which is a measure of the after-tax returns that would be required by an investor in a particular class of assets, weighted by the value of each asset relative to the total value of all assets purchased. Because the value ascribed to this asset was completed on a relative fair value basis, an increase or decrease of the interest rate by 1% would not have a significant impact on the allocation of the consideration.
Promissory Note Related to Revenue Sharing Obligation
As indicated previously, effective November 7, 2011 Amylin and Lilly entered into the Termination Agreement to terminate their collaboration for exenatide and resolve the outstanding litigation between the companies. Amylin also issued a secured promissory note to Lilly under which Amylin agreed to make future revenue sharing payments to Lilly in an amount equal to 15 percent of global net sales of exenatide products until Amylin has made aggregate payments to Lilly of $1.2 billion plus accrued interest, the Revenue Sharing Obligation, or the RSO. In the event Amylin receives upfront or milestone payments from a third party in connection with an agreement with respect to exenatide products, Amylin is obligated to make payments on the RSO equal to 20% of such upfront or milestone payments. The RSO was valued using the income approach utilizing cash flow analyses projecting expected net sales for exenatide products over the life cycle and was calculated using a discount rate of 14.4%. In determining the selected discount rate, we considered a number of factors, the most significant of which were the RSO stated rate negotiations, third-party market participant expectations on a similar instrument, our ratings on existing debt, the increased level of the borrowing, our ability to pay and various other market considerations.
We believe that the interest rate on this instrument is one of the most sensitive estimate overall in our accounting for the Termination Agreement with Lilly. If we used a rate that was either 1% higher or 1% lower it would have had the impact of changing overall consideration by approximately $45 million dollars and a corresponding change to the value assigned to reacquired rights of which $20 million more or less would have been charged to expense for net costs associated with reacquisition of economic interest in exenatide.
These estimates and assumptions are judgmental in nature and have a significant impact on the determination of the fair value of the RSO as of the transaction date, the amount of the related debt discount and the associated interest expense over the life of the RSO.
Convertible Senior Notes
During 2009, we adopted new authoritative guidance that significantly impacts the accounting for our convertible senior notes issued in June 2007 by requiring us to account separately for the liability and equity components of the notes. The liability component is measured so the effective interest expense associated with the notes reflects the issuer’s borrowing rate at the date of issuance for similar debt instruments without the conversion feature. The difference between the cash proceeds associated with the notes and this estimated fair value is recorded as a debt discount and amortized to interest expense over the life of the notes.
38
Determining the fair value of the liability component requires the use of accounting estimates and assumptions. These estimates and assumptions are judgmental in nature and could have a significant impact on the determination of the liability component and, in effect, the associated interest expense. According to the guidance, the carrying amount of the liability component is determined by measuring the fair value of a similar liability that does not have an associated equity component. If no similar liabilities exist, estimates of fair value are primarily determined using assumptions that market participants would use in pricing the liability component, including market interest rates, credit standing, yield curves and volatilities.
Loss Protection Liability
In connection with the Termination Agreement, Amylin agreed to reimburse Lilly for exenatide-related losses incurred by Lilly OUS during the period of the OUS Transition. The liability was accounted for as a financial instrument and was recorded at the fair value of Amylin’s obligations. The liability was valued using a probability weighted analysis of expected payments and such payments were discounted to present value using a discount rate of 20%. The selection of the discount rate considered a number of factors, including credit spreads for companies with credit rating similar to Amylin, the risks associated with the forecasted losses provided by Lilly, the relatively short-term nature of the OUS Transition Period Loss Protection and the possibility that Amylin could take control of the OUS operations from Lilly sooner than December 31, 2013. Significant inputs to the valuation include the following:
| | • | | financial projections for the OUS markets which were provided to Amylin’s management by Lilly; |
| | • | | a variety of scenarios under which management estimated the extent to which these financial projections would be achieved, and the resulting payouts of the amounts that could be made for the twelve months ended December 31, 2012 and 2013. |
The greatest driver of variability in this liability is the estimated losses that will be subject to the guarantee. This liability is carried at fair value and is remeasured each reporting period.
Recently Issued Accounting Pronouncements
For a discussion of recently issued accounting pronouncements, refer to the section titled “Recently Issued Accounting Standards” within Note 1.Summary of Significant Accounting Policies to our Consolidated Financial Statements.
Results of Operations
Net Product Sales
Net product sales for the years ended December 31, 2011, 2010 and 2009 were $621.6 million, $651.1 million and $754.0 million, respectively, and consisted of sales of BYETTA and SYMLIN, less allowances for product returns, rebates and wholesaler chargebacks, wholesaler discounts, and prescription vouchers.
The following table provides information regarding net product sales (in millions):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
BYETTA | | $ | 517.7 | | | $ | 559.3 | | | $ | 667.6 | |
SYMLIN | | | 103.9 | | | | 91.8 | | | | 86.4 | |
| | | | | | | | | | | | |
| | $ | 621.6 | | | $ | 651.1 | | | $ | 754.0 | |
| | | | | | | | | | | | |
The decrease in net product sales for BYETTA for the year ended December 31, 2011 as compared to the same period in 2010, and for the year ended December 31, 2010 as compared to the same period in 2009, primarily reflects reduced prescription demand due to new entrants to the market, partially offset by higher prices.
The increase in net product sales for SYMLIN for the year ended December 31, 2011 compared to the same period in 2010, and for the year ended December 31, 2010 as compared to the same period in 2009, primarily reflects higher prices, partially offset by reduced prescription demand.
Sales of our products in future periods may be impacted by numerous factors, including potential competition, the success of our commercial launch of BYDUREON in the U.S., regulatory matters, economic factors and other environmental factors.
39
Revenues under Collaborative Agreements
The following table summarizes the components of revenues under collaborative agreements for the years ended December 31, 2011, 2010 and 2009 (in millions):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Amortization of up-front payments | | $ | 10.3 | | | $ | 7.5 | | | $ | 4.4 | |
Recognition of milestone payments | | | 15.0 | | | | 10.2 | | | | — | |
Royalty revenues | | | 3.8 | | | | — | | | | — | |
| | | | | | | | | | | | |
| | $ | 29.1 | | | $ | 17.7 | | | $ | 4.4 | |
| | | | | | | | | | | | |
Amortization of up-front payments for the year ended December 31, 2011 consists of amounts earned pursuant to our obesity collaboration with Takeda and amortization of deferred collaborative revenue associated with an upfront payment we received from Lilly in 2008 in connection with the Exenatide Once Weekly Supply Agreement, or the Supply Agreement. During the third quarter of 2011 we began to perform services under the Supply Agreement, therefore amortization of the deferred collaborative revenue commenced. As described in Note 2 of the notes to our Consolidated Financial Statements, in connection with the Termination Agreement, the deferred collaborative revenue balance was discharged effective November 7, 2011 and there will be no future revenue recognition of related collaborative revenue. For the year ended December 31, 2010, amortization of up-front payments consists entirely of amounts earned pursuant to our obesity collaboration with Takeda, while amortization of up-front payments for the year ended December 31, 2009 consists of amounts earned pursuant to our collaboration with Lilly, for which amortization ended in 2009, and our collaboration with Takeda, for which amortization began in late 2009.
Milestone payments for the year ended December 31, 2011 consist of a $15 million milestone payment received from Lilly in connection with the July 2011 commercial launch of BYDUREON in the European Union. Milestone payments for the year ended December 31, 2010 consisted primarily of a $10 million milestone earned as a result of Lilly’s launch of BYETTA in Japan in late 2010. There were no milestone payments in 2009.
Royalty revenues represent royalties earned on the gross margin of sales of exenatide outside the United States. The cumulative gross margin threshold for sales of exenatide outside the United States was achieved at the end of the first quarter of 2011 therefore we did not record royalties during 2010 or 2009.
In future periods, we expect that revenues under collaborative agreements will consist of continued amortization of the $75 million up-front payment received from Takeda upon signing of our collaboration agreement in 2009. This up-front payment is being amortized ratably over a ten year period representing the estimated development period of the compounds subject to the Takeda collaboration agreement. Additionally, we expect to continue to earn royalties on product sales of BYETTA and BYDUREON outside of the United States until commercialization of all exenatide markets outside the United States is transitioned from Lilly to Amylin, which by contract occurs no later than December 31, 2013. In the event the OUS transition does not occur by December 31, 2013, the agreement contains various provisions with respect to future rights and obligations of each party that extend beyond December 31, 2013.
40
Costs and Expenses
The following table provides information regarding our costs and expenses (in millions):
| | | | | | | | | | | | |
| | | Year ended
December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Cost of Goods Sold | | $ | 48.4 | | | $ | 61.7 | | | $ | 83.0 | |
Gross margin % | | | 92 | % | | | 91 | % | | | 89 | % |
Selling, general and administrative | | $ | 271.2 | | | $ | 276.9 | | | $ | 330.5 | |
Research and development | | $ | 161.2 | | | $ | 183.1 | | | $ | 198.4 | |
Collaborative profit sharing | | $ | 222.5 | | | $ | 257.1 | | | $ | 302.9 | |
Net costs to reacquire economic interest in exenatide products | | $ | 431.6 | | | | — | | | | — | |
Restructuring | | $ | 7.2 | | | $ | 16.8 | | | $ | 17.0 | |
Cost of Goods Sold
Cost of goods sold is comprised primarily of manufacturing costs associated with BYETTA and SYMLIN sales during the period. The gross margin for the year ended December 31, 2011 improved compared to the same period of 2010. The improvement reflects higher net sales prices and lower unit costs driven by operation efficiencies and production volumes. The gross margin for the year ended December 31, 2010 improved compared to the same period of 2009. The improvement in 2010, as compared to 2009, primarily reflects higher net sales prices partially offset by slightly higher per unit costs. Annual fluctuations in gross margins may be influenced by production volumes, product mix, pricing and the level of sales allowances.
Selling, General and Administrative Expenses
The decrease of $5.7 million in selling, general and administrative expense during the year ended December 31, 2011 as compared to the same period of 2010 primarily reflects reduced business infrastructure spending resulting from continued efforts to drive efficiencies in the business, offset by higher expenses due to BYDUREON pre-launch activities. The decrease in 2010 compared to 2009 primarily reflects lower sales force and business infrastructure expenses following a 2009 restructuring of our sales force, and continued efforts to drive efficiencies in our cost structure.
In future periods, we expect that selling, general and administrative expenses will increase as a result of the Termination Agreement since we obtained exclusive rights to commercialize exenatide products in the United States.
Research and Development Expenses
Our research and development costs are comprised of salaries and bonuses, benefits, non-cash stock-based compensation, license fees, milestones under license agreements, costs paid to third-party contractors to perform research, conduct clinical trials, and develop drug materials and delivery devices; and associated overhead expenses and facilities costs. Reimbursed research and development costs under collaborative arrangements are recorded as a reduction to research and development costs. We charge direct internal and external program costs to the respective development programs. We also incur indirect costs that are not allocated to specific programs because such costs benefit multiple development programs and allow us to increase our pharmaceutical development capabilities. These consist primarily of facilities costs and other internal shared resources related to the development and maintenance of systems and processes applicable to all of our programs.
Our research and development efforts are focused on diabetes, obesity and other diseases. We also maintain an active discovery research program. In diabetes, we have three approved products, BYDUREON, BYETTA and SYMLIN. In obesity, we plan to evaluate certain assets with our partner Takeda as potential candidates for the treatment of obesity and related indications under the terms of our collaboration with Takeda. As part of this program, we intend to conduct additional clinical trials of our drug candidates, or combinations of drug candidates.
41
The following table sets forth information regarding our research and development expenses for our major projects for the years ended December 31, 2011, 2010 and 2009 (in millions):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010(1) | | | 2009(1) | |
Diabetes(2) | | $ | 77.8 | | | $ | 96.9 | | | $ | 87.0 | |
Obesity(3) | | | 28.3 | | | | 12.9 | | | | 26.0 | |
Research and early-stage programs | | | 14.9 | | | | 21.2 | | | | 24.8 | |
Indirect costs | | | 40.2 | | | | 52.1 | | | | 60.6 | |
| | | | | | | | | | | | |
| | $ | 161.2 | | | $ | 183.1 | | | $ | 198.4 | |
| | | | | | | | | | | | |
| (1) | Research and development expenses for the years ended December 31, 2010 and 2009 has been revised to conform with the current presentation. |
| (2) | Research and development expenses for our diabetes program consist primarily of costs associated with BYETTA and BYDUREON which are shared by Lilly pursuant to our collaboration agreement. Cost-sharing payments received from Lilly are recorded as a reduction to research and development and were $79.3 million, $72.6 million and $66.6 million for the years ended December 31, 2011, 2010 and 2009. |
| (3) | Research and development expenses for our obesity program include costs associated with our collaboration agreement with Takeda. Cost-sharing payments received from Takeda are recorded as a reduction to research and development and were $11.6 million, $16.7 million and $1.5 million for the years ended December 31, 2011, 2010 and 2009. |
Research and development expenses decreased $21.9 million for the year ended December 31, 2011 for the same period in 2010. The decrease primarily reflects the favorable disposition of certain cost-sharing disputes with Lilly, partially offset by increased spending on our metreleptin lipodystrophy development program.
Research and development expenses decreased $15.3 million for the year ended December 31, 2010 for the same period in 2009. The $15.3 million decrease primarily reflects decreased development expenses for our obesity programs, driven by development expense cost-sharing with Takeda. The decrease in obesity development expenses is partially offset by increased BYDUREON expenses associated with manufacturing readiness, a cardiovascular outcomes study and development activities for the exenatide suspension formulation.
In future periods, we expect that research and development expenses will increase as a result of the Termination Agreement since we obtained exclusive rights to develop exenatide products in the United States.
Collaborative Profit-Sharing
Collaborative profit-sharing was $222.5 million, $257.1 million and $302.9 million for the years ended December 31, 2011, 2010 and 2009, respectively, and consists of Lilly’s 50% share of the gross margin for BYETTA sales in the United States. As further described in Note 2 of the footnotes to our Consolidated Financial Statements, as a result of the Termination Agreement, for the twelve months ended December 31, 2011, collaborative profit sharing reflects approximately eleven months of such sharing with Lilly while the twelve months ended December 31, 2010 and 2009 both reflect a full twelve months of such sharing.
42
Net costs to reacquire economic interest in exenatide products
The net costs to reacquire economic interest in exenatide products relate to the November 7, 2011 Termination Agreement. The costs are comprised of the following (in millions):
| | | | |
| | | Year ended
December 31, | |
| | | 2011 | |
Cost to reacquire US unapproved products | | $ | 461.6 | |
Foregone milestone payments under collaboration agreement and other settled amounts | | | (58.0 | ) |
Transaction costs related to the reacquisition of exenatide product rights | | | 8.3 | |
Loss on fair value adjustment for loss protection liability | | | 15.7 | |
Amortization of intangible asset for US exenatide rights | | | 4.0 | |
| | | | |
| | $ | 431.6 | |
| | | | |
Refer to Note 2 of our Consolidated Financial Statements for a discussion of these costs.
Restructuring
The following table sets forth the components of the restructuring charges recognized for the years ended December 31, 2011, 2010 and 2009 (in millions):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Facility related charges | | $ | 1.8 | | | $ | 5.8 | | | $ | 5.6 | |
Employee separation costs | | | 5.1 | | | | 3.1 | | | | 10.9 | |
Asset impairments | | | 0.3 | | | | 7.4 | | | | — | |
Other restructuring charges | | | — | | | | 0.5 | | | | 0.5 | |
| | | | | | | | | | | | |
| | $ | 7.2 | | | $ | 16.8 | | | $ | 17.0 | |
| | | | | | | | | | | | |
During the year ended December 31, 2011 we reduced our workforce and recorded restructuring charges of $7.2 million consisting of employee separation costs, facilities-related charges, asset impairment charges and other direct incremental charges.
During the year ended December 31, 2010 we reduced our workforce and recorded restructuring charges of $16.8 million consisting of employee separation costs, facilities-related charges, asset impairment charges and other direct incremental charges.
During the year ended December 31, 2009, we announced a restructuring of our sales force to merge our existing primary care and specialty sales forces into a single organization and recorded restructuring charges of $17.0 million consisting of employee separation costs and facilities related charges.
We have substantially completed all of the above activities included in the restructuring plan and all costs associated with the restructurings were incurred during the years ended December 31, 2011, 2010 and 2009.
Interest and Other Expense, net
The following table provides information regarding our interest and other expense, net (in millions):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Interest and other income | | $ | 1.9 | | | $ | 2.7 | | | $ | 7.8 | |
Interest and other expense | | | (27.0 | ) | | | (28.1 | ) | | | (19.3 | ) |
Loss on impairment of investments | | | — | | | | (0.2 | ) | | | (1.4 | ) |
Accretion expense on promissory note related to RSO | | | (11.0 | ) | | | — | | | | — | |
Loss on fair value adjustments | | | (15.8 | ) | | | — | | | | — | |
| | | | | | | | | | | | |
Total interest and other expense, net | | $ | (51.9 | ) | | $ | (25.6 | ) | | $ | (12.9 | ) |
| | | | | | | | | | | | |
43
Interest and other income consist primarily of interest income from investment of cash and investments. The decrease in 2011 compared to 2010 primarily reflects lower average investment balances and lower interest rates in 2011 as compared to 2010. The decrease in 2010 compared to 2009 primarily reflects lower average investment balances and lower interest rates in 2010 compared to 2009.
Interest and other expense consist primarily of interest expense on our long-term debt obligations, exclusive of the promissory note related to the RSO entered into in connection with the Termination Agreement, including amortization of debt discounts. The decrease in 2011 compared to 2010 primarily reflects an increase in interest capitalized on our Ohio manufacturing facility in 2011. The increase in 2010 compared to 2009 primarily reflects a decrease in interest capitalized on our Ohio manufacturing facility in 2010.
We did not recognize any loss on impairment of investments for the year ended December 31, 2011. At December 31, 2011, gross unrealized losses on our short-term investments were $0.1 million, all of which we determined to be temporary. We recognized a loss on impairment of investments of $0.2 million for the year ended December 31, 2010. The loss represents credit-related losses associated with one security in our portfolio and was based upon the amortized cost basis and the observed market prices for the security. At December 31, 2010, gross unrealized losses on our short-term investments were $0.1 million, all of which we determined to be temporary. We recognized a loss on impairment of investments of $1.4 million for the year ended December 31, 2009. The loss represents credit-related losses associated with two securities in our portfolio and was based upon the amortized cost basis and the observed market prices for the securities.
The accretion expense on the promissory note related to the RSO relates to the Termination Agreement, and was entered into effective November 7, 2011 therefore there is no such interest during 2010 and 2009.
The loss fair value adjustment relates to a fair value adjustment recorded on a derivative instrument embedded in the promissory note and provides Amylin with the ability to discharge all or a portion of the obligation under certain circumstances (see Note 2 and Note 8 to the Consolidated Financial Statements for additional information).
Net Loss
Our net loss for the year ended December 31, 2011 was $543.4 million compared to $152.3 million in 2010 and $186.3 million in 2009. The increase in our net loss in 2011 compared to 2010 reflects $431.6 million of costs associated with the reacquisition of the economic interest in exenatide products from Lilly. Excluding the effect to these costs, our net loss for 2011 improved in comparison to 2010 as a result of the decreased expenses discussed above. The decrease in our net loss in 2010 compared to 2009 primarily reflects the decreased expenses discussed above.
We may incur operating losses for the next few years. Our ability to reach profitability in the future will be heavily dependent upon the amount of product sales that we achieve for BYDUREON, BYETTA and SYMLIN. Our ability to achieve profitability in the future will also depend on our ability to control our operating expenses, including costs associated with the commercialization of BYDUREON, expenses associated with the continued commercialization of BYETTA and SYMLIN, and expenses associated with our research and development programs, including our obesity and our early-stage development programs and related support infrastructure. Our operating results may fluctuate from quarter to quarter as a result of differences in the timing of expenses incurred and revenues recognized.
Liquidity and Capital Resources
Since our inception, we have financed our operations primarily through public sales and private placements of our common and preferred stock, debt financings, payments received pursuant to our exenatide collaboration with Lilly and our obesity collaboration with Takeda, reimbursement of SYMLIN development expenses through earlier collaboration agreements, and since the second quarter of 2005, through product sales of BYETTA and SYMLIN.
At December 31, 2011, we had $204.1 million in cash, cash equivalents and short-term investments, compared to $442.7 million at December 31, 2010. We have demonstrated strong financial discipline over the last few years and we are committed to continuing to manage our expenses closely in-line with expected revenues. We will continue to aggressively manage our expenses to minimize the amount of cash we use for operating activities. We have $575 million of convertible debt that matures in 2014 and a $165 million loan facility from Lilly that matures in 2016. Accordingly, we are evaluating opportunities to refinance this existing indebtedness from time to time. If we require additional financing in the future, we cannot assure you that it will be available to us on favorable terms, or at all. Although we have previously been successful in obtaining financing through our debt and equity securities offerings, there can be no assurance that we will be able to do so in the future.
44
Our operating activities provided cash of $88.3 million, used cash of $38.7 million and provided cash of $19.9 million for the years ended December 31, 2011, 2010 and 2009, respectively.
Cash provided by operating activities for the year ended December 31, 2011 was primarily impacted by cash provided due to decreases in accounts receivable and inventories of $9.2 million and $9.2 million, respectively, and increases in accounts payable and accrued liabilities, accrued compensation and deferred collaborative profit sharing of $18.4 million, $27.4 million and $15.5 million, respectively. The decrease in accounts receivable is primarily due to lower product sales. The decrease in inventories largely reflects reductions in raw materials and finished goods inventory due to the timing and volume of production for BYETTA and SYMLIN, offset by increases in BYDUREON inventories. The increase in accounts payable and accrued liabilities primarily relates to the timing of payments . The increase in accrued compensation is due to accruals for bonuses; our management did not pay the corporate bonus for the year ended December 31, 2010. The increase in deferred collaborative profit sharing reflects payments due to us from Lilly for its 60% share of the capital expenditures we have made for the BYDUREON pen device. These sources of cash are partially offset by a use of cash resulting from a decrease in the payable to our former collaborative partner of $12.9 million. The payable to collaborative partner has declined as a result of the termination of our collaboration with Lilly.
Our cash used by our operating activities for the year ended December 31, 2010 includes a net loss of $152.3 million, $186.3 million of which is comprised of non-cash items consisting primarily of stock-based compensation, depreciation and amortization and restructuring charges. Cash provided by operating activities in the year ended December 31, 2009 included an up-front payment of $75.0 million from Takeda in connection with our collaboration with them.
The improvement in operating cash flows for the twelve months ended December 31, 2011 compared to the twelve months ended December 31, 2010 is largely due to improvements in cash flows from working capital changes resulting from our efforts to manage our expenses. As we prepare for the United States commercial launch of BYDUREON we expect our cash requirements for working capital will increase. Working capital changes may fluctuate from quarter to quarter due to timing of inventory and other current asset purchases and the timing of payment of accounts payable, accrued compensation and other current liabilities.
