UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
(Mark One)
x QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the quarterly period ended September 30, 2016
Or
¨ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from ________________________
Commission file number: 001-36333
Bio-Path Holdings, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | | 87-0652870 |
| (State or other jurisdiction of | | (I.R.S. Employer |
| incorporation or organization) | | Identification No.) |
4710 Bellaire Boulevard, Suite 210, Bellaire, Texas 77401
(Address of principal executive offices)
Registrant’s telephone no., including area code: (832) 742-1357
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yesx No¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yesx No¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer¨ | Accelerated filerx |
| Non-accelerated filer¨ (Do not check if a smaller reporting company) | Smaller reporting company¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes¨ Nox
At November 2, 2016, the Company had 95,645,224 outstanding shares of common stock, par value $0.001 per share.
Unless the context requires otherwise, references in this Quarterly Report on Form 10-Q to “we,” “our,” “us,” “the Company” and “Bio-Path” refer to Bio-Path Holdings, Inc. and its wholly-owned subsidiary. Bio-Path Holdings, Inc.’s wholly-owned subsidiary, Bio-Path, Inc., is sometimes referred to herein as “Bio-Path Subsidiary.”
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Quarterly Report on Form 10-Q contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Forward-looking statements can be identified by words such as “anticipate,” “expect,” “intend,” “plan,” “believe,” “seek,” “estimate,” “project,” “goal,” “strategy,” “future,” “likely,” “may,” “should,” “will” and variations of these words and similar references to future periods, although not all forward-looking statements contain these identifying words. Forward-looking statements are neither historical facts nor assurances of future performance. Instead, they are based on our current beliefs, expectations and assumptions regarding the future of our business, future plans and strategies, projections, anticipated events and trends, the economy and other future conditions. Because forward-looking statements relate to the future, they are subject to inherent risks, uncertainties and changes in circumstances, including those discussed in “Item 1A. Risk Factors” to Part I of our Annual Report on Form 10-K as of the fiscal year ended December 31, 2015, and in other reports or documents we file with the U.S. Securities and Exchange Commission (“SEC���). As a result, our actual results may differ materially from those expressed or forecasted in the forward-looking statements, and you should not rely on such forward-looking statements. Please refer to “Item 1A. Risk Factors” to Part I of our Annual Report on Form 10-K as of the fiscal year ended December 31, 2015, and other reports or documents we file with the SEC for a discussion of risks and factors that could cause our actual results and financial condition to differ materially from those expressed or forecasted in this Quarterly Report on Form 10-Q.
Any forward-looking statement made by us in this Quarterly Report on Form 10-Q is based only on information currently available to us and speaks only as of the date on which it is made. We undertake no obligation to publicly update any forward-looking statement, whether as a result of new information, future developments or otherwise. However, you should carefully review the risk factors set forth in other reports or documents we file from time to time with the SEC.
TABLE OF CONTENTS
PART I - FINANCIAL INFORMATION
ITEM 1. CONSOLIDATED FINANCIAL STATEMENTS
BIO-PATH HOLDINGS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share data)
(Unaudited)
| | | As of September 30, | | | As of December 31, | |
| | | 2016 | | | 2015 | |
| | | | | | | |
| Assets | | | | | | | | |
| | | | | | | | | |
| Current assets | | | | | | | | |
| Cash | | $ | 11,299 | | | $ | 8,854 | |
| Prepaid drug product for testing | | | 196 | | | | 560 | |
| Other current assets | | | 912 | | | | 179 | |
| | | | | | | | | |
| Total current assets | | | 12,407 | | | | 9,593 | |
| | | | | | | | | |
| Fixed assets | | | | | | | | |
| Furniture, fixtures & equipment | | | 148 | | | | 123 | |
| Less accumulated depreciation | | | (82 | ) | | | (51 | ) |
| | | | 66 | | | | 72 | |
| Other assets | | | | | | | | |
| Technology licenses | | | 2,500 | | | | 2,500 | |
| Less accumulated amortization | | | (1,531 | ) | | | (1,410 | ) |
| | | | 969 | | | | 1,090 | |
| | | | | | | | | |
| Total Assets | | $ | 13,442 | | | $ | 10,755 | |
| | | | | | | | | |
| Liabilities & Shareholders’ Equity | | | | | | | | |
| | | | | | | | | |
| Current liabilities | | | | | | | | |
| Accounts payable | | | 118 | | | | 54 | |
| Accrued expense | | | 748 | | | | 883 | |
| | | | | | | | | |
| Total current liabilities | | | 866 | | | | 937 | |
| | | | | | | | | |
| Warrant liability | | | 3,199 | | | | - | |
| | | | | | | | | |
| Total Liabilities | | | 4,065 | | | | 937 | |
| | | | | | | | | |
| Shareholders’ equity | | | | | | | | |
| Preferred stock, $.001 par value 10,000,000 shares authorized, no shares issued and outstanding | | | - | | | | - | |
| Common stock, $.001 par value, 200,000,000 shares authorized 95,645,224 and 89,762,872 shares issued and outstanding as of 9/30/16 and 12/31/15, respectively | | | 96 | | | | 90 | |
| Additional paid in capital | | | 40,044 | | | | 35,112 | |
| Accumulated deficit | | | (30,763 | ) | | | (25,384 | ) |
| | | | | | | | | |
| Total shareholders’ equity | | | 9,377 | | | | 9,818 | |
| | | | | | | | | |
| Total Liabilities & Shareholders’ Equity | | $ | 13,442 | | | $ | 10,755 | |
SEE ACCOMPANYING NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
BIO-PATH HOLDINGS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
(Unaudited)
| | | Three Months Ended September 30, | | | Nine Months Ended September 30, | |
| | | 2016 | | | 2015 | | | 2016 | | | 2015 | |
| | | | | | | | | | | | | |
| Revenue | | $ | - | | | $ | - | | | $ | - | | | $ | - | |
| | | | | | | | | | | | | | | | | |
| Operating expenses | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Research and development | | | 2,313 | | | | 992 | | | | 4,520 | | | | 2,149 | |
| General and administrative | | | 681 | | | | 520 | | | | 2,287 | | | | 1,873 | |
| | | | | | | | | | | | | | | | | |
| Total operating expenses | | | 2,994 | | | | 1,512 | | | | 6,807 | | | | 4,022 | |
| | | | | | | | | | | | | | | | | |
| Net operating loss | | $ | (2,994 | ) | | $ | (1,512 | ) | | $ | (6,807 | ) | | $ | (4,022 | ) |
| | | | | | | | | | | | | | | | | |
| Other income | | | | | | | | | | | | | | | | |
| Change in fair value of warrant liability | | | 1,420 | | | | - | | | | 1,420 | | | | - | |
| Interest income | | | 4 | | | | 4 | | | | 8 | | | | 14 | |
| | | | | | | | | | | | | | | | | |
| Total other income | | | 1,424 | | | | 4 | | | | 1,428 | | | | 14 | |
| | | | | | | | | | | | | | | | | |
| Net loss | | $ | (1,570 | ) | | $ | (1,508 | ) | | $ | (5,379 | ) | | $ | (4,008 | ) |
| | | | | | | | | | | | | | | | | |
| Net loss per share, basic and diluted | | $ | (0.02 | ) | | $ | (0.02 | ) | | $ | (0.06 | ) | | $ | (0.04 | ) |
| | | | | | | | | | | | | | | | | |
| Basic and diluted weighted average number of common shares outstanding | | | 95,645 | | | | 89,763 | | | | 91,724 | | | | 89,763 | |
SEE ACCOMPANYING NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
BIO-PATH HOLDINGS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
(Unaudited)
| | | Nine Months Ended September 30, | |
| | | 2016 | | | 2015 | |
| | | | | | | |
| Cash flow from operating activities | | | | | | | | |
| | | | | | | | | |
| Net loss | | $ | (5,379 | ) | | $ | (4,008 | ) |
| | | | | | | | | |
| Adjustments to reconcile net loss to net cash used in operating activities | | | | | | | | |
| Amortization | | | 121 | | | | 121 | |
| Depreciation | | | 31 | | | | 31 | |
| Stock-based compensation | | | 550 | | | | 283 | |
| Change in fair value of warrant liability | | | (1,420 | ) | | | - | |
| (Increase) decrease in assets | | | | | | | | |
| Prepaid drug product for testing | | | 364 | | | | (250 | ) |
| Other current assets | | | (733 | ) | | | (100 | ) |
| Increase (decrease) in liabilities | | | | | | | | |
| Accounts payable and accrued expenses | | | (71 | ) | | | (81 | ) |
| | | | | | | | | |
| Net cash used in operating activities | | | (6,537 | ) | | | (4,004 | ) |
| | | | | | | | | |
| Cash flow from investing activities | | | | | | | | |
| | | | | | | | | |
| Purchase furniture, fixtures & equipment | | | (25 | ) | | | - | |
| | | | | | | | | |
| Net cash used in investing activities | | | (25 | ) | | | - | |
| | | | | | | | | |
| Cash flow from financing activities | | | | | | | | |
| | | | | | | | | |
| Net proceeds from sale of common stock | | | 9,007 | | | | - | |
| | | | | | | | | |
| Net cash provided by financing activities | | | 9,007 | | | | - | |
| | | | | | | | | |
| Net increase (decrease) in cash | | | 2,445 | | | | (4,004 | ) |
| | | | | | | | | |
| Cash, beginning of period | | | 8,854 | | | | 13,859 | |
| | | | | | | | | |
| Cash, end of period | | $ | 11,299 | | | $ | 9,855 | |
SEE ACCOMPANYING NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
BIO-PATH HOLDINGS, INC.
