United States
Securities and Exchange Commission
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
| x | Annual Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the fiscal year ended December 31, 2013
or
| ¨ | Transition Report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the transition period from to
Commission file number 001-32954
CLEVELAND BIOLABS, INC.
(Exact name of registrant as specified in its charter)
| | |
| DELAWARE | | 20-0077155 |
(State or other jurisdiction of
incorporation or organization) | | (I.R.S. Employer
Identification No.) |
| | |
| 73 High Street, Buffalo, NY 14203 | | (716) 849-6810 |
| (Address of principal executive offices) | | Telephone No. |
Securities Registered Pursuant to Section 12(b) of the Act:
| | |
Title of each class | | Name of each exchange which registered |
| Common Stock, par value $0.005 per share | | NASDAQ Capital Market |
Securities Registered Pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | | |
Large accelerated filer | | ¨ | | Accelerated filer | | x |
Non-accelerated filer | | ¨ | | Smaller reporting company | | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the voting and non-voting common equity held by non-affiliates, computed by reference to the price at which the common equity was last sold or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed second fiscal quarter was $65,171,586. There were 50,919,820 shares of common stock outstanding as of March 14, 2014.
DOCUMENTS INCORPORATED BY REFERENCE
The definitive proxy statement relating to the registrant’s 2014 Annual Meeting of Stockholders is incorporated by reference in Part III to the extent described therein.
Cleveland BioLabs, Inc.
Form 10-K
For the Fiscal Year Ended December 31, 2013
INDEX
i
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements that involve risks and uncertainties. Forward-looking statements give our current expectations of forecasts of future events. All statements other than statements of current or historical fact contained in this annual report, including statements regarding our future financial position, business strategy, new products, budgets, liquidity, cash flows, projected costs, regulatory approvals or the impact of any laws or regulations applicable to us, and plans and objectives of management for future operations, are forward-looking statements. The words “anticipate,” “believe,” “continue,” “should,” “estimate,” “expect,” “intend,” “may,” “plan,” “project,” “will,” and similar expressions, as they relate to us, are intended to identify forward-looking statements.
We have based these forward-looking statements on our current expectations about future events. While we believe these expectations are reasonable, such forward-looking statements are inherently subject to risks and uncertainties, many of which are beyond our control. Our actual future results may differ materially from those discussed here for various reasons. When you consider these forward-looking statements, you should keep in mind these risk factors and other cautionary statements in this annual report including in Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in Item 1A “Risk Factors.”
Given these risks and uncertainties, you are cautioned not to place undue reliance on such forward-looking statements. The forward-looking statements included in this report are made only as of the date hereof. We do not undertake any obligation to update any such statements or to publicly announce the results of any revisions to any of such statements to reflect future events or developments. When used in the report, unless otherwise stated or the context otherwise requires, the terms “Cleveland BioLabs” and “CBLI” refer to Cleveland BioLabs, Inc., but not its consolidated subsidiaries and “the Company,” “we,” “us” and “our” refer to Cleveland BioLabs, Inc. together with its consolidated subsidiaries.
ii
PART I
Item 1. Business
GENERAL OVERVIEW
We are an innovative drug development company seeking to develop first-in-class pharmaceuticals designed to address diseases with significant unmet medical need. We combine our proven scientific expertise and our depth of knowledge about our products’ mechanisms of action into a passion for developing drugs to save lives. Our programs are focused on the implementation of novel pharmacological approaches to control cell death. Our proprietary drug candidates act via unique mechanisms and targets to kill cancer and protect healthy cells. We conduct business in the United States and the Russian Federation and have worldwide development and commercialization rights to all of our product candidates, subject to certain financial obligations to our current licensors. Our lead product candidates are Entolimod, which we are developing as a radiation countermeasure and an oncology drug, and Curaxin CBL0137, our lead oncology product candidate. We also have an additional clinical stage program and multiple innovative projects in different stages of preclinical drug development (see “Product Development Pipeline – Other Compounds”).
Entolimod, our most advanced product candidate, is a Toll-like receptor 5, or TLR5, agonist, which we are developing as a radiation countermeasure for prevention of death from Acute Radiation Syndrome, or ARS, and as an oncology drug. We believe that Entolimod is the most efficacious radiation countermeasure currently in development. Following is a summary of the clinical development of Entolimod to date and regulatory status:
Entolimod is being developed under the U.S. Food & Drug Administration’s, or FDA’s, Animal Efficacy Rule, or the Animal Rule, for the indication of reducing the risk of death following total body irradiation (see “Government Regulation – Animal Rule”). We have completed two dose escalation clinical studies designed to evaluate the safety, pharmacokinetics and pharmacodynamics in a total of 150 healthy volunteers. Administration of Entolimod was not associated with irreversible harm at any of the doses evaluated in these two studies. We have completed a Good Laboratory Practices, or GLP, randomized, blinded, placebo-controlled, pivotal study designed to evaluate the dose-dependent effect of Entolimod on survival and biomarker induction in 179 non-human primates exposed to 7.2 Gy total body irradiation when Entolimod or placebo were administered at 25 hours after radiation exposure. We have completed a GLP, randomized, open-label, placebo-controlled, pivotal study designed to evaluate the dose-dependent effect of Entolimod on biomarker induction in 160 non-irradiated non-human primates. We were granted Fast Track and Orphan Drug designations by the FDA. We plan to meet with the FDA regarding our human dose-conversion and our intent to submit a pre-Emergency Use Application, or pre-EUA, and, if appropriate after such meeting, plan to submit a pre-EUA in 2014.
Additionally, we are conducting a Phase 1 open-label, dose-escalation trial of Entolimod in patients with advanced cancer in the United States. In 2014, we plan to initiate, in the Russian Federation, a placebo-controlled, randomized trial of Entolimod in healthy volunteers to define optimal innate stimulatory dose and to support the safety database for our radiation countermeasure development program.
Curaxin CBL0137, our lead oncology product candidate, acts through a novel mechanism enabling this compound to simultaneously target three molecular pathways within cancer cells. We believe that CBL0137 has the potential to be a broadly-marketed cancer treatment that will address the unmet needs of treating diseases such as glioblastoma, lymphoma and treatment resistant neuroblastoma in children. CBL0137 inhibits Nuclear Factor kappa-B, or NF-kB, and Heat Shock Factor Protein-1, or HSF-1, transcription factors that are essential for viability of many types of tumors and activates tumor suppressor protein p53 by modulating intracellular localization and activity of chromatin remodeling complex Facilitates Chromatin Transcription, or FACT. CBL0137 has demonstrated reproducible anti-tumor effects in animal models of colon, breast, renal, pancreatic, head and neck and prostate cancers, melanoma, non-small cell lung cancer, glioblastoma, lymphoma, leukemia and neuroblastoma. We are currently enrolling two Phase 1 trials of CBL0137: (i) a multi-center, single agent, dose escalation study evaluating oral administration of CBL0137 in subjects with advanced solid tumors that are
1
resistant or refractory to standard of care treatment in the Russian Federation; and (ii) a multi-center, single agent dose escalation study in the United States, evaluating intravenous administration of CBL0137 in patients with metastatic or unresectable advanced solid cancers and lymphomas in the United States. We are conducting parallel evaluation of oral and intravenous routes of administration and continuous low-dose versus interrupted high-dose schedules to reduce our developmental risk by fully characterizing the clinical pharmacology of CBL0137.
CORPORATE INFORMATION
We were incorporated in Delaware in June 2003 as a spin-off company from the Cleveland Clinic Foundation, or CCF. We exclusively license our founding intellectual property from CCF. In 2007, we relocated the company to Buffalo, New York and became affiliated with Roswell Park Cancer Institute, or RPCI, through technology licensing and research collaboration relationships. Our common stock is listed on the NASDAQ Capital Market under the symbol “CBLI.”
Our principal executive offices are located at 73 High Street, Buffalo, New York 14203, and our telephone number at that address is (716) 849-6810.
The CBLI logo and CBLI product names are proprietary trade names of CBLI or its subsidiaries. We may indicate U.S. trademark registrations and U.S. trademarks with the symbols “®” and “TM”, respectively. Other third-party logos and product/trade names are registered trademarks or trade names of their respective owners.
PRODUCT DEVELOPMENT PIPELINE
Our product development programs arise from internally developed and in-licensed intellectual property from our innovation partners, CCF and RPCI. For instance, Entolimod emerged from our strategic licensing arrangement with CCF and CBL0137 emerged from our internal research efforts. In building our product development pipeline, we have intentionally pursued drug targets with applicability across multiple therapeutic areas and indications. This approach gives us multiple product opportunities and ensures that our success is not dependent on any single product or indication.
Our primary product development programs and their respective development stages are illustrated below:

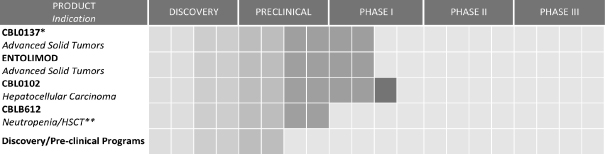
| * | Lead product development program |
| ** | HSCT means hematopoietic stem cell transplant |
2
Our product development efforts were initiated by discoveries related to apoptosis, a tightly regulated form of cell death that can occur in response to internal stresses or external events such as exposure to radiation or toxic chemicals. Apoptosis is a major determinant of the tissue damage that occurs in a variety of medical conditions involving ischemia, or temporary loss of blood flow, such as cerebral stroke, heart attack and acute renal failure. In addition, apoptotic loss of cells of the hematopoietic, or HP, system and gastrointestinal, or GI, tract is largely responsible for the acute lethality of high-dose radiation exposure. On the other hand, apoptosis is also an important protective mechanism that allows the body to eliminate defective cells such as those with cancer-forming potential.
We have developed novel strategies to target the molecular mechanisms controlling apoptotic cell death for therapeutic benefit. These strategies take advantage of the fact that tumor and normal cells respond to apoptosis-inducing stresses differently due to tumor-specific defects in cellular signaling pathways such as inactivation of p53 (a pro-apoptosis regulator) and constitutive activation of NF-kB (a pro-survival regulator).
Thus, we designed two oppositely-directed general therapeutic concepts:
| | (a) | Temporary and reversible suppression of apoptosis in normal cells to protect healthy tissues from stress-induced damage using compounds we categorize as Protectans, which include Entolimod and CBLB612; and |
| | (b) | Reactivation of apoptosis in tumor cells to eliminate cancer using compounds we categorize as Curaxins, which include CBL0137 and CBL0102. |
Entolimod Biodefense Indication
Our lead Protectan product candidate is Entolimod, an engineered derivative of theSalmonella flagellin protein that was designed to retain its specific TLR5-activating capacity while increasing its stability, reducing its immunogenicity and enabling high-yield production. We are developing Entolimod for dual indications: (i) as a radiation countermeasure for prevention of death from ARS, which we refer to as a Biodefense Indication; and (ii) as an oncology drug (discussed in “Product Development Pipeline – Other Programs”).
The market for radiation countermeasures grew dramatically following the September 11, 2001 terrorist attacks and the subsequent use of anthrax in a biological attack in the United States. Terrorist activities worldwide have continued in the intervening years and the possibility of chemical, biological, radiation and nuclear attacks continues to represent a perceived threat for governments world-wide. In addition to the U.S. government, we believe the potential markets for the sale of radiation countermeasures include U.S. and foreign state and local governments, including both defense and public health agencies, non-governmental organizations and multinational companies, transportation and security companies, healthcare providers, hospitals and clinics, and nuclear power facilities.
Acute high-dose whole body or significant partial body radiation exposure induces massive apoptosis of cells of the HP system and GI tract, which leads to ARS, a potentially fatal condition for which there are currently no FDA-approved treatments. The threat of ARS is primarily limited to emergency/defense scenarios and is significant given the possibility of nuclear/radiological accidents, warfare or terrorist incidents. The scale of possible exposure (number of people affected) has been estimated by the U.S. government to be in the range of 500,000 based on a modeled 10-kiloton device detonation in New York City. And we believe the current lack of approved efficacious treatments to deal with such an event makes Entolimod a compelling product candidate. It is not feasible or ethical to test the efficacy of Entolimod as a radiation countermeasure in humans. Therefore, we are developing Entolimod under the FDA’s Animal Rule guidance (see “Government Regulation – Animal Rule”). The Animal Rule authorizes the FDA to rely on data from animal studies to provide evidence of a product’s effectiveness under circumstances where there is a reasonably well-understood mechanism for the activity of the product. Under these requirements, and with the FDA’s prior agreement, medical countermeasures, like Entolimod, may be approved for use in humans based on evidence of effectiveness derived from appropriate animal studies, evidence of safety derived from studies in humans and any additional supporting data.
3
In 2014, we plan to meet with the FDA regarding human dose-conversion and our intent to file a pre-EUA. If appropriate after such meeting, we will submit a pre-EUA using the human dose of Entolimod that we determined through our proprietary dose conversion methodology, which utilizes the data from our pivotal non-human primate studies and our clinical studies of Entolimod in healthy volunteers. If authorized, pre-EUA status will allow Entolimod to be sold into the National Stockpile and used under a state of emergency. Such authorization is not equivalent to full licensure through approval of a biologic license application, or BLA, but precedes full licensure, and, importantly, would position Entolimod for potential sales in advance of full licensure in the United States. We further believe pre-EUA status will position us to explore sales opportunities with foreign governments.
Our pivotal efficacy study conducted in 179 non-human primates demonstrated with a high degree of statistical significance that injection of a single dose of Entolimod given to rhesus macaques 25 hours after exposure to a 70% lethal dose of total body irradiation improved animal survival by nearly three-fold compared to the control group. Dose-dependence of Entolimod’s efficacy was demonstrated with doses above the minimal efficacious dose establishing a plateau at approximately 75% survival at 60 days after irradiation, as compared to 27.5% survival in the placebo-treated group.
Our pivotal study conducted in 160 non-human primates established the dose-dependent effect of Entolimod on biomarkers for efficacy in non-irradiated non-human primates.
Our clinical studies of Entolimod in 150 healthy human subjects demonstrated the safety profile of Entolimod and established the dose-dependent effect of Entolimod on efficacy biomarkers in humans. In these studies, and in our currently ongoing Phase 1 oncology study, transient decrease in blood pressure and elevation of liver enzymes were observed along with transient mild to moderate flu-like syndrome. Such effects are the most common adverse events and they are linked to up-regulation of cytokines that are also biomarkers for efficacy.
The FDA has granted Fast Track status to Entolimod (see “Government Regulation – Fast Track Designation”) and Orphan Drug status for prevention of death following a potentially lethal dose of total body irradiation during or after a radiation disaster (see “Government Regulation – Orphan Drug Designation”).
We have worldwide development and commercialization rights to Entolimod.
Curaxin CBL0137
Our lead Curaxin product candidate is CBL0137, a small molecule with a multi-targeted mechanism of action that may be broadly useful for treatment of many different types of cancer with greater efficacy and substantially lower risk for patients developing drug resistance than conventional chemotherapeutic agents. CBL0137 inhibits NF-kB and HSF-1 transcription factors that are essential for viability of many tumor types of tumors and activates tumor suppressor protein p53 by modulating intracellular localization and activity of chromatin remodeling complex FACT. CBL0137 has been shown to be efficacious in pre-clinical models of colon, breast, renal, pancreatic, head and neck and prostate cancers, melanoma, non-small cell lung cancer, glioblastoma, lymphoma, leukemia and neuroblastoma.
We are currently enrolling patients in a Phase 1 multi-center, single agent, dose escalation study of an oral administration of CBL0137 in subjects with advanced solid tumors that are resistant or refractory to the standard of care in the Russian Federation. We are also currently enrolling patients in a Phase 1 multi-center, single agent, dose escalation study of an intravenous administration of CBL0137 in subjects with metastatic or unresectable advanced solid cancers and lymphomas in the United States. These studies are designed to evaluate safety, pharmacokinetics, and document any objective tumor response. The exploratory objectives of these trials are to examine (i) the relationship between tumor expression of FACT and response to CBL0137, and (ii) the effect of CBL0137 on the expression of biomarkers in peripheral blood mononuclear cells, or PBMCs, and on soluble
4
factors in serum. We are conducting parallel evaluation of oral and intravenous routes of administration and continuous low-dose versus interrupted high-dose schedules to reduce our developmental risk by fully characterizing the clinical pharmacology of CBL0137.
Our majority-owned subsidiary, Incuron, holds worldwide development and commercialization rights to CBL0137.
Other Programs
In addition to our Entolimod and CBL0137 development programs, we have an additional clinical stage program and multiple earlier stage development programs. The most advanced of these programs and projects are described below.
Curaxin CBL0102, a non-proprietary molecule that is a relative of 9-aminoacridine, a compound that is the core structure of many existing drugs. CBL0102 is a Quinacrine, a compound with a long history of use in humans as a treatment for malaria, osteoarthritis and autoimmune disorders. Quinacrine was not, however, previously used as an anti-cancer agent. In 2008, we completed a Phase 2 study in 31 patients with late stage, hormone refractory (androgen-independent) prostate cancer that had not responded to or relapsed following previous hormonal therapy and/or chemotherapy. The study results showed that one patient had a partial response, while 50% of the patients exhibited a decrease or stabilization in the rate of prostate cancer progression. CBL0102 was well-tolerated and there were no serious adverse events attributed to the drug. Therefore, the trial provided indications of anti-cancer activity and demonstrated safety for CBL0102 treatment for the cancer patients who were in the trial. In late 2013, we completed a Phase 1 safety and tolerability study of CBL0102 in patients with liver metastases from solid tumors of epithelial origin or primary advanced hepatic carcinoma for which standard therapy had failed or did not exist in the Russian Federation and we are in the process of finalizing the reports for this study. An investigator-initiated Phase 1/2 trial evaluating the tolerability of CBL0102 in combination with erlotinib in patients with stage 3B-4 non-small cell lung cancer is currently enrolling patients at the Case Comprehensive Cancer Center. The FDA has granted CBL0102 Orphan Drug status for treatment of hepatocellular carcinoma. Our majority owned subsidiary, Incuron, holds worldwide development and commercialization rights to CBL0102.
Entolimodis an engineered derivative of theSalmonella flagellin protein that was designed to retain its specific TLR5-activating capacity while increasing its stability, reducing its immunogenicity and enabling high-yield production. In addition to developing Entolimod as a radiation countermeasure for prevention of death from ARS, we are also developing Entolimod as an oncology drug. We believe that Entolimod has the potential to treat cancer by activating the innate and adaptive immune response in patients. In preclinical studies, Entolimod produced tissue-specific activation of innate immune responses via interaction with its receptor, TLR5, and the liver was identified as a primary mediator of Entolimod activity. Entolimod has also been shown to have a direct cytotoxic effect on tumors expressing TLR5 in animal models. Evaluations of local administration of Entolimod in organs expressing TLR5, such as the bladder, have also been performed in pre-clinical models. We are currently enrolling patients in a Phase 1, open-label, dose-escalation study designed to evaluate the safety, pharmacokinetics, pharmacodynamics and clinical activity of Entolimod in advanced cancer patients. In the second half of 2014 we plan to initiate a placebo-controlled, randomized trial of Entolimod in healthy subjects to define an optimal immunostimulatory dose. The study will be performed in the Russian Federation as the first of two planned studies under a 149 million ruble matching funds development contract that we received in October 2013 from the Ministry of Industry and Trade of the Russian Federation, or MPT (see Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations”).
Protectan CBLB612 is a proprietary compound based upon a natural activator of another tissue-specific component of the innate immune system, the TLR2/TLR6 heterodimeric receptor. CBLB612 is a pharmacologically optimized synthetic molecule that structurally mimics naturally occurring lipopeptides of Mycoplasma (a genus of parasitic bacteria) and activates NF-kB pro-survival and immunoregulatory signaling
5
pathways via specific binding to TLR2 on a subset of body tissues and cell types that express this receptor. Preclinical studies have shown that the efficacy of CBLB612 exceeds that of granulocyte colony-stimulating factor, or G-CSF (Amgen’s Neupogen®), the market-leading drug used for stimulation of white blood cell regeneration. CBLB612’s hematopoietic stem cell, or HSC, stimulatory activity outweighed that of G-CSF when the drugs were administered either as monotherapies, in either mice or non-human primates, or in combination with Plerixafor (Genzyme’s Mozobil®, a chemokine receptor antagonist approved by the FDA as an HSC mobilizer). However, the highest degree of HSC mobilization, 12-fold greater than that induced by the current clinical standard ofG-CSF+Plerixafor, was observed when CBLB612 was added to that combination. The strong synergistic effect of this triple drug combination provides further support for development of CBLB612 as a valuable stem cell mobilizing agent. In 2014, we plan to initiate a Phase 1, single-center, blind, placebo-controlled, single ascending dose study in the Russian Federation to evaluate the safety and tolerability of CBLB612 in healthy volunteers and measure response of various HP stem and progenitor cell types in order to gain a preliminary estimate of the drug’s HSC stimulatory efficacy under a 139 million ruble matching funds development contract that we received in July 2012 from MPT (see Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations”). We have licensed CBLB612 to Zhejiang Hisun Pharmaceutical Co., Ltd. for the territories of China, Taiwan, Hong Kong and Macau. We have rest-of-world development and commercialization rights to CBLB612.
Mobilan is our most advanced discovery/pre-clinical stage program. Mobilan is a nanoparticle-formulated recombinant non-replicating adenovirus that directs expression of TLR5 and its agonistic ligand, flagellin. In pre-clinical studies, delivery of Mobilan to tumor cells results in constitutive autocrine TLR5 signaling and strong activation of the innate immune system with subsequent development of adaptive anti-tumor immune responses. Mobilan is in the pre-clinical stage of development as a universal anti-cancer therapy. In 2014, we plan to file an IND in the Russian Federation under a 149 million ruble matching funds development contract that we received in October 2013 from MPT (see Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations”). Our majority owned subsidiary, Panacela, holds worldwide development and commercialization rights to Mobilan.
STRATEGIC PARTNERSHIPS
Since our inception, strategic alliances and collaborations have been integral to our business. We have leveraged the experience, contacts and knowledge of our founders to engage funding partners in the Russian Federation and to develop and maintain academic-corporate innovation partnerships with CCF and RPCI. Through these partnerships we have collaborated with world-class scientists to develop our novel technologies and accessed non-traditional funding sources, including federal and foreign government contracts and project-oriented funding to support the development of certain of our technologies. We have received project-oriented funding from Russian Federation based venture funds BioProcess Capital Partners, or BCP, and Open Joint Stock Company “Rusnano”, or Rusnano, through the formation of our majority owned subsidiaries, Incuron and Panacela, both of which are co-located in the Russian Federation and the United States. We believe that these companies, as well as our wholly-owned subsidiary BioLab 612, may benefit from programs supporting domestic pharmaceutical industry development in the Russian Federation as well as the relative ease of enrolling patients as compared to western markets. We have negotiated exclusive licenses to rights in each of our technologies from CCF and RPCI.
BioProcess Capital Partners
In December 2009, we entered into our Incuron joint venture with BCP to develop Curaxin compounds for treatment of oncological diseases. According to the terms of the agreement, we transferred rights in the Curaxin molecules to the new joint venture company, Incuron, in which BCP agreed to contribute an aggregate of 549,497,000 Russian rubles (approximately $17.2 million as noted below) to support development of the compounds. As of December 31, 2013, Incuron had received from BCP payments of 369,570,000 Russian rubles (approximately $11.7 million) and BCP will make the balance of its contribution of 179,927,000 Russian rubles
6
(approximately $4.9 million, based on the March 11, 2014 exchange rate) upon the achievement of predetermined development milestones, which are expected in early 2014. As of December 31, 2013, we had a 59.2% ownership interest in Incuron. After the remaining contractual investments, CBLI expects to own approximately 47% of Incuron. CBLI has an option to maintain a majority ownership stake by investing $3.0 million in Incuron within 60 days of the last contribution by BCP.
Rusnano
In October 2011, we entered into our Panacela joint venture with Rusnano to carry out a complete cycle of development and commercialization in the Russian Federation for the treatment of oncological, infectious or other diseases. We invested $3.0 million in Panacela preferred shares and warrants, and, together with certain third-party owners, assigned and/or provided exclusive licenses, as applicable, to Panacela to provide Panacela with worldwide development and commercialization rights to five preclinical product candidates in exchange for Panacela common shares. Rusnano invested $9.0 million in Panacela preferred shares and warrants. In 2013, Rusnano loaned Panacela $1.5 million through a convertible term loan, or the Panacela Loan, and revised their original investment agreement to provide that Rusnano may invest an additional $15.5 million at their option and to remove the predetermined development milestones and timelines for further investment. As of December 31, 2013, we had an ownership stake of approximately 54.6% in Panacela. However, we may have a less than majority ownership interest in Panacela if Panacela raises additional capital through dilutive financing with a third-party or by Rusnano exercising their options to invest an additional $15.5 million, exercising their warrants in Panacela or converting the Panacela Loan into additional shares of Panacela preferred stock, which may be at a discounted price based upon the terms of the Panacela Loan.
Cleveland Clinic Foundation
In July 2004, CBLI entered into an exclusive license agreement with CCF, or the CCF License, pursuant to which CBLI was granted an exclusive license to CCF’s research base underlying our therapeutic platform including Entolimod, CBLB612, Curaxin CBL0102, Mobilan and several earlier stage compounds that are not currently material to our business. In consideration for the CCF License, we agreed to issue CCF common stock and make certain milestone, royalty and sublicense royalty payments as described below.
The CCF License requires milestone payments, which may be credited against future royalties owed to CCF, as described in the table below. CBLI has also agreed to make milestone payments of up to approximately $6.5 million for each Panacela Product that achieves certain developmental and regulatory milestones, provided that if CBLI or an affiliate of CBLI and CCF jointly own the Panacela Product, the milestone amounts will be reduced by 50%.
| | | | | | | | |
Milestone Description | | For Products Limited to
Biodefense Uses | | | For All Other Products
(Maximum amount)* | |
For any IND filing for a product | | $ | 50,000 | | | $ | 50,000 | |
For any product entering Phase II clinical trials or similar registration | | | 100,000 | | | | 250,000 | |
For any product entering Phase III clinical trials | | | — | | | | 700,000 | |
For any product license application, BLA or NDA Filing for a product | | | 350,000 | | | | 1,500,000 | |
Upon regulatory approval permitting any product to be sold to the commercial market | | | 1,000,000 | | | | 4,000,000 | |
| * | Maximum amounts listed for achievement of milestone in United States. If milestones are reached in another country first, milestone payments will be prorated for certain products under the license based on the market size for the product in such country as that market relates to the then current U.S. market. |
7
The CCF license requires royalty payments of (a) 2% of net sales of any product candidate under a licensed patent solely owned by CCF; and (b) 1% of net sales of any product candidate under a licensed patent that is jointly owned by CCF and CBLI or an affiliate of CBLI. Further, if CBLI receives upfront sublicense fees or sublicense royalty payments for sublicenses granted by CBLI to third parties for any licensed patents solely owned by CCF, CBLI will pay CCF (i) 35% of such fees if the sublicense is granted prior to filing an IND application, (ii) 20% of such fees if the sublicense is granted after an IND filing but prior to final approval of the Product License Application or NDA, or (iii) 10% of such fees if the sublicense is granted after final approval of the relevant Product License Application or NDA, provided that such sublicense fees shall not be less than 1% of net sales. The above sublicense fees and sublicense royalty payments are reduced by 50% if CCF and CBLI or an affiliate of CBLI jointly own the licensed patent.
Through December 31, 2013, CBLI had paid CCF $150,000 for milestone payments on products limited to biodefense uses, and $400,000 for all other products.
CCF may terminate the CCF License upon a material breach by us, as specified in the agreement. However, we may avoid such termination if we cure the breach within 90 days of receipt of a termination notice. CBLI may terminate the CCF License in its entirety or any specific patent licensed under the agreement by giving at least 90 days written notice of such termination to CCF. The agreement will, subject to certain exceptions, automatically terminate with respect to a licensed product if CCF does not receive a royalty payment for more than one-year after the payment of royalties has begun.
Roswell Park Cancer Institute
We have entered into a number of agreements with RPCI relating to the licensure and development of our product candidates including:
| | • | | Two exclusive license and option agreements effective December 2007 and September 2011; |
| | • | | Various sponsored research agreements; and |
| | • | | Clinical trial agreements for the conduct of the Phase 1 Entolimod oncology study and the Phase 1 CBL0137 intravenous administration study. |
In December 2007, CBLI entered into an agreement with RPCI pursuant to which CBLI has an option to exclusively license any technological improvements to our foundational technology developed by RPCI for the term of the agreement. We believe our option to license additional technology under the agreement potentially provides us with access to technology that may supplement our product pipeline in the future. In consideration for this option and exclusive license, we agreed to make certain milestone, royalty and sublicense royalty payments. Additionally, RPCI may terminate the license upon a material breach by us. However, we may avoid such termination if we cure the breach within 90 days of receipt of a termination notice. The license does not have a specified term; however, as each patent covered by this license agreement expires, the royalties to be paid on each product relating to the licensed patent shall cease.
In September 2011, Panacela entered into an agreement with RPCI, or the Panacela-RPCI License, to exclusively license certain rights Panacela Products, including Mobilan and several earlier stage compounds that are not currently material to our business, and to non-exclusively license certain know-how relating to the aforementioned product candidates for the limited purposes of research and development and regulatory, export and other government filings. Additionally, under the Panacela-RPCI License, Panacela has a right to exclusively license (i) any technological improvements to the Panacela Products developed by RPCI before September 2016, and (ii) any technology jointly developed by Panacela and RPCI. In consideration for the Panacela-RPCI License, Panacela agreed to issue RPCI common stock and to make certain milestone, royalty and sublicense royalty payments as described below.
8
The Panacela-RPCI License requires milestone payments for developmental and regulatory milestones reached in the United States of up to approximately $2.5 million for each Panacela Product that achieves certain developmental and regulatory milestones. Additionally, Panacela will owe additional payments of up to approximately $275,000 for each other country where a licensed Panacela Product achieves similar milestones. Through December 31, 2013, Panacela had not made any milestone payments to RPCI related to the above mentioned license agreement.
The Panacela-RPCI License requires royalty payments on net sales based on percentages in the low single digits. In addition, if Panacela sublicenses any of the licensed Panacela Products, Panacela will owe sublicensing fees ranging from 5% to 15% of fees received from sublicense by Panacela or an affiliate depending upon whether or not an IND has been filed or final approval of the relevant NDA has been obtained for such licensed product.
As each patent covered by the Panacela-RPCI License expires, the license agreement will terminate as to such patent. In addition, the license agreement will terminate with respect of the licensed know-how after 20 years. RPCI may terminate the license upon a material breach by us, as specified in the agreement. However, we may avoid such termination if we cure the breach within 90 days of receipt of a termination notice (or 30 days if notice relates to non-payment of amounts due to RPCI). Panacela may terminate the license agreement in whole or as to any specific patent licensed under the agreement by giving at least 60 days written notice of such termination to RPCI. The agreement will, subject to certain exceptions, automatically terminate with respect to a licensed Panacela Product if Panacela fails to market, promote and otherwise exploit the licensed technology so that RPCI does not receive a royalty payment during any 12-month period after the first commercial sale of such licensed product.
We have also entered into a number of sponsored research agreements with RPCI pursuant to which we have sponsored research to be conducted by RPCI. Under these agreements, we own any invention that is described in our research plan, co-own any inventions not described in our research plan that are made by Dr. Andrei Gudkov, our Chief Scientific Officer who is also the Senior Vice President of Basic Science at RPCI, and RPCI owns any other inventions not described in our research plan. We further have a right to exclusively license RPCI’s ownership in any invention developed under such sponsored research agreements that are owned by RPCI. These agreements with RPCI expire in 2014, although we expect to enter into similar future arrangements.