Our investing activities used cash of $126.8 million, provided cash of $159.7 million and used cash of $116.1 million years ended December 31, 2011, 2010 and 2009, respectively. Investing activities in 2011 included cash paid to reacquire the economic interest in exenatide products of $253.0 million. Additionally, investing activities for all three years consisted of purchases and sales of short-term investments and purchases of property, plant and equipment. Purchases of property, plant and equipment decreased to $54.5 million from $91.1 million in 2010, and decreased from $152.1 million in 2009 to $91.1 million in 2010. The decreases in both 2011 and 2010 were as expected and primarily reflect a reduction of costs associated with our manufacturing facility for BYDUREON, offset by costs incurred in connection with the BYDUREON pen device. Through December 31, 2011, we have incurred $216.4 million in capital expenditures associated with the BYDUREON pen device and incurred total combined capital expenditures for the manufacturing facility and the pen device of $859.2 million. We have billed Lilly $103.4 million for its share of expenditures for the pen device, all of which has been received to date, $16.8 million of which was received during 2011 and is included in cash used for operating activities as discussed above. We expect that our capital expenditures will continue to decrease in 2012 and will be principally focused on strategically investing in exenatide life cycle management, We will continue to evaluate potential additional investments in our Ohio manufacturing facility during the product life cycle for BYDUREON.
Financing activities used cash of $26.2 million, $77.3 million and $20.3 million in the years ended December 31, 2011, 2010 and 2009, respectively. Financing activities in 2011 include the repayment of our $200 million of 2.5% convertible senior notes. In October 2008, we entered into a loan agreement with Lilly pursuant to which Lilly made available to us a $165 million unsecured line of credit, and in May 2011 we drew $165 million on this line of credit, or the Lilly Loan. On November 7, 201, in connection with the Termination Agreement, we amended and restated the agreement and extended the maturity to June 30, 2016. Additionally, during the year ended December 31, 2011, we received proceeds from the exercise of stock options and proceeds from employee stock purchase plan of $8.8 million. Financing activities in 2010 include $93.8 million in scheduled repayments of our note payable, which was paid in full during the year, offset by proceeds from the exercise of stock options and proceeds from employee stock purchase plan. Financing activities in 2009 include $31.3 million in repayments of our note payable partially offset by proceeds from the exercise of stock options and proceeds from our employee stock purchase plan.
At December 31, 2011, our outstanding debt includes $575 million of the 2007 Notes due June 15, 2014. The 2007 Notes are currently convertible into a total of up to 9.4 million shares of our common stock at approximately $61.07 per share and are not redeemable at our option. Additionally, the $165 million Lilly Loan is due June 30, 2016 and the $1.2 billion related to the promissory note related to the RSO matures December 31, 2036.
45
As of December 31, 2011 we had $10.3 million of letters of credit outstanding which are secured by $10.5 million of restricted cash pursuant to a Letter of Credit and Cash Collateral Agreement entered into in December 2011.
Use of Non-GAAP Financial Measures and Reconciliations to GAAP Results
We use certain non-GAAP financial measures, which exclude stock-based compensation, amortization of intangible assets, restructuring and acquisition-related charges and infrequently occurring gains and losses in our calculation of operating income or loss. This non-GAAP financial measure is intended to approximate our operating cash flows before working capital changes and should be considered in addition to, not as a substitute for, measures of our financial performance or liquidity prepared in accordance with GAAP. Our non-GAAP financial measures may be defined differently from time to time and may be defined differently than similar terms used by other companies, and accordingly, care should be exercised in understanding how we define our non-GAAP financial measures.
Our management uses the non-GAAP financial measures to gain an understanding of the Company’s comparative operating performance (when comparing such results with previous periods) and future prospects and excludes these items from its internal financial statements for purposes of its internal budgets and financial goals. These non-GAAP financial measures are used by our management in their financial and operating decision-making because management believes they reflect the Company’s ongoing business in a manner that allows meaningful period-to-period comparisons. Our management believes that these non-GAAP financial measures provide useful information to investors and others (a) in understanding and evaluating our current operating performance and future prospects in the same manner as management does, if they so choose, and (b) in comparing in a consistent manner our current financial results with our past financial results.
Our non-GAAP operating income for the year ended December 31, 2011 was $25.7 million compared to non-GAAP operating loss of $4.4 million and $58.7 million for the same periods in 2010 and 2009, respectively.
A reconciliation of reported GAAP operating net loss to non-GAAP operating income (loss) excluding non-cash items is provided in the tables that follow (in thousands, unaudited):
| | | | | | | | | | | | |
| | | Twelve months ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
GAAP operating loss | | $ | (491,459 | ) | | $ | (126,743 | ) | | $ | (173,347 | ) |
| | | |
Revenue sharing obligation note payments required on net BYETTA sales | | | (6,088 | ) | | | — | | | | — | |
Stock-based compensation | | | 32,472 | | | | 36,022 | | | | 43,762 | |
Other non-cash compensation | | | 16,527 | | | | 19,735 | | | | 20,161 | |
Depreciation and amortization | | | 47,373 | | | | 57,335 | | | | 38,198 | |
Amortization of deferred revenue and other credits | | | (11,930 | ) | | | (7,500 | ) | | | (4,426 | ) |
Net costs to reacquire economic interest in exenatide products | | | 431,587 | | | | — | | | | — | |
Restructuring | | | 7,190 | | | | 16,780 | | | | 16,980 | |
| | | | | | | | | | | | |
Non-GAAP operating income (loss) | | $ | 25,672 | | | $ | (4,371 | ) | | $ | (58,672 | ) |
| | | | | | | | | | | | |
46
The following table summarizes our contractual obligations and maturity dates as of December 31, 2011 (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | | Payments Due by Period | |
Contractual Obligations | | Total | | | Less
than 1
year | | | 1 - 3
years | | | 3 - 5
years | | | More
than 5
years | |
Long-term debt obligations, including interest | | $ | 2,033,140 | | | $ | 87,635 | | | $ | 783,165 | | | $ | 333,945 | | | $ | 828,395 | |
Inventory purchase obligations | | | 91,284 | | | | 90,244 | | | | 1,040 | | | | — | | | | — | |
Construction contracts | | | 5,300 | | | | 5,300 | | | | — | | | | — | | | | — | |
Operating leases | | | 166,954 | | | | 28,186 | | | | 57,697 | | | | 41,382 | | | | 39,689 | |
| | | | | | | | | | | | | | | | | | | | |
Total(1) | | $ | 2,296,678 | | | $ | 211,365 | | | $ | 841,902 | | | $ | 375,327 | | | $ | 868,084 | |
| | | | | | | | | | | | | | | | | | | | |
| (1) | Excludes long-term obligation of $8.2 million related to deferred compensation, the payment of which is subject to elections made by participants that are subject to change. |
In addition, under certain license and collaboration agreements we are required to pay royalties and/or milestone payments upon the successful development and commercialization of related products. We expect to make development milestone payments up to $7.3 million associated with licensing agreements in the next 12 months. Additional milestones of up to approximately $235.0 million could be paid over the next two to seven years if development and commercialization of all our early stage programs continue and are successful. The majority of these milestones relate to potential future regulatory approvals and subsequent sales thresholds. Given the inherent risk in pharmaceutical development, it is highly unlikely that we will ultimately make all of these milestone payments; however, we would consider these payments as positive because they would signify that the related products are successfully moving through development and commercialization.
Our future capital requirements will depend on many factors, including: the amount of product sales we achieve for BYDUREON, BYETTA and SYMLIN; costs associated with the commercialization of BYDUREON and the continued commercialization of BYETTA and SYMLIN; costs associated with the operation of our BYDUREON manufacturing facility; costs of potential licenses or acquisitions; the potential need to repay existing indebtedness; our ability to receive or need to make milestone payments; our ability, and the extent to which we establish collaborative arrangements for exenatide products outside the United States, for SYMLIN or any of our product candidates; progress in our research and development programs and the magnitude of these programs; costs involved in preparing, filing, prosecuting, maintaining, enforcing or defending our patents; competing technological and market developments; and costs of manufacturing, including costs associated with establishing our own manufacturing capabilities or obtaining and validating additional manufacturers of our products; and scale-up costs for our drug candidates.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that are currently or reasonably likely to be material to our consolidated financial position or results of operations.
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk |
We invest our excess cash primarily in United States Government securities, securities of agencies sponsored by the US Government, asset-backed securities, and debt instruments of financial institutions and corporations with investment-grade credit ratings. These instruments have various short-term maturities. We do not utilize derivative financial instruments, derivative commodity instruments or other market risk sensitive instruments, positions or transactions in any material fashion. Accordingly, we believe that, while the instruments held are subject to changes in the financial standing of the issuer of such securities, we are not subject to any material risks arising from changes in interest rates, foreign currency exchange rates, commodity prices, equity prices or other market changes that affect market risk sensitive investments. Our debt is not subject to significant swings in valuation as interest rates on our debt are fixed. The fair value of our 2007 Notes at December 31, 2011 was approximately $514.3 million. A hypothetical 1% adverse move in interest rates along the entire interest rate yield curve would not materially affect the fair value of our financial instruments that are exposed to changes in interest rates.
47
| Item 8. | Financial Statements and Supplementary Data |
The financial statements and supplemental data required by this item are set forth at the pages indicated in Part IV, Item 15(a)(1) of this annual report.
| Item 9. | Changes In and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of our disclosure controls and procedures as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Based on this evaluation, our principal executive officer and our principal financial officer concluded that our disclosure controls and procedures were effective as of December 31, 2011.
Our management does not expect that our disclosure controls and procedures or our internal controls over financial reporting will prevent all potential error and fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, or misstatements due to error, if any, within the Company have been detected. While we believe that our disclosure controls and procedures and internal control over financial reporting are and have been effective, we intend to continue to examine and refine our disclosure controls and procedures and internal control over financial reporting and to monitor ongoing developments in these areas.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework inInternal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2011. The effectiveness of our internal control over financial reporting as of December 31, 2011 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report which is included herein.
Changes in Internal Control Over Financial Reporting
There has been no change in our internal control over financial reporting in our most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
48
Report of Independent Registered Public Accounting Firm on Internal Control
Over Financial Reporting
The Board of Directors and Stockholders of Amylin Pharmaceuticals, Inc.
We have audited Amylin Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Amylin Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Amylin Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2011, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Amylin Pharmaceuticals, Inc. as of December 31, 2011 and 2010, and the related consolidated statements of operations, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2011 of Amylin Pharmaceuticals, Inc. and our report dated February 22, 2012 expressed an unqualified opinion thereon.
San Diego, California
February 22, 2012
49
PART III
| Item 9B. | Other Information |
None.
| Item 10. | Directors, Executive Officers and Corporate Governance |
The information required by this item with respect to directors is incorporated by reference from the information under the captions “Election of Directors,” “Section 16(a) Beneficial Ownership Reporting Compliance,” and “Code of Business Conduct and Ethics” contained in the proxy statement to be filed with the SEC pursuant to Regulation 14A in connection with our 2011 annual meeting of stockholders. The information required by this item with respect to executive officers appears under Part I of this annual report on Form 10-K under the caption “Business—Executive Officers.”
| Item 11. | Executive Compensation |
The information required by this item is incorporated by reference to the information under the captions “Compensation of Directors,” “Executive Compensation,” “Report of the Compensation Committee of the Board of Directors on Executive Compensation,” and “Compensation Committee Interlocks and Insider Participation” contained in the proxy statement to be filed with the SEC pursuant to Regulation 14A in connection with our 2011 annual meeting of stockholders.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Shareholder Matters |
The information required by this item is incorporated by reference to the information under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” contained in the proxy statement to be filed with the SEC pursuant to Regulation 14A in connection with our 2011 annual meeting of stockholders.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this item is incorporated by reference to the information under the captions “Election of Directors” and “Certain Transactions” contained in the proxy statement to be filed with the SEC pursuant to Regulation 14A in connection with our 2011 annual meeting of stockholders.
| Item 14. | Principal Accountant Fees and Services |
The information required by this item is incorporated by reference to the information under the caption contained in “Ratification of Selection of Independent Registered Public Accounting Firm” contained in the proxy statement to be filed with the SEC pursuant to Regulation 14A in connection with our 2011 annual meeting of stockholders.
50
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
| (a)(1) | Index to Consolidated Financial Statements |
The financial statements required by this item are submitted in a separate section beginning on page F-1 of this annual report.
| (a)(2) | Financial Statement Schedules: The following Schedule is filed as part of this annual report on Form 10-K: |
All other schedules have been omitted because they are not applicable or required, or the information required to be set forth therein is included in the Consolidated Financial Statements or notes thereto.
| (a)(3) | Index to Exhibits—See Item 15(b) below. |
| | | | |
Exhibit
Footnote | | Exhibit
Number | | |
| | |
| (1) | | 3.1 | | Amended and Restated Certificate of Incorporation of the Registrant. |
| | |
| (3) | | 3.2 | | Fourth Amended and Restated Bylaws of the Registrant. |
| | |
| (6) | | 3.3 | | Certificate of Amendment of Amended and Restated Certificate of Incorporation of the Registrant. |
| | |
| (21) | | 3.4 | | Certificate of Amendment of Amended and Restated Certificate of Incorporation of the Registrant. |
| | |
| | 4.1 | | Reference is made to Exhibits 3.1 - 3.4. |
| | |
| (8) | | 4.2 | | Rights Agreement, dated June 17, 2002, between the Registrant and American Stock Transfer & Trust Company. |
| | |
| (8) | | 4.3 | | Certificate of Designation of Series A Junior Participating Preferred Stock. |
| | |
| (11) | | 4.4 | | First Amendment to Rights Agreement, dated December 13, 2002, between the Registrant and American Stock Transfer & Trust Company. |
| | |
| (20) | | 4.5 | | Indenture, dated as of June 8, 2007, between Registrant and The Bank of New York Trust Company, N.A. (as Trustee). |
| | |
| | 4.6 | | Secured Promissory Note, dated November 7, 2011, from Registrant to Eli Lilly and Company. |
| | |
| | 4.7 | | Security Agreement, dated November 7, 2011, and First Amendment thereto, dated January 5, 2012, among Registrant, Registrant’s wholly-owned subsidiary, Amylin Ohio LLC, other grantor parties thereto, and Eli Lilly and Company.* |
| | |
| | 4.8 | | Subsidiary Guarantee Agreement, dated November 7, 2011, by Registrant’s wholly-owned subsidiary, Amylin Ohio LLC and each of the other parties thereto in favor of Eli Lilly and Company. |
| | |
| | 4.9 | | Amended and Restated Loan Agreement, dated November 7, 2011, between Registrant and Eli Lilly and Company. |
| | |
| | 4.10 | | Amended and Restated Promissory Note, dated November 7, 2011, from Registrant to Eli Lilly and Company. |
| | |
| (1) | | 10.1 | | Form of Indemnity Agreement entered into between the Registrant and its directors and officers.† |
| | |
| (25) | | 10.2 | | Registrant’s Amended and Restated 2001Employee Stock Purchase Plan.† |
| | |
| (24) | | 10.3 | | Registrant’s Non-Employee Directors’ Stock Option Plan (the “Directors’ Plan”).† |
| | |
| (4)(2) | | 10.4 | | Patent and Technology License Agreement, Consulting Agreement and Nonstatutory Stock Option Agreement, dated October 1, 1996, between the Registrant and Dr. John Eng. |
| | |
| (5) | | 10.5 | | Registrant’s Directors’ Deferred Compensation Plan.† |
| | |
| (26) | | 10.6 | | Registrant’s Directors’ Plan Stock Option Agreement, as amended.† |
51
| | | | |
Exhibit
Footnote | | Exhibit
Number | | |
| | |
| (7)(2) | | 10.7 | | Development and License Agreement, dated May 15, 2000, between the Registrant and Alkermes Controlled Therapeutics II, Inc. |
| | |
| (23) | | 10.8 | | Registrant’s Amended and Restated Officer Change in Control Severance Benefit Plan.† |
| | |
| (17) | | 10.9 | | Registrant’s Amended and Restated 2001 Equity Incentive Plan.† |
| | |
| (9) | | 10.10 | | Form of Registrant’s 2001 Equity Incentive Plan Officer Stock Option Agreement, as amended.† |
| | |
| (9) | | 10.11 | | Form of Registrant’s 2001 Equity Incentive Plan Stock Option Agreement, as amended.† |
| | |
| (10)(2) | | 10.12 | | Manufacturing Agreement, dated May 12, 2003, between Registrant and Lonza Braine, S.A., formerly UCB S.A. |
| | |
| (12)(2) | | 10.13 | | Exenatide Manufacturing Agreement, dated October 1, 2003, between Registrant and Mallinckrodt Inc. |
| | |
| (12)(2) | | 10.14 | | Commercial Supply Agreement for Exenatide, dated December 23, 2003, between Registrant and Bachem, Inc. |
| | |
| (13)(2) | | 10.15 | | Commercial Supply Agreement, dated February 14, 2005, between Registrant and Baxter Pharmaceutical Solutions LLC. |
| | |
| | 10.16 | | Summary Description of Registrant’s Named Executive Officer Oral At-Will Employment Agreements.† |
| | |
| (14) | | 10.17 | | Description of Registrant’s Executive Cash Bonus Plan.† |
| | |
| (16)(2) | | 10.18 | | Amendment to Development and License Agreement, dated October 24, 2005, between Registrant and Alkermes Controlled Therapeutics II. |
| | |
| (15)(2) | | 10.19 | | Commercial Supply Agreement for Pramlintide, dated June 28, 2005, between Registrant and Bachem, Inc. |
| | |
| (18)(2) | | 10.20 | | Commercial Supply Agreement, dated October 12, 2006, between Registrant and Wockhardt UK (Holdings) Ltd. |
| | |
| (19) | | 10.21 | | Employment Agreement, dated March 7, 2007, between Registrant and Daniel M. Bradbury.† |
| | |
| (30) | | 10.22 | | Registrant’s 2001 Non-Qualified Deferred Compensation Plan.† |
| | |
| (22) | | 10.23 | | First Amendment to Exenatide Manufacturing Agreement, dated January 6, 2006, between Registrant and Mallinckrodt Inc. |
| | |
| (22)(2) | | 10.24 | | Amended and Restated Commercial Supply Agreement, dated April 1, 2008, between Registrant and Wockhardt UK (Holdings) Ltd. |
| | |
| (22)(2) | | 10.25 | | Third Amendment to Supply Agreement, dated January 1, 2008, between Registrant and Mallinckrodt Inc. |
| | |
| (3) | | 10.26 | | Amendment to Employment Agreement, dated December 3, 2008, between Registrant and Daniel M. Bradbury.† |
| | |
| (23)(2) | | 10.27 | | Amendment to Commercial Supply Agreement, dated December 8, 2008, between Registrant and Baxter Pharmaceutical Solutions LLC. |
| | |
| (23)(2) | | 10.28 | | Amendment to the Amended and Restated Commercial Supply Agreement, dated January 23, 2009, between Registrant and Wockhardt UK (Holdings) Ltd. |
| | |
| (25) | | 10.29 | | Registrant’s 2009 Equity Incentive Plan.† |
| | |
| (25) | | 10.30 | | Registrant’s 2009 Equity Incentive Plan Officer Stock Option Agreement.† |
| | |
| (25) | | 10.31 | | Registrant’s 2009 Equity Incentive Plan Stock Option Agreement.† |
| | |
| (26)(2) | | 10.32 | | License, Development and Commercialization Agreement, dated October 30, 2009, between Registrant and Takeda Pharmaceutical Company Limited. |
| | |
| (26)(2) | | 10.33 | | Side Letter Agreement, dated October 30, 2009, between Registrant and Takeda Pharmaceutical Company Limited. |
| | |
| (27) | | 10.34 | | Registrant’s 2009 Equity Incentive Plan Restricted Stock Unit Award Agreement.† |
| | |
| (27)(2) | | 10.35 | | Amendment to Exenatide Manufacturing Agreement, dated January 8, 2010, between Registrant and Mallinckrodt Inc. |
| | |
| (28) | | 10.36 | | Amendment to Development and License Agreement, dated July 13, 2006, between Registrant and Alkermes Controlled Therapeutics Inc. II. |
52
| | | | |
Exhibit
Footnote | | Exhibit
Number | | |
| | |
| (29) | | 10.37 | | Second Amendment to Commercial Supply Agreement for Exenatide, dated April 29, 2010, between Registrant and Bachem Inc. |
| | |
| (29)(2) | | 10.38 | | Third Amendment to Commercial Supply Agreement for Exenatide, dated September 20, 2010, between Registrant and Bachem Inc. |
| | |
| (30) | | 10.39 | | Registrant’s Form of Restricted Stock Award Agreement.† |
| | |
| (30)(2) | | 10.40 | | Second Amendment to Amended and Restated Commercial Supply Agreement, dated November 1, 2010, between Registrant and Wockhardt UK (Holdings) Ltd. |
| | |
| (31)(2) | | 10.41 | | Amendment to Exenatide Manufacturing Agreement, dated February 14, 2011, between Registrant and Mallinckrodt Inc. |
| | |
| | 10.42 | | Exenatide Supply Agreement, dated July 31, 2007, between Registrant’s wholly-owned subsidiary, Amylin Ohio LLC, and Lonza Ltd. and Lonza Sales Ltd, and amendments thereto.* |
| | |
| | 10.43 | | Supply Agreement, dated December 19, 2007, between Registrant’s wholly-owned subsidiary, Amylin Ohio LLC, and Alkermes, Inc. and amendments thereto.* |
| | |
| | 10.44 | | Third Amendment to Amended and Restated Commercial Supply Agreement, dated November 1, 2011, between Registrant and Wockhardt UK (Holdings) Ltd.* |
| | |
| | 10.45 | | Second Amendment to Commercial Supply Agreement, dated January 1, 2012, between Registrant and Baxter Pharmaceuticals Solutions LLC.* |
| | |
| | 10.46 | | Settlement and Termination Agreement, dated November 7, 2011, between Registrant and Eli Lilly and Company.* |
| | |
| | 10.47 | | Amended and Restated Exenatide Once Weekly Supply Agreement, dated November 7, 2011, between Registrant and Eli Lilly and Company.* |
| | |
| | 10.48 | | Amended and Restated Device and Finished EQW Product Manufacturing Agreement, dated November 7, 2011, between Registrant and Eli Lilly and Company.* |
| | |
| | 21.1 | | Subsidiaries of Registrant. |
| | |
| | 23.1 | | Consent of Independent Registered Public Accounting Firm. |
| | |
| | 24.1 | | Power of Attorney. Reference is made to page 56. |
| | |
| | 31.1 | | Certification of Principal Financial Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended. |
| | |
| | 31.2 | | Certification of Principal Executive Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended. |
| | |
| | 32.1 | | Certifications Pursuant to U.S.C. Section 1350, As Adopted Pursuant to Section 906 of the Public Company Accounting Reform and Investor Protection Act of 2002. |
| | |
| | 101 | | The following financial statements and footnotes from the Amylin Pharmaceuticals, Inc. Annual Report on Form 10-K for the fiscal year ended December 31, 2011 are formatted in Extensible Business Reporting Language (XBRL): (i) Consolidated Balance Sheets; (ii) Consolidated Statements of Operations; (iii) Consolidated Statements of Stockholders’ Equity; (iv) Consolidated Statements of Cash Flows; and (v) the notes to the consolidated financial statements. |
| † | Indicates management or compensatory plan or arrangement required to be identified pursuant to Item 15(c). |
| * | Confidential treatment has been requested with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission. |
| (1) | Filed as an exhibit to the Registrant’s Registration Statement on Form S-1 (No. 33-44195) or amendments thereto and incorporated herein by reference. |
| (2) | Confidential Treatment has been granted by the Securities and Exchange Commission with respect to portions of this agreement. |
| (3) | Filed as an exhibit on Form 8-K dated December 8, 2008, and incorporated herein by reference. |
53
| (4) | Filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 1996, and incorporated herein by reference. |
| (5) | Filed as an exhibit to the Registrant’s Registration Statement on Form S-8 (No. 333-61660) or amendments thereto and incorporated herein by reference. |
| (6) | Filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2001, and incorporated herein by reference. |
| (7) | Filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2000, and incorporated herein by reference. |
| (8) | Filed as an exhibit on Form 8-K dated June 18, 2002, and incorporated herein by reference. |
| (9) | Filed as an exhibit to Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2003, and incorporated herein by reference. |
| (10) | Filed as an exhibit to Amendment 1 to Registrant’s Quarterly Report on Form 10-Q/A for the quarter ended September 30, 2003, and incorporated herein by reference. |
| (11) | Filed as an exhibit to Registrant’s Annual Report on Form 10-K for the fiscal year ended December 31, 2002, and incorporated herein by reference. |
| (12) | Filed as an exhibit to Amendment 1 to Registrant’s Annual Report on Form 10-K/A for the fiscal year ended December 31, 2003, and incorporated herein by reference. |
| (13) | Filed as an exhibit to Registrant’s Annual Report on Form 10-K for the fiscal year ended December 31, 2004, and incorporated herein by reference. |
| (14) | Filed on Form 8-K dated December 7, 2011, and incorporated herein by reference. |
| (15) | Filed as an exhibit to Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2005, and incorporated herein by reference. |
| (16) | Filed as an exhibit to Registrant’s Annual Report on Form 10-K for the fiscal year ended December 31, 2005, and incorporated herein by reference. |
| (17) | Filed as an exhibit on Form 8-K dated May 30, 2008 and incorporated herein by reference. |
| (18) | Filed as an exhibit to Registrant’s Annual Report on Form 10-K for the fiscal year ended December 31, 2006, and incorporated herein by reference. |
| (19) | Filed as an exhibit to Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2007, and incorporated herein by reference. |
| (20) | Filed as an exhibit on Form 8-K dated June 8, 2007, and incorporated herein by reference. |
| (21) | Filed as an exhibit to Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2007, and incorporated herein by reference. |
| (22) | Filed as an exhibit to Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2008, and incorporated herein by reference. |
| (23) | Filed as an exhibit to Registrant’s Annual Report on Form 10-K for the fiscal year ended December 31, 2008, and incorporated herein by reference. |
54
| (24) | Filed as an exhibit to Registrant’s Quarterly report on form 10-Q for the quarter ended March 31, 2009, and incorporated herein by reference. |
| (25) | Filed as an exhibit on Form 8-K dated June 10, 2009, and incorporated herein by reference. |
| (26) | Filed as an exhibit to Registrant’s Annual Report on Form 10-K for the fiscal year ended December 31, 2009, and incorporated herein by reference. |
| (27) | Filed as an exhibit to Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010, and incorporated herein by reference. |
| (28) | Filed as an exhibit to Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2010, and incorporated herein by reference. |
| (29) | Filed as an exhibit to Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2010, and incorporated herein by reference. |
| (30) | Filed as an exhibit to Registrant’s Annual Report on Form 10-K for the fiscal year ended December 31, 2010, and incorporated herein by reference. |
| (31) | Filed as an exhibit to Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2011, and incorporated herein by reference. |
Note on trademarks used in this report:
Amylin®, BYETTA® and SYMLIN® and BYDUREON™ are trademarks of Amylin Pharmaceuticals, Inc. Other brands, names and trademarks contained in this Annual Report on Form 10-K are the property of their respective owners.