Notes to the Unaudited Consolidated Financial Statements
for the Period Ended September 30, 2016
Unless the context requires otherwise, references in these Notes to the Unaudited Consolidated Financial Statements to “we,” “our,” “us,” “the Company” and “Bio-Path” refer to Bio-Path Holdings, Inc. and its subsidiary. Bio-Path Holdings, Inc.’s wholly-owned subsidiary, Bio-Path, Inc., is sometimes referred to herein as “Bio-Path Subsidiary.”
The accompanying interim financial statements have been prepared with the instructions to Form 10-Q pursuant to the rules and regulations of the Securities and Exchange Commission (“SEC”) and, therefore, do not include all information and footnotes necessary for a complete presentation of the Company’s financial position, results of operations, cash flows, and stockholders’ equity in conformity with generally accepted accounting principles. In the opinion of management, all adjustments considered necessary for a fair presentation of the results of operations and financial position have been included and all such adjustments are of a normal recurring nature. The unaudited quarterly financial statements should be read in conjunction with the audited financial statements and notes thereto included in the Annual Report on Form 10-K of the Company as of and for the fiscal year ended December 31, 2015. The results of operations for the period ended September 30, 2016, are not necessarily indicative of the results for a full-year period.
| 1. | Organization and Business |
The Company is a clinical and preclinical stage oncology focused antisense drug development company utilizing a novel technology that achieves systemic delivery for target specific protein inhibition for any gene product that is over-expressed in disease. The Company’s drug delivery and antisense technology, called DNAbilize™, is a platform that uses P-ethoxy, which is a deoxyribonucleic acid (DNA) backbone modification that is intended to protect the DNA from destruction by the body’s enzymes when circulating in vivo, incorporated inside of a neutral charged lipid bilayer. The Company believes this combination allows for high efficiency loading of antisense DNA into non-toxic, cell-membrane-like structures for delivery of the antisense drug substance into cells. In vivo, the DNAbilize™ delivered antisense drug substances are systemically distributed throughout the body to allow for reduction or elimination of proteins in blood diseases and solid organs.
Using DNAbilize™ as a platform for drug development and manufacturing, we currently have two antisense drug candidates in development to treat a total of five different disease indications. Our lead drug candidate, Liposomal Grb2 (“BP1001”), targets the protein Grb2 and has entered the efficacy portion of a Phase II clinical trial for acute myeloid leukemia (AML) and is preparing to enter the safety segment of a Phase II clinical trial for blast phase and accelerated phase chronic myelogenous leukemia (CML). BP1001 is also in preclinical studies for solid tumors, including triple negative breast cancer and inflammatory breast cancer.
The Company’s second drug candidate, Liposomal Bcl2 (“BP1002”), targets the protein Bcl2, which is responsible for driving cell proliferation in up to 60% of all cancers. BP1002 is in preparation for an Investigational New Drug application.
Bio-Path Subsidiary was founded in May 2007 as a Utah corporation. In February 2008, Bio-Path Subsidiary completed a reverse merger with Ogden Golf Co. Corporation, a public company traded over the counter that had no current operations. The name of Ogden Golf was changed to Bio-Path Holdings, Inc. and the directors and officers of Bio-Path, Inc. became the directors and officers of Bio-Path Holdings, Inc. The Company’s operations to date have been limited to organizing and staffing the Company, acquiring, developing and securing its technology and undertaking product development for a limited number of product candidates.
In June 2015, the Company established an “at the market” (“ATM”) program through which it may offer and sell up to $25.0 million of its common stock from time to time, at Bio-Path’s discretion, through an investment banking firm, acting as sales agent. Sales of Bio-Path common stock under the ATM program will be made directly on or through the NASDAQ Capital Market, among other methods. Pursuant to the Securities Purchase Agreement (as defined below), the Company is subject to certain restrictions on its ability to offer and sell shares of common stock under the ATM program. As of September 30, 2016, the Company has not offered or sold any shares of its common stock under the ATM program.
On June 29, 2016, the Company entered into the Securities Purchase Agreement with certain healthcare focused institutional investors pursuant to which the Company agreed to sell an aggregate of 5,882,352 shares of the Company’s common stock and warrants to purchase up to 2,941,176 shares of the Company’s common stock for gross proceeds of approximately $10.0 million (the “2016 Registered Direct Offering”). The 2016 Registered Direct Offering closed on July 5, 2016. The net proceeds to the Company from the 2016 Registered Direct Offering, after deducting the placement agent’s fees and expenses and the Company’s offering expenses, and excluding the proceeds, if any, from the exercise of the warrants issued in the offering, was approximately $9.3 million. These proceeds were partially offset by additional financing costs incurred during the period of $0.3 million. The Company has made an accounting policy election related to the treatment of offering costs. This election will result in the Company recording costs in the same manner dictated by the instrument attributable for incurring these costs.
As of September 30, 2016, Bio-Path had $11.3 million in cash on hand.
As the Company has not begun its planned principal operations of commercializing a product candidate, the Company’s activities are subject to significant risks and uncertainties, including the potential requirement to secure additional funding, the outcome of the Company’s clinical trials, and failing to operationalize the Company’s current drug candidates before another company develops similar products.
| 2. | Recent Accounting Pronouncements |
In May 2014, the FASB issued Accounting Standards Update No. 2014-09,Revenue from Contracts with Customers.The new standard provides comprehensive guidance for recognizing revenue as goods or services are delivered to the customer in an amount that is expected to be earned from those same goods or services. The new standard is effective for fiscal years beginning after December 15, 2016, including interim periods within those fiscal years. Management is currently evaluating the impact of future adoption of the new standard on the Company’s consolidated financial statements.
In February 2016, the FASB issued Accounting Standards Update No. 2016-02,Leases. The new standard establishes a right-of-use (“ROU”) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the income statement. The new standard is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. Management is currently evaluating the impact of future adoption of the new standard on the Company’s consolidated financial statements.
In March 2016, the FASB issued Accounting Standards Update No. 2016-09,Stock Compensation.The new standard simplifies certain aspects of the accounting for share-based payment award transactions by allowing entities to continue to use current GAAP by estimating the number of awards that are expected to vest or, alternatively, entities can elect to account for forfeitures as they occur. The new standard is effective for fiscal years beginning after December 15, 2016, including interim periods within those fiscal years. Management is currently evaluating the impact of future potential adoption of the new standard on the Company’s consolidated financial statements.
Management has reviewed all other recently issued pronouncements and has determined they will have no material impact on the Company’s consolidated financial statements.
| 3. | Prepaid Drug Product for Testing |
Advance payments, including nonrefundable amounts, for goods or services that will be used or rendered for future clinical development activities are deferred and capitalized. Such amounts will be recognized as an expense as the related goods are delivered or the related services are performed. The Company incurred installments to its contract drug manufacturing and raw material suppliers totaling $0.6 million in late 2015 pursuant to drug supply contracts for the manufacture and delivery of the Company’s lead drug product for testing in a Phase II clinical trial. This amount was carried on the Balance Sheet as of December 31, 2015 at cost as Prepaid Drug Product for Testing. The Company recognized certain expenses and incurred additional installment costs during 2016, with advanced payments totaling $0.2 million, which are carried on the Balance Sheet as of September 30, 2016 as Prepaid Drug Product for Testing (See Note 11).
As of September 30, 2016, Other Current Assets included prepaid expenses of $0.9 million, comprised primarily of prepayments made to the Company’s clinical research organization for our Phase II clinical trial for BP1001 in AML. As of December 31, 2015, Other Current Assets included prepaid expenses of $0.2 million.
As of September 30, 2016, Current Liabilities included accounts payable of $0.1 million, comprised primarily of amounts owed for manufacturing development and testing services, audit fees and legal fees. By the first week of November 2016, the September 30, 2016 amounts included in accounts payable had been substantially paid. As of December 31, 2015, Current Liabilities included accounts payable of $0.1 million.
As of September 30, 2016, Current Liabilities included accrued expense of $0.7 million for clinical trial expenses, preclinical studies, legal fees, accrued vacation and employee bonus accrual. As of December 31, 2015, Current Liabilities included accrued expense of $0.9 million.
In connection with the 2016 Registered Direct Offering, the Company issued warrants to purchase up to 2,941,176 shares of the Company’s common stock to certain healthcare focused institutional investors, as well as warrants to purchase up to 250,000 shares of the Company’s common stock to H.C. Wainwright & Co., LLC and its designees as compensation for its services as the placement agent (collectively, the “2016 Warrants”). The 2016 Warrants contain a provision for net cash settlement in the event of certain fundamental transactions involving the Company (defined in the 2016 Warrants to include, among other things, the Company’s approval and consummation of a merger with another entity, the Company’s approval and consummation of the sale of all or substantially all of the Company’s assets or the occurrence of certain other change of control transactions).
