As filed with the Securities and Exchange Commission on October 10, 2018
Registration No. 333-227400
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT
NO. 2
TO
FORM S-1
REGISTRATION STATEMENT
UNDER THE SECURITIES ACT OF 1933

Synthetic Biologics, Inc.
(Exact name of Registrant as specified in its charter)
| Nevada | 2834 | 13-3808303 |
| (State or other jurisdiction of | (Primary Standard Industrial | (I.R.S. Employer |
| incorporation or organization) | Classification Code Number) | Identification Number) |
9605 Medical Center Drive, Suite 270
Rockville, Maryland 20850
(301) 417-4364
(Address, including zip code, and telephone number, including area code, of Registrant’s principal executive offices)
Steven A. Shallcross
Interim Chief Executive Officer and
Chief Financial Officer
Synthetic Biologics, Inc.
9605 Medical Center Drive, Suite 270
Rockville, Maryland 20850
(301) 417-4364
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Leslie Marlow, Esq. Hank Gracin, Esq. Patrick J. Egan, Esq. The Chrysler Building 405 Lexington Avenue, 26th Floor New York, New York 10174 (212) 907-6457 | Oded Har-Even, Esq. Robert V. Condon III, Esq. Zysman, Aharoni, Gayer and Sullivan & Worcester LLP 1633 Broadway New York, NY 10019 (212) 660-5000 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box.þ
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act of 1933, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act of 1933, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act of 1933, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.¨
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act of 1934.
| Large accelerated filer | ¨ | Accelerated filer | þ |
| Non-accelerated filer | ¨ | Smaller reporting company | ¨ |
| Emerging growth company | ¨ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act.¨
CALCULATION OF REGISTRATION FEE
| Title of each class of securities to be registered (1) | Proposed maximum aggregate offering price(1)(2)(3) | Amount of registration fee(4) | ||||||
| Class A Units consisting of:(5) | $ | 23,000,000 | $ | 2,787.60 | ||||
| (i) Common Stock, par value $0.001 par value | ||||||||
| (ii) Warrants to purchase shares of Common Stock (6) | ||||||||
| Class B Units consisting of:(5) | $ | 23,000,000 | $ | 2,787.60 | ||||
| (i) Series B Convertible Preferred Stock, par value $0.001 per share | ||||||||
| (ii) Warrants to purchase Common Stock (6) | ||||||||
| (iii) Common Stock issuable upon conversion of the Series B Convertible Preferred Stock(6) | ||||||||
| Common Stock issuable upon exercise of Warrants (7) | $ | 55,200,000 | $ | 6,690.24 | ||||
| Total | $ | 101,200,000 | $ | 12,265.44 | (8) | |||
| (1) | Estimated solely for the purpose of calculating the amount of the registration fee pursuant to Rule 457(o) of the Securities Act of 1933, as amended (the “Securities Act”). |
| (2) | Pursuant to Rule 416, the securities being registered hereunder include such indeterminate number of additional securities as may be issued after the date hereof as a result of stock splits, stock dividends or similar transactions. |
| (3) | Includes shares of common stock the underwriters have the option to purchase solely to cover over-allotments, if any. |
| (4) | Calculated under Section 6(b) of the Securities Act as .00012120 of the proposed maximum aggregate offering price. |
| (5) | The proposed maximum offering price of the Class A Units proposed to be sold in the offering will be reduced on a dollar-for-dollar basis on the offering price of any Class B Units offered and sold in the offering, and as such the proposed aggregate maximum offering price of the Class A Units and Class B Units if any, is $23,000,000. |
| (6) | No additional registration fee is payable pursuant to Rule 457(i) under the Securities Act. |
| (7) | The Warrants are exercisable at a per share exercise price equal to 120% of the public offering price of one share of Common Stock. The proposed maximum aggregate public offering price of the shares of Common Stock issuable upon exercise of the Warrants was calculated to be $55,200,000, which is equal to 120% of $46,000,000. |
| (8) | A filing fee of $8,641.56 was previously paid. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities, nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
| PRELIMINARY PROSPECTUS | Subject to completion | dated October 10, 2018 |
Up to 9,523,809 Class A Units Consisting of Shares of Common Stock and Warrants
Up to 20,000 Class B Units Consisting of Series B Convertible Preferred Stock and Warrants
9,523,809 Shares of Common Stock Underlying the Series B Convertible Preferred Stock and
9,523,809 Shares of Common Stock Underlying the Warrants

We are offering up to 9,523,809 Class A Units, each Class A Unit consisting of one share of our common stock and one warrant to purchase one share of our common stock at a price of 120% of the public offering price of the Class A Units. Each warrant will be exercisable upon issuance and will expire three years from date of issuance. The shares of common stock and warrants that are part of a Class A Unit are immediately separable and will be issued separately in this offering. We are also offering the shares of common stock issuable upon exercise of warrants sold in Class A Units.
We are also offering to each purchaser whose purchase of Class A Units in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering, the opportunity in lieu of purchasing Class A Units, to purchase Class B Units. Each Class B Unit will consist of one share of Series B Convertible Preferred Stock, or the Series B Preferred, with a stated value of $1,000 and convertible into shares of our common stock at the public offering price of the Class A Units, together with the equivalent number of warrants as would have been issued to such purchaser of Class B Units if they had purchased Class A Units based on the public offering price. The Series B Preferred do not generally have any voting rights unless and until converted into shares of common stock. The shares of Series B Preferred and warrants that are part of a Class B Unit are immediately separable and will be issued separately in this offering. The number of shares of our common stock outstanding after this offering will fluctuate depending on how many Class B Units are sold in this offering and whether and to what extent holders of Series B Preferred shares convert their shares to common stock. We are also offering the shares of common stock issuable upon exercise of warrants sold in Class B Units and upon conversion of the Series B Preferred. For each Class B Unit we sell, the number of Class A Units we are offering will be decreased on a dollar-for-dollar basis.Because we will issue a common stock purchase warrant as part of the Class A Unit or Class B unit, the number of warrants sold in this offering will not change as a result of the change in the mix of Class A Units and Class B Units.
Our common stock is listed on the NYSE American under the symbol “SYN”. On October 8, 2018, the last reported sale price of our common stock on the NYSE American was $2.10 per share. The public offering price of the Class A Units will be determined between us, the underwriters and investors based on market conditions at the time of pricing, and may be at a discount to the current market price of our common stock. Therefore, the recent market price used throughout this prospectus may not be indicative of the final offering price. The public offering price of the Class B Units will be $1,000 per unit. There is no established trading market for the warrants or the Series B Preferred and we do not expect a market to develop. In addition, we do not intend to apply for the listing of the warrants or the Series B Preferred on any national securities exchange or other trading market. Without an active trading market, the liquidity of the warrants and the Series B Preferred will be limited.
Investing in our securities involves risk. See “Risk Factors” beginning on page 15 of this prospectus for a discussion of information that should be considered in connection with an investment in our securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| Per Class A Unit | Per Class B Unit | Total | ||||||||||
| Public offering price | $ | $ | $ | |||||||||
| Underwriting discounts and commissions(1) | $ | $ | $ | |||||||||
| Proceeds, before expenses, to us | $ | $ | $ | |||||||||
| (1) | We have also agreed to reimburse the underwriters for certain expenses incurred in connection with this offering. See “Underwriting” beginning on page 47 of this prospectus for a description of the compensation payable to the underwriters. |
We have granted a 45-day option to the representative of the underwriters to purchase additional shares of common stock and/or additional warrants in amounts up to 15% of the common stock, warrants and/or common stock issuable upon conversion of the Series B Preferred included in the Class B Units sold in the offering, solely to cover over-allotments, if any.
We expect that delivery of the securities offered hereby against payment will be made on or about , 2018.
A.G.P.
The date of this prospectus is , 2018.
TABLE OF CONTENTS
You should rely only on the information contained in this prospectus and any free writing prospectus that we have authorized for use in connection with this offering. Neither we nor the underwriters have authorized anyone to provide you with information that is different. We are offering to sell, and seeking offers to buy, the securities covered hereby only in jurisdictions where offers and sales are permitted. The information in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or any sale of the securities covered hereby. Our business, financial condition, results of operations and prospects may have changed since that date. We are not, and the underwriters are not, making an offer of these securities in any jurisdiction where the offer is not permitted. You should also read and consider the information in the documents to which we have referred you under the caption “Where You Can Find Additional Information” in the prospectus. In addition, this prospectus contains summaries of certain provisions contained in some of the documents described herein, but reference is made to the actual documents for complete information. All of the summaries are qualified in their entirety by the actual documents. Copies of some of the documents referred to herein have been filed, will be filed or will be incorporated by reference as exhibits to the registration statement of which this prospectus is a part, and you may obtain copies of those documents as described below under the heading “Where You Can Find Additional Information.”
For investors outside the United States: Neither we nor any of the underwriters have taken any action that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the securities covered hereby and the distribution of this prospectus outside of the United States.
This prospectus includes statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. We believe that the data obtained from these industry publications and third-party research, surveys and studies are reliable. We are ultimately responsible for all disclosure included in this prospectus.
Except where the context requires otherwise, in this prospectus the “Company,” “Synthetic Biologics,” “Synthetic,” “we,” “us” and “our” refer to Synthetic Biologics, Inc., a Nevada corporation, and, where appropriate, its wholly owned subsidiaries.
This summary highlights information contained in other parts of this prospectus or incorporated by reference into this prospectus from our filings with the Securities and Exchange Commission, or SEC, listed in the section of the prospectus entitled “Incorporation of Certain Documents by Reference.” Because it is only a summary, it does not contain all of the information that you should consider before purchasing our securities in this offering and it is qualified in its entirety by, and should be read in conjunction with, the more detailed information appearing elsewhere or incorporated by reference into this prospectus. You should read the entire prospectus, the registration statement of which this prospectus is a part, and the information incorporated by reference herein in their entirety, including the “Risk Factors” and our financial statements and the related notes incorporated by reference into this prospectus, before purchasing our securities in this offering.
Our Business
Overview
We are a late-stage clinical company focused on developing therapeutics designed to preserve the microbiome to protect and restore the health of patients. Our lead candidates poised for Phase 3 development are: (1) SYN-004 (ribaxamase) which is designed to protect the gut microbiome from the effects of certain commonly used intravenous (IV) beta-lactam antibiotics for the prevention ofC. difficile infection (CDI), overgrowth of pathogenic organisms and the emergence of antimicrobial resistance (AMR), and (2) SYN-010 which is intended to reduce the impact of methane-producing organisms in the gut microbiome to treat an underlying cause of irritable bowel syndrome with constipation (IBS-C). Our preclinical pursuits include an oral formulation of the enzyme intestinal alkaline phosphatase (IAP) to treat both local GI and systemic diseases as well as monoclonal antibody therapies for the prevention and treatment of pertussis, and novel discovery stage biotherapeutics for the treatment of phenylketonuria (PKU).
Product Pipeline:
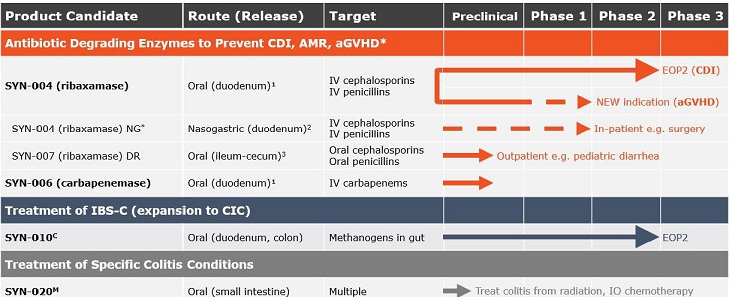
¹Designed to degrade excess antibiotic excreted into the GI tract before the antibiotic reaches the colon and causes dysbiosis.
²For use in patients who can’t swallow the capsule or its contents.
³Designed to degrade non-absorbed antibiotic remaining in the GI tract before the antibiotic reaches the colon and causes dysbiosis.
C - License and collaboration with Cedars-Sinai Medical Center
M - Scientific collaboration with Massachusetts General Hospital
* Development of nasogastric tube (NG) dosing is based on guidance and recommendations by physicians, surgeons, KOLs, our expert steering committee and the U.S. Food and Drug Administration (FDA).
| 3 |
Summary of Clinical and Preclinical Programs
| Therapeutic Area | Product Candidate | Status | ||
| Prevention of CDI, overgrowth of pathogenic organisms and AMR (Degrade IV beta-lactam antibiotics) | SYN-004 (ribaxamase) (oral enzyme) | ● Reported supportive Phase 1a/1b data (1Q 2015)
● Reported supportive topline data from two Phase 2a clinical trials (4Q 2015 & 2Q 2016)
● Initiated Phase 2b proof-of-concept clinical trial (3Q 2015)
● Received USAN approval of the generic name “ribaxamase” for SYN-004 (July 2016)
● Completed Enrollment of Phase 2b proof-of-concept clinical trial (3Q 2016)
● Awarded contract by the CDC (4Q 2016)
● Announced positive topline data from Phase 2b proof-of-concept clinical trial, including achievement of primary endpoint of significantly reducing CDI (1Q 2017)
● Announced additional results from Phase 2b proof-of-concept clinical trial demonstrating SYN-004 (ribaxamase) protected and maintained the naturally occurring composition of gut microbes from antibiotic-mediated dysbiosis in treated patients (2Q 2017)
● Announced additional results from Phase 2b proof-of-concept clinical trial funded by a contract awarded by the CDC, demonstrating that SYN-004 (ribaxamase) prevented significant change to the presence of certain AMR genes in the gut resistome of patients receiving SYN-004 compared to placebo (3Q 2017)
● Presented additional supportive results regarding several exploratory endpoints from Phase 2b proof-of-concept clinical trial designed to evaluate SYN-004’s (ribaxamase) ability to protect the gut microbiome from opportunistic bacterial infections and prevent the emergence of antimicrobial resistance (AMR) in the gut microbiome (4Q 2017)
● Reached preliminary agreement with the FDA on key elements of a proposed Phase 3 clinical trial program, including de-coupled co-primary endpoints designed to evaluate efficacy separate from safety in a patient population being treated with a representative selection of IV-beta-lactam antibiotics (1H 2018)
● End of Phase 2 meeting with FDA held to solidify remaining elements of planned Phase 3 clinical trial (3Q 2018)
● Expect results from End of Phase 2 meeting with FDA (4Q 2018)
● Clarified market/partner needs and identified potential additional indications for SYN-004 in specialty patient populations such as allogenic hematopoietic cell transplant recipients
● Plan to initiate clinical trial(s) (2H 2019) which may include a broad Phase 3 clinical trial and/or Phase 1/2 clinical trial(s) in a specialty population leading to a subsequent Phase 3 clinical trial |
| Treatment of IBS-C | SYN-010 (oral modified-release lovastatin lactone) | ● Collaboration with Cedars-Sinai Medical Center (“CSMC”)
● Reported supportive topline data from two Phase 2 clinical trials (4Q 2015 & 1Q 2016)
● Received Type C meeting responses from FDA regarding late-stage aspects of clinical pathway (2Q 2016)
● Presented detailed data supporting previously reported positive topline data from two Phase 2 clinical trials at DDW (May 2016) |
| 4 |
● Held End of Phase 2 meeting with FDA (July 2016)
● Confirmed key elements of Pivotal Phase 2b/3 clinical trial design pursuant to consultations with FDA (1Q 2017) | ||||
● Announced issuance of key U.S. composition of matter patent providing important intellectual property protection in the U.S until at least 2035 (Q2 2018)
● Entered into agreement with CSMC for an investigator-sponsored Phase 2 clinical study of SYN-010 to evaluate SYN-010 dose response and inform Phase 3 clinical development (Q3 2018)
● Anticipate dosing first patient in the Phase 2b investigator sponsored clinical study during Q4 2018
● Anticipate data readout from the Phase 2b investigator sponsored clinical study during 2H 2019 | ||||
| Prevention of CDI, overgrowth of pathogenic organisms and AMR (Degrade IV carbapenem antibiotics) | SYN-006 (oral enzyme) | ● Identified P2A as a potent carbapenemase that is stable in the GI tract
● Manufactured and formulated research lot for oral delivery (2017)
● Demonstrated microbiome protection in a pig model of ertapenem administration (Q1 2018) | ||
| Prevention of CDI, overgrowth of pathogenic organisms and AMR (Degrade oral beta-lactam antibiotics) | SYN-007 (oral enzyme) | ● Preclinical work ongoing to expand the utility of SYN-004 (ribaxamase) for use with oral beta-lactam antibiotics
● Presented supportive data from canine animal model at the Microbiome World Congress, America (Q4 2017)
● Reported supportive data from a second canine animal model demonstrating that when co-administered with oral amoxicillin and oral Augmentin, oral SYN-007 did not interfere with systemic absorption of antibiotics but did diminish microbiome damage associated with these antibiotics (2Q 2018) | ||
| Preserve gut barrier, treat local GI inflammation, restore gut microbiome | SYN-020 (oral IAP enzyme) | ● Generated high expressing manufacturing cell lines for intestinal alkaline phosphatase (IAP) (1H 2017)
● Identified downstream process and tablet formulations (2H 2017)
● Identified three potential clinical indications in areas of unmet medical need including, enterocolitis associated with radiation therapy, enterocolitis associated with checkpoint inhibitor therapy for cancer, and microscopic colitis (2H 2018)
● Ongoing preclinical efficacy studies
● Anticipated IND filing (Q4 2019)
● Plan to initiate Phase 1 clinical trial (Q1 2020) | ||
| Prevention and treatment of pertussis | SYN-005 (monoclonal antibody therapies) | ● Reported supportive preclinical research findings (2014)
● The University of Texas at Austin (“UT Austin”) received a grant from the Bill and Melinda Gates Foundation to support a preclinical study to evaluate the prophylactic capability of SYN-005 (4Q 2015)
● Reported supportive preclinical data demonstrating hu1B7, a component of SYN-005, provided protection from pertussis for five weeks in neonatal non-human primate study (Q2 2017)
● Reported supportive preclinical data demonstrating that an extended half-life version of hu1B7, a component of SYN-005, provided protection from pertussis for five weeks in a non-human neonatal primate study (Q4 2017)
● Collaborations with Intrexon and UT Austin |
| 5 |
Our Microbiome-Focused Pipeline
Our SYN-004 (ribaxamase) and SYN-010 programs are focused on protecting the healthy function of the gut microbiome, or gut flora, which is composed of billions of microbial organisms including a natural balance of both “good” beneficial species and potentially “bad” pathogenic species. When the natural balance or normal function of these microbial species is disrupted, a person’s health can be compromised. All of our programs are supported by our growing intellectual property portfolio. We are maintaining and building our patent portfolio through: filing new patent applications; prosecuting existing applications; and licensing and acquiring new patents and patent applications. Our plan remains focused on the advancement of our two late-stage clinical programs. We continue to actively manage resources in preparation for the advancement of our two late-stage microbiome-focused clinical programs, including our pursuit of opportunities that will allow us to establish the clinical infrastructure and financial resources necessary to successfully initiate and complete this plan.
SYN-004 (ribaxamase) — Prevention of C. difficile infections (CDI), overgrowth by pathogenic organisms, and the emergence of antimicrobial resistance (AMR)
SYN-004 (ribaxamase) is a proprietary oral 75 mg capsule prophylactic therapy designed to degrade certain IV beta-lactam antibiotics excreted into the GI tract and maintain the natural balance of the gut microbiome to prevent CDI, reduce overgrowth of pathogenic organisms, and suppress the emergence of antimicrobial-resistant organisms. Published clinical literature has also suggested that preventing microbiome damage caused by IV beta-lactam antibiotics excreted into the GI tract may have potential therapeutic benefit as a means of preventing acute graft-vs-host disease in hematopoietic cell transplant patients. SYN-004 (ribaxamase) is a beta-lactamase enzyme intended to be administered as two-75 mg capsules which, when released in the proximal small intestine, can degrade beta-lactam antibiotics in the GI tract without altering systemic antibiotic levels. Beta-lactam antibiotics are a mainstay in hospital infection management and include the commonly used penicillin and cephalosporin classes of antibiotics.
In November 2012, we acquired a series of oral beta-lactamase enzymes (P1A, P2A and P3A) and related assets targeting the prevention of CDI, the leading healthcare-associated infection that generally occurs secondary to treatment with IV antibiotics from Prev ABR LLC. The acquired assets include a pre-Investigational New Drug (IND) package for P3A, Phase 1 and Phase 2 clinical data for P1A, manufacturing processes and data, and a portfolio of issued and pending U.S. and foreign patents intended to support an IND and Biologics License Application (BLA) with the FDA. Utilizing this portfolio of assets, we developed a proprietary, second generation oral beta-lactamase enzyme product candidate that we now refer to as SYN-004 or by its generic name “ribaxamase”.
Compared to the first generation oral enzyme candidate of P1A, we believe that the second generation candidate, SYN-004 (ribaxamase), will have activity against a broader spectrum of beta-lactam antibiotics, including both penicillins and certain cephalosporins. Due to the structural similarities between P1A and SYN-004 (ribaxamase), and based on previous discussions with the FDA, certain preclinical data collected on P1A was used in support of an IND application for SYN-004 (ribaxamase).
Specifically, P1A had been evaluated in four Phase 1 and one Phase 2 clinical trials conducted in Europe. In total, 112 patients and 143 healthy volunteers participated in these studies.
C. difficile
C.difficile is the leading type of hospital acquired infection and is frequently associated with IV beta-lactam antibiotic treatment. According to a paper published in BMC Infectious Diseases (Desai K et al. BMC Infect Dis. 2016; 16: 303) the economic cost of CDI was approximately $5.4 billion in 2016 ($4.7 billion in healthcare settings; $725 million in the community) in the U.S., mostly due to hospitalizations. CDI is a rising global hospital acquired infection (HAI) problem in which the toxins produced byC. difficile bacteria result inC. difficile associated diarrhea (CDAD), and in the most serious cases, pseudomembranous colitis (severe inflammation of the lower GI tract) that can lead to death. The CDC identifiedC. difficile as an “urgent public health threat,” particularly given its resistance to many drugs used to treat other infections. CDI is a major unintended risk associated with the prophylactic or therapeutic use of IV antibiotics, which may alter the natural balance of microflora that normally protect the GI tract, leading toC. difficile overgrowth and infection. Other risk factors for CDI include hospitalization, prolonged length of stay (estimated at 7 days), underlying illness, and immune-compromising conditions including the administration of chemotherapy and advanced age. In addition, approximately 20% of patients who have been diagnosed with CDI experience a recurrence of CDI within one to three months.
| 6 |
Limitations of Current Treatments and Market Opportunity
CDI is a widespread and often drug resistant infectious disease. According to an article published in the New England Journal of Medicine (Leffler DA et al. N Engl J Med 2015; 372:1539-1548), it is estimated that 453,000 patients are infected withC. difficile annually in the U.S., and it has been reported that approximately 29,000 patients die due to CDI-associated complications each year. Controlling the spread of CDI has proven challenging, as theC. difficile spores are easily transferred to patients via normal contact with healthcare personnel and with inanimate objects. There is currently no vaccine or approved product for the prevention of primary (incident) CDI.
