Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K/A
(Amendment No. 1)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2006
Commission file number000-28401
MAXYGEN, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 77-0449487 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
515 Galveston Drive
Redwood City, California 94063
(Address of principal executive offices)
Registrant’s telephone number, including area code:
(650) 298-5300
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Name of Each Exchange on Which Registered | |
| Common Stock, $0.0001 par value | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 ofRegulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of thisForm 10-K or any amendment to thisForm 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” inRule 12b-2 of the Exchange Act.
Large accelerated filer o Accelerated filer þ Non-accelerated filer o
Indicate by check mark whether the registrant is a shell company (as defined inRule 12b-2 of the Exchange Act). Yes o No þ
As of June 30, 2006, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the voting stock held by non-affiliates, computed by reference to the closing price for the common stock as quoted by the Nasdaq Global Stock Market as of that date, was approximately $160,482,000. Shares of common stock held by each executive officer and director and by each person who owned 5% or more of the outstanding common stock have been excluded as such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of February 28, 2007, there were 36,669,560 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Certain portions of the registrant’s proxy statement for the 2007 Annual Meeting of Stockholders (hereinafter referred to as the “2007 Proxy Statement”) are incorporated by reference into Part III of this report.
TABLE OF CONTENTS
| Business | 1 | |||||||
| Exhibits, Financial Statement Schedules | 15 | |||||||
| 18 | ||||||||
| EXHIBIT 31.1 | ||||||||
| EXHIBIT 31.2 | ||||||||
i
Table of Contents
EXPLANATORY NOTE:
This amendment to the registrant’s Annual Report on Form10-K for the year ended December 31, 2006, originally filed with the Securities and Exchange Commission (“SEC”) on March 14, 2007, is being filed solely to supplement disclosure in Item 1 of Part I of the original Form10-K under the caption “Other Assets and Areas of Research — Investment in Avidia Inc.” regarding the terms of the Cross License Agreement, dated as of July 16, 2003, between Maxygen, Inc. and Amgen Mountain View Inc. (as successor to Avidia, Inc.), which was filed as Exhibit 10.27 to the original Form10-K. This amendment is being filed in connection with the resolution of comments received from the SEC regarding an application for confidential treatment of the Cross License Agreement. This amendment does not reflect events occurring after the filing of the original Form10-K, and is not intended to modify or update the disclosures therein in any way, except as specifically noted above.
Forward Looking Statements
This report contains forward-looking statements within the meaning of federal securities laws that relate to future events or our future financial performance. In some cases, you can identify forward-looking statements by terminology such as “may,” “can,” “will,” “should,” “could,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “intend,” “potential” or “continue” or the negative of these terms or other comparable terminology. Examples of these forward-looking statements include, but are not limited to, statements regarding the following:
| • | our ability to develop products suitable for commercialization; | |
| • | our predicted development and commercial timelines for any of our potential products, including the timing of any commencement of clinical trials of any product candidate and the progress of such clinical trials; | |
| • | our liquidity and future financial performance; | |
| • | the establishment, development and maintenance of any manufacturing or collaborative relationships; | |
| • | the effectiveness of our MolecularBreeding directed evolution platform and other technologies and processes; | |
| • | our ability to protect our intellectual property portfolio and rights; | |
| • | our ability to identify and develop new potential products; | |
| • | the attributes of any products we may develop; and | |
| • | our business strategies and plans. |
These statements are only predictions. Risks and uncertainties and the occurrence of other events could cause actual results to differ materially from these predictions. Important factors that could cause our actual results to differ materially from the forward-looking statements we make in this report are set forth in this report, including the factors described in the section entitled “Item 1A — Risk Factors.”
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. Moreover, neither we nor any other person assumes responsibility for the accuracy and completeness of these statements. Other than as required by applicable law, we disclaim any obligation to update or revise any forward-looking statement contained in this report as a result of new information or future events or developments.
ii
Table of Contents
PART I
| Item 1 | BUSINESS |
Overview
We are a biotechnology company committed to the discovery and development of improved next-generation protein pharmaceuticals for the treatment of disease and serious medical conditions. Four of our next-generation product candidates are currently in pre-clinical or clinical development:
| • | MAXY-G34, a granulocyte colony stimulating factor, or G-CSF, for the treatment of neutropenia; | |
| • | MAXY-alpha, an interferon alpha product for the treatment of hepatitis C virus, or HCV, infection; | |
| • | MAXY-VII, a factor VII product for the treatment of uncontrolled bleeding in trauma, intracerebral hemorrhage and other indications; and | |
| • | MAXY-gamma, an interferon gamma product for the treatment of idiopathic pulmonary fibrosis and other indications. |
2006 Highlights
During 2006:
| • | We initiated a Phase I clinical trial in the United States to evaluate Maxy-G34. | |
| • | F. Hoffmann-La Roche Ltd., or Roche, initiated a Phase Ia clinical trial in New Zealand to evaluate MAXY-alpha and we received a $2 million milestone payment from Roche for the commencement of the trial. | |
| • | We achieved a manufacturing process development milestone in our MAXY-VII program and received a $5 million milestone payment from Roche for the achievement of the milestone. | |
| • | We received a $17.8 million cash payment for our equity interests in Avidia, Inc., which was acquired by Amgen Inc. | |
| • | We expanded the scope of exclusive licenses previously granted to Codexis, Inc. to our MolecularBreeding directed evolution platform for certain applications relating to energy, including biofuels, in exchange for the right to receive a percentage of revenues received by Codexis for the sale of energy products and the use of processes in the energy field. |
Business Strategy
Our goal is to develop and commercialize improved and proprietary versions of currently marketed or clinically validated therapeutic proteins. To achieve this objective, we are pursuing the following key strategies:
| • | Advance the Development of Our Lead Product Candidates. Our primary focus is the advancement of our lead product candidate, MAXY-G34, through clinical development. We initiated a Phase I clinical trial in the United States for our lead MAXY-G34 product candidate in the third quarter of 2006 and plan to commence Phase IIa clinical trials in the first half of 2007. Roche has an exclusive license to develop and commercialize our MAXY-alpha product candidates for the treatment of HCV infection. Roche initiated a Phase Ia clinical trial of MAXY-alpha in New Zealand at the end of 2006 and has informed us that a Phase Ib clinical trial of MAXY-alpha in HCV patients will begin in 2007. In December 2005, we entered into an agreement with Roche relating to the co-development and commercialization of our MAXY-VII product candidates for acute bleeding indications. In March 2007, we received notice from Roche that Roche has elected to terminate this agreement, effective April 12, 2007. | |
| • | Create Value with Improved Next-Generation Protein Drugs. Our research and development efforts focus on seeking to make improved and proprietary versions of currently marketed therapeutic proteins that address significant market opportunities. We identify currently marketed or clinically validated protein products that require or could benefit from improvements to address existing medical needs in the treatment |
1
Table of Contents
| of disease and serious medical conditions, and use our proprietary MolecularBreeding directed molecular evolution platform and other technologies to improve these products. We commit resources to only those product candidates that we believe are commercially viable. |
| • | Increase Value, Reduce Risk. By using our technologies to improve the properties of currently marketed or clinically validated therapeutic proteins that have significant commercial value, we believe we can potentially create novel and proprietarybest-in-class products that take advantage of known utility, development paths and markets. We believe that this approach may result in reduced risk and enhanced chances of commercial success as compared to the development of novel pharmaceutical products directed at unvalidated targets. | |
| • | Establish a Portfolio of Next-Generation Therapeutic Proteins. In addition to our existing product candidates, we have an ongoing discovery effort focused on next-generation protein or protein subunit pharmaceuticals. Our goal is to leverage discoveries in our research programs or in-licensed product candidates to extend and expand our product pipeline. By broadening our product portfolio, we hope to increase the probability of clinical and commercial success and reduce our exposure to the impact of any one product failure. | |
| • | Establish Strategic Collaborations to Advance our Product Pipeline and Leverage Our Development Resources. Part of our strategy has been to establish strategic collaborations that enable us to retain a large portion of the eventual value from our product candidates. We may enter into strategic collaborations at various stages in our research and development process that will allow us to further diversify our product development risks, reduce costs, access the complementary skills and infrastructure possessed by our partners and accelerate the development and commercialization of our product candidates. | |
| • | Manage Our Financial Resources. Fiscal discipline and pragmatic allocation of our resources are key components of our strategy. We focus our financial resources on those functions that should enhance our ability to generate improved next-generation product candidates and rapidly advance these new product candidates through pre-clinical and clinical development. |
Product Pipeline
The following table summarizes the status of our product pipeline:
| Development | Commercialization | |||||||
Product Candidate | Indication | Responsibilities | Rights | Status | ||||
| MAXY-G34 | Neutropenia | Maxygen | Maxygen | Phase I; plan to commence Phase IIa clinical trial in first half of 2007. | ||||
| MAXY-alpha | Hepatitis C | Roche | Roche | Phase Ia; Roche expects to commence a Phase Ib clinical trial in 2007. | ||||
| MAXY-VII | Uncontrolled bleeding | Maxygen* | Maxygen* | Pre-clinical** | ||||
| MAXY-gamma | Idiopathic Pulmonary Fibrosis | InterMune | InterMune | Pre-clinical |
| * | Maxygen and Roche are currently parties to an agreement relating to the co-development and commercialization of our MAXY-VII product candidates for acute bleeding indications. On March 13, 2007, we received notice from Roche that Roche has elected to terminate this agreement, effective April 12, 2007. Upon termination of the agreement, all rights to our MAXY-VII product candidates will revert to Maxygen. | |
| ** | “Pre-clinical” means process development, productscale-up, formulation and further testing in animals, including toxicology, pharmacokinetics and pharmacodynamics. |
2
Table of Contents
MAXY-G34
Our MAXY-G34 product candidate was designed to be an improved next-generation granulocyte colony stimulating factor, or G-CSF, for the treatment of neutropenia. G-CSF is a natural protein that works by stimulating the body’s bone marrow to produce more white blood cells.
