UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 20-F
(Mark One)
☐ | REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2020
OR
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
OR
☐ | SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report
Commission file number: 001-39131
LIMINAL BIOSCIENCES INC.
(Exact name of Registrant as specified in its charter)
Canada
(Jurisdiction of incorporation or organization)
440 Armand-Frappier Boulevard, Suite 300
Laval, H7V 4B4, Québec
H7V 4B4
+1 450 781 0115
(address of principal executive offices)
Bruce Pritchard
Chief Executive Officer
Liminal BioSciences Inc.
440 Armand-Frappier Boulevard,
Suite 300
Laval, Québec
H7V 4B4
Tel: +1 450 781 0115
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered, pursuant to Section 12(b) of the Act
Title of each class | | Trading Symbol(s) | | Name of each exchange on which registered |
Common Shares, no par value | | LMNL | | The Nasdaq Stock Market LLC |
Securities registered or to be registered pursuant to Section 12(g) of the Act. None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act. None
Indicate the number of outstanding shares of each of the issuer’s classes of capital stock or common stock as of the close of business covered by the annual report.
Common Shares, no par value: 29,943,839 common shares outstanding as of December 31, 2020
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ☐ Accelerated filer ☐ Non-accelerated filer ☒ Emerging growth company ☒
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards † provided pursuant to Section 13(a) of the Exchange Act. ☐
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under section 404(b) of the Sarbanes-Oxley Act (15 U.S.C 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☐ No ☒
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
U.S. GAAP ☐ | | International Financial Reporting Standards as issued by the International Accounting Standards Board ☒ | | Other ☐ |
If “Other” has been checked in response to the previous question indicate by check mark which financial statement item the registrant has elected to follow. Item 17 ☐ Item 18 ☐
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
TABLE OF CONTENTS
i
ii
INTRODUCTION
Unless the context otherwise requires, all references in this Annual Report on Form 20-F, or the Annual Report, to “Liminal,” “company,” “we,” “us” and “our” refer to Liminal BioSciences Inc. and, where appropriate, our subsidiaries.
Our fiscal year ends on December 31. This Annual Report includes our audited consolidated financial statements as of December 31, 2020 and 2019 and for the years ended December 31, 2020, 2019 and 2018, which are prepared in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board, or IFRS. None of our financial statements were prepared in accordance with generally accepted accounting principles in the United States, or U.S. GAAP.
Except for the executive and directors’ compensation (Item 6B) which is presented in U.S. Dollars (unless otherwise specified), our financial information is presented in Canadian Dollars and all references in this Annual Report to “$” means Canadian Dollars and all references to “US$” means U.S. Dollars. For the convenience of the reader, in this Annual Report, unless otherwise indicated, translations from Canadian Dollars into U.S. Dollars were made at the rate of $1.00 to US$0.7442, which is the average rate for the 2020 fiscal year, (2019 average rate: $1.00=US$ 0.7525). Translations from Great Britain Pounds or GBP into U.S. Dollars were made at the rate of 1 GPB to US$ 1.2861 which is the average rate for the 2020 fiscal year (2019 average rate: 1.00 GBP = US$ 1.2747). Such U.S. Dollar amounts are not necessarily indicative of the amounts of U.S. Dollars that could actually have been purchased upon exchange of Canadian Dollars or GBP at the dates indicated.
We have made rounding adjustments to some of the figures included in this Annual Report. Accordingly, numerical figures shown as totals in some tables may not be an arithmetic aggregation of the figures that preceded them.
Unless otherwise indicated, all financial information included in this Annual Report has been retroactively adjusted to give effect to the 1000-for-1 consolidation of our common shares effected on July 5, 2019.
This Annual Report includes registered and unregistered trademarks such as Liminal BioSciences™ and Ryplazim® which are protected under applicable intellectual property laws and are the property of Liminal. Solely for convenience, our trademarks referred to in this Annual Report and in other publicly filed documents may appear without the ® or ™ symbol, but such references are not intended to indicate, in any way, that we will not assert our rights to the fullest extent under applicable law. All other trademarks used in this Annual Report are the property of their respective owners.
We are incorporated under the laws of Canada. Substantially all of our assets are located outside the United States. In addition, several of our directors and officers are nationals and/or residents of countries other than the United States, and all or a substantial portion of such persons’ assets may be located outside the United States. As a result, it may be difficult for investors to effect service of process within the United States upon us or such persons or to enforce against them or against us, judgments obtained in United States courts, including judgments predicated upon the civil liability provisions of the securities laws of the United States or any state thereof. In addition, investors should not assume that the courts of Canada (i) would enforce judgments of U.S. courts obtained in actions against us, our officers or directors, or other said persons, predicated upon the civil liability provisions of the U.S. federal securities laws or other laws of the United States or (ii) would enforce, in original actions, liabilities against us or such directors, officers or experts predicated upon the United States federal securities laws or any securities or other laws of any state or jurisdiction of the United States.
In addition, there is doubt as to the applicability of the civil liability provisions of U.S. federal securities law to original actions instituted in Canada. It may be difficult for an investor, or any other person or entity, to assert U.S. securities laws claims in original actions instituted in Canada.
iii
Special Note Regarding Forward-Looking Statements
This Annual Report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, that are based on our management’s beliefs and assumptions and on information currently available to our management. These statements are “forward-looking” because they represent our expectations, intentions, plans and beliefs about our business and the markets we operate in and on various estimates and assumptions based on information available to our management at the time these statements are made. For example, forward-looking statements around financial performance and revenues are based on financial modelling undertaken by our management. This financial modelling takes into account revenues that are uncertain. It also includes forward-looking revenues from transactions based on probability. In assessing probability, management considers the status of negotiations for any revenue generating transactions, and the likelihood, based on the probability of income, that associated costs will be incurred. Management then ranks the probabilities in such a way that only those revenues deemed highly or reasonably likely to be secured are included in the projections.
All statements other than statements of historical facts may be forward-looking statements. Without limiting the generality of the foregoing, words such as “may”, “will”, “expect”, “believe”, “anticipate”, “intend”, “could”, “might”, “would”, “should”, “estimate”, “continue”, “plan” or “pursue”, “seek”, “project”, “predict”, “potential” or “targeting” or the negative of these terms, other variations thereof, comparable terminology or similar expressions, are intended to identify forward-looking statements although not all forward-looking statements contains these terms and phrases.
Forward-looking statements are provided for the purposes of assisting you in understanding us and our business, operations, prospects and risks at a point in time in the context of historical and possible future developments and therefore you are cautioned that such information may not be appropriate for other purposes. Actual events or results may differ materially from those anticipated in these forward-looking statements if known or unknown risks affect our business, or if estimates or assumptions turn out to be inaccurate. In particular, forward-looking statements included in this Annual Report include, without limitation, statements with respect to:
| • | our ability to develop, manufacture and successfully commercialize value-added pharmaceutical products; |
| • | our ability to obtain required regulatory approvals; |
| • | the availability of funds and resources to pursue research and development projects; |
| • | the successful and timely completion of our clinical trials; |
| • | our ability to take advantage of business opportunities in the pharmaceutical industry; |
| • | a potential strategic transaction for our plasma-derived therapeutics business that we may pursue, in whole or in part, including a potential divestment or sale of non-core assets; |
| • | our reliance on key personnel, collaborative partners and other third parties; |
| • | the validity and enforceability of our patents and proprietary technology; |
| • | expectations regarding our ability to raise capital; |
| • | the use of certain hazardous materials; |
| • | the availability and sources of raw materials; |
| • | our manufacturing capabilities; |
| • | the value of our intangible assets; |
| • | negative operating cash flow; |
| • | the outcome of any current or pending litigation against us; |
iv
| • | uncertainties related to the regulatory process and approvals; |
| • | increasing data security costs; |
| • | costs related to environmental safety regulations; |
| • | competing drugs, as well as from current and future competitors; |
| • | developing products for the indications we are targeting; |
| • | market acceptance of our product candidates by patients and healthcare professionals; |
| • | availability of third-party coverage and adequate reimbursement; |
| • | general changes in economic or market conditions; |
| • | volatility of our share price; and |
| • | other risks and uncertainties, including those listed in this section of this Annual Report F titled “Item 3.D—Risk Factors.” |
You should refer to the section of this Annual Report on Form 20-F titled “Item 3.D—Risk Factors” for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report on Form 20-F will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this Annual Report on Form 20-F and the documents that we reference in this Annual Report on Form 20-F and have filed as exhibits to this Annual Report on Form 20-F completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
This Annual Report on Form 20-F contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third-party information. While we believe the market position, market opportunity and market size information included in this Annual Report on Form 20-F is generally reliable, such information is inherently imprecise.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this Annual Report on Form 20-F, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete. Our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements.
v
PART I
Item 1.Identity of Directors, Senior Management and Advisers.
Not applicable.
Item 2.Offer Statistics and Expected Timetable.
Not applicable.
A.Selected Financial Data
This item is no longer required as we have elected to early adopt the changes to Item 301 of Regulation S-K contained in SEC Release No. 33-10890.
B.Capitalization and Indebtedness
Not applicable.
C.Reasons for the Offer and Use of Proceeds
Not applicable.
D.Risk Factors
Our business faces significant risks. You should carefully consider all of the information set forth in this Annual Report and in our other filings with the United States Securities and Exchange Commission, or the SEC, including the following risk factors which we face and which are faced by our industry. Our business, financial condition or results of operations could be materially adversely affected by any of these risks. This report also contains forward-looking statements that involve risks and uncertainties. Our results could materially differ from those anticipated in these forward-looking statements, as a result of certain factors including the risks described below and elsewhere in this Annual Report and our other SEC filings. See “Special Note Regarding Forward-Looking Statements” above.
Summary of Selected Risks Associated with Our Business
Risks Related to Our Financial Position
| • | We will require additional funding and may not be able to raise the capital necessary to continue as a going concern or to complete the research and development of our pipeline of product candidates and their commercialization, if approved. |
| • | We are not profitable and may never achieve profitability. |
| • | The market conditions or our business performance may prevent us from having access to the public markets in the future. |
Risks Related to the Development and Commercialization of our Product Candidates
| • | Our commercial success depends largely on the development and commercialization of our product candidates. |
| • | We do not have the required regulatory approval to commercialize our product candidates and cannot guarantee that we will obtain such regulatory approval. We may also be required to conduct post-approval clinical trials as a condition to licensing a product. |
| • | We may fail to achieve our publicly-announced milestones or not achieve publicly-announced milestones in due time. |
1
| • | Interim top-line and preliminary results from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures, which could result in material changes in the final data. |
Risks Related to Regulatory Approval of Our Product Candidates and Other Legal Compliance Matters
| • | Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions. |
| • | Even if we obtain marketing approvals for our product candidates, the terms of approvals and ongoing regulation of our products may limit how we manufacture and market our products and compliance with such requirements may involve substantial resources, which could materially impair our ability to generate revenue. |
| • | Some of our employees in our manufacturing facility are unionized, as employees may be in manufacturing facilities of our suppliers. While unionized facilities are subject to the risk of labour disruptions from time to time, we cannot predict whether or when any labour disruption may arise, or how long such a disruption could last. |
Risks Related to Our Business Operations and Our Reliance on Third Parties
| • | The success of our product candidates is influenced by our collaborations with our partners and any adverse developments in our relationship with our partners could materially harm our business. |
| • | Our business could be adversely affected by the effects of health epidemics, including the global COVID-19 pandemic. |
| • | Our product candidates are complex and difficult to manufacture, which could cause production problems that result in delays in our development or commercialization. We also rely in whole or in part on third parties for the manufacture and supply of our product candidates and such reliance may adversely affect us if the third parties are unable to fulfill their obligations. |
| • | If we breach any of the agreements under which we license rights to our product candidates or technology from third parties, we could lose license rights that are important to our business. |
| • | We may not be successful in entering into strategic agreements with third parties for the divestment of, or other strategic transaction with respect to our plasma-derived therapeutics business, and related non-core assets, in whole or in part, which may result in a shut down of the plasma-derived therapeutics’ business. A shut-down of our plasma-derived therapeutics business may adversely affect our short-term cash position and expose us to increased liabilities. |
Risks Related to our Intellectual Property
| • | Our failure to protect our intellectual property may have a material adverse effect on our ability to develop and commercialize our product candidates. |
| • | If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose intellectual property rights that are important to our business. |
| • | We may not be able to protect our intellectual property rights throughout the world. |
| • | The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of Canada and the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. |
Risks Related to Our Common Shares and Our Status as a Public Company
| • | The price of our common shares historically has been volatile. This volatility, and the sale of substantial amounts of our common shares, could adversely affect the price of our common shares. |
2
| • | All of our debt obligations, and any future indebtedness we may incur, could lead to adverse consequences and will have priority over our common shares with respect to payment in the event of a liquidation, dissolution or winding up. |
| • | We are a “controlled company” within the meaning of the applicable Nasdaq listing rules and, as a result, qualify for exemptions from certain corporate governance requirements. If we rely on these exemptions, you will not have the same protections afforded to shareholders of companies that are subject to such requirements. |
| • | We are an “emerging growth company” and as a result of the reduced disclosure and governance requirements applicable to emerging growth companies, our common shares may be less attractive to investors. |
| • | We qualify as a foreign private issuer and, as a result, we will not be subject to U.S. proxy rules and will be subject to Exchange Act reporting obligations that permit less detailed and frequent reporting than that of a U.S. domestic public company. |
| • | U.S. investors may be unable to enforce certain judgments. |
Risks Related to Our Financial Position
We will require additional funding and may not be able to raise the capital necessary to continue as a going concern or to complete the research and development of our pipeline of product candidates and their commercialization, if approved.
As of December 31, 2020, we had approximately $45.1 million of cash and cash equivalents. This will not provide us with sufficient funds to continue maintaining our operating activities, even at low spending levels, for the next 12 months.
Additionally, we will require additional funding and may not be able to raise the capital necessary to continue and complete the research and development of our product candidates and their commercialization, if approved. We have historically generated revenues, but have never achieved or maintained profitability, and will need additional financing in order to continue our activities.
Our future capital requirements will depend on many factors, including:
| • | the progress, results and costs of laboratory testing, manufacturing, preclinical and clinical development for our current product candidates; |
| • | the scope, progress, results and costs of preclinical development, laboratory testing and clinical trials of other product candidates that we may pursue; |
| • | the ability to close a strategic transaction for our plasma-derived therapeutics business that we may pursue, in whole or in part, including a potential divestment or sale of non-core assets; |
| • | the development requirements of other product candidates that we may pursue; |
| • | the timing and amounts of any milestone or royalty payments we may be required to make under future license agreements; |
| • | the costs of building out our infrastructure, including hiring additional clinical, regulatory, quality control and manufacturing personnel; |
| • | the costs, timing and outcome of regulatory review of our product candidates, including potential regulatory delays related to the COVID-19 pandemic; |
| • | the costs and timing of future commercialization activities, including product manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval; |
3
| • | the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims; and |
| • | the extent to which we acquire or in-license other product candidates and technologies. |
In the past, we have been financed in part through the Restructuring Transactions (as defined below), debt and public equity offerings and we may effect additional debt or equity offerings to raise capital, the size of which cannot be predicted. The issuance and sales of substantial amounts of equity or other securities, or the perception that such issuances and sales may occur, could adversely affect the market price of our common shares. In addition, to the extent that we raise additional capital by issuing equity securities, our existing shareholders’ ownership may experience substantial dilution, and the terms of these securities may include liquidation or other preferences that adversely affect shareholder rights. Equity and debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as redeeming our shares, making investments, incurring additional debt, making capital expenditures or declaring dividends.
The incurrence of additional indebtedness could result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants therein, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely affect our ability to conduct our business.
If we raise additional capital through collaborations, strategic alliances or third-party licensing arrangements, we may have to relinquish valuable rights to our intellectual property, future revenue streams, research programs or product candidates, or grant licenses on terms that may not be favorable to us. If we are unable to raise additional capital through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts, or grant rights to develop and market product candidates that we would otherwise develop and market ourselves.
There is no assurance that sufficient financing will be available when needed to allow us to continue as a going concern. The perception that we may not be able to continue as a going concern may also make it more difficult to operate our business due to concerns about our ability to meet our contractual obligations. Our ability to continue as a going concern is contingent upon, among other factors, obtaining alternate financing. We cannot provide any assurance that we will be able to raise additional capital on acceptable terms, if at all, particularly if the COVID-19 pandemic continues to disrupt global financial markets, reducing our ability to access capital, which could in the future negatively affect our liquidity.
If we are unable to secure additional capital, we may be required to curtail our research and development initiatives and take additional measures to reduce costs in order to conserve our cash in amounts sufficient to sustain operations and meet our obligations. These measures could cause significant delays in our preclinical, clinical and regulatory efforts, which are critical to the realization of our business plan. The accompanying consolidated financial statements do not include any adjustments that may be necessary should we be unable to continue as a going concern. Such adjustments could be material.
We are not profitable and may never achieve profitability.
We are not profitable and may never achieve profitability. We have been reporting losses since our inception. We will need to generate significant revenues to achieve profitability. There is no guarantee that we will succeed in commercializing our product candidates, controlling our expenses and developing additional product candidates, and, therefore, we may never become profitable.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. These net losses will adversely impact our shareholders’ deficit and net assets and may fluctuate significantly from quarter to quarter and year to year. We anticipate that our expenses will increase substantially as we:
| • | continue our ongoing and planned research and development of fezagepras and other product candidates under development; |
| • | conduct preclinical studies and clinical trials for other current and future product candidates; |
| • | seek to discover and develop additional product candidates and further expand our clinical product pipeline; |
4
| • | seek regulatory approvals for any product candidates that successfully complete clinical trials; |
| • | continue to scale up manufacturing capacity with the aim of securing sufficient quantities to meet our capacity requirements for clinical trials and potential commercialization; |
| • | develop, maintain, expand and protect our intellectual property portfolio; |
| • | acquire or in-license other product candidates and technologies; |
| • | hire additional clinical, quality control, regulatory and manufacturing personnel; |
| • | add clinical, operational, financial and management information systems and personnel, including personnel to support our product development and planned future commercialization efforts; |
| • | establish sales, marketing and distribution infrastructure to commercialize any product candidate for which we may obtain regulatory approval; and |
| • | expand our operations in the United Kingdom, Canada and other geographies. |
To become and remain profitable, we must succeed in developing and eventually commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing preclinical studies and clinical trials of our product candidates, obtaining regulatory approval, manufacturing, marketing and selling any products for which we may obtain regulatory approval, as well as discovering and developing additional product candidates. We may never succeed in these activities and, even if we do, may never generate revenue that is significant enough to achieve profitability.
Because of the numerous risks and uncertainties associated with the development, manufacture, delivery and commercialization of drugs, we are unable to accurately predict the timing or amount of expenses or when, or if, we will be able to achieve profitability. If we are required by regulatory authorities to perform studies in addition to those currently expected, or if there are any delays in the initiation and completion of our clinical trials or the development of any of our product candidates, our expenses could increase and profitability could be further delayed.
Even if we achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable could impair our ability to raise capital, expand our business, maintain our research and development efforts or continue our operations.
The market conditions or our business performance may prevent us from having access to the public markets in the future.
The market conditions or our business performance may prevent us from having access to the public markets in the future. In such a case, we will have to use other means of financing, such as issuing debt instruments or entering into private financing agreements, which may not be available to us on favorable terms and conditions or at all. These debt instruments may contain terms and conditions (e.g. covenants, etc.) which may be challenging or difficult for us to respect, may be breached or trigger default provisions. Accordingly, we may be required to compensate counterparties, for costs and losses incurred as a result of various events, including breaches of representations and warranties, covenants, claims that may arise during the terms of said debt instruments or as a result of litigation that may be suffered by counterparties. If adequate funding is not available to us, we may be required to delay, reduce or eliminate our research and development of new product candidates, our clinical trials or our marketing and commercialization efforts to launch and distribute products, if any.
5
Risks Related to the Development and Commercialization of our Product Candidates
Our commercial success depends largely on the development and commercialization of our product candidates.
Our commercial success depends largely on the development and commercialization of our product candidates. Our failure to do so will have a material adverse effect on us. Our focus has been on development activities for our lead product candidate fezagepras and finding strategic alternatives, which may include a divestment or other strategic transaction for our plasma-derived therapeutics business, including investigative product Ryplazim and related non-core assets, in which we have invested a significant portion of our financial resources and time, and which may result in a shutdown of our plasma-derived therapeutics business. Our other compounds are at earlier stages of development.
The time required to obtain marketing approval by the U.S. Food and Drug Administration, or FDA, and other regulatory authorities is unpredictable but typically takes many years following the commencement of preclinical studies and clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. For example, in response to our biologics license application, or BLA, which we submitted to the FDA seeking approval for use of Ryplazim for the treatment of congenital plasminogen deficiency, in April 2018 the FDA denied approval of the BLA in its current form and issued a complete response letter, or CRL. In addition, approval policies, regulations, or the type and amount of preclinical and clinical data, and the chemistry, manufacturing and controls, necessary to gain approval may change during the course of a product candidate’s research and development and may vary among jurisdictions. It is possible that none of our existing product candidates or any future product candidates will ever obtain regulatory approval.
Our ability to generate revenues in the future is primarily dependent on the commercialization and/or partnering of our product candidates. There can be no guarantee of commercialization of these product candidates as they remain in development. Our ability to successfully commercialize any product candidates will depend on numerous factors including, without limitation:
| • | the timing, progress, costs, and results of ongoing and planned clinical trials; |
| • | the willingness of the FDA and other regulatory agencies to grant approvals for the conduct of our clinical trials and acceptance of our preclinical studies as the basis for the review and approval of our product candidates; |
| • | the timing, outcome and costs of seeking and obtaining regulatory approvals from the FDA and other regulatory agencies; |
| • | market acceptance of our products, if any, by the medical community, patients and reimbursement by third-party payors (such as governmental health administration authorities and private health coverage insurers); |
| • | the timing and cost of establishing successful marketing and sales force or the entering into a commercial agreement with a partner for the marketing and sale of our products, if any; |
| • | the timing and costs of maintaining manufacturing plants and supply agreements of our product candidates for clinical trials and other studies and, if approved, for commercial supply; |
| • | our ability to meet milestones under agreements with third parties and to enter into collaborative agreements with third parties for the development and commercialization of our product candidates; |
| • | an increase in the number of competitors in the same market; |
| • | our ability to obtain coverage and adequate reimbursement from third-party payors and patients' willingness to pay out-of-pocket in the absence of such coverage and adequate reimbursement; |
| • | the cost to maintain, expand, defend and enforce our intellectual property portfolio and our ability to effectively prosecute and protect our intellectual property and avoid patent infringement; |
| • | the costs of acquiring, licensing or investing in additional businesses, products or technologies; and |
6
| • | any other condition, obligation or requirement that may arise, all of which may delay our capacity to generate revenues and will adversely materially affect our financial conditions and operating results. |
Many of these factors are beyond our control, including the time needed to adequately complete clinical testing and the regulatory submission process. It is possible that none of our product candidates will ever obtain regulatory approval, even if we expend substantial time and resources seeking such approval. If we do not achieve one or more of these factors in a timely manner or at all, or any other factors impacting the successful development of biopharmaceutical products, we could experience significant delays or an inability to successfully develop our product candidates, which would materially harm our business.
We do not have the required regulatory approval to commercialize our product candidates and cannot guarantee that we will obtain such regulatory approval. We may also be required to conduct post-approval clinical trials as a condition to licensing a product.
We do not have the required regulatory approval to commercialize our product candidates and cannot guarantee that we will obtain such regulatory approval. The commercialization of our product candidates first requires the approval of the regulatory agencies in each of the countries where we intend to sell our product candidates. In order to obtain the required approvals, we must demonstrate, following preclinical studies and clinical trials, the safety, efficacy and quality of a product and that it can be manufactured in compliance with current good manufacturing practices, or cGMP, and required specifications. There can be no guarantee that we will succeed in obtaining regulatory approval from the FDA and the regulatory approvals of agencies in other countries to sell our product candidates. Our product candidates are still subject to clinical trials and if the results of such trials are not positive, we may not be in a position to make any filing to obtain the mandatory regulatory approval or we may have to perform additional clinical or product validation studies on any of our product candidates until the results support the safety and efficacy of such product, therefore incurring additional delays and costs. The filing of new drug applications, or NDA, or BLAs is complex and we rely in part on third-party service providers or consultants to help us perform these tasks.
The obtaining of regulatory approval is subject to the discretion of regulatory agencies. Therefore, even if we have obtained positive results relating to the safety and efficacy of a product, a regulatory agency may not accept such results as being conclusive and allow us to sell our product candidates in a given country. Furthermore, the obtaining of regulatory approval is subject to the review and inspection of our manufacturing facility or manufacturing facilities of our third-party contract manufacturers and the product’s manufacturing process, including product batch validation and quality controls; these facilities and processes must comply with FDA cGMP regulations. A regulatory agency may require that additional tests on the safety and efficacy of a product or changes in the manufacturing facility or manufacturing process be conducted prior to granting approval, if any.
While fezagepras and Ryplazim have been granted beneficial designations from health regulatory entities, our development strategy may also include the use of additional expedited pathways, such as through the accelerated or contingent approval pathway. Depending on results of the preclinical and clinical trials in our other product candidates, we may also pursue such status for those candidates. There is no certainty that our product candidates will qualify for breakthrough therapy, orphan drug or additional beneficial designations, nor can we assume that the clinical data obtained from trials of our product candidates will be sufficient to qualify for any expedited approval program.
The FDA or a comparable regulatory authority may require more information, including additional preclinical or clinical data to support approval, including data that would require us to perform additional preclinical studies, clinical trials, or both, or modify our manufacturing processes, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program. If we change our manufacturing processes, we may be required to conduct additional clinical trials or other studies, which also could delay or prevent approval of our product candidates. If we were to obtain approval, regulatory authorities may approve any of our product candidates for fewer indications than we request (including failing to approve the most commercially promising indications), may impose warnings and restrictions on prescription and distribution, may grant approval contingent on the performance of costly post-marketing clinical trials or other post-marketing commitments, or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate.
7
Even if a product candidate were to successfully obtain marketing approval from the FDA or other comparable regulatory authorities in other jurisdictions, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, or may be subject to burdensome post-approval study or risk management requirements. If we are unable to obtain regulatory approval for one of our product candidates in one or more jurisdictions, or any approval contains significant limitations, we may not be able to obtain sufficient funding to continue the development of that product or generate revenue attributable to that product candidate. Also, any regulatory approval of our current or future product candidates, once obtained, may be withdrawn.
In response to our BLA, which we submitted to the FDA seeking approval for use of Ryplazim for the treatment of congenital plasminogen deficiency in April 2018, the FDA denied approval of the BLA in its current form and issued a CRL. A CRL is issued by the FDA when the review of an application is complete, and questions remain that preclude approval of the BLA in its current form. The FDA comments in the CRL focused on chemistry, manufacturing and controls deficiencies in our BLA. The FDA requires that we revisit analytical methods, including revalidation of certain data and improvement of statistical strength of analytical method validation. We have to engage a contract research and development organization, or CRO, which will act as our U.S. agent. There can be no assurance that our BLA will be approved by the FDA, or that any of our product candidates will ever be approved for commercialization.
The manufacture, marketing and sale of our products, if any, will be subject to ongoing and extensive governmental regulation in the country in which we intend to market our products.
The manufacture, marketing and sale of our products, if any will be subject to ongoing and extensive governmental regulation in the country in which we intend to market our products. For instance, if we obtain marketing approval for one product candidate in the United States, the marketing of this product will be subject to extensive regulatory requirements administered by the FDA and other regulatory bodies, such as adverse event reporting requirements as well as marketing and promotional restrictions and obligations, which among other things, prohibit the promotion of a product for uses not approved by the FDA as reflected in the product’s approved labeling. The manufacturing facilities for our product will also be subject to continual review and periodic inspection and approval of manufacturing modifications. Manufacturing facilities are subject to inspections by the FDA and must comply with the FDA’s cGMP regulations. Failure to comply with any of these post-approval requirements can result in a series of sanctions, including withdrawal of the right to market a product. The failure to obtain, or a delay in obtaining, approval from the FDA or other regulatory bodies may postpone our capacity to generate revenues and adversely materially affect our financial conditions and operating results.
Ryplazim requires a manufacturing process that is complex and involves biological intermediates that may be susceptible to contamination and variations in yield.
Plasma is a raw material that is susceptible to damage and contamination and may contain human pathogens, any of which would render the plasma unsuitable for further manufacturing. For instance, contamination or improper storage of plasma by us or third-party suppliers may require us to destroy some of our raw material. If unsuitable plasma is not identified and discarded prior to its release to our manufacturing processes, it may be necessary to discard intermediate or finished product made from that plasma or to recall any finished product released to the market.
The manufacture of Ryplazim is an extremely complex process of bioseparation, purification, filling and finishing. Once we have manufactured Ryplazim, it must be handled carefully and kept at appropriate temperatures. Our failure, or the failure of third parties that supply, ship or distribute our product candidates, to properly care for Ryplazim may require that such product candidates be destroyed.
Contamination of our product candidates could cause investors, patients or other third parties with whom we conduct business to lose confidence in the reliability of our manufacturing procedures, or to be in breach of agreements, which could adversely affect our sales and profits. In addition, faulty or contaminated product candidates that are unknowingly distributed could result in patient harm, threaten the reputation of our product candidates and expose us to product liability damages and claims.
8
Due to the nature of plasma, there will be variations in the biologic properties of the plasma we collect or purchase for bioseparation and purification that may result in fluctuations in the obtainable yield, even if cGMP regulations are followed. Lower yields may limit production of Ryplazim or our other product candidates due to capacity constraints.
Our ability to continue to produce plasma-derived products depends on the safety of our plasma supply, testing by third parties and manufacturing and regulatory processes against transmittable diseases. Our failure to comply with these processes and regulations could have a material adverse effect on our results of operations and financial condition.
Despite overlapping safeguards, including the screening of donors and other steps to remove or inactivate viruses and other infectious disease causing agents, the risk of transmissible disease through blood plasma products cannot be entirely eliminated. For example, since plasma-derived therapeutics involves the use and purification of human plasma, there has been concern raised about the risk of transmitting human immunodeficiency virus, HIV, prions, West Nile virus, H1N1 virus or “swine flu” and other blood-borne pathogens through plasma-derived products. In addition, we are required to comply with the cGMP regulations to ensure the safety, purity, and potency of blood and blood components. The public health objective in testing human blood donations for evidence of relevant transfusion-transmitted infections and in notifying donors is to prevent the transmission of relevant transfusion-transmitted infections. For example, in the event that an infected blood unit is released for transfusion, we are subject to the “lookback” requirements, which are intended to help ensure the continued safety of the blood supply by providing necessary information to consignees of blood and blood components and appropriate notification of recipients of blood components that are at increased risk of having received an infected unit of blood. Failure to continue to produce plasma-derived products in compliance with relevant regulations could have a material adverse effect on our results of operations and financial condition.
We could become supply-constrained and our financial performance would suffer if we cannot obtain adequate quantities of FDA-approved human source plasma that meet proper specifications.
In order for plasma to be used in the manufacturing of Ryplazim, the individual centers at which the plasma is collected must be licensed by the FDA and approved by the regulatory authorities of any country in which we may wish to commercialize our products, as applicable. When we open a new plasma center, and on an ongoing basis after licensure, it must be inspected by the FDA for compliance with cGMP and other regulatory requirements. Therefore, even if we are able to construct new plasma collection centers, in addition to the Amherst, New York center, to complement our Winnipeg, Manitoba, Canada plasma collection center, an unsatisfactory inspection could prevent a new center from being licensed or risk the suspension or revocation of an existing license.
We may not have adequate quantities of compliant plasma to manufacture Ryplazim. Therefore, we are reliant on the purchase of plasma from third parties to manufacture Ryplazim. We can give no assurances that appropriate plasma will be available to us on commercially reasonable terms, or at all, to manufacture Ryplazim. In order to maintain a plasma center’s license, its operations must continue to conform to cGMP and other regulatory requirements. In the event that we determine that plasma was not collected in compliance with cGMP, we may be unable to use and may ultimately destroy plasma collected from that center. Additionally, if non-compliance in the plasma collection process is identified after the impacted plasma has been pooled with compliant plasma from other sources, entire plasma pools, in-process intermediate materials and our supply of Ryplazim could be impacted.
We plan to increase our supplies of plasma for use in the manufacturing processes through increased purchases of plasma from third-party suppliers as well as collections from our existing Winnipeg plasma collection center and Amherst plasma collection center. This strategy is dependent upon our ability to maintain a cGMP compliant environment in our plasma facility and to expand production and attract donors to our facility. If we misjudge the readiness of a center for an FDA inspection, we may lose credibility with the FDA and cause the FDA to more closely examine all of our operations. Such additional scrutiny could materially hamper our operations and our ability to increase plasma collections. Our ability to expand production and increase our plasma collection center to more efficient production levels may be affected by changes in the economic environment and population in selected regions where we operate our current or future plasma collection centers, by the entry of competitive plasma centers into regions where we operate such centers, by misjudging the demographic potential of individual regions where we expect to expand production and attract new donors, by unexpected facility related challenges or by unexpected management challenges at selected plasma facilities held by us from time to time.
9
Our product candidates may cause serious and unexpected side effects, which could jeopardize our ability to obtain marketing approval for, and continue marketing of, our product candidates.
As for all pharmaceutical products, the use of our product candidates sometimes produces undesirable side effects or adverse reactions or events, or adverse events. Known adverse events of a number of our product candidates include allergic or anaphylactic reactions including shock and the transmission of infective agents. Further, the use of certain product candidates sometimes produces additional adverse events.
In addition, the use of our product candidates may be associated with serious and unexpected adverse events, or with less serious reactions at a greater than expected frequency. This may be especially true when our product candidates are used in critically ill patient populations. When these unexpected events are reported to us, we must undertake a thorough investigation to determine causality and implications for product safety. These events must also be specifically reported to the applicable regulatory authorities. If our evaluation concludes, or regulatory authorities perceive, that there is an unreasonable risk associated with the product, we would be obligated to withdraw the impacted lot(s) of that product. Furthermore, an unexpected adverse event caused by a new product may be recognized only after extensive use of the product, which could expose us to product liability risks, enforcement action by regulatory authorities and damage to our reputation.
Once we produce a product, we rely on physicians to prescribe and administer it as we have directed and for the indications described on the labeling. Although physicians, in the practice of medicine, may prescribe approved drugs for unapproved indications, pharmaceutical companies are prohibited from marketing or promoting their drug products for uses outside the approved label, a practice known as off-label promotion. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label may be subject to significant liability. In addition, the off-label use of our products, if any, may increase the risk of product liability claims and produce results such as reduced efficacy or other adverse effects, and the reputation of our products, if any, in the marketplace may suffer.