According to IMS Health Incorporated,* in 2016, 227 million doses of SYN-004 (ribaxamase)-addressable intravenous Penicillin and Cephalosporin antibiotics were administered in the United States which may contribute to the onset of CDI. Additional data derived from IMS Health Incorporated states that in 2016, the worldwide market for SYN-004 (ribaxamase)-addressable intravenous beta-lactam antibiotics was approximately 7.5 billion doses, which may represent a multi-billion dollar opportunity for us. According to the CDC reportAntibiotic Resistance Threats in the United States, 2013, at least 2 million people in the U.S. each year acquire serious infections with bacteria that are resistant to one or more of the antibiotics designed to treat those infections, which results in an estimated $20 billion in excess direct healthcare costs.
| * | This information is an estimate derived from the use of information under license from the following IMS Health Incorporated information service: IMS Health Analytics for the full year 2016. IMS expressly reserves all rights, including rights of copying, distribution, and republication. |
Clinical Update
On April 23, 2018, we announced that we had reached preliminary agreement with the FDA on key elements of a proposed clinical trial program for our planned Phase 3 clinical trial for ribaxamase. In accordance with recommendations and guidance received from the FDA, we expect the Phase 3 trial to evaluate the efficacy and safety of ribaxamase as separate, co-primary endpoints in a patient population being treated with a representative selection of intravenous (IV) beta-lactam antibiotics, which will include ceftriaxone and piperacillin/tazobactam. The inclusion of more than one beta-lactam antibiotic in this trial is intended to evaluate the potential utility of ribaxamase for co-administration with a greater number of cephalosporin and penicillin beta-lactam antibiotics. The proposed Phase 3 clinical trial discussed with the FDA will comprise a global, event-driven clinical trial with a fixed maximum number of patients and will seek to evaluate the efficacy and safety of ribaxamase in a broader patient population by enrolling patients with a variety of underlying infections. We expect the primary efficacy endpoint of the proposed Phase 3 trial will be the reduction in the incidence of CDI in the ribaxamase treatment group compared to placebo. We have also reached preliminary agreement with the FDA to evaluate mortality risk as the primary safety endpoint for this trial, which will be separate from the primary efficacy endpoint of reduction of the incidence of CDI. The designation of efficacy and safety as separate and decoupled endpoints is critical for clinical studies of this nature, where the underlying population is projected to have a comparatively high incidence of safety events that may significantly dilute the smaller number of CDI events.
We plan to continue collaborative discussions with the FDA to solidify the remaining details of the proposed Phase 3 clinical trial program during an anticipated End of Phase 2 meeting with the FDA in the third quarter of 2018. In parallel with clinical and regulatory efforts, we have recently completed a Health Economics Outcomes Research study, which was conducted to generate key insights on how we can expect Health Care Practitioners, or HCPs, to evaluate patient access for ribaxamase while also providing a framework for potential reimbursement strategies. After evaluating findings from the study, and after extensive discussions with pharmaceutical companies, physicians, research institutions and clinical development groups worldwide, we believe that there is significant potential value in exploring the development of SYN-004 (ribaxamase) in a more narrow patient population where the incidence of the disease endpoint is high and the clinical development may be less costly. One potential narrow patient population for SYN-004 could be allogenic hematopoietic cell transplant (HCT) recipients, who have a very high risk of CDI, VRE colonization and potentially fatal bacteremia, and acute-graft-vs-host disease. Published literature has demonstrated a strong association between these adverse outcomes and microbiome damage caused by IV beta-lactam antibiotics in these patients. Further examination and discussions with key opinion leaders (KOLs) who are experts in allogenic HCT are ongoing to evaluate a potential clinical development pathway forward for SYN-004 in such a narrow, specialty patient population.
Contingent on potential interest from prospective partners and/or appropriate funding, we anticipate initiating the Phase 3 clinical trial currently under discussion with the FDA in 2H 2019 which will evaluate SYN-004 (ribaxamase) effects on CDI in a broad and diverse patient population. In parallel, discussions with KOLs are ongoing to determine if further investigation in the form of a potential Phase 1 and/or Phase 2 clinical trial(s) evaluating SYN-004 (ribaxamase) in a specialized patient population such as allogenic HCT patients may also and/or alternatively be pursued in 2H 2019. If it is determined that the clinical advancement of SYN-004 is more favorable and significantly less costly in a specialized patient population, we may elect to prioritize and pursue this strategy in advance of pursuing the broader, Phase 3 clinical program currently under discussion with the FDA. If approved by the FDA, SYN-004 (ribaxamase) would be the first available drug designed to prevent primary Clostridium difficile infection by protecting the gut microbiome from antibiotic-mediated dysbiosis.
| 7 |
SYN-010 — Treatment of Irritable Bowel Syndrome with Constipation (IBS-C)
SYN-010 is our proprietary, modified-release formulation of lovastatin lactone that is intended to reduce methane production by certain microorganisms (M. smithii) in the gut while minimizing disruption to the microbiome. Methane produced byM. smithii is an underlying cause of pain, bloating and constipation associated with IBS-C, and published reports have associated higher intestinal methane production with increased constipation severity in IBS-C patients. SYN-010 is intended to act primarily in the intestinal lumen while avoiding systemic absorption, thereby targeting the major cause of IBS-C, not just the patient’s symptoms.
In December 2013, through our subsidiary Synthetic Biomics, Inc. (SYN Biomics), we entered into a worldwide exclusive license agreement with Cedars-Sinai Medical Center (CSMC) and acquired the rights to develop products for therapeutic and prophylactic treatments of acute and chronic diseases, including the development of SYN-010 to target IBS-C. We licensed from CSMC a portfolio of intellectual property comprised of several U.S. and foreign patents and pending patent applications for various fields of use, including IBS-C, obesity and diabetes. An investigational team, led by Mark Pimentel, M.D. at CSMC, discovered that these products may reduce the production of methane gas by certain GI microorganisms.
We believe SYN-010 may reduce the impact of methane producing organisms on IBS-C.
Irritable Bowel Syndrome
IBS is a functional GI disorder characterized by gas, abdominal pain, bloating and diarrhea or constipation, or alternating episodes of both. The illness affects both men and women; however, two-thirds of diagnosed sufferers are women. The onset of IBS can begin anytime from adolescence to adulthood. Four bowel patterns may be seen with IBS including: IBS-C (constipation predominant), IBS-D (diarrhea predominant), IBS-M (mixed diarrhea and constipation) and IBS-U (unsubtyped). According to GlobalData’s IBS — Global Drug Forecast and Market Analysis to 2023 (December 2014 ), the prevalence of IBS in adults in the United States, Europe and Japan was expected to be 41.1 million in 2016, and it has been reported that up to 20 percent of all IBS patients have IBS-C. Extensive studies conducted by Dr. Pimentel and collaborators have shown that overproduction of methane gas is directly associated with bloating, pain and constipation in IBS-C patients. Investigators at CSMC have discovered that inhibiting intestinal methane production may reverse constipation associated with IBS-C, and may be beneficial in treating other major diseases such as obesity, insulin resistance and type 2 diabetes.
Limitations of Current Treatments and Market Opportunity
Currently, the FDA approved therapies for the treatment of IBS-C include prescription and over-the-counter laxatives, which provide patients with temporary symptomatic relief and often cause diarrhea, but are not designed to and do not treat the underlying cause of pain, bloating and constipation associated with IBS-C. Additionally, these same therapies may come with undesirable safety side-effect profiles, the most common of which is diarrhea. As a result, these therapies have struggled to find adoption in several key markets, including Europe. We believe this presents an important opportunity for SYN-010. Towards the end of 2017, we engaged outside consultants to evaluate the potential regulatory pathway towards EMA marketing approval. According to IMS Health Analytics, U.S. sales in 2016 for IBS-C and Chronic Idiopathic Constipation (CIC) therapeutics as well as OTC laxatives/products were approximately $2.5 billion, representing a constant annual growth rate (CAGR) of 19% from 2012.
Clinical Update
On September 5, 2018, we entered into an agreement with CSMC for an investigator-sponsored Phase 2 clinical study of SYN-010 to be co-funded by us and CSMC (the “Study”).
The Study will provide further evaluation of the efficacy and safety of SYN-010, our modified-release reformulation of lovastatin lactone, which is exclusively licensed to us by CSMC. SYN-010 is designed to reduce methane production by certain microorganisms (M. smithii)in the gut to treat an underlying cause of irritable bowel syndrome with constipation (IBS-C). The data from this study will provide additional insights into potential SYN-010 clinical efficacy, including dose response and microbiome effects, ideally solidifying existing clinical outcomes data, and potentially simplifying Phase 3 clinical development.
The Study will be conducted out of the Pimentel Laboratory at CSMC and is expected to be a 12-week, placebo-controlled, double-blind, randomized clinical trial to evaluate two dose strengths of oral SYN-010 (21 mg and 42 mg) in approximately 150 patients diagnosed with IBS-C. The investigator-sponsored Study will be led by the gastrointestinal microbiota researcher Ruchi Mathur, M.D., director of Metabolism, Clinical Research and Administrative Operations at the Medically Associated Science and Technology (MAST) Program at CSMC. The Study is expected to begin enrollment during the fourth quarter of 2018, contingent upon approval of the clinical study protocol by the CSMC Institutional Review Board.
The primary objective for the Study will be to determine the efficacy of SYN-010, measured as an improvement from baseline in the weekly average number of complete spontaneous bowel movements (CSBMs) during the 12-week treatment period for SYN-010 21 mg and 42 mg daily doses relative to placebo. Secondary efficacy endpoints for both dose strengths of SYN-010 are expected to measure changes from baseline in abdominal pain, bloating, stool frequency as well as the use of rescue medication relative to placebo. Exploratory outcomes include Adequate Relief and quality of life measures using the well-validated EQ-5D-5L and PAC-SYM patient questionnaires.
| 8 |
We expect that CSMC will dose the first patient in the investigator-sponsored Phase 2b clinical study in Q4 2018. A data readout from this clinical trial study is anticipated in 2H 2019.
Allowance of Key U.S. Patent
On May 1, 2018, the United States Patent and Trademark Office (USPTO) issued U.S. Patent No. 9,956,292 which includes claims related to composition of matter for the use of anti-methanogenic compositions to treat IBS-C. The patent will provide key intellectual property protection in the U.S. for SYN-010 and will expire no later than 2035.
Research Programs
Infectious disease outbreaks are increasing while intervention options are declining due to widespread multidrug-resistant bacteria, increasing numbers of immuno-compromised patients (e.g., the elderly and cancer patients) and the isolation of new pathogens.
SYN-007 — Prevention of CDI, overgrowth of pathogenic organisms and the emergence of antimicrobial resistance (AMR)
SYN-007 is a specially formulated version of SYN-004 (ribaxamase) designed to degrade orally administered beta-lactam antibiotics to protect the gut microbiome from antibiotic-mediated dysbiosis. SYN-007 is formulated for release in the distal small intestine to allow systemic absorption of the oral antibiotic while still providing protection upstream of the colon and to the gut microbiome. SYN-007 is designed for patients who have been administered SYN-004 (ribaxamase) in combination with intravenous beta-lactam antibiotics and who are then transferred to an oral beta-lactam antibiotic, thereby extending gut microbiome protection from antibiotic-mediated dysbiosis. Data from a recent canine study completed during the second half of 2017 demonstrated that, when co-administered with oral amoxicillin, oral SYN-007 did not interfere with amoxicillin absorption and did demonstrate protection of the gut microbiome. The data from this canine study were presented during recent microbiome conferences in Q4 2017 and Q1 2018. A second canine study was completed during Q2 2018 in which oral SYN-007 was co-administered with oral amoxicillin and oral Augmentin. Again, SYN-007 did not interfere with systemic absorption of the antibiotics but did diminish the microbiome damage associated with these antibiotics.
SYN-006 — Prevention of CDI, overgrowth of pathogenic organisms and the emergence of antimicrobial resistance (AMR)
The second pipeline product, termed SYN-006, has the potential to further expand the utility of our SYN-004 (ribaxamase) program to a broader spectrum of IV beta-lactam antibiotics in the GI tract to include carbapenem antibiotics. Carbapenems are broad-spectrum beta-lactam antibiotics that have been shown to significantly damage the gut microbiome, incur a high risk forC. difficile infection, and enable GI overgrowth with multidrug resistant organisms. Carbapenems are frequently a last line of defense antibiotic, therefore the emergence and spread of carbapenem resistance presents an urgent threat. SYN-006 is a carbapenemase designed to degrade intravenous (IV) carbapenem antibiotics within the GI tract to maintain the natural balance of the gut microbiome for the prevention of CDI, overgrowth of pathogenic organisms and the emergence of antimicrobial resistance (AMR). It is anticipated that, by protecting the gut microbiome from exposure to carbapenem antibiotics, SYN-006 may potentially diminish the spread of such resistance. At the ID Week 2017 conference, we presented a poster demonstrating SYN-006’s broad activity against four carbapenem antibiotics as well as efficacy in a canine model. The poster also showed data from a porcine model indicating that the carbapenem, ertapenem, potently damaged gut microbiomes and mediated expansion of antibiotic resistance genes in the GI tract. More recently, we successfully formulated SYN-006 for oral delivery and evaluated it in a porcine efficacy model in conjunction with IV ertapenem. The data, presented at a clinical conference during the first quarter of 2018, demonstrated that SYN-006 did not interfere with serum levels of ertapenem and did diminish antibiotic-mediated dysbiosis.
SYN-005 — Pertussis (Whooping Cough)
The SYN-005 program is developing monoclonal antibodies both as a prophylaxis and a treatment for pertussis.Bordetella pertussis (B. pertussis) is a gram-negative bacterium that infects the upper respiratory tract, causing uncontrollable and violent coughing. Antibiotic treatment does not have a major effect on the course of pertussis. While such treatment can eliminate theB. pertussis bacteria from the respiratory tract, it does not neutralize the pertussis toxin. Infants with pertussis often require hospitalization in pediatric intensive care units, frequently requiring mechanical ventilation. The incidence of pertussis is increasing due to the declining effectiveness of the acellular vaccine introduced in the 1990s, exposure of unvaccinated and under-vaccinated individuals including infants who are not yet fully vaccinated and exposure of individuals whose immunity has diminished over time.
According to the Centers for Disease Control and Prevention (CDC) , there were 24.1 million cases of whooping cough worldwide in 2014, and it is estimated thatB. pertussisinfection caused up to 167,700 deaths in children younger than 5 years in 2014.
| 9 |
Intrexon Collaboration and The University of Texas at Austin Agreement
In August 2012, we entered into a worldwide exclusive channel collaboration with Intrexon develop monoclonal antibody (mAb) therapies for the treatment of certain infectious diseases not adequately addressed by existing therapies. In December 2012, we initiated mAb development for the prevention and treatment of pertussis focusing on toxin neutralization. Unlike antibiotics, we are developing a mAb therapy to target and neutralize the pertussis toxin as a prophylaxis for high-risk newborns and in order to shorten the course, diminish the long-term complications, and reduce the mortality rate in infected infants.
To further the development of this potential therapy for pertussis, we entered into an agreement with UT Austin to license the rights to certain research and pending patents related to pertussis antibodies. These research efforts are being conducted at the Cockrell School of Engineering in the laboratory of Associate Professor, Jennifer A. Maynard, Ph.D., the Laurence E. McMakin, Jr. Centennial Faculty Fellow in the McKetta Department of Chemical Engineering. Dr. Maynard brings to the project her expertise in defining the key neutralizing epitopes of pertussis toxin to optimize the potential efficacy of antibody therapeutics.
Preclinical Development
Working with our collaborator, Intrexon, and our academic collaborator, UT Austin, we have established a humanized mAb product candidate, SYN-005, designed to neutralize pertussis toxin, a major cause of pertussis-mediated infant morbidity and mortality. The two humanized mAbs, hu1B7 and hu11E6, bound tightly to the toxin and potently neutralized the toxin. In addition, the antibodies, individually or in combination, were highly efficacious in a murine model of pertussis in which they completely mitigated elevations of the white blood cell count that is characteristic of the illness.
In April 2014, and again in September 2014, we received positive preclinical research findings of SYN-005 for the treatment of pertussis in three non-human primate studies (n = 19). In the latter two pertussis studies in particular, SYN-005 rapidly stopped the rise in white blood cell count that is characteristic of the disease and accelerated its return to baseline.
In September 2014, we received U.S. Orphan Drug Designation from the FDA for SYN-005 for the treatment of pertussis.
In October 2015, the Bill & Melinda Gates Foundation awarded a grant to UT Austin to generate preclinical proof-of-concept data in the neonatal non-human primate model to test the hypothesis that antibody administration at birth may have a role in the prevention of pertussis.
In December 2015, the non-human primate prophylaxis study was initiated by UT Austin to determine if administration of hu1B7, one component of SYN-005, at two days of age could protect animals from a subsequent pertussis infection. On April 19, 2017, we announced supportive preclinical data demonstrating hu1B7 provided five weeks of protection from pertussis in neonatal non-human primates. Control animals (n=6), infected withBordetella pertussis (B. pertussis) at five weeks of age, demonstrated marked elevations in white blood cell counts and most exhibited behavioral signs of pertussis, including coughing and diminished activity. In contrast, the experimental animals (n=7), who were treated with hu1B7 at two days of age and then infected five weeks later, had significantly lower peak white blood cell counts (p=0.004) that remained within the normal range or were only slightly elevated. Importantly, all seven of the animals that received prophylactic hu1B7 appeared healthy and none exhibited any behavioral signs of pertussis. Building on this early success, we performed preclinical testing of a modified version of hu1B7 that has the potential to extend the plasma half-life. The modified hu1B7 achieved higher plasma levels at five weeks than the parental hu1B7 antibody and was efficacious in preventing clinical pertussis. The extended half-life antibody has the potential to substantially reduce the required dose and cost for prophylaxis for application in the Developing World. This current study expands the potential clinical utility beyond treatment to also include prophylaxis.
SYN-020 — Oral Intestinal Alkaline Phosphatase
SYN-020 is in the preclinical development stage. SYN-020 is being developed as a modified-release oral dosage form of intestinal alkaline phosphatase (IAP). IAP is an endogenous enzyme expressed in the upper GI tract that functions as a broadly acting phosphatase that generally serves to maintain GI homeostasis and promote commensal microbiota. In animal models, IAP is anti-inflammatory, tightens the gut barrier to diminish “leaky gut,” and accelerates gut microbiome recovery from antibiotic-mediated dysbiosis. Published reports have demonstrated efficacy for several indications with oral IAP in many animal models including colitis, antibiotic-mediated dysbiosis, and metabolic syndrome as well as in a pilot human clinical trial with ulcerative colitis patients.
| 10 |
Limitations of Current Treatments and Clinical Update
Despite its therapeutic potential, clinical application of an oral IAP product has been hindered by inefficient manufacturing with a high cost of goods. We have established manufacturing processes with the potential to yield product with a cost of goods which we believe to be suitable for commercialization. Recent advances include cell lines that express up to 3 grams/L along with a chromatographic downstream process and potential tablet formulations. We are currently optimizing these technologies and pursuing animal efficacy studies. During Q2 2018, we completed several preclinical animal studies that support the clinical utility of SYN-020 for multiple gastrointestinal disorders. We are currently evaluating and establishing strategies to advance IAP to and through clinical trials for several novel indications, including enterocolitis associated with radiation therapy for cancer and checkpoint inhibitor therapy for cancer and microscopic colitis, all of which have unmet medical needs and span a range of market sizes. Importantly, we believe that with a small capital commitment, we can begin moving SYN-020 towards an IND. We are targeting filing an IND during Q4 2019 and commencing a Phase 1 clinical trial during Q1 2020.
SYN-200 — Treatment of Phenylketonuria (PKU)
PKU is a genetic disease that begins at birth characterized by a deficiency in the liver enzyme that breaks down the essential amino acid phenylalanine (Phe), a building block of proteins normally obtained through the foods we eat. As a result, Phe accumulates in the body, becoming toxic and leading to serious health consequences, including profound mental retardation, brain damage, mental illness, behavioral problems, seizures, tremors, limited cognitive ability and hyperactivity. If left untreated, the most severe form of PKU leads to permanent cognitive damage. PKU affects more than 14,000 people in the U.S. and 50,000 people in developed nations globally. There is no existing cure for PKU, requiring patients to maintain a life-long treatment program and a carefully controlled diet.
Intrexon Collaboration
In August 2015, we initiated the SYN-200 discovery program for development and commercialization of novel biotherapeutics for the treatment of patients with PKU pursuant to an exclusive channel collaboration with Intrexon. We intend to utilize Intrexon’s ActoBiotics platform to provide a proprietary method of delivering therapeutic protein to the GI tract through food-grade microbes. This program is in the discovery stage.
Company History
Our predecessor, Sheffield Pharmaceuticals, Inc., was incorporated in 1986, and in 2006 engaged in a reverse merger with Pipex Therapeutics, Inc., a Delaware corporation formed in 2001. After the merger, we changed our name to Pipex Pharmaceuticals, Inc., and in October 2008 we changed our name to Adeona Pharmaceuticals, Inc. On October 15, 2009, we engaged in a merger with a wholly owned subsidiary for the purpose of reincorporating in the State of Nevada. After reprioritizing our focus on the emerging area of synthetic biologics and entering into our first collaboration with Intrexon, we amended our Articles of Incorporation to change our name to Synthetic Biologics, Inc. on February 15, 2012.