Neutropenia is a severe decrease in neutrophil cell counts in the blood. Neutrophils are a specific type of blood cell that plays an important role in the defense against bacterial infections. Neutropenia is a common side effect of chemotherapeutic treatments for many forms of cancer, including breast cancer, lung cancer, lymphomas and leukemias. Neutropenia is also seen in a variety of other medical conditions and is sometimes caused by therapies administered to treat a disease, such as in patients treated for HCV infection. Neutropenic patients contract bacterial infections more easily and often, some of which can be life threatening. Further, and most importantly, chemotherapy treatment for neutropenic patients may be reduced or delayed, which can result in cancer progression.
MAXY-G34 may help the body make white blood cells more quickly than the products currently approved for the treatment of neutropenia, Neupogen and Neulasta, which could make it an attractive alternative for both doctors and patients. In multiple pre-clinical animal models, we have generated data that suggest that our MAXY-G34 product candidate reduces the duration of neutropenia by clinically relevant periods (approximately 25% shorter duration of neutropenia) when compared to the currently marketed products. This may help protect patients from chemotherapy and radiation therapy-related infections, shorten the duration of hospital stays and help keep patients on schedule for their cancer treatments.
Market Opportunity. Neupogen, a first-generation G-CSF product, and Neulasta, a second-generationG-CSF product, currently dominate the market to treat chemotherapy and radiation-induced neutropenia. Worldwide sales of G-CSF products were approximately $3.9 billion in 2006.
Development Status. In the third quarter of 2006, we initiated a Phase I clinical trial in the United States for our lead MAXY-G34 product candidate in healthy male and female volunteers. The preliminary results of the Phase I clinical trial indicate that MAXY-G34 was generally safe and well tolerated throughout the study, at all doses, which ranged from 10 to 150 µg/kg of MAXY G-34. All doses tested in this Phase I trial increased the neutrophil levels as compared to the placebo controls. In subjectfollow-up 30 days after the administration of MAXY-G34, no antibodies were detected in any clinical trial participants. As expected based on the results of pre-clinical animal studies, MAXY-G34 demonstrated a prolonged half-life compared to Neulasta. The median half-life observed for MAXY-G34 in healthy volunteers in the Phase I clinical trial was approximately 2.3 times the median terminal half-life for Neulasta in healthy volunteers as previously reported. At the 60, 100 and 150 µg/kg doses of MAXY-G34, low but detectable levels of MAXY-G34 were observed at the end of the21-day collection period.
We have commenced additional Phase I studies to conduct further dosing studies and to obtain further data on certain biological markers. We expect that the data obtained from these additional studies may allow us to determine whether to evaluate MAXY-G34 for use in certain indications in addition to chemotherapy-induced neutropenia.
In the first half of 2007, we expect to commence a Phase IIa clinical trial of MAXY-G34 in cancer patients. We plan to conduct such clinical trials at multiple sites in Eastern Europe. In this Phase IIa clinical trial, cancer patients will be given a single dose of MAXY-G34 therapy per chemotherapy cycle, with each patient receiving multiple chemotherapy cycles.
We currently retain all rights to our MAXY-G34 product candidates.
MAXY-alpha
Our MAXY-alpha product candidates are designed to be superior next-generation interferon alpha products for the treatment of HCV and Hepatitis B virus, or HBV, infections. Alpha interferon is a natural protein that is produced by many cell types, including T-cells and B-cells, macrophages, fibroblasts, endothelial cells, osteoblasts and others, and is an important component of the anti-viral response, stimulating both macrophages and natural killer (NK) cells.
HCV infection causes chronic inflammation in the liver. In a majority of patients, HCV infection can persist for decades and eventually lead to cirrhosis, liver failure and liver cancer. HCV infection represents a significant
3
Table of Contents
medical problem worldwide. According to the World Health Organization, HCV is responsible for up to 76% of all liver cancer cases and two thirds of all liver transplants in the developed world. The U.S. Centers for Disease Control, or CDC, estimates that approximately 4.1 million people in the United States, or 1.6% of the U.S. population, have been infected with HCV, 3.2 million of whom are chronically infected with HCV and at risk of developing liver cirrhosis or liver cancer. The World Health Organization estimates that approximately 180 million people, or 3% of the world’s population, are infected with HCV worldwide, 130 million of whom are chronic HCV carriers. The World Health Organization also estimates that three to four million persons are newly infected each year, 70% of whom will develop chronic hepatitis.
Currently, there is no vaccine available to prevent HCV infection. The standard treatment for HCV infection is a combination of pegylated interferon and ribavirin, a small molecule. Approximately 50% of patients infected with HCV genotype I, the most common HCV genotype in the United States, fail to show a long-term sustained response to the standard treatment. As a result, we believe that new safe and effective treatment options for HCV infection are needed. We are working in collaboration with our partner, Roche, to develop a potentially superior next-generation interferon alpha product that is more effective at treating HCV infection. Using our proprietary MolecularBreeding directed molecular evolution platform and other technologies, we have created novel interferon alpha variants that we believe may have improved clinical properties as compared to current alpha interferon products. The lead novel interferon alpha molecules have been pegylated to obtain the desired biological half-life and dosing convenience of currently approved interferon alpha products.
Although various third parties are currently developing small molecules intended for the treatment of HCV infection, we believe that interferon alpha will remain an important product in the treatment of HCV in combination with small molecules. In addition, we believe that our pegylated MAXY-alpha product candidate may have the potential to treat a larger percentage of the 40% to 50% of HCV patients who are not effectively treated with existing drugs.
Market Opportunity. Based on currently available market data, worldwide sales of drugs for the treatment of HCV, including pegylated interferon alpha and ribavirin, were approximately $3.0 billion in 2006. Sales of HCV drugs are expected to grow due to improved market penetration through improvements in therapies, increased awareness and improved diagnosis rates.
Development Status. In 2003, we entered into a broad strategic alliance with Roche, a market leader in interferon alpha therapies, to develop novel improved interferon alpha and beta products for HCV and HBV infections and a wide range of other indications. See “Our Strategic Collaborations — Roche — MAXY-alpha.” Roche initiated a Phase Ia clinical trial in New Zealand for our lead MAXY-alpha product candidate at the end of 2006 and has informed us that they plan to commence a Phase Ib clinical trial for MAXY-alpha in 2007.
Outside the areas of the treatment of HCV and HBV infections, we retain the right to develop novel interferon alpha product candidates specifically tailored for other indications, including infectious diseases, oncology, inflammatory diseases and autoimmune diseases.
MAXY-VII
Our MAXY-VII product candidates are designed to be superior next-generation factor VII products to treat uncontrolled bleeding in emergency situations, such as trauma, intracerebral hemorrhage, or ICH, certain surgeries and other acute indications. Factor VII is a natural protein with a pivotal role in blood coagulation and clotting. Blood clotting factors, such as factors VII, VIII and IX, have been used for many years to control bleeding episodes.
