Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Washington, D.C. 20549
Form 10-K
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF
THE SECURITIES EXCHANGE ACT OF 1934
THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2007
Commission file number000-28401
MAXYGEN, INC.
(Exact name of registrant as specified in its charter)
(Exact name of registrant as specified in its charter)
Delaware | 77-0449487 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
515 Galveston Drive
Redwood City, California 94063
(Address of principal executive offices)
Redwood City, California 94063
(Address of principal executive offices)
Registrant’s telephone number, including area code:
(650) 298-5300
(650) 298-5300
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Name of Each Exchange on Which Registered | |
| Common Stock, $0.0001 par value | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 ofRegulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of thisForm 10-K or any amendment to thisForm 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” inRule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer o | Accelerated filer þ | Non-accelerated filer o (Do not check if a smaller reporting company) | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined inRule 12b-2 of the Exchange Act). Yes o No þ
As of June 30, 2007, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the voting stock held by non-affiliates, computed by reference to the closing price for the common stock as quoted by the Nasdaq Global Stock Market as of that date, was approximately $191,723,000. Shares of common stock held by each executive officer and director and by each person who owned 5% or more of the outstanding common stock have been excluded as such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of February 29, 2008, there were 37,005,680 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Certain portions of the registrant’s proxy statement for the 2008 Annual Meeting of Stockholders (hereinafter referred to as the “2008 Proxy Statement”) are incorporated by reference into Part III of this report.
TABLE OF CONTENTS
This report and the disclosures herein include, on a consolidated basis, the business and operations of Maxygen, Inc. and its wholly-owned subsidiaries, Maxygen ApS and Maxygen Holdings Ltd. For the two months ended February 28, 2005, the operations of Codexis, Inc. are also included. On February 28, 2005, our voting interests in Codexis fell below 50% and, from such date, the financial position and results of operations of Codexis are no longer consolidated with our financial position and results of operations. We instead reflect our investment in Codexis under the equity method of accounting. In this report, “Maxygen,” the “Company,” “we,” “us” and “our” refer to such consolidated entities, unless, in each case, the context indicates that the disclosure applies only to a named subsidiary.
We own or have rights to various copyrights, trademarks and trade names used in our business, including Maxygen®, MaxyScan® and MolecularBreeding.tm The following, which may appear in this document, are registered or other trademarks owned by the following companies: Neupogen® (Amgen Inc.); Neulasta® (Amgen Inc.); NovoSeven® (Novo Nordisk A/S); Orencia® (Bristol-Myers Squibb Company); PEG-Intron® (Schering Corporation) and Aranesp® (Amgen Inc.). Other service marks, trademarks and trade names referred to in this report, and in the documents incorporated by reference in this report, are the property of their respective owners. The use of the word “partner” and “partnership” does not mean a legal partner or legal partnership.
i
Table of Contents
Forward Looking Statements
This report contains forward-looking statements within the meaning of federal securities laws that relate to future events or our future financial performance. In some cases, you can identify forward-looking statements by terminology such as “may,” “can,” “will,” “should,” “could,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “intend,” “potential” or “continue” or the negative of these terms or other comparable terminology. Examples of these forward-looking statements include, but are not limited to, statements regarding the following:
| • | our ability to develop products suitable for commercialization; | |
| • | our predicted development and commercial timelines for any of our potential products, including the timing and jurisdiction of any regulatory filing or submission relating to the clinical development of any product candidate, the timing of any commencement of clinical trials of any product candidate and the progress or status of such clinical trials; | |
| • | our liquidity and future financial performance; | |
| • | the establishment, development and maintenance of any manufacturing or collaborative relationships; | |
| • | the effectiveness of our MolecularBreeding directed evolution platform and other technologies and processes; | |
| • | our ability to protect our intellectual property portfolio and rights; | |
| • | our ability to identify and develop new potential products; | |
| • | the attributes of any products we may develop; and | |
| • | our business strategies and plans. |
These statements are only predictions. Risks and uncertainties and the occurrence of other events could cause actual results to differ materially from these predictions. Important factors that could cause our actual results to differ materially from the forward-looking statements we make in this report are set forth in this report, including the factors described in the section entitled “Item 1A — Risk Factors.”
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. Moreover, neither we nor any other person assumes responsibility for the accuracy and completeness of these statements. Other than as required by applicable law, we disclaim any obligation to update or revise any forward-looking statement contained in this report as a result of new information or future events or developments.
ii
Table of Contents
PART I
| Item 1 | BUSINESS |
Overview
We are a biotechnology company committed to the discovery and development of improved next-generation protein pharmaceuticals for the treatment of disease and serious medical conditions. We began operations in March 1997 with the mission to develop important commercial products through the use of biotechnology. Since then, we have established a focus in human therapeutics, particularly on the development of optimized protein pharmaceuticals. Three of our next-generation product candidates are currently in clinical or preclinical development:
| • | MAXY-G34, a pegylated, granulocyte colony stimulating factor (G-CSF) product for the treatment of neutropenia; | |
| • | MAXY-VII, a factor VIIa product for the treatment of hemophilia and possibly acute bleeding conditions; and | |
| • | MAXY-4, a CTLA-4-Ig product for the treatment of rheumatoid arthritis and other immune or autoimmune diseases. |
In addition to our clinical and preclinical stage product candidates, we have other research stage programs and assets outside of our core business, including research on certain vaccine programs.
We use our technology platform, MolecularBreeding directed evolution, along with PEGylation, glycosylation, rational design and mutagenesis, in an effort to create improved, proprietary proteins. Our MolecularBreeding technology platform, also referred to as DNA shuffling, can be used to create new versions of any known protein. We believe that once a desired improvement has been identified, the chance of being able to create a novel protein with that desired improvement is high.
Major Developments in 2007
In July 2007, we commenced a Phase IIa clinical trial of MAXY-G34 in breast cancer patients in Eastern Europe. We announced positive progress in this clinical trial in January 2008, including that MAXY-G34 showed clinical activity at the lowest dose level of 10 µg/kg and that no immunogenicity or serious adverse events were observed.
In May 2007, we announced our plans to proceed with the clinical development of our lead MAXY-VII product candidate for hemophilia. We currently plan to file our first clinical trial application (CTA) in the first half of 2008 and commence a first in human clinical trial in hemophilia patients in the second half of 2008.
In November 2007, we announced the preclinical development of our most recent product candidate,MAXY-4, for the treatment of rheumatoid arthritis. We plan to conductin vivoproof-of-concept preclinical studies with our MAXY-4 product candidates and select one or more lead product candidates in 2008.
During 2007, we recognized $8.3 million in revenue under our license agreement with Codexis, Inc., or Codexis, including approximately $7.5 million recognized in the fourth quarter of 2007 in connection with an expanded collaboration agreement between Royal Dutch Shell plc and Codexis for the development of new enzymes to convert biomass to fuel.
In December 2007, we licensed our proprietary dengue virus antigen technology to sanofi pasteur, the vaccines division of the sanofi-aventis Group, for development and worldwide commercialization of a second generation dengue vaccine. We received an upfront fee and are eligible to receive up to an additional $23.0 million of event-based payments under the agreement, as well as royalties on any product sales.
Also during 2007, collaboration agreements with F. Hoffmann-La Roche Ltd., or Roche, for two of our leading product candidates were terminated. In April 2007, Roche terminated the collaboration agreement for ourMAXY-VII product candidates due to the inability of the parties to de-risk the program in preclinical testing by failing to establish an animal model suitable to demonstrate cessation of acute bleeding in a trauma setting. In
1
Table of Contents
November 2007, we agreed with Roche to terminate the collaboration agreement for our MAXY-alpha product candidates based on preliminary observations made in a Phase Ia clinical trial.
Finally, in November 2007, we implemented a plan to consolidate our research and development activities at our U.S. facilities in Redwood City, California in an effort to reduce costs and increase overall operational efficiency across our research, preclinical, clinical and regulatory activities. The consolidation has resulted in the cessation of research and development operations at Maxygen ApS, our wholly owned subsidiary in Denmark. As a result of the consolidation, we have recorded charges of approximately $5.2 million relating to one-time termination benefits for the affected employees of Maxygen ApS and related expenses and we expect to incur additional costs of approximately $1.0 million relating to the consolidation and expect to complete the activities related to this consolidation during the second quarter of 2008.
Business Strategy
Our goal is to develop and commercialize improved and proprietary versions of currently marketed or clinically validated therapeutic proteins. To achieve this objective, we are pursuing the following key strategies:
| • | Advance the Development of Our Lead Product Candidates. Our primary focus is the advancement of our lead product candidate, MAXY-G34, through clinical development and the commencement of clinical development for our MAXY-VII product candidate for hemophilia. We initiated a Phase I clinical trial in the United States for our lead MAXY-G34 product candidate in the third quarter of 2006 and commenced a Phase IIa clinical trial in breast cancer patients in July 2007. We currently plan to file our first clinical trial application (CTA) for our lead MAXY-VII product candidate in the first half of 2008 and expect to commence clinical trials in hemophilia patients in 2008. We also plan to conductin vivo proof-of-concept preclinical studies with our MAXY-4 product candidates and select one or more lead product candidates in 2008. | |
| • | Create Value with Improved Next-Generation Protein Drugs. Our research and development efforts focus on seeking to make improved and proprietary versions of currently marketed therapeutic proteins that address significant market opportunities. We continue to identify new product opportunities based upon currently marketed or clinically validated protein products that require or could benefit from improvements to address existing medical needs in the treatment of disease and serious medical conditions, and use our proprietary MolecularBreeding directed molecular evolution platform and other technologies in an effort to improve these products. We commit resources to only those product candidates that we believe are scientifically and commercially viable. | |
| • | Increase Value, Reduce Risk. By using our technologies to improve the properties of currently marketed or clinically validated therapeutic proteins that have significant commercial value, we believe we can potentially create novel and proprietarybest-in-class products that take advantage of known utility, development paths and markets. We believe that this approach may result in reduced risk and enhanced chances of commercial success as compared to the development of novel pharmaceutical products directed at unvalidated targets. | |
| • | Establish a Portfolio of Next-Generation Therapeutic Proteins. In addition to our existing product candidates, we have an ongoing discovery effort focused on next-generation protein pharmaceuticals. Our goal is to leverage discoveries in our research programs or in-licensed product candidates to extend and expand our product pipeline. By broadening our product portfolio, we hope to increase the probability of clinical and commercial success and reduce the risks arising from the impact of any one product failure. | |
| • | Establish Strategic Collaborations to Advance our Product Pipeline and Leverage Our Development Resources. Part of our strategy has been to establish strategic collaborations with leading biopharmaceutical corporations that enable us to retain a large portion of the eventual value from our product candidates. We may enter into strategic collaborations at various stages in our research and development process that will allow us to further diversify our product development risks, reduce costs, access the complementary skills and infrastructure possessed by our partners and accelerate the development and commercialization of our product candidates. |
2
Table of Contents
| • | Manage Our Financial Resources. Fiscal discipline and pragmatic allocation of our resources are key components of our strategy. We focus our financial resources on those functions that should enhance our ability to generate improved next-generation product candidates and rapidly advance these new product candidates through preclinical and clinical development. |
Product Pipeline
The following table summarizes the status of our product pipeline:
| Development | ||||||||
| Responsibilities/ | ||||||||
| Commercialization | Commercialization | |||||||
Product Candidate | Indication | Rights | Rights | Status | ||||
| MAXY-G34 | Neutropenia | Maxygen | Maxygen | Phase IIa | ||||
| MAXY-VII | Hemophilia | Maxygen | Maxygen | Preclinical;* plan to file clinical trial application (CTA) in the first half of 2008 | ||||
| MAXY-4 | Rheumatoid Arthritis | Maxygen | Maxygen | Preclinical;* plan to conductin vivo proof-of-concept preclinical studies and select lead candidate(s) in 2008 |
| * | “Preclinical” means candidate selection, process development, productscale-up, formulation and further testing in animals, including toxicology, pharmacokinetics and pharmacodynamics. |
MAXY-G34
Our lead MAXY-G34 product candidate has been designed to be an improved next-generation pegylated, granulocyte colony stimulating factor, or G-CSF, for the treatment of neutropenia. G-CSF is a natural protein that functions by stimulating the body’s bone marrow to produce more white blood cells.
Neutropenia is a severe decrease in neutrophil cell counts in the blood. Neutropenia is a common side effect of chemotherapeutic treatments for many forms of cancer, including breast cancer, lung cancer, lymphomas and leukemias. Neutropenic patients are at increased risk of contracting bacterial infections, some of which can be life threatening. Further, and most importantly, neutropenic patients may receive reduced or delayed chemotherapy treatment, which can result in cancer progression.
Treatment with MAXY-G34 may help the body make white blood cells more rapidly and for a more sustainable period than the products currently approved for the treatment of neutropenia, Neupogen and Neulasta, which could make it an attractive alternative for both doctors and patients. In multiple preclinical animal models, we have generated data that suggest that our MAXY-G34 product candidate reduces the duration of severe neutropenia by clinically relevant periods (approximately 25% shorter duration of neutropenia) when compared to the currently marketed products. If these results translate into the human clinical setting, MAXY-G34 may help protect patients from chemotherapy and radiation therapy-related febrile neutropenia and infections, shorten the duration of hospital stays and help keep patients on schedule for their cancer treatments.
Market Opportunity. Neupogen, a first-generation G-CSF product, and Neulasta, a second-generation pegylated G-CSF product, currently dominate the market to treat chemotherapy and radiation-induced neutropenia. Worldwide sales of G-CSF products were approximately $4.8 billion in 2007.
Development Status. The initial Phase I clinical trial of MAXY-G34 was initiated in August 2006 in the United States. This study in healthy volunteers was designed to determine safety, pharmacokinetics, pharmacodynamics and immunogenicity of a single subcutaneous injection of MAXY-G34. The study initially investigated
3
Table of Contents
doses ranging from 10 to 150 µg/kg of MAXY-G34 with a subsequent study extension to investigate the 5 µg/kg dose of MAXY-G34 in a total of 47 subjects, of which 40 received MAXY-G34. The results of the Phase I clinical trial with a single dose of MAXY-G34 indicated that the drug was generally safe and well tolerated throughout the study, at all doses from 5 to 150 µg/kg of MAXY-G34. All doses tested in this Phase I trial increased the neutrophil levels as compared to the placebo controls. The half-life observed for MAXY-G34 in the initial Phase I clinical trial was approximately 2.3 times the terminal half-life for Neulasta in healthy volunteers, as estimated from the historical literature for Neulasta. No antibodies were detected in any clinical trial participants 30 and 90 days after the administration of MAXY-G34. The adverse events observed were similar in frequency and type to those previously reported for currently marketed G-CSF products. The most common adverse event observed was bone pain. Low but detectable levels of MAXY-G34 were observed at the end of the21-day collection period across the dose range studied.
Preliminary data from a separate Phase I study that examined MAXY-G34 at a 100 µg/kg dose or a 6mg fixed dose of Neulasta in healthy volunteers were generally consistent with the results previously obtained in the initial Phase I study (which did not involve a Neulasta cohort). Six volunteers were examined in each group. These results further indicated that both drugs were generally safe and well tolerated in healthy volunteers. Both MAXY-G34 and Neulasta increased the neutrophil levels and both drugs led to a substantial increase in the level of CD34+ cells in blood. Based on comparisons of the area under the curve for neutrophils and CD34+, at the single doses of MAXY-G34 and Neulasta being compared, MAXY-G34 administration resulted in numerically larger neutrophil levels, while Neulasta administration resulted in numerically larger numbers of CD34+ progenitor cells released. No inferences can be reliably drawn regarding these observed differences, in light of the small number of subjects in this study. In this study, the half-life observed for MAXY-G34 was approximately 1.3 times that observed for Neulasta. No antibodies binding to MAXY-G34 were detected in this trial at any time up to 90 days post-administration of MAXY-G34.
In July 2007, we commenced a Phase IIa clinical trial of MAXY-G34 in breast cancer patients in Eastern Europe. In this Phase IIa clinical trial, patients are being administered a single dose of MAXY-G34 therapy per three week chemotherapy cycle, with each patient receiving six cycles of TAC (docetaxel, adriamycin and cyclophosphamide) chemotherapy. The study intends to investigate safety, efficacy and pharmacokinetics of MAXY-G34 across the dose range of 10 µg/kg to 100 µg/kg with recommendations for dose-escalation provided by an independent Data Safety Monitoring Board, or DSMB.
Initial observations from the Phase IIa trial were reported in January 2008 and included the following:
| • | Patients in the initial 10 µg/kg MAXY-G34 cohort met the safety criterion for duration of neutropenia (less than or equal to five days of severe neutropenia), and, as a result, none of the MAXY-G34 treated patients needed to be switched to Neulasta. In breast cancer patients receiving TAC chemotherapy without G-CSF support, severe neutropenia has been reported to last approximately seven days. | |
| • | No serious adverse events or immunogenicity issues were noted for the initial cohort of patients on 10 µg/kg of MAXY-G34. Data available from multiple patients after multiple doses revealed no binding antibodies, and drug response in all patients had been sustained. | |
| • | The DSMB overseeing the trial approved escalation to the second dose level of 30 µg/kg MAXY-G34. The first cohort of patients at this level has been enrolled and commenced treatment. | |
| • | No serious adverse events or drug-related grade 3 or 4 adverse events were reported in any patient receiving MAXY-G34. A total of 23 doses of MAXY-G34 had been delivered to patients in the 10 µg/kg and 30 µg/kg cohorts. |
On February 12, 2008, it was reported that the DSMB had met and approved escalation to the third dose level of 60 µg/kg of MAXY-G34 and we have enrolled the first cohort of patients at this dose level.
We currently retain all rights to our MAXY-G34 product candidate.
4
Table of Contents
MAXY-VII
Our lead MAXY-VII product candidate has been designed to be a superior next-generation factor VIIa product to treat hemophilia and, potentially, acute bleeding indications. Factor VIIa is a natural protein with a pivotal role in blood coagulation and clotting.
Hemophilia is an inherited genetic disease defined by a lack of one or more proteins (clotting factors) required to clot the blood. Hemophilia A, in which patients lack clotting Factor VIII, accounts for the majority of patients. The second most common type of hemophilia is Hemophilia B, in which patients lack Factor IX. The first line of treatment for these patients is to replace the missing clotting factor with recombinant or plasma-derived Factor VIII and Factor IX. Some patients develop antibodies, also referred to as inhibitors, to these replacement factors, and are subsequently treated with recombinant Factor VIIa.
NovoSeven, a recombinant human Factor VII product of Novo Nordisk A/S approved for the treatment of hemophilia patients with inhibitors, is the only Factor VII product currently approved for human use. Our lead MAXY-VII product candidate has been designed to deliver greater potency, improved efficacy, a longer circulating half-life and an overall improved therapeutic index in the treatment of uncontrolled bleeding compared to NovoSeven. Studies in preclinical animal models have demonstrated that MAXY-VII may be more effective than NovoSeven at treating uncontrolled bleeding.
Novo Nordisk A/S is currently conducting clinical trials of NovoSeven for various indications associated with uncontrolled bleeding, including trauma and surgical bleeds.
Market Opportunity. NovoSeven is currently approved for the treatment of hemophilia patients who have developed antibodies to replacement factor VIII or factor IX therapy. Sales of NovoSeven in 2007 were approximately $1.1 billion. The use of recombinant Factor VII-based products for the treatment of new indications, such as severe bleeding in trauma, is also forecasted to represent significant new market opportunities for next-generation recombinant Factor VII products.
Development Status. We currently plan to file our first clinical trial application (CTA) for our leadMAXY-VII product candidate in the first half of 2008 and expect to commence a first in human clinical trial in hemophilia patients in 2008.
In December 2005, we entered into an agreement with Roche relating to the development and commercialization of these product candidates for acute bleeding indications. Roche terminated this agreement in April 2007 due to the inability of the parties to de-risk the program in preclinical testing by failing to establish an animal model suitable to demonstrate cessation of acute bleeding in a trauma setting. Under the terms of the agreement, we had agreed with Roche to share the costs of worldwide research and development activities in connection with the development of the MAXY-VII product candidates. We were to lead early stage clinical development of these product candidates and Roche was to lead late stage clinical development, with exclusive worldwide rights to commercialize the MAXY-VII products subject to the agreement. In the United States, we had the option to co-fund marketing activities in exchange for increased royalty amounts. Under the agreement, we received an upfront fee of $8 million from Roche and, in 2006, we received a $5 million payment from Roche for the achievement of a manufacturing process development milestone. We were eligible to receive additional milestone payments of up to $82 million upon the achievement of further development milestones, as well as royalties on product sales. As a result of the termination of this agreement, we recognized approximately $5.6 million of deferred revenue in 2007 relating to the upfront fee received from Roche in 2005. Subsequent to the termination of this agreement, all rights related to MAXY-VII were returned to us and we have since advanced preclinical development of our leadMAXY-VII product candidate for the treatment of hemophilia.
We currently retain all rights to our MAXY-VII product candidates.
MAXY-4
Our MAXY-4 product candidates are designed to be superior, next-generation CTLA-4 Ig therapeutics for the treatment of a broad array of autoimmune disorders, including rheumatoid arthritis. These candidates are designed
5
Table of Contents
to block the co-stimulation of T cells, a subset of white blood cells that are known to be involved in the pathogenesis of autoimmunity.
Rheumatoid arthritis, or RA, is a chronic autoimmune disease characterized by chronic pain and disability of the peripheral joints. RA affects approximately 1% of the world’s population and its incidence is about twice as frequent among women than it is among men. Biologic therapeutics available for RA focus upon the greater than 4 million moderate-to-severe patients diagnosed with this severely debilitating condition in the developed world.
We have utilized our MolecularBreeding directed evolution technology to prepare our MAXY-4 candidates that have demonstrated improved potency in several preclinical assays as compared to Orencia (which is indicated for reducing signs and symptoms, inducing major clinical response, slowing the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more disease-modifying, anti-rheumatic drugs) and belatacept (currently under development by Bristol-Myers Squibb Company for organ transplant therapy).
Market Opportunity. In 2007, worldwide sales of protein therapeutics used for the treatment of RA were greater than $10 billion. In 2007, sales of Orencia, which was launched in the United States during 2006 and in the European Union during 2007, were $231 million and are expected to exceed $1 billion by 2012. In addition to the RA marketplace, future commercial opportunities for our MAXY-4 program may include other autoimmune diseases, such as Crohn’s disease, systemic lupus erythematosus, psoriasis, transplant rejection and ulcerative colitis.
Development Status. We plan to completein vivoproof-of-concept preclinical studies of our MAXY-4 product candidates and select one or more lead candidates in 2008.
We currently retain all rights to our MAXY-4 product candidates.
Other Assets and Areas of Research
In addition to our development stage product candidates, we have other, earlier stage programs in preclinical research and assets outside of our core business, including vaccine research and an investment in Codexis, a biotechnology company focused on developing biocatalytic process technologies for pharmaceutical, energy and industrial chemical applications.
Vaccines
We believe that our proprietary technologies have the potential to transform the design and development of vaccines through the optimization of properties that allow for the generation of broad and strong immune responses. We currently have an active program to advance the research for development of a preventative HIV vaccine. Our vaccine research program is fully funded by research grants from the National Institutes of Health, or NIH. In 2005, the NIH awarded us two competitive grants, including $11.7 million over approximately five years as part of the HIV Research and Development, or HIVRAD, program. The HIVRAD grant provides funds for the use of our MolecularBreeding directed evolution platform to generate novel HIV-1 antigens potentially capable of inducing broad antibody responses to multiple strains of the HIV-1 virus. The NIH also awarded us a Phase I grant in 2005 and two additional grants in 2006 totaling $500,000 from the NIH Small Business Innovation Research, or SBIR, program to fund investigations into the effect on immunogenicity of secondary modifications to a specific HIV-1 envelope protein. As part of the SBIR program, the NIH also awarded us one grant totaling $1.0 million over two years for work on vaccines for equine encephalitis. We are currently working in collaboration with Monogram Biosciences, Inc., Aldevron LLC and the Scripps Research Institute with respect to these government-funded projects.
We were also awarded a one-year contract of $2.4 million in 2005 from the Department of Defense to fund work to develop a high-throughput HIV vaccine-screening platform. This grant, which was renewed in 2006 for $2.4 million, expired in October 2007.
In addition, in December 2007, we licensed our proprietary dengue virus antigen technology to sanofi pasteur, the vaccines division of the sanofi-aventis Group. Under the terms of the agreement, we have transferred to sanofi
6
Table of Contents
pasteur a portfolio of preclinical dengue antigens for development and worldwide commercialization of a second generation dengue vaccine. We received an upfront fee and are eligible to receive up to an additional $23.0 million of event-based payments under the agreement, as well as royalties on any product sales.
Codexis, Inc.
We have a minority investment in Codexis. We formed Codexis in January 2002 as a wholly owned subsidiary to operate our former chemicals business. Our voting rights in Codexis were reduced below 50% in the first quarter of 2005 and, as a result, Codexis is no longer consolidated in our financial statements. As of December 31, 2007, we owned approximately 8.9 million shares of Codexis common stock and various classes of Codexis preferred stock convertible into common stock, equal to approximately 25% of the issued and outstanding capital stock of Codexis. The convertible preferred stock of the most recent Codexis financing is senior in liquidation preference to our shares and was sold at a price of $8.50 per share. We are not obligated to fund the operations or other capital requirements of Codexis. For more information regarding our investment in Codexis, see Note 1 of the Notes to Consolidated Financial Statements.
In 2002, we entered into a license agreement with Codexis pursuant to which we granted to Codexis certain exclusive rights to our MolecularBreeding directed evolution platform for certain small molecule pharmaceutical, energy and industrial chemical applications. Under the agreement, Codexis is also eligible to acquire certain additional rights for use in limited subfields based upon predetermined research expenditure requirements. In partial consideration for the rights granted to Codexis under the license agreement, we received shares of Codexis common and preferred stock. In 2006, we amended the license agreement to expand the scope of the license to include certain applications relating to energy, including biofuels. In consideration of this expanded license, we were granted the right to receive a percentage of all consideration received by Codexis from a third party, including license fees, milestone payments, royalties and the purchase of equity securities (subject to certain limitations) and research funding (in excess of a specified base rate), that relates to the use of the licensed rights for the development or commercialization of certain products or processes in the energy field. We are also entitled to a percentage of net sales of any energy product that Codexis commercializes directly. We are also entitled to a percentage of any assets, rights or interests Codexis may obtain through an affiliate that engages in a line of business relating to such products or processes. The license agreement does not otherwise provide for the payment by us or to us of any amounts for license fees, milestone payments or royalties. The license agreement expires upon the last to expire of the patent rights licensed under the agreement but may be terminated earlier by us under certain circumstances, including a material breach by Codexis of certain obligations under the license agreement. Following the termination or expiration of the license agreement, Codexis would retain a license for an additional fifty (50) years to certain rights and materials transferred by us to Codexis under the agreement.
During 2007, we recognized $8.3 million in revenue from Codexis under this license agreement, including approximately $7.5 million in connection with an expanded collaboration agreement between Royal Dutch Shell plc and Codexis for the development of new enzymes to convert biomass to fuel.
Avidia Inc.
In July 2003, we established Avidia, Inc. (formerly Avidia Research Institute), or Avidia, a biotechnology company focused on developing products based on a new class of subunit proteins, together with two third-party investors. We received equity interests in Avidia through our initial contribution of technology and funding and our participation in subsequent preferred stock financings by Avidia. In October 2006, Amgen Inc. completed the acquisition of Avidia. As a result of the acquisition, we received cash proceeds of approximately $17.8 million (before $140,000 of income taxes) in exchange for our equity interests in Avidia and may receive up to an additional $1.4 million in cash, contingent upon the development of certain Avidia products by Amgen Inc. This contingent amount was reduced from $2.8 million based on the discontinuation by Amgen Inc. of certain development activities. For more information regarding the sale of our investment in Avidia, see Note 12 of the Notes to Consolidated Financial Statements.
Under a cross license agreement that we entered into with Avidia at the time of Avidia’s formation, we have exclusive and non-exclusive license rights to use certain Avidia technology to develop and commercialize therapeutic
7
Table of Contents
products directed to certain specific targets, including CD40, CD-40 ligand, CTLA-4, CD28, B.7.1 (CD80), B.7.2 (CD86), p40 (subunit of IL-12 and IL-23), TPO, any interferonand/or interferon receptor, TPO/IL3 and TNF/IL-1. The cross license agreement does not provide for the payment by us or to us of any amounts for license fees, milestone payments or royalties. The cross license agreement has a twenty-five (25) year term, but may terminate earlier based on the expiration of the patent rights licensed under the agreement. Following the expiration of the cross license agreement, the licenses granted under the agreement become perpetual on acountry-by-country basis.
Under the cross license agreement, Avidia also granted us certain limited options to acquire additional licenses to develop and commercialize other therapeutic products researched by Avidia. The exercise of an option would require us to enter into a separate product license agreement for any such product with Amgen Mountain View Inc. (as successor to Avidia) on pre-agreed terms that would include the payment by us to Amgen Mountain View Inc. of royalties based on net sales of the products subject to the product license agreement and milestone payments based upon our achievement of certain regulatory and clinical milestones for such product, up to an aggregate of $19.8 million. To date, we have not entered into a product license agreement for any such product.
MAXY-alpha
Our MAXY-alpha product candidates have been designed to be superior next-generation interferon alpha products for the treatment of HCV and Hepatitis B virus, or HBV, infections and other indications, including infectious diseases, oncology, inflammatory diseases and autoimmune diseases.
In 2003, we entered into a broad strategic alliance with Roche, a market leader in interferon alpha therapies, to develop novel improved interferon alpha and beta products for HCV and HBV infections and a wide range of other indications. Roche initiated a Phase Ia trial of MAXY-alpha for the treatment of HCV in November 2006 and, in November 2007, we agreed with Roche to discontinue the clinical development of MAXY-alpha and terminate the collaboration agreement based on preliminary observations made during the Phase Ia trial, including an unexpected reduction of pharmacodynamic and pharmacokinetic effects of MAXY-alpha. Under the agreement, Roche had exclusively licensed from us worldwide commercialization rights to specific novel interferon product candidates for the treatment of HCV and HBV infections. We received an initial payment and full research and development funding for the first two years of the agreement. In addition, we received $9 million of milestone payments from Roche during the term of the agreement. In addition to royalties on any product sales, we would have also been eligible to receive additional milestone payments of up to $50 million. In connection with the termination of the collaboration agreement, all rights to the MAXY-alpha product candidates have reverted back to us. At present, we have no plans for the further development of our MAXY-alpha product candidates.
MAXY-gamma
Our MAXY-gamma product candidates are next-generation versions of interferon gamma. In 2001, we granted an exclusive license to InterMune, Inc., or InterMune, to develop and market our MAXY-gamma product candidates for all human therapeutic indications. Based on InterMune’s decision to discontinue the development of our MAXY-gamma product candidates, we agreed with InterMune to terminate the license agreement, effective July 31, 2007. Under the agreement, we received upfront license fees and full research funding during the research phase of the collaboration, which was completed in June 2004, and would have been eligible to receive development and commercialization milestone payments that could have exceeded $60 million, in addition to royalties on product sales. As a result of the termination of this license agreement, all rights licensed to InterMune under the agreement have reverted to us. At present, we have no plans for the further development of our MAXY-gamma product candidates.
8
Table of Contents
Our Technologies
MolecularBreeding Directed Evolution Technologies
Our MolecularBreeding directed evolution technologies mimic the natural events of evolution. First, genes are subjected to DNAShuffling recombination technologies, generating a diverse library of gene variants. Second, our proprietary MaxyScan screening platform selects individual proteins from the gene variants in the library. The proteins that show improvements in the desired characteristics become the initial lead candidates. After confirmation of activity, the initial lead candidates are then used as the genetic starting material for additional rounds of shuffling. Once the level of improvement needed for the particular protein pharmaceutical is achieved, the group of product candidates is evaluated to select one or more product candidates for development.
The MolecularBreeding Directed Evolution Process
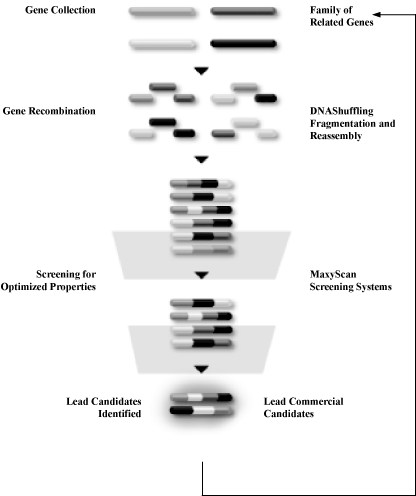
Step One: DNAShuffling Recombination Technologies
Our DNAShuffling recombination technologies work as follows: a single gene or multiple genes are cleaved into fragments and recombined, creating a population of new gene variants. The new genes created by DNAShuffling are then selected for one or more desired characteristics. This selection process yields a population of genes that becomes the starting point for the next cycle of recombination. As with classical breeding of plants and animals, this process is repeated until genes expressing the desired properties are identified.
9
Table of Contents
DNAShuffling can be used to evolve properties that are coded for by single genes, multiple genes or entire genomes. By repeating the process, DNAShuffling ultimately generate libraries with a high percentage of genes that have the desired function. Due to the high quality of these libraries, a relatively small number of screening tests need to be performed to identify gene variants with the desired commercial qualities. This process can reduce the cost and time associated with identifying multiple potential products.
Step Two: MaxyScan Screening
The ability to screen or select for a desired improvement in function is essential to the effective development of an improved gene or protein. As a result, we have invested significant resources in developing automated, stringent, rapid screens and selection formats.
