UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
Form 10-K
| | | |
| (Mark One) | | |
þ | | ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | | For the fiscal year ended: December 31, 2008 |
o | | TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
COMMISSION FILE NUMBER000-23017
ECHO THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
| | | |
| DELAWARE | | 41-1649949 |
(State or other jurisdiction of
incorporation or organization) | | (I.R.S. Employer
Identification No.) |
| | | |
| 10 Forge Parkway, Franklin, Massachusetts | | 02038 |
| (Address of principal executive offices) | | (Zip Code) |
(Registrant’s telephone number, including area code)
(508) 553-8850
Securities registered pursuant to Section 12(b) of the Act:
None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $.01 par value per share
Indicate by check mark if the registrant is well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 ofRegulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of thisForm 10-K or any amendment to thisForm 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule12b-2 of the Exchange Act. (Check one):
| | | | | | | |
Large accelerated filer o | | Accelerated filer o | | Non-accelerated filer o | | Smaller reporting company þ |
| | | | | | | |
| | | (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined inRule 12b-2 of the Act). Yes o No þ
The approximate aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant as of June 30, 2008, based upon the closing price of such stock on that date, was approximately $36,548,000.
The number of shares of the registrant’s common stock outstanding as of April 8, 2009 was 19,741,838.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement (the “Proxy Statement”) to be filed with the Securities and Exchange Commission relative to the issuer’s 2009 Annual Meeting of Shareholders are incorporated by reference into Part III of thisForm 10-K.
ECHO THERAPEUTICS, INC.
2008 Annual Report onForm 10-K
TABLE OF CONTENTS
In this report, the “Company”, “Echo,” “we,” “us,” and “our” refer to Echo Therapeutics, Inc., formerly known as Sontra Medical Corporation. “Common Stock” refers to Echo’s Common Stock, $0.01 par value.
We own or have rights to various copyrights, trademarks and trade names used in our business, including the following: Symphonytm tCGM System, Preludetm SkinPrep System, SonoPrep® System, AzoneTStm and Durhalievetm.
This Annual Report onForm 10-K contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, and Section 27A of the Securities Act of 1933, as amended. For this purpose, any statements contained herein that are not statements of historical fact may be deemed to be forward-looking statements. Without limiting the foregoing, the words “believes,” “anticipates,” “plans,” “expects” and similar expressions are intended to identify forward-looking statements. The important factors discussed under the caption “Risk Factors” in Item 1.A of this report, among others, could cause actual results to differ materially from those indicated by forward-looking statements made herein and presented elsewhere by management. Such forward-looking statements represent management’s current expectations and are inherently uncertain. Investors are warned that actual results may differ from management’s expectations. Echo does not undertake any obligation to update forward-looking statements.
PART I
Overview
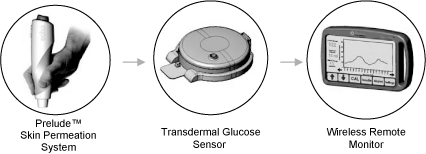
We are a medical device and specialty pharmaceutical company. We are developing a non-invasive, wireless, transdermal continuous glucose monitoring (tCGM) system for use in clinical settings and for people with diabetes together with a wide range of transdermal reformulations of specialty pharmaceutical products previously approved by the United States Food and Drug Administration (FDA).
Our Symphonytm tCGM System is a next-generation, non-invasive (needle-free), wireless tCGM system designed to provide reliable, on-demand blood glucose data conveniently, continuously and cost-effectively. Symphony incorporates our patented feedback mechanism for optimal skin permeation control and our continuous transdermal sensor to detect glucose trends, for controlling complications associated with blood glucose levels that stray outside of a medically recommended target range. With Symphony, we are focused on changing the current standard of care paradigm of invasive (needle-based), episodic blood glucose testing with our needle-free tCGM technology designed to improve patient compliance to frequent glucose testing and achieve better overall glucose control. All existing FDA-approved continuous glucose monitoring (CGM) systems are needle-based, requiring insertion of a glucose sensor into the patient’s skin, which gives rise to risks of infection, inflammation or bleeding at the insertion site. Symphony is a non-invasive tCGM system that does not require insertion of its glucose sensor and thus does not give rise to the risks associated with needle-based CGM systems.
Our specialty pharmaceuticals pipeline is based on our proprietary AzoneTStm transdermal drug reformulation technology. We believe that, despite their commercial success in large, chronic disease markets, certain FDA-approved products with safety, efficacyand/or patient comfort and convenience issues that limit or prohibit their full commercial potential are amenable to our AzoneTS reformulation technology focused on improved dermal penetration. We are leveraging our AzoneTS dermal penetration technology to engineer and develop a broad range of novel topical reformulations of commercially successful, FDA-approved products, generally in accordance with the FDA’s Section 505(b)(2) guidelines. Our lead AzoneTS product candidate is Durhalievetm, an AzoneTS topical reformulation of triamcinolone acetonide. Durhalieve is covered by our New Drug Application (NDA) on file with the FDA for treatment of corticosteroid-responsive dermatoses. We believe Durhalieve also has the potential to be a needle-free treatment for keloid scarring.
Symphonytm tCGMSystem
Our lead medical device program is focused on our Symphony tCGM System, a next-generation, non-invasive (needle-free), wireless, transdermal continuous glucose monitoring system designed to provide reliable, real-time blood glucose data conveniently, continuously and cost-effectively. Symphony incorporates our proprietary skin permeation, transdermal sensor, wireless transmission and data display technologies.
3
SymphonytmSkin Permeation Technology
Our skin permeation technologies include both gentle abrasion and an ultrasound device. The key feature of our skin permeation technology is our patented feedback mechanism to achieve optimal and pain-free skin preparation for our transdermal sensing technologies.
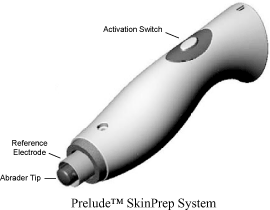
PreludetmSkinPrep System
We are developing our Preludetm SkinPrep System as a safe, effective, easy-to-use and low-cost transdermal skin permeation device for our Symphony tCGM System to enhance the flow of interstitial fluids and molecules across the protective membrane of the stratum corneum, the outmost protective layer of the skin. Prelude incorporates our patented skin permeation control technology into a comfortable, hand-held device used to prepare a small area of the skin for our non-invasive sensor components of our tCGM system. We have successfully used Prelude in internal studies and a pilot clinical study during calendar 2008 in a diabetes home-use setting (seeSymphony Diabetes Home Use Pilot Studies). We plan to use Prelude in the remaining pilot and pivotal clinical studies necessary to commercialize our Symphony tCGM System.
SonoPrep® System
Our SonoPrep System also is a skin permeation system that incorporates our patented skin permeation control technology and utilizes ultrasound as a skin permeation medium to enhance the flow of interstitial fluids and molecules across the protective membrane of the stratum corneum, to our continuous glucose sensors. Our SonoPrep skin preparation system was used in several pilot clinical studies of our Symphony tCGM System conducted in several hospital critical care studies and diabetes home use studies through March of 2008.
Skin Permeation Control Feedback Technology
The proprietary feedback control mechanism in our skin permeation system consists of software, microprocessor controlled circuit and measuring electrodes. While the system is in operation, the circuit also measures the real-time electrical conductivity of the test skin site. The system turns off automatically when the conductivity measurement reaches the appropriate stopping point as identified by the software, thus producing individualized, optimal skin permeation. As a result, the system only removes the outmost layer of the epidermis, the stratum corneum, a layer of about 0.01 mm in thickness consisting of only dry, dead skin cells. With the advantages of our proprietary feedback control mechanism, we believe the skin permeation process is safe, effective and pain-free.
SymphonytmTransdermal Glucose Sensor Technology
The transdermal glucose sensor component of our Symphony tCGM System consists of an electrochemical glucose sensor, a hydrogel layer, a potentiostat and a short-range RF transmitter. Sensor readings are transmitted to a remote monitor/receiver where data are processed and displayed to the user.
4
When the Symphony sensor is placed on a properly permeated skin site, the hydrogel not only can establish and maintain continuous fluid paths through the skin, but it can also act as a reliable reservoir for the sensing chemistry to receive glucose flux and convert it into hydrogen peroxide. The hydrogen peroxide is consumed by the electrochemical sensor and continuous electrical signal is generated in the process.
The diffusion gradient and sensor consumption of glucose are a function of the blood glucose concentration in the subpapillary vessels beneath the epidermis. As a result, the sensor generates continuous current that is proportional to the blood glucose concentration. The first calibration is made when the sensor stabilizes, followed by subsequent calibrations depending on the applications. Due to our advanced hydrogel chemistry and the well-controlled skin permeation process, the glucose flux detected by the sensor can provide reliable continuous glucose readings for 24 hours.
SymphonytmWireless Transmitter
The Symphony transmitter assembly consists of coupling hydrogel/chemistry, sensor electrodes and integrated electronics. To preserve data integrity and save power, the assembly digitizes, stores and transmits discrete, coded signals via a wireless link to the Symphony receiving monitor. Once the sensor is connected and adhered to the skin, the transmitter switches on automatically and starts to broadcast data once every minute. A medical grade adhesive keeps the transmitter in place and a hermetic seal protects the chemistry and electronics from fluid invasion. Disposable components of the transmitter assembly are the hydrogel and the sensor electrodes. In our current prototype design, our Symphony transmitter is reusable, but we believe it can also be disposable depending on its application. Based upon our pilot clinical studies to date, we believe our Symphony transmitter assembly can be applied to the forearm, upper arm, chest, upper back, abdomen and many other sites on the body with tangible soft tissue.
SymphonytmMonitor
Our Symphony monitor receives digitally coded data from the transmitter assembly and then decodes the data once every minute. Once a reference blood glucose value is entered manually (a calibration) after a one hourwarm-up period, the decoded data are converted to blood glucose reading by a built-in data algorithm. The Symphony monitor displays the date, time of day, sensor current, blood glucose reading and rate of increase or decrease, amount of time the transmitter has been switched on, battery status, and any alarm or error modes.
A membrane key pad allows the user to interface with the monitor. The keys provide for on/off, data entry, calibration, alarm silencing, and monitor resetting. Both the key pad and the LCD display are back lighted to support full operation in the dark. The monitor also contains an audible alarm, a RF receiver, a programming port, a rechargeable battery pack and a USB data port for computerized record keeping or in-depth data analysis. We expect our Symphony monitor to offer several display options to the user, including a graphical presentation of blood glucose level over time, and to allow for programmable alarm thresholds at prescribed blood glucose limits to help the user identify impending hyperglycemia and hypoglycemia.
SymphonytmMarket Opportunities
Importance of Continuous Blood Glucose Monitoring
We believe that continuous blood glucose monitoring can be an important part of a diabetes patient’s daily disease management program. Continuous blood glucose monitoring can help plan diabetes treatment, guide day-to-day choices about diet, exercise and insulin use, and avoid unwanted low blood glucose (hypoglycemia) and high blood glucose (hyperglycemia) events and the complications that they can cause. Blood glucose levels are affected by many factors such as the carbohydrate and fat content of food, exercise, stress, illness, and variability in insulin absorption among others; therefore, it is often challenging for diabetes patients to avoid frequent and unpredictable excursions above or below normal glucose levels. Patients are often unaware that their glucose levels are either too high or too low, resulting in their inability to tightly control their glucose levels and prevent the complications associated with unwanted glucose excursions.
In an attempt to achieve and maintain blood glucose levels within a desired range, diabetes patients must measure their glucose levels. The ADA recommends that patients test their blood glucose levels at least three or four
5
times per day; however, despite evidence that intensive glucose management reduces the long-term complications associated with diabetes, industry sources estimate that people with diabetes test, on average, less than twice per day. We believe our Symphony tCGM System has the potential to improve patient compliance to frequent glucose testing, achieve better glucose control and make a positive impact on overall day-to-day diabetes management.
Hospital Critical Care Market
We believe Symphony has the potential to offer a non-invasive, wireless, tCGM solution for use in the rapidly emerging hospital critical care market. A primary cause of infection in critically ill patients is hyperglycemia which is a result of insulin resistance and total parenteral nutrition. Clinical studies have demonstrated that intensive insulin therapy to maintain tight glycemic control significantly reduces patient mortality, complications and infection rates, as well as hospital stays, services and overall hospital costs.
Regular monitoring of blood glucose levels is rapidly becoming a necessary procedure performed by hospital critical care personnel to achieve tight glycemic control and ensure improved patient outcomes. In a recent survey by Boston Biomedical Consultants of more than 60 hospital critical care unit managers and nurse clinicians in the United States, more than 90% of those surveyed acknowledged the benefits of tight glycemic control protocols in the hospital critical care setting. We believe tight glycemic control protocols are becoming the new standard of care in hospital critical care units across the United States, for patients with and without diabetes.
Today, standard practice by critical care nurses is to measure blood glucose at the patient’s bedside periodically. We believe that a CGM system such as Symphony will save valuable nursing time and expense by avoiding the need for frequent blood glucose sampling, in addition to providing more clinically relevant, real-time glucose level and trending information needed to develop better control algorithms for insulin administration.
SymphonytmHospital Critical Care Pilot Studies
In December 2006, we completed our initial hospital critical care pilot study at the Surgical Critical Care Unit of Tufts Medical Center (Tufts) using our ultrasound skin permeation system on patients for a total of 24 hours, including during and after cardiovascular surgery. There were two phases to this pilot study. The initial phase provided our technical team and Tufts physicians and nurses with valuable hands-on experience with Symphony and highlighted necessary technical and clinical implementation modifications. After making these modifications, we completed the second phase of the study. In the second phase of the study, a total of 147 reference blood glucose measurements from 8 subjects were collected and analyzed with a proprietary data processing algorithm we developed for our July 2006 diabetes home use study.
The primary statistical tools used to evaluate the performance of Symphony were Clarke Error Grid analysis and Mean Absolute Relative Difference (MARD). The Clarke Error Grid analysis was designed to evaluate the performance of glucometers and is used as an analytical tool to assess performance of continuous glucose monitors. The Clarke Error Grid is a plot of all data pairs categorized into five discrete areas: A, B, C, D and E. The A and B areas are the most clinically desirable zones and D and E are the least clinically desirable zones. Devices with a higher combined A and B percentage (closer to 100%) and lower combined D and E percentage (closer to 0) are considered to have better performance. Monitor performance is generally considered acceptable if at least ninety-five percent (95%) of the data points fall within the A/B region, along with negligible or no D/E points. MARD is an error calculation tool that measures the average relative difference between Symphony and the reference measurements, on a percentage basis. A low MARD error, below 20%, is consistent with an accurate device.
Clarke Error Grid analysis of data from the second phase of the study showed that Symphony had 100% of the data in the combined A and B areas with approximately 86% in the “A” region, 14% in the “B” region, and 0% in the “C” and “D” regions. The MARD for the study was approximately 11%. We believe the results of the second phase of this study showed that, despite significant adverse conditions typically associated with open heart surgery and recovery from such surgery in the complex hospital critical care setting, Symphony accurately and reliably measured blood glucose readings continuously for 24 hours.
In March 2008, we announced results from our second pilot study using our ultrasound-based Symphony system in the hospital critical setting at Tufts. The pilot study was designed to evaluate the performance of
6
Symphony, including our newly-improved biosensor technology incorporating proprietary hydrogel chemistry. The study enrolled 25 adult patients scheduled for elective cardiac surgery. The study included both intra-operative and post-operative continuous monitoring of blood glucose levels. Two biosensors were applied to each subject, one prior to surgery and one after surgery. Both Symphony sensors remained on the patient for 24 hours. Blood glucose levels were monitored per the established protocol of Tufts. The participating subjects and the Tufts medical staff were blinded to data collected by the Symphony monitor. In this trial, the continuous data were compared to reference measurements from blood analyzers, glucometers and lab results based on the Tufts glucose monitoring protocols. Those reference measurements were paired with the Symphony results through a data analysis algorithm.
Using approximately 1200 hours of continuous data from Symphony and 482 reference blood glucose measurements from 25 subjects in the study, Clarke Error Grid analysis of the study data showed that Symphony had over 97% of the data in the combined A and B areas with approximately 70% in the “A” region, 27% in the “B” region, and less than 3% in the “D” region. The MARD for the study was approximately 16%. A total of 49 Symphony sensors were used during the study. There were no Symphony “out-of-the-box” system failures and no adverse events, indicating strong reliability of Symphony for applications in the hospital critical care setting. We believe the objective measurements of the accuracy and clinical relevance of Symphony are equivalent or superior to that of the published clinical results of other FDA-approved, commercialized CGM systems for the 24 hour time period measured.
Diabetes Home Use Market
Diabetes is a chronic and life-threatening disease caused by the body’s inability to produce or properly use insulin, a key hormone the body uses to manage glucose, which fuels the cells in the body. Insulin regulates the uptake of sugar from the blood into the cells. When glucose builds up in the blood instead of going into cells, it can cause the cells to become starved for energy and, over time, damage the eyes, kidneys, nerves or heart. Although not all of the causes of diabetes are known, genetics and lifestyle factors such as obesity and lack of exercise appear to play important roles. According to the American Diabetes Association (ADA), about 21 million people in the United States, or approximately seven percent (7%) of the population, have diabetes, including over 6 million people who remain unaware that they have the disease. In addition, before people develop Type 2 diabetes (discussed below), they usually have “pre-diabetes,” or blood glucose levels that are higher than normal but not yet high enough to be diagnosed as diabetes. According to the ADA, there are 54 million people in the United States who have pre-diabetes.
When blood glucose levels are high, diabetes patients often administer insulin to reduce their blood glucose level. Unfortunately, insulin administration can reduce blood glucose levels below the normal range, causing hypoglycemia. In cases of severe hypoglycemia, diabetes patients risk severe and acute complications, such as loss of consciousness or death. Due to the drastic nature of acute complications associated with hypoglycemia, many diabetes patients are afraid of sharply reducing their blood glucose levels and often remain in a hyperglycemic state, exposing themselves to long-term complications of that condition.
Diabetes is typically classified into two major groups: Type 1 and Type 2. Type 1 diabetes usually develops in children and young adults and is characterized by the body’s inability to produce insulin. People with Type 1 diabetes rely on frequent administration of insulin to regulate their blood glucose levels. Type 2 diabetes, by far the most common form of the disease worldwide, results when either the body does not produce enough insulin or cells in the body ignore the insulin produced and become insulin-resistant. People with Type 2 diabetes often require diet and nutrition management, exercise, oral medications or insulin administration to regulate their blood glucose levels.
According to the ADA, the cost of diabetes care in the United States in 2007 was more than $174 billion, including $116 billion in excess medical expenditures attributed to diabetes and $58 billion in reduced national productivity. The ADA estimates that people with diabetes, on average, have medical expenditures that are approximately 2.3 times higher than the expenditures would be in the absence of diabetes and that approximately $1 in $10 healthcare dollars is attributed to diabetes. A significant portion of overall diabetes care costs, approximately $7 billion according to industry sources, is attributable to costs associated with monitoring blood glucose levels, and that market segment is projected to grow substantially by 2010 as patients and their physicians seek ways to manage glucose levels more effectively.
7
SymphonytmDiabetes Home Use Pilot Studies
We completed our first diabetes home use pilot study in April 2003. The study was conducted using our first generation, ultrasound-based skin permeation system and our first generation glucose flux sensor prototypes. Twenty glucose flux sensors (two per patient) were placed over 20 ultrasound-treated skin sites of 10 adult subjects with Type 1 or Type 2 diabetes. Data were collected for eight to nine hours. As a control, blood glucose was measured from an intravenous catheter or finger-stick blood withdrawn every twenty minutes. Data comparing blood glucose measurements to data from the glucose flux sensor showed a meaningful correlation (average r = 0.84). The accuracy of the data from this study demonstrated the technical feasibility of our first-generation Symphony system in the diabetes home use market. In November 2004, we completed a second pilot study that included 12 adult participants with either Type 1 or Type 2 diabetes with glucose clamping. Three glucose sensors were applied to each participant, allowing over 2,000 glucose measurements to be collected. Data from this study showed a strong correlation (r = 0.90) between our transdermal sensor and reference blood glucose measurements.
In July 2006, we completed our first pilot clinical study using our current generation Symphony system, which includes our second generation SonoPrep® ultrasound-based skin permeation device and improved single-use glucose transdermal sensors, on 10 patients with diabetes at an independent clinic. The sensors were coupled with a miniature analyzer which sent digitized data wirelessly to a monitor for data processing and display. The glucose sensor signal was referenced to finger-stick blood glucose meter readings. A total of 222 data points from this study were collected. The results showed that our transdermal sensor could accurately measure continuous blood glucose readings for 12 hours with a single point calibration after only a one hourwarm-up period.
Since 2006, it has been our goal to develop a low-cost and convenient device to replace SonoPrep as the skin permeation system. In early 2008, Preludetm was developed based on microdermabrasion technology coupled with our patented feedback mechanism to control the skin permeation process. In July 2008, our Symphony system using Prelude was successfully tested on ten (10) patients with diabetes in a clinical research facility simulating home care settings. In this extensive24-hour study the patients’ venous blood samples were taken from intravenous lines at15-minute intervals and were used to obtain reference blood glucose (BG) values with YSI glucose analyzers. A total of 17 Symphony datasets and 1274 reference BG samples were analyzed. It was found from this study that the overall MARD was 13.8%. Clarke Error Grid Analysis showed 98.8% in zones A+B with 77.4% in A and 0.5% and 0.7% in zones C and D respectively. No pain, skin irritation or other adverse events were observed during the study. From this study we have demonstrated that needle-free, transdermal continuous glucose monitoring can be achieved using Prelude as the skin permeation system. Performance of Symphony tCGM was comparable to published performance metrics for currently marketed CGM devices.
We believe that incorporating our Preludetm SkinPrep System as the skin permeation component of our Symphony system will offer diabetes patients a painless easy-to-use and cost effective solution for frequently monitoring of their blood glucose levels. Our sensor technology advances for the diabetes home use market are expected to include simplified application, extended use life and significantly improved overall ease of use. We expect these advancements to reduce system costs are needed for applicable reimbursement models.
Specialty Pharmaceuticals
Skin is composed of three primary layers: the epidermis, an outermost layer which protects the body from the environment; the dermis, a middle layer which regulates temperature and provides the epidermis with blood and nutrients; and the innermost subcutaneous layer, which is primarily composed of fat and collagen. Individual components and layers of the skin (the dermis, epidermis and subcutaneous) present different degrees of resistance to the diffusion of drug molecules. For example, the stratum corneum, which is the outermost portion of the epidermis, is highly keratinized, and is responsible for keeping water in the body and keeping other harmful chemicals and pathogens out, giving skin its property of being a natural barrier to infection. However, this also provides a challenge to getting typical small molecule drugs across the outermost layers of the skin and into the blood.
Successful transdermal absorption of small molecule drugs encompasses a complex series of processes through which the drug moves from an applied formulation on the surface of the skin, through its successively deeper layers into the vasculature and capillary circulation of the dermis and subcutaneous layers. By reversibly
8
disrupting the barrier function of the stratum corneum, penetration enhancers allow drug molecules to diffuse more readily through the stratum corneum into viable layers of the skin, so that they can find their way into the capillary circulation.
Rather than acting as a drug carrier, non-polar enhancers such as our AzoneTS preferentially partition into lipid regions of the skin, disrupting and fluidizing these structures. We believe AzoneTS increases the permeability of the skin, possibility by partitioning directly into lipid bilayers, as well as the intercellular lipid layers of the stratum corneum.
AzoneTStmTransdermal Drug Reformulation Technology
AzoneTS is a nontoxic, nonirritating skin penetration enhancer that is intended to enable topical application of FDA-approved prescription medications, including pharmaceutical products that previously could only be administered systemically. AzoneTS increases lipid membrane fluidity in the stratum corneum, thereby decreasing the diffusion resistance to topically applied therapeutics. AzoneTS is a chemical combination of pyrolidone and decylmethyl sulfoxide, both of which alone act as weak penetration enhancers. Our proprietary synergistic chemical combination enables AzoneTS to be a highly effective skin penetration enhancer at low concentration levels. When combined with AzoneTS, we believe the penetration of numerous commercially successful, FDA-approved drugs can be substantially improved from two to more than twenty fold. We believe that, despite their commercial success in large, chronic markets, many FDA-approved products with safety, efficacyand/or patient comfort and convenience issues that limit or prohibit their full commercial potential are amenable to our AzoneTS reformulation technology focused on improved dermal penetration.
We expect that our AzoneTS new product opportunities will be AzoneTS reformulations of FDA-approved products or active pharmaceutical ingredients (APIs), and, when developed pursuant to the FDA’s Section 505(b)(2) guidelines, will have a significantly reduced nonclinical testing burden compared to new chemical entities (NCEs) developed under the FDA’s Section 505(b)(1) requirements. We expect to be able to leverage our extensive AzoneTS nonclinical Drug Master File (DMF), as well as the established safety database of the FDA-approved products we reformulate with AzoneTS. We believe the main nonclinical safety studies that the FDA will require us to perform for our AzoneTS reformulation product candidates under a Section 505(b)(2) will be pharmacokinetic studies and local and eye irritancy studies. However, additional studies may be required.
There are several factors that will ultimately determine the cost and time associated with developing new AzoneTS-based products. These include:
| | |
| | • | ease of reformulation of the API with AzoneTS; |
| |
| | • | how close the current route of administration is to the proposed route; |
| |
| | • | the level of additional toxicology information required by the FDA; and |
| |
| | • | clinical burden needed to show adequate safety and efficacy of the new product. |
Durhalievetm
Our most advanced AzoneTS drug candidate is Durhalieve, an AzoneTS formulation of triamcinolone acetonide, a widely-used, medium potency corticosteroid approved by the FDA for treatment of corticosteroid-responsive dermatoses. We have filed a NDA with the FDA seeking market approval for Durhalieve for treatment of corticosteroid-responsive dermatoses. We are now working to satisfy the FDA’s remaining manufacturing requirements necessary to secure FDA approval of Durhalieve. In addition to Durhalieve for the treatment of corticosteroid-responsive dermatoses, we are preparing to file an Investigational New Drug (IND) application with the FDA to initiate Phase 2 clinical development of Durhalieve as a treatment for keloid scarring.
If Durhalieve is approved by the FDA for treatment of corticosteroid-responsive dermatoses, we believe we will be well-positioned to offer the pharmaceutical industry a highly efficient and relatively low cost, advanced topical alternative for several FDA-approved oral and injectable specialty pharmaceutical products.
9
AzoneTS Section 505(b)(2) Development Strategy
We are leveraging our AzoneTS transdermal drug reformulation technology to improve the dermal penetration of a wide variety of FDA-approved products in accordance with the FDA’s Section 505(b)(2) guidelines. Our Section 505(b)(2) development strategy for our AzoneTS drug candidates is based on our belief that the existence of adequate nonclinical and well-controlled clinical data regarding each FDA-approved product reformulated with AzoneTS will enable us to forego substantially all costly and time-consuming nonclinical and Phase 1 clinical development activities typically associated with development of a new chemical entity and allow us to enter directly into Phase 2 clinical development. Accordingly, we believe our strategic focus on securing FDA approval of Durhalieve and leveraging that approval and our AzoneTS reformulation platform to develop improved transdermal formulations of well-developed and widely-used FDA-approved products could significantly reduce the time, cost and clinical risk typically associated with development and commercialization of new chemical entities.
The benefits of a 505(b)(2) regulatory and development strategy include:
| | |
| | • | smaller patient exposure database; |
| |
| | • | reliance upon the established safety database of the API’s; |
| |
| | • | reduction of data to support efficacy; |
| |
| | • | minimum requirement of one adequate and well-controlled study compared to two for a NCE; |
| |
| | • | limited if any requirement for additional nonclinical studies; |
| |
| | • | reliance upon previous findings of nonclinical safety for the API’s and the AzoneTS Type V nonclinical DMF; and |
| |
| | • | 3-year period of marketing exclusivity (in addition to existing patent protection) if the NDA contains reports of new clinical studies other than bioavailability. |
Additional AzoneTS Drug Candidates
We are conducting reformulation feasibility studies involving several additional AzoneTS drug candidates. We plan to submit an IND application for clinical development of each AzoneTS drug candidate that successfully completes our reformulation feasibility program.
4/48 Partnering Program
We believe our AzoneTS platform offers the pharmaceutical industry a sustainable source of clinical stage product candidate opportunities. We intend to leverage our AzoneTS platform and our strategic relationship with Cato Research, a global contract research organization, to develop and commercialize a broad pipeline of new drug candidates through internal programs and strategic collaborations with leading pharmaceutical and biotechnology companies. Our AzoneTS 4/48 Program contemplates a product candidate pipeline-to-NDA development model including development costs of approximately $4 million and total development time of approximately 48 months in accordance with the FDA’s Section 505(b)(2) guidelines.
Prelude for Transdermal Drug Delivery
We believe our Preludetm SkinPrep System will have potential to be positioned in the transdermal drug delivery market. We expect the localized disruption of the stratum corneum created by the Prelude System will potentially allow transdermal drug delivery, possibly including drugs that have low and high molecular weights. In addition, Prelude has the potential to provide a safe and cost effective skin permeation process for the delivery of topical anesthetic creams to allow rapid delivery of medications across the epidermis. We believe our Prelude skin permeation process has the potential to increase skin permeation up to 100 times greater than untreated skin, perhaps making it possible to deliver a wide array of large molecule drugs.
10
SonoPrep for Transdermal Drug Delivery
In February 2004, we received 510(k) marketing clearance from the FDA for use of our ultrasound-based SonoPrep skin permeation system in electrophysiology applications. In August 2004, we received 510(k) marketing clearance from the FDA to market SonoPrep for use with over-the-counter 4% topical lidocaine for dermal anesthesia. In the third quarter of 2007, we determined that SonoPrep, as a stand-alone product opportunity, was not commercially relevant to our long-term strategic plan. Therefore, we ceased actively marketing and selling the product. Although we are not currently marketing SonoPrep for its FDA-cleared indication, SonoPrep remained a relevant skin permeation device during 2008 for certain clinical studies, exclusively relating to continuous glucose monitoring.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, promotion, advertising, distribution, marketing and export and import of products such as those we are developing. In the United States, pharmaceuticals, biologics and medical devices are subject to rigorous FDA regulation. Federal and state statutes and regulations govern, among other things, the testing, manufacture, safety, efficacy, labeling, storage, export, record keeping, approval, marketing, advertising and promotion of our potential products. The information that must be submitted to the FDA in order to obtain approval to market a new drug varies depending on whether the drug is a new product whose safety and effectiveness has not previously been demonstrated in humans, or a drug whose active ingredient(s) and certain other properties are the same as those of a previously approved drug. Drugs follow either the new drug application or biologics license application routes. Product development and approval within this regulatory framework takes a number of years and involves significant uncertainty combined with the expenditure of substantial resources.
FDA Approval Process for Medical Devices
Generally, medical devices require FDA approval or clearance before they may be marketed. There are two review procedures by which a product may receive such approval or clearance. Some products may qualify for clearance under a pre-market notification, or 510(k) procedure, in which the manufacturer provides to the FDA a pre-market notification that it intends to begin marketing the product, and demonstrates to the FDA’s satisfaction that the product is substantially equivalent to a legally marketed device. A product is considered substantially equivalent if it has the same intended use, and also has either the same technological characteristics (as defined in the Food, Drug and Cosmetic Act), or if the product has different technological characteristics, the information submitted in the pre-market notification demonstrates that the product is as safe and effective as a legally marketed device and does not raise different questions of safety and effectiveness than a legally marketed device. Marketing may commence when the FDA issues a clearance letter. If a medical device does not qualify for the 510(k) procedure, the FDA must approve a pre-market approval application (PMA) before marketing can begin. PMA applications must demonstrate, among other matters, that the medical device is safe and effective. The PMA approval process is typically more comprehensive than the 510(k) process, and usually requires pre-clinical and extensive clinical studies. Further, before the FDA will approve a PMA, the manufacturer must pass an inspection demonstrating its compliance with the requirements of the FDA’s quality system regulations. FDA requests for additional studies during the review period are not uncommon and can significantly delay approvals.