The CDC estimates that 160,000 deaths occur in the United States each year as a direct result of trauma (injury), and cite unintentional trauma as the leading cause of death for people under age 45. Uncontrolled bleeding is believed to account for approximately half of all trauma-related deaths that occur within the first 48 hours after injury. There is currently no effective medical therapy for uncontrolled bleeding other than physical interventions, such as surgery and blood transfusions.
NovoSeven, a recombinant human factor VII product of Novo Nordisk A/S approved in the United States and Europe for the treatment of hemophilia, is the only factor VII product currently approved for any indication. Our MAXY-VII product candidates have been designed to deliver improved efficacy, a longer circulating half-life and
4
Table of Contents
an overall improved therapeutic index compared to NovoSeven. Studies in pre-clinical animal models have demonstrated that MAXY-VII may be more effective than NovoSeven at treating uncontrolled bleeding.
Novo Nordisk A/S has been conducting clinical trials of NovoSeven for acute indications, including ICH and trauma. In February 2007, Novo Nordisk A/S announced that NovoSeven missed the primary endpoint of improvement in mortality and severe disability at day 90 in a Phase III clinical trial to treat ICH and that it will not seek regulatory approval of NovoSeven for the treatment of this indication. Novo Nordisk A/S is currently conducting clinical trials of NovoSeven for trauma and other acute bleeding indications.
Market Opportunity. NovoSeven is currently approved in the United States and Europe only for the treatment of hemophilia patients who have become resistant to factor VIII therapy due to the development of antibodies. Sales of NovoSeven in 2006 were approximately $980 million. The use of recombinant factor VII-based products for the treatment of new indications, such as severe bleeding in trauma, surgery and ICH, are forecasted to represent significant market opportunities for next-generation recombinant factor VII products. Analysts have estimated that the immediate market for an approved factor-VII based therapy for ICH alone could be as large as $500 million annually.
Development Status. In December 2005, we entered into a co-development and commercialization agreement with Roche relating to the development and commercialization of our portfolio of next-generation recombinant factor VII products for multiple indications. See “Our Strategic Collaborations — Roche — MAXY-VII.” In 2006, we achieved a manufacturing process development milestone in our MAXY-VII program and received a $5 million milestone payment from Roche. On March 13, 2007, we received notice from Roche that Roche has elected to terminate this agreement, effective April 12, 2007, due to the inability of the parties to establish an animal model intended to provide pre-clinical de-risking of the program.
In light of Roche’s notice to us regarding the termination of the co-development and commercialization agreement for our MAXY-VII product candidates, and the recent announcement by Novo Nordisk A/S regarding the results of its clinical trials of NovoSeven for ICH, we are currently evaluating our plans for the continued development of our MAXY-VII product candidates for acute bleeding indications and hemophilia.
We have retained all rights for the development and commercialization of factor VII products for hemophilia and, as of April 12, 2007, the effective date of the termination of our co-development and commercialization agreement with Roche, all other rights to our MAXY-VII variants will revert to us.
MAXY-gamma
Our MAXY-gamma product candidates are next-generation versions of interferon gamma that may be useful for the treatment of a variety of diseases, including idiopathic pulmonary fibrosis, or IPF, HCV, tuberculosis and meningitis. We believe that interferon gamma may help in the prevention of excessive scarring, or fibrosis, of organs such as the liver and the lung. Interferon gamma is a naturally occurring protein in the human body that plays a role in the activation of the immune system to fight infectious pathogens. Interferon gamma has a wide spectrum of biologic effects, including anti-infective, anti-viral, anti-fibrotic, anti-proliferative, immunomodulatory and chemosensitization activities.
IPF is an inflammatory disease that results in severe fibrosis of the lungs. In time, this fibrosis can build up to the point where the lungs are unable to provide oxygen to the tissues of the body. The average survival rate of a patient with IPF is four to six years after diagnosis.
Our MAXY-gamma product candidates are designed to have improved efficacy and a less-frequent dosing regimen over Actimmune, the currently marketed interferon gamma product of InterMune, Inc., or InterMune. Actimmune is marketed for the treatment of severe, malignant osteopetrosis and chronic granulomatous disease (CGD) and, until recently, was being developed by InterMune for the treatment of IPF.
Market Opportunity. We believe that the development of a next-generation version of interferon gamma to treat IPF may represent a significant market opportunity.
Development Status. We have established a collaboration with InterMune in which InterMune has an exclusive license to develop and market our MAXY-gamma product candidates for all human therapeutic
5
Table of Contents
indications. See “Our Strategic Collaborations — InterMune — MAXY-gamma.” InterMune has not informed us whether it intends to continue to advance these product candidates into clinical development during 2007. In March 2007, InterMune announced that it was discontinuing development of Actimmune for IPF. If InterMune elects to cease development of our MAXY-gamma product candidates, all rights to our MAXY-gamma product candidates would revert back to us.
Other Assets and Areas of Research
In addition to our development stage product candidates, we have other earlier stage programs in pre-clinical research, and assets outside of our core business, including vaccine research and investments in other biotechnology companies.
Vaccines
We believe that our proprietary technologies have the potential to transform the design and development of vaccines through the optimization of properties that allow for the generation of broad and strong immune responses. We currently have an active program to advance the research for development of a preventative HIV vaccine. Our vaccine research program is being funded by research grants from the National Institutes of Health, or NIH, and the U.S. Department of Defense. In 2005, the NIH awarded us two competitive grants, including $11.7 million over approximately five years as part of the HIV Research and Development, or HIVRAD, program, and a Phase I grant from the NIH Small Business Innovation Research, or SBIR, program. We were also awarded a one-year contract of $2.4 million in 2005 from the Department of Defense for HIV vaccine discovery. In 2006, as part of the SBIR program, the NIH awarded us two additional grants totaling $500,000 as part of the HIV research program and one grant totaling $1.0 million over two years for work on vaccines for equine encephalitis.
The HIVRAD grant provides funds for the use of our MolecularBreeding directed evolution platform to generate novel HIV-1 antigens potentially capable of inducing broad antibody responses to multiple strains of the HIV-1 virus. Three of the SBIR awards fund investigations into the effect on immunogenicity of secondary modifications to a specific HIV-1 envelope protein and one of the SBIR awards granted in 2006, totaling $1.0 million over two years, will fund research on the development of vaccines for equine encephalitis. The grant from the Department of Defense will fund work to develop a high-throughput HIV vaccine screening platform. We are currently working in collaboration with Monogram Biosciences, Inc., Aldevron LLC and the Scripps Research Institute with respect to these government-funded projects.
Investment in Codexis, Inc.
We have a minority investment in Codexis, Inc., or Codexis, a biotechnology company focused on developing innovative biotechnology-based solutions for improving the manufacture of existing small molecule pharmaceutical products. We formed Codexis in January 2002 as a wholly-owned subsidiary to operate our former chemicals business. Our voting rights in Codexis were reduced below 50% in the first quarter of 2005 and, as a result, Codexis is no longer consolidated in our financial statements. In 2006, we expanded the scope of exclusive licenses previously granted to Codexis, Inc. to our MolecularBreeding directed evolution platform for certain applications relating to energy, including biofuels, in exchange for the right to receive a percentage of revenues received by Codexis for the sale of energy products and the use of processes in the energy field. As of December 31, 2006, our ownership in Codexis was approximately 32%, based upon the voting rights of the issued and outstanding shares of the common and preferred stock of Codexis. We are not obligated to fund the operations or other capital requirements of Codexis. For more information regarding our investment in Codexis, see Note 1 of the Notes to Consolidated Financial Statements.
Investment in Avidia Inc.
In July 2003, we established Avidia, Inc. (formerly Avidia Research Institute), or Avidia, a biotechnology company focused on developing products based on a new class of subunit proteins, together with two third-party investors. We received equity interests in Avidia through our initial contribution of technology and funding and our participation in subsequent preferred stock financings by Avidia. On October 24, 2006, Amgen Inc. completed the
6
Table of Contents
acquisition of Avidia. As a result of the acquisition, we received approximately $17.8 million in cash in exchange for our equity interests in Avidia and may receive up to an additional $2.8 million in cash, contingent upon the development of certain Avidia products by Amgen Inc. For more information regarding the sale of our investment in Avidia, see Note 13 of the Notes to Consolidated Financial Statements.