55
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | |
| | AMYLIN PHARMACEUTICALS, INC. |
| | |
| Date: February 22, 2011 | | | | |
| | |
| | By: | | /s/ DANIEL M. BRADBURY Daniel M. Bradbury,
President and Chief Executive Officer |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Daniel M. Bradbury and Mark G. Foletta, and each of them, as his or her true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him or her and in his or her name, place, and stead, in any and all capacities, to sign any and all amendments to this Report, and any other documents in connection therewith, and to file the same, with all exhibits thereto, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them or their or his substitute or substituted, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| | | | |
Signatures | | Title | | Date |
| | |
/s/ DANIEL M. BRADBURY Daniel M. Bradbury | | President and Chief Executive Officer (Principal Executive Officer) | | February 22, 2012 |
| | |
/s/ MARK G. FOLETTA Mark G. Foletta | | Senior Vice President, Finance and Chief Financial Officer (Principal Financial Officer) | | February 22, 2012 |
| | |
/s/ LAURA M. CLAGUE Laura M. Clague | | Vice President, Finance and Corporate Controller (Principal Accounting Officer) | | February 22, 2012 |
| | |
/S/ PAULO F. COSTA Paulo F. Costa | | Chairman of the Board | | February 22, 2012 |
| | |
/s/ ADRIAN ADAMS Adrian Adams | | Director | | February 22, 2012 |
| | |
/s/ TERESA BECK Teresa Beck | | Director | | February 22, 2012 |
| | |
/s/ M. KATHLEEN BEHRENS M. Kathleen Behrens | | Director | | February 22, 2012 |
56
| | | | |
Signatures | | Title | | Date |
| | |
/s/ PAUL N. CLARK Paul N. Clark | | Director | | February 22, 2012 |
| | |
Alexander J. Denner | | Director | | |
| | |
/s/ KARIN EASTHAM Karin Eastham | | Director | | February 22, 2012 |
| | |
/s/ JAMES R. GAVIN III, M.D., PHD. James R. Gavin III, M.D., PhD. | | Director | | February 22, 2012 |
| | |
/s/ JAY S. SKYLER, M.D. Jay S. Skyler, M.D., MACP | | Director | | February 22, 2012 |
| | |
/s/ JOSEPH P. SULLIVAN Joseph P. Sullivan | | Director | | February 22, 2012 |
57
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Amylin Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Amylin Pharmaceuticals, Inc. as of December 31, 2011 and 2010, and the related consolidated statements of operations, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2011. Our audits also included the financial statement schedule listed in the Index at Item 15(a)(2). These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Amylin Pharmaceuticals, Inc., at December 31, 2011 and 2010, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2011, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Amylin Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 22, 2012 expressed an unqualified opinion thereon.
San Diego, California
February 22, 2012
F-1
AMYLIN PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
(in thousands, except per share data)
| | | | | | | | |
| | | December 31, | |
| | | 2011 | | | 2010 | |
| ASSETS | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 99,859 | | | $ | 164,521 | |
Short-term investments | | | 104,206 | | | | 278,142 | |
Restricted cash | | | 10,519 | | | | 15,000 | |
Accounts receivable, net | | | 45,489 | | | | 54,645 | |
Inventories, net | | | 111,959 | | | | 118,629 | |
Other current assets | | | 49,158 | | | | 45,458 | |
| | | | | | | | |
Total current assets | | | 421,190 | | | | 676,395 | |
Property, plant and equipment, net | | | 831,162 | | | | 811,745 | |
Intangible asset related to reacquired economic interest, net | | | 273,842 | | | | — | |
Economic interest in exenatide products to be reacquired as a business | | | 327,697 | | | | — | |
Other long-term assets | | | 16,308 | | | | 43,289 | |
| | | | | | | | |
| | $ | 1,870,199 | | | $ | 1,531,429 | |
| | | | | | | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 21,035 | | | $ | 23,920 | |
Accrued compensation | | | 66,776 | | | | 43,231 | |
Payable to former collaborative partner | | | 29,140 | | | | 42,060 | |
Restructuring liability, current portion | | | 8,405 | | | | 6,532 | |
Promissory note related to revenue sharing obligation, current portion | | | 63,552 | | | | — | |
Deferred revenue, current portion | | | 7,500 | | | | 7,500 | |
Convertible senior notes, current portion | | | — | | | | 200,000 | |
Other current liabilities | | | 99,609 | | | | 78,352 | |
| | | | | | | | |
Total current liabilities | | | 296,017 | | | | 401,595 | |
Deferred revenue, net of current portion | | | 51,250 | | | | 58,750 | |
Long-term deferred credit | | | — | | | | 125,000 | |
Deferred collaborative profit-sharing | | | — | | | | 87,848 | |
Restructuring liability, net of current portion | | | 23,877 | | | | 28,764 | |
Convertible senior notes, net of current portion | | | 496,037 | | | | 468,697 | |
Note payable | | | 155,064 | | | | — | |
Promissory note related to revenue sharing obligation, net of current portion | | | 924,306 | | | | — | |
Other long-term obligations, net of current portion | | | 62,393 | | | | 17,292 | |
Commitments and contingencies | | | | | | | | |
Stockholders’ equity (deficit): | | | | | | | | |
Preferred stock, $.001 par value, 7,500 shares authorized, none issued and outstanding at December 31, 2011 and 2010 | | | — | | | | — | |
Common stock, $.001 par value, 450,000 shares authorized, 146,289 and 144,100 issued and outstanding at December 31, 2011 and 2010 | | | 146 | | | | 144 | |
Additional paid-in capital | | | 2,506,013 | | | | 2,444,266 | |
Accumulated deficit | | | (2,643,579 | ) | | | (2,100,180 | ) |
Accumulated other comprehensive loss | | | (1,325 | ) | | | (747 | ) |
| | | | | | | | |
Total stockholders’ equity (deficit) | | | (138,745 | ) | | | 343,483 | |
| | | | | | | | |
| | $ | 1,870,199 | | | $ | 1,531,429 | |
| | | | | | | | |
See accompanying notes to consolidated financial statements.
F-2
AMYLIN PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share data)
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Revenues: | | | | | | | | | | | | |
Net product sales | | $ | 621,570 | | | $ | 651,113 | | | $ | 753,993 | |
Revenues under collaborative agreements | | | 29,108 | | | | 17,700 | | | | 4,426 | |
| | | | | | | | | | | | |
Total revenues | | | 650,678 | | | | 668,813 | | | | 758,419 | |
Costs and expenses: | | | | | | | | | | | | |
Cost of goods sold | | | 48,376 | | | | 61,687 | | | | 82,999 | |
Selling, general and administrative | | | 271,224 | | | | 276,879 | | | | 330,486 | |
Research and development | | | 161,215 | | | | 183,083 | | | | 198,440 | |
Collaborative profit-sharing | | | 222,545 | | | | 257,127 | | | | 302,861 | |
Net costs associated with reacquisition of economic interest in exenatide products | | | 431,587 | | | | — | | | | — | |
Restructuring | | | 7,190 | | | | 16,780 | | | | 16,980 | |
| | | | | | | | | | | | |
Total costs and expenses | | | 1,142,137 | | | | 795,556 | | | | 931,766 | |
| | | | | | | | | | | | |
Operating loss | | | (491,459 | ) | | | (126,743 | ) | | | (173,347 | ) |
Interest and other expense, net | | | (51,940 | ) | | | (25,570 | ) | | | (12,909 | ) |
| | | | | | | | | | | | |
Net loss | | $ | (543,399 | ) | | $ | (152,313 | ) | | $ | (186,256 | ) |
| | | | | | | | | | | | |
Net loss per share—basic and diluted | | $ | (3.73 | ) | | $ | (1.06 | ) | | $ | (1.32 | ) |
| | | | | | | | | | | | |
Shares used in computing net loss per share, basic and diluted | | | 145,730 | | | | 143,525 | | | | 140,702 | |
| | | | | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-3
AMYLIN PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
For the years ended December 31, 2011, 2010 and 2009
(in thousands)
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Common stock | | | Additional | | | Accumulated | | | Accumulated
other
comprehensive | | | Total
stockholders’ | |
| | | Shares | | | Amount | | | paid-in capital | | | deficit | | | (loss) income | | | equity(deficit) | |
Balance at December 31, 2008 | | | 137,623 | | | $ | 138 | | | $ | 2,291,762 | | | $ | (1,761,611 | ) | | $ | (11,012 | ) | | $ | 519,277 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | — | | | | — | | | | — | | | | (186,256 | ) | | | — | | | | (186,256 | ) |
Unrealized gain on available-for-sale securities | | | — | | | | — | | | | — | | | | — | | | | 9,332 | | | | 9,332 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (176,924 | ) |
Issuance of common stock upon exercise of options, net | | | 599 | | | | 1 | | | | 4,556 | | | | — | | | | — | | | | 4,557 | |
Issuance of common stock for other employee benefit plans | | | 1,280 | | | | 1 | | | | 11,508 | | | | — | | | | — | | | | 11,509 | |
Issuance of common stock for employee stock ownership plan | | | 2,245 | | | | 2 | | | | 20,248 | | | | — | | | | — | | | | 20,250 | |
Employee stock-based compensation | | | — | | | | — | | | | 43,762 | | | | — | | | | — | | | | 43,762 | |
Non-employee stock-based compensation | | | — | | | | — | | | | 103 | | | | — | | | | — | | | | 103 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2009 | | | 141,747 | | | | 142 | | | | 2,371,939 | | | | (1,947,867 | ) | | | (1,680 | ) | | | 422,534 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | — | | | | — | | | | — | | | | (152,313 | ) | | | — | | | | (152,313 | ) |
Unrealized gain on available-for-sale securities | | | — | | | | — | | | | — | | | | — | | | | 933 | | | | 933 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (151,380 | ) |
Issuance of common stock upon exercise of options, net | | | 701 | | | | — | | | | 9,182 | | | | — | | | | — | | | | 9,182 | |
Issuance of common stock for other employee benefit plans | | | 772 | | | | 1 | | | | 11,168 | | | | — | | | | — | | | | 11,169 | |
Issuance of common stock for employee stock ownership plan | | | 880 | | | | 1 | | | | 15,848 | | | | — | | | | — | | | | 15,849 | |
Employee stock-based compensation | | | — | | | | — | | | | 36,022 | | | | — | | | | — | | | | 36,022 | |
Non-employee stock-based compensation | | | — | | | | — | | | | 107 | | | | — | | | | — | | | | 107 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2010 | | | 144,100 | | | | 144 | | | | 2,444,266 | | | | (2,100,180 | ) | | | (747 | ) | | | 343,483 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | — | | | | — | | | | — | | | | (543,399 | ) | | | — | | | | (543,399 | ) |
Unrealized loss on available-for-sale securities | | | — | | | | — | | | | — | | | | — | | | | (578 | ) | | | (578 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (543,977 | ) |
Issuance of common stock upon exercise of options, net | | | 384 | | | | — | | | | 3,230 | | | | — | | | | — | | | | 3,230 | |
Issuance of common stock for other employee benefit plans | | | 760 | | | | 1 | | | | 9,546 | | | | — | | | | — | | | | 9,547 | |
Issuance of common stock for employee stock ownership plan | | | 1,045 | | | | 1 | | | | 16,456 | | | | — | | | | — | | | | 16,457 | |
Employee stock-based compensation | | | — | | | | — | | | | 32,472 | | | | — | | | | — | | | | 32,472 | |
Non-employee stock-based compensation | | | — | | | | — | | | | 43 | | | | — | | | | — | | | | 43 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2011 | | | 146,289 | | | $ | 146 | | | $ | 2,506,013 | | | $ | (2,643,579 | ) | | $ | (1,325 | ) | | $ | (138,745 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-4
AMYLIN PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| | | | | | | | | | | | |
| | | Years ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Operating activities: | | | | | | | | | | | | |
Net loss | | $ | (543,399 | ) | | $ | (152,313 | ) | | $ | (186,256 | ) |
Adjustments to reconcile net loss to net cash (used for) provided by operating activities: | | | | | | | | | | | | |
Reacquired economic rights for exenatide | | | 403,598 | | | | — | | | | — | |
Interest expense and debt discount accretion on promissory note related to revenue sharing obligation | | | 10,984 | | | | — | | | | — | |
Change in fair value of assets and liabilities carried at fair value | | | 31,412 | | | | — | | | | — | |
Depreciation and amortization | | | 47,373 | | | | 57,335 | | | | 38,197 | |
Amortization of debt discount and debt issuance costs | | | 14,110 | | | | 13,579 | | | | 8,209 | |
Benefit plan compensation settled in stock | | | 16,527 | | | | 19,735 | | | | 20,161 | |
Employee stock-based compensation | | | 32,472 | | | | 36,022 | | | | 43,762 | |
Amortization of deferred revenue and other deferred credits | | | (11,930 | ) | | | (7,500 | ) | | | (4,336 | ) |
Loss on impairment of investments | | | — | | | | 198 | | | | 1,377 | |
Restructuring | | | 645 | | | | 769 | | | | 494 | |
Other non-cash expenses | | | 4,232 | | | | 6,212 | | | | 10,548 | |
Changes in operating assets and liabilities: | | | | | | | | | | | | |
Accounts receivable | | | 9,156 | | | | 6,087 | | | | 1,637 | |
Inventories | | | 9,172 | | | | 9,255 | | | | 16,123 | |
Other current assets | | | 12,833 | | | | 5,353 | | | | (35,749 | ) |
Long-term prepaid assets | | | 3,048 | | | | (811 | ) | | | (8,754 | ) |
Accounts payable and accrued liabilities | | | 18,372 | | | | (25,021 | ) | | | 5,574 | |
Accrued compensation | | | 27,431 | | | | (27,765 | ) | | | 11,009 | |
Payable to collaborative partner | | | (12,920 | ) | | | (7,585 | ) | | | (10,825 | ) |
Deferred revenue | | | — | | | | — | | | | 75,000 | |
Deferred collaborative profit-sharing | | | 15,510 | | | | 33,278 | | | | 54,570 | |
Restructuring liability | | | (3,014 | ) | | | 3,316 | | | | (15,252 | ) |
Other assets and liabilities, net | | | 2,690 | | | | (8,867 | ) | | | (5,566 | ) |
| | | | | | | | | | | | |
Net cash (used for) provided by operating activities | | | 88,302 | | | | (38,723 | ) | | | 19,923 | |
Investing activities: | | | | | | | | | | | | |
Purchases of short-term investments | | | (599,623 | ) | | | (573,626 | ) | | | (794,008 | ) |
Sales and maturities of short-term investments | | | 773,514 | | | | 841,296 | | | | 832,900 | |
Decrease/(increase) of restricted cash | | | 4,481 | | | | (15,000 | ) | | | — | |
Purchases of property, plant and equipment | | | (54,454 | ) | | | (91,132 | ) | | | (152,051 | ) |
Cash paid to reacquire economic rights for exenatide products, including transaction costs | | | (252,999 | ) | | | — | | | | — | |
Decrease (increase) in other long-term assets | | | 2,296 | | | | (1,869 | ) | | | (2,913 | ) |
| | | | | | | | | | | | |
Net cash provided by (used for) investing activities | | | (126,785 | ) | | | 159,669 | | | | (116,072 | ) |
Financing activities: | | | | | | | | | | | | |
Proceeds from issuance of common stock, net | | | 8,821 | | | | 16,500 | | | | 10,961 | |
Proceeds from long-term loan payable | | | 165,000 | | | | — | | | | — | |
Repayment of convertible debt | | | (200,000 | ) | | | — | | | | — | |
Repayment of notes payable | | | — | | | | (93,750 | ) | | | (31,250 | ) |
| | | | | | | | | | | | |
Net cash used for financing activities | | | (26,179 | ) | | | (77,250 | ) | | | (20,289 | ) |
| | | | | | | | | | | | |
Increase (decrease) in cash and cash equivalents | | | (64,662 | ) | | | 43,696 | | | | (116,438 | ) |
Cash and cash equivalents at beginning of year | | | 164,521 | | | | 120,825 | | | | 237,263 | |
| | | | | | | | | | | | |
Cash and cash equivalents at end of year | | $ | 99,859 | | | $ | 164,521 | | | $ | 120,825 | |
| | | | | | | | | | | | |
Supplemental disclosures of cash flow information: | | | | | | | | | | | | |
Interest paid, net of interest capitalized | | $ | 11,501 | | | $ | 15,483 | | | $ | 13,218 | |
Interest capitalized | | $ | 26,795 | | | $ | 25,904 | | | $ | 33,280 | |
Non-cash interest capitalized to property, plant and equipment | | $ | 15,248 | | | $ | 14,383 | | | $ | 17,676 | |
Receivable arising from sale of property, plant and equipment | | $ | — | | | $ | 6,500 | | | $ | — | |
Non-cash dispositions of property, plant and equipment | | $ | 296 | | | $ | 7,398 | | | $ | — | |
Property, plant and equipment additions in other current liabilities | | $ | 1,228 | | | $ | 513 | | | $ | 10,123 | |
Non-cash investing activities: | | | | | | | | | | | | |
Intangible asset related to reacquired economic interest | | $ | 274,849 | | | $ | — | | | $ | — | |
Economic interest in exenatide products to be acquired as a business | | $ | 327,697 | | | $ | — | | | $ | — | |
Liabilities settled in connection with the termination of a collaboration | | $ | 223,928 | | | $ | — | | | $ | — | |
Derivative asset-options embedded in promissory note related to revenue sharing obligation | | $ | 23,441 | | | $ | — | | | $ | — | |
Loss protection liability valued at fair value | | $ | 30,758 | | | $ | — | | | $ | — | |
Non-cash financing activities: | | | | | | | | | | | | |
Promissory note related to revenue sharing obligation incurred to reacquire economic interest in exenatide | | $ | 976,821 | | | $ | — | | | $ | — | |
Shares contributed as employer 401(k) match | | $ | 3,956 | | | $ | 3,851 | | | $ | 5,105 | |
Shares contributed to employee stock ownership plan | | $ | 16,457 | | | $ | 15,849 | | | $ | 20,250 | |
See accompanying notes to consolidated financial statements.
F-5
AMYLIN PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Summary of Significant Accounting Policies
Organization
Amylin Pharmaceuticals, Inc. (referred to as we, us, or Amylin) is a biopharmaceutical company engaged in the discovery, development and commercialization of drug candidates for the treatment of diabetes, obesity and other diseases. We were incorporated in Delaware on September 29, 1987.
Basis of Presentation
Our consolidated financial statements include the accounts of our wholly owned subsidiaries, Amylin Ohio, LLC, and Amylin Investments, LLC. All significant intercompany transactions and balances have been eliminated in consolidation.