In addition, a number of other FDA requirements apply to medical device manufacturers and distributors. Device manufacturers must be registered and their products listed with the FDA and certain adverse events and product malfunctions must be reported to the FDA. The FDA also prohibits an approved or cleared device from being marketed for unapproved or uncleared uses. Our product labeling, promotion and advertising will be subject to continuing FDA regulation. Manufacturers must comply with the FDA’s quality system regulation, which establishes extensive requirements for quality control and manufacturing procedures. The FDA periodically inspects facilities to ascertain compliance with these and other requirements. Thus, manufacturers and distributors must continue to spend time, money and effort to maintain compliance. Failure to comply with the applicable regulatory requirements may subject us to a variety of administrative and judicially imposed sanctions, including withdrawal of an approval or clearance, warning letters, product recalls, product seizures, total or partial suspension
11
of production or distribution, injunctions, and civil and criminal penalties against us or our officers, directors or employees. Failure to comply with regulatory requirements could have a material adverse effect on our business, financial condition and results of operations.
In order to obtain marketing clearance for our Symphony tCGM System, we will be required to file a PMA that demonstrates the safety and effectiveness of the product.
FDA Approval Process for Pharmaceuticals Products
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FD&CA, and implementing regulations. The steps required before a drug may be marketed in the United States include:
| | |
| | • | completion of preclinical laboratory tests, animal studies and formulation studies under the FDA’s current good laboratory practices; |
| |
| | • | submission to the FDA of an investigational new drug for human clinical testing, which must become effective before human clinical trials may begin and must include independent institutional review board approval at each clinical site before the trial is initiated; |
| |
| | • | performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product for each indication; |
| |
| | • | submission to the FDA of a new drug application; |
| |
| | • | satisfactory completion of an FDA Advisory Committee review, if applicable; |
| |
| | • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current Good Manufacturing Practices (GMP), or current GMP, to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
| |
| | • | FDA review and approval of the new drug application. |
Preclinical tests include laboratory evaluations of product chemistry, toxicity, and formulation, as well as animal studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of the investigational new drug. An investigational new drug will automatically become effective 30 days after receipt by the FDA, unless before that time, the FDA raises concerns or questions about issues such as the conduct of the trials as outlined in the IND application. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. Submission of an IND application may not result in the FDA allowing clinical trials to commence.
Clinical trials involve the administration of the investigational product to human subjects under specific protocols and the supervision of qualified investigators. Each clinical protocol must be submitted to the FDA as part of the IND application, and an institutional review board at each site where the study is conducted must also approve the proposed study.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined. Phase 1 trials usually involve the initial introduction of the investigational drug into humans to evaluate the product candidate’s safety, dosage tolerance and pharmacodynamics and, if possible, to gain an early indication of its effectiveness. Phase 2 trials usually involve controlled trials in a limited patient population to evaluate dosage tolerance and appropriate dosage, identify possible adverse effects and safety risks and evaluate the preliminary efficacy of the drug candidate for specific indications. Phase 3 trials usually further evaluate clinical efficacy and test further for safety in an expanded patient population. Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within any specified period, if at all. Furthermore, the FDA or we may suspend or terminate clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
Assuming successful completion of the required clinical testing, the results of the preclinical studies and of the clinical studies, together with other detailed information, including information on the chemistry, manufacture and
12
control criteria of the product, are submitted to the FDA in the form of a new drug application or biologics license application requesting approval to market the product candidate for one or more indications. The FDA reviews a new drug application to determine, among other things, whether a product candidate is safe and effective for its intended use and whether its manufacturing is current GMP-compliant to assure and preserve the product candidate’s identity, strength, quality and purity. The FDA reviews a biologics license application to determine, among other things, whether the product candidate is safe, pure and potent and the facility in which it is manufactured, processed, packed or held meets standards designed to assure the product candidate’s continued safety, purity and potency. The FDA may refuse to accept and review insufficiently complete applications.
Before approving a NDA, the FDA will inspect the facility or the facilities at which the product candidate is manufactured. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable; it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The FDA may limit the indications for use or place other conditions on any approvals that could restrict the commercial application of the products. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval.
Post-Approval Requirements
If we obtain regulatory approval of a product candidate, we will be required to comply with a number of post-approval requirements. For example, as a condition of approval of a new drug application, the FDA may require post-marketing testing and surveillance to monitor the product’s safety or efficacy.
In addition, holders of an approved NDA are required to report certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling for their products. Also, quality control and manufacturing procedures must continue to conform to current GMP after approval. The FDA periodically inspects manufacturing facilities to assess compliance with current GMP, which imposes certain procedural, substantive and recordkeeping requirements. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with current good manufacturing practice and other aspects of regulatory compliance. We use, and will continue to use in at least the near term, third-party manufacturers to produce our products in clinical and commercial quantities. Future FDA inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution or require substantial resources to correct.
Discovery of problems with a product or failure to comply with the applicable FDA requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include a clinical hold on or termination of studies, the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, restriction on marketing, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Also, new government requirements may be established that could delay or prevent regulatory approval of our products under development.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements
13
governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Related Matters
From time to time, federal legislation is drafted, introduced and passed in the United States that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or FDA regulations, guidance or interpretations changed, or what the impact of such changes, if any, may be.
Research and Development
A major portion of our operating expenses to date is related to our research and development (R&D) activities. R&D expenses generally consist of internal salaries and related costs, independent R&D costs, costs associated with collaborative R&D and in-licensing arrangements. A substantial portion of our development expense is third party expense relating to product engineering and feasibility testing and conducting nonclinical and clinical trials. R&D expenses were approximately $3,191,000 and $1,306,000, for the years ended December 31, 2008 and 2007, respectively. In connection with the acquisition of Durham Pharmaceuticals Ltd. (d/b/a Echo Therapeutics, Inc.) in September 2007, we purchased R&D in the amount of approximately $6,995,000. We intend to maintain our strong commitment to R&D as an essential component of our product development efforts. Licensed or acquired technology developed by third parties is an additional source of potential products.
Manufacturing
We do not have, and do not intend to establish in the near term, any of our own manufacturing capability for our medical device products, or pharmaceutical product candidates, or their active pharmaceutical ingredients, or the capability to package any products we may sell in the future. We intend to rely on a number of third-party manufacturers and suppliers to manufacture our medical devices and to produce and supply the active pharmaceutical ingredients and drug products that we require to meet the preclinical research and clinical study requirements.
We currently do not have long-term supply contracts with any of our third-party manufacturers and suppliers. Our third-party manufacturers are subject to the FDA’s current GMP requirements and other rules and regulations prescribed by domestic and foreign regulatory authorities. We depend on our third-party suppliers and manufacturers for continued compliance with current GMP requirements and applicable foreign standards.
If our product candidates are approved, we believe that qualified suppliers and manufacturers for such medical device products and pharmaceutical product candidates will be available in the future, at a reasonable cost to us, although there can be no assurance that this will be the case.
Competition
The medical device and pharmaceutical industries are intensely competitive. Many companies, universities, and research organizations developing competing product candidates have greater resources and significantly greater experience in financial, research and development, manufacturing, marketing, sales, distribution, and technical regulatory matters than we have. In addition, many competitors have greater name recognition and more extensive collaborative relationships. Our competitors could commence and complete clinical testing of their product candidates, obtain regulatory approvals, and begin commercial-scale manufacturing of their products faster than we are able to for our products. They could develop products that would render our product candidates, and those of our collaborators, obsolete and noncompetitive. Our competitors may develop more effective or more affordable products or achieve earlier patent protection or product commercialization than we do.
We expect that any products that we develop will compete primarily on the basis of product efficiency, safety, patient convenience, reliability, availability and price. However, there can be no assurance that we will successfully
14
develop technologies and products that are more effective, safer, more convenient, more reliable, more available or more affordable than those being developed by its current and future competitors. Please see “Risks related to competition” in Item 1A of this Report.
Intellectual Property
We maintain a comprehensive portfolio of intellectual property that we consider to be of material importance in protecting our technologies. We have pursued a course of developing and acquiring patents and patent rights and licensing technology. Our success depends primarily on our ability to establish and maintain the proprietary nature of our technology through the patent process and to license third-party patents and patent applications necessary to develop our products. In order to protect our proprietary technologies, we also rely on a combination of trademark, copyright and trade secret protection, as well as confidentiality agreements with employees, consultants and third parties.
Our intellectual property covers skin permeation control for enhanced drug delivery and transdermal diagnostics in the field of medical devices, and reformulations of topical products in the field of specialty pharmaceuticals. These patents provide significant protection from new entrants, and we focus our patent coverage only on aspects of our technologies that we believe will be significant and that could provide barriers to entry for our competition worldwide. Our success depends to a significant degree upon our ability to develop proprietary products and technologies and to obtain patent coverage for such products and technologies. We intend to file patent applications covering newly developed products and technologies.
Pursuant to a license agreement entered into with MIT in June 1998, we had an exclusive, worldwide license to certain patent rights related to the use of ultrasound to enhance skin permeability for applications in transdermal diagnostics and drug delivery. Due to the successful development of Preludetm SkinPrep System and discontinued use of ultrasound for skin permeation in 2008, we terminated this licensing agreement as of January 1, 2009.
Durhalieve’s pharmaceutical formulation and product manufacturing process are protected in the United States by a patent that expires in September 2019. Protection for Durhalieve outside of the United States extends for varying periods in accordance with the legal life of patents in the countries in which they are issued. The protection afforded, which differs from country to country, depends upon the type of patent, its scope of coverage and the availability of legal remedies in that country. This patent and its foreign equivalents also protect several other AzoneTS based products that we are currently developing. In addition to patent protection for our AzoneTS based products, certain intellectual property regarding detailed safety, toxicology and manufacturing for AzoneTS is carefully maintained in Drug Master Files on file with the FDA and not subject to the public domain.
Employees
As of December 31, 2008, we had eleven (11) employees and two (2) independent contractor arrangements with consultants. Of this group of employees and consultants, five (5) are involved with finance and administration and eight (8) are involved with research and development, and clinical and regulatory matters. In addition to these individuals, we utilize outside contract engineering and contract manufacturing firms to support our operations. We have engaged a clinical research organization and several consulting firms involved with regulatory strategy and clinical trial planning. We believe that with sufficient funding during the next 12 months, we will increase the number of employees in the areas of clinical research and testing, customer development and product marketing.
As of December 31, 2007, we had ten (10) employees and six (6) independent contractor arrangements with consultants. Of this group of employees and consultants, five (5) were involved with finance and administration and eleven (11) were involved with research and development, and clinical and regulatory matters.
Company Information
Echo Therapeutics, Inc., a Minnesota corporation, (“Echo-MN”) was formed through the merger of Sontra Medical Corporation (“Sontra”) and Durham Pharmaceuticals Ltd. (doing business as Echo Therapeutics, Inc.) on September 14, 2007 (the “Merger”).
15
Effective October 8, 2007, Sontra changed its name to Echo Therapeutics, Inc. (“Echo”). Our principal executive offices are located at 10 Forge Parkway, Franklin, Massachusetts 02038, and our telephone number is(508) 553-8850.
On June 9, 2008, Echo-MN entered into an Agreement and Plan of Merger (the “Merger Agreement”) with a wholly-owned subsidiary of the same name, Echo Therapeutics, Inc., a Delaware corporation (“Echo-DE”), in order to change Echo-MN’s state of incorporation from Minnesota to Delaware (the “Merger”). The Merger Agreement and Merger were approved by Echo-MN’s shareholders at Echo-MN’s Annual Meeting of the Shareholders on May 20, 2008. Pursuant to the Merger Agreement, Echo-MN merged with and into Echo-DE and Echo-DE is the surviving corporation (the “Company”). Each share of common stock, par value $0.01 per share, of Echo-MN that was issued and outstanding immediately prior to the Merger was converted into one issued and outstanding share of common stock, par value $0.01 per share, of the Company (“Common Stock”), so that the holders of all of the issued and outstanding shares of common stock of Echo-MN immediately prior to the Merger became the holders of Common Stock of the Company when the Merger became effective.
We make our annual reports onForm 10-K, quarterly reports onForm 10-Q, current reports onForm 8-K and amendments to those reports available through our website, free of charge, as soon as reasonably practicable after we file such material with, or furnish it to the SEC. Our OTC Bulletin Board symbol is “ECTE” and our corporate website is www.echotx.com. The contents of our website are not part of this Report, and our internet address is included in this document as an inactive textual reference only.
Risk Factors
We operate in a rapidly changing environment that involves a number of risks, some of which are beyond our control. Forward-looking statements in this document and those made from time to time by us through our senior management are made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. Forward-looking statements concerning the expected future revenues or earnings or concerning projected plans, performance, or development of products and services, as well as other estimates related to future operations are necessarily only estimates of future results and there can be no assurance that actual results will not materially differ from expectations. Forward-looking statements represent management’s current expectations and are inherently uncertain. We do not undertake any obligation to update forward-looking statements. Factors that could cause actual results to differ materially from results anticipated in forward-looking statements include, but are not limited to, the following:
If we fail to raise additional capital, we will be unable to continue our business.
Our development efforts to date have consumed and will continue to require substantial amounts of capital in connection with research and development of our tCGM system and specialty pharmaceuticals programs. As we conduct more advanced product development of our Symphony tCGM System and AzoneTS reformulation pipeline, including Durhalieve, we will need significant funding to complete our product development programs and to pursue product commercialization. Additionally, our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern. Our ability to continue our business and our research, development and commercialization activities associated with our product pipeline is highly dependent on our ability to obtain additional sources of financing.
The terms of our Secured Note financing entered into in March 2008 tightly restrict our ability to incur additional indebtedness. Our Senior Convertible Notes also contain restrictions on our ability to incur further debt. Accordingly, our previous financings restrict the manner in which we can pursue additional financing. Any future equity financing, if available, may result in substantial dilution to existing shareholders.
Any failure by us to timely procure additional financing or investment adequate to fund our ongoing operations, including planned product development initiatives, clinical studies and commercialization efforts, will have material adverse consequences on our business operations and as a result, on our consolidated financial condition, results of operations and cash flows. To the extent that we attempt to raise additional funds through third
16
party collaborationsand/or licensing arrangements, we may be required to relinquish some rights to our technologies or products currently in various stages of development, or grant licenses or other rights on terms that are not favorable to us. We may not be successful in entering into any new collaboration or license agreement that results in material revenues. Additionally, we do not expect that the revenues generated from these arrangements would be sufficient alone to continue or expand our research or development activities and otherwise sustain our operation in the near term, if at all.
Our indebtedness under our Secured Notes is secured by substantially all of our assets, including our rights to our Symphonytm tCGM System and AzoneTStmtransdermal drug reformulation technology. If an event of default occurs under our Secured Notes, the holder of the Secured Notes may foreclose on our assets.
In connection with the issuance of our Secured Notes in March 2008, we granted the holder of the Secured Notes a security interest in substantially all of our assets. Our subsidiaries also guaranteed our obligations under the Secured Notes. The events of default under the Secured Notes include:
| | |
| | • | our failure to make a payment when due or payable; |
| |
| | • | a breach or notice of intent to breach of any material term, covenant or condition in the Secured Note or any of the transaction documents and such breach is not cured within five business days after notice; |
| |
| | • | any false, incorrect or breach in any material respect of any material representation or warranty; |
| |
| | • | the default of more than $25,000 of any other of our indebtedness that causes such amount to become due and payable; or |
| |
| | • | our bankruptcy (whether voluntary or involuntary) or general assignment for the benefit of its creditors. |
The occurrence of an event of default under our Secured Notes could result in an event of default under our Senior Convertible Notes as well. Upon the occurrence of an event of default under the terms of the Secured Notes, the holder of the Secured Notes may enforce its rights as a secured party and we may lose all or a portion of our assets, be forced to materially reduce our business activities, or cease operations entirely.
A failure to maintain a sufficient level of stockholders’ equity could result in an event of default and seizure of our assets.
The terms of our agreement with the holder of the Secured Notes includes a provision that we maintain stockholders’ equity at all times equal to or in excess of $6.5 million. Generally, stockholders’ equity is defined in the agreement as the aggregate value of all cash and cash equivalents, a portion of any accounts receivable, all inventory (valued at cost multiplied by 0.80 and excluding the portion of all work in process inventory that is in excess of 10% of the total inventory) and all other real and personal property (including intangible assets) and equipment, net of depreciation, minus the aggregate amount of all of our liabilities (excluding the impact of the warrants issued in connection with the Secured Notes).
We have historically incurred significant operating losses and expect to continue doing so in the foreseeable future. Costs associated with further development of our products and clinical programs will increase our operating expenses. Accordingly, without additional new equity capital to offset expected continuing losses, we will be at a further risk of failing to comply with the stockholders’ equity covenant as we continue to operate our business.
Furthermore, our ability to comply with the stockholders’ equity requirement would be further jeopardized if we experience an impairment of our intangible and long-lived assets. We are required to test periodically our intangible and long-lived assets, including the intangible assets associated with the ETI Acquisition, to determine if impairment has occurred. If such assets are deemed impaired, an impairment loss would be recognized equal to the amount by which the carrying amount exceeds the fair value of the assets. An impairment loss would cause stockholders’ equity to decline and could cause us to fail to comply with the requirement that we maintain stockholders’ equity equal to or in excess of $6.5 million (excluding the impact of the warrants issued in connection with the Secured Notes).
17
While the terms of our agreement with the holder of the Secured Notes provide for a thirty day grace period in the event that we fail to maintain a sufficient level of stockholders’ equity, a failure to satisfy this obligation would give the holder of the Secured Notes the right to declare an event of default and foreclose on our assets. Accordingly, a failure to maintain the required level of stockholders’ equity could result in a loss of all or a portion of our assets, force us to materially reduce our business activities, or cease operations entirely.
The terms of our Secured Notes requiring that we maintain a level of stockholders’ equity could impede or prohibit our ability to develop our products.
Costs that we incur in connection with the development of our products will increase our operating expenses. An increase in our operating expenses, without offsetting additional equity capital, will decrease our stockholders’ equity. Accordingly, without the receipt of substantial additional equity capital, we may need to delay or halt the development of any or all of our products in order to maintain sufficient stockholders’ equity so as to remain in compliance with the terms of our agreement with the holders of the Secured Notes. The failure to timely pursue product development could further delay our ability to commercialize our products and increase the costs associated with developing our products. Any delay or halt to our development plans could have a material adverse effect on our operations and our prospects for future commercialization of our products.
We have a history of operating losses, and we expect our operating losses to continue for the foreseeable future and we may not continue as a going concern.
We have generated limited revenue and have had operating losses since our inception, including a net loss of approximately $10,596,000 for the year ended December 31, 2008. As of December 31, 2008, we had an accumulated deficit of approximately $58,282,000. Losses have resulted principally from costs incurred in connection with our research and development activities and from general and administrative costs associated with our operations. The audit report from Wolf & Company, P.C., our independent registered public accounting firm, relating to our 2008 financial statements contains Wolf’s opinion that our recurring losses from operations, significant accumulated deficit, significant working capital deficit and our failure to raise sufficient capital to fund our operations, raise substantial doubt about our ability to continue as a going concern. We also expect to experience negative cash flows for the foreseeable future as we fund our operating losses and capital expenditures. This will result in decreases in our working capital, total assets and shareholders’ equity, which may not be offset by future funding. It is possible that we will never generate sufficient revenue to achieve and sustain profitability. Even if we achieve profitability, we may not be able to sustain or increase profitability. We expect our operating losses to continue and increase for the foreseeable future as we continue to expend substantial resources to conduct research and development, seek to obtain regulatory approval for our Symphony tCGM System, Durhalieve and our other AzoneTS pipeline candidates, identify and secure collaborative partnerships, and manage and execute our obligations in possible strategic collaborations. These losses, among other things, have had and will continue to have an adverse effect on our stockholders’ equity.
Our principal shareholders own a significant percentage of our stock and will be able to exercise significant influence.
Our principal shareholders own a significant percentage of our outstanding capital stock. Accordingly, these shareholders may be able to determine the composition of a majority of our Board of Directors, retain the voting power to approve certain matters requiring shareholder approval, and continue to have significant influence over our affairs. This concentration of ownership could have the effect of delaying or preventing a change in our control. Furthermore, so long as the purchasers in our January 2007 strategic private placement own 20% of the shares purchased in that transaction, the purchasers have the right to designate one director for election to our Board of Directors. The candidate is designated by the purchaser holding at least a majority of the shares of common stock purchased in the January 2007 financing, and such holder is currently Sherbrooke Partners, LLC, a beneficial owner of approximately 8.71% of our outstanding common stock as of March 25, 2009. Additionally, this significant concentration of share ownership may adversely affect the trading price of our common stock because investors often perceive disadvantages in owning stock in companies with a concentration of ownership.
18
Our future success is dependent upon successful collaborations with strategic partners.
Our future success is dependent upon our ability to selectively enter into and maintain collaborative arrangements with third parties for technology research and development, clinical testing, product development and sales and marketing. If we are unable to enter into any additional development agreements or collaborative arrangements with strategic partners, we will be required to internally fund all of our product development activities, significantly increasing business risk and capital requirements in the development, clinical testing, manufacturing, marketing and commercialization of new products. We could also encounter significant delays in introducing products into markets or find that the development, manufacture or sale of proposed products in such markets is adversely affected by the absence of those collaborative arrangements.
The process of establishing collaborative partners is difficult, time-consuming and involves significant uncertainty. Discussions with potential collaborators may not lead to the establishment of new collaborative relationships on favorable terms, if at all. If successful in establishing a collaborative agreement, such agreement may never result in the successful development of products or the generation of significant revenue. Any such agreements could limit our flexibility in pursuing alternatives for the development or commercialization of our products. Even if we were to enter into additional collaborative arrangements with third parties, there can be no assurance that our financial condition or results of operations will significantly improve.
The trading price of our common stock entails additional regulatory requirements, which may negatively affect such trading price.
Our common stock is currently listed on the OTC Bulletin Board, anover-the-counter electronic quotation service, which stock currently trades below $5.00 per share and the trading price of our common stock may continue to be below $5.00 per share. As a result of this price level, trading in our common stock would be subject to the requirements of certain rules promulgated under the Securities Exchange Act of 1934, as amended (the “Exchange Act”). The additional burdens imposed upon broker-dealers by such requirements may discourage broker-dealers from effecting transactions in our common stock. As a consequence, the market liquidity of our common stock could be severely affected or limited by these regulatory requirements.
Our stock price has been volatile and may fluctuate in the future.
The trading price of our common stock may fluctuate significantly. This price may be influenced by many factors, including:
| | |
| | • | our financial condition, performance and prospects; |
| |
| | • | the depth and liquidity of the market for our common stock; |
| |
| | • | our ability to enter into successful collaborative arrangements with strategic partners for research and development, clinical testing, and sales and marketing; |
| |
| | • | sales by selling shareholders of shares issued and issuable in connection with our private placements in 2003, 2004, 2006, 2007 and 2008; |
| |
| | • | investor perception of us and the industry in which we operate; |
| |
| | • | general financial and other market conditions; and |
| |
| | • | domestic and international economic conditions. |
Public stock markets have experienced extreme price and trading volume volatility, particularly in the technology and life sciences sectors of the market. This volatility has significantly affected the market prices of securities of many technology companies for reasons frequently unrelated to or disproportionately impacted by the operating performance of these companies. These broad market fluctuations may adversely affect the market price of our common stock. In addition, fluctuations in our stock price may have made our stock attractive to momentum, hedge or day-trading investors who often shift funds into and out of stocks rapidly, exacerbating price fluctuations in either direction particularly when viewed on a quarterly basis.
19
Securities we issue to fund our operations could dilute or otherwise adversely affect our shareholders.
We need to raise substantial additional funds through public or private equity financings to fund our operations. If we raise funds by issuing equity securities, the percentage ownership of current shareholders will be significantly reduced and the new equity securities may have rights senior to those of the shares of our common stock. We may not obtain sufficient financing on terms that are favorable to current investors or us. We may delay, limit or eliminate some or all of our proposed operations if adequate funds are not available.
Risks related to our discovery, development and commercialization of new medical products
Our medical device products are based on new technologies and are in early stages of development, and may not be successfully developed or achieve market acceptance.
Most of our products under development have a high risk of failure because they are based on new technologies and are in the early stages of development. To date, we have tested the feasibility of our Prelude technology for various applications, including continuous glucose monitoring and certain anesthetic applications. We have also tested the feasability of our Symphony tCGM System technology for the monitoring of glucose on a continuous basis. However, to develop additional products or additional uses, substantial expenditures will be required, including for feasibility studies, pre-clinical studies, prototype development and clinical testing. Projected costs for such development are difficult to estimate and they may change and increase frequently.
Our success is dependent on further developing new and existing products and obtaining favorable results from pre-clinical studies and clinical trials and satisfying regulatory standards and approvals required for the market introduction of such products, including skin permeation methods and our tCGM system. There can be no assurance that we will not encounter unforeseen problems in the development of our transdermal continuous glucose monitoring and skin permeation technologies, or that we will be able to successfully address the problems that do arise. There can be no assurance that any of our potential products will be successfully developed, proven safe and efficacious in clinical trials, meet applicable regulatory standards, be capable of being produced in commercial quantities at acceptable costs, or be eligible for third-party reimbursement from governmental or private insurers. Even if we successfully develop new products, there can be no assurance that such products will be successfully marketed or achieve market acceptance, or that expected markets will develop for such products. If any of our development programs are not successfully completed, required regulatory approvals or clearances are not obtained, or potential products for which approvals or clearances are obtained are not commercially successful, our business, financial condition and results of operations would be materially adversely affected.
In addition, because our products are based on new technologies, they are subject to lengthy sales cycles and may take substantial time and effort to achieve market acceptance, especially at hospitals, which typically have a lengthy and rigorous approval process for adopting new technologies.
Our future success may be dependent in part upon successful development of our continuous glucose monitor for the hospital intensive care unit and critical care unit markets.
We have completed the initial prototypes and have conducted several pilot human clinical studies in2006-2008 at a leading Boston-area hospital, with a member of our Clinical Advisory Board serving as principal investigator. Although we believe the clinical rationale exists for our tCGM system for the ICU market, there can be no assurance that such a market will be established, or that we will be able to successfully develop a product that will prove effective for this market or gain market acceptance should such a market develop. Our tCGM product development process may take several years and will require substantial capital outlays. If the ICU market does not develop as we expect, or if we are unable to successfully develop a tCGM product for such markets on a timely basis and within cost constraints, then our business and financial results will be materially adversely affected.
Our pharmaceutical products are in various stages of development and may not be successfully developed or obtain regulatory approval.
The product candidates in our Azonetm pipeline are at various stages of developments. We have filed a NDA covering Durhalievetm, our most advanced Azonetm product candidate, for corticosteroid responsive dermatoses,
20
with the FDA and we now seeking to satisfy FDA manufacturing requirements necessary to secure market approval. We believe that the failure to advance Durhalievetm beyond its current stage of development will have a material adverse effect on our prospects and on our business, financial condition and results of operations.
Several of our Azonetm pipeline programs involve topical reformulations of FDA-approved products. We must successfully complete nonclinical development activities, including reformulation development, process development and manufacturing activities, with respect to these product candidates before we can commence late-stage clinical development necessary to obtain regulatory approval. These nonclinical development activities, as well as the clinical trials associated with each AzoneTS reformulation development program, are expensive, uncertain and will take several years to complete and may not yield results that support further clinical development or product approvals.
Conducting clinical studies for any of our product candidates for approval in the United States requires filing an IND and reaching agreement with the FDA on clinical protocols, finding appropriate clinical sites and clinical investigators, securing approvals for such studies from the institutional review board at each such site, manufacturing clinical quantities of drug candidates, supplying drug product to clinical sites and enrolling sufficient numbers of participants. We cannot guarantee that we will be able to successfully accomplish these activities or all of the other activities necessary to initiate and complete clinical trials. As a result, our nonclinical and clinical development programs may be extended, delayed or terminated, and we may be unable to obtain regulatory approvals or successfully commercialize our product candidates.
Our results to date provide no basis for predicting whether any of our product candidates will be safe or effective, or receive regulatory approval.
Most of our AzoneTS-related product candidates are in early stages of reformulation development and the likelihood of success of each product candidate is highly speculative. To date, the data supporting our AzoneTS drug development programs and new chemical entities are derived solely from laboratory and nonclinical studies and limited early stage clinical trials. Additional clinical trials in humans may not show that our AzoneTS-reformulated drug candidates are safe and effective, in which event we may need to further reformulate or abandon development of one or more of our current product candidates. It is impossible to predict when or if our early-stage AzoneTS pipeline products will prove effective or safe in humans or will receive regulatory approval. These AzoneTS-reformulated compounds may not demonstrate in patients the chemical and pharmacological properties ascribed to them in laboratory studies, or early stage clinical trials, and they may interact with human biological systems or other drugs in unforeseen, ineffective or harmful ways. If we are unable to discover or successfully develop drugs that are effective and safe in humans, we will not have a viable business.
We may not be able to gain market acceptance of our product candidates, which would prevent us from becoming profitable.
Even if our product candidates receive regulatory approval, we cannot be certain that any of our product candidates will gain market acceptance among physicians, patients, healthcare payers, hospitals, pharmaceutical companies and the medical community in general. The degree of market acceptance will depend on a number of factors, including:
| | |
| | • | Our establishment and demonstration to the medical community of the clinical efficacy and safety of our product candidates; |
| |
| | • | Our ability to create products that are superior to alternatives currently on the market; and |
| |
| | • | Our ability to establish in the medical community the potential advantage of our treatments over alternative treatment methods. |
These and other factors combined with obtaining regulatory approvals will not guarantee future revenues. Sales of medical products largely depend on the reimbursement of patients’ medical expenses by government healthcare programs and private health insurers. Governments and private insurers closely examine medical products to determine whether they should be covered by reimbursement and if so, the level of reimbursement that will apply. We cannot be certain that third party payers will sufficiently reimburse sales of our products, or enable us
21
to sell our products at profitable prices. Similar concerns could also limit the reimbursement amounts that health insurers or government agencies in other countries are prepared to pay for our products. In many countries where we plan to market our products, including countries in Europe, Japan and Canada, the pricing of prescription drugs is controlled by the government or regulatory agencies. Government or other regulatory agencies in these countries could determine that the pricing for our products should be based on prices of other commercially available drugs for the same disease, rather than allowing us to market our products at a premium as new drugs.
Sales of medical products also depend on physicians’ willingness to prescribe the treatment, which is likely to be based on a determination by these physicians that the products are safe, therapeutically effective and cost-effective. We cannot predict whether physicians, other healthcare providers, government agencies or private insurers will determine that our products are safe, therapeutically effective and cost effective relative to competing treatments.
We have limited manufacturing capabilities and manufacturing personnel, and if our manufacturing capabilities are insufficient to produce, or have produced by third-parties, an adequate supply of our pharmaceutical product candidates in nonclinical, clinical or commercial quantities, our development and commercialization activities will be limited and our business will be harmed.
To date, our pharmaceutical product candidates have been manufactured in small, research-scale quantities by us and for us by third party contract manufacturers for nonclinical studies and clinical trials. If any of our product candidates is approved by the FDA or other regulatory agencies for commercial sale, we will need to manufacture it in larger quantities. We intend to continue to use third parties to manufacture our products for research and development purposes and, if our products are approved, for commercial purposes. Our third party manufacturers may not be able to successfully increase the manufacturing capacity for any of our product candidates in a timely or efficient manner, or at all. If we are unable to successfully increase the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in the supply of the product candidate. Our failure or the failure of our third party manufacturers to comply with the FDA’s GMP and to pass inspections of the manufacturing facilities by the FDA or other regulatory agencies could seriously harm our business.
