Filed Pursuant to Rule 424(b)(3)
Registration Statement No. 333-180562
November 15, 2012
PROSPECTUS SUPPLEMENT NO. 10
SYNTHETIC BIOLOGICS, INC.
112,573 Shares of Common Stock
This prospectus supplement amends and supplements our prospectus, dated July 26, 2012 relating to the resale, from time to time, of up to 112,573 shares of common stock of Synthetic Biologics, Inc. upon the exercise of warrants issued in July 2011 at an exercise price of $1.00 per share and warrants sold in our July 2010 offering at an exercise price of $1.32 per share. We will receive proceeds if the warrants are exercised for cash; to the extent we receive such proceeds, they will be used for working capital purposes.
Our common stock became eligible for trading on the NYSE MKT October 16, 2008. Our common stock is eligible for quotation on the NYSE MKT under the symbol “SYN”. The closing price of our stock on November 14, 2012 was $2.09.
This prospectus supplement is being filed to include the information set forth in the Quarterly Report on Form 10-Q filed on November 14, 2012, which is set forth below. This prospectus supplement should be read in conjunction with the prospectus dated July 26, 2012, supplement no. 1 dated August 9, 2012, prospectus supplement no. 2 dated August 15, 2012, prospectus supplement no. 3 dated August 15, 2012, prospectus supplement no. 4 dated September 12, 2012, prospectus supplement no. 5 dated October 9, 2012, prospectus supplement no. 6 dated October 17, 2012, prospectus supplement no. 7 dated November 1, 2012, prospectus supplement no. 8 dated November 14, 2012; and prospectus supplement no. 9 dated November 15, 2012 which are to be delivered with this prospectus supplement.
Investing in our securities involves a high degree of risk. See “Risk Factors” beginning on page 4 of the original prospectus for more information.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus or the prospectus to which it relates is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this Prospectus Supplement No. 10 is November 15, 2012.
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-Q
(Mark One)
| S | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the quarterly period ended September 30, 2012 |
OR
| £ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES ACT OF 1934 |
| For the transition period from ____________ to ____________ |
Commission File Number: 1-12584
SYNTHETIC BIOLOGICS, INC.
(Name of small business issuer in its charter)
| Nevada | 13-3808303 | |
| (State or other jurisdiction of incorporation or organization) | (IRS Employer Identification Number) | |
| 617 Detroit Street, Suite 100 | ||
| Ann Arbor, MI | 48104 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code:
(734) 332-7800
Securities registered pursuant to Section 12(b) of the Act:
Common Stock, $0.001 par value per share
Securities registered pursuant to Section 12(g) of the Act:
None.
(Title of Class)
Indicate by check mark whether the issuer: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated file, a non-accelerated file, or a smaller reporting company. See the definitions of “large accelerated filer, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer ¨ |
| Non-Accelerated filer ¨ | Smaller reporting company x |
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
As of November 12, 2012, the registrant had 43,697,748 shares of common stock outstanding.
SYNTHETIC BIOLOGICS, INC.
FORM 10-Q
TABLE OF CONTENTS
| Page | |||
| PART I. FINANCIAL INFORMATION | |||
| Item 1. | Financial Statements (Unaudited) | 3 | |
| Consolidated Balance Sheets as of September 30, 2012 and December 31, 2011 | 3 | ||
| Consolidated Statements of Operations for the three and nine months ended September 30, 2012 and 2011 | 4 | ||
| Consolidated Statements of Cash Flows for the nine months ended September 30, 2012 and 2011 | 5 | ||
| Notes to Consolidated Financial Statements | 6 | ||
| Item 2. | Management’s Discussion and Analysis of Financial Information and Results of Operations | 13 | |
| Item 3. | Quantitative and Qualitative Disclosures About Market Risks | 23 | |
| Item 4. | Controls and Procedures | 23 | |
| PART II. OTHER INFORMATION | |||
| Item 1. | Legal Proceedings | 24 | |
| Item 1A. | Risk Factors | 24 | |
| Item 2. | Unregistered Sales of Equity Securities and Use of Proceeds | 34 | |
| Item 3. | Defaults Upon Senior Securities | 34 | |
| Item 4. | Mine Safety Disclosures | 34 | |
| Item 5. | Other Information | 34 | |
| Item 6. | Exhibits | 35 | |
| SIGNATURES | 36 | ||
| GLOSSARY | 37 | ||
| 2 |
PART I.—FINANCIAL INFORMATION
ITEM 1. FINANCIAL STATEMENTS
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Balance Sheets
(In thousands, except share data)
| Assets | September 30, 2012 | December 31, 2011 | ||||||
| (Unaudited) | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | 4,577 | $ | 6,678 | ||||
| Accounts receivable - net | 122 | 405 | ||||||
| Other | 151 | 16 | ||||||
| Assets of discontinued operations | — | 23 | ||||||
| Total Current Assets | 4,850 | 7,122 | ||||||
| Property and equipment, net | 249 | 323 | ||||||
| Long-term note receivable | 700 | — | ||||||
| Deposits and other assets | 27 | 31 | ||||||
| Total Assets | $ | 5,826 | $ | 7,476 | ||||
| Liabilities and Stockholders' Equity | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | 390 | $ | 388 | ||||
| Accrued liabilities | 27 | 29 | ||||||
| Total Current Liabilities | 417 | 417 | ||||||
| Total Liabilities | 417 | 417 | ||||||
| Stockholders' Equity: | ||||||||
| Preferred stock, $0.001 par value; 10,000,000 shares authorized, | ||||||||
| none issued and outstanding | — | — | ||||||
| Common stock, $0.001 par value; 100,000,000 shares authorized, | ||||||||
| 33,477,020 issued and 33,395,538 outstanding in 2012 and | ||||||||
| 31,374,002 issued and 31,292,520 outstanding in 2011 | 33 | 31 | ||||||
| Additional paid-in capital | 62,251 | 58,901 | ||||||
| Accumulated deficit | (56,875 | ) | (51,873 | ) | ||||
| Total Stockholders' Equity | 5,409 | 7,059 | ||||||
| Total Liabilities and Stockholders' Equity | $ | 5,826 | $ | 7,476 | ||||
| See accompanying notes to unaudited consolidated financial statements. |
| 3 |
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Statements of Operations
(In thousands, except share data)
(Unaudited)
| Three months ended September 30, | Nine months ended September 30, | |||||||||||||||
| 2012 | 2011 | 2012 | 2011 | |||||||||||||
| Operating Costs and Expenses: | ||||||||||||||||
| General and administrative | $ | 1,073 | $ | 582 | $ | 3,717 | $ | 2,339 | ||||||||
| Research and development | 763 | 289 | 1,696 | 801 | ||||||||||||
| Total Operating Costs and Expenses | 1,836 | 871 | 5,413 | 3,140 | ||||||||||||
| Loss from Continuing Operations | (1,836 | ) | (871 | ) | (5,413 | ) | (3,140 | ) | ||||||||
| Other Income (Expense): | ||||||||||||||||
| Warrant expense | — | — | — | (1,492 | ) | |||||||||||
| Change in fair value of warrant liability | — | (165 | ) | — | (242 | ) | ||||||||||
| Other income | 10 | 6 | 22 | 55 | ||||||||||||
| Total Other Income (Expense), net | 10 | (159 | ) | 22 | (1,679 | ) | ||||||||||
| Net Loss from Continuing Operations | (1,826 | ) | (1,030 | ) | (5,391 | ) | (4,819 | ) | ||||||||
| Net Income (Loss) from Discontinued Operations | (104 | ) | (68 | ) | 389 | (145 | ) | |||||||||
| Net Loss and Comprehensive Loss | $ | (1,930 | ) | $ | (1,098 | ) | $ | (5,002 | ) | $ | (4,964 | ) | ||||
| Net Income (Loss) Per Share - Basic and Dilutive: | ||||||||||||||||
| Continuing operations | $ | (0.05 | ) | $ | (0.04 | ) | $ | (0.16 | ) | $ | (0.18 | ) | ||||
| Discontinued operations | — | — | 0.01 | — | ||||||||||||
| Net Loss Per Share | $ | (0.05 | ) | $ | (0.04 | ) | $ | (0.15 | ) | $ | (0.18 | ) | ||||
| Weighted average number of shares outstanding | ||||||||||||||||
| during the period - Basic and Dilutive | 33,383,226 | 28,089,492 | 32,801,415 | 27,075,730 | ||||||||||||
See accompanying notes to unaudited consolidated financial statements
| 4 |
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Statements of Cash Flows
(In thousands)
(Unaudited)
| Nine months ended September 30, | ||||||||
| 2012 | 2011 | |||||||
| Cash Flows From Operating Activities: | ||||||||
| Net loss | $ | (5,002 | ) | $ | (4,964 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activites: | ||||||||
| Stock-based compensation | 1,303 | 483 | ||||||
| Stock option modification expense | — | 398 | ||||||
| Stock issued as employee compensation | — | 76 | ||||||
| Stock issued for consulting fees | — | 165 | ||||||
| Warrant expense | — | 1,492 | ||||||
| Change in fair value of warrant liability | — | 242 | ||||||
| Depreciation | 51 | 133 | ||||||
| Provision for uncollectible accounts receivable | 269 | 254 | ||||||
| Gain on sale of discontinued operations | (677 | ) | — | |||||
| Loss on sale of equipment | — | 6 | ||||||
| Impairment on loss of equipment | 30 | — | ||||||
| Gain on the settlement of accounts payable | — | (63 | ) | |||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | 14 | (411 | ) | |||||
| Other current assets | (135 | ) | 220 | |||||
| Deposits and other assets | 4 | 58 | ||||||
| Accounts payable | 2 | (40 | ) | |||||
| Accrued liabilities | (2 | ) | (169 | ) | ||||
| Liabilities of discontinued operations | — | (24 | ) | |||||
| Net Cash Used In Operating Activities | (4,143 | ) | (2,144 | ) | ||||
| Cash Flows From Investing Activities: | ||||||||
| Purchase of short term investments | — | (2,866 | ) | |||||
| Purchase of capital equipment | (7 | ) | — | |||||
| Proceeds from the sale of equipment | — | 1 | ||||||
| Net Cash Used In Investing Activities | (7 | ) | (2,865 | ) | ||||
| Cash Flows From Financing Activities: | ||||||||
| Proceeds from issuance of common stock for stock option exercises | 94 | 8 | ||||||
| Proceeds from issuance of common stock for warrant exercises | 1,955 | — | ||||||
| Proceeds from issuance of common stock , net offering costs $296 | — | 6,961 | ||||||
| Net Cash Provided By Financing Activities | 2,049 | 6,969 | ||||||
| Net increase (decrease) in cash | (2,101 | ) | 1,960 | |||||
| Cash at beginning of period | 6,678 | 2,649 | ||||||
| Cash and cash equivalents at end of period | $ | 4,577 | $ | 4,608 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for interest | $ | — | $ | — | ||||
| Cash paid for taxes | $ | — | $ | — | ||||
See accompanying notes to unaudited consolidated financial statements
| 5 |
Synthetic Biologics, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
(Unaudited)
1. Organization
Synthetic Biologics, Inc. (the “Company” or “Synthetic Biologics”), formerly Adeona Pharmaceuticals, Inc., is a biotechnology company focused on the development of synthetic biologics and innovative medicines for serious infections and diseases. The Company is developing a series of monoclonal antibodies (mAbs) for the treatment of certain infectious diseases and a synthetic DNA-based therapy for the treatment of pulmonary arterial hypertension (PAH). In addition, Synthetic Biologics is developing a product candidate to treat relapsing-remitting multiple sclerosis (MS) and cognitive dysfunction in MS; designing a clinical development pathway for the treatment of amyotrophic lateral sclerosis (ALS); and, has partnered the development of a treatment for fibromyalgia. The Company is also evaluating additional in-licensing opportunities.
| Therapeutic Area | Product Candidate | Status | ||
| Acinetobacter infection | SYN-001 | Discovery; Collaboration with Intrexon | ||
| (monoclonal antibody) | ||||
| Infectious disease* | SYN-002 | Discovery; Collaboration with Intrexon | ||
| (monoclonal antibody) | ||||
| Infectious disease* | SYN-003 | Discovery; Collaboration with Intrexon | ||
| (monoclonal antibody) | ||||
| PAH | SYN-PAH | Preclinical; Collaboration with Intrexon | ||
| (synthetic DNA-based therapy) | ||||
| Relapsing-remitting MS | Trimesta (oral estriol) | Patients fully enrolled in Phase II clinical trial; dosing and monitoring underway | ||
| Cognitive dysfunction in MS | Trimesta (oral estriol) | Patient enrollment underway in Phase II clinical trial | ||
| ALS | AEN-100 | Clinical trial design ongoing | ||
| (gastroretentive zinc acetate) | ||||
| Fibromyalgia | Effirma | Partnered with Meda AB | ||
| (oral flupirtine) |
*Infectious disease targets to be disclosed in the future.
2. Basis of Presentation
The accompanying consolidated financial statements have been prepared pursuant to the rules and regulations of Securities and Exchange Commission (“SEC”) for interim financial information. Accordingly they do not include all of the information and notes required by U.S. GAAP for complete financial statements. The accompanying consolidated financial statements include all adjustments, composed of normal recurring adjustments, considered necessary by management to fairly state our results of operations, financial position and cash flows. The operating results for the interim periods are not necessarily indicative of results that may be expected for any other interim period or for the full year. These consolidated financial statements should be read in conjunction with the consolidated financial statements and notes thereto included in our Annual Report on Form 10-K/A for the year ended December 31, 2011 (“2011 Form 10-K”) as filed with the SEC. The interim results for the three and nine months ended September 30, 2012, are not necessarily indicative of results for the full year.
The consolidated financial statements are prepared in conformity with U.S. GAAP, which requires the use of estimates, judgments and assumptions that affect the amounts of assets and liabilities at the reporting date and the amounts of revenue and expenses in the periods presented. We believe that the accounting estimates employed are appropriate and the resulting balances are reasonable; however, due to the inherent uncertainties in making estimates actual results could differ from the original estimates, requiring adjustments to these balances in future periods.
| 6 |
3. Discontinued Operations of Adeona Clinical Laboratory and Note Receivable
On March 8, 2012, the Company sold all of its interest in Adeona Clinical Laboratory, LLC (the “Lab”) to Hartlab, LLC, an entity controlled by the Lab’s former owner. In connection with the sale of the Lab, the consideration received was (i) the immediate assignment of the Lab’s outstanding accounts receivable up through the date of closing, plus (ii) $700,000 payable pursuant to the terms of a two-year promissory note bearing interest at 5.7% per annum secured by all of the assets of the Lab. The note and all unpaid interest are due on March 1, 2014.
In accordance with ASC Topic 205-20 “Presentation of Financial Statements—Discontinued Operations ” (ASC 205-20), the Company determined that all the criteria had been met and classified the Lab as discontinued operations and its results of operations, financial position and cash flows are separately reported for all periods presented. The assets of the discontinued operations are presented separately under the caption “Assets of discontinued operations” in the accompanying Consolidated Balance Sheets at September 30, 2012, and December 31, 2011, and consist of the following(in thousands):
| September 30, 2012 | December 31, 2011 | |||||||
| Assets of discontinued operations: | ||||||||
| Property and equipment, net | $ | — | $ | 23 | ||||
| Total assets | $ | — | $ | 23 | ||||
The summarized statement of operations data for Adeona Clinical Laboratory for the three and nine months ended September 30, 2012 and 2011 are as follows(in thousands):
| Three months ended September 30, | Nine months ended September 30, | |||||||||||||||
| 2012 | 2011 | 2012 | 2011 | |||||||||||||
| Laboratory fees, net | $ | — | $ | 293 | $ | 115 | $ | 972 | ||||||||
| Operating Costs and Expenses: | ||||||||||||||||
| General and administrative | 104 | 100 | 287 | 311 | ||||||||||||
| Cost of laboratory services | — | 261 | 116 | 806 | ||||||||||||
| Total operating costs and expenses | 104 | 361 | 403 | 1,117 | ||||||||||||
| Income (loss) from discontinued operations | (104 | ) | (68 | ) | (288 | ) | (145 | ) | ||||||||
| Other Income: | ||||||||||||||||
| Gain on sale of Adeona Clinical Laboratory | — | — | 677 | — | ||||||||||||
| Net income (loss) from discontinued operations | $ | (104 | ) | $ | (68 | ) | $ | 389 | $ | (145 | ) | |||||
4. Fair Value of Financial Instruments
The fair value accounting standards define fair value as the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is determined based upon assumptions that market participants would use in pricing an asset or liability. Fair value measurements are rated on a three-tier hierarchy as follows:
| · | Level 1 inputs: Quoted prices (unadjusted) for identical assets or liabilities in active markets; |
| · | Level 2 inputs: Inputs, other than quoted prices included in Level 1 that are observable either directly or indirectly; and |
| · | Level 3 inputs: Unobservable inputs for which there is little or no market data, which require the reporting entity to develop its own assumptions. |
If the inputs used to measure fair value fall in different levels of the fair value hierarchy, the hierarchy level is based upon the lowest level of input that is significant to the fair value measurement.
Cash and cash equivalents include money market accounts and mutual funds of $4.5 million and $5.0 million as of September 30, 2012 and December 31, 2011, respectively, that are measured using Level 1 inputs.
5. Selected Balance Sheet Information
Accounts receivable consisted of the following at September 30, 2012 and December 31, 2011(in thousands):
| September 30, 2012 | December 31, 2011 | |||||||
| Accounts receivable | $ | 682 | $ | 692 | ||||
| Bad debt allowance - customer | (560 | ) | (287 | ) | ||||
| Accounts receivable, net | $ | 122 | $ | 405 | ||||
Property and Equipment consisted of the following at September 30, 2012, and December 31, 2011(in thousands) :
| September 30, 2012 | December 31, 2011 | |||||||
| Manufacturing equipment | $ | 335 | $ | 400 | ||||
| Computer and office equipment | 32 | 159 | ||||||
| Laboratory equipment | 133 | 136 | ||||||
| Total | 500 | 695 | ||||||
| Less accumulated depreciation | (251 | ) | (372 | ) | ||||
| Property and equipment, net | $ | 249 | $ | 323 | ||||
Depreciation expense for the nine months ended September 30, 2012 and 2011 was approximately $51,000 and $133,000, respectively.
| 7 |
6. Stock-Based Compensation
During 2001, Pipex Therapeutics’ Board of Directors and stockholders adopted the 2001 Stock Incentive Plan (the “2001 Stock Plan”). This plan was assumed by Pipex in the October 2006 merger with Sheffield. As of the date of the merger, there were 1,489,353 options issued and outstanding under the 2001 plan. The total number of shares of stock with respect to which stock options and stock appreciation rights may be granted to any one employee of the Company or a subsidiary during any one-year period under the 2001 plan shall not exceed 250,000. All awards pursuant to the 2001 Stock Plan shall terminate upon the termination of the grantee’s employment for any reason. Awards include options, restricted shares, stock appreciation rights, performance shares and cash-based awards (the “Awards”). The 2001 Stock Plan contains certain anti-dilution provisions in the event of a stock split, stock dividend or other capital adjustment, as defined in the plan. The 2001 Stock Plan provides for a Committee of the Board to grant awards and to determine the exercise price, vesting term, expiration date and all other terms and conditions of the awards, including acceleration of the vesting of an award at any time. As of September 30, 2012, there were 1,066,007 options issued and outstanding under the 2001 Stock Plan.
On March 20, 2007, the Company’s Board of Directors approved the Company’s 2007 Stock Incentive Plan (the “2007 Stock Plan”) for the issuance of up to 2,500,000 shares of common stock to be granted through incentive stock options, nonqualified stock options, stock appreciation rights, dividend equivalent rights, restricted stock, restricted stock units and other stock-based awards to officers, other employees, directors and consultants of the Company and its subsidiaries. This plan was approved by stockholders on November 2, 2007. The exercise price of stock options under the 2007 Stock Plan is determined by the compensation committee of the Board of Directors, and may be equal to or greater than the fair market value of the Company’s common stock on the date the option is granted. The total number of shares of stock with respect to which stock options and stock appreciation rights may be granted to any one employee of the Company or a subsidiary during any one-year period under the 2001 plan shall not exceed 250,000. Options become exercisable over various periods from the date of grant, and generally expire ten years after the grant date. As of September 30, 2012, there are 912,739 options issued and outstanding under the 2007 Stock Plan.
On November 2, 2010, the Board of Directors and stockholders adopted the 2010 Stock Incentive Plan (“2010 Stock Plan”) for the issuance of up to 3,000,000 shares of common stock to be granted through incentive stock options, nonqualified stock options, stock appreciation rights, dividend equivalent rights, restricted stock, restricted stock units and other stock-based awards to officers, other employees, directors and consultants of the Company and its subsidiaries. The exercise price of stock options under the 2010 Stock Plan is determined by the compensation committee of the Board of Directors, and may be equal to or greater than the fair market value of the Company’s common stock on the date the option is granted. Options become exercisable over various periods from the date of grant, and generally expire seven to ten years after the grant date. As of September 30, 2012, there are 2,280,000 options issued and outstanding under the 2010 Stock Plan.
In the event of an employee’s termination, the Company will cease to recognize compensation expense for that employee. There is no deferred compensation recorded upon initial grant date, instead, the fair value of the stock-based payment is recognized ratably over the stated vesting period.
The Company has applied fair value accounting for all stock-based payment awards since inception. The fair value of each option or warrant granted is estimated on the date of grant using the Black-Scholes option-pricing model. The Black-Scholes assumptions used in the months ended September 30, 2012 and 2011 are as follows:
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||||||
| 2012 | 2011 | 2012 | 2011 | |||||||||||||
| Exercise price | $1.96 – $2.27 | $0.64 – $0.87 | $1.70 – $2.47 | $0.64 – $2.22 | ||||||||||||
| Expected dividends | 0% | 0% | 0% | 0% | ||||||||||||
| Expected volatility | 153% | 177% – 180% | 108% – 174% | 177% – 188% | ||||||||||||
| Risk free interest rates | 0.63% – 0.65% | 1.40% – 2.17% | 0.37% – 1.98% | 1.40% – 3.58% | ||||||||||||
| Expected life options | 5 years – 10 years | 7 years | 5 years – 10 years | 5 years – 7 years | ||||||||||||
| Expected forfeitures | 0% | 0% | 0% | 0% | ||||||||||||
| 8 |
The Company records stock-based compensation based upon the stated vested provisions in the related agreements, with recognition of expense recorded on the straight line basis over the term of the related agreement. The vesting provisions for these agreements have various terms as follows:
| ● | immediate vesting, | ||
| ● | one-half vesting immediately and the remainder over three years | ||
| ● | monthly over three years, | ||
| ● | quarterly over three years, | ||
| ● | annually over three years, | ||
| ● | one-third immediate vesting and remaining annually over two years, | ||
| ● | one-eighth immediate vesting with remaining vesting over two years, | ||
| ● | one-half immediate vesting with remaining vesting over nine months; and | ||
| ● | one-quarter immediate vesting with the remaining over three years. | ||
During the nine months ended September 30, 2012, the Company granted 1,840,000 options to employees and consultants having a fair value of approximately $4.0 million based upon the Black-Scholes option pricing model. During the same period of 2011, the Company granted 377,002 options to employees having a fair value of approximately $475,000 based upon the Black-Scholes option pricing model.
A summary of stock option activities as of September 30, 2012, and for the year ended December 31, 2011, is as follows:
| Number of Options Outstanding | Weighted Average Exercise Price per Share | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||||||
| Balance – December 31, 2010 | 2,539,091 | $ | 1.32 | |||||||||||||
| Granted | 557,002 | 1.26 | ||||||||||||||
| Exercised | (23,333 | ) | 0.57 | $ | 20,000 | |||||||||||
| Forfeited or expired | (93,750 | ) | 0.59 | |||||||||||||
| Balance – December 31, 2011 | 2,979,010 | 1.34 | ||||||||||||||
| Granted | 1,840,000 | 2.24 | ||||||||||||||
| Exercised | (334,851 | ) | 0.28 | $ | 625,000 | |||||||||||
| Forfeited or expired | (225,413 | ) | 2.37 | |||||||||||||
| Balance – September 30, 2012 – outstanding | 4,258,746 | $ | 1.76 | 6.77 years | $ | 1,980,000 | ||||||||||
| Balance – September 30, 2012 – exercisable | 2,620,784 | $ | 1.56 | 5.85 years | $ | 1,660,000 | ||||||||||
The weighted-average grant-date fair value of options granted during the nine months ended September 30, 2012 and 2011 was $2.20 and $1.26, respectively.
During the nine months ended September 30, 2012 and 2011, 334,851 and 10,000 stock options were exercised, respectively.
The Company recognized $395,000 and $60,000 in stock-based compensation expense relating to stock options during the three months ended September 30, 2012 and 2011, respectively, and $1.3 million and $894,000 during the nine months ended September 30, 2012 and 2011, respectively.
As of September 30, 2012, total unrecognized stock-based compensation expense related to stock options was $3.3 million, which is expected to be expensed through August 2015.
7. Stock Purchase Warrants
On March 15, 2012, the Company entered into a consulting agreement for a financial communications program, for a period of twelve months that began on February 20, 2012. As compensation for such program, the consultant is paid a monthly fee and will be issued a performance warrant exercisable for 250,000 shares of the Company’s common stock based on achievement of certain stock price milestones. Upon initiation of the program, 50,000 of the performance warrants will vest. The performance warrant is exercisable for a period of two years from the date of issuance for an exercise price equal to the price of the Company’s common stock on the date of execution. The expense recorded for the nine months ended September 30, 2012 approximated $63,000 and was estimated using the Monte Carlo valuation model. The assumptions used by the Company are summarized in the following table:
| 9 |
| Exercise price | $ | 2.20 | ||
| Expected dividends | 0 | % | ||
| Expected volatility | 110 | % | ||
| Risk free interest rate | 0.26 | % | ||
| Expected life of warrant | 2 years |
On December 20, 2011, the Company entered into a consulting agreement for financial advisory services, for a period of twelve months. As compensation for such services, the consultant is paid a monthly fee and on February 2, 2012, was issued a warrant exercisable for 100,000 shares of the Company’s common stock. The warrant is exercisable upon issuance for a period of five years from the date of issue at an exercise price equal to the price of the Company’s common stock on the date of issue. The fair value of the warrant approximated $200,000 and was measured using the Black-Scholes valuation model. The assumptions used by the Company are summarized in the following table:
| Exercise price | $ | 1.14 | ||
| Expected dividends | 0 | % | ||
| Expected volatility | 174 | % | ||
| Risk free interest rate | 0.71 | % | ||
| Expected life of warrant | 5 years |
On April 6, 2011, the Company entered into a Common Stock Purchase Agreement with an institutional investor. As part of this agreement, the Company issued a warrant to purchase 844,391 shares of common stock. The warrants have an exercise price of $1.00 and a life of fifteen months. The warrants vested immediately and all warrants were exercised.
On January 28, 2011, the Company entered into a Common Stock Purchase Agreement with three institutional investors. As part of this agreement, the Company issued warrants to purchase 1,428,572 shares of common stock. The warrants have an exercise price of $1.40 and a life of fifteen months. The warrants vested immediately and all warrants were exercised.
On July 2, 2010, the Company entered into a Common Stock Purchase Agreement with a single investor. As part of this agreement, the Company issued warrants to purchase 60,606 shares of common stock to the placement agent, or its permitted assigns. The warrants have an exercise price of $1.32 and a life of five years. The warrants vested on January 1, 2011 and expire December 31, 2015. Since these warrants were granted as part of an equity raise, the Company has treated them as a direct offering cost. The result of the transaction has no affect to equity. As of September 30, 2012, there were 18,182 warrants outstanding.
A summary of warrant activities as of September 30, 2012, and for the year ended December 31, 2011, is as follows:
| Warrants | Weighted Average Exercise Price | |||||||
| Balance – December 31, 2010 | 1,131,078 | $ | 3.49 | |||||
| Granted | 2,272,963 | 1.25 | ||||||
| Exercised | (15,615 | ) | 1.03 | |||||
| Expired | (129,240 | ) | 2.08 | |||||
| Balance – December 31, 2011 | 3,259,186 | 1.99 | ||||||
| Granted | 350,000 | 1.90 | ||||||
| Exercised | (1,768,167 | ) | 1.11 | |||||
| Cancelled by cashless exercise | (516,917 | ) | 1.40 | |||||
| Balance – September 30, 2012 – outstanding | 1,324,102 | $ | 3.25 | |||||
| Balance – September 30, 2012 – exercisable | 1,124,102 | $ | 3.44 | |||||
The Company recognized $1,000 and $165,000 in compensation expense relating to stock purchase warrants for the three months ended September 30, 2012 and 2011, respectively, and $271,000 and $1.7 million for the nine months ended September 30, 2012 and 2011, respectively.
| 10 |
The warrants outstanding as of September 30, 2012, are as follows:
| Exercise Price | Warrants Outstanding | Warrants Exercisable | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value | ||||||||||||||
| $ | 1.14 | 100,000 | 100,000 | 4.34 years | $ | 94,000 | ||||||||||||
| $ | 1.32 | 18,182 | 18,182 | 3.25 years | 14,000 | |||||||||||||
| $ | 2.20 | 250,000 | 50,000 | 1.45 years | - | |||||||||||||
| $ | 2.22 | 517,257 | 517,257 | 4.16 years | - | |||||||||||||
| $ | 3.30 | 61,207 | 61,207 | 2.66 years | - | |||||||||||||
| $ | 3.75 | 50,000 | 50,000 | 3.38 years | - | |||||||||||||
| $ | 6.36 | 327,456 | 327,456 | 0.11 years | - | |||||||||||||
| 1,324,102 | 1,124,102 | 2.58 years | $ | 108,000 | ||||||||||||||
8. Stockholders’ Equity
During the nine months ended September 30, 2012, the Company issued 334,851 shares of common stock, in connection with the exercise of stock options, for proceeds of approximately $94,000. The Company also issued 1,768,167 shares of common stock in connection with the exercise of warrants, for proceeds of approximately $2.0 million.
9. Recent Accounting Pronouncements
There were no accounting standards or interpretations issued or recently adopted that are expected to have a material impact on the Company’s financial position, operations, or cash flows.
10. Subsequent Events
Intrexon Collaboration for Infectious Diseases
On October 16, 2012, a closing was held for the transaction previously announced on August 8, 2012 between the Company and Intrexon Corporation (“Intrexon”). The Company issued 3,552,210 shares of Company common stock, $0.001 par value, which issuance is also deemed paid in partial consideration for the execution and delivery of the Exclusive Channel Collaboration Agreement, dated August 6, 2012, between the Company and Intrexon. The offer and issuance of such shares of common stock have not been registered under the Securities Act of 1933, as amended (the “Securities Act”), and therefore may not be offered or sold in the United States absent registration or an applicable exemption from registration requirements. For this issuance, the Company is relying on the exemption from federal registration under Section 4(2) and Regulation D of the Securities Act, based on the Company’s belief that the offer and sale of such shares of common stock does not involve a public offering as Intrexon is an “accredited investor” as defined under Section 501 promulgated under the Securities Act and no general solicitation has been involved in the offering. The Company will record research and development expense of $7.6 million, the fair value of these shares based on the quoted closing trading price of $2.15 per share.
Private Placement Financing
On October 25, 2012, the Company entered into a Stock Purchase Agreement (the “Purchase Agreement”) with certain accredited investors (the “Purchasers”), pursuant to which the Company agreed to sell to the Purchasers in a private placement an aggregate of 6,750,000 shares of the Company’s common stock at a price per share of $1.60 (the “Common Shares”) for aggregate gross proceeds of $10.8 million and net proceeds of $10.1 million (the “Offering”). On October 30, 2012, the Company completed the Offering. The Company intends to use the net proceeds from the Offering to develop its monoclonal antibody and synthetic DNA programs through its Exclusive Channel Collaborations with Intrexon Corporation, and for general corporate purposes, including the execution of its business plan and expansion of its pipeline.
In connection with the Offering, the Company also entered into a registration rights agreement with certain of the Purchasers (the “Registration Rights Agreement”). The Registration Rights Agreement requires that the Company file a registration statement (the “Initial Registration Statement”) with the Securities and Exchange Commission (the “SEC”) within forty-five (45) days of the closing date of the Offering (the “Filing Date”) for the resale by the Purchasers of all of the Common Shares owned by such Purchasers and all shares of Common Stock issuable upon any stock split, dividend or other distribution, recapitalization or similar event with respect thereto (the “Registrable Securities”). The Initial Registration Statement must be declared effective by the SEC within ninety (90) days of the closing date of the Offering (the “Effectiveness Date”) subject to certain adjustments. Upon the occurrence of certain events (each an “Event”), including, but not limited to, that the Initial Registration Statement is not filed prior to the Filing Date, the Company will be required to pay to each of the Purchasers liquidated damages of 1.5% of their aggregate purchase price upon the date of the Event and then monthly thereafter until the Event is cured. In no event will the aggregate amount of liquidated damages payable to each of the Purchasers exceed in the aggregate 10% of the aggregate purchase price paid by such Purchaser for the Registrable Securities.
In connection with the Offering, the Company also entered into an agreement with a certain Purchaser that is an affiliate of Intrexon Corporation (the “Joinder Agreement”) pursuant to which such Purchaser agreed to be bound by the terms of and join Intrexon Corporation as a party to its registration rights agreement with the Company entered into in connection with the Exclusive Channel Collaboration Agreement between the Company and Intrexon Corporation dated August 6, 2012.
