Use these links to rapidly review the document
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K/A
(Amendment No. 1)
| (Mark One) | |
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE FISCAL YEAR ENDED DECEMBER 27, 2006 | |
OR | |
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE TRANSITION PERIOD FROM TO | |
Commission File Number 0-24320
TAPESTRY PHARMACEUTICALS, INC.
(Exact name of Registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) | 84-1187753 (I.R.S. Employer Identification No.) |
4840 Pearl East Circle, Suite 300W
Boulder, Colorado 80301
(Address of principal executive office, including zip code)
(303) 516-8500
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange of Which Registered | |
|---|---|---|
| Common Stock, par value $0.0075 per share | NASDAQ Capital Market | |
| Preferred Share Purchase Rights | NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer (See the definition of "accelerated filer" and "large accelerated filer" in Rule 12b-2 of the Exchange Act).
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer ý |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant was $13,599,512 as of June 28, 2006 (the last business day of the registrant's second fiscal quarter in 2006). For purposes of making this calculation, the registrant has defined affiliates as including Special Situations Fund III, L.P. ("SSF") and all directors and executive officers and related parties thereto. SSF and certain other stockholders are beneficial owners of more than 10% of the registrant's outstanding common stock. However, only SSF is included as an affiliate because only it has a contractual right to designate persons for election to the board of directors. SSF has not exercised that right.
As of March 28, 2007, the registrant had 16,374,395 shares of Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
None.
2
This Amendment No. 1 to the Annual Report on Form 10-K (this "Amendment") amends the previously filed Annual Report of Tapestry Pharmaceuticals, Inc. ("we," "Tapestry" or the "Company") on Form 10-K for the year ended December 27, 2006 (the "Original Annual Report") to incorporate the information included herein and conform the information to the disclosure contained in our Registration Statement on Form S-1 as filed with the Securities and Exchange Commission on April 23, 2007. Other than as set forth herein, we have not undertaken to update any information provided in the Original Annual Report. This amendment does not reflect events occurring after the date of the Original Annual Report nor does it modify or update the disclosure contained in the Original Annual Report other than as reflected in the Registration Statement.
Item 1
Special Note Regarding Forward Looking Statements
This report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. These forward-looking statements include, but are not limited to, statements concerning our plans to continue development of TPI 287 and our current product candidates; develop an oral formulation of TPI 287; commence Phase II clinical trials of TPI 287 in the first half of 2007 and thereafter; increase clinical spending; seek regulatory approvals; address certain markets; and raise additional capital, as well as our statement as to the adequacy of our capital to fund our operations and capital expenditures for the next 12 months. In some cases, these statements may be identified by terminology such as "may," "will," "should," "expect," "plan," "anticipate," "believe," "estimate," "predict," "potential," or "continue" or the negative of such terms and other comparable terminology. These statements involve known and unknown risks and uncertainties that may cause our or our industry's results, levels of activity, performance or achievements to be materially different from those expressed or implied by the forward-looking statements. Factors that may cause or contribute to such differences include, among other things, those discussed under the captions "Business," "Risk Factors" and "Management's Discussion and Analysis of Financial Condition and Results of Operations." Forward- looking statements not specifically described above also may be found in these and other sections of this report.
3
Overview
We are a biopharmaceutical company focused on the development of proprietary therapies for the treatment of cancer. Our core capabilities are deriving and developing drug candidates from natural products. We are currently devoting substantially all of our efforts to the development of TPI 287, a proprietary next generation taxane for the treatment of multiple cancer indications. Taxanes comprise a class of drugs derived from natural products that are used in the treatment of various forms of cancer.
We recently completed dosing in a Phase I clinical trial of TPI 287 and expect to complete dosing in another Phase I clinical trial during the second half of 2007. In addition, we recently commenced enrollment in a Phase II clinical trial of TPI 287 in patients with an advanced form of prostate cancer called hormone refractory prostate cancer, or HRPC. This is our first of several planned Phase II clinical trials of TPI 287 in multiple cancer indications. In our clinical trials to date, we have evaluated intravenous, or IV, formulations of TPI 287. We plan to use an IV formulation in our currently planned Phase II clinical trials. We also are developing an oral formulation of TPI 287 and plan to initiate a Phase Ib/II pharmacokinetic clinical trial of TPI 287 in the summer of 2007. In this clinical trial, we plan to orally administer an IV formulation of TPI 287 to evaluate the drug's activity and oral bioavailability in humans. We hold all worldwide commercial rights for TPI 287.
In preclinical studies, TPI 287 has exhibited the following differentiating characteristics that we believe could lead to advantages compared to currently marketed taxanes:
- •
- higher activity in tumors that have developed resistance to taxane therapy following treatment with chemotherapy drugs;
- •
- higher activity in particular tumor types that are innately resistant to taxane therapy;
- •
- the ability to cross the blood-brain barrier;
- •
- oral bioavailability and activity; and
- •
- activity when used in combination with therapeutic agents that are not currently approved for use in combination with taxanes.
According to the American Cancer Society, cancer is the second leading cause of death in the United States, exceeded only by heart disease, and accounts for approximately one in every four deaths. Approximately 560,000 Americans are expected to die of cancer in 2007. The National Institutes of Health estimated the direct medical cost of cancer was $78.2 billion in 2006. Taxanes are well established as part of the standard of care for many tumor types and are expected to continue to be so for the foreseeable future. Some of the best-known taxanes are Bristol-Myers Squibb's Taxol® (paclitaxel), Sanofi-Aventis' Taxotere® (docetaxel) and Abraxis BioScience's Abraxane® (paclitaxel), which had combined worldwide sales in 2006 of $2.9 billion, based on the average exchange rate during 2006.
Scientific Background
Cancer is characterized by rapid, uncontrolled cell division resulting in the growth of an abnormal mass of cells generally referred to as a malignant tumor. Cancerous tumors can arise in almost any tissue or organ of the body, and cancer cells, if not eradicated, can spread, or metastasize, throughout the body. As these tumors grow, they cause damage to the surrounding tissue and organs and typically result in death if left untreated. Cancer is believed to occur as a result of a number of hereditary and environmental factors. These factors cause genetic changes that affect the ability of cells to regulate their growth and differentiation.
4
The most common methods of treating patients with cancer are surgery, radiation and drug therapy. A cancer patient often receives treatment with a combination of these methods. Surgery and radiation therapy are particularly effective in patients in which the disease is localized. Physicians generally use systemic anticancer drugs, referred to as chemotherapy drugs, in situations in which the cancer has metastasized. The goal of chemotherapy is to damage and kill cancer cells or to interfere with the molecular and cellular processes that control the development, growth and survival of cancer cells. In many cases, chemotherapy entails the administration of several different drugs in combination.
The most common approach to pharmacological cancer treatment has been to develop drugs, referred to as cytotoxic drug therapies, that kill rapidly proliferating cancer cells. These drugs have been very effective in the treatment of some cancers. However, cytotoxic drug therapies typically act in an indiscriminate manner, killing healthy as well as cancerous cells. As a result, side effects can be wide-ranging, including immune system compromise, nausea, vomiting, diarrhea, sores of the mouth and digestive tract, hair loss and damage to peripheral nerve tissue and heart tissue. Due to their toxicity, many cytotoxic drugs have a narrow dose range above which the toxicity causes unacceptable or even fatal levels of damage and below which the drugs are not effective in eradicating cancer cells. The key challenge with cytotoxic therapies is to develop therapies that kill the cancer cells while limiting damage to healthy cells. In contrast, many recently approved cancer therapies are cytostatic, meaning that they are intended to stop cancerous tumors from growing as opposed to killing cancer cells. However, these cytostatic therapies generally are only approved for use in combination with traditional cytotoxic therapies.
Taxanes are part of a larger class of cytotoxic compounds known as microtubule spindle poisons. These compounds work by altering or attacking a structural component of cells known as microtubules, which play an important role in the cellular life cycle. In normal cells, microtubules act as an intracellular transport system. During cell division, microtubules must be stabilized to ensure a proper alignment of chromosomes before a cell may separate into two cells. Taxanes bind to microtubules of actively dividing cells and prevent cells from completing the normal cell division process. This arrests the cellular life cycle and eventually triggers a process known as apoptosis in which affected cells self-destruct.
In addition to typical toxic side effects exhibited by cytotoxic drug therapies, existing taxanes are susceptible to a biological process called multiple drug resistance, or MDR. MDR generally refers to the resistance of tumor cells to several unrelated drugs after an initial exposure to a single chemotherapy drug. A particular family of proteins that bind to foreign molecules and transport them outside the cell is believed to be responsible for MDR. The majority of patients undergoing chemotherapy with currently approved taxanes develop resistance to therapy. Although there are many possible mechanisms for such resistance, MDR is believed to be the predominant cause of developed resistance in patients being treated with taxanes and other cytotoxic therapies.
Historically, most chemotherapy drugs have only been available through IV administration. In particular, all of the currently approved taxanes are available only through IV administration. However, a number of recently approved cancer drugs can be administered orally, such as Hoffmann-La Roche's Xeloda®, which is approved for the treatment of specific types of colon, rectal and breast cancer, and Novartis' Gleevec®, which is approved for the treatment of leukemia and specific types of gastrointestinal tumors. We believe that an effective, orally administered taxane could allow more effective delivery of therapy through optimization of drug level exposure over time. In addition, an oral formulation could be more convenient for patients and would allow more convenient combination with other orally administered drugs, provide more flexibility in dosing regimens and result in lower overall cost of administration than an IV therapy.
5
Our Strategy
Our goal is to become a leading biopharmaceutical company in the development and commercialization of proprietary therapies for the treatment of cancer. Key elements of our strategy to achieve this goal are to:
- •
- Advance the clinical development of an IV formulation of TPI 287 for multiple cancer indications. Our ongoing and planned Phase II clinical trials are designed to evaluate whether TPI 287 is effective in tumors that have developed resistance to taxane therapy and in tumor types that are innately resistant to taxane therapy. We recently commenced enrollment in a Phase II clinical trial in patients with HRPC and also plan to conduct Phase II clinical trials in patients with gliomas, pancreatic cancer and other MDR tumors.
- •
- Pursue clinical development of the oral formulation of TPI 287. In our preclinical studies, TPI 287 exhibited significant oral bioavailability and activity in animal models. Accordingly, we are developing an oral formulation of TPI 287 and plan to initiate a Phase Ib/II pharmacokinetic clinical trial of TPI 287 in the summer of 2007. In this clinical trial, we plan to orally administer an IV formulation of TPI 287 to evaluate the drug's activity and oral bioavailability in humans.
- •
- Establish strategic alliances with leading pharmaceutical and biotechnology companies. We currently plan to retain development and commercialization rights to TPI 287 in the United States and the European Union until we complete our ongoing and currently planned Phase II clinical trials of an IV formulation. After the completion of these Phase II clinical trials, we plan to establish strategic alliances with leading pharmaceutical and biotechnology companies in the United States and the European Union. In addition, we may seek collaborators in markets in the Asia-Pacific region before the completion of our Phase II clinical trials if the initial results of our Phase II clinical trials of an IV formulation and the results of our Phase Ib/II pharmacokinetic clinical trial of orally administered TPI 287 are favorable. We expect that any strategic alliance that we enter into will provide us with access to the therapeutic area expertise and development and commercialization resources of our collaborators, as well as augment our financial resources.
- •
- Strategically in-license or acquire attractive development candidates. We intend to expand our product pipeline through strategically in-licensing or acquiring product candidates for the treatment of cancer. We believe that this approach will enable us to avoid much of the cost, time and risk of drug discovery while focusing critical resources on the development of promising drug candidates.
- •
- Establish specialized sales and marketing capabilities. We may retain U.S. marketing and sales rights or co-promotion rights for our product candidates for which we receive regulatory approval in markets in which we believe it is possible to gain access through a focused, specialized sales force. For markets in which we believe a large sales force is required to gain access, and for markets outside the United States, we plan to commercialize products for which we obtain regulatory approval through a variety of collaboration and distribution arrangements with other pharmaceutical and biotechnology companies.
TPI 287
We are developing TPI 287 as a proprietary therapy for multiple cancer indications. We recently completed dosing in a Phase I clinical trial of TPI 287 and expect to complete dosing in a second
6
Phase I clinical trial during the second half of 2007. The following table summarizes key information about our additional ongoing and planned clinical development efforts relating to TPI 287.
| INDICATION | STATUS | TIMING | |||
|---|---|---|---|---|---|
| IV Administration | |||||
Hormone refractory prostate cancer | Phase II clinical trial in process | Completion expected in the first half of 2008 | |||
Glioblastoma multiforme | Phase II clinical trial of TPI 287 as a single agent planned | Expected to begin in the summer of 2007 | |||
Multiple primary CNS tumors | Phase Ib/II clinical trial of TPI 287 in combination with temozolomide planned | Expected to begin in the second half of 2007 | |||
Pancreatic cancer | Phase II clinical trial planned | Expected to begin in the second half of 2007 | |||
Other drug resistant tumors, possibly including lung and breast cancers | Phase II clinical trial in development | To be determined | |||
Oral Administration | |||||
Multiple cancer indications | Phase Ib/II pharmacokinetic clinical trial of oral administration of an IV formulation planned | Expected to begin in the summer of 2007 | |||
Undetermined tumor type | Phase I/II dose escalating clinical trial of an oral formulation in development | To be determined | |||
Structure and Characteristics
We have designed TPI 287 in a manner that makes the molecule more lipophilic, or fat soluble, than other taxanes. In addition, a component of the molecule is designed to be hydrophilic, or water soluble. We believe these structural characteristics contribute to a higher binding affinity to an important element of microtubules known as tubulin. The solubility profile of TPI 287 also enhances its ability to diffuse across cell membranes and to bind to lipophilic components inside of cells, such as tubulin. We believe these properties may help to improve the distribution of TPI 287 throughout body tissues compared to other taxanes and reduce the ability of MDR proteins to bind to TPI 287. A reduced binding affinity with MDR proteins may be associated with activity in tumors that overexpress MDR proteins. We believe these structural characteristics also contribute to TPI 287's observed oral bioavailability and ability to cross the blood-brain barrier in preclinical studies.
Preclinical Testing
We have completed an initial series of tests in an artificial environment outside of a living organism, referred to asin vitro tests. Thesein vitro tests were designed to assess the ability of TPI 287 to inhibit tumor growth. In addition, we have completed tests within living organisms, referred to asin
7
vivo tests. Thesein vivo tests were designed to investigate whether TPI 287 had differentiating characteristics that could lead to advantages compared to currently marketed therapies.
In Vitro Studies
In preclinical testing, TPI 287 inhibited tumor cell growth in a number ofin vitro cell lines, including cell lines that either have developed resistance to taxane therapy or that are innately resistant to taxane therapy.
The table below sets forth the amount of TPI 287 required inin vitro tests in the tumor types listed to inhibit the growth of half of the tumor cells of the applicable tumor type, which is referred to in the oncology industry as IC50, together with the IC50 of a comparator drug that is commonly used to treat each of these tumor types. A tumor listed as "resistant" indicates that the tumor cell line has demonstrated MDR and is resistant to taxane therapy. The last column in the table lists a cytotoxicity index that is calculated by dividing the IC50 of the comparator drug by the IC50 of TPI 287. The cytotoxicity index is a measure of the relative potency of TPI 287 and the comparator drug in inhibiting cell growth in the applicable tumor type. A cytotoxicity index of between 0.1 and 10 generally indicates comparable potency between TPI 287 and the comparator drug.
In onein vitro study, we measured the IC50 of TPI 287 and compared the results to the observed IC50 for paclitaxel in two well characterized human breast cancer cell lines. One of these breast cancer cell lines is sensitive to taxanes and the other is resistant to taxanes. As shown in the first two rows of the table below, in these studies, TPI 287 was approximately 200 times more potent than paclitaxel in the resistant cell line and had comparable potency to paclitaxel in the taxane sensitive cell line.
| Tumor Cell Line Origin | TPI 287 IC50 | Comparator | Comparator IC50 | Cytotoxicity Index | ||||
|---|---|---|---|---|---|---|---|---|
| Breast (resistant) | 0.0100 | Paclitaxel | 2.0 | 200 | ||||
| Breast | 0.0006 | Paclitaxel | 0.0003 | 0.5 | ||||
Colon (resistant) | 0.0095 | Irinotecan | 0.05 | 5.3 | ||||
Liver (resistant) | 0.1000 | Oxaliplatin | 5.5 | 55 | ||||
Lung | 0.0046 | Docetaxel | 0.0072 | 1.6 | ||||
Neuroblastoma (resistant) | 0.0034 | Vincristine | 13.4 | 3900 | ||||
| Neuroblastoma | 0.0049 | Vincristine | 17.7 | 3600 | ||||
Pancreas | 0.0763 | Gemcytabine | 0.124 | 1.6 | ||||
Prostate (resistant) | 0.0050 | Cisplatin | 1.4 | 280 | ||||
Uterine (resistant) | 0.0100 | Doxorubicin | 0.8 | 80 | ||||
| Uterine | 0.0053 | Doxorubicin | 0.2 | 38 |
In Vivo Studies
We have conducted the following types ofin vivo studies:
- •
- subcutaneous xenograft studies in which we implanted human tumor cells into the flanks of mice that were specially bred not to reject foreign tissue, referred to as nude mice;
- •
- orthotopic xenograft studies in which we implanted human tumor cells into the same tissue of nude mice in which the tumor originated in humans; and
- •
- studies in which we measured drug absorption and distribution in target tissues in rodents, referred to as pharmacokinetic studies.
8
In thesein vivo studies, TPI 287 exhibited differentiating characteristics that we believe could lead to advantages compared to currently marketed taxanes.
Activity in Resistant Tumors
In subcutaneous mouse xenograft studies, TPI 287 reduced the rate of tumor growth in both taxane resistant and taxane sensitive cancer cell lines. In these studies, TPI 287 exhibited activity in tumor types that have developed resistance to taxane therapy following treatment with chemotherapy drugs or are innately resistant to taxane therapy, including cell lines derived from breast, colon and neuroblastoma tumors.
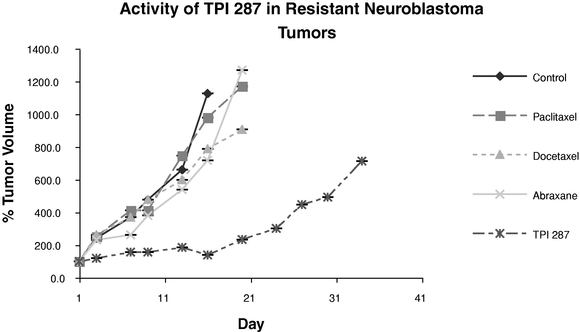
In one study, we used subcutaneous mouse xenograft models to measure the activity of TPI 287 in comparison to select cytotoxic agents in resistant neuroblastoma tumors. We monitored these tumors until they reached a predetermined size. We then treated the mice with equally toxic doses of TPI 287, docetaxel, paclitaxel and Abraxane® and compared the results to a control group. We evaluated the ability of the test agents to inhibit tumor growth by measuring the tumor size over time.
The chart below shows the average percentage change in tumor volume for each group of eight mice over the duration of the study. On average, TPI 287 was substantially more active in this study than the other cytotoxic agents, although variability among test subjects was significant at some measurement points. The shallower curve of the group treated with TPI 287 indicates greater inhibition of tumor growth than in any of the groups treated with comparative agents or the control group.
Crossing the Blood-Brain Barrier
In pharmacokinetic studies performed on mice and rats, TPI 287 exhibited the ability to cross the blood-brain barrier. In these studies, we administered TPI 287 intravenously in a number of test subjects. We then measured the concentration of the drug in the blood and the brain at predetermined times over the following 96 hours.
9
We found that shortly after IV administration of TPI 287, the highest concentration of the drug in the brain was more than twice as high as the highest concentration of TPI 287 in the blood plasma. We also found that TPI 287's total exposure in the brain over the 96 hours of the study was nearly four times its total exposure in a similar volume of blood plasma over the same period of time. Finally, we observed that, on average, TPI 287 remained present in the brains of the test subjects nearly two and a half times longer than it remained present in blood plasma.
We believe these characteristics indicate the potential to use TPI 287 to treat primary and metastatic tumors of the central nervous system, or CNS.
Oral Bioavailability and Activity
In a subcutaneous mouse xenograft study, we observed oral bioavailability and activity of TPI 287 in glioblastome multiforme, or GBM, an aggressive primary brain tumor. In this study, we measured the activity of TPI 287 on GBM tumors. We monitored these tumors until they reached a predetermined size. We then treated the mice with an IV formulation of TPI 287 administered orally and compared the results to a control group. We evaluated the ability of TPI 287 to inhibit tumor growth by measuring the tumor size over time. The chart below shows the average percentage change in tumor volume in each group over the duration of the study.
On average, TPI 287 was substantially more active in this study than the control group. The shallower curve of the group treated with TPI 287 indicates inhibition of tumor growth relative to the control group. This suggests that orally administered TPI 287 was bioavailable and active in this tumor model. No taxane is currently approved either for oral administration or for the treatment of GBM.

Activity in Combination Therapy
In orthotopic mouse xenograft studies, TPI 287 exhibited activity when used in combination with temozolomide, an approved treatment for GBM. In these studies, we implanted human GBM tumor cells into the brains of nude mice. We then treated the mice with either TPI 287, temozolomide or a combination of TPI 287 and temozolomide and compared the results to a control group. Rather than measuring tumor growth or inhibition, we monitored the survival rate of the mice over time.
10
In one study, 100% of the untreated control mice died within 14 days, while 100% of the mice treated with a combination of TPI 287 and temozolomide survived for 45 days. At 100 days, 10% of the mice treated with temozolomide alone had survived, while 30% of the mice treated with a combination of TPI 287 and temozolomide had survived. In this study, on average, mice treated with temozolomide alone survived 380% longer than the control group, while mice treated with a combination of TPI 287 and temozolomide survived 650% longer than the control group.
The table below sets forth the following results from this study:
- •
- the median survival time for mice in each group, which is a measure of the time at which 50% of the mice in each group had died;
- •
- the median survival benefit for mice in each group, which is a measure of the percentage difference between the median survival time for each group compared to the control group; and
- •
- the percentage of mice in each group that had survived at 100 days.
| | Control | TPI 287 | Temozolomide | Temozolomide plus TPI 287 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Median survival time (days) | 10 | 14.5 | 48 | 75 | |||||
| Median survival benefit (% increase) | — | 45 | % | 380 | % | 650 | % | ||
| 100 day survival rate | 0 | % | 0 | % | 10 | % | 30 | % |
A p-value is a measure of statistical significance and indicates the probability that the result obtained in a study is due to chance rather than a causal relationship between measures. A p-value of 0.05 or less generally indicates statistical significance. In this study, the median survival benefit of the mice treated with a combination of TPI 287 and temozolomide compared to the median survival benefit of the mice treated with temozolomide alone had a p-value of 0.046, suggesting that there is a less than five in 100 chance that the statistical observation was caused by random chance.
Toxicology
In preparation for clinical development of TPI 287, we conducted toxicology studies in mice, rats and dogs to determine the maximum tolerated dose, which we refer to as MTD, for each animal species. In each of these studies, we observed side effects similar to those of other cytotoxic drugs. The initial dose in our Phase I clinical trials was determined by taking approximately one-tenth of the MTD in dogs, or seven milligrams per square meter of surface area of the subject.
Phase I Clinical Development
We recently completed dosing in a Phase I clinical trial of TPI 287 and expect to complete dosing in another Phase I clinical trial during the second half of 2007. In our Phase I clinical trials, we have administered TPI 287 in patients with a wide range of tumor types, including tumors of the breast, prostate, kidney, colon, rectum and cervix, as well as cancer of the bone. We also expect to begin clinical trials on oral delivery of TPI 287 in the summer of 2007.
IV Administration
In May 2005, we began our first Phase I trial, which is ongoing. This trial is an open label, multi-center, dose-escalation trial designed to determine the MTD and evaluate the safety profile of TPI 287 administered intravenously once per week for three weeks in a four week treatment cycle. This trial is also designed to measure pharmacokinetic and pharmacodynamic profiles of TPI 287 in patients with tumors that have recurred or stopped responding to therapy. We continue treatment until the patient reaches a dose-limiting toxicity, demonstrates progressive disease, develops significant other illnesses or
11
is taken off the study based on a decision by the patient or the clinical investigator. This trial is nearing completion and our preliminary data show side effects for TPI 287 similar to those of other cytotoxic drugs. We expect to present the data from this trial at a conference in June 2007.
In January 2006, we began our second Phase I trial, with an alternative dosing schedule from our initial Phase I trial. This trial was an open label, multi-center, dose-escalation trial designed to determine the MTD and evaluate the safety profile of TPI 287 administered intravenously once every three weeks. The trial was also designed to determine the pharmacokinetic and pharmacodynamic profiles of TPI 287, as well as explore the anti-tumor activity, in patients with tumors that have recurred or stopped responding to therapy. We continued treatment until the patient reached a dose-limiting toxicity, demonstrated progressive disease, developed significant other illnesses or was taken off the study based on a decision by the patient or the clinical investigator. We expect to present the data from this trial at a conference in June 2007.
In addition, we plan to begin a Phase Ib/II clinical trial to evaluate the safety of IV administration of TPI 287 in combination with temozolomide in patients with primary CNS tumors. We expect this study to begin in the second half of 2007.
Oral Administration
We believe that an effective, orally administered taxane could allow more effective delivery of therapy through optimization of drug level exposure over time. In addition, an oral formulation could be more convenient for patients and would allow more convenient combination with other orally administered drugs, provide more flexibility in dosing regimens and result in lower overall cost of administration than an IV therapy.
TPI 287 has exhibited significant oral bioavailability and activity in animal models and we plan to begin to investigate its bioavailability and activity in humans. We expect to begin a Phase Ib/II pharmacokinetic clinical trial using an IV formulation of TPI 287 administered orally in the summer of 2007. We intend to compare the pharmacokinetics in patients treated with an IV formulation of TPI 287 orally and intravenously. This will be a randomized trial that will involve approximately eight patients per group in a patient population with multiple tumor types that have not responded to standard therapy. Each patient will either receive an initial oral dose of TPI 287 followed one week later by an IV dose or an initial IV dose of TPI 287 followed one week later by an oral dose. After an IV dose of TPI 287 in the third week of the trial, all patients will continue to receive TPI 287 once per week for three weeks in a four week treatment cycle.
In this study, we will measure drug absorption and distribution in target tissues. We expect to begin this trial in the summer of 2007.
Phase II Clinical Development
Overview. Our Phase II clinical development program will focus on generating data relating to what we believe to be the differentiating characteristics of TPI 287 that could lead to advantages compared to currently marketed taxanes. We first intend to investigate the activity of TPI 287 in patients with tumors that are resistant to taxane therapy. We recently commenced enrollment in a Phase II clinical trial in patients with HRPC and also plan to conduct Phase II clinical trials in patients with primary cancers of the CNS that are generally protected by the blood-brain barrier, including GBM, and in patients with pancreatic cancer. We currently expect to begin each of these clinical trials before the end of 2007. We are also planning additional trials in other resistant tumor types, which may include breast and lung tumors.
As a result of ongoing data collection, evaluation of new information and input from our investigators, advisory boards and key opinion leaders, we may terminate or modify the design of any of
12
our proposed Phase II clinical trials. In addition, we may consider conducting additional Phase II clinical trials in other tumor types as a result of clinical information from ongoing or completed clinical trials.
- •
- Disease overview and current treatments. Prostate cancer forms in the prostate, a gland in the male reproductive system located near the bladder. Prostate cancer primarily afflicts men over the age of 40. While the five year survival rate of patients with prostate cancer is nearly 100%, the five year survival rate of patients with advanced stage prostate cancer is approximately 33%. If diagnosed early, prostate cancer can be completely cured in most men through surgery or radiation treatment. In later stages, prostate cancer is typically treated with hormones to block stimulation of cancer growth. However, if the disease progresses to HRPC despite hormone therapy, more aggressive therapy, such as chemotherapy, may be necessary. The taxane docetaxel is currently the most widely prescribed chemotherapy drug for the treatment of advanced stage prostate cancer, although the anthracycline mitoxantrone is also approved for this indication. These drugs are most commonly administered together with prednisone, a corticosteroid.
