Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K/A
Amendment No. 2
| [X] | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For Fiscal Year Ended December 31, 2002
or
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT 1934 |
From the transition period from to
Commission file number 000-31255
ISTA PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 33-0511729 | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization) | Identification No.) |
15279 Alton Parkway, Suite 100, Irvine, California 92618
(Address of principal executive offices)
(949) 788-6000
(Registrant’s telephone number)
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $.001 par value
(Title of Class)
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15 (d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K [ ]
Indicate by check mark whether the registrant is an accelerated filer (as defined in Exchange Act Rule 12b-2). Yes [ ] No [X]
As of June 28, 2002, the aggregate market value of the Registrant’s voting and non-voting common equities held by non-affiliates was approximately $11,525,000.
As of February 18, 2003 there were 13,291,017 shares of Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
None.
TABLE OF CONTENTS
| PART I | ||||||||
| Item 1: Business | ||||||||
| PART IV | ||||||||
| Item 15: Exhibits, Financial Statement Schedules and Reports on Form 8-K | ||||||||
| SIGNATURES | ||||||||
| EXHIBIT 10.30 | ||||||||
| EXHIBIT 10.33 | ||||||||
| EXHIBIT 99.1 | ||||||||
| EXHIBIT 99.2 | ||||||||
Table of Contents
EXPLANATORY NOTE
This Amendment No. 2 to the Annual Report on Form 10-K/A for ISTA Pharmaceuticals, Inc. for the fiscal year ended December 31, 2003 (the “Report”) supplements the Report by revising certain information contained Item 1 of Amendment No. 1 and also files Exhibits 10.30 and 10.33. No further revisions have been made to the previously filed Report.
i
Table of Contents
PART I
Item 1: Business
Overview
ISTA Pharmaceuticals, Inc. (the “Company” or “ISTA”) was founded to discover, develop and market new remedies for diseases and conditions of the eye. Some of the products that we are developing involve the use of highly purified formulations of hyaluronidase. Our lead ovine hyaluronidase-based product candidate, Vitrase®, is a proprietary drug we are developing for the treatment of vitreous hemorrhage and diabetic retinopathy. We are also developing products that are designed to treat glaucoma, hyphema, and ocular inflammation.
In December 2001, we announced our strategic plan to transition from a development organization to a fully-integrated, specialty pharmaceutical company, with a primary focus on ophthalmology. We intend to execute this plan by continuing to advance products currently under development, and by acquiring complementary products, either already marketed or in late-stage development.
In March 2002, we announced the preliminary results for our Phase III clinical studies of Vitrase® for the treatment of vitreous hemorrhage. The data from the two Phase III studies did not show a statistically significant improvement in the primary (surrogate) endpoint. However, further analysis has shown that for the 55 IU dose of Vitrase®, in both studies, there was a clinically meaningful and statistically significant decrease in the density of vitreous hemorrhage and a statistically significant difference in the proportion of patients with an improvement in best-corrected visual acuity at one and two months, which extended to the three-month post-treatment visit in the study conducted outside North America. Based on these improvements in visual function, and our review and discussions with the Food and Drug Administration (“FDA”) regarding the data from the two studies, we submitted the clinical section and the chemistry, manufacturing and controls (“CMC”) section of the New Drug Application (“NDA”) for Vitrase® for the treatment of vitreous hemorrhage in October 2002. The FDA accepted those sections of the Vitrase® NDA for review in December 2002, along with the pre-clinical pharmacology and toxicology section that was submitted in January 2002. On April 3, 2003, the FDA issued an “approvable” letter in which the FDA cited issues primarily related to the sufficiency of the efficacy data submitted with the NDA for Vitrase®. The FDA requested additional analysis of the existing data and an additional confirmatory clinical study based upon that analysis. We intend to discuss with the FDA the comments contained in the “approvable” letter and, based upon those discussions, determine the next appropriate steps in the review and approval process of Vitrase®.
In May 2002, we acquired substantially all of the assets of AcSentient, Inc. The assets include United States marketing rights for a new formulation of timolol, which we have named ISTALOL™, a beta-blocking agent for treating glaucoma. AcSentient previously acquired rights to this compound from Senju Pharmaceutical Co., Ltd. Senju is headquartered in Osaka, Japan and has a diverse portfolio including ophthalmic, central nervous system, cardiovascular, circulatory, gastro-intestinal, respiratory, oncology and dermatological products. Senju submitted the NDA for ISTALOL™ to the FDA in September 2002. That NDA included data from pre-clinical studies conducted in Japan, combined with data from a Phase I clinical study conducted in the United States and a multi-center Phase III clinical study recently completed in the United States. The assets acquired from AcSentient also included United States product rights to bromfenac, a topical non-steroidal anti-inflammatory compound for the treatment of ocular inflammation, and worldwide marketing rights for Caprogel®, a novel compound for the treatment of hyphema, or bleeding into the front of the eye. AcSentient previously acquired the rights to these two compounds from Senju and the Eastern Virginia School of Medicine, respectively. Under the terms of the transaction, we have assumed payment and other obligations and responsibility for final development of bromfenac and Caprogel®.
We are currently initiating a Phase III study of bromfenac in the United States. Assuming successful completion of our clinical studies, we anticipate submitting an NDA for bromfenac in early 2004. We are currently conducting feasibility studies for the commercialization of Caprogel®. Once completed, and if these studies yield promising results, we intend to pursue further clinical development of Caprogel® consistent with such studies’ results. However, the timing and scope for our development of Caprogel® may change based on our assessment, from time to time, of this product candidate's market potential, other product opportunities and our corporate priorities.
1
Table of Contents
We anticipate incurring additional research and development expenses in connection with the final development of bromfenac and Caprogel®, and if these compounds are approved for sale in the United States, we expect to incur significant marketing and sales expenses. In addition, we will also be responsible for certain milestone payments to a third party in connection with the marketing approval in the United States for these compounds.
On November 19, 2002, the Company consummated a private investment in public equity (or “PIPE”) transaction, involving a private placement of 10,526,306 shares of its common stock for the aggregate purchase price of approximately $40.0 million, or $3.80 per share, and warrants to purchase up to 1,578,946 shares of its common stock for an exercise price of $3.80 per share. Investors in the PIPE transaction included persons and entities affiliated with The Sprout Group, Sanderling Venture Partners, Investor Growth Capital Limited, Gund Investment Corporation, KBL Healthcare, MDS Capital, and Ontario Teachers’ Pension Plan Board. In addition, $4.0 million of promissory notes previously issued to several of the same investors in ISTA’s September 2002 bridge financing were converted into 1,052,620 shares of ISTA common stock, based upon the conversion price of $3.80 per share, concurrently with the consummation of the PIPE transaction. We believe this funding will enable us to execute on our plans to complete development work on our late-stage product candidates and introduce these products to the ophthalmic marketplace if they are approved by the FDA.
We also took significant steps in 2002 to scale up ISTA’s manufacturing and marketing capabilities, in order to further enhance our ability to execute our strategic plan. We entered into two key manufacturing agreements for the supply of finished product from Cardinal Health and Bausch & Lomb Incorporated. We also added two senior management executives to bolster our operations and marketing capabilities: Thomas A. Mitro, Vice President, Sales and Marketing, and Kirk McMullin, Vice President, Operations. In March 2003 we added Lauren P. Silvernail as our Chief Financial Officer and Vice President, Corporate Development.
Anatomy of the Eye
The human eye is approximately one inch in diameter and functions much like a camera. The eye incorporates a lens system (the cornea and the lens) that focuses light, a variable aperture system (the iris) that controls the amount of light passing through the eye and a film (the retina) that records the image. The cornea, lens and iris operate to focus light rays on the retina, which contains the receptors that transmit images through the optic nerve to the brain.
The cavity between the lens and the retina is filled with the vitreous humor, a clear, gel-like substance. The vitreous humor is nearly solid in children and undergoes a natural transition to liquid as one ages.
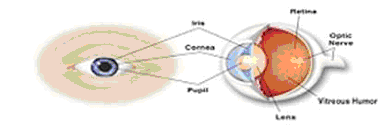
| Cornea: | The clear, transparent outer portion of the front of the eye that provides most of the eye’s focusing power. | |
| Iris: | The colored part of the eye that helps control the amount of light that enters the eye. | |
| Pupil: | The dark hole in the middle of the iris through which light enters the eye. | |
| Lens: | The transparent structure inside the eye (behind the cornea and iris) that also focuses light rays onto the retina. | |
| Vitreous humor: | The clear, gel-like substance that fills the back of the eye between the lens and the retina. | |
| Retina: | The nerve layer that lines the back of the eye. The retina senses light and transmits impulses that are sent through the optic nerve to the brain. | |
| Optic nerve: | The nerve that connects the eye to the brain and carries the impulses formed by the retina. |
2
Table of Contents
Hyaluronidase
The term hyaluronidase describes a group of naturally occurring enzymes that can digest certain forms of carbohydrate molecules called proteoglycans. The primary role of hyaluronidase is to digest proteoglycans such as hyaluronan, hyaluronic acid and chondroitin sulphate, substances that are part of various connective tissues in the body, including connective tissues in the eye. Physicians have used bovine-derived hyaluronidase safely and extensively for over 50 years to enhance the absorption of drugs.
In the eye, pharmacological applications of hyaluronidase exploit the properties of this enzyme to modify the structure of the eye for therapeutic purposes. In our development work, we have focused on applications of ovine-derived hyaluronidase to take advantage of its ability to digest proteoglycans to treat a variety of eye diseases and conditions, including vitreous hemorrhage, diabetic retinopathy and corneal opacification. For treatment of serious conditions of the eye, or ophthalmic indications, only highly purified and specially formulated ovine hyaluronidase can be used. Hyaluronidase that is less pure or formulated with preservatives has been shown to be dangerous to the eye and may cause blindness.
Product Development Programs
We have five product candidates in clinical development. The following is a summary of our clinical product candidates:
| Product | Indication | Development Status | Marketing Rights | |||
| Vitrase® | Vitreous hemorrhage | NDA Reviewed and FDA “Approvable” Letter Received | Allergan, Otsuka | |||
| Vitrase® | Diabetic retinopathy | Phase IIa | Allergan, Otsuka | |||
| ISTALOL™ | Glaucoma | NDA Submission | — | |||
| Accepted for Review | ||||||
| Bromfenac | Ocular pain and | Phase III | — | |||
| inflammation | ||||||
| Caprogel® | Hyphema | Feasibility Study Ongoing | — |
Vitrase®
We are developing Vitrase®, a proprietary formulation of ovine hyaluronidase, for the treatment of vitreous hemorrhage and diabetic retinopathy. When injected into the vitreous humor, Vitrase® breaks down the proteoglycan matrix, causing the vitreous humor to liquefy. We believe that this also results in the separation of the vitreous humor from the retina and that, together, these effects are beneficial for the treatment of vitreous hemorrhage and diabetic retinopathy. Vitrase® is administered directly into the vitreous humor through a single-dose injection. The procedure is performed in several minutes in an ophthalmologist’s office and is virtually painless due to the application of a topical anesthetic.
Vitreous Hemorrhage
A vitreous hemorrhage occurs when retinal blood vessels rupture and bleed into the vitreous humor. These hemorrhages result from leakage from abnormal, weak blood vessels and are associated with diabetic retinopathy, trauma and other factors. The immediate consequence of a vitreous hemorrhage is a reduction in the amount of light that can pass through the normally clear vitreous humor to the retina. The effects of a hemorrhage can be limited to a few dark spots in vision or, in the case of a severe vitreous hemorrhage, can result in completely obscured vision. Depending on the severity of the vitreous hemorrhage, it may take several months or significantly longer for the body to reabsorb the blood and for the patient to regain vision. In addition to obstructing the patient’s vision, a vitreous hemorrhage often prevents physicians from seeing into the back of the eye to diagnose or treat the cause of the hemorrhage. If extensive or repeated bleeding occurs, fibrous tissue or scarring can form on the retina, which can lead to a detachment of the retina and permanent vision loss or blindness.