Additionally, if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such products, including during any long-term follow-up observation period recommended or required for patients who receive treatment using our products, a number of potentially significant negative consequences could result, including:
| • | regulatory authorities may withdraw approvals of such product; |
| • | regulatory authorities may require additional warnings on the label; |
| • | we may be required to create a Risk Evaluation and Mitigation Strategy or similar risk management plan, which could include a medication guide outlining the risks of such side effects for distribution to patients, a communication plan for healthcare providers and/or other elements to assure safe use; |
| • | we could be sued and held liable for harm caused to patients; and |
| • | our reputation may suffer. |
Any of the foregoing could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
Clinical trials may not demonstrate a clinical benefit of our product candidates.
Clinical trials may not demonstrate a clinical benefit of our product candidates. Positive results from preclinical studies and early clinical trials should not be relied upon as evidence that later stage, large scale or controlled clinical trials will succeed. We will be required to demonstrate with substantial evidence through well-controlled clinical trials that our product candidates are safe and effective for use in a diverse population before we can seek regulatory approvals for their commercial sale. Success in early clinical trials does not mean that future clinical trials will be successful because product candidates in later stage clinical trials may fail to demonstrate sufficient safety and efficacy to the satisfaction of regulatory authorities despite having progressed through initial clinical trials.
10
We may experience numerous unforeseen events prior to, during or as a result of clinical trials that could delay or prevent our ability to receive marketing approval or commercialize any of our product candidates, including:
| • | the FDA or other comparable regulatory authority may disagree as to the number, design or implementation of our clinical trials, or may not interpret the results from clinical trials as we do; |
| • | regulators or institutional review boards, or IRBs, may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
| • | we may not reach agreement on acceptable terms with prospective clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different clinical trial sites; |
| • | clinical trials of our product candidates may produce negative or inconclusive results; |
| • | we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs; |
| • | the number of patients required for clinical trials of our product candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate, participants may drop out of these clinical trials at a higher rate than we anticipate or we may fail to recruit eligible patients to participate in a trial; |
| • | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; |
| • | regulators may issue a clinical hold, or regulators or IRBs may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks; |
| • | the cost of clinical trials of our product candidates may be greater than we anticipate; |
| • | the FDA or other comparable regulatory authorities may fail to approve our manufacturing processes or facilities; |
| • | the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; |
| • | our product candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators or IRBs to suspend or terminate the clinical trials; and |
| • | the approval policies or regulations of the FDA or other comparable regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
To the extent that the results of the trials are not satisfactory for the FDA or regulatory authorities in other countries or jurisdictions to approve our BLA, new drug application, or NDA, or other comparable applications, the commercialization of our product candidates may be significantly delayed, or we may be required to expend significant additional resources, which may not be available to us, to conduct additional trials in support of potential approval of our product candidates.
Even after the completion of Phase 3 clinical trials, regulatory authorities may disagree with our clinical trial design and our interpretation of data and may require us or our partners to conduct additional clinical trials to demonstrate the efficacy of our product candidates. There is no assurance that a regulatory authority will issue a biologics license or manufacturing authorization. Regulatory authorities may determine that our product candidates, the manufacturing process or the manufacturing facilities do not meet applicable requirements to ensure the continued safety, purity and potency of our product candidates.
11
Our product candidates could cause undesirable and potentially serious side effects during clinical trials that could delay or prevent their regulatory approval or commercialization.
Our product candidates could cause undesirable and potentially serious side effects during clinical trials that could delay or prevent their regulatory approval or commercialization. Undesirable side effects caused by any of our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by regulatory authorities for any or all targeted indications. This, in turn, could prevent us from commercializing our product candidates and generating revenues from their sale. In addition, if our product candidates receive marketing approval and we or others later identify undesirable side effects caused by the product:
| • | regulatory authorities may withdraw their approval of the product; |
| • | we may be required to recall the product, change the way the product is administered, conduct additional clinical trials or change the labelling of the product; |
| • | a product may become less competitive and product sales may decrease; or |
| • | our reputation may suffer. |
Any one or a combination of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product, which in turn could delay or prevent us from generating revenues from the sale of the affected product.
In addition, as with the results of any statistical sampling, we cannot be sure that all side effects of our product candidates may be uncovered in clinical trials, and it may be the case that only with a significantly larger number of patients exposed to the product candidate for a longer duration, may a more complete safety profile be identified.
Failure to recruit and enroll patients for clinical trials may cause the development of our product candidates to be delayed.
Failure to recruit and enroll patients for clinical trials may cause the development of our product candidates to be delayed. We may encounter delays or rejections in recruiting and enrolling enough patients to complete clinical trials. Patient enrollment depends on many factors, including:
| • | the patient eligibility criteria defined in the protocol; |
| • | the number of patients with the disease or condition being studied; |
| • | the understanding of risks and benefits of the product candidate in the trial; |
| • | clinicians’ and patients’ perceptions as to the potential advantages of the product candidate being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating or drugs that may be used off-label for these indications; |
| • | the size and nature of the patient population who meet inclusion criteria; |
| • | the proximity of patients to study sites; |
| • | the design of the clinical trial; |
| • | clinical trial investigators’ ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| • | competing clinical trials for similar therapies or other new therapeutics not involving T cell-based immunotherapy; |
| • | our ability to obtain and maintain patient consents; |
12
| • | the risk that patients enrolled in clinical trials will drop out of the clinical trials before completion of their treatment; and |
| • | the impact of public health epidemics or pandemics, such as the ongoing COVID-19 pandemic. |
In particular, some of our clinical trials are designed to enroll patients with characteristics that are found in a very small population. In addition, since the number of qualified clinical investigators is limited, we expect to conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which could further reduce the number of patients who are available for our clinical trials in these clinical trial sites.
Delays in patient enrollment may result in increased costs or may affect the timing or outcome of the planned clinical trials, which could prevent completion of these clinical trials and adversely affect our ability to advance the development of our product candidates. In addition, many of the factors that may lead to a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
We do not know whether any of our ongoing or planned clinical trials will proceed or be completed on schedule, or at all.
We do not know whether any of our ongoing or planned clinical trials will proceed or be completed on schedule, or at all. The commencement of our planned clinical trials could be substantially delayed or prevented by several factors, including:
| • | limited number of, and competition for, suitable patients with the indications required for enrollment in our clinical trials; |
| • | limited number of, and competition for, suitable sites to conduct our clinical trials; |
| • | delay or failure to obtain FDA or non-U.S. regulatory agencies’ approval or agreement to commence a clinical trial; |
| • | delay or failure to obtain sufficient supplies of the product candidate for our clinical trials; |
| • | delay or failure to reach agreement on acceptable clinical trial agreement terms or clinical trial protocols with prospective sites or investigators; and |
| • | delay or failure to obtain IRB approval to conduct a clinical trial at a prospective site. |
The completion of any clinical trial could also be substantially delayed or prevented by several factors, including:
| • | slower than expected rates of patient recruitment and enrollment; |
| • | failure of patients to complete the clinical trial; |
| • | unforeseen safety issues; |
| • | lack of efficacy evidenced during any clinical trial; |
| • | termination of any clinical trial by one or more clinical trial sites; |
| • | inability or unwillingness of patients or medical investigators to follow clinical trial protocols; |
| • | failure by clinical research organization to comply with applicable laws and regulations; |
| • | inability to monitor patients adequately during or after treatment; and |
| • | introduction of competitive product candidates that may impede our ability to retain patients in any clinical trial. |
13
Clinical trials may be suspended or terminated at any time by the FDA, other regulatory authorities, the IRB overseeing the clinical trial at issue, any of our clinical trial sites with respect to that site, or us. Any failure or significant delay in completing any clinical trial for our product candidates could materially harm our financial results and the commercial prospects for our product candidates. In addition, the impact of public health epidemics or pandemics, such as the ongoing COVID-19 pandemic, which is currently having a global impact, may delay or prevent patients from enrolling or from receiving treatment in accordance with the protocol and the required timelines, which could delay our clinical trials, or otherwise prevent us from completing our clinical trials on our planned timelines or at all, and harm our ability to obtain approval for such product candidate. For example, in connection with the COVID-19 pandemic, in May 2020 we recently terminated treatment in an open-label rollover, single-arm Phase 2 clinical trial of fezagepras for the treatment of Alström syndrome which was started in October 2017 as a result of the re-deployment of clinical staff because of the current COVID-19 pandemic.
We may not be successful in our efforts to build a pipeline of product candidates.
We intend to develop additional product candidates. However, we may not be able to develop product candidates that are safe and effective, or which compare favorably with other commercially available alternatives. Even if we are successful in continuing to build our pipeline and developing product candidates, the potential product candidates that we identify may not be suitable for clinical development, including as a result of lack of safety, lack of tolerability, lack of efficacy or other characteristics that indicate that they are unlikely to be products that will receive marketing approval, achieve market acceptance or obtain reimbursements from third-party payors. We cannot provide any assurance that we will be able to successfully advance any of these additional product candidates through the development process. Our research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development or commercialization for many reasons, including the following:
| • | we may not be successful in building a platform to identify additional product candidates; |
| • | we may not be able or willing to assemble sufficient resources to acquire or discover additional product candidates; |
| • | our product candidates may not succeed in preclinical or clinical testing; |
| • | we may fail to obtain regulatory approval to initiate a clinical trial, to enter into clinical trial agreements with study sites and principal investigators, obtain the relevant ethics committee approvals to initiate the clinical trials or fail or be delayed to recruit and enroll subjects; |
| • | a product candidate may on further study be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria; |
| • | competitors may develop alternatives that render our product candidates obsolete or less attractive; |
| • | product candidates we develop may nevertheless be covered by third-parties’ patents or other exclusive rights; |
| • | the market for a product candidate may change during our development program so that the continued development of that product candidate is no longer reasonable; |
| • | we may revisit our corporate strategy and corporate priorities and no longer pursue the development, manufacture or commercialization of certain product candidates that require substantial technical, financial and human resources; |
| • | we may be unable to find qualified contract manufacturing organizations or contract research organizations or delays or inability to obtain timely regulatory approvals; |
| • | a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
| • | a product candidate may not be accepted as safe and effective by patients, the medical community or third-party payors, if applicable. |
14
If any of these events occur, we may be forced to abandon our development efforts for a program or programs, or we may not be able to identify, discover, develop or commercialize additional product candidates, which would have a material adverse effect on our business and could potentially cause us to cease operations.
Even if we receive FDA or other regulatory approval to market our product candidates, we cannot assure that any such product candidates will be commercialized, widely accepted in the marketplace or as effective or more effective than other commercially available alternatives. Further, because of our limited financial and managerial resources, we are required to focus our research programs on certain product candidates and on specific diseases. As a result, we may fail to capitalize on viable commercial products or profitable market opportunities, be required to forego or delay pursuit of opportunities with other product candidates or other diseases that may later prove to have greater commercial potential, or relinquish valuable rights to such product candidates through divestment, sale, collaboration, licensing, royalty or other strategic alternative arrangements in cases in which it would have been advantageous for us to retain sole development, manufacture and/or commercialization rights.
If we do not successfully develop, manufacture and commercialize product candidates or collaborate with others to do so, we will not be able to obtain product revenue in future periods, which could significantly harm our financial position.
We are exploring investing efforts into building a drug discovery platform but these efforts may not be successful.
We intend to develop a drug discovery platform that will be focused on exploring structural biology and drug design by utilizing new tools and technologies, which may include artificial intelligence and machine learning, in the drug discovery process with the goal to identify novel biologic targets that may have been previously undruggable and potentially develop product candidates for these novel targets.
However, we may not be successful in our development of a drug discovery platform, as developing a drug discovery platform to identify new product candidates requires substantial technical, financial and human resources. Even if we are successful, the potential product candidates that we identify may not be suitable for clinical development, including as a result of lack of safety, lack of tolerability or other characteristics that indicate that they are unlikely to be products that will receive marketing approval, achieve market acceptance or obtain reimbursements from third-party payors. If we are unable to develop our drug discovery platform and identify suitable compounds for preclinical and clinical development, we may not be able to obtain sufficient product revenues in future periods, which could significantly harm our financial position.
We face competition and the development of new product candidates by other companies could materially adversely affect our business and our product candidates.
We face competition and the development of new product candidates by other companies could materially adversely affect our business and our product candidates. The biopharmaceutical and pharmaceutical industries are highly competitive, and we must compete with pharmaceutical companies, biotechnology companies, academic and research institutions as well as governmental agencies for the development and commercialization of product candidates. Some of these competitors develop product candidates in the indications in which we are involved and could be considered direct or indirect competitors.
In the indications currently being studied by us for development, there may exist companies that are at a more advanced stage of developing a product to treat those same diseases. Some of these competitors have capital resources, research and development personnel and facilities that are superior to ours. Mergers and acquisitions in the biotechnology and pharmaceutical industries may also result in even more resources being concentrated among a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical studies, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. In addition, some competitors are more experienced than we are in the commercialization of medical products and already have a sales force in place to launch new products. Consequently, they may be able to develop alternative forms of medical treatment which could compete with our product candidates and commercialize them more rapidly and effectively than we can.
15
If the market opportunities for any of our product candidates we may develop are smaller than we believe they are, our revenue may be adversely affected and our business may suffer. Moreover, because the target patient populations we are seeking to treat are small, and the addressable patient population even smaller, we must be able to successfully identify patients and capture a significant market share to achieve profitability and growth.
We focus our research and product development on treatments for rare indications. Given the small number of patients who have the diseases that we are targeting, it is critical to our ability to grow and become profitable that we continue to successfully identify patients with these rare diseases. Our projections of both the number of people who have these diseases, as well as the subset of people with these diseases who have the potential to benefit from treatment with our product candidates we may develop, are based on our beliefs and estimates. These estimates have been derived from a variety of sources, including the scientific literature, surveys of clinics, patient foundations or market research that we conducted, and may prove to be incorrect or contain errors. New studies may change the estimated incidence or prevalence of these diseases. The number of patients may turn out to be lower than expected. The effort to identify patients with diseases we seek to treat is in early stages, and we cannot accurately predict the number of patients for whom treatment might be possible. Additionally, the potentially addressable patient population for our product candidates we may develop may be limited or may not be amenable to treatment with our other product candidates we may develop, and new patients may become increasingly difficult to identify or gain access to, which would adversely affect our results of operations and our business. Further, even if we obtain significant market share for our product candidates we may develop, because the potential target populations are very small, we may never achieve profitability despite obtaining such significant market share.
We depend on our key personnel to research, develop, manufacture and bring new products to the market and the loss of key personnel or the inability to attract highly qualified individuals could have a material adverse effect on our business and growth potential.
We depend on our key personnel to research, develop, manufacture and bring new products to the market and the loss of key personnel or the inability to attract highly qualified individuals could have a material adverse effect on our business and growth potential. Our mission is to discover or acquire novel therapeutic product candidates targeting unmet medical needs in attractive specialty markets. The achievement of this mission requires qualified scientific and management personnel. The loss of scientific personnel or of members of management could have a material adverse effect on our business. In addition, our growth is and will continue to be dependent, in part, on our ability to retain and hire qualified scientific personnel. There can be no guarantee that we will be able to continue to retain our current employees or will be able to attract qualified personnel and members of management to pursue our business plan.
We may fail to achieve our publicly-announced milestones or not achieve publicly-announced milestones in due time.
We may fail to achieve publicly-announced milestones or may not achieve our publicly-announced milestones in due time. From time to time, we publicly announce the timing of the occurrence of certain events. These statements are forward-looking and are based on management’s best estimate relating to the occurrence of such events. However, the events and actual timing of such events may differ from what has been publicly disclosed. These variations may occur as a result of a series of events, including the nature of the results obtained during a clinical trial or during a research phase, problems with a supplier or contract manufacturing organization or any other event having the effect of delaying or otherwise impacting the milestone publicly announced. Our policy on forward-looking information does not consist in updating such information if the publicly disclosed timeline varies, unless otherwise required to do so by law. Any variation in the timing of certain events having the effect of postponing or otherwise postponing such events could have an adverse material effect on our business plan, financial conditions or operating results.
16
We may develop and conduct trials of fezagepras in combination therapy with other therapies, which exposes us to additional risks.
We may develop and conduct trials of fezagepras both as a monotherapy and in combination with nintedanib and/or pirfenidone and may develop future product candidates that involve combination therapy with one or more currently approved therapies. Even if any product candidate we develop were to receive marketing approval or be commercialized for use in combination with other existing therapies, we would continue to be subject to the risks that the FDA or similar regulatory authorities outside of the United States could revoke approval of the therapy used in combination with our product candidate or that safety, efficacy, manufacturing or supply issues could arise with these existing therapies. This could result in our own products being removed from the market or being less successful commercially.
Interim top-line and preliminary results from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures, which could result in material changes in the final data.
From time to time, we may publish interim top-line or preliminary results from our clinical trials. Interim results from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Preliminary or top-line results also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are available. Differences between preliminary or interim data and final data could significantly harm our business prospects and may cause the trading price of our common stock to fluctuate significantly.
If we are unable to enter into divestment, sales, marketing and distribution, licensing agreements or other strategic alternative transactions with third parties, our product candidates, if and when they are approved, may not be successfully launch or commercialized.
We currently do not have commercialization capabilities, such that we are unable to commercialize any product candidate for which we may obtain regulatory approval. To achieve commercial success for any product candidate for which we may obtain marketing approval, we will need to enter into distribution or licensing agreements or other alternative strategic transaction, which may involve divestment of certain assets, with third parties to commercialize and deliver our product candidates to patients and healthcare providers.
We would have to pursue collaborative arrangements regarding the sales and marketing of our products. However, we may not be successful in entering into arrangements with third parties to sell, market and distribute our product candidates or may be unable to do so on terms that are favorable to us, or if we are able to do so, that they would be effective and successful in commercializing our products. In addition, we would have limited control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our product candidates effectively. If we do not establish sales, marketing and distribution capabilities successfully in collaboration with third parties, we will not be successful in commercializing our product candidates in the United States or elsewhere.
The development, manufacture and commercialization of product candidates could expose us to liability claims which could exceed our insurance coverage.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. In addition, a product liability claim could tarnish our reputation, whether or not such claims are covered by insurance or are with or without merit. Such liability claims may, amongst other things, result in:
| • | decreased demand for any product candidates or drugs that we may develop; |
| • | injury to our reputation and significant negative media attention; |
| • | withdrawal of clinical trial participants; |
17
| • | clinical trial hold or product recall; |
| • | substantial monetary awards paid to trial participants or patients; |
| • | reduced resources of our management to pursue our business strategy; and |
| • | the inability to commercialize any products that we may develop. |
The development, manufacture and commercialization of product candidates could expose us to liability claims which could exceed our insurance coverage. A risk of product liability claims is inherent in the development and commercialization of human therapeutic products. Product liability insurance is very expensive and offers limited protection. A product liability claim against us could potentially be greater than the coverage offered and, therefore, have a material adverse effect upon us and our financial position.
If our competitors develop and market products that are safer, more effective or more convenient or less expensive than our products, if any, our commercial opportunity will be reduced or eliminated.
Our commercial opportunity with respect to our products, if approved, will be reduced or eliminated if our competitors develop and market products that are more effective, have fewer side effects, are more convenient or are less expensive than our products, if any. Our competitors include large, fully-integrated pharmaceutical companies and more established biotechnology and medical device companies, many of which have significant resources and expertise in research and development, manufacturing, testing, obtaining regulatory approvals and marketing. It is possible that competitors will succeed in developing technologies that are safer, more effective, more convenient or with a lower cost of goods and price than those in our product candidates, or that would render our technology obsolete or non-competitive. See “—Risks Related to our Intellectual Property” for additional risks relating to our intellectual property protection from generics entering into our commercial markets.
We may not be successful in achieving cost of goods at commercial scale that provide for an attractive margin.
We have not yet established manufacturing capacity at sufficient commercial scale and may not have the financial, technical or human resources to do so, or we may overestimate cost reductions from economies of scale that can be realized with our manufacturing processes. We may be unable to manage the cost of goods for our product candidates to levels that will allow for a margin in line with our expectations and return on investment if and when those product candidates are commercialized.
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or have a greater likelihood of success.
Because we have limited financial and management resources, we focus on research programs and product candidates that we identify for specific indications. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through divestment, sale, collaboration, licensing, royalty or other strategic alternative arrangements in cases in which it would have been more advantageous for us to retain sole development, manufacture and/or commercialization rights to such product candidate.
18
Risks Related to Regulatory Approval of Our Product Candidates and Other Legal Compliance Matters
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, but a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing approval of a product candidate, comparable regulatory authorities in other jurisdictions must also approve the manufacturing, marketing and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the United States, including additional preclinical studies or clinical trials as clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval.
Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our products in certain countries. If we fail to comply with the regulatory requirements in international markets and/or to receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market potential of our product candidates will be harmed.
Even if we obtain marketing approvals for our product candidates, the terms of approvals and ongoing regulation of our products may limit how we manufacture and market our products and compliance with such requirements may involve substantial resources, which could materially impair our ability to generate revenue.
Even if marketing approval of a product candidate is granted, an approved product and its manufacturer and marketer are subject to ongoing review and extensive regulatory requirements for manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, sampling, and recordkeeping, including the potential to conduct costly post- marketing studies or clinical trials and surveillance to monitor the safety or efficacy of the product. We must also comply with requirements concerning advertising and promotion for any of our product candidates for which we obtain marketing approval. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved labeling. Thus, we will not be able to promote any products we develop for indications or uses for which they are not approved. In addition, manufacturers of approved products and those manufacturers’ facilities are required to comply with extensive regulatory requirements of the FDA and other regulatory authorities, including ensuring that quality control and manufacturing procedures conform to cGMP and other comparable regulations and standards, which include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation and reporting requirements. We or our suppliers could be subject to periodic unannounced inspections by the FDA or other regulatory authorities to monitor and ensure compliance with cGMP.
Accordingly, assuming we receive marketing approval for one or more of our product candidates, we and our suppliers will continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production, product surveillance and quality control. If we are not able to comply with post- approval regulatory requirements, we could have the marketing approvals for our products withdrawn by regulatory authorities and our ability to market any future products could be limited, which could adversely affect our ability to achieve or sustain profitability.
Thus, the cost of compliance with post-approval regulations may have a negative effect on our operating results and financial condition.
19
Any product candidate for which we obtain marketing approval could be subject to post-marketing restrictions or recall or withdrawal from the market, and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our product candidates, when and if any of them are approved.
The FDA and other federal and state agencies, including the U.S. Department of Justice, or DOJ, closely regulate compliance with all requirements governing prescription drug products, including requirements pertaining to marketing and promotion of products in accordance with the provisions of the approved labeling and manufacturing of products in accordance with cGMP requirements. The FDA and DOJ impose stringent restrictions on manufacturers’ communications regarding off-label use and if we do not market our products for their approved indications, or if other of our marketing claims are deemed false or misleading, we may be subject to enforcement action. Violations of such requirements may lead to investigations alleging violations of the Food, Drug and Cosmetic Act and other statutes, including the False Claims Act and other federal and state health care fraud and abuse laws as well as state consumer protection laws.
Our failure to comply with all regulatory requirements, including cGMP, and later discovery of previously unknown adverse events or other problems with our products, manufacturers or manufacturing processes, may yield various results, including:
| • | litigation involving patients taking our products; |
| • | restrictions on such products, manufacturers or manufacturing processes; |
| • | restrictions on the labeling or marketing of a product; |
| • | restrictions on product distribution or use; |
| • | requirements to conduct post-marketing studies or clinical trials; |
| • | warning or untitled letters; |
| • | withdrawal of the products from the market; |
| • | refusal to approve pending applications or supplements to approved applications that we submit; |
| • | fines, restitution or disgorgement of profits or revenue; |
| • | suspension or withdrawal of marketing approvals; |
| • | suspension of any ongoing clinical trials; |
| • | damage to relationships with any potential collaborators; |
| • | unfavorable press coverage and damage to our reputation; |
| • | refusal to permit the import or export of our products; |
| • | injunctions or the imposition of civil or criminal penalties. |
Noncompliance by us or any collaborator with regulatory requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties. Similarly, failure to comply with regulatory requirements regarding the protection of personal information can also lead to significant penalties and sanctions.
Noncompliance with EU requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, also can result in significant financial penalties. Similarly, failure to comply with the European Union’s requirements regarding the protection of personal information can also lead to significant penalties and sanctions.
20
If any of these events occurs, our ability to sell such product may be impaired, and we may incur substantial additional expense to comply with regulatory requirements, which could adversely affect our business, financial condition and results of operations.
Our employees, independent contractors, principal investigators, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee fraud or other misconduct or failure to comply with applicable regulatory requirements. Misconduct by employees and independent contractors, such as principal investigators, consultants, commercial partners, and vendors, could include failures to comply with regulations of the FDA and other comparable regulatory authorities, to provide accurate information to such regulators, to comply with manufacturing standards we have established, to comply with healthcare fraud and abuse laws, to report financial information or data accurately or to disclose unauthorized activities to us. In particular, sales, marketing and other business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of business activities, including, but not limited to, research, manufacturing, distribution, pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee and independent contractor misconduct could also involve the improper use of individually identifiable information, including, without limitation, information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation.
In addition, federal procurement laws impose substantial penalties for misconduct in connection with government contracts and require certain contractors to maintain a code of business ethics and conduct. It is not always possible to identify and deter employee and independent contractor misconduct, and any precautions we take to detect and prevent improper activities may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws. If any such actions are instituted against us, those actions could have a significant impact on our business, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgement of profits, imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, or other government supported healthcare in other jurisdictions, contractual damages, reputational harm, diminished profits and future earnings, additional reporting or oversight obligations if we become subject to a corporate integrity agreement or other agreement to resolve allegations of noncompliance with the law and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate.
Some of our employees in our manufacturing facility are unionized, as employees may be in manufacturing facilities of our suppliers. While unionized facilities are subject to the risk of labour disruptions from time to time, we cannot predict whether or when any labour disruption may arise, or how long such a disruption could last.
We are exposed to significant labour disruptions which could lead to a lengthy shutdown of our or our customers’ and/or our suppliers’ production lines, which could have a material adverse effect on our operations and profitability. In January 2021, the Quebec’s Tribunal Administratif du Travail has approved the unionization request made by the production technicians and team leaders of our manufacturing facility in Laval, Quebec, Canada. This group represents approximately 25 employees. The process to negotiate a first bargaining agreement between this group and the Company is expected to start during the first few months of 2021.
Our current and future operations are subject to extensive healthcare laws, regulation and enforcement and failure to comply with those laws could have a material adverse effect on our results of operations and financial condition.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any product we may develop and any product for which we obtain marketing approval. Our current and future operations expose us to broadly applicable healthcare laws and regulations that may affect the business or financial arrangements and relationships through which we research, market, sell and distribute our products, if any. The laws that may affect our operations in the United States include, but are not limited to:
21
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
| • | federal civil and criminal false claims laws, including the False Claims Act, which can be enforced by private individuals through civil whistleblower or qui tams actions, and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which, among other things, created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, which imposes certain requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform services for them that involve the use, or disclosure of, individually identifiable health information, and their covered subcontractors, relating to the privacy, security, and transmission of individually identifiable health information; |
| • | the Physician Payments Sunshine Act, enacted as part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or, collectively, ACA, which requires certain manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the Centers for Medicare & Medicaid Services, or CMS, information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members, and beginning in 2022, will require applicable manufacturers to report information regarding payments and transfers of value provided during the previous year to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, anesthesiologist assistants, and certified nurse-midwives; and |
| • | foreign and state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require product manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; state and local laws that require the registration of pharmaceutical sales representatives; and state and foreign laws governing the privacy and security of health information in certain circumstances, including General Data Protection Regulations, or GDPR, many of which differ from each other in significant ways, thus complicating compliance efforts. |
The scope of these laws and our lack of experience in establishing the compliance programs necessary to comply with this complex and evolving regulatory environment increases the risks that we may violate the applicable laws and regulations. If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to significant penalties, including civil, criminal, and administrative penalties, damages, fines, disgorgement, imprisonment, the curtailment or restructuring of our operations, diminished profits and future earnings, the exclusion from participation in federal and state healthcare programs, additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, any of which could materially adversely affect our ability to operate our business and our financial results.
22
Third-party payors, including governmental health administration authorities, private health insurers and other organizations, may not provide coverage and adequate reimbursement for our products, if any, and related treatment.
Even if we obtain approvals from the FDA or other comparable regulatory agencies and are able to initiate commercialization of our clinical-stage product candidates or any other product candidates we develop, the product candidate may not achieve market acceptance among physicians, patients, hospitals, including pharmacy directors, and third-party payors and, ultimately, may not be commercially successful. Market acceptance of our products, if any, is uncertain and depends on a variety of factors, some of which are not under our control. Our ability to commercialize our products, if any, with success will depend on a variety of factors. One of these is the extent to which coverage and adequate reimbursement for our products, if any, and related treatment will be made available by third-party payors, such as governmental health administration authorities, managed-care organizations, private health coverage insurers and other organizations. Obtaining and maintaining coverage and adequate reimbursement for a product is time-consuming and a costly process that could require us to provide supporting scientific, clinical and cost effectiveness data for the use of a product. There can be no guarantee our data will be positive enough for third-party payors to cover and reimburse one of our products, if any.
If third-party payors fail to provide coverage and adequate reimbursement rates for our product candidates, our revenues and potential for profitability will be reduced. Our product revenues will depend principally upon the coverage and reimbursement rates established by third-party payors. These third-party payors are increasingly challenging the price and examining the cost effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status, if any, of newly approved drugs, pharmaceutical products or product indications. We may need to conduct post-marketing clinical trials in order to demonstrate the cost-effectiveness of our products, if any. Such clinical trials may require us to dedicate a significant amount of management time and financial and other resources. If reimbursement of such product is unavailable or limited in scope, our revenues could be reduced. Moreover, the determination of the price of certain drugs in orphan disease indications could be even more difficult to make due to lack of comparable products. Further, no uniform policy for coverage and reimbursement exists in the United States, and coverage and reimbursement can differ significantly from payor to payor. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates, but also have their own methods and approval process apart from Medicare determinations. Our inability to promptly obtain coverage and adequate reimbursement rates from third-party payors for products that we may develop and for which we obtain regulatory approval could have a material and adverse effect on our business, financial condition, results of operations and prospects.
Other factors that will have an impact on the acceptance of our products, if any, include:
| • | acceptance of the products by physicians and patients as safe and effective treatments; |
| • | the effectiveness of our sales and marketing efforts (or those of our commercial partner); |
| • | storage requirements and ease of administration; |
| • | prevalence and severity of side effects; and |
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
The trend toward managed healthcare in the United States, the growth of organizations such as HMOs and MCOs, and legislative proposals to reform healthcare and government insurance programs could significantly influence the purchase of pharmaceutical products, resulting in lower prices and a reduction in product demand.
23
In the United States and some foreign jurisdictions there have been, and continue to be, several legislative and regulatory changes and proposed reforms of the healthcare system to contain costs, improve quality, and expand access to care. In the United States, there have been and continue to be a number of healthcare-related legislative initiatives that have significantly affected the pharmaceutical industry. For example, the ACA was passed in March 2010, and substantially changed the way healthcare is financed by both governmental and private insurers, and continues to significantly impact the U.S. pharmaceutical industry.
There have been executive judicial and Congressional challenges to certain aspects of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively the ACA.
For example, the Tax Cuts and Jobs Act of 2017, or the Tax Act included a provision that repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” In addition, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminated the health insurer tax. Further, the Bipartisan Budget Act of 2018, among other things, amended the ACA, effective January 1, 2019, to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole”. On December 14, 2018, a Texas U.S. District Court Judge ruled that the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as part of the Tax Act. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. The U.S. Supreme Court is currently reviewing the case, although it is unknown when a decision will be made. Further, although the U.S. Supreme Court has not yet ruled on the constitutionality of the ACA, on January 28, 2021, President Biden issued an executive order to initiate a special enrollment period from February 15, 2021 through May 15, 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructs certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. It is unclear how the Supreme Court ruling, other such litigation, and the healthcare reform measures of the Biden administration will impact the ACA.
Moreover, there has been heightened governmental scrutiny in the United States of pharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. Such scrutiny has resulted in several recent Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products.
24
At the federal level, the Trump administration used several means to propose or implement drug pricing reform, including through federal budget proposals, executive orders and policy initiatives. For example, on July 24, 2020 and September 13, 2020, the Trump administration announced several executive orders related to prescription drug pricing that seek to implement several of the administration’s proposals. As a result, the FDA released a final rule on September 24, 2020, effective November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. Further, on November 20, 2020, the U.S. Department of Health & Human Services, or HHS, finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Medicare Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The implementation of the rule has been delayed by the Biden administration from January 1, 2022 to January 1, 2023 in response to ongoing litigation. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a new safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers, the implementation of which have also been delayed pending review by the Biden administration until March 22, 2021. On November 20, 2020, CMS issued an interim final rule implementing the Trump administration’s Most Favored Nation executive order, which would tie Medicare Part B payments for certain physician-administered drugs to the lowest price paid in other economically advanced countries, effective January 1, 2021. On December 28, 2020, the U.S. District Court in Northern California issued a nationwide preliminary injunction against implementation of the interim final rule. It is unclear whether the Biden administration will work to reverse these measures or pursue similar policy initiatives. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Current and future healthcare legislation could have a significant impact on our business. There is uncertainty with respect to the impact these changes, if any, may have, and any changes likely will take time to unfold. In addition, it is possible that additional governmental action is taken to address the COVID-19 pandemic. For example, on April 18, 2020, CMS announced that qualified health plan issuers under the ACA may suspend activities related to the collection and reporting of quality data that would have otherwise been reported between May and June 2020 given the challenges healthcare providers are facing responding to the COVID-19 pandemic. Any additional federal or state healthcare reform measures could limit the amounts that third-party payors will pay for healthcare products and services, and, in turn, could significantly reduce the projected value of certain development projects and reduce our profitability.
We are subject to U.S. and certain foreign export and import controls, sanctions, embargoes, anti-corruption laws, and anti-money laundering laws and regulations. Compliance with these legal standards could impair our ability to compete in domestic and international markets. We can face criminal liability and other serious consequences for violations, which can harm our business.
We are subject to export control and import laws and regulations, including the U.S. Export Administration Regulations, U.S. Customs regulations, various economic and trade sanctions regulations administered by the U.S. Treasury Department’s Office of Foreign Assets Controls, the Foreign Corrupt Practices Act, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, and other state and national anti-bribery and anti-money laundering laws in the countries in which we conduct activities. Anti-corruption laws are interpreted broadly and prohibit companies and their employees, agents, contractors, and other collaborators from authorizing, promising, offering, or providing, directly or indirectly, improper payments or anything else of value to recipients in the public or private sector. We may engage third parties to develop, manufacture or sell our products, if any, outside the United States, to conduct clinical trials, and/or to obtain necessary permits, licenses, patent registrations, and other regulatory approvals. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities, and other organizations. We can be held liable for the corrupt or other illegal activities of our employees, agents, contractors, and other collaborators, even if we do not explicitly authorize or have actual knowledge of such activities. Any violations of the laws and regulations described above may result in substantial civil and criminal fines and penalties, imprisonment, the loss of export or import privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm, and other consequences.