- •
- Market opportunity. Prostate cancer is the most common non-skin cancer among men in the United States, with over 1.0 million men currently diagnosed with the disease, and the second leading cause of cancer deaths in men in the United States. The American Cancer Society estimates that 218,890 men will be diagnosed with prostate cancer and that 27,050 men will die from the disease in the United States in 2007.
- •
- Trial Design. The primary objective of this clinical trial is to evaluate the response rate to TPI 287 in patients with HRPC who have had prior treatment with a taxane. The trial is designed as an open-label, multi-center trial with two groups of approximately 40 patients each. One group is comprised of patients who have progressive prostate cancer but who have previously received taxane therapy for over three months. We assume these patients have taxane-sensitive tumors that have developed taxane resistance. The second group is comprised of patients who have progressive prostate cancer but who have previously received taxane therapy for three months or less. We assume these patients have not responded to prior taxane therapy. In this trial, we are administering TPI 287 once every three weeks. We are measuring the response of the patients to treatment with TPI 287. We define a response as either a reduction in the level of the patients' prostate-specific antigen, or PSA, or by a protocol referred to as the "Response Evaluation Criteria in Solid Tumors" for patients with disease that can be measured by radiographic studies, such as a CT scan, or through magnetic resonance imaging. We are conducting this trial in two stages. In each group, we will evaluate the first 22 patients to determine if there is a beneficial effect in some patients before deciding whether to enroll the next 18 patients. We are also evaluating each patient's time to progression while on TPI 287, the duration of patient response to TPI 287 in those patients who have a response and the effect of TPI 287 on pain intensity in patients who reported pain at the beginning of the study. We will continue to monitor any adverse events related to the use of TPI 287 in this target patient population. In addition, we will evaluate original tumor biopsies and blood samples for genetic and protein markers to help to understand patients' response to TPI 287. We expect to complete this trial in the first half of 2008.
Prostate Cancer
Glioblastoma Multiforme and Other Primary CNS Tumors
- •
- Disease overview and current treatments. Gliomas are tumors that form in the supportive tissue of the brain. GBM is the most common type of primary CNS tumor and is among the most difficult to treat. The five year survival rate of patients with GBM tumors is approximately 3%. Treatment of GBM usually involves surgery, which is typically followed by a combination of
13
- •
- Market opportunity and current treatments. The American Cancer Society estimates that 20,500 people will be diagnosed with some form of primary CNS tumor and that 12,740 people will die from these tumors in the United States in 2007. Approximately 20% of all primary CNS tumors are GBM tumors.
- •
- Trial Design. We intend to conduct both a Phase II clinical trial in which we will administer TPI 287 alone and a Phase Ib/II clinical trial of TPI 287 in combination with temozolomide.
- •
- Phase II. The primary objective of this trial is to evaluate the six-month progression free survival, or PFS, of patients with GBM that has recurred or progressed following prior radiation and temozolomide therapy. PFS refers to the length of time between the start of study treatment and documentation of tumor progression. The trial is designed as an open-label, multi-center trial involving up to 50 patients. In this trial, we will administer TPI 287 once per week for three weeks in a four week treatment cycle. We will also evaluate any response to treatment with TPI 287. We will continue to evaluate adverse events related to the use of TPI 287 in this target patient population. We expect to begin this trial in the summer of 2007.
- •
- Phase Ib/II. The primary objective of this trial is to determine the MTD of the combination of TPI 287 and temozolomide in the treatment of patients with primary CNS tumors, which may include GBM, anaplastic astrocytomas and oligodendrogliomas that have recurred or progressed following prior temozolamide therapy. The trial is designed as an open-label, multi-center dose escalation trial involving up to 60 patients. We will also monitor the pharmacokinetics of both TPI 287 and temozolamide and evaluate any possible drug interactions by pharmacokinetic changes or safety effects. We will also evaluate any responses to treatment with TPI 287 in combination with temozolomide. We expect to begin this trial in the second half of 2007.
radiation and chemotherapy with temozolomide. Additional chemotherapy strategies may include use of carmustine. Agents that interfere with the development of blood vessels rather than tumors themselves, or anti-angiogenesis agents, have shown promise in combination with chemotherapy in studies conducted by third parties.
Pancreatic Cancer
- •
- Disease overview and current treatments. Pancreatic cancer is a tumor that forms in the pancreas, a small, thin gland located behind other organs in the abdomen. Pancreatic cancer is difficult to detect and diagnose due to a lack of noticeable symptoms of the disease in its early stages. The one year survival rate of patients with all stages of pancreatic cancer is approximately 26% and the five year survival rate is approximately 5%. For newly diagnosed patients who are eligible for surgical treatment of the disease, surgery is typically followed by a combination of radiation and chemotherapy. Patients whose disease has become inoperable because of extensive local spread of the disease receive a combination of radiation and chemotherapy. Advanced stage patients with more widespread or metastatic disease are generally treated with chemotherapy alone. In patients with inoperable cancer, therapy is only palliative, which means that it only slows the disease's progress rather than providing a cure. Gemcitabine is approved as the initial chemotherapy for patients with inoperable pancreatic cancer. Other drugs are only marginally effective, including fluoropyrimidine and Tarceva® (erlotinib).
- •
- Market opportunity. The American Cancer Society estimates that 37,170 people will be diagnosed with pancreatic cancer and that 33,370 people will die from the disease in the United States in 2007. Pancreatic cancer is the fourth leading cause of cancer deaths in the United States.
14
- •
- Trial Design. The primary objective of this trial is to evaluate the PFS in patients with pancreatic cancer on therapy with either TPI 287 or fluoropyrimidine. Patients must have tumors that have recurred or progressed following prior therapy using gemcitabine either alone or in combination with other agents. The trial is designed as a randomized, multi-center trial with two groups of approximately 100 patients each. We will administer TPI 287 once per week for three weeks in a four week treatment cycle. If we observe a specified number of deaths or instances of progression of the disease in patients, we will discontinue the trial if there is no statistically significant difference in PFS between TPI 287 and fluoropyrimidine. We will also evaluate any response to treatment with TPI 287 and will compare the adverse events in each target patient population. We expect to begin this trial in the second half of 2007.
Intellectual Property
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, technology and know-how, to operate without infringing the proprietary rights of others and to prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. We also rely on trade secrets, know-how, continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position.
As of April 18, 2007, our patent rights relating to TPI 287 consist of six U.S. patent applications, as well as foreign counterparts to some of these patent applications. These applications disclose the composition of matter of TPI 287, the formulation of TPI 287 and the method of use of TPI 287 in the treatment of cancer. U.S. patents generally have a term of 20 years from the filing date of the earliest non-provisional application.
Our patent position, like that of many biopharmaceutical companies, is generally uncertain and involves complex legal and factual questions. Our ability to maintain and solidify our proprietary position for our technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We cannot assure you that any of our patent applications will result in the issuance of any patents. We may rely, in some circumstances, on trade secrets to protect our technology. Despite our efforts to protect our proprietary technology and processes, trade secrets can be difficult to protect.
In March 2007, we received an office action from the U.S. Patent and Trademark Office relating to one of our patent applications that makes claims to the composition of matter of TPI 287. In this action, the patent examiner rejected all of the pending claims of our patent application. We are preparing a response to this office action, which we expect to file before September 2007. We cannot predict the ultimate outcome of this office action. If the arguments in our response are not successful, some or all of the original claims in our patent application may have to be narrowed or may not issue at all. If this occurs, this patent application may not result in an issued patent or the patent, if issued, may not provide significant protection or competitive advantage with regard to TPI 287.
For a full discussion of the risks relating to our intellectual property, see "Risk Factors—Risks Relating to Our Intellectual Property."
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, approval, manufacturing, labeling, post-approval monitoring and reporting, packaging, promotion, storage, advertising, distribution, marketing and export and import of pharmaceutical products such as those we are developing. The process of obtaining regulatory approvals and the subsequent compliance with all
15
applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
United States Government Regulation
In the United States, the information that must be submitted to the U.S. Food and Drug Administration, or FDA, in order to obtain approval to market a new drug varies depending upon whether the drug is a new product the safety and efficacy of which have not previously been demonstrated in humans or a drug with active ingredients and certain other properties that are the same as those of a previously approved drug. A company with a product with safety and efficacy that have not previously been demonstrated in humans will be required to file a new drug application, or NDA.
The NDA Approval Process. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act and implementing regulations. Failure to comply with the applicable FDA requirements at any time during the product development process, approval process or after approval may result in administrative or judicial sanctions. These sanctions could include the FDA's imposition of a clinical hold on trials, refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
The steps required before a drug product may be marketed in the United States generally include:
- •
- completion of preclinical laboratory tests, animal studies and formulation studies under the FDA's good laboratory practices regulations;
- •
- submission to the FDA of an investigational new drug application, or IND, for human clinical testing, which must become effective before human clinical trials may begin and which must include independent Institutional Review Board, or IRB, approval at each clinical site before the trials may be initiated;
- •
- performance of adequate and well-controlled clinical trials in accordance with good clinical practices to establish the safety and efficacy of the product for each indication for which approval is sought;
- •
- submission to the FDA of an NDA;
- •
- review by an FDA Advisory Committee, if applicable;
- •
- satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practices, or cGMP, to assure that the facility or facilities, methods and controls are adequate to assure the product's identity, strength, quality, purity and stability; and
- •
- FDA review and potential approval of the NDA.
Preclinical tests include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and pharmacological activity of the product. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. Preclinical testing will often continue after the IND is submitted. The IND must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about the conduct of the trials as outlined in the IND. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. If these concerns or questions are unresolved, the FDA may choose to not allow the
16
clinical trials to begin. In other words, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each site at which the study is conducted must approve the protocol and any amendments. All research subjects must provide their informed consent in writing.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined. Phase I trials usually involve the initial introduction of the investigational drug into healthy volunteers to evaluate the product's safety, dosage tolerance and pharmacokinetics and, if possible, to gain an early indication of its effectiveness.
Phase II trials usually involve controlled trials in a limited patient population afflicted with a specified disease to:
- •
- evaluate dosage tolerance and optimal dosage;
- •
- identify possible adverse effects and safety risks; and
- •
- provide a preliminary evaluation of the efficacy of the drug for the specific indications being studied.
Phase II trials are sometimes denoted by companies as Phase IIa or Phase IIb trials. Phase IIa trials typically represent the first human clinical trial of a drug candidate in a smaller patient population and are designed to provide earlier information on drug safety and efficacy. Phase IIb trials typically involve larger numbers of patients or longer durations of therapy and may involve comparison with placebo, standard treatments or other active comparators.
Phase III trials usually further evaluate clinical efficacy and test further for safety in an expanded patient population. Phase III trials usually involve comparison with placebo, standard treatments or other active comparators. These trials are intended to establish the overall risk-benefit profile of the product and provide an adequate basis for physician labeling. Phase III trials are usually larger, more time consuming, more complex and more costly than Phase I and Phase II trials.
Phase I, Phase II and Phase III testing may not be completed successfully within any specified period, if at all. Furthermore, the FDA, an IRB or we may suspend or terminate clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of research if the research is not being conducted in accordance with the IRB's requirements or if the research has been associated with unexpected serious harm to patients.
Assuming successful completion of the required clinical testing, the results of the preclinical studies and of the clinical trials, together with other detailed information, including information on the chemistry, manufacture and composition of the product, are submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. In most cases, the NDA must be accompanied by a substantial user fee. The FDA will initially review the NDA for completeness before it accepts the NDA for filing. The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency's threshold determination that the NDA is sufficiently complete to permit a substantive review. After the NDA is accepted for filing, the FDA reviews the NDA to determine, among other things, whether a product is safe and effective for its intended use and whether the product is being manufactured in accordance with cGMP to assure and preserve the product's identity, strength, quality, purity and stability.
17
Under the Pediatric Research Equity Act of 2003, or PREA, NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted.
Before approving an NDA, the FDA will inspect the facility or the facilities at which the product is manufactured. The FDA will not approve the product unless cGMP compliance is satisfactory. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable, it may refuse to approve the NDA or issue a not approvable letter. A not approvable letter outlines the deficiencies in the submission and often requires additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. Data obtained from clinical trials are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The FDA may limit the indications for use or place other conditions on any approvals that could restrict the commercial application of the products. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Requirements. After regulatory approval of a product is obtained, we are required to comply with a number of post-approval requirements. For example, as a condition of approval of an NDA, the FDA may require post marketing testing, including Phase IV trials, and surveillance to monitor the product's safety or efficacy. In addition, holders of an approved NDA are required to report certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling for their products. The FDA strictly regulates the promotional claims that may be made about prescription drug products. In particular, a drug may not be promoted for uses that are not approved by the FDA as reflected in the product's approved labeling. In addition, the FDA requires substantiation of any claims of superiority of one product over another, including that such claims be proven by adequate and well-controlled head-to-head clinical trials. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes certain procedural, substantive and recordkeeping requirements. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our product candidates. Future FDA inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product, including new safety risks, or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing. Newly discovered or developed safety or effectiveness data may require changes to a product's approved labeling, including the addition of new warnings and contraindications and may require the conduct of further clinical investigations to support
18
such changes. Also, new government requirements, including those resulting from new legislation, may be established that could delay or prevent regulatory approval of our products under development.
Regulation Outside the United States
In addition to regulations in the United States, we will be subject to a variety of regulations in other jurisdictions governing clinical trials, marketing, commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of countries outside the United States before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement, among other things, vary greatly from country to country.
To obtain regulatory approval of a drug under European Union regulatory systems, we may submit marketing authorizations either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for medicines produced by certain biotechnological processes and optional for those which are highly innovative, provides for the grant of a single marketing authorization that is valid for all European Union member states. All marketing authorizations for products designated as orphan drugs must be granted in accordance with the centralized procedure. The decentralized procedure provides for approval by one or more other, or concerned, member states of an assessment of an application performed by one member state, known as the reference member state. Under this procedure, an applicant submits an application, or dossier, and related materials including a draft summary of product characteristics, and draft labeling and package leaflet, to the reference member state and concerned member states. The reference member state prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state's assessment report, each concerned member state must decide whether to approve the assessment report and related materials. If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to the public health, the disputed points may eventually be referred to the European Commission, whose decision is binding on all member states.
Pharmaceutical Pricing and Reimbursement
In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third party payors. Third party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. These third party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare product candidates. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. Our product candidates may not be considered cost-effective. Adequate third party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
In 2003, the United States government enacted legislation providing a partial prescription drug benefit for Medicare recipients, which became effective at the beginning of 2006. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive marketing approval. However, to obtain payments under this program, we will be required to sell products to Medicare recipients through drug procurement organizations operating pursuant to this legislation. These organizations will negotiate prices for our products, which are likely to be lower than we might otherwise obtain. Federal, state and local governments in the United States continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs.
19
Future legislation could limit payments for pharmaceuticals such as the drug candidates that we are developing.
Different pricing and reimbursement schemes exist in other countries. In the European Community, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on managed care in the United States has increased and will continue to increase the pressure on pharmaceutical pricing.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. While we believe that our technologies, knowledge, experience and scientific resources provide us with competitive advantages, we face potential competition from many different sources, including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies and other private and public research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
Many of our competitors may have significantly greater financial resources and expertise than we do in research and development, preclinical testing, conducting clinical trials, obtaining regulatory approvals, manufacturing and marketing and distribution of approved products. Smaller and other early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to or necessary for our programs or advantageous to our business.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer or more effective, have fewer side effects, are more convenient or are less expensive than any products that we may develop. Our competitors may also obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours. In addition, our ability to compete may be affected because in some cases insurers or third party payors seek to encourage the use of generic products. In particular, generic versions of cancer treatments are widely available. This may have the effect of making branded products less attractive, from a cost perspective, to buyers. Several companies are also developing improved taxane analogues including Bristol-Myers Squibb Company, which is developing BMS 184476 and BMS 275183, and Sanofi-Aventis, which is developing XRP 6258 and XRP 9881. Companies developing or selling taxane products include Abbott Laboratories, Abraxis BioScience, Bristol-Myers Squibb, Daiichi Pharmaceutical Co., Sanofi-Aventis and Wyeth, each of which has significantly greater resources than we do.
20
If TPI 287 is approved for the cancer indications that we are investigating, it will compete with currently marketed drugs and potentially with other product candidates that are currently in development for the same indications. The competition for TPI 287 includes the following:
| Indication | Primary Competitors | |
|---|---|---|
| Prostate cancer | Taxol® (paclitaxel) Taxotere® (docetaxel) Abraxane® (paclitaxel) Generic forms of paclitaxel | |
| CNS tumors | Temodar® (temozolomide) BiCNU® (carmustine) | |
| Pancreatic cancer | Gemzar® (gemcitabine) Xeloda® (5-fluorouracil) Tarceva® (erlotinib) |
The key competitive factors affecting the success of our product candidates are likely to be efficacy, safety, convenience and ease of administration, breadth of approved indications, price and availability of reimbursement.
Manufacturing
We do not currently own or operate manufacturing facilities for the production of clinical or commercial quantities of TPI 287. We currently rely, and expect to continue to rely, on third parties for the manufacture of our product candidates, including TPI 287, and any products that we may develop or acquire, other than small amounts of compounds that we synthesize ourselves for preclinical testing. To date, we have obtained our supply of the bulk drug substance for TPI 287 from one third-party manufacturer. We have engaged a second manufacturer to provide the fill and finish services for an IV formulation of TPI 287 that we are using in our ongoing clinical trials. We obtain our supplies of TPI 287 from these third party manufacturers pursuant to short term agreements that include specific supply timelines and volume expectations. If either of these manufacturers should become unavailable to us for any reason, we believe that there are a number of potential replacements, although we might incur some delay in identifying and qualifying such replacements. We believe that we currently have sufficient quantities of the TPI 287 bulk drug substance on hand to allow us to conduct our currently planned clinical trials for the next 12 months.
TPI 287 is an organic compound of low molecular weight referred to as a small molecule. Small molecules generally are known for their ease of synthesis and reasonable cost of their starting materials. TPI 287 is manufactured in reliable and reproducible synthetic processes from readily available starting materials. Although one of the intermediate raw materials for TPI 287 is manufactured by a contractor using proprietary technology, we believe that we have purchased a sufficient quantity of this intermediate raw material to allow us to produce enough TPI 287 bulk drug substance to complete our currently planned Phase II clinical trials. The chemistry of TPI 287 is amenable to scale up and does not require unusual equipment in the manufacturing process. We expect to continue to develop drug candidates that can be produced cost-effectively at contract manufacturing facilities. For example, we have identified multiple vendors capable of performing fill and finish services for an oral formulation of TPI 287.
Employees
As of December 2006, we had 32 full-time employees and six part-time employees, including a total of nine employees with Ph.D. or M.D. degrees. Of our workforce, 23 employees are engaged in drug development. We believe that our relations with our employees are good. None of our employees
21
is represented by labor unions or covered by collective bargaining agreements. In addition, we contract with outside consultants for services relating to our drug development programs.
Properties
The following table sets forth information regarding our leased facilities:
| Location | Approximate Square Feet | Use | Lease Expiration Date | |||
|---|---|---|---|---|---|---|
| Boulder, Colorado | 29,000 | Future office and laboratory space(1) | November 1, 2012 | |||
| Boulder, Colorado | 16,000 | Office and laboratory space | February 1, 2008 | |||
| Roseland, New Jersey | 2,300 | Office space | December 31, 2008 | |||
| Boulder, Colorado | 2,121 | Laboratory space | September 30, 2007 | |||
| New York, New York | 850 | Office space | Month-to-month |
- (1)
- In March 2007, we entered into a five year lease covering this space, into which we plan to consolidate our Colorado-based administrative and research and development facilities. We plan to take possession on November 1, 2007 in order to make necessary renovations.
We also own five acres of undeveloped land in Longmont, Colorado which we account for as other assets.
We believe that these existing facilities are adequate to meet current foreseeable requirements or that suitable additional or substitute space will be available on commercially reasonable terms.
Legal Proceedings
We currently are in arbitration through the American Arbitration Association with the assignor of certain patents and patent applications relating to pharmaceutical formulations containing Vitamin E. The arbitration began in November 2005 in Boulder, Colorado. We licensed these patent applications in 1998. The licensor claims that we have failed in our obligation to develop the technology and is demanding the patents be returned. We have denied this claim and have alleged that the licensor committed fraud in inducing us to enter into the license agreement. We are seeking unspecified damages against the licensor.
We agreed to employ Donald H. Picker, Ph.D., as our president in December 2006. Prior to joining our company, Dr. Picker had served as Executive Vice President of Research and Development of Callisto Pharmaceuticals, Inc. In December 2006, Callisto filed a complaint in the Supreme Court of New York, County of New York, relating to our employment of Dr. Picker. The suit names Tapestry and two of our officers, Leonard Shaykin and Kai Larson, as defendants. In its complaint, Callisto alleges breaches of a confidentiality agreement between Callisto and Tapestry and interference with Dr. Picker's contractual relationship with Callisto. Callisto seeks unspecified actual and punitive damages. We believe these claims are without merit and we are vigorously defending against them.
Dr. Steven K. Carter, a member of our board of directors, is also a director of Callisto and was deposed in connection with this suit. We have reimbursed Dr. Carter's costs in connection with this deposition.
22
You should carefully consider the following risk factors related to our current business operations before making a decision to invest in our common stock. Additional risks of which we are not yet aware or that we currently believe are immaterial may also adversely impair us or our operations. If any of the events or circumstances described in the following risk factors actually occurs, our business may suffer, the trading price of common stock could decline, and you may lose all or part of your investment.
Risks Related to Our Business
We are currently devoting substantially all of our efforts to the development of one product candidate, TPI 287, which is in an early stage of clinical development. If we are unable to successfully develop and commercialize TPI 287, or experience significant delays in doing so, our business, financial condition and results of operations will be materially harmed.
We are currently devoting substantially all of our efforts to the development of TPI 287. Our ability to generate product revenues, which we do not expect for at least the next several years, if ever, depends on the successful development and eventual commercialization of TPI 287. However, TPI 287 is in an early stage of clinical development. We recently completed dosing in a Phase I clinical trial of TPI 287 and expect to complete dosing in another Phase I clinical trial during the second half of 2007. In addition, we recently commenced enrollment in a Phase II clinical trial of TPI 287 in patients with an advanced form of prostate cancer called hormone refractory prostate cancer, or HRPC. We also expect to initiate a Phase Ib/II pharmacokinetic clinical trial of TPI 287 in the summer of 2007 to evaluate the drug's activity and oral bioavailability in humans. The success of TPI 287 will depend on several factors, including the following:
- •
- successful completion of clinical trials;
- •
- receiving and maintaining regulatory approval from the U.S. Food and Drug Administration, or FDA, and similar regulatory authorities outside the United States;
- •
- establishing and maintaining commercial manufacturing arrangements with third party manufacturers;
- •
- commercial sales of TPI 287, whether alone or in collaboration with others;
- •
- of the product by patients, the medical community and third party payors;
- •
- from other therapies; and
- •
- continued acceptable safety profile of TPI 287 following approval.
Because of our focus on one product candidate, if TPI 287 does not prove successful in clinical trials or is not commercialized because we have insufficient resources for continued development for any other reason, we may be required to suspend or discontinue our operations and you could lose your entire investment.
We have incurred significant losses since our inception. We expect to incur losses for the foreseeable future and may never achieve or maintain profitability.
We have incurred significant operating losses since our inception in 1991. Our net loss from continuing operations was $16.6 million in 2006, $17.2 million in 2005 and $21.6 million in 2004. We have an accumulated deficit of $123.9 million as of December 27, 2006. To date, we have financed our operations primarily with the net proceeds of public offerings of common stock and private placements of equity securities, with proceeds from the exercise of warrants and options and with debt. We also have funded our capital requirements with the proceeds of the sale of our paclitaxel business to Mayne
23
Pharma (USA) Inc. in December 2003. Since we sold our paclitaxel business to Mayne Pharma, we have devoted substantially all of our efforts to research and development activities, including clinical trials. We have not completed development of any drugs. We expect to continue to incur significant and increasing operating losses for at least the next several years. We anticipate that our expenses will increase substantially as we:
- •
- continue the clinical development of TPI 287;
- •
- subject to the successful completion of clinical trials, seek regulatory approvals for TPI 287 and potentially other product candidates;
- •
- establish a sales and marketing infrastructure to commercialize products for which we may obtain regulatory approval; and
- •
- add operational, financial and management information systems and personnel, including personnel to support our product development efforts.
To become and remain profitable, we must succeed in developing and eventually commercializing drugs with significant market potential. This will require us to be successful in a range of challenging activities, including discovering, in-licensing or acquiring product candidates, successfully completing preclinical testing and clinical trials of our product candidates, obtaining regulatory approval for these product candidates and manufacturing, marketing and selling those products for which we may obtain regulatory approval. We are only in the preliminary stages of development of TPI 287, our only current product candidate. We may never succeed in these activities and may never generate revenues that are significant enough to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the market price of our common stock and could impair our ability to raise capital, expand our business, diversify our product offerings or continue our operations. A decline in the market price of our common stock could also cause you to lose all or a part of your investment.
We will need substantial additional funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts.
We expect our research and development expenses to increase in connection with our ongoing activities, particularly as we continue the clinical development of TPI 287. In addition, subject to obtaining regulatory approval of TPI 287 or any other product candidate, we expect to incur significant commercialization expenses for product sales, marketing, securing commercial quantities of product from our manufacturers and distribution. We will need substantial additional funding and may be unable to raise capital when needed or on attractive terms, which would force us to delay, reduce or eliminate our research and development programs or commercialization efforts.
We believe that our existing cash and cash equivalents and short-term investments, will be sufficient to fund our operations through the end of 2007. Our future capital requirements will depend on many factors, including:
- •
- the progress and results of our clinical trials of TPI 287;
- •
- the number and development requirements of other product candidates that we pursue, including the scope, progress, results and costs of preclinical development, laboratory testing and clinical trials of any other product candidate;
- •
- the costs, timing and outcome of regulatory review;
- •
- the costs of commercialization activities, including product marketing, sales and distribution;
- •
- the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims;
24
- •
- the extent to which we acquire or invest in new businesses, products and technologies;
- •
- our ability to establish and maintain collaborations; and
- •
- changes in facility or staffing requirements.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through public or private equity offerings and debt financings, corporate collaboration and licensing arrangements. If we raise additional funds by issuing equity securities, our stockholders will experience dilution. Debt financing, if available, may involve agreements that include covenants restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. Any debt financing or additional equity that we raise may contain terms, such as liquidation and other preferences, that are not favorable to us or our stockholders. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
Our short history as a drug development company may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
We are an early stage drug development company. Since we sold our paclitaxel business to Mayne Pharma in 2003, our operations have been limited to undertaking preclinical studies and limited clinical trials of our product candidates. Since we began devoting substantially all of our efforts to the development of TPI 287, we have not demonstrated our ability to successfully complete large-scale clinical trials, obtain regulatory approvals, manufacture a commercial scale product, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer history as a drug development company.
In addition, as a new business, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors. We will need to transition from having a development focus to becoming capable of supporting commercial activities. We may not be successful in such a transition.
If we are not successful in establishing and maintaining development and commercialization collaborations for our product candidates, we may have to reduce or delay our product development and commercialization efforts and increase our expenditures.
For each of our product candidates, we plan to evaluate the merits of retaining commercialization rights for ourselves or selectively entering into strategic alliances with leading pharmaceutical and biotechnology companies to assist us in advancing our development programs. Other than arrangements with academic scientists and institutions that have provided us basic scientific research capabilities, we have not entered into any such arrangements to date.
If we enter into any of these strategic alliances, they may provide us with access to the therapeutic area expertise and the development and commercialization resources of our collaborators, as well as augment our financial resources.