Griffin Securities, Inc. (the “Placement Agent”) served as the placement agent for the Offering. In consideration for services rendered as the Placement Agent in the Offering, the Company agreed to (i) pay to the Placement Agent cash commissions equal to 6.0% of the gross proceeds received in the Offering, (ii) issue to the Placement Agent, or its designee, a five-year warrant to purchase up to 635,855 shares of the Company’s common stock with an exercise price of $1.60 per share (the “Placement Agent Warrant”) and (iii) reimburse the Placement Agent for its reasonable actual out-of-pocket expenses incurred in connection with the Offering, including reasonable legal fees and disbursements. The Placement Agent Warrant also provides for the same registration rights and obligations, and is subject to certain limitations, as set forth in the Registration Rights Agreement with respect to the Common Shares underlying such warrant.
| 11 |
The fair value of the warrant approximated $1.4 million and was measured using the Black-Scholes valuation model. The assumptions used by the Company are summarized in the following table:
| Exercise price | $1.60 | ||
| Expected dividends | 0% | ||
| Expected volatility | 153% | ||
| Risk free interest rate | 0.82% | ||
| Expected life of warrant | 5 years |
Agreement to Acquire C. difficile Clinical-stage Program
On November 8, 2012, the Company entered into an Asset Purchase Agreement (the “Agreement”) with Prev ABR LLC (“Prev”), pursuant to which the Company has the right to acquire theC. diff program assets of Prev, including pre-Investigational New Drug (IND) package, Phase I and Phase II clinical data, manufacturing process data and all issued and pending U.S. and international patents. Pursuant to the Agreement, the Company paid Prev an initial cash payment of $100,000 upon execution of the Agreement and subject to closing conditions anticipated to occur within 30 days, the Company will pay an additional payment $135,000 in cash and 625,000 unregistered shares of the Company’s common stock to Prev. In addition, upon the achievement of the milestones set forth below, Prev may be entitled to receive additional consideration payable 50% in cash and 50% in stock of the Company, subject to Prev’s option to receive the entire payment in shares of the Company’s stock, with the exception of the first milestone payments to be paid in cash: (i) upon commencement of an IND; (ii) upon commencement of a Phase I clinical trial; (iii) upon commencement of a Phase II clinical trial; (iv) upon commencement of a Phase III clinical trial; (v) upon Biologic License Application (BLA) filing in the U.S. and for territories outside of the U.S. (as defined in the Agreement); and, (vi) upon BLA approval in the U.S. and upon approval in territories outside the-U.S. The Agreement and stock issuances are subject to prior approval of the NYSE MKT, LLC. The Agreement is subject to certain due diligence obligations and no royalties are payable to Prev under the Agreement.
The Agreement provides for termination prior to closing: (i) upon the mutual agreement of the parties; (ii) by Prev if the closing has not occurred within thirty (30) days of the execution of the Agreement; provided that such failure to close is not due to the failure of Prev to fulfill its obligations under the Agreement or Prev has not been the cause of such failure, or (iii) by the Company at any time. If the Agreement is terminated by the Company then the Company shall be entitled to receive a refund of half of its initial cash payment, in addition to any fees paid by the Company on behalf of Prev and if such termination is due to the failure of Prev to fulfill its obligations under the Agreement or a breach of a representation or warranty of Prev then the Company shall be entitled to a refund of the entire cash payment in addition to any fees paid by the Company on behalf of Prev.
The Agreement also provides that Prev has a right to the return to it of all assets acquired by the Company under the Agreement if on or prior to the date that is (i) thirty (30) months after the execution of the Agreement, the Company has not initiated toxicology studies in non-rodent models or (ii) thirty six (36) months have not filed an IND under the program related to the assets and such failure is not due to action or inaction of Prev or breach of its representations or warranties or covenants or if there is a change of control as defined in the Agreement and after such change of control the assets are not further developed; provided however that such thirty (30) and thirty six (36) month periods can be extended by the Company for an additional twelve (12) months upon payment of a cash milestone payment.
| 12 |
ITEM 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL INFORMATION AND RESULTS OF OPERATIONS
The following discussion should be read in conjunction with the attached unaudited consolidated financial statements and notes thereto, and with our audited consolidated financial statements and notes thereto for the fiscal year ended December 31, 2011, found in our Annual Report on Form 10-K/A. In addition to historical information, the following discussion contains forward-looking statements that involve risks, uncertainties and assumptions. Where possible, we have tried to identify these forward looking statements by using words such as “anticipate,” “believe,” “intends,” or similar expressions. Our actual results could differ materially from those anticipated by the forward-looking statements due to important factors and risks including, but not limited to, those set forth under “Risk Factors” in this 10-Q and as applicable in Part I, Item 1A of our Annual Report on Form 10-K/A.
Overview
We are a biotechnology company focused on the development of synthetic biologics and innovative medicines for serious infections and diseases. We are developing a series of monoclonal antibodies (mAbs) for the treatment of certain infectious diseases and a synthetic DNA-based therapy for the treatment of pulmonary arterial hypertension (PAH). In addition, we are developing a product candidate to treat relapsing-remitting multiple sclerosis (MS) and cognitive dysfunction in MS; designing a clinical development pathway for the treatment of amyotrophic lateral sclerosis (ALS); and, have partnered the development of a treatment for fibromyalgia. We are also evaluating additional in-licensing opportunities.
Product Pipeline:
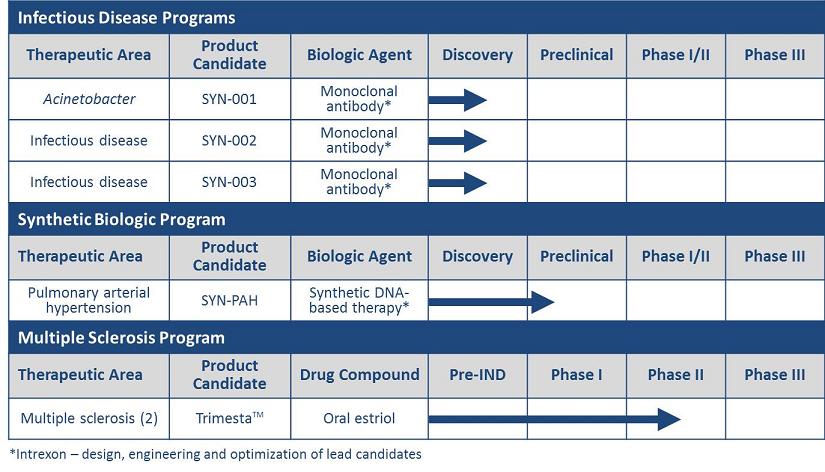
Infectious Disease Programs:
| • | In August 2012, we entered into a second worldwide exclusive channel collaboration with Intrexon Corporation (Intrexon) through which we intend to develop and commercialize a series of mAb therapies for the treatment of certain infectious diseases not adequately addressed by existing therapies. Utilizing Intrexon’s comprehensive suite of proprietary technologies, including the mAbLogix™ and LEAP™ platforms, we intend to target three infectious disease indications as part of the Intrexon collaboration. In September 2012, we initiated efforts to develop our first mAb therapy for the treatment of acinetobacter infections. Many strains ofAcinetobacter are multidrug-resistant and pose an increasing global threat to hospitalized patients, wounded military personnel and those affected by natural disasters. A treatment for acinetobacter infections represents a multi-billion dollar market opportunity.
(mAbLogix™ and LEAP™ are registered trademarks of Intrexon Corporation)
|
| 13 |
Synthetic Biologic Program:
| • | Our synthetic DNA-based product candidate is intended to treat PAH, a serious life-threatening lung disease. This product is designed to deliver DNA that encodes a therapeutic protein called prostacyclin synthase (PGIS) locally to the pulmonary arteries of PAH patients via a single procedure, and, via an oral daily pill, control the long-term local expression of such therapeutic protein. We are developing this initial product candidate pursuant a global exclusive channel collaboration that we entered into with Intrexon in November 2011. As part of this collaboration, we have access to Intrexon's UltraVector® platform and RheoSwitch Therapeutic System® for this product application. We anticipate that by continuously producing and delivering prostacyclin directly where it is needed, in the pulmonary arteries of PAH patients, this product candidate may overcome the dose limiting side effects of systemic prostacyclin treatments for PAH, a mainstay of PAH treatment. According to GlobalData, the global market for PAH treatments is estimated to exceed $3.6 billion by 2015. |
(UltraVector® and RheoSwitch Therapeutic System® are registered trademarks of Intrexon Corporation)
Multiple Sclerosis Programs:
| • | TrimestaTM (oral estriol) is being developed as an oral once-daily treatment for relapsing-remitting MS in women. Patient enrollment of 164 patients is complete in this randomized, double-blind, placebo-controlled Phase II clinical trial being conducted at 15 centers in the U.S. Patients are being dosed and monitored for two years. This clinical trial is supported by grants exceeding $8 million, which should be sufficient to fund the trial through completion. Current sales of injectable disease-modifying therapies for MS are estimated at $8.9 billion annually. According to various reports, sales of oral disease-modifying therapies for MS, of which Trimesta™, if and when approved, would be in such class, are anticipated to grow from $500 million in 2010 to $5 billion annually by 2017. |
| • | TrimestaTM is also being developed for the treatment of cognitive dysfunction in female MS patients. In January 2012, patient enrollment began in a randomized, double-blind, placebo-controlled Phase II clinical trial being conducted at University of California, Los Angeles (UCLA). Patient recruitment and enrollment into this trial is ongoing. The majority of the costs of this trial are being funded by grants from foundations and charitable organizations and we have pledged approximately $500,000 to UCLA to partially fund this trial payable over three years. An estimated 50-65% of MS patients are expected to develop disabilities due to cognitive dysfunction and there is currently no approved treatment. |
Other Programs:
| • | AEN-100 (gastroretentive zinc acetate) is a novel formulation of zinc acetate that may be used for the treatment of ALS, also known as Lou Gehrig’s disease. Previous investigator studies have suggested that alterations in the handling and disposition of zinc ions in the brain may be important in the initiation and development of ALS. We are currently collaborating with the investigator and, based on feedback from the United States Food & Drug Administration (FDA), intend to design a clinical development pathway for AEN-100 in the treatment of ALS. There is only one approved therapy for ALS, the efficacy of which is considered to be marginal. Based on an estimated annual price of $10,000 per ALS patient, we estimate that the total market potential in the U.S. is $300 million. |
| • | EffirmaTM (flupirtine) is being developed for the treatment of fibromyalgia by Meda AB (Meda), a multi-billion dollar international pharmaceutical company. On May 6, 2010, we entered into a sublicense agreement with Meda covering all of our patents’ rights on the use of flupirtine for fibromyalgia in the U.S., Canada and Japan. According to Meda’s 2011 Annual Report filed in May 2012, flupirtine for fibromyalgia is currently in Phase II development. The sublicense agreement provides that all ongoing and future development costs are to borne by Meda and we are entitled to receive certain payments if milestones are achieved and royalties on sales. Based on an estimated annual price of $1,200 per fibromyalgia patient, we estimate that the total market potential in the U.S. is $6 billion. |
Recent Developments
On November 18, 2011, we entered into a Channel Agreement with Intrexon that governs an “exclusive channel collaboration” arrangement in which we intend to use Intrexon’s technology directed towards the production of PGIS, through the use ofin vivo conditionally regulated embedded controllable bioreactors for the treatment of PAH. The Channel Agreement establishes committees comprised of our and Intrexon representatives that will govern activities related to the PAH program in the areas of project establishment, chemistry, manufacturing and controls, clinical and regulatory matters, commercialization efforts and intellectual property.
| 14 |
As partial consideration for execution of the Channel Agreement, we entered into a Stock Purchase Agreement with Intrexon pursuant to which we issued to Intrexon a number of shares of our common stock equal to 9.995% of the number of shares of our common stock issued and outstanding following and giving effect to such issuance (the “First Tranche”) at a purchase price equal to the $0.001 par value of such shares, which issuance was deemed paid in partial consideration for the execution and delivery of the Channel Agreement. We also agreed to issue additional shares of our common stock to Intrexon upon dosing of the first patient in a Phase II clinical trial sponsored by us in the U.S., or similar study as the parties may agree in a country other than the U.S.
On December 21, 2011, we announced that the Board of Directors had taken several actions to prioritize our focus on our entry into the emerging field of synthetic biology. In connection with the change in business focus on March 8, 2012, we entered into a Membership Interest Purchase Agreement, and certain related agreements, pursuant to which we sold all of our interest in the Adeona Clinical Laboratory (the “Lab”) to Hartlab, LLC, an entity controlled by the Lab’s former owner, in consideration for (i) the immediate assignment of the Lab’s outstanding accounts receivable up through the date of closing, plus (ii) Seven Hundred Thousand Dollars ($700,000) payable pursuant to the terms of a two-year non-recourse promissory note secured by all of the assets of the Lab.See Note 3 to the Notes to the Consolidated Financial Statements – Discontinued Operations of Adeona Clinical Laboratory and Note Receivable.
On February 15, 2012, upon stockholder approval, we amended our Articles of Incorporation to change our name to Synthetic Biologics, Inc. Our common stock continues trade on the NYSE MKT (formerly the NYSE Amex and American Stock Exchange), under the symbol “SYN”. Prior to this time and since October 16, 2008, our name was Adeona Pharmaceuticals, Inc. and we traded on the NYSE MKT stock exchange under the symbol “AEN”. We are incorporated in the State of Nevada. We continue to maintain our principal executive offices in Ann Arbor, MI, and are currently located at 617 Detroit Street, Suite 100, Ann Arbor, MI 48104.
On August 6, 2012, we expanded our relationship with Intrexon and entered into the Channel Agreement with Intrexon that governs an “exclusive channel collaboration” arrangement in which we will use Intrexon’s technology relating to the identification, design and production of human antibodies and DNA vectors for the development and commercialization of a series of monoclonal antibody therapies for the treatment of certain serious infectious diseases (the “Program”). The Channel Agreement establishes committees comprised of our and Intrexon representatives that will govern activities related to the Program in the areas of project establishment, chemistry, manufacturing and controls, clinical and regulatory matters, commercialization efforts and intellectual property.
On October 16, 2012, a closing was held for the transaction previously announced in August 2012 between ourselves and Intrexon. Pursuant to the terms of a Stock Issuance Agreement with Intreson, we issued 3,552,210 shares of our common stock, $0.001 par value, which issuance is also deemed paid in partial consideration for the execution and delivery of the Exclusive Channel Collaboration Agreement, dated August 6, 2012, between ourselves and Intrexon. We also agreed to register the shares issued to Intrexon in accordance with the First Amendment to Registration Rights Agreement.
On October 25, 2012, we entered into a Stock Purchase Agreement (the “Purchase Agreement”) with certain accredited investors (the “Purchasers”), pursuant to which we agreed to sell to the Purchasers in a private placement an aggregate of 6,750,000 shares of our common stock at a price per share of $1.60 (the “Common Shares”) for aggregate gross proceeds of $10.8 million and net proceeds of $10.1 million (the “Offering”). On October 30, 2012, we completed the Offering. We intend to use the net proceeds from the Offering to develop our monoclonal antibody and synthetic DNA programs through our Exclusive Channel Collaborations with Intrexon Corporation, and for general corporate purposes, including the execution of our business plan and expansion of our pipeline.
In connection with the Offering, we also entered into a registration rights agreement with certain of the Purchasers (the “Registration Rights Agreement”). The Registration Rights Agreement requires that we file a registration statement (the “Initial Registration Statement”) with the Securities and Exchange Commission (the “SEC”) within forty-five (45) days of the closing date of the Offering (the “Filing Date”) for the resale by the Purchasers of all of the Common Shares owned by such Purchasers and all shares of Common Stock issuable upon any stock split, dividend or other distribution, recapitalization or similar event with respect thereto (the “Registrable Securities”). The Initial Registration Statement must be declared effective by the SEC within ninety (90) days of the closing date of the Offering (the “Effectiveness Date”) subject to certain adjustments. Upon the occurrence of certain events (each an “Event”), including, but not limited to, that the Initial Registration Statement is not filed prior to the Filing Date, we will be required to pay to each of the Purchasers liquidated damages of 1.5% of their aggregate purchase price upon the date of the Event and then monthly thereafter until the Event is cured. In no event will the aggregate amount of liquidated damages payable to each of the Purchasers exceed in the aggregate 10% of the aggregate purchase price paid by such Purchaser for the Registrable Securities.
| 15 |
In connection with the Offering, we also entered into an agreement with a certain Purchaser that is an affiliate of Intrexon Corporation (the “Joinder Agreement”) pursuant to which such Purchaser agreed to be bound by the terms of and join Intrexon Corporation as a party to its registration rights agreement with us entered into in connection with the Exclusive Channel Collaboration Agreement between ourselves and Intrexon Corporation dated August 6, 2012.
Griffin Securities, Inc. (the “Placement Agent”) served as the placement agent for the Offering. In consideration for services rendered as the Placement Agent in the Offering, we agreed to (i) pay to the Placement Agent cash commissions equal to 6.0% of the gross proceeds received in the Offering, (ii) issue to the Placement Agent, or its designee, a five-year warrant to purchase up to 635,855 shares of our common stock with an exercise price of $1.60 per share (the “Placement Agent Warrant”) and (iii) reimburse the Placement Agent for its reasonable actual out-of-pocket expenses incurred in connection with the Offering, including reasonable legal fees and disbursements. The Placement Agent Warrant also provides for the same registration rights and obligations, and is subject to certain limitations, as set forth in the Registration Rights Agreement with respect to the Common Shares underlying such warrant.
On November 8, 2012, we entered into an Asset Purchase Agreement (the “Agreement”) with Prev ABR LLC (“Prev”), pursuant to which we have the right to acquire theC. diff program assets of Prev, including pre-Investigational New Drug (IND) package, Phase I and Phase II clinical data, manufacturing process data and all issued and pending U.S. and international patents. Pursuant to the Agreement, we paid Prev an initial cash payment of $100,000 upon execution of the Agreement and subject to closing conditions anticipated to occur within 30 days, we will pay an additional payment $135,000 in cash and 625,000 unregistered shares of our common stock to Prev. In addition, upon the achievement of the milestones set forth below, Prev may be entitled to receive additional consideration payable 50% in cash and 50% in our stock, subject to Prev’s option to receive the entire payment in shares of our stock, with the exception of the first milestone payments to be paid in cash: (i) upon commencement of an IND; (ii) upon commencement of a Phase I clinical trial; (iii) upon commencement of a Phase II clinical trial; (iv) upon commencement of a Phase III clinical trial; (v) upon Biologic License Application (BLA) filing in the U.S. and for territories outside of the U.S. (as defined in the Agreement); and, (vi) upon BLA approval in the U.S. and upon approval in territories outside the-U.S. The Agreement and stock issuances are subject to prior approval of the NYSE MKT, LLC. The Agreement is subject to certain due diligence obligations and no royalties are payable to Prev under the Agreement.