25
Risks Related to our Business Operations and our Reliance on Third Parties
The success of our product candidates is influenced by our collaborations with our partners and any adverse developments in our relationship with our partners could materially harm our business.
The success of our product candidates is influenced by our collaborations with our partners. Any adverse developments in our relationship with our partners could materially harm our business. We are subject to a number of risks associated with any collaboration that could be entered into for the development of our product candidates, including the risk that these collaborators may terminate the relevant agreement(s) upon the occurrence of certain specified events, including a material breach by us of any of our obligations under the respective agreements. If any of our partners terminate or breach our agreements with them, or otherwise fail to complete their obligations in a timely manner, it may have a detrimental effect on our financial position by reducing or eliminating the potential for us to receive technology access and license fees, milestones and royalties, reimbursement of development costs, as well as possibly requiring us to devote additional efforts and incur costs associated with pursuing internal development of product candidates. Furthermore, if our partners do not prioritize and commit sufficient resources to programs associated with our product candidates or collaboration product candidates, we or our partners may be unable to commercialize these product candidates, which would limit our ability to generate revenue and become profitable.
We may not be able successful in entering into strategic agreements with third parties for the divestment of our plasma-derived therapeutics business, and related non-core assets, in whole or in part, which may result in a shut down of the plasma-derived therapeutics’ business, which may adversely affect our short term cash position and expose us to increased liabilities.
We have announced that our plasma-derived therapeutics business is no longer aligned with our new corporate strategy and that we are re-focusing our corporate strategy and resources on our small molecule therapeutics business, including our lead drug candidate fezagepras, to become more streamlined with a singular focus on our core research capabilities and emerging pipeline. We have not made any decisions related to any specific strategic alternatives for our plasma-derived therapeutics business and there can be no assurance of any successful outcome, or the timing or form of these efforts. Should these efforts be unsuccessful, we may close our plasma-derived therapeutics related operations. A closure of these operations may adversely affect our short-term cash position and expose us to increased liabilities, including liabilities related to the early termination of contractual obligations.
We have entered, and may in the future enter into, collaboration agreements with third parties for the development and commercialization of our product candidates, which may adversely affect our ability to generate revenue.
We have entered into and may seek to enter into additional collaborations or partnerships with third parties for the development, manufacture and/or potential commercialization of our product candidates. Should we seek to collaborate with a third party with respect to a prospective development program, we may not be able to locate a suitable partner or to enter into an agreement on commercially reasonable terms or at all. Even if we succeed in securing partners for the development and commercialization of our product candidates, we have limited control over the time and resources that our partners may dedicate to the development and commercialization of our product candidates. These collaborations pose a number of risks, including the following:
| • | partners may not have sufficient resources or decide not to devote the necessary resources due to internal constraints such as budget limitations, lack of human resources or a change in strategic focus; |
| • | partners may believe our intellectual property is not valid or is unenforceable or the product candidate infringes on the intellectual property rights of others; |
| • | partners may dispute their responsibility to conduct development and commercialization activities pursuant to the applicable collaboration, including the payment of related costs or the division of any revenue; |
| • | partners may decide to pursue a competitive product developed outside of the collaboration arrangement; |
| • | partners may not be able to obtain, or believe they cannot obtain, the necessary regulatory approvals; or |
26
| • | partners may delay the development or commercialization of our product candidates in favor of developing or commercializing another party’s product candidate. |
Thus, collaboration agreements may not lead to development, manufacture, regulatory approval or successful commercialization of product candidates in the most efficient manner or at all. Some collaboration agreements are terminable without cause on short notice. Once a collaboration agreement is signed, it may not lead to regulatory approval and commercialization of a product candidate. We also face competition in seeking out partners. If we are unable to secure new collaborations that achieve the collaborator’s objectives and meet our expectations, we may be unable to advance our product candidates and may not generate meaningful revenue.
Our business could be adversely affected by the effects of health epidemics, including the global COVID-19 pandemic.
In connection with the COVID-19 pandemic, governments have implemented significant measures, including closures, quarantines, travel restrictions and other social distancing directives, intended to control the spread of the virus. To the extent that these restrictions remain in plan, additional prevention and mitigation measures are implemented in the future, or there is uncertainty about the effectiveness of these or any other measures to contain or treat COVID-19, our ability to continue to operate our plasma collection centers and our third-party contract manufacturing organizations’ ability to manufacture and deliver on a timely basis our product candidates and our ability to advance our research and development programs and business and corporate development activities may be limited. Such restrictions, limitations or other events may result in a period of business and manufacturing disruption, and in reduced operations, any of which could materially affect our business, financial condition and results of operations.
The continued spread of COVID-19, which has caused a broad impact globally, may materially affect us economically. The economic impact brought by, and the duration of, COVID-19, are uncertain and fluid, and the impact of this or any future widespread pandemic could result in significant disruption of global financial markets, reducing our ability to access capital, which could in the future negatively affect our liquidity. In addition, any recession or market correction in connection with the spread of COVID-19 could materially affect our business and the value of our common shares.
The continued spread of COVID-19 globally has impacted certain of our clinical trials, and could also adversely affect our planned clinical trial operations, including our ability to initiate the trials on the expected timelines and recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 if an outbreak occurs in their geography. Further, the COVID-19 pandemic could result , in delays in our clinical trials due to prioritization of hospital resources toward the outbreak, restrictions in travel, potential unwillingness of patients to enroll in trials at this time, or the inability of patients to comply with clinical trial protocols to the extent that quarantines or travel restrictions impede patient movement or interrupt healthcare services. In addition, we rely on independent clinical investigators, contract research organizations and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our preclinical studies and clinical trials, and the COVID-19 pandemic may affect their ability to devote sufficient time and resources to our programs or to travel to sites to perform work for us. For example, in connection with the COVID-19 pandemic, in May 2020 we terminated treatment in an open-label rollover, single-arm Phase 2 clinical trial of fezagepras for the treatment of Alström syndrome which was started in October 2017 as a result of the re-deployment of the clinical trial site clinical staff because of the current COVID-19 pandemic.
Additionally, COVID-19 may also result in delays in receiving approvals from local and foreign regulatory authorities, including the FDA’s review and approval of our Biologics License Application, or BLA, for Ryplazim, delays in regulatory inspections, including delays caused by travel restrictions, delays in necessary interactions with local and foreign regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees, and refusals to accept data from clinical trials conducted in these affected geographies. For example, the FDA has suspended its inspections outside of the United States which are not considered mission critical due to the COVID-19 outbreak, which may impact the FDA’s product application reviews.
27
The COVID-19 pandemic continues to evolve. The extent to which COVID-19 may impact our business, operations and clinical trials will depend on future developments, including the duration of the outbreak, travel restrictions and social distancing in Canada, in the United Kingdom, the United States and other countries, the effectiveness of actions taken in Canada, the United Kingdom, the United States and other countries to contain and treat the disease, and the extent to which Canada, the United Kingdom, the United States and additional countries are required to move to complete lock-down status. The ultimate long-term impact of the COVID-19 pandemic is highly uncertain and cannot be predicted with confidence.
We may rely on third-party suppliers of services to conduct preclinical studies and clinical trials and the failure by such third parties to comply with their obligations may delay the studies and trials, as applicable, and/or have an adverse effect on our development program.
We may rely on third-party suppliers of services to conduct preclinical studies and clinical trials and the failure by such third parties to comply with their obligations may delay the studies and trials, as applicable, and/or have an adverse effect on our development program. We have limited resources to conduct preclinical studies and clinical trials and may rely on third-party suppliers of services to conduct our studies and trials. If our third-party suppliers of services become unavailable for any reason, including as a result of the failure to comply with the rules and regulations governing the conduct of preclinical studies and clinical trials, operational failures, such as equipment failures or unplanned facility shutdowns, damage from any event, including fire, flood and earthquake, and impacts from public health crises, business restructuring or insolvency, or if they fail to perform their contractual obligations pursuant to the terms of the agreements entered into with us, such as failing to do the testing, compute the data or complete the reports further to the testing, we may incur delays in connection with the planned timing of our submission of an investigative new drug application, or IND, clinical trial application, or CTA, NDA or BLA submission. If the damage to any of our third-party suppliers of services is extensive or if, for any reason, such suppliers do not operate in compliance with Good Clinical Practices or are unable or refuse to perform their contractual obligations, we will need to find alternative third-party suppliers of services.
If we are required to change or select new third-party suppliers of services, the timing of the work related to preclinical studies and/or clinical trials could be delayed since the number of competent and reliable third-party suppliers to conduct preclinical and clinical work in compliance with good laboratory practice is limited. Any selection of new third-party suppliers to carry out work related to preclinical studies and clinical trials will be time-consuming and will result in additional delays in receiving data, analysis and reports from such third-party suppliers which, in turn, will delay the obtaining of regulatory approval to commercialize our product candidates. Furthermore, such delays could increase our expenditures to develop a product and materially adversely affect our operating results and financial condition.
Our product candidates are complex and difficult to manufacture, which could cause production problems that result in delays in our development or commercialization. We also rely in whole or in part on third parties for the manufacture and supply of our product candidates and such reliance may adversely affect us if the third parties are unable to fulfill their obligations.
We currently manufacture our product candidate Ryplazim in drug substance, ourselves at our facility in Canada and the drug substance is filled finished in finished product form by our contract manufacturer in the United States. Our product candidate fezagepras is manufactured by third party contract manufacturers. The manufacturing process we and the third parties with whom we contract use to produce our product candidates is complex, and several factors could cause production interruptions, including equipment malfunctions, facility contamination, raw material shortages or contamination, natural disasters, disruption in utility services, human error or disruptions in the operations of our suppliers.
28
If our in-house and third-party manufacturing capabilities become unavailable to us for any reason, including as a result of the failure to comply with cGMP regulations, manufacturing problems or other operational failures, such as equipment failures or unplanned facility shutdowns required to comply with cGMP, damage from any event, including fire, flood and earthquake, and impacts from public health crises, business restructuring or insolvency, we may be delayed in manufacturing product candidates and could be unable to meet the regulatory requirements of the FDA or other regulatory agencies to obtain market approval for our product candidates or be unable to manufacture and supply product for commercialization. Any such event could delay the supply of a product to conduct clinical trials and, if a product has reached commercialization, could prevent the supply of the product and adversely affect our revenues. If the damage to manufacturing facilities is extensive, we will need to find alternative manufacturing capacity. For example, restarting our in-house manufacturing or, alternatively, the selection of an alternative third-party manufacturer will be time-consuming and costly. Qualification of a new third-party manufacturer, if available, will include an assessment of the capacity of such third-party manufacturer to produce the quantities that may be requested from time to time by us, the manufacturing process and its compliance with cGMP and typically takes approximately 18 months. In addition, the third-party manufacturer will have to familiarize itself with our technology. Any delay in finding an alternative third-party manufacturer of a product, or recommencing our in-house manufacturing of such product, could result in a shortage of the product, delay clinical trial programs and the filing for regulatory approval of a product, and deprive us of potential product revenues.
We expect that we will have to continue to rely in whole or in part on third parties for the manufacture and supply of our product candidates and such reliance may adversely affect us if the third parties are unable to fulfill their obligations. We do not have the resources, facilities or experience to manufacture all of our product candidates on our own, which requires us to shift more production to third-party manufacturers. We may need to rely more heavily on third parties to manufacture and supply product candidates for clinical trials and we may rely on third parties for some time to manufacture and supply large quantities of product for commercial sales. Our reliance on third-party manufacturers will expose us to a number of risks, including failure to perform their contractual obligations under agreements with us, such as a failure to deliver the quantities requested on a timely basis.
In connection with the CRL we received in April 2018 with respect to Ryplazim, we have revisited our manufacturing methodologies to address concerns raised by the FDA, including the implementation and validation of additional analytical assays and controls in the manufacturing process. Such changes may cause problems and delays at our production facility and there can be no assurances that we will be successful in implementing the new procedures or that our manufacturing facility will be approved by the FDA. See “—We do not have the required regulatory approval to commercialize our product candidates and cannot guarantee that we will obtain such regulatory approval. We received a complete response letter from the FDA regarding Ryplazim, declining to approve the existing BLA in its current form. We may also be required to conduct post-approval clinical trials as a condition to licensing a product.” above.
Our revenues and operating results may also be negatively affected by any increase in the cost of goods that is not favorable to us.
As a company based outside of the United States, our business is subject to economic, political, regulatory and other risks associated with international operations.
As a company based in Canada with operations in the United Kingdom and in the United States, our business is subject to risks associated with conducting business internationally. Many of our suppliers and clinical trial relationships are located outside the United States. Accordingly, our future results could be harmed by a variety of factors, including:
| • | economic weakness, including inflation, or political instability in particular non-U.S. economies and markets; |
| • | differing and changing regulatory requirements for product approvals; |
| • | differing jurisdictions could present different issues for securing, maintaining or obtaining freedom to operate in such jurisdictions; |
| • | potentially reduced protection for intellectual property rights; |
29
| • | difficulties in compliance with different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations; |
| • | changes in non-U.S. regulations and customs, tariffs and trade barriers; |
| • | changes in a specific country’s or region’s political or economic environment; |
| • | trade protection measures, import or export licensing requirements or other restrictive actions by governments; |
| • | differing reimbursement regimes in certain non-U.S. markets; |
| • | negative consequences from changes in tax laws; |
| • | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
| • | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| • | litigation or administrative actions resulting from claims against us by current or former employees or consultants individually or as part of class actions, including claims of wrongful terminations, discrimination, misclassification or other violations of labor law or other alleged conduct; |
| • | difficulties associated with staffing and managing international operations, including differing labor relations; |
| • | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| • | business interruptions resulting from geo-political actions, including war and terrorism, or natural disasters and public health crises, such as the COVID-19 pandemic which is currently having a global impact. |
We may not receive the full payment of all milestones or royalty payments pursuant to the agreements entered into with third parties.
We may not receive the full payment of all milestones or royalty payments pursuant to the agreements entered or to be entered into with third parties and, consequently, our financial conditions and operating results could be adversely impacted. We have entered into license agreements and other forms of agreements with third parties regarding the development and commercialization of some of our technologies and product candidates. These agreements generally require that the third party pays to us certain amounts upon the attainment of various milestones and possibly include royalties on the sale of the developed product, if approved. There can be no guarantee that we will receive the payments described in those agreements since the development of the product candidates may be cancelled if the research does not yield positive results. Under such circumstances, we would not receive royalties as well. Even if the development of a product yields positive results, all of the risks described herein with respect to the obtaining of regulatory approval are applicable. Finally, if there occurs a disagreement between us and the third party, the payment relating to the attainment of milestones or of royalties may be delayed. The occurrence of any of those circumstances could have a material adverse effect on our financial condition and operating results.
30
If we breach any of the agreements under which we license rights to our product candidates or technology from third parties, we could lose license rights that are important to our business.
If we breach any of the agreements under which we license rights to our product candidates or technology from third parties, we could lose license rights that are important to our business. We license the development, manufacturing and commercialization rights for certain product candidates, and could, potentially, enter into similar licenses for other product candidates in the future. Under these licenses, we are subject to various obligations, including royalty and milestone payments, annual maintenance fees, limits on sublicensing, insurance obligations and the obligation to use commercially reasonable best efforts to develop and exploit the licensed technology. If we fail to comply with any of these obligations or otherwise breach these agreements, our licensors may have the right to terminate the license in whole or in part or to terminate the exclusive nature of the license. Loss of any of these licenses or the exclusivity rights provided therein could harm our financial condition and operating results.
We may be subject to damages resulting from claims that we, or our employees, collaborators or consultants, have wrongfully used or disclosed alleged trade secrets of third parties.
We may be subject to damages resulting from claims that we, or our employees, collaborators or consultants, have wrongfully used or disclosed alleged trade secrets of third parties. Many of our employees were previously employed, and certain of our consultants are currently employed, at universities, public institutions, biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that we, or these employees or consultants, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of these current or former employers. Litigation may be necessary to defend against these claims.
If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. We may be subject to claims that employees of our partners or licensors of technology licensed by us have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. We may become involved in litigation to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel.
Our internal information technology systems, or those of our third-party vendors, collaborators or other contractors or consultants, may fail or suffer security breaches, which could result in a significant disruption of our product development programs, give rise to significant liability, subject us to costly and protracted litigation, cause significant reputational harm and our ability to operate our business effectively.
We are increasingly dependent upon information technology systems, infrastructure, and data to operate our business. In the ordinary course of business, we collect, store, and transmit confidential information (including but not limited to intellectual property, proprietary business information, and personal information). It is critical that we do so in a secure manner to maintain the confidentiality and integrity of such information. We also have outsourced elements of our operations to third parties, and as a result we manage a number of third-party vendors and other contractors and consultants who have access to our confidential information.
31
Our internal information technology systems and those of our current and any future third-party vendors, collaborators and other contractors or consultants may be vulnerable to a variety of disruptive elements, including cyber-attacks by malicious third parties (including the deployment of computer viruses, harmful malware, ransomware, denial-of-service attacks, social engineering, and other means to affect service reliability and threaten the confidentiality, integrity, and availability of information), unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. In particular, the risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity, and sophistication of attempted attacks and intrusions from around the world have increased, and phishing and social engineering attacks have increased in particular in connection with the COVID-19 pandemic. Previously, we experienced a phishing incident where one employee was convinced to purchase credit cards and provide the numbers of such credit cards to the attackers. We may not be able to anticipate all types of security threats, and we may not be able to implement preventive measures effective against all such security threats. The techniques used by cyber criminals change frequently, may not be recognized until launched, and can originate from a wide variety of sources, including outside groups such as external service providers, organized crime affiliates, terrorist organizations, or hostile foreign governments or agencies. While we have not experienced any significant system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations or a loss of, or damage to, our data or applications, or those of our third-party vendors and other collaborators, contractors and consultants, it could result in a disruption of our development programs and our business operations, whether due to a loss of our trade secrets or other proprietary information, significant delays or setbacks in our research, or other similar disruptions. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur significant liability, our competitive position could be harmed, our reputation could be damaged, and the further development and commercialization of our product candidates could be delayed. In addition, any such event that leads to unauthorized access, use, or disclosure of personal information, including personal information regarding our patients or employees, could compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, and otherwise subject us to liability under laws and regulations that protect the privacy and security of personal information. The costs related to significant security breaches or disruptions could be material and exceed the limits of the cybersecurity insurance we maintain against such risks. If the information technology systems of our third-party vendors and other collaborators, contractors and consultants become subject to disruptions or security breaches, we may be exposed to material liability and have insufficient recourse against such third parties and we may have to expend significant resources to mitigate the impact of such an event, and to develop and implement protections to prevent future events of this nature from occurring.
We may be subject to environmental remediation obligations or other obligations under environmental laws and regulations and climate change could exacerbate certain of the threats facing our business.
We may be subject to environmental remediation obligations or other obligations under environmental laws and regulations and climate change could exacerbate certain of the threats facing our business. We are subject to laws and regulations concerning the environment, safety matters, regulation of chemicals and product safety in the countries where we operate our business. These requirements include regulation of the handling, manufacture, transportation, use and disposal of materials, including the discharge of pollutants into the environment. In the normal course of our business, hazardous substances may be released into the environment, which could cause environmental or property damage or personal injuries, and which could subject us to remediation obligations regarding contaminated soil and groundwater or potential liability for damage claims.
In addition, global climate change could exacerbate certain of the threats facing our business, including the business continuity depends on how well we protect our facilities and equipment. Several areas of our operations further raise environmental considerations, such as greenhouse gas emissions and disposal of hazardous residual materials. Failure to recognize and adequately respond to changing governmental and public expectations on environmental matters could result in fines, missed opportunities, additional regulatory scrutiny or harm to our brand and reputation which could potentially have an adverse effect on our business and financial results.
32
Risks Related to our Intellectual Property
Our failure to protect our intellectual property may have a material adverse effect on our ability to develop and commercialize our product candidates.
Our failure to protect our intellectual property may have a material adverse effect on our ability to develop and commercialize our product candidates. We will be able to protect our intellectual property rights from unauthorized use by third parties only to the extent that our intellectual property rights are covered by valid and enforceable patents or are effectively maintained as trade secrets. We try to protect our intellectual property by filing patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. Because the patent position of pharmaceutical companies involves complex legal and factual questions, the issuance, scope and enforceability of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. If our patents are invalidated or found to be unenforceable, we will lose the ability to exclude others from making, using or selling the inventions claimed. Moreover, an issued patent does not guarantee us the right to use the patented technology or commercialize a product using that technology. Third parties may have blocking patents that could be used to prevent us from developing our product candidates, selling our products, if any, or commercializing our patented technology. Thus, patents that we own may not allow us to exploit the rights conferred by our intellectual property protection. Moreover, our pending patent applications may not result in patents being issued. Even if issued, they may not be issued with claims sufficiently broad to protect our product candidates and technologies or may not provide us with a competitive advantage against competitors with similar product candidates or technologies. Furthermore, others may independently develop product candidates or technologies similar to those that we have developed or discover our trade secrets. In addition, the laws of many countries do not protect intellectual property rights to the same extent as the laws of Canada, Europe and the United States, and those countries may also lack adequate rules and procedures for defending intellectual property rights effectively. There can be no guarantee that we will receive patents in countries where we file patent applications for our product candidates. As a result, the validity and enforceability of our patents cannot be predicted with certainty. In addition, we cannot guarantee that:
| • | we or our licensors were the first to make the inventions covered by each of our issued patents and pending patent applications; |
| • | we or our licensors were the first to file patent applications for these inventions; |
| • | others will not independently develop similar or alternative technologies or duplicate any of our or our licensors’ technologies; |
| • | any of our or our licensors’ pending patent applications will result in issued patents; |
| • | that there are no prior public disclosures that could invalidate our or our licensors' patents, as the case may be; |
| • | that there are not unpublished applications or patent applications maintained in secrecy that may later issue with claims covering our product candidates; |
| • | that others will not circumvent our owned or in-licensed patents; |
| • | our owned or in-licensed issued patents will provide us with any competitive advantages or that it will not be narrowed in scope or be held invalid or unenforceable as a result of legal challenges by third parties; |
| • | any of our or our licensors’ patents will be valid or enforceable; |
| • | any patents issued to us or our licensors and collaboration partners will provide us with any competitive advantages, or will not be challenged by third parties; |
| • | we will develop or in-license additional proprietary technologies that are patentable; or |
| • | the patents of others will not have an adverse effect on our business. |
33
We rely on trade secrets, know-how and technology, which are not protected by patents, to maintain our competitive position.
We also rely on trade secrets, know-how and technology, which are not protected by patents, to maintain our competitive position. We try to protect this information by entering into confidentiality undertakings with parties that have access to it, such as our current and prospective suppliers, employees and consultants. Any of these parties may breach the undertakings and disclose the confidential information to our competitors. Enforcing a claim that a third party illegally obtained and is using trade secrets is expensive and time consuming and the outcome is unpredictable. In addition, it could divert management’s attention. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our competitive position could be harmed.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose intellectual property rights that are important to our business.
We may need to obtain additional licenses from others to advance our research and development activities or allow the commercialization of our product candidates. Our license agreements impose, and we expect that future license agreements will impose, various development, diligence, commercialization, and other obligations on us. Our licensors might conclude that we have materially breached our obligations under such license agreements and might therefore terminate the license agreements, thereby removing or limiting our ability to develop and commercialize product candidates and technology covered by these license agreements. If these in-licenses are terminated, or if the underlying patents fail to provide the intended exclusivity, competitors or other third parties would have the freedom to seek regulatory approval of, and to market, product candidates identical to ours and we may be required to cease our development and commercialization of any related product. In addition, the agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. Disputes may arise regarding intellectual property subject to a licensing agreement, including:
| • | the scope of rights granted under the license agreement and other interpretation-related issues; |
| • | the extent to which our product candidates, technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; |
| • | the sublicensing of patent and other rights under our collaborative development relationships; |
| • | our diligence obligations under the license agreement and what activities satisfy those diligence obligations; |
| • | the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and |
| • | the priority of invention of patented technology. |
We may not be able to protect our intellectual property rights throughout the world.
We may not be able to protect our intellectual property rights throughout the world. Filing, prosecuting and defending patents on all of our product candidates and products, if any, when and if we have any, in every jurisdiction would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we or our licensors have not obtained patent protection to develop our own product candidates. These product candidates may compete with our product candidates, when and if we have any, and may not be covered by any of our or our licensors’ patent claims or other intellectual property rights.
34
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of Canada and the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of Canada and the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biotechnology and/or pharmaceuticals, which could make it difficult for us to stop the infringement of our patents. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the USPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and/or applications. We have systems in place to remind us to pay these fees, and we employ an outside firm and rely on our outside counsel to pay these fees due to non-U.S. patent agencies. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business.
35
Patent protection for our product candidates or products, if any, may expire before we are able to maximize their commercial value, which may subject us to increased competition and reduce or eliminate our opportunity to generate product revenue.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Patent protection for our product candidates or products, if any, may expire before we are able to maximize their commercial value, which may subject us to increased competition and reduce or eliminate our opportunity to generate product revenue. The patents for our product candidates have varying expiration dates and, when these patents expire, we may be subject to increased competition and may not be able to recover our development costs. In some of the larger economic territories, such as Canada, the United States and Europe, patent term extension/restoration may be available to compensate for time taken during aspects of the product candidate’s regulatory review. However, we cannot be certain that an extension will be granted, or if granted, what the applicable time period or the scope of patent protection afforded during any extended period will be. In addition, even though some regulatory agencies may provide some other form of exclusivity for a product candidate under its own laws and regulations, we may not be able to qualify the product candidate or obtain the exclusive time period.
If we are unable to obtain patent term extension/restoration or some other exclusivity, we could be subject to increased competition and our opportunity to establish or maintain product revenue could be substantially reduced or eliminated. Furthermore, we may not have sufficient time to recover our development costs prior to the expiration of our Canadian and non-Canadian patents.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and may be unable to protect our rights to, or use of, our technology.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and may be unable to protect our rights to, or use of, our technology. If we choose to go to court to restrain a third party from using the inventions claimed in our patents or licensed patents, that individual or corporation has the right to ask the court to rule that these patents are invalid and/or should not be enforced against that third party. These lawsuits are expensive and would consume time and other resources even if we were successful in stopping the infringement of these patents. In addition, there is a risk that the court will decide that these patents are invalid or unenforceable and that we do not have the right to refrain the third party from using the inventions. There is also the risk that, even if the validity of these patents is upheld, the court will refuse to grant a decision or judgment in our favor on the ground that such other party’s activities do not infringe our rights.
If we wish to use the technology or compound claimed in issued and unexpired patents owned by a third party, we will need to obtain a license from such third party, enter into litigation to challenge the validity or enforceability of the patents or incur the risk of litigation in the event that the owner asserts that we infringed its patents. The failure to obtain a license to technology or the failure to challenge an issued patent that we may require to develop or commercialize our product candidates may have a material adverse impact on our operating results and financial condition.
If a third party asserts that we infringed their patents or other proprietary rights, we could face a number of risks that could seriously harm our results of operations, financial condition and competitive position, including:
| • | patent infringement and other intellectual property claims, which would be costly and time consuming to defend, whether or not the claims have merit, and which could delay the regulatory approval process and divert management’s attention from our business; |
| • | substantial damages for past infringement (including treble damages and attorneys’ fees for willful infringement), which we may have to pay if a court determines that our product candidates or technologies infringe a competitor’s patent or other proprietary rights; |
36
| • | a court prohibiting us from selling or licensing our technologies or future drugs unless the third party licenses its patents or other proprietary rights to us on commercially reasonable terms, which it is not required to do; and |
| • | if a license is available from a third party, we may have to pay substantial royalties or lump sum payments or grant cross licenses to its patents or other proprietary rights to obtain that license. |
The biopharmaceutical industry has produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our product candidates or methods of use either do not infringe the patent claims of the relevant patent and/or that the patent claims are invalid, and we may not be able to do this. Proving invalidity during a court proceeding, in particular, is difficult since it requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
Canadian patent laws as well as the laws of some foreign jurisdictions provide for provisional rights in published patent applications beginning on the date of publication, including the right to obtain reasonable royalties, if a patent is subsequently issued and certain other conditions are met. We cannot predict the outcome of any invalidity challenge. Alternatively, it is possible that we may determine it prudent to seek a license from the patent holder to avoid potential litigation and other potential disputes. We cannot be sure that a license would be available to us on acceptable terms, or at all.
Because some patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in Canada and many foreign jurisdictions are typically not published until 18 months after filing, and because publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our licensors’ issued patents or our pending applications or our licensors’ pending applications, or that we or our licensors were the first to invent the technology.
Patent applications filed by third parties that cover technology similar to our technology may have priority over our or our licensors’ patent applications and could further require us to obtain rights to issued patents covering such technologies. If another party files a U.S. patent application on an invention similar to ours, we may elect to participate in or be drawn into an interference or derivation proceeding at the U.S. Patent and Trademark Office, or USPTO, to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful, resulting in a loss of our U.S. patent position with respect to such inventions.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations. We cannot predict whether third parties will assert these claims against us or against our licensors, or whether those claims will harm our business. If we are forced to defend against these claims, whether they are with or without any merit, whether they are resolved in favor of or against us or our licensors, we may face costly litigation and diversion of management’s attention and resources. As a result of these disputes, we may have to develop costly non-infringing technology, or enter into licensing agreements. These agreements, if necessary, may be unavailable on terms acceptable to us, if at all, which could seriously harm our business or financial condition. Even if a license is offered to us, it may be non-exclusive and our competitors may gain access to the same technology.
37
Our commercial success depends, in part, on our ability not to infringe on third-parties’ patents and other intellectual property rights. Our capacity to commercialize our product candidates will depend, in part, on the non-infringement of third-parties’ patents and other intellectual property rights. The biopharmaceutical and pharmaceutical industries have produced a multitude of patents and it is not always clear to participants, including us, which patents cover various types of products or methods of use. The scope and breadth of patents is subject to interpretation by the courts and such interpretation may vary depending on the jurisdiction where the claim is filed and the court where such claim is litigated. The holding of patents by us for our product candidates and our applications does not guarantee that we are not infringing on other third-parties’ patents and there can be no guarantee that we will not be in violation of third-parties’ patents and other intellectual property rights. Patent analysis for non-infringement is based in part on a review of publicly available databases. Although we review from time to time certain databases to conduct patent searches, we do not have access to all databases. It is also possible that some of the information contained in the databases has not been reviewed by us or was found to be irrelevant at the time the searches were conducted. In addition, because patents take years to be issued, there may be currently pending applications that we are unaware of which may later be issued. As a result of the foregoing, there can be no guarantee that we will not violate third-party patents. Because of the difficulty in analyzing and interpreting patents, there can be no guarantee that a third party will not assert that we infringe upon any of its patents or any of its other intellectual property rights.
There is no guarantee that we will not become involved in litigation. Litigation with any third party, even if the allegations are without merit, is expensive, time-consuming and will divert management’s attention from the daily execution of our business plan. Litigation implies that a portion of our financial assets would be used to sustain the costs of litigation instead of being allocated to further the development of our business plan. If we are involved in patent infringement litigation, we will need to demonstrate that our product candidates do not infringe the patent claims of the relevant patent, that the patent claims are invalid or that the patent is unenforceable. If we were found liable for infringement of third-parties’ patents or other intellectual property rights, we could be required to enter into royalty or licensing agreements on terms and conditions that may not be favorable to us, and/or pay damages, including up to treble damages (but only if found liable of willful infringement) and/or cease the development and commercialization of our product candidates. Any finding that we are guilty of patent infringement could materially adversely affect our business, financial conditions and operating results.
There may be issued patents that we are unaware of that our product candidates may infringe, or patents that we believe we do not infringe but could be found to be infringing.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
Changes in either the patent laws or interpretation of the patent laws in the United States or other jurisdictions could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. For example, prior to March 2013, the first to invent a claimed invention was entitled to a patent in the United States, assuming that other requirements for patentability were met, while outside the United States, the first to file a patent application was entitled to the patent. After March 2013, under the Leahy-Smith America Invents Act (or the America Invents Act), enacted in September 2011, the United States transitioned to a first inventor to file system in which, assuming that other requirements for patentability are met, the first inventor to file a patent application is entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. The America Invents Act also included a number of significant changes that affect the way patent applications are prosecuted and that provide additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including post-grant review, inter parties review, and derivation proceedings. These changes have increased the uncertainties related to patent prosecution and patent challenges.
In addition, the patent positions of companies in the development and commercialization of pharmaceuticals are particularly uncertain. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. This combination of events has created uncertainty with respect to the validity and enforceability of patents, once obtained. The laws and regulations governing patents could change in unpredictable ways depending on future actions by the U.S. Congress, the federal courts, the USPTO, and the legislative, judicial, and regulatory branches of other jurisdictions in which we may seek patent protection, and such changes could have a material adverse effect on our existing patent portfolio and our ability to protect and enforce our intellectual property in the future.
38
Risks Related to our Common Shares and our Status as a Public Company
The price of our common shares historically has been volatile. This volatility, and the sale of substantial amounts of our common shares, could adversely affect the price of our common shares.
The market prices for the securities of biopharmaceutical companies, including us, have historically been volatile. The market has from time to time experienced extreme price and volume fluctuations that are unrelated to the operating performance of any particular company.
The trading price of our common shares could continue to be subject to wide fluctuations in price in response to various factors, many of which are beyond our control, including the results and adequacy of our preclinical studies and clinical trials, as well as those of its collaborators, or its competitors; other evidence of the safety or effectiveness of our product candidates or those of our competitors; announcements of technological innovations or new product candidates by us or our competitors; governmental regulatory actions; developments with collaborators; developments (including litigation) concerning patent or other proprietary rights of us or our competitors; concern as to the safety of our product candidates; period-to-period fluctuations in operating results; changes in estimates of our performance by securities analysts; market conditions for biotechnology stocks in general; geo-political actions, including war and terrorism, or natural disasters and public health crises, such as the COVID-19 pandemic which is currently having a global impact; and other factors not within our control could have a significant adverse impact on the market price of our securities, regardless of our operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been instituted. A class action suit against us could result in substantial costs, potential liabilities and the diversion of management’s attention and resources.
While our common shares are listed on Nasdaq, an active and liquid trading market may not be sustained. If an active public market is not sustained, it may be difficult for shareholders to sell common shares at a price that is attractive, or at all. Additionally, the listing of our common shares on Nasdaq are conditional upon our ability to maintain the applicable continued listing requirements of Nasdaq. Failure to maintain the applicable continued listing requirements of the Nasdaq could result in our common shares being delisted from Nasdaq. Nasdaq may review a listing and delist securities based on the review of the financial condition of an issuer. If the market price of our common shares declines on Nasdaq and it becomes doubtful that we can continue as a going concern, Nasdaq may commence a remedial review process that could lead to the delisting of our common shares.
We expect to issue common shares in the future. Holders of stock options and warrants may elect to exercise their options or warrants into common shares depending on the stock price. Future issuances of common shares, or the perception that such issuances are likely to occur, could affect the prevailing trading prices of our common shares. Future issuances of our common shares could result in substantial dilution to our shareholders. In addition, the existence of warrants may encourage short selling by market participants.