Patients who seek medical care for a vitreous hemorrhage often visit a physician, who then refers them to a retinal specialist. Treatment options for patients with a vitreous hemorrhage are limited. Currently, there is no drug treatment for vitreous hemorrhage and most retinal specialists initially recommend a “watchful waiting” period, during which the attending physician provides no medical treatment in the hope that the hemorrhage will clear on its own. The risks related to watchful waiting may include continued bleeding and, if caused by diabetic retinopathy, disease progression during the time it takes for the blood to clear on its own, if at all.
3
Table of Contents
An alternative to watchful waiting is a surgical procedure called a vitrectomy, in which the vitreous humor and hemorrhage are surgically removed and replaced with a balanced salt solution. There are serious risks associated with a vitrectomy, including both cataract formation and possible loss of vision associated with retinal detachment. These risks contribute to the limited use of vitrectomy as an initial treatment option for vitreous hemorrhage patients.
We believe that a substantial opportunity exists to establish Vitrase® as the initial therapy for a vitreous hemorrhage. Vitrase®, when injected into a blood-filled vitreous humor, promotes clearance by causing the vitreous humor to liquefy and the blood to settle to the bottom of the eye. Vitrase® also stimulates the cells responsible for engulfing and breaking down the blood, accelerating the reabsorption of the blood. This clears the path for light to reach the retina enabling the patient to regain vision. In addition, clearing the hemorrhage permits the retinal specialist to visualize, diagnose and treat the underlying cause of the vitreous hemorrhage.
Hemorrhage density can vary significantly between patients who experience vitreous hemorrhage, but even a mild hemorrhage indicates the existence of a serious problem. Because of the absence of a validated and generally accepted medical definition of the various densities of vitreous hemorrhage, we classify a vitreous hemorrhage as either mild, moderate or severe depending on the density of the vitreous hemorrhage as observed by the physician:
| • | mild vitreous hemorrhage is characterized by trace blurring of retinal blood vessels | ||
| • | moderate vitreous hemorrhage is characterized by partial obscuration of retinal blood vessels and/or the optic nerve | ||
| • | severe vitreous hemorrhage is characterized by complete obscuration of retinal blood vessels and/or the optic nerve |
Market Opportunity. Based on data compiled by Business Genetics, Inc., we believe that approximately 450,000 cases of vitreous hemorrhage occur each year in the United States, a total of 400,000 cases occur each year in the five largest European markets and 190,000 cases occur each year in Japan. Approximately 60% of all of these cases are due to diabetic retinopathy, 15% are due to trauma and 25% are due to other factors. As a result of the natural history of the disease, we believe Vitrase®, if approved, is unlikely to be used in all cases of vitreous hemorrhage. We believe that approximately half of all cases are candidates for treatment using Vitrase®.
Clinical/Regulatory Status. In October 1998, the FDA granted fast-track designation for Vitrase® for the treatment of vitreous hemorrhage. The FDA’s provision for “fast-track” designated drugs, such as Vitrase®, provides for early submission of completed sections of the NDA. In January 2002, as a part of our rolling NDA for Vitrase® for the treatment of vitreous hemorrhage, we submitted the pre-clinical pharmacology and toxicology section.
We have completed two Phase III trials of Vitrase® for the treatment of vitreous hemorrhage. These trials were prospective, randomized, parallel, placebo-controlled and double-masked studies. We conducted one of the trials, our North America trial, in the United States, Mexico and Canada with an enrollment of 750 patients. We conducted the other trial in Europe, Brazil, Australia and South Africa with an enrollment of 556 patients. Patients enrolled in the studies will continue to be monitored in 2003. We contracted with CroMedica Global, Inc. in connection with the North America trial and Covance Clinical Periapproval Services, Inc. in connection with the Europe, Brazil, Australia and South Africa trial to provide certain management and support services, including initiation visits with prospective candidates, site management, data accumulation, drug supply management and site audits.
In both studies, we enrolled patients who had both a vitreous hemorrhage that had been present for at least one month and a Best Corrected Visual Acuity (“BCVA”) of less than 20/200 at initial screening. After enrollment, patients were randomly assigned to either a test group or a control group. Patients in the test group received either a 7.5 (North America only), 55 or 75 international unit, or IU, injection of Vitrase®. Patients in the control group received a saline injection. Treatment success in both studies was defined by the clearance of the vitreous hemorrhage sufficient to allow for the occurrence of any one of the following, which must have occurred within three months following treatment with Vitrase®:
| • | panretinal laser photocoagulation surgery to slow or stop the cause of the vitreous hemorrhage | ||
| • | other surgical treatment not specifically indicated for the clearance of the vitreous hemorrhage (for example, vitrectomy to enable treatment of retinal detachment) | ||
| • | documented medical evidence that the clinical cause of the vitreous hemorrhage has been resolved without the need for further therapy |
4
Table of Contents
Additionally, at each study visit and specifically at months one, two and three following treatment, BCVA was assessed using an eye chart in both studies.
In March 2002, we announced the preliminary results for our Phase III clinical studies of Vitrase® for the treatment of vitreous hemorrhage. The data from the two Phase III studies did not show a statistically significant improvement in the primary (surrogate) endpoint. However, further analysis has shown that for the 55 IU dose of Vitrase®, in both studies, there was a clinically meaningful and statistically significant decrease in the density of vitreous hemorrhage and a statistically significant difference in the proportion of patients with an improvement in best-corrected visual acuity at one and two months, which extended to the three-month post-treatment visit in the study conducted outside North America. Based on these improvements in visual function, and our review and discussions with the Food and Drug Administration (“FDA”) regarding the data from the two studies, we submitted the clinical section and the chemistry, manufacturing and controls (“CMC”) section of the New Drug Application (“NDA”) for Vitrase® for the treatment of vitreous hemorrhage in October 2002. The FDA accepted those sections of the Vitrase® NDA for review in December 2002, along with the pre-clinical pharmacology and toxicology section that was submitted in January 2002. On April 3, 2003, the FDA issued an “approvable” letter in which the FDA cited issues primarily related to the sufficiency of the efficacy data submitted with the NDA for Vitrase®. The FDA requested additional analysis of the existing data and an additional confirmatory clinical study based upon that analysis. We intend to discuss with the FDA the comments contained in the “approvable” letter and, based upon those discussions, determine the next appropriate steps in the review and approval process of Vitrase®.
Diabetic Retinopathy
Abnormal changes and/or damage to the blood vessels in the eye due to diabetes are known as diabetic retinopathy. Diabetic retinopathy is a progressive disease consisting of two stages, nonproliferative and proliferative. Nonproliferative diabetic retinopathy is the first stage of diabetic retinopathy and occurs when the retinal blood vessels swell and leak fluid and small amounts of blood into the eye. Under current practice, physicians do not generally treat patients at the nonproliferative stage of the disease because there is no effective treatment available. If untreated, nonproliferative diabetic retinopathy will likely progress into the more dangerous, proliferative stage of diabetic retinopathy. Proliferative diabetic retinopathy occurs when normal retinal blood vessels become obstructed and new, abnormal blood vessels begin to grow, or proliferate, on the surface of the retina or into the vitreous humor. The growth of these new, abnormal blood vessels creates a dangerous condition because they are weak and grow beyond the supporting structure of the retina. These blood vessels are prone to bleed and hemorrhage, and can lead to serious problems, including retinal tears and retinal detachment, both of which can severely impair vision and cause blindness. There is no cure for diabetic retinopathy.
The most common treatment for patients with proliferative diabetic retinopathy is panretinal laser photocoagulation. In this treatment, a laser makes hundreds of tiny burns to the retina to reduce the growth of the abnormal blood vessels into the vitreous humor. Panretinal laser photocoagulation surgery has been shown to be effective in slowing the progression of proliferative diabetic retinopathy. Panretinal laser photocoagulation surgery frequently leads to increased loss of night vision and can make night driving more difficult. Also, after panretinal laser photocoagulation surgery, peripheral, or side vision, is often not as good as before the surgery.
We believe that Vitrase® can treat diabetic retinopathy at the nonproliferative stage. Following injection into the vitreous humor, Vitrase® acts to separate the vitreous humor from the retina, thereby limiting growth of retinal blood vessels into the vitreous humor. We believe that Vitrase® achieves this by breaking down the proteoglycan component of the substance that binds the vitreous humor to the retina and by liquefying the vitreous humor. This process allows the vitreous humor to detach from the retina. Retinal specialists consider this detachment to be beneficial to diabetic retinopathy patients because it may delay the progression of the disease.
Market Opportunity. Diabetes continues to be a major healthcare problem in the United States and is projected to continue growing rapidly in many regions outside the United States. Eye disease is commonly associated with diabetes, and the risk of blindness to individuals with diabetes is 25 times greater than in the general population. Of the nearly eight million individuals in the United States diagnosed with diabetes, four to six million have some form of diabetic retinopathy. The majority of individuals with diabetic retinopathy are in the nonproliferative stage of the disease. We believe that these people are potential candidates for treatment using Vitrase®.
Clinical/Regulatory Status. We have completed a 60 patient pilot Phase IIa clinical trial in Mexico City to evaluate the safety and efficacy of a single-dose injection of Vitrase® to cause a detachment of the vitreous humor from the retina and the impact on slowing the progression of diabetic retinopathy over a one-year period. This trial was a prospective, randomized, placebo controlled, double masked study. Patients with nonproliferative diabetic retinopathy were randomly assigned to one of four groups; a control group who
5
Table of Contents
received a saline injection, a test group who received a 75 IU injection of Vitrase®, a test group who received a 0.3 ml injection of a gas called sulphur-hexafluroride or SF6, and a test group who received a 75 IU injection of Vitrase® and a 0.3 ml injection of SF6 at 4 weeks following Vitrase® injection if a complete detachment of the vitreous humor from the retina had not occurred.
Interim study results at 16 weeks post-treatment demonstrated that complete detachment of the vitreous humor from the retina was documented in 60% of eyes treated with a single dose of Vitrase®. This is compared with only 6% of patients in the saline control group. In the study’s two additional treatment groups, detachment of the vitreous humor from the retina was documented in 53% of eyes treated with SF6 gas and in 50% of eyes treated with a combination of Vitrase® and SF6 gas.
To assess and measure the progression of diabetic retinopathy for study patients, seven-field fundus photographs (photographs of the retina) were taken at approximately 16 weeks following study entry and again 12 months later. The fundus photographs were reviewed in a masked fashion by an independent reading center to determine an Early Treatment for Diabetic Retinopathy Study (ETDRS) retinopathy severity scale score assigned for each patient’s study eye. These ETDRS retinopathy severity scores were then evaluated to identify any changes in the patients’ diabetic retinopathy over the one-year evaluation period. Of the study’s 60 patients, complete sets of fundus photographs were available, analyzed and scored for 46 patients.
Although the study was not designed to demonstrate statistical significance for progression of diabetic retinopathy among study groups, evaluation of ETDRS retinopathy severity scores did highlight trends for slowing the progress of diabetic retinopathy. Of the 46 patients with fundus photographs that were analyzed and scored, 67% of patients treated with Vitrase® (10 of 15) had stable ETDRS scores, indicating no measurable progression of diabetic retinopathy over the one-year evaluation period. This compares to stable ETDRS scores for the one-year evaluation period of 38% of patients treated with saline (6 of 16); 40% of patients treated with SF6 gas (6 of 15) and 43% of patients treated with Vitrase® plus SF6 gas (6 of 14). Worsening of ETDRS scores, indicating a progression of diabetic retinopathy over the one-year evaluation period was seen in 13% of patients treated with Vitrase® (2 of 15); 38% of patients treated with saline (6 of 16); 20% of patients treated with SF6 gas (3 of 15) and 21% of patients treated with Vitrase® plus SF6 gas (3 of 14).