We entered into an asset transfer and clinical trial agreement with RPCI for the conduct, by RPCI, of our Phase 1 clinical trial to evaluate the safety and pharmacokinetic profile of Entolimod in patients with advanced cancers and a clinical trial agreement for RPCI to conduct, as one site in a multi-site trial, our Phase 1 clinical trial to evaluate the safety, pharmacokinetics and pharmacodynamics of intravenous administration of CBL0137 in patients with metastatic or unresectable advanced solid cancers and lymphomas. Either party may terminate these agreements upon 30 days’ notice to the other party.
INTELLECTUAL PROPERTY
Our intellectual property consists of patents, trademarks, trade secrets and know-how. Our ability to compete effectively depends in large part on our ability to obtain patents for our technologies and products, maintain trade secrets, operate without infringing the rights of others and prevent others from infringing our proprietary rights. We will be able to protect our proprietary technologies from unauthorized use by third parties only to the extent that they are covered by valid and enforceable patents, or are effectively maintained as trade secrets. As a result, patents or other proprietary rights are an essential element of our business. Our patent portfolio includes patents and patent applications with claims directed to compositions of matter, pharmaceutical formulations and methods of use. Some of our issued patents, and the patents that may be issued based on our patent applications, may be eligible for patent life extension under the Drug Price Competition and Patent Term Restoration Act of 1984 in the United States, supplementary protection certificates in the European Union or similar mechanisms in other countries or territories. The following are the patent positions relating to our product candidates as of December 31, 2013.
9
In the United States, we have 10 issued or allowed patents relating to our clinical-stage programs expiring on various dates between 2024 and 2030 as well as numerous pending patent applications and foreign counterpart patent filings which relate to our proprietary technologies. These patents and patent applications include claims directed to compositions of matter and methods of use.
We have 7 issued U.S. patents covering Entolimod, which expire between 2024 and 2029. These patents include method of use claims relating to our biodefense indication, reducing effects of chemotherapy and treatment of reperfusion injuries. In addition, we have pending U.S. patent applications related to compositions of matter and oncology methods of use, which, if issued, will expire between 2025 and 2032.
We have 1 issued U.S. patent covering CBL0137, which expires in 2030. This patent includes method of use claims relating to apoptosis induction along with inhibition of adaptive heat shock response. In addition, we have a pending U.S. patent application that includes CBL0137 composition of matter and method of use claims, which, if issued, will expire in 2029.
We have 2 issued U.S. patents covering CBLB612 and related agents, which expire between 2026 and 2027. These patents include composition of matter and methods of use claims. In addition, we have a pending U.S. patent application that includes method of use claims relating to increasing mobility of hematopoietic stem cells, which, if issued, will expire in 2028.
We have 1 pending U.S. patent application covering CBL0102, which, if issued, will expire in 2025. This patent includes method of use claims related to treatment of liver cancer.
In addition, as of December 31, 2013, we had more than a hundred additional patents and patent applications filed worldwide. Any patents that may issue from our pending patent applications would expire between 2024 and 2035, excluding patent term extensions. These patents and patent applications disclose compositions of matter and methods of use.
Our policy is to seek patent protection for the inventions that we consider important to the development of our business. We intend to continue to file patent applications to protect technology and compounds that are commercially important to our business, and to do so in countries where we believe it is commercially reasonable and advantageous to do so. We also rely on trade secrets to protect our technology where patent protection is deemed inappropriate or unobtainable. We protect our proprietary technology and processes, in part, by confidentiality agreements with our employees, consultants, collaborators and contractors.
RESEARCH AND DEVELOPMENT
In 2013, we transferred 26 laboratory and preclinical employee positions to Buffalo BioLabs, LLC, or BBL, an entity owned in part by our Chief Scientific Officer and director, Dr. Andrei Gudkov, to enable us to better focus our on clinical development activities. In connection with this transaction, we entered into a Master Services Agreement with BBL, pursuant to which BBL agreed to perform laboratory and preclinical research services for us. As of December 31, 2013, our research and development group, including Russian-based personnel, consisted of 18 individuals. Our research and development focuses on management of outsourced preclinical research, clinical trials and manufacturing technologies. We invested $19.5 million, $22.5 million and $22.8 million in research and development in the years ended December 31, 2013, 2012 and 2011, respectively.
SALES AND MARKETING
We currently do not have marketing, sales or distribution capabilities. We do, however, currently have worldwide development and commercialization rights for products arising out of substantially all of our programs. In order to commercialize any of these drugs, if and when they are approved for sale, we will need to enter into partnerships for the commercialization of the approved product(s) or develop the necessary marketing, sales and distribution capabilities.
10
COMPETITION
The biotechnology and biopharmaceutical industries are characterized by rapid technological developments and intense competition. This competition comes from both biotechnology and major pharmaceutical companies. Many of these companies have substantially greater financial, marketing and human resources than we do, including, in some cases, considerably more experience in clinical testing, manufacturing and marketing of pharmaceutical products. There are also academic institutions, governmental agencies and other research organizations that are conducting research in areas in which we are working. They may also develop products that may be competitive with our product candidates, either on their own or through collaborative efforts. We expect to encounter significant competition for any products we develop. Our product candidates’ competitive position among other biotechnology and biopharmaceutical companies will be based on, among other things, time to market, patent position, product efficacy, safety, reliability, availability, patient convenience, delivery devices and price. Additionally, competitive products may have superior safety or efficacy, be manufactured less expensively, or have better concept of operations, or CONOPs, usability for biodefense products. In these cases, we may not be able to commercialize our product candidates or achieve a competitive position in the market. This would adversely affect our business.
Specifically, the competition for Entolimod, CBL0137 and our other product candidates includes the following:
Entolimod Biodefense Indication
Product candidates for treatment of ARS face significant competition for U.S. government funding for both development and procurement of medical countermeasures and must satisfy government procurement requirements for biodefense products. Currently there are no FDA-approved drugs for the efficacious treatment of ARS. However, we are aware of a number of companies also developing radiation countermeasures to treat the effects of ARS including Aeolus Pharmaceuticals, Araim Pharmaceuticals, Inc., Cellerant Therapeutics, Exponential Biotherapies Inc., Humanetics Corporation, ImmuneRegen BioSciences, Inc., Neumedicines, Inc., Onconova Therapeutics, Inc., Pluristem Therapeutics, RxBio, Inc., Soligenix, Inc., and the University of Arkansas Medical Sciences Centers. Although their approaches to treatment of ARS are different, we compete with these companies for U.S. government development funding and may ultimately compete with them for U.S. and foreign government purchase and stockpiling of radiation countermeasures. Additionally, our ability to sell to the government also can be influenced by indirect competition from other products, such as Neupogen® (Amgen, Inc.) and potassium iodide, both of which were recently purchased for use as a radiation countermeasure.
Curaxins CBL0137 and CBL0102
Chemotherapy is a large cancer drug category. These treatments are the foundation for treatment of all cancer types and used in most combination regimens. Drugs in this category include, among others, irinotecan, carboplatin, taxanes and doxorubicin. These drugs act on various cell division pathways and ultimately cause cell death. This cell division pathway may not always be specific to the cancer cell but often effects normal cells such as red blood cells, white blood cells and other healthy tissues. Although these drugs as a treatment category in general carry higher toxicities than targeted therapies, they are nonetheless an important drug category for improving patient survival.
Entolimod Oncology Program
The number of cancer therapies is extremely large, numbering in the thousands. Immunotherapies and targeted therapies are primary drivers of growth in this segment. Examples of marketed drugs in these categories include: Avastin® (Roche) for a range of solid tumors including colorectal, lung, breast, renal and gastric cancers, Rituximab® (Roche) for CD20 positive, B-cell non-Hodgkin’s lymphoma and Arzerra® (GSK) and Campath® (Bayer Healthcare Pharmaceuticals) for CD20 positive chronic lymphocytic leukemia; Yervoy® (Bristol-Myers Squibb) for melanoma, Herceptin® (Roche) for human epidermal growth factor receptor-2, or HER-2, positive
11
tumors, Gleevec® (Novartis) for Philadelphia chromosome tumor mutations, Erbitux® (Eli Lilly) and Iressa® (AstraZeneca) for epidermal growth factor receptor, or EGRF, expressing tumors and Zelboraf® (Genentech) for BRAF mutated tumors.
CBLB612
Stem cell mobilization is a significant therapeutic category within oncology. G-CSF, marketed as Neupogen® (Amgen, Inc.), is the current standard against which all other mobilization agents for stem cells are measured. Its primary use was established in cancer patients with neutropenia (low white blood cells) due to chemotherapy. In recent years a long-acting release formulation of G-CSF, Neulasta® (Amgen, Inc.), was approved and is prescribed to approximately 50% of U.S. cancer patients with neutropenia. However, Neupogen® is still widely prescribed due to stronger reimbursement and is more often used in Europe. Mozobil® (Genzyme Corporation) is a more recent FDA approved drug designed to help increase the number of stem cells collected from a patient’s blood before being transplanted back into the body after chemotherapy.
MANUFACTURING
Our product candidates are biologics and small molecules that can be readily synthesized by processes that we have developed. We do not own or operate manufacturing facilities for the production of our product candidates for pre-clinical, clinical or commercial quantities. We rely on third-party manufacturers, and in most cases only one third-party, to manufacture critical raw materials, drug substance and final drug product for our research, pre-clinical development and clinical trial activities. Commercial quantities of any drugs we seek to develop will have to be manufactured in facilities and by processes that comply with the FDA and other regulations, and we plan to rely on third parties to manufacture commercial quantities of products we successfully develop.
GOVERNMENT REGULATION
Government authorities in the U.S. and in other countries, regulate the research, development, testing, manufacture, packaging, storage, record-keeping, promotion, advertising, distribution, marketing, quality control, labeling and export and import of pharmaceutical products such as those that we are developing. We cannot provide assurance that any of our product candidates will prove to be safe or effective, will receive regulatory approvals or will be successfully commercialized.
U.S. Drug Development Process
In the U.S., the FDA regulates drugs and drug testing under the Federal Food, Drug, and Cosmetic Act and in the case of biologics, also under the Public Health Service Act. Our product candidates must follow an established process before they may be marketed in the U.S.:
| | • | | Preclinical laboratory and animal tests performed in compliance with Good Laboratory Practices, or GLP; |
| | • | | Development of manufacturing processes which conform to current Good Manufacturing Practices, or cGMP; |
| | • | | Submission and acceptance of an IND application which must become effective before human clinical trials may begin; |
| | • | | Performance of adequate and well-controlled human clinical trials in compliance with current Good Clinical Practices, or cGCP, to establish the safety and efficacy of the proposed drug for its intended use; provided, however, that for Entolimod development under the Animal Rule, we are required to perform pivotal animal studies in compliance with GLP to establish efficacy; |
| | • | | Submission to and review and approval by the FDA of a NDA or BLA prior to any commercial sale or shipment of a product; |
12
Nonclinical testing.Nonclinical testing includes laboratory evaluation of a product candidate, its chemistry, formulation, safety and stability, as well as animal studies to assess the potential safety and efficacy of the product candidate. The conduct of the nonclinical tests must comply with federal regulations and requirements including cGMP and GLP. Prior to the initiation of GLP animal studies, including our pivotal studies for development of Entolimod under the Animal Rule, an Institutional Animal Care and Use Committee, or IACUC, at each testing site must review and approve each study protocol and any amendments thereto.
We must submit to the FDA the results of nonclinical studies, which may include laboratory evaluations and animal studies, together with manufacturing information and analytical data, and the proposed clinical protocol for the first clinical trial of the drug as part of an IND. An IND is a request for FDA authorization to administer an investigational drug to humans. Such authorization must be secured prior to the interstate shipment and administration of any new drug that is not the subject of an approved NDA or BLA. Nonclinical tests and studies can take several years to complete, and despite completion of those tests and studies, the FDA may not permit clinical testing to begin.
The IND process.The FDA requires a 30-day waiting period after the submission of each IND application before clinical trials may begin. This waiting period is designed to allow the FDA to review the IND to determine whether human research subjects will be exposed to unreasonable health risks. At any time during this 30-day period or at any time thereafter, the FDA may raise concerns or questions about the conduct of the trials as outlined in the IND and impose a “clinical hold” that may affect one or more specific studies or all studies conducted under the IND. In the case of a clinical hold, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials placed on hold can begin or continue. The IND application process may be extremely costly and could substantially delay development of our products. Moreover, positive results of preclinical animal tests do not necessarily indicate positive results in clinical trials.
Prior to the initiation of clinical studies, each clinical protocol must be submitted to the IND and to an independent Institutional Review Board, or IRB, at each medical site proposing to conduct the clinical trial. The IRB must review and approve each study protocol, and any amendments thereto, and study subjects must sign an informed consent. Protocols include, among other things, the objectives of the study, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor patient safety. Progress reports of work performed in support of IND studies must be submitted at least annually to the FDA. Reports of serious and unexpected adverse events must be submitted to the FDA and the investigators in a timely manner.
Clinical trials.Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| | • | | Phase 1: The drug is introduced into healthy human subjects or patients (in the case of certain inherently toxic products for severe or life-threatening diseases such as cancer) and tested for safety, dosage tolerance, absorption, distribution, metabolism and excretion. |
| | • | | Phase 2: Involves studies in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| | • | | Phase 3: Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to establish the overall risk-benefit ratio of the product and provide, if appropriate, an adequate basis for product labeling. |
We cannot be certain that we will successfully complete any phase of clinical testing of our product candidates within any specific time period, if at all. Clinical testing must meet requirements of IRB oversight, informed consent and cGCP. The FDA, the sponsor, or the IRB at each institution at which a clinical trial is being performed may suspend a clinical trial at any time for various reasons, including a belief that the subjects are being exposed to an unacceptable health risk.
13
During the development of a new drug, sponsors are given an opportunity to meet with the FDA at certain points. These meetings typically occur prior to submission of an IND, at the end of Phase 2 and before NDA or BLA submission. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and FDA to reach agreement on the next phase of development. Sponsors typically use the end-of-Phase 2 meeting to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trial that they believe will support approval of the new drug.
The NDA or BLA process. If clinical trials are successful, the next step in the drug regulatory approval process is the preparation and submission to the FDA of an NDA or BLA, as applicable. The NDA or BLA, as applicable, is a vehicle through which drug sponsors formally propose that the FDA approve a new pharmaceutical for marketing and sale in the U.S. The NDA or BLA, as applicable, must contain a description of the manufacturing process and quality control methods, as well as results of preclinical tests, toxicology studies, clinical trials and proposed labeling, among other things. A substantial user fee must also be paid with the application, unless an exemption applies. Every newly marketed pharmaceutical must be the subject of an approved NDA or BLA.
Upon submission of an NDA or BLA, the FDA will make a threshold determination of whether the application is sufficiently complete to permit review, and, if not, will issue a refuse-to-file letter. If the application is accepted for filing, the FDA will attempt to review and take action on the application in accordance with performance goal commitments the FDA has made in connection with the prescription drug user fee law in effect at that time. Current timing commitments under the user fee law vary depending on whether an NDA or BLA is for a priority drug or not, and in any event are not a guarantee that an application will be approved or even acted upon by any specific deadline. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the NDA or BLA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved, but the FDA is not bound by the recommendation of an advisory committee. The FDA may deny or delay approval of applications that do not meet applicable regulatory criteria or if the FDA determines that the data do not adequately establish the safety and efficacy of the drug. In addition, the FDA may approve a product candidate subject to the completion of post-marketing studies, commonly referred to as Phase 4 trials, to monitor the effect of the approved product. The FDA may also grant approval with restrictive product labeling, or may impose other restrictions on marketing or distribution such as the adoption of a special risk management plan. The FDA has broad post-market regulatory and enforcement powers, including the ability to issue warning letters, levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products, and withdraw approvals.
Manufacturing and post-marketing requirements. If approved, a pharmaceutical may only be marketed in the dosage forms and for the indications approved in the NDA or BLA, as applicable. Special requirements also apply to any samples that are distributed in accordance with the Prescription Drug Marketing Act. The manufacturers of approved products and their manufacturing facilities are subject to continual review and periodic inspections by the FDA and other authorities where applicable, and must comply with ongoing requirements, including the FDA’s cGMP requirements. Once the FDA approves a product, a manufacturer must provide certain updated safety and efficacy information, submit copies of promotional materials to the FDA, and make certain other required reports. Product and labeling changes, as well as certain changes in a manufacturing process or facility or other post-approval changes, may necessitate additional FDA review and approval. Failure to comply with the statutory and regulatory requirements subjects the manufacturer to possible legal or regulatory action, such as untitled letters, warning letters, suspension of manufacturing, seizure of product, voluntary recall of a product, injunctive action or possible criminal or civil penalties. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval. Because we intend to contract with third parties for manufacturing of our products, our ability to control third party compliance with FDA requirements will be limited to contractual remedies and rights of inspection. Failure of third party manufacturers to comply with cGMP or other FDA requirements applicable to our products may result in, among other things, total or partial suspension of production, failure of the government to grant approval for marketing, and withdrawal, suspension,
14
or revocation of marketing approvals. With respect to post-market product advertising and promotion, the FDA imposes a number of complex regulations on entities that advertise and promote pharmaceuticals, which include, among others, standards for direct-to-consumer advertising, promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses.
The FDA’s policies may change, and additional government regulations may be enacted which could prevent or delay regulatory approval of our potential products. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or abroad.
Animal Rule
In 2002, the FDA amended its requirements applicable to BLAs/NDAs to permit the approval of certain drugs and biologics that are intended to reduce or prevent serious or life-threatening conditions based on evidence of safety from clinical trial(s) in healthy subjects and effectiveness from appropriate animal studies when human efficacy studies are not ethical or feasible. These regulations, which are known as the “Animal Rule”, authorize the FDA to rely on animal studies to provide evidence of a product’s effectiveness under circumstances where there is a reasonably well-understood mechanism for the activity of the agent. Under these requirements, and with the FDA’s prior agreement, drugs used to reduce or prevent the toxicity of chemical, biological, radiological or nuclear substances may be approved for use in humans based on evidence of effectiveness derived from appropriate animal studies and any additional supporting data. Products evaluated for under this rule must demonstrate effectiveness through pivotal animal studies, which are generally equivalent in design and robustness to Phase 3 clinical studies. The animal study endpoint must be clearly related to the desired benefit in humans and the information obtained from animal studies must allow for selection of an effective dose in humans. Safety under this rule is established under preexisting requirements, including safety studies in both animals (toxicology) and humans. Products approved under the Animal Rule are subject to additional requirements including post-marketing study requirements, restrictions imposed on marketing or distribution or requirements to provide information to patients.
We intend to utilize the Animal Rule in seeking marketing approval for Entolimod as a radiation countermeasure because we cannot ethically expose humans to lethal doses of radiation. Other countries may not at this time have established criteria for review and approval of these types of products outside their normal review process, i.e. there is no “Animal Rule” equivalent in countries other than the U.S., but some may have similar policy objectives in place for these product candidates. Given the nature of nuclear and radiological threats, we do not believe that the lack of established criteria for review and approval of these types of products in other countries will significantly inhibit us from pursuing sales of Entolimod to foreign countries.
All data obtained from the pre-clinical studies and clinical trials of Entolimod, in addition to detailed information on the manufacture and composition of the product, would be submitted in a BLA to the FDA for review and approval for the manufacture, marketing and commercial shipments of Entolimod.
15
Emergency Use Authorization
The Commissioner of the FDA, under delegated authority from the Secretary of the Department of Health and Human Services, or HHS, may, under certain circumstances, issue an Emergency Use Authorization, or EUA, that would permit the use of an unapproved drug product or unapproved use of an approved drug product. Before an EUA may be issued, the Secretary must declare an emergency based on one of the following grounds:
| | • | | a determination by the Secretary of Department of Homeland Security that there is a domestic emergency, or a significant potential for a domestic emergency, involving a heightened risk of attack with a specified biological, chemical, radiological or nuclear agent or agents; |
| | • | | a determination by the Secretary of the Department of Defense, or DoD, that there is a military emergency, or a significant potential for a military emergency, involving a heightened risk to United States military forces of attack with a specified biological, chemical, radiological, or nuclear agent of agents; or |
| | • | | a determination by the Secretary of HHS of a public health emergency that effects or has the significant potential to affect, national security, and that involves a specified biological, chemical, radiological, or nuclear agent or agents, or a specified disease or condition that may be attributable to such agent or agent. |
In order to be the subject of an EUA, the FDA Commissioner must conclude that, based on the totality of scientific evidence available, it is reasonable to believe that the product may be effective in diagnosing, treating or preventing a disease attributable to the agents described above; that the product’s potential benefits outweigh its potential risks; and that there is no adequate, approved alternative to the product.
Although an EUA cannot be issued until after an emergency has been declared by the Secretary of HHS, the FDA strongly encourages an entity with a possible candidate product, particularly one at an advanced stage of development, to contact the FDA center responsible for the candidate product before a determination of actual or potential emergency. Such an entity may submit a request for consideration that includes data to demonstrate that, based on the totality of scientific evidence available, it is reasonable to believe that the product may be effective in diagnosing, treating, or preventing the serious or life-threatening disease or condition. This is called a pre-EUA submission and its purpose is to allow FDA review considering that during an emergency, the time available for the submission and review of an EUA request may be severely limited. In 2014, we plan to meet with the FDA regarding human dose-conversion of Entolimod and, if appropriate after such meeting, submit a pre-EUA in order to inform and expedite the FDA’s issuance of an EUA, should one become necessary in the event of an emergency. The FDA does not have review deadlines with respect to pre-EUA submissions. Additionally, if we submit a pre-EUA, there is no guarantee that the FDA will agree that Entolimod meets the criteria for EUA, or, if they do agree, that such agreement by the FDA will lead to procurement by the U.S. or other governments or further development funding.
Public Readiness and Emergency Preparedness Act
The Public Readiness and Emergency Preparedness Act, or PREP Act, provides immunity for manufacturers from all claims under state or federal law for “loss” arising out of the administration or use of a “covered countermeasure.” However, injured persons may still bring a suit for “willful misconduct” against the manufacturer under some circumstances. “Covered countermeasures” include security countermeasures and “qualified pandemic or epidemic products”, including products intended to diagnose or treat pandemic or epidemic disease, such as pandemic vaccines, as well as treatments intended to address conditions caused by such products. For these immunities to apply, the Secretary of HHS must issue a declaration in cases of public health emergency or “credible risk” of a future public health emergency. Since 2007, the Secretary of HHS has issued 8 declarations and six amendments under the PREP Act to protect countermeasures that are necessary to prepare the nation for potential pandemics or epidemics from liability.
16
Fast Track Designation
Entolimod has been granted Fast Track designation by the FDA for reducing the risk of death following total body irradiation. The FDA’s Fast Track designation program is designed to facilitate the development and review of new drugs, including biological products that are intended to treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs for the conditions. Fast Track designation applies to a combination of the product and the specific indication for which it is being studied. Thus, it is the development program for a specific drug for a specific indication that receives Fast Track designation. The sponsor of a product designated as being in a Fast Track drug development program may engage in early communication with the FDA, including timely meetings and early feedback on clinical trials and may submit portions of an NDA or BLA on a rolling basis rather than waiting to submit a complete application. Products in Fast Track drug development programs also may receive priority review or accelerated approval, under which an application may be reviewed within six months after a complete NDA or BLA is accepted for filing or sponsors may rely on a surrogate endpoint for approval, respectively. The FDA may notify a sponsor that its program is no longer classified as a Fast Track development program if the Fast Track designation is no longer supported by emerging data or the designated drug development program is no longer being pursued. Receipt of Fast Track Status does not guarantee that we will experience a faster development process, review or approval as compared to conventional FDA procedures or that we will qualify or be able to take advantage of the FDA’s expedited review procedures.
Orphan Drug Designation
Entolimod and CBL0102 have been granted Orphan Drug designation by the FDA for prevention of death following a potentially lethal dose of total body irradiation and treatment of hepatocellular carcinoma, respectively. Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition which is defined as one affecting fewer than 200,000 individuals in the United States or more than 200,000 individuals where there is no reasonable expectation that the product development cost will be recovered from product sales in the United States. Orphan drug designation must be requested before submitting an NDA or BLA and does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
If an orphan drug-designated product subsequently receives the first FDA approval for the disease for which it has such designation, the product will be entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication, except in very limited circumstances for seven years as compared to five years for a standard new drug approval. As referenced above, we have received Orphan Drug status for two of our products. We intend to seek Orphan Drug status for our other products as appropriate, but an Orphan Drug designation may not provide us with a material commercial advantage.
Foreign Regulation
In addition to regulations in the United States, we are and will be subject to a variety of foreign regulations governing clinical trials and will be subject to a variety of foreign regulation governing commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. Other countries, at this time, do not have an equivalent to the Animal Rule and, as a result, do not have established criteria for review and approval of these types of products outside their normal review process, but some countries may have similar policy objectives in place for these product candidates.
17
As in the United States, the European Union may grant orphan drug status for specific indications if the request is made before an application for marketing authorization is made. The European Union considers an orphan medicinal product to be one that affects less than five of every 10,000 people in the European Union. A company whose application for orphan drug designation in the European Union is approved is eligible to receive, among other benefits, regulatory assistance in preparing the marketing application, protocol assistance and reduced application fees. Orphan drugs in the European Union also enjoy economic and marketing benefits, including up to ten years of market exclusivity for the approved indication, unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan designated product.
Our activities in Russia, through our subsidiaries, are regulated by the Ministry of Health and Social Development of the Russian Federation, or Minsotsrazvitiye. This federal executive authority is responsible for developing state policies as well as normative and legal regulations in the healthcare and pharmaceutical industries, including policies and regulations regarding the quality, efficacy and safety of pharmaceutical products. In addition, the Federal Service on Surveillance in Healthcare and Social Development, or Roszdravnadzor, is the subordinate executive authority to Minsotzrazvitiye, which, among other things (i) performs control and surveillance of certain activities, including pre-clinical and clinical trials and checks for compliance with state standards for medical products and pharmaceutical activities; (ii) issues licenses for the manufacture of drug products and pharmaceutical activities; (iii) grants allowance for clinical trials, use of new medical technologies and import and export of medical products, including import of products for use in clinical trials; and (iv) reviews and grants or denies registrations of medical products for commercial sale in Russia. The principal statute that governs our activities in Russia is the Federal Law of the Russian Federation from 12 April 2010 No. 61-FZ “On the [Use and Circulation] of Medicines”. This law regulates the research, development, testing, pre-clinical and clinical studies, governmental registration, quality control, manufacture, storage, transporting, export and import, licensing, advertisement, sale, transfer, utilization and destruction of medical products within the Russian Federation. All medical products must be registered in Russia and comply with stringent safety and quality controls and testing. In addition to Law No. 61-FZ, we are subject to a number of other laws and orders that regulate our activities in Russia relating to our drug development activities, taxation, corporate existence, labor laws and other areas. In particular, the existence, legal relations and transactions effected by our Russian subsidiaries are governed by the federal law No. 14-FZ “On Companies with Limited Liability”, which was enacted on February 8, 1998 and amended on November 30, 2011. Pursuant to this law, each subsidiary must hold an annual general meeting of its participants no later than four months after the end of each fiscal year, at which time, among other things, the annual financial results are reviewed and adopted. There are also equity holder and other approval requirements applicable to large transactions and affiliated transactions. Additionally, under the applicable Russian labor code, our Russian subsidiaries must enter into employment contracts with each employee, afford them at least 28 paid vacation days, limit the working week to 40 hours per week and follow the code’s specific procedures and safeguards that serve to protect an employee’s rights in the event the employee in Russia is terminated.
EMPLOYEES
As of March 14, 2014, we had 44 employees, 23 of whom are located in the U.S. and 21 of whom are located outside of the U.S.
ENVIRONMENT
We have made, and will continue to make, expenditures for environmental compliance and protection. Expenditures for compliance with environmental laws and regulations have not had, and are not expected to have, a material effect on our capital expenditures, results of operations, or competitive position.
18
AVAILABLE INFORMATION
Our internet website address is http://www.cbiolabs.com/. Through our website, we make available, free of charge, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, any amendments to those reports, proxy and registration statements, and all of our insider Section 16 reports, as soon as reasonably practicable after such material is electronically filed with, or furnished to, the U.S. Securities and Exchange Commission, or the SEC. These SEC reports can be accessed through the “Investors” section of our website. The information found on our website is not part of this or any other report we file with, or furnish to, the SEC. Paper copies of our SEC reports are available free of charge upon request in writing to Corporate Secretary, Cleveland BioLabs, Inc. 73 High Street, Buffalo NY 14203. The content on any website referred to in this Form 10-K is not incorporated by reference into this Form 10-K unless expressly noted.
Item 1A. Risk Factors
RISKS RELATING TO OUR FINANCIAL POSITION AND NEED FOR ADDITIONAL FINANCING
We will require substantial additional financing in order to meet our business objectives.
Since our inception, most of our resources have been dedicated to the pre-clinical and clinical development of our product candidates. In particular, we are currently conducting multiple clinical trials of our product candidates, each of which will require substantial funds to complete. We believe that we will continue to expend substantial resources for the foreseeable future developing our pre-clinical and clinical product candidates. These expenditures will include costs associated with research and development, conducting pre-clinical and clinical trials, obtaining regulatory approvals and products from third-party manufacturers, as well as marketing and selling any products approved for sale. In addition, other unanticipated costs may arise. Because the outcome of our planned and anticipated clinical trials is highly uncertain, we cannot reasonably estimate the actual amounts of capital necessary to successfully complete the development and commercialization of our product candidates.
As of December 31, 2013, and after giving effect to our equity raise in January 2014, our cash, cash equivalents and short-term investments amounted to $16.8 million. We believe that our existing cash, cash equivalents, and marketable securities will allow us to fund our operating plan into the first quarter of 2015.
Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceutical products, we are unable to estimate the exact amounts of our total capital requirements. Our future capital requirements depend on many factors, including:
| | • | | the number and characteristics of the product candidates we pursue; |
| | • | | the scope, progress, results and costs of researching and developing our product candidates, and conducting pre-clinical and clinical trials; |
| | • | | the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates; |
| | • | | the cost of commercialization activities for any of our product candidates that are approved for sale, including marketing, sales and distribution costs; |
| | • | | the cost of manufacturing our product candidates and any products we successfully commercialize; |
| | • | | our ability to establish and maintain strategic partnerships, licensing or other arrangements and the financial terms of such agreements; |
| | • | | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs and the outcome of such litigation; |
| | • | | whether we realize the full amount of any projected cost savings associated with our strategic restructuring; |
| | • | | the occurrence of a breach or event of default under our loan agreement with Hercules or under any other agreements with third parties; |
19
| | • | | the success of any pre-EUA submission we make with the FDA; and |
| | • | | the timing, receipt and amount of sales of, or royalties on, our future products, if any. |
In addition, it is possible that Hercules Technology II, L.P., or Hercules, could take the position that the decision of Biomedical Advanced Research and Development Authority of the U.S. Department of Health and Human Services, or BARDA, to terminate negotiations of our proposal constitutes a material adverse effect under our loan and security agreement with Hercules, under which we had $6.6 million in liability as of December 31, 2013, including a $550,000 end of term charge on the loan. Such determination by Hercules could trigger a repayment of all principal and interest due under the loan, as well as the prepayment charge under the loan, unless Hercules waives such event of default.
If our available cash and cash equivalents are insufficient to satisfy our liquidity requirements, or if we identify additional opportunities to do so, we may seek to sell additional equity or debt securities or obtain additional credit facilities. The sale of additional equity or convertible debt securities may result in additional dilution to our stockholders. If we raise additional funds through the issuance of debt securities or preferred stock or through additional credit facilities, these securities and/or the loans under credit facilities could provide for rights senior to those of our common stock and could contain covenants that would restrict our operations. Furthermore, any funds raised through collaboration and licensing arrangements with third parties may require us to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us. In any such event, our business prospects, financial condition and results of operations could be materially adversely affected.