All of our debt obligations, and any future indebtedness we may incur, could lead to adverse consequences and will have priority over our common shares with respect to payment in the event of a liquidation, dissolution or winding up.
In any liquidation, dissolution or winding up of us, our common shares would rank below all debt claims against us, including our secured indebtedness held by SALP. In addition, any convertible or exchangeable securities or other equity securities currently outstanding or that we may issue in the future may have rights, preferences and privileges more favorable than those of our common shares. In addition, if we are unable to repay, refinance or restructure our indebtedness when payment is due, the lenders could proceed against any collateral granted to them to secure such indebtedness or force us into bankruptcy or liquidation. As a result, holders of our common shares may not be entitled to receive any payment or other distribution of assets upon the liquidation or dissolution until after our obligations to our debt holders and holders of equity securities that rank senior to our common shares have been satisfied.
39
We are a “controlled company” within the meaning of the applicable Nasdaq listing rules and, as a result, qualify for exemptions from certain corporate governance requirements. If we rely on these exemptions, you will not have the same protections afforded to shareholders of companies that are subject to such requirements.
We believe SALP will continue to control a majority of the voting power of our outstanding common shares for the foreseeable future. As a result, we will be a “controlled company” within the meaning of applicable Nasdaq listing rules. Under these rules, a company of which more than 50% of the voting power for the election of directors is held by an individual, group or another company is a “controlled company.” For so long as we remain a “controlled company,” we may elect not to comply with certain corporate governance requirements, including the requirements:
| • | that a majority of the board of directors consists of independent directors; |
| • | for an annual performance evaluation of the HR and compensation committee; and |
| • | that we have a HR and compensation committee that is composed entirely of independent directors with a written charter addressing the committee’s purpose and responsibilities. |
We may continue to use all or some of these exemptions in the future. As a result, you may not have the same protections afforded to shareholders of companies that are subject to all of the Nasdaq corporate governance requirements.
We expect that SALP will continue to own a significant percentage of our common shares and will be able to exert significant control over matters subject to shareholder approval.
SALP is currently our majority shareholder and we expect that SALP will continue to be our controlling shareholder in the future. As of the date hereof, SALP beneficially owns approximately 66.38% of the voting power of our outstanding share capital, and although we have registered for resale all of SALP’s shares, we believe SALP will continue to remain a controlling shareholder for the foreseeable future. Therefore, SALP will have the ability to substantially influence us and exert significant control through this ownership position. For example, SALP may be able to control elections of directors, issuance of equity, including to our employees under equity incentive plans, amendments of our organizational documents, or approval of any merger, amalgamation, sale of assets or other major corporate transaction. SALP’s interests may not always coincide with our corporate interests or the interests of other shareholders, and it may exercise its voting and other rights in a manner with which you may not agree or that may not be in the best interests of our other shareholders. Further, there may be changes to the management or ownership of SALP that could impact SALP’s interests in a way that may not coincide with our corporate interests or the interests of other shareholders. So long as SALP continues to own a majority of our equity, it will continue to be able to strongly influence and effectively control our decisions.
Our organizational and ownership structure may create significant conflicts of interests.
Our organizational and ownership structure involves a number of relationships that may give rise to certain conflicts of interest between us and minority holders of our common shares, on the one hand, and SALP and its owners, on the other hand. Certain of our directors who have been nominated to our board by SALP have economic exposure to the financial performance of SALP and its investments, including our shares, and, accordingly, their interests may be aligned with SALP’s interests, which may not always coincide with our corporate interests or the interests of our other shareholders. Further, our other shareholders may not have visibility into the economic exposure of such directors to the financial performance of SALP’s investments, including our shares, which may change at any time through acquisition, disposition, dilution, or otherwise. Any change in the economic exposure of directors nominated to our board by SALP to the financial performance of SALP and its investments, including our shares, could impact the interests of those holders.
In addition, SALP is entitled to nominate two directors for election to our board of directors. While any directors nominated by SALP must be the subject of a favorable recommendation of our HR and compensation committee and must act in accordance with their fiduciary duties to our other shareholders, such directors’ interests may not coincide with the interests of our other shareholders.
40
In addition, we are party to certain related party agreements with SALP. SALP and its partners, as well as directors nominated by SALP, may have interests which differ from our interests or those of the minority holders of our common shares. These agreements are, and any future material transaction between us and SALP or any other affiliate of SALP will be, subject to our related party transaction procedures and the standards of conduct contained in our Code of Ethics and Business Conduct under which a director is expected to declare his or her interest in any transaction or agreement in which he or she may have a material interest, and as circumstances warrant, abstain from voting on the approval of any such transaction or agreement. To the extent we fail to appropriately deal with any such related-party transaction or conflict of interest, it could negatively impact our reputation and ability to raise additional funds and the willingness of counterparties to do business with us, all of which could have an adverse effect on our business, financial condition, results of operations, and cash flows.
If we fail to implement and maintain effective internal controls over financial reporting, our ability to produce accurate financial statements on a timely basis could be impaired.
As a public company in the United States, we are subject to reporting obligations under U.S. securities laws, including the Sarbanes-Oxley Act. Section 404(a) of the Sarbanes-Oxley Act, or Section 404(a), requires that management assess and report annually on the effectiveness of our internal control over financial reporting and identify any material weaknesses in our internal control over financial reporting. Although Section 404(b) of the Sarbanes-Oxley Act, or Section 404(b), requires our independent registered public accounting firm to issue an annual report that addresses the effectiveness of our internal control over financial reporting, we have opted to rely on the exemptions provided in the JOBS Act, and consequently will not be required to comply with SEC rules that implement Section 404(b) until such time as we are no longer an emerging growth company.
The presence of material weaknesses could result in financial statement errors which, in turn, could lead to errors in our financial reports or delays in our financial reporting, which could require us to restate our operating results or result in our auditors issuing a qualified audit report. In order to establish and maintain effective disclosure controls and procedures and internal control over financial reporting, we will need to expend significant resources and provide significant management oversight. Developing, implementing and testing changes to our internal control may require specific compliance training of our directors and employees, entail substantial costs in order to modify our existing accounting systems, take a significant period of time to complete and divert management’s attention from other business concerns. These changes may not, however, be effective in establishing and maintaining adequate internal controls.
If either we are unable to conclude that we have effective internal control over financial reporting or, at the appropriate time, our independent auditors are unwilling or unable to provide us with an unqualified report on the effectiveness of our internal control over financial reporting as required by Section 404(b), investors may lose confidence in our operating results, the price of our common shares could decline and we may be subject to litigation or regulatory enforcement actions. In addition, if we are unable to meet the requirements of Section 404, we may not be able to remain listed on the Nasdaq.
We will incur significantly increased costs as a result of operating as a reporting company whose common shares are publicly traded in the United States and is unable to use the Multijurisdictional Disclosure System, and our management will be required to devote substantial time to new compliance initiatives.
As a public company whose shares are publicly traded in the United States and reporting under the Exchange Act and is no longer eligible to report under the Multijurisdictional Disclosure System, we incur significant legal, accounting and other expenses that we did not incur previously. These expenses will likely be even more significant after we no longer qualify as an emerging growth company. The Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of the Nasdaq and other applicable securities rules and regulations impose various requirements on public companies in the United States, including the establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our senior management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect that these rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance, which in turn could make it more difficult for us to attract and retain qualified senior management personnel or members for our board of directors.
41
However, these rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
Pursuant to Section 404 of the Sarbanes-Oxley Act, or Section 404, we will be required to furnish a report by our senior management on our internal control over financial reporting. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To prepare for eventual compliance with Section 404, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude, within the prescribed timeframe or at all, that our internal control over financial reporting is effective as required by Section 404.
Compliance with laws and regulations affecting public companies may cause us to incur additional expenses and may also increase the compliance risks associated with such changes, which may have a material adverse effect on our financial condition and operating results.
The occurrence of any changes in the laws and regulations applicable to public companies, including any changes to the existing disclosure obligations under applicable Canadian securities laws and regulations and related rules and policies, may cause us to incur additional expenses associated with the assessment of the impacts of such changes as well as a result of the implementation and monitoring of its compliance obligations, including any new internal processes and controls that need to be implemented or reporting requirements as a result of such changes.
Any changes in the laws and regulations affecting public companies may also increase the compliance risks associated with such changes, which could result in enforcement actions, penalties or lawsuits, which may have a material adverse effect on our financial condition and operating results.
Any such increased compliance risks may make it more difficult for us to comply with our indemnification obligations and to secure appropriate directors and officers’ liability insurance policies or may result in a significant increase in the cost to secure appropriate insurance coverage. We may not be able to afford a significant increase in the costs to secure appropriate directors and officers’ liability insurance coverage and may have to secure reduced coverage limits or settle for a higher retention amount for indemnifiable losses, which may not cover the claims against our past, present or future directors for which we are bound to indemnify our directors and officers. Any reduced limit in the insurance coverage or increase of the retention amount for indemnifiable losses may result in a difficulty to attract and retain experienced and qualified directors to serve on our board of directors and officers.
42
We are an “emerging growth company” and as a result of the reduced disclosure and governance requirements applicable to emerging growth companies, our common shares may be less attractive to investors.
We are an “emerging growth company” as defined in the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404, exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. As an emerging growth company, we are able to report only two years of financial results and selected financial data compared to three and five years, respectively, for comparable data reported by other public companies. We may take advantage of these exemptions until we are no longer an emerging growth company. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier, including if the aggregate market value of our common shares held by non-affiliates exceeds $700 million as of the end of our second fiscal quarter before that time, in which case we would no longer be an emerging growth company as of the following December 31st (the last day of our fiscal year). We cannot predict if investors will find our common shares less attractive because we may rely on these exemptions. If some investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and the price of our common shares may be more volatile.
Under Section 107(b) of the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
We qualify as a foreign private issuer and, as a result, we will not be subject to U.S. proxy rules and will be subject to Exchange Act reporting obligations that permit less detailed and frequent reporting than that of a U.S. domestic public company.
We report under the Exchange Act as a non-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including (i) the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; (ii) the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and (iii) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K upon the occurrence of specified significant events. In addition, foreign private issuers are not required to file their annual report on Form 20-F until 120 days after the end of each fiscal year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year.
Foreign private issuers also are exempt from Regulation FD, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
If we lose our status as a foreign private issuer, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and Nasdaq rules. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the cost we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time consuming and costly. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our board of directors.
43
U.S. investors may be unable to enforce certain judgments.
We are a company existing under the CBCA. Most of our directors and officers are residents of Canada or otherwise reside outside the United States, and substantially all of our assets are located outside the United States. As a result, it may be difficult to effect service within the United States upon us or upon some of our directors and officers. Execution by U.S. courts of any judgment obtained against us or any of our directors or officers in U.S. courts may be limited to assets located in the United States. It may also be difficult for holders of securities who reside in the United States to realize in the United States upon judgments of U.S. courts predicated upon civil liability of us and our directors and executive officers under the U.S. federal securities laws. There may be doubt as to the enforceability in Canada against non-U.S. entities or their controlling persons, directors and officers who are not residents of the United States, in original actions or in actions for enforcement of judgments of U.S. courts, of liabilities predicated solely upon U.S. federal or state securities laws.
We may be a passive foreign investment company, which may result in adverse U.S. federal income tax consequences for U.S. Holders of our common shares.
Generally, if for any taxable year 75% or more of our gross income is passive income, or at least 50% of the average quarterly value of our assets are held for the production of, or produce, passive income, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. Although we do not believe that we were a PFIC for the year ending December 31, 2020, our determination is based on an interpretation of complex provisions of the law, which are not addressed in a significant number of administrative pronouncements or rulings by the Internal Revenue Service. Accordingly, there can be no assurance that our conclusions regarding our status as a PFIC for the 2020 taxable year will not be challenged by the Internal Revenue Service and, if challenged, upheld in appropriate proceedings. In addition, because PFIC status is determined on an annual basis and generally cannot be determined until the end of the taxable year, there can be no assurance that we will not be a PFIC for the current or future taxable years. If we are characterized as a PFIC, our shareholders who are U.S. holders may suffer adverse tax consequences, including the treatment of gains realized on the sale of our common shares as ordinary income, rather than as capital gain, the loss of the preferential rate applicable to dividends received on our common shares by individuals who are U.S. Holders, and the addition of interest charges to the tax on such gains and certain distributions. A U.S. shareholder of a PFIC generally may mitigate these adverse U.S. federal income tax consequences by making a “qualified electing fund” election, or, to a lesser extent, a “mark to market” election. However, we do not intend to provide the information necessary for U.S. Holders to make qualified electing fund elections if we are classified as a PFIC.
We may be impacted by certain taxes in multiple jurisdictions.
We are a multinational corporation with operations in multiple jurisdictions. As a result, we need to be compliant with the tax laws and regulations of Canadian federal, provincial and local governments, the United States, the United Kingdom and other international jurisdictions. This includes transfer pricing laws and regulations between many of these jurisdictions. Significant judgment is required in determining our provision for income taxes and claims for investment tax credits related to qualifying Scientific Research and Experimental Development expenditures, both in Canada and in foreign jurisdictions. Various internal and external factors may have favorable or unfavorable effects on future provisions for income taxes and our effective income tax rate. These factors may include, but are not limited to, changes in tax laws, regulations and/or tax rates, audits by tax authorities, changing interpretations of existing tax laws or regulations, changes in estimates of prior years’ items and changes in future levels of research and development, or R&D, spending. Furthermore, new accounting pronouncements or new interpretations of existing accounting pronouncements may have a material impact on our effective income tax rate.
We may be impacted by certain tax treatments applicable to various revenue streams in different tax jurisdictions. We are subject to withholding taxes on certain of our revenue streams. The withholding tax rates that are used are based on the interpretation of specific tax acts and related treaties. If a tax authority has a different interpretation from us, it could potentially impose additional taxes, interest and/or penalties. These additional costs would potentially reduce the amount of revenue we ultimately receive and retain. From time-to-time, we have implemented reorganization transactions to improve or simplify our overall tax structure. Challenges from a tax authority having a different interpretation of the effect of these transactions could potentially have a materially adverse impact on us, both in the form of additional taxes, interest and/or penalties that may be assessed and additional costs that we may incur in contesting such assessments.
44
We do not intend to pay dividends in the foreseeable future.
We have never declared or paid any dividends on our common shares. We intend, for the foreseeable future, to retain our future earnings, if any, to finance our commercial activities, further research and the expansion of our business. As a result, the return on an investment in our common shares will likely depend upon any future appreciation in value, if any, and on a shareholder’s ability to sell our common shares. The payment of future dividends, if any, will be reviewed periodically by our board of directors and will depend upon, among other things, conditions then existing including earnings, financial conditions, cash on hand, financial requirements to fund our commercial activities, development and growth, and other factors that our board of directors may consider appropriate in the circumstances.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our share price and trading volume could decline.
The trading market for our common shares will depend in part on the research and reports that securities or industry analysts publish about us or our business. If few securities analysts commence coverage of us, or if industry analysts cease coverage of us, the trading price for our common shares would be negatively affected. If one or more of the analysts who cover us downgrade our common shares or publish inaccurate or unfavorable research about our business, our common shares price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for our common shares could decrease, which might cause the price of our common shares and trading volume to decline.
Item 4.Information on the Company.
A.History and Development of the Company
We were incorporated on October 14, 1994 under the Canada Business Corporations Act, or the CBCA, under the name Innovon Life Sciences Holdings Limited. We changed our name to “Prometic Life Sciences Inc.” on May 19, 1998 and subsequently changed our name to “Liminal BioSciences Inc.” on October 3, 2019. On July 28, 1998, our common shares began trading on the Toronto Stock Exchange, or the TSX, under the trading symbol “PLI” and, on October 7, 2019, began trading under the trading symbol “LMNL.” On November 18, 2019, our common shares began trading on the Nasdaq Global Market, or the Nasdaq, under the trading symbol “LMNL.” On August 5, 2020, we voluntarily delisted our common shares from the TSX.
Our head and registered office is located at 440 Armand-Frappier Blvd., Suite 300, Laval, Québec, Canada H7V 4B4. The telephone number of our principal executive office is 1-450-781-0115 and our corporate website is www.liminalbiosciences.com. The information contained on our website is not incorporated by reference into this Annual Report, and you should not consider any information contained on, or that can be accessed through, our website as part of this Annual Report or in deciding whether to purchase our common shares. The SEC maintains an Internet website that contains reports and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov.
In April and June 2019, we entered into a series of related arrangements to restructure our outstanding indebtedness, reduce our interest and certain other payment obligations, and raise sufficient cash to build a robust balance-sheet for the next phase of our development.
| • | $114.4 million aggregate gross proceeds were raised through a combination of a private placement offering of our common shares, or the Private Placement, followed by an equity rights offering, or the Rights Offering, to our shareholders at a price of $15.21 per common share, or the Transaction Price; |
| • | Approximately $229 million of the outstanding debt owned by Structured Alpha LP, or SALP, was converted into our common shares at the Transaction Price, comprising all but $10 million of our outstanding debt held by SALP, or the Debt Conversion; and |
| • | the adjustment of the per warrant exercise price of certain of our outstanding common share purchase warrants held by SALP to the Transaction Price, or the Warrant Repricing. |
These transactions will be referred to collectively in this Annual Report as the Restructuring Transactions. Following the Restructuring Transactions, SALP became our majority and controlling shareholder.
45
Our actual capital expenditures for the years ended December 31, 2018, 2019 and 2020 amounted to $5.7 million, $1.0 million and $1.1 million respectively. These capital expenditures primarily consisted of investments in our biotherapeutics and bioproduction facilities in connection with the development of Ryplazim® and expenses related to the opening of a new plasma collection center in the United States. We expect our capital expenditures to be consistent with our previous expenditures, as we continue to advance our research and development programs and grow our operations.
B.Business Overview
We are a clinical-stage biopharmaceutical company focused on the discovery and development of novel small molecule drug candidates for the treatment of patients suffering from respiratory fibrotic diseases and other fibrotic or inflammatory diseases that have a high unmet medical need. We have a deep understanding of certain biological targets and pathways that have been implicated in the fibrotic process, including fatty acid receptors such as free fatty acid receptor 1, FFAR1 (also known as G-protein-coupled receptor 40, or GPR40), a related receptor (G-protein-coupled receptor 84, or GPR84) and peroxisome proliferator-activated receptors, or PPARs.
In preclinical studies, we observed that targeting these receptors promoted normal tissue regeneration and scar resolution, including preventing the progression of, and reversing established fibrosis. We have generated preclinical data that we believe demonstrate proof-of-mechanism of fezagepras for the treatment of respiratory, liver and kidney disorders associated with chronic or severe fibrosis. Our research program is focused on fibrotic, inflammatory and metabolic conditions in patients with respiratory, liver or renal disease.
Our lead small molecule product candidate, fezagepras (also known as PBI-4050), is being developed for the treatment of idiopathic pulmonary fibrosis, or IPF. Fezagepras is an anti-inflammatory and anti-fibrotic small molecule designed to modulate the activity of multiple receptors, including GPR40 and PPAR alpha. Fezagepras has been observed to regulate several cell types involved in the fibrotic pathway: macrophages, fibroblasts/myofibroblasts and epithelial cells. We have observed that fezagepras regulated fibrotic and inflammatory markers in rodent and normal human fibroblasts, idiopathic pulmonary fibrosis patient fibroblasts, human epithelial cells and in rodent macrophages.
In December 2020, we initiated a Phase 1 clinical trial to evaluate multiple ascending doses of fezagepras in healthy volunteers. The data from this Phase 1 clinical trial will help define dose levels and regimen for the design of a global Phase 2 clinical trial of fezagepras in IPF that we plan to initiate in the first half of 2022. IPF, is a chronic lung disease characterized by a progressive and irreversible decline in lung function when lung tissue becomes damaged, stiff, and scarred. As tissue scarring progresses, transfer of oxygen into the bloodstream is increasingly impaired, leading to irreversible loss of lung function, as well as high morbidity and mortality rates.
In addition, we expect to initiate preparatory work for an anticipated Phase 1b/2a clinical trial of fezagepras in the US for patients with high triglyceride levels (hypertriglyceridemia) in 2022. This clinical trial is expected to enable the definition of future development for fezagepras in other indications.
Fezagepras has been granted Orphan Drug Designation by the U.S. Food and Drug Administration, or FDA, and the European Medicines Agency, or EMA, for the treatment of IPF and Alström syndrome. Fezagepras has also been granted a Promising Innovative Medicine, or PIM, designation by the UK Medicines and Healthcare products Regulatory Agency, or MHRA, for the treatment of IPF and Alström syndrome.
As part of our drug discovery platform, we are developing a selective GPR84 antagonist candidate that we believe could be used as monotherapy or in combination with other approved drugs. GPR84 is a pro-inflammatory target primarily expressed on cells associated with the immune system and its expression levels increase significantly during periods of inflammatory stress. Inhibition of GPR84 can inhibit neutrophil and macrophage migration and reduce cytokine release. Our GPR84 antagonist program is currently at the pre-clinical stage. Pending the outcome of our preclinical research, and successful identification of a pre-clinical drug candidate, we plan to initiate a pre-clinical IND enabling program to support a First-in-Human Phase 1 single ascending dose clinical trial of such GPR-84 drug candidate in healthy volunteers for safety and tolerability.
46
We are also developing an oral, selective OXER1 antagonist candidate. OXER1 is a GPCR that is highly selective for 5-oxo-ETE, believed to be one of the most potent human eosinophil chemo-attractants. Migration of eosinophils to body sites including the lungs and intestines is mediated by eosinophil chemo-attractants such as 5-oxo-ETE. Eosinophils play a key role in Type 2 inflammation-driven diseases, including respiratory diseases and gastro-intestinal diseases. Our OXER1 antagonist discovery program is currently at the preclinical stage.
We have developed our plasma-derived drug Ryplazim® (plasminogen), or Ryplazim, a highly purified glu-plasminogen derived from human plasma that acts as a plasminogen replacement therapy for patients deficient in plasminogen protein. We developed Ryplazim for the treatment of signs and symptoms associated with congenital plasminogen deficiency, or C-PLGD, a rare disorder associated with abnormal accumulation or growth of fibrin-rich pseudomembranous lesions on mucous membranes. Left untreated, the resulting internal lesions may impair organ function whilst external lesions may require frequent surgical removal. Both impact quality of life. Congenital plasminogen deficiency is caused by mutations in plasminogen, the gene coding for production of the zymogen plasminogen. We resubmitted a BLA to the FDA, in the third quarter of 2020, based on the results from our open-label Phase 2/3 clinical trial of Ryplazim for the treatment of congenital plasminogen deficiency completed in October 2018. The Prescription Drug User Fee Act, as amended, or PDUFA, target action date is June 5, 2021.
We have been engaged in a process to find a commercialization partner for Ryplazim, which has to date not resulted in an executable transaction. We have commenced a process to evaluate potential strategic alternatives for our plasma-derived therapeutics business aimed at minimizing our cash burn. These alternatives may result in a divestment, in whole or in part, of the plasma-derived therapeutics business and/or other non-core assets, or in other courses of action including but not limited to other strategic transactions or the closure of our Ryplazim related operations. We believe that such change in our business strategy will allow us to become more streamlined and with a singular focus on our core research capabilities and emerging pipeline. Ryplazim was granted Orphan Drug Designation and the Rare Pediatric Disease Designation by the FDA for the treatment of congenital plasminogen deficiency. Pending the outcome of the discussions on strategic alternatives for our plasma therapeutics business, if our BLA for Ryplazim is approved by the FDA, we may be also eligible to receive a Pediatric Rare Disease Priority Review Voucher, or Priority Review Voucher, or PRV, from the FDA. If we receive regulatory approval on Ryplazim, and if we receive a PRV for Ryplazim, we anticipate seeking to monetize any such PRV in 2021.
As further described above under Risk Factors—"We will require additional funding and may not be able to raise the capital necessary to continue as a going concern or to complete the research and development of our pipeline of product candidates and their commercialization, if approved”, and we do not have sufficient funds to continue maintaining our operating activities, even at low spending levels, for the next 12 months. Our December 31, 2020 financial statements have been prepared on a going concern basis.
Our Team
We have assembled a team of employees with experience across the spectrum of drug discovery and development who have experience developing products. We are led by Bruce Pritchard, our Chief Executive Officer, who brings over 25 years of experience acting as an executive and director of companies in the biotechnology, and life sciences sectors.
47
Our Pipeline
The following table sets forth our product candidates and their current development stages.
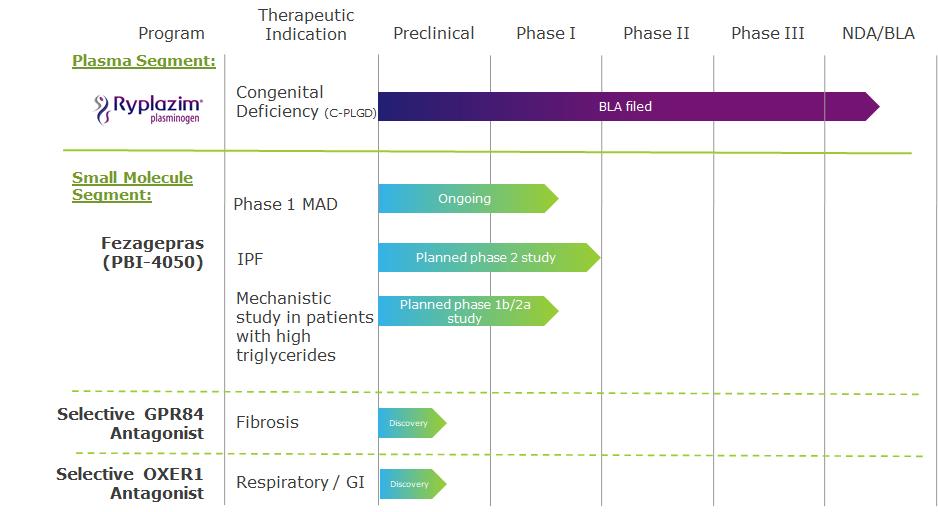
Program Therapeutic Indication Preclinical, Phase I Phase II Phase III NDA/BLA plasma segment Ryplazim plasminogen Congenital Deficiency (C-PLGD) BLA filed ongoing planned phase 2 study planned phase 1b/2a study discovery Small Molecule Segment Fezagepras PBI-4050 MAD IPF Mechanism study in patients with high triglycerides Selective GPR84 Antagonist Fibrosis Selective OXER1 Antagonist Respiratory / GI
Our Strategy
We believe that we have a novel, differentiated approach to treating the complex biology of fibrotic disease in multiple organ systems; a strategy to build a broader portfolio of novel small molecule compounds over time; and the opportunities for collaboration in our small molecule development programs. The key elements of our strategy include:
| • | Advancing the clinical development of our lead product candidate: |
We are developing fezagepras for the treatment of patients with diseases related to fibrosis, including pulmonary fibrosis. We have generated preclinical data that we believe demonstrated proof-of-mechanism of fezagepras for the treatment of respiratory, liver and kidney disorders associated with chronic or severe fibrosis. We have initiated an additional Phase 1 clinical trial to evaluate multiple ascending doses and dosing regimen of fezagepras in healthy volunteers expected to be followed by a Phase 2 clinical trial of fezagepras in IPF anticipated to be started in the first half of 2022.
We believe that the data from the Phase 1 clinical trial will help define dose levels and regimen for future clinical trials of fezagepras, including our planned Phase 2 clinical trial of fezagepras in IPF and the Phase 1b/2a trial in high triglyceride patients, subject to the current COVID-19 pandemic.
48
| • | Leveraging our drug discovery platform and knowledge base to develop an early-stage drug portfolio: |
Our early-stage drug development efforts are focused on expanding our research and development pipeline both for the treatment of diseases associated with fibrosis and other inflammatory diseases. Accordingly, we first intend to develop oral, selective GPR84 antagonists to treat fibrosis, with a first focus on respiratory disease, and develop an early-stage pipeline of novel compounds for the treatment of respiratory, liver and kidney diseases by leveraging our own drug discovery platform, entering into collaborations with third parties or in-licensing or acquiring new compounds and technologies. GPR84, a free fatty acid receptor is a pro-inflammatory target primarily expressed on cells associated with the immune system (neutrophils and monocyte or macrophages) and its expression levels increase significantly during periods of inflammatory stress by inhibiting the macrophage driven inflammatory conditions. We believe a GPR84 antagonist has the potential to be used in both monotherapy and in combination with one or more of our other product candidates in development or with therapeutics developed by third parties. Our GPR84 antagonist discovery program is currently at the pre-clinical stage. Pending the outcome of our preclinical research, and successful identification of a pre-clinical drug candidate, we plan to initiate a pre-clinical IND enabling program to support a First-in-Human Phase 1 single ascending dose clinical trial of such GPR84 drug candidate in healthy volunteers for safety and tolerability.
In addition to GRP84, we intend to develop a novel selective OXER1 antagonist candidate. OXER1 is a GPCR that is highly selective for 5-oxo-ETE, believed to be one of the most potent human eosinophil chemo-attractants. Eosinophils play a key role in Type 2 inflammation-driven diseases, including respiratory diseases and gastrointestinal diseases. Our OXER1 antagonist discovery program is currently at the pre-clinical stage. Pending the outcome of our preclinical research, we plan to nominate a preclinical product candidate for our OXER1 antagonist program in the second half of 2021.
Our Small Molecule Program
Our research program is focused on inflammatory, fibrotic and metabolic conditions in patients with respiratory, liver or renal disease.
Fibrosis
Fibrosis is an aberrant response of the body to tissue injury that may be caused by trauma, inflammation, infection, cell injury or cancer. The normal response to injury involves the activation of cells that produce collagen and other components of the extracellular matrix, or ECM, that are part of the healing process. This healing process helps to fill in tissue voids created by the injury or damage, segregate infections or cancer, and provide strength to the recovering tissue. Under normal circumstances, where the cause of the tissue injury is limited, the scarring process is self-limited and the scar resolves to approximate normal tissue architecture. However, in certain disease states, this process is prolonged and excessively active, resulting in progressive tissue scarring, or fibrosis, which can cause organ dysfunction and failure as well as, in the case of certain cancers, promote tumor progression.
Excess levels of various profibrotic cytokines and growth factors are associated with fibrosis. Innate inflammatory cells increase the abundance of myofibroblasts, a cell type that drives wound healing, and stimulates them to deposit ECM proteins such as collagen at the site of tissue injury. In the case of normal healing of a limited tissue injury, myofibroblasts eventually die by programmed cell death, or apoptosis, and the fibrous scarring process recedes. Accordingly, we believe there is a need for therapies that could effectively target pathophysiological pathways involved in fibrosis.
49
Fezagepras
We are developing fezagepras, our lead small molecule product candidate, primarily for the treatment of pulmonary fibrosis. We also anticipate developing fezagepras for the treatment of other diseases characterized by fibrosis. Fezagepras is an anti-inflammatory and anti-fibrotic small molecule designed to modulate the activity of multiple receptors, including FFAR1 and PPAR alpha. Both FFAR1 and PPAR alpha have been shown to have an impact on metabolic control and to play a role in the pathogenesis of diseases. Further, third-party data suggests that FFAR1 plays a role in glucose control, while PPAR alpha appears to modulate lipids. Given their long association with the control of metabolic disease and the association between metabolic and fibrotic processes, the PPAR agonist class has a strong development rationale in fibrotic disease. Despite this rationale, historical efforts to develop PPAR agonists for the treatment of fibrotic disease faced challenges either in terms of efficacy or safety and tolerability, depending on the PPAR agonist subtype. More recently, however, an enhanced understanding of both the PPAR class and fibrotic disease had led to the development of several next-generation PPAR agonists, which each interact with the target in a differentiated manner from previous efforts, which has renewed interest in the class as potential anti-fibrotic agents.
Our research has focused on the role of these receptors on the pathogenesis of fibrosis in multiple organs. Fezagepras has demonstrated anti-inflammatory and anti-fibrotic activity in more than 30 different animal models, including models of lung fibrosis, chronic kidney disease, diabetic kidney disease, liver fibrosis, heart fibrosis, and scleroderma.
Fezagepras has been observed to regulate several cell types involved in the fibrotic pathway: macrophages, fibroblasts/myofibroblasts and epithelial cells. We have observed that fezagepras regulates fibrotic and inflammatory markers in rodent and normal human fibroblasts, IPF patient fibroblasts, human epithelial cells and in rodent macrophages. We believe the modulating effects of fezagepras on known aspects of the fibrosis process suggest it has the potential to treat fibrosis-related diseases, for which there is a substantial unmet medical need. We are planning to further investigate its effect on patients with idiopathic pulmonary fibrosis (IPF).
Fezagepras has been administered to healthy subjects, patients with IPF, cystic fibrosis, Alström and type 2 diabetic patients with metabolic syndrome as single daily doses up to 1200mg. Based on our new understanding of the mechanism of action of fezagepras and the fact that fezagepras has a relatively short half-life, we believe this means that fezagepras needs to be given more frequently than once a day and maybe at a higher total daily dose as well.
Fezagepras has been evaluated in more than 30 different animal models involving six organ systems (lung models, heart models, kidney models, liver models, pancreas models and skin models), and has consistently shown activity in all models. Given the wide-ranging preclinical data, we believe that fezagepras represents a differentiated and unique approach to treating a complex condition such as fibrosis. We believe that fezagepras has broad applicability in multiple therapeutic categories for the treatment of other diseases characterized by fibrosis. We continue to review the potential broader use of fezagepras in other areas such as skin fibrosis and ocular fibrosis.
Fezagepras has been granted Orphan Drug Designation by the FDA and the European Medicines Agency (EMA) for the treatment of IPF and Alström syndrome. Fezagepras has also been granted a Promising Innovative Medicine (PIM) designation by the UK Medicines and Healthcare products Regulatory Agency (MHRA) for the treatment of IPF and Alström syndrome.
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis, or IPF, is a rare chronic lung disease characterized by a progressive and irreversible decline in lung function when lung tissue becomes damaged, stiff, and scarred. As tissue scarring progresses, transfer of oxygen into the bloodstream is increasingly impaired, leading to irreversible loss of lung function, as well as high morbidity and mortality rates.
Patients with IPF experience debilitating symptoms, including shortness of breath and difficulty performing routine functions, such as walking and talking. Other symptoms include chronic dry, hacking cough, fatigue, weakness, discomfort in the chest, loss of appetite and weight loss.
50
IPF usually occurs in adult individuals over 40 years of age, particularly in those with a history of cigarette smoking, and affects men more often than women. IPF affects approximately 200,000 people in the United States and Europe with about 75,000 new cases diagnosed annually. Approximately 40,000 people with IPF die each year. The five-year mortality rate for patients with IPF is estimated to range from 50% to 70% of those affected.
Limitations of Current Treatment Options for Idiopathic Pulmonary Fibrosis
Treatment options for patients with IPF are limited. Two products, nintedanib (OFEV® - Boehringer-Ingelheim) and pirfenidone (Esbriet® - Roche), are approved in most geographies for the treatment of IPF. However, neither demonstrated the ability to stabilize lung function in clinical trials, but rather were approved based upon their ability to slow the rate of decline in lung function. In addition, the tolerability profile of each drug is problematic for chronic usage as they are both associated with side effects (particularly GI-related), which have limited their use. Accordingly, we believe there remains a substantial unmet medical need in IPF.