The continued development of Vitrase® for the treatment of diabetic retinopathy will be dependent upon a number of factors including, among others, the FDA’s evaluation of the Vitrase® NDA for the treatment of vitreous hemorrhage, the successful completion of any additional clinical trials for the diabetic retinopathy indication and the continuing assessment of the market opportunity for this indication as compared to other product opportunities we may be pursuing at the time.
ISTALOL™
ISTALOL™ is the Company’s once daily, topical solution of timolol, a beta-blocking agent for the treatment of glaucoma. The product was developed by Senju in Japan. In May 2002, as part of the AcSentient asset acquisition, ISTA acquired marketing rights for ISTALOL™ in the United States.
Glaucoma is a disease that gradually reduces eyesight without warning and often without symptoms. Vision loss is caused by damage to the optic nerve. It was once thought that high intraocular pressure (“IOP”) was the main cause of this damage. It is now believed that other factors must also be involved since people with “normal” IOP can experience vision loss from glaucoma. Glaucoma is a chronic disease that must be treated for life. Currently, its causes are not well understood and there is no cure.
The most common form of glaucoma, Primary Open Angle Glaucoma, affects about three million Americans. It happens when the eye’s drainage canals become clogged over time. The IOP rises because the correct amount of fluid cannot drain out of the eye. With open angle glaucoma, the entrances to the drainage canals are clear and should be working correctly. The clogging problem occurs inside the drainage canals. ISTALOL™ is a beta-blocker that improves drainage of aqueous humor from the eyeball.
Market Opportunity. According to prescription data compiled by IMS Health, we estimate that the United States pharmaceutical market for the treatment of glaucoma exceeds $1.1 billion per year. As such, ISTALOL™ represents an important opportunity for the Company, with potential for excellent market penetration within a few years after product launch.
Clinical/Regulatory Status. Senju submitted an NDA for ISTALOL™ to the FDA in September 2002 and was accepted for review in November 2002. The NDA is based on data from a Phase I clinical study and a multi-center Phase III clinical trial conducted in the United States, as well as results from preclinical studies and Phase I and Phase II studies conducted in Japan.
6
Table of Contents
In the clinical trials, ISTALOL™ has shown efficacy and safety comparable to timolol maleate, which is the leading beta-blocker to treat glaucoma in the United States. Advantages of ISTALOL™ include enhanced corneal penetration and once-daily administration. Third party formulations of timolol currently on the market are twice-daily solutions or gel formulations, which are known to cause blurring of patients’ vision.
Bromfenac
Bromfenac is a topical non-steroidal anti-inflammatory compound for the treatment of ocular inflammation. The product was developed by Senju in Japan. Senju launched bromfenac in Japan in 2000 and its rapid sales growth is principally due to its superior potency and twice-daily dosing regimen as compared to the requirement of four doses-per-day for most other anti-inflammatory products on the Japanese market. In May 2002, as part of our AcSentient asset acquisition, ISTA acquired marketing rights for bromfenac in the United States.
Market Opportunity. According to prescription data compiled by IMS Health, we estimate that the current global ophthalmic anti-inflammatory and allergies markets to be approximately $500 million and $630 million per year, respectively.
Clinical/Regulatory Status. Phase I, Phase II and Phase III clinical studies of bromfenac have been completed in Japan and the product has been approved and was launched in 2000 in Japan. We are currently initiating a Phase III study of bromfenac in the United States. Assuming successful completion of our clinical studies, we anticipate submitting an NDA for bromfenac in early 2004.
Caprogel®
Caprogel®, a topical gel form of amniocaproic acid, is a new compound for treating hyphema, which typically results from trauma to the eye. The product was initially licensed from the Eastern Virginia School of Medicine. In May 2002, as part of our AcSentient asset acquisition, ISTA acquired worldwide marketing rights for Caprogel®.
Hyphema is a term used to describe bleeding in the anterior chamber, the space between the cornea and the iris, of the eye. It occurs when blood vessels in the iris bleed and leak into the clear aqueous fluid. Hyphemas are usually characterized by pooling of blood in the anterior chamber that may be visible to the naked eye. The red blood cells of very small hyphemas are visible only with magnification. Even the slightest amount of blood in the anterior chamber will cause decreased vision when mixed in the clear aqueous fluid.
Some of the symptoms of hyphema are decreased vision, depending on the amount of blood in the eye, pool of blood in the anterior chamber and elevated intraocular pressure. A doctor will assess visual acuity, measure intraocular pressure and examine the eye with a split lamp microscope and ophthalmoscope.
The treatment is dependent on the cause and severity of the hyphema. Frequently, the blood is reabsorbed over a period of days to weeks. During this time, the doctor will carefully monitor the intraocular pressure for signs of the blood preventing normal flow of the aqueous through the eye’s angle structures. If the eye pressure becomes elevated, eye drops may be prescribed to control it. The pupils are also evaluated to rule out damage to the iris.
In some cases, a procedure is performed to irrigate the blood from the anterior chamber to prevent secondary complications such as glaucoma and bloodstains on the cornea.
Market Opportunity. Based on data compiled by the Matson Jack Group, we believe that Hyphema affects an estimated 50,000 patients per year in the United States and currently there is no available pharmaceutical agent approved for its treatment.
Clinical/Regulatory Status. We are currently conducting feasibility studies for the commercialization of Caprogel®. Once completed, and if these studies yield promising results, we intend to pursue further clinical development of Caprogel® consistent with such studies’ results. However, the timing and scope for our development of Caprogel® may change based on our assessment, from time to time, of this product candidate’s market potential, other product opportunities and our corporate priorities. Caprogel® has received an orphan drug designation for the treatment of hypherma from the FDA, which may result in ISTA receiving a seven year market exclusivity privilege
7
Table of Contents
We continually evaluate new opportunities for late-stage or currently marketed complementary product candidates and, if and when appropriate, intend to pursue such opportunities through further product acquisitions and related development activities. ISTA’s ability to execute on such opportunities in some circumstances will be dependant upon our ability to raise additional capital on commercially reasonable terms.
Collaboration With Allergan
In March 2000, we began a collaboration with Allergan, Inc. with respect to the development and commercialization of Vitrase® worldwide (other than in Mexico until April 2004 and Japan) for certain ophthalmic uses, including a license agreement for the marketing, sale and distribution of Vitrase®, a supply agreement for Vitrase® and a stock purchase agreement for $10.0 million of our Series D preferred stock. These shares of Series D Preferred Stock were subsequently converted into 102,407 shares of our common stock. Allergan is a leading provider of eye care and specialty pharmaceutical products throughout the world. A joint operating committee has been constituted and consists of an equal number of members from each company who will oversee development, regulatory and marketing activities with respect to Vitrase®, as more fully described below.
Under the terms of our agreements with Allergan:
| • | Development. We are responsible for all product development, preclinical studies and clinical trials in support of marketing approvals of Vitrase® for the treatment of vitreous hemorrhage in the United States and Europe. We are also responsible for all preclinical studies and clinical trials to demonstrate the safety and efficacy of Vitrase® for the treatment of diabetic retinopathy. | ||
| • | Regulatory Approvals. We are responsible for applying for and obtaining regulatory approval of Vitrase® in the United States and in the European Union. Allergan will be responsible for applying for and obtaining regulatory approvals of Vitrase® in markets outside the United States and the European Union where it deems appropriate, other than Mexico (until April 2004) and Japan. | ||
| • | Manufacturing. We are responsible for the manufacture of Vitrase® and, if approved, for supplying all of Allergan’s requirements for Vitrase® during the term of the license agreement. | ||
| • | Marketing. In the United States, Allergan will be responsible for the overall management of marketing, sale and distribution activities for Vitrase® through its established sales and marketing organization. Under the terms of the license agreement, we will employ medical specialists in the United States to assist in physician training and usage development. In all markets outside the United States, except Mexico and Japan, Allergan will be solely responsible for the marketing, sale and distribution of Vitrase®. | ||
| • | Milestone Payments. Allergan has agreed to pay us up to $35.0 million in milestone payments based on our achievement of specified regulatory and development objectives with respect to Vitrase® for the treatment of vitreous hemorrhage and diabetic retinopathy. To date, we have not earned any milestone payments from Allergan and we cannot guarantee that we will receive any future milestone payments. | ||
| • | Profit Sharing and Royalties. In the United States, we will share profits on the sale of Vitrase® with Allergan on a 50/50 basis during the term of the license agreement. In all markets outside the United States, except Mexico (until April 2004) and Japan, we will receive a royalty on all sales of Vitrase® by Allergan. | ||
| • | Term and Termination. Allergan’s license to market, sell and distribute Vitrase® in the United States will expire ten full calendar years following the date of its first commercial sale, at which time all commercial rights for Vitrase® in the United States will revert to us. Allergan’s obligation to pay royalties will terminate on a country-by-country basis upon the latest of 10 full calendar years following the date of the first commercial sale in each particular country and the expiration date of the last-to-expire licensed patent relating to Vitrase® in that country. Allergan may terminate the license agreement at any time with three months notice to us. We may terminate the license agreement on a country-by-country basis in certain countries if Allergan fails to commercialize Vitrase® within 12 months of regulatory approval in that country. | ||
| • | Board of Director Representation and Visitation Rights. Allergan has the right to request that we nominate an Allergan representative to our Board of Directors. In the event that the Allergan nominee is not elected to the Board of Directors, |
8
Table of Contents
| Allergan has certain visitation rights, which include the designation of an Allergan representative to attend and observe all of our Board of Director meetings. |
Collaboration With Otsuka
In December 2001, we began a collaboration with Otsuka Pharmaceutical Co., Ltd. with respect to the commercialization of Vitrase® in Japan for certain ophthalmic uses, including a license agreement for the clinical development, regulatory approval, marketing, sale and distribution of Vitrase®, a supply agreement for Vitrase® and a securities purchase agreement for 84,567 shares of our common stock for the aggregate purchase price of $4.0 million. Otsuka is part of the Otsuka Group, headquartered in Tokyo, Japan and has a diverse portfolio including ophthalmic, central nervous system, cardiovascular, circulatory, gastro-intestinal, respiratory, oncological and dermatological products.
