EXHIBIT 99.1
Unless the context indicates otherwise, the terms “we,” “us” and “our” refer to Geron Corporation, a Delaware corporation, and its subsidiary on a consolidated basis. This Exhibit 99.1 includes trademarks, service marks and trade names owned by us or other companies. All trademarks, service marks and trade names included in this Exhibit 99.1 are the property of their respective owners.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Exhibit 99.1 contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. These statements relate to future events or to our future operating or financial performance and involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. Forward-looking statements may include, but are not limited to, statements about:
•the therapeutic potential of imetelstat and its expected uses and benefits;
•our plans to submit a New Drug Application, or NDA, and a marketing authorization application, or MAA, for imetelstat in Low or Intermediate-1 risk myelodysplastic syndromes, or lower risk MDS, and the anticipated timing thereof;
•the progress, timing, magnitude, scope and costs of clinical development, manufacturing and commercialization of imetelstat, including the number of indications which are or may in the future be pursued, subject to clearances and approvals by the U.S. Food and Drug Administration, or FDA, and other regulatory authorities;
•the continued enrollment in, and the continued conduct and completion of, IMpactMF, our Phase 3 clinical trial in relapsed/refractory myelofibrosis, or MF, IMproveMF, our Phase 1 combination clinical trial in frontline Intermediate-2 or High Risk MF, and IMpress, an investigator-led Phase 2 clinical trial in Intermediate-2 or High-Risk myelodysplastic syndromes and acute myeloid leukemia;
•the achievement and timing of clinical trial events related to IMpactMF, IMproveMF and IMpress, including the completion of patient enrollment and anticipated timing of data availability;
•other potential planned future clinical development plans for imetelstat;
•our plans to market and sell imetelstat, upon regulatory approval or clearance, in the U.S. and in Europe, whether alone or with new collaborative partners, and the anticipated timing thereof;
•our ability to obtain and maintain potential new collaborative arrangements to assist us in the development and potential commercialization of imetelstat, especially outside the U.S.;
•the availability of coverage and adequate third-party reimbursement for imetelstat, if any;
•our ability to consistently and reproducibly manufacture imetelstat;
•our ability to meaningfully reduce manufacturing costs of imetelstat;
•the impacts of the ongoing COVID-19 global pandemic, macroeconomic conditions, such as rising inflation rates, uncertain credit and global financial markets and supply chain disruptions, and geopolitical events, such as the conflict between Russia and Ukraine and related sanctions, on the foregoing;
•our ability to establish and maintain agreements with third-party service providers to support the development of imetelstat;
•the size and timing of expenditures and whether there are unanticipated expenditures;
•our ability to manage the anticipated growth of our business, including our ability to recruit and retain key personnel to support the development and potential future commercialization of imetelstat;
1
•our estimates and expectations regarding the sufficiency of our cash resources, our cash resource conservation efforts;
•our estimates regarding the extent of our ability to progress our imetelstat program with our existing capital resources;
•our ability to protect our intellectual property and the duration of any such protection, and operate our business without infringing upon the intellectual property rights of others;
•the implementation of our corporate strategy;
•our future financial and operating performance;
•our anticipated use of our existing capital resources; and
•our plans, objectives, expectations and intentions and any other statements that are not historical fact.
In some cases, you can identify forward-looking statements by terms such as “may,” “plan,” “intend,” “will,” “should,” “could,” “would,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,” “potential” and similar expressions intended to identify forward-looking statements. These statements reflect our current views with respect to future events, are based on assumptions and are subject to risks and uncertainties. Given these risks and uncertainties, you should not place undue reliance on these forward-looking statements. We discuss many of these risks, uncertainties and other factors in greater detail under the heading “Risk Factors” below. Also, these forward-looking statements represent our estimates and assumptions only as of the date of this Exhibit 99.1. Unless required by law, we undertake no obligation to update or revise any forward-looking statements to reflect new information or future events or developments. Thus, you should not assume that our silence over time means that actual events are bearing out as expressed or implied in such forward-looking statements. We qualify all of the forward-looking statements in this Exhibit 99.1 by these cautionary statements.
*********
Company Overview
Summary
We are a late-stage biopharmaceutical company pursuing therapies with the potential to extend and enrich the lives of patients living with hematologic malignancies. Our investigational first-in-class telomerase inhibitor, imetelstat, harnesses Nobel Prize winning science in a treatment that may alter the underlying course of these diseases.
Our lead indication for imetelstat is in Low or Intermediate-1 risk myelodysplastic syndromes, or lower risk MDS. In January 2023, we reported positive top-line results from our IMerge Phase 3 clinical trial. The trial met its primary endpoint of 8-week transfusion independence rate and a key secondary endpoint of 24-week transfusion independence rate, demonstrating highly statistically significant (i.e., P<0.001 for both) and clinically meaningful benefits in imetelstat versus placebo. Furthermore, statistically significant and clinically meaningful efficacy results were observed in the trial across key subtypes, including patients who were ringed sideroblast positive, or RS positive, and ringed sideroblast negative, or RS negative; patients with high and very high baseline transfusion burden; and patients classified as Low or Intermediate-1 risk according to the International Prognostic Scoring System, or IPSS.
Based on the positive top-line data from IMerge Phase 3 and the prior IMerge Phase 2, we plan to submit a New Drug Application, or NDA, to the U.S. Food and Drug Administration, or FDA, in the U.S. in mid-2023 and a marketing authorization application, or MAA, in Europe in the second half of 2023 for the use of imetelstat in adult patients with lower risk MDS. If the NDA is accepted for filing and imetelstat is approved for commercialization by the FDA within the timelines we expect, we anticipate commercial launch of imetelstat in lower risk MDS in the U.S. could occur in the first half of 2024. In Europe, we anticipate review of the planned MAA, if validated by the European Medicines Agency, or EMA, could take approximately 14 months and, if approved, we anticipate that the commercial launch of imetelstat in lower risk MDS in Europe could occur by the end of 2024.
2
We believe that the positive top-line data from IMerge Phase 3 and IMerge Phase 2, as well as our prior Phase 2 clinical trial of imetelstat in patients with Intermediate-2 or High-Risk myelofibrosis who have relapsed after or are refractory to treatment with a janus associate kinase inhibitor, or JAK inhibitor, or relapsed/refractory MF, provide strong evidence that imetelstat targets telomerase to inhibit the uncontrolled proliferation of malignant stem and progenitor cells enabling recovery of bone marrow and normal blood cell production, which suggest potential disease-modifying activity. We believe this potential for disease modification could differentiate imetelstat from currently approved treatments in myeloid hematologic malignancies. Accordingly, in addition to lower risk MDS, we are developing imetelstat for the treatment of several myeloid hematologic malignancies with the following ongoing clinical trials:
•IMpactMF, a Phase 3 clinical trial in relapsed/refractory MF with overall survival, or OS, as the primary endpoint, that currently is enrolling patients. Based on our planning assumptions for enrollment and event (death) rates in the trial, we expect the interim analysis for OS in IMpactMF may occur in 2024, and the final analysis may occur in 2025. Because these analyses are event-driven and it is uncertain whether actual rates for enrollment and events will reflect current planning assumptions, the results may be available at different times than currently expected.
•IMproveMF, a Phase 1 combination clinical trial in frontline Intermediate-2 or High-Risk myelofibrosis, or frontline MF, that currently is enrolling patients and the first patient was dosed in April 2021; and
•IMpress, an investigator-led Phase 2 clinical trial in Intermediate-2 or High-Risk myelodysplastic syndromes, or higher risk MDS, and acute myeloid leukemia, or AML, with the initial clinical site planned to open in the first quarter of 2023.
Imetelstat Regulatory Designations
Imetelstat has been granted Fast Track designations by the FDA for development in two indications: (1) for the treatment of adult patients with transfusion-dependent anemia due to lower risk MDS, who do not have a deletion 5q chromosomal abnormality, also known as non-del(5q), and who are refractory or resistant to treatment with an erythropoiesis stimulating agent, or ESA (i.e., the treatment population in IMerge Phase 3); and (2) for the treatment of adult patients with Intermediate-2 or High-Risk MF whose disease has relapsed after or is refractory to JAK inhibitor treatment (i.e., the treatment population in IMpactMF). The FDA granted orphan drug designation to imetelstat in June 2015 for the treatment of MF and for the treatment of MDS in December 2015, and the EMA granted orphan drug designation in December 2015 to imetelstat for the treatment of MF and in July 2020 for the treatment of MDS. In October 2021, we gained access to the Innovative Licensing and Access Pathway, or ILAP, in the United Kingdom, or U.K., through the receipt of an Innovation Passport for imetelstat to treat lower risk MDS.
Stage-Gated Milestone-Driven Global Commercial Plans for Imetelstat
If imetelstat is approved in lower risk MDS for marketing by regulatory authorities, we plan to commercialize imetelstat ourselves in the U.S. and may seek commercialization partners for territories outside of the U.S. We have therefore developed a commercial pre-launch and potential launch plan that is driven by the achievement of certain regulatory milestones, such as FDA acceptance for filing of our planned NDA, as well as EMA validation of our planned EU MAA submission. We are conducting pre-commercial preparations for the U.S., such as: enhancing and/or establishing company processes and systems to support a potential commercial launch, refining our market research in lower risk MDS, engaging in marketing and commercial access/reimbursement preparatory efforts, as well as executing on long-lead time activities, such as selecting a third-party logistics provider, completing state licensing requirements and hiring personnel to support potential sales, marketing and commercial operations. In light of the positive top-line IMerge Phase 3 results, we continue to evaluate our strategy for the potential launch and commercialization of imetelstat in Europe. Based on our internal estimates of pricing and addressable patient populations in 2030 and if regulatory authorities approve imetelstat for marketing in lower risk MDS and relapsed/refractory MF, we believe the combined potential peak market opportunity in the U.S. and key European markets for imetelstat is approximately $3.0 billion, of which lower risk MDS represents approximately $1.2 billion and relapsed/refractory MF represents approximately $1.8 billion.
3
IMerge: Ongoing Phase 2/3 Clinical Trial in Lower Risk MDS
Trial Design
IMerge is a two-part Phase 2/3 clinical trial evaluating imetelstat (7.5 mg/kg dose administered as a two-hour intravenous infusion every four weeks) in transfusion dependent lower risk MDS patients who are relapsed after or refractory to prior treatment with an ESA. To be eligible for IMerge, patients are required to be transfusion dependent, defined as requiring at least four units of packed red blood cells, or RBCs, over an eight-week period during the 16 weeks prior to entry into the trial.
IMerge Phase 3 is a double-blind, 2:1 randomized, placebo-controlled clinical trial that, based on discussions with U.S. and European regulatory authorities, was designed to support, if successful, the registration of imetelstat in lower risk MDS. The trial enrolled patients with lower risk transfusion dependent MDS who were relapsed, or refractory to, or ineligible for ESA, had not received prior treatment with either a hypomethylating agent, or HMA, or lenalidomide and were non-del(5q). IMerge Phase 3 is being conducted at 118 medical centers globally in 17 countries in North America, Europe, Middle East and Asia.
The primary efficacy endpoint of IMerge Phase 3 is the rate of red blood cell transfusion independence, or RBC-TI, lasting at least eight weeks, defined as the proportion of patients without any RBC transfusions during any consecutive eight weeks since entry to the trial, or 8-week TI. Key secondary endpoints for IMerge Phase 3 include the rate of RBC-TI lasting at least 24 weeks, or 24-week TI, and the rate of hematologic improvement erythroid, or HI-E, which is a rise in hemoglobin of at least 1.5 g/dL above the pretreatment level for at least eight weeks or a reduction of at least four units of RBC transfusions over eight weeks compared with the prior RBC transfusion burden. Other secondary endpoints include the time to and duration of RBC-TI; the proportion of patients achieving Complete Response, or CR, or Partial Response, or PR, according to the 2006 International Working Group, or IWG, criteria for MDS; the proportion of patients requiring RBC transfusions and the transfusion burden; the proportion of patients requiring the use of myeloid growth factors and the dose; assessments of the change in the patients’ quality of life using several validated instruments; as well as an assessment of OS, and time to progression to AML.
Positive Top-Line Results from IMerge Phase 3
In January 2023, we reported positive top-line results from IMerge Phase 3. A total of 178 patients were enrolled in IMerge Phase 3, with patients randomized on a 2:1 basis to imetelstat (n=118) or placebo (n=60), and represents the intent-to-treat population in the trial. The population for the analysis of safety data is based on 177 patients (imetelstat, n=118, placebo, n=59) because one patient in the placebo arm was enrolled, but did not receive any treatment. The trial met its primary endpoint of 8-week TI rate and key secondary endpoint of 24-week TI rate, among others, demonstrating statistically significant and clinically meaningful results with imetelstat versus placebo with no new safety signals and safety results consistent with prior imetelstat clinical trials. Key patient participation information is as follows:
| | |
| Imetelstat (n=118) | Placebo (n=60) |
Median time on study, months (range) | 19.5 (1.4-36.2) | 17.5 (0.7-34.3) |
Median time on treatment, months (range) | 7.8 (0.03-32.5) | 6.5 (0.03-26.7) |
Median time on treatment for 8-week TI responders, months (range) | 17.2 (1.8-32.5) | 15.0 (4.1-25.8) |
Median treatment, cycles (range) | 8 (1-34) | 8 (1-30) |
Median treatment cycles for 8-week TI responders, months (range) | 18 (3-34) | 17 (3-29) |
Key baseline demographics and disease characteristics of patients in IMerge Phase 3, summarized below, demonstrate the broad range of lower risk MDS subtypes, and patients with high disease burden, enrolled in the trial:
4
| | |
| (n=118) | (n=60) |
Age, years, median (range) | 71.5 (44-87) | 73.0 (39-85) |
WHO 2001 category, n (%) | | |
RS+ | 73 (61.9) | 37 (61.7) |
RS- | 44 (37.3) | 23 (38.3) |
RBC transfusion burden, units/8 weeks, median (range) | 6 (4-33) | 6 (4-13) |
4 - 6 units / 8 weeks, n (%) | 62 (52.5) | 33 (55.0) |
>6 units / 8 weeks, n (%) | 56 (47.5) | 27 (45.0) |
IPSS risk category, n (%) | | |
Low | 80 (67.8) | 39 (65.0) |
Intermediate-1 | 38 (32.2) | 21 (35.0) |
Prior luspatercept use, n (%)* | 7 (5.9) | 4 (6.7) |
Pre-treatment hemoglobin**, median (range), g/dL | 7.9 (5.3-10.1) | 7.8 (6.1-9.2) |
_________________
* There were an insufficient number of patients (imetelstat: 7/118; placebo: 4/60) who were previously treated with luspatercept enrolled in IMerge Phase 3 to draw conclusions about the effect of imetelstat treatment in such patients. Of these patients, no imetelstat-treated patients and one placebo patient achieved 8-week TI.
** Pretreatment hemoglobin is defined as the average of all hemoglobin values in the eight weeks prior to the first dose date, excluding values that were within 14 days after transfusion (thus considered to be influenced by transfusion).
After a median follow-up time of 18 months for all patients in the trial, the following chart is the status of patients as of October 13, 2022, the clinical cut-off date for top-line results.
| | |
| Imetelstat (n=118) | Placebo (n=59) |
Treatment ongoing, n (%) | 27 (22.9) | 14 (23.7) |
Treatment discontinued, n (%) | 91 (77.1) | 45 (76.3) |
Lack of efficacy | 28 (23.7) | 25 (42.4) |
Adverse event | 19 (16.1) | 0 |
Cytopenias | 11 (9.3) | 0 |
Unrelated | 8 (6.8) | 0 |
Disease relapse after initial response on study | 17 (14.4) | 1 (1.7) |
Patient decision | 16 (13.6) | 10 (16.9) |
Progressive disease | 7 (5.9) | 5 (8.5) |
AML progression | 2 (1.7) | 1 (1.7) |
Investigator decision | 2 (1.7) | 2 (3.4) |
Death* | 1 (0.8) | 2 (3.4) |
Lost to follow up | 1 (0.8) | 0 |
_________________
* On imetelstat treatment arm, patient death: neutropenic sepsis not related to drug after ~two-year treatment duration
On placebo treatment arm, patient deaths: (1) COVID-19 and (1) heart valve issue
Efficacy results for the primary 8-week TI endpoint and 24-week TI secondary endpoint summarized below illustrate the depth and durability of transfusion independence demonstrated with high statistical significance for imetelstat versus placebo in IMerge Phase 3.
| | | |
| Imetelstat (n=118) | Placebo (n=60) | P-value * |
5
| | | |
Primary endpoint: 8-week TI, n (%) | 47 (39.8) | 9 (15.0) | <0.001 |
95% confidence interval | (30.9, 49.3) | (7.1, 26.6) | |
Secondary endpoint: 24-week TI, n (%) | 33 (28.0) | 2 (3.3) | <0.001 |
95% confidence interval | (20.1, 37.0) | (0.4, 11.5) | |
_________________
* Cochran Mantel Haenszel test stratified for prior RBC transfusion burden (≤6 units or >6 units of RBCs/8 weeks) and baseline IPSS risk score (Low or Intermediate-1)
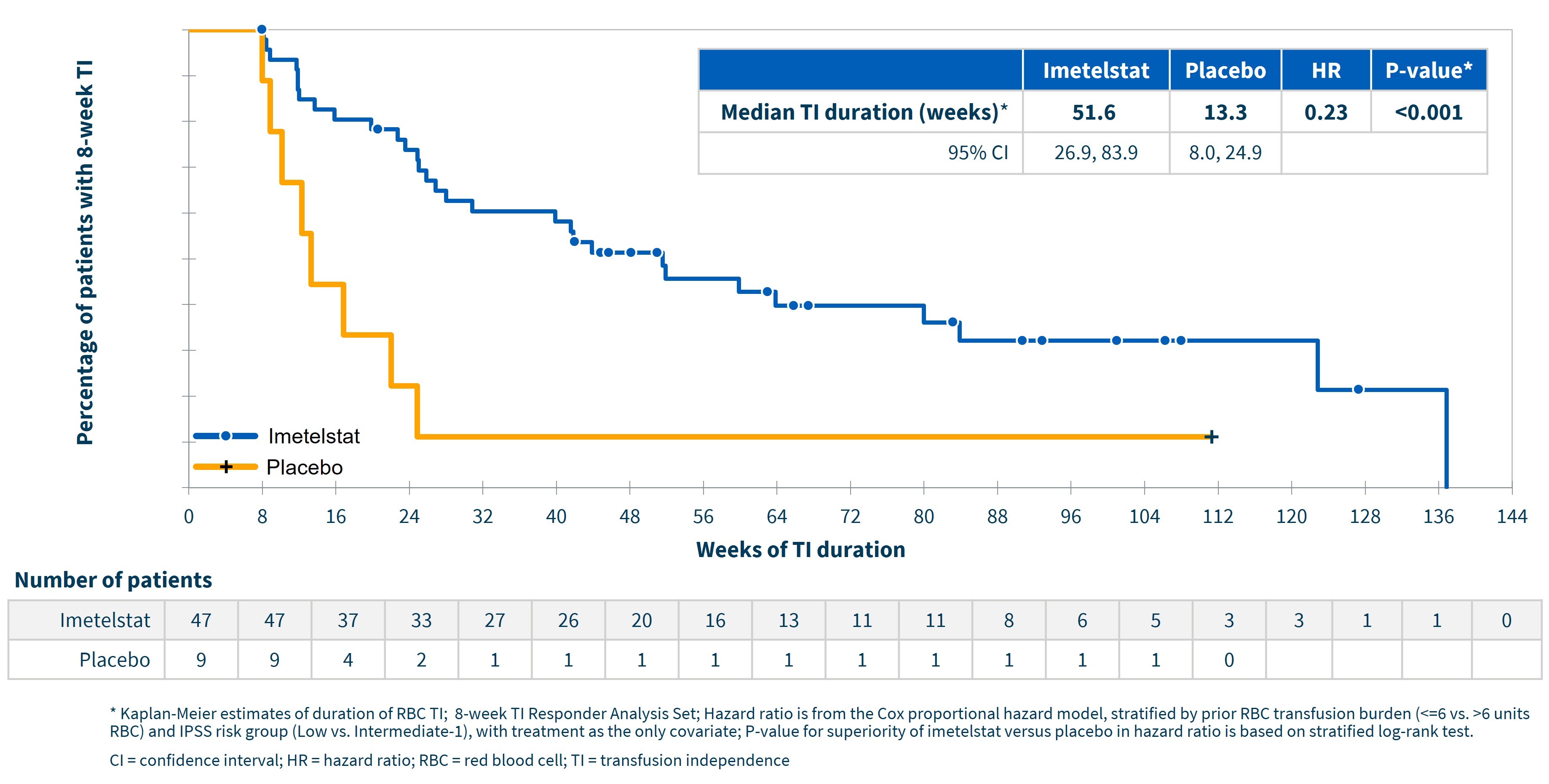
Highly statistically significant (p<0.001; hazard ratio 0.23) durable transfusion independence was achieved with median TI duration approaching one year (95% CI (weeks): 26.9, 83.9) for imetelstat and approximately 13 weeks for placebo (95% CI (weeks): 8.0, 24.9), using Kaplan Meier estimates, as shown in the following graph. Approximately 83% of patients achieving 8-week TI had a single continuous TI period. For imetelstat patients achieving 24-week TI, the median TI duration was 80.0 weeks (95% CI (weeks): 51.6, not estimable (NE)). In addition, approximately 70% of imetelstat patients who achieved 8-week TI continued to achieve 24-week TI.
6
In addition, there was an increasing magnitude of benefit for imetelstat versus placebo with longer time intervals, as shown in the graph below.
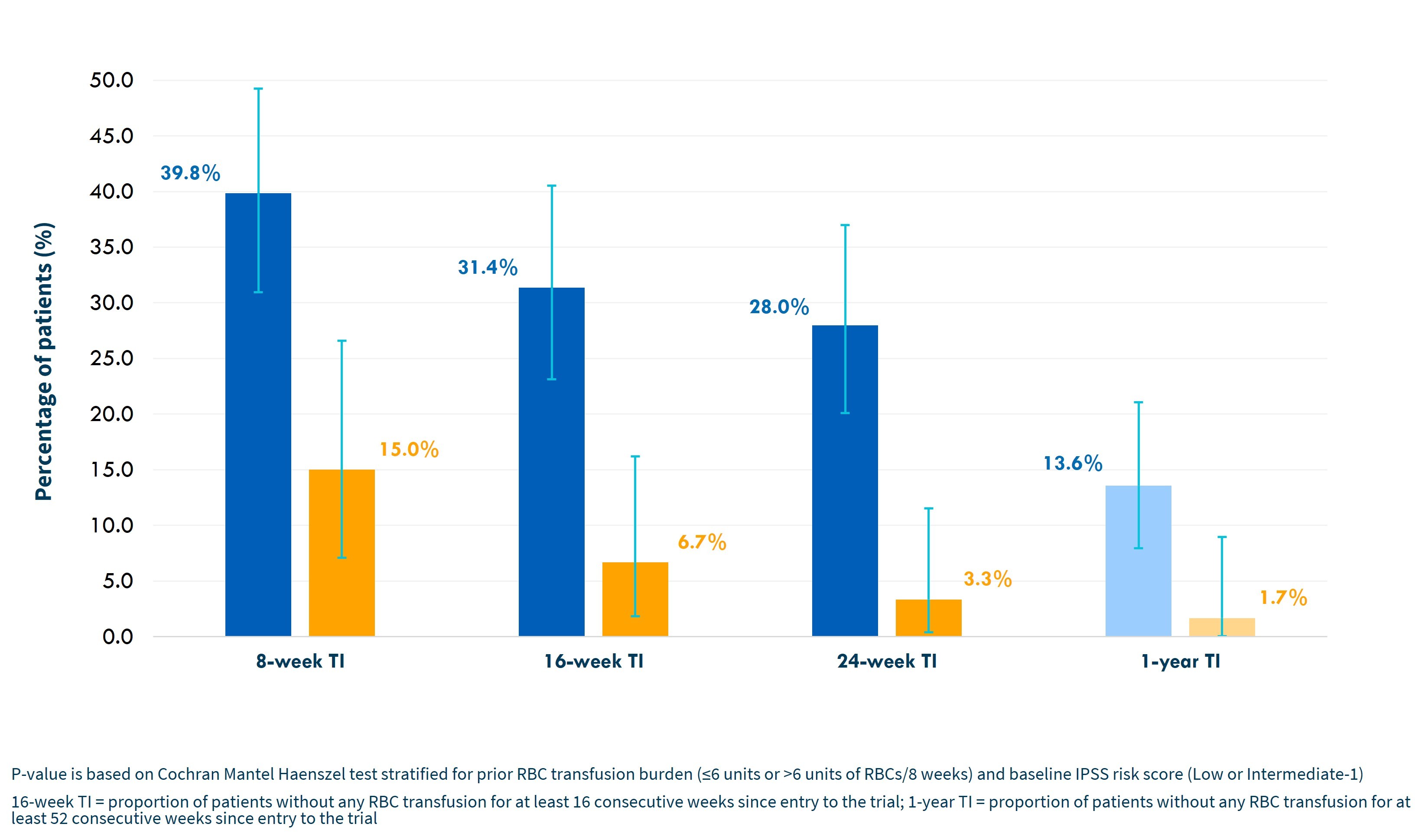
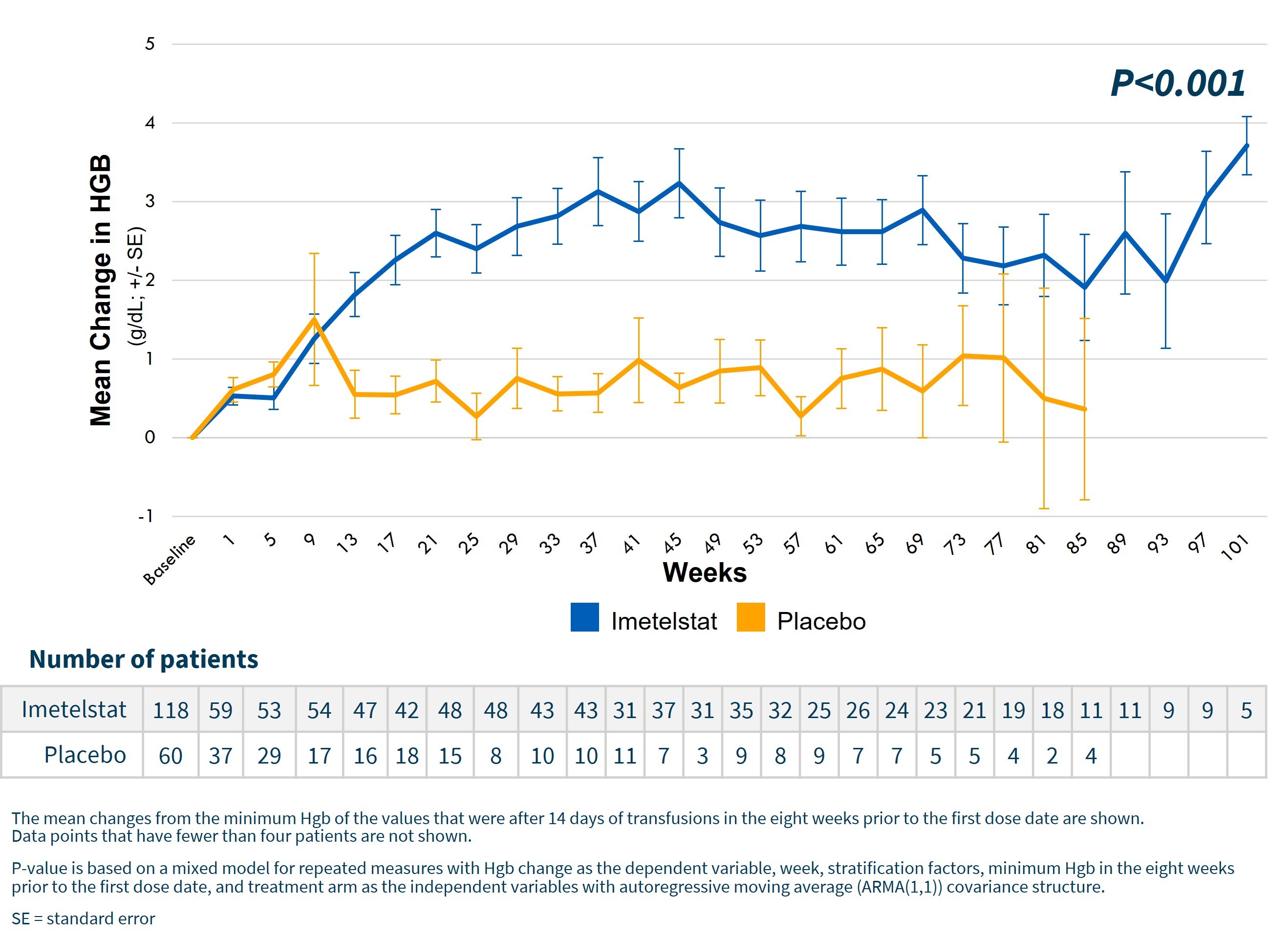
Highly statistically significant (p<0.001) increase in hemoglobin levels over time for imetelstat patients as shown in the graph below. For patients achieving 8-week TI, median increases in hemoglobin were 3.6 g/dL for imetelstat and 0.8 g/dL for placebo. The peak hemoglobin reached for these patients was 11.3 g/dL for imetelstat and 8.9 g/dL for placebo.
7
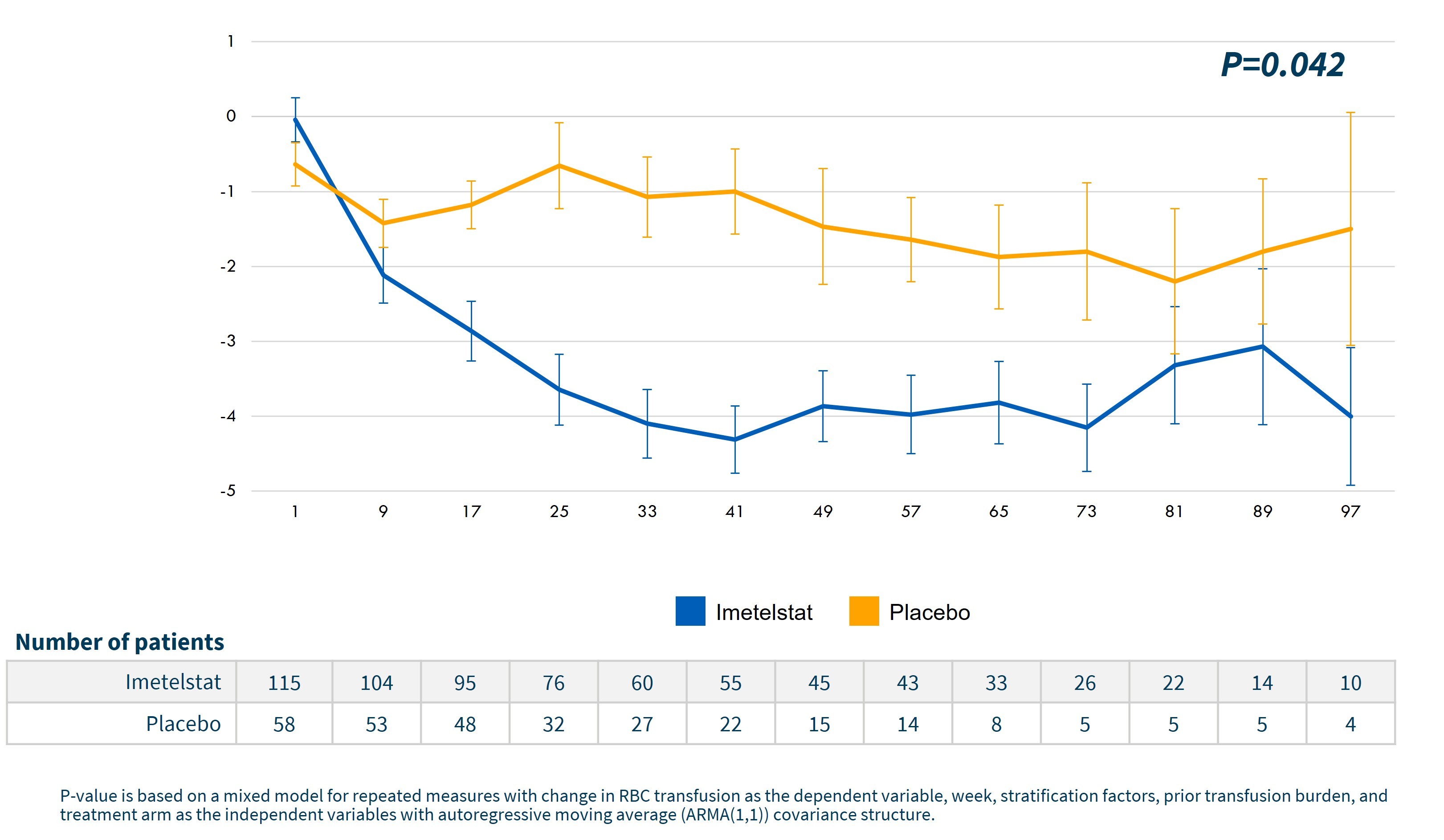
Corresponding to the increases in hemoglobin, a statistically significant decrease in the number of RBC units transfused was achieved for imetelstat treated patients versus placebo, as shown in the graph below.