We believe that the commercial potential of drug treatments for IPF is well-established. In 2019, worldwide reported sales for Esbriet® and Ofev® totalled approximately $3 billion representing around 30% growth from the reported sales in 2018.
If our drug candidate fezagepras is approved and successfully commercialised to treat IPF, we believe that it could compete with both Esbriet® and Ofev®, along with potential additional competitor products that may reach the market ahead of fezagepras. In particular, the Roche/Promedior asset, PRM-151 and the Fibrogen asset, pamrevlumab, which are currently in Phase 3 programs in IPF. Pamrevlumab is an injectable monoclonal antibody, which, we believe, may be more expensive and less convenient than small molecule oral drugs such as fezagepras. United Therapeutics is planning to begin a Phase 3 IPF trial with treprostinil (already approved for treatment of pulmonary arterial hypertension), in 2021. There are also numerous Phase 2 programs underway including two Bristol Myers Squibb assets (BMS-986278 and CC-90001), the Galecto Bio asset, TD-139, the Boehringer-Ingelheim asset, BI-1015550 and Galapagos NV’s GLPG1205, a selective GPR84 antagonist. We may also face competition from these programs in terms of enrolment of patients into our planned clinical trials.
Completed Open Label Phase 2 Clinical Trial
In January 2017, we completed an open-label, single arm, exploratory, observational Phase 2 trial of fezagepras, both as a monotherapy and in combination with either nintedanib or pirfenidone, in 41 patients with IPF. The primary endpoints of this trial were safety and tolerability. Nine patients received 800 mg doses of fezagepras alone, 16 patients received 800 mg oral doses of fezagepras in combination with nintedanib and 16 patients received 800 mg doses of fezagepras in combination with pirfenidone, with 40 subjects completing the trial as planned (n= 9, 16 and 15) respectively; all administered orally on a daily basis for 12 weeks. The full results of this trial were published in the European Respiratory Journal in 2018.
We also completed a single-center, single-arm, open-label Phase 2 trial evaluating fezagepras in 12 subjects with Alström syndrome who were 16 years of age or older. Patients received a total oral daily dose of 800 mg of fezagepras for an initial 24 weeks with continuation for a further 36 or 48 weeks. The primary objective of the trial was to evaluate the safety and tolerability of fezagepras in this patient population. Standard assessments of safety included adverse events, clinical laboratory tests, vital signs, physical examination and electrocardiograms.
Following completion of our Phase 2 clinical trial of fezagepras in patients with Alström syndrome, we initiated an open-label, single-arm, multi-center rollover Phase 2 clinical trial evaluating the long-term safety and tolerability of fezagepras in nine patients who completed the end-of-treatment visit for the initial Phase 2 trial. Each patient was administered a total daily dose of 800 mg of fezagepras. In connection with the COVID-19 pandemic, we ended the treatment under this rollover Phase 2 clinical trial. Nine patients have completed more than two years of treatment with fezagepras, and six of which have completed more than three years of treatment with fezagepras.
51
The fezagepras clinical development program includes 8 completed studies, and 3 studies which were terminated early, not due to safety reasons. These included studies in healthy adult subjects (n=94); adult subjects with stable renal impairment (n=8); adult subjects with IPF (n=41); adult subjects with type 2 diabetes mellitus and metabolic syndrome (n=103 and n=26); adults with cystic fibrosis (n=7) and subjects with ALMS (n=12). In these studies, doses of up to 800 mg once daily were administered for a maximum of 72 weeks and doses up to 1200 mg once daily for 12 weeks. No deaths occurred in these studies. While 12 SAEs were reported, only one of which was thought to be related to the administration of the study drug and all resolved. The most common TEAEs were headache, diarrhea, insomnia, hypoglycemia, upper respiratory tract infection and somnolence. Most TEAEs were mild in severity and resolved. No other clinically significant observations related to study drug administration were reported in any study.
Preclinical Data
Antifibrotic Activity of Fezagepras Observed in Animal Models of Lung Fibrosis
We used a mouse model of bleomycin-induced lung fibrosis to evaluate the effect of fezagepras on histological lesions and inflammatory and fibrotic markers. In this model, female mice were treated with bleomycin (0.025 U) via intratracheal administration on Day 0. On Day 7, 28 mice were randomized into three groups according to their bleomycin-induced body weight loss. Mice were treated orally with fezagepras (100 mg/kg and 200 mg/kg) or vehicle from Day 7 through Day 20. On day 21, mice were euthanized. We observed a dose-dependent reduction in histological lesions of the lung.
Observed Antifibrotic Activity of Fezagepras in a Mouse Model of Bleomycin-Induced Lung Fibrosis
| |
Parameter | Finding |
Lung lesions | Fezagepras reduced histological lesions of lung fibrosis in a dose-dependent manner |
Inflammatory and fibrotic markers | Fezagepras (200 mg/kg) significantly (p ≤ 0.05) decreased the mRNA expression of CTGF, collagen 1 and IL-23p19. |
CTGF = connective tissue growth factor; IL = interleukin; mRNA = messenger RNA.
Effect of Fezagepras on Bleomycin-Induced Histological Lesions

52
Fezagepras and Pirfenidone
We also used the mouse model of bleomycin-induced lung fibrosis to evaluate the effect of 200 mg/kg doses of fezagepras, 400 mg/kg doses of pirfenidone, and the same doses of fezagepras in combination with pirfenidone on histological lesions and inflammatory and fibrotic markers. In this preclinical study, mice were randomized into three groups according to their bleomycin-induced body weight loss and treated orally with either fezagepras, pirfenidone or a combination of both compounds from Days 7 to 20. Only the animals that recovered their body weight loss by Day 20 were used for data analysis. Mice were euthanized on Day 21. Based on the Ashcroft score, a validated measure of histological lesions of lung fibrosis, fezagepras showed a statistically significant reduction in lung fibrosis compared to the bleomycin control group. We also observed a reduction in the histological lesion score of the mice treated with pirfenidone, but the reduction was not statistically significant compared to the bleomycin control group. Fezagepras also decreased the overexpression of pro-inflammatory and fibrotic markers compared to pirfenidone. We also observed a statistically significant reduction in histological lesions of the lung in the mice treated with fezagepras alone and in the mice treated with fezagepras in combination with pirfenidone.
Observed Antifibrotic Activity of Fezagepras in a Mouse Model of Bleomycin-Induced Lung Fibrosis: Fezagepras and Pirfenidone
| |
Parameter | Finding |
Toxicity | No toxicity was reported with fezagepras. Pirfenidone induced severe vertigo that was reduced in the combination therapy (fezagepras + pirfenidone). |
Bronchoalveolar fluid | All treatments (fezagepras; pirfenidone; and fezagepras + pirfenidone) significantly (p < 0.05) reduced CTGF level. |
Only the combination therapy significantly (p = 0.0424) decreased IL-1β protein level. |
Only the combination therapy significantly (p < 0.05) decreased TNF-α protein level. |
Lung lesions | Only fezagepras and the combination therapy significantly (p < 0.05) reduced histological lesions; pirfenidone had no effect. |
Fezagepras and the combination therapy significantly (p < 0.001) decreased the lesion score determined by the Ashcroft’s score;a pirfenidone alone had a statistically insignificant effect (p = 0.0538). |
All treatments significantly (p ≤ 0.05) reduced the percentage of leukocyte infiltration. |
Inflammatory and fibrotic markers | Fezagepras and the combination therapy significantly (p < 0.05) reduced the percentage of collagen in leukocyte infiltration. |
All treatments significantly (p < 0.05) decreased the overexpression of collagen, fibronectin, IL-23p19, IL-6, and TGF-β. Only fezagepras and the combination therapy significantly (p < 0.05) decreased CTGF and SPARC mRNA expression. Only the combination therapy significantly (p < 0.05) reduced collagen 3 and MMP2 mRNA expression. |
All treatments significantly (p < 0.05) increased IFN-γ protein level in the lung. |
Fezagepras and the combination therapy significantly (p < 0.05) increased IL‑12p40 and IL-1β protein levels in the lung. |
All treatments significantly (p < 0.01) increased the content of TNF-α protein level. |
53
CTGF = connective tissue growth factor; IFN-γ = interferon gamma; IL = interleukin; MMP = matrix metalloproteinase; mRNA = messenger RNA; SPARC = matricellular protein secreted protein acidic and rich in cysteine; TGF-β = transforming growth factor beta; TNF-α = tumor necrosis factor alpha.
a Ashcroft’s score: Briefly, the entire fields of each lung section were read by a blinded examiner, and each field was visually graded from 0 to 8. Criteria for grading lung fibrosis were as follows: Grade 0= normal lung; Grade 1= minimal fibrous thickening of alveolar or bronchiolar walls; Grade 3= moderate thickening of walls without obvious damage to lung architecture; Grade 5= increased fibrosis with definitive damage to lung structure and formation of fibrous bands or small fibrous masses; Grade 7= severe distortion of structure and large fibrous area; Grade 8= total fibrous obliteration of lung fields.
Observed Effect of Fezagepras and Pirfenidone on Bleomycin-Induced Histological Lesions
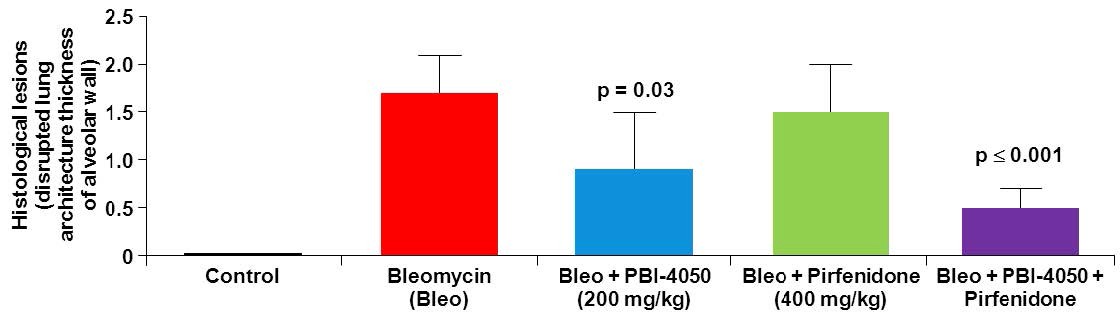
PBI-4050 + Pirfenidone p = 0.03 p ≤ 0.001
Histological lesions (disrupted lung architecture thickness of alveolar wall) Control Bleomycin (B
Fezagepras and Nintedanib
We used a mouse model of bleomycin-induced pulmonary fibrosis to evaluate the effect of 200 mg/kg doses of fezagepras, 60 mg/kg doses of nintedanib and 200 mg/kg doses of fezagepras in combination with 60 mg/kg doses of nintedanib on inflammatory and fibrotic markers. In this preclinical study, mice were randomized into three groups according to their bleomycin-induced body weight loss and treated orally with either fezagepras, nintedanib or a combination of both compounds from Days 7 to 20. Mice were euthanized on Day 21. We observed a statistically significant reduction in inflammatory and fibrotic markers in lung tissue in the mice treated with fezagepras alone and in the mice treated with fezagepras in combination with nintedanib.
54
Observed Antifibrotic Activity of fezagepras in a Mouse Model of Bleomycin-Induced Lung Fibrosis: Fezagepras and Nintedanib
| |
Parameter | Finding |
Bronchoalveolar fluid | All treatments (fezagepras; nintedanib; and fezagepras + nintedanib) significantly (p ≤ .05) reduced CTGF. Only fezagepras and the combination therapy significantly (p < 0.05) reduced MCP-1 levels. |
Inflammatory and fibrotic markers | All treatments induced a significant (p < 0.05) decrease of MCP-1 level and iNOS mRNA expression in lung tissue. |
Only fezagepras and the combination therapy significantly (p < 0.05) reduced the percentage of collagen in inflamed lesions and in total lung tissue. |
Only fezagepras significantly (p < 0.05) decreased the mRNA expression of collagen and fibronectin in lung tissue. |
Fezagepras significantly (p < 0.05) reduced TNF-α, IL-6, and CTGF mRNA expression in lung tissue. Nintedanib and the combination therapy induced a significant (p < 0.05) reduction of IL-6 and CTGF. |
CTGF = connective tissue growth factor; IL = interleukin; iNOS = inducible nitric oxide synthase; MCP = monocyte chemotactic protein; mRNA = messenger RNA; TNF-α = tumor necrosis factor alpha.
Our Selective G-protein coupled receptor 84 (GPR84) Antagonist Program
We are developing a selective GPR84 antagonist candidate that we believe could be used as monotherapy or in combination with other approved drugs. GPR84 is a pro-inflammatory target primarily expressed on cells associated with the immune system and its expression levels increase significantly during periods of inflammatory stress. Inhibition of GPR84 can inhibit neutrophil and macrophage migration and reduce cytokine release.
However, GPR84 expression is not restricted to cells in the immune system; it is also expressed in tissues such as the lung, brain, heart, muscle, colon, kidney, liver, intestine and adipose. Through its role in inflammation, GPR84 may be a mediator of the relationship between inflammation, obesity and diabetes. Rodent models suggest that GPR84 expression is up-regulated in adipocytes in response to TNF-α released from infiltrating macrophages, and that this in turn can lead to a down-regulation in adiponectin expression in adipocytes. Adiponectin is known to have anti-diabetic, anti-inflammatory, and anti-atherogenic effects, and it also functions as an insulin sensitizer.
We believe we have a strong understanding and well-established, in-house expertise in GPR84 biology. Our prior research has shown that GPR84-deficient mice develop reduced fibrosis in a model of kidney fibrosis (adenine-induced chronic kidney disease). We have also shown that GPR84 mRNA is overexpressed in various acute and chronic kidney models such as 5/6-nephrectomy (Nx)-induced chronic kidney disease, doxorubicin (DOX)-induced nephropathy and in adenine-induced tubulointerstitial injury.
Our GPR84 antagonist discovery program is currently at the pre-clinical stage. Pending the outcome of our preclinical research, and successful identification of a pre-clinical drug candidate, we plan to initiate a pre-clinical IND enabling program to support a First-in-Human Phase 1 single ascending dose clinical trial of our GPR84 drug candidate in healthy volunteers for safety and tolerability. Galapagos Therapeutics has reported positive topline results with GLPG1205in IPF patients in its Phase 2 PINTA proof-of-concept clinical trial. The GLPG1205 program is the only other GPR84 antagonist development program to our knowledge.
55
Fezagepras for the Treatment of Fibrosis Associated with Other Indications
Fibrosis Associated with Non-Alcoholic Steatohepatitis (NASH)
Non-alcoholic fatty liver, or NAFLD, is a condition that is estimated to affect 75 million people in the United States due to the obesity epidemic and is the manifestation of metabolic disease in the liver. NASH is a progressed state of NAFLD, where the chronic injury suffered by the liver due to the excess fatty deposits associated with NAFLD trigger inflammation and fibrosis. Though only a small percentage of NAFLD patients progress to NASH, the sheer number of NAFLD patients has made NASH the most common cause of severe liver disease worldwide. NASH and its associated co-morbidities, such as fibrosis, remain a major unmet medical need with treatment offering little recourse.
Though the biologic mechanisms underlying the pathogenesis of NASH are not fully characterized, the current understanding describes excess lipids present in the liver ultimately leading to hepatoxic injury, followed by inflammation and fibrosis with an associated decline in liver function. Current research implicates multiple pathways for both the initial lipid accumulation and the dysregulated healing response.
There are no currently approved therapies for NAFLD, NASH or associated fibrosis, although one potential therapy (Ocaliva®, developed by Intercept Pharmaceuticals) is currently under FDA review. Current treatment options are limited to diet and lifestyle modifications that may control or reduce the amount of excess fat deposits in the liver.
Observed Antifibrotic Activity of fezagepras Following High Fat Diet (HFD), a Model of Metabolic Syndrome
Obesity and its resulting metabolic disturbances are health threats and a leading cause of a host of diseases, including non-alcoholic fatty liver disease. In preclinical studies, C57BL/6 mice were fed with either a standard diet or a high fat diet, or HFD, for 14 weeks. These mice were then divided into five groups as follows: standard diet, HFD + vehicle, and HFD + fezagepras (200 mg/kg, oral once a day), GPR84-/- + vehicle, and GPR84-/- + fezagepras (200 mg/kg, oral once a day) and treated for an additional six or 10 weeks.
Blood biochemistry, serum insulin level, liver, and white adipose tissue, or WAT, gene expression for markers of inflammation, fibrosis, adipokines, PPARs, fatty acid metabolism, energy expenditure and glucose transporter and histology assessment of these tissues were performed.
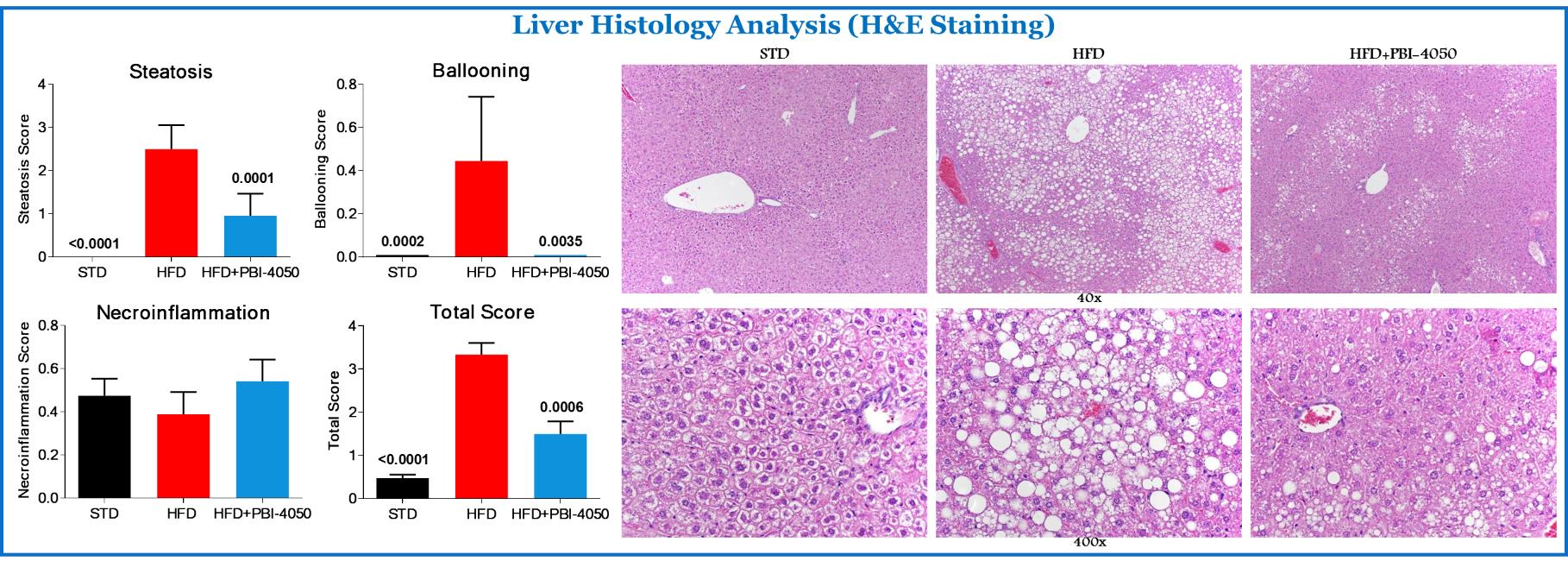
Our Selective Oxo-eicosanoid receptor 1 (OXER1) Antagonist Program
We are developing an oral, selective OXER1 antagonist candidate. OXER1 is a GPCR that is highly selective for 5-oxo-ETE, believed to be one of the most potent human eosinophil chemo-attractants. Migration of eosinophils to body sites including the lungs and intestines is mediated by eosinophil chemo-attractants such as 5-oxo-ETE. Eosinophils play a key role in Type 2 inflammation-driven diseases, including respiratory diseases and gastro-intestinal diseases.
56
Eosinophilic-related diseases represent a significant area of unmet need in global health. Several biologics have been approved for the treatment of eosinophil-related diseases. Several of the approved monoclonal antibody treatments for severe eosinophilic asthma are currently in clinical trials aimed at expanding their indications to include other eosinophilic disorders. Compared to approved biologics, small molecule OXE receptor antagonists may offer a promising and potentially more cost-effective option for treatment of eosinophilic-driven disorders. Compared to approved biologics, small molecule OXE receptor antagonists may offer a promising and potentially more cost-effective option for treatment of eosinophilic-driven disorders.
Our OXER1 antagonist program is based on the research of Dr. William Powell, Professor Emeritus in the Department of Medicine at McGill University, working in collaboration with Dr. Joshua Rokach of the Florida Institute of Technology.
In addition to our experienced in-house medicinal chemistry group, we have a strong understanding and well-established, in-house expertise in GPCR biology.
Our OXER1 antagonist program is currently at the pre-clinical stage. Pending the outcome of our preclinical research, we plan to nominate a preclinical product candidate for our OXER1 antagonist program in the second half of 2021.
Our Plasma-Derived Therapeutics Program
Ryplazim
We have developed our plasma-derived investigative drug, Ryplazim® (plasminogen), or Ryplazim, a highly purified glu-plasminogen derived from human plasma that acts as a plasminogen replacement therapy for the treatment of signs and symptoms associated with congenital plasminogen deficiency, or C-PLGD.
Plasminogen and Congenital Plasminogen Deficiency
Plasminogen is a naturally occurring protein that is synthesized by the liver and circulates in the blood. Activated plasminogen, plasmin, is a fundamental component of the fibrinolytic system and is the main enzyme involved in the lysis of blood clots and clearance of extravasated fibrin. Plasminogen is therefore vital in wound healing, cell migration, tissue remodeling, angiogenesis and embryogenesis. Patients may be born with the inability to produce sufficient plasminogen naturally, a condition referred to as congenital plasminogen deficiency, or suffer an acute or acquired deficiency following a trauma or an illness. Patients with congenital plasminogen deficiency experience an accumulation of fibrin growths or lesions on mucosal surfaces throughout the body. Many cases are first diagnosed in the pediatric population, and if left untreated, disease manifestations may be organ-compromising. Peer-reviewed publications report that the condition may have a prevalence of 1.6 cases per million globally. Proprietary data sources and analyses involving the U.S. population suggest that the number of people potentially affected by plasminogen deficiency in the United States may be greater than these early epidemiological estimates. Congenital plasminogen deficiency requires lifelong therapy to avoid recurrence of lesions. There are currently no approved therapies for the treatment of congenital plasminogen deficiency.
The most common and visible lesion associated with plasminogen deficiency is ligneous conjunctivitis, or LC, which is characterized by thick, woody (ligneous) growths on the conjunctiva of the eye, and if left untreated, can lead to corneal damage and blindness. Ligneous growths tend to recur after surgical excision, thereby requiring multiple surgeries. While ligneous conjunctivitis is the most common lesion, congenital plasminogen deficiency is a multi-systemic disease that can also affect the ears, tracheobronchial tree, genitourinary tract, and gingiva. Tracheobronchial lesions can result in respiratory failure. Hydrocephalus has also been reported in children with severe hypoplasminogenemia, apparently related to the deposition of fibrin in the cerebral ventricular system. Patients who suffer from plasminogen deficiency also have impaired post-surgical wound healing.
Patients with plasminogen deficiency may be born with the inability to produce sufficient plasminogen naturally, a condition referred to as congenital plasminogen deficiency, or suffer an acute or acquired deficiency following a trauma or an illness. Commercially available diagnostics are used to diagnose patients with plasminogen deficiency and genealogical tracking is also available given the autosomal recessive natures of the disease.
57
Our Solution for Congenital Plasminogen Deficiency: Ryplazim
Ryplazim is a highly purified glu-plasminogen derived from human plasma that acts as a plasminogen replacement therapy for patients with congenital plasminogen deficiency, a rare inherited disorder caused by a mutation of the PLG gene. Ryplazim is a highly purified glu-pasminogen that acts as a replacement therapy for activated plasminogen, a fundamental component of the fibrinolytic system and the main enzyme involved in the lysis of blood clots, clearance of extravasated fibrin and other vital physical processes.
Completed Phase 2/3 Clinical Trial
In a pivotal Phase 2/3 clinical trial of Ryplazim for the treatment of congenital plasminogen deficiency, 15 patients (six children and nine adults) were administered 6.6 mg/kg intravenous doses of Ryplazim every two to four days for 48 weeks. The primary efficacy endpoint was clinical success, defined as 50% of the subjects achieving a greater than 50% improvement in lesion number/size or functionality impact from baseline. The primary pharmacokinetic endpoint was an increase in trough plasminogen activity levels by at least an absolute 10% above baseline at 12 weeks. Ryplazim met both its primary efficacy and pharmacokinetic endpoints following the intravenous administration of Ryplazim to fifteen patients for 48 weeks, as we observed a reduction in visible and non-visible lesions of greater than 50% in all patients at 48 weeks and at least a 10% improvement from baseline trough plasminogen levels at 12 weeks.
Ryplazim was well tolerated in both children and adults at 48 weeks. No serious adverse events were reported and no patient permanently discontinued treatment during the pivotal Phase 2/3 clinical trial of Ryplazim due to an adverse event. Most adverse events related to the completed Phase 2/3 clinical trial were mild or moderate in severity and deemed by the investigators to be unrelated to the study drug. Two patients had adverse events of severe intensity. One of the patients in the Phase 2/3 clinical trial reported anxiety, nausea, fatigue, arthralgia, back pain, dizziness, paresthesia and flushing after her twentieth infusion, which were resolved after temporary treatment discontinuation without re-emergence with restart of treatment. The other patient in this trial had severe back pain that resolved three days later. The most common adverse events observed in patients in this trial were headache and nasopharyngitis. Several patients in the trial had adverse events (including epistaxis, hematuria and dysmenorrhea) and/or laboratory abnormalities (including blood in urine and elevated D-dimer levels) consistent with physiologic trial drug activity, specifically increased fibrinolytic capacity with lesion dissolution. Minor bleeding events of hemorrhage (eye, skin, uterine, and vaginal), epistaxis, hematuria, cervical and oral discharge occurred in a few patients in this trial in areas near the lesion sites or associated with urinary excretion. No additional analyses were performed on any of these secretions. These findings are consistent with abnormal urinalysis findings of blood and protein in urine, which became macroscopic with urinary tract lesion lysis. Gross hematuria was not persistent or continuous. In addition, there were no clinically significant findings for vital signs or viral testing. Anti-plasminogen antibodies were not detected in any patient in this trial.
BLA Resubmission and Complete Response Letter
We resubmitted a BLA to the FDA, in the third quarter of 2020, based on the results from our open-label Phase 2/3 clinical trial of Ryplazim for the treatment of congenital plasminogen deficiency completed in October 2018. The PDUFA target action date is June 5, 2021.
We have been engaged in a process to find a commercialization partner for Ryplazim, which has to date not resulted in an executable transaction. We have commenced a process to evaluate potential strategic alternatives for our plasma-derived therapeutics business aimed at minimizing our cash burn. These alternatives may result in a divestment, in whole or in part, of the plasma-derived therapeutics business and/or other non-core assets, or in other courses of action including but not limited to other strategic transactions or the closure of our Ryplazim related operations. We believe that such change in our business strategy will allow us to become more streamlined and with a singular focus on our its core research capabilities and emerging pipeline.
Ryplazim was granted Orphan Drug Designation and the Rare Pediatric Disease Designation by the FDA for the treatment of congenital plasminogen deficiency. Pending the successful outcome of the discussions on strategic alternatives for our plasma therapeutics business, if our BLA for Ryplazim is approved by the FDA, we may be also eligible to receive a PRV from the FDA. If we receive regulatory approval on Ryplazim, and if we receive a PRV for Ryplazim, we anticipate seeking to monetize any such PRV in 2021.
58
Manufacturing, Plasma Collection and Processing
We rely and expect to continue to rely for the foreseeable future, on third-party contract development manufacturing organizations, or CDMOs, to produce our small molecule product candidates for preclinical and clinical testing, as well as for commercial manufacture if our product candidates receive marketing approval. We require that our CDMOs produce bulk drug substances and finished drug products in accordance with current Good Manufacturing Practices, or cGMPs, and all other applicable laws and regulations. We maintain agreements with our manufacturers that include confidentiality and intellectual property provisions to protect our proprietary rights related to our product candidates.
We have engaged CDMOs to manufacture supply of certain of our product candidates for preclinical, clinical and commercial use. Additional CDMOs are used to fill finish, label, package and distribute drug product for preclinical and clinical use. We do not currently have arrangements in place for redundant supply. As our development programs expand and we build new process efficiencies, we expect to continually evaluate this strategy with the objective of satisfying demand for registration trials and, if approved, the manufacture, sale and distribution of commercial products.
Our wholly-owned subsidiary, Prometic Bioproduction Inc., or PBP, operates our plasma processing facility in Laval, Québec, Canada, where we manufacture Ryplazim in accordance with cGMPs to be used in our clinical trials and “compassionate use” programs. “Compassionate Use” is the use outside of a clinical trial of an investigational, or not approved, product candidate when patient enrollment in a clinical trial is not possible, typically due to patient ineligibility or a lack of ongoing clinical trials.
We also have a plasma collection center in Winnipeg, Manitoba, Canada, which is an FDA and Health Canada licensed, EMA compliant and International Quality Plasma Program certified plasma collection facility located in close proximity to Emergent’s Winnipeg-based cGMP-compliant manufacturing facility. We believe that the plasma collection center allows for strategic sourcing of plasma for the purification of Ryplazim using our plasma-protein purification platform. Our wholly-owned subsidiary, Prometic Plasma Resources Inc., or PPR, also sells specialty plasma and red blood cells to third parties in the ordinary course. PPR is responsible for supplying and purchasing our plasma requirements. In 2021, we received a BLA approval from the FDA for the approval of our U.S.-based plasma collection center, located in Amherst, New York and received a Clinical Laboratory License from the State of New York. The U.S. collection center is operated by our wholly-owned subsidiary, Prometic Plasma Resources (USA) Inc. In 2020, we have also leveraged our plasma collection capabilities to commence collecting blood plasma from recovered COVID-19 patients to be potentially used in the manufacture of hyperimmune immunoglobulins by third parties for clinical trial purposes. In January 2021, we announced that we were exploring potential strategic alternatives for our plasma-derived therapeutics’ business, which may include the sale or divestment of non-core assets related to such plasma-derived business.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary rights. While we believe that our product candidates, technology, knowledge, experience and scientific resources provide us with competitive advantages, we compete in the highly competitive markets and face significant competition from many sources, including pharmaceutical and biotechnology companies, as well as academic institutions, governmental agencies and private and public research institutions.
We compete in the segments of the biotechnology, pharmaceutical and other related industries that develop and market therapies for the treatment of disorders associated with fibrosis. There are many other companies, including large biotechnology and pharmaceutical companies, that have commercialized and/or are developing therapies for the same therapeutic areas that our product candidates target. There are two approved products available for the treatment of IPF: Boehringer Ingelheim (Ofev / nintedanib) and Roche (Esbriet / pirfenidone). In addition, there are several product candidates in development for IPF including two product candidates already in Phase 3 clinical trials: FibroGen Inc. (pamrevlumab), and Promedior (recently acquired by Roche), PRM-151. In February 2021, Galapagos NV and Gilead announced the discontinuation of the ISABELA Phase 3 trials of GLPG1690 in IPF. Other PPAR agonists that are in clinical development for the treatment of fibrotic and metabolic indications include lanifibranor (Inventiva S.A.), pemafibrate (Kowa Pharmaceuticals) and seladelpar (Cymabay Therapeutics, Inc.).
59
Many of the companies against which we are competing or against which we may compete in the future, either alone or with their strategic collaborators, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved drugs than we do. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies or universities and research institutions. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and enrolling patients for our clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
We could see a reduction or elimination of our commercial opportunity if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. The availability of reimbursement from government and other third-party payors will also significantly affect the pricing and competitiveness of our products. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
License, Manufacturing, Supply and Royalty Agreements
Adsorbent Supply Agreement-
We currently have an adsorbent supply agreement, dated June 18, 2019, with PBL (now Astrea Bioseparations Ltd) for the supply by PBL to us of affinity adsorbents used in the manufacturing activities for plasma-derived products, including our proprietary affinity adsorbents used in the manufacturing of Ryplazim. The price for the adsorbents was detailed in the agreement and is subject to increase in accordance with the agreement, which does not contain any minimum purchase requirements. The agreement has an initial ten-year term with subsequent five-year terms, unless not renewed by us or earlier terminated or not renewed by us in accordance with its terms.
Plasma Purchase Agreements-
We currently have a plasma purchase agreement, dated November 6, 2015, with Interstate Blood Bank, Inc., or IBBI, for the supply by IBBI to us of plasma used in the manufacturing of Ryplazim. The price for the plasma is detailed in the agreement and is subject to increase or decrease in accordance with the agreement. We have agreed to certain annual minimum purchase amounts. The agreement expires on December 31, 2021, unless earlier terminated by either party in accordance with its terms.
We also have a plasma purchase agreement, dated June 6, 2017, with Grifols Worldwide Operations Limited, or Grifols, for the supply by Grifols to us of plasma used in the manufacturing of Ryplazim. The price for the plasma is detailed in the agreement and is subject to increase in accordance with the agreement. We have agreed to certain annual minimum purchase amounts. The agreement has an initial term until December 31, 2022, with an option to mutually renew the agreement for additional one-year terms, unless earlier terminated by either party in accordance with its terms.
Supply Agreements-
We currently have a master services agreement, or Emergent MSA, dated May 15, 2015, with Cangene Corporation d/b/a Emergent BioSolutions, or Emergent, for the provision of manufacturing services in quantities specified in statement of works. We have agreed to certain annual minimum commitment amounts. The Emergent MSA has an initial fifteen-year term, unless earlier terminated in accordance with its terms.
Royalty Stream Purchase Agreement with Structured Alpha-
In April 2018, we entered into a royalty stream purchase agreement with SALP, our lender under that certain loan agreement dated November 30, 2017. Pursuant to the royalty stream purchase agreement, SALP holds the right to receive a low single digit royalty on net sales, licensing revenues, and joint venture revenues attributable to certain of our proprietary intellectual property rights, or royalty income. During a specified period, we have the right to repurchase the royalty income for the fair market value of SALP’s interest under the royalty stream
60
purchase agreement, which is determined on a net present value basis by considering the remaining life of the associated patents (and any relevant extensions or additional exclusivity periods that may be associated with the associated patents or products) and associated cash flows, all as determined pursuant to a valuation performed by a mutually agreed upon third party. The agreement expires upon the earlier of the mutual agreement of the parties and the later of expiration on a country-by-country basis of the last applicable patent right and any applicable data or regulatory exclusivity in such country. The obligation under the royalty stream purchase agreement is secured by all of our assets until the expiry of the last patent anticipated in 2033. Either party may terminate the agreement upon an uncured material breach by the other party.