Under the terms of our agreements with Otsuka:
| • | Clinical Development. Otsuka is responsible for all preclinical studies and clinical trials in support of marketing approval of Vitrase® for the treatment of vitreous hemorrhage in Japan. Otsuka is also responsible for all preclinical studies and clinical trials for regulatory approvals for additional indications including the treatment of diabetic retinopathy. | ||
| • | Regulatory Approvals. Otsuka is responsible for applying for and obtaining regulatory approval of Vitrase® in Japan. Otsuka will also be responsible for obtaining National Health Insurance pricing approval for Vitrase® from the Japanese Ministry of Health. We will be responsible for providing Otsuka with copies of preclinical and clinical data and study reports and other documents in connection with our regulatory filings for Vitrase® in the United States. | ||
| • | Manufacturing. We are responsible for the manufacture of Vitrase® and for the supply of all of Otsuka’s requirements for clinical trials in Japan. If approved, we will also be responsible for supplying all of Otsuka’s commercial product requirements for Vitrase® during the term of the license agreement. | ||
| • | Marketing. Otsuka will be responsible for the overall management of marketing, sales and distribution activities for Vitrase® in Japan through its established sales and marketing organization. | ||
| • | Milestone Payments. Otsuka paid us a non-refundable license fee of $5,000,000 as part of the license agreement. Otsuka has also agreed to pay us a milestone payment of $10,000,000 upon regulatory approval of Vitrase® for the treatment of vitreous hemorrhage in Japan. To date, we have not earned this milestone payment from Otsuka and we cannot guarantee that we will receive this milestone payment in the future. | ||
| • | Commercial Purchase of Vitrase®. Otsuka is required to purchase Vitrase® from us at a percentage of the annual National Health Insurance price as established for Vitrase® in Japan during the term of the license agreement, unless we are unable to supply Vitrase® to Otsuka. If we are unable to supply Vitrase® to Otsuka and Otsuka, or a third party, is required to manufacture Vitrase® to meet Otsuka’s supply requirements, we will only receive a royalty on sales of Vitrase® by Otsuka. | ||
| • | Term and Termination. Our agreements with Otsuka will terminate upon the latest of 15 full calendar years following the date of the first commercial sale of Vitrase® in Japan and the expiration date of the last-to-expire licensed patent relating to Vitrase® in Japan. Otsuka may terminate the agreements at any time with six months notice to us. We may terminate the agreements if Otsuka fails to submit an application for regulatory approval of Vitrase® in Japan within 12 months of completing all necessary clinical trials and the initial meeting with the Japanese Ministry of Health to review the clinical trial results. | ||
Collaboration With Senju
In May 2002, the Company expanded its late-stage product portfolio, acquiring three promising therapeutic products with ophthalmic applications from AcSentient. The costs associated with this acquisition were approximately $1.7 million. ISTALOL™ and bromfenac were products developed by Senju. As a result of this acquisition, we began a collaboration with Senju including individual license agreements for the commercialization of ISTALOL™ and bromfenac in the United States. These license agreements were originally executed between Senju and AcSentient. The full rights and obligations of AcSentient under both license agreements were transferred to ISTA as a part of the acquisition agreement between ISTA and AcSentient, with such transfer approved by Senju.
9
Table of Contents
Under the terms of our agreements with Senju:
| • | Clinical Development. Senju is responsible for completing development activities for ISTALOL™. ISTA is responsible for completing development activities for bromfenac. Assuming successful completion of our clinical studies, we anticipate submitting an NDA for bromfenac in early 2004. | ||
| • | Regulatory Approvals. Senju was responsible for preparing and submitting an NDA to the FDA for ISTALOL™. Senju submitted this NDA in September 2002, and the FDA accepted it for review in November 2002. ISTA is responsible for the preparation and submission of an NDA to the FDA for bromfenac following the successful conclusion of the Phase III clinical study. All follow-up activities with the FDA for both ISTALOL™ and bromfenac will be handled by ISTA. | ||
| • | Manufacturing. ISTA will be responsible for the manufacture of ISTALOL™ and bromfenac. | ||
| • | Marketing. ISTA will be responsible for the overall management of marketing, sales and distribution activities for ISTALOL™ and bromfenac in the United States. | ||
| • | Milestone Payments. ISTA will be required to pay to Senju non-refundable milestone payments of up to $3,500,000, if all such milestones, relating to the development process and regulatory approval of both ISTALOL™ and bromfenac, are accomplished. | ||
| • | Term and Termination. The license agreements with Senju will terminate upon the last-to-expire licensed patent in the United States relating to ISTALOL™ and bromfenac, respectively. Neither party is entitled to terminate the agreements without cause. | ||
Collaboration With the Eastern Virginia School of Medicine
Caprogel® was initially licensed by AcSentient from the Eastern Virginia School of Medicine. In May 2002, as part of our AcSentient asset acquisition, ISTA acquired worldwide marketing rights for Caprogel®. The full rights and obligations of AcSentient under the Eastern Virginia School of Medicine license agreement were assigned to ISTA as a part of the acquisition agreement between ISTA and AcSentient, with such transfer approved by the Eastern Virginia School of Medicine.
Under the terms of our agreements with the Eastern Virginia School of Medicine:
| • | Clinical Development. ISTA is responsible for completing development activities for Caprogel®. We are currently conducting feasibility studies for the commercialization of Caprogel®. Once completed, and if these studies yield promising results, we intend to pursue further clinical development of Caprogel® consistent with such studies results. However, the timing and scope for our development of Caprogel® may change based on our assessment, from time to time, of this product candidate's market potential, other product opportunities and our corporate priorities. | ||
| • | Regulatory Approvals. ISTA is responsible for the preparation and submission of an NDA to the FDA for Caprogel® assuming the successful conclusion of our clinical study efforts. All follow-up activities with the FDA for Caprogel® will be handled by ISTA. | ||
| • | Manufacturing. ISTA will be responsible for the manufacture of Caprogel®. | ||
| • | Marketing. ISTA will be responsible for the overall management of marketing, sales and distribution activities for Caprogel® on a worldwide basis. | ||
| • | Royalties. The Eastern Virginia School of Medicine will receive royalty payments on all sales of Caprogel® by ISTA worldwide. | ||
| • | Term and Termination. The license agreement with the Eastern Virginia School of Medicine will terminate upon the last-to-expire licensed patent in the United States relating to Caprogel®. ISTA may terminate the license agreement with the Eastern Virginia School of Medicine upon three months prior written notice. | ||
Research and Development
Since our inception, we have made substantial investments in research and development. During the years ended December 31, 2002, 2001 and 2000, we spent $14.8 million, $15.8 million and $16.2 million, respectively, on research and development activities.
10
Table of Contents
We plan to focus our near-term research and development efforts on the continued development of the products in our current development pipeline, which include Vitrase®, ISTALOL™, bromfenac and Caprogel®. Building on this pipeline, ISTA’s goal is to become a fully-integrated specialty pharmaceutical company by developing or acquiring complementary products, either already marketed or in late stage development. Some acquired products may require additional research and development activities prior to regulatory approval and commercialization.
Patents and Proprietary Rights
Our success will depend in part on our ability to obtain patent protection for our inventions, to preserve our trade secrets and to operate without infringing the proprietary rights of third parties. Our strategy is to actively pursue patent protection in the United States and foreign jurisdictions for technology that we believe to be proprietary and that offers a potential competitive advantage for our inventions. To date, we have filed six U.S. patent applications for Vitrase® technologies, two of which have been issued claiming uses of ovine hyaluronidase to clear vitreous hemorrhage, U.S. Patent Nos. 6,039,943 (expiring on August 24, 2018) and 5,866,120 (expiring on January 1, 2015), one of which is pending seeking to claim an additional use of ovine hyaluronidase to clear vitreous hemorrhage and one of which is pending seeking to claim a use of ovine hyaluronidase to treat other vitreoretinal disorders.
In addition to patents, we rely on trade secrets and proprietary know-how. We seek protection of these trade secrets and proprietary know-how, in part, through confidentiality and proprietary information agreements. We make efforts to require our employees, directors, consultants and advisors, outside scientific collaborators and sponsored researchers, other advisors and other individuals and entities to execute confidentiality agreements upon the start of employment, consulting or other contractual relationships with us. These agreements provide that all confidential information developed or made known to the individual or entity during the course of the relationship is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees and some other parties, the agreements provide that all inventions conceived by the individual will be our exclusive property. These agreements may not provide meaningful protection for or adequate remedies to protect our technology in the event of unauthorized use or disclosure of information. Furthermore, our trade secrets may otherwise become known to, or be independently developed by, our competitors.
We also file trademark applications to protect the names of our products. These applications may not mature to registration and may be challenged by third parties.
Competition
The markets for therapies that treat diseases and conditions of the eye are subject to intense competition and technological change. Many companies, including major pharmaceutical companies and specialized biotechnology companies, are engaged in activities similar to ours. Such companies include Allergan, Inc., Alcon Laboratories, Inc., Bausch & Lomb Incorporated, CIBA Vision (a unit of Novartis AG), Pharmacia/Pfizer and Eli Lilly and Company. Many of these companies have substantially greater financial and other resources, larger research and development staffs and more extensive marketing and manufacturing organizations than ours. Many of these companies have significant experience in preclinical testing, clinical trials and other parts of the regulatory approval process.
Should we be successful in acquiring or in-licensing currently marketed ophthalmic products, we will be subject to intense competition from major pharmaceutical companies who have extensive marketing and distribution organization and substantially greater financial resources than ours.
We are not aware of any other drug candidates in clinical trials for the treatment of vitreous hemorrhage or hyphema. Eli Lilly and Company is currently conducting clinical trials for the use of a systemic drug to treat diabetic retinopathy, and several companies are working on drugs and systems to help control diabetes and the consequences of diabetes, including diabetic retinopathy. In addition, numerous companies are working on alternate therapies for ocular inflammation and glaucoma.
Our success will depend, in part, on our ability to:
| • | demonstrate the safety and efficacy of our products | ||
| • | obtain regulatory approval in a timely manner | ||
| • | demonstrate potential advantages over alternative treatment methods |
11
Table of Contents
| • | effectively market and distribute our products, either through our collaborators or by establishing and enhancing our own resources in these areas | ||
| • | obtain reimbursement coverage from insurance companies and other third-party payers | ||
| • | demonstrate cost-effectiveness | ||
| • | obtain patent protection and effectively enforce our patent rights |
Marketing and Sales
Upon receipt of applicable regulatory approvals, we plan to market and distribute Vitrase® in the United States and all international markets, except Mexico (until April 2004) and Japan, through our collaboration with Allergan. We have a distribution agreement with Laboratorios Sophia S.A. de C.V. providing for the marketing, sales and distribution of Vitrase® in Mexico until April 2004. Pursuant to our agreement with Laboratorios Sophia we are entitled to 75% of the final retail price for all sales of Vitrase® by Laboratorios Sophia. The agreement with Laboratorios Sophia may not be terminated by either party without cause. We have an agreement with Otsuka providing for the development and commercialization of Vitrase® in Japan. In the United States, the primary target market for Vitrase® will initially be retinal specialists to whom most patients with vitreous hemorrhage are referred.
We plan to market and distribute ISTALOL™, bromfenac and Caprogel® in the United States through our own resources. We will therefore need to establish our own sales, marketing and distribution capabilities. We will need to devote significant financial and management resources to develop such sales, marketing and distribution capabilities.
Third-party Reimbursement
In the United States, physicians, hospitals and other healthcare providers that purchase pharmaceutical products generally rely on third-party payers, principally private health insurance plans and Medicare and, to a lesser extent, Medicaid, to reimburse all or part of the cost of the product and procedure for which the product is being used. We expect that patients with a vitreous hemorrhage who are candidates for Vitrase® treatment will include, primarily due to demographic factors, patients with health insurance coverage provided by Medicare and private insurers, including Medicare health maintenance organizations. Currently, a Medicare reimbursement code has been established for the intravitreal injection of a pharmaceutical agent, which, we believe, will be appropriate for physician billing for a Vitrase® injection. Hospitals and physicians are reimbursed separately for drugs. Typically, the Health Care Financing Administration, or HCFA, the governmental agency responsible for Medicare reimbursement policy, does not issue national coverage guidelines for individual drugs. Drug specific coverage policies are primarily developed by individual health insurance companies following Medicare’s criteria for drug coverage, which include, among other requirements, that the drug be FDA approved, be used in connection with a physician service and be medically reasonable for the treatment of an illness or injury. While reimbursement may be available under existing payment codes for miscellaneous injectable drugs, each Medicare health insurance provider reviews such reimbursement requests separately. Widespread and uniform reimbursement for our injectable drug products will require the establishment of a specific reimbursement code for the injectable drug, which is issued by HCFA following review of an application by the manufacturer. To support our applications for reimbursement coverage with Medicare and other major third-party payers, we intend to use data from clinical trials, including Phase III and Phase IV clinical trials, to demonstrate the clinical utility and potential economic benefits of using Vitrase® for the treatment of vitreous hemorrhage. The lack of satisfactory reimbursement for our drug products will limit their widespread use and lower potential product revenues.
Reimbursement systems in international markets vary significantly by country and, within some countries, by region. Reimbursement approvals must be obtained on a country-by-country basis. In many foreign markets, including markets in which we anticipate selling our products, the pricing of prescription pharmaceuticals is subject to government pricing control. In these markets, once marketing approval is received, pricing negotiations could take another six to twelve months or longer. As in the United States, the lack of satisfactory reimbursement or inadequate government pricing of our products will limit their widespread use and lower potential product revenues.