Furthermore, as shown in the table below, statistically significant 8-week RBC-TI was observed with imetelstat versus placebo across lower risk MDS subtypes (p<0.05) and similar 8-week RBC-TI was observed for imetelstat within each subtype category in comparison to the overall population.
| | | | |
| Imetelstat, n (%) | Placebo, n (%) | Difference (95% CI) | P-value* |
Overall | 47/118 (39.8) | 9/60 (15.0) | 24.8 (9.9, 36.9) | <0.001 |
WHO category | | | | |
RS+ | 33/73 (45.2) | 7/37 (18.9) | 26.3 (5.9, 42.2) | 0.016 |
RS- | 14/44 (31.8) | 2/23 (8.7) | 23.1 (-1.3, 40.6) | 0.038 |
Transfusion burden | | | | |
4-6 units | 28/62 (45.2) | 7/33 (21.2) | 23.9 (1.9, 41.4) | 0.027 |
>6 units | 19/56 (33.9) | 2/27 (7.4) | 26.5 (4.7, 41.8) | 0.023 |
IPSS risk category | | | | |
Low | 32/80 (40.0) | 8/39 (20.5) | 19.5 (-0.1, 35.2) | 0.034 |
Intermediate-1 | 15/38 (39.5) | 1/21 (4.8) | 34.7 (8.8, 52.4) | 0.004 |
_________________
* Cochran Mantel Haenszel test stratified for prior RBC transfusion burden (≤6 units or >6 units of RBCs/8 weeks) and baseline IPSS risk score (Low or Intermediate-1)
Specifically for the RS+ and RS- subtypes, statistically significant improvement in both 8-week and 24-week TI was achieved with imetelstat versus placebo, as shown in the table below.
| | | | | | |
| RS+ | RS- |
| Imetelstat (n=73) | Placebo (n=37) | P-value* | Imetelstat (n=44) | Placebo (n=23) | P-value* |
8-week TI responders, n (%) | 33 (45.2) | 7 (18.9) | 0.016 | 14 (31.8) | 2 (8.7) | 0.038 |
Median duration of TI*, weeks (95% CI) | 46.9 (25.9, 83.9) | 16.9 (8.0, 24.9) | 0.035 | 51.6 (11.9, NE) | 11.2 (10.1, NE) | 0.062 |
24-week TI responders, n (%) | 24 (32.9) | 2 (5.4) | 0.003 | 9 (20.5) | 0 (0.0) | 0.019 |
Median duration of TI*, weeks (95% CI) | 80.0 (41.6, NE) | NE (24.9, NE) | 0.808 | 122.9 (25.0, NE) | NE (NE, NE) | NE |
8
_________________
* Kaplan-Meier estimates of duration of RBC TI; 8-week/24-week TI Responder Analysis Set; P-value for TI rate is based on Cochran Mantel Haenszel test stratified for prior RBC transfusion burden (≤6 units or >6 units of RBCs/8 weeks) and baseline IPSS risk score (Low or Intermediate-1); P-value for duration of TI is based on stratified log-rank test.
NE = not estimable
The IMerge Phase 3 protocol specified use of the International Working Group, or IWG, 2006 criteria to measure HI-E and was not statistically significant (p=0.112) for imetelstat versus placebo. The current IWG 2018 HI-E criteria places greater emphasis on durability by measuring response for ≥16 weeks, rather than ≥8 weeks. Under these new 2018 IWG criteria, a highly statistically significant (p<0.001) HI-E rate was achieved for imetelstat versus placebo. See the table below for HI-E response under both 2018 and 2006 IWG criteria.
| | | |
| Imetelstat (n=118) | Placebo (n=60) | P-value * |
HI-E per IWG 2018, n (%) | 50 (42.4) | 8 (13.3) | <0.001 |
95% CI, % | (33.3, 51.8) | (5.9, 24.6) | |
16-week TI, n (%) | 37 (31.4) | 4 (6.7) | <0.001 |
95% CI, % | (23.1, 40.5) | (1.9, 16.2) | |
Transfusion reduction by ≥50%/16 weeks | 51 (43.2) | 9 (15.0) | <0.001 |
95% CI, % | (34.1, 52.7) | (7.1, 26.6) | |
HI-E per IWG 2006, n (%) | 75 (63.6) | 31 (51.7) | 0.112 |
95% CI, % | (54.2, 72.2) | (38.4, 64.8) | |
≥1.5 g/dL increase in Hgb ≥8 weeks, n (%) | 40 (33.9) | 6 (10.0) | <0.001 |
95% CI, % | (25.4, 43.2) | (3.8, 20.5) | |
Transfusion reduction by ≥4U/8 weeks, n (%) | 71 (60.2) | 30 (50.0) | 0.175 |
95% CI, % | (50.8, 69.1) | (36.8, 63.2) | |
_________________
* Cochran Mantel Haenszel test stratified for prior RBC transfusion burden (≤6 units or >6 units of RBCs/8 weeks) and baseline IPSS risk score (Low or Intermediate-1)
9
Clinical and molecular evidence supporting the potential for disease modification with imetelstat includes a one-year median TI duration for imetelstat 8-week TI responders, a median rise of 3.6 g/dL in hemoglobin levels in those same patients and >50% variant allele frequency decreases in SF3B1, TET2, DNMT3A and ASXL1 mutations, as shown in the graph below.
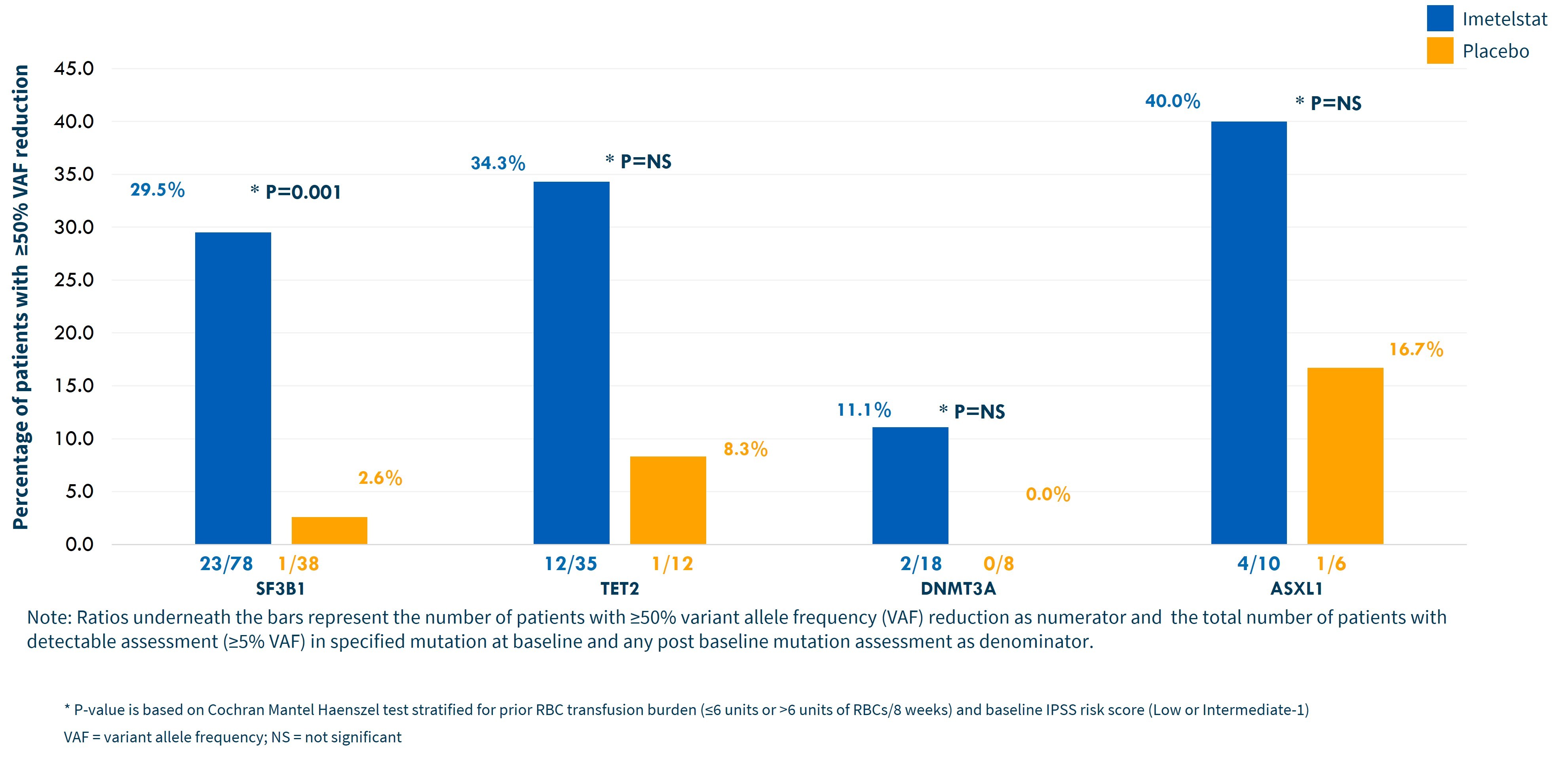
The safety results in IMerge Phase 3 were consistent with prior clinical trials of imetelstat in hematologic malignancies, and no new safety signals were identified. The most frequent non-hematologic toxicities occurring in ≥10% of patients on either imetelstat or placebo arms are listed in the table below. Grade 3 elevations in liver function tests, or LFTs, on imetelstat were short in duration (median < 2 weeks) and more than 80% resolved to Grade 2 or lower within 4 weeks, with no cases of liver test elevations consistent with Hy’s Law or Drug Induced Liver Injury.
| | | | |
AE, n (%) | Imetelstat (n=118) | Placebo (n=59) |
| All Grades | Grade 3/4 | All Grades | Grade 3/4 |
Asthenia | 22 (18.6) | 0 | 8 (13.6) | 0 |
COVID-19* | 21 (17.8) | 2 (1.7) | 9 (15.2) | 3 (5.1) |
Peripheral edema | 13 (11.0) | 0 | 8 (13.6) | 0 |
Headache | 15 (12.7) | 1 (0.8) | 3 (5.1) | 0 |
Diarrhea | 14 (11.9) | 1 (0.8) | 7 (11.9) | 1 (1.7) |
Alanine aminotransferase increased | 14 (11.9) | 3 (2.5) | 4 (6.8) | 2 (3.4) |
Hyperbilirubinemia | 11 (9.3) | 1 (0.8) | 6 (10.2) | 1 (1.7) |
Constipation | 9 (7.6) | 0 | 7 (11.9) | 0 |
Pyrexia | 9 (7.6) | 2 (1.7) | 7 (11.9) | 0 |
_________________
* Includes COVID-19, asymptomatic COVID-19, COVID-19 pneumonia
| | | | |
LFT Lab Abnormality, n (%) | Imetelstat (n=118) | Placebo (n=59) |
| All Grades | Grade 3 | All Grades | Grade 3 |
10
| | | | |
Alanine Aminotransferase (ALT*) | 46 (39.3) | 4 (3.4) | 22 (37.3) | 3 (5.1) |
Alkaline Phosphatase (ALP) | 53 (44.9) | 0 | 7 (11.9) | 0 |
Aspartate Aminotransferase (AST) | 57 (48.3) | 1 (0.8) | 13 (22.0) | 1 (1.7) |
Bilirubin | 46 (39.0) | 1 (0.8) | 23 (39.0) | 1 (1.7) |
_________________
* n=117 for ALT imetelstat patients
The most frequent hematologic toxicities are listed in the table below.
| | | | |
AE, n (%) | Imetelstat (n=118) | Placebo (n=59) |
| All Grades | Grade 3/4 | All Grades | Grade 3/4 |
Thrombocytopenia | 89 (75.4) | 73 (61.9) | 6 (10.2) | 5 (8.5) |
Neutropenia | 87 (73.7) | 80 (67.8) | 4 (6.8) | 2 (3.4) |
Anemia | 24 (20.3) | 23 (19.5) | 6 (10.2) | 4 (6.8) |
Leukopenia | 12 (10.2) | 9 (7.6) | 1 (1.7) | 0 |
Clinical consequences from cytopenias were similar between imetelstat and placebo groups as shown in the table below.
| | |
Event, n (%) | Imetelstat (n=118) | Placebo (n=59) |
Grade >3 bleeding events* | 3 (2.5) | 1 (1.7) |
Grade >3 infections+ | 13 (11.0) | 8 (13.6) |
Grade febrile neutropenia** | 1 (0.8) | 0 |
_________________
* No ≥Grade 3 bleeding events in the setting of Grade 3/4 thrombocytopenia; on imetelstat: two patients with Grade 4 gastrointestinal bleeding, unrelated and resolved and one Grade 3 hematuria, unrelated and resolved
+ On imetelstat: three patients with Grade 3/4 infections in setting of Grade 3/4 neutropenia; all three were sepsis and resolved with only one considered related
** Occurred at day 33, lasted 8 days; assessed by investigator as possibly related to imetelstat; patient subsequently achieved TI >40 weeks and remains on treatment at data cut-off
Furthermore, as shown in the table below, the median duration of cytopenias was shorter for imetelstat versus placebo and the resolution to Grade 2 or lower was higher for imetelstat versus placebo.
| | |
| Imetelstat+ | Placebo |
Thrombocytopenia events* | | |
Median duration, weeks, (range) | 1.4 (0.1-12.6) | 2.0 (0.3-11.6) |
Resolved within <4 weeks, % | 86.3 | 44.4 |
Neutropenia events* | | |
Median duration, weeks, (range) | 1.9 (0-15.9) | 2.2 (1.0-4.6) |
Resolved within <4 weeks, % | 81.0 | 50.0 |
_________________
11
+ 18% of imetelstat treated patients received a median of 1 platelet transfusions; 35% of imetelstat treated patients received growth factor support
* Analysis performed for patients who experienced Grade 3/4 cytopenias. Resolution determined by return to Grade 2 or lower
Planned Next Steps to Advance Imetelstat in Lower Risk MDS Toward Potential Commercialization
We plan to present additional data from IMerge Phase 3 at medical meetings later this year, including data relating to potential correlation of decreases in mutation burden and abnormal cytogenetic clones with clinical responses, patient reported outcomes, hTERT and telomerase activity biomarker data and continued follow-up of durability of transfusion independence. These data, which are not yet available to Geron, will not be available to you prior to investing in this offering.
We believe the depth, breadth and durability of transfusion independence reported from IMerge Phase 3 to date, as well as from IMerge Phase 2, provide strong support for our planned regulatory submissions in the U.S. and in the EU for the use of imetelstat in patients with lower risk MDS. We therefore plan to submit an NDA in the U.S. in mid-2023 and an MAA in Europe in the second half of 2023 for the use of imetelstat in adult patients with lower risk MDS. With Fast Track designation for imetelstat from the FDA for the treatment of adult patients with transfusion-dependent anemia due to lower risk MDS, non-del(5q), and who are refractory or resistant to treatment with an ESA (i.e., the treatment population in IMerge Phase 3), the FDA granted our request for rolling submission of the NDA. If the NDA is accepted for filing and approved within the timelines we expect, we anticipate that the commercial launch of imetelstat in lower risk MDS could occur in the U.S. in the first half of 2024. In Europe, we anticipate review of the planned MAA, if validated by the EMA could take approximately 14 months and, if approved, we anticipate that the commercial launch of imetelstat in lower risk MDS in Europe could occur by the end of 2024.
IMpactMF: Ongoing Phase 3 Clinical Trial in Relapsed/Refractory MF
IMpactMF, our Phase 3 clinical trial in relapsed/refractory MF, is an open label, 2:1 randomized, controlled clinical trial designed to evaluate imetelstat (9.4 mg/kg administered by intravenous infusion over two hours every three weeks) in approximately 320 patients. Patients relapsed after or refractory to a JAK inhibitor are defined as having an inadequate spleen response or symptom response after treatment with a JAK inhibitor for at least six months, including an optimal dose of a JAK inhibitor for at least two months. The best available therapy, or BAT, control arm of IMpactMF excludes the use of JAK inhibitors. With respect to the trial design for IMpactMF, the FDA urged us to consider adding a third dosing arm to assess a lower dose and/or a more frequent dosing schedule that might improve the planned trial’s chance of success by identifying a less toxic regimen and/or more effective spleen response, one of the trial’s secondary endpoints. Based on data from IMbark, we believe that testing a lower dose regimen would likely result in a lower median OS, which is the trial’s primary endpoint, in the imetelstat treatment arm. We believe existing data also suggest that lowering the dose would not result in a clinically meaningful reduction in toxicity. For these reasons, we therefore determined not to add a third dosing arm to the trial design, and the FDA did not object to our proposed imetelstat dose and schedule of 9.4 mg/kg every three weeks.
The primary efficacy endpoint for IMpactMF is OS. Key secondary endpoints include symptom response; spleen response; progression free survival; complete remission, partial remission or clinical improvement, as defined by the International Working Group for Myeloproliferative Neoplasms Research and Treatment criteria; duration of response; safety; pharmacokinetics; and patient reported outcomes. Currently, we have selected 165 of the planned 180 sites to participate in IMpactMF across North America, South America, Europe, Australia and Asia.
IMpactMF is designed with >85% power to detect a 40% reduction in the risk of death (hazard ratio=0.60; one-sided alpha=0.025). The final analysis for OS is planned to be conducted after more than 50% of the patients planned to be enrolled in the trial have died (referred to as an event). An interim analysis of OS, in which the alpha spend is expected to be approximately 0.01, is planned to be conducted after approximately 70% of the total projected number of events (deaths) for the final analysis have occurred.
12
Current Status of IMpactMF
IMpactMF opened for patient screening and enrollment in December 2020. As of December 31, 2022, we had 140 of the 165 selected sites open for patient enrollment, and we are continuing to select additional sites. The first patient was dosed in April 2021. Given the uncertain and unpredictable impact of the COVID-19 pandemic on our clinical trial activities, including the constraints on clinical site personnel resources due to other competing trials in MF at the sites where IMpactMF is planned to be conducted, under current planning assumptions, we expect IMpactMF to be fully enrolled in 2024. Based on our planning assumptions for enrollment and event (death) rates in the trial, we expect the interim analysis for OS in IMpactMF may occur in 2024 and the final analysis may occur in 2025. Because these analyses are event-driven and it is uncertain whether actual rates for enrollment and events will reflect current planning assumptions, the results may be available at different times than currently expected. At the interim analysis, if the pre-specified statistical OS criterion is met, then we expect such data may potentially support the registration of imetelstat in relapsed/refractory MF. Subject to protocol-specified stopping rules for futility, if the pre-specified OS criterion is not met at the interim analysis, the trial will continue to the final analysis, which is expected to occur approximately one year later.
The timing and achievement of either or both of the planned analyses depend on numerous factors, including delays or interruptions related to the effects of the COVID-19 pandemic or geopolitical events. In addition, our ability to enroll, conduct and complete IMpactMF depends on whether we can obtain and maintain the relevant clearances from regulatory authorities and other institutions to enroll, conduct and complete the trial, and our ability to raise additional capital following this offering if and when needed in order to complete the trial.
Improvement in Overall Survival and Potential Disease-Modifying Activity Observed in IMbark Phase 2
The IMbark Phase 2 clinical trial was designed to evaluate two dosing regimens of imetelstat (either 4.7 mg/kg or 9.4 mg/kg administered by intravenous infusion every three weeks) in patients with relapsed/refractory MF. The co-primary efficacy endpoints for IMbark were spleen response rate, defined as the proportion of patients who achieve a reduction of at least 35% in spleen volume as assessed by imaging, and symptom response rate, defined as the proportion of patients who achieve a reduction of at least 50% in Total Symptom Score, at 24 weeks. Key secondary endpoints were OS and safety.
We previously reported efficacy and safety results from the IMbark Phase 2 clinical trial, including median OS of 28.1 months for patients on the high dose arm of the study, which is almost twice the reported median OS of 14 – 16 months in medical literature. To evaluate this potential benefit, we conducted a post-hoc analysis of OS for patients treated with imetelstat 9.4 mg/kg in IMbark compared to OS calculated from real world data, or RWD, collected at the Moffitt Cancer Center for patients who had discontinued treatment with ruxolitinib, a JAK inhibitor, and who were subsequently treated with BAT. To make a comparison between the IMbark data and RWD, a cohort from the real-world dataset was identified that closely matched the IMbark patients, using guidelines for inclusion and exclusion criteria as defined in the IMbark clinical protocol, such as platelet count and spleen size. Calculations from two propensity score analysis approaches resulted in a median OS of 30.7 months for the imetelstat-treated patients from IMbark, which is more than double the median OS of 12.0 months using RWD for patients treated with BAT. These analyses also showed a 65% – 67% lower risk of death for the imetelstat-treated patients vs. BAT-treated patients. We believe these analyses suggest potentially longer OS for imetelstat-treated relapsed/refractory MF patients in IMbark, compared to BAT in closely-matched patients from RWD. However, comparative analyses between RWD and our clinical trial data have several limitations. For instance, the analyses create a balance between treatment groups with respect to commonly available covariates, but do not take into account the unmeasured and unknown covariates that may affect the outcomes of the analyses. Potential biases are introduced by factors which include, for example, the selection of the patients included in the analyses, misclassification in the matching process, the small sample size, and estimates that may not represent the outcomes for the true treated patient population. For these and other reasons, such comparative analyses and any conclusions from such analyses should be considered carefully and with caution, and should not be relied upon as demonstrative or otherwise predictive or indicative of any current or potential future clinical trial results of imetelstat in relapsed/refractory MF, including IMpactMF.
In IMbark, patients also experienced other positive clinical outcomes, including symptom improvement, spleen reduction and bone marrow fibrosis improvement. In June 2020, we reported correlation analyses from
13
IMbark that showed a trend of longer OS in patients who achieved symptom response, spleen volume reductions and improved bone marrow fibrosis, in a dose-dependent manner. Furthermore, the reductions in the variant allele frequency of key driver mutations in MF and the improvement in bone marrow fibrosis observed in IMbark have also been correlated to the improvement in OS. We believe the improvement in bone marrow fibrosis, potential survival benefit, molecular data and correlations from IMbark provide strong evidence of the potential for disease modification with imetelstat, which we believe differentiates imetelstat from currently approved treatments for MF.
The safety results observed in IMbark were consistent with prior clinical trials of imetelstat in hematologic malignancies, and no new safety signals were identified. In the 9.4 mg/kg arm, reversible and manageable Grade 3/4 thrombocytopenia and neutropenia were reported in 24/59 patients (41%) and 19/59 patients (32%), respectively, without significant clinical consequences. 1/59 patients (2%) had Grade 3 febrile neutropenia. 3/59 patients (5%) had Grade 3/4 bleeding. 6/59 patients (10%) had Grade 3/4 infections. Furthermore, more than 70% of the observed Grade 3/4 cytopenias resolved to Grade 2 or lower by laboratory assessment within four weeks.
Exploratory Clinical Trials of Imetelstat in Additional Indications
IMproveMF: Phase 1 Combination Clinical Trial in Frontline Myelofibrosis (Frontline MF)
IMproveMF is a two-part Phase 1 clinical trial evaluating imetelstat in combination with ruxolitinib in patients with frontline MF. The trial is designed to use a Bayesian Optimal Interval design to test various doses of imetelstat in an escalating dose sequence with a defined number of patients per dosing arm. Escalation to the next higher dosing arm will only occur if the prior dose is tolerable to the patients. The primary objective of the first part of IMproveMF is to identify a recommended dosing regimen for further evaluation. Up to 20 patients are expected to be enrolled into the first part of IMproveMF, or IMproveMF Part 1. The first patient was dosed in IMproveMF in August 2022.
Upon identification of a tolerable dosing regimen for the combination treatment of imetelstat and ruxolitinib, the second part of IMproveMF, or IMproveMF Part 2, is planned to evaluate the efficacy and further evaluate the safety of that dosing regimen. Under IMproveMF Part 2, the primary endpoints are safety and symptom response rate, defined as the proportion of patients who achieve a >50% reduction in Total Symptom Score at 24 weeks. Secondary endpoints include change in fibrosis; spleen response rate, defined as the proportion of patients who achieve a >35% reduction in spleen volume from baseline as assessed by imaging; and the number of patients achieving complete remission, partial remission or clinical improvement, as defined by the International Working Group for Myeloproliferative Neoplasms Research and Treatment criteria. Up to 20 patients are expected to be enrolled into the IMproveMF Part 2.
IMpress: Investigator-Led Phase 2 Clinical Trial in Higher Risk Myelodysplastic Syndromes (Higher Risk MDS) and Acute Myeloid Leukemia (AML)
In collaboration with investigators in Germany, France and Australia, we are supporting IMpress, an investigator-led study of imetelstat in patients with higher risk MDS and relapsed/refractory AML, post-treatment with a hypomethylating agent, or HMA. IMpress is an open-label, single arm, Phase 2 clinical trial being conducted at three clinical sites. The first site is planned to open in the first quarter of 2023. The primary endpoint is overall response rate per criteria from the 2018 International Working Group for MDS and the European LeukemiaNet for AML. Secondary endpoints include safety, duration of response, progression-free survival and overall survival. In addition, pending the results of IMpress, we plan to support a Phase 1/2 investigator-led study in relapsed/refractory AML using a combination approach of imetelstat and venetoclax or azacitidine.
Research Programs
Preclinical Lymphoid Hematologic Malignancy Program
Academic research data suggests that certain lymphoid hematologic malignancies have higher telomerase activity and shorter telomeres when compared to normal healthy cells. Thus, we believe a telomerase inhibition approach may find utility in this disease setting.
14
Based on this scientific hypothesis, we initiated a preclinical research project with MD Anderson Cancer Center to determine the potential application of imetelstat in lymphoid hematologic malignancies. Preliminary results from this research were published in Blood in November 2022. Based on early results, we plan to collaborate further with MD Anderson Cancer Center to conduct preclinical research to assess the potential therapeutic effect of imetelstat in lymphoid malignancies.
Next Generation Telomerase Inhibitor Discovery Program
We have initiated a discovery program to identify a lead compound as a potential next generation oral telomerase inhibitor. If such a compound is identified, we plan to conduct preclinical experiments that may serve as a basis for potential future clinical testing. Discovery research is an uncertain and unpredictable process. As such, the timing and nature of any results from this discovery effort are difficult to forecast. If we select a lead compound from this discovery program, we expect to provide an update on our efforts at that time.
********
Risk Factor Summary
Below is a summary of material factors that make an investment in our securities speculative or risky. Importantly, this summary does not address all of the risks and uncertainties that we face. You should understand that it is not possible to predict or identify all such factors. Consequently, you should not consider this summary to be a complete discussion of all potential risks or uncertainties that may substantially impact our business. Additional discussion of the risks and uncertainties summarized in this risk factor summary, as well as other risks and uncertainties that we face, are described under the heading “Risk Factors” below, and this summary is qualified in its entirety by that discussion. Moreover, we operate in a competitive and rapidly changing environment. New factors emerge from time to time and it is not possible to predict the impact of all of these factors on our business, financial condition or results of operations. You should consider carefully the risks and uncertainties described under the heading “Risk Factors” below.
•We are wholly dependent on the success of our sole product candidate, imetelstat.
•If we fail to demonstrate sufficient safety and efficacy data from IMerge Phase 3 to the satisfaction of the FDA or similar international regulatory authorities, additional clinical testing may be required before we can seek regulatory approval for imetelstat in lower risk MDS and begin commercialization, if at all, any of which could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our business and commercial prospects.
•Any suspension of or delays in IMpactMF, including due to the effects of macroeconomic conditions such the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession, could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our commercial prospects.
•Any termination of IMpactMF would have a material adverse effect on our business, business prospects and the future of imetelstat.
•Imetelstat may continue to cause, or have attributed to it, undesirable or unintended side effects or other adverse events that could further delay or prevent the commencement and/or completion of clinical trials for imetelstat, further delay or prevent its regulatory approval, or limit its commercial potential.
•If IMpactMF fails to demonstrate safety and effectiveness to the satisfaction of the FDA or similar regulatory authorities in other countries or does not otherwise produce positive results, we would incur additional costs, experience delays in completing or ultimately fail in completing the development and commercialization of imetelstat in relapsed/refractory MF, which would have a material adverse effect on our business, business prospects and the future of imetelstat.
15
•Clinical drug development involves a lengthy and expensive process with uncertain timelines and uncertain outcomes, and results of earlier stage clinical trials and non-clinical studies may not be predictive of future results.
•We rely on third parties to conduct our clinical trials and their failure to perform could have a material adverse effect on our business that might cause us to cease operations.
•Our failure to obtain regulatory approval for imetelstat in the U.S. would have a material adverse effect on our business that would likely cause us to cease operations.
•If imetelstat is approved for marketing and commercialization and we are unable to establish sales, marketing and distribution capabilities, or obtain coverage and adequate third-party payor reimbursement, we will be unable to successfully commercialize imetelstat.
•If we are not successful in commercializing imetelstat, we will not be able to achieve our projections for future revenue, if any.
•We rely on third parties to manufacture and supply imetelstat, and may be unable to ensure that we have adequate quantities of imetelstat that meet specifications that may be approved or required by regulatory authorities, and timelines necessary for current and potential future clinical trials and potential commercial uses.
•Regulatory inspections of third-party manufacturers may identify deficiencies in manufacturing processes or facilities which could impact the ability of third-party manufacturers to produce and deliver products, including imetelstat.
•The COVID-19 pandemic has affected and continues to affect our ability to conduct clinical trial activities, causing delays in IMpactMF, IMproveMF, and our investigator-led clinical trial, IMpress, in AML and higher risk MDS. Additionally, the COVID-19 pandemic may delay and disrupt regulatory activities and our manufacturing and supply chain and have other adverse effects on our business and operations.
•In the absence of potential proceeds from exercises of currently outstanding warrants and potential drawdowns under our term loan facility, or Loan Agreement, with Hercules Capital Inc., or Hercules, and Silicon Valley Bank, or SVB, we will require substantial additional funding following this offering to further advance the imetelstat program, including through the completion of IMpactMF, IMproveMF and IMpress, as well as to conduct the clinical, regulatory and potential commercialization activities necessary to potentially bring imetelstat to market in relapsed/refractory MF and any other future indications, and our need for additional funds may arise sooner than planned. We cannot predict with any certainty whether and to what extent the outstanding warrants will be exercised for cash, or the timing or availability of additional funds under the Loan Agreement, if at all. The global economic slowdown, as well as inflation, rising interest rates and the prospects for recession, could materially and adversely affect our ability to raise additional capital, which could negative affect our liquidity, our business and business prospects and the future of imetelstat. If we are unable to raise this capital when needed, we would be forced to delay, reduce or eliminate our research and development activities and other operations or commercialization efforts which would have a material adverse effect on our business that might cause us to cease operations. Raising additional capital may subject us to unfavorable terms, cause dilution to our existing stockholders, restrict our operations, or require us to relinquish certain rights to imetelstat.