Corporate Information
Our executive offices are located at 9605 Medical Center Drive, Suite 270, Rockville, Maryland 20850. Our telephone number is (301) 417-4364, and our website address iswww.syntheticbiologics.com. The information contained on our website is not part of, and should not be construed as being incorporated by reference into this prospectus supplement.
| 11 |
| Class A Units offered by us | Up to 9,523,809 Class A Units. Each Class A Unit will consist of one share of our common stock and a warrant to purchase one share of our common stock at an exercise price equal to 120% of the public offering price of the Class A Unit. The Class A Units will not be certificated and the shares of common stock and warrant that are part of such unit will be immediately separable and will be issued separately in this offering. Assuming no exercise of the over-allotment option and we sell all Class A Units (and no Class B Units) being offered in this offering at an assumed public offering price of $ 2.10 per share (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018), we would issue in this offering an aggregate of 9,523,809 shares of our common stock and warrants to purchase 9,523,809 shares of our common stock. The actual offering price per each Class A Unit will be negotiated between us and the underwriters based on the trading of our common stock prior to the offering, among other things, and may be at a discount to the current market price. We are also offering the shares of common stock issuable upon exercise of warrants sold in Class A Units. |
Assumed Public Offering Price | $2.10 per Class A Unit. |
| Class B Units offered by us | Up to 20,000 Class B Units. We are also offering to each purchaser whose purchase of Class A Units in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the holder, 9.99%) of our outstanding common stock immediately following the consummation of this offering, the opportunity to purchase, if the purchaser so chooses, Class B Units, in lieu of Class A Units. Each Class B Unit will consist of one share of our Series B Preferred, with a stated value of $1,000 and convertible into shares of our common stock, at the public offering price of the Class A Units, together with an equivalent number of warrants as would have been issued to such purchaser if they had purchased Class A Units based on the public offering price of the Class A Units. The Series B Preferred do not generally have any voting rights but are convertible into shares of common stock. The Class B Units will not be certificated and the shares of Series B Preferred and warrants that are part of such unit will be immediately separable and will be issued separately in this offering. We are also offering the shares of common stock issuable upon exercise of warrants sold in Class B Units and upon conversion of the Series B Preferred. For each Class B Unit we sell, the number of Class A Units we are offering will be decreased on a dollar-for-dollar basis. Because we will issue a warrant as part of each Unit, the number of warrants sold in this offering will not change as a result of a change in the mix of the Units sold. |
Public Offering Price Per | $1,000 per Class B Unit. |
| Warrants offered by us | Each warrant included in the Units will have an exercise price of 120% of the public offering price of the Class A Units, will be exercisable upon issuance and will expire three years from the date of issuance. Each warrant will be exercisable to purchase one share of our common stock. No fractional shares of common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, we will round up to the next whole share. The warrants also provide that in the event of a fundamental transaction we are required to cause any successor entity to assume our obligations under the warrants. In addition, the holder of the warrant will be entitled to receive upon exercise of the warrant the kind and amount of securities, cash or property that the holder would have received had the holder exercised the warrant immediately prior to such fundamental transaction. This prospectus also relates to the offering of the shares of common stock issuable upon exercise of the warrants. Subject to certain exceptions, the warrants provide for adjustment of the exercise price, which initially will be 120% of the public offering price of the Class A Units, if we or any of our subsidiaries, as applicable, sell or grant any right to reprice, or otherwise dispose of or issue (or announce any offer, sale, grant or any option to purchase or other disposition) any shares of our common stock or common stock equivalents, at an effective price per share that is less than the exercise price then in effect (such lower price, the “Base Share Price” and such issuances collectively, a “Dilutive Issuance”). In the event a Dilutive Issuance occurs, the exercise price shall be reduced to equal the Base Share Price. |
| 12 |
| Over-allotment option | We have granted the underwriters a 45-day option to purchase up to 1,428,571 additional shares of common stock at an assumed offering price of $2.10 per share and/or additional warrants to purchase up to an additional 1,428,571 shares of our common stock from us at a price of $0.01 per warrant, to cover over-allotments, if any, of the shares of common stock, shares of common stock issuable upon conversion of the Series B Preferred and warrants comprising the Units. |
| Common stock to be outstanding after the offering | 16,732,129 shares of our common stock (at an assumed public offering price of $2.10 per share which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) and assumes that no shares of Series B Preferred are sold in this offering and that none of the warrants are exercised. If the underwriters’ over-allotment option is exercised in full, the total number of shares of common stock outstanding immediately after this offering would be 18,160,700 (at an assumed public offering price of $2.10 per share which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) and assuming all shares of Series B Preferred sold in this offering convert to common stock and that none of the warrants are exercised). This prospectus also includes the shares of our common stock issuable upon conversion of the Series B Preferred and exercise of the warrants. |
| Series B Convertible Preferred Stock | The Series B Preferred will be convertible into shares of our common stock (subject to adjustment as provided in the related certificate of designation of preferences, rights and limitations) at any time at the option of the holder, at a conversion price equal to the public offering price of the Class A Units. See “Description of Securities We Are Offering— Preferred Stock — Series B Convertible Preferred Stock” for a discussion of the terms of the Series B Preferred. |
| 13 |
| Use of Proceeds | We intend to use the net proceeds, if any, from the sales of securities offered by this prospectus to fund our and our subsidiaries’ preclinical and clinical programs, (including, but not limited to,provide approximately $5.0-$7.0 million in funding for manufacturing scale-up activities to progress SYN-004 towards a potential Phase 3 clinical trial (broad indication) and/or initiate a Phase 1/2 clinical trial(s) in a specialty population, approximately $7.5 million in funding for preclinical development and related manufacturing activities for our IND and Phase 1 clinical trial for our SYN-020 program and required milestone payments) and for working capital and general corporate purposes, including, to acquire, license or invest in complementary businesses, technologies, product candidates or other intellectual property. We have broad discretion in determining how the proceeds of this offering will be used, and our discretion is not limited by the aforementioned possible uses. Our board of directors believes the flexibility in application of the net proceeds is prudent. See “Use of Proceeds.” |
| Risk Factors | See the section entitled “Risk Factors” beginning on page 15 of this prospectus for a discussion of factors you should carefully consider before deciding to invest in our securities. |
| Market symbol and trading | Our common stock is listed on the NYSE American under the symbol “SYN”. There is no established trading market for the Series B Preferred or warrants and we do not expect a market to develop. In addition, we do not intend to apply for the listing of the Series B Preferred or warrants on any national securities exchange or other trading market. Without an active trading market, the liquidity of the Series B Preferred and warrants will be limited. |
The number of shares of common stock shown above to be outstanding after this offering is based on 7,208,320 shares outstanding as of October 8, 2018, and assumes the issuance and sale of9,523,809 Class A Units in this offering and no Class B Units.
Unless we indicate otherwise, all information in this prospectus is as of October 8, 2018 and:
| · | reflects a one-for-thirty-five reverse stock split of our issued and outstanding shares of common stock, options and warrants effected on August 10, 2018 and the corresponding adjustment of all common stock prices per share and stock option and warrant exercise prices per share and preferred stock conversion ratios without taking into account fractional shares which are rounded up to the nearest whole number; |
| · | assumes no exercise by theunderwriters of their over-allotment option; |
| · | excludes 634,921 shares of our common stock issuable upon conversion of outstanding shares of preferred stock; |
| · | excludes 347,765 shares of our common stock issuable upon exercise of outstanding options under our equity incentive plans at a weighted-average exercise price of $54.19 per share; |
| · | excludes 915,854 shares of our common stock reserved for issuance upon the exercise of outstanding warrants with a weighted-average exercise price of $75.16 per share and assumes no exercise of the warrants issued in this offering; |
| · | assumes no shares of Series B Preferred are sold in this offering; and |
| · | excludes 170,674 shares of our common stock that are reserved for equity awards that may be granted under our equity incentive plans. |
To the extent we sell any Class B Units in this offering, the same aggregate number of common stock equivalents resulting from this offering would be convertible under the Series B Preferred issued as part of the Class B Units.
| 14 |
An investment in our securities involves a high degree of risk. You should carefully consider the risks and uncertainties described below together with all of the other information contained or incorporated by reference in this prospectus, including our consolidated financial statements and the related notes, before making a decision to invest in our securities. You should also consider the risks, uncertainties and assumptions discussed under Item 1A, “Risk Factors,” in Part I of our Annual Report on Form 10-K for the year ended December 31, 2017 and Item 1A, “Risk Factors,” in Part II of our Quarterly Report on Form 10-Q for the quarter ended June 30, 2018 and any updates or other risks contained in other filings that we may make with the SEC after the date of this prospectus, all of which are incorporated herein by reference, and may be amended, supplemented or superseded from time to time by other reports we file with the SEC in the future and any additional prospectus supplement. If any of these risks actually occur, our business, results of operations and financial condition could suffer. In that case, the market price of our common stock could decline, and you may lose all or part of your investment.
RISKS RELATED TO THIS OFFERING
Investors will experience immediate and substantial dilution in the book value per share of the securities purchased in this offering.
Investors purchasing securities in this offering will incur immediate and substantial dilution in net tangible book value per share of our common stock. After giving effect to the sale of9,523,809 Class A Units, at an assumed public offering price of $2.10 per Class A Unit (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) assuming no sale of any Class B Units and after deducting the estimated underwriting discount and estimated offering expenses payable by us, purchasers of our Class A units in this offering will incur immediate dilution of $0.10 per share in the net tangible book value of the common stock they acquire. For a further description of the dilution that investors in this offering will experience, see “Dilution”.
In addition, to the extent that outstanding stock options or warrants or preferred stock (including the exercise of any warrants) have been or may be exercised or converted or other shares issued, you may experience further dilution.
Our management will have broad discretion over the use of proceeds from this offering and may not use the proceeds effectively.
Our management will have broad discretion over the use of proceeds from this offering. The net proceeds from this offering will be used to fund our and our subsidiaries’ preclinical and clinical programs (including, but not limited to, provide approximately $5.0-$7.0 in funding for manufacturing scale-up activities to progress SYN-004 towards a potential Phase 3 (broad indication) clinical trial and/or initiate a Phase1/2 clinical trial(s) in a specialty population, approximately $7.5 million in funding for preclinical development and related manufacturing activities in preparation for our IND and Phase 1 clinical trial for our SYN-020 program and required milestone payments) and for working capital and general corporate purposes, including, to acquire, license or invest in complementary businesses, technologies, product candidates or other intellectual property. Our management will have considerable discretion in the application of the net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. The net proceeds may be used for corporate purposes that do not improve our operating results or enhance the value of our common stock.
Even if this offering is successful, we will need to raise additional capital in the future to continue operations, which may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate our product development efforts or other operations.
We have had recurring losses from operations, negative operating cash flow and an accumulated deficit. We do not generate any cash from operations and must raise additional funds in order to continue operating our business. We expect to continue to fund our operations primarily through equity and debt financings in the future. Our cash requirements may vary from those now planned depending upon numerous factors, including the result of future research and development activities. We expect our expenses to increase in connection with our ongoing activities, particularly as we continue research and development activities and initiate and conduct clinical trials of, and seek marketing approval for, our product candidates. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution. We expect that our existing cash together with the proceeds from this offering, will be sufficient to meet our anticipated cash requirements for the next twelve months. We will, however, require additional financing in order to complete our planned Phase 3 clinical trial for SYN-004 and/or our planned Phase 2b/3 clinical trial for SYN-010. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. There are no other commitments by any person for future financing. Our securities may be offered to other investors at a price lower than the price per share offered to current stockholders, or upon terms which may be deemed more favorable than those offered to current stockholders. In addition, the issuance of securities in any future financing may dilute an investor's equity ownership and have the effect of depressing the market price for our securities. Moreover, we may issue derivative securities, including options and/or warrants, from time to time, to procure qualified personnel or for other business reasons. The issuance of any such derivative securities, which is at the discretion of our board of directors, may further dilute the equity ownership of our stockholders. No assurance can be given as to our ability to procure additional financing, if required, and on terms deemed favorable to us. To the extent additional capital is required and cannot be raised successfully, we may then have to limit our then current operations and/or may have to curtail certain, if not all, of our business objectives and plans.
| 15 |
There is no established market for the Series B Preferred or warrants being offered in this offering.
There is no established trading market for the Series B Preferred or warrants and we do not expect a market to develop. In addition, we do not intend to apply for the listing of the Series B Preferred or warrants on any national securities exchange or other trading market. Without an active trading market, the liquidity of the Series B Preferred or warrants will be limited.
Holders of Series B Preferred will have limited voting rights.
Except with respect to certain material changes in the terms of the Series B Preferred and certain other matters and except as may be required by Nevada law, holders of Series B Preferred will have no voting rights. Holders of Series B Preferred will have no right to vote for any members of our board of directors.
The warrants are speculative and holders of the warrants will not have rights of common stockholders until such warrants are exercised.
The warrants being offered do not confer any rights of common stock ownership on their holders, such as voting rights or the right to receive dividends, but rather merely represent the right to acquire shares of common stock at a fixed price for a limited period of time. Specifically, commencing on the date of issuance, holders of the warrants may exercise their right to acquire the common stock and pay an exercise price per share equal to 120% of the public offering price, or $2.52 per share (assuming a public offering price of $2.10 per Class A Unit, which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) prior to three years from the date of issuance, after which date any unexercised warrants will expire and have no further value. Moreover, there can also be no assurance that the market price of the common stock will ever equal or exceed the exercise price of the warrants, and consequently, whether it will ever be profitable for holders of the warrants to exercise the warrants.
The proceeds received from the exercise of the warrants issued in this offering on a cash basis could be decreased upon the occurrence of certain events, which could result in a decrease in our stock price and have a dilutive effect on our existing stockholders.
The warrants being offered in this offering contain a provision that, subject to certain exceptions, resets the exercise price of such warrants if at any time while such warrants are outstanding we sell or issue (or are deemed to sell or issue) shares of our common stock or rights, warrants, options or other securities or debt convertible, exercisable or exchangeable for shares of our common stock at a price below the then current exercise price per share for such warrants. In the event of future price resets, (1) proceeds that we will receive upon exercise of any warrants for cash will be reduced, and (2) the number of shares of our common stock that are subject to such warrants increase so that the aggregate purchase price payable applicable to the exercise of the warrants after the reset of the exercise price is the same as the aggregate purchase price payable immediately prior to the reset. Any future resets to the exercise price of the warrants issued in this offering will have a further dilutive effect on our existing stockholders and could result in a decrease in our stock price.
RISKS RELATING TO OUR BUSINESS
We will need to raise additional capital to operate our business and our failure to obtain funding when needed may force us to delay, reduce or eliminate our development programs or commercialization efforts.
During the six months ended June 30, 2018, our operating activities used net cash of approximately $10.4 million and as of June 30, 2018 our cash and cash equivalents were $7.1 million. With the exception of the three months ended June 30, 2010, we have experienced significant losses since inception and have a significant accumulated deficit. As of June 30, 2018, our accumulated deficit totaled approximately $200.8 million on a consolidated basis. We expect to incur additional operating losses in the future and therefore expect our cumulative losses to increase. With the exception of the quarter ended June 30, 2010, and limited laboratory revenues from Adeona Clinical Laboratory, which we sold in March 2012, we have generated very minimal revenues. We do not expect to derive revenue from any source in the near future until we or our potential partners successfully commercialize our products. We expect our expenses to increase in connection with our anticipated activities, particularly as we continue research and development, initiate and conduct clinical trials, and seek marketing approval for our product candidates. Until such time as we receive approval from the FDA and other regulatory authorities for our product candidates, we will not be permitted to sell our products and therefore will not have product revenues from the sale of products. For the foreseeable future we will have to fund all of our operations and capital expenditures from equity and debt offerings, cash on hand, licensing and collaboration fees and grants, if any.
We will need to raise additional capital to fund our operations and meet our current timelines and we cannot be certain that funding will be available on acceptable terms on a timely basis, or at all. Based on our current plans, our cash and cash equivalents together with the proceeds of this offering will not be sufficient to complete our planned Phase 3 clinical trial for SYN-004 or our planned Phase2b/3 clinical trial for SYN-010. Any additional sources of financing will likely involve the issuance of our equity or debt securities, which will have a dilutive effect on our stockholders. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience significant dilution. Any debt financing, if available, may involve restrictive covenants that may impact our ability to conduct our business. A failure otherwise to raise additional funds when needed in the future could result in us being unable to complete planned preclinical and clinical trials or obtain approval of our product candidates from the FDA and other regulatory authorities. In addition, we could be forced to delay, discontinue or curtail product development, forego sales and marketing efforts, and forego licensing in attractive business opportunities. Our ability to raise capital through the sale of securities may be limited by the rules of the SEC and NYSE American that place limits on the number and dollar amount of securities that may be sold. There can be no assurances that we will be able to raise the funds needed, especially in light of the fact that our ability to sell securities registered on our registration statement on Form S-3 will be limited until such time the market value of our voting securities held by non-affiliates is $75 million or more. We also may be required to seek collaborators for our product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available.
| 16 |
We expect to continue to incur significant operating and capital expenditures.
Other than with respect to the three months ended December 31, 2017 and June 30, 2010, we have a history of losses and we have incurred, and will continue to incur, substantial losses and negative operating cash flow. Even if we succeed in developing and commercializing one or more of our product candidates, we may still incur substantial losses for the foreseeable future and may not sustain profitability. We expect that our pivotal Phase 2b/3 and Phase 3 clinical trials will enroll a greater number of patients than our prior clinical trials and will be more costly than our prior clinical trials. In addition, we anticipate a need for additional employees as we undertake later stage clinical trials. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will substantially increase in the foreseeable future as we do the following:
| · | continue to undertake preclinical development and pivotal clinical trials for our product candidates, including SYN-010 and SYN-004 (ribaxamase); |
| · | seek regulatory approvals for our product candidates; |
| · | develop our product candidates for commercialization; |
| · | implement additional internal systems and infrastructure; |
| · | license or acquire additional technologies; |
| · | lease additional or alternative office facilities; |
| · | manufacture product for clinical trials; and |
| · | hire additional personnel, including members of our management team. |
We may experience negative cash flow for the foreseeable future as we fund our development and clinical programs with capital expenditures. As a result, we will need to generate significant revenues in order to achieve and maintain profitability. We may not be able to generate these revenues or achieve profitability in the future. Our failure to achieve or maintain profitability could negatively impact the value of our common stock and underlying securities.
We currently have no significant source of revenue and may never generate significant revenue. Currently, we have no products approved for commercial sale.
Our ability to generate revenue depends heavily on:
| · | our ability to raise additional capital on a timely basis to continue to fund our clinical trials; |
| · | demonstration in current and future clinical trials that our lead product candidates, SYN-010 for the treatment of IBS-C and SYN-004 (ribaxamase) for the prevention ofC. difficile, are safe and effective; |
| · | our ability to seek and obtain regulatory approvals, including with respect to the indications we are seeking; |
| · | successful manufacture and commercialization of our product candidates; and |
| · | market acceptance of our products. |
All of our existing product candidates are in various stages of development and will require extensive additional clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before they could provide us with any revenue. As a result, even if we successfully develop, achieve regulatory approval and commercialize our products, we may be unable to generate revenue for many years, if at all. We do not anticipate that we will generate revenue from product sales for at least several years, if at all. If we are unable to generate revenue from product sales, we will not become profitable, and we may be unable to continue our operations.
| 17 |
Our consolidated financial statements have been prepared assuming that we will continue as a going concern.
Our consolidated financial statements as of December 31, 2017 have been prepared under the assumption that we will continue as a going concern for the next twelve months. In addition, our independent registered public accounting firm has issued a report that includes an explanatory paragraph referring to our recurring losses from operations and expressing substantial doubt in our ability to continue as a going concern without additional capital becoming available. Our ability to continue as a going concern is dependent upon our ability to obtain additional equity or debt financing, attain further operating efficiencies, reduce expenditures, and, ultimately, to generate revenue. Our consolidated financial statements as of December 31, 2017 did not include any adjustments that might result from the outcome of this uncertainty.
Our research and development efforts may not succeed in developing commercially successful products and technologies, which may limit our ability to achieve profitability. We are largely dependent on the success of our lead product candidates, SYN-004 (ribaxamase) and SYN-010, which require significant additional clinical testing before we can seek regulatory approval and we cannot be certain that these product candidates will receive regulatory approval or be successfully commercialized.
We must continue to explore opportunities that may lead to new products and technologies. To accomplish this, we must commit substantial efforts, funds, and other resources to research and development. A high rate of failure is inherent in the research and development of new products and technologies. Any such expenditures that we make will be made without any assurance that our efforts will be successful. Failure can occur at any point in the process, including after significant funds have been invested.
The success of our business currently depends on our development, approval and commercialization of our lead product candidates, SYN-004 and SYN-010, which are our only two product candidates for which we have conducted clinical trials. Even though we are pursuing a registration pathway for each of these product candidates based on specific FDA input, there are many uncertainties known and unknown that may affect the outcome of future clinical trials. All of our product candidates, including SYN-004 and SYN-010, will require additional clinical and non-clinical development, regulatory review and approval in multiple jurisdictions, substantial investment, access to sufficient commercial manufacturing capacity and significant marketing efforts before we can generate any revenue from product sales. Regardless of whether our clinical trials are deemed to be successful, promising new product candidates may fail to reach the market or may only have limited commercial success because of efficacy or safety concerns, failure to achieve positive clinical outcomes, inability to obtain necessary regulatory approvals or satisfy regulatory criteria, limited scope of approved uses, excessive costs to manufacture, the failure to establish or maintain intellectual property rights, or infringement of the intellectual property rights of others. Failure to obtain regulatory approvals of SYN-004 or SYN-010 in a timely manner would have a material adverse impact on our business. Even if we successfully develop SYN-010, SYN-004 or other new products or enhancements, they may be quickly rendered obsolete by changing customer preferences, changing industry standards, or competitors’ innovations. Innovations may not be quickly accepted in the marketplace because of, among other things, entrenched patterns of clinical practice or uncertainty over third-party reimbursement. We cannot state with certainty when or whether any of our products under development will be launched, whether we will be able to develop, license, or otherwise acquire drug candidates or products, or whether any products will be commercially successful. Failure to launch successful new products or new indications for existing products may cause our products to become obsolete, which may limit our ability to achieve profitability.
We are actively seeking and may form or seek strategic alliances or enter into additional licensing arrangements in the future, and we may not realize the benefits of such alliances or licensing arrangements.
We are actively seeking and may form or seek strategic alliances, create joint ventures or collaborations or enter into additional licensing arrangements with third parties that we believe will complement or augment our development and commercialization efforts with respect to our product candidates and any future product candidates that we may develop. Any of these relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute our existing stockholders or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for our product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy. If we license products or businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture. We cannot be certain that, following a strategic transaction or license, we will achieve the revenue or specific net income that justifies such transaction. Any delays in entering into new strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates in certain geographies for certain indications, which would harm our business prospects, financial condition and results of operations.
| 18 |
We may not be able to retain rights licensed to us by others to commercialize key products and may not be able to establish or maintain the relationships we need to develop, manufacture, and market our products.
In addition to our own patent applications, we also currently rely on licensing agreements with third party patent holders/licensors for our products. We have an exclusive license agreement with CSMC relating to our IBS-C program. This agreement requires us or our sublicensee to use our best efforts to commercialize each of the technologies as well as meet certain diligence requirements and timelines in order to keep the license agreement in effect. In the event we or our sublicensee are not able to meet our diligence requirements, we may not be able to retain the rights granted under our agreement or renegotiate our arrangement institution on reasonable terms, or at all. If the license were to terminate and we were to lose the right to commercialize our products, our business opportunity would be adversely affected. Furthermore, we currently have very limited product development capabilities, and limited marketing or sales capabilities. For us to research, develop, and test our product candidates, we would need to contract with outside researchers, in most cases those parties that did the original research and from whom we have licensed the technologies. Our ECC agreements with Intrexon provide that Intrexon may terminate an agreement if we do not perform certain specified requirements, including developing therapies considered superior. Our agreement with UT Austin allows the UT Austin to terminate its agreement if we fail to comply with the terms of the agreement. Our agreement with CSMC allows CSMC to terminate its agreement if we fail to comply with the terms of the agreement.
We can give no assurances that any of our issued patents licensed to us or any of our other patent applications will provide us with significant proprietary protection or be of commercial benefit to us. Furthermore, the issuance of a patent is not conclusive as to its validity or enforceability, nor does the issuance of a patent provide the patent holder with freedom to operate without infringing the patent rights of others.
We will incur additional expenses in connection with our arrangements with Intrexon, our development of SYN-004, SYN 010 and SYN-020, and our agreement with CSMC.
Pursuant to our ECC agreements with Intrexon, we are responsible for future research and development expenses of product candidates developed under our collaboration, the effect of which has and will continue to increase the level of our overall research and development expenses going forward. Our agreements with CSMC requires that we initiate certain studies and file or have accepted an NDA within a certain amount of time, each of which are costly and will require additional expenditures. Although all manufacturing, preclinical studies and human clinical trials are expensive and difficult to design and implement, costs associated with the manufacturing, research and development of biologic product candidates are generally greater in comparison to small molecule product candidates. We have added additional personnel to support our ECC agreements with Intrexon, and research and development of our candidates, SYN-004, SYN-010 and SYN-020. In addition, we have commenced or intend to commence manufacturing of SYN-004, SYN-010 and SYN-020 material to support our planned preclinical and clinical studies which will require us to incur additional expenses.
Because our biologic programs are relatively new, we have only recently assumed development responsibility and costs associated with such programs. In addition, because development activities in collaboration with Intrexon are determined pursuant to joint steering committees comprised of Intrexon and ourselves and we have limited product development experience, future development costs associated with these programs may be difficult to anticipate and exceed our expectations. Our actual cash requirements may vary materially from our current expectations for a number of other factors that may include, but are not limited to, unanticipated technical challenges, changes in the focus and direction of our development activities or adjustments necessitated by changes in the competitive landscape in which we operate. If we are unable to continue to financially support such collaborations due to our own working capital constraints, we may be forced to delay our activities. If we are unable to obtain additional financing on terms acceptable to us or at all, we may be forced to seek licensing partners or discontinue development.