Government Regulation
Our pharmaceutical products are subject to extensive government regulation in the United States. If we distribute our products abroad, these products will also be subject to extensive foreign government regulation. In the United States, the FDA regulates
12
Table of Contents
pharmaceutical products. FDA regulations govern the testing, manufacturing, advertising, promotion, labeling, sale and distribution of our products.
The FDA approval process for drugs includes:
| • | preclinical studies | ||
| • | submission of an Investigational NDA for clinical trials | ||
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product | ||
| • | submission of a NDA | ||
| • | review of the NDA | ||
| • | inspection of the facilities used in the manufacturing of the drug to assess compliance with the current Good Manufacturing Practices regulations |
The NDA includes comprehensive and complete descriptions of the preclinical testing, clinical trials, and the chemical, manufacturing and control requirements of a drug that enables the FDA to determine the drug’s safety and efficacy. An NDA must be submitted by us, and filed and approved by the FDA before any of our drugs can be marketed commercially in the United States.
The FDA testing and approval process requires substantial time, effort and money. We cannot assure you that any approval will ever be granted.
Preclinical studies include laboratory evaluation of the product, as well as animal studies to assess the potential safety and effectiveness of the product. These studies must be performed according to good laboratory practices. The results of the preclinical studies, together with manufacturing information and analytical data, are submitted to the FDA as part of the Investigational New Drug Application (“IND”). Clinical trials may begin 30 days after the IND is received, unless the FDA raises concerns or questions about the conduct of the clinical trials. If concerns or questions are raised, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed. We cannot assure you that submission of an IND will result in authorization to commence clinical trials. Nor can we assure you that if clinical trials are approved, that data will result in marketing approval.
Clinical trials involve the administration of the product that is the subject of the trial to volunteers or patients under the supervision of a qualified principal investigator. Furthermore, each clinical trial must be reviewed and approved by an independent institutional review board at each institution at which the study will be conducted. The institutional review board will consider, among other things, ethical factors, the safety of human subjects and the possible liability of the institution. Also, clinical trials must be performed according to good clinical practices. Good clinical practices are enumerated in FDA regulations and guidance documents.
Clinical trials typically are conducted in three sequential phases: Phases I, II and III, with Phase IV studies conducted after approval and generally required for drugs subject to accelerated approval regulations. These phases may overlap. In Phase I clinical trials, the drug is usually tested on healthy volunteers to determine:
| • | safety | ||
| • | any adverse effects | ||
| • | dosage tolerance | ||
| • | absorption | ||
| • | metabolism | ||
| • | distribution | ||
| • | excretion |
13
Table of Contents
| • | other drug effects |
In Phase II clinical trials, the drug is usually tested on a limited number of afflicted patients to evaluate the efficacy of the drug for specific, targeted indications, determine dosage tolerance and optimal dosage, identify possible adverse effects and safety risks. In Phase III clinical trials, the drug is usually tested on a larger number of patients, in an expanded patient population and at multiple clinical sites. The FDA may require that we suspend clinical trials at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk.
In Phase IV clinical trials or other post-approval commitments, additional studies and patient follow-up are conducted to gain experience from the treatment of patients in the intended therapeutic indication. Additional studies and follow-up are also conducted to document a clinical benefit where drugs are approved under accelerated approval regulations and based on surrogate endpoints. In clinical trials, surrogate endpoints are alternative measurements of the symptoms of a disease or condition that are substituted for measurements of observable clinical symptoms. Failure to promptly conduct Phase IV clinical trials and follow-up could result in expedited withdrawal of products approved under accelerated approval regulations.
The facilities, procedures, and operations of our contract manufacturers must be determined to be adequate by the FDA before approval of product manufacturing. Manufacturing facilities are subject to inspections by the FDA for compliance with current Good Manufacturing Practices (“cGMP”), licensing specifications, and other FDA regulations before and after an NDA has been approved. Foreign manufacturing facilities are also subject to periodic FDA inspections or inspections by foreign regulatory authorities. Among other things, the FDA may withhold approval of NDAs or other product applications of a facility if deficiencies are found at the facility. Vendors that supply us finished products or components used to manufacture, package and label products are subject to similar regulation and periodic inspections.
Following such inspections, the FDA may issue notices on Form 483 and Warning Letters that could cause us to modify certain activities identified during the inspection. A Form 483 notice is generally issued at the conclusion of a FDA inspection and lists conditions the FDA investigators believe may violate cGMP or other FDA regulations. FDA guidelines specify that a Warning Letter be issued only for violations of “regulatory significance” for which the failure to adequately and promptly achieve correction maybe expected to result in an enforcement action.
Failure to comply with FDA and other governmental regulations can result in fines, unanticipated compliance expenditures, recall or seizure of products, total or partial suspension of production and/or distribution, suspension of the FDA ‘s review of NDAs, injunctions and criminal prosecution. Any of these actions could have a material adverse effect on us.
Food and Drug Administration Modernization Act of 1997
The Food and Drug Administration Modernization Act of 1997 was enacted, in part, to ensure the availability of safe and effective drugs, biologics and medical devices by expediting the FDA review process for new products. The Modernization Act establishes a statutory program for the approval of fast-track products. The fast-track provisions essentially codify the FDA’s accelerated approval regulations for drugs and biologics. A fast-track product is defined as a new drug or biologic intended for the treatment of a serious or life-threatening condition that demonstrates the potential to address unmet medical needs for this condition. Under the new fast-track program, the sponsor of a new drug or biologic may request the FDA to designate the drug or biologic as a fast-track product at any time during the clinical development of the product. The Modernization Act specifies that the FDA must determine if the product qualifies for fast-track designation within 60 days of receipt of the sponsor’s request. Fast-track designated products may qualify for accelerated approval and priority review, or review within six months. Accelerated approval will be subject to:
| • | post-approval studies and follow-up to validate the surrogate endpoint or confirm the effect on the clinical endpoint | ||
| • | prior review of all promotional materials |
If a preliminary review of the clinical data suggests that the product is effective, the FDA may initiate review of sections of an application for fast-track designation for a product before the application is complete. This rolling review is available if the applicant provides a schedule for submission of remaining information and pays applicable user fees. However, the time period specified in the Prescription Drug User Fees Act, which governs the time period goals the FDA has committed for reviewing an application, does not begin until the complete application is submitted.
14
Table of Contents
In October 1998, the FDA granted our application for fast-track designation for Vitrase® for the treatment of vitreous hemorrhage. During 2002 we submitted three sections of the Vitrase® NDA as they were completed. The pre-clinical pharmacology and toxicology section was submitted in January 2002. The clinical section and the chemistry, manufacturing and controls (“CMC”) section were submitted in October 2002. The FDA accepted those sections of the Vitrase® NDA for review in December 2002, along with the pre-clinical pharmacology and toxicology section that was submitted in January 2002. On April 3, 2003, the FDA issued an “approvable” letter in which the FDA cited issues primarily related to the sufficiency of the efficacy data submitted with the NDA for Vitrase®. The FDA requested additional analysis of the existing data and an additional confirmatory clinical study based upon that analysis. We intend to discuss with the FDA the comments contained in the “approvable” letter and, based upon those discussions, determine the next appropriate steps in the review and approval process of Vitrase®.
One of our product candidates, Caprogel®, for the treatment of hyphema, has been designated by the FDA as an orphan drug. An orphan drug is defined in the 1984 amendments of the Orphan Drug Act as a drug intended to treat a condition affecting fewer than 200,000 persons in the United States. The Orphan Drug Act has numerous incentives including:
| • | seven years of exclusive marketing upon FDA approval | ||
| • | tax credit for clinical research expense | ||
| • | grant support for investigation of rare disease treatments | ||
| • | user fee waiver |
International
For marketing outside the United States, we also are subject to foreign regulatory requirements governing human clinical trials and marketing approval for pharmaceutical products. The requirements governing the conduct of clinical trials, product approval, pricing and reimbursement vary widely from country to country. Whether or not FDA approval has been obtained, approval of a product by the comparable regulatory authorities of foreign countries must be obtained before manufacturing or marketing the product in those countries. The approval process varies from country to country and the time required for such approvals may differ substantially from that required for FDA approval. We cannot assure you that clinical trials conducted in one country will be accepted by other countries or that approval in one country will result in approval in any other country. For clinical trials conducted outside the United States, the clinical stages are generally comparable to the phases of clinical development established by the FDA.
Manufacturing
Ovine hyaluronidase, the active pharmaceutical ingredient used in Vitrase®, is sourced from ovine testes and processed in several stages to produce a highly purified raw material for formulation. We have a supply agreement with Biozyme Laboratories Limited (Biozyme) for cGMP-grade ovine hyaluronidase for use in ophthalmic applications. The ovine hyaluronidase is lyophilized, or freeze dried, by Biozyme and delivered to our contract manufacturer for formulation and filling of dose specific vials. Vitrase® is currently required to be stored under refrigerated conditions prior to its use. We have entered into an agreement with R. P. Scherer West, Inc. (dba SP Pharmaceuticals, Inc.) to manufacture commercial quantities of Vitrase®. R. P. Scherer West is a wholly owned subsidiary of Cardinal Health. Under the terms of the agreement, Cardinal Health Sterile Technologies, a member of Cardinal Health’s Pharmaceutical Technologies & Services group, will manufacture, package and perform certain manufacturing quality assurance and quality control tests on the product in accordance with specifications provided by us. Currently, Biozyme and Cardinal Health are our sole sources for ovine hyaluronidase and finished product, respectively. We are seeking additional manufacturing sources for these products.
We have supply agreements with Bausch & Lomb Incorporated to manufacture commercial quantities of ISTALOL™ and bromfenac. Under the terms of the agreements, Bausch & Lomb will manufacture, package and perform certain manufacturing quality assurance and quality control tests on the two products in accordance with specifications provided by the Company. Currently, Bausch & Lomb is our sole source for ISTALOL™ and bromfenac. We are seeking additional manufacturing sources for these products.
15
Table of Contents
Human Resources
As of February 18, 2003, we had 39 full-time employees. Approximately 32 of our employees are involved in research and clinical development activities. Six of our employees hold Ph.D. or M.D. degrees and eight other employees hold other advanced degrees. Our employees do not have a collective bargaining agreement. We consider our relations with our employees to be good.
General Information
We incorporated in California in February 1992 as Advanced Corneal Systems, Inc. In March 2000, we changed our name to ISTA Pharmaceuticals, Inc. and we reincorporated in Delaware in August 2000. Our corporate headquarters and principal research laboratories are located at 15279 Alton Parkway, Building 100, Irvine, California 92618, and our telephone number is (949) 788-6000. Vitrase®, ISTALOL™, Caprogel®, ISTA, ISTA Pharmaceuticals and the ISTA logo are our trademarks.
We maintain an Internet website at www.istavision.com where our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports are available without charge, as reasonably practicable following the time they are filed with or furnished to the SEC.
RISK FACTORS
In addition to other information included in this report, the following factors should be considered in evaluating our business and future prospects:
Risks Related to Our Company
If we do not receive and maintain regulatory approvals for our product candidates, we or our marketing partners will not be able to commercialize our products, which would substantially impair our ability to generate revenues and materially harm our business and financial condition.
None of our product candidates has received regulatory approval from the FDA. Approval from the FDA is necessary to manufacture and market pharmaceutical products in the United States. Many other countries including the major European countries and Japan have similar requirements.
The NDA process is extensive, time-consuming and costly, and there is no guarantee that regulatory authorities will approve NDAs of any of our product candidates. We have submitted NDAs which are currently pending before the FDA for both Vitrase® and for ISTALOL™. Moreover, we depend on the assistance of Senju Pharmaceuticals for obtaining regulatory approval for ISTALOL™.