Assignment Agreement with Innovon-
In October 2001, we and our subsidiary Prometic BioSciences Inc. (now Liminal R&D BioSciences Inc., or LBRD) entered into an assignment agreement with Innovon Pharmaceuticals Inc., or Innovon, and Pierre Laurin pursuant to which Innovon and Pierre Laurin assigned to LBRD and us, all rights, title and interests in certain defined compounds (the “2001 Agreement”).
In consideration for the assignment by Innovon and Pierre Laurin, we are obligated to pay Innovon a low single digit percentage of any consideration received by us in connection with a license or grant of rights to third parties with respect to such assigned defined compounds. Additionally, we are obligated to pay a less than one percent royalty on net sales of such assigned compounds. At any time after a certain period, in the event that (i) we fail to develop or commercialize an assigned compound (as defined and covered by the 2001 Agreement, as amended (“Compounds”)), for an unreasonable period of time, (ii) we elect to cease further development or commercialization of an assigned Compound or (iii) of our insolvency, we are obligated to reassign the applicable Compound to Innovon and Pierre Laurin subject to certain conditions, including payment by Innovon and Pierre Laurin of a consideration to be negotiated between the parties and payable in the form of royalties.
The 2001 Agreement expires, on an assigned Compound-by-assigned Compound basis, on the last sale of the applicable assigned Compound, or upon the occurrence of any Event of Default, which includes, subject to applicable laws, insolvency, bankruptcy, an arrangement with creditors, a restructuring, protective administration or liquidation of the Company. Innovon and/or Pierre Laurin may also terminate the 2001 Agreement upon our breach that remains uncured for a certain number of days.
On December 14, 2015, Innovon and Pierre Laurin consented to the assignment and transfer our worldwide intellectual property rights related to the assigned Compounds, except for our Canadian rights, to our subsidiary Prometic Pharma SMT Limited (now known as Liminal BioSciences Limited).
Amended and Restated License Agreement with The Royal Institution for the Advancement of Learning/ McGill University and Florida Institute of Technology, Inc.-
Concurrent with the acquisition of Fairhaven Pharmaceuticals Inc., or Fairhaven, in July 2020, Fairhaven entered into an amended and restated license agreement with The Royal Institution for the Advancement of Learning/McGill University and Florida Institute of Technology, Inc. (the “Licensors”) effective as of May 16, 2018. Pursuant to the amended and restated license agreement, the Licensors granted us worldwide, exclusive royalty-bearing right to use and practice certain licensed patents covering 5-oxo-ETE receptor antagonist compounds and indole analogs as 5-oxo-ETE receptor antagonists and methods of use thereof (the “Licensed Patents”) and know-how, with the exclusive right to develop, modify, adapt, improve and customize certain tangible devices, materials, services or processes, in the course of exploitation which, in the absence of the amended and restated license agreement, would infringe one or more of the issued or pending claims of Licensed Patents (the “Licensed Products”), and the right to make, have made, commercialize, exploit, reproduce, market, sell, rent, distribute, lease or otherwise transfer, import and export certain Licensed Products, contingent upon our material compliance with the terms of the Amended and Restated License Agreement. We expect to utilize the Licensed Patents and know-how licensed from the Licensors in connection with the development of our pre-clinical OXER1 antagonist program. Under the Amended and Restated License Agreement, we are obliged to pay a low single digit royalty to the Licensors based on a percentage of net sales of the Licensed Products. The Amended and Restated License Agreement terminates upon the last to expire, or become abandoned, Licensed Patent(s), whether by statute or otherwise, unless it earlier terminates by operation of law or by acts of the parties in accordance with the terms of the Amended and Restated License Agreement.
61
License Agreement and Contract Manufacturing Agreement with Hematech-
In May 2012, we entered into an amended and restated license agreement with Hematech Biotherapeutics Inc., or Hematech, pursuant to which we granted Hematech an exclusive license under certain of our technology to develop, manufacture and commercialize plasma-derived products in Taiwan and a co-exclusive license under such technology to develop, manufacture and commercialize plasminogen products anywhere in the world other than China as well as a contract manufacturing agreement pursuant to which we have the exclusive right to obtain supply of plasma-derived products from Hematech for use outside of Taiwan. Currently, Hematech is not manufacturing plasma-derived products. Subsequently, in December 2016, we entered into an amendment to the license agreement pursuant to which we re-obtained our exclusive rights to the plasminogen products around the world.
Under the license agreement, we are entitled to receive a low single digit royalty on net sales of plasma-derived products in Taiwan. In connection with our re-obtaining rights to our plasminogen products we issued to Hematech 1,683 common shares and are obligated to pay a mid-single digit royalty on net sales of plasminogen products for congenital deficiency up to December 16, 2020 and subject to both a minimum and cap in the low seven figures.
In January 2021, the parties entered into a Termination Agreement, whereby the parties have agreed to terminate the license agreement, manufacturing agreement and all associated agreements effective upon final payment of the above-mentioned royalty on or before August 15, 2021.
Intellectual Property
We own and control the intellectual property utilized in the vast majority of our technologies, products and potential product candidates, giving us the option to develop and eventually commercialize these products in various geographies, to develop new formulations, and to select CDMOs and CROs of our choice. Our intellectual property rights include our trademarks, patents and patent applications, regulatory dossiers, manufacturing, and process know-how. Our intellectual property portfolio has been built in large part from in-house technology and product research and development over the past 20 years as well as strategic relationships and joint ventures.
Our approach regarding our intellectual property portfolio is to file and/or license patents and patent applications as appropriate and to seek to obtain patent protection in at least the major pharmaceutical markets, including the United States, major European countries and Japan. We also rely on trade secrets, proprietary unpatented information, trademarks, and contractual arrangements to protect our technology and enhance our competitive position. We currently have a patent estate comprised of owned and in-licensed patents and patent applications. Our patent portfolio includes patents and patent applications claiming compounds, pharmaceutical compositions, nutraceuticals, processes, and methods for treating diseases, disorders, or conditions.
Fezagepras
Our fezagepras program is covered by a patent portfolio that we own and that is comprised of issued patents, as well as allowed and pending patent applications, related to fezagepras and related derivative compounds, salt forms and formulations of the same, and methods of making and using the same therapeutically. The portfolio is comprised of patents granted in Canada, United States, Europe, China, Japan, Russia and other countries, including granted U.S. patents with claims directed to the fezagepras compound and its use in treating various diseases including inflammatory diseases and renal disorders. These patents are expected to expire no sooner than 2030, excluding any patent term adjustments or extensions. We also own a patent family with claims directed to methods of treating pulmonary fibrosis (including idiopathic pulmonary fibrosis, or IPF), liver fibrosis, cardiac fibrosis or skin fibrosis with fezagepras or related derivatives. Patents in this family have been granted in Canada, United States, Europe, China, Japan, Russia and other countries and are expected to expire no sooner than 2034, excluding any patent term adjustments or extensions.
62
Plasminogen or Ryplazim
Our Ryplazim program is covered by a wholly-owned patent portfolio comprised of two issued U.S. patents, as well as allowed and pending patent applications, related to the Ryplazim clinical formulation and methods of use. Specifically, two patents have been granted in the United States and one patent has been granted in Japan, and corresponding patent applications are pending in Canada, United States, Europe, China, Russia and other countries with claims directed to the current formulation. The current patents and any patents granted from the pending applications are expected to expire in 2035, excluding any patent term adjustments or extensions. A second patent family is granted in Russia and pending in Canada, United States, Europe, China, Japan, and other countries with claims directed to the use of a plasminogen dosage regimen for restoring and maintaining physiological plasminogen levels in plasminogen-deficient patients. The Russian patent and any patents that may be granted from the pending patent applications are expected to expire in 2036, absent any patent term adjustments or extensions. We also have wholly-owned pending patent applications related to the use of plasminogen to treat conditions associated with PAI-1 overexpression which are expected to expire in 2038.
Our Trademark Portfolio
RYPLAZIM is our registered trademark, in the United States, Canada, Europe, Japan and Turkey used in association with plasminogen for the treatment of plasminogen congenital deficiency in those countries, if approval is received from the relevant regulatory authorities.
LIMINAL BIOSCIENCES is our trademark currently registered in Australia, China and pending registration in Canada and in several other countries. This mark is our name and is used in association with several goods and services, namely pharmaceutical research and development, and pharmaceutical preparations and compositions being developed for the treatment of fibrosis and other indications.
We also have unregistered trademark rights in certain countries in which we operate, where trademark rights arise from use, rather than registration.
Other Intellectual Property Portfolio
Our portfolio of intellectual property contains additional trademarks, pending trademark registrations and domain names associated with our trademarks and pending trademark applications.
Our Policy on Intellectual Property
Our intellectual property practice is to keep all information relating to proprietary compounds, inventions, improvements, trade secrets, know-how and continuing technological innovation confidential and, where practicable, file patent and trademark applications. In particular, as part of our intellectual property protection practice, we, where we deem practicable and commercially reasonable:
| • | file patent applications for any new and patentable invention, development or improvement in the United States and in other countries; |
| • | prosecute pending patent applications in conformity with applicable patent laws and in a manner that covers our activities; |
| • | file trademark applications in countries of interest for our trademarks; |
| • | register domain names whose addresses include our trademark names; and |
| • | maintain our intellectual property rights by paying government fees as may be necessary to ensure such rights remain in force. |
63
Government Regulation
In the United States, pharmaceutical and biological products are subject to extensive regulation by the FDA under the Federal Food, Drug, and Cosmetic Act, or FDCA, and the Public Health Service Act, or PHSA. The FDCA, PHSA, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending BLAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
We cannot market a drug or biological product in the United States until the product candidate has received FDA approval. The steps required before a new drug or biologic may be marketed in the United States generally include the following:
| • | completion of extensive nonclinical laboratory tests, animal studies, and formulation studies in accordance with the FDA's Good Laboratory Practice, or GLP, regulations; |
| • | submission to the FDA of an Investigational New Drug, or IND, for human clinical testing, which must become effective before human clinical trials may begin; |
| • | performance of adequate and well-controlled human clinical trials in accordance with Good Clinical Practice, or GCP, requirements to establish the safety and efficacy of the product for each proposed indication; |
| • | submission to the FDA of a BLA, in the case of biological product candidates or a New Drug Application, or NDA in the case of small molecule drug candidates, after completion of all pivotal clinical trials; |
| • | satisfactory completion of an FDA inspection of sites involved in our clinical trials; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the active pharmaceutical ingredient, or API, and finished product are produced and tested to assess compliance with cGMPs; and |
| • | FDA review and approval of the BLA prior to any commercial marketing or sale of the product in the United States. |
Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Nonclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the nonclinical tests must comply with federal regulations and requirements, including GLP regulations. The results of nonclinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls and a proposed clinical trial protocol. Long-term nonclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin. If the FDA raises concerns or questions about the conduct of the trial, such as whether human research subjects will be exposed to an unreasonable health risk, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed.
Clinical trials involve the administration of the investigational new biologic to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted in compliance with federal regulations, including GCP requirements, as well as under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol and subsequent protocol amendments must be submitted to the FDA as part of the IND.
64
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board, or IRB, for approval at each site at which the clinical trial will be conducted. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB's requirements, or may impose other conditions.
U.S. Biopharmaceutical Products Development Process
Before testing any product candidate, the product candidate enters the nonclinical testing stage. Nonclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of the nonclinical tests must comply with federal regulations and requirements including GLPs.
The clinical study sponsor must submit the results of the nonclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some nonclinical testing typically continues after the IND is submitted. An IND is an exemption from the FDCA that allows an unapproved product to be shipped in interstate commerce for use in an investigational clinical trial and a request for FDA authorization to administer an investigational product to humans. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA requests certain changes to a protocol before the study can begin, or the FDA places the clinical study on a clinical hold within that 30-day time period. The FDA may also impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, studies may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such studies.
Clinical trials involve the administration of the product candidate to healthy volunteers or subjects under the supervision of qualified investigators, generally physicians not employed by or under the study sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical study, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical study will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research subjects provide informed consent. Further, each clinical study must be reviewed and approved by an independent IRB, at or servicing each institution at which the clinical study will be conducted. An IRB is charged with protecting the welfare and rights of study participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical study subject or his or her legal representative and must monitor the clinical study until completed. Additionally, some trials are overseen by an independent group of qualified experts organized by the trial sponsor, known as a data safety monitoring board or committee.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| • | Phase 1. The product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| • | Phase 2. The product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| • | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. |
65
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the biological product has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the characteristics of the product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHSA emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
After the completion of clinical trials, FDA approval of a BLA or NDA must be obtained before commercial marketing of the product. The BLA or NDA must include results of product development, laboratory and animal studies, human studies, information on the manufacture and composition of the product, proposed labeling and other relevant information. In addition, under the Pediatric Research Equity Act, or PREA, a BLA or NDA or supplement must contain data to assess the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any biological product for an indication for which orphan designation has been granted. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, as amended, or PDUFA, each BLA or NDA must be accompanied by a significant user fee. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs or NDAs for product candidates designated as orphan drugs, unless the product candidate also includes a non-orphan indication.
66
Within 60 days following submission of the application, the FDA reviews the application submitted to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any application that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA or NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review to determine, among other things, whether the proposed product is safe, potent, and effective, for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, safety, strength, quality, potency and purity. The FDA may refer applications for novel products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. FDA also will determine whether a Risk Evaluation and Mitigation Strategy, or REMS, is necessary to assure the safe use of the product. If the FDA concludes a REMS is needed, the sponsor must submit a proposed REMS; the FDA will not approve the application without a REMS, if required.
The FDA also offers a number of expedited development and review programs for qualifying product candidates. Priority review designation for BLA is assigned by FDA, if at all, at the time of acceptance of a BLA for a product that treats a serious or life-threatening condition and, if approved, would be a significant improvement in safety or effectiveness relative to available therapies.
Before approving an application, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, the FDA will typically inspect one or more clinical trial sites to assure that the clinical trials were conducted in compliance with IND study requirements and GCP requirements. To assure cGMP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA or NDA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. If the agency decides not to approve the BLA or NDA in its present form, the FDA will issue a complete response letter that usually describes all of the specific deficiencies identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. In addition, the FDA may require post marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to further assess a biological product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized. As a condition for approval, the FDA may also require additional nonclinical testing as a Phase 4 commitment.
One of the performance goals agreed to by the FDA under the PDUFA is to review standard BLAs and NDAs in ten months from filing and priority BLAs and NDAs in six months from filing, whereupon a review decision is to be made. The FDA does not always meet its PDUFA goal dates and its review goals are subject to change from time to time. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
67
Maintaining substantial compliance with applicable federal, state, and local statutes and regulations requires the expenditure of substantial time and financial resources. Rigorous and extensive FDA regulation of biopharmaceutical products continues after approval, particularly with respect to cGMP. Manufacturers of our products are required to comply with applicable requirements in the cGMP regulations, including quality control and quality assurance and maintenance of records and documentation. Following approval, the manufacturing facilities are subject to biennial inspections and such inspections may result in an issuance of FDA Form 483 deficiency observations or a warning letter, which can lead to plant shutdown and other more serious penalties and fines. Prior to the institution of any manufacturing changes, a determination needs to be made whether FDA approval is required in advance. If not done in accordance with FDA expectations, the FDA may restrict supply and may take further action. Annual product reports are required to be submitted annually. Other post-approval requirements applicable to biological products, include reporting of cGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information, and complying with electronic record and signature requirements. After a BLA is approved, the product also may be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan designation, or ODD, to a drug or biologic intended to treat a rare disease or condition, defined as a disease or condition with either a patient population of fewer than 200,000 individuals in the United States, or a patient population greater of than 200,000 individuals in the United States when there is no reasonable expectation that the cost of developing and making available the drug or biologic in the United States will be recovered from sales in the United States for that drug or biologic. ODD must be requested before submitting a BLA or NDA. After the FDA grants ODD, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.
If a product that has received ODD and subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including a full BLA or NDA, to market the same biologic or drug for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or if FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of ODD are tax credits for certain research and a waiver of the application user fee.
A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received ODD. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Rare Pediatric Disease Designation
The Rare Pediatric Disease Priority Review Voucher Program, or the PRV Program, or the PRV Program, is intended to incentivize pharmaceutical companies to develop drugs for rare pediatric diseases. A company that obtains approval of an NDA or a BLA for a designated rare pediatric disease may be eligible for a PRV from the FDA, which may be redeemed to obtain priority review for a subsequent NDA or BLA by the owner of such PRV. A PRV is fully transferable and can be sold to any company, who in turn can redeem the PRV for priority review of a marketing application in six months, compared to the standard timeframe of approximately ten months. The Congress has approved a four-year extension of the PRV Program on December 21, 2020. A drug that receives a Rare Pediatric Disease Designation before September 30, 2024 continues to be eligible for a PRV if the drug is approved before September 30, 2026. Extension beyond these dates will require further Congressional action.
68
Other Healthcare Laws and Compliance Requirements
Pharmaceutical companies are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which they conduct their business. Such laws include, without limitation: the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, to induce, or in return for, either the referral of an individual, or the purchase or recommendation of an item or service for which payment may be made under any federal healthcare program; federal civil and criminal false claims laws, including the civil False Claims Act, and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment to the federal government, including federal healthcare programs, that are false or fraudulent; the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal statutes which prohibit, among other things, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters, and which, as amended by the Health Information for Economic and Clinical Health Act of 2009, or HITECH, also imposes certain requirements on HIPAA covered entities and their business associates, and their covered subcontractors relating to the privacy, security and transmission of individually identifiable health information; the U.S. federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to annually report to the federal government, information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members, and, beginning in 2022, will require applicable manufacturers to report information regarding payments and transfers of value provided during the previous year to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, anesthesiologist assistants, and certified nurse-midwives; and U.S. state and foreign law equivalents of each of the above federal laws, which, in some cases, differ from each other in significant ways, and may not have the same effect, thus complicating compliance efforts. If their operations are found to be in violation of any of such laws or any other governmental regulations that apply, they may be subject to significant penalties, including, without limitation, civil, criminal and administrative penalties, damages, fines, exclusion from government-funded healthcare programs, such as Medicare and Medicaid or similar programs in other countries or jurisdictions, integrity oversight and reporting obligations to resolve allegations of non-compliance, disgorgement, imprisonment, contractual damages, reputational harm, diminished profits and the curtailment or restructuring of our operations.
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical or biological product for which we obtain regulatory approval. Sales of any product depend, in part, on the extent to which such product will be covered by third-party payors, such as federal, state, and foreign government healthcare programs, commercial insurance and managed healthcare organizations, and the level of reimbursement for such product by third-party payors. Decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. As there is no uniform policy of coverage and reimbursement for drug products among third‑party payors in the United States, coverage and reimbursement policies for drug products can differ significantly from payor to payor. There may be significant delays in obtaining coverage and reimbursement as the process of determining coverage and reimbursement is often time‑consuming and costly which will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage or adequate reimbursement will be obtained. It is difficult to predict at this time what government authorities and third‑party payors will decide with respect to coverage and reimbursement for our drug products. For products administered under the supervision of a physician, obtaining coverage and adequate reimbursement may be particularly difficult because of the higher prices often associated with such drugs. Additionally, separate reimbursement for the product itself or the treatment or procedure in which the product is used may not be available, which may impact physician utilization.
69
In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Third-party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost effectiveness of pharmaceutical or biological products, medical devices and medical services, in addition to questioning safety and efficacy. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit sales of any product. Decreases in third-party reimbursement for any product or a decision by a third-party payor not to cover a product could reduce physician usage and patient demand for the product.
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of reform proposals to change the healthcare system. There is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by federal and state legislative initiatives, including those designed to limit the pricing, coverage, and reimbursement of pharmaceutical and biopharmaceutical products, especially under government-funded health care programs, and increased governmental control of drug pricing.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or, collectively, ACA, was signed into law, which substantially changed the way healthcare is financed by both governmental and private insurers in the United States, and significantly affected the pharmaceutical industry. The ACA contains a number of other provisions of particular import to the pharmaceutical and biotechnology industries, including, but not limited to, those governing enrollment in federal healthcare programs, a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. Since its enactment, there have been executive, judicial, Congressional, and executive branch challenges to certain aspects of the ACA. For example, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminated the health insurer tax. In addition, the Tax Cuts and Jobs Act of 2017, or the Tax Act, was enacted, which, among other things, removed penalties for not complying with ACA’s individual mandate to carry health insurance. Since the enactment of the Tax Act, there have been additional amendments to certain provisions of the ACA. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the Tax Act, the remaining provisions of the ACA are invalid as well. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. The U.S. Supreme Court is currently reviewing the case, although it is unknown when a decision will be made. Further, although the U.S. Supreme Court has not yet ruled on the constitutionality of the ACA, on January 28, 2021, President Biden issued an executive order to initiate a special enrollment period from February 15, 2021 through May 15, 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructs certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. It is unclear how the Supreme Court ruling, other such litigation, and the healthcare reform measures of the Biden administration will impact the ACA.
70
Other legislative changes have been proposed and adopted since the ACA was enacted, including aggregate reductions of Medicare payments to providers of 2% per fiscal year and reduced payments to several types of Medicare providers, which will remain in effect through 2030 with the exception of a temporary suspension from May 1, 2020 through March 31, 2021 unless additional Congressional action is taken. Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the federal level, the Trump administration used several means to propose or implement drug pricing reform, including through federal budget proposals, executive orders and policy initiatives. For example, on July 24, 2020 and September 13, 2020, the Trump administration announced several executive orders related to prescription drug pricing that seek to implement several of the administration’s proposals. As a result, the FDA released a final rule on September 24, 2020, effective November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. Further, on November 20, 2020, the U.S. Department of Health & Human Services, or HHS, finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Medicare Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The implementation of the rule has been delayed by the Biden administration from January 1, 2022 to January 1, 2023 in response to ongoing litigation. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a new safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers, the implementation of which have also been delayed pending review by the Biden administration until March 22, 2021. On November 20, 2020, the Centers for Medicare & Medicaid Services, or CMS, issued an interim final rule implementing the Trump administration’s Most Favored Nation executive order, which would tie Medicare Part B payments for certain physician-administered drugs to the lowest price paid in other economically advanced countries, effective January 1, 2021. On December 28, 2020, the U.S. District Court in Northern California issued a nationwide preliminary injunction against implementation of the interim final rule. It is unclear whether the Biden administration will work to reverse these measures or pursue similar policy initiatives. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, it is possible that additional governmental action is taken to address the COVID-19 pandemic.
Product Development
We have made significant investments over the last twenty years in the development of our proprietary technologies, our small molecule and plasma-derived therapeutics platforms and the product candidates arising therefrom. These investments and in-house development strategy have allowed us to be flexible in our approaches and adaptive when needed as well as retaining control over intellectual property rights and the potential commercial upside thereon. Furthermore, it allows us to develop the necessary skill sets internally on both the development of manufacturing processes as well as product development (preclinical and clinical) in various disease indications. Notwithstanding the foregoing, we believe that it is important to have a balance between in-house product development and outsourcing same or partnering such activities. Finally, pursuing the development and commercialization phase in partnership with other companies (especially for specific indications and/or geographic regions) is also interesting for us because it provides continuous external validation of our technology and possibilities of short and long term revenues from fees collected at the initiation of the partnership as well as via milestones payments and royalty streams.
Environmental Protection
We produce a certain amount of chemical waste in our R&D and manufacturing activities that is managed in accordance with applicable environmental protection standards by companies that specialize in hazardous waste management. Our research laboratories generate hazardous waste that is also removed by companies that specialize in hazardous waste management, in accordance with strict internal procedures and applicable regulatory requirements. Compliance with such requirements is not expected to have a significant effect on our competitive position.
71
C.Organizational Structure
We are structured as a parent company with separate operating entities which all are directly or indirectly controlled by us.
The following chart indicates:
| (i) | the name of the operating subsidiaries and their acronym; |
| (ii) | jurisdiction of incorporation of our operating subsidiaries; and |
| (iii) | voting interest (expressed as a percentage) beneficially owned, controlled or directed by us in each subsidiary. |

Structured Alpha LP (“SALP”) Cayman Island Liminal BioSciences Inc. (“Liminal”) Canada Liminal R&D Biosciences Inc. (“LBRD”) Canada 100% Prometic Plasma Resources Inc. (“PPR”) Canada 100% Prometic Plasma Resources (USA) Inc. (“PPR USA”) Delaware, USA 100% Prometic Bioproduction Inc. * (“PBP”) Canada 100% NantPro Biosciences, LLC (“NantPro”) Delaware, USA 73% Telesta Therapeutics Inc. (“TST”) Canada 100% Prometic Biotherapeutics Inc. * (“PBT”) Delaware, USA 100% Liminal BioSciences Holdings Limited (“LBH”) UK 100% Liminal Biosciences Limited * (“LBL”) UK 100% Fairhaven Pharmaceuticals Inc. (“Fairhaven”) 100% Pathogen Removal and Diagnostic Technologies Inc. (“PRDT”) 77%
* Subsidiary that meet the definition of significant subsidiaries for the financial year ended December 31, 2020.
D.Property, Plants and Equipment.
We lease our principal executive offices, operational offices, manufacturing plant(s), plasma collection centers and laboratory space, which consists of approximately 9,110 square meters, in Laval, Quebec, Canada; Sawston, United Kingdom; Winnipeg, Manitoba, Canada; Rockville, Maryland and Amherst, New York, United States. The lease for our principal executive office in Laval expires on October 31, 2025. The lease for our office in Cambridge, United Kingdom expires on November 12, 2024. The lease for our plasma collection center in Winnipeg expires on February 28, 2023, while the lease for our plasma collection center in Amherst, New York expires on December 1, 2034. The leases for our manufacturing plant and laboratory space, both located in Laval, both expire on October 31, 2025. We believe our current facilities are sufficient to meet our needs. If we need to add new facilities or expand existing facilities as we add employees, we believe that suitable additional space will be available to accommodate any such expansion of our operations.
Item 4A.Unresolved Staff Comments.
Not applicable.
72
Item 5.Operating and Financial Review and Prospects.
Overview
We are a clinical-stage biopharmaceutical company focused on the discovery and development of novel small molecule drug candidates for the treatment of patients suffering from respiratory fibrotic diseases and other fibrotic or inflammatory diseases that have high unmet medical need. We have a deep understanding of certain biological targets and pathways that have been implicated in the fibrotic process, including fatty acid receptors such as free fatty acid receptor 1, FFAR1 (also known as G-protein-coupled receptor 40, or GPR40), a related receptor (G‑protein-coupled receptor 84, or GPR84), and peroxisome proliferator-activated receptors, or PPARs.
Our lead small molecule product candidate, fezagepras (also known as PBI-4050), is being developed for the treatment of idiopathic pulmonary fibrosis, or IPF.
As part of our plasma-derived therapeutic program, we have developed our plasma-derived drug Ryplazim®, a highly purified glu-plasminogen derived from human plasma that acts as a plasminogen replacement therapy for patients deficient in plasminogen protein.
Financial Performance
Under item 5, amounts in tables are expressed in thousands of Canadian dollars, except per share amounts which are in Canadian dollars.
On July 5, 2019, we performed a 1000 to 1 share consolidation of our issued equity instruments including common shares, warrants, options and restricted stock units, or RSU. The quantities and per unit prices presented in the 20-F have been retroactively adjusted to give effect to the share consolidation.
On November 25, 2019, we completed a disposition of all our shares in Prometic Bioseparations Ltd. or PBL to Gamma Biosciences GP LLC, a subsidiary of KKR & Co. As a result of this transaction, we no longer retain any interest in PBL and its subsidiary Prometic Manufacturing Inc. or PMI and have ceased to consolidate these entities in our consolidated financial statements as of the date of the disposal. Our interest in PBL and PMI has been presented separately as “Discontinued Operations” in the current and comparative results, in accordance with the guidance under IFRS 5, Non-Current Asset Held for Sale and Discontinued Operations. Unless otherwise indicated, all financial information represents results from continuing and discontinuing operations.
Financial operations overview
Revenue
Revenues include revenues from plasma sales, royalty revenues, revenues from the rendering of research and development services and rental revenues.
Cost of sales and other production expenses
Cost of sales and other production expenses includes the cost of the inventory sold, as well as non-capitalizable overhead related to commercial inventory and inventory write-downs. Government grants credits for eligible salaries and rent at our Winnipeg plasma collection center reduce the cost of production.
Research and development expenses
Research and development or R&D expenses comprise the costs to manufacture the plasma-derived product candidates (only Ryplazim® since 2019) used in pre-clinical studies, clinical trials, and supplied to clinical trial patients and certain other patients in connection with expanded access programs, including on a named patient basis and via a compassionate use programs until Ryplazim® is commercially approved and available, if ever. It also comprises the costs for the development of our production processes for Ryplazim® in preparation of the resubmission of the BLA, which was resubmitted in September 2020, and in preparation for commercial readiness of our manufacturing site. It also includes the cost of product candidates used in our small molecule clinical trials such as fezagepras, the cost of external consultants supporting the clinical trials and pre-clinical studies, employee compensation and other operating expenses involved in research and development activities. Government grants credits for eligible R&D salaries and rent in Canada reduce the R&D expenses.
73
Administration, selling and marketing expenses
Administration, selling and marketing expenses mainly consist of salaries and benefits related to our executive, finance, human resources, business development, legal, intellectual property, and information technology support functions. Professional fees reported under administrative expenses mainly include legal fees, accounting fees, audit fees and fees for taxation advisory. It also includes operating expenses such as insurance costs, office expenses, and travel costs pertaining to administration, selling and marketing activities. Government grants credits for eligible administrative salaries and rent in Canada are also included in administration, selling and marketing expenses.
Selling and marketing expenses include costs associated with managing our commercial activities as we prepare for our first commercial launch.
Loss (gain) on foreign exchange
Gain or loss on foreign exchange includes the effects of foreign exchange variations on monetary assets and liabilities denominated in foreign currencies between the rates at which they were initially recorded at in the functional currency at the date of the transaction and when they are retranslated at the functional currency spot rate of exchange at the reporting date. All differences are included in the consolidated statement of operations.
Finance costs
Finance costs mainly includes interest expense from the long-term debt and from the lease liabilities following the adoption of IFRS 16, Leases or IFRS 16 and banking charges. Finance costs also includes financing transaction cost associated with financial instruments carried at fair value through profit or loss. Finance costs are presented net of interest income which primarily results from the interest earned on the cash and cash equivalents we hold.
Loss (gain) on extinguishments of liabilities
When the terms of our long-term debt are modified significantly, the then existing debt is considered extinguished and the carrying amount of the debt before modification is derecognized, and the fair value of the modified debt is recognized. The difference is recorded as a loss (gain) on extinguishment of liabilities. Deferred financing fees, if any, carried on the statement of financial position that pertain to the pre-modified debt are expensed immediately and are also included in the loss or gain.
Change in fair value of financial instruments measured at fair value through profit or loss
Fair value increases and decreases on financial instruments measured at fair value through profit or loss are presented here. Over the past three years, this caption includes the changes in fair values of an investment inconvertible debt and the warrant liability.
Impairment losses
Impairment losses includes impairments recorded on long-lived assets, including but not limited to capital assets, right-of-use assets and intangible assets.
Share of losses of an associate
Our pro rata share of the losses incurred by an associate are recognized in the profit and loss. An associate is an entity over which we exercise significant influence.
Income tax expense
Income tax expense includes the current tax expense that will be payable to or collectable from the taxation authorities in the various jurisdiction in which we operate. This includes the U.K. small and medium enterprise R&D tax credits we were eligible for until 2018 inclusively. Income tax expense also includes deferred income tax expense and recoveries. Deferred income tax assets are recognized to the extent that it is probable that future tax profits will allow the deferred tax assets to be recovered.
74
Discontinued operations
Following the sale of two of our subsidiaries previously included in our bioseparations segment on November 25, 2019, we have restated the prior periods to remove the impact of those operations from all lines in the financial statements (revenues, cost of sales and production cost, R&D and administration, selling and marketing being the lines most impacted) and have reclassified those results to the net income from discontinued operations line in the financial statement. The amounts showing as net income from discontinued operations do not equal the results reported in prior periods for the bioseparation segment since the ownership of one subsidiary that was part of this segment was not sold and since certain of the corporate expenses that were previously allocated to the segment were not reclassified in the results of discontinued operations if those cost remained going forward. The gain on the sale of the subsidiaries is presented distinctly.
In item 5, sections A and B, we have omitted the discussion regarding our financial condition and results of operations for the year ended December 31, 2019 compared to the year ended December 31, 2018. For the discussions of the comparisons between the 2019 and 2018 periods, readers must refer to our 20-F filed with the SEC on March 20, 2020.
