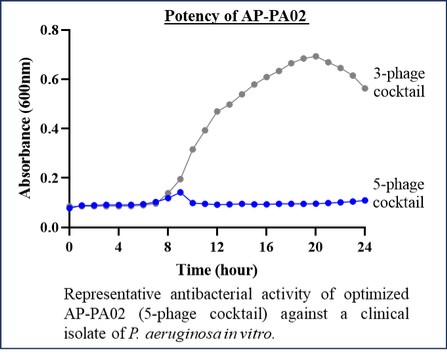
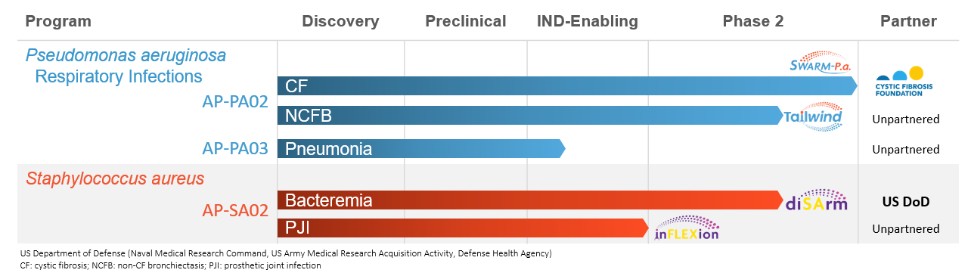
Free signup for more
- Track your favorite companies
- Receive email alerts for new filings
- Personalized dashboard of news and more
- Access all data and search results
Content analysis
?| Positive | ||
| Negative | ||
| Uncertain | ||
| Constraining | ||
| Legalese | ||
| Litigious | ||
| Readability |
H.S. sophomore Avg
|
|
New words:
adversarial, alleging, architecture, Attorney, Biden, biometric, Birx, blinded, Blueprint, burdensome, certainty, codified, collateral, Command, commerce, composed, concentrated, concentration, condensed, conspiracy, constrain, correlation, curve, cyber, Deborah, deception, deceptive, deployment, destruction, detrimental, deviation, difficulty, disaggregated, disagree, dismemberment, dissuade, Distinguish, Driven, drought, economy, energy, erode, Erroneously, exacerbated, extortion, forfeited, formulary, freedom, ft, FTC, gathered, gathering, GILTI, globe, gut, hacking, hardware, hereof, herewith, HHS, hostile, improper, Index, inhibit, injunction, Inline, Instance, intelligence, intrusion, inventorship, lag, Linkbase, lodge, low, macroeconomic, magnitude, maker, McConnell, mid, military, misappropriated, misappropriation, misuse, movement, narrowly, nationwide, Naval, negate, noncapitalizable, Nonmonetary, Northern, nuclear, obsolete, omitted, online, opt, outsource, overhead, Palestine, parent, pivotal, plant, plausibility, plausible, pose, PPACA, predominately, presidential, prone, provoke, quantified, ranked, ratifying, receivership, reexamination, repetitive, resubstitution, retaliatory, risky, Rychlik, safeguard, safeguarding, Schema, sector, segment, sick, Simplification, sophisticated, sophistication, specialty, spectrum, speculative, sq, stead, stemming, strong, supervisory, tampering, Taxonomy, tertiary, theft, thereunto, thing, tighter, titer, treble, trickery, undergone, undersigned, underwritten, undistributed, unemployment, unenforceable, unintentional, unpatented, uptake, USPTO, vacation, variation, virtue, wall, wellbeing, wound, XBRL
Removed:
acceleration, accessibility, Accountancy, accountant, accreted, accretion, acquire, acted, adequacy, adjournment, adjudged, adjust, administering, admitted, advancement, Advocacy, affiliated, affirmatively, Affordability, aforementioned, aggressively, AgilVax, Aid, Alabama, Allen, allocate, Amgen, analyst, Anatomic, Appeal, appointing, Ariad, arm, Armed, Army, Arthur, Article, aseptic, assemble, asserted, Assistant, assisting, assured, attainment, attend, attended, Attending, attribute, AU, Aviragen, avoid, Barbara, Bayshore, bear, bio, Biochemistry, Biogen, biomedical, biopharma, Birmingham, blank, blocking, borrowed, borrower, bowel, Brain, breached, Brexit, Bristol, budgeting, built, Burlingame, call, capitalization, caption, car, career, catastrophic, CDPH, Central, certified, CFU, chair, chairman, chairperson, challenger, chance, Chapter, Charitable, check, Chicago, circumvented, clear, cloud, CMC, Coalition, College, collegiality, collusion, Cologne, column, combine, compound, confidence, consultant, contractually, convincing, copy, Coronado, correct, counsel, counseling, CPA, curtailed, customer, DAC, decade, dedication, deducting, delegate, delete, deliberation, Deloitte, Dendritic, denominator, deputization, derivative, derived, designee, designing, desire, detected, diplomatic, direction, disallowance, discontinue, discussing, disposing, dispositive, disqualifying, diversion, division, dubbed, efficiency, electrical, elimination, enact, encouraged, enforceable, enjoined, enjoy, enterococcal, Enzon, EPS, Evercore, evergreen, evidenced, evolve, exceeded, exchangeable, exclude, exculpation, exercisable, existed, existence, exiting, Exploring, facilitating, faculty, fellow, fiduciary, fifty, filling, Fisher, formal, founder, frame, fringe, front, frustrate, fullest, furnish, GenApex, Genicon, Gillette, Graduate, grew, guarantee, Haven, head, heading, headquartered, hedge, hematopoietic, Hertz, Highway, home, honor, hospitalization, hosting, Hybritech, immigration, immunology, impacting, improbable, improving, inaccurate, inappropriate, incentivized, inconclusive, indemnification, indemnified, indemnify, indemnitee, indemnitor, influenza, Inhibitex, instability, insured, intentional, invalidity, investigate, investigative, investigator, involuntary, IPR, IRA, ISI, issuable, iv, job, joining, JP, Kramer, landscape, Laureate, lawsuit, Likewise, listing, litigated, litigating, lived, living, locate, LP, makeup, matching, Max, measuring, membership, merging, Miami, microbiology, misconduct, MIT, MMA, Modernization, modifying, molecular, month, necessarily, newer, Nobel, nominee, noncompetitive, notify, nucleic, numerator, objective, obligate, occupy, oncology, opposed, OSHA, override, Parasitic, partnership, patented, Pathology, Patrick, Paul, Paycheck, payout, pecuniary, penalty, perception, permissible, personalized, pertaining, pertinent, petition, PEW, Pharma, pioneer, Pittsburgh, Planck, PMA, postdoctoral, postponement, PPP, practiced, PREA, preclude, preferred, preparing, preponderance, preside, pressure, presumption, prevailing, printed, proactively, procedural, Professor, profitably, promised, promissory, promoting, promptly, promulgation, proposing, proximity, PTO, publish, PUMA, purified, purifying, qualify, quarantine, Ralph, reached, reallocate, reallocated, reallocation, recall, recommend, recommended, recommending, recording, recuse, reducing, Reed, reelection, reflecting, regional, rejoined, reliable, remained, remedy, rental, repealed, replaced, reproduce, Resident, resort, retainer, retired, retiring, Retrovirology, revised, riskier, Robin, Roche, Rockefeller, rotation, Rush, Samsung, Santa, Sarissa, science, Scientist, SciStem, Seattle, securing, server, serving, session, SGI, shape, shift, shorter, significance, sound, Southern, Southwestern, spanning, speak, specification, spent, Squibb, staffing, standing, stay, Steinman, Steve, structured, studying, sublease, subscription, sued, Suite, supermajority, systemically, tail, Takeda, taxation, Taxing, TCJA, terminable, terrorism, Thermo, thought, tied, titled, Touche, traditionally, tranche, transit, translational, travel, traveling, Trega, turned, UCLA, unconsolidated, undesirable, unexercisable, unforeseen, unintended, unintentionally, unlawful, unreliable, unrest, unsecured, unused, USDA, Valley, variability, Vaxart, venture, veteran, viable, viewed, violated, vulnerable, Walter, Wellesley, withdrawing, Women, word
Filing tables
Filing exhibits
Related press release
ARMP similar filings
Filing view
External links