substance called globotriaosylceramide, or Gb3, in blood vessel walls throughout their body. The ultimate consequences of Gb3 deposition range from episodes of pain and impaired peripheral sensation to end-organ failure, particularly of the kidneys, but also of the heart and the cerebrovascular system. Fabry disease occurs in one person per 40,000 to 60,000 males.
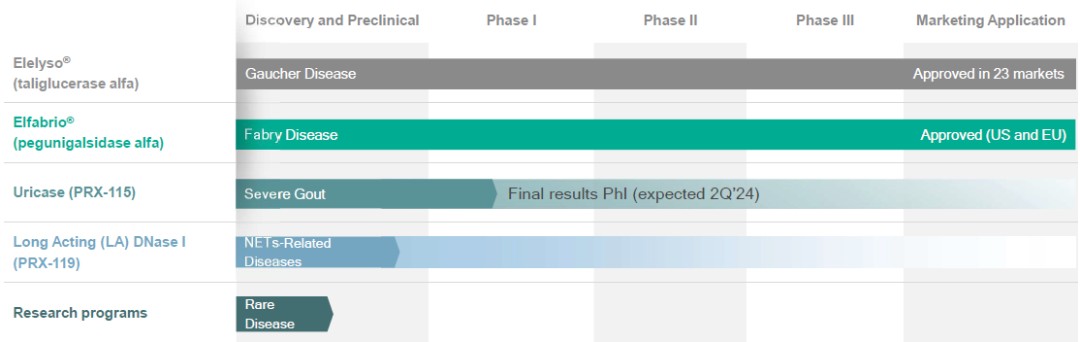
The global market for Fabry disease, that includes Sanofi’s Fabrazyme®, Shire’s (acquired by Takeda Pharmaceutical Company Limited) Replagal®, and Amicus Therapeutics’ Galafold®, among others, was approximately $2.0 billion in 2022, is forecasted to be approximately $2.0 billion in 2023 and is forecasted to grow at a CAGR of approximately 13% from 2022-2028.
In preclinical studies, PRX-102 showed significantly longer half-life due to higher enzyme stability, enhanced activity in Fabry disease affected organs leading to reduction of the accumulated substrate and reduced immunogenicity, which together can potentially lead to improved efficacy through increased substrate clearance and significantly lower formation of anti-drug antibodies, or ADAs. In 2015, we completed a phase I/II clinical trial of PRX-102, which was a worldwide, multi-center, open-label, dose ranging study designed to evaluate the safety, tolerability, pharmacokinetics, immunogenicity and efficacy parameters of PRX-102 in adult patients with Fabry disease. Our phase III clinical program, which has now been completed, included the following three separate studies:
The BALANCE study (PB-102-F20), a pivotal 24-month, randomized, double blind, active control study of PRX-102 in adult Fabry patients with deteriorating renal function that was designed to evaluate the safety and efficacy of 1 mg/kg of PRX-102 administered every two weeks compared to agalsidase beta;
The BRIDGE study (PB-102-F30), a 12-month open-label, single arm switch-over study evaluating the safety and efficacy of PRX-102\, 1 mg/kg infused every two weeks, in up to 22 Fabry patients previously treated with agalsidase alfa for at least two years and on a stable dose for at least six months; and
The BRIGHT study (PB-102-F50), a multicenter, multinational open-label, switch-over study designed to evaluate the safety, efficacy and pharmacokinetics of treatment with 2 mg/kg of PRX-102 administered every four weeks for 52 weeks (a total of 14 infusions). The 2 mg/kg every four weeks dosage was not approved by the EMA or the FDA.
Patients who completed the BALANCE, BRIDGE and BRIGHT studies, and the extension of the phase I/II study, were offered the opportunity to continue PRX-102 treatment in one of two long-term open-label extension studies. Overall, 126 subjects who participated in our PRX-102 clinical program have opted, with the advice of the treating physician, to enroll in one of our long-term, open label, extension studies of PRX-102. Such extension studies include 97 patients in the 1 mg/kg every two weeks extension study (PB-102-F60) with a total cumulative exposure of approximately 480 patient years (10 subjects who completed an extension study from the phase I/II study, 18 subjects who completed the BRIDGE study; 69 subjects who completed the BALANCE study), and 29 subjects who completed the BRIGHT study, in the 2 mg/kg every four weeks extension study (PB-102-F51) with a total cumulative exposure of approximately 145 patient years. Two of such subjects are being treated with 1 mg/kg every two weeks. As of March 1, 2023, sponsorship of the two extension studies was transferred to Chiesi, and Chiesi is now administering the open-label extension studies.
A BLA for Elfabrio for the treatment of adult patients with Fabry disease was first submitted to the FDA on May 27, 2020 under the FDA’s Accelerated Approval pathway, but resulted in a CRL. The BLA was resubmitted to the FDA on November 9, 2022.
The MAA was submitted to the EMA on February 7, 2022, after the October 8, 2021 meeting we held, together with Chiesi, with the Rapporteur and Co-Rapporteur of the EMA regarding PRX-102.
The FDA publicly released the internal review documents for Elfabrio (pegunigalsidase alfa-iwxj) injection BLA 761161. These documents provide previously unavailable additional information regarding the basis for the FDA’s May 2023 approval decision. In particular, the FDA determined that substantial evidence of effectiveness for Elfabrio in Fabry patients was established with one adequate and well-controlled study (Study PB-102-F01/02) with confirmatory evidence provided by the BALANCE study (also referred to as Study PB-102-F20). The FDA review team also concluded that the BALANCE study met its primary efficacy endpoint, which assessed the annualized rate of change in eGFR (estimated glomerular filtration rate) over 104 weeks.
However, the FDA also determined that the results from the BALANCE study did not support a non-inferiority claim to the comparator product due to the lack of data to support a non-inferiority margin.
In February 2020, we, together with Chiesi, announced an agreement with the FDA for the Initial Pediatric Study Plan (iPSP) for PRX-102, which is intended to be initiated post-marketing approval. The joint announcement was made after completion of discussions with the FDA and receipt of confirmation from the FDA in an official “Agreement Letter” which outlines an agreed-upon