UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2013
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES ACT OF 1934 |
For the transition period from______________________ to____________________________
Commission File Number: 1-12584

SYNTHETIC BIOLOGICS, INC.
(Name of small business issuer in its charter)
| Nevada | 13-3808303 |
| (State or other jurisdiction of incorporation or organization) | (IRS Employer Identification Number) |
| 155 Gibbs Street, Suite 412 | |
| Rockville, MD | 20850 |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code:
(734) 332-7800
| Securities registered pursuant to Section 12(b) of the Act: | Name of each exchange on which registered |
| (Title of Class) | |
| Common Stock, $0.001 par value per share | NYSE MKT |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the issuer: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of issuer’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every interactive data file required to be submitted and posted pursuant to Rule 405 of Regulation S-T (section 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).Yes x No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated file, a non-accelerated file, or a smaller reporting company. See the definitions of “large accelerated filer, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |
| Non-accelerated filer | ¨ | Smaller reporting company | x | |
| (Do not check if a smaller reporting company) | ||||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant as of June 28, 2013, was approximately $47.1 million based on $1.72, the price at which the registrant’s common stock was last sold on that date.
As of March 27, 2014, the issuer had 58,276,556 shares of common stock outstanding.
Documents incorporated by reference: None.
SYNTHETIC BIOLOGICS, INC.
FORM 10-K
TABLE OF CONTENTS
| Page | |||
| PART I. | |||
| Item 1. | Business | 3 | |
| Item 1A. | Risk Factors | 19 | |
| Item 1B. | Unresolved Staff Comments | 28 | |
| Item 2. | Properties | 28 | |
| Item 3. | Legal Proceedings | 28 | |
| Item 4. | Mine Safety Disclosures | 28 | |
| PART II. | |||
| Item 5. | Market for Registrant’s Common Equity Related Stockholder Matters and Issuer Purchases of Equity Securities | 29 | |
| Item 6. | Selected Financial Data | 29 | |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 30 | |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | 37 | |
| Item 8. | Financial Statements and Supplementary Data | 38 | |
| Item 9. | Changes in and Discussions with Accountants on Accounting and Financial Disclosure | 59 | |
| Item 9A. | Controls and Procedures | 59 | |
| Item 9B. | Other Information | 59 | |
| PART III. | |||
| Item 10. | Directors, Executive Officers and Corporate Governance | 60 | |
| Item 11. | Executive Compensation | 62 | |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 65 | |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence | 66 | |
| Item 14. | Principal Accountant Fees and Services | 67 | |
| PART IV. | |||
| Item 15. | Exhibits and Financial Statement Schedules | 68 | |
| SIGNATURES | 72 | ||
| GLOSSARY | |||
| 2 |
PART I
Forward-Looking Statements
Certain of the matters discussed within this report include forward-looking statements on our current expectations and projections about future events. In some cases you can identify forward-looking statements by terminology such as “may,” “should,” “potential,” “continue,” “expects,” “anticipates,” “intends,” “plans,” “believes,” “estimates,” and similar expressions. These statements are based on our current beliefs, expectations, and assumptions and are subject to a number of risks and uncertainties, many of which are difficult to predict and generally beyond our control, that could cause actual results to differ materially from those expressed, projected or implied in or by the forward-looking statements. Such risks and uncertainties include the risks noted under “Item 1A Risk Factors.” We do not undertake any obligation to update any forward-looking statements. Unless the context requires otherwise, references to “we,” “us,” “our,” and “Synthetic Biologics,” refer to Synthetic Biologics, Inc. and its subsidiaries.
Item 1. Business
We are a biotechnology company focused on the development of novel anti-infective biologic and drug candidates targeting specific pathogens that cause serious infections and other diseases. We are developing an oral treatment to reduce the impact of methane producing organisms on constipation-predominant irritable bowel syndrome (C-IBS), an oral biologic to protect the gastrointestinal (GI) microflora from the effects of intravenous (IV) antibiotics for the prevention ofClostridium difficile (C. diff) infection, a series of monoclonal antibodies (mAbs) for the treatment of Pertussis andAcinetobacter infections, and a biologic targeted at the prevention and treatment of a root cause of a subset of IBS. In addition, we have two legacy programs. We are developing an oral estriol drug for the treatment of relapsing-remitting multiple sclerosis (MS) and cognitive dysfunction in MS. We have also partnered the development of a treatment for fibromyalgia.
Product Pipeline:

Summary of Pathogen-Specific Anti-Infective Biologic and Drug Programs:
| · | C-IBS: In December 2013, through our majority-owned subsidiary, Synthetic Biomics, Inc., we entered into a worldwide exclusive license agreement with Cedars-Sinai Medical Center (CSMC) for the right to develop products for therapeutic and prophylactic treatments for acute and chronic diseases. An investigational team led by Mark Pimentel, M.D. at CSMC has discovered that these products are intended to target the production of methane gas by certain pathogenic gastrointestinal (GI) microorganisms that are perceived as the underlying cause of gas, pain and constipation associated with C-IBS, as well as diseases such as obesity and type 2 diabetes. Initially we will focus on the development of an oral treatment to reduce the impact of methane producing organisms on C-IBS. We intend to initiatein vivo/pharmacokinetic/pharmacodynamic studies in the first half of 2014, and to initiate a Phase II clinical trial during the second half of 2014 under an Investigational New Drug application (IND). |
| 3 |
| · | C. diff infections: We are in preclinical development of a novel second-generation oral enzyme drug candidate, SYN-004, for co-administration with commonly used IV antibiotics intended to prevent the development of severe effects of C. diff infections.C. diff infections are a leading cause of hospital acquired infections (HAIs), that generally occur secondary to treatment with IV antibiotics. Designed to be given orally to protect the gut while certain IV beta-lactam antibiotics (penicillins and cephalosporins) fight the primary infection, SYN-004 is believed to have a similar profile to its first-generation predecessor, which demonstrated favorable protection of the gut flora (microbiome) during treatment with certain penicillins, with the potentially added ability to act against a broader spectrum of IV beta-lactam antibiotics. Beta-lactam antibiotics are a mainstay in hospital infection management and include the commonly used penicillin and cephalosporin classes of antibiotics. Approximately 14.4 million patients are administered "SYN-004 target" IV beta-lactam antibiotics annually, representing an estimated target market for SYN-004 of 117.6 million beta-lactam doses purchased by U.S. hospitals. The addressable market for SYN-004 is significant. Currently there are no approved treatments designed to protect the microbiome from the damaging effects of IV antibiotics. This worldwide opportunity could represent a multi-billion dollar market.* We intend to initiate Phase Ia and Ib clinical trials during the second half of 2014. |
| * | This information is an estimate derived from the use of information under license from the following IMS Health Incorporated information service: CDM Hospital database for full year 2012. IMS expressly reserves all rights, including rights of copying, distribution and republication. |
| • | Pertussis: In December 2012, in collaboration with Intrexon Corporation (NYSE: XON) (Intrexon), we initiated development of a mAb therapy for the treatment of Pertussis infections, more commonly known as whooping cough. We are developing a mAb therapy, SYN-005, designed to target and neutralize the pertussis toxin, in order to reduce the mortality rate in infants and potentially shorten the duration of chronic cough in afflicted adults. To further the development of this potential therapy for Pertussis, we entered into an agreement with The University of Texas at Austin to license the rights to certain research and pending patents related to pertussis antibodies. According to the World Health Organization, each year, B. pertussis infection causes an estimated 300,000 deaths worldwide, primarily among young, unvaccinated infants. As part of our IND-enabling studies, we initiated a pilot large animal study in the first quarter of 2014 utilizing the antibody combination in a non-human primate model. This model, in addition to the murine model, is supportive of the development of a pertussis therapeutic. We are currently planning a confirmatory follow-up large animal study, and expect to report topline results during the second quarter of 2014. |
| • | Acinetobacter infections: In September 2012, in collaboration with Intrexon, we initiated efforts to develop a mAb therapy for the treatment of Acinetobacter infections. Many strains of Acinetobacter are multidrug-resistant and pose an increasing global threat to hospitalized patients, wounded military personnel and those affected by natural disasters. A treatment for Acinetobacter infections represents a billion dollar market opportunity. The generation of a panel of antibodies is ongoing. |
| • | IBS: In December 2013, in collaboration with Intrexon, and partially utilizing the intellectual property optioned from CSMC, we announced we intend to develop biologic approaches targeted at the prevention, and acute and chronic treatment of a subset of IBS pathologies specifically caused by auto-antibodies. |
Summary of Multiple Sclerosis Program:
| • | TrimestaTM (oral estriol) is being developed as an oral once-daily treatment for relapsing-remitting MS in women. Patient enrollment is complete in this two-year, randomized, double-blind, placebo-controlled Phase II clinical trial being conducted at 15 centers in the United States. The primary endpoint is relapse rate at two years. This investigator-initiated trial evaluating our drug candidate, Trimesta™, is supported by grants awarded to the University of California, Los Angeles (UCLA) exceeding $8.0 million, which should be sufficient to fund the trial through completion. Annual worldwide sales of current MS therapies are estimated at $14.1 billion. Top-line results are scheduled to be presented at the American Academy of Neurology’s Annual Meeting in April 2014 by the lead principal investigator, Rhonda Voskuhl, MD. |
| • | Trimesta is also being developed for the treatment of cognitive dysfunction in female MS patients. This 12-month randomized, double-blind, placebo-controlled Phase II clinical trial is being conducted at UCLA. The primary endpoint is the effect on cognitive function as assessed by Paced Auditory Serial Addition Test (PASAT). Patient enrollment is ongoing. The majority of the costs of this trial are being funded by grants from foundations and charitable organizations and we have pledged approximately $500,000 to UCLA to partially fund this trial payable over three years. An estimated 50-65% of MS patients are expected to develop disabilities due to cognitive dysfunction and there is currently no approved treatment. |
| 4 |
Summary of Fibromyalgia Program:
| • | Effirma™ (flupirtine) is being developed for the treatment of fibromyalgia by Meda AB (Meda), a multi-billion dollar international pharmaceutical company. On May 6, 2010, we entered into a sublicense agreement with Meda covering all of our patents’ rights on the use of flupirtine for fibromyalgia in the United States, Canada and Japan. The sublicense agreement provides that all ongoing and future development costs are to be borne by Meda and we are entitled to receive certain payments if milestones are achieved and royalties on sales. According to Meda’s 2012 Year-End Report filed in February 2013, Meda has received the go-ahead from the United States Food and Drug Administration (FDA) to conduct a Phase II proof of concept study for the treatment of fibromyalgia. Meda also announced that the randomized, double-blind, placebo and active-controlled study of patients with fibromyalgia will be conducted at 25 clinics in the United States Based on an estimated annual price of $1,200 per fibromyalgia patient, we estimate that the total market potential in the United States is $6.0 billion. |
Pipeline Programs and Therapeutic Areas
Pathogen-Specific Anti-Infective Biologic and Drug Programs
We are a biotechnology company focused on the development of novel anti-infective biologic and drug candidates targeting specific pathogens that cause serious infections and other diseases. Infectious disease outbreaks are increasing while intervention options are declining due to widespread multidrug-resistant bacteria, increasing numbers of immuno-compromised patients (e.g., the elderly and cancer patients), and the isolation of new pathogens. We are developing an oral treatment to reduce the impact of methane producing organisms on C-IBS, an oral biologic to protect the gastrointestinal microflora from the effects of IV antibiotics for the prevention ofC. diff infection, a series of monoclonal antibodies for the treatment of Pertussis andAcinetobacter infections, and a biologic targeted at the prevention and treatment of a root cause of a subset of IBS.
Several of our programs are focused on protecting the microbiome, or our gut flora, which is home to millions of bacteria, composed of a natural balance of both “good” beneficial bacteria and “bad” pathogenic bacteria. When that natural balance of all of these bacteria is disrupted, a person’s health is compromised.
C-IBS:
Irritable Bowel Syndrome (IBS) is a functional GI disorder characterized by gas, abdominal pain, bloating and diarrhea or constipation, or alternating episodes of both. According to reports published by The International Foundation for Functional Gastrointestinal Disorders (IFFGD),IBS affects an estimated 10 to 15 percent of the population, or as many as 40 million Americans. The illness affects both men and women; two-thirds of diagnosed sufferers are women. The onset of IBS can begin anytime from adolescence to adulthood.Four bowel patterns may be seen with IBS, including: C-IBS (constipation predominant), D-IBS (diarrhea predominant), M-IBS (mixed diarrhea and constipation) and A-IBS (alternating diarrhea and constipation).
It has been reported that one-third of all IBS patients have C-IBS.Current FDA-approved therapies for the treatment of C-IBS includeAMITIZA® (lubiprostone) and LINZESS® (linaclotide). Prescription and over-the-counter laxatives are also used by C-IBS patients for symptomatic relief. According to GlobalData, sales of approved drugs to treat C-IBS in seven major markets are projected to reach $1.3 billion by 2018.
C-IBS: Acquisition of Clinical-Stage Program
In December 2013, we entered into a worldwide exclusive license agreement with CSMC for the right to develop products for therapeutic and prophylactic treatments for acute and chronic diseases. We licensed and optioned from CSMC a portfolio of intellectual property comprised of several U.S. and international patents and pending patent applications for various fields of use, including C-IBS, obesity and diabetes. An investigational team led by Mark Pimentel, M.D. at CSMC has discovered that these products are intended to target the production of methane gas by certain pathogenic gastrointestinal microorganisms that are perceived as the underlying cause of gas, pain and constipation associated with C-IBS, as well as diseases such as obesity and type 2 diabetes. Initially we will focus on the development of an oral treatment to reduce the impact of methane producing organisms on C-IBS.
IBS: Gas Producing Organisms Background
In the 1990’s, research showed that IBS patients (over a given time) produced five times more gas than did people without IBS. Since the only source of those gases was bacterial, the initial presumption was that IBS patients had excessive bacteria in the colon. Subsequent studies showed that IBS patients had excessive quantities of gas in the small bowel; these data were the catalyst for studying small bowel bacteria in IBS. Normally the small intestine contains a very small quantity of bacteria. In published studies, indirect measures of small bowel bacteria suggest that 84% of IBS sufferers have excessive quantities of bacteria typically found in the colon.
The CSMC investigational team led by Dr. Pimentel is researching a recent theory that defines IBS as a bacterial disease. Gut microflora that should normally be confined to the large intestine inappropriately colonize the small intestine. This process is referred to as small intestine bacterial overgrowth (SIBO), which results in gas, bloating, abdominal pain and altered stool habits characterized by IBS.
| 5 |
C-IBS: Methane Producing Organisms Background
Further research by the CSMC investigational team led by Dr. Pimentel is focused on the C-IBS patient population. The theory that defines this patient set is that the constipation associated with C-IBS is due to an infectious disease. Overgrowth of certain gut microflora may lead to overproduction of methane gas resulting in pain, bloating and constipation. CSMC investigators have discovered that inhibiting intestinal methane production may treat the underlying cause of major diseases, including constipation associated with C-IBS.
C-IBS: Preclinical and Clinical Development
Ongoing efforts led by Dr. Pimentel include formulating and testing non-antibiotic FDA-approved oral drug candidates for ultimate product registration via potential expedited pathways. Such candidates are intended for the specific elimination of methane gas production within the intestines, with the goal of having little or no unintended impact on a patient's normal intestinal microflora. Initially we will focus on the development of an oral treatment to reduce the impact of methane producing organisms on C-IBS.
We intend to initiatein vivo/pharmacokinetic/pharmacodynamic studies in the first half of 2014, and to initiate a Phase II clinical trial during the second half of 2014 under an IND.
C. difficile:
According to the Agency for Healthcare Research and Quality, aggregate costs associated withC.diff infection (CDI)-related stays in the hospital were $8.2 billion in the U.S. during 2009. CDI is a rising global HAI problem in which the toxins produced by C. difficile bacteria result in diarrhea antibiotic-associated diarrhea (AAD), and in the most serious cases, pseudomembranous colitis (erosion of the lower GI tract) that can lead to death. The Centers for Disease Control and Prevention (CDC) recently identified C. diffas an “urgent public health threat,” particularly given its resistance to many drugs used to treat other infections. CDI is a major, unintended risk associated with the prophylactic or therapeutic use of IV antibiotics, which may alter the natural balance of microflora that normally protect the GI tract, leading to C. difficile overgrowth and infection. Other risk factors for CDI include hospitalization, prolonged length of stay, underlying illness, immune-compromising conditions including the administration of chemotherapy, and advanced age.
CDI is a widespread and often drug resistant infectious disease, and it is estimated that 1.1 million patients are infected with C. diff annually in the U.S.*, and it has been reported that 30,000 patients die with a C. diff infection each year. CDI has surpassed methicillin-resistant staphylococcus aureus (MRSA) as the most frequent infection acquired in the hospital. Controlling the spread of CDI has proven challenging, as the C. difficile spores are easily transferred to patients via normal contact with healthcare personnel and other inanimate objects. There is currently no vaccine or approved product for the prevention of C. diff infection.
| * | This information is an estimated derived from the use of information under license from the following IMS Health Incorporated information service: CDM Hospital database for full year 2012. IMS expressly reserves all rights, including rights of copying, distribution and republication. |
C. difficile: Acquisition of Clinical-Stage Program
In November 2012, we acquired a series of oral beta-lactamase enzymes (P1A, P2A and P3A) and related assets targeting the prevention of CDI, the leading cause of HAIs that generally occurs secondary to treatment with IV antibiotics. The acquired assets include a pre-IND package for P3A (SYN-004), Phase I and Phase II clinical data for P1A, manufacturing processes and data, and a portfolio of issued and pending U.S. and international patents intended to support an IND and Biologics License Application (BLA) with the FDA. Utilizing this portfolio of assets, we intend to develop a proprietary oral beta-lactamase enzyme product candidate, SYN-004, previously known as P3A. When co-administered with certain IV beta-lactam antibiotics, it is expected that SYN-004 can degrade the antibiotic that is excreted in the GI tract, thus preserving the natural balance of the patient's microflora, and preventing opportunistic infections including CDI. Beta-lactam antibiotics are a mainstay in hospital infection management and include the commonly used penicillin and cephalosporin classes of antibiotics. Approximately 14.4 million patients are administered "SYN-004 target" IV beta-lactam antibiotics annually, representing an estimated target market for SYN-004 of 117.6 million beta-lactam doses purchased by U.S. hospitals. The addressable market is significant and currently there are no approved treatments designed to protect the microbiome from the damaging effects of IV antibiotics. This worldwide opportunity could represent a multi-billion dollar market.*
| * | This information is an estimated derived from the use of information under license from the following IMS Health Incorporated information service: CDM Hospital database for full year 2012. IMS expressly reserves all rights, including rights of copying, distribution and republication. |
C. difficile: Oral Enzyme Background
We acquired a series of oral beta-lactamase enzymes. Beta-lactamase enzymes have the ability to degrade beta-lactam antibiotics that may be excreted into the GI tract. P1A (the first generation candidate) showed acceptable safety and tolerability in a Phase I study. In addition, two Phase II clinical studies demonstrated that P1A had the ability to preserve GI microflora in hospitalized patients treated with intravenous ampicillin or the combination of piperacillin and tazobactam.
| 6 |
C. difficile: Preclinical and Clinical Development
Compared to the first generation oral enzyme candidate, P1A, we believe that the second generation candidate, SYN-004 (formerly P3A), will have activity against a broader spectrum of beta-lactam antibiotics, including both penicillins and certain cephalosporins. Due to the structural similarities between P1A and SYN-004, and based on previous discussions with the FDA, it is anticipated that certain preclinical data collected on P1A may be used in support of an IND for our new product candidate, SYN-004.
In October 2013, we initiated manufacturing of SYN-004 material to support our planned preclinical and clinical studies. We intend to initiate Phase Ia and Ib clinical trials during the second half of 2014.
Monoclonal Antibodies:
Monoclonal Antibodies for Infectious Diseases
Acting as the body's army, antibodies are proteins, generally found in the bloodstream, that provide immunity in detecting and destroying pathogens, such as viruses and bacteria and their associated toxins. MAbs can also be designed and produced as therapeutic agents, utilizing protein engineering and recombinant production technologies. The mAbs being developed under our collaboration with Intrexon are intended to supplement a patient's own immune system by providing the means to specifically and rapidly neutralize and/or clear specific pathogens and toxins of interest in a process known as “passive immunity”. Many pathogens that cause infectious diseases are innately resistant to, or over time have developed increased resistance to, antibiotics and other drugs.
Intrexon Collaboration: Monoclonal Antibodies for Infectious Diseases
In August 2012, we entered into a worldwide exclusive channel collaboration (“Second ECC”) with Intrexon through which we intend to develop a series of mAb therapies for the treatment of certain infectious diseases not adequately addressed by existing therapies. Utilizing Intrexon’s comprehensive suite of proprietary technologies, including the mAbLogixTM platform for rapid discovery of fully human mAbs and the LEAPTM cell processing station, our initial efforts will target three infectious disease indications.*** We also have the option to target an additional five infectious disease indications under this collaboration. To date, we have initiated development of a mAb therapy for the treatment of Pertussis and Acinetobacter infections.
***mAbLogixTM and LEAPTM are registered trademarks of Intrexon Corporation
Bordetella pertussis (B. pertussis) is a gram-negative bacterium that infects the upper respiratory tract, causing uncontrollable, and violent coughing. Antibiotic treatment does not have a major effect on the course of Pertussis, because while it can eliminate the B. pertussis bacteria from the respiratory tract, it does not neutralize the pertussis toxin. Infants with Pertussis often require hospitalization in pediatric intensive care units, frequently requiring mechanical ventilation. Pertussis in adults generally leads to a chronic cough referred to as the "cough of 100 days." The incidence of Pertussis is increasing due to a less effective acellular vaccine introduced in the 1990s, exposure of unvaccinated and under-vaccinated individuals including infants who are not yet fully vaccinated, exposure of individuals whose immunity has diminished over time, as well as asymptomatic carriers.
According to the World Health Organization there are 50 million cases of whooping cough andB. pertussis infection causes an estimated 300,000 deaths each year worldwide, primarily among young, unvaccinated infants. Recent news reports throughout the U.S. indicate that the pertussis vaccine introduced in the 1990s does not provide long-term protection and, as a result, whooping cough cases have increased to a 60-year high. There is no approved treatment for Pertussis, and antibiotic treatment does not have a major effect on the course of Pertussis, because while it can eliminate theB. pertussis bacteria from the respiratory tract, it does not neutralize the pertussis toxin.
Pertussis: Intrexon Collaboration and The University of Texas at Austin Agreement
In December 2012, we initiated mAb development for the treatment of Pertussis focusing on toxin neutralization pursuant to our August 2012 collaboration with Intrexon. Unlike antibiotics, we are developing a mAb therapy, SYN-005, to target and neutralize the pertussis toxin, in order to reduce the mortality rate in infants and shorten the duration of chronic cough in afflicted adults.
To further the development of this potential therapy for pertussis, we have entered into an agreement with The University of Texas at Austin to license the rights to certain research and pending patents related to pertussis antibodies. These research efforts are being conducted at the Cockrell School of Engineering in the laboratory of Assistant Professor, Jennifer A. Maynard, Ph.D., the Laurence E. McMakin, Jr. Centennial Faculty Fellow in the McKetta Department of Chemical Engineering. Dr. Maynard brings to the project her expertise in defining the key neutralizing epitopes of pertussis toxin to optimize the potential efficacy of antibody therapeutics.
| 7 |
Pertussis: Preclinical and Clinical Development
Working with our collaborator, Intrexon, and our academic collaborator, The University of Texas at Austin, we have established a combination of two humanized antibodies designed to neutralize pertussis toxin, a major cause of pertussis-mediated infant morbidity and mortality. Benchtop studies demonstrated high affinity binding to the toxin, as well as potent neutralization of the toxin. In addition, the antibodies were highly efficacious in a murine model of pertussis in which they completely mitigated elevations of the white blood cell count that is characteristic of the illness.
As part of our IND-enabling studies, we initiated a pilot large animal study in the first quarter of 2014 utilizing the antibody combination in a non-human primate model, which in addition to the murine model, is supportive of the development of a pertussis therapeutic. We are currently planning a confirmatory follow-up large animal study, and expect to report topline results during the second quarter of 2014.
Manufacturing of antibodies for nonclinical development is underway. We are filing patents to strengthen our intellectual property position around pertussis antibodies. In addition, we intend to file an orphan drug application for the pertussis indication.
Acinetobacter Infections:
Acinetobacter baumanii is a difficult to treat pathogen due to its rapid and well-established development of resistance to most antibiotics, making it a multidrug-resistant pathogen. In addition, as a biofilm-forming pathogen, Acinetobacter baumanii has the ability to survive up to twice as long as non-biofilm-forming pathogens. In the U.S., Acinetobacter baumanii has been reported to be the cause of up to 2.6% of hospital acquired infections, 1.3% of bloodstream infections and 7% of ICU respiratory tract infections, and more than half of the Acinetobacter baumanii isolates are multidrug-resistant. According to published articles, mortality rates associated withAcinetobacter infections as high as 43% are reported in hospitals and ICU settings. While Acinetobacter baumanii is a well-documented pathogen in the hospital setting, this pathogen also poses an increasing danger to wounded servicemen and women in military treatment centers and to those treated in trauma centers following natural disasters.
A treatment for Acinetobacter infections represents a billion dollar market opportunity.
Acinetobacter: Intrexon Collaboration
In August 2012, we initiated a mAb discovery and development program for Acinetobacter infections pursuant to our August 2012 collaboration with Intrexon. Discovery efforts for the development of a mAb are currently underway.
IBS:
Existing IBS therapies, which are primarily focused on supportive care, are unlikely to address the treatment needs of the patient population with auto-antibodies, an underlying immune-specific pathology. Through our collaboration with Intrexon, we intend to address the unmet medical need in these patients with personalized medicine and target the root causes of a subset of IBS-associated pathologies.
IBS: Intrexon Collaboration
In December 2013, in collaboration with Intrexon, and partially utilizing the intellectual property optioned from CSMC, we announced an intent to develop biologic approaches targeted at the prevention, and acute and chronic treatment of a subset of IBS pathologies specifically caused by auto-antibodies.
We intend to utilize intellectual property optioned from CSMC. According to an increasing body of recent work conducted by CSMC, a subset of IBS cases appear to be causally initiated by one or more encounters with acute infectious gastroenteritis, such as the foodborne illness, Campylobacter jejuni. CSMC has identified a novel autoimmune target for this subset of IBS cases because of the development of cross-reacting antibodies between a bacterial toxin and a protein important for controlling GI motility. This program is in the discovery stage.
Multiple Sclerosis Program
Relapsing-Remitting MS:
MS is a progressive neurological disease in which the body loses the ability to transmit messages along the central nervous system, leading to pain, loss of muscle control, paralysis, cognitive impairment and in some cases death. According to the National Multiple Sclerosis Society (NMSS), more than 2.3 million people worldwide (approximately 400,000 patients in the U.S. of which approximately 65% are women) have been diagnosed with MS. The diagnosis is typically made in young adults, ages 20 to 50. According to the NMSS, approximately 85% of MS patients are initially diagnosed with the relapsing-remitting form, and 10-15% with other progressive forms.
There are nine FDA-approved therapies for the treatment of relapsing-remitting MS: Betaseron®, Rebif®, Avonex®, Copaxone®, Tysabri®, Gilenya®, Extavia®, Aubagio® and TecfideraTM. Many of these therapies provide only a modest benefit for patients with relapsing-remitting MS. All of these drugs except Gilenya®, Aubagio® and TecfideraTM require frequent (daily, weekly & monthly) injections (or infusions) on an ongoing basis and can be associated with unpleasant side effects (such as flu-like symptoms) and high rates of non-compliance among users. Despite the availability of therapies for the treatment of relapsing-remitting MS, the disease is highly underserved and exacts a heavy personal and economic toll. Annual worldwide sales of current MS therapies are estimated at $14.1 billion.
Relapsing-Remitting MS: Background
Research has shown that pregnant women with MS tend to experience a spontaneous reduction of disease symptoms during pregnancy, particularly in the third trimester. The PRIMS (Pregnancy In MS) study published in 1998, a landmark observational clinical study published in the New England Journal of Medicine followed 254 women with MS during 269 pregnancies and for up to one year after delivery. The PRIMS study demonstrated that relapse rates were significantly reduced by 71% (p < 0.001) through the third trimester of pregnancy compared to pre-pregnancy-rates, and that relapse rates increased by 120% (p < 0.001) during the first three months after birth (post-partum) and then return to pre-pregnancy rates. It has been hypothesized that the female hormone, estriol, produced by the placenta during pregnancy, plays a role in “fetal immune privilege”, a process that prevents a mother’s immune system from attacking and rejecting the fetus. The maternal levels of estriol increase linearly through the third trimester of pregnancy until birth, whereupon it abruptly returns to low circulating levels. The anti-autoimmune effects of estriol are thought to be responsible for the therapeutic effects of pregnancy on MS.
| 8 |
Rhonda Voskuhl, M.D., Director, UCLA MS program, UCLA Department of Neurology, has found that plasma levels of estriol achieved during pregnancy have potent immunomodulatory effects. She further postulated and tested in a pilot clinical study that oral doses of estriol may have a therapeutic benefit when administered to non-pregnant female MS patients by, in essence, mimicking the spontaneous reduction in relapse rates seen in MS patients during pregnancy.
Estriol has been approved and marketed for over 40 years throughout Europe and Asia for the oral treatment of post-menopausal symptoms. It has never been approved by the U.S. FDA for any indication.
Relapsing-Remitting MS: Clinical Development
Trimesta (oral estriol) is being developed for the treatment of relapsing-remitting MS in women. An investigator-initiated, 10-patient, 22-month, single-agent, crossover clinical trial to study the therapeutic effects of 8 mg. of oral Trimesta taken daily in non-pregnant female relapsing-remitting MS patients was completed in the U.S. The total volume and number of gadolinium-enhancing lesions were measured by brain magnetic resonance imaging (an established neuroimaging measure of disease activity in MS). Over the next three months of treatment with Trimesta, the median total enhancing lesion volumes decreased by 79% (p = 0.02) and the number of lesions decreased by 82% (p = 0.09). They remained decreased during the next 3 months of treatment, with lesion volumes decreased by 82% (p = 0.01), and numbers decreased by 82% (p =0.02). Following a six-month drug holiday during which the patients were not on any drug therapies, median lesion volumes and numbers returned to near baseline pretreatment levels. Trimesta therapy was reinitiated during a four-month retreatment phase of this clinical trial. The relapsing-remitting MS patients again demonstrated a decrease in enhancing lesion volumes of 88% (p = 0.008) and a decrease in the number of lesions by 48% (p = 0.04) compared with original baseline scores.
A Phase II randomized, double-blind, placebo-controlled clinical trial is currently underway at 15 centers in the U.S. under the direction of Lead Principal Investigator, Dr. Rhonda Voskuhl. The purpose of this clinical trial is to evaluate whether 8 mg of oral Trimesta taken daily over a two year period will reduce the rate of relapses in a large population of female patients with relapsing-remitting MS. Investigators are administering either Trimesta or matching placebo, in addition to a standard of care, glatiramer acetate injections (Copaxone®), an FDA-approved therapy for MS, to women between the ages of 18 to 50 who have been recently diagnosed with relapsing-remitting MS. Relapse rates at two years is the primary endpoint in this clinical trial being run under an investigator-initiated IND.
Patients in this Phase II relapsing-remitting MS trial completed their final 24-month visit during January 2014. Dr. Rhonda Voskuhl is scheduled to present topline results at the American Academy of Neurology's 66th Annual Meeting in Philadelphia at the end of April 2014.
With over $8 million in grant funding awarded to UCLA to date, from organizations such as the National Institutes for Health and the National Multiple Sclerosis Society, the ongoing Trimesta clinical trial should be funded to its completion.
Relapsing-Remitting MS: Patents
In March 2014, we announced that the U.S. Patent & Trademark Office issued U.S. Patent No. 8,658,627 entitled,Pregnancy Hormone Combination for Treatment of Autoimmune Diseases, to the Regents of the University of California. The patent includes claims to the use of our drug candidate, Trimesta™ (oral estriol), in conjunction with a gestagen for the treatment of multiple sclerosis (MS) and other autoimmune diseases. The patent also includes a claim for the administration of Trimesta™, a gestagen and a third standard of care MS agent, such as glatiramer acetate injection (Copaxone®), interferon beta-1a (Avonex®, Rebif®), interferon beta-1b (Betaseron®, Extavia®) or sphingosine-1-phosphate receptor modulator (Gilenya®).