The Agreement provides for termination prior to closing: (i) upon the mutual agreement of the parties; (ii) by Prev if the closing has not occurred within thirty (30) days of the execution of the Agreement; provided that such failure to close is not due to the failure of Prev to fulfill its obligations under the Agreement or Prev has not been the cause of such failure, or (iii) by us at any time. If the Agreement is terminated by us then we shall be entitled to receive a refund of half of its initial cash payment, in addition to any fees paid by us on behalf of Prev and if such termination is due to the failure of Prev to fulfill its obligations under the Agreement or a breach of a representation or warranty of Prev then we shall be entitled to a refund of the entire cash payment in addition to any fees paid by us on behalf of Prev.
The Agreement also provides that Prev has a right to the return to it of all assets acquired by us under the Agreement if on or prior to the date that is (i) thirty (30) months after the execution of the Agreement, we have not initiated toxicology studies in non-rodent models or (ii) thirty six (36) months have not filed an IND under the program related to the assets and such failure is not due to action or inaction of Prev or breach of its representations or warranties or covenants or if there is a change of control as defined in the Agreement and after such change of control the assets are not further developed; provided however that such thirty (30) and thirty six (36) month periods can be extended by us for an additional twelve (12) months upon payment of a cash milestone payment.
To date, we have financed our operations primarily through public and private sales of our common stock, and we expect to continue to seek to obtain the required capital in a similar manner. We have incurred an accumulated deficit of $56.9 million through September 30, 2012. We cannot provide any assurance that we will be able to achieve profitability on a sustained basis, if at all, obtain the required funding, obtain the required regulatory approvals, or complete additional corporate partnering or acquisition transactions.
Pipeline Programs and Therapeutic Areas
Infectious Disease Programs
We are pursuing the development of treatments for infectious diseases. Infectious disease outbreaks are increasing while intervention options declining due to widespread multidrug-resistant pathogens, increasing numbers of immuno-compromised patients and the discovery of new pathogens.
Infectious diseases are caused by organisms that are typically invisible to the naked eye, such as bacteria, viruses, toxins, parasites or fungi. Many microorganisms settle in and on our bodies; normally they are harmless or even helpful, but under certain circumstances they may cause disease. An infectious disease is termed contagious if it can easily be spread, directly or indirectly, from one person to another. Some infectious diseases, however, are transmitted via bites from insects or animals, while others are acquired by consuming contaminated food or water, along with other exposures in the environment.
Intrexon Collaboration for Infectious Diseases
In August 2012, we entered into a second worldwide exclusive channel collaboration with Intrexon through which we intend to develop a series of mAb therapies for the treatment of certain infectious diseases not adequately addressed by existing therapies. Utilizing Intrexon’s comprehensive suite of proprietary technologies, including the mAbLogix™ platform for rapid discovery of fully human mAbs and the LEAP™ cell processing station, our initial efforts will target three infectious disease indications. We also have the option to target an additional five infectious disease indications under this collaboration.
Monoclonal Antibodies for Infectious Diseases
Acting as the body's army, antibodies are proteins, generally found in the bloodstream, that provide immunity in detecting and destroying pathogens, such as viruses and bacteria and their associated toxins. MAbs can also be designed and produced as therapeutic agents, utilizing protein engineering and recombinant production technologies. The mAbs being developed under the Synthetic Biologics’ collaboration with Intrexon are intended to supplement a patient's own immune system by providing the means to specifically and rapidly neutralize and/or clear specific pathogens and toxins of interest in a process known as “passive immunity”. Many pathogens that cause infectious diseases are innately resistant to, or over time have developed increased resistance to, antibiotics and other drugs. We intend to utilize Intrexon’s comprehensive suite of proprietary mAb design and recombinant protein production technologies to efficiently create potent candidate mAbs for human testing and use to specifically treat certain infectious diseases for which current therapies are unavailable or inadequate.
| 16 |
First Infectious Disease Target: Acinetobacter
In September 2012, we initiated efforts to develop our first mAb therapy for the treatment of acinetobacter infections under our collaboration with Intrexon.Acinetobacter is a difficult to treat pathogen due to its rapid and well-established resistance to most antibiotics, making it a multidrug-resistant pathogen. In addition, as a biofilm-forming pathogen,Acinetobacter has the ability to survive up to twice as long as non-biofilm-forming pathogens. In the U.S.,Acinetobacter has been reported to be the cause of up to 2.6% of hospital acquired infections, 1.3% of bloodstream infections and 7% of ICU respiratory tract infections, and more than half of theAcinetobacter isolates are multidrug-resistant. Patients with infections caused byAcinetobacterhave been reported havingmortality rates as high as 43%in the hospital and in the ICU. WhileAcinetobacter is a well-documented pathogen in the hospital setting, this pathogen also poses an increasing danger to wounded servicemen and women in military treatment centers and to those treated in trauma centers following natural disasters.
A treatment for acinetobacter infections represents a multi-billion dollar market opportunity.
Synthetic Biologic Program
We are engaged in the emerging field of synthetic biology directed for the purpose of developing new human therapeutic products. Synthetic biology is an emerging field that combines molecular biology and automation to design, optimize and construct new biological systems and functions. These technologies utilize a combination of automated processes including, DNA sequencing, computer-aided design, DNA synthesis, fabrication of modular transgenes and high throughput testing to create and optimize biologic products.
Intrexon Collaboration for a Synthetic DNA-Based Therapy for PAH
In November 2011, we entered into a collaboration with Intrexon for the design, optimization and development of a synthetic DNA-based therapeutic product candidate utilizing Intrexon’s proprietary technologies for the treatment of PAH. Synthetic DNA-based therapeutics comprise constructs of DNA that can be administered to patients via a single procedure. Once introduced, they are intended to continuously produce therapeutic proteinsin vivo in a controllable and localized fashion for up to a period of years.
An important feature of our synthetic DNA-based product candidate for PAH being developed in collaboration with Intrexon may be the incorporation of its proprietary technologies: the UltraVector® platform for design, construction, and testing of genetic components and the RheoSwitch Therapeutic System®. The RheoSwitch Therapeutic System® is intended to provide unprecedented control of therapeutic protein expression through the use of a highly specific orally available activating ligand that can be taken by patients on a daily basis as one or more pills. In this way, the levels ofin vivo protein expression may be adjusted from time to time by treating physicians through simple dose adjustment of the oral activating ligand. Such system also provides an important safety mechanism not previously available in gene therapy clinical trials since in the absence of taking an oral pill, protein expression would not be expected to occur.
PAH
PAH is a progressive, disabling and life-threatening disorder characterized by abnormally high blood pressure (hypertension) in the pulmonary artery, the blood vessel that carries blood from the heart to the lungs. Hypertension occurs when most of the very small arteries throughout the lungs narrow in diameter, therefore constricting blood flow through the lungs. The constriction of blood flow causes the pressure to increase in the pulmonary artery and in the right ventricle (the heart chamber that pumps blood into the pulmonary artery). Signs and symptoms of PAH take place when the increased pressure cannot overcome the constriction and there is insufficient blood flow to the body. Shortness of breath during exertion and fainting spells are the most common early symptoms of PAH. Despite current treatments, PAH generally has a very poor outcome and is associated with high rates of mortality within three to five years of diagnosis.
Synthetic DNA-Based Therapeutic for PAH
Our initial synthetic DNA-based therapeutic product candidate is intended for the treatment of PAH, a serious life-threatening lung disease. This product candidate is designed to deliver DNA that encodes a therapeutic protein called prostacyclin synthase (PGIS) locally to the pulmonary arteries of PAH patients via a single pulmonary catheter procedure and via an oral daily pill, control the long-term local expression of such therapeutic protein.
We are developing this initial product candidate in collaboration with Intrexon. Under the collaboration, we intend to utilize Intrexon's advanced transgene engineering platform for the controlled, precise and continuousin vivo cellular production of PGIS. PGIS is a specific effector enzyme that regulates the production of prostacyclin, a potent mediator of arterial dilation that also prevents smooth muscle proliferation and arterial wall thickening. PGIS expression is decreased in the lungs of PAH patients and deficiency in prostacyclin production is strongly implicated in PAH. We anticipate that by continuously producing and delivering prostacyclin directly where it is needed, in the pulmonary arteries of PAH patients via PGIS, this product candidate may overcome the dose limiting side effects of systemic prostacyclin-based treatments for PAH. While systemic prostacyclin-based treatments for PAH are currently a mainstay of PAH therapy, their considerable systemic side effects limit their dose and ultimate long-term utility.
The global market potential for the treatment of PAH is estimated to be up to $3.6 billion by 2015, according to GlobalData, Pulmonary Arterial Hypertension (PAH) – Drug Pipeline Analysis and Market Forecasts for 2016.
Multiple Sclerosis Program
We are developing our product candidate, TrimestaTM (oral estriol), to treat relapsing-remitting MS and cognitive dysfunction in MS.
| 17 |
Relapsing-Remitting MS in Women
MS is a progressive neurological disease in which the body loses the ability to transmit messages along central nervous system nerve cells, leading to a loss of muscle control, paralysis, cognitive impairment and in some cases death. According to the National Multiple Sclerosis Society (NMSS), currently, more than 2.5 million people worldwide (approximately 400,000 patients in the U.S. of which 70% are estimated to be women) have been diagnosed with MS. Young adults, ages 20 to 50, and two to three times as many women than men are predominantly diagnosed with MS. According to the NMSS, approximately 85% of MS patients are initially diagnosed with the relapsing-remitting form, compared to 10-15% with other progressive forms.
There are currently eight FDA approved therapies for the treatment of relapsing-remitting MS: Betaseron®, Rebif®, Avonex®, Novantrone®, Copaxone®, Tysabri® , Gilenya® and Extavia®. These therapies provide only a modest benefit for patients with relapsing-remitting MS and therefore serve to only delay progression of the disease. All of these drugs except Gilenya® require frequent (daily, weekly & monthly) injections (or infusions) on an ongoing basis and can be associated with unpleasant side effects (such as flu-like symptoms), high rates of non-compliance among users, and eventual loss of efficacy due to the appearance of resistance in approximately 30% of patients. Despite the availability of multiple FDA-approved therapies for the treatment of relapsing-remitting MS, the disease is highly underserved and exacts a heavy economic toll.
Current sales of injectable disease-modifying therapies for MS are estimated at $8.9 billion annually. According to various reports, sales of oral disease-modifying therapies for MS, of which Trimesta, if and when approved, would be in such class, are anticipated to grow from $500 million in 2010 to in excess of $5 billion annually by 2017.
Relapsing-Remitting MS: Background
It has been scientifically documented that pregnant women with certain autoimmune diseases experience a spontaneous reduction of disease symptoms during pregnancy, particularly in the third trimester. The PRIMS (Pregnancy In MS) study, a landmark clinical study published in theNew England Journal of Medicine followed 254 women with MS during 269 pregnancies and for up to one year after delivery. The PRIMS study demonstrated that relapse rates were significantly reduced by 71% (p < 0.001) through the third trimester of pregnancy compared to pre-pregnancy-rates, and that relapse rates increased by 120% (p < 0.001) during the first three months after birth (post-partum) before returning to pre-pregnancy rates. It has been hypothesized that the female hormone, estriol, produced by the placenta during pregnancy, plays a role in “fetal immune privilege”, a process that prevents a mother’s immune system from attacking and rejecting her fetus. Maternal levels of estriol increase in a linear fashion through the third trimester of pregnancy until birth, whereupon they abruptly return to low circulating levels. The anti-autoimmune effects of estriol are thought to be responsible for the therapeutic effects of pregnancy on MS.
Rhonda Voskuhl, M.D., Director, UCLA MS program, UCLA Department of Neurology, has found that pregnancy levels of estriol have potent immunomodulatory effects. She further postulated and tested in pilot clinical studies that oral doses of estriol may have a therapeutic benefit when administered to non-pregnant female MS patients by, in essence, mimicking the spontaneous reduction in relapse rates seen in MS patients during pregnancy.
Estriol has been approved and marketed for over 40 years throughout Europe and Asia for the oral treatment of post-menopausal symptoms. It has never been approved by the U.S. FDA for any indication.
Relapsing-Remitting MS: Clinical Development
Our Trimesta (oral estriol) drug candidate is for the treatment of relapsing-remitting MS in women. An investigator-initiated, 10-patient, 22-month, single-agent, crossover clinical trial to study the therapeutic effects of 8 mg of oral Trimesta taken daily in non-pregnant female relapsing-remitting MS patients was completed in the U.S. The total volume and number of gadolinium-enhancing lesions were measured by brain magnetic resonance imaging (an established neuroimaging measurement of disease activity in MS). Over the next three months of treatment with Trimesta, the median total enhancing lesion volumes decreased by 79% (p = 0.02) and the number of lesions decreased by 82% (p = 0.09). They remained decreased during the next 3 months of treatment, with lesion volumes decreased by 82% (p = 0.01), and numbers decreased by 82% (p =0.02). Following a six-month drug holiday during which the patients were not on any drug therapies, median lesion volumes and numbers returned to near baseline pretreatment levels. Trimesta therapy was reinitiated during a four-month retreatment phase of this clinical trial. The relapsing-remitting MS patients again demonstrated a decrease in enhancing lesion volumes of 88% (p = 0.008) and a decrease in the number of lesions by 48% (p = 0.04) compared with original baseline scores.
A Phase II randomized, double-blind, placebo-controlled clinical trial is currently underway at 15 centers in the U.S. The purpose of this clinical trial is to study whether 8 mg of oral Trimesta taken daily over a two year period will reduce the rate of relapses in a large population of female patients with relapsing-remitting MS. Investigators are administering either Trimesta or matching placebo, in addition to a standard of care, glatiramer acetate (Copaxone®) injections, an FDA-approved therapy for MS, to women between the ages of 18 to 50 who have been recently diagnosed with relapsing-remitting MS. Relapse rates at two years is the primary endpoint in this clinical trial being run under an investigator-initiated IND. As of January 23, 2012, 164 patients have been enrolled in the clinical trial and the trial enrollment has been closed. The patients will be dosed and monitored for two years.
With over $8 million in grant funding to date, the ongoing Trimesta clinical trial should be funded to its completion.
| 18 |
Cognitive Dysfunction in MS
According to the NMSS and the Multiple Sclerosis Society of Canada publication,Hold that Thought! Cognition and MS , it is fairly common for people with MS to complain of problems remembering things, finding the right words, concentrating on a task or something they are reading, or following a conversation. These are all cognitive symptoms of MS. Of those affected by MS, 50-65% have cognitive dysfunction issues. Despite the fact that most symptoms are mild to moderate, they can have a significant impact on a person’s ability to normally function. The overall cognitive dysfunction can be described as a reduction in mental “sharpness.”
The major areas of cognition that can be dysfunctional include what are termed complex attention and executive functions. Complex attention involves multitasking, the speed with which information can be processed, learning and memory, and perceptual skills; executive functions include problem solving, organizational skills, the ability to plan, and word finding. Just as the nature, frequency, and severity of MS-related physical problems can widely vary, not all people with MS will display these cognitive issues, and no two people will experience exactly the same types or severity of problems.
Cognitive Dysfunction in MS: Background
In the investigator-initiated, 10-patient, 22-month, single-agent, crossover clinical trial conducted by Dr. Rhonda Voskuhl, a statistically significant 14% improvement from baseline in Paced Auditory Serial Addition Test (PASAT) cognitive testing scores (p = 0.04) was observed in relapsing-remitting MS patients after six months of Trimesta therapy. PASAT is a routine cognitive test performed in patients with a wide variety of neuropsychological disorders such as MS. The PASAT scores are expressed as a mean percent change from baseline.
Cognitive Dysfunction in MS: Clinical Development
Our Trimesta (oral estriol) drug candidate is also being developed for the treatment of cognitive dysfunction in female MS patients. This randomized, double-blind, placebo-controlled Phase II clinical trial to evaluate Trimesta’s potential neuroprotective and therapeutic effect on cognitive dysfunction in female MS patients is currently enrolling relapsing-remitting or secondary-progressive female MS patients at UCLA. Up to 64 patients between the ages of 18 and 50 will be randomized 1:1 into the treatment and placebo groups. Dr. Voskuhl will administer either oral Trimesta or a matching placebo, in addition to any FDA-approved MS treatment. Each patient will be dosed and monitored for one year after being enrolled. The primary endpoint in this clinical trial being run under an investigator-initiated IND application is expected to be improvement in PASAT cognitive testing scores versus matching placebo. We and a private foundation have pledged to equally support this new clinical trial, and we will also provide Trimesta drug supply. The trial also received contributions from several other supporters. Patient recruitment and enrollment into this trial is ongoing.
Other Programs
ALS
ALS, also known as Lou Gehrig’s disease, is a devastating progressive neurodegenerative disease that affects the motor nerve cells in the brain and the spinal cords. It is estimated that as many as 30,000 Americans may have the disease at any given time. The progressive degeneration of the motor neurons in ALS eventually leads to the death of the patient. Motor neurons reach from the brain to the spinal cord and from the spinal cord to the muscles throughout the body. When motor neurons die, the ability of the brain to initiate and control muscle movement is lost. With voluntary muscle action progressively affected, patients in the later stages of the disease may become totally paralyzed. While non-invasive ventilation and gastrostomy tubes prolong life by 6-12 months, the average lifespan from time of symptom onset is 2-5 years. Currently, RILUTEK® is the only FDA-approved drug for ALS. RILUTEK is an N-methyl d-aspartate (NMDA) receptor antagonist and has been shown to prolong life in patients with ALS by 3 months. Presently, there is no cure for ALS.