•We have incurred significant losses and negative cash flows from operations since our inception and anticipate that we will continue to incur significant expenses and losses for the foreseeable future.
•Our level of indebtedness and debt service obligations could adversely affect our financial condition, and may make it more difficult for us to fund our operations.
•If we are unable to obtain and maintain sufficient intellectual property protection for imetelstat for an adequate amount of time, or if the scope of the intellectual property protection is not sufficiently broad, our competitors could develop and commercialize products similar or identical to imetelstat, and our ability to successfully commercialize imetelstat may be adversely affected.
16
•If competitors develop products, product candidates or technologies that are superior to or more cost-effective than imetelstat, this would significantly impact the development and commercial viability of imetelstat; severely and adversely affect our financial results, business and business prospects and the future of imetelstat; and might cause us to cease operations.
•We and certain of our officers have been named as defendants in two pending securities class action lawsuits and seven shareholder derivative lawsuits. These lawsuits, and potential similar or related lawsuits, could result in substantial damages, divert management’s time and attention from our business, and have a material adverse effect on our results of operations. These lawsuits, and any other lawsuits to which we are subject, will be costly to defend or pursue and are uncertain in their outcome.
•We are subject to legal and contractual obligations related to privacy and information security. Our actual or perceived failure, or that of third parties upon which we rely, to comply with such obligations could harm our business.
•Additionally, if our information technology systems or data, or those of third parties upon which we rely, are or were compromised, we could experience adverse consequences.
•Changes in and failures to comply with privacy and data protection obligations may adversely affect our business, operations and financial performance.
********
17
RISK FACTORS
Our business involves significant risks, some of which are described below. You should carefully consider the following risks, as well as the other information contained in our most recent Annual Report on Form 10-K, our most recent Quarterly Report on Form 10-Q, including our financial statements and the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in our updated business overview summary provided above. The occurrence of any of the events or developments described below could have a material adverse effect on our business, results of operations, financial condition, prospects and stock price. In such an event, the market price of our common stock could decline, and you may lose all or part of your investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business operations and the market price of our common stock.
RISKS RELATED TO THE DEVELOPMENT OF IMETELSTAT
Our future success depends solely on imetelstat, our only product candidate, and we cannot be certain that we will be able to continue to develop imetelstat or advance imetelstat to subsequent clinical trials, or that we will be able to receive regulatory approval for or to commercialize imetelstat, on a timely basis or at all.
Imetelstat is our sole product candidate, upon whose success we are wholly dependent. We do not currently have any other products or product candidates. Our ability to develop imetelstat and launch it commercially is subject to significant risks and uncertainties, including, obtaining regulatory approval from the to the U.S. Food and Drug Administration, or FDA, and the European Medicines Agency, or EMA, for commercializing imetelstat in lower risk MDS, as well as, among other things, our ability to:
•submit a New Drug Application, or NDA, to the FDA in the U.S., and a marketing authorization application, or MAA to the EMA in the European Union, or the EU, that is accepted and/or validated for filing by the respective regulatory agency;
•obtain from the FDA and EMA their respective determinations that the regulatory submissions are sufficient to support regulatory approval to commercialize imetelstat in lower risk MDS, without the requirement for additional pre-approval clinical trials or further testing or development commitments, if at all, any of which could result in increased costs to us, delay or limit our ability to generate revenue;
•obtain sufficient safety and efficacy data from IMpactMF to support any application for regulatory approval in relapsed/refractory MF, without clinically meaningful safety issues, side effects or dose-limiting toxicities related to imetelstat that may negatively impact its benefit-risk profile, whether or not in the same indications or therapeutic areas;
•ascertain that the use of imetelstat does not result in significant systemic or organ toxicities, including hepatotoxicity, or other safety issues resulting in an unacceptable benefit-risk profile;
•obtain substantial additional capital in order to enable us to conduct our operations and further advance the imetelstat program, including through the completion of IMpactMF, IMproveMF and IMpress, as well as to conduct the clinical, regulatory and potential commercialization activities necessary to potentially bring imetelstat to market in relapsed/refractory MF and any other future indications;
•develop clinical plans for, and successfully commence, conduct and complete potential future clinical trials of imetelstat;
•generate sufficient safety and efficacy data from ongoing and potential future clinical trials of imetelstat that provide a positive benefit-risk profile to support the continued and future development of imetelstat;
•obtain and maintain required regulatory clearances and approvals to enable continued clinical development, as well as potential commercialization, of imetelstat;
•enter into and maintain arrangements with third parties to provide services needed to further research and develop imetelstat, including maintaining the agreements with our contract research organizations, or CROs, or to manufacture imetelstat, in each case at commercially reasonable costs;
•recruit and retain sufficient qualified and experienced personnel to support the development and potential commercialization of imetelstat in the U.S., including to enroll, conduct and complete current
18
and potential future clinical trials of imetelstat, and to provide internal capabilities for sales, marketing, distribution and other functions to support the potential commercialization of imetelstat in the U.S.;
•enter into and maintain arrangements with third parties to provide services needed to support the potential commercialization of imetelstat for territories outside of the U.S. in compliance with applicable laws;
•achieve acceptance of imetelstat, if approved, by patients and the relevant medical communities;
•compete effectively with other approved treatments in lower risk MDS;
•obtain appropriate coverage and reimbursement levels for the cost of imetelstat from governmental authorities, private health insurers and other third-party payors; and
•obtain, maintain and enforce adequate intellectual property and regulatory exclusivity for imetelstat both in the U.S. and globally.
If we are not able to successfully achieve the above-stated goals and overcome other challenges that we may encounter in the research, development, manufacturing and potential commercialization of imetelstat, we may be forced to abandon our development and/or commercialization of imetelstat, which would severely harm our business, prospects and our ability to raise additional capital, and might cause us to cease operations.
Our current and potential future clinical trials of imetelstat could be interrupted, delayed, terminated or abandoned for a variety of reasons, including due to the effects of macroeconomic conditions such as the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession, which could severely and adversely affect our financial results, business and business prospects, and the future of imetelstat.
Currently, the active clinical trials of imetelstat are IMpactMF and IMproveMF, and the investigator-led clinical trial, IMpress, and we are also retaining remaining patients in the treatment or follow-up phase of IMerge Phase 3. The conduct and completion of IMpactMF, IMproveMF and IMpress could be interrupted, delayed or abandoned for a variety of reasons, including due to the effects of macroeconomic conditions such as the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession. In particular, the fluidity and dynamic nature of the COVID-19 pandemic precludes any firm estimates as to the ultimate effect COVID-19 will have on our current and potential future clinical trials, our operations and our business, all of which depend on the continued worldwide progress toward managing this health crisis. Although vaccine distribution, including booster shots, is being conducted in many countries, the emergence of COVID-19 variants and subvariants, and the resurgence of COVID-19 cases in many parts of the world cause further uncertainty and unpredictability on clinical trial activities, including clinical site initiations, patient screening and enrollment, as well as constraints on available sites and site personnel. For instance, the pace of enrollment for IMerge Phase 3 was slower than planned due to the COVID-19 pandemic, and we have experienced a similar effect on our other clinical trials, and we may face difficulties in retaining patients in the treatment or follow-up phases of our clinical trials. Site personnel resources for IMpactMF and IMproveMF remain constrained in the countries where we plan to conduct the trials, due to the negative impact of COVID-19, as well as a number of competing trials in MF and other oncology indications. Based on assumptions for enrollment and event (death) rates in the trial, we expect the interim analysis for OS for IMpactMF may occur in 2024 and the final analysis may occur in 2025. Because these analyses are event-driven and it is uncertain whether actual rates for enrollment and events will reflect current planning assumptions, the results may be available at different times than currently expected. Such macroeconomic conditions have and may continue to cause delays and suspensions in clinical trial activities at clinical sites. In additional, we may also experience clinical trial failures or delays related to:
•overcoming patient recruitment and enrollment challenges and operational delays related to opening new clinical sites, and conducting and completing IMpactMF, IMproveMF and IMpress due to the effects of the COVID-19 pandemic, while also competing with clinical trials for other investigational drugs in the same patient population;
•clinical sites electing to terminate their participation in any of our clinical trials, which would likely have a detrimental effect on patient enrollment;
19
•any inability to successfully retain patients in IMpactMF, including completing the planned interim analysis for IMpactMF;
•difficulties in patient recruitment and enrollment in IMproveMF;
•patient recruitment, enrollment, or retention, clinical site initiation, or retention problems associated with civil or political unrest or military conflicts around the world, including specifically the current military conflict between Ukraine and Russia;
•a higher number of patients being required for clinical trials, or higher than expected patient drop out rates;
•obtaining and/or maintaining regulatory clearances in the U.S. or other countries to conduct clinical trials, such as obtaining or maintaining regulatory clearances to commence, conduct or modify current or potential future clinical trials of imetelstat, in a timely manner, or at all, which could, for example, prevent us from, or result in substantial delays in, conducting or completing IMpactMF, IMproveMF and IMpress, or commencing potential future clinical trials of imetelstat;
•maintaining the investigational new drug applications, or INDs, and equivalent submissions in other countries for imetelstat without such INDs and/or equivalent submissions in other countries being placed on full or partial clinical hold, suspended or subject to other requirements by the FDA or other similar international regulatory authorities;
•contracting with a sufficient number of clinical trial sites to conduct current and potential future clinical trials, and ensuring that such contracts contain all necessary terms and conditions required by applicable laws, including providing for valid mechanisms to engage in cross-border data transfers, as well as identifying, recruiting and training suitable clinical investigators, especially given the constraints caused by the COVID-19 pandemic, and other competing clinical trials in MF and other oncology indications;
•obtaining or accessing necessary clinical data in accordance with appropriate clinical or quality practices and regulatory requirements, in a timely and accurate manner to ensure complete data sets;
•responding to safety findings, recommendations or conclusions by the internal data safety review committees, independent data monitoring committees and/or hepatic expert committees of current and potential future clinical trials of imetelstat based on emerging data occurring during such clinical trials, such as significant systemic or organ toxicities, including severe cytopenias, hepatotoxicity, fatal bleeding with or without any associated thrombocytopenia, or reduced platelet count, patient injury or death, or other safety issues, resulting in an unacceptable benefit-risk profile;
•use of trial endpoints that inherently require prolonged periods of clinical observation or analysis of the resulting data to determine trial outcomes;
•manufacturing sufficient quantities that meet our specifications and timelines of imetelstat, or other clinical trial materials, in a manner that meets the quality standards of the FDA and other similar international regulatory authorities, and responding to any disruptions to drug supply, clinical trial materials or quality issues that may arise, including as a result of (a) limitations in available manufacturing capacity due to obligations to manufacture and distribute vaccines to address the COVID-19 pandemic; (b) temporary or permanent shut down of contract manufacturing facilities due to violations of good manufacturing practices, or GMP, regulations or other applicable requirements; (c) infections or cross-contaminations of product candidates in the manufacturing process; (d) or capacity limitations;
•ensuring the ability to manufacture and supply imetelstat at acceptable costs for potential future clinical trials of imetelstat and potential commercial uses;
•obtaining sufficient quantities of any study-related treatments, materials (including best available therapy, or BAT, comparator products, placebo or combination therapies) or ancillary supplies, including in light of challenges and delays that may arise from the effects of macroeconomic conditions like the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession;
20
•obtaining acceptance by regulatory authorities of any manufacturing changes for imetelstat, as well as successfully implementing any such manufacturing changes;
•complying with current and future regulatory requirements, policies or guidelines, including domestic and international laws and regulations pertaining to fraud and abuse, transparency, and the privacy and security of health information;
•reaching agreement on acceptable terms and on a timely basis, if at all, with collaborators, physician investigators, vendors and other third parties located in the U.S. or jurisdictions in other countries, including our CROs, laboratory service providers and clinical trial sites, on all aspects of clinical development and collaborating with them successfully, including with respect to challenges and delays that have arisen and may continue to arise from the effects of the COVID-19 pandemic;
•third-party clinical investigators or our CROs losing the licenses or permits necessary to perform our clinical trials, not performing our clinical trials according to our anticipated schedule or consistent with the clinical trial protocol, good clinical practices, or GCP, or regulatory requirements, or not performing data collection or analyses in a timely or accurate manner;
•third-party contractors becoming debarred, disqualified or suspended or otherwise penalized by the FDA or other similar international regulatory authorities for violations of applicable regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or all of the data produced by such contractors in support of any applications for regulatory approval;
•obtaining timely review and clearances by regulatory authorities for any clinical protocol amendments, modifications to our manufacturing process which may be sought for current and potential future clinical trials of imetelstat, including responding to questions or comments from these authorities in a timely and adequate manner, which could, for example, prevent us from conducting or completing IMpactMF, IMproveMF or IMpress, or commencing potential future clinical trials of imetelstat; and
•obtaining institutional review board or ethics committee approvals for clinical trial protocols or protocol amendments, including any future refinements to the trial designs we may seek for IMpactMF, IMproveMF or IMpress, or as a result of changes in regulatory requirements and policies, which could, for example, prevent us from conducting or completing IMpactMF, IMproveMF or IMpress, and commencing potential future clinical trials of imetelstat.
We could also encounter delays if a clinical trial is suspended or terminated. Clinical trials may be suspended or terminated due to a number of factors, including:
•failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols;
•inspection of the clinical trial operations or trial site by the FDA or similar international regulatory authorities resulting in the imposition of a clinical hold;
•safety issues or adverse side effects;
•failure to demonstrate a benefit from using a drug; or
•changes in governmental regulations or administrative actions.
Failures or delays with respect to any of the aforementioned events could adversely affect our ability to conduct or complete IMpactMF, IMproveMF or IMpress, or to commence, conduct and complete potential future clinical trials of imetelstat, which could increase development costs, or interrupt, further delay or halt our development or potential commercialization of imetelstat, any of which could severely and adversely affect our financial results, business and business prospects, and the future of imetelstat.
21
Further difficulties retaining patients in IMerge Phase 3 and enrolling or retaining patients in IMpactMF, IMproveMF and the investigator-led clinical trial IMpress, whether as a result of the effects of macroeconomic conditions like the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession, or for any other reasons, could further delay or otherwise adversely affect our clinical development and commercialization activities, which would cause our business and business prospects to be severely harmed.
The timely completion of a clinical trial in accordance with its protocol depends, among other things, on the ability to enroll a sufficient number of patients who remain in the trial until its conclusion. Further challenges in retaining patients in IMerge Phase 3 and screening, enrolling and retaining patients in IMpactMF, IMproveMF and IMpress, whether as a result of the effects of due to the effects of macroeconomic conditions like the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession, or for any other reasons, may further delay our conduct of such trials, or cause them to be discontinued. For example, we have clinical trial sites in Ukraine, Russia and nearby European countries, and have experienced, and may continue to experience, delays and suspensions in clinical trial activities at clinical sites in Ukraine and Russia due to the current civil or political unrest conditions, including delays in clinical site initiations, patient screening and enrollment, as well as constraints on available sites and site personnel.
Although we reported positive top-line results from IMerge Phase 3 in January 2023, retaining remaining patients in the treatment or follow-up phase would allow us to continue to assess the longer-term durability of RBC-TI responses. Therefore, if we experience difficulties in retaining such patients, whether due to the effects of macroeconomic conditions like the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession, or for any other reasons, our ability to assess the longer-term durability of RBC-TI responses would be adversely affected. The retention of patients in IMerge Phase 3 and the enrollment and retention of patients in IMpactMF, IMproveMF and IMpress, depend on many factors, such as:
•our ability to identify and screen patients who meet the patient eligibility criteria specified in the protocol;
•the size of the patient population required for analysis of the trial’s primary endpoint;
•the proximity of patients to trial sites, and patients’ willingness and ability to travel to trial sites for treatment or monitoring during the COVID-19 pandemic or civil or political unrest, such as the military conflict between Ukraine and Russia;
•the design of the trial;
•our ability to recruit and retain clinical trial investigators with the appropriate competencies and experience;
•clinicians’ and patients’ perceptions of the potential advantages of imetelstat, both in relation to other available therapies, including new drugs that have been approved or may be approved for the indications being investigated, and as a result of data reported from previous or current clinical trials of imetelstat, and their willingness to participate in clinical trials of imetelstat;
•monitoring patients adequately during and after treatment;
•the ability to obtain and maintain patient consents;
•the risk that disease progression will result in death or clinical deterioration before the patient can enroll in a clinical trial of imetelstat, or before sufficient data has been collected from such patient, such that any data collected from the patient does not contribute in a meaningful way to the interpretation of the results of the clinical trial in which the patient is enrolled; and
•the risk that patients enrolled in any imetelstat clinical trial will drop out of the trial before completion, due to lack of efficacy, adverse side effects, investigator decision, progressive disease, site restrictions due to the effects of macroeconomic conditions like the COVID-19 pandemic, or civil or political unrest
22
or military conflicts around the world, such as the military conflict between Ukraine and Russia, alternate treatments being approved for the indication, or personal issues.
In addition, IMpactMF has competed and will continue to compete with, and earlier stage clinical trials of imetelstat, such as IMproveMF and IMpress, will compete with, other clinical trials for product candidates that are in the same therapeutic areas with imetelstat, and such trials may also be conducted at the same clinical sites. This competition is reducing the number of clinical sites and hospital staff available to participate in IMpactMF, IMproveMF and IMpress, as well as the number and type of patients available to enroll or remain in current and potential future imetelstat clinical trials. Moreover, because imetelstat represents a departure from more commonly used methods for cancer treatment, potential patients and their doctors may be inclined to use conventional therapies, rather than enroll patients into imetelstat clinical trials, or may decide not to enroll, or may not recommend enrollment, in IMpactMF, IMproveMF or IMpress, based on efficacy and safety results reported to date and that may be reported in the future.
Furthermore, if imetelstat is approved for commercialization, we will need to complete substantial preparations to be ready for any potential future commercialization of imetelstat. The development of an in-house marketing and sales force or entering into an arrangement with a third party for the commercialization of imetelstat outside of the U.S. will require significant capital expenditures, management resources and time, and may have an adverse effect on the timely completion of IMpactMF, IMproveMF and IMpress.
Delays caused by the effects of macroeconomic effects, like the COVID-19 pandemic or civil or political unrest or military conflicts around the world, such as the current military conflict between Ukraine and Russia, or other factors in patient enrollment, or the inability to retain or treat patients, have resulted in and may in the future result in further increased costs due to extended timelines and other factors, and may lead to incomplete data sets, or adversely affect the timing or outcome of current and potential future clinical trials of imetelstat which could delay or prevent the commencement, conduct or completion of these trials and adversely affect the clinical development, as well as the timing or outcome of the potential commercialization of imetelstat. Such occurrences would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat.
Imetelstat may continue to cause, or have attributed to it, undesirable or unintended side effects or other adverse events that could further delay or prevent the commencement and/or completion of clinical trials for imetelstat, further delay or prevent its regulatory approval, or limit its commercial potential.
Imetelstat may continue to cause, or have attributed to it, undesirable or unintended side effects or other adverse events affecting its safety or efficacy that could interrupt, further delay or halt current or potential future clinical trials of imetelstat, such as IMerge Phase 3, IMpactMF, IMproveMF and IMpress. In this regard, adverse events and dose-limiting toxicities observed in previous and ongoing clinical trials of imetelstat include:
•hematologic toxicities, such as profound and/or prolonged thrombocytopenia or neutropenia, including one case of febrile neutropenia after prolonged myelosuppression with intracranial hemorrhage resulting in patient death, which the investigator assessed as possibly related to imetelstat, as well as reversible Grade 4 febrile neutropenia;
•bleeding events, with or without thrombocytopenia, including reversible Grade 3/4 bleeding events;
•hepatotoxicity and liver function test abnormalities, as well as hepatic failure;
•gastrointestinal events;
•muscular and joint pain;
•infusion-related reactions.
23
If patients in any clinical trials of imetelstat, including IMerge Phase 3, IMpactMF, IMproveMF, IMpress or any potential future clinical trials of imetelstat, experience similar or more severe adverse events, or new or unusual adverse events, or if the FDA or other similar international regulatory authorities determine that efficacy and safety data in current or potential future clinical trials of imetelstat do not support an adequate benefit-risk profile to justify continued treatment of patients, then the FDA or other similar international regulatory authorities may again place one or more of the INDs for imetelstat on clinical hold, as occurred in March 2014. If this were to occur, there would be a significant delay in, or possible termination of, such clinical trial or all the imetelstat clinical trials and any potential commercialization efforts, which might cause us to cease operations. For example, we recently became aware of a case in our IMpactMF clinical trial of a patient with myelofibrosis associated with underlying progressive bone marrow failure, who died from febrile neutropenia, pulmonary hemorrhage and bilateral pneumonia, which, at the time of reporting, the investigator related to imetelstat. If such toxicities or other safety issues in any clinical trial of imetelstat are determined by us, the FDA or similar international regulatory authorities to result in an unacceptable benefit-risk profile, then:
•additional information supporting the benefit-risk profile of imetelstat may be requested by the FDA or similar international regulatory authorities and if any such information supplied by us, or by our former collaboration partner, is not deemed acceptable, current clinical trials of imetelstat could be suspended, terminated, or placed on clinical hold by the FDA or similar international regulatory authorities;
•the ability to retain enrolled patients in current clinical trials may be negatively affected, resulting in incomplete data sets and the inability to adequately assess the benefit-risk profile of imetelstat in a specific patient population; or
•additional, unexpected clinical trials or non-clinical studies may be required to be conducted.
Further, clinical trials by their nature examine the effect of a potential therapy in a sample of the potential future patient population. As such, clinical trials conducted with imetelstat, to date and in the future, may not uncover all possible adverse events that patients treated with imetelstat may experience. Because remaining patients in IMerge Phase 3, IMpactMF and IMproveMF continue to receive imetelstat treatment, additional or more severe toxicities or safety issues, including additional non-serious or serious adverse events and dose-limiting toxicities, may be observed as patient treatment continues and more data become available. In addition, because additional data are being generated from these trials, the benefit-risk profile of imetelstat will continue to be assessed, including the risk of hepatotoxicity, severe cytopenias, fatal bleeding with or without any associated thrombocytopenia, patient injury or death, and any other severe adverse effects that may be associated with life-threatening clinical outcomes.
The occurrence of any of the aforementioned events could interrupt, further delay, or halt, any development, and as a result, impact or preclude the potential commercialization of imetelstat, as well as increase costs to develop imetelstat, which would have a severe adverse effect on our results of operations, financial condition and ability to raise additional capital, business prospects and the future of imetelstat, any of which might cause us to cease operations.
The design of a clinical trial can determine whether its results will support regulatory approval of a product, and flaws in the trial design may not become apparent until the clinical trial is well advanced or during the approval process after the trial is completed.
A trial design that is considered appropriate for regulatory approval includes a sufficiently large sample size with appropriate statistical power, as well as proper control of bias, to allow a meaningful interpretation of the results. The preliminary results of imetelstat clinical trials with smaller sample sizes can be disproportionately influenced by the impact the treatment had on a few individuals, which limits the ability to generalize the results across a broader community, making the trial results of clinical trials with smaller sample sizes less reliable than trials with a larger number of patients. As a result, there may be less certainty that imetelstat will achieve a statistically significant effect in any future clinical trials.
For example, we shortened the follow-up period after the last patient has been enrolled from 15 months to 12 months to enable an earlier clinical cut-off date for the primary analysis in IMerge Phase 3. Although we reported positive top-line results from IMerge Phase 3 in January 2023, our decision to shorten the follow-up period after the last patient has been enrolled may result in further clinical responses that could occur after the 12-month clinical
24
cut-off date being excluded from the primary analysis. The exclusion of this future data from the primary analysis could reduce the overall efficacy results of the trial, including durability of transfusion independence, which could limit or prevent marketing approval of imetelstat in lower risk MDS by the FDA or similar international regulatory authorities, cause them not to approve imetelstat at all or require additional clinical trials or further testing prior to granting any regulatory approval to market imetelstat in lower risk MDS.
Moreover, with respect to the trial design for IMpactMF, the FDA urged us to consider adding a third dosing arm to the trial to assess a lower dose and/or a more frequent dosing schedule that might improve the trial’s chance of success by identifying a less toxic regimen and/or more effective spleen response, one of the trial’s secondary endpoints. Based on data from IMbark, we believe that testing a lower dose regimen would likely result in a lower median OS, which is the trial’s primary endpoint, in the imetelstat treatment arm. Existing data also suggest that lowering the dose would not result in a clinically meaningful reduction in toxicity, and for these reasons we therefore determined not to add a third dosing arm to the trial design, and the FDA did not object to our proposed imetelstat dose and schedule of 9.4 mg/kg every three weeks. Our belief may ultimately be incorrect. Therefore, our failure to add a third dosing arm could result in a failure to maintain regulatory clearance from the FDA and similar international regulatory authorities, could result in the trial’s failure, or could otherwise delay, limit or prevent marketing approval of imetelstat in relapsed/refractory MF by the FDA or similar international regulatory authorities.
Results and data we disclosed from prior non-clinical studies and clinical trials may not predict success in later clinical trials, and we cannot assure you that any ongoing or future clinical trials of imetelstat will lead to similar results and data that could potentially enable us to obtain any regulatory approvals.
Success in non-clinical testing and early clinical trials, including Phase 2 clinical trials, such as IMbark, does not ensure that later clinical trials will be successful, nor does it predict final clinical trial results. In addition, even though we reported positive top-line results from IMerge Phase 3 in January 2023, this does not ensure that any other clinical trials of imetelstat, including IMpactMF, IMproveMF and IMpress, will be successful. We cannot be certain that any of the prior, current or potential future clinical trials of imetelstat will generate sufficient, consistent or adequate efficacy and safety data demonstrating a positive benefit-risk profile, which would be necessary to obtain regulatory approval to market imetelstat in any indication. Imetelstat in later stages of clinical trials may fail to show the desired benefit-risk profile despite having progressed through non-clinical studies and initial clinical trials. Many companies in the biopharmaceutical industry have frequently suffered significant setbacks in later clinical trials, even after achieving promising results in earlier non-clinical studies or clinical trials.
In IMbark, we reported a median overall survival of 19.9 months and 28.1 months for the 4.7 mg/kg and 9.4 mg/kg dosing arms, respectively, in relapsed/refractory MF patients. In general, Phase 3 clinical trials with larger numbers of patients or longer durations of therapy may fail to replicate efficacy and safety results observed in earlier clinical trials, such as IMbark, and if this were to occur with IMpactMF, this would adversely affect future development prospects of imetelstat, and as a result, impact the potential commercialization of imetelstat, which would substantially impair our ability to raise additional capital.
Furthermore, non-clinical and clinical data are often susceptible to varying interpretations and analyses. In some instances, there can be significant variability between different clinical trials of imetelstat due to numerous factors, including changes in trial procedures set forth in trial protocols, differences in the size and type of patient populations, and changes in and adherence to the dosing regimens. For example, complete and partial remissions were observed in an investigator-sponsored pilot study of imetelstat conducted at Mayo Clinic in MF patients, or the Pilot Study. However, similar activity was not observed in the MF patients enrolled in IMbark, as shown by the one partial remission observed in the IMbark primary analysis. We believe that differences in the IMbark study design when compared to the Pilot Study design, such as more restrictive patient enrollment criteria requiring either documented objective lack of response to a JAK inhibitor or evidence of progressive disease while on treatment with a JAK inhibitor, may have contributed to the data observed in IMbark differing significantly from data reported from the Pilot Study, but we cannot assure you that any future clinical trials of imetelstat in relapsed/refractory MF will yield results comparable to IMbark or the Pilot Study. In addition, the potential improvement in survival observed in the 9.4 mg/kg dosing arm in IMbark will need to be further assessed in IMpactMF, and similar results, including potential improvement in survival, if any, with respect to any patient population or patient population subgroup, may not be observed in IMpactMF. Likewise, although the statistical analyses comparing IMbark data to
25
closely matched real world data, or RWD, published in the September 2021 issue of the Annals of Hematology, suggest potentially favorable OS for in relapsed/refractory MF patients treated with imetelstat, compared to BAT using closely matched patients’ RWD, such comparative analyses between RWD and our clinical trial data have several limitations. For instance, the analyses create a balance between treatment groups with respect to commonly available covariates, but do not take into account the unmeasured and unknown covariates that may affect the outcomes of the analyses. Potential biases are introduced by factors which include, for example, the selection of the patients included in the analyses, misclassification in the matching process, the small sample size, and estimates that may not represent the outcomes for the true treated patient population. For these and other reasons, such comparative analyses and any conclusions from such analyses should be considered carefully and with caution, and should not be relied upon as demonstrative or otherwise predictive or indicative of any current or potential future clinical trial results of imetelstat in relapsed/refractory MF, including IMpactMF.
Failure to achieve results supporting a positive benefit-risk profile in current or potential future imetelstat clinical trials would interrupt, further delay, or halt, any development, and as a result, potential commercialization of imetelstat, which would have a severe adverse effect on our results of operations, financial condition and ability to raise additional capital, business prospects and the future of imetelstat.
Interim, “snapshot,” “top-line,” and preliminary data or statistical analyses from clinical trials that we announce or publish from time-to-time may change as more patient data become available, may be more positive than the final data, and are subject to audit and verification procedures that could result in material changes in the final data. Thus, such preliminary data should be considered carefully and with caution and not relied upon as indicative of future clinical results.
From time-to-time, preliminary or interim safety and efficacy data from previous and current imetelstat clinical trials have been reported or announced by us, clinical investigators or our former collaboration partner. Preliminary data is based on a preliminary analysis of then available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As such, preliminary or interim results may not be reproduced in any current or potential future clinical trials of imetelstat, and thus should be considered carefully and with caution, and not relied upon as indicative of future clinical results. Additional or updated safety and efficacy data from current or potential future clinical trials of imetelstat may result in a benefit-risk profile that does not justify the continued development of imetelstat in a particular patient population, or at all. Any data reported from IMpactMF may materially differ from and be less positive than data previously reported from IMbark. Thus, reported data should be considered carefully and with caution, and not relied upon as indicative of future clinical results. Such additional data could result in a lower benefit-risk profile than initially expected, which could hinder the potential success of IMpactMF, IMproveMF or IMpress, or cause us to abandon further development of imetelstat entirely.
In January 2023, we announced positive top-line results from IMerge Phase 3. Such top-line results may differ from future results of the same study, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Top-line data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, top-line data, including from IMerge Phase 3, should be viewed with caution until the final data are available. Material adverse differences in final data, compared to preliminary or interim data, could severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, including the potential commercialization of imetelstat, and might cause us to cease operations.
The research and development of imetelstat is subject to numerous risks and uncertainties.
The science and technology of telomere biology, telomerase and our proprietary oligonucleotide chemistry are relatively new. There is no precedent for the successful commercialization of a therapeutic product candidate based on these technologies. Significant research and development activities will be necessary to further develop imetelstat, our sole product candidate, and may take years to accomplish, if at all.
26
Because of the significant scientific, regulatory and commercial challenges that must be overcome to successfully research, develop and commercialize imetelstat, the development of imetelstat in myeloid hematologic malignancies, including MDS and MF, may be further delayed or abandoned, even after significant resources have been expended on it. Examples of such situations include:
•in September 2012, the discontinuation of our Phase 2 clinical trial of imetelstat in metastatic breast cancer;
•in April 2013, the discontinuation of our development of imetelstat in solid tumors with short telomeres;
•in March 2014, the full clinical hold placed by the FDA on imetelstat clinical trials;
•in the third quarter of 2016, closure of the 4.7 mg/kg dosing arm in IMbark to new patient enrollment and suspension of enrollment in the 9.4 mg/kg dosing arm in IMbark because an insufficient number of patients in the 9.4 mg/kg dosing arm met the protocol defined interim efficacy criteria at 12 weeks;
•in the third quarter of 2017, expansion of IMerge Phase 2 to enroll additional lower risk MDS patients in a target patient population; and
•in September 2018, our former collaboration partner’s decision to terminate its imetelstat collaboration agreement with us.