Settlement and Termination Agreement
On November 7, 2011, Amylin entered into a Settlement and Termination Agreement, or the Termination Agreement, with Eli Lilly & Company, or Lilly, to terminate our alliance for exenatide and resolve the outstanding litigation between the companies. Note 2 further describes the terms of the Termination Agreement and Amylin’s related accounting treatment.
Use of Estimates
The preparation of the consolidated financial statements in conformity with US GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Revenue Recognition
Net Product Sales
We sell BYETTA® (exenatide) injection for the treatment of type 2 diabetes and SYMLIN® (pramlintide acetate) injection for the treatment of type 1 and type 2 diabetes primarily to wholesale distributors, who, in turn, sell to retail pharmacies and government entities. Product sales are recognized when delivery of the products has occurred, title has passed to the customer, the selling price is fixed or determinable, collectability is reasonably assured and we have no further obligations. We record product sales net of allowances for product returns, rebates, wholesaler chargebacks, wholesaler discounts, and prescription vouchers at the time of sale and report product sales net of such allowances. We must make significant judgments in determining these allowances. If actual results differ from our estimates, we will be required to make adjustments to these allowances in the future.
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act (PPACA) as amended by the Health Care and Education Reconciliation Act. There are a number of provisions in the new legislation that will impact the pharmaceutical industry through increased discounts and an expansion of government funded insurance programs. Beginning in January 2011, drug manufacturers provide a discount of 50 percent of the patient’s cost of branded prescription drugs for Medicare Part D participants who are in the “donut hole” (the coverage gap in Medicare prescription drug coverage). Our rebate allowance includes an accrual for our estimated share of the donut hole costs associated with product sales made through December 31, 2011 and was calculated using historical Part D utilization information provided by the Center for Medicare and Medicaid Services and third party market research data. The rebate allowance provided each quarter will vary depending upon estimated utilization rates.
We record all US BYETTA and SYMLIN product sales. For the first eleven months of the year ended December 31, 2011, and for the twelve months ended December 31, 2010 and 2009, with respect to BYETTA, we have determined that we are qualified as a principal based on our responsibilities under our contracts with Lilly, which include manufacture of product for sale in the US, responsibility for establishing pricing in the US, distribution, ownership of product inventory and credit risk from customers. As further described in Note 2, as a result of the Termination Agreement full responsibility for the commercialization of exenatide in the US was transferred to Amylin effective November 30, 2011.
F-6
Revenues Under Collaborative Agreements
Revenues under collaborative agreements consist of the amortization of product and technology license fees, milestone payments and royalties earned. Upfront product and technology license fees under multiple-element arrangements are deferred and recognized over the period of such services or performance if such arrangements require on-going services or performance. Non-refundable amounts received for substantive milestones are recognized upon achievement of the milestone. Any amounts received prior to satisfying these revenue recognition criteria are recorded as deferred revenue. Royalty revenue is earned on annual gross margins for all exenatide products sold outside of the US and is recorded based upon gross margins for such sales.
Collaborative Profit-Sharing
Collaborative profit-sharing represents Lilly’s 50% share of the gross margin for BYETTA sales in the US (see Note 4). As further described in Note 2, as a result of the Termination Agreement, for the twelve months ended December 31, 2011, collaborative profit sharing reflects approximately eleven months of such sharing with Lilly while the twelve months ended December 31, 2010 and 2009 both reflect a full twelve months of such sharing.
Shipping and Handling Costs
Shipping and handling costs incurred for product shipments are included in cost of goods sold in the accompanying consolidated statements of operations.
Research and Development Expenses
Research and development costs are expensed as incurred and include salaries and bonuses, benefits, non-cash stock-based compensation, license fees, milestone payments due under license agreements, costs paid to third-party contractors to perform research, conduct clinical trials, and develop drug materials and delivery devices; and associated overhead and facilities costs. Reimbursed research and development costs under collaborative arrangements are recorded as a reduction to research and development expenses and are recognized in the period in which the related costs are incurred. Clinical trial costs, including costs associated with third-party contractors, are a significant component of research and development expenses. Invoicing from third- party contractors for services performed can lag several months. We accrue the costs of services rendered in connection with such activities based on our estimate of management fees, site management and monitoring costs, and data management costs. Actual clinical trial costs may differ from estimates and are adjusted in the period in which they become known.
Concentrations of Risk
We rely on third-party manufacturers for the production of certain of our products and drug candidates. If our third-party manufacturers are unable to continue manufacturing our products and/or drug candidates, or if we lose one of our sole source suppliers used in our manufacturing processes, we may not be able to meet market demand for our products and could be materially and adversely affected.
We have a collaboration agreement with Takeda Pharmaceutical Company Limited, or Takeda, for the development and commercialization of pharmaceutical products for obesity and related indications. Under this agreement Takeda provides funding for development and will provide funding for commercialization expenses. If Takeda is unable to perform these activities or were to terminate our collaboration with them, we would likely need to find a third party collaborator to continue developing our obesity program, which we may be unable to do.
We are also subject to credit risk from our accounts receivable related to product sales. We sell our products in the United States primarily to wholesale distributors. Our top four customers represented approximately 96% of net product sales in 2011 and 96% of the accounts receivable balance at December 31, 2011. We evaluate the credit worthiness of our customers and generally do not require collateral. We have not experienced any material losses on uncollectible accounts receivable to date.
Net product sales for the years ended December 31, 2011, 2010 and 2009 were $621.6 million, $651.1 million and $754.0 million, respectively, and consisted of sales of BYETTA and SYMLIN, less allowances for product returns, rebates and wholesaler chargebacks, wholesaler discounts, and prescription vouchers.
F-7
The following table provides information regarding net product sales by product (in millions):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
BYETTA | | $ | 517.7 | | | $ | 559.3 | | | $ | 667.6 | |
SYMLIN | | | 103.9 | | | | 91.8 | | | | 86.4 | |
| | | | | | | | | | | | |
| | $ | 621.6 | | | $ | 651.1 | | | $ | 754.0 | |
| | | | | | | | | | | | |
Three of our customers each accounted for more than 10% of total net product sales for the year ended December 31, 2011. Four of our customers each accounted for more than 10% of total net product sales for the year ended December 31, 2010, and three of our customers each accounted for more than 10% of total net product sales for the year ended December 31, 2009. The following table summarizes the percent of our total net product sales that were attributed to each of these four customers (as a % of net product sales):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
McKesson Corporation | | | 38 | % | | | 36 | % | | | 38 | % |
Cardinal Health, Inc. | | | 35 | % | | | 35 | % | | | 35 | % |
Medco Health Solutions | | | 14 | % | | | 14 | % | | | 12 | % |
Amerisourcebergen Drug Corporation | | | * | % | | | 10 | % | | | * | % |
We invest our excess cash in accordance with our investment policy; our investments include US Government securities, securities of agencies sponsored by the US Government, asset-backed securities, mortgage-backed securities, debt instruments of financial institutions and corporations with investment- grade credit ratings. We mitigate credit risk by maintaining a well diversified portfolio and limiting the amount of investment exposure as to institution, maturity and investment type. Financial instruments that potentially subject us to significant credit risk consist principally of cash equivalents and short-term investments.
Cash and Cash Equivalents
We consider instruments with a maturity date of less than 90 days from the date of purchase to be cash equivalents.
Restricted Cash
Restricted cash relates to cash that is pledged as collateral for letters of credit issued by us, primarily in connection with office leases, pursuant to an Amended and Restated Letter of Credit and Cash Collateral Agreement entered into in December 2011.
Fair Value Measurements
We have certain financial assets and liabilities recorded at fair value which have been classified as Level 1, 2 or 3 within the fair value hierarchy as described in the accounting standards for fair value measurements.
The authoritative guidance for fair value measurements defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or the most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Market participants are buyers and sellers in the principal market that are (i) independent, (ii) knowledgeable, (iii) able to transact, and (iv) willing to transact. The guidance prioritizes the inputs used in measuring fair value into the following hierarchy:
| | |
| Level 1 | | Quoted prices (unadjusted) in active markets for identical assets or liabilities; |
| |
| Level 2 | | Inputs other than quoted prices included within Level 1 that are either directly or indirectly observable; and |
| |
| Level 3 | | Unobservable inputs in which little or no market activity exists, therefore requiring an entity to develop its own assumptions about the assumptions that market participants would use in pricing. |
F-8
The following table summarizes the assets and liabilities measured at fair value on a recurring basis (in thousands):
| | | | | | | | | | | | | | | | |
| | | Fair value measurements as of
December 31, 2011 | |
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | | | | | | | | |
Cash and cash equivalents | | $ | 99,859 | | | $ | 62,100 | | | $ | 37,759 | | | $ | — | |
Short-term investments | | | 104,206 | | | | 63,188 | | | | 41,018 | | | | — | |
Restricted cash | | | 10,519 | | | | 10,519 | | | | — | | | | — | |
Deferred compensation plan assets | | | 8,192 | | | | 8,192 | | | | — | | | | — | |
Embedded derivative (2) | | | 7,690 | | | | — | | | | — | | | | 7,690 | |
| | | | | | | | | | | | | | | | |
| | $ | 230,466 | | | $ | 143,999 | | | $ | 78,777 | | | $ | 7,690 | |
| | | | | | | | | | | | | | | | |
Liabilities: | | | | | | | | | | | | | | | | |
Deferred compensation plan liabilities | | $ | 8,192 | | | $ | 8,192 | | | $ | — | | | $ | — | |
Loss protection liability carried at fair value (3) | | | 46,419 | | | | — | | | | — | | | | 46,419 | |
| | | | | | | | | | | | | | | | |
| | $ | 54,611 | | | $ | 8,192 | | | $ | — | | | $ | 46,419 | |
| | | | | | | | | | | | | | | | |
| |
| | | Fair value measurements as of
December 31, 2010 | |
| | | Total | | | Level 1 | | | Level 2(1) | | | Level 3 | |
Assets: | | | | | | | | | | | | | | | | |
Cash and cash equivalents | | $ | 164,521 | | | $ | 95,090 | | | $ | 69,431 | | | $ | — | �� |
Short-term investments | | | 278,142 | | | | 141,767 | | | | 136,375 | | | | — | |
Restricted cash | | | 15,000 | | | | 15,000 | | | | — | | | | — | |
Deferred compensation plan assets | | | 8,520 | | | | 8,520 | | | | — | | | | — | |
| | | | | | | | | | | | | | | | |
| | $ | 466,183 | | | $ | 260,377 | | | $ | 205,806 | | | $ | — | |
| | | | | | | | | | | | | | | | |
Liabilities: | | | | | | | | | | | | | | | | |
Deferred compensation plan liabilities | | | 8,520 | | | | 8,520 | | | | — | | | | — | |
| | | | | | | | | | | | | | | | |
| | $ | 8,520 | | | $ | 8,520 | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | | | | |
| (1) | During 2011, the Company changed how it categorizes amounts within the fair value hierarchy and thus, the amounts now reported as Level 2 fair value instruments at December 31, 2010 were previously shown as Level 1 and have been reclassified. |
| (2) | The embedded derivative is associated with the promissory note related to the Revenue Sharing Obligation, or RSO, issued in connection with the Termination Agreement (see Note 2). The embedded derivative represents embedded options which would allow Amylin to discharge all or a portion of the obligation under certain circumstances (see Note 8 for the terms of the RSO). The fair value of the embedded derivative is calculated using a probability weighted expected return method, or PWERM. Significant inputs to the valuation include the following: |
| | • | | Management’s estimates of total revenue of exenatide products over the period in which the RSO is to be repaid; |
| | • | | A variety of scenarios under which management estimated the probability that BYDUREONTM (exenatide for extended-release injectable suspension) would be approved by the US Food and Drug Administration , or the FDA, prior to June 30, 2014; |
| | • | | A variety of scenarios under which all exenatide products would be removed from certain geographic markets for safety or efficacy reasons and remain unsalable for four consecutive years. |
| (3) | The loss protection liability arose in connection with the Termination Agreement (see Note 2) and relates to payments required to be made prior to the final transfer of the markets outside the United States from Lilly to Amylin. The maximum that could be paid under the loss protection liability is $60.0 million over the period December 1, 2011 and December 31, 2013. The fair value of the loss protection liability is calculated using a PWERM. Significant inputs to the valuation include the following: |
| | • | | Financial projections for markets outside the United States, or the OUS markets, which were provided to Amylin’s management by Lilly; |
| | • | | A variety of scenarios under which management estimated the extent to which these financial projections would be achieved, and the resulting payouts of the amounts that could be made for the twelve months ended December 31, 2012 and 2013. |
F-9
There were no transfers between fair value measurement levels during the years ended December 31, 2011 and 2010, respectively.
The following table presents a reconciliation of the assets and liabilities measured at fair value on a quarterly basis using significant unobservable inputs (Level 3) from January 1, 2011 to December 31, 2011 (in thousands):
| | | | |
| | | Derivative
Asset –
Embedded
Options | |
Assets: | | | | |
Embedded derivative: | | | | |
Balance at January 1, 2011 | | $ | — | |
Initial recognition of asset at fair value, November 2011 | | | 23,441 | |
Adjustment to fair value charged to expense | | | (15,751 | ) |
| | | | |
Balance at December 31, 2011 | | $ | 7,690 | |
| | | | |
| |
| | | Loss Protection
Liability carried at
fair value | |
Liabilities: | | | | |
Loss protection liability carried at fair value: | | | | |
Balance at January 1, 2011 | | $ | — | |
Initial recognition of liability at fair value, November 2011 | | | 30,758 | |
Adjustment to fair value charged to expense | | | 15,661 | |
| | | | |
Balance at December 31, 2011 | | $ | 46,419 | |
| | | | |
The fair value adjustment related to the embedded derivative represents the change in the fair value of the embedded option that relates to BYDUREON approval. As of December 31, 2011 we reviewed the status of the FDA regulatory approval process related to BYDUREON and re-assessed the likelihood that BYDUREON would receive FDA approval, which resulted in a decrease in the value of the related derivative asset.
The fair value adjustment related to the loss protection liability arose based upon new information we received regarding the amount of estimated loss protection we would be likely to pay to Lilly under this obligation.
Short-Term Investments
Our short-term investments, consisting principally of debt securities, are classified as available-for-sale, are stated at fair value and consist of both Level 1 and Level 2 financial instruments in the fair value hierarchy. We base the fair value of our Level 1 financial instruments that are in active markets using quoted market prices for identical instruments. Our Level 1 financial instruments include money market funds and mutual fund investments. We obtain the fair value of our Level 2 financial instruments, which are not in active markets, from a primary professional pricing source using quoted market prices for identical or comparable instruments, rather than direct observations of quoted prices in active markets. Fair value obtained from this professional pricing source can also be based on pricing models whereby all significant observable inputs, including maturity dates, issue dates, settlement date benchmark yields, reported trades, broker-dealer quotes, issue spreads, benchmark securities, bids, offers or other market related data, are observable or can be derived from or corroborated by observable market data for substantially the full term of the asset. We validate the quoted market prices provided by our primary pricing service by comparing the fair values of our Level 2 investment portfolio balance provided by our primary pricing service against the fair values of our Level 2 investment portfolio balance provided by our investment managers.
Unrealized holding gains or losses on these securities are included in other comprehensive loss in equity, net of related tax effects. The amortized cost of debt securities in this category is adjusted for amortization of premiums and accretion of discounts to maturity. For investments in mortgage-backed securities, amortization of premiums and accretion of discounts are recognized in interest income using the interest method, adjusted for anticipated prepayments as applicable. Estimates of expected cash flows are updated periodically and changes are recognized in the calculated effective yield prospectively as appropriate. Such amortization is included in interest income. Realized gains and losses are included in interest income and declines in value judged to be other-than-temporary on available-for-sale securities are included in impairment loss on investments. In assessing potential impairment of our short-term investments, we evaluate the impact of interest rates, potential prepayments on mortgage-backed securities, changes in credit quality, the length of time and extent to which the market value has been less than cost, and our intent and ability not to sell the security in order to allow for an anticipated recovery in fair value. The cost of securities sold is based on the specific-identification method.
F-10
Accounts Receivable
Trade accounts receivable are recorded net of allowances for cash discounts for prompt payment, doubtful accounts, product returns and chargebacks. Allowances for rebate discounts and distribution fees are included in other current liabilities in the accompanying consolidated balance sheets. Estimates for allowances for doubtful accounts are determined based on existing contractual obligations, historical payment patterns and individual customer circumstances. The allowance for doubtful accounts was $2.1 million and $1.0 million at December 31, 2011 and 2010, respectively.
Inventories, net
Inventories are stated at the lower of cost or market (net realizable value) and net of a valuation allowance for potential excess or obsolete material, with no reserve and $1.4 million at December 31, 2011 and December 31, 2010, respectively. Cost is determined by the first-in, first-out method.
Raw materials consist of bulk drug material for BYETTA, SYMLIN and BYDUREON. Work-in-process inventories consist of in-process BYETTA cartridges, in-process SYMLIN cartridges and in-process vials for BYDUREON. Finished goods inventories consist of BYETTA drug product in a disposable pen/cartridge delivery system and finished SYMLIN drug product in a disposable pen/cartridge delivery system.
We expense costs relating to the purchase and production of pre-approval inventories for which the sole use is pre-approval products as research and development expense in the period incurred until such time as we believe future commercialization is probable and future economic benefit is expected to be realized. Beginning in the fourth quarter of 2011 we began capitalizing pre-approval inventory specific to BYDUREON based upon management’s judgment of probable future commercial use and net realizable value. As of December 31, 2011, we have capitalized $45.1 million of pre-approval inventories. As disclosed in Note 14, on January 27, 2012 the FDA approved BYDUREON for commercial sale in the United States.
Property, Plant and Equipment
Property, plant and equipment is recorded at cost. Depreciation is computed using the straight-line method over the estimated useful lives of the assets as follows:
| | |
| Land improvements | | 10 years |
| Laboratory equipment | | 5 to 10 years |
| Leasehold improvements | | Lesser of the lease term or the useful life |
| Production equipment | | 10 years |
| Office equipment, furniture and computer software | | 3 to 5 years |
| Buildings | | 5 to 40 years |
We recorded depreciation expense of $49.4 million, $57.3 million, and $37.8 million in the years ended December 31, 2011, 2010 and 2009, respectively.
Interest costs incurred during the construction of major capital projects are capitalized until the underlying asset is ready for its intended use, at which point the interest cost is amortized as depreciation expense over the estimated useful life of the asset.
FDA validation costs, which to date relate to our manufacturing facility for BYDUREON, are capitalized as part of the effort required to acquire and construct long-lived assets, including readying them for their initial intended use, and are amortized over the estimated useful life of the asset.
We record impairment losses on property, plant and equipment used in operations when events and circumstances indicate that assets might be impaired and the undiscounted cash flows estimated to be generated by those assets are less than the carrying amount of those assets. We are subject to regulatory requirements with respect to our currently approved products and product candidates that can result in us not obtaining approval for product candidates in development or even discontinuance of the ability to sell our existing products. Therefore, we must regularly evaluate our ability to realize assets associated with our products and product candidates, including our BYDUREON manufacturing facility. As of December 31, 2011 there are no indicators of impairment associated with such assets. We also record assets to be disposed of at the lower of their carrying amount or fair value less cost to sell. For the years ended December 31, 2011, 2010 and 2009, we recorded $0.2 million, $7.4 million and $0 million, respectively, in asset impairments related to impaired leasehold improvements
F-11
associated with facility leases we will no longer use in our operations as part of our restructuring discussed in Note 5. While we have a history of operating and cash flow losses, we believe the expected future cash flows to be received support the carrying value of our long-lived assets and accordingly, we have not recognized any material impairment losses as of December 31, 2011, other than the impaired leasehold improvements noted above.
Investments in Unconsolidated Entities
We use the equity method of accounting for investments in other companies that are not controlled by us and in which our interest is generally between 20% and 50% of the voting shares or we have significant influence over the entity, or both. Our share of the income or losses of these entities is included in interest and other expense. As of December 31, 2011, we have no investments in unconsolidated entities. As of December 31, 2010 the net book value of such assets totaled $2.3 million and was included in other long-term assets. During the year ended December 31, 2011 we did not record any equity method investee gains or losses. We recorded $3.4 million and $4.0 million of equity method investee losses during the years ended December 31, 2010 and 2009, respectively. During the year ended December 31, 2010 we recognized an impairment loss of $1.7 million on one of our equity method investments. We recognized the impairment loss after assessing the financial and technical performance of the entity in which the investment was made as well as the entity’s ability to raise additional capital in significantly deteriorated financial markets to fund ongoing operations. There were no such impairments during the years ended December 31, 2011 or December 31, 2009.
Intangible assets
Our intangible assets consist of the consideration allocated to the reacquired economic interest in the US for BYETTA. This intangible asset is being amortized over the expected economic use of the asset. Our amortization policy reflects the pattern by which the economic benefits of the intangible assets are consumed, and that pattern is reliably determinable.
| | | | | | | | | | | | | | | | |
| | | Estimated | | | Gross | | | | | | | |
| | | Amortization Period | | | Carrying | | | Accumulated | | | Intangible | |
| | | (years) | | | Amount | | | Amortization | | | Assets, Net | |
Intangible Assets Subject to Amortization: | | | | | | | | | | | | | | | | |
Intangible asset related to reacquired economic interest in the US for BYETTA | | | 13 | | | $ | 277,848 | | | $ | (4,006 | ) | | $ | 273,842 | |
| | | | | | | | | | | | | | | | |
For the year ended December 31, 2011 total expense related to the amortization of intangible assets was $4.0 million. For the years ended December 31, 2010 and 2009 there was no amortization expense associated with intangible asset amortization.
The estimated annual amortization of intangible assets for the next five years is shown in the following table (in thousands). Actual amortization expense reported in future periods could differ from these estimates.
| | | | |
| Twelve months ended December 31, | | Estimated
Amortization
Expense | |
2012 | | $ | 31,122 | |
2013 | | | 26,069 | |
2014 | | | 26,224 | |
2015 | | | 24,191 | |
2016 | | | 21,373 | |
Thereafter: | | | 144,863 | |
| | | | |
| | $ | 273,842 | |
| | | | |
F-12
Net Loss Per Share
Basic and diluted net loss applicable to common stock per share is computed using the weighted average number of common shares outstanding during the period. Shares used in calculating basic and diluted net loss per common share exclude the following common share equivalents (in thousands):
| | | | | | | | | | | | |
| | | Years ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Antidilutive options and awards to purchase common stock | | | 388 | | | | 1,432 | | | | 209 | |
Antidilutive shares underlying convertible senior notes | | | 11,091 | | | | 15,238 | | | | 15,238 | |
| | | | | | | | | | | | |
| | | 11,479 | | | | 16,670 | | | | 15,447 | |
| | | | | | | | | | | | |
In future periods, if we report net income and the common share equivalents for our convertible senior notes are dilutive, the common stock equivalents will be included in the weighted average shares computation and interest expense related to the notes will be added back to net income to calculate diluted earnings per share.