In April 2013, we announced that the U.S. Patent & Trademark Office issued U.S. Patent No. 8,372,826 entitled, Estriol Therapy for Multiple Sclerosis and Other Autoimmune Diseases, to the Regents of the University of California which includes claims to the use of our drug candidate, Trimesta™ (oral estriol), in combination with glatiramer acetate injection (Copaxone®). According to Teva Pharmaceutical Industries Ltd.’s Form 20-F for the year ended December 31, 2012, filed with the SEC on February 12, 2013. Copaxone® is the number one selling drug for multiple sclerosis with approximately $4 billion in annual sales. Currently marketed exclusively by Teva Pharmaceutical Industries Ltd., Copaxone® is expected to face generic competition as certain patent terms begin to expire in 2014.
Through our wholly owned subsidiary, we hold the exclusive worldwide license to issued U.S. Patents 8,658,627, 8,372,826 and 6,936,599 and pending patents for multiple sclerosis and other autoimmune diseases covering the uses of our drug candidate, Trimesta™.
| 9 |
Cognitive Dysfunction in MS:
According to the NMSS and the Multiple Sclerosis Society of Canada publication, Hold that Thought! Cognition and MS, it is fairly common for people with MS to complain of cognitive difficulties, such as remembering things, finding the right words and the ability to concentrate. Among MS patients, 50-65% have some degree of cognitive dysfunction.
The major areas of cognition that may be affected include complex attention and executive functions. Complex attention involves multitasking, the speed with which information can be processed, learning and memory, and perceptual skills; executive functions include problem solving, organizational skills, the ability to plan, and word finding. Just as the nature, frequency, and severity of MS-related physical problems can widely vary, not all people with MS will have cognitive dysfunction, and no two people will experience exactly the same type or severity.
Cognitive Dysfunction in MS: Background
In the investigator-initiated, 10-patient, 22-month, single-agent, crossover clinical trial conducted by Dr. Rhonda Voskuhl, a statistically significant 14% improvement from baseline in the PASAT cognitive testing scores (p = 0.04) was observed in relapsing-remitting MS patients after six months of Trimesta therapy. PASAT is a routine cognitive test performed in patients with a wide variety of neuropsychological disorders such as MS. The PASAT scores are expressed as a mean percent change from baseline.
Cognitive Dysfunction in MS: Clinical Development
Our Trimesta (oral estriol) drug candidate is also being developed for the treatment of cognitive dysfunction in female MS patients. This randomized, double-blind, placebo-controlled Phase II clinical trial to evaluate Trimesta’s potential neuroprotective and therapeutic effect on cognitive dysfunction in female MS patients is currently enrolling relapsing-remitting or secondary-progressive female MS patients at UCLA. Up to 64 patients between the ages of 18 and 50 will be randomized 1:1 into the treatment and placebo groups. Dr. Voskuhl will administer either oral Trimesta or a matching placebo, in addition to any FDA-approved MS treatment. Each patient will be dosed and monitored for one year after being enrolled. The primary endpoint in this clinical trial being run under an investigator-initiated IND application is expected to be improvement in PASAT cognitive testing scores versus matching placebo. We and a private foundation have pledged to equally support this new clinical trial, and we will also provide Trimesta drug supply. The trial also received contributions from several other supporters. Patient recruitment and enrollment into this trial is ongoing.
Fibromyalgia Program
Fibromyalgia is a chronic and debilitating condition characterized by widespread pain and stiffness throughout the body, often accompanied by severe fatigue, insomnia and alterations in mood. According to the National Fibromyalgia Association, fibromyalgia affects an estimated 3-6% of the population worldwide, including an estimated 10 million people in the U.S. There are presently three FDA products approved for the treatment of fibromyalgia - Lyrica®, Cymbalta® and Savella®.
Based on an estimated annual price of $1,200 per fibromyalgia patient, we estimate that the total market potential in the U.S. is $6 billion.
Fibromyalgia: Meda Corporate Partnership
On May 6, 2010, we entered into a sublicense agreement with Meda, a multi-billion dollar international pharmaceutical company, pursuant to which Meda assumed all future development costs and may commercialize flupirtine, a molecular entity with a unique mode of action for the treatment of fibromyalgia in the U.S. As consideration for such sublicense, we received an up-front payment of $2.5 million and are entitled to milestone payments of $5.0 million upon the FDA’s acceptance of the New Drug Application (NDA) for flupirtine for fibromyalgia and $10.0 million upon FDA approval of such NDA. Pursuant to the sublicense agreement, we will also receive a 7% royalty on net sales of flupirtine for fibromyalgia in the U.S., Canada and Japan, with such royalties being shared equally with our licensor, McLean Hospital, a Harvard teaching hospital.
Flupirtine is approved and marketed by Meda and its distributors in Europe and other countries for indications other than fibromyalgia and has been prescribed to millions of patients worldwide. We believe that such substantial human experience with flupirtine should greatly assist the FDA in its evaluation of the safety of flupirtine upon review of an NDA of flupirtine for fibromyalgia.
Fibromyalgia: Clinical Development
Our Effirma (flupirtine) drug candidate for the treatment of fibromyalgia, has been partnered to Meda (see “Fibromyalgia: Meda Corporate Partnership” section above). Effirma is a selective neuronal potassium channel opener that also has N-methyl-D-aspartic (NMDA) receptor antagonist properties. Effirma is a non-opioid, non-NSAID, non-steroidal, analgesic. Preclinical data and clinical experience suggest that Effirma should also be effective for neuropathic pain since it acts in the central nervous system via a mechanism of action distinguishable from most marketed analgesics. Effirma is especially attractive because it operates through non-opiate pain pathways, exhibits no known abuse potential, and lacks withdrawal effects. In addition, no tolerance to its antinocioceptive effects has been observed. One common link between neuroprotection, nocioception and Effirma may be the N-methyl-D-aspartic acid glutamate system, a major receptor subtype for the excitotoxic neurotransmitter, glutamate. Effirma has strong inhibitory actions on N-methyl-D-aspartic acid-mediated neurotransmission. Flupirtine was originally developed by Asta Medica (subsequently acquired by Meda) and has been approved and is marketed by Meda in Europe since 1984, as well as other countries, for the treatment of pain. It has never been approved by the FDA for any indication.
| 10 |
According to Meda’s 2012 Year-End Report filed in February 2013, Meda has received the go-ahead from the FDA to conduct a Phase II proof of concept study for the treatment of fibromyalgia. Meda also announced that the randomized, double-blind, placebo and active-controlled study of patients with fibromyalgia will be conducted at 25 clinics in the U.S.
Intellectual Property
Our goal is to (a) obtain, maintain, and enforce patent protection for our products, formulations, processes, methods, and other proprietary technologies, (b) preserve our trade secrets, and (c) operate without infringing on the proprietary rights of other parties, worldwide. We seek, where appropriate, the broadest intellectual property protection for product candidates, proprietary information, and proprietary technology through a combination of contractual arrangements and patents. Below is a description of our license and development agreements relating to our product candidates.
Cedars-Sinai Medical Center License and Options Agreements
On December 5, 2013, through our newly formed, majority owned subsidiary, Synthetic Biomics, Inc. (“SYN Biomics”), we entered into a worldwide exclusive license agreement (the “CSMC License Agreement”) and option agreement (the “CSMC Option Agreement”) with CSMC for the right to develop, manufacture, use, and sell products for the human and veterinary therapeutic and prophylactic treatments for acute and chronic diseases. An investigational team lead by Mark Pimentel, M.D. at CSMC has discovered that these products are intended to target certain pathogenic GI microorganisms that are perceived as an underlying cause of diseases such as C-IBS, obesity and type 2 diabetes. The portfolio of intellectual property licensed to SYN Biomics under the CSMC License Agreement includes nine issued U.S. patents, one issued European patent validated in 18 countries, one issued European patent validated in three countries, two issued Australian patents, and one issued Japanese patent as well as 15 pending U.S. and international patent applications for most fields of use and modalities (subject to certain agreed-upon exceptions); two pending U.S. patent applications are optioned to SYN Biomics under the CSMC Option Agreement.
Under the terms of the CSMC License Agreement we issued 291,569 unregistered shares of our common stock to CSMC, as payment of an initial license fee and patent reimbursement fees of $150,000 and $220,000, respectively. The parties also entered into a Stock Purchase Agreement with respect to such stock issuance and other issuances of unregistered shares of our common stock that may be issued to CSMC in lieu of cash, including license fees, milestone payments, expense reimbursements and option fees under the CSMC License Agreement or CSMC Option Agreement. Any and all such stock issuances by us shall be subject to the prior approval of the NYSE MKT, LLC. The CSMC License Agreement also provides that commencing on the second anniversary of the CSMC License Agreement, SYN Biomics will pay an annual maintenance fee, which payment shall be creditable against annual royalty payments owed under the CSMC License Agreement. In addition to royalty payments which are a percentage of Net Sales (as defined in the CSMC License Agreement) of Licensed Products (as defined in the CSMC License Agreement) and Licensed Technology products (as defined in the CSMC License Agreement), SYN Biomics is obligated to pay CMSC a percentage of any non-royalty sublicense revenues, as well as additional consideration upon the achievement of the following milestones (the first two of which are payable in cash or unregistered shares of our stock at our option): (i) successful Phase I trial completion of the first Licensed Product or first Licensed Technology Product; (ii) successful Phase II trial completion of the first Licensed Product or first Licensed Technology Product; (iii) initiation of Phase III dosing for each additional indication of a Licensed Product or Licensed Technology Product; (iv) successful Phase III trial completion for each Licensed Product and each Licensed Technology Product; (v) the FDA’s acceptance of a New Drug Application for each Licensed Product and each Licensed Technology Product; (vi) regulatory approval for each Licensed Product and each Licensed Technology Product; and (vii) the first commercial sale of each Licensed Product and each Licensed Technology Product. The stock issuances are subject to prior approval of the NYSE MKT, LLC.
Prior to the execution of the CSMC License Agreement, SYN Biomics issued shares of common stock of SYN Biomics to each of CSMC and Mark Pimentel, M.D. (the primary inventor of the intellectual property), representing 11.5% and 8.5%, respectively, of the outstanding shares of SYN Biomics (the “SYN Biomics Shares”). The Stock Purchase Agreements for the SYN Biomics Shares provide for certain anti-dilution protection until such time as an aggregate of $3.0 million in proceeds from equity financings are received by SYN Biomics as well as a right, under certain circumstances in the event that the SYN Biomics Shares are not then freely tradeable, and subject to NYSE MKT, LLC approval, as of the 18 and 36 month anniversary date of the effective date of the Stock Purchase Agreements, for each of CSMC and the Dr. Pimentel to exchange up to 50% of their SYN Biomics shares for unregistered shares of our common stock, with the rate of exchange based upon the relative contribution of the valuation of SYN Biomics to the public market valuation of us at the time of each exchange. The Stock Purchase Agreements also provide for tag-along rights in the event of the sale by us of our shares of SYN Biomics.
| 11 |
The CSMC License Agreement terminates: (i) automatically if SYN Biomics enters into a liquidating bankruptcy or other specified bankruptcy event or if the performance of any term, covenant, condition or provision of the CSMC License Agreement will jeopardize the licensure of CMSC, its participation in certain reimbursement programs, its full accreditation by the Joint Commission of Accreditation of Healthcare Organizations or any similar state organizations, its tax exempt status or is deemed illegal; (ii) upon 30 days notice from CMSC if SYN Biomics fails to make a payment or use commercially reasonable efforts to exploit the patent rights; (iii) upon 60 days notice from CMSC if SYN Biomics fails to cure any breach or default of any material obligations under the CSMC License Agreement; or (iv) upon 90 days notice from SYN Biomics if CMCS fails to cure any breach or default of any material obligations under the CSMC License Agreement. SYN Biomics also has the right to terminate the License Agreement without cause upon 6 months notice to CSMC; however, upon such termination, SYN Biomics is obligated to pay a termination fee with the amount of such fee reduced: (i) if such termination occurs after an IND submission to the FDA but prior to completion of a Phase II clinical trial, (ii) reduced further if such termination occurs after completion of Phase II clinical trial but prior to completion of a Phase III clinical trial; and (iii) reduced to zero if such termination occurs after completion of a Phase III clinical trial.
Pursuant to the terms of the CSMC Option Agreement, SYN Biomics has a period of six months to negotiate an exclusive license to develop, manufacture, use, and sell biologic products relating to the prevention, acute treatment and chronic treatment of irritable bowel syndrome or other indications utilized or derived from certain optioned patent applications, pending completion of certain limited testing of technology embodied in the patent applications. Under terms of the CSMC Option Agreement we issued 43,342 shares of our unregistered stock to CSMC, as payment of a non-refundable option fee of $55,000. In addition, SYN Biomics has the right to extend the option period for an additional six months, for an additional non-refundable extension fee of $25,000, payable in unregistered shares of our common stock having a market value of 110% of such amount, subject to approval of NYSE MKT, LLC, or in cash. At any time during the 6 or 12 month option period (if so extended) SYN Biomics has the right to exercise the option and negotiate an exclusive license to the optioned patent applications, which shall provide for: (i) a $50,000 license issue fee plus reimbursement of patent expenses incurred by CSMC prior to the exclusive license, payable to CSMC in unregistered shares of our stock having a market value of 110% of such amount, subject to approval of the NYSE MKT, LLC, or in cash, (ii) the same milestone payments, royalties and sublicense fees as are payable under the CSMC License Agreement dated December 5, 2013 for separately licensed intellectual property, and (iii) such other customary terms and conditions CSMC typically includes in its license agreements.
In collaboration with Intrexon, and partially utilizing the intellectual property optioned and/or licensed from CSMC described in the CSMC Option Agreement, we intend to develop biologic approaches for the prevention, acute and chronic treatment of a subset of IBS pathologies specifically caused by auto-antibodies. During the option period, SYN Biomics, we and Intrexon will seek to create and test a variety of biologic candidates for the treatment of a subset of IBS cases. This biologic program has been selected as the third target under our Second ECC with Intrexon dated August 6, 2012 (see “Infectious Disease Collaboration with Intrexon” below).
The University of Texas at Austin License Agreement and Sponsored Research Agreement
On December 19, 2012, we entered into a Patent License Agreement (the “Texas License Agreement”) with The University of Texas at Austin (the “University”) for the exclusive license of the right to use, develop, manufacture, market and commercialize certain research and patents related to pertussis antibodies developed in the lab of Dr. Jennifer A. Maynard, Assistant Professor of Chemical Engineering. The Texas License Agreement provides that the University is entitled to payment of past patent expenses, an annual payment of $50,000 per year commencing on the effective date through December 31, 2014 and a $25,000 payment on December 31, 2015 and milestone payments of $50,000 upon commencement of Phase I Clinical Trials, $100,000 upon commencement of Phase III Clinical Trials, $250,000 upon NDA submission in the United States, $100,000 upon European Medicines Agency approval and $100,000 upon regulatory approval in an Asian country. In addition, the University is entitled to a running royalty upon Net Product Sales and Net Service Sales (as defined in the Texas License Agreement). The License Agreement terminates upon the expiration of the patent rights (as defined in the Texas License Agreement); provided, however that the Texas License Agreement is subject to early termination by us in our discretion and by the University for a breach of the Texas License Agreement by us.
In connection with the Texas License Agreement, we also entered into a Sponsored Research Agreement (the “Sponsored Research Agreement”) with the University pursuant to which the University will perform certain research work related to pertussis under the direction of Dr. Jennifer Maynard and we will obtain certain rights to patents and technology developed during the course of such research. All inventions conceived during such research shall be subject to the Texas License Agreement. The Sponsored Research Agreement may be renewed annually, in our sole discretion, after the first year for two additional one year terms with a fixed fee for the first year of $303,287 and for the second and third years, if renewed, a fixed fee of $316,438 and $328,758 respectively, all payable in quarterly installments. If renewed by us after the first year for the remaining two years, the research shall be performed from the effective date of the Sponsored Research Agreement until December 31, 2015; provided, however, the Sponsored Research Agreement is subject to early termination upon the written agreement of the parties, a default in the material obligations under the Sponsored Research Agreement which remain uncured for sixty days after receipt of notice, automatically upon our bankruptcy or insolvency and by us in our sole discretion at any time after the one year anniversary of the date of execution thereof upon no less than 90 days notice. Upon termination prior to December 31, 2014, we shall only be responsible for payment of expenses that do not exceed the fixed annual amount and are incurred prior to the termination date and non-cancellable expenses committed to be expended by the University prior to the termination date for the lesser of the remainder of their appointment in the case of salaries and December 31, 2014. Upon a termination after December 31, 2014 or due to a breach by the University, we shall only be responsible for all reasonable expenses that do not exceed the fixed annual amount and that are incurred by the University prior to the termination date for services performed prior to the termination date.
| 12 |
Oral Enzyme for C. difficile Program Acquisition Agreement
On November 8, 2012, we entered into an Asset Purchase Agreement (the “Prev Agreement”) with Prev ABR LLC (“Prev”), and subsequently closed the transaction on November 28, 2012. Pursuant to the Prev Agreement we acquired the C. difficile program assets of Prev, including pre-IND package for P3A (SYN-004), Phase I and Phase II clinical data for P1A, manufacturing processes and data, and a portfolio of issued and pending U.S. and international patents intended to support an IND and BLA with the FDA. Pursuant to the Prev Agreement, we paid Prev an initial cash payment of $100,000 upon execution of the Prev Agreement and at closing paid an additional cash payment of $135,000 and issued 625,000 unregistered shares of our common stock to Prev. In addition, upon the achievement of the milestones set forth below, Prev may be entitled to receive additional consideration payable 50% in cash and 50% in our stock, subject to Prev’s option to receive the entire payment in shares of our stock, with the exception of the first milestone payments to be paid in cash: (i) upon commencement of an IND; (ii) upon commencement of a Phase I clinical trial; (iii) upon commencement of a Phase II clinical trial; (iv) upon commencement of a Phase III clinical trial; (v) upon Biologic License Application (BLA) filing in the U.S. and for territories outside of the U.S. (as defined in the Prev Agreement); and (vi) upon BLA approval in the U.S. and upon approval in territories outside the U.S. The future stock issuances are subject to prior approval of the NYSE MKT, LLC. No royalties are payable to Prev under the Prev Agreement.
The Prev Agreement also provides that Prev has a right to the return to it of all assets acquired by us under the Prev Agreement if on or prior to the date that is (i) 30 months after the execution of the Prev Agreement, we have not initiated toxicology studies in non-rodent models or (ii) 36 months have not filed an IND under the program related to the assets and such failure is not due to action or inaction of Prev or breach of its representations or warranties or covenants or if there is a change of control as defined in the Prev Agreement and after such change of control the assets are not further developed; provided however that such 30 and 36 month periods can be extended by us for an additional 12 months upon payment of a cash milestone payment.
Infectious Disease Collaboration with Intrexon
On August 6, 2012, we expanded our relationship with Intrexon and entered into the Second ECC with Intrexon that governs a “channel collaboration” arrangement in which we will use Intrexon’s technology relating to the identification, design and production of human antibodies and DNA vectors for the development and commercialization of a series of monoclonal antibody therapies for the treatment of certain serious infectious diseases the “Program”) for the treatment of eight specific target infectious disease indications (the “Field”). Initially, our development efforts will target three infectious diseases within the Field. Within the first two years of the collaboration, we have the right to exchange our initial three targets on a one-for-one basis with any of the other five targeted infectious diseases in the Field at no additional cost. We also have the option, within such two year period, to choose to develop any or all of the other five target diseases in the Field, upon payment of the additional consideration described below. Such license is exclusive with respect to any clinical development, selling, offering for sale or other commercialization of our products within the Field (“Synthetic Products”), and otherwise is non-exclusive. We may not sublicense the rights described without Intrexon’s written consent. Under the Second ECC, and subject to certain exceptions, we are responsible for, among other things, the performance of the Program including the development, commercialization and manufacturing of products.
Subject to certain expense allocations and other offsets provided in the Second ECC, we will pay Intrexon royalties on annual net sales of the Synthetic Products, calculated on a Synthetic Product-by-Synthetic Product basis. We have likewise agreed to pay Intrexon a percentage of quarterly revenue obtained from a sublicensor in the event of a sublicensing arrangement.
We may voluntarily terminate the Second ECC upon 90 days written notice to Intrexon. Intrexon may also terminate the Second ECC if we elect not to pursue the development of a Program identified by Intrexon that is a “Superior Therapy” as defined in the Second ECC upon 60 days notice unless we remedy the circumstances giving rise to the termination during such notice period. Each party has the right to terminate the agreement upon 60 days notice if the other party commits a material breach of the Second ECC, subject to certain cure periods.
Upon termination of the Second ECC, we may continue to develop and commercialize any Synthetic Product that, at the time of termination satisfies one of the following:
| • | is being commercialized by us, |
| • | has received regulatory approval, |
| • | is a subject of an application for regulatory approval that is pending before the applicable regulatory authority, |
| • | is a subject of at least a Phase II or Phase III clinical trial if such termination is by Intrexon due to a material breach by us of the Second ECC or by us upon 60 days notice after the first 18 months. |
Our obligation to pay the royalties described above with respect to these “retained” products will survive termination of the Second ECC.
On October 16, 2012, we issued 3,552,210 shares of our Common Stock as consideration in connection with the Second ECC and the related Stock Issuance Agreement with Intrexon that we entered into on August 6, 2012 (the “Second Stock Issuance Agreement”).
| 13 |
We also agreed upon the filing of an IND application with the FDA for a Synthetic Product, or alternatively the filing of the first equivalent regulatory filing with a foreign regulatory agency (both as applicable, the “IND Milestone Event”), to pay Intrexon either (i) $2.0 million in cash, or (ii) that number of shares of Common Stock (the “IND Milestone Shares”) having a fair market value equaling $2.0 million where such fair market value is determined using published market data of the share price for Common Stock at the close of market on the business day immediately preceding the date of public announcement of attainment of the IND Milestone Event.
Upon the first to occur of either first commercial sale of a Synthetic Product in a country or the granting of the regulatory approval of that Synthetic Product (both as applicable, the “Approval Milestone Event”), we agreed to pay to Intrexon either (i) $3.0 million in cash, or (ii) that number of shares of Common Stock (the “Approval Milestone Shares”) having a fair market value equaling $3.0 million where such fair market value is determined using published market data of the share price for Common Stock at the close of market on the business day immediately preceding the date of public announcement of attainment of the Approval Milestone Event.
We also agreed that we will pay an optional and varying fee whereby we remit a payment, in cash or equity at our sole discretion, to Intrexon calculated as a multiple of the number of targets in excess of three total that we desire to elect (the “Field Expansion Fee”). The Field Expansion Fee must be paid completely in either Common Stock or cash, and will comprise either (i) $2.0 million in cash for each target in excess of three total that we elect, or (ii) that number of shares of Common Stock (the “Field Expansion Fee Shares”) having a fair market value equaling $2.0 million for each such target that we elect in excess of three where such fair market value is determined using published market data establishing the volume-weighted average price for a share of Common Stock over the 30 day period immediately preceding the date of the Field Expansion Fee Closing.
In connection with the transactions contemplated by the Second Stock Issuance Agreement, and pursuant to the First Amendment to Registration Rights Agreement (the “First Amendment to Registration Rights Agreement”) executed and delivered by the parties at the closing, we filed a “resale” registration statement registering the resale of certain of the shares issued under the Second Stock Issuance Agreement.
McLean Hospital Exclusive License Agreement and Meda AB Sublicense Agreement
In 2005, as amended in 2007 and 2010, we entered into an exclusive license agreement with the McLean Hospital, a Harvard University teaching hospital, relating to U.S. Patent No. 6,610,324 and its foreign equivalents, entitled “Flupirtine in the treatment of fibromyalgia and related conditions.” Pursuant to this agreement, we paid an upfront fee and back patent costs of approximately $62,000 and agreed to pay McLean royalties on net sales of oral flupirtine equal to 3.5% of net sales of oral flupirtine for indications covered by the issued patents, reduced to 1.75% if we have a license to other intellectual property covering those indications. In addition, we agreed to use our best efforts to commercialize oral flupirtine for the therapeutic uses embodied in the patent applications. Furthermore, we agreed to reimburse McLean Hospital all future patent costs and pay the following milestone payments: $150,000 upon the initiation of a pivotal Phase III clinical trial of oral flupirtine; $300,000 upon the filing of an NDA for oral flupirtine; and $600,000 upon FDA approval of oral flupirtine. The due diligence requirements of the exclusive license agreement were amended in April of 2010 and further amended by a Non-Disturbance Agreement that was signed with McLean Hospital, Meda and us. The agreement remains in effect until the later of (i) the date all issued patents and filed patent applications within the Patent Rights (as defined in the agreement) expire or are abandoned and (ii) one year after the last Commercial Sale (as defined in the agreement) for which royalty is due or ten years after expiration or abandonment date set forth in clause (i) above, whichever is earlier. We have the right to terminate the agreement at any time upon 90 days notice. In addition, McLean may terminate the agreement (i) upon 10 days notice for nonpayment unless payment is made within such 10 days, (ii) immediately upon written notice if we fail to maintain required insurance or become insolvent, make an assignment for the benefit of creditors or petition for bankruptcy is filed for or against us or (ii) if we, our affiliates or our sublicensees default in performance of their obligations under the agreement and such default is not cured within 60 days.
Effective May 6, 2010, we entered into a Sublicense Agreement with Meda AB of Sweden. Pursuant to this agreement, Meda has been granted an exclusive sublicense to all of our patents covering the use of oral flupirtine for fibromyalgia. These patents have been issued in the U.S. and are pending in Canada and Japan (the “Territory”). This agreement provides that Meda will assume all future development costs for the commercialization of oral flupirtine for fibromyalgia. As consideration for this sublicense, we received an up-front payment of $2.5 million upon execution of this agreement and are entitled to milestone payments of $5.0 million upon filing of an NDA with the FDA for oral flupirtine for fibromyalgia and $10.0 million upon marketing approval. This agreement also provides that we are entitled to receive royalties of 7% of net sales of oral flupirtine approved for the treatment of fibromyalgia covered by issued patent claims in the Territory. Pursuant to the terms of this agreement with our university licensor, we are obligated to share half of the royalties we receive with the university licensor, McLean Hospital, and we were obligated to pay them $375,000 upon receipt of an upfront payment, which we did pay in May 2010 when we received the payment from Meda. The agreement continues in effect country by country until the earlier of the expiration of the Royalty Period (as defined in the agreement) or the termination of the McLean license. Meda has the right to terminate the agreement at any time upon 90 days notice. In addition, a party may terminate the agreement upon 30 days notice if the other party breached material obligations and such breach is not cured within a period of time set forth in the agreement. The parties also have the right to terminate the agreement upon 60 days notice in the event of the filing by a party of a bankruptcy petition, the filing of an involuntary petition not dismissed within 60 days, a party proposes a written agreement of composition or extension of its debt, a party becomes Insolvent (as defined in the agreement), liquidates, dissolves, ceases to conduct business or makes an assignment for the benefit of creditors. Upon a termination, all licenses revert to us.
| 14 |
The Regents of University of California License Agreement
In July 2005, we were granted an exclusive worldwide license agreement with the Regents of the University of California (the “Regents”) relating to issued U.S. Patent Nos. 6,936,599, 8,372,826 and 8,658,627 and pending patent applications covering the uses of the drug candidate Trimesta (oral estriol), which has been subsequently amended. Pursuant to this agreement, we paid an upfront license fee and reimbursed patent expenses totaling approximately $61,000 and agreed to pay a license fee of $25,000 during 2006. We also agreed to pay annual maintenance fees, milestone payments totaling $750,000 that are payable on filing an NDA, and on approval of an NDA with the FDA, an additional $750,000 payable upon the first achievement of $50.0 million in annual sales while covered by a validly issued U.S. patent as well as a 4% royalty on net sales of Trimesta covered by the licensed patents. We may be permitted to partially pay milestone payments in the form of equity. The duration of this agreement is from the effective date of July 11, 2005 until the last-to-expire patent in Regent’s Patent Rights, or until the last patent application licensed under this agreement is abandoned and no patent in Regent’s Patent Rights ever issues. We have the right to terminate this agreement at any time and termination will be effective 90 days after the effective date of the termination notice. The Regents may terminate the agreement with a written notice of default if we violate or fail to perform any material term or covenant of this agreement including failure within three years from the successful completion of the ongoing clinical trial of estriol for relapsing-remitting MS being conducted by Dr. Rhonda Voskuhl as principal investigator, to initiate a Phase III clinical trial, or within 17 years of the effective date of the agreement to complete the commercial sale of a product for human therapeutics for the treatment of autoimmune diseases, including MS. However, we have 60 days after the effective date of the notice of default to repair the default.
Manufacturing
We utilize contract manufacturing firms to produce our investigational products Trimesta and SYN-004 in accordance with “current good manufacturing processes” (cGMP) guidelines outlined by the FDA.
Research and Development
During the years ended December 31, 2013 and 2012, we incurred $6.5 million and $12.3 million, respectively, in research and development expenses.
Goverment Regulation
In the U.S., the formulation, manufacturing, packaging, storing, labeling, promotion, advertising, distribution and sale of our products are subject to regulation by various governmental agencies, including (1) the FDA, (2) the Federal Trade Commission, or FTC, (3) the Consumer Product Safety Commission, or CPSC, (4) the U.S. Department of Agriculture, or USDA. Our proposed activities may also be regulated by various agencies of the states, localities and foreign countries in which our proposed products may be manufactured, distributed and sold. The FDA, in particular, regulates the formulation, manufacture and labeling of over-the-counter, or OTC drugs, prescription drugs, conventional foods, dietary supplements, and cosmetics such as those that we intend to distribute. FDA regulations require us and our suppliers to meet relevant cGMP regulations for the preparation, packing, labeling, and storage of all drugs and foods.
Any products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing FDA regulation, including record-keeping requirements, reporting of adverse experiences, submitting periodic reports, drug sampling and distribution requirements, manufacturing or labeling changes, record-keeping requirements, and compliance with FDA promotion and advertising requirements. Drug manufacturers and their subcontractors are required to register their facilities with the FDA and state agencies, and are subject to periodic unannounced inspections for GMP compliance, imposing procedural and documentation requirements upon us and third-party manufacturers. Failure to comply with these regulations could result, among other things, in suspension of regulatory approval, recalls, suspension of production or injunctions, seizures, or civil or criminal sanctions. We cannot be certain that we or our present or future subcontractors will be able to comply with these regulations.
The FDA regulates prescription drug labeling and promotion activities. The FDA actively enforces regulations prohibiting the marketing of products for unapproved uses. The FDA permits the promotion of drugs for unapproved uses in certain circumstances, subject to stringent requirements. We and our product candidates are subject to a variety of state laws and regulations which may hinder our ability to market our products. Whether or not FDA approval has been obtained, approval by foreign regulatory authorities must be obtained prior to commencing clinical trials, and sales and marketing efforts in those countries. These approval procedures vary in complexity from country to country, and the processes may be longer or shorter than that required for FDA approval. We may incur significant costs to comply with these laws and regulations now or in the future.
The FDA, comparable foreign regulators and state and local pharmacy regulators impose substantial requirements upon clinical development, manufacture and marketing of pharmaceutical products. These and other entities regulate research and development and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising, and promotion of our products. The drug approval process required by the FDA under the Food, Drug, and Cosmetic Act generally involves:
| · | preclinical laboratory and animal tests; |
| · | submission of an IND, prior to commencing human clinical trials; |
| · | adequate and well-controlled human clinical trials to establish safety and efficacy for intended use; |
| · | submission to the FDA of a New Drug Application (NDA) or BLA; and |
| · | FDA review and approval of an NDA or BLA. |
| 15 |
The testing and approval process requires substantial time, effort, and financial resources, and we cannot be certain that any approval will be granted on a timely basis, if at all.
Preclinical tests include laboratory evaluation of the product candidate, its chemistry, formulation and stability, and animal studies to assess potential safety and efficacy. Certain preclinical tests must be conducted in compliance with good laboratory practice regulations. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring them to be replicated. In some cases, long-term preclinical studies are conducted concurrently with clinical studies.