Although we recently received an “approvable” letter from the FDA with respect to ISTA’s NDA for Vitrase® for the treatment of vitreous hemorrhage, the FDA has requested additional analysis of the existing data and an additional confirmatory clinical study based upon that analysis. There can be no assurances that the FDA will approve our NDA for Vitrase®, even if we decide to and are successfully able to undertake the further analysis and clinical testing requested by the FDA.
Clinical testing of pharmaceutical products is also a long, expensive and uncertain process. Even if initial results of preclinical studies or clinical trial results are positive, we may obtain different results in later stages of drug development, including failure to show desired safety and efficacy. Our bromfenac product, a topical non-steroidal anti-inflammatory compound for the treatment of ocular inflammation is currently undergoing clinical trials. The clinical trials of any of our product candidates could be unsuccessful, which would prevent us from obtaining regulatory approval and commercializing the product.
FDA approval can be delayed, limited or not granted for many reasons, including, among others:
| • | FDA officials may not find a product candidate safe or effective to merit an approval | ||
| • | FDA officials may not find that the data from preclinical testing and clinical trials justifies approval, or they may require additional studies that would make it commercially unattractive to continue pursuit of approval |
16
Table of Contents
| • | the FDA might not approve our manufacturing processes or facilities, or the processes or facilities of our contract manufacturers or raw material suppliers | ||
| • | the FDA may change its approval policies or adopt new regulations | ||
| • | the FDA may approve a product candidate for indications that are narrow or under conditions that place our product at a competitive disadvantage, which may limit our sales and marketing activities or otherwise adversely impact the commercial potential of a product |
If the FDA does not approve our products on commercially viable terms or we terminate development of any of our products due to difficulties encountered in the regulatory approval process, it will have a material adverse impact on our business and we will be dependent on the development of our other product candidates and/or our ability to successfully acquire other products and technologies.
In addition, we intend to market our products, and perhaps have certain of our products manufactured, in foreign countries. The process of obtaining approvals in foreign countries is subject to delay and failure for similar reasons.
If our product candidates are approved by the FDA but do not gain market acceptance, our business will suffer because we might not be able to fund future operations.
A number of factors may affect the market acceptance of Vitrase®, ISTALOL™, bromfenac, Caprogel® or any other products we develop or acquire in the future, including, among others:
| • | the price of our products relative to other therapies for the same or similar treatments | ||
| • | the perception by patients, physicians and other members of the health care community of the effectiveness and safety of our products for their prescribed treatments | ||
| • | our ability to fund sales and marketing departments | ||
| • | the effectiveness of our sales and marketing efforts |
If our products do not gain market acceptance we may not be able to fund future operations, including the development or acquisition of new product candidates and/or our sales and marketing efforts for our approved products.
If we are unable to sufficiently develop our sales, marketing and distribution capabilities or enter into agreements with third parties to perform these functions, we will not be able to commercialize products.
We currently are in the process of developing our sales, marketing and distribution capabilities. However, our current capabilities in these areas are limited. In order to commercialize any products, we must internally develop substantial sales, marketing and distribution capabilities, or establish collaborations or other arrangements with third parties to perform these services. We do not have extensive experience in these areas, and we may not be able to establish adequate in-house sales, marketing and distribution capabilities or engage and effectively manage relationships with third parties to perform any or all of such services. For example, we intend to rely on Allergan to commercialize Vitrase® in the United States, and Otsuka Pharmaceuticals, Co. Ltd. to market Vitrase® in Japan. To the extent that we enter into co-promotion or other licensing arrangements, our product revenues are likely to be lower than if we directly marketed and sold our products, and any revenues we receive will depend upon the efforts of third parties, whose efforts may not be successful.
We have a history of net losses and negative cash flow, and we may never achieve or maintain profitability.
We have only a limited operating history upon which you can evaluate our business. We have never been profitable, and we might never become profitable. As of December 31, 2002, our accumulated deficit was $122.4 million, including a net loss of approximately $23.0 million for the year ended December 31, 2002. We have not generated any revenue from product sales to date, and we may never generate revenues from product sales in the future. Even if we do achieve significant revenues from product sales, we expect to incur significant operating losses over the next several years. As of December 31, 2002, we had approximately $35.7 million in cash and short-term investments and working capital of $33.0 million. We anticipate that our existing capital resources will enable us to
17
Table of Contents
fund operations for at least the next twelve months. We may be required to raise additional capital in the future. If we are required to raise additional capital in the future there can be no assurance that the additional financing will be available on favorable terms.
We may be unable to execute our strategic plan to transition to a fully-integrated specialty pharmaceutical company, which could have a material adverse impact on our business and financial condition.
Our strategy to acquire or in-license ophthalmic pharmaceutical products, either currently marketed or in late-stage development, will be dependent upon a number of factors including identifying such acquisition opportunities, successfully negotiating favorable terms with third parties for the acquisition or licensing of such products and our ability to raise additional capital to facilitate the acquisition or licensing of such products. Furthermore, we have limited sales, marketing and distribution capabilities to support the marketing of any products, and we do not have experience in managing third-party manufacturers of any products in commercial quantities. In addition, if we acquire or in-license late-stage development products, our ability to successfully commercialize such products will also be dependent on our ability to successfully complete development of such products, including obtaining the necessary regulatory approvals. For example, we have in-licensed U.S. marketing rights to ISTALOL™ and bromfenac from Senju Pharmaceutical, and we have in-licensed worldwide rights to Caprogel® from the Eastern Virginia School of Medicine. Senju is responsible for the development of ISTALOL™, including obtaining the necessary regulatory approvals in the United States. The Company is responsible for the further development of bromfenac and Caprogel®.
We may not be able to identify any product acquisition opportunities or be successful in negotiating favorable terms for any such product acquisitions. Should we be successful in acquiring or licensing any products, we will need to establish and enhance our sales, marketing, distribution and manufacturing capabilities, each of which will require substantial financial and management resources. Our failure to establish effective sales, marketing, distribution and manufacturing capabilities on a timely basis would adversely affect our ability to commercialize any acquired products. If we are unable to execute our strategic plan to transition to a fully-integrated specialty pharmaceutical company on a timely basis, our ability to generate revenues would be substantially impaired which would materially harm our business and financial condition.
If we have problems with our contract manufacturers, our product development and commercialization efforts could be delayed or stopped.
We have entered into a master services agreement with R.P Scherer West, Inc. (which was subsequently acquired by Cardinal Health), for the manufacture of commercial quantities, if approved, of our Vitrase® finished product. We have also entered into a manufacturing services agreement with Bausch and Lomb for the manufacture of commercial quantities, if approved, of our ISTALOL™ and bromfenac products. To date, we have needed these products only in amounts sufficient for clinical trials. Before any contract manufacturer can produce commercial quantities of a product, we must demonstrate to the FDA’s satisfaction that the product source for commercial quantities is substantially equivalent to the supply of the product used in our clinical trials. Such demonstration may include the requirement to conduct additional clinical trials. In addition, the manufacturing facilities of all of our contract manufacturers must comply with current Good Manufacturing Practice regulations, which the FDA strictly enforces. Moreover, the facilities of the contract manufacturer must undergo and pass pre-approval inspection by the FDA before any of our products can be approved for manufacture. We cannot assure you that Cardinal Health or Bausch and Lomb will be able, as applicable, to develop processes necessary to produce substantially equivalent product or that regulatory authorities will approve them as a manufacturer. Failure to develop necessary production processes or receive regulatory approval could delay or stop our efforts to develop and commercialize our product candidates.
Our strategic partners may not perform their duties under our agreements, in which case our ability to commercialize our products may be significantly impaired.
We have entered into collaborations with Allergan, Inc. and Otsuka Pharmaceuticals, Co. Ltd. relating to Vitrase®, and with Senju Pharmaceuticals relating to ISTALOL™ and bromfenac. If we obtain regulatory approval for Vitrase® in the United States and Europe, we will be dependent on Allergan for the commercialization of Vitrase® in the United States and Europe. We will depend on Otsuka for obtaining regulatory approval of Vitrase® in Japan, and if such approval is obtained, we will be dependent upon Otsuka for the commercialization of Vitrase® in Japan. We will also be dependent on Senju Pharmaceuticals for obtaining regulatory approval for ISTALOL™ in the United States. The amount and timing of resources that Allergan, Otsuka, and Senju dedicate to our collaborations is not within our control. Accordingly, any breach or termination of our agreements by these collaborators could delay or stop the development and/or commercialization of our product candidates. Our collaborative partners may change their strategic focus, terminate our agreements, or pursue alternative technologies. Although our agreements with Allergan, Otsuka and Senju contain reciprocal terms providing that neither we nor they may develop products that directly compete in the same form with the
18
Table of Contents
products involved in the collaboration, there can be no assurances that our collaborators will not develop competing products in different forms or products that compete indirectly with our products. Accordingly, unfavorable developments in our relationship with our strategic partners could have a significant adverse effect on us and our financial condition.
If we have problems with our sole source suppliers, our product development and commercialization efforts for our product candidates could be delayed or stopped.
Some materials used in the Company’s products are currently sourced from a single source. Biozyme Laboratories, Ltd. is currently our only source for highly purified ovine hyaluronidase, which is the active ingredient in Vitrase®. If approved, commercial quantities of Vitrase® will be supplied by Cardinal Health as the sole source. ISTALOL™ and bromfenac, if approved, will be supplied by a single source, Bausch and Lomb. The Company has not established and may not be able to establish arrangements with additional suppliers for these ingredients or products. Difficulties in our relationship with our sole source suppliers or delays or interruptions in such suppliers’ supply of our requirements could limit our ability to provide sufficient quantities of our products, on a timely basis, for clinical trials and, if our products are approved, could limit commercial sales, which would have a material adverse effect on our business and financial condition. While we are currently pursuing additional sources for these products and materials, our success in establishing such additional supply arrangements cannot be assured.
We depend on our Chief Executive Officer, Vicente Anido, Jr., Ph.D., to execute our strategic plan to transition to a fully-integrated specialty pharmaceutical company.
Our success largely depends on the skills, experience and efforts of our Chief Executive Officer, Vicente Anido, Jr., Ph.D. We have entered into a written employment agreement with Dr. Anido that can be terminated at any time by the Company or by Dr. Anido. In the event Dr. Anido’s employment is terminated, other than voluntarily or for cause, Dr. Anido will receive nine months of salary as severance compensation. In the event Dr. Anido’s employment is terminated after a change of control of the Company (such as a merger where the Company is bought by another entity, or a sale of substantially all of the assets of the Company), other than voluntarily or for cause, then Dr. Anido’s stock options will immediately vest and become exercisable in full, and Dr. Anido will receive twenty-four months of salary as severance compensation. In addition, we do not maintain “key person” life insurance policies covering Dr. Anido. The loss of Dr. Anido would jeopardize our ability to execute our strategic plan and materially harm our business.
Risks Related to Our Industry
Our suppliers and manufacturers are subject to regulation by the FDA and other agencies, and if they do not meet their commitments, we would have to find substitute suppliers or manufacturers, which could delay the supply of our products to market.
Regulatory requirements applicable to pharmaceutical products make the substitution of suppliers and manufacturers costly and time consuming. We have no internal manufacturing capabilities and are, and expect to be in the future, entirely dependent on contract manufacturers and suppliers for the manufacture of our products and for their active ingredients. The disqualification of these suppliers through their failure to comply with regulatory requirements could negatively impact our business because the delays and costs in obtaining and qualifying alternate suppliers (if such alternative suppliers are available, which we cannot assure) could delay clinical trials or otherwise inhibit our ability to bring approved products to market, which would have a material adverse affect on our business and financial condition.
We may be required to initiate or defend against legal proceedings related to intellectual property rights, which may result in substantial expense, delay and/or cessation of our development and commercialization of our products.