Derivative Financial Instruments
The Company’s promissory note related to the RSO payable to Lilly (see Notes 2 and 8) contains an embedded feature that allows Amylin to discharge the obligation under certain circumstances. Specifically, the RSO could be fully discharged in the event (i) BYDUREON is not approved by June 30, 2014 or (ii) in the event all exenatide products are removed from either the US market, the OUS market or worldwide, for four consecutive years for safety or efficacy reasons. In accordance with applicable authoritative guidance for derivative instruments, we have bifurcated the embedded options from the RSO and are accounting for them as derivative assets. These embedded options were initially recorded as derivative assets at their fair value, defined as Level 3 in the fair value hierarchy.
From time to time we mitigate certain financial exposures, including currency risk and interest rate risk, through a controlled program of risk management that includes the use of derivative financial instruments. Derivatives are recorded on the balance sheet at fair value, with changes in value being recorded in interest and other income and interest and other expense.
We recognized unrealized losses on derivative financial instruments of $15.8 million for the year ended December 31, 2011, realized gains on derivative financial instruments of $2.8 million for the year ended December 31,2010 and unrealized gains of $1.9 million on derivative financial instruments for the year ended December 31, 2009.
The following table summarizes the fair value and balance sheet classification of our derivative financial instruments as of December 31, 2011 and 2010 (in thousands):
| | | | | | | | | | | | |
| | | December 31, 2011 | | December 31, 2010 |
| | | Fair Value | | | Balance sheet
location | | Fair Value | | | Balance sheet
location |
Embedded derivative (tied to FDA approval of BYDUREON) | | $ | 5,249 | | | Other current
assets | | $ | — | | | Other current
assets |
Embedded derivative (tied to safety or efficacy issues associated with exenatide products) | | | 2,441 | | | Other long-term
assets | | | — | | | Other long-term
assets |
| | | | | | | | | | | | |
Total derivative assets | | $ | 7,690 | | | | | $ | — | | | |
| | | | | | | | | | | | |
Comprehensive Loss
Comprehensive loss includes net loss and unrealized gains and losses on investments net of related tax effects. We disclose the accumulated balance of other comprehensive loss as a separate component of stockholder’s equity.
F-13
Accounting for Stock-Based Compensation
Stock-based compensation expense for equity-classified awards, principally related to stock options, restricted stock units, or RSUs, and stock purchase rights under the Employee Stock Purchase Plan, or ESPP, is measured at the grant date, based on the estimated fair value of the award and is recognized over the employees requisite service period. We use the Black-Scholes-Merton option-pricing model to estimate the fair value of stock options granted and stock purchase rights under the ESPP. The assumptions used for the specified reporting periods and the resulting estimates of weighted-average estimated fair value per share of options granted and employee stock purchase rights during those periods are as follows:
| | | | | | | | | | | | |
| | | Years ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Volatility—stock options | | | 60.4 | % | | | 60.9 | % | | | 80.9 | % |
Volatility—stock purchase rights | | | 78.7 | % | | | 49.6 | % | | | 97.9 | % |
Expected life in years—stock options | | | 4.3 | | | | 4.2 | | | | 4.3 | |
Expected life in years—stock purchase rights | | | 0.5 | | | | 0.5 | | | | 0.5 | |
Risk-free interest rate—stock options | | | 2.0 | % | | | 2.7 | % | | | 1.6 | % |
Risk-free interest rate—stock purchase rights | | | 0.1 | % | | | 0.2 | % | | | 0.4 | % |
Dividend yield | | | — | % | | | — | % | | | — | % |
Weighted average estimated fair value per share of options granted | | $ | 7.28 | | | $ | 9.10 | | | $ | 5.73 | |
Weighted average estimated fair value per share of stock purchase rights granted | | $ | 4.75 | | | $ | 5.77 | | | $ | 4.04 | |
We estimate volatility based upon the historical volatility of our common stock for a period corresponding to the expected term of our employee stock options and the implied volatility of market-traded options on our common stock with various maturities between six months and two years. The determination to use implied volatility in addition to historical volatility was based upon the availability of actively traded options on our common stock and our assessment that the addition of implied volatility is more representative of future stock price trends than historical volatility alone.
The expected life of our employee stock options represents the weighted-average period of time that options granted are expected to be outstanding in consideration of historical exercise patterns and the assumption that all outstanding options will be exercised at the mid-point of the then current date and their maximum contractual term.
The risk-free interest rates are based on the yield curve of US Treasury strip securities in effect at the time of grant for periods corresponding with the expected life of our employee stock options. We have never paid dividends and do not anticipate doing so for the foreseeable future. Accordingly, we have assumed no dividend yield for purposes of estimating the fair value of our stock-based payments to employees.
The fair values of RSUs are estimated based on the market price of our common stock on the date of grant. The weighted-average estimated fair values of employee RSUs granted during 2011 and 2010 were $14.13 and $14.33, respectively. No RSUs were granted in 2009.
Stock-based compensation expense recognized is based on awards ultimately expected to vest, and therefore is reduced by expected forfeitures. We estimate forfeitures based upon historical forfeiture rates, and will adjust our estimate of forfeitures if actual forfeitures differ, or are expected to differ, from such estimates. Changes in estimated forfeitures will be recognized through a cumulative adjustment in the period of the change and will also impact the amount of stock-based compensation expense in future periods.
Total employee non-cash stock-based compensation expense by operating statement classification is presented below (in thousands):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Selling, general and administrative expenses | | $ | 21,716 | | | $ | 24,053 | | | $ | 28,351 | |
Research and development expenses | | | 10,756 | | | | 11,969 | | | | 15,411 | |
| | | | | | | | | | | | |
Total | | $ | 32,472 | | | $ | 36,022 | | | $ | 43,762 | |
| | | | | | | | | | | | |
Stock-based compensation expense capitalized as part of inventory and fixed assets was negligible and did not impact our reported cash flows for the years ended December 31, 2011, 2010 and 2009.
F-14
In addition to the stock-based compensation discussed above, we also record non-cash expense associated with our Employee Stock Ownership Plan, or ESOP, and our 401(k) plan. The breakdown of non-cash ESOP and 401(k) expense by operating statement classification is presented below (in thousands):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Selling, general and administrative expenses | | $ | 9,809 | | | $ | 11,349 | | | $ | 11,609 | |
Research and development expenses | | | 6,718 | | | | 8,386 | | | | 8,552 | |
| | | | | | | | | | | | |
Total | | $ | 16,527 | | | $ | 19,735 | | | $ | 20,161 | |
| | | | | | | | | | | | |
Recently Issued Accounting Pronouncements
In December 2011, the FASB issued ASU 2011-11, Balance Sheet (Topic 210): Disclosures about Offsetting Assets and Liabilities (ASU 2011-11). This newly issued accounting standard requires an entity to disclose both gross and net information about instruments and transactions eligible for offset in the statement of financial position as well as instruments and transactions executed under a master netting or similar arrangement and was issued to enable users of financial statements to understand the effects or potential effects of those arrangements on its financial position. This ASU is required to be applied retrospectively and is effective for fiscal years, and interim periods within those years, beginning on or after January 1, 2013. As this accounting standard only requires enhanced disclosure, the adoption of this standard is not expected to have an impact our financial position or results of operations.
In June 2011, the FASB issued ASU No. 2011-05, “Comprehensive Income (Topic 220): Presentation of Comprehensive Income ” (ASU 2011-05). This newly issued accounting standard (1) eliminates the option to present the components of other comprehensive income as part of the statement of changes in stockholders’ equity; (2) requires the consecutive presentation of the statement of net income and other comprehensive income; and (3) requires an entity to present reclassification adjustments on the face of the financial statements from other comprehensive income to net income. The amendments in this ASU do not change the items that must be reported in other comprehensive income or when an item of other comprehensive income must be reclassified to net income nor do the amendments affect how earnings per share is calculated or presented. In December 2011, the FASB issued ASU No. 2011-12, Deferral of the Effective Date for Amendments to the Presentation of Reclassifications of Items Out of Accumulated Other Comprehensive Income in Accounting Standards Update No. 2011-05 , which defers the requirement within ASU 2011-05 to present on the face of the financial statements the effects of reclassifications out of accumulated other comprehensive income on the components of net income and other comprehensive income for all periods presented. During the deferral, entities should continue to report reclassifications out of accumulated other comprehensive income consistent with the presentation requirements in effect prior to the issuance of ASU 2011-05. These ASUs are required to be applied retrospectively and are effective for fiscal years, and interim periods within those years, beginning after December 15, 2011, which for Amylin means January 1, 2012. As these accounting standards do not change the items that must be reported in other comprehensive income or when an item of other comprehensive income must be reclassified to net income, the adoption of these standards is not expected to have an impact on our financial position or results of operations.
2. Settlement and Termination of Lilly Collaboration
As indicated in Note 1, effective November 7, 2011 Amylin and Lilly entered enter into the Termination Agreement to terminate their collaboration for exenatide and resolve the outstanding litigation between the companies. As part of the agreement, the parties agreed to transition full responsibility for the worldwide development and commercialization of exenatide to Amylin, starting in the United States, or US, on November 30, 2011 and progressing to all markets by the end of 2013. In the event the OUS transition does not occur by December 31, 2013, our transition agreement with Lilly contains various provisions with respect to future rights and obligations of each party that extend beyond December 31, 2013.
Under the terms of the new agreement, Amylin agreed to make a one-time, upfront payment to Lilly of $250 million. Amylin also agreed to make future revenue sharing payments to Lilly in an amount equal to 15 percent of global net sales of exenatide products until Amylin has made aggregate payments to Lilly of $1.2 billion plus accrued interest. In connection with this revenue sharing obligation, Amylin issued a secured promissory note in the amount of $1.2 billion, the terms of which are further described in Note 8. If Amylin’s investigational once weekly version of exenatide, BYDUREON, has not received FDA approval prior to June 30, 2014, Amylin’s revenue sharing obligations will terminate, and Amylin shall thereafter pay Lilly 8 percent of global net sales of exenatide products. With the FDA approval of BYDUREON on January 27, 2012 (see Note 14), this option for the debt to be discharged and replaced by an 8% royalty has expired. In addition, the note, or a portion of the note, would be fully discharged if all exenatide products are withdrawn from either the US market, all of Europe or both for four consecutive years for safety or efficacy reasons. In the event Amylin receives upfront or milestone payments from a third party in connection with an agreement with respect to exenatide products, Amylin is
F-15
obligated to make payments on the RSO equal to 20% of such upfront or milestone payments. Amylin will also pay a $150 million milestone to Lilly contingent upon FDA approval of a once monthly suspension version of exenatide that is currently in Phase 2. The companies also agreed that the maturity date for the $165 million line of credit that Amylin drew from Lilly in May 2011, or the Lilly Note Payable will be extended from the second quarter of 2014 to the second quarter of 2016; no other terms of the Lilly Note Payable were changed.
Lilly’s involvement in the US commercial operations ceased on November 30, 2011. Regarding the OUS markets, Lilly is to transfer responsibility for commercialization of BYETTA injection and BYDUREON to Amylin or its designee by December 31, 2013, which is the expected end of the OUS Transition period. Amylin will provide input to Lilly regarding their plans to manage and ultimately transfer the OUS markets to Amylin during the OUS Transition period and has guaranteed to reimburse Lilly for any losses they may incur pertaining to the OUS exenatide-related activities during that period, up to a total of $60 million. In addition to the consideration specifically outlined in the Termination Agreement, the total consideration was adjusted for a settlement of pre-existing relationships.
We evaluated whether the Termination Agreement should be accounted for as a single transaction or as a transaction that consists of separate elements relating to the OUS operations and US operations. Because the OUS operations and US operations (1) have independent economic value and substance, (2) could be purchased or sold on an individual basis and (3) qualify as a business combination and the reacquisition of previously shared economic interests through the termination of a contract, respectively, we determined that the OUS operations and US operations should be accounted for as separate elements. The OUS operation constitutes a business, as defined by ASC 805, “Business Combinations”, and the transition of the OUS operations will be accounted for as a business combination when control transfers from Lilly to Amylin. With respect to the contract termination, certain aspects of the US operations represent the reacquisition of a previously shared economic interest that qualify as an asset for developed products and the amounts relating to the unapproved products are expensed. The transaction also included the modification of the Lilly Note Payable and the settlement of pre-existing relationships, which resulted in an adjustment to the consideration transferred as described below.
The consideration transferred consists of the following:
| | | | |
| | | In 000’s | |
Upfront payment, in cash | | $ | 250,000 | |
Promissory note related to the RSO, at fair value | | | 976,821 | |
Loss protection liability related to the OUS Transition period | | | 30,758 | |
Settlement of pre-existing relationships, foregone milestone payments under collaboration agreement and other settled amounts, at fair value | | | (159,885 | ) |
| | | | |
Total consideration paid | | $ | 1,097,694 | |
Derivative asset embedded in promissory note related to RSO | | | (23,441 | ) |
Consideration paid to extending the maturity on Note Payable to Lilly | | | (10,112 | ) |
| | | | |
Net consideration related to exenatide rights reacquired in the US and to be acquired as a business OUS | | $ | 1,064,141 | |
| | | | |
The components of the consideration are described below:
| | • | | The RSO represents the fair value of Amylin’s obligation to make payments to Lilly in the amount of 15 percent of global net sales of exenatide products until an aggregate amount of $1.2 billion plus accrued interest is made. The RSO was valued using the income approach utilizing cash flow analyses projecting expected net sales for exenatide products over the life cycle. |
| | • | | The loss protection liability related to the OUS market is a financial instrument recorded at fair value and represents Amylin’s obligation to reimburse Lilly for exenatide-related losses in the OUS markets during the OUS Transition period. The liability was valued using a PWERM and the resulting estimated liability was discounted to present value. Significant inputs to the valuation are described in Note 1 under the headingFair Value Measurements. This liability is a Level 3 financial instrument. |
| | • | | The settlement of pre-existing relationships, foregone milestone payments under collaboration agreement and other settled amounts, at fair value relate primarily to payments Lilly made to Amylin in connection with our former collaboration which were recorded as deferred liabilities (see Note 5). To reflect the settlement of the pre-existing relationship between Amylin and Lilly, the unamortized portion of the deferred liabilities as of November 7, 2011 was removed from the balance sheet and allocated as a reduction of the consideration for the transaction. Additionally, included herein are foregone milestone payments under the collaboration agreement and other settled amounts primarily representing milestones associated with the commercialization of exenatide that Amylin was entitled to receive from Lilly under the former collaboration. Under the Termination Agreement, Amylin will forego the receipt of these milestones. The fair value of these foregone milestones was calculated using a PWERM and such payments were discounted to present value. Significant inputs to the valuation included a variety of scenarios under which management estimated the probability that BYDUREON would be approved by (1) the FDA prior to June 30, 2014 and (2) the regulatory agency of a certain OUS market for which Lilly had an obligation to pay Amylin a commercialization milestone. |
F-16
| | • | | The derivative asset embedded in the promissory note represents Amylin’s ability to discharge all or a portion of the obligation under certain circumstances (see Note 8 for the terms of the RSO). The fair value of the embedded derivative is calculated using a PWERM. Significant inputs to the valuation are described in Note 1 under the headingFair Value Measurements. This asset is a Level 3 financial instrument. |
| | • | | The consideration paid to modify the loan by extending the maturity on the note payable to Lilly represents the amount Amylin is deemed to have paid in connection with the extension of the maturity date of such note payable from May 23, 2014 to June 30, 2016. All other financial terms of the note are unchanged, including the interest rate of 5.51%, which is considered to be a below market interest rate. The deemed payment was valued using a discounted cash flow model which calculated the difference between the value of the additional interest payments that Amylin would have made at a market rate during the period of the debt extension (i.e., between May 23, 2014 and June 30, 2016) and the stated rate. |
The $150 million milestone payment which is payable upon FDA approval of once-monthly exenatide is considered to be contingent consideration for accounting purposes and was allocated between the US and OUS operations on a relative fair value basis as of the transaction date, with $103.5 million ascribed to the reacquired rights from the US contract termination and $46.5 million to the OUS business to be acquired. This contingent consideration was not included in the allocation of the consideration for either the US reacquired rights resulting from the termination of a contract or the OUS business for the following reasons:
| | • | | The US reacquired rights arising from the termination of a contract follows asset acquisition accounting which means that the contingent consideration arrangement will be recognized when the contingency is resolved and the consideration is paid or becomes payable. |
| | • | | The OUS operations will be accounted for as a business combination when control transfers from Lilly to Amylin; since the business combination has not yet been consummated, nothing has been recorded in connection with the OUS portion of the contingent consideration. |
In addition to the components of consideration summarized above, a total of $11.3 million of transaction costs were incurred in connection with the transaction. The transaction costs were allocated between the US reacquired rights for approved and unapproved products arising from the termination of a contract and the OUS business to be acquired. This resulted in total capitalized transaction costs of $3.0 million for the reacquired rights on approved products in the US and $8.3 million of transaction costs expensed as operating expense.
We have allocated the consideration based upon the relative fair values relating to the reacquired rights in the US arising from the termination of a contract and the OUS operations to be acquired as a business as of November 7, 2011 as follows:
| | | | |
| | | Total
Consideration
Allocated
(in 000’s) | |
US reacquired rights resulting from the termination of a contract: | | | | |
Intangible asset related to reacquired economic interest in the US for BYETTA | | $ | 274,849 | |
Reacquired economic interest in unapproved exenatide products in the US | | | 461,595 | |
| | | | |
Total allocated to US reacquired rights: | | | 736,444 | |
OUS operations to be acquired as a business: | | | | |
Economic interest in exenatide products to be reacquired as a business | | | 327,697 | |
| | | | |
Total consideration allocated | | $ | 1,064,141 | |
| | | | |
| | • | | The intangible asset related to the reacquired economic interest in the US for BYETTA represents the fair value of Lilly’s former share of the BYETTA US gross margin and was based upon estimated future cash flows for BYETTA, using the multi-period excess earnings discounted cash flow method. The intangible asset will be amortized over the expected economic use of the asset. |
F-17
| | • | | The reacquired economic interest in unapproved exenatide products in the US does not meet the definition of an asset given the uncertainty surrounding their future economic benefit and therefore these costs were expensed. The amount represents our anticipated increase in the US estimated cash flows for the unapproved products and was derived using the multi-period excess earnings discounted cash flow method to value the technology. |
| | • | | The economic interest in exenatide products to be reacquired as a business relates to the economic interests in the OUS markets which will be acquired upon completion of the OUS transition, anticipated to be no later than December 31, 2013, and represents the prepaid acquisition price. The amount allocated to this asset represents the OUS estimated cash flows for BYETTA, BYDUREON and exenatide products currently in development and were estimated using the multi-period excess earnings discounted cash flow method to value the economic rights. Because the OUS markets represent a business consisting of long-term assets to be acquired under ASC 805, the value ascribed to this asset is recorded as a long-term asset. |
The following table reconciles the amount allocated to reacquired economic interest in unapproved exenatide products in the US (i.e., the second item in the table above) to the net costs associated with reacquisition of economic interest in exenatide products, as reported for the year ended December 31, 2011 on the Consolidated Statements of Operations:
| | | | |
| | | Year ended
December 31, 2011
(in 000’s) | |
Cost to reacquire economic interest in unapproved exenatide products in the US | | $ | 461,595 | |
Foregone milestone payments under collaboration agreement and other settled amounts | | | (57,995 | ) |
Transaction costs related to the reacquisition of exenatide product rights | | | 8,320 | |
Loss on fair value adjustment for loss protection liability | | | 15,661 | |
Amortization of intangible asset for US exenatide rights | | | 4,006 | |
| | | | |
Net costs associated with reacquisition of economic interest in exenatide products | | $ | 431,587 | |
| | | | |
3. Investments
The following is a summary of our short-term investments as of December 31, 2011 and 2010 (in thousands):
| | | | | | | | | | | | | | | | |
| | | Available-for-Sale Securities | |
| | | Amortized
Cost | | | Gross
Unrealized
Gains(1) | | | Gross
Unrealized
Losses(1) | | | Estimated
Fair
Value | |
December 31, 2011 | | | | | | | | | | | | | | | | |
Obligations of US Government-sponsored enterprises | | $ | 40,914 | | | $ | 3 | | | $ | (45 | ) | | $ | 40,872 | |
Corporate debt securities | | | 62,466 | | | | 890 | | | | (22 | ) | | | 63,334 | |
| | | | | | | | | | | | | | | | |
Total | | $ | 103,380 | | | $ | 893 | | | $ | (67 | ) | | $ | 104,206 | |
| | | | | | | | | | | | | | | | |
December 31, 2010 | | | | | | | | | | | | | | | | |
US Treasury securities | | $ | 46,234 | | | $ | 4 | | | $ | — | | | $ | 46,238 | |
Obligations of US Government-sponsored enterprises | | | 37,826 | | | | 191 | | | | (50 | ) | | | 37,967 | |
Corporate debt securities | | | 191,680 | | | | 743 | | | | (23 | ) | | | 192,400 | |
Asset backed securities | | | 1,531 | | | | 6 | | | | — | | | | 1,537 | |
| | | | | | | | | | | | | | | | |
Total | | $ | 277,271 | | | $ | 944 | | | $ | (73 | ) | | $ | 278,142 | |
| | | | | | | | | | | | | | | | |
| (1) | Other comprehensive loss included unrealized losses of $1.6 million and $1.1 million on investments underlying our 2001 Non-Qualified Deferred Compensation Plan at December 31, 2011 and 2010, respectively. | |
The gross realized gains on sales of available-for-sale securities totaled approximately $0.2 million, $0.2 million and $0.7 million and the gross realized losses totaled $0.2 million, $0 million and $0.6 million for the years ended December 31, 2011, 2010 and 2009, respectively.
Contractual maturities of short-term investments at December 31, 2011 were as follows (in thousands):
| | | | |
| | | Fair Value | |
Due within 1 year | | $ | 65,034 | |
After 1 but within 5 years | | | 36,774 | |
After 5 but within 10 years | | | — | |
After 10 years | | | 2,398 | |
| | | | |
Total | | $ | 104,206 | |
| | | | |
F-18
For purposes of these maturity classifications, the final maturity date is used for securities not due at a single maturity date, specifically mortgage-backed securities, which are included in Obligations of US Government-sponsored enterprises in the table above, and asset-backed securities.