We will submit the preclinical test results, together with manufacturing information and analytical data, to the FDA as part of an IND, which must become effective before we begin human clinical trials. The IND automatically becomes effective 30 days after filing, unless the FDA raises questions about conduct of the trials outlined in the IND and imposes a clinical hold, in which case, the IND sponsor and FDA must resolve the matters before clinical trials can begin. It is possible that our submission may not result in FDA authorization to commence clinical trials.
Clinical trials must be supervised by qualified investigators in accordance with good clinical practice (GCP) regulations, which include informed consent requirements. Each study must be approved and monitored by the appropriate IRBs which are periodically informed of the study’s progress, adverse events and changes in research. Annual updates are submitted to the FDA and more frequently if certain serious adverse events occur.
Human clinical trials of drug candidates typically have three sequential phases that may overlap:
Phase I: The drug is initially tested in healthy human subjects or patients for safety, dosage tolerance, absorption, metabolism, distribution, and excretion.
Phase II: The drug is studied in a limited patient population to identify possible adverse effects and safety risks, determine efficacy for specific diseases and establish dosage tolerance and optimal dosage.
Phase III: When Phase II evaluations demonstrate that a dosage range is effective with an acceptable safety profile, Phase III trials to further evaluate dosage, clinical efficacy and safety, are undertaken in an expanded patient population, often at geographically dispersed sites.
We cannot be certain that we will successfully complete Phase I, Phase II, or Phase III testing of our product candidates within any specific time period, if at all. Furthermore, the FDA, an Investigational Review Board (IRB) or the IND sponsor may suspend clinical trials at any time on various grounds, including a finding that subjects or patients are exposed to unacceptable health risk. Concurrent with these trials and studies, we also develop chemistry and physical characteristics data and finalize a manufacturing process in accordance with cGMP requirements. The manufacturing process must conform to consistency and quality standards, and we must develop methods for testing the quality, purity, and potency of the final products. Appropriate packaging is selected and tested, and chemistry stability studies are conducted to demonstrate that the product does not undergo unacceptable deterioration over its shelf-life. Results of the foregoing are submitted to the FDA as part of a NDA (or BLA in case of biologic products) for marketing and commercial shipment approval. The FDA reviews each NDA or BLA submitted and may request additional information.
Once the FDA accepts the NDA or BLA for filing, it begins its in-depth review. The FDA has substantial discretion in the approval process and may disagree with our interpretation of the data submitted or identify new concerns. The process may be significantly extended by requests for new information or clarification of information already submitted. As part of this review, the FDA may refer the application to an advisory committee, typically a panel of clinicians. Manufacturing establishments often are inspected prior to NDA or BLA approval to assure compliance with GMPs and with manufacturing commitments made in the application.
Submission of an NDA or BLA with clinical data requires payment of a fee. In return, the FDA assigns a goal of ten months for issuing its “complete response,” in which the FDA may approve or deny the NDA or BLA, or require additional clinical data. Even if these data are submitted, the FDA may ultimately decide the NDA or BLA does not satisfy approval criteria. If the FDA approves the NDA or BLA, the product becomes available for marketing. Product approval may be withdrawn if regulatory compliance is not maintained or safety problems occur. The FDA may require post-marketing studies, also known as phase IV studies, as a condition of approval, and requires surveillance programs to monitor approved products that have been commercialized. The agency has the power to require changes in labeling or prohibit further marketing based on the results of post-marketing surveillance.
Satisfaction of these and other regulatory requirements typically takes several years, and the actual time required may vary substantially based upon the type, complexity and novelty of the product. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures on our activities. We cannot be certain that the FDA or other regulatory agencies will approve any of our products on a timely basis, if at all. Success in preclinical or early-stage clinical trials does not assure success in later-stage clinical trials. Data obtained from preclinical and clinical activities are not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. Even if a product receives regulatory approval, the approval may be significantly limited to specific indications or uses.
Even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Delays in obtaining, or failures to obtain regulatory approvals would have a material adverse effect on our business.
| 16 |
The FDA’s policies may change, and additional government regulations may be enacted which could prevent or delay regulatory approval of our potential products. Increased attention to the containment of health care costs worldwide could result in new government regulations materially adverse to our business. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
Competitive Environment
The pharmaceutical and biotechnology industries are characterized by rapidly evolving technology and intense competition. Our competitors include major multi-national pharmaceutical companies and biotechnology companies developing both generic and proprietary therapies to treat serious diseases. Many of these companies are well-established and possess technical, human, research and development, financial, and sales and marketing resources significantly greater than ours. In addition, many of our potential competitors have formed strategic collaborations, partnerships and other types of joint ventures with larger, well established industry competitors that afford these companies potential research and development and commercialization advantages in the therapeutic areas we are currently pursuing.
Academic research centers, governmental agencies and other public and private research organizations are also conducting and financing research activities which may produce products directly competitive to those being developed by us. In addition, many of these competitors may be able to obtain patent protection, obtain FDA and other regulatory approvals and begin commercial sales of their products before us.
Our History
Our predecessor, Sheffield Pharmaceuticals, Inc., was incorporated in 1986, and in 2006 engaged in a reverse merger with Pipex Therapeutics, Inc., a publicly-traded Delaware corporation formed in 2001. After the merger, we changed our name to Pipex Pharmaceuticals, Inc., and in October 2008 we changed our name to Adeona Pharmaceuticals, Inc. On October 15, 2009, we engaged in a merger with a wholly owned subsidiary for the purpose of reincorporating in the State of Nevada. After reprioritizing our focus on the emerging area of synthetic biologics and entering into our first collaboration with Intrexon, we amended our Articles of Incorporation to change our name to Synthetic Biologics, Inc. on February 15, 2012.
| 17 |
Employees
As of March 27, 2014, we employed approximately fourteen individuals, nine of whom are full-time employees. A significant number of our management and professional employees have had prior experience with pharmaceutical, biotechnology or medical product companies. None of our employees are covered by collective bargaining agreements, and management considers relations with our employees to be good.
Properties
Our principal executive offices are located at 155 Gibbs Street, Suite 412, Rockville, Maryland 20850. We also maintain an administrative and finance office located at 617 Detroit St., Ste. 100, Ann Arbor, Michigan 48104.
Available Information
Additional information about Synthetic Biologics is contained at our website,www.syntheticbiologics.com. Information on our website is not incorporated by reference into this report. We make available on our website our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K as soon as reasonably practicable after those reports are filed with the SEC. The following Corporate Governance documents are also posted on our website: Code of Conduct, Code of Ethics for Financial Management and the Charters for the Audit Committee, Compensation Committee and Nominations Committee of the Board of Directors. Our phone number is (734) 332-7800 and our facsimile number is (734) 332-7878.
| 18 |
Item 1A. Risk Factors
Investing in our common stock involves a high degree of risk. In addition to the risks related to our business set forth in this Form 10-K and the other information included and incorporated by reference in this Form 10-K, you should carefully consider the risks described below before purchasing our common stock. Additional risks, uncertainties and other factors not presently known to us or that we currently deem immaterial may also impair our business operations.
RISKS RELATING TO OUR BUSINESS
We will need to raise additional capital to operate our business.
With the exception of the three months ended June 30, 2010, we have experienced significant losses since inception and have a significant accumulated deficit. We expect to incur additional operating losses in the future and therefore our cumulative losses to increase. To date, other than the licensing fee we received from Meda AB for the development and commercialization of Effirma (flupirtine) for fibromyalgia in the U.S., Canada and Japan and limited laboratory revenues from Adeona Clinical Laboratory, which we sold in March 2012, we have generated very minimal revenues. Inasmuch as our sole source of revenue (with the exception of the Meda licensing fee) has been our laboratory revenue and our laboratory was sold in March 2012, we do not expect to derive revenue from any source in the near future until we or our partners successfully commercialize our products. As of December 31, 2013, our accumulated deficit totaled approximately $81.2 million on a consolidated basis. Until such time as we receive approval from the FDA and other regulatory authorities for our product candidates, we will not be permitted to sell our products and therefore will not have product revenues from the sale of products. For the foreseeable future we will have to fund all of our operations and capital expenditures from equity and debt offerings, cash on hand, licensing fees and grants. If our current cash, cash equivalents and short-term investments are not sufficient to sustain our operations, we will need to seek additional sources of financing and such additional financing may not be available on favorable terms, if at all. If we do not succeed in raising additional funds on acceptable terms, we may be unable to complete planned preclinical and clinical trials or obtain approval of our product candidates from the FDA and other regulatory authorities. In addition, we could be forced to delay, discontinue or curtail product development, forego sales and marketing efforts, and forego licensing in attractive business opportunities. Any additional sources of financing will likely involve the issuance of our equity or debt securities, which will have a dilutive effect on our stockholders.
We have not been able to sustain profitability.
Other than with respect to the three months ended June 30, 2010, we have a history of losses and we have incurred, and will continue to incur, substantial losses and negative operating cash flow. Even if we succeed in developing and commercializing one or more of our product candidates, we may still incur substantial losses for the foreseeable future and may not sustain profitability. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will substantially increase in the foreseeable future as we do the following:
| · | continue to undertake preclinical development and clinical trials for our product candidates; |
| · | expand our research activities with Intrexon relating to monoclonal antibodies for infectious diseases; |
| · | seek regulatory approvals for our product candidates; |
| · | develop our product candidates for commercialization; |
| · | implement additional internal systems and infrastructure; |
| · | lease additional or alternative office facilities; and |
| · | hire additional personnel, including members of our management team. |
We may experience negative cash flow for the foreseeable future as we fund our technology development with capital expenditures. As a result, we will need to generate significant revenues in order to achieve and maintain profitability. We may not be able to generate these revenues or achieve profitability in the future. Our failure to achieve or maintain profitability could negatively impact the value of our common stock and underlying securities.
Our research and development efforts may not succeed in developing commercially successful products and technologies, which may limit our ability to achieve profitability.
We must continue to explore opportunities that may lead to new products and technologies. To accomplish this, we must commit substantial efforts, funds, and other resources to research and development. A high rate of failure is inherent in the research and development of new products and technologies. Any such expenditures that we make will be made without any assurance that our efforts will be successful. Failure can occur at any point in the process, including after significant funds have been invested.
Regardless of whether our clinical trials are deemed to be successful, promising new product candidates may fail to reach the market or may only have limited commercial success because of efficacy or safety concerns, failure to achieve positive clinical outcomes, inability to obtain necessary regulatory approvals or satisfy regulatory criteria, limited scope of approved uses, excessive costs to manufacture, the failure to establish or maintain intellectual property rights, or infringement of the intellectual property rights of others. Even if we successfully develop new products or enhancements, they may be quickly rendered obsolete by changing customer preferences, changing industry standards, or competitors' innovations. Innovations may not be quickly accepted in the marketplace because of, among other things, entrenched patterns of clinical practice or uncertainty over third-party reimbursement. We cannot state with certainty when or whether any of our products under development will be launched, whether we will be able to develop, license, or otherwise acquire drug candidates or products, or whether any products will be commercially successful. Failure to launch successful new products or new indications for existing products may cause our products to become obsolete, which may limit our ability to achieve profitability.
| 19 |
The technology on which our channel partnering arrangement with Intrexon is based on early stage technology.
On August 8, 2012, we announced an exclusive channel collaboration with Intrexon relating to the design, production, testing and commercialization of monoclonal antibodies for the treatment of certain infectious diseases. Although monoclonal antibody therapeutics are well established in the biotechnology and pharmaceutical sectors, their use for the treatment of infectious disease is extremely limited. In order for monoclonal antibodies to be effective for infectious diseases, they must not only properly target the organism of interest (or its toxins), but may also need to overcome defenses and forms of resistance of such organisms. To accomplish this may require the use of more than one specific monoclonal antibody, and mixtures of different monoclonal antibodies, which may create additional unforeseen complications, including increased manufacturing complexity and expense. In order to be competitive, monoclonal antibodies will be required to be produced at a low enough cost of goods in order to be profitably marketed. We have very limited development and manufacturing experience in the field of monoclonal antibodies and infectious disease. We cannot assure that any monoclonal antibody candidates will provide satisfactoryin vitro andin vivo nonclinical results sufficient to warrant the expense of cGMP manufacture and clinical testing in human clinical trials.
We may not generate additional revenue from our relationships with our corporate collaborators.
On May 6, 2010, we entered into a sublicense agreement with Meda AB whereby we may receive milestone payments totaling $17.5 million (including an upfront payment of $2.5 million that has already been received), plus royalties on our flupirtine program. There can be no assurance that Meda AB will successfully develop flupirtine for fibromyalgia in the U.S., Canada or Japan that would allow us to receive such additional $15.0 million in milestone payments and royalties on sales in connection with such agreement. The successful achievement of the various milestones set forth in the sublicense agreement is not within our control and we will be dependent upon Meda AB for achievement of such milestones. According to Meda’s 2012 Year-End Report filed in February 2013, Meda has received the go-ahead from the FDA to conduct a Phase II proof of concept study for the treatment of fibromyalgia. There can be no assurance that Meda will initiate or successfully complete such planned study.
We have experienced several management changes.
We have had significant changes in management in the past few years. Jeffrey Riley was appointed Chief Executive Officer and President on February 3, 2012. Effective February 6, 2012, C. Evan Ballantyne was appointed Chief Financial Officer. James S. Kuo, M.D., served as Chief Executive Officer and President from February 6, 2010 until February 3, 2012. Changes in our key positions, as well as additions of new personnel and departures of existing personnel, can be disruptive, might lead to additional departures of existing personnel and could have a material adverse effect on our business, operating results, financial results and internal controls over financial reporting.
We may not be able to retain rights licensed to us by others to commercialize key products and may not be able to establish or maintain the relationships we need to develop, manufacture, and market our products.
In addition to our own patent applications, we also currently rely on licensing agreements with third party patent holders/licensors for our products. We have an exclusive license agreement with the McLean Hospital relating to the use of flupirtine to treat fibromyalgia which was sublicensed to Meda AB, an exclusive license agreement with the Regents of the University of California relating to our Trimesta technology, and an exclusive license agreement with CSMC relating to our C-IBS program. Each of these agreements requires us or our sublicensee to use our best efforts to commercialize each of the technologies as well as meet certain diligence requirements and timelines in order to keep the license agreement in effect. In the event we or our sublicensee are not able to meet our diligence requirements, we may not be able to retain the rights granted under our agreements or renegotiate our arrangement with these institutions on reasonable terms, or at all. Furthermore, we currently have very limited product development capabilities, and limited marketing or sales capabilities. For us to research, develop, and test our product candidates, we would need to contract with outside researchers, in most cases those parties that did the original research and from whom we have licensed the technologies. Our Second ECC with Intrexon provides that Intrexon may terminate the agreement if we do not perform certain specified requirements, including developing therapies considered superior. Our agreement with The University of Texas allows the University to terminate its agreement if we fail to comply with the terms of the agreement. Our agreement with Prev provides Prev with the right to the return of the assets if we do not perform certain requirements. Our agreement with CSMC allows CSMC to terminate its agreement if we fail to comply with the terms of the agreement.
We can give no assurances that any of our issued patents licensed to us or any of our other patent applications will provide us with significant proprietary protection or be of commercial benefit to us. Furthermore, the issuance of a patent is not conclusive as to its validity or enforceability, nor does the issuance of a patent provide the patent holder with freedom to operate without infringing the patent rights of others.
| 20 |
We will incur additional expenses in connection with the Second ECC arrangement with Intrexon and our agreements with Prev and CSMC.
Pursuant to the Second ECC with Intrexon, we are responsible for future research and development expenses of product candidates developed under our collaboration, the effect of which has and will continue to increase the level of our overall research and development expenses going forward. Our agreements with Prev and CSMC require that we initiate certain studies and file or have accepted an NDA within a certain amount of time, each of which are costly and will require additional expenditures. Although all manufacturing, preclinical studies and human clinical trials are expensive and difficult to design and implement, costs associated with the manufacturing, research and development of biologic product candidates are generally greater in comparison to small molecule product candidates. We have added additional personnel and expect to add additional personnel to support the Second ECC with Intrexon, and research and development of our candidates, SYN-004 and SYN-010. In addition, we have commenced manufacturing of SYN-004 material to support our planned preclinical and clinical studies which will require us to incur additional expenses.
Because our biologic programs are relatively new, we have only recently assumed development responsibility and costs associated with such programs. In addition, because development activities in collaboration with Intrexon are determined pursuant to a joint steering committees comprised of Intrexon and ourselves and we have limited experience, future development costs associated with this program may be difficult to anticipate and exceed our expectations. Our actual cash requirements may vary materially from our current expectations for a number of other factors that may include, but are not limited to, unanticipated technical challenges, changes in the focus and direction of our development activities or adjustments necessitated by changes in the competitive landscape in which we operate. If we are unable to continue to financially support such collaborations due to our own working capital constraints, we may be forced to delay our activities. If we are unable to obtain additional financing on terms acceptable to us or at all, we may be forced to seek licensing partners or discontinue development.
Developments by competitors may render our products or technologies obsolete or non-competitive.
Companies that currently sell or are developing both generic and proprietary products to treat multiple sclerosis include: Abbott Biotherapeutics Corporation, Bayer Health Care, Biogen Idec, Genzyme, GlaxoSmithKline Pharmaceuticals, Merck & Co., Pfizer, Novartis, Sanofi, Teva Pharmaceuticals, Momenta/Sandoz, Mylan and Synthon. Companies that currently sell or are developing proprietary products for the prevention and treatment of serious infections and other diseases include: Actelion, Astra-Zeneca, Basilea, Cubist, Forest Laboratories, GlaxoSmithKline Pharmaceuticals, Ironwood Pharmaceuticals, Merck & Co., Merus, Pfizer, Symphogen, Takeda Pharmceuticals and The Medicines Company. Many of our competitors have significant financial and human resources. The infectious disease market is highly competitive with many generic and proprietary intravenous and oral formulations available to physicians and their patients. For our monoclonal antibodies, we currently do not expect to be able to deliver our infectious disease candidates via the oral route and may thus be limited to the in-patient and/or acute treatment setting. In addition, academic research centers may develop technologies that compete with our Trimesta, flupirtine and SYN-004 technologies. Should clinicians or regulatory authorities view alternative therapeutic regiments as more effective than our products, this might delay or prevent us from obtaining regulatory approval for our products, or it might prevent us from obtaining favorable reimbursement rates from payers, such as Medicare, Medicaid, hospitals and private insurers.
We operate in a highly competitive environment.
The pharmaceutical and biotechnology industries, including the monoclonal antibody industry, are characterized by rapidly evolving technology and intense competition. Our competitors include major multi-national pharmaceutical companies and biotechnology companies developing both generic and proprietary therapies to treat serious diseases. Many of these companies are well-established and possess technical, human, research and development, financial, and sales and marketing resources significantly greater than ours. In addition, many of our potential competitors have formed strategic collaborations, partnerships and other types of joint ventures with larger, well established industry competitors that afford these companies potential research and development and commercialization advantages in the therapeutic areas we are currently pursuing.
Academic research centers, governmental agencies and other public and private research organizations are also conducting and financing research activities which may produce products directly competitive to those being developed by us. In addition, many of these competitors may be able to obtain patent protection, obtain FDA and other regulatory approvals and begin commercial sales of their products before us.
Competitors could develop and/or gain FDA approval of our product candidates for a different indication.
Since we do not have composition of matter patent claims for flupirtine and estriol, others may obtain approvals for other uses of these products that are not covered by our issued or pending patents. For example, the active ingredients in both Effirma (flurpirtine) and Trimesta (oral estriol) have been approved for marketing in overseas countries for different uses. Other companies, including the original developers or licensees or affiliates may seek to develop Effirma or Trimesta or their respective active ingredient(s) for other uses in the U.S. or any country we are seeking approval for. We cannot provide any assurances that any other company may obtain FDA approval for products that contain flupirtine and estriol in various formulations or delivery systems that might adversely affect our ability or the ability of Meda to develop and market these products in the U.S. We are aware that other companies have intellectual property protection using the active ingredients and have conducted clinical trials of flupirtine and estriol for different applications than what we are developing. Many of these companies may have more resources than us. We cannot provide any assurances that our products will be FDA-approved prior to our competitors.
| 21 |
If a product containing our active ingredients is already marketed or if the FDA approves other products containing our active ingredients in the future to treat indications, physicians may elect to prescribe and substitute a competitor’s products to treat the diseases for which we are intending to commercialize; this is commonly referred to as “off-label” use. While under FDA regulations a competitor is not allowed to promote off-label uses of its product, the FDA does not regulate the practice of medicine and, as a result, cannot direct physicians to select certain products for their patients. Consequently, we might be limited in our ability to prevent off-label use of a competitor’s product to treat the diseases we are intending to commercialize, even if we have issued method of use patents for that indication. If we are not able to obtain and enforce our patents, if any, or otherwise receive orphan drug protection, a competitor could develop and commercialize similar products for the same indications that we are pursuing. We cannot provide any assurances that a competitor will not obtain FDA approval for a product that contains the same active ingredients as our products.
We rely on method patents and patent applications and various regulatory exclusivities to protect some of our product candidates and our ability to compete may be limited or eliminated if we are not able to protect our products.
Our competitiveness may be adversely affected if we are unable to protect our proprietary technologies. We do not have composition of matter patents for Trimesta or Effirma, or their respective active ingredients estriol and flupirtine. We rely on issued patent and pending patent applications for use of Trimesta to treat MS (issued U.S. Patent Nos. 6,936,599, 8,372,826 and 8,658,627) and various other therapeutic indications, which have been exclusively licensed to us. We have exclusively licensed an issued patent for the treatment of fibromyalgia with flupirtine, which we have sublicensed to Meda AB.
The patent positions of pharmaceutical companies are uncertain and may involve complex legal and factual questions. We may incur significant expenses in protecting our intellectual property and defending or assessing claims with respect to intellectual property owned by others. Any patent or other infringement litigation by or against us could cause us to incur significant expenses and divert the attention of our management.
Others may file patent applications or obtain patents on similar technologies or compounds that compete with our products. We cannot predict how broad the claims in any such patents or applications will be, and whether they will be allowed. Once claims have been issued, we cannot predict how they will be construed or enforced. We may infringe intellectual property rights of others without being aware of it. If another party claims we are infringing their technology, we could have to defend an expensive and time consuming lawsuit, pay a large sum if we are found to be infringing, or be prohibited from selling or licensing our products unless we obtain a license or redesign our product, which may not be possible.
We also rely on trade secrets and proprietary know-how to develop and maintain our competitive position. Some of our current or former employees, consultants, scientific advisors, current or prospective corporate collaborators, may unintentionally or willfully disclose our confidential information to competitors or use our proprietary technology for their own benefit. Furthermore, enforcing a claim alleging the infringement of our trade secrets would be expensive and difficult to prove, making the outcome uncertain. Our competitors may also independently develop similar knowledge, methods, and know-how or gain access to our proprietary information through some other means.
We may fail to retain or recruit necessary personnel, and we may be unable to secure the services of consultants.
As of March 27, 2014, we employed approximately fourteen individuals, nine of whom are full-time employees. We have also engaged clinical consultants to advise us on our clinical programs and regulatory consultants to advise us on our dealings with the FDA and other foreign regulatory authorities. We have been and will be required to retain additional consultants and employees in order to fulfill our obligations under the Second ECC with Intrexon, our development obligations under our agreement with Prev and our agreement with CSMC. Our future performance will depend in part on our ability to successfully integrate newly hired officers into our management team and our ability to develop an effective working relationship among senior management.
Certain of our directors, scientific advisors, and consultants serve as officers, directors, scientific advisors, or consultants of other biopharmaceutical or biotechnology companies that might be developing competitive products to ours. Other than corporate opportunities, none of our directors are obligated under any agreement or understanding with us to make any additional products or technologies available to us. Similarly, we can give no assurances, and we do not expect and stockholders should not expect, that any biomedical or pharmaceutical product or technology identified by any of our directors or affiliates in the future would be made available to us other than corporate opportunities. We can give no assurances that any such other companies will not have interests that are in conflict with our interests.
Losing key personnel or failing to recruit necessary additional personnel would impede our ability to attain our development objectives. There is intense competition for qualified personnel in the drug and biologic development areas, and we may not be able to attract and retain the qualified personnel we would need to develop our business.
| 22 |
We rely on independent organizations, advisors, and consultants to perform certain services for us, including handling substantially all aspects of regulatory approval, clinical management, manufacturing, marketing, and sales. We expect that this will continue to be the case. Such services may not always be available to us on a timely basis when we need them.
If the parties we depend on for supplying substance raw materials for our product candidates and certain manufacturing-related services do not timely supply these products and services in sufficient quality or quantity, it may delay or impair our ability to develop, manufacture and market our product candidates.
We rely on suppliers for the substance raw materials of our product candidates and third parties for certain manufacturing-related services to produce material that meets appropriate content, quality and stability standards and use in clinical trials of our products and, after approval, for commercial distribution. With the exception of FUJIFILM Diosynth Biotechnologies UK Limited, our manufacturer for SYN-004, we have not yet established cGMP manufacturers for our biologic and drug candidates. To succeed, clinical trials require adequate supplies of study material, which may be difficult or uneconomical to procure or manufacture and there can be no assurance that we will successfully procure such study material. We and our suppliers and vendors may not be able to (i) produce our study material to appropriate standards for use in clinical studies, (ii) perform under any definitive manufacturing, supply or service agreements with us, or (iii) remain in business for a sufficient time to successfully produce and market our product candidates. If we do not maintain important manufacturing and service relationships, we may fail to find a replacement supplier or required vendor or develop our own manufacturing capabilities which could delay or impair our ability to obtain regulatory approval for our products and substantially increase our costs or deplete profit margins, if any. If we do find replacement manufacturers and vendors, we may not be able to enter into agreements with them on terms and conditions favorable to us and, there could be a substantial delay before a new facility could be qualified and registered with the FDA and foreign regulatory authorities.
Clinical trials are very expensive, time-consuming, and difficult to design and implement.
Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time-consuming. We estimate that clinical trials of our product candidates would take at least several years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. Commencement and completion of clinical trials may be delayed by several factors, including:
| · | obtaining an IND application with the FDA to commence clinical trials; |
| · | identification of, and acceptable arrangements with, one or more clinical sites; |
| · | obtaining IRB approval to commence clinical trials; |
| · | unforeseen safety issues; |
| · | determination of dosing; |
| · | lack of effectiveness during clinical trials; |
| · | slower than expected rates of patient recruitment; |
| · | inability to monitor patients adequately during or after treatment; |
| · | inability or unwillingness of medical investigators to follow our clinical protocols; and |
| · | unwillingness of the FDA or IRBs to permit the clinical trials to be initiated. |
In addition, we, IRBs or the FDA may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if IRBs or the FDA finds deficiencies in our submissions or conduct of our trials.
The results of our clinical trials may not support our product candidate claims and the results of preclinical studies and completed clinical trials are not necessarily predictive of future results.
To date, long-term safety and efficacy have not yet been demonstrated in clinical trials for any of our product candidates. Favorable results in our early studies or trials may not be repeated in later studies or trials. Even if our clinical trials are initiated and completed as planned, we cannot be certain that the results will support our product candidate claims. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. Furthermore, success of our predecessor with P1A, does not ensure success of SYN-004. We cannot be sure that the results of later clinical trials would replicate the results of prior clinical trials and preclinical testing nor that they would satisfy the requirements of the FDA or other regulatory agencies. Clinical trials may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses. Any such failure could cause us or our sublicensee to abandon a product candidate and might delay development of other product candidates. Preclinical and clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization. Any delay in, or termination of, our clinical trials would delay our obtaining FDA approval for the affected product candidate and, ultimately, our ability to commercialize that product candidate.
| 23 |
We depend on third parties, including researchers and sublicensees, who are not under our control.
Since we have in-licensed some of our product candidates, have sublicensed a product candidate and have collaboration agreements for the development of other product candidates, we depend upon our sublicensee and independent investigators and scientific collaborators, such as universities and medical institutions or private physician scientists, to advise us and to conduct our preclinical and clinical trials under agreements with us. These collaborators are not our employees and we cannot control the amount or timing of resources that they devote to our programs or the timing of their procurement of clinical-trial data or their compliance with applicable regulatory guidelines. Should any of these scientific inventors/advisors or those of our sublicensee become disabled or die unexpectedly, or should they fail to comply with applicable regulatory guidelines, we or our sublicensee may be forced to scale back or terminate development of that program. They may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking those programs ourselves. Failing to devote sufficient time and resources to our drug-development programs, or substandard performance and failure to comply with regulatory guidelines, could result in delay of any FDA applications and our commercialization of the drug candidate involved.
These collaborators may also have relationships with other commercial entities, some of which may compete with us. Our collaborators assisting our competitors could harm our competitive position. For example, we are highly dependent on scientific collaborators for our Trimesta development program and our C-IBS development program. Specifically, all of the clinical trials have been conducted under investigator-sponsored IND applications, not corporate-sponsored INDs. We have sometimes experienced difficulty in collecting data generated from these investigator-sponsored clinical trials for our programs. We cannot provide any assurances that we will not experience any additional delays in the future.
We are also highly dependent on government and private grants to fund certain of our clinical trials for our product candidates. For example, Trimesta (oral estriol) has received grants totaling over $8.0 million, predominantly from the Southern California Chapter of the NMSS and the National Institutes of Health which funds a majority of the ongoing clinical trial in relapsing-remitting MS for women. Although we believe that the grant funding received to date is sufficient to complete the current clinical trial based upon current cost estimates, if we experience any additional unanticipated costs or require further clinical trials, and our scientific collaborator is unable to maintain or receive additional grants, we might be forced to scale back or terminate the development of this product candidate. We will also need to cross reference our IND with the inventor/IND holder for this program should we elect to file our own corporate IND for our Trimesta (oral estriol) program. The on-going and future development and commercialization of Effirma (flupirtine) for fibromyalgia is the responsibility of Meda AB and no assurance can be given that Meda will gain the FDA’s acceptance of the NDA or obtain NDA approval from the FDA of flupirtine for fibromyalgia.
With respect to our product candidates in collaboration with Intrexon, we are dependent upon Intrexon’s synthetic biology facilities and capabilities as we have no such facilities and capabilities of our own. We are also reliant on their vectors, monoclonal antibody discovery, production cell line development and know-how. If any of the foregoing were to become inaccessible or terminated, it would be difficult for us to develop and commercialize our synthetic biologic product candidates.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights, as well as costs associated with lawsuits.
If any other person files patent applications, or is issued patents, claiming technology also claimed by us in pending applications, we may be required to participate in interference proceedings in the U.S. Patent and Trademark Office to determine priority of invention. We, or our licensors, may also need to participate in interference proceedings involving our issued patents and pending applications of another entity.
The intellectual property environment in the monoclonal antibody field is particularly complex, constantly evolving and highly fragmented. We have not conducted freedom-to-use patent searches on all aspects of our product candidates or potential product candidates, and we may be unaware of relevant patents and patent applications of third parties. In addition, the freedom-to-use patent searches that have been conducted may not have identified all relevant issued patents or pending patents. We cannot provide assurance that our proposed products in this area will not ultimately be held to infringe one or more valid claims owned by third parties which may exist or come to exist in the future or that in such case we will be able to obtain a license from such parties on acceptable terms.
We cannot guarantee that the practice of our technologies will not conflict with the rights of others. In some foreign jurisdictions, we could become involved in opposition proceedings, either by opposing the validity of another’s foreign patent or by persons opposing the validity of our foreign patents.
We may also face frivolous litigation or lawsuits from various competitors or from litigious securities attorneys. The cost to us of any litigation or other proceeding relating to these areas, even if deemed frivolous or resolved in our favor, could be substantial and could distract management from our business. Uncertainties resulting from initiation and continuation of any litigation could have a material adverse effect on our ability to continue our operations.
If we infringe the rights of others we could be prevented from selling products or forced to pay damages.
If our products, methods, processes, and other technologies are found to infringe the proprietary rights of other parties, we could be required to pay damages, or we may be required to cease using the technology or to license rights from the prevailing party. Any prevailing party may be unwilling to offer us a license on commercially acceptable terms.
| 24 |
RISKS RELATING TO OUR STOCK
We will seek to raise additional funds in the future, which may be dilutive to stockholders or impose operational restrictions.
We expect to seek to raise additional capital in the future to help fund development of our proposed products. If we raise additional capital through the issuance of equity or of debt securities, the percentage ownership of our current stockholders will be reduced. We may also enter into strategic transactions, issue equity as part of license issue fees to our licensors, compensate consultants or settle outstanding payables using equity that may be dilutive. Our stockholders may experience additional dilution in net book value per share and any additional equity securities may have rights, preferences and privileges senior to those of the holders of our common stock.