ALS: Background
There are multiple lines of scientific research that suggest a potential benefit of zinc therapy for ALS patients, including:
| ● | The use of zinc therapy for ALS patients is supported in animal models of ALS. Approximately 2% of ALS diagnoses are associated with a mutation in the copper/zinc superoxide dismutase (SOD1) gene. In ALS mutant SOD1 animal models, zinc supplementation has been shown to delay death. |
| ● | Genetic mutations affecting the ability of a protein known as copper/zinc SOD1 to properly bind zinc are associated with the familial form of ALS. |
| ● | Zinc is an important modifier of glutamate toxicity, a neurotransmitter linked to cell death in ALS patients. |
ALS: Clinical Development
Preparations are underway to design a clinical development pathway for our proprietary drug candidate, AEN-100, a gastroretentive, sustained-release zinc-based tablet, under an investigator-initiated IND application. Manufacturing and stability studies of AEN-100 are ongoing. In July 2012, the investigator, PNA Center for Neurological Research (PNA), held a meeting with the Neurology Products Division of the FDA to discuss the intended clinical development plan; FDA recommendations from that meeting will be incorporated into the development pathway.
| 19 |
In November 2011, PNA reported top-line results from its pilot Phase I/II open label, three month safety study of oral high dose zinc therapy in ALS. The clinical study met its primary outcome as no safety issues related to zinc therapy were observed. In addition, an average decrease in the monthly rate of disease progression was observed in the ALS patients on zinc therapy, compared to published historical controls, as well as compared to the average monthly rate of disease progression of the subjects prior to enrollment in the study. AEN-100 is not the same zinc formulation utilized by PNA in its previously completed Phase I/II safety study of zinc for ALS, and PNA intends to conduct a Phase I study of AEN-100 in normal volunteers prior to initiating the intended Phase II clinical trial in ALS patients.
Fibromyalgia
Fibromyalgia is a chronic and debilitating condition characterized by widespread pain and stiffness throughout the body, often accompanied by severe fatigue, insomnia and mood symptoms. Fibromyalgia affects an estimated 3-6% of the population worldwide, including an estimated 10 million people in the U.S. There are presently three FDA products approved for this indication in the U.S. – Lyrica®, Cymbalta® and Savella®. Flupirtine is differentiated from these products in that it employs a unique mode of action.
Based on an estimated annual price of $1,200 per fibromyalgia patient, we estimate that the total market potential in the U.S. is $6 billion.
Fibromyalgia:Meda Corporate Partnership
On May 6, 2010, we entered into a sublicense agreement with Meda, a multi-billion dollar international pharmaceutical company, pursuant to which Meda assumed all future development costs and may commercialize flupirtine for fibromyalgia in the U.S. As consideration for such sublicense, we received an up-front payment of $2.5 million and are entitled to milestone payments of $5 million upon the FDA’s acceptance of the New Drug Application (NDA) for flupirtine for fibromyalgia and $10 million upon FDA approval of such NDA. Pursuant to the sublicense agreement, we will also receive a 7% royalty on net sales of flupirtine for fibromyalgia in the U.S., Canada and Japan, with such royalties being shared equally with our licensor, McLean Hospital, a Harvard teaching hospital.
Flupirtine is approved and marketed by Meda and its distributors in Europe and other countries for indications other than fibromyalgia and has been prescribed to millions of patients worldwide. We believe that such substantial human experience with flupirtine should greatly assist the FDA in its evaluation of the safety of flupirtine upon review of an NDA of flupirtine for fibromyalgia. According to Meda’s 2011 Annual Report filed in May 2012, flupirtine for fibromyalgia is in Phase II development.
Fibromyalgia:Clinical Development
Our Effirma (flupirtine) product candidate for the treatment of fibromyalgia, has been partnered to Meda (see “Fibromyalgia: Meda Corporate Partnership” section above). Effirma is a selective neuronal potassium channel opener that also has NMDA receptor antagonist properties. Effirma is a non-opioid, non-NSAID, non-steroidal, analgesic. Preclinical data and clinical experience suggest that Effirma should also be effective for neuropathic pain since it acts in the central nervous system via a mechanism of action distinguishable from most marketed analgesics. Effirma is especially attractive because it operates through non-opiate pain pathways, exhibits no known abuse potential, and lacks withdrawal effects. In addition, no tolerance to its antinocioceptive effects has been observed. One common link between neuroprotection, nocioception and Effirma may be the N-methyl-D-aspartic acid glutamate system, a major receptor subtype for the excitotoxic neurotransmitter, glutamate. Effirma has strong inhibitory actions on N-methyl-D-aspartic acid-mediated neurotransmission. Flupirtine was originally developed by Asta Medica (subsequently acquired by Meda) and has been approved and is marketed by Meda in Europe since 1984, as well as other countries, for the treatment of pain. It has never been approved by the FDA for any indication.
Critical Accounting Policies
The consolidated financial statements are prepared in conformity with U.S. GAAP, which require the use of estimates, judgments and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses in the periods presented. We believe that the accounting estimates employed are appropriate and resulting balances are reasonable; however, due to inherent uncertainties in making estimates actual results could differ from the original estimates, requiring adjustments to these balances in future periods. The critical accounting estimates that affect the consolidated financial statements and the judgments and assumptions used are consistent with those described in the MD&A section in our 2011 Form 10-K/A.
Results of Operations
Three Months Ended September 30, 2012 and 2011
General and Administrative Expenses
| 20 |
General and administrative expenses increased to $1.1 million for the three months ended September 30, 2012, from $582,000 for the three months ended September 30, 2011. This increase of approximately 84% is primarily the result of additional employee costs, expanded investor relation activities and legal fees. The charge relating to stock-based compensation expense was $279,000 for the three months ended September 30, 2012, compared to $51,000 for the three months ended September 30, 2011.
Research and Development Expenses
Research and development expenses increased to $763,000 for the three months ended September 30, 2012, from $289,000 for the three months ended September 30, 2011. This increase of approximately 164% is primarily the result of additional employee costs and increased program costs associated with our expanded pipeline. Research and development expenses also include a charge relating to stock-based compensation expense of $116,000 for the three months ended September 30, 2011, compared to $6,000 for the three months ended September 30, 2011.
Other Income (Expense), Net
Other income was $10,000 for the three months ended September 30, 2012 and $6,000 for the three months ended September 30, 2011.
Loss from Continuing Operations
Our loss from continuing operations was $1.8 million, or $0.05 per common share for the three months ended September 30, 2012, compared to a net loss of $1.0 million, or $0.04 per common share for the three months ended September 30, 2011.
Loss from Discontinued Operations
Our loss from discontinued operations was $104,000, or $0.00 per common share for the three months ended September 30, 2012, compared to a net loss of $68,000, or $0.00 per common share for the three months ended September 30, 2011. On March 8, 2012, we entered into a Membership Interest Purchase Agreement, and certain related agreements, pursuant to which we sold all of our interest in the Lab to Hartlab, LLC. This resulted in the classification of the Lab as discontinued operations.See Note 3 – Discontinued Operations of Adeona Clinical Laboratory and Note Receivable for summarized statement of operations data for the three months ended September 30, 2012 and 2011.
Nine Months Ended September 30, 2012 and 2011
General and Administrative Expenses
General and administrative expenses increased to $3.7 million for the nine months ended September 30, 2012, from $2.3 million for the nine months ended September 30, 2011. This increase of 59% is primarily the result of additional employee costs, expanded investor relation activities and outside legal fees related to various Securities and Exchange Commission issues related to Berman & Company P.A.’s failure to follow proper partner rotation procedures. The charge relating to share-based compensation expense was $1.1 million for the nine months ended September 30, 2012, compared to $861,000 for the nine months ended September 30, 2011. The stock-based compensation for the nine months ended September 30, 2011 includes a charge of $398,000 relating to the modification of certain stock options, prior to expiration, held by a member of the Board of Directors.
Research and Development Expenses
Research and development expenses increased to $1.7 million for the nine months ended September 30, 2012, from $801,000 for the nine months ended September 30, 2011. This increase of 112% is primarily the result of additional employee costs and increased program costs associated with our expanded pipeline, including the initiation of preclinical and clinical programs. Research and development expenses also include a charge relating to share-based compensation expense of $238,000 for the nine months ended September 30, 2012, compared to $20,000 for the nine months ended September 30, 2011.
Other Income (Expense), Net
Other income was $22,000 for the nine months ended September 30, 2012, compared to other expense of $1.7 million for the nine months ended September 30, 2011. Other expense for the nine months ended September 30, 2011, includes $1.7 million relating to the estimated fair value of the warrants associated with the January 2011 and April 2011 financings, adjusted for the change in their fair value at September 30, 2011.
Loss from Continuing Operations
Our loss from continuing operations was $5.4 million, or $0.16 per common share for the nine months ended September 30, 2012, compared to a net loss of $4.8 million, or $0.18 per common share for the nine months ended September 30, 2011.
| 21 |
Income (Loss) from Discontinued Operations
Our income from discontinued operations was $389,000, or $0.01 per common share for the nine months ended September 30, 2012, compared to a net loss of $145,000, or $0.00 per common share for the nine months ended September 30, 2011. On March 8, 2012, we entered into a Membership Interest Purchase Agreement, and certain related agreements, pursuant to which we sold all of our interest in the Lab to Hartlab, LLC. This resulted in the classification of the Lab as discontinued operations.See Note 3 – Discontinued Operations of Adeona Clinical Laboratory and Note Receivable for summarized statement of operations data for the nine months ended September 30, 2012 and 2011.
Liquidity and Capital Resources
We have financed our operations since inception primarily through proceeds from equity financings and various private financings, primarily involving private sales of our common stock and other equity securities, corporate partnering license fees and from the proceeds from the sale of our common stock under registration statements on Forms S-1 and S-3, a private placement financing, laboratory testing revenues and miscellaneous equipment sales.
Our cash totaled $4.6 million as of September 30, 2012, a decrease of $2.1 million from December 31, 2011. During the nine months ended September 30, 2012, the primary sources of cash were proceeds from the stock option exercises of $94,000 and warrant exercises of $2.0 million. The primary use of cash during the nine months ended September 30, 2012 was for working capital requirements.
On October 31, 2012, we completed a private placement financing of 6,750,000 shares of our common stock at a price per share of $1.60 for aggregate gross proceeds of $10.8 million and net proceeds of $10.1 million. Our cash at October 31, 2012 was approximately $14.2 million.
Our continued operations will primarily depend on our ability to raise additional capital from various sources including equity and debt financings, as well as, license fees from potential corporate partners and joint ventures. Such additional funds may not become available on acceptable terms and there can be no assurance that any additional funding that we do obtain will be sufficient to meet our needs in the long term. We will continue to fund operations from cash on hand and through the similar sources of capital previously described. We can give no assurances that any additional capital that we are able to obtain will be sufficient to meet our needs.
Current and Future Financing Needs
We have incurred an accumulated deficit of approximately $56.9 million as of September 30, 2012. With the exception of the three months ended June 30, 2010, we have incurred negative cash flow from operations since we started our business. We have spent, and expect to continue to spend, substantial amounts in connection with implementing our business strategy, including our planned product development efforts, our clinical trials, and our research and discovery efforts.
Based on our current plans, we believe that our cash, which includes aggregate gross proceeds of $10.8 million from our recent financing, will be sufficient to enable us to meet our planned operating needs for at least the next 12 months.
However, the actual amount of funds we will need to operate is subject to many factors, some of which are beyond our control. These factors include the following:
| · | the progress of our research activities; | |
| · | the number and scope of our research programs; | |
| · | the progress of our preclinical and clinical development activities; | |
| · | the progress of the development efforts of parties with whom we have entered into research and development agreements; | |
| · | costs associated with additional clinical trials of our product candidates; | |
| · | our ability to maintain current research and development licensing arrangements and to establish new research and development and licensing arrangements; | |
| · | our ability to achieve our milestones under licensing arrangements | |
| · | the costs involved in prosecuting and enforcing patent claims and other intellectual property rights; and | |
| · | the costs and timing of regulatory approvals. |
We have based our estimate on assumptions that may prove to be wrong. We may need to obtain additional funds sooner or in greater amounts than we currently anticipate. Potential sources of financing include strategic relationships, public or private sales of our shares or debt and other sources. We may seek to access the public or private equity markets when conditions are favorable due to our long-term capital requirements. We do not have any committed sources of financing at this time, and it is uncertain whether additional funding will be available when we need it on terms that will be acceptable to us, or at all. If we raise funds by selling additional shares of common stock or other securities convertible into common stock, the ownership interest of our existing stockholders will be diluted. Our recent loss of S-3 eligibility due to the failure of Berman & Company, P.A. to follow proper partner rotation procedures may also negatively affect our ability to raise capital. If we are not able to obtain financing when needed, we may be unable to carry out our business plan. As a result, we may have to significantly limit our operations and our business, financial condition and results of operations would be materially harmed.
| 22 |
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Synthetic Biologics, Inc. is a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and is not required to provide the information required under this item.
ITEM 4. CONTROLS AND PROCEDURES
(a) Evaluation of disclosure controls and procedures
Pursuant to Rule 13a-15(b) under the Securities Exchange Act of 1934 (“Exchange Act”), the Company carried out an evaluation, with the participation of the Company’s management, including the Company’s Chief Executive Officer (“CEO”) and Chief Financial Officer (“CFO”), of the effectiveness of the Company’s disclosure controls and procedures (as defined under Rule 13a-15(e) under the Exchange Act) as of the end of the period covered by this report. Based upon that evaluation, the Company’s CEO and CFO concluded that the Company’s disclosure controls and procedures are effective as of September 30, 2012 to ensure that information required to be disclosed by the Company in the reports that the Company files or submits under the Exchange Act, is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to the Company’s management, including the Company’s CEO and CFO, as appropriate, to allow timely decisions regarding required disclosure.
(b) Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act) that occurred during our fiscal quarter ended September 30, 2012, that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
| 23 |
PART II—OTHER INFORMATION
ITEM 1. LEGAL PROCEEDINGS
None.
ITEM 1A. RISK FACTORS
The following information updates, and should be read in conjunction with, the information disclosed in Part 1, Item 1A, “Risk Factors,” of our Annual Report on Form 10-K/A for the fiscal year ended December 31, 2011, which was filed with the Securities and Exchange Commission on May 11, 2012.
RISKS RELATING TO OUR BUSINESS
We will need to raise additional capital to operate our business.
With the exception of the three months ended June 30, 2010, we have experienced significant losses since inception and have a significant accumulated deficit. We expect to incur additional operating losses in the future and therefore our cumulative losses to increase. To date, other than the licensing fee we received from Meda AB for the development and commercialization of Effirma (flupirtine) for fibromyalgia in the U.S., Canada and Japan and limited laboratory revenues from Adeona Clinical Laboratory, which we have recently sold, we have generated very minimal revenues. Inasmuch as our sole source of revenue (with the exception of the Meda licensing fee) has been our laboratory revenue and our laboratory was sold recently, we do not expect to derive revenue from any source in the near future until we or our partners successfully commercialize our products. As of September 30, 2012, our accumulated deficit totaled approximately $56.9 million on a consolidated basis. Until such time as we receive approval from the FDA and other regulatory authorities for our product candidates, we will not be permitted to sell our products and therefore will not have product revenues from the sale of products. For the foreseeable future we will have to fund all of our operations and capital expenditures from equity and debt offerings, cash on hand, licensing fees and grants. If our current cash, cash equivalents and short-term investments are not sufficient to sustain our operations, we will need to seek additional sources of financing and such additional financing may not be available on favorable terms, if at all. Our recent loss of S-3 eligibility due to the failure of Berman & Company, P.A. to follow proper partner rotation procedures may also negatively affect our ability to raise capital. If we do not succeed in raising additional funds on acceptable terms, we may be unable to complete planned preclinical and clinical trials or obtain approval of our product candidates from the FDA and other regulatory authorities. In addition, we could be forced to delay, discontinue or curtail product development, forego sales and marketing efforts, and forego licensing in attractive business opportunities. Any additional sources of financing will likely involve the issuance of our equity or debt securities, which will have a dilutive effect on our stockholders.
We have not been able to sustain profitability.
Other than with respect to the three months ended June 30, 2010, we have a history of losses and we have incurred and continue to incur substantial losses and negative operating cash flow. Even if we succeed in developing and commercializing one or more of our product candidates, we may still incur substantial losses for the foreseeable future and may not sustain profitability. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will substantially increase in the foreseeable future as we do the following:
| ● | continue to undertake preclinical development and clinical trials for our product candidates; | |
| ● | expand our research activities with Intrexon relating to monoclonal antibodies for infectious diseases; | |
| ● | seek regulatory approvals for our product candidates; | |
| ● | develop our product candidates for commercialization; | |
| ● | implement additional internal systems and infrastructure; | |
| ● | lease additional or alternative office facilities; and | |
| ● | hire additional personnel, including members of our management team. |
We may experience negative cash flow for the foreseeable future as we fund our technology development with capital expenditures. As a result, we will need to generate significant revenues in order to achieve and maintain profitability. We may not be able to generate these revenues or achieve profitability in the future. Our failure to achieve or maintain profitability could negatively impact the value of our common stock and underlying securities.