The following table shows the gross unrealized losses and fair value of our investments with unrealized losses that are not deemed to be other-than-temporarily impaired, aggregated by investment category and length of time that individual securities have been in a continuous unrealized loss position, at December 31, 2011 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Less than 12 Months | | | 12 Months or Greater | | | Total | |
| | | Fair
Value | | | Unrealized
Losses | | | Fair
Value | | | Unrealized
Losses | | | Fair
Value | | | Unrealized
Losses | |
Obligations of U.S Government- sponsored enterprises | | $ | 32,061 | | | $ | (35 | ) | | $ | 1,966 | | | $ | (10 | ) | | $ | 34,027 | | | $ | (45 | ) |
Corporate debt securities | | | 41,565 | | | | (22 | ) | | | — | | | | — | | | | 41,565 | | | | (22 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
| | $ | 73,626 | | | $ | (57 | ) | | $ | 1,966 | | | $ | (10 | ) | | $ | 75,592 | | | $ | (67 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
During the year ended December 31, 2009 we recognized a $1.4 million other-than-temporary impairment loss for credit-related losses associated with two securities in our portfolio. The impairment loss was based upon the difference between the amortized cost basis and the observed market prices for the securities.
The unrealized losses on our remaining investments are due in most instances to the increased volatility in the markets impacting the classes of securities we invest in and are not due to deterioration in credit ratings. Our investments have a short effective duration, and since we have the ability and intent not to sell these investments until a recovery of fair value, which may be maturity, we do not consider these investments to be other-than-temporarily impaired at December 31, 2011.
4. Other Financial Information
Inventories consist of the following (in thousands):
| | | | | | | | | | | | |
| | | December 31, 2011 | |
| | | Commercial | | | Pre-approval(1) | | | Total | |
Raw materials | | $ | 43,125 | | | $ | 31,668 | | | $ | 74,793 | |
Work-in-process | | | 16,703 | | | | 13,452 | | | | 30,155 | |
Finished goods | | | 7,011 | | | | — | | | | 7,011 | |
| | | | | | | | | | | | |
| | $ | 66,839 | | | $ | 45,120 | | | $ | 111,959 | |
| | | | | | | | | | | | |
| |
| | | December 31, 2010 | |
| | | Commercial | | | Pre-approval | | | Total | |
Raw materials | | $ | 90,183 | | | $ | — | | | $ | 90,183 | |
Work-in-process | | | 19,051 | | | | — | | | | 19,051 | |
Finished goods | | | 9,395 | | | | — | | | | 9,395 | |
| | | | | | | | | | | | |
| | $ | 118,629 | | | $ | — | | | $ | 118,629 | |
| | | | | | | | | | | | |
| (1) | Pre-approval inventories include raw materials and work-in-process for the manufacture of BYDUREON. As indicated in Note 14, on January 27, 2012 we announced that we received FDA approval for BYDUREON. |
Inventories are stated at the lower of cost or market (net realizable value) and net of a valuation allowance for potential excess or obsolete material of $0 and $1.4 million at December 31, 2011 and 2010, respectively. Raw materials consist of bulk drug material for BYETTA, SYMLIN and BYDUREON. Work-in-process inventories consist of in-process BYETTA cartridges, in-process SYMLIN cartridges and in-process BYDUREON vials. Finished goods inventories consist of BYETTA drug product in a disposable pen/cartridge delivery system, finished SYMLIN drug product in a disposable pen/cartridge delivery system.
F-19
Other current assets consist of the following (in thousands):
| | | | | | | | |
| | | At December 31, | |
| | | 2011 | | | 2010 | |
Prepaid expenses | | $ | 24,669 | | | $ | 16,272 | |
Interest and other receivables | | | 16,297 | | | | 20,631 | |
Other current assets | | | 8,192 | | | | 8,555 | |
| | | | | | | | |
| | $ | 49,158 | | | $ | 45,458 | |
| | | | | | | | |
Property, plant and equipment consist of the following (in thousands):
| | | | | | | | |
| | | At December 31, | |
| | | 2011 | | | 2010 | |
Land and land improvements | | $ | 13,853 | | | $ | 13,853 | |
Laboratory equipment | | | 51,511 | | | | 54,710 | |
Leasehold improvements | | | 73,902 | | | | 73,381 | |
Production equipment | | | 141,287 | | | | 140,949 | |
Office equipment, furniture and computer software | | | 75,998 | | | | 87,588 | |
Buildings | | | 245,722 | | | | 245,396 | |
Construction in progress | | | 437,032 | | | | 370,058 | |
| | | | | | | | |
| | | 1,039,305 | | | | 985,935 | |
Less accumulated depreciation and amortization | | | (208,143 | ) | | | (174,190 | ) |
| | | | | | | | |
| | $ | 831,162 | | | $ | 811,745 | |
| | | | | | | | |
Construction in progress consists of costs associated with our manufacturing facility for BYDUREON, which is currently under construction in Ohio (see Note 4), and costs associated with the BYDUREON pen device. During 2011 we placed into service certain general plant assets and during 2010 we placed into service the filling line, warehouse and certain general plant assets.
Other current liabilities consist of the following (in thousands):
| | | | | | | | |
| | | At December 31, | |
| | | 2011 | | | 2010 | |
Accrued rebate discounts | | $ | 45,862 | | | $ | 41,094 | |
Accrued expenses | | | 43,458 | | | | 30,603 | |
Other current liabilities | | | 10,289 | | | | 6,655 | |
| | | | | | | | |
| | $ | 99,609 | | | $ | 78,352 | |
| | | | | | | | |
5. Collaborative Agreements
We have entered into various collaborative agreements which provide us with rights to develop, produce and market products using certain know-how, technology and patent rights maintained by our collaborative partners. Terms of the various collaboration agreements may require us to make or receive milestone payments upon the achievement of certain product research and development objectives and pay or receive royalties on future sales, if any, of commercial products resulting from the collaboration.
Amounts due from our collaborative partners related to development activities are generally reflected as an increase to or a reduction of research and development expenses and amounts due to or from our collaborative partners related to sharing of commercialization expenses are generally reflected as an increase to or reduction of selling, general and administrative expenses. Milestone payments and up-front payments received are generally reflected as collaborative revenue as discussed above in Note 1, and milestone payments and up-front payments made are generally recorded as research and development expenses if the payments relate to drug candidates that have not yet received regulatory approval. Milestone payments and up-front payments made related to approved drugs will generally be capitalized and amortized to cost of goods sold over the economic life of the product. Royalties received are generally reflected as collaborative revenues and royalties paid are generally reflected as cost of goods sold.
For collaborations with commercialized products, if we are the principal we record revenue and the corresponding operating costs in their respective line items within our statement of operations based on the nature of the shared expenses. If we are not the principal (which is the case for our sales of exenatide products to Lilly for sale outside the United States), we record operating costs as a reduction of revenue. The principal is the party who is responsible for delivering the product or service to the customer, has latitude with establishing price and has the risks and rewards of providing product or service to the customer, including inventory and credit risk.
F-20
Collaboration with Eli Lilly and Company
In September 2002, we and Lilly entered into a Collaboration Agreement for the global development and commercialization of exenatide, or the Lilly Agreement. The Lilly Agreement was amended in 2006 and in 2009. As described in Note 2, on November 7, 2011 we and Lilly entered into the Termination Agreement to terminate our collaboration for exenatide and resolve the outstanding litigation between the companies. Under the agreement, the companies completed the US Transition on November 30, 2011, or the US Transition Date and the OUS Transition will occur no later than December 31, 2013. In the event the OUS transition does not occur by December 31, 2013, the agreement contains various provisions with respect to future rights and obligations of each party that extend beyond December 31, 2013.
The Lilly Agreement includes BYETTA, our twice-daily formulation of exenatide for the treatment of type 2 diabetes, and any sustained release formulations of exenatide such as BYDUREON, our once-weekly formulation of exenatide for the treatment of type 2 diabetes, which received FDA approval on January 27, 2012. Under the terms of the Lilly Agreement, operating profits from products sold in the United States were shared equally between us and Lilly through November 30, 2011, which is the date Lilly’s involvement in the United States commercial operations ceased. Lilly was responsible for 53% of shared exenatide global development and commercialization expenses that generate utility both in the United States and outside the United States through the US Transition Date; we were responsible for 47% of these expenses. Lilly is responsible for 100% of all exenatide development and commercialization expenses that generate utility predominantly outside of the US until the OUS Transition is complete. The Lilly Agreement provides for tiered royalties payable to us by Lilly based upon the annual gross margin for all OUS exenatide product sales, including any long-acting release formulations. Royalty payments for OUS exenatide product sales commenced during the second quarter of 2011 upon the achievement of a one-time cumulative gross margin threshold.
At the commencement of the Lilly Agreement, Lilly made initial non-refundable payments to us totaling $80 million, of which $50 million was amortized to revenues under collaborative agreements prior to 2004. The remaining $30 million was amortized to revenues ratably over a seven-year period which ended in 2009 and represented our estimate of the period of our performance of significant development activities under the agreement.
Under the Lilly Agreement, Lilly also agreed to make milestone payments contingent upon the commercial launch of exenatide in selected territories throughout the world, including both twice-daily and sustained release formulations. From the inception of the Lilly Agreement, the total commercial milestone payments earned and recorded as revenue through December 31, 2011 totaled $65 million, of which $15 million was earned upon the July 2011 launch of BYDUREON in the European Union.
In October 2008, we and Lilly entered into an Exenatide Once Weekly Supply Agreement, or the Supply Agreement, pursuant to which we agreed to supply commercial quantities of BYDUREON for sale in the US. Under the terms of the Supply Agreement, Lilly made a cash payment of $125 million to us, which represents an amount to compensate us for the estimated past and future cost of carrying Lilly’s share of the capital investment made in our manufacturing facility in Ohio. Lilly’s share of the capital investment would have otherwise been charged to them when we allocated product costs to them for products produced at the facility through our existing cost sharing arrangement. In addition to this cash payment, we intended to recover Lilly’s share of the capital investment in the facility through an allocation of depreciation expense in cost of goods as discussed below. Under the terms of the Supply Agreement, we agreed not to charge Lilly for its share of the interest costs capitalized to the facility or any future financing cost that may be related to financing the facility. The $125 million payment is comprised of the following two components:
| | • | | A reimbursement to us of Lilly’s share of interest costs capitalized to the facility. These interest costs were to be credited to Lilly in the form of a reduction of the cost of goods sold for BYDUREON as incurred under the Supply Agreement. The deferred credit associated with this balance was to be amortized to collaborative profit sharing over the estimated life of the underlying assets. As of November 7, 2011 the unamortized deferred credit associated with capitalized interest totaled $72.2 million. |
| | • | | Deferred collaborative revenue for services we would have provided to Lilly under the Supply Agreement. The deferred collaborative revenue was being amortized to revenues under collaborative agreements ratably over the economic useful life of the BYDUREON product. As of November 7, 2011 the unamortized deferred collaborative revenue balance totaled $48.3 million. |
F-21
In connection with the Termination Agreement, the Supply Agreement was amended and restated effective November 7, 2011, or the Amended and Restated Supply Agreement, pursuant to which Lilly will pay Amylin a fixed price for product supplied under the agreement and Amylin no longer has an obligation to reimburse Lilly for cost of goods sold for BYDUREON as incurred, nor does Amylin have an obligation to provide collaborative services to Lilly under the Supply Agreement. Under the terms of the Amended and Restated Supply Agreement, we are required to manufacture BYDUREON intended for commercial sale by Lilly in jurisdictions outside the United States through the OUS Transition period.
In May 2009, we and Lilly entered into a joint supply agreement for a BYDUREON pen device, or the Device Agreement. Through the effective date of the Termination Agreement we collaborated with Lilly in the development of a BYDUREON a dual chamber cartridge pen device. We and Lilly shared the capital and development costs and the total capital investment through November 7, 2011 was allocated approximately 60% to Lilly and 40% to us, with Lilly funding its share as the capital expenditures were incurred. Through December 31, 2011, we have incurred $216.4 million in capital expenditures associated with the BYDUREON pen device, which amount is included in construction in progress. Through November 7, 2011 Lilly has paid $103.4 million for these expenditures. Capital reimbursements from Lilly, which were included in deferred collaborative profit-sharing in the accompanying consolidated balance sheet, were being deferred and were to be amortized to collaborative profit sharing for Lilly’s share of the depreciation included in cost of goods sold for the BYDUREON pen device, as incurred. In connection with the Termination Agreement, the Device Agreement was amended and restated effective November 7, 2011 pursuant to which Amylin will pay Lilly a fixed price for product supplied under the agreement and Amylin no longer has an obligation to reimburse Lilly for these deferred costs.
Both the Supply Agreement and the Device Agreement were evaluated for settlement of a pre-existing relationship as of November 7, 2011 and the consideration allocated to the Termination Agreement was adjusted by the amount of the unamortized balance of the $125 million payment made by Lilly and the total capital expenditures paid to Amylin by Lilly; see further discussion in Note 2.
The following is a summary of activity related to our collaboration with Lilly and the location in the consolidated statements of operations (in thousands):
| | | | | | | | | | | | | | |
| | | Classification within
Consolidated Statements of | | Year Ended December 31, | |
Activity | | Operations | | 2011 | | | 2010 | | | 2009 | |
Royalty revenue | | Revenues under collaborative agreements | | $ | 3,845 | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | | |
Milestone payments | | Revenues under collaborative agreements | | $ | 15,000 | | | $ | 10,000 | | | $ | — | |
| | | | | | | | | | | | | | |
Amortization of up-front payments | | Revenues under collaborative agreements | | $ | 2,763 | | | $ | — | | | $ | 3,086 | |
| | | | | | | | | | | | | | |
Gross margin cost-sharing | | Collaborative profit sharing | | $ | (222,545 | ) | | $ | (257,127 | ) | | $ | (302,861 | ) |
| | | | | | | | | | | | | | |
Development expense cost-sharing payments received from Lilly for BYETTA and BYDUREON development expense | | Reduction of research and development expense | | $ | 79,288 | | | $ | 72,555 | | | $ | 66,571 | |
| | | | | | | | | | | | | | |
Cost-sharing payments due to Lilly for shared sales force expenses, marketing expenses and other commercial or operational support | | Increase to selling, general and administrative expense | | $ | (10,177 | ) | | $ | (25,388 | ) | | $ | (5,251 | ) |
| | | | | | | | | | | | | | |
Collaboration with Alkermes, Inc.
In May 2000 we entered into a development and license agreement with Alkermes Controlled Therapeutics, Inc. II, or Alkermes, a subsidiary of Alkermes, Inc., a company specializing in the development of products based on proprietary drug delivery technologies. The development and license agreement, or the Alkermes Agreement, was amended in 2005 and provides for Alkermes to assist us in the development, manufacture and commercialization of BYDUREON. Under the terms of the Alkermes Agreement, Alkermes has transferred to us its technology for manufacturing BYDUREON. We are responsible for manufacturing BYDUREON for commercial sale. In exchange, Alkermes is entitled to receive funding for research and development and may earn future milestone payments upon achieving specified development and commercialization goals. Alkermes will also receive royalties on any future product sales.
In addition to the collaboration agreement, Alkermes is also supplying us with the polymer materials required for the commercial manufacture of BYDUREON under a Supply Agreement dated December 29, 2007.
F-22
Collaboration with Takeda Pharmaceutical Company, Ltd
On October 30, 2009, we and Takeda Pharmaceutical Company Limited, or Takeda, entered into a License, Development and Commercialization Agreement, or the Takeda Agreement, pursuant to which the companies will co-develop and commercialize pharmaceutical products containing compounds specified in the Takeda Agreement for the treatment of human indications including, but not limited to, (i) weight management and/or obesity, (ii) glycemic control and (iii) cardiovascular disease. We received a one-time, nonrefundable cash payment of $75 million from Takeda in connection with the execution of the Takeda Agreement. We recorded the up-front payment as deferred revenue in our consolidated balance sheets and will recognize the revenue over the estimated development period of ten years. As of December 31, 2011 deferred revenue associated with the Takeda Agreement equaled $58.8 million, of which $51.3 million is classified as long-term.
The following is a summary of activity related to the Takeda Agreement and the location in the consolidated statements of operations (in thousands):
| | | | | | | | | | | | | | |
| | | Classification within
Consolidated Statements of | | Year Ended December 31, | |
Activity | | Operations | | 2011 | | | 2010 | | | 2009 | |
Amortization of up-front payments | | Revenues under collaborative agreements | | $ | 7,500 | | | $ | 7,500 | | | $ | 1,250 | |
| | | | | | | | | | | | | | |
Cost-sharing payments due from Takeda for shared development expenses | | Reduction of research and development expense | | $ | 11,642 | | | $ | 16,697 | | | $ | 1,530 | |
| | | | | | | | | | | | | | |
6. Restructuring
During 2011 we reduced our workforce, referred to as the 2011 Restructuring, and recorded restructuring charges of $7.2 million consisting of employee separation costs, facilities-related charges, asset impairment charges and other direct incremental charges.
During 2010 we reduced our workforce, referred to as the 2010 Restructuring, and recorded restructuring charges of $16.8 million consisting of employee separation costs, facilities-related charges, asset impairments charges and other direct incremental charges.
In 2009, we announced a restructuring of our sales force, or the 2009 Restructuring. We recorded restructuring charges of $17.0 million during the year ended December 31, 2009 consisting of employee separation costs, facilities related charges and other direct incremental charges.
The costs associated with the 2011, 2010 and 2009 Restructuring activities were reported as a separate line item in the accompanying Consolidated Statement of Operations for each year and were comprised of the following components (in thousands):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | Accruals | | | Non-cash
items | | | Total | |
2009 Activity | | | | | | | | | | | | |
Facilities related charges | | $ | 5,391 | | | $ | 213 | | | $ | 5,604 | |
Employee separation costs | | | 10,629 | | | | 281 | | | | 10,910 | |
Other restructuring charges | | | 466 | | | | — | | | | 466 | |
| | | | | | | | | | | | |
Total restructuring charges for the year ended December 31, 2009: | | $ | 16,486 | | | $ | 494 | | | $ | 16,980 | |
| | | | | | | | | | | | |
2010 Activity | | | | | | | | | | | | |
Facilities related charges | | $ | 12,388 | | | $ | (6,629 | ) | | $ | 5,759 | |
Employee separation costs | | | 3,156 | | | | — | | | | 3,156 | |
Asset impairments | | | — | | | | 7,398 | | | | 7,398 | |
Other restructuring charges | | | 467 | | | | — | | | | 467 | |
| | | | | | | | | | | | |
Total restructuring charges for the year ended December 31, 2010: | | $ | 16,011 | | | $ | 769 | | | $ | 16,780 | |
| | | | | | | | | | | | |
2011 Activity | | | | | | | | | | | | |
Facilities related charges | | $ | 1,787 | | | $ | — | | | $ | 1,787 | |
Employee separation costs | | | 5,108 | | | | — | | | | 5,108 | |
Asset impairments | | | (399 | ) | | | 645 | | | | 246 | |
Other restructuring charges | | | 49 | | | | — | | | | 49 | |
| | | | | | | | | | | | |
Total restructuring charges for the year ended December 31, 2011: | | $ | 6,545 | | | $ | 645 | | | $ | 7,190 | |
| | | | | | | | | | | | |
F-23
Facilities related charges recorded during the year ended December 31, 2009 consisted of additional estimated losses associated with the facility leases we ceased using in 2008. An additional loss of $5.6 million was recorded and was based upon recently executed sub-lease agreements and a related reassessment of current market conditions.
Facilities related charges recorded as part of the 2010 Restructuring included estimated losses associated with certain facility leases in our San Diego campus which we no longer use in our operations and which we ceased using in the year ended December 31, 2010 of $ 3.2 million. Additionally, we recorded additional estimated losses associated with the facility leases we ceased using in 2008 of $2.6 million based upon recently executed sub-lease agreements and a related reassessment of current market conditions. The facilities related charge recorded during the year ended December 31, 2010 is net of a non-cash credit related to the reversal of deferred rent associated with the leases.
In 2011, we recorded additional estimated losses associated with the facility leases we ceased using in 2008 and 2010 of $1.8 million based upon recently executed sub-lease agreements and a related reassessment of current market conditions.
We expect to incur approximately $8.9 million of accretion expense over the remaining term of the leases, which have expiration dates from 2015 to 2018.
Employee separation costs for the 2009, 2010 and 2011 Restructurings consist primarily of severance costs. Asset impairments recorded as part of the 2010 Restructuring primarily relate to impaired leasehold improvements associated with the facility leases discussed above. The asset impairment costs recorded as part of the 2011 Restructuring Other restructuring charges consist of incremental direct costs associated with the 2009, 2010 and 2011 Restructurings.
The following table sets forth activity in the restructuring liability (in thousands):
| | | | | | | | | | | | | | | | |
| | | Employee
separation costs | | | Facilities
related charges | | | Other
restructuring
charges | | | Total | |
Balance at December 31, 2009 | | $ | — | | | $ | 31,980 | | | $ | — | | | $ | 31,980 | |
Accruals | | | 3,156 | | | | 12,388 | | | | 467 | | | | 16,011 | |
Payments | | | (3,156 | ) | | | (11,744 | ) | | | (467 | ) | | | (15,367 | ) |
Accretion of sub-lease expense | | | — | | | | 2,672 | | | | — | | | | 2,672 | |
| | | | | | | | | | | | | | | | |
Balance at December 31, 2010 | | $ | — | | | $ | 35,296 | | | $ | — | | | $ | 35,296 | |
| | | | | | | | | | | | | | | | |
Accruals | | $ | 5,108 | | | $ | 1,787 | | | $ | (350 | ) | | $ | 6,545 | |
Payments | | | (2,963 | ) | | | (10,219 | ) | | | 350 | | | | (12,832 | ) |
Accretion of sub-lease expense | | | — | | | | 3,273 | | | | — | | | | 3,273 | |
| | | | | | | | | | | | | | | | |
Balance at December 31, 2011 | | $ | 2,145 | | | $ | 30,137 | | | $ | — | | | $ | 32,282 | |
| | | | | | | | | | | | | | | | |
We have substantially completed all of the activities included in the restructuring plans for 2008, 2009 and 2010. All of the costs associated with the 2011, 2010 and 2009 Restructuring activities were incurred during the years ended December 31, 2011, 2010 and 2009, respectively.
7. Commitments and Contingencies
Lease Commitments
We lease our facilities under operating leases, with various terms, the majority of which expire between 2015 and 2019. The minimum annual rent on our facilities is subject to increases based on stated rental adjustment terms of certain leases, taxes, insurance and operating costs. For financial reporting purposes, rent expense is recognized on a straight-line basis over the term of the leases. Accordingly, rent expense recognized in excess of rent paid is reflected as deferred rent. Deferred rent totaled $5.7 million and $6.0 million at December 31, 2011 and 2010, respectively, of which $4.9 million and $5.3 million is included in other long-term obligations, net of current portion in the accompanying Consolidated Balance Sheets at December 31, 2011 and 2010, respectively. Certain of our facility leases contain incentives in the form of reimbursement from the landlord for a portion of the costs of leasehold improvements we have incurred. These incentives are
F-24
recognized as a reduction of rental expense on a straight-line basis over the term of the respective leases. Unamortized leasehold improvement incentives totaled $3.5 million and $4.1 million for the years ended December 31, 2011 and 2010, respectively, of which $3.0 million and $3.5 million is included in other long-term obligations, net of current portion in the accompanying consolidated balance sheets at December 31, 2011 and 2010, respectively. In connection with certain restructuring activities, discussed in Note 6, we have subleased certain facility leases. As of December 31, 2011, the total minimum rentals to be received in the future under noncancelable subleases is $32.7 million.