We have developed screening tests that can measure the production of proteins or small molecules in culture without significant purification steps or specific test reagents, thereby eliminating time-consuming steps required for traditional screening tests. We are also focusing on the development of reliable, cell-based screening tests that are predictive of specific functions relevant to our human therapeutics programs. Accordingly, we continue to develop new screening approaches and technologies. Our approach is to create multitiered screening systems where we use a less sensitive screening test as a first screen to quickly select proteins with the desired characteristics, followed by a more sensitive screening test to confirm value in these variants and to select for final lead product candidates. Unlike approaches that create random diversity, our MolecularBreeding directed evolution platform produces potentially valuable libraries of gene variants with a predominance of active genes with the desired function. As a result of capturing the natural process of sexual recombination with our proprietary DNAShuffling methods, we are also able to generate gene variants with the desired characteristic at a frequency 5 to 10-fold higher than combinatorial chemistry, rational design or other directed evolution methods. This allows us to use complex biological screens and formats as a final screening test, as relatively few proteins need to be screened to detect an improvement in the starting gene activity. Furthermore, this allows us to focus on developing screens that generate a broader range of information that is more responsive to commercial and clinical concerns. This separates us from many of our potential competitors who invest significant time and money to screen billions of compounds per day. While we have the capability to screen billions of compounds per day, we generally need to screen far fewer, on the order of 10,000 candidates per day or less. Some of our screening capabilities include mass spectrometry, in vivo animal assays, bioassays, immunochemical assays, chemical assays, and biochemical assays.
We have access to multiple sources of genetic starting material. In addition to the wealth of publicly available genetic sequence information, we have typically been able to access our collaborators’ proprietary genes for use outside their specific fields of interest. Furthermore, we are able to inexpensively obtain our own genetic starting material or information, either through our own in-house efforts or through collaborations with third parties. This information and such materials when coupled with our DNAShuffling recombination technologies, can provide a virtually infinite amount of new, proprietary gene variants some of which may have potential commercial value.
Other Technologies
In addition to our proprietary MolecularBreeding directed evolution platform, we have acquired capabilities with regard to several complementary technologies potentially useful for the development of protein-based pharmaceuticals. Two examples of the tools that we use to post-translationally modify protein drugs are pegylation and glycosylation technologies. Over the last few years, glycosylation and pegylation have been validated technically and commercially through the successes of drugs, such as the pegylated interferons (Pegasys and PEG-Intron) and Aranesp, a hyper-glycosylated erythropoeitin. These post-translational modifications of proteins have been demonstrated to change the pharmacokinetics and pharmacodynamics of certain protein drugs. In addition, these modifications can change the solubility, bioavailability and immunogenicity profile of protein drugs.
Intellectual Property and Technology Licenses
Pursuant to a technology transfer agreement we entered into with Affymax Technologies N.V. and Glaxo Group Limited (each of which was then a wholly-owned subsidiary of what is now GlaxoSmithKline plc), we were assigned all the patents, applications and know-how related to our MolecularBreeding directed evolution platform,
10
Table of Contents
subject to certain internal research rights retained by GlaxoSmithKline plc. Affymetrix, Inc. retains an exclusive, royalty-free license under some of the patents and patent applications previously owned by Affymax for use in the diagnostics and research supply markets for specific applications. In addition, Affymax assigned jointly to us and to Affymetrix a family of patent applications relating to circular PCR techniques.
We have an extensive patent portfolio including over 80 issued U.S. patents and over 50 foreign patents relating to our proprietary MolecularBreeding directed evolution platform. Additionally, we have over 20 pending U.S. patent applications and over 50 pending foreign counterpart applications relating to our MolecularBreeding directed evolution platform and specialized screening technologies, and the application of these technologies to the development of protein pharmaceuticals and other industries, including agriculture, vaccines, gene therapy and chemicals.
Our expanding patent estate provides us with an increasingly broad and unique platform from which to create and potentially improve protein-based therapeutic products. Patents owned by us or for which we have exclusive licenses cover a broad range of activities surrounding recombination-based directed molecular evolution including:
| • | methods for template-based gene recombination to produce chimeric genes, including use of single or double-stranded templates; | |
| • | methods for recombining nucleic acid segments produced by incomplete nucleic acid chain extension reactions to produce chimeric genes, including the staggered extension process (StEP); | |
| • | methods utilizing reiterative screening or only a single cycle of screening; | |
| • | methods of combining any mutagenesis technique with DNA recombination methods to produce new chimeric genes; | |
| • | methods using synthesized nucleic acid fragments; | |
| • | in vivo and in vitro recombination methods of the above, in a variety of formats; | |
| • | methods of screening directed evolution libraries; | |
| • | methods for ligation- and single-stranded template-based recombination and reassembly; | |
| • | mutagenesis, including codon and gene site saturation mutagenesis, used in conjunction with recombination and reassembly; | |
| • | cell-based recombination methods; and | |
| • | fluorescence-, bioluminescence-, and nutrient-based screening methods, including the use of ultra-high throughput FACS-based methods for screening diverse variants. |
Such patents reinforce our preeminent position as an industry leader in recombination-based directed molecular evolution technologies for the preparation of chimeric genes for commercial applications.
In addition to the patents that we own directly, we have also exclusively licensed patent rights and technology for specific uses from Novozymes A/S, the California Institute of Technology, the University of Washington, GGMJ Technologies, L.L.C. and the University of Minnesota. These licenses give us rights to an additional 21 issued U.S. patents, 13 granted foreign patents and over 20 pending U.S. and foreign counterpart applications.
We have also received from Affymax (when it was a subsidiary of what is now GlaxoSmithKline plc) a worldwide, non-exclusive license to certain Affymax patent applications and patents related to technology for displaying multiple diverse proteins on the surface of bacterial viruses.
As part of our confidentiality and trade secret protection procedures, we enter into confidentiality agreements with our employees, consultants and potential collaborative partners. Despite these precautions, third parties or former employees could obtain and use information regarding our technologies without authorization, or develop similar technology independently. It is difficult for us to monitor unauthorized use of our proprietary methods and information. Effective protection of intellectual property rights is also unavailable or limited in some foreign countries. The efforts that we take to protect our proprietary information and rights may be inadequate to protect
11
Table of Contents
such information and rights. Our competitors could independently develop similar technology or design around any patents or other intellectual property rights we hold.
In July 2005, our European Patent 0752008, covering our first generation directed molecular evolution technologies, was the subject of an opposition proceeding before the European Patent Office. All claims of the patent were upheld as valid with minor amendments. We appealed this decision to the Appeals Division, which decided in a December 2007 hearing to maintain the patent with claims slightly broader than those maintained in the opposition proceeding. In October 2005, the Australian Patent Office found, in an opposition proceeding regarding our Australian patent application No. 703264 that corresponds to European Patent 0752008, 89 of our claims to be patentable as presented. In February 2006, our European Patent 0876509, covering one embodiment of our second-generation directed molecular evolution technologies, was the subject of an opposition proceeding before the European Patent Office. The opposition board revoked the patent on the grounds of lack of inventive step. We appealed this decision to the appeals board of the European Patent Office and in February 2008, the Appeals Division reversed the decision of the opposition board and maintained the patent with all its claims as originally issued.
Product Patents
Our patent portfolio consists of the following issued patents and pending patent applications for each of our primary product candidates:
| • | For our MAXY-G34 product candidates, we have three U.S. patents, 16 pending U.S. applications, 10 foreign patents and 42 pending foreign applications. | |
| • | For our MAXY-VII product candidates, we have one U.S. patent, 21 pending U.S. applications, eight foreign patents and 49 pending foreign applications. We also have exclusive licenses to two U.S. patents, ten pending U.S. applications and four pending foreign applications. | |
| • | For our MAXY-4 product candidates, we have one pending U.S. application. |
Manufacturing
We rely on third party manufacturers and collaborators to produce our compounds for clinical purposes and may do so for commercial production of any drug candidates that are approved for marketing. We have established agreements with Rentschler Biotechnologie GmbH, or Rentschler, in Germany for the manufacture of supplies of our MAXY-G34 product candidates for Phase I and II clinical trials and for the manufacture of supplies of our MAXY-VII product candidates for preclinical studies and Phase I and II clinical trials.
We believe that our current contract manufacturing agreement with Rentschler will be sufficient to accommodate clinical trials of our MAXY-G34 and MAXY-VII product candidates through Phase I and II clinical trials. However, we will need to secure additional manufacturing arrangements with contract manufacturers or develop our own manufacturing capability to meet our future needs, which would require significant capital investment.
Competition
Any products that we develop will compete in highly competitive markets. We face competition from large pharmaceutical and biopharmaceutical companies, such as Eli Lilly and Company, Pfizer, Inc., Genentech, Inc., Novo Nordisk A/S, Bristol-Myers Squibb Company, Schering-Plough Corporation and Amgen Inc., and from smaller biotechnology companies, such as Human Genome Sciences, Inc., Neose Technologies, Inc., CoGenesys, Inc., Zymogenetics, Inc., Inspiration Biopharmaceuticals, Inc. and Catalyst Biosciences, Inc. In the vaccines field, we face competition from biotechnology companies, such as Corixa Corporation (now a subsidiary of GlaxoSmithKline plc) and Vical Corporation, as well as large pharmaceutical companies, including GlaxoSmithKline plc, sanofi-aventis, Novartis AG, Pfizer Inc. and Merck & Co., Inc.
Many of our potential competitors, either alone or together with their collaborative partners, have substantially greater financial, technical and personnel resources than we do, and there can be no assurance that they will not succeed in developing technologies and products that would render our technologies and products or those of a
12
Table of Contents
collaborator obsolete or noncompetitive. In addition, many of our competitors have significantly greater experience than we do in developing products, undertaking preclinical testing and clinical trials, obtaining FDA and other regulatory approvals of products, and manufacturing and marketing products.
We are a leader in the field of directed molecular evolution. We are aware that other companies, including Verenium Corporation, Xencor, Inc. and Nautilus Biotech, have alternative methods for obtaining and generating genetic diversity or use mutagenesis techniques to produce genetic diversity. Academic institutions such as the California Institute of Technology, Pennsylvania State University, the University of California and the University of Washington are also working in this field. We have licensed certain patents from certain of these institutions. This field is highly competitive and companies and academic and research institutions are actively seeking to develop technologies that could be competitive with our technologies.
We are aware that other companies, organizations and persons have described technologies that appear to have some similarities to our patented proprietary technologies. We monitor publications and patents that relate to directed molecular evolution to be aware of developments in the field and evaluate appropriate courses of action in relation to these developments.
Research and Development Expenses
The majority of our operating expenses to date have been related to research and development. Our research and development expenses were $59.9 million in 2007, $49.1 million in 2006 and $41.9 million in 2005 (including research and development expenses attributable to Codexis in 2005). Additional information required by this item is incorporated herein by reference to “Research and Development Expenses” in Note 1 of the Notes to Consolidated Financial Statements. We intend to maintain our strong commitment to research and development as an essential component of our product development effort. We may also license technology or products developed by third parties to obtain access to additional potential products.
Geographic Distribution
We have operations in two geographic locations, the United States and Denmark. In November 2007, we implemented a plan to consolidate our research and development activities at our U.S. facilities. The consolidation has resulted in the cessation of our research and development operations in Denmark, although we may continue to conduct limited administrative activities through Maxygen ApS for the foreseeable future. In addition, certain of our licensors are based outside the United States. Additional information required by this item is incorporated herein by reference to “Segment Information — Geographic Information” in Note 11 of the Notes to Consolidated Financial Statements.
Segment Reporting
We operate our business in one segment, human therapeutics. Prior to February 28, 2005, the date on which Codexis ceased to be our consolidated subsidiary, we operated our business in two segments, human therapeutics and chemicals. Additional information required by this item is incorporated herein by reference to “Segment Information” in Note 11 of the Notes to Consolidated Financial Statements.
Government Regulation
We are subject to regulation by the FDA and comparable regulatory agencies in foreign countries with respect to the development and commercialization of products resulting from our drug discovery activities. The FDA and comparable regulatory bodies in other countries currently regulate therapeutic proteins and related pharmaceutical products as biologics. Biologics are subject to extensive pre- and post-market regulation by the FDA, including regulations that govern the collection, testing, manufacture, safety, efficacy, potency, labeling, storage, record keeping, advertising, promotion, sale and distribution of the products.
The time required for completing testing and obtaining approvals of our product candidates is uncertain but will take several years and approvals will only be obtained if our product candidates are shown to be safe and efficacious in clinical trials. Any delay in testing may hinder product development. In addition, we may encounter
13
Table of Contents
delays in product development or rejections of product applications due to changes in FDA or foreign regulatory policies during the period of product development and testing. Failure to comply with regulatory requirements may subject us to, among other things, civil penalties and criminal prosecution; restrictions on product development and production; suspension, delay or withdrawal of approvals; and the seizure or recall of products. The lengthy process of obtaining regulatory approvals and ensuring compliance with appropriate statutes and regulations requires the expenditure of substantial resources. Any delay or failure, by us to obtain regulatory approvals could adversely affect our ability to commercialize product candidates and generate sales revenue. Such delays or failures could also impact our likelihood of receiving milestone and royalty payments under any future collaborative arrangement.
Under FDA regulations, the clinical testing program required for marketing approval of a new drug typically involves three sequential phases, which may overlap.
| • | Phase I: Studies are conducted in normal, healthy human volunteers or patients to determine safety, dosage tolerance, absorption, metabolism, distribution and excretion. If possible, Phase I studies may also be designed to gain early evidence of effectiveness. | |
| • | Phase II: Studies are conducted in small groups of patients afflicted with a specific disease to determine dosage tolerance and optimal dosage, to gain preliminary evidence of efficacy, and to determine the common short-term side effects and risks associated with the substance being tested. | |
| • | Phase III: Involves large-scale studies conducted in disease-afflicted patients to provide statistical evidence of efficacy and safety and to provide an adequate basis for physician labeling. |
To date, neither we nor any corporate collaborator has successfully completed all stages of clinical development for any of our product candidates. If we (or a corporate collaborator) are unable to continue or successfully complete clinical trials of MAXY-G34 or any of our other product candidates, or decide not to continue clinical trials for a particular indication, we will not be able to seek or obtain regulatory approval for commercialization of the applicable product candidate for the relevant indication.
Phase I, Phase II or Phase III clinical testing may not be completed successfully within any specific period of time, if at all, with respect to any of our potential products. Furthermore, we, an institutional review board, the FDA or other regulatory bodies may deny approval for conducting a clinical trial or temporarily or permanently suspend a clinical trial at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
FDA marketing approval is only applicable in the United States. Marketing approval in foreign countries is subject to the regulations of those countries. The approval procedures vary among countries and can involve additional testing. The requirements for approval and the time required to obtain approval may differ from that required for FDA approval.
Although there are some centralized procedures for filings in the European Union countries, in general each country has its own procedures and requirements, and compliance with these procedures and requirements may be expensive and time-consuming. Accordingly, there may be substantial delays in obtaining required approvals from foreign regulatory authorities after the relevant applications are filed, if approvals are ultimately received at all.
Environmental Regulation
We seek to comply with all applicable statutory and administrative requirements concerning environmental quality. We have made, and will continue to make, expenditures for environmental compliance and protection. Expenditures for compliance with environmental laws have not had, and are not expected to have, a material effect on our capital expenditures, results of operation or competitive position.
Employees
As of March 3, 2008, we had 96 employees.
14
Table of Contents
Corporate Background and History
We began operations in 1997 to commercialize technologies originally conceived at Affymax Research Institute, then a subsidiary of what is now GlaxoSmithKline plc. We were incorporated in Delaware on May 7, 1996 and began operations in March 1997. Our principal executive offices are located at 515 Galveston Drive, Redwood City, CA 94063. Our telephone number is(650) 298-5300.
Our operations were originally focused on the application of our MolecularBreeding directed evolution platform and other technologies to the development of multiple products in a broad range of industries, including human therapeutics, chemicals and agriculture. In August 2000, to complement and expand our human therapeutics operations, we established our Danish subsidiary, Maxygen ApS, through the acquisition of ProFound Pharma A/S, a privately held Danish biotechnology company. In 2002, we formed two wholly owned subsidiaries, Codexis, Inc. and Verdia, Inc., to operate our chemicals and agriculture businesses.
Over the past several years, we have shifted our primary focus to the development of next-generation protein pharmaceuticals. Accordingly, in 2004, we sold Verdia to Pioneer Hi-Bred International, Inc., a wholly owned subsidiary of E.I. du Pont de Nemours and Company, for $64 million in cash. Codexis received financing from third party investors and operated as independent subsidiary beginning in September 2002 and, in February 2005, our voting rights in Codexis were reduced below 50%. In addition, in November 2007, we implemented a plan to consolidate our research and development activities at our U.S. facilities in Redwood City, California. The consolidation has resulted in the cessation of research and development operations at Maxygen ApS and the elimination of all employment positions at that site. We expect to complete the activities related to this consolidation during the second quarter of 2008.
Available Information
Our web site is located at www.maxygen.com. We make available free of charge, on or through our web site, our annual, quarterly and current reports, and any amendments to those reports, as soon as reasonably practicable after electronically filing or furnishing such reports with the Securities and Exchange Commission, or SEC. Information contained on our web site is not part of this report.
| Item 1A | RISK FACTORS |
This report contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of factors both in and out of our control, including the risks faced by us described below and elsewhere in this report.
You should carefully consider the risks described below, together with all of the other information included in this report, in considering our business and prospects. The risks and uncertainties described below are not the only ones facing our company. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations. If any of the following risks actually occur, our business could be harmed. In such case, the trading price of our common stock could decline, and you may lose all or part of your investment.
We have a history of net losses. We expect to continue to incur net losses and may not achieve or maintain profitability.
We have incurred net losses since our inception, including net losses of $49.3 million in 2007, $16.5 million in 2006 and $18.4 million in 2005. As of December 31, 2007, we had an accumulated deficit of $270.0 million. We expect to incur losses and negative cash flow from operating activities for at least the next several years and may never achieve profitability. To date, we have derived substantially all our revenues from collaborations, license agreements and grants and expect to derive a substantial majority of our revenue from such sources for at least the next several years. Revenues from such sources are uncertain because such agreements and grants generally have fixed terms and may be terminated under certain conditions, and because our ability to secure future agreements will depend upon our ability to address the needs of potential future collaborators. We expect to spend significant amounts to fund the development of our product candidates. As a result, we expect that our operating expenses will
15
Table of Contents
exceed revenues in the near term and we do not expect to achieve profitability during the next several years, if at all. If the time required for us to achieve profitability is longer than we anticipate, we may not be able to continue our business.
We are an early stage company deploying unproven technologies. If we do not develop commercially successful products, we may be forced to cease operations.
You must evaluate us in light of the uncertainties and complexities affecting an early stage biotechnology company. We may not be successful in the commercial development of products. Successful products will require significant investment and development, including clinical testing, to demonstrate their safety and effectiveness before their commercialization. To date, companies in the biotechnology industry have developed and commercialized only a limited number of biological products. We have not proven our ability to develop or commercialize any products. We, alone or in conjunction with corporate collaborators, will need to conduct a substantial amount of additional development before any regulatory authority will approve any of our potential products. This research and development may not indicate that our products are safe and effective, in which case regulatory authorities may not approve them. Problems are frequently encountered in connection with the development and utilization of new and unproven technologies, and the competitive environment in which we operate could limit our ability to develop commercially successful products.
The prospects of our current product candidates are highly uncertain. In particular, ongoing clinical trials of our lead MAXY-G34 product candidate may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing for this product candidate or cease our trials, which could adversely affect our business and cause a significant drop in our stock price.
There is a substantial risk that our drug discovery and development efforts may not result in the development of any commercially successful products. Our lead MAXY-G34 product candidate is currently in Phase IIa clinical trials in breast cancer patients for the treatment of neutropenia. While we announced positive progress in this clinical trial in January 2008, the results of preliminary studies and early-stage clinical trials do not necessarily predict the results of later-stage clinical trials, including the safety and efficacy profiles of any particular drug candidate. Moreover, in our industry, most product candidates fail before entering clinical trials or in clinical trials and most products that commence clinical trials are not approved for use in humans and never reach the market. Accordingly, negative or inconclusive results from ongoing or future clinical trials of MAXY-G34 could lead us to cease the development of this product candidate and decide not to advance this product candidate into later stage clinical trials, such as a Phase IIb trial or pivotal Phase III trials. We may decide to cease further development of the product candidate for a variety of reasons, including evidence that MAXY-G34 may not meaningfully reduce the period or risk of neutropenia following chemotherapy, may remain in circulation longer than desired or cause adverse side effects. In addition, regardless of the clinical properties of MAXY-G34, we may cease development of this product candidate at any time if we determine that we cannot afford the costs of further developing this product candidate ourselves and we are unable to enter into a collaborative or other arrangement with a third party to fund the further development and commercialization of this product candidate.
Even if MAXY-G34 successfully completes clinical trials and is approved for marketing in the United States or other countries, it will need to compete with other G-CSF drugs then on the market. The ability of MAXY-G34 to be successful in the market will depend on a variety of factors, including, for example, whether MAXY-G34 is clinically differentiated from other G-CSF drugs, the scope and limitations of the label approved by regulators for the use of MAXY-G34, the price of MAXY-G34, reimbursement decisions by third parties with regard toMAXY-G34, and the effort and success of marketing activities undertaken with regard to MAXY-G34.
We are aware that Amgen Inc. and other third parties have a number of issued patents that claim certain G-CSF compositions and their use. Amgen Inc. and other third parties also have pending patent applications that are directed at certain G-CSF compositions and their use and these applications could result in issued patents. For example, we recently became aware that the U.S Patent and Trademark Office allowed claims relating to G-CSF variants in a patent application owned by Amgen Inc. No patent has yet issued on this application but may issue later in 2008. The owners of issued patents, such as Amgen, Inc., could elect to commence a patent infringement suit
16
Table of Contents
against us with regard to MAXY-G34 in the courts or before the International Trade Commission. While we believe that we would have good defenses to any such suit, the outcome of patent litigation is necessarily uncertain and we could be forced to expend significant resources in the defense of any such suit, and we may not prevail. If the outcome of any such suit or action was unfavorable to us, we might have to pay significant damages to the patent owner, and if any patents found to be infringed had not expired, we could be enjoined from commercializing or importing MAXY-G34.
Drug development is a long, expensive and uncertain process and may not result in the development of any commercially successful products.
The development of human therapeutic products is long and uncertain. Most product candidates fail before entering clinical trials or in clinical trials. Moreover, most products that commence clinical trials are not approved for use in humans and never reach the market. In addition, due to the nature of human therapeutic research and development, the expected timing of product development, initiation of clinical trials and the results of such development and clinical trials are uncertain and subject to change at any point. Such uncertainty, which exists even for product candidates that appear promising based on earlier data, may result in research or development delays, clinical trial delays and failures, product candidate failures and delays in regulatory action or approval. Such delays or failures could reduce or eliminate our revenue by delaying or terminating the potential development and commercialization of our product candidates and could drastically reduce the price of our stock and our ability to raise capital. Without sufficient capital, we could be forced to reduce or cease our operations.
All of our product candidates are subject to the risks of failure inherent in drug development. Preclinical studies may not yield results that would satisfactorily support the filing of an investigational new drug application (IND) with respect to our drug candidates, and the results of preclinical studies do not necessarily predict the results of clinical trials. Moreover, the available animal models may be unsuitable for assessing our potential products for one or more indications, increasing the risk that animal models may not provide accurate or meaningful data as to the suitability or advantages of our potential products as treatments for the diseases or medical conditions of interest. Similarly, early-stage clinical trials may not predict the results of later-stage clinical trials, including the safety and efficacy profiles of any particular drug candidate. In addition, there can be no assurance that the design of our clinical trials will result in obtaining the desired efficacy data to support regulatory approval. Even if we believe the data collected from clinical trials of our drug candidates are promising, such data may not be sufficient to support approval by the U.S. Food and Drug Administration (FDA) or any foreign regulatory agency, which could delay, limit or prevent regulatory approval of our drug candidates. The FDA and similar regulatory agencies determine the type and amount of data necessary to obtain approval of any drug candidate, and as a result of new data or changes in the policies or practices of such agencies, the type and amount of data required for approval may change in the period between the start of product development and the completion of clinical trials.
Any failure or substantial delay in successfully completing clinical trials, obtaining regulatory approval and commercializing any of our current or future product candidates could severely harm our business.
The development of our product candidates, which is based on modifications to natural human proteins, may be subject to substantial delays, increased development costs, reduced market potential for any resulting product or the termination of the affected development program by us or a collaborator, each of which could adversely affect our business.
We design our product candidates to confer what we believe will be improved biological properties as compared to one or more currently marketed products. As a result, our product candidates differ from currently marketed drugs in ways that we expect will be beneficial. However, the impact of the modifications that we make in our product candidates may not be fully apparent in preclinical testing and may only be discovered in clinical testing. Such altered properties may render a product candidate unsuitable or less beneficial than expected for one or more diseases or medical conditions of possible interest or make the product candidate unsuitable for further development. For example, our products may be found to be more immunogenic than the corresponding natural human proteins or demonstrate undesirable pharmokinetic or pharmodynamic properties. For a particular product
17
Table of Contents
candidate, this may lead to the redirection of the development strategy which could result in substantial delays, increased development costs, decreased likelihood of obtaining regulatory approval, and reduced market potential for any resulting product. This also could result in the termination of the development of the affected product candidate. In either case, such results could adversely affect our business.
In addition, we or a collaborator may determine that certain preclinical or clinical product candidates or programs do not have sufficient therapeutic or commercial potential to warrant further advancement for a particular indication or all indications, and may elect to terminate a program for such indications or product candidates at any time. For example, we may conclude that a product candidate is not differentiated in a meaningful way from existing products, or that the costs of seeking to establish that a product candidate is differentiated would be prohibitive, or that the market size for a differentiated product with the attributes of a particular product candidate does not justify the expense and risk of further development. If we terminate a preclinical or clinical program in which we have invested significant resources, our financial condition and results of operations may be adversely affected, as we will have expended resources on a program that will not provide a return on our investment and we will have missed the opportunity to have allocated those resources to potentially more productive uses.
In particular, the failure of our MAXY-G34 or MAXY-VII product candidates in clinical development could have a material adverse impact on our business. Termination of either program may also cause the price of our stock to drop significantly.
Our clinical development strategy, which relies on third party contract research organizations, exposes us to additional risk.
We do not have the ability to independently conduct clinical trials for our product candidates in the United States and other countries, and therefore rely on third parties, such as contract research organizations, to assist us in designing our clinical trials, preparing documents for submission to regulatory authorities, obtaining regulatory approval to conduct clinical trials, enrolling qualified patients, conducting our clinical trials, and analyzing the results of such trials. If these third parties do not successfully carry out their contractual duties, do not conduct the clinical trials in accordance with planned deadlines and the approved protocol and regulatory requirements, or are unable to manage the conduct of our clinical trials effectively in compliance with FDA and other regulatory requirements, it could adversely impact the results obtained in such trials and delay the progress or completion of clinical trials, regulatory submissions and commercialization of our potential products. In any such case, we may be affected by increased costs and delays or both, which may harm our business.
Our revenues, expenses and operating results are subject to fluctuations that may cause our stock price to decline.
Our revenues, expenses and operating results have fluctuated in the past and are likely to do so in the future. These fluctuations could cause our stock price to fluctuate significantly or decline. Some of the factors that could cause our revenues, expenses and operating results to fluctuate include:
| • | the termination of research and development contracts with collaborators or government research grants, which may not be renewed or replaced; | |
| • | the success rate of our development or discovery efforts leading to milestones and royalties under future collaboration arrangements, if any; | |
| • | the timing of licensing fees or the achievement of milestones under new or existing licensing and collaborative arrangements; | |
| • | the timing of expenses, particularly with respect to contract manufacturing, preclinical studies and clinical trials; | |
| • | the timing and willingness of any future collaborators to commercialize our products, which would result in royalties to us; and | |
| • | general and industry specific economic conditions, which may affect the research and development expenditures of any future collaborator. |
18
Table of Contents
In addition, a large portion of our expenses is relatively fixed, including expenses for facilities, equipment and personnel. Accordingly, if revenues fluctuate unexpectedly due to unexpected expiration of government research grants, failure to obtain anticipated new contracts or other factors, we may not be able to immediately reduce our operating expenses, which could significantly harm our operating results for a particular fiscal period.
Due to the possibility of fluctuations in our revenues and expenses, we believe that quarter-to-quarter comparisons of our operating results are not a good indication of our future performance. Our operating results in some quarters may not meet the expectations of stock market analysts and investors. In that case, our stock price would likely decline.
Our current and future product candidates could take a long time to complete clinical development, may fail in clinical development, or may never gain approval, which could reduce or eliminate our revenue by delaying or terminating the potential commercialization of our product candidates.
The conduct of clinical trials for a single product candidate is a time-consuming, expensive and uncertain process and typically requires years to complete. In July 2007, we initiated a Phase IIa clinical trial in Eastern Europe for our MAXY-G34 product candidate for the treatment of chemotherapy-induced neutropenia. Thus, our most advanced product candidate is now only in the early stages of clinical trials.
Our product candidates or potential product candidates may produce undesirable toxicities and adverse effects in preclinical studies. Such toxicities or adverse effects could delay or prevent the filing of an IND with respect to such product candidates or potential product candidates. In clinical trials, administering any of our product candidates to humans may produce undesirable toxicities or side effects. These toxicities or side effects could interrupt, delay, suspend or terminate clinical trials of our product candidates and could result in the FDA or other regulatory authorities denying approval of our product candidates for any or all targeted indications. Indications of potential adverse effects or toxicities which may occur in clinical trials and which we believe are not significant during the course of such trials may later turn out to actually constitute serious adverse effects or toxicities when a drug has been used in large populations or for extended periods of time.
Although MAXY-G34 has demonstrated properties in preclinical and early clinical testing indicating that it may have advantages as compared to currently marketed drugs, the results from preclinical testing in vitro and animal models, as well as early, small scale clinical trials, often are not predictive of results obtained in larger later stage clinical trials designed to prove safety and efficacy. For example, after promising preclinical and early clinical data from our lead MAXY-alpha product candidate, clinical trials of this product candidate were terminated after an unexpected reduction of the pharmacodynamic and pharmacokinetic effects were observed and antibodies binding to MAXY-alpha were identified in a Phase I trial. As a result, there can be no assurances that clinical trials of any of our current or future product candidates will be completed or produce sufficient safety and efficacy data necessary to obtain regulatory approval or result in a marketed product.
In addition, the timing of the commencement, continuation or completion of clinical trials may be subject to significant delays, or a clinical trial may be suspended or delayed by us, a collaborator, the FDA or other foreign governmental agencies for various reasons, including:
| • | deficiencies in the conduct of the clinical trials; | |
| • | negative or inconclusive results from the clinical trials that necessitate additional clinical studies; | |
| • | difficulties or delays in identifying and enrolling patients who meet trial eligibility criteria; | |
| • | delays in obtaining or maintaining required approvals from institutions, review boards or other reviewing entities at clinical sites; | |
| • | inadequate supply or deficient quality of product candidate materials necessary for the conduct of the clinical trials; | |
| • | the occurrence of unacceptable toxicities or properties or unforeseen adverse side effects, especially as compared to currently approved drugs intended to treat the same indications; | |
| • | our lack of financial resources to continue the development of a product candidate; |
19
Table of Contents
| • | future legislation or administrative action or changes in FDA policy or the policy of foreign regulatory agencies during the period of product development, clinical trials and FDA regulatory review; or | |
| • | other reasons that are internal to the business of a collaborative partner, which it may not share with us. |
As a result of these risks and other factors, we may conduct lengthy and expensive clinical trials of MAXY-G34 or our other current or future product candidates, only to learn that a particular product candidate has failed to demonstrate sufficient safety or efficacy necessary to obtain regulatory approval for one or more therapeutic indications, has failed to demonstrate clinically relevant differentiation of our products from currently marketed products, does not offer therapeutic or other improvements compared to other marketed drugs, has unforeseen adverse side effects or does not otherwise demonstrate sufficient potential to make the commercialization of the product worthwhile. Any failure or substantial delay in successfully completing clinical trials, obtaining regulatory approval and commercializing our product candidates could severely harm our business.
Our potential products are subject to a lengthy and uncertain regulatory process and may never gain approval. If our potential products are not approved, we will not be able to commercialize those products.
The FDA must approve any therapeutic product or vaccine before it can be marketed in the United States. Other countries also require approvals from regulatory authorities comparable to the FDA before products can be marketed in the applicable country. Before we can file biologic license application (BLA) with the FDA or other regulatory entity, the product candidate must undergo extensive testing, including animal studies and human clinical trials, which can take many years and require substantial expenditures. Data obtained from such testing may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval.
Because our potential products involve the application of new technologies and may be based upon new therapeutic approaches, they may be subject to substantial review by government regulatory authorities and these authorities may grant regulatory approvals more slowly for our products than for products using more conventional technologies. Neither the FDA nor any other regulatory authority has approved any therapeutic product candidate developed with our MolecularBreeding directed evolution platform for commercialization in the United States or elsewhere. We, or a collaborator, may not be able to conduct clinical testing or obtain the necessary approvals from the FDA or other regulatory authorities for our products.
Regulatory approval of a BLA is never guaranteed, and the approval process typically takes several years and is extremely expensive. The FDA and other regulatory agencies also have substantial discretion in the drug approval process. Despite the time and expense exerted, failure can occur at any stage and we could encounter problems that cause us to abandon clinical trials or to repeat or perform additional preclinical testing and clinical trials. The number and focus of preclinical studies and clinical trials that will be required for approval from the FDA and other regulatory agencies varies depending on the drug candidate, the disease or condition that the drug candidate is designed to address, and the regulations applicable to any particular drug candidate. The FDA and other regulatory agencies can delay, limit or deny approval of a drug candidate for many reasons, including:
| • | a drug candidate may not be safe or effective; | |
| • | regulatory officials may not find the data from preclinical testing and clinical trials sufficient; | |
| • | the FDA and other regulatory agencies might not approve our third-party manufacturer’s processes or facilities; or | |
| • | the FDA or other regulatory agencies may change its approval policies or adopt new regulations. |
Even if we receive regulatory approval to sell a product, the approved label for a product may entail limitations on the indicated uses for which we can market a product. For example, even if MAXY-G34 is approved by the FDA, if we are not able to obtain broad labeling for this product allowing approved use with multiple chemotherapy regimens for multiple cancers, MAXY-G34 may not be adopted by hospital formularies or otherwise have limited commercial success which could have a significant adverse impact on our business. Further, once regulatory approval is obtained, a marketed product and its manufacturer are subject to continued review, and discovery of previously unknown problems or side effects associated with an approved product or the discovery of previously unknown problems with the manufacturer may result in restrictions on the product, the manufacturer or the
20
Table of Contents
manufacturing facility, including withdrawal of the product from the market. In certain countries, regulatory agencies also set or approve prices.