Due to this provision and in accordance with ASC 480-10 (FASB Statement 150, Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity), the 2016 Warrants were classified as a liability and recorded at fair value using the Binomial Lattice Model. The estimated fair value of the Warrant Liability for the 2016 Warrants on the closing date, July 5, 2016, was $4.6 million. As of September 30, 2016, the fair value of the Warrant Liability was $3.2 million. The net change in fair value of $1.4 million during the quarter is shown as other income on the Company’s Consolidated Statements of Operations. The Company will continue to measure the fair value of the 2016 Warrants each quarter until they are exercised or expire and any change will be adjusted accordingly on the Company’s financial statements.
| 8. | Fair Value Measurements |
In accordance with ASC 820, the Company uses various inputs to measure the 2016 Warrants on a recurring basis to determine the fair value of the liability. ASC 820 also establishes a hierarchy categorizing inputs into three levels used to measure and disclose fair value. The hierarchy gives the highest priority to quoted prices available in active markets and the lowest priority to unobservable inputs. An explanation of each level in the hierarchy is described below:
Level 1 – Unadjusted quoted prices in active markets for identical instruments that are accessible by the Company on the measurement date
Level 2 – Quoted prices in markets that are not active or inputs which are either directly or indirectly observable
Level 3 – Unobservable inputs for the instrument requiring the development of assumptions by the Company
The following table summarizes the Company’s 2016 Warrants measured at fair value within the hierarchy on a recurring basis as of September 30, 2016:
| | | Fair Value Measurements at
September 30, 2016
(in thousands) | |
| | | | Level 1 | | | | Level 2 | | | | Level 3 | | | | Total | |
| Liabilities: | | | | | | | | | | | | | | | | |
| Warrant liability | | $ | - | | | $ | - | | | $ | 3,199 | | | $ | 3,199 | |
The Company did not have the 2016 Warrants at December 31, 2015.
The following table summarizes changes to the fair value of the Level 3 2016 Warrants for the nine months ended September 30, 2016:
| | | Fair Value of
Warrant Liability | |
| | | (in thousands) | |
| Balance at December 31, 2015 | | $ | - | |
| Issuance | | | 4,619 | |
| Change in fair value | | | (1,420 | ) |
| Balance at September 30, 2016 | | $ | 3,199 | |
The Company utilized the Binomial Lattice Model for estimating the fair value of the 2016 Warrants using the following assumptions as of September 30, 2016:
| | | As of
September 30,
2016 |
| Risk-free interest rate | | | 1.14 | % |
| Expected volatility | | | 104 | % |
| Expected term in years | | | 5.3 | |
| Dividend yield | | | - | % |
Stockholders’ Equity totaled $9.4 million as of September 30, 2016 compared to $9.8 million as of December 31, 2015. There were 95,645,224 shares of common stock issued and outstanding as of September 30, 2016. There were no preferred shares outstanding as of September 30, 2016.
| 10. | Stock-Based Compensation and Warrants |
The Plan - In 2007, the Company adopted the First Amended 2007 Stock Incentive Plan, as amended (the “Plan”). The Plan provides for the grant of Incentive Stock Options, Nonqualified Stock Options, Restricted Stock Awards, Restricted Stock Unit Awards, Performance Awards and other stock-based awards, or any combination of the foregoing to the Company’s key employees, non-employee directors and consultants. Under the Plan, the exercise price is determined by the Board of Directors or the compensation committee of the Board of Directors, and for options intended to qualify as qualified incentive stock options, may not be less than the fair market value as determined by the closing stock price at the date of the grant. Each option and award shall vest and expire as determined by the Board of Directors or the compensation committee. Options expire no later than ten years from the date of grant. All grants provide for accelerated vesting if there is a change of control, as defined in the Plan.
Stock-based compensation expense was $0.2 million and $0.1 million for the three months ended September 30, 2016 and September 30, 2015, respectively. Of these amounts, stock-based compensation expense for personnel involved in the Company’s general and administrative activities for both the three months ended September 30, 2016 and September 30, 2015 was $0.1 million. Stock-based compensation expense for personnel involved in the Company’s research and development activities for the three months ended September 30, 2016 and September 30, 2015 was $0.1 million and $36,000, respectively.
Stock-based compensation expense was $0.6 million and $0.3 million for the nine months ended September 30, 2016 and September 30, 2015, respectively. Of these amounts, stock-based compensation expense for personnel involved in the Company’s general and administrative activities for the nine months ended September 30, 2016 and September 30, 2015 was $0.3 million and $0.2 million, respectively. Stock-based compensation expense for personnel involved in the Company’s research and development activities for the nine months ended September 30, 2016 and September 30, 2015 was $0.2 million and $0.1 million, respectively.
The Company utilized the Black-Scholes valuation model for estimating the fair value of the stock options granted, with the following weighted average assumptions for options granted in the nine months ended September 30, 2016 and 2015:
| | | 2016 | | 2015 |
| Risk-free interest rate | | | 1.37 | % | | | 1.65 | % |
| Expected volatility | | | 109 | % | | | 138 | % |
| Expected term in years | | | 6.1 | | | | 6.1 | |
| Dividend yield | | | - | % | | | - | % |
The following summary represents option activity under the Company’s stock-based compensation plan for the nine months ended September 30, 2016:
| | | | | | Weighted | |
| | | | | | Average | |
| | | | | | Exercise | |
| | | Options | | | Price | |
| | | (in thousands) | | | | |
| Outstanding at December 31, 2015 | | | 5,752 | | | $ | 1.05 | |
| Granted | | | 1,300 | | | | 2.55 | |
| Outstanding at September 30, 2016 | | | 7,052 | | | | 1.33 | |
| Exercisable at September 30, 2016 | | | 5,420 | | | $ | 1.00 | |
As of September 30, 2016, the aggregate intrinsic value of outstanding stock options was $2.4 million. The aggregate intrinsic value represents the total pretax intrinsic value (the difference between the Company’s closing stock price on September 30, 2016 and the exercise price, multiplied by the number of in-the-money options) that would have been received by the option holders had all option holders exercised their options on September 30, 2016. This amount changes based on the fair market value of the Company’s stock. As of December 31, 2015, the aggregate intrinsic value of outstanding stock options was $2.0 million.
As of September 30, 2016, unamortized stock-based compensation expense for all outstanding options was $2.1 million, which is expected to be recognized over a weighted average vesting period of 3.1 years.
Warrants- There were no warrants for services granted during the three months ended September 30, 2016. The Company had 10,000 warrants for services outstanding as of September 30, 2016 with a weighted average exercise price of $0.90. The warrants issued in connection with the sale of units of common stock were for cash value received and as such were not grants of compensation-based warrants.
| 11. | Commitments and Contingencies |
Technology License –The Company has negotiated an exclusive license agreement (the “License Agreement”) with The University of Texas, MD Anderson Cancer Center (“MD Anderson”) to develop drug delivery technology for antisense and siRNA drug products. The License Agreement requires, among other things, the Company to reimburse MD Anderson for ongoing patent expense and an annual license maintenance fee. The annual license maintenance fee attributable to the License Agreement totaling $0.1 million was included in Current Liabilities as of December 31, 2015 and was paid in April 2016.
Operating Leases – In April 2014, the Company entered into a lease agreement for a larger office space, which it occupied as of August 2014. The remaining lease payments due under this lease as of September 30, 2016 are $0.2 million.
In April 2016, the Company entered into a three-year lease agreement for lab space located in Bellaire, Texas. The term of lease began on May 1, 2016 and terminates on April 30, 2019 and will require Bio-Path to pay $2,500 per month over the term of the lease. The remaining lease payments due under this lease as of September 30, 2016 are $0.1 million.
Drug Supplier Project Plan – Bio-Path has a project plan agreement with a producer of the Company’s drug product for the manufacture and delivery of four batches of final drug product, three of which have been delivered to the Company as of September 30, 2016 and the fourth batch is expected to complete production in early 2017. As of September 30, 2016, the remaining commitment for these batches requires the Company to pay $0.1 million when the drug product from the fourth batch is completed and delivered. In addition, the Company has entered into an agreement with its drug substance provider for two batches of material to be used in the final drug product supplier plan with a remaining commitment totaling $0.1 million. In September 2016, the Company entered into an agreement with its drug substance provider for four batches of material that is expected to be delivered to the Company in the first quarter 2017. The commitment for the four batches of drug substance material total $0.9 million. The amounts paid for manufacture of the Company’s Grb2 drug substance and BP1001 drug product that have not been expensed totals $0.2 million and is carried on the balance sheet as of September 30, 2016 as Prepaid Drug Product for Testing (See Note 3). Commitments to the drug substance and drug product manufacturers for manufacturing development of Bio-Path’s second drug product candidate total $0.1 million. The balance of drug supplier commitments totaling $0.2 million is for assay development and manufacturing and supplier development.
Service Agreement –On September 30, 2016, the Company entered into a service agreement with a preclinical stage biotechnology company in connection with a development project involving our DNAbilize™ technology, pursuant to which we agreed to perform certain evaluation services in exchange for $50,000. While the agreement was entered into during the quarter ended September 30, 2016, due to the nature of the agreement, revenue will be recorded and recognized as services are performed. As of September 30, 2016, no services had been performed and, accordingly, no revenue was recorded or recognized during the period.