We may require additional capital beyond our currently forecasted amounts and additional funds may not be available when we need them, on terms that are acceptable to us, or at all. In particular, the decline in the market price of our common stock could make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem appropriate. In addition, the variable rate clauses associated in many of our stock purchase agreements that prohibit certain types of capital raising activities for certain periods of time and pledge of assets in our loan and security agreement with Hercules may inhibit our ability to attract future investors and/or lenders. Additionally, our corporate structure, including the ownership of several of our product candidates in our non-wholly owned subsidiaries, may deter third parties from entering into collaboration and licensing arrangements with us. If we fail to raise sufficient additional financing, on terms and dates acceptable to us, we may not be able to continue our operations and the development of our product candidates, and may be required to reduce staff, reduce or eliminate research and development, slow the development of our product candidates, outsource or eliminate several business functions or shut down operations.
We have a history of operating losses. We expect to continue to incur losses and may not continue as a going concern.
We incurred net losses of approximately $20.1 million, $22.4 million and $5.2 million for the years ended December 31, 2013, 2012 and 2011, respectfully. We expect significant losses to continue for the next few years as we spend substantial sums on the continued research and development of our proprietary product candidates, and there is no certainty that we will ever become profitable as a result of these expenditures. As a result of losses that will continue throughout our development stage, we may exhaust our financial resources and be unable to complete the development of our product candidates.
Our ability to become profitable depends primarily on the following factors:
| | • | | our ability to obtain adequate sources of continued financing; |
| | • | | our ability to obtain approval for, and if approved, to successfully commercialize our product candidates; |
| | • | | our ability to successfully enter into license, development or other partnership agreements with third-parties for the development and/or commercialization of one or more of our product candidates; |
20
| | • | | our R&D efforts, including the timing and cost of clinical trials; and |
| | • | | our ability to enter into favorable alliances with third-parties who can provide substantial capabilities in clinical development, manufacturing, regulatory affairs, sales, marketing and distribution. |
Even if we successfully develop and market our product candidates, we may not generate sufficient or sustainable revenue to achieve or sustain profitability.
We may be unable to service our existing debt due to lack of cash flow, which could lead to default.
In September 2013, we entered into a loan and security agreement with Hercules Technology II, L.P., or Hercules, under which we borrowed $6.0 million. The current interest rate is 10.45%, with the initial 12 months of the facility requiring interest only payments and the following 30 months requiring interest and principal payments. The loan matures on January 1, 2017. Since entering into the agreement with Hercules, we have been making monthly interest-only payments to Hercules of approximately $54,000 per month and plan to continue making such payments until November 2014 when our payments will increase to approximately $228,000 per month, with a principal and interest payment of approximately $907,000 due in January 2017. As of December 31, 2013, the outstanding principal owed to Hercules was $6.0 million. Additionally, upon termination of the loan, we will also owe Hercules an end-of-term fee of $550,000. We granted Hercules a first priority security interest in substantially all of our assets, with the exception of our intellectual property, where the security interest is limited to proceeds of intellectual property.
If we do not make the required payments when due, either at maturity, or at applicable installment payment dates, or if we breach the agreement, default under the agreement by having a material adverse event happen to the business of the Company or become insolvent, Hercules could elect to declare all amounts outstanding together with all accrued and unpaid interest and penalties, to be immediately due and payable. In order to continue our planned operations and satisfy our debt obligations with Hercules, we will need to raise additional capital in the future. Additional capital may not be available on terms acceptable to us, or at all. Even if we were able to repay the full amount in cash, any such repayment could leave us with little or no working capital for our business. If we are unable to repay these amounts, Hercules will have a first claim on our assets pledged under the loan agreement. If Hercules should attempt to foreclose on the collateral, there may not be any assets remaining for distribution to shareholders after repayment in full of such secured indebtedness. Any default under the loan agreement and resulting foreclosure would have a material adverse effect on our financial condition and our ability to continue our operations.
Additionally, in September 2013, our majority owned subsidiary Panacela entered into a $1.5 million Convertible Loan Agreement with Rusnano, or the Rusnano Loan, and is required to pay all unpaid principal and interest under the loan in September 2015. The loan may be converted into shares of Panacela stock at any time at Rusnano’s option or will automatically convert upon certain financing events. In the event Panacela defaults on the loan and such default is not cured, Rusnano shall have the right to exercise a Warrant to purchase shares of Cleveland BioLabs common stock equal to 69.2% of the outstanding amount remaining unpaid under the Rusnano Loan at the time of exercise, divided by the exercise price of $1.694 per share.
Our ability to use our net operating loss carryforwards may be limited.
As of December 31, 2013, we had federal net operating loss carryforwards, or NOLs, of $109.9 million to offset future taxable income, which begin to expire if not utilized by 2023. Under the provisions of the Internal Revenue Code, substantial changes in our ownership, in certain circumstances, will limit the amount of NOLs that can be utilized annually in the future to offset taxable income. In particular, section 382 of the Internal Revenue Code imposed limitations on a company’s ability to use NOLs if a company experiences a more than 50% ownership change over a three-year period. If we are limited in our ability to use our NOLs in future years in which we have taxable income, we will pay more taxes than if we were able to utilize our NOLs fully. A full valuation allowance has been recorded against our deferred tax assets, including the net operating loss carryforwards, as we believe it is more likely than not we will be unable to realize the benefit of these assets.
21
RISKS RELATED TO PRODUCT DEVELOPMENT
We may not be able to successfully and timely develop our products.
Our product candidates range from ones currently in the research stage to ones currently in the clinical stage of development and all require further testing to determine their technical and commercial viability. Our success will depend on our ability to achieve scientific, clinical and technological advances and to translate such advances into reliable, commercially competitive products on a timely basis. In addition, the success of our subsidiaries will depend on their ability to meet developmental milestones in a timely manner or to fulfill certain other development requirements under contractual agreements, which are pre-requisites to their receipt of additional funding from their non-controlling interest holders or the government agency funding their government contracts. Products that we may develop are not likely to be commercially available for several years. The proposed development schedules for our products may be affected by a variety of factors, including, among others, technological difficulties, proprietary technology of others, the government approval process, the availability of funds and changes in government regulation, many of which will not be within our control. Any delay in the development, introduction or marketing of our products could result either in such products being marketed at a time when their cost and performance characteristics would not be competitive in the marketplace or in the shortening of their commercial lives. In light of the long-term nature of our projects and the unproven technology involved, we may not be able to complete successfully the development or marketing of any products.
We may fail to develop and commercialize some or all of our products successfully or in a timely manner because:
| | • | | pre-clinical study or clinical trial results may show the product to be less effective than desired (e.g., the study failed to meet its primary objectives) or to have harmful or problematic side effects; |
| | • | | we fail to receive the necessary regulatory approvals or there is a delay in receiving such approvals. Among other things, such delays may be caused by slow enrollment in clinical studies, length of time to achieve study endpoints, additional time requirements for data analysis or a pre-EUA, NDA or BLA preparation, discussions with the FDA, an FDA request for additional pre-clinical or clinical data or unexpected safety or manufacturing issues; |
| | • | | they fail to conform to a changing standard of care for the diseases they seek to treat; |
| | • | | they are less effective or more expensive than current or alternative treatment methods; |
| | • | | of manufacturing costs, pricing or reimbursement issues, or other factors that make the product not economically feasible; or |
| | • | | proprietary rights of others and their competing products and technologies may prevent our product from being commercialized. |
Our collaborative relationships with third parties could cause us to expend significant resources and incur substantial business risk with no assurance of financial return.
We anticipate substantial reliance upon strategic collaborations for marketing and commercialization of our product candidates and we may rely even more on strategic collaborations for R&D of our product candidates. Our business depends on our ability to sell drugs to both government agencies and to the general pharmaceutical market. Offering Entolimod for its biodefense indication use to government agencies may require us to develop new sales, marketing or distribution capabilities beyond those already existing in the Company and we may not be successful in selling Entolimod for its biodefense indication use in the United States or in foreign countries despite our efforts. Selling oncology drugs will require a more significant infrastructure. We plan to sell oncology drugs through strategic partnerships with pharmaceutical companies. If we are unable to establish or manage such strategic collaborations on terms favorable to us in the future, our revenue and drug development may be limited. To date, we have not entered into any strategic collaboration with a third party capable of
22
providing these services and we can make no guarantee that we will be able to enter into a strategic collaboration in the future. In addition, we have not yet marketed or sold any of our product candidates or entered into successful collaborations for these services in order to ultimately commercialize our product candidates. We also rely on third-party collaborations with our manufacturers. Manufacturers producing our product candidates must follow current Good Manufacturing Practice, or cGMP, regulations enforced by the FDA and foreign equivalents.
Establishing strategic collaborations is difficult and time-consuming. Our discussion with potential collaborators may not lead to the establishment of collaborations on favorable terms, if at all. Potential collaborators may reject collaborations based upon their assessment of our financial, regulatory or intellectual property position. Even if we successfully establish new collaborations, these relationships may never result in the successful development or commercialization of our product candidates or the generation of sales revenue. In addition to the extent that we enter into collaborative arrangements, our drug revenues are likely to be lower than if we directly marketed and sold any drugs that we may develop.
We will not be able to commercialize our product candidates if our pre-clinical development efforts are not successful, our clinical trials do not demonstrate safety or our clinical trials or animal studies do not demonstrate efficacy.
Before obtaining required regulatory approvals for the commercial sale of any of our product candidates, we must conduct extensive pre-clinical testing and clinical trials to demonstrate that our product candidates are safe and clinical or animal trials to demonstrate the efficacy of our product candidates. Pre-clinical and clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. Success in pre-clinical testing and early clinical trials does not ensure that later clinical trials or animal efficacy studies will be successful and interim results of a clinical trial or animal efficacy study does not necessarily predict final results. In addition, we must outsource our clinical trials and a majority of our animal studies required to obtain regulatory approval of our products. We are not certain that we will successfully or promptly finalize agreements for the conduct of these studies. Delay in finalizing such agreements would delay the commencement of our pre-clinical and clinical studies, such as animal efficacy studies for Entolimod’s biodefense indication and clinical trials of Entolimod, CBL0102 and CBL0137 for oncology indications. In addition, we are seeking FDA agreement on the scope and design of our pivotal animal efficacy and human safety program for Entolimod’s biodefense indication. Delay in agreement with the FDA on this program will delay conduct of the pivotal animal efficacy and human safety studies.
Agreements with contract research organizations, or CROs, and study investigators, for clinical or animal testing and with other third parties for data management services place substantial responsibilities on these parties, which could result in delays in, or termination of, our clinical trials if these parties fail to perform as expected. For example, if any of our clinical trial sites fail to comply with Good Clinical Practices or our pivotal animal studies fail to comply with Good Laboratory Practices we may be unable to use the data generated at those sites. In these studies, if contracted CROs or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to our protocols or for other reasons, our clinical or animal studies may be extended, delayed or terminated, and we may be unable to obtain regulatory approval for or successfully commercialize our product candidates.
Our clinical trial operations will be subject to regulatory inspections at any time. If regulatory inspectors conclude that we or our clinical trial sites are not in compliance with applicable regulatory requirements for conducting clinical trials, we or they may receive warning letters or other correspondence detailing deficiencies and we will be required to implement corrective actions. If regulatory agencies deem our responses to be inadequate, or are dissatisfied with the corrective actions that we or our clinical trial sites have implemented, our clinical trials may be temporarily or permanently discontinued, we may be fined, we or our investigators may be the subject of an enforcement action, the government may refuse to approve our marketing applications or allow us to manufacture or market our products or we may be criminally prosecuted.
23
In addition, a failure of one or more of our clinical trials or animal studies can occur at any stage of testing and such failure could have a material adverse effect on our ability to generate revenue and could require us to reduce the scope of or discontinue our operations. We may experience numerous unforeseen events during, or as a result of, pre-clinical testing and the clinical trial or animal study process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates, including:
| | • | | Regulators or institutional review boards, or IRB, may not authorize us to commence a clinical trial or conduct a clinical trial at a prospective trial site or an institutional animal care and use committee, or IACUC, may not authorize us to commence an animal study at a prospective study site; |
| | • | | We may decide, or regulators may require us, to conduct additional pre-clinical testing or clinical trials, or we may abandon projects that we expect to be promising, if our pre-clinical tests, clinical trials or animal efficacy studies produce negative or inconclusive results; |
| | • | | We might have to suspend or terminate our clinical trials if the participants are being exposed to unacceptable safety risks; |
| | • | | Regulators or IRBs may require that we hold, suspend or terminate clinical development for various reasons, including noncompliance with regulatory requirements or if it is believed that the clinical trials present an unacceptable safety risk to the patients enrolled in our clinical trials; |
| | • | | The cost of our clinical trials or animal studies could escalate and become cost prohibitive; |
| | • | | Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the product not commercially viable; |
| | • | | We may not be successful in recruiting a sufficient number of qualifying subjects for our clinical trials or certain animals used in our animal studies or facilities conducting our studies may not be available at the time that we plan to initiate a study; and |
| | • | | The effects of our product candidates may not be the desired effects, may include undesirable side effects, or the product candidates may have other unexpected characteristics; |
| | • | | Our collaborators that conduct our clinical or pivotal animal studies could go out of business and not be available for FDA inspection when we submit our product for approval. |
Even if we or our collaborators complete our animal studies and clinical trials and receive regulatory approval, it is possible that a product may be found to be ineffective or unsafe due to conditions or facts that arise after development has been completed and regulatory approvals have been obtained. In this event, we may be required to withdraw such product from the market. To the extent that our success will depend on any regulatory approvals from government authorities outside of the United States that perform roles similar to that of the FDA, uncertainties similar to those stated above will also exist.
Our majority-owned subsidiaries have significant non-controlling interest holders and, as such, are not operated solely for our benefit.
As of December 31, 2013, we owned 59.2% of the equity interests in Incuron and 54.6% of the equity interests in Panacela. Additionally, we anticipate that Incuron will receive their last funding tranche from BioProcess Capital Partners in early 2014 and that following such investment our ownership interest in Incuron will fall below 50% if we do not invest $3 million of additional funds. Although these subsidiaries are currently majority-owned by us and are consolidated in our results, they have significant non-controlling interest holders, each of which are funds regulated by the Russian Federation government. As such, we share ownership and management of our subsidiaries with one or more parties who may not have the same goals, strategies, priorities, or resources as we do.
In each of our majority-owned subsidiaries, both we and our co-owners have certain rights in respect of such subsidiaries. Our majority-owned subsidiaries provide the right to each party to designate certain of the board
24
members and certain decisions in respect of these subsidiaries may not be made without a supermajority vote of the equity holders or the consent of all of the equity holders. The right to transfer ownership interests in our majority-owned subsidiaries is restricted by provisions such as rights of first refusal and tag along and drag along rights. In addition, the use of funds and other matters are subject to monitoring and oversight by both groups of equity holders. Furthermore, we are required to pay more attention to our relationship with our co-owners as well as with the subsidiaries, and if a co-owner changes, our relationship may be materially adversely affected. These various restrictions may lead to additional organizational formalities as well as time-consuming procedures for sharing information and making decisions. In addition, the benefits from a successful joint venture are shared among the co-owners, so that we would not receive all the benefits from our successful joint ventures.
The co-owners of our majority-owned subsidiaries are required to make additional payments to the subsidiaries to finance their operations. Such additional contributions are dependent on the satisfaction of various developmental milestones by our majority-owned subsidiaries, which may not be achieved within set time periods, and if such contributions or investments are not achieved, may result in a material adverse effect in our business, financial condition and results of operations.
If parties on whom we rely to manufacture our product candidates do not manufacture them in satisfactory quality, in a timely manner, in sufficient quantities or at an acceptable cost, clinical development and commercialization of our product candidates could be delayed.
We do not own or operate manufacturing facilities. Consequently, we rely on third parties as sole suppliers of our product candidates. We do not expect to establish our own manufacturing facilities and we will continue to rely on third-party manufacturers to produce supplies for pre-clinical, clinical and pivotal animal studies and for commercial quantities of any products or product candidates that we market or may supply to our collaborators. Our dependence on third parties for the manufacture of our product candidates may adversely affect our ability to develop and commercialize any product candidates on a timely and competitive basis.
To date, our product candidates have only been manufactured in quantities sufficient for pre-clinical studies and initial clinical trials. We rely on a single collaborator for production of each of our product candidates. For a variety of reasons, dependence on any single manufacturer may adversely affect our ability to develop and commercialize our product candidates on a timely and competitive basis. In addition, our current contractual arrangements alone may not be sufficient to guarantee that we will be able to procure the needed supplies as we complete clinical development and/or enter commercialization.
Additionally, in connection with our application for commercial approvals and if any product candidate is approved by the FDA or other regulatory agencies for commercial sale, we will need to procure commercial quantities from qualified third-party manufacturers. We may not be able to contract for increased manufacturing capacity for any of our product candidates in a timely or economic manner or at all. A significant scale-up in manufacturing may require additional validation studies and commensurate financial investments by the contract manufacturers. If we are unable to successfully increase the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage of supply, which could limit our sales and could initiate regulatory intervention to minimize the public health risk.
Other risks associated with our reliance on contract manufacturers include the following:
| | • | | Contract manufacturers may encounter difficulties in achieving volume production, quality control and quality assurance and also may experience shortages in qualified personnel and obtaining active ingredients for our product candidates. |
| | • | | If, for any circumstance, we are required to change manufacturers, we could be faced with significant monetary and lost opportunity costs with switching manufacturers. Furthermore, such change may take a significant amount of time. The FDA and foreign regulatory agencies must approve these manufacturers in advance. This requires prior approval of regulatory submissions as well as successful completion of pre-approval inspections to ensure compliance with FDA and foreign regulations and standards. |
25
| | • | | Contract manufacturers are subject to ongoing periodic, unannounced inspection by the FDA and state and foreign agencies or their designees to ensure strict compliance with cGMP and other governmental regulations and corresponding foreign standards. We do not have control over compliance by our contract manufacturers with these regulations and standards. Our contract manufacturers may not be able to comply with cGMP and other FDA requirements or other regulatory requirements outside the United States. Failure of contract manufacturers to comply with applicable regulations could result in delays, suspensions or withdrawal of approvals, seizures or recalls of product candidates and operating restrictions, any of which could significantly and adversely affect our business. |
| | • | | Contract manufacturers may breach the manufacturing agreements that we have with them because of factors beyond our control or may terminate or fail to renew a manufacturing agreement based on their own business priorities at a time that is costly or inconvenient to us. |
Changes to the manufacturing process during the conduct of clinical trials or after marketing approval also require regulatory submissions and the demonstration to the FDA or other regulatory authorities that the product manufactured under the new conditions complies with cGMP requirements. These requirements especially apply to moving manufacturing functions to another facility. In each phase of investigation, sufficient information about changes in the manufacturing process must be submitted to the regulatory authorities and may require prior approval before implementation with the potential of substantial delay or the inability to implement the requested changes.
RISKS RELATING TO REGULATORY APPROVAL
We may not be able to obtain regulatory approval in a timely manner or at all and the results of clinical trials may not be favorable.
The testing, marketing and manufacturing of any product for use in the United States will require approval from the FDA. We cannot predict with any certainty the amount of time necessary to obtain FDA approval and whether any such approval will ultimately be granted. Pre-clinical studies and clinical trials may reveal that one or more products are ineffective or unsafe, in which event, further development of such products could be seriously delayed, terminated or rendered more expensive. Moreover, obtaining approval for certain products may require testing on human subjects of substances whose effects on humans are not fully understood or documented.
In addition, we expect to rely on an FDA regulation known as the “Animal Rule” to obtain approval for Entolimod’s biodefense indication. The Animal Rule permits the use of animal efficacy studies together with human clinical safety trials to support an application for marketing approval of products when human efficacy studies are neither ethical nor feasible. These regulations are relatively new and we have limited experience in the application of these rules to the product candidates that we are developing. As such, we cannot predict the time required for them to confirm the relevant rules, or the scope thereof. Additionally, we may submit an application with the FDA for pre- EUA, so that Entolimod may be used in an emergency situation. If and when we provide the FDA with the data to support a pre-EUA for Entolimod in the event of a radiation emergency we cannot guarantee that the FDA will review the data in a timely manner, or that, when the data is reviewed, that the FDA will accept the data. The FDA may decide that our data are insufficient for pre-EUA or BLA approval and require additional pre-clinical, clinical or other studies, refuse to approve our products, or place restrictions on our ability to commercialize those products. If we are not successful in completing the development, licensure and commercialization of Entolimod for its biodefense indication use, or if we are significantly delayed in doing so, our business will be materially harmed.
The receipt of FDA approval may be delayed for reasons other than the results of pre-clinical studies and clinical trials. For example, in 2011, the IND application for Entolimod’s biodefense indication was transferred within the FDA from the Division of Biologic Oncology Products, or DBOP, to the Division of Medical Imaging Products, or DMIP. As a result of this transfer, we requested and participated in nine meetings with DMIP during
26
2011-2013 to review the product mechanisms of action, safety profile and preliminary estimation of an effective human dose. DMIP has agreed on the scope and design of the proposed pivotal animal efficacy program and has acknowledged that specific cytokines do play an important role in Entolimod’s mechanism of action and, as such, can be used as biomarkers for animal-to-human dose-conversion. DMIP has also provided advice on the design of the remaining program needed for BLA submission. However, we are still in the process of reaching an agreement with FDA on the certain elements of the design of our remaining clinical studies for Entolimod. There can be no guarantee that we will reach a satisfactory agreement in a timely manner, or at all, or that DMIP may request any additional information related to our pre-clinical or clinical programs.
Delays in obtaining FDA or any other necessary regulatory approvals of any proposed product or the failure to receive such approvals would have an adverse effect on our ability to develop such product, the product’s potential commercial success and/or on our business, prospects, financial condition and results of operations.
Failure to obtain regulatory approval in international jurisdictions could prevent us from marketing our products abroad.
We intend to market our product candidates, including specifically the product candidates being developed by our subsidiaries, in the United States, the Russian Federation and other countries and regulatory jurisdictions. In order to market our product candidates in the United States, Russia and other jurisdictions, we must obtain separate regulatory approvals in each of these countries and territories. The procedures and requirements for obtaining marketing approval vary among countries and regulatory jurisdictions and can involve additional clinical trials or other tests. In addition, we do not have in-house experience and expertise regarding the procedures and requirements for filing for and obtaining marketing approval for drugs in countries outside of the United States, Europe and Japan and may need to engage and rely upon expertise of third parties when we file for marketing approval in countries outside of the United States, Europe and Japan. Also, the time required to obtain approval in markets outside of the United States may differ from that required to obtain FDA approval, while still including all of the risks associated with obtaining FDA approval. We may not be able to obtain all of the desirable or necessary regulatory approvals on a timely basis, if at all. Approval by a regulatory authority in a particular country or regulatory jurisdiction, such as the FDA in the United States or the Roszdravnadzor in Russia, does not ensure approval by a regulatory authority in another country.
We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our product candidates in any or all of the countries or regulatory jurisdictions in which we desire to market our product candidates. At this time, other countries do not have an equivalent to the Animal Rule and, as a result, such countries do not have established criteria for review and approval for this type of product outside their normal review process. Specifically, because such other countries do not have an equivalent to the Animal Rule, we may not be able to file for or receive regulatory approvals for Entolimod’s biodefense indication outside the United States based on our animal efficacy and human safety data.
The Fast Track designation for Entolimod may not actually lead to a faster development or regulatory review or approval process.
We have obtained a “Fast Track” designation from the FDA for Entolimod’s biodefense indication. However, we may not experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may withdraw our Fast Track designation if the FDA believes that the designation is no longer supported by data from our clinical development program. Our Fast Track designation does not guarantee that we will qualify for or be able to take advantage of the FDA’s expedited review procedures or that any application that we may submit to the FDA for regulatory approval will be accepted for filing or ultimately approved.
27
Any pre-EUA submission we make to the FDA may not be successful and even if such submission is successful it may not accelerate BLA approval of Entolimod or result in any purchase by the U.S. government for this product.
In 2014, we plan to meet with the FDA regarding human dose-conversion of Entolimod and, if appropriate after such meeting, submit a pre-EUA in order to inform and expedite the FDA’s issuance of an EUA, should one become necessary in the event of an emergency. The FDA does not have review deadlines with respect to pre-EUA submissions and, therefore, the timing of any approval of a pre-EUA submission is uncertain. If we submit a pre-EUA, the FDA may decide not to accept the data or decide that our data are insufficient for pre-EUA and require additional pre-clinical, clinical or other studies, refuse to approve our products, or place restrictions on our ability to commercialize those products. An acceptance of our pre-EUA submission does not guarantee, and may not accelerate, BLA approval of Entolimod as a radiation countermeasure. Further, even if our pre-EUA submission is authorized, there is no guarantee that such authorization will lead to procurement by the U.S. or other governments or any additional development funding. If we are not successful in partnering Entolimod or completing the development, licensure and commercialization of Entolimod for its biodefense indication use, or if we are significantly delayed in doing so, our business may be materially harmed.
Even if our drug candidates obtain regulatory approval, we will be subject to on-going government regulation.
Even if our drug candidates obtain regulatory approval, our products will be subject to continuing regulation by the FDA, including record keeping requirements, submitting periodic reports to the FDA, reporting of any adverse experiences with the product and complying with Risk Evaluation and Mitigation Strategies and drug sampling and distribution requirements. In addition, updated safety and efficacy information must be maintained and provided to the FDA. We or our collaborative partners, if any, must comply with requirements concerning advertising and promotional labeling, including the prohibition against promoting and non-FDA approved or “off-label” indications or products. Failure to comply with these requirements could result in significant enforcement action by the FDA, including warning letters, orders to pull the promotional materials and substantial fines.
After FDA approval of a product, the discovery of problems with a product or its class, or the failure to comply with requirements may result in restrictions on a product, manufacturer, or holder of an approved marketing application. These include withdrawal or recall of the product from the market or other voluntary or FDA-initiated action that could delay or prevent further marketing. Newly discovered or developed safety or effectiveness data, including from other products in a therapeutic class, may require changes to a product’s approved labeling, including the addition of new warnings and contraindications. Also, the FDA requires post-market clinical testing of products approved under the Animal Rule at the time of a declared emergency and may require post-market clinical testing of other products. They may also require surveillance to monitor the product’s safety or efficacy to evaluate long-term effects. It is also possible that rare but serious adverse events not seen in our drug candidates may be identified after marketing approval. This could result in withdrawal of our product from the market.
Compliance with post-marketing regulations may be time-consuming and costly and could delay or prevent us from generating revenue from the commercialization of our drug candidates.
If physicians and patients do not accept and use our drugs, we will not achieve sufficient product revenues and our business will suffer.
Even if we gain marketing approval of our drug candidates, government purchasers, physicians and/or patients may not accept and use them. Acceptance and use of these products may depend on a number of factors including:
| | • | | Perceptions by members of the government healthcare community, including physicians, about the safety and effectiveness of our drugs; |
| | • | | Published studies demonstrating the safety and effectiveness of our drugs; |
28
| | • | | Adequate reimbursement for our products from payors; and |
| | • | | Effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any. |
The failure of our drugs, if approved for marketing, to gain acceptance in the market would harm our business and could require us to seek additional financing.
RISKS RELATED TO OUR DEPENDENCE ON U.S. GOVERNMENT CONTRACTS AND GRANTS
If we are unable to procure additional government funding, we may not be able to fund future R&D and implement technological improvements, which would materially harm our financial conditions and operating results.
In 2013, we received 26.8% of our revenues from government contract and grant development work in connection with grants from the DoD. In 2012 and 2011, we received 34.8% and 87.6% of our revenues from U.S. government contract and grant development work.
These revenues have funded some of our personnel and other R&D and General and Administrative costs and expenses. Our current grants and contracts with the U.S. government expire in March 2014. It is possible that we may not choose to apply for or, if we do apply, be able to procure new grants and contracts that provide sufficient funding, or any funding at all. In addition, the finalization of new contracts and grants may require a significant time from the initial request and negotiations for such contracts and grants are subject to a significant amount of uncertainty.
For example, in May 2011, we announced that we had concluded advanced stages of contract negotiation with BARDA for the funding of certain development activities relating to Entolimod’s biodefense indication in our 2010 proposal to BARDA. BARDA indicated that further contract-related negotiations would require clarification of the development path for Entolimod’s biodefense indication with the FDA, which is in the process of actively reviewing our IND application for Entolimod. BARDA indicated that we might resubmit an updated proposal upon confirmation from the FDA that they do not have any objections to us proceeding with our development plan as a result of this review. We received a confirmatory letter from the FDA in late 2011 and submitted a white paper to BARDA under its currently open Broad Agency Announcement, or the BAA. In April 2012, we announced that BARDA had declined to invite the Company to submit a full proposal pursuant to the white paper submitted. After further discussions with both the FDA and BARDA, we announced in October 2012, that the Company had submitted a proposal to BARDA under the BAA for the remaining development steps needed for FDA licensure of Entolimod as a medical radiation countermeasure. In January 2014, we announced that BARDA had terminated negotiations of our proposal due to lack of availability of funds. If and when we do submit additional funding proposals to BARDA or other U.S. or foreign government agencies there is no assurance that such agencies will make a positive decision with regard to funding our proposal(s) or award a contract (if one is awarded) in a timely manner.
If we are unable to obtain sufficient grants and contracts on a timely basis or if our existing grants and contracts are not funded, our ability to fund future R&D would be diminished, which would negatively impact our ability to compete in our industry and could materially and adversely affect our business, financial condition and results of operations.
Our future business may be harmed as a result of the government contracting process as it involves risks not present in the commercial marketplace.
We expect that a significant portion of the business that we will seek in the near future will be under government contracts or subcontracts, both U.S. and foreign, which may be awarded through competitive bidding. Competitive bidding for government contracts presents a number of risks that are not typically present in the commercial contracting process, which may include:
| | • | | The need to devote substantial time and attention of management and key employees to the preparation of bids and proposals for contracts that may not be awarded to us; |
29
| | • | | The need to accurately estimate the resources and cost structure that will be required to perform any contract that we might be awarded; |
| | • | | The risk that the government will issue a request for proposal to which we would not be eligible to respond; |
| | • | | The risk that third parties may submit protests to our responses to requests for proposal that could result in delays or withdrawals of those requests for proposal; |
| | • | | The expenses that we might incur and the delays that we might suffer if our competitors protest or challenge contract awards made to us pursuant to competitive bidding and the risk that any such protest or challenge could result in the resubmission of bids based on modified specifications, or in termination, reduction or modification of the awarded contract; and |
| | • | | The risk that review of our proposal or award of a contract or an option to an existing contract could be significantly delayed for reasons including, but not limited to, the need for us to resubmit our proposal or limitations on available funds due to government budget cuts. |
The U.S. government may choose to award future contracts for the supply of medical radiation countermeasures to our competitors instead of to us. If we are unable to win particular contracts, or if the government chooses not to fully exercise all options under contracts awarded to us, we may not be able to operate in the market for products that are provided under those contracts for a number of years. If we are unable to consistently win new contract awards and have the options under our existing contracts exercised over an extended period, or if we fail to anticipate all of the costs and resources that will be required to secure such contract awards, our growth strategy and our business, financial condition and operating results could be materially adversely affected.
The market for U.S. and other government funding is highly competitive.
Our biodefense product candidate, Entolimod, faces significant competition for U.S. government funding for both development and procurement of medical countermeasures for biological, chemical and nuclear threats, diagnostic testing systems and other emergency preparedness countermeasures. In addition, we may not be able to compete effectively if our products and product candidates do not satisfy procurement requirements of the U.S. government with respect to biodefense products. Our opportunities to succeed in this industry could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects, are more convenient or are less expensive than any products that we may develop.