We are substantially controlled by our current officers, directors, and principal stockholders.
Currently, our directors, executive officers, and principal stockholders beneficially own a substantial number of shares of our common stock. As a result, they will be able to exert substantial influence over the election of our Board of Directors and the vote on issues submitted to our stockholders. Our executive officers and directors beneficially owned approximately 1.7 million shares of our common stock, including stock options and warrants exercisable within 60 days of March 27, 2014. Through Intrexon Corporation and NRM VII Holdings I, LLC, Randal J. Kirk indirectly, beneficially owns approximately 12.3 million shares of our common stock. Steve H. Kanzer, directly, and through Accredited Venture Capital, LLC, indirectly, beneficially owns approximately 7.8 million shares of our common stock. Our executive officers, directors and principal stockholders together beneficially owned approximately 21.7 million shares of our common stock, including the stock options and warrants exercisable within 60 days of March 27, 2014. Because our common stock has from time to time been “thinly traded”, the sale of a substantial number of shares by our executive officers, directors and principal stockholders would have an adverse effect on the market for our stock and our share price.
Our shares of common stock are from time to time thinly traded, so stockholders may be unable to sell at or near ask prices or at all if they need to sell shares to raise money or otherwise desire to liquidate their shares.
Our common stock has from time to time been “thinly-traded,” meaning that the number of persons interested in purchasing our common stock at or near ask prices at any given time may be relatively small or non-existent. This situation is attributable to a number of factors, including the fact that we are a small company that is relatively unknown to stock analysts, stock brokers, institutional investors and others in the investment community that generate or influence sales volume, and that even if we came to the attention of such persons, they tend to be risk-averse and would be reluctant to follow an unproven company such as ours or purchase or recommend the purchase of our shares until such time as we became more seasoned and viable. As a consequence, there may be periods of several days or more when trading activity in our shares is minimal or non-existent, as compared to a seasoned issuer which has a large and steady volume of trading activity that will generally support continuous sales without an adverse effect on share price. We cannot give stockholders any assurance that a broader or more active public trading market for our common shares will develop or be sustained, or that current trading levels will be sustained.
We cannot assure you that the common stock will be liquid or that it will remain listed on the NYSE MKT.
We cannot assure you that we will be able to maintain the continued listing standards of the NYSE MKT. The NYSE MKT requires companies to meet certain continued listing criteria including certain minimum stockholders' equity as outlined in the NYSE MKT Exchange Company Guide. We may not be able to maintain such minimum stockholders' equity and/or issue additional equity securities in exchange for cash or other assets, if available, to maintain certain minimum stockholders' equity required by the NYSE MKT. If we are delisted from the NYSE MKT then our common stock will trade, if at all, only on the over-the-counter market, such as the OTC Bulletin Board securities market, and then only if one or more registered broker-dealer market makers comply with quotation requirements. In addition, delisting of our common stock could depress our stock price, substantially limit liquidity of our common stock and materially adversely affect our ability to raise capital on terms acceptable to us, or at all. Delisting from the NYSE MKT could also have other negative results, including the potential loss of confidence by suppliers and employees, the loss of institutional investor interest and fewer business development opportunities. In order to remain listed on NYSE MKT, we are required to maintain a minimum stockholders’ equity of $6.0 million.
There may be issuances of shares of preferred stock in the future.
Although we currently do not have preferred shares outstanding, the Board of Directors could authorize the issuance of a series of preferred stock that would grant holders preferred rights to our assets upon liquidation, the right to receive dividends before dividends would be declared to common stockholders, and the right to the redemption of such shares, possibly together with a premium, prior to the redemption of the common stock. To the extent that we do issue preferred stock, the rights of holders of common stock could be impaired thereby, including without limitation, with respect to liquidation.
Our failure to fulfill all of our registration requirements may cause us to suffer liquidated damages, which may be very costly.
Pursuant to the terms of the registration rights agreement that we entered into with Intrexon and an affiliated entity, we are required to file a registration statement with respect to securities issued and to maintain the effectiveness of such registration statement. The failure to do so could result in the payment of damages by us. There can be no assurance that we will be able to maintain the effectiveness of any registration statement, and therefore there can be no assurance that we will not incur damages with respect to such agreements.
| 25 |
Stockholders may experience future dilution as a result of future equity offerings.
In order to raise additional capital, we may in the future offer additional shares of our common stock or other securities convertible into or exchangeable for our common stock at prices that may not be the same as the price per share in this offering. We may sell shares or other securities in any other offering at a price per share that is less than the price per share paid by existing stockholders, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders. The price per share at which we sell additional shares of our common stock, or securities convertible or exchangeable into common stock, in future transactions may be higher or lower than the price per share paid by existing stockholders.
We do not intend to pay dividends in the foreseeable future.
We have never paid cash dividends on our common stock. We currently intend to retain our future earnings, if any, to finance the operation and growth of our business and currently do not plan to pay any cash dividends in the foreseeable future. If we do not pay dividends, our common stock may be less valuable because a return on your investment will only occur if the market price of our common stock price appreciates.
Resales of our common stock in the public market by our stockholders may cause the market price of our common stock to fall.
We may issue common stock from time to time in connection with future offerings. Any issuance from time to time of new shares of our common stock, or our ability to issue shares of common stock in future offerings, could result in resales of our common stock by our current stockholders concerned about the potential dilution of their holdings. In turn, these resales could have the effect of depressing the market price for our common stock.
RISKS RELATED TO OUR INDUSTRY
If we do not obtain the necessary regulatory approvals in the U.S. and/or other countries we will not be able to sell our product candidates.
We cannot assure you that we will receive the approvals necessary to commercialize any of our product candidates or any product candidates we acquire or develop in the future. We will need FDA approval to commercialize our product candidates in the U.S. and approvals from the FDA-equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. We will be required to conduct clinical trials that will be costly. We cannot predict whether our clinical trials will demonstrate the safety and efficacy of our product candidates or if the results of any clinical trials will be sufficient to advance to the next phase of development or for approval from the FDA. We also cannot predict whether our research and clinical approaches will result in drugs or therapeutics that the FDA considers safe and effective for the proposed indications. The FDA has substantial discretion in the drug approval process. The approval process may be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals may prevent or delay commercialization of, and our ability to derive product revenues from, our product candidates; and diminish any competitive advantages that we may otherwise believe that we hold.
Even if we comply with all FDA requests, the FDA may ultimately reject one or more of our NDAs or BLAs. We may never obtain regulatory clearance for any of our product candidates. Failure to obtain FDA approval of any of our product candidates will severely undermine our business by leaving us without a saleable product, and therefore without any source of revenues, until another product candidate can be developed. There is no guarantee that we will ever be able to develop or acquire another product candidate.
In addition, the FDA may require us to conduct additional pre-clinical and clinical testing or to perform post-marketing studies, as a condition to granting marketing approval of a product. The results generated after approval could result in loss of marketing approval, changes in product labeling, and/or new or increased concerns about the side effects or efficacy of a product. The FDA has significant post-market authority, including the explicit authority to require post-market studies and clinical trials, labeling changes based on new safety information, and compliance with FDA-approved risk evaluation and mitigation strategies. The FDA’s exercise of its authority has in some cases resulted, and in the future could result, in delays or increased costs during product development, clinical trials and regulatory review, increased costs to comply with additional post-approval regulatory requirements and potential restrictions on sales of approved products.
In foreign jurisdictions, we must also receive approval from the appropriate regulatory authorities before we can commercialize any products, which can be time consuming and costly. Foreign regulatory approval processes generally include all of the risks associated with the FDA approval procedures described above. There can be no assurance that we will receive the approvals necessary to commercialize our product candidate for sale outside the United States.
If the FDA approves any of our product candidates, the labeling, manufacturing, packaging, adverse event reporting, storage, advertising, promotion and record-keeping for our products will be subject to ongoing FDA requirements and continued regulatory oversight and review. Our drug manufacturers and subcontractors that we retain will comply with FDA and other regulations. We may also be subject to additional FDA post-marketing obligations. If we are not able to maintain regulatory compliance, we may not be permitted to market our product candidates and/or may be subject to product recalls, seizures, suspension of regulatory approval, suspension of production, injunctions or civil or criminal sanctions. The subsequent discovery of previously unknown problems with any marketed product, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the product, and could include withdrawal of the product from the market.
| 26 |
We do not have a guarantee of patent term restoration and marketing exclusivity of the ingredients for our drugs even if we are granted FDA approval of our products.
The U.S. Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman) permits the FDA to approve Abbreviated New Drug Applications (ANDAs) for generic versions of innovator drugs, as well as NDAs with less original clinical data, and provides patent restoration and exclusivity protections to innovator drug manufacturers. The ANDA process permits competitor companies to obtain marketing approval for drugs with the same active ingredient and for the same uses as innovator drugs, but does not require the conduct and submission of clinical studies demonstrating safety and efficacy. As a result, a competitor could copy any of our drugs and only need to submit data demonstrating that the copy is bioequivalent to gain marketing approval from the FDA. Hatch-Waxman requires a competitor that submits an ANDA, or otherwise relies on safety and efficacy data for one of our drugs, to notify us and/or our business partners of potential infringement of our patent rights. We and/or our business partners may sue the company for patent infringement, which would result in a 30-month stay of approval of the competitor’s application. The discovery, trial and appeals process in such suits can take several years. If the litigation is resolved in favor of the generic applicant or the challenged patent expires during the 30-month period, the stay is lifted and the FDA may approve the application. Hatch-Waxman also allows competitors to market copies of innovator products by submitting significantly less clinical data outside the ANDA context. Such applications, known as “505(b)(2) NDAs” or “paper NDAs,” may rely on clinical investigations not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use and are subject to the ANDA notification procedures described above.
The law also permits restoration of a portion of a product’s patent term that is lost during clinical development and NDA review, and provides statutory protection, known as exclusivity, against FDA approval or acceptance of certain competitor applications. Restoration can return up to five years of patent term for a patent covering a new product or its use to compensate for time lost during product development and regulatory review. The restoration period is generally one-half the time between the effective date of an IND and submission of an NDA, plus the time between NDA submission and its approval (subject to the five-year limit), and no extension can extend total patent life beyond 14 years after the drug approval date. Applications for patent term extension are subject to U.S. Patent and Trademark Office (USPTO) approval, in conjunction with FDA. Approval of these applications takes at least nine months, and there can be no guarantee that it will be given at all.
Hatch-Waxman also provides for differing periods of statutory protection for new drugs approved under an NDA. Among the types of exclusivity are those for a “new molecular entity” and those for a new formulation or indication for a previously-approved drug. If granted, marketing exclusivity for the types of products that we are developing, which include only drugs with innovative changes to previously-approved products using the same active ingredient, would prohibit the FDA from approving an ANDA or 505(b)(2) NDA relying on our safety and efficacy data for three years. This three-year exclusivity, however, covers only the innovation associated with the original NDA. It does not prohibit the FDA from approving applications for drugs with the same active ingredient but without our new innovative change. These marketing exclusivity protections do not prohibit the FDA from approving a full NDA, even if it contains the innovative change.
| 27 |
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
We currently rent approximately 1,200 square feet of office space in Rockville, Maryland for monthly rent of $3,522, and we rent approximately 1,600 square feet of office space in Ann Arbor, Michigan for monthly rent of $2,767. We believe our current offices will be adequate for the foreseeable future.
Item 3. Legal Proceedings
None.
Item 4. Mine Safety Disclosures
Not applicable.
| 28 |
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchase of Equity Securities
Our common stock has traded on the NYSE MKT, LLC under the symbol “SYN” since February 16, 2012. Prior to this time, our common stock traded under the symbol “AEN” since October 16, 2008. The following table states the range of the high and low sales prices of our common stock for each of the calendar quarters during the years ended December 31, 2013 and December 31, 2012. These quotations represent inter-dealer prices, without retail mark-up, markdown, or commission, and may not represent actual transactions. The last price of our common stock as reported on the NYSE MKT on March 27, 2014 was $2.60 per share. As of March 27, 2014, there were approximately 350 stockholders of record of our common stock. This number does not include beneficial owners from whom shares are held by nominees in street name.
| High | Low | |||||||
| YEAR ENDED DECEMBER 31, 2013 | ||||||||
| Fourth quarter | $ | 1.85 | $ | 1.00 | ||||
| Third quarter | $ | 1.71 | $ | 1.42 | ||||
| Second quarter | $ | 1.83 | $ | 1.38 | ||||
| First quarter | $ | 2.00 | $ | 1.65 | ||||
| YEAR ENDED DECEMBER 31, 2012 | ||||||||
| Fourth quarter | $ | 2.43 | $ | 1.60 | ||||
| Third quarter | $ | 2.41 | $ | 1.80 | ||||
| Second quarter | $ | 2.25 | $ | 1.51 | ||||
| First quarter | $ | 2.80 | $ | 1.27 | ||||
Dividend Policy
We have never paid any cash dividends on our common stock to date, and do not anticipate paying such cash dividends in the foreseeable future. Whether we declare and pay dividends is determined by our Board of Directors at their discretion, subject to certain limitations imposed under Nevada corporate law. The timing, amount and form of dividends, if any, will depend on, among other things, our results of operations, financial condition, cash requirements and other factors deemed relevant by our Board of Directors.
Equity Compensation Plan Information
See Item 12 - Executive compensation for equity compensation plan information.
Recent Sales of Unregistered Securities
In December 2013, under the terms of the Cedars-Sinai Medical Center (CSMC) License Agreement, we issued 291,569 shares of our common stock to CSMC as payment of an initial license fee and patent reimbursement fees. In addition, in December 2013, under the terms of the CMSC Option Agreement, we issued 43,342 shares of our common stock to CMSC as payment of a non-refundable option fee in accordance with the terms of the CMSC Option Agreement. These issuances were not registered under the Securities Act of 1933, as amended (the “Securities Act’) and were deemed to be exempt from registration under the Securities Act in reliance upon Section 4(a)(2) as transactions by an issuer not involving any public offering. The recipient of the securities represented their its intention to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof, and appropriate legends were placed upon the stock certificates issued in these transactions.
All other sales of unregistered securities have been previously reported.
Item 6. Selected Financial Data
Not applicable because we are a smaller reporting company.
| 29 |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion of our financial condition and results of operations should be read in conjunction with the audited financial statements and notes thereto for the year ended December 31, 2013 found in this report. In addition to historical information, the following discussion contains forward-looking statements that involve risks, uncertainties and assumptions. Where possible, we have tried to identify these forward looking statements by using words such as “anticipate,” “believe,” “intends,” or similar expressions. Our actual results could differ materially from those anticipated by the forward-looking statements due to important factors and risks including, but not limited to, those set forth under “Risk Factors” in Part I, Item 1A of this Report.
Overview
We are a biotechnology company focused on the development of novel anti-infective biologic and drug candidates targeting specific pathogens that cause serious infections and other diseases. We are developing an oral treatment to reduce the impact of methane producing organisms on constipation-predominant irritable bowel syndrome (C-IBS), an oral biologic to protect the gastrointestinal (GI) microflora from the effects of intravenous (IV) antibiotics for the prevention ofClostridium difficile (C. diff) infection, a series of monoclonal antibodies (mAbs) for the treatment of Pertussis andAcinetobacter infections, and a biologic targeted at the prevention and treatment of a root cause of a subset of IBS. In addition, we have two legacy programs. We are developing an oral estriol drug for the treatment of relapsing-remitting multiple sclerosis (MS) and cognitive dysfunction in MS. We have also partnered the development of a treatment for fibromyalgia.
Product Pipeline:
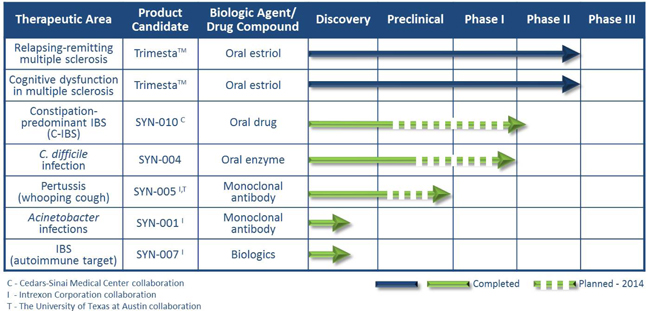
Summary of Pathogen-Specific Anti-Infective Biologic and Drug Programs:
| · | C-IBS: In December 2013, through our majority-owned subsidiary, Synthetic Biomics, Inc., we entered into a worldwide exclusive license agreement with Cedars-Sinai Medical Center (CSMC) for the right to develop products for therapeutic and prophylactic treatments for acute and chronic diseases. An investigational team led by Mark Pimentel, M.D. at CSMC has discovered that these products are intended to target the production of methane gas by certain pathogenic gastrointestinal (GI) microorganisms that are perceived as the underlying cause of gas, pain and constipation associated with C-IBS, as well as diseases such as obesity and type 2 diabetes. Initially we will focus on the development of an oral treatment to reduce the impact of methane producing organisms on C-IBS. We intend to initiatein vivo/pharmacokinetic/pharmacodynamic studies in the first half of 2014, and to initiate a Phase II clinical trial during the second half of 2014 under an Investigational New Drug application (IND). |
| · | C. diff infections: We are in preclinical development of a novel second-generation oral enzyme drug candidate, SYN-004, for co-administration with commonly used IV antibiotics intended to prevent the development of severe effects of C. diff infections.C. diff infections are a leading cause of hospital acquired infections (HAIs), that generally occur secondary to treatment with IV antibiotics. Designed to be given orally to protect the gut while certain IV beta-lactam antibiotics (penicillins and cephalosporins) fight the primary infection, SYN-004 is believed to have a similar profile to its first-generation predecessor, which demonstrated favorable protection of the gut flora (microbiome) during treatment with certain penicillins, with the potentially added ability to act against a broader spectrum of IV beta-lactam antibiotics. Beta-lactam antibiotics are a mainstay in hospital infection management and include the commonly used penicillin and cephalosporin classes of antibiotics. Approximately 14.4 million patients are administered "SYN-004 target" IV beta-lactam antibiotics annually, representing an estimated target market for SYN-004 of 117.6 million beta-lactam doses purchased by U.S. hospitals. The addressable market for SYN-004 is significant. Currently there are no approved treatments designed to protect the microbiome from the damaging effects of IV antibiotics. This worldwide opportunity could represent a multi-billion dollar market.* We intend to initiate Phase Ia and Ib clinical trials during the second half of 2014. |
| 30 |
| * | This information is an estimate derived from the use of information under license from the following IMS Health Incorporated information service: CDM Hospital database for full year 2012. IMS expressly reserves all rights, including rights of copying, distribution and republication. |
| • | Pertussis: In December 2012, in collaboration with Intrexon Corporation (NYSE: XON) (Intrexon), we initiated development of a mAb therapy for the treatment of Pertussis infections, more commonly known as whooping cough. We are developing a mAb therapy, SYN-005, designed to target and neutralize the pertussis toxin, in order to reduce the mortality rate in infants and shorten the duration of chronic cough in afflicted adults. To further the development of this potential therapy for Pertussis, we entered into an agreement with The University of Texas at Austin to license the rights to certain research and pending patents related to pertussis antibodies. According to the World Health Organization, each year, B. pertussis infection causes an estimated 300,000 deaths worldwide, primarily among young, unvaccinated infants. As part of our IND-enabling studies, we initiated a pilot large animal study in the first quarter of 2014 utilizing the antibody combination in a non-human primate model. This model, in addition to the murine model, is supportive of the development of a pertussis therapeutic. We are currently planning a confirmatory follow-up large animal study, and expect to report topline results during the second quarter of 2014. |
| • | Acinetobacter infections: In September 2012, in collaboration with Intrexon, we initiated efforts to develop a mAb therapy for the treatment of Acinetobacter infections. Many strains of Acinetobacter are multidrug-resistant and pose an increasing global threat to hospitalized patients, wounded military personnel and those affected by natural disasters. A treatment for Acinetobacter infections represents a billion dollar market opportunity. The generation of a panel of antibodies is ongoing. |
| • | IBS: In December 2013, in collaboration with Intrexon, and partially utilizing the intellectual property optioned from CSMC, we intend to develop biologic approaches targeted at the prevention, and acute and chronic treatment of a subset of IBS pathologies specifically caused by auto-antibodies. |
Summary of Multiple Sclerosis Program:
| • | Trimesta™ (oral estriol) is being developed as an oral once-daily treatment for relapsing-remitting MS in women. Patient enrollment is complete in this two-year, randomized, double-blind, placebo-controlled Phase II clinical trial being conducted at 15 centers in the United States. The primary endpoint is relapse rate at two years. This investigator-initiated trial evaluating our drug candidate, Trimesta™, is supported by grants awarded to the University of California, Los Angeles (UCLA) exceeding $8.0 million, which should be sufficient to fund the trial through completion. Annual worldwide sales of current MS therapies are estimated at $14.1 billion. Top-line results are scheduled to be presented at the American Academy of Neurology’s Annual Meeting in April 2014 by the lead principal investigator, Rhonda Voskuhl, MD. |
| • | Trimesta is also being developed for the treatment of cognitive dysfunction in female MS patients. This 12-month randomized, double-blind, placebo-controlled Phase II clinical trial is being conducted at UCLA. The primary endpoint is the effect on cognitive function as assessed by Paced Auditory Serial Addition Test (PASAT). Patient enrollment is ongoing. The majority of the costs of this trial are being funded by grants from foundations and charitable organizations and we have pledged approximately $500,000 to UCLA to partially fund this trial payable over three years. An estimated 50-65% of MS patients are expected to develop disabilities due to cognitive dysfunction and there is currently no approved treatment. |
| 31 |
Summary of Fibromyalgia Program:
| • | EffirmaTM (flupirtine) is being developed for the treatment of fibromyalgia by Meda AB (Meda), a multi-billion dollar international pharmaceutical company. On May 6, 2010, we entered into a sublicense agreement with Meda covering all of our patents’ rights on the use of flupirtine for fibromyalgia in the United States, Canada and Japan. The sublicense agreement provides that all ongoing and future development costs are to be borne by Meda and we are entitled to receive certain payments if milestones are achieved and royalties on sales. According to Meda’s 2012 Year-End Report filed in February 2013, Meda has received the go-ahead from the United States Food and Drug Administration (FDA) to conduct a Phase II proof of concept study for the treatment of fibromyalgia. Meda also announced that the randomized, double-blind, placebo and active-controlled study of patients with fibromyalgia will be conducted at 25 clinics in the United States Based on an estimated annual price of $1,200 per fibromyalgia patient, we estimate that the total market potential in the United States is $6.0 billion. |
Recent Developments
On December 11, 2013, we completed a firm commitment underwritten public offering of 13,225,000 shares of our common stock at a closing price of $1.00 per share for gross proceeds of $13.2 million. We paid direct offering costs of $1.0 million.
On December 5, 2013, through our newly formed, majority owned subsidiary, Synthetic Biomics, Inc. (“SYN Biomics”), we entered into a worldwide exclusive license agreement (the “CSMC License Agreement”) and option agreement (the “CSMC Option Agreement”) with CSMC for the right to develop, manufacture, use, and sell products for the human and veterinary therapeutic and prophylactic treatments for acute and chronic diseases. An investigational team lead by Mark Pimentel, M.D. at CSMC has discovered that these products are intended to target certain pathogenic GI microorganisms that are perceived as an underlying cause of diseases such as C-IBS, obesity and type 2 diabetes. The portfolio of intellectual property licensed to SYN Biomics under the License Agreement includes nine issued U.S. patents, one issued European patent validated in 18 countries, one issued European patent validated in three countries, two issued Australian patents, and one issued Japanese patent as well as 13 pending U.S. and international patent applications for most fields of use and modalities (subject to certain agreed-upon exceptions); two pending U.S. patent applications are optioned to SYN Biomics under the Option Agreement.”
Under the CSMC License Agreement we issued 291,569 unregistered shares of our common stock to CSMC, as payment of an initial license fee and patent reimbursement fees of $150,000 and $220,000, respectively. The parties also entered into a Stock Purchase Agreement with respect to such stock issuance and other issuances of our unregistered shares of common stock that may be issued to CSMC in lieu of cash, including license fees, milestone payments expense reimbursements and options fees under the CSMC License Agreement or CSMC Option Agreement. Any and all such stock issuances by us shall be subject to the prior approval of the NYSE MKT, LLC. The CSMC License Agreement also provides that commencing on the second anniversary of the CSMC License Agreement, SYN Biomics will pay an annual maintenance fee, which payment shall be creditable against annual royalty payments owed under the CSMC License Agreement. In addition to royalty payments which are a percentage of Net Sales (as defined in the CSMC License Agreement) of Licensed Products (as defined in the CSMC License Agreement) and Licensed Technology products (as defined in the CSMC License Agreement), SYN Biomics is obligated to pay CMSC a percentage of any non-royalty sublicense revenues as well as additional consideration upon the achievement of the following milestones (the first two of which are payable in cash or our unregistered shares of stock at our option): (i) successful Phase I trial completion of the first Licensed Product or first Licensed Technology Product; (ii) successful Phase II trial completion of the first Licensed Product or first Licensed Technology Product; (iii) initiation of Phase III dosing for each additional indication of a Licensed Product or Licensed Technology Product; (iv) successful Phase III trial completion for each Licensed Product and each Licensed Technology Product; (v) the FDA’s acceptance of a New Drug Application for each Licensed Product and each Licensed Technology Product; (vi) regulatory approval for each Licensed Product and each Licensed Technology Product; and (vii) the first commercial sale of each Licensed Product and each Licensed Technology Product. The stock issuances are subject to prior approval of the NYSE MKT, LLC. The CSMC License Agreement automatically terminates upon the occurrence of certain events and SYN Biomics has the right to terminate the CSMC License Agreement without cause, upon six months notice to CSMC however, upon such termination, SYN Biomics is obligated to pay a termination fee with the amount of such fee reduced: (i) if such termination occurs after an IND submission to the FDA but prior to completion of a Phase II clinical trial; (ii) reduced further if such termination is after completion of Phase II clinical trial but prior to completion of a Phase III clinical trial; and (iii) reduced to zero if such termination occurs after completion of a Phase III clinical trial.
Prior to the execution of the License Agreement, we issued shares of common stock of SYN Biomics to each of CSMC and Mark Pimentel, M.D. (the primary inventor of the intellectual property), representing 11.5% and 8.5%, respectively, of the outstanding shares of SYN Biomics (the “SYN Biomics Shares”). The Stock Purchase Agreements for the SYN Biomics Shares provide for certain anti-dilution protection until such time as an aggregate of $3.0 million in proceeds from equity financings are received by SYN Biomics as well as a right, under certain circumstances in the event that the SYN Biomics Shares are not then freely tradeable, and subject to NYSE MKT, LLC approval, as of the 18 and 36 month anniversary date of the effective date of the Stock Purchase Agreements, for each of CSMC and the Dr. Pimentel to exchange up to 50% of their SYN Biomics shares for unregistered shares of our common stock, with the rate of exchange based upon the relative contribution of the valuation of SYN Biomics to our public market valuation at the time of each exchange. The Stock Purchase Agreements also provide for tag-along rights in the event of the sale by us of our shares of SYN Biomics.
| 32 |
Pursuant to the terms of the CSMC Option Agreement, SYN Biomics has a period of six months to negotiate an exclusive license to develop, manufacture, use, and sell biologic products relating to the prevention, acute treatment and chronic treatment of IBS or other indications utilized or derived from certain optioned patent applications pending completion of certain limited testing of technology embodied in the patents applications. Under terms of the CSMC Option Agreement we issued 43,342 shares of our unregistered stock to CSMC, as payment of a non-refundable option fee of $55,000. In addition, SYN Biomics has the right to extend the option period for an additional six months, for an additional non-refundable extension fee of $25,000, payable in our unregistered shares of common stock having a market value of 110% of such amount, subject to approval of NYSE MKT, LLC, or in cash. At any time during the 6 or 12 month option period (if so extended) SYN Biomics has the right to exercise the option and negotiate an exclusive license to be optioned patent applications, which shall provide for (i) a $50,000 license issue fee plus reimbursement of patent expenses incurred by CSMC prior to the exclusive license payable to CSMC in our unregistered shares of stock having a market value of 110% of such amount, subject to approval of the NYSE MKT, LLC, or in cash, (ii) the same milestone payments, royalties and sublicense fees as are payable under the CSMC License Agreement, and (iii) such other customary terms and conditions CSMC typically includes in its license agreements.
In collaboration with Intrexon, and partially utilizing the intellectual property optioned or licensed from CSMC described in the CSMC Option Agreement, we and SYN Biomics intend to develop biologic approaches for the prevention, and acute and chronic treatment of a subset of IBS pathologies specifically caused by auto-antibodies. During the option period, we, SYN Biomics and Intrexon will seek to create and test a variety of biologic candidates for the treatment of IBS. This biologic program has been selected as the third target under our Exclusive Channel Collaboration Agreement with Intrexon dated August 6, 2012.
On July 3, 2013, we entered into a Controlled Equity OfferingSM Sales Agreement with Cantor Fitzgerald & Co. pursuant to which we may offer and sell shares of our common stock in an at-the-market public offering (the “ATM”) for up to $15.0 million of shares of our common stock from time to time through Cantor Fitzgerald & Co., acting as agent. As of the date of this filing, we have not sold any shares under the ATM. We amended the Controlled Equity Offering SM Sales Agreement on December 10, 2013 to limit our ability to sell shares of our common stock under such agreement to the lesser of $15.0 million or the amount that we can sell under General Instruction I.B.6 of Form S-3, if still applicable, after this offering. We will not use the ATM unless and until we file an updated prospectus supplement reflecting the number or dollar amount of shares which we may sell under the ATM after taking into account the foregoing amendment, but only if such amount is less than $15.0 million.
| 33 |
Since our inception in January 2001, our efforts and resources have been focused primarily on acquiring and developing our product candidates, our clinical trials, raising capital and recruiting personnel. As of June 30, 2010, we emerged from the development stage after entering into a sublicense agreement with Meda AB and receiving an up-front payment of $2.5 million. We consider this sublicense agreement to be an indication that we commenced our principal operations.
To date, we have financed our operations primarily through public and private sales of our common stock, and we expect to continue to seek to obtain the required capital in a similar manner. We have incurred an accumulated deficit of $81.2 million through December 31, 2013. We cannot provide any assurance that we will be able to achieve profitability on a sustained basis, if at all, obtain the required funding, obtain the required regulatory approvals, or complete additional corporate partnering or acquisition transactions.
Critical Accounting Policies
The preparation of our consolidated financial statements in accordance with U.S. generally accepted accounting principles (GAAP) requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, net revenues and expenses, and related disclosures. We believe our estimates and assumptions are reasonable; however, actual results and the timing of the recognition of such amounts could differ from these estimates.
There are accounting policies that we believe are significant to the presentation of our consolidated financial statements. The most significant accounting policies relate to stock-based compensation, revenue recognition and accounts receivable.
Stock-Based Compensation
Calculating stock-based compensation expense requires the input of highly subjective assumptions. We apply the Black-Scholes option pricing model to determine the fair value of our stock options. Inherent in this model are assumptions related to expected stock-price volatility, option life, risk-free interest rate and dividend yield. We estimate the volatility of our common stock at the date of grant based on historical volatility. We estimate the expected life of our option using the contractual term of the option. The risk-free interest rate is based on the U.S. Treasury zero-coupon yield curve on the grant date for a maturity similar to the expected life of the options. The dividend rate is based on our historical rate, which we anticipate to remain at zero. The assumptions used in calculating the fair value of stock options represent our best estimates, however these estimates involve inherent uncertainties and the application of management judgment. As a result, if factors change and different assumptions are used, the stock-based compensation expense could be materially different in the future. In addition, we are required to estimate the expected forfeiture rate and only recognize expense for those stock options expected to vest over the service period.
| 34 |
Revenue Recognition
We record revenue when all of the following have occurred: (1) persuasive evidence of an arrangement exists, (2) the service is completed without further obligation, (3) the sales price to the customer is fixed or determinable, and (4) collectability is reasonably assured. We recognize milestone payments or upfront payments that have no contingencies as revenue when payment is received.