We may not be able to negotiate collaboration or other alternative arrangements with these other companies for the development or commercialization of our product candidates on acceptable terms, or at all. We face, and will continue to face, significant competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement. We may not be successful in our efforts, if any, to establish and implement collaborations or other alternative arrangements. The terms of any collaborations or other arrangements that we
25
establish may not be favorable to us. If we are not able to establish collaboration arrangements, we may have to reduce or delay further development of some of our programs, increase our planned expenditures and undertake development and commercialization activities at our own expense.
Any collaboration that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. It is likely that our collaborators will have significant discretion in determining the efforts and resources that they will apply to these collaborations. For example, it is possible that:
- •
- collaborators may not pursue further development and commercialization of compounds resulting from collaborations or may elect not to renew research and development programs;
- •
- collaborators may delay clinical trials, underfund a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require the development of a new formulation of a product candidate for clinical testing;
- •
- a collaborator with marketing and distribution rights to one or more of our products may not commit enough resources to the marketing and distribution of our products, limiting our potential revenues from the commercialization of these products; and
- •
- disputes may arise delaying or terminating the research, development or commercialization of our product candidates, or result in significant legal proceedings.
We also expect to be subject to additional risks in future collaboration agreements we may enter into, including the following:
- •
- collaboration agreements are likely to be for fixed terms and subject to termination by our collaborators in the event of a material breach or lack of scientific progress by us;
- •
- our collaborators are likely to have the first right to maintain or defend our intellectual property rights and, although we would likely have the right to assume the maintenance and defense of our intellectual property rights if our collaborators do not, our ability to do so may be compromised by our collaborators' acts or omissions;
- •
- our collaborators may use our intellectual property rights in such a way as to cause litigation that could jeopardize or invalidate our intellectual property rights or expose us to potential liability; and
- •
- our collaborators may disclose our trade secrets to third parties without our authorization.
Collaborations with pharmaceutical companies and other third parties often are terminated or allowed to expire by the other party. Such terminations or expirations would adversely affect us financially and could harm our business reputation.
The results of preclinical studies and early stage clinical trials do not ensure success in later stage clinical trials, the results of clinical trials of a product candidate for particular indications are not necessarily indicative of the results of clinical trials of the product candidate in other indications and interim results from a clinical trial are not necessarily indicative of the successful outcome of that trial.
We do not currently have any products that have received regulatory approval, and our product development efforts are at an early stage. The outcome of preclinical studies and early clinical trials may not accurately predict the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. For example, the results that we observed in our preclinical studies of TPI 287 may not be indicative of successful results in our ongoing and planned Phase I and Phase II clinical trials of TPI 287 in multiple cancer indications. Many drug candidates have failed to show efficacy or safety in humans even after promising preclinical study results.
26
Even if our early phase clinical trials are successful, we will need to conduct additional clinical trials in a larger number of patients taking the drug for longer periods before we are able to seek approvals from the FDA and similar regulatory authorities outside the United States to market and sell a product candidate. Similarly, even if clinical trials of a product candidate are successful in one indication, clinical trials of that product candidate for other indications may be unsuccessful. For example, although we recently commenced enrollment in a Phase II clinical trial of TPI 287 for HRPC, we also plan to conduct clinical trials of TPI 287 for tumors of the central nervous system and pancreatic cancer. As such, the results from our ongoing Phase II clinical trials of TPI 287 may not necessarily be indicative of the results we may obtain in future clinical trials of TPI 287 in other cancer indications. Furthermore, even if clinical trials of one formulation of a drug are successful, clinical trials for other formulations may be unsuccessful. For example, the results from our ongoing Phase II clinical trials of an IV formulation of TPI 287 may not accurately predict the results we obtain in future clinical trials of an oral formulation of TPI 287. If we are not successful in commercializing TPI 287 in any of the cancers that we are investigating or as an oral formulation, or are significantly delayed in doing so, our business will be materially harmed and our stock price may decline.
If our preclinical studies do not produce positive results or if our clinical trials do not demonstrate safety and efficacy in humans, we may experience delays, incur additional costs and ultimately be unable to commercialize our product candidates.
Before obtaining regulatory approval for the sale of our product candidates, we must conduct, at our own expense, extensive preclinical tests to demonstrate the safety of our product candidates in animals and clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Preclinical and clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, preclinical testing and the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates, including:
- •
- regulators or institutional review boards may not authorize us to commence or continue a clinical trial or conduct a clinical trial at a prospective trial site;
- •
- our preclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional preclinical testing or clinical trials, or we may abandon projects that we expect to be promising;
- •
- the number of patients required for our clinical trials may be larger than we anticipate, enrollment in our clinical trials may be slower than we anticipate, or participants may drop out of our clinical trials at a higher rate than we anticipate;
- •
- we or our third party contractors may fail to comply with regulatory requirements;
- •
- our third party contractors may fail to meet their contractual obligations to us in a timely manner or at all;
- •
- we might have to suspend or terminate our clinical trials if the participants are being exposed to unacceptable health risks;
- •
- regulators or institutional review boards may require that we hold, suspend or terminate clinical research for various reasons, including safety risks or noncompliance with regulatory requirements;
- •
- the cost of our clinical trials may be greater than we anticipate;
- •
- the supply or quality of our product candidates, components of our product candidates or other materials necessary to conduct our clinical trials may be insufficient or inadequate because we
27
- •
- our product candidates may not show the desired level of efficacy, may include undesirable side effects or may have other unexpected characteristics.
do not currently have any agreements with third party manufacturers for the long-term commercial supply of any of our product candidates; and
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete our clinical trials or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
- •
- be delayed in obtaining marketing approval for our product candidates;
- •
- not be able to obtain marketing approval;
- •
- obtain approval for indications that are not as broad as intended; or
- •
- have the product removed from the market after obtaining marketing approval.
Our product development costs also will increase if we experience delays in testing or approvals. We do not know whether any preclinical studies or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, if at all. Significant preclinical or clinical trial delays also could shorten the patent protection period during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to commercialize our products or product candidates.
If we observe serious adverse events or identify inappropriate side effects during the development of our product candidates, we may need to abandon or delay our development of the applicable product candidate.
Our product candidates may produce serious unforeseen adverse events, have undesirable side effects or have other characteristics that are unexpected. As a result, we might need to interrupt, delay or halt clinical trials of our product candidates. We may suspend or terminate clinical trials at any time. Regulators or institutional review boards may require that we hold, suspend or terminate clinical trials for various reasons, including a finding that participants are being exposed to unacceptable health risks. In addition, if other pharmaceutical companies announce that they have observed frequent unforeseen adverse events or safety issues in their trials involving products or product candidates similar to, or competitive with, our product candidates, we could encounter delays in the timing of our clinical trials or difficulties in obtaining the approval of our product candidates. The public perception of our product candidates might also be adversely affected, which could adversely affect our business, financial condition and results of operations, even if the concern relates to another company's product or product candidate.
The commercial success of TPI 287 or any other product candidates that we may develop will depend upon the degree of market acceptance by physicians, patients, healthcare payors and others in the medical community.
Any products that we bring to the market, including TPI 287 if it receives marketing approval, may not gain market acceptance by physicians, patients, healthcare payors and others in the medical community. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenues and we may not become profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, including:
- •
- the prevalence and severity of any side effects;
- •
- the efficacy and potential advantages over alternative treatments;
- •
- the ability to offer our product candidates for sale at competitive prices;
28
- •
- relative convenience and ease of administration;
- •
- the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
- •
- the strength of marketing and distribution support; and
- •
- sufficient third party coverage or reimbursement.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our product candidates, we may be unable to generate product revenues.
We do not have a sales or marketing organization and have no experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any approved product, we must either develop a sales and marketing organization or outsource these functions to third parties. We plan to retain United States marketing and sales rights or co-promotion rights for our product candidates for which we receive regulatory approval in markets in which we believe it is possible to gain access through a focused, specialized sales force. For markets in which we believe a large sales force is required to gain access, and for markets outside the United States, we plan to commercialize products for which we obtain regulatory approval through a variety of collaboration and distribution arrangements with other pharmaceutical and biotechnology companies.
There are risks involved with establishing our own sales and marketing capabilities, as well as in entering into arrangements with third parties to perform these services. For example, developing a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or prohibited as a result of FDA requirements or other reasons, we would incur related expenses too early relative to the product launch. This may be costly, and our investment would be lost if we cannot retain our sales and marketing personnel. In addition, even if we establish our own sales force and marketing capabilities, our sales force and marketing teams may not be successful in commercializing our products.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
The development and commercialization of new drugs is highly competitive. We face competition with respect to TPI 287 and any products we may seek to develop or commercialize in the future from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide.
If TPI 287 is approved for the cancer indications that we are currently investigating, it will compete principally with the following:
- •
- Bristol-Myers Squibb's Taxol®, Sanofi-Aventis' Taxotere®, Abraxis BioScience's Abraxane® and generic forms of paclitaxel in the treatment of prostate cancer;
- •
- Schering-Plough's Temodar® (temozolomide) and Bristol-Myers Squibb's BiCNU® (carmustine) in the treatment of CNS tumors; and
- •
- Eli Lilly's Gemzar® (gemcitabine), Hoffmann-La Roche's Xeloda® (5-fluorouracil) and OSI Pharmaceuticals' Tarceva® (erlotinib) in the treatment of pancreatic cancer.
Potential competitors also include academic institutions, government agencies and other private and public research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, product development, manufacturing and commercialization.
29
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer or more effective, have fewer side effects, are more convenient or are less expensive than any products that we may develop. Our competitors may also obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours. In addition, our ability to compete may be negatively affected because in some cases insurers or third party payors seek to encourage the use of generic products. This may make branded products less attractive to buyers due to higher costs.
We believe that many competitors are attempting to develop therapeutics for many of our target indications, including academic institutions, government agencies, public and private research organizations, large pharmaceutical companies and smaller more focused companies. We are aware of numerous product candidates in clinical development for each of the cancer indications that we are investigating.
Many of our competitors may have significantly greater financial resources and expertise than we do in the following:
- •
- research and development;
- •
- preclinical testing and conducting clinical trials;
- •
- obtaining regulatory approvals;
- •
- manufacturing; and
- •
- marketing and distribution.
Smaller and other early stage development companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical companies or other organizations. In addition, other companies and institutions compete with us in recruiting and retaining highly qualified scientific and management personnel and in establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to or necessary for our programs or advantageous to our business.
If we are unable to obtain adequate reimbursement from governments or third party payors for any products that we may develop or if we are unable to obtain acceptable prices for those products, our revenues and prospects for profitability will suffer.
Our revenues and profits will depend heavily upon the availability of adequate reimbursement for the use of our approved product candidates from governmental and other third party payors, both in the United States and in other markets. Reimbursement by a third party payor may depend upon a number of factors, including the third party payor's determination that use of a product is:
- •
- a covered benefit under its health plan;
- •
- safe, effective and medically necessary;
- •
- appropriate for the specific patient;
- •
- cost-effective; and
- •
- neither experimental nor investigational.
Obtaining reimbursement approval for a product from each government or other third party payor is a time consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to each payor. We may not be able to provide data sufficient to gain acceptance with respect to reimbursement. Even when a payor determines that a product is eligible for reimbursement, the payor may impose coverage limitations that preclude
30
payment for some uses that are approved by the FDA or comparable authorities. In addition, there is a risk that full reimbursement may not be available for high priced products. Moreover, eligibility for coverage does not imply that any product will be reimbursed in all cases or at a rate that allows us to make a profit or even cover our costs. Interim payments for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. A primary trend in the United States healthcare industry and elsewhere is toward cost containment.
We expect recent changes in the Medicare program and increasing emphasis on managed care to continue to put pressure on pharmaceutical product pricing. In 2003, the U.S. government enacted legislation providing a partial prescription drug benefit for Medicare recipients, which became effective at the beginning of 2006. However, to obtain payments under this program, we will be required to sell products to Medicare recipients through drug procurement organizations operating pursuant to this legislation. These organizations will negotiate prices for our products, which are likely to be lower than those we might otherwise obtain. Federal, state and local governments in the United States continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs. Future legislation could limit payments for the product candidates that we are developing.
Recent proposed legislation may permit re-importation of drugs from foreign countries into the United States, including foreign countries where the drugs are sold at lower prices than in the United States, which could force us to lower the prices at which we sell our products and impair our ability to derive revenues from these products.
Legislation has been introduced in the U.S. Congress that, if enacted, would permit more widespread re-importation of drugs from foreign countries into the United States. This could include re-importation from foreign countries where the drugs are sold at lower prices than in the United States. Such legislation, or similar regulatory changes, could lead to a decrease in the price we receive for any approved products, which, in turn, could impair our ability to generate revenues. Alternatively, in response to legislation such as this, we might elect not to seek approval for or market our products in foreign jurisdictions in order to minimize the risk of re-importation, which could also reduce the revenue we generate from our product sales.
Governments outside the United States tend to impose strict price controls, which may adversely affect our revenues, if any.
In some countries, particularly the countries of the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely affected.
Risks Related to Our Dependence on Third Parties
Use of third parties to manufacture our product candidates may increase the risk that we will not have sufficient quantities of our product candidates or be able to obtain such quantities at an acceptable cost, and clinical development and commercialization of our product candidates could be delayed, prevented or impaired.
We do not currently own or operate manufacturing facilities for the production of clinical or commercial quantities of our product candidates. We have limited personnel with experience in drug manufacturing and we lack the resources and the capabilities to manufacture any of our product candidates on a clinical or commercial scale.
31
We currently rely, and expect to continue to rely, on third parties for the manufacture of our product candidates and any products that we may develop, other than small amounts of compounds that we synthesize for preclinical testing. To date, we have obtained our supply of the bulk drug substance for TPI 287 from one third-party manufacturer. We have engaged a second manufacturer to provide the fill and finish services for an IV formulation of TPI 287 that we are using in our ongoing clinical trials. We do not have any agreements with third party manufacturers for the long term commercial supply of any of our product candidates. We obtain our supplies of TPI 287 from these third party manufacturers pursuant to short term agreements that include specific supply timelines and volume expectations. If any of these manufacturers should become unavailable to us for any reason, we may be delayed in identifying and qualifying such replacements.
Reliance on third party manufacturers entails risks to which we would not be subject if we manufactured product candidates or products ourselves, including:
- •
- reliance on the third party for regulatory compliance and quality control and assurance;
- •
- the possible breach of the manufacturing agreement by the third party because of factors beyond our control; and
- •
- the possible termination or nonrenewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us.
Our manufacturers may not be able to comply with current good manufacturing practice, or cGMP, regulations or other regulatory requirements or similar regulatory requirements outside the United States. We and our manufacturers are subject to unannounced inspections by the FDA, state regulators and similar regulators outside the United States. Our failure, or the failure of our third party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our product candidates.
Our product candidates and any products that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that are both capable of manufacturing for us and willing to do so. If the third parties that we engage to manufacture product for our preclinical tests and clinical trials should cease to continue to do so for any reason, we likely would experience delays in advancing these trials while we identify and qualify replacement suppliers and we may be unable to obtain replacement supplies on terms that are favorable to us. In addition, if we are not able to obtain adequate supplies of our product candidates or the drug substances used to manufacture them, it will be more difficult for us to develop our product candidates and compete effectively.
Our current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our future profit margins and our ability to develop product candidates and commercialize any products that receive regulatory approval on a timely and competitive basis.
We rely on third parties to conduct our clinical trials and those third parties may not perform satisfactorily, including failing to meet established deadlines for the completion of such trials.
We do not independently conduct clinical trials for our product candidates. We rely on third parties, such as contract research organizations, clinical data management organizations, medical institutions and clinical investigators, to perform this function. Our reliance on these third parties for clinical development activities reduces our control over these activities. We are responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to
32
as Good Clinical Practices, for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, regulatory approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
We also rely on other third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our existing or future distributors could delay clinical development or regulatory approval of our product candidates or commercialization of our products, producing additional losses and depriving us of potential product revenue.
We may not be successful in our efforts to in-license or acquire attractive development candidates.
We may attempt to enhance our product pipeline through strategically in-licensing or acquiring product candidates for the treatment of cancer. However, we may be unable to license or acquire suitable product candidates from third parties for a number of reasons. In particular, the licensing and acquisition of pharmaceutical products is competitive. A number of more established companies are also pursuing strategies to license or acquire products in the cancer market. These established companies may have a competitive advantage over us due to their size, cash resources or greater clinical development and commercialization capabilities. Other factors that may prevent us from licensing or otherwise acquiring suitable product candidates include the following:
- •
- we may be unable to license or acquire the relevant technology on terms that would allow us to make an appropriate return from the product;
- •
- companies that perceive us to be their competitors may be unwilling to assign or license their product rights to us; or
- •
- we may be unable to identify suitable products or product candidates within our areas of expertise.
If we in-license product candidates in the future and fail to comply with our obligations in any such license with a third party, we could lose license rights that are important to our business.
We expect that any licenses that we enter into the future will provide us rights to third party intellectual property that is important to our business. We expect that future licenses will impose various development and commercialization, milestone payment, royalty, sublicensing, patent protection and maintenance, insurance and other obligations on us. If we fail to comply with these obligations or otherwise breach the license agreement, the licensor may have the right to terminate the license in whole, terminate the exclusive nature of the license or bring a claim against us for damages. Any such termination or claim would likely prevent or impede our ability to market any product that is covered by the licensed patents. Even if we contest any such termination or claim and are ultimately successful, our stock price could suffer. In addition, upon any termination of a license agreement, we may be required to license to the licensor any related intellectual property that we developed.
We may be unable to attract and retain the qualified employees we need to be successful.
We are highly dependent on members of our staff that lead or play critical roles in our research and development efforts. We require highly qualified and trained scientists with the necessary skills to develop our product candidates. Recruiting and retaining qualified technical and managerial personnel
33
will also be critical to our success. We face intense competition for these professionals from other companies in our industry and the turnover rate for these professionals can be high. The loss of any of these persons, or our inability to recruit additional personnel necessary to our business, could substantially impair our research and development efforts and impede our ability to develop and commercialize any of our products. In addition, we rely on other consultants and advisors to assist us in formulating our research and development strategy. Some have consulting or other advisory arrangements with other entities that may conflict or compete with their obligations to us.
We expect to expand our development, regulatory and sales and marketing capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of drug development, regulatory affairs and sales and marketing. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources and the inexperience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
Our use of hazardous materials exposes us to the risk of material environmental liabilities. We also incur substantial costs to comply with environmental and occupational safety laws regulating the use of hazardous materials. If we violate these laws, we would be subject to significant fines, liabilities or other adverse consequences.
We use radioactive materials and other hazardous or biohazardous substances in our research and development activities. As a result, we are potentially subject to material liabilities related to personal injuries or property damages that may be caused by the spread of radioactive contamination or by other hazardous substance releases or exposures at, or from, our facilities. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, we cannot completely eliminate the risk of accidental contamination or injury from these materials. Decontamination costs associated with radioactivity releases, other clean-up cost, and related damages or liabilities could be significant and could harm our business. The cost of this liability could exceed our resources, and we do not maintain liability insurance for these risks.
We are required to comply with increasingly stringent laws and regulations governing environmental protection and workplace safety, including requirements governing the handling, storage, use and disposal of radioactive and other hazardous substances and wastes, and laboratory operating and safety procedures. These laws and regulations can impose substantial fines and criminal sanctions for violations. Maintaining compliance with these laws and regulations with regard to our operations could require substantial additional resources. These costs could limit our ability to conduct operations in a cost-effective manner.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop. Product liability claims might be brought against us by clinical trial patients, consumers or health care providers or by pharmaceutical companies or others selling our
34
products. If we complete clinical testing for our product candidates and receive regulatory approval to market our products, the FDA will require us to include extensive warnings, precautions and other risk and safety information in the label for our products that, among other things, identify the known potential adverse effects and the patients who should not receive our product. These warnings may not be deemed adequate, and physicians and patients may not comply with these warnings.
If we cannot successfully defend ourselves against such claims, we may incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
- •
- decreased demand for any product candidates or products that we may develop;
- •
- injury to our reputation;
- •
- withdrawal of clinical trial participants;
- •
- costs to defend the related litigation;
- •
- substantial monetary awards to trial participants or patients;
- •
- loss of revenue; and
- •
- the inability to commercialize any products that we may develop.
We have product liability insurance that covers our clinical trials up to a $5.0 million annual aggregate limit and subject to a per claim deductible. We cannot predict all of the possible harms or side effects that may result from the testing and use of our product candidates. As a result, the amount of insurance coverage we currently hold, or that we may obtain, may not be adequate to protect us from any liabilities. We may require increased liability coverage as our product candidates' advance in clinical trials. In addition, we intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval for any products. Further, insurance coverage is increasingly expensive, and we do not know whether we will be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. A successful product liability claim brought against us in excess of our insurance coverage or a product recall could adversely affect our business, results of operations and financial condition.
If our internal control over financial reporting is not considered effective, our business could be materially harmed and our stock price could decline.
Beginning in 2007, Section 404 of the Sarbanes-Oxley Act of 2002 requires us to evaluate the effectiveness of our internal control over financial reporting as of the end of each fiscal year and to include a management report assessing the effectiveness of our internal control over financial reporting in our annual report on Form 10-K for that fiscal year. Beginning in 2008, Section 404 also requires our independent registered public accounting firm to attest to, and report on, management's assessment of our internal control over financial reporting.
Any system of controls, however well designed and operated, can provide only reasonable, and not absolute, assurance that the objectives of the system are met. In addition, the design of any control system is based in part upon certain assumptions about the likelihood of future events. Because of these and other inherent limitations of control systems, there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, regardless of how remote. For example, in our Quarterly Report on Form 10-Q/A for the quarter ended September 27, 2006, we disclosed that we did not maintain effective control over the completeness and accuracy of the supporting schedule of weighted average shares outstanding and that as a result it was necessary to restate our Consolidated Statements of Operations included in that report. We cannot assure you that we or our independent registered public accounting firm will not identify additional material weaknesses in our internal control systems in the future. If additional material weaknesses in our
35
internal control systems are detected, our internal control over financial reporting may not be considered effective and we may experience a loss of public confidence, which could have an adverse effect on our business and on the market price of our common stock.
Risks Related to Regulatory Approval of Our Product Candidates
If we are not able to obtain required regulatory approvals, we will not be able to commercialize our product candidates, and our ability to generate revenue will be materially impaired.
Our product candidates and the activities associated with their development and commercialization, including their testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Failure to obtain regulatory approval for a product candidate will prevent us from commercializing the product candidate. We have not received regulatory approval to market any of our product candidates in any jurisdiction. We have only limited experience in filing and prosecuting the applications necessary to gain regulatory approvals and expect to rely on third party contract research organizations to assist us in this process. Securing FDA approval requires the submission of extensive preclinical and clinical data and supporting information to the FDA for each therapeutic indication to establish the product candidate's safety and efficacy. Securing FDA approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the FDA. Our future products may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining regulatory approval or prevent or limit commercial use.
The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review criteria for each submitted product application, may cause delays in the approval or rejection of an application. The FDA has substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies. In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent regulatory approval of a product candidate. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable.
Any product for which we obtain marketing approval could be subject to restrictions or withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our products, when and if any of them are approved.
Any product for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to continual requirements of and review by the FDA and comparable regulatory authorities. These requirements include submissions of safety and other post-marketing information and periodic reports, registration requirements, cGMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians and recordkeeping. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Later discovery of previously unknown problems with our products, manufacturers or manufacturing processes, including new safety risks, or failure to comply with regulatory requirements, may result in actions such as:
- •
- restrictions on such products, manufacturers or manufacturing processes;
36
- •
- warning letters;
- •
- withdrawal of the products from the market;
- •
- refusal to approve pending applications or supplements to approved applications that we submit;
- •
- product recalls;
- •
- fines;
- •
- suspension or withdrawal of regulatory approvals;
- •
- refusal to permit the import or export of our products;
- •
- product seizures; or
- •
- injunctions or the imposition of civil or criminal penalties.
Failure to obtain regulatory approval in international jurisdictions would prevent us from marketing our products abroad, and regulatory approvals in some international jurisdictions can be conditioned on broad license grants.
We intend to have our products marketed outside the United States. With respect to some of our product candidates, we expect that a future collaborator will have responsibility to obtain regulatory approval outside the United States, and we will rely on our collaborator to obtain these approvals. The approval procedure varies among countries and can involve requirements for additional testing. The time required to obtain approval may differ from that required to obtain FDA approval. The regulatory approval process outside the United States may include all of the risks associated with obtaining FDA approval, as well as risks attributable to the satisfaction of local regulation in foreign jurisdictions. In addition, in many countries outside the United States, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. We may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. We and our collaborators may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market.
Recently, some foreign jurisdictions have required biopharmaceutical companies to grant broad licenses to domestic manufacturers as a condition to providing regulatory approval to market and sell within the applicable jurisdiction. If we were forced to grant a broad license to our products as a condition to selling within the applicable jurisdiction, our revenues and profitability would be materially impaired.
In addition, we, along with our collaborators or subcontractors, may not employ, in any capacity, persons who have been debarred under the FDA's Application Integrity Policy. Employment of such a debarred person, even if inadvertently, may result in delays in the FDA's review or approval of our product candidates or the rejection of data developed with the involvement of such person.
Risks Related to Our Intellectual Property
Our success is dependent on obtaining and defending patents and proprietary technology.
Our success in commercializing, producing and marketing products and technologies in the future depends, in large part, on our ability to obtain and maintain proprietary protection of the intellectual property related to our technologies and products, both in the United States and other countries, and to operate without infringing the proprietary rights of third parties. We will be able to protect our
37
proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies are covered by valid and enforceable patents. The patent positions of biotechnology companies, including our patent positions, are generally uncertain and involve complex legal and factual questions.
We do not know whether any of our pending or future patent applications will result in the issuance of patents. In addition, we cannot predict the breadth of claims that may be allowed and issued to us for patents related to our technologies or products, if any. Before a patent is issued, its coverage can be significantly narrowed, either in the United States or abroad. To the extent patents have been issued or will be issued, some of these patents are subject to further proceedings that may limit their scope and once patents have been issued, we cannot predict how the claims will be construed or enforced. It is not possible to determine which patents may provide significant proprietary protection or competitive advantage, or which patents may be circumvented or invalidated. Furthermore, patents already issued to us, or patents that may be issued on our pending applications, may become subject to dispute, including interference proceedings in the United States to determine priority of invention. If our currently issued patents are invalidated or if the claims of those patents are narrowed, our ability to prevent competitors from marketing products that are currently protected by those patents could be reduced or eliminated. We could then face increased competition resulting in reduced market share, prices and profit.
In addition, the laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States, and many companies have encountered significant problems in protecting and defending their proprietary rights in foreign jurisdictions. For example, methods of treating humans are not patentable in many countries outside of the United States.
Our patents may not afford us protection against competitors with similar technology. Because there is a lengthy time between when a patent application is filed and when it is issued, and because publications of discoveries in the scientific literature often lag behind actual discoveries, we cannot be certain that we were the first to make the inventions claimed in our patent applications or that we were the first to file for protection of the inventions set forth in these patent applications. We may also incur substantial costs in asserting claims against, and defending claims asserted against us by, third parties to prevent the infringement of our patents and proprietary rights by others. Participation in such infringement proceedings may adversely affect our business and financial condition, even if the eventual outcome is favorable.
In March 2007, we received an office action from the U.S. Patent and Trademark Office relating to one of our patent applications that makes claims to the composition of matter of TPI 287. In this action, the patent examiner rejected all of the pending claims of our patent application. We are preparing a response to this office action, which we expect to file before September 2007. We cannot predict the ultimate outcome of this office action. If the arguments in our response are not successful, some or all of the original claims in our patent application may have to be narrowed or may not issue at all. If this occurs, this patent application may not result in an issued patent or the patent, if issued, may not provide significant protection or competitive advantage with regard to TPI 287.
Litigation or third party claims of intellectual property infringement could require us to spend substantial time and money defending against any such claim and adversely affect our ability to develop and commercialize our products.