Under a cross license agreement that we entered into with Avidia at the time of Avidia’s formation, we have exclusive and non-exclusive license rights to use certain Avidia technology to develop and commercialize therapeutic products directed to certain specific targets, including CD40,CD-40 ligand,CTLA-4, CD28, B.7.1 (CD80), B.7.2 (CD86), p40 (subunit ofIL-12 andIL-23), TPO, any interferon and/or interferon receptor,TPO/IL3 andTNF/IL-1. The cross license agreement does not provide for the payment by us or to us of any amounts for license fees, milestone payments or royalties. The cross license agreement has atwenty-five (25) year term, but may terminate earlier based on the expiration of the patent rights licensed under the agreement. Following the expiration of the cross license agreement, the licenses granted under the agreement become perpetual on acountry-by-country basis.
Under the cross license agreement, Avidia also granted us certain limited options to acquire additional licenses to develop and commercialize other therapeutic products researched by Avidia. The exercise of an option would require us to enter into a separate product license agreement for any such product with Amgen Mountain View Inc. (as successor to Avidia) on pre-agreed terms that would include the payment by us to Amgen Mountain View Inc. of royalties based on net sales of the products subject to the product license agreement and milestone payments based upon our achievement of certain regulatory and clinical milestones for such product, up to an aggregate of $19.8 million.
Our Technologies
MolecularBreeding
Our MolecularBreeding directed evolution technologies mimic the natural events of evolution. First, genes are subjected to DNAShuffling recombination technologies, generating a diverse library of gene variants. Second, our proprietary MaxyScan screening systems select individual proteins from the gene variants in the library. The proteins that show improvements in the desired characteristics become the initial lead candidates. After confirmation of activity, the initial lead candidates are then used as the genetic starting material for additional rounds of shuffling. Once the level of improvement needed for the particular protein pharmaceutical is achieved, the group of lead candidates is considered for advancement as a product candidate for development.
7
Table of Contents
The MolecularBreeding Directed Evolution Process
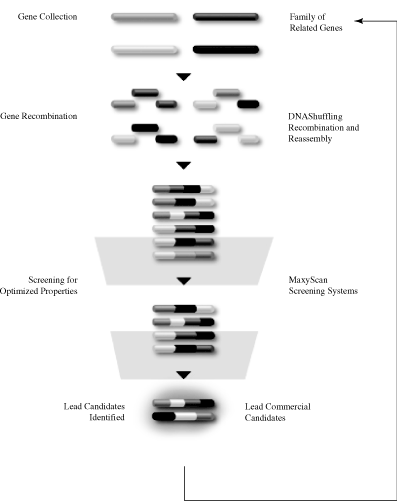
Step One: DNAShuffling Recombination Technologies
Our DNAShuffling recombination technologies work as follows: a single gene or multiple genes are cleaved into fragments and recombined, creating a population of new gene variants. The new genes created by DNAShuffling are then selected for one or more desired characteristics. This selection process yields a population of genes that becomes the starting point for the next cycle of recombination. As with classical breeding of plants and animals, this process is repeated until genes expressing the desired properties are identified.
DNAShuffling can be used to evolve properties that are coded for by single genes, multiple genes or entire genomes. By repeating the process, DNAShuffling ultimately generate libraries with a high percentage of genes that have the desired function. Due to the high quality of these libraries, a relatively small number of screening tests need to be performed to identify gene variants with the desired commercial qualities. This process can reduce the cost and time associated with identifying multiple potential products.
Step Two: MaxyScan Screening
The ability to screen or select for a desired improvement in function is essential to the effective development of an improved gene or protein. As a result, we have invested significant resources in developing automated, stringent, rapid screens and selection formats.
8
Table of Contents
We have developed screening tests that can measure the production of proteins or small molecules in culture without significant purification steps or specific test reagents, thereby eliminating time-consuming steps required for traditional screening tests. We are also focusing on the development of reliable, cell-based screening tests that are predictive of specific functions relevant to our human therapeutics programs. Accordingly, we continue to develop new screening approaches and technologies. Our approach is to create multitiered screening systems where we use a less sensitive screening test as a first screen to quickly select proteins with the desired characteristics, followed by a more sensitive screening test to confirm value in these variants and to select for final lead product candidates. Unlike approaches that create random diversity, our MolecularBreeding directed evolution platform produces potentially valuable libraries of gene variants with a predominance of active genes with the desired function. As a result of capturing the natural process of sexual recombination with our proprietary DNAShuffling methods, we are also able to generate gene variants with the desired characteristic at a frequency 5 to 10-fold higher than combinatorial chemistry, rational design or other directed evolution methods. This allows us to use complex biological screens and formats as a final screening test, as relatively few proteins need to be screened to detect an improvement in the starting gene activity. Furthermore, this allows us to focus on developing screens that generate a broader range of information that is more responsive to commercial and clinical concerns. This separates us from many of our potential competitors who invest significant time and money to screen billions of compounds per day. While we have the capability to screen billions of compounds per day, we generally need to screen far fewer, on the order of 10,000 candidates per day or less. Some of our screening capabilities include mass spectrometry, in vivo animal assays, bioassays, immunochemical assays, chemical assays, and biochemical assays.
We have access to multiple sources of genetic starting material. In addition to the wealth of publicly available genetic sequence information, we are typically able to access our collaborators’ proprietary genes for use outside their specific fields of interest. Furthermore, we are able to inexpensively obtain our own genetic starting material or information, either through our own in-house efforts or through collaborations with third parties. This information and such materials when coupled with our DNAShuffling recombination technologies, can provide a virtually infinite amount of new, proprietary gene variants some of which may have potential commercial value.
Other Technologies
In addition to our proprietary MolecularBreeding directed evolution platform, we have acquired capabilities with regard to several complementary technologies potentially useful for the development of protein-based pharmaceuticals. Two examples of the tools that we use to post-translationally modify protein drugs are pegylation and glycosylation technologies. Over the last few years, glycosylation and pegylation have been validated technically and commercially through the successes of drugs, such as the pegylated interferons (Pegasys and PEG-Intron) and Aranesp, a hyper-glycosylated erythropoeitin. These post-translational modifications of proteins have been demonstrated to change the pharmacokinetics and pharmacodynamics of certain protein drugs. In addition these modifications can change the solubility, bioavailability and immunogencity profile of protein drugs.
Our Strategic Collaborations
Roche
MAXY-alpha. In May 2003, we established a broad alliance with Roche to develop our MAXY-alpha product candidates for a wide range of indications. Roche exclusively licensed from us worldwide commercialization rights to specific novel interferon product candidates for the treatment of HCV and HBV infections. We received an initial payment and full research and development funding for the first two years of the collaboration. In addition, we are eligible to receive milestone payments based on the achievement of certain development milestones, royalties based on product sales, and option fees. This agreement also provides us and Roche with the option to expand the collaboration to develop other novel interferon alpha and beta products specifically tailored for indications outside of HCV and HBV infections, including oncology, autoimmune diseases, inflammatory diseases, and other infectious diseases, such as HIV. We retain the right to develop such products, while Roche may elect to acquire worldwide license and commercialization rights to these product candidates on pre-agreed terms. We have the option to co-fund development in the United States of any product to which Roche acquires a license in exchange for profit sharing or an increased royalty rate. The funded research term of this collaboration ended in December 2005. In November 2006, Roche initiated a Phase Ia clinical trial in New Zealand to evaluate our MAXY-alpha product
9
Table of Contents
candidates and we received a $2 million milestone payment from Roche. Roche has informed us that they plan to commence a Phase Ib clinical trial for MAXY-alpha in 2007.
MAXY-VII. In December 2005, we established an alliance with Roche to co-develop our MAXY-VII product candidates for the treatment of uncontrolled bleeding in multiple indications, including trauma and intracerebral hemorrhage. We received an up-front fee of $8 million from Roche and, in 2006, we received a $5 million milestone payment from Roche for our achievement of a manufacturing process development milestone. On March 13, 2007, we received notice from Roche that Roche will terminate this agreement, effective April 12, 2007, due to the inability of the parties to establish an animal model intended to provide pre-clinical de-risking of the program. In light of Roche’s notice to us regarding the termination of the co-development and commercialization agreement for our MAXY-VII product candidates, and the recent announcement by Novo Nordisk A/S regarding the results of its clinical trials of NovoSeven for ICH, we are currently evaluating our plans for the continued development of our MAXY-VII product candidates for acute bleeding indications and hemophilia. We retained all rights for development and commercialization for next-generation novel recombinant factor VII products for the treatment of hemophilia and, as of April 12, 2007, the effective date of the termination of our co-development and commercialization agreement with Roche, all other rights to our MAXY-VII variants will revert to us.