License Revenues
Our licensing agreements may contain multiple elements, such as non-refundable up-front fees, payments related to the achievement of particular milestones and royalties. Fees associated with substantive at risk performance-based milestones are recognized as revenue upon completion of the scientific or regulatory event specified in the agreement. When we have substantive continuing performance obligations under an arrangement, revenue is recognized over the performance period of the obligations using a time-based proportional performance approach. Under the time-based method, revenue is recognized over the arrangement’s estimated performance period based on the elapsed time compared to the total estimated performance period. Revenue recognized at any point in time is limited to the amount of non-contingent payments received or due. When we have no substantive continuing performance obligations under an arrangement, we recognize revenue as the related fees become due.
Revenues from royalties on third-party sales of licensed technologies are generally recognized in accordance with the contract terms when the royalties can be reliably determined and collectability is reasonably assured.
Research and Development Costs
We expense research and development costs associated with developmental products not yet approved by the FDA to research and development expense as incurred. Research and development costs consist primarily of license fees (including upfront payments), milestone payments, manufacturing costs, salaries, stock-based compensation and related personnel costs, fees paid to consultants and outside service providers for laboratory development, legal expenses resulting from intellectual property prosecution and other expenses relating to the design, development, testing and enhancement of our product candidates.
Results of Operations
Year Ended December 31, 2013 and 2012
General and Administrative Expenses
General and administrative expenses increased to $5.8 million for the year ended December 31, 2013, from $5.0 million for the year ended December 31, 2012. This increase of 16% is primarily the result of bad debt expense of $763,000 associated with the determination that the note receivable and interest receivable from the sale of Adeona Clinical Laboratory is uncollectible. See Note 3 - Discontinued Operations of Adeona Clinical Laboratory and Note Receivable. The charge relating to stock-based compensation expense was $1.3 million for the year ended December 31, 2013, compared to $1.5 million for the year ended December 31, 2012.
Research and Development Expenses
Research and development expenses decreased to $6.5 million for the year ended December 31, 2013, from $12.3 million for the year ended December 31, 2012. This decrease of 47% is primarily the result of recording the fair value ($7.8 million) of the common stock issued to Intrexon as consideration for the Exclusive Channel Collaboration Agreement and the fair value ($1.2 million) of the common stock issued for the acquisition of the C. diff program assets of Prev ABR LLC for the year ended December 31, 2012. These were non-cash charges. The decrease in research and development costs for the year ended December 31, 2013 was off-set by increases in additional employee costs and increases in program costs associated with our infectious disease programs. The charge relating to share-based compensation expense was $375,000 for the year ended December 31, 2013, compared to $400,000 for the year ended December 31, 2012.
Other Income
Other income was $21,000 for the year ended December 31, 2013, compared to other income of $15,000 for the year ended December 31, 2012.
| 35 |
Loss from Continuing Operations
Our loss from continuing operations for the year ended December 31, 2013, was $12.3 million, or $0.27 per common share, compared to $17.3 million, or $0.50 per common share for the year ended December 31, 2012.
Income (Loss) from Discontinued Operations
There was no income or loss from discontinued operations for the year ended December 31, 2013. Our income from discontinued operations was $216,000, or $0.01 per common share for the year ended December 31, 2012. On March 8, 2012, we entered into a Membership Interest Purchase Agreement, and certain related agreements, pursuant to which we sold all of our interest in the Lab to Hartlab, LLC. This resulted in the classification of the Lab as discontinued operations. See Note 3 - Discontinued Operations of Adeona Clinical Laboratory and Note Receivable for summarized statement of operations data for the years ended December 31, 2013 and 2012.
Liquidity and Capital Resources
We have financed our operations since inception primarily through proceeds from equity financings, corporate partnering license fees, laboratory revenues and miscellaneous equipment sales.
On December 11, 2013, we completed a firm commitment underwritten public offering of 13,225,000 shares of our common stock at a closing price of $1.00 for gross proceeds of $13.2 million. We paid direct offering costs of $1.0 million.
Our cash totaled $14.6 million at December 31, 2013, an increase of $4.6 million from December 31, 2012. During the year ended December 31, 2013, the primary sources of cash were net proceeds from the issuances of common stock in a firm commitment underwritten public offering of $12.2 million and stock option exercises of $231,000. The primary use of cash during the year ended December 31, 2013 was for working capital requirements.
Our cash totaled $10.0 million at December 31, 2012, an increase of $3.3 million from December 31, 2011. During the year ended December 31, 2012, the primary sources of cash were net proceeds from the issuance of common stock in a private placement financing of $10.2 million and stock option exercises of $127,000 and warrant exercises of $2.0 million. The primary use of cash during the year ended December 31, 2012 was for working capital requirements.
As of March 27, 2014, our cash balance was approximately $11.3 million.
Our continued operations will primarily depend on whether we are able to generate revenues and profits through partnerships, joint ventures and/or raise additional funds through various potential sources, such as license fees from a potential corporate partner, equity and debt financing. Such additional funds may not become available on acceptable terms and there can be no assurance that any additional funding that we do obtain will be sufficient to meet our needs in the long term. We will continue to fund operations from cash on hand and through the similar sources of capital previously described. We can give no assurances that any additional capital that we are able to obtain will be sufficient to meet our needs.
Current and Future Financing Needs
We have incurred an accumulated deficit of $81.2 million through December 31, 2013. With the exception of the quarter ended June 30, 2010, we have incurred negative cash flow from operations since we started our business. We have spent, and expect to continue to spend, substantial amounts in connection with implementing our business strategy, including our planned product development efforts, our clinical trials, and our research and discovery efforts.
Based on our current plans, we believe that our cash will be sufficient to enable us to meet our planned operating needs for at least the next 12 months.
However, the actual amount of funds we will need to operate is subject to many factors, some of which are beyond our control. These factors include the following:
| · | the progress of our research activities; |
| · | the number and scope of our research programs; |
| · | the progress of our preclinical and clinical development activities; |
| · | the progress of the development efforts of parties with whom we have entered into research and development agreements; |
| · | our ability to maintain current research and development licensing arrangements and to establish new research and development and licensing arrangements; |
| · | our ability to achieve our milestones under licensing arrangements; |
| · | the costs involved in prosecuting and enforcing patent claims and other intellectual property rights; and |
| · | the costs and timing of regulatory approvals. |
| 36 |
We have based our estimate on assumptions that may prove to be wrong. We may need to obtain additional funds sooner or in greater amounts than we currently anticipate. Potential sources of financing include strategic relationships, public or private sales of our shares or debt and other sources. We may seek to access the public or private equity markets when conditions are favorable due to our long-term capital requirements. We do not have any committed sources of financing at this time, and it is uncertain whether additional funding will be available when we need it on terms that will be acceptable to us, or at all. If we raise funds by selling additional shares of common stock or other securities convertible into common stock, the ownership interest of our existing stockholders will be diluted. If we are not able to obtain financing when needed, we may be unable to carry out our business plan. As a result, we may have to significantly limit our operations and our business, financial condition and results of operations would be materially harmed.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Not applicable because we are a smaller reporting company.
| 37 |
Item 8. Financial Statements and Supplemental Data
| Page | |
| Reports of Independent Registered Public Accounting Firm | 39 |
| Consolidated Balance Sheets | 40 |
| Consolidated Statements of Operations | 41 |
| Consolidated Statements of Equity | 42 |
| Consolidated Statements of Cash Flows | 43 |
| Notes to Consolidated Financial Statements | 44 |
| 38 |
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Synthetic Biologics, Inc.
Rockville, Maryland
We have audited the accompanying consolidated balance sheets of Synthetics Biologics, Inc. as of December 31, 2013 and 2012 and the related consolidated statements of operations, equity, and cash flows for the years then ended. These financial statements are the responsibility of Synthetic Biologics, Inc.’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Synthetic Biologics, Inc. at December 31, 2013 and 2012, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
| /s/ BDO USA, LLP | |
| BDO USA, LLP | |
| Troy, Michigan | |
| March 31, 2014 |
| 39 |
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Balance Sheets
(In thousands except share amounts)
| December 31, 2013 | December 31, 2012 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | 14,625 | $ | 9,954 | ||||
| Prepaid expenses and other current assets | 1,591 | 2,509 | ||||||
| Total Current Assets | 16,216 | 12,463 | ||||||
| Property and equipment, net | 37 | 223 | ||||||
| Long-term note receivable | - | 700 | ||||||
| Deposits and other assets | 4 | 37 | ||||||
| Total Assets | $ | 16,257 | $ | 13,423 | ||||
| Liabilities and Equity | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | 142 | $ | 395 | ||||
| Accrued liabilities | 885 | - | ||||||
| Total Current Liabilities | 1,027 | 395 | ||||||
| Total Liabilities | 1,027 | 395 | ||||||
| Commitments and Contingencies | - | - | ||||||
| Equity: | ||||||||
| Preferred stock, $0.001 par value; 10,000,000 shares authorized, none issued and outstanding | - | - | ||||||
| Common stock, $0.001 par value; 100,000,000 shares authorized, 58,295,808 issued and 58,214,326 outstanding and 44,444,230 issued and 44,362,748 outstanding | 58 | 44 | ||||||
| Additional paid-in capital | 96,430 | 81,925 | ||||||
| Accumulated deficit | (81,258 | ) | (68,941 | ) | ||||
| Total Synthetic Biologics, Inc. and Subsidiaries Equity | 15,230 | 13,028 | ||||||
| Non-controlling interest | - | - | ||||||
| Total Equity | 15,230 | 13,028 | ||||||
| Total Liabilities and Equity | $ | 16,257 | $ | 13,423 | ||||
See accompanying notes to consolidated financial statements
| 40 |
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Statements of Operations
(In thousands, except share and per share amounts)
| For the twelve months ended December 31, | ||||||||
| 2013 | 2012 | |||||||
| Operating Costs and Expenses: | ||||||||
| General and administrative | $ | 5,832 | $ | 5,012 | ||||
| Research and development | 6,507 | 12,287 | ||||||
| Total Operating Costs and Expenses | 12,339 | 17,299 | ||||||
| Loss from Operations | (12,339 | ) | (17,299 | ) | ||||
| Other Income (Expense): | ||||||||
| Interest income | 33 | 33 | ||||||
| Other expense | (12 | ) | (18 | ) | ||||
| Total Other Income | 21 | 15 | ||||||
| Loss from Continuing Operations | (12,318 | ) | (17,284 | ) | ||||
| Income from Discontinued Operations | - | 216 | ||||||
| Net Loss | (12,318 | ) | (17,068 | ) | ||||
| Net Loss Attributable to Non-controlling Interest | (1 | ) | - | |||||
| Net Loss Attributable to Synthetic Biologics, Inc. and Subsidiaries | $ | (12,317 | ) | $ | (17,068 | ) | ||
| Net Income (Loss) Per Share - Basic and Dilutive: | ||||||||
| Continuing operations | $ | (0.27 | ) | $ | (0.50 | ) | ||
| Discontinued operations | - | 0.01 | ||||||
| Net Income (Loss) Per Share Attributable to Synthetic Biologics, Inc. and Subsidiaries | $ | (0.27 | ) | $ | (0.49 | ) | ||
| Weighted average number of shares outstanding during the period - Basic and Dilutive | 45,667,813 | 34,896,592 | ||||||
See accompanying notes to consolidated financial statements
| 41 |
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Statements of Equity
(In thousands, except share amounts)
| Common Stock $0.001 Par Value | ||||||||||||||||||||||||
| Shares | Amount | APIC | Accumulated Deficit | Non-Controlling Interest | Total Equity | |||||||||||||||||||
| Balance at December 31, 2011 | 31,292,520 | $ | 31 | $ | 58,901 | $ | (51,873 | ) | $ | - | $ | 7,059 | ||||||||||||
| Stock-based compensation | - | - | 1,852 | - | - | 1,852 | ||||||||||||||||||
| Issuance of common stock for exclusive channel collaboration agreement | 3,552,210 | 3 | 7,811 | - | - | 7,814 | ||||||||||||||||||
| Issuance of common stock for acquisition of program assets | 625,000 | 1 | 1,168 | - | - | 1,169 | ||||||||||||||||||
| Issuance of common stock for stock options and warrants exercised | 2,143,018 | 2 | 2,080 | - | - | 2,082 | ||||||||||||||||||
| Issuance of common stock, net of issuance costs of $680 | 6,750,000 | 7 | 10,113 | - | - | 10,120 | ||||||||||||||||||
| Net loss | - | - | - | (17,068 | ) | - | (17,068 | ) | ||||||||||||||||
| Balance at December 31, 2012 | 44,362,748 | 44 | 81,925 | (68,941 | ) | - | 13,028 | |||||||||||||||||
| Stock-based compensation | - | - | 1,669 | - | - | 1,669 | ||||||||||||||||||
| Issuance of common stock, net of issuance costs of $1,031 | 13,225,000 | 13 | 12,180 | - | - | 12,193 | ||||||||||||||||||
| Stock issued for exercise of stock options | 291,667 | 1 | 231 | - | - | 232 | ||||||||||||||||||
| Stock issued for license agreement | 334,911 | - | 425 | - | - | 425 | ||||||||||||||||||
| Issuance of stock | - | - | - | - | 1 | 1 | ||||||||||||||||||
| Net loss | - | - | - | (12,317 | ) | (1 | ) | (12,318 | ) | |||||||||||||||
| Balance at December 31, 2013 | 58,214,326 | $ | 58 | $ | 96,430 | $ | (81,258 | ) | $ | - | $ | 15,230 | ||||||||||||
See accompanying notes to consolidated financial statements
| 42 |
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Statements of Cash Flows
(In thousands)
| For twelve months ended December 31, | ||||||||
| 2013 | 2012 | |||||||
| Cash Flows From Operating Activities: | ||||||||
| Net loss | $ | (12,318 | ) | $ | (17,068 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activites: | ||||||||
| Stock-based compensation | 1,669 | 1,851 | ||||||
| Stock issued for exclusive channel collaboration agreement | - | 7,815 | ||||||
| Stock issued for acquisition of program assets | - | 1,169 | ||||||
| Stock issued for license agreement | 425 | - | ||||||
| Depreciation | 43 | 63 | ||||||
| Provision for uncollectible accounts receivable | - | 442 | ||||||
| Write-off of uncollectible note and interest receivable | 763 | - | ||||||
| Loss on sale of equipment | 58 | - | ||||||
| Impairment loss on equipment | 121 | 47 | ||||||
| Gain on sale of discontinued operations | - | (677 | ) | |||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | - | 14 | ||||||
| Prepaid expenses and other current assets | 855 | (2,544 | ) | |||||
| Deposits and other assets | 33 | (6 | ) | |||||
| Accounts payable | (253 | ) | 7 | |||||
| Accrued liabilities | 885 | (29 | ) | |||||
| Net Cash Used In Operating Activities | (7,719 | ) | (8,916 | ) | ||||
| Cash Flows From Investing Activities: | ||||||||
| Purchase of property and equipment | (36 | ) | (10 | ) | ||||
| Net Cash Used In Investing Activities | (36 | ) | (10 | ) | ||||
| Cash Flows From Financing Activities: | ||||||||
| Proceeds from issuance of common stock for stock option and warrants exercises | 232 | 2,082 | ||||||
| Proceeds from issuance of common stock, net issuance costs $1,031 and $680, respectively | 12,193 | 10,120 | ||||||
| Cash received from issuance of stock to non-controlling interest | 1 | - | ||||||
| Net Cash Provided By Financing Activities | 12,426 | 12,202 | ||||||
| Net increase in cash and cash equivalents | 4,671 | 3,276 | ||||||
| Cash and cash equivalents at beginning of period | 9,954 | 6,678 | ||||||
| Cash and cash equivalents at end of period | $ | 14,625 | $ | 9,954 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for interest | $ | - | $ | - | ||||
| Cash paid for taxes | $ | - | $ | - | ||||
See accompanying notes to consolidated financial statements
| 43 |
Synthetic Biologics, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
December 31, 2013 and 2012
1. Organization and Nature of Operations and Basis of Presentation
Description of Business
Synthetic Biologics, Inc. (the “Company” or “Synthetic Biologics”) is a biotechnology company focused on the development of novel anti-infective biologic and drug candidates targeting specific pathogens that cause serious infections and other diseases. The Company is developing an oral treatment to reduce the impact of methane producing organisms on constipation-predominant irritable bowel syndrome (C-IBS), an oral biologic to protect the GI microflora from the effects of IV antibiotics for the prevention ofC. diff infection, a series of monoclonal antibodies for the treatment of Pertussis andAcinetobacter infections, and a biologic targeted at the prevention and treatment of a root cause of a subset of IBS. In addition, the Company has two legacy programs. The Company is developing an oral estriol drug for the treatment of relapsing-remitting multiple sclerosis (MS) and cognitive dysfunction in MS. The Company has also partnered the development of a treatment for fibromyalgia.
| Therapeutic Area | Product Candidate | Status | ||
| Relapsing-remitting MS | Trimesta (oral estriol) | Topline results to be presented by lead principal investigator at American Academy of Neurology Annual Meeting in April 2014 | ||
| Cognitive dysfunction in MS | Trimesta (oral estriol) | Patient enrollment underway in Phase II clinical trial | ||
| Constipation-predominant irritable bowel syndrome (C-IBS) | SYN-010 (oral compound) | Planning forin vivo studies underway; Intend to initiate Phase II clinical trial during 2nd half of 2014; Collaboration with Cedars-Sinai Medical Center | ||
| C. difficile infection prevention | SYN-004 (oral enzyme) | SYN-004, second generation candidate in preclinical; Intend to initiate Phase Ia and Ib clinical trials during 2nd half of 2014 | ||
| Pertussis | SYN-005 (monoclonal antibody) | Preclinical; Intend to report additional topline results from large animal study during 2nd quarter of 2014; Collaborations with Intrexon and The University of Texas at Austin | ||
| Acinetobacter infection | SYN-001 (monoclonal antibody) | Discovery; Collaboration with Intrexon | ||
| IBS | SYN-007 (biologic) | Discovery; Collaboration with Intrexon | ||
| Fibromyalgia | Effirma (oral flupirtine) | Partnered with Meda AB |
In addition, the Company is seeking development partners for its zinc-based intellectual property and assets including, AEN-100.
Basis of Presentation and Corporate Structure
As of December 31, 2013, the Company had eight subsidiaries, Pipex Therapeutics, Inc. (“Pipex Therapeutics”), Effective Pharmaceuticals, Inc. (“EPI”), Solovax, Inc. (“Solovax”), CD4 Biosciences, Inc. (“CD4”), Epitope Pharmaceuticals, Inc. (“Epitope”), Healthmine, Inc. (“Healthmine”), Putney Drug Corp. (“Putney”) and Synthetic Biomics, Inc. (“Synthetic Biomics”). As of December 31, 2013, Pipex Therapeutics, EPI, Healthmine and Putney are wholly owned, and Solovax, CD4, Epitope and Synthetic Biomics are majority-owned.
| 44 |
For financial reporting purposes, the outstanding common stock of the Company is that of Synthetic Biologics, Inc. All statements of operations, equity and cash flows for each of the entities are presented as consolidated. All subsidiaries were formed under the laws of the State of Delaware on January 8, 2001, except for EPI, which was incorporated in Delaware on December 12, 2000, Epitope which was incorporated in Delaware in January of 2002, Putney which was incorporated in Delaware in November of 2006, Healthmine which was formed in Delaware in December of 2007 and Synthetic Biomics which was incorporated in Nevada in December of 2013.
On March 8, 2012, the Company sold all of its interest in Adeona Clinical Laboratory, LLC (the “Lab”) to Hartlab, LLC, an entity controlled by the Lab’s former owner, in consideration for (i) the immediate assignment of the Lab’s outstanding accounts receivable up through the date of closing, plus (ii) $700,000 payable pursuant to the terms of a two-year non-recourse promissory note secured by all the assets of the Lab. Accordingly, this business has been presented in the consolidated financial statements as discontinued operations. During September 2013, the note receivable and associated interest receivable were deemed uncollectible. Accordingly, the Company recorded bad debt expense of $763,000. This transaction is described in more detail in Note 3 - Discontinued Operations of Adeona Clinical Laboratory.
2. Summary of Significant Accounting Policies
Principles of Consolidation
All inter-company transactions and accounts have been eliminated in consolidation.
Use of Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the U.S.A. requires management to make estimates and assumptions that affect the reported amounts reported in the consolidated financial statements and accompanying notes. Such estimates and assumptions impact, among others, the following: the estimated useful lives for property and equipment, fair value of warrants and stock options granted for services or compensation, respectively, estimates of the probability and potential magnitude of contingent liabilities, and the valuation allowance for deferred tax assets due to continuing and expected future operating losses.
Making estimates requires management to exercise significant judgment. It is at least reasonably possible that the estimate of the effect of a condition, situation or set of circumstances that existed at the date of the consolidated financial statements, which management considered in formulating its estimate could change in the near term due to one or more future confirming events. Accordingly, actual results could differ from those estimates.
Accounts Receivable and Allowance for Doubtful Accounts
Accounts receivable were reported at realizable value, net of allowances for doubtful accounts, which were estimated and recorded in the period the related revenue was recorded. The Company estimated and reviewed the collectability of its receivables based on a number of factors, including the period they were outstanding. Historical collection and payer reimbursement experience was an integral part of the estimation process related to allowances for doubtful accounts associated with Adeona Clinical Laboratory. In addition, the Company regularly assessed the state of its billing operations in order to identify issues, which impacted the collectability of these receivables or reserve estimates. Revisions to the allowances for doubtful accounts estimates were recorded as an adjustment to bad debt expense. Receivables deemed uncollectible were charged against the allowance for doubtful accounts.
Non-controlling Interest
The Company’s non-controlling interest is accounted for under ASC 810,Consolidation(“ASC 810”) and represents the minority shareholder’s ownership interest related to the Company’s subsidiary, Synthetic Biomics, Inc. In accordance with ASC 810, the Company reports its non-controlling interest in subsidiaries as a separate component of equity in the Consolidated Balance Sheets and reports both net loss attributable to the non-controlling interest and net loss attributable to the Company’s common shareholders on the face of the consolidated Statements of Operations. The Company’s equity interest in Synthetic Biomics, Inc. is 80% and the non-controlling stockholder’s interest is 20%. This is reflected in the Consolidated Statements of Equity.
Revenue Recognition
The Company records revenue when all of the following have occurred: (1) persuasive evidence of an arrangement exists, (2) the service is completed without further obligation, (3) the sales price to the customer is fixed or determinable, and (4) collectability is reasonably assured. The Company recognizes milestone payments or upfront payments that have no contingencies as revenue when payment is received. For the year ended December 31, 2013 the Company did not report any revenues. For the year ended December 31, 2012, the Company’s only stream of revenue was laboratory revenue. Laboratory revenues are a component of discontinued operations for the year ended December 31, 2012. See Note 3 - Discontinued Operations of Adeona Clinical Laboratory.
| 45 |
License Revenues
The Company’s licensing agreements may contain multiple elements, such as non-refundable up-front fees, payments related to the achievement of particular milestones and royalties. Fees associated with substantive at risk performance-based milestones are recognized as revenue upon completion of the scientific or regulatory event specified in the agreement. When the Company has substantive continuing performance obligations under an arrangement, revenue is recognized over the performance period of the obligations using a time-based proportional performance approach. Under the time-based method, revenue is recognized over the arrangement’s estimated performance period based on the elapsed time compared to the total estimated performance period. Revenue recognized at any point in time is limited to the amount of non-contingent payments received or due. When the Company has no substantive continuing performance obligations under an arrangement, it recognizes revenue as the related fees become due.
Revenues from royalties on third-party sales of licensed technologies are generally recognized in accordance with the contract terms when the royalties can be reliably determined and collectibility is reasonably assured. To date, the Company has not received any royalty revenues.
In 2010, the Company entered into the Meda Agreement for the development and commercialization of Effirma (oral flupirtine) for fibromyalgia. As consideration for the sublicense, the Company received an up-front payment of $2.5 million upon execution of the Meda Agreement. This payment was recorded as license revenue in 2010. Pursuant to the Company’s license agreement with McLean Hospital, the Company paid 15% of the $2.5 million payment ($375,000), that was netted against the revenues received from Meda AB. The Company is also entitled to additional milestone payments of $5.0 million upon filing of an NDA with the U.S. FDA for oral flupirtine for fibromyalgia and $10.0 million upon marketing approval. The Meda Agreement also provides that the Company is entitled to receive net royalties of 7% of net sales of oral flupirtine approved for the treatment of fibromyalgia covered by issued patent claims in the U.S. and Japan. The Meda Agreement provides that Meda AB will assume all future development costs for the commercialization of oral flupirtine for fibromyalgia. Pursuant to the terms of the Company’s agreement with McLean Hospital, the Company is obligated to pay half of all future royalties the Company receives. Future milestone payments will be recorded as revenue when payment is received as there are no future deliverables, and it is non-refundable.
Laboratory Revenues
The Company primarily recognized revenue for services rendered upon completion of the testing process. Billing for services reimbursed by third-party payers, including Medicare and Medicaid, were recorded as revenues, net of allowances for differences between amounts billed and the estimated receipts from such payers.
The Company maintained a sales allowance to compensate for the difference in its billing practices and insurance company reimbursements. In determining this allowance, the Company looked at several factors, the most significant of which was the average difference between the amount charged and the amount reimbursed by insurance carriers over the prior 12 months, otherwise known as the yearly average adjustment amount. The allowance taken was the averaged yearly average adjustment amount for these prior period multiplied by the period’s actual gross sales to determine the actual sales allowance for each period. See Note 3 - Discontinued Operations of Adeona Clinical Laboratory.
Risks and Uncertainties
The Company's operations could be subject to significant risks and uncertainties including financial, operational and regulatory risks and the potential risk of business failure. The global economic crisis has caused a general tightening in the credit markets, lower levels of liquidity, increases in the rates of default and bankruptcy, and extreme volatility in credit, equity and fixed income markets. These conditions may not only limit our access to capital, but also make it difficult for our customers, our vendors and us to accurately forecast and plan future business activities.
Cash and Cash Equivalents
Cash and cash equivalents include cash and highly liquid short-term investments with original maturities of three months or less.
| 46 |
Property and Equipment
Property and equipment is recorded at cost and depreciated or amortized using the straight-line method over the estimated useful life of the asset or the underlying lease term for leasehold improvements, whichever is shorter. The estimated useful life by asset description is noted in the following table.
| Asset Description | Estimated Useful Life | |
| Office equipment and furniture | 3-5 years | |
| Laboratory equipment | 7-10 years | |
| Manufacturing equipment | 10 years | |
| Leasehold improvements and fixtures | Lesser of estimated useful or life of lease |
Depreciation expense was approximately $43,000 and $63,000 for the years ended December 31, 2013 and 2012, respectively. When assets are disposed of, the cost and accumulated depreciation are removed from the accounts. Repairs and maintenance are charged to expense as incurred.
During 2013 and 2012, the Company reviewed property and equipment for impairment and determined that certain items were impaired due to obsolescence. As a result of this review, the Company recorded an impairment loss of approximately $121,000 and $47,000 for the years ended December 31, 2013 and 2012, respectively.
Long-Lived Assets
The Company reviews its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. If such an event or change in circumstances occurs and potential impairment is indicated because the carrying values exceed the estimated future undiscounted cash flows of the asset, the Company will measure the impairment loss as the amount by which the carrying value of the asset exceeds its fair value.
Net Income (Loss) per Share
Net income (loss) per share is computed by dividing net income (loss) by the weighted average number of common shares outstanding. Diluted income (loss) per share is computed by dividing net income (loss) by the weighted average number of common shares outstanding including the effect of common share equivalents. All common equivalent shares were anti-dilutive, at December 31, 2013 and December 31, 2012, as such there is no separate computation for diluted loss per share. The number of options and warrants for the purchase of common stock, that were excluded from the computations of net loss per common share for the year ended December 31, 2013 were 3,909,580 and 1,632,501, respectively, and for the year ended December 31, 2012 were 4,453,746 and 1,632,501, respectively.
Research and Development Costs
The Company expenses research and development costs associated with developmental products not yet approved by the FDA to research and development expense as incurred. Research and development costs consist primarily of license fees (including upfront payments), milestone payments, manufacturing costs, salaries, stock-based compensation and related personnel costs, fees paid to consultants and outside service providers for laboratory development, legal expenses resulting from intellectual property prosecution and other expenses relating to the design, development, testing and enhancement of the Company’s product candidates.
Fair Value of Financial Instruments
The fair value accounting standards define fair value as the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is determined based upon assumptions that market participants would use in pricing an asset or liability. Fair value measurements are rated on a three-tier hierarchy as follows:
| · | Level 1 inputs: Quoted prices (unadjusted) for identical assets or liabilities in active markets; |
| · | Level 2 inputs: Inputs, other than quoted prices included in Level 1 that are observable either directly or indirectly; and |
| · | Level 3 inputs: Unobservable inputs for which there is little or no market data, which require the reporting entity to develop its own assumptions. |
In many cases, a valuation technique used to measure fair value includes inputs from multiple levels of the fair value hierarchy described above. The lowest level of significant input determines the placement of the entire fair value measurement in the hierarchy.
| 47 |
The carrying amounts of the Company’s short-term financial instruments, including cash and cash equivalents, other current assets, accounts payable and accrued liabilities, approximate fair value due to the relatively short period to maturity for these instruments.
Cash and cash equivalents include money market accounts and mutual funds of $11.0 million and $8.6 million as of December 31, 2013 and December 31, 2012, respectively, that are measured using Level 1 inputs.
Stock-Based Payment Arrangements
Generally, all forms of stock-based payments, including stock option grants, warrants, restricted stock grants and stock appreciation rights are measured at their fair value on the awards’ grant date typically using a Black-Scholes pricing model, based on the estimated number of awards that are ultimately expected to vest. Stock-based compensation awards issued to non-employees for services rendered are recorded at either the fair value of the services rendered or the fair value of the stock-based payment, whichever is more readily determinable and are re-measured over the corresponding vesting period. The expense resulting from stock-based payments is recorded in research and development expense or general and administrative expense in the consolidated statement of operations, depending on the nature of the services provided.
Income Taxes
The Company accounts for income taxes in accordance with accounting guidance now codified as FASB ASC Topic 740-10 which requires that the Company recognize deferred tax liabilities and assets based on the differences between the financial statement carrying amounts and the tax bases of assets and liabilities, using enacted tax rates in effect in the years the differences are expected to reverse. Deferred income tax benefit (expense) results from the change in net deferred tax assets or deferred tax liabilities. A valuation allowance is recorded when it is more likely than not that some or all deferred tax assets will not be realized.
In addition, accounting guidance now codified as FASB ASC Topic 740-10 requires management to assess the need to accrue or disclose uncertain tax positions for proposed potential adjustments from various federal and state authorities who regularly audit the Company in the normal course of business. In making these assessments, management must often analyze complex tax laws of multiple jurisdictions. The Company records the related interest expense and penalties, if any, as tax expense in the tax provision. At December 31, 2013 and 2012, respectively, the Company did not record any liabilities for uncertain tax positions.
Recent Accounting Pronouncements
There were no accounting standards or interpretations issued or recently adopted that are expected to have a material impact on the Company’s financial position, operations, or cash flows.
3. Discontinued Operations of Adeona Clinical Laboratory and Note Receivable
On March 8, 2012, the Company sold all of its interest in Adeona Clinical Laboratory, LLC (the “Lab”) to Hartlab, LLC, an entity controlled by the Lab’s former owner. In connection with the sale of the Lab, the consideration received was (i) the immediate assignment of the Lab’s outstanding accounts receivable up through the date of closing, plus (ii) $700,000 payable pursuant to the terms of a two-year promissory note bearing interest at 5.7% per annum secured by all of the assets of the Lab. The note and all unpaid interest are due on March 1, 2014. During the year ended December 31, 2013, the note receivable and associated interest receivable were deemed uncollectible. Accordingly, the Company recorded bad debt expense of $763,000.
In accordance with ASC Topic 205-20 “Presentation of Financial Statements-Discontinued Operations” (ASC 205-20), the Company determined that all the criteria for reporting a discontinued operation had been met. Accordingly, the Lab has been classified as a discontinued operation and its results of operations, financial position and cash flows are separately reported for all periods presented.