ITEM 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
When you read this Item of this Quarterly Report on Form 10-Q, it is important that you also read the unaudited financial statements and related notes included elsewhere in this Quarterly Report on Form 10-Q and our audited financial statements and notes thereto included in our Annual Report on Form 10-K as of the fiscal year ended December 31, 2015. This Quarterly Report on Form 10-Q contains forward-looking statements that involve risks and uncertainties, such as statements of our plans, objectives, expectations, and intentions. We use words such as “anticipate,” “estimate,” “plan,” “project,” “continuing,” “ongoing,” “expect,” “believe,” “intend,” “may,” “will,” “should,” “could,” and similar expressions to identify forward-looking statements. Our actual results could differ materially from those anticipated in these forward-looking statements for many reasons, including the matters discussed in “Item 1A. Risk Factors” to Part I of our Annual Report on Form 10-K as of the fiscal year ended December 31, 2015, and other risks and uncertainties discussed in filings made with the SEC. See “Cautionary Note Regarding Forward-Looking Statements” in this Quarterly Report on Form 10-Q for additional discussion regarding risks associated with forward-looking statements.
Overview
We are a clinical and preclinical stage oncology focused antisense drug development company utilizing a novel technology that achieves systemic delivery for target specific protein inhibition for any gene product that is over-expressed in disease. Our drug delivery and antisense technology, called DNAbilize™, is a platform that uses P-ethoxy, which is a deoxyribonucleic acid (DNA) backbone modification that is intended to protect the DNA from destruction by the body’s enzymes when circulatingin vivo, incorporated inside of a neutral charged lipid bilayer. We believe this combination allows for high efficiency loading of antisense DNA into non-toxic, cell-membrane-like structures for delivery of the antisense drug substance into cells.In vivo, the DNAbilize™ delivered antisense drug substances are systemically distributed throughout the body to allow for reduction or elimination of proteins in blood diseases and solid organs.
Using DNAbilize™ as a platform for drug development and manufacturing, we currently have two antisense drug candidates in development to treat a total of five different disease indications. Our lead drug candidate, Liposomal Grb2 (“BP1001”), targets the protein Grb2 and has entered the efficacy portion of a Phase II clinical trial for acute myeloid leukemia (AML) and is preparing to enter the safety segment of a Phase II clinical trial for blast phase and accelerated phase chronic myelogenous leukemia (CML). BP1001 is also in preclinical studies for solid tumors, including triple negative breast cancer (TNBC) and inflammatory breast cancer (IBC).
Our second drug candidate, Liposomal Bcl2 (“BP1002”), targets the protein Bcl2, which is responsible for driving cell proliferation in up to 60% of all cancers. BP1002 is in preparation for an Investigational New Drug (IND) application.
We currently maintain an exclusive license agreement (the “License Agreement”) with The University of Texas, MD Anderson Cancer Center (“MD Anderson”), under which we license from MD Anderson the delivery technology platform and BP1001 and BP1002. We are developing antisense drug candidates to treat cancer and autoimmune disorders where targeting a single protein may be advantageous and result in reduced adverse effects as compared to small molecule inhibitors with off-target and non-specific effects. We have composition of matter and method of use intellectual property for the manufacture of neutral charged DNA-liposome complexes.
On June 29, 2016, we entered into a securities purchase agreement (the “Securities Purchase Agreement”) with certain healthcare focused institutional investors pursuant to which we agreed to sell an aggregate of 5,882,352 shares of our common stock and warrants to purchase up to 2,941,176 shares of our common stock for gross proceeds of approximately $10.0 million. The offering closed on July 5, 2016. The net proceeds to the Company from the offering, after deducting the placement agent’s fees and expenses and our offering expenses, and excluding the proceeds, if any, from the exercise of the warrants issued in the offering, was approximately $9.3 million.
As of September 30, 2016, we had an accumulated deficit of $30.8 million. Our net loss was $1.6 million and $1.5 million for the three months ended September 30, 2016 and 2015, respectively. Our net loss was $5.4 million and $4.0 million for the nine months ended September 30, 2016 and 2015, respectively. We expect to continue to incur significant operating losses and we anticipate that our losses may increase substantially as we expand our drug development programs and commercialization efforts. To achieve profitability, we must enter into license or development agreements with third parties, or successfully develop and obtain regulatory approval for one or more of our drug candidates and effectively commercialize any drug candidates we develop. In addition, if we obtain regulatory approval of one or more of our drug candidates, we expect to incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution. There can be no assurance that we will be able to raise additional capital when needed or on terms that are favorable to us, if at all. Even if we succeed in developing and commercializing one or more of our drug candidates, we may not be able to generate sufficient revenue and we may never be able to achieve or sustain profitability.
Basic Technical Information
Ribonucleic acid (RNA) is a biologically significant type of molecule consisting of a chain of nucleotide units. Each nucleotide consists of a nitrogenous base, a ribose sugar, and a phosphate. Although similar in some ways to DNA, RNA differs from DNA in a few important structural details. RNA is transcribed from DNA by enzymes called RNA polymerases and is generally further processed by other enzymes. RNA is central to protein synthesis. DNA carries the genetic information of a cell and consists of thousands of genes. Each gene serves as a recipe on how to build a protein molecule. Proteins perform important tasks for the cell functions or serve as building blocks. The flow of information from the genes determines the protein composition and thereby the functions of the cell.
The DNA is situated in the nucleus of the cell, organized into chromosomes. Every cell must contain the genetic information and the DNA is therefore duplicated before a cell divides (replication). When proteins are needed, the corresponding genes are transcribed into RNA (transcription). The RNA is first processed so that non-coding parts are removed (processing) and is then transported out of the nucleus (transport). Outside the nucleus, the proteins are built based upon the code in the RNA (translation).
Our basic drug development concept is to block the expression of proteins that cause disease. RNA is essential in the process of creating proteins. We intend to develop drugs and drug delivery systems that work by delivering short strands of DNA material (antisense DNA) that are inserted into a cell to block the production of proteins associated with disease.
Antisense DNA therapeutics is the field of designing short DNA sequences that are complementary to an RNA for a protein of interest with the intention of inhibiting the production of the targeted protein. The DNA will find the matching RNA and form a complex. The complexed RNA will not have access to the protein-making machinery, which prevents the cell from translating it into a protein. Thus, protein production is turned off and levels of the targeted protein are reduced in the cell. This gene-specific process of controlling protein expression has led to great interest in using antisense DNA to shut off the production of proteins involved in disease. Antisense therapeutics have been in development for over 20 years; however, there have been many challenges to antisense therapeutics that have prevented or reduced the successful distribution and transfer of DNA into cells. Of all delivery methods in use today, we believe only DNAbilize™ has the potential to overcome the most common challenges associated with antisense therapeutics.
Overview of Drug Candidates and Delivery Technology
BP1001
BP1001 is targeted at the protein Grb2. Antisense inhibition of Grb2 interrupts the signals between mutated and activated receptors that connect to a well-known cancer associated switch called Ras protein. Inhibition of Grb2 does not cause cell death and thus does not result in adverse events typically observed with receptor inhibitors or Ras pathway inhibitors. We believe that BP1001 has the potential to be an ideal combination for any number of cancer therapeutics where the Ras pathway is aberrantly activated and patient fitness is a major concern.
We have completed our Phase I clinical trials for BP1001 for indications for AML, CML, MDS and Acute Lymphoblastic Leukemia (ALL). We are currently prioritizing our efforts on AML and CML and have begun the Phase Ib/Phase II clinical trials for these indications. Priorities for additional indications, including MDS or ALL, are expected to be addressed in the future as the results of our Phase II and work in solid tumors progresses.
Indications for Acute Myeloid Leukemia (AML) and Chronic Myelogenous Leukemia (CML)
AML – Background and Common Treatments. AML is the rapid accumulation of immature myeloid cells in the blood, resulting in a drop of the other cell types such as red blood cells and platelets. The expansion of immature monocytes leaves the patient unable to fight infection. If AML is left untreated, it usually results in death within three months. AML incidence increases with age, with more than 50% of the cases in people age 60 or older. AML is the most common acute leukemia in adults, and the National Cancer Institute estimates that approximately 20,000 new cases occur each year. The cure rate is between 5-15% in older adults, and those who cannot receive the standard course of chemotherapy have an average survival rate of five to ten months. The standard induction therapy for AML is Cytarabine with anthracycline. Of those patients who are able to receive the standard induction therapy, about 75% will likely relapse. AML is an area of high unmet need for both the relapsed and the de novo elderly population who are typically ineligible for induction therapy.
CML – Background and Common Treatments. CML is characterized by expansion in the blood and bone marrow of mature myeloid cells and their precursors. It can show no symptoms and is often detected during a routine blood test. If left untreated, after several years it will progress to an accelerated phase and eventually blast crisis where it becomes an acute leukemia. With the introduction of drugs such as Gleevec, the life expectancy of patients treated in the chronic phase has been significantly improved, and only 1-1.5% of patients ever go into blast crisis. However, for those patients who do progress into blast crisis, there are currently few treatment options. Myeloid cells in blast crisis have accumulated genetic abnormalities that resist traditional treatment methods that kill leukemic cells. Patients in blast crisis have an average survival rate of seven to eleven months. New treatments for this critical population are necessary.
BP1001 Development and Treatment for AML and CML. Our lead liposome delivered antisense drug candidate, BP1001, has been clinically tested in patients having AML, CML, MDS and ALL in a Phase I trial. During the Phase I trial, 80% of the evaluable patients had refractory or relapsed AML, having failed at least six prior therapies. In our study, 83% of patients showed decreased circulating blasts and anti-leukemic activity and eight patients stabilized for extended treatments.