Developments by competitors may render our products or technologies obsolete or non-competitive.
Companies that currently sell or are developing proprietary products for the prevention and treatment ofC. difficile infection include: Actelion Pharmaceutical Ltd., Merck & Co. Inc., Merus B.V., Pfizer Inc., and Sanofi S.A. Companies that currently sell or are developing proprietary products for IBS-C include: Actavis plc, Ironwood Pharmaceuticals, Inc., Synergy Pharmaceuticals Inc., and Takeda Pharmaceutical Company Limited. Companies that currently sell or are developing proprietary products for pertussis include: GlaxoSmithKline plc, MitsubishiTanabe Pharma Corporation and Sanofi S.A. Companies that sell or are developing products for the treatment of PKU include: BioMarin Pharmaceutical Inc., Codexis, Inc. and Synlogic, Inc. Many of our competitors have significant financial and human resources. The infectious disease market is highly competitive with many generic and proprietary intravenous and oral formulations available to physicians and their patients. For our monoclonal antibodies, we currently do not expect to be able to deliver our infectious disease candidates via the oral route and may thus be limited to the in-patient and/or acute treatment setting. In addition, academic research centers may develop technologies that compete with our SYN-004, SYN-010, SYN-005, SYN-020 products and our other technologies. Should clinicians or regulatory authorities view alternative therapeutic regiments as more effective than our products, this might delay or prevent us from obtaining regulatory approval for our products, or it might prevent us from obtaining favorable reimbursement rates from payers, such as Medicare, Medicaid, hospitals and private insurers.
| 19 |
We operate in a highly competitive environment.
The pharmaceutical and biotechnology industries, including the monoclonal antibody industry, are characterized by rapidly evolving technology and intense competition. Our competitors include major multi-national pharmaceutical companies and biotechnology companies developing both generic and proprietary therapies to treat serious diseases. Many of our competitors have drugs that have already been commercialized and therefore benefit from being first to market their products. Many of these companies are well-established and possess technical, human, research and development, financial, and sales and marketing resources significantly greater than ours. In addition, many of our potential competitors have formed strategic collaborations, partnerships and other types of joint ventures with larger, well established industry competitors that afford these companies potential research and development and commercialization advantages in the therapeutic areas we are currently pursuing.
Academic research centers, governmental agencies and other public and private research organizations are also conducting and financing research activities which may produce products directly competitive to those being developed by us. In addition, many of these competitors may be able to obtain patent protection, obtain FDA and other regulatory approvals and begin commercial sales of their products before us. These competitors will compete with us in product sales as well as recruitment and retention of qualified scientific and management personnel, establishment of clinical trial sites and patient enrollment for clinical trials, as well as in the acquisition of technologies and technology licenses complementary to our programs or advantageous to our business.
Competitors could develop and/or gain FDA approval of our product candidates for a different indication.
Many of our competitors may have more resources than us. We cannot provide any assurances that our products will be FDA approved prior to those of our competitors. We are subject to the risk that products containing our active ingredients that are already marketed to treat other indications, or future FDA approved products containing our active ingredients that are marketed to treat other indications, may be prescribed by physicians, or that physicians may substitute a competitor’s products, to treat the diseases for which we are intending to commercialize; this is commonly referred to as “off-label” use. While under FDA regulations a competitor is not allowed to promote off-label uses of its product, the FDA does not regulate the practice of medicine and, as a result, cannot direct physicians to select certain products for their patients. Consequently, we might be limited in our ability to prevent off-label use of a competitor’s product to treat the diseases we are intending to commercialize, even if we have issued method of use patents for that indication. If we are not able to obtain and enforce our patents, if any, or otherwise receive orphan drug protection, a competitor could develop and commercialize similar products for the same indications that we are pursuing. We cannot provide any assurances that a competitor will not obtain FDA approval for a product that contains the same active ingredients as our products.
If the parties we depend on for supplying substance raw materials for our product candidates and certain manufacturing-related services do not timely supply these products and services in sufficient quality or quantity, it may delay or impair our ability to develop, manufacture and market our product candidates.
We rely on suppliers for the substance raw materials of our product candidates and third parties for manufacturing-related services to produce material that meets appropriate content, quality and stability standards and use in clinical trials of our products and, after approval, for commercial distribution. To succeed, clinical trials require adequate supplies of study material, which may be difficult or uneconomical to procure or manufacture and there can be no assurance that we will successfully procure such study material or even if procured, that we can do so in quantities and in a timely manner to allow our clinical trials to proceed as planned. We and our suppliers and vendors may not be able to (i) produce our study material to appropriate standards for use in clinical studies, (ii) perform under any definitive manufacturing, supply or service agreements with us, or (iii) remain in business for a sufficient time to successfully produce and market our product candidates. If we do not maintain important manufacturing and service relationships, we may fail to find a replacement supplier or required vendor or manufacturer which could delay or impair our ability to obtain regulatory approval for our products and substantially increase our costs or deplete profit margins, if any. If we do find replacement manufacturers and vendors, we may not be able to enter into agreements with them on terms and conditions favorable to us and, there could be a substantial delay before a new facility could be qualified and registered with the FDA and foreign regulatory authorities.
The third-party manufacturers of the active pharmaceutical ingredient (API) and drug product for our lead product candidates, SYN-010 and SYN-004, are established cGMP manufacturers. For all other therapeutic areas we have not yet established cGMP manufacturers for our biologic and drug candidates. We currently have manufacturers for each of our lead product candidates as well as our SYN-020 program, however, we believe additional manufacturers are available, if any of our manufacturers were to limit or terminate production or otherwise fail to meet the quality or delivery requirements needed to satisfy the supply commitments, the process of locating and qualifying alternate sources could require up to several months, during which time our production could be delayed. Any curtailment in the availability of SYN-004 or SYN-010 could have a material adverse effect on our business, financial position and results of operations. In addition, because regulatory authorities must generally approve raw material sources for pharmaceutical products, changes in raw material suppliers may result in production delays or higher raw material costs.
| 20 |
The manufacture of our product candidates requires significant expertise and manufacturers may encounter difficulties in production, particularly in scaling up production. These problems include difficulties with production costs and yields, quality control, including stability of the product and quality assurance testing, shortages of qualified personnel, as well as compliance with federal, state and foreign regulations. We may experience longer than expected lead times with respect to the manufacture of SYN-004 (ribaxamase), which may result from the increase in manufacturing scale necessary to conduct our anticipated Phase 3 clinical trial(s) and result in trial delays. In addition, any delay or interruption in the supply of clinical trial supplies could delay the completion of our clinical trials, increase the costs associated with conducting our clinical trials and, depending upon the period of delay, require us to commence new clinical trials at significant additional expense or to terminate a clinical trial.
We are responsible for ensuring that each of our contract manufacturers comply with the cGMP requirements of the FDA and other regulatory authorities from which we seek to obtain product approval. While we oversee compliance, we do not have control over our manufacturers and their compliance with regulatory requirements. These requirements include, among other things, quality control, quality assurance and the maintenance of records and documentation. The approval process for NDAs includes a review of the manufacturer’s compliance with cGMP requirements. We are responsible for regularly assessing a contract manufacturer’s compliance with cGMP requirements through record reviews and periodic audits and for ensuring that the contract manufacturer takes responsibility and corrective action for any identified deviations.
A failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. Furthermore, if our manufacturers fail to deliver the required commercial quantities on a timely basis and at commercially reasonable prices, we may be unable to meet demand for any approved products and would lose potential revenues.
We may not be able to manufacture our product candidates in commercial quantities, which would prevent us from commercializing our product candidates.
To date, our product candidates have been manufactured in small quantities for preclinical studies and clinical trials. If any of our product candidates is approved by the FDA or comparable regulatory authorities in other countries for commercial sale, we will need to manufacture such product candidate in larger quantities. We may not be able to increase successfully the manufacturing capacity for any of our product candidates in a timely or economic manner, or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. If we are unable to increase successfully the manufacturing capacity for a product candidate, the clinical trials as well as the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in supply. Our product candidates require precise, high quality manufacturing. Our failure to achieve and maintain these high quality manufacturing standards in collaboration with our third-party manufacturers, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could harm our business, financial condition and results of operations.
If we do not obtain the necessary regulatory approvals in the U.S. and/or other countries we will not be able to sell our product candidates.
We cannot assure you that we will receive the approvals necessary to commercialize any of our product candidates or any product candidates we acquire or develop in the future. We will need FDA approval to commercialize our product candidates in the U.S. and approvals from the FDA-equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. We will be required to conduct clinical trials that will be costly. We cannot predict whether our clinical trials will demonstrate the safety and efficacy of our product candidates or if the results of any clinical trials will be sufficient to advance to the next phase of development or for approval from the FDA. We also cannot predict whether our research and clinical approaches will result in drugs or therapeutics that the FDA considers safe and effective for the proposed indications. The FDA has substantial discretion in the drug approval process. The approval process may be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals may prevent or delay commercialization of, and our ability to derive product revenues from our product candidates; and diminish any competitive advantages that we may otherwise believe that we hold.
Even if we comply with all FDA requests, the FDA may ultimately reject one or more of our NDAs or BLAs. We may never obtain regulatory clearance for any of our product candidates. Failure to obtain FDA approval of any of our product candidates will severely undermine our business by leaving us without a saleable product, and therefore without any source of revenues, until another product candidate can be developed. There is no guarantee that we will ever be able to develop or acquire another product candidate.
In addition, the FDA may require us to conduct additional pre-clinical and clinical testing or to perform post-marketing studies, as a condition to granting marketing approval of a product. The results generated after approval could result in loss of marketing approval, changes in product labeling, and/or new or increased concerns about the side effects or efficacy of a product. The FDA has significant post-market authority, including the explicit authority to require post-market studies and clinical trials, labeling changes based on new safety information, and compliance with FDA-approved risk evaluation and mitigation strategies. The FDA’s exercise of its authority has in some cases resulted, and in the future could result, in delays or increased costs during product development, clinical trials and regulatory review, increased costs to comply with additional post-approval regulatory requirements and potential restrictions on sales of approved products.
| 21 |
In foreign jurisdictions, we must also receive approval from the appropriate regulatory authorities before we can commercialize any products, which can be time consuming and costly. Foreign regulatory approval processes generally include all of the risks associated with the FDA approval procedures described above. There can be no assurance that we will receive the approvals necessary to commercialize our product candidate for sale outside the United States.
If the FDA approves any of our product candidates, the labeling, manufacturing, packaging, adverse event reporting, storage, advertising, promotion and record-keeping for our products will be subject to ongoing FDA requirements and continued regulatory oversight and review. Our drug manufacturers and subcontractors that we retain will be required to comply with FDA and other regulations. We may also be subject to additional FDA post-marketing obligations. If we are not able to maintain regulatory compliance, we may not be permitted to market our product candidates and/or may be subject to product recalls, seizures, suspension of regulatory approval, suspension of production, injunctions or civil or criminal sanctions. The subsequent discovery of previously unknown problems with any marketed product, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the product, and could include withdrawal of the product from the market.
Clinical trials are very expensive, time-consuming, and difficult to design and implement.
Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time-consuming. We estimate that clinical trials for our product candidates would take at least several years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. Commencement and completion of clinical trials may be delayed by several factors, including:
| · | obtaining an IND application with the FDA to commence clinical trials; |
| · | identification of, and acceptable arrangements with, one or more clinical sites; |
| · | obtaining IRB approval to commence clinical trials; |
| · | unforeseen safety issues; |
| · | determination of dosing; |
| · | lack of effectiveness during clinical trials; |
| · | slower than expected rates of patient recruitment; |
| · | inability to monitor patients adequately during or after treatment; |
| · | inability to obtain supply of our drug candidate in a timely manner; |
| · | inability or unwillingness of medical investigators to follow our clinical protocols; and |
| · | unwillingness of the FDA or IRBs to permit the clinical trials to be initiated. |
In addition, we, IRBs or the FDA may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if IRBs or the FDA finds deficiencies in our submissions or conduct of our trials.
| 22 |
The results of our clinical trials may not support our product candidate claims and the results of preclinical studies and completed clinical trials are not necessarily predictive of future results.
To date, long-term safety and efficacy have not yet been demonstrated in clinical trials for any of our product candidates. Favorable results in our early studies or trials may not be repeated in later studies or trials. Even if our clinical trials are initiated and completed as planned, we cannot be certain that the results will support our product candidate claims. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. Success of our predecessor P1A clinical product or positive topline data from our previous SYN-004 Phase 1 and Phase 2 clinical trials, does not ensure success of SYN-004, and positive topline data for our SYN-010 Phase 2 clinical trials does not ensure success of SYN-010. Furthermore, the FDA could determine that SYN-004 has not demonstrated safety and require additional clinical trials and safety data, despite positive results from our SYN-004 Phase 2b clinical trial and the determination by clinical sites investigators and an independent third party that the adverse events that occurred in the group that received SYN-004 in our Phase 2b clinical trial were not drug related. We cannot be sure that the results of later clinical trials would replicate the results of prior clinical trials and preclinical testing nor that they would satisfy the requirements of the FDA or other regulatory agencies. Clinical trials may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety issues, notwithstanding promising results in earlier trials. Most product candidates that commence clinical trials are never approved as products. Any such failure could cause us or our sublicensee to abandon a product candidate and might delay development of other product candidates. Preclinical and clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization. Any delay in, or termination of, our clinical trials would delay our obtaining FDA approval for the affected product candidate and, ultimately, our ability to commercialize that product candidate.
If we encounter difficulties enrolling patients in our clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
Delays in patient enrollment may result in increased cost or may adversely affect timing or outcome of planned clinical trials, which could prevent completion of these trials and adversely affect our ability to advance the development of our product candidates.
Delays in clinical testing could result in increased costs to us and delay our ability to generate revenue.
We may experience delays in clinical testing of our product candidates. We do not know whether planned clinical trials will begin on time, will need to be redesigned or will be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including delays in obtaining regulatory approval to commence a clinical trial, in securing clinical trial agreements with prospective sites with acceptable terms, in obtaining institutional review board approval to conduct a clinical trial at a prospective site, in recruiting patients to participate in a clinical trial or in obtaining sufficient supplies of clinical trial materials. Manufacturing considerations for SYN-004 (ribaxamase) and our other product candidates may include an expected several month lead time following a decision to commence any clinical trial(s) and capacity considerations of our third-party contract manufacturers to provide clinical supply of SYN-004 or our other product candidates could cause delays in clinical trials. Many factors affect patient enrollment, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the clinical trial, competing clinical trials and new drugs approved for the conditions we are investigating. Clinical investigators will need to decide whether to offer their patients enrollment in clinical trials of our product candidates versus treating these patients with commercially available drugs that have established safety and efficacy profiles. Any delays in completing our clinical trials will increase our costs, slow down our product development and timeliness and approval process and delay our ability to generate revenue.
Patients who are administered our product candidates may experience unexpected side effects or other safety risks that could cause a halt in their clinical development, preclude approval of our product candidates or limit their commercial potential.
Our clinical trials may be suspended at any time for a number of reasons. We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to the clinical trial patients. In addition, the FDA or other regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the clinical trial patients. For example, the FDA could determine that SYN-004 has not demonstrated safety and require additional clinical trials and safety data, despite positive results from our SYN-004 Phase 2b clinical trial and the determination by clinical sites investigators and an independent third party that the adverse events that occurred in the group that received SYN-004 in our Phase 2b clinical trial were not drug related.
Administering any product candidate to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical trials of our product candidates and could result in the FDA or other regulatory authorities denying further development or approval of our product candidates for any or all targeted indications. Ultimately, some or all of our product candidates may prove to be unsafe for human use. Moreover, we could be subject to significant liability if any volunteer or patient suffers, or appears to suffer, adverse health effects as a result of participating in our clinical trials. Any of these events could prevent us from achieving or maintaining market acceptance of our product candidates and could substantially increase commercialization costs.
| 23 |
An NDA submitted under Section 505(b)(2) subjects us to the risk that we may be subject to a patent infringement lawsuit that would delay or prevent the review or approval of our product candidate.
We plan to submit SYN-010 to the FDA for approval under Section 505(b)(2) of the FDCA. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies that were not conducted by, or for, the applicant and on which the applicant has not obtained a right of reference. The 505(b)(2) application would enable us to reference published literature and/or the FDA’s previous findings of safety and effectiveness for the branded reference drug. For NDAs submitted under Section 505(b)(2) of the FDCA, the patent certification and related provisions of the Hatch-Waxman Act apply. In accordance with the Hatch-Waxman Act, such NDAs may be required to include certifications, known as paragraph IV certifications, that certify that any patents listed in the Patent and Exclusivity Information Addendum of the FDA’s publication, Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book, with respect to any product referenced in the 505(b)(2) application, are invalid, unenforceable or will not be infringed by the manufacture, use or sale of the product that is the subject of the 505(b)(2) NDA.
Under the Hatch-Waxman Act, the holder of patents that the 505(b)(2) application references may file a patent infringement lawsuit after receiving notice of the paragraph IV certification. Filing of a patent infringement lawsuit against the filer of the 505(b)(2) applicant within 45 days of the patent owner’s receipt of notice triggers a one-time, automatic, 30-month stay of the FDA’s ability to approve the 505(b)(2) NDA, unless patent litigation is resolved in the favor of the paragraph IV filer or the patent expires before that time. Accordingly, we may invest a significant amount of time and expense in the development of one or more product candidates only to be subject to significant delay and patent litigation before such product candidates may be commercialized, if at all. In addition, a 505(b)(2) application will not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired. The FDA may also require us to perform one or more additional clinical studies or measurements to support the change from the branded reference drug, which could be time consuming and could substantially delay our achievement of regulatory approvals for such product candidates. The FDA may also reject our future 505(b)(2) submissions and require us to file such submissions under Section 505(b)(1) of the FDCA, which would require us to provide extensive data to establish safety and effectiveness of the drug for the proposed use and could cause delay and be considerably more expensive and time consuming. These factors, among others, may limit our ability to successfully commercialize our product candidates.
Our product candidates, if approved for sale, may not gain acceptance among physicians, patients and the medical community, thereby limiting our potential to generate revenues.
If one of our product candidates is approved for commercial sale by the FDA or other regulatory authorities, the degree of market acceptance of any approved product by physicians, healthcare professionals and third-party payors and our profitability and growth will depend on a number of factors, including:
| · | demonstration of safety and efficacy; |
| · | changes in the practice guidelines and the standard of care for the targeted indication; |
| · | relative convenience and ease of administration; |
| · | the prevalence and severity of any adverse side effects; |
| · | budget impact of adoption of our product on relevant drug formularies; |
| · | the availability, cost and potential advantages of alternative treatments, including less expensive generic drugs; |
| · | pricing, reimbursement and cost effectiveness, which may be subject to regulatory control; |
| · | effectiveness of our or any of our partners’ sales and marketing strategies; |
| · | the product labeling or product insert required by the FDA or regulatory authority in other countries; and |
| · | the availability of adequate third-party insurance coverage or reimbursement. |
| 24 |
If any product candidate that we develop does not provide a treatment regimen that is as beneficial as, or is perceived as being as beneficial as, the current standard of care or otherwise does not provide patient benefit, that product candidate, if approved for commercial sale by the FDA or other regulatory authorities, likely will not achieve market acceptance. Our ability to effectively promote and sell any approved products will also depend on pricing and cost-effectiveness, including our ability to produce a product at a competitive price and our ability to obtain sufficient third-party coverage or reimbursement. If any product candidate is approved but does not achieve an adequate level of acceptance by physicians, patients and third-party payors, our ability to generate revenues from that product would be substantially reduced. In addition, our efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources, may be constrained by FDA rules and policies on product promotion, and may never be successful.
We depend on third parties, including researchers and sublicensees, who are not under our control. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to seek or obtain regulatory approval for or commercialize our product candidates.
Since we have in-licensed some of our product candidates, have sublicensed a product candidate and have collaboration agreements for the development of other product candidates, we depend upon our sublicensee and independent investigators and scientific collaborators, such as universities and medical institutions or private physician scientists, to advise us and to conduct our preclinical and clinical trials under agreements with us. These collaborators are not our employees and we cannot control the amount or timing of resources that they devote to our programs or the timing of their procurement of clinical-trial data or their compliance with applicable regulatory guidelines. Should any of these scientific inventors/advisors or those of our sublicensee become disabled or die unexpectedly, or should they fail to comply with applicable regulatory guidelines, we or our sublicensee may be forced to scale back or terminate development of that program. They may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking those programs ourselves. Failing to devote sufficient time and resources to our drug-development programs, or substandard performance and failure to comply with regulatory guidelines, could result in delay of any FDA applications and our commercialization of the drug candidate involved.
These collaborators may also have relationships with other commercial entities, some of which may compete with us. Our collaborators assisting our competitors could harm our competitive position. For example, we are highly dependent on scientific collaborators for our IBS-C development program, each of whom are employed by third parties.
With respect to our product candidates in collaboration with Intrexon, we are dependent upon Intrexon’s synthetic biology facilities and capabilities as we have no such facilities and capabilities of our own. We are also reliant on their vectors, monoclonal antibody discovery, production cell line development and know-how.
With respect to our product candidate for pertussis in collaboration with University of Texas at Austin, we are dependent on its research laboratories as we have no such facilities or capabilities of our own. If any of the foregoing were to become inaccessible or terminated, it would be difficult for us to develop and commercialize our synthetic biologic product candidates.
We have in the past and expect to have in the future agreements with third-party contract research organizations (CROs), under which we have delegated to the CROs the responsibility to coordinate and monitor the conduct of our SYN-004 and SYN-010 clinical trials and to manage data for our clinical programs. We, our CROs and our clinical sites are required to comply with current Good Clinical Practices, or cGCPs, regulations and guidelines issued by the FDA and by similar governmental authorities in other countries where we are conducting clinical trials. We have an ongoing obligation to monitor the activities conducted by our CROs and at our clinical sites to confirm compliance with these requirements. In the future, if we, our CROs or our clinical sites fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA may require us to perform additional clinical trials before approving our marketing applications. In addition, our clinical trials must be conducted with product produced under cGMP regulations, and will require a large number of test subjects. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process. If our CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, our financial results and the commercial prospects for our product candidates would be harmed, our costs could increase, and our ability to generate revenue could be delayed.
We currently have no marketing, sales or distribution organization and have no experience in marketing products as a company. If we are unable to establish marketing and sales capabilities or enter into agreements with third parties to market and sell our product candidates, we may not be able to generate product revenue.
We currently have no marketing, sales or distribution capabilities and have no experience in marketing products. We may develop an in-house marketing organization and sales force, which will require significant capital expenditures, management resources and time. We will have to compete with other pharmaceutical and biotechnology companies to recruit, hire, train and retain marketing and sales personnel.
| 25 |
If we are unable or decide not to establish internal sales, marketing and distribution capabilities, we will pursue collaborative arrangements regarding the sales and marketing of our products; however, there can be no assurance that we will be able to establish or maintain such collaborative arrangements. Any revenue we receive will depend upon the efforts of such third parties, which may not be successful. We may have little or no control over the marketing and sales efforts of such third parties and our revenue from product sales may be lower than if we had commercialized our product candidates ourselves. We also face competition in our search for third parties to assist us with the sales and marketing efforts of our product candidates.
There can be no assurance that we will be able to develop in-house sales and distribution capabilities or establish or maintain relationships with third-party collaborators to commercialize any product in the United States or overseas.