The summarized statement of operations data for Adeona Clinical Laboratory for the years ended December 31, 2013 and December 31, 2012 are as follows (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Laboratory fees, net | $ | - | $ | 115 | ||||
| Operating Costs and Expenses: | ||||||||
| General and administrative | - | 466 | ||||||
| Cost of laboratory services | - | 110 | ||||||
| Total operating costs and expenses | - | 576 | ||||||
| Loss from discontinued operations | - | (461 | ) | |||||
| Other Income: | ||||||||
| Gain on sale of Adeona Clinical Laboratory | - | 677 | ||||||
| Income (loss) from discontinued operations | $ | - | $ | 216 | ||||
| 48 |
4. Selected Balance Sheet Information
Prepaid expenses and other current assets (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Intrexon prepaid research and development expenses - See Note 10 | $ | 1,361 | $ | 2,412 | ||||
| Prepaid insurances | 177 | 35 | ||||||
| Prepaid expenses | 53 | 45 | ||||||
| Other receivables | - | 17 | ||||||
| Total | $ | 1,591 | $ | 2,509 | ||||
The Intrexon prepaid research and development expenses are classified as a current asset. The Company may terminate the arrangement at any time and receive a cash refund of the remaining balance minus any amounts owed to Intrexon. The Company anticipates that the majority of the prepaid will be applied to research and development goods and services during 2014.
Property and equipment (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Manufacturing equipment | $ | - | $ | 297 | ||||
| Computer and office equipment | 45 | 35 | ||||||
| Software | 11 | - | ||||||
| Laboratory equipment | - | 133 | ||||||
| 56 | 465 | |||||||
| Less accumulated depreciation | (19 | ) | (242 | ) | ||||
| Total | $ | 37 | $ | 223 | ||||
Accrued liabilities (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Accrued manufacturing costs | $ | 662 | $ | - | ||||
| Accrued vendor payments | 220 | - | ||||||
| Compensation | 3 | - | ||||||
| Total | $ | 885 | $ | - | ||||
5. Stock-Based Compensation
Stock Incentive Plan
During 2001, the Company’s Board of Directors and stockholders adopted the 2001 Stock Incentive Plan (the “2001 Stock Plan”). The total number of shares of stock with respect to which stock options and stock appreciation rights may be granted to any one employee of the Company or a subsidiary during any one-year period under the 2001 Stock Plan shall not exceed 250,000. All awards pursuant to the 2001 Stock Plan shall terminate upon the termination of the grantee’s employment for any reason. Awards include options, restricted shares, stock appreciation rights, performance shares and cash-based awards (the “Awards”). The 2001 Stock Plan contains certain anti-dilution provisions in the event of a stock split, stock dividend or other capital adjustment, as defined in the plan. The 2001 Stock Plan provides for a Committee of the Board to grant awards and to determine the exercise price, vesting term, expiration date and all other terms and conditions of the awards, including acceleration of the vesting of an award at any time. As of December 31, 2013, there were 953,507 options issued and outstanding under the 2001 Stock Plan.
| 49 |
On March 20, 2007, the Company’s Board of Directors approved the 2007 Stock Incentive Plan (the “2007 Stock Plan”) for the issuance of up to 2,500,000 shares of common stock to be granted through incentive stock options, nonqualified stock options, stock appreciation rights, dividend equivalent rights, restricted stock, restricted stock units and other stock-based awards to officers, other employees, directors and consultants of the Company and its subsidiaries. This plan was approved by stockholders on November 2, 2007. The exercise price of stock options under the 2007 Stock Plan is determined by the compensation committee of the Board of Directors, and may be equal to or greater than the fair market value of the Company’s common stock on the date the option is granted. The total number of shares of stock with respect to which stock options and stock appreciation rights may be granted to any one employee of the Company or a subsidiary during any one-year period under the 2007 plan shall not exceed 250,000. Options become exercisable over various periods from the date of grant, and generally expire ten years after the grant date. As of December 31, 2013, there were 443,573 options issued and outstanding under the 2007 Stock Plan.
On November 2, 2010, the Board of Directors and stockholders adopted the 2010 Stock Incentive Plan (“2010 Stock Plan”) for the issuance of up to 3,000,000 shares of common stock to be granted through incentive stock options, nonqualified stock options, stock appreciation rights, dividend equivalent rights, restricted stock, restricted stock units and other stock-based awards to officers, other employees, directors and consultants of the Company and its subsidiaries. The exercise price of stock options under the 2010 Stock Plan is determined by the compensation committee of the Board of Directors, and may be equal to or greater than the fair market value of the Company’s common stock on the date the option is granted. Options become exercisable over various period from the date of grant, and generally expire ten years after the grant date. As of December 31, 2013, there were 2,512,500 options issued and outstanding under the 2010 Stock Plan.
In the event of an employee’s termination, the Company will cease to recognize compensation expense for that employee. There is no deferred compensation recorded upon initial grant date, instead, the fair value of the stock-based payment is recognized ratably over the stated vesting period.
On October 22, 2013, the stockholders approved and adopted an amendment to the Company’s 2010 Incentive Stock Plan to increase the number of shares of Company’s common stock reserved for issuance under the Plan from 3,000,000 to 6,000,000.
The Company has applied fair value accounting for all share based payment awards since inception. The fair value of each option or warrant granted is estimated on the date of grant using the Black-Scholes option pricing model. The Black-Scholes assumptions used in the years ended December 31, 2013 and 2012 are as follows:
| Year ended December 31, | ||||
| 2013 | 2012 | |||
| Exercise price | $1.64 - $1.74 | $1.69 - $2.47 | ||
| Expected dividends | 0% | 0% | ||
| Expected volatility | 141% - 154% | 108% - 174% | ||
| Risk fee interest rate | 0.77% - 2.54% | 0.37% - 1.98% | ||
| Expected life of option | 5 – 10 years | 5 - 10 years | ||
| Expected forfeitures | 0% | 0% | ||
The Company records stock-based compensation based upon the stated vested provisions in the related agreements. The vesting provisions for these agreements have various terms as follows:
| · | immediate vesting, |
| · | half vesting immediately and the remainder over three years, |
| · | quarterly over three years, |
| · | annually over three years, |
| · | one-third immediate vesting and remaining annually over two years, |
| · | one half immediate vesting with remaining vesting over nine months, |
| · | one quarter immediate vesting with the remaining over three years |
| · | one quarter immediate vesting with the remaining over 33 months; and |
| · | monthly over three years. |
During 2013, the Company granted 222,500 options to employees and directors having an approximate fair value of $350,000 based upon the Black-Scholes option pricing model. During 2012, the Company granted 2,075,000 options to employees and directors having an approximate fair value of $4.5 million based upon the Black-Scholes option pricing model.
Stock-based compensation expense included in general and administrative expenses and research and development expenses relating to stock options issued employees for the years ended December 31, 2013 and 2012 were $1.3 million and $1.4 million, respectively. Stock-based compensation expense included in general and administrative expenses and research and development expenses relating to stock options issued to consultants for the years ended December 31, 2013 and 2012 were $324,000 and $216,000, respectively.
| 50 |
A summary of stock option activities for the year ended December 31, 2013 and for the year ended December 31, 2012, is as follows:
| Weighted | ||||||||||||||||
| Average | ||||||||||||||||
| Weighted | Remaining | Aggregate | ||||||||||||||
| Average Exercise | Contractual | Intrinsic | ||||||||||||||
| Options | Price | Life | Value | |||||||||||||
| Balance - December 31, 2011 | 2,979,010 | $ | 1.34 | 6.01 years | $ | - | ||||||||||
| Granted | 2,075,000 | $ | 2.21 | |||||||||||||
| Exercised | (374,851 | ) | $ | 0.34 | $ | 661,000 | ||||||||||
| Forfeited | (225,413 | ) | $ | 2.37 | ||||||||||||
| Balance - December 31, 2012 | 4,453,746 | $ | 1.78 | 6.43 years | $ | 1,308,000 | ||||||||||
| Granted | 222,500 | $ | 1.69 | |||||||||||||
| Exercised | (291,666 | ) | $ | 0.79 | $ | 71,000 | ||||||||||
| Forfeited | (475,000 | ) | $ | 2.30 | ||||||||||||
| Balance - December 31, 2013 - outstanding | 3,909,580 | $ | 1.78 | 5.59 years | $ | 785,000 | ||||||||||
| Balance - December 31, 2013 - exercisable | 3,153,537 | $ | 1.69 | 5.24 years | $ | 773,000 | ||||||||||
| Grant date fair value of options granted - 2013 | $ | 350,000 | ||||||||||||||
| Weighted average grant date fair value - 2013 | $ | 1.57 | ||||||||||||||
| Grant date fair value of options granted - 2012 | $ | 4,468,000 | ||||||||||||||
| Weighted average grant date fair value - 2012 | $ | 2.15 | ||||||||||||||
The options outstanding and exercisable at December 31, 2013 are as follows:
| Options Outstanding | Options Exercisable | |||||||||||||||||||
| Weighted | Weighted Average | Weighted | Weighted Average | |||||||||||||||||
| Range of | Average | Remaining | Average | Remaining | ||||||||||||||||
| Exercise Price | Options | Exercise Price | Contractual Life | Options | Exercise Price | Contractual Life | ||||||||||||||
| $0.09 - $2.00 | 2,093,196 | $ | 1.29 | 4.73 years | 1,901,737 | $ | 1.25 | 4.64 years | ||||||||||||
| $2.01 - $3.00 | 1,770,560 | 2.28 | 6.65 years | 1,205,976 | 2.26 | 6.25 years | ||||||||||||||
| $3.01 - $6.00 | 45,824 | 5.24 | 3.62 years | 45,824 | 5.24 | 3.62 years | ||||||||||||||
| $0.09 - $6.00 | 3,909,580 | $ | 1.78 | 5.59 years | 3,153,537 | $ | 1.69 | 5.24 years | ||||||||||||
The options outstanding and exercisable at December 31, 2012 are as follows:
| Options Outstanding | Options Exercisable | |||||||||||||||||||
| Weighted | Weighted Average | Weighted | Weighted Average | |||||||||||||||||
| Range of | Average | Remaining | Average | Remaining | ||||||||||||||||
| Exercise Price | Options | Exercise Price | Contractual Life | Options | Exercise Price | Contractual Life | ||||||||||||||
| $0.09 - $2.00 | 2,162,362 | $ | 1.78 | 5.76 years | 1,801,187 | $ | 1.14 | 5.52 years | ||||||||||||
| $2.01 - $3.00 | 2,212,227 | 2.26 | 7.15 years | 1,011,533 | 2.17 | 6.14 years | ||||||||||||||
| $3.01 - $6.00 | 79,157 | 4.87 | 4.49 years | 79,157 | 4.87 | 4.49 years | ||||||||||||||
| $0.09 - $6.00 | 4,453,746 | $ | 1.78 | 6.43 years | 2,891,877 | $ | 1.60 | 5.71 years | ||||||||||||
The following is a summary of the Company’s non-vested stock options at December 31, 2013:
| Weighted Average | ||||||||
| Unvested | Grant | |||||||
| Stock Options | Date Fair Value | |||||||
| Non-vested - December 31, 2012 | 1,561,869 | $ | 2.11 | |||||
| Granted | 222,500 | 1.69 | ||||||
| Vested/Exercised | (883,882 | ) | 1.93 | |||||
| Forfeited/Cancelled | (144,444 | ) | 2.27 | |||||
| Non-vested - December 31, 2013 | 756,043 | $ | 2.17 | |||||
| Weighted average remaining period for vesting | 1.30 years | |||||||
As of December 31, 2013, total unrecognized stock-based compensation expense related to stock options was $1.6 million, which is expected to be expensed through October 2016.
| 51 |
FASB’s guidance for stock-based payments requires cash flows from excess tax benefits to be classified as a part of cash flows from financing activities. Excess tax benefits are realized tax benefits from tax deductions for exercised options in excess of the deferred tax asset attributable to stock compensation costs for such options. The Company did not record any excess tax benefits in 2013 or 2012. Cash received from option exercises under the Company’s stock-based compensation plans for the years ended December 31, 2013 and 2012 was $231,000 and $127,000, respectively.
Stock Warrants
On October 25, 2012, the Company entered into a Common Stock Purchase Agreement with certain accredited investors. As part of this agreement, the Company issued warrants to purchase 635,855 shares of common stock to the placement agent, or its permitted assigns. The warrants have an exercise price of $1.60 and a life of five years. The warrants vested immediately and expire October 25, 2017. Since these warrants were granted as part of an equity raise, the Company has treated them as a direct offering cost. The result of the transaction has no affect to equity. As of December 31, 2013, all the warrants were outstanding.
On March 15, 2012, the Company entered into a consulting agreement for a financial communications program, for a period of 12 months that began on February 20, 2012, which was extended to March 14, 2014. As compensation for such program, the consultant is paid a monthly fee and will be issued a performance warrant exercisable for 250,000 shares of the Company’s common stock based on achievement of certain stock price milestones through March 14, 2013. In March 2013, the Company extended the period to which the milestones could be achieved to March 14, 2014. Upon initiation of the program, 50,000 of the performance warrants vested. The performance warrant is exercisable for a period of two years from the date of issuance for an exercise price equal to the price ($2.20 per share) of the Company’s common stock on the date of execution (March 15, 2012). The expense recorded for the years ended December 31, 2013 and December 31, 2012 approximated $0 and $63,000, respectively, and was estimated using the Monte Carlo valuation model. The assumptions used by the Company are summarized in the following table:
| Exercise price | $ | 2.20 | ||
| Expected dividends | 0 | % | ||
| Expected volatility | 110 | % | ||
| Risk free interest rate | 0.26 | % | ||
| Expected life of warrant | 2 years |
On December 20, 2011, the Company entered into a consulting agreement for financial advisory services, for a period of 12 months. As compensation for such services, the consultant is paid a monthly fee and on February 2, 2012, was issued a warrant exercisable for 100,000 shares of the Company’s common stock. The warrant is exercisable upon issuance for a period of five years from the date of issue at an exercise price equal to the price of the Company’s common stock on the date of issue. The fair value of the warrant approximated $200,000 and was measured using the Black-Scholes valuation model. All of this expense was recorded in the year ended December 31, 2012. The assumptions used by the Company are summarized in the following table:
| Exercise price | $ | 1.14 | ||
| Expected dividends | 0 | % | ||
| Expected volatility | 174 | % | ||
| Risk free interest rate | 0.71 | % | ||
| Expected life of warrant | 5 years |
A summary of warrant activity for the Company for the year ended December 31, 2012 and for the year ended December 31, 2013 is as follows:
| Weighted Average | ||||||||
| Number of Warrants | Exercise Price | |||||||
| Balance at December 31, 2011 | 3,259,186 | $ | 1.95 | |||||
| Granted | 985,855 | 1.71 | ||||||
| Exercised | (1,768,167 | ) | 1.11 | |||||
| Forfeited | (844,373 | ) | 3.32 | |||||
| Balance as December 31, 2012 | 1,632,501 | 1.99 | ||||||
| Granted | - | - | ||||||
| Exercised | - | - | ||||||
| Forfeited | - | - | ||||||
| Balance as December 31, 2013 | 1,632,501 | $ | 1.99 | |||||
There was no stock-based compensation expense included in general and administrative expenses relating to warrants issued to consultants for the year ended December 31, 2013. Stock-based compensation expense included in general and administrative expenses relating to warrants issued to consultants for the year ended December 31, 2012 was $271,000.
| 52 |
A summary of all outstanding and exercisable warrants as of December 31, 2013 is as follows:
| Exercise Price | Warrants Outstanding | Warrants Exercisable | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value | ||||||||||||||
| $ | 1.14 | 100,000 | 100,000 | 3.09 years | $ | 43,000 | ||||||||||||
| $ | 1.32 | 18,182 | 18,182 | 2.00 years | $ | 4,000 | ||||||||||||
| $ | 1.60 | 635,855 | 635,855 | 3.82 years | $ | - | ||||||||||||
| $ | 2.20 | 250,000 | 50,000 | 0.20 years | $ | - | ||||||||||||
| $ | 2.22 | 517,257 | 517,257 | 2.91 years | $ | - | ||||||||||||
| $ | 3.30 | 61,207 | 61,207 | 1.41 years | $ | - | ||||||||||||
| $ | 3.75 | 50,000 | 50,000 | 2.13 years | $ | - | ||||||||||||
| $ | 1.99 | 1,632,501 | 1,432,501 | 2.77 years | $ | 47,000 | ||||||||||||
Options of Subsidiary
As of December 31, 2013, Epitope, a majority-owned subsidiary of Synthetic Biologics, has 50,000 stock options outstanding and 40,000 stock options exercisable. These stock options have an exercise price of $0.001 and a remaining contractual life of 4.50 years.
6. Stockholders’ Equity
Year Ended December 31, 2012
On October 31, 2012, the Company completed a private placement financing of 6,750,000 shares of the Company’s common stock at a closing price of $1.60 for gross proceeds of $10.8 million. The Company paid direct offering costs of $680,000. See Note 5 regarding warrants granted with this offering.
During the year ended December 31, 2012, the Company issued 374,851 shares of common stock, in connection with the exercise of stock options, for proceeds of approximately $127,000. The Company also issued 1,768,167 shares of common stock in connection with the exercise of warrants, for proceeds of approximately $2.0 million. The Company also issued 3,552,210 shares of common stock as consideration for a second Exclusive Channel Collaboration Agreement with Intrexon, having a fair value of $7.8 million ($2.20 per share), based on the quoted closing trading price. These shares are subject to the registration rights agreement as described in Note 7. In connection with the private placement that was completed on October 31, 2012, the Company also entered into an agreement with NRM VII Holdings I, LLC, an affiliate of Intrexon that acquired 3,125,000 shares of the Company’s common stock in the private placement, pursuant to which NRM VII Holdings I, LLC agreed to be bound by the terms of and join Intrexon as a party to its registration rights agreement as described in Note 7. In addition, the Company issued 625,000 shares of common stock as consideration for the acquisition of the C. diff program assets of Prev ABR LLC, having a fair value of $1.2 million ($1.87 per share), based on the quoted closing trading price.
Year Ended December 31, 2013
On December 11, 2013, the Company completed a firm commitment underwritten public offering of 13,225,000 shares of the Company’s common stock at a closing price of $1.00 for gross proceeds of $13.2 million. The Company paid direct offering costs of $1.0 million.
During the year ended December 31, 2013, the Company issued 291,667 shares of common stock, in connection with the exercise of stock options, for proceeds of approximately $231,000. The Company also issued 334,911 shares of common stock in consideration for entering into worldwide exclusive license and option agreements with Cedars-Sinai Medical Center (“CSMC”), having a fair value of $425,000 ($1.27 per share), based on the average of prior 10 days quoted closing trading price.
On July 3, 2013, the Company entered into a Controlled Equity OfferingSM Sales Agreement with Cantor Fitzgerald & Co. pursuant to which it may offer and sell shares of the Company’s common stock in an at-the-market public offering (the “ATM”) for up to $15.0 million of shares of the Company’s common stock from time to time through Cantor Fitzgerald & Co., acting as agent. As of the date of this filing, the Company has not sold any shares under the ATM. The Company amended the Controlled Equity Offering SM Sales Agreement on December 10, 2013 to limit its ability to sell shares of the Company’s common stock under such agreement to the lesser of $15.0 million or the amount that the Company can sell under General Instruction I.B.6 of Form S-3, if still applicable, after this offering. The Company will not use the ATM unless and until it files an updated prospectus supplement reflecting the number or dollar amount of shares which it may sell under the ATM after taking into account the foregoing amendment, but only if such amount is less than $15.0 million.
7. License, Collaborative and Employment Agreements and Commitments
License and Collaborative Agreements
As described below, the Company has entered into several license and collaborative agreements for the right to use research, technology and patents. Some of these license and collaborative agreements may contain milestones. The specific timing of such milestones cannot be predicted and are dependent on future developments as well as regulatory actions which cannot be predicted with certainty (including actions which may never occur). Further, under the terms of certain licensing agreements, the Company may have the obligation to pay certain milestones contingent upon the achievement of specific levels of sales. Due to the long-range nature of such commercial milestone amounts, they are neither probable at this time nor predictable and consequently are not included in this disclosure.
| 53 |
On December 5, 2013, the Company, through its newly formed, majority owned subsidiary, Synthetic Biomics, Inc. (“SYN Biomics”) entered into a worldwide exclusive license agreement (the “License Agreement”) and option agreement (the “Option Agreement”) with CSMC for the development of new treatment approaches to target non-bacterial intestinal microorganism life forms known as archaea that are associated with intestinal methane production and chronic diseases such as irritable bowel syndrome (IBS), obesity and type 2 diabetes, through its newly formed subsidiary, Synthetic Biomics, Inc. As part of the terms of the License Agreement the Company issued 291,569 unregistered shares of Company common stock to CSMC, with the approval of the NYSE MKT, LLC, for the initial license fee and patent reimbursement fee of $150,000 and $220,000, respectively. The License Agreement also provides that commencing on the second anniversary of the License Agreement, SYN Biomics will pay an annual maintenance fee, which payment shall be creditable against annual royalty payments owed under the License Agreement. In addition to royalty payments which are a percentage of Net Sales (as defined in the License Agreement) of Licensed Products (as defined in the License Agreement) and Licensed Technology products (as defined in the License Agreement), SYN Biomics is obligated to pay CMSC a percentage of any non-royalty sublicense revenues, as well as additional consideration upon the achievement of milestones (the first two of which are payable in cash or unregistered shares of Company stock at the Company’s option).
The License Agreement terminates: (i) automatically if SYN Biomics enters into a liquidating bankruptcy or other specified bankruptcy event or if the performance of any term, covenant, condition or provision of the License Agreement will jeopardize the licensure of CMSC, its participation in certain reimbursement programs, its full accreditation by the Joint Commission of Accreditation of Healthcare Organizations or any similar state organizations, its tax exempt status or is deemed illegal; (ii) upon 30 days notice from CMSC if SYN Biomics fails to make a payment or use commercially reasonable efforts to exploit the patent rights; (iii) upon 60 days notice from CMSC if SYN Biomics fails to cure any breach or default of any material obligations under the License Agreement; or (iv) upon 90 days notice from SYN Biomics if CMCS fails to cure any breach or default of any material obligations under the License Agreement. SYN Biomics also has the right to terminate the License Agreement without cause upon six months notice to CSMC; however, upon such termination, SYN Biomics is obligated to pay a termination fee with the amount of such fee reduced: (i) if such termination occurs after an Investigational New Drug submission to the FDA but prior to completion of a Phase II clinical trial, (ii) reduced further if such termination occurs after completion of Phase II clinical trial but prior to completion of a Phase III clinical trial; and (iii) reduced to zero if such termination occurs after completion of a Phase III clinical trial.
Pursuant to the terms of the Option Agreement, SYN Biomics has a period of six months to negotiate an exclusive license to develop, manufacture, use, and sell biologic products relating to the prevention, acute treatment and chronic treatment of irritable bowel syndrome or other indications utilized or derived from certain optioned patent applications, pending completion of certain limited testing of technology embodied in the patent applications. Per the terms of the Option Agreement, the Company issued 43,342 shares of unregistered Company stock, as payment of a $50,000 non-refundable option fee upon execution of the agreement, with the approval of the NYSE MKT, LLC. In addition, SYN Biomics has the right to extend the option period for an additional six months, for an additional non-refundable extension fee of $25,000, payable in unregistered shares of the Company’s common stock having a market value of 110% of such amount, subject to approval of NYSE MKT, LLC, or in cash. At any time during the 6 or 12 month option period, if so extended, SYN Biomics has the right to exercise the option and negotiate an exclusive license to the optioned patent applications, which shall provide for: (i) a $50,000 license issue fee plus reimbursement of patent expenses incurred by CSMC prior to the exclusive license, payable to CSMC in unregistered shares of Company stock having a market value of 110% of such amount, subject to approval of the NYSE MKT, LLC, or in cash, (ii) the same milestone payments, royalties and sublicense fees as are payable under the License Agreement dated December 5, 2013 for separately licensed intellectual property, and (iii) such other customary terms and conditions CSMC typically includes in its license agreements.
Prior to the execution of the CSMC License Agreement, SYN Biomics issued shares of common stock of SYN Biomics to each of CSMC and Mark Pimentel, M.D. (the primary inventor of the intellectual property), representing 11.5% and 8.5%, respectively, of the outstanding shares of SYN Biomics (the “SYN Biomics Shares”). The Stock Purchase Agreements for the SYN Biomics Shares provide for certain anti-dilution protection until such time as an aggregate of $3.0 million in proceeds from equity financings are received by SYN Biomics as well as a right, under certain circumstances in the event that the SYN Biomics Shares are not then freely tradable, and subject to NUSE MKT, LLC approval, as of the 18 and 36 month anniversary date of the effective date of the Stock Purchase Agreements, for each of CSMC and the Dr. Pimentel to exchange up to 50% of their SYN Biomics shares for unregistered share of the Company’s common stock, with the rate of exchange based upon the relative contribution of the valuation of SYN Biomics to the public market valuation of us at the time of each exchange. The Stock Purchase Agreements also provide for tag-along rights in the event of the sale by the Company of its shares of SYN Biomics.
On December 19, 2012, the Company entered into a License Agreement with The University of Texas at Austin (the “University”) for the exclusive license of the right to use, develop, manufacture, market and commercialize certain research and patents related to pertussis antibodies. The License Agreement provides that the University is entitled to payment of past patent expenses, an annual payment of $50,000 per year commencing on the effective date through December 31, 2014 and a $25,000 payment on December 31, 2015 and milestone payments of $50,000 upon commencement of Phase I clinical trials, $100,000 upon commencement of Phase III clinical trials, $250,000 upon NDA submission in the U.S., $100,000 upon European Medicines Agency approval and $100,000 upon regulatory approval in an Asian country. In addition, the University is entitled to a running royalty upon net sales (as defined in the License Agreement). The License Agreement terminates upon the expiration of the patent rights (as defined in the License Agreement); provided, however that the License Agreement is subject to early termination by the Company in its discretion and by the University for a breach of the License Agreement by the Company.
In connection with the License Agreement, the Company and the University also entered into a Sponsored Research Agreement pursuant to which the University will perform certain research work related to pertussis. The Sponsored Research Agreement may be renewed annually, in the sole discretion of the Company, after the first year for two additional one year terms with a fixed fee for the first year of $303,287 and for the second and third years, if renewed, a fixed fee of $316,438 and $328,758 respectively, all payable in quarterly installments. If renewed by the Company after the first year for the remaining two years, the research shall be performed from the effective date of the Sponsored Research Agreement until December 31, 2015; provided, however, the Sponsored Research Agreement is subject to early termination upon the written agreement of the parties, a default in the material obligations under the Research Agreement which remain uncured for sixty days after receipt of notice, automatically upon the Company’s bankruptcy or insolvency and by the Company in its sole discretion at any time after the one year anniversary of the date of execution thereof upon no less than 90 days notice. Upon termination prior to December 31, 2014, the Company shall only be responsible for payment of expenses that do not exceed the fixed annual amount and are incurred prior to the termination date and non-cancellable expenses committed to be expended by the University prior to the termination date for the lesser of the remainder of their appointment in the case of salaries and December 31, 2014. Upon a termination after December 31, 2014 or due to a breach by the University, the Company shall only be responsible for all reasonable expenses that do not exceed the fixed annual amount and that are incurred by the University prior to the termination date for services performed prior to the termination date.
| 54 |
On November 28, 2012, a closing was held for the transaction contemplated by the Prev Agreement the Company entered into with Prev ABR LLC (“Prev”), pursuant to which it acquired the C. diff program assets of Prev, including pre-Investigational New Drug (IND) package, Phase I and Phase II clinical data, manufacturing process data and all issued and pending U.S. and international patents. Pursuant to the Prev Agreement, the Company paid Prev an initial cash payment of $100,000 upon execution of the Prev Agreement and at closing paid an additional cash payment of $135,000 and issued 625,000 unregistered shares of our common stock to Prev. See Note 6. In addition, upon the achievement of the milestones set forth below, Prev may be entitled to receive additional consideration payable 50% in cash and 50% in our stock, subject to Prev’s option to receive the entire payment in shares of our stock, with the exception of the first milestone payments to be paid in cash: (i) upon commencement of an IND; (ii) upon commencement of a Phase I clinical trial; (iii) upon commencement of a Phase II clinical trial; (iv) upon commencement of a Phase III clinical trial; (v) upon Biologic License Application (BLA) filing in the U.S. and for territories outside of the U.S. (as defined in the Prev Agreement); and (vi) upon BLA approval in the U.S. and upon approval in territories outside the-U.S. The Prev Agreement also provides that Prev has a right to the return to it of all assets acquired by the Company under the Prev Agreement if on or prior to the date that is (i) 30 months after the execution of the Prev Agreement, the Company has not initiated toxicology studies in non-rodent models or (ii) 36 months have not filed an IND under the program related to the assets and such failure is not due to action or inaction of Prev or breach of its representations or warranties or covenants or if there is a change of control as defined in the Prev Agreement and after such change of control the assets are not further developed; provided however that such 30 and 36 month periods can be extended by the Company for an additional 12 months upon payment of a cash milestone payment. No milestones were achieved or such payments made during the year ended December 31, 2013.
On August 6, 2012, the Company expanded its relationship with Intrexon and entered into an exclusive channel collaboration (“Second ECC”) with Intrexon that governs an “exclusive channel collaboration” arrangement in which the Company will use Intrexon’s technology relating to the identification, design and production of human antibodies and DNA vectors for the development and commercialization of a series of monoclonal antibody therapies for the treatment of certain serious infectious diseases. Pursuant to the terms of the Second Stock Issuance Agreement with Intrexon, which was approved by the Company’s stockholders on October 5, 2012, the Company issued 3,552,210 shares of its common stock, $0.001 par value, which issuance is also deemed paid in consideration for the execution and delivery of the Second ECC, dated August 6, 2012, between the Company and Intrexon. The fair value of this transaction was $7.8 million and was charged to research and development expense for the year ended December 31, 2012, in accordance with the Company’s accounting policy. In connection with the transactions contemplated by the Second Stock Issuance Agreement, and pursuant to the First Amendment to Registration Rights Agreement (the “First Amendment to Registration Rights Agreement”) executed and delivered by the parties at the closing, which was declared effective on May 5, 2013. The Company filed a “resale” registration statement registering the resale of certain of the shares issued under the Second Stock Issuance Agreement.
Subject to certain expense allocations and other offsets provided in the Second ECC, the Company will pay Intrexon royalties on annual net sales of the Synthetic Products, calculated on a Synthetic Product-by-Synthetic Product basis. The Company has likewise agreed to pay Intrexon a percentage of quarterly revenue obtained from a sublicensor in the event of a sublicensing arrangement. No such payments were made during the year ended December 31, 2013.
The Company also agreed upon the filing of an IND application with the FDA for a Synthetic Product, or alternatively the filing of the first equivalent regulatory filing with a foreign regulatory agency (both as applicable, the “IND Milestone Event”), to pay Intrexon either (i) $2.0 million in cash, or (ii) that number of shares of Common Stock (the “IND Milestone Shares”) having a fair market value equaling $2.0 million where such fair market value is determined using published market data of the share price for Common Stock at the close of market on the business day immediately preceding the date of public announcement of attainment of the IND Milestone Event.
Upon the first to occur of either first commercial sale of a Synthetic Product in a country or the granting of the regulatory approval of that Synthetic Product (both as applicable, the “Approval Milestone Event”), the Company agreed to pay to Intrexon either (i) $3.0 million in cash, or (ii) that number of shares of Common Stock (the “Approval Milestone Shares”) having a fair market value equaling $3.0 million where such fair market value is determined using published market data of the share price for Common Stock at the close of market on the business day immediately preceding the date of public announcement of attainment of the Approval Milestone Event.