Further delay, suspension or abandonment of our development of imetelstat in myeloid hematologic malignancies, including with respect to our IMpactMF, IMproveMF and IMpress clinical trials, could have a material adverse effect on the future of imetelstat and our business prospects, including the potential commercialization of imetelstat in indications other than lower risk MDS.
We rely on third parties to conduct our current and potential future clinical trials of imetelstat. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to continue the development of, obtain regulatory approval for, or commercialize imetelstat.
We do not have the ability to independently conduct clinical trials. Therefore, we rely on medical institutions, clinical investigators, contract laboratories and other third parties, such as CROs, service providers, vendors, suppliers and consultants, to conduct clinical trials of imetelstat. The third parties we contract with for execution of our current and potential future clinical trials of imetelstat play a critical role in the conduct of these trials and the subsequent collection and analysis of data. However, these third parties are not our employees, and except for contractual duties and obligations, we have limited ability to control their performance, or the amount or timing of resources that they devote to imetelstat. For example, we have retained CROs to support our imetelstat clinical development activities, and any failure by our CROs to perform their contractual obligations, whether due to the effects of the COVID-19 pandemic or otherwise, or disputes with our CROs about the quality of their performance or other matters, could further delay or halt our imetelstat clinical development activities including current or future imetelstat clinical trials. These third parties may also have relationships with other commercial entities, some of which may compete with us. Under certain circumstances, these third parties may terminate their agreements with us without cause and upon immediate written notice.
Although we rely on third parties to conduct our imetelstat clinical trials, including IMerge Phase 3, IMpactMF and IMproveMF, we remain responsible for ensuring that each clinical trial is conducted in accordance with its investigational plan and protocol, and applicable laws. Moreover, the FDA and similar international regulatory authorities require us to comply with GCP regulations and standards for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the rights, integrity and confidentiality of patients participating in clinical trials are protected, including being adequately informed of the potential risks. Regulatory authorities enforce these GCP requirements through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs fail to comply with applicable GCP requirements, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, or similar international regulatory authorities, may require us to perform additional clinical trials before approving any application for approval. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP or other applicable regulations. In addition, our clinical trials must be conducted with imetelstat produced under applicable GMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials,
27
which would further delay the process for any regulatory approval. Our ability to comply with these regulations and standards may be contingent upon activities conducted by third parties, and if they fail to perform in accordance with contractual obligations and legal requirements, our development of imetelstat may be interrupted, further delayed or halted. Any failures by us or third parties noted above would have a severe adverse effect on our results of operations, financial condition and ability to raise additional capital, business prospects and the future of imetelstat, including the potential commercialization of imetelstat, any of which might cause us to cease operations.
We also are required to register imetelstat clinical trials that we sponsor and post the results of certain completed clinical trials on certain government-sponsored databases, such as ClinicalTrials.gov, within certain timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Furthermore, the execution of clinical trials and the subsequent compilation and analysis of the data produced, including the interim and final analyses for IMpactMF, requires coordination among various parties. In order for these functions to be carried out effectively and efficiently, it is imperative that these parties communicate and coordinate with one another. If the quality or accuracy of the clinical data obtained, compiled or analyzed by third parties is compromised due to their failure to adhere to our clinical trial protocols, GCP or GMP requirements, or for any other reason, we may need to enter into new arrangements with alternative third parties, which would cause delay, and could be difficult, costly or impossible. If third parties conducting our clinical trials do not perform their contractual duties or obligations, experience work stoppages, do not meet expected deadlines, terminate their agreements with us or need to be replaced, our clinical trials may be extended, delayed or terminated, or may be unsuccessful or need to be repeated, which could have a material adverse effect on our business, including the potential commercialization of imetelstat, and might cause us to cease operations.
If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative third parties or do so on commercially reasonable terms. Switching or adding CROs, investigators and other third parties involves additional costs and delays because of the time it takes to finalize a contract with a new CRO and for their commencement of work. As a result, delays can occur, which could materially impact our ability to meet our desired clinical development timelines. The COVID-19 pandemic and public health safety measures taken in response have also had a significant impact on our CROs and other third parties. Although we carefully manage our relationships with our CROs, investigators and other third parties, we and any of these third parties may nonetheless encounter challenges or delays in the future, which could have a material and adverse impact on our business, business prospects and the future of imetelstat.
In addition, certain principal investigators for our clinical trials serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA. The FDA may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected conduct of the trial. The FDA may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of any applications for approval by the FDA and may ultimately lead to the denial of approval of imetelstat.
We will not control the conduct of current or any potential future investigator-led clinical trials, and data from such trials could show marginal efficacy and/or clinically relevant safety concerns related to imetelstat resulting in an unfavorable benefit-to-risk assessment that could impact our ongoing clinical trials or development program for imetelstat.
We will not control the design or administration of the investigator-led clinical trial, IMpress, or any potential future investigator-led trials, nor the submission, approval or maintenance of any IND or foreign equivalent required to conduct these clinical trials. In addition, we will not have control over the timing and reporting of the data from any such investigator-led clinical trials. A delay in the timely completion of or reporting of data from any potential future investigator-led clinical trial could have a material adverse effect on our ability to further develop imetelstat or to advance imetelstat to subsequent clinical trials.
Investigator-led clinical trials may be conducted under less rigorous clinical standards than those used in company-sponsored clinical trials. Accordingly, regulatory authorities may closely scrutinize the data collected from these investigator-led clinical trials. In addition, any potential future investigator-led clinical trials could show
28
marginal efficacy and/or clinically relevant safety concerns that could delay the further clinical development or marketing approval of imetelstat for any indication. To the extent that the results of any potential future investigator-led clinical trials raise safety or other concerns regarding imetelstat, regulatory authorities may question the results of such investigator-led clinical trials, or question the results of IMerge Phase 3, IMpactMF, and IMproveMF. Safety concerns arising from any potential future investigator-led clinical trials could result in partial or full clinical holds being placed on the imetelstat INDs by the FDA or other similar international regulatory authorities, as occurred in March 2014, which would further delay or prevent us from advancing imetelstat into further clinical development and cause us to discontinue our development of imetelstat, which would severely harm our business and prospects, including the potential commercialization of imetelstat, and could potentially cause us to cease operations.
Risks Related to Regulatory APPROVAL and Commercialization of Imetelstat
Our inability to obtain and maintain regulatory clearances and approvals to continue the clinical development of, and to potentially commercialize, imetelstat, would severely and adversely affect imetelstat’s future value, and our business and business prospects, and might cause us to cease operations.
Federal, state and local governments in the U.S. and governments in other countries have significant regulations in place that govern drug research and development and may prevent us from successfully conducting development efforts or potentially commercializing imetelstat. Delays in obtaining or failure to maintain regulatory clearances and approvals, or limitations in the scope of such clearances or approvals, could:
•impede or halt our activities and plans for clinical development and commercialization;
•significantly harm the commercial potential of imetelstat;
•impose additional development costs;
•diminish any competitive advantages that may have been available to us; or
•further delay or preclude any revenue we may receive from the future commercialization of imetelstat, if any.
In addition, with respect to the trial design for IMpactMF, the FDA urged us to consider adding a third dosing arm to the trial to assess a lower dose and/or a more frequent dosing schedule that might improve the trial’s chance of success by identifying a less toxic regimen and/or more effective spleen response, one of the trial’s secondary endpoints. Based on data from IMbark, we believe that testing a lower dose regimen would likely result in a lower median OS, which is the trial’s primary endpoint, in the imetelstat treatment arm. Existing data also suggest that lowering the dose would not result in a clinically meaningful reduction in toxicity, and for these reasons we therefore determined not to add a third dosing arm to the trial design and the FDA did not object to our proposed imetelstat dose and schedule of 9.4 mg/kg every three weeks. Our belief may ultimately be incorrect. Therefore, our failure to add a third dosing arm could result in a failure to maintain regulatory clearance from the FDA and similar international regulatory authorities, could result in the trial’s failure, or could otherwise delay, limit or prevent marketing approval of imetelstat for relapsed/refractory MF by the FDA or similar international regulatory authorities.
Furthermore, in IMerge Phase 3 we shortened the follow-up period after the last patient has been enrolled from 15 months to 12 months to enable an earlier clinical cut-off date for the primary analysis. Although we reported positive top-line results from IMerge Phase 3 in January 2023, our decision to shorten the follow-up period after the last patient has been enrolled may result in further clinical responses that could occur after the 12-month clinical cut-off date being excluded from the primary analysis. The exclusion of this future data from the primary analysis could reduce the overall efficacy results, including durability of transfusion independence, which could otherwise delay, limit or prevent marketing approval of imetelstat in lower risk MDS by the FDA or similar international regulatory authorities or require additional clinical trials and further testing prior to granting any regulatory approval to market imetelstat in lower risk MDS.
29
Even though we reported positive top-line results from IMerge Phase 3 in January 2023, those results are not necessarily be predictive of imetelstat activity in other indications and for other pivotal trials that may be needed to support any application to the FDA or similar international regulatory authorities for such other indications, such as from IMpactMF. We may therefore fail to further develop or commercialize imetelstat, which would severely and adversely affect our business and business prospects, and might cause us to cease operations.
If we are unable to prepare and timely submit the planned NDA for imetelstat in lower risk MDS, and to successfully obtain regulatory approval for and commercialize imetelstat, or experience significant delays in doing so, our business will be materially harmed.
The process of obtaining marketing approvals, both in the U.S. and in other countries, is lengthy, expensive and uncertain. It may take many years to obtain approval, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Of the large number of drugs in development, only a small percentage complete the regulatory approval process and are successfully commercialized. In addition, the lengthy review process as well as the unpredictability of future clinical trial results may result in a delay in obtaining, or our failure to obtain, regulatory approval for imetelstat in lower risk MDS, relapsed/refractory MF, or any other indication, which would significantly harm our business, business prospects, including the potential commercialization of imetelstat, and the future value of imetelstat and might cause us to cease operations.
Securing marketing approval requires the submission of extensive non-clinical and clinical data and supporting information to regulatory authorities for each therapeutic indication to establish the product candidate’s safety and efficacy, as well as information about the product manufacturing process and any inspections of manufacturing facilities conducted by regulatory authorities through the filing of an NDA in the U.S. and an MAA in Europe. As a company, we have not previously submitted an NDA to the FDA or similar applications to comparable international regulatory authorities for imetelstat. The preparation of an NDA requires a great deal of effort and expertise, and if we do not secure the necessary resources and retain personnel having the requisite expertise to prepare and submit an NDA, the filing of any NDA would be delayed. Further, if we submit an NDA, there can be no assurance that it will be accepted by the FDA. If the FDA determines after an initial review of the NDA that the data included in the application is insufficient and not ready for formal consideration, we could receive a “refuse to file” notice. The FDA also has substantial discretion in the approval process.
While we reported positive top-line results from IMerge Phase 3 in lower risk MDS, and while we believe that these results and our assessment of the positive benefit-risk profile of imetelstat, combined with data from our Phase 2 clinical trials, are supportive of planned regulatory submissions in the U.S. and in the EU for imetelstat in lower risk MDS, regulatory authorities in those jurisdictions may disagree with our interpretation of the data and may require additional clinical testing before we can seek regulatory approval and begin commercialization of imetelstat, if at all, any of which could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our commercial prospects. There is no guarantee that we will obtain regulatory approval or be able to commence commercialization on the timeline we are planning or at all.
Imetelstat must receive all relevant regulatory approvals before it may be marketed in the U.S. or other countries. Regulatory authorities have substantial discretion in the approval process and can delay, limit or deny approval of imetelstat or require us to conduct additional non-clinical or clinical testing or abandon a program for many reasons, including:
•disagreement with the design or implementation of our clinical trials, including our statistical analysis of trial results;
•failure to demonstrate to the FDA or similar international regulatory authorities that imetelstat’s efficacy results, including duration of response, is acceptable;
•unfavorable benefit-to-risk assessment, in the case of marginal efficacy and/or clinically relevant safety concerns, for any proposed indication;
•serious and unexpected drug-related side effects experienced by participants in our clinical trials or by individuals using drugs similar to imetelstat;
30
•disagreement with our interpretation of data from non-clinical studies or clinical trials;
•failure to collect data from clinical trials of imetelstat meeting the level of integrity or statistical or clinical significance required by the FDA or similar international regulatory authorities, or a determination such data is not sufficient to support the submission of an NDA, MAA, or other submission, or to obtain regulatory approval in the U.S., the EU or elsewhere;
•deficiencies in our clinical trial operations or the clinical trial operations of trial sites;
•identification of critical issues as a result of a pre-approval health authority inspection that could negatively impact the integrity of data in an NDA or MAA and lead to a rejection by the FDA and similar international health authorities;
•errors or deficiencies in the conduct of the imetelstat program prior to its transition to us by our former collaborator, and/or in the transition of the imetelstat program to us by our former collaborator;
•unwillingness or inability by our former collaborator to provide information requested by the FDA or similar international regulatory authorities regarding the time period when our former collaborator was responsible for the imetelstat program;
•a determination by the FDA or similar international regulatory authorities that the appropriate indication for commercial use of imetelstat is narrower or more restrictive than anticipated;
•failure to satisfy the requirement to develop a risk evaluation and mitigation strategy, or REMS, for the U.S. and a risk management plan for the EU including post-marketing studies, as a potential condition to approval;
•disagreement regarding the formulation, labeling and/or the specifications for imetelstat;
•a determination by the FDA or similar international regulatory authorities that the manufacturing processes, test procedures and specifications applicable to the manufacture of imetelstat, or the facilities of the third-party manufacturers with which we contract for clinical and commercial supplies of imetelstat are inadequate, or failure by such third-party manufacturers to maintain compliance with the regulatory and other requirements established by the FDA or similar international regulatory authorities;
•the failure of the quality or stability of imetelstat to meet acceptable regulatory standards;
•the FDA or similar international regulatory authorities may lack resources or be delayed in conducting pre-approval inspections due to reasons related to COVID-19 or otherwise;
•we or any third-party service providers may be unable to demonstrate compliance with current good manufacturing practices, or cGMPs, and/or good clinical practices, GCPs, to the satisfaction of the FDA or similar international regulatory authorities;
•changes in regulatory policies or approval processes, or potential reduction of unmet medical need with the entry of competitive therapies to the market, could render our clinical efficacy or safety data insufficient for approval; or
•political factors surrounding the approval process, such as government shutdowns, political instability or global pandemics, such as COVID-19.
Furthermore, in recent years, there has been increased public and political scrutiny on the FDA and similar international regulatory authorities with respect to the approval process for new drugs, and as a result regulatory authorities may apply more stringent regulatory standards, especially regarding drug safety, when reviewing regulatory submissions for new drugs.
Even if we believe we have complied with all of the regulatory requirements to receive marketing approval for imetelstat, we may not obtain marketing approval for reasons that we do not currently predict. If we fail to obtain regulatory approval for imetelstat, we will have no commercialized products and correspondingly no revenue.
31
Any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render imetelstat not commercially viable, which would harm imetelstat’s future value and our business and business prospects. In addition, obtaining regulatory approval is a lengthy, expensive and uncertain process. For example, following the result of a referendum in 2016, the U.K. left the EU on January 31, 2020, commonly referred to as Brexit, and its withdrawal from the EU was completed on December 31, 2020. The withdrawal of the U.K. from the EU has resulted in uncertainty in relation to the regulatory process in the U.K., and for Europe could potentially result in a delay in the review of regulatory submissions which could also lead to less efficient, more expensive, and potentially lengthier regulatory review processes for companies like us, who may seek to obtain regulatory approval for drug products in the EU or the U.K. Such regulatory changes in the U.K. or elsewhere could adversely affect and/or delay our ability to obtain approval of, and market and sell, imetelstat in the U.S. or other countries.
Regulatory authorities may also not approve the labeling claims that are necessary or desirable for the successful commercialization of a drug, such as imetelstat. For example, future regulatory clearances, if any, that we might obtain for imetelstat may be limited to fewer or narrower indications than we might request, or may be granted subject to the performance of post-marketing studies, which may impose further requirements or restrictions on the distribution or use of imetelstat, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria, and requiring treated patients to enroll in a registry. These limitations and restrictions may limit the size of the market for imetelstat and affect reimbursement by third-party payors. Future regulatory clearances, if any, may be limited to a smaller patient population, or may require a different drug formulation or a different manufacturing process, than we might in the future decide to seek.
In addition, failure by our former collaborator to comply with applicable regulatory guidelines prior to our assumption of sponsorship of the imetelstat program could result in administrative or judicially imposed sanctions on us, including warning letters, civil and criminal penalties, injunctions, product seizures or detention, product recalls, total or partial suspension of manufacturing activities, and the potential refusal to approve any NDAs.
Any delay in obtaining or failure to obtain required approvals of imetelstat, or limitations on any regulatory approval that we might receive in the future, if any, could reduce the potential commercial use of imetelstat, and potential market demand for imetelstat and therefore result in decreased revenue for us from any commercialization of imetelstat, any of which would severely and adversely affect our financial results and ability to raise additional capital, the price of our common stock, our business and business prospects, including the potential commercialization of imetelstat, and the future of imetelstat, and might cause us to cease operations.
If imetelstat is approved for commercialization and we are unable to establish sales, marketing and distribution capabilities or enter into agreements with third parties to commercialize imetelstat, we will be unable to successfully commercialize imetelstat if and when it is approved.
We will need to complete substantial preparations to be ready for any potential future commercialization of imetelstat, and currently we have no sales, marketing or distribution capabilities and no experience in marketing products. To advance imetelstat to potential marketing approval, we will be required to complete our commercialization preparatory activities, and continue to incur related expenses, before we obtain any marketing approval. These activities will include, among other things, the development of an in-house marketing and sales force, which will require significant capital expenditures, management resources and time. We will have to compete with other companies to recruit, hire, train and retain qualified marketing and sales personnel. If we are unable to adequately prepare for the potential future commercialization of imetelstat, we may not be able to generate product revenue if marketing authorization is obtained.
There are risks involved with both establishing our own sales, marketing and distribution capabilities and entering into arrangements with third parties to perform these services. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of imetelstat for which we recruit a sales and marketing force and establish distribution capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses, which would be costly. Even if imetelstat is approved in lower risk MDS and we are able to establish our own sales and marketing capabilities, imetelstat will be a newly-marketed drug. As a result, we will be required to expend
32
significant time and resources to train sales personnel in commercializing imetelstat. If we are unable to effectively train sales personnel and equip them with compliant and effective materials, our efforts to successfully commercialize imetelstat could be adversely affected, which would negatively impact our business, business prospects and the future value of imetelstat.
Factors that may inhibit our efforts to commercialize imetelstat on our own include:
•our inability to recruit, train and retain adequate numbers of effective sales, marketing, distribution, coverage or reimbursement, customer service, medical affairs and other support personnel;
•our inability to equip sales personnel with compliant and effective materials, including medical and sales literature to help them educate physicians regarding the indications we are targeting and imetelstat, if approved;
•the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to prescribe imetelstat;
•the inability of reimbursement professionals to negotiate arrangements for formulary access, reimbursement and other acceptance by payors;
•the lack of complementary medicines to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines;
•the inability to price imetelstat at a sufficient price point to ensure an adequate and attractive level of profitability;
•unforeseen costs and expenses associated with creating an independent sales and marketing organization;
•our inability to maintain existing supply arrangements, or to establish new supply arrangements with third-party suppliers and contract manufacturers to ensure sufficient commercial supplies;
•our inability to obtain and maintain patent protection, trade secret protection and regulatory exclusivity, both in the U.S. and in other countries;
•lack of an acceptable safety profile following any regulatory approval; and
•our inability to compete effectively with other therapies.
If we enter into arrangements with third parties to perform commercialization services like sales, marketing and distribution, we will be reliant on the efforts of such third parties, and our sales revenue from sales of imetelstat or the profitability from such sales to us are likely to be lower than if we were to market and sell imetelstat ourselves. In addition, we may not be successful in entering into arrangements with third parties to commercialize imetelstat or may be unable to do so on terms that are favorable to us. In entering into third-party commercialization arrangements, any revenue we receive will depend upon the efforts of the third parties, and we cannot assure you that such third parties will establish adequate commercialization capabilities or devote the necessary resources and attention to commercialize imetelstat effectively. We also face competition in our search for third parties to assist us with the commercialization efforts of imetelstat.
Our inability to successfully establish commercialization capabilities for imetelstat, if we receive regulatory approval to do so, would severely and adversely affect our financial results, business and business prospects, including the potential commercialization of imetelstat, and the future of imetelstat.
If acceptable prices or adequate reimbursement for imetelstat is not obtained, the use of imetelstat could be severely limited.
33
The ability to successfully commercialize imetelstat, if approved, will depend significantly on obtaining acceptable prices and the availability of coverage and adequate reimbursement to the patient from third-party payors. Government payors, such as the Medicare and Medicaid programs, and other third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and the reimbursement levels. Assuming we obtain coverage for imetelstat by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. If imetelstat is approved for commercial sale, patients are unlikely to use it unless coverage is provided, and reimbursement is adequate to cover all or a significant portion of its cost. Therefore, coverage and adequate reimbursement will be critical to new product acceptance.
Government authorities and other third-party payors are developing increasingly sophisticated methods of controlling healthcare costs, such as by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices as a condition of coverage, are using restrictive formularies and preferred drug lists to leverage greater discounts in competitive classes, and are challenging the prices charged for medical products. Further, no uniform policy requirement for coverage and reimbursement for drug products exists among third-party payors in the U.S. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of imetelstat to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
We cannot be sure that coverage and reimbursement will be available for imetelstat, if approved for commercial sale, and, if reimbursement is available, what the level of reimbursement will be. There may also be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or similar international regulatory authorities. Coverage and reimbursement may impact the demand for, or the price of imetelstat, if marketing approval is obtained. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not successfully commercialize imetelstat, even if marketing approval is obtained, which would negatively impact our business and business prospects.
Although orphan drug designation has been granted to imetelstat for the treatment of MF and MDS in the U.S. and in the EU, these designations may not be maintained, which would eliminate the benefits associated with orphan drug designation, including the potential for market exclusivity, which would likely result in decreased sales revenue from commercialization of imetelstat, if any, and would likely harm our business and business prospects.
The FDA granted orphan drug designation to imetelstat in June 2015 for the treatment of MF and for the treatment of MDS in December 2015, and the EMA granted orphan drug designation in December 2015 to imetelstat for the treatment of MF and in July 2020 for the treatment of MDS. The designation of imetelstat as an orphan drug does not guarantee that any regulatory authority will accelerate regulatory review of, or ultimately approve, imetelstat, nor does it limit the ability of any regulatory authority to grant orphan drug designation to product candidates of other companies that treat the same indications as imetelstat prior to imetelstat receiving any exclusive marketing approval.
We may lose orphan drug exclusivity for certain reasons, including if the FDA or EMA determines that the request for orphan drug designation was materially defective or if we cannot ensure sufficient quantities of imetelstat to meet the needs of patients with MF or MDS. Failure to maintain orphan designation status in the EU at the time of submitting the MAA, or failure to complete the agreed pediatric plan, would lead to the loss of the additional two-year exclusivity period.
Even if we maintain orphan drug exclusivity for imetelstat, the exclusivity may not effectively protect imetelstat from all competition because different drugs with different active moieties can be approved for the same condition. Even after an orphan drug product is approved, the FDA or EMA can subsequently approve a different drug with the same active moiety for the same condition, if the FDA or EMA concludes that the later drug is safer, more effective, or makes a major contribution to patient care. The occurrence of any of these events could result in decreased sales of imetelstat, should it ever receive marketing approval, and may harm our business and business
34
prospects. In addition, orphan drug designation will neither shorten the development time nor regulatory review time for imetelstat, and it does not give imetelstat any advantage in the regulatory review or approval process.
A Fast Track designation by the FDA, such as the Fast Track designations received for imetelstat for MDS and MF, does not guarantee marketing approval and may not lead to a faster development, regulatory review or approval process.
In October 2017, the FDA granted Fast Track designation to imetelstat for the treatment of adult patients with transfusion-dependent low red blood cell counts, or anemia, due to non-del(5q) lower risk MDS, and who are refractory or resistant to treatment with an ESA. In September 2019, the FDA granted Fast Track designation to imetelstat for the treatment of adult patients with relapsed/refractory MF.
Fast Track designation provides opportunities for frequent interactions with FDA review staff, as well as eligibility for priority review, if relevant criteria are met, and rolling review of the sponsor’s NDA. Fast Track designation is intended to facilitate and expedite development and review of an NDA to address unmet medical needs in the treatment of serious or life-threatening conditions. However, Fast Track designation does not accelerate conduct of clinical trials or mean that the regulatory requirements are less stringent, nor does it ensure that any imetelstat NDA will be approved or that any approval will be granted within any particular timeframe. In addition, the FDA may withdraw Fast Track designation for any indication if it believes that the designation is no longer supported by data emerging from the imetelstat clinical development program.
The Innovation Passport designation from the United Kingdom regulatory authorities does not guarantee marketing approval and may not lead to a faster development, regulatory review or approval process.
In October 2021, we gained access to the ILAP through the receipt of an Innovation Passport for imetelstat to treat lower risk MDS. The ILAP is a new program sponsored by the Medicines and Healthcare products Regulatory Agency, or MHRA, in the U.K., post-Brexit. The objective of this new licensing and access pathway is to reduce the time to market and enable earlier patient access for innovative medicines. The Innovation Passport is the first prescribed entry point in the ILAP process. Key benefits of being within ILAP include a potential 150-day accelerated assessment and rolling review of an MAA, as well as opportunities for frequent interactions with the review staff at the MHRA and its partner agencies to discuss imetelstat’s development, regulatory and reimbursement plans.
Although the goal of ILAP and the Innovation Passport is to reduce the time to market and enable earlier patient access, it does not accelerate conduct of clinical trials or mean that the regulatory requirements are less stringent, nor does it ensure that any imetelstat MAA will be approved or that any approval will be granted within any particular timeframe. Despite receiving Innovation Passport designation, we may decide to delay or forego the commercialization of imetelstat in the U.K.
Failure to achieve continued compliance with government regulations could delay or halt potential commercialization of imetelstat.
Approved products and their manufacturers are subject to continual review, and discovery of previously unknown problems with a product or its manufacturer may result in restrictions on the product or manufacturer, including import restrictions, seizure and withdrawal of the product from the market. If approved for commercial sale, future sales of imetelstat will be subject to government regulation related to numerous matters, including the processes of:
•advertising and promoting;
35
If, and to the extent that, we are unable to comply with these regulations, our ability to earn potential revenue from the commercialization of imetelstat, if any, would be materially and adversely impacted.
In addition, if imetelstat causes serious or unexpected side effects or is associated with other safety risks after receiving marketing approval, a number of potential significant negative consequences could result, including, but not limited to:
•regulatory authorities may withdraw their approval of imetelstat;
•we may be required to recall imetelstat, seek to change the way it is administered, conduct additional clinical trials or change the labeling of the product;
•regulatory authorities may require revisions to the labeling of imetelstat,, including limitations on approved uses or the addition of further warnings, contraindications or other safety information, or may impose restrictions on distribution in the form of REMS in connection with approval, if any;
•we may experience manufacturing delays and supply disruptions if regulatory inspectors identify regulatory noncompliance by third party manufacturers requiring remediation;
•imetelstat may be rendered less competitive and sales may decrease;
•our reputation may suffer generally both among clinicians and patients;
•we may be exposed to potential lawsuits and associated legal expenses, including costs of resolving claims;
•the FDA or similar international regulatory authorities may refuse to approve pending applications or supplements to approved applications filed by us, or may suspend or revoke license approvals; or
•we may be required to change or stop ongoing clinical trials of imetelstat, which would negatively impact the development of imetelstat for other potential indications.
Any of these events could prevent us from achieving or maintaining market acceptance for imetelstat or could substantially increase the costs and expenses of commercializing imetelstat, which in turn could delay or prevent us from generating any revenues from the sale of the imetelstat.
Moreover, the FDA strictly regulates the promotional claims that may be made about drug products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. The FDA and other agencies actively enforce regulations prohibiting the promotion of any drug product for off-label uses. If we were found to have improperly promoted off-label use of imetelstat, we would be subject to significant civil, criminal and administrative penalties, which would inhibit our ability to commercialize imetelstat and generate revenue, require us to expend significant time and resources in response, and generate negative publicity. Enforcement actions include, among others:
•adverse regulatory inspection findings;
•fines, warning letters, or untitled letters;
•voluntary or mandatory product recalls or public notification or medical product safety alerts to healthcare professionals;
•restrictions on, or prohibitions against, marketing imetelstat;
•restrictions on, or prohibitions against, importation or exportation of imetelstat;
•suspension of review or refusal to approve pending applications or supplements to approved applications;
•exclusion from participation in government-funded healthcare programs;
•exclusion from eligibility for the award of government contracts for imetelstat;
•suspension or withdrawal of product approvals;
36
•civil and criminal penalties and fines.
The imposition of any of these penalties or other commercial limitations would severely and adversely affect our financial results, business and business prospects, including the potential commercialization of imetelstat, and the future of imetelstat, and might cause us to cease operations.
If, in the future, we seek regulatory approval to market imetelstat internationally, we may experience a variety of risks that would materially adversely affect our business.
If, in the future, we seek regulatory approval of imetelstat outside of the U.S., and if the necessary approvals are obtained, we will be subject to additional risks related to operating in countries outside of the U.S., including:
•foreign regulatory approvals, if any, may take longer and be more costly to obtain than approvals in the U.S., due to differing regulatory requirements in foreign countries, such as the lack of pathways for accelerated drug approval;
•regulatory authorities outside of the U.S. may disagree with the design, implementation or results of our clinical trials or our interpretation of data from nonclinical studies or clinical trials;
•approval policies or regulations of regulatory authorities outside of the U.S. may significantly change in a manner rendering our clinical data insufficient for potential approval;
•the COVID-19 pandemic may negatively impact our ability to produce imetelstat and conduct clinical trials in countries outside of the U.S.;
•we may experience unexpected changes in tariffs, trade barriers, price and exchange controls and other regulatory requirements;
•general economic weakness, including inflation, rising interest rates, the prospect of a recession or civil or political instability in particular economies and markets outside of the U.S., including as a result of the conflict between Russia and Ukraine;
•risks of potential noncompliance with legal requirements applicable to privacy, data protection, information security and other matters;
•risks of potential noncompliance with tax, employment, immigration and labor laws for employees living or traveling abroad;
•increased taxes outside of the U.S., including withholding of payroll taxes;
•significant foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country;
•difficulties staffing and managing operations outside of the U.S.;
•complexities associated with managing multiple payor reimbursement regimes and government payors in foreign countries;
•workforce uncertainty in countries where labor unrest is more common than in the U.S.;
•potential liability under the Foreign Corrupt Practices Act of 1977 or comparable regulations outside of the U.S.;
•challenges enforcing our contractual and intellectual property rights, especially in those countries outside of the U.S. that do not respect and protect intellectual property rights to the same extent as the U.S.;
37
•production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
•business interruptions resulting from geopolitical actions, including war and terrorism including the conflict between Russia and Ukraine.
These and other risks associated with international operations may materially adversely affect our ability to attain or maintain profitable operations.
We are also subject to numerous regulatory requirements outside of the U.S. governing, among other things, the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The regulatory approval process varies among countries and may include all of the risks associated with FDA approval described above as well as risks attributable to the satisfaction of local regulations in jurisdictions outside of the U.S. Moreover, the time required to obtain approval may differ from that required to obtain FDA approval. Approval by the FDA does not ensure approval by regulatory authorities outside the U.S. and vice versa.