Even if our products are approved, if doctors decide not to prescribe SYN-010 or hospitals decide not to prescribe SYN-004, we may be unable to generate sufficient revenue to sustain our business.
To increase awareness and adoption of our products once approved, we and our collaborators will need to educate doctors and hospitals on the benefits and value of our products through published papers, presentations at scientific conferences and one-on-one education sessions. In addition, we and our collaborators will need to assure doctors of our ability to obtain and maintain adequate reimbursement coverage from third-party payors. We and our collaborators may need to hire additional commercial, scientific, technical, sales and marketing and other personnel to support this process. If our educational efforts fail and medical practitioners do not decide to prescribe our products in sufficient volume, we may be unable to generate sufficient revenue to sustain our business. In addition, factors outside of our control, such as insurance reimbursement are expected to influence market acceptance of our products. Accordingly, even if we receive regulatory approval for the use of our products, we may not be successful in generating revenue from the sale of our products.
Reimbursement may not be available for our product candidates, which would impede sales.
Market acceptance and sales of our product candidates may depend on coverage and reimbursement policies and health care reform measures. Decisions about formulary coverage as well as levels at which government authorities and third-party payers, such as private health insurers and health maintenance organizations, reimburse patients for the price they pay for our products as well as levels at which these payors pay directly for our products, where applicable, could affect whether we are able to commercialize these products. We cannot be sure that reimbursement will be available for any of our products. Also, we cannot be sure that coverage or reimbursement amounts will not reduce the demand for, or the price of, our products. If coverage and reimbursement are not available or are available only at limited levels, we may not be able to commercialize our products.
In recent years, officials have made numerous proposals to change the health care system in the United States. These proposals include measures that would limit or prohibit payments for certain medical treatments or subject the pricing of drugs to government control. In addition, in many foreign countries, particularly the countries of the European Union, the pricing of prescription drugs is subject to government control. If our products are or become subject to government regulation that limits or prohibits payment for our products, or that subjects the price of our products to governmental control, we may not be able to generate revenue, attain profitability or commercialize our products.
As a result of legislative proposals and the trend towards managed health care in the United States, third-party payors are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement of new drugs. They may also impose strict prior authorization requirements and/or refuse to provide any coverage of uses of approved products for medical indications other than those for which the FDA has granted market approvals. As a result, significant uncertainty exists as to whether and how much third-party payors will reimburse patients for their use of newly-approved drugs, which in turn will put pressure on the pricing of drugs.
Healthcare reform measures could hinder or prevent our product candidates’ commercial success.
The U.S. government and other governments have shown significant interest in pursuing continued healthcare reform. Any government-adopted reform measures could adversely impact the pricing of healthcare products and services in the United States or internationally and the amount of reimbursement available from governmental agencies or other third party payors. The continuing efforts of the U.S. and foreign governments, insurance companies, managed care organizations and other payors of health care services to contain or reduce health care costs may adversely affect our ability to set prices for our products which we believe are fair, and our ability to generate revenues and achieve and maintain profitability.
| 26 |
New laws, regulations and judicial decisions, or new interpretations of existing laws, regulations and decisions, that relate to healthcare availability, methods of delivery or payment for products and services, or sales, marketing or pricing, may limit our potential revenue, and we may need to revise our research and development programs. The pricing and reimbursement environment may change in the future and become more challenging due to several reasons, including policies advanced by the current executive administration in the United States, new healthcare legislation or fiscal challenges faced by government health administration authorities. Specifically, in both the United States and some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the health care system in ways that could affect our ability to sell our products profitably.
If product liability lawsuits are successfully brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability lawsuits related to the testing of our product candidates, and will face an even greater risk if we sell our product candidates commercially. Currently, we are not aware of any anticipated product liability claims with respect to our product candidates. In the future, an individual may bring a liability claim against us if one of our product candidates causes, or merely appears to have caused, an injury. If we cannot successfully defend ourselves against the product liability claim, we may incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| · | decreased demand for our product candidates; |
| · | injury to our reputation; |
| · | withdrawal of clinical trial participants; |
| · | costs of related litigation; |
| · | initiation of investigations by regulators; |
| · | substantial monetary awards to patients or other claimants; |
| · | distraction of management’s attention from our primary business; |
| · | product recalls; |
| · | loss of revenue; and |
| · | the inability to commercialize our product candidates. |
We have clinical trial liability insurance. We intend to expand our insurance coverage to include the sale of commercial products if marketing approval is obtained for our product candidates. Our current insurance coverage may prove insufficient to cover any liability claims brought against us. In addition, because of the increasing costs of insurance coverage, we may not be able to maintain insurance coverage at a reasonable cost or obtain insurance coverage that will be adequate to satisfy liabilities that may arise.
We rely on patent applications and various regulatory exclusivities to protect some of our product candidates and our ability to compete may be limited or eliminated if we are not able to protect our products.
The patent positions of pharmaceutical companies are uncertain and may involve complex legal and factual questions. We may incur significant expenses in protecting our intellectual property and defending or assessing claims with respect to intellectual property owned by others. Any patent or other infringement litigation by or against us could cause us to incur significant expenses and divert the attention of our management.
Others may file patent applications or obtain patents on similar technologies or compounds that compete with our products. We cannot predict how broad the claims in any such patents or applications will be, and whether they will be allowed. Once claims have been issued, we cannot predict how they will be construed or enforced. We may infringe intellectual property rights of others without being aware of it. If another party claims we are infringing their technology, we could have to defend an expensive and time consuming lawsuit, pay a large sum if we are found to be infringing, or be prohibited from selling or licensing our products unless we obtain a license or redesign our product, which may not be possible.
We also rely on trade secrets and proprietary know-how to develop and maintain our competitive position. Some of our current or former employees, consultants, scientific advisors, current or prospective corporate collaborators, may unintentionally or willfully disclose our confidential information to competitors or use our proprietary technology for their own benefit. Furthermore, enforcing a claim alleging the infringement of our trade secrets would be expensive and difficult to prove, making the outcome uncertain. Our competitors may also independently develop similar knowledge, methods, and know-how or gain access to our proprietary information through some other means.
| 27 |
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights, as well as costs associated with lawsuits.
If any other person files patent applications, or is issued patents, claiming technology also claimed by us in pending applications, we may be required to participate in interference proceedings in the U.S. Patent and Trademark Office to determine priority of invention. We, or our licensors, may also need to participate in interference proceedings involving our issued patents and pending applications of another entity.
The intellectual property environment in the monoclonal antibody field is particularly complex, constantly evolving and highly fragmented. We have not conducted freedom-to-use patent searches on all aspects of our product candidates or potential product candidates, and we may be unaware of relevant patents and patent applications of third parties. In addition, the freedom-to-use patent searches that have been conducted may not have identified all relevant issued patents or pending patents. We cannot provide assurance that our proposed products in this area will not ultimately be held to infringe one or more valid claims owned by third parties which may exist or come to exist in the future or that in such case we will be able to obtain a license from such parties on acceptable terms.
We cannot guarantee that the practice of our technologies will not conflict with the rights of others. In some foreign jurisdictions, we could become involved in opposition proceedings, either by opposing the validity of another’s foreign patent or by persons opposing the validity of our foreign patents.
We may also face frivolous litigation or lawsuits from various competitors or from litigious securities attorneys. The cost to us of any litigation or other proceeding relating to these areas, even if deemed frivolous or resolved in our favor, could be substantial and could distract management from our business. Uncertainties resulting from initiation and continuation of any litigation could have a material adverse effect on our ability to continue our operations.
If we infringe the rights of others we could be prevented from selling products or forced to pay damages.
If our products, methods, processes, and other technologies are found to infringe the proprietary rights of other parties, we could be required to pay damages, or we may be required to cease using the technology or to license rights from the prevailing party. Any prevailing party may be unwilling to offer us a license on commercially acceptable terms.
We do not have a guarantee of patent term restoration and marketing exclusivity of the ingredients for our drugs even if we are granted FDA approval of our products.
The U.S. Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman) permits the FDA to approve Abbreviated New Drug Applications (ANDAs) for generic versions of innovator drugs, as well as NDAs with less original clinical data, and provides patent restoration and exclusivity protections to innovator drug manufacturers. The ANDA process permits competitor companies to obtain marketing approval for drugs with the same active ingredient and for the same uses as innovator drugs, but does not require the conduct and submission of clinical studies demonstrating safety and efficacy. As a result, a competitor could copy any of our drugs and only need to submit data demonstrating that the copy is bioequivalent to gain marketing approval from the FDA. Hatch-Waxman requires a competitor that submits an ANDA, or otherwise relies on safety and efficacy data for one of our drugs, to notify us and/or our business partners of potential infringement of our patent rights. We and/or our business partners may sue the company for patent infringement, which would result in a 30-month stay of approval of the competitor’s application. The discovery, trial and appeals process in such suits can take several years. If the litigation is resolved in favor of the generic applicant or the challenged patent expires during the 30-month period, the stay is lifted and the FDA may approve the application. Hatch-Waxman also allows competitors to market copies of innovator products by submitting significantly less clinical data outside the ANDA context. Such applications, known as Section 505(b)(2) NDAs may rely on clinical investigations not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use and are subject to the ANDA notification procedures described above.
The law also permits restoration of a portion of a product’s patent term that is lost during clinical development and NDA review, and provides statutory protection, known as exclusivity, against FDA approval or acceptance of certain competitor applications. Restoration can return up to five years of patent term for a patent covering a new product or its use to compensate for time lost during product development and regulatory review. The restoration period is generally one-half the time between the effective date of an IND and submission of an NDA, plus the time between NDA submission and its approval (subject to the five-year limit), and no extension can extend total patent life beyond 14 years after the drug approval date. Applications for patent term extension are subject to U.S. Patent and Trademark Office (USPTO) approval, in conjunction with FDA. Approval of these applications takes at least nine months, and there can be no guarantee that it will be given at all.
| 28 |
Hatch-Waxman also provides for differing periods of statutory protection for new drugs approved under an NDA. Among the types of exclusivity are those for a “new chemical entity” and those for a new formulation or indication for a previously-approved drug. If granted, marketing exclusivity for the types of products that we are developing, which include only drugs with innovative changes to previously-approved products using the same active ingredient, would prohibit the FDA from approving an ANDA or 505(b)(2) NDA relying on our safety and efficacy data for three years. This three-year exclusivity, however, covers only the innovation associated with the original NDA. It does not prohibit the FDA from approving applications for drugs with the same active ingredient but without our new innovative change. These marketing exclusivity protections do not prohibit the FDA from approving a full NDA, even if it contains the innovative change.
The technology on which our channel partnering arrangements with Intrexon are based on early stage technology.
On August 8, 2012, we announced an exclusive channel collaboration with Intrexon relating to the design, production, testing and commercialization of human recombinant monoclonal antibodies for the treatment of certain infectious diseases. Although monoclonal antibody therapeutics are well established in the biotechnology and pharmaceutical sectors, their use for the treatment of infectious disease is extremely limited. In order for monoclonal antibodies to be effective for infectious diseases, they must not only properly target the organism of interest (or its toxins), but may also need to overcome defenses and forms of resistance of such organisms. To accomplish this may require the use of more than one specific monoclonal antibody, and mixtures of different monoclonal antibodies, which may create additional unforeseen complications, including increased manufacturing complexity and expense. In order to be competitive, monoclonal antibodies will be required to be produced at a low enough cost of goods in order to be profitably marketed. We have very limited development and manufacturing experience in the field of monoclonal antibodies and infectious disease. We cannot assure that any monoclonal antibody candidates will provide satisfactoryin vitro andin vivo nonclinical results sufficient to warrant the expense of cGMP manufacture and clinical testing in human clinical trials.
On August 10, 2015, we expanded our relationship with Intrexon and entered into an ECC that governs a “channel collaboration” arrangement in which we intend to use Intrexon’s technology for development of biotherapeutic products for the treatment of PKU in humans. The strategy is to orally deliver a bacterium,Lactococcus lactis, that has been engineered to efficiently degrade phenylalanine in the GI tract to prevent phenylalanine absorption into the blood. The strategy is supported by data from rodent studies. The extent to which the data translate to large animal models and to a human therapeutic remains unknown. While genetically-modified versions ofLactococcus lactis have been tested in human clinical trials for other indications, the regulatory paths for recombinant bacterial products have not been fully established.
We do not expect to generate any additional revenue from our sublicense with Meda AB due to recent developments in Europe.
On May 6, 2010, we entered into a sublicense agreement with Meda AB whereby we were given the right to receive certain milestone payments totaling $17.5 million (including an upfront payment of $2.5 million that was received in 2010), plus certain royalties on our flupirtine program. Meda AB informed us that due to the decision of the European Medicines Agency (EMA) to limit the use of flupirtine for long-term pill and systemic use, it has postponed its planned fibromyalgia clinical trials in the U.S. Therefore, we do not expect that the various milestones set forth in the sublicense agreement will be achieved by Meda AB, or that Meda AB will develop flupirtine for fibromyalgia in the U.S., Canada or Japan and accordingly we do not expect to receive any additional milestone payments or royalties on sales in connection with the sublicense agreement.
We may fail to retain or recruit necessary personnel, and we may be unable to secure the services of consultants.
As of September 30, 2018, we employed 25 individuals, 24 of whom are full-time employees. We have also engaged clinical consultants to advise us on our clinical programs and regulatory consultants to advise us on our dealings with the FDA and other foreign regulatory authorities. We have been and will be required to retain additional consultants and employees in order to fulfill our obligations under the ECC agreements with Intrexon, our development of SYN 010 and SYN-004 and our agreement with CSMC. Our future performance will depend in part on our ability to successfully integrate newly hired officers into our management team and our ability to develop an effective working relationship among senior management.
Certain of our directors, scientific advisors, and consultants serve as officers, directors, scientific advisors, or consultants of other biopharmaceutical or biotechnology companies that might be developing competitive products to ours. Other than corporate opportunities, none of our directors are obligated under any agreement or understanding with us to make any additional products or technologies available to us. Similarly, we can give no assurances, and we do not expect and stockholders should not expect, that any biomedical or pharmaceutical product or technology identified by any of our directors or affiliates in the future would be made available to us other than corporate opportunities. We can give no assurances that any such other companies will not have interests that are in conflict with our interests.
| 29 |
Losing key personnel or failing to recruit necessary additional personnel would impede our ability to attain our development objectives. There is intense competition for qualified personnel in the drug and biologic development areas, and we may not be able to attract and retain the qualified personnel we would need to develop our business.
We rely on independent organizations, advisors, and consultants to perform certain services for us, including handling substantially all aspects of regulatory approval, clinical management, manufacturing, marketing, and sales. We expect that this will continue to be the case. Such services may not always be available to us on a timely basis when we need them.
We expect to increase the size of our organization, and we may experience difficulties in managing growth.
We are a small company with 25 employees as of September 30, 2018. To continue our clinical trials and commercialize our product candidates, we will need to expand our employee base for managerial, operational, financial and other resources. Future growth will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain and integrate additional employees. Our future financial performance and our ability to commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to:
| · | manage development efforts effectively; | |
| · | manage our commercialization activities effectively; | |
| · | integrate additional management, administrative, manufacturing and sales and marketing personnel; | |
| · | maintain sufficient administrative, accounting and management information systems and controls; and | |
| · | hire and train additional qualified personnel. |
We may not be able to accomplish these tasks, and our failure to accomplish any of them could harm our financial results and impact our ability to achieve development milestones.
Our management team may invest or spend the proceeds of our prior offerings and future offerings in ways with which you may not agree or in ways which may not yield a significant return.
Our management will have broad discretion over the use of proceeds from our offerings. The net proceeds from our offerings, including sales made under the sales agreement that we entered into on August 5, 2016 with FBR Capital Markets & Co. now known as B. Riley FBR, Inc. (the “B. Riley FBR Sales Agreement”), will be used primarily for general corporate purposes, which may include, among other things, for clinical trials for our product candidates, paying general and administrative expenses and accounts payable, increasing our working capital, funding research and development and funding capital expenditures. We may also use a portion of the net proceeds for licensing or acquiring intellectual property to incorporate into our products and product candidates or our research and development programs and to in-license, acquire or invest in complementary businesses or products, although we have no commitments or agreements with respect to any such licenses, acquisitions or investments as of the date of this filing supplement. Our management will have considerable discretion in the application of the net proceeds, and investors will not have the opportunity, as part of their investment decision, to assess whether the proceeds are being used appropriately. The net proceeds may be used for corporate purposes that do not increase our operating results or enhance the value of our common stock. The failure of our management to use funds effectively could have a material adverse effect on our business, cause the market price of our common stock to decline and impair the commercialization of our products and/or delay the development of our product candidates. Pending their use, we may invest the net proceeds from this offering in short-term, investment-grade, interest-bearing instruments and U.S. government securities. These investments may not yield a favorable return to our stockholders.
RISKS RELATING TO OUR SECURITIES
We cannot assure you that our common stock will be liquid or that it will remain listed on the NYSE American. A failure to regain compliance with the NYSE American stockholders equity listing requirements or failure to continue to meet the other listing requirements could result in a de-listing of our common stock.
Our common stock is listed on the NYSE American. The NYSE American’s listing standards generally mandate that we meet certain requirements relating to stockholders’ equity, stock price, market capitalization, aggregate market value of publicly held shares and distribution requirements. We cannot assure you that we will be able to maintain the continued listing standards of the NYSE American. The NYSE American requires companies to meet certain continued listing criteria including a minimum stockholders’ equity of $6.0 million if an issuer has sustained losses from continuing operations and/or net losses in its five most recent years, as outlined in the NYSE American Company Guide. At June 30, 2018, we had stockholders’ deficit of $208.8 million. The NYSE American Company Guide also states that the NYSE normally will not consider removing from listing securities of an issuer with total value of market capitalization of at least $50.0 million and 1,100,000 shares publicly held, a market value of publicly held shares of at least $15.0 million and 400 round lot shareholders. Although we have more than 1,100,000 shares publicly held and 400 round lot shareholders, our stock price is volatile and, during the first two quarters of 2018, the price of our common stock experienced a sustained decrease resulting in a period where our market capitalization fell below $50.0 million. Our market capitalization is currently below $50.0 million.
| 30 |
On March 7, 2018, we announced that we received written communication from the NYSE American stating we were no longer in compliance with certain continued listing standards as set forth in the NYSE American Company Guide. Specifically, based on our annual report on Form 10-K for the year ended December 31, 2017, and filed with the SEC on February 22, 2018, we are below compliance with Part 10, Section 1003(iii) of the NYSE American Company Guide since we reported a stockholders’ deficit of $1.5 million and net losses in five of our most recent fiscal years as of December 31, 2017. On April 3, 2018, we submitted a plan of compliance to the NYSE American outlining our plan to regain compliance with certain continued listing standards as set forth in Part 10, Section 1003(iii) of the NYSE American Company Guide by September 2, 2018, the conclusion of the compliance plan period. There can be no assurance that we can regain compliance with the listing standard of the NYSE American, or that the NYSE American will continue to list our common stock if we regain compliance, or if we continue to fail to maintain the minimum stockholders’ equity. In addition, in the future we may not be able to maintain such minimum stockholders’ equity and/or issue additional equity securities in exchange for cash or other assets, if available, to maintain certain minimum stockholders’ equity required by the NYSE American. If we are delisted from the NYSE American then our common stock will trade, if at all, only on the over-the-counter market, such as the OTC Bulletin Board securities market, and then only if one or more registered broker-dealer market makers comply with quotation requirements. If our common stock is delisted from the NYSE American due to our failure to regain compliance with the listing standards by the end of the compliance period or for any other reason, and the market value of our shares of common stock held by non-affiliates remains below $15 million, we will likely no longer be eligible to sell common stock pursuant to the B. Riley FBR Sales Agreement or otherwise utilize our shelf registration statement. In addition, delisting of our common stock could depress our stock price, substantially limit liquidity of our common stock and materially adversely affect our ability to raise capital on terms acceptable to us, or at all. Delisting from the NYSE American could also have other negative results, including the potential loss of confidence by suppliers and employees, the loss of institutional investor interest and fewer business development opportunities. On May 18, 2018 we received notification from the NYSE American that NYSE Regulation has reviewed our plan of compliance and determined to accept the plan and grant a plan period through September 2, 2019. NYSE Regulation staff will review our company periodically for compliance with the initiatives outlined in the plan. If we are not in compliance with the continued listing standards by September 2, 2019 or if we do not make progress consistent with the plan during the plan period, NYSE Regulation staff will initiate delisting proceeding as appropriate.
If our common stock falls below $0.20 per share on a 30-trading-day average it will become subject to the continued listing evaluation and follow-up procedures set forth in Section 1009 of the NYSE American Company Guide which could, among other things, result in initiation of immediate delisting procedures. In the event that we were to fail to meet the requirements of NYSE American per share price requirement or stockholders equity requirement and we could not timely cure such deficiency, our listing could become subject to NYSE American continued listing evaluation and follow-up procedures, which could result in delisting procedures. Based on the low stock price on July 28, 2018, our board of directors approved aone-for-thirty-fiveproportionate reverse stock split of our authorized number of shares of common stock and our outstanding number of shares of common stock that we effected on August 10, 2018. However, there can be no assurance that the reverse stock split will result in a sustained higher stock price that will allow us to meet the NYSE American stock price listing requirements or that the reverse stock split will not inhibit our ability to seek equity financing as a remedy to regain compliance with NYSE American stockholders’ equity requirements.
Holders of our warrants issued in our October 2014 offering, and our November 2016 offering, and our Series A Preferred Stock have no rights as common stockholders until they exercise their warrants or convert their Series A Preferred Stock and acquire our common stock.
Until the holders of the warrants we issued in our October 2014 offering and our November 2016 offering and the holders of our Series A Preferred Stock acquire shares of our common stock by exercising their warrants or converting their Series A Preferred Stock, respectively, the holders have no rights as a stockholder with respect to the shares of common stock underlying their securities. Upon exercise of the warrants or conversion of the Series A Preferred Stock, the holders will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after the exercise date.
Because there is no established public trading market for the October 2014 or November 2016 warrants or the Series A Preferred Stock we issued, the liquidity of each such security is limited. We do not expect a market to develop, nor do we intend to apply to list the October 2014 or November 2016 warrants or the Series A Preferred Stock on any securities exchange. Upon exercise of the October 2014 or November 2016 warrants and conversion of the Series A Preferred Stock, our stockholders will experience dilution.
| 31 |
The fundamental change purchase feature of the warrants we issued in our November 2016 offering may delay or prevent an otherwise beneficial attempt to take over our company.
The terms of the November 2016 warrants require us to offer to purchase the warrants for cash in the event of a fundamental change, as defined. This feature may have the effect of delaying or preventing a takeover of our company that would otherwise be beneficial to investors.
Warrants are a risky investment. Holders of outstanding warrants may not be able to recover the investment in the warrants, and the warrants may expire worthless.
Whether our outstanding warrants will have any value will depend on the market conditions for, and the price of, our common stock, which conditions will depend on factors related and unrelated to the success of our clinical development program, and cannot be predicted at this time.
If our common stock price does not increase to an amount sufficiently above the exercise prices of the warrants during the periods the warrants are exercisable, holders of warrants will be unable to recover any of their investment in the warrants. In fact, the warrants issued in November 2016 that had an exercise price of $60.20 (post reverse stock split) expired unexercised because their exercise price was above the common stock trading price. There can be no assurance that any of the factors that could impact the trading price of our common stock will result in the trading price increasing to an amount that will exceed the exercise price or the price required for holders of warrants to achieve a positive return on their investment in the warrants.