InterMune
MAXY-gamma. In October 2001, we entered into a license and collaboration agreement with InterMune to develop and commercialize our novel, next-generation interferon gamma products. Under the terms of this agreement, InterMune has exclusive rights to develop the next-generation interferon gamma products created by us, and retains exclusive worldwide commercialization rights for all human therapeutic indications. We received up-front license fees and full research funding during the research phase of the collaboration, which was completed in June 2004. We remain eligible to receive development and commercialization milestone payments that could exceed $60 million, in addition to royalties on product sales. InterMune has not informed us whether it intends to continue to advance these product candidates into clinical development during 2007. In March 2007, InterMune announced that it was discontinuing development of Actimmune for IPF. If InterMune elects to cease development of our MAXY-gamma product candidates, all rights to our MAXY-gamma product candidates would revert back to us.
Intellectual Property and Technology Licenses
Pursuant to a technology transfer agreement we entered into with Affymax Technologies N.V. and Glaxo Group Limited (each of which was then a wholly-owned subsidiary of what is now GlaxoSmithKline plc), we were assigned all the patents, applications and know-how related to our MolecularBreeding directed evolution platform, subject to certain internal research rights retained by GlaxoSmithKline plc. Affymetrix, Inc. retains an exclusive, royalty-free license under some of the patents and patent applications previously owned by Affymax for use in the diagnostics and research supply markets for specific applications. In addition, Affymax assigned jointly to us and to Affymetrix a family of patent applications relating to circular PCR techniques.
We have an extensive patent portfolio including over 80 issued U.S. patents and over 50 foreign patents relating to our proprietary MolecularBreeding directed evolution platform. Counterpart applications of the U.S. patents are pending in other major industrialized countries. Additionally, we have over 20 pending U.S. patent applications and over 100 pending foreign and international counterpart applications relating to our MolecularBreeding directed evolution platform and specialized screening technologies, and the application of these technologies to the development of protein pharmaceuticals and other industries, including agriculture, vaccines, gene therapy and chemicals.
Our expanding patent estate provides us with an increasingly broad and unique platform from which to create and potentially improve protein-based therapeutic products. Patents owned by us or for which we have exclusive licenses cover a broad range of activities surrounding recombination-based directed molecular evolution including:
| • | methods for template-based gene recombination to produce chimeric genes, including use of single or double-stranded templates; |
10
Table of Contents
| • | methods for recombining nucleic acid segments produced by incomplete nucleic acid chain extension reactions to produce chimeric genes, including the staggered extension process (StEP); | |
| • | methods utilizing reiterative screening or only a single cycle of screening; | |
| • | methods of combining any mutagenesis technique with DNA recombination methods to produce new chimeric genes; | |
| • | methods using synthesized nucleic acid fragments; | |
| • | in vivo and in vitro recombination methods of the above, in a variety of formats; | |
| • | methods of screening directed evolution libraries; | |
| • | methods for ligation- and single-stranded template-based recombination and reassembly; | |
| • | mutagenesis, including codon and gene site saturation mutagenesis, used in conjunction with recombination and reassembly; | |
| • | cell-based recombination methods; and | |
| • | fluorescence-, bioluminescence-, and nutrient-based screening methods, including the use of ultra-high throughput FACS-based methods for screening diverse variants. |
Such patents reinforce our preeminent position as an industry leader in recombination-based directed molecular evolution technologies for the preparation of chimeric genes for commercial applications.
In addition to the patents that we own directly, we have also exclusively licensed patent rights and technology for specific uses from Novozymes A/S, the California Institute of Technology, the University of Washington, GGMJ Technologies, L.L.C. and the University of Minnesota. These licenses give us rights to an additional 21 issued U.S. patents, 13 granted foreign patents and over 30 pending U.S. and foreign counterpart applications.
We have also received from Affymax (when it was a subsidiary of what is now GlaxoSmithKline plc) a worldwide, non-exclusive license to certain Affymax patent applications and patents related to technology for displaying multiple diverse proteins on the surface of bacterial viruses.
As part of our confidentiality and trade secret protection procedures, we enter into confidentiality agreements with our employees, consultants and potential collaborative partners. Despite these precautions, third parties or former employees could obtain and use information regarding our technologies without authorization, or develop similar technology independently. It is difficult for us to monitor unauthorized use of our proprietary methods and information. Effective protection of intellectual property rights is also unavailable or limited in some foreign countries. The efforts that we take to protect our proprietary information and rights may be inadequate to protect such information and rights. Our competitors could independently develop similar technology or design around any patents or other intellectual property rights we hold.
In July 2005, our European Patent 0752008, covering our first generation directed molecular evolution technologies, was the subject of an opposition proceeding before the European Patent Office. All claims of the patent were upheld as valid with minor amendments. In October 2005, the Australian Patent Office found, in an opposition proceeding regarding our Australian patent application No. 703264 that corresponds to European Patent 0752008, 89 of our claims to be patentable as presented. In February 2006, our European Patent 0876509, covering one embodiment of our second-generation directed molecular evolution technologies, was the subject of an opposition proceeding before the European Patent Office. The opposition board revoked the patent on the grounds of lack of inventive step. We have appealed this decision to the appeals board of the European Patent Office. The decision of the appeals board will likely be issued in 2008.
11
Table of Contents
Product Patents
Our patent portfolio consists of the following issued patents and pending patent applications for each of our primary product candidates:
| • | For our MAXY-G34 product candidates, we have three U.S. patents, 16 pending U.S. applications, six foreign patents and 30 pending foreign or international applications. | |
| • | For our MAXY-alpha product candidates, we have 20 pending U.S. applications and 47 pending foreign or international applications. Certain of these applications are jointly held by us and our collaborative partner, Roche. | |
| • | For our MAXY-VII product candidates, we have one U.S. patent, 21 pending U.S. applications, three foreign patents and 40 pending foreign or international applications. We also have exclusive licenses to two U.S. patents, ten pending U.S. applications and four pending foreign or international applications. | |
| • | For our MAXY-gamma product candidates, we have two U.S. patents, six pending U.S. applications, six foreign patents and 37 pending foreign or international applications. |
Manufacturing
We rely on third party manufacturers and collaborators to produce our compounds for clinical purposes and may do so for commercial production of any drug candidates that are approved for marketing. We have established agreements with Rentschler Biotechnologie GmbH, or Rentschler, in Germany for the manufacture of supplies of our MAXY-G34 product candidates for Phase I and II clinical trials and for the manufacture of supplies of our MAXY-VII product candidates for pre-clinical studies and Phase I and II clinical trials.
We believe that our current contract manufacturing agreement with Rentschler will be sufficient to accommodate clinical trials of our MAXY-G34 product candidates through Phase I and II clinical trials. However, we will need to secure additional manufacturing arrangements with contract manufacturers or develop our own manufacturing capability to meet our future needs, which would require significant capital investment.
Competition
Any products that we develop will compete in highly competitive markets. We face competition from large pharmaceutical and biopharmaceutical companies, such as Eli Lilly and Company, Pfizer, Inc., Genentech, Inc., Novo Nordisk A/S, Schering-Plough Corporation and Amgen Inc., and from smaller biotechnology companies, such as Human Genome Sciences, Inc., Neose Technologies, Inc., Zymogenetics, Inc. and InterMune, Inc. In the vaccines field, we face competition from biotechnology companies, such as Corixa Corporation (now a subsidiary of GlaxoSmithKline plc) and Vical Corporation, as well as large pharmaceutical companies, including GlaxoSmithKline plc, Sanofi-Aventis, Novartis AG, Pfizer Inc. and Merck & Co., Inc.