In Europe, the Clinical Trials Regulation, which came into effect in January 2022, introduced substantial changes in how clinical trials are authorized in the European Economic Area, or EEA, enabling sponsors to submit a single application to run a clinical trial in several European countries. The objectives of the new regulation include consistent rules for conducting trials throughout the European Union, consistent data standards and adverse events listing, and consistent information on the authorization status. Information on the conduct and results of each clinical trial carried out in the EU will be made publicly available. Commencing in January 2023, clinical trial sponsors will need to use the Clinical Trials Information System, or CTIS, to apply to start a new clinical trial in the EEA; and from January 2025, clinical trials in the EEA will need to comply with the Clinical Trials Regulation.
In addition, a new pan-European clinical trial data information database has been created that will be complementary to the database established for pharmacovigilance (Regulation (EC) No 726/2004 with respect to centrally authorized medicinal products). In addition, Commission Implementing Regulation (EU) No 520/2012 outlines the practical implications for marketing authorization holders, national competent authorities, and the EMA. Also, Commission Delegated Regulation (EU) No 357/2014 on post-authorization efficacy studies specifies the situations in which such studies may be required. Post-authorization efficacy studies may be required where concerns relating to some aspects of efficacy of the medicinal product are identified and can be resolved only after the medicinal product has been marketed, or where the understanding of the disease, the clinical methodology or the use of the medicinal product under real-life conditions indicate that previous efficacy evaluations might have to be revised significantly.
Brexit is also expected to disrupt the operation of pre- and post-authorization clinical trial infrastructure. The rules around GMP and pharmacovigilance in the U.K. currently remain similar to the EU requirements. However, the Falsified Medicines Directive will not apply in Great Britain though it is likely that the U.K. will implement a procedure to minimize the risk of falsified medicines.
Uncertainty in the regulatory framework and future legislation could lead to disruption in the execution of international multi-center clinical trials, the monitoring of adverse events through pharmacovigilance programs, the evaluation of the benefit-risk profiles of new medicinal products, and determination of marketing authorization across different jurisdictions. Changes to existing regulations may add considerably to the development lead time to marketing authorization and commercialization of products in the EU and increase our costs. We cannot predict the impact of such changes and future regulation on our business or the results of our operations.
We may be subject to requests for access to imetelstat. Demand for compassionate use of imetelstat could strain our resources, delay our drug development activities, negatively impact our regulatory approval or commercial activities, and result in losses.
We are developing imetelstat to treat life-threatening hematologic malignancies for which there are currently limited therapeutic options. Other companies in our field have been the target of campaigns requesting access to unapproved drugs. If we experience similar request for access campaigns, we may experience significant disruption to our business which could result in losses. We are a small company with limited resources, and any unanticipated
38
trials or access programs resulting from requests for access could deplete our drug supply, increase our capital expenditures, reduce the availability of potentially eligible clinical trial participants, and otherwise divert our resources from our primary goals.
In addition, legislation referred to as “Right to Try” laws have been introduced at the local and national levels, which are intended to give patients access to unapproved therapies. New and emerging legislation regarding expanded access to unapproved drugs for life-threatening illnesses could negatively impact our business in the future. Either activism or legislation related to requests for access may require us to initiate an unanticipated expanded access program or to make imetelstat more widely available sooner than anticipated.
Patients who receive access to unapproved drugs through compassionate use or expanded access programs have life-threatening illnesses and generally have exhausted all other available therapies. The risk for serious adverse events, including those which may be unrelated to imetelstat, in this patient population is high and could have a negative impact on the safety profile of imetelstat, which could cause significant delays or an inability to successfully commercialize imetelstat and could materially harm our business. In addition, if, in order to perform the controlled clinical trials required for potential regulatory approval and successful commercialization of imetelstat, we do not provide compassionate use access or expanded access programs in response to requests for access from patients in the U.S. or elsewhere in the world, we may receive adverse publicity or experience other disruptions. Should we agree to provide compassionate use access or decide to initiate an expanded access program, we could experience adverse publicity or other disruptions related to potential participants in such programs. Similarly, we could experience adverse publicity or other disruptions if we were to restructure or pause any compassionate use and/or expanded access program after initiating such a program or after the provision of our product through compassionate access to an individual patient or patients.
Risks Related to Manufacturing Imetelstat
Failure by us to establish and/or maintain a manufacturing supply chain to appropriately and adequately supply imetelstat for future clinical and commercial uses would result in a further delay in or cessation of clinical trials and a delay in our ability to obtain regulatory approvals of imetelstat, and affect our ability to commercialize imetelstat, and our business and business prospects could be severely harmed, and we could cease operations.
The manufacture of imetelstat must comply with applicable regulatory standards for current and potential future clinical trials and potential commercial uses. The process of manufacturing imetelstat is complex and remains subject to several risks, including:
•the ability to scale-up and attain sufficient production yields with appropriate quality control and quality assurance to meet the needs of our clinical trials and potential future market demand, and to establish commercial supply agreements;
•reliance on third-party manufacturers and suppliers, whose efforts we do not control;
•supply chain issues, including the timely availability and shelf life requirements of raw materials and other supplies, any of which may be impacted by a number of factors, including the effects of macroeconomic conditions such as the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia;
•shortage of qualified personnel; and
•regulatory acceptance and compliance with regulatory requirements, which are less well-defined for oligonucleotide products than for small molecule drugs and vary in each country where imetelstat might be sold or used.
As a result of these and other risks, we may be unable to establish and/or maintain a manufacturing infrastructure and supply chain capable of providing imetelstat for IMerge Phase 3, IMpactMF, IMproveMF and IMpress, and potential future commercial uses, which would delay or result in a cessation of such current or potential future clinical trials of imetelstat. Occurrence of any such events would further delay or preclude any applications for regulatory approval and therefore further delay or preclude our ability to earn revenue from the
39
commercialization, if any, of imetelstat, which would severely and adversely affect our financial results, business and business prospects, and might cause us to cease operations.
If third parties that manufacture imetelstat fail to perform as needed, then the clinical and commercial supply of imetelstat will be limited, and we may be unable to conduct or complete current or potential future clinical trials of imetelstat or to commercialize imetelstat in the future.
Our imetelstat manufacturing supply chain relies, and will continue to rely, solely upon third-party manufacturers to perform certain process development or other technical and scientific work with respect to imetelstat, as well as to supply starting materials and manufacture drug substance and drug product. While we have established arrangements with third parties for the manufacture of imetelstat, our manufacturing supply chain is highly specialized, and as such we are reliant upon a small group of third-party manufacturers to supply starting materials, drug substance and drug product. Failure by such third-party manufacturers to perform in a timely manner and in compliance with all regulatory requirements, or at all, could further delay, perhaps substantially, or preclude our ability to pursue imetelstat development on our own, increase our costs and otherwise negatively affect our financial results, business and business prospects. In this regard, a recent FDA inspection of one of our third-party manufacturers identified certain deficiencies in the manufacturer’s processes and facilities which, while not directly related to the production of imetelstat, could impact the manufacturer’s ability to produce and deliver products, including imetelstat, if not remediated by the manufacturer, and could lead to delays or shortages in drug supply, or the inability to manufacture or ship drug supply necessary for non-clinical and clinical activities, and commercialization. In addition, we may not be able to obtain imetelstat from third-party manufacturers on acceptable terms, or at all. We expect to rely on third-party manufacturers to produce and deliver sufficient quantities of imetelstat and other materials to support clinical trials on a timely basis and to comply with applicable regulatory requirements. We do not have direct control over these third-party personnel or operations. Reliance on these third-party manufacturers is subject to numerous risks, including:
•being unable to contract with suitable third-party manufacturers, including for potential commercial supply of imetelstat, because the number of potential manufacturers is limited;
•delays and disruptions experienced by third-party manufacturers due to the effects of the COVID-19 pandemic, which have adversely impacted and could continue to adversely impact the ability of such parties to fulfill their contractual obligations to us;
•capacity limitations and scheduling constraints experienced by third-party manufacturers due to scheduling and other commitments, and queued manufacturing activities in contracted facilities;
•potential shortages of available manufacturing capacity or consumable manufacturing supplies at third-party manufacturers, due to obligations to manufacture and distribute vaccines to address the spread of COVID-19; and we anticipate that other delays, or potential shortages of consumable manufacturing supplies, may continue throughout 2023;
•requirements by regulatory authorities to validate and qualify significant activities for any current or replacement manufacturer, which could involve new testing and compliance inspections;
•the inability to execute timely contracts with third-party manufacturers and suppliers on acceptable terms, or at all;
•the inability of third-party manufacturers to timely formulate and manufacture imetelstat or to produce or ship imetelstat in the quantities or of the quality required to meet clinical and commercial needs, whether due to the effects of the COVID-19 pandemic or any other reasons;
•the possible mislabeling by third-party manufacturers of clinical supplies, potentially resulting in the wrong dose amounts being supplied or active drug or comparator not being properly identified;
•decisions by third-party manufacturers to exit the contract manufacturing business during the time required to supply clinical trials or to successfully produce, store and distribute imetelstat to meet commercial needs;
40
•compliance by third-party manufacturers with GMP standards mandated by the FDA and state agencies and other government regulations corresponding to similar international regulatory authorities, including any deficiencies identified during regulatory inspections, such as those identified in a recent FDA inspection of one of our third-party manufacturers;
•breach or termination of manufacturing or supply contracts;
•inadequate storage or maintenance at contracted facilities resulting in theft or spoilage; and
•natural disasters that affect contracted facilities.
Each of these risks could lead to delays or shortages in drug supply, or the inability to manufacture or ship drug supply necessary for non-clinical and clinical activities, and commercialization. For example, manufacturing delays could adversely impact the conduct or completion of imetelstat clinical trials, such as IMerge Phase 3, IMpactMF, IMproveMF and IMpress, or commencement of potential future clinical trials of imetelstat, or preclude or delay potential future commercial sales, which could severely and adversely affect our financial results, business and business prospects, and the future of imetelstat.
In addition, third-party manufacturers and/or any other manufacturers may need to make substantial investments to enable sufficient capacity increases and cost reductions, and to implement those regulatory and compliance standards necessary for successful Phase 3 clinical trials and commercial production of imetelstat. These third-party manufacturers may not be willing or able to achieve such capacity increases, cost reductions, or regulatory and compliance standards, and even if they do, such achievements may not be at commercially reasonable costs. Changing manufacturers may be prolonged and difficult due to inherent technical complexities and because the number of potential manufacturers is limited. It may be difficult or impossible for us to find a replacement manufacturer on acceptable terms, or at all.
It may not be possible to manufacture imetelstat at costs or scales necessary to conduct clinical trials or potential future commercialization activities.
Oligonucleotides are relatively large molecules produced using complex chemistry, and the cost of manufacturing an oligonucleotide like imetelstat is greater than the cost of making typical small molecule drugs. Therefore, imetelstat for clinical use is more expensive to manufacture than most other treatments currently available today or that may be available in the future. Similarly, the cost of manufacturing imetelstat for commercial use will need to be significantly lower than current costs in order for imetelstat to become a commercially successful product. We may not be able to enter into suitable commercial supply agreements, or to achieve sufficient scale increases or cost reductions necessary for successful commercial production of imetelstat. Failure to achieve necessary cost reductions could result in decreased sales or reduced gross margins, if any, for us, which would materially and adversely affect our financial results, business and business prospects, and the future of imetelstat.
RISKS RELATED TO COVID-19
The effects of the ongoing COVID-19 global pandemic have negatively impacted, and will likely continue to negatively impact, our business and healthcare resources around the world, including a significant number of current and planned clinical sites involved with IMpactMF, IMproveMF and IMpress.
Our business and business prospects, our financial condition and ability to raise additional capital, and the future of imetelstat generally could be materially and adversely affected by the effects of the ongoing global COVID-19 pandemic. The ongoing COVID-19 pandemic and public health safety measures taken in response to COVID-19 have had a significant impact, both direct and indirect, on businesses, as significant reductions in business-related activities have occurred, clinical development and regulatory activities have been curtailed, delayed or suspended and supply chains have been disrupted. We have allowed voluntary access to our offices in California and New Jersey to employees who have been vaccinated. While almost all of our employees continue to work remotely without any significant disruption to our business, the effects of our policies regarding remote working may negatively impact productivity, disrupt our business and continue to delay our imetelstat development program and clinical trial timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. In addition, our increased reliance
41
on personnel working remotely could increase our cybersecurity risk, create data accessibility concerns and make us more susceptible to communication disruptions, any of which could adversely impact our business operations. These and similar, and perhaps more severe, disruptions in our operations could continue to negatively impact our business and business prospects, our financial condition and the future of imetelstat.
Due to the effects of the COVID-19 pandemic, we have had, and expect to continue to have, or we may potentially have in the future, disruptions and/or delays in our imetelstat development program, including with respect to our ability to:
•open trial sites for screening and enrollment;
•screen, enroll and assess patients;
•retain enrolled patients in our clinical trials;
•ensure patient visits to clinical sites and laboratories;
•conduct monitoring visits;
•manufacture and/or supply study drug or other supplies;
•report trial results; or
•interact with regulators or other important agencies due to limitations in employee resources or otherwise.
Restrictions on travel, availability of site personnel, and diversion of hospital staff and resources to COVID-19 patients, have disrupted our clinical trial operations, as well as patient recruitment in many areas, resulting in a slowdown in patient enrollment and/or deviations from or disruptions in key clinical trial activities, such as opening, initiating and monitoring clinical trial sites. Although vaccine distribution, including booster shots, is being conducted in many countries, the emergence of COVID-19 variants and subvariants, and the resurgence of COVID-19 cases in parts of the world, causes further uncertainty and unpredictability in clinical trial activities, including clinical site initiations, patient screening and enrollment. Like many other biopharmaceutical companies, we have experienced and continue to experience delays in clinical site initiations and patient screening and enrollment in our clinical trials, IMerge Phase 3, IMpactMF and IMproveMF, due to the COVID-19 pandemic, which have impacted our trial operations. Even though we completed patient enrollment in IMerge Phase 3, the pace of enrollment was slower than planned.
For IMpactMF, in addition to the negative impact of COVID-19, site personnel resources remain constrained in the countries where we planned to conduct the trial due to the number of competing trials in MF and other oncology indications. As such, we have experienced and expect to continue to experience disruption in clinical trial activities and delays in enrollment, as well as constraints on available sites and site personnel. Based on assumptions for enrollment and event (death) rates in the trial, we expect the interim analysis for OS for IMpactMF may occur in 2024 and the final analysis may occur in 2025. Because these analyses are event-driven and it is uncertain whether actual rates for enrollment and events will reflect current planning assumptions, the results may be available at different times than currently expected.
The continuing effects of the COVID-19 pandemic could cause further disruptions to our clinical development timelines, including continued delays in enrollment and clinical trial site initiation in IMpactMF, IMproveMF and IMpress, and other disruptions that could severely impact our business and the imetelstat development program, including those resulting from:
•new, continued or heightened difficulties in opening clinical trial sites for patient screening and enrollment and recruiting clinical site investigators and clinical site staff;
•continued or heightened delays or difficulties caused by missed patient visits to clinical sites and laboratories, and uncertainty how the FDA will view deviations from clinical protocols caused by the effects of the COVID-19 pandemic;
42
•potential refusal by the FDA to accept data, including from clinical trials in affected geographies or failure to comply with updated FDA guidance and expectations related to the conduct of clinical trials during the COVID-19 pandemic;
•continued or heightened delays or disruptions in clinical trial activities, including executing clinical site contracts, due to reduced availability of personnel at CROs, clinical site legal groups and vendors, or for any other reasons;
•substantial reduction of healthcare resources available for the conduct of clinical trials, including the temporary postponement of clinical trial activities at certain hospitals serving as our clinical trial sites and diversion of hospital staff away from the conduct of our clinical trials, such as those experienced by us to date;
•interruption of, or delays in receiving, supplies of imetelstat from our third-party manufacturers due to among other things, staffing shortages, production slowdowns or stoppages, shipping delays, shortages in raw materials or laboratory supplies because of ongoing efforts to address the pandemic, limitations in available capacity at contract manufacturing vendors or drug distribution service providers due to obligations to manufacture and distribute vaccines to address the spread of COVID-19, disruptions in supply chain and production systems and import/export complications;
•interruption of key clinical trial activities, such as clinical trial site data monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others or interruption of clinical trial subject visits and study procedures (particularly any procedures that may be deemed non-essential), which may impact the integrity of subject data and clinical study endpoints;
•loss of potential and recruited patients in clinical trials due to clinical site COVID-19 activities, desire of patients to avoid frequent visits to hospitals because of potential increased exposure to COVID-19, or loss of life of patients due to COVID-19;
•increased costs for clinical trial activities due to delays or disruptions in opening sites, screening and enrolling patients or treating and following patients, whether as a result of the effects of the COVID-19 pandemic or for any other reasons, which would require further additional capital that may not be available; and
•limitations on employee resources that would otherwise be focused on the conduct of our clinical trials, product development, manufacturing, potential commercialization activities and general company operations, including because of sickness of employees or their families, the desire of employees to avoid contact with large groups of people, an increased reliance on working from home, or mass transit disruptions.
These and other factors arising from the effects of the COVID-19 pandemic could further adversely impact our ability to enroll, conduct and complete IMpactMF, IMproveMF and IMpress and any potential future clinical trials of imetelstat, and could otherwise materially and adversely affect our business and business prospects, our financial condition and the future of imetelstat.
In addition, we rely on third-party CROs and other third parties to assist us with clinical trial activities. The COVID-19 pandemic has also had a significant impact on our CROs and other vendors, and we cannot guarantee that they will continue to perform their contractual duties in a timely and satisfactory manner as a result of the COVID-19 pandemic. Also, absenteeism by governmental employees or the focus of regulatory authorities’ efforts and attention on the approval of other therapeutics or other activities related to COVID-19 could likewise impact the timeliness of regulatory authority responses and the processing of regulatory submissions for imetelstat. In any event, if the effects of the COVID-19 pandemic become more severe, we may experience more significant disruptions to our clinical development timelines, which would materially and adversely affect our business and business prospects, our financial condition and ability to raise additional capital, and the future of imetelstat.
While at this time we believe that we have sufficient drug supply for potential commercialization activities necessary to potentially bring imetelstat to market in lower risk MDS in the U.S., as well as for clinical use in IMerge Phase 3, IMpactMF, IMproveMF and IMpress, we could experience disruptions to our supply chain, as well as delays or limitations in our ability to obtain sufficient materials for the manufacture of imetelstat for our current
43
and potential future clinical trials. Such disruptions could adversely affect our ability to conduct ongoing and potential future clinical trials of imetelstat. For example, some of our suppliers of certain materials used in the production of imetelstat are located in countries that were or are heavily affected by the COVID-19 pandemic. In these countries, shipping delays, closures and other restrictions resulting from the COVID-19 pandemic in the region could disrupt our supply chain or limit our ability to obtain sufficient materials for the manufacture of imetelstat. In addition, we may experience limitations in available capacity at contract manufacturers or drug suppliers, or potential shortages of consumable manufacturing supplies, due to obligations to manufacture and distribute vaccines to address the spread of COVID-19. We anticipate other delays, or potential shortages of consumable manufacturing supplies due to the COVID-19 pandemic may continue throughout 2023.
The effects of the COVID-19 pandemic, as well as broader economic conditions, including inflation, rising interest rates and the prospects for recession, have increased market volatility and could result in a significant long-term disruption of global financial markets, reducing or eliminating our ability to raise additional capital. In the absence of potential proceeds from exercises of currently outstanding warrants and potential drawdowns under the Loan Agreement, we will require substantial additional funding to further advance the imetelstat program, including through the completion of IMpactMF, IMproveMF and IMpress, as well as to conduct the clinical, regulatory and potential commercialization activities necessary to potentially bring imetelstat to market in relapsed/refractory MF and any other future indications. We cannot predict with any certainty whether and to what extent the outstanding warrants will be exercised for cash, or the timing of availability of additional funds under the Loan Agreement, if at all. In addition, the global economic slowdown caused by, among other things, the COVID-19 pandemic and the military conflict between Ukraine and Russia, as well as inflation, rising interest rates and the prospects for recession, could materially and adversely affect our business and the value of our common stock. As a result of these factors, our ability to raise additional capital may be impaired which could negatively affect our liquidity, our business and business prospects, and the future of imetelstat.
The extent to which the COVID-19 pandemic impacts our business, our regulatory and clinical development activities, clinical supply chain and other business operations, as well as the value of and market for our common stock, will depend on future developments that are highly uncertain and cannot be predicted with confidence at this time. Such developments include continued spread of COVID-19 variants and subvariants in the U.S. and other countries and the potential emergence of new variants or additional sub-variants that may prove especially contagious or virulent, the ultimate duration and severity of the pandemic, travel restrictions, quarantines, social distancing, shipping delays and business closure requirements in the U.S. and in other countries, and the effectiveness of vaccination programs and other actions taken globally to treat and manage this health crisis. Accordingly, we do not yet know the full extent of potential delays or impacts on our business, our regulatory and clinical development activities, clinical supply chain and other business operations or the global economy as a whole. However, these effects could materially and adversely affect our business and business prospects, our financial condition and ability to raise additional capital, and the future of imetelstat. In addition, to the extent the effects of the COVID-19 pandemic adversely affect our business and financial condition, they may also have the effect of heightening many of the other risks and uncertainties described elsewhere under the heading “Risk Factors”.
Risks Related to Our Financial Position and Need For Additional Financing
Our failure to obtain substantial additional capital would force us to further delay, reduce or eliminate development of imetelstat in current and any potential future clinical trials of imetelstat, and our potential future imetelstat commercialization efforts, any of which would severely and adversely affect our financial results, business and business prospects, and might cause us to cease operations.
Successful drug development and commercialization requires significant amounts of capital. Based on our current operating plan, our expectations regarding the timing of the submission, review and approval of our NDA in the U.S. for the use of imetelstat in adult patients with lower risk MDS, and our anticipated use of the net proceeds from our recently-announced public offering of securities, if completed, we believe that our existing cash, cash equivalents, restricted cash and current and noncurrent marketable securities, together with the anticipated net proceeds from our recently-announced public offering of securities, if completed, will be sufficient to fund our projected operating requirements until the middle of 2024, which includes preparatory activities for the potential U.S. commercial launch of imetelstat in lower risk MDS. Assuming future proceeds of up to $109.2 million from
44
potential cash exercises of currently outstanding warrants and potential drawdowns of up to $75.0 million available under the Loan Agreement (which is subject to our achievement of certain clinical and regulatory milestones and satisfaction of certain capitalization and other requirements to our existing capital resources, as well as approval by an investment committee comprised of Hercules and SVB for the final $25.0 million tranche), we believe that our existing cash, cash equivalents, restricted cash and current and noncurrent marketable securities, together with the anticipated net proceeds from our recently-announced public offering of securities, if completed, could be sufficient to fund our projected operating requirements until the end of 2025, which would include the anticipated completion of the final analysis for IMpactMF in 2025. In the absence of future proceeds from potential cash exercises of currently outstanding warrants and potential drawdowns under the Loan Agreement, we will require substantial additional funding to further advance the imetelstat program, including through the completion of IMpactMF, IMproveMF and the investigator-led trial IMpress, as well as conducting the clinical, regulatory and potential commercialization activities necessary to potentially bring imetelstat to market in relapsed/refractory MF and any other future indications, and our need for additional funds may arise sooner than planned.
We cannot predict with any certainty whether and to what extent the outstanding warrants will be exercised for cash, or the timing or availability of additional funds under the Loan Agreement, if at all. For example, holders of our outstanding warrants may not exercise any portion of a warrant if, after giving effect to such exercise, the holder would own more than 9.99% of our outstanding common stock immediately after exercise, which percentage may be changed at the holder's election upon 61 days' notice to us subject to the terms of the warrants. In addition, in the event that a registration statement registering the issuance of the shares of common stock underlying our purchase warrants is not effective or available for the issuance of such shares to such holder at the time of exercise, a holder of our purchase warrants may only exercise such purchase warrants through a “cashless exercise,” in which case, the holder would receive upon such exercise, the net number of shares of common stock determined according to the formula set forth in the purchase warrant, and we would not receive any proceeds from the exercise of such purchase warrants. As a result, we may not receive our anticipated proceeds from certain of our outstanding warrants. In addition, drawdowns under the Loan Agreement are subject to our achievement of certain clinical and regulatory milestones and satisfaction of certain capitalization and other requirements to our existing capital resources, as well as approval by an investment committee comprised of Hercules and SVB for the final $25.0 million tranche. Even if we receive the future proceeds in full from the potential cash exercise of outstanding warrants and potential drawdowns under the Loan Agreement, we may still require additional funding. If adequate funds are not available on a timely basis, if at all, we may be unable to pursue further development, including conducting and completing IMpactMF, IMproveMF and IMpress, or commencing, conducting or completing potential future clinical trials of imetelstat, or pursuing potential commercialization of imetelstat, which would severely harm our business and we might cease operations. In addition, our ability to commercialize imetelstat in the U.S., if regulatory approval is granted, depends on us being able to establish sales and marketing capabilities which we may be unable to do in a timely manner or at all.
Because the outcome of any clinical activities and/or regulatory approval process is highly uncertain, we cannot reasonably estimate whether any development activities we may undertake will succeed, whether we will be able to commercialize imetelstat, and we may never recoup our investment in any imetelstat development, which would adversely affect our financial condition and our business and business prospects, and might cause us to cease operations. In addition, our plans and timing expectations will be further delayed or interrupted by macroeconomic conditions, such as if COVID-19 or pandemic conditions worsen, creating further limitations on our clinical trial or commercial preparatory activities, or could be disrupted by civil or political unrest or military conflicts around the world, such as the current military conflict between Ukraine and Russia. Further, our future capital requirements are difficult to forecast and will depend on many factors, including:
•the accuracy of the assumptions underlying our estimates for our capital needs;
•the scope, progress, timing, magnitude and costs of clinical development, manufacturing and potential commercialization of imetelstat, including the number of indications being pursued, subject to clearances and approvals by the FDA and similar international regulatory authorities;
•the scope, progress, duration, results and costs of current clinical trials, including IMerge Phase 3, IMpactMF, IMproveMF and IMpress, and any potential future clinical trials of imetelstat, as well as non-clinical studies and assessments of imetelstat;
45
•delays or disruptions in opening sites, screening and enrolling patients or treating and following patients, in IMpactMF, IMproveMF, IMpress, or any potential future clinical trials of imetelstat, whether as a result of the effects of macroeconomic conditions like the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession;
•the costs, timing and outcomes of regulatory reviews or other regulatory actions related to imetelstat, such as obtaining and maintaining regulatory clearances and approvals to continue clinical development of imetelstat in current and potential future clinical trials, as well as to commence potential commercialization of imetelstat in the U.S. and in other countries;
•the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims;
•the costs of manufacturing imetelstat, including our ability to achieve any meaningful reduction in manufacturing costs;
•the costs of multiple third-party vendors and service providers, including our CROs and third-party manufacturers, to pursue the development, manufacturing and potential commercialization of imetelstat;
•our ability to establish, enforce and maintain collaborative or other strategic arrangements for research, development, clinical testing and manufacturing of imetelstat and potential future commercialization and marketing;
•our efforts to enhance operational, financial and management processes and systems that will be required for future development and commercialization of imetelstat, and our ability to successfully recruit and retain additional key personnel to support the development and potential future commercialization of imetelstat;
•our ability to successfully market and sell imetelstat, if imetelstat receives future regulatory approval or clearance, in the U.S. and other countries, and the associated costs;
•the costs and timing necessary to build a sales force in the U.S. and potentially other countries to market and sell imetelstat, should it receive regulatory approval, to the extent that such sales, marketing, manufacturing and distribution are not the responsibility of any collaborator;
•the sales price for imetelstat, if any;
•the availability of coverage and adequate third-party reimbursement for imetelstat, if any;
•the extent to which we acquire or in-license other drugs and technologies, or to which we out-license imetelstat;
•the extent to which we acquire or invest in businesses, products or technologies, although we currently have no commitments or agreements relating to any of these types of transactions;
•the extent to which we are able to enter into strategic partnerships, collaborations and alliances or licensing arrangements with third parties including for the commercialization of imetelstat in certain global regions;
•our ability to establish and maintain collaborations on favorable terms, if at all;
•the success of any collaborations that we may enter into with third parties;
•expenses associated with settlement of the pending securities class action lawsuits, and the ongoing derivative lawsuits, as well as any other potential litigation;
•the extent and scope of our general and administrative expenses, including expenses associated with potential future litigation;
•our level of indebtedness and associated debt service obligations;
•the costs of maintaining and operating facilities in California and New Jersey, telecommunications and administrative oversight, as well as higher expenses for travel;
46
•broader economic conditions, including inflation, rising interest rates and the prospects for recession, that may reduce our ability to access debt capital or financing on preferable terms, which may adversely affect future capital requirements and forecasts;
•the costs of enabling our personnel to work remotely, including providing supplies, equipment and technology necessary for them to perform their responsibilities; and
•the proceeds, if any, of cash exercises of our currently outstanding warrants.
Until we can generate a sufficient amount of revenue from imetelstat to finance our cash requirements, which we may never achieve, we expect to finance future cash needs through a combination of public or private equity offerings, debt financings, collaborations, strategic alliances, licensing arrangements and other marketing and distribution arrangements, which may not be possible. Availability of such financing sources may be negatively impacted by any further delays in reporting results from IMpactMF or investors’ perception of top-line results from IMerge Phase 3, despite our interpretation of such data being positive, as well as factors such as the global economic slowdown caused by inflation, rising interest rates and the prospects for recession.
Additional financing through public or private debt or equity financings, including pursuant to the 2020 Sales Agreement with B. Riley Securities; the remaining tranches of up to $75.0 million available under the Loan Agreement, which is subject to the achievement of certain clinical and regulatory milestones and satisfaction of certain capitalization and other requirements, as well as approval by an investment committee comprised of Hercules and SVB for the final $25.0 million tranche; capital lease transactions or other financing sources, may not be available on acceptable terms, or at all. We may be unable to raise equity capital, or may be forced to do so at a stock price or on other terms that could result in substantial dilution of ownership for our stockholders. The receptivity of the public and private debt and equity markets to proposed financings has been substantially affected by uncertainty in the general economic, market and political climate due to the effects of macroeconomic conditions like the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession, and may in the future be affected by other factors which are unpredictable and over which we have no control. In this regard, the effects of the COVID-19 pandemic have increased market volatility and could result in a significant long-term disruption of global financial markets, which could reduce or eliminate our ability to raise additional funds through financings, and could negatively impact the terms upon which we may raise those funds. Similarly, these macroeconomic conditions have created extreme volatility and disruption in the capital markets and is expected to have further global economic consequences. If the equity and credit markets deteriorate, including as a result of macroeconomic conditions like the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession, it may make any necessary debt or equity financing more difficult to obtain in a timely manner or on favorable terms, more costly or more dilutive. If we are unable to raise additional capital or establish alternative collaborative arrangements with third-party collaborative partners for imetelstat, the development and potential commercialization of imetelstat may be further delayed, altered or abandoned, which might cause us to cease operations.
In addition, we may seek additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. Due to uncertainty in the general economic, market and political climate, we may determine that it is necessary or appropriate to raise additional funds proactively to meet longer-term anticipated operating plans. To the extent that we raise additional capital following our recently-announced public offering of securities through the sale of equity or convertible debt securities, including pursuant to the 2020 Sales Agreement, your ownership interest as a stockholder may be diluted, and the terms may include liquidation or other preferences that materially and adversely affect your rights as a stockholder. In addition, we have borrowed, and in the future may borrow, additional capital from institutional and commercial banking sources to fund imetelstat development and our future growth, including pursuant to our Loan Agreement or potentially pursuant to new arrangements with different lenders. We may borrow funds on terms under agreements, such as the Loan Agreement, that include restrictive covenants, including covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. Moreover, if we raise additional funds through alliance, collaborative or licensing arrangements with third parties, we may have to relinquish valuable rights to imetelstat or our technologies or grant licenses on terms that are not favorable to us.