U.S. government agencies have special contracting requirements, which create additional risks.
We have historically entered into contracts with various U.S. government agencies. Due to these contracts with government agencies, we are subject to various federal contract requirements. Future sales to U.S. government agencies will depend, in part, on our ability to meet these requirements, certain of which we may not be able to satisfy.
U.S. government contracts typically contain unfavorable termination provisions and are subject to audit by the government at its sole discretion even after the end of the period of performance under the contract, which subjects us to additional risks. These risks include the ability of the U.S. government to unilaterally:
| | • | | Suspend or prevent us for a set period of time from receiving new contracts or extending existing contracts based on violations or suspected violations of laws or regulations; |
| | • | | Terminate our existing contracts; |
| | • | | Reduce the scope and value of our existing contracts; |
| | • | | Audit and object to our contract-related costs and fees, including allocated indirect costs; |
| | • | | Control and potentially prohibit the export of our products; and |
| | • | | Change certain terms and conditions in our contracts. |
30
Pursuant to our government contracts, we are generally permitted to retain title to any patentable invention or discovery made while performing the contract. However, the U.S. government is generally entitled to receive a non-exclusive, non-transferable, irrevocable, paid-up license to the subject inventions throughout the world. In addition, our government contracts generally provide that the U.S. government retains unlimited rights in the technical data produced under such government contract.
Our business could be adversely affected by a negative audit by the U.S. government.
As a U.S. government contractor, we may become subject to periodic audits and reviews by U.S. government agencies such as the Defense Contract Audit Agency, or the DCAA. These agencies review a contractor’s performance under its contracts, cost structure and compliance with applicable laws, regulations and standards. The DCAA also reviews the adequacy of, and a contractor’s compliance with, its internal control systems and policies, including the contractor’s purchasing, property, estimating, compensation and management information systems. Any costs found to be improperly allocated to a specific contract will not be reimbursed, which such costs already reimbursed must be refunded.
Based on the results of these audits, the U.S. government may adjust our contract-related costs and fees, which have already been paid to us, including allocated indirect costs. In addition, if an audit or review uncovers any improper or illegal activity, we may be subject to civil and criminal penalties and administrative sanctions, including termination of our contracts, forfeiture of profits, suspension of payments, fines and suspension or prohibition from doing business with the U.S. government. We could also suffer serious harm to our reputation if allegations of impropriety were made against us. In addition, under U.S. government purchasing regulations, some of our costs, including most financing costs, amortization of intangible assets, portions of our R&D costs and some marketing expenses, may not be reimbursable or allowed under our contracts. Further, as a U.S. government contractor, we may become subject to an increased risk of investigations, criminal prosecution, civil fraud, whistleblower lawsuits and other legal actions and liabilities to which purely private sector companies are not.
RISKS RELATING TO OUR INTELLECTUAL PROPERTY
We rely upon licensed patents to protect our technology. We may be unable to obtain or protect such intellectual property rights and we may be liable for infringing upon the intellectual property rights of others.
Our ability to compete effectively will depend on our ability to maintain the proprietary nature of our technologies and the proprietary technology of others with which we have entered into licensing agreements. We have entered into five separate exclusive license agreements to license our product candidates that are not owned by us and some product candidates are covered by up to three separate license agreements. Pursuant to these license agreements we maintain patents and patent applications covering our product candidates. We do not know whether any of these patent applications that are still in the approval process will ultimately result in the issuance of a patent with respect to the technology owned by us or licensed to us. The patent position of pharmaceutical or biotechnology companies, including ours, is generally uncertain and involves complex legal and factual considerations. The standards that the United States Patent and Trademark Office use to grant patents are not always applied predictably or uniformly and can change. There is also no uniform, worldwide policy regarding the subject matter and scope of claims granted or allowable in pharmaceutical or biotechnology patents. Accordingly, we do not know the degree of future protection for our proprietary rights or the breadth of claims that will be allowed in any patents issued to us or to others.
Our technology may be found in the future to infringe upon the rights of others or be infringed upon by others. In such a case, others may assert infringement claims against us, and should we be found to infringe upon their patents, or otherwise impermissibly utilize their intellectual property, we might be forced to pay damages, potentially including treble damages, if we are found to have willfully infringed on such parties’ patent rights. Furthermore, parties making claims against us may be able to obtain injunctive or other equitable relief which could effectively block our ability to further develop, commercialize and sell products. In addition to any
31
damages we might have to pay, we may be required to obtain licenses from the holders of this intellectual property, enter into royalty agreements, or redesign our products so as not to utilize this intellectual property, each of which may prove to be uneconomical or otherwise impossible. Conversely, we may not always be able to successfully pursue our claims against others that infringe upon our technology and the technology exclusively licensed by us or developed with our collaborative partners. Thus, the proprietary nature of our technology or technology licensed by us may not provide adequate protection against competitors.
Moreover, the cost to us of any litigation or other proceeding relating to our patents and other intellectual property rights, even if resolved in our favor, could be substantial and the litigation would divert our management’s efforts and our resources. Uncertainties resulting from the initiation and continuation of any litigation could limit our ability to continue our operations.
If we fail to comply with our obligations under our license agreement with third parties, we could lose our ability to develop our product candidates.
The manufacture and sale of any products developed by us may involve the use of processes, products or information, the rights to certain of which are owned by others. Although we have obtained exclusive licenses for our product candidates from CCF, RPCI and CCIA with regard to the use of patent applications as described above and certain processes, products and information of others, these licenses could be terminated or expire during critical periods and we may not be able to obtain licenses for other rights that may be important to us, or, if obtained, such licenses may not be obtained on commercially reasonable terms. Furthermore, some of our product candidates require the use of technology licensed from multiple third parties, each of which is necessary for the development of such product candidates. If we are unable to maintain and/or obtain licenses, we may have to develop alternatives to avoid infringing upon the patents of others, potentially causing increased costs and delays in product development and introduction or precluding the development, manufacture, or sale of planned products. Additionally, the patents underlying any licenses may not be valid and enforceable. To the extent any products developed by us are based on licensed technology, royalty payments on the licenses will reduce our gross profit from such product sales and may render the sales of such products uneconomical.
Our current exclusive licenses impose various development, royalty, diligence, record keeping, insurance and other obligations on us. If we breach any of these obligations and do not cure such breaches within the relevant cure period, the licensor may have the right to terminate the license, which could result in us being unable to develop, manufacture and sell products that are covered by the licensed technology or enable a competitor to gain access to the licensed technology.
In addition, while we cannot currently determine the dollar amount of the royalty and other payments we will be required to make in the future under the license agreements, if any, the amounts may be significant. The dollar amount of our future payment obligations will depend on the technology and intellectual property we use in products that we successfully develop and commercialize, if any.
If we are not able to protect and control our unpatented trade secrets, know-how and other technology, we may suffer competitive harm.
We also rely on a combination of trade secrets, know-how, technology and nondisclosure and other contractual agreements and technical measures to protect our rights in the technology. However, trade secrets are difficult to protect and we rely on third parties to develop our products and thus must share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements will typically restrict the ability of our collaborators, advisors, employees and consultants to publish data potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these
32
agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication. If any trade secret, know-how or other technology not protected by a patent or intellectual property right were disclosed to, or independently developed by, a competitor, our business, financial condition and results of operations could be materially adversely affected.
RISKS RELATING TO OUR INDUSTRY AND OTHER EXTERNAL FACTORS
The biopharmaceutical market in which we compete is highly competitive.
The biopharmaceutical industry is characterized by rapid and significant technological change. Our success will depend on our ability to develop and apply our technologies in the design and development of our product candidates and to establish and maintain a market for our product candidates. In addition, there are many companies, both public and private, including major pharmaceutical and chemical companies, specialized biotechnology firms, universities and other research institutions engaged in developing pharmaceutical and biotechnology products. Many of these companies have substantially greater financial, technical, research and development resources and human resources than us. Competitors may develop products or other technologies that are more effective than those that are being developed by us or may obtain FDA or other governmental approvals for products more rapidly than us. If we commence commercial sales of products, we still must compete in the manufacturing and marketing of such products, areas in which we have no experience.
Our growth could be limited if we are unable to attract and retain key personnel and consultants.
We have limited experience in filing and prosecuting regulatory applications to obtain marketing approval from the FDA or other regulatory authorities. The loss of services of one or more of our key employees or consultants could have a negative impact on our business or our ability to expand our research, development and clinical programs. We depend on our scientific and clinical collaborators and advisors, all of whom have outside commitments that may limit their availability to us. In addition, we believe that our future success will depend in large part upon our ability to attract and retain highly skilled scientific, managerial and marketing personnel, particularly as we expand our activities in clinical trials, the regulatory approval process, external partner solicitations and sales and manufacturing. We routinely enter into consulting agreements with our scientific and clinical collaborators and advisors, opinion leaders and heads of academic departments in the ordinary course of our business. We also enter into contractual agreements with physicians and institutions who recruit patients into our clinical trials on our behalf in the ordinary course of our business. In addition, as a result of our 2013 corporate restructuring and workforce reductions, we may face additional challenges in retaining our existing senior management and key employees and recruiting new employees to join our company as our business needs change. We face significant competition for this type of personnel and for employees from other companies, research and academic institutions, government entities and other organizations. We cannot predict our success in hiring or retaining the personnel we require for continued growth.
We may be subject to damages resulting from claims that we, our employees, or our consultants have wrongfully used or disclosed alleged trade secrets of their former employers.
We engage as employees and consultants individuals who were previously employed at other biotechnology or pharmaceutical companies, including at competitors or potential competitors. Although no claims against us are currently pending, we may become subject to claims that we or our employees have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and distract management.
We may incur substantial liabilities from any product liability and other claims if our insurance coverage for those claims is inadequate.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face an even greater risk if the product candidates are sold commercially. An individual
33
may bring a product liability claim against us if one of the product candidates causes, or merely appears to have caused, an injury. If we cannot successfully defend ourselves against the product liability claim, we will incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
| | • | | Decreased demand for our product candidates; |
| | • | | Injury to our reputation; |
| | • | | Withdrawal of clinical trial participants; |
| | • | | Costs of related litigation; |
| | • | | Diversion of our management’s time and attention; |
| | • | | Substantial monetary awards to patients or other claimants; |
| | • | | The inability to commercialize product candidates; and |
| | • | | Increased difficulty in raising required additional funds in the private and public capital markets. |
We currently have product liability insurance and intend to expand such coverage from coverage for clinical trials to include the sale of commercial products if marketing approval is obtained for any of our product candidates. However, insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage that will be adequate to satisfy any liability that may arise.
From time to time, we may also become subject to litigation, such as stockholder derivative claims or securities fraud claims, which could involve our directors and officers as defendants. We currently have director and officer, or D&O, insurance to cover such risk exposure for our directors and officers. Our bylaws require us to indemnify our current and past directors and officers from reasonable expenses related to the defense of any action arising from their service to us. Our certificate of incorporation and by-laws include provisions to indemnify the directors and officers to the fullest extent permitted by the Delaware General Corporation Law, including circumstances under which indemnification is otherwise discretionary. If our D&O insurance is insufficient to cover all such expenses for all directors and officers, we would be obligated to cover any shortfall, which may be substantial. Such expenditure could have a material adverse effect on our results of operation, financial condition and liquidity. Further, if D&O insurance becomes prohibitively expensive to maintain in the future, we may be unable to renew such insurance on economic terms or unable renew such insurance at all. The lack of D&O insurance may make it difficult for us to retain and attract talented and skilled directors and officers to serve our company, which could adversely affect our business.
We have been named as a defendant in a lawsuit that could result in substantial costs and divert management’s attention.
We have been named as a defendant in a lawsuit initiated earlier this year that generally alleges we misrepresented the state of our funding negotiations with BARDA during the period leading up to the sale of our common stock and warrants in January 2014, and as a result, the plaintiffs were harmed when our stock price declined following the announcement that BARDA had terminated negotiations with us. The complaint asserts claims under Section 10(b) of the Securities Exchange Act of 1934 and SEC Rule 10b-5, as well as claims for fraudulent inducement, breach of contract, and indemnification. Any conclusion of these matters in a manner adverse to us would have a material adverse effect on our financial condition and business. For example, we could incur substantial costs not covered by our directors’ and officers’ liability insurance, suffer a significant adverse impact on our reputation and divert management’s attention and resources from other priorities, including the execution of business plans and strategies that are important to our ability to grow our business, any of which could have a material adverse effect on our business. In addition, any of these matters could require payments that are not covered by our available directors’ and officers’ liability insurance, which could have a material adverse effect on our operating results or financial condition. Additional similar lawsuits might be filed.
34
Our former laboratories used certain chemical and biological agents and compounds that may be deemed hazardous and we were subject to various safety and environmental laws and regulations. Our prior compliance with these laws and regulations may result in significant costs, which could materially reduce our ability to become profitable.
Until late 2013, we had laboratories that used hazardous materials, including chemicals and biological agents and compounds that could be dangerous to human health and safety or the environment. As appropriate, we stored these materials and wastes resulting from their use at our laboratory facility pending their ultimate use or disposal. We contracted with a third party to properly dispose of these materials and wastes. We were subject to a variety of federal, state and local laws and regulations governing the use, generation, manufacture, storage, handling and disposal of these materials and wastes. We may incur significant costs if we unknowingly failed to comply with environmental laws and regulations.
Political or social factors may delay or impair our ability to market our products.
Entolimod is being developed to treat radiation sickness, which is a disease that may be caused by terrorist acts. The political and social responses to terrorism have been highly charged and unpredictable. Political or social pressures may delay or cause resistance to bringing our products to market or limit pricing of our products, which would harm our business. Changes to favorable laws, such as the Project BioShield Act, could have a material adverse effect on our ability to generate revenue and could require us to reduce the scope of or discontinue our operations.
We hope to receive funding from U.S. or foreign government agencies for the development of Entolimod and our products. Changes in government budgets and agendas, however, have previously resulted in termination of our contract negotiations and may, in the future, result in future funding being decreased and de-prioritized, government contracts contain provisions that permit cancellation in the event that funds are unavailable to the government agency. Furthermore, we cannot be certain of the timing of any future funding and substantial delays or cancellations of funding could result from protests or challenges from third parties. If the U.S. government fails to continue to adequately fund R&D programs, we may be unable to generate sufficient revenues to continue development of Entolimod or continuation of our other operations. Similarly, if our pre-EUA submission for Entolimod is authorized by the FDA or we develop another product candidate that is approved by the FDA, but the U.S. government does not place sufficient orders for this product, our future business may be harmed.
Failure to comply with the United States Foreign Corrupt Practices Act and similar foreign laws could subject us to penalties and other adverse consequences.
We are required to comply with the United States Foreign Corrupt Practices Act, or FCPA, which prohibits U.S. companies from engaging in bribery or other prohibited payments to foreign officials for the purpose of obtaining or retaining business. Foreign companies, including some that may compete with us, are not subject to these prohibitions. Furthermore, foreign jurisdictions in which we operate may have laws that are similar to the FCPA to which we are or may become subject. This may place us at a significant competitive disadvantage. Corruption, extortion, bribery, pay-offs, theft and other fraudulent practices may occur from time to time in the foreign markets where we conduct business. Although we inform our personnel that such practices are illegal, we can make no assurance that our employees or other agents will not engage in illegal conduct for which we might be held responsible. If our employees or other agents are found to have engaged in such practices, we could suffer severe penalties and other consequences that may have a material adverse effect on our business, financial condition and results of operations.
The FCPA also obligates companies whose securities are listed in the United States to comply with certain accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries and to devise and maintain an adequate system of internal accounting controls for international operations.
35
Compliance with the FCPA and similar foreign anti-bribery laws is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, such anti-bribery laws present particular challenges in the biotech or pharmaceutical industry, because, in many countries, hospitals are operated by the government and doctors and other hospital employees may be considered foreign officials.
Our business is subject to changing regulations for corporate governance and public disclosure that has increased both our costs and the risk of noncompliance.
Each year, under Section 404 of the Sarbanes-Oxley Act, we are required to evaluate our internal controls systems in order to allow management to report on our internal controls as required by and to permit our independent registered public accounting firm to attest to our internal controls. As a result, we have incurred and will continue to incur additional expenses and divert our management’s time to comply with these regulations. In addition, if we cannot continue to comply with the requirements of Section 404 in a timely manner, we might be subject to sanctions or investigation by regulatory and quasi-governmental authorities, such as the SEC, the Public Company Accounting Oversight Board, or The NASDAQ Stock Market. Any such action could adversely affect our financial results and the market price of our common stock.
In addition, stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business.
RISKS RELATING TO OUR SECURITIES
The price of our common stock has been and could remain volatile, which may in turn expose us to securities litigation.
The market price of our common stock has historically experienced and may continue to experience significant volatility. From January 2013 through December 2013, the market price of our common stock, which is listed on the NASDAQ Capital Market, fluctuated from a high of $2.28 per share in the first quarter of 2013 to a low of $0.97 in the fourth quarter of 2013. Additionally, since December 2013 our stock has further fluctuated to a low of $0.63 per share. The listing of our common stock on the NASDAQ Capital Market does not assure that a meaningful, consistent and liquid trading market will exist, and in recent years, the market has experienced extreme price and volume fluctuations that have particularly affected the market prices of many smaller companies like us. Our common stock is thus subject to this volatility in addition to volatility caused by the occurrence of industry and company specific events. Factors that could cause fluctuations include, but are not limited to, the following:
| | • | | Our progress in developing and commercializing our products; |
| | • | | Price and volume fluctuations in the overall stock market from time to time; |
| | • | | Fluctuations in stock market prices and trading volumes of similar companies; |
| | • | | Actual or anticipated changes in our earnings or fluctuations in our operating results or in the expectations of securities analysts; |
| | • | | General economic conditions and trends; |
| | • | | Major catastrophic events; |
| | • | | Sales of large blocks of our stock; |
| | • | | Departures of key personnel; |
| | • | | Changes in the regulatory status of our product candidates, including results of our pre-clinical studies and clinical trials; |
| | • | | Status of contract and funding negotiations relating to our product candidates; |
36
| | • | | Events affecting our collaborators; |
| | • | | Announcements of new products or technologies, commercial relationships or other events by us or our competitors; |
| | • | | Regulatory developments in the U.S. and other countries; |
| | • | | Failure of our common stock to be listed or quoted on the NASDAQ Capital Market, other national market system or any national stock exchange; |
| | • | | Changes in accounting principles; and |
| | • | | Discussion of us or our stock price by the financial and scientific press and in online investor communities. |
As a result of the volatility of our stock price, we could be subject to securities litigation, which could result in substantial costs and divert management’s attention and company resources from our business.
Issuance of additional equity may adversely affect the market price of our stock.
We are currently authorized to issue 160,000,000 shares of common stock and 10,000,000 of preferred stock. As of December 31, 2013, 45,182,114 shares of our common stock were issued and outstanding and we had issued warrants to purchase 10,534,545 shares and had granted 5,564,833 options. On January 16, 2014, we issued an additional 5,737,706 shares of common stock and warrants to purchase 5,909,838 shares of common stock, which included 172,132 warrants issued to the placement agent. To the extent the shares of common stock are issued or options and warrants are exercised, holders of our common stock will experience dilution.
In the event of any future issuances of equity securities or securities convertible into or exchangeable for, common stock, holders of our common stock may experience dilution. Furthermore, our outstanding warrants contain provisions that, in certain circumstances, could result in the number of shares of common stock issuable upon the exercise of such warrants to increase and/or the exercise price of such warrants to decrease.
Moreover, our board of directors is authorized to issue preferred stock without any action on the part of our stockholders. Our board of directors also has the power, without stockholder approval, to set the terms of any such preferred stock that may be issued, including voting rights, conversion rights, dividend rights, preferences over our common stock with respect to dividends or if we liquidate, dissolve or wind up our business and other terms. If we issue preferred stock in the future that has preference over our common stock with respect to the payment of dividends or upon our liquidation, dissolution or winding up, or if we issue preferred stock with voting rights that dilute the voting power of our common stock, the market price of our common stock could decrease. Any provision permitting the conversion of any such preferred stock into our common stock could result in significant dilution to the holders of our common stock.
We also consider from time to time various strategic alternatives that could involve issuances of additional shares of common stock or shares of preferred stock, including but not limited to acquisitions and business combinations.
We have no plans to pay dividends on our common stock and investors may not receive funds without selling their common stock.
We have not declared or paid any cash dividends on our common stock, nor do we expect to pay any cash dividends on our common stock for the foreseeable future. We currently intend to retain any additional future earnings to finance our operations and growth and, therefore, we have no plans to pay cash dividends on our common stock at this time. Any future determination to pay cash dividends on our common stock will be at the discretion of our board of directors and will be dependent on our earnings, financial condition, operating results, capital requirements, any contractual restrictions, regulatory and other restrictions on the payment of dividends by our subsidiaries to us and other factors that our board of directors deems relevant.
37
Accordingly, investors may have to sell some or all of their common stock in order to generate cash from your investment. Investors may not receive a gain on their investment when they sell our common stock and may lose the entire amount of their investment.
Provisions in our charter documents and Delaware law may inhibit a takeover or impact operational control of our company, which could adversely affect the value of our common stock.
Our certificate of incorporation and bylaws, as well as Delaware corporate law, contain provisions that could delay or prevent a change of control or changes in our management that a stockholder might consider favorable. These provisions include, among others, prohibiting stockholder action by written consent, advance notice for raising business or making nominations at meetings of stockholders and the issuance of preferred stock with rights that may be senior to those of our common stock without stockholder approval. These provisions would apply even if a takeover offer may be considered beneficial by some of our stockholders. If a change of control or change in management is delayed or prevented, the market price of our common stock could decline.
RISKS RELATED TO CONDUCTING BUSINESS IN THE RUSSIAN FEDERATION
Political, economic and governmental instability in Russia could materially adversely affect our operations and financial results.
Political and economic relations between Russia and the United States, two of the jurisdictions in which we operate, are complex. Political, ethnic, religious, historical and other differences have, on occasion, given rise to tensions. The current situation in Ukraine and Crimea along with the response of the Russian and United States governments to this situation, have the potential to materially adversely affect our operations in Russia. In connection with the current situation in Ukraine, the United States has ordered sanctions against Russian and Crimean officials. While we do not anticipate that the current sanctions will materially affect our business, if further sanctions are ordered by the United States or other international interests, such sanction may materially adversely affect out operations in Russia.
These current events may negatively affect the Russian economy and have negatively affected the value of the Russian ruble relative to the U.S. dollar. Continuing fluctuations in the rates at which the U.S. dollar are exchanged into Russian rubles may result in both foreign currency transaction and translation losses. We are subject to exchange rate fluctuations as (i) CBLI exchanges dollar-denominated funds into ruble-denominated funds in order to conduct operations of our Russian-based subsidiary BioLab 612, (ii) Panacela, Incuron and BioLab 612 use their ruble-denominated funds to pay for services under dollar-denominated contracts, including payments to CBLI for services we provide to our subsidiaries, and (iii) the US dollar equivalent of ruble denominated assets and liabilities fluctuate from period-to-period causing us to record foreign currency translation adjustments which are reflected as a change in other comprehensive income (loss). As the dollar strengthens or weakens relative to the ruble, our ruble-denominated revenue and expenses decline or increase respectively, when translated into U.S. Dollars for financial reporting purposes. Should exchange rates in effect at the time of this filing as compared to early 2014 and 2013, continue throughout the year, we expect the exchange rates to reduce our revenues and expenses in 2014 compared to 2013, and we would also record other comprehensive losses on our ruble denominated assets and liabilities when translated into the US dollar. Additionally, the purchasing power of US dollar denominated services is reduced, such as those being provided in the US for Incuron’s Phase 2 trial of the intravenous application of CBL0137.
Even before the current events mentioned above, and since the early 1990s, Russia has sought to transform from a one-party state with a centrally planned economy to a democracy with a market economy. As a result of the sweeping nature of various reforms and the failure of some of them, the political system of Russia remains vulnerable to popular dissatisfaction, including demands for autonomy from particular regional and ethnic groups. Current and future changes in the Russian government, major policy shifts or lack of consensus between various branches of the government and powerful economic groups could disrupt or reverse economic and regulatory reforms. Furthermore, the Russian economy is vulnerable to market downturns and economic
38
slowdowns elsewhere in the world, and has experienced periods of considerable instability. Although the Russian economy showed positive trends until 2008, including annual increases in the gross domestic product, a relatively stable currency, strong domestic demand, rising real wages and a reduced rate of inflation, these trends were interrupted by the global financial crisis in late 2008, in which Russia experienced adverse economic and financial effects including a substantial decrease in the growth rate of gross domestic product, depreciation of local currency and a decline in domestic and international demand for its products and services. Economic instability in Russia could materially adversely affect our business, financial condition and results of operations.
Emerging markets, such as Russia, are subject to greater risks than more developed markets and financial turmoil in Russia could disrupt our business.
Investors in emerging markets, such as Russia, should be aware that these markets are subject to greater risks than more developed markets, including significant economic risks. For example, the Russian economy has periodically experienced high rates of inflation. According to The World Bank and Bloomberg, the annual inflation rate in Russia, as measured by the consumer price index, was 8.4% in 2011 and 5.1% in 2012. Periods of higher inflation may slow economic growth. Inflation also is likely to increase some of our costs and expenses including the costs for our subsidiaries to conduct business operations, including any outsourced product testing costs.
Prospective investors in our common stock should note that emerging markets are subject to rapid change and that the information set out in this Annual Report on Form 10-K about our operations in Russia may become outdated relatively quickly.
The legal system in Russia can create an uncertain environment for business activity, which could materially adversely affect our business and operations in Russia.
The legal framework to support a market economy remains new and in flux in Russia and, as a result, its legal system can be characterized by: inconsistencies between and among laws and governmental, ministerial and local regulations, orders, decisions, resolutions and other acts; gaps in the regulatory structure resulting from the delay in adoption or absence of implementing regulations; selective enforcement of laws or regulations, sometimes in ways that have been perceived as being motivated by political or financial considerations; limited judicial and administrative guidance on interpreting legislation; relatively limited experience of judges and courts in interpreting recent commercial legislation; a perceived lack of judicial and prosecutorial independence from political, social and commercial forces; inadequate court system resources; a high degree of discretion on the part of the judiciary and governmental authorities; and underdeveloped bankruptcy procedures that are subject to abuse.
In addition, as is true of civil law systems generally, judicial precedents generally have no binding effect on subsequent decisions. Not all legislation and court decisions in Russia are readily available to the public or organized in a manner that facilitates understanding. Enforcement of court orders can in practice be very difficult. All of these factors make judicial decisions difficult to predict and effective redress uncertain. Additionally, court claims and governmental prosecutions may be used in furtherance of what some perceive to be political or commercial aims.
In February 2014, a new law came into force amending the Russian Constitution and merging the Higher Arbitrary court with the Higher Court, creating a new Higher Court. As of the date of this Annual Report, the new Higher Court is in the process of being established. We cannot predict the effect of this merger on judicial practice and the Russian judicial system.
The untested nature of much of recent legislation in Russia and the rapid evolution of its legal system may result in ambiguities, inconsistencies and anomalies in the application and interpretation of laws and regulations. Any of these factors may affect our ability to enforce our rights under our contracts or to defend ourselves against claims by others, or result in our being subject to unpredictable requirements. These uncertainties also extend to
39
property rights and the expropriation or nationalization of any of our entities, their assets or portions thereof, potentially without adequate compensation, could materially adversely affect our business, financial condition and results of operations.
Changes in the tax system in Russia or the arbitrary or unforeseen application of existing rules could materially adversely affect our financial condition and results of operations.
There have been significant changes to the taxation system in Russia in recent years as the authorities have gradually replaced legislation regulating the application of major taxes such as corporate income tax, value added tax, corporate property tax and other taxes with new legislation. Tax authorities in Russia have also been aggressive in their interpretation of tax laws and their many ambiguities, as well as in their enforcement and collection activities. Technical violations of contradictory laws and regulations, many of which are relatively new and have not been subject to extensive application or interpretation, can lead to penalties. High-profile companies can be particularly vulnerable to aggressive application of unclear requirements. Many companies must negotiate their tax bills with tax inspectors who may demand higher taxes than applicable law appears to provide. Our Russian subsidiaries’ tax liabilities may become greater than the estimated amount that they have expensed to date and paid or accrued on the balance sheets, particularly if the tax benefits currently received in Russia are changed or removed. Any additional tax liability, as well as any unforeseen changes in tax laws, could materially adversely affect our future results of operations, financial condition or cash flows in a particular period.
In October 2006, the Supreme Arbitration Court of Russia issued a ruling that introduced the concept of an “unjustified tax benefit,” which is a benefit that may be disallowed for tax purposes. Specific examples cited by the court include benefits obtained under transactions lacking a business purpose (i.e., when the only purpose of a deal or structure is to derive tax benefits). The tax authorities have actively sought to apply this concept when challenging tax positions taken by taxpayers. Although the intention of the ruling was to combat tax abuse, in practice there is no assurance that the tax authorities will not seek to apply this concept in a broader sense than may have been intended by the court. In addition, the tax authorities and the courts have indicated a willingness to interpret broadly the application of criminal responsibility for tax violations.
The tax systems in Russia impose additional burdens and costs on our operations there and complicate our tax planning and related business decisions. For example, the tax environment in Russia has historically been complicated by contradictions in Russian tax law and tax laws are unclear in areas such as the deductibility of certain expenses. This uncertainty could result in a greater than expected tax burden and potentially exposes us to significant fines and penalties and enforcement measures, despite our best efforts at compliance. These factors raise the risk of a sudden imposition of arbitrary or onerous taxes on our operations in these countries. This could materially adversely affect our financial condition and results of operations.
Selective or arbitrary government action may have an adverse effect on our business and the value of our common stock.
Government authorities have a high degree of discretion in Russia and have at times exercised their discretion selectively or arbitrarily, without hearing or prior notice, and sometimes in a manner that is influenced by political or commercial considerations. The government also has the power, in certain circumstances, to interfere with the performance of, nullify or terminate contracts. Selective or arbitrary actions have included withdrawal of licenses, sudden and unexpected tax audits, criminal prosecutions and civil actions. Federal and local government entities have also used common defects in documentation as pretexts for court claims and other demands to invalidate and/or to void transactions, apparently for political purposes. We cannot assure you that regulators, judicial authorities or third parties will not challenge our compliance with applicable laws, decrees and regulations in Russia. Selective or arbitrary government action could have a material adverse effect on our business and on the value of our common stock.
40
Shareholder liability under Russian legislation could cause us to become liable for the obligations of our subsidiaries.
The Russian Civil Code and the Law on Limited Liability Companies generally provide that shareholders in a Russian limited liability company are not liable for the obligations of the company and bear only the risk of loss of their investment. This may not be the case, however, when one person, an effective parent, is capable of determining decisions made by another, an effective subsidiary. The effective parent bears joint and several responsibilities for transactions concluded by the effective subsidiary in carrying out these decisions in certain circumstances.
In addition, a parent is secondarily liable for an effective subsidiary’s debts if an effective subsidiary becomes insolvent or bankrupt as a result of the action or inaction of the parent. This is the case no matter how the parent’s capability to determine decisions of the effective subsidiary arises. For example, this liability could arise through ownership of voting securities or by contract. In these instances, other shareholders of the effective subsidiary may claim compensation for the effective subsidiary’s losses from the parent that caused the effective subsidiary to act or fail to act, knowing that such action or inaction would result in losses. Accordingly, in CBLI’s position as a parent, it could be liable in some cases for the debts of its effective subsidiaries. Although the total indebtedness of CBLI’s effective subsidiaries in Russia is currently immaterial, it is possible that CBLI could face material liability in this regard in the future, which could materially adversely affect our business and our results of operations.