Many of our potential competitors, either alone or together with their collaborative partners, have substantially greater financial, technical and personnel resources than we do, and there can be no assurance that they will not succeed in developing technologies and products that would render our technologies and products or those of our collaborators obsolete or noncompetitive. In addition, many of our competitors have significantly greater experience than we do in:
| • | developing products; | |
| • | undertaking pre-clinical testing and clinical trials; | |
| • | obtaining FDA and other regulatory approvals of products; and | |
| • | manufacturing and marketing products. |
We are a leader in the field of directed molecular evolution. We are aware that other companies, including Diversa Corporation, Xencor, Inc. and Nautilus Biotech, have alternative methods for obtaining and generating genetic diversity or use mutagenesis techniques to produce genetic diversity. Academic institutions such as the California Institute of Technology, Pennsylvania State University and the University of Washington are also
12
Table of Contents
working in this field. We have licensed certain patents from certain of these institutions. This field is highly competitive and companies and academic and research institutions are actively seeking to develop technologies that could be competitive with our technologies.
We are aware that other companies, organizations and persons have described technologies that appear to have some similarities to our patented proprietary technologies. We monitor publications and patents that relate to directed molecular evolution to be aware of developments in the field and evaluate appropriate courses of action in relation to these developments.
Research and Development Expenses
The majority of our operating expenses to date have been related to research and development. Our research and development expenses from continuing operations were $49.1 million in 2006, $41.9 million in 2005 and $53.6 million in 2004 (including research and development expenses attributable to Codexis in 2004 and 2005). Additional information required by this item is incorporated herein by reference to “Research and Development Expenses” in Note 1 of the Notes to Consolidated Financial Statements. We intend to maintain our strong commitment to research and development as an essential component of our product development effort. We may also license technology or products developed by third parties to obtain access to additional potential products.
Geographic Distribution
We have operations in two geographic locations, the United States and Denmark. In addition, certain of our collaborators are based outside the United States. Additional information required by this item is incorporated herein by reference to “Segment Information — Geographic Information” in Note 12 of the Notes to Consolidated Financial Statements.
Segment Reporting
We operate our business in one segment, human therapeutics. Prior to February 28, 2005, the date on which Codexis ceased to be our consolidated subsidiary, we operated our business in two segments, human therapeutics and chemicals. Additional information required by this item is incorporated herein by reference to “Segment Information” in Note 12 of the Notes to Consolidated Financial Statements.
Government Regulation
We are subject to regulation by the FDA and comparable regulatory agencies in foreign countries with respect to the development and commercialization of products resulting from our drug discovery activities. The FDA and comparable regulatory bodies in other countries currently regulate therapeutic proteins and related pharmaceutical products as biologics. Biologics are subject to extensive pre- and post-market regulation by the FDA, including regulations that govern the collection, testing, manufacture, safety, efficacy, potency, labeling, storage, record keeping, advertising, promotion, sale and distribution of the products.
The time required for completing testing and obtaining approvals of our product candidates is uncertain but will take several years and approvals will only be obtained if our product candidates are shown to be safe and efficacious in clinical trials. Any delay in testing may hinder product development. In addition, we may encounter delays in product development or rejections of product applications due to changes in FDA or foreign regulatory policies during the period of product development and testing. Failure to comply with regulatory requirements may subject us to, among other things, civil penalties and criminal prosecution; restrictions on product development and production; suspension, delay or withdrawal of approvals; and the seizure or recall of products. The lengthy process of obtaining regulatory approvals and ensuring compliance with appropriate statutes and regulations requires the expenditure of substantial resources. Any delay or failure, by us or our corporate partners, to obtain regulatory approvals could adversely affect our ability to commercialize product candidates, receive milestone and royalty payments and generate sales revenue.
Under FDA regulations, the clinical testing program required for marketing approval of a new drug typically involves three sequential phases, which may overlap.
13
Table of Contents
| • | Phase I: Studies are conducted on normal, healthy human volunteers to determine safety, dosage tolerance, absorption, metabolism, distribution and excretion. If possible, Phase I studies may also be designed to gain early evidence of effectiveness. | |
| • | Phase II: Studies are conducted on small groups of patients afflicted with a specific disease to determine dosage tolerance and optimal dosage, to gain preliminary evidence of efficacy, and to determine the common short-term side effects and risks associated with the substance being tested. | |
| • | Phase III: Involves large-scale studies conducted on disease-afflicted patients to provide statistical evidence of efficacy and safety and to provide an adequate basis for physician labeling. |
In 2006, Phase I clinical trials were initiated for our lead MAXY-G34 and MAXY-alpha product candidates. To date, neither we nor our corporate collaborators have successfully completed clinical trials for any of our product candidates. If we or our corporate collaborators are unable to continue or successfully complete clinical trials of MAXY-G34, MAXY-alpha or any of our other product candidates, or decide not to continue clinical trials for a particular indication, we will not be able to seek or obtain regulatory approval for commercialization of the applicable product candidate for the relevant indication.
Phase I, Phase II or Phase III clinical testing may not be completed successfully within any specific period of time, if at all, with respect to any of our potential products. Furthermore, we, an institutional review board, the FDA or other regulatory bodies may suspend a clinical trial at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
FDA marketing approval is only applicable in the United States. Marketing approval in foreign countries is subject to the regulations of those countries. The approval procedures vary among countries and can involve additional testing. The requirements for approval and the time required to obtain approval may differ from that required for FDA approval.
Although there are some centralized procedures for filings in the European Union countries, in general each country has its own procedures and requirements, and compliance with these procedures and requirements may be expensive and time-consuming. Accordingly, there may be substantial delays in obtaining required approvals from foreign regulatory authorities after the relevant applications are filed, if approvals are ultimately received at all.
Environmental Regulation
We seek to comply with all applicable statutory and administrative requirements concerning environmental quality. We have made, and will continue to make, expenditures for environmental compliance and protection. Expenditures for compliance with environmental laws have not had, and are not expected to have, a material effect on our capital expenditures, results of operation or competitive position.
Employees
As of January 31, 2007, we had 151 employees, 50 of whom hold Ph.D., Ph.D. equivalent or M.D. degrees and 101 of whom were engaged in full-time research and development activities. We plan to expand our corporate development programs and hire additional staff if additional corporate collaborations are established or if existing corporate collaborations are expanded. We continue to search for qualified individuals with interdisciplinary training and flexibility to address the various aspects and applications of our technologies. None of our employees is represented by a labor union, and we consider our employee relations to be good.
Corporate Background and History
We began operations in 1997 to commercialize technologies originally conceived at Affymax Research Institute, then a subsidiary of what is now GlaxoSmithKline plc. We were incorporated in Delaware on May 7, 1996 and began operations in March 1997. Our principal executive offices are located at 515 Galveston Drive, Redwood City, CA 94063. Our telephone number is(650) 298-5300.
Our operations were originally focused on the application of our MolecularBreeding directed evolution platform and other technologies to the development of multiple products in a broad range of industries, including
14
Table of Contents
human therapeutics, chemicals and agriculture. In August 2000, to complement and expand our human therapeutics operations, we established our Danish subsidiary, Maxygen ApS, through the acquisition of ProFound Pharma A/S, a privately held Danish biotechnology company. In 2002, we formed two wholly owned subsidiaries, Codexis, Inc. and Verdia, Inc., to operate our chemicals and agriculture businesses.
Over the past several years, we have shifted our primary focus to the development of next-generation protein pharmaceuticals. Accordingly, in 2004, we sold Verdia to Pioneer Hi-Bred International, Inc., a wholly-owned subsidiary of E.I. du Pont de Nemours and Company, for $64 million in cash. Codexis received financing from third party investors and operated as independent subsidiary beginning in September 2002 and, in February 2005, our voting rights in Codexis were reduced below 50%. As a result, we no longer consolidate Codexis in our financial statements and instead reflect our investment in Codexis under the equity method of accounting.
Available Information
Our web site is located at www.maxygen.com. We make available free of charge, on or through our web site, our annual, quarterly and current reports, and any amendments to those reports, as soon as reasonably practicable after electronically filing or furnishing such reports with the Securities and Exchange Commission, or SEC. Information contained on our web site is not part of this report.