Our success also depends in part on our ability to avoid infringing patents and proprietary rights of third parties and breaching any licenses that we may enter into with regard to any future products. There are many pharmaceutical and chemical patents and applications being filed, published, and issued frequently throughout the world. Some of these patents and applications contain disclosures and claims that are similar to technologies and products that we are using and developing. Some of these
38
patents and disclosures contain claims and disclosures that are difficult to interpret. It is possible that a third party may own or control issued patents, or patent applications or in the future may file, patent applications covering technologies or products we are developing.
If our technology, products or activities are deemed to infringe the other companies' rights, we could be subject to damages or be prevented from using the technology or selling the product that is infringing other companies' rights, or we could be required to obtain licenses to use that technology or sell the product. If patents covering technologies required by our operations are issued to others, we may have to rely on licenses from third parties, which may not be available on commercially reasonable terms, if at all. We could be required to pay substantial license fees or royalties or both. Even if we are able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. Third parties may accuse us of employing their proprietary technology without authorization. In addition, third parties may obtain patents that relate to our technologies and claim that our use of such technologies infringes their patents, even if we have received patent protection for our technology. These claims could require us to incur substantial costs and could have a material adverse effect on us, regardless of the merit of the claims, including the following:
- •
- the diversion of management and technical personnel in defending against any such claims or enforcing our patents either in civil litigation or in a proceeding in the U.S. Patent and Trademark Office, either of which could be expensive and time consuming;
- •
- paying a large sum for damages if we are found to infringe;
- •
- being prohibited from selling or licensing our products or product candidates unless and until we obtain a license from the patent holder, who may refuse to grant us a license or who may only agree to do so on unfavorable terms;
- •
- redesigning our products or product candidates so they do not infringe on the patent holder's technology if we are unable to obtain a license, which, even if possible, could require substantial additional capital and could significantly delay commercialization while we attempt to design around the patents or rights infringed;
- •
- incurring substantial cost in defending ourselves and indemnifying any future collaborator in patent infringement or proprietary rights violation actions brought against them relating to their development and commercialization of our product candidates; and
- •
- incurring substantial cost in indemnifying the investors in our 2006 private placement in the event that any intellectual property infringement is deemed to be a breach of the purchase agreement for the private placement.
We may be required to obtain rights to proprietary technologies that are required to further develop our business and that may not be available or may be costly.
Our development programs may require the use of multiple products or technologies proprietary to other parties. Third party suppliers may not be able to furnish us with a supply of these products sufficient to satisfy our requirements. We may not be able to obtain additional licenses we may need in the future on terms acceptable to us. Our inability to obtain any one or more of these licenses, on commercially reasonable terms, if at all, or to circumvent the need for any such license, could cause significant delays and cost increases and materially affect our ability to develop and commercialize our product candidates. In connection with our efforts to obtain rights to these proprietary technologies, we may find it necessary to convey rights to our technology to others. Some of our products may require the use of multiple proprietary technologies. Consequently, we may be required to make cumulative royalty payments to several third parties. These cumulative royalties could become commercially
39
prohibitive. We may not be able to successfully negotiate the amounts of these royalties on terms acceptable to us.
We may rely in part on third party licenses for access to intellectual property for our product candidates. We expect that any such future licenses will impose various development and commercialization, milestone payment, royalty, sublicensing, patent protection and maintenance, insurance and other obligations on us. If we fail to comply with these obligations or otherwise breach the license agreement, the licensor may have the right to terminate the license in whole, terminate the exclusive nature of the license or bring a claim against us for damages. Any such termination or claim would likely prevent or impede our ability to market any product that is covered by the licensed patents. Even if we contest any such termination or claim and are ultimately successful, our stock price could suffer. In addition, upon any termination of a license agreement, we may be required to license to the licensor any related intellectual property that we developed.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patented technology, we rely upon unpatented proprietary technology, processes and know-how. We seek to protect our unpatented proprietary information in part by confidentiality agreements with our employees, consultants and third parties. These agreements may be breached and we may not have adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently developed by competitors. If we are unable to protect the confidentiality of our proprietary information and know-how, competitors may be able to use this information to develop products that compete with our products, which could adversely impact our business.
Risks Related to Ownership of Our Common Stock
Our principal stockholders will maintain the ability to control all matters submitted to stockholders for approval.
A small number of investors have acquired shares of our common stock and warrants to purchase shares of our common stock that, in the aggregate, represent a majority of our capital stock. As a result, if these stockholders were to choose to act together, they would be able to control all matters submitted to our stockholders for approval, as well as our management and affairs. For example, these stockholders, if they choose to act together, will control the election of directors and approval of any merger, consolidation or sale of all or substantially all of our assets. This concentration of voting power could delay or prevent an acquisition of our company on terms that other stockholders may desire. In addition, one of these stockholders, Special Situations Fund III, L.P., has the right, which it has not exercised, to designate up to two persons for election to our board of directors.
The investors who acquired shares of our common stock and warrants in our 2006 private placement have contractual preemptive rights to acquire their pro rata share of any common stock issued by us. If these stockholders do not waive their preemptive rights or do not otherwise cooperate with us in effecting future offerings, we will be unlikely to be able to complete any of those offerings.
Under the terms of the purchase agreement for our 2006 private placement, we must first offer to sell to each investor that continues to hold at least 50% of the shares of common stock acquired by that investor in our 2006 private placement such investor's pro rata share of any common stock or securities exercisable or exchangeable for or convertible into common stock for a fifteen business day period before we can make an offer of such securities to others. An investor's pro rata share is calculated by reference to the number of shares that continue to be held that were acquired in our 2006 private placement or upon the exercise of warrants and the number of shares of our common
40
stock then issued and outstanding. We believe it will be difficult or impossible to complete a public or private offering if we are required to comply with the preemptive rights granted to the investors under the purchase agreement.
The warrants issued in our 2006 private placement provide for the reduction of the exercise price of the warrants if we issue common stock for a consideration per share less than the then current exercise price of the warrants. If we issue common stock for a consideration per share less than $2.40 after taking in to account the underwriting discount, if applicable, the exercise price of the warrants will be reduced and the number of shares issuable upon exercise of the warrants will be increased.
Under the terms of the warrants issued in our 2006 private placement, the exercise price of the warrants is to be reduced, subject to certain exceptions, if we issue shares of common stock or certain options or rights to acquire common stock for no consideration or for consideration per share less than the exercise price of the warrants in effect immediately prior to the time of such issue or sale. The warrants specify a formula for a weighted average adjustment in the exercise price that takes into account the number of shares of common stock outstanding, the number of shares issued below the exercise price and the aggregate consideration received upon such issuance. Under the warrants, if the exercise price is decreased, the number of shares issuable upon exercise is correspondingly increased. The exercise price of the warrants is currently $2.40 per share. Accordingly, in general, if we issue common stock for a consideration per share of less than $2.40, after taking into account any applicable underwriting discount, the exercise price of the warrants will be correspondingly reduced. Any such reduction in exercise price would result in additional dilution to the holders of our common stock.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital will be required in the future to continue our research, development and clinical efforts. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell common stock in one or more transactions at prices and in a manner we determine from time to time. Stockholders who hold our common stock or purchase stock in any of these offerings may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders.
During 2006, our board of directors and stockholders adopted our 2006 equity incentive plan, which initially reserved 6,577,106 shares of common stock for issuance thereunder. Immediately following any issuance of common stock by us during the three year period ending April 4, 2009, other than issuances of common stock upon exercise of the warrants issued to the investors in our 2006 private placement, the number of shares available for issuance under the our 2006 equity incentive plan will be increased to 20% of the number of fully diluted shares of our common stock immediately after such issuance, including all shares of common stock issuable upon exercise of then outstanding options or warrants, less the number of shares of common stock subject to existing options under our other stock option and equity incentive plans, up to a maximum of 8,177,106 shares. As a result of common stock issuances during 2006, the number of shares available under our 2006 equity incentive plan increased to 6,622,935.
Sales of a substantial number of shares in the future may impact the market price of our common stock.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through public or private equity offerings and debt financings, corporate collaboration and licensing arrangements. Any future sales of a substantial number of shares of our common stock in the public market, either by us or by investors, or the perception that such sales may occur, could depress the market price of our common stock. We are unable to predict the effect that future sales may have
41
on the then prevailing market price of our common stock. Pursuant to a registration rights agreement entered into between us and the investors in our 2006 private placement, we registered with the Securities and Exchange Commission, or SEC, the resale of shares of common stock acquired in the private placement and shares of common stock issuable upon exercise of the warrants. We must, among other things, keep this registration statement effective with the SEC for the resale of such shares of common stock. While we cannot determine precisely the total number of shares of common stock remaining for sale under the registration statement, we estimate based upon publicly available information that up to approximately 23,656,559 shares remain available for sale, including shares that may be acquired upon the exercise of outstanding warrants.
If we fail to continue to meet all applicable NASDAQ Capital Market requirements and NASDAQ determines to delist our common stock, the delisting could adversely affect the market liquidity of our common stock and the market price of our common stock could decline.
Our common stock is listed on the NASDAQ Capital Market. In order to maintain that listing, we must satisfy minimum financial and other requirements. On February 25, 2005, we received notice from the NASDAQ Stock Market, Inc. that our common stock had not met the $1.00 per share minimum bid price requirement for 30 consecutive business days and that, if we were unable to demonstrate compliance with this requirement during the applicable grace periods, our common stock would be delisted after that time. On February 6, 2006, we effected a one-for-ten reverse stock split of our common stock to regain compliance with this listing requirement. Since this time, the closing bid price of our common stock has remained above $1.00 in compliance with the minimum bid price requirement.
It is possible that the minimum bid price of our common stock could fall below the required level or that we would otherwise fail to satisfy another NASDAQ requirement for continued listing of our common stock. For example, we could fail to maintain compliance with the NASDAQ Capital Market listing requirements if we did not maintain minimum stockholder equity of at least $2.5 million as a result of continuing losses.
If we fail to continue to meet all applicable NASDAQ Capital Market requirements in the future and NASDAQ determines to delist our common stock, the delisting could adversely affect the market liquidity of our common stock and the market price of our common stock could decline. Such delisting could also adversely affect our ability to obtain financing for the continuation of our operations and could result in the loss of confidence by investors, suppliers and employees.
Provisions in our charter documents and under Delaware law could discourage, delay or prevent an acquisition of us, which may be beneficial to our stockholders, and may prevent or delay attempts by our stockholders to replace or remove our current management.
Our certificate of incorporation and bylaws provide that our board of directors is divided into three classes, each consisting, as nearly as possible, of one-third of the total number of directors, with each class having a three-year term. The number of our directors may be changed only by a resolution of our board of directors and directors can be removed only for cause by the vote of the holders of at least 80% of the voting power of all of our capital stock, voting together as a single class. Stockholders may take action only at a stockholders' meeting and not by written consent. Certain provisions of our certificate of incorporation and bylaws, including the provisions providing for a classified board of directors, may not be amended without the vote of at least 80% of the voting power of all of our capital stock entitled to vote generally in the election of directors, voting together as a single class. Our bylaws provide that stockholders wishing to nominate a director at an annual meeting or at a special meeting called for the purpose of electing directors or to bring business before any meeting of stockholders must comply with strict advance written notice provisions. Our bylaws also provide that
42
special meetings of stockholders may be called only by the chairman of our board of directors, or certain of our officers, or by resolution of our directors.
These provisions of our certificate of incorporation and our bylaws could discourage potential acquisition proposals and could delay or prevent a change in control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions are intended to enhance the likelihood of continuity and stability in the composition of our board of directors and in the policies formulated by our board of directors. We also intended these provisions to discourage certain types of transactions that may involve an actual or threatened change of control. We designed these provisions to reduce our vulnerability to unsolicited acquisition proposals and to discourage certain tactics that may be used in proxy contests. These provisions, however, could also have the effect of discouraging others from making a tender offer for our shares. As a consequence, they also may inhibit fluctuations in the market price of our shares that could result from actual or rumored takeover attempts. Because our board of directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. We are permitted to issue shares of our preferred stock without stockholder approval upon such terms as our board of directors determines. Therefore, the rights of the holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of our preferred stock that may be issued in the future. In addition, the issuance of preferred stock could have a dilutive effect on the holdings of our current stockholders.
We are also subject to provisions of Delaware law that prohibit us from engaging in any business combination with any "interested stockholder," meaning generally that a stockholder who beneficially owns more than 15% of our outstanding voting stock cannot acquire us for a period of three years from the date of the transaction in which the person acquired more than 15% of our outstanding voting stock, unless various conditions are met, such as approval of the transaction by our board of directors.
Our stockholder rights plan could prevent a change in control of our company in instances in which some stockholders may believe a change in control is in their best interests.
In December 2006, our board of directors adopted a stockholder rights plan to replace our stockholder rights plan that expired in November 2006 at the end of its ten-year term. Our plan may have the effect of discouraging, delaying or preventing a merger or acquisition of us that is beneficial to our stockholders by diluting the ability of a potential acquirer to acquire us. Pursuant to the terms of our plan, when a person or group, except under certain circumstances, acquires 15% or more of our outstanding common stock or 10 business days after announcement of a tender or exchange offer for 15% or more of our outstanding common stock, the rights, other than rights held by the person or group who has acquired or announced an offer to acquire 15% or more of our outstanding common stock, would generally become exercisable for shares of our common stock at a discount. Because the potential acquirer's rights would not become exercisable for our shares of common stock at a discount, the potential acquirer would suffer substantial dilution and may lose its ability to acquire us. In addition, the existence of the plan itself may deter a potential acquirer from acquiring us. As a result, either by operation of the plan or by its potential deterrent effect, mergers and acquisitions of us that our stockholders may consider in their best interests may not occur, including transactions in which stockholders might otherwise receive a premium for their shares.
Because the investors in our 2006 private placement own a substantial percentage of our outstanding common stock, our stockholder rights plan provides that such investors and their respective affiliates will be exempt from the stockholder rights plan, unless an investor and its affiliates acquire, after April 4, 2006, more than 1% of our then issued and outstanding common stock, not including the
43
shares of common stock issued to the investors in the private placement or shares of common stock issued upon exercise of the warrants issued to the investors in the financing.
Because we do not expect to pay dividends in the foreseeable future, you must rely on stock appreciation for any return on your investment.
We have paid no cash dividends on our common stock to date, and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. As a result, we do not expect to pay any cash dividends in the foreseeable future, and payment of cash dividends, if any, will also depend on our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors. Furthermore, we may in the future become subject to contractual restrictions on, or prohibitions against, the payment of dividends. Accordingly, the success of your investment in our common stock will likely depend entirely upon any future appreciation. There is no guarantee that our common stock will appreciate in value or even maintain the price at which you purchased your shares, and you may not realize a return on your investment in our common stock.
If our stock price is volatile, purchasers of our common stock could incur substantial losses.
Our stock price is likely to be volatile. The stock market in general and the market for biotechnology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may not be able to sell their common stock at or above the price at which they purchased their stock. The market price for our common stock may be influenced by many factors, including:
- •
- results of clinical trials of our product candidates or those of our competitors;
- •
- regulatory or legal developments in the United States and other countries;
- •
- variations in our financial results or those of companies that are perceived to be similar to us;
- •
- changes in the structure of healthcare payment systems;
- •
- market conditions in the pharmaceutical and biotechnology sectors and issuance of new or changed securities analysts' reports or recommendations;
- •
- general economic, industry and market conditions; and
- •
- the other factors described in this "Risk Factors" section.
44
The following table sets forth information regarding our leased facilities:
| Location | Approximate Square Feet | Use | Lease Expiration Date | |||
|---|---|---|---|---|---|---|
| Boulder, Colorado | 29,000 | Future office and laboratory space(1) | November 1, 2012 | |||
| Boulder, Colorado | 16,000 | Office and laboratory space | February 1, 2008 | |||
| Roseland, New Jersey | 2,300 | Office space | December 31, 2008 | |||
| Boulder, Colorado | 2,121 | Laboratory space | September 30, 2007 | |||
| New York, New York | 850 | Office space | Month-to-month |
- (1)
- In March 2007, we entered into a five year lease covering this space, into which we plan to consolidate our Colorado-based administrative and research and development facilities. We plan to take possession on November 1, 2007 in order to make necessary renovations.
We also own five acres of undeveloped land in Longmont, Colorado which we account for as other assets.
We believe that these existing facilities are adequate to meet current foreseeable requirements or that suitable additional or substitute space will be available on commercially reasonable terms.
We currently are in arbitration through the American Arbitration Association with the assignor of certain patents and patent applications relating to pharmaceutical formulations containing Vitamin E. The arbitration began in November 2005 in Boulder, Colorado. We licensed these patent applications in 1998. The licensor claims that we have failed in our obligation to develop the technology and is demanding the patents be returned. We have denied this claim and have alleged that the licensor committed fraud in inducing us to enter into the license agreement. We are seeking unspecified damages against the licensor.
We agreed to employ Donald H. Picker, Ph.D., as our president in December 2006. Prior to joining our company, Dr. Picker had served as Executive Vice President of Research and Development of Callisto Pharmaceuticals, Inc. In December 2006, Callisto filed a complaint in the Supreme Court of New York, County of New York, relating to our employment of Dr. Picker. The suit names Tapestry and two of our officers, Leonard Shaykin and Kai Larson, as defendants. In its complaint, Callisto alleges breaches of a confidentiality agreement between Callisto and Tapestry and interference with Dr. Picker's contractual relationship with Callisto. Callisto seeks unspecified actual and punitive damages. We believe these claims are without merit and we are vigorously defending against them.
Dr. Steven K. Carter, a member of our board of directors, is also a director of Callisto and was deposed in connection with this suit. We have reimbursed Dr. Carter's costs in connection with this deposition.
45
Item 5
Market for Registrant's Common Equity, Related Stockholder Matters
and Issuer Purchases of Equity Securities
Market Information
Our common stock is listed on the Nasdaq Capital Market, where it trades under the symbol "TPPH." We implemented a one-for-ten reverse split of our common stock effective for trading on February 6, 2006. All share and per share amounts for all periods presented reflect this reverse stock split. The following table sets forth, for the periods indicated, the high and low closing sale prices for our common stock for the fiscal years ended December 27, 2006 and December 28, 2005:
| | | High | Low | |||||
|---|---|---|---|---|---|---|---|---|
| 2006 | Fourth Quarter | $ | 2.22 | $ | 1.47 | |||
| Third Quarter | 3.60 | 2.12 | ||||||
| Second Quarter | 4.38 | 2.35 | ||||||
| First Quarter | 4.70 | 2.30 | ||||||
| 2005 | Fourth Quarter | $ | 4.00 | $ | 2.60 | |||
| Third Quarter | 5.50 | 3.50 | ||||||
| Second Quarter | 6.60 | 4.50 | ||||||
| First Quarter | 12.20 | 6.50 | ||||||
On April 20, 2007, the last sale price of our common stock on the Nasdaq Capital Market was $1.90 per share.
Stockholders
As of April 20, 2007, we had 348 stockholders of record.
Dividends
To date, we have not paid any dividends on our common stock. We intend to retain future earnings, if any, to finance the operation and expansion of our business and, therefore, we do not anticipate paying any cash dividends on our common stock in the foreseeable future, if at all.
Securities Authorized for Issuance Under Equity Compensation Plans
See Item 12 regarding securities authorized for issuance under our equity compensation plans.
Sales of Unregistered Securities
On December 12, 2006, we issued 30,000 shares of common stock to Schwartz Communications, Inc. under the terms of a consulting agreement. The shares were issued in reliance upon an exemption provided by Section 4(2) of the Securities Act of 1933, as amended, and Regulation D thereunder relative to sales by an issuer not involving any public offering.
46
ITEM 7
Management's Discussion and Analysis of
Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations contains information that management believes is relevant to an assessment and understanding of our results of operations. Some of the statements set forth below constitute forward-looking statements that are based upon current expectations. In some cases, you can identify forward-looking statements by terminology such as "may," "will," "could," "would," "should," "expect," "intend," "plan," "anticipate," "believe," "estimate," "predict," "project," "potential," "continue," or "ongoing" or the negative of these terms or other comparable terminology. Forward-looking statements involve risks and uncertainties. Our actual results and the timing of events could differ materially from those discussed in our forward-looking statements as a result of many factors, including those set forth under "Risk Factors" and elsewhere in this report. You should read this discussion together with our financial statements and the related notes.
General
We are a biopharmaceutical company focused on the development of proprietary therapies for the treatment of cancer. Our core capabilities are deriving and developing drug candidates from natural products. We are currently devoting substantially all of our efforts to the development of TPI 287, a proprietary next generation taxane for the treatment of multiple cancer indications. Taxanes comprise a class of drugs derived from natural products that are used in the treatment of various forms of cancer.
We recently completed dosing in one Phase I clinical trial of TPI 287 and expect to complete dosing in another Phase I clinical trial during the second half of 2007. In addition, we recently commenced enrollment in a Phase II clinical trial of TPI 287 in patients with an advanced form of prostate cancer called hormone refractory prostate cancer, or HRPC. This is our first of several planned Phase II clinical trials of TPI 287 in multiple cancer indications. In our clinical trials to date, we have evaluated intravenous, or IV, formulations of TPI 287. We plan to use an IV formulation in our currently planned Phase II clinical trials. We also are developing an oral formulation of TPI 287 and plan to initiate a Phase Ib/II pharmacokinetic clinical trial of TPI 287 in the summer of 2007. In this clinical trial, we plan to orally administer an IV formulation of TPI 287 to evaluate the drug's activity and oral bioavailability in humans. We hold all worldwide commercial rights for TPI 287.
We intend to expand our product pipeline through strategically in-licensing or acquiring product candidates that complement our business. This may involve the examination of individual molecules, classes of compounds or technologies in cancer as well as other therapeutic areas. Our acquisitions of new product candidates or technologies may also involve the acquisition of, or merger with, other companies.
We have incurred significant operating losses since our inception in 1991. Our net loss from continuing operations was $16.6 million in 2006, $17.2 million in 2005 and $21.6 million in 2004. We have an accumulated deficit of $123.9 million as of December 27, 2006. We expect to incur significant and increasing operating losses for at least the next several years, until such time, if ever, as we are able to generate sufficient sales to support our development operations, including the research and development activity discussed above.
To become and remain profitable, we must succeed in developing and eventually commercializing drugs with significant market potential. This will require us to be successful in a range of challenging activities, including discovering, in-licensing or acquiring product candidates, successfully completing preclinical testing and clinical trials of our product candidates, obtaining regulatory approval for these product candidates and manufacturing, marketing and selling those products for which we may obtain regulatory approval. We are only in the preliminary stages of development of TPI 287, our only current product candidate. We may never succeed in these activities and may never generate revenues that are significant enough to achieve profitability.
47
Private Placement
On April 5, 2006 we sold an aggregate of 12,750,000 shares of our common stock and warrants to purchase up to 12,750,000 shares of our common stock in a private placement for a total of $25.5 million, not including any proceeds that might be received upon exercise of the warrants. We received approximately $23.8 million net of the placement agent fees and other expenses in this private placement. The warrants currently have an exercise price of $2.40 per share. Under the terms of these warrants, the exercise price may be reduced if we issue shares of common stock or certain options or rights to acquire common stock for no consideration or for consideration per share less than the exercise price of the warrants in effect immediately prior to the time of such issue or sale. The warrants specify a formula for a weighted average adjustment in the exercise price that takes into account the number of shares of common stock outstanding, the number of shares issued below the exercise price and the aggregate consideration received upon such issuance.
Under the warrants, if the exercise price is decreased, the number of shares issuable upon exercise is correspondingly increased. Accordingly, if we issue common stock for a consideration per share of less than $2.40, after taking into account any applicable underwriting discount, the exercise price of the warrants will be correspondingly reduced. Any such reduction in exercise price would result in additional dilution to the holders of our common stock.
We may call up to 20% of the outstanding warrants at a redemption price of $0.0075 per share of common stock underlying the warrants during any three month period if certain conditions are satisfied, including the trading price of our common stock exceeding $4.80 for 20 consecutive trading days, upon 30 days prior notice. During the notice period, the holders would be entitled to exercise the warrant or any portion of it. Up to half of these warrants may be exercised on a cashless or net exercise basis. There can be no assurance, however, that we will receive any funds from the exercise of warrants.
In addition, we issued warrants to purchase 50,000 shares of common stock to a financial advisor and issued warrants to purchase 100,000 shares of common stock to an outside consultant as a finders' fee on substantially similar terms as the warrants issued in this private placement.
In connection with the private placement, we entered into a registration rights agreement in which we agreed to make the requisite filings with the Securities and Exchange Commission to achieve and substantially maintain the effectiveness of a registration statement covering shares sold in the private placement, as well as shares underlying warrants issued in this private placement. If we fail to maintain effectiveness of that registration statement through as late as April 6, 2013, subject to our right to suspend use of the registration statement in certain circumstances, we will have to pay liquidated damages to investors in an amount equal to 1.5% of the amount invested by them for each 30 day period during which the registration statement should have been effective.
We are required to use the net proceeds from this private placement to fund the development of TPI 287 in accordance with a budget for calendar years 2006 and 2007 that was adopted by our board of directors before the closing of the private placement. Any change to this budget will require the approval of a majority of the independent members of our board of directors.
Research and Development
Our current business is focused on research and development of proprietary therapies for the treatment of cancer. We were previously engaged in development activities related to our paclitaxel business, which we sold to Mayne Pharma (USA) Inc. in 2003, genomic technologies, which we discontinued in 2004, and Huntington's Disease, which we ceased in 2006. Costs relating to genomic technologies, excluding our Huntington's Disease program, and the paclitaxel business, are aggregated
48
in discontinued operations. During the last three fiscal years, we have incurred the following expenses related to research and development projects:
| | 2004 | 2005 | 2006 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | (in thousands) | |||||||||
| Oncology | $ | 12,474 | $ | 9,818 | $ | 10,476 | ||||
| Huntington's Disease | 1,030 | 812 | (87 | ) | ||||||
| Discontinued operations | 4,242 | 393 | 59 | |||||||
| $ | 17,746 | $ | 11,023 | $ | 10,448 | |||||
We expect research and development, which includes the cost of our clinical development of TPI 287, to continue to be the most significant expense for our business for the foreseeable future. Our research and development activity is subject to change as we develop a better understanding of our projects and their prospects.
In May 2005, we began our initial Phase I clinical trial, which is ongoing. This trial is designed to determine TPI 287's maximum tolerated dose, or MTD, and evaluate the safety and anti-tumor activity and the pharmacokinetic and pharmacodynamic profiles of TPI 287 in eligible patients with recurrent or refractory malignancies. This trial is nearing completion, and we expect to present the data from this trial at a conference in June 2007. In January 2006, we began our second Phase I trial with an alternative dosing regimen from our initial Phase I trial, which we recently completed. This trial is designed to determine TPI 287's MTD and evaluate the safety and anti-tumor activity and the pharmacokinetic and pharmacodynamic profiles of TPI 287 in eligible patients with recurrent or refractory malignancies. We expect to present the data from this trial at a conference in June 2007.
We cannot be sure that we will be able to achieve our goals relating to these programs. We also cannot estimate the cost necessary to complete these programs, the timing of material net cash inflows from these programs, or whether we will ever recognize revenues from these programs. Continued development of these programs is dependent upon raising additional capital. We cannot be certain that we will be able to obtain capital on acceptable terms, or at all.
Results of Operations
Year Ended December 27, 2006 Compared to Year Ended December 28, 2005
Research and Development Expense. Research and development expenses from continuing operations for 2006 were $10.4 million, as compared to $10.6 million in 2005, a decrease of $241,000. Oncology related research and development expenditures increased by $657,000 to $10.5 million for 2006. In 2006, we ceased our Huntington's Disease research. As a result, our spending on research related to Huntington's Disease decreased by $899,000 from $812,000 in 2005 to a credit of $87,000 for 2006. The decrease in expenses related to the Huntington's Disease program was a result of a reversal of laboratory fees owed to a research university as well as a reduction in work force. We do not expect to incur additional expenses related to this program in the future.