Phase I Clinical Trials
The Phase I clinical trial was a dose-escalating study to determine the safety and tolerance of escalating doses of BP1001. The study determined an optimal biologically active dose for further development. The pharmacokinetics of BP1001 in patients from the study are being evaluated. In addition, patient blood samples from the trial were tested using a new assay developed by us to measure down-regulation of the target protein, the critical scientific data that demonstrated the delivery technology does in fact successfully deliver the antisense drug substance to the cell and across the cell membrane into the interior of the cell where expression of the target protein is blocked. The clinical trial was conducted at MD Anderson.
The original IND granted by the U.S. Food and Drug Administration (FDA) in March 2010 allowed us to proceed with a Phase I clinical trial having five cohorts culminating in a maximum dose of 50 mg/m2. However, in November 2012, we announced that since there had been no evidence of significant toxicity from treatment of patients with BP1001, we requested the FDA to allow higher dosing in patients. The principal investigator for the clinical trial, in consultation with our management team, advised us that with the absence of any real toxicity barriers, we should continue to evaluate higher doses of BP1001. The absence of significant toxicity provided a significant opportunity for us to test higher doses in patients in order to find a dose that provides maximum potential benefit and duration of anti-leukemia effect. These actions were approved and a revised protocol was submitted allowing higher dosing. We announced in October 2014 that we completed Cohort 6, successfully treating three patients at a dose 90 mg/m2. There has been no evidence of significant toxicity from treatment of patients with BP1001 in our Phase I clinical trial.
An important outcome for the Phase I clinical trial is the ability to assess for the first time the performance of our delivery technology platform in human patients. We have developed two new assays to be able to provide scientific proof of concept of the delivery technology. The first involves a novel detection method for the drug substance in blood samples to assess the pharmacokinetics of the drug. The second involves a method to measure down-regulation of the target protein in a patient blood sample that was achieved. The latter measurement provides critical proof that DNAbilize™ neutral liposome delivery technology delivered the drug substance to the cell and was able to transport it across the cell membrane into the interior to block cellular production of the Grb2 protein.
In this regard, in August 2013 we announced that our DNAbilize™ liposomal delivery technology achieved a major milestone in the development of antisense therapeutics based on a scientific assay confirming that treating patients with our drug candidate BP1001 inhibits the Grb2 disease-causing target protein in patients with blood cancers. Inhibition of the disease-causing protein has the effect of down regulating the disease. This will allow for BP1001 to be used potentially in combination with current frontline treatments. This discovery also points to the potential use of a liposomal antisense treatment as a standalone treatment to transform and manage a disease that has a disease-causing protein as a chronic disorder. This accomplishment is a potentially significant breakthrough for antisense therapeutics, whose development, to date, as a class of therapeutics has been severely limited by a lack of a systemic delivery mechanism that can safely distribute the drug throughout the body and deliver the antisense drug substance across the cell membrane into the interior of the cell. Further, we expect that scientific proof of principle for DNAbilize™ may lead to licensing and business development opportunities, supporting our business model.
The principal investigator for the Phase I clinical trial is a leading expert in the treatment of CML, AML, MDS and ALL. Because the results of the first trial produced unexpected and clinically interesting results in some patients, the principal investigator prepared an abstract of the results of the first cohort that was accepted for presentation at the American Society of Hematology (ASH) annual meeting in December 2011. Results that demonstrated potential anti-leukemia benefits in treated patients were included in the presentation. Subsequently, in 2013 the principal investigator prepared an abstract of updated information on the results of the clinical trial through Cohort 5, which was accepted for presentation at the ASH annual meeting in December 2013. Highlights (which have been updated to include patients from Cohort 6) of the presentation prepared by the principal investigator for the meeting included:
Data from the Phase I Clinical Trial
| | · | Among 20 evaluable patients, 15 demonstrated anti-leukemia activity with reduction in peripheral or bone marrow blasts from baseline. |
| | · | Five patients demonstrated transient improvement and/or stable disease, three of whom received a total of five cycles each. |
| | · | Two patients, in addition to achieving market blast percentage declines, also experienced transient improvements in leukemia cutis lesions. |
Disease Stabilization in MDS and AML
| | · | Two patients with MDS, a 53-year-old male and a 72-year-old female, both achieved disease stabilization and continued therapy for five cycles before disease progression. |
| | · | A 54-year-old HIV positive male with AML achieved stable disease and marked reduction in peripheral blasts, continuing therapy for five cycles before disease progression. |
Experience in CML-Blast Phase
| | · | Patient with myeloid blast crisis of CML. |
| | · | Prior therapies consisted of: imatinib, dastinib, nilotinib, DCC-2036, Cytarabine + Fludarabine + Dasatinib + Gemtuzumab, PHA-739358, Clofarabine + Dasatinib. |
| | · | Upon start of BP1001, patient showed a significant reduction in blasts from 81% to 5%, but due to leptomeningeal disease progression discontinued therapy before full cycle. |
Inhibition of Target Grb2 Protein
| | · | Grb2 levels were compared to baseline prior to treatment. |
| | · | By end of treatment, BP1001 decreased Grb2 in 11 out of 13 samples (85%) tested (average reduction 50%). |
The Phase I clinical trial is typically ended when a maximum tolerated dose (MTD) is encountered. However, due to the lack of toxicity of the drug, a MTD was not observed. As a result, an optimal biological dose was determined and we completed Cohort 6 of our Phase I clinical trial. It is noted, however, that the lack of toxicity is a major advantage for the drug candidate BP1001 since it allows higher levels of drug to be administered to the patient, increasing the potential therapeutic benefit.
In April 2015, we received orphan drug designation by the FDA for BP1001 in AML. Orphan drug status provides Bio-Path with seven years of exclusivity after receiving formal marketing approval, as well as additional development incentives. The FDA grants this designation to certain drugs that target diseases affecting fewer than 200,000 people in the United States. Previously, we received orphan drug designation for BP1001 for CML. In October 2016, BP1001 received orphan drug designation for AML in the European Union (EU) from the European Medicines Agency (EMA). To receive orphan drug designation from the EMA, a therapy must be intended for the treatment of a life-threatening or chronically debilitating rare condition with a prevalence of less than five in 10,000 in the EU. Orphan drug designation provides incentives designed to facilitate development including fee reductions for protocol assistance, scientific advice and importantly, may provide up to ten years of market exclusivity in the EU following product approval.
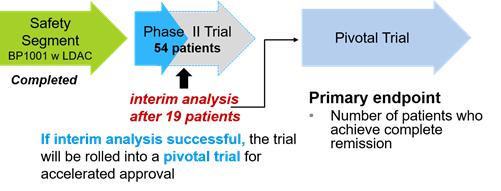
Phase II Clinical Trials
On February 9, 2015, we announced that we began enrollment into the combination therapy Phase Ib clinical trial, the safety segment of the Phase II clinical trial, for BP1001 in patients with AML. The combination therapy Phase Ib clinical trial consisted of two dosing cohorts of BP1001 (60 mg/m2 and 90 mg/m2) to test the safety profile of treating AML patients with BP1001 in combination with low dose Ara C (LDAC). Patients ineligible for intensive induction therapy are currently treated only with LDAC.
On October 9, 2015, we announced the completion of Cohort 7, the first dosing cohort of the Phase Ib clinical trial, consisting of a 60 mg/m2 dose of BP1001 in combination with LDAC. On March 3, 2016, we announced the completion of Cohort 8, the second dosing cohort of the Phase Ib clinical trial, consisting of a 90 mg/m2 dose of BP1001 in combination with LDAC. On June 6, 2016, we announced data from the safety segment of the Phase II combination therapy of BP1001 and LDAC showed no dose limiting toxicities. Of the six evaluable patients, four patients completed more than two cycles of treatment, three patients achieved complete remission and two patients achieved partial remission. Pharmacokinetics of BP1001 demonstrated a half-life at 60 mg/m2 of 30 hours, significantly better than the 90 mg/m2 dose. The final analysis of these data, along with the demonstrated reductions in bone marrow blasts, suggested that 60 mg/m2 was the appropriate dose for use in the Phase II trial. Administratively, this required us to reformat documents for the Phase II trial with the 60 mg/m2 dose and resubmit for approvals with the FDA and site Institutional Review Boards, requiring additional time prior to starting the Phase II trial.
On November 2, 2016, we announced that the first patient in the efficacy portion of the Phase II trial was dosed. The full trial design includes approximately 54 evaluable patients with an interim analysis to be performed after 19 patients are treated with the combination. In the event the interim results exceed the primary endpoint in the number of patients that meet or exceed statistically determined thresholds, we may seek to convert the trial into a registration trial for accelerated approval. The multi-site trial will be conducted at leading cancer centers, among them are Weill Medical College of Cornell University, Baylor Scott & White Health, The University of Kansas and MD Anderson.