Our majority-owned Russian subsidiaries can be forced into liquidation on the basis of formal noncompliance with certain legal requirements.
Our subsidiaries operate in Russia primarily through Incuron, BioLab 612, and the wholly-owned Russian subsidiary of Panacela, all of which were organized under the laws of the Russian Federation. Certain provisions of Russian law may allow a court to order the liquidation of a locally organized legal entity on the basis of its formal noncompliance with certain requirements during formation, reorganization or during its operations. Additionally, Russian corporate law allows the government to liquidate a company if its net assets fall below a certain threshold. Similarly, there have also been cases in Russia in which formal deficiencies in the establishment process of a legal entity or noncompliance with provisions of law have been used by courts as a basis for liquidation of a legal entity. Weaknesses in the legal systems of Russia create an uncertain legal environment, which makes the decisions of a court or a governmental authority difficult, if not impossible, to predict. If involuntary liquidation of either of the aforementioned entities were to occur, such liquidation could materially adversely affect our financial condition and results of operations.
Crime and corruption could disrupt our ability to conduct our business.
Political and economic changes in Russia in recent years have resulted in significant dislocations of authority. The local and international press has reported the existence of significant organized criminal activity, particularly in large metropolitan centers. In addition, the local and international press has reported high levels of corruption, including the bribing of officials for the purpose of initiating investigations by government agencies. Press reports have also described instances in which state officials have engaged in selective investigations and prosecutions to further the interests of the state and individual officials, as well as private businesses, including competitors and corporate raiders. Corruption in Russia is pervasive and, in some cases, is worsening. The government in Russia has recently pursued a campaign against corruption. However, there is no assurance that such laws or other laws enacted elsewhere will be applied with any effectiveness by the local authorities and the continuing effects of corruption, money laundering and other criminal activity could have a negative effect on the Russian economy and could materially adversely affect our business in Russia.
Item 1B. Unresolved Staff Comments
None.
41
Item 2. Description of Properties
Our corporate headquarters is located at 73 High Street, Buffalo, New York 14203. We have approximately 32,000 square feet of laboratory and office space under a twelve-year lease through June of 2019 with successive two-year renewals, of which 10,135 square feet has been subleased through the end of the lease period to BBL. Either party upon 90 days written notice to the other party can terminate the sublease. This space serves as the corporate headquarters and primary research facilities for us and U.S. corporate headquarters for Incuron and Panacela. In addition, we have less than 3,600 square feet under lease outside of the United States expiring at varying times through November 2014. We do not own any real property.
Item 3. Legal Proceedings
On February 20, 2014, Sabby Healthcare Volatility Master Fund, Ltd. and Sabby Volatility Warrant Master Fund, Ltd., two purchasers in the January 2014 sale of securities, brought suit in the U.S. District Court for the Southern District of New York against the Company in an action captioned Sabby Healthcare Volatility Master Fund, Ltd. v. Cleveland Biolabs, Inc., No. 14-cv-1055 (S.D.N.Y.). The plaintiffs allege that the Company misrepresented the state of its funding negotiations with BARDA during the period leading up to the sale of the Securities, and as a result, the plaintiffs were harmed when the Company’s stock price declined following the announcement that BARDA had terminated negotiations with the Company. The complaint asserts claims under Section 10(b) of the Securities Exchange Act of 1934 and SEC Rule 10b-5, as well as claims for fraudulent inducement, breach of contract, and indemnification. The plaintiffs seek $2 million, plus interest, attorney’s fees, and litigation costs.
In addition to the above stated litigation, from time to time we may be involved in lawsuits, claims and legal proceedings, including commercial and contract disputes, employment matters, product liability claims, environmental liabilities and intellectual property disputes.
Item 4. Mine Safety Disclosure
None.
42
PART II
Item 5: Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
STOCK EXCHANGE LISTING
Our common stock trades on The NASDAQ Capital Market under the symbol “CBLI.” We have not paid dividends on our common stock. We currently intend to retain all future income for use in the operation of our business and for future stock repurchases and, therefore, we have no plans to pay cash dividends on our common stock at this time.
STOCK PRICES
The following table sets forth the range of high and low sale prices on The NASDAQ Capital Market, for each quarter during 2013 and 2012. On March 14, 2014, the last reported sale price of our common stock was $0.67 per share.
| | | | | | | | |
2013 | | High | | | Low | |
First Quarter | | $ | 2.28 | | | $ | 1.30 | |
Second Quarter | | $ | 2.26 | | | $ | 1.43 | |
Third Quarter | | $ | 1.84 | | | $ | 1.46 | |
Fourth Quarter | | $ | 1.68 | | | $ | 0.97 | |
| | |
2012 | | High | | | Low | |
First Quarter | | $ | 4.06 | | | $ | 2.45 | |
Second Quarter | | $ | 2.57 | | | $ | 1.15 | |
Third Quarter | | $ | 2.95 | | | $ | 1.31 | |
Fourth Quarter | | $ | 2.76 | | | $ | 1.23 | |
STOCKHOLDERS
As of December 31, 2013, there were approximately 33 stockholders of record of our common stock. Because many of our shares are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of beneficial stockholders represented by these record holders.
UNREGISTERED SALE OF SECURITIES
During 2013, as consideration for consulting services provided, we issued an aggregate of 175,000 shares of our common stock to various consultants without registration in reliance on the exemptions afforded by Section 4(2) of the Securities Act of 1933, as amended, or the Securities Act, based in part on the representations made by such consultants.
On September 30, 2013, pursuant to the Loan and Security Agreement between us, our wholly owned subsidiary, Biolab 612, LLC, and Hercules Technology II, L.P., or Hercules, we issued to Hercules a warrant to purchase 156,250 shares of our common stock at an exercise price of $1.60 per share. The exercise price of the warrant may be adjusted downward if during the one-year period following the closing date, we sell and issue shares of common stock or convertible stock in a transaction not registered under the Securities Act at a price per share less than the exercise price. The warrant will expire five years from the date of the grant. We issued the warrant in reliance on the exemption from registration provided for under Section 4(2) of the Securities Act. We relied on the exemption from registration provided for under Section 4(2) of the Securities Act based in part on the representations made by Hercules, including the representations with respect to Hercules’ status as an accredited investor, as such term is defined in Rule 501(a) of the Securities Act, and Hercules’ investment intent with respect to the warrant and the underlying shares of common stock.
43
ISSUER PURCHASES OF EQUITY SECURITIES
We made no repurchases of our securities during the year ended December 31, 2013.
FIVE-YEAR COMPARATIVE STOCK PERFORMANCE GRAPH
The following graph compares the yearly percentage change in the cumulative total stockholder return on our common stock with the cumulative total return on the NASDAQ Composite Index and NASDAQ Biotechnology Index over the past five years. The comparisons assume $100 was invested on December 31, 2008 in our common stock, in the NASDAQ Composite Index and the NASDAQ Biotechnology Index, and assumes reinvestment of dividends, if any.

| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | Year Ended December 31, | |
| | | | | 2008 | | | 2009 | | | 2010 | | | 2011 | | | 2012 | | | 2013 | |
Cleveland BioLabs, Inc. | | Return % | | | | | | | 55.40 | | | | 118.13 | | | | (60.39 | ) | | | (53.50 | ) | | | (12.03 | ) |
| | Cum $ | | $ | 100.00 | | | $ | 155.40 | | | $ | 338.97 | | | $ | 134.27 | | | $ | 62.44 | | | $ | 54.93 | |
| | | | | | | |
Nasdaq Composite Total Returns | | Return % | | | | | | | 45.32 | | | | 18.02 | | | | (0.83 | ) | | | 17.45 | | | | 40.12 | |
| | Cum $ | | $ | 100.00 | | | $ | 145.32 | | | $ | 171.50 | | | $ | 170.08 | | | $ | 199.76 | | | $ | 279.90 | |
| | | | | | | |
NASDAQ Biotechnology Index | | Return % | | | | | | | 15.63 | | | | 15.01 | | | | 11.81 | | | | 31.91 | | | | 65.61 | |
| | Cum $ | | $ | 100.00 | | | $ | 115.63 | | | $ | 132.98 | | | $ | 148.69 | | | $ | 196.13 | | | $ | 324.80 | |
The stock price performance shown in this performance graph is not necessarily indicative of future price performance. Historical stock price information used to prepare this graph and table was obtained from NASDAQ OMX, a source we believe is reliable. However, we are not responsible for any errors or omissions in such information.
The material in this section is being furnished and shall not be deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act or otherwise subject to the liability of that section, nor shall the material in this section be deemed to be incorporated by reference in any registration statement or other document filed with the SEC under the Securities Act of 1933, except to the extent we specifically and expressly incorporate it by reference into such filing.
44
Item 6: Selected Financial Data
The following selected financial data has been derived from our audited financial statements. The information below is not necessarily indicative of the results of future operations and should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and Item 1A, “Risk Factors,” and the financial statements and related notes thereto included in Item 8, “Financial Statements and Supplementary Data” of this Form 10-K, in order to fully understand factors that may affect the comparability of the information presented below:
SELECTED FINANCIAL DATA
| | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, | |
(in thousands, except per share data) | | 2013 | | | 2012 | | | 2011 | | | 2010 | | | 2009 | |
Consolidated statements of operations data: | | | | | | | | | | | | | | | | | | | | |
Revenues: | | | | | | | | | | | | | | | | | | | | |
Government contract or grant | | $ | 8,488 | | | $ | 3,571 | | | $ | 8,790 | | | $ | 15,332 | | | $ | 12,696 | |
Commercial | | | — | | | | — | | | | — | | | | — | | | | 1,650 | |
| | | | | | | | | | | | | | | | | | | | |
Total revenues | | | 8,488 | | | | 3,571 | | | | 8,790 | | | | 15,332 | | | | 14,346 | |
Operating expenses (1) | | | 31,564 | | | | 33,617 | | | | 33,895 | | | | 26,069 | | | | 20,729 | |
Loss from operations | | | (23,076 | ) | | | (30,047 | ) | | | (25,105 | ) | | | (10,737 | ) | | | (6,383 | ) |
Other income (expense): | | | | | | | | | | | | | | | | | | | | |
Change in value of warrant liability | | | 2,864 | | | | 7,702 | | | | 19,822 | | | | (16,012 | ) | | | (6,268 | ) |
Other income (expense) | | | 83 | | | | (70 | ) | | | 53 | | | | 77 | | | | (175 | ) |
| | | | | | | | | | | | | | | | | | | | |
Total other income (expense) | | | 2,947 | | | | 7,632 | | | | 19,875 | | | | (15,935 | ) | | | (6,443 | ) |
| | | | | | | | | | | | | | | | | | | | |
Net loss | | | (20,129 | ) | | | (22,415 | ) | | | (5,230 | ) | | | (26,672 | ) | | | (12,826 | ) |
| | | | | |
Net loss attributable to noncontrolling interests | | | 2,866 | | | | 4,180 | | | | 1,216 | | | | 306 | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Net loss attributable to Cleveland BioLabs, Inc. | | | (17,263 | ) | | | (18,234 | ) | | | (4,014 | ) | | | (26,366 | ) | | | (12,826 | ) |
Dividends on convertible preferred stock | | | — | | | | — | | | | — | | | | — | | | | 616 | |
| | | | | | | | | | | | | | | | | | | | |
Net loss available to common stockholders | | $ | (17,263 | ) | | $ | (18,234 | ) | | $ | (4,014 | ) | | $ | (26,366 | ) | | $ | (13,442 | ) |
| | | | | | | | | | | | | | | | | | | | |
Net loss per share, basic and diluted | | $ | (0.38 | ) | | $ | (0.49 | ) | | $ | (0.12 | ) | | $ | (1.01 | ) | | $ | (0.82 | ) |
| | | | | | | | | | | | | | | | | | | | |
| |
| | | December 31, | |
(in thousands) | | 2013 | | | 2012 | | | 2011 | | | 2010 | | | 2009 | |
Consolidated balance sheet data: | | | | | | | | | | | | | | | | | | | | |
Cash and cash equivalents | | $ | 10,048 | | | $ | 25,652 | | | $ | 22,873 | | | $ | 10,919 | | | $ | 963 | |
Short-term investments | | | 306 | | | | 2,634 | | | | 5,520 | | | | 459 | | | | — | |
Total current assets | | | 11,157 | | | | 29,406 | | | | 31,010 | | | | 17,751 | | | | 4,735 | |
Total assets | | | 14,696 | | | | 32,010 | | | | 32,127 | | | | 19,887 | | | | 6,554 | |
Capital leases (current & noncurrent) | | | 91 | | | | 169 | | | | — | | | | — | | | | — | |
Long-term debt (current & noncurrent) | | | 7,473 | | | | — | | | | — | | | | — | | | | — | |
Stockholder’s equity (deficit) | | | 1,581 | | | | 20,486 | | | | 22,245 | | | | (12,500 | ) | | | (6,800 | ) |
| (1) | Operating expenses in 2013, 2012, 2011, 2010 and 2009 included employee stock-based compensation costs of $1.5, $2.5, $4.0, $6.7 million and $3.7 million, net of tax. |
45
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
This management’s discussion and analysis of financial condition and results of operations and other portions of this filing contains forward-looking information that involves risks and uncertainties. Our actual results could differ materially from those anticipated by the forward-looking information. Factors that may cause such differences include, but are not limited to, availability and cost of financial resources, results of our R&D efforts and clinical trials, product demand, market acceptance and other factors discussed in this annual report in Item 1A, “Risk Factors” and the Company’s other Securities and Exchange Commission, or SEC, filings. The following discussion should be read in conjunction with our financial statements and the related notes included elsewhere in this filing.
OVERVIEW
We are an innovative drug development company seeking to develop first-in-class pharmaceuticals designed to address diseases with significant unmet medical need. Our lead product candidates are Entolimod, which we are developing as a radiation countermeasure and an oncology drug, and Curaxin CBL0137, our lead oncology product candidate. We conduct business in the United States and in the Russian Federation through several legal entities, some of which are majority-owned in collaboration with financial partners. See Item 1, “Business” for more information on our product candidates and our strategic partnerships. We refer to Cleveland BioLabs, Inc. or CBLI, along with our wholly-owned subsidiary BioLab 612, LLC, or BioLab 612, as CBLI Stand-alone. We refer to CBLI Stand-alone, in combination with, our majority-owned subsidiaries Incuron and Panacela, as CBLI Consolidated.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect our reported amounts of assets, liabilities, revenues and expenses.
On an ongoing basis, we evaluate our estimates and judgments, including those related to accrued expenses, income taxes, stock-based compensation, investments and in-process research and development. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the reported amounts of revenues and expenses that are not readily apparent from other sources. Actual results may differ from these estimates.
We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our financial statements.
Revenue Recognition
Revenue is recognized when persuasive evidence of an arrangement exists, delivery has occurred, the fee is fixed and determinable, collectability is reasonably assured, contractual obligations have been satisfied and title and risk of loss have been transferred to the customer. We generate our revenue from two different types of contractual arrangements: cost-reimbursable grants and contracts and fixed-price grants and contracts. Costs consist primarily of actual internal labor charges, subcontractor and material costs incurred, plus an allocation of fringe benefits, overhead and general and administrative expenses, based on the terms of the contract.
Revenues on cost-reimbursable grants and contracts are recognized in an amount equal to the costs incurred during the period, plus an estimate of the applicable fee earned. The estimate of the applicable fee earned is determined by reference to the contract: if the contract defines the fee in terms of risk-based milestones and specifies the fees to be earned upon the completion of each milestone, then the fee is recognized when the related milestones are earned. Otherwise, we compute fee income earned in a given period by using a proportional performance method based on costs incurred during the period as compared to total estimated project costs and application of the resulting fraction to the total project fee specified in the contract.
46
Revenues on fixed-price grants and contracts are recognized using a percentage-of-completion method, which uses assumptions and estimates, as appropriate. These assumptions and estimates are developed in coordination with the principal investigator performing the work under the fixed-price grants to determine levels of accomplishments throughout the life of the grant.
Stock-Based Compensation
We expense all share-based awards to employees and consultants, including grants of stock options and shares, based on their estimated fair value at the date of grant. Costs of all share-based payments are recognized over the requisite service period that an employee or consultant must provide to earn the award (i.e., the vesting period) and allocated to the functional operating expense associated with that employee or consultant.
Fair Value of Financial Instruments
The carrying value of cash and cash equivalents, accounts receivable, short-term investments, accounts payable and accrued expenses approximates fair value due to the relatively short maturity of these instruments. Common stock warrants, which are classified as liabilities, are recorded at their fair market value as of each reporting period.
The measurement of fair value requires the use of techniques based on observable and unobservable inputs. Observable inputs reflect market data obtained from independent sources, while unobservable inputs reflect our market assumptions. The inputs create the following fair value hierarchy:
| | • | | Level 1 – Quoted prices for identical instruments in active markets. |
| | • | | Level 2 – Quoted prices for similar instruments in active markets; quoted prices for identical or similar instruments in markets that are not active; and model-derived valuations where inputs are observable or where significant value drivers are observable. |
| | • | | Level 3 – Instruments where significant value drivers are unobservable to third parties. |
We use the Black-Scholes model to determine the fair value of certain common stock warrants on a recurring basis and classify such warrants in Level 3. The Black-Scholes model utilizes inputs consisting of: (i) the closing price of our common stock; (ii) the expected remaining life of the warrants; (iii) the expected volatility using a weighted-average of historical volatilities of CBLI and a group of comparable companies; and (iv) the risk-free market rate.
As of December 31, 2013, we held approximately $1.5 million in accrued expenses classified as Level 3 securities for warrants to purchase common stock and for compensatory stock options not yet issued.
Income Taxes
Determining the consolidated provision for income tax expense, deferred tax assets and liabilities and related valuation allowance, if any, involves judgment. On an on-going basis, we evaluate whether a valuation allowance is needed to reduce our deferred income tax assets to an amount that is more likely than not to be realized. The evaluation process includes assessing historical and current results in addition to future expected results. Upon determining that we would be able to realize our deferred tax assets, an adjustment to the deferred tax valuation allowance would increase income in the period we make such determination.
Revenue
Our revenue originates from grants and contracts from both United States federal and state government sources and Russian Federation government sources. U.S. federal grants and contracts are provided to advance research and development for product candidates that are of interest for potential sale to DoD or BARDA. State grants are usually designed to stimulate economic activity. Russian government contracts are provided to develop the biotechnology and pharmaceutical industries in Russia.
47
Research and Development Expenses
Research and development, or R&D, costs are expensed as incurred. Advance payments are deferred and expensed as performance occurs. R&D costs include the cost of our personnel consisting of salaries, incentive and stock-based compensation, out-of-pocket pre-clinical and clinical trial costs usually associated with contract research organizations, drug product manufacturing and formulation and a pro-rata share of facilities expense and other overhead items.
General and Administrative Expenses
General and administrative, or G&A, functions include executive management, finance and administration, government affairs and regulations, corporate development, human resources, legal and compliance. The specific costs include the cost of our personnel consisting of salaries, incentive and stock-based compensation, out-of-pocket costs usually associated with attorneys (both corporate and intellectual property), bankers, accountants and other advisors and a pro-rata share of facilities expense and other overhead items.
Other Income and Expenses
Other income and expenses primarily consists of interest income on our investments, changes in the market value of our derivative financial instruments and foreign currency transaction gains or losses.
YEAR ENDED DECEMBER 31, 2013 COMPARED TO YEAR ENDED DECEMBER 31, 2012
Revenue
Revenue increased by $4.9 million to $8.5 million for the year ended December 31, 2013 from $3.6 million for the year ended December 31, 2012, representing an increase of 138%. This increase consisted of increases of $1.0 million from U.S. government contracts and $3.9 million from Russian government contracts, primarily due to increased development activities under Russian government contracts, including two new contracts from MPT that were awarded in the fourth quarter of 2013 for development of an oncology application of Entolimod and Mobilan.
The following table sets forth details regarding the sources of our government grant and contract revenue:
| | | | | | | | | | | | | | |
Funding Source | | Program | | 2013 | | | 2012 | | | Variance | |
DoD | | MCS Contract (1) | | $ | 1,511,812 | | | $ | 1,113,830 | | | $ | 397,982 | |
MPT | | CBLB622 Pre-clinical (2) | | | 1,065,454 | | | | 888,686 | | | | 176,768 | |
MPT | | CBLB502 Colorectal Cancer (2) | | | 937,499 | | | | — | | | | 937,499 | |
DoD | | DTRA Contract | | | 765,096 | | | | 130,149 | | | | 634,947 | |
| | | | | | | | | | | | | | |
| | | | | 4,279,861 | | | | 2,132,665 | | | | 2,147,196 | |
Skolkovo Foundation | | Curaxin research (2) | | | 2,060,080 | | | | 488,781 | | | | 1,571,299 | |
MPT | | Xenomycins Pre-clinical (2) | | | 1,210,526 | | | | 949,264 | | | | 261,262 | |
MPT | | Mobilan Pre-clinical (2) | | | 937,499 | | | | — | | | | 937,499 | |
| | | | | | | | | | | | | | |
| | | | $ | 8,487,966 | | | $ | 3,570,710 | | | $ | 4,917,256 | |
| | | | | | | | | | | | | | |
| (1) | The Medical Countermeasure Systems, or MCS, Contract was formerly known as the Chemical Biological Medical Systems-Medical Identification and Treatment Systems, or CBMS-MITS Contract. |
| (2) | The contracts received from Russian government entities are denominated in Russian Rubles (RUR). The revenue above was calculated using average exchange rates for the periods presented. |
48
We anticipate our revenue over the next year will continue to be derived primarily from government grants and contracts. The following table sets forth information regarding our currently active contracts:
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | As of December 31, 2013 | |
Funding Source | | Program | | Total Award
Value | | | Funded Award
Value | | | Cumulative
Revenue
Recognized | | | Funded
Backlog | | | Unfunded
Backlog | |
MPT | | CBLB612 Pre-clinical (1) | | $ | 4,325,111 | | | $ | 2,974,635 | | | $ | 1,954,140 | | | $ | 1,020,495 | | | $ | 1,350,476 | |
MPT | | Entolimod Colorectal Cancer (1) | | | 4,584,090 | | | | 3,368,050 | | | | 937,499 | | | | 2,430,551 | | | | 1,216,040 | |
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | 8,909,201 | | | | 6,342,685 | | | | 2,891,639 | | | | 3,451,046 | | | | 2,566,516 | |
Skolkovo Foundation | | Curaxin research (1) | | | 4,706,983 | | | | 4,706,983 | | | | 3,637,545 | | | | 1,069,438 | | | | — | |
MPT | | Xenomycins Pre-clinical (1) | | | 4,558,260 | | | | 3,381,940 | | | | 2,159,790 | | | | 1,222,150 | | | | 1,176,320 | |
MPT | | Mobilan Pre-clinical (1) | | | 4,584,090 | | | | 3,368,050 | | | | 937,499 | | | | 2,430,551 | | | | 1,216,040 | |
| | | | | | | | | | | | | | | | | | | | | | |
| | | | $ | 22,758,534 | | | $ | 17,799,658 | | | $ | 9,626,473 | | | $ | 8,173,185 | | | $ | 4,958,876 | |
| | | | | | | | | | | | | | | | | | | | | | |
| (1) | The contract values above are calculated based on the cumulative revenue recognized to date plus our backlog valued at the December 31, 2013 exchange rate. Since December 31, 2013, the Russian Ruble exchange rate has increased from $32.2792 to $36.2618 as of March 11, 2014. Based on the March 11, 2014 exchange rate, the funded backlog value decreased from $8.2 million to $7.4 million and the unfunded backlog value decreased from $5.0 million to $4.5 million. |
Research and Development Expenses
R&D expenses decreased by $3.0 million to $19.5 million for the year ended December 31, 2013 from $22.5 million for the year ended December 31, 2012, representing a decrease of 13%. This net decrease primarily reflected decreases of $2.7 million related to Entolimod’s biodefense indication, as the development in 2013 focused on a less expensive, non-irradiated non-human primate study, and $1.6 million related to a narrowed scope of development for the compounds under development by Panacela. These decreases were partially offset by an increase of $1.2 million related to our Curaxin spending, primarily due to the initiation of a clinical trial in the United States for CBL0137. The following table sets forth our R&D expenses by drug candidate:
| | | | | | | | | | | | |
| | | 2013 | | | 2012 | | | Variance | |
Entolimod for Biodefense Applications | | $ | 9,337,962 | | | $ | 11,986,020 | | | $ | (2,648,058 | ) |
CBLB612 | | | 1,149,098 | | | | 1,039,832 | | | | 109,266 | |
Entolimod for Oncology Indications | | | 628,797 | | | | 605,365 | | | | 23,432 | |
| | | | | | | | | | | | |
| | | 11,115,857 | | | | 13,631,217 | | | | (2,515,360 | ) |
Curaxins | | | 4,459,854 | | | | 3,276,866 | | | | 1,182,988 | |
Panacela product candidates | | | 3,950,239 | | | | 5,593,722 | | | | (1,643,483 | ) |
| | | | | | | | | | | | |
Total research & development expenses | | $ | 19,525,950 | | | $ | 22,501,805 | | | $ | (2,975,855 | ) |
| | | | | | | | | | | | |
On September 30, 2013, we transferred 26 laboratory and pre-clinical employee positions to BBL an entity owned in part by our Chief Scientific Officer and director, Dr. Andrei Gudkov, to enable us to better focus on clinical development activities. In connection with this transition, we entered into a Master Services Agreement, or MSA, with BBL, pursuant to which BBL agreed to perform laboratory and pre-clinical research services for us. We plan to engage BBL for pre-clinical research services in the future. The prices for these services are based on an evaluation of detailed proposals prepared for us by BBL to support the activities to be conducted.
In connection with the above described restructuring, we recorded $0.1 million for one-time termination costs, $0.1 million for an idle facilities reserve and $0.3 million for an impairment loss as R&D expense for the year ended December 31, 2013. In addition, we recognized $0.4 million in R&D expense related to BBL during the year ended December 31, 2013 to support pre-clinical activities. As a result of the restructuring, we estimate saving $2.5 million in payroll related expenses on an annual basis, which will be offset by any pre-clinical activities that we contract to BBL going forward.
49
General and Administrative Expenses
G&A costs increased by $0.9 million to $12.0 million for the year ended December 31, 2013 from $11.1 million for the year ended December 31, 2012, representing an increase of 8%. This net increase was primarily attributable to increases of $1.0 million related to our Russian-based subsidiaries, $0.4 million in corporate legal and intellectual property fees and $0.4 million due to a reduction in incentive tax refunds. These increases were partially offset by decreases of $0.7 million in business development expenses and $0.2 million in non-cash stock-based compensation.
Other Income and Expenses
Other income decreased by $4.7 million to $2.9 million for the year ended December 31, 2013 from $7.6 million for the year ended December 31, 2012, representing a decrease of 61%. The change in the fair market value of our stock yielded a change in the fair market value of our accrued warrant liability, which was the primary reason for this decrease.
YEAR ENDED DECEMBER 31, 2012 COMPARED TO YEAR ENDED DECEMBER 31, 2011
Revenue
Revenue decreased by $5.2 million to $3.6 million for the year ended December 31, 2012 from $8.8 million for the year ended December 31, 2011, representing a decrease of 59%. This decrease consisted of a reduction in $4.1 million of U.S. government contracts and grant revenues and $2.3 million of revenue from the NY State/RPCI Sponsored Research Agreement. These decreases were partially offset by an increase of $1.2 million in revenues from our Russian government grants.
The following table sets forth details regarding the sources of our government grant and contract revenue:
| | | | | | | | | | | | | | |
Funding Source | | Program | | 2012 | | | 2011 | | | Variance | |
DoD | | MCS Contract (1) | | $ | 1,113,830 | | | $ | 3,684,142 | | | $ | (2,570,312 | ) |
MPT | | CBLB622 Pre-clinical (2) | | | 888,686 | | | | — | | | | 888,686 | |
DoD | | DTRA Contract | | | 130,149 | | | | 1,462,417 | | | | (1,332,268 | ) |
NY State/RPCI | | Sponsored Research Agreement | | | — | | | | 2,317,218 | | | | (2,317,218 | ) |
HHS | | BARDA Contract | | | — | | | | 237,748 | | | | (237,748 | ) |
| | | | | | | | | | | | | | |
| | | | | 2,132,665 | | | | 7,701,525 | | | | (5,568,860 | ) |
MPT | | Xenomycins Pre-clinical (2) | | | 949,264 | | | | — | | | | 949,264 | |
Skolkovo Foundation | | Curaxin research (2) | | | 488,781 | | | | 1,088,684 | | | | (599,903 | ) |
| | | | | | | | | | | | | | |
| | | | $ | 3,570,710 | | | $ | 8,790,209 | | | $ | (5,219,499 | ) |
| | | | | | | | | | | | | | |
| (1) | The MCS Contract was formerly known as the CBMS-MITS Contract. |
| (2) | The contracts received from Russian government entities are denominated in Russian Rubles (RUR). The revenue above was calculated using average exchange rates for the periods presented. |
50
Research and Development Expenses
R&D expenses decreased by $0.3 million to $22.5 million for the year ended December 31, 2012 from $22.8 million for the year ended December 31, 2011, representing a decrease of 1%. The following table sets forth our R&D expenses for 2012 and 2011 by drug candidate, which illustrate a reduced development effort for Entolimod’s biodefense indication offset mainly by increased development efforts for Panacela compounds as Panacela became fully operational in the fourth quarter of 2011 and its first full year of operations was 2012.
| | | | | | | | | | | | |
| | | 2012 | | | 2011 | | | Variance | |
Entolimod for Biodefense Applications | | $ | 11,986,020 | | | $ | 17,294,937 | | | $ | (5,308,917 | ) |
CBLB612 | | | 1,039,832 | | | | 481,371 | | | | 558,461 | |
Entolimod for Oncology Indications | | | 605,365 | | | | 260,777 | | | | 344,588 | |
General | | | — | | | | 872,455 | | | | (872,455 | ) |
| | | | | | | | | | | | |
| | | 13,631,217 | | | | 18,909,540 | | | | (5,278,323 | ) |
Panacela product candidates | | | 5,593,722 | | | | 748,497 | | | | 4,845,225 | |
Curaxins | | | 3,276,866 | | | | 3,130,850 | | | | 146,016 | |
| | | | | | | | | | | | |
Total research & development expenses | | $ | 22,501,805 | | | $ | 22,788,887 | | | $ | (287,082 | ) |
| | | | | | | | | | | | |
General and Administrative Expenses
G&A expenses remained relatively unchanged for the years ended December 31, 2012 and 2011 at $11.1 million per year. Significant variances between periods included a $1.4 million increase in G&A costs associated with subsidiaries that were not active in the same periods in 2011, a $1.0 million increase in business development expenses and a $0.4 million increase in miscellaneous G&A costs, offset by a decrease of $1.2 million due to a non-cash charge regarding a change in estimates for patents costs recorded in 2011.
Other Income and Expenses
Other income decreased by $12.2 million to $7.6 million for the year ended December 31, 2012 from $19.8 million for the year ended December 31, 2011, a change of 62%. The change in the fair market value of our stock yielded a change in the fair market value of our accrued warrant liability which was the primary reason for this decrease.