The increase in oncology related expenses was primarily due to an increase in compensation expense of $1.0 million, an increase in clinical trial costs of $292,000, an increase in supply costs of $403,000, an increase in legal fees of $173,000 and an increase in insurance expenses of $173,000, which were partially offset by reductions in outside manufacturing costs of $1.2 million associated with preparing the active pharmaceutical ingredient and drug product for clinical trials of TPI 287. We expect our oncology related expenses to increase in 2007 and 2008 in connection with our ongoing and planned Phase I and Phase II clinical trials of TPI 287.
The increase in compensation related expenses for 2006 was primarily related to non-cash compensation expense of $1.3 million for 2006, as compared to $36,000 for 2005, resulting from the
49
implementation of SFAS 123(R) and the modifications of vested stock options. In addition, in the first quarter of 2006, we recorded a $646,000 charge in connection with the termination of the employment of our executive vice president and secretary. These increases were partially offset by lower salary expense due to staffing reductions that we implemented in July 2005. We expect our compensation expenses to increase in 2007 and 2008 as a result of an increase in our headcount to support our increased clinical development activities, as well as an increase in non-cash equity compensation expense as a result of stock options issued to new research and development personnel.
General and Administrative Expense. General and administrative expense from continuing operations for 2006 was $7.0 million, an increase of $1.3 million from 2005. This increase was primarily due to higher compensation related expenses of $1.5 million and costs related to our board of directors of $231,000, which were partially offset by a reduction in insurance expenses of $174,000, a reduction in legal expenses of $68,000 and a reduction in outside service costs related to investor relations of $133,000. The increase in compensation related expenses, including costs related to our board of directors, was primarily related to non-cash compensation expense of $2.0 million for 2006, as compared to $11,000 for 2005, resulting from the implementation of SFAS 123(R), partially offset by lower salary expense due to the staffing reductions in July 2005. We expect our general and administrative expenses for 2007 to remain similar to 2006 levels.
Interest and Other Income. Interest and other income of $1.3 million for 2006 increased by $565,000 from 2005. The increase was primarily attributable to higher interest income due to higher investment balances and higher effective interest rates earned in 2006 compared to 2005.
Interest and Other Expense. Interest and other expense was $518,000 for 2006, a decrease of $39,000 from 2005. The decrease is attributable to our repurchase of notes payable to TL Ventures V L.P. and TL Ventures V Interfund L.P. in November 2006.
Impairment Charges. Impairment charges for 2005 of $1.1 million were related to a reduction in the fair market value of our investment in ChromaDex of $963,000 and a reduction in the fair market value of our land of $104,000. There were no impairment charges in 2006.
Discontinued Operations. Loss from discontinued operations was $59,000 for 2006, as compared to $358,000 in 2005, a decrease of $299,000. This decrease was primarily a result of our discontinuation of our genomics business in 2005. Our only expense related to this business in 2006 was compensation related expense of $57,000 primarily related to our implementation of SFAS 123(R).
Year Ended December 28, 2005 Compared to Year Ended December 29, 2004
Research and Development Expense. Research and development expenses from continuing operations for 2005 were $10.6 million, as compared to $13.5 million in 2004, a decrease of $2.9 million. This decrease was primarily due to decreased preclinical development activities, and consisted of a reduction in outside toxicology expense of $1.2 million and a reduction in contract manufacturing expense of $1.8 million. Reductions in compensation and consulting related expenses, which were primarily due to the staffing reductions in July 2005, were partially offset by payments under an employment agreement between us and a former founder to his estate following his death in August 2005.
General and Administrative Expense. General and administrative expense from continuing operations for 2005 was $5.6 million, a decrease of $2.2 million from 2004. This decrease was primarily due to a reduction in compensation related expenses of $868,000 as a result of our staffing reductions in July 2005, a reduction in legal expenses of $352,000, a reduction in insurance expenses of $206,000, a reduction in consulting and outside service expenses of $467,000 and a reduction in rent and occupancy expense of $108,000.
50
Interest and Other Income. Interest and other income of $731,000 for 2005 increased by $37,000 from $694,000 in 2004. The increase was primarily attributable to a refund of income taxes in 2005 in the amount of $114,000 and a gain on sale of investments of $28,000, offset by lower interest income of $92,000 as a result of lower investment balances in 2005 compared to 2004.
Interest and Other Expense. Interest and other expense was $557,000 for 2005, a decrease of $390,000 from 2004. The decrease is attributable to a partial repayment of our notes payable to TL Ventures V L.P. and TL Ventures V Interfund L.P. in February 2005 in connection with the restructuring of our notes owed to them.
Impairment Charges. Impairment charges for 2005 of $1.1 million were related to a reduction in the fair market value of our investment in ChromaDex of $963,000 and a reduction in the fair market value of land of $104,000. There where no impairment charges from continuing operations in 2004.
Discontinued Operations. Loss from discontinued operations was $358,000 for 2005, as compared to $2.6 million in 2004, a decrease of $2.2 million. The loss in 2005 was due to the remaining activity related to the discontinuation of our genomics business. The loss in 2004 was primarily due to a loss of $5.7 million related to the discontinuation of our genomics business, which was partially offset by $3.0 million of proceeds from the settlement of litigation against Mylan Laboratories related to the paclitaxel business that we sold in 2003.
Research and development expense included in discontinued operations was $393,000 for 2005, as compared to $4.2 million for 2004, a decrease of $3.8 million. The decrease was primarily attributable to the majority of the costs associated with the discontinuation of our genomics business being accounted for in 2004. The 2005 expenses primarily consisted of patent legal costs of $185,000 and costs related to the closure of our Delaware facility in the amount of $129,000.
There were no general and administrative expenses included in discontinued operations in 2005 or 2004.
Liquidity and Capital Resources
Our capital requirements for operations have been, and will continue to be, significant. As of December 27, 2006, we had a working capital balance of $20.2 million, as compared to a working capital balance of $11.6 million as of December 28, 2005. To date, we have financed our operations primarily with the net proceeds of public offerings of common stock and private placements of equity securities, with proceeds from the exercise of warrants and options and with debt. We also have funded our capital requirements with the proceeds of the sale of our paclitaxel business to Mayne Pharma in 2003.
We expect our capital needs to increase in 2007 as compared to 2006 due, in part, to increases in expenses related to our clinical studies and to the capital requirements associated with our new laboratory space.
Reverse Stock Split and NASDAQ Listing. Our common stock is listed on the NASDAQ Capital Market. In order to maintain that listing, we must satisfy minimum financial and other requirements. On February 25, 2005, we received notice from the NASDAQ Stock Market, Inc. that our common stock had not met the $1.00 per share minimum bid price requirement for 30 consecutive business days and that, if we were unable to demonstrate compliance with this requirement during the applicable grace periods, our common stock would be delisted after that time. On February 6, 2006, we effectuated a one-for-ten reverse stock split of our common stock to regain compliance with this listing requirement. Since this time, the closing bid price of our common stock has remained above $1.00 in compliance with the minimum bid price requirement.
Notes Payable. On November 17, 2006, we repurchased two non-interest bearing promissory notes issued by us to TL Ventures V L.P. and TL Ventures V Interfund L.P. in February 2005. These
51
promissory notes had an outstanding principal balance of $3.0 million, and we repurchased them for an aggregate payment of $2.7 million in cash. The repurchase of these notes did not have a material impact on our financial results.
Private Placement. On April 5, 2006 we sold an aggregate of 12,750,000 shares of our common stock and warrants to purchase up to 12,750,000 shares of our common stock in a private placement for a total of $25.5 million, not including any proceeds that might be received upon exercise of the warrants. We received approximately $23.8 million net of the placement agent fees and other expenses in this private placement. The warrants currently have an exercise price of $2.40 per share. We may call up to 20% of the outstanding warrants at a redemption price of $0.0075 per share of common stock underlying the warrants during any three-month period if certain conditions are satisfied, including the trading price of our common stock exceeding $4.80 for 20 consecutive trading days, upon 30 days prior notice. During the notice period, the holders would be entitled to exercise the warrant or any portion of it. Up to half of these warrants may be exercised on a cashless or net exercise basis. There can be no assurance, however, that we will receive any funds from the exercise of warrants.
Liquidity. We have no revenue and we have incurred significant operating losses since our inception. Our net loss from continuing operations was $16.7 million in 2006, $17.5 million in 2005 and $24.2 million in 2004. We have an accumulated deficit of $123.9 million as of December 27, 2006. As of December 27, 2006, we had cash and short-term investments totaling approximately $22.5 million. Our capital requirements for research and development, including the cost of clinical trials, have been and will continue to be significant.
We believe that our existing cash and cash equivalents and short-term investments, will be sufficient to enable us to fund our operations through the end of 2007. We expect our research and development expenses to increase in connection with our ongoing activities, particularly as we continue the clinical development of TPI 287. In addition, subject to obtaining regulatory approval of TPI 287 or any other product candidate, we expect to incur significant commercialization expenses for product sales, marketing, securing commercial quantities of product from our manufacturers and distribution. We will need substantial additional funding and may be unable to raise capital when needed or on attractive terms, which would force us to delay, reduce or eliminate our research and development programs or commercialization efforts. See "Risk Factors—Risks Related to Our Business".
Working Capital and Cash Flow. Cash and cash equivalents and short-term investments were $22.5 million at December 27, 2006 and $14.1 million at December 28, 2005, an increase of $8.4 million. For the year ending December 27, 2006, net cash used in operating activities was $11.8 million. Net cash used in investing activities was $8.7 million for the year ended December 27, 2006 primarily due to net purchases of investments. Net cash provided by financing operations was $20.2 million for the year ended December 27, 2006 year due to our private placement during the second quarter, offset by the payment of notes payable in conjunction with our repurchase of notes payable to TL Ventures V L.P. and TL Ventures V Interfund L.P. in November of 2006.
Our cash used in operating activities was primarily used to advance our product development efforts and for general corporate purposes. The majority of our future cash expenditures are expected to continue to be used to advance our product development programs including clinical trials of TPI 287, as well as for general corporate purposes.
Capital Expenditures. We spent $290,000 during 2006 for capital projects. We expect capital expenditures to increase during 2007. We expect the primary focus of capital spending during 2007 to be in support of our research and development activities as well as to fund additional equipment related to an anticipated consolidation of our administrative and research and development facilities in our new laboratory space.
Net Operating Loss Carryforwards. As of December 27, 2006, we had approximately $113.7 million of net operating loss carryforwards, which we refer to as NOLs, to offset future taxable income. The
52
Internal Revenue Code contains provisions that may limit the use of NOLs and tax credits available for use in any given year upon the occurrence of certain events, including significant changes in ownership interest. A change in ownership of a company within a three-year period results in an annual limitation on our ability to utilize our NOLs and tax credit carryforwards from tax periods prior to the ownership change. As a result of our 2006 private placement, which is considered to be a significant change in ownership for tax purposes, our NOL and tax credit carryforwards as of December 27, 2006 are subject to significant limitations. We may be subject to additional limitation in connection with this offering. We believe that we will not be able to use a significant portion of our NOLs and tax credits to offset our income in the future.
Business Development Activities. We intend to expand our product pipeline through strategically in-licensing or acquiring product candidates that complement our business. This may involve the examination of individual molecules, classes of compounds or technologies in cancer as well as other therapeutic areas. Our acquisitions of new product candidates or technologies may involve the acquisition of, or merger with other companies. Such transactions could materially affect our capital requirements.
Critical Accounting Policies
We have identified certain accounting policies as critical to our business operations and the understanding of our results of operations. The impact and any associated risks related to these policies on our business operations are discussed throughout this report where such policies affect our reported and expected financial results. For a detailed discussion on the application of these and other accounting policies, see note 1 to our consolidated financial statements.
Long-Lived Assets Policy: In accordance with SFAS No. 144, "Accounting for the Impairment of Long-Lived Assets," we review the carrying amount of long-lived assets when facts and circumstances suggest they may be impaired. If this review indicates long-lived assets will not be recoverable as determined based on the undiscounted cash flow estimated to be generated by these assets, we reduce the carrying amount of these long-lived assets to estimated fair value or discounted cash flow, as appropriate.
In the second quarter of 2005, we recognized an impairment on the value of land of $104,000 and in the third quarter recognized an impairment of $963,000 on the value of our investment in ChromaDex. In 2004, we recognized an impairment loss of $205,000 associated with the gene isolation and service business due to our decision to discontinue our efforts in that business and our inability to find a buyer for the assets. Also in 2004, as part of our preparation of the financial statements, we recognized an impairment loss of $1.2 million in connection with the closure of our genomics division. These 2004 impairments are included in discontinued operations. Such impairment losses were the only impairment charges of long-lived assets recorded in the fiscal years ended December 28, 2005 and December 29, 2004. There was no impairment of assets in 2006.
Stock Based Compensation: On December 29, 2005, we adopted SFAS No. 123 (revised 2004), "Share-Based Payment," which we refer to as SFAS 123(R). SFAS 123(R) requires us to measure and recognize compensation expense for all share-based payment awards made to our employees and directors based on estimated fair values. SFAS 123(R) supersedes our previous accounting under Accounting Principles Board Opinion No. 25, "Accounting for Stock Issued to Employees." Under the provisions of this opinion and its related interpretations, no compensation expense was recognized with respect to the grant to employees of options to purchase our common stock when such stock options were granted with exercise prices equal to or greater than market value of the underlying common stock on the date of grant.
We adopted SFAS 123(R) using the modified prospective transition method, which requires the application of the accounting standard as of December 29, 2005, the first day of our 2006 fiscal year.
53
Our consolidated financial statements as of and for the fiscal year ended December 27, 2006 reflect the impact of SFAS 123(R). In accordance with the modified prospective transition method, our consolidated financial statements for prior periods have not been restated to reflect, and do not include, the impact of SFAS 123(R).
Stock-based compensation expense recognized under SFAS 123(R) for the fiscal year ended December 27, 2006 was $3.4 million, which increased basic and diluted loss per share from continuing operations by $0.26. Included in stock-based compensation expense for the fiscal year ended December 27, 2006, is a one time non-cash fixed period charge of $381,000, as a result of the modification of vested options on April 4, 2006 as discussed in "Note 8. Equity Incentive Plans" in our consolidated financial statements. The stock-based compensation expense is calculated on a straight-line basis over the vesting periods of the related options. This charge had no impact on our reported cash flows.
Investments: Short-term investments consist of commercial paper, investment grade government agency, auction rate, and corporate debt securities due within one year. Investments with maturities beyond one year may be classified as short-term based on their highly liquid nature and because such investments represent the investment of cash that is available for current operations. We record our investments in auction rate securities at cost, which approximates fair market value. In accordance with ARB 43, "Restatement and Revision of Accounting Research Bulletins," despite the long-term nature of their stated contractual maturities, we have the ability and the intent to liquidate investments in auction rate securities within six months and therefore we have classified these investments as short term. We classify our investments as available-for-sale, and we report them at market value as of the balance sheet date. We recognize interest income when it is earned. We report unrealized gains and losses as a separate component of stockholder's equity until we sell the security or we determine a decline in fair value is other than temporary.
We account for our investment in ChromaDex under the cost method. Under the cost method, we carry the investment at cost and we adjust only for other-than-temporary declines in fair value, distributions of earnings or additional investments. See "Note 12. Investment in ChromaDex, Inc." in our consolidated financial statements for further information on ChromaDex.
Future Contractual Obligations
The table below summarizes our future contractual obligations at December 27, 2006:
| | Total | Less than 1 year | 1-3 years | 4-5 years | After 5 years | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | (in thousands) | ||||||||||||
| Capital lease | $ | 250 | $ | 130 | $ | 120 | — | — | |||||
| Operating leases | 162 | 151 | 11 | — | — | ||||||||
| Total | $ | 412 | $ | 281 | $ | 131 | — | — | |||||
In February 2007, we leased 2,300 square feet of office space in Roseland, New Jersey for research and development support. The lease runs through December 31, 2008 and requires monthly rental payments of $4,500.
In March 2007, we entered into a five year lease covering approximately 29,000 square feet of office and laboratory space in Boulder, Colorado. We plan to consolidate our Colorado-based administrative and research and development facilities into this space in the fourth quarter of 2007. We plan to take possession of this space in November 2007 to make the necessary renovations.
We have employment agreements with a number of key employees. These employment agreements contain provisions regarding the termination of employment including termination of employment associated with a change in control. These termination benefits range from 100% to 300% of the
54
employee's base salary for the prior 12-month period immediately preceding the termination date, payment of any accrued but unpaid salary, bonus and vacation and payment of up to 100% of the prior year bonus or 75% of base salary for the prior 12-month period.
Impact of Recent Accounting Pronouncements
On June 9, 2005, the FASB issued SFAS No. 154, "Accounting Changes and Error Corrections." SFAS No. 154 replaces APB Opinion No. 20, "Accounting Changes," and SFAS No. 3, "Reporting Accounting Changes in Interim Financial Statements," and changes the requirements for the accounting for and reporting of a change in accounting principle. SFAS No. 154 applies to all voluntary changes in accounting principle. It also applies to changes required by an accounting pronouncement in the unusual instance that the pronouncement does not include specific transition provisions. SFAS No. 154 must be adopted for accounting changes and corrections of errors made in fiscal years beginning after December 15, 2005. The adoption of SFAS No. 154 did not have a material impact on our financial results.
In July 2006, the Financial Accounting Standards Board issued FASB Interpretation 48,"Accounting for Uncertainty in Income Taxes: an interpretation of FASB Statement No. 109." Interpretation 48, which clarifies Statement 109, "Accounting for Income Taxes," establishes the criterion that an individual tax position has to meet for some or all of the benefits of that position to be recognized in our financial statements. On initial application, Interpretation 48 will be applied to all tax positions for which the statute of limitations remains open. Only tax positions that meet the more-likely-than-not recognition threshold at the adoption date will be recognized or continue to be recognized. The cumulative effect of applying Interpretation 48 will be reported as an adjustment to retained earnings at the beginning of the period in which it is adopted. Interpretation 48 is effective for fiscal years beginning after December 15, 2006. The adoption of Interpretation 48 did not have a material impact on our financial results.
In September 2006, the FASB issued SFAS No. 157, "Fair Value Measurements." This standard defines fair value, establishes a framework for measuring fair value in accounting principles generally accepted in the United States, and expands disclosure about fair value measurements. This pronouncement applies to other accounting standards that require or permit fair value measurements. Accordingly, this statement does not require any new fair value measurement. This statement is effective for fiscal years beginning after November 15, 2007, and interim periods within those fiscal years. We will be required to adopt SFAS No. 157 in the first quarter of fiscal year 2008. Management has not yet determined the impact of adopting this statement.
Quantitative and Qualitative Disclosures About Market Risk
We currently invest our excess cash balances in money market accounts, and short-term investments that are subject to interest rate risk. The amount of interest income we earn on these funds will decline if interest rates decline. Our investments are subject to a loss of principal with an increase in interest rates if sold prior to their maturity. However, due to the short-term nature of the majority of our investments, the high credit quality of our portfolio and our ability to hold our investments until maturity, an immediate change in interest rates would not have a material impact on our financial position, results of operations or cash flows.
55
Item 10
Directors, Executive Officers and Corporate Governance
Executive Officers and Directors
Our executive officers and directors and their respective ages and positions as of March 28, 2007 are as follows:
| Name | Age | Position | ||
|---|---|---|---|---|
| Leonard P. Shaykin | 63 | Chairman of our Board and Chief Executive Officer | ||
Donald H. Picker | 61 | President | ||
Martin Batt | 64 | Senior Vice President and Chief Operating Officer | ||
Gordon H. Link, Jr. | 53 | Senior Vice President and Chief Financial Officer | ||
Kai P. Larson | 42 | Vice President and General Counsel | ||
Stephen K. Carter | 68 | Director | ||
George M. Gould | 69 | Director | ||
Arthur H. Hayes, Jr. | 72 | Director | ||
Elliot M. Maza | 51 | Director | ||
The Honorable Richard N. Perle | 64 | Director | ||
Patricia A. Pilia | 57 | Director | ||
Robert E. Pollack | 66 | Director |
Our executive officers are appointed by and serve at the discretion of our board of directors. There are no family relationships between any of our directors or executive officers.
Leonard P. Shaykin has served as the Chairman of our board of directors since 1993 and as our Chief Executive Officer since 1999. In 1995, Mr. Shaykin founded Shaykin & Co., LLC, a private investment and management company. Prior to founding Shaykin & Co., Mr. Shaykin was a managing partner of Adler & Shaykin, an investment partnership organized to sponsor management leveraged buyouts. Prior to that, Mr. Shaykin was Vice President, Director and a member of the investment committee of Citicorp Venture Capital, Ltd. and Citicorp Capital Investors, Inc., the venture capital and equity investment subsidiaries of Citicorp and Citibank. Mr. Shaykin is a trustee of The Jackson Laboratory, a not-for-profit genetic research institute; a member of the board of directors of Trireme Systems, Ltd., a private company that provides integrated tracking and surveillance technology solutions for the commercial and security needs of businesses and government agencies; a member of the board of the American Friends of the University of Sussex, Brighton, UK; and a member of the advisory board of the American Center for Democracy. Mr. Shaykin received a B.A. and an M.A. from the University of Chicago and an M.B.A. from the University of Chicago Graduate School of Business.
Donald H. Picker, Ph.D., has served as our President since January 2007. Prior to joining us, from May 2003 to December 2006, Mr. Picker was Executive Vice President of Research and Development of Callisto Pharmaceuticals, Inc. From 1999 to 2003, Mr. Picker was Chief Executive Officer and President and a member of the board of directors of Synergy Pharmaceuticals Inc. Prior to that, Mr. Picker served as President and Chief Operating Officer of LXR Biotechnology Inc. and as Senior Vice President of Research and Development at Genta Inc. Mr. Picker is a member of the board of directors of Xenomics Inc., a public DNA diagnostic company he co-founded in 2004. He also
56
co-founded Fermavir Pharmaceuticals, Inc., a public biopharmaceutical company specializing in infectious diseases, in 2005. Mr. Picker received a B.S. in chemistry from Brooklyn Polytechnic Institute and a Ph.D. in organic chemistry from the State University of New York at Albany.
Martin Batt has served as our Senior Vice President, Chief Operating Officer since April 2005. Previously, he served as our Vice President, Chief Operating Officer from July 2004 to April 2005 and as our Chief Information Officer from 2002 to July 2004. Prior to joining us, Mr. Batt was a partner in the consulting firm of Grisanti, Galef & Goldress, Inc., which specializes in operating and reviving distressed companies by providing leadership in senior executive positions. Prior to that, Mr. Batt served in various information technology positions at U. S. Steel Corporation. Mr. Batt received a B.S. from Point Park College.
Gordon H. Link, Jr. has served as our Senior Vice President and Chief Financial Officer since 2002. Previously, he served as the President of our Genomics Division from 2000 to 2002 and as our Vice President and Chief Financial Officer from 1993 to 2002. Prior to joining us, Mr. Link served as Corporate Controller of Synergen, Inc., Treasurer of the Syntex-Synergen Neuroscience Joint Venture, Treasurer of Synergen Development Corporation and Audit Manager with Deloitte & Touche USA LLP. He is a certified public accountant (inactive) and a certified management accountant. Mr. Link received a B.S. from Rensselaer Polytechnic Institute and a B.A. in accounting from Metropolitan State College. He also attended the graduate school of the University of Denver.
Kai P. Larson, Esq., has served as our Vice President and General Counsel since 1999. Previously, he served as our Director of Legal Affairs from 1994 to 1999. Prior to joining us, Mr. Larson was as an attorney in the New York office of Kirkland & Ellis LLP. Mr. Larson received a B.A. from Brigham Young University and a J.D. from Columbia University School of Law.
Stephen K. Carter, M.D., has served as a member of our board of directors since March 2004. He has been a consultant in the pharmaceutical industry since 1997. Dr. Carter is a member of the board of directors of Alfacell Corporation, Callisto Pharmaceuticals, Inc., Celator Technologies, Inc., Cytogen Corporation, Emisphere Technologies, Inc. and Vion Pharmaceuticals, Inc. In addition, Dr. Carter has served as consultant to and Senior Vice President of Clinical and Regulatory Affiars at SUGEN Inc. He has also served as Senior Vice President, Research and Development at Boehringer Ingelheim Pharmaceuticals, Inc. and Senior Vice President, Worldwide Clinical Research and Development at Bristol-Myers Squibb Co. Dr. Carter is a former Deputy Director at the National Cancer Institute's Division of Cancer Treatment and is a member of the American Society of Clinical Oncology. Dr. Carter received an A.B. from Columbia College and an M.D. from New York Medical College.
George M. Gould, Esq., has served as a member of our board of directors since January 2003. He has served as Of Counsel to the law firm of Gibbons P.C. since 1996. Mr. Gould is a member of the board of directors of Angiogenex, Inc. and Supratek Pharma Inc. Mr. Gould has also served as a Senior Vice President of PharmaGenics, Inc. and Vice President, Licensing & Corporate Development and Chief Patent Counsel for Hoffmann-La Roche Inc. Mr. Gould received a B.A. in organic chemistry from The Johns Hopkins University, a J.D. from Columbia University School of Law and an L.L.M. from New York University School of Law. He also attended the New York University Graduate School of Chemistry.
Arthur H. Hayes, Jr., M.D., has served as a member of our board of directors since 1996. He was President and Chief Operating Officer of MediScience Associates, Inc., a pharmaceutical consulting company, from 1991 until December 2005. In addition, Dr. Hayes has served as a Professor of Medicine at New York Medical College and Pennsylvania State University College of Medicine; Commissioner of the United States Food and Drug Administration; President and Chief Executive Officer and a member of the board of directors of EM Pharmaceuticals, Inc.; Provost and Dean at New York Medical College; Director of the Institute of Human Values in Medical Ethics, International Health; and Director and Chairman of the Department of Biomedical Sciences. Dr. Hayes has also
57
held several other posts with Pennsylvania State University, including Dean of Admissions and Director of the Division of Clinical Pharmacology. Dr. Hayes currently serves on the board of directors of Myriad Genetics, Inc. and Celgene Corporation. Dr. Hayes received a B.A. from Santa Clara University and an M.S. from Oxford University, where he was a Rhodes Scholar. Dr. Hayes received an M.D. from Cornell University Medical College and attended Cornell's Graduate School of Medical Sciences, Department of Pharmacology.
Elliot M. Maza, has served as a member of our board of directors since December 2004. Since May 2006, Mr. Maza has served as President and Chief Financial Officer of Intellect Neurosciences, Inc., a biopharmaceutical company specializing in the research and development of drugs to treat Alzheimer's disease and other major disorders of the central nervous system. From December 2003 to May 2006, Mr. Maza was Chief Financial Officer of Emisphere Technologies, Inc., a public biopharmaceutical company specializing in oral drug delivery. From March 1999 to December 2003, he was a partner at Ernst & Young LLP. Prior to that, Mr. Maza held various positions at Goldman Sachs & Co., J.P. Morgan Securities, Inc. and the law firm of Sullivan & Cromwell LLP. He is a certified public accountant. Mr. Maza received a B.A. in accounting from Touro College and a J.D. from the University of Pennsylvania Law School.
The Honorable Richard N. Perle has served as a member of our board of directors since July 2000. He has served as a fellow at the American Enterprise Institute since 1987. Mr. Perle is also a director of Autonomy, plc. From 1981 to 1987, Mr. Perle served as the United States Assistant Secretary of Defense for International Security Policy. He received a B.A. from the University of Southern California and an M.A. from Princeton University. Mr. Perle also attended the London School of Economics and completed various fellowships at Princeton University, the Ford Foundation and the American Council of Learned Societies.
Patricia A. Pilia, Ph.D., has served as a member of our board of directors since our inception in 1991. Her term expires at our 2007 annual meeting of stockholders and she will not be standing for reelection. She is a co-founder of our company as well as one of our predecessor companies, Pacific Biotechnology, Inc. In addition, she was an officer from 1991 until February 2006, serving in various capacities including Executive Vice President, Vice President of BioResearch and Toxicology, Secretary, and the head of various departments. Prior to joining us, Dr. Pilia served as Assistant Professor of Pathology in the Colleges of Medicine, Dental Medicine and Graduate Studies at the Medical University of South Carolina and as the Assistant Director of the Immunopathology Diagnostic and Research Laboratories. Dr. Pilia is a member of the board of trustees of Rosemont College and the Academic Affairs Committee as well as Vice Chairman of the Development Committee. Currently, she is a member of the board of directors and consults for Regulus Pharmaceuticals, Inc., a regulatory consulting firm, and Sequoyah Consulting, a pharmaceutical and business development company. Dr. Pilia also sits on the advisory board of AZOS, an artificial intelligence and communications development company. Dr. Pilia received a B.S. from Boston University and a M.S. and a Ph.D. from the Medical University of South Carolina.