The Company also agreed that it will pay an optional and varying fee whereby the Company remits a payment, in cash or equity at our sole discretion, to Intrexon calculated as a multiple of the number of targets in excess of three total that the Company desires to elect (the “Field Expansion Fee”). The Field Expansion Fee must be paid completely in either Common Stock or cash, and will comprise either (i) $2.0 million in cash for each target in excess of three total that the Company elects, or (ii) that number of shares of Common Stock (the “Field Expansion Fee Shares”) having a fair market value equaling $2.0 million for each such target that the Company elects in excess of three where such fair market value is determined using published market data establishing the volume-weighted average price for a share of Common Stock over the 30 day period immediately preceding the date of the Field Expansion Fee Closing. No milestones were achieved or such payments were made during the year ended December 31, 2013.
| 55 |
On November 18, 2011, the Company entered into the Initial Channel Agreement (“Initial ECC”) with Intrexon that governs an “exclusive channel collaboration” arrangement in which the Company initially intended to use Intrexon’s technology directed towards the production of PGIS. As consideration for execution of the Initial ECC, the Company entered into the Initial Stock Purchase Agreement with Intrexon pursuant to which the Company issued to Intrexon a number of shares of our common stock equal to 9.995% of the number of shares of the Company’s common stock issued and outstanding following and giving effect to such issuance at a purchase price equal to the $0.001 par value of such shares, which issuance was deemed paid in consideration for the execution and delivery of the Initial ECC which was terminated on April 16, 2013. In connection with the transactions contemplated by the Stock Purchase Agreement, and pursuant to the Registration Rights Agreement executed and delivered by the Company to Intrexon, the Company agreed to file a “resale” registration statement registering the resale of the First Tranche Shares within 120 days of the closing date of such issuance. The registration statement registering such shares was declared effective on April 13, 2012.
During December 2012, the Company paid Intrexon a prepayment of research and development expenses of $2.5 million for research and development goods and services to be provided in the future and has been recorded on the Company’s consolidated balance sheets in prepaid expenses and other current assets. Related research and development expenses of $1.0 million and $87,000 were recorded against this prepayment for the years ended December 31, 2013 and 2012, respectively. At December 31, 2013, the Intrexon prepayment of research and development expenses was $1.4 million.
In September of 2005, the Company entered into a three-year research agreement with the University of Michigan. Pursuant to that agreement, the Company sponsored research of approximately $460,000 per year. On March 20, 2008, the Company terminated the agreement. On March 24, 2009, the Company entered into a payment plan with the University of Michigan to pay the outstanding balance of $197,000. The Company agreed to pay $5,000 per month, the balance was paid in full July 2012.
Employment Agreements
Effective February 3, 2012, Jeffrey Riley was appointed to serve as the Company’s Chief Executive Officer and President. In connection with his appointment, Mr. Riley entered into a three-year employment agreement with the Company (the “Riley Employment Agreement”). Pursuant to the Riley Employment Agreement, Mr. Riley will be entitled to an annual base salary of $348,000 and will be eligible for discretionary performance and transactional bonus payments. Additionally, Mr. Riley was granted options to purchase 750,000 shares of the Company’s common stock with an exercise price equal to the per share market price on the date of issue. These options will vest pro rata, on a monthly basis, over 36 months. The Company measured the fair value of the stock options at approximately $1.7 million using a Black-Scholes valuation model.
Effective February 6, 2012, C. Evan Ballantyne was appointed the Company’s Chief Financial Officer. In connection with his appointment, Mr. Ballantyne entered into a three-year employment agreement with the Company (the “Ballantyne Employment Agreement”). Pursuant to the Ballantyne Employment Agreement, Mr. Ballantyne will be entitled to an annual base salary of $298,000 and will be eligible for discretionary performance and transactional bonus payments. Additionally, Mr. Ballantyne was granted options to purchase 425,000 shares of our common stock with an exercise price equal to our per share market price on the date of issue. These options will vest pro rata, on a monthly basis, over 36 months. The Company measured the fair value of the stock options at approximately $1 million using a Black-Scholes valuation model.
Other Commitments
As of December 31, 2013, amounts due for license and sponsored research agreements are as follows, excluding potential milestone payments which are contingent upon the occurence of future events (in thousands):
| Year Ending December 31, | ||||
| 2014 | $ | 378 | ||
| 2015 | 336 | |||
| 2016 | 10 | |||
| 2017 | 10 | |||
| 2018 | 10 | |||
| Total | $ | 744 |
| 56 |
Operating Lease
During 2012, the Company entered into a one year operating lease for office space in Ann Arbor, Michigan. In March 2013, this lease was amended to extend the term of the lease to December 31, 2014, for annual lease payments of $33,000. In March 2012, the Company also entered into a one year operating lease that may be renewed for two additional terms of one year, for office space in Rockville, Maryland, for annual lease payments of $42,000. This lease was extended in March 2013 for a term of one year. The Maryland office lease may be terminated with 60 days written notice.
During the years ended December 31, 2013 and 2012, the Company recognized rent expense of $68,000 and $123,000, respectively.
8. Stock Repurchase Program
On April 3, 2009, the Company’s Board of Directors approved a Stock Repurchase Program authorizing the Company to repurchase, from time-to-time and through December 31, 2009, up to $1 million of its common stock, up to a maximum of four million shares at prices of up to $5 per share. As of December 31, 2013, the Company had repurchased 81,482 shares for approximately $50,000 ($0.61 per share), based upon the quoted closing trading price. These treasury shares are not included in the computation of earnings (loss) per share and are deemed to be canceled and retired. The Company had no stock repurchases during the years ended December 31, 2013 and 2012.
9. Income Taxes
There was no income tax expense for the years ended December 31, 2013 and 2012 due to the Company’s net losses.
The Company’s tax expense differs from the “expected” tax expense for the years ended December 31, 2013 and 2012 (computed by applying the Federal Corporate tax rate of 34% to loss before taxes and 3.96% for Michigan State Corporate taxes, the blended rate used was 37.96%), as follows (in thousands):
| 2013 | 2012 | |||||||
| Computed “expected” tax benefit - Federal | $ | (4,188 | ) | $ | (5,803 | ) | ||
| Computed “expected” tax benefit - State | (488 | ) | (676 | ) | ||||
| Meals, entertainment and other | 6 | 5 | ||||||
| Non-deductible stock-based compensation | 511 | 518 | ||||||
| Realized loss on debt securities | - | - | ||||||
| Change in valuation allowance | 4,159 | 5,956 | ||||||
| $ | - | $ | - | |||||
The effects of temporary differences that gave rise to significant portions of deferred tax assets at December 31, 2013 and 2012 are as follows (in thousands):
| Deferred tax assets: | 2013 | 2012 | ||||||
| Stock issued for services | $ | 441 | $ | 318 | ||||
| Bad debt - change in allowance | 1,099 | 1,099 | ||||||
| Stock issued for acquisition of program | 652 | 444 | ||||||
| Stock issued for license agreement | 2,707 | 2,989 | ||||||
| Net operating loss carry-forward | 16,168 | 12,058 | ||||||
| Total gross deferred tax assets | 21,067 | 16,908 | ||||||
| Less valuation allowance | (21,067 | ) | (16,908 | ) | ||||
| Net deferred tax assets | $ | - | $ | - | ||||
At December 31, 2013, the Company has a net operating loss carry-forward of approximately $42.8 million available to offset future taxable income expiring through 2033. However, utilization of these net operating losses may be limited due to potential ownership changes under Section 382 of the Internal Revenue Code. The prior year deferred tax assets were not appropriately stated. However, they have been corrected in the table above. Management does not believe this has a material impact on the consolidated financial statements for the year ended December 31, 2012.
The valuation allowance at December 31, 2012 was approximately $16.9 million. The net change in valuation allowance during the year ended December 31, 2013 was an increase of approximately $4.2 million. In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred income tax assets will not be realized. The ultimate realization of deferred income tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred income tax liabilities, projected future taxable income, and tax planning strategies in making this assessment. Based on consideration of these items, management has determined that enough uncertainty exists relative to the realization of the deferred income tax asset balances to warrant the application of a full valuation allowance as of December 31, 2013.
| 57 |
ASC 740-10 “Accounting for Uncertain Tax Positions” prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return and also provides guidance on de-recognition, classification, interest and penalties, accounting in interim periods, disclosure, and transition.
As of December 31, 2013 and 2012, the Company had no unrecognized tax benefits and no adjustments to liabilities or operations were required under ASC 740-10. The Company’s practice was and continues to be to recognize interest and penalty expenses related to uncertain tax positions in income tax expense, which was zero for the years ended December 31, 2013 and 2012. The Company files United States federal and various state income tax returns.
The Company is routinely subject to examinations by taxing authorities in these various jurisdictions. The Company’s U.S. tax matters for the years 2000 through 2013 remain subject to examination by the Internal Revenue Service due to the Company’s NOL carryforwards. The Company’s U. S. tax matters remain subject to examination by various state and local tax jurisdictions due to our NOL carryforwards.
The Company does not anticipate that it is reasonably possible that unrecognized tax benefits as of December 31, 2013 will significantly change within the next 12 months.
10. Related Party Transactions
From January 2, 2012 through September 30, 2012, Steve H. Kanzer was engaged as the Company’s Interim Director of its Biologics Division. In connection with his appointment, Mr. Kanzer entered into a six month employment agreement with the Company on a full time basis (the “Kanzer Employment Agreement”), which may be extended for an additional three months upon consent of the parties. Pursuant to the Kanzer Employment Agreement, Mr. Kanzer was entitled to a base salary of $90,000 for the term, healthcare coverage pursuant to the Company’s healthcare insurance plan, reimbursement for certain relocation expenses and rent expense. The Kanzer Employment Agreement also included confidentiality obligations and inventions assignments by Mr. Kanzer. Mr. Kanzer was not entitled to severance pay upon termination of his employment. On October 1, 2012, the Kanzer Employment Agreement was amended (the “Amended Kanzer Employment Agreement”) and Mr. Kanzer was engaged as the Company Licensing Associate. In connection with this appointment, Mr. Kanzer entered into a two year agreement with the Company on a part time basis (2.5 days per week). Pursuant to the Amended Kanzer Employment Agreement, Mr. Kanzer was entitled to a base salary of $150,000 for the term and healthcare coverage pursuant to the Company’s healthcare insurance plans. Effective December 5, 2013, Mr. Kanzer was appointed CEO and President of the Company’s majority owned subsidiary, Synthetic Biomics, Inc. In connection with this appointment, Mr. Kanzer was entitled to an annual base salary of $195,000. On February 26, 2014, Mr. Kanzer resigned as CEO and President of Synthetic Biomics, Inc. and as a member of the Company’s Board of Directors.
In August 2012, the Company entered into a Second ECC with Intrexon and issued 3,552,210 shares of common stock as consideration, having a fair value of $7.8 million ($2.20 per share), based on the quoted closing trading price on October 5, 2012. In November 2011, the Company entered into an Initial ECC with Intrexon and issued 3,123,558 shares of common stock as consideration, having a fair value of $1.7 million ($0.54 per share), based on the quoted closing trading price. In connection with the November 2011 and August 2012 Exclusive Channel Agreements, the Company paid Intrexon approximately $2.9 million during 2012, including a prepayment of research and development expenses of $2.5 million for research and development goods and services to be provided in the future which has been recorded on the Company’s balance sheet in prepaid expense as described in Note 4. In October 2012, the Company consummated its October 2012 Private Placement and entered into a stock purchase agreement with several investors, including NRM VII Holdings I, LLC, an entity affiliated with Intrexon. Randal J. Kirk, directly and through certain affiliates, has voting and dispositive power over a majority of the outstanding capital of Intrexon Corporation, and controls NRM VII Holdings I, LLC. Mr. Kirk disclaims beneficial ownership of the shares held by Intrexon Corporation and NRM VII Holdings I, LLC, except to the extent of any pecuniary interest therein.
| 58 |
Item 9. Changes In and Discussions with Accountants on Accounting and Financial Disclosures
Not applicable.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
The Company has adopted and maintains disclosure controls and procedures that are designed to provide reasonable assurance that information required to be disclosed in the reports filed under the Exchange Act, such as this Form 10-K, is collected, recorded, processed, summarized and reported within the time periods specified in the rules of the SEC. The Company’s disclosure controls and procedures are also designed to ensure that such information is accumulated and communicated to management to allow timely decisions regarding required disclosure. As required under Exchange Act Rule 13a-15, the Company’s management, including the Chief Executive Officer and Chief Financial Officer, has conducted an evaluation of the effectiveness of disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based upon that evaluation, the Company’s Chief Executive Officer and Chief Financial Officer concluded that the Company’s disclosure controls and procedures are effective to ensure that information required to be disclosed by the Company in the reports that the Company files or submits under the Exchange Act, is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to the Company’s management, including the Company’s Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Management’s Report on Internal Control Over Financial Reporting
The Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Exchange Act Rule 13a-15. The Company’s internal control over financial reporting is designed to provide reasonable assurance to the Company’s management and Board of Directors regarding the preparation and fair presentation of published financial statements. Management conducted an assessment of the Company’s internal control over financial reporting based on the framework and criteria established by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (1992). Based on the assessment, management concluded that, as of December 31, 2013, the Company’s internal control over financial reporting is effective based on those criteria.
The Company’s management, including its Chief Executive Officer and Chief Financial Officer, does not expect that the Company’s disclosure controls and procedures and its internal control processes will prevent all error and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of error or fraud, if any, within the Company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that the breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the control. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and may not be detected. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Changes in Internal Control
There has been no change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act) that occurred during our fiscal year ended December 31, 2013 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
This Annual Report on Form 10-K does not include an attestation report of the Company’s registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the Company’s registered public accounting firm pursuant to rules of the SEC that permit the Company to provide only management’s report in this Annual Report on Form 10-K.
Item 9B. Other Information
None.
| 59 |
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Below is certain information regarding our directors and executive officers.
| Name | Age | Position | ||
| Jeffrey Riley | 51 | Chief Executive Officer, President and Director | ||
| C. Evan Ballantyne | 54 | Chief Financial Officer | ||
| Jeffrey J. Kraws | 49 | Chairman | ||
| Scott L. Tarriff | 54 | Director | ||
| Jeffrey Wolf, J.D. | 50 | Director |
Jeffrey Riley. Mr. Riley, a member of the Synthetic Biologics’ Board of Directors since March 2010 and Chairman of the Board from November 2011 to May 2012, was appointed as the Company’s President and Chief Executive Officer in February 2012. Since November 2009 until January 2012, Mr. Riley served as the Managing Director of 526 Ventures, a life science-focused consulting firm with a commercial and transactional focus, and from April 2009 until February 2012 he was the business officer of Ruga Corporation, a Stanford University spin-out oncology drug discovery company focused on targeting tumor adaptive responses. From January 2005 until January 2010, Mr. Riley was a member of the advisory board and a venture partner of Queensland Biocapital Fund, an Australia-based venture fund. Mr. Riley has held senior corporate and commercial development positions with multiple venture-backed biotech companies. In these positions, he was responsible for raising equity and negotiating alliances including in-licensing, out-licensing, distribution agreements, technology acquisitions and research agreements with large pharmaceutical companies and government agencies. Mr. Riley's pharmaceutical experience includes commercial management and mergers and acquisition roles for Pfizer and SmithKline Beecham. Additionally, Mr. Riley served as CFO and VP Corporate Development for Nichols Institute Diagnostics, later acquired by Corning and spun out as Quest Diagnostics. Mr. Riley’s education includes: a B.S. degree from Boise State University, coursework at UCSF/Berkeley in drug discovery/development and participation in a dual-degree graduate program, an M.B.A./M.I.M. sponsored by Arizona State University and the Thunderbird School of Global Management.
Mr. Riley brings to the Board extensive knowledge of the pharmaceutical industry. Having served in senior corporate positions in biotech and pharmaceutical companies he has a vast knowledge of the industry. His business experience provides him with a broad understanding of the operational, financial and strategic issues facing public companies.
C. Evan Ballantyne. Mr. Ballantyne joined Synthetic Biologics as its Chief Financial Officer in February 2012. He also serves as the Company's Corporate Secretary and Treasurer. From 2006 until its acquisition in April 2011, Mr. Ballantyne served as Executive Vice President and Chief Financial Officer of Clinical Data, Inc., a publicly-traded biopharmaceutical company which was acquired by Forest Laboratories, Inc. for $1.3 billion. While at Clinical Data, he was instrumental in leading corporate financings totaling approximately $220 million as well as a number of acquisition and divestitures totaling $116 million. Mr. Ballantyne has also served as Chief Financial Officer of a number of private medical technology companies, including Avedro and ZymeQuest. Earlier in his career, he served as Vice President and Chief Operating Officer for ACNielsen Europe Middle East & Africa and held the Chief Financial Officer position as well for two years. There, Mr. Ballantyne was responsible for all aspects of operations, strategic planning and finance in more than 45 countries for a corporation with 9,700 employees. He also helped lead the company's successful ISO certification process. He began his career at the Dun & Bradstreet Corporation where he held several senior financial positions. Mr. Ballantyne earned a BA from the University of Western Ontario, and took a post-graduate degree in Business Administration with Honors from the University of Windsor.
Jeffrey J. Kraws. Mr. Kraws has been a director since January of 2006, and was appointed independent, non-executive Chairman of the Board in May 2012. Since 2003, Mr. Kraws has served Chief Executive Officer and co-founder of Crystal Research Associates, and since February 2012, he has served as partner and co-founder of TopHat Capital, LLC. Well known and respected on Wall Street, Mr. Kraws has received some of the most prestigious awards in the industry. Among other awards, he was given a “5-Star Rating” in 2001 by Zacks and was ranked the number one analyst among all pharmaceutical analysts for stock performance in 2001 by Starmine.com. Prior to founding Crystal Research Associates, Mr. Kraws served as co-president of The Investor Relations Group (IRG), a firm representing primarily under-followed, small-capitalization companies. Previously, Mr. Kraws served as a managing director of healthcare research for Ryan Beck & Co. and as director of research/senior pharmaceutical analyst and managing director at Gruntal & Co., LLC (prior to its merger with Ryan Beck & Company). Mr. Kraws served as managing director of the healthcare research group and senior pharmaceutical analyst at First Union Securities (formerly EVEREN Securities); as senior U.S. pharmaceutical analyst for the Swedish-Swiss conglomerate Asea Brown Boveri; and as managing director and president of the Brokerage/Investment Banking operation of ABB Aros Securities, Inc. He also served as senior pharmaceutical analyst at Nationsbanc Montgomery Securities, BT Alex Brown & Sons, and Buckingham Research. Mr. Kraws also has industry experience, having been responsible for competitive analysis within the treasury group at Bristol-Myers-Squibb Company. During 2006 through February of 2007, Mr. Kraws served as our Vice President of Business Development, on a part-time basis. Since December 2013, Mr. Kraws serves on the board of directors of Saleen Automotive, Inc. (OTC: SLNN). He holds an M.B.A. from Cornell University and a B.S. degree from State University of New York-Buffalo. During 2006 through February of 2007, Mr. Kraws served as our Vice President of Business Development, on a part-time basis.
| 60 |
Mr. Kraws brings a strong business background to Synthetic Biologics, having worked as a pharmaceutical analyst for over 22 years. Mr. Kraws brings to the Board significant strategic, business and financial experience related to the business and financial issues facing pharmaceutical companies. Mr. Kraws has a broad understanding of the operational, financial and strategic issues facing pharmaceutical companies. Through his services as the Company’s Vice President of Business Development during 2006 and a part of 2007, he developed extensive knowledge of Synthetic Biologics’ business.
Scott L. Tarriff. Mr. Tarriff has been a director since February 3, 2012. Since January 2007 he has served as a director and Chief Executive Officer of Eagle Pharmaceuticals, Inc. a publicly traded, hospital specialty company. Eagle Pharmaceuticals, Inc. (NASDAQ: EGRX) is focused on developing branded parenteral products through the application of various in-licensed drug delivery technologies. Prior to forming Eagle Pharmaceuticals, Inc., Mr. Tarriff was president and chief executive officer of Par Pharmaceutical Companies, Inc. Mr. Tarriff joined Par Pharmaceutical Companies, Inc., in 1998 as executive vice president. Mr. Tarriff was named president and Chief Executive Officer of Par Pharmaceutical, Inc., the company’s principal operating subsidiary, in 2001, and was elected to the company’s Board of Directors in 2002. In September 2003, he was appointed President and Chief Executive Officer of Par Pharmaceutical Companies, Inc. Mr. Tarriff joined Par Pharmaceutical Companies, Inc. following a 12-year career at Bristol-Meyers Squibb. From 2009 until 2011, Mr. Tarriff served as a director of Clinical Data, Inc. He received his M.B.A. from Rider College and his undergraduate degree from Pennsylvania State University.
Mr. Tarriff brings to our Board of Directors significant knowledge of and experience in the pharmaceutical and medical industries. He has extensive business, managerial, executive and leadership experience that further qualify him to serve as a member of the Board and a valuable understanding of the role played by the Board of Directors acquired through service on the boards of many companies. He has had a long and successful career in top executive leadership positions with leading, publicly traded pharmaceutical companies including Par Pharmaceuticals Companies, Inc. and Bristol-Myers Squibb.
Jeffrey Wolf, J.D. Mr. Wolf, a director since 2006, has substantial experience in creating, financing, nurturing and growing new ventures based upon breakthrough research and technology. In August 2008, Mr. Wolf founded Heat Biologics, Inc. (NASDAQ: HTBX), a publicly traded company engaged in research and development of drugs focused on combating cancer and other diseases. Since April 2010, Mr. Wolf has served as the Chief Executive Officer and Chairman of the Board of Heat Biologics, Inc. Prior to founding Heat Biologics, Inc., from June 1997 to March 2011, Mr. Wolf has served as managing director at Seed-One Ventures, LLC a venture firm focused on launching and growing exceptional healthcare companies from the ground up. Since founding Seed-One, Mr. Wolf has founded and run several medical companies. Mr. Wolf’s start-ups include Avigen, a San Francisco-based gene therapy company where he was a co-founder and director; TyRx Pharma, a Princeton-based company focused on the development of bio-compatible polymers where he was a co-founder and Chairman; EluSys Therapeutics, a New Jersey company focused on the development of novel technology to remove blood-borne pathogens where he was a co-founder, Chairman and Chief Executive Officer; and GenerationOne, a Miami-based company focused on mobile-based collaborative care, where he was the founder, Chairman and Chief Executive Officer. Mr. Wolf received his M.B.A. from Stanford Business School, his J.D. from New York University School of Law and his B.A. from the University of Chicago, where he graduated with honors in Economics. Mr. Wolf serves as a director of several Seed-One portfolio companies.
Mr. Wolf has extensive knowledge of the industry and in particular research and development. His legal and business background provide him with a broad understanding of the legal, operational, financial and strategic issues facing Synthetic Biologics. Having served as a board member on other public company boards, Mr. Wolf has an extensive understanding of the operational, financial and strategic issues facing public companies.
Directors’ Term of Office
Directors will hold office until the next annual meeting of stockholders and the election and qualification of their successors. Officers are elected annually by our Board of Directors and serve at the discretion of the Board of Directors.
Audit Committee
The Audit Committee is comprised of Mr. Wolf and Mr. Tarriff. The Audit Committee is responsible for recommending our independent public accounting firm and reviewing management’s actions in matters relating to audit functions. The Committee reviews with our independent public accountants the scope and results of the audit engagement and the system of internal controls and procedures. The Committee also reviews the effectiveness of procedures intended to prevent violations of laws. The Committee also reviews, prior to publication, our reports on Form 10-K and Form 10-Q. Our Board has determined that all audit committee members are independent under applicable SEC regulations. Our Board of Directors has determined that both Mr. Wolf and Mr. Tarriff qualify as “audit committee financial experts” as that term is used in Section 407 of Regulation S-K. Our Audit Committee charter is located on our websitewww.syntheticbiologics.com.
Compensation Committee
Our Compensation Committee consists of Mr. Wolf and Mr. Kraws. This committee performs several functions, including reviewing all forms of compensation provided to our executive officers, directors, consultants and employees, including stock compensation. Our Board has determined that all compensation committee members are independent under applicable SEC regulations. Our Compensation Committee charter is located on our websitewww.syntheticbiologics.com.
| 61 |
Nominations Committee
Our Nominations Committee consists of Mr. Kraws and Mr. Wolf. This committee performs several functions, including identifying qualified individuals to become members of the Board and recommending appointments to the Board and appointment of executive officers. The committee seeks individuals who have an inquisitive and objective perspective, practical wisdom and mature judgment, and the talent and expertise to understand, and provide sound and prudent guidance with respect to, our activities, operations and interests. Candidates must also be individuals who have the highest personal and professional integrity, who have demonstrated exceptional ability and judgment, and who are likely to be the most effective, in conjunction with the other members of the Board, in collectively serving the long-term interests of stockholders. Our Board has determined that all nominations committee members are independent under applicable SEC regulations. Our Nominations Committee charter is located on our website www.syntheticbiologics.com.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act of 1934 requires our executive officers, directors and persons who beneficially own more than 10 percent of a registered class of the Synthetic Biologics’ equity securities, to file with the SEC initial reports of ownership and reports of changes in ownership of our common stock. Such officers, directors and persons are required by SEC regulation to furnish us with copies of all Section 16(a) forms that they file with the SEC.
Based solely on a review of the copies of such forms that were received by us, or written representations from certain reporting persons that no Form 5s were required for those persons, we are not aware of any failures to file reports or report transactions in a timely manner during the year ended December 31, 2013.
Code of Ethics
We have long maintained a Code of Conduct which is applicable to all of our directors, officers and employees. In addition, we have adopted a Code of Ethics for Financial Management which applies to our Chief Executive Officer, Chief Financial Officer, Treasurer and Controller. Each of these codes is posted on our website atwww.syntheticbiologics.com.
Item 11. Executive Compensation
Summary Compensation Table
The following table summarizes all compensation awarded to, earned by or paid to Jeffrey Riley and C. Evan Ballantyne, during the fiscal years ended December 31, 2013 and 2012.
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Options Awards($) (1) | All Other Compensation ($) (2) | Total ($) | ||||||||||||||||||
| Jeffrey Riley, President and CEO (3) | 2013 | $ | 348,000 | $ | - | $ | - | $ | 16,000 | $ | 364,000 | |||||||||||||
| 2012 | $ | 314,000 | $ | 52,000 | $ | 1,716,000 | $ | 7,000 | $ | 2,089,000 | ||||||||||||||
| C. Evan Ballantyne, CFO (4) | 2013 | $ | 298,000 | $ | - | $ | - | $ | 16,000 | $ | 314,000 | |||||||||||||
| 2012 | $ | 267,000 | $ | 44,000 | $ | 1,044,000 | $ | 7,000 | $ | 1,362,000 | ||||||||||||||
| (1) | Amount reflects the grant date fair value of the named executive officer’s stock options, calculated in accordance with FASB ASC Topic 718. For a discussion of the assumptions used in calculating these values, see Note 5 to our consolidated financial statements |
| (2) | The all other compensation column is comprised of the portion of medical, dental and vision premiums paid by us on behalf of our named executive officer. These benefits are offered to all full-time Synthetic Biologics employees. |
| (3) | Mr. Riley was appointed as the Company’s President and Chief Executive Officer on February 3, 2012. |
| (4) | Mr. Ballantyne was appointed as the Company’s Chief Financial Officer on February 6, 2012. |
| 62 |
Outstanding Equity Awards at Fiscal Year End
The table below reflects all outstanding equity awards made to each of the named executive officers that are outstanding at December 31, 2013. We currently grant stock-based awards pursuant to our 2010 Stock Incentive Plan (the “2010 Stock Plan”) and have outstanding awards under our 2001 Stock Incentive Plan (the “2001 Stock Plan”) and 2007 Stock Incentive Plan (the “2007 Stock Plan”).
| Name | Grant Date (1) | Number of Securities Underlying Unexercised Options Exercisable | Number of Securities Underlying Unexercised Options Unexercisable | Option Exercise Price ($) | Option Expiration Date | |||||||||||||
| Jeffrey Riley | 02/03/12 | (2) | 458,333 | 291,667 | $ | 2.30 | 02/03/22 | |||||||||||
| 11/17/11 | (3) | 100,000 | - | $ | 0.49 | 11/17/18 | ||||||||||||
| 01/05/11 | 25,000 | - | $ | 1.50 | 01/05/18 | |||||||||||||
| 12/01/10 | 8,333 | - | $ | 0.74 | 12/01/20 | |||||||||||||
| 03/03/10 | 25,000 | - | $ | 0.87 | 03/03/20 | |||||||||||||
| C. Evan Ballantyne | 02/06/12 | (2) | 259,722 | 165,278 | $ | 2.47 | 02/06/22 | |||||||||||
| (1) | Unless otherwise noted, options vest immediately on the date of grant. |
| (2) | These options will vest pro rata, on a monthly basis, over 36 months. |
| (3) | These options vest 12,500 shares immediately and the remainder in equal quarterly installments for seven quarters commencing on February 17, 2012. |
Employment Agreements
On February 3, 2012, Jeffrey Riley was appointed to serve as our Chief Executive Officer and President, and on February 6, 2012, C. Evan Ballantyne was appointed to serve as our Chief Financial Officer. The following are summaries of the agreements that were executed in connection with these appointments.
Jeffrey Riley Employment Agreement
In connection with his appointment, Mr. Riley entered into a three-year employment agreement with us (the “Riley Employment Agreement”). Pursuant to the Riley Employment Agreement, Mr. Riley will be entitled to an annual base salary of $348,000 and will be eligible for discretionary performance and transactional bonus payments. Additionally, Mr. Riley was granted options to purchase 750,000 shares of our common stock with an exercise price equal to our per share market price on the date of issue. These options will vest pro rata, on a monthly basis, over 36 months. The Riley Employment Agreement also includes confidentiality obligations and inventions assignments by Mr. Riley.
If Mr. Riley’s employment is terminated for any reason, he or his estate as the case may be, will be entitled to receive the accrued base salary, vacation pay, expense reimbursement and any other entitlements accrued by him to the extent not previously paid (the “Accrued Obligations”); provided,however, that if his employment is terminated (1) by us without Just Cause (as defined in the Riley Employment Agreement) or by Mr. Riley for Good Reason (as defined in the Riley Employment Agreement) then in addition to paying the Accrued Obligations, (x) we shall continue to pay his then current base salary and continue to provide benefits at least equal to those which were provided at the time of termination for a period of six months and (y) he shall have the right to exercise any vested options until the earlier of the expiration of the severance or the expiration of the term of the option, or (2) by reason of his death or Disability (as defined in the Riley Employment Agreement), then in addition to paying the Accrued Obligations, he would have the right to exercise any vested options until the expiration of the term of the option. In such event, if Mr. Riley commenced employment with another employer and becomes eligible to receive medical or other welfare benefits under another employer-provided plan, the medical and other welfare benefits to be provided by us as described herein will terminate.
C. Evan Ballantyne Employment Agreement
Effective February 6, 2012, C. Evan Ballantyne was appointed our Chief Financial Officer. In connection with his appointment, Mr. Ballantyne entered into a three-year employment agreement with us (the “Ballantyne Employment Agreement”). Pursuant to the Ballantyne Employment Agreement, Mr. Ballantyne will be entitled to an annual base salary of $298,000 and will be eligible for discretionary performance and transactional bonus payments. Additionally, Mr. Ballantyne was granted options to purchase 425,000 shares of our common stock with an exercise price equal to our per share market price on the date of issue. These options will vest pro rata, on a monthly basis, over 36 months. The Ballantyne Employment Agreement also includes confidentiality obligations and inventions assignments by Mr. Ballantyne.
| 63 |
If Mr. Ballantyne’s employment is terminated for any reason, he or his estate as the case may be, will be entitled to receive the accrued base salary, vacation pay, expense reimbursement and any other entitlements accrued by him to the extent not previously paid (the “Accrued Obligations”); provided,however, that if his employment is terminated (1) by us without Just Cause (as defined in the Ballantyne Employment Agreement) or by Mr. Ballantyne for Good Reason (as defined in the Ballantyne Employment Agreement) then in addition to paying the Accrued Obligations, (i) we shall continue to pay his then current base salary and continue to provide benefits at least equal to those which were provided at the time of termination for a period of six months and (ii) he shall have the right to exercise any vested options until the earlier of the expiration of the severance or the expiration of the term of the option, or (2) by reason of his death or Disability (as defined in the Ballantyne Employment Agreement), then in addition to paying the Accrued Obligations, he would have the right to exercise any vested options until the expiration of the term of the option. In such event, if Mr. Ballantyne, commenced employment with another employer and becomes eligible to receive medical or other welfare benefits under another employer-provided plan, the medical and other welfare benefits to be provided by us as described herein will terminate.