We rely on patents to protect our intellectual property rights. The strength of this protection, however, is uncertain. In particular, it is not certain that:
| • | our patents and pending patent applications use technology that we invented first | ||
| • | we were the first to file patent applications for these inventions | ||
| • | others will not independently develop similar or alternative technologies or duplicate our technologies | ||
| • | any of our pending patent applications will result in issued patents |
19
Table of Contents
We currently have 56 patent applications pending. In the event any patents are issued to us as a result of such applications, there can be no assurance that such patents will provide a basis for commercially viable products, will provide us with any competitive advantages, or will not face third party challenges or be the subject of further proceedings limiting their scope or enforceability.
We may become involved in interference proceedings in the U.S. Patent and Trademark Office to determine the priority of our inventions. In addition, costly litigation could be necessary to protect our patent position. We also rely on trademarks to protect the names of our products. These trademarks may be challenged by others. If we enforce our trademarks against third parties, such enforcement proceedings may be expensive. We also rely on trade secrets, unpatented proprietary know-how and continuing technological innovation that we seek to protect with confidentiality agreements with employees, consultants and others with whom we discuss our business. Disputes may arise concerning the ownership of intellectual property or the applicability or enforceability of these agreements, and we might not be able to resolve these disputes in our favor.
The Company licenses patents held by the Eastern Virginia School of Medicine (for Caprogel®) and Senju Pharmaceutical (for ISTALOL™ and bromfenac). The license agreements with these licensors provide that the licensor is responsible for prosecution, maintenance, protection and defense of such patents, at the licensor’s cost with respect to the agreements with Senju, and at our cost with respect to the agreement with the Eastern Virginia School of Medicine. If the licensor chooses not to protect its own patent rights, we may not be able to take actions to secure our related product marketing rights.
In addition to protecting our own intellectual property rights, third parties may assert patent, trademark or copyright infringement or other intellectual property claims against us based on what they believe are their own intellectual property rights. We may be required to pay substantial damages, including but not limited to treble damages, for past infringement if it is ultimately determined that our products infringe a third party’s intellectual property rights. Even if infringement claims against us are without merit, defending a lawsuit takes significant time, may be expensive and may divert management’s attention from other business concerns. Further, we may be stopped from developing, manufacturing or selling our products until we obtain a license from the owner of the relevant technology or other intellectual property rights. If such a license is available at all, it may require us to pay substantial royalties or other fees.
If we do not receive third-party reimbursement, our products may not be accepted in the market.
Our ability to earn sufficient returns on our products will depend in part on the extent to which reimbursement for our products and related treatments will be available from government health administration authorities, private health insurers, managed care organizations and other healthcare providers.
Third-party payers are increasingly attempting to limit both the coverage and the level of reimbursement of new drug products to contain costs. Consequently, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. If we succeed in bringing one or more of our product candidates to market, third-party payers may not establish adequate levels of reimbursement for our products, which could limit their market acceptance.
We face intense competition and rapid technological change that could result in products that are superior to the products we are developing.
We have numerous competitors in the United States and abroad, including, among others, major pharmaceutical and specialized biotechnology firms, universities and other research institutions that may be developing competing products. Such competitors may include Allergan, Inc., Alcon Laboratories, Inc., Bausch & Lomb Incorporated, CIBA Vision (a unit of Novartis AG), Pharmacia/Pfizer and Eli Lilly and Company. These competitors may develop technologies and products that are more effective or less costly than our current or future product candidates or that could render our technologies and product candidates obsolete or noncompetitive. Many of these competitors have substantially more resources and product development, manufacturing and marketing experience and capabilities than we do. In addition, many of our competitors have significantly greater experience than we do in undertaking preclinical testing and clinical trials of pharmaceutical product candidates and obtaining FDA and other regulatory approvals of products and therapies for use in healthcare.
We are exposed to product liability claims, and insurance against these claims may not be available to us on reasonable terms.
We currently maintain clinical trial liability insurance with per occurrence and aggregate coverage limits of $5 million. The coverage limits of our insurance policies may be inadequate to protect us from any liabilities we might incur in connection with
20
Table of Contents
clinical trials or the sale of our products. Product liability insurance is expensive and in the future may not be available on commercially acceptable terms, or at all. A successful claim or claims brought against us in excess of our insurance coverage could materially harm our business and financial condition.
Risks Relating to Our Stock
Our stock price is subject to significant volatility.
During the two most recent fiscal years and up until the present, the daily closing price per share of our common stock has ranged from a high of $133.75 per share to a low of $2.60 per share. Our stock price has been and may continue to be subject to significant volatility. The following factors, in addition to other risks and uncertainties described in this section and elsewhere in this report, may cause the market price of our common stock to fall:
| • | the scope, outcome and timeliness of any governmental, court or other regulatory action that may involve us (including, without limitation, the scope, outcome or timeliness of any inspection or other action of the FDA) | ||
| • | the availability to us, on commercially reasonable terms, of third party sourced products and materials | ||
| • | developments concerning proprietary rights, including the ability of third parties to assert patents or other intellectual property rights against us which, among other things, could cause a delay or disruption in the development, manufacture, marketing or sale of our products | ||
| • | competitors announcing products under development or new commercial products | ||
| • | competitors publicity regarding actual or potential products under development | ||
| • | period-to-period fluctuations in our financial results | ||
| • | economic and other external factors, including disasters and other crises |
We participate in a highly dynamic industry, which often results in significant volatility in the market price of our common stock irrespective of company performance. Fluctuations in the price of our common stock may be exacerbated by conditions in the healthcare and technology industry segments or conditions in the financial markets generally.
Trading in our stock over the last 12 months has been limited, so investors may not be able to sell as much stock as they want at prevailing prices.
The average daily trading volume in our common stock for the twelve-month period ending December 31, 2002 was approximately 1,872 shares, and the average daily number of transactions was approximately 65 for the same period. If limited trading in our stock continues, it may be difficult for investors to sell their shares in the public market at any given time at prevailing prices.
Future sales of shares of our common stock, including sales of shares following the expiration of “lock-up” arrangements, may negatively affect our stock price.
As a result of our recent PIPE financing transaction, the PIPE investors received approximately 11.6 million shares of our common stock. The shares of common stock issued in connection with the PIPE transaction represented approximately 87% of our common stock. In connection with our PIPE financing transaction, we also issued warrants to the PIPE investors that are exercisable for the purchase of up to an aggregate of 1,578,946 shares of our common stock based upon a purchase price of $3.80 per share. The exercise of these warrants could result in significant dilution to shareholders at the time of exercise.
The PIPE investors also entered into “lock-up” agreements with the Company that impose restrictions on the ability of the PIPE investors to sell or otherwise dispose of the shares of our common stock that they received in the PIPE transaction. These “lock-up” restrictions expire on May 19, 2003. We will file a registration statement on Form S-3 to cover the shares issued to the PIPE investors and issuable upon conversion of the warrants. In the future, we may issue additional options, warrants or other derivative securities convertible into our Common Stock.
21
Table of Contents
Sales of substantial amounts of shares of our common stock, or even the potential for such sales, could lower the market price of our common stock and impair our ability to raise capital through the sale of equity securities.
Concentration of ownership could delay or prevent change in control or otherwise influence or control most matters submitted to our stockholders.
Our directors, officers, and principal stockholders together control approximately 89.0% of our voting securities, a concentration of ownership that could delay or prevent a change in control. Our executive officers and directors beneficially own approximately 2.5% of our voting securities and our 5% or greater stockholders beneficially own approximately 86.5% of our voting securities. These stockholders, if acting together, would be able to influence and possibly control most matters submitted for approval by our stockholders, including the election of directors, delaying or preventing a change of control, and the consideration of transactions in which stockholders might otherwise receive a premium for their shares over then-current market prices.
Our stockholder rights plan, provisions in our charter documents, and Delaware law may inhibit a takeover of the Company, which could limit the price investors might be willing to pay in the future for our common stock, and could entrench management.
We have a stockholder rights plan that may have the effect of discouraging unsolicited takeover proposals, thereby entrenching current management and possibly depressing the market price of our common stock. The rights issued under the stockholder rights plan would cause substantial dilution to a person or group that attempts to acquire us on terms not approved in advance by our board of directors. In addition, our charter and bylaws contain provisions that may discourage unsolicited takeover proposals that stockholders may consider to be in their best interests. These provisions include:
| • | a classified board of directors, | ||
| • | the ability of the board of directors to designate the terms of and issue new series of preferred stock, | ||
| • | advance notice requirements for nominations for election to the board of directors, and | ||
| • | special voting requirements for the amendment of our charter and bylaws. | ||
We are also subject to anti-takeover provisions under Delaware law, each of which could delay or prevent a change of control. Together these provisions and the rights plan may make more difficult the removal of management and may discourage transactions that otherwise could involve payment of a premium over prevailing market prices for common stock.