Our research and development efforts may not succeed in developing commercially successful products and technologies, which may limit our ability to achieve profitability.
We must continue to explore opportunities that may lead to new products and technologies. To accomplish this, we must commit substantial efforts, funds, and other resources to research and development. A high rate of failure is inherent in the research and development of new products and technologies. Any such expenditures that we make will be made without any assurance that our efforts will be successful. Failure can occur at any point in the process, including after significant funds have been invested.
| 24 |
Regardless of whether our clinical trials are deemed to be successful, promising new product candidates may fail to reach the market or may only have limited commercial success because of efficacy or safety concerns, failure to achieve positive clinical outcomes, inability to obtain necessary regulatory approvals or satisfy regulatory criteria, limited scope of approved uses, excessive costs to manufacture, the failure to establish or maintain intellectual property rights, or infringement of the intellectual property rights of others. Even if we successfully develop new products or enhancements, they may be quickly rendered obsolete by changing customer preferences, changing industry standards, or competitors' innovations. Innovations may not be quickly accepted in the marketplace because of, among other things, entrenched patterns of clinical practice or uncertainty over third-party reimbursement. We cannot state with certainty when or whether any of our products under development will be launched, whether we will be able to develop, license, or otherwise acquire drug candidates or products, or whether any products will be commercially successful. Failure to launch successful new products or new indications for existing products may cause our products to become obsolete, which may limit our ability to achieve profitability.
The technology on which our channel partnering arrangements with Intrexon is based on early stage technology.
We have an exclusive channel collaboration arrangement with Intrexon that contemplates the use of Intrexon’s transgene engineering platform technology and regulatory control technology for thein vivo cellular production of PGIS, a specific effector enzyme that regulates the production of prostacyclin. Such technologies have a limited history of use in the design and development of human therapeutic product candidates and may therefore involve unanticipated risks or delays.
On August 8, 2012, we announced an additional exclusive channel collaboration with Intrexon relating to the design, production, testing and commercialization of monoclonal antibodies for the treatment of certain infectious diseases. Although monoclonal antibody therapeutics are well established in the biotechnology and pharmaceutical sectors, their use for the treatment of infectious disease is extremely limited. In order for monoclonal antibodies to be effective for infectious diseases, they must not only properly target the organism of interest (or its toxins), but may also need to overcome defenses and forms of resistance of such organisms. To accomplish this may require the use of more than one specific monoclonal antibody, and mixtures of different monoclonal antibodies, which may create additional unforeseen complications, including increased manufacturing complexity and expense. In order to be competitive, monoclonal antibodies will be required to be produced at a low enough cost of goods in order to be profitably marketed. We have very limited development and manufacturing experience in the field of monoclonal antibodies and infectious disease. We cannot assure that any monoclonal antibody candidates will provide satisfactory in vitro and in vivo nonclinical results sufficient to warrant the expense of cGMP manufacture and clinical testing in human clinical trials.
DNA-based therapy has not yet been proven to be successful.
The FDA has not yet approved any human DNA-based therapy product for sale. The field of DNA-based therapy, also referred to as gene therapy or gene transfer, is experimental and has not yet proven successful in many clinical trials. Clinical trials with DNA-based therapy have encountered a multitude of significant technical problems in the past, including, unintended integration with host DNA, poor levels of protein expression, transient protein expression, viral overload, immune reactions to either viral capsids utilized to deliver DNA, DNA itself, proteins expressed or cells transfected with DNA. There can be no assurance that our preclinical animals studies or human clinical trials will be successful or that we will receive the regulatory approvals necessary to initiate such studies. To the extent that we utilize viral constructs or other systems to deliver our DNA-based therapies and the same or similar delivery systems demonstrate unanticipated and/or unacceptable side effects in preclinical or clinical trials conducted by ourselves or others we may be forced to, or elect to, discontinue development of such product candidates.
We may not generate additional revenue from our relationships with our corporate collaborators.
On May 6, 2010, we entered into a sublicense agreement with Meda AB whereby we may receive milestone payments totaling $17.5 million (including an upfront payment of $2.5 million that has already been received), plus royalties on our flupirtine program. There can be no assurance that Meda AB will successfully develop flupirtine for fibromyalgia in the U.S., Canada or Japan that would allow us to receive such additional $15 million in milestone payments and royalties on sales in connection with such agreement. The successful achievement of the various milestones set forth in the sublicense agreement is not within our control and we will be dependent upon Meda AB for achievement of such milestones. According to Meda’s 2011 Annual Report filed in May 2012, flupirtine for fibromyalgia is in Phase II development.
We have experienced several management changes.
We have had significant changes in management in the past few years. Jeffrey Riley was appointed Chief Executive Officer and President on February 3, 2012. Effective February 6, 2012, C. Evan Ballantyne was appointed Chief Financial Officer. James S. Kuo, M.D., served as Chief Executive Officer and President from February 6, 2010 until February 3, 2012. Changes in our key positions, as well as additions of new personnel and departures of existing personnel, can be disruptive, might lead to additional departures of existing personnel and could have a material adverse effect on our business, operating results, financial results and internal controls over financial reporting.
| 25 |
We may not be able to retain rights licensed to us by others to commercialize key products and may not be able to establish or maintain the relationships we need to develop, manufacture, and market our products.
In addition to our own patent applications, we also currently rely on licensing agreements with third party patent holders/licensors for our products. We have an exclusive license agreement with the McLean Hospital relating to the use of flupirtine to treat fibromyalgia which was sublicensed to Meda AB and an exclusive license agreement with the Regents of the University of California relating to our Trimesta technology. Each of these agreements requires us or our sublicensee to use our best efforts to commercialize each of the technologies as well as meet certain diligence requirements and timelines in order to keep the license agreement in effect. In the event we or our sublicensee are not able to meet our diligence requirements, we may not be able to retain the rights granted under our agreements or renegotiate our arrangement with these institutions on reasonable terms, or at all. Furthermore, we currently have very limited product development capabilities, and limited marketing or sales capabilities. For us to research, develop, and test our product candidates, we would need to contract with outside researchers, in most cases those parties that did the original research and from whom we have licensed the technologies. Our exclusive channel collaboration agreements with Intrexon provide that Intrexon may terminate such agreement if we do not perform certain specified requirements, including developing therapies considered superior.
We can give no assurances that any of our issued patents licensed to us or any of our other patent applications will provide us with significant proprietary protection or be of commercial benefit to us. Furthermore, the issuance of a patent is not conclusive as to its validity or enforceability, nor does the issuance of a patent provide the patent holder with freedom to operate without infringing the patent rights of others.
We will incur additional expenses in connection with our exclusive channel collaboration arrangements with Intrexon.
Pursuant to our exclusive channel collaborations with Intrexon, we are responsible for future research and development expenses of product candidates developed under each such collaboration, the effect of which has and will continue to increase the level of our overall research and development expenses going forward. Although all manufacturing, preclinical studies and human clinical trials are expensive and difficult to design and implement, costs associated with the manufacturing, research and development of biologic product candidates are generally greater in comparison to small molecule product candidates. We have added additional personnel and expect to add additional personnel to support our exclusive channel collaborations with Intrexon.
Because our collaborations with Intrexon are relatively new, we have only recently assumed development responsibility and costs associated with such programs. In addition, because development activities are determined pursuant to a joint steering committees comprised of Intrexon and ourselves and we have limited experience, future development costs associated this program may be difficult to anticipate and exceed our expectations. Our actual cash requirements may vary materially from our current expectations for a number of other factors that may include, but are not limited to, unanticipated technical challenges, changes in the focus and direction of our development activities or adjustments necessitated by changes in the competitive landscape in which we operate. If we are unable to continue to financially support such collaborations due to our own working capital constraints, we may be forced to delay our activities. If we are unable to obtain additional financing on terms acceptable to us or at all, we may be forced to seek licensing partners or discontinue development.
Developments by competitors may render our products or technologies obsolete or non-competitive.
Companies that currently sell or are developing both generic and proprietary products to treat serious diseases include: Actelion Pharmaceuticals, Bayer Health Care, Biogen Idec, Eli Lilly & Co., Genzyme, GlaxoSmithKline Pharmaceuticals, Merck & Co., Pfizer, Novartis, Teva Pharmaceuticals and United Therapeutics. Companies that currently sell or are developing both generic and proprietary products to treat infectious diseases include: MedImmune, Pfizer, Cubist, Optimer Pharmaceuticals, Symphogen, Merus, GlaxoSmithKline Pharmaceuticals, Merck & Co. and Novartis. Many of our competitors have significant financial and human resources. The pulmonary arterial hypertension market is highly competitive and several different product classes currently compete in this space, including prostacyclin-based therapies, endothelin receptor antagonists and phosphodiesterase type 5 inhibitors. Prostacyclin-based therapies for PAH are available in a number of delivery formats, including intravenous, subcutaneous and inhaled routes and an oral prostacyclin-based product candidate is currently under NDA review in the U.S. The infectious disease market is highly competitive with many generic and proprietary intravenous and oral formulations available to physicians and their patients. As monoclonal antibodies, we currently do not expect to be able to deliver our infectious disease candidates via the oral route and may thus be limited to the in-patient and/or acute treatment setting. In addition, academic research centers may develop technologies that compete with our Trimesta, sustained-release zinc preparation - AEN-100, and flupirtine technologies. Should clinicians or regulatory authorities view these therapeutic regiments as more effective than our products, this might delay or prevent us from obtaining regulatory approval for our products, or it might prevent us from obtaining favorable reimbursement rates from payers, such as Medicare, Medicaid and private insurers.
We operate in a highly competitive environment.
The pharmaceutical and biotechnology industries, including the monoclonal antibody industry, are characterized by rapidly evolving technology and intense competition. Our competitors include major multi-national pharmaceutical companies and biotechnology companies developing both generic and proprietary therapies to treat serious diseases. Many of these companies are well-established and possess technical, human, research and development, financial, and sales and marketing resources significantly greater than ours. In addition, many of our potential competitors have formed strategic collaborations, partnerships and other types of joint ventures with larger, well established industry competitors that afford these companies potential research and development and commercialization advantages in the therapeutic areas we are currently pursuing.
| 26 |
Academic research centers, governmental agencies and other public and private research organizations are also conducting and financing research activities which may produce products directly competitive to those being developed by us. In addition, many of these competitors may be able to obtain patent protection, obtain FDA and other regulatory approvals and begin commercial sales of their products before us.
Competitors could develop and/or gain FDA approval of our products for a different indication.
Since we do not have composition of matter patent claims for flupirtine, estriol or zinc acetate, others may obtain approvals for other uses of these products that are not covered by our issued or pending patents. For example, the active ingredients in both Effirma (flurpirtine) and Trimesta (oral estriol) have been approved for marketing in overseas countries for different uses and an oral immediate release form of zinc is approved in the U.S. and Europe for the treatment of Wilson’s disease. Other companies, including the original developers or licensees or affiliates may seek to develop Effirma or Trimesta or their respective active ingredient(s) for other uses in the U.S. or any country we are seeking approval for. We cannot provide any assurances that any other company may obtain FDA approval for products that contain flupirtine, estriol or zinc in various formulations or delivery systems that might adversely affect our ability or the ability of Meda to develop and market these products in the U.S. We are aware that other companies have intellectual property protection using the active ingredients and have conducted clinical trials of flupirtine, estriol and zinc for different applications than what we are developing. Many of these companies may have more resources than us. We cannot provide any assurances that our products will be FDA-approved prior to our competitors.
If a product containing our active ingredients is already marketed or if the FDA approves other products containing our active ingredients in the future to treat indications, physicians may elect to prescribe and substitute a competitor’s products to treat the diseases for which we are intending to commercialize; this is commonly referred to as “off-label” use. While under FDA regulations a competitor is not allowed to promote off-label uses of its product, the FDA does not regulate the practice of medicine and, as a result, cannot direct physicians to select certain products for their patients. Consequently, we might be limited in our ability to prevent off-label use of a competitor’s product to treat the diseases we are intending to commercialize, even if we have issued method of use patents for that indication. If we are not able to obtain and enforce our patents, if any, or otherwise receive orphan drug protection in the case of ALS, a competitor could develop and commercialize similar products for the same indications that we are pursuing. We cannot provide any assurances that a competitor will not obtain FDA approval for a product that contains the same active ingredients as our products.
We rely on method patents and patent applications and various regulatory exclusivities to protect some of our product candidates and our ability to compete may be limited or eliminated if we are not able to protect our products.
Our competitiveness may be adversely affected if we are unable to protect our proprietary technologies. We do not have composition of matter patents for Trimesta or Effirma, or their respective active ingredients estriol and flupirtine. We rely on issued patent and pending patent applications for use of Trimesta to treat MS (issued U.S. Patent No. 6,936,599) and various other therapeutic indications, which have been exclusively licensed to us. We have exclusively licensed an issued patent for the treatment of fibromyalgia with flupirtine, which we have sublicensed to Meda AB.
Our AEN-100 drug candidate (gastroretentive zinc acetate) is the subject of U.S. and international pending patent applications, such as published U.S. patent application Ser. No. 11/621,962 and corresponding international applications that claim priority to January 10, 2006 as well as additional patent applications. On October 26, 2011, we received a final rejection letter with regard to U.S. patent application Ser. No. 11/621,962. On February 15, 2012, we filed a Request for Continued Examination.
The patent positions of pharmaceutical companies are uncertain and may involve complex legal and factual questions. We may incur significant expense in protecting our intellectual property and defending or assessing claims with respect to intellectual property owned by others. Any patent or other infringement litigation by or against us could cause us to incur significant expense and divert the attention of our management.
Others may file patent applications or obtain patents on similar technologies or compounds that compete with our products. We cannot predict how broad the claims in any such patents or applications will be, and whether they will be allowed. Once claims have been issued, we cannot predict how they will be construed or enforced. We may infringe intellectual property rights of others without being aware of it. If another party claims we are infringing their technology, we could have to defend an expensive and time consuming lawsuit, pay a large sum if we are found to be infringing, or be prohibited from selling or licensing our products unless we obtain a license or redesign our product, which may not be possible.
We also rely on trade secrets and proprietary know-how to develop and maintain our competitive position. Some of our current or former employees, consultants, scientific advisors, current or prospective corporate collaborators, may unintentionally or willfully disclose our confidential information to competitors or use our proprietary technology for their own benefit. Furthermore, enforcing a claim alleging the infringement of our trade secrets would be expensive and difficult to prove, making the outcome uncertain. Our competitors may also independently develop similar knowledge, methods, and know-how or gain access to our proprietary information through some other means.
We may fail to retain or recruit necessary personnel, and we may be unable to secure the services of consultants.
As of November 10, 2012, we had twelve employees. We have also engaged clinical consultants to advise us on our clinical programs and regulatory consultants to advise us on our dealings with the FDA and other foreign regulatory authorities. We have been and will be required to retain additional consultants and employees in order to fulfill our obligations under our exclusive channel collaborations with Intrexon. Our future performance will depend in part on our ability to successfully integrate newly hired officers into our management team and our ability to develop an effective working relationship among senior management.
| 27 |
Certain of our directors, scientific advisors, and consultants serve as officers, directors, scientific advisors, or consultants of other biopharmaceutical or biotechnology companies that might be developing competitive products to ours. Other than corporate opportunities, none of our directors are obligated under any agreement or understanding with us to make any additional products or technologies available to us. Similarly, we can give no assurances, and we do not expect and stockholders should not expect, that any biomedical or pharmaceutical product or technology identified by any of our directors or affiliates in the future would be made available to us other than corporate opportunities. We can give no assurances that any such other companies will not have interests that are in conflict with our interests.
Losing key personnel or failing to recruit necessary additional personnel would impede our ability to attain our development objectives. There is intense competition for qualified personnel in the drug-development field, and we may not be able to attract and retain the qualified personnel we would need to develop our business.
We rely on independent organizations, advisors, and consultants to perform certain services for us, including handling substantially all aspects of regulatory approval, clinical management, manufacturing, marketing, and sales. We expect that this will continue to be the case. Such services may not always be available to us on a timely basis when we need them.
If the parties we depend on for supplying our drug substance raw materials and certain manufacturing-related services do not timely supply these products and services, it may delay or impair our ability to develop, manufacture and market our products.
We rely on suppliers for our drug substance raw materials and third parties for certain manufacturing-related services to produce material that meets appropriate content, quality and stability standards and use in clinical trials of our products and, after approval, for commercial distribution. We have not yet established a cGMP manufacturer for neither our DNA-based nor monoclonal antibody therapies. Our AEN-100 product candidate has limited stability data to date and is the subject of ongoing stability studies. To succeed, clinical trials require adequate supplies of drug substance and drug product, which may be difficult or uneconomical to procure or manufacture. We and our suppliers and vendors may not be able to (i) produce our drug substance or drug product to appropriate standards for use in clinical studies, (ii) perform under any definitive manufacturing, supply or service agreements with us, or (iii) remain in business for a sufficient time to successfully produce and market our product candidates. If we do not maintain important manufacturing and service relationships, we may fail to find a replacement supplier or required vendor or develop our own manufacturing capabilities which could delay or impair our ability to obtain regulatory approval for our products and substantially increase our costs or deplete profit margins, if any. If we do find replacement manufacturers and vendors, we may not be able to enter into agreements with them on terms and conditions favorable to us and, there could be a substantial delay before a new facility could be qualified and registered with the FDA and foreign regulatory authorities.