During the period while we are engaged in product development, the policies of the FDA and foreign regulatory entities may change and additional government laws or regulations may be enacted that could prevent or delay regulatory approval of our drug candidates. If we are not able to maintain regulatory compliance, we might not obtain approval of our products or be permitted to market our products. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. In this regard, legislation has been proposed in the United States but not yet enacted into law that would define a regulatory approval process for protein drugs that are similar to already marketed protein drugs.
Our manufacturing strategy, which relies on third-party manufacturers, exposes us to additional risks.
We do not currently have the resources, facilities or experience to manufacture any product candidates or potential products ourselves. Completion of any clinical trials and any commercialization of our products will require access to, or development of, manufacturing facilities that meet FDA standards or other regulatory requirements to manufacture a sufficient supply of our potential products. We currently depend on third parties for the scale up and manufacture of our product candidates for preclinical and clinical purposes. If our third party manufacturer is unable to manufacture preclinical or clinical supplies in a timely manner, or is unable or unwilling to satisfy our needs or FDA or other regulatory requirements, it could delay clinical trials, regulatory submissions and commercialization of our potential products, entail higher costs and possibly result in our being unable to sell our products. In addition, technical problems or other manufacturing delays could delay the advancement of potential products into preclinical or clinical trials, delay or prevent us from achieving development milestones under a collaborative agreement or result in the termination of development of particular product candidates, adversely affecting our revenues and product development timetable, which in turn could adversely affect our business and our stock price.
There are a limited number of contract manufacturers that are suitable for the manufacture of protein pharmaceuticals in compliance with current Good Manufacturing Practices (GMP) requirements and there is often limited access to such facilities. If we are unable to enter into agreements with qualified manufacturers that will provide us with our product candidates in a timely manner and at an acceptable cost, the development or commercialization of a potential product could be delayed, which would adversely affect our business.
With regard to our MAXY-G34 product candidate, we obtain polyethylene glycol (PEG) for use in making such product from Nektar Therapeutics AL, Corporation (formerly Shearwater Polymers, Inc.), a subsidiary of Nektar Therapeutics. If Nektar fails or is unable to timely supply us with PEG that meets our product needs, then we could encounter delays in the development or commercialization of MAXY-G34, which in turn could adversely affect our business and our stock price.
In addition, failure of any third party manufacturers or us to comply with applicable regulations, including pre- or post-approval inspections and the current GMP requirements of the FDA or other comparable regulatory agencies, could result in sanctions being imposed on us. These sanctions could include fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our products, delay, suspension or withdrawal of approvals, license revocation, product seizures or recalls, operational restrictions and criminal prosecutions, any of which could significantly and adversely affect our business.
The manufacturing of our product candidates presents technological, logistical and regulatory risks, each of which may adversely affect our potential revenues.
The manufacturing and manufacturing development of pharmaceuticals, and, in particular, biologicals, are technologically and logistically complex and heavily regulated by the FDA and other governmental authorities. The
21
Table of Contents
manufacturing and manufacturing development of our product candidates present many risks, including, but not limited to, the following:
| • | before we can obtain approval of any of our product candidates for the treatment of a particular disease or condition, we must demonstrate to the satisfaction of the FDA and other governmental authorities that the drug manufactured for commercial use is comparable to the drug manufactured for clinical trials and that the manufacturing facility complies with applicable laws and regulations; | |
| • | it may not be technically feasible to scale up an existing manufacturing process to meet demand or suchscale-up may take longer than anticipated; and | |
| • | failure to comply with strictly enforced GMP regulations and similar foreign standards may result in delays in product approval or withdrawal of an approved product from the market. |
Any of these factors could delay any clinical trials, regulatory submissions or commercialization of our product candidates, entail higher costs and result in our being unable to effectively sell any products.
We may need additional capital in the future. If additional capital is not available, we may have to curtail or cease operations.
We anticipate that existing cash and cash equivalents and income earned thereon, together with anticipated revenues from existing license agreements and grants, will enable us to maintain our currently planned operations for at least the next twelve months. However, our current plans and assumptions may change, and our capital requirements may increase in future periods depending on many factors, including payments received under our license agreements and government grants, the progress and scope of our research and development projects, the extent to which we advance products into and through clinical trials with our own resources, the effect of any acquisitions, and the filing, prosecution and enforcement of patent claims. Changes may also occur that would consume available capital resources significantly sooner than we expect.
We have no committed sources of capital and do not know whether additional financing will be available when needed, or, if available, that the terms will be favorable to us or our stockholders. If additional funds are not available, we may be forced to delay or terminate research or preclinical development programs, clinical trials or the commercialization of products, if any, resulting from our technologies, curtail or cease operations or obtain funds through collaborative and licensing arrangements that may require us to relinquish commercial rights or potential markets, or grant licenses on terms that are not favorable to us. If adequate funds are not available, we will not be able to successfully execute our business plan or continue our business.
If we are unable to enter into or maintain future collaboration arrangements for any of our product candidates, we may not be able to effectively develop and market some of our products.
Since we do not currently possess the resources necessary to develop and commercialize multiple products, or the resources to complete all approval processes that may be required for these potential products, we have generally sought to enter into collaborative arrangements to fund the development of new product candidates for specific indications and to develop and commercialize potential products. We are not currently party to a collaboration arrangement with respect to any of our primary product candidates and, if we are unable to enter into any new collaboration arrangements, or if any future collaboration arrangements are not maintained, our potential products may not be commercialized.
We have limited or no control over the resources that a collaborator may devote to the development and commercialization of our potential products. A collaborator may elect not to develop potential products arising out of a collaborative arrangement or not to devote sufficient resources to the development, manufacture, marketing or sale of these products. Further, a collaborator may not perform its obligations as expected and may delay the development or commercialization of a product candidate, terminate its agreement with us, or breach or otherwise fail to conduct its collaborative activities successfully and in a timely manner. If any of these events occur, we may not be able to develop or commercialize our potential products.
22
Table of Contents
In April 2007, Roche terminated its agreement with us relating to the co-development and commercialization of our MAXY-VII product candidates for acute bleeding indications due to the inability of the parties to de-risk the program in preclinical testing by failing to establish an animal model suitable to demonstrate cessation of acute bleeding in a trauma setting. In November 2007, we agreed with Roche to terminate the collaboration agreement for our MAXY-alpha product candidates due to preliminary observations of an unexpected reduction of pharmacodynamic and pharmacokinetic effects of MAXY-alpha during a Phase Ia clinical trial. We currently intend to advance one of our MAXY-VII product candidates into clinical trials for the treatment of hemophilia, and we are currently evaluating our plans for the continued development of our MAXY-VII product candidates for acute bleeding indications and the continued development of our MAXY-alpha product candidates for all indications. However, the termination of these agreements may make it more difficult or impossible for us to enter into an agreement with another third party for the development or commercialization of these product candidates for such indications. For example, if we are unable to enter into a new collaboration or licensing arrangement for the development of MAXY-VII, we may elect to discontinue further development of MAXY-VII for acute indications. If, on the other hand, we decide to use our own resources to continue the development of these product candidates for acute indications, as well as for hemophilia, our operating expenses could increase substantially, which may harm our business.
Any conflicts with a collaborator could harm our business.
An important part of our strategy involves conducting proprietary research programs. As a result, we may pursue opportunities in fields that could conflict with a future collaborator. Moreover, disagreements with a collaborator could develop over rights to our intellectual property. Any conflict with a collaborator could reduce our ability to obtain future collaboration agreements and negatively impact our relationship with a future collaborator, which could reduce our revenues.
In addition, a collaborator may market products intended to treat the medical conditions that our product candidates are planned to be used to treat, and could become our competitors in the future. For example, a collaborator could develop and commercialize competing products, fail to rapidly develop our product candidates, fail to obtain timely regulatory approvals for product commercialization, terminate their agreements with us prematurely, or fail to devote sufficient resources to allow the development and commercialization of our products. Any of these circumstances could harm our product development efforts. We have limited ability to prevent actions by any future collaborator that could have any adverse impact on the development and commercialization of our related product candidates.
Any inability to adequately protect our proprietary technologies could harm our competitive position.
Our success will depend in part on our ability to obtain patents and maintain adequate protection of our intellectual property for our technologies and products in the United States and other countries. If we do not adequately protect our intellectual property, competitors may be able to practice our technologies and erode our competitive advantage. The laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States, and many companies have encountered significant problems in protecting their proprietary rights in these foreign countries. These problems can be caused by, for example, a lack of rules and processes allowing for meaningfully defending intellectual property rights.
We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies are covered by valid and enforceable patents or are effectively maintained as trade secrets. The patent positions of biopharmaceutical and biotechnology companies, including our patent positions, are often uncertain and involve complex legal and factual questions. We apply for patents covering our technologies and potential products as we deem appropriate. However, we may not obtain patents on all inventions for which we seek patents, and any patents we obtain may be challenged and may be narrowed in scope or extinguished as a result of such challenges. Our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from practicing our technologies or from developing competing products. Enforcement of our patents against infringers could require us to expend significant amounts with no assurance that we would be successful in any litigation. Others may independently develop similar or alternative technologies or design around our patented
23
Table of Contents
technologies or products. In addition, others may challenge or invalidate our patents or our patents may fail to provide us with any competitive advantages.
Recently, the U.S. Patent and Trademark Office adopted new rules that were to become effective on November 1, 2007, regarding processes for obtaining patents in the United States. However, the U.S. District Court for the Eastern District of Virginia issued a preliminary injunction preventing implementation of the new rules until a consolidated lawsuit challenging the rules is resolved. The new rules are numerous and complex and their impact, as well as the resolution of the injunction and pending lawsuit, is still uncertain. The new rules, if made effective, generally are expected to make it more difficult for patent applicants to obtain patents, especially with regard to biotechnology products and processes. Although we do not believe that the rule changes, if made effective, would likely have a material adverse impact with regard to our MAXY-G34 or MAXY-VII programs, it may be more difficult to obtain patent protection in the United States for any future product candidates.
We also rely upon trade secret protection for our confidential and proprietary information. We have taken security measures to protect our proprietary information. These measures may not provide adequate protection for our trade secrets or other proprietary information. We seek to protect our proprietary information by entering into confidentiality agreements with employees, collaborators and consultants. Nevertheless, employees, collaborators or consultants may still disclose or misuse our proprietary information, and we may not be able to meaningfully protect our trade secrets. In addition, others may independently develop substantially equivalent proprietary information or techniques or otherwise gain access to our trade secrets.
Litigation or other proceedings or third party claims of intellectual property infringement could require us to spend time and money and could require us to shut down some of our operations.
Our ability to develop products depends in part on not infringing patents or other proprietary rights of third parties, and not breaching any licenses that we have entered into with regard to our technologies and products. In particular, others have obtained patents, and have filed, and in the future are likely to file, patent applications that may issue as patents that cover genes or gene fragments or corresponding proteins or peptides that we may wish to utilize to develop, manufacture and commercialize our product candidates. There are often multiple patents owned by third parties that cover particular proteins and related nucleic acids that are of interest to us in the development of our product candidates. For example, we are aware that Amgen, Inc. and others have issued patents and pending patent applications relating to G-CSF, and that Novo Nordisk A/S and others have issued patents and pending patent applications relating to Factor VII. To the extent that these patents, or patents that may issue in the future, cover methods or compositions that we wish to use in developing, manufacturing or commercializing our product candidates, and such use by us or on our behalf would constitute infringement of an issued valid patent claim, we would need to obtain a license from the proprietor of the relevant patent rights, which may not be available to us on acceptable terms, if at all.
Third parties may assert that we are employing their proprietary technology without authorization. In particular, our efforts to develop improved, next-generation protein pharmaceuticals could lead to allegations of patent infringement by the parties that hold patents covering other versions of such proteins or methods of making and using such proteins. In addition, third parties that do not have patents that currently cover our activities may obtain such patents in the future and then claim that our activities or product candidates infringe these patents. We could incur substantial costs and diversion of the time and attention of management and technical personnel in defending ourselves against any of these claims or enforcing our patents or other intellectual property rights against others. Furthermore, parties making claims against us may be able to obtain injunctive or other equitable relief that could effectively block our ability to further develop, commercialize and sell products. In addition, in the event of a successful claim of infringement against us, we may be required to pay damages and obtain one or more licenses from third parties. We may not be able to obtain these licenses at a reasonable cost, if at all. In that event, we could encounter delays in product introductions while we attempt to develop alternative methods or products, or be required to cease commercializing affected products.
We monitor the public disclosures of other companies operating in our industry regarding their technological development efforts. If we determine that these efforts violate our intellectual property or other rights, we intend to take appropriate action, which could include litigation. Any action we take could result in substantial costs and
24
Table of Contents
diversion of management and technical personnel. Furthermore, the outcome of any action we take to protect our rights may not be resolved in our favor.
Budget or cash constraints may force us to delay or terminate our efforts to develop certain products and could prevent us from executing our business plan, meeting our stated timetables and commercializing our potential products as quickly as possible.
Because we are an emerging company with limited resources, and because the research and development of pharmaceuticals is a long and expensive process, we must regularly assess the most efficient allocation of our research and development resources. Accordingly, we may choose to delay or terminate our research and development efforts for a promising product candidate to allocate those resources to another program, which could cause us to fall behind our timetables for development and prevent us from commercializing product candidates as quickly as possible. As a result, we may not be able to fully realize the value of some of our product candidates in a timely manner, since they will be delayed in reaching the market, or may not reach the market at all.
We are continuing our efforts to contain costs and continue to believe that strict cost containment in the near term is essential if our current funds are to be sufficient to allow us to continue our currently planned operations. We assess market conditions on an ongoing basis and plan to take appropriate actions as required. However, we may not be able to effectively contain our costs and achieve an expense structure commensurate with our business activities and revenues. As a result, we could have inadequate levels of cash for future operations or for future capital requirements, which could significantly harm our ability to operate the business.
Our revenues are primarily derived from government grants and license agreements, and our inability to maintain these grants and agreements or establish and maintain new collaborations, license agreements or grants would adversely impact our revenues, financial position and results of operation.
Our collaboration agreements with Roche for our MAXY-alpha and MAXY-VII product candidates were terminated in 2007 and we currently have two government grants and a license agreement with Codexis that we expected to generate revenue in 2008. We expect that substantially all of our revenue for the foreseeable future will result from government grants and this license agreement. If the government grants or the license agreement is materially amended or terminated and we are unable to enter into new collaboration or license agreements or obtain new grants, our revenues, financial position and results of operations would be materially adversely affected.
Other biological products may compete with our products.
If approved for sale by regulatory authorities, our next-generation protein therapeutics will likely compete with already approved earlier-generation products based on the same protein. In addition, as the patent protection for such earlier-generation protein products expires, we expect that additional products with amino acid sequences identical or substantially similar to those of the earlier-generation protein products that have lost patent protection will also enter the marketplace, and compete with such earlier generation protein products and our products. This competition may be intense, with success determined by product attributes, price and marketing power. The availability of such similar products may result in price erosion for all products of the class and could lead to limits on reimbursement for our products by third party payors.
With regard to our MAXY-G34 product candidate, we expect Neulasta and Neupogen (from Amgen, Inc.) to compete with MAXY-G34, if commercialized. In addition, we are aware that Neose Technologies, Inc. and CoGenesys, Inc. are developing G-CSF products based on naturally occurring human G-CSF.
With regard to our MAXY-VII product candidate, we expect NovoSeven (from Novo Nordisk A/S) to compete with MAXY-VII, if commercialized. In addition, we are aware that Novo Nordisk, Neose Technologies Inc., Inspiration Biopharmaceuticals, Inc. and Catalyst Biosciences, Inc. have announced that they are developing Factor VIIa based products that, if marketed, could compete with MAXY-VII.
With regard to our MAXY-4 product candidates, we expect Orencia (from Bristol Myers Squibb Company) to compete with MAXY-4, if commercialized. In addition, we are aware that Bristol Myers Squibb Company is also developing Belatacept that, if marketed, could compete with MAXY-4.
25
Table of Contents
The Committee for Medicinal Products for Human Use (CHMP) of the European Agency for the Evaluation of Medicinal Products (EMEA) has adopted guidelines for assessing the comparability of biosimilar products including G-CSF. The basis for such approvals in the European Union will be proof of comparability of the new protein drug to the prior drug, which will require clinical studies of the biosimilar protein drug.
In the United States, there is presently no legislation that specifically addresses the regulatory process for approval of biosimilar protein drugs, and to date only a biosimilar human growth hormone and certain insulin products have been approved by the FDA under a new drug application (NDA) in accordance with Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act. However, legislation has been introduced into both the U.S. Senate and House of Representatives that addresses the development path and requirements for biosimilar protein drugs. It is not clear whether such legislation will be enacted into law, and if passed, what the substance of such legislation will be. However, any law that permits the approval of biosimilars would likely lead to the eventual introduction of biosimilar protein products in the United States, which could result in increased competition for all forms of a particular therapeutic protein.
Many potential competitors who have greater resources and experience than we do may develop products and technologies that make ours obsolete.
The biotechnology industry is characterized by rapid technological change, and the area of gene research is a rapidly evolving field. Our future success will depend on our ability to maintain a competitive position with respect to technological advances. Rapid technological or product development by others may result in our products and technologies becoming obsolete.
As a company that is focused on next-generation protein therapeutic products, we face, and will continue to face, intense competition from both large and small biotechnology companies, as well as academic and research institutions and government agencies, that are pursuing competing technologies for modifying DNA and proteins. These companies and organizations may develop technologies that are alternatives to our technologies. Further, our competitors in the protein optimization field, including companies that have developed and commercialized prior versions of protein therapeutic products, may be more effective at implementing their technologies to develop commercial products. Some of these competitors have entered into collaborations with leading companies within our target markets to produce commercial products. In addition, therapeutic products that are small molecules may be developed by our competitors that could reduce or displace the market for our protein therapeutic products. Small molecule drugs are often less expensive and easier to administer than protein therapeutics and therefore would have competitive advantages if they were developed and shown to be safe and effective for the indication that our product candidates are targeting.
Even if approved by the FDA or a comparable foreign regulatory agency, any products that we develop through our technologies will compete in multiple, highly competitive markets may fail to achieve market acceptance, which would impair our ability to become profitable. Most of the companies and organizations competing with us in the markets for such products have greater capital resources, research and development and marketing staff and facilities and capabilities, and greater experience in modifying DNA and proteins, obtaining regulatory approvals, manufacturing products and marketing. Accordingly, our competitors may be able to develop technologies and products more easily, which would render our technologies and products and those of a collaborator obsolete and noncompetitive.
In addition, if any of our drug candidates are approved for commercial sale, they will need to compete with other products intended to treat the same disease, including the marketed versions of the protein therapeutic drug that we have sought to improve, and possibly including other variant versions of such drug, and generic bioequivalent or biosimilar versions of such drugs, and small molecule drugs. Such competition may be intense and lead to price reductions for all forms of a particular therapeutic protein. Moreover, any adverse developments related to a currently marketed version of the protein therapeutic drug that we have sought to improve or a generic bioequivalent or biosimilar version of such drug may have a significant adverse impact on the continued development or future commercialization and marketing of our related product candidates and could cause us to change our development plans or discontinue further development of such product candidates. If we are unable to market and commercialize our product successfully, our business would be adversely affected.
26
Table of Contents
Legislative actions, new accounting pronouncements and higher compliance costs may adversely impact our future financial position and results of operations.
Future changes in financial accounting standards may cause adverse, unexpected earnings fluctuations and may adversely affect our reported results of operations. For example, our implementation of Statement of Financial Accounting Standard No. 123 (revised 2004), “Share-Based Payment,” or SFAS 123(R) in 2006 had a material impact on our consolidated results of operations and net loss per share for the years ended December 31, 2006 and 2007 and is expected to have a material impact on our results of operations in the future. The continued impact of expensing stock-based compensation will depend in part upon the timing and amount of future equity compensation awards. New accounting pronouncements and varying interpretations of such pronouncements have occurred with frequency in the recent past and may occur in the future. In addition, we may make changes in our accounting policies in the future.
In addition, compliance with changing regulations regarding corporate governance and public disclosure may also result in additional expenses. Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002 and related SEC regulations and Nasdaq Global Market listing requirements, have often created uncertainty for companies such as ours and compliance costs generally have increased as a result of this uncertainty and other factors. We are committed to maintaining high standards of corporate governance and public disclosure. As a result, we intend to invest all reasonably necessary resources to comply with evolving standards, and this investment may result in increased general and administrative expenses and cause a diversion of management time and attention from revenue-generating activities to compliance activities.
If we do not attract and retain key employees, our business could be impaired.
To be successful and achieve our objectives, we must attract and retain qualified scientific and management personnel. If we are unsuccessful in attracting and retaining qualified personnel, particularly at the management level, our business could be impaired. We have been successful in hiring and retaining key personnel in the past; however, we face significant competition for experienced, management level personnel. Although we believe have been successful in attracting and retaining qualified personnel, competition for experienced management personnel and scientists from numerous companies and academic and other research institutions may limit our ability to do so in the future on acceptable terms. Failure to attract and retain personnel could prevent us from pursuing collaborations or developing our products or core technologies.
The operation of international locations may increase operating expenses and divert management attention.
Since 2000, we have conducted certain of our operations through Maxygen ApS, our Danish subsidiary. Although we are currently in the final stages of consolidating our operations in the United States, we will continue to conduct certain limited operations through Maxygen ApS until the consolidation is complete and may continue to conduct limited administrative activities through Maxygen ApS for the foreseeable future. As a result, we will continue to face certain risks related to the operation of a foreign subsidiary. Operation as an international entity requires additional management attention and resources. As long as we continue to operate internationally, we are subject to risks of doing business internationally, including compliance with foreign regulatory and legal requirements; difficulties in staffing and managing foreign operations; currency exchange risks; and potentially adverse tax consequences.
In addition, the consolidation of our operations has required us to transfer certain assets and functions from our Danish facility to our U.S. facility and hire additional employees in the United States. Any failure or delay in effectively transferring such assets or functions or hiring and integrating additional employees in the United States could hinder or delay the development of product candidates and research programs, which could adversely affect our business. We may also incur other material charges not currently contemplated due to events that may occur as a result of, or associated with, the consolidation that could adversely affect our financial position and results of operations.
27
Table of Contents
Acquisitions could result in dilution, operating difficulties and other harmful consequences.
If appropriate opportunities present themselves, we may acquire businesses or technologies that complement our capabilities. The process of integrating any acquisition may create unforeseen operating difficulties and expenditures and is itself risky. The areas where we may face difficulties include:
| • | diversion of management time (both ours and that of the acquired company) from focus on operating the businesses to issues of integration during the period of negotiation through closing and further diversion of such time after closing; | |
| • | decline in employee morale and retention issues resulting from changes in compensation, reporting relationships, future prospects, or the direction of the business; | |
| • | the need to integrate each company’s accounting, management information, human resource and other administrative systems to permit effective management and the lack of control if such integration is delayed or not implemented; and | |
| • | the need to implement controls, procedures and policies appropriate for a larger public company in companies that before acquisition had been smaller, private companies. |
We do not have extensive experience in managing this integration process. Moreover, the anticipated benefits of any or all of these acquisitions may not be realized.
Future acquisitions could result in potentially dilutive issuances of equity securities, the incurrence of debt, contingent liabilities or amortization expenses related to intangible assets, any of which could harm our business or adversely affect our results of operations. Future acquisitions may require us to obtain additional equity or debt financing, which may not be available on favorable terms or at all. Even if available, this financing may be dilutive.
Our stock price has been, and may continue to be, extremely volatile, and an investment in our stock could decline in value.
The trading prices of life science company stocks in general, and ours in particular, have experienced significant price fluctuations in the last several years. During 2007, the price of our common stock on the Nasdaq Global Market ranged from $6.12 to $12.41. The valuations of many life science companies without product revenues and earnings, including ours, are based on valuation standards such as price to sales ratios and progress in product development or clinical trials. Trading prices based on these valuations may not be sustained. Any negative change in the public’s perception of the prospects of biotechnology or life science companies could depress our stock price regardless of our results of operations. Other broad market and industry factors may decrease the trading price of our common stock, regardless of our performance. In addition, our stock price could be subject to wide fluctuations in response to factors including the following:
| • | our failure to meet our publicly announced revenueand/or expense projectionsand/or product development timetables; | |
| • | adverse or inconclusive results or delays in preclinical development or clinical trials; | |
| • | any entry into or material amendment or termination of a collaborative or license agreement; | |
| • | any decisions to discontinue or delay development programs or clinical trials; | |
| • | announcements of new technological innovations or new products by us or our competitors; | |
| • | conditions or trends in the biotechnology and life science industries; | |
| • | changes in the market valuations of other biotechnology or life science companies; | |
| • | developments in domestic and international governmental policy or regulations; | |
| • | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; | |
| • | changes in general economic, political and market conditions, such as recessions, interest rate changes, terrorist acts and other factors; |
28
Table of Contents
| • | developments in or challenges relating to our patent or other proprietary rights, including lawsuits or proceedings alleging patent infringement based on the development, manufacturing or commercialization of our product candidates; and | |
| • | sales of our common stock or other securities in the open market. |
In the past, stockholders have often instituted securities class action litigation after periods of volatility in the market price of a company’s securities. If a stockholder files a securities class action suit against us, we could incur substantial legal fees and our management’s attention and resources would be diverted from operating our business to respond to the litigation.
Substantial sales of shares may adversely impact the market price of our common stock.
If our stockholders sell substantial amounts of our common stock, including shares issued upon the exercise of outstanding options, the market price of our common stock may decline. Our common stock trading volume is low and thus the market price of our common stock is particularly sensitive to trading volume. Our low trading volume may also make it more difficult for us to sell equity or equity related securities in the future at a time and price that we deem appropriate. Significant sales of our common stock may adversely impact the then-prevailing market price of our common stock.
If we or a collaborator receives regulatory approval for one of our drug candidates, we will be subject to ongoing FDA obligations and continued regulatory review, and we may also be subject to additional FDA post-marketing obligations, all of which may result in significant expense and limit our ability to commercialize our potential drugs.
Any regulatory approvals that we or a collaborator receives for one of our product candidates may also be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for potentially costly post-marketingfollow-up studies. In addition, if the FDA or a foreign regulatory agency approves any of our drug candidates, the labeling, packaging, adverse event reporting, storage, advertising, promotion, and record keeping for the product will be subject to extensive regulatory requirements. The subsequent discovery of previously unknown problems with the product, including adverse events of unanticipated severity or frequency, may result in restriction on the marketing of the product, and could include withdrawal of the drug from the market.
We may be subject to costly product liability claims and may not have adequate insurance.
Because we conduct clinical trials in humans, we face the risk that the use of our product candidates will result in adverse effects. We currently maintain product liability insurance for our clinical trials, however, such liability insurance may not be adequate to fully cover any liabilities that arise from clinical trials of our product candidates. We may not have sufficient resources to pay for any liabilities resulting from a claim excluded from, or beyond the limit of, our insurance coverage.
We currently have no product marketing capabilities.
We plan to commercialize products resulting from our proprietary programs either directly or through licensing to other companies or co-promotion with other companies. We have no experience in marketing, and we currently do not have the resources or capability to market products. In order for us to commercialize these products directly, we would need to develop, or obtain through outsourcing arrangements, the capability to market and sell products, which could require significant capital investment. We do not have these capabilities, and we may not be able to develop or otherwise obtain the requisite marketing and sales capabilities. If we are unable to successfully commercialize products resulting from our proprietary research efforts, we will continue to incur losses.
The coverage and reimbursement status of newly approved drugs is uncertain and failure to obtain adequate coverage and reimbursement could limit our ability to market any drugs we may develop and decrease our ability to generate revenue.
There is significant uncertainty related to the coverage and reimbursement of newly approved drugs. The commercial success of our potential drugs in both domestic and international markets is substantially dependent on whether third-party coverage and reimbursement is available for the ordering of our potential drugs by the medical profession for use by their patients. Medicare, Medicaid, health maintenance organizations and other third-party
29
Table of Contents
payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new drugs, and, as a result, they may not cover or provide adequate payment for our potential drugs. They may not view our potential drugs as cost-effective and reimbursement may not be available to consumers or may not be sufficient to allow our potential drugs to be marketed on a competitive basis. Likewise, legislative or regulatory efforts to control or reduce healthcare costs or reform government healthcare programs could result in lower prices or rejection of our potential drugs. Changes in coverage and reimbursement policies or healthcare cost containment initiatives that limit or restrict reimbursement for our drugs may cause any revenue from a potential drug to decline.
Some of our existing stockholders can exert control over us, and may not make decisions that are in the best interests of all stockholders.
As of December 31, 2007, our executive officers and directors, together with GlaxoSmithKline plc, controlled approximately 22% of our outstanding common stock. As a result, these stockholders, if they act together, and GlaxoSmithKline plc, which owns approximately 18% of our outstanding common stock, by itself, could exert a significant degree of influence over our management and affairs and over matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. In addition, this concentration of ownership may delay or prevent a change in control of our company and might affect the market price of our common stock, even when a change may be in the best interests of all stockholders. In addition, the interests of this concentration of ownership may not always coincide with our interests or the interests of other stockholders and accordingly, they could cause us to enter into transactions or agreements that we would not otherwise consider. This concentration of ownership could also depress our stock price.
Our facilities in California are located near an earthquake fault, and an earthquake or other types of natural disasters or resource shortages could disrupt our operations and adversely affect our results.
Our U.S. facilities are located in our corporate headquarters in Redwood City, California near active earthquake zones. We do not have a formal business continuity or disaster recovery plan, and in the event of a natural disaster, such as an earthquake or localized extended outages of critical utilities or transportation systems, we could experience a significant business interruption.
| Item 1B | UNRESOLVED STAFF COMMENTS |
Not applicable.
| Item 2 | PROPERTIES |
As of March 1, 2008, we leased an aggregate of 56,980 square feet of office and laboratory facilities in Redwood City, California. Our leases expire on February 28, 2009 and include options to extend for an additional one-year term. We also lease an aggregate of 26,275 square feet of office and laboratory facilities in Horsholm, Denmark. The lease for our Danish facilities is scheduled to be terminated as of May 31, 2008 in connection with the cessation of research and development operations at Maxygen ApS.
We believe that our existing facilities are adequate to meet our needs for the immediate future. We believe that we can accommodate future growth, if any, by leasing additional or alternative space. For additional information regarding our lease obligations, see Note 7 of the Notes to Consolidated Financial Statements.
| Item 3 | LEGAL PROCEEDINGS |
The information included in Note 10 of the Notes to Consolidated Financial Statements in Part II — Item 8 of this report is incorporated herein by reference.
| Item 4 | SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS |
Not applicable.
30
Table of Contents
Part II
| Item 5 | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock has been traded on the Nasdaq Global Market under the symbol “MAXY” since December 16, 1999. During the last two fiscal years, through December 31, 2007, the high and low sale prices for our common stock, as reported on the Nasdaq Global Market, were as follows:
| High | Low | |||||||
| Year ended December 31, 2007 | ||||||||
| First Quarter | $ | 12.41 | $ | 10.00 | ||||
| Second Quarter | 11.94 | 8.33 | ||||||
| Third Quarter | 9.56 | 6.49 | ||||||
| Fourth Quarter | 8.94 | 6.12 | ||||||
| Year ended December 31, 2006 | ||||||||
| First Quarter | $ | 8.95 | $ | 7.45 | ||||
| Second Quarter | 8.44 | 7.01 | ||||||
| Third Quarter | 8.78 | 6.78 | ||||||
| Fourth Quarter | 10.98 | 7.97 | ||||||
Holders
As of February 29, 2008, there were 226 holders of record of our common stock, one of which is Cede & Co., a nominee for Depository Trust Company (“DTC”). All of the shares of common stock held by brokerage firms, banks and other financial institutions as nominees for beneficial owners are deposited into participant accounts at DTC, and therefore, are considered to be held of record by Cede & Co. as one stockholder.
Dividends
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain future earnings, if any, for development of our business and, therefore, do not anticipate that we will declare or pay cash dividends on our capital stock in the foreseeable future.