We may not be able to obtain and maintain the third party relationships that are necessary to develop, commercialize and manufacture some or all of our product candidates.
We expect to depend on collaborators, partners, licensees, contract research organizations, manufacturers and other third parties to support our efforts to develop and commercialize our product candidates, to manufacture clinical and commercial scale quantities of our product candidates and products and to market, sell, and distribute any products we successfully develop.
We cannot guarantee that we will be able to successfully negotiate agreements for or maintain relationships with collaborators, partners, licensees, clinical investigators, manufacturers and other third parties on favorable terms, if at all. If we are unable to obtain or maintain these agreements, we may not be able to clinically develop, formulate, manufacture, obtain regulatory approvals for or commercialize our product candidates, which will in turn adversely affect our business.
We expect to expend substantial management time and effort to enter into relationships with third parties and, if we successfully enter into such relationships, to manage these relationships. We must attract and retain collaborative partners and to develop technologies and pipeline candidates that meet the requirements of prospective collaborative partners. In addition, substantial amounts of our expenditures will be paid to third parties in these relationships.
However, we cannot control the amount or timing of resources our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will fulfill their obligations to us under these arrangements in a timely fashion, if at all. In addition, our contract partners will generally have the right to abandon research projects and terminate applicable agreements, including funding obligations, prior to or upon the expiration of agreed upon research terms. There can be no assurance that we will be successful in establishing multiple future collaborative arrangements on acceptable terms
22
or at all, that current or future collaborative partners will not terminate funding before completion of projects, that our existing or future collaborative arrangements will result in successful product commercialization or that we will derive any revenues from such arrangements. Some or all of our collaborative partners may receive co-exclusive rights to market products incorporating our technology in a particular field of use or a particular territory. The grant of such rights will limit our alternatives in commercializing certain of our products and technologies.
We have no experience in sales, marketing and distribution and may have to enter into agreements with third parties to perform these functions, which could prevent us from successfully commercializing our product candidates.
We currently have no sales, marketing or distribution capabilities. To commercialize our product candidates, we must either develop our own sales, marketing and distribution capabilities, which will be expensive and time consuming, or make arrangements with third parties to perform these services for us. If we enter into third party arrangements, the third parties may not be capable of successfully selling any of our product candidates. If we decide to market any of our products on our own, we will have to commit significant resources to developing a marketing and sales force and supporting distribution capabilities. If we decide to enter into arrangements with third parties for performance of these services, we may find that they are not available on terms acceptable to us, or at all. If we are not able to establish and maintain successful arrangements with third parties or build our own sales and marketing infrastructure, we may not be able to commercialize our product candidates which would adversely affect our business and financial condition.
We may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable pharmaceutical product candidates.
We have limited technical, managerial and financial resources to determine which of our product candidates should proceed to initial clinical trials, later stage clinical development and potential commercialization. We may make incorrect determinations. Our decisions to allocate our research and development, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of viable commercial products and may divert resources from better opportunities. Similarly, our decisions to delay or terminate drug development programs may also be incorrect and could cause us to miss valuable opportunities.
If we are not able to retain our current senior management team and our scientific and medical advisors or continue to attract and retain qualified scientific, technical and business personnel, our business will suffer.
We are dependent on the members of our management team and our network of clinical, regulatory, scientific and technical advisors for our business success. An important element of our strategy is to take advantage of the research and development expertise of our current management and to utilize the unique expertise of our advisors. We currently have employment agreements with all of our senior executive officers. Our employment agreements with our senior executive officers are terminable on short notice or no notice and provide for severance and change of control benefits. The loss of any one of our senior executive officers could result in a significant loss in the knowledge and experience that we, as an organization, possess and could cause significant delays, or outright failure, in the development and further commercialization of our product candidates.
Disputes under key agreements or conflicts of interest with our scientific advisors or clinical investigators could delay or prevent development or commercialization of our product candidates.
Any agreements we have or may enter into with third parties, such as collaboration, license, formulation supplier, manufacturing, clinical research organization or clinical trial agreements, may give rise to disputes regarding the rights and obligations of the parties. Disagreements could develop over rights to ownership or use of intellectual property, the scope and direction of research and development, the approach for regulatory approvals or commercialization strategy. We intend to conduct research programs in a range of therapeutic areas, but our pursuit of these opportunities could result in conflicts with the other parties to these agreements that may be developing or selling pharmaceuticals or conducting other activities in these same therapeutic areas. Any disputes or commercial conflicts could lead to the termination of our agreements, delay progress of our product development programs,
23
compromise our ability to renew agreements or obtain future agreements, lead to the loss of intellectual property rights or result in costly litigation.
We collaborate with outside scientific advisors and collaborators at academic and other institutions that assist us in our research and development efforts. Our scientific advisors are not our employees and may have other commitments that limit their availability to us. If a conflict of interest between their work for us and their work for another entity arises, we may lose their services.
We may have challenges in managing our outside contractors for product and regulatory accomplishments.
We rely heavily upon and have relationships with outside contractors and consultants with expertise in drug development, regulatory strategy, manufacturing and other matters. These parties are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us. We have limited control over the activities of consultants and outside contractors and, except as otherwise required by our collaboration and consulting agreements, can expect only limited amounts of their time to be dedicated to our activities. If any third party with whom we have or enter into a relationship is unable or refuses to contribute to projects on which we need their help, our ability to generate advances in our technologies and develop our product candidates could be significantly harmed.
Risks related to regulatory approvals
None of our product candidates has received regulatory approvals. If we are unable to obtain regulatory approvals to market one or more of our product candidates, our business will be adversely affected.
Most of our pharmaceutical product candidates are in early stages of development, and we do not expect these product candidates to be commercially available in the foreseeable future, if at all. Our product candidates are subject to strict regulation by regulatory authorities in the United States and in other countries. In particular, human pharmaceutical therapeutic product candidates are subject to rigorous preclinical and clinical testing and other requirements by the FDA in the United States and similar authorities in other countries in order to demonstrate safety and efficacy. Data obtained from preclinical studies and clinical trials are subject to varying interpretations that could delay, limit or prevent regulatory approval, and failure to comply with regulatory requirements or inadequate manufacturing processes are examples of other problems that could prevent approval. Because our product candidates involve or are expected to involve the application of new technologies or are based upon a new therapeutic approach, they may be subject to substantial additional review by various governmental regulatory authorities, and, as a result, the process of obtaining regulatory approvals for them may proceed more slowly than for product candidates based upon more conventional technologies. In addition, we may encounter delays or rejections due to additional government regulation from future legislation, administrative action or changes in the FDA policy. We do not know whether regulatory agencies will grant approval for any of our product candidates. Even if we complete preclinical studies and clinical trials successfully, we may not be able to obtain regulatory approvals or we may not receive approvals to make claims about our products that we believe to be necessary to effectively market our products.
We cannot market any product candidate until we have completed all necessary preclinical studies and clinical trials and have obtained the necessary regulatory approvals. Outside the United States, our ability to market any of our potential products is dependent upon receiving marketing approvals from the appropriate regulatory authorities. These foreign regulatory approval processes include all of the risks associated with the FDA approval process described above plus additional risks. If we are unable to receive regulatory approvals, we will be unable to commercialize our product candidates, and our business may fail.
If we are unable to successfully complete the preclinical studies or clinical trials necessary to support PMA or NDA submissions to the FDA, we may be unable to commercialize our tCGM system or AzoneTS products under development, which could impair our financial position.
Before submitting a PMA application to the FDA for our tCGM system or a NDA for an AzoneTS product candidate, we must successfully complete preclinical studies and clinical trials that we believe will demonstrate that our product is safe and effective. Product development, including preclinical studies and clinical trials, is a long,
24
expensive and uncertain process and is subject to delays and failure at any stage. Furthermore, the data obtained from the studies and trial may be inadequate to support approval of a PMA or NDA as the case may be. With respect to our medical device programs, while we have in the past obtained, and may in the future obtain, an Investigational Device Exemption, or IDE, prior to commencing clinical trials for our tCGM systems, FDA approval of an IDE application permitting us to conduct testing does not mean that the FDA will consider the data gathered in the trial to be sufficient to support approval of a PMA application, even if the trial’s intended safety and efficacy endpoints are achieved.
The commencement or completion of any of our clinical trials may be delayed or halted, or be inadequate to support approval of a PMA or NDA as the case may be, for numerous reasons, including, but not limited to, the following:
| | |
| | • | the FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial, or place a clinical trial on hold; |
| |
| | • | patients do not enroll in clinical trials at the rate we expect; |
| |
| | • | patients do not comply with trial protocols; |
| |
| | • | patientfollow-up is not at the rate we expect; |
| |
| | • | patients experience adverse side effects; |
| |
| | • | patients die during a clinical trial, even though their death may not be related to our products; |
| |
| | • | institutional review boards, or IRBs, and third-party clinical investigators may delay or reject our trial protocol; |
| |
| | • | third-party clinical investigators decline to participate in a trial or do not perform a trial on our anticipated schedule or consistent with the investigator agreements, clinical trial protocol, good clinical practices or other FDA or IRB requirements; |
| |
| | • | third-party organizations do not perform data collection, monitoring and analysis in a timely or accurate manner or consistent with the clinical trial protocol or investigational or statistical plans; |
| |
| | • | regulatory inspections of our clinical trials or manufacturing facilities may, among other things, require us to undertake corrective action or suspend or terminate our clinical trials; |
| |
| | • | changes in governmental regulations or administrative actions; |
| |
| | • | the interim or final results of the clinical trial are inconclusive or unfavorable as to safety or efficacy; and |
| |
| | • | the FDA concludes that our trial design is inadequate to demonstrate safety and efficacy. |
The results of preclinical studies do not necessarily predict future clinical trial results, and predecessor clinical trial results may not be repeated in subsequent clinical trials. Additionally, the FDA may disagree with our interpretation of the data from our preclinical studies and clinical trials, or may find the clinical trial design, conduct or results inadequate to prove safety or efficacy, and may require us to pursue additional pre-clinical studies or clinical trials, which could further delay the approval of our products. If we are unable to demonstrate the safety and efficacy of our products in our clinical trials, we will be unable to obtain regulatory approval to market our products. The data we collect from our current clinical trials, our pre-clinical studies and other clinical trials may not be sufficient to support FDA approval.
We depend on clinical investigators and clinical sites to enroll patients in our clinical trials and other third parties to manage the trials and to perform related data collection and analysis, and, as a result, we may face costs and delays that are outside of our control.
We rely on clinical investigators and clinical sites to enroll patients in our clinical trials and other third parties to manage the trial and to perform related data collection and analysis. However, we may not be able to control the amount and timing of resources that clinical sites may devote to our clinical trials. If these clinical investigators and clinical sites fail to enroll a sufficient number of patients in our clinical trials or fail to ensure compliance by patients
25
with clinical protocols or fail to comply with regulatory requirements, we will be unable to complete these trials, which could prevent us from obtaining regulatory approvals for our products. Our agreements with clinical investigators and clinical sites for clinical testing place substantial responsibilities on these parties and, if these parties fail to perform as expected, our trials could be delayed or terminated. If these clinical investigators, clinical sites or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated, or the clinical data may be rejected by the FDA, and we may be unable to obtain regulatory approval for, or successfully commercialize, our products.
The regulatory approval process is costly and lengthy and we may not be able to successfully obtain all required regulatory approvals.
The preclinical development, clinical trials, manufacturing, marketing and labeling of medical devices and specialty pharmaceuticals are all subject to extensive regulation by numerous governmental authorities and agencies in the United States and other countries. We must obtain regulatory approval for each of our product candidates before marketing or selling any of them. It is not possible to predict how long the approval processes of the FDA or any other applicable federal or foreign regulatory authority or agency for any of our products will take or whether any such approvals ultimately will be granted. The FDA and foreign regulatory agencies have substantial discretion in the medical device and drug approval process, and positive results in preclinical testing or early phases of clinical studies offer no assurance of success in later phases of the approval process. Generally, preclinical and clinical testing of products can take many years and require the expenditure of substantial resources, and the data obtained from these tests and trials can be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. If we encounter significant delays in the regulatory process that result in excessive costs, this may prevent us from continuing to develop our product candidates. Any delay in obtaining, or failure to obtain, approvals could adversely affect the marketing of our products and our ability to generate product revenue. The risks associated with the approval process include:
| | |
| | • | failure of our product candidates to meet a regulatory entity’s requirements for safety, efficacy and quality; |
| |
| | • | limitation on the indicated uses for which a product may be marketed; |
| |
| | • | unforeseen safety issues or side effects; and |
| |
| | • | governmental or regulatory delays and changes in regulatory requirements and guidelines. |
Even if we receive regulatory approvals for marketing our product candidates, if we fail to comply with continuing regulatory requirements, we could lose our regulatory approvals, and our business would be adversely affected.
If we obtain regulatory agency approval for a new product, this approval may entail limitations on the indicated uses for which it can be marketed that could limit the potential commercial use of the product. Additionally, approved products are subject to continual review, and discovery of previously unknown problems with a product or its manufacturer may result in restrictions on the product of any commercially viable product will be subject to government regulation from several standpoints including the processes of manufacturing, advertising and promoting, selling and marketing, labeling and distribution. Enforcement actions resulting from our failure to comply with government and regulatory requirements could result in fines, suspension of approvals, withdrawal of approvals, product recalls, product seizures, mandatory operating restrictions, criminal prosecution, civil penalties and other actions that could impair the manufacturing, marketing and sale of our potential products and our ability to conduct our business.
26
Even if we are able to obtain regulatory approvals for any of our product candidates, if they exhibit harmful side effects after approval, our regulatory approvals could be revoked or otherwise negatively impacted, and we could be subject to costly and damaging product liability claims.
Even if we receive regulatory approval for any of our product candidates, we will have tested them in only a small number of patients during our clinical trials. If our applications for marketing are approved and more patients begin to use our product, new risks and side effects associated with our products may be discovered. As a result, regulatory authorities may revoke their approvals. We may be required to conduct additional clinical trials, make changes in labeling of our product, reformulate our product or make changes and obtain new approvals for our and our suppliers’ manufacturing facilities. We might have to withdraw or recall our products from the marketplace. We may also experience a significant drop in the potential sales of our product if and when regulatory approvals for such product are obtained, experience harm to our reputation in the marketplace or become subject to lawsuits, including class actions. Any of these results could decrease or prevent any sales of our approved product or substantially increase the costs and expenses of commercializing and marketing our product.
Failure to obtain regulatory and pricing approvals in foreign jurisdictions could delay or prevent commercialization of our products abroad.
If we succeed in developing any products, we intend to market them in Europe, Japan, Canada and other foreign jurisdictions. In order to do so, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval abroad may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval and additional risks associated with requirements particular to those foreign jurisdictions where we will seek regulatory approval of our products. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. We and our collaborators may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market outside the United States. The failure to obtain these approvals could materially adversely affect our business, financial condition and results of operations.
Healthcare reform measures could adversely affect our business.
The efforts of governmental and third-party payers to contain or reduce the costs of healthcare may adversely affect the business and financial condition of pharmaceutical companies. In the United States and in foreign jurisdictions there have been, and we expect that there will continue to be, a number of legislative and regulatory proposals aimed at changing the healthcare system. For example, in some countries other than the United States, pricing of prescription drugs is subject to government control, and we expect proposals to implement similar controls in the United States to continue. The pendency or approval of such proposals could result in a decrease in our common stock price or limit our ability to raise capital or to enter into collaborations or license rights to our products.
New federal legislation may increase the pressure to reduce prices of pharmaceutical products paid for by Medicare, which could adversely affect our revenues, if any.
The Medicare Prescription Drug Improvement and Modernization Act of 2003, or MMA, expanded Medicare coverage for drug purchases by the elderly and disabled beginning in 2006. The new legislation uses formularies, preferred drug lists and similar mechanisms that may limit the number of drugs that will be covered in any therapeutic class or reduce the reimbursement for some of the drugs in a class.
As a result of the expansion of legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. Legislation that would permit the federal government to negotiate drug prices directly with manufacturers under the Medicare prescription drug programs is currently under consideration. These cost reduction initiatives could decrease the coverage and price that we receive for our products in the future and could seriously harm our business. While the MMA applies only to drug benefits
27
for Medicare beneficiaries, private payers often follow Medicare coverage policy and payment limitations in setting their own reimbursement systems, and any limits on or reductions in reimbursement that occur in the Medicare program may result in similar limits on or reductions in payments from private payers.
New federal laws or regulations on drug importation could make lower cost versions of our future products available, which could adversely affect our revenues, if any.
Both federal and state governments in the United States and governments in other countries continue to propose and pass legislation designed to contain or reduce the cost of healthcare. Legislation and regulations affecting the pricing of pharmaceuticals and other medical products may be adopted before any of our potential products are approved for marketing. Cost control initiatives could decrease the price that we receive for any product candidate we may develop in the future. In addition, third-party payors are increasingly challenging the price and cost-effectiveness of medical products and services and any of our potential products may ultimately not be considered cost-effective by these third parties. Any of these initiatives or developments could materially harm our business.
The prices of some drugs and medical devices are lower in other countries than in the United States because of government regulation and market conditions. Under current law, importation of drugs into the United States is generally not permitted unless the drugs are approved in the United States and the entity that holds that approval consents to the importation. Various proposals have been advanced to permit the importation of drugs from other countries to provide lower cost alternatives to the products available in the United States. In addition, the MMA requires the Secretary of Health and Human Services to promulgate regulations for drug reimportation from Canada into the United States under some circumstances, including when the drugs are sold at a lower price than in the United States.
If the laws or regulations are changed to permit the importation of drugs or medical devices into the United States in circumstances that are currently not permitted, such a change could have an adverse effect on our business by making available lower priced alternatives to our future products.
If we fail to obtain adequate reimbursement for our product candidates, the use of our potential products could be severely limited.
Our ability to successfully commercialize our product candidates will depend significantly on our ability to obtain acceptable prices and the availability of reimbursement to the patient from third-party payors. Significant uncertainty exists as to the reimbursement status of newly-approved healthcare products. If our potential products are not considered cost-effective or if we fail to generate adequate third-party reimbursement for the users of our potential products and treatments, then we may be unable to maintain price levels sufficient to realize an appropriate return on our investment in product development. In both the United States and other markets, sales of our potential products, if any, will depend in part on the availability of reimbursement from third-party payors, examples of which include government health administration authorities, private health insurers, health maintenance organizations, and pharmacy benefit management companies.
Risks related to competition
We operate in highly competitive medical device and specialty pharmaceuticals markets and face competition from large, well-established companies with significantly more resources, and, as a result, we may not be able to compete effectively.
The industry in which we operate is intensely competitive. Many companies, universities, and research organizations developing competing product candidates have greater resources and significantly greater experience in financial, research and development, manufacturing, marketing, sales, distribution, and technical regulatory matters than we have. In addition, many competitors have greater name recognition and more extensive collaborative relationships. Our competitors could commence and complete clinical testing of their product candidates, obtain regulatory approvals, and begin commercial-scale manufacturing of their products faster than we are able to for our products. They could develop products that would render our product candidates, and those of our collaborators, obsolete and noncompetitive. Our competitors may develop more effective or more affordable
28
products or achieve earlier patent protection or product commercialization than us. If we are unable to compete effectively against these companies, then we may not be able to commercialize our product candidates or achieve a competitive position in the market. This would adversely affect our ability to generate revenues.
The markets for glucose monitoring devices and specialty pharmaceuticals are intensely competitive, subject to rapid change and significantly affected by new product introductions and other market activities of industry participants. With respect to our Symphony tCGM System, we expect to compete directly with Roche Disetronic, a division of Roche Diagnostics; LifeScan, Inc., a division of Johnson & Johnson; the MediSense and TheraSense divisions of Abbott Laboratories and Bayer Corporation, each of which manufactures and markets products for the single-point finger stick device market. Collectively, we believe these companies currently account for substantially all of the worldwide sales of self-monitored glucose testing systems. Several companies are developing or currently marketing short-term continuous glucose monitoring products that will compete directly with our Symphony tCGM System. To date, Abbott, Cygnus, DEXCOM and Medtronic have received approval from the FDA for continuous glucose monitors. We believe that the product originally developed and marketed by Cygnus is no longer actively marketed. Most of the companies developing or marketing competing glucose monitoring devices and specialty pharmaceuticals are publicly traded or divisions of publicly-traded companies, and these companies enjoy several competitive advantages, including:
| | |
| | • | significantly greater name recognition; |
| |
| | • | established relations with healthcare professionals, customers and third-party payors; |
| |
| | • | established distribution networks; |
| |
| | • | additional lines of products, and the ability to offer rebates or bundle products to offer higher discounts or incentives to gain a competitive advantage; |
| |
| | • | greater experience in conducting research and development, manufacturing, clinical trials, obtaining regulatory approval for products and marketing approved products; and |
| |
| | • | greater financial and human resources for product development, sales and marketing, and patent litigation. |
As a result, we may not be able to compete effectively against these companies or their products.
No continuous glucose monitoring system has yet received FDA clearance as a replacement forsingle-point finger stick devices, and our Symphonytm tCGM System may never be approved for that indication.
We do not expect our Symphony tCGM System to eliminate the need for single-point finger stick devices in the foreseeable future and it may not ever be approved by the FDA for that indication. At present, there is no established precedent for FDA approval of a continuous glucose monitoring system as a replacement for single-point finger stick devices. If any of our competitors were to obtain replacement claim labeling for a continuous glucose monitoring system, our Symphony tCGM System may not be able to compete effectively against that system and our business would suffer.
Technological breakthroughs in the glucose monitoring market could render our products obsolete.
The glucose monitoring market is subject to rapid technological change and product innovation. Our products are based on our proprietary technology, but a number of companies and medical researchers are pursuing new technologies for the monitoring of glucose levels. FDA approval of a commercially viable continuous glucose monitor or sensor produced by one of our competitors could significantly reduce market acceptance of our system. Several of our competitors are in various stages of developing continuous glucose monitors or sensors, including non-invasive and invasive devices, and the FDA has approved four of these competing products. In addition, the National Institutes of Health and other supporters of diabetes research are continually seeking ways to prevent, cure or improve treatment of diabetes. Therefore, our products may be rendered obsolete by technological breakthroughs in diabetes monitoring, treatment, prevention or cure.
29
Risks related to products liability exposure
We may have significant product liability exposure which may harm our business and our reputation.
We face exposure to product liability and other claims if our product candidates, products or processes are alleged to have caused harm. These risks are inherent in the testing, manufacturing, and marketing of human therapeutic products. Although we currently maintain product liability insurance, we may not have sufficient insurance coverage, and we may not be able to obtain sufficient coverage at a reasonable cost, if at all. Our inability to obtain product liability insurance at an acceptable cost or to otherwise protect against potential product liability claims could prevent or inhibit the commercialization of any products or product candidates that we develop. If we are sued for any injury caused by our products, product candidates or processes, our liability could exceed our product liability insurance coverage and our total assets. Claims against us, regardless of their merit or potential outcome, may also generate negative publicity or hurt our ability to obtain physician endorsement of our products or expand our business.
Risks related to our intellectual property
Our inability to adequately protect our intellectual property could allow our competitors and others to produce products based on our technology, which could substantially impair our ability to compete.
Our success and our ability to compete is dependent, in part, upon our ability to maintain the proprietary nature of our technologies. We rely on a combination of patent, copyright and trademark law, and trade secrets and nondisclosure agreements to protect our intellectual property. However, such methods may not be adequate to protect us or permit us to gain or maintain a competitive advantage. Our patent applications may not issue as patents in a form that will be advantageous to us, or at all. Our issued patents, and those that may issue in the future, may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing related products.
To protect our proprietary rights, we may in the future need to assert claims of infringement against third parties to protect our intellectual property. The outcome of litigation to enforce our intellectual property rights in patents, copyrights, trade secrets or trademarks is highly unpredictable, could result in substantial costs and diversion of resources, and could have a material adverse effect on our financial condition and results of operations regardless of the final outcome of such litigation. In the event of an adverse judgment, a court could hold that some or all of our asserted intellectual property rights are not infringed, invalid or unenforceable, and could award attorney fees.
Despite our efforts to safeguard our unpatented and unregistered intellectual property rights, we may not be successful in doing so or the steps taken by us in this regard may not be adequate to detect or deter misappropriation of our technology or to prevent an unauthorized third party from copying or otherwise obtaining and using our products, technology or other information that we regard as proprietary. Additionally, third parties may be able to design around our patents. Furthermore, the laws of foreign countries may not protect our proprietary rights to the same extent as the laws of the United States. Our inability to adequately protect our intellectual property could allow our competitors and others to produce products based on our technology, which could substantially impair our ability to compete.
We are subject to claims of infringement or misappropriation of the intellectual property rights of others, which could prohibit us from commercializing our product candidates, require us to obtain licenses from third parties or to develop non-infringing alternatives, and subject us to substantial monetary damages and injunctive relief.
As is generally the case in the medical device and pharmaceutical industries in which we operate, third-parties may, in the future, assert infringement or misappropriation claims against us with respect to our current or future products. Whether a product infringes a patent involves complex legal and factual issues, the determination of which is often uncertain. Therefore, we cannot be certain that we have not infringed the intellectual property rights of such third parties or others. Our competitors may assert that our product candidates, technologiesand/or the methods we employ in the use of our tCGM systemsand/or AzoneTS pipeline candidates are covered by U.S. or
30
foreign patents held by them. This risk is exacerbated by the fact that there are numerous issued patents and pending patent applications relating to self-monitored glucose testing systems in the medical technology field. Because patent applications may take years to issue, there may be applications now pending of which we are unaware that may later result in issued patents that our products infringe. There could also be existing patents of which we are unaware that one or more components of our system may inadvertently infringe. As the number of competitors in the market for continuous glucose monitoring systems and specialty pharmaceuticals grows, the possibility of inadvertent patent infringement by us or a patent infringement claim against us increases.
Any infringement or misappropriation claim could cause us to incur significant costs, could place significant strain on our financial resources, divert management’s attention from our business and harm our reputation. If the relevant patents were upheld as valid and enforceable and we were found to infringe, we could be prohibited from selling our product that is found to infringe unless we could obtain licenses to use the technology covered by the patent or are able to design around the patent. We may be unable to obtain a license on terms acceptable to us, if at all, and we may not be able to redesign our products to avoid infringement. Even if we are able to redesign our products to avoid an infringement claim, we may not receive FDA approval for such changes in a timely manner or at all. A court could also order us to pay compensatory damages for such infringement, plus prejudgment interest and could, in addition, treble the compensatory damages and award attorney fees. These damages could be substantial and could harm our reputation, business, financial condition and operating results. A court also could enter orders that temporarily, preliminarily or permanently enjoin us and our customers from making, using, selling or offering to sell, or could enter an order mandating that we undertake certain remedial activities. Depending on the nature of the relief ordered by the court, we could become liable for additional damages to third parties.
We lease approximately 13,000 square feet of research-scale manufacturing, laboratory and office space in a single facility located in Franklin, Massachusetts under a lease expiring March 31, 2010. We have the option to extend the lease for an additional one (1) year period. We have never engaged in real estate investment activities and we have no current plans to do so.
| |
| ITEM 3. | LEGAL PROCEEDINGS. |
We are not a party to any legal proceedings.
PART II
| |
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES. |
Our common stock is currently traded through theOver-The-Counter Bulletin Board (OTCBB) and is quoted under the symbol “ECTE”. The following table sets forth the range of high and low closing prices for our common stock for the periods indicated as reported by the OTCBB in 2007 and 2008. The number of common shareholders of record of Echo Therapeutics, Inc. as of March 25, 2009 was 122.
| | | | | | | | | |
Fiscal Year Ended December 31, 2007 | | High | | | Low | |
| |
| First Quarter | | $ | 1.55 | | | $ | 0.22 | |
| Second Quarter | | $ | 2.40 | | | $ | 1.04 | |
| Third Quarter | | $ | 2.74 | | | $ | 2.10 | |
| Fourth Quarter | | $ | 2.14 | | | $ | 1.25 | |
| | | | | | | | | |
Fiscal Year Ended December 31, 2008 | | High | | | Low | |
| |
| First Quarter | | $ | 1.90 | | | $ | 1.10 | |
| Second Quarter | | $ | 2.14 | | | $ | 1.00 | |
| Third Quarter | | $ | 1.94 | | | $ | 0.66 | |
| Fourth Quarter | | $ | 0.81 | | | $ | 0.35 | |
31
| | |
| * | | The quotations reflect inter-dealer prices, without retailmark-up, mark-down or commission, and may not represent actual transactions. |
We have never paid or declared any cash or other dividends on our common stock. Furthermore, we are generally restricted by the terms of our outstanding indebtedness from making dividend payments to holders of our capital stock.
Information regarding our equity compensation plans and the securities authorized for issuance hereunder is set forth in Item 11 below.
Effective December 31, 2008, Cato Research Ltd., a vendor, agreed to convert approximately $140,000 of our obligations to them into 318,905 shares of our restricted common stock, $.01 par value.
We did not repurchase any shares of common stock during the year ended December 31, 2008.
During 2008, warrants to purchase 726,250 shares of our common stock were exercised voluntarily, providing gross proceeds of approximately $114,000.
On January 30, 2007, we repurchased all outstanding shares of our Series A Preferred Stock for approximately $73,000 (see Note 9 to our consolidated financial statements), as was required by our Second Amended and Restated Articles of Incorporation, as amended. At the time of the repurchase, we issued to the holders a common stock dividend in the amount of 10,487 shares of common stock, valued at $3,440, representing the amount of the accrued dividend for the period July 1, 2006 through January 30, 2007.
| |
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS. |
The following discussion of our consolidated financial condition and results of operations should be read in conjunction with the financial statements and the related notes thereto included elsewhere in thisForm 10-K. The matters discussed herein contain forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, and Section 27A of the Securities Act of 1933, as amended, which involve risks and uncertainties. All statements other than statements of historical information provided herein may be deemed to be forward-looking statements. Without limiting the foregoing, the words “believes”, “anticipates”, “plans”, “expects” and similar expressions are intended to identify forward-looking statements. Factors that could cause actual results to differ materially from those reflected in the forward-looking statements include, but are not limited to, those discussed in “Risk Factors” and elsewhere in this report and the risks discussed in our other filings with the SEC. Readers are cautioned not to place undue reliance on these forward-looking statements, which reflect management’s analysis, judgment, belief or expectation only as of the date hereof. We undertake no obligation to publicly revise these forward-looking statements to reflect events or circumstances that arise after the date hereof.
Organization
Echo Therapeutics, Inc. was formed through the merger of a wholly-owned subsidiary of Sontra Medical Corporation and Durham Pharmaceuticals Ltd. (doing business as Echo Therapeutics, Inc.) in September 2007. Previously, Durham Pharmaceuticals Ltd. was a majority-owned subsidiary of Cato BioVentures. Effective October 8, 2007, Sontra Medical Corporation, a Minnesota corporation, changed its name to Echo Therapeutics, Inc. (“Echo-MN”).
On June 9, 2008, Echo-MN entered into an Agreement and Plan of Merger (the “Merger Agreement”) with its wholly-owned subsidiary of the same name, Echo Therapeutics, Inc., a Delaware corporation (“Echo-DE”), in order to change Echo-MN’s state of incorporation from Minnesota to Delaware (the “Merger”). The Merger Agreement and Merger were approved by Echo-MN’s shareholders at Echo-MN’s Annual Meeting of the Shareholders on May 20, 2008. Pursuant to the Merger Agreement, Echo-MN merged with and into Echo-DE and Echo-DE is the surviving corporation.