If successful large-scale manufacturing of DNA-based products is not possible, we or our collaborators may be unable to manufacture enough of our product candidates to achieve regulatory approval or market our DNA-based products.
Few companies to date have demonstrated successful large-scale manufacturing of DNA-based products, including those that have had significantly more resources than us and it is anticipated that significant challenges will be faced in the scale-up of our manufacturing process for commercial production. There are a limited number of contract manufacturers qualified to perform large-scale manufacturing of DNA-based products. We or our collaborators may be unable to manufacture commercial-scale quantities of DNA-based products or receive appropriate government approvals on a timely basis or at all. Failure to successfully manufacture or obtain appropriate government approvals on a timely basis or at all would prevent us from achieving our business objectives.
Clinical trials are very expensive, time-consuming, and difficult to design and implement.
Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time-consuming. We estimate that clinical trials of our product candidates would take at least several years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. Commencement and completion of clinical trials may be delayed by several factors, including:
| ● | obtaining an IND application with the FDA to commence clinical trials; | |
| ● | identification of, and acceptable arrangements with, one or more clinical sites; | |
| ● | obtaining IRB approval to commence clinical trials; | |
| ● | unforeseen safety issues; | |
| ● | determination of dosing; | |
| ● | lack of effectiveness during clinical trials; | |
| ● | slower than expected rates of patient recruitment; | |
| ● | inability to monitor patients adequately during or after treatment; | |
| ● | inability or unwillingness of medical investigators to follow our clinical protocols; and | |
| ● | unwillingness of the FDA or IRBs to permit the clinical trials to be initiated. |
| 28 |
In addition, we, IRBs or the FDA may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if IRBs or the FDA finds deficiencies in our submissions or conduct of our trials.
The results of our clinical trials may not support our product candidate claims and the results of preclinical studies and completed clinical trials are not necessarily predictive of future results.
To date, long-term safety and efficacy have not yet been demonstrated in clinical trials for any of our diagnostic product candidates. Favorable results in our early studies or trials may not be repeated in later studies or trials. Even if our clinical trials are initiated and completed as planned, we cannot be certain that the results will support our product candidate claims. Success in preclinical testing and Phase II clinical trials does not ensure that later Phase II or Phase III clinical trials will be successful. We cannot be sure that the results of later clinical trials would replicate the results of prior clinical trials and preclinical testing. In particular, the limited results that we have obtained for our diagnostic tests may not predict results from studies in larger numbers of subjects drawn from more diverse populations over a longer period of time. Clinical trials may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses. Any such failure could cause us or our sublicensee to abandon a product candidate and might delay development of other product candidates. Preclinical and clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization. Any delay in, or termination of, our clinical trials would delay our obtaining FDA approval for the affected product candidate and, ultimately, our ability to commercialize that product candidate.
We depend on third parties, including researchers and sublicensees, who are not under our control.
Since we have in-licensed some of our product candidates, have sublicensed a product candidate and have collaboration agreements for the development of other product candidates, we depend upon our sublicensee and independent investigators and scientific collaborators, such as universities and medical institutions or private physician scientists, to conduct our preclinical and clinical trials under agreements with us. These collaborators are not our employees and we cannot control the amount or timing of resources that they devote to our programs or the timing of their procurement of clinical-trial data or their compliance with applicable regulatory guidelines. Should any of these scientific inventors/advisors or those of our sublicensee become disabled or die unexpectedly, or should they fail to comply with applicable regulatory guidelines, we or our sublicensee may be forced to scale back or terminate development of that program. They may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking those programs ourselves. Failing to devote sufficient time and resources to our drug-development programs, or substandard performance and failure to comply with regulatory guidelines, could result in delay of any FDA applications and our commercialization of the drug candidate involved.
These collaborators may also have relationships with other commercial entities, some of which may compete with us. Our collaborators assisting our competitors at our expense could harm our competitive position. For example, we are highly dependent on scientific collaborators for our Trimesta development program. Specifically, all of the clinical trials have been conducted under physician-sponsored IND applications, not corporate-sponsored INDs. Generally, we have experienced difficulty in collecting data generated from these physician-sponsored clinical trials for our programs. We cannot provide any assurances that we will not experience any additional delays in the future.
We are also highly dependent on government and private grants to fund certain of our clinical trials for our product candidates. For example, Trimesta (oral estriol) has received grants totaling over $8 million, predominantly from the Southern California Chapter of the NMSS and the National Institutes of Health which funds a majority of the ongoing clinical trial in relapsing-remitting MS for women. Although we believe that the grant funding received to date is sufficient to complete the current clinical trial based upon current cost estimates, if we experience any additional unanticipated costs or require further clinical trials, and our scientific collaborator is unable to maintain or receive additional grants, we might be forced to scale back or terminate the development of this product candidate. We will also need to cross reference our IND with the inventor/IND holder for this program should we elect to file our own corporate IND for our Trimesta (oral estriol) program. The on-going and future development and commercialization of Effirma (flupirtine) for fibromyalgia is the responsibility of Meda AB and no assurance can be given that Meda will gain the FDA’s acceptance of the NDA or obtain NDA approval from the FDA of flupirtine for fibromyalgia.
Our AEN-100 program for ALS is reliant on the investigator-initiated IND of PNA. The planned clinical trial that we intend to conduct with PNA is still the subject of further protocol development. In addition, we may need to conduct additional clinical or non-clinical studies to support a New Drug Application or to support further clinical trials. Any additional studies of AEN-100 may produce unanticipated and unacceptable safety, tolerability or bioavailability results that may substantially delay further development work of the planned clinical trial.
With respect to our synthetic biologic product candidates, we are dependent upon Intrexon’s synthetic biology facilities and capabilities as we have no such facilities and capabilities of our own. We are also reliant on their vector engineering platform, gene expression switch technology, monoclonal antibody discovery, production cell line development and know-how. If any of the foregoing were to become inaccessible or terminated, it would be difficult for us to develop and commercialize our synthetic biologic product candidates.
| 29 |
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights, as well as costs associated with lawsuits.
If any other person files patent applications, or is issued patents, claiming technology also claimed by us in pending applications, we may be required to participate in interference proceedings in the U.S. Patent and Trademark Office to determine priority of invention. We, or our licensors, may also need to participate in interference proceedings involving our issued patents and pending applications of another entity.
The intellectual property environment in the area of DNA-based therapeutics is particularly complex, constantly evolving and highly fragmented. Other companies and institutions have issued patents and have filed or will file patent applications that may issue into patents that cover or attempt to cover genes, vectors, cell lines, and methods of making and using DNA and DNA-based therapy products used in, or similar to our product candidate, and technologies. The same is true of the monoclonal antibody field in terms of methods of producing monoclonal antibodies for human use. We have not conducted freedom-to-use patent searches on all aspects of our product candidates or potential product candidates, and we may be unaware of relevant patents and patent applications of third parties. In addition, the freedom-to-use patent searches that have been conducted may not have identified all relevant issued patents or pending patents. We cannot provide assurance that our proposed products in this area will not ultimately be held to infringe one or more valid claims owned by third parties which may exist or come to exist in the future or that in such case we will be able to obtain a license from such parties on acceptable terms.
We cannot guarantee that the practice of our technologies will not conflict with the rights of others. In some foreign jurisdictions, we could become involved in opposition proceedings, either by opposing the validity of another’s foreign patent or by persons opposing the validity of our foreign patents.
We may also face frivolous litigation or lawsuits from various competitors or from litigious securities attorneys. The cost to us of any litigation or other proceeding relating to these areas, even if deemed frivolous or resolved in our favor, could be substantial and could distract management from our business. Uncertainties resulting from initiation and continuation of any litigation could have a material adverse effect on our ability to continue our operations.
If we infringe the rights of others we could be prevented from selling products or forced to pay damages.
If our products, methods, processes, and other technologies are found to infringe the proprietary rights of other parties, we could be required to pay damages, or we may be required to cease using the technology or to license rights from the prevailing party. Any prevailing party may be unwilling to offer us a license on commercially acceptable terms.
RISKS RELATING TO OUR STOCK
We will seek to raise additional funds in the future, which may be dilutive to stockholders or impose operational restrictions.
We expect to seek to raise additional capital in the future to help fund development of our proposed products. If we raise additional capital through the issuance of equity or of debt securities, the percentage ownership of our current stockholders will be reduced. We may also enter into strategic transactions, issue equity as part of license issue fees to our licensors, compensate consultants or settle outstanding payables using equity that may be dilutive. Our stockholders may experience additional dilution in net book value per share and any additional equity securities may have rights, preferences and privileges senior to those of the holders of our common stock.
We are substantially controlled by our current officers, directors, and principal stockholders.
Currently, our directors, executive officers, and principal stockholders beneficially own a substantial number of shares of our common stock. As a result, they will be able to exert substantial influence over the election of our Board of Directors and the vote on issues submitted to our stockholders. Our executive officers and directors beneficially owned approximately 9.0 million shares of our common stock, including stock options and warrants exercisable within 60 days of October 31, 2012. Our executive officers, directors and principal stockholders together beneficially owned approximately 18.8 million shares of our common stock, including the stock options and warrants exercisable within 60 days of October 31, 2012. Because our common stock has from time to time been “thinly traded”, the sale of a substantial number of shares by our executive officers, directors and principal stockholders would have an adverse effect on the market for our stock and our share price.
Our shares of common stock are from time to time thinly traded, so stockholders may be unable to sell at or near ask prices or at all if they need to sell shares to raise money or otherwise desire to liquidate their shares.
Our common stock has from time to time been “thinly-traded,” meaning that the number of persons interested in purchasing our common stock at or near ask prices at any given time may be relatively small or non-existent. This situation is attributable to a number of factors, including the fact that we are a small company that is relatively unknown to stock analysts, stock brokers, institutional investors and others in the investment community that generate or influence sales volume, and that even if we came to the attention of such persons, they tend to be risk-averse and would be reluctant to follow an unproven company such as ours or purchase or recommend the purchase of our shares until such time as we became more seasoned and viable. As a consequence, there may be periods of several days or more when trading activity in our shares is minimal or non-existent, as compared to a seasoned issuer which has a large and steady volume of trading activity that will generally support continuous sales without an adverse effect on share price. We cannot give stockholders any assurance that a broader or more active public trading market for our common shares will develop or be sustained, or that current trading levels will be sustained.
| 30 |
We cannot assure you that the common stock will be liquid or that it will remain listed on the NYSE MKT.
We cannot assure you that we will be able to maintain the continued listing standards of the NYSE MKT (formerly the NYSE Amex and the American Stock Exchange). The NYSE MKT requires companies to meet certain continued listing criteria including certain minimum stockholders' equity and equity prices per share as outlined in the NYSE MKT Exchange Company Guide. We may not be able to maintain such minimum stockholders' equity or prices per share or may be required to effect a reverse stock split to maintain such minimum prices and/or issue additional equity securities in exchange for cash or other assets, if available, to maintain certain minimum stockholders' equity required by the NYSE MKT. If we are delisted from the NYSE MKT then our common stock will trade, if at all, only on the over-the-counter market, such as the OTC Bulletin Board securities market, and then only if one or more registered broker-dealer market makers comply with quotation requirements. In addition, delisting of our common stock could depress our stock price, substantially limit liquidity of our common stock and materially adversely affect our ability to raise capital on terms acceptable to us, or at all. Delisting from the NYSE MKT could also have other negative results, including the potential loss of confidence by suppliers and employees, the loss of institutional investor interest and fewer business development opportunities. In order to remain listed on NYSE MKT, we are required to maintain a minimum stockholders’ equity of $6 million. The net proceeds of $10.1 million from our recent financing increased our stockholders’ equity well above the minimum requirement of $6 million. However, we could be subject to certain additional regulatory requirements or additional inquiries from the NYSE MKT, because our stockholders’ equity at September 30, 2012 was $5.4 million.
There may be issuances of shares of preferred stock in the future.
Although we currently do not have preferred shares outstanding, the Board of Directors could authorize the issuance of a series of preferred stock that would grant holders preferred rights to our assets upon liquidation, the right to receive dividends before dividends would be declared to common stockholders, and the right to the redemption of such shares, possibly together with a premium, prior to the redemption of the common stock. To the extent that we do issue preferred stock, the rights of holders of common stock could be impaired thereby, including without limitation, with respect to liquidation.
Our failure to fulfill all of our registration requirements may cause us to suffer liquidated damages, which may be very costly.
The Registration Rights Agreement that we entered into in connection with the Offering requires that we file the Initial Registration Statement with the SEC on or prior to the Filing Date and that the Initial Registration Statement is declared effective by the SEC on or prior to the Effectiveness Date subject to certain adjustments. Upon the occurrence of certain events (each an “Event”), including, but not limited to, that the Initial Registration Statement is not filed prior to the Filing Date or declared effective prior to the Effectiveness Date, the Company will be required to pay to each of the Purchasers liquidated damages of 1.5% of their aggregate purchase price upon the date of the Event and then monthly thereafter until the Event is cured. In no event will the aggregate amount of liquidated damages payable to each of the Purchasers exceed in the aggregate 10% of the aggregate purchase price paid by such Purchaser. In addition, pursuant to the terms of the registration rights agreement that we entered into with Intrexon and an affiliated entity, we are required to file a registration statement with respect to securities issued to them within a certain time period and maintain the effectiveness of such registration statement. The failure to do so could result in the payment of damages by us. There can be no assurance as to when the Initial Registration Statement will be declared effective or that we will be able to maintain the effectiveness of any registration statement, and therefore there can be no assurance that we will not incur penalties or damages with respect to such agreements.
RISKS RELATED TO OUR INDUSTRY
We are subject to government regulation, compliance with which can be costly and difficult.
In the U.S., the formulation, manufacturing, packaging, storing, labeling, promotion, advertising, distribution and sale of our products are subject to regulation by various governmental agencies, including (1) the FDA, (2) the Federal Trade Commission, or FTC, (3) the Consumer Product Safety Commission, or CPSC, (4) the U.S. Department of Agriculture, or USDA. Our proposed activities may also be regulated by various agencies of the states, localities and foreign countries in which our proposed products may be manufactured, distributed and sold. The FDA, in particular, regulates the formulation, manufacture and labeling of over-the-counter, or OTC drugs, prescription drugs, conventional foods, dietary supplements, and cosmetics such as those that we intend to distribute. FDA regulations require us and our suppliers to meet relevant cGMP regulations for the preparation, packing, labeling, and storage of all drugs and foods.
Any products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing FDA regulation, including record-keeping requirements, reporting of adverse experiences, submitting periodic reports, drug sampling and distribution requirements, manufacturing or labeling changes, record-keeping requirements, and compliance with FDA promotion and advertising requirements. Drug manufacturers and their subcontractors are required to register their facilities with the FDA and state agencies, and are subject to periodic unannounced inspections for GMP compliance, imposing procedural and documentation requirements upon us and third-party manufacturers. Failure to comply with these regulations could result, among other things, in suspension of regulatory approval, recalls, suspension of production or injunctions, seizures, or civil or criminal sanctions. We cannot be certain that we or our present or future subcontractors will be able to comply with these regulations.
The FDA regulates prescription drug labeling and promotion activities. The FDA actively enforces regulations prohibiting the marketing of products for unapproved uses. The FDA permits the promotion of drugs for unapproved uses in certain circumstances, subject to stringent requirements. We and our product candidates are subject to a variety of state laws and regulations which may hinder our ability to market our products. Whether or not FDA approval has been obtained, approval by foreign regulatory authorities must be obtained prior to commencing clinical trials, and sales and marketing efforts in those countries. These approval procedures vary in complexity from country to country, and the processes may be longer or shorter than that required for FDA approval. We may incur significant costs to comply with these laws and regulations now or in the future.
We intend to develop our zinc candidate, AEN-100, as a drug and intend to file an IND with the FDA in order to conduct necessary clinical trials to support new medical claims and ultimately file one or more NDA with respect to such products which would subject us to time, expense and uncertainty associated with achieving approval of such NDA by the FDA.
The FDA, comparable foreign regulators and state and local pharmacy regulators impose substantial requirements upon clinical development, manufacture and marketing of pharmaceutical products. These and other entities regulate research and development and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising, and promotion of our products. The drug approval process required by the FDA under the Food, Drug, and Cosmetic Act generally involves:
| 31 |
| ● | preclinical laboratory and animal tests; | |
| ● | submission of an IND, prior to commencing human clinical trials; | |
| ● | adequate and well-controlled human clinical trials to establish safety and efficacy for intended use; | |
| ● | submission to the FDA of an NDA or Biologics License Application (BLA); and | |
| ● | FDA review and approval of an NDA or BLA. |
The testing and approval process requires substantial time, effort, and financial resources, and we cannot be certain that any approval will be granted on a timely basis, if at all.
Preclinical tests include laboratory evaluation of the product candidate, its chemistry, formulation and stability, and animal studies to assess potential safety and efficacy. Certain preclinical tests must be conducted in compliance with good laboratory practice regulations. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring them to be replicated. In some cases, long-term preclinical studies are conducted concurrently with clinical studies.
We will submit the preclinical test results, together with manufacturing information and analytical data, to the FDA as part of an IND, which must become effective before we begin human clinical trials. The IND automatically becomes effective 30 days after filing, unless the FDA raises questions about conduct of the trials outlined in the IND and imposes a clinical hold, in which case, the IND sponsor and FDA must resolve the matters before clinical trials can begin. It is possible that our submission may not result in FDA authorization to commence clinical trials.