22
Table of Contents
PART IV
Item 15:Exhibits, Financial Statement Schedules and Reports on Form 8-K
| (3) | Exhibits |
| Exhibit Number | Description | |||
| 3.1 | Restated Certificate of Incorporation of registrant (1) | |||
| 3.2 | Certificate of Correction to Restated Certificate of Incorporation (1) | |||
| 3.3 | Certificate of Correction to Restated Certificate of Incorporation (2) | |||
| 3.3 | Bylaws of the registrant, as amended (3) | |||
| 3.4 | Certificate of Amendment to Bylaws of the registrant (1) | |||
| 4.1 | Specimen common stock certificate (3) | |||
| 4.2 | Preferred Stock Rights Agreement, dated as of December 31, 2001, between the registrant and Mellon Investor Services LLC (4) | |||
| 4.3 | First Amendment to the Preferred Stock Rights Agreement with Mellon Investor Services LLC, dated November 18, 2002 (5) | |||
| 10.1 | Amended and Restated Investors Rights Agreement dated as of March 29, 2000 (3) | |||
| 10.2 | 1993 Stock Plan and forms of agreements thereunder (3) | |||
| 10.3 | 2000 Stock Plan, as amended, and forms of agreements thereunder (3) | |||
| 10.4 | 2000 Employee Stock Purchase Plan, as amended (3) | |||
| 10.5 | Form of Indemnification Agreement with executive officers and directors (3) | |||
| 10.6 | Clinical Development Agreement between Covance, Inc. and the registrant dated as of October 28, 1998, as amended (3) | |||
| 10.7 | Agreement between CroMedica Global Inc. and the registrant as of September 8, 1998 (3) | |||
| 10.8 | Agreement between CroMedica Global Inc. and the registrant as of May 19, 1999 (3) | |||
| 10.9 | Lease between the registrant and Aetna Life Insurance Company dated September 13, 1996 for leased premises located at Suite 100, 15279 Alton Parkway, Irvine, California (3) | |||
| 10.10 | Distributor Agreement between Laboratorios Sophia S.A. de C.V. and the registrant as of April 23, 1998 (3) | |||
| 10.11 | Supply Agreement between Biozyme Laboratories, Ltd. and the registrant as of September 23, 1999 (3) | |||
| 10.12 | License Agreement between Allergan Sales, Inc., Allergan Sales, Ltd. and the registrant as of March 29, 2000 (3),(6) | |||
| 10.13 | Supply Agreement between Allergan Sales, Inc., Allergan Sales, Ltd. and the registrant as of March 29, 2000 (3),(6) | |||
| 10.14 | Amendment No. 1 to Contract Clinical Research Agreement with CroMedica Global, Inc., dated May 19, 1999 (7) | |||
23
Table of Contents
| Exhibit Number | Description | |||
| 10.15 | Amendment No. 2 to Contract Clinical Research Agreement with CroMedica Global, Inc., dated May 19, 1999 (7) | |||
| 10.16 | License Agreement between Otsuka Pharmaceutical Co., Ltd., and the registrant, dated as of December 13, 2001 (6),(8) | |||
| 10.17 | Supply Agreement between Otsuka Pharmaceutical Co., Ltd., and the registrant, dated as of December 13, 2001 (6),(8) | |||
| 10.18 | Securities Purchase Agreement between Otsuka Pharmaceutical Co., Ltd., and the registrant, dated as of December 13, 2001 (6),(8) | |||
| 10.19 | Registration Rights Agreement between Otsuka Pharmaceutical Co., Ltd., and the registrant, dated as of December 13, 2001 (6),(8) | |||
| 10.20 | Amendment No. 3 to Contract Clinical Research Agreement with CroMedica Global, Inc., dated December 5, 2001 (9) | |||
| 10.21 | Executive Employment Agreement between Vicente Anido, Jr., Ph.D. and the registrant, dated December 21, 2001 (9) | |||
| 10.22 | Stand-Alone Stock Option Agreement between Vicente Anido, Jr., Ph.D. and the registrant, dated December 21, 2001 (9) | |||
| 10.23 | Transition and Release Agreement between Edward H. Danse and the registrant, dated January 2, 2002 (9) | |||
| 10.24 | Facility Lease Amendment No. 1 with Alton Plaza Property, Inc. (9) | |||
| 10.25 | Facility Lease Amendment No. 2 with Alton Plaza Property, Inc. (9) | |||
| 10.26 | Asset Purchase and Sale Agreement with AcSentient, Inc., dated May 3, 2002 (10) | |||
| 10.27 | Master Services Agreement with R.P. Scherer West, Inc. doing business as SP Pharmaceuticals, dated June 7, 2002 (11) | |||
| 10.28 | Form of Change in Control Severance Agreement with certain officers (12) | |||
| 10.29 | Form of Change in Control Severance Agreement with (Thomas A. Mitro and Kirk McMullin.) (12) | |||
| 10.30 | Note and Warrant Purchase Agreement between the registrant and Investors, dated September 19, 2002 | |||
| 10.31 | Form of Senior Secured Convertible Promissory Note issued under the Note and Warrant Purchase Agreement (13) | |||
| 10.32 | Form of Warrant issued under the Note and Warrant Purchase Agreement (13) | |||
| 10.33 | Common Stock and Warrant Purchase Agreement between the registrant and Investors, dated September 19, 2002 | |||
| 10.34 | Form of Warrant to be issued under the Common Stock and Warrant Purchase Agreement (13) | |||
| 10.35 | Individual Non-Qualified Stock Option Agreement between Thomas A. Mitro and the registrant, dated July 1, 2002 (14) | |||
| 10.36 | Individual Non-Qualified Stock Option Agreement between Kirk McMullin and the registrant, dated August 5, 2002 (14) | |||
| 10.37 | Bausch & Lomb Pharmaceuticals, Inc. Contract Manufacturing Supply Agreement with registrant, dated November 25, 2002 (1), (6) | |||
| 10.38 | Bausch & Lomb Pharmaceuticals, Inc. Contract Manufacturing Supply Agreement with registrant, dated February 6, 2003 (1), (6) | |||
| 10.39 | License Agreement between the Eastern Virginia School of Medicine, and AcSentient, Inc., dated as of January 29, 2002 (6), (15) | |||
| 10.40 | Addendum between the Eastern Virginia School of Medicine, and AcSentient, Inc., dated as of April 30, 2002 (6), (15) | |||
| 10.41 | Letter regarding consent to assignment of License Agreement from AcSentient, Inc. to the Eastern Virginia School of Medicine, dated April 26, 2002 (15) | |||
24
Table of Contents
| Exhibit Number | Description | |||
| 10.42 | License Agreement between Senju Pharmaceutical Co., Ltd. and AcSentient, Inc., dated March 7, 2002 (6), (15) | |||
| 10.43 | Agreement between Senju Pharmaceutical Co., Ltd. and AcSentient, Inc., dated April 17, 2002 (6), (15) | |||
| 10.44 | Series D Preferred Stock Purchase Agreement between Allergan Pharmaceuticals (Ireland) Ltd., Inc., and registrant, dated as of March 29, 2000 (3) | |||
| 10.45 | Amendment to Bromfenac License Agreement between Senju Pharmaceutical Co., Ltd., and registrant, dated August 13, 2002 (6), (15) | |||
| 10.46 | Amendment to Timolol Agreement between Senju Pharmaceutical Co., Ltd., and registrant, dated August 13, 2002 (6), (15) | |||
| 21.1 | Subsidiaries of the registrant (1) | |||
| 99.1 | President and Chief Executive Officer’s Certification Pursuant to Section 1350, Chapter 63 of Title 18 of the United States Code, as adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |||
| 99.2 | Chief Financial Officer and Vice President, Corporate Development’s Certification Pursuant to Section 1350, Chapter 63 of Title 18 of the United States Code, as adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |||
| (1) | Incorporated by reference to the registrant’s report on Form 10-K for the year ended December 31, 2002, filed with the Commission on March 7, 2003. | |
| (2) | Incorporated by reference from the registrant’s report on Form 8-K filed with the Commission on April 9, 2003. | |
| (3) | Incorporated by reference to the registrant’s Registration Statement on Form S-1 (File No. 333-34120). | |
| (4) | Incorporated by reference to the registrant’s report on Form 8-A filed with the Commission on January 22, 2002. | |
| (5) | Incorporated by reference from the registrant’s Registration Statement on Form 8-A12G/A filed with the Commission on November 19, 2002. | |
| (6) | Confidential treatment requested as to certain portions. | |
| (7) | Incorporated by reference to the registrant’s report on Form 10-K for the year ended December 31, 2000, filed with the Commission on March 30, 2001. | |
| (8) | Incorporated by reference from the registrant’s report on Form 8-K filed with the Commission on January 2, 2002. | |
| (9) | Incorporated by reference from the registrant’s report on Form 10-K for the year ended December 31, 2001, filed with the Commission on April 1, 2002. | |
| (10) | Incorporated by reference from the registrant’s report on Form 8-K filed with the Commission on May 6, 2002. | |
| (11) | Incorporated by reference from the registrant’s report on Form 8-K filed with the Commission on July 9, 2002. | |
| (12) | Incorporated by reference from the registrant’s report on Form 10-Q for the period ended June 30, 2002 filed with the Commission on August 14, 2002. | |
| (13) | Incorporated by reference from the registrant’s report on Form 8-K filed with the Commission September 25, 2002. |
25
Table of Contents
| (14) | Incorporated by reference from the registrant’s Registration Statement on Form S-8 filed with the Commission on February 18, 2003. | |
| (15) | Incorporated by reference from the registrant’s report on Form 10-K/A for the period ended December 31, 2002 filed with the Commission on April 30, 2003. |
26
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Amendment No. 2 to its Annual Report on Form 10-K/A to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Irvine, State of California, on May 14, 2003.
| By: | /s/ VICENTE ANIDO, JR. | |
| Vicente Anido, Jr., Ph.D. | ||
| President and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this Form 10-K has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ VICENTE ANIDO, JR. | ||||
| President, Chief Executive Officer and Director | May 14, 2003 | |||
| Vicente Anido, Jr., Ph.D. | ||||
| /s/ LAUREN P. SILVERNAIL | Chief Financial Officer and Vice President, | |||
| Corporate Development | May 14, 2003 | |||
| Lauren P. Silvernail | ||||
| /s/ ROBERT G. MCNEIL * | ||||
| Chairman of the Board | May 14, 2003 | |||
| Robert G. McNeil, Ph.D. | ||||
| /s/ JEFFREY L. EDWARDS * | ||||
| Director | May 14, 2003 | |||
| Jeffrey L. Edwards | ||||
| /s/ BENJAMIN F. MCGRAW III * | ||||
| Director | May 14, 2003 | |||
| Benjamin F. McGraw III | ||||
| /s/ PETER BARTON HUTT * | ||||
| Director | May 14, 2003 | |||
| Peter Barton Hutt | ||||
| /s/ KATHLEEN D. LAPORTE * | ||||
| Director | May 14, 2003 | |||
| Kathleen D. LaPorte | ||||
| /s/ LIZA PAGE NELSON * | ||||
| Director | May 14, 2003 | |||
| Liza Page Nelson | ||||
| /s/ RICHARD C. WILLIAMS * | ||||
| Director | May 14, 2003 | |||
| Richard C. Williams | ||||
| /s/ WAYNE I. ROE * | ||||
| Wayne I. Roe | Director | May 14, 2003 |
| *By: | /s/ VICENTE ANIDO, JR. |
| Vicente Anido, Jr. (Attorney-in-fact) | |
27
Table of Contents
CERTIFICATION OF CHIEF EXECUTIVE OFFICER
I, Vicente Anido, Jr., Ph.D., certify that:
1. I have reviewed this annual report on Form 10-K, as amended, of ISTA Pharmaceuticals, Inc.;
2. Based on my knowledge, this annual report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this annual report;
3. Based on my knowledge, the financial statements, and other financial information included in this annual report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this annual report;
4. The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-14 and 15d-14) for the registrant and have:
| a. designed such disclosure controls and procedures to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this annual report is being prepared; | ||
| b. evaluated the effectiveness of the registrant’s disclosure controls and procedures as of a date within 90 days prior to the filing date of this annual report (the “Evaluation Date”); and | ||
| c. presented in this annual report our conclusions about the effectiveness of the disclosure controls and procedures based on our evaluation as of the Evaluation Date; |
5. The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation, to the registrant’s auditors and the audit committee of registrant’s board of directors (or persons performing the evaluation functions):
| a. all significant deficiencies in the design or operation of internal controls which could adversely affect the registrant’s ability to record, process, summarize and report financial data and have identified for the registrant’s auditors any material weaknesses in internal controls; and | ||
| b. any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal controls; and |
6. The registrant’s other certifying officer and I have indicated in this annual report whether there were significant changes in internal controls or in other factors that could significantly affect internal controls subsequent to the date of our most recent evaluation, including any corrective actions with regard to significant deficiencies and material weaknesses.
Date: May 14, 2003
/s/ VICENTE ANIDO, JR., Ph.D
Vicente Anido, Jr., Ph.D.
President and Chief Executive Officer
28
Table of Contents
CERTIFICATION OF CHIEF FINANCIAL OFFICER
I, Lauren P. Silvernail, certify that:
1. I have reviewed this annual report on Form 10-K, as amended, of ISTA Pharmaceuticals, Inc.;
2. Based on my knowledge, this annual report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this annual report;
3. Based on my knowledge, the financial statements, and other financial information included in this annual report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this annual report;
4. The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-14 and 15d-14) for the registrant and have:
| a. designed such disclosure controls and procedures to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this annual report is being prepared; | ||
| b. evaluated the effectiveness of the registrant’s disclosure controls and procedures as of a date within 90 days prior to the filing date of this annual report (the “Evaluation Date”); and | ||
| c. presented in this annual report our conclusions about the effectiveness of the disclosure controls and procedures based on our evaluation as of the Evaluation Date; |
5. The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation, to the registrant’s auditors and the audit committee of registrant’s board of directors (or persons performing the evaluation functions):
| a. all significant deficiencies in the design or operation of internal controls which could adversely affect the registrant’s ability to record, process, summarize and report financial data and have identified for the registrant’s auditors any material weaknesses in internal controls; and | ||
| b. any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal controls; and |
6. The registrant’s other certifying officer and I have indicated in this annual report whether there were significant changes in internal controls or in other factors that could significantly affect internal controls subsequent to the date of our most recent evaluation, including any corrective actions with regard to significant deficiencies and material weaknesses.
Date: May 14, 2003
/s/ Lauren P. Silvernail
Lauren P. Silvernail
Chief Financial Officer and Vice
President, Corporate Development
Lauren P. Silvernail
Chief Financial Officer and Vice
President, Corporate Development
29