The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. As of
32
December 31, 2008, the Company had cash of approximately $243,000, total debt of approximately $2,543,000 and an accumulated deficit of approximately $58,282,000. Through December 31, 2008, the Company has not been able to generate sufficient sales from its operations to cover its costs and operating expenses. Although the Company has been able to obtain unsecured and secured debt and issue securities through a series of private placements to raise capital in order to fund its operations, it is not known whether the Company will be able to continue this practice, or be able to obtain other types of financing to meet its cash operating expenses. This, in turn, raises substantial doubt about the Company’s ability to continue as a going concern. Subsequent to December 31, 2008, the Company received gross proceeds from its ongoingSeries A-2 Preferred Stock and Warrant Financing of approximately $350,000 (see Note 14). Additional financing is necessary to fund operations in 2009 and beyond. Management is currently pursuing additional private equity financing, and such financing is expected to be completed during 2009. However, no assurances can be given as to the success of these plans. The financial statements do not include any adjustments that might result from the outcome of these uncertainties.
Operating Plan
We are a medical device and specialty pharmaceutical company. We are developing a non-invasive, wireless, transdermal continuous glucose monitoring (tCGM) system for people with diabetes and for use in a clinical setting (initially in acute care), together with transdermal reformulations of specialty pharmaceutical products previously approved by the FDA.
We are principally involved with product development and clinical studies for our tCGM System. Our Symphony tCGM System is a next-generation, non-invasive (needle-free), wireless tCGM system designed to provide reliable, continuous blood glucose data conveniently and cost-effectively. Presently, we have completed development of our prototype tCGM System, including our proprietary skin permeation system and have engaged several development, engineering and manufacturing firms to assist us with all necessary final development efforts in connection with our plan to obtain regulatory approval through the FDA. We have completed several clinical studies over the past two years for the use of our tCGM System in acute care (hospital) and diabetes home use environments. We have engaged a consulting firm to evaluate our regulatory strategy for obtaining marketing approval from the FDA of our tCGM System. In order to complete our product development, clinical development programs and to obtain regulatory approval for our tCGM System, we will be required to raise substantial additional financing.
A continuous glucose monitoring system will improve disease management for people with diabetes and those in acute care environments. A product that meets or exceeds the development requirements has the potential to leap frog the competition in all point of care (POC) glucose testing market segments. Our technology and product engineering in development can be applied to an acute care monitoring focus and a patient home-use monitoring system for Diabetics. Much of the technology for these markets will be developed in parallel. There is a significant unmet clinical need for a continuous glucose monitor in an acute care setting and eventually on a hospital-wide basis. Presently, hospitals are not using CGM’s in hospital environments and are limited to using hand-held glucometers and labs to monitor one-point-in-time glucose readings under a pre-determined protocol. Additionally, the acute care monitoring product has fewer market barriers and technology risks than the Diabetic patient home use product, in addition to a potentially shortened regulatory path. Consequently, our strategic development focus is initially with the clinical setting monitoring product; however, technology evolution and clinical awareness achieved with this first product focus will support the development and regulatory approval process for the patient home-use monitoring system for Diabetics. This product strategy supports our technology/product evolution and our successful, to date, stepping stone regulatory approval strategy.
Our specialty pharmaceuticals pipeline, resulting from our acquisition of Durham Pharmaceuticals, Ltd. in September 2007, is based on our proprietary AzoneTStm transdermal drug reformulation technology. We believe that, despite their commercial success in large, chronic markets, many FDA-approved products with safety, efficacyand/or patient comfort and convenience issues that limit or prohibit their full commercial potential are amenable to our AzoneTS reformulation technology focused on improved dermal penetration. We are leveraging our AzoneTS dermal penetration technology to engineer and develop a broad range novel topical reformulations of commercially successful, FDA-approved products, generally in accordance with the FDA’s Section 505(b)(2) guidelines. Our lead AzoneTS product candidate is Durhalievetm, an AzoneTS topical reformulation of triamcinolone acetonide.
33
Durhalieve is covered by our New Drug Application (NDA) on file with the FDA for treatment of corticosteroid-responsive dermatoses. Also, we believe that Durhalieve has the potential to be an effective topical (needle-free) treatment for keloid scarring.
We have engaged a clinical research organization to advise and manage the final product development requirements for its Durhalieve drug candidate. Presently, we are working to satisfy the FDA’s remaining manufacturing and clinical study requirements necessary to secure FDA approval of Durhalieve.
Research and Development
A significant portion of our research and development expenses includes salaries paid to personnel and outside consultants and service providers, as well as for the cost of materials used in research and development, clinical studies, prototype manufacturing and related information technology and the allocation of facilities costs.
Following the acquisition of the AzoneTS-based technology, we have continued to advance the development programs for Durhalieve for the treatment of corticosteroid-responsive dermatoses and for the early stage AzoneTS reformulation drug candidates. Unfortunately, due to financial constraints, we have been unable to advance our AzoneTS product development programs as rapidly as we had originally anticipated and none of the specialty pharmaceutical development programs have been completed. If we are unable to complete our current planned development schedule for Durhalieve, then we may incur additional expenses to commercialize Durhalieve, which additional expenses cannot be estimated at this time. Should Durhalieve not receive FDA approval, then we may be required to write-off all or a portion of the intangible assets acquired as an impairment charge based on the specific facts and circumstances that will be evaluated at a future date.
We made a strategic decision to focus on the completion and approval of our Symphony tCGM System because we concluded that this product is the most advanced product (as to development effort) in our pipeline and, we believe, the regulatory path to FDA approval may be a more efficient process than our specialty pharmaceutical product candidates.
Critical Accounting Policies and Estimates
Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period.
On an ongoing basis, we evaluate our estimates and judgments for all assets and liabilities, including those related to stock-based compensation. We base our estimates and judgments on historical experience, current economic and industry conditions and on various other factors that are believed to be reasonable under the circumstances. This forms the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our consolidated financial statements.
Our significant accounting policies are described in “Item 7 — Financial Statements — Note 2 — Summary of Significant Accounting Policies.” We believe the critical accounting policies discussed below are those most important for an understanding of our financial condition and results of operations and require our most difficult, subjective or complex judgments.
Intangible Assets and Other Long-Lived Assets —We record intangible assets at acquisition date fair value. As a policy, we amortize our intangible assets using the straight-line method over their estimated useful lives, as follows: patents and licenses, two to 20 years; definite-lived core and developed technology, five to 25 years; and other intangible assets, over various periods. In connection with the ETI Acquisition, intangible assets related to contractual arrangements and technology are amortized over estimated useful lives of three (3) and eight (8) years, respectively, on a straight-line basis.
34
We use the income approach to determine the fair values of capitalizable purchased research and development. This approach calculates fair value by estimating the estimated purchased after-tax cash flows attributable to an in-process project over its useful life and then discounting these after-tax cash flows back to a present value. We base revenue assumptions on estimates of relevant market sizes, expected market growth rates, expected trends in technology and expected product introductions by competitors. In arriving at the value of the in-process projects, we consider, among other factors: the in-process projects’ stage of completion; the complexity of the work completed as of the acquisition date; the costs already incurred; the projected costs to complete; the contribution of core technologies and other acquired assets; the expected introduction date and the estimated useful life of the technology. We base the discount rate used to arrive at a present value as of the date of acquisition on the time value of money and medical technology investment risk factors. For the in-process projects that we acquired in connection with the ETI Acquisition, we used ranges of risk- adjusted discount rates to discount our projected cash flows of 17.8 percent to 27.1 percent. We believe that the estimated capitalizable purchased research and development amounts so determined represent the fair value at the date of the ETI Acquisition and do not exceed the amount a third party would pay for the projects.
Purchased Research and Development —Our purchased research and development represents the value of in-process projects that have not yet reached technological feasibility, have no alternative future uses and for which there are no reliable estimates of future cash flows. We expense the value attributable to these in-process projects at the time of the acquisition. If the projects are not successful or completed in a timely manner, we may not realize the financial benefits expected for these projects or for the acquisition as a whole. The purchased research and development relates to seven (7) new pharmaceutical product candidates using our AzoneTS reformulation technology related to the acquisition of ETI. These product candidates are in early stages of product development, and technological feasibility has not been achieved. There are no alternative uses of these product candidates and no reliable estimates of future cash flows for them. Accordingly, we determined that we may not realize any financial benefits, and therefore no capitalizable value was assigned to these product candidates in connection with the acquisition of ETI. The amount of purchased research and development in the Consolidated Statement of Operations for the year ended December 31, 2007 represented the excess value of the acquisition value of approximately $16.9 million over the fair value of the identifiable intangible assets and other assets of approximately $10.0 million.
Accounting for Impairment and Disposal of Long-Lived Assets —In accordance with SFAS No. 144, Accounting for the Impairment and Disposal of Long-Lived Assets, we review intangible assets subject to amortization quarterly to determine if any adverse conditions exist or a change in circumstances has occurred that would indicate impairment or a change in the remaining useful life. Conditions that would indicate impairment and trigger an impairment assessment include, but are not limited to, a significant adverse change in legal factors or business climate that could affect the value of an asset, or an adverse action or assessment by a regulator. If the carrying value of an asset exceeds its undiscounted cash flows, we write-down the carrying value of the intangible asset to its fair value in the period identified.
For purposes of this analysis, we estimate our cash flows using a projection period not exceeding ten years, market size based on estimated prescription volume, estimated market share, average pricing, estimated costs to complete product development, operating expenses and a blended tax rate. Generally, cash flow forecasts for purposes of cash impairment analysis are prepared on a consistent basis and using a consistent methodology as those prepared for the income approach used to initially estimate the intangible asset’s fair value.
If the carrying value of assets is determined not to be recoverable, we record an impairment loss equal to the excess of the carrying value over the fair value of the assets. Our estimate of fair value is based on the best information available to us, in the absence of quoted market prices.
We generally calculate fair value as the present value of estimated future cash flows that we expect to generate from the asset using the income approach as described above. Significant estimates included in the discounted cash flow analysis as consistent with those described above are used except that we introduce a risk-adjusted discount rate. The risk-adjusted discount rate is estimated using a weighted-average cost of capital analysis. If the estimate of an intangible asset’s remaining useful life is changed, we amortize the remaining carrying value of the intangible
35
asset prospectively over the revised remaining useful life. For other long-lived assets, we evaluate quarterly whether events or circumstances have occurred that indicate that the carrying value of these assets may be impaired.
Share-based Payments — We record share-based payments at fair value. The grant date fair value of awards to employees and directors, net of expected forfeitures, is recognized as expense in the statement of operations over the requisite service period. The fair value of options is calculated using the Black-Scholes option pricing model. This option valuation model requires input of assumptions including the volatility of our stock price, the expected life of the option and the risk-free interest rate. We estimate the volatility of our stock price using historical prices. We estimate the expected life of our option using the average of the vesting period and the contractual term of the option. The estimated forfeiture rate is based on historical forfeiture information as well as subsequent events occurring prior to the issuance of the financial statements. Because our stock options have characteristics significantly different from those of traded options, and because changes in the input assumptions can materially affect the fair value estimate, the existing model may not necessarily provide a reliable single measure of fair value of our stock options.
We believe that full consideration has been given to all relevant circumstances that we may be subject to, and the consolidated financial statements accurately reflect our best estimate of the results of operations, financial position and cash flows for the periods presented.
Results of Operations
As of December 31, 2008, we had eleven (11) employees and used the services of two (2) independent contractors. Of this group of employees and contractors, five (5) are involved with finance and administration and eight (8) are involved with research and development, and clinical and regulatory matters. In addition to these individuals, we utilize outside contract engineering and contract manufacturing firms to support our operations. We have engaged a clinical research organization and several consulting firms involved with regulatory strategy and clinical trial planning. We believe that with sufficient funding during the next 12 months, we will increase the number of employees in the areas of clinical research and testing, customer development and product marketing.
As of December 31, 2007, we had ten (10) employees and used the services of six (6) independent contractors. Of this group of employees and contractors, five (5) were involved with finance and administration and eleven (11) were involved with research and development, and clinical and regulatory matters.
We conduct our operations in leased facilities and have agreed to a lease through March 2010 with an option for an extension of an additional year. Our property and equipment does not include manufacturing machinery and is limited to laboratory testing equipment, office furniture and computer systems (network hardware and software and employee desk top systems). We do not anticipate any significant purchases or sales of property and equipment during the next 12 months.
Comparison of the years ended December 31, 2008 and 2007
Licensing Revenue — Licensing revenue for the year ended December 31, 2008 was zero compared to approximately $46,000 for the year ended December 31, 2007. License revenue was realized through an agreement with the Horticulture and Food Research Institute of New Zealand Limited (“HortResearch”) during the period November 2005 to November 2007 for the license of our ultrasonic skin permeation technology. Under the agreement, we received $100,000 and were required to perform certain training and consulting services over a one year period. In November 2007, HortResearch did not renew its license option.
Product Revenue and Cost of Product Revenue — We determined in the third quarter of 2007 that marketing and selling our SonoPrep System as a stand-alone product opportunity was not commercially relevant to our long-term strategic plan; therefore, we ceased actively marketing and selling the product. We recorded no product revenue or cost of product revenue in the year ended December 31, 2008 versus recording approximately $12,000 of revenue with a cost of product revenue of approximately $2,000 for the year ended December 31, 2007.
Research and Development Expenses — Research and development expenses increased by approximately $1,885,000 to approximately $3,191,000 for the year ended December 31, 2008 from approximately $1,306,000 for the year ended December 31, 2007. Research and development expenses increased primarily as a result of our
36
increased product and clinical development with our Symphony and Prelude Systems. Research and development expenses amounted to approximately 46% of total operating expenses during the year ended December 31, 2008, and included product development expenses for our Prelude skin permeation technology, product development for our Symphony transdermal continuous glucose monitoring (tCGM) system, clinical study expenses (including contract research organizations) associated with our tCGM system in hospital critical care and ambulatory settings, regulatory consulting expenses related to our medical devices and specialty pharmaceutical products, and research expenses related to our specialty pharmaceutical product candidate, Durhalieve. Product development and clinical expenses included in research and development expenses represented approximately 96% and 4%, respectively, of research and development expenses for the year ended December 31, 2008. During 2008 the Company conducted a critical care trial at a Boston hospital, conducted a diabetes home use clinical trial at a research clinic and completed numerous in-house clinical studies. We engaged several engineering and product development firms during 2008 for the purposes of developing our next generation Symphony and Prelude devices to be used for obtaining marketing approval with the FDA.
Product development and clinical expenses for the year ended December 31, 2007 included in research and development expenses represented prototype development and internal testing of our tCGM system and our next generation skin permeation technology and a pilot study of our tCGM system in a hospital critical care setting. Product development and clinical expenses amounted to approximately 94% and 6%, respectively, of research and development expenses for the year ended December 31, 2007.
Purchased Research and Development Expenses —As result of the ETI Acquisition in September 2007, we recognized approximately $7.0 million as purchased research and development expense in the year ended December 31, 2007 for projects with no alternative use and for which there were no reliable estimates of future cash flows.
Selling, General and Administrative Expenses — Selling, general and administrative expenses decreased by approximately $865,000 to approximately $3,736,000 for the year ended December 31, 2008 from approximately $4,601,000 for the year ended December 31, 2007. The net decrease was due to the combination of: a decrease of approximately $900,000 in share-based compensation expenses from $2,725,000 in 2007 to $1,825,000 in 2008; a decrease of $260,000 in officers’ bonuses from 2007 to 2008;, and an increase in 2008 compared with 2007 in investor relations costs, additional public company costs related to legal, accounting, printing and media and expanded capital raising costs. Selling, general and administrative expenses represented 54% and 36% of total operating expenses during the years ended December 31, 2008 and 2007, respectively. Share-based compensation expenses are non-cash charges relating to the fair value of restricted common stock, options and warrants to purchase our common stock issued to employees, directors and certain service providers. We are not engaged in selling activities and accordingly, general and administrative expenses relate to salaries and benefits for our executive, financial and administrative staff, public company costs related to investor relations, legal, accounting, printing and media costs, capital-raising costs, including those associated with travel, legal and accounting services, and costs for general operations.
Other Income (Expense) — Interest income was approximately $25,000 for the year ended December 31, 2008 compared to interest income of approximately $56,000 for the year ended December 31, 2007, a decrease of $31,000. The decrease in interest income for the year ended December 31, 2008 was primarily attributable to our lower average amount of cash equivalents and short term investments on hand during 2008 compared to 2007.
Interest Expense — Interest expense was approximately $1,050,000 for the year ended December 31, 2008, compared to interest expense of approximately $436,000 for the year ended December 31, 2007, an increase of $614,000. The increase in interest expense is due principally to an increase in the amount of financings through the use of convertible and secured note arrangements. Bridge Notes in the amount of approximately $1,325,000 issued on September 14, 2007 were either exchanged for Senior Convertible Notes or converted into shares of our common stock in February 2008. Senior Convertible Notes in the amount of approximately $2,078,000 were exchanged for Series A Convertible Preferred Stock on September 30, 2008. Secured Senior Notes were issued on March 24, April 24, June 2, and June 24, 2008 with a principal amount in the aggregate of approximately $2,200,000. In connection with these financings, we incurred non-cash interest expense of approximately $1,037,000 related primarily to the amortization of discounts on notes payable, the amortization of deferred financing costs and interest
37
satisfied through the issuance of promissory notes. Interest expense of approximately $9,000 for the year ended December 31, 2007 related to an outstanding note payable on equipment financing secured in 2005. The entire outstanding note payable on equipment financing was paid in full in September 2007.
Derivative Loss — Due to certain requirements to obtain and maintain an effective registration statement covering the shares of common stock underlying the Imperium Warrants, we originally determined that the Imperium Warrants did not meet the requirements for classification as equity as described in EITF IssueNo. 00-19,“Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock.” As a result, the fair value of the Imperium Warrants was recorded as a derivative liability which resulted in the recognition of a derivative loss upon issuance in the amount of approximately $569,000 in March 2008. As of May 14, 2008, the Company and Imperium executed Amendment No. 1 to the Registration Rights Agreement and, as a result, the Imperium Warrants were no longer required to be recorded as a derivative liability. The derivative liability was adjusted to fair value at May 14, 2008 with the net increase in the fair value of approximately $18,000 being recorded as an additional derivative loss.
Loss on Extinguishment of Debt — We determined that the terms of the Senior Convertible Notes are deemed “substantially different,” as described in EITF IssueNo. 96-19, “Debtor’s Accounting for a Modification or Exchange of Debt Instruments,” from the terms of the Bridge Notes based on the change in the fair value of the embedded conversion features. As a result, we recorded the Senior Convertible Notes issued in exchange for the Bridge Notes at fair value on the date of issuance and recorded a loss on extinguishment of debt of approximately $586,000. The fair value of the warrants issued to the holders of the Bridge Notes upon conversion into the Senior Convertible Notes, which we estimated to be approximately $626,000, was also included in the loss on extinguishment of debt. The difference between the fair value and the face value of the Senior Convertible Notes is being accreted to interest expense over the term of the notes. On September 30, 2008, the Company exchanged an aggregate principal amount of $2,077,868 of its Senior Convertible Notes, including notes for amounts related to interest expense, for 1,539,161 shares of Series A Preferred Stock and five-year warrants to purchase 153,912 shares of the Company’s common stock at an exercise price of $1.00 per share. The Company recorded an extinguishment loss of approximately $845,000 in connection with this exchange.
Net Loss — As a result of the factors described above, we had a net loss of approximately $10,596,000 for the year ended December 31, 2008 compared to approximately $13,226,000 for the year ended December 31, 2007.
Liquidity and Capital Resources
We have financed our operations since inception primarily through private sales of our common stock and preferred stock, the issuance of convertible promissory notes and secured promissory notes, and cash received in connection with exercises of common stock purchase options and warrants. As of December 31, 2008, we had approximately $243,000 of cash and cash equivalents, with no other short term investments.
Net cash used in operating activities was approximately $3,945,000 for the year ended December 31, 2008. The use of cash in operating activities was primarily attributable to the net loss of approximately $10,596,000 for the year ended December 31, 2008, offset by non-cash expenses of approximately $587,000 for derivative losses, approximately $192,000 for depreciation and amortization, approximately $1,825,000 for share-based compensation expense, approximately $2,057,000 for debt extinguishment losses due to promissory note modifications and exchanges, and approximately $1,037,000 in non-cash interest expense relating to the Senior Convertible Promissory Notes and Secured Senior Promissory Notes. An increase in accounts payable provided approximately $933,000 of cash. Changes in other current assets and current liabilities used net cash of approximately $227,000.
Net cash used in investing activities was approximately $29,000 for the year ended December 31, 2008. The cash was used to purchase property and equipment
Net cash provided by financing activities was approximately $3,023,000 for the year ended December 31, 2008. The source of cash was primarily attributable to the proceeds from Secured Senior Convertible Promissory Notes of $2,000,000, proceeds from the exercise of warrants and options of approximately $114,000, proceeds from the issuance of the Senior Convertible Promissory Notes of $700,000 and proceeds from the sale ofSeries A-1
38
preferred stock of approximately $705,000. Approximately $496,000 was used to pay various deferred financing costs that are amortized to interest expense over the life of the Promissory Notes.
At December 31, 2008, we had outstanding warrants to purchase 4,432,828 shares of common stock at exercise prices ranging from $0.21 to $50.00.
As of April 8, 2009, we had cash and cash equivalents of approximately $310,000.
Our management continues to aggressively pursue additional financing from existing relationships (prior shareholders, investors and lenders) and from new investors. In 2008, we engaged an investment banking firm to assist us with these efforts. Our ability to incur additional indebtedness is limited due to the need to obtain consent of the holder under the terms of our Secured Notes. In addition, our agreement with the holder of the Secured Notes contains a number of restrictive covenants, including a requirement to maintain stockholders’ equity, as defined, equal to or in excess of $6.5 million. In order to advance our product development, clinical programs and financing activities, we expect our monthly operating costs associated with salaries and benefits, consulting costs, legal costs and other working capital costs to increase. Accordingly, we have relied primarily on raising equity capital in order to continue our operations in order to achieve our business objectives while continuing to satisfy the restrictive covenants of the Secured Notes, and we plan to continue to do so in 2009.
For additional information about our operating plan with respect to our products and product development, please see our Business discussion in Item 1 above.
The current economic conditions have had a significant impact on our ability to raise necessary capital to fund our product development and clinical programs in accordance with our original projected level of operations. We believe that uncertainties in the financial markets have limited the availability of financing for us. During 2009, our product development, clinical programs and FDA meetings and communication plans will be conducted to the extent possible based on available funding from investors. Without sufficient funding for our programs, our progress to obtain regulatory approval for our medical device product Symphony tCGM System and our specialty pharmaceutical candidate Durhalieve may be delayed.
We have the ability to manage our costs aggressively and increase our operating efficiencies while continuing our current level of product development and clinical programs thereby maximizing the time available to complete an equity financing. Delays in obtaining an equity financing could cause us to delay or halt our product development and clinical programs. Although we believe that the required financing can be completed, there can be no guarantee that additional equity capital will be available on terms favorable to us, if at all.
Even if we are successful in raising additional debt or equity capital during 2009, we will still be required to raise substantial additional capital in the future to fund our research and development programs, commercialize our product candidates and achieve profitability. Our ability to fund our future operating requirements will depend on many factors, including the following:
| | |
| | • | our ability to obtain funding from third parties, including any future collaborative partners, on reasonable terms; |
| |
| | • | our progress on research and development programs; |
| |
| | • | the time and costs required to gain regulatory approvals; |
| |
| | • | the costs of manufacturing, marketing and distributing our products, if successfully developed and approved; |
| |
| | • | the costs of filing, prosecuting and enforcing patents, patent applications, patent claims and trademarks; |
| |
| | • | the status of competing products; and |
| |
| | • | the market acceptance and third-party reimbursement of our products, if successfully developed and approved. |
Discount and Credit Arrangement with Cato Research —In connection with the ETI Acquisition, we entered into a Strategic Master Services Agreement with Cato Research (“Cato Master Agreement”) to provide us with contract research and development services and regulatory advice (“CRO Services”). The Cato Master Agreement
39
has an initial one year term, and is automatically renewable for additional one-year terms unless either party provides sixty days’ notice prior to the expiration of any one year term. The Cato Master Agreement provides an initial $80,000 credit, of which $38,400 and $41,600 was used in 2008 and 2007, respectively, for CRO Services and an ongoing 25% discount on all CRO Services rendered on a time and materials basis during the term of the agreement.
January 2007 Strategic Private Placement — On January 30, 2007, we issued and sold in a strategic private placement an aggregate of 6,600,000 shares of common stock at a purchase price per share of $0.10, for an aggregate purchase price of $660,000. We also issued warrants to purchase an aggregate of 1,650,000 shares of our common stock for an exercise price of $0.21 per share. The warrants have a term of two years and contain customary provisions for adjustment to the exercise price in the event of stock splits, combinations and dividends and weighted average anti-dilution provisions that provide for an adjustment to the then effective exercise price (and number of shares of common stock issuable upon exercise) upon certain dilutive issuances of equity securities. In addition, if the per share market value (as defined in the warrants) of the common stock for any twenty (20) consecutive trading days equals or exceeds $0.63 per share, then we may, with the prior written consent of the warrant holders, redeem the unexercised portion of the warrants in cash at a price equal to the number of shares of common stock that remain subject to the warrant multiplied by $0.001. During 2007, Warrants to purchase 312,500 shares of our common stock were exercised voluntarily, providing proceeds of $64,702.
In December 2007, we requested from the holders of these warrants the right to redeem for $0.001 per share the then outstanding warrants to purchase 1,506,250 shares of our common stock issued in our January 2007 strategic private placement. In connection with this request, no warrants were redeemed during 2007 and warrants to purchase 168,750 shares of our common stock were exercised voluntarily for gross proceeds to us of approximately $35,000.
During 2008 and through March 25, 2009, no warrants have been redeemed and warrants to purchase 738,750 shares (including warrants to purchase 193,750 shares that were exercised through a cash-less exercise provision) of our common stock were exercised voluntarily for gross proceeds to us of approximately $114,100. As of January 30, 2009, warrants to purchase 62,500 shares of common stock expired and warrants to purchase 536,250 shares of common stock were modified to extend their contractual life though January 30, 2014.
June and July 2007 Private Placements — During June and July 2007, we completed a total of three closings of a private placement of our common stock which provided us with aggregate gross proceeds of $1,815,000 (net proceeds were approximately $1,641,000) (the “June 2007 Financing”). The investors in the June 2007 Financing purchased 1,815,000 shares of our common stock at a per share purchase price of $1.00 and received five-year warrants to purchase an aggregate of 544,500 shares of common stock at a per share exercise price of $1.40 (the “June 2007 Warrants”) . We have the right to terminate the June 2007 Warrants, upon fifteen (15) days’ notice, in the event (i) the closing bid price of our common stock for twenty-two (22) consecutive trading days is equal to or greater than $3.00 per share and (ii) we have registered for resale the shares of common stock underlying the June 2007 Warrants, provided that the registered holder has the right to exercise the June 2007 Warrant at any time prior to such termination. The June 2007 Warrants contain customary provisions for adjustment to the exercise price in the event of stock splits, combinations and dividends. The June 2007 Warrants also contain weighted average anti-dilution provisions that provide for an adjustment to the then effective exercise price if we issue equity securities without consideration or for consideration less than $1.00 per share.
Senior Promissory Bridge Notes — On September 14, 2007, we completed a private placement of unsecured Senior Promissory Bridge Notes (the “Bridge Notes”) in the aggregate principal amount of $1,325,000 to strategic institutional and individual accredited investors. The Bridge Notes were scheduled to be due September 15, 2008 and accrue interest at a rate of 10% per annum, with interest payable upon maturity. The terms of the Bridge Notes provided that if we engaged in a subsequent equity or equity linked financing or a combination of equity financings resulting in gross proceeds totaling at least $2,500,000 on or before December 15, 2007, inclusive of the Bridge Notes (a “Qualified Financing”), then the Bridge Notes would be converted automatically into the equity securities issued in the Qualified Financing. The terms of the Bridge Notes reflected a 20% premium in the event of an exchange such that upon an automatic exchange of the Bridge Notes in any Qualified Financing, the holders of the
40
Bridge Notes would be deemed to have tendered an amount equal to 120% of the outstanding principal and interest of the Bridge Notes in exchange for the equity securities issued to such holders in the Qualified Financing.
In 2008, all Bridge Notes were converted either to common stock or to Senior Convertible Notes (seeSenior Convertible Notesbelow).
Senior Convertible Notes — On February 11, 2008, we completed an approximately $2,300,000 private financing with Montaur Capital through Platinum Long Term Growth VII, LLC and certain other select institutional and strategic investors of unsecured senior convertible notes, due February 12, 2011 (the “Senior Convertible Notes”) and warrants. The $2,292,459 in aggregate principal amount of Senior Convertible Notes issued in the financing bear interest annually at a rate of 8.0% per annum and will provide investors with the right to convert principal into shares of our common stock at $1.35 per share. The conversion price is subject to weighted average anti-dilution protection, excluding certain customary exceptions.
Additionally, the investors received warrants to purchase 849,059 shares of common stock at an exercise price of $1.00 per share (as of December 31, 2008) for a term of five years. The warrants provide for full anti-dilution protection to the holders and allow for cashless exercise.
In connection with the financing, certain holders in the principal amount of $1,275,000 of Bridge Notes exchanged their Bridge Notes at 120% of the outstanding principal and interest as payment toward the purchase price of the Senior Convertible Notes purchased by such holders. Accordingly, we issued Senior Convertible Notes in the financing in the aggregate principal balance of $1,592,459 to the former holders of the Bridge Notes upon their surrender of the Bridge Notes, and we received gross cash proceeds in the amount of $700,000 in connection with the financing. One holder in the principal amount of $50,000 of our Bridge Notes converted the principal and accrued interest into 52,041 shares of our common stock, at a $1.00 conversion price as provided in the terms of the Bridge Notes.
Interest on the Senior Convertible Notes is payable quarterly. We have the right to repay the principal amount of the Senior Convertible Notes in cash, in whole or in part, prior to maturity, and cash or shares of common stock in an amount equal to the amount of interest that would have otherwise accrued from the date of prepayment to either the earlier of (1) six months after such prepayment or (ii) the maturity date, subject to certain restrictions.
For so long as at least 25% of the principal amount of Senior Convertible Notes are outstanding, we may not permit any of our subsidiaries to incur certain additional indebtedness (excluding certain indebtedness the principal amount of which cannot exceed $5,000,000, subject to certain restrictions) without the prior written consent of the holders of at least a majority of the aggregate principal amount of the Senior Convertible Notes outstanding.
Our subsidiary, Sontra Medical, Inc. agreed to guarantee our obligations under the Senior Convertible Notes pursuant to a separate guaranty agreements. Additionally, for so long as any Senior Convertible Notes or warrants issued in connection with those notes remain outstanding, we agree that we will not, or permit our subsidiaries to, declare or pay any dividends or make any distribution to any holders of common stock or purchase or acquire any of its common stock or equity securities.
We paid Burnham Hill Partners $162,000 in connection with the Senior Convertible Note financing and provided to it and its designees warrants to purchase an aggregate of 175,013 shares of common stock at an exercise price of $1.49 per share.
2008 Secured Note and Warrant Financing —On March 24, 2008, we entered into a Secured Note financing with Imperium Master Fund, Ltd. (“Imperium”), providing for, at our option, the issuance of up to an aggregate $2,000,000 of original issue discount senior secured notes (the “Secured Notes”) in four equal tranches, together with warrants (the “Imperium Warrants”) to purchase up to 653,728 shares of our common stock at an exercise price of $1.94 per share as of December 31, 2008. On March 24, 2008, we drew down the initial $500,000 in gross proceeds and issued the Imperium Warrants upon execution of the agreement. We also issued additional Secured Notes and drew down $500,000 in gross proceeds on each of April 24, 2008, June 2, 2008 and June 24, 2008, completing the financing for $2,000,000.
We are not required to make monthly cash payments of principal and interest under our Secured Notes. Instead, the outstanding principal of each Secured Note will accrete in value at an annual rate of 10%, compounded monthly,
41
resulting in a total principal amount of approximately $552,357 due for each Secured Note at maturity. If, however, we complete an equity issuance in one or more series of transactions totaling $5,000,000 (a “Qualified Issuance”), then the aggregate amount due for each Secured Note will be reduced from $552,357 to $546,903 and the annual accretion value will be reduced from 10% to 9%.