31
Table of Contents
Company Stock Price Performance(1)
The following graph shows the cumulative total stockholder return of an investment of $100 in cash on December 31, 2002 through December 31, 2007 for (i) our common stock, (ii) the Nasdaq Composite Index and (iii) the Nasdaq Biotechnology Index. All values assume reinvestment of the full amount of all dividends. Stockholder returns over the indicated period should not be considered indicative of future stockholder returns.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among Maxygen, Inc., The NASDAQ Composite Index
And The NASDAQ Biotechnology Index
Among Maxygen, Inc., The NASDAQ Composite Index
And The NASDAQ Biotechnology Index
| * | $100 invested on 12/31/02 in stock or index-including reinvestment of dividends. |
Fiscal year ending December 31.
| Total Return Analysis | ||||||||||||||||||||||||
| 12/31/2002 | 12/31/2003 | 12/31/2004 | 12/31/2005 | 12/31/2006 | 12/31/2007 | |||||||||||||||||||
| Maxygen, Inc. | 100.00 | 139.50 | 167.85 | 98.56 | 141.34 | 105.38 | ||||||||||||||||||
| Nasdaq Composite Index | 100.00 | 149.75 | 164.64 | 168.60 | 187.83 | 205.22 | ||||||||||||||||||
| Nasdaq Biotechnology Index | 100.00 | 146.95 | 164.05 | 185.29 | 183.09 | 186.22 | ||||||||||||||||||
| (1) | The material in this section is not “soliciting material,” is not deemed “filed” with the SEC and is not to be incorporated by reference in any of our filings under the Securities Act or the Exchange Act whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. |
32
Table of Contents
| Item 6 | SELECTED FINANCIAL DATA |
The following selected financial information is derived from our audited consolidated financial statements. When you read this selected financial data, it is important that you also read the historical financial statements and related notes included in this report, as well as the section of this report entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” Historical results are not necessarily indicative of future results. Historical results include the consolidated operations of Codexis, Inc. for all periods through February 28, 2005. After that date, we account for Codexis, Inc. under the equity method of accounting.
| Year Ended December 31, | ||||||||||||||||||||
| 2003 | 2004 | 2005 | 2006 | 2007 | ||||||||||||||||
| (In thousands, except per share data) | ||||||||||||||||||||
Consolidated Statements of Operations Data: | ||||||||||||||||||||
| Collaborative research and development revenue | $ | 20,573 | $ | 14,333 | $ | 11,594 | $ | 20,544 | $ | 10,232 | ||||||||||
| Revenue from related party | — | — | — | — | 8,286 | |||||||||||||||
| Grant revenue | 2,282 | 1,942 | 2,907 | 4,477 | 4,639 | |||||||||||||||
| Total revenues | 22,855 | 16,275 | 14,501 | 25,021 | 23,157 | |||||||||||||||
| Operating expenses: | ||||||||||||||||||||
| Research and development | 45,949 | 53,586 | 41,904 | 49,130 | 59,851 | |||||||||||||||
| General and administrative | 11,831 | 14,435 | 13,221 | 17,559 | 14,951 | |||||||||||||||
| Restructuring charge | — | — | — | — | 5,212 | |||||||||||||||
| Amortization of goodwill and other intangible assets | 698 | — | — | — | — | |||||||||||||||
| Total operating expenses | 58,478 | 68,021 | 55,125 | 66,689 | 80,014 | |||||||||||||||
| Loss from operations | (35,623 | ) | (51,746 | ) | (40,624 | ) | (41,668 | ) | (56,857 | ) | ||||||||||
| Interest income and other income (expense), net | 5,253 | 4,055 | 5,572 | 8,524 | 7,542 | |||||||||||||||
| Equity in net loss of minority investee | (500 | ) | (1,395 | ) | — | (1,000 | ) | — | ||||||||||||
| Gain on sale of equity investment(1) | — | — | — | 17,662 | — | |||||||||||||||
| Loss from continuing operations | (30,870 | ) | (49,086 | ) | (35,052 | ) | (16,482 | ) | (49,315 | ) | ||||||||||
| Discontinued operations: | ||||||||||||||||||||
| Loss from discontinued operations | (1,586 | ) | (2,769 | ) | — | — | — | |||||||||||||
| Gain on sale of discontinued operations (net of taxes and transaction costs) | — | 61,197 | — | — | — | |||||||||||||||
| Income (loss) from discontinued operations | (1,586 | ) | 58,428 | — | — | — | ||||||||||||||
| Cumulative effect adjustment(2) | — | — | 16,616 | — | — | |||||||||||||||
| Net income (loss) | (32,456 | ) | 9,342 | (18,436 | ) | (16,482 | ) | (49,315 | ) | |||||||||||
| Subsidiary preferred stock accretion | (1,279 | ) | (1,000 | ) | (167 | ) | — | — | ||||||||||||
| Income (loss) applicable to common stockholders | $ | (33,735 | ) | $ | 8,342 | $ | (18,603 | ) | $ | (16,482 | ) | $ | (49,315 | ) | ||||||
| Basic and diluted income (loss) per share: | ||||||||||||||||||||
| Continuing operations | $ | (0.89 | ) | $ | (1.40 | ) | $ | (0.98 | ) | $ | (0.46 | ) | $ | (1.34 | ) | |||||
| Discontinued operations | $ | (0.05 | ) | $ | 1.66 | $ | — | $ | — | $ | — | |||||||||
| Cumulative effect adjustment | $ | — | $ | — | $ | 0.46 | $ | — | $ | — | ||||||||||
| Applicable to common stockholders | $ | (0.98 | ) | $ | 0.24 | $ | (0.52 | ) | $ | (0.46 | ) | $ | (1.34 | ) | ||||||
| Shares used in basic and diluted per share calculations | 34,519 | 35,176 | 35,765 | 36,046 | 36,787 | |||||||||||||||
| (1) | The gain on sale of equity investment in the year ended December 31, 2006 resulted from the net gain on the disposal of our investment in Avidia (see Note 1 of Notes to Consolidated Financial Statements). | |
| (2) | The cumulative effect adjustment in the year ended December 31, 2005 resulted from the deconsolidation of Codexis, Inc. as of February 28, 2005 (see Note 1 of Notes to Consolidated Financial Statements). |
33
Table of Contents
| December 31, | ||||||||||||||||||||
| 2003 | 2004 | 2005 | 2006 | 2007 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
Consolidated Balance Sheet Data: | ||||||||||||||||||||
| Cash, cash equivalents and investments | $ | 191,868 | $ | 232,893 | $ | 188,323 | $ | 182,876 | $ | 145,813 | ||||||||||
| Working capital | 115,724 | 211,999 | 152,230 | 175,356 | 138,171 | |||||||||||||||
| Total assets | 234,069 | 263,105 | 214,523 | 205,647 | 172,709 | |||||||||||||||
| Non-current portion of equipment financing | — | 1,751 | — | — | — | |||||||||||||||
| Minority interest | 21,210 | 32,180 | — | — | — | |||||||||||||||
| Accumulated deficit | (195,128 | ) | (185,786 | ) | (204,222 | ) | (220,704 | ) | (270,019 | ) | ||||||||||
| Total stockholders’ equity | 198,224 | 211,341 | 197,344 | 189,799 | 153,494 | |||||||||||||||
QUARTERLY FINANCIAL DATA
| Quarter Ended | ||||||||||||||||
| March 31, | June 30, | Sept. 30, | Dec. 31, | |||||||||||||
| (In thousands, except per share data) | ||||||||||||||||
| (Unaudited) | ||||||||||||||||
2007 | ||||||||||||||||
| Collaborative research and development revenue | $ | 7,597 | $ | 874 | $ | 5 | $ | 1,756 | ||||||||
| Revenue from related party | 556 | — | 34 | 7,696 | ||||||||||||
| Grant revenue | 939 | 1,207 | 964 | 1,529 | ||||||||||||
| Total revenues | 9,092 | 2,081 | 1,003 | 10,981 | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 14,600 | 16,343 | 13,570 | 15,338 | ||||||||||||
| General and administrative | 4,331 | 3,528 | 3,951 | 3,141 | ||||||||||||
| Restructuring charge | — | — | — | 5,212 | ||||||||||||
| Total operating expenses | 18,931 | 19,871 | 17,521 | 23,691 | ||||||||||||
| Loss from operations | (9,839 | ) | (17,790 | ) | (16,518 | ) | (12,710 | ) | ||||||||
| Interest income and other income (expense), net | 2,174 | 2,118 | 1,814 | 1,436 | ||||||||||||
| Net income (loss) | $ | (7,665 | ) | $ | (15,672 | ) | $ | (14,704 | ) | $ | (11,274 | ) | ||||
| Basic and diluted income (loss) per share | $ | (0.21 | ) | $ | (0.43 | ) | $ | (0.40 | ) | $ | (0.31 | ) | ||||
| Shares used in basic and diluted per share calculations | 36,586 | 36,791 | 36,872 | 36,898 | ||||||||||||
34
Table of Contents
| Quarter Ended | ||||||||||||||||
| March 31, | June 30, | Sept. 30, | Dec. 31, | |||||||||||||
| (In thousands, except per share data) | ||||||||||||||||
| (Unaudited) | ||||||||||||||||
2006 | ||||||||||||||||
| Collaborative research and development revenue | $ | 4,067 | $ | 7,950 | $ | 3,201 | $ | 5,326 | ||||||||
| Grant revenue | 898 | 1,355 | 1,040 | 1,184 | ||||||||||||
| Total revenues | 4,965 | 9,305 | 4,241 | 6,510 | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 13,262 | 10,212 | 12,020 | 13,636 | ||||||||||||
| General and administrative | 4,318 | 4,065 | 4,547 | 4,629 | ||||||||||||
| Total operating expenses | 17,580 | 14,277 | 16,567 | 18,265 | ||||||||||||
| Loss from operations | (12,615 | ) | (4,972 | ) | (12,326 | ) | (11,755 | ) | ||||||||
| Interest income and other income (expense), net | 1,893 | 1,931 | 2,267 | 2,433 | ||||||||||||
| Equity in net loss of minority investee | — | (342 | ) | (658 | ) | — | ||||||||||
| Gain on sale of equity investment(1) | — | — | — | 17,662 | ||||||||||||
| Net income (loss) | $ | (10,722 | ) | $ | (3,383 | ) | $ | (10,717 | ) | $ | 8,340 | |||||
| Basic and diluted income (loss) per share | $ | (0.30 | ) | $ | (0.09 | ) | $ | (0.30 | ) | $ | 0.23 | |||||
| Shares used in basic and diluted per share calculations | 35,973 | 36,024 | 36,078 | 36,109 | ||||||||||||
| (1) | The gain on sale of equity investment in the year ended December 31, 2006 resulted from the net gain on the disposal of our investment in Avidia (see Note 1 of Notes to Consolidated Financial Statements). |
| Item 7 | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following discussion should be read in conjunction with our consolidated financial statements and the related notes and other financial information appearing elsewhere in this report. This report contains forward-looking statements that involve risks and uncertainties. Our actual results may differ materially from those indicated in forward-looking statements. See “Forward-Looking Statements” and “Risk Factors.”
Overview
We are a biotechnology company committed to the discovery and development of improved next-generation protein pharmaceuticals for the treatment of disease and serious medical conditions. We began operations in March 1997 with the mission to develop important commercial products through the use of biotechnology. Since then, we have established a focus in human therapeutics, particularly on the development of optimized protein pharmaceuticals. Three of our next-generation product candidates are currently in clinical or preclinical development: MAXY-G34, a pegylated, granulocyte colony stimulating factor (G-CSF) product for the treatment of neutropenia; MAXY-VII, a factor VIIa product for the treatment of hemophilia and possibly acute bleeding conditions; and MAXY-4, a CTLA-4-Ig product for the treatment of rheumatoid arthritis and other immune or autoimmune diseases.
In addition to our clinical and preclinical stage product candidates, we have other research stage programs and assets outside of our core business, including research on certain vaccine programs.
We use our technology platform, MolecularBreeding directed evolution, along with PEGylation, glycosylation, rational design and mutagenesis, in an effort to create improved, proprietary proteins. Our MolecularBreeding technology platform, also referred to as DNA shuffling, can be used to create new versions of any known protein. We believe that once a desired improvement has been identified, the chance of being able to create a novel protein with that desired improvement is high.
35
Table of Contents
To date, we have generated revenues from collaborations with pharmaceutical, chemical and agriculture companies and from government grants. However, over the past several years, we have strategically shifted our focus to pharmaceutical products and believe this is an important step in building long-term value in our company. Revenues from our collaboration agreements were $10.2 million, $20.5 million and $11.6 million in 2007, 2006 and 2005, respectively. During 2007, we also recorded $8.3 million in revenue from related party under our license agreement with Codexis. These revenues reflect amounts due to us from payments received by Codexis under its collaboration arrangement with Shell Oil Products US that began in November 2006 and $7.5 million recognized in the fourth quarter of 2007 in connection with an expanded collaboration agreement between Royal Dutch Shell plc and Codexis for the development of new enzymes to convert biomass to fuel. We expect our total revenues to decrease in 2008 compared to 2007, primarily due to the loss of collaborative research and development revenue under our co-development agreement with Roche for our MAXY-VII product candidates, which Roche terminated in April 2007. We also expect much lower revenue in 2008 under our license agreement with Codexis since a significant portion of the revenue we recorded under this agreement in 2007 related to a one-time upfront payment that Codexis received from Shell. Due to the nature of our research, our revenue may fluctuate substantially from year to year, based on the completion of new licensing or collaborative agreements and the achievement of development related milestones. As a result, due to the uncertain nature of the events generating the revenue, we cannot predict with any certainty whether we will enter into new collaboration arrangements or receive any future milestone payments or royalty payments under such collaborations or whether any independent research effort or future collaboration will ultimately result in a commercial product.
We have incurred significant operating losses from continuing operations since our inception. As of December 31, 2007, our accumulated deficit was $270.0 million. We have invested heavily in establishing our proprietary technologies. Our research and development expenses for 2007 were $59.9 million, compared to $49.1 million in 2006 and $41.9 million in 2005 (including $2.5 million of research and development expenses attributable to Codexis, our former chemicals segment, in 2005). We expect to incur additional operating losses over at least the next several years and may never achieve profitability.
We continue to maintain a strong cash position to fund our expanded product development efforts, with cash, cash equivalents and marketable securities totaling $145.8 million as of December 31, 2007.
Major Developments in 2007
In July 2007, we commenced a Phase IIa clinical trial of MAXY-G34 in breast cancer patients in Eastern Europe. We announced positive progress in this clinical trial in January 2008, including that MAXY-G34 showed clinical activity at the lowest dose level of 10 µg/kg and that no immunogenicity or serious adverse events were observed.
In May 2007, we announced our plans to proceed with the clinical development of our lead MAXY-VII product candidate for hemophilia. We currently plan to file our first clinical trial application (CTA) in the first half of 2008 and commence a first in human clinical trial in hemophilia patients in the second half of 2008.
In November 2007, we announced the preclinical development of our most recent product candidate,MAXY-4, for the treatment of rheumatoid arthritis. We plan to conductin vivoproof-of-concept preclinical studies with our MAXY-4 product candidates and select one or more lead product candidates in 2008.
During 2007, we recognized approximately $8.3 million in revenue under our license agreement with Codexis, including $7.5 million recognized in the fourth quarter of 2007 in connection with an expanded collaboration agreement between Royal Dutch Shell plc and Codexis for the development of new enzymes to convert biomass to fuel.
In December 2007, we licensed our proprietary dengue virus antigen technology to sanofi pasteur, the vaccines division of the sanofi-aventis Group for development and worldwide commercialization of a second-generation vaccine. We received an upfront fee and are eligible to receive up to an additional $23.0 million of event-based payments under the agreement, as well as royalties on any product sales.
In April 2007, Roche terminated the collaboration agreement for our MAXY-VII product candidates due to the inability of the parties to de-risk the program in preclinical testing by failing to establish an animal model suitable to
36
Table of Contents
demonstrate cessation of acute bleeding in a trauma setting. We entered into this agreement with Roche in December 2005. Revenues under this agreement constituted a substantial portion of our total revenues in 2006 and 2007 and we expected this agreement to provide a substantial portion of our revenue for the next several years. In addition to an upfront fee, revenues earned as net reimbursement of our research and development activities under the agreement and a $5 million milestone payment received in 2006, we were also eligible to receive additional milestone payments under this agreement of up to $82 million upon the achievement of development and other milestones, as well as royalties on any product sales.
In November 2007, we agreed with Roche to terminate the collaboration agreement for our MAXY-alpha product candidates due to preliminary observations of an unexpected reduction of pharmacodynamic and pharmacokinetic effects of MAXY-alpha during a Phase Ia clinical trial. We entered into this agreement with Roche in May 2003. We received an initial payment and full research and development funding for the first two years of this agreement. In addition, we received $9 million of milestone payments from Roche during the term of the agreement and we were eligible to receive additional milestone payments of up to $50 million, as well as royalties on any product sales.
Also in November 2007, we implemented a plan to consolidate our research and development activities at our U.S. facilities in Redwood City, California in an effort to reduce costs and increase overall operational efficiency across our research, preclinical, clinical and regulatory activities. The consolidation has resulted in the cessation of research and development operations at Maxygen ApS, our wholly owned subsidiary in Denmark. As a result of the consolidation, we have recorded charges of approximately $5.2 million relating primarily to one-time termination benefits for the affected employees of Maxygen ApS and related expenses and we expect to incur additional costs of approximately $1.0 million relating to the consolidation and expect to complete the activities related to this consolidation during the second quarter of 2008.
Background
We began operations in 1997 to commercialize technologies originally conceived at Affymax Research Institute, then a subsidiary of what is now GlaxoSmithKline plc. Our operations were originally focused on the application of our MolecularBreeding directed evolution platform and other technologies to the development of multiple products in a broad range of industries, including human therapeutics, chemicals and agriculture. In August 2000, to complement and expand our human therapeutics operations, we established our Danish subsidiary, Maxygen ApS, through the acquisition of ProFound Pharma A/S, a privately held Danish biotechnology company. In 2002, prior to our focus on human therapeutics, we established two industrial subsidiaries, Codexis, Inc., or Codexis, and Verdia, Inc., or Verdia.
We established Codexis to focus on the development of biocatalysis and fermentation processes and advanced small-molecule pharmaceutical intermediaries for the pharmaceutical industry. Codexis received financing from third party investors and operated as an independent subsidiary beginning in September 2002. In February 2005, our voting rights in Codexis were reduced below 50%. As a result, we no longer consolidate the financial position and results of operations of Codexis with our financial results as of such date and instead account for Codexis under the equity method of accounting. To reflect what our basis in Codexis would have been under equity accounting, we recorded a cumulative effect adjustment of $16.6 million in the first quarter of 2005, which reduced our net loss in 2005 and brought our investment basis in Codexis to zero as of February 28, 2005. At December 31, 2007, we had an equity interest in Codexis of approximately 25% of its outstanding capital stock. We are not obligated to fund the operations or other capital requirements of Codexis.
We established Verdia to focus on the development of processes and products for the agricultural industry. On July 1, 2004, we completed the sale of Verdia to Pioneer Hi-Bred International, Inc., a wholly-owned subsidiary of E.I. du Pont de Nemours and Company, for cash proceeds of $64.0 million.
In July 2003, we established Avidia Inc. (formerly Avidia Research Institute), or Avidia, together with third-party investors. Avidia was formed as a spin-out of Maxygen to focus on the development of a new class of subunit proteins as therapeutic products. We also received equity interests in Avidia through our initial contribution of technology and funding and our participation in subsequent preferred stock financings of Avidia. On October 24, 2006, Amgen Inc. completed the acquisition of Avidia and Avidia became a wholly owned subsidiary of Amgen Inc.
37
Table of Contents
At the time of the acquisition of Avidia by Amgen Inc., our basis in Avidia was zero. As a result of the acquisition, we received cash proceeds of approximately $17.8 million (before $140,000 of income taxes) in the fourth quarter of 2006 in exchange for our equity interests in Avidia and may receive up to an additional $1.4 million in cash, contingent upon the development of certain Avidia products by Amgen Inc. This contingent amount was reduced from $2.8 million based on the discontinuation by Amgen Inc. of certain development activities. Under an agreement that we entered into with Avidia at the time of Avidia’s formation, we have retained certain exclusive and non-exclusive rights to use Avidia technology to develop and commercialize products directed to certain specific targets.
As discussed above, we implemented a plan in November 2007 to consolidate our research and development activities at our U.S. facilities in Redwood City, California resulting in the cessation of research and development operations at Maxygen ApS.
For the purposes of this report, our continuing operations consist of the results of Maxygen, Inc. and its wholly-owned subsidiaries, Maxygen ApS (Denmark) and Maxygen Holdings Ltd. (Cayman Islands), as well as the results of Codexis through February 28, 2005.
Critical Accounting Policies and Estimates
General
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make judgments, estimates and assumptions in the preparation of our consolidated financial statements and accompanying notes (see Note 1 of Notes to Consolidated Financial Statements). Actual results could differ from those estimates. We believe the following are our critical accounting policies, including those that reflect the more significant judgments, estimates and assumptions we make in the preparation of our consolidated financial statements.
Goodwill and Intangible Impairment
Goodwill and other intangible assets are generally evaluated on an individual acquisition or market basis at least annually whenever events or changes in circumstances indicate that such assets are impaired or the estimated useful lives are no longer appropriate. We review our intangible assets (including goodwill) for impairment at least annually based on estimated future discounted cash flows attributable to the assets and other factors to determine the fair value of the respective assets. In the event such cash flows are not expected to be sufficient to recover the recorded value of the assets, the assets will be written down to their estimated fair values. No impairment charges were recorded in 2005, 2006 or 2007.
The valuation in connection with the initial purchase price allocation and the ongoing evaluation for impairment of goodwill and intangible assets requires significant management estimates and judgment. The purchase price allocation process requires management estimates and judgment as to expectations for various products and business strategies. If any of the significant assumptions differ from the estimates and judgments used in the purchase price allocation, this could result in different valuations for goodwill and intangible assets. Once it is established, we must test goodwill annually for impairment using a two-step process as required by Statement of Financial Accounting Standard (SFAS) No. 142 “Goodwill and Other Intangible Assets,” (SFAS 142). In addition, in certain circumstances, we must assess if goodwill should be tested for impairment between annual tests. Intangible assets with definite useful lives must be tested for impairment in accordance with SFAS No. 144 “Accounting for the Impairment or Disposal of Long-Lived Assets.” When we conduct our impairment tests for goodwill and intangibles, factors that are considered important in determining whether impairment might exist include existing product portfolio, product development cycle, development expenses, potential royalties and product sales, costs of goods and selling expenses and overall product lifecycle. Any changes in key assumptions about the business and its prospects, or changes in market conditions or other external events, could result in an impairment charge and such a charge could have a material adverse effect on our consolidated results of operations.
38
Table of Contents
Source of Revenue and Revenue Recognition Policy
We recognize revenues from collaboration agreements as earned upon our achievement of the performance requirements of the agreements. Our corporate collaboration agreements have generally provided for research funding for a specified number of full-time equivalent researchers working in defined research programs. Revenue related to these payments is earned as the related research work is performed. In addition, a collaborator may make technology advancement payments that are intended to fund further development of our core technology, as opposed to a defined research program. Payments received that are related to future performance are deferred and recognized as revenue as the performance requirements are achieved. Such payments are recognized ratably over the related research and development period.
Revenue related to performance milestones is recognized based upon the achievement of the milestones, as defined in the respective agreements. Substantive, at-risk incentive milestones, if any, are recognized as revenue upon achievement of the incentive milestone event when we have no future performance obligations related to the payment and we judge the event to be the culmination of a separate earnings process. Incentive milestone payments are triggered either by the results of our research efforts or by events external to us, such as regulatory approval to market a product. We receive royalties from licensees, which are based on sales to third parties of licensed products. Royalties are recorded as earned in accordance with the contract terms when third-party results can be reliably measured and collectibility is reasonably assured.
Non-refundable upfront payments received in connection with research and development collaboration agreements, including license fees, and technology advancement funding that is intended for the development of our core technologies, are deferred upon receipt and recognized as revenue over the relevant research and development periods specified in the agreement. Under arrangements where we expect our research and development obligations to be performed evenly over the specified period, the upfront payments are recognized on a straight-line basis over the period. Under arrangements where we expect our research and development obligations to vary significantly from period to period, we recognize the upfront payments based upon the actual amount of research and development efforts incurred relative to the amount of our total expected effort. In cases where the planned levels of research services fluctuate substantially over the research term, this requires us to make critical estimates in both the remaining time period and the total expected costs of our obligations and, therefore, a change in the estimate of total costs to be incurred or in the remaining time period could have a significant impact on the revenue recognized in future periods.
The determination of separate units of accounting in arrangements involving multiple deliverables as required under EITF IssueNo. 00-21, “Revenue Arrangements with Multiple Deliverables,” requires management to exercise judgment as to whether the delivered item has stand alone value to the collaborator and to estimate whether there is objective and reliable evidence of fair value for the undelivered items. Collaborative agreements also may contain multiple deliverables that require management to determine whether or not the deliverables are separate units of accounting.
Revenue from license agreements for which no further performance obligations exist is recognized as revenue on the earlier of when payments are received or when the amount can be reliably measured and collectibility is reasonably assured.
Revenue related to grant agreements with various government agencies is recognized as the related research and development expenses are incurred, and when these research and development expenses are within the prior approved funding amounts. Certain grant agreements provide an option for the government to audit the amount of research and development expenses, both direct and indirect, that have been submitted to the government agency for reimbursement. We believe the overhead rates we used to calculate our indirect research and development expenses are within the contractual guidelines of allowable costs and are reasonable estimates of our indirect expenses incurred through the term of the agreements.
Our sources of potential revenues for the next several years are likely to be license payments and research funding under existing and possible future government research grants and may include research funding and milestone payments under possible future collaborative arrangements. See Note 3 of Notes to Consolidated Financial Statements.
39
Table of Contents
Accounting for Clinical Trial Costs
We charge research and development costs, including clinical study costs, to expense when incurred, consistent with SFAS No. 2,“Accounting for Research and Development Costs.” Clinical study costs are a significant component of research and development expenses. Most of our clinical studies are performed by a third-party contract research organization (CRO). The clinical trials generally have three distinctive stages plus pass through costs:
| • | start-up — initial setting up of the trial; | |
| • | enrollment of patients in the trial; and | |
| • | close down and reporting of the trial. |
We review the list of expenses for the trial from the original signed agreements and separate them into what we perceive asstart-up, enrollment or closedown expenses of the clinical trial. The start up costs, which usually occur within a few months after the contract has been executed and include costs, such as study initiation, site recruitment, regulatory applications and investigator meetings, are expensed ratably over the start up period. The site management, study management, medical and safety monitoring and data management expenses are calculated on a per patient basis and expensed ratably over the treatment period beginning on the date that the patient enrolls. The close down and reporting expenses are expensed ratably over the close out period of time. Pass through costs, including the costs of the drugs, are expensed as incurred.
In general, our service agreements permit us to terminate at will where we would continue to be responsible for payment of all services completed (or pro-rata completed) at the time of notice of termination, plus any non-cancellable expenses that have been entered into by the CRO on our behalf.
Restructuring Charge
In November 2007, we implemented a plan to consolidate our research and development activities at our U.S. facilities in Redwood City, California. The consolidation has resulted in the cessation of research and development operations at Maxygen ApS, our wholly owned subsidiary in Denmark. In connection with the consolidation, we have recorded estimated expenses for severance and outplacement costs and other restructuring costs. In accordance with SFAS No. 146,“Accounting for Costs Associated with Exit or Disposal Activities” (SFAS 146), costs associated with restructuring activities generally have been recognized when they are incurred rather than at the date of a commitment to an exit or disposal plan. However, in the case of leases, the expense is estimated and accrued when the property is vacated or at the point when we cease to use the leased equipment. Given the significance of, and the timing of the execution of such activities, this process is complex and involves periodic reassessments of estimates made at the time the original decisions were made, including estimating the salvage value of equipment consistent with abandonment date. In addition, post-employment benefits accrued for workforce reductions related to restructuring activities are accounted for under SFAS No. 112,“Employers’ Accounting for Postemployment Benefits” (SFAS 112). A liability for post-employment benefits is recorded when payment is probable, the amount is reasonably estimable, the obligation is attributable to employees’ services already rendered and the obligation relates to rights that have vested or accumulated. We continually evaluate the adequacy of the remaining liabilities under this restructuring. Although we believe that these estimates accurately reflect the costs of our restructuring plans, actual results may differ, thereby requiring us to record additional provisions or reverse a portion of such provisions.
In connection with the cessation of research and development operations at Maxygen ApS, we have determined that the remaining useful lives of certain of the fixed assets at Maxygen ApS have been shortened to three months for equipment and to six months for leasehold improvements at November 30, 2007. The remaining book value of these assets is depreciated over the shorter estimated remaining useful lives and the depreciation expense is reflected in research and development expenses in the Consolidated Statements of Operations.
40
Table of Contents
Stock-Based Compensation Expense
Beginning on January 1, 2006, we began accounting for stock options and shares purchased under our Employee Stock Purchase Plan, or ESPP, under the provisions of SFAS No. 123(R), “Share-Based Payment,” (SFAS 123(R)), which requires the recognition of the fair value of equity-based compensation. We estimate the fair value of stock options and ESPP shares using the Black-Scholes-Merton option valuation model. This model requires the input of subjective assumptions in implementing SFAS 123(R), the most significant of which are our estimates of the expected volatility of the market price of our stock and the expected term of each award. We estimate expected volatility and future stock price trends based on a combination of historical and implied volatilities. When establishing an estimate of the expected term of an award, we consider the vesting period for the award, our historical experience of employee stock option exercises (including forfeitures), the expected volatility, and a comparison to relevant peer group data. The assumptions used in calculating the fair value of share-based payment awards represent management’s best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As a result, if factors change and we use different assumptions, our stock-based compensation expense could be significantly different from what we have recorded in the current period.
We have adopted SFAS 123(R) using the modified prospective transition method. Under this transition method, compensation cost recognized during the year ended December 31, 2006 includes: (a) compensation cost for all share-based payments granted prior to, but not yet vested as of, January 1, 2006, based on the grant date fair value estimated in accordance with the original provisions of SFAS No. 123, “Accounting For Stock-Based Compensation” (SFAS 123), amortized on a graded vesting basis over the options’ vesting period, and (b) compensation cost for all share-based payments granted subsequent to January 1, 2006, based on the grant date fair value estimated in accordance with the provisions of SFAS 123(R) amortized on a straight-line basis over the options’ vesting period. Under this method of implementation, no restatement of prior periods has been made. Prior to our implementation of SFAS 123(R), we accounted for stock options and ESPP shares under the provisions of Accounting Principles Board Opinion No. 25, “Accounting for Stock Issued to Employees,” or APB 25, and related interpretations and made pro forma footnote disclosures as required by SFAS No. 148, “Accounting For Stock-Based Compensation — Transition and Disclosure,” which amended SFAS 123. Since the exercise price of all options granted was not below the fair market price of the underlying common stock on the grant date, prior to our implementation of SFAS 123(R) we generally recognized no equity-based compensation expense in our Consolidated Statements of Operations. Accordingly, there was no stock-based compensation expense related to employee stock options recognized during 2005. See Note 1 of Notes to Consolidated Financial Statements under the caption “Stock-Based Compensation” for a further discussion.
Stock-based compensation expense recognized under SFAS 123(R) in the Consolidated Statements of Operations for the year ended December 31, 2007 was $5.8 million related to employee stock options, $666,000 related to consultant stock options, $80,000 related to the ESPP and $287,000 included in the restructuring charge, and for the year ended December 31, 2006 was $5.7 million related to employee stock options, $914,000 related to consultant stock options and $87,000 related to the ESPP. As a result of the application of SFAS 123(R), our net loss for the years ended December 31, 2006 and 2007 increased by $6.7 million and $5.8 million, respectively. SFAS 123(R) also increased basic and fully diluted loss per share from continuing operations by $0.19 and $0.16 for the years ended December 31, 2006 and 2007, respectively. SFAS 123(R) did not have an impact on cash flows from operations during the years ended December 31, 2006 and 2007.
In connection with the grant of stock options to consultants, we recorded stock compensation expense of $102,000 in 2005, $914,000 in 2006 and $666,000 in 2007. Stock compensation expense in connection with the grant of stock options to consultants included in research and development expense was $102,000 in 2005, $12,000 in 2006 and $236,000 in 2007. Stock compensation expense in connection with the grant of stock options to consultants included in general and administrative expense was none in 2005, $902,000 in 2006 and $430,000 in 2007.
In connection with the cessation of our operations in Denmark and the consolidation of our operations in the United States, we recorded stock compensation expense of $287,000 in 2007 as part of the restructuring charge. This expense results from the extension of the exercise period of certain stock options held by affected employees of Maxygen ApS, as required under Danish law in connection with the termination of such employees.
41
Table of Contents
Results of Operations
Revenues
Our revenues have been derived primarily from collaboration and license agreements and government research grants. Total revenues were $23.2 million in 2007, $25.0 million in 2006 and $14.5 million in 2005. The decrease in revenue from 2006 to 2007 and the increase in revenue from 2005 to 2006 resulted primarily from changes in revenues recognized under our prior collaboration agreement with Roche for our MAXY-VII product candidates, as discussed below.
Revenues from our collaboration agreements were $10.2 million in 2007, $20.5 million in 2006 and $11.6 million in 2005. Roche was the only collaborative partner that contributed significantly to our collaborative research and development revenues during 2005, 2006 and 2007. The initial funded research term of our collaboration with Roche for our MAXY-alpha product candidates ended in December 2005 and the collaboration agreement was terminated in November 2007. In December 2005, we entered into a new collaboration with Roche for co-development and commercialization of our MAXY-VII product candidates for acute bleeding indications. In April 2007, Roche terminated this agreement due to the inability of the parties to de-risk the program in preclinical testing by failing to establish an animal model suitable to demonstrate cessation of acute bleeding in a trauma setting.
The decrease in collaborative research and development revenue of $10.3 million from 2006 to 2007 was primarily due to the loss of collaborative research and development revenue under the co-development and commercialization agreement with Roche for our MAXY-VII product candidates. For 2007, our collaborative research and development revenue included $2.9 million earned as net reimbursement of our research and development activities prior to the termination of this agreement. The termination of this agreement also caused us to accelerate the recognition of $5.6 million of deferred revenue in the first half of 2007 relating to the $8.0 million non-refundable upfront license fee received from Roche in 2005. Revenue in 2007 also includes a $1.5 million non-refundable license fee received from sanofi pasteur.
The increase in collaborative research and development revenue of $9.0 million from 2005 to 2006 (including $1.5 million of revenues attributable to the consolidation of Codexis through February 28, 2005) was primarily due to collaborative research and development revenue from the collaboration with Roche for our MAXY-VII product candidates. Our revenues for 2006 relating to this collaboration consisted primarily of a $5.0 million milestone payment, $11.1 million earned as net reimbursement of our research and development activities and $2.4 million related to the amortization of the non-refundable upfront license fee we received from Roche in December 2005. Our revenues for 2006 also include a $2.0 million milestone payment we received from Roche as a result of the initiation by Roche of clinical trials of our MAXY-alpha product candidate.
In 2007, we recognized $8.3 million in revenue from related party under our license agreement with Codexis. These revenues reflect amounts due to us from payments received by Codexis under its collaboration arrangement with Shell Oil Products US that began in November 2006 and $7.5 million recognized in the fourth quarter of 2007 in connection with an expanded collaboration agreement between Royal Dutch Shell plc and Codexis for the development of new enzymes to convert biomass to fuel.
Revenues from government research grants were $4.6 million in 2007, $4.5 million in 2006 and $2.9 million in 2005. The increase in grant revenue of $1.6 million from 2005 to 2006 and $0.1 million from 2006 to 2007 primarily reflects an increase in activity due to the beginning of three new government grant projects in the third quarter of 2005. In 2007, revenues from government research grants also include $2.2 million from a contract with the U.S. Department of Defense for HIV vaccine discovery that expired in October 2007.
We expect our revenues for 2008 to decrease compared to 2007, due primarily to the loss of collaborative research and development revenue under the co-development and commercialization agreement with Roche for our MAXY-VII product candidates. We also expect much lower revenue in 2008 under our license agreement with Codexis since a significant portion of the revenue recorded under this agreement in 2007 related to the one-time upfront payment that Codexis received from Shell. Our revenue may fluctuate substantially from year to year based on the completion of new licensing or collaborative agreements and our receipt of development related milestones, royalties and other payments under such agreements. However, we cannot predict with any certainty whether we
42
Table of Contents
will enter into any new licensing or collaborative agreements or receive any milestone, royalty or other payments under any existing or future licensing, collaboration or other agreements or whether any particular collaboration or research effort will ultimately result in a commercial product.