We may not have the funds necessary to fulfill our obligation to repurchase the November 2016 warrants.
Under certain circumstances, if an extraordinary transaction (as defined in the warrant agreement) occurs, holders of the warrants issued in November 2016 may require us to repurchase the remaining unexercised portion of such warrants for an amount of cash equal to the value of the warrant as determined in accordance with the Black Scholes option pricing model and the terms of the warrants. Our ability to repurchase the warrants depends on our ability to generate cash flow in the future. To some extent, this is subject to general economic, financial, competitive, legislative and regulatory factors and other factors that are beyond our control. We cannot assure you that we will maintain sufficient cash reserves or that our business will generate cash flow from operations at levels sufficient to permit us to repurchase the warrants.
The issuance of shares of common stock upon conversion of the Series A Preferred Stock would reduce the relative voting power of holders of our common stock, would dilute the ownership of such holders and may adversely affect the market price of our common stock.
The conversion of the Series A Preferred Stock to common stock would dilute the ownership interest of existing holders of our common stock, and any sales in the public market of the common stock issuable upon conversion of the Series A Preferred Stock could adversely affect prevailing market prices of our common stock. Sales by such holders of a substantial number of shares of our common stock in the public market, or the perception that such sales might occur, could have a material adverse effect on the price of our common stock.
The holders of shares of the Series A Preferred Stock may exercise significant influence over us.
The holders of the Series A Preferred Stock will own approximately 9% of our shares of common stock on a fully diluted as-converted basis based on the number of shares of common stock outstanding as of the date hereof.
In addition, under the terms of the Certificate of Designation that governs the Series A Preferred Stock, the Series A Preferred Stock generally ranks, with respect to liquidation, dividends and redemption, senior to other securities (including our common stock and Series B Preferred) and, so long as any shares of Series A Preferred Stock remain outstanding, the approval of the holders of a majority of the Series A Preferred Stock outstanding at the time of approval is required in order for us to, among other things, (i) alter or change adversely the powers, preferences or rights given to the Series A Preferred Stock or alter or amend the Certificate of Designation; (ii) amend our Articles of Incorporation or bylaws in any manner that adversely affects any powers, preferences or rights of the Series A Preferred Stock; (iii) authorize or create any series or class of stock ranking as to redemption, distribution of assets upon a Liquidation Event (as defined in the Certificate of Designation) or dividends senior to, or otherwisepari passu with, the Series A Preferred Stock; (iv) declare or make any dividends other than dividend payments on the Series A Preferred Stock or other distributions payable solely in common stock; (v) authorize any increase in the number of shares of Series A Preferred Stock or issue any additional shares of Series A Preferred Stock; or (vi) enter into any agreement with respect to any of the foregoing.
| 32 |
The holders of Series A Preferred Stock have rights, preferences and privileges that are not held by, and are preferential to, the rights of our common stockholders and holders of our Series B Preferred Stock.
Upon our liquidation, dissolution or winding up, the holders of the Series A Preferred Stock will be entitled to receive out of our assets, in preference to the holders of the common stock and any junior preferred stock (including the Series B Preferred offered hereby), an amount per share equal to the greater of (i) the sum of the Accreted Value (as defined in the Certificate of Designation) plus an amount equal to all accrued or declared and unpaid dividends on the Series A Preferred Stock that have not previously been added to the Accrued Value, or (ii) the amount that such shares would have been entitled to receive if they had converted into common stock immediately prior to such liquidation, dissolution or winding up. In addition, upon consummation of a specified change of control transaction, each holder of Series A Preferred Stock will be entitled to have us redeem the Series A Preferred Stock at a price specified in the Certificate of Designation. These provisions may make it more costly for a potential acquirer to engage in a business combination transaction with us. Provisions that have the effect of discouraging, delaying or preventing a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock and could also affect the price that some investors are willing to pay for our common stock. If there are insufficient assets to pay in full such amounts, then the available assets will be ratably distributed to the holders of the Series A Preferred Stock in accordance with the respective amounts that would be payable on such shares if all amounts payable thereon were paid in full. This will reduce the remaining amount of our assets, if any, available to distribute to holders of our common stock. The holders of Series A Preferred Stock also have a preferential right to receive cumulative dividends on the Accreted Value of each share of Series A Preferred Stock at an initial rate of 2% per annum, compounded quarterly.
In addition, the holders of the Series A Preferred Stock also have certain redemption and conversion rights.
Our obligations to the holders of Series A Preferred Stock could limit our ability to obtain additional financing or increase our borrowing costs, which could have an adverse effect on our financial condition. These preferential rights could also result in divergent interests between the holders of shares of the Series A Preferred Stock and holders of our common stock and Series B Preferred.
The redemption right of the holders of the Series A Preferred Stock may delay or prevent an otherwise beneficial change of control transaction or result in a depletion of our cash in order to satisfy the redemption right of the holders Series A Preferred Stock.
The terms of the Series A Preferred Stock provide the holders with the right to require us to redeem the stock upon a change of control for cash in the event of a fundamental change, as defined. This feature may have the effect of delaying or preventing a change of control that would otherwise be beneficial to investors or depleting our cash.
The market price of our common stock has been and may continue to be volatile and adversely affected by various factors.
The market price of our common stock could fluctuate significantly in response to various factors and events, including:
| · | our ability to execute our business plan; |
| · | operating results below expectations; |
| · | announcements concerning product development results, including clinical trial results, or intellectual property rights of others; |
| · | litigation or public concern about the safety of our potential products; |
| · | our issuance of additional securities, including debt or equity or a combination thereof, necessary to fund our operating expenses; |
| · | announcements of technological innovations or new products by us or our competitors; |
| · | loss of any strategic relationship; |
| · | industry developments, including, without limitation, changes in healthcare policies or practices or third-party reimbursement policies; |
| · | economic and other external factors effecting U.S. or Global equity markets; |
| · | period-to-period fluctuations in our financial results; and |
| · | whether an active trading market in our common stock develops and is maintained. |
| 33 |
In addition, the securities markets have from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These market fluctuations may also materially and adversely affect the market price of our common stock.
Our articles of incorporation and bylaws and Nevada law may have anti-takeover effects that could discourage, delay or prevent a change in control, which may cause our stock price to decline.
Our articles of incorporation, as amended, our amended and restated bylaws and Nevada law could make it more difficult for a third party to acquire us, even if closing such a transaction would be beneficial to our stockholders. The board of directors could authorize the issuance of an additional series of preferred stock that would grant holders preferred rights to our assets upon liquidation, special voting rights, the right to receive dividends before dividends would be declared to common stockholders, and the right to the redemption of such shares, possibly together with a premium, prior to the redemption of the common stock. To the extent that we do issue additional preferred stock, the rights of holders of common stock could be impaired thereby, including without limitation, with respect to liquidation.
Provisions of our articles of incorporation, as amended and our amended and restated bylaws may also prevent or frustrate attempts by our stockholders to replace or remove our management. In particular, our articles of incorporation, as amended, and amended and restated bylaws, among other things:
| · | provide the board of directors with the ability to alter the bylaws without stockholder approval; and |
| · | provide that vacancies on the board of directors may be filled by a majority of directors in office, although less than a quorum. |
Our failure to fulfill all of our registration requirements may cause us to suffer liquidated damages, which may be very costly.
Pursuant to the terms of the registration rights agreement that we entered into with Intrexon and an affiliated entity, we were required to file a registration statement with respect to securities issued and are required to maintain the effectiveness of such registration statement. The failure to do so could result in the payment of damages by us. There can be no assurance that we will be able to maintain the effectiveness of any registration statement, and therefore there can be no assurance that we will not incur damages with respect to such agreements.
Pursuant to the terms of the registration rights agreement that we entered into with holders of our Series A Preferred Stock, we are required to file a registration statement with respect to the securities issued to them upon their request within certain time periods and are required to maintain the effectiveness of such registration statement. The failure to do so could result in the payment of damages by us. There can be no assurance that we will be able to meet the required filing deadlines or maintain the effectiveness of any registration statement, and therefore there can be no assurance that we will not incur damages with respect to such agreements.
We do not intend to pay dividends in the foreseeable future.
We have never paid cash dividends on our common stock. We currently intend to retain our future earnings, if any, to finance the operation and growth of our business and currently do not plan to pay any cash dividends in the foreseeable future. If we do not pay dividends, our common stock may be less valuable because a return on your investment will only occur if the market price of our common stock price appreciates. Our Series A Preferred Stockholders rank senior to our common stockholders with respect to dividends and, subject to any senior rights of the Series A Preferred Stock, the holders of the Series B Preferred will participate, on an as-if-converted-to-common stock basis, in any dividends to the holders of common stock.
Resales of our common stock in the public market by our stockholders may cause the market price of our common stock to fall.
We may issue common stock from time to time in connection with future offerings. Any issuance from time to time of new shares of our common stock, or our ability to issue shares of common stock in future offerings, could result in resales of our common stock by our current stockholders concerned about the potential dilution of their holdings. In turn, these resales could have the effect of depressing the market price for our common stock.
The shares of common stock offered under the B. Riley FBR Sales Agreement may be sold in “at the market” offerings, and investors who buy shares at different times will likely pay different prices.
Investors who purchase shares that are sold under the B. Riley FBR Sales Agreement at different times will likely pay different prices, and so may experience different outcomes in their investment results. We will have discretion, subject to market demand, to vary the timing, prices, and numbers of shares sold, and there is no minimum or maximum sales price. Investors may experience declines in the value of their shares as a result of share sales made at prices lower than the prices they paid.
| 34 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference herein contain forward-looking statements, including statements regarding the progress and timing of our product development, the goals of our development activities, estimates of the potential markets for our product candidates, estimates of the capacity of manufacturing and other facilities to support our products, our expected future revenues, operations and expenditures and projected cash needs. The forward-looking statements are contained principally in the sections entitled “Prospectus Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business” of this prospectus and the documents incorporated by reference. These statements relate to future events of our financial performance and involve known and unknown risks, uncertainties and other factors that could cause our actual results, levels of activity, performance or achievement to differ materially from those expressed or implied by these forward-looking statements. Those risks and uncertainties include, among others:
| · | our ability to implement our business plan; |
| · | our ability to raise additional capital to meet our liquidity needs; |
| · | our ability to generate sufficient proceeds from this offering; |
| · | our ability to generate product revenues; |
| · | our ability to achieve profitability; |
| · | our ability to satisfy U.S. (including the FDA), and international regulatory requirements; |
| · | our ability to obtain market acceptance of our products; |
| · | our ability to compete in the market; |
| · | our ability to advance our clinical trials; |
| · | our ability to fund, design and implement clinical trials; |
| · | our ability to demonstrate that our product candidates are safe for human use and effective for indicated uses; |
| · | our ability to gain acceptance of physicians and patients for use of our products; |
| · | our dependency on third-party researchers and manufacturers and licensors; |
| · | our ability to establish and maintain strategic partnerships, including for the distribution of products; |
| · | our ability to attract and retain sufficient, qualified personnel; |
| · | our ability to obtain or maintain patents or other appropriate protection for the intellectual property; |
| · | our dependency on the intellectual property licensed to us or possessed by third parties; |
| · | our ability to adequately support future growth; |
| · | our ability to maintain our NYSE American listing; and |
| · | potential product liability or intellectual property infringement claims. |
Forward-looking statements include all statements that are not historical facts. In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,” “potential,” or the negative of those terms, and similar expressions and comparable terminology intended to identify forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. These forward-looking statements represent our estimates and assumptions only as of the date of this prospectus and, except as required by law, we undertake no obligation to update or review publicly any forward-looking statements, whether as a result of new information, future events or otherwise after the date of this prospectus. You should read this prospectus, the documents incorporated by reference in this prospectus, the documents referenced in this prospectus and the documents filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
| 35 |
We estimate that the net proceeds of this offering will be approximately $18.0 million, or approximately $20.8 million if the underwriters exercise in full their over-allotment option, assuming a public offering price of $2.10 per Class A Unit (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) and $1,000 per Class B Unit, after deducting the estimated underwriting discount and estimated offering expenses payable by us, and excluding the proceeds, if any, from the exercise of the warrants. The public offering price per Class A Unit will be determined between us, the underwriters and investors based on market conditions at the time of pricing, and may be at a discount to the current market price of our common stock. We will only receive additional proceeds from the exercise of the warrants issuable in connection with this offering if such warrants are exercised at their exercise price of 120% of the public offering price of the Class A Units and the holders of such warrants pay the exercise price in cash upon such exercise and do not utilize the cashless exercise provision of the warrants.
We intend to use the net proceeds, if any, from the sales of securities offered by this prospectus to fund our and our subsidiaries’ preclinical and clinical programs (including, but not limited to, provide approximately $5.0-$7.0 million in funding for manufacturing scale-up activities to progress SYN-004 towards a potential Phase 3 clinical trial (broad indication) and/or initiate a Phase 1/2 clinical trial(s) in a specialty population, approximately $7.5 million in funding for preclinical development and related manufacturing activities in preparation for our IND and Phase 1 clinical trial for our SYN-020 program and required milestone payments) and for working capital and general corporate purposes, including, to acquire, license or invest in complementary businesses, technologies, product candidates or other intellectual property. We have broad discretion in determining how the proceeds of this offering will be used, and our discretion is not limited by the aforementioned possible uses. Our board of directors believes the flexibility in application of the net proceeds is prudent.
The expected use of proceeds from this offering represent our current intentions based on present plans and business conditions. The amounts and timing of our actual expenditures will depend on numerous factors, including our development and commercialization efforts, as well as the amount of cash used in our operations. We therefore cannot estimate with certainty the amount of net proceeds to be used for the purposes described above. We may find it necessary or advisable to reallocate the net proceeds of this offering within the categories listed above or to use the net proceeds for other purposes. Accordingly we will have broad discretion in the application of the net proceeds. Pending the uses described above, we plan to invest the net proceeds from this offering in short-term, investment-grade, interest-bearing securities.
| 36 |
The following table sets forth our capitalization as of June 30, 2018:
| · | on an actual basis, adjusted to reflect the reverse stock split of one-for-thirty-five effective August 10, 2018; |
| · | on a pro forma basis to give effect to the issuance of 3,485,483 shares of common stock for which we received net proceeds of $11.8 million from July 1, 2018 through and immediately prior to the date of this prospectus but does not reflect reductions in cash subsequent to June 30, 2018 as a result of expenses incurred in the ordinary course of business; and |
| · | on a pro forma as adjusted basis to give effect to (i) the issuance of 3,485,483 shares of common stock for which we received net proceeds of $11.8 million from July 1, 2018 through and immediately prior to the date of this prospectus but does not reflect reductions in cash subsequent to June 30, 2018 as a result of expenses incurred in the ordinary course of business and (ii) the sale of9,523,809 Class A Units in this offering at the assumed public offering price of $2.10 per Class A Unit (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018), after deducting underwriting discounts and commissions and other estimated offering expenses payable by us. The pro forma as adjusted basis assumes no sale of Class B Units and excludes the proceeds, if any, from the exercise of any warrants issued in this offering. |
This Capitalization table should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our historical financial statements and notes to those financial statements that are incorporated by reference in this prospectus.
| As of June 30, 2018 (in thousands) | ||||||||||||
| Actual | Pro Forma | Pro Forma As Adjusted | ||||||||||
| Cash and cash equivalents | $ | 7,129 | $ | 18,886 | $ | 36,886 | ||||||
| Series A convertible preferred stock | 12,173 | 12,173 | 12,173 | |||||||||
| Common stock, $0.001 par value; 35,000,000 shares authorized, 3,722,837, shares issued and outstanding actual; Preferred Stock, $0.001 par value, 10,000,000 shares authorized, no shares issued and outstanding | 130 | 7 | 17 | |||||||||
| Additional paid-in capital | 194,186 | 205,940 | 223,930 | |||||||||
| Accumulated deficit | (200,803 | ) | (200,803 | ) | (200,803 | ) | ||||||
| Total Synthetic Biologics, Inc. and Subsidiaries Equity (Deficit) | (6,487 | ) | 5,144 | 23,144 | ||||||||
| Non-controlling interest | (1,940 | ) | (1,940 | ) | (1,940 | ) | ||||||
| Total Stockholders’ Equity (Deficit) | (8,427 | ) | 3,204 | 21,204 | ||||||||
| Total Capitalization | $ | (8,427 | ) | $ | 3,204 | $ | 21,204 | |||||
Unless we indicate otherwise, all information in this Capitalization section is as of October 8, 2018 and:
| · | reflects a one-for-thirty-five reverse stock split of our issued and outstanding shares of common stock, options and warrants effected onAugust 10, 2018 and the corresponding adjustment of all common stock prices per share and stock option and warrant exercise prices per share and conversion ratios without taking into account fractional shares which are rounded up to the nearest whole number; |
| · | assumes no exercise by theunderwriters of their over-allotment option; |
| · | excludes shares of our common stock issuable upon conversion of outstanding shares of Series A preferred stock; |
| · | excludes 347,765 shares of our common stock issuable upon exercise of outstanding options under our equity incentive plans at a weighted-average exercise price of $54.19 per share; |
| · | excludes 915,854 shares of our common stock reserved for issuance upon the exercise of outstanding warrants with a weighted-average exercise price of $75.16 per share and assumes no exercise of the warrants issued in this offering; |
| · | assumes no shares of Series B Preferred are sold in this offering; and | |
| · | excludes 170,674 shares of our common stock that are reserved for equity awards that may be granted under our equity incentive plans. |
To the extent we sell any Class B Units in this offering, the same aggregate number of common stock equivalents resulting from this offering would be convertible under the Series B Preferred issued as part of the Class B Units.
Each increase (decrease) of 250,000 Class A Units to be purchased at $2.10 per unit (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) would increase or (decrease) additional paid-in capital, total stockholders’ deficit and total capitalization by approximately $0.5 million, assuming the offering price remains at $2.10 and after deducting estimated underwriters’ discounts and commissions and estimated offering expenses payable by us. Each increase (decrease) of 500,000 Class A Units to be purchased at $2.10 per unit (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) would increase or (decrease) additional paid-in capital, total stockholders’ equity (deficit) and total capitalization by approximately $1.0 million, assuming the offering price remains at $2.10 and after deducting estimated underwriters’ discounts and commissions and estimated offering expenses payable by us.
A $0.25 increase (decrease) in the assumed public offering price of $2.10 per Class A Unit (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) would result in an incremental increase (decrease) in each of our additional paid-in capital, total stockholders’ equity (deficit) and total capitalization on a pro forma as adjusted basis by approximately $2.2 million, assuming that the number of Class A Units sold by us as set forth on the cover page of this prospect remains the same and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. A $0.50 increase (decrease) in the assumed public offering price of $2.10 per Class A Units (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) would result in an incremental increase (decrease) in each of our additional paid-in capital, total stockholders’ equity (deficit) and total capitalization on a pro forma as adjusted basis by approximately $4.4 million, assuming that the number of Class A Units sold by us as set forth on the cover page of this prospect remains the same and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
| 37 |
If you invest in our securities in this offering, you will experience dilution to the extent of the difference between the public offering price per Class A Unit in this offering and our as adjusted net tangible book value per share immediately after this offering assuming no value is attributed to the warrants, and the warrants are accounted for and classified as equity. This calculation does not reflect any dilution associated with the sale and exercise of the warrants.
Our net tangible book value on June 30, 2018 was approximately $3.7 million, or $1.01 per share. “Net tangible book value” is total assets minus the sum of liabilities and intangible assets. “Net tangible book value per share” is net tangible book value divided by the total number of shares outstanding.
Our pro forma net tangible book value as of June 30, 2018 was $15.5 million or $2.15 per share of common stock, based upon 7,208,320 shares outstanding, after giving effect to issuances of 3,485,483 shares of common stock for which we received net proceeds of $11.8 million from July 1, 2018 through and immediately prior to the date of this prospectus but does not reflect reductions in cash subsequent to June 30, 2018 as a result of expenses incurred in the ordinary course of business. After giving effect to the sale by us of 9,523,809 Class A units in this offering at a public offering price of $2.10 per Class A Unit, (which was the last reported sale price of our common stock on the NYSE American of October 8, 2018) and no Class B Units, and after deducting the estimated underwriting discount and estimated offering expenses payable by us, our pro forma as adjusted net tangible book value as of June 30, 2018 would have been approximately $33.5 million, or approximately $2.00 per share. This represents an immediate decrease in pro forma as adjusted net tangible book value of $0.15 per share to existing stockholders and an immediate dilution of $0.10 per share to new investors purchasing Class A Units in this offering. The following table illustrates this per share dilution:
| Assumed public offering price per Class A Unit | $ | 2.10 | ||||||
| Pro forma net tangible book value per share as of June 30, 2018 | $ | 2.15 | ||||||
| Decrease in pro forma net tangible book value per share after this offering | $ | (0.15 | ) | |||||
| Pro forma as adjusted net tangible book value per share after giving effect to this offering | $ | 2.00 | ||||||
| Dilution per share to investors purchasing our common stock in this offering | $ | 0.10 |
The information above and below assumes that no shares of Series B Preferred are sold in this offering. The information above assumes that the underwriters do not exercise their over-allotment option. If the underwriters exercise their over-allotment option in full, the pro forma as adjusted net tangible book value would be $2.00 per share, representing an immediate decrease to existing stockholders of $0.15 per share and an immediate dilution of $0.10 per share to new investors.
To the extent we sell any Class B Units in this offering, the same aggregate number of common stock equivalents resulting from this offering would be convertible under the Series B Preferred issued as part of the Class B Units.
A $0.25 increase (decrease) in the assumed public offering price of $2.10 per Class A Unit (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) would result in an incremental increase (decrease) in our pro forma net tangible book value of approximately $2.2 million or approximately $0.13 per share, and would result in an incremental increase (decrease) in the dilution to new investors of approximately $0.12 per share, assuming that the number of Class A Units sold by us remains the same and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. A $0.50 increase (decrease) in the assumed public offering price of $2.10 per Class A Unit (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018) would result in an incremental increase (decrease) in our pro forma net tangible book value of approximately $4.4 million or approximately $0.26 per share, and would result in an incremental increase (decrease) in the dilution to new investors of approximately $0.24 per share, assuming that the number of Class A Units sold by us remains the same and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
We may also increase or decrease the number of Class A Units we are offering from the assumed number of Class A Units set forth above. An increase (decrease) of 250,000 in the assumed number of Class A Units sold by us in this offering would result in an incremental increase (decrease) in our pro forma net tangible book value of approximately $0.5 million or approximately $0.00 per share, and would result in an incremental increase (decrease) in the dilution to new investors of approximately $0.00 per share, assuming that the assumed public offering price of the common stock remains the same and after deducting the estimated underwriting discount and estimated offering expenses payable by us. An increase (decrease) of 500,000 in the assumed number of Class A Units sold by us in this offering would result in an incremental increase (decrease) in our pro forma net tangible book value of approximately $1.0 million or approximately $0.00 per share and would result in an incremental increase (decrease) in the dilution to new investors of approximately $0.00 per share, assuming that the assumed public offering price of the common stock remains the same and after deducting the estimated.