Each Secured Note is due twelve months after the date of the issuance, provided, however, that if we completed a Qualified Issuance by October 31, 2008, we had a right to extend the maturity date of each Secured Note to 24 months after the date of the issuance. We did not complete a Qualified Issuance by October 31, 2008. We have the right to repay the principal amount of the Secured Notes in cash, in whole, but not in part, prior to maturity at a premium of 1.02 times the unpaid principal plus any other amount due under the Secured Notes.
In addition, we agreed to certain covenants, including a prohibition on an ability to incur future indebtedness (subject to certain exceptions) or make any dividend or payment to holders of our capital stock (other than shares of the class of stock held by such recipient), and a requirement that we maintain $6,500,000 in stockholders’ equity (excluding any impact of the issuance of the Imperium Warrants to stockholders’ equity) for so long as the Secured Notes remain outstanding. We also agreed that we may not redeem our Senior Convertible Notes as long as the Secured Notes remain outstanding, unless we receive at least $10 million in gross proceeds from an issuance or series of related issuances of equity securities.
Our subsidiary, Sontra Medical, Inc., guaranteed our obligations under the Secured Notes. Additionally, the Secured Notes are secured by all of our assets.
The Imperium Warrants have a term of five years and are immediately exercisable at an exercise price of $1.89 per share (as of April 8, 2009). The Imperium Warrants provide for weighted average exercise price and number of warrant shares anti-dilution protection upon future issuances or deemed issuances (subject to customary exceptions) below the exercise price.
In connection with the issuance of the Secured Notes and Imperium Warrants, on March 24, 2008, we entered into a registration rights agreement pursuant to which we have agreed to file a registration statement with the Securities and Exchange Commission covering the resale of the common stock issuable upon exercise of the Imperium Warrants within 60 days after issuance of the securities. We also agreed to use our best efforts to cause the registration statement to become effective under the Securities Act as soon as practicable after the filing of the registration statement, but no later than 180 days after issuance of the Imperium Warrants. As of May 14, 2008, we entered into Amendment No. 1 to the Registration Rights Agreement and eliminated the requirement for registration of the underlying securities, but granted the holders of the Imperium Warrants piggy-back registration rights with respect to certain registration statement filings by us in the future.
2008 Series A Preferred Stock Exchange Agreement —On September 30, 2008, we entered into an Exchange Agreement (the “Exchange Agreement”) with substantially all of the holders of the Senior Convertible Notes (collectively, the “Investors”). The Investors are the holders of an aggregate of $1,980,212 in principal amount of the Senior Convertible Notes and an aggregate $97,656 in principal amount of additional notes issued as interest on the Senior Convertible Notes (collectively, the “Notes”), for a total aggregate of $2,077,868 in principal amount of Notes constituting all of the issued and Senior Convertible Notes, except for the aggregate $318,475 in principal amount of notes held by Gemini Master Fund Ltd. (“Gemini”), who was not a party to the Exchange Agreement. Accrued interest of $19,201 payable to Gemini for the year ended December 31, 2008 was paid by issuing additional notes. The Senior Convertible Notes held by Gemini remain outstanding with original terms. The Gemini Notes are convertible into shares of the Company’s common stock at the option of the holder at a price per share of $1.35, subject to adjustment for stock splits, combinations or similar events and subject to customary weighted average anti-dilution adjustments.
Pursuant to the terms of the Exchange Agreement, the Company issued and delivered to the Investors, in exchange for the cancellation of the Notes, 1,539,161 shares of the Series A Convertible Preferred Stock, plus 14 fractional shares to be settled in cash at $1.00 per share, and five-year warrants to purchase 153,912 shares of common stock at an exercise price of $1.00 per share, subject to adjustment for stock splits, combinations or similar events.
42
2008 Vendor Obligation Conversion —As of December 31, 2008, the Company issued 318,905 shares of its Common Stock to Cato in settlement of a payable in the amount of $140,318.
2009Series A-2 Preferred Stock Financing —On March 6, March 13, and April 8, 2009, we entered into an Amended and Restated Stock and Warrant Purchase Agreement with certain select institutional and strategic accredited investors (the“A-2 Investors”) in connection with a private placement transaction (the“A-2 Financing”) in which theA-2 Investors purchased an aggregate of 700,000 shares of ourSeries A-2 Convertible Preferred Stock at a per share price of $.50 (the“A-2 Shares”) and received warrants to purchase a number of shares of our Common Stock equal to (i) thirty-five percent (35%) or (ii) for investments of $250,000 or more, fifty percent (50%) of the number ofA-2 Shares purchased by eachA-2 Investor at an exercise price per share equal to $.75. Each share ofSeries A-2 Preferred Stock is initially convertible into one share of Common Stock, subject to adjustment for stock splits, combinations or similar events. As of April 8, 2009, theA-2 Financing provided us with gross proceeds of $350,000. We intend to use the net proceeds for working capital and general corporate purposes.
2008Series A-1 Preferred Stock Financing —On October 28, 2008 and October 31, 2008, we entered into a Stock and Warrant Purchase Agreement with certain select institutional and strategic accredited investors (the“A-1 Investors”) in connection with a private placement transaction (the“A-1 Financing”) in which theA-1 Investors purchased an aggregate of 766,667 shares of ourSeries A-1 Convertible Preferred Stock at a per share price of $1.00 (the“A-1 Shares”) and received warrants to purchase a number of shares of the our Common Stock equal to thirty-five percent (35%) of the number ofA-1 Shares purchased by eachA-1 Investor at an exercise price per share equal to $1.50. Each share ofSeries A-1 Preferred Stock is initially convertible into one share of common stock, subject to adjustment for stock splits, combinations or similar events. TheA-1 Financing provided us with gross proceeds of $766,667. We intend to use the net proceeds for working capital and general corporate purposes.
As of December 31, 2008, the Company satisfied 8% quarterly dividend commitments for its Series A Shares andSeries A-1 Shares with the issuance of additional preferred stock of 31,033 shares and 11,469 shares, respectively.
We conduct our operations in leased facilities and have agreed to a lease through March 2010 with an option for an extension of an additional year. Our property and equipment does not include manufacturing machinery and is limited to laboratory testing equipment, office furniture and computer systems (network hardware and software and employee desk top systems). We do not anticipate any significant purchases or sales of property and equipment during the next 12 months.
We anticipate that no hiring of additional employees will occur until sufficient funding has been obtained.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements, including derivative instruments that have or are reasonably likely to have a current or future material effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources. We have certain warrants and options outstanding but we do not expect to receive any material proceeds from the exercise of these instruments unless and until the trading price of our common stock is significantly greater than the applicable exercise prices of the options and warrants and following any necessary registering of underlying securities.
Effect of Inflation and Changes in Prices
Management does not believe that inflation and changes in price will have a material effect on our operations.
43
| |
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA. |
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Echo Therapeutics, Inc. Consolidated Financial Statements
| | | | | |
| | | Page | |
| |
| | | F-1 | |
| | | F-2 | |
| | | F-3 | |
| | | F-4 | |
| | | F-5 | |
| | | F-7 | |
44
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors of
Echo Therapeutics, Inc.
Franklin, Massachusetts
We have audited the accompanying consolidated balance sheets of Echo Therapeutics, Inc. as of December 31, 2008 and 2007, and the related consolidated statements of operations, changes in stockholders’ equity and cash flows for the years then ended. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall consolidated financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Echo Therapeutics, Inc. as of December 31, 2008 and 2007, and the results of its operations and its cash flows for the years then ended in conformity with U.S. generally accepted accounting principles.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations, has a significant accumulated deficit, has a significant working capital deficit and has been unable to raise sufficient capital to fund its operations. This raises substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ WOLF & COMPANY, P.C.
Boston, Massachusetts
April 9, 2009
F-1
Echo Therapeutics, Inc.
(Formerly Sontra Medical Corporation)
Consolidated Balance Sheets
| | | | | | | | | |
| | | As of, | |
| | | December 31,
| | | December 31,
| |
| | | 2008 | | | 2007 | |
| |
ASSETS |
| Current Assets: | | | | | | | | |
| Cash and cash equivalents | | $ | 242,867 | | | $ | 1,193,163 | |
| Prepaid expenses and other current assets | | | 17,132 | | | | 25,263 | |
| | | | | | | | | |
| Total current assets | | | 259,999 | | | | 1,218,426 | |
| | | | | | | | | |
| Property and Equipment, at cost: | | | | | | | | |
| Computer equipment | | | 242,495 | | | | 233,946 | |
| Office and laboratory equipment | | | 609,029 | | | | 588,498 | |
| Furniture and fixtures | | | 14,288 | | | | 14,288 | |
| Manufacturing equipment | | | 129,320 | | | | 197,888 | |
| Leasehold improvements | | | 177,768 | | | | 177,768 | |
| | | | | | | | | |
| | | | 1,172,900 | | | | 1,212,388 | |
| Less-Accumulated depreciation and amortization | | | (1,105,330 | ) | | | (1,100,507 | ) |
| | | | | | | | | |
| Net property and equipment | | | 67,570 | | | | 111,881 | |
| | | | | | | | | |
| Other Assets: | | | | | | | | |
| Restricted cash | | | 10,250 | | | | 10,250 | |
| Intangible assets, net of accumulated amortization | | | 9,827,154 | | | | 9,945,486 | |
| Deferred financing costs, net of accumulated amortization | | | 141,373 | | | | — | |
| Deposits and other assets | | | 2,000 | | | | 2,000 | |
| | | | | | | | | |
| Total other assets | | | 9,980,777 | | | | 9,957,736 | |
| | | | | | | | | |
| Total assets | | $ | 10,308,346 | | | $ | 11,288,043 | |
| | | | | | | | | |
| |
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
| Current Liabilities: | | | | | | | | |
| Accounts payable | | $ | 1,177,719 | | | $ | 382,308 | |
| Current portion of notes payable, net of discounts | | | 2,054,062 | | | | 386,458 | |
| Accrued expenses | | | 174,768 | | | | 451,136 | |
| | | | | | | | | |
| Total current liabilities | | | 3,406,549 | | | | 1,219,902 | |
| | | | | | | | | |
| Notes Payable, net of current portion and discounts | | | 300,467 | | | | — | |
| | | | | | | | | |
| Commitments | | | | | | | | |
| Stockholders’ Equity: | | | | | | | | |
| Convertible Preferred Stock, $0.01 par value, authorized 10,000,000 and 7,000,000 shares at December 31, 2008 and 2007, respectively: | | | | | | | | |
| Series A, authorized 2,636,363 shares and none, issued and outstanding 1,570,194 shares and none, at December 31, 2008 and 2007, respectively (preference in liquidation of $2,119,762 at December 31, 2008) | | | 15,702 | | | | — | |
Series A-1, authorized 2,000,000 and none, issued and outstanding 778,136 shares and none, at December 31, 2008 and 2007, respectively (preference in liquidation of $778,136 at December 31, 2008) | | | 7,781 | | | | — | |
| Common stock, $0.01 par value, authorized 60,000,000 shares, issued and outstanding 19,095,838 shares and 17,870,804 shares at December 31, 2008 and 2007, respectively | | | 190,960 | | | | 178,710 | |
| Additional paid-in capital | | | 64,668,550 | | | | 57,575,593 | |
| Accumulated deficit | | | (58,281,663 | ) | | | (47,686,162 | ) |
| | | | | | | | | |
| Total stockholders’ equity | | | 6,601,330 | | | | 10,068,141 | |
| | | | | | | | | |
| Total liabilities and stockholders’ equity | | $ | 10,308,346 | | | $ | 11,288,043 | |
| | | | | | | | | |
See report of independent registered public accounting firm and notes to the consolidated financial statements.
F-2
Echo Therapeutics, Inc.
(Formerly Sontra Medical Corporation)
Consolidated Statements of Operations
| | | | | | | | | |
| | | For the Years Ended
| |
| | | December 31, | |
| | | 2008 | | | 2007 | |
| |
| Revenue: | | | | | | | | |
| Product revenue | | $ | — | | | $ | 12,120 | |
| Licensing revenue | | | — | | | | 45,833 | |
| | | | | | | | | |
| Total revenue | | | — | | | | 57,953 | |
| | | | | | | | | |
| Operating Expenses: | | | | | | | | |
| Cost of product revenue | | | — | | | | 1,556 | |
| Research and development | | | 3,191,060 | | | | 1,306,424 | |
| Purchased research and development | | | — | | | | 6,994,578 | |
| Selling, general and administrative | | | 3,735,640 | | | | 4,600,627 | |
| | | | | | | | | |
| Total operating expenses | | | 6,926,700 | | | | 12,903,185 | |
| | | | | | | | | |
| Loss from operations | | | (6,926,700 | ) | | | (12,845,232 | ) |
| | | | | | | | | |
| Other Income (Expense): | | | | | | | | |
| Interest income | | | 24,641 | | | | 55,635 | |
| Interest expense | | | (1,049,953 | ) | | | (435,904 | ) |
| Loss on extinguishment of debt | | | (2,056,773 | ) | | | — | |
| Derivative loss | | | (586,716 | ) | | | — | |
| | | | | | | | | |
| Other income (expense), net | | | (3,668,801 | ) | | | (380,269 | ) |
| | | | | | | | | |
| Net loss | | | (10,595,501 | ) | | | (13,225,501 | ) |
Accretion of dividend on Series A andA-1 | | | | | | | | |
| Convertible Preferred Stock | | | (53,371 | ) | | | (483 | ) |
| | | | | | | | | |
| Net loss applicable to common shareholders | | $ | (10,648,872 | ) | | $ | (13,225,984 | ) |
| | | | | | | | | |
| Net loss per common share, basic and diluted | | $ | (0.57 | ) | | $ | (1.12 | ) |
| | | | | | | | | |
| Basic and diluted weighted average common shares outstanding | | | 18,608,905 | | | | 11,769,951 | |
| | | | | | | | | |
See report of independent registered public accounting firm and notes to the consolidated financial statements.
F-3
Echo Therapeutics, Inc.
(Formerly Sontra Medical Corporation)
Consolidated Statements of Changes in Stockholders’ Equity
Years Ended December 31, 2008 and 2007
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Convertible
| | | | | | | | | | | | | | | | |
| | | Preferred Stock | | | Common Stock | | | Additional
| | | | | | Total
| |
| | | Number of
| | | Carrying
| | | Number of
| | | Carrying
| | | Paid-in
| | | Accumulated
| | | Stockholders’
| |
| | | Shares | | | Value | | | Shares | | | Value | | | Capital | | | Deficit | | | Equity | |
| |
| Balance December 31, 2006 | | | 73,334 | | | $ | 76,291 | | | | 2,776,192 | | | $ | 27,762 | | | $ | 34,822,306 | | | $ | (34,460,661 | ) | | $ | 465,698 | |
| Dividend paid on Series A preferred stock | | | — | | | | (3,440 | ) | | | 10,487 | | | | 105 | | | | 3,335 | | | | — | | | | — | |
| Accretion of Series A preferred stock dividend | | | — | | | | 483 | | | | — | | | | — | | | | (483 | ) | | | — | | | | — | |
| Repurchase of Series A preferred stock | | | (73,334 | ) | | | (73,334 | ) | | | — | | | | — | | | | — | | | | — | | | | (73,334 | ) |
| Exercise of stock warrants to purchase common stock | | | — | | | | — | | | | 312,500 | | | | 3,126 | | | | 61,576 | | | | — | | | | 64,702 | |
| Exercise of stock options | | | — | | | | — | | | | 5,000 | | | | 50 | | | | 1,300 | | | | — | | | | 1,350 | |
| Issuance of common stock in private placement, net of issuance costs of $241,674 | | | — | | | | — | | | | 8,415,000 | | | | 84,150 | | | | 2,149,176 | | | | — | | | | 2,233,326 | |
| Issuance of common stock and warrants for placement agent fees (fair value of $241,539) | | | — | | | | — | | | | 54,750 | | | | 548 | | | | (548 | ) | | | — | | | | — | |
| Issuance of common stock in connection with ETI acquisition | | | — | | | | — | | | | 6,250,000 | | | | 62,500 | | | | 15,500,000 | | | | — | | | | 15,562,500 | |
| Fair value of warrants issued in connection with ETI acquisition | | | — | | | | — | | | | — | | | | — | | | | 989,215 | | | | — | | | | 989,215 | |
| Share-based payments — options, net of forfeitures | | | — | | | | — | | | | — | | | | — | | | | 2,609,811 | | | | — | | | | 2,609,811 | |
| Share-based payments — restricted stock for services, net of forfeitures | | | — | | | | — | | | | 46,875 | | | | 469 | | | | 114,905 | | | | — | | | | 115,374 | |
| Beneficial Conversion Feature — Senior Notes | | | — | | | | — | | | | — | | | | — | | | | 1,325,000 | | | | — | | | | 1,325,000 | |
| Net Loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (13,225,501 | ) | | | (13,225,501 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance December 31, 2007 | | | — | | | | — | | | | 17,870,804 | | | | 178,710 | | | | 57,575,593 | | | | (47,686,162 | ) | | | 10,068,141 | |
| Issuance of Common Stock for conversion of Bridge Notes | | | — | | | | — | | | | 52,041 | | | | 520 | | | | 51,521 | | | | — | | | | 52,041 | |
| Issuance of Common Stock in settlement of accounts payable and accrued expenses | | | — | | | | — | | | | 318,905 | | | | 3,189 | | | | 137,129 | | | | — | | | | 140,318 | |
| Intrinsic value of conversion feature on Bridge Notes upon conversion to Senior Convertible Notes | | | — | | | | — | | | | — | | | | — | | | | (212,328 | ) | | | — | | | | (212,328 | ) |
| Beneficial Conversion Feature — Senior Convertible Notes | | | — | | | | — | | | | — | | | | — | | | | 121,320 | | | | — | | | | 121,320 | |
| Issuance of Series A Preferred | | | 1,539,161 | | | | 15,392 | | | | — | | | | — | | | | 2,062,476 | | | | — | | | | 2,077,868 | |
Issuance ofSeries A-1 Preferred, net of issuance cost of $63,333 | | | 766,667 | | | | 7,667 | | | | — | | | | — | | | | 697,667 | | | | — | | | | 705,334 | |
| Dividends on Preferred Stock | | | 42,502 | | | | 424 | | | | — | | | | — | | | | (433 | ) | | | — | | | | (9 | ) |
| Exercise of January 2007 PIPE Warrants | | | — | | | | — | | | | 705,873 | | | | 7,059 | | | | 107,020 | | | | — | | | | 114,079 | |
| Exercise of Stock Options | | | — | | | | — | | | | 9,465 | | | | 95 | | | | (95 | ) | | | — | | | | — | |
| Warrants issued to Bridge Note holders upon conversion to Senior Convertible Notes | | | — | | | | — | | | | — | | | | — | | | | 626,480 | | | | — | | | | 626,480 | |
| Warrants issued to Senior Convertible Note holders | | | — | | | | — | | | | — | | | | — | | | | 219,838 | | | | — | | | | 219,838 | |
| Fair value of warrants issued in connection with exchange of Senior Convertible Notes for Series A Preferred Stock | | | — | | | | — | | | | — | | | | — | | | | 106,182 | | | | — | | | | 106,182 | |
| Fair value of warrants reclassified from derivative liability | | | — | | | | — | | | | — | | | | — | | | | 919,593 | | | | — | | | | 919,593 | |
| Fair value of warrants issued for services | | | — | | | | — | | | | — | | | | — | | | | 185,667 | | | | — | | | | 185,667 | |
| Share-based payments — restricted stock for services, net of forfeitures | | | — | | | | — | | | | 138,750 | | | | 1,387 | | | | 246,113 | | | | — | | | | 247,500 | |
| Share-based payments — options and restricted stock, net of forfeitures | | | — | | | | — | | | | — | | | | — | | | | 1,824,807 | | | | — | | | | 1,824,807 | |
| Net Loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (10,595,501 | ) | | | (10,595,501 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance December 31, 2008 | | | 2,348,330 | | | $ | 23,483 | | | | 19,095,838 | | | $ | 190,960 | | | $ | 64,668,550 | | | $ | (58,281,663 | ) | | $ | 6,601,330 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
See report of independent registered public accounting firm and the notes to the consolidated financial statements.
F-4
Echo Therapeutics, Inc
(Formerly Sontra Medical Corporation)
Consolidated Statements of Cash Flows
| | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2008 | | | 2007 | |
| |
Cash Flows From Operating Activities: | | | | | | | | |
| Net loss | | $ | (10,595,501 | ) | | $ | (13,225,501 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | |
| Depreciation and amortization | | | 191,723 | | | | 151,571 | |
| Share-based compensation | | | 1,824,807 | | | | 2,725,185 | |
| Purchased research and development | | | — | | | | 6,994,578 | |
| Fair value of common stock and warrants issued for services | | | 247,500 | | | | — | |
| Derivative loss | | | 586,716 | | | | — | |
| Loss on extinguishment of debt | | | 2,056,773 | | | | — | |
| Non-cash interest expense | | | 1,037,019 | | | | 386,458 | |
| Changes in assets and liabilities: | | | | | | | | |
| Inventory | | | — | | | | 1,556 | |
| Prepaid expenses and other current assets | | | 8,131 | | | | (8,317 | ) |
| Accounts payable | | | 933,049 | | | | 336,491 | |
| Deferred revenue | | | — | | | | (45,833 | ) |
| Accrued expenses | | | (234,858 | ) | | | 140,771 | |
| | | | | | | | | |
| Net cash used in operating activities | | | (3,944,641 | ) | | | (2,543,041 | ) |
| | | | | | | | | |
Cash Flows from Investing Activities: | | | | | | | | |
| Cash paid for Acquisition of Durham | | | — | | | | (219,815 | ) |
| Purchase of property and equipment | | | (29,080 | ) | | | (19,425 | ) |
| Decrease(Increase) in restricted cash | | | — | | | | 9,699 | |
| | | | | | | | | |
| Net cash used in investing activities | | | (29,080 | ) | | | (229,541 | ) |
| | | | | | | | | |
Cash Flows From Financing Activities | | | | | | | | |
| Repurchase of Series A preferred stock | | | — | | | | (73,334 | ) |
Proceeds from the sale ofSeries A-1 preferred stock | | | 705,334 | | | | — | |
| Proceeds from the sale of common stock, net of expenses | | | — | | | | 2,233,326 | |
| Proceeds from Secured Senior Convertible Notes and Warrants | | | 2,000,000 | | | | — | |
| Proceeds from Senior Convertible Promissory Notes | | | 700,000 | | | | 1,325,000 | |
| Deferred financing costs | | | (495,979 | ) | | | — | |
Dividends paid on Series A andA-1 preferred stock | | | (9 | ) | | | — | |
| Payments on equipment note payable | | | — | | | | (144,316 | ) |
| Proceeds from exercise of stock options | | | — | | | | 1,350 | |
| Proceeds from the exercise of warrants | | | 114,079 | | | | 64,702 | |
| | | | | | | | | |
| Net cash provided by financing activities | | | 3,023,425 | | | | 3,406,728 | |
| | | | | | | | | |
| Net (Decrease) Increase in Cash and Cash Equivalents | | | (950,296 | ) | | | 634,146 | |
| Cash and Cash Equivalents, beginning of period | | | 1,193,163 | | | | 559,017 | |
| | | | | | | | | |
| Cash and Cash Equivalents, end of period | | $ | 242,867 | | | $ | 1,193,163 | |
| | | | | | | | | |
See report of independent registered public accounting firm and the notes to the consolidated financial statements.
F-5
Echo Therapeutics, Inc
(Formerly Sontra Medical Corporation)
Consolidated Statements of Cash Flows
| | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2008 | | | 2007 | |
| |
| Supplemental Disclosure of Cash Flow Information and Non Cash Financing Cash paid for interest | | $ | 9,831 | | | $ | 10,041 | |
| | | | | | | | | |
Accretion of dividend on Series A andA-1 Convertible Preferred Stock | | $ | 53,371 | | | $ | 483 | |
| | | | | | | | | |
| Fair value of common stock issued for Durham acquisition | | $ | — | | | $ | 15,562,500 | |
| | | | | | | | | |
| Fair value of warrants issued for costs of Durham acquisition | | $ | — | | | $ | 989,215 | |
| | | | | | | | | |
| Fair value of common stock and warrants issued for equity financing costs | | $ | 166,901 | | | $ | 241,539 | |
| | | | | | | | | |
| Beneficial conversion feature on Senior Convertible Notes | | $ | 121,320 | | | $ | 1,325,000 | |
| | | | | | | | | |
| Common stock issued for conversion of Senior Convertible Notes and accrued interest | | $ | 52,041 | | | $ | — | |
| | | | | | | | | |
| Fair value of warrants issued for deferred financing costs | | $ | 185,667 | | | $ | — | |
| | | | | | | | | |
| Excess of fair value of the Series A Convertible Preferred Stock over the carrying value of the Senior Convertible Notes included in extinguishment loss | | $ | 738,578 | | | $ | — | |
| | | | | | | | | |
| Fair value of warrants issued in connection with Senior Convertible Notes included in extinguishment loss | | $ | 626,480 | | | $ | — | |
| | | | | | | | | |
| Adjustment to fair value of Bridge Notes exchanged for Senior Convertible notes included in extinguishment loss | | $ | 585,533 | | | $ | — | |
| | | | | | | | | |
| Fair value of warrants issued in connection with exchange of Senior Convertible Notes for preferred stock included in extinguishment loss | | $ | 106,182 | | | $ | — | |
| | | | | | | | | |
| Intrinsic value of the conversion feature of the Bridge Notes when converted to Senior Convertible Notes recorded as a reduction of additional paid-in capital | | $ | 212,328 | | | $ | — | |
| | | | | | | | | |
| Relative fair value of warrants issued with Secured Notes | | $ | 219,838 | | | $ | — | |
| | | | | | | | | |
| Reclassification of derivative liability to additional paid-in capital | | $ | 919,593 | | | $ | — | |
| | | | | | | | | |
| Senior Convertible Notes issued for accrued interest | | $ | 116,875 | | | $ | — | |
| | | | | | | | | |
| Conversion of Senior Convertible Notes to Series A preferred stock | | $ | 2,077,868 | | | $ | — | |
| | | | | | | | | |
| Issuance of common stock in settlement of accounts payable and accrued expenses | | $ | 140,318 | | | $ | — | |
| | | | | | | | | |
See report of independent registered public accounting firm and the notes to the consolidated financial statements.
F-6
(1) ORGANIZATION, BASIS OF PRESENTATION AND GOING CONCERN
Organization and Basis of Presentation
Echo Therapeutics, Inc. was formed through the merger of Sontra Medical Corporation (“Sontra”) and Durham Pharmaceuticals Ltd. (doing business as Echo Therapeutics, Inc. (“ETI”)) in September 2007 (the “Merger”). Previously, ETI was a majority-owned subsidiary of Cato BioVentures (“Cato”). Effective October 8, 2007, Sontra changed its name to Echo Therapeutics, Inc. (the “Company”).
On June 9, 2008, Echo-MN entered into an Agreement and Plan of Merger (the “Merger Agreement”) with a wholly-owned subsidiary of the same name, Echo Therapeutics, Inc., a Delaware corporation (“Echo-DE”), in order to change Echo-MN’s state of incorporation from Minnesota to Delaware (the “Merger”). The Merger Agreement and Merger were approved by Echo-MN’s shareholders at Echo-MN’s Annual Meeting of the Shareholders on May 20, 2008. Pursuant to the Merger Agreement, Echo-MN merged with and into Echo-DE and Echo-DE is the surviving corporation (the “Company”).
Each share of common stock, par value $0.01 per share, of Echo-MN that was issued and outstanding immediately prior to the Merger was converted into one issued and outstanding share of common stock, par value $0.01 per share, of the Company (“Common Stock”), so that the holders of all of the issued and outstanding shares of common stock of Echo-MN immediately prior to the Merger became the holders of Common Stock of the Company when the Merger became effective.
The Company is a medical device and specialty pharmaceutical company. We are developing a non-invasive, wireless, transdermal continuous glucose monitoring (tCGM) system for use in hospital critical care units and for people with diabetes and a wide range of transdermal reformulations of specialty pharmaceutical products previously approved by the United States Food and Drug Administration (FDA).
The accompanying consolidated financial statements include the accounts of the Company and its wholly-owned subsidiary, Sontra Medical, Inc., a Delaware corporation. All significant inter-company balances and transactions have been eliminated in consolidation.
Going Concern
The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. As of December 31, 2008, the Company had cash of approximately $243,000, a working capital deficit of approximately $3,147,000 and an accumulated deficit of approximately $58,282,000. Through December 31, 2008, the Company has not been able to generate sufficient revenue from its operations to cover its costs and operating expenses. Although the Company has been able to obtain unsecured and secured debt and issue securities through a series of private placements to raise capital in order to fund its operations, it is not known whether the Company will be able to continue this practice, or be able to obtain other types of financing to meet its cash operating expenses. This, in turn, raises substantial doubt about the Company’s ability to continue as a going concern. Subsequent to December 31, 2008, the Company received gross proceeds from its ongoingSeries A-2 Preferred Stock and Warrant Financing of approximately $350,000 (see Note 14). Management is currently pursuing additional private equity financing, and such financing is expected to be completed during 2009. However, no assurances can be given as to the success of these plans. The financial statements do not include any adjustments that might result from the outcome of these uncertainties.
(See report of independent registered public accounting firm.)
F-7
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| |
| (2) | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
The accompanying financial statements reflect the application of the following accounting policies:
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make judgments, estimates and assumptions that affect the amounts reported in the Company’s consolidated financial statements and accompanying notes. Actual results could differ materially from those estimates. The Company considers the valuation of intangible assets, the recoverability of long-lived assets, the realizability of deferred tax assets and the fair value of share-based payments issued to be material accounting estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments with maturities of ninety days or less to be cash equivalents. Cash equivalents consisted of money market funds as of December 31, 2008 and 2007. The Company maintains its cash in bank deposit accounts which, at times, may exceed the federally insured limits. Restricted cash represents a security deposit on the Company’s leased offices.
Intangible Assets and Other Long-Lived Assets
The Company records intangible assets at the acquisition date fair value. As a policy, the Company amortizes its intangible assets using the straight-line method over their estimated useful lives, as follows: patents and licenses, two to 20 years; definite-lived core and developed technology, five to 25 years; and other intangible assets over various periods. In connection with the ETI Acquisition, intangible assets related to contractual arrangements and technology are amortized over estimated useful lives of three (3) and eight (8) years, respectively, on a straight-line basis. The estimated useful life of intangible assets begins when an asset is expected to start contributing directly or indirectly to future cash flows.
The Company uses the income approach to determine the fair values of its capitalizable technology related intangible assets. This approach calculates fair value by estimating the estimated purchased after-tax cash flows over the assets’ useful life and then discounting these after-tax cash flows back to a present value. The Company bases its revenue assumptions on estimates of relevant market sizes, expected market growth rates, expected trends in technology and expected product introductions by competitors. In arriving at the value of the in-process pharmaceutical products, the Company considers, among other factors: the in-process pharmaceutical products’ stage of completion; the complexity of the work completed as of the acquisition date; the costs already incurred; the projected costs to complete; the contribution of core technologies and other acquired assets; the expected introduction date and the estimated useful life of the technology. The Company bases the discount rate used to arrive at a present value as of the date of acquisition on the time value of money and medical technology investment risk factors. For the in-process pharmaceutical products the Company acquired in connection with its ETI Acquisition, the Company used ranges of risk-adjusted discount rates to discount its projected cash flows of 17.8 percent to 27.1 percent. The Company believes that the estimated technology related intangible assets so determined represent the fair value at the date of the ETI Acquisition and do not exceed the amount a third party would pay for the projects.