Prior to our deconsolidation of Codexis, as of February 28, 2005, we operated as two segments, human therapeutics and chemicals. Revenues for each operating segment were derived from our collaboration agreements and government research grants and were categorized based on the industry of the product or technology under development. Results of Codexis through February 28, 2005 are shown as our chemicals segment. After February 28, 2005, we have operated as one segment, human therapeutics. The following table presents revenues for each operating segment (in thousands):
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| Human therapeutics | $ | 12,991 | $ | 25,021 | $ | 23,157 | ||||||
| Chemicals (through February 28, 2005) | 1,510 | — | — | |||||||||
| Total revenue | $ | 14,501 | $ | 25,021 | $ | 23,157 | ||||||
The changes in revenue for our human therapeutics segment from 2005 to 2006 and from 2006 to 2007 are discussed above.
Research and Development Expenses
Our research and development expenses consist primarily of research consultants and external collaborative research expenses (including contract manufacturing and clinical trial expenses), salaries and benefits, facility costs, supplies and depreciation. Research and development expenses were $59.9 million in 2007, $49.1 million in 2006 and $41.9 million in 2005 (including $2.5 million of research and development expenses attributable to Codexis in 2005).
The increases in our research and development expenses of $10.8 million from 2006 to 2007 and $9.8 million from 2005 to 2006 (excluding $2.5 million of research and development expenses attributable to Codexis in 2005) were primarily related to increased external expenses associated with the development of our product candidates, including expenses related to clinical trials of our MAXY-G34 product candidates, the manufacture of MAXY-G34 and MAXY-VII product for clinical trials and increased salaries and benefits. For 2006, our implementation of SFAS 123(R) also increased our research and development expenses compared to 2005.
Stock compensation expenses included in research and development expenses increased from $2.1 million in 2006 to $3.0 million in 2007, primarily as a result of the issuance of stock options to new U.S. employees that we hired to expand our clinical regulatory organization and to replace capabilities previously performed in Denmark. Stock compensation expenses included in research and development expenses increased from $115,000 in 2005 to $2.1 million in 2006, primarily as a result of our implementation of SFAS 123(R). See Note 1 of Notes to Consolidated Financial Statements under the caption “Stock-Based Compensation.”
We do not track fully burdened research and development costs by project. However, we do estimate, based on full-time equivalent personnel effort, the percentage of research and development efforts (as measured in hours incurred, which approximates costs) undertaken for projects funded by collaborators and government grants, on the one hand, and projects funded by us, on the other hand. To approximate research and development expenses by funding category, the number of hours expended in each category has been multiplied by the approximate cost per hour of research and development effort and added to project-specific external costs. In the case where a collaborative partner is sharing the research and development costs, the expenses for that project are allocated proportionately between the collaborative projects funded by third parties and internal projects. We believe that presenting our research and development expenses in these categories will provide our investors with meaningful information on how our resources are being used.
43
Table of Contents
The following table presents our approximate research and development expenses by funding category (in thousands):
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| Collaborative projects funded by third parties(1) | $ | 4,146 | $ | 9,906 | $ | 2,713 | ||||||
| Government grants | 2,369 | 4,215 | 5,106 | |||||||||
| Internal projects | 35,389 | 35,009 | 52,032 | |||||||||
| Total | $ | 41,904 | $ | 49,130 | $ | 59,851 | ||||||
| (1) | Research and development expenses related to collaborative projects funded by third parties may be less than the reported revenues due to the amortization of non-refundable upfront payments, as well as a portion of the collaborative research and development revenue that is charged for general and administrative expenses. |
Our product development programs are at an early stage and may not result in any marketed products. Product candidates that may appear promising at early stages of development may not reach the market for a number of reasons. Product candidates may be found ineffective or cause harmful side effects during clinical trials, may take longer to pass through clinical trials than had been anticipated, may fail to receive necessary regulatory approvals, may prove impracticable to manufacture in commercial quantities at reasonable costs and with acceptable quality and may be barred from commercialization if they are found to infringe or otherwise violate a third party’s intellectual property rights. In addition, competitors may develop superior competing products. Furthermore, it is uncertain which of our internally developed product candidates will be subject to future collaborative arrangements. The participation of a collaborative partner may accelerate the time to completion and reduce the cost to us of a product candidate or it may delay the time to completion and increase the cost to us due to the alteration of our existing strategy. The risks and uncertainties associated with our research and development projects are discussed more fully in the section of this report entitled “Item 1A — Risk Factors.” Because of these risks and uncertainties, we cannot predict when or whether we will successfully complete the development of any of our product candidates or the ultimate product development cost in any particular case.
We expect that our research and development costs will decrease in 2008 over 2007, due to savings realized in connection with the cessation of our operations in Denmark and the consolidation of our operations in the United States. Any decrease in our research and development costs will be partially offset by an increase in research and development costs resulting from advancement of our product candidates through clinical and preclinical development, including the continued clinical development of our MAXY-G34 product candidate and the planned clinical development of our MAXY-VII product candidates. We expect to continue to devote substantial resources to research and development and we expect research and development expenses to increase over the next several years if we are successful in advancing our product candidates into and through clinical trials. To the extent we out-license our product candidates prior to commencement of clinical trials or collaborate with others with respect to clinical trials, increases in research and development expenses may be reduced or avoided. We intend to manage the level of our expenditures for research and development, including clinical trials, to balance advancing our product candidates against maintaining adequate cash resources for our operations. In addition, the application of SFAS 123(R) had a material impact on our consolidated results of operations and net loss per share for the years ended December 31, 2006 and 2007 and is expected to have a material impact on our consolidated results of operations and net loss per share in the future. The continued impact of SFAS 123(R) will depend on, among other things, the levels of share-based payments granted in the future. See Note 1 of Notes to Consolidated Financial Statements under the caption “Stock-Based Compensation.”
General and Administrative Expenses
Our general and administrative expenses consist primarily of personnel costs for finance, legal, general management, business development and human resources, as well as stock compensation, insurance premiums and professional expenses, such as external expenditures for legal and accounting services. General and administrative expenses were $15.0 million in 2007, $17.6 million in 2006 and $13.2 million in 2005. General and administrative stock compensation expense was $3.5 million in 2007, $4.6 million in 2006 and $68,000 in 2005.
44
Table of Contents
The decrease of $2.6 million in general and administrative expenses from 2006 to 2007 was primarily due to $1.1 million lower stock compensation expense resulting from consultant options granted in April 2006 which were fully amortized by April 2007 and an increased number of unvested options being cancelled in 2007 compared to 2006. The decrease also reflects lower expenditures on external consultants and market analysis. The increase in general and administrative expenses of $4.3 million from 2005 to 2006 was primarily due to the increase in stock compensation expense included in general and administrative expenses resulting from our implementation of SFAS 123(R) and higher stock compensation expense resulting from consultant options. This increase was offset in part by reduced personnel costs resulting from terminations and the deconsolidation of Codexis in 2005.
Our general and administrative expenses during 2008 should be consistent with general and administrative expenses for 2007, depending on, among other things, the levels of share-based payments granted in 2008, the use of external consultants and market analysis, and expenditures for legal and accounting services.
Restructuring Charge
In 2007, we recognized a charge of $5.2 million resulting from the cessation of operations at Maxygen ApS, our Danish subsidiary, and the consolidation of our operations in the United States. This charge primarily includes post-employment and one-time termination benefits for the affected employees of Maxygen ApS and other costs associated with exit activities. This charge also includes stock compensation expense of approximately $287,000. This expense results from the extension of the exercise period of certain stock options held by affected employees of Maxygen ApS, as required under Danish law in connection with the termination of such employees. We expect to recognize additional costs of approximately $1.0 million in the first half of 2008 relating to facilities closure and lease termination expenses. See Note 14 of the Notes to Consolidated Financial Statements for further discussion of this matter.
Interest Income and Other Income (Expense), Net
Interest income and other income (expense), net represents income earned on our cash, cash equivalents and marketable securities and currency transaction gains or losses related to the funding of our Danish subsidiary, Maxygen ApS. Interest income and other income (expense), net was $7.5 million in 2007, $8.5 million in 2006 and $5.6 million in 2005. Included in these amounts are foreign exchange losses of $1.1 million in 2007, $137,000 in 2006 and $537,000 in 2005. The decrease from 2006 to 2007 reflects higher foreign exchange losses in 2007 partially offset by higher interest income resulting from higher interest rates on a lower investment base. The increases in interest income and other income (expense), net from 2005 to 2006 reflect higher interest income reflecting higher interest rates on lower average balances of cash, cash equivalents and marketable securities, plus a decrease in foreign exchange losses.
Equity in Losses of Minority Investee
Equity in losses of minority investee reflects our share of the net loss of Codexis. In May 2006, we purchased $600,000 of secured subordinated convertible promissory notes and, in August 2006, the notes and accrued interest were converted into Codexis preferred stock and we purchased approximately $400,000 of additional preferred stock. Subsequent to our investments in May and August 2006, we recorded losses of $1.0 million under the equity method of accounting and as of December 31, 2006, we had recorded losses equal to our investment basis in Codexis. We are not obligated to fund the operations or other capital requirements of Codexis. As of December 31, 2007, our equity interest in Codexis was approximately 25% of its outstanding capital stock.
Gain on Sale of Equity Investment
On October 24, 2006, Amgen Inc. completed the acquisition of Avidia and Avidia became a wholly owned subsidiary of Amgen Inc. As a result of the acquisition, we received cash proceeds of approximately $17.8 million (before $140,000 of income taxes) in the fourth quarter of 2006 in exchange for our equity interests in Avidia and may receive up to an additional $1.4 million in cash, contingent upon the development of certain Avidia products by Amgen Inc. This contingent amount was reduced from $2.8 million based on the discontinuation by Amgen Inc. of certain development activities. We reported an income tax provision of $140,000 attributable to federal alternative
45
Table of Contents
minimum taxes as a result of the gain on sale of our equity interests in Avidia. Accordingly, we recorded a gain net of taxes on disposal of this investment of approximately $17.7 million in the fourth quarter of 2006. See Notes 1 and 12 of the Notes to Consolidated Financial Statements.
Cumulative Effect Adjustment
Codexis was formed in January 2002 and financed by us and several other investors in September and October of 2002. Until February 28, 2005, we recognized 100% of the operating results of Codexis, even though we only owned a majority of the voting interests in Codexis. At such time, we had recorded cumulative losses of Codexis in the amount of $26.4 million, which was in excess of our investment basis of $9.8 million. On February 28, 2005, our voting rights in Codexis were reduced below 50%. As a result, we no longer consolidate the financial position and results of operations of Codexis with our financial results as of such date and instead account for Codexis under the equity method of accounting. To reflect what our basis in Codexis would have been under equity accounting, we recorded a non-recurring cumulative effect adjustment of $16.6 million in the first quarter of 2005 to bring our investment basis in Codexis to zero as of February 28, 2005. This cumulative effect adjustment does not have any tax consequences.
Subsidiary Preferred Stock Accretion
In 2002, Codexis sold $25 million of Codexis series B redeemable convertible preferred stock to investors, of which $5 million was purchased by us and $20 million was purchased by several other investors. In connection with the redemption rights of the Codexis series B stockholders, we recorded accretion of the redemption premium for the series B redeemable convertible preferred stock, excluding the shares owned by us, in the amount of $167,000 for the year ended December 31, 2005. The accretion was recorded as subsidiary preferred stock accretion on the Consolidated Statements of Operations and as a reduction of additional paid-in capital and an increase to minority interest on the Consolidated Balance Sheets. No accretion was recorded for the years ended December 31, 2006 and 2007. Any obligation to make redemption payments is solely an obligation of Codexis and any payments are to be made solely from assets of Codexis. Since we no longer consolidate the financial position of Codexis, as of February 28, 2005, we no longer recognized accretion for the Codexis redemption premium. We also no longer reflect amounts as minority interest on the Consolidated Balance Sheets. We recorded a $2.3 million adjustment to additional paid-in capital in the three month period ended March 31, 2005 to eliminate the reduction of additional paid-in capital that had resulted from Codexis’ preferred stock accretion prior to February 28, 2005.
Provision for Income Taxes
No income tax expense was recorded from continuing operations for the years ended December 31, 2007 and 2005. Income tax expense from continuing operations for the year ended December 31, 2006 was $140,000. In 2006, we reported an income tax provision of $140,000 attributable to federal alternative minimum taxes as a result of the gain on the sale of our equity interests in Avidia. This amount has been netted against the gain on sale of our equity interests in Avidia and is reflected in gain on sale of equity investment in the Consolidated Statements of Operations. For 2007 and 2005, there was no provision for U.S. federal, U.S. state, or foreign income taxes as we incurred operating losses for all jurisdictions or had sufficient operating losses to carry forward and to reduce any operating income to zero.
Deferred tax assets and the associated valuation allowance increased by $5.5 million in 2007 due primarily to increases in federal and state net operating loss carryforwards and deferred taxes related to deductible stock option compensation, offset in part by the use of foreign net operating loss carryforwards. Deferred tax assets and the associated valuation allowance increased by $5.6 million in 2006 due primarily to increases in state and foreign net operating loss carryforwards and deferred taxes related to deductible stock option compensation, offset in part by the use of federal net operating loss carryforwards and a reduction in capitalized research and development costs due to a reduction in state tax rate.
As of December 31, 2007, we had net operating loss carryforwards for federal income tax purposes of approximately $37.1 million, which expire in the years 2022 through 2027, and federal research and development tax credit carryforwards of approximately $2.5 million, which expire in the years 2012 through 2027. As of
46
Table of Contents
December 31, 2007, we had net operating loss carryforwards for state income tax purposes of approximately $59.3 million that expire in the years 2015 through 2017 and state research and development tax credits of approximately $2.5 million that have no expiration date. As of December 31, 2007, we had net operating loss carryforwards for foreign income tax purposes of approximately $6.8 million that have no expiration date.
Utilization of our net operating losses and credits may be subject to substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code of 1986, as amended, and similar state provisions. Such an annual limitation could result in the expiration of the net operating losses and credits before utilization. See Note 9 of the Notes to Consolidated Financial Statements.
Recent Accounting Pronouncements
On June 27, 2007, the Financial Accounting Standards Board (FASB) ratified the consensus reached by the FASB Emerging Issues Task Force on IssueNo. 07-3, “Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities”(EITF 07-3).EITF 07-3 requires entities to defer income statement recognition of non-refundable advance payments for research and development activities, such as upfront non-refundable payments to contract research organizations, if the contracted party has not yet performed activities related to the upfront payment. Amounts deferred are to be recognized by the contracting company as expense when the research and development activities are performed. The application ofEITF 07-3 is effective for interim or annual reporting periods in fiscal years beginning after December 15, 2007. Earlier application ofEITF 07-3 is not permitted. Companies are required to report the effects of applyingEITF 07-3 prospectively for new contracts entered into after the effective date ofEITF 07-3. We do not expect the application ofEITF 07-3 to have material impact on our consolidated results of operations and financial condition.
In February 2007, the FASB issued SFAS No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities — Including an amendment of FASB Statement No. 115” (SFAS 159). SFAS 159 allows entities the option to measure eligible financial instruments at fair value as of specified dates. Such election, which may be applied on an instrument by instrument basis, is typically irrevocable once elected. SFAS 159 is effective for fiscal years beginning after November 15, 2007, and early application is allowed under certain circumstances. We currently are determining whether fair value accounting is appropriate for any of our eligible items and cannot estimate the impact, if any, which SFAS 159 will have on our consolidated results of operations and financial condition.
In September 2006, the FASB issued SFAS No. 157, “Fair Value Measurements” (SFAS 157). SFAS 157 provides guidance for using fair value to measure assets and liabilities. It also responds to investors’ request for expanded information about the extent to which companies measure assets and liabilities at fair value, the information used to measure fair value and the effect of fair value measurements on earnings. SFAS 157 applies whenever other standards require (or permit) assets or liabilities to be measured at fair value, and does not expand the use of fair value in any new circumstances. SFAS 157 is effective for financial statements issued for fiscal years beginning after November 15, 2007. We do not expect the application of SFAS 157 to have material impact on our consolidated results of operations and financial condition.
Liquidity and Capital Resources
Since inception, we have financed our continuing operations primarily through private placements and public offerings of equity securities, research and development funding from collaborators and government grants. In addition, as a result of the acquisition of Avidia by Amgen Inc., we received cash proceeds of approximately $17.8 million (before $140,000 of income taxes) in the fourth quarter of 2006 in exchange for our equity interests in Avidia and, on July 1, 2004, we received cash proceeds of $64.0 million from the sale of Verdia, our former agriculture subsidiary and the sole component of our agriculture segment. As of December 31, 2007, we had $145.8 million in cash, cash equivalents and marketable securities.
Net cash used in operating activities was $41.4 million in 2007, $22.1 million in 2006 and $29.9 million in 2005. Uses of cash in operating activities were primarily to fund losses from continuing operations. The $19.3 million increase in cash used in operating activities from 2006 to 2007 and the $7.8 million decrease in cash used in operating activities from 2005 to 2006 primarily relate to the timing of payments received from Roche
47
Table of Contents
under our prior collaboration agreements. For 2005, net cash used in operating activities includes a one-time upfront payment of $8.0 million related to our MAXY-VII program, which we recorded as deferred revenue. For 2006, net cash used in operating activities includes the receipt of $12.0 million related to various milestone payments under our collaboration agreements with Roche, $5.0 million of which related to our MAXY-alpha program and was recorded as revenue and accounts receivable in 2005. Operating expenses in 2006 also included incremental non-cash stock compensation expense of $5.7 million compared to 2005.
Net cash provided by investing activities was $66.5 million in 2007, $40.7 million in 2006 and $16.3 million in 2005. The cash provided during 2005, 2006 and 2007 was primarily related to maturities of available-for-sale securities in excess of purchases, offset in 2005 by the $2.6 million used by Codexis to acquire Julich Fine Chemicals GmbH in February 2005. In addition, in 2006, we received cash proceeds of $17.8 million (before $140,000 of income taxes) from the sale of our equity interests in Avidia. The majority of additions of property and equipment in 2005 related to Codexis’ investment in its bioprocessing facility. We expect to continue to make investments in the purchase of property and equipment to support our operations. We may use a portion of our cash to acquire or invest in businesses, products or technologies, or to obtain the right to use such technologies.
Net cash provided by financing activities was $5.1 million in 2007, $1.0 million in 2006 and $2.2 million in 2005. The cash provided during 2005, 2006 and 2007 relate to proceeds from the sale of common stock in connection with our ESPP and the exercise of stock options by employees. The cash provided during 2005 also included proceeds of equipment loans entered into by Codexis, net of repayments of such loans.
In accordance with FASB Statement No. 52, “Foreign Currency Translation,” the functional currency for our Danish operations was its local currency through November 30, 2007. However, as the result of our plan to consolidate our research and development activities at our U.S. facilities in Redwood City, California and cease operations at Maxygen ApS, we have reevaluated the functional currency for our Danish operations and determined that it is the U.S. dollar after November 30, 2007. The effects of foreign exchange rate changes on the translation of the local currency financial statements into U.S. dollars are reported as a component of accumulated other comprehensives loss on the Consolidated Balance Sheets. The effect of exchange rate changes on cash and cash equivalents was an increase of $382,000 in 2007, a reduction of $55,000 in 2006 and an increase of $276,000 in 2005.
The following are contractual commitments as of December 31, 2007 associated with lease obligations and purchase obligations (in thousands):
| Payments Due by Period | ||||||||||||||||||||
| Less | More | |||||||||||||||||||
| than 1 | 1-3 | 4-5 | than 5 | |||||||||||||||||
Contractual Obligations | Total | Year | Years | Years | Years | |||||||||||||||
| Operating lease obligations | $ | 1,863 | $ | 1,664 | $ | 199 | $ | — | $ | — | ||||||||||
| Purchase obligations | 6,583 | 5,043 | 1,540 | — | — | |||||||||||||||
| Total | $ | 8,446 | $ | 6,707 | $ | 1,739 | $ | — | $ | — | ||||||||||
As of December 31, 2007, we are eligible to receive up to $23.0 million in potential event based payments from sanofi pasteur, the vaccines division of the sanofi-aventis Group, under our existing license agreement relating to the development of a vaccine for the dengue virus. We also received an upfront fee and may earn royalties on future product sales, if any.
We believe that our current cash, cash equivalents, short-term investments and long-term investments, together with funding expected to be received from licensors and government grants, will be sufficient to satisfy our anticipated cash needs for working capital and capital expenditures for at least the next twelve months. However, it is possible that we will seek additional financing within this timeframe. We may raise additional funds through public or private financing, collaborative relationships or other arrangements. Additional funding, if sought, may not be available on terms favorable to us. Further, any additional equity financing may be dilutive to stockholders, and debt financing, if available, may involve restrictive covenants. Our failure to raise capital when needed may harm our business and operating results.
48
Table of Contents
| Item 7A | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We are exposed to market risks, including changes in interest rates and foreign currency exchange. To mitigate some foreign currency exchange rate risk, we from time to time enter currency forward contracts. We do not use derivative financial instruments for speculative or trading purposes.
Interest Rate and Market Risk
The primary objective of our investment activities is to preserve principal while at the same time maximizing yields without significantly increasing risk. To achieve this objective, we maintain our portfolio of cash equivalents, short-term and long-term investments in a variety of securities, including corporate obligations and money market funds. All investments and substantially all cash and cash equivalents are held in U.S. currency, with approximately 1.8% of cash and cash equivalents held in Danish kroner at December 31, 2007. As of December 31, 2007, 100% of our total portfolio was scheduled to mature in one year or less.
The following table represents the fair value balance of our cash, cash equivalents, short-term and long-term investments that are subject to interest rate risk by average interest rates as of December 31, 2007 (dollars in thousands):
| Expected Maturity | ||||||||
| 2008 | 2009 | |||||||
| Cash and cash equivalents | $ | 77,130 | $ | — | ||||
| Average interest rates | 4.85 | % | — | |||||
| Short-term investments | $ | 68,683 | $ | — | ||||
| Average interest rates | 4.31 | % | — | |||||
| Long-term investments | $ | — | $ | — | ||||
| Average interest rates | — | — | ||||||
We did not hold derivative instruments intended to mitigate interest rate risk as of December 31, 2007, and we have never held such instruments in the past. If market interest rates were to increase by 100 basis points, or 1%, from December 31, 2007 levels, the fair value of our portfolio would decline by approximately $1.4 million. The modeling technique used measures the change in fair values arising from an immediate hypothetical shift in market interest rates and assumes ending fair values include principal plus accrued interest.
Due to the recent adverse developments in the credit markets, we may experience reduced liquidity with respect to some of our investments. These investments are generally held to maturity, which averages one year or less and may not exceed two years. However, if the need arose to liquidate such securities before maturity, we may experience losses on liquidation. As of December 31, 2007, we held $68.7 million of commercial debt securities, with an average time to maturity of 99 days. To date we have not experienced any liquidity issues with respect to these securities, but should such issues arise, we may be required to hold some, or all, of these securities until maturity. We believe that, even allowing for potential liquidity issues with respect to these securities, our remaining cash and cash equivalents and the maturities of short-term investments will be sufficient to meet our anticipated cash needs for at least the next twelve months. We have the ability and intent to hold our debt securities to maturity when they will be redeemed at full par value.
49
Table of Contents
Foreign Currency Exchange Risk
A portion of our operations previously consisted of research and development activities performed in Denmark by our wholly-owned subsidiary, Maxygen ApS. The functional currency of our Danish operations was its local currency through November 30, 2007. However, in November 2007, we implemented a plan to consolidate our research and development activities at our U.S. facilities in Redwood City, California, resulting in the cessation of research and development operations at Maxygen ApS. As a result, we have reevaluated the functional currency for our Danish operations and determined that it is the U.S. dollar after November 30, 2007. In 2007, excluding stock compensation expenses, approximately 52% of our operating expenses related to Maxygen ApS. As a result, our financial results may be affected by changes in the foreign currency exchange rates of the Danish kroner and the euro. A decrease in the value of the U.S. dollar against the Danish kroner or the euro will result in an increase of our reported operating expenses. To protect against reductions in value and the volatility of future cash flows caused by changes in foreign currency exchange rates, we from time to time enter into cash flow hedging arrangements. Currency forward contracts are utilized in these hedging arrangements. Our hedging arrangements are intended to reduce, but may not always eliminate, the impact of foreign currency exchange rate movements. Gains and losses on these foreign currency investments are generally offset by corresponding losses and gains on the related hedging instruments, resulting in negligible net exposure to us on the amounts hedged.
At December 31, 2007, we had foreign currency contracts outstanding in the form of forward exchange contracts totaling $8.8 million. At December 31, 2006, we had no foreign currency contracts outstanding in the form of forward exchange contracts. During 2007, we recognized $77,000 in foreign exchange losses from hedge contracts and these losses were included with interest and other income, net. During 2006, we recognized $43,000 in foreign exchange losses from hedge contracts and these losses were included with operating expenses.
50
Table of Contents
| Item 8 | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of
Maxygen, Inc.
We have audited the accompanying consolidated balance sheets of Maxygen, Inc. as of December 31, 2006 and 2007, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2007. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Maxygen, Inc. at December 31, 2006 and 2007, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2007, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 1 to the consolidated financial statements, as of January 1, 2006, Maxygen, Inc. changed its method of accounting for stock-based compensation.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Maxygen, Inc.’s internal control over financial reporting as of December 31, 2007, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 7, 2008 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
Palo Alto, California
March 7, 2008
51
Table of Contents
MAXYGEN, INC.
CONSOLIDATED BALANCE SHEETS
| December 31, | ||||||||
| 2006 | 2007 | |||||||
| (In thousands, except share and per share data) | ||||||||
ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 46,504 | $ | 77,130 | ||||
| Short-term investments | 133,384 | 68,683 | ||||||
| Related party receivable | — | 7,493 | ||||||
| Accounts receivable and other receivables | 4,099 | 1,383 | ||||||
| Prepaid expenses and other current assets | 3,133 | 2,683 | ||||||
| Total current assets | 187,120 | 157,372 | ||||||
| Property and equipment, net | 3,262 | 3,060 | ||||||
| Goodwill | 12,192 | 12,192 | ||||||
| Long-term investments | 2,988 | — | ||||||
| Deposits and other long-term assets | 85 | 85 | ||||||
| Total assets | $ | 205,647 | $ | 172,709 | ||||
LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 2,435 | $ | 2,871 | ||||
| Accrued compensation | 4,708 | 6,880 | ||||||
| Accrued restructuring charges | — | 4,413 | ||||||
| Accrued project costs | 1,407 | 3,787 | ||||||
| Other accrued liabilities | 1,547 | 1,250 | ||||||
| Deferred revenue | 1,527 | — | ||||||
| Taxes payable | 140 | — | ||||||
| Total current liabilities | 11,764 | 19,201 | ||||||
| Non-current deferred revenue | 4,066 | — | ||||||
| Other long-term liabilities | 18 | 14 | ||||||
| Commitments and contingencies (Notes 7 and 10) | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred stock, $0.0001 par value, 5,000,000 shares authorized, no shares issued and outstanding at December 31, 2006 and 2007 | — | — | ||||||
| Common stock, $0.0001 par value, 100,000,000 shares authorized, 36,157,910 and 36,926,566 shares issued and outstanding at December 31, 2006 and 2007, respectively | 4 | 4 | ||||||
| Additional paid-in capital | 411,195 | 423,541 | ||||||
| Accumulated other comprehensive loss | (696 | ) | (32 | ) | ||||
| Accumulated deficit | (220,704 | ) | (270,019 | ) | ||||
| Total stockholders’ equity | 189,799 | 153,494 | ||||||
| Total liabilities and stockholders’ equity | $ | 205,647 | $ | 172,709 | ||||
See accompanying notes.
52
Table of Contents
MAXYGEN, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| (In thousands, except per share data) | ||||||||||||
| Collaborative research and development revenue | $ | 11,594 | $ | 20,544 | $ | 10,232 | ||||||
| Revenue from related party | — | — | 8,286 | |||||||||
| Grant revenue | 2,907 | 4,477 | 4,639 | |||||||||
| Total revenues | 14,501 | 25,021 | 23,157 | |||||||||
| Operating expenses: | ||||||||||||
| Research and development | 41,904 | 49,130 | 59,851 | |||||||||
| General and administrative | 13,221 | 17,559 | 14,951 | |||||||||
| Restructuring charge | — | — | 5,212 | |||||||||
| Total operating expenses | 55,125 | 66,689 | 80,014 | |||||||||
| Loss from operations | (40,624 | ) | (41,668 | ) | (56,857 | ) | ||||||
| Interest income and other income (expense), net | 5,572 | 8,524 | 7,542 | |||||||||
| Equity in net loss of minority investee | — | (1,000 | ) | — | ||||||||
| Gain on sale of equity investment | — | 17,662 | — | |||||||||
| Cumulative effect adjustment | 16,616 | — | — | |||||||||
| Net loss | (18,436 | ) | (16,482 | ) | (49,315 | ) | ||||||
| Subsidiary preferred stock accretion | (167 | ) | — | — | ||||||||
| Loss applicable to common stockholders | $ | (18,603 | ) | $ | (16,482 | ) | $ | (49,315 | ) | |||
| Basic and diluted net loss per share: | ||||||||||||
| Continuing operations | $ | (0.98 | ) | $ | (0.46 | ) | $ | (1.34 | ) | |||
| Cumulative effect adjustment | $ | 0.46 | — | — | ||||||||
| Applicable to common stockholders | $ | (0.52 | ) | $ | (0.46 | ) | $ | (1.34 | ) | |||
| Shares used in basic and diluted net loss per share calculations | 35,765 | 36,046 | 36,787 | |||||||||
See accompanying notes.
53
Table of Contents
MAXYGEN, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
| Accumulated | ||||||||||||||||||||||||
| Other | ||||||||||||||||||||||||
| Additional | Comprehensive | Total | ||||||||||||||||||||||
| Common Stock | Paid-In | Income | Accumulated | Stockholders’ | ||||||||||||||||||||
| Shares | Amount | Capital | (Loss) | Deficit | Equity | |||||||||||||||||||
| (In thousands, except share and per share data) | ||||||||||||||||||||||||
| Balance at January 1, 2005 | 35,636,333 | $ | 3 | $ | 399,314 | $ | (2,190 | ) | $ | (185,786 | ) | $ | 211,341 | |||||||||||
| Issuance of common stock upon exercise of options for cash and for services rendered | 198,487 | — | 805 | — | — | 805 | ||||||||||||||||||
| Issuance of common stock under employee stock purchase plan | 37,966 | — | 299 | — | — | 299 | ||||||||||||||||||
| Issuance of common stock under 401(k) employer matching contribution | 49,392 | — | 350 | — | — | 350 | ||||||||||||||||||
| Stock compensation expense for consultant options, and fully vested stock options for services rendered | — | — | 102 | — | — | 102 | ||||||||||||||||||
| Subsidiary preferred stock accretion | — | — | (167 | ) | — | — | (167 | ) | ||||||||||||||||
| Modification of employee stock options | — | — | 81 | — | — | 81 | ||||||||||||||||||
| Deconsolidation of Codexis, Inc. | — | — | 2,336 | — | — | 2,336 | ||||||||||||||||||
| Components of comprehensive loss: | ||||||||||||||||||||||||
| Net loss | — | — | — | — | (18,436 | ) | (18,436 | ) | ||||||||||||||||
| Currency translation adjustment | — | — | — | 645 | — | 645 | ||||||||||||||||||
| Change in unrealized gain(loss) on available-for-sale securities | — | — | — | (12 | ) | — | (12 | ) | ||||||||||||||||
| Comprehensive loss | (17,803 | ) | ||||||||||||||||||||||
| Balance at December 31, 2005 | 35,922,178 | 3 | 403,120 | (1,557 | ) | (204,222 | ) | 197,344 | ||||||||||||||||
| Issuance of common stock upon exercise of options for cash and for services rendered | 150,985 | 1 | 698 | — | — | 699 | ||||||||||||||||||
| Issuance of common stock under employee stock purchase plan | 46,815 | — | 309 | — | — | 309 | ||||||||||||||||||
| Issuance of common stock under 401(k) employer matching contribution | 37,932 | — | 325 | — | — | 325 | ||||||||||||||||||
| Stock compensation expense for consultant options | — | — | 914 | — | — | 914 | ||||||||||||||||||
| Stock based compensation expense under SFAS 123(R) | — | — | 5,829 | — | — | 5,829 | ||||||||||||||||||
| Components of comprehensive loss: | — | — | — | — | — | — | ||||||||||||||||||
| Net loss | — | — | — | — | (16,482 | ) | (16,482 | ) | ||||||||||||||||
| Currency translation adjustment | — | — | — | 331 | — | 331 | ||||||||||||||||||
| Change in unrealized gain (loss) on available-for-sale securities | — | — | — | 530 | — | 530 | ||||||||||||||||||
| Comprehensive loss | (15,621 | ) | ||||||||||||||||||||||
| Balance at December 31, 2006 | 36,157,910 | 4 | 411,195 | (696 | ) | (220,704 | ) | 189,799 | ||||||||||||||||
| Issuance of common stock upon exercise of options for cash and for services rendered | 670,018 | — | 4,755 | — | — | 4,755 | ||||||||||||||||||
| Issuance of common stock under employee stock purchase plan | 54,383 | — | 387 | — | — | 387 | ||||||||||||||||||
| Issuance of common stock under 401(k) employer matching contribution | 44,255 | — | 368 | — | — | 368 | ||||||||||||||||||
| Stock compensation expense for consultant options | — | — | 560 | — | — | 560 | ||||||||||||||||||
| Stock based compensation expense under SFAS 123(R) | — | — | 6,171 | — | — | 6,171 | ||||||||||||||||||
| Modification of a term of an employee stock option | — | — | 105 | — | — | 105 | ||||||||||||||||||
| Components of comprehensive loss: | ||||||||||||||||||||||||
| Net loss | — | — | — | — | (49,315 | ) | (49,315 | ) | ||||||||||||||||
| Currency translation adjustment | — | — | — | 382 | — | 382 | ||||||||||||||||||
| Change in unrealized gain (loss) on available-for-sale securities | — | — | — | 282 | — | 282 | ||||||||||||||||||
| Comprehensive loss | — | — | — | — | — | (48,651 | ) | |||||||||||||||||
| Balance at December 31, 2007 | 36,926,566 | $ | 4 | $ | 423,541 | $ | (32 | ) | $ | (270,019 | ) | $ | 153,494 | |||||||||||
See accompanying notes.