We lease vehicles for our field force under operating leases, with lease terms up to four years, of which the first year is non-cancellable. Minimum future payments for the non-cancellable term of these leases are $0.3 million at December 31, 2011.
Minimum future annual obligations for facility and vehicle operating leases for years ending after December 31, 2011 are as follows (in thousands):
| | | | |
2012 | | $ | 28,186 | |
2013 | | | 28,506 | |
2014 | | | 29,191 | |
2015 | | | 20,847 | |
2016 | | | 20,534 | |
Thereafter | | | 39,689 | |
| | | | |
Total minimum lease payments | | $ | 166,953 | |
| | | | |
Rent expense for the years ended December 31, 2011, 2010 and 2009, was $10.6 million, $12.2 million and $12.5 million, respectively.
Other Commitments
We have committed to make potential future milestone payments to third parties as part of in-licensing and development programs primarily related to research and development agreements. Potential future payments generally become due and payable only upon the achievement of certain developmental, regulatory or commercial milestones, such as achievement of regulatory approval, successful development and commercialization of products, and subsequent product sales. Because the achievement of these milestones is neither probable nor reasonably estimable, we have not recorded a liability on the balance sheet for any such contingencies.
As of December 31, 2011, if all such milestones are successfully achieved, the potential future milestone and other contingency payments due under certain contractual agreements are approximately $242.3 million in aggregate, of which $7.3 million is expected to be paid over the next twelve months.
We have committed to make future minimum payments to third parties for certain inventories in the normal course of business. The minimum contractual purchase commitments total $91.3 million as of December 31, 2011.
As of December 31, 2011, commitments associated with capital investments on the BYDUREON pen device are $5.3 million.
F-25
8. Indebtedness
Our indebtedness is summarized as follows:
| | | | | | | | |
| | | December 31,
2011 | | | December 31,
2010 | |
Current portion: | | | | | | | | |
Promissory note related to revenue sharing obligation | | $ | 63,552 | | | $ | — | |
Convertible senior notes-Due April 15, 2011 | | | — | | | | 200,000 | |
| | | | | | | | |
Current portion of indebtedness | | $ | 63,552 | | | $ | 200,000 | |
| | | | | | | | |
Non-current portion: | | | | | | | | |
Convertible Senior notes, net of debt discount | | $ | 496,037 | | | $ | 468,697 | |
Notes payable, net of debt discount | | | 155,064 | | | | — | |
Promissory note related to revenue sharing obligation | | | 924,306 | | | | — | |
| | | | | | | | |
Non-current portion of indebtedness | | $ | 1,575,407 | | | $ | 468,697 | |
| | | | | | | | |
The following is a summary description of our indebtedness as of December 31, 2011:
Promissory Note Related to Revenue Sharing Obligation
As described in Note 2, in connection with the Termination Agreement, effective November 7, 2011 we entered into a secured promissory note with Lilly ( previously defined as the RSO), under which we agreed to pay to Lilly a principal sum of $1.2 billion, plus interest. Repayments on the Secured Promissory Note are determined based upon the quarterly net sales of exenatide products The RSO has a scheduled maturity of December 31, 2036, however we may prepay all or any portion of the balance without penalty.
The following table summarizes the principal amount of the liability component (including accrued interest), the unamortized discount and net carrying amount of the RSO as of December 31, 2011 and 2010 (in thousands):
| | | | | | | | |
| | | December 31,
2011 | | | December 31,
2010 | |
Promissory note related to revenue sharing obligation —Due December 31, 2036 | | | | | | | | |
Principal amount, including accrued interest | | $ | 1,209,109 | | | $ | — | |
Unamortized debt discount | | | (221,251 | ) | | | — | |
| | | | | | | | |
Net carrying amount | | | 987,858 | | | | — | |
Less current portion | | | (63,552 | ) | | | — | |
| | | | | | | | |
Non-current portion | | $ | 924,306 | | | $ | — | |
| | | | | | | | |
The significant terms of the RSO are summarized below.
Interest accruals.Interest on the RSO accrues and compounds in an amount equal to 2.295% of the total RSO balance outstanding on the last day of the calendar quarter (with an annual effective rate of 9.5%). Interest is not payable in cash when it accrues, but is instead added to the then outstanding principal amount of the RSO on the last day of each quarter. During the period commencing December 1, 2011 and including December 31, 2011, interest accrued and compounded in an amount equal to 0.759% of the RSO balance as of December 1, 2011.
Debt discount amortization.The debt discount is being amortized to interest expense over the expected term of the RSO at an effective interest rate of 14.4% for the year ended December 31, 2011.
Calculation of payment amounts.Amylin is required to make quarterly payments on the RSO in an amount equal to 15% of net sales for exenatide products for the immediate preceding calendar quarter. For the period commencing with December 1, 2011 through and including December 31, 2013, Amylin is obligated to make payments on the RSO equal to the greater of (i) 15% of net sales for exenatide products in the United States and, if control of an OUS jurisdiction has transitioned to Amylin from Lilly, net sales for exenatide products in those OUS jurisdictions, for the immediately preceding calendar quarter and (ii) 15% of 80% of a product revenue forecast that was mutually agreed upon by both Amylin and Lilly. In the event Amylin receives upfront or milestone payments from a third party in connection with an agreement with respect
F-26
to exenatide products, Amylin is obligated to make payments on the RSO equal to 20% of such upfront or milestone payments. The minimum annual RSO payments Amylin could be required to make for the years ended December 31, 2012 and 2013 is $61.3 million and $86.5 million, respectively.
Provisions related to repayment.The repayment terms for the RSO contain provisions whereby the obligation under the RSO could change as described in Note 2. The RSO could be fully discharged in the event (i) BYDUREON is not approved by June 30, 2014 and (ii) in the event all exenatide products are removed from the US, all of Europe or both for safety or efficacy reasons and continuing for a period of four years. As discussed in Note 2, these options were accounted for as embedded derivatives. As indicated in Note 14, the FDA approved BYDUREON on January 27, 2012 therefore the provision related to BYDUREON approval has expired. The provision related to removal of exenatide products from the market for safety or efficacy reasons is considered to have a very low likelihood of occurrence. See theFair Value Measurementsdiscussion in Note 1, regarding the considerations associated with valuing these embedded derivatives as of December 31, 2011.
Security Agreement and Events of Default.In connection with the Termination Agreement, on November 7, 2011, Amylin, Amylin Ohio LLC and Lilly entered into a Security Agreement pursuant to which Amylin and Amylin Ohio LLC granted to Lilly, as collateral to secure the RSO, a security interest in intellectual property relating to the exenatide products, U. S. regulatory approvals relating to the exenatide products, certain third party license agreements, certain deposit accounts into which counterparties of such license agreements are required to make payments, certain third party supply agreements, inventory and a supply agreement for BYDUREON entered into between Amylin’s wholly-owned subsidiary Amylin Ohio LLC and Amylin, collectively referred to as the Collateral. On November 7, 2011, Amylin Ohio LLC and Lilly entered into a Subsidiary Guarantee Agreement, or the Guarantee, pursuant to which Amylin Ohio LLC provided a guarantee of the Secured Obligations to Lilly. Any Amylin affiliate that owns Collateral is required to become a grantor under the Security Agreement and guarantee the RSO.
Under the terms of the promissory note an event of default would occur if Amylin fails to make RSO payments in accordance with the terms of the Termination Agreement, upon the occurrence of a bankruptcy or insolvency of Amylin or any of its affiliates party to the Security Agreement or providing a guarantee, if certain representations and warranties of Amylin and Amylin Ohio LLC are not true and correct in any material respects when made, or if Amylin breaches certain assignment provisions in the Termination Agreement, Note Agreement or Security Agreement. Upon the occurrence and continuance of an event of default under the Note, Lilly may declare all outstanding amounts under the Note due and payable and may exercise its rights with respect to the Collateral under the Security Agreement. The sole recourse in respect of the Secured Obligations under the Note, the Security Agreement and the Guarantee is limited to the Collateral. These features are not significant to the accounting for this instrument.
Convertible Senior Notes
The following table summarizes the principal amount of the liability component, the unamortized discount and net carrying amount of our convertible senior notes (in thousands):
| | | | | | | | |
| | | December 31,
2011 | | | December 31,
2010 | |
2004 Notes—Due April 15, 2011 | | | | | | | | |
Principal amount | | $ | ��� | | | $ | 200,000 | |
| | | | | | | | |
2007 Notes—Due June 15, 2014 | | | | | | | | |
Principal amount | | | 575,000 | | | | 575,000 | |
Unamortized debt discount | | | (78,963 | ) | | | (106,303 | ) |
| | | | | | | | |
Net carrying amount | | | 496,037 | | | | 468,697 | |
| | | | | | | | |
Total convertible senior notes, net | | | 496,037 | | | | 668,697 | |
Less current portion | | | — | | | | (200,000 | ) |
| | | | | | | | |
Non-current portion | | $ | 496,037 | | | $ | 468,697 | |
| | | | | | | | |
In April 2004, we issued an aggregate principal amount of $200 million of 2.5% convertible senior notes in a private placement, referred to as the 2004 Notes. The 2004 Notes matured and were repaid in full on April 15, 2011.
In June 2007, we issued the 2007 Notes in a private placement, which have an aggregate principal amount of $575 million, and are due June 15, 2014. The 2007 Notes are senior unsecured obligations and rank equally with all other existing and future senior unsecured debt. The 2007 Notes bear interest at 3.0% per year, payable in cash semi-annually, and are initially convertible into a total of up to 9.4 million shares of common stock at a conversion price of $61.07 per share, subject to the customary adjustment for stock dividends and other dilutive transactions. In addition, if a “fundamental change” (as defined in the associated indenture agreement) occurs prior to the maturity date, we will in some cases increase
F-27
the conversion rate for a holder of notes that elects to convert our notes in connection with such fundamental change. The maximum conversion rate is 22.9252 ($43.62 per share), which would result in a maximum issuance 13.2 million shares of common stock if all holders converted at the maximum conversion rate.
The 2007 Notes will be convertible into shares of our common stock unless we elect net-share settlement. If we elect net-share settlement, we will satisfy the accreted value of the obligation in cash and will satisfy the excess of conversion value over the accreted value in shares of our common stock based on a daily conversion value, determined in accordance with the associated indenture agreement, calculated on a proportionate basis for each day of the relevant 20-day observation period. Holders may convert the 2007 Notes only in the following circumstances and to the following extent: (1) during the five business-day period after any five consecutive trading day period (the measurement period) in which the trading price per note for each day of such measurement period was less than 97% of the product of the last reported sale price of our common stock and the conversion rate on each such day; (2) during any calendar quarter after the calendar quarter ending March 31, 2007, if the last reported sale price of our common stock for 20 or more trading days in a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter exceeds 130% of the applicable conversion price in effect on the last trading day of the immediately preceding calendar quarter; (3) upon the occurrence of specified events; and (4) the 2007 Notes will be convertible at any time on or after April 15, 2014 through the scheduled trading day immediately preceding the maturity date.
Subject to certain exceptions, if we experience a “designated event” (as defined in the associated indenture agreement) including a “fundamental change,” including if a majority of our Board of Directors ceases to be composed of a majority of the existing directors or other individuals approved by a majority of the existing directors, holders of the 2007 Notes will, for the duration of the notes, have the option to require us to repurchase all or any portion of their 2007 Notes. The designated event repurchase price will be 100% of the principal amount of the 2007 Notes to be purchased plus any accrued interest up to but excluding the relevant repurchase date. We will pay cash for all notes so repurchased. We may not redeem the Notes prior to maturity. The 2007 Notes have been registered under the Securities Act of 1933, as amended, to permit registered resale of the 2007 Notes and of the common stock issuable upon conversion of the 2007 Notes. The 2007 Notes pay interest in cash, semi-annually in arrears on June 15 and December 15 of each year, which began on December 15, 2007.
Since we have the option to elect net-share settlement upon conversion of the 2007 Notes, we account for the 2007 Notes in accordance with the authoritative guidance for accounting for convertible debt instruments that may be settled in cash upon conversion. In accordance with such guidance, we allocated $185.6 million of the fair value of the convertible debt instruments to equity. Additionally, of the total debt issuance costs of $16.3 million incurred in connection with the issuance of the 2007 Notes, $5.3 million was allocated to equity, resulting in total revised debt issuance costs of $11.0 million, and recorded a debt discount of $180.3 million. The carrying amount of the equity component of the 2007 Notes was $180.3 million at both December 31, 2011 and 2010. The debt discount and issuance costs are being amortized to interest expense over the term of the 2007 Notes, approximately two years of which remain as of December 31, 2011. The effective interest rate on the net carrying value of the 2007 Notes was 9.3% for the years ended December 31, 2011, 2010 and 2009.
The net book value of debt issuance costs as of December 31, 2011 and 2010 was $3.9 million and $5.4 million, respectively. Amortization expense associated with these debt issuance costs was $1.6 million for each of the years ended December 31, 2011, 2010 and 2009. The fair value of the 2007 Notes, determined by observed market prices within the Level 1 hierarchy, was $514.3 million and $501.7 million at December 31, 2011 and 2010 respectively.
Notes Payable
In October 2008, we and Lilly entered into a loan agreement pursuant to which Lilly made available to us a $165 million unsecured line of credit. In May 2011 we drew $165 million from this facility, referred to as the Note Payable to Lilly. The interest rate on the Note Payable to Lilly is fixed at 5.51% and is due and payable quarterly in arrears on the first business day of each quarter and the initial maturity date was May 23, 2014. In connection with the Termination Agreement, the Note Payable to Lilly was amended and restated effective November 7, 2011 (“the Amended Note Payable to Lilly”). Under the terms of the Amended Note Payable to Lilly, the maturity date was extended to June 30, 2016; the interest rate on the Amended Lilly Loan remains fixed at 5.51% and interest continues to be due and payable quarterly in arrears on the first business day of each quarter. As indicated in Note 2, a portion of the consideration paid in connection with the Termination Agreement represents the value Amylin received in connection with the extension of the maturity date of the Lilly Loan at a below market interest rate. The value representing the fee paid to extend the note at a below market interest rate was recorded as a discount on the Lilly Loan and will be accreted to interest expense over the remaining life of the loan.
In 2007, we entered into a $140 million credit agreement with Bank of America, N.A., or “Bank of America”, as administrative agent, collateral agent and letter of credit issuer, Silicon Valley Bank and RBS Asset Finance, Inc., as syndication agents, and Comerica Bank and BMO Capital Markets Financing, Inc., as documentation agents. The credit
F-28
agreement provided for a $125 million term loan and a $15 million revolving credit facility. The revolving credit facility also provided for the issuance of letters of credit and foreign exchange hedging up to the $15 million borrowing limit. Both loans had a final maturity date of December 21, 2010. In December 2010 the term loan was paid in full and foreign exchange hedging contracts were terminated.
Our domestic subsidiaries, Amylin Ohio LLC and Amylin Investments LLC, were co-borrowers under the credit agreement. The loans under the revolving credit facility were collateralized by substantially all of our (including the two domestic subsidiaries) assets (other than intellectual property and certain other excluded collateral). The term loan was repayable on a quarterly basis, with no payments due quarters one through four, 6.25% of the outstanding principal due quarters five through eleven, and 56.25% of the outstanding principal due in quarter 12. Interest on the term loan was paid quarterly on the unpaid principal balance at 1.75% above the three month London Interbank Offered Rate. We incurred debt issuance costs of $1.7 million in connection with the credit agreement, which were amortized to interest expense on a straight-line basis over the term of the credit agreement and were fully amortized as of December 31, 2011 and 2010. Amortization expense associated with these debt issuance costs was $0, $0.5 million and $0.6 million for the years ended December 31, 2011, 2010 and 2009, respectively.
In connection with the execution of the term loan, we entered into an interest rate swap with an initial notional amount of $125 million in 2007 that resulted in a fixed rate of 5.717%. The interest rate swap matured on December 21, 2010 therefore the instrument had no fair value as of December 31, 2010. For the year ended December 31, 2010, the recognized gain on the interest rate swap agreement was $2.8 million. For the year ended December 31, 2009, the recognized gain on the interest rate swap was $2.2 million. Recognized gains and losses on the interest rate swap are included in interest and other expense.
In December 2011, we entered into an Amended and Restated Letter of Credit and Cash Collateral Agreement with Bank of America which provides for the issuance of letters of credit under which we agreed to deliver cash collateral to Bank of America in the amount of $10.5 million. As of December 31, 2011 we had issued $10.3 million of standby letters of credit, primarily in connection with office leases.
Maturities
Maturities on long-term debt for each of the next five years as of December 31, 2011 are as follows (in thousands):
| | | | |
| | | December 31,
2011 | |
2012 | | $ | 61,295 | |
2013 | | | 86,454 | |
2014 | | | 652,655 | |
2015 | | | 77,655 | |
2016 | | | 242,655 | |
The annual payments on the RSO vary depending upon net sales of exenatide products. The maturities for 2012 and 2013 include the minimum contractual RSO payments required under the Termination Agreement. Given the uncertainty regarding future net sales levels of exenatide, the RSO maturities for 2014 through 2016 were calculated assuming total exenatide net sales in each of those years remains equal to the 2011 net BYETTA sales.
9. Stockholders’ Equity
Stock-based Compensation Plans
Stock Options and Restricted Stock Units
We have two plans under which we currently grant stock options, restricted stock or restricted stock units: the 2009 Equity Incentive Plan, or the 2009 Plan, and the 2003 Non-Employee Directors’ Stock Option Plan, or the 2003 Directors’ Plan. The 2009 Plan replaced the 2001 Stock Option Plan, or the 2001 Plan. Options granted under the 2001 Plan remain outstanding until exercised or cancelled. Under the 2003 Directors’ Plan, non-qualified stock options and restricted stock may be granted to our non-employee directors. The 2003 Directors’ Plan provides for automatic stock option grants to non-employee directors upon their initial appointment or election to our Board of Directors which are issued from shares authorized under the 2009 Plan. Both the 2009 Plan and the 2003 Directors’ Plan provide that the number of shares of common stock available for issuance shall be reduced by a ratio of 1.5-to-1.0 for each share of common stock issued pursuant to a restricted stock award.
F-29
To date, stock-based compensation awards under the 2001 Plan, the 2003 Directors’ Plan and the 2009 Plan consist of incentive and non-qualified stock options and restricted stock units. Stock options granted under each of the plans must have an exercise price equal to at least 100% of the fair market value of our common stock on the date of grant and generally vest over four years. Stock options granted prior to October 10, 2007 have a maximum contractual term of ten years and stock options granted after October 10, 2007 have a maximum contractual term of seven years. Restricted stock units granted under each of the plans are generally performance based awards, and vest upon the achievement of defined performance targets. At December 31, 2011, an aggregate of 27.3 million shares were reserved for future issuance under our stock option plans, of which 7.7 million shares were available for future grants.
The following table summarizes our stock option activity and related information for the year ended December 31, 2011:
| | | | | | | | | | | | | | | | |
| | | Shares
(thousands) | | | Weighted-Average
Exercise Price | | | Weighted-
Average
Remaining
Contractual
Term (years) | | | Aggregate Intrinsic
Value (thousands) | |
Options outstanding at December 31, 2010 | | | 17,505 | | | $ | 24.19 | | | | | | | | | |
Granted | | | 3,199 | | | $ | 14.82 | | | | | | | | | |
Exercised | | | (430 | ) | | $ | 8.54 | | | | | | | | | |
Cancelled/Forfeited | | | (2,734 | ) | | $ | 24.30 | | | | | | | | | |
| | | | | | | | | | | | | | | | |
Options outstanding at December 31, 2011 | | | 17,540 | | | $ | 22.84 | | | | 4.08 | | | $ | 3,972 | |
| | | | | | | | | | | | | | | | |
Options exercisable at December 31, 2011 | | | 12,765 | | | $ | 25.67 | | | | 3.56 | | | $ | 2,706 | |
| | | | | | | | | | | | | | | | |
Options vested and expected to vest at December 31, 2011 | | | 17,540 | | | $ | 22.84 | | | | 4.08 | | | $ | 3,972 | |
| | | | | | | | | | | | | | | | |
The total intrinsic value of stock options exercised was $1.5 million, $4.0 million and $2.8 million during the years ended December 31, 2011, 2010 and 2009, respectively. We received cash from the exercise of stock options of $3.2 million, $9.2 million and $4.6 million during the years ended December 31, 2011, 2010 and 2009, respectively. We did not record any tax benefits related to the exercise of employee stock options due to our net loss position. Upon option exercise, we issue new shares of our common stock.
At December 31, 2011, total unrecognized estimated non-cash, stock-based compensation expense related to nonvested stock options granted prior to that date was $26.3 million, with a weighted-average amortization period of 2.3 years. We record non-cash, stock-based compensation expense for options with pro-rata vesting on a straight-line basis over the awards’ vesting period.
The following table summarizes our restricted stock unit activity for the year ended December 31, 2011:
| | | | | | | | |
| | | Shares
(thousands) | | | Weighted-Average
Grant Date
Fair Value | |
Restricted stock units outstanding at December 31, 2010 | | | 1,107 | | | $ | 14.30 | |
Granted | | | 497 | | | $ | 14.13 | |
Vested | | | (25 | ) | | $ | 14.10 | |
Cancelled/Forfeited | | | (215 | ) | | $ | 14.25 | |
| | | | | | | | |
Restricted stock units outstanding at December 31, 2011 | | | 1,364 | | | $ | 14.16 | |
| | | | | | | | |
The fair value of the restricted stock units is based on the market value of our stock on the date of grant. Compensation expense, net of the effect of estimated forfeitures, is recognized ratably over the expected vesting period. At December 31, 2011, total unrecognized estimated non-cash, stock based compensation expense related to nonvested restricted stock units outstanding as of that date was $3.2 million, with a weighted-average amortization period of 1.1 years.
F-30
Employee Stock Purchase Plan
Our 2001 Employee Stock Purchase Plan, or the 2001 Purchase Plan, enables participants to contribute up to 15% of their eligible compensation for the purchase of our common stock at the lower of 85% of the fair market value of our common stock (i) on the employee’s enrollment date or (ii) the purchase date. The terms of any offerings under the 2001 Purchase Plan are established by the Compensation and Human Resources Committee of the Board of Directors. In April 2010, the Compensation and Human Resources Committee approved a series of four consecutive six-month offerings commencing on September 1, 2010. As of December 31, 2011, 0.5 million shares were reserved for future issuance under the 2001 Purchase Plan.
The total intrinsic value of purchase rights exercised was $1.0 million, $3.6 million and $1.1 million during the years ended December 31, 2011, 2010 and 2009, respectively. At December 31, 2011, total unrecognized non-cash, compensation expense for nonvested purchase rights granted prior to that date was $0.4 million, with a weighted-average amortization period of 0.2 years.