Compensation of Directors
The following table sets forth information for the fiscal year ended December 31, 2013 regarding the compensation of our directors who at December 31, 2013 were not also named executive officers.
| Name | Fees Earned or Paid in Cash | Option Awards(1) (3) | Other Compensation | Total | ||||||||||||
| Jeffrey J. Kraws (2) | $ | 150,000 | $ | 40,000 | $ | - | $ | 190,000 | ||||||||
| Scott Tarriff | $ | 24,000 | $ | 80,000 | $ | - | $ | 104,000 | ||||||||
| Jeffrey Wolf | $ | 24,000 | $ | 80,000 | $ | - | $ | 104,000 | ||||||||
| Steve H. Kanzer (4) | $ | - | $ | - | $ | - | $ | - | ||||||||
| (1) | The amounts in the “Option awards” column reflect the dollar amounts of the grant date fair value for the financial statement reporting purposes for stock options for the fiscal year ended December 31, 2013 in accordance with ASC 718. The fair value of the options was determined using the Black-Scholes model. |
| (2) | Mr. Kraws was appointed as our independent, non-executive Chairman of the Board in May 2012. Pursuant to his agreement he receives annual compensation of $150,000. |
| (3) | As of December 31, 2013, the following are the outstanding aggregate number of option awards held by each of our then directors who were not also named executive officers: |
| Name | Option Awards (#) | |||
| Steve H. Kanzer | 304,391 | |||
| Jeffrey J. Kraws | 412,105 | |||
| Scott Tarriff | 75,000 | |||
| Jeffrey Wolf | 133,332 | |||
| (4) | On February 26, 2014, Mr. Kanzer resigned as CEO and President of Synthetic Biomics, Inc. and as a member of our Board of Directors. |
In October 2013, director compensation for independent members was approved at $4,500 per board meeting that they attend in person and $2,000 per telephonic board meeting. In addition, we grant independent members of the Board of Directors upon appointment 25,000 stock options to purchase shares of our common stock at an exercise price equal to the fair market value of the our common stock on the date of grant, and an additional 25,000 stock options each year. Directors who are employees, or who are compensated for board service pursuant to a separate agreement or arrangement, do not receive additional compensation for attending board, committee and stockholder meetings. We also reimburse directors for travel and other out-of-pocket expenses incurred in attending Board of Director and committee meetings.
| 64 |
Item 12. Security Ownership of Certain Beneficial Owners
The following table sets forth information, as of March 27, 2014, or as otherwise set forth below, with respect to the beneficial ownership of our common stock (i) all persons know to us to be the beneficial owners of more than 5% of the outstanding shares of our common stock, (ii) each of our directors and our executive officer named in the Summary Compensation Table, and (iii) all of our directors and our executive officer as a group.
Principal Stockholders’ Table
| Shares Owned (1) | ||||||||
| Name and Address of Beneficial Ownership (2) | Number of Shares | Percentage of Shares (3) | ||||||
| Accredited Venture Capital, LLC (4) | 7,086,380 | 12.16 | % | |||||
| Intrexon Corporation (5) | 8,675,768 | 14.89 | % | |||||
| NRM VII Holding I, LLC (5) | 3,625,000 | 6.22 | % | |||||
| C. Evan Ballantyne (6) | 318,750 | * | ||||||
| Steve H. Kanzer (7) | 7,766,017 | 13.26 | % | |||||
| Jeffrey J. Kraws (8) | 412,105 | * | ||||||
| Jeffrey Riley (9) | 731,233 | 1.24 | % | |||||
| Scott L. Tarriff (10) | 75,000 | * | ||||||
| Jeffrey Wolf (11) | 133,332 | * | ||||||
| Randal J. Kirk(12) | 12,300,768 | 21.11 | % | |||||
| All officers and directors as a group (5 persons) | 1,670,420 | 2.77 | % | |||||
* represents less than 1% of our common stock
| (1) | The address for each beneficial owner except Accredited Venture Capital, LLC, Steve H. Kanzer, Intrexon Corporation, NRM VII Holdings I, LLC and Randal J. Kirk is 155 Gibbs Street, Suite 412, Rockville, Maryland 20850.The address for Accredited Venture Capital, LLC and Steve H. Kanzer is 9045 LaFontana Blvd, Suite 238, Boca Raton, Florida 33434. The address for Intrexon Corporation and NRM VII Holdings I, LLC is 20358 Seneca Meadows Pkwy, Germantown, Maryland 20876 and the address for Mr. Kirk is The Governor Tyler, 1881 Grove Avenue, Radford, Virginia 24141. |
| (2) | Beneficial ownership is determined in accordance with SEC rules and generally includes voting or investment power with respect to securities. Except as indicated in the footnotes to the table, to the knowledge of the Company, the persons named in the table have sole voting and investment power with respect to all shares of common stock shown as beneficially owned by them, subject to community property laws, where applicable. Pursuant to the rules of the SEC, the number of shares of our common stock deemed outstanding includes shares issuable pursuant to options held by the respective person or group that are currently exercisable or may be exercised within 60 days of March 27, 2014. | |
| (3) | As of March 27, 2014, we had 58,276,556 shares of common stock outstanding. |
| (4) | Consists of 7,086,380 shares of common stock issued to Accredited Venture Capital, LLC. Pharmainvestors, LLC is the managing member of Accredited Venture Capital, LLC, and Mr. Kanzer is the managing member of Pharmainvestors, LLC. As such, Mr. Kanzer may be considered to have control over the voting and disposition of the shares registered in the name of Accredited Venture Capital, LLC, and therefore, such shares are also included in the shares listed as held by Mr. Kanzer. Mr. Kanzer disclaims beneficial ownership of those shares, except to the extent of his pecuniary interest. |
| (5) | Share ownership information is based on information contained in a schedule 13D filed with the SEC on December 19, 2013. Does not include additional shares that have not yet been earned but may in the future be earned under the terms of agreements with Intrexon Corporation. |
| (6) | Includes 318,750 shares issuable upon exercise of options held by Mr. Ballantyne that are exercisable within the 60-day period following March 27, 2013. Does not include an additional 106,250 shares issuable upon exercise of options held by Mr. Ballantyne that are not exercisable within the 60-day period following March 27, 2014. |
| (7) | Includes 7,086,380 shares of common stock issued to Accredited Venture Capital, LLC and 304,391 shares issuable upon exercise of options held by Mr. Kanzer that are exercisable within the 60-day period following March 27, 2014. |
| (8) | Includes 412,105 shares issuable upon exercise of options held by Mr. Kraws that are exercisable within the 60-day period following March 27, 2014. |
| (9) | Includes 720,833 shares issuable upon exercise of options held by Mr. Riley that are exercisable within the 60-day period following March 27, 2012. Does not include an additional 187,500 shares issuable upon exercise of options held by Mr. Riley that are not exercisable within the 60-day period following March 27, 2014. |
| 65 |
| (10) | Includes 75,000 shares issuable upon exercise of options held by Mr. Tarriff that are exercisable within the 60-day period following March 27, 2014. |
| (11) | Includes 133,332 shares issuable upon exercise of options held by Mr. Wolf that are exercisable within the 60-day period following March 27, 2014. |
| (12) | Share ownership information is based on information contained in a schedule 13D filed with the SEC on December 19, 2013. All such shares are held by Intrexon Corporation and NRM VII Holdings I, LLC. Mr. Kirk, directly and through certain affiliates, has voting and dispositive power over a majority of the outstanding capital of Intrexon Corporation, and controls NRM VII Holdings I, LLC. Mr. Kirk disclaims beneficial ownership of the shares held by Intrexon Corporation and NRM VII Holdings I, LLC, except to the extent of any pecuniary interest therein. |
Equity Compensation Plan Information
The following table sets forth information about the securities authorized for issuance under our equity compensation plans for the fiscal year ended December 31, 2013.
| Plan Category | Number of Securities to be Issued Upon Exercise of Outstanding Options | Weighted- Average Exercise Price of Outstanding Options | Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans | |||||||||
| Equity compensation plans approved by stockholders: | ||||||||||||
| 2001 Stock Incentive Plan | 953,507 | $ | 1.44 | 60,190 | ||||||||
| 2007 Stock Incentive Plan | 443,573 | $ | 1.55 | 533,101 | ||||||||
| 2010 Stock Incentive Plan | 2,512,500 | $ | 1.95 | 3,484,030 | ||||||||
| Equity compensation plans not approved by stockholder | N/A | N/A | N/A | |||||||||
| Total | 3,909,580 | $ | 1.78 | 4,077,321 | ||||||||
Item 13. Certain Relationships and Related Transactions, and Director Independence
Pursuant to our charter, our Audit Committee reviews on an on-going basis for potential conflicts of interest, and approve if appropriate, all our “Related Party Transactions” as required by Section 120 of the NYSE MKT Company Guide. For purposes of the Audit Committee Charter, “Related Party Transactions” shall mean those transactions required to be disclosed pursuant to SEC Regulation S-K, Item 404.
From January 2, 2012 through September 30, 2012, Steve H. Kanzer was engaged as our Interim Director of our Biologics Division. In connection with his appointment, Mr. Kanzer entered into a six month employment agreement with us on a full time basis (the “Kanzer Employment Agreement”), which may be extended for an additional three months upon consent of the parties. Pursuant to the Kanzer Employment Agreement, Mr. Kanzer was entitled to a base salary of $90,000 for the term, healthcare coverage pursuant to our healthcare insurance plan, reimbursement for certain relocation expenses and rent expense. The Kanzer Employment Agreement also includes confidentiality obligations and inventions assignments by Mr. Kanzer. Mr. Kanzer was not entitled to severance pay upon termination of his employment. On October 1, 2012, the Kanzer Employment Agreement was amended (the “Amended Kanzer Employment Agreement”) and Mr. Kanzer was engaged as our Licensing Associate. In connection with this appointment, Mr. Kanzer entered into a two year agreement with us on a part time basis (2.5 days per week). Pursuant to the Amended Kanzer Employment Agreement, Mr. Kanzer was entitled to a base salary of $150,000 for the term and healthcare coverage pursuant to our healthcare insurance plans. Effective December 5, 2013, Mr. Kanzer was appointed CEO and President of our majority owned subsidiary, Synthetic Biomics, Inc. In connection with this appointment, Mr. Kanzer was entitled to an annual base salary of $195,000 for the term and healthcare coverage pursuant to our healthcare insurance plans. On February 26, 2014, Mr. Kanzer resigned as CEO and President of Synthetic Biomics, Inc. and as a member of our Board of Directors.
The Board of Directors has determined that Mr. Kraws, Mr. Tarriff and Mr. Wolf are independent directors.
| 66 |
Item 14. Principal Accountant Fees and Services
Independent Registered Public Accounting Firm Fees and Services
The following table sets forth the aggregate fees including expenses billed to us for the years ended December 31, 2013 and 2012 by BDO USA, LLP.
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Audit Fees and Expenses(1) | $ | 215,000 | $ | 134,000 | ||||
| $ | 215,000 | $ | 134,000 | |||||
| (1) | Audit fees and expenses were for professional services rendered for the audit and reviews of the consolidated financial statements of the Company, professional services rendered for issuance of consents and assistance with review of documents filed with the SEC. |
The Audit Committee has adopted procedures for pre-approving all audit and non-audit services provided by the independent registered public accounting firm, including the fees and terms of such services. These procedures include reviewing detailed back-up documentation for audit and permitted non-audit services. The documentation includes a description of, and a budgeted amount for, particular categories of non-audit services that are recurring in nature and therefore anticipated at the time that the budget is submitted. Audit Committee approval is required to exceed the pre-approved amount for a particular category of non-audit services and to engage the independent registered public accounting firm for any non-audit services not included in those pre-approved amounts. For both types of pre-approval, the Audit Committee considers whether such services are consistent with the rules on auditor independence promulgated by the SEC and the PCAOB. The Audit Committee also considers whether the independent registered public accounting firm is best positioned to provide the most effective and efficient service, based on such reasons as the auditor’s familiarity with our business, people, culture, accounting systems, risk profile, and whether the services enhance our ability to manage or control risks and improve audit quality. The Audit Committee may form and delegate pre-approval authority to subcommittees consisting of one or more members of the Audit Committee, and such subcommittees must report any pre-approval decisions to the Audit Committee at its next scheduled meeting. All of the services provided by the independent registered public accounting firm were pre-approved by your Audit Committee.
| 67 |
PART IV
Item 15. Exhibits and Financial Statement Schedules and Reports on Form 8-K
| (a)(1) | The following financial statements are included in this Annual Report on Form 10-K for the fiscal years ended December 31, 2013 and 2012. |
| 1. | Independent Registered Public Accounting Firm |
| 2. | Consolidated Balance Sheets as of December 31, 2013 and 2012 |
| 3. | Consolidated Statements of Operations for the years ended December 31, 2013 and 2012 |
| 4. | Consolidated Statements of changes in Stockholders’ Equity for the years ended December 31, 2013 and 2012 |
| 5. | Consolidated Statements of Cash Flows for the years ended December 31, 2013 and 2012 |
| 6. | Notes to Consolidated Financial Statements |
| (a)(2) | All financial statement schedules have been omitted as the required information is either inapplicable or included in the Consolidated Financial Statements or related notes. |
| (a)(3) | The following exhibits are either filed as part of this report or are incorporated herein by reference: |
| 1.1 | Controlled Equity OfferingSMSales Agreement, dated December 11, 2013, between Synthetic Biologics, Inc. and Cantor Fitzgerald & Co. (Incorporated by reference to Exhibit 1.2 to the Registrant’s Registration Statement on Form S-3 filed on July 3, 2013.) | |
| 1.2 | Underwriting Agreement, dated December 11, 2013, between Synthetic Biologics, Inc. and Aegis Capital Corp.-12-13-13 (Incorporated by reference to Exhibit 1.1 to the Registrant’s Current Report on Form 8-K filed on December 13, 2013.) | |
| 1.3 | Amendment No.1 to Controlled Equity OfferingSMSales Agreement, dated December 11, 2013, between Synthetic Biologics, Inc. and Cantor Fitzgerald & Co. (Incorporated by reference to Exhibit 1.2 to the Registrant’s Current Report on Form 8-K filed on December 13, 2013.) |
| 3.1 | Certificate of Incorporation, as amended (Incorporated by reference to (i) Exhibit 3.1 of the Registrant’s Current Report on Form 8-K filed October 16, 2008, (ii) Exhibit 3.1 of the Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2001 filed August 14, 2001 and (iii) Exhibits 3.1, 4.1 and 4.2 of the Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 1998 filed August 14, 1998.) |
| 3.2 | Articles of Merger (Incorporated by reference to Exhibit 3.1 of the Registrant’s Current Report on Form 8-K filed October 19, 2009.) |
| 3.3 | Certificate of Merger filed with the Secretary of State of Delaware (Incorporated by reference to Exhibit 3.2 of the Registrant’s Current Report on Form 8-K filed October 19, 2009.) |
| 3.4 | Articles of Incorporation filed with the Nevada Secretary of State (Incorporated by reference to Exhibit 3.3 of the Registrant’s Current Report on Form 8-K filed October 19, 2009.) |
| 3.5 | By-Laws (Incorporated by reference to (i) Exhibit 3.4 of the Registrant’s Current Report on Form 8-K filed October 19, 2009 and (ii) Exhibit 3.1 of the Registrant’s Current Report on Form 8-K filed June 3, 2010.) |
| 3.6 | Amended and Restated Bylaws Adopted and Effective October 31, 2011 (Incorporated by reference to Exhibit 3.1 of the Registrant’s Current Report on Form 8-K filed November 2, 2011.) |
| 3.7 | Certificate of Amendment to Articles of Incorporation (Incorporated by reference to Exhibit 3.1 of the Registrant’s Current Report on Form 8-K filed February 16, 2012.) |
| 4.1 | Form of Warrant Certificate (Incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K filed December 1, 2006.) |
| *4.2 | 2001 Stock Incentive Plan (Incorporated by reference to Exhibit 4.1 of the Registrant’s Registration Statement on Form S-8 filed January 18, 2008.) |
| *4.3 | 2007 Stock Incentive Plan (Incorporated by reference to Exhibit 4.2 of the Registrant’s Registration Statement on Form S-8 filed January 18, 2008.) |
| *4.4 | 2010 Stock Incentive Plan (Incorporated by reference to Exhibit 4.1 of the Registrant’s Registration Statement on Form S-8 filed November 29, 2010.) |
| 4.5 | Form of Warrant Certificate issued to Enclave Capital LLC (Incorporated by reference to Exhibit 4.1 of the Registrant’s Form 8-K filed July 6, 2010.) |
| 4.6 | Form of Warrant to Purchase Common Stock issued January 2011(Incorporated by reference to Exhibit 10.3 of the Registrant’s Current Report on Form 8-K filed February 2, 2011.) |
| 4.7 | Form of Warrant to Purchase Common Stock issued April 2011(Incorporated by reference to Exhibit 10.3 of the Registrant’s Current Report on Form 8-K filed April 6, 2011.) |
| 4.8 | Form of Exchange Warrant to Purchase Common Stock issued in exchange of the Warrant issued April 2011 (Incorporated by reference to Exhibit 4.1 of the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2011 filed August 15, 2011.) |
| 4.9 | Form of Exchange Warrant to Purchase Common Stock issued in exchange of the Warrant issued February 2011 (Incorporated by reference to Exhibit 4.2 of the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2011 filed August 15, 2011.) |
| 4.10 | Form of Warrant to Purchase Common Stock issued February 2012 (Incorporated by reference to Exhibit 4.10 of the Registrant’s Annual Report on Form 10-K for the year ended December 3, 2011 filed March 30, 2012.) |
| 68 |
| 4.11 | Form of Warrant to Purchase Common Stock issued to Griffin Securities, Inc. on October 30, 2012 (Incorporated by reference to Exhibit 10.4 of the Registrant’s Current Report on Form 8-K filed October 31, 2012.) |
| 4.12 | Form of Warrant Certificate issued to Redington, Inc. (Incorporated by reference to Exhibit 4.13 of the Registrant’s Registration Statement on Form S-1 filed on December 13, 2012.) | |
| 4.13 | Specimen Stock Certificate(Incorporated by reference to Exhibit 4.1 to the Registrant’s Registration Statement on Form S-3 filed on July 3, 2013.) | |
| 4.14 | Amended and Restated 2010 Stock Incentive Plan (Incorporated by reference to Exhibit 4.1 to the Registrant’s Registration Statement on Form S-8 filed on November 15, 2013.) |
| 10.1 | Unit Purchase Agreement (Incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed December 1, 2006.) |
| 10.2 | License Agreement between The Regents of the University of California and Epitope Pharmaceuticals, Inc. (Incorporated by reference to Exhibit 10.22 of the Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2008 filed August 14, 2008.) |
| *10.3 | Form of Director/Officer Indemnification Agreement (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed January 6, 2009.) |
| *10.4 | Warrant Cancellation and Registration Rights Agreement between Accredited Adventures Capital LLC and Adeona (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed January 20, 2009.) |
| 10.5 | Stock Purchase Agreement with Neil O. Colwell and Connie Colwell (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed April 16, 2009.) |
| 10.6 | Agreement and Plan of Reincorporation Merger (Incorporated by reference to Exhibit 1.1 of the Registrant’s Current Report on Form 8-K filed October 19, 2009.) |
| *10.7 | Employment Agreement with James S. Kuo, M.D., (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed February 9, 2010.) |
| 10.8 | Sublicense Agreement between Meda AB, Adeona Pharamaceuticals, Inc. and Pipex Therapeutics, Inc. (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed May 11, 2010.) |
| 10.9 | Non-Disturbance Agreement among Pipex Therapeutics, Inc., Mclean Hospital Corp and Meda AB (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed May 11, 2010.) |
| 10.10 | Placement Agent Agreement with Enclave Capital LLC (Incorporated by reference to Exhibit 1.1 of the Registrant’s Current Report on Form 8-K filed July 6, 2010.) |
| 10.11 | Common Stock Purchase Agreement with Seaside 88,LP (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed July 6, 2010.) |
| 10.12 | Agreement with Chardan Capital Markets, LLC (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed February 2, 2011.) |
| 10.13 | Securities Purchase Agreement with investors (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed February 2, 2011.) |
| 10.14 | McLean Hospital Corporation Exclusive License Agreement (Incorporated by reference to Exhibit 10.21 of the Registrant’s Annual Report on Form 10-K filed March 31, 2011.) |
| 10.15 | Agreement with Chardan Capital Markets, LLC (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed April 6, 2011.) |
| 10.16 | Securities Purchase Agreement with investors (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed April 6, 2011.) |
| 10.17 | Exchange Agreement with respect to Warrant issued April 2011(Incorporated by reference to Exhibit 10.1 of the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2011 filed August 15, 2011.) |
| 10.18 | Exchange Agreement with respect to Warrant issued February 2011(Incorporated by reference to Exhibit 10.2 of the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2011 filed August 15, 2011.) |
| 10.19 | Stock Purchase Agreement with Intrexon Corporation (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed November 21, 2011.) |
| 10.20 | Registration Rights Agreement with Intrexon Corporation (Incorporated by reference to Exhibit 10.3 of the Registrant’s Current Report on Form 8-K filed November 21, 2011.) |
| *10.21 | Employment Agreement with Jeffrey Riley (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed February 6, 2012.) |
| 69 |
| 10.22 | Consulting Agreement with Dr. James Kuo (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed February 6, 2012.) |
| *10.23 | Employment Agreement with C. Evan Ballantyne (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed February 7, 2012.) |
| *10.24 | Employment Agreement with Steve H. Kanzer (Incorporated by reference to Exhibit 10.30 of the Registrant’s Annual Report on Form 10-K for the year ended December 3, 2011 filed March 30, 2012.) |
| 10.25 | Membership Interest Purchase Agreement by and among Synthetic Biologics, Inc., Hartlab LLC, and Adeona Clinical Laboratory, LLC, dated as of March 7, 2012 (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed March 12, 2012.) |
| 10.26 | Pledge and Security Agreement between Synthetic Biologics, Inc. and Hartlab, LLC dated as of March 7, 2012 (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed March 12, 2012.) |
| 10.27 | Non-Recourse Promissory Note between Synthetic Biologics, Inc. and Hartlab, LLC dated as of March 7, 2012 (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed March 12, 2012.) |
| 10.28 | Financial Advisory Agreement with Griffin Securities, Inc. (Incorporated by reference to Exhibit 10.34 of the Registrant’s Annual Report on Form 10-K for the year ended December 3, 2011 filed March 30, 2012.) |
| 10.29 | Exclusive Channel Collaboration Agreement with Intrexon Corporation (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed August 9, 2012.) |
| 10.30 | Stock Purchase Agreement with Intrexon Corporation (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed August 9, 2012.) |
| 10.31 | First Amendment to Registration Rights Agreement between Synthetic Biologics, Inc. and Intrexon Corporation (Incorporated by reference to Exhibit 10.3 of the Registrant’s Current Report on Form 8-K filed August 9, 2012.) |
| 10.32 | Stock Purchase Agreement dated October 25, 2012 with investors (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed October 31, 2012.) |
| 10.33 | Registration Rights Agreement dated October 25, 2012 with investors (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed October 31, 2012.) |
| 10.34 | Joinder Agreement by and among Synthetic Biologics, Inc., NRM VII Holdings I, LLC and Intrexon Corporation (Incorporated by reference to Exhibit 10.3 of the Registrant’s Current Report on Form 8-K filed October 31, 2012.) |
| 10.35 | Asset Purchase Agreement dated November 8, 2012 between Synthetic Biologics, Inc. and Prev ABR LLC (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed November 13, 2012.) |
| *10.36 | Amendment to Employment Agreement dated October 1, 2012 with Steve H. Kanzer (Incorporated by reference to Exhibit 10.42 of the Registrant’s Registration Statement on Form S-1 filed December 13, 2012.) |
| 10.37 | Patent License Agreement dated December 19, 2012 between Synthetic Biologics, Inc. and The University of Texas at Austin (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed December 21, 2012.) |
| 10.38 | Sponsored Research Agreement dated December 19, 2012 between Synthetic Biologics, Inc. and The University of Texas at Austin (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed December 21, 2012.) |
| 10.39 | Exclusive License Agreement with The Regents of The University of California (Incorporated by reference to Exhibit 10.45 of the Registrant’s Annual Report on Form 10-K filed April 16, 2013.) |
| 10.40 | First Amendment to Exclusive License Agreement with The Regents of The University of California (Incorporated by reference to Exhibit 10.46 of the Registrant’s Annual Report on Form 10-K filed April 16, 2013.) |
| 10.41 | Second Amendment to Exclusive License Agreement with The Regents of The University of California (Incorporated by reference to Exhibit 10.47 of the Registrant’s Annual Report on Form 10-K filed April 16, 2013.) |
| 10.42 | Third Amendment to Exclusive License Agreement with The Regents of The University of California (Incorporated by reference to Exhibit 10.48 of the Registrant’s Annual Report on Form 10-K filed April 16, 2013.) |
| 10.43 | Fourth Amendment to Exclusive License Agreement with The Regents of The University of California (Incorporated by reference to Exhibit 10.49 of the Registrant’s Annual Report on Form 10-K filed April 16, 2013.) | |
| 10.44 | Exclusive License Agreement between Synthetic Biologics, Inc., Synthetic Biomics, Inc. and Cedars-Sinai Medical Center dated December 5, 2013 (Incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on December 10, 2013.) | |
| 10.45 | Exclusive Option Agreement between Synthetic Biologics, Inc., Synthetic Biomics, Inc. and Cedars-Sinai Medical Center dated December 5, 2013 (Incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K filed on December 10, 2013.) | |
| 10.46 | Stock Purchase Agreement between Synthetic Biologics, Inc., Synthetic Biomics, Inc. and Cedars-Sinai Medical Center dated December 5, 2013(Incorporated by reference to Exhibit 10.3 to the Registrant’s Current Report on Form 8-K filed on December 10, 2013.) | |
| 10.47 | Stock Purchase Agreement between Synthetic Biologics, Inc., Synthetic Biomics, Inc. and Mark Pimentel dated December 5, 2013 (Incorporated by reference to Exhibit 10.4 to the Registrant’s Current Report on Form 8-K filed on December 10, 2013.) | |
| 10.48 | Stock Purchase Agreement between Synthetic Biologics, Inc., Synthetic Biomics, Inc. and Cedars-Sinai Medical Center dated December 5, 2013(Incorporated by reference to Exhibit 10.5 to the Registrant’s Current Report on Form 8-K filed on December 10, 2013.) | |
| 10.49 | First Amendment to Exclusive License Agreement. (1) |
| 21 | List of Subsidiaries (1) |
| 23.1 | Consent of Independent Registered Public Accounting Firm (BDO USA, LLP) (1) |
| 31.1 | Certification of Jeffrey Riley, Chief Executive Officer, pursuant to Rule 13a-14(a)/15d-14(a) (1) |
| 31.2 | Certification of C. Evan Ballantyne, Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) (1) |
| 32.1 | Certification of Jeffrey Riley, Chief Executive Officer pursuant to Section 1350 of the Sarbanes-Oxley Act of 2002 (1) |
| 32.2 | Certification C. Evan Ballantyne, Chief Financial Officer pursuant to Section 1350 of the Sarbanes-Oxley Act of 2002 (1) |
| 70 |
**+101.INS XBRL Instance Document
**+101.SCH XBRL Taxonomy Extension Schema Document
**+101.CAL XBRL Taxonomy Extension Calculation Linkbase Document
**+101.DEF XBRL Taxonomy Extension Definition Linkbase Document
**+101.LAB XBRL Taxonomy Extension Label Linkbase Document
**+101.PRE XBRL Taxonomy Extension Presentation Linkbase Document
(1) Filed herewith.
* Management contract or compensatory plan or arrangement required to be identified pursuant to Item 15(a)(3) of this report
** As provided in Rule 406T of Regulation S-T, this information is deemed furnished and not filed for purposes of Sections 11 and 12 of the Securities Act of 1933, as amended, and Section 18 of the Securities Exchange Act of 1934, as amended.
| 71 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned.
| SYNTHETIC BIOLOGICS, INC. | |||
| By: | /s/ Jeffrey Riley | ||
| Jeffrey Riley | |||
| Chief Executive Officer and Director | |||
| (Principal Executive Officer) | |||
| Date: March 31, 2014 | |||
| By: | /s/ C. Evan Ballantyne | ||
| C. Evan Ballantyne | |||
| Chief Financial Officer | |||
| (Principal Financial and Principal Accounting Officer) | |||
| Date: March 31, 2014 | |||
Pursuant to the requirements of the Securities Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| By: | /s/ Jeffrey Riley | |||
| Date: March 31, 2014 | Jeffrey Riley | |||
| Chief Executive Officer and Director | ||||
| (Principal Executive Officer) | ||||
| Date: March 31, 2014 | By: | /s/ Jeffrey J. Kraws | ||
| Jeffrey J. Kraws | ||||
| Chairman | ||||
| Date: March 31, 2014 | By: | /s/ Scott L. Tarriff | ||
| Scott L. Tarriff | ||||
| Director | ||||
| Date: March 31, 2014 | By: | /s/ Jeffrey Wolf | ||
| Jeffrey Wolf | ||||
| Director | ||||
| 72 |
GLOSSARY
| Term | Definition | |
| Adverse Event | Any adverse change in health or “side-effect” that occurs in a person participating in a clinical trial, from the time they consent to joining the trial until a pre-specified period of time after their treatment has been completed. | |
| BLA - Biologics License Application | An application in the U.S. through which biologic sponsors formally propose that the FDA approve a new biologic for sale and marketing. | |
| Clinical Study/Trial | A research study that is conducted to find out if a treatment or procedure is safe and/or effective in humans. | |
| Controlled Clinical Trial | A clinical study that compares patients receiving a specific treatment to patients receiving an alternate treatment for the condition of interest. The alternate treatment may be another active treatment, standard of care for the condition and/or a placebo (inactive) treatment. | |
| Double-blinded Study/Trial | Both the participant and the researcher are unaware of who is receiving the active treatment or the placebo. | |
| FDA - Food & Drug Administration | The U.S. government agency that ensures that medicines, medical devices, prescription medical foods and radiation-emitting consumer products are safe and effective. Authorized by Congress to enforce the Federal Food, Drug, and Cosmetic Act and several other public health laws, the agency monitors the manufacture, import, transport, storage, and sale of $1 trillion worth of goods annually. | |
| GMP - Good Manufacturing Practice | Regulations that require that manufacturers, processors, and packagers of drugs, medical devices, some food, and blood take proactive steps to ensure that their products are consistently produced, pure, and stable. GMP regulations require a quality approach to manufacturing, enabling companies to minimize or eliminate instances of contamination, mix-ups, and errors. | |
| Monoclonal Antibodies (mAbs) | Acting as the body's army, antibodies are proteins, generally found in the bloodstream, that provide immunity in detecting and destroying pathogens, such as viruses and bacteria and their associated toxins. | |
| IND - Investigational New Drug | An application in the U.S. submitted to the FDA for a new drug or biologic that, if allowed, will be used in a clinical trial. | |
| IRB - Institutional Review Board | A committee designated to formally approve, monitor, and review biomedical research at an institution involving human studies. Institutional Review Boards aim to protect the rights and welfare of the research subjects. | |
| NDA - New Drug Application | An application in the U.S. through which drug sponsors formally propose that the FDA approve a new pharmaceutical for sale and marketing. | |
| Open-label Clinical Study/Trial | A trial in which both the treating physician and the patient know they are receiving the experimental treatment. | |
| Phase I Clinical Trial | A Phase I trial represents an initial study in a small group of patients to primarily test for safety. |
| 73 |
| Phase II Clinical Trial | A Phase II trial represents a study in a larger number of patients to assess the safety and efficacy of a product. | |
| Phase III Clinical Trial | Phase III trials are initiated to establish safety and efficacy in an expanded patient population and at multiple clinical trial sites and are generally larger than trials in earlier phases of development. | |
| Placebo | An inactive pill or liquid. Many studies compare an active drug to a placebo to determine whether any changes seen during the study can be attributed to the active drug. | |
| Principal Investigator | This is the study director who is ultimately responsible for the conduct of the study. | |
| Prospective Clinical Study/Trial | A clinical study/trial in which participants are identified and then followed throughout the study going forward in time. | |
| Protocol | A clinical study/trial’s plan - includes the schedule of tests, requirements for participation, procedures, and medications. | |
| Randomized Study/Trial | Participants in a study are assigned by chance to either one or more of the active treatment group(s) or the placebo group. | |
| Single-blinded Study/Trial | One party, either the participant or the researcher, does not know if the participant is taking the active treatment or the placebo. | |
| Study/Trial Coordinator | Staff member who is often the primary contact for research participants and coordinates their care and evaluations throughout the study. |
| 74 |