54
Table of Contents
MAXYGEN, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
Operating activities | ||||||||||||
| Net loss | $ | (18,436 | ) | $ | (16,482 | ) | $ | (49,315 | ) | |||
| Cumulative effect adjustment | (16,616 | ) | — | — | ||||||||
| Net loss from continuing operations | (35,052 | ) | (16,482 | ) | (49,315 | ) | ||||||
| Adjustments to reconcile net loss from continuing operations to net cash used in operating activities: | ||||||||||||
| Depreciation and amortization | 3,530 | 2,294 | 1,671 | |||||||||
| Equity in losses of minority investee | — | 1,000 | — | |||||||||
| Gain on sale of equity investment | — | (17,662 | ) | — | ||||||||
| Non-cash stock compensation | 423 | 6,154 | 6,644 | |||||||||
| Common stock issued and stock options granted to consultants for services rendered and for certain technology rights | 110 | 914 | 560 | |||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Related party receivable | — | — | (7,493 | ) | ||||||||
| Accounts receivable and other receivables | (5,003 | ) | 1,977 | 2,716 | ||||||||
| Prepaid expenses and other current assets | 17 | 783 | 450 | |||||||||
| Deposits and other assets | 25 | 249 | — | |||||||||
| Accounts payable | 902 | 895 | 436 | |||||||||
| Accrued compensation | (583 | ) | 385 | 2,172 | ||||||||
| Accrued restructuring charges | — | — | 4,413 | |||||||||
| Accrued program termination costs | (2,209 | ) | — | — | ||||||||
| Accrued project costs | 1,183 | 224 | 2,380 | |||||||||
| Other accrued liabilities | 159 | (383 | ) | (301 | ) | |||||||
| Taxes payable | (921 | ) | 140 | (140 | ) | |||||||
| Deferred revenue | 7,542 | (2,592 | ) | (5,593 | ) | |||||||
| Net cash used in operating activities | (29,877 | ) | (22,104 | ) | (41,400 | ) | ||||||
Investing activities | ||||||||||||
| Purchases of available-for-sale securities | (188,635 | ) | (146,046 | ) | (179,619 | ) | ||||||
| Maturities of available-for-sale securities | 209,767 | 171,587 | 247,590 | |||||||||
| Investment in minority investee | — | (1,000 | ) | — | ||||||||
| Acquisition of property and equipment | (2,240 | ) | (1,488 | ) | (1,469 | ) | ||||||
| Cash used in acquisition, net of cash acquired | (2,617 | ) | — | — | ||||||||
| Proceeds from the sale of Avidia | — | 17,662 | — | |||||||||
| Net cash provided by investing activities | 16,275 | 40,715 | 66,502 | |||||||||
Financing activities | ||||||||||||
| Repayments under equipment financing obligation | (115 | ) | — | — | ||||||||
| Borrowings under equipment financing obligation | 1,229 | — | — | |||||||||
| Proceeds from issuance of common stock | 1,102 | 1,008 | 5,142 | |||||||||
| Net cash provided by financing activities | 2,216 | 1,008 | 5,142 | |||||||||
| Codexis related net adjustment | (2,365 | ) | — | — | ||||||||
| Effect of exchange rate changes on cash and cash equivalents | 276 | (55 | ) | 382 | ||||||||
| Net increase (decrease) in cash and cash equivalents | (13,475 | ) | 19,564 | 30,626 | ||||||||
| Cash and cash equivalents at beginning of period | 40,415 | 26,940 | 46,504 | |||||||||
| Cash and cash equivalents at end of period | $ | 26,940 | $ | 46,504 | $ | 77,130 | ||||||
Supplemental Cash Flow Information | ||||||||||||
| Non-cash transactions: | ||||||||||||
| Codexis common stock issued in acquisition | $ | (188 | ) | $ | — | $ | — | |||||
| Cash paid during the period for interest | $ | 41 | $ | 15 | $ | — | ||||||
| Cash paid during the period for income taxes | $ | 912 | $ | — | $ | 140 | ||||||
See accompanying notes.
55
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| 1. | Summary of Significant Accounting Policies |
Organization and Principles of Consolidation
Maxygen, Inc. (the “Company”) was incorporated under the laws of the State of Delaware on May 7, 1996. The Company is a biotechnology company committed to the discovery and development of improved next-generation protein pharmaceuticals for the treatment of disease and serious medical conditions. The Company began operations in March 1997 with the mission to develop important commercial products through the use of biotechnology. Since then, the Company has established a focus in human therapeutics, particularly on the development and commercialization of optimized protein pharmaceuticals.
The Company will require additional financial resources to complete the development and commercialization of its product candidates. The Company’s management may finance the Company’s operations through issuances of equity securities, collaborative research and development arrangements, government grants, or debt financing.
The consolidated financial statements include the amounts of the Company and its wholly-owned subsidiaries, Maxygen ApS (Denmark) (“Maxygen ApS”), which was acquired by the Company in August 2000, and Maxygen Holdings Ltd. (Cayman Islands) (“Maxygen Holdings”). For the two months ended February 28, 2005, the results of operations of Codexis, Inc. (“Codexis”), a biotechnology company focused on developing biocatalytic process technologies for pharmaceutical, energy and industrial chemical applications, are also included in the Consolidated Financial Statements. Subsequent to February 28, 2005, the Company’s investment in Codexis is reflected using the equity method of accounting.
The Company’s ownership in Codexis as of December 31, 2004 was approximately 51.4%, based upon the voting rights of the issued and outstanding shares of Codexis common and preferred stock. In accordance with Emerging Issues Task Force Consensus96-16, “Investor Accounting for an Investee When the Investor Has a Majority of the Voting Interest but the Minority Stockholder or Stockholders Have Certain Approval or Veto Rights”(“EITF 96-16”) and paragraph 1 of Accounting Research Bulletin No. 51, “Consolidated Financial Statements” (“ARB 51”), the Company has included 100% of the net losses of Codexis in the determination of the Company’s consolidated net loss. As a result of the issuance of Codexis common stock in connection with the acquisition by Codexis of Julich Fine Chemicals GmbH and certain other matters that occurred in the first quarter of 2005, the Company’s voting rights of the issued and outstanding shares of Codexis common and preferred stock were reduced below 50%. As a result, as of February 28, 2005, the date upon which such rights fell below 50%, the Company no longer consolidates the financial results of Codexis. In accordance with APB 18, “The Equity Method of Accounting for Investments in Common Stock,” the Company accounts for its investment in Codexis under the equity method of accounting after February 28, 2005.
The Company holds shares of Codexis series B redeemable convertible preferred stock. In connection with the redemption rights of the Codexis series B stockholders, the Company has recorded accretion of the redemption premium for the series B redeemable convertible preferred stock, excluding shares owned by the Company, in the amount of $167,000 for the year ended December 31, 2005. The accretion is recorded as subsidiary preferred stock accretion on the Consolidated Statements of Operations and as a reduction of additional paid-in capital and an increase to minority interest on the Consolidated Balance Sheets. No accretion was recorded for the years ended December 31, 2006 and 2007. Any obligation to make redemption payments is solely an obligation of Codexis and any payments are to be made solely from assets of Codexis.
As of December 31, 2005, the Company had recorded losses equal to its investment basis in Codexis. In May 2006, the Company purchased $600,000 of secured subordinated convertible promissory notes and, in August 2006, the notes and accrued interest were converted into Codexis preferred stock and the Company purchased approximately $400,000 of additional preferred stock. Subsequent to its investments in May and August 2006, the Company recorded losses of $1.0 million in 2006 under the equity method of accounting and, at December 31, 2006, had recorded losses equal to its investment basis in Codexis. The Company’s investment basis in Codexis as of December 31, 2006 and 2007 was zero. The Company is not obligated to fund the operations or other capital
56
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
requirements of Codexis. As of December 31, 2007, the Company’s equity interest in Codexis was approximately 25%.
Until March 31, 2005, the Company’s investment in Avidia Inc. (“Avidia”) was accounted for under the equity method of accounting and the Company’s share of Avidia’s results was recorded to the extent of the Company’s accounting basis in Avidia as a component of equity in net loss of minority investee in the Consolidated Statements of Operations. After March 31, 2005, the Company’s investment in Avidia was accounted for under the cost method of accounting. As of December 31, 2005, the Company had recorded losses equal to its investment basis in Avidia.
On October 24, 2006, Amgen Inc. (“Amgen”) completed the acquisition of Avidia. At the time of the acquisition of Avidia by Amgen, the Company’s basis in Avidia was zero. As a result of the acquisition, the Company received cash proceeds of approximately $17.8 million (before $140,000 of income taxes) in the fourth quarter of 2006 in exchange for its equity interests in Avidia and may receive up to an additional $1.4 million in cash, contingent upon the development of certain Avidia products by Amgen. This contingent amount was reduced from $2.8 million based on the discontinuation by Amgen Inc. of certain development activities. Accordingly, the Company recorded a gain on disposal of this investment of approximately $17.8 million in the fourth quarter of 2006. Any additional gain as a result of the contingent amounts potentially payable to the Company by Amgen will be recognized only if and when the contingency is satisfied. See Note 12.
Cumulative Effect Adjustment
Codexis was formed in January 2002 and financed by the Company and several other independent investors. In accordance withEITF 96-16 and ARB 51, through February 28, 2005, the Company consolidated 100% of the operating results of Codexis, even though it only owned a majority of the voting interests in Codexis. From March 2002 through February 28, 2005, the Company had recorded cumulative losses of Codexis of $26.4 million, which was in excess of the Company’s investment basis of $9.8 million. On February 28, 2005, the Company’s voting rights in Codexis were reduced below 50%. As a result, the Company no longer consolidates the financial position and results of operations of Codexis with the Company’s financial results as of such date and instead accounts for Codexis under the equity method of accounting. To reflect what the Company’s basis in Codexis would have been under equity accounting as required byEITF 96-16, the Company recorded a cumulative effect adjustment of $16.6 million in the first quarter of 2005 to bring its investment basis in Codexis to zero as of February 28, 2005. This cumulative effect adjustment does not have any tax consequences.
Foreign Currency Translation
The functional currency of Maxygen ApS was the Danish kroner through November 30, 2007. Assets and liabilities of Maxygen ApS were translated at current exchange rates. Revenues and expenses were translated at average exchange rates in effect during the period. Gains and losses from currency translation were included in accumulated other comprehensive loss. However, as the result of the Company’s plan to consolidate its research and development activities at its U.S. facilities in Redwood City, California and cease operations at Maxygen ApS, the Company has reevaluated the functional currency for its Danish operations and determined that it is the U.S. dollar after December 1, 2007. After December 1, 2007, nonmonetary assets and liabilities which are denominated in currencies other than the U.S. dollar have been remeasured into U.S. dollars at historical exchange rates beginning with the November 30, 2007 exchange rates. Translation adjustments from prior periods will continue to remain in accumulated other comprehensive loss. Currency transaction gains or losses are included in interest income and other income (expense), net.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires the Company’s management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
57
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Reclassifications
Stock compensation expenses of $183,000 in the year ended December 31, 2005 have been reclassified within operating expenses (into either research and development expenses or general and administrative expenses) in the Consolidated Statements of Operations as a result of the Company’s implementation of Statement of Financial Accounting Standards (“SFAS”) No. 123(R), “Share-Based Payment” (“SFAS 123(R)”), effective January 1, 2006. Previously, stock compensation expense had been presented separately on the face of the Consolidated Statements of Operations.
Cash, Cash Equivalents and Investments
The Company considers all highly liquid investments with original maturity dates of three months or less, as of the date of purchase, to be cash equivalents. Cash equivalents include marketable debt securities, government and corporate debt obligations. Short and long-term investments include government and corporate debt obligations.
The Company’s management determines the appropriate classification of debt securities as current or non-current at the time of purchase and reevaluates such designation as of each balance sheet date. The Company’s debt securities are classified as available-for-sale and are carried at estimated fair value in cash equivalents and investments. Unrealized gains and losses are reported as accumulated other comprehensive income (loss) in stockholders’ equity. The amortized cost of debt securities in this category is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization is included in interest income and other income (expense), net. Realized gains and losses on available-for-sale securities and declines in value deemed to be other than temporary, if any, are included in interest income and other income (expense), net. The cost of securities sold is based on the specific identification method. Interest and dividends on securities classified as available-for-sale are included in interest income and other income (expense), net.
Derivatives and Financial Instruments
The Company addresses certain financial exposures through a program of risk management that includes the use of derivative financial instruments. The Company generally enters into foreign currency forward exchange contracts that expire within eighteen months to reduce the effects of fluctuating foreign currency exchange rates on forecasted cash requirements.
The Company accounts for derivative instruments under the provisions of SFAS No. 133, “Accounting for Derivative Instruments and Hedging Activities” (“SFAS 133”), as amended, which requires that all derivative instruments be reported on the balance sheet at fair value and establishes criteria for designation and evaluating effectiveness of hedging relationships.
Derivatives that are designated as foreign currency cash flow hedges are recognized on the balance sheet at their fair value. Changes in the fair value of derivatives that are highly effective as, and that are designated and qualify as, foreign currency cash flow hedges are recorded in other comprehensive income until the associated hedged transaction impacts earnings. Changes in the fair value of derivatives that are ineffective are recorded as interest income and other income (expense), net in the period of change.
Under hedge accounting, the Company documents all relationships between hedging instruments and hedged items, as well as its risk-management objective and strategy for undertaking various hedge transactions. The Company also assesses, both at the hedge’s inception and on an ongoing basis, whether the derivatives that are used in hedging transactions are highly effective in offsetting changes in fair values or cash flows of hedged items. When it is determined that a derivative is not highly effective as a hedge or that it has ceased to be a highly effective hedge, the Company discontinues hedge accounting prospectively.
The purpose of the hedging activities is to minimize the effect of foreign currency exchange rate movements on the cash flows related to the Company’s funding of Maxygen ApS and payments to vendors in Europe. To date,
58
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
foreign currency contracts are denominated in Danish kroner and euros. At December 31, 2007, the Company had foreign currency contracts outstanding in the form of forward exchange contracts totaling $8.8 million. These contracts were entered into in December of 2007 to cover a substantial portion of the disbursements scheduled for the first quarter of 2008. Because of the short duration of less than 90 days, the Company made the decision to not designate these contracts as cash flow hedges and will therefore recognize changes in their fair value as interest income and other income (expense), net in the period of change. During 2007, the Company recognized $77,000 in foreign exchange losses from hedge contracts and these losses were included with interest and other income, net. The Company had no foreign currency contracts outstanding at December 31, 2006.
As a matter of policy, the Company only enters into contracts with counterparties that have at least an “A” (or equivalent) credit rating. The counterparties to these contracts are major financial institutions. Exposure to credit loss in the event of nonperformance by any of the counterparties is limited to only the recognized, but not realized, gains attributable to the contracts. Management believes risk of loss is remote and in any event would not be material. Costs associated with entering into such contracts have not been material to the Company’s financial results. The Company does not utilize derivative financial instruments for trading or speculative purposes.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to a concentration of credit risk consist of investments and accounts receivable. The Company is exposed to credit risks in the event of default by the financial issuers or collaborators to the extent of the amount recorded on the balance sheet. A portion of the Company’s accounts receivable balance at December 31, 2006 and 2007 consisted of balances due from government agencies. Each grant agreement is subject to funding approvals by the U.S. government. Certain grant agreements provide an option for the government to audit the amount of research and development expenses, both direct and indirect, that have been submitted to the government agency for reimbursement. The Company does not require collateral or other security to support the financial instruments subject to credit risk.
Property and Equipment
Property and equipment, including the cost of purchased software, are stated at cost, less accumulated depreciation. Depreciation is provided using the straight-line method over the estimated useful life of the assets (generally three to five years). Leasehold improvements are amortized over the shorter of the lease term or the estimated useful life of the assets.
In November 2007, the Company implemented a plan to consolidate its organization to reduce costs and increase overall operational efficiency across its research, preclinical, clinical and regulatory activities. The consolidation has resulted in the cessation of research and development operations at Maxygen ApS, the Company’s wholly owned subsidiary in Denmark, and the elimination of all employment positions at that site. In connection with the cessation of operations at Maxygen ApS, the Company has determined that the remaining useful lives of certain of the fixed assets at Maxygen ApS have been shortened to three months for equipment and to six months for leasehold improvements at November 30, 2007. The remaining book value of these assets is depreciated over the shorter estimated remaining useful lives and the depreciation expense is reflected in research and development expenses on the Consolidated Statements of Operations.
Goodwill and Other Intangible Assets
In connection with the acquisition of Maxygen ApS in 2000, the Company allocated $26.2 million to goodwill and other intangible assets. Prior to adoption of SFAS No. 142 “Goodwill and Other Intangible Assets” (“SFAS 142”) in 2002, the Company amortized a portion of the goodwill each year. As of December 31, 2001, the net goodwill balance was $12.2 million. Beginning on January 1, 2002, goodwill is no longer amortized and goodwill and other intangible assets are generally evaluated on an individual acquisition or market basis at least annually whenever events or changes in circumstances indicate that such assets are impaired or the estimated useful
59
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
lives are no longer appropriate. In accordance with SFAS 142, the Company reviews its intangible assets (including goodwill) for impairment at least annually based on estimated future discounted cash flows attributable to the assets and other factors to determine the fair value of the respective assets. In the event such cash flows are not expected to be sufficient to recover the recorded value of the assets, the assets will be written down to their estimated fair values. No impairment charges were recorded in 2005, 2006 or 2007.
The valuation in connection with the initial purchase price allocation and the ongoing evaluation for impairment of goodwill and intangible assets requires significant management estimates and judgment. The purchase price allocation process requires management estimates and judgment as to expectations for various products and business strategies. If any of the significant assumptions differ from the estimates and judgments used in the purchase price allocation, this could result in different valuations for goodwill and intangible assets. Once it is established, the Company must test goodwill annually for impairment using a two-step process as required by SFAS 142. In addition, in certain circumstances, the Company must assess if goodwill should be tested for impairment between annual tests. Intangible assets with definite useful lives must be tested for impairment in accordance with Statement of Financial Accounting Standard No. 144 “Accounting for the Impairment or Disposal of Long-Lived Assets.” When the Company conducts its impairment tests for goodwill and intangibles, factors that are considered important in determining whether impairment might exist include existing product portfolio, product development cycle, development expenses, potential royalties and product sales, costs of goods and selling expenses and overall product lifecycle. Any changes in key assumptions about the business and its prospects, or changes in market conditions or other external events, could result in an impairment charge and such a charge could have a material adverse effect on the Company’s consolidated results of operations.
Long-Lived Assets
The Company reviews long-lived assets including intangible assets with finite lives for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable, such as a significant industry downturn, significant decline in the market value of the Company, or significant reductions in projected future cash flows. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition is less than its carrying amount. The amount of the impairment loss, if any, is determined using discounted cash flows. In assessing the recoverability of long-lived assets, including intangible assets, the Company must make assumptions regarding estimated future cash flows and other factors to determine the fair value of the respective assets.
Revenue Recognition
The Company has generally recognized revenue from multiple element arrangements under collaborative research agreements, including license payments, research and development services, milestones, and royalties. Revenue arrangements with multiple deliverables are accounted for under the provisions of Staff Accounting Bulletin No. 104 and Emerging Issues Task Force IssueNo. 00-21, “Revenue Arrangements with Multiple Deliverables,” and are divided into separate units of accounting if certain criteria are met, including whether the delivered item has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items in the arrangement. The consideration the Company receives is allocated among the separate units of accounting based on their respective fair values, and the applicable revenue recognition criteria are considered separately for each of the separate units.
Non-refundable upfront payments received in connection with collaboration agreements, including license fees, and technology advancement funding that is intended for the development of the Company’s core technologies, are deferred upon receipt and recognized as revenue over the relevant research and development periods specified in the agreement. Under arrangements where the Company expects its research and development obligations to be performed evenly over the specified period, the upfront payments are recognized on a straight-line basis over the period. Under arrangements where the Company expects its research and development obligations to
60
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
vary significantly from period to period, the Company recognizes the upfront payments based upon the actual amount of research and development efforts incurred relative to the amount of the total expected effort to be incurred by the Company. In cases where the planned levels of research services fluctuate substantially over the research term, this requires the Company to make critical estimates in both the remaining time period and the total expected costs of its obligations and, therefore, a change in the estimate of total costs to be incurred or in the remaining time period could have a significant impact on the revenue recognized in future periods.
Revenue related to collaborative research payments from a collaborator is recognized as research services are performed over the related funding periods for each contract. Under these agreements, the Company is typically required to perform research and development activities as specified in the respective agreement. Generally, the payments received are not refundable and are based on a contractual cost per full-time equivalent employee working on the project. Under certain collaborative research and development agreements, the Company and the collaborative partner may agree to share in the costs of research and development. In periods where the Company incurs more costs than the collaborative partner, payments from the collaborative partner are included in collaborative research and development revenues and, in periods where the collaborative partner incurs more expenses than the Company, the Company’s payments to the collaborative partner are included in research and development expenses. Research and development expenses (including associated general and administrative expenses) under the collaborative research agreements approximate or exceed the research funding revenue recognized under such agreements over the term of the respective agreements. Deferred revenue may result when the Company does not incur the required level of effort during a specific period in comparison to funds received under the respective contracts.
Payments received relating to substantive, at-risk incentive milestones, if any, are recognized as revenue upon achievement of the incentive milestone event because the Company has no future performance obligations related to the payment. Incentive milestone payments may be triggered either by the results of the Company’s research efforts or by events external to the Company, such as regulatory approval to market a product.
The Company is eligible to receive royalties from licensees, which are typically based on sales of licensed products to third parties. Royalties are recorded as earned in accordance with the contract terms when third party sales can be reliably measured and collectibility is reasonably assured.
Revenue from license agreements for which no further performance obligations exist are recognized as revenue on the earlier of when payments are received or the amount can be reliably measured and collectibility is reasonably assured.
The Company has been awarded grants from various government agencies. The terms of these grant agreements range from one to five years with various termination dates, the last of which is July 2008 for existing agreements. Revenue related to these grant agreements is recognized as the related research and development expenses are incurred.
Research and Development Expenses
Research and development expenses consist of costs incurred for both Company-sponsored and collaborative research and development activities. These costs include direct and research-related overhead expenses, which include salaries and other personnel-related expenses, facility costs, supplies and depreciation of facilities and laboratory equipment, as well as research consultants and the cost of funding research at universities and other research institutions, and are expensed as incurred. Costs to acquire technologies that are utilized in research and development and that have no alternative future use are expensed when incurred. See Note 4 for detail regarding the Company’s sponsored license and research agreements.
The Company does not track fully burdened research and development costs by project. However, the Company does estimate, based on full-time equivalent personnel effort, the percentage of research and development efforts (as measured in hours incurred, which approximates costs) undertaken for projects funded by the
61
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Company’s collaborators and government grants, on the one hand, and projects funded by the Company, on the other hand. To approximate research and development expenses by funding category, the number of hours expended in each category has been multiplied by the approximate cost per hour of research and development effort and added to project-specific external costs. In the case where a collaborative partner is sharing the research and development costs, the expenses for that project are allocated proportionately between the collaborative projects funded by third parties and internal projects. The Company believes that presenting its research and development expenses in these categories will provide its investors with meaningful information on how the Company’s resources are being used.
The following table presents the Company’s approximate research and development expenses by funding category (in thousands):
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| Collaborative projects funded by third parties(1) | $ | 4,146 | $ | 9,906 | $ | 2,713 | ||||||
| Government grants | 2,369 | 4,215 | 5,106 | |||||||||
| Internal projects | 35,389 | 35,009 | 52,032 | |||||||||
| Total | $ | 41,904 | $ | 49,130 | $ | 59,851 | ||||||
| (1) | Research and development expenses related to collaborative projects funded by third parties are less than the reported revenues due to the amortization of non-refundable upfront payments, as well as a portion of the collaborative research and development revenue that is charged for general and administrative expenses. |
Accounting for Clinical Trial Costs
The Company charges research and development costs, including clinical study costs, to expense when incurred, consistent with SFAS No. 2,“Accounting for Research and Development Costs.” Clinical study costs are a significant component of research and development expenses. Most of the Company’s clinical studies are performed by a third-party contract research organization (CRO). The clinical trials generally have three distinctive stages plus pass through costs:
| • | start-up — initial setting up of the trial; | |
| • | enrollment of patients in the trial; and | |
| • | close down and reporting of the trial. |
The Company reviews the list of expenses for the trial from the original signed agreements and separates them into what it perceives asstart-up, enrollment or closedown expenses of the clinical trial. The start up costs, which usually occur within a few months after the contract has been executed and include costs, such as study initiation, site recruitment, regulatory applications and investigator meetings, are expensed ratably over the start up period. The site management, study management, medical and safety monitoring and data management expenses are calculated on a per patient basis and expensed ratably over the treatment period beginning on the date that the patient enrolls. The close down and reporting expenses are expensed ratably over the close out period of time. Pass through costs, including the costs of the drugs, are expensed as incurred.
In general, the Company’s service agreements permit us to terminate at will where we would continue to be responsible for payment of all services completed (or pro-rata completed) at the time of notice of termination, plus any non-cancellable expenses that have been entered into by the CRO on the Company’s behalf.
62
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Restructuring Charge
In November 2007, the Company implemented a plan to consolidate its research and development activities at its U.S. facilities in Redwood City, California. The consolidation has resulted in the cessation of research and development operations at Maxygen ApS, the Company’s wholly owned subsidiary in Denmark. In connection with the consolidation, the Company has recorded estimated expenses for severance and outplacement costs and other restructuring costs. In accordance with SFAS No. 146,“Accounting for Costs Associated with Exit or Disposal Activities” (“SFAS 146”), generally costs associated with restructuring activities have been recognized when they are incurred rather than at the date of a commitment to an exit or disposal plan. However, in the case of leases, the expense is estimated and accrued when the property is vacated or at the point when we cease to use the leased equipment. Given the significance of, and the timing of the execution of such activities, this process is complex and involves periodic reassessments of estimates made at the time the original decisions were made, including estimating the salvage value of equipment consistent with abandonment date. In addition, post-employment benefits accrued for workforce reductions related to restructuring activities are accounted for under SFAS No. 112,“Employer’s Accounting for Post-employment Benefits” (“SFAS 112”). A liability for post-employment benefits is recorded when payment is probable, the amount is reasonably estimable, the obligation is attributable to employees’ services already rendered and the obligation relates to rights that have vested or accumulated. The Company continually evaluates the adequacy of the remaining liabilities under this restructuring. Although the Company believes that these estimates accurately reflect the costs of its restructuring plans, actual results may differ, thereby requiring the Company to record additional provisions or reverse a portion of such provisions.
Stock-Based Compensation
As of December��31, 2007, the Company had five stock option plans: the 2006 Equity Incentive Plan (the “2006 Plan”); the 1997 Stock Option Plan (the “1997 Plan”); the 1999 Nonemployee Directors Stock Option Plan; the 2000 International Stock Option Plan; and the 2000 Non-Officer Stock Option Plan. These stock plans generally provide for the grant of stock options to employees, directorsand/or consultants. The 2006 Plan, which has replaced the 1997 Plan as to future awards, also provides for the grant of additional equity-based awards, including stock appreciation rights, restricted stock, restricted stock units, performance shares, performance units and dividend equivalents. In connection with stockholder approval of the 2006 Plan, the 1997 Plan was terminated as to future awards. The Company also has an Employee Stock Purchase Plan (“ESPP”) that enables eligible employees to purchase Company common stock.
Effective January 1, 2006, the Company adopted the provisions of SFAS 123(R), which requires companies to recognize the cost of employee services received in exchange for awards of equity instruments based upon the grant-date fair value of those awards. The fair value of stock options and ESPP shares is estimated using the Black-Scholes-Merton option valuation model. This model requires the input of subjective assumptions in applying SFAS 123(R), including expected stock price volatility, estimated life and estimated forfeitures of each award. The Company has adopted SFAS 123(R) using the modified prospective transition method. Under this transition method, compensation cost recognized during the years ended December 31, 2006 and December 31, 2007 includes: (a) compensation cost for all share-based payments granted prior to, but not yet vested as of, January 1, 2006, based on the grant date fair value estimated in accordance with the original provisions of SFAS No. 123, “Accounting for Stock-Based Compensation,” (“SFAS 123”) amortized on a graded vesting basis over the options’ vesting period, and (b) compensation cost for all share-based payments granted subsequent to January 1, 2006, based on the grant-date fair value estimated in accordance with the provisions of SFAS 123(R) amortized on a straight-line basis over the options’ vesting period. Under this method of implementation, no restatement of prior periods has been made.
Stock-based compensation expense recognized under SFAS 123(R) in the Consolidated Statements of Operations for the year ended December 31, 2007 was $5.8 million related to employee stock options, $666,000 related to consultant stock options, $80,000 related to the ESPP. Restructuring charges for 2007 included
63
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
$287,000 of stock-based compensation expense related to the extension of the exercise period of certain stock options held by affected employees of Maxygen ApS, as required under Danish law in connection with the termination of such employees. Stock-based compensation expense recognized under SFAS 123(R) for the year ended December 31, 2006 was $5.7 million related to employee stock options, $914,000 related to consultant stock options and $87,000 related to the ESPP. As a result of its application of SFAS 123(R), the Company’s net loss for the years ended December 31, 2006 and 2007 increased by $6.7 million and $5.8 million, respectively. SFAS 123(R) also increased basic and fully diluted loss per share from continuing operations by $0.19 and $0.16 for the years ended December 31, 2006 and 2007, respectively. The application of SFAS 123(R) did not have an impact on cash flows from operations during the years ended December 31, 2006 and 2007.
Prior to January 1, 2006, the Company measured compensation expense for its employee equity-based compensation plans using the intrinsic value method under the provisions of Accounting Principles Board Opinion No. 25, “Accounting for Stock Issued to Employees” and related interpretations. As the exercise price of all options granted under these plans was not below the fair market price of the underlying common stock on the grant date, no equity-based compensation cost was recognized in the Consolidated Statements of Operations under the intrinsic value method.
Stock Options
The exercise price of each stock option equals the closing market price of the Company’s stock on the date of grant. Most options are scheduled to vest over four years and all options expire no later than 10 years from the grant date. The fair value of each option grant is estimated on the date of grant using the Black-Scholes-Merton option pricing model. This model was developed for use in estimating the value of publicly traded options that have no vesting restrictions and are fully transferable. The Company’s employee stock options have characteristics significantly different from those of publicly traded options.
As part of its adoption of SFAS 123(R), the Company also examined its historical pattern of option exercises in an effort to determine if there were any discernable activity patterns based on certain employee populations. From this analysis, the Company identified no discernable activity patterns other than the employee populations for its U.S. and Danish operations. The Company uses the Black-Scholes-Merton option pricing model to value the options for each of the employee populations. The weighted average assumptions used in the model for each employee population are outlined in the following table:
| Year Ended December 31, | ||||||||
| 2006 | 2007 | |||||||
| Danish | Danish | |||||||
| U.S. Employees | Employees | U.S. Employees | Employees | |||||
| Expected dividend yield | 0% | 0% | 0% | 0% | ||||
| Risk-free interest rate range — Options | 4.30% to 5.11% | 4.34% to 5.17% | 3.81% to 4.90% | 4.76% to 4.97% | ||||
| Risk-free interest rate range — ESPP | 3.20% to 5.05% | — | 4.74% to 5.09% | — | ||||
| Expected life — Options | 5.13 years | 2.4 years | 5.70 years | 2.4 years | ||||
| Expected life — ESPP | 0.48 years to 0.92 years | — | 0.50 years to 0.94 years | — | ||||
| Expected volatility — Options | 52.43% to 56.42% | 44.38% to 45.78% | 50.93% to 52.96% | 44.88% to 45.99% | ||||
| Expected volatility — ESPP | 42.97% to 51.05% | — | 44.76% to 48.31% | — | ||||
The computation of the expected volatility assumption used in the Black-Scholes-Merton calculations for new grants is based on a combination of historical and implied volatilities. When establishing the expected life assumption, the Company reviews annual historical employee exercise behavior of option grants with similar vesting periods.
64
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
A summary of the changes in stock options outstanding under the Company’s equity-based compensation plans during the year ended December 31, 2007 is presented below:
| Weighted- | ||||||||||||||||
| Weighted- | Average | |||||||||||||||
| Average | Remaining | |||||||||||||||
| Exercise | Contractual | Aggregate | ||||||||||||||
| Price per | Term | Intrinsic | ||||||||||||||
| Shares | Share | (In Years) | Value | |||||||||||||
| (In thousands) | ||||||||||||||||
| Options outstanding at December 31, 2006 | 10,686,460 | $ | 13.13 | |||||||||||||
| Granted | 1,960,730 | 10.00 | ||||||||||||||
| Exercised | (670,018 | ) | 7.10 | |||||||||||||
| Canceled | (1,035,651 | ) | 10.59 | |||||||||||||
| Expired | (49,751 | ) | 8.11 | |||||||||||||
| Options outstanding at December 31, 2007 | 10,891,770 | $ | 13.20 | 5.81 | $ | 4,003 | ||||||||||
| Options vested and expected to vest at December 31, 2007 | 10,666,320 | $ | 13.28 | 5.76 | $ | 3,958 | ||||||||||
| Options exercisable at December 31, 2007 | 7,899,125 | $ | 14.66 | 4.83 | $ | 3,445 | ||||||||||
The intrinsic value of options exercised during the years ended December 31, 2005, 2006 and 2007 was $855,000, $594,000 and $2.5 million, respectively. The estimated fair value of shares vested during the years ended December 31, 2005, 2006 and 2007 was $11.4 million, $11.1 million and $12.9 million, respectively. The weighted average grant date fair value of options granted during the year ended December 31, 2007 was $10.00. At December 31, 2007, the Company had $11.4 million of total unrecognized compensation expense, net of estimated forfeitures, related to stock options that will be recognized over the weighted average remaining vesting period of 2.50 years. Cash received from stock option exercises and purchases under the ESPP was $5.1 million during the year ended December 31, 2007.