47
We cannot assure you that our existing capital resources, together with the anticipated net proceeds from our recently-announced public offering of securities, if completed, future interest income, future proceeds from potential cash exercises of currently outstanding warrants and potential future sales of our common stock, including under the 2020 Sales Agreement with B. Riley Securities or potential future drawdowns, if available, of the remaining up to $75.0 million under the Loan Agreement (which is subject to the achievement of certain clinical and regulatory milestones and satisfaction of certain capitalization and other requirements, as well as approval by an investment committee comprised of Hercules and SVB for the final $25.0 million tranche), will be sufficient to fund our operating plans. In this regard, under the Loan Agreement, our ability to draw down any funds is subject to our achievement of certain clinical and regulatory milestones and satisfaction of certain capitalization requirements and other conditions, including, with respect to the final $25.0 million tranche of the Loan Agreement, approval by an investment committee comprised of Hercules and SVB. Without such approval, we will not be eligible to draw down any funds under the final tranche, and we will need to obtain additional or alternative financing to advance our development of imetelstat.
We currently have no source of product revenue and may never become profitable.
Although in the past we have received license and other payments under former license and collaboration agreements, we do not currently have any material revenue-generating license or collaboration agreements, have no products approved for commercialization and have never generated any revenue from product sales. In addition, we are incurring and have incurred operating losses every year since our operations began in 1990, except for one. As of September 30, 2022, our accumulated deficit was approximately $1.4 billion. Losses have resulted principally from costs incurred in connection with our research and development activities and from general and administrative costs associated with our operations.
Substantially all of our revenues to date have been payments under collaboration agreements and milestones, royalties and other revenues from our licensing arrangements. Our license agreements related to our hTERT technology have expired or been terminated due to expiration of the underlying hTERT patents, and will not generate any further revenues. We have no ongoing collaborations related to imetelstat and have no current plans to enter into any corporate collaboration, partnership or license agreements that result in revenues, although we may seek a collaborative partner or partners, at an appropriate time, to assist us in the potential development and commercialization of imetelstat, especially outside the U.S., and to provide funding for such activities.
We also expect to experience increased negative cash flow for the foreseeable future as we fund our operations and imetelstat clinical development activities and research programs continue, and we prepare for potential commercialization of imetelstat. This will result in decreases in our working capital, total assets and stockholders’ equity. Further, we may be unable to replenish our working capital by future financings. We will need to generate significant revenues to achieve consistent future profitability. We may never achieve consistent future profitability. Even if we do become profitable in the future, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to achieve consistent future profitability could negatively impact the market price of our common stock and our ability to sustain operations.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Our net operating loss carryforwards attributable to tax years beginning before January 1, 2018 could expire unused and be unavailable to offset future income tax liabilities. Under the Tax Cuts and Jobs Act of 2017, or the Tax Act, as modified by the Coronavirus Aid, Relief and Economic Security Act, or CARES Act, federal net operating losses incurred in taxable years beginning after December 31, 2017, can be carried forward indefinitely, but the deductibility of such federal net operating losses in taxable years beginning after December 31, 2020, is limited to 80% of taxable income. It is uncertain if and to what extent various states will conform to the Tax Act or the CARES Act.
Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” generally defined as a greater than 50‑percentage‑point cumulative change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes (such as research and development tax credits) to offset its post-change taxable income or taxes may be limited. Changes
48
in our stock ownership, some of which are outside of our control, may have resulted in, or other future changes (including as a result of our recently-announced public offering of securities, if completed) could result in, an ownership change. If a limitation were to apply, utilization of a portion of our domestic net operating loss and tax credit carryforwards could be limited in future periods. In addition, a portion of the carryforwards may expire before being available to reduce future income tax liabilities which could adversely impact our financial position. At the state level, there may be periods during which the use of net operating loss carryforwards is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
Risks Related to our Indebtedness
Our level of indebtedness and debt service obligations could adversely affect our financial condition, and may make it more difficult for us to fund our operations.
As of September 30, 2022, the total outstanding principal amount under the Loan Agreement was $50.0 million. The tranches for the remaining $75.0 million available to us under the Loan Agreement are as follows: (a) the first remaining tranche of $20.0 million is available within 30 days of the achievement of certain clinical and financial milestones until September 15, 2023, subject to the achievement of such milestones; (b) the second remaining tranche of $10.0 million is available from January 1, 2023 until December 15, 2023, subject to the achievement of certain clinical and regulatory milestones, and satisfaction of certain other requirements; (c) the third remaining tranche of $20.0 million is available from September 15, 2023 until September 15, 2024, subject to the achievement of certain clinical and regulatory milestones, and satisfaction of certain capitalization requirements; and (d) the final remaining tranche of $25.0 million is available through December 31, 2024, subject to approval by an investment committee comprised of Hercules and SVB. Without the achievement of the required clinical and regulatory milestones and satisfaction of certain capitalization and other requirements, we will not be eligible to draw funds under the first three remaining tranches. If we do not receive investment committee approval, we will not be eligible to draw funds under the final remaining tranche under the Loan Agreement. If we are not able to draw additional funds under the Loan Agreement, we will need to obtain additional or alternative financing to advance our development of imetelstat. Such additional or alternative financing may not be available on attractive terms, if at all, and could be more costly for us to obtain. In addition, before we would consider drawing down any of the remaining tranches under the Loan Agreement, if available, we must first satisfy ourselves that we will have access to future alternate sources of capital, such as from the equity capital markets or debt capital markets, in order to repay any additional principal borrowed, which we may be unable to do, in which case, our liquidity and ability to fund our operations may be substantially impaired. As a result, our development and potential commercialization of imetelstat and other research and development programs could be significantly delayed, which would materially adversely affect our business, business prospects, financial condition and operating results.
All obligations under the Loan Agreement are secured by substantially all of our existing property and assets, excluding intellectual property, which is subject to a negative pledge. This indebtedness may create additional financing risk for us, particularly if our business or prevailing financial market conditions are not conducive to paying off or refinancing the outstanding debt obligations at maturity. If we are able to draw down any of the remaining tranches under the Loan Agreement, our indebtedness will increase, which would further increase our risk of being unable to pay off or refinance our outstanding debt obligations at maturity. Our indebtedness could also have important negative consequences, including:
•we will need to repay the indebtedness by making payments of interest and principal, which will reduce the amount of cash available to finance our operations, our research and development efforts and other general corporate activities; and
•our failure to comply with the obligations of our affirmative and restrictive covenants in the Loan Agreement could result in an event of default that, if not cured or waived, would accelerate our obligation to repay this indebtedness, and Hercules and SVB could seek to enforce its security interest in the assets securing such indebtedness.
To the extent additional debt is added to our current debt levels, the risks described above could increase.
49
The terms of the Loan Agreement place restrictions on our operating and financial flexibility.
The Loan Agreement imposes operating and other restrictions on us. Such restrictions will affect, and in many respects limit or prohibit, our ability and the ability of any future subsidiaries to, among other things:
•dispose of certain assets;
•change our line of business;
•engage in mergers, acquisitions or consolidations;
•incur additional indebtedness;
•pay dividends and make contributions or repurchase our capital stock; and
•engage in certain transactions with affiliates.
The Loan Agreement, as recently amended in June 2022, also contains financial covenants. Beginning June 1, 2022 and prior to receiving potential regulatory approval for imetelstat, if any, we are required to maintain a minimum cash balance in an amount equal to the greater of: 50% of the outstanding principal amount under the Loan Agreement or $30.0 million. Under the Loan Agreement, if we enter into certain licensing transactions, this cash covenant requirement would increase to $35.0 million. After the potential regulatory approval for imetelstat, if any, the minimum cash requirement may be satisfied through one of the following three options, as elected by us: (a) maintaining a cash balance in an amount not less than 40% of the outstanding principal amount under the Loan Agreement; (b) maintaining a cash balance in an amount not less than 25% of the outstanding principal amount under the Loan Agreement, if our market cap is or exceeds $750.0 million; or (c) maintaining six month net product revenues of at least 70% of net product revenues forecasted by us, should any potential regulatory approval for imetelstat be obtained. The breach of any of these restrictive covenants or any other terms of the Loan Agreement would accelerate our obligation to repay our indebtedness under the Loan Agreement, which could have a material adverse effect on our business, business prospects and financial position.
We may not have cash available in an amount sufficient to enable us to make interest or principal payments on our indebtedness when due.
Our ability to make scheduled payments on or to refinance our indebtedness depends on our future performance and ability to raise additional sources of cash, which is subject to economic, financial, competitive and other factors beyond our control. If we are unable to generate sufficient cash to service our debt, we may be required to adopt one or more alternatives, such as selling assets, restructuring our debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. If we desire to refinance our indebtedness, our ability to do so will depend on the state of the capital and lending markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
Failure to satisfy our current and future debt obligations under the Loan Agreement could result in an event of default. In addition, the Loan Agreement includes customary affirmative and negative covenants and other events of default, the occurrence and continuance of which provide Hercules and SVB with the right to demand immediate repayment of all principal and unpaid interest under the Loan Agreement, and to exercise remedies against us and the collateral securing the Loan Agreement. These events of default include, among other things:
•insolvency, liquidation, bankruptcy or similar events;
•failure to observe any covenant or secured obligation under the Loan Agreement, which failure, in most cases, is not cured within 15 days;
•occurrence of an event that could reasonably be expected to have a material adverse effect on our business, operations, properties, assets or financial condition;
•material misrepresentations;
50
•occurrence of any default under any other agreement involving indebtedness in excess of specified amounts, or the occurrence of a default under any agreement that could reasonably be expected to have a material adverse effect on us; and
•certain money judgments being entered against us or any portion of our assets are attached or seized.
In the event of default, Hercules and SVB could accelerate all of the amounts due under the Loan Agreement. Under such circumstances, we may not have enough available cash or be able to raise additional funds through equity or debt financings to repay such indebtedness at the time of such acceleration. In that case, we may be required to delay, limit, reduce or terminate imetelstat development or potential commercialization efforts or grant to others rights to develop and market imetelstat. Hercules and SVB could also exercise their rights to take possession and dispose of the collateral securing the Loan Agreement, which collateral includes substantially all of our property other than intellectual property. Our business, financial condition and results of operations could be materially adversely affected as a result of any of these events.
Risks Related to Protecting Our Intellectual Property
If we are unable to obtain and maintain sufficient intellectual property protection for imetelstat for an adequate amount of time, or if the scope of the intellectual property protection is not sufficiently broad, our competitors could develop and commercialize products similar or identical to imetelstat, and our ability to successfully commercialize imetelstat may be adversely affected.
Protection of our proprietary technology is critically important to our business. Our success and the success of our planned future development and commercialization of imetelstat will depend on our ability to protect our technologies and imetelstat through patents and other intellectual property rights. Our success will depend in part on our ability to obtain, maintain, enforce, and extend our patents and maintain trade secrets, both in the U.S. and in other countries.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the U.S. and in other countries. Such challenges may result in loss of exclusivity or in patent claims being narrowed, invalidated, or held unenforceable, which could limit our ability to stop others from using or commercializing imetelstat or our technology and/or limit the duration of the patent protection for imetelstat and our technology. In the event that we are unsuccessful in obtaining, maintaining, enforcing and extending our patents and other intellectual property rights or having our licensors maintain the intellectual property rights we have licensed, the value of imetelstat and/or our technologies will be adversely affected, and we may not be able to further develop or potentially commercialize imetelstat.
While we have method-of-use patents that protect the use of imetelstat for the treatment of certain diseases, this type of patent does not prevent a generic competitor from making and marketing a product that is identical to imetelstat for an indication that is outside the scope of our approved use after our composition of matter patents or their patent term extensions have expired. Moreover, even if competitors do not actively promote their product for our approved indications, physicians may prescribe or use these generic products “off-label,” which would result in decreased sales for us.
Loss or impairment of our intellectual property rights related to imetelstat might further delay or halt ongoing or potential future clinical trials of imetelstat and any applications for regulatory approval, and therefore might further delay or preclude any future development or commercialization of imetelstat by us. Further, if imetelstat is approved for commercial sale, such loss of intellectual property rights could impair our ability to exclude others from commercializing products similar or identical to imetelstat and therefore result in decreased sales for us. Occurrence of any of these events would materially and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.
51
Obtaining and maintaining our patent rights depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for noncompliance with these requirements.
The U.S. Patent and Trademark Office, or the Patent Office, and various governmental patent agencies in other countries require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In addition, periodic maintenance fees, renewal fees, annuity fees and various other government fees on patents and/or patent applications will have to be paid to the Patent Office and various governmental patent agencies in other countries over the lifetime of our owned and licensed patents and/or patent applications and any patent rights we may own or license in the future. Maintaining such compliance may be impacted by the COVID-19 pandemic and the military conflict between Ukraine and Russia. Noncompliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, nonpayment of fees and failure to properly legalize and submit formal documents. In many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. There are situations, however, in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. For example, we have issued and pending patents and patent applications in Ukraine and Russia, and if we are unable to submit responses to governmental patent agencies or make payments related to such patents and patent applications in a timely manner due to the military conflict in the region, these patents or patent applications may be irrevocably lost. In such an event, potential competitors might be able to enter the market with imetelstat or similar products, and this circumstance could harm our financial condition, business and business prospects and the future of imetelstat. In addition, if we are responsible for patent prosecution and maintenance of patent rights in-licensed to us or jointly owned with us, any of the foregoing could expose us to liability to the applicable patent owner or patent co-owner.
Patent terms may be inadequate to protect our competitive position on imetelstat for an adequate amount of time.
Patents have a limited lifespan. In the U.S., the natural expiration of a patent is generally 20 years after its first effective nonprovisional filing date. Given the amount of time required for the development, testing and regulatory review of imetelstat, patents protecting imetelstat (e.g., patents claiming imetelstat and/or components thereof, methods of use, or methods of making) might expire before imetelstat is commercialized. As a result, our intellectual property may not provide us with sufficient rights to exclude others from commercializing products similar or identical to imetelstat.
In the U.S., the Hatch-Waxman Act permits one patent per approved product to receive a patent term extension of up to five years beyond its normal expiration. The length of the patent term extension is typically calculated as one half of the clinical trial period plus the entire period of time during the review of the NDA by the FDA, minus any time of delay by us during these periods. There is also a limit on the patent term extension to a term that is no greater than fourteen years from drug approval. One of our owned or licensed U.S. patents may be eligible for patent term extension under the Hatch-Waxman Act.
Similar extensions are also available in certain countries and territories outside the U.S., such as in Japan, and in Europe as Supplementary Protection Certificates, or SPCs. If we select and are granted a patent term extension on a recently filed and issued patent, we may not receive the full benefit of a possible patent term extension, if at all. We might also not be granted a patent term extension at all, because of, for example, failure to apply within the applicable period, failure to apply prior to the expiration of relevant patents or otherwise failure to satisfy any of the numerous applicable requirements. Moreover, the applicable authorities, including the FDA and the Patent Office in the U.S., and any equivalent regulatory authorities in other countries, may not agree with our assessment of whether such extensions are available, may refuse to grant extensions to our patents, or may grant more limited extensions than we request. If we fail to apply for applicable patent term extensions or adjustments, we will have a more limited time during which we can enforce our granted patent rights. Should we seek a patent term extension, we may not be granted any such patent term extension and/or the applicable time period of such patent term extension could be less than five years. Moreover, in some countries, including the U.S., the scope of protection for claims under such patent term extensions, if any, does not extend to the full scope of the claims but is limited to the product composition as approved and, for a method of treatment patent, is limited to the approved indication. Thus, for example, if we do not receive a patent term extension for our U.S. composition of matter patent for imetelstat, as
52
approved by the regulatory authorities, our U.S. composition of matter patent will expire in December 2025. If we do not have sufficient patent life to protect imetelstat, our financial results, business and business prospects, and the future of imetelstat would be materially and adversely affected, which might cause us to cease operations.
If regulatory approval of imetelstat occurs after a patent has expired in a country that does not allow interim patent term extensions, as is the case in many countries and territories including Europe, we will be unable to obtain any patent term extension of that expired patent, and the duration of our patent rights may be limited. If we do not receive marketing approval and submit a request for an SPC before our patents expire in the European Economic Area, or EEA, where we have imetelstat composition of matter patents, our imetelstat composition of matter patents will expire in September 2024. In all other countries outside the U.S. and the EEA where we have imetelstat composition of matter patents, either: (a) extension of patent term is not available, and the patents will expire in September 2024, or (b) we may not have marketing authorization in those countries in sufficient time to file an extension of patent term before our composition of matter patents expire in September 2024. If we do not have sufficient patent life to protect imetelstat, our financial results, business and business prospects, and the future of imetelstat would be materially and adversely affected, which might cause us to cease operations.
Also, there are regulations for the listing of patents in the Approved Drug Products with Therapeutic Equivalence Evaluations, or the Orange Book. If we submit a patent for listing in the Orange Book, the FDA may decline to list the patent, or a manufacturer of generic drugs may challenge the listing. If imetelstat is approved and an appropriate patent covering imetelstat is not listed in the Orange Book or is subsequently removed from the Orange Book, a manufacturer of generic drugs would not be required to provide advance notice to us of any abbreviated NDA filed with the FDA to obtain permission to sell a generic version of imetelstat. Any of the foregoing could harm our competitive position, business, financial condition, results of operations and prospects.
Changes in U.S. or international patent law or interpretations of such patent laws could diminish the value of our patents in general, thereby impairing our ability to protect our technologies and imetelstat.
The patent positions of pharmaceutical and biopharmaceutical companies, including ours, are highly uncertain and involve complex legal and technical questions. In particular, legal principles for biotechnology and pharmaceutical patents in the U.S. and in other countries are evolving, and the extent to which we will be able to obtain patent coverage to protect our technologies and imetelstat, or enforce or defend issued patents, is uncertain.
The U.S. has enacted and implemented wide-ranging patent reform legislation, including the Leahy-Smith America Invents Act, or the AIA, signed into law on September 16, 2011. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Depending on actions by Congress, the federal courts, and the Patent Office, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents or patents that we might obtain in the future. Similarly, changes in patent law and regulations in other countries or jurisdictions or changes in the governmental bodies that enforce them or changes in how the relevant governmental authority enforces patent laws or regulations may weaken our ability to obtain new patents or to enforce our existing patents or patents that we may obtain in the future. Occurrence of these events and/or significant impairment of our imetelstat patent rights would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, which might cause us to cease operations.
As a result of the AIA, in March 2013, the U.S. transitioned to a first-inventor-to-file system under which, assuming the other requirements for patentability are met, the first inventor to file a patent application is entitled to the patent. However, since the publication of discoveries in scientific or patent literature tends to lag behind actual discoveries by at least several months and sometimes several years, we are not able to be certain upon filing that the persons or entities that we name as inventors or applicants in our patent applications were the first to invent the inventions disclosed therein, or the first to file patent applications for these inventions. Thus, our ability to protect our patentable intellectual property depends, in part, on our ability to be the first to file patent applications with respect to our inventions, or inventions that were developed by our former collaboration partner and assigned to us, for the future development, commercialization and manufacture of imetelstat. As a result, if we are not the first-inventor-to-file, we may not be able to obtain patents for discoveries that we otherwise would consider patentable and that we consider to be significant to the future success of imetelstat. Delay in the filing of a patent application
53
for any purpose, including further development or refinement of an invention, may result in the risk of loss of patent rights.
Following the result of a referendum in 2016, the U.K. left the EU on January 31, 2020, commonly referred to as Brexit. The impact of the withdrawal of the U.K. from the EU will not be known for some time, which could lead to a period of uncertainty relating to our ability to obtain and maintain SPCs of imetelstat based on our U.K. patents and our ability to establish and maintain European trademarks in the U.K. In 2012, the European Patent Package, or EU Patent Package, regulations were passed with the goal of providing for a single pan-European Unitary Patent, and a new European Unified Patent Court, or UPC, for litigation of European patents. It is possible that implementation of the EU Patent Package will occur in the first half of 2023. If the EU Patent Package is ratified and in effect, all European patents, including those issued prior to ratification, would by default automatically fall under the jurisdiction of the UPC and allow for the possibility of obtaining pan-European injunctions. Under the EU Patent Package as currently proposed, once the UPC is established, patent holders are permitted to “opt out” of the UPC on a patent-by-patent basis during an initial seven year period after the EU Patent Package is ratified. Owners of traditional European patent applications who receive notice of grant after the EU Patent Package is ratified could either accept a Unitary Patent or validate the patent nationally and file an opt-out demand. The EU Patent Package may increase the uncertainties and costs surrounding the enforcement or defense of our issued European patents and pending applications. The full impact on future European patent filing strategy and the enforcement or defense of our issued European patents in member states and/or the UPC is not known.
Challenges to our owned or licensed patent rights would result in costly and time-consuming legal proceedings that could prevent or limit development of imetelstat.
Our patents or those patent rights we have licensed, including patent rights that we may seek with respect to inventions made by past or future collaborators, may be challenged through administrative or judicial proceedings, which could result in the loss of important patent rights. For example, where more than one party seeks U.S. patent protection for the same technology in patent applications that are subject to the law before the implementation of the AIA, the Patent Office may declare an interference proceeding in order to ascertain the party to which the patent should be issued. Patent interferences are typically complex, highly contested legal proceedings, subject to appeal. They are usually expensive and prolonged and can cause significant delay in the issuance of patents. Our pending patent applications or our issued patents, or those we have licensed and may license from others, may be drawn into interference proceedings or be challenged through post-grant review procedures or litigation, any of which could delay or prevent the issuance of patents, or result in the loss of issued patent rights. We may not be able to obtain from our past or future collaborators the information needed to support our patent rights which could result in the loss of important patent rights.
Under the AIA, interference proceedings between patent applications filed on or after March 16, 2013, have been replaced with other types of proceedings, including derivation proceedings. The AIA also includes post-grant review procedures subjecting U.S. patents to post-grant review procedures similar to European oppositions, such as inter partes review, or IPR, covered business method post-grant reviews and other post-grant reviews. This applies to all of our U.S. patents and those we have licensed and may license from others, even those issued before March 16, 2013. Because of a lower evidentiary standard necessary to invalidate a patent claim in Patent Office proceedings compared to the evidentiary standard in U.S. federal court, a third-party could potentially provide evidence in a Patent Office proceeding sufficient for the Patent Office to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third-party could attempt to use the Patent Office procedures to invalidate patent claims that would not have been invalidated if first challenged by the third-party as a defendant in a district court action. U.S. patents owned or licensed by us may therefore be subject to post-grant review procedures, as well as other forms of review and re-examination. In addition, the IPR process under the AIA permits any person, whether they are accused of infringing the patent at issue or not, to challenge the validity of certain patents. As a result, entities associated with hedge funds have challenged valuable pharmaceutical patents through the IPR process. Significant impairment of our imetelstat patent rights would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, which might cause us to cease operations.
Certain jurisdictions, such as Europe, New Zealand and Australia, permit oppositions to be filed against granted patents or patents proposed to be granted. Because we seek to enable potential global commercialization of
54
imetelstat, securing both proprietary protection and freedom to operate outside of the U.S. is important to our business. Opposition proceedings require significant time and costs, and if we are unsuccessful or are unable to commit these types of resources to protect our imetelstat patent rights, we could lose our patent rights and we could be prevented or limited in the development and commercialization of imetelstat.
As more groups become engaged in scientific research and product development in the areas of telomerase biology and hematologic malignancies, the risk of our patents, or patents that we have in-licensed, being challenged through patent interferences, derivation proceedings, IPRs, post-grant proceedings, oppositions, re-examinations, litigation or other means will likely increase. For example, litigation may arise as a result of our decision to enforce our patent rights against third parties. Challenges to our patents through these procedures would be extremely expensive and time-consuming, even if the outcome was favorable to us. An adverse outcome in a patent dispute could severely harm our ability to further develop or commercialize imetelstat, or could otherwise have a material adverse effect on our business, and might cause us to cease operations, by:
•causing us to lose patent rights in the relevant jurisdiction(s);
•subjecting us to litigation, or otherwise preventing us from commercializing imetelstat in the relevant jurisdiction(s);
•requiring us to obtain licenses to the disputed patents;
•forcing us to cease using the disputed technology; or
•requiring us to develop or obtain alternative technologies.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting, maintaining, defending and enforcing patents for imetelstat and our technologies in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the U.S. are less extensive than those in the U.S. The requirements for patentability may differ in certain countries, particularly in developing countries; thus, even in countries where we do pursue patent protection, there can be no assurance that any patents will issue with claims that cover imetelstat and our technologies. There can be no assurance that we will obtain or maintain patent rights inside or outside the U.S. under any future license agreements. In addition, the laws of some countries outside the U.S. do not protect intellectual property rights to the same extent as federal and state laws in the U.S. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the U.S., even in jurisdictions where we pursue patent protection, or from selling or importing products made using our inventions in and into the U.S. or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not pursued and obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the U.S. These products may compete with imetelstat and our technologies and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in jurisdictions outside the U.S. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those relating to biotechnology and pharmaceutical products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. For example, many countries outside the U.S. have compulsory licensing laws under which a patent owner must grant licenses to third parties. Proceedings to enforce our patent rights, even if obtained, in jurisdictions outside the U.S. could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. While we intend to protect our intellectual property rights in major markets for imetelstat, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market imetelstat. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we own and potentially develop in the future.
55
We may be subject to infringement claims that are costly to defend, and such claims may limit our ability to use disputed technologies and prevent us from pursuing research, development, manufacturing or commercialization of imetelstat.
The commercial success of imetelstat will depend upon our ability to research, develop, manufacture, market and sell imetelstat without infringing or otherwise violating the intellectual property and other proprietary rights of third parties. There is considerable intellectual property litigation in the biotechnology and pharmaceutical industries, and many pharmaceutical companies, including potential competitors, have substantial patent portfolios. Since we cannot be aware of all intellectual property rights potentially relating to imetelstat and its uses, we do not know with certainty that imetelstat, or the intended commercialization thereof, does not and will not infringe or otherwise violate any third-party’s intellectual property. For example, we are aware that certain third parties have or may be prosecuting patents and patent estates that may relate to imetelstat, and while we believe these patents will expire before imetelstat is able to be commercialized and/or that these patents are invalid and/or would not be infringed by the manufacture, use or sale of imetelstat, it is possible that the owner(s) of these patents will assert claims against us in the future.
In the event our technologies infringe the rights of others or require the use of discoveries and technologies controlled by third parties, we may be prevented from pursuing research, development, manufacturing or commercialization of imetelstat, or may be required to obtain unblocking licenses from such third parties, develop alternative non-infringing technologies, which we may not be able to do at an acceptable cost or on acceptable terms, or at all, or cease the development of imetelstat. If we are unable to resolve an infringement claim successfully, we could be subject to an injunction that would prevent us from potentially commercializing imetelstat and could also require us to pay substantial damages. In addition, while our past collaboration agreements have terminated, we are still subject to indemnification obligations to certain collaborators, including with respect to claims of third-party patent infringement.
In addition to infringement claims, in the future we may also be subject to other claims relating to intellectual property, such as claims that we have misappropriated the trade secrets of third parties. Provided that we are successful in continuing the development of imetelstat, we expect to see more efforts by others to obtain patents that are positioned to cover imetelstat. Our success therefore depends significantly on our ability to operate without infringing patents and the proprietary rights of others.
We may become aware of discoveries and technologies controlled by third parties that are advantageous or necessary to further develop or manufacture imetelstat. Under such circumstances, we may initiate negotiations for licenses to other technologies as the need or opportunity arises. We may not be able to obtain a license to a technology required to pursue the research, development, manufacture or commercialization of imetelstat on commercially favorable terms, or at all, or such licenses may be terminated on certain grounds, including as a result of our failure to comply with any material obligations under such licenses. If we do not obtain a necessary license or if such a license is terminated, we may need to redesign such technologies or obtain rights to alternative technologies, which may not be possible, and even if possible, could cause further delays in the development efforts for imetelstat and could increase the development and/or production costs of imetelstat. In cases where we are unable to license necessary technologies, we could be subject to litigation and prevented from pursuing research, development, manufacturing or commercialization of imetelstat, which would materially and adversely impact our business. Failure by us to obtain rights to alternative technologies or a license to any technology that may be required to pursue research, development, manufacturing or commercialization of imetelstat would further delay current and potential future clinical trials of imetelstat and any applications for regulatory approval, impair our ability to sell imetelstat, if approved, and therefore result in decreased sales of imetelstat for us. Occurrence of any of these events would materially and adversely affect our business, and might cause us to cease operations.
We are seeking registered trademarks for a commercial trade name for imetelstat in the U.S. and jurisdictions outside of the U.S. and failure to secure such registrations could adversely affect our business.
We are seeking registration of trademarks for a potential commercial trade name for imetelstat in the U.S. and other jurisdictions outside of the U.S. During trademark registration proceedings, we may receive rejections. Although we are given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the U.S. Patent and Trademark Office and in comparable agencies in many jurisdictions outside of the
56
U.S., third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. Moreover, any name we propose to use for imetelstat in the U.S. and Europe must be approved by the FDA and the EMA respectively, regardless of whether we have registered it, or applied to register it, as a trademark. Both the FDA and the EMA typically conduct a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA or the EMA rejects all of our proposed proprietary product names, we may be required to expend additional time and resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA and the EMA.
We may become involved in disputes with past or future collaborator(s) over intellectual property inventorship, ownership or use, and publications by us, or by investigators, scientific consultants, research collaborators or others. Such disputes could impair our ability to obtain patent protection or protect our proprietary information, which, in either case, could have a significant impact on our business.
Inventions discovered under research, material transfer or other collaboration agreements may become jointly owned by us and the other party to such agreements in some cases, and may be the exclusive property of either party in other cases. Under some circumstances, it may be difficult to determine who invents and owns a particular invention, or whether it is jointly owned, and disputes can arise regarding inventorship, ownership and use of those inventions. These disputes could be costly and time-consuming, and an unfavorable outcome could have a significant adverse effect on our business if we are not able to protect or license rights to these inventions. In addition, clinical trial investigators, scientific consultants and research collaborators generally have contractual rights to publish data and other proprietary information, subject to review by the trial sponsor. Publications by us, or by investigators, scientific consultants, previous employees, research collaborators or others, either with permission or in contravention of the terms of their agreements with us or with our past or future collaborators, may impair our ability to obtain patent protection or protect proprietary information which would have a material adverse effect on our business, and might cause us to cease operations.
Much of the information and know-how that is critical to our business is not patentable, and we may not be able to prevent others from obtaining this information and establishing competitive enterprises.
We rely on trade secrets to protect our proprietary technology, especially in circumstances in which we believe patent protection is not appropriate or available. We attempt to protect our proprietary technology in part by confidentiality agreements with our employees, consultants, collaborators and contractors. However, we cannot provide assurance that these agreements will not be breached, that we would have adequate remedies for any breach, or that our trade secrets will not otherwise become known or be independently discovered by competitors, any of which would harm our business significantly.
In May 2016, the Defend Trade Secrets Act of 2016, or the DTSA, was enacted, providing a federal cause of action for misappropriation of trade secrets. Under the DTSA, an employer may not collect enhanced damages or attorney fees from an employee or contractor in a trade secret dispute brought under the DTSA, unless certain advanced provisions are observed. We cannot provide assurance that our existing agreements with employees and contractors contain notice provisions that would enable us to seek enhanced damages or attorneys’ fees in the event of any dispute for misappropriation of trade secrets brought under the DTSA.
Risks Related to Managing Our Growth and Other Business Operations
We may be unable to successfully retain or recruit key personnel to support the development and potential future commercialization of imetelstat or to otherwise successfully manage our growth.
Our ability to successfully develop imetelstat in the future and to potentially commercialize imetelstat depends to a significant extent on the skills, experience and efforts of our executive officers and key members of our staff. In addition, we need to recruit, maintain, motivate and integrate additional personnel with expertise and experience in clinical science, biostatistics, clinical operations, pharmacovigilance, quality, manufacturing, regulatory affairs, medical affairs, legal affairs, market access, pricing, commercial operations, sales, and marketing, to enable us to further develop and potentially commercialize imetelstat.