Liquidity and Capital Resources
We incurred net losses of $135.6 million from our inception through December 31, 2013. Historically, we have not generated, and do not expect to generate, revenue from sales of product candidates in the immediate future. Since our founding in 2003, we have funded our operations through a variety of means:
| | • | | Through December 31, 2013, CBLI has raised $107.8 million of net equity capital, including amounts received from the exercise of options and warrants. CBLI has also received $5.8 million in net proceeds from the issuance of long-term debt instruments; |
| | • | | On January 16, 2014, CBLI received net proceeds of $6.4 million resulting from the issuance of 5,737,706 shares of common stock and warrants to purchase 5,909,838 shares of common stock, which included warrants to purchase 172,132 shares of common stock issued to the placement agent; |
| | • | | DoD and BARDA have funded grants and contracts totaling $44.6 million for the development of Entolimod as a radiation countermeasure; |
| | • | | Entities affiliated with the Russian Federation have awarded us contracts totaling $22.8 million through a series of awards of over $4.0 million each. All awards are valued based on revenue recognized to date, with the remaining backlog valued at the December 31, 2013 exchange rate. These contracts include a requirement for us to contribute matching funds, which we have satisfied or expect to satisfy with both the value of developed intellectual property at the time of award, incurred development |
51
| | expenses and future expenses. At December 31, 2013, $17.8 million of the awards were funded; $10.5 million was received, of which $1.1 million remains as deferred revenue. We expect to recognize the remaining funding from 2014 to 2015; |
| | • | | We have been awarded $4.0 million in grants and contracts not described above, all of which was recognized at December 31, 2013; |
| | • | | Incuron was formed to develop and commercialize our Curaxin product line, namely two compounds CBL0137 and CBL0102. BCP committed to contribute up to $16.7 million (based on the current exchange rate) of funding as development milestones were accomplished. To date, Incuron has received $11.7 million of funding from BCP. BCP’s remaining capital contribution of $5.0 million is due upon completion of certain developmental milestones, which we believe will occur in 2014. |
| | • | | Panacela was formed to develop and commercialize several pre-clinical compounds. Rusnano contributed $9.0 million at formation, a $1.5 million convertible loan and has an option to contribute up to $15.5 million of additional funding. CBLI contributed $3.0 million plus intellectual property at formation and has an option to contribute additional capital based on agreed-upon terms. |
At December 31, 2013, we had cash, cash equivalents and short-term investments of $10.4 million. Of that total, $2.4 million was restricted for the use of our majority-owned subsidiaries, leaving $8.0 million available to CBLI Stand-alone. We had an additional $2.9 million of restricted cash held for performance bonds in connection with our Russian contracts, which are classified as a long-term asset.
Operating Activities
Net cash used in operations increased by $2.5 million to $23.1 million for the year ended December 31, 2013 from $20.6 million for the year ended December 31, 2012. After adjusting for non-cash items, our net loss decreased by $6.0 million between the periods. This increase in operating cash flows was offset by an $8.5 million change in our working capital, primarily attributable to changes in deferred revenue received in 2012.
Investing Activities
Net cash provided by investing activities decreased by $0.8 million to $0.6 million for the year ended December 31, 2013 from $1.4 million for the year ended December 31, 2012. This change was primarily attributable to a reduction in short-term investment activity.
Financing Activities
Cash provided by financing activities decreased by $14.3 million to $7.3 million for the year ended December 31, 2013, from $21.5 million for the year ended December 31, 2012. This net decrease was comprised of a decrease in the amount of cash raised through the issuance of common stock of $15.7 million and a decrease in the amount of cash raised by our majority-owned subsidiaries of $5.9 million. These decreases were partially offset by debt issuance proceeds of $7.3 million.
Other
We have incurred cumulative net losses and expect to incur additional losses related to our research and development activities. We do not have commercial products and have limited capital resources. We will need additional funds to complete the development of our product candidates. Our plans with regard to these matters may include seeking additional capital through a combination of government contracts, collaborative agreements, strategic alliances, research grants and equity and debt financing. There is no assurance that we will be successful in obtaining additional financing on commercially reasonable terms or that we will be able to secure funding from anticipated government contracts and grants.
52
We believe that our funds as of December 31, 2013, combined with the net proceeds of $6.4 million from our January 2014 public offering and the cash flows from existing government grants and contracts, will be sufficient to fund our projected operating requirements into the first quarter of 2015. Our success is dependent upon commercializing our research and development programs and our ability to obtain adequate future financing. There can be no assurance that we will be able to obtain future financing or, if obtained, what the terms of such future financing may be, or that any amount that we are able to obtain will be adequate to support our working capital requirements until we achieve profitable operations. If we are unable to raise adequate capital and/or achieve profitable operations, future operations might need to be scaled back or discontinued. The financial statements do not include any adjustments relating to the recoverability of the carrying amount of recorded assets and liabilities that might result from the outcome of these uncertainties.
Contractual Obligations and Purchase Commitments
The following table summarizes our contractual obligations and purchase commitments as of December 31, 2013:
| | | | | | | | | | | | | | | | | | | | |
| | | Payments Due by Period | |
| | | Total | | | Less than
1 Year | | | 1-3
Years | | | 4-5
Years | | | More than
5 Years | |
Long-term debt obligations | | $ | 9,972,228 | | | $ | 931,727 | | | $ | 7,575,243 | | | $ | 1,465,258 | | | $ | — | |
Capital lease obligations | | | 98,953 | | | | 91,341 | | | | 7,612 | | | | — | | | | — | |
Operating lease obligations | | | 2,104,568 | | | | 471,925 | | | | 699,498 | | | | 742,097 | | | | 191,048 | |
Purchase obligations (1) | | | 1,024,477 | | | | 1,024,477 | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Total | | $ | 13,200,226 | | | $ | 2,519,470 | | | $ | 8,282,353 | | | $ | 2,207,355 | | | $ | 191,048 | |
| | | | | | | | | | | | | | | | | | | | |
| (1) | At December 31, 2013, we had approximately $1.0 million in committed purchase obligations with fixed and determinable terms. In addition to the above, we are party to several agreements to perform (i) clinical trials for Entolimod for patients with advanced cancer and the oral and intravenous administrations of CBL0137; and (ii) long-term stability testing of our product candidates. These agreements are contingent on future events, e.g. the rate of patient accrual, the duration of testing and sample testing and validation. These agreements can be cancelled by either party, in which case we would generally be liable for costs and expenses incurred during the closeout or winding down period. As such these contingent obligations, totaling approximately $3.9 million, are excluded in the table above. |
In addition to the above listed commitments, we may be required to match up to approximately $13.1 million in development funding over the next three years to fully realize the funding of our Russian grants.
Off-Balance Sheet Arrangements
The Company does not have any off-balance sheet arrangements except for the operating lease obligations described in Note 12, “Commitments and Contingencies,” of the consolidated financial statements.
Item 7A: Quantitative and Qualitative Disclosures about Market Risk
Our exposure to market risk is primarily confined to our cash and cash equivalents, short-term investments and certain warrants we account for as derivative instruments. Because of the short-term maturities of our cash and cash equivalents and short-term investments, we do not believe that an increase in market rates would have a significant impact on the realized value of our cash or short-term investments. At December 31, 2013, approximately 62% of our cash, cash equivalents and short-term investments were denominated in U.S. dollars and the balance of 38% was denominated in Russian Rubles. A 10% increase or decrease in the value Russian
53
Ruble exchange rate would have resulted in a corresponding increase or decrease in the value of our year-end cash, cash equivalents and short-term investments balance of approximately $0.4 million. Changes in our stock price could have a significant impact on the fair market value of certain warrants we have issued, which could result in an adverse or positive impact on our results of operations. At December 31, 2013, a 10% increase or decrease in our stock price would have resulted in a corresponding increase or decrease of approximately $0.3 million in our accrued warrant liability.
Item 8: Financial Statements and Supplementary Data
54

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of Cleveland BioLabs, Inc. and Subsidiaries
We have audited the accompanying consolidated balance sheets of CLEVELAND BIOLABS, INC. AND SUBSIDIARIES as of December 31, 2013 and 2012, and the related consolidated statements of operations, comprehensive income (loss), stockholders’ equity (deficit), and cash flows for each of the years in the three-year period ended December 31, 2013. We also have audited Cleveland BioLabs and Subsidiaries’ internal control over financial reporting as of December 31, 2013, based on criteria established inInternal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Cleveland BioLabs and Subsidiaries’ management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on these consolidated financial statements and an opinion on the company’s internal control over financial reporting based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
55
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Cleveland BioLabs, Inc. and Subsidiaries as of December 31, 2013 and 2012, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2013 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, Cleveland BioLabs, Inc. and Subsidiaries maintained, in all material respects, effective internal control over financial reporting as of December 31, 2013, based on criteria established inInternal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
MEADEN & MOORE, LTD.
Certified Public Accountants
Cleveland, Ohio
March 17, 2014
56
CLEVELAND BIOLABS, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
| | | | | | | | |
| | | December 31, | |
| | | 2013 | | | 2012 | |
| ASSETS | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 10,048,466 | | | $ | 25,652,083 | |
Short-term investments | | | 305,538 | | | | 2,633,944 | |
Accounts receivable | | | 458,391 | | | | 41,896 | |
Other current assets | | | 344,386 | | | | 1,078,040 | |
| | | | | | | | |
Total current assets | | | 11,156,781 | | | | 29,405,963 | |
| | |
Equipment, net | | | 457,912 | | | | 986,553 | |
Restricted cash | | | 2,921,724 | | | | 1,577,920 | |
Other long-term assets | | | 159,224 | | | | 39,597 | |
| | | | | | | | |
Total assets | | $ | 14,695,641 | | | $ | 32,010,033 | |
| | | | | | | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 794,397 | | | $ | 1,523,875 | |
Accrued expenses | | | 2,445,446 | | | | 2,410,592 | |
Deferred revenue | | | 1,069,438 | | | | 3,314,918 | |
Accrued warrant liability | | | 1,241,311 | | | | 4,105,659 | |
Current portion of note payable | | | 351,527 | | | | — | |
Current portion of capital lease obligation | | | 83,634 | | | | 71,679 | |
| | | | | | | | |
Total current liabilities | | | 5,985,753 | | | | 11,426,723 | |
| | |
Noncurrent portion of capital lease obligation | | | 7,522 | | | | 97,602 | |
Long-term debt | | | 7,121,388 | | | | — | |
| | |
Commitments and contingencies | | | — | | | | — | |
| | | | | | | | |
Total liabilities | | | 13,114,663 | | | | 11,524,325 | |
| | |
Stockholders’ equity: | | | | | | | | |
Preferred stock, $.005 par value; 10,000,000 shares authorized, 0 shares issued and outstanding as of December 31, 2013 and December 31, 2012, respectively | | | — | | | | — | |
Common stock, $.005 par value; 160,000,000 shares authorized, 45,182,114 shares issued and outstanding as of December 31, 2013; 80,000,000 shares authorized, 44,730,445 issued and outstanding as of December 31, 2012 | | | 225,911 | | | | 223,653 | |
Additional paid-in capital | | | 125,508,471 | | | | 123,864,830 | |
Accumulated other comprehensive income | | | 307,339 | | | | 546,473 | |
Accumulated deficit | | | (135,564,666 | ) | | | (118,301,789 | ) |
| | | | | | | | |
Total Cleveland BioLabs, Inc. stockholders’ (deficit) equity | | | (9,522,945 | ) | | | 6,333,167 | |
Noncontrolling interest in stockholders’ equity | | | 11,103,923 | | | | 14,152,541 | |
| | | | | | | | |
Total stockholders’ equity | | | 1,580,978 | | | | 20,485,708 | |
| | | | | | | | |
Total liabilities and stockholders’ equity | | $ | 14,695,641 | | | $ | 32,010,033 | |
| | | | | | | | |
See Notes to Consolidated Financial Statements
57
CLEVELAND BIOLABS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
| | | | | | | | | | | | |
| | | For the Years Ended December 31, | |
| | | 2013 | | | 2012 | | | 2011 | |
Revenues: | | | | | | | | | | | | |
Grants and contracts | | $ | 8,487,966 | | | $ | 3,570,710 | | | $ | 8,790,209 | |
| | | |
Operating expenses: | | | | | | | | | | | | |
Research and development | | | 19,525,950 | | | | 22,501,805 | | | | 22,788,887 | |
General and administrative | | | 12,038,775 | | | | 11,115,511 | | | | 11,106,493 | |
| | | | | | | | | | | | |
Total operating expenses | | | 31,564,725 | | | | 33,617,316 | | | | 33,895,380 | |
| | | | | | | | | | | | |
Loss from operations | | | (23,076,759 | ) | | | (30,046,606 | ) | | | (25,105,171 | ) |
| | | |
Other income: | | | | | | | | | | | | |
Interest and other income (expense) | | | 83,127 | | | | (70,015 | ) | | | 53,659 | |
Change in value of warrant liability | | | 2,864,348 | | | | 7,701,981 | | | | 19,821,787 | |
| | | | | | | | | | | | |
Total other income | | | 2,947,475 | | | | 7,631,966 | | | | 19,875,446 | |
| | | | | | | | | | | | |
Net loss | | | (20,129,284 | ) | | | (22,414,640 | ) | | | (5,229,725 | ) |
| | | | | | | | | | | | |
Net loss attributable to noncontrolling interests | | | 2,866,407 | | | | 4,180,498 | | | | 1,216,055 | |
| | | | | | | | | | | | |
Net loss attributable to Cleveland BioLabs, Inc. | | $ | (17,262,877 | ) | | $ | (18,234,142 | ) | | $ | (4,013,670 | ) |
| | | | | | | | | | | | |
Net loss available to common stockholders per share of common stock, basic and diluted | | $ | (0.38 | ) | | $ | (0.49 | ) | | $ | (0.12 | ) |
| | | | | | | | | | | | |
Weighted average number of shares used in calculating net loss per share, basic and diluted | | | 45,002,823 | | | | 37,388,847 | | | | 32,561,743 | |
| | | | | | | | | | | | |
See Notes to Consolidated Financial Statements
58
CLEVELAND BIOLABS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
| | | | | | | | | | | | |
| | | For the Years Ended December 31, | |
| | | 2013 | | | 2012 | | | 2011 | |
Net loss including noncontrolling interests | | $ | (20,129,284 | ) | | $ | (22,414,640 | ) | | $ | (5,229,725 | ) |
Other comprehensive income (loss): | | | | | | | | | | | | |
Foreign currency translation adjustment | | | (421,345 | ) | | | 797,558 | | | | 153,815 | |
| | | | | | | | | | | | |
Comprehensive loss including noncontrolling interests | | | (20,550,629 | ) | | | (21,617,082 | ) | | | (5,075,910 | ) |
Comprehensive loss attributable to noncontrolling interests | | | 3,048,618 | | | | 3,844,800 | | | | 1,177,397 | |
| | | | | | | | | | | | |
Comprehensive loss attributable to Cleveland BioLabs, Inc. | | $ | (17,502,011 | ) | | $ | (17,772,282 | ) | | $ | (3,898,513 | ) |
| | | | | | | | | | | | |
See Notes to Consolidated Financial Statements
59
CLEVELAND BIOLABS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2013 | | | 2012 | | | 2011 | |
Cash flows from operating activities: | | | | | | | | | | | | |
Net income (loss) | | $ | (20,129,284 | ) | | $ | (22,414,640 | ) | | $ | (5,229,725 | ) |
Adjustments to reconcile net income (loss) to net cash used in operating activities: | | | | | | | | | | | | |
Depreciation | | | 376,213 | | | | 479,595 | | | | 512,366 | |
Amortization | | | 94,995 | | | | — | | | | 13,147 | |
Unrealized currency loss on short-term investments | | | — | | | | 52,726 | | | | — | |
(Gain) loss on equipment disposal | | | (3,222 | ) | | | 18,997 | | | | — | |
Impairment loss on property and equipment | | | 293,162 | | | | — | | | | — | |
Noncash compensation | | | 1,473,449 | | | | 2,535,217 | | | | 4,044,858 | |
Warrant issuance costs | | | — | | | | 244,857 | | | | 150,827 | |
Change in value of warrant liability | | | (2,864,348 | ) | | | (7,701,981 | ) | | | (19,821,787 | ) |
Patent costs | | | — | | | | — | | | | 1,481,318 | |
Changes in operating assets and liabilities: | | | | | | | | | | | | |
Accounts receivable | | | (416,495 | ) | | | 1,736,199 | | | | 3,641,491 | |
Other current assets | | | 697,966 | | | | (182,428 | ) | | | 123,197 | |
Other long-term assets | | | (29,732 | ) | | | (6,414 | ) | | | (919 | ) |
Accounts payable | | | (713,502 | ) | | | 609,522 | | | | (348,282 | ) |
Deferred revenue | | | (2,062,332 | ) | | | 3,238,124 | | | | (2,317,218 | ) |
Accrued expenses | | | 180,483 | | | | 740,723 | | | | 845,337 | |
| | | | | | | | | | | | |
Net cash used in operating activities | | | (23,102,647 | ) | | | (20,649,503 | ) | | | (16,905,390 | ) |
Cash flows from investing activities: | | | | | | | | | | | | |
Purchase of short-term investments | | | — | | | | (5,220,781 | ) | | | (5,520,000 | ) |
Sale of short-term investments | | | 2,197,940 | | | | 8,312,120 | | | | 434,835 | |
Purchase of equipment | | | (139,491 | ) | | | (178,271 | ) | | | (655,553 | ) |
Increase in restricted cash | | | (1,497,740 | ) | | | (1,541,366 | ) | | | — | |
Investment in patents | | | — | | | | — | | | | (326,171 | ) |
| | | | | | | | | | | | |
Net cash provided by (used in) investing activities | | | 560,709 | | | | 1,371,702 | | | | (6,066,889 | ) |
Cash flows from financing activities: | | | | | | | | | | | | |
Issuance of common stock, net of offering costs | | | — | | | | 15,675,727 | | | | 21,946,801 | |
Net proceeds from issuance of debt | | | 7,327,675 | | | | — | | | | — | |
Noncontrolling interest capital contribution to Incuron, LLC | | | — | | | | 5,893,557 | | | | 2,340,374 | |
Noncontrolling interest capital contribution to Panacela Labs, Inc. | | | — | | | | — | | | | 9,000,066 | |
Exercise of options | | | 12,392 | | | | 2,375 | | | | 532,408 | |
Repayment of capital lease obligation | | | (78,125 | ) | | | (52,410 | ) | | | — | |
Exercise of warrants | | | — | | | | — | | | | 949,793 | |
| | | | | | | | | | | | |
Net cash provided by financing activities | | | 7,261,942 | | | | 21,519,249 | | | | 34,769,442 | |
Effect of exchange rate change on cash and cash equivalents | | | (323,621 | ) | | | 538,046 | | | | 156,889 | |
| | | | | | | | | | | | |
Increase (decrease) in cash and cash equivalents | | | (15,603,617 | ) | | | 2,779,494 | | | | 11,954,052 | |
Cash and cash equivalents at beginning of period | | | 25,652,083 | | | | 22,872,589 | | | | 10,918,537 | |
| | | | | | | | | | | | |
Cash and cash equivalents at end of period | | $ | 10,048,466 | | | $ | 25,652,083 | | | $ | 22,872,589 | |
| | | | | | | | | | | | |
Supplemental disclosure of cash flow information: | | | | | | | | | | | | |
Cash paid during the period for interest | | $ | 128,811 | | | $ | 23,708 | | | $ | — | |
Supplemental schedule of noncash financing activities: | | | | | | | | | | | | |
Fair value of warrants issued in connection with debt | | $ | 117,999 | | | $ | — | | | $ | — | |
Equipment acquired through lease financing | | $ | — | | | $ | 221,690 | | | $ | — | |
Conversion of warrant liability to equity upon warrant exercise | | $ | — | | | $ | — | | | $ | 995,428 | |
Noncash financing costs on common stock offering | | $ | — | | | $ | — | | | $ | 207,905 | |
Noncash warrant issuance costs | | $ | — | | | $ | — | | | $ | 19,361 | |
See Notes to Consolidated Financial Statements
60
CLEVELAND BIOLABS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY
(UNAUDITED)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Common Stock | | | Additional
Paid-in
Capital | | | Accumulated
Other
Comprehensive
Income (Loss) | | | Accumulated
Deficit | | | Noncontrolling
Interests | | | Total | |
| | | Shares | | | Amount | | | | | | |
Balance at December 31, 2010 | | | 28,959,176 | | | | 144,796 | | | $ | 80,241,717 | | | $ | (30,544 | ) | | $ | (96,053,977 | ) | | $ | 3,197,873 | | | $ | (12,500,135 | ) |
Issuance of common stock net of offering costs of $1,619,638 | | | 5,872,500 | | | | 29,363 | | | | 21,840,999 | | | | — | | | | — | | | | — | | | | 21,870,362 | |
Allocation of financing proceeds to fair value of warrants | | | — | | | | — | | | | (2,525,175 | ) | | | — | | | | — | | | | — | | | | (2,525,175 | ) |
Noncontrolling interest capital contribution to Incuron, LLC | | | — | | | | — | | | | 176,092 | | | | — | | | | — | | | | 2,164,282 | | | | 2,340,374 | |
Noncontrolling interest capital contribution to Panacela Labs, Inc. | | | — | | | | — | | | | — | | | | — | | | | — | | | | 9,000,066 | | | | 9,000,066 | |
Stock based compensation | | | 308,850 | | | | 1,544 | | | | 6,656,742 | | | | — | | | | — | | | | — | | | | 6,658,286 | |
Exercise of options | | | 190,255 | | | | 951 | | | | 531,457 | | | | — | | | | — | | | | — | | | | 532,408 | |
Exercise of warrants | | | 281,411 | | | | 1,407 | | | | 1,943,813 | | | | — | | | | — | | | | — | | | | 1,945,220 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | (4,013,670 | ) | | | (1,216,055 | ) | | | (5,229,725 | ) |
Foreign currency translation | | | — | | | | — | | | | — | | | | 115,157 | | | | — | | | | 38,658 | | | | 153,815 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2011 | | | 35,612,192 | | | | 178,061 | | | $ | 108,865,645 | | | $ | 84,613 | | | $ | (100,067,647 | ) | | $ | 13,184,824 | | | $ | 22,245,496 | |
| | | | | | | |
Issuance of common stock net of offering costs of $1,129,916 | | | 8,525,000 | | | | 42,625 | | | | 15,877,959 | | | | — | | | | — | | | | — | | | | 15,920,584 | |
Allocation of financing proceeds to fair value of warrants | | | — | | | | — | | | | (4,521,681 | ) | | | — | | | | — | | | | — | | | | (4,521,681 | ) |
Noncontrolling interest capital contribution to Incuron, LLC | | | — | | | | — | | | | 1,081,040 | | | | — | | | | — | | | | 4,812,517 | | | | 5,893,557 | |
Stock based compensation | | | 592,003 | | | | 2,961 | | | | 2,559,498 | | | | — | | | | — | | | | — | | | | 2,562,459 | |
Exercise of options | | | 1,250 | | | | 6 | | | | 2,369 | | | | — | | | | — | | | | — | | | | 2,375 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | (18,234,142 | ) | | | (4,180,498 | ) | | | (22,414,640 | ) |
Foreign currency translation | | | — | | | | — | | | | — | | | | 461,860 | | | | — | | | | 335,698 | | | | 797,558 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2012 | | | 44,730,445 | | | $ | 223,653 | | | $ | 123,864,830 | | | $ | 546,473 | | | $ | (118,301,789 | ) | | $ | 14,152,541 | | | $ | 20,485,708 | |
Stock based compensation | | | 441,988 | | | | 2,210 | | | | 1,513,299 | | | | — | | | | — | | | | — | | | | 1,515,509 | |
Exercise of options | | | 9,681 | | | | 48 | | | | 12,343 | | | | — | | | | — | | | | — | | | | 12,391 | |
Allocation of debt proceeds to fair value of warrants | | | — | | | | — | | | | 117,999 | | | | — | | | | — | | | | — | | | | 117,999 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | (17,262,877 | ) | | | (2,866,407 | ) | | | (20,129,284 | ) |
Foreign currency translation | | | — | | | | — | | | | — | | | | (239,134 | ) | | | — | | | | (182,211 | ) | | | (421,345 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2013 | | | 45,182,114 | | | $ | 225,911 | | | $ | 125,508,471 | | | $ | 307,339 | | | $ | (135,564,666 | ) | | $ | 11,103,923 | | | $ | 1,580,978 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
See Notes to Consolidated Financial Statements
61
CLEVELAND BIOLABS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Description of Business
Cleveland BioLabs, Inc. is an innovative drug development company seeking to develop first-in-class pharmaceuticals designed to address diseases with significant unmet medical need. Our programs are focused on the implementation of novel pharmacological approaches to control cell death. As used throughout these consolidated financial statements, the terms “Cleveland BioLabs” and “CBLI” refer to Cleveland BioLabs, Inc., but not its consolidated subsidiaries and “the Company,” “we,” “us” and “our” refer to Cleveland BioLabs, Inc. together with its consolidated subsidiaries.
CBLI was incorporated in Delaware in June 2003 and is headquartered in Buffalo, New York. CBLI has one wholly-owned operating subsidiary, BioLab 612, LLC, or BioLab 612, which began operations in 2012. CBLI has two majority-owned operating subsidiaries, Incuron, LLC, or Incuron, and Panacela Labs, Inc., or Panacela, which were formed in 2010 and 2011, respectively. Additionally, Panacela has a wholly-owned operating subsidiary, Panacela Labs, LLC.
2. Summary of Significant Accounting Policies
Basis of Presentation and Consolidation
The accompanying consolidated financial statements include the accounts of CBLI and its subsidiaries. All significant intercompany balances and transactions have been eliminated in consolidation. These financial statements have been prepared on the accrual basis in accordance with accounting principles generally accepted in the United States, or GAAP.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
Of the $10 million and $25.7 million of cash and cash equivalents at December 31, 2013 and December 31, 2012, respectively, $3.5 million and $13.0 million, respectively, consisted of highly liquid investments with maturities of 90 days or less when purchased. These investments consist of commercial paper, short-term debt securities, time deposits and investments in money market funds with commercial banks and financial institutions. As of December 31, 2013, $2.0 million of the Company’s cash and cash equivalents were restricted to the use of its majority-owned subsidiaries, leaving $8.0 million available for general use.
Short-Term Investments
The Company’s short-term investments are classified as held to maturity given the intent and ability to hold the investments to maturity. Accordingly, these investments are carried at amortized cost. Short-term investments classified as held-to-maturity consisted of certificates of deposit with maturity dates beyond three months and less than one year. As of December 31, 2013, all of the Company’s short-term investments were restricted for use by its majority-owned subsidiaries.
62
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to a significant concentration of credit risk primarily consist of cash and cash equivalents and short-term investments. The Company maintains cash balances with financial institutions in excess of insured limits. The Company does not believe it is exposed to significant credit risk due to the financial position of the depository institutions in which those deposits are held.
As of December 31, 2013, the Company held 36% of its cash and cash equivalents and 100% of its short-term investments in accounts located outside of the United States.
As of March 11, 2014, the Russian Ruble exchange rate increased from $32.2792 to $36.2618, resulting in a decrease of $350,557 to the Company’s cash and cash equivalents and $29,765 to the Company’s short-term investment balances as compared to December 31, 2013.
Significant Customers and Accounts Receivable
Grant and contract revenue from the United States government accounted for 26.8%, 34.8% and 87.6% of total revenue for the years ended December 31, 2013, 2012 and 2011, respectively. The Company anticipates that its sources of revenue in the near future will be from development contracts from the United States and Russian federal governments, however there is no guarantee that this revenue stream will continue in the future.
Grant and contract revenue received by our subsidiaries from Russian government agencies accounted for 73.2%, 65.2% and 12.4% of total revenues for the years ended December 31, 2013, 2012 and 2011, respectively.
Accounts receivable consist of amounts due under contracts with certain government and foreign entities. The Company extends unsecured credit to its government customers under normal trade agreements and contracted terms, which generally require payment within 30 days.
Management estimates an allowance for doubtful accounts that is based upon management’s review of delinquent accounts and an assessment of the Company’s historical evidence of collections. There were no allowances for doubtful accounts as of December 31, 2013 and 2012, as the collection history from the Company’s customers indicated that collection was probable.
Equipment
Equipment is stated at cost, net of accumulated depreciation. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts and any resulting gain or loss is credited or charged to operations. Repair and maintenance costs are expensed as incurred.
Equipment is depreciated using the straight-line method over the estimated useful lives of the respective assets as follows:
| | | | |
Asset Category | | Estimated Useful Life
(in Years) | |
Laboratory equipment | | | 5 | |
Furniture and fixtures | | | 5 | |
Computer equipment | | | 3 | |
Impairment of Long-Lived Assets
Long-lived assets to be held and used are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amounts of the assets or related asset group may not be recoverable. Determination of recoverability is based on an estimate of discounted future cash flows resulting from the use of the asset. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the asset or asset group, the carrying amount of the asset is written down to its estimated net realizable value.
63
In connection with the restructuring discussed in Note 8, “Restructuring,” the Company entered into an equipment lease agreement with Buffalo BioLabs, or BBL, effective January 1, 2014 for the equipment used by the employees transitioned to BBL. The estimated fair value of those assets using a discounted cash flow approach was $243,000. Comparing this fair value to the carrying amount of the equipment as of December 31, 2013 of $536,000 resulted in a $293,000 write-down of the assets, which was included in research and development expenses for the year ended December 31, 2013.
Restricted Cash
Restricted cash at December 31, 2013 includes certificates of deposit denominated in Russian Rubles and posted by Panacela and BioLab 612 as collateral for performance guarantees for their contracts with the Ministry of Industry and Trade of the Russian Federation. The guarantees require Panacela and BioLab 612 to satisfactorily perform their statements of work under the contracts. Both Panacela and BioLab 612 anticipate receiving the full proceeds of their deposits at the completion of the contracts, which for each contract is more than a year away. As a consequence, all of the Company’s restricted cash is classified as a noncurrent asset.
Intellectual Property
Costs related to filing and pursuing patent applications are recognized as general and administrative expenses as incurred, since the recoverability of such expenditures is uncertain. Upon marketability approval by the U.S. Food and Drug Administration, or FDA, or a respective foreign governing body, such costs will be capitalized and depreciated over the expected life of the related patent.
During the year-ended December 31, 2011, the Company performed its periodic review of capitalized patent costs and incorporated a more restrictive standard of capitalization widely utilized in the biotechnology industry, which includes a prerequisite of the FDA marketability approval as one of several factors needed to justify the continued capitalization of costs associated with securing patents. Given that the Company is currently developing requisite data towards submission to the FDA of biological license and new drug applications in support of its existing product candidates, capitalized patent costs of approximately $1,500,000 were expensed during the year ended December 31, 2011. This item has been treated as a change in estimate in the accompanying financial statements.
Deferred Revenue
Deferred revenue represents cash received under grants and contracts in excess of the revenue recognizable through the end of the respective financial reporting period. The revenue associated with these advances will be recognized in future periods as the applicable costs are incurred.
Accrued Warrant Liability
Certain warrants are accounted for as derivative instruments in accordance with the Financial Accounting Standards Board Accounting Standards Codification, or the Codification, on derivatives and hedging as the warrant holders, under certain change of control situations, could require settlement in cash. As such, the warrants were initially recorded as liabilities based on their fair values on the date of issuance. Subsequent changes in the value of the warrants are recorded in the statements of operations as “Change in value of warrant liability.”
The Company’s remaining outstanding warrants were treated as equity upon issuance and continue to be treated as equity since they did not contain any mandatory redemption features or other provisions that would require a different classification of these warrant instruments outside of permanent equity.
64
Foreign Currency Translation
The Russian ruble is the functional currency of our foreign subsidiaries, which are all located in the Russian Federation. Assets and liabilities of these subsidiaries are translated into U.S. dollars at the period-end exchange rate. Income and expense items are translated at the average exchange rates during the period. The net effect of this translation is recorded in the consolidated financial statements as accumulated other comprehensive income (loss).
Revenue Recognition
The Company generates grant and contract revenue from two different types of contractual arrangements: cost reimbursable grants and contracts and fixed-price grants and contracts. Costs consist primarily of internal labor charges, subcontractors and materials, as well as an allocation of fringe benefits, overhead and general and administrative expenses, based on the terms of the contract. Under cost reimbursable grants and contracts, revenue is recognized during the period that the associated research and development costs are incurred. Under fixed-price grants and contracts, revenue is recognized using the percentage-of-completion method. The assumptions and estimates used in determination of the percentage-of-completion are developed in coordination with the principal investigator performing the work.