The following table summarizes outstanding and exercisable options at December 31, 2007:
| Options Outstanding | Options Exercisable | |||||||||||||||||||
| Weighted-Average | ||||||||||||||||||||
| Number of | Remaining | Number of | ||||||||||||||||||
| Range of | Shares | Contractual Life | Weighted-Average | Shares | Weighted-Average | |||||||||||||||
Exercise Prices | Outstanding | (In Years) | Exercise Price | Outstanding | Exercise Price | |||||||||||||||
| $ 0.30 - $ 7.00 | 1,220,881 | 5.79 | $ | 5.80 | 950,262 | $ | 5.48 | |||||||||||||
| $ 7.08 - $ 7.40 | 1,136,207 | 5.25 | $ | 7.33 | 1,106,207 | $ | 7.34 | |||||||||||||
| $ 7.47 - $ 7.89 | 1,495,890 | 7.29 | $ | 7.72 | 781,461 | $ | 7.71 | |||||||||||||
| $ 7.92 - $10.33 | 1,686,565 | 7.54 | $ | 8.97 | 906,666 | $ | 9.24 | |||||||||||||
| $10.41 - $10.76 | 1,512,245 | 6.23 | $ | 10.59 | 1,057,844 | $ | 10.57 | |||||||||||||
| $10.94 - $12.44 | 1,698,123 | 6.46 | $ | 11.90 | 954,826 | $ | 11.95 | |||||||||||||
| $12.99 - $17.20 | 933,191 | 3.28 | $ | 13.86 | 933,191 | $ | 13.86 | |||||||||||||
| $17.41 - $53.75 | 779,002 | 2.79 | $ | 35.88 | 779,002 | $ | 35.88 | |||||||||||||
| $56.88 - $59.81 | 429,666 | 2.40 | $ | 57.17 | 429,666 | $ | 57.17 | |||||||||||||
| 10,891,770 | 5.81 | $ | 13.20 | 7,899,125 | $ | 14.66 | ||||||||||||||
Employee Stock Purchase Plan
Under the ESPP, eligible employees may purchase common stock at a discount, through payroll deductions, during defined offering periods. The price at which stock is purchased under the ESPP is equal to 85% of the lower
65
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
of (i) the fair market value of the common stock on the first day of the offering period or (ii) the fair market value of the common stock on the purchase date. During the year ended December 31, 2007, 54,383 shares of common stock were purchased pursuant to the ESPP. The weighted average fair value of purchase rights granted during the year ended December 31, 2007 was $0.48. Compensation expense is calculated using the fair value of the employees’ purchase rights under the Black-Scholes-Merton model. For the year ended December 31, 2007, ESPP compensation expense was $80,000.
Pro Forma Information under SFAS 123 for Periods Prior to 2006
Prior to January 1, 2006, the Company followed the disclosure-only provisions of SFAS 123. The following table illustrates the effect on net loss and loss per common share for the year ended December 31, 2005 if the fair value recognition provisions of SFAS 123, as amended, had been applied to options granted under the Company’s equity-based employee compensation plans. For purposes of this pro forma disclosure, the estimated value of the options is recognized over the options’ vesting periods. If the Company had recognized the expense of equity programs in the Consolidated Statements of Operations, additional paid-in capital would have increased by a corresponding amount, net of applicable taxes.
| Year Ended | ||||
| December 31, | ||||
| 2005 | ||||
| (In thousands) | ||||
| Net loss, as reported | $ | (18,436 | ) | |
| Add: Stock based employee compensation cost included in the determination of net loss, as reported | — | |||
| Deduct: Total stock based employee compensation expense determined under the fair value-based method for all awards | (5,867 | ) | ||
| Pro forma net loss | $ | (24,303 | ) | |
| Net loss per common share: | ||||
| Basic and diluted — as reported | $ | (0.52 | ) | |
| Basic and diluted — pro forma | $ | (0.68 | ) | |
For purposes of the weighted average estimated fair value calculations, the fair value of each stock option grant is estimated on the date of grant using the Black-Scholes-Merton option pricing model and the following assumptions:
| Year Ended | ||
| December 31, | ||
| 2005 | ||
| Expected dividend yield | 0% | |
| Risk-free interest rate range — Options | 3.71% to 4.39% | |
| Risk-free interest rate range — ESPP | 1.23% to 3.38% | |
| Expected life — Options | 4 years | |
| Expected life — ESPP | 0.7 years | |
| Expected volatility | 0.5 |
Based on the Black-Scholes-Merton option pricing model, the weighted average estimated fair value of employee stock option grants was $5.28 per share for the year ended December 31, 2005 and the weighted average fair value of purchase rights granted under the ESPP was $2.01 per share for the year ended December 31, 2005.
66
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Valuation and Expense Information under SFAS 123(R)
For the years ended December 31, 2006 and 2007, stock-based compensation expense related to employee stock options and employee stock purchases under SFAS 123(R) and stock-based compensation expense related to consultant stock options was allocated as follows (in thousands):
| Year Ended | Year Ended | |||||||
| December 31, | December 31, | |||||||
| 2006 | 2007 | |||||||
| Research and development | $ | 2,128 | $ | 3,000 | ||||
| General and administrative | 4,615 | 3,550 | ||||||
| Restructuring charge | — | 287 | ||||||
| Stock-based compensation expense before income taxes | 6,743 | 6,837 | ||||||
| Income tax benefit | — | — | ||||||
| Total stock-based compensation expense after income taxes | $ | 6,743 | $ | 6,837 | ||||
There was no capitalized stock-based employee compensation cost as of December 31, 2007. There were no recognized tax benefits during the year ended December 31, 2007.
Income (Loss) Per Common Share
Basic and diluted income (loss) per common share has been computed using the weighted-average number of shares of common stock outstanding during the period.
The following table presents the calculation of basic and diluted income (loss) per common share (in thousands, except per share data):
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| Loss applicable to common stockholders | $ | (18,603 | ) | $ | (16,482 | ) | $ | (49,315 | ) | |||
| Basic and diluted: | ||||||||||||
| Weighted-average shares of common stock outstanding | 35,765 | 36,046 | 36,787 | |||||||||
| Less: weighted-average shares subject to repurchase | — | — | — | |||||||||
| Weighted-average shares used in computing basic and diluted income (loss) per share | 35,765 | 36,046 | 36,787 | |||||||||
| Basic and diluted net loss per common share | $ | (0.52 | ) | $ | (0.46 | ) | $ | (1.34 | ) | |||
The total number of shares excluded from the calculations of diluted income net loss per share, prior to application of the treasury stock method for options, was approximately 9,401,599 at December 31, 2005, 10,686,460 at December 31, 2006 and 10,891,770 at December 31, 2007.
67
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Accumulated Other Comprehensive Income (Loss)
Accumulated other comprehensive income (loss) is primarily comprised of net unrealized gains or losses on available-for-sale securities, foreign currency translation adjustments and changes in foreign currency contracts. The components of accumulated other comprehensive loss are as follows (in thousands):
| December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| Unrealized gain on available-for-sale securities | $ | — | $ | 46 | $ | 220 | ||||||
| Unrealized losses on available-for-sale securities | (592 | ) | (108 | ) | — | |||||||
| Foreign currency translation adjustments | (579 | ) | (634 | ) | (252 | ) | ||||||
| Changes in foreign currency contracts | (386 | ) | — | — | ||||||||
| Accumulated other comprehensive loss | $ | (1,557 | ) | $ | (696 | ) | $ | (32 | ) | |||
Recent Accounting Pronouncements
On June 27, 2007, the Financial Accounting Standards Board (“FASB”) ratified the consensus reached by the FASB Emerging Issues Task Force on IssueNo. 07-3, “Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities”(“EITF 07-3”).EITF 07-3 requires entities to defer income statement recognition of non-refundable advance payments for research and development activities, such as upfront non-refundable payments to contract research organizations, if the contracted party has not yet performed activities related to the upfront payment. Amounts deferred are to be recognized by the contracting company as expense when the research and development activities are performed. The application ofEITF 07-3 is effective for interim or annual reporting periods in fiscal years beginning after December 15, 2007. Earlier application ofEITF 07-3 is not permitted. Companies are required to report the effects of applyingEITF 07-3 prospectively for new contracts entered into after the effective date ofEITF 07-3. The Company does not expect the application ofEITF 07-3 to have material impact on its consolidated results of operations and financial condition.
In February 2007, the FASB issued SFAS No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities — Including an amendment of FASB Statement No. 115” (“SFAS 159”). SFAS 159 allows entities the option to measure eligible financial instruments at fair value as of specified dates. Such election, which may be applied on an instrument by instrument basis, is typically irrevocable once elected. SFAS 159 is effective for fiscal years beginning after November 15, 2007, and early application is allowed under certain circumstances. The Company currently is determining whether fair value accounting is appropriate for any of its eligible items and cannot estimate the impact, if any, which SFAS 159 will have on its consolidated results of operations and financial condition.
In September 2006, the FASB issued SFAS No. 157, “Fair Value Measurements” (“SFAS 157”). SFAS 157 provides guidance for using fair value to measure assets and liabilities. It also responds to investors’ request for expanded information about the extent to which companies measure assets and liabilities at fair value, the information used to measure fair value and the effect of fair value measurements on earnings. SFAS 157 applies whenever other standards require (or permit) assets or liabilities to be measured at fair value, and does not expand the use of fair value in any new circumstances. SFAS 157 is effective for financial statements issued for fiscal years beginning after November 15, 2007. The Company does not expect the application of SFAS 157 to have material impact on its consolidated results of operations and financial condition.
68
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| 2. | Cash Equivalents and Investments |
The Company’s cash equivalents and investments as of December 31, 2007 were as follows (in thousands):
| Gross | Gross | |||||||||||||||
| Amortized | Unrealized | Unrealized | Estimated | |||||||||||||
| Cost | Gains | Losses | Fair Value | |||||||||||||
| Money market funds | $ | 77,130 | $ | — | $ | — | $ | 77,130 | ||||||||
| Commercial paper | 43,704 | 211 | — | 43,915 | ||||||||||||
| Corporate bonds | 13,569 | 5 | — | 13,574 | ||||||||||||
| U.S. government agency securities | 11,190 | 4 | — | 11,194 | ||||||||||||
| Total | 145,593 | 220 | — | 145,813 | ||||||||||||
| Less amounts classified as cash equivalents | (77,130 | ) | — | — | (77,130 | ) | ||||||||||
| Total investments | $ | 68,463 | $ | 220 | $ | — | $ | 68,683 | ||||||||
The Company’s cash equivalents and investments as of December 31, 2006 were as follows (in thousands):
| Gross | Gross | |||||||||||||||
| Amortized | Unrealized | Unrealized | Estimated | |||||||||||||
| Cost | Gains | Losses | Fair Value | |||||||||||||
| Money market funds | $ | 45,258 | $ | — | $ | — | $ | 45,258 | ||||||||
| Commercial paper | ||||||||||||||||
| Corporate bonds | 20,082 | 2 | (7 | ) | 20,077 | |||||||||||
| U.S. government agency securities | 57,459 | 16 | (101 | ) | 57,374 | |||||||||||
| Commercial paper | 60,139 | 28 | — | 60,167 | ||||||||||||
| Total | 182,938 | 46 | (108 | ) | 182,876 | |||||||||||
| Less amounts classified as cash equivalents | (46,504 | ) | — | — | (46,504 | ) | ||||||||||
| Total investments | $ | 136,434 | $ | 46 | $ | (108 | ) | $ | 136,372 | |||||||
Realized gains or losses on the maturity of available-for-sale securities for 2005, 2006 and 2007 were insignificant. The change in unrealized holding gains (losses) on available-for-sale securities included in accumulated other comprehensive income (loss) were unrealized losses of $12,000 in 2005, unrealized gains of $530,000 in 2006 and unrealized gains of $282,000 in 2007.
At December 31, 2007, the contractual maturities of investments were as follows (in thousands):
| Amortized | Estimated | |||||||
| Cost | Fair Value | |||||||
| Due within one year | $ | 68,463 | $ | 68,683 | ||||
| Due after one year through two years | — | — | ||||||
| $ | 68,463 | $ | 68,683 | |||||
| 3. | Collaborative Agreements |
During 2005, 2006 and 2007, the Company recognized revenue from nine collaborative agreements. These agreements typically include, or included, upfront licensing fees, technology advancement fees and research funding, as well as the potential to earn milestone payments and royalties on future product sales or cost savings. The agreements generally require, or required, the Company to devote a specified number of full-time equivalent employees to the research efforts over defined research terms ranging from three to five years. Total revenue
69
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
recognized under these collaboration agreements was $11.6 million in 2005, $20.5 million in 2006 and $10.2 million in 2007.
The following table represents the percentage of the Company’s total revenue that has been recognized pursuant to the Company’s largest non-grant collaborators:
| December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| Partner A | 69.4 | % | 82.0 | % | 37.0 | % | ||||||
No other collaborator has comprised more than 10% of revenue in any period presented. The collaboration agreements that generated revenue in 2005, 2006and/or 2007 are summarized below, organized by segment.
Human Therapeutics
Roche (MAXY-VII)
In December 2005, the Company formed a strategic alliance with F. Hoffmann-La Roche Ltd. (“Roche”) to collaborate on the global development and commercialization of the Company’s portfolio of next-generation recombinant factor VII products. Factor VII is a natural protein with a pivotal role in blood coagulation and clotting. The collaboration focused on the development of lead candidates for the treatment of uncontrolled bleeding in trauma and intracerebral hemorrhage.
Under the terms of the collaboration, the Company and Roche had agreed to share certain costs of worldwide research and development activities in connection with the development of the factor VII product candidates. The Company had agreed to lead early stage clinical development of these product candidates and Roche had agreed to lead late stage clinical development. Roche had been granted exclusive worldwide rights to commercialize the factor VII products subject to the collaboration and the Company had retained rights for the development of factor VII products for hemophilia.
The Company received an upfront fee of $8.0 million in 2005 and received $5.0 million in 2006 for the achievement of a preclinical milestone. Roche elected to terminate this agreement in April 2007.
Roche (MAXY-alpha)
In May 2003, the Company formed a broad strategic alliance with Roche to collaborate on the global development and commercialization of the Company’s portfolio of next-generation interferon alpha and beta variants for a wide range of indications. The collaboration had been initially focused on the development of lead candidates for the treatment of hepatitis C virus (“HCV”) and hepatitis B virus (“HBV”) infections.
Roche had licensed from the Company worldwide commercialization rights to specific novel interferon product candidates for the treatment of HCV and HBV infection. The Company received an initial payment and research and development funding for the first two years of the collaboration. In addition, the Company had been eligible to receive milestone payments and royalties based on product sales. The funded research term of this collaboration ended in December 2005. In 2006, Roche initiated a Phase Ia clinical trial in New Zealand to evaluate our MAXY-alpha product candidate and the Company received a $2.0 million milestone payment in connection with the commencement of such clinical trials. The Company and Roche agreed to terminate this agreement in November 2007.
Chemicals (Codexis)
Our former chemicals segment was operated by Codexis. Effective February 28, 2005, the Company’s voting interests in Codexis fell below 50% and, from such time, Codexis ceased to be a consolidated subsidiary of the Company. The Company instead accounts for Codexis under the equity method of accounting from that date.
70
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
During 2005 (through February 28, 2005), Codexis received approximately $1.5 million of research and development fundingand/or other fees under collaboration agreements with Teva Pharmaceutical Industries Ltd., Pfizer, Inc., Cargill, Incorporated, Rio Tinto Corporation plc, and Eli Lilly and Company. The Company assigned some of these agreements to Codexis in connection with the capitalization of Codexis in March 2002. See Note 11.
| 4. | Technology Licenses and Research Agreements |
The Company has entered into several research agreements to fund research at universities and other collaborators. These agreements are generally cancelable by either party upon written notice and may be extended by mutual consent of both parties. Research and development expenses are recognized as the related services are performed, generally ratably over the period of the service. Expenses under these agreements were approximately $1.5 million in 2005, $3.1 million in 2006 and $4.1 million in 2007.
| 5. | Properties and Equipment |
Property and equipment consisted of the following (in thousands):
| December 31, | ||||||||
| 2006 | 2007 | |||||||
| Leasehold improvements | $ | 3,982 | $ | 4,158 | ||||
| Machinery and laboratory equipment | 15,592 | 16,645 | ||||||
| Computer equipment and software | 2,806 | 2,973 | ||||||
| Furniture and fixtures | 1,218 | 1,244 | ||||||
| 23,598 | 25,020 | |||||||
| Less accumulated depreciation and amortization | (20,336 | ) | (21,960 | ) | ||||
| Property and equipment, net | $ | 3,262 | $ | 3,060 | ||||
| 6. | Equipment Financing |
For the two months ended February 28, 2005, Codexis borrowed $1.2 million and repaid $115,000 under an equipment financing agreement it entered into with a financing company in 2004 for up to $4.8 million of equipment purchases.
| 7. | Commitments |
The Company has entered into various operating leases for its facilities and certain computer equipment and material contracts. The leases expire on various dates through 2009. The facilities leases also include scheduled rent increases. The scheduled rent increases are recognized on a straight-line basis over the term of the leases. The material contracts expire on various dates through 2010.
Minimum annual rental commitments under operating leases are as follows (in thousands):
Year Ending December 31, | ||||
| 2008 | $ | 6,707 | ||
| 2009 | 1,716 | |||
| 2010 | 23 | |||
| Thereafter | — | |||
| $ | 8,446 | |||
Total rent expense was $2.3 million in 2005, $2.0 million in 2006 and $2.2 million in 2007.
71
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| 8. | Stockholders’ Equity |
Codexis Redeemable Convertible Preferred Stock
In connection with the redemption rights of the Codexis series B stockholders, the Company recorded accretion of the redemption premium for the series B redeemable convertible preferred stock, excluding the shares owned by the Company, in the amount of $167,000 for the year ended December 31, 2005. The accretion is recorded as subsidiary preferred stock accretion on the Consolidated Statements of Operations and as a reduction of additional paid-in capital and an increase to minority interest on the Consolidated Balance Sheets. The Company recorded a $2.3 million adjustment to additional paid-in capital in 2005 to eliminate the reduction of additional paid-in capital that had resulted from Codexis’ preferred stock accretion prior to February 28, 2005, the date Codexis ceased to be a consolidated subsidiary of the Company.
Maxygen Preferred Stock
The Company is authorized, subject to limitations prescribed by Delaware law, to provide for the issuance of preferred stock in one or more series, to establish from time to time the number of shares included within each series, to fix the rights, preferences and privileges of the shares of each wholly unissued series and any qualifications, limitations or restrictions thereon, and to increase or decrease the number of shares of any such series (but not below the number of shares of such series then outstanding) without any further vote or action by the stockholders.
401(k) Savings Plan
The Company has a savings plan that qualifies as a deferred salary arrangement under Section 401(k) of the Internal Revenue Code (the “401(k) Plan”). Under the 401(k) Plan, participating employees may defer a percentage (not to exceed 100%) of their eligible pretax earnings up to the Internal Revenue Service’s annual contribution limit. All employees on the United States payroll of the Company age 18 years or older are eligible to participate in the 401(k) Plan. The Company has not been required to contribute to the 401(k) Plan, but beginning in 2001 elected to match contributions of its participating employees in an amount up to a maximum of the lesser of (i) 50% of the employee’s 401(k) yearly contribution or (ii) 6% of the employee’s yearly base salary. The matching contribution is made in the form of newly issued shares of Company common stock as of each June 30 and December 31. All matching contributions vest immediately. The Company may discontinue such matching contributions at any time. The fair value of the Company’s matching contribution to the 401(k) Plan was $350,000 in 2005, $325,000 in 2006 and $368,000 in 2007.
2006 Equity Incentive Plan
The Company’s stockholders approved the 2006 Equity Incentive Plan (the “2006 Plan”) on May 30, 2006. The 2006 Plan replaced the Company’s 1997 Stock Option Plan (the “1997 Plan”). The 2006 Plan provides for the grant of stock options (both nonstatutory and incentive stock options), stock appreciation rights, restricted stock, restricted stock units, performance shares, performance units and dividend equivalents to employees (including officers), directors and consultants of the Company and its subsidiaries and affiliates. No equity awards may be granted under the 2006 Plan after February 7, 2016. The maximum term of the options granted under the 2006 Plan is ten years. Options granted under the 2006 Plan vest and become exercisable pursuant to a vesting schedule determined by the administrator of the plan, generally over a four-year period at a rate of 25% at the end of the first year and monthly for the three years thereafter. The 2006 Plan does not provide for annual increases in the number of shares available for issuance under the 2006 Plan. At December 31, 2007, 5,319,151 shares remained available for future awards under the 2006 Plan.
72
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
1997 Stock Option Plan
The Company’s stockholders originally approved the 1997 Plan on March 30, 1997. The 1997 Plan, which was scheduled to expire in March 2007, was replaced by the 2006 Plan. The maximum term of the options granted under the 1997 Plan is ten years. Options granted under the 1997 Plan vest and become exercisable pursuant to a vesting schedule determined by the administrator of the plan, generally over a four-year period at a rate of 25% at the end of each year for grants made prior to January 1, 2002 and for grants made after January 1, 2002, over a four-year period at a rate of 25% at the end of the first year and monthly for the three years thereafter. In addition, a number of grants made in 2003 vest over three years, 16.67% on July 1, 2003 and monthly for the two and a half years thereafter. In connection with the stockholder approval of the 2006 Plan, shares available for future awards under the 1997 Plan were transferred to the 2006 Plan and the 1997 Plan was terminated as to future awards. As a result, no shares remained available for future awards under the 1997 Plan at December 31, 2007.
1999 Nonemployee Directors Stock Option Plan
The Company’s stockholders approved the 1999 Nonemployee Directors Stock Option Plan (the “Directors’ Plan”) on December 14, 1999. There are a total of 300,000 shares of common stock reserved for issuance under the Directors’ Plan. Each nonemployee director is automatically granted a nonstatutory stock option to purchase 20,000 shares of common stock on the date upon which such person first becomes a director. At the first board meeting immediately following each annual stockholders’ meeting, each non-employee director is automatically granted a nonstatutory option to purchase 5,000 shares of common stock. The exercise price of options under the Directors’ Plan is equal to the fair market value of the common stock on the date of grant. Options have a term of ten years. Generally, each initial grant under the Directors’ Plan vests as to 25% of the shares subject to the option at the end of each year. Each subsequent grant generally vests in full one year after the date of grant. The Directors’ Plan will terminate in September 2009, unless terminated earlier in accordance with the provisions of the Directors’ Plan. At December 31, 2007, 65,000 shares remained available for future option grants under the Directors’ Plan.
2000 International Stock Option Plan
The board of directors adopted the 2000 International Stock Option Plan (the “International Plan”) on April 10, 2000 and amended it on March 1, 2001. The International Plan has not been approved by the Company’s stockholders, as no such approval is required. Under the International Plan, the board of directors may issue nonqualified stock options to employees, directors and consultants ofnon-U.S. subsidiaries of the Company. No options may be granted under the International Plan after April 10, 2010. Under the International Plan, nonstatutory options may be granted at prices not lower than 85% of fair value at the date of grant (except in the case of replacement options in the context of acquisitions), as determined by the board of directors. The maximum term of the options granted under the International Plan is ten years. Options granted under the International Plan vest and become exercisable pursuant to a vesting schedule determined by the administrator of the plan. The International Plan also provides for annual increases in the number of shares available for issuance on the first day of each year equal to 0.6% of the outstanding shares on the date of the annual increase, or a lower amount determined by the board of directors. At December 31, 2007, 2,016,918 shares remained available for future option grants under the International Plan. As a result of the cessation of research and development operations at Maxygen ApS, the Company plans to discontinue the International Plan as to future awards.
2000 Non-Officer Employee Stock Option Plan
The board of directors adopted the 2000 Non-Officer Stock Option Plan (the “Non-Officer Plan”) on December 6, 2000. The Non-Officer Plan has not been approved by the Company’s stockholders, as no such approval is required. Under the Non-Officer Plan, the board of directors may issue nonqualified stock options to employees (other than executive officers and stockholders owning 10% or more of the Company’s common stock) and consultants of the Company or any of its affiliates. No options may be granted under the Non-Officer Plan after
73
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 6, 2010. Under the Non-Officer Plan, nonstatutory options may be granted at prices not lower than 85% of fair value at the date of grant (except in the case of replacement options in the context of acquisitions), as determined by the board of directors. The maximum term of the options granted under the Non-Officer Plan is ten years. Options granted under the Non-Officer Plan vest and become exercisable pursuant to a vesting schedule determined by the administrator of the plan, generally over a four-year period at a rate of 25% at the end of each year for grants made prior to January 1, 2002, or for grants made after January 1, 2002, over a four-year period at a rate of 25% at the end of the first year and monthly for the three years thereafter. In addition, a number of grants made in 2003 vest over three years, 16.67% on July 1, 2003 and monthly for the two and a half years thereafter. The Non-Officer Plan provides for annual increases in the number of shares available for issuance on the first day of each year equal to the greater of (i) 250,000 shares and (ii) 0.7% of the outstanding shares on the date of the annual increase, or a lower amount determined by the board of directors. At December 31, 2007, 596,192 shares remained available for future option grants under the Non-Officer Plan.
Activity under the 2006 Plan, the 1997 Plan, the Directors’ Plan, the International Plan and the Non-Officer Plan (collectively, the “Plans”) was as follows:
| Options Outstanding | ||||||||||||
| Weighted-Average | ||||||||||||
| Exercise | ||||||||||||
| Shares | Number of | Price per | ||||||||||
| Available | Shares | Share | ||||||||||
| Balance at January 1, 2005 | 6,736,161 | 8,910,881 | $ | 17.19 | ||||||||
| Shares authorized | 1,889,270 | — | — | |||||||||
| Options granted | (2,132,290 | ) | 2,132,290 | $ | 9.34 | |||||||
| Options exercised | — | (198,487 | ) | $ | 4.06 | |||||||
| Options canceled | 1,443,085 | (1,443,085 | ) | $ | 14.31 | |||||||
| Balance at December 31, 2005 | 7,936,226 | 9,401,599 | $ | 16.13 | ||||||||
| Shares authorized | 1,903,875 | — | — | |||||||||
| Options granted | (2,157,836 | ) | 2,157,836 | $ | 7.81 | |||||||
| Options exercised | — | (150,903 | ) | $ | 4.62 | |||||||
| Options canceled | 708,422 | (709,922 | ) | $ | 38.57 | |||||||
| Options expired | 12,150 | (12,150 | ) | $ | 12.18 | |||||||
| Balance at December 31, 2006 | 8,402,837 | 10,686,460 | $ | 13.13 | ||||||||
| Shares authorized | 469,752 | — | — | |||||||||
| Options granted | (1,960,730 | ) | 1,960,730 | $ | 10.00 | |||||||
| Options exercised | — | (670,018 | ) | $ | 7.10 | |||||||
| Options canceled | 1,035,651 | (1,035,651 | ) | $ | 10.59 | |||||||
| Options expired | 49,751 | (49,751 | ) | $ | 8.11 | |||||||
| Balance at December 31, 2007 | 7,997,261 | 10,891,770 | $ | 13.20 | ||||||||
1999 Employee Stock Purchase Plan
The Company’s stockholders approved the 1999 Employee Stock Purchase Plan (the “ESPP”) on December 14, 1999. The ESPP is intended to qualify as an employee stock purchase plan under Section 423 of the Internal Revenue Code. A total of 400,000 shares of the Company’s common stock were initially reserved for issuance under the ESPP. The ESPP permits eligible employees to purchase common stock at a discount, but only through payroll deductions, during defined offering periods. The price at which stock is purchased under the ESPP is equal to 85% of the lower of (i) the fair market value of the common stock on the first day of the offering period or (ii) the fair market value of the common stock on the purchase date. In addition, the ESPP provides for annual
74
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
increases in the number of shares available for issuance under the purchase plan on the first day of each year, beginning January 1, 2001, equal to the lesser of 200,000 shares, 0.75% of the outstanding shares on the date of the annual increase, or a lower amount determined by the board of directors. The ESPP will terminate in September 2019, unless terminated earlier in accordance with the provisions of the ESPP. The initial offering period commenced on December 16, 1999 and the first purchase under the ESPP occurred on September 29, 2000 when 67,540 shares of common stock were purchased. In 2005, 2006 and 2007, 37,966 shares, 46,815 shares and 54,383 shares of common stock were purchased pursuant to the ESPP, respectively. The weighted average fair value of purchase rights granted during the year was $2.01 in 2005, $1.92 in 2006 and $0.48 in 2007. At December 31, 2007, 1,193,804 shares remained available for purchase under the ESPP.
Fair value disclosures
Options granted to consultants are periodically re-valued as they vest in accordance with SFAS 123(R) and FASB Emerging Issues Task Force on IssueNo. 96-18, “Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services,” using aBlack-Scholes-Merton model and the following weighted-average assumptions for 2005: estimated volatility of 0.60, risk-free interest rates of 3.78% to 3.99%, no dividend yield, and an expected life of 4.0 years; for 2006: estimated volatility of 0.52 to 0.54, risk-free interest rates of 4.52% to 5.15%, no dividend yield, and an expected life of 5.13 years; and for 2007: estimated volatility of 0.51 to 0.53, risk-free interest rates of 4.54% to 4.92%, no dividend yield, and an expected life of 5.70 years. The Company recorded total compensation expense of $102,000 in 2005, $914,000 in 2006 and $666,000 in 2007 related to the Black-Scholes-Merton revaluation of options grants to consultants. Stock compensation expense relating to the acceleration of stock options was immaterial for 2005. There was no stock compensation expense relating to the acceleration of stock options for 2006 or 2007.
Common Stock
At December 31, 2007, the Company had reserved shares of common stock for future issuance as follows:
| 2006 Equity Incentive Plan | 6,041,949 | |||
| 2000 Non-Officer Employee Stock Option Plan | 3,993,003 | |||
| 2000 International Stock Option Plan | 3,168,369 | |||
| 1999 Employee Stock Purchase Plan | 1,193,804 | |||
| 1999 Nonemployee Directors Stock Option Plan | 300,000 | |||
| 1997 Stock Option Plan | 5,385,710 | |||
| 20,082,835 | ||||
| 9. | Income Taxes |
Worldwide income (loss) from continuing operations before provision for income taxes consists of the following (in thousands):
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| United States | $ | (7,445 | ) | $ | 1,807 | $ | (15,155 | ) | ||||
| Foreign | (27,607 | ) | (18,289 | ) | (34,160 | ) | ||||||
| Loss from continuing operations | $ | (35,052 | ) | $ | (16,482 | ) | $ | (49,315 | ) | |||
No income tax expense was recorded from continuing operations for the years ended December 31, 2007 and 2005. Income tax expense from continuing operations for the years ended December 31, 2006 was $140,000. In 2006, the Company utilized prior year federal net operating loss carry forwards to reduce the federal taxable income
75
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
to zero for regular tax purposes. However, for federal purposes, the Company is subject to alternative minimum tax and has reported an income tax provision of $140,000 in 2006. This amount has been netted against the gain on sale of the Company’s equity interests in Avidia and is reflected in gain on sale of equity investment in the Consolidated Statements of Operations
During 2006, the Company’s total deferred tax assets increased due to increases in state and foreign net operating losses and deferred taxes related to deductible stock option compensation, offset in part by the use of federal net operating loss carryforwards and a reduction in capitalized research and development costs due to a reduction in state tax rate. During 2007, the Company’s total deferred tax assets increased by $5.5 million due primarily to increases in federal and state net operating loss carryforwards and deferred taxes related to deductible stock option compensation, offset in part by the use of foreign net operating loss carryforwards.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets are as follows (in thousands):
| December 31, | ||||||||
| 2006 | 2007 | |||||||
| Net operating loss carryforwards | $ | 15,104 | $ | 17,824 | ||||
| Research credits | 5,575 | 5,335 | ||||||
| Capitalized research | 1,776 | 2,067 | ||||||
| Investment in subsidiary | 4,014 | 4,119 | ||||||
| Stock based compensation | 5,065 | 7,406 | ||||||
| Other | 5,316 | 5,587 | ||||||
| Total deferred tax assets | 36,850 | 42,338 | ||||||
| Valuation allowance | (36,850 | ) | (42,338 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
The valuation allowance increased by $5.6 million in 2006 and increased by $5.5 million in 2007. In assessing the realizability of deferred tax assets, the Company considered whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The Company considered future earnings, future taxable income, and the scheduled reversal of deferred taxes in making this assessment. Based on this assessment, the deferred tax assets have been fully offset by a valuation allowance at December 31, 2006 and 2007.
As of December 31, 2007, the Company had net operating loss carryforwards for federal income tax purposes of approximately $37.1 million, which expire in the years 2022 through 2027 and federal research and development tax credit carryforwards of approximately $2.5 million, which expire in the years 2012 through 2027. As of December 31, 2007, the Company had net operating loss carryforwards for state income tax purposes of approximately $59.3 million that expire in the years 2015 through 2017 and state research and development tax credits of approximately $2.5 million that have no expiration date. As of December 31, 2007, the Company had net operating loss carryforwards for foreign income tax purposes of approximately $6.8 million that have no expiration date.
Approximately $4.3 million of the valuation allowance for deferred tax assets relates to benefits of stock options deductions that, when recognized, will be allocated directly to additional paid-in capital.
Utilization of the Company’s net operating loss carryforwards may be subject to substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. Such an annual limitation could result in the expiration of the net operating loss carryforwards before utilization.
76
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The Company has not provided for U.S. federal income taxes on all of thenon-U.S. subsidiaries’ undistributed earnings as of December 31, 2007 because such earnings are intended to be indefinitely reinvested. Upon distribution of those earnings in the form of dividends or otherwise, the Company would be subject to applicable U.S. federal and state income taxes.