Shares Reserved for Future Issuance
The following shares of common stock are reserved for future issuance at December 31, 2011 (in thousands):
| | | | |
Stock Option Plans | | | 27,289 | |
Employee Stock Purchase Plan | | | 547 | |
Convertible Senior Notes | | | 9,416 | |
| | | | |
| | | 37,252 | |
| | | | |
Shareholder Rights Plan
In June 2002, we adopted a Preferred Share Purchase Rights Plan (the “Rights Plan”). The Rights Plan provides for a dividend distribution of one preferred stock purchase right (a “Right”) for each outstanding share of our common stock, par value $0.001 per share, held of record at the close of business on June 28, 2002. The Rights are not currently exercisable. Under certain conditions involving an acquisition or proposed acquisition by any person or group of 15% or more of our common stock, the Rights permit the holders (other than the 15% holder) to purchase one one-hundredth of a share of Series A Junior Participating Preferred Stock, par value $0.001 per share (the “Preferred Shares”) at a price of $100 per one one-hundredth of a Preferred Share, subject to adjustment. Each one one-hundredth of a share of Preferred Shares has designations and powers, preferences and rights and the qualifications, limitations and restrictions which make our value approximately equal to the value of one share of our common stock. Under certain conditions, the Rights are redeemable by our Board of Directors in whole, but not in part, at a price of $0.001 per Right.
10. Benefit Plans
Defined Contribution 401(k) Plan
We have a defined contribution 401(k) plan for the benefit of all eligible employees. Active employees who are at least 18 years old and are not otherwise disqualified under the terms of the 401(k) plan are eligible to participate. Discretionary matching contributions are based on a percentage of employee contributions and are funded by newly issued shares of our common stock. Participants vest in the employer matching contributions over four years of service, at 25% for one year or more of service but less than two years, at 50% for two years or more of service but less than three years, at 75% for three years or more of service but less than four years, and 100% for four or more years of service. Any forfeitures of non-vested amounts shall be used to pay administrative plan expenses, to restore any rehired employees who previously forfeited their non-vested balance under certain circumstances, or shall be used to reduce future employer contributions and shall be allocated to the participant accounts. We recorded expense of $3.2 million, $3.8 million and $3.9 million for matching contributions in the years ended December 31, 2011, 2010 and 2009, respectively.
Deferred Compensation Plans
In August 1997, we adopted a Non-Employee Directors’ Deferred Compensation Plan (the “Directors’ Deferral Plan”) that permits participating non-employee directors to elect, on an annual basis, to defer all or a portion of their cash compensation in a deferred stock account, pursuant to which the deferred fees are credited in the form of phantom shares of our common stock, based on the market price of the stock at the time the fees are earned. Deferred amounts are valued at the fair market value of our common stock at each reporting date and are included in accrued compensation in the accompanying consolidated balance sheets. Upon termination of service the director’s account is settled in either cash or stock, at our discretion. In connection with this plan we recorded expense of $0.8 million, $0.8 million and $0.9 million for the years ended December 31, 2011, 2010 and 2009, respectively.
F-31
We adopted a Deferred Compensation Plan in April 2001 which allows officers to defer up to 80% and directors to defer up to 100% of their eligible annual compensation. The trust assets, consisting of primarily cash, mutual funds and equity securities are recorded at current market prices. The company-owned assets are placed in a “rabbi trust” and are included in other current assets in the accompanying consolidated balance sheets. The trust assets had a fair value of $8.2 million and $8.5 million at December 31, 2011 and 2010, respectively, including unrealized losses of approximately $1.6 million and $1.1 million at December 31, 2011 and 2010, respectively. Unrealized gains and losses on the trust assets are included in accumulated other comprehensive loss in the accompanying consolidated balance sheets. The corresponding liability was $8.2 million and $8.5 million at December 31, 2011 and 2010, respectively, of which $7.7 million and $7.8 million are included in other long-term liabilities, net of current portion in the accompanying consolidated balance sheets at December 31, 2011 and 2010, respectively. The current portion of the corresponding liability is included in accrued compensation in the accompanying consolidated balance sheets at December 31, 2011 and 2010. Total contributions to this plan, consisting solely of compensation deferred by participants, were $0.7 million, $1.5 million and $1.7 million for the years ended December 31, 2011, 2010 and 2009, respectively.
Employee Stock Ownership Plan
In December 2007, we adopted the Employee Stock Ownership Plan, or ESOP. Active employees who are at least 18 years old and have met minimum service requirements and are not otherwise disqualified under the terms of the ESOP are eligible to participate. Each participant has an ESOP account into which we make mandatory annual contributions in the form of shares of our common stock equal to 10% of a participant’s plan year eligible compensation. We may make additional discretionary contributions for any plan year, and total contributions are limited to the lesser of 100% of a participant’s plan year eligible compensation and limitations established by the Internal Revenue Service Code (IRS Code). A participant’s eligible compensation primarily includes wages and bonus.
Participants vest in their accounts over four years of service, at 25% for one year or more of service but less than two years, at 50% for two years or more of service but less than three years, at 75% for three years or more of service but less than four years, and 100% for four years or more of service. Any forfeitures of non-vested amounts shall be used to restore any rehired employees who previously forfeited their non-vested balance under certain circumstances, or shall be used to reduce future employer contributions and shall be allocated to the participant accounts. Distributions generally are made only upon termination of employment and as necessary by regulatory requirements.
Shares committed to be released or that have been allocated to participant accounts are treated as outstanding shares for calculating earnings per share. The ESOP held 4.1 million shares at December 31, 2011, of which 0.4 million were unvested and had a fair value of $4.8 million. We recorded ESOP expense of $13.3 million, $15.9 million and $16.3 million for the years ended December 31, 2011, 2010 and 2009, respectively, for our contribution.
11. Interest and other expense, net
The following table summarizes the components of interest and other expense, net for the three years ended December 31, 2011 (in thousands):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Interest and other income | | $ | 1,901 | | | $ | 2,698 | | | $ | 7,768 | |
Interest and other expense | | | (38,090 | ) | | | (28,070 | ) | | | (19,300 | ) |
Loss on fair value adjustments | | | (15,751 | ) | | | — | | | | — | |
Loss on impairment of investments | | | — | | | | (198 | ) | | | (1,377 | ) |
| | | | | | | | | | | | |
Total interest and other expense, net | | $ | (51,940 | ) | | $ | (25,570 | ) | | $ | (12,909 | ) |
| | | | | | | | | | | | |
The following table summarizes the interest expense we capitalized associated with construction in progress for the three years ended December 31, 2011 (in thousands):
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Coupon interest expense | | $ | 11,547 | | | $ | 11,521 | | | $ | 15,604 | |
Non-cash interest from debt discount | | | 15,248 | | | | 14,383 | | | | 17,676 | |
| | | | | | | | | | | | |
Total capitalized interest | | $ | 26,795 | | | $ | 25,904 | | | $ | 33,280 | |
| | | | | | | | | | | | |
F-32
12. Litigation
From time to time in the ordinary course of business, we become involved in various lawsuits, claims and proceedings relating to the conduct of our business, including those pertaining to product liability, patent infringement and employment claims. As of December 31, 2011, we and Lilly were involved in product liability cases involving plaintiffs in various courts in the United States. Additionally, plaintiffs who previously filed cases have subsequently dismissed their cases or claims without prejudice. These cases have been brought by individuals who allege they have used BYETTA. They generally seek compensatory and punitive damages for alleged injuries, consisting primarily of pancreatitis, and in some cases, wrongful death. Most of the cases are pending in California state court, where the Judicial Council has granted our petition for a “coordinated proceeding” for all California state court cases alleging harm allegedly as a result of BYETTA use. We also have received notice from plaintiff’s counsel of additional claims by individuals who have not filed suit. While we cannot reasonably predict the outcome of any lawsuit, claim or proceeding, we and Lilly intend to vigorously defend these matters. However, if we are unsuccessful in our defense, these matters could result in a material adverse impact to our financial position and results of operations.
13. Income Taxes
We have net deferred tax assets relating primarily to net operating loss carryforwards. Subject to certain limitations, these deferred tax assets may be used to offset taxable income in future periods. Since we have been unprofitable since inception and the likelihood of future profitability is not assured, we have provided a valuation allowance for most of these deferred tax assets in our consolidated balance sheets at December 31, 2011 and 2010, respectively. If we determine that we are able to realize a portion or all of these deferred tax assets in the future, we will record an adjustment to increase their recorded value and a corresponding adjustment to increase income or additional paid in capital, as appropriate, in that same period.
We recognize the impact of a tax position in our financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. We provide estimates for unrecognized tax benefits which relate primarily to issues common among corporations in our industry. We apply a variety of methodologies in making these estimates which include advice from industry and subject experts, evaluation of public actions taken by the Internal Revenue Service and other taxing authorities, as well as our own industry experience. If our estimates are not representative of actual outcomes, our results could be materially impacted.
The provision (benefit) for income taxes includes the following (in thousands):
| | | | | | | | | | | | |
| | | Years ended December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Current (benefit) provision: | | | | | | | | | | | | |
Federal | | $ | | | | $ | (514 | ) | | $ | (2,027 | ) |
State | | | 35 | | | | 36 | | | | 34 | |
Foreign | | | — | | | | — | | | | — | |
| | | | | | | | | | | | |
Total current (benefit) provision | | | 35 | | | | (478 | ) | | | (1,993 | ) |
Deferred (benefit) provision: | | | | | | | | | | | | |
Federal | | | — | | | | — | | | | — | |
State | | | 26 | | | | 26 | | | | 26 | |
Foreign | | | — | | | | — | | | | — | |
| | | | | | | | | | | | |
Total deferred provision | | | 26 | | | | 26 | | | | 26 | |
| | | | | | | | | | | | |
Total (benefit) provision | | $ | 61 | | | $ | (452 | ) | | $ | (1,967 | ) |
| | | | | | | | | | | | |
These amounts are included in interest and other expense in the accompanying consolidated statements of operations.
The 2010 current Federal income tax benefit reflects the operation of the intraperiod tax allocation rules under which a tax benefit is provided in continuing operations to offset a tax provision recorded directly to other comprehensive income related to current unrealized gains on investment securities available for sale. The 2009 current Federal income tax benefit reflects refundable research credits and the refund of alternative minimum taxes from the carryback of net operating losses. The Housing and Economic Recovery Act of 2008 (P.L. 110-289), enacted on July 30, 2008, and extended through 2009 by the American Recovery and Reinvestment Act of 2009, provided for the acceleration of a portion of unused pre-2006 research credits and alternative minimum tax credits in lieu of claiming the 50% bonus depreciation allowance enacted in the Economic Stimulus Act of 2008.
F-33
Deferred income taxes reflect the temporary differences between the carrying amounts of assets and liabilities for financial statement purposes and the amounts used for income tax purposes and the net tax effects of net operating loss and credit carryforwards. Significant components of our deferred tax assets as of December 31, 2011 and 2010 are shown below (in thousands). A valuation allowance of $868.1 million was recognized at December 31, 2011 to offset the deferred tax assets, as realization of such assets has not met the more likely than not threshold required under current accounting guidance. Included in the gross deferred tax assets below are pre-January 1, 2006 stock option deductions that, when recognized, are estimated to increase additional paid in capital by $21.7 million.
| | | | | | | | |
| | | 2011 | | | 2010 | |
Deferred tax assets: | | | | | | | | |
Net operating loss carryforwards | | $ | 515,963 | | | $ | 465,192 | |
Research tax credits | | | 65,978 | | | | 60,918 | |
Capitalized research and development expenses | | | 37,913 | | | | 50,489 | |
Accrued expenses | | | 83,921 | | | | 71,984 | |
Deferred revenue | | | 23,470 | | | | 71,046 | |
Reacquisition of economic interest in exenatide products | | | 159,937 | | | | — | |
| | |
Stock-based compensation expense | | | 36,381 | | | | 32,944 | |
Other, net | | | 17,807 | | | | 16,576 | |
| | | | | | | | |
Total deferred tax assets | | | 941,370 | | | | 769,149 | |
Valuation allowance for deferred tax assets | | | (868,145 | ) | | | (704,119 | ) |
| | | | | | | | |
Total deferred tax assets after valuation allowance | | | 73,225 | | | | 65,030 | |
| | | | | | | | |
Deferred tax liabilities: | | | | | | | | |
Convertible debt discount | | | (29,333 | ) | | | (39,489 | ) |
Fixed assets | | | (42,875 | ) | | | (24,501 | ) |
Other, net | | | — | | | | — | |
| | | | | | | | |
Total deferred tax liabilities | | | (72,208 | ) | | | (63,990 | ) |
| | | | | | | | |
Net deferred tax asset after valuation allowance | | $ | 1,017 | | | $ | 1,040 | |
| | | | | | | | |
The net deferred tax assets are included in other long-term assets in the accompanying consolidated balance sheets.
Following is a summary of our Federal net operating loss carryforwards, Federal research tax credit carryforwards and California net operating loss carryforwards at December 31, 2011 (in thousands):
| | | | | | | | | | | | |
| | | Federal net operating
loss carryforwards | | | California net
operating loss
carryforwards | | | Federal research
and development
tax credit
carryforwards | |
Expiring within one year | | $ | 402 | | | $ | — | | | $ | 1,881 | |
After 1 but within 5 years | | | — | | | | 415,782 | | | | — | |
After 5 but within 10 years | | | 177,820 | | | | 105,174 | | | | 8,715 | |
After 10 years | | | 1,363,175 | | | | 132,804 | | | | 56,788 | |
| | | | | | | | | | | | |
| | $ | 1,541,397 | | | $ | 653,760 | | | $ | 67,384 | |
| | | | | | | | | | | | |
We experienced changes in control that triggered the limitations of Section 382 of the Internal Revenue Code on our net operating loss carryforwards. The Section 382 limitations were immaterial to our total net operating losses and are reflected in the net operating loss of approximately $1.5 billion presented above.
At December 31, 2011, we had Federal net operating loss carryforwards of approximately $1.5 billion, which begin to expire at the end of 2012. We also have California net operating loss carryforwards of $653.8 million, which begin to expire at the end of 2014, and other state net operating loss carryforwards of approximately $339.8 million, which begin to expire at the end of 2012. The difference between the Federal and California tax loss carryforwards is attributable to the capitalization of research and development expenses for California tax purposes, the prior years’ limitation on California loss carryforwards and apportionment of losses to other states. We have Federal research tax credit carryforwards of $67.4 million, which began to expire at the end of 2011, and California research tax credit carryforwards of $29.8 million, which carry forward indefinitely.
F-34
The reconciliation between our effective tax rate and the federal statutory rate is as follows:
| | | | | | | | | | | | |
| | | Tax rate for the years ended
December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Federal statutory rate applied to net loss before income tax (benefit) provision | | | (35.0 | )% | | | (35.0 | )% | | | (35.0 | )% |
State taxes | | | (0.1 | )% | | | (3.5 | )% | | | 2.4 | % |
Research and development tax credits | | | (0.8 | )% | | | (0.9 | )% | | | 0.1 | % |
Stock-based compensation | | | 1.4 | % | | | 5.6 | % | | | 6.4 | % |
Expiring federal net operating losses | | | 2.5 | % | | | 5.0 | % | | | 5.4 | % |
Reacquisition of economic interest in exenatide products | | | 2.1 | % | | | — | | | | — | |
Increase in valuation allowance | | | 30.2 | % | | | 27.8 | % | | | 17.1 | % |
Other | | | (0.2 | )% | | | 0.7 | % | | | 2.6 | % |
| | | | | | | | | | | | |
Effective tax rate | | | 0.1 | % | | | (0.3 | )% | | | (1.0 | )% |
| | | | | | | | | | | | |
The state tax effects during 2009 include a 5.4% increase in the effective state tax rate (fully offset by a decrease in valuation allowance) relating to a decrease to certain deferred tax assets as a result of a California tax law change that was enacted in February 2009. This change allows an elective single sales factor for state apportionment for taxable years beginning on or after January 1, 2011. We expect to benefit from the California single sales factor election for apportioning income for years 2011 and beyond. As a result of our anticipated election of the single sales factor, we have re-measured our deferred tax assets taking into account the expected reduced California apportionment factor under the elective single sales factor.
Because we adopted the provisions of fair value method of accounting for stock-based compensation arrangements effective January 1, 2006, we recognize excess tax benefits associated with stock-based compensation directly to stockholders’ equity only when realized. Accordingly, deferred tax assets are not recognized for net operating loss carryforwards resulting from excess tax benefits occurring from January 1, 2006 onward. A windfall tax benefit occurs when the actual tax benefit realized upon an employee’s disposition of a stock- based award exceeds any existing deferred tax asset associated with the award. At December 31, 2011, deferred tax assets do not include $45.6 million of excess tax benefits from stock-based compensation.
Income taxes paid during the years ended December 31, 2011, 2010 and 2009 totaled $22 thousand, $28 thousand and $43 thousand, respectively.
The reconciliation of the total amounts of unrecognized tax benefits at the beginning and end of the years ended December 31, 2011, 2010 and 2009 is as follows (in thousands):
| | | | | | | | | | | | |
| | | December 31, | |
| | | 2011 | | | 2010 | | | 2009 | |
Reconciliation of unrecognized tax benefits: | | | | | | | | | | | | |
Unrecognized tax benefits related to reductions in tax losses and credits as of the beginning of the year | | $ | 44,329 | | | $ | 36,305 | | | $ | 22,441 | |
Increase (decrease) in unrecognized tax benefits related to reductions in tax losses and credits as a result of tax positions taken during a prior period | | | 775 | | | | (262 | ) | | | 2,752 | |
Increase in unrecognized tax benefits related to reductions in tax losses and credits as a result of tax positions taken during the current period | | | 14,419 | | | | 8,286 | | | | 11,112 | |
| | | | | | | | | | | | |
Unrecognized tax benefits related to reductions in tax losses and credits as of the end of the year | | $ | 59,523 | | | $ | 44,329 | | | $ | 36,305 | |
| | | | | | | | | | | | |
The balance of unrecognized tax benefits at December 31, 2011 of $59.5 million are tax benefits that, if recognized, would not affect our effective tax rate as long as they remain subject to a full valuation allowance. The net effect on the deferred tax assets and corresponding decrease in the valuation allowance at December 31, 2011, 2010 and 2009 resulting from unrecognized tax benefits is $51.3 million, $36.1 million and $28.3 million, respectively. We have not recognized any accrued interest and penalties related to unrecognized tax benefits during the years ended December 31, 2011, 2010 and 2009. We will elect a treatment for interest and penalties when they occur. We are subject to taxation in the United States and
F-35
various state jurisdictions. Effectively all of our historical tax years are subject to examination by the Internal Revenue Service and various state jurisdictions due to the generation of net operating loss and credit carryforwards. We do not foresee any material changes to unrecognized tax benefits within the next twelve months.
14. Subsequent Event
On January 27, 2012 Amylin announced that the US FDA approved BYDUREON, the first once-weekly treatment for type 2 diabetes. BYDUREON is a glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes in multiple clinical settings.
15. Quarterly Financial Data (Unaudited)
The following financial information reflects all normal recurring adjustments, which are, in the opinion of management, necessary for a fair statement of the results of the interim periods. Summarized quarterly data for fiscal 2011 and 2010 are as follows (in thousands, except per share data):
| | | | | | | | | | | | | | | | |
| | | For the quarters ending | |
| | | March 31 | | | June 30 | | | September 30 | | | December 31 | |
2011: | | | | | | | | | | | | | | | | |
Net product sales | | $ | 150,839 | | | $ | 154,789 | | | $ | 155,075 | | | $ | 160,867 | |
Revenues under collaborative agreements | | | 1,875 | | | | 3,276 | | | | 19,889 | | | | 4,068 | |
Gross profit from product sales | | | 138,295 | | | | 142,946 | | | | 143,404 | | | | 148,549 | |
Restructuring | | | 2,858 | | | | 126 | | | | 2,499 | | | | 1,707 | |
Net costs associated with reacquisition of economic interest in exenatide products | | | — | | | | — | | | | — | | | | 431,587 | |
Net loss | | | (37,324 | ) | | | (31,408 | ) | | | (13,196 | ) | | | (461,471 | ) |
Basic and diluted net loss per share(1) | | $ | (0.26 | ) | | $ | (0.22 | ) | | $ | (0.09 | ) | | $ | (3.15 | ) |
2010: | | | | | | | | | | | | | | | | |
Net product sales | | $ | 172,261 | | | $ | 162,511 | | | $ | 154,026 | | | $ | 162,315 | |
Revenues under collaborative agreements | | | 1,875 | | | | 1,875 | | | | 2,075 | | | | 11,875 | |
Gross profit from product sales | | | 151,759 | | | | 148,049 | | | | 141,346 | | | | 148,272 | |
Restructuring | | | — | | | | 3,424 | | | | 6,028 | | | | 7,328 | |
Net loss | | | (38,203 | ) | | | (44,196 | ) | | | (50,732 | ) | | | (19,182 | ) |
Basic and diluted net loss per share(1) | | $ | (0.27 | ) | | $ | (0.31 | ) | | $ | (0. 35 | ) | | $ | (0.13 | ) |
| (1) | Net loss per share is computed independently for each of the quarters presented. Therefore, the sum of the quarterly per-share calculations will not necessarily equal the annual per share calculation. |
F-36
Schedule II
AMYLIN PHARMACEUTICALS, INC
Schedule II: Valuation Accounts
(in thousands)
| | | | | | | | | | | | | | | | |
| | | Balance at
beginning of
period | | | Additions | | | Deductions | | | Balance at
end of
period | |
Year ended December 31, 2011 | | | | | | | | | | | | | | | | |
Inventory reserve | | $ | 1,373 | | | | — | | | | 1,372 | | | $ | 1 | |
| | | | | | | | | | | | | | | | |
Accounts receivable allowances(1) | | $ | 21,227 | | | | 49,719 | | | | 45,994 | | | $ | 24,952 | |
| | | | | | | | | | | | | | | | |
Year ended December 31, 2010 | | | | | | | | | | | | | | | | |
Inventory reserve | | $ | 297 | | | | 1,710 | | | | 634 | | | $ | 1,373 | |
| | | | | | | | | | | | | | | | |
Accounts receivable allowances(1) | | $ | 20,295 | | | | 48,337 | | | | 47,405 | | | $ | 21,227 | |
| | | | | | | | | | | | | | | | |
Year ended December 31, 2009 | | | | | | | | | | | | | | | | |
Inventory reserve | | $ | 5,101 | | | | 1,007 | | | | 5,811 | | | $ | 297 | |
| | | | | | | | | | | | | | | | |
Accounts receivable allowances(1) | | $ | 15,041 | | | | 44,783 | | | | 39,529 | | | $ | 20,295 | |
| | | | | | | | | | | | | | | | |
| (1) | Allowances for prompt payment, product returns, doubtful accounts and chargebacks. |
F-37