Clinical trials must be supervised by a qualified investigator in accordance with good clinical practice (GCP) regulations, which include informed consent requirements. An independent IRB at each medical center reviews and approves and monitors the study, and is periodically informed of the study’s progress, adverse events and changes in research. Progress reports are submitted annually to the FDA and more frequently if adverse events occur.
Human clinical trials of drug candidates typically have three sequential phases that may overlap:
Phase I: The drug is initially tested in healthy human subjects or patients for safety, dosage tolerance, absorption, metabolism, distribution, and excretion.
Phase II: The drug is studied in a limited patient population to identify possible adverse effects and safety risks, determine efficacy for specific diseases and establish dosage tolerance and optimal dosage.
Phase III: When Phase II evaluations demonstrate that a dosage range is effective with an acceptable safety profile, Phase III trials to further evaluate dosage, clinical efficacy and safety, are undertaken in an expanded patient population, often at geographically dispersed sites.
We cannot be certain that we will successfully complete Phase I, Phase II, or Phase III testing of our product candidates within any specific time period, if at all. Furthermore, the FDA, an IRB or the IND sponsor may suspend clinical trials at any time on various grounds, including a finding that subjects or patients are exposed to unacceptable health risk. Concurrent with these trials and studies, we also develop chemistry and physical characteristics data and finalize a manufacturing process in accordance with good manufacturing practice (GMP) requirements. The manufacturing process must conform to consistency and quality standards, and we must develop methods for testing the quality, purity, and potency of the final products. Appropriate packaging is selected and tested, and chemistry stability studies are conducted to demonstrate that the product does not undergo unacceptable deterioration over its shelf-life. Results of the foregoing are submitted to the FDA as part of a NDA (or BLA in case of biologic products) for marketing and commercial shipment approval. The FDA reviews each NDA or BLA submitted and may request additional information.
Once the FDA accepts the NDA or BLA for filing, it begins its in-depth review. The FDA has substantial discretion in the approval process and may disagree with our interpretation of the data submitted. The process may be significantly extended by requests for additional information or clarification regarding information already provided. As part of this review, the FDA may refer the application to an appropriate advisory committee, typically a panel of clinicians. Manufacturing establishments often are inspected prior to NDA or BLA approval to assure compliance with GMPs and with manufacturing commitments made in the application.
Submission of an NDA or BLA with clinical data requires payment of a fee. In return, the FDA assigns a goal of ten months for issuing its “complete response,” in which the FDA may approve or deny the NDA or BLA, or require additional clinical data. Even if these data are submitted, the FDA may ultimately decide the NDA or BLA does not satisfy approval criteria. If the FDA approves the NDA or BLA, the product becomes available for physicians prescription. Product approval may be withdrawn if regulatory compliance is not maintained or safety problems occur. The FDA may require post-marketing studies, also known as phase IV studies, as a condition of approval, and requires surveillance programs to monitor approved products that have been commercialized. The agency has the power to require changes in labeling or prohibit further marketing based on the results of post-marketing surveillance.
| 32 |
Satisfaction of these and other regulatory requirements typically takes several years, and the actual time required may vary substantially based upon the type, complexity and novelty of the product. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures on our activities. We cannot be certain that the FDA or other regulatory agencies will approve any of our products on a timely basis, if at all. Success in preclinical or early-stage clinical trials does not assure success in later-stage clinical trials. Data obtained from preclinical and clinical activities are not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. Even if a product receives regulatory approval, the approval may be significantly limited to specific indications or uses.
Even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Delays in obtaining, or failures to obtain regulatory approvals would have a material adverse effect on our business.
The FDA’s policies may change, and additional government regulations may be enacted which could prevent or delay regulatory approval of our potential products. Increased attention to the containment of health care costs worldwide could result in new government regulations materially adverse to our business. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
We do not have a guarantee of patent restoration and marketing exclusivity of the ingredients for our drugs even if we are granted FDA approval of our products.
The U.S. Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman) permits the FDA to approve Abbreviated New Drug Applications (ANDAs) for generic versions of innovator drugs, as well as NDAs with less original clinical data, and provides patent restoration and exclusivity protections to innovator drug manufacturers. The ANDA process permits competitor companies to obtain marketing approval for drugs with the same active ingredient and for the same uses as innovator drugs, but does not require the conduct and submission of clinical studies demonstrating safety and efficacy. As a result, a competitor could copy any of our drugs and only need to submit data demonstrating that the copy is bioequivalent to gain marketing approval from the FDA. Hatch-Waxman requires a competitor that submits an ANDA, or otherwise relies on safety and efficacy data for one of our drugs, to notify us and/or our business partners of potential infringement of our patent rights. We and/or our business partners may sue the company for patent infringement, which would result in a 30-month stay of approval of the competitor’s application. The discovery, trial and appeals process in such suits can take several years. If the litigation is resolved in favor of the generic applicant or the challenged patent expires during the 30-month period, the stay is lifted and the FDA may approve the application. Hatch-Waxman also allows competitors to market copies of innovator products by submitting significantly less clinical data outside the ANDA context. Such applications, known as “505(b)(2) NDAs” or “paper NDAs,” may rely on clinical investigations not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use and are subject to the ANDA notification procedures described above.
The law also permits restoration of a portion of a product’s patent term that is lost during clinical development and NDA review, and provides statutory protection, known as exclusivity, against FDA approval or acceptance of certain competitor applications. Restoration can return up to five years of patent term for a patent covering a new product or its use to compensate for time lost during product development and regulatory review. The restoration period is generally one-half the time between the effective date of an IND and submission of an NDA, plus the time between NDA submission and its approval (subject to the five-year limit), and no extension can extend total patent life beyond 14 years after the drug approval date. Applications for patent term extension are subject to U.S. Patent and Trademark Office (USPTO) approval, in conjunction with FDA. Approval of these applications takes at least nine months, and there can be no guarantee that it will be given at all.
Hatch-Waxman also provides for differing periods of statutory protection for new drugs approved under an NDA. Among the types of exclusivity are those for a “new molecular entity” and those for a new formulation or indication for a previously-approved drug. If granted, marketing exclusivity for the types of products that we are developing, which include only drugs with innovative changes to previously-approved products using the same active ingredient, would prohibit the FDA from approving an ANDA or 505(b)(2) NDA relying on our safety and efficacy data for three years. This three-year exclusivity, however, covers only the innovation associated with the original NDA. It does not prohibit the FDA from approving applications for drugs with the same active ingredient but without our new innovative change. These marketing exclusivity protections do not prohibit the FDA from approving a full NDA, even if it contains the innovative change.
| 33 |
ITEM 2. UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS
On October 16, 2012, a closing was held for the transaction previously announced on August 8, 2012 between us and Intrexon. We issued 3,552,210 shares of our common stock, $0.001 par value, which issuance is also deemed paid in partial consideration for the execution and delivery of the Exclusive Channel Collaboration Agreement, dated August 6, 2012, between us and Intrexon. The offer and issuance of such shares of common stock have not been registered under the Securities Act of 1933, as amended, and therefore may not be offered or sold in the United States absent registration or an applicable exemption from registration requirements. For this issuance, we are relying on the exemption from federal registration under Section 4(2) and Regulation D of the Securities Act, based on our belief that the offer and sale of such shares of common stock does not involve a public offering as Intrexon is an “accredited investor” as defined under Section 501 promulgated under the Securities Act and no general solicitation has been involved in the offering.
On October 30, 2012, we issued 6,750,000 shares of our common stock at a price per share of $1.60 for aggregate gross proceeds of $10.8 million and net proceeds of $10.1 million to certain accredited investors. In connection with the offering, we also issued to the placement agent, and its designees, a five-year warrant to purchase up to 635,855 shares of our common stock with an exercise price of $1.60 per share. The offer and issuance of such shares of common stock, the warrants and the shares of common stock underlying such warrants have not been registered under the Securities Act of 1933, as amended, and therefore may not be offered or sold in the United States absent registration or an applicable exemption from registration requirements. For these issuances, we are relying on the exemption from federal registration under Section 4(2) and Regulation D of the Securities Act, based on our belief that the offer and sale of such shares of common stock, the warrants and the shares of common stock underlying such warrants do not involve a public offering as the purchasers of the common stock and placement agent are “accredited investors” as defined under Section 501 promulgated under the Securities Act and no general solicitation has been involved in the offering.
ITEM 3. DEFAULTS UPON SENIOR SECURITIES
None.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
ITEM 5. OTHER INFORMATION
None.
| 34 |
ITEM 6. EXHIBITS
| 31.1 | Certification of Principal Executive Officer pursuant to Rule 13a-14(a)/15d-14(a) * |
| 31.2 | Certification of Principal Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) * |
| 32.1 | Certification of Principal Executive Officer pursuant to Section 1350 of the Sarbanes-Oxley Act of 2002 * |
| 32.2 | Certification of Principal Financial Officer pursuant to Section 1350 of the Sarbanes-Oxley Act of 2002 * |
| EX-101.INS | XBRL Instance Document * | |
| EX-101.SCH | XBRL Taxonomy Extension Schema * | |
| EX-101.CAL | XBRL Taxonomy Extension Calculation Linkbase * | |
| EX-101.DEF | XBRL Taxonomy Extension Definition Linkbase * | |
| EX-101.LAB | XBRL Taxonomy Extension Label Linkbase * | |
| EX-101.PRE | XBRL Taxonomy Extension Presentation Linkbase * |
*Filed herewith.
| 35 |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned.
| SYNTHETIC BIOLOGICS, INC. | ||
| By: | /s/ Jeffrey Riley | |
| Jeffrey Riley | ||
| President and Chief Executive Officer | ||
| (Principal Executive Officer) | ||
| Date: November 14, 2012 | ||
| By: | /s/ C. Evan Ballantyne | |
| C. Evan Ballantyne | ||
| Chief Financial Officer | ||
| (Principal Financial Officer) | ||
| Date: November 14, 2012 | ||
| 36 |
GLOSSARY
| Term | Definition | |
| Adverse Event | Any adverse change in health or “side-effect” that occurs in a person participating in a clinical trial, from the time they consent to joining the trial until a pre-specified period of time after their treatment has been completed. | |
| AEN-100 (zinc) | Synthetic Biologics’ proprietary, once-daily, gastroretentive, sustained-release oral tablet formulation of zinc acetate. | |
| Bioavailability | The quantity or fraction of the ingested dose that is absorbed by the body. | |
| BLA - Biologics License Application | An application in the U.S. through which biologic sponsors formally propose that the FDA approve a new biologic for sale and marketing. | |
| Clinical Study/Trial | A research study that is conducted to find out if a treatment or procedure is safe and/or effective in humans. | |
| Controlled Clinical Trial | A clinical study that compares patients receiving a specific treatment to patients receiving an alternate treatment for the condition of interest. The alternate treatment may be another active treatment, standard of care for the condition and/or a placebo (inactive) treatment. | |
| Double-blinded Study/Trial | Both the participant and the researcher are unaware of who is receiving the active treatment or the placebo. | |
| Effirma (flupirtine) | Proposed tradename of Synthetic Biologics’ centrally-acting investigational oral drug for the treatment of fibromyalgia syndrome | |
| FDA - Food & Drug Administration | The U.S. government agency that ensures that medicines, medical devices, prescription medical foods and radiation-emitting consumer products are safe and effective. Authorized by Congress to enforce the Federal Food, Drug, and Cosmetic Act and several other public health laws, the agency monitors the manufacture, import, transport, storage, and sale of $1 trillion worth of goods annually. | |
| Gastroretentive | Medications designed to be retained in the upper gastrointestinal system. | |
| GMP - Good Manufacturing Practice | Regulations that require that manufacturers, processors, and packagers of drugs, medical devices, some food, and blood take proactive steps to ensure that their products are consistently produced, pure, and stable. GMP regulations require a quality approach to manufacturing, enabling companies to minimize or eliminate instances of contamination, mix-ups, and errors. | |
| Monoclonal Antibodies (mAbs) | Acting as the body's army, antibodies are proteins, generally found in the bloodstream, that provide immunity in detecting and destroying pathogens, such as viruses and bacteria and their associated toxins. | |
| IND - Investigational New Drug | An application in the U.S. submitted to the FDA for a new drug or biologic that, if allowed, will be used in a clinical trial. | |
| IRB - Institutional Review Board | A committee designated to formally approve, monitor, and review biomedical research at an institution involving human studies. Institutional Review Boards aim to protect the rights and welfare of the research subjects. | |
| NDA - New Drug Application | An application in the U.S. through which drug sponsors formally propose that the FDA approve a new pharmaceutical for sale and marketing. | |
| Open-label Clinical Study/Trial | A trial in which both the treating physician and the patient know they are receiving the experimental treatment. | |
| Phase I Clinical Trial | A Phase I trial represents an initial study in a small group of patients to primarily test for safety. |
| 37 |
| Phase II Clinical Trial | A Phase II trial represents a study in a larger number of patients to assess the safety and efficacy of a product. | |
| Phase III Clinical Trial | Phase III trials are initiated to establish safety and efficacy in an expanded patient population and at multiple clinical trial sites and are generally larger than trials in earlier phases of development. | |
| Placebo | An inactive pill or liquid. Many studies compare an active drug to a placebo to determine whether any changes seen during the study can be attributed to the active drug. | |
| Principal Investigator | This is the study director who is ultimately responsible for the conduct of the study. | |
| Prospective Clinical Study/Trial | A clinical study/trial in which participants are identified and then followed throughout the study going forward in time. | |
| Protocol | A clinical study/trial’s plan — includes the schedule of tests, requirements for participation, procedures, and medications. | |
| Randomized Study/Trial | Participants in a study are assigned by chance to either one or more of the active treatment group(s) or the placebo group. | |
| Single-blinded Study/Trial | One party, either the participant or the researcher, does not know if the participant is taking the active treatment or the placebo. | |
| Study/Trial Coordinator | Staff member who is often the primary contact for research participants and coordinates their care and evaluations throughout the study. | |
| Synthetic Biology | Synthetic biology is an emerging field that combines molecular biology and automation to design, optimize and construct new biological systems and functions.
These technologies utilize a combination of automated processes including, DNA sequencing, computer-aided design, DNA synthesis, fabrication of modular transgenes and high throughput testing to create and optimize biologic products. | |
| Trimesta (oral estriol) | Proposed tradename of Synthetic Biologics’ investigational oral drug for the treatment of relapsing- remitting MS and cognitive dysfunction in MS. |
| 38 |
EXHIBIT 31.1
CERTIFICATION OF PRINCIPAL EXECUTIVE OFFICER
PURSUANT TO RULE 13a-14 OR RULE 15d-14 OF THE SECURITIES EXCHANGE ACT OF 1934,
AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Jeffrey Riley, certify that:
| 1. | I have reviewed this quarterly report on Form 10-Q of Synthetic Biologics, Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant's other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant's other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| Date: November 14, 2012 | By: | /s/ Jeffrey Riley |
| Name: Jeffrey Riley | ||
| Title: President and Chief Executive Officer | ||
| (Principal Executive Officer) |
EXHIBIT 31.2
CERTIFICATION OF PRINCIPAL FINANCIAL OFFICER
PURSUANT TO RULE 13a-14 OR RULE 15d-14 OF THE SECURITIES EXCHANGE ACT OF 1934,
AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, C. Evan Ballantyne, certify that:
| 1. | I have reviewed this quarterly report on Form 10-Q of Synthetic Biologics, Inc.; |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
| 4. | The registrant's other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
| a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
| b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
| c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
| d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
| 5. | The registrant's other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
| a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
| b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
| Date: November 14, 2012 | By: | /s/ C. Evan Ballantyne |
| Name: C. Evan Ballantyne | ||
| Title: Chief Financial Officer | ||
| (Principal Financial Officer) |
EXHIBIT 32.1
CERTIFICATION PRINCIPAL EXECUTIVE OFFICER
PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
Pursuant to 18 U.S.C. § 1350, as created by Section 906 of the Sarbanes-Oxley Act of 2002, the undersigned officer of Synthetic Biologics, Inc. (the “Registrant”) hereby certifies, to such officer’s knowledge, that:
| (1) | the accompanying Quarterly Report on Form 10-Q of the Registrant for the quarter ended September 30, 2012 (the “Report”) fully complies with the requirements of Section 13(a) or Section 15(d), as applicable, of the Securities Exchange Act of 1934, as amended; and |
| (2) | the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Registrant. |
| Date: November 14, 2012 | ||
| By: | /s/ Jeffrey Riley | |
| Name: Jeffrey Riley | ||
| Title: President and Chief Executive Officer | ||
| (Principal Executive Officer) | ||
EXHIBIT 32.2
CERTIFICATION PRINCIPAL FINANCIAL OFFICER
PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
Pursuant to 18 U.S.C. § 1350, as created by Section 906 of the Sarbanes-Oxley Act of 2002, the undersigned officer of Synthetic Biologics, Inc. (the “Registrant”) hereby certifies, to such officer’s knowledge, that:
| (1) | the accompanying Quarterly Report on Form 10-Q of the Registrant for the quarter ended September 30, 2012 (the “Report”) fully complies with the requirements of Section 13(a) or Section 15(d), as applicable, of the Securities Exchange Act of 1934, as amended; and |
| (2) | the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Registrant. |
| Date: November 14, 2012 | ||
| By: | /s/ C. Evan Ballantyne | |
| Name: C. Evan Ballantyne | ||
| Title: Chief Financial Officer | ||
| (Principal Financial Officer) | ||