A reconciliation of income taxes at the statutory federal income tax rate to income taxes included in the Consolidated Statements of Operations is as follows (in thousands):
| December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| U.S. federal taxes (benefit) | ||||||||||||
| At statutory rate | $ | (12,269 | ) | $ | (5,720 | ) | $ | (17,260 | ) | |||
| State taxes (net of federal) | (1,965 | ) | (533 | ) | (1,850 | ) | ||||||
| Alternative minimum taxes | — | 140 | — | |||||||||
| Stock related deductions | — | (208 | ) | (33 | ) | |||||||
| Unbenefited foreign losses | 10,120 | 5,610 | 14,314 | |||||||||
| Other | (801 | ) | (232 | ) | (242 | ) | ||||||
| Unbenefited losses | 4,915 | 1,083 | 5,071 | |||||||||
| Total | $ | — | $ | 140 | $ | — | ||||||
Effective January 1, 2007, the Company adopted the provisions of FASB Interpretation No. 48, “Accounting for Uncertainty in Income Taxes — an interpretation of FASB Statement No. 109” (“FIN 48”). FIN 48 prescribes a recognition threshold and a measurement attribute for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For those benefits to be recognized, a tax position must be more-likely-than-not to be sustained upon examination by taxing authorities. There was not a material impact on the Company’s consolidated financial position and results of operations as a result of the adoption of the provisions of FIN 48. At December 31, 2007, the Company had a liability for unrecognized tax benefits of approximately $5.6 million (none of which, if recognized, would favorably affect the Company’s effective tax rate). The Company does not believe there will be any material changes in its unrecognized tax positions over the next twelve months.
Interest and penalty costs related to unrecognized tax benefits, if any, are classified as a component of interest income and other income (expense), net in the accompanying Consolidated Statements of Operations. The Company, however, did not recognize any interest and penalty expense related to unrecognized tax benefits for the year ended December 31, 2007.
The Company files income tax returns in the U.S. federal jurisdiction and various state jurisdictions. The Company is subject to U.S. federal and state income tax examination for calendar tax years ending 1997 through 2007. Additionally, the Company is subject to various international tax examinations for the calendar tax years ending 2002 through 2007. Danish tax authorities are currently auditing the Company’s Danish tax filings for the years 2003 through 2006. The Company does not believe that there will be any material tax exposure as a result of this audit.
77
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows:
| 2007 | ||||
| Balance at January 1, 2007 | $ | 5,590 | ||
| Increases (decrease) related to prior year tax positions | — | |||
| Increases related to current year tax positions | — | |||
| Settlements | — | |||
| Reductions due to lapse of applicable statute of limitations | — | |||
| Balance at December 31, 2007 | $ | 5,590 | ||
| 10. | Litigation |
In December 2001, a lawsuit was filed in the U.S. District Court for the Southern District of New York against the Company, its chief executive officer, Russell Howard, and its chief financial officer at the time of the initial public offering, Simba Gill, together with certain underwriters of the Company’s initial public offering and secondary public offering of common stock. The complaint, which alleges claims under Sections 11, 12(a)(2) and 15 of the Securities Act of 1933 and Section 10(b) of the Securities Exchange Act of 1934, is among the so-called “laddering” cases that have been commenced against over 300 companies that had public offerings of securities in 1999 and 2000. The complaint has been consolidated with other laddering claims in a proceeding styledIn re Initial Public Offering Securities Litigation, No. 21 MC 92 (SAS), pending before the Honorable Shira A. Scheindlin. In February 2003, the court dismissed the Section 10(b) claim against Drs. Howard and Gill; the remainder of the case remains pending.
In June 2003, the Company agreed to the terms of a tentative settlement agreement along with other defendant issuers inIn re Initial Public Offering Securities Litigation. The tentative settlement provides that the insurers of the 309 defendant issuers will pay to the plaintiffs $1 billion, less any recovery of damages the plaintiffs receive from the defendant underwriters. If the plaintiffs received over $5 billion in damages from the defendant underwriters, the Company would be entitled to reimbursement of various expenses incurred by it as a result of the litigation. As part of the tentative settlement, the Company would assign to the plaintiffs “excess compensation claims” and certain other of its claims against the defendant underwriters based on the alleged actions of the defendant underwriters. The settlement was subject to acceptance by a substantial majority of defendants and execution of a definitive settlement agreement. The settlement was also subject to approval of the Court, which cannot be assured. On February 15, 2005, the Court tentatively approved the proposed settlement, conditioned upon the parties altering the proposed settlement to comply with the Private Securities Litigation Reform Act’s settlement discharge provision. The settlement did not contemplate any settlement payments by the Company.
On December 5, 2006, the U.S. Second Circuit Court of Appeals reversed the District Court’s ruling certifying the consolidated cases as class actions. On April 9, 2007, the Court of Appeals denied a motion for a rehearing en banc. On June 22, 2007, the parties submitted a Stipulation and Proposed Order to terminate the settlement agreement, which the District Court so ordered on June 25, 2007. The parties are now considering alternative options.
If an alternative settlement agreement is not reached, and an action proceeds against the Company based on the facts alleged in the above referenced proceeding, the Company intends to defend the lawsuit vigorously. The Company believes the lawsuit against it and its officers is without merit. If the outcome of the litigation is adverse to the Company and if the Company is required to pay significant damages, its business would be significantly harmed.
In a related matter, on July 30, 2007, the Company received a demand letter, addressed to its board of directors, from counsel for a purported stockholder concerning alleged violations by unspecified persons and entities of Section 16(b) of the Securities Exchange Act of 1934 Act in connection with the Company’s initial public offering. On October 5, 2007, a complaint was filed in the U.S. District Court for the Western District of Washington against
78
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
certain underwriters of the Company’s initial public offering of common stock. The complaint also named the Company as a nominal defendant. An amended complaint was filed on February 28, 2008. The complaint alleges claims under Sections 16(b) of the Securities Exchange Act of 1934 in connection with the Company’s initial public offering in 1999. The Company is evaluating this matter, but does not believe that these claims will have a material adverse effect on its business or financial statements.
The Company is not currently a party to any other material pending legal proceedings.
From time to time, the Company becomes involved in claims and legal proceedings that arise in the ordinary course of its business. The Company does not believe that the resolution of these claims will have a material adverse effect on its financial statements.
| 11. | Segment Information |
On February 28, 2005, the Company’s voting interests in Codexis fell below 50% and from such time Codexis ceased to be a consolidated subsidiary of the Company. Accordingly, the results of operations of Codexis are included in the consolidated results presented below only until February 28, 2005. Thereafter the Company’s investment in Codexis is reflected under the equity method of accounting. The Company has determined its reportable operating segments based upon how the business is managed and operated. The accounting policies of the segments described above are the same as those described in Note 1. Corporate administrative costs are generally allocated based on headcount.
Segment Earnings
| Human | Chemicals | Maxygen, Inc. | ||||||||||
| Therapeutics | (Codexis) | Consolidated | ||||||||||
| (In thousands) | ||||||||||||
2005 | ||||||||||||
| Segment loss | $ | (39,116 | ) | $ | (1,325 | ) | $ | (40,441 | ) | |||
| Stock compensation | (183 | ) | — | (183 | ) | |||||||
| Interest income and other income (expense), net | 5,552 | 20 | 5,572 | |||||||||
| Loss from continuing operations | (33,747 | ) | (1,305 | ) | (35,052 | ) | ||||||
| Cumulative effect of change in accounting principle | 16,616 | — | 16,616 | |||||||||
| Subsidiary preferred stock accretion | — | (167 | ) | (167 | ) | |||||||
| Loss applicable to common stockholders | $ | (17,131 | ) | $ | (1,472 | ) | $ | (18,603 | ) | |||
2006 | ||||||||||||
| Segment loss | $ | (41,668 | ) | $ | — | $ | (41,668 | ) | ||||
| Interest income and other income (expense), net | 8,524 | — | 8,524 | |||||||||
| Equity in losses of minority investee | (1,000 | ) | — | (1,000 | ) | |||||||
| Gain on sale of equity investment | 17,662 | — | 17,662 | |||||||||
| Loss applicable to common stockholders | $ | (16,482 | ) | $ | — | $ | (16,482 | ) | ||||
2007 | ||||||||||||
| Segment loss | $ | (56,857 | ) | $ | — | $ | (56,857 | ) | ||||
| Interest income and other income (expense), net | 7,542 | — | 7,542 | |||||||||
| Loss applicable to common stockholders | $ | (49,315 | ) | $ | — | $ | (49,315 | ) | ||||
In 2005, depreciation expense for the two segments was $3,268,000 for human therapeutics and $262,000 for chemicals. The operations of Codexis are not included in the consolidated operations of the Company after February 28, 2005. In 2006 and 2007, depreciation expense was $2,294,000 and $1,671,000, respectively.
79
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Segment Revenue
Revenues for each operating segment are derived from the Company’s collaboration agreements and government grants and are categorized based on the industry of the product or technology under development. On February 28, 2005, the Company’s voting interests in Codexis fell below 50% and from such time Codexis ceased to be a consolidated subsidiary of the Company. Thus, the revenues of Codexis are included in the consolidated results presented below only until February 28, 2005 and thereafter the Company’s investment in Codexis is reflected under the equity method of accounting. The following table presents revenues for each reporting segment (in thousands):
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
| Human therapeutics | $ | 12,991 | $ | 25,021 | $ | 23,157 | ||||||
| Chemicals (through February 28, 2005) | 1,510 | — | — | |||||||||
| Total revenue | $ | 14,501 | $ | 25,021 | $ | 23,157 | ||||||
Identifiable Assets and Acquisition of Property and Equipment
During 2006 and 2007, the Company operated its business in one segment, Human Therapeutics. Accordingly, the identifiable assets at December 31, 2006 and 2007 of $205.6 million and $172.7 million, respectively, relate to Human Therapeutics and the acquisition of property and equipment for the years ended December 31, 2006 and 2007 was $1.5 million, all of which related to human therapeutics.
Human Therapeutics
On February 28, 2005, the Company’s voting interests in Codexis fell below 50% and from such time Codexis ceased to be a consolidated subsidiary of the Company. Accordingly, the results of operations of Codexis are
80
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
included in the consolidated results presented in the Consolidated Statements of Operations only until February 28, 2005. Presented below are the results of operations for the human therapeutics business on a stand-alone basis:
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
Revenues | ||||||||||||
| Collaborative research and development revenue | $ | 10,084 | $ | 20,544 | $ | 10,232 | ||||||
| Revenue from related party | — | — | 8,286 | |||||||||
| Grant revenue | 2,907 | 4,477 | 4,639 | |||||||||
| Total revenues | 12,991 | 25,021 | 23,157 | |||||||||
Operating Expenses: | ||||||||||||
| Research and development | 39,370 | 49,130 | 59,851 | |||||||||
| General and administrative | 12,920 | 17,559 | 14,951 | |||||||||
| Restructuring charge | — | — | 5,212 | |||||||||
| Total operating expenses | 52,290 | 66,689 | 80,014 | |||||||||
| Loss from operations | (39,299 | ) | (41,668 | ) | (56,857 | ) | ||||||
| Interest income and other income (expense), net | 5,552 | 8,524 | 7,542 | |||||||||
| Equity in losses of minority investee | — | (1,000 | ) | — | ||||||||
| Gain on sale of equity investment | — | 17,662 | — | |||||||||
| Loss from continuing operations | (33,747 | ) | (16,482 | ) | (49,315 | ) | ||||||
| Cumulative effect adjustment | 16,616 | — | — | |||||||||
| Net loss | $ | (17,131 | ) | $ | (16,482 | ) | $ | (49,315 | ) | |||
Geographic Information
The Company’s primary country of operation is the United States, its country of domicile. Revenues are attributed to countries based on the location of collaborators. Long-lived assets include property and equipment and intangible assets.
| Year Ended December 31, | ||||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
Revenues | ||||||||||||
| United States | $ | 13,768 | $ | 25,021 | $ | 22,904 | ||||||
| Denmark | 86 | — | — | |||||||||
| Australia | 513 | — | — | |||||||||
| Other foreign countries | 134 | — | 253 | |||||||||
| Total revenue | $ | 14,501 | $ | 25,021 | $ | 23,157 | ||||||
81
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| December 31, | ||||||||
| 2006 | 2007 | |||||||
| (In thousands) | ||||||||
Long-Lived Assets | ||||||||
| United States | $ | 40,055 | $ | 40,593 | ||||
| Denmark | 9,721 | 11,238 | ||||||
| Total long-lived assets | $ | 49,776 | $ | 51,831 | ||||
Major Customers
Major customers (consisting of the Company’s non-grant collaborators) that represent more than 10% of total Company revenue are presented in the following table:
| 2005 | 2006 | 2007 | ||||||||||
Human Therapeutics | ||||||||||||
| Partner A | 69.4 | % | 82.0 | % | 37.0 | % | ||||||
12. Avidia Inc. (formerly Avidia Research Institute)
On July 15, 2003, the Company and a third party investor formed Avidia Inc. As of December 31, 2005, the Company had an equity interest of approximately 13% in Avidia, based upon the voting rights of the issued and outstanding shares of Avidia’s common and preferred stock. Until March 31, 2005, the Company’s investment in Avidia was accounted for under the equity method of accounting and the Company’s share of Avidia’s results was recorded to the extent of the Company’s accounting basis in Avidia as a component of equity in net loss of minority investee in the Consolidated Statements of Operations. In April 2005, Avidia issued additional equity securities, which lowered the Company’s ownership below 20%. Since the Company did not have the ability to exercise significant influence over Avidia’s operating and financial policies, after March 31, 2005, the Company’s investment in Avidia was accounted for under the cost method of accounting. As of December 31, 2005, the Company had recorded losses equal to its investment basis.
On October 24, 2006, Amgen Inc. (“Amgen”) completed the acquisition of Avidia. As a result of the acquisition, the Company received cash proceeds of approximately $17.8 million (before income taxes of $140,000) in the fourth quarter of 2006 in exchange for its equity interests in Avidia and may receive up to an additional $1.4 million in cash, contingent upon the development of certain Avidia products by Amgen. This contingent amount was reduced from $2.8 million based on the discontinuation by Amgen Inc. of certain development activities. Accordingly, the Company recorded a gain on the exchange of its equity interest in Avidia of approximately $17.8 million in the fourth quarter of 2006. Any additional gain as a result of the contingent amounts potentially payable to the Company by Amgen will be recognized only if and when the contingency is satisfied.
| 13. | Guarantees and Indemnifications |
In November 2002, the FASB issued Interpretation No. 45, “Guarantor’s Accounting and Disclosure Requirements for Guarantees, including Indirect Guarantees of Indebtedness of Others” (“FIN 45”). FIN 45 requires that upon issuance of a guarantee, the guarantor must recognize a liability for the fair value of the obligations it assumes under that guarantee.
As permitted under Delaware law and in accordance with the Company’s Bylaws, the Company indemnifies its officers and directors for certain events or occurrences, subject to certain limits, while the officer or director is or was serving at the Company’s request in such capacity. The indemnification agreements with the Company’s officers and directors terminate upon termination of their employment, but the termination does not affect claims
82
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
for indemnification relating to events occurring prior to the effective date of termination. The maximum amount of potential future indemnification is unlimited; however, the Company’s director and officer insurance policy reduces the Company’s exposure and may enable the Company to recover a portion of any future amounts paid. The Company believes that the fair value of these indemnification agreements is minimal. Accordingly, the Company has not recorded any liabilities for these agreements as of December 31, 2007.
In addition, the Company customarily agrees in the ordinary course of its business to indemnification provisions in its collaboration agreements, in various agreements involving parties performing services for the Company in the ordinary course of business, and in its real estate leases. With respect to lease agreements, the indemnification provisions typically apply to claims asserted against the landlord relating to personal injury or property damage caused by the Company, to violations of law by the Company or to certain breaches of the Company’s contractual obligations. The indemnification provisions appearing in the Company’s collaboration agreements are similar, but in addition provide some limited indemnification for its collaborator in the event of third party claims alleging infringement of certain intellectual property rights. In each of the cases above, the indemnification obligation generally survives the termination of the agreement for some extended period, although the obligation typically has the most relevance during the contract term and for a short period of time thereafter. The maximum potential amount of future payments that the Company could be required to make under these provisions is generally unlimited. The Company has purchased insurance policies covering personal injury, property damage and general liability that reduce its exposure for indemnification and would enable it in many cases to recover a portion of any future amounts paid. The Company has never paid any material amounts to defend lawsuits or settle claims related to these indemnification provisions. Accordingly, the Company believes the estimated fair value of these indemnification arrangements is minimal. Accordingly, the Company has not recorded any liabilities for these agreements as of December 31, 2007.
| 14. | Restructuring Charges |
In November 2007, the Company implemented a plan to consolidate its organization to reduce costs and increase overall operational efficiency across its research, preclinical, clinical and regulatory activities. The consolidation has resulted in the cessation of research and development operations at Maxygen ApS, the Company’s wholly owned subsidiary in Denmark, and the elimination of all employment positions at that site. As a result of these actions, a charge of $5.2 million was recorded in the year ended December 31, 2007. The restructuring charge, which includes approximately $287,000 of non-cash stock compensation, is related to severance and other benefits for the Company’s Danish employees. The activity in the restructuring accrual related to the action described above was as follows (in thousands):
| As at December 31, | ||||||||||||||||||||||||
| Fiscal Year | Fiscal Year | Balance at | 2007 | |||||||||||||||||||||
| 2007 Cash | 2007 Non-Cash | Cash | December 31, | Total Costs to | Total Expected | |||||||||||||||||||
| Charges | Charges | Payments | 2007 | Date | Costs | |||||||||||||||||||
| Employee severance and other benefits charges | $ | 5,212 | $ | 287 | $ | (512 | ) | $ | 4,413 | $ | 5,212 | $ | 5,212 | |||||||||||
| Contract termination and other associated costs | — | — | — | — | — | 1,000 | ||||||||||||||||||
| $ | 5,212 | $ | 287 | $ | (512 | ) | $ | 4,413 | $ | 5,212 | $ | 6,212 | ||||||||||||
The Company expects to incur additional costs of approximately $1.0 million relating to closure of the facility, disposing of remaining fixed assets and terminating various leases, including the building lease. The Company expects to complete the activities related to this consolidation during the first half of 2008.
83
Table of Contents
MAXYGEN, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
In June 2005, the Company implemented a plan to consolidate its organization to increase focus on development of its pharmaceutical product candidates. The restructuring plan included a reduction of approximately 16% of the Company’s personnel over the following four months. The Company recorded severance charges of $807,000 during 2005 in connection with the restructuring. The restructuring balances are comprised entirely of cash charges and are included in operating expenses on the Consolidated Statements of Operations as follows (in thousands):
| Charges | ||||
| Severance costs | ||||
| Research and development | $ | 471 | ||
| General and administrative | 336 | |||
| Total | $ | 807 | ||
| 15. | Related Party Transactions |
On April 1, 2006, the Company entered into a two-year consulting agreement with Waverley Associates, Inc. (“Waverley”), a private investment firm for which Mr. Isaac Stein is the president and sole stockholder. Mr. Stein also currently serves as chairman of the Company’s board of directors. Pursuant to the terms of the consulting agreement, the Company agreed to pay consulting fees to Waverley of $24,166 per month during the term of the consulting agreement and granted Mr. Stein an option to purchase 250,000 shares of the Company’s common stock at an exercise price of $8.28 per share. The option vested and became fully exercisable in May 2007. For the years ended December 31, 2006 and 2007, the Company recognized stock-based compensation expense under SFAS 123(R) in the Consolidated Statements of Operations related to stock options of $6.7 million and $6.8 million, respectively, of which approximately $902,000 in 2006 and $430,000 in 2007 is attributable to the option granted to Mr. Stein. For the years ended December 31, 2006 and 2007, total expense under this arrangement, including cash payments, was approximately $1.1 million and $720,000, respectively. In December 2007, the Company’s Board of Directors approved the extension of the term of the consulting agreement for an additional one-year period (until March 2009).
In December 2006, the Company expanded the scope of exclusive licenses previously granted to Codexis to its MolecularBreeding directed evolution platform for certain applications relating to energy, including biofuels. Under the license agreement, as amended, the Company is entitled to receive a percentage of all consideration received by Codexis from a third party, including license fees, milestone payments, royalties and the purchase of equity securities (subject to certain limitations) and research funding (in excess of a specified base rate), that relates to the use of the licensed rights for the development or commercialization of certain products or processes in the energy field. In November 2006, Codexis entered into a collaboration agreement with Shell Oil Products US to explore enhanced methods of converting biomass to biofuels and, in November 2007, Codexis entered into an expanded collaboration agreement with Royal Dutch Shell plc. Accordingly, during 2007, the Company recognized approximately $8.3 million from Codexis as a result of revenues received by Codexis under its collaboration agreement with Shell. The payments from Codexis are reflected as revenue from related party in the Consolidated Statements of Operations.
84
Table of Contents
| Item 9 | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
Not applicable.
| Item 9A | CONTROLS AND PROCEDURES |
Evaluation of Controls and Procedures
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we evaluated the effectiveness of the design and operation of our disclosure controls and procedures (as defined inRules 13a-15(e) and15d-15(e) under the Securities Exchange Act of 1934 (“the Exchange Act”)) as of the end of the period covered by this report. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures are effective in reaching a reasonable level of assurance that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time period specified in the Securities and Exchange Commission’s rules and forms.
Changes in Internal Control
There has been no change in our internal controls over financial reporting (as such term is defined inRules 13a-15(f) and15d-15(f) under the Exchange Act) during our most recently completed fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal controls over financial reporting.
Annual Report on Internal Control Over Financial Reporting
Company management is responsible for establishing and maintaining adequate internal control over financial reporting. Management assessed the effectiveness of our internal control over financial reporting as of December 31, 2007. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in “Internal Control — Integrated Framework.” Based on the assessment using those criteria, management believes that, as of December 31, 2007, our internal control over financial reporting was effective.
Ernst & Young LLP, an independent registered public accounting firm, assessed the effectiveness of our internal controls over financial reporting as of December 31, 2007 and has issued an unqualified opinion. Their report appears below.
Limitations on the Effectiveness of Controls
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures and internal controls over financial reporting will prevent all error and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system will be met. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
85
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of
Maxygen, Inc.
We have audited Maxygen Inc.’s internal control over financial reporting as of December 31, 2007, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Maxygen, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included above under the caption Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Maxygen, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2007, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Maxygen, Inc. as of December 31, 2006 and 2007, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2007 of Maxygen, Inc. and our report dated March 7, 2008 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Palo Alto, California
March 7, 2008
86
Table of Contents
| Item 9B | OTHER INFORMATION |
Not applicable.
PART III
| Item 10 | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
We have adopted a written code of ethics that applies to our senior financial officers, including our principal executive officer, principal financial officer and principal accounting officer. We have posted the text of such code of ethics on our website (www.maxygen.com). We intend to satisfy the disclosure requirement of Item 5.05 ofForm 8-K regarding an amendment to, or a waiver from, a provision of our code of ethics that applies to our principal executive officer, principal financial officer, or principal accounting officer by posting such information on our website.
The remaining information required by this item is incorporated by reference from the sections captioned “Election of Directors,” “Executive Officers,” “Section 16(a) Beneficial Ownership Reporting Compliance” and “Board of Directors’ Meetings and Committees — Audit Committee” contained in the 2008 Proxy Statement.
| Item 11 | EXECUTIVE COMPENSATION |
The information required by this item is incorporated by reference from the sections captioned “Executive Compensation,” “Director Compensation,” “Compensation Committee Report” and “Compensation Committee Interlocks and Insider Participation” contained in the 2008 Proxy Statement.
| Item 12 | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this item is incorporated by reference from the section captioned “Security Ownership of Certain Beneficial Owners and Management” contained in the 2008 Proxy Statement.
| Item 13 | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this item is incorporated by reference from the sections captioned “Related Party Transactions” and “Board of Directors’ Meetings and Committees” contained in the 2008 Proxy Statement.
| Item 14 | PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information required by this item is incorporated by reference from the section captioned “Ratification of Selection of Independent Registered Public Accounting Firm” contained in the 2008 Proxy Statement.
87
Table of Contents
PART IV
| Item 15 | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
15(a)(1) Financial Statements. The following documents are being filed as part of this report:
| Page | ||||
| 51 | ||||
| 52 | ||||
| 53 | ||||
| 54 | ||||
| 55 | ||||
| 56 | ||||
15(a)(2) Financial Statement Schedules. Financial statement schedules have been omitted because they are either presented elsewhere, are inapplicable or are immaterial as defined in the instructions.
15(a)(3) Exhibits.
See attached Exhibit Index.
88
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
MAXYGEN, INC.
| By: /s/ Russell J. Howard |
Russell J. Howard
Chief Executive Officer
March 7, 2008
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Michael Rabson and Lawrence W. Briscoe or either of them, his or her true and lawfulattorneys-in-fact and agents, with full power of substitution and re-substitution, for him or her and in his or her name, place and stead, in any and all capacities to sign any and all amendments to this Report onForm 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or either of them, or their, his or her substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||||
/s/ Russell J. Howard Russell J. Howard | Chief Executive Officer and Director (Principal Executive Officer) | March 7, 2008 | ||||
/s/ Lawrence W. Briscoe Lawrence W. Briscoe | Chief Financial Officer (Principal Financial and Accounting Officer) | March 7, 2008 | ||||
/s/ Isaac Stein Isaac Stein | Chairman of the Board | March 7, 2008 | ||||
/s/ M.R.C. Greenwood M.R.C. Greenwood | Director | March 7, 2008 | ||||
/s/ Louis G. Lange Louis G. Lange | Director | March 7, 2008 | ||||
/s/ Ernest Mario Ernest Mario | Director | March 7, 2008 | ||||
/s/ Gordon Ringold Gordon Ringold | Director | March 7, 2008 | ||||
/s/ James R. Sulat James R. Sulat | Director | March 7, 2008 | ||||
89
Table of Contents
EXHIBIT INDEX
| Incorporation by Reference | Filed | |||||||||||||||||||||||
Exhibit No. | Description of Exhibit | Form | SEC File No. | Exhibit | Filing Date | Herewith | ||||||||||||||||||
| 3 | .1 | Amended and Restated Certificate of Incorporation | 10-Q | 000-28401 | 3 | .1 | 8/14/2000 | |||||||||||||||||
| 3 | .2 | Amended and Restated Bylaws | 8-K | 000-28401 | 3 | .1 | 9/07/2007 | |||||||||||||||||
| 4 | .1 | Specimen Common Stock Certificate | S-1 | 333-89413 | 4 | .1 | 11/22/1999 | |||||||||||||||||
| *10 | .1 | Form of Executive Officer and Director | S-1 | 333-89413 | 10 | .7 | 10/20/1999 | |||||||||||||||||
| Indemnification Agreement | ||||||||||||||||||||||||
| *10 | .2 | Form of Executive Officer Change of Control Agreement | 10-Q | 000-28401 | 10 | .1 | 8/14/2001 | |||||||||||||||||
| *10 | .2.1 | Form of Amendment No. 1 to Executive Officer Change of Control Agreement | 10-K | 000-28401 | 10 | .3 | 3/27/2003 | |||||||||||||||||
| *10 | .2.2 | Form of Amendment No. 2 to Executive Officer Change of Control Agreement | 8-K | 000-28401 | 10 | .3 | 6/30/2006 | |||||||||||||||||
| *10 | .3 | 1997 Stock Option Plan, as amended, with applicable option agreement | 10-Q | 000-28401 | 10 | .1 | 8/14/2002 | |||||||||||||||||
| *10 | .4 | Form of Amendment to Stock Option Agreements | 8-K | 000-28401 | 10 | .2 | 6/30/2006 | |||||||||||||||||
| *10 | .5 | 1999 Nonemployee Directors Stock Option Plan, as amended, with applicable option agreement | 10-Q | 000-28401 | 10 | .3 | 8/14/2001 | |||||||||||||||||
| *10 | .5 | 1999 Employee Stock Purchase Plan, as amended | 10-K | 000-28401 | 10 | .11 | 3/21/2001 | |||||||||||||||||
| *10 | .6 | 2000 International Stock Option Plan, as amended, with applicable option agreement | 10-K | 000-28401 | 10 | .6 | 3/25/2002 | |||||||||||||||||
| 10 | .7 | 2000 Non-Officer Stock Option Plan, as amended, with applicable option agreement | S-8 | 333-57486 | 99 | .3 | 3/23/2001 | |||||||||||||||||
| *10 | .8 | 2006 Equity Incentive Plan (including related form of stock option agreement) | 8-K | 000-28401 | 10 | .4 | 6/30/2006 | |||||||||||||||||
| *10 | .8.1 | Form of Restricted Stock Unit Award Agreement under 2006 Equity Incentive Plan | S-8 | 333-138898 | 4 | .2 | 11/22/2006 | |||||||||||||||||
| 10 | .9 | Lease, dated as of October 21, 1998, between Metropolitan Life Insurance Company and Maxygen, Inc. | S-1 | 333-89413 | 10 | .4 | 10/20/1999 | |||||||||||||||||
| 10 | .9.1 | First Amendment to Lease, dated as of February 26, 1999, by and between Metropolitan Life Insurance Company and Maxygen, Inc. | S-1 | 333-89413 | 10 | .5 | 10/20/1999 | |||||||||||||||||
| 10 | .9.2 | Second Amendment to Lease, dated as of October 24, 2000, by and between Metropolitan Life Insurance Company and Maxygen, Inc. | 10-K | 000-28401 | 10 | .6 | 3/21/2001 | |||||||||||||||||
| 10 | .9.3 | Third Amendment to Lease, dated October 22, 2003, by and between Metropolitan Life Insurance Company and Maxygen, Inc. | 10-K | 000-28401 | 10 | .15 | 3/12/2004 | |||||||||||||||||
90
Table of Contents
| Incorporation by Reference | Filed | |||||||||||||||||||||||
Exhibit No. | Description of Exhibit | Form | SEC File No. | Exhibit | Filing Date | Herewith | ||||||||||||||||||
| 10 | .9.4 | Fourth Amendment to Lease dated December 15, 2004 by and between Metropolitan Life Insurance Company and Maxygen, Inc. | 10-K | 000-28401 | 10 | .13 | 3/15/2005 | |||||||||||||||||
| 10 | .9.5 | Fifth Amendment to Lease dated as of August 24, 2006, by and between Metropolitan Life Insurance Company and Maxygen, Inc. | 8-K | 000-28401 | 10 | .2 | 8/25/2006 | |||||||||||||||||
| 10 | .10 | Lease, dated December 15, 2004, between Metropolitan Life Insurance Company and Maxygen, Inc. | 10-K | 000-28401 | 10 | .14 | 3/15/2005 | |||||||||||||||||
| 10 | .10.1 | First Amendment to Lease, dated as of August 24, 2006, by and between Metropolitan Life Insurance Company and Maxygen, Inc. | 8-K | 000-28401 | 10 | .1 | 8/25/2006 | |||||||||||||||||
| 10 | .11 | Lease Agreement, dated May 5, 2000, between ProFound Pharma A/S and The Science Park in Horsholm | 10-Q | 000-28401 | 10 | .1 | 11/14/2000 | |||||||||||||||||
| 10 | .12+ | Technology Transfer Agreement, dated March 14, 1997 (effective March 1, 1998), among Maxygen, Inc., Affymax Technologies N.V. and Glaxo Group Limited, as amended | S-1 | 333-89413 | 10 | .3 | 12/15/1999 | |||||||||||||||||
| *10 | .13 | Description of 2006 Executive Officer Cash Bonus Plan | 8-K | 000-28401 | 10 | .1 | 6/30/2006 | |||||||||||||||||
| *10 | .14 | Letter Agreement (re tax equalization payments), dated November 20, 2006, between Elliot Goldstein and Maxygen, Inc. | 10-K | 000-28401 | 10 | .25 | 3/14/2007 | |||||||||||||||||
| 10 | .15+ | Cross License Agreement, dated as of July 16, 2003, between Maxygen, Inc. and Amgen Mountain View Inc. (as successor to Avidia, Inc.) | 10-K | 000-28401 | 10 | .27 | 3/14/2007 | |||||||||||||||||
| 10 | .16+ | Amended and Restated Exclusive License Agreement, dated July 10, 2006 (effective as of April 1, 2006), between Regents of the University of Minnesota and Maxygen, Inc. | 10-Q | 000-28401 | 10 | .6 | 8/07/2006 | |||||||||||||||||
| *10 | .17 | Consulting Agreement, between the Company and Waverley Associates, Inc., dated as of April 1, 2006 | 8-K | 000-28401 | 10 | .1 | 4/04/2006 | |||||||||||||||||
| *10 | .18 | Consulting Agreement, between the Company and Michael Rabson, effective as of April 1, 2008 | 8-K | 000-28401 | 10 | .1 | 3/04/2008 | |||||||||||||||||
| *10 | .18.1 | Letter Agreement (re extension of Consulting Agreement), between the Company and Waverley Associates, Inc., dated as of December 19, 2007 | X | |||||||||||||||||||||
| 10 | .19+ | License Agreement, dated as of March 28, 2002, between the Company and Codexis, Inc. | X | |||||||||||||||||||||
91
Table of Contents
| Incorporation by Reference | Filed | |||||||||||||||||||||||
Exhibit No. | Description of Exhibit | Form | SEC File No. | Exhibit | Filing Date | Herewith | ||||||||||||||||||
| 10 | .19.1+ | Amendment No. 1 to License Agreement, dated as of September 13, 2002, between the Company and Codexis, Inc. | X | |||||||||||||||||||||
| 10 | .19.2 | Amendment No. 2 to License Agreement, dated as of October 1, 2002, between the Company and Codexis, Inc. | X | |||||||||||||||||||||
| 10 | .19.3+ | Amendment No. 3 to License Agreement, dated as of August 22, 2006, between the Company and Codexis, Inc. | X | |||||||||||||||||||||
| *10 | .20 | Description of Non-Employee Director Compensation | X | |||||||||||||||||||||
| 21 | .1 | List of Subsidiaries | X | |||||||||||||||||||||
| 23 | .1 | Consent of Independent Registered Public Accounting Firm | X | |||||||||||||||||||||
| 24 | .1 | Power of Attorney (included on signature page) | X | |||||||||||||||||||||
| 31 | .1 | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | X | |||||||||||||||||||||
| 31 | .2 | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | X | |||||||||||||||||||||
| 32 | .1 | Certification of Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | X | |||||||||||||||||||||
| * | Management contract or compensatory plan or arrangement. | |
| + | Confidential treatment has been granted, or requested, with respect to portions of the exhibit. A complete copy of the agreement, including the redacted terms, has been separately filed with the Securities and Exchange Commission. |
92