PART IV
| Item 15 | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
15(a)(3) Exhibits.
| Exhibit | ||||
Number | Description of Exhibit | |||
| 2 | .1 | Exchange Agreement, dated as of April 12, 2000, by and among Maxygen, Inc., Maxygen Holdings Ltd., ProFound Pharma A/S and the shareholders of ProFound Pharma A/S(1) | ||
| 2 | .2 | Amendment No. 1 to the Exchange Agreement, dated as of July 31, 2000, by and among Maxygen, Inc., Maxygen Holdings Ltd., ProFound Pharma A/S and the shareholders of ProFound Pharma A/S(1) | ||
| 2 | .3 | Agreement and Plan of Merger, dated June 2, 2004, by and among Pioneer Hi-Bred International, Inc., Tango Merger Sub, Inc., Maxygen, Inc. and Verdia, Inc.(2) | ||
| 3 | .1 | Amended and Restated Certificate of Incorporation(3) | ||
| 3 | .2 | Amended and Restated Bylaws(3) | ||
| 4 | .1 | Specimen Common Stock Certificate (Exhibit 4.1 to Amendment No. 1)(4) | ||
| *10 | .1 | Form of Executive Officer and Director Indemnification Agreement (Exhibit 10.7)(4) | ||
| *10 | .2 | Form of Executive Officer Change of Control Plan (Exhibit 10.1)(5) | ||
| *10 | .3 | Form of Amendment No. 1 to Executive Officer Change of Control Plan (Exhibit 10.3)(6) | ||
| *10 | .4 | Form of Amendment No. 2 to Executive Officer Change of Control Plan (Exhibit 10.3)(7) | ||
| *10 | .5 | 1997 Stock Option Plan, as amended, with applicable option agreement(8) | ||
| *10 | .6 | Form of Amendment to Stock Option Agreements (Exhibit 10.2)(7) | ||
| *10 | .7 | 1999 Nonemployee Directors Stock Option Plan, as amended, with applicable option agreement (Exhibit 10.3)(5) | ||
| *10 | .8 | 1999 Employee Stock Purchase Plan, as amended (Exhibit 10.11)(9) | ||
| *10 | .9 | 2000 International Stock Option Plan, as amended, with applicable option agreement (10) | ||
| *10 | .10 | 2000 Non-Officer Stock Option Plan, as amended, with applicable option agreement (11) | ||
| *10 | .11 | 2006 Equity Incentive Plan (including related form of stock option agreement) (Exhibit 10.4)(7) | ||
| *10 | .12 | Form of Restricted Stock Unit Award Agreement under 2006 Equity Incentive Plan (12) | ||
| 10 | .13 | Lease, dated as of October 21, 1998, between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.4)(4) | ||
15
Table of Contents
| Exhibit | ||||
Number | Description of Exhibit | |||
| 10 | .14 | First Amendment to Lease, dated as of February 26, 1999, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.5)(4) | ||
| 10 | .15 | Second Amendment to Lease, dated as of October 24, 2000, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.6)(9) | ||
| 10 | .16 | Third Amendment to Lease, dated October 22, 2003, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.15) (13) | ||
| 10 | .17 | Fourth Amendment to Lease dated December 15, 2004 by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.13) (14) | ||
| 10 | .18 | Fifth Amendment to Lease dated as of August 24, 2006, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.2) (15) | ||
| 10 | .19 | Lease, dated December 15, 2004, between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.14) (14) | ||
| 10 | .20 | First Amendment to Lease, dated as of August 24, 2006, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.1) (15) | ||
| 10 | .21 | Lease Agreement, dated May 5, 2000, between ProFound Pharma A/S and The Science Park in Horsholm (16) | ||
| 10 | .22+ | Technology Transfer Agreement, dated March 14, 1997 (effective March 1, 1998), among Maxygen, Inc., Affymax Technologies N.V. and Glaxo Group Limited, as amended (Exhibit 10.3 to Amendment No. 2)(4) | ||
| *10 | .23 | Description of 2006 Executive Officer Cash Bonus Plan (Exhibit 10.1)(7) | ||
| *10 | .24 | Description of Goldstein Reimbursement Arrangement (17) | ||
| *10 | .25# | Letter Agreement (re tax equalization payments), dated November 20, 2006, between Elliot Goldstein and Maxygen, Inc. | ||
| 10 | .26+ | Co-Development and Commercialization Agreement, dated as of December 9, 2005, amongHoffman-La Roche, Inc., F. Hoffman-La Roche Ltd. and Maxygen Holdings Ltd. (Exhibit 10.23) (18) | ||
| 10 | .27#+ | Cross License Agreement, dated as of July 16, 2003, between Maxygen, Inc. and Amgen Mountain View Inc. (as successor to Avidia, Inc.) | ||
| 10 | .28+ | Amended and Restated Exclusive License Agreement, dated July 10, 2006 (effective as of April 1, 2006), between Regents of the University of Minnesota and Maxygen, Inc. (19) | ||
| *10 | .29 | Consulting Agreement, dated as of January 12, 2006 (effective January 1, 2006), between Balkrishan Gill and Maxygen, Inc. (Exhibit 10.24) (18) | ||
| *10 | .30 | Consulting Agreement, between the Company and Waverley Associates, Inc., dated as of April 1, 2006 (20) | ||
| 21 | .1# | List of Subsidiaries | ||
| 23 | .1# | Consent of Independent Registered Public Accounting Firm | ||
| 24 | .1# | Power of Attorney | ||
| 31 | .1 | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | ||
| 31 | .2 | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | ||
| 32 | .1# | Certification of Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | ||
| * | Management contract or compensatory plan or arrangement. | |
| # | Previously filed with Maxygen’s Annual Report on Form10-K for the year ended December 31, 2006, as filed with the Commission on March 14, 2007. | |
| + | Confidential treatment has been granted, or requested, with respect to portions of the exhibit. A complete copy of the agreement, including the redacted terms, has been separately filed with the Securities and Exchange Commission. |
16
Table of Contents
| (1) | Incorporated by reference to the corresponding exhibit to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on August 15, 2000. | |
| (2) | Incorporated by reference to Exhibit 2.1 to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on July 2, 2004. | |
| (3) | Incorporated by reference to the corresponding exhibit to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended June 30, 2000, filed with the Securities and Exchange Commission on August 14, 2000. | |
| (4) | Incorporated by reference to the indicated exhibit to Maxygen’s Registration Statement onForm S-1, as amended(No. 333-89413) initially filed with the Securities and Exchange Commission on October 20, 1999. | |
| (5) | Incorporated by reference to the indicated exhibit to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended June 30, 2001, filed with the Securities and Exchange Commission on August 14, 2001. | |
| (6) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2002, filed with the Securities and Exchange Commission on March 27, 2003. | |
| (7) | Incorporated by reference to the indicated exhibit to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on June 30, 2006). | |
| (8) | Incorporated by reference to exhibit 10.1 to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended June 30, 2002, filed with the Securities and Exchange Commission on August 14, 2002. | |
| (9) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2000, filed with the Securities and Exchange Commission on March 21, 2001. | |
| (10) | Incorporated by reference to exhibit 10.6 to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2001, filed with the Securities and Exchange Commission on March 25, 2002. | |
| (11) | Incorporated by reference to the exhibit 99.3 to Maxygen’s Registration Statement onForm S-8(No. 333-57486) filed with the Securities and Exchange Commission on March 23, 2001. | |
| (12) | Incorporated by reference to the exhibit 4.2 to Maxygen’s Registration Statement onForm S-8(No. 333-138898) filed with the Securities and Exchange Commission on November 22, 2006. | |
| (13) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2003, filed with the Securities and Exchange Commission on March 12, 2004. | |
| (14) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2004, filed with the Securities and Exchange Commission on March 14, 2005. | |
| (15) | Incorporated by reference to the indicated exhibit to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on August 25, 2006. | |
| (16) | Incorporated by reference to exhibit 10.1 to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended September 30, 2000, filed with the Securities and Exchange Commission on November 14, 2000. | |
| (17) | Incorporated by reference to Exhibit 10.1 to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on February 7, 2005. | |
| (18) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2005, filed with the Securities and Exchange Commission on March 15, 2006. | |
| (19) | Incorporated by reference to Exhibit 10.6 to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended June 30, 2006, filed with the Securities and Exchange Commission on August 7, 2006. | |
| (20) | Incorporated by reference to Exhibit 10.1 to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on April 4, 2006. |
17
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
MAXYGEN, INC.