Research and Development
Research and development costs are expensed as incurred. Research and development costs primarily consist of salaries, fringe benefits, materials and related expenses for personnel and facility expenses. Other research and development expenses include fees paid to consultants and outside service providers, the costs of materials used in clinical trials and research and development and stock-based compensation.
Accounting for Stock-Based Compensation
The 2006 Equity Incentive Plan, as amended, or the “Plan”, authorizes CBLI to grant (i) options to purchase common stock, (ii) restricted or unrestricted stock units, and (iii) stock appreciation rights, so long as the exercise or grant price of each are at least equal to the fair market value of the stock on the date of grant. At the 2012 annual meeting of stockholders, an amendment to increase the maximum number of shares of common stock reserved for issuance under the Plan was approved, and as of December 31, 2013, an aggregate of 10.0 million shares of common stock were authorized for issuance under the Plan, of which a total of approximately 2.1 million shares of common stock remained available for future awards. A single participant cannot be awarded more than 400,000 shares annually. Awards granted under the Plan have a contractual life of no more than 10 years. The terms and conditions of equity awards (such as price, vesting schedule, term and number of shares) under the Plan are specified in an award document, and approved by compensation committee of the CBLI’s board of directors.
The Company utilizes the Black-Scholes valuation model for estimating the fair value of all stock options granted. Set forth below are the assumptions used in valuing the stock options granted and a discussion of the Company’s methodology for developing each of the assumptions used:
| | | | | | | | | | | | |
| | | For the year ended December 31, | |
| | | 2013 | | | 2012 | | | 2011 | |
Risk-free interest rate | | | .02 - 1.92 | % | | | .65 - 1.49 | % | | | .96 - 2.61 | % |
Expected dividend yield | | | 0 | % | | | 0 | % | | | 0 | % |
Expected life | | | 5 - 7.3 Years | | | | 5 - 6 Years | | | | 5 - 6 Years | |
Expected volatility | | | 78.62 - 89.66 | % | | | 86.58 - 92.60 | % | | | 84.28 - 92.38 | % |
“Risk-free interest rate” means the range of U.S. Treasury rates with a term that most closely resembles the expected life of the option as of the date the option is granted.
65
“Expected dividend yield” means the Company does not pay regular dividends on its common stock and does not anticipate paying any dividends in the foreseeable future.
“Expected life” means the period of time that options granted are expected to remain outstanding, based wholly on the use of the simplified (safe harbor) method. The simplified method is used because the Company does not yet have adequate historical exercise information to estimate the expected life the options granted.
“Expected volatility” means a measure of the amount by which a financial variable, such as share price, has fluctuated (historical volatility) or is expected to fluctuate (implied volatility) during a period. Expected volatility is based on the Company’s historical volatility and incorporates the volatility of the common stock of comparable companies when the expected life of the option exceeds the Company’s trading history.
In June 2013, CBLI’s stockholders approved the 2013 Employee Stock Purchase Plan, or ESPP, which provides a means by which eligible employees of CBLI, and certain designated related corporations may be given an opportunity to purchase shares of CBLI common stock. As of December 31, 2013, there are 2.1 million shares of common stock reserved for purchase under the ESPP. The number of shares reserved for purchase under the ESPP increases on January 1 of each calendar year by the lesser of (i) 10% of the total number of shares of common stock outstanding on December 31 of the preceding year, or (ii) 200,000 shares of common stock. The ESPP allows employees to use up to 15% of their compensation to purchase shares of common stock at an amount equal to 85% of the fair market value of the our common stock on the offering date or the purchase date, whichever is less.
Income taxes
No income tax expense was recorded for the years ended December 31, 2013, 2012 and 2011, as the Company did not have taxable income for any of the years presented. A full valuation allowance has been recorded against the Company’s net deferred tax asset.
Earnings/(loss) per share
Basic net income (loss) per share of common stock excludes dilution for potential common stock issuances and is computed by dividing net income (loss) by the weighted average number of shares outstanding for the period. Diluted net income (loss) per share reflects the potential dilution that could occur if securities or other contracts to issue common stock were exercised or converted into common stock. Diluted net loss per share is identical to basic net loss per share as potentially dilutive securities have been excluded from the calculation of diluted net loss per common share because the inclusion of such securities would be antidilutive.
The Company has excluded the following outstanding warrants and options from the calculation of diluted net loss per share because all such securities were antidilutive for the periods presented:
| | | | | | | | | | | | |
| | | As of December 31, | |
Common Equivalent Securities | | 2013 | | | 2012 | | | 2011 | |
Preferred Shares | | | — | | | | — | | | | — | |
Warrants | | | 10,534,245 | | | | 10,377,995 | | | | 12,564,193 | |
Options | | | 5,564,833 | | | | 5,016,966 | | | | 4,117,979 | |
| | | | | | | | | | | | |
Total | | | 16,099,078 | | | | 15,394,961 | | | | 16,682,172 | |
| | | | | | | | | | | | |
Comprehensive Income (Loss)
The Company applies the Codification on comprehensive income (loss) that requires disclosure of all components of comprehensive income (loss) on an annual and interim basis. Comprehensive income (loss) is defined as the change in equity of a business enterprise during a period from transactions and other events and circumstances from non-owner sources.
66
Recently Issued Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board or other standard setting bodies that are adopted by the Company as of the specified effective date. Unless otherwise discussed, the Company believes that the impact of recently issued standards that are not yet effective will not have a material impact on its financial position or results of operations upon adoption.
3. Fair Value Measurements
The Company measures and records cash equivalents and warrant liabilities at fair value in the accompanying financial statements. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability, an exit price, in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value include:
| | • | | Level 1 – Observable inputs for identical assets or liabilities such as quoted prices in active markets; |
| | • | | Level 2 – Inputs other than quoted prices in active markets that are either directly or indirectly observable; and |
| | • | | Level 3 – Unobservable inputs in which little or no market data exists, which are therefore developed by the Company using estimates and assumptions that reflect those that a market participant would use. |
The following tables represent the Company’s fair value hierarchy for its financial assets and liabilities measured at fair value on a recurring basis as of December 31, 2013 and December 31, 2012:
| | | | | | | | | | | | | | | | |
| | | As of December 31, 2013 | |
| | | Level 1 | | | Level 2 | | | Level 3 | | | Total | |
Liabilities: | | | | | | | | | | | | | | | | |
Accrued warrant liability | | $ | — | | | $ | — | | | $ | 1,241,311 | | | $ | 1,241,311 | |
Compensatory stock options not yet issued (1) | | | — | | | | — | | | | 309,450 | | | | 309,450 | |
| | | | | | | | | | | | | | | | |
Total liabilities | | $ | — | | | $ | — | | | $ | 1,550,761 | | | $ | 1,550,761 | |
| | | | | | | | | | | | | | | | |
| |
| | | As of December 31, 2012 | |
| | | Level 1 | | | Level 2 | | | Level 3 | | | Total | |
Liabilities: | | | | | | | | | | | | | | | | |
Accrued warrant liability | | $ | — | | | $ | — | | | $ | 4,105,659 | | | $ | 4,105,659 | |
| | | | | | | | | | | | | | | | |
Total liabilities | | $ | — | | | $ | — | | | $ | 4,105,659 | | | $ | 4,105,659 | |
| | | | | | | | | | | | | | | | |
| (1) | Included in accrued expenses in the accompanying consolidated balance sheets. |
The Company has certain warrants that could require settlement in cash if a fundamental transaction occurs, as defined in the respective agreements. These agreements specify the amount due to warrant holders is based on the Black-Scholes pricing model. The following are the assumptions used to measure the accrued warrant liability at December 31, 2013 and 2012, which were determined in a manner consistent with that described for grants of options to purchase common stock as set forth in Note 2, “Summary of Significant Accounting Policies”:
| | | | | | | | |
| | | December 31, 2013 | | | December 31, 2012 | |
Stock Price | | $ | 1.17 | | | $ | 1.33 | |
Exercise Price | | $ | 1.60 - 5.00 | | | $ | 1.60 - 5.00 | |
Term in years | | | 0.59 - 1.91 | | | | 1.09 - 2.41 | |
Volatility | | | 42.52 - 76.03 | % | | | 82.75 - 95.91 | % |
Annual rate of quarterly dividends | | | 0 | % | | | 0 | % |
Discount rate- bond equivalent yield | | | .08 - .36 | % | | | .17 - .29 | % |
67
The following are the assumptions used to measure the compensatory stock options not yet issued at December 31, 2013:
| | | | |
| | | December 31, 2013 | |
Stock price | | $ | 0.72 | |
Term in years | | | 5 | |
Volatility | | | 75.68 | % |
Annual rate of quarterly dividends | | | 0 | % |
Discount rate – bond equivalent yield | | | 1.52 | % |
The following table sets forth a summary of changes in the fair value of the Company’s Level 3 fair value measurements for the year ended December 31, 2013 and 2012:
| | | | | | | | |
| | | Year Ended December 31, 2013 | |
| | | Accrued Warrant
Liability | | | Compensatory
Stock Options
Issued After Year
End | |
Beginning Balance | | $ | 4,105,659 | | | $ | — | |
Total (gains) or losses, realized and unrealized, included in earnings (1)(2) | | | (2,864,348 | ) | | | — | |
Estimates and other changes in fair value | | | — | | | | 309,450 | |
| | | | | | | | |
Balance, December 31, 2013 | | $ | 1,241,311 | | | $ | 309,450 | |
| | | | | | | | |
| |
| | | Year Ended December 31, 2012 | |
| | | Accrued Warrant
Liability | | | Compensatory
Stock Options
Issued After Year
End | |
Beginning Balance | | $ | 7,285,959 | | | $ | 378,750 | |
Total (gains) or losses, realized and unrealized, included in earnings (1)(2) | | | (7,701,981 | ) | | | 51,823 | |
Issuances | | | 4,521,681 | | | | — | |
Settlements | | | — | | | | (430,573 | ) |
| | | | | | | | |
Balance, December 31, 2012 | | $ | 4,105,659 | | | $ | — | |
| | | | | | | | |
| (1) | Unrealized gains or losses related to the accrued warrant liability were included as change in value of accrued warrant liability. There were no realized gains or losses for the years ended December 31, 2013 and 2012. |
| (2) | Expenses recorded for compensatory stock options not yet issued are included in research and development expense and general and administrative expense. |
Separate disclosure is required for assets and liabilities measured at fair value on a recurring basis, as documented above, from those measured at fair value on a nonrecurring basis. As of December 31, 2013 and 2012, the Company had no assets or liabilities that were measured at fair value on a nonrecurring basis.
68
The Company considers the accrued warrant liability and compensatory stock options not yet issued to be Level 3 because some of the inputs into the measurements are neither directly or indirectly observable. Both the accrued warrant liability and compensatory stock options not yet issued use management’s estimate for the expected term, which is based on the safe harbor method as historical exercise information over the term of each security is not readily available. Additionally, the number of compensatory options awarded involves an estimate of management’s performance in relation to the targets set forth in the Company’s annual Executive Compensation Plan. The following table summarizes the unobservable inputs into the fair value measurements:
| | | | | | | | | | | | |
| | | December 31, 2013 | |
Description | | Fair Value | | | Valuation Technique | | Unobservable Input | | Range | |
Compensatory stock options not yet issued | | $ | 309,450 | | | Black-scholes pricing model | | Expected term | | | 5 | |
| | | | | | | | Quantity of options | | | 696,000 | |
| | | | |
Accrued warrant liability | | | 1,241,311 | | | Black-scholes pricing model | | Expected term | | | 0.59 - 1.91 | |
| | | | | | | | | | | | |
| | $ | 1,550,761 | | | | | | | | | |
| | | | | | | | | | | | |
Management believes the value of the accrued warrant liability and compensatory stock options are more sensitive to changes in the Company’s stock price at the end of the respective reporting period as opposed to changes in the expected term. At December 31, 2013, a 10% increase in the expected term of the Company’s warrants measured using the Black-Scholes pricing model would increase the warrant liability by approximately 22%, while a 10% decrease in the expected term would decrease the warrant liability by approximately 16%. A 10% increase in the Company’s stock price would result in an increase in the accrued warrant liability of approximately 32%, while a 10% decrease in the stock price would decrease the warrant liability by approximately 26%. At December 31, 2013, a 10% increase or decrease in the expected term of the Company’s compensatory stock options not yet issued would increase or decrease the amount accrued by approximately 6%, while a 10% increase or decrease in the stock price would increase or decrease the amount accrued by approximately 10%.
The carrying amounts of the Company’s remaining financial instruments, which include cash, short-term investments, accounts receivable and accounts payable, approximate their fair values due to their short maturities.
4. Equipment
The following table summarizes the Company’s gross equipment costs for the years ended December 31, 2013 and 2012:
| | | | | | | | |
| | | As of December 31, | |
| | | 2013 | | | 2012 | |
Lab equipment | | $ | 1,083,463 | | | $ | 1,903,533 | |
Computer equipment | | | 332,386 | | | | 335,264 | |
Furniture | | | 528,807 | | | | 523,665 | |
| | | | | | | | |
| | | 1,944,656 | | | | 2,762,462 | |
Less accumulated depreciation | | | (1,486,744 | ) | | | (1,775,909 | ) |
| | | | | | | | |
Equipment, net | | $ | 457,912 | | | $ | 986,553 | |
| | | | | | | | |
5. Noncontrolling Interests
On May 31, 2012, Bioprocess Capital Partners, LLC, or BCP, the noncontrolling interest holder in Incuron, contributed approximately 194.0 million Russian rubles (approximately $5.9 million) to Incuron, which increased its ownership percentage to 40.8% and decreased CBLI’s ownership percentage to 59.2%, which is the ownership percentage at December 31, 2013.
69
On January 20, 2011 and March 14, 2011, BCP contributed 68.0 million Russian Rubles (approximately $2.3 million) and 1.73 million Russian Rubles (approximately $0.1 million), respectively, to Incuron, increasing their ownership percentage from 16.1% to 24.2% and decreasing CBLI’s ownership percentage from 83.9% to 75.8%.
The following quantifies the effects of changes in CBLI’s ownership interest in Incuron, on CBLI’s equity for the years ending December 31, 2013, 2012 and 2011:
| | | | | | | | | | | | |
| | | For the Year Ending December 31, | |
| | | 2013 | | | 2012 | | | 2011 | |
Net loss attributable to CBLI | | $ | (17,262,877 | ) | | $ | (18,234,142 | ) | | $ | (4,013,670 | ) |
Increase in CBLI’s additional paid-in capital due to the issuance of additional membership interests to the noncontrolling interest of Incuron | | | — | | | | 1,081,040 | | | | 176,092 | |
| | | | | | | | | | | | |
Change from net income attributable to CBLI and issuance of additional membership interests to the noncontrolling interest of Incuron | | $ | (17,262,877 | ) | | $ | (17,153,102 | ) | | $ | (3,837,578 | ) |
| | | | | | | | | | | | |
On October 4, 2011, CBLI consummated the transactions contemplated by the Investment Agreement, dated as of September 19, 2011, or the Investment Agreement, with Open Joint Stock Company “Rusnano”, or Rusnano, to provide funding to Panacela to carry out a complete cycle of development, research, performance of clinical trials, production and sales of a line of pharmaceutical drugs for the treatment of oncological, infectious or other diseases.
Pursuant to the Investment Agreement, (i) CBLI invested $3.0 million in Panacela preferred shares and warrants, and, together with certain third-party owners, assigned and/or provided exclusive licenses, as applicable, to Panacela in respect of certain intellectual property in exchange for Panacela common shares, and (ii) Rusnano invested $9.0 million in Panacela preferred shares and warrants, with an additional $17.0 million available for investment. $1.5 million of the $17.0 million was invested in the form of a convertible loan, discussed in Note 6, “Debt,” leaving $15.5 million currently available to be invested at Rusnano’s option. At December 31, 2013, CBLI had an ownership stake of 54.6% in Panacela.
The Panacela preferred shares are convertible into common shares at any time following issuance. The conversion price is equal to the preferred share issuance price of $1,057 per share, subject to proportional adjustment for any stock split, stock dividend, reclassification or similar event with respect to the Panacela common shares. The preferred shares are automatically convertible into common shares upon the occurrence of a qualifying public offering of Panacela, carry no redemption rights and have the ability to vote and participate in dividends on a basis consistent with common shareholders.
The Panacela warrants provide CBLI and Rusnano with an option to increase their respective investments at two and four years following the initial investment. The warrants are exercisable into Panacela preferred shares at an exercise price equal to 20% or 40% above the preferred stock issuance price of $1,057 per share, subject to proportional adjustment for any stock split, stock dividend, reclassification or similar event with respect to the Panacela common shares. The warrants issued to CBLI and Rusnano that had an exercise price equal to 20% above the preferred stock issuance price expired in October 2013.
The preferred shares and warrant instruments have been classified as permanent equity instruments by Panacela. The value assigned to the preferred shares and warrants was based on their relative fair value at the date of issuance. The resultant embedded beneficial conversion feature relating to the preferred shares was considered a deemed dividend, and since Panacela had an accumulated deficit, had no impact on the Panacela statement of stockholders’ equity.
70
6. Debt
On September 30, 2013, CBLI and BioLab 612 entered into a Loan and Security Agreement, or the Loan Agreement, with Hercules Technology II, L.P., or Hercules, pursuant to which we issued a $6 million note and received net proceeds of $5.9 million. The loan bears interest at the greater of (i) 10.45% per annum or (ii) 10.45% plus the prevailing prime rate minus 4.25%. The loan matures on January 1, 2017, and requires interest-only payments for the initial 12 months and principal and interest payments in 27 monthly installments thereafter.
In connection with the Loan Agreement, CBLI granted a first priority lien in substantially all of CBLI’s assets (exclusive of intellectual property). The Loan Agreement also contains representations and warranties by CBLI and Hercules, indemnification provisions in favor of Hercules, customary covenants (including limitations on other indebtedness, liens, acquisitions, investments and dividends, but excluding any financial covenants), and events of default (including payment defaults, breaches of covenants, material adverse events and events leading to bankruptcy or insolvency). Prepayment of the loan is subject to a penalty rate applied to the balance of the secured obligation and ranges from 1% to 3% based on the date the loan is prepaid. Additionally, Hercules has a right to participate in subsequent private placements of CBLI equity securities at the same price, terms and conditions as other investors, up to an aggregate amount of $1 million.
As additional consideration for the loan, CBLI issued Hercules a five-year warrant to purchase 156,250 shares of CBLI common stock at an exercise price of $1.60 per share. The Company recorded the fair value of the warrant of $117,999 as equity and as a discount to the carrying value of the loan. The fair value of the warrant was calculated using the Black-Scholes option pricing model with the following assumptions:
| | | | |
Risk-free interest rate | | | 0.48 | % |
Expected dividend yield | | | 0 | % |
Expected life (years) | | | 2.5 | |
Expected volatility | | | 82.29 | % |
Reflected as a discount to the loan are the following items: the warrant, a $100,000 facility fee CBLI paid to Hercules which was deducted from the gross proceeds of the loan, and a $550,000 payment that is due upon full repayment of the loan or on the maturity date, whichever occurs sooner. In connection with the closing of the loan, CBLI incurred $102,000 in debt issuance costs, primarily related to legal fees, which are included in non-current assets in our consolidated balance sheet. CBLI will amortize the loan discounts and debt issuance costs to interest expense over the term of the loan using the effective interest rate method, which approximates 16.6%. Additionally, the $550,000 end-of-term charge is also reflected as a long-term liability in conjunction with the $6 million note.
The Loan Agreement triggered a reduction in the exercise price of the Company’s warrants issued in March 2010 from $2.00 to $1.60 per share.
The following schedule shows the payments for principal and the end of term charge on the Hercules loan by calendar year:
| | | | |
2014 | | $ | 351,527 | |
2015 | | | 2,246,215 | |
2016 | | | 2,495,163 | |
2017 | | | 1,457,095 | |
| | | | |
Total | | $ | 6,550,000 | |
| | | | |
On September 3, 2013, Panacela entered into a Master Agreement, or the Panacela Loan, with Rusnano and CBLI pursuant to which Panacela issued a $1,530,000 note to Rusnano. The Panacela Loan bears interest at a
71
rate of 16.3% per annum and matures on September 10, 2015, at which time Panacela must repay all unpaid principal and accrued interest. Prior to March 10, 2015, the loan is mandatorily convertible into shares of Panacela preferred stock at a conversion price of $1,057 per share if Panacela completes a qualified financing in accordance with the terms of the Panacela Loan. Subsequent to March 10, 2015, Rusnano has the option to convert the unpaid principal plus interest into shares of Panacela preferred stock at a conversion price of $1,057 per share, or if Panacela has a qualified financing event, at a discounted price of 0.75 times the purchase price per share.
In connection with the Panacela Loan, CBLI issued Rusnano a warrant that has an exercise period that begins upon an event of default on the Panacela Loan and expires on December 31, 2016. Upon an event of default, Rusnano has the option to assign 69.2% of the unpaid principal and interest under the Panacela Loan to CBLI in exchange for shares of CBLI common stock at a price of $1.694 per share.
For the year ended December 31, 2013, included in interest and other income (expense) are $256,970 and $81,142 of interest expense related to the Hercules Loan Agreement and Panacela Loan, respectively.
7. Stockholders’ Equity
In October 2012, CBLI completed a public offering of 7,500,000 units at a price of $2.00 per unit, with each unit consisting of one share of CBLI common stock, par value $0.005 per share, and one warrant to purchase 0.5 of a share of CBLI common stock at an exercise price of $3.00 per whole share, or the 2012 Offering. The shares of CBLI common stock and the warrants issued in the 2012 Offering were issued separately. Under the terms of the related underwriting agreement, CBLI granted the Underwriters an option, exercisable for 30 days, to purchase up to an additional 1,125,000 shares of CBLI common stock (with an over-allotment price of $1.995 per share) and/or additional warrants to purchase up to 562,500 shares of CBLI common stock (with an over-allotment price of $0.0094 for each warrant to purchase a whole share) to cover over-allotments, if any. The underwriter exercised their option in part by purchasing 1,025,000 shares and 562,500 warrants of the over-allotment option within 30 days of the closing.
Certain warrants issued during the 2012 offering contain provisions that could require the Company to settle the warrants in cash and, accordingly, were originally recorded as a liability in the amount of $4,521,681 determined by the Black-Scholes valuation model with the following assumptions:
| | | | |
Stock price | | $ | 2.48 | |
Exercise price | | $ | 3.00 | |
Term in years | | | 2.51 | |
Volatility | | | 79.70 | % |
Annual rate of quarterly dividends | | | — | |
Discount rate- bond equivalent yield | | | 0.35 | % |
The 2012 Offering triggered a reduction in the exercise price of the Company’s warrants issued in March 2010 from $4.00 to $2.00 per share.
In June 2011, the Company issued 5,872,500 shares of its common stock and warrants to purchase a total of 2,936,250 shares of its common stock for gross proceeds of $23.5 million. The common stock and warrants were sold in units, at a price of $4.00 per unit, with each unit consisting of: (i) one share of common stock; (ii) a warrant to purchase 0.25 of a share of common stock, with an exercise price of $4.50 per share; and (iii) a warrant to purchase 0.25 of a share of common stock, with an exercise price of $5.00 per share. In addition, the placement agent and the financial advisor also collectively received warrants to purchase up to 176,175 shares of common stock. In the event of stock splits, stock dividends, combinations of shares and similar recapitalization transactions, the number of shares issuable and the exercise price associated with all warrants issued in this transaction may be adjusted. At December 31, 2011, all outstanding warrants were exercisable.
72
Certain warrants issued in June 2011 contain provisions that could require the Company to settle the warrants in cash, and accordingly, were originally recorded as a liability in the amount of $2,525,175 determined by the Black-Scholes valuation model with the following assumptions:
| | | | |
Stock price | | $ | 4.45 | |
Exercise price | | $ | 5.00 | |
Term in years | | | 2.50 | |
Volatility | | | 69.36 | % |
Annual rate of quarterly dividends | | | — | |
Discount rate- bond equivalent yield | | | 0.53 | % |
The following table sets forth the changes in the number of warrants outstanding for the periods presented, exclusive of the warrants issued to Rusnano in connection with their loan to Panacela as those warrants are not exercisable until an event of default which has not occurred:
| | | | | | | | | | | | |
| | | Number of
Warrants | | | Weighted Average
Exercise Price | | | Number of Common
Shares Exercisable Into | |
Outstanding at December 31, 2011 | | | 10,121,219 | | | $ | 3.76 | | | | 12,564,193 | |
Granted | | | 4,312,500 | | | | 3.00 | | | | 4,312,500 | |
Forfeited, Canceled | | | (4,055,724 | ) | | | 5.07 | | | | (6,498,698 | ) |
| | | | | | | | | | | | |
Outstanding at December 31, 2012 | | | 10,377,995 | | | | 2.94 | | | | 10,377,995 | |
Granted | | | 156,250 | | | | 1.60 | | | | 156,250 | |
| | | | | | | | | | | | |
Outstanding at December 31, 2013 | | | 10,534,245 | | | | 2.70 | | | | 10,534,245 | |
| | | | | | | | | | | | |
The following table sets forth the details of the outstanding warrants as of December 31, 2013:
| | | | | | | | |
Expiration Date | | Current
Exercise Price | | | Number of
Warrants | |
March 2, 2015 | | $ | 1.60 | | | | 935,385 | |
June 17, 2015 | | | 5.00 | | | | 176,175 | |
February 12, 2016 | | | 1.60 | | | | 929,826 | |
March 19, 2016 | | | 1.60 | | | | 1,921,795 | |
March 26, 2016 | | | 1.60 | | | | 634,189 | |
June 22, 2016 | | | 5.00 | | | | 1,468,125 | |
October 24, 2017 | | | 3.00 | | | | 4,312,500 | |
September 30, 2018 | | | 1.60 | | | | 156,250 | |
| | | | | | | | |
| | | | | | | 10,534,245 | |
| | | | | | | | |
The January 2014 equity offering, as described in Note 14, “Subsequent Events”, triggered a reduction in the exercise price of certain warrants. All warrants in the table above that have an exercise price of $1.60 now have an exercise price of $1.22, except for the 156,250 warrants that expire on September 30, 2018.
73
Equity Incentive Plan
The following is a summary of option award activity under the Plan for the year ended December 31, 2013:
| | | | | | | | | | | | | | | | |
| | | Year Ended December 31, 2013 | |
| | | Total Stock Options
Outstanding | | | Weighted Average
Exercise Price per
Share | | | Nonvested Stock
Options | | | Weighted Average
Grant Date Fair
Value per Share | |
December 31, 2012 | | | 5,016,916 | | | $ | 4.54 | | | | 404,500 | | | $ | 2.30 | |
Granted | | | 903,775 | | | | 1.67 | | | | 903,775 | | | | 1.21 | |
Vested | | | — | | | | — | | | | (513,296 | ) | | | 1.73 | |
Exercised | | | (9,681 | ) | | | 1.28 | | | | — | | | | — | |
Forfeited, Canceled | | | (346,177 | ) | | | 3.53 | | | | (200,500 | ) | | | 1.21 | |
| | | | | | | | | | | | | | | | |
December 31, 2013 | | | 5,564,833 | | | | 4.14 | | | | 594,479 | | | | 1.50 | |
| | | | | | | | | | | | | | | | |
The following is a summary of outstanding stock options under the Plan as of December 31, 2013:
| | | | | | | | |
| | | Stock Options
Outstanding | | | Vested Stock
Options | |
Quantity | | | 5,564,833 | | | | 4,970,354 | |
Weighted-average exercise price | | $ | 4.14 | | | $ | 4.40 | |
Weighted Average Remaining Contractual Term (in Years) | | | 6.75 | | | | 6.47 | |
Intrinsic value | | $ | 11,592 | | | $ | 11,592 | |
For the years ended December 31, 2013, 2012 and 2011, the Company granted 903,775, 1,132,383, and 1,459,393 stock options, respectively, with a weighted-average grant date fair value of $1.21, $1.32 and $4.04, respectively. For the years ended December 31, 2013, 2012 and 2011, the total fair value of options vested was $887,603, $1,549,888 and $5,381,855, respectively. The total intrinsic value of options exercised for the years ended December 31, 2013, 2012 and 2011 was $5,736, $1,485 and $818,723, respectively.
As of December 31, 2013, total compensation cost not yet recognized related to non-vested stock options was $445,647. The Company expects to recognize this cost over a weighted average period of 0.76 years.
8. Restructuring
On September 30, 2013, we transferred 26 laboratory and pre-clinical employee positions to BBL, an entity owned in part by our Chief Scientific Officer and director, Dr. Andrei Gudkov, to enable us to better focus on clinical development activities. In connection with this transition, we entered into a Master Services Agreement, or MSA, with BBL, pursuant to which BBL agreed to perform laboratory and pre-clinical research services for us. We plan to engage BBL for pre-clinical research services in the future. Such services are discussed further in Note 9, “Significant Alliances and Related Parties.”
As a result of the above described restructuring, we recorded $101,000 for one-time employee transition costs, $112,000 for an idle facilities reserve and $293,000 for an impairment loss on research equipment. All of these costs were recorded as R&D expense during the year ended December 31, 2013. As of December 31, 2013, an accrual of $100,000 related to these restructuring costs was included in accrued expenses.
9. Significant Alliances and Related Parties
Roswell Park Cancer Institute
The Company has entered into several agreements with Roswell Park Cancer Institute, or RPCI, including: various sponsored research agreements, an exclusive license agreement and clinical trial agreements for the conduct of the Phase 1 Entolimod oncology study and the Phase 1 CBL0137 intravenous administration study. Additionally, the Company’s Chief Scientific Officer is the Senior Vice President of Basic Research at RPCI.
74
The Company recognized $0, $0 and $2,317,218 of revenue from RPCI during the years ended December 31, 2013, 2012 and 2011, respectively. The Company incurred $2,323,078, $3,876,073 and $2,689,503 in expense to RPCI related to research grants and agreements for the years ended December 31, 2013, 2012 and 2011, respectively. The Company had $172,295 and $900,300 included in accounts payable owed to RPCI at December 31, 2013 and 2012, respectively. In addition, the Company had $491,397 and $553,644 in accrued expenses payable to RPCI at December 31, 2013 and 2012, respectively.
Cleveland Clinic Foundation
CBLI has entered into an exclusive license agreement, or the License, with the Cleveland Clinic Foundation, or CCF, pursuant to which CBLI was granted an exclusive license to CCF’s research base underlying our therapeutic platform and certain product candidate in development by Panacela. CBLI has the primary responsibility to fund all newly developed patents; however, CCF retains ownership of those patents covered by the agreement. CBLI also has agreed to use commercially diligent efforts to bring one or more products to market as soon as practical, consistent with sound and reasonable business practices and judgments. In consideration for the CCF License, CBLI agreed to issue CCF common stock and make certain milestone, royalty and sublicense royalty payments. Milestone payments, which may be credited against future royalties, amounted to $0, $100,000 and $100,000 for the years ended December 31, 2013, 2012 and 2011, respectively. No royalty or sublicense royalty payments were made to CCF in the years ended December 31, 2013, 2012 and 2011, respectively.
The Company also recognized $7,516, $4,804 and $2,558 as research and development expense to CCF for the years ended December 31, 2013, 2012 and 2011, respectively. The Company did not have any liabilities to CCF at December 31, 2013 and 2012.