In accordance with SFAS No. 144,Accounting for the Impairment and Disposal of Long-Lived Assets, the Company reviews intangible assets subject to amortization quarterly to determine if any adverse conditions exist or a change in circumstances has occurred that would indicate impairment or a change in the remaining useful life. Conditions that would indicate impairment and trigger an impairment assessment include, but are not limited to, a significant adverse change in legal factors or business climate that could affect the value of an asset, or an adverse
(See report of independent registered public accounting firm.)
F-8
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
action or assessment by a regulator. If the carrying value of an asset exceeds its undiscounted cash flows, the Company writes-down the carrying value of the intangible asset to its fair value in the period identified.
The Company generally calculates fair value as the present value of estimated future cash flows that the Company expects to generate from the asset using a risk-adjusted discount rate. If the estimate of an intangible asset’s remaining useful life is changed, the Company amortizes the remaining carrying value of the intangible asset prospectively over the revised remaining useful life. For other long-lived assets, the Company evaluates quarterly whether events or circumstances have occurred that indicate that the carrying value of these assets may be impaired. No impairment losses were recorded in the years ended December 31, 2008 or 2007.
In reviewing the long-lived assets as of December 31, 2008, the Company concluded that there were no events or changes in circumstances that would indicate that the carrying value of the long-lived assets, including the Intangible Assets relating to our acquisition of ETI, may not be recoverable. The Company has determined that the estimated life for the intangible assets related to the acquisition of ETI is eight (8) years as of December 31, 2008.
Purchased Research and Development
The Company’s purchased research and development expense in 2007 represents the value of in-process projects that have not yet reached technological feasibility, have no alternative future uses and for which there are no reliable estimates of future cash flows as of the date of acquisition. The primary basis for determining the technological feasibility of these projects is obtaining regulatory approval to market the underlying products in an applicable geographic region. The Company expensed the value attributable to these in-process projects at the time of the acquisition. If the projects are not successful or completed in a timely manner, the Company may not realize the financial benefits expected for these projects or for the acquisition as a whole.
Depreciation and Amortization
The Company provides for depreciation and amortization by charges to operations for the cost of assets using the straight-line method based on the estimated useful lives of the related assets, as follows:
| | | |
Asset Classification | | Estimated Useful Life |
| |
| Computer equipment | | 3 years |
| Office and laboratory equipment | | 3-5 years |
| Furniture and fixtures | | 7 years |
| Manufacturing equipment | | 5 years |
| Leasehold improvements | | Life of lease |
Share-Based Payments
The Company applies the provisions of SFAS No. 123(R), Share-Based Payment, which is a revision of SFAS No. 123, Accounting for Stock-Based Compensation. Under SFAS No. 123(R), the Company recognizes compensation costs resulting from the issuance of stock-based awards to employees and directors as an expense in the statement of operations over the service period based on a measurement of fair value for each stock award.
The Company’s policy is to grant employee and director stock options with an exercise price equal to the fair value of the Company’s common stock at the date of grant.
SFAS No. 123(R) permits public companies to adopt one of two transition methods: a “modified prospective” approach or a “modified retrospective” approach. Under the modified prospective approach, compensation cost is recognized beginning with the effective date of SFAS 123(R) for all share-based payments granted after the
(See report of independent registered public accounting firm.)
F-9
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
effective date of SFAS No. 123(R) and for all awards granted to employees prior to the effective date of SFAS No. 123(R) that remain unvested on the effective date. The Company adopted the modified prospective approach.
Derivative Instruments
The Company generally does not use derivative instruments to hedge exposures to cash-flow or market risks; however, certain warrants to purchase common stock that are indexed to the Company’s common stock are classified as liabilities when the Company is not permitted to settle the instruments in unregistered shares. In such instances, in accordance with EITF IssueNo. 00-19, net-cash settlement is assumed for financial reporting purposes, even when the terms of the underlying contracts do not provide for a net-cash settlement. Such financial instruments are initially recorded at relative fair value with subsequent changes in fair value charged (credited) to operations in each reporting period. If the Company subsequently obtains the ability to settle the instruments in unregistered shares, the instruments are reclassified to equity at their fair value.
On March 24, 2008, the Company entered into a registration rights agreement pursuant to which the Company agreed to file a registration statement with the Securities and Exchange Commission covering the resale of the common stock issuable upon exercise of warrants issued to Imperium Master Fund, Ltd. (“Imperium”) within 60 days after issuance of the securities. The Company also agreed to use its best efforts to cause the registration statement to become effective under the Securities Act as soon as practicable after filing the registration statement, but in no event later than 180 days after issuance of the Imperium Warrants. As of May 14, 2008, the Company and Imperium executed Amendment No. 1 to the Registration Rights Agreement to eliminate these registration requirements and, as a result, the related warrants were no longer required to be recorded as a derivative liability (see Note 7).
Concentration of Credit Risk
SFAS No. 105,Disclosure of Information about Financial Instruments with Off-Balance-Sheet Risk and Financial Instruments with Concentrations of Credit Risk, requires disclosure of any significant off-balance-sheet risks and credit risk concentrations. The Company has no significant off-balance-sheet risk. Financial instruments, which subject the Company to credit risk, principally consist of cash and cash equivalents. The Company mitigates its risk by maintaining the majority of its cash and equivalents with high-quality financial institutions.
Financial Instruments
SFAS No. 107,Disclosures about Fair Value of Financial Instruments, requires disclosure about fair value of financial instruments. The estimated fair market value of the Company’s financial instruments, which include cash and cash equivalents, restricted cash and accounts payable, approximates their carrying value due to the short-term nature of these instruments and their market terms. The Company determined that is was impracticable to estimate the fair value of the Senior Convertible Notes and Senior Secured Notes at December 31, 2008 since a quoted market price is not available and because the Company has not obtained or developed a valuation model to make the estimate, and the cost of obtaining an independent valuation is excessive considering the materiality of the instruments to the Company.
Net Loss per Common Share
Basic and diluted net loss per share of the Company’s common stock is presented in conformity with SFAS No. 128,Earnings per Share. For the periods presented, options, warrants and convertible securities were anti-dilutive and excluded from diluted loss per share calculations. Accordingly, basic and diluted net loss per share
(See report of independent registered public accounting firm.)
F-10
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
of common stock has been computed by dividing the net loss applicable to common stockholders in each period by the weighted average number of shares of common stock outstanding during such period.
Segment Information
SFAS No. 131,Disclosures about Segments of an Enterprise and Related Information, established standards for reporting information regarding operating segments and for related disclosures about products and services and geographical areas. Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker, or decision making group, in making decisions regarding resource allocation and assessing performance. To date, the Company has viewed its operations and manages its business as principally one operating segment, which is development of specialty pharmaceutical drugs and transdermal diagnostics and drug delivery products for sale to the medical market. As of December 31, 2008 and 2007, all of the Company’s assets were located in the United States.
Research and Development Expenses
The Company charges research and development expenses to operations as incurred. Research and development expenses primarily consist of salaries and related expenses for personnel and outside consulting services. Other research and development expenses include the costs of materials and inventory supplies used in research and development, prototype manufacturing, clinical studies, related information technology and an allocation of facilities costs.
Income Taxes
The Company accounts for federal and state income taxes in accordance with SFAS No. 109, Accounting for Income Taxes. Under SFAS No. 109, deferred tax assets and liabilities are recognized based upon temporary differences between the financial statement and the tax basis of assets and liabilities. Deferred income taxes are based upon prescribed rates and enacted laws applicable to periods in which differences are expected to reverse. SFAS No. 109 requires that a valuation allowance be recorded when it is more likely than not that some portion or all of the deferred tax assets will not be realized. Accordingly, since the Company cannot be assured of realizing the deferred tax asset, a full valuation allowance has been provided.
The Company is primarily subject to U.S. federal and Massachusetts state income tax. Tax years subsequent to 2004 remain open to examination by U.S. federal and state tax authorities.
The Company’s policy is to recognize interest and penalties related to income tax matters in income tax expense. As of December 31, 2008 and 2007, the Company had no accruals for interest or penalties related to income tax matters.
Revenue Recognition
Product revenue is recognized when persuasive evidence of an arrangement exists in the form of a signed non-cancelable purchase order, the product is shipped, the selling price is fixed and determinable, and collection is reasonably assured. Licensing revenue is recognized over the term of the licensing agreement as the Company meets its contractual obligations. The Company defers licensing revenue if a performance obligation exists.
Reclassifications
Certain comparative amounts have been reclassified to correspond with the current year’s presentation.
(See report of independent registered public accounting firm.)
F-11
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Recent Accounting Pronouncements
In December 2007, the Financial Accounting Standards Board (FASB) issued Statement No. 141 (revised) (No. 141 R), “Business Combinations.” This Statement replaces FASB Statement No. 141, and applies to all business entities that previously used the pooling-of-interests method of accounting for some business combinations. Under Statement No. 141R, an acquirer is required to recognize, at fair value, the assets acquired, liabilities assumed, and any non-controlling interest in the entity acquired at the acquisition date. Further, it requires that acquisition costs and expected restructuring costs be recognized separately from the acquisition, and that the acquirer, in a business combination executed in stages, recognizes the identifiable assets and liabilities as well as the non-controlling interest in the entity acquired, at the full amounts of their fair values. SFAS No. 141R also requires an acquirer to recognize the assets acquired and liabilities assumed arising from contractual contingencies as of the acquisition date. Also under this statement, an acquirer is required to recognize contingent consideration as of the acquisition date and eliminates the concept of negative goodwill and requires gain recognition in instances in which the fair value of the identifiable net assets exceeds the fair value of the consideration plus any non-controlling interest in the entity acquired as of the acquisition date. SFAS No. 141R makes significant amendments to other Statements and other authoritative guidance, and applies prospectively to business combinations on or after the acquiring entities first fiscal year that begins after December 15, 2008, which is fiscal year 2009 for the Company. It may not be applied prior to that date.
In September 2006, the FASB issued Statement No. 157, “Fair Value Measurements.” This Statement defines fair value, establishes a framework for measuring fair value in accordance with generally accepted accounting principles, and expands disclosures about fair value measurements. The definition of fair value retains the exchange price notion in earlier definitions of fair value. This Statement clarifies that the exchange price is the price in an orderly transaction between market participants to sell the asset or transfer the liability in the market in which the reporting entity would transact for the asset or liability, that is, the principal or most advantageous market for the asset or liability. Emphasis is placed on fair value being a market-based measurement, not an entity-specific measurement, and therefore a fair value measurement should be determined based on the assumptions that market participants would use in pricing the asset or liability. As a basis for considering these market participant assumptions, a fair value hierarchy has been established to distinguish between (1) market participant assumptions developed based on market data obtained from sources independent of the reporting entity (observable inputs) and (2) the reporting entity’s own assumptions about market participant assumptions developed based on the best information available in the circumstances (unobservable inputs). In February 2008, the FASB issued a Staff Position which delays the effective date of Statement No. 157 for non-financial assets and non-financial liabilities, except for items that are recognized or disclosed at fair value in the financial statements on a recurring basis, to fiscal years beginning after November 15, 2008. The Company adopted this statement, except for items covered by the Staff Position, as of January 1, 2008 and the adoption did not have a material impact on the consolidated financial statements.
In March of 2008, the FASB issued Statement No. 161, “Disclosures about Derivative Instruments and Hedging Activities, an amendment of FASB Statement No. 133.” This Statement changes the disclosure requirements for derivative instruments and hedging activities. Entities are required to provide enhanced disclosures about (a) how and why an entity uses derivative instruments, (b) how derivative instruments and related hedged items are accounted for under SFAS 133 and its related interpretations, and (c) how derivative instruments and related hedged items affect an entity’s financial position, financial performance, and cash flows. This Statement is effective for fiscal years and interim periods beginning after November 15, 2008 and is not expected to have a material impact on the consolidated financial statements of the Company.
In June 2008, the FASB ratified EITF IssueNo. 07-5, “Determining Whether an Instrument (or an Embedded Feature) Is Indexed to an Entity’s Own Stock”(EITF 07-5).EITF 07-5 provides that an entity should use a two step
(See report of independent registered public accounting firm.)
F-12
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
approach to evaluate whether an equity-linked financial instrument (or embedded feature) is indexed to its own stock, including evaluating the instrument’s contingent exercise and settlement provisions.EITF 07-5 is effective for fiscal years beginning after December 15, 2008. The consensus must be applied to outstanding instruments as of the beginning of the fiscal year in which the consensus is adopted and should be treated as a cumulative-effect adjustment to the opening balance of retained earnings. Early adoption is not permitted. On January 1, 2009, the Company will reclassify approximately $1,100,000, representing the fair value of all warrants with antidilution provisions, from additional paid-in capital to derivative liability.
The Company completed the acquisition of Durham (the “ETI Acquisition”) on September 14, 2007. ETI was a development-stage company focused on a broad portfolio of advanced topical formulations of well-established, FDA-approved products using its proprietary AzoneTStm dermal penetration technology platform. ETI’s lead AzoneTS-based product is Durhalievetm, an advanced formulation of triamcinolone adetonide for the treatment of corticosteroid-responsive dermatoses. Durhalieve is covered by a pending New Drug Application on file with the FDA.
The ETI Acquisition was accounted for as a purchase of assets, rather than a business combination, as ETI did not meet the definition of a “business”, using the purchase method. The Company paid approximately $16,929,000 to complete the ETI Acquisition, consisting of approximately $1,306,000 related to transaction fees (including the fair value of warrants of approximately $989,000 issued to an investment banking firm), $60,000 paid in cash to Durham Pharmaceuticals, LLC, an affiliate of ETI, and $15,562,500 of the Company’s common stock. The estimated fair value of the 6,250,000 shares of common stock issued in the ETI Acquisition at $2.49 per share was based on the average closing stock price for a period of several days before and after the date the acquisition agreement was reached and announced. The total purchase price consideration for the ETI Acquisition is as follows:
| | | | | |
| Consideration: | | | | |
| Cash | | $ | 60,000 | |
| Common stock | | | 15,562,500 | |
| | | | | |
| Total consideration | | | 15,622,500 | |
| | | | | |
| Transaction costs: | | | | |
| Fair value of warrants | | | 989,215 | |
| Cash (including $157,000 included in accrued expenses at December 31, 2007) | | | 316,815 | |
| | | | | |
| Total transaction costs | | | 1,306,030 | |
| | | | | |
| Total acquisition cost | | $ | 16,928,530 | |
| | | | | |
(See report of independent registered public accounting firm.)
F-13
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The transaction costs above include legal, accounting, asset valuation consultants, corporate services, and investment banker fees with details as follows:
| | | | | |
Category | | Cost | |
| |
| Investment banker fees | | $ | 150,000 | |
| Legal expenses | | | 130,967 | |
| Asset valuation consultants | | | 27,600 | |
| Accountants | | | 8,000 | |
| Corporate services fees | | | 248 | |
| | | | | |
| Total | | $ | 316,815 | |
| | | | | |
The Company allocated the costs of the ETI Acquisition to the assets acquired and the liabilities assumed using all information reasonably available to the Company. The purchase price allocation for the ETI Acquisition is as set out below:
| | | | | |
| Current assets acquired | | $ | 3,952 | |
| Purchased research and development | | | 6,994,578 | |
| Technology related intangible assets | | | 9,625,000 | |
| Contract related intangible asset | | | 355,000 | |
| Current liabilities assumed | | | (50,000 | ) |
| | | | | |
| | | $ | 16,928,530 | |
| | | | | |
The fair value of intangible assets was based on valuations using an income approach, with estimates and assumptions determined by management using all information reasonably available. A range of discount rates of between 17.8% and 27.1% was used in this approach, which the Company believes to be commensurate with the stage of development and the uncertainties in the economic estimates determined. The technology related intangible assets represent the AzoneTS patents, the AzoneTS Drug Master Files that include all technical and clinical data, and an estimated fair value of two (2) commercializable indications for the drug known as Durhalieve. The contract related intangible asset represents favorable discount terms of a contract with Cato Research, a global contract research and development organization and an affiliate of Cato BioVentures, which holds a significant ownership interest in the Company as a result of the ETI Acquisition. None of the amortization of intangible assets acquired is expected to be deductible for income tax purposes.
The amount allocated to purchased research and development relates to the value associated with the on-going development of seven (7) new pharmaceutical product candidates all using the Company’s AzoneTS reformulation technology. These product candidates are in early stages of product development; therefore, technological feasibility has not yet been determined There are no projected alternative uses of these product candidates and no reliable estimates of future cash flows from them. Accordingly, approximately $7.0 million of purchased in-process research and development was expensed on the date of the ETI Acquisition.
(See report of independent registered public accounting firm.)
F-14
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
As of December 31, 2008, intangible assets related to the ETI Acquisition are as follows:
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | 2008 | | | 2007 | |
| | | Estimated
| | | | | | Accumulated
| | | | | | | |
| | | Life | | | Cost | | | Amortization | | | Net | | | Net | |
| |
| Contract related intangible asset: | | | | | | | | | | | | | | | | | | | | |
| Cato Research discounted contract | | | 3 years | | | $ | 355,000 | | | $ | 152,846 | | | $ | 202,154 | | | $ | 320,486 | |
| | | | | | | | | | | | | | | | | | | | | |
| Technology related intangible assets: | | | | | | | | | | | | | | | | | | | | |
| Patents for the AzoneTS-based product candidates and formulation | | | 8 years | | | | 1,305,000 | | | | — | | | | 1,305,000 | | | | 1,305,000 | |
| Drug Master Files containing formulation, clinical and safety documentation used by the FDA | | | 8 years | | | | 1,500,000 | | | | — | | | | 1,500,000 | | | | 1,500,000 | |
| In-process pharmaceutical products for two(2) indications | | | 8 years | | | | 6,820,000 | | | | — | | | | 6,820,000 | | | | 6,820,000 | |
| | | | | | | | | | | | | | | | | | | | | |
| Total technology related intangible assets | | | | | | | 9,625,000 | | | | — | | | | 9,625,000 | | | | 9,625,000 | |
| | | | | | | | | | | | | | | | | | | | | |
| Total | | | | | | $ | 9,980,000 | | | $ | 152,846 | | | $ | 9,827,154 | | | $ | 9,945,486 | |
| | | | | | | | | | | | | | | | | | | | | |
The technology related intangible assets are being amortized on a straight line basis over approximately 8 years beginning with the start of its useful life, which the Company has estimated to be 2011, and the contract related intangible asset over approximately 3 years beginning with the date of acquisition. Amortization expense was approximately $118,300 and $34,500 for the years ended December 31, 2008 and 2007, respectively, and is included in research and development in the Statement of Operations.
Estimated amortization expense for each of the next five years is as follows:
| | | | | |
| | | Estimated
| |
| | | Amortization
| |
Year Ending December 31: | | Expense | |
| |
| 2009 | | $ | 118,000 | |
| 2010 | | | 84,000 | |
| 2011 | | | 1,203,000 | |
| 2012 | | | 1,203,000 | |
| 2013 | | | 1,203,000 | |
(See report of independent registered public accounting firm.)
F-15
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| |
| (5) | OPERATING LEASE COMMITMENTS |
The Company leases approximately 13,000 square feet of manufacturing, laboratory and office space in a single facility located in Franklin, Massachusetts under a lease expiring March 31, 2010. The Company has the option to extend the lease for an additional one (1) year period by providing notice to the landlord by September 30, 2009. Future minimum lease payments under this operating lease are approximately as follows:
| | | | | |
| | | Amount | |
| |
| For the years ended December 31, | | | | |
| 2009 | | $ | 193,000 | |
| 2010 | | | 49,000 | |
| | | | | |
| Total | | $ | 242,000 | |
| | | | | |
The Company’s facilities lease expense was approximately $172,000 and $159,000 for the years ended December 31, 2008 and 2007, respectively.
The Company also had a non-cancellable operating lease related to its office equipment that expired in December 2008. The office equipment is being leased on a month-to-month basis going forward.
| |
| (6) | PATENT LICENSE AGREEMENT |
On December 31, 2008, the Company had a patent license agreement with the Massachusetts Institute of Technology (MIT) that granted Sontra Medical, Inc, a wholly-owned subsidiary of the Company, an exclusive right and license to certain existing and future MIT patents that relate to ultrasound technology. Under the agreement, the Company was obligated to pay MIT a minimum annual license maintenance fee of $25,000 to be creditable towards the payment of royalties. As a result of the progress made in development of other skin permeation technology, the Company chose not to continue its license agreement with MIT after December 31, 2008.
(See report of independent registered public accounting firm.)
F-16
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Notes payable at December 31, 2008 and 2007 consisted of the following:
| | | | | | | | | |
| | | 2008 | | | 2007 | |
| |
| Senior Notes: | | | | | | | | |
| Senior promissory bridge notes | | $ | — | | | $ | 1,325,000 | |
| Unamortized discount | | | — | | | | (938,542 | ) |
| | | | | | | | | |
| | | | — | | | | 386,458 | |
| | | | | | | | | |
| Senior Convertible Notes: | | | | | | | | |
| Senior Convertible notes | | | 331,448 | | | | — | |
| Unamortized Discount | | | (30,981 | ) | | | — | |
| | | | | | | | | |
| | | | 300,467 | | | | — | |
| | | | | | | | | |
| Senior Secured Notes: | | | | | | | | |
| Senior Secured notes | | | 2,209,426 | | | | — | |
| Unamortized Discounts | | | (155,364 | ) | | | — | |
| | | | | | | | | |
| | | | 2,054,062 | | | | — | |
| | | | | | | | | |
| Total notes payable | | | 2,354,529 | | | | 386,458 | |
| Less current portion of notes payable | | | 2,054,062 | | | | 386,458 | |
| | | | | | | | | |
| Notes Payable, net of current portion | | $ | 300,467 | | | $ | — | |
| | | | | | | | | |
Equipment Note Payable
In May 2005, the Company entered into a note payable agreement with a third-party lender for financing equipment purchases in the amount of $237,541. The note was repayable over a four year term and the Company was obligated to make monthly interest and principal payments of $6,017. Interest accrued at an annual rate of 10.39% and the note was secured by certain property and equipment of the Company. Interest expense related to this note was none and $9,063 for the years ended December 31, 2008 and 2007, respectively. On September 12, 2007, the Company repaid the entire outstanding balance of the note payable, or approximately $70,000.
2007 and 2008 Senior Promissory Bridge Notes and Senior Convertible Notes
On September 14, 2007, the Company completed a private placement of unsecured Senior Promissory Bridge Notes (the “Bridge Notes”) in the aggregate principal amount of $1,325,000 to Montaur Capital through Platinum Long Term Growth VII, LLC and to other strategic institutional and individual accredited investors. The Bridge Notes were scheduled to be due September 15, 2008 and accrued interest at a rate of 10% per annum, with interest payable upon maturity. If the Company had completed a subsequent equity or equity-linked financing or a combination of equity financings resulting in gross proceeds to the Company totaling at least $2,500,000 on or before December 15, 2007, inclusive of the Bridge Notes (a “Qualified Financing”), then the Bridge Notes would have converted automatically into the equity securities issued in the Qualified Financing. The terms of the Bridge Notes reflected a 20% premium in the event of an exchange such that upon an automatic exchange of the Bridge Notes in any Qualified Financing, the holders of the Bridge Notes would be deemed to have tendered an amount equal to 120% of the outstanding principal and interest of the Bridge Notes in exchange for the equity securities issued to such holders in the Qualified Financing. A Qualified Financing was not completed by December 31, 2007.
(See report of independent registered public accounting firm.)
F-17
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Accordingly, the holders of the Bridge Notes had one demand registration right and the Bridge Notes became (1) convertible into shares of common stock at a ratio determined by dividing the outstanding principal and interest of each Bridge Note by a price per share equal to the price per share of the Company’s most recent equity or equity-linked financing or (2) exchangeable at a 20% premium for securities issued in any other subsequent equity or equity-linked financing.
On February 11, 2008, the Company completed an approximately $2.3 million private financing with substantially all of the holders of the Bridge Notes. The $2,292,459 in aggregate principal amount of unsecured Senior Convertible Notes issued in the financing (the “Senior Convertible Notes”) bear interest annually at a rate of 8% per annum and provide the holders with the right to convert principal into shares of the Company’s common stock at $1.35 per share. The conversion price is subject to weighted average anti-dilution protection, excluding certain customary exceptions. The Senior Convertible Notes have a three-year term and the Company may elect to make payments of interest in cash, additional notes, or stock.
Additionally, the investors received warrants to purchase 849,059 shares of common stock at an exercise price of $1.00 per share as of December 31, 2008 for a term of five years. The warrants provide for full anti-dilution price protection to the holders and allow for cashless exercise.
In connection with the Senior Convertible Note financing, certain holders representing $1,275,000 face value of Bridge Notes exchanged their Bridge Notes at 120% of the outstanding principal and interest of the Bridge Notes as payment toward the purchase price of the Senior Convertible Notes purchased by such holders. Accordingly, the Company issued notes in the Senior Convertible Note financing in the aggregate principal balance of $1,592,459 to the former holders of the Bridge Notes upon their surrender of the Bridge Notes, and the Company received gross cash proceeds in the amount of $700,000 in connection with the financing.
One holder of a Bridge Note with a face value of $50,000 converted the principal and accrued interest of such Bridge Note into 52,041 shares of the Company’s common stock, using a $1.00 conversion price as provided in the terms of the Bridge Notes. No Bridge Notes remained outstanding at December 31, 2008.
The Company has determined that the terms of the Senior Convertible Notes are deemed “substantially different”, as described in EITF IssueNo. 96-19, “Debtor’s Accounting for a Modification or Exchange of Debt Instruments”, from the terms of the Bridge Notes based on the change in the fair value of the embedded conversion features. As a result, the Company recorded the Senior Convertible Notes issued in exchange for the Bridge Notes at fair value on the date of issuance and recorded a loss on extinguishment of approximately $586,000. An amount equal to the intrinsic value of the conversion feature of the Bridge Notes as measured on the date of exchange for the Senior Convertible Notes, or approximately $213,000, was recorded as a reduction in additional paid-in capital in accordance with EITF IssueNo. 98-5, “Accounting for Convertible Securities with Beneficial Conversion Features or Contingently Adjustable Conversion Ratios”. The fair value of the warrants issued to the holders of the Bridge Notes upon conversion into the Senior Convertible Notes, which the Company estimated to be approximately $626,000, was recorded as additional paid-in capital and also included in the loss on extinguishment. The difference between the fair value and the face value of the Senior Convertible Notes is being accreted to interest expense over the term of the notes.
The new cash proceeds from the Senior Convertible Notes of $700,000 were allocated between the notes and the warrants issued in the Senior Convertible Note financing on a relative fair value basis. Approximately $220,000 of the proceeds was allocated to the warrants and recorded as additional paid-in capital and a discount on the Senior Convertible Notes. The Company determined that the effective conversion price, after allocation of the proceeds, resulted in a beneficial conversion feature of approximately $121,000 which was recorded as additional paid-in capital and a further discount on the Senior Convertible Notes. The discounts on the Senior Convertible Notes are being accreted to interest expense over the term of the notes.
(See report of independent registered public accounting firm.)
F-18
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Interest expense related to the Bridge Notes for the years ended December 31, 2008 and 2007, including amortization of discounts, was $197,000 and $425,000, respectively. Interest expense related to the Senior Convertible Notes for the years ended December 31, 2008 and 2007, including amortization of discounts and deferred financing costs, was approximately $356,000 and $0, respectively. During the year ended December 31, 2008, accrued interest of approximately $117,000 was satisfied by the issuance of additional Senior Convertible Notes.
2008 Senior Secured Note and Warrant Financing
On March 24, 2008, the Company entered into a secured note financing agreement with Imperium, providing for, at the Company’s option, the issuance of up to an aggregate $2,000,000 of original issue discount senior secured notes (the “Secured Notes”) in up to four equal tranches, together with warrants (the “Imperium Warrants”) to purchase up to 653,728 shares of common stock at an exercise price of $1.94 per share as of December 31, 2008. On March 24, 2008, the Company drew down the initial $500,000 in gross proceeds and issued the Imperium Warrants upon execution of the agreement. The Company also issued additional Secured Notes and drew down $500,000 in gross proceeds on each of April 24, 2008, June 2, 2008 and June 24, 2008, completing the financing for $2,000,000.
The Company is not required to make monthly cash payments of principal and interest under the Secured Notes. Instead, the outstanding principal of each Secured Note will accrete in value at an annual rate of 10%, compounded monthly, resulting in a total principal amount of approximately $552,357 due for each Secured Note at maturity. If, however, the Company completes an equity issuance in one or more series of transactions totaling $5,000,000 (a “Qualified Issuance”), then the aggregate amount due for each Secured Note will be reduced from $552,357 to $546,903 and the annual accretion value will be reduced from 10% to 9%.
Each Secured Note is due twelve months after the date of its issuance; provided, however, that if the Company completes a Qualified Issuance by October 31, 2008, the Company has a right to extend the maturity date of each Secured Note to 24 months after the date of its issuance. As of October 31, 2008, the Company had not closed on a Qualified Issuance. The Company has the right to repay the principal amount of the Secured Notes in cash, in whole, but not in part, prior to maturity at a premium of 1.02 times the unpaid principal plus any other amount due under the Secured Notes.
Events of default under the Secured Notes include: (1) failure to make a payment when due or payable; (2) a breach of or notice of intent to breach any material term, covenant or condition in the Secured Note or any of the transaction documents and such breach is not cured within five business days after notice; (3) any false, incorrect or breach in any material respect of any material representation or warranty made by the Company in the transaction documents; (4) the default of more than $25,000 of any of the Company’s other indebtedness that causes such debt to become due and payable; or (5) a bankruptcy (whether voluntary or involuntary) or general assignment for the benefit of the Company’s creditors. All amounts outstanding under the Secured Notes, plus an amount equal to the product of 1.10 and all amounts outstanding under the Secured Notes, become due and payable upon the occurrence of an event of default or upon a change in control (as defined in the Secured Notes).
In addition, the Company has agreed to certain covenants, including a prohibition on the ability to incur future indebtedness (subject to certain exceptions) or make any dividend or payment to holders of its capital stock (other than shares of the class of stock held by such recipient), and a requirement that the Company maintain $6.5 million in stockholders’ equity (excluding any impact of the issuance of the Imperium Warrants to stockholders’ equity) for so long as the Secured Notes remain outstanding. The Company also agreed that it may not redeem its Senior Convertible Notes (discussed above) as long as the Secured Notes remain outstanding, unless it receives at least $10 million in gross proceeds from an issuance or series of related issuances of equity securities.
The Company’s subsidiary, Sontra Medical, Inc., has guaranteed the obligations under the Secured Notes. Additionally, the Secured Notes are secured by all of the Company’s assets.
(See report of independent registered public accounting firm.)
F-19
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The Imperium Warrants have a term of five years and are immediately exercisable at an exercise price of $1.94 per share as of December 31, 2008. The Imperium Warrants provide for weighted average anti-dilution protection upon future issuances or deemed issuances (subject to customary exceptions) below the exercise price. The Imperium Warrants also allow for cashless exercise unless the shares underlying the warrants are registered for resale on a registration statement filed under the Securities Act of 1933, as amended (the “Securities Act”).
In connection with the issuance of the Secured Notes and Imperium Warrants, on March 24, 2008, the Company and Imperium entered into a Registration Rights Agreement (the “Registration Rights Agreement”) pursuant to which the Company agreed to file a registration statement with the SEC covering the resale of the common stock issuable upon exercise of the Imperium Warrants within 60 days after issuance of the securities. The Company also agreed to use its best efforts to cause the registration statement to become effective under the Securities Act as soon as practicable after the filing of the registration statement, but in no event later than 180 days after issuance of the Imperium Warrants. As of May 14, 2008, the Company and Imperium executed Amendment No. 1 to the Registration Rights Agreement that eliminated the requirement for registration of the underlying securities, but granted the holders of the Imperium Warrants piggy-back registration rights with respect to certain registration statement filings by the Company in the future.