75
A.Operating Results
Comparison of years ended December 31, 2020, 2019 and 2018
The consolidated statements of operations for the year ended December 31, 2020 compared to the corresponding periods in 2019 and 2018 are presented in the following tables:
| | Year ended December 31 | | Change | |
| | 2020 | | | 2019 | | | 2018 | | | 2020 vs 2019 | | | 2019 vs 2018 | |
Revenues | | $ | 3,317 | | | $ | 4,904 | | | $ | 24,633 | | | $ | (1,587 | ) | | $ | (19,729 | ) |
| | | | | | | | | | | | | | | | | | | | |
Expenses | | | | | | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | 2,033 | | | | 2,763 | | | | 25,707 | | | | (730 | ) | | | (22,944 | ) |
Research and development expenses | | | 56,826 | | | | 75,114 | | | | 84,858 | | | | (18,288 | ) | | | (9,744 | ) |
Administration, selling and marketing expenses | | | 38,552 | | | | 45,283 | | | | 29,448 | | | | (6,731 | ) | | | 15,835 | |
Loss (gain) on foreign exchange | | | (668 | ) | | | (1,451 | ) | | | 4,696 | | | | 783 | | | | (6,147 | ) |
Finance costs | | | 8,982 | | | | 14,056 | | | | 22,041 | | | | (5,074 | ) | | | (7,985 | ) |
Loss (gain) on extinguishments of liabilities | | | (79 | ) | | | 92,374 | | | | (33,626 | ) | | | (92,453 | ) | | | 126,000 | |
Change in fair value of financial instruments measured at fair value through profit or loss | | | (850 | ) | | | (1,140 | ) | | | 1,000 | | | | 290 | | | | (2,140 | ) |
Impairment losses | | | 20,859 | | | | 12,366 | | | | 149,952 | | | | 8,493 | | | | (137,586 | ) |
Share of losses of an associate | | | — | | | | — | | | | 22 | | | | — | | | | (22 | ) |
Net loss from continuing operations before taxes | | $ | (122,338 | ) | | $ | (234,461 | ) | | $ | (259,465 | ) | | $ | 112,123 | | | $ | 25,004 | |
Income tax expense (recovery) from continuing operations: | | | | | | | | | | | | | | | | | | | | |
Current | | | (136 | ) | | | (348 | ) | | | (5,822 | ) | | | 212 | | | | 5,474 | |
Deferred | | | (65 | ) | | | 111 | | | | (13,815 | ) | | | (176 | ) | | | 13,926 | |
| | | (201 | ) | | | (237 | ) | | | (19,637 | ) | | | 36 | | | | 19,400 | |
Net loss from continuing operations | | $ | (122,137 | ) | | $ | (234,224 | ) | | $ | (239,828 | ) | | $ | 112,087 | | | $ | 5,604 | |
| | | | | | | | | | | | | | | | | | | | |
Discontinued operations, net of taxes | | | | | | | | | | | | | | | | | | | | |
Gain on sale of subsidiaries | | | 3,380 | | | | 26,346 | | | | — | | | | (22,966 | ) | | | 26,346 | |
Net income from discontinued operations | | | — | | | | 1,125 | | | | 1,932 | | | | (1,125 | ) | | | (807 | ) |
Net loss | | $ | (118,757 | ) | | $ | (206,753 | ) | | $ | (237,896 | ) | | $ | 87,996 | | | $ | 31,143 | |
| | | | | | | | | | | | | | | | | | | | |
Net income (loss) attributable to: | | | | | | | | | | | | | | | | | | | | |
Non-controlling interests - continuing operations | | | (832 | ) | | | (1,044 | ) | | | (42,530 | ) | | | 212 | | | | 41,486 | |
Owners of the parent | | | | | | | | | | | | | | | | | | | | |
- Continuing operations | | | (121,305 | ) | | | (233,180 | ) | | | (197,298 | ) | | | 111,875 | | | | (35,882 | ) |
- Discontinued operations | | | 3,380 | | | | 27,471 | | | | 1,932 | | | | (24,091 | ) | | | 25,539 | |
| | | (117,925 | ) | | | (205,709 | ) | | | (195,366 | ) | | | 87,784 | | | | (10,343 | ) |
Net loss | | $ | (118,757 | ) | | $ | (206,753 | ) | | $ | (237,896 | ) | | $ | 87,996 | | | $ | 31,143 | |
| | | | | | | | | | | | | | | | | | | | |
Income (loss) per share | | | | | | | | | | | | | | | | | | | | |
Attributable to the owners of the parent basic and diluted: | | | | | | | | | | | | | | | | | | | | |
From continuing operations | | $ | (4.96 | ) | | $ | (14.52 | ) | | $ | (238.28 | ) | | $ | 9.55 | | | $ | 223.76 | |
From discontinued operations | | | 0.14 | | | | 1.71 | | | | 2.33 | | | | (1.57 | ) | | | (0.62 | ) |
Total loss per share | | $ | (4.83 | ) | | $ | (12.81 | ) | | $ | (235.95 | ) | | $ | 7.98 | | | $ | 223.14 | |
Weighted average number of outstanding shares (in thousands) | | | 24,438 | | | | 16,062 | | | | 828 | | | | 8,376 | | | | 15,234 | |
76
Revenues
The following tables provides the breakdown of total revenues from continuing operations by source of revenue for the year ended December 31, 2020 compared to the corresponding periods in 2019 and 2018:
| | Year ended December 31 | | Change | |
| | 2020 | | | 2019 | | | 2018 | | | 2020 vs 2019 | | | 2019 vs 2018 | |
Revenues from the sale of goods | | $ | 2,593 | | | $ | 4,734 | | | $ | 23,874 | | | $ | (2,141 | ) | | $ | (19,140 | ) |
Other revenues | | | 724 | | | | 170 | | | | 759 | | | | 554 | | | | (589 | ) |
| | $ | 3,317 | | | $ | 4,904 | | | $ | 24,633 | | | $ | (1,587 | ) | | $ | (19,729 | ) |
Revenues in 2020, 2019 and 2018 were mainly driven by sales of plasma.
The decrease of $2.1 million in revenues from the sale of goods during the year ended December 31, 2020 compared to the corresponding period in 2019 is mainly due to a $2.0 million reduction in sales of specialty plasma and of $0.4 million of normal source plasma in 2020 as a result of a reduction in the collection of plasma partially caused by the COVID measures implemented at our collection centers and by the timing of these sales which can vary from period to period. These reductions were offset by the sales of $0.2 million of COVID plasma in the year ended December 31, 2020. Other revenues, which are mainly comprised of royalty and rental revenues, increased by $0.6 million for year ended December 31, 2020 compared to the corresponding period in 2019 mainly due to the royalty revenues earned on third party product sales, products that were previously sold by our former bioseparations segment.
Cost of sales and other production expenses
Cost of sales and other production expenses during the year ended December 31, 2020 decreased by $0.7 million compared to the corresponding period in 2019 mainly due to the decrease in sales of specialty and normal source plasma. This was partially offset by an increase in operating costs from our plasma collection center in Winnipeg that are allocated to other production expenses instead of R&D costs in 2019 as we were doing more development work in the plasma-derived therapeutic segment in prior years.
Research and development expenses
The R&D expenses for the year ended December 31, 2020 compared to the same periods in 2019 and 2018, broken down into its two main components, are presented in the following tables:
| | Year ended December 31 | | Change | |
| | 2020 | | | 2019 | | | 2018 | | | 2020 vs 2019 | | | 2019 vs 2018 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | $ | 27,533 | | | $ | 37,044 | | | $ | 38,667 | | | $ | (9,511 | ) | | $ | (1,623 | ) |
Other research and development expenses | | | 29,293 | | | | 38,070 | | | | 46,191 | | | | (8,777 | ) | | | (8,121 | ) |
Total research and development expenses | | $ | 56,826 | | | $ | 75,114 | | | $ | 84,858 | | | $ | (18,288 | ) | | $ | (9,744 | ) |
Since we have not entered the commercial production stage for Ryplazim® or for our small molecules, the cost of manufacturing or purchasing these product candidates was classified as R&D expenses.
Our R&D manufacturing costs for the plasma-derived therapeutics segment comprises those of our Laval plant where Ryplazim® is produced and the costs of the contract development and manufacturing organization, or CDMO, we have an agreement with to increase our production capacity for Ryplazim®, while the small molecule product candidates are manufactured by third parties.
77
The manufacturing and purchase cost of these product candidates for the year ended December 31, 2020 decreased by $9.5 million compared to the corresponding period in 2019. This change was mainly driven by a reduction of materials expensed for R&D purposes of $4.4 million as a result of managements’ outlook of the usage of the inventory to supply clinical trial patients as the expected timeline for the BLA resubmission was further out in 2019. This included a particularly high expensing of inventory on hand during 2019 as clinical batch runs were produced. The expensing of materials in 2020 is lower due to a combination of a reduction of clinical trial patients in our clinical trial programs and the reduction in the time period we expect to supply those patients.
Additionally, during the year ended December 31, 2020 we recognized credits, which further reduced our production costs, of $4.1 million and $0.4 million representing the amount we are eligible to under the Canada Emergency Wage Subsidy, or CEWS government grant, and the Canada Emergency Rent Subsidy program, or CERS government grant, respectively, two grant programs created in 2020 by the Canadian government in response to the COVID-19 pandemic. We also recognized $1.3 million in R&D tax credit in the year ended December 31, 2020 compared to a reversal of R&D tax credit of $1.0 million in the comparative period following the resolution of R&D tax credit uncertainties regarding the eligibility of certain expenses from 2014 to 2019, upon conclusion of an audit by the taxation authorities in 2020. These decreases were partially offset by an increase of $0.5 million related to consulting fees incurred in order to prepare for the resubmission of the BLA and the FDA audit of this resubmission.
A key criterion for being eligible for the CEWS is to have experienced a reduction in revenues in accordance with specific benchmarks and options as defined under the program. The variation in revenue is generally one that is determined each month by comparing the current year with the equivalent month in 2019. As such, we may be eligible only for certain periods depending on the variation of revenue for each month. Under this program, the subsidy generally represented up to 75% of eligible employees’ insurable renumeration, subject to certain criteria and limitations. Since its creation, the program has been subject to multiple modifications and extensions, under which the program is now announced to be in existence until June 2021. Recent modifications provide for a clarification of the rules of application for the first quarter of 2021 with more updates to come for later periods. The CERS government grant also sets out eligibility conditions and subsidy rates which are generally similar to those adopted for the CEWS program, and as such our eligibility can also vary from month to month.
Other R&D expenses decreased by $8.8 million during the year ended December 31, 2020 compared to the corresponding period in 2019. This decrease was driven by a reduction of $3.8 million in share-based payments compensation expense recognized during the year ended December 31, 2020 compared to the corresponding period in 2019, due to the changes made to our long-term equity incentive plan explained further below under “share-based payments expense”. This decrease is also explained by the decrease in payroll and related expenses of $3.3 million due to a reduction in our workforce and to variations in the expense related to our short-term incentive plan or STIP, in this case a decline compared to the comparative period, and the recognition of $1.0 million for the CEWS government grant credit. The decrease was also due to a reduction of $1.2 million in operating expense as traveling and related expenses were significantly reduced due to the COVID-19 Pandemic and a decrease in depreciation and amortization expense of $1.2 million mainly caused by the lower carrying amount of our capital and intangible assets following impairments recorded during the year ended December 31, 2019 These decreases were offset by reductions in Québec R&D tax credits during the year ended December 31, 2020, which resulted in an increased R&D expense of $1.2 million, and by the increase of $1.5 million in fees for consultant services related to our small molecules therapeutics operations, as we relied more on consultants to perform some of the tasks previously performed by employees.
Administration, selling and marketing expenses
The decrease of $6.7 million in administration, selling and marketing expenses during the year ended December 31, 2020 compared to the corresponding period in 2019 was mainly attributable to a reduction of $11.4 million in share-based payments compensation expense, a decrease of $4.3 million in payroll and related expenses mainly cause by the reduction in the workforce, to a decline in the expense related to the STIP, and the recognition of $1.5 in credits pertaining to the CEWS government grant. This decrease was partially offset by an increase of $11.0 million in directors’ and officers’ insurance cost resulting from our listing on the Nasdaq Stock Market LLC, or the Nasdaq, and the recognition of a $2.2 million expense pertaining to the additional warrants issued following an amendment to the private placement agreement completed in November 2020.
78
Share-based payments expense
Share-based payments compensation expense represents the expense recorded as a result of share options and RSU issued to employees and board members. The table below, shows the share-based payments compensation expense recorded in continuing and discontinuing operations. This expense has been recorded as follows in the consolidated statements of operations:
| | Year ended December 31 | | Change | |
| | 2020 | | | 2019 | | | 2018 | | | 2020 vs 2019 | | | 2019 vs 2018 | |
Cost of sales and other production expenses | | $ | 40 | | | $ | 107 | | | $ | 299 | | | $ | (67 | ) | | $ | (192 | ) |
Research and development expenses | | | 2,946 | | | | 7,137 | | | | 2,295 | | | | (4,191 | ) | | | 4,842 | |
Administration, selling and marketing expenses | | | 3,248 | | | | 14,786 | | | | 4,128 | | | | (11,538 | ) | | | 10,658 | |
| | $ | 6,234 | | | $ | 22,030 | | | $ | 6,722 | | | $ | (15,796 | ) | | $ | 15,308 | |
The above table includes the share-based payment expense included in both continuing and discontinued operations. The share-based payment expense from discontinued operations was $489 and $322 for the years ended December 31, 2019 and 2018 respectively.
During 2019, we made significant changes to our long-term equity incentive plan to ensure alignment with performance and building shareholder value, and attraction and retention of key employees to drive our future growth. The following important changes were made:
| • | the cancellation in June 2019 and August 2019 of the outstanding share options for active employees in return for the issuance of new vested options having an exercise price reflecting the share price at the time of the grant subject to stockholder approval; |
| • | the modification of the outstanding performance-based RSU into time-vesting RSU; and |
| • | the issuance of the 2019 annual stock option grant to employees and executives. The vesting terms have been changed from those set in the recent years, especially at the executive level; a portion of the executive grants vested immediately while the overall vesting period was extended up to a period of 6 years. |
Some of these changes triggered an immediate or accelerated recognition of share-based compensation expense resulting in an impact of approximately $ 14.9 million on the results during the quarter ended June 30, 2019. Further details of these changes and their accounting impact are provided in Note 18b to the consolidated financial statements for the year ended December 31, 2020.
Finance costs
The finance costs decreased by $5.1 million during the year ended December 31, 2020 compared to the corresponding period in 2019 reflecting principally the reduction in interest expense on the long-term debt mainly to a lower average debt level than the previous year despite the long-term debt balance at December 31, 2020 of $40.5 million being higher by $31.7 million from the prior year-end. The average debt balance was higher in 2019 reflecting the important debt level we had up until the debt restructuring that took place on April 23, 2019.
The adoption of the new lease standard IFRS 16 at the beginning of 2019, under which lease liabilities are recognized in the consolidated statement of financial position for the discounted value of the future lease payments at initial adoption and with interest expense recognized over the term of each lease, contributes to the increase of finance costs from 2019 and onwards compared to previous years. The new standard was adopted using the modified retrospective approach and as such, the 2018 figures are not restated. Previously, the embedded interest component in each lease payment was recognized as part of the lease expense included in the various functions presented in the statement of operations such as cost of sales and other production expenses, R&D, and administration, selling and marketing. The interest expense on the lease liabilities from continuing operations for the years ended December 31, 2019 was $6.4 million and partially offset by the decline in interest expense from long-term debt and the amortization of the credit facility fees. During the year ended December 31, 2020, the interest expense on the lease liabilities from continuing operations was $6.0 million.
79
Loss (gain) on extinguishments of liabilities
The gain on extinguishment of liabilities was $0.1 million for the year ended December 31, 2020 compared to a loss on extinguishment of liabilities of $92.4 million in 2019. The loss on extinguishment of liabilities in the year ended December 31, 2019 is principally due to us concluding a debt restructuring agreement on April 23, 2019 with our major creditor, SALP where the debt, subsequently referred to as the first term loan was reduced to $10.0 million plus interest due, in exchange for the issuance by us of 15,050,312 of common share to SALP. The details of the computation of the gain and loss on extinguishments of liabilities are presented in note 16 of the consolidated financial statements for the year ended December 31, 2020.
Change in fair value of financial instruments measured at fair value through profit or loss
On November 3, 2020, as part of the consideration for the private placement, we issued 6,315,788 warrants that expire on November 3, 2025 with an exercise price initially set at US$5.50. On November 25, 2020, we issued an additional 1,578,946 warrants with the same terms and conditions. These warrants do not meet the definition of an equity instrument and are treated as a warrant liability which is measured at fair value through profit and loss on a recurring basis. The change in fair value of the warrant liability from the various issuance dates to December 31, 2020 recognized in the consolidated statement of operations during the year ended December 31, 2020 was a gain of $0.9 million.
In November 2018, as part of the modification of the terms of our four loan agreements discussed above, we issued Warrant #9 to SALP. These warrants did not meet the definition of an equity instrument and were treated as a warrants liability which was measured at fair value through profit and loss on a recurring basis. The change in fair value of this different warrant liability, recognized in the consolidated statements of operations during the year ended December 31, 2019 was a gain of $1.1 million.
Impairment losses
2020
During the year ended December 31, 2020, we recorded impairments on our assets totalling $20.9 million mainly due to the impairments explained below.
At the end of 2020, in reviewing our portfolio of compounds in the small molecule therapeutics segment, we identified impairment indicators for certain patents. One of the patent families concerned a molecule that had entered a phase 1 clinical trial in 2019 that was subsequently discontinued after the review of the pharmacokinetic data for the first three cohorts obtained. Additional pre-clinical studies conducted in 2020 to further our understanding of the mechanism of action, or MOA, led to findings that the MOA included engaging a receptor which has been known in other products which engage the same receptor to occasionally cause undesirable side effects. Subsequently, we decided that the preclinical and clinical development activities associated with demonstrating that such molecule did not induce such side effects would be both time-consuming and costly and therefore the future development has been suspended. Another patent family impaired concerned another molecule that is licensed for development with a third party, whose research and development work we believe to be delayed from the agreed upon timelines and is unlikely to perform significant development in the near future. Further, the development of another compound was deprioritized as we wish to prioritize development of our lead compound fezagepras, as well as GPR84 and OXER1 drug candidates, which led to the impairment of the related patents. These small molecules patents were written down to their net recoverable amount of $nil, as both the fair value less costs of disposal, or FVLCD and the value in use were determined to be insignificant, resulting in an impairment of $1.2 million for the year ended December 31, 2020.
80
Subsequent to December 31, 2020, we announced that we have undertaken an evaluation of potential alternatives aimed at minimizing the plasma-derived therapeutics segment cash burn which may result in divestment in whole or part of this business, or other courses of action including but not limited to the closure of the Ryplazim® related operations, in order to focus our resources on the small molecules segment.
As the capital, intangible and ROU assets in the Ryplazim® CGU were no longer to be used as originally planned, we proceeded to review them for impairment and writing them down to their net recoverable value determined using the FVLCD based on a market approach. The Ryplazim® CGU includes the assets involved in production, R&D and commercialization activities relating to the Ryplazim® product candidate that has yet to receive regulatory approval for commercialization. The Ryplazim® CGU evaluation excluded the assets pertaining to the plasma collection activities since these can generate distinct cash inflows and could potentially be divested separately from the Ryplazim® assets. The plasma collection assets were not considered impaired.
The FVLCD was calculated using a discounted cash flow model for one year and a terminal value of $58.1 million using a post-tax discount rate of 7.75% and is considered a level 3 computation in the fair value hierarchy under IFRS 13, Fair value measurement. As part of this valuation exercise, we needed to make several key assumptions which affected the cash inflows and outflows considered in the model. The significant estimates used in determining the FVLCD are disclosed further under “Critical Accounting Policies and Estimates”.
As a result of this exercise, the Company recorded an impairment of $666 on capital assets, $18,553 on ROU assets and $480 on intangible assets, representing an aggregate impairment of $19,698 on the plasma-derived therapeutic assets for the year ended December 31, 2020.
2019
During the year 2019, we evaluated our intellectual property and the related market opportunities in the context of our financial situation and made further decisions about the areas we would or would not pursue.
One of these decisions affecting our plasma-derived therapeutics segment was to no longer pursue other indications relating to the human-plasma protein plasminogen. We ceased all R&D activities in the plasma-derived therapeutics segment not relating to Ryplazim®. As a result, our long-term production forecasts for plasminogen were reduced and one of our planned manufacturing facilities and a technical transfer facility were determined to be no longer required. We also decided to close our R&D facility in Rockville, MD by the end of 2020. Consequently, the capital and intangible assets in the plasma-derived therapeutics segment that were no longer to be used as originally planned were reviewed for impairment and written-down to their net recoverable value determined as the FVLCD using a market approach. We assessed the resale value of the property, plant and equipment, the licenses and patents in their present condition, less cost of disposal and consequently, recorded an impairment of $7.1 million and $4.5 million on capital assets and intangible assets, respectively, for the year ended December 31, 2019.
In reviewing our portfolio of compounds in the small molecule therapeutics segment, we identified compounds that where not within the areas of fibrosis in which we intend to focus and evaluated the net recoverable value of those related patents as nil, determined as the FVLCD using a market approach. An impairment on intangible assets of $0.6 million was recognized for the year ended December 31, 2019.
As a result of the sale of two of our subsidiaries previously included in our bioseparations segment, some intellectual property including patents that we retained are no longer expected to be developed. We evaluated the net recoverable value of those patents as nil, using a fair value less cost of disposal using a market approach. An impairment on intangible assets of $0.1 million was recognized for the year ended December 31, 2019.
81
Net loss from continuing operations
The net loss from continuing operations decreased by $112.1 million during the year ended December 31, 2020 compared to the corresponding period in 2019. This decrease is mainly explained by the following:
| • | the decrease in the loss on extinguishment of liabilities of $92.5 million, related to the debt restructuring that occurred during the second quarter of 2019, |
| • | the decrease in the share-based payments expense of $15.8 million related to the significant changes made to the Company’s long-term equity incentive plan in June 2019, |
| • | a decrease in R&D expenditures excluding share-based payment expenses of approximately $14.1million, |
| • | the decrease in finance cost of $5.1 million for the year ended December 31, 2020 reflecting the lower average levels of debt since the April 23, 2019 debt restructuring; and |
| • | the increase in impairment losses of $8.5 million due to the 2020 impairment on plasma-derived assets discussed above. |
Net income from discontinued operations
Following the sale of our interests in PBL and PMI in November 2019, the results of the two subsidiaries are presented separately as discontinued operations in our 2019 and 2018 results. The net income from discontinued operations for the year ended December 31, 2019 and 2018 represented a net income of $1.1 million and $1.9 million, respectively. The sale of the subsidiaries generated a gain of $26.3 million in the year ended December 31, 2019, the year of the sale, and a gain of $3.4 million in the year ended December 31, 2020 as an additional amount of proceeds was received upon resolution of a taxation matter. Details on the gains recorded are broken down into the main components in the following table:
Year ended December 31 | | | | 2020 | | | 2019 | |
Fair value of the consideration received and receivable: | | $ | 3,380 | | | $ | 51,927 | |
Less: | | | | | | | |
Carrying amount of net assets sold | | — | | | | (22,015 | ) |
Transaction costs | | — | | | | (5,015 | ) |
Add: Reclassification of foreign currency translation reserve from other comprehensive income into the statement of operations | | — | | | | 1,449 | |
Gain on sale of subsidiaries (income tax $nil) | | $ | 3,380 | | | $ | 26,346 | |
82
Comparison of quarters ended December 31, 2020, 2019 and 2018
The consolidated statements of operations for the quarter ended December 31, 2020 compared to the same periods in 2019 and 2018 are presented in the following tables:
| | Quarter ended December 31 | | Change | |
| | 2020 | | | 2019 | | | 2018 | | | 2020 vs 2019 | | | 2019 vs 2018 | |
Revenues | | $ | 1,035 | | | $ | 1,050 | | | $ | 3,379 | | | $ | (15 | ) | | $ | (2,329 | ) |
| | | | | | | | | | | | | | | | | | | | |
Expenses | | | | | | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | 445 | | | | 528 | | | | 3,359 | | | | (83 | ) | | | (2,831 | ) |
Research and development expenses | | | 11,636 | | | | 17,253 | | | | 19,191 | | | | (5,617 | ) | | | (1,938 | ) |
Administration, selling and marketing expenses | | | 9,070 | | | | 10,278 | | | | 10,165 | | | | (1,208 | ) | | | 113 | |
Bad debt expense | | | — | | | | — | | | | — | | | | — | | | | — | |
Loss (gain) on foreign exchange | | | 42 | | | | (205 | ) | | | 3,819 | | | | 247 | | | | (4,024 | ) |
Finance costs | | | 3,264 | | | | 1,858 | | | | 6,554 | | | | 1,406 | | | | (4,696 | ) |
Gain on extinguishments of liabilities | | | — | | | | — | | | | (34,904 | ) | | | — | | | | 34,904 | |
Change in fair value of financial instruments measured at fair value through profit or loss | | | (850 | ) | | | — | | | | 1,000 | | | | (850 | ) | | | (1,000 | ) |
Impairment losses | | | 20,859 | | | | 12,366 | | | | 149,952 | | | | 8,493 | | | | (137,586 | ) |
Net loss from continuing operations before taxes | | $ | (43,431 | ) | | $ | (41,028 | ) | | $ | (155,757 | ) | | $ | (2,403 | ) | | $ | 114,729 | |
Income tax expense (recovery) from continuing operations: | | | | | | | | | | | | | | | | | | | | |
Current | | | 5 | | | | (1,587 | ) | | | (1,887 | ) | | | 1,592 | | | | 300 | |
Deferred | | | (65 | ) | | | 111 | | | | (11,725 | ) | | | (176 | ) | | | 11,836 | |
| | | (60 | ) | | | (1,476 | ) | | | (13,612 | ) | | | 1,416 | | | | 12,136 | |
Net loss from continuing operations | | $ | (43,371 | ) | | $ | (39,552 | ) | | $ | (142,145 | ) | | $ | (3,819 | ) | | $ | 102,593 | |
| | | | | | | | | | | | | | | | | | | | |
Discontinued operations, net of taxes | | | | | | | | | | | | | | | | | | | | |
Gain on sale of subsidiaries | | | 3,380 | | | | 26,346 | | | | — | | | | (22,966 | ) | | | 26,346 | |
Net income(loss) from discontinued operations | | | — | | | | (1,303 | ) | | | 831 | | | | 1,303 | | | | (2,134 | ) |
Net loss | | $ | (39,991 | ) | | $ | (14,509 | ) | | $ | (141,314 | ) | | $ | (25,482 | ) | | $ | 126,805 | |
| | | | | | | | | | | | | | | | | | | | |
Net income (loss) attributable to: | | | | | | | | | | | | | | | | | | | | |
Non-controlling interests - continuing operations | | | (268 | ) | | | (155 | ) | | | (38,361 | ) | | | (113 | ) | | | 38,206 | |
Owners of the parent | | | | | | | | | | | | | | | | | | | | |
- Continuing operations | | | (43,103 | ) | | | (39,397 | ) | | | (103,784 | ) | | | (3,706 | ) | | | 64,387 | |
- Discontinued operations | | | 3,380 | | | | 25,043 | | | | 831 | | | | (21,663 | ) | | | 24,212 | |
| | | (39,723 | ) | | | (14,354 | ) | | | (102,953 | ) | | | (25,369 | ) | | | 88,599 | |
Net loss | | $ | (39,991 | ) | | $ | (14,509 | ) | | $ | (141,314 | ) | | $ | (25,482 | ) | | $ | 126,805 | |
| | | | | | | | | | | | | | | | | | | | |
Income (loss) per share | | | | | | | | | | | | | | | | | | | | |
Attributable to the owners of the parent basic and diluted: | | | | | | | | | | | | | | | | | | | | |
From continuing operations | | $ | (1.58 | ) | | $ | (1.69 | ) | | $ | (125.04 | ) | | $ | 0.11 | | | $ | 123.35 | |
From discontinued operations | | | 0.12 | | | | 1.07 | | | | 1.00 | | | | (0.95 | ) | | | 0.07 | |
Total loss per share | | $ | (1.45 | ) | | $ | (0.62 | ) | | $ | (124.04 | ) | | $ | (0.84 | ) | | $ | 123.42 | |
Weighted average number of outstanding shares (in thousands) | | | 27,333 | | | | 23,313 | | | | 830 | | | | 4,020 | | | | 22,483 | |
83
Revenues from continuing operations, cost of sales and other production expenses
The following tables provides the breakdown of total revenues from continuing operations by source for the quarter ended December 31, 2020 compared to the corresponding periods in 2019 and 2018:
| | Quarter ended December 31 | | Change | |
| | 2020 | | | 2019 | | | 2018 | | | 2020 vs 2019 | | | 2019 vs 2018 | |
Revenues from the sale of goods | | $ | 751 | | | $ | 1,016 | | | $ | 3,332 | | | $ | (265 | ) | | $ | (2,316 | ) |
Other revenues | | | 284 | | | | 34 | | | | 47 | | | | 250 | | | | (13 | ) |
| | $ | 1,035 | | | $ | 1,050 | | | $ | 3,379 | | | $ | (15 | ) | | $ | (2,329 | ) |
The decrease of $0.3 million in the revenues from the sale of goods during the quarter ended December 31, 2020 compared to the corresponding period in 2019 is mainly due to the reduction in sales of specialty plasma.
Cost of sales and other production expenses during the quarter ended December 31, 2020 decreased by $0.1 million mainly due to the reduction in the revenues from the sale of goods.
Research and development expenses
The R&D expenses for the quarter ended December 31, 2020 compared to the same periods in 2019 and 2018, broken down into its two main components, are presented in the following table:
| | Quarter ended December 31 | | Change | |
| | 2020 | | | 2019 | | | 2018 | | | 2020 vs 2019 | | | 2019 vs 2018 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | $ | 5,867 | | | $ | 6,592 | | | $ | 10,463 | | | $ | (725 | ) | | $ | (3,871 | ) |
Other research and development expenses | | | 5,769 | | | | 10,661 | | | | 8,728 | | | | (4,892 | ) | | | 1,933 | |
Total research and development expenses | | $ | 11,636 | | | $ | 17,253 | | | $ | 19,191 | | | $ | (5,617 | ) | | $ | (1,938 | ) |
The manufacturing and purchase cost of product candidates for the quarter ended December 31, 2020 decreased by $0.7 million compared to the corresponding period in 2019, mainly due to a decrease in payroll and related expenses of $1.0 million due to a reduction in our workforce and a decrease in our short‑term incentive plan or STIP expense, the recognition of a credit of $1.3 million related to the CEWS government grant as explained above and the recognition of $1.3 million in R&D tax credit in the quarter ended December 31, 2020 compared to a reversal of R&D tax credit of $1.2 million in the comparative period, following the resolution of R&D tax credit uncertainties regarding the eligibility of certain expenses from 2014 to 2019 upon conclusion of an audit by the taxation authorities in 2020. These decreases were offset by an increase of $4.5 million in materials expensed.
Other R&D expenses during the quarter ended December 31, 2020 decreased by $4.9 million compared to the corresponding period in 2019 mainly due to a decrease in payroll and related expenses of $2.8 million due to a reduction in our workforce and in our short-term incentive plan or STIP expense and to a reduction of $1.0 million in share-based payments expenses due to the impact of the resignation of our chief executive officer during the fourth quarter of 2020.
Administration, selling and marketing expenses
The decrease of $1.2 million in administration, selling and marketing expenses during the quarter ended December 31, 2020 compared to the corresponding period in 2019 was mainly attributable to a reduction of $2.7 million in share-based payment expense mainly due to the impact of the resignation of our former chief executive officer during the quarter ended December 31, 2020, a decrease of $1.5 million in payroll and related expenses mainly cause by the reduction in the workforce and to a decline in the expense related to the STIP, and the recognition of $0.5 million in credits pertaining to the CEWS government grant. This decrease was partially offset by an increase of $1.7 million in directors’ and officers’ insurance cost and the recognition of a $2.2 million expense pertaining to the additional warrants issued following an amendment to the private placement agreement completed in November 2020.
84
Share-based payments expense
Share-based payments expense represents the expense recorded as a result of stock options and RSU issued to employees and board members. This expense has been recorded as follows in the consolidated statements of operations:
| | Quarter ended December 31 | | Change | |
| | 2020 | | | 2019 | | | 2018 | | | 2020 vs 2019 | | | 2019 vs 2018 | |
Cost of sales and other production expenses | | $ | 10 | | | $ | 12 | | | $ | 128 | | | $ | (2 | ) | | $ | (116 | ) |
Research and development expenses | | | (67 | ) | | | 963 | | | | 1,008 | | | | (1,030 | ) | | | (45 | ) |
Administration, selling and marketing expenses | | | (905 | ) | | | 1,995 | | | | 2,603 | | | | (2,900 | ) | | | (608 | ) |
| | $ | (962 | ) | | $ | 2,970 | | | $ | 3,739 | | | $ | (3,932 | ) | | $ | (769 | ) |
The above table includes the share-based payment expense included in both continuing and discontinued operations.
Share-based payments expenses decreased by $3.9 million during the quarter ended December 31, 2020 compared to the corresponding period in 2019 mainly due to the accounting impact on the estimated forfeitures for the unvested stock options held by the former CEO following his resignation.
Finance costs
Finance costs increased by $1.4 million for the quarter ended December 31, 2020 compared to the corresponding period reflecting higher interest expense due to an increased debt level following the issuance of secured convertible debentures in July 2020 and of the second term loan, as we drew down our full line of credit with SALP in September 2020.
Change in fair value of financial instruments measured at fair value through profit or loss
During the quarter ended December 31, 2020, we recorded a $0.9 million loss on the increase in fair value of the warrant liability between the time of the issuance of the November 2020 warrants and December 31, 2020.
Impairment losses
During the quarter ended December 31, 2020, we recorded an impairment of $20.9 million mainly due to impairments on certain of our ROU assets related to the Ryplazim® CGU whereas in the corresponding period of 2019, we recorded impairments totalling $12.4 million as a result of us narrowing our focus in the plasma-derived therapeutics segment to the production of Ryplazim for congenital plasminogen deficiency, resulting in the impairment of certain capital and intangible assets. Further details are provided in note 24 of the consolidated financial statements for the year ended December 31, 2020.
Net loss from continuing operations
The net loss from continuing operations increased by $2.4 million during the quarter ended December 31, 2020 compared to the corresponding period in 2019. This was mainly driven by the reduction in R&D expenses of $5.6 million and the increase in impairment losses of $8.5 million.
85
Discontinued operations, net of taxes
The results from discontinued operations have been separated into two components to distinguish the gain we made upon the sale of the business from the results from its operations.
There was no net income from discontinued operations in 2020 whereas in 2019, there was the equivalent of approximately 11 months of operations of the operations that were sold in November 2019.
During the quarters ended December 31, 2020 and 2019, we realized a gain upon the sale of the bioseparations operations which was determined as follows:
Quarter ended December 31 | | | | 2020 | | | 2019 | |
Fair value of the consideration received and receivable: | | $ | 3,380 | | | $ | 51,927 | |
Less: | | | | | | | |
Carrying amount of net assets sold | | — | | | | (22,015 | ) |
Transaction costs | | — | | | | (5,015 | ) |
Add: Reclassification of foreign currency translation reserve from other comprehensive income into the statement of operations | | — | | | | 1,449 | |
Gain on sale of subsidiaries (income tax $nil) | | $ | 3,380 | | | $ | 26,346 | |
The gain recorded during the quarter ended December 31, 2019 is higher reflecting the sale of the business in that period whereas the gain recorded during the quarter ended December 31, 2020 represents additional proceeds received upon resolution of a taxation matter.
Segmented information analysis
Comparison of years ended December 31, 2020, 2019 and 2018
The loss and the net loss before income taxes from continuing operations for each segment for the years ended December 31, 2020, 2019 and 2018 are presented in the following tables.