Indications for Triple Negative Breast Cancer (TNBC) and Inflammatory Breast Cancer (IBC)
TNBC and IBC – Background and Common Treatments. Approximately 15 to 20% of breast cancers fall into the category of triple-negative. TNBC tumors do not express estrogen receptors, progesterone receptors and low human epidermal growth factor receptor 2 (HER2). These negative results mean that the growth of the cancer is not supported by the hormones estrogen and progesterone, or by the presence of HER2 receptors. Therefore, TNBC does not respond to hormonal therapy or therapies that target HER2 receptors. In addition, TNBC tumors are very aggressive. IBC often presents as TNBC and is a rare and very aggressive disease in which cancer cells block lymph vessels in the skin of the breast. This type of breast cancer is called “inflammatory” because the breast often looks swollen and red, or “inflamed.” IBC accounts for 2 to 5% of all breast cancers. IBC tumors are very aggressive and are frequently hormone receptor negative, which means hormone therapies may not be effective. The five-year survival rate for IBC is approximately 40% versus approximately 87% for all breast cancers combined, making IBC a priority area for development of new treatments. The current treatment regimen includes radiation, chemotherapy and surgery. A lack of targeted treatments for these types of breast cancer has led to development of new therapeutics currently in clinical trials. Because of the aggressiveness of these cancers, a systemic treatment is needed. BP1001 represents a systemic treatment that targets an important pathway for TNBC and IBC cell growth and has potential to be integral for the treatment of these diseases.
BP1001 Development and Treatment for TNBC and IBC. In July 2013, we announced that we were initiating preclinical testing of BP1001 for TNBC and IBC. Our plan is to develop BP1001 as a targeted therapy against TNBC and IBC. Our treatment goals are two-pronged: the first is to develop BP1001 as a tumor reduction agent in combination with other approved drugs in preoperative settings for TNBC and IBC patients, and the second is to develop BP1001 as a drug to treat and control or eliminate cancer metastasis in TNBC and IBC patients. Both of these treatment goals address high need situations for patients. Once the preclinical studies are completed, we believe that the observations that we learned from the original Phase I trial will help us increase of the speed of progress for such Phase I trial in TNBC and IBC, as the toxicity profile of BP1001 is currently well-established.
Indications for Other Solid Tumors (e.g., Lymphoma, Colon, Thyroid, and Head and Neck Cancers)
Cancers of colon, thyroid, head and neck, and lymphoma are solid tumors which utilize the same signaling pathway as TNBC and IBC, which involve the Grb2 protein. It has been proposed that BP1001 may have clinical efficacy in these indications due to the overlapping similarity of the mechanisms of their growth and proliferation. As our program for BP1001 continues to develop, it is anticipated that these indications will be assessed in preclinical research.
BP1002
BP1002, also known by its scientific name as Liposomal Bcl2, is our second liposome delivered antisense drug candidate. BP1002 is intended to target the lymphoma and certain solid tumor markets. Clinical targets for BP1002 include lymphoma, breast cancer, colon cancer, prostate cancer and leukemia. We believe that BP1002 has the potential to treat 40 to 60% of solid tumors.
Bcl2 is a protein that is involved in regulating apoptosis, or programmed cell death. Apoptosis is a physiologic mechanism of cell turnover by which cells actively commit suicide in response to aberrant external signals. Over-expression of Bcl2 prevents the induction of apoptosis in response to cellular insults such as treatment with chemotherapeutic agents. Bcl2 is over-expressed in more than 90% of follicular B-cell non-Hodgkin’s lymphoma due to a chromosomal rearrangement and is the key factor in the initiation of this malignancy. Bcl2 is also overexpressed in a wide variety of solid tumors (it is estimated to be over-expressed in 40% of cancers). For example, Bcl2 over-expression has been associated with the progression of prostate cancer from hormone dependence to hormone independence and may contribute to the relative drug resistant phenotype typically observed in hormone independent prostate cancer.
Non-Hodgkin’s Lymphomas –Background and Common Treatments. There are 56,000 new cases of non-Hodgkin’s lymphoma (NHL) per year, with approximately 30% being follicular lymphoma (FL) and approximately 60% being the more aggressive diffuse large B cell lymphoma (DLBCL) type. A consensus on front-line treatment for FL has not been established as many factors are taken into account in the treatment approach (e.g., age, stage of disease, cell surface markers). Rituximab is a treatment of choice for the majority of lymphomas and is typically used in combination with other chemotherapy agents or as a maintenance treatment.
BP1002 – Development and Treatment for FL, DLBCL, MALT, MCL AND BL. On December 22, 2014, we announced that we initiated development of BP1002 as a treatment for FL. We intend to file a new IND to begin clinical testing of BP1002 in patients with multiple types of lymphoma. We anticipate that the Phase I trial will be open to refractory and relapsed patients with FL and other sub-types of NHL, including DLBCL, mucosa-associated lymphoid tissue (MALT), mantle cell lymphoma (MCL) and Burkitt’s lymphoma (BL).
Treatments of varying efficacy exist for FL and DLBCL; however, due to the wide variety of subtypes of this disease, a frontline approach is lacking. Bcl2 is over-expressed in 85% of patients due to a translocation between chromosomes 18 and 14, a hallmark of the disease. Therapies that directly and specifically block or inhibit protein synthesis of Bcl2 could be transformative in this indication. Toxicity in competing therapeutics using small molecule inhibitors of Bcl2 occurs due to non-specificity of the inhibitors. Bcl2 is part of a large family of proteins. Small molecule inhibitors developed against it typically bind to more than one member of the family. This leads to unexpected off-target adverse effects. A previous attempt at a Bcl2 antisense by Genta Inc. failed to show an improvement in remission or overall survival rates. This antisense was a phosphorothioate DNA with dose-limiting toxicity and it also did not have a lipid delivery mechanism to aid in prevention of clearance by the liver, reducing the levels of antisense reaching diseased cells. We believe that BP1002 overcomes the failures of previous attempts at inhibiting Bcl2 by specifically interrupting the protein expression of one protein and not a family of necessary proteins and does so without inherent toxicity. With BP1002, more drug substance can reach the circulating lymphocytes so that the cancer cells can be treated with a therapeutically relevant dose. We believe BP1002 provides a new tool for cancer treatment for not just lymphomas, but also many cancers for which Bcl2 expression is driving cell proliferation. The introduction of a new, non-toxic, and specific Bcl2 inhibitor could be a major advance in cancer therapeutics.
DNAbilize™
DNAbilize™ technology is available for out-licensing. We intend to apply our drug delivery technology template to new disease-causing protein targets as a means to develop new liposomal antisense drug candidates. A new product identification template was recently approved that defines a process of scientific, preclinical, commercial and intellectual property evaluation of potential new drug candidates for inclusion into our drug product development pipeline. A significant amount of capital is expected to be allocated to in-license promising protein targets that can be developed as new liposomal antisense drug candidates. As we expand, we will look at indications where a systemic delivery is needed and antisense can be used to slow, reverse, or cure a disease, either alone or in combination with another drug.
We are interested in pursuing a wide-ranging, proactive licensing program to include co-development of specific liposomal antisense drug candidates, sub-licensing our delivery template for outside development of liposomal antisense drug candidates or out-licensing a partially-developed drug candidate for final development and marketing.
Company History and Available Information
We were originally incorporated in May 2000 as a Utah corporation under the name Ogden Golf Co. Corporation, but terminated our retail golf store operations in December 2006. In February 2008, we completed a reverse merger with Bio-Path Subsidiary. The name of Ogden Golf Co. Corporation was changed to Bio-Path Holdings, Inc. and the directors and officers of Bio-Path Subsidiary became the directors and officers of Bio-Path Holdings, Inc. On March 10, 2014, our common stock ceased trading on the OTCQX and commenced trading on the NASDAQ Capital Market under the ticker symbol “BPTH.” Effective December 31, 2014, we changed our state of incorporation from Utah to Delaware through a statutory conversion pursuant to the Utah Revised Business Corporation Act and the Delaware General Corporation Law. Our principal executive offices are located at 4710 Bellaire Boulevard, Suite 210, Bellaire, Texas 77401, and our telephone number is (832) 742-1357.
Recent Accounting Pronouncements
See Note 2 to the Unaudited Consolidated Financial Statements for a discussion of the impact of a new accounting standards update on the Company’s consolidated financial statements.
Financial Operations Overview
Revenue
We have not generated significant revenues to date. Our ability to generate revenues from our drug candidates, which we do not expect will occur for many years, if ever, will depend heavily on the successful development and eventual commercialization of our drug candidates.
Research and development expenses
Research and development expenses consist of costs associated with our research activities, including the development of our drug candidates. Our research and development expenses consist of:
| · | expenses related to research and development personnel, including salaries, benefits, travel and stock-based compensation; |
| | · | external research and development expenses incurred under arrangements with third parties, such as contract research organizations, clinical sites, manufacturing organizations and consultants; |
| | · | license fees, including maintenance fees and patent expense paid to MD Anderson in connection with the License Agreement; and |
| | · | costs of materials used during research and development activities. |
Costs and expenses that can be clearly identified as research and development are charged to expense as incurred in accordance with generally accepted accounting policies (“GAAP”). Advance payments, including nonrefundable amounts, for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts will be recognized as an expense as the related goods are delivered or the related services are performed. If the goods will not be delivered, or services will not be rendered, then the capitalized advance payment is charged to expense.