Consultants
CBLI has entered into a consulting agreement with our Chief Scientific Officer, Dr. Andrei Gudkov. The Company incurred $202,843, $200,695 and $186,224 for consulting services in the years ending December 31, 2013, 2012 and 2011, respectively. The Company incurred $0, $0 and $24,476 in bonuses for the years ending December 31, 2013, 2012 and 2011, respectively. The Company incurred $164,661, $32,659 and $109,137 in non-cash, stock based compensation expense for the years ending December 31, 2013, 2012 and 2011, respectively.
The Company had $62,059 and $28,245 for consulting services included in accrued expenses at December 31, 2013 and 2012, respectively. The Company had $66,692 and $24,476 included in accrued bonuses at December 31, 2013 and 2012, respectively.
Dr. Gudkov has equity interests in other entities, not including BBL, that are unaffiliated with the Company. The Company recognized $29,607, $0 and $0 as research and development expense to these entities for the years ended December 31, 2013, 2012 and 2011, respectively. As of December 31, 2013 and 2012, there were no amounts owed to these entities. During the years ended December 31, 2013 and 2012, the Company recognized other income from these entities of $217,347 and $407,395, respectively. The Company held no accounts receivable from these entities as of December 31, 2013 and 2012.
Buffalo BioLabs
Pursuant to the MSA that was executed with BBL (see Note 8, “Restructuring”), the Company recognized $419,284 as research and development expense for the year ended December 31, 2013, and included $111,356 in accounts payable at December 31, 2013. We also received $64,362 and $6,500 from BBL for sublease and other income for the years ended December 31, 2013 and 2012, respectively.
75
10. Income Taxes
The Company accounts for income taxes using the asset and liability method. Deferred taxes are determined by calculating the future tax consequences attributable to differences between the financial accounting and tax bases of existing assets and liabilities. A valuation allowance is recorded against deferred tax assets when, in the opinion of management, it is more likely than not that the Company will not be able to realize the benefit from its deferred tax assets.
The Company files income tax returns, as prescribed by the national, state and local jurisdictions in which it operates. The Company’s uncertain tax positions are related to tax years that remain subject to examination and are recognized in the financial statements when the recognition threshold and measurement attributes are met. Interest and penalties related to tax deficiencies and uncertain tax positions are recorded as income tax expense.
Income (loss) from continuing operations consists of the following:
| | | | | | | | | | | | |
| | | For the Year Ended December 31, | |
| | | 2013 | | | 2012 | | | 2011 | |
US operations | | $ | (13,505,266 | ) | | $ | (14,317,608 | ) | | $ | (1,775,053 | ) |
Foreign operations | | | (6,624,018 | ) | | | (8,097,032 | ) | | | (3,454,672 | ) |
| | | | | | | | | | | | |
| | $ | (20,129,284 | ) | | $ | (22,414,640 | ) | | $ | (5,229,725 | ) |
| | | | | | | | | | | | |
The provision for income taxes charged to continuing operations is $0 for all periods presented.
Deferred tax assets (liabilities) were comprised of the following as of the periods presented below:
| | | | | | | | | | | | |
| | | As of December 31, | |
| | | 2013 | | | 2012 | | | 2011 | |
Deferred tax assets: | | | | | | | | | | | | |
Operating loss carryforwards | | $ | 44,120,000 | | | $ | 37,642,000 | | | $ | 28,972,000 | |
Accrued expenses | | | 8,861,000 | | | | 8,576,000 | | | | 7,778,000 | |
Tax credit carryforwards | | | 3,186,000 | | | | 2,921,000 | | | | 2,537,000 | |
Intellectual property | | | 4,846,000 | | | | 3,377,000 | | | | 1,604,000 | |
Outside tax basis difference in affiliate | | | 2,825,000 | | | | 1,616,000 | | | | 1,378,000 | |
Equipment | | | 388,000 | | | | 237,000 | | | | 156,000 | |
Other | | | — | | | | 4,000 | | | | 4,000 | |
| | | | | | | | | | | | |
Total deferred tax assets | | | 64,226,000 | | | | 54,373,000 | | | | 42,429,000 | |
Deferred tax liabilities | | | — | | | | — | | | | — | |
Net deferred tax asset | | | 64,226,000 | | | | 54,373,000 | | | | 42,429,000 | |
Valuation allowance | | | (64,226,000 | ) | | | (54,373,000 | ) | | | (42,429,000 | ) |
| | | | | | | | | | | | |
| | $ | — | | | $ | — | | | $ | — | |
| | | | | | | | | | | | |
The provision for income taxes differs from the amount of income tax determined by applying the applicable U.S. statutory federal income tax rate to the pretax loss from continuing operations as a result of the following differences:
| | | | | | | | | | | | |
| | | For the Year Ended December 31, | |
| | | 2013 | | | 2012 | | | 2011 | |
Tax at the U.S. statutory rate | | $ | (6,810,000 | ) | | $ | (7,621,000 | ) | | $ | (1,778,000 | ) |
Change in value of warrant liability | | | (974,000 | ) | | | (2,619,000 | ) | | | (6,739,000 | ) |
Stock option expenses | | | — | | | | — | | | | (140,000 | ) |
Valuation allowance | | | 7,723,000 | | | | 10,204,000 | | | | 8,639,000 | |
Other | | | 61,000 | | | | 36,000 | | | | 18,000 | |
| | | | | | | | | | | | |
| | $ | — | | | $ | — | | | $ | — | |
| | | | | | | | | | | | |
76
At December 31, 2013, the Company has U.S. federal net operating loss carryforwards of approximately $109,919,000, which begin to expire if not utilized by 2023, and approximately $3,313,000 of tax credit carryforwards that begin to expire if not utilized by 2024. The Company also has U.S. state net operating loss carryforwards of approximately $99,930,000, which begin to expire if not utilized by 2027 and state tax credit carryforwards of approximately $318,000, which begin to expire if not utilized by 2022.
The Company files U.S. federal tax returns, along with various state and foreign income tax returns. All federal, state and foreign tax returns for the years ended December 31, 2012, 2011 and 2010 are still open for examination.
The following presents a roll-forward of the unrecognized tax benefits and the associated interest and penalties:
| | | | | | | | |
| | | Unrecognized
Tax Benefits | | | Interest
and Penalties | |
Balance at January 1, 2012 | | $ | 407,000 | | | $ | — | |
| | |
Prior year tax position | | | — | | | | — | |
Current year tax position | | | — | | | | — | |
Deferred tax position | | | 30,000 | | | | — | |
Settlements with tax authorities | | | — | | | | — | |
Expiration of the statute of limitations | | | — | | | | — | |
| | | | | | | | |
Balance at December 31, 2012 | | | 437,000 | | | | — | |
| | |
Prior year tax position | | | — | | | | — | |
Current year tax position | | | — | | | | — | |
Deferred tax position | | | 8,000 | | | | — | |
Settlements with tax authorities | | | — | | | | — | |
Expiration of the statute of limitations | | | — | | | | — | |
| | | | | | | | |
Balance at December 31, 2013 | | $ | 445,000 | | | $ | — | |
| | | | | | | | |
CBLI received New York State incentive tax credit refunds of $62,000, $532,000 and $367,000 during 2013, 2012 and 2011, respectively. These refundable tax credits were based on the Company’s research and development activities, real estate tax payments, employment levels and equipment purchases. Since there is no state tax liability or refund of prior year tax payments, these refundable tax credits were recorded against operating expenses in the year of receipt, instead of being recorded as an income tax benefit.
11. Employee Benefit Plan
CBLI maintains an active defined contribution retirement plan for its employees, referred to herein as the Benefit Plan. All employees satisfying certain service requirements are eligible to participate in the Benefit Plan. The Company makes matching cash contributions each payroll period, up to 4% of employees’ contributions. The Company’s expense relating to the Benefit Plan was $196,257, $201,510 and $182,669 for the years ended December 31, 2013, 2012 and 2011, respectively.
12. Commitments and Contingencies
The Company has entered into various agreements with third parties and certain related parties in connection with the research and development activities of its existing product candidates as well as discovery efforts on potential new product candidates. These agreements include fixed obligations to sponsor research and development activities and minimum royalty payments for licensed patents. These amounts do not include any additional amounts that the Company may be required to pay under its license agreements upon the achievement of scientific, regulatory and commercial milestones, including milestones such as the submission of an IND to
77
the FDA and the first commercial sale of the Company’s products in various countries. As of December 31, 2013 the Company is uncertain as to whether any of these contingent events will become realized. The Company is also party to five agreements that require it to make milestone payments, pay royalties on net sales of the Company’s products, and make payments on sublicense income received by the Company relating to certain products. There were no milestone payments or royalties on net sales accrued for any of these agreements as of December 31, 2013 and 2012 as none were due.
From time-to-time, the Company may have certain contingent liabilities that arise in the ordinary course of business. The Company accrues for liabilities when it is probable that future expenditures will be made and such expenditures can be reasonably estimated. For all periods presented, the Company is not a party to any pending material litigation or other material legal proceedings.
The Company has entered into agreements with three key executives who, if terminated by the Company without cause as described in these agreements, would be entitled to severance pay.
As of December 31, 2013, the Company had unconditional purchase obligations totaling $1,024,477 for goods and services, all of which the Company anticipates to incur during 2014.
Capital Lease
In December 2011, the Company entered into a capital lease for scientific equipment in the amount of $304,673. The terms of the lease required an upfront payment of $82,983 and monthly payments of $7,616 for 36 months once the lease term began in March 2012. Principal payments under the capital lease obligation were $78,125 and $52,410 for the years ended December 31, 2013 and 2012, respectively. Interest payments under the capital lease obligation were $20,827 and $23,708 for the years ended December 31, 2013 and 2012, respectively. As of December 31, 2012, accumulated depreciation for the leased equipment was $50,779. As of December 31, 2013, the Company recognized an impairment loss of $105,491 related to this leased asset as the anticipated discounted future cash flows resulting from the lease with BBL exceeded the carrying value of the asset, and as such, the Company adjusted the gross cost and carrying value down to $87,468, resulting in $0 accumulated depreciation as of December 31, 2013.
As of December 31, 2013, future minimum future lease payments under capital leases are as follows:
| | | | |
2014 | | $ | 91,341 | |
2015 | | | 7,612 | |
| | | | |
Total minimum lease payments | | | 98,953 | |
Interest expense related to future periods | | | 7,797 | |
| | | | |
Present value of minimum lease payments | | | 91,156 | |
Less: current portion | | | 83,634 | |
| | | | |
Non-current portion | | $ | 7,522 | |
| | | | |
Operating Leases
The Company leases laboratory facilities and office facilities at various locations with expiration dates ranging from 2014 to 2019. The Company recognizes rent expense on a straight-line basis over the term of the related operating leases. For the years ended December 31, 2013, 2012 and 2011, total rent expense related to the Company’s operating leases was $511,029, $459,150 and $396,667, respectively.
78
As of December 31, 2013, future minimum payments under operating leases are as follows:
| | | | |
2014 | | $ | 471,925 | |
2015 | | | 344,580 | |
2016 | | | 354,918 | |
2017 | | | 365,565 | |
2018 | | | 376,532 | |
2019 | | | 191,048 | |
| | | | |
Total minimum lease payments | | $ | 2,104,568 | |
| | | | |
13. Quarterly Financial Data (Unaudited)
The following is a summary of the quarterly consolidated results of operations for the years ended December 31, 2013 and December 31, 2012:
| | | | | | | | | | | | | | | | |
| | | For the Quarter Ended | |
| | | March 31, 2013 | | | June 30, 2013 | | | September 30, 2013 | | | December 31, 2013 | |
Revenues | | $ | 1,367,472 | | | $ | 1,613,262 | | | $ | 1,635,600 | | | $ | 3,871,632 | |
Loss from Operations | | | (7,447,515 | ) | | | (6,776,618 | ) | | | (5,856,468 | ) | | | (2,996,158 | ) |
Net Income (Loss) | | | (10,787,148 | ) | | | (3,886,421 | ) | | | (4,610,961 | ) | | | (844,754 | ) |
Net Income (Loss) Attributable to Cleveland BioLabs, Inc. | | | (9,764,323 | ) | | | (3,042,111 | ) | | | (4,091,196 | ) | | | (365,247 | ) |
Basic Earnings (Loss) Per Share Available for Common Shareholders | | $ | (0.22 | ) | | $ | (0.07 | ) | | $ | (0.09 | ) | | $ | (0.01 | ) |
Fully Diluted Earnings (Loss) Per Share Available for Common Shareholders | | $ | (0.22 | ) | | $ | (0.07 | ) | | $ | (0.09 | ) | | $ | (0.01 | ) |
Weighted Average Shares Outstanding, Basic | | | 44,826,576 | | | | 44,948,591 | | | | 45,061,274 | | | | 45,170,129 | |
Weighted Average Shares Outstanding, Diluted | | | 44,826,576 | | | | 44,948,591 | | | | 45,061,274 | | | | 45,170,129 | |
| | | | | | | | | | | | | | | | |
| | | For the Quarter Ended | |
| | | March 31, 2012 | | | June 30, 2012 | | | September 30, 2012 | | | December 31, 2012 | |
Revenues | | $ | 931,397 | | | $ | 258,237 | | | $ | 219,575 | | | $ | 2,161,501 | |
Loss from Operations | | | (7,481,875 | ) | | | (9,161,724 | ) | | | (7,841,541 | ) | | | (5,561,466 | ) |
Net Income (Loss) | | | (6,398,894 | ) | | | (5,906,870 | ) | | | (12,315,676 | ) | | | 2,206,800 | |
Net Income (Loss) Attributable to Cleveland BioLabs, Inc. | | | (5,387,146 | ) | | | (5,078,684 | ) | | | (10,877,836 | ) | | | 3,109,524 | |
Basic Earnings (Loss) Per Share Available for Common Shareholders | | $ | (0.15 | ) | | $ | (0.14 | ) | | $ | (0.30 | ) | | $ | 0.07 | |
Fully Diluted Earnings (Loss) Per Share Available for Common Shareholders | | $ | (0.15 | ) | | $ | (0.14 | ) | | $ | (0.30 | ) | | $ | 0.07 | |
Weighted Average Shares Outstanding, Basic | | | 35,657,563 | | | | 35,745,675 | | | | 35,879,245 | | | | 42,236,226 | |
Weighted Average Shares Outstanding, Diluted | | | 35,657,563 | | | | 35,745,675 | | | | 35,879,245 | | | | 42,565,945 | |
14. Subsequent Events
On January 16, 2014, the Company completed a public offering of 5,737,706 shares of the Company’s common stock at a price of $1.22 per share, resulting in net proceeds of approximately $6.4 million after deducting for placement agent fees and offering expenses. In connection with the offering, the Company issued 2,868,853 Series A Warrants and 2,868,853 Series B Warrants to the purchasers. Each Series A Warrant has an exercise price of $1.22 per share, and will become exercisable six months following the date of issuance and expire five
79
years from the date of issuance. Each Series B Warrant has an exercise price of $1.22 per share, and will become exercisable six months following the date of issuance and expire 18 months from the date of issuance. In addition to the warrants issued to the purchasers, the Company also issued 86,066 of Series A Warrants and 86,066 of Series B Warrants to the placement agent as compensation for completing the offering. The warrants to the placement agent have the same terms, including exercise price, as the warrants issued to investors. The offering also triggered a reduction in the exercise price of 4,421,195 of the Company’s warrants from $1.60 to $1.22.
On February 20, 2014, Sabby Healthcare Volatility Master Fund, Ltd. and Sabby Volatility Warrant Master Fund, Ltd., two purchasers in the January 2014 sale of securities, brought suit in the U.S. District Court for the Southern District of New York against the Company in an action captioned Sabby Healthcare Volatility Master Fund, Ltd. v. Cleveland Biolabs, Inc., No. 14-cv-1055 (S.D.N.Y.). The plaintiffs allege that the Company misrepresented the state of its funding negotiations with BARDA during the period leading up to the sale of the Securities, and as a result, the plaintiffs were harmed when the Company’s stock price declined following the announcement that BARDA had terminated negotiations with the Company. The complaint asserts claims under Section 10(b) of the Securities Exchange Act of 1934 and SEC Rule 10b-5, as well as claims for fraudulent inducement, breach of contract, and indemnification. The plaintiffs seek $2 million, plus interest, attorney’s fees, and litigation costs.
80
Item 9: Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A: Controls and Procedures
Effectiveness of Disclosure
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, as of December 31, 2013. Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2013, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective to assure that information required to be declared by us in reports that we file or submit under the Exchange Act is (1) recorded, processed, summarized and reported within the periods specified in the SEC’s rules and forms, and (2) accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with the participation of management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework inInternal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework inInternal Control – Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2013.
The effectiveness of our internal control over financial reporting as of December 31, 2013 has been audited by Meaden & Moore, Ltd., an independent registered public accounting firm, as stated in their report which appears herein.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting during our fourth fiscal quarter ended December 31, 2013 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 9B: Other Information
None.
81
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Management and Corporate Governance Matters” and “Section 16(a) Beneficial Ownership Reporting Compliance” in the Company’s Proxy Statement for the 2014 Annual Meeting of Stockholders.
Item 11. Executive Compensation
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Executive Officer and Director Compensation,” “Compensation Discussion and Analysis,” “Management and Corporate Governance Matters – Compensation Committee Interlocks and Insider Participation,” and “Compensation Committee Report” in the Company’s Proxy Statement for the 2014 Annual Meeting of Stockholders.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” in the Company’s Proxy Statement for the 2014 Annual Meeting of Stockholders.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Certain Relationships and Related Person Transactions” and “Management and Corporate Governance Matters” in our Proxy Statement for the 2014 Annual Meeting of Stockholders.
Item 14. Principal Accounting Fees and Services
The response to this item is incorporated by reference from the discussion responsive thereto under the caption “Independent Registered Public Accounting Firm” in the Company’s Proxy Statement for the 2014 Annual Meeting of Stockholders.
82
PART IV
Item 15. Exhibits, Financial Statement Schedules
The following exhibits are incorporated herein by reference or attached hereto.
| | |
| Exhibit No. | | Identification of Exhibit |
| |
| 3.1 | | Restated Certificate of Incorporation filed with the Secretary of State of Delaware on March 18, 2010 (Incorporated by reference to Form 10-K for the year ended December 31, 2009, filed on March 22, 2010). |
| |
| 3.2 | | Certificate of Amendment to the Restated Certificate of Incorporation, filed with the Secretary of State of Delaware on June 20, 2013 (Incorporated by reference to Form 10-Q for the period ended June 30, 2013, filed on August 9, 2013). |
| |
| 3.3 | | Second Amended and Restated By-Laws (Incorporated by reference to Form 8-K filed on December 5, 2007). |
| |
| 4.1 | | Form of Common Stock Purchase Warrant (Series D Transaction) (Incorporated by reference to Form 8-K filed on March 30, 2009). |
| |
| 4.2 | | Form of Common Stock Purchase Warrant (Private Placement closed on March 2, 2010) (Incorporated by reference to Form 8-K/A filed on February 26, 2010). |
| |
| 4.3 | | Form of Series F Warrants (Incorporated by reference to Form 8-K filed on June 21, 2011). |
| |
| 4.4 | | Form of Warrant Agreement by and between Cleveland BioLabs, Inc. and Continental Stock Transfer & Trust Company (Incorporated by reference to Form 8-K filed on October 22, 2012). |
| |
| 4.5 | | Warrant to Purchase Common Stock issued to Open Joint Stock Company “Rusnano” (Incorporated by reference to Form 8-K filed on September 5, 2013). |
| |
| 4.6 | | Warrant Agreement, dated September 30, 2013, between Cleveland BioLabs, Inc. and Hercules Technology II, L.P. (Incorporated by reference to Form 10-Q for the period ended September 30, 2013, filed on November 8, 2013). |
| |
| 4.7 | | Form of Series A/B Warrant to Purchase Common Stock (Incorporated by reference to Form 8-K filed on January 15, 2014). |
| |
| 10.1.1 | | Exclusive License Agreement by and between The Cleveland Clinic Foundation and Cleveland BioLabs, Inc., effective as of July 1, 2004 (Incorporated by reference to Amendment No. 1 to Registration Statement on Form SB-2 filed on April 25, 2006 (File No. 333-131918)). |
| |
| 10.1.2 | | Second Amendment to Exclusive License Agreement, dated September 22, 2011, by and between The Cleveland Clinic Foundation and the registrant (Incorporated by reference to Form 10-Q for the period ended September 30, 2011, filed on November 9, 2011). † |
| |
| 10.2.1 | | Employment Agreement by and between Cleveland BioLabs, Inc. and Dr. Yakov Kogan, dated August 1, 2004 (Incorporated by reference to Amendment No. 1 to Registration Statement on Form SB-2 filed on April 25, 2006 (File No. 333-131918)). |
| |
| 10.2.2 | | Amendment to Employment Agreement by and between Cleveland BioLabs, Inc. and Dr. Yakov Kogan, dated as of December 31, 2008 (Incorporated by reference to Form 10-K for the year ended December 31, 2008, filed on March 30, 2009). |
| |
| 10.3 | | Employment Agreement made as of April 4, 2013 and effective as of April 1, 2013 by and between Cleveland BioLabs, Inc. and Jean Viallet (Incorporated by reference to Form 8-K filed on April 9, 2013). |
83
| | |
| Exhibit No. | | Identification of Exhibit |
| |
| 10.4.1 | | Cleveland BioLabs, Inc. Equity Incentive Plan (Incorporated by reference to Proxy Statement on Schedule 14A filed on April 1, 2008). |
| |
| 10.4.2 | | First Amendment to Cleveland BioLabs, Inc. Equity Incentive Plan (Incorporated by reference to Form 8-K filed on June 9, 2010). |
| |
| 10.4.3 | | Second Amendment to Cleveland BioLabs, Inc. Equity Incentive Plan (Incorporated by reference to Form 8-K filed on June 15, 2012). |
| |
| 10.4.4 | | Form of Stock Award Grant Agreement (Incorporated by reference to Form 8-K filed on June 15, 2012). |
| |
| 10.4.5 | | Form of Non-Qualified Stock Option Agreement (Incorporated by reference to Form 8-K filed on June 15, 2012). |
| |
| 10.5 | | Cleveland Biolabs, Inc. 2013 Employee Stock Purchase Plan (Incorporated by reference to Form 8-K filed on June 20, 2013). |
| |
| 10.6.1 | | Contract (W9113M-10-C-0088), effective as of September 15, 2010, between Cleveland BioLabs, Inc. and the U.S. Army Space and Missile Defense Command/Army Forces Strategic Command (the “2010 DoD Contract”) (Incorporated by reference to Form 10-Q for the period ended September 30, 2010, filed on November 15, 2010). |
| |
| 10.6.2 | | Amendment of Solicitation/Modification of Contract No. 1, effective as of September 17, 2010, to the 2010 DoD Contract (Incorporated by reference to Form 10-Q for the period ended September 30, 2010, filed on November 15, 2010). |
| |
| 10.6.3 | | Amendment of Solicitation/Modification of Contract No. 2, effective as of June 23, 2011, to the 2010 DoD Contract (Incorporated by reference to Form 8-K filed on June 29, 2011). |
| |
| 10.7 | | Process Development and Manufacturing Agreement between Cleveland BioLabs, Inc. and SynCo Bio Partners B.V., effective as of August 31, 2006 (Incorporated by reference to Form 8-K filed on October 25, 2006). |
| |
| 10.8 | | Form of Securities Purchase Agreement (Incorporated by reference to Form 8-K filed on March 30, 2009). |
| |
| 10.9 | | Form of Registration Rights Agreement (Incorporated by reference to Form 8-K filed on March 30, 2009). |
| |
| 10.10 | | Amendment and Waiver Agreement, dated March 20, 2009 (Incorporated by reference toForm 8-K filed on March 30, 2009). |
| |
| 10.11 | | Form of Amendment and Reaffirmation Agreement (Incorporated by reference to Form 8-K filed on March 30, 2009). |
| |
| 10.12 | | License Agreement between Cleveland BioLabs, Inc. and Zhejiang Hisun Pharmaceutical Co., Ltd., dated September 3, 2009 (Incorporated by reference to Form 8-K filed on September 9, 2009). |
| |
| 10.13.1 | | Participation Agreement, dated December 30, 2009, by and between Cleveland BioLabs, Inc. and Bioprocess Capital Partners, LLC (Incorporated by reference to Form 8-K filed on January 5, 2010). |
| |
| 10.13.2 | | First Amendment to Participation Agreement, dated April 13, 2010, by and between Cleveland BioLabs, Inc. and Bioprocess Capital Partners, LLC (Incorporated by reference to Form 10-Q for the period ended June 30, 2010, filed on August 16, 2010). |
| |
| 10.14.1 | | Securities Purchase Agreement dated February 25, 2010 (Incorporated by reference to Form 8-K filed on February 26, 2010). |
84
| | |
| Exhibit No. | | Identification of Exhibit |
| |
| 10.14.2 | | Form of Amendment to Securities Purchase Agreement, dated December 23, 2010, among the Company and the amending purchasers identified on the signature pages thereto (Incorporated by reference to Form 8-K filed on December 29, 2010). |
| |
| 10.15.1 | | Consulting Agreement, dated January 1, 2010, between Cleveland BioLabs, Inc. and Andrei Gudkov (Incorporated by reference to Form 8-K filed on June 13, 2011). |
| |
| 10.15.2 | | First Amendment to Consulting Agreement, dated June 10, 2011, between Cleveland BioLabs, Inc. and Andrei Gudkov (Incorporated by reference to Form 8-K filed on June 13, 2011). |
| |
| 10.16 | | Form of Securities Purchase Agreement, dated June 17, 2011, by and between Cleveland BioLabs, Inc. and the investors in the Offering (Incorporated by reference to Form 8-K filed on June 21, 2011). |
| |
| 10.17 | | Employment Agreement, dated August 4, 2011, between Cleveland BioLabs, Inc. and C. Neil Lyons (Incorporated by reference to Form 8-K filed on August 4, 2011). |
| |
| 10.18.1 | | Investment Agreement, dated September 19, 2011, by and among Panacela Labs, Inc., Cleveland BioLabs, Inc. and Open Joint Stock Company Rusnano (Incorporated by reference to Form 10-Q for the period ended September 30, 2011, filed on November 9, 2011). |
| |
| 10.18.2 | | Amendment No. 1 to Investment Agreement, dated September 3, 2013, by and among Panacela Labs, Inc., Cleveland BioLabs, Inc. and Open Joint Stock Company Rusnano. |
| |
| 10.19 | | Exclusive License and Option Agreement, dated September 23, 2011, by and between Health Research, Inc., Roswell Park Institute Division, Roswell Park Cancer Institute Corporation, and Panacela Labs, Inc (Incorporated by reference to Form 10-Q for the period ended September 30, 2011, filed on November 9, 2011). † |
| |
| 10.20 | | Amended and Restated Exclusive Sublicense Agreement, dated September 23, 2011, by and between Cleveland BioLabs, Inc. and Panacela Labs, Inc. (Incorporated by reference to Form 10-Q for the period ended September 30, 2011, filed on November 9, 2011). |
| |
| 10.21 | | Assignment Agreement, dated September 23, 2011, by and between Cleveland BioLabs, Inc., and Panacela Labs, Inc. (Incorporated by reference to Form 10-Q for the period ended September 30, 2011, filed on November 9, 2011). |
| |
| 10.22 | | Master Agreement dated September 3, 2013 by and among Panacela Labs, Inc., Cleveland Biolabs, Inc. and Open Joint Stock Company “Rusnano” (Incorporated by reference to Form 8-K filed on September 5, 2013). |
| |
| 10.23 | | Convertible Loan Agreement dated September 3, 2013 between Open Joint Stock Company “Rusnano” and Panacela Labs, Inc. (Incorporated by reference to Form 8-K filed on September 5, 2013). |
| |
| 10.24 | | Master Services Agreement, dated October 14, 2014, between Buffalo BioLabs, LLC and Cleveland BioLabs, Inc. (Incorporated by reference to Form 8-K filed on October 18, 2013). |
| |
| 10.25 | | Loan and Security Agreement, dated September 30, 2013, by and among Cleveland BioLabs, Inc., Biolab 612, LLC and Hercules Technology II, L.P. (Incorporated by reference to Form 10-Q for the period ended September 30, 2013, filed on November 8, 2013). |
| |
| 10.26 | | Letter Agreement, dated January 9, 2014, by and among Cleveland BioLabs, Inc. and H.C. Wainwright & Co., LLC (Incorporated by reference to Form 8-K filed on January 15, 2014). |
| |
| 10.27 | | Securities Purchase Agreement, dated January 14, 2014, by and among Cleveland BioLabs, Inc. and the Purchasers set forth therein (Incorporated by reference to Form 8-K filed on January 15, 2014). |
85
| | |
| Exhibit No. | | Identification of Exhibit |
| |
| 10.28 | | Annual Executive Bonus Plan. |
| |
| 10.29 | | 2012 Long-term Executive Compensation Plan (Incorporated by reference to Form 8-K filed on June 15, 2012). |
| |
| 14.1 | | Code of Ethics for Senior Executives and Financial Officers (Incorporated by reference toForm 8-K filed on June 15, 2012). |
| |
| 23.1 | | Consent of Meaden & Moore, Ltd. |
| |
| 31.1 | | Rule 13a-14(a)/15d-14(a) Certification of Yakov Kogan |
| |
| 31.2 | | Rule 13a-14(a)/15d-14(a) Certification of C. Neil Lyons |
| |
| 32.1 | | Section 1350 Certification. |
| |
| 101.1 | | The following financial statements and supplementary data are filed as a part of this annual report on Form 10-K for the quarter and year ended December 31, 2013: (i) Consolidated Balance Sheets at December 31, 2013 and 2012; (ii) Consolidated Statements of Operations for years ended December 31, 2013, 2012, and 2011; (iii) Consolidated Statements of Stockholders’ Equity for period from December 31, 2010 to December 31, 2013; (iv) Consolidated Statements of Cash Flows for years ended December 31, 2013, 2012, and 2011; and (v) Notes to Consolidated Financial Statements as blocks of text. * |
| * | Pursuant to Rule 406T of Regulation S-T, the Interactive Data Files on Exhibit 101 hereto are deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities and Exchange Act of 1934, as amended, and otherwise are not subject to liability under those sections. |
| † | Confidential treatment has been requested from the Securities and Exchange Commission as to certain portions of this document. |
86
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| | | | | | |
| | | | CLEVELAND BIOLABS, INC. |
| | | |
Dated: March 17, 2014 | | | | By: | | /s/ YAKOV KOGAN |
| | | | | | Yakov Kogan |
| | | | | | Chief Executive Officer |
| | |
| | | | CLEVELAND BIOLABS, INC. |
| | | |
Dated: March 17, 2014 | | | | By: | | /s/ C. NEIL LYONS |
| | | | | | C. Neil Lyons |
| | | | | | Chief Financial Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons in the capacities and on the dates indicated.
| | | | |
Signature | | Title | | Date |
| | |
/S/ Yakov Kogan Yakov Kogan | | Chief Executive Officer and Director (Principal Executive Officer) | | March 17, 2014 |
| | |
/S/ C. Neil Lyons C. Neil Lyons | | Chief Financial Officer (Principal Financial and Accounting Officer) | | March 17, 2014 |
| | |
/S/ David C. Hohn David C. Hohn | | Director | | March 17, 2014 |
| | |
/S/ James J. Antal James J. Antal | | Director | | March 17, 2014 |
| | |
/S/ Julia R. Brown Julia R. Brown | | Director | | March 17, 2014 |
| | |
/S/ Paul DiCorleto Paul DiCorleto | | Director | | March 17, 2014 |
| | |
/S/ Anthony Principi Anthony Principi | | Director | | March 17, 2014 |
| | |
/S/ Randy Saluck Randy Saluck | | Director | | March 17, 2014 |
| | |
/S/ Andrei Gudkov Andrei Gudkov | | Director | | March 17, 2014 |
87