Due to certain requirements to obtain and maintain an effective registration statement covering the shares underlying the Imperium Warrants, the Company originally determined that the Imperium Warrants did not meet the requirements for classification as equity as described in EITF IssueNo. 00-19, “Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock” as of the issuance date. As a result, the fair value of the Imperium Warrants was recorded as a derivative liability which resulted in the recognition of a derivative loss upon issuance in the amount of approximately $569,000. As a result of the execution of Amendment No. 1 to the Registration Rights Agreement, the Company determined that the Imperium Warrants met the requirements for classification as equity as described in EITF IssueNo. 00-19 as of May 14, 2008. The derivative liability was adjusted to fair value as of May 14, 2008, resulting in an additional derivative loss of approximately $18,000. The remaining derivative liability of $919,593 was reclassified to additional paid-in capital. The derivative loss for the year ended December 31, 2008 amounted to approximately $587,000.
Interest expense related to the Secured Notes in the year ended December 31, 2008, including amortization of discounts and deferred financing costs, was approximately $492,000.
2008 Exchange Agreement
On September 30, 2008, the Company entered into an Exchange Agreement (the “Exchange Agreement”) with substantially all of the holders of the Senior Convertible Notes (collectively, the “Investors”). The Investors are the holders of an aggregate of $1,980,212 in principal amount of the Senior Convertible Notes and an aggregate $97,656 in principal amount of additional notes issued as interest on the Senior Convertible Notes (collectively, the “Notes”), for a total aggregate of $2,077,868 in principal amount of Notes constituting all of the issued and Senior Convertible Notes, except for the aggregate $318,475 in principal amount of notes held by Gemini Master Fund Ltd. (“Gemini”), who was not a party to the Exchange Agreement. Accrued interest of $19,201 payable to Gemini for the year ended December 31, 2008 was paid by issuing additional notes. The Senior Convertible Notes held by Gemini remain outstanding with original terms. The Gemini Notes are convertible into shares of the Company’s common stock at the option of the holder at a price per share of $1.35, subject to adjustment for stock splits, combinations or similar events and subject to customary weighted average anti-dilution adjustments.
Pursuant to the terms of the Exchange Agreement, the Company issued and delivered to the Investors, in exchange for the cancellation of the Notes, 1,539,161 shares of a newly authorized Series A Convertible Preferred
(See report of independent registered public accounting firm.)
F-20
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Stock (see Note 9) and five-year warrants to purchase 153,912 shares of common stock at an exercise price of $1.00 per share, subject to adjustment for stock splits, combinations or similar events.
As the Senior Convertible Notes did not contain the right to convert into Series A Convertible Preferred Stock, the Company accounted for the exchange as an extinguishment of the Senior Convertible Notes. As a result, the difference between the fair values of the Series A Convertible Preferred Stock issued over the carrying value of the Senior Convertible Notes exchanged of $738,578 was recorded as an extinguishment loss in the year ended December 31, 2008. The fair value of the warrants issued of $106,182 was also recorded as an extinguishment loss in the year ended December 31, 2008.
| |
| (8) | FAIR VALUES OF ASSETS AND LIABILITIES |
Effective January 1, 2008, the Company adopted SFAS No. 157, “Fair Value Measurements”, which provides a framework for measuring fair value under GAAP.
In accordance with SFAS 157, the Company groups its financial assets and financial liabilities measured at fair value in three levels, based on the markets in which the assets and liabilities are traded and the reliability of the assumptions used to determine fair value.
Level 1 — Quoted prices in active markets for identical assets or liabilities. Level 1 assets and liabilities include debt and equity securities that are traded in an active exchange market. Valuations are obtained from readily available pricing sources for market transactions involving identical assets or liabilities.
Level 2 — Observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. For example, Level 2 assets and liabilities may include debt securities with quoted prices that are traded less frequently than exchange-traded instruments.
Level 3 — Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. Level 3 assets and liabilities include financial instruments whose value is determined using pricing models, discounted cash flow methodologies, or similar techniques, as well as instruments for which the determination of fair value requires significant management judgment or estimation. This category includes, for example, certain private equity investments and long-term derivative contracts.
The only asset or liability measured at fair value on a recurring basis is the Company’s derivative liability and it is included in Level 3 in the fair value hierarchy. As a result of the execution of Amendment No. 1 to the Registration Rights Agreement as described in Note 7, the remaining liability balance was classified as additional paid-in capital as of May 14, 2008 and the derivative liability was eliminated at that time.
The table below presents the changes in Level 3 derivative liability measured at fair value on a recurring basis.
| | | | | |
| | | Year Ended
| |
| | | December 31,
| |
| | | 2008 | |
| |
| Balance as of January 1, 2008 | | $ | — | |
| Total realized/unrealized loss included in net loss | | | 17,349 | (1) |
| Purchases, sales, issuances and settlements | | | 902,244 | |
| Reclassification to equity | | | (919,593 | ) |
| | | | | |
| Balance as of December 31, 2008 | | $ | — | |
| | | | | |
| | |
| (1) | | Included in derivative loss on the statement of operations for the year ended December 31, 2008. |
(See report of independent registered public accounting firm.)
F-21
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Also, the Company may be required, from time to time, to measure certain other financial assets at fair value on a nonrecurring basis. These adjustments to fair value usually result from application of lower-of-cost-or-market accounting or write-downs of individual assets. There were no such adjustments in the year ended December 31, 2008.
| |
| (9) | SERIES A CONVERTIBLE PREFERRED STOCK |
Effective upon the Company’s reincorporation in Delaware on June 9, 2008, the Company is authorized in its Certificate of Incorporation to issue up to 10,000,000 shares of preferred stock with the rights, preferences and privileges to be fixed by the Board of Directors.
Series A Preferred Stock — Echo-MN
On January 30, 2007, the Company repurchased all outstanding shares of Series A Preferred Stock for approximately $73,000. In conjunction with the dividend on the Echo-MN Series A Convertible Preferred Stock, the Company accreted dividends of $483 and paid annual dividends of $3,440 in the form of 10,487 shares of common stock for the year ended December 31, 2007.
Series A Preferred Stock and Warrant Financing
On September 29, 2008, the Board of Directors authorized the creation and issuance of a new class of Series A Convertible Preferred Stock. The Board of Directors authorized the issuance of up to 2,636,363 shares of Series A Convertible Preferred Stock (“Series A Stock”) with the rights, preferences and privileges described below.
Pursuant to the terms of the Certificate of Designation, Preference and Rights of Series A Convertible Preferred Stock, each share of Series A Stock is initially convertible into one share of common stock, subject to adjustment for stock splits and combinations or similar events. The Series A Stock will pay a quarterly dividend, at an annual rate of 8%, which is payable in cash or in kind at the option of the Company. Each holder of Series A Stock may convert its Series A Stock at any time following issuance of the Series A Stock. The Series A Stock has no voting power, except as otherwise required under Delaware General Corporate Law. The Series A Stock is not redeemable.
In the event that the Company liquidates, dissolves, or winds up its affairs (each, a “Liquidation Event”), the holders of Series A Stock will be entitled to receive (subject to the rights of any securities designated as senior to the Series A Stock) a liquidation preference equal to the greater of (i) $1.35 per share or (ii) the amount that would be distributed in such Liquidation Event on the number of shares of common stock issuable upon conversion of the Series A Stock. The Series A Stock ranks pari passu with theSeries A-1 Stock (see below) andSeries A-2 Stock (see Note 14). The Company cannot create or issue any security senior to the Series A Stock without the approval of the holders of the majority of the outstanding Series A Stock.
In connection with the 2008 Exchange Agreement (see Note 7), the Company issued and delivered to certain Senior Convertible Note holders, in exchange for the cancellation of their Notes, 1,539,161 shares of Series A Stock and five-year warrants to purchase 153,912 shares of common stock at an exercise price of $1.00 per share, subject to adjustment for stock splits, combinations or similar events.
In conjunction with the dividend on the new Series A Stock, Echo-DE accreted dividends of $41,899 for the year ended December 31, 2008. The accreted dividends were satisfied through the issuance of 31,033 shares of Series A Preferred Stock and $5 in the form of cash relating to fractional shares. The accretion and settlement of the Series A Stock dividend had no net effect on additional paid-in capital other than to the extent of the par value of the Series A Stock issued and cash paid.
(See report of independent registered public accounting firm.)
F-22
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Series A-1 Preferred Stock and Warrant Financing
On October 15, 2008, the Board of Directors authorized the creation and issuance of a new class ofSeries A-1 Convertible Preferred Stock. The Board of Directors authorized the issuance of up to 2,000,000 shares ofSeries A-1 Convertible Preferred Stock(“Series A-1 Stock”) with the rights, preferences and privileges described below.
Pursuant to the terms of the Certificate of Designation, Preference and Rights ofSeries A-1 Convertible Preferred Stock, each share ofSeries A-1 Stock is initially convertible into one share of Common Stock, subject to adjustment for stock splits and combinations or similar events. TheSeries A-1 Stock will pay a quarterly dividend, at an annual rate of 8%, which is payable in cash or in kind at the option of the Company. Each Investor may convert its Shares at any time following issuance of the Shares. TheSeries A-1 Stock is not redeemable.
Each holder ofSeries A-1 Stock shall have the right, exercisable on an all or none basis, to participate in the Company’s first equity offering or series of equity-linked offerings to occur after the date of the Financing that yields gross proceeds to the Company of at least $2,000,000 (the “Qualified Offering”) on the same terms and conditions as offered by the Company to the other purchasers of such securities issued and sold by the Company in the Qualified Offering (the “Additional Securities”), except that the consideration for each holder’s participation in the Qualified Offering shall be the surrender of 100% of such holder’s shares ofSeries A-1 Stock in exchange for Additional Securities with a purchase price equal to an aggregate of 115% of the Liquidation Preference of theSeries A-1 Stock surrendered by such holder.
In the event that the Company liquidates, dissolves, or winds up its affairs (each, a “Liquidation Event”), the holders ofSeries A-1 Stock will be entitled to receive (subject to the rights of any securities designated as senior to theSeries A-1 Stock) a liquidation preference equal to the greater of (i) $1.00 per share or (ii) the amount that would be distributed in such Liquidation Event on the number of shares of common stock issuable upon conversion of theSeries A-1 Stock. TheSeries A-1 Stock ranks pari passu with the Series A Stock (see above) andSeries A-2 Stock (see Note 14). The Company cannot create or issue any security senior to theSeries A-1 Stock without the approval of the holders of the majority of the aggregate outstanding Series A andSeries A-1 Stock.
On October 28, 2008 and October 31, 2008, the Company entered into a Stock and Warrant Purchase Agreement (the “Purchase Agreement”) with strategic institutional and accredited investors (the “Investors”) in connection with the Company’s private placement (the“A-1 Financing”) of 766,667 shares of itsSeries A-1 Convertible Preferred Stock (the “Shares”) at a price of $1.00 per share together with warrants to purchase 268,333 shares of the Company’s Common Stock, $0.01 par value (the “Common Stock”) which is equal to thirty-five percent (35%) of the number of Shares purchased by each Investor (the “Warrants”) in theA-1 Financing. The Company received gross proceeds of $766,667 from theA-1 Financing. The Company estimated the fair value of the warrants to be approximately $148,200 which was recorded as both a debit and credit to additional paid-in capital.
Pursuant to the Purchase Agreement, the Company issued Warrants to the Investors to purchase up to 268,333 shares of Common Stock. The Warrants are immediately exercisable at a price per share of $1.50, subject to adjustment for stock splits, combinations or similar events, and will expire no later than October 1, 2013. The Warrants allow for cashless exercise. In addition, the Company has the option to redeem the Warrants, in whole but not in part, upon satisfaction of certain conditions, including (i) the availability of an effective registration statement or Rule 144 exemption for any resale by the holder, (ii) the shares of Common Stock trading at a price per share in excess of 200% of the then-applicable exercise price for ten (10) trading days out of a period of fifteen (15) consecutive trading days prior to the redemption, and (iii) an average daily trading volume during such fifteen (15) consecutive trading days of at least 50,000 shares of Common Stock. Finally, an exercise under the Warrants may not result in the holder beneficially owning more than 4.99% or 9.99%, as applicable, of all of the
(See report of independent registered public accounting firm.)
F-23
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Common Stock outstanding at the time; provided, however, that a holder may waive the foregoing provision upon sixty-one (61) days’ advance written notice to the Company.
In connection with the Financing, the Company retained Burnham Hill Partners (“BHP”) as its placement agent. The Company agreed to pay to BHP for its services as follows: (a) a cash fee equal to 8% of the gross proceeds of the Financing; and (b) warrants to acquire a number of shares of Common Stock of the Company equal to 10% of the number of as-converted Shares issued to Investors in theA-1 Financing at a per share exercise price of $1.10. The Company also agreed to pay reasonable out of pocket expenses of BHP incurred in connection with theA-1 Financing in an amount not to exceed $5,000. These costs, totaling $63,333, were recorded as stock issuance costs and net against the proceeds from theA-1 Financing.
In conjunction with the dividend on theSeries A-1 Stock, Echo-DE accreted dividends of $11,473 for the year ended December 31, 2008. In the year ended December 31, 2008, Echo-DE paid annual dividends of $11,469 in the form of 11,469 shares ofSeries A-1 Stock and $4 in the form of cash for fractional shares. The accretion and settlement of the Series A Stock dividend had no net effect on additional paid-in capital other than to the extent of the par value of the Series A Stock issued and cash paid.
The Company has authorized 60,000,000 shares of common stock, $0.01 par value per share, of which 19,095,838 and 17,870,804 shares were issued and outstanding as of December 31, 2008 and 2007, respectively.
ETI Acquisition
In connection with the ETI Acquisition, each outstanding share of ETI common stock was canceled, extinguished and converted automatically into 0.4356 shares of the Company’s common stock. The Company issued an aggregate of 6,250,000 shares of its common stock with a fair value of approximately $15,562,500 to the former ETI shareholders in connection with the ETI Acquisition.
Burnham Hill Partners Investment Banking and Financial Advisory Agreements
BHP was engaged to advise the Company in connection with the ETI Acquisition and certain financing activities. The Company issued BHP five-year warrants to purchase 425,000 shares of Company common stock at an exercise price of $2.18 (as of December 31, 2008) per share upon consummation of the ETI Acquisition, and agreed to pay BHP $150,000 in cash upon the Company’s receipt of $2,500,000, inclusive of the Senior Notes, in connection with a Qualified Financing. The fair value of the BHP warrants was estimated to be approximately $989,000 and the amount was included in the purchase price of ETI because the cost was directly related to the consummation of the ETI Acquisition. The warrants include a weighted average anti-dilution provision if the issuance or deemed issuance price is less than the exercise price for any sale or deemed sale of the Company’s common stock. In 2008, BHP agreed to reduce their cash fee of $150,000 to $120,000 reducing expenses in the period.
Effective July 25, 2007, the Company engaged BHP as its non-exclusive financial advisor for a period of six months. For these financial advisory services, BHP received $50,000 in cash, and the Company issued BHP five-year warrants to purchase 60,000 shares of the Company’s common stock at an exercise price of $1.63 per share. The fair value of these BHP warrants was estimated to be approximately $133,000 and was included in selling, general and administrative expenses. The warrants include a weighted average anti-dilution provision if the issuance or deemed issuance price is less than the exercise price for any sale or deemed sale of the Company’s common stock.
(See report of independent registered public accounting firm.)
F-24
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
On February 11, 2008, the Company provided warrants to purchase 175,013 shares of the Company’s common stock at an exercise price of $1.49 per share to Burnham Hill Partners, a division of Pali Capital, Inc., for a financing fee in connection with the Senior Convertible Note financing. The fair value of these services has been determined to be approximately $186,000 and has been recorded as a deferred financing cost with amortization over a three year period.
In connection with the October 2008Series A-1 Preferred Stock financing (see Note 9), the Company retained BHP as its placement agent. The Company agreed to pay to BHP for its services as follows: (a) a cash fee equal to 8% of the gross proceeds of theA-1 Financing; and (b) warrants to acquire a number of shares of Common Stock of the Company equal to 10% of the number of as-converted Shares issued to Investors in theA-1 Financing at a per share exercise price of $1.10. The Company also agreed to pay reasonable out of pocket expenses of BHP incurred in connection with theA-1 Financing in an amount not to exceed $5,000. These costs, including the fair value of the warrants, will be recorded as stock issuance costs and net against the proceeds from theA-1 Financing. As a result of theA-1 Financing, the Company paid BHP $61,333 and provided warrants to purchase 26,833 shares of common stock with an exercise price of $1.10 per share. The fair value of these warrant shares was determined to be approximately $15,000 as of February 11, 2008 which was recorded as both a debit and credit to additional paid-in capital as a stock issuance cost.
As of January 30, 2009, the Company agreed to a financial advisory agreement with BHP that resulted in the repricing and term extension of the BHP and affiliate warrants to $.55 per share (see Note 14).
2008 Private Placements of Common Stock
During 2008, warrants to purchase 726,250 shares of our common stock were exercised voluntarily, providing gross proceeds of approximately $114,000. Total common shares of 705,873 were issued in connection with these exercises as certain warrants were exercised through a cashless exercise provision allowed in the holder’s warrant agreement.
During 2008, the Company issued 150,000 shares of fully-vested, unregistered shares of common stock with a fair value of $247,500 to a consultant for investor relations services.
As of December 31, 2008, the Company issued 318,905 shares of its Common Stock to Cato in settlement of a payable in the amount of $140,318.
2007 Private Placements of Common Stock
On January 3, 2007, the Company entered into a definitive common stock and warrant purchase agreement with Sherbrooke Partners, LLC (“Sherbrooke”), certain other accredited investors and certain members of the Company’s board of directors and management team to issue 6,000,000 shares of common stock for $0.10 per share and two-year warrants to purchase 1,500,000 shares of common stock at an exercise price of $0.21 per share (the closing price of the Company’s common stock on the NASDAQ Capital Market as of December 29, 2006) in exchange for $600,000 (the “January 2007 Financing”). On January 30, 2007, the Company closed a $660,000 (includes a 10% over-allotment) common stock and warrant financing with Sherbrooke, certain other accredited investors and certain members of the board of directors and management. The Company issued 6,600,000 shares of common stock for $0.10 per share and two-year warrants to purchase 1,650,000 shares of common stock at an exercise price of $0.21 per share in the January 2007 Financing (the “January 2007 Warrants”).
The January 2007 Warrants expire two years from the date of the closing of the January 2007 Financing and contain customary provisions for adjustment to the exercise price in the event of stock splits, combinations and dividends. The January 2007 Warrants also contain weighted average anti-dilution provisions that provide for an
(See report of independent registered public accounting firm.)
F-25
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
adjustment to the then effective exercise price (and number of shares of common stock issuable upon exercise) upon certain dilutive issuances by the Company of equity securities. In addition, if the per share market value (as defined in the January 2007 Warrants) of the Company’s common stock for any twenty (20) consecutive trading days equals or exceeds $0.63 per share, then the Company may, with the prior written consent of the January 2007 Warrant holders, redeem the unexercised portion of the January 2007 Warrants in cash at a price equal to the number of shares of common stock that remain subject to the January 2007 Warrant multiplied by $0.001. On January 30, 2009, in connection with a financial advisory agreement with BHP, the original term of warrants to purchase 536,250 shares of common stock was extended through January 30, 2014 (see Note 14).
In June and July 2007, the Company completed a total of three closings of a private placement of its common stock which provided the Company with aggregate gross proceeds of $1,815,000 (net proceeds were approximately $1,641,000) (the “June 2007 Financing”). The investors in the June 2007 Financing purchased 1,815,000 shares of the Company’s common stock at a per share purchase price of $1.00 and received five-year warrants to purchase an aggregate of 544,500 shares of common stock at a per share exercise price of $1.40 (the “June 2007 Warrants”) . The Company has the right to terminate the June 2007 Warrants, upon fifteen (15) days’ notice, in the event (i) the closing bid price of the Company’s Common Stock for twenty-two (22) consecutive trading days is equal to or greater than $3.00 per share and (ii) the Company has registered for resale the shares of Common Stock underlying the June 2007 Warrants, provided however, that each registered holder has the right to exercise the June 2007 Warrants at any time prior to such termination. The June 2007 Warrants contain customary provisions for adjustment to the exercise price in the event of stock splits, combinations and dividends. The June 2007 Warrants also contain weighted average anti-dilution provisions that provide for an adjustment to the then effective exercise price if the Company issues equity securities without consideration or for consideration less than $1.00 per share.
In connection with warrants and restricted stock issued to Legend (defined below) in connection with the June 2007 Financing, the total fair value of these instruments of $170,913 was recorded, net of the par value of the common stock, as both a debit and credit to additional paid-in capital.
The Company used the net proceeds of the June 2007 Financing for product development, funding of clinical trials, possible acquisitions or licensing of technologies or businesses, working capital and general corporate purposes.
Legend Merchant Group Placement Agent and Financial Advisory Agreements
In connection with the June 2007 Financing, the Company retained Legend Merchant Group, Inc. (“Legend”) as placement agent. The Company agreed to pay Legend for its services (a) a cash fee equal to 7% of the aggregate gross proceeds raised by the Company in the June 2007 Financing from investors introduced to the Company by Legend, excluding the proceeds from any June 2007 Warrant exercises, (b) shares of unregistered Common Stock of the Company equal to 5% of the number of shares issued to investors introduced to the Company by Legend and (c) warrants to acquire a number of shares of common stock of the Company equal to 5% of the number of shares issued to investors introduced to the Company by Legend at a per share exercise price equal to $1.40. Legend received fees and compensation only on funds raised by Legend from new investors in the Company (i.e., only those parties, and affiliates, who had not previously participated in a financing transaction with the Company and who were not introduced by Sherbrooke Partners (and its affiliates), management or members of the Board of Directors of the Company). The Company also agreed to pay up to $15,000 of reasonable legal counsel fees of Legend in connection with the June 2007 Financing.
For its services as placement agent in the June 2007 Financing, the Company paid Legend $76,650 in cash, 54,750 shares of common stock of the Company with a fair value of $123,763 and five-year warrants to purchase of
(See report of independent registered public accounting firm.)
F-26
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
54,750 shares of Company common stock at an exercise price of $1.40 per share (the “Legend Warrants”) with a fair value of $117,776.
The Company is not required to register for resale under the Securities Act the shares of common stock issued to the investors and Legend in the June 2007 Financing or the shares issuable upon the exercise of the June 2007 Warrants and Legend Warrants. However, the Company did agree with Legend that it would provide piggyback registration rights to the investors and Legend with respect to the securities purchased in the June 2007 Financing, subject to customary exceptions.
On April 4, 2007, the Company entered into an Advisory Agreement with Legend, pursuant to which the Company issued 75,000 shares of its common stock with a fair value of $116,250 to Legend for advisory services rendered. The Company recorded the fair value of these shares as selling, general and administrative expense.
Common Stock Reserves
As of December 31, 2008, the Company had the following reserves established for the future issuance of common stock as follows:
| | | | | |
| Reserve for exercise of warrants | | | 4,432,828 | |
| Reserve for the issuance of restricted stock grants | | | 292,875 | |
| Reserve for the issuance of stock options outside of a qualified plan | | | 2,900,000 | |
| Reserve for the exercise of stock options | | | 3,518,138 | |
| | | | | |
| Total Reserves | | | 11,143,841 | |
| | | | | |
In 1997, the Company adopted its 1997 Long-Term Incentive and Stock Option Plan (the “1997 Plan”). Pursuant to the 1997 Plan, the Company’s Board of Directors (or committeesand/or executive officers delegated by the Board of Directors) may grant incentive and nonqualified stock options to the Company’s employees, officers, directors, consultants and advisors. The Company has reserved an aggregate of 150,000 shares of its common stock for issuance upon exercise of options granted under the 1997 Plan. As of December 31, 2008, there were options to purchase an aggregate of 25,000 shares of common stock outstanding under the 1997 Plan and 116,546 shares available for future option grants under the 1997 Plan.
In connection with the ChoiceTel Merger in year 2002, the Company assumed all outstanding options under the 1999 Sontra Medical, Inc. Stock Option and Incentive Plan (the “1999 Plan”). The Company may not grant any additional options under the 1999 Plan. The Company assumed options to purchase an aggregate of 86,567 shares of common stock under the 1999 Plan. As of December 31, 2008, there were options to purchase an aggregate of 5,780 shares of common stock outstanding under the 1999 Plan and none available for future grants.
In March 2003, the Company’s stockholders approved its 2003 Stock Option and Incentive Plan (the “2003 Plan”). Pursuant to the 2003 Plan, the Company’s Board of Directors (or committeesand/or executive officers delegated by the Board of Directors) may grant incentive and nonqualified stock options, restricted stock and other stock-based awards to the Company’s employees, officers, directors, consultants and advisors. On May 22, 2007, the shareholders of the Company increased the number of shares authorized for issuance under the 2003 Plan by 1,000,000 shares. As of December 31, 2008, the maximum aggregate number of shares that may be authorized for issuance under the 2003 Plan for all periods is 1,600,000. As of December 31, 2008, there were restricted shares of common stock and options to purchase an aggregate of 1,013,750 shares of Common Stock outstanding under the 2003 Plan and 573,250 shares available for future grants under the 2003 Plan.
(See report of independent registered public accounting firm.)
F-27
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
On May 20, 2008, the Company’s shareholders approved the Echo Therapeutics, Inc. 2008 Equity Incentive Plan (the “2008 Plan”) at the Company’s Annual Meeting of Shareholders. The 2008 Plan provides for grants of incentive stock options to employees and nonqualified stock options and restricted stock to employees, consultants and non-employee directors of the Company. As of December 31, 2008, the number of shares authorized for issuance under the 2008 Plan was 1,700,000 shares and no grants had been made under the 2008 Plan.
Share-Based Compensation
For options and employee restricted stock issued and outstanding during the year ended December 31, 2008 and 2007, the Company recorded additional paid-in capital and non-cash compensation expense of $1,824,807 and $2,609,811, respectively, each net of estimated forfeitures.
The fair value of each option award is estimated on the date of grant using the Black-Scholes option pricing model that uses the assumptions noted in the following table. Expected volatilities are based on historical volatility of the Company’s common stock using historical periods consistent with the expected term of the options. The Company uses historical data, as well as subsequent events occurring prior to the issuance of the financial statements, to estimate option exercise and employee termination within the valuation model. The expected term of options granted under the Company’s stock plans, all of which qualify as “plain vanilla,” is based on the average of the contractual term (generally 10 years) and the vesting period (generally 36 — 42 months) as permitted under SEC Staff Accounting Bulletin No. 107 and 110. The risk-free rate is based on the yield of a U.S. Treasury security with a term consistent with the option. Restricted stock grants are valued based on the closing market price for the Company’s common stock on the grant date.
The assumptions used principally for options granted to employees in the years ended December 31, 2008 and 2007 were as follows:
| | | | | |
| | | 2008 | | 2007 |
| |
| Risk-free interest rate | | 2.25% - 4.11% | | 4.11% - 4.72% |
| Expected dividend yield | | — | | — |
| Expected term (employee grants) | | 6 - 6.75 years | | 6 - 6.75 years |
| Forfeiture rate (excluding fully vested options) | | 11% - 33% | | 11% - 33% |
| Expected volatility | | 151% - 156% | | 118% - 164% |
A summary of option activity under the Company’s stock plans and options granted to officers of the Company outside any plan as of December 31, 2008 and changes during the year then ended is presented below:
| | | | | | | | | | | | | | | | | |
| | | | | | | | | Weighted-
| | | | |
| | | | | | Weighted-
| | | Average
| | | | |
| | | | | | Average
| | | Remaining
| | | Aggregate
| |
| | | | | | Exercise
| | | Contractual
| | | Intrinsic
| |
Options | | Shares | | | Price | | | Term | | | Value | |
| |
| Outstanding at January 1, 2008 | | | 4,066,271 | | | $ | 1.99 | | | | | | | | | |
| Granted | | | 265,000 | | | $ | 1.52 | | | | | | | | | |
| Exercised | | | (66,666 | ) | | $ | 1.39 | | | | | | | | | |
| Forfeited or expired | | | (320,075 | ) | | $ | 3.27 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding at December 31, 2008 | | | 3,944,530 | | | $ | 1.87 | | | | 8.76 years | | | $ | 33,950 | |
| | | | | | | | | | | | | | | | | |
| Exercisable at December 31, 2008 | | | 2,661,959 | | | $ | 1.92 | | | | 8.69 years | | | $ | 29,698 | |
| | | | | | | | | | | | | | | | | |
(See report of independent registered public accounting firm.)
F-28
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The weighted-average grant-date fair value of options granted during the year ended December 31, 2008 was $1.52 per share. Share-based compensation expense recognized in the year ended December 31, 2008 was $181,031 for options granted during the year ended December 31, 2008. The intrinsic value of options exercised in the year ended December 31, 2008 was $15,333. The Company received cash proceeds of zero and $1,350 from the exercise of options in the years ended December 31, 2008 and 2007, respectively. As of December 31, 2008, there was approximately $1,508,992 of total unrecognized compensation expense related to non-vested share-based option compensation arrangements.
As of December 31, 2008, the Company had outstanding restricted stock grants (including 13,125 shares issued under the 2003 Plan) amounting to 292,875 shares at a weighted-average grant-date fair value of $1.74 per share. Of the outstanding restricted stock grants, 279,750 shares have not been registered. A summary of the status of the Company’s nonvested restricted stock grants as of December 31, 2008, and changes during the year ended December 31, 2008, is presented below:
| | | | | | | | | |
| | | | | | Weighted-
| |
| | | | | | Average
| |
| | | | | | Grant-Date
| |
Nonvested Shares | | Shares | | | Fair Value | |
| |
| Nonvested at January 1, 2008 | | | 16,875 | | | $ | 1.77 | |
| Granted | | | — | | | | — | |
| Vested | | | (1,875 | ) | | $ | 1.77 | |
| Forfeited | | | (11,250 | ) | | $ | 1.77 | |
| | | | | | | | | |
| Nonvested at December 31, 2008 | | | 3,750 | | | $ | 1.77 | |
| | | | | | | | | |
Share-based compensation recognized in the year ended December 31, 2008 related to restricted stock grants was $5,808.
As of December 31, 2008, there was approximately $9,425 of total unrecognized compensation expense related to non-vested share-based restricted stock arrangements granted under the Company’s stock plans.
The Company determines the fair value of warrants issued using the Black-Scholes option pricing model. The assumptions used in connection with the Black-Scholes option pricing model for common stock warrants granted to BHP during the years ended December 31, 2008 and 2007 were as follows:
| | | | | |
| | | 2008 | | 2007 |
| |
| Risk-free interest rate | | 3.34% | | 4.72% |
| Expected dividend yield | | — | | — |
| Expected term (contractual term) | | 5 years | | 5 years |
| Forfeiture rate | | 0% | | 0% |
| Expected volatility | | 155% | | 159% - 161% |
Expected volatilities are based on historical volatility of the Company’s common stock using historical periods consistent with the expected term of the warrant. The risk-free rate is based on the yield of a U.S. Treasury security with a term consistent with the warrant.
The warrants issued in the years ended December 31, 2008 and 2007 generally have a term of five (5) years, a non-redeemable feature and a cashless exercise provision. Certain warrants have a standard weighted average anti-dilution protection and piggy back registration rights.
(See report of independent registered public accounting firm.)