57
We face intense competition for qualified individuals from numerous pharmaceutical, biopharmaceutical and biotechnology companies, as well as academic and other research institutions, and competition in our geographic regions is particularly intense. The substantial risks and uncertainties related to our development and potential commercialization of imetelstat and the risks and uncertainties regarding our future business viability could have an adverse impact on our ability to retain and recruit qualified personnel. We may also face higher than expected personnel costs in order to attract new personnel due to shortages in qualified applicants, or to maintain our current management and personnel due to the increased number of opportunities in the biotechnology sector. If we are unable to successfully retain, motivate and incentivize our existing personnel, or to attract, assimilate and retain other highly qualified personnel in the future on acceptable terms, our ability to further develop and potentially commercialize imetelstat will be impaired, and our business and the price of our common stock would be adversely impacted.
In addition, our personnel are currently performing their duties in multiple jurisdictions, and if we are unable or fail to comply with employment, tax, benefits and other laws in such jurisdictions, we may face penalties, fines or litigation. Further, if members of our management and other key personnel in critical functions across our organization are unable to perform their duties or have limited availability due to the effects of the COVID-19 pandemic, we may not be able to execute on our business strategy and/or our operations may be negatively impacted.
Our future financial performance and our ability to develop, manufacture and commercialize imetelstat will depend, in part, on our ability to effectively manage any future growth. Our management may have to divert financial and other resources, as well as devote a substantial amount of time, to managing growth activities, such as enhancing operational, financial and management processes and systems. If we do not effectively manage the expansion of our operations, we could experience weaknesses in our infrastructure and ability to comply with applicable legal and regulatory requirements and regulations, operational mistakes or shortcomings, loss of business opportunities, loss of employees and reduced productivity among remaining employees. The expansion of our operations also could lead to significant costs and could delay the execution of our business plans or disrupt our current operations. Our ineffective performance in managing any such future growth would negatively impact our business prospects.
As our operations continue to expand, we expect that we will need to manage new and additional relationships with various service providers, vendors, suppliers and other third parties, as well as a workforce in multiple countries, jurisdictions and locations. For example, in September 2021, we established a subsidiary in the U.K. to accommodate our growing workforce in that location. Our business needs and the expansion of our workforce may require us to establish additional business offices or entities in additional jurisdictions outside of the U.S., including additional subsidiaries, or to retain third parties to manage employment-related matters in new countries, jurisdictions and locations. Because the legal and regulatory requirements related to the operation and maintenance of such entities, and the employment of personnel in such countries, jurisdictions and regions is multi-national and complex, we may be unable to effectively operate and maintain such entities, or be unable to attract and retain ex-U.S. personnel, which could lead to significant costs and could delay the execution of our business plans or disrupt our current and future operations. If we fail to achieve key development goals, our abilities to grow as a company, and to further develop and potentially commercialize imetelstat, could be prevented or hindered, and our business and business prospects would be severely harmed, which might cause us to cease operations.
Notwithstanding our research and discovery efforts, we expect imetelstat to remain our sole product candidate for the foreseeable future. If we are unable to successfully develop and commercialize imetelstat, our business and business prospects would be severely harmed, which might cause us to cease operations.
Other than imetelstat, we do not currently have any other product candidates. While we recently initiated a discovery program to identify a lead compound as a potential next generation oral telomerase inhibitor, our discovery efforts are at an early stage and may not be successful. In this regard, internal discovery efforts to identify new product candidates require substantial technical, financial and human resources, and the outcome of those efforts are uncertain and unpredictable. In addition, these discovery efforts may initially show promise in identifying a potential product candidate, yet fail to yield a product candidate for clinical development for a number of reasons, including where the research methodology used may not be successful in identifying a potential product candidate, or where a potential product candidate may, on further study, be shown to have inadequate efficacy, harmful side
58
effects, suboptimal pharmaceutical profile or other characteristics suggesting that it is unlikely to be an effective product. Furthermore, in addition to research and development risks, any potential lead compounds identified during discovery may not be patentable, and therefore unsuitable for further development. Likewise, our research efforts to evaluate imetelstat in lymphoid hematologic malignancies may not be successful. In any event, notwithstanding our research and discovery efforts, we remain and expect to continue remain wholly reliant upon the development of imetelstat, our sole product candidate, for the foreseeable future. If we are unable to successfully develop and commercialize imetelstat, our business and business prospects would be severely harmed, which might cause us to cease operations. Similarly, if we are unable to discover and develop new product candidates or to develop imetelstat in lymphoid hematologic malignancies through our research and discovery efforts, our business and business prospects would be harmed.
If we seek to establish potential future collaborative arrangements for imetelstat, we may be unable to establish such collaborative arrangements on acceptable terms, or at all, and may have to delay, alter or abandon our imetelstat development and commercialization plans.
We intend to develop imetelstat broadly for hematologic malignancies, and to potentially commercialize, market and sell imetelstat in the U.S. We may seek a collaborative partner or partners, at an appropriate time, to assist us in the potential development and commercialization of imetelstat, especially outside the U.S., and to provide funding for such activities. We face significant competition in seeking appropriate collaborative partners, and these potential collaborative arrangements are complex and time consuming to negotiate, document and implement. Our ability to seek and establish potential collaborative arrangements may be impacted by the effects of the COVID-19 pandemic on our clinical trial activities and the resulting delays in reporting any results from IMpactMF, as well as the period of the patent term for our intellectual property portfolio and market exclusivity for imetelstat. We may not be able to establish collaborative arrangements on acceptable terms, or at all. In this regard, collaborative arrangements with third parties may require us to relinquish material rights, including revenue from potential commercialization, or assume material ongoing development obligations that we would have to fund or otherwise support.
In any event, we are unable to predict when, if ever, we will enter into any collaborative arrangements because of the numerous risks and uncertainties associated with establishing collaborative arrangements. Moreover, given the significant risks and uncertainties regarding the future imetelstat development program, potential collaborative partners may be reluctant to enter into new collaborative arrangements with us, or may only be willing to do so on terms that are not favorable to us. As a result, we may not be successful in finding a collaborative partner or partners on favorable terms, if at all. If we are unable to negotiate collaborative arrangements, we may have to:
•delay or curtail the additional development of imetelstat;
•further delay or abandon the potential commercialization of imetelstat outside of the U.S.;
•reduce the scope of potential future sales or marketing activities; or
•increase our expenditures and undertake development or commercialization activities at our own expense, which will require substantial additional capital than our current resources.
In order to advance the imetelstat program, including through completion of IMpactMF, IMproveMF and IMpress, or to commence, conduct and complete potential future clinical trials of imetelstat, and in the absence of potential proceeds from exercises of currently outstanding warrants and potential drawdowns under the Loan Agreement, we will require substantial additional funding. However, we cannot predict with any certainty whether and to what extent the outstanding warrants will be exercised for cash, or the timing or availability of additional funds under the Loan Agreement, if at all. In addition, if we elect to increase our expenditures to fund imetelstat development or commercialization activities outside the U.S., we will be required to substantially increase our personnel resources and we will need to obtain substantial further capital, which may not be available to us on acceptable terms, or at all. If we are unable to raise substantial additional capital if and when needed, we will not be able to further advance the imetelstat program, including through the completion of IMpactMF, IMproveMF and IMpress, as well as to conduct the clinical, regulatory and potential commercialization activities necessary to potentially bring imetelstat to market in relapsed/refractory MF and any other future indications to generate product revenues. Establishing the infrastructure necessary to further develop, commercialize, market and sell imetelstat worldwide will require substantial resources and may divert the attention of our management and key personnel and negatively impact our imetelstat development or commercialization efforts in the U.S.
59
We currently have no products approved for commercial sale, and we have not yet demonstrated an ability to obtain marketing approvals for any product candidates, which makes it difficult to assess our future viability.
We have never derived any revenue from the sales of any products. Our operations to date have been limited to organizing and staffing our company, acquiring, developing and securing our technology, undertaking non-clinical studies and clinical trials of imetelstat and past product candidates that we have subsequently discontinued, and engaging in research and development under collaboration agreements. We have not yet demonstrated an ability to obtain regulatory approvals for commercialization activities, formulate and manufacture commercial-scale products, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, for these and other reasons discussed elsewhere in these risk factors, it is difficult to predict our future success and the viability of our business and the imetelstat program.
We have business operations in the United Kingdom which exposes us to additional costs and risks.
Our business operations in the U.K. subject us to certain additional costs and risks associated with doing business outside the U.S., including:
•the increased complexity and costs inherent in managing international operations in geographically disparate locations;
•challenges of complying with diverse regulatory, financial and legal requirements, which are subject to change at any time;
•potentially adverse tax consequences, including changes in applicable tax laws and regulations;
•potentially costly trade laws, tariffs, export quotas, custom duties or other trade restrictions, and any changes to them;
•compliance with tax, employment, immigration and labor laws for employees living or traveling abroad;
•liabilities for activities of, or related to, our international operations;
•challenges inherent in efficiently managing employees in diverse geographies, including the need to adapt systems, policies, benefits and compliance programs to differing labor and other regulations;
•natural disasters, political and economic instability, including terrorism and civil and political unrest, outbreak of health epidemics, including the evolving COVID-19 pandemic, and the resulting global economic and social impacts;
•workforce uncertainty in countries where labor unrest is more common than in the U.S.; and
•compliance with the United Kingdom Bribery Act 2010, or UK Bribery Act, and similar antibribery and anticorruption laws in other jurisdictions, and the Foreign Corrupt Practices Act, including its books and records provisions and its anti-bribery provisions, including by failing to maintain accurate information and control over sales and distributors’ activities.
In addition, our international operations in the U.K. expose us to fluctuations in currency exchange rates between the British pound and the U.S. dollar. Given the volatility of currency exchange rates, there is no assurance that we will be able to effectively manage currency transaction and/or conversion risks. To date, we have not entered into derivative instruments to offset the impact of foreign exchange fluctuations, which fluctuations could have an adverse effect on our financial condition and results of operations.
We may not be able to obtain or maintain sufficient insurance on commercially reasonable terms or with adequate coverage against potential liabilities in order to protect ourselves against product liability claims or claims related to clinical trial conduct, or claims related to data protection.
Our business exposes us to potential product liability and other risks that are inherent in the testing, manufacturing and marketing of human therapeutic and diagnostic products. We may become subject to product liability claims or claims related to clinical trial conduct, including if the use of imetelstat is alleged to have injured patients, such as injuries alleged to arise from any hepatotoxicity or hemorrhagic event associated with the use of
60
imetelstat. We currently have limited clinical trial liability insurance, and we may not be able to maintain this type of insurance for any of our current or potential future clinical trials of imetelstat. In addition, this type of insurance may become too expensive for us to afford because of the highly risky and uncertain nature of clinical trials generally and the high cost of insurance for our business activities. We may be unable to obtain or maintain clinical trial insurance in all of the jurisdictions where we conduct current or potential future clinical trials. In addition, business liability, product liability and cybersecurity insurance are becoming increasingly expensive, particularly for biotechnology and pharmaceutical companies, and the pool of insurers offering insurance coverage to biotechnology and pharmaceutical companies generally is becoming smaller, making it more difficult to obtain insurance for our business activities at a reasonable price, or at all. Being unable to obtain or maintain product liability, clinical trial liability, cybersecurity or other insurance for our business activities in the future on acceptable terms or with adequate coverage against potential liabilities would have a material adverse effect on our business, and could cause us to cease our development of imetelstat.
We and certain of our officers have been named as defendants in two pending securities class action lawsuits and seven shareholder derivative lawsuits. These lawsuits, and potential similar or related lawsuits, could result in substantial damages, divert management’s time and attention from our business, and have a material adverse effect on our results of operations. These lawsuits, and any other lawsuits to which we are subject, will be costly to defend or pursue and are uncertain in their outcome.
Securities-related class action lawsuits and/or derivative lawsuits have often been brought against companies, including biotechnology and biopharmaceutical companies, that experience volatility in the market price of their securities. This risk is especially relevant for us because we often experience significant stock price volatility in connection with our product development activities.
Between January 23, 2020 and March 5, 2020, three securities class action lawsuits were filed against us and certain of our officers. One of the lawsuits was voluntarily dismissed on March 19, 2020. The other two lawsuits, filed in the U.S. District Court, or the Court, for the Northern District of California, or the Northern District, were consolidated by the Court on May 14, 2020, and on August 20, 2020, the lead plaintiffs filed a consolidated class action complaint. The consolidated class action complaint alleges violations of the Securities Exchange Act of 1934, as amended, or the Exchange Act, in connection with allegedly false and misleading statements made by us related to IMbark during the period from March 19, 2018, to September 26, 2018. The consolidated class action complaint alleges, among other things, that we violated Sections 10(b) and 20(a) of the Exchange Act and SEC Rule 10b-5 by failing to disclose facts related to the alleged failure of IMbark to meet the two primary endpoints of the trial, spleen response rate and Total Symptom Score, and that our stock price dropped when such information was disclosed. The plaintiffs in the consolidated class action complaint seek damages and interest, and an award of reasonable costs, including attorneys’ fees. On October 22, 2020, lead plaintiffs filed an amended consolidated class action complaint. We filed a motion to dismiss the amended consolidated class action complaint on November 23, 2020. On April 12, 2021, the Court granted in part and denied in part our motion to dismiss. Our answer to the amended consolidated class action complaint was filed on May 13, 2021. On September 30, 2021, lead plaintiffs filed their motion for class certification, and on April 2, 2022, the Court granted the lead plaintiffs’ motion for class certification. On September 2, 2022, the parties agreed to a settlement and entered into a Stipulation and Agreement of Settlement, or the Stipulation, which is subject to court approval. On October 13, 2022, the Court preliminarily approved the parties’ settlement, permitted notice to be distributed to the class members, and scheduled a final approval hearing for March 30, 2023. Final approval of the settlement is subject to a number of conditions and contingencies out of our control. There can be no guarantee that all of these conditions and contingencies will occur. Should a material condition or contingency to the settlement fail to occur, one or both of the parties to the settlement may exercise their right to terminate the settlement agreement.
Between April 23, 2020 and June 8, 2021, seven shareholder derivative actions were filed, naming as defendants certain of our current officers and certain current and former members of our board. Of these actions, or the Derivative Lawsuits, two were filed in the Northern District, two were filed in the Court of Chancery of the State of Delaware, two were filed in the U.S. District Court for the District of Delaware, and one was filed in the Superior Court of California for the County of San Mateo, respectively. The plaintiffs in the Derivative Lawsuits allege breach of fiduciary duty and/or violations of Section 14 of the Exchange Act, based on the same underlying facts as the consolidated class action lawsuit described above. The plaintiffs seek damages, corporate governance reforms,
61
equitable relief, restitution, and an award of reasonable costs, including attorneys’ fees. The status of the seven Derivative Lawsuits is currently as follows:
•On July 2, 2021, we filed a motion to dismiss the consolidated shareholder derivative actions filed in the Court of Chancery of the State of Delaware, or the Chancery Court Derivative Lawsuits. On September 1, 2021, the plaintiffs filed a consolidated amended complaint in the Chancery Court Derivative Lawsuits. On October 12, 2021, we filed our motion to dismiss the consolidated amended complaint. The Court of Chancery of the State of Delaware heard oral argument on the motion on February 15, 2022, and, on June 22, 2022, issued an order staying its decision on our motion to dismiss until after final resolution of the consolidated class action lawsuit described above. On December 21, 2022, the parties in the Chancery Court Derivative Lawsuits entered into a Stipulation of Settlement, or the Derivative Stipulation, that, subject to final approval by the Court of Chancery of the State of Delaware, will resolve the Chancery Court Derivative Lawsuits;
•The consolidated shareholder derivative actions filed in the U.S. District Court for the District of Delaware have been stayed pending the ruling on our motion to dismiss the Chancery Court Derivative Lawsuits. On December 21, 2022, the parties in the consolidated District of Delaware derivative actions entered into the Derivative Stipulation, that, subject to final approval by the Court of Chancery of the State of Delaware, will resolve the consolidated District of Delaware derivative actions;
•The consolidated shareholder derivative actions filed in the Northern District were initially stayed through the ruling on our motion to dismiss in the consolidated class action lawsuit described above and then subsequently were stayed through the ruling on the lead plaintiffs’ motion for class certification in the consolidated class action lawsuit. Subsequent to the grant of class certification in the consolidated class action lawsuit, on May 3, 2022, the Northern District entered an order providing plaintiffs until June 7, 2022, to file an amended complaint. On June 7, 2022, plaintiffs filed an amended shareholder derivative complaint. On July 6, 2022, the Northern District entered an order staying the consolidated shareholder derivative actions filed in the Northern District until the earlier of either a public announcement of a settlement in the consolidated class action lawsuit or a final, non-appealable judgment in the consolidated class action lawsuit. The stay has subsequently been extended on a number of occasions and the case is currently stayed through December 31, 2022. On December 21, 2022, the parties in the consolidated derivative actions in the Northern District entered into the Derivative Stipulation, that, subject to final approval by the Court of Chancery of the State of Delaware, will resolve the consolidated derivative actions in the Northern District; and
•Our motion to dismiss the shareholder derivative action pursuant to the forum selection clause in our amended and restated bylaws was filed in the Superior Court of California for the County of San Mateo on August 5, 2021. At the hearing on the motion to dismiss on November 2, 2021, the court granted our motion to dismiss and stayed the case until April 19, 2022. At the case management conference on April 19, 2022, the court continued the stay until June 14, 2022. At the case management conference on June 14, 2022, the court continued the stay until December 13, 2022. On December 13, 2022, the court dismissed the action without prejudice.
It is possible that additional lawsuits will be filed, or allegations received from stockholders, with respect to these same or other matters and also naming us and/or our officers and directors as defendants. Such lawsuits and any other related lawsuits are subject to inherent uncertainties, and the actual defense and disposition costs will depend upon many unknown factors. The outcome of such lawsuits is necessarily uncertain. We could be forced to expend significant resources in the defense of the pending lawsuits and any additional lawsuits, and we may not prevail. In addition, we have and may continue to incur substantial legal fees and costs in connection with such lawsuits. As discussed in Note 5 on Contingencies and Uncertainties in Notes to Condensed Consolidated Financial Statements of our quarterly report on Form 10-Q for the quarter ended September 30, 2022, we recorded the total settlement amount for the Stipulation on our condensed consolidated balance sheet as of September 30, 2022. We currently are not able to estimate the possible additional costs to us, if any, from these matters, and we cannot be certain how long it may take to resolve the pending lawsuits or the possible amount of any damages or legal costs that we may be required to pay. Monitoring, initiating and defending against legal actions is time-consuming for our management, is likely to be expensive and may detract from our ability to fully focus our internal resources on our business activities. We could be forced to expend significant resources in the settlement or defense of the pending
62
lawsuits and any potential future lawsuits, and we may not prevail in such lawsuits. Additionally, we may not be successful in having any such lawsuits dismissed or settled within the limits of our insurance coverage.
We have not established any reserve for any potential liability relating to the pending lawsuits or any potential future lawsuits, other than for the total settlement amount under the Stipulation. It is possible that we could, in the future, incur judgments or enter into settlements of claims for monetary damages. A decision adverse to our interests in the pending lawsuits, or in similar or related litigation, could result in the payment of substantial damages, or possibly fines, and could have a material adverse effect on our business, our stock price, cash flow, results of operations and financial condition.
We may be subject to third-party litigation, and such litigation would be costly to defend or pursue and uncertain in its outcome.
Our business may bring us into conflict with our licensees, licensors, or others with whom we have contractual or other business relationships, or with our competitors or others whose interests differ from ours. We may experience employment-related disputes as we seek to expand our personnel resources. We may become involved in performance or other disputes with the CROs we have retained to support our imetelstat clinical development activities, or with other third parties such as service providers, vendors, manufacturers, suppliers or consultants, which could result in a further delay or cessation of current and potential future clinical trials and otherwise significantly further delay our ability to develop imetelstat. If we are unable to resolve those conflicts on terms that are satisfactory to all parties, we may become involved in litigation brought by or against us.
Lawsuits are subject to inherent uncertainties, and defense and disposition costs depend upon many unknown factors. Despite the availability of insurance, we may incur substantial legal fees and costs in connection with litigation. Lawsuits could result in judgments against us that require us to pay damages, enjoin us from certain activities, or otherwise negatively affect our legal or contractual rights, which could have a significant adverse effect on our business. In addition, the inherent uncertainty of such litigation could lead to increased volatility in our stock price and a decrease in the value of our stockholders’ investment in our securities.
We are subject to U.S. and certain foreign export and import controls, sanctions, embargoes, anti-corruption laws, and anti-money laundering laws and regulations. Compliance with these legal standards could impair our ability to compete in domestic and international markets. We can face criminal liability and other serious consequences for violations, which can harm our business.
We are subject to export control and import laws and regulations, including the U.S. Export Administration Regulations, U.S. Customs regulations, various economic and trade sanctions regulations administered by the U.S. Treasury Department’s Office of Foreign Assets Controls, the U.S. Foreign Corrupt Practices Act of 1977, as amended, or FCPA, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, and other state and national anti-bribery and anti-money laundering laws in the countries in which we conduct activities. Anti-corruption laws are interpreted broadly and prohibit companies and their employees, agents, contractors, and other collaborators from authorizing, promising, offering, or providing, directly or indirectly, improper payments or anything else of value to recipients in the public or private sector. We may engage third parties to sell our products outside the United States, to conduct clinical trials, and/or to obtain necessary permits, licenses, patent registrations, and other regulatory approvals. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities, and other organizations. We can be held liable for the corrupt or other illegal activities of our employees, agents, contractors, and other collaborators, even if we do not explicitly authorize or have actual knowledge of such activities. Any violations of the laws and regulations described above may result in substantial civil and criminal fines and penalties, imprisonment, the loss of export or import privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm, and other consequences.
63
Risks Related to Competitive Factors
If competitors develop products, product candidates or technologies that are superior to or more cost-effective than imetelstat, this would significantly impact the development and commercial viability of imetelstat, which would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.
The pharmaceutical and biotechnology industries are characterized by intense and dynamic competition with rapidly advancing technologies and a strong emphasis on proprietary products. While we believe our proprietary oligonucleotide chemistry; experience with the biological mechanisms related to imetelstat, telomeres and telomerase; clinical data to date indicating potential disease-modifying activity with imetelstat treatment; and knowledge and expertise around the development of potential treatments for myeloid hematologic malignancies provide us with competitive advantages, we face competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions, governmental agencies, and public and private research institutions. Imetelstat will compete, if approved, with other products and therapies that currently exist, are being developed or will in the future be developed, some of which we may not currently be aware of.
If approved for commercial sale for the treatment of lower risk MDS, imetelstat would compete against a number of currently existing therapies, including ESAs and other hematopoietic growth factors that are indicated for anemia; immunomodulators, such as Revlimid (lenalidomide) by Celgene Corporation, a Bristol-Myers Squibb Corporation, or Celgene; hypomethylating agents, such as Vidaza (azacitidine) by Celgene and manufacturers of generic azacitidine; Dacogen (decitabine) by Otsuka America Pharmaceutical, Inc. and other manufacturers in the U.S. and Janssen in the EU; Inqovi (oral combination of decitabine and cedazuridine) by Astex Pharmaceuticals, Inc., or Astex; and Reblozyl (luspatercept), a TGF-beta inhibitor, by Acceleron Pharma, Inc., or Acceleron (acquired by Merck & Co., Inc., or Merck, in November 2021), in collaboration with Celgene.
Other therapies currently in Phase 3 development in lower risk MDS, some of which may obtain regulatory approval earlier than imetelstat include roxadustat, a hypoxia-inducible factor prolyl hydroxylase inhibitor, by FibroGen, Inc; Onureg (oral azacytidine) by Bristol-Myers Squibb Corporation, or BMS; and Hengqu (hetrombopag), an oral nonpeptide thrombopoietin receptor agonist, by Jiangsu Hengrui Pharmaceuticals Co., Ltd.
In addition, there are multiple Phase 1 and Phase 2 clinical trials of other agents being developed for lower risk MDS, including but not limited to: LB‐100, a PP2A inhibitor, by Lixte Biotechnology Holdings, Inc.; bemcentinib, an AXL inhibitor, by BerGenBio ASA; H3B‐8800, a spliceosome inhibitor, by H3 Biomedicine, Inc.; KER-050, a TGF-beta inhibitor, by Keros Therapeutics, Inc., or Keros Therapeutics; TP-0184, an inhibitor of ALK2 or ACVR1 kinase, by Sumitomo Dainippon Pharma Oncology, Inc; ilginatinib (NS-018), a JAK2 inhibitor, by NS Pharma, Inc., a U.S. subsidiary of Nippon Shinyaku Co., Ltd., or NS Pharma; RVT-2001, a SF3B1 modulator, by Roivant Sciences, Ltd.; sabatolimab (MBG453), a TIM-3 inhibitor, by Novartis AG; a lower dose of ASTX727, an oral formulation of decitabine and cedazuridine, referred to as ASTX727 LD, by Astex; ASTX030, an oral formulation of azacitidine and cedazuridine, by Astex; R289, an oral inhibitor of interleukin receptor-associated kinases 1 and 4, or IRAK1/4, by Rigel Pharmaceuticals, Inc.; a combination treatment regimen of luspatercept and lenalidomide by BMS; and HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, by BMS and etavopivat, an oral, small molecule activator of erythrocyte pyruvate kinase (PKR) by Forma Therapeutics, Inc., a Novo Nordisk Company.
If approved for commercial sale for the treatment of MF, imetelstat would compete against currently approved JAK inhibitors: Jakafi (ruxolitinib) by Incyte Corporation, or Incyte, and Inrebic (fedratinib) by Celgene, as well as a kinase inhibitor, Vonjo (pacritinib), by CTI Biopharma Corp., which was approved in February 2022 for the treatment of adults with Intermediate or High-Risk primary or secondary myelofibrosis with a platelet count below 50 × 109/L. Other treatment modalities for MF include hydroxyurea for the management of splenomegaly, leukocytosis, thrombocytosis and constitutional symptoms; splenectomy and splenic irradiation for the management of splenomegaly and co-existing cytopenias; chemotherapy and pegylated interferon. Drugs for the treatment of MF-associated anemia include ESAs, androgens, danazol, corticosteroids, thalidomide and lenalidomide.
Other therapies currently in Phase 3 development in MF, some of which may obtain regulatory approval earlier than imetelstat, include momelotinib, a JAK inhibitor, by GlaxoSmithKline plc; or momelotinib plus
64
AZD5153, a BET inhibitor by GlaxoSmithKline plc; pelabresib (CPI-0610), a BET inhibitor, by MorphoSys AG; navitoclax, a BCLXL, BCL-2 and BCLW inhibitor, by AbbVie, Inc.; and parsaclisib, a PI3K delta inhibitor, by Incyte. Other approaches for MF currently under investigation that could compete with imetelstat in the future include luspatercept; zinpentraxin alfa (RG6354, formerly PRM-151), an anti-fibrosis antibody, by F. Hoffmann-La Roche, Ltd.; LCL-161, an inhibitor of apoptosis protein (IAP), by Novartis; KRT-232, an inhibitor of MDM2, by Kartos Therapeutics, Inc.; GB2064, a LOXL2 inhibitor, by Galecto Biotech; elraglusib (9-ING-41), a glycogen synthase kinase-3 beta inhibitor, by Actuate Therapeutics, Inc.; XPOVIO (selinexor), a nuclear export inhibitor, by Karyopharm Therapeutics, Inc.; TL-895, an oral tyrosine kinase inhibitor, by Telios Pharma, Inc.; IMG-7289, a LSD1 inhibitor, by Imago Biosciences, Inc.; APG-1252, a dual BCL-2/BCL-XL inhibitor, by Ascentage Pharma; ilginatinib (NS-018), a JAK2 inhibitor by NS Pharma; DISC-0974, a monoclonal antibody against hemojuvelin (HJV) by DISC Management Inc.; KER-050 in combination with ruxolitinib, by Keros Therapeutics; CK0804, an allogeneic T-regulatory cell agent, by Cellenkos, Inc. in collaboration with Incyte; and a mutated-CALR vaccine, a peptide-based vaccine, from the Icahn School of Medicine at Mount Sinai.
Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. We anticipate increased competition in the future as new companies explore treatments for myeloid hematologic malignancies, which may significantly impact the commercial viability of imetelstat. Academic institutions, government agencies and other public and private research organizations may also conduct research, seek patent protection and establish collaborative arrangements for research, clinical development and marketing of products similar to imetelstat. These companies and institutions compete with us in recruiting and retaining qualified development and management personnel as well as in acquiring technologies complementary to the imetelstat program.
Many of our competitors, either alone or with their strategic partners, could have substantially greater financial, technical and human resources than we do and significantly greater experience in obtaining FDA and other regulatory approvals of treatments and commercializing those treatments. We believe that the commercial success of imetelstat is subject to a number of factors, including:
•product efficacy and safety;
•method of product administration;
•the timing and scope of regulatory consents;
•status of coverage and level of reimbursement;
•level of generic competition;
•patent position, including potentially dominant patent positions of others.
As a result of the foregoing, competitors may develop more commercially desirable or affordable products than imetelstat, or achieve earlier patent protection or product commercialization than we may be able to achieve with imetelstat. Competitors have developed, or are in the process of developing, technologies that are, or in the future may be, competitive to imetelstat. Some of these products may have an entirely different approach or means of accomplishing therapeutic effects similar or superior to those that may be demonstrated by imetelstat. Competitors may develop products that are safer, more effective, or less costly than imetelstat, or more convenient to administer to patients and, therefore, present a serious competitive threat to imetelstat. In addition, competitors may price their products below what we may determine to be an acceptable price for imetelstat, may receive better third-party payor coverage and/or reimbursement, or may be more cost-effective than imetelstat. Such competitive products or activities by competitors may render imetelstat obsolete, which may cause us to cease any further development or future commercialization of imetelstat, which would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.
65
To be commercially successful, imetelstat must be accepted by the healthcare community, which can be very slow to adopt or unreceptive to new technologies and products.
Even if approved for marketing, imetelstat may not achieve market acceptance, or the potential worldwide or U.S. revenue we believe may be possible, since hospitals, physicians, patients or the medical community in general may decide not to accept and utilize imetelstat. If approved for commercial sale, imetelstat will compete with a number of conventional and widely accepted drugs and therapies manufactured and marketed by major pharmaceutical companies. The degree of market acceptance of imetelstat will depend on a number of factors, including:
•the clinical indications for which imetelstat is approved, if any;
•the country and/or regions within which imetelstat is approved, if any;
•the establishment and demonstration to the medical community of the clinical efficacy and safety of imetelstat;
•the ability to demonstrate that imetelstat is superior to alternatives on the market at the time;
•the ability to establish in the medical community the potential advantages of imetelstat over alternative treatment methods, including with respect to efficacy, safety, cost or route of administration;
•the willingness of medical professionals to prescribe, and patients to use, imetelstat, or to continue to use imetelstat;
•the publication of unfavorable safety or efficacy data concerning imetelstat by third parties or us;
•restrictions on use of imetelstat in combination with other products;
•the label and promotional claims allowed by the FDA or similar international regulatory authorities for imetelstat, if any, including usage for only certain indications and any limitations or warnings about the prevalence or severity of any side effects;
•the timing of market introduction of imetelstat as well as competitive products, including sequencing of available products;
•the effectiveness of sales, marketing and distribution support for imetelstat, particularly during the remote COVID-19 environment;
•the extent to which imetelstat is approved for inclusion on formularies in hospitals and managed care organizations;
•the pricing of imetelstat, both in absolute terms and relative to alternative treatments;
•the availability of coverage and adequate reimbursement by government and third-party payors; and
•the willingness of patients to pay out-of-pocket in the absence of coverage by third-party payors, including governmental authorities.