| By: | /s/ Russell J. Howard |
Russell J. Howard
Chief Executive Officer
June 25, 2007
18
Table of Contents
EXHIBIT INDEX
| Exhibit | ||||
Number | Description of Exhibit | |||
| 2 | .1 | Exchange Agreement, dated as of April 12, 2000, by and among Maxygen, Inc., Maxygen Holdings Ltd., ProFound Pharma A/S and the shareholders of ProFound Pharma A/S(1) | ||
| 2 | .2 | Amendment No. 1 to the Exchange Agreement, dated as of July 31, 2000, by and among Maxygen, Inc., Maxygen Holdings Ltd., ProFound Pharma A/S and the shareholders of ProFound Pharma A/S (1) | ||
| 2 | .3 | Agreement and Plan of Merger, dated June 2, 2004, by and among Pioneer Hi-Bred International, Inc., Tango Merger Sub, Inc., Maxygen, Inc. and Verdia, Inc.(2) | ||
| 3 | .1 | Amended and Restated Certificate of Incorporation(3) | ||
| 3 | .2 | Amended and Restated Bylaws(3) | ||
| 4 | .1 | Specimen Common Stock Certificate (Exhibit 4.1 to Amendment No. 1) (4) | ||
| *10 | .1 | Form of Executive Officer and Director Indemnification Agreement (Exhibit 10.7)(4) | ||
| *10 | .2 | Form of Executive Officer Change of Control Plan (Exhibit 10.1)(5) | ||
| *10 | .3 | Form of Amendment No. 1 to Executive Officer Change of Control Plan (Exhibit 10.3)(6) | ||
| *10 | .4 | Form of Amendment No. 2 to Executive Officer Change of Control Plan (Exhibit 10.3)(7) | ||
| *10 | .5 | 1997 Stock Option Plan, as amended, with applicable option agreement(8) | ||
| *10 | .6 | Form of Amendment to Stock Option Agreements (Exhibit 10.2)(7) | ||
| *10 | .7 | 1999 Nonemployee Directors Stock Option Plan, as amended, with applicable option agreement (Exhibit 10.3)(5) | ||
| *10 | .8 | 1999 Employee Stock Purchase Plan, as amended (Exhibit 10.11)(9) | ||
| *10 | .9 | 2000 International Stock Option Plan, as amended, with applicable option agreement(10) | ||
| *10 | .10 | 2000 Non-Officer Stock Option Plan, as amended, with applicable option agreement(11) | ||
| *10 | .11 | 2006 Equity Incentive Plan (including related form of stock option agreement) (Exhibit 10.4)(7) | ||
| *10 | .12 | Form of Restricted Stock Unit Award Agreement under 2006 Equity Incentive Plan(12) | ||
| 10 | .13 | Lease, dated as of October 21, 1998, between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.4)(4) | ||
| 10 | .14 | First Amendment to Lease, dated as of February 26, 1999, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.5)(4) | ||
| 10 | .15 | Second Amendment to Lease, dated as of October 24, 2000, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.6)(9) | ||
| 10 | .16 | Third Amendment to Lease, dated October 22, 2003, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.15)(13) | ||
| 10 | .17 | Fourth Amendment to Lease dated December 15, 2004 by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.13)(14) | ||
| 10 | .18 | Fifth Amendment to Lease dated as of August 24, 2006, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.2)(15) | ||
| 10 | .19 | Lease, dated December 15, 2004, between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.14)(14) | ||
| 10 | .20 | First Amendment to Lease, dated as of August 24, 2006, by and between Metropolitan Life Insurance Company and Maxygen, Inc. (Exhibit 10.1)(15) | ||
| 10 | .21 | Lease Agreement, dated May 5, 2000, between ProFound Pharma A/S and The Science Park in Horsholm (16) | ||
| 10 | .22+ | Technology Transfer Agreement, dated March 14, 1997 (effective March 1, 1998), among Maxygen, Inc., Affymax Technologies N.V. and Glaxo Group Limited, as amended (Exhibit 10.3 to Amendment No. 2) (4) | ||
| *10 | .23 | Description of 2006 Executive Officer Cash Bonus Plan (Exhibit 10.1)(7) | ||
| *10 | .24 | Description of Goldstein Reimbursement Arrangement(17) | ||
19
Table of Contents
| Exhibit | ||||
Number | Description of Exhibit | |||
| *10 | .25# | Letter Agreement (re tax equalization payments), dated November 20, 2006, between Elliot Goldstein and Maxygen, Inc. | ||
| 10 | .26+ | Co-Development and Commercialization Agreement, dated as of December 9, 2005, amongHoffman-La Roche, Inc., F. Hoffman-La Roche Ltd. and Maxygen Holdings Ltd. (Exhibit 10.23)(18) | ||
| 10 | .27#+ | Cross License Agreement, dated as of July 16, 2003, between Maxygen, Inc. and Amgen Mountain View Inc. (as successor to Avidia, Inc.) | ||
| 10 | .28+ | Amended and Restated Exclusive License Agreement, dated July 10, 2006 (effective as of April 1, 2006), between Regents of the University of Minnesota and Maxygen, Inc.(19) | ||
| *10 | .29 | Consulting Agreement, dated as of January 12, 2006 (effective January 1, 2006), between Balkrishan Gill and Maxygen, Inc. (Exhibit 10.24)(18) | ||
| *10 | .30 | Consulting Agreement, between the Company and Waverley Associates, Inc., dated as of April 1, 2006 (20) | ||
| 21 | .1# | List of Subsidiaries | ||
| 23 | .1# | Consent of Independent Registered Public Accounting Firm | ||
| 24 | .1# | Power of Attorney | ||
| 31 | .1 | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | ||
| 31 | .2 | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | ||
| 32 | .1# | Certification of Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | ||
| * | Management contract or compensatory plan or arrangement. | |
| # | Previously filed with Maxygen’s Annual Report on Form10-K for the year ended December 31, 2006, as filed with the Commission on March 14, 2007. | |
| + | Confidential treatment has been granted, or requested, with respect to portions of the exhibit. A complete copy of the agreement, including the redacted terms, has been separately filed with the Securities and Exchange Commission. | |
| (1) | Incorporated by reference to the corresponding exhibit to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on August 15, 2000. | |
| (2) | Incorporated by reference to Exhibit 2.1 to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on July 2, 2004. | |
| (3) | Incorporated by reference to the corresponding exhibit to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended June 30, 2000, filed with the Securities and Exchange Commission on August 14, 2000. | |
| (4) | Incorporated by reference to the indicated exhibit to Maxygen’s Registration Statement onForm S-1, as amended(No. 333-89413) initially filed with the Securities and Exchange Commission on October 20, 1999. | |
| (5) | Incorporated by reference to the indicated exhibit to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended June 30, 2001, filed with the Securities and Exchange Commission on August 14, 2001. | |
| (6) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2002, filed with the Securities and Exchange Commission on March 27, 2003. | |
| (7) | Incorporated by reference to the indicated exhibit to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on June 30, 2006). | |
| (8) | Incorporated by reference to exhibit 10.1 to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended June 30, 2002, filed with the Securities and Exchange Commission on August 14, 2002. |
20
Table of Contents
| (9) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2000, filed with the Securities and Exchange Commission on March 21, 2001. | |
| (10) | Incorporated by reference to exhibit 10.6 to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2001, filed with the Securities and Exchange Commission on March 25, 2002. | |
| (11) | Incorporated by reference to the exhibit 99.3 to Maxygen’s Registration Statement onForm S-8(No. 333-57486) filed with the Securities and Exchange Commission on March 23, 2001. | |
| (12) | Incorporated by reference to the exhibit 4.2 to Maxygen’s Registration Statement onForm S-8(No. 333-138898) filed with the Securities and Exchange Commission on November 22, 2006. | |
| (13) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2003, filed with the Securities and Exchange Commission on March 12, 2004. | |
| (14) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2004, filed with the Securities and Exchange Commission on March 14, 2005. | |
| (15) | Incorporated by reference to the indicated exhibit to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on August 25, 2006. | |
| (16) | Incorporated by reference to exhibit 10.1 to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended September 30, 2000, filed with the Securities and Exchange Commission on November 14, 2000. | |
| (17) | Incorporated by reference to Exhibit 10.1 to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on February 7, 2005. | |
| (18) | Incorporated by reference to the indicated exhibit to Maxygen’s Annual Report onForm 10-K (FileNo. 000-28401) for the year ended December 31, 2005, filed with the Securities and Exchange Commission on March 15, 2006. | |
| (19) | Incorporated by reference to Exhibit 10.6 to Maxygen’s Quarterly Report onForm 10-Q (FileNo. 000-28401) for the quarter ended June 30, 2006, filed with the Securities and Exchange Commission on August 7, 2006. | |
| (20) | Incorporated by reference to Exhibit 10.1 to Maxygen’s Current Report onForm 8-K (FileNo. 000-28401) filed with the Securities and Exchange Commission on April 4, 2006. |
21