We expect research and development expenses associated with the completion of the associated clinical trials to be substantial and to increase over time. The successful development of our drug candidates is highly uncertain. At this time, we cannot reasonably estimate or know the nature, timing and estimated costs of the efforts that will be necessary to complete development of our drug candidates or the period, if any, in which material net cash inflows from our drug candidates may commence. This is due to the numerous risks and uncertainties associated with developing drugs, including the uncertainty of:
| | · | the rate of progress, results and costs of completion of ongoing clinical trials of our drug candidates; |
| | · | the size, scope, rate of progress, results and costs of completion of any potential future clinical trials and preclinical trials of our drug candidates that we may initiate; |
| | · | competing technological and market developments; |
| | · | the performance of third-party manufacturers and suppliers; |
| | · | the ability of our drug candidates, if they receive regulatory approval, to achieve market success; and |
| | · | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our drug candidates. |
A change in the outcome of any of these variables with respect to the development of a drug candidate could mean a significant change in the costs and timing associated with the development of that drug candidate. For example, if the FDA or other regulatory authority were to require us to conduct clinical trials beyond those which we currently anticipate will be required for the completion of clinical development of a drug candidate or if we experience significant delays in enrollment in any clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development.
General and administrative expenses
Our general and administrative expenses consist primarily of salaries and benefits for management and administrative personnel, professional fees for legal, accounting and other services, travel costs and facility-related costs such as rent, utilities and other general office expenses.
Results of Operations
Comparisons of the Three Months Ended September 30, 2016 to the Three Months Ended September 30, 2015
Research and Development Expense. Our research and development expense was $2.3 million for the three months ended September 30, 2016, an increase of $1.3 million compared to the three months ended September 30, 2015. The increase in research and development expense was primarily due to the release of drug material for our Phase II clinical trial for BP1001 in AML and associated clinical trial costs. The following table sets forth our research and development expenses (in thousands):
| | | Three Months Ended | |
| | | September 30, | |
| | | 2016 | | | 2015 | |
| Research and development expense | | $ | 2,204 | | | $ | 956 | |
| Non-cash stock-based compensation expense | | | 109 | | | | 36 | |
| Total research and development expense | | $ | 2,313 | | | $ | 992 | |
General and Administrative Expense. Our general and administrative expense for the three months ended September 30, 2016 was $0.7 million, an increase of $0.2 million compared to the three months ended September 30, 2015. The increase in general and administrative expense was primarily due to increased audit fees, stock-based compensation expense and salaries and benefits expense. The following table sets forth our general and administrative expenses (in thousands):
| | | Three Months Ended | |
| | | September 30, | |
| | | 2016 | | | 2015 | |
| General and administrative expense | | $ | 558 | | | $ | 457 | |
| Non-cash stock-based compensation expense | | | 123 | | | | 63 | |
| Total general and administrative expense | | $ | 681 | | | $ | 520 | |
Change in Fair Value of Warrant Liability. The change in fair value of the warrant liability for the three months ended September 30, 2016 resulted in non-cash income of $1.4 million.
Net Loss. Our net loss for the three months ended September 30, 2016 was $1.6 million, an increase of $0.1 million compared the three months ended September 30, 2015. Net loss per share, both basic and diluted, was $0.02 per share for the three months ended September 30, 2016 and September 30, 2015. Net loss per share is calculated using the weighted average number of shares of common stock outstanding during the applicable periods and excludes stock options and warrants because they are antidilutive.
Comparisons of the Nine Months Ended September 30, 2016 to the Nine Months Ended September 30, 2015
Research and Development Expense. Our research and development expense was $4.5 million for the nine months ended September 30, 2016, an increase of $2.4 million compared to the nine months ended September 30, 2015. The increase in research and development expense was primarily due to the release of drug material for our Phase II clinical trial for BP1001 in AML and associated clinical trial costs as well as increased salaries and benefits expense. The following table sets forth our research and development expenses (in thousands):
| | | Nine Months Ended | |
| | | September 30, | |
| | | 2016 | | | 2015 | |
| Research and development expense | | $ | 4,274 | | | $ | 2,052 | |
| Non-cash stock-based compensation expense | | | 246 | | | | 97 | |
| Total research and development expense | | $ | 4,520 | | | $ | 2,149 | |
General and Administrative Expense. Our general and administrative expense for the nine months ended September 30, 2016 was $2.3 million, an increase of $0.4 million compared to the nine months ended September 30, 2015. The increase in general and administrative expense was primarily due to increased third party fees related to corporate communications, stock-based compensation expense and salaries and benefits expense. The following table sets forth our general and administrative expenses (in thousands):
| | | Nine Months Ended | |
| | | September 30, | |
| | | 2016 | | | 2015 | |
| General and administrative expense | | $ | 1,983 | | | $ | 1,687 | |
| Non-cash stock-based compensation expense | | | 304 | | | | 186 | |
| Total general and administrative expense | | $ | 2,287 | | | $ | 1,873 | |
Change in Fair Value of Warrant Liability. The change in fair value of the warrant liability for the nine months ended September 30, 2016 resulted in non-cash income of $1.4 million.
Net Loss. Our net loss for the nine months ended September 30, 2016 was $5.4 million, an increase of $1.4 million compared the nine months ended September 30, 2015. Net loss per share, both basic and diluted, was $0.06 per share for the nine months ended September 30, 2016 compared to $0.04 per share for the nine months ended September 30, 2015. Net loss per share is calculated using the weighted average number of shares of common stock outstanding during the applicable periods and excludes stock options and warrants because they are antidilutive.
Liquidity and Capital Resources
Overview
We have not generated significant revenues to date. Since our inception, we have funded our operations primarily through public and private offerings of our capital stock and other securities. We expect to finance our foreseeable cash requirements through cash on hand, cash from operations, debt financings and public or private equity offerings. Additionally, we may seek collaborations and license arrangements for our drug candidates. We may seek to access the public or private equity markets whenever conditions are favorable. We currently have no lines of credit or other arranged access to debt financing.
We had a cash balance of $11.3 million as of September 30, 2016 compared to a cash balance of $8.9 million as of December 31, 2015. We believe that our available cash at September 30, 2016 will be sufficient to fund our liquidity and capital expenditure requirements for at least the next 12 months.
Cash Flows
Operating Activities. Net cash used in operating activities for the nine months ended September 30, 2016 was $6.5 million. Net cash used in operating activities consisted primarily of the net loss for the period of $5.4 million, a non-cash decrease in fair value of the warrant liability of $1.4 million, an increase in other current assets of $0.7 million and a decrease in current liabilities of $0.1 million. These were partially offset by non-cash stock-based compensation expense of $0.6 million, a decrease in prepaid drug product for testing of $0.4 million and technology license amortization expense of $0.1 million.
Investing Activities. Net cash used in investing activities for the nine months ended September 30, 2016 consisted of a fixed asset purchase totaling $25,000.
Financing Activities. Net cash provided by financing activities for the nine months ended September 30, 2016 was $9.0 million. Net cash provided by financing activities consisted of net proceeds of $9.3 million from the registered direct offering described below, which closed on July 5, 2016. These proceeds were partially offset by additional financing costs incurred during the period of $0.3 million.
2014 Shelf Registration
On November 5, 2013, we filed a shelf registration statement on Form S-3 with the SEC, which was declared effective by the SEC on January 13, 2014 (the “Shelf Registration Statement”). The Shelf Registration Statement was filed to register the offering and sale of up to $100.0 million of our common stock, preferred stock, warrants to purchase common stock or preferred stock or any combination thereof, either individually or in units. The foregoing does not constitute an offer to sell or the solicitation of an offer to buy securities, and shall not constitute an offer, solicitation or sale in any jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of that jurisdiction.
“At the Market” Offering
On June 24, 2015, we entered into a Controlled Equity OfferingSM Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co. (“Cantor Fitzgerald”), as sales agent, pursuant to which we may offer and sell, from time to time, through Cantor Fitzgerald shares of our common stock. Sales of shares of common stock under the Sales Agreement will be made pursuant to the Shelf Registration Statement and a related prospectus supplement filed with the SEC on June 25, 2015, for an aggregate offering price of up to $25.0 million. Under the Sales Agreement, Cantor Fitzgerald may sell shares by any method deemed to be an “at the market” offering as defined in Rule 415 under the Securities Act or any other method permitted by law, including in privately negotiated transactions. We will pay Cantor Fitzgerald a commission of 3.4% of the aggregate gross proceeds from each sale of shares under the Sales Agreement and have agreed to provide Cantor Fitzgerald with customary indemnification and contribution rights. We have also agreed to reimburse Cantor Fitzgerald for certain specified expenses. Pursuant to the Securities Purchase Agreement, we are subject to certain restrictions on our ability to offer and sell shares of our common stock under the Sales Agreement. As of September 30, 2016, we have not offered or sold any shares of common stock under the Sales Agreement.
2016 Registered Direct Offering
On June 29, 2016, we entered into the Securities Purchase Agreement with certain healthcare focused institutional investors pursuant to which we agreed to sell an aggregate of 5,882,352 shares of our common stock and warrants to purchase up to 2,941,176 shares of our common stock for gross proceeds of approximately $10.0 million (the “2016 Registered Direct Offering”). The 2016 Registered Direct Offering closed on July 5, 2016. The net proceeds to the Company from the 2016 Registered Direct Offering, after deducting the placement agent’s fees and expenses and our offering expenses, and excluding the proceeds, if any, from the exercise of the warrants issued in the offering, was approximately $9.3 million.
Future Capital Requirements
We expect to continue to incur significant operating expenses in connection with our ongoing activities, including conducting clinical trials, manufacturing and seeking regulatory approval of our drug candidates, BP1001 and BP1002. Accordingly, we will continue to require substantial additional capital to fund our projected operating requirements. Such additional capital may not be available when needed or on terms favorable to us. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our current and future operating plan. There can be no assurance that we will be able to continue to raise additional capital through the sale of our securities in the future. Our future capital requirements may change and will depend on numerous factors, which are discussed in detail in “Item 1A. Risk Factors” to Part I of our Annual Report on Form 10-K as of the fiscal year ended December 31, 2015.