Robert E. Pollack, Ph.D., has served as a member of our board of directors since July 2000. He is currently a Professor of Biological Sciences, Adjunct Professor of Environmental, Ecological and Evolutionary Biology and Lecturer in Psychiatry at the Center for Psychoanalytic Training and Research at Columbia University. In addition, Dr. Pollack is a Director of the Center for the Study of Science and Religion at Columbia University and an Adjunct Professor of Science and Religion at Union Theological Seminary. He has served as Professor of Biological Sciences at Columbia University and Dean of Columbia College. He received the Alexander Hamilton Medal from Columbia University and has held a Guggenheim Fellowship. Dr. Pollack currently serves on the advisory board of the John Templeton Foundation and as a Senior Consultant for the Director, Program of Dialogue on Science, Ethics and Religion, American Association for the Advancement of Science. Dr. Pollack received a B.A. in physics from Columbia University and a Ph.D. in biology from Brandeis University.
58
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act of 1934 (the "1934 Act") requires the Company's directors and executive officers, and persons who own more than ten percent of a registered class of the Company's equity securities, to file with the SEC initial reports of ownership and reports of changes in ownership of common stock and other equity securities of the Company. Officers, directors and greater than ten percent stockholders are required by SEC regulation to furnish the Company with copies of all Section 16(a) forms they file.
To the Company's knowledge, based solely on a review of the copies of such reports furnished to the Company and written representations that no other reports were required, during the fiscal year ended December 27, 2006, all Section 16(a) filing requirements applicable to its officers, directors and greater than ten percent beneficial owners were complied with except as follows: one report, covering one transaction, was filed late by Martin Batt; one report, covering one transaction, was filed late by Dr. Stephen K. Carter; one report, covering one transaction, was filed late by George M. Gould; one report, covering one transaction, was filed late by Kai P. Larson; one report, covering one transaction, was filed late by Gordon H. Link, Jr.; one report, covering one transaction, was filed late by Elliot M. Maza; one report, covering one transaction, was filed late by Richard N. Perle; one report, covering one transaction, was filed late by Patricia A. Pilia; one report, covering one transaction, was filed late by Robert E. Pollack; and one report, covering one transaction, was filed late by Leonard P. Shaykin. In addition, initial reports of ownership were filed late by Matthew J. Majoros, our corporate controller, Baker Biotech Capital III (GP), LLC, Baker Bros. Capital (GP), LLC, Baker Biotech Capital II (Z)(GP), LLC, Baker Biotech Capital III (Z)(GP), LLC and 14159 Capital (GP), LLC.
Audit Committee
We have a standing audit committee established in accordance with applicable Securities Exchange Act rules. Our audit committee is currently composed of Mr. Maza (Chair), Mr. Gould, and Mr. Perle, each of whom our board of directors has determined is independent under SEC rules and Nasdaq listing standards. Our board of directors has determined that Mr. Maza qualifies as an "audit committee financial expert," as that term is defined in Item 407(d)(5)(ii) of Regulation S-K.
Stockholder Recommendations for Board Nominees
We did not implement any changes to our process for stockholder recommendations of director nominees during 2006.
Code Of Ethics
We have adopted a Code of Ethics and Business Conduct that applies to all officers, directors and employees. The Code of Ethics and Business Conduct is available on our website at www.tapestrypharma.com. We intend to disclose any substantive amendments to our Code of Ethics and Business Conduct, and any waiver from a provision of this Code of Ethics and Business Conduct granted to our Principal Executive Officer, Principal Financial Officer or Principal Accounting Officer, on our website within five business days following such amendment or waiver and in any required filings with the SEC. The information contained on or connected to our Internet website is not incorporated by reference into this report and should not be considered part of this or any other report that we file with or furnish to the SEC.
59
Compensation Discussion and Analysis
Overview
The compensation committee of our board of directors is responsible for implementing an executive compensation strategy that will aid us in achieving our goals. To this end, the compensation committee seeks to award compensation in a manner that will motivate and reward management, while remaining consistent with our stage of development. In addition, the compensation committee understands that compensation policies affect our ability to recruit and retain qualified employees. To meet these objectives, the compensation committee determinates the allocation of total compensation among a mix of salary, bonus and equity incentives for each executive officer based on the criteria described below.
The Compensation Review Process
The compensation committee uses a number of criteria when determining executive compensation. The compensation committee evaluates our performance as a company, the individual performance of the applicable executive and compensation benchmarks at comparable companies. Historically, compensation decisions have been made in the fourth quarter of each year, and have taken into account the factors described below and our overall financial condition at such time.
Company Performance. As a development stage biopharmaceutical company with no commercial products, our primary goal is to advance TPI 287 and any other product candidates we may develop or acquire through the clinical trial process and obtain the regulatory approvals necessary to permit it to be marketed. Because we currently have no revenue, another important goal is to secure funding for the further development of these product candidates. The compensation committee assesses the performance of management with respect to these goals through regular activities of our board of directors and its committees. For example, members of the compensation committee also serve as members and chairs of committees of our board of directors, including the research and development committee, the audit committee and any ad hoc committees charged with tasks such as finance. As such, they attend regular meetings of our board of directors and its committees and interact with persons both inside and outside of our company, including our accountants, consultants, attorneys and advisors. Members are then able to report back to the entire compensation committee regarding the performance of management based upon their knowledge gained through these activities. The compensation committee may at the beginning of, during, or at the end of a given year in its discretion set particular milestones related to the achievement of our goals during that year and evaluate our performance in light of such milestones.
For 2006, we achieved a number of key performance milestones that were identified by the members of the compensation committee during the course of the year, including the successful closing of our 2006 private placement, substantial progress in Phase I clinical trials and preparing for the initiation of Phase II clinical trials. In addition to achieving these milestones, we made important advancements in process development, formulation development and discovery and preclinical development of additional drug candidates and implemented significant cost-cutting measures. Consequently, the compensation committee believes that our progress during 2006 was consistent with the expectations of our board of directors and represents a significant achievement by management.
Individual Performance. We would be unable to accomplish any of our corporate goals without the contributions of individual executive officers. The compensation committee strives to motivate and reward individual contribution to our efforts and accomplishments. It directly assesses the performance of our chief executive officer and his contribution to our activities. For example, the compensation
60
committee meets periodically with the chief executive officer throughout the year to discuss his and our company's activities, goals and performance. Additionally, the compensation committee meets in executive session to discuss the chief executive officer's performance and compensation related to such performance. When making executive compensation decisions for other executive officers, the compensation committee relies upon an informal performance assessment and compensation recommendation from the chief executive officer, as well as interactions members of the committee have with those officers. After considering these factors, the compensation committee then uses its discretion in making compensation decisions.
Benchmarking. The compensation committee reviews annually executive compensation reports, such as the Radford Biotechnology Survey published by Aon Consulting, Inc., which summarize compensation practices in the biotechnology and life sciences industries. These reports also contain information about base salary and bonuses for comparable companies in the biotechnology and life sciences industries. The compensation committee uses this information to assess the competitive compensation environment and to compare proposed compensation awards for our executives officers against compensation paid by other companies.
Elements of Executive Compensation
To meet the objectives for our compensation program, the compensation committee determines the allocation of total compensation among a mix of base salary, cash bonus and equity incentives for each executive officer.
Base Salary. The compensation committee sets base salaries for our executive officers after reviewing compensation at comparable companies based on the compensation reports described above. For 2007, base salaries for our five most highly compensated executive officers were not increased from their 2006 levels.
Bonuses. We pay annual bonuses to our executive officers. The annual bonuses are intended to compensate our executive officers for their performance over the past year and are awarded at the discretion of the compensation committee. When determining the amount of an individual officer's bonus, the compensation committee takes into account its assessment of that individual's contribution to our efforts and achievement of corporate goals as well as our financial condition. During 2006, the executive officers spent the majority of their time and efforts on our drug development efforts, reducing of drug manufacturing costs and obtaining financing. Given the substantial completion of our Phase I clinical trials, the preparations undertaken to initiate Phase II clinical trials and the successful completion of our 2006 private placement, the compensation committee awarded bonuses for each executive officer who served as such during the entire year.
Equity-based Incentives. We have a long history of granting equity-based incentives to management. The compensation committee believes that providing management with equity incentives is an effective method to motivate them to contribute to the long-term value of our common stock. As such, equity-based incentives align the interests of management and our stockholders and provide an incentive for future performance. Traditionally, the compensation committee has favored stock options over other forms of equity compensation due to the more favorable tax treatment for employees.
In late 2005 and early 2006, we faced a confluence of circumstances that prompted the compensation committee to propose action with regard to both past and future stock option grants. Most of our outstanding options were granted and priced at a time when we were focused on the paclitaxel manufacturing business, which proved to be much less profitable than anticipated. With the approval of our stockholders, we sold the paclitaxel manufacturing business in 2003. Since the sale, we have transitioned from a raw material manufacturer into a biopharmaceutical company focused on developing proprietary drugs. As a result, our goals, challenges, risks and operations changed
61
significantly. Moreover, the trading price of our common stock decreased significantly after the sale of the paclitaxel business. Because almost all of our outstanding stock options had exercise prices much higher than the trading price of our common stock, our stock options no longer provided significant incentive for option holders.
As a result of these circumstances, the compensation committee determined that it would be appropriate to reduce the exercise price of most outstanding stock options to an amount more likely to provide an incentive to option holders. In addition, the compensation committee decided to establish a new equity incentive plan from which options could be granted for the benefit of employees in the future. On February 8, 2006, our board of directors approved, subject to stockholder approval, the amendment of options held by then current directors, officers, employees and consultants under our existing equity incentive plans, provided these individuals continued to provide services to us, to reduce the exercise price of these options to $4.02, which was the average of the closing sale prices of our common stock on the NASDAQ Capital Market on the fourth through eighth trading days following our announcement that we had entered into an agreement for a private placement of our common stock. We received stockholder approval of both the repricing and an amendment to our bylaws to permit the repricing on April 4, 2006. While the shares issued in our 2006 private placement transaction did not vote on the repricing or bylaw amendment, our agreement with the investors in the private placement expressly permitted the repricing.
In 2006, our board of directors adopted and our stockholders approved our 2006 equity incentive plan that made available a number of shares equal to 20% of the number of shares of our common stock, calculated on a fully diluted basis, as permitted by the terms of our 2006 private placement. The compensation committee also granted new options to purchase shares of common stock to certain of our employees, including our executive officers under our 2006 equity incentive plan. Options granted under our 2006 equity incentive plan will fully vest and become exercisable five years after the grant date and expire ten years after the grant date. Each option is subject to accelerated vesting based on and subject to future increases in the trading price of our common stock. The compensation committee chose to implement this accelerated vesting arrangement so as to more closely align the interests of our stockholders and management by ensuring that management will only enjoy the benefits of vesting acceleration if the stockholders have been rewarded by an increase in the value of their shares.
We have a share retention policy that has been applicable to our key employees, executive officers and directors since 2003. The purpose is to align more closely the interests of these persons with those of our stockholders. In accordance with the share retention policy, these persons may not sell more than 50% of vested shares granted pursuant to an option or restricted share grant while still employed by us or serving as a director. The policy applies to grants of options and restricted stock made after the date the policy was adopted. The compensation committee may waive compliance with the policy if compliance would create a significant hardship for any option recipient.
Compensation of Executive Officers
Chief Executive Officer. The compensation for Leonard Shaykin, our Chief Executive Officer, was based upon the same procedures and criteria as described above relating to executive compensation in general. In determining Mr. Shaykin's total compensation, the compensation committee subjectively evaluated such factors as his performance and contribution to the attainment of our corporate goals. Our 2006 corporate goals were generally to progress in our drug development efforts related to TPI 287 and to secure significant financing to support such continued progress. In 2006, Mr. Shaykin was primarily responsible for directing the development of TPI 287. In addition, Mr. Shaykin was primarily responsible for locating investors and negotiating the terms of our 2006 private placement. The compensation committee considers Mr. Shaykin's contribution to be significant given the difficult nature of the equity markets at the time our 2006 private placement was effected and the challenges in obtaining financing for early stage biopharmaceutical companies.
62
The compensation committee believes that Mr. Shaykin's contribution to our company and its activities has been and continues to be vital and substantial. Consequently, the compensation committee awarded Mr. Shaykin a cash bonus of $350,000 for 2006. Furthermore, the compensation committee, subject to stockholder approval, amended Mr. Shaykin's outstanding stock options to reduce their exercise price and also granted new stock options under our 2006 equity incentive plan. As a result of these actions, options to acquire 122,500 shares of common stock were repriced and options to acquire 1,492,112 shares of common stock were granted to Mr. Shaykin during 2006.
In determining the number of stock options to be granted, the compensation committee determined that an appropriate equity-based award to a chief executive officer for a development stage company would be structured so as to bring such executive's fully diluted ownership within a range of 5% to 10% of outstanding shares. Consequently, the compensation committee granted to Mr. Shaykin stock options under our 2006 equity incentive plan such that he holds directly or has the right to acquire approximately 7% of the shares of our common stock determined on a fully diluted basis.
The compensation committee has reviewed all components of our chief executive officer compensation, including salary, bonus and equity-based incentive compensation and determined that the chief executive officer's total compensation, including any potential payouts due in the case of the severance and change-in-control scenarios discussed below, is reasonable.
Other Executive Officers. In considering compensation for the other executive officers, the compensation committee relied on informal performance assessments by our Chief Executive Officer, interactions with the executives and recommendations from our Chief Executive Officer. The compensation committee recognizes that our senior management team played a significant role in achieving our important milestones during 2006, particularly our successful 2006 private placement and substantial completion of our Phase I clinical trials. The compensation committee therefore rewarded them with the bonuses and equity compensation as set forth in the Summary Compensation Table.
Severance Benefits
We have employment agreements with Mr. Shaykin, Mr. Picker, Mr. Batt, Mr. Link and Mr. Larson that provide for payments to and continuation of specified benefits for each such employee if their employment is terminated by us without cause, or by the employee for good reason. The maximum cash amounts that would be paid to each such employee under his employment agreement in the circumstances described are as follows: Mr. Shaykin, $1,387,500; Mr. Picker, $300,000; Mr. Batt, $270,000; Mr. Link $660,000; and Mr. Larson, $605,000. In connection with the appointment of Mr. Picker as our President, a question arose as to whether Mr. Batt's duties changed in a way that would permit him to terminate his employment for good reason. To resolve this issue and as an inducement to Mr. Batt to continue his employment with us until at least December 31, 2008, we amended Mr. Batt's employment agreement to provide that the termination of the agreement by Mr. Batt at any time on or after December 31, 2008 would constitute good reason for purposes thereof and thereby entitle Mr. Batt to the receipt of his full severance benefits under his employment agreement, including accelerated vesting of outstanding stock options.
The employment agreements with Mr. Shaykin, Mr. Link and Mr. Larson also provide that in the event we undergo or anticipate a change of control, including the sale of substantially all of our assets, and either we terminate these individual's employment without cause or the individual terminates his employment for good reason, then we must make certain payments. Our 2006 private placement was deemed a change of control under our employment agreements with Mr. Shaykin, Mr. Link and Mr. Larson. Each of these executives waived, subject to specified conditions, any requirement that a minimum annual bonus be paid following our 2006 private placement. If the conditions to the waiver are not satisfied, the failure to be paid the minimum annual bonus would constitute good reason for the employee to terminate his employment and receive the benefits described above.
63
We terminated the employment of Patricia Pilia with our company in February 2006 without cause. Under the terms of her employment agreement, we paid Dr. Pilia $646,250, representing 275% of her base salary, plus approximately $39,400 for accrued but unpaid salary and vacation. We will also pay for health and other benefits for up to 18 months following termination of her employment as required by the agreement. Dr. Pilia remains one of our directors until our 2007 annual meeting of stockholders, and she will not be standing for reelection.
A full description of the severance and change of control arrangements that we have entered into with each of our executive officers is set forth below under "Potential Payments Upon Termination or Change of Control." We believe these severance and change of control benefits are an essential element of our executive compensation package and assist us in recruiting and retaining talented individuals.
Employee Benefits
We provide standard employee benefits to all of our employees. Benefits available to executive and nonexecutive employees include health insurance, vacation, disability insurance, life insurance and participation in our 401(k) and employee stock ownership plans and employee stock option program. The cost of participation in our benefits programs is borne partially by the employees, including executives. The compensation committee believes that providing these standard employee benefits is necessary in order to attract and retain qualified employees.
Perquisites
We do not provide significant perquisites or personal benefits to any employees, including executive officers, and have no current plans to do so.
Summary Compensation Table
The following table sets forth information regarding compensation earned during the fiscal year ended December 27, 2006, by our Chief Executive Officer, our Chief Financial Officer, our two other executive officers who were serving as such at the end of the fiscal year ended December 27, 2006 and one former executive officer. We refer to these individuals as our named executive officers.
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Option Awards ($)(1) | All Other Compensation ($)(2) | Total ($) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Leonard P. Shaykin Chairman of our Board and Chief Executive Officer | 2006 | $ | 370,000 | $ | 350,000 | $ | 1,066,057 | (3) | $ | 26,856 | $ | 1,812,913 | |||||
Martin M. Batt Senior Vice President and Chief Operating Officer | 2006 | 270,000 | 140,000 | 296,054 | (4) | 26,856 | 732,910 | ||||||||||
Gordon H. Link Senior Vice President and Chief Financial Officer | 2006 | 240,000 | 133,333 | 384,470 | (5) | 26,856 | 784,659 | ||||||||||
Kai P. Larson Vice President and General Counsel | 2006 | 220,000 | 113,333 | 251,153 | (6) | 26,856 | 611,342 | ||||||||||
64
Patricia A. Pilia Former Executive Vice President and Secretary | 2006 | 75,557 | — | 269,394 | (7) | 679,208 | (8) | 1,024,159 |
- (1)
- The amounts in the "Option Awards" column reflect the aggregate compensation cost for the fiscal year ended December 27, 2006 for stock options, including those repriced during the year, and are in each case calculated in accordance with SFAS 123(R) using the modified prospective transition method and a Black-Scholes valuation model disregarding the estimate of forfeitures related to service-based vesting conditions. See note 1 to our consolidated financial statements for a discussion of assumptions made in determining the grant date fair value and compensation cost of our option awards.
- (2)
- Except as set forth in footnote 8 below, the amounts in the "All Other Compensation" column reflect contributions of common stock to our employee stock ownership plan valued at fair market value as of the date of the contribution for each of our named executive officers.
- (3)
- Includes compensation expense of $130,862 as a result of the repricing of options to acquire 122,500 shares of common stock with a weighted average exercise price per share of $42.77 held by Mr. Shaykin to an exercise price of $4.02 per share.
- (4)
- Includes compensation expense of $4,996 as a result of the repricing of options to acquire 21,500 shares of common stock with a weighted average exercise price per share of $16.19 held by Mr. Batt to an exercise price of $4.02 per share.
- (5)
- Includes compensation expense of $70,538 as a result of the repricing of options to acquire 72,000 shares of common stock with a weighted average exercise price per share of $44.65 held by Mr. Link to an exercise price of $4.02 per share.
- (6)
- Includes compensation expense of $38,922 as a result of the repricing of options to acquire 43,416 shares of common stock with a weighted average exercise price per share of $49.70 held by Mr. Larson to an exercise price of $4.02 per share.
- (7)
- Includes compensation expense of $138,138 as a result of the repricing of options to acquire 127,267 shares of common stock with a weighted average exercise price per share of $37.82 held by Dr. Pilia to an exercise price of $4.02 per share.
- (8)
- Includes a $646,250 severance payment, representing 275% of Dr. Pilia's base salary, and $5,428 in reimbursement of health, disability and life insurance benefits in connection with our termination of Dr. Pilia's employment without cause as of February 23, 2006 and $26,755 in fees paid to Dr. Pilia in her capacity as a member of our board of directors after such termination. Dr. Pilia remains one of our directors until our 2007 annual meeting of stockholders. She will not be standing for reelection.
Grants of Plan-Based Awards
All options granted to our named executive officers are non-qualified stock options. The following table sets forth for the fiscal year ended December 27, 2006 information regarding grants of plan-based awards to the named executive officers. The options were granted under our 2006 equity incentive plan, except as noted.
65
| Name | Grant Date | Number of Securities Underlying Options (#) | Exercise or Base Price of Option Awards ($/Sh) | Closing Sale Price on Grant Date ($/Sh) | Grant Date Fair Value of Option Awards ($)(1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Leonard P. Shaykin | 10/5/2006 4/4/2006 4/4/2006 | 630,000 1,492,112 122,500 | (4) | $ | 2.10 4.02 4.02 | (2) (3) (3) | $ | 1.78 3.75 3.75 | $ | 937,944 4,749,840 134,890 | |||
Martin M. Batt | 10/5/2006 4/4/2006 4/4/2006 | 250,000 497,482 21,500 | (4) | 2.10 4.02 4.02 | (2) (3) (3) | 1.78 3.75 3.75 | 372,200 1,583,634 7,501 | ||||||
Gordon H. Link | 10/5/2006 4/4/2006 4/4/2006 | 250,000 331,653 72,000 | (4) | 2.10 4.02 4.02 | (2) (3) (3) | 1.78 3.75 3.75 | 372,200 1,055,751 73,196 | ||||||
Kai P. Larson | 10/5/2006 4/4/2006 4/4/2006 | 100,000 187,243 43,416 | (4) | 2.10 4.02 4.02 | (2) (3) (3) | 1.78 3.75 3.75 | 148,880 596,051 41,358 | ||||||
Patricia A. Pilia | 6/22/2006 4/4/2006 4/4/2006 | 1,950 19,163 127,267 | (4) | 2.62 4.02 4.02 | (3) (3) | 2.62 3.75 3.75 | 4,364 61,002 140,574 | ||||||
- (1)
- The amounts in the "Grant Date Fair Value of Option Awards" column reflect the grant date fair value of stock options during the fiscal year ended December 27, 2006, including those repriced during the year, calculated in accordance with SFAS 123(R) using the modified prospective transition method and a Black-Scholes valuation model disregarding the estimate of forfeitures related to service based vesting conditions. See note 1 to our consolidated financial statements included at the end of this prospectus for a discussion of assumptions made in determining the grant date fair value of our option awards.
- (2)
- The exercise price of stock options granted on October 5, 2006, was set by the compensation committee of our board of directors at the greater of $2.10 per share or the closing sale price of our common stock in recognition of the fact that the trading price was near historic lows.
- (3)
- In accordance with the terms of our 2006 private placement, the exercise price of these stock options was established as the average of the closing sale prices of our common stock on the NASDAQ Capital Market on the fourth through eighth trading day following our public annoucement of that transaction.
- (4)
- Each of these grants represents multiple stock options for each named executive officer under our 1994 long-term performance incentive plan, 1998 stock incentive plan and 2004 equity incentive plan that were repriced effective April 4, 2006 by amending the exercise price of the original options as described under "Stock Option Repricing."
Stock Option Repricing
On February 8, 2006, our board of directors approved, subject to stockholder approval, the amendment of certain outstanding options to purchase shares of common stock under our existing equity incentive plans, including all options held by those persons who were then serving as directors and executive officers. Our stockholders approved such amendment on April 4, 2006. The exercise price of each repriced option was reduced to $4.02 per share as of the date of stockholder approval. We repriced options to purchase a total of 626,568 shares of common stock outstanding under all of our
66
equity compensation plans. Prior to the repricing, these options had exercise prices ranging from $4.20 to $112.50.
Outstanding Equity Awards
The following table sets forth information regarding outstanding equity awards at December 27, 2006, for the named executive officers.
Outstanding Equity Awards at December 27, 2006
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | |||||
|---|---|---|---|---|---|---|---|---|---|
| Leonard P. Shaykin | — | 630,000 | (1) | $ | 2.10 | 10/5/2016 | |||
| — | 1,492,112 | (2)(3) | 4.02 | 4/4/2016 | |||||
| 13,889 | (4) | 11,111 | (2)(4)(5) | 4.02 | 9/2/2013 | ||||
| 18,750 | (4) | — | 4.02 | 6/21/2011 | |||||
| 18,750 | (4) | — | 4.02 | 4/16/2011 | |||||
| 7,194 | (4) | — | 4.02 | 9/13/2010 | |||||
| 4,806 | (4) | — | 4.02 | 6/27/2010 | |||||
| 20,000 | (4) | — | 4.02 | 8/17/2009 | |||||
| 8,000 | (4) | — | 4.02 | 10/5/2008 | |||||
| 20,000 | (4) | — | 4.02 | 3/27/2008 | |||||
Martin M. Batt | — | 250,000 | (1) | 2.10 | 10/5/2016 | ||||
| — | 497,482 | (2)(3) | 4.02 | 4/4/2016 | |||||
| 2,000 | (4) | 6,000 | (2)(4)(6) | 4.02 | 10/28/2014 | ||||
| 3,334 | (4) | 2,666 | (2)(4)(5) | 4.02 | 9/2/2013 | ||||
| 7,500 | (4) | — | 4.02 | 8/21/2012 | |||||
Gordon Link | — | 250,000 | (1) | $ | 2.10 | 10/5/2016 | |||
| — | 331,653 | (2)(3) | 4.02 | 4/4/2016 | |||||
| 2,000 | (4) | 6,000 | (2)(4)(6) | 4.02 | 10/28/2014 | ||||
| 6,111 | (4) | 4,889 | (2)(4)(5) | 4.02 | 9/2/2013 | ||||
| 12,500 | (4) | — | 4.02 | 6/21/2011 | |||||
| 12,500 | (4) | — | 4.02 | 4/16/2011 | |||||
| 4,797 | (4) | — | 4.02 | 9/13/2010 | |||||
| 3,203 | (4) | — | 4.02 | 6/27/2010 | |||||
| 10,000 | (4) | — | 4.02 | 7/23/2009 | |||||
| 5,000 | (4) | — | 4.02 | 10/5/2008 | |||||
| 5,000 | (4) | — | 4.02 | 3/27/2008 | |||||
Kai P. Larson | — | 100,000 | (1) | 2.10 | 10/5/2016 | ||||
| — | 187,243 | (2)(3) | 4.02 | 4/4/2016 | |||||
| 1,500 | (4) | 4,500 | (2)(4)(6) | 4.02 | 10/28/2014 | ||||
| 6,111 | (4) | 4,889 | (2)(4)(5) | 4.02 | 9/2/2013 | ||||
| 6,666 | (4) | — | 4.02 | 4/16/2011 | |||||
| 10,000 | (4) | — | 4.02 | 6/21/2011 | |||||
| 3,597 | (4) | — | 4.02 | 9/13/2010 | |||||
| 2,403 | (4) | — | 4.02 | 6/27/2010 | |||||
| 2,500 | (4) | — | 4.02 | 7/23/2009 | |||||
| 1,250 | (4) | — | 4.02 | 10/5/2008 | |||||
67
Patricia Pilia | — | 1,950 | (7) | 2.62 | 6/22/2016 | ||||
| — | 19,163 | (2)(3) | 4.02 | 4/4/2016 | |||||
| 1,500 | (4) | 4,500 | (2)(4)(6) | 4.02 | 10/28/2014 | ||||
| 6,111 | (4) | 4,889 | (2)(4)(5) | 4.02 | 9/2/2013 | ||||
| 10,000 | (4) | — | 4.02 | 6/21/2011 | |||||
| 10,000 | (4) | — | 4.02 | 4/16/2011 | |||||
| 5,995 | (4) | — | 4.02 | 9/13/2010 | |||||
| 4,005 | (4) | — | 4.02 | 6/27/2010 | |||||
| 12,500 | (4) | — | 4.02 | 8/17/2009 | |||||
| 6,000 | (4) | — | 4.02 | 10/5/2008 | |||||
| 7,600 | (4) | — | 4.02 | 3/27/2008 | |||||
| 1,667 | (4)(8) | — | 4.02 | 9/2/2013 | |||||
| 2,500 | (4)(8) | — | 4.02 | 4/16/2011 | |||||
| 5,995 | (4)(8) | — | 4.02 | 9/13/2010 | |||||
| 4,005 | (4)(8) | — | 4.02 | 6/27/2010 | |||||
| 20,000 | (4)(8) | — | 4.02 | 8/17/2009 | |||||
| 8,000 | (4)(8) | — | 4.02 | 10/5/2008 | |||||
| 12,000 | (4)(8) | — | 4.02 | 3/27/2008 |
- (1)
- These options fully vest and become exercisable on October 5, 2011, subject to accelerated vesting as follows:1/6 when the 20 trading day average of the per share closing sale price of our common stock equals or exceeds $2.73;1/6 when such average equals or exceeds $3.36;1/6 when such average equals or exceeds $3.99;1/6 when such average equals or exceeds $4.62;1/6 when such average equals or exceeds $5.25; and1/6 when such average equals or exceeds $6.30.