F-29
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
At December 31, 2008, the Company had the following outstanding warrants:
| | | | | | | | | | | | | |
| | | Number of
| | | | | | | |
| | | Shares
| | | Exercise
| | | | |
| | | Exercisable | | | Price | | | Date of Expiration | |
| |
| Granted to investors and placement agent in private placement | | | 118,620 | | | $ | 24.50 | | | | 12/8/2009 | |
| Granted to investor in former subsidiary | | | 15,000 | | | $ | 50.00 | | | | 2/23/2010 | |
| Granted to investors and placement agent in private placement | | | 476,830 | | | $ | 5.80 | | | | 3/16/2016 | |
| Granted to investors in private placement | | | 611,250 | | | $ | 0.21 | | | | 1/30/09 | |
| Granted to investors and placement agent in private placement | | | 599,250 | | | $ | 1.40 | | | | 6/15-7/16/2012 | |
| Granted to financial investment advisor | | | 60,000 | | | $ | 1.58 | | | | 7/25/2012 | |
| Granted to financial advisor in connection with an acquisition | | | 425,000 | | | $ | 2.18 | | | | 9/14/2012 | |
| Granted to financial investment advisor | | | 175,013 | | | $ | 1.49 | | | | 2/11/2013 | |
| Granted to financial investment advisor | | | 26,833 | | | $ | 1.10 | | | | 10/31/2013 | |
| Granted to investors in private placement | | | 849,059 | | | $ | 1.00 | | | | 2/11/2013 | |
| Granted to investors in private placement | | | 653,728 | | | $ | 1.94 | | | | 3/14/2013 | |
| Granted to investors in private placement of preferred stock | | | 153,912 | | | $ | 1.00 | | | | 9/30/2013 | |
| Granted to investors in private placement of preferred stock | | | 268,333 | | | $ | 1.50 | | | | 10/28-10/31/2013 | |
| | | | | | | | | | | | | |
| Total | | | 4,432,828 | | | | | | | | | |
| | | | | | | | | | | | | |
| Weighted average exercise price | | | | | | $ | 2.57 | | | | | |
| | | | | | | | | | | | | |
| Weighted average duration in years | | | | | | | | | | | 3.21 years | |
| | | | | | | | | | | | | |
A summary of warrant activity in the year ended December 31, 2008 is as follows:
| | | | | | | | | |
| | | | | | Weighted-
| |
| | | | | | Average
| |
| | | | | | Exercise
| |
Warrants | | Shares | | | Price | |
| |
| Outstanding at January 1, 2008 | | | 3,574,459 | | | $ | 4.66 | |
| Granted | | | 2,231,878 | | | $ | 1.40 | |
| Exercised | | | (726,250 | ) | | $ | 0.21 | |
| Forfeited or expired | | | (647,259 | ) | | $ | 6.08 | |
| | | | | | | | | |
| Outstanding at December 31, 2008 | | | 4,432,828 | | | $ | 2.57 | |
| | | | | | | | | |
Subsequent to December 31, 2008, certain warrant terms were modified (see Note 14).
No provision or benefit for federal or state income taxes has been recorded, as the Company has incurred a net loss for all periods presented, and has provided a valuation allowance against its deferred tax assets.
(See report of independent registered public accounting firm.)
F-30
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
At December 31, 2008, the Company had federal net operating loss carryforwards of approximately $43,800,000, which will expire in varying amounts beginning in 2018. The Company also had research and development tax credit carryforwards of approximately $827,000 which will begin to expire in 2018 unless previously utilized. The United States Tax Reform Act of 1986 contains provisions that may limit the Company’s net operating loss carryforwards available to be used in any given year in the event of significant changes in the ownership interests of significant stockholders, as defined.
Significant components of the Company’s net deferred tax asset are as follows:
| | | | | | | | | |
| | | December 31, | |
| | | 2008 | | | 2007 | |
| |
| Deferred Tax Assets/(Liabilities) | | | | | | | | |
| Net operating loss carryforwards | | $ | 16,842,000 | | | $ | 14,656,000 | |
| Research credit carryforward | | | 827,000 | | | | 834,000 | |
| Acquired intangible assets, net | | | (3,785,000 | ) | | | (3,800,000 | ) |
| Other temporary differences | | | 132,000 | | | | 154,000 | |
| | | | | | | | | |
| Total deferred tax assets, net | | | 14,016,000 | | | | 11,844,000 | |
| Valuation allowance | | | (14,016,000 | ) | | | (11,844,000 | ) |
| | | | | | | | | |
| Net deferred tax asset | | $ | — | | | $ | — | |
| | | | | | | | | |
The Company’s valuation allowance increased by $2,172,000 in 2008 and decreased by $1,895,000 in 2007. SFAS No. 109 requires that a valuation allowance be recorded when it is more likely than not that some portion or all of the net deferred tax assets will not be realized. Since the Company cannot be assured of realizing the net deferred tax asset, a full valuation allowance has been provided.
Income taxes computed using the federal statutory income tax rate differs from the Company’s effective tax rate primarily due to the following:
| | | | | | | | | |
| | | December 31, | |
| | | 2008 | | | 2007 | |
| |
| Benefit at Statutory rate | | $ | 3,708,000 | | | $ | 4,629,000 | |
| State Tax Benefit | | | 344,000 | | | | 430,000 | |
| IPR&D | | | — | | | | (2,448,000 | ) |
| Other Permanent Items | | | (1,930,000 | ) | | | (1,137,000 | ) |
| R&D Credits | | | 50,000 | | | | 100,000 | |
| | | | | | | | | |
| Benefit at Effective Rate | | | 2,172,000 | | | | 1,574,000 | |
| Acquired intangible assets | | | — | | | | (3,800,000 | ) |
| Acquired net operating loss carryforward | | | — | | | | 331,000 | |
| Changes in Valuation Allowance | | | (2,172,000 | ) | | | 1,895,000 | |
| | | | | | | | | |
| | | $ | — | | | $ | — | |
| | | | | | | | | |
(See report of independent registered public accounting firm.)
F-31
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Series A-2 Preferred Stock and Warrant Financing
On February 24, 2009, the Board of Directors authorized the creation and issuance of a new class ofSeries A-2 Convertible Preferred Stock. The Board of Directors authorized the issuance of up to 4,400,000 shares ofSeries A-2 Convertible Preferred Stock(“Series A-2 Stock”) with the rights, preferences and privileges described below.
Pursuant to the terms of the Certificate of Designation, Preference and Rights ofSeries A-2 Convertible Preferred Stock, each share ofSeries A-2 Stock is initially convertible into one share of Common Stock, subject to adjustment for stock splits, combinations or similar events and subject to customary anti-dilution provisions. TheSeries A-2 Stock will pay a quarterly dividend, at an annual rate of 8%, which is payable in cash or in kind at the option of the Company. Each Investor may convert its Shares at any time following issuance of the Shares. TheSeries A-2 Stock is not redeemable.
Each holder ofSeries A-2 Stock shall have the right, exercisable on an all or none basis, to participate in the Company’s first equity offering or series of equity-linked offerings to occur after the date of the Financing that yields gross proceeds to the Company of at least $2,000,000 (the “Qualified Offering”) on the same terms and conditions as offered by the Company to the other purchasers of such securities issued and sold by the Company in the Qualified Offering (the “Additional Securities”), except that the consideration for each holder’s participation in the Qualified Offering shall be the surrender of 100% of such holder’s shares ofSeries A-2 Stock in exchange for Additional Securities with a purchase price equal to an aggregate of 115% of the Liquidation Preference of theSeries A-2 Stock surrendered by such holder. The liquidation preferences of theSeries A-2 Stock are consistent with theSeries A-1 and Series A Stock and theSeries A-2 Stock is not redeemable.
In the event that the Company liquidates, dissolves, or winds up its affairs (each, a “Liquidation Event”), the holders ofSeries A-2 Stock will be entitled to receive (subject to the rights of any securities designated as senior to theSeries A-2 Stock) a liquidation preference equal to the greater of (i) $.50 per share or (ii) the amount that would be distributed in such Liquidation Event on the number of shares of common stock issuable upon conversion of theSeries A-2 Stock. TheSeries A-2 Stock ranks pari passu with the Series A Stock andSeries A-1 Stock (see Note 9). The Company cannot create or issue any security senior to theSeries A-2 Stock without the approval of the holders of the majority of the aggregate outstanding Series A,Series A-1 Stock andSeries A-2 Stock.
Subsequent to December 31, 2008, the Company entered into an Amended and Restated Stock and Warrant Purchase Agreement (the“A-2 Purchase Agreement”) with certain select institutional and accredited investors (the “Investors”) in connection with the Company’s private placement (the“A-2 Financing”) of 700,000 shares of itsSeries A-2 Convertible Preferred Stock (the“Series A-2 Stock”) at a price of $0.50 per share together with warrants to purchase a number of shares of the Company’s Common Stock equal to (i) thirty-five percent (35%) or (ii) for investments of $250,000 or more, fifty percent (50%) of the number of shares ofSeries A-2 Stock purchased by each Investor (the“A-2 Warrants”) in theA-2 Financing. The Company has received gross proceeds of $350,000 from theA-2 Financing and theA-2 Financing is still ongoing.
Pursuant to theA-2 Purchase Agreement, the Company issuedA-2 Warrants to the Investors to purchase up to 320,000 shares of Common Stock. TheA-2 Warrants are immediately exercisable at a price per share of $0.75, subject to adjustment for stock splits, combinations or similar events, and will expire no later than April 30, 2014. TheA-2 Warrants allow for cashless exercise. In addition, the Company has the option to redeem theA-2 Warrants, in whole but not in part, upon satisfaction of certain conditions, including (i) the availability of an effective registration statement or Rule 144 exemption for any resale by the holder, (ii) the shares of Common Stock trading at a price per share in excess of 200% of the then-applicable exercise price for ten (10) trading days out of a period of fifteen (15) consecutive trading days prior to the redemption, and (iii) an average daily trading volume during such fifteen (15) consecutive trading days of at least 50,000 shares of Common Stock. Finally, an exercise under the
(See report of independent registered public accounting firm.)
F-32
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Warrants may not result in the holder beneficially owning more than 4.99% or 9.99%, as applicable, of all of the Common Stock outstanding at the time; provided, however, that a holder may waive the foregoing provision upon sixty-one (61) days’ advance written notice to the Company.
Re-pricing of Warrants to Financial Advisor and Affiliates
On January 30, 2009, the Company entered into an engagement letter with BHP pursuant to which BHP will provide financial advisory services to the Company (the “Advisory Agreement”) only as requested by the Company from time to time. In connection with BHP’s engagement under the Advisory Agreement, all warrants held by BHPand/or its registered employees on January 30, 2009 (the “Amended Warrants”) were amended with a term of five (5) years from the date of the Agreement and an exercise price equal to the lower of (i) $0.55 or (ii) the current exercise price of such warrants. Additionally, if and when the Company first requests financial advisory services from BHP pursuant to the Agreement, the Company shall issue to BHPand/or its registered assignees or designees an aggregate of 360,000 warrants in substantially the same form as the Amended Warrants.
On February 19, 2009, BHP and the Company amended the Advisory Agreement, effective as of January 30, 2009, to reflect their original intention to amend warrants held by BHPand/or its registered employees and affiliates of BHPand/or its registered employees on January 30, 2009. As a result, a total of 560,118 shares were repriced to $0.55 per share on January 30, 2009. The closing price of the Company’s stock on January 30, 2009 was $0.30. The excess of the fair value of the modified warrants over the fair value of the original warrants immediately prior to modification, or approximately $14,000 will be charged to expense as of the date of the modification.
Grant of Restricted Stock and Re-pricing of Certain Nonqualified Stock Options
On February 20, 2009, the Compensation Committee of the Board of Directors of the Company approved a reduction of the exercise price of options for certain officers of the Company to purchase an aggregate of 1,300,000 shares of the Company’s common stock to $0.55 per share (the “Repricing”). Prior to the repricing, the exercise price of such options was $1.39 per share. The Company also repriced options held by certain employees and consultants for the purchase of 550,000 shares of Common Stock with original exercise prices of between $0.61 and $2.25 to an exercise price of $0.40. All other terms of the options remained the same. In addition, the Compensation Committee also approved the cancellation of stock options for certain officers of the Company to purchase 1,250,000 shares of the Company’s common stock that were granted on September 14, 2007 at an exercise price per share equal to $2.39. The excess of the fair value of the modified options over the fair value of the original options immediately prior to modification, or approximately $35,000, will be charged to expense over the remaining vesting period of the options.
Further, the Compensation Committee granted an aggregate of 637,250 restricted shares of the Company’s common stock with a fair value of approximately $255,000 to certain employees and officers of the Company (the “Restricted Share Grant”) pursuant to Restricted Stock Agreements under the Company’s 2008 Plan. Subject to the terms and conditions of the Restricted Stock Agreements, the restricted shares will vest upon the first to occur of (i) FDA approval of the Company’s Symphony tCGM System; or (ii) the sale of all or substantially all of the assets of the Company or all or substantially all of the outstanding capital stock of the Company in exchange for Liquid Proceeds. For the purposes of the Restricted Share Grants, “Liquid Proceeds” means (a) cash; (b) securities which can be sold immediately on NYSE or NASDAQ; (c) securities which are or will be registered such that they can be sold upon on NYSE or NASDAQ upon termination of alock-up period not to exceed one hundred eighty (180) days; or (d) or a combination of cash and the foregoing securities. The Restricted Share Grant was effected under Section 4(2) of the Securities Act.
(See report of independent registered public accounting firm.)
F-33
ECHO THERAPEUTICS, INC.
(Formerly Sontra Medical Corporation)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Grant of Warrants to Vendor
Effective March 2, 2008, the Company’s board of directors approved the issuance of warrants to purchase 50,000 shares of the Company’s common stock at an exercise price of $0.70 per share to a vendor for engineering services. The warrants have a five year term. The fair value of the services has been determined to be approximately $13,400.
Grant of Warrants to Service Provider
Effective March 15, 2009, the Company’s board of directors approved the issuance to a service provider of warrants to purchase 60,000 shares of the Company’s common stock at an exercise price of $0.60 per share in consideration for the interest-free deferral of amounts currently owed, and amounts to be invoiced, for consulting services. The warrants have a five year term. The warrants vest at 20,000 shares immediately and an additional 10,000 shares vest on each of October 1, 2009, January 1, 2010, April 1, 2010 and July 1, 2010 unless the Company pays all outstanding invoices due to the service provider in full prior to any such vesting date. The fair value of the warrants has been determined to be approximately $24,500 and will be recognized over the vesting period.
Extension of Maturity Date for Senior Secured Note
On March 23, 2009, the Company and Imperium Master Fund, Ltd. (“Imperium”) entered into an amendment agreement (the “Amendment”) pursuant to which the Company and Imperium agreed to extend the maturity date of the Original Issue Discount Senior Secured Note issued by the Company to Imperium on March 24, 2008 (the “Note”) to April 24, 2009. The Note, which was issued by the Company to Imperium pursuant to a Securities Purchase and Loan Agreement between the Company and Imperium dated as of March 24, 2008 (as amended to date, the “Loan Agreement”), originally had a maturity date of March 24, 2009. The purpose of the maturity date extension is to provide the Company and Imperium sufficient time to amend the Loan Agreement, the Note and all other senior secured notes issued by the Company thereunder (collectively, the “Notes”), to provide for, subject to certain conditions, a one-year extension of the maturity of the Notes.
Re-pricing of Warrants to Investor
On April 6, 2009, the Company executed a letter agreement pursuant to which it agreed to change the exercise price of all outstanding warrants for the purchase of 950,085 shares of Common Stock with exercise prices between $1.50 and $1.00 to $.75 for an investor and its subsidiaries.
(See report of independent registered public accounting firm.)
F-34
| |
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None.
| |
| ITEM 9A(T). | CONTROLS AND PROCEDURES. |
We have carried out an evaluation, under the supervision and the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined inRules 13a-15(e) and15d-15(e) under the Securities Exchange Act of 1934, as amended (the “Securities Act”)), as of December 31, 2008. Based upon that evaluation, our principal executive officer and principal financial officer concluded that, as of the end of that period, our disclosure controls and procedures are effective in providing reasonable assurance that (a) the information required to be disclosed by us in the reports that we file or submit under the Securities Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and (b) such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. In designing and evaluating our disclosure controls and procedures, our management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Changes in Internal Control over Financial Reporting
We evaluate the effectiveness of our internal control over financial reporting in order to comply with Section 404 of the Sarbanes-Oxley Act of 2002. Section 404 requires us to evaluate annually the effectiveness of our internal controls over financial reporting as of the end of each fiscal year, and to include a management report assessing the effectiveness of our internal control over financial reporting in all annual reports. We have not made any changes in our internal control over financial reporting during our fourth fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined inRules 13a-15(f) and15d-15(f) under the Securities Act as a process designed by, or under the supervision of, a company’s principal executive and principal financial officers and effected by a company’s board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that:
| | |
| | • | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; |
| |
| | • | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and |
| |
| | • | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
45
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2008. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control — Integrated Framework.
Following our assessment, management has concluded that, as of December 31, 2008, our internal control over financial reporting is effective based on those criteria.
This annual report does not include an attestation report of the company’s registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the company’s registered public accounting firm pursuant to temporary rules of the Securities and Exchange Commission that permit the company to provide only management’s report in this annual report.
PART III
| |
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE. |
Incorporated by reference to the portions of our Definitive Proxy Statement entitled “Election of Directors,” “Directors and Executive Officers,” “The Board of Directors and its Committees,” “Audit Committee Financial Expert” and “Section 16(a) Beneficial Ownership Reporting Compliance.”
We have adopted a Code of Business Conduct and Ethics that applies to all of our directors, officers and employees, including our principal executive officer and principal financial officer. A copy of our Code of Business Conduct and Ethics is filed with or incorporated by reference in this report, and is also posted to our website atwww.echotx.com.
| |
| ITEM 11. | EXECUTIVE COMPENSATION. |
Incorporated by reference to the portions of our Definitive Proxy Statement entitled “Executive Compensation,” “Outstanding Equity Awards at Fiscal Year-End” and “Director Compensation.”
| |
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
Incorporated by reference to the portion of our Definitive Proxy Statement entitled “Securities Ownership of Certain Beneficial Owners and Management.”
| |
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE. |
Incorporated by reference to the portions of our Definitive Proxy Statement entitled “Certain Relationships and Related Transaction,” “Independence of Members of Board of Directors” and “The Board of Directors and its Committees.”
| |
| ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES. |
Incorporated by reference to the portions of our Definitive Proxy Statement entitled “Independent Registered Public Accounting Firm” and “Audit Committee Policy on Pre-Approval of Services of Independent Registered Public Accounting Firm.”
PART IV
| |
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES. |
The Exhibits listed in the Exhibit Index immediately preceding such Exhibits are filed with or incorporated by reference in this report.
46
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
ECHO THERAPEUTICS, INC.
| | |
| | By: | /s/ Patrick T. Mooney |
Name: Patrick T. Mooney, M.D.
| | |
| | Title: | Chief Executive Officer |
Date: April 13, 2009
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities indicated on April 13, 2009.
| | | |
By: /s/ Patrick T. Mooney | | By: /s/ Shawn K. Singh |
| Name: Patrick T. Mooney, M.D. | | Name: Shawn K. Singh, J.D. |
Title: Chief Executive Officer and Chairman
of the Board
(Principal Executive Officer) | | Title: President and Director |
| | | |
| By: /s/ Walter W. Witoshkin | | By: /s/ Robert S. Langer |
| Name: Walter W. Witoshkin | | Name: Robert S. Langer, Sc.D |
| Title: Director | | Title: Director |
| | | |
| By: /s/ Vincent D. Enright | | By: /s/ Harry G. Mitchell |
| Name: Vincent D. Enright | | Name: Harry G. Mitchell |
| Title: Director | | Title: Chief Operating Officer,
Chief Financial Officer and Treasurer
(Principal Financial Officer and
Principal Accounting Officer) |
47
EXHIBIT INDEX
| | | | | |
Exhibit
| | |
Number | | Description of Document |
| |
| | 2 | .1 | | Agreement and Plan of Merger and Reorganization by and among the Registrant, Durham Pharmaceuticals Ltd. (d/b/a Echo Therapeutics, Inc.) and Durham Pharmaceuticals Acquisition Co. dated as of September 14, 2007 is incorporated herein by reference to Exhibit 2.1 of the Registrant’s Current Report onForm 8-K dated September 14, 2007. |
| | 2 | .2 | | Asset Purchase Agreement by and between the Registrant and DP Pharmaceuticals, LLC dated as of September 14, 2007 is incorporated herein by reference to Exhibit 2.2 of the Registrant’s Current Report onForm 8-K dated September 14, 2007. |
| | 2 | .3 | | Agreement and Plan of Merger, dated June 9, 2008, between Echo Therapeutics, Inc., a Minnesota corporation, and Echo Therapeutics, Inc., a Delaware corporation, is incorporated herein by reference to Exhibit 2.1 to the Registrant’s Current Report onForm 8-K dated June 9, 2008. |
| | 2 | .4 | | Certificate of Ownership and Merger merging Echo Therapeutics, Inc., a Minnesota corporation, with and into Echo Therapeutics, Inc., a Delaware corporation, is incorporated herein by reference to Exhibit 2.2 to the Registrant’s Current Report onForm 8-K dated June 9, 2008. |
| | 2 | .5 | | Articles of Merger merging Echo Therapeutics, Inc., a Minnesota corporation, with and into Echo Therapeutics, Inc., a Delaware corporation, is incorporated herein by reference to Exhibit 2.3 to the Registrant’s Current Report onForm 8-K dated June 9, 2008. |
| | 3 | .1 | | Certificate of Incorporation of the Registrant is incorporated herein by reference to Exhibit 3.1 to the Registrant’s Current Report onForm 8-K dated June 9, 2008. |
| | 3 | .2 | | Bylaws of the Registrant is incorporated herein by reference to Exhibit 3.2 to the Registrant’s Current Report onForm 8-K dated June��9, 2008. |
| | 4 | .1 | | Form of Warrant to Purchase Common Stock is incorporated herein by reference to Exhibit 4.1 to the Registrant’s Current Report onForm 8-K dated September 30, 2008. |
| | 4 | .2 | | Form of Warrant to Purchase Common Stock is incorporated herein by reference to Exhibit 4.1 to the Registrant’s Current Report onForm 8-K dated October 28, 2008. |
| | 10 | .1* | | 2003 Stock Option and Incentive Plan, as amended, is incorporated herein by reference to Appendix I of the Registrant’s Definitive Schedule 14A filed on April 17, 2007. |
| | 10 | .2* | | Form of Restricted Stock Agreement for use under the Registrant’s 2003 Stock Option and Incentive Plan is incorporated herein by reference to Exhibit 99.1 to the Registrant’s Current Report onForm 8-K dated August 14, 2006. |
| | 10 | .3* | | 1999 Stock Option and Incentive Plan is incorporated herein by reference to Exhibit 10.31 of the Registrant’s Registration Statement onForm S-4 filed on April 24, 2002. |
| | 10 | .4* | | 1997 Long-Term Incentive and Stock Option Plan, as amended, is incorporated herein by reference to Exhibit 10.3 of the Registrant’s Quarterly Report onForm 10-QSB for the period ended June 30, 2002. |
| | 10 | .5 | | Lease Agreement between the Registrant and Forge Park Investors LLC dated January 24, 2003 is incorporated herein by reference to Exhibit 10.13 to the Registrant’s Annual Report onForm 10-K for the fiscal year ended December 31, 2002. |
| | 10 | .6 | | Patent License Agreement (Exclusive) between Sontra Medical, Inc. and the Massachusetts Institute of Technology dated as of June 30, 1998 is incorporated herein by reference to Exhibit 10.39 of the Registrant’s Registration Statement onForm S-4 filed on April 24, 2002. |
| | 10 | .7 | | Form of Common Stock Purchase Warrant is incorporated herein by reference to Appendix E to the Registrant’s Definitive Schedule 14A filed September 8, 2003. |
| | 10 | .8 | | Form of Placement Agent Common Stock Purchase Warrant is incorporated herein by reference to Exhibit 99.4 to the Registrant’s Registration Statement onForm S-3 filed on October 15, 2003. |
| | 10 | .9 | | Common Stock and Warrant Purchase Agreement dated as of December 8, 2004 by and among the Registrant and the investors listed on Schedule 1 thereto is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated December 8, 2004. |
| | 10 | .10 | | Form of Common Stock Purchase Warrant is incorporated herein by reference to Exhibit 10.2 to the Registrant’s Current Report onForm 8-K dated December 8, 2004. |
48
| | | | | |
Exhibit
| | |
Number | | Description of Document |
| |
| | 10 | .11 | | Common Stock and Warrant Purchase Agreement dated as of March 7, 2006 by and among the Registrant and the investors listed on Schedule 1 thereto is incorporated herein by reference to Exhibit 99.1 to the Registrant’s Current Report onForm 8-K dated March 7, 2006. |
| | 10 | .12 | | Form of Common Stock Purchase Warrant is incorporated herein by reference to Exhibit 99.2 to the Registrant’s Current Report onForm 8-K dated March 7, 2006. |
| | 10 | .13 | | Common Stock and Warrant Purchase Agreement dated as of January 2, 2007 by and among the Registrant, Sherbrooke Partners, LLC and the purchasers named therein is incorporated herein by reference to Exhibit 10.1 of the Registrant’s Current Report onForm 8-K dated January 1, 2007. |
| | 10 | .14 | | Form of Warrant to Purchase Shares of Common Stock is incorporated herein by reference to Exhibit 10.2 of the Registrant’s Current Report onForm 8-K dated January 1, 2007. |
| | 10 | .15 | | Form of Subscription Agreement is incorporated herein by reference to Exhibit 10.1 of the Registrant’s Current Report onForm 8-K dated June 15, 2007. |
| | 10 | .16 | | Form of Warrant to Purchase Shares of Common Stock is incorporated herein by reference to Exhibit 10.2 of the Registrant’s Current Report onForm 8-K dated June 15, 2007. |
| | 10 | .17 | | Strategic Master Services Agreement by and between the Registrant and Cato Research Ltd. dated September 14, 2007 is incorporated herein by reference to Exhibit 10.1 of the Registrant’s Current Report onForm 8-K dated September 14, 2007. |
| | 10 | .18* | | Employment Agreement by and between the Registrant and Patrick T. Mooney dated as of September 14, 2007 is incorporated herein by reference to Exhibit 10.4 to the Registrant’s Current Report onForm 8-K dated September 14, 2007 (including Nonqualified Stock Option Agreement). |
| | 10 | .19* | | Employment Agreement by and between the Registrant and Shawn K. Singh dated as of September 14, 2007 is incorporated herein by reference to Exhibit 10.5 to the Registrant’s Current Report onForm 8-K dated September 14, 2007 (including Nonqualified Stock Option Agreement). |
| | 10 | .20* | | Employment Agreement by and between the Registrant and Harry G. Mitchell dated as of September 14, 2007 is incorporated herein by reference to Exhibit 10.6 to the Registrant’s Current Report onForm 8-K dated September 14, 2007 (including Nonqualified Stock Option Agreement). |
| | 10 | .21 | | Form of Warrant to Purchase Shares of Common Stock is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Quarterly Report onForm 10-QSB for the period ended September 30, 2007. |
| | 10 | .22* | | Form of Nonqualified Stock Option Agreement is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated December 22, 2007. |
| | 10 | .23 | | First Amendment to Lease dated February 11, 2008 by and between the Registrant and CRP-2 Forge, LLC, as successor in interest to Forge Park Investors LLC is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated March 13, 2008. |
| | 10 | .24 | | Note and Warrant Purchase Agreement by and among the Registrant and the Purchasers named therein dated as of February 11, 2008 is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated February 11, 2008. |
| | 10 | .25 | | Guaranty dated as of February 11, 2008 executed by Sontra Medical, Inc. is incorporated herein by reference to Exhibit 10.2 to the Registrant’s Current Report onForm 8-K dated February 11, 2008. |
| | 10 | .26 | | Guaranty dated as of February 11, 2008 executed by Echo Therapeutics, Inc. is incorporated herein by reference to Exhibit 10.3 to the Registrant’s Current Report onForm 8-K dated February 11, 2008. |
| | 10 | .27 | | Form of Senior Convertible Promissory Note is incorporated herein by reference to Exhibit 10.4 to the Registrant’s Current Report onForm 8-K dated February 11, 2008. |
| | 10 | .28 | | Form of Warrant is incorporated herein by reference to Exhibit 10.5 to the Registrant’s Current Report onForm 8-K dated February 11, 2008. |
| | 10 | .29 | | Securities Purchase and Loan Agreement by and among the Registrant and the Purchaser named therein dated as of March 24, 2008 is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated March 24, 2008. |
| | 10 | .30 | | Form of Original Issue Discount Senior Secured Note is incorporated herein by reference to Exhibit 10.2 to the Registrant’s Current Report onForm 8-K dated March 24, 2008. |
49
| | | | | |
Exhibit
| | |
Number | | Description of Document |
| |
| | 10 | .31 | | Form of Warrant is incorporated herein by reference to Exhibit 10.3 to the Registrant’s Current Report onForm 8-K dated March 24, 2008. |
| | 10 | .32 | | Guaranty dated as of March 24, 2008 executed by Echo Therapeutics, Inc., Sontra Medical, Inc., Imperium Advisers, LLC, and Imperium Master Fund, Ltd. is incorporated herein by reference to Exhibit 10.4 to the Registrant’s Current Report onForm 8-K dated March 24, 2008. |
| | 10 | .33 | | Security Agreement by and among the Registrant, Imperium Advisers, LLC, Imperium Master Fund, Ltd., Echo Therapeutics, Inc. and Sontra Medical, Inc. dated as of March 24, 2008 is incorporated herein by reference to Exhibit 10.5 to the Registrant’s Current Report onForm 8-K dated March 24, 2008. |
| | 10 | .34 | | Registration Rights Agreement by and among the Registrant and the Purchaser dated as of March 24, 2008 is incorporated herein by reference to Exhibit 10.6 to the Registrant’s Current Report onForm 8-K dated March 24, 2008. |
| | 10 | .35* | | Nonqualified Stock Option Agreement by and between the Registrant and Vincent D. Enright dated as of March 25, 2008 is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated March 25, 2008. |
| | 10 | .36 | | Dermatology Right of First Offer Agreement between the Registrant and Cato BioVentures dated as of April 21, 2008 is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated April 21, 2008. |
| | 10 | .37 | | Amendment No. 1 to Registration Rights Agreement by and among the Registrant and Imperium Master Fund, Ltd. dated as of May 14, 2008 is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated May 14, 2008. |
| | 10 | .38* | | 2008 Equity Incentive Plan is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated April 20, 2008. |
| | 10 | .39 | | Exchange Agreement between the Registrant and the Investors named therein dated as of September 30, 2008 is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated September 30, 2008. |
| | 10 | .40 | | Stock and Warrant Purchase Agreement between the Registrant and the Purchasers named therein dated as of October 28, 2008 is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated October 28, 2008. |
| | 10 | .41 | | Letter agreement between the Registrant and Burnham Hill Partners LLC dated January 30, 2009 is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated January 30, 2009. |
| | 10 | .42 | | Letter agreement between the Registrant and Imperium Master Fund, Ltd. dated March 23, 2009 is incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K dated March 23, 2009. |
| | 14 | | | Code of Business Conduct and Ethics of the Registrant (filed herewith). |
| | 21 | | | Subsidiaries of the Registrant (filed herewith). |
| | 23 | | | Consent of Wolf & Company, P.C. (filed herewith). |
| | 31 | .1 | | Certification of the Chief Executive Officer and Chairman of the Board pursuant toRule 13a-14(a) orRule 15d-14(a) of the Securities and Exchange Act of 1934 as adopted under Section 302 of the Sarbanes-Oxley Act of 2002. |
| | 31 | .2 | | Certification of the Chief Operating Officer, Chief Financial Officer and Treasurer pursuant toRule 13a-14(a) orRule 15d-14(a) of the Securities and Exchange Act of 1934 as adopted under Section 302 of the Sarbanes-Oxley Act of 2002. |
| | 32 | .1 | | Certification of the Chief Executive Officer and Chairman of the Board pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| | 32 | .2 | | Certification of the Chief Operating Officer, Chief Financial Officer and Treasurer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| | |
| * | | Management contract or compensatory plan or arrangement filed in response to Item 13 ofForm 10-K. |
50