The foregoing discussion and table do not take into account further dilution to new investors that could occur upon the exercise of outstanding options or warrants or conversion of our outstanding Series A Preferred Stock. In addition, we may choose to raise additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance of these securities could result in further dilution to our stockholders.
| 38 |
MARKET FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
Market Information
Our common stock has traded on the NYSE American under the symbol “SYN” since February 16, 2012. Prior to February 16, 2012, our common stock traded under the symbol “AEN” since October 16, 2008. The following table states the range of the high and low sales prices of our common stock for the year ended December 31, 2016, and the year ended December 31, 2017 and the first and second fiscal quarter of 2018 and for the third quarter through October 8, 2018 (as adjusted to reflect the one-for-thirty-five reverse stock split effective August 10, 2018). These quotations represent inter-dealer prices, without retail mark-up, markdown, or commission, and may not represent actual transactions. The last price of our common stock as reported on the NYSE American on October 8, 2018 was $2.10 per share. As of October 8, 2018, there were approximately 347 stockholders of record of our common stock. This number does not include beneficial owners from whom shares are held by nominees in street name.
On August 1, 2018, we announced a reverse stock split of our shares of common stock at a ratio of one-for-thirty-five. The reverse stock split took effect at 11 p.m. (Eastern Time) on August 10, 2018, and our common stock began to trade on a post-split basis at the market open on August 13, 2018. When the reverse stock split became effective, every 35 shares of our issued and outstanding common stock were combined into one share of common stock. Effecting the reverse stock split reduced the number of issued and outstanding common stock from approximately 132,969,743 shares to approximately 3,799,136. It also subsequently adjusted outstanding options issued under our equity incentive plan, outstanding warrants to purchase common stock and our outstanding preferred stock.
| High | Low | |||||||
| YEAR ENDED DECEMBER 31, 2016 | ||||||||
| First Quarter | $ | 82.60 | $ | 35.35 | ||||
| Second Quarter | $ | 95.55 | $ | 57.40 | ||||
| Third Quarter | $ | 66.85 | $ | 54.95 | ||||
| Fourth Quarter | $ | 61.95 | $ | 26.60 | ||||
| YEAR ENDED DECEMBER 31, 2017 | ||||||||
| First Quarter | $ | 36.05 | $ | 20.65 | ||||
| Second Quarter | $ | 26.25 | $ | 14.35 | ||||
| Third Quarter | $ | 36.75 | $ | 16.10 | ||||
| Fourth Quarter | $ | 33.60 | $ | 17.50 | ||||
| YEAR ENDED DECEMBER 31, 2018 | ||||||||
| First Quarter | $ | 21.00 | $ | 9.80 | ||||
| Second Quarter | $ | 12.95 | $ | 6.65 | ||||
| Third Quarter (through October 8, 2018) | $ | 9.10 | $ | 1.86 | ||||
| 39 |
We have never paid cash dividends on our common stock. Moreover, we do not anticipate paying periodic cash dividends on our common stock for the foreseeable future. We intend to use all available cash and liquid assets in the operation and growth of our business, subject to terms of any preferred stock or debt securities. Any future determination about the payment of dividends will be made at the discretion of our board of directors and will be subject to the rights of any outstanding preferred stock and will depend upon our earnings, if any, capital requirements, operating and financial conditions and on such other factors as our board of directors deems relevant. The Series A Preferred Stock ranks senior to the shares of our common stock with respect to dividend rights and holders of Series A Preferred Stock are entitled to a cumulative dividend at the rate of 2.0% per annum, payable quarterly in arrears, as set forth in the Certificate of Designation of Series A Preferred Stock.
| 40 |
Authorized Capital
Our authorized capital currently consists of 200 million shares of common stock, par value $0.001 per share, and 10 million shares of preferred stock, par value $0.001 per share. As of October 8, 2018, 7,208,320 shares of common stock were issued and outstanding, and 120,000 shares of preferred stock were issued and outstanding.
Common Stock
We may issue shares of our common stock from time to time. We currently have authorized 200,000,000 million shares of common stock, par value $.001 per share. We may offer shares of common stock alone or underlying the registered securities convertible into or exercisable for our common stock.
Voting. The holders of our common stock are entitled to one vote for each share held of record on all matters submitted to a vote of the stockholders, including the election of directors, and do not have cumulative voting rights. Accordingly, the holders of a majority of the shares of our common stock entitled to vote in any election of directors can elect all of the directors standing for election.
Dividends. Subject to preferences that may be applicable to any then outstanding preferred stock, the holders of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds.
Liquidation. In the event of our liquidation, dissolution or winding up, holders of our common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities, subject to the satisfaction of any liquidation preference granted to the holders of any then outstanding shares of preferred stock.
Rights and Preferences. The holders of our common stock have no preemptive, conversion or subscription rights, and there are no redemption or sinking fund provisions applicable to our common stock. The rights, preferences and privileges of the holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of our preferred stock that we may designate and issue in the future.
Fully Paid and Nonassessable. All of our outstanding shares of common stock are, and the shares of common stock to be issued under this prospectus will be, fully paid and nonassessable.
In this prospectus, we have summarized certain general features of our common stock under “Description of Our Securities—Common Stock”. We urge you, however, to read the applicable prospectus supplement (and any related free writing prospectus that we may authorize to be provided to you) related to any common stock being offered.
Preferred Stock
Our board of directors has the authority, without action by our stockholders, to designate and issue up to 10 million shares of preferred stock in one or more series or classes and to designate the rights, preferences and privileges of each series or class, which may be greater than the rights of our common stock. It is not possible to state the actual effect of the issuance of any shares of preferred stock upon the rights of holders of our common stock until our board of directors determines the specific rights of the holders of the preferred stock. However, the effects might include:
| ● | restricting dividends on our common stock; |
| ● | diluting the voting power of our common stock; |
| ● | impairing liquidation rights of our common stock; or |
| ● | delaying or preventing a change in control of us without further action by our stockholders. |
| 41 |
The board of directors’ authority to issue preferred stock without stockholder approval could make it more difficult for a third-party to acquire control of our company, and could discourage such attempt. We have no present plans to issue any shares of preferred stock.
Series A Preferred
We had 120,000 shares of Series A Preferred Stock outstanding as of October 8, 2018.
The Series A Preferred Stock ranks senior to the shares of our common stock, and any other class or series of stock issued by us with respect to dividend rights, redemption rights and rights on the distribution of assets on our voluntary or involuntary liquidation, dissolution or winding up. Holders of Series A Preferred Stock are entitled to a cumulative dividend at the rate of 2.0% per annum, payable quarterly in arrears, as set forth in the Certificate of Designation of Series A Preferred Stock classifying the Series A Preferred Stock. The Series A Preferred Stock is convertible at the option of the holders at any time into shares of common stock at a conversion price of $18.90 per share (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018), subject to certain customary anti-dilution adjustments.
Any conversion of Series A Preferred Stock may be settled by us in shares of common stock only.
The holder’s ability to convert the Series A Preferred Stock into common stock is subject to (i) a 19.99% blocker provision to comply with NYSE American Listing Rules, (ii) if so elected by the holder, a 4.99% blocker provision that will prohibit beneficial ownership of more than 4.99% of our outstanding shares common stock or voting power at any time, and (iii) applicable regulatory restrictions.
In the event of our liquidation, dissolution or winding-up, holders of the Series A Preferred Stock are entitled to a preference on liquidation equal to the greater of (i) an amount per share equal to the stated value plus any accrued and unpaid dividends on such share of Series A Preferred Stock (the “Accreted Value”), and (ii) the amount such holders would receive in such liquidation if they converted their shares of Series A Preferred Stock (based on the Accreted Value and without regard to any conversion limitation) into shares of the common stock immediately prior to any such liquidation, dissolution or winding-up (the greater of (i) and (ii), is referred to as the “Liquidation Value”).
Except as otherwise required by law, the holders of Series A Preferred Stock have no voting rights, other than customary protections against adverse amendments and issuance ofpari passu or senior preferred stock. Upon certain change of control events involving our company, we will be required to repurchase all of the Series A Preferred Stock at a redemption price equal to the greater of (i) the Accreted Value and (ii) the amount that would be payable upon a change of control (as defined in the Certificate of Designation) in respect of common stock issuable upon conversion of such share of Series A Preferred Stock if all outstanding shares of Series A Preferred Stock were converted into common stock immediately prior to the change of control.
On or at any time after (i) the VWAP (as defined in the Certificate of Designation) for at least 20 trading days in any 30 trading day period is greater than $70.00 (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018), subject to adjustment in the case of stock split, stock dividends or the like we have the right, after providing notice not less than 6 months prior to the redemption date, to redeem, in whole or in part, on a pro rata basis from all holders thereof based on the number of shares of Series A Preferred Stock then held, the outstanding Series A Preferred Stock, for cash, at a redemption price per share of Series A Preferred Stock of $7,875.00 (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018), subject to appropriate adjustment in the event of any stock dividend, stock split, combination or other similar recapitalization with respect to the Series A Preferred Stock, or (ii) the five year anniversary of the issue date, we have the right to redeem, in whole or in part, on a pro rata basis from all holders thereof based on the number of shares of Series A Preferred Stock then held, the outstanding Series A Preferred Stock, for cash, at a redemption price per share equal to the Liquidation Value.
Warrants
As of October 8, 2018, we had issued and outstanding warrants to purchase a total of 915,857 shares of our common stock outstanding at a weighted-average price of $75.16 (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018 without taking into account fractional shares which are rounded up to the nearest whole number).
On November 18, 2016, we completed a public offering of 714,286 shares of common stock (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018) in combination with accompanying warrants to purchase an aggregate of 1,428,571 shares of the common stock(as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018), of which warrants to purchase 714,286 (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018 without taking into account fractional shares) shares of common stock are outstanding (the “Series A Warrants”). The per share exercise price of the Series A Warrants is $50.05 (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018) subject to further adjustment as specified in the warrant agreements. The Series A Warrants may be exercised at any time until the four-year anniversary of the issuance date. The warrants include a provision that if we were to enter into a certain transaction, as defined in the agreement, the warrants would be purchased from the holder for cash.
| 42 |
On October 10, 2014, we issued 14,059,616 units at a price of $1.47 per unit to certain institutional investors in a registered direct offering, each unit consisted of one share of our common stock and a warrant to purchase 0.5 shares of common stock. The warrants, exercisable for an aggregate of 200,852 (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018 without taking into account fractional shares) shares of common stock, have an exercise price of $61.25 per share (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018) and a life of five years. The warrants vested immediately and expire on October 10, 2019.
Options
As of October 8, 2018, options to purchase an aggregate of 347,765 (as adjusted to reflect the 1-for-35 reverse stock split effected August 10, 2018 without taking into account fractional shares) shares of common stock were outstanding under our equity incentive plans.
Stockholder Registration Rights
We are party to a registration rights agreement (the “Registration Rights Agreement”) that provides the holder of the Series A Preferred Stock with certain registration rights. Pursuant to the terms of the Registration Rights Agreement, we agreed to file a registration statement covering resales of the shares of common stock issuable upon conversion of the Series A Preferred Stock with the SEC within 60 days following receipt of a request at any time (as long as the requestor beneficially owns at least ten percent (10%) of our common stock then outstanding or is otherwise deemed our affiliate) and to use reasonable best efforts to have the registration statement declared effective within 120 days following receipt of such request.
We have agreed to pay certain penalties if the registration statement is not declared effective by the SEC on or before the required deadline. After that deadline and until such time as the registration statement is declared effective (or until we are no longer required to cause the registration statement to be declared effective), we will be required to pay additional liquidated damages.
Pursuant to the terms of the registration rights agreement that we entered into with Intrexon and an affiliated entity, we were required to file a registration statement with respect to securities issued and are required to maintain the effectiveness of such registration statement. The failure to do so could result in the payment of damages by us. The registration statement was declared effective on April 29, 2013.
Anti-Takeover Effects of Certain Provisions of our Articles of Incorporation and Bylaws
Our Articles of Incorporation, as amended, and amended and restated bylaws contain certain provisions that may have anti-takeover effects, making it more difficult for or preventing a third party from acquiring control of the Company or changing its board of directors and management. According to our Amended and Restated Bylaws and Articles of Incorporation, neither the holders of our common stock nor the holders of any preferred stock we may issue in the future have cumulative voting rights in the election of our directors. The lack of cumulative voting makes it more difficult for other stockholders to replace our board of directors or for a third party to obtain control of our company by replacing its board of directors.
Authorized but Unissued Shares
Our authorized but unissued shares of common stock and preferred stock will be available for future issuance without stockholder approval. We may use additional shares for a variety of purposes, including future public offerings to raise additional capital, to fund acquisitions and as employee compensation. The existence of authorized but unissued shares of common stock and preferred stock could render more difficult or discourage an attempt to obtain control of us by means of a proxy contest, tender offer, merger or otherwise.
| 43 |
Anti-Takeover Effects of Nevada Law
Business Combinations
The “business combination” provisions of Sections 78.411 to 78.444, inclusive, of the Nevada Revised Statute (the “NRS”) generally prohibit a Nevada corporation with at least 200 stockholders from engaging in various “combination” transactions with any interested stockholder for a period of two years after the date of the transaction in which the person became an interested stockholder, unless the transaction is approved by the board of directors prior to the date the interested stockholder obtained such status or the combination is approved by the board of directors and thereafter is approved at a meeting of the stockholders by the affirmative vote of stockholders representing at least 60% of the outstanding voting power held by disinterested stockholders, and extends beyond the expiration of the two-year period, unless:
| · | the combination was approved by the board of directors prior to the person becoming an interested stockholder or the transaction by which the person first became an interested stockholder was approved by the board of directors before the person became an interested stockholder or the combination is later approved by a majority of the voting power held by disinterested stockholders; or |
| · | if the consideration to be paid by the interested stockholder is at least equal to the highest of: (a) the highest price per share paid by the interested stockholder within the two years immediately preceding the date of the announcement of the combination or in the transaction in which it became an interested stockholder, whichever is higher, (b) the market value per share of common stock on the date of announcement of the combination and the date the interested stockholder acquired the shares, whichever is higher, or (c) for holders of preferred stock, the highest liquidation value of the preferred stock, if it is higher. |
A “combination” is generally defined to include mergers or consolidations or any sale, lease exchange, mortgage, pledge, transfer, or other disposition, in one transaction or a series of transactions, with an “interested stockholder” having: (a) an aggregate market value equal to 5% or more of the aggregate market value of the assets of the corporation, (b) an aggregate market value equal to 5% or more of the aggregate market value of all outstanding shares of the corporation, (c) 10% or more of the earning power or net income of the corporation, and (d) certain other transactions with an interested stockholder or an affiliate or associate of an interested stockholder.
In general, an “interested stockholder” is a person who, together with affiliates and associates, owns (or within two years, did own) 10% or more of a corporation’s voting stock. The statute could prohibit or delay mergers or other takeover or change in control attempts and, accordingly, may discourage attempts to acquire our company even though such a transaction may offer our stockholders the opportunity to sell their stock at a price above the prevailing market price.
Control Share Acquisitions
The “control share” provisions of Sections 78.378 to 78.3793, inclusive, of the NRS apply to “issuing corporations” that are Nevada corporations with at least 200 stockholders, including at least 100 stockholders of record who are Nevada residents, and that conduct business directly or indirectly in Nevada. The control share statute prohibits an acquirer, under certain circumstances, from voting its shares of a target corporation’s stock after crossing certain ownership threshold percentages, unless the acquirer obtains approval of the target corporation’s disinterested stockholders. The statute specifies three thresholds: one-fifth or more but less than one-third, one-third but less than a majority, and a majority or more, of the outstanding voting power. Generally, once an acquirer crosses one of the above thresholds, those shares in an offer or acquisition and acquired within 90 days thereof become “control shares” and such control shares are deprived of the right to vote until disinterested stockholders restore the right. These provisions also provide that if control shares are accorded full voting rights and the acquiring person has acquired a majority or more of all voting power, all other stockholders who do not vote in favor of authorizing voting rights to the control shares are entitled to demand payment for the fair value of their shares in accordance with statutory procedures established for dissenters’ rights.
A corporation may elect to not be governed by, or “opt out” of, the control share provisions by making an election in its articles of incorporation or bylaws, provided that the opt-out election must be in place on the 10th day following the date an acquiring person has acquired a controlling interest, that is, crossing any of the three thresholds described above. We have not opted out of the control share statutes, and will be subject to these statutes if we are an “issuing corporation” as defined in such statutes.
The effect of the Nevada control share statutes is that the acquiring person, and those acting in association with the acquiring person, will obtain only such voting rights in the control shares as are conferred by a resolution of the stockholders at an annual or special meeting. The Nevada control share law, if applicable, could have the effect of discouraging takeovers of our company.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Corporate Stock Transfer, Inc. The transfer agent’s address is 3200 Cherry Creek South Drive, Suite 430, Denver, Colorado 80209. The transfer agent for any series of preferred stock that we may offer under this prospectus will be named and described in the prospectus supplement for that series.
Listing on the NYSE American
Our common stock is listed on the NYSE American under the symbol “SYN”.
| 44 |
DESCRIPTION OF SECURITIES WE ARE OFFERING
We are offering up to 9,523,809 Class A Units, assuming no exercise of the over-allotment option. We are also offering to each purchaser whose purchase of Class A Units in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering, the opportunity in lieu of purchasing Class A Units, to purchase Class B Units. Each Class B Unit will consist of one share of Series B Preferred with a stated value of $1,000 and convertible into shares of our common stock at the public offering price of the Class A Units, together with the equivalent number of warrants as would have been issued to such purchaser of Class B Units if they had purchased Class A Units based on the public offering price. For each Class B Unit we sell, the number of Class A Units we are offering will be decreased on a dollar-for-dollar basis. Because we will issue a warrant as part of each Unit, the number of warrants sold in this offering will not change as a result of a change in the mix of the Units sold. The number of shares of our common stock outstanding after this offering will fluctuate depending on how many Class B Units are sold in this offering and whether and to what extent holders of Series B Preferred shares convert their shares to common stock. We are also offering the shares of common stock issuable upon exercise of warrants sold in Class B Units and upon conversion of the Series B Preferred.
Common Stock
The material terms and provisions of our common stock and each other class of our securities which qualifies or limits our common stock are described under the caption “Description of Our Securities” in this prospectus.
Preferred Stock
Pursuant to the terms of our articles of incorporation, our board of directors has the authority to issue preferred stock in one or more classes or series and to fix the designations, powers, preferences and rights, and the qualifications, limitations or restrictions thereof, including dividend rights, conversion right, voting rights, terms of redemption, liquidation preferences and the number of shares constituting any class or series, without further vote or action by the stockholders.
Series B Convertible Preferred Stock
The following is a summary of the material terms of the Series B Preferred. This summary is not complete. The following summary of the terms and provisions of the Series B Preferred is qualified in its entirety by reference to the Certificate of Designation of the Series B Preferred, the form of which has been filed as an exhibit to the registration statement of which this prospectus is a part.
General.Our board of directors has designated up to shares of the 10,000,000 authorized shares of preferred stock as Series B Convertible Preferred Stock. When issued, the shares of Series B Preferred will be validly issued, fully paid and non-assessable. Each share of Series B Preferred will have a stated value of $1,000 per share.
Rank.The Series B Preferred will rank junior to the Series A Preferred Stock and on parity to our common stock.
Conversion.Each share of Series B Preferred is convertible into shares of our common stock (subject to adjustment as provided in the related certificate of designation of preferences, rights and limitations) at any time at the option of the holder at a conversion price equal to the stated value of the Series B Preferred of $1,000 divided by the public offering price of the Class A Units in this offering. Holders of Series B Preferred will be prohibited from converting Series B Preferred into shares of our common stock if, as a result of such conversion, the holder, together with its affiliates, would own more than 4.99% of the total number of shares of our common stock then issued and outstanding. However, any holder may increase or decrease such percentage to any other percentage not in excess of 9.99% upon at least 61 days’ prior notice from the holder to us.
Liquidation Preference. Subject to the senior rights of the Series A Preferred Stock, in the event of our liquidation, dissolution or winding-up, holders of Series B Preferred will be entitled to receive if the Series B Preferred were fully converted into shares of our common stock at the conversion price (disregarding for such purposes any conversion limitations) which amounts shall be paidpari passu with all holders of common stock.
Voting Rights. Shares of Series B Preferred will generally have no voting rights, except as required by law and except that the affirmative vote of the holders of a majority of the then outstanding shares of Series B Preferred is required to, (a) alter or change adversely the powers, preferences or rights given to the Series B Preferred, (b) amend our articles of incorporation or other charter documents in any manner that materially adversely affects any rights of the holders, (c) increase the number of authorized shares of Series B Preferred, or (d) enter into any agreement with respect to any of the foregoing.
Dividends.Shares of Series B Preferred will not be entitled to receive any dividends, unless and until specifically declared by our board of directors. Subject to any senior rights of the Series A Preferred Stock, the holders of the Series B Preferred will participate, on an as-if-converted-to-common stock basis, in any dividends to the holders of common stock.
Redemption.We are not obligated to redeem or repurchase any shares of Series B Preferred. Shares of Series B Preferred are not otherwise entitled to any redemption rights or mandatory sinking fund or analogous fund provisions.
| 45 |
Exchange Listing. We do not plan on making an application to list the Series B Preferred on the NYSE American, any other national securities exchange or other nationally recognized trading system.
Warrants
The following summary of certain terms and provisions of the warrants that are being offered hereby is not complete and is subject to, and qualified in its entirety by, the provisions of the warrant agent agreement, the form of which is filed as an exhibit to the registration statement of which this prospectus forms a part. Prospective investors should carefully review the terms and provisions of the form of warrant agent agreement for a complete description of the terms and conditions of the common warrants.
Form. The warrants will be issued in electronic book entry form. The form of warrant is filed as an exhibit to this registration statement.
Exercisability. The warrants are exercisable at any time after their original issuance and will expire on the third anniversary of the original issuance date. The warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice and by payment in full in immediately available funds for the number of shares of common stock purchased upon such exercise. If at the time of exercise, there is no effective registration statement registering, or no current prospectus available for, the issuance of the shares of common stock to the holder, then the common warrant may only be exercised through a cashless exercise, in which case the holder would receive upon such exercise the net number of shares of common stock determined according to the formula set forth in the warrant. No fractional shares of common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, we will pay the holder an amount in cash equal to the fractional amount multiplied by the fair market value of any such fractional shares.
Exercise Limitations. Under the warrants, we may not effect the exercise of any warrant, and a holder will not be entitled to exercise any portion of any warrant, which, upon giving effect to such exercise, would cause (i) the aggregate number of shares of our common stock beneficially owned by the holder (together with its affiliates) to exceed [4.99%/9.99%] of the number of shares of our common stock outstanding immediately after giving effect to the exercise, or (ii) the combined voting power of our securities beneficially owned by the holder (together with its affiliates) to exceed [4.99%/9.99%] of the combined voting power of all of our securities then outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the warrants. However, any holder may increase or decrease such percentage to any other percentage not in excess of 9.99% upon at least 61 days’ prior notice from the holder to us.
Exercise Price. The exercise price per whole share of our common stock purchasable upon the exercise of the warrants is 120% of the public offering price of the Class A Units, or $2.52 per share, based on an assumed offering price of $2.10 per Class A Unit (which was the last reported sale price of our common stock on the NYSE American on October 8, 2018). The exercise price of the warrants is subject to appropriate adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting our common stock and also upon any distributions of assets, including cash, stock or other property to our stockholders. Subject to certain exceptions, the warrants provide for adjustment of the exercise price if we or any of our subsidiaries, as applicable, sell or grant any right to reprice, or otherwise dispose of or issue (or announce any offer, sale, grant or any option to purchase or other disposition) any shares of our common stock or common stock equivalents, at an effective price per share that is less than the exercise price then in effect (such lower price, the “Base Share Price” and such issuances collectively, a “Dilutive Issuance”). In the event a Dilutive Issuance occurs, the exercise price shall be reduced to equal the Base Share Price.