The established use of conventional products competitive with imetelstat may limit or preclude the potential for imetelstat to receive market acceptance upon any commercialization. We may be unable to demonstrate any therapeutic or economic advantage for imetelstat compared to established or standard-of-care therapies, or newly developed therapies, for myeloid hematologic malignancies. Third-party payors may decide that any potential benefit that imetelstat may provide to clinical outcomes in myeloid hematologic malignancies is not adequate to justify the costs of treatment with imetelstat. If the healthcare community does not accept imetelstat for any of the foregoing reasons, or for any other reasons, our ability to further develop or potentially commercialize imetelstat may be negatively impacted or precluded altogether, which would seriously and adversely affect our business and business prospects.
66
If the market opportunities for imetelstat are smaller than we believe, our potential revenue may be adversely affected, and our business may suffer.
Our initial focus for imetelstat development has been on the lead indications, lower risk MDS and relapsed/refractory MF. The addressable patient populations, if imetelstat is approved in those indications, are based on our estimates. These estimates, which have been derived from a variety of sources, including scientific literature, surveys of clinics, patient foundations and market research, may prove to be incorrect. Further, new information from us or others may change the estimated incidence or prevalence of those indications.
Any regulatory approval of imetelstat would be limited to the therapeutic indications examined in our clinical trials and as determined by the FDA and similar international regulatory authorities, which would not permit us to market imetelstat for any other indications not expressly approved by those regulatory authorities. Additionally, the potentially addressable patient population for imetelstat may not ultimately be amenable to treatment with imetelstat. Even if we receive regulatory approval for imetelstat, such approval could be conditioned upon label restrictions that materially limit the addressable patient population.
Our market opportunity may also be limited by the pricing we are able to achieve for imetelstat, if approved, the quality and expiration of our intellectual property rights and licenses, and future competitor treatments that enter the market. If any of our estimates prove to be inaccurate, the market opportunities for imetelstat that we or any potential future collaborative partners develop could be significantly diminished which would have a material adverse impact on our business and business prospects.
The adoption of health policy changes and healthcare reform in the U.S. may adversely affect our business and financial results.
In the U.S. and some jurisdictions outside the U.S., there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could impact our business. For example, in response to the COVID-19 pandemic, the CARES Act was signed into law in March 2020. The CARES Act is aimed at providing emergency assistance and healthcare for individuals, families and businesses affected by the COVID-19 pandemic and generally supporting the U.S. economy. Generally, there has been increasing legislative and enforcement interest in the U.S. with respect to drug pricing, including specialty drug pricing practices, in light of the rising cost of prescription drugs and biologics. Specifically, there have been U.S. Congressional inquiries and federal and state legislative activity designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the price of drugs under Medicare, and reform government program reimbursement methodologies for drugs and biologics. In July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to Biden’s executive order, on September 9, 2021, the Department of Health and Human Services, or HHS, released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. Further, the Inflation Reduction Act of 2022, or the IRA, signed into law by President Biden on August 16, 2022, includes several provisions to lower prescription drug costs for people with Medicare and reduce drug spending by the federal government. Some of the specific provisions under the IRA that could impact us include:
•Requirement that the federal government negotiate prices for certain single-source drugs covered under Medicare Part B and D with the highest total spending, beginning in 2026; and
•Requirement that drug companies pay rebates to Medicare if prices rise faster than inflation for drugs used by Medicare beneficiaries, beginning in 2023.
Further, the Biden administration released an additional executive order on October 14, 2022, directing HHS to submit a report within 90 days on how the Center for Medicare and Medicaid Innovation can be further leveraged to test new models for lowering drug costs for Medicare and Medicaid beneficiaries. We expect that additional state and federal healthcare reform measures may be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could affect pricing for imetelstat if it is approved.
67
Moreover, the U.S. and some jurisdictions in other countries are considering or have enacted legislative and regulatory proposals to contain healthcare costs, as well as to improve quality and expand access. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the ACA was signed into law, which included a number of provisions of importance to the biopharmaceutical industry. There have been judicial and Congressional challenges to certain aspects of the ACA, and it is possible that the ACA will be subject to judicial or Congressional challenges in the future. We expect that other healthcare reform measures that may be adopted in the future may result in more rigorous coverage criteria and lower reimbursement, and additional downward pressure on the price that may be charged for imetelstat.
If future legislation were to impose direct governmental price controls and access restrictions, it could have a significant adverse impact on our business and financial results. Managed care organizations, as well as Medicaid and other government agencies, continue to seek price discounts. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biologic product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, to encourage importation from other countries and bulk purchasing. Due to the volatility in the current economic and market dynamics, we are unable to predict the impact of any unforeseen or unknown legislative, regulatory, payor or policy actions, which may include cost containment and healthcare reform measures. Such policy actions could have a material adverse impact on future worldwide sales of imetelstat, if approved.
Cost control initiatives also could decrease the price that we may receive for imetelstat in the future. If imetelstat is not considered cost-effective or adequate third-party reimbursement for the users of imetelstat cannot be obtained, then we may be unable to maintain price levels sufficient to realize an appropriate return on our investment in imetelstat. Any of these events would severely and adversely affect our financial results, business and business prospects, and might cause us to cease operations.
If we fail to comply with federal, state and international healthcare laws, including fraud and abuse, transparency, and health information privacy and security laws, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected.
Our business operations and current and future arrangements with investigators, healthcare professionals, consultants, third-party payors and customers, may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations, including federal and state fraud and abuse laws, including anti-kickback and false claims laws; data privacy and security laws, including the Health Insurance Portability and Accountability Act, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH; and transparency laws related to payments and/or other transfers of value made to physicians, other healthcare professionals and teaching hospitals. These laws may constrain the business or financial arrangements and relationships through which we conduct our operations, including how we research, market, sell and distribute imetelstat, if marketing approval is obtained. For details regarding the restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate see Item 1 “Business—Government Regulation— Fraud and Abuse, Data Privacy and Security, and Transparency Laws and Regulations” in our Annual Report on Form 10-K for the year ended December 31, 2021.
Federal and state enforcement bodies have increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. If our operations are found to be in violation of any of these or any other healthcare and privacy-related regulatory laws that may apply to us, we may be subject to significant penalties, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgement, imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, reputational harm, diminished profits and future earnings, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations. Defending against any such actions can be costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired.
68
Our employees, independent contractors, principal investigators, clinical trial sites, contract research organizations, consultants or vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, principal investigators, clinical trial sites, CROs, consultants or vendors may engage in fraudulent or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violate the FDA’s or similar international regulatory authorities’ regulations, including those laws requiring the reporting of true, complete and accurate information; manufacturing standards; healthcare fraud and abuse laws and regulations; or laws that require the true, complete and accurate reporting of financial information or data. Specifically, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements.
Activities subject to these laws also involve the improper use or misrepresentation of information obtained in the course of clinical trials or creating fraudulent data in our non-clinical studies or clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter misconduct by our employees and third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Additionally, we are subject to the risk that a person could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished potential profits and future earnings, and curtailment of our operations, any of which could adversely affect our business, financial condition, results of operations or prospects.
RISKS RELATED TO INFORMATION TECHNOLOGY SYSTEMS,
DATA SECURITY AND DATA PRIVACY
If our information technology systems or data, or those of third parties upon which we rely, are or were compromised, we could experience adverse consequences resulting from such compromise, including, but not limited to, regulatory investigations and actions; litigation; fines and penalties; a disruption of our business operations such as our clinical trials; reputational harm; loss of revenue and profits; and other adverse consequences.
In the ordinary course of our business, we (and third parties upon which we rely) may collect, receive, store, process, use, transfer, make accessible, protect, secure, dispose of, transmit, disclose, or otherwise process (commonly known as processing) proprietary, confidential, and sensitive data, including personal data (such as health-related data and participant study related data), intellectual property, and trade secrets (collectively, sensitive information). In addition, we rely on third-party service providers to establish and maintain appropriate information technology and data security protections over the information technology systems they provide us to operate our critical business systems, including cloud-based infrastructure and systems, employee email, and data storage and management systems. However, except for contractual duties and obligations, we have limited ability to control their safeguards and actions related to such matters, and these third parties may not have adequate information security measures in place. We may share or receive sensitive information with or from third parties. In particular, the COVID-19 pandemic has caused us to modify our business and information technology practices, including that most of our employees continue to work remotely. Additionally, the prevalent use of mobile devices that access our sensitive information increases the risk of breaches.
Our information technology systems, including in our remote work environment as a result of the COVID-19 pandemic, and those of the third parties upon which we rely, are potentially vulnerable to evolving threats. These threats are prevalent, continue to increase, and come from a variety of sources such as “hackers,” threat or internal bad actors, personnel (such as through theft, error or misuse), sophisticated nation states and nation-state-supported actors. These threats include, but are not limited to, social-engineering attacks, malicious code or malware, unauthorized intrusions, denial-of-service attacks, personnel misconduct or errors, ransomware attacks, supply-chain
69
attacks, software bugs, computer viruses, server malfunctions, software, hardware or data center failures, loss of data or other information technology assets, natural disasters, terrorism, war, and telecommunication and electrical failures. In particular, ransomware attacks are becoming increasingly prevalent and severe and can lead to significant interruptions in operations, loss of data and income, reputational harm, and diversion of funds. If we were to experience such an attack, extortion payments might alleviate the negative impact of a ransomware attack, but we might be unwilling or unable to make such payments due to, for example, applicable laws or regulations prohibiting such payments. Similarly, supply-chain attacks and attacks on clinical trial sites as well as regulatory and health authorities have increased in frequency and severity, and we cannot guarantee that third parties and infrastructure in our supply chain or our third-party partners’ supply chains, or of clinical trial sites and regulatory and health authorities, have not been compromised or that they do not contain exploitable defects or bugs that could result in a breach of or disruption to our information technology systems (including those related to imetelstat) or the third-party information technology systems that support us and the services provided to us. These threats may result in unauthorized, unlawful or accidental loss, corruption, access, modification, destruction, alteration, acquisition or disclosure of sensitive information, such as clinical trial data or information, intellectual property, proprietary business data and personal data. The costs to us to attempt to protect against such breaches could be significant, including potentially requiring us to modify our business (including non-clinical and clinical trial activities), and while we have implemented security measures designed to protect our information technology systems and to identify and remediate vulnerabilities, such measures may not be successful. We may be unable in the future to detect vulnerabilities in our information technology systems because such threats and techniques change frequently, are sophisticated in nature, and may not be detected until after a security incident has occurred.
If we or third parties upon which we rely experience or are perceived to have experienced a breach, we may experience adverse consequences. These consequences may include: government enforcement actions (for example, investigations, fines, penalties, audits, and inspections), interruptions in our operations, including disruption of our imetelstat development program, interruptions or restrictions on processing sensitive data (which could result in delays in obtaining, or our inability to obtain, regulatory approvals and significantly increase our costs to recover or reproduce the data), reputational harm, litigation (including class action claims), indemnification obligations, negative publicity, monetary fund diversions, financial loss, and other harms. In addition, such a breach may require notification of the breach to relevant stakeholders. Such disclosures are costly, and the disclosure or the failure to comply with such requirements could lead to adverse consequences.
Many of our contracts with relevant stakeholders include obligations relating to the safeguard of sensitive information, and a breach could lead to claims against us by such stakeholders. There can be no assurance that the limitations of liability in our contracts would be enforceable or adequate or would otherwise protect us from liabilities, damages, or claims relating to our data privacy and security obligations. In addition, failure to maintain effective internal accounting controls related to data security breaches and cybersecurity in general could impact our ability to produce timely and accurate financial statements and could subject us to regulatory scrutiny.
We are subject to stringent and changing obligations related to data privacy and security. Our actual or perceived failure to comply with such obligations could lead to regulatory investigations and actions; litigation; fines and penalties; disruptions to our business operations; reputational harm; loss of revenue and profits; and other adverse business impacts.
In the ordinary course of business, we process personal data and other sensitive data, including proprietary and confidential business data, trade secrets, intellectual property, data we collect about trial participants in connection with clinical trials, and sensitive third-party data. We are therefore subject to or affected by numerous data privacy and security obligations, such as various federal, state, local and foreign laws, regulations, guidances, industry standards, external and internal privacy and security policies, contracts, and other obligations governing the processing of personal data by us and on our behalf. These obligations may change, are subject to differing interpretations and may be inconsistent among jurisdictions or conflict. The global data protection landscape is rapidly evolving, and implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future. This evolution may create uncertainty in our business; affect us or our collaborators’, service providers’ and contractors’ ability to operate in certain jurisdictions or to collect, store, transfer, use and share personal data; necessitate the acceptance of more onerous obligations in our contracts; result in liability; or impose additional costs on us. The cost of compliance with these laws, regulations and guidances is high and is likely to increase in the future. These obligations may necessitate changes to our information technologies, systems, and
70
practices and to those of any third parties that process personal data on our behalf. In addition, these obligations may require us to change our business model.
Outside the U.S., an increasing number of laws, regulations, and industry standards apply to data privacy and security. For example, the European Union’s General Data Protection Regulation (GDPR) (EU) 2016/679, or the EU GDPR, imposes strict requirements on the processing of personal data. Under the EU GDPR, government regulators may impose temporary or definitive bans on data processing, as well as fines of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater.
Certain jurisdictions, including the EU and the U.K., have enacted data localization laws and cross-border personal data transfer laws, which would make it more difficult to transfer information across jurisdictions (such as transferring or receiving personal data that originates in the EU or in jurisdictions in other countries). Existing mechanisms that facilitate cross-border personal data transfers may change or be invalidated. The processing of sensitive personal data, such as physical health conditions, is a topic of active interest among regulators from other countries. As we expand into countries and jurisdictions outside the U.S., we may be subject to additional laws and regulations that may affect how we conduct business. For example, absent appropriate safeguards or other circumstances, the EU GDPR generally restricts the transfer of personal data to countries outside of the European Economic Area, or EEA, that the European Commission considers do not provide an adequate level of data privacy and security, such as the U.S.
To address this, the European Commission has released a set of “Standard Contractual Clauses” that are designed to be a valid mechanism by which entities can transfer personal data out of the EEA to jurisdictions that the European Commission has not found to provide an adequate level of protection. Currently, these Standard Contractual Clauses are a valid mechanism to transfer personal data outside of the EEA. The Standard Contractual Clauses, however, require parties that rely upon that legal mechanism to comply with additional obligations, such as conducting transfer impact assessments to determine whether additional security measures are necessary to protect the at-issue personal data. Moreover, due to potential legal challenges, there exists some uncertainty regarding whether the Standard Contractual Clauses will remain a valid mechanism for transfers of personal data out of the EEA. Addressing this uncertainty, on October 7, 2022, President Biden signed an executive order implementing the EU-U.S. Data Privacy Framework, to facilitate the cross-border transfer of personal information from the EU to the U.S. in compliance with the EU GDPR. The European Commission will conduct a determination as to whether the U.S. commitments meet GDPR's adequacy standard for the transfer of personal data, a process anticipated to take about six months. Once ratified by the European Commission, participation in the revised framework will require that companies self-certify their adherence with the U.S. Department of Commerce. In addition, the U.K. similarly restricts personal data transfers outside of those jurisdictions to countries such as the U.S. that do not provide an adequate level of personal data protection.
If we cannot implement a valid compliance mechanism for cross-border data transfers, we may face increased exposure to regulatory actions, substantial fines, and injunctions against processing or transferring personal data from Europe or elsewhere. The GDPR has increased our responsibility and potential liability in relation to personal data that we process or control compared to prior EU law, including in clinical trials, which could divert management’s attention and increase our cost of doing business; limit our ability to collaborate with parties that are subject to European and other data privacy and security laws; or require us to increase our personal data processing capabilities and infrastructure in Europe and/or elsewhere at significant expense.
Likewise, we expect that there will continue to be new proposed laws, regulations and industry standards relating to privacy and data protection in the U.S. For example, HIPAA, as amended by HITECH, imposes specific requirements relating to the privacy, security, and transmission of individually identifiable health data. Additionally, the California Consumer Privacy Act of 2018, or CCPA, imposes obligations on businesses to which it applies. These obligations include, but are not limited to, providing specific disclosures in privacy notices and affording California residents certain rights related to their personal data. The CCPA allows for statutory fines for noncompliance (up to $7,500 per violation). While the CCPA contains limited exceptions for clinical trial data, the CCPA’s implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future. It is anticipated that the California Privacy Rights Act of 2020, or CPRA, will expand the CCPA. For example, the CPRA establishes a new California Privacy Protection Agency to implement and enforce the CPRA, which could increase the risk of an enforcement action. Other states have also enacted data privacy laws. For
71
example, Virginia passed the Consumer Data Protection Act, and Colorado passed the Colorado Privacy Act, both of which differ from the CPRA and become effective in 2023. If we become subject to new data privacy laws, at the state level, the risk of enforcement action against us could increase because we may become subject to additional obligations, and the number of individuals or entities that can initiate actions against us may increase (including individuals, via a private right of action, and state actors).
Although we endeavor to comply with all applicable data privacy and security obligations, we may at times fail to do so or may be perceived to have failed to do so. Moreover, despite our efforts, we may not be successful in achieving compliance if our personnel or third parties upon whom we rely fail to comply with such obligations, which could negatively impact our business operations and compliance posture. For example, any failure by a third-party processor, including our clinical trial sites, to comply with applicable law, regulations, or contractual obligations could result in adverse effects, including inability to operate our business and proceedings against us by governmental entities or others. If we fail, or are perceived to have failed, to address or comply with data privacy and security obligations, we could face significant consequences. These consequences may include, but are not limited to, government enforcement actions (e.g., investigations, fines, penalties, audits, inspections, and similar activities); litigation (including class-related claims); additional reporting requirements and/or oversight; bans on processing personal data; orders to destroy or not use personal data; and imprisonment of company officials. Any of these events could have a material adverse effect on our reputation, business, or financial condition, including but not limited to: interruptions or stoppages in our business operations (including, as relevant, our clinical trials if any); inability to process personal data or to operate in certain jurisdictions; limited ability to develop or commercialize imetelstat; expenditure of time and resources to defend any claim or inquiry; adverse publicity; or revision or restructuring of our operations. Moreover, clinical trial participants or research subjects about whom we or our vendors obtain information, as well as the providers who share this information with us, may contractually limit our ability to use and disclose the information.
General Risk FACTORs
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our CROs, suppliers, and other contractors and consultants, could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, public health pandemics or epidemics (including, for example, the ongoing COVID-19 pandemic), civil or political unrest or military conflicts around the world (such as the current military conflict between Ukraine and Russia), terrorism, insurrection or war, and other natural or man-made disasters or business interruptions. Furthermore, other events, such as the armed conflict between Russia and Ukraine, could lead to sanctions, embargoes, supply shortages, regional instability, geopolitical shifts, cyberattacks, other retaliatory actions, and adverse effects on macroeconomic conditions, currency exchange rates, and financial markets, which could adversely impact our operations and financial results, as well as those of third parties with whom we conduct business. There is a risk that one or more of our CROs, suppliers, and other contractors and consultants might not survive an economic downturn. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
Our ability to develop and potentially commercialize imetelstat could be disrupted if our operations or those of our CROs and other contractors or consultants are affected by geopolitical events, man-made or natural disasters or other business interruptions. Our corporate headquarters are located in California near major earthquake faults and fire zones. The ultimate impact on us, our CROs, contractors and consultants, and our general infrastructure of being located near major earthquake faults and fire zones and being consolidated in certain geographical areas is unknown, but our operations and financial condition could suffer in the event of a major earthquake, fire or other natural disaster.
Changes in tax laws or regulations that are applied adversely to us or our customers may have a material adverse effect on our business, cash flow, financial condition or results of operations.
New income, sales, use, excise or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could affect the tax treatment of our domestic and foreign sales and earnings. Any new taxes could
72
adversely affect our domestic and international business operations and our business and financial condition. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, the Tax Act, as modified by the CARES Act, significantly revised the Code, and recently enacted federal tax legislation made additional changes. Future guidance from the U.S. Internal Revenue Service and other tax authorities with respect to such legislation may adversely affect us, and certain aspects of such legislation could be repealed or modified in the future, which could have an adverse effect on us. For example, the recently enacted Inflation Reduction Act of 2022, or the Inflation Reduction Act, includes provisions that will impact the U.S. federal income taxation of corporations, including imposing a minimum tax on the book income of certain large corporations and an excise tax on certain corporate stock repurchases that would be imposed on the corporation repurchasing such stock. It is uncertain if and to what extent various states will conform to the Tax Act, the CARES Act, the Inflation Reduction Act or any newly enacted federal tax legislation. Changes in corporate tax rates, the realization of net deferred tax assets relating to our U.S. operations, the taxation of earnings from other countries, and the deductibility of expenses under the Tax Act or future tax reform legislation could have a material impact on the value of our deferred tax assets, could result in significant one-time charges in the current or future taxable years, and could increase our future U.S. tax expense.
Failure to achieve and maintain effective internal controls in accordance with Section 404 of the Sarbanes-Oxley Act of 2002 could have a material adverse effect on our business and stock price.
Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, requires that we establish and maintain an adequate internal control structure and procedures for financial reporting. Our Annual Reports on Form 10-K must contain an annual assessment by management of the effectiveness of our internal control over financial reporting and must include disclosure of any material weaknesses in internal control over financial reporting that we have identified. In the past, our independent registered public accounting firm provided an opinion annually on the effectiveness of our internal control over financial reporting. As a smaller reporting company, we are no longer subject to this requirement.
The requirements of Section 404 are ongoing and also apply to future years. We expect that our internal control over financial reporting will continue to evolve as our business develops. Although we are committed to continue to improve our internal control processes and we will continue to diligently and vigorously review our internal control over financial reporting in order to ensure compliance with Section 404 requirements, any control system, regardless of how well designed, operated and evaluated, can provide only reasonable, not absolute, assurance that its objectives will be met. Therefore, we cannot assure you that material weaknesses or significant deficiencies will not exist or otherwise be discovered in the future, particularly in light of our increased reliance on personnel working remotely as a result of the COVID-19 pandemic. If material weaknesses or other significant deficiencies occur, such weaknesses or deficiencies could result in misstatements of our results of operations, restatements of our financial statements, a decline in our stock price, or other material adverse effects on our business, reputation, results of operations, financial condition or liquidity.
Risks Related to Our Common Stock and Financial Reporting
Historically, our stock price has been extremely volatile and your investment may suffer a decline in value.
Historically, our stock price has been extremely volatile. Between January 1, 2013 and December 31, 2022, our stock has traded as high as $7.79 per share and as low as $0.75 per share. Between January 1, 2022 and December 31, 2022, the price has ranged between a high of $3.06 per share and a low of $0.99 per share. The significant market price fluctuations of our common stock have been due to and may in the future be influenced by a variety of factors, including:
•announcements regarding our ability to timely submit applications for regulatory approvals of imetelstat in lower risk MDS;
•regulatory approval or non-approval of imetelstat, specific label indications for or restrictions, warnings or limitations in its use, or delays in the regulatory review process;
73
•announcements regarding the research and development of imetelstat, or adverse efficacy or safety results of, further delays in the commencement, enrollment or conduct of, discontinuation of, or further modifications or refinements to any current clinical trials of imetelstat, including IMerge Phase 3 or IMpactMF, IMproveMF and IMpress, or for potential future clinical trials of imetelstat, for any reason, or our inability, for any reason, to successfully continue the development of imetelstat;
•obtaining substantial additional capital on commercially reasonable terms, necessary to further advance the imetelstat program, including through the completion of IMpactMF, IMproveMF and IMpress, as well as to conduct the clinical, regulatory and potential commercialization activities necessary to potentially bring imetelstat to market in relapsed/refractory MF and other future indications;
•timeliness of preliminary, interim or final clinical trial data expected to be reported with respect to current or potential future clinical trials of imetelstat, and investor perceptions thereof;
•not receiving timely regulatory clearances or approvals in any jurisdiction, whether within or outside of the U.S. to continue clinical development of imetelstat in relapsed/refractory MF, lower risk MDS or any additional myeloid hematologic malignancies in a timely manner or at all;
•changes in laws or regulations applicable to imetelstat, including but not limited to clinical trial requirements for approval;
•announcements regarding the safety of imetelstat and partial or full clinical holds placed on the imetelstat INDs by the FDA or similar international regulatory authorities, or other regulatory developments related to imetelstat;
•the successful completion of any clinical trials, regulatory approval and commercialization of imetelstat for one or more label expansion indications;
•announcements of technological innovations, new commercial products, or clinical progress or lack thereof by us, potential future collaborative partners or our competitors;
•adverse developments concerning our manufacturers, including our inability to obtain adequate product supply for imetelstat or inability to do so at acceptable prices;
•our failure to launch and commercialize imetelstat on the timelines anticipated, or at all;
•the size and growth of our lead indications, lower risk MDS and relapsed/refractory MF;
•announcements concerning imetelstat proprietary rights;
•the terms and timing of any future collaboration agreements for the development and potential commercialization of imetelstat that we may establish;
•announcements of significant acquisitions, strategic partnerships, collaborations, joint ventures or capital commitments by us or our competitors;
•our ability to acquire or in-license new product candidates to grow our pipeline;
•the demand in the market for our common stock;
•fluctuations in our operating results;
•increased or continuing operating losses;
•general domestic and international market conditions or market conditions relating to the biopharmaceutical and pharmaceutical industries, especially given the volatility caused by macroeconomic conditions like the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession;
•perceptions of the biotechnology and pharmaceutical industry by the public, legislature, regulators and the investment community;
•comments by securities analysts or other third parties, including blogs, articles and other media;
74
•our failure to meet the estimates and projections of the investment community or that we may otherwise provide to the public;
•publication of research reports about us or our industry, or positive or negative recommendations or withdrawal of research coverage by securities analysts;
•large stockholders increasing or exiting their position in our common stock or an increase in the short interest in our common stock;
•changes in the market valuations of similar companies;
•announcements of or developments concerning pending and potential future litigation;
•disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies;
•actions instituted by activist shareholders or others;
•the issuance of common stock to partners, vendors or investors to raise additional capital;
•other events or factors that are beyond our control; and
•the occurrence of any other risks and uncertainties discussed under the heading “Risk Factors.”
Stock prices and trading volumes for many biopharmaceutical companies fluctuate widely for a number of reasons, including factors which may be unrelated to their businesses or results of operations, such as media coverage, statements made on message boards and social media forums, legislative and regulatory measures and the activities of various interest groups or organizations. In addition to the risk factors described in this section, overall market volatility, as well as general domestic or international economic, market and political conditions, including those resulting from the effects of macroeconomic conditions like the COVID-19 pandemic, civil or political unrest or military conflicts around the world, such as the military conflict between Ukraine and Russia, inflation, rising interest rates or prospects of a recession, could materially and adversely affect the market price of our common stock and the return on your investment in our securities.
In addition, as further discussed in the Risk Factor above entitled “We and certain of our officers have been named as defendants in two pending securities class action lawsuits and seven shareholder derivative lawsuits. These lawsuits, and potential similar or related lawsuits, could result in substantial damages, divert management’s time and attention from our business, and have a material adverse effect on our results of operations. These lawsuits, and any other lawsuits to which we are subject, will be costly to defend or pursue and are uncertain in their outcome”, we and one of our officers have been named as defendants in two securities class action lawsuits. In addition, certain of our current officers and current and former board members have been named as defendants in the Derivative Lawsuits filed in the Northern District, the Court of Chancery of the State of Delaware, the District Court for the District of Delaware and the California Superior Court for the County of San Mateo, respectively. Such lawsuits have often been instituted against companies, including us, whose securities have experienced periods of volatility in market price. The pending lawsuits and any lawsuits brought against us in the future could result in substantial costs, which would hurt our financial condition and results of operations and divert management’s attention and resources, which could result in delays of IMpactMF, IMproveMF and IMpress, and/or could preclude or delay potential future clinical trials, or could preclude or delay commercialization efforts.
The sale of a substantial number of shares may adversely affect the market price of our common stock.
As of September 30, 2022, we had 675,000,000 shares of common stock authorized for issuance and 381,214,326 shares of common stock outstanding. Additionally, as of September 30, 2022, we had reserved 200,777,933 shares of our common stock for future issuance pursuant to our stock option and equity incentive plans and outstanding warrants.
Future sales of our common stock or the perception that such sales could occur, or the issuance of common stock to fund our operations and imetelstat development, including pursuant to the 2020 Sales Agreement with B. Riley Securities or upon the potential exercise of currently outstanding warrants, could cause immediate dilution and adversely affect the market price of our common stock. The sale or issuance of our securities, as well as the existence of outstanding stock options and shares of common stock reserved for issuance under our stock option and
75
equity incentive plans and outstanding warrants, also may adversely affect the terms upon which we are able to obtain additional capital through the sale of equity securities, which could negatively affect the market price of our common stock and the return on stockholders’ investment.
Our undesignated preferred stock may inhibit potential acquisition bids; this may adversely affect the market price of our common stock and the voting rights of holders of our common stock.
Our certificate of incorporation provides our board of directors with the authority to issue up to 3,000,000 shares of undesignated preferred stock and to determine or alter the rights, preferences, privileges and restrictions granted to or imported upon these shares without further vote or action by our stockholders. The issuance of shares of preferred stock may delay or prevent a change in control transaction without further action by our stockholders. As a result, the market price of our common stock may be adversely affected.
In addition, if in the future, we issue preferred stock that has preference over our common stock with respect to the payment of dividends or upon our liquidation, dissolution or winding up, or if we issue preferred stock with voting rights that dilute the voting power of our common stock, the rights of holders of our common stock or the market price of our common stock could be adversely affected.
Provisions in our charter, bylaws and Delaware law may inhibit potential acquisition bids for us, which may prevent holders of our common stock from benefiting from what they believe may be the positive aspects of acquisitions and takeovers.
Provisions of our charter documents and bylaws may make it substantially more difficult for a third-party to acquire control of us and may prevent changes in our management, including provisions that:
•prevent stockholders from taking actions by written consent;
•divide the board of directors into separate classes with terms of office that are structured to prevent all of the directors from being elected in any one year; and
•set forth procedures for nominating directors and submitting proposals for consideration at stockholders’ meetings.
Provisions of Delaware law may also inhibit potential acquisition bids for us or prevent us from engaging in business combinations. In addition, we have individual severance agreements with our executive officers and a company-wide severance plan, either of which could require a potential acquirer to pay a higher price. Either collectively or individually, these provisions may prevent holders of our common stock from benefiting from what they may believe are the positive aspects of acquisitions and takeovers, including the potential realization of a higher rate of return on their investment from these types of transactions.
Our amended and restated bylaws provide that the Court of Chancery of the State of Delaware will be the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, or employees.
Our amended and restated bylaws provide that, unless we consent to the selection of an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for:
•any derivative action or proceeding brought on our behalf;
•any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees to us or to our stockholders;
•any action asserting a claim arising pursuant to any provision of the General Corporation Law of the State of Delaware, our certificate of incorporation, or our bylaws; or
76
•any action asserting a claim governed by the internal affairs doctrine.
While the exclusive forum provisions in our bylaws do not apply to lawsuits brought to enforce a duty or liability created by the Exchange Act or the Securities Act of 1933, as amended, or any claim for which the federal courts have exclusive jurisdiction, these provisions may nonetheless limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our current or former directors, officers, or other employees, which may discourage such lawsuits against us and our current or former directors, officers, and other employees. Alternatively, if a court were to find the exclusive forum provisions contained in our bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could have a material and adverse impact on our business and our financial condition.
We do not intend to pay cash dividends on our common stock in the foreseeable future.
We do not anticipate paying cash dividends on our common stock in the foreseeable future. Any payment of cash dividends will depend upon our financial condition, results of operations, capital requirements and other factors, and will be at the discretion of our board of directors. In addition, the terms of our Loan Agreement prevent us from paying dividends and any future debt agreements may continue to preclude us from paying dividends. As a result, capital appreciation, if any, of our common stock will be the sole source of gain for our stockholders for the foreseeable future.
77