Off-Balance Sheet Arrangements
As of September 30, 2016, we did not have any material off-balance sheet arrangements.
Critical Accounting Policies
The preparation of financial statements in conformity with GAAP in the United States has required the management of the Company to make assumptions, estimates and judgments that affect the amounts reported in the financial statements, including the notes thereto, and related disclosures of commitments and contingencies, if any. We consider our critical accounting policies to be those that require the more significant judgments and estimates in the preparation of financial statements. Our significant accounting policies are discussed in Note 2 to our consolidated financial statements included in our Annual Report on Form 10-K as of the year ended December 31, 2015.
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk. We had cash and cash equivalents of approximately $11.3 million as of September 30, 2016. Although this cash account is subject to fluctuations in interest rates and market conditions, no significant gain or loss on this account is expected to be recognized in earnings. We do not invest in derivative securities.
Capital Market Risk. We have not generated significant revenues to date and depend on funds raised through other sources to fund our operations. One source of funding is through future debt or equity offerings. Our ability to raise funds in this manner depends upon capital market forces affecting our stock price.
ITEM 4. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
It is management’s responsibility to establish and maintain adequate disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Disclosure controls and procedures are controls and other procedures of a company that are designed to ensure that information required to be disclosed by the company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to management, including the company’s principal executive and principal financial officers, as appropriate, to allow timely decisions regarding required disclosure.
Our management, including our Chief Executive Officer (who is also our Chief Financial Officer), has reviewed and evaluated the effectiveness of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, as of the end of the period covered by this Quarterly Report on Form 10-Q. Following this review and evaluation, our management determined that as of the end of the period covered by this Quarterly Report on Form 10-Q, our disclosure controls and procedures are effective to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms, and is accumulated and communicated to management, including our Chief Executive Officer and our Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the period covered by this Quarterly Report on Form 10-Q that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
PART II - OTHER INFORMATION
ITEM 1. LEGAL PROCEEDINGS
None.
ITEM 1A. RISK FACTORS
There were no material changes from the risk factors previously disclosed under “Item 1A. Risk Factors” to Part I of our Annual Report on Form 10-K as of the fiscal year ended December 31, 2015.
ITEM 2. UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS
On July 5, 2016, we issued warrants to purchase up to 250,000 shares of our common stock in a private placement to H.C. Wainwright & Co., LLC and its designees as compensation for its services as a placement agent in connection with the 2016 Registered Director Offering (the “Placement Agent Warrants”). Subject to certain ownership limitations, the Placement Agent Warrants become exercisable on January 5, 2017, have a term of five years after they become exercisable and have an exercise price of $2.46 per share of common stock.
The Placement Agent Warrants and the shares issuable upon exercise of the Placement Agent Warrants were not registered under the Securities Act and were issued pursuant to an exemption from registration under Section 4(a)(2) of the Securities Act.
ITEM 3. DEFAULTS UPON SENIOR SECURITIES
None.
ITEM 4. MINE SAFETY DISCLOSURES
None.
ITEM 5. OTHER INFORMATION
None.
ITEM 6. EXHIBITS
Exhibit
No. | | Description of Exhibit |
| 2.1 | | Agreement and Plan of Merger and Reorganization dated September 27, 2007, by and among the Company, Biopath Acquisition Corp., a Utah corporation and wholly owned subsidiary of the registrant, and Bio-Path, Inc., a Utah corporation (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on September 27, 2007). |
| 3.1 | | Certificate of Incorporation (incorporated by reference to Exhibit 3.3 to the Company’s Current Report on Form 8-K filed on January 6, 2015). |
| 3.2 | | Bylaws (incorporated by reference to Exhibit 3.4 to the Company’s Current Report on Form 8-K filed on January 6, 2015). |
| 3.3 | | Articles of Merger relating to the merger of Biopath Acquisition Corp. with and into Bio-Path, Inc. (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on February 19, 2008). |
| 3.4 | | Certificate of Conversion (incorporated by reference to Exhibit 3.2 to the Company’s Current Report on Form 8-K filed on January 6, 2015). |
| 3.5 | | Articles of Transfer (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on January 6, 2015). |
| 4.1 | | Form of Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Company’s Annual Report on Form 10-K filed on March 16, 2015). |
| 4.2 | | Warrant Agreement, dated April 25, 2008, by and between the Company and Randeep Suneja, M.D. (incorporated by reference to Exhibit 4.2 to the Company’s Annual Report on Form 10-K filed on March 31, 2014). |
| 4.3 | | Form of Warrant issued to Maxim Group LLC, Sabby Healthcare Volatility Master Fund, Ltd. and Sabby Volatility Warrant Master Fund, Ltd. (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on January 21, 2014). |
| 4.4 | | Form of Warrant issued to certain investors (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on June 29, 2016). |
| 4.5 | | Form of Warrant issued to H.C. Wainwright & Co., LLC and certain of its designees (incorporated by reference to Exhibit 4.5 to the Company’s Quarterly Report on Form 10-Q filed on August 9, 2016). |
| 31* | | Certification of Principal Executive Officer and Principal Financial Officer pursuant to Exchange Act Rules 13a-14 and 15d-14, as adopted pursuant to Section 302 Sarbanes Oxley Act of 2002. |
| 32* | | Certification of Principal Executive Officer and Principal Financial Officer pursuant to Section 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act of 2002. |
| 101.INS* | | XBRL Instance Document |
| 101.SCH* | | XBRL Taxonomy Extension Schema Document |
| 101.CAL* | | XBRL Taxonomy Extension Calculation Linkbase Document |
| 101.DEF* | | XBRL Taxonomy Extension Definition Linkbase Document |
| 101.LAB* | | XBRL Taxonomy Extension Label Linkbase Document |
| 101.PRE* | | XBRL Taxonomy Extension Presentation Linkbase Document |
SIGNATURE
In accordance with the requirements of the Exchange Act, the Company has caused this Quarterly Report on Form 10-Q to be signed on its behalf by the undersigned thereunto duly authorized.
| Dated: November 9, 2016 | BIO-PATH HOLDINGS, INC. |
| | |
| | |
| | By | /s/ Peter H. Nielsen |
| | | Peter H. Nielsen |
| | | President |
| | | Chief Executive Officer |
| | | (Principal Executive Officer) |
| | | Chief Financial Officer |
| | | (Principal Financial Officer) |
EXHIBIT INDEX
Exhibit
No. | | Description of Exhibit |
| 2.1 | | Agreement and Plan of Merger and Reorganization dated September 27, 2007, by and among the Company, Biopath Acquisition Corp., a Utah corporation and wholly owned subsidiary of the registrant, and Bio-Path, Inc., a Utah corporation (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on September 27, 2007). |
| 3.1 | | Certificate of Incorporation (incorporated by reference to Exhibit 3.3 to the Company’s Current Report on Form 8-K filed on January 6, 2015). |
| 3.2 | | Bylaws (incorporated by reference to Exhibit 3.4 to the Company’s Current Report on Form 8-K filed on January 6, 2015). |
| 3.3 | | Articles of Merger relating to the merger of Biopath Acquisition Corp. with and into Bio-Path, Inc. (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on February 19, 2008). |
| 3.4 | | Certificate of Conversion (incorporated by reference to Exhibit 3.2 to the Company’s Current Report on Form 8-K filed on January 6, 2015). |
| 3.5 | | Articles of Transfer (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on January 6, 2015). |
| 4.1 | | Form of Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Company’s Annual Report on Form 10-K filed on March 16, 2015). |
| 4.2 | | Warrant Agreement, dated April 25, 2008, by and between the Company and Randeep Suneja, M.D. (incorporated by reference to Exhibit 4.2 to the Company’s Annual Report on Form 10-K filed on March 31, 2014). |
| 4.3 | | Form of Warrant issued to Maxim Group LLC, Sabby Healthcare Volatility Master Fund, Ltd. and Sabby Volatility Warrant Master Fund, Ltd. (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on January 21, 2014). |
| 4.4 | | Form of Warrant issued to certain investors (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on June 29, 2016). |
| 4.5 | | Form of Warrant issued to H.C. Wainwright & Co., LLC and certain of its designees (incorporated by reference to Exhibit 4.5 to the Company’s Quarterly Report on Form 10-Q filed on August 9, 2016). |
| 31* | | Certification of Principal Executive Officer and Principal Financial Officer pursuant to Exchange Act Rules 13a-14 and 15d-14, as adopted pursuant to Section 302 Sarbanes Oxley Act of 2002. |
| 32* | | Certification of Principal Executive Officer and Principal Financial Officer pursuant to Section 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act of 2002. |
| 101.INS* | | XBRL Instance Document |
| 101.SCH* | | XBRL Taxonomy Extension Schema Document |
| 101.CAL* | | XBRL Taxonomy Extension Calculation Linkbase Document |
| 101.DEF* | | XBRL Taxonomy Extension Definition Linkbase Document |
| 101.LAB* | | XBRL Taxonomy Extension Label Linkbase Document |
| 101.PRE* | | XBRL Taxonomy Extension Presentation Linkbase Document |