Transferability. Subject to applicable laws, the warrants may be offered for sale, sold, transferred or assigned without our consent.
Exchange Listing. We do not plan on applying to list the warrants on the NYSE American, any other national securities exchange or any other nationally recognized trading system.
Fundamental Transactions. In the event of a fundamental transaction, as described in the warrants and generally including any reorganization, recapitalization or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding common stock, or any person or group becoming the beneficial owner of 50% of the voting power represented by our outstanding common stock, the holders of the common warrants will be entitled to receive upon exercise of the warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the warrants immediately prior to such fundamental transaction without regard to any limitations on exercise contained in the warrants. In the event of a fundamental transaction, we are required to cause any successor entity to assume all of our obligations under the warrants.
Right as a Stockholder. Except by virtue of such holder’s ownership of shares of our common stock, the holder of a warrant does not have the rights or privileges of a holder of our common stock, including any voting rights, until the holder exercises the warrant.
| 46 |
We have entered into an underwriting agreement, dated , 2018, with A.G.P., acting as the representative of the several underwriters named below. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to the underwriters, and the underwriters have severally agreed to purchase, the number of Units, provided below opposite their respective names.
| Underwriters | Number of Class A Units | Number of Class B Units | ||||||
| A.G.P./Alliance Global Partners | ||||||||
| Total | ||||||||
The underwriters are offering the Units subject to their acceptance of the Units from us and subject to prior sale. The underwriting agreement provides that the obligations of the several underwriters to pay for and accept delivery of the Units offered by this prospectus are subject to the approval of certain legal matters by their counsel and to certain other conditions. The underwriter is obligated to purchase all of the Units if any of the securities are purchased, other than those shares covered by the over-allotment option to purchase additional securities described below.
Discount, Commissions and Expenses
The underwriters have advised us that they propose to offer the Units to the public at the public offering price set forth on the cover page of this prospectus and to certain dealers at that price less a concession not in excess of $ per Unit. The underwriters may allow, and certain dealers may reallow, a discount from the concession not in excess of $ per Unit to certain brokers and dealers. After this offering, the public offering price, concession and reallowance to dealers may be changed by the representative. No such change shall change the amount of proceeds to be received by us as set forth on the cover page of this prospectus. The Units are offered by the underwriters as stated herein, subject to receipt and acceptance by them and subject to their right to reject any order in whole or in part. The underwriters have informed us that they do not intend to confirm sales to any accounts over which they exercise discretionary authority.
The following table shows the underwriting discount payable to the underwriters by us in connection with this offering.
| Total | ||||||||||||||||
| Per Class A Unit | Per Class B Unit | Without Over- Allotment | With Over- Allotment | |||||||||||||
| Public offering price | $ | $ | $ | $ | ||||||||||||
| Underwriting discount(1) | $ | $ | $ | $ | ||||||||||||
| Proceeds, before expenses, to us | $ | $ | $ | $ | ||||||||||||
We have agreed to reimburse the underwriters for certain out-of-pocket expenses not to exceed $150,000 in the aggregate without our consent which shall not be unreasonably withheld. We estimate that expenses payable by us in connection with this offering, including reimbursement of the underwriters out-of-pocket expenses, but excluding the underwriting discount referred to above, will be approximately $ .
| (1) | The underwriter will receive a discount of 7% to the public offering price with respect to any Class A Units or Class B Units purchased in this offering by investors. |
Option To Purchase Additional Shares and Warrants
We have granted to the underwriters an over-allotment option exercisable not later than 45 days after the date of this prospectus to purchase up to additional shares of common stock (15% of the shares included in the Class A Units sold in this offering and the shares of common stock issuable upon conversion of the Series B Preferred included in Class B Units sold in the offering) and/or warrants to purchase a maximum of shares of common stock from us to cover over-allotments, if any. If the underwriters exercise all or part of this option, they will purchase such common stock covered by the option at the public offering price per Class A Unit, minus once cent and the warrants covered by this option at a price of one cent per warrant, in each case less the underwriting discounts and commissions. If this option is exercised in full, the total offering price to the public will be approximately $ million and the total net proceeds, after expenses, to us will be approximately $ million
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act and liabilities arising from breaches of representations and warranties contained in the underwriting agreement, or to contribute to payments that the underwriters may be required to make in respect of those liabilities.
| 47 |
Determination of Offering Price
The actual offering price of the securities we are offering will be negotiated between us and the underwriter based on the trading of our shares of common stock prior to the offering, among other things, and may be at a discount to the current market price.
Lock-up Agreements
We, our officers and directors have agreed, subject to limited exceptions, for a period of 90 days after the date of the underwriting agreement, not to offer, sell, contract to sell, pledge, grant any option to purchase, make any short sale or otherwise dispose of, directly or indirectly any shares of common stock or any securities convertible into or exchangeable for our common stock either owned as of the date of the underwriting agreement or thereafter acquired without the prior written consent of the representative. The representative may, in its sole discretion and at any time or from time to time before the termination of the lock-up period, without notice, release all or any portion of the securities subject to lock-up agreements.
Price Stabilization, Short Positions and Penalty Bids
The underwriter may engage in syndicate covering transactions, stabilizing transactions and penalty bids or purchases for the purpose of pegging, fixing or maintaining the price of our shares of common stock:
| · | Syndicate covering transactions involve purchases of securities in the open market after the distribution has been completed in order to cover syndicate short positions. Such a naked short position would be closed out by buying securities in the open market. A naked short position is more likely to be created if the underwriter is concerned that there could be downward pressure on the price of the securities in the open market after pricing that could adversely affect investors who purchase in the offering. |
| · | Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specific maximum. |
| · | Penalty bids permit the underwriter to reclaim a selling concession from a syndicate member when the securities originally sold by the syndicate member are purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions. |
These syndicate covering transactions, stabilizing transactions and penalty bids may have the effect of raising or maintaining the market prices of our securities or preventing or retarding a decline in the market prices of our securities. As a result, the price of our shares of common stock may be higher than the price that might otherwise exist in the open market. Neither we nor the underwriter make any representation or prediction as to the effect that the transactions described above may have on the price of our shares of common stock. These transactions may be effected on the NYSE American, in the over-the-counter market or on any other trading market and, if commenced, may be discontinued at any time.
In connection with this offering, the underwriter also may engage in passive market making transactions in our shares of common stock in accordance with Regulation M during a period before the commencement of offers or sales of our shares of common stock in this offering and extending through the completion of the distribution. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for that security. However, if all independent bids are lowered below the passive market maker’s bid that bid must then be lowered when specific purchase limits are exceeded. Passive market making may stabilize the market price of the securities at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
Neither we nor the underwriter make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the prices of our securities. In addition, neither we nor the underwriter make any representation that the underwriter will engage in these transactions or that any transactions, once commenced, will not be discontinued without notice.
Electronic Distribution
This prospectus in electronic format may be made available on websites or through other online services maintained by one or more of the underwriters, or by their affiliates. Other than this prospectus in electronic format, the information on any underwriter’s website and any information contained in any other website maintained by an underwriter is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or any underwriter in its capacity as underwriter, and should not be relied upon by investors.
Other
From time to time, certain of the underwriters and/or their affiliates have provided, and may in the future provide, various investment banking and other financial services for us for which services they have received and, may in the future receive, customary fees. In the course of their businesses, the underwriters and their affiliates may actively trade our securities or loans for their own account or for the accounts of customers, and, accordingly, the underwriters and their affiliates may at any time hold long or short positions in such securities or loans. Except for services provided in connection with this offering, no underwriter has provided any investment banking or other financial services to us during the 180-day period preceding the date of this prospectus and we do not expect to retain any underwriter to perform any investment banking or other financial services for at least 90 days after the date of this prospectus.
| 48 |
Notice to Investors in Canada
The securities may be sold only to purchasers purchasing, or deemed to be purchasing, as principal that are accredited investors, as defined in National Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are permitted clients, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale securities must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements of applicable securities laws.
Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies for rescission or damages if this prospectus (including any amendment thereto) contains a misrepresentation, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s province or territory for particulars of these rights or consult with a legal advisor.
Pursuant to section 3A.3 (or, in the case of securities issued or guaranteed by the government of a non-Canadian jurisdiction, section 3A.4) of National Instrument 33-105 Underwriting Conflicts (“NI 33-105”), the underwriters are not required to comply with the disclosure requirements of NI 33-105 regarding underwriter conflicts of interest in connection with this offering.
Notice to Investors in the United Kingdom
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a “Relevant Member State”) an offer to the public of any securities which are the subject of the offering contemplated by this prospectus may not be made in that Relevant Member State except that an offer to the public in that Relevant Member State of any such securities may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
(a) to legal entities which are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities;
(b) to any legal entity which has two or more of: (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than €43,000,000; and (3) an annual net turnover of more than €50,000,000, as shown in its last annual or consolidated accounts;
(c) by the underwriters to fewer than 100 natural or legal persons (other than qualified investors as defined in the Prospectus Directive); or
(d) in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of these securities shall result in a requirement for the publication by the issuer or the underwriters of a prospectus pursuant to Article 3 of the Prospectus Directive.
For the purposes of this provision, the expression an “offer to the public” in relation to any of the securities in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any such securities to be offered so as to enable an investor to decide to purchase any such securities, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State and the expression “Prospectus Directive” means Directive 2003/71/EC and includes any relevant implementing measure in each Relevant Member State.
Each underwriter has represented, warranted and agreed that:
(a) it has only communicated or caused to be communicated and will only communicate or cause to be communicated any invitation or inducement to engage in investment activity (within the meaning of section 21 of the Financial Services and Markets Act 2000 (the FSMA)) received by it in connection with the issue or sale of any of the securities in circumstances in which section 21(1) of the FSMA does not apply to the issuer; and
(b) it has complied with and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the securities in, from or otherwise involving the United Kingdom.
European Economic Area
In particular, this document does not constitute an approved prospectus in accordance with European Commission’s Regulation on Prospectuses no. 809/2004 and no such prospectus is to be prepared and approved in connection with this offering. Accordingly, in relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (being the Directive of the European Parliament and of the Council 2003/71/EC and including any relevant implementing measure in each Relevant Member State) (each, a Relevant Member State), with effect from and including the date on which the Prospectus Directive is implemented in that Relevant Member State (the Relevant Implementation Date) an offer of securities to the public may not be made in that Relevant Member State prior to the publication of a prospectus in relation to such securities which has been approved by the competent authority in that Relevant Member State or, where appropriate, approved in another Relevant Member State and notified to the competent authority in that Relevant Member State, all in accordance with the Prospectus Directive, except that it may, with effect from and including the Relevant Implementation Date, make an offer of securities to the public in that Relevant Member State at any time:
| · | to legal entities which are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities; |
| · | to any legal entity which has two or more of: (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than €43,000,000; and (3) an annual net turnover of more than €50,000,000, as shown in the last annual or consolidated accounts; or |
| · | in any other circumstances which do not require the publication by the Issuer of a prospectus pursuant to Article 3 of the Prospectus Directive. |
For the purposes of this provision, the expression an “offer of securities to the public” in relation to any of the securities in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and the securities to be offered so as to enable an investor to decide to purchase or subscribe for the securities, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State. For these purposes the securities offered hereby are “securities.”
| 49 |
Gracin & Marlow, LLP, New York, New York will pass upon certain legal matters related to the issuance and sale of the pre-funded warrants offered on our behalf and Parsons Behle & Latimer, Reno, Nevada will pass upon certain legal matters relating to the issuance and sale of the common stock offered hereby on our behalf. Zysman, Aharoni, Gayer and Sullivan & Worcester LLP, is acting as counsel to the underwriters in this offering.
The financial statements as of December 31, 2017 and 2016 and for the years then ended and management’s assessment of the effectiveness of internal control over financial reporting as of December 31, 2017 incorporated by reference in this Prospectus and in the Registration Statement have been so incorporated in reliance on the reports of BDO USA, LLP, an independent registered public accounting firm (the reports on the financial statements contain an explanatory paragraph regarding the Company’s ability to continue as a going concern), incorporated by reference in this Prospectus and in the Registration Statement, given on the authority of said firm as experts in auditing and accounting.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the Securities and Exchange Commission (the “SEC”) a registration statement on Form S-1 under the Securities Act with respect to the securities offered by this prospectus. This prospectus, which is part of the registration statement, omits certain information, exhibits, schedules and undertakings set forth in the registration statement. For further information pertaining to us and the securities offered hereby, reference is made to the registration statement and the exhibits and schedules to the registration statement. Statements contained in this prospectus as to the contents or provisions of any documents referred to in this prospectus are not necessarily complete, and in each instance where a copy of the document has been filed as an exhibit to the registration statement, reference is made to the exhibit for a more complete description of the matters involved.
You may read and copy all or any portion of the registration statement without charge at the public reference room of the SEC at 100 F Street, N.E., Washington, D.C. 20549. Copies of the registration statement may be obtained from the SEC at prescribed rates from the public reference room of the SEC at such address. You may obtain information regarding the operation of the public reference room by calling 1-800-SEC-0330. In addition, registration statements and certain other filings made with the SEC electronically are publicly available through the SEC’s website atwww.sec.gov. The registration statement, including all exhibits and amendments to the registration statement, has been filed electronically with the SEC. You may also read all or any portion of the registration statement and certain other filings made with the SEC on our website atwww.syntheticbiologics.com. The information contained in, and that can be accessed through, our website is not incorporated into and is not part of this prospectus.
We are subject to the information and periodic reporting requirements of the Exchange Act and, accordingly, are required to file annual reports containing financial statements audited by an independent public accounting firm, quarterly reports containing unaudited financial data, current reports, proxy statements and other information with the SEC. You will be able to inspect and copy such periodic reports, proxy statements and other information at the SEC’s public reference room, the website of the SEC referred to above, and our website atwww.syntheticbiologics.com. Except for the specific incorporated reports and documents listed above, no information available on or through our website shall be deemed to be incorporated in this prospectus or the registration statement of which it forms a part.
INCORPORATION OF CERTAIN DOCUMENTS BY REFERENCE
The SEC allows us to “incorporate by reference” certain information that we will file with it which means that we can disclose important information to you by referring you to those documents instead of having to repeat the information in this prospectus. The later information that we file with the SEC will automatically update and supersede this information. We incorporate by referencethe documents listed below and any future filings madewith the SEC under Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act(Commission File No. 001-35994)) after (i) the date of this initial registration statement and prior to effectiveness of this registration statement and (ii) the date of this prospectus and before the completion of the offering of the securities included in this prospectus,however, we will not incorporate by reference any documents or portions thereof that are not deemed “filed” with the SEC, or any information furnished pursuant to Items 2.02 or 7.01 of Form 8-K or related exhibits furnished pursuant to Item 9.01 of Form 8-K:
| · | Our annual report on Form 10-K for the fiscal year ended December 31, 2017 filed with the SEC on February 22, 2018 (File No. 001-12584); |
| · | Our quarterly reports on Form 10-Q for the quarters ended March 31, 2018 and June 30, 2018 filed with the SEC on May 8, 2018 and August 8, 2018, respectively (File No. 001-12584); |
| 50 |
| · | Our current reports on Form 8-K (File No. 001-12584) filed with the SEC on January 8, 2018, March 7, 2018; April 23, 2018, May 7, 2018, May 22, 2018; August 1, 2018, August 13, 2018, September 6, 2018, September 6, 2018, September 26, 2018, October 2, 2018 and October 10, 2018; |
| · | Our definitive proxy statement on Schedule 14A filed with the SEC on August 14, 2018 (File No. 001-12584); and |
| · | The description of our common stock set forth in our registration statement on Form 8-A12B, filed with the SEC on June 20, 2007 (File No. 000-12584). |
We will provide to each person, including any beneficial owner, to whom a prospectus is delivered, a copy of any or all of the reports or documents that we incorporate by reference in this prospectus contained in the registration statement (except exhibits to the documents that are not specifically incorporated by reference) at no cost to you, by writing or calling us at the following address and telephone number:
Synthetic Biologics, Inc.
9605 Medical Center Drive, Suite 270, Suite 12
Rockville, Maryland 20850
(301) 417-4364
Information about us is available at our website atwww.syntheticbiologics.com. Except for the specific incorporated reports and documents listed above, no information available on or through our website shall be deemed to be incorporated in this prospectus or the registration statement of which it forms a part. Any statement contained in this registration statement or in a document incorporated or deemed to be incorporated by reference in this registration statement shall be deemed to be modified or superseded for purposes of this registration statement to the extent that a statement contained in this registration statement or in any other subsequently filed document which also is or is deemed to be incorporated by reference in this registration statement modifies or supersedes that statement. Any statement so modified or superseded shall not be deemed, except as so modified or superseded, to constitute a part of this registration statement.
| 51 |

Up to 9,523,809 Class A Units Consisting of Shares of Common Stock and Warrants
Up to 20,000 Class B Units Consisting of Series B Convertible Preferred Stock and Warrants
9,523,809 Shares of Common Stock Underlying the Series B Convertible Preferred Stock and
9,523,809 Shares of Common Stock Underlying the Warrants
PROSPECTUS
A.G.P.
, 2018
Through and including , 2018 (25 days after commencement of this offering), all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to their unsold allotments or subscriptions.
PART II - INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 13. OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION
We estimate that expenses in connection with the distribution described in this registration statement (other than fees and commissions charged by the underwriters) will be as set forth below. We will pay all of the expenses with respect to the distribution, and such amounts, with the exception of the SEC registration fee and the Financial Industry Regulatory Authority, Inc. (“FINRA”) filing fee, are estimates.
| SEC registration fee | $ | 12,266 | ||
| FINRA filing fee | 15,680 | |||
| Accounting fees and expenses | 70,000 | |||
| Printing fees | 10,000 | |||
| Legal fees and expenses | 300,000 | |||
| Underwriters’ out-of-pocket expenses | 150,000 | |||
| Marketing fees | 2,500 | |||
| Other (including transfer agent and warrant agent fees) | 39,554 | |||
| Total | $ | 600,000 |
ITEM 14. INDEMNIFICATION OF DIRECTORS AND OFFICERS
Section 78.138 of the Nevada Revised Statute provides that a director or officer is not individually liable to the corporation or its stockholders or creditors for any damages as a result of any act or failure to act in his capacity as a director or officer unless it is proven that (1) his act or failure to act constituted a breach of his fiduciary duties as a director or officer and (2) his breach of those duties involved intentional misconduct, fraud or a knowing violation of law.
This provision is intended to afford directors and officers protection against and to limit their potential liability for monetary damages resulting from suits alleging a breach of the duty of care by a director or officer. As a consequence of this provision, stockholders of our company will be unable to recover monetary damages against directors or officers for action taken by them that may constitute negligence or gross negligence in performance of their duties unless such conduct falls within one of the foregoing exceptions. The provision, however, does not alter the applicable standards governing a director’s or officer’s fiduciary duty and does not eliminate or limit the right of our company or any stockholder to obtain an injunction or any other type of non-monetary relief in the event of a breach of fiduciary duty.
The Registrant’s Articles of Incorporation, as amended, and amended and restated bylaws provide for indemnification of directors, officers, employees or agents of the Registrant to the fullest extent permitted by Nevada law (as amended from time to time). Section 78.7502 of the Nevada Revised Statute provides that such indemnification may only be provided if the person acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, the best interest of the Registrant and, with respect to any criminal action or proceeding, had no reasonable cause to behave his conduct was unlawful.
In any underwriting agreement we enter into in connection with the sale of the securities being registered hereby, the underwriters will agree to indemnify, under certain conditions, us, our directors, our officers and persons who control us, within the meaning of the Securities Act, against certain liabilities.
ITEM 15. RECENT SALES OF UNREGISTERED SECURITIES
The following information sets forth certain information with respect to all securities which we have sold during the last three years.
On January 24, 2018, we issued to a consultant as a partial payment for services, a warrant to purchase 714 shares of our common stock at an exercise price of $18.55 per share (as adjusted for the reverse split). The issuance of the warrant was exempt from registration provisions of the Securities Act under the exemption provided for by the Section 4(a)(2) thereof for transactions not involving a public offering.
On September 11, 2017, the Company issued 120,000 shares of its Series A Convertible Preferred Stock to an accredited investor in accordance with the terms of the Purchase Agreement. The offer and issuance of the Series A Convertible Preferred Stock and the shares of Common Stock issuable upon conversion of the Series A Convertible Preferred Stock have not been registered under the Securities Act of 1933, as amended (the “Securities Act”), and therefore may not be offered or sold in the United States absent registration or an applicable exemption from registration requirements. For this issuance, the Company is relying on the exemption from federal registration under Section 4(a)(2) of the Securities Act based on the Company’s belief that the offer and sale of such securities does not involve a public offering as the investor is an “accredited investor” as defined under Section 501 promulgated under the Securities Act.
| II-1 |
ITEM 16. EXHIBITS
| * | Filed herewith |
| ** | Previously filed |
| II-2 |
ITEM 17. UNDERTAKINGS
(a) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933, as amended (the “Securities Act”);
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement.
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
Provided, however, that Paragraphs (a)(1)(i), (ii), and (iii) of this section do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Commission by the registrant pursuant to section 13 or section 15(d) of the Securities Exchange Act of 1934 that are incorporated by reference in the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initialbona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act to any purchaser: If the registrant is subject to Rule 430C (§230.430C of this chapter), each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A (§230.430A of this chapter), shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
| II-3 |
(5) That, for the purpose of determining liability under the Securities Act to any purchaser in the initial distribution of the securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424 (§230.424 of this chapter);
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
(b) The undersigned registrant hereby undertakes that, for purposes of determining any liability under the Securities Act, each filing of the registrant’s annual report pursuant to section 13(a) or section 15(d) of the Securities Exchange Act of 1934 (and, where applicable, each filing of an employee benefit plan’s annual report pursuant to section 15(d) of the Securities Exchange Act of 1934) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(c) The undersigned registrant hereby undertakes to supplement the prospectus, after the expiration of the subscription period, to set forth the results of the subscription offer, the transactions by the underwriters during the subscription period, the amount of unsubscribed securities to be purchased by the underwriters, and the terms of any subsequent reoffering thereof. If any public offering by the underwriters is to be made on terms differing from those set forth on the cover page of the prospectus, a post-effective amendment will be filed to set forth the terms of such offering.
(d) Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Company pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
(e) For the purpose of determining any liability under the Securities Act, the registrant will treat the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant under Rule 424(b)(1), or (4), or 497(h) under the Securities Act as part of this registration statement as of the time the Commission declared it effective.
(f) For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
| II-4 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the Registrant has duly caused this Amendment No. 2 to Registration Statement on Form S-1 to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Rockville, State of Maryland, October 10, 2018.
| SYNTHETIC BIOLOGICS, INC. | ||
| By: | /s/ Steven A. Shallcross | |
| Name: | Steven A. Shallcross | |
| Title: | Interim Chief Executive Officer and Chief Financial Officer | |
Pursuant to the requirements of the Securities Act 1933, this report has been signed by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| /s/ Steven A. Shallcross | Interim Chief Executive Officer and Chief Financial Officer | ||||
| Steven A. Shallcross | (Principal Executive Officer and Principal Financial and Accounting Officer) | October 10, 2018 | |||
| * | Chairman | October 10, 2018 | |||
| Jeffrey J. Kraws | |||||
| * | Director | October 10, 2018 | |||
| Scott L. Tarriff | |||||
| * | Director | October 10, 2018 | |||
| Jeffrey Wolf | |||||
| *By: | /s/ Steven A. Shallcross | ||||
| Steven A. Shallcross | |||||
| Attorney-in-Fact | |||||
| II-5 |