- (2)
- The vesting of these options will be accelerated as follows:1/6 when the 20 trading day average of the per share closing sale price of our common stock equals or exceeds $5.23;1/6 when such average equals or exceeds $6.43;1/6 when such average equals or exceeds $7.64;1/6 when such average equals or exceeds $8.84;1/6 when such average equals or exceeds $10.05; and1/6 when such average equals or exceeds $12.06.
- (3)
- These options fully vest and become exercisable on April 4, 2011, subject to accelerated vesting as specified in footnote 2.
- (4)
- Each of these grants was repriced effective April 4, 2006 by amending the exercise price of the original grant as described under "Stock Option Repricing."
- (5)
- These options fully vest and become exercisable on September 2, 2008, subject to accelerated vesting as specified in footnote 2.
- (6)
- These options fully vest and become exercisable on October 28, 2009, subject to accelerated vesting as specified in footnote 2.
- (7)
- These options fully vest and become exercisable on June 22, 2007.
- (8)
- These options were initially granted to Sterling Ainsworth, formerly our chief executive officer, and transferred to Dr. Pilia following Dr. Ainsworth's death in 2005.
Option Exercises and Stock Vested
For the fiscal year ended December 27, 2006, none of our named executive officers acquired any shares of our common stock by exercise of a stock option or by vesting of a stock award.
68
Pension Benefits
None of our named executive officers participates in or has account balances in nonqualified defined benefit plans sponsored by us.
Nonqualified Deferred Compensation
None of our named executive officers participates in or has account balances in nonqualified defined contribution plans or other deferred compensation plans maintained by us.
Potential Payments Upon Termination or Change of Control
We have entered into employment agreements with our named executive officers that require us to make the payments described below upon termination of employment.
Leonard Shaykin
Pursuant to the terms of the employment agreement that we have entered into with Mr. Shaykin, if we terminate Mr. Shaykin's employment without cause or Mr. Shaykin terminates his employment for good reason, Mr. Shaykin will be entitled to the following benefits: a payment equal to the greater of 100% of his prior year's bonus or 75% of his base annual salary; a payment equal to 300% of his base annual salary; a payment equal to accrued, unpaid salary and bonus through the date of termination; and health and welfare benefits as in effect immediately prior to termination for a maximum of 18 months following termination. As defined in Mr. Shaykin's employment agreement, "good reason" includes, along with other events, our board of directors' failure to grant, in each calendar year after a change of control occurs or is anticipated, a minimum annual bonus at least equal to the average of the three years' prior annual bonuses, if such a failure is in anticipation of or following a change of control. If Mr. Shaykin had been terminated without cause or resigned for good reason on December 27, 2006, Mr. Shaykin would have received from us a lump sum payment of $1,737,500, which includes Mr. Shaykin's declared but unpaid 2006 annual bonus of $350,000, and been entitled to health and welfare benefits in an amount of up to approximately $23,000, based on the current premiums paid by us with respect to such policies as of the termination date.
The sale of our paclitaxel business to Mayne Pharma in 2003 may be deemed to have been a sale of substantially all our assets. In connection with the sale, Mr. Shaykin provided us with a written waiver of any requirement that a minimum annual bonus be paid to him insofar as the sale of the paclitaxel business could be construed to constitute a change of control pursuant to his employment agreement. Similarly, our 2006 private placement could also be construed to constitute a change of control. Mr. Shaykin also has provided a written waiver to us which waives, subject to specified conditions, any requirement that a minimum annual bonus be paid to him related to the private placement. On February 23, 2006, we amended Mr. Shaykin's employment agreement to conform to the requirements of Section 409A of the Internal Revenue Code of 1986, as amended, or the Code, and of the related proposed regulations issued by the Treasury Department. As a result of the amendment, any payment of severance benefits to Mr. Shaykin will likely be delayed until six months and one day following his termination of employment, rather than being due immediately upon termination.
Gordon Link
Pursuant to the terms of the employment agreement that we have entered into with Mr. Link, if we terminate Mr. Link's employment without cause or Mr. Link terminates his employment for good reason, Mr. Link will be entitled to the following benefits: a payment equal to the greater of 100% of his prior year's bonus or 75% of his base annual salary; a payment equal to 200% of his base annual salary; a payment equal to accrued, unpaid salary and bonus through the date of termination; and health and welfare benefits as in effect immediately prior to termination for a maximum of 18 months
69
following termination. As defined in Mr. Link's employment agreement, "good reason" includes, along with other events, our board of directors' failure to grant, in each calendar year after a change of control occurs or is anticipated, a minimum annual bonus at least equal to the average of the three years' prior annual bonuses, if such a failure is in anticipation of or following a change of control. If Mr. Link had been terminated without cause or resigned for good reason on December 27, 2006, Mr. Link would have received from us a lump sum payment of $793,333, which includes Mr. Link's declared but unpaid 2006 annual bonus of $133,333, and been entitled to health and welfare benefits in an amount of up to approximately $21,000, based on the current premiums paid by us with respect to such policies as of the termination date.
The sale of our paclitaxel business to Mayne Pharma in 2003 may be deemed to have been a sale of substantially all of our assets. In connection with the sale, Mr. Link provided us with a written waiver of any requirement that a minimum annual bonus be paid to him insofar as the sale of the paclitaxel business could be construed as a change in control pursuant to his employment agreement. Similarly, our 2006 private placement could be construed to constitute a change of control. Mr. Link also has provided a written waiver to us which waives, subject to specified conditions, any requirement that a minimum annual bonus be paid to him related to the private placement. On February 23, 2006, we amended the employment agreement with Mr. Link to conform to the requirements of Section 409A of the Code and of the related proposed regulations issued by the Treasury Department. As a result of the amendment, any payment of severance benefits to Mr. Link will likely be delayed until six months and one day following his termination of employment, rather than being due immediately upon termination.
Kai Larson
Pursuant to the terms of the employment agreement that we have entered into with Mr. Larson, if we terminate Mr. Larson's employment without cause or Mr. Larson terminates his employment for good reason, Mr. Larson will be entitled to the following benefits: a payment equal to the greater of 100% of his prior year's bonus or 75% of his base annual salary; a payment equal to 200% of his base annual salary; a payment equal to accrued, unpaid salary and bonus through the date of termination; a payment equal to accrued, unpaid salary and bonus through the date of termination; and health and welfare benefits as in effect immediately prior to termination for a maximum of 18 months following termination. As defined in Mr. Larson's employment agreement, "good reason" includes, along with other events, our board of directors' failure to grant, in each calendar year after a change of control occurs or is anticipated, a minimum annual bonus at least equal to the average of the three years' prior annual bonuses, if such a failure is in anticipation of or following a change of control. If Mr. Larson had been terminated without cause or resigned for good reason on December 27, 2006, Mr. Larson would have received from us a lump sum payment of $718,333, which includes Mr. Larson's declared but unpaid 2006 annual bonus of $113,333, and been entitled to health and welfare benefits in an amount of up to approximately $21,000, based on the current premiums paid by us with respect to such policies as of the termination date.
The sale of our paclitaxel business to Mayne Pharma in 2003 may be deemed to have been a sale of substantially all of our assets. In connection with the sale, Mr. Larson provided us with a written waiver of any requirement that a minimum annual bonus be paid to him insofar as the sale of the paclitaxel business could be construed as a change in control pursuant to his employment agreement. Similarly, our 2006 private placement could be construed to constitute a change of control. Mr. Larson also has provided a written waiver to us which waives, subject to specified conditions, any requirement that a minimum annual bonus be paid to him related to the private placement. On February 23, 2006, we amended the employment agreement with Mr. Larson to conform to the requirements of Section 409A of the Code, and of the related proposed regulations issued by the Treasury Department. As a result of the amendment, any payment of severance benefits to Mr. Larson will likely be delayed
70
until six months and one day following his termination of employment, rather than being due immediately upon termination.
Martin Batt
Pursuant to the employment agreement that we have entered into with Mr. Batt, if we terminate Mr. Batt's employment without cause or Mr. Batt terminates his employment for good reason Mr. Batt will be entitled to the following benefits: a payment equal to 100% of Mr. Batt's base annual salary; and payment equal to accrued, unpaid salary through the date of termination. In addition, Mr. Batt would be entitled to reimbursement of health insurance premiums for a maximum of 18 months following termination and accelerated vesting of his outstanding stock option awards. As defined in Mr. Batt's employment agreement, "good reason" includes, along with other events, the termination of the employment agreement by Mr. Batt on or after December 31, 2008. If Mr. Batt had been terminated without cause or resigned for good reason on December 27, 2006, Mr. Batt would have received from us a lump sum payment of $270,000, received accelerated vesting of 756,148 stock options and been entitled to health insurance benefits in an amount of up to approximately $15,000, based on the current premiums paid with respect to such policy as of the termination date.
Patricia Pilia
On February 23, 2006, we gave notice to Dr. Pilia of termination of her employment without cause. Consequently, in satisfaction of the terms of her employment agreement, we paid Dr. Pilia $646,250, representing 275% of her base salary, and approximately $39,400 for accrued but unpaid salary and vacation. We are responsible for paying for health and certain other benefits for 18 months following termination as set forth in her employment agreement.
On March 22, 2006, we and Dr. Pilia entered into a settlement agreement in connection with the termination of her employment. This settlement agreement provides that we will take no action to remove Dr. Pilia from her position as a member of our board of directors and will allow her to serve the remainder of her current term until our 2007 annual meeting of stockholders. In addition, Dr. Pilia shall also continue to serve as a member of the research and development committee for the remainder of her term. Pursuant to the settlement agreement, we agreed to compensate Dr. Pilia for her board and committee service consistent with that provided for other non-employee directors. The settlement agreement confirms the severance and benefits payments we are required to make to Dr. Pilia following termination of her employment. It also provides that Dr. Pilia will not be in violation of certain non-compete provisions of her employment agreement if she becomes involved in the development or marketing of paclitaxel or docetaxel; that she will not make any public disclosure designed to discourage any person from doing business with us; and that she will provide assistance for compensation in connection with actions taken by her during the period she was employed by us or served as a member of our board of directors. The settlement agreement also provides for mutual releases by us and Dr. Pilia.
Compensation of Directors
Stock Option Grants. In January 2006, our board of directors adopted, subject to stockholder approval, our 2006 equity incentive plan. In conjunction with the adoption of our 2006 equity incentive plan, and subject to stockholder approval, our board of directors granted options to purchase 30,000 shares of common stock to each non-employee director, totaling 180,000 shares in aggregate. Stockholders approved our 2006 equity incentive plan and the grant on April 4, 2006. Under our 2006 equity incentive plan, our non-employee directors are eligible to receive automatic and discretionary grants of options to purchase our common stock. All such options are to be exercisable at an exercise price equal to the fair market value of the common stock on the date of grant and are subject to vesting.
71
We automatically grant options to purchase 1,500 shares of common stock to each non-employee director who either is elected or reelected as a member of our board of directors at an annual meeting of stockholders or otherwise appointed as a member of our board of directors in accordance with our bylaws. Similarly, non-employee directors who continue service as a member of our board of directors after an annual meeting of stockholders at which such individual is not subject to re-election also receive such a grant. In each case, the automatic grant occurs on the business day next following each such annual meeting or appointment.
Non-employee directors who serve as the chairs of our committees receive an additional automatic grant of stock options each year. Specifically, we automatically grant options to purchase 1,500 shares of common stock to the chairs of the audit, compensation, nominating and corporate governance and research and development committees of our board of directors, effective on the business day next following our annual meeting of stockholders.
Non-employee directors who serve on our research and development committee receive an automatic grant to purchase 1,000 shares of common stock upon their initial appointment to the committee. Non-employee directors who continue to serve on the committee after an annual meeting of stockholders automatically receive an additional grant to purchase 450 shares of common stock.
Other Compensation. We pay to non-employee directors $3,000 for each regular board of directors meeting and $500 for each special board of directors meeting attended. In addition, we pay directors serving on committees of our board of directors (other than the research and development committee) for attendance at each committee meeting as follows: $1,000 for the committee chair and $500 for non-chair committee members. We pay members of the research and development committee $2,500 for attendance at research and development committee meetings that are not held concurrently with regularly scheduled meetings of our board of directors. We also reimburse directors for costs incurred in attending meetings of our board of directors and its committees. We pay non-employee directors an annual retainer of $10,000, payable quarterly, the chair of the audit committee an additional annual retainer of $10,000, and the co-chairs of the research and development committee an additional annual retainers $40,000 per year.
The following table sets forth for the fiscal year ended December 27, 2006 information with respect to the compensation paid to all of our non-employee directors:
| Name | Fees Earned or Paid in Cash ($) | Option Awards ($)(1) | Total ($) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Stephen K. Carter, M.D. | $ | 66,000 | $ | 19,229 | $ | 85,229 | |||
| George M. Gould, Esq. | 29,500 | 22,746 | 52,246 | ||||||
| Arthur P. Hayes, Jr., M.D. | 34,500 | 30,918 | 65,418 | ||||||
| Elliot M. Maza | 41,000 | 23,349 | 64,349 | ||||||
| Richard N. Perle | 26,500 | 30,075 | 56,575 | ||||||
| Patricia A. Pilia, Ph.D.(2) | 26,755 | 10,969 | 37,724 | ||||||
| Robert E. Pollack, Ph.D. | 68,000 | 31,954 | 99,954 | ||||||
- (1)
- The amounts in the "Option Awards" column reflect the aggregate compensation cost to the Company for the fiscal year ended December 27, 2006 for stock options, including those repriced during the year, and are in each case calculated in accordance with SFAS 123(R) using the modified prospective transition method and a Black-Scholes valuation model disregarding the estimate of forfeitures related to service based vesting conditions. See note 1 to our consolidated financial statements for a discussion of assumptions made in determining the grant date fair value
72
and compensation cost of our option awards. The table below sets forth each option award by director and the full grant date fair value of each award granted or repriced.
| Director | Grant Date | Number of Securities Underlying Options (#) | Exercise Price of Option Awards ($/Sh) | Grant Date Fair Value of Option Award ($) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Stephen K. Carter, M.D.(a) | 6/22/2006 4/4/2006 4/4/2006 | 1,500 30,000 4,350 | * | $ | 2.62 4.02 4.02 | $ | 3,357 95,499 1,153 | |||
| George M. Gould, Esq.(b) | 6/22/2006 4/4/2006 4/4/2006 | 3,000 30,000 6,000 | * | 2.62 4.02 4.02 | 6,714 95,499 967 | |||||
| Arthur P. Hayes, Jr., M.D.(c) | 6/22/2006 4/4/2006 4/4/2006 | 1,950 30,000 12,750 | * | 2.62 4.02 4.02 | 4,364 95,499 11,072 | |||||
| Elliot M. Maza(d) | 6/22/2006 4/4/2006 4/4/2006 | 3,000 30,000 4,000 | * | 2.62 4.02 4.02 | 6,714 95,499 263 | |||||
| Richard N. Perle(e) | 6/22/2006 4/4/2006 4/4/2006 | 1,500 30,000 13,000 | * | 2.62 4.02 4.02 | 3,357 95,499 12,031 | |||||
| Patricia A. Pilia, Ph.D.(f) | 6/22/2006 4/4/2006 | 1,950 19,163 | 2.62 4.02 | 4,364 61,002 | ||||||
| Robert E. Pollack, Ph.D.(g) | 6/22/2006 4/4/2006 4/4/2006 | 3,000 30,000 12,450 | * | 2.62 4.02 4.02 | 6,714 95,499 12,314 | |||||
- *
- Each of these grants represents multiple stock options for each director that were repriced effective April 4, 2006 by amending the exercise price of the original options as described under "Stock Option Repricing."
- (a)
- As of December 27, 2006, Dr. Carter held options to purchase 4,350 shares of our common stock that were exercisable, options to purchase 1,500 shares that fully vest and become exercisable on June 22, 2007 and options to purchase 30,000 shares that fully vest and become exercisable on April 4, 2011, subject to accelerated vesting as follows:1/6 when the 20 trading day average of the per share closing sale price of the common stock equals or exceeds $5.23;1/6 when such average equals or exceeds $6.43;1/6 when such average equals or exceeds $7.64;1/6 when such average equals or exceeds $8.84;1/6 when such average equals or exceeds $10.05; and1/6 when such average equals or exceeds $12.06.
- (b)
- As of December 27, 2006, Mr. Gould held options to purchase 6,000 shares of our common stock that were exercisable, options to purchase 3,000 shares that fully vest and become exercisable on June 22, 2007 and options to purchase 30,000 shares that fully vest and become exercisable on April 4, 2011, subject to accelerated vesting on the same basis as options granted to Dr. Carter as specified in footnote (a) above.
- (c)
- As of December 27, 2006, Dr. Hayes held options to purchase 12,750 shares of our common stock that were exercisable, options to purchase 1,950 shares that fully vest and become exercisable on June 22, 2007 and options to purchase 30,000 shares that fully vest and become exercisable on April 4, 2011, subject to accelerated vesting on the same basis as options granted to Dr. Carter as specified in footnote (a) above.
73
- (d)
- As of December 27, 2006, Mr. Maza held options to purchase 4,000 shares of our common stock that were exercisable, options to purchase 3,000 shares that fully vest and become exercisable on June 22, 2007 and options to purchase 30,000 shares that fully vest and become exercisable on April 4, 2011, subject to accelerated vesting on the same basis as options granted to Dr. Carter as specified in footnote (a) above.
- (e)
- As of December 27, 2006, Mr. Perle held options to purchase 13,000 shares of our common stock that were exercisable, options to purchase 1,500 shares that fully vest and become exercisable on June 22, 2007 and options to purchase 30,000 shares that fully vest and become exercisable on April 4, 2011, subject to accelerated vesting on the same basis as options granted to Dr. Carter as specified in footnote (a) above.
- (f)
- For information about options held by Dr. Pilia, see the "Outstanding Equity Awards at December 27, 2006" table above.
- (g)
- As of December 27, 2006, Dr. Pollack held options to purchase 12,450 shares of our common stock that were exercisable, options to purchase 3,000 shares that fully vest and become exercisable on June 22, 2007 and options to purchase 30,000 shares that fully vest and become exercisable on April 4, 2011, subject to accelerated vesting on the same basis as options granted to Dr. Carter as specified in footnote (a) above.
- (2)
- Dr. Pilia served as our Executive Vice President and Secretary until February 23, 2006, when her employment was terminated without cause. Dr. Pilia continued to serve as a member of our board of directors after such termination. Her term expires at our 2007 annual meeting of stockholders, and she will not be standing for reelection. All compensation for Dr. Pilia for fiscal 2006, including for her service as a director after the termination of her employment is reflected in the summary compensation table. The amounts in this table reflect compensation only for her service as a director.
Compensation Committee Interlocks and Insider Participation
During the year ended December 27, 2006, our compensation committee consisted of Dr. Pollack (chair), Dr. Carter, Mr. Maza and Mr. Gould. Dr. Pollack currently serves as the chair of our compensation committee. None of our executive officers serve as a member of the board of directors or of the compensation committee, or other committee serving a similar function, of any entity that has one or more executive officers who serve on our board of directors or compensation committee. None of the members of our compensation committee has ever been an employee of our company.
74
Equity Compensation Plan Information
The following table sets forth certain information as of December 27, 2006 concerning our common stock that may be issued upon the exercise of options or the purchases of restricted stock under all of our equity compensation plans approved by stockholders and equity compensation plans not approved by stockholders:
| Plan Category | Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted-average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuances under equity compensation plans (excluding securities reflected in column (a)) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | (a) | (b) | (c) | |||||||
| Equity compensation plans approved by security holders: | ||||||||||
| 2006 Equity Incentive Plan | 5,109,629 | $ | 3.40 | 1,503,306 | ||||||
| 2004 Equity Incentive Plan | 93,200 | $ | 4.33 | — | ||||||
| 2004 Non-Employee Directors' Stock Option Plan | 15,350 | $ | 4.20 | — | ||||||
| 1994 Long-Term Performance Incentive Plan | 510,269 | $ | 6.88 | — | ||||||
| Total Approved Plans | 5,728,448 | $ | 3.73 | 1,503,306 | ||||||
Equity compensation plans not approved by security holders: | ||||||||||
| Non-plan | 50 | $ | 4.02 | — | ||||||
| 1998 Stock Option Plan | 76,795 | $ | 11.29 | — | ||||||
| Total Unapproved Plans | 76,845 | $ | 11.28 | — | ||||||
| Total Plans | 5,805,293 | $ | 3.83 | 1,503,306 | ||||||
Compensation Committee Report(1)
The Compensation Committee has reviewed and discussed with management the Compensation Discussion and Analysis ("CD&A") contained in this proxy statement. Based on this review and discussion, the Compensation Committee has recommended to the Board of directors that the CD&A be incorporated into our Annual Report on Form 10-K for the fiscal year ended December 27, 2006.
| Robert E. Pollack, Ph.D., Chair Stephen K. Carter, M.D. George M. Gould, Esq. Elliot M. Maza |
- (1)
- The material in this report is not "soliciting material," is furnished to, but not deemed "filed" with, the Commission and is not deemed to be incorporated by reference in any filing of the Company under the Securities Act or the Exchange Act, other than the Company's Annual Report on Form 10-K, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
75
Item 12
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth information regarding the beneficial ownership of our common stock as of March 28, 2007 by:
- •
- each person who is known by us to beneficially own more than 5% of our common stock;
- •
- each of our directors;
- •
- each of the named executive officers; and
- •
- all of our directors and executive officers as a group.
Percentage ownership is based on 16,374,395 shares outstanding as of March 28, 2007. Beneficial ownership is calculated based on SEC requirements and includes voting or investment power with respect to our common stock. All shares of our common stock subject to outstanding options or warrants currently exercisable or exercisable within 60 days after March 28, 2007 are deemed to be outstanding and beneficially owned by the person holding the option or warrant for the purpose of calculating the percentage ownership of that person, but are not deemed to be outstanding for calculating the percentage ownership of any other person. Unless otherwise indicated below, each stockholder named in the table has sole voting and investment power with respect to all shares beneficially owned, subject to applicable community property laws. Unless otherwise indicated below, the address of each individual listed in the table is c/o Tapestry Pharmaceuticals, Inc., 4840 Pearl East Circle, Suite 300W, Boulder, CO 80301. This table is based upon information supplied by officers, directors and principal stockholders and Schedules 13D and 13G and Forms 3 and 4 filed with the SEC.
| Name of Beneficial Owner(1) | Number of Shares Beneficially Owned | Percentage of Shares Beneficially Owned | |||
|---|---|---|---|---|---|
| 5% Stockholders | |||||
| Special Situations Fund III, L.P. 527 Madison Avenue Suite 2600 New York, NY 10022 | 4,956,953 | (1) | 30.3 | % | |
| Tang Capital Partners, LP 4401 Eastgate Mall San Diego, CA 92121 | 2,143,727 | (2) | 13.1 | ||
| Baker Brothers Life Science Capital LLC 667 Madison Avenue 17th Floor New York, NY 10021 | 2,000,000 | (3) | 12.2 | ||
| Capital Ventures International One Capitol Place, P.O. Box 1787 GT Grand Cayman, Cayman Islands, B.W.I. | 1,709,630 | (4) | 9.999 | ||
| Biotechnology Value Fund, L.P. 900 North Michigan Avenue, Suite 1100 Chicago, Illinois, 60611 | 1,672,485 | (5) | 9.999 | ||
| Fort Mason Master, L.P. Four Embarcadero Center, Suite 2050 San Francisco, CA 94111 | 1,651,690 | (6) | 9.999 | ||
| Xmark JV Investment Partners, LLC 301 Tresser Boulevard, Suite 1320 Stamford, CT 06901 | 1,000,000 | (7) | 5.8 | ||
76
Directors and Executive Officers | |||||
| Leonard P. Shaykin | 213,952 | (8) | 1.3 | % | |
| Stephen K. Carter, M.D. | 4,350 | (9) | * | ||
| George M. Gould | 6,000 | (10) | * | ||
| Arthur H. Hayes, Jr. | 12,750 | (11) | * | ||
| Elliot M. Maza | 4,000 | (12) | * | ||
| The Honorable Richard N. Perle | 14,300 | (13) | * | ||
| Patricia A. Pilia | 141,237 | (14) | * | ||
| Robert E. Pollack | 12,560 | (15) | * | ||
| Donald H. Picker | — | (16) | * | ||
| Martin M. Batt | 36,432 | (17) | * | ||
| Kai P. Larson | 61,081 | (18) | * | ||
| Gordon H. Link, Jr. | 95,651 | (19) | * | ||
| All Directors and Executive Officers as a Group (12 persons) | 602,313 | (20) | 3.6 |
- *
- Less than 1%
- (1)
- Consists of 1,250,367 shares of our common stock and warrants to purchase 1,200,000 shares of our common stock held by Special Situations Fund III, Q.P. L.P.; 423,824 shares of our common stock and warrants to purchase 350,000 shares of our common stock held by Special Situations Life Science Fund L.P.; 372,963 shares of our common stock and warrants to purchase 350,000 shares of our common stock held by Special Situations Cayman Fund L.P.; and 509,799 shares of our common stock and warrants to purchase 500,000 shares of our common stock held by Special Situations Private Equity Fund L.P. MGP Advisers Limited Partnership, or MGP, is the general partner of and investment adviser to Special Situations Fund III, Q.P., L.P. AWM Investment Company, Inc., or AWM, is the general partner of MGP and the general partner of and investment adviser to Special Situations Cayman Fund L.P. MG Advisers, L.L.C., or MG, is the general partner of and investment adviser to Special Situations Private Equity Fund, L.P. LS Advisers, L.L.C., or LS, is the general partner of and investment adviser to Special Situations Life Sciences Fund, L.P. Austin W. Marxe and David M. Greenhouse are the principal owners of MGP, AWM, LS and MG. Accordingly, they are principally responsible for the selection, acquisition and disposition of the portfolios securities by the investment advisers on behalf of their funds. Messrs. Marxe and Greenhouse disclaim beneficial ownership of these securities except to the extent of their pecuniary interest therein, if any.
- (2)
- Consists of 1,783,350 shares of our common stock and warrants to purchase 1,705,000 shares of common stock held by Tang Capital Partners, L.P.; 51,858 shares of our common stock and warrants to purchase 35,000 shares of common stock held by Kevin C. Tang as Custodian for Julian Kong Tang; 60,688 shares of our common stock and warrants to purchase 50,000 shares of common stock held by Kevin C. Tang as Custodian for Justin Lee Tang; 15,000 shares of our common stock and warrants to purchase 15,000 shares of common stock held by Kevin C. Tang as Custodian for Noa Young Tang; 150,000 shares of our common stock and warrants to purchase 150,000 shares of common stock held by Kevin Tang and Haeyoung Tang as Trustees for The Tang Family Trust Dated 8-27-02, or the Tang Family Trust; 27,190 shares of common stock held by Tang Advisors LLC Profit Sharing Plan; 15,759 shares of our common stock and warrants to purchase 10,000 shares of common stock held by IRA FBO Chang L. Kong; 15,155 shares of our common stock and warrants to purchase 10,000 shares of common stock held by IRA FBO Chung W. Kong; and 25,000 shares of our common stock and warrants to purchase 25,000 shares of common stock held by IRA FBO Kevin Tang DB Securities Inc. The warrants may not be exercised to the extent
77
that the holder and its affiliates would beneficially own more than 9.999% of our outstanding common stock. This restriction can be waived by the holder upon 61 days written notice. As a result of this restriction, the warrants currently are not exercisable for any shares of common stock. This number of shares will change depending upon changes in the outstanding shares or waiver of the restriction. Kevin C. Tang is the sole manager of Tang Capital Management, LLC, which is the general partner of Tang Capital Partners, L.P. Accordingly, Mr. Tang and Tang Capital Management share voting and investment power over the securities beneficially owned by Tang Capital Partners, L.P. Mr. Tang disclaims beneficial ownership of the securities except to the extent of his pecuniary interest therein. Mr. Tang and Haeyoung K. Tang share voting and investment power over the securities beneficially owned by The Tang Family Trust. Mr. Tang exercises sole voting and investment power over the securities held by Mr. Tang as custodian for his minor children. Mr. Tang disclaims beneficial ownership of the securities except to the extent of his pecuniary interest therein, if any.
- (3)
- Consists of 1,844,485 shares of our common stock and warrants to purchase 1,844,485 shares of our common stock held by Baker Brothers Life Sciences, L.P.; 113,874 shares of our common stock and warrants to purchase 113,874 shares of our common stock held by 14159, L.P.; and 41,641 shares of our common stock and warrants to purchase 41,641 shares of our common stock held by Baker Bros. Investments, II, L.P. The warrants may not be exercised to the extent that the holder and its affiliates would beneficially own more than 9.999% of our outstanding common stock. This restriction can be waived by the holder upon 61 days written notice. As a result of this restriction, the warrants currently are not exercisable for any shares of common stock. This number of shares will change depending upon changes in the outstanding shares or waiver of the restriction. Baker Brothers Life Sciences Capital (GP), LLC is the general partner of Baker Brothers Life Sciences Capital, L.P., which in turn is the general partner of Baker Brothers Life Sciences, L.P. 14159 Capital (GP), LLC is the sole general partner of 14159 Capital, L.P., which in turn is the sole general partner of 14159, L.P. Baker Bros. Capital (GP), LLC is the sole general partner of Baker Bros. Capital, L.P., which in turn is the sole general partner of Baker Bros. Investments II, L.P. Felix J. Baker and Julian C. Baker are the controlling members of Baker Brothers Life Sciences Capital (GP), LLC, 14159 Capital (GP), LLC and Baker Bros. Capital (GP), LLC. They exercise shared voting and investment power of the securities beneficially owned by these entities. Messrs. Baker and Baker disclaim beneficial ownership of such securities, except to the extent of their pecuniary interest therein, if any.
- (4)
- Consists of 986,019 shares of our common stock held by Capital Ventures International, or CVI. CVI also holds a warrant to purchase 1,000,000 shares of our common stock. The warrant may not be exercised to the extent that the holder and its affiliates would beneficially own more than 9.999% of our outstanding common stock. This restriction can be waived by the holder upon 61 days written notice. As a result of this restriction, the warrant currently is exercisable for up to an aggregate of 723,611 shares of common stock. This number of shares will change depending upon changes in the outstanding shares or waiver of the restriction. Heights Capital Management, Inc., the investment advisor of CVI, has discretionary authority to vote and dispose of the shares held by CVI and may be deemed to be the beneficial owner of these shares.
- (5)
- Consists of 285,635 shares of our common stock and warrants to purchase 336,000 shares of our common stock held by Biotechnology Value Fund, L.P.; 199,055 shares of our common stock and warrants to purchase 229,800 shares of our common stock held by Biotechnology Value Fund II, L.P.; 751,671 shares of our common stock and warrants to purchase 833,100 shares of our common stock held by BVF Investments, L.L.C.; and 84,239 shares and warrants to purchase 101,100 shares of our common stock held by Investments 10, L.L.C The warrants may not be exercised to the extent that the holder and its affiliates would beneficially own more than 9.999% of our outstanding common stock. This restriction can be waived by the holder upon 61 days written
78
notice. As a result of this restriction, the warrants currently are exercisable for up to an aggregate of 351,858 shares of common stock. This number of shares will change depending upon changes in the outstanding shares or waiver of the restriction. Pursuant to the operating agreement of BVF Investments, L.L.C., BVF Partners, L.P. is authorized, among other things, to invest the funds of Ziff Asset Management, L.P., the majority member of BVF Investments, L.L.C., in shares of our common stock and to vote and exercise dispositive power over those shares of the common stock. BVF Partners, L.P. and BVF Inc. share voting and dispositive power over shares of the common stock beneficially owned by Biotechnology Value Fund, L.P., Biotechnology Value Fund II, L.P., BVF Investments, L.L.C, and those owned by Investment 10, L.L.C., on whose behalf BVF Partners, L.P. acts as an investment manager. Accordingly, BVF Partners, L.P. and BVF Inc. have beneficial ownership of all of the shares of the common stock owned by such parties.
- (6)
- Consists of 1,507,356 shares of our common stock beneficially owned by Fort Mason Master, L.P. and Fort Mason Partners, L.P. These entities also hold warrants to purchase 1,408,650 and 91,350 shares of our common stock, respectively, for an aggregate of 1,500,000 shares. Such warrants, however, may not be exercised to the extent that the holder and its affiliates would beneficially own more than 9.999% of our outstanding common stock. This restriction can be waived by the holder upon 61 days written notice. As a result of this restriction, the warrants currently are exercisable for 144,154 shares of common stock. This number of shares will change depending upon changes in the outstanding shares or waiver of the restriction. Fort Mason Capital, LLC serves as the general partner of Fort Mason Master, L.P. and Fort Mason Partners, L.P. and, in such capacity, exercises sole voting and investment authority with respect to the shares of record owned by Fort Mason Master, L.P. and Fort Mason Partners, L.P. Daniel German serves as the sole managing member of Fort Mason Capital, LLC. Fort Mason Capital, LLC and Mr. German each disclaim beneficial ownership of such shares, except to the extent of its or his pecuniary interest therein, if any.
- (7)
- Consists of warrants to purchase 500,000 shares of our common stock held by Xmark JV Investment Partners, LLC, warrants to purchase 225,000 shares of our common stock held by Xmark Opportunity Fund, L.P. and warrants to purchase 275,000 shares of our common stock held by Xmark Opportunity Fund, Ltd. Xmark Opportunity Partners, LLC possesses sole power to vote and direct the disposition of all of these securities. Xmark Capital Partners, LLC is the Managing Member of Xmark Opportunity Partners, LLC. Mitchell D. Kaye and David C. Cavalier are the Chief Executive Officer and Chief Operating Officer, respectively, of Xmark Capital Partners, LLC. As such, they share voting and investment authority with respect to these warrants held by Xmark JV Investment Partners, LLC, Xmark Opportunity Fund, L.P. and Xmark Opportunity Fund, Ltd. Messrs. Kaye and Cavalier disclaim beneficial ownership of the warrants except to the extent of their pecuniary interest therein, if any.
- (8)
- Consists of 62,462 shares of our common stock held by Mr. Shaykin, 122,500 shares issuable upon exercise of options granted to Mr. Shaykin under the 1994 plan, and 21,490 shares beneficially owned through our Employee Stock Ownership Plan, or the ESOP plan, as of December 31, 2006. It also includes 7,500 shares of common stock held in private foundation for which Mr. Shaykin exercises voting control. Mr. Shaykin disclaims beneficial ownership of such shares.
- (9)
- Consists of 3,050 shares of our common stock issuable upon exercise of options granted to Dr. Carter under the 1994 plan and 1,300 shares issuable upon exercise of options granted under the 2004 directors' plan.
- (10)
- Consists of 3,000 shares of our common stock issuable upon exercise of options granted to Mr. Gould under the 1994 plan and 3,000 shares issuable upon exercise of options granted under the 2004 directors' plan.
79
- (11)
- Consists of 11,000 shares of our common stock issuable upon exercise of options granted to Dr. Hayes under the 1994 plan and 1,750 shares issuable upon exercise of options granted under the 2004 directors' plan.
- (12)
- Consists of 4,000 shares of our common stock issuable upon exercise of options granted to Mr. Maza under the 2004 directors' plan.
- (13)
- Consists of 1,300 shares of our common stock held by Mr. Perle, 11,000 shares issuable upon exercise of options granted to Mr. Perle under the 1994 plan and 2,000 shares issuable upon exercise of options granted under the 2004 directors' plan.
- (14)
- Consists of 16,971 shares of our common stock held by Dr. Pilia, 67,100 shares issuable upon exercise of options granted to Dr. Pilia under the 1994 plan, 3,000 shares issuable upon exercise of options granted to Dr. Pilia under the 2004 equity incentive plan, and 54,167 shares issuable upon exercise of options granted to Sterling Ainsworth under the 1994 plan and subsequently transferred to Dr. Pilia.
- (15)
- Consists of 110 shares of our common stock held by Dr. Pollack, 9,150 shares issuable upon exercise of options granted to Dr. Pollack under the 1994 plan and 3,300 shares issuable upon exercise of options granted under the 2004 directors' plan.
- (16)
- Mr. Picker joined our company in January 2007 and is not currently the beneficial owner of any shares. He holds options to purchase 444,085 shares of our common stock, none of which is exercisable within 60 days of March 28, 2007.
- (17)
- Consists of 2,012 shares of our common stock held by Mr. Batt, 6,000 shares issuable upon the exercise of options granted to Mr. Batt under the 1994 plan, 4,000 shares issuable upon the exercise of options granted under the 2004 plan, 7,500 shares issuable upon the exercise of options granted under the 1998 plan and 16,921 shares beneficially owned through our ESOP plan as of December 31, 2006.
- (18)
- Consists of 37,416 shares of our common stock issuable upon the exercise of options granted to Mr. Larson under the 1994 plan, 3,000 shares issuable upon the exercise of options granted under the 2004 equity incentive plan and 20,666 shares beneficially owned through our ESOP plan as of December 31, 2006.
- (19)
- Consists of 6,153 shares of our common stock held by Mr. Link, 64,000 shares issuable upon the exercise of options granted to Mr. Link under the 1994 plan, 4,000 shares issuable upon the exercise of options granted under the 2004 equity incentive plan and 21,499 shares beneficially owned through our ESOP plan as of December 31, 2006.
- (20)
- Consists of shares described in the notes above, as applicable.
Item 13
Certain Relationships and Related Transactions, and Director Independence
TRANSACTIONS WITH RELATED PERSONS
Transactions with Regulus Pharmaceutical Consulting, Inc.
Patricia Pilia, Ph.D., a member of our board of directors, is a consultant with Regulus Pharmaceutical Consulting, Inc., which provides regulatory consulting services to us. Dr. Pilia holds an option entitling her to purchase up to 10% of the outstanding stock of Regulus, subject to vesting requirements. As compensation for consulting services provided to us, we paid Regulus $65,000 during 2006. Dr. Pilia did not provide any of the consulting services referred to above.
80
Registration Rights Agreement
In connection with our 2006 private placement, we entered into a registration rights agreement with the investors and registered the resale of shares of common stock held or to be acquired upon the exercise of warrants held by such investors.
Policies and Procedures for Review and Approval of Conflicting Activities
Our written Code of Ethics and Business Conduct, or our Code, governs situations where conflicts of interests arise, including related-person transactions. In particular, it states that no employee shall, directly or indirectly, engage in, or have any interest, financial or otherwise, in any other business enterprise which interferes or is likely to interfere with the employee's independent exercise of judgment in our company's best interest. Under our Code, a conflict of interest exists when an employee is involved in an activity:
- •
- the operations of which are in conflict with a present or prospective activity of our company, including research and development;
- •
- which provides products or services directly to, or purchases products or services from, our company;
- •
- which subjects the employee to unreasonable time demands that prevent the employee from devoting proper attention to his or her responsibilities to our company;
- •
- which is so operated that the employee's involvement with the outside business activity will reflect adversely upon our company; or
- •
- other situations in which the interest in question is such as to bring it within the area of potential conflict of interest.
In such situations, our Code requires our officers to seek authorization from the audit committee prior to entering into any related-person transaction. Material related-person transactions that are approved by the audit committee and that involve any officer or director are also to be publicly disclosed as required by applicable law.
Pursuant to our Code, non-employee members of our board of directors may have various business, financial, scientific or other relationships with existing or potential collaborators, suppliers or competitors. Any actual or potential conflicts of interest relating to any of these relationships of our non-employee directors that have been disclosed to our board of directors shall not be considered violations of our Code. However, if our board of directors affirmatively determines that any such relationship is inconsistent with the director's responsibilities, it shall so advise the director and the director shall terminate the relationship as promptly as practical.
Item 14
Principal Accountant Fees and Services
Information Regarding our Independent Registered Public Accounting Firm
Audit Fees. During the fiscal years ended December 28, 2005 and December 27, 2006, the aggregate fees billed by Grant Thornton LLP for the audit of our financial statements for such fiscal years, the reviews of our interim financial statements, and Sarbanes-Oxley Section 404 attestation services were $160,000 and $126,000, respectively.
Audit-Related Fees. During the fiscal years ended December 28, 2005 and December 27, 2006, the aggregate fees billed by Grant Thornton for audit related services were $6,000 and $2,200, respectively. These fees related to assistance with our registration statements.
81
Tax Fees. During the fiscal years ended December 28, 2005 and December 27, 2006, Grant Thornton did not perform any tax compliance, tax advice or tax planning services for us.
All Other Fees. During the fiscal years ended December 28, 2005 and December 27, 2006, Grant Thornton did not perform any other services for us.
Audit Committee Pre-Approval Policy. In accordance with our Audit Committee Charter, all audit services and permitted non-audit related services to be performed by Grant Thornton for us must be pre-approved by our Audit Committee, subject to ade minimis exception permitted by Section 10A(i)(1)(B) of the Securities Exchange Act of 1934, as amended. The Audit Committee may form and delegate authority to subcommittees consisting of one or more members when appropriate, including the authority to grant pre-approvals of audit and permitted non-audit services, provided that decisions of such subcommittee to grant pre-approvals shall be presented to the full Audit Committee at its next scheduled meeting. During 2005 and 2006, all services billed by Grant Thornton were pre-approved by the Audit Committee in accordance with this policy.
82
Item 15
Exhibits and Financial Statement Schedules
| Exhibit Number | Description of Exhibit | |
|---|---|---|
| 3.1 | Second Amended and Restated Certificate of Incorporation of the Company, as amended February 3, 2006(1) | |
3.2 | Amended and Restated Bylaws of the Company as amended through April 4, 2006(13) | |
3.3 | Certificate of Designation of Series C Junior Participating Preferred Stock(3) | |
4.1 | Common Stock Certificate(1) | |
4.2 | Rights Agreement dated December 12, 2006 between the Company and American Stock Transfer and Trust Company, as Rights Agent(3) | |
4.3 | The Certificate of Incorporation and Bylaws of the Company are included as Exhibits 3.1 - 3.2 | |
10.1** | Company's Amended and Restated 1994 Long-Term Performance Incentive Plan, as amended through March 4, 2002(2) | |
10.2** | Company's Amended and Restated 1998 Stock Incentive Plan, as amended through October 15, 2002(2) | |
10.3** | Company's 2004 Equity Incentive Plan, as amended and restated effective June 10, 2005(4) | |
10.4** | Form of Stock Option Agreement for certain options granted under the Company's 2004 Equity Incentive Plan(8) | |
10.5** | Company's 2004 Non Employee Director's Stock Option Plan(5) | |
10.6** | Form of Stock Option Agreement for options granted under the Company's 2004 Non-Employee Directors' Stock Option Plan(8) | |
10.7** | Company's 2006 Equity Incentive Plan(15) | |
10.8** | Form of Stock Option Agreement for options granted under the Company's 2006 Equity Incentive Plan(12) | |
10.9** | Employment Agreement effective October 1, 2001 between the Company and Leonard Shaykin(6) | |
10.10** | Employment Agreement effective October 1, 2001 between the Company and Patricia Pilia(6) | |
10.11** | Employment Agreement effective October 1, 2001 between the Company and Gordon Link(6) | |
10.12** | Employment Agreement effective October 1, 2001 between the Company and Kai Larson(6) | |
10.13** | Employment Agreement effective October 28, 2005 between the Company and Martin Batt(4) | |
10.14** | Employment Agreement effective January 2, 2007 between the Company and Donald H. Picker Ph.D.(13) | |
83
10.15** | Form of waiver agreement signed by Patricia A. Pilia, Gordon Link and Kai P. Larson on September 10, 2003 and by Leonard P. Shaykin on September 12, 2003 (together with Schedule required by Instruction 2 to Item 601 Regulation S-K)(2) | |
10.16** | Settlement Agreement dated March 22, 2006 between the Company and Patricia A. Pilia(10) | |
10.17** | Amendment to Employment Agreement executed by the Company and Patricia Pilia dated March 22, 2006(10) | |
10.18** | Form of Amendment to Employment Agreement executed by the Company and Leonard Shaykin, Gordon Link and Kai Larson dated February 27, 2006 (together with Schedule required by Instruction 2 to Item 601 of Regulation S-K)(9) | |
10.19** | Amended and Restated Employment Agreement dated March 28, 2007 between the Company and Martin Batt(14) | |
10.20 | Form of Director and Officer Indemnification Agreement signed by the Company and each of Martin M. Batt, George Gould, Esq., Arthur Hull Hayes, Jr., M.D., Kai Larson, Gordon H. Link, Jr., Patricia A. Pilia, Ph.D., The Honorable Richard N. Perle, Robert E. Pollack, Ph.D., and Leonard P. Shaykin on the dates set forth on the Schedule previously filed and incorporated herein by reference, which Schedule is amended to include the Director and Officer Indemnification Agreement signed by Stephen Carter, M.D. on March 7, 2004 and Elliot Maza on December 14, 2004(7) | |
10.21 | Purchase Agreement, dated as of February 2, 2006, by and among the Company, Special Situations Cayman Fund, L.P. Special Situations Fund L.P., Special Situations Fund II Q.P. L.P., Special Situations Life Sciences Fund III L.P., Special Situations Private Equity Fund L.P., Tang Capital Partners, LP, Baker Biotech Fund II (Z) L.P., Baker Biotech Fund III L.P., Baker Biotech Fund III (Z), L.P., Baker Bros. Investments II, L.P., 14159, L.P., Biotechnology Value Fund, L.P., Biotechnology Value Fund I, L.P., BVF Investments, L.L.C., Investments 10, L.L.C., Fort Mason Master, LP, Fort Mason Partners, LP, Capital Ventures International, Merlin BioMed Long Term Appreciation, LP, Merlin BioMed Offshore Master Fund, Versant Capital Management LLC, Xmark JV Investment Partners, LLC, Xmark Opportunity Fund, L.P., Xmark Opportunity Fund, Ltd. and the other parties signatory thereto(1) | |
10.22 | Registration Rights Agreement, dated April 5, 2006 entered into by and among the Company and the Purchasers(11) | |
10.23 | Form of Warrant, dated April 5, 2006 between the Company and the Purchasers (together with schedule prepared in accordance with Instruction 2 to Item 601 of Regulation S-K)(11) | |
10.24 | Lease Agreement, dated March 15, 2007, between the Company and 2945 Wilderness Place, LTD(15) | |
10.25** | Salaries and Bonuses of Named Executive Officers(13) | |
10.26** | Compensation of Directors(15) | |
21.1 | List of Subsidiaries(15) | |
24.1 | Power of Attorney(13) | |
31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934, as amended* | |
84
31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934, as amended* |
- *
- Filed herewith
- **
- A management compensation plan
- (1)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated February 2, 2006 (File No. 0-24320)
- (2)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K for the year ended December 31, 2003 (File No. 0-24320)
- (3)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated December 14, 2006 (File No. 0-24320)
- (4)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K for the year ended December 28, 2005 (File No. 0-24320)
- (5)
- Incorporated herein by reference to the Company's Quarterly Report on Form 10-Q for the period ending June 30, 2004 (File No. 0-24320)
- (6)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K for the year ended December 31, 2001 (File No. 0-24320)
- (7)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K/A for the year ended December 31, 2002, filed on August 8, 2003 (File No. 0-24320)
- (8)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated December 14, 2004 (File No. 0-24320)
- (9)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated February 23, 2006 (File No. 0-24320)
- (10)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated March 22, 2006 (File No. 0-24320)
- (11)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated April 4, 2006 (File No. 0-24320)
- (12)
- Incorporated herein by reference to the Company's Quarterly Report on Form 10-Q for the period ending March 29, 2006 (File No. 0-24320)
- (13)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K for the year ended December 27, 2006 (File No. 0-24320)
- (14)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated March 28, 2007 (File No. 0-24320)
- (15)
- Incorporated herein by reference to the Company's Registration Statement on Form S-1 dated April 23, 2007 (File No. 333-142301)
85
Pursuant to the requirements of Section 13 of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| TAPESTRY PHARMACEUTICALS, INC. | ||||
By: | /s/ GORDON H. LINK, JR. Gordon H. Link, Jr. Senior Vice President and Chief Financial Officer | April 26, 2007 | ||
86
Exhibits
| Exhibit Number | Description of Exhibit | |
|---|---|---|
| 3.1 | Second Amended and Restated Certificate of Incorporation of the Company, as amended February 3, 2006(1) | |
3.2 | Amended and Restated Bylaws of the Company as amended through April 4, 2006(13) | |
3.3 | Certificate of Designation of Series C Junior Participating Preferred Stock(3) | |
4.1 | Common Stock Certificate(1) | |
4.2 | Rights Agreement dated December 12, 2006 between the Company and American Stock Transfer and Trust Company, as Rights Agent(3) | |
4.3 | The Certificate of Incorporation and Bylaws of the Company are included as Exhibits 3.1 - 3.2 | |
10.1** | Company's Amended and Restated 1994 Long-Term Performance Incentive Plan, as amended through March 4, 2002(2) | |
10.2** | Company's Amended and Restated 1998 Stock Incentive Plan, as amended through October 15, 2002(2) | |
10.3** | Company's 2004 Equity Incentive Plan, as amended and restated effective June 10, 2005(4) | |
10.4** | Form of Stock Option Agreement for certain options granted under the Company's 2004 Equity Incentive Plan(8) | |
10.5** | Company's 2004 Non Employee Director's Stock Option Plan(5) | |
10.6** | Form of Stock Option Agreement for options granted under the Company's 2004 Non-Employee Directors' Stock Option Plan(8) | |
10.7** | Company's 2006 Equity Incentive Plan(15) | |
10.8** | Form of Stock Option Agreement for options granted under the Company's 2006 Equity Incentive Plan(12) | |
10.9** | Employment Agreement effective October 1, 2001 between the Company and Leonard Shaykin(6) | |
10.10** | Employment Agreement effective October 1, 2001 between the Company and Patricia Pilia(6) | |
10.11** | Employment Agreement effective October 1, 2001 between the Company and Gordon Link(6) | |
10.12** | Employment Agreement effective October 1, 2001 between the Company and Kai Larson(6) | |
10.13** | Employment Agreement effective October 28, 2005 between the Company and Martin Batt(4) | |
10.14** | Employment Agreement effective January 2, 2007 between the Company and Donald H. Picker Ph.D.(13) | |
10.15** | Form of waiver agreement signed by Patricia A. Pilia, Gordon Link and Kai P. Larson on September 10, 2003 and by Leonard P. Shaykin on September 12, 2003 (together with Schedule required by Instruction 2 to Item 601 Regulation S-K)(2) | |
10.16** | Settlement Agreement dated March 22, 2006 between the Company and Patricia A. Pilia(10) | |
10.17** | Amendment to Employment Agreement executed by the Company and Patricia Pilia dated March 22, 2006(10) | |
10.18** | Form of Amendment to Employment Agreement executed by the Company and Leonard Shaykin, Gordon Link and Kai Larson dated February 27, 2006 (together with Schedule required by Instruction 2 to Item 601 of Regulation S-K)(9) | |
10.19** | Amended and Restated Employment Agreement dated March 28, 2007 between the Company and Martin Batt(14) | |
10.20 | Form of Director and Officer Indemnification Agreement signed by the Company and each of Martin M. Batt, George Gould, Esq., Arthur Hull Hayes, Jr., M.D., Kai Larson, Gordon H. Link, Jr., Patricia A. Pilia, Ph.D., The Honorable Richard N. Perle, Robert E. Pollack, Ph.D., and Leonard P. Shaykin on the dates set forth on the Schedule previously filed and incorporated herein by reference, which Schedule is amended to include the Director and Officer Indemnification Agreement signed by Stephen Carter, M.D. on March 7, 2004 and Elliot Maza on December 14, 2004(7) | |
10.21 | Purchase Agreement, dated as of February 2, 2006, by and among the Company, Special Situations Cayman Fund L.P., Special Situations Fund L.P., Special Situations Fund III Q.P. L.P., Special Situations Life Sciences Fund III L.P., Special Situations Private Equity Fund L.P., Tang Capital Partners, LP, Baker Biotech Fund II (Z) L.P., Baker Biotech Fund III L.P., Baker Biotech Fund III (Z), L.P., Baker Bros. Investments II, L.P., 14159, L.P., Biotechnology Value Fund, L.P., Biotechnology Value Fund II, L.P., BVF Investments, L.L.C., Investments 10, L.L.C., Fort Mason Master, LP, Fort Mason Partners, LP, Capital Ventures International, Merlin BioMed Long Term Appreciation, LP, Merlin BioMed Offshore Master Fund, Versant Capital Management LLC, Xmark JV Investment Partners, LLC, Xmark Opportunity Fund, L.P., Xmark Opportunity Fund, Ltd. and the other parties signatory thereto(1) | |
10.22 | Registration Rights Agreement, dated April 5, 2006 entered into by and among the Company and the Purchasers(11) | |
10.23 | Form of Warrant, dated April 5, 2006 between the Company and the Purchasers (together with schedule prepared in accordance with Instruction 2 to Item 601 of Regulation S-K)(11) | |
10.24 | Lease Agreement, dated March 15, 2007, between the Company and 2945 Wilderness Place, LTD(15) | |
10.25** | Salaries and Bonuses of Named Executive Officers(13) | |
10.26** | Compensation of Directors(15) | |
21.1 | List of Subsidiaries(15) | |
24.1 | Power of Attorney(13) | |
31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934, as amended* | |
31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934, as amended* |
- *
- Filed herewith
- **
- A management compensation plan.
- (1)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated February 2, 2006 (File No. 0-24320)
- (2)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K for the year ended December 31, 2003 (File No. 0-24320)
- (3)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated December 14, 2006 (File No. 0-24320)
- (4)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K for the year ended December 28, 2005 (File No. 0-24320)
- (5)
- Incorporated herein by reference to the Company's Quarterly Report on Form 10-Q for the period ending June 30, 2004 (File No. 0-24320)
- (6)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K for the year ended December 31, 2001 (File No. 0-24320)
- (7)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K/A for the year ended December 31, 2002, filed on August 8, 2003 (File No. 0-24320)
- (8)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated December 14, 2004 (File No. 0-24320)
- (9)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated February 23, 2006 (File No. 0-24320)
- (10)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated March 22, 2006 (File No. 0-24320)
- (11)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated April 4, 2006 (File No. 0-24320)
- (12)
- Incorporated herein by reference to the Company's Quarterly Report on Form 10-Q for the period ending March 29, 2006 (File No. 0-24320)
- (13)
- Incorporated herein by reference to the Company's Annual Report on Form 10-K for the year ended December 27, 2006 (File No. 0-24320)
- (14)
- Incorporated herein by reference to the Company's Current Report on Form 8-K dated March 28, 2007 (File No. 0-24320)
- (15)
- Incorporated herein by reference to the Company's Registration Statement on Form S-1 dated April 23, 2007 (File No. 333-142301)