For the year ended December 31, 2020 | | Small molecule therapeutics | | | Plasma- derived therapeutics | | | Reconciliation to statement of operations | | | Total | |
Revenues | | $ | 5 | | | $ | 2,599 | | | $ | 713 | | | $ | 3,317 | |
| | | | | | | | | | | | | | | | |
Expenses | | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | — | | | | 1,905 | | | | 128 | | | | 2,033 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | | 106 | | | | 27,427 | | | | — | | | | 27,533 | |
R&D - Other expenses | | | 13,107 | | | | 16,239 | | | | (53 | ) | | | 29,293 | |
Administration, selling and marketing expenses | | | 3,269 | | | | 6,532 | | | | 28,751 | | | | 38,552 | |
Segment loss | | $ | (16,477 | ) | | $ | (49,504 | ) | | $ | (28,113 | ) | | $ | (94,094 | ) |
Gain on foreign exchange | | | | | | | | | | | | | | | (668 | ) |
Finance costs | | | | | | | | | | | | | | | 8,982 | |
Gain on extinguishments of liabilities | | | | | | | | | | | | | | | (79 | ) |
Change in fair value of financial instruments measured at fair value through profit or loss | | | | | | | | | | | | | | | (850 | ) |
Impairment losses | | | | | | | | | | | | | | | 20,859 | |
Net loss before income taxes from continuing operations | | | | | | | | | | | | | | $ | (122,338 | ) |
Other information | | | | | | | | | | | | | | | | |
Depreciation and amortization | | $ | 1,007 | | | $ | 6,735 | | | $ | 705 | | | $ | 8,447 | |
Share-based payment expense | | | 2,153 | | | | 558 | | | | 3,523 | | | | 6,234 | |
86
For the year ended December 31, 2019 | | Small molecule therapeutics | | | Plasma- derived therapeutics | | | Reconciliation to statement of operations | | | Total | |
Revenues | | $ | 34 | | | $ | 4,736 | | | $ | 134 | | | $ | 4,904 | |
| | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | — | | | | 2,633 | | | | 130 | | | | 2,763 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | | 132 | | | | 37,107 | | | | (195 | ) | | | 37,044 | |
R&D - Other expenses | | | 15,419 | | | | 22,366 | | | | 285 | | | | 38,070 | |
Administration, selling and marketing expenses | | | 4,709 | | | | 8,368 | | | | 32,206 | | | | 45,283 | |
Segment loss | | $ | (20,226 | ) | | $ | (65,738 | ) | | $ | (32,292 | ) | | $ | (118,256 | ) |
Gain on foreign exchange | | | | | | | | | | | | | | | (1,451 | ) |
Finance costs | | | | | | | | | | | | | | | 14,056 | |
Loss on extinguishments of liabilities | | | | | | | | | | | | | | | 92,374 | |
Change in fair value of financial instruments measured at fair value through profit or loss | | | | | | | | | | | | | | | (1,140 | ) |
Impairment losses | | | | | | | | | | | | | | | 12,366 | |
Net loss before income taxes from continuing operations | | | | | | | | | | | | | | $ | (234,461 | ) |
Other information | | | | | | | | | | | | | | | | |
Depreciation and amortization | | $ | 779 | | | $ | 7,400 | | | $ | 679 | | | $ | 8,858 | |
Share-based payment expense | | | 4,782 | | | | 4,390 | | | | 12,369 | | | | 21,541 | |
For the year ended December 31, 2018 | | Small molecule therapeutics | | | Plasma- derived therapeutics | | | Reconciliation to statement of operations | | | Total | |
Revenues | | $ | — | | | $ | 24,521 | | | $ | 112 | | | $ | 24,633 | |
| | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | — | | | | 25,297 | | | | 410 | | | | 25,707 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | | 1,692 | | | | 37,107 | | | | (132 | ) | | | 38,667 | |
R&D - Other expenses | | | 14,234 | | | | 31,727 | | | | 230 | | | | 46,191 | |
Administration, selling and marketing expenses | | | 3,522 | | | | 10,393 | | | | 15,533 | | | | 29,448 | |
Segment loss | | $ | (19,448 | ) | | $ | (80,003 | ) | | $ | (15,929 | ) | | $ | (115,380 | ) |
Loss on foreign exchange | | | | | | | | | | | | | | | 4,696 | |
Finance costs | | | | | | | | | | | | | | | 22,041 | |
Gain on extinguishments of liabilities | | | | | | | | | | | | | | | (33,626 | ) |
Share of losses of an associate | | | | | | | | | | | | | | | 22 | |
Impairment losses | | | | | | | | | | | | | | | 149,952 | |
Change in fair value of financial instruments measured at fair value through profit or loss | | | | | | | | | | | | | | | 1,000 | |
Net loss before income taxes from continuing operations | | | | | | | | | | | | | | $ | (259,465 | ) |
Other information | | | | | | | | | | | | | | | | |
Depreciation and amortization | | $ | 480 | | | $ | 3,644 | | | $ | 415 | | | $ | 4,539 | |
Share-based payment expense | | | 1,270 | | | | 1,524 | | | | 3,606 | | | | 6,400 | |
As mentioned previously, the amounts for depreciation and amortization expense during 2019 and 2020 have increased for all segments since the adoption of IFRS 16 captures part of the lease cost as depreciation of right-of-use assets.
87
Small molecule therapeutics segment
The loss for the small molecule therapeutics segment was $16.5 million during the year ended December 31, 2020 compared to $20.2 million during the corresponding period in 2019, representing a decrease in segment loss of $3.7 million. This decrease is mainly due to a decrease in other R&D expenses of $2.3 million and of administration, selling and marketing of $1.4 million.
The decrease in R&D – other expenses is explained by a number of variances, including a reduction in share-based payments expense of $1.0 million, the recognition of a credit of $1.0 million related to the CEWS government grant, a reduction in clinical trials of $1.2 million and a decrease in payroll and related expenses of $4.1 million due to a reduction in our workforce and to variations in the pay-out related to our STIP. These decreases were partially offset by an increase in preclinical study expenses of $2.1 million primarily for GPR84, the increase in professional fees of $1.6 million and the reduction in R&D tax credits of $1.2 million as in 2019, we utilized previously unrecognized non-refundable Canadian Federal R&D tax credits resulting in additional R&D credits of $1.3 million being recorded.
The administration, selling and marketing expenses decrease was mainly due to a reduction of $1.6 million in share-based payments expense.
Plasma-derived therapeutic segment
The revenues for the plasma-derived therapeutics segment are generated from the sales of specialty plasma and normal source plasma to third parties, although the revenues from the latter have been minimal during the last two years. The revenues for the plasma-derived therapeutics segment were $2.6 million for the year ended December 31, 2020 compared to $4.7 million for the corresponding period in 2019, representing a decrease of $2.1 million mainly due to lower specialty plasma sales.
The manufacturing cost of plasma-derived product candidates to be used in clinical trials and for the development of our production processes of $27.4 million during the year ended December 31, 2020 decreased by $9.7 million from $37.1 million in the previous period. This change was mainly driven by a reduction of materials expensed for R&D purposes of $4.4 million. The higher expensing of materials in 2019 reflected managements’ outlook regarding the usage of the inventories to supply clinical trial patients in light of the anticipated timeline for BLA resubmission. The expensing of materials in 2020 is lower due to a combination of a reduction of patients in our compassionate use programs and the reduction in the time period we expect to supply those patients. Additionally, we recognized a credit of $4.1 million relating to the CEWS grant. We also recognized $1.3 million in R&D tax credit in the year ended December 31, 2020 compared to a reversal of R&D tax credit of $1.0 million in the comparative period following the resolution of R&D tax credit uncertainties regarding the eligibility of certain expenses from 2014 to 2019, upon conclusion of an audit by the taxation authorities in 2020. These decreases were partially offset by an increase of $0.6 million related to professional fees and operating expenses incurred to prepare for the FDA audit in connection with the resubmission of the BLA.
R&D - other expenses were $16.2 million for the year ended December 31, 2020, a decrease of $6.1 million from the $22.4 million for the year ended December 31, 2019. The decrease is results from a reduction in payroll and related expenses of $1.1 million mainly due a reduction of our workforce in our R&D facility in Rockville, MD and a reduction of $3.1 million related to share-based payments expense.
Administration, selling and marketing expenses at $6.5 million for the year ended December 31, 2020 decreased by $1.8 million from $8.4 million for the year ended December 31, 2019 as a result of a reduction in payroll and related expenses of $1.5 million due to reduction in workforce.
The loss for the plasma-derived therapeutic segment was $49.5 million for the year ended December 31, 2020 compared to $65.7 million during the corresponding period in 2019, representing a decrease of $16.2 million in segment loss. The decrease was mainly driven by the overall reduction in the manufacturing cost of Ryplazim® to be used in clinical trials and development of our production process of $9.7 million and the reduction of $6.1 million in R&D - other expenses as explained above.
88
Comparison of quarters ended December 31, 2020, 2019 and 2018
The loss for each segment and the net loss before income taxes from continuing operations for the quarters ended December 31, 2020, 2019 and 2018 are presented in the following tables:
| | Small | | | Plasma- | | | Reconciliation | | | | | |
| | molecule | | | derived | | | to statement | | | | | |
For the quarter ended December 31, 2020 | | therapeutics | | | therapeutics | | | of operations | | | Total | |
Revenues | | $ | 3 | | | $ | 756 | | | $ | 276 | | | $ | 1,035 | |
| | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | — | | | | 392 | | | | 53 | | | | 445 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | | 49 | | | | 5,818 | | | | — | | | | 5,867 | |
R&D - Other expenses | | | 3,033 | | | | 2,881 | | | | (145 | ) | | | 5,769 | |
Administration, selling and marketing expenses | | | 927 | | | | 1,029 | | | | 7,114 | | | | 9,070 | |
Segment loss | | $ | (4,006 | ) | | $ | (9,364 | ) | | $ | (6,746 | ) | | $ | (20,116 | ) |
Loss on foreign exchange | | | | | | | | | | | | | | | 42 | |
Finance costs | | | | | | | | | | | | | | | 3,264 | |
Change in fair value of financial instruments measured at FVPL | | | | | | | | | | | | | | | (850 | ) |
Impairment losses | | | | | | | | | | | | | | | 20,859 | |
Net loss before income taxes from continuing operations | | | | | | | | | | | | | | $ | (43,431 | ) |
Other information | | | | | | | | | | | | | | | | |
Depreciation and amortization | | $ | 260 | | | $ | 1,784 | | | $ | 183 | | | $ | 2,227 | |
Share-based payment expense | | | 756 | | | | 175 | | | | (1,893 | ) | | | (962 | ) |
For the quarter ended December 31, 2019 | | Small molecule therapeutics | | | Plasma- derived therapeutics | | | Reconciliation to statement of operations | | | Total | |
Revenues | | | 1 | | | | 1,015 | | | | 34 | | | | 1,050 | |
| | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | — | | | | 493 | | | | 35 | | | | 528 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | | 78 | | | | 6,511 | | | | 3 | | | | 6,592 | |
R&D - Other expenses | | | 5,062 | | | | 5,444 | | | | 155 | | | | 10,661 | |
Administration, selling and marketing expenses | | | 1,268 | | | | 2,418 | | | | 6,592 | | | | 10,278 | |
Segment loss | | $ | (6,407 | ) | | $ | (13,851 | ) | | $ | (6,751 | ) | | $ | (27,009 | ) |
Gain on foreign exchange | | | | | | | | | | | | | | | (205 | ) |
Finance costs | | | | | | | | | | | | | | | 1,858 | |
Impairment losses | | | | | | | | | | | | | | | 12,366 | |
Net loss before income taxes from continuing operations | | | | | | | | | | | | | | $ | (41,028 | ) |
Other information | | | | | | | | | | | | | | | | |
Depreciation and amortization | | $ | 206 | | | $ | 1,899 | | | $ | 217 | | | $ | 2,322 | |
Share-based payment expense | | | 525 | | | | 562 | | | | 1,658 | | | | 2,745 | |
89
For the quarter ended December 31, 2018 | | Small molecule therapeutics | | | Plasma- derived therapeutics | | | Reconciliation to statement of operations | | | Total | |
Revenues | | $ | — | | | $ | 3,352 | | | $ | 27 | | | $ | 3,379 | |
| | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | — | | | | 3,230 | | | | 129 | | | | 3,359 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | | (59 | ) | | | 10,508 | | | | 14 | | | | 10,463 | |
R&D - Other expenses | | | 2,587 | | | | 6,033 | | | | 108 | | | | 8,728 | |
Administration, selling and marketing expenses | | | 700 | | | | 2,120 | | | | 7,345 | | | | 10,165 | |
Segment loss | | $ | (3,228 | ) | | $ | (18,539 | ) | | $ | (7,569 | ) | | $ | (29,336 | ) |
Loss on foreign exchange | | | | | | | | | | | | | | | 3,819 | |
Finance costs | | | | | | | | | | | | | | | 6,554 | |
Gain on extinguishments of liabilities | | | | | | | | | | | | | | | (34,904 | ) |
Impairment losses | | | | | | | | | | | | | | | 149,952 | |
Change in fair value of financial instruments measured at fair value through profit or loss | | | | | | | | | | | | | | | 1,000 | |
Net loss before income taxes from continuing operations | | | | | | | | | | | | | | $ | (155,757 | ) |
Other information | | | | | | | | | | | | | | | | |
Depreciation and amortization | | $ | 130 | | | $ | 920 | | | $ | 160 | | | $ | 1,210 | |
Share-based payment expense | | | 691 | | | | 735 | | | | 2,182 | | | | 3,608 | |
Small molecule therapeutics segment
The segment loss for the small molecule therapeutics segment was $4.0 million during the quarter ended December 31, 2020 compared to $6.4 million during the corresponding period in 2019, representing a decrease in segment loss of $2.4 million. This decrease in loss is partially explained by a reduction $2.0 million in R&D – other expenses, mainly due to reduction in payroll and related expenses also declined by $3.1 million, partially offset by an increase in preclinical studies expenses by 0.8 million.
Plasma-derived therapeutic segment
The revenues for the plasma-derived therapeutics segment were $0.8 million for the year ended December 31, 2020 compared to $1.0 million for the corresponding period in 2019, representing a decrease of $0.3 million mainly due to lower specialty plasma sales.
The cost of sales and production expenses decreased by $0.1 million during the quarter ended December 31, 2020 compared to the corresponding period in 2019 mainly due to the reduction in the volume of sales of specialty plasma as the margin remained stable.
The manufacturing cost of plasma-derived product candidates to be used in clinical trials and for the development of our production processes of $5.8 million during the quarter ended December 31, 2020 decreased by $0.7 million from $6.5 million in the previous period. This decrease was mainly driven by the increase in R&D tax credit of $2.5 million due to the recognition of $1.3 million in R&D tax credit in the quarter ended December 31, 2020 compared to a reversal of R&D tax credit of $1.2 million in the comparative period following the resolution of R&D tax credit uncertainties regarding the eligibility of certain expenses from 2014 to 2019, upon conclusion of an audit by the taxation authorities in 2020. The decrease is also due to a decrease in payroll and related expenses of $1.0 million and the recognition of a credit of $1.3 million related to the CEWS government grant. This was partially offset by an increase of materials expensed for R&D purposes of $4.5 million.
90
Other R&D expenses at $2.9 million for the quarter ended December 31, 2020 decreased by $2.6 million from $5.4 million for the quarter ended December 31, 2019 due to a reduction of $0.3 million in share-based payments and a reduction in payroll and other related expenses of $1.1 million as a result of the reduction of headcount in our R&D facility in Rockville, MD, a reduction in the expense related to the STIP and this being partially offset by an increase in support provided by employees working in the small molecules segment and Corporate.
Administration, selling and marketing expenses at $1.0 million during the quarter ended December 31, 2020 decreased by $1.4 million from $2.4 million for the quarter ended December 31, 2019 mainly due to reduction of workforce in our R&D facility in Rockville, MD.
The loss for the plasma-derived therapeutic segment was $9.4 million for the quarter ended December 31, 2020 compared to $13.9 million during the corresponding period in 2019, representing a decrease of $4.5 million in segment loss. The decrease was mainly driven by the overall reduction in R&D – other expenses of $2.6 million and of administration, selling and marketing expenses of $1.4 million.
Selected annual information
The following table presents selected audited annual information for the years ended December 31, 2020, 2019 and 2018.
| | | | | | 2020 | | | 2019 | | | 2018 | |
Revenues | | | | | | $ | 3,317 | | | | 4,904 | | | $ | 24,633 | |
Net loss from continuing operations attributable to owners of the parent | | | | | | | (121,305 | ) | | | (233,180 | ) | | | (197,298 | ) |
Net loss from continuing operations per share attributable to owners of the parent (basic and diluted) | | | | | | | (4.96 | ) | | | (14.52 | ) | | | (238.28 | ) |
Total assets | | | | | | | 117,784 | | | | 165,098 | | | | 102,892 | |
Total long-term financial liabilities | | | | | | $ | 78,785 | | | $ | 38,721 | | | $ | 126,965 | |
Revenues from the sales of goods decreased by $1.6 million in 2020 mainly due to decrease in specialty plasma sales and decreased by $19.1 million in 2019 mainly due to the fact that the 2018 revenues include $22.9 million in sales of excess normal source plasma in 2018.
The net loss from continuing operations attributable to the owners of the parent, defined as the amount attributable to the shareholders of Liminal Biosciences, decreased significantly by $111.9 million from 2019 to 2020 due mainly to the following: 1) to the recognition of a loss on extinguishment of liabilities in the comparative 2019 period of $92.4 million and a reduction of $5.1 million of finance cost following the debt restructuring in April 2019, and 2) the decrease in share-based payments expense of approximately $14.1 million. This was partially offset by the increase in impairment losses of $8.5 million due to an impairment in plasma-derived assets in the current period.
The net loss from continuing operations attributable to the owner of the parent increased by $35.9 million from 2018 to 2019. This increase was mainly due to the following: 1) the recognition of a gain on extinguishments of liabilities of $33.6 million following the modifications to the US$ credit facility and loans we had with SALP in November 2018 compared to the loss on extinguishment of liabilities of $92.4 million following the debt restructuring, reflecting an increase in losses of $126.0 million, 2) a decline In R&D expenses by $8.7 million from the previous year 3) an increase in financing cost by $14.2 million, 4) the recording of impairment loss on the Nantpro licence in 2018 (net of the portion attributable to the non-controlling interest) of $102.9 million in 2018 compared to $12.4 million in 2019, decreasing the impairment loss by $95.0 million.
The net loss from continuing operations per share attributable to the owners of the parent on a basic and diluted basis reflects the changes in the net loss from continuing operations attributable to the owner of the parent but also the increase in the number of common shares outstanding from year to year and was significantly impacted by the number of shares issued in April 2019 upon a debt restructuring transaction and the issuance of equity following private placements. The weighted average number of shares increased from 828 thousand common shares in 2018, to 16,062 thousand common shares in 2019 then to 24,438 thousand common shares in 2020 causing the loss per share amounts to be significantly lower in 2019 and in 2020.
91
Total assets decreased by $47.3 million from $165.1 million at December 31, 2019 to $117.8 million at December 31, 2020 mainly due to a reduction in cash and cash equivalents of $16.2 million, a reduction of income tax receivable of $9.2 million as prior year claims were received and no new claims are being recorded as we are no longer eligible for U.K. R&D tax credit, and a reduction in the long-term assets following the impairment recorded on certain of the plasma-derived therapeutic assets in 2020. Total assets increased by $62.2 million from $102.9 million at December 31, 2018 to $165.1 million at December 31, 2019 mainly due to recognition of the right-of-use assets following the adoption of IFRS 16 and a higher cash and cash equivalents balance at December 31, 2019 by $53.9 million.
Long-term financial liabilities increased by $40.1 million at December 31, 2020 from December 31, 2019, mainly due our drawdown on the non-revolving line of credit of $29.1 million on September 14, 2020 and to the November 2020 warrants recognized as a warrant liability having a balance of $11.6 million at December 31, 2020. Long-term financial liabilities decreased by $88.2 million at December 31, 2019 from December 31, 2018, mainly due to the restructuring of the long-term debt on April 23, 2019, which was partially offset by the recording of the long-term portion of lease liabilities following the adoption of IFRS 16 on January 1, 2019.
Summary of consolidated quarterly results
The following table presents selected quarterly financial information for the last eight quarters:
| | 2020 | | 2019 | |
| | Q4 | | | Q3 | | | | | Q2 | | | | | Q1 | | | | | Q4 | | | | | Q3 | | | | | Q2 | | | Q1 | |
Revenues | | $ | 1,035 | | | $ | 639 | | | | | $ | 540 | | | | | $ | 1,103 | | | | | $ | 1,050 | | | | | $ | 828 | | | | | $ | 762 | | | $ | 2,264 | |
R&D expenses | | | 11,636 | | | | 12,408 | | | | | | 15,797 | | | | | | 16,985 | | | | | | 17,253 | | | | | | 18,101 | | | | | | 22,289 | | | | 17,471 | |
Administration, selling and marketing expenses | | | 9,070 | | | | 8,959 | | | | | | 9,851 | | | | | | 10,672 | | | | | | 10,278 | | | | | | 9,865 | | | | | | 18,045 | | | | 7,095 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Element attributable to the owners of the parent: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss from continuing operations | | | (43,103 | ) | | | (23,098 | ) | | | | | (27,760 | ) | | | | | (27,344 | ) | | | | | (39,397 | ) | | | | | (29,521 | ) | | | | | (135,846 | ) | | | (28,416 | ) |
Net income (loss) from discontinued operations | | | 3,380 | | | | - | | | | | | - | | | | | | - | | | | | | 25,043 | | | | | | (81 | ) | | | | | 2,229 | | | | 280 | |
Basic & diluted earning per share from continuing operations | | | (1.58 | ) | | | (0.98 | ) | | | | | (1.19 | ) | | | | | (1.17 | ) | | | | | (1.69 | ) | | | | | (1.27 | ) | | | | | (8.26 | ) | | | (33.59 | ) |
Basic & diluted earning per share from discontinuing operations | | | 0.12 | | | | - | | | | | | - | | | | | | - | | | | | | 1.07 | | | | | | - | | | | | | 0.14 | | | | 0.33 | |
Our revenues are mainly driven by the timing of our specialty plasma sales which may vary from quarter to quarter however generally represent a stable revenue stream. R&D expenses have steadily declined since the second quarter of 2019, mainly due to the reduction in manufacturing costs for Ryplazim used in R&D as well as the reduction of payroll and related expenses following workforce reductions and recognition of the CEWS government grant. Administration, selling and marketing expenses remain generally stable except for the peak in the second quarter of 2019 as various changes were made to our long-term equity incentive plans which resulted in a significant expense taken in that particular quarter.
Net loss from continuing operations variation over the last eight quarters is mainly explained by R&D expenses and administration, selling and marketing expenses as discussed above. Net loss from continuing operations during the second quarter of 2019 was significantly higher due to the loss on extinguishment of liability of $92.3 million due to the debt restructuring. The net loss from continuing operations in the fourth quarters of 2019 and 2020 were higher due to the recognition of impairment losses, mainly for our plasma-derived therapeutics segment, of $12.4 million and $20.9 million respectively.
92
Net income from discontinuing operations represents the activities from our former Bioseparations segment divested in November 2019 and includes a gain on the sale of $26.3 million. The fourth quarter of 2020 includes an additional gain of $3.4 million as we received additional proceeds following the resolution of uncertain tax positions for one of the subsidiaries sold.
The basic and diluted loss per share from continuing operations decreased significantly between the first and second quarter of 2019 mainly due to the increased number of shares outstanding following the debt restructuring in the second quarter of 2019. For the subsequent quarters, the basic and diluted loss per share from continuing operations is generally declining reflecting the decreasing trend of the net loss from continuing operations.
Acquisition of Fairhaven Pharmaceuticals Inc.
Pursuant to a share purchase agreement, or SPA, dated July 17, 2020, we acquired 100% of the issued and outstanding common shares of Fairhaven, a company with a preclinical research program of small molecule antagonists. In payment of the initial amount of $3.6 million due upon closing of the acquisition, we issued 202,308 common shares recorded at a fair value of $3.4 million based on the closing price of our common shares at the date of the transaction. Upon achievement of certain pre-determined research and development milestones prior to the fifth anniversary of the closing date of the acquisition, additional payments in the form of common shares totaling up to $4.4 million may become due.
As Fairhaven did not meet the definition of a business under IFRS 3, "Business Combinations", the acquisition has been accounted for as an asset acquisition essentially resulting in the recognition of an intangible asset representing the licensing rights acquired. Refer to note 5 to the consolidated financial statements for the year ended December 31, 2020 for the complete details regarding the accounting for this transaction.
Outstanding share data
We are authorized to issue an unlimited number of common shares. At March 22, 2021, 29,943,839 common shares, 1,870,212 options to purchase common shares, 4,192 restricted share units and 8,067,469 warrants to purchase common shares were issued and outstanding.
Transactions between related parties (as defined per IAS 24)
Balances and transactions between our subsidiaries, which are related parties, have been eliminated on consolidation and are not reported. These transactions have been recorded at the exchange amount, meaning the amount agreed to between the parties.
Following the debt modification on November 14, 2018, we assessed whether SALP, the holder of the debt, had gained significant influence for accounting purposes, despite holding less than 20% of voting rights. We deemed that qualitative factors were significant enough to conclude that the holder of the debt had gained significant influence over us and had become a related party. SALP subsequently became our majority shareholder, or our parent entity, following the debt restructuring completed on April 23, 2019.
All material transactions with SALP are disclosed in notes 15, 16, 17a, 19a, 19c and 29 in the consolidated financial statements for the year ended December 31, 2020. The key transactions with our parent entity mainly pertain to financing transactions and are for significant amounts. Related party transactions with SALP include:
| • | the issuance of loans, sometimes in combination with warrants in exchange of cash; |
| • | the recording and payment of interest on the loans with SALP with cash; |
| • | the modification of the terms of warrants held by SALP in payment for modifications to the terms of the loans; |
| • | the extinguishment of loans in exchange for the issuance of common shares (debt restructuring). |
| • | the issuance of common shares, sometimes in combination with warrants in exchange for cash; and |
| • | the reimbursement of professional fee expenses. |
93
In addition to our transactions with our parent, a former CEO had a share purchase loan outstanding in the amount of $0.4 million at December 31, 2018 with the Company. The loan bore interest at prime plus 1% and had a maturity date of the earlier of (i) March 31, 2019 or (ii) 30 days preceding a targeted Nasdaq or New York Stock Exchange listing date of our common shares. As part of the settlement agreement concluded in April 2019 with the former CEO, common shares held in escrow as security for a share purchase loan of $0.4 million to the former CEO were released and the loan extinguished in exchange for the receipt of a payment of $137,000, representing the fair value of the shares at the time of the settlement.
Changes in accounting policies
We have applied the accounting policies used in the annual consolidated financial statements in a consistent manner with those applied by us in our December 31, 2019 and 2018 audited annual consolidated financial statements except for the amendments to certain accounting standards which are relevant to us and were adopted as of January 1, 2019 as described below.
IFRS 16, Leases or IFRS 16
IFRS 16 replaces IAS 17, Leases or IAS 17. IFRS 16 provides a single lessee accounting model, requiring the recognition of assets and liabilities for all leases, unless the lease term is less than 12 months, or the underlying asset has a low value. IFRS 16 substantially carries forward the lessor accounting in IAS 17 with the distinction between operating leases and finance leases being retained.
Effective January 1, 2019, we adopted IFRS 16 using the modified retrospective approach and accordingly the information presented for 2018 has not been restated. The cumulative effect of initially applying the standard is recognized at the date of initial application. The current and long-term portions of operating and finance lease inducements and obligations presented in the statement of financial position at December 31, 2018, reflect the accounting treatment under IAS 17 and related interpretations.
We elected to use the transitional practical expedient allowing the standard to be applied only to contracts that were previously identified as leases under IAS 17 and IFRIC 4, Determining whether an arrangement contains a lease at the date of initial application. We applied the definition of a lease under IFRS 16 to contracts entered into or changed on or after January 1, 2019.
We also elected to record right-of-use assets for leases previously classified as operating leases under IAS 17 based on the corresponding lease liability, adjusted for prepaids or liabilities existing at the date of the transition that relate to the lease. When measuring lease liabilities, we discounted lease payments using its incremental borrowing rate at January 1, 2019. The weighted average discount rate applied to the total lease liabilities recognized on transition was 18.54%. For leases that were previously classified as finance leases under IAS 17, the carrying amount of the right-of-use asset and the lease liability at the date of adoption was established as the carrying amount of the lease asset classified in capital assets and the finance lease obligation at December 31, 2018. These assets and liabilities are grouped under right-of-use assets and lease liabilities as of January 1, 2019 and IFRS 16 applies to these leases as of that date.
In addition, we elected to apply the practical expedient to account for leases for which the lease term ends within 12 months of the date of initial application as short-term leases for which it is not required to recognize a right-of-use asset and a corresponding lease liability. We also elected to not apply IFRS 16 when the underlying asset in a lease is of low value.
We have elected, for the class of assets related to the lease of building space, not to separate non-lease components from lease components, and instead account for each lease component and any associated non-lease components as a single lease component.
94
The table below shows which line items of the consolidated financial statements were affected by the adoption of IFRS 16 and the impact. There was no net impact on the deficit.
| | | | | | | | As reported | | | Adjustments | | | Balance | |
| | | | | | | | as at | | | for the | | | as at | |
| | | | | | | | December 31, | | | transition | | | January 1, | |
| | | | | | | | 2018 | | | to IFRS 16 | | | 2019 | |
Assets | | | | | | | | | | | | | | | | | | |
Prepaids | | | | | | | | $ | 1,452 | | | $ | (84 | ) | | $ | 1,368 | |
Capital assets | | | | | | | | | 41,113 | | | | (1,043 | ) | | | 40,070 | |
Right-of-use assets | | | | | | | | | — | | | | 39,149 | | | | 39,149 | |
| | | | | | | | | | | | | | | | | | |
Liabilities | | | | | | | | | | | | | | | | | | |
Accounts payable and accrued liabilities | | | | | | | | $ | 31,855 | | | $ | (2,499 | ) | | $ | 29,356 | |
Current portion of lease liabilities | | | | | | | | | — | | | | 8,575 | | | | 8,575 | |
Long-term portion of lease liabilities | | | | | | | | | — | | | | 34,126 | | | | 34,126 | |
Long-term portion of operating and finance lease inducements and obligations | | | | | | | | | 1,850 | | | | (1,850 | ) | | | — | |
Other long-term liabilities | | | | | | | | | 5,695 | | | | (330 | ) | | | 5,365 | |
Prior to adopting IFRS 16, the total minimum operating lease commitments as at December 31, 2018 were $75.0 million. The decrease between the total of the minimum lease payments set out in Note 29 of the audited annual consolidated financial statements for the year ended December 31, 2018 and the total lease liabilities recognized on adoption of $42.7 million was principally due to the effect of discounting on the minimum lease payments. The amount also decreased slightly due to the fact that certain costs that are contractually committed under lease contracts, but which do not qualify to be accounted for as a lease liability, such as variable lease payments not tied to an index or rate, were previously included in the lease commitment table whereas they are not included in the calculation of the lease liabilities. These impacts were partially offset by the inclusion of lease payments beyond minimum commitments relating to reasonably certain renewal periods that had not yet been exercised as at December 31, 2018 which effect is to increase the liability. Right-of-use assets at transition have been measured at an amount equal to the corresponding lease liabilities, adjusted for any prepaid or accrued rent relating to that lease.
The consolidated statement of operations for the year ended December 31, 2019 and onwards were impacted by the adoption of IFRS 16 as the recording of depreciation of the right-of-use assets continues to be recorded in the same financial statement line items as it was previously while the implicit financing component of leasing agreements is now recorded under finance costs. The impact is not simply in the form of a reclassification but also in terms of measurement, which are significantly affected by the discount rates used and whether we have included renewal periods when calculating the lease liability.
The consolidated cash flow statement for the year ended December 31, 2019 and onwards were also impacted since the cash flows attributable to the lease component of the lease agreements are now shown as payments of principal and interest on lease liabilities which are now part of cash flows from financing activities.
IFRIC 23, Uncertainty over income tax treatments or IFRIC 23
IFRIC 23 clarifies how the recognition and measurement requirements of IAS 12 – Income Taxes are applied where there is uncertainty over income tax treatments. The Interpretation is effective for annual periods beginning on or after January 1, 2019 and was adopted on that date. We assessed the impact of this Interpretation and concluded that it had no impact on the amounts recorded in its consolidated statements of financial position on the date of adoption.
95
Amendments to IFRS 3, Business Combinations or IFRS 3
The amendments to IFRS 3 clarifies the definition of a business and includes an optional concentration test to determine whether an acquired set of activities and assets is a business. These amendments were adopted on January 1, 2020 and are applied prospectively to acquisitions made on or after this date.
New Standards and interpretations not yet adopted
The IFRS accounting standards, amendments, and interpretations that we reasonably expects may have a material impact on our disclosures, our financial position or our results of operations when applied at a future date are as follows:
Amendment to IFRS 16, Leases or IFRS 16 for COVID-19-Related Rent Concessions - IFRS 16 has been revised to incorporate an amendment issued by the IASB in May 2020. The amendment permits lessees not to assess whether particular COVID-19-related rent concessions are lease modifications and, instead, account for those rent concessions as if they were not lease modifications. In addition, the amendment to IFRS 16 provides specific disclosure requirements regarding COVID-19-related rent concessions. The amendment is effective for annual reporting periods beginning on or after June 1, 2020 and earlier application is permitted. Presently, we have not benefited from COVID-19 related rent concessions.
Amendments to IAS 37, Provisions, Contingent Liabilities and Contingent Assets or IAS 37 - IAS 37 has been revised to specify which costs an entity includes in determining the cost of fulfilling a contract for the purpose of assessing whether the contract is onerous. The amendments are effective for annual reporting periods beginning on or after January 1, 2022. The cumulative effect of initially applying the amendment, if any, will be recorded as an adjustment to the opening retained earnings and comparative periods will not be restated. Earlier application is permitted.
Amendment to IFRS 9 Financial Instruments or IFRS 9 - IFRS 9 has been revised to clarify the fees an entity includes when assessing whether the terms of a new or modified financial liability are substantially different from the terms of the original financial liability. The amendment is effective for annual reporting periods beginning on or after January 1, 2022 and is to be applied to financial liabilities that are modified after the date of adoption. Earlier application is permitted.
Amendments to IAS 1, Presentation of Financial Statements or IAS 1 - IAS 1 has been revised to clarify how to classify debt and other liabilities as current or non-current. The amendments help to determine whether, in the statement of financial position, debt and other liabilities with an uncertain settlement date should be classified as current (due or potentially due to be settled within one year) or non-current. The amendments also include clarifying the classification requirements for debt an entity might settle by converting it into equity. The amendments are applicable retrospectively and is effective for annual reporting periods beginning on or after January 1, 2023 with earlier application permitted.
At the present time, we do not expect the amendments to IFRS 16, IAS 37, IFRS 9 and IAS 1 will have a significant effect on its financial statements when these amendments are adopted by us. This assessment may change as we approach the various dates of adoption, as additional amendments may be issued and new transactions occur.
Critical Accounting Policies and Estimates
Our financial statements are prepared in accordance with IFRS. Some of the accounting methods and policies used in preparing our financial statements under IFRS are based on complex and subjective assessments by our management or on estimates based on past experience and assumptions deemed realistic and reasonable based on the facts and circumstances concerned. The actual value of our assets, liabilities and shareholders’ equity and of our EPS could differ from the value derived from these estimates if conditions changed and these changes had an impact on the assumptions adopted. We believe that the most significant management judgments and assumptions in the preparation of our financial statements are described below. We have also provided the critical accounting policies. See Note 2 to our consolidated financial statements for the year ended December 31, 2020 for a description of our other significant accounting policies.
96
Critical accounting policies
Financial instruments
Recognition and derecognition
Financial instruments are recognized in our consolidated statement of financial position when we become a party to the contractual obligations of the instrument. On initial recognition, financial instruments are recognized at their fair value plus, in the case of financial instruments not at fair value through profit or loss (“FVPL”), transaction costs that are directly attributable to the acquisition or issue of financial instruments. Financial assets are subsequently derecognized when payment is received in cash or other financial assets or if the debtor is discharged of its liability.
A financial liability is derecognized when the obligation under the liability is discharged or cancelled or expires. When an existing liability is replaced by another from the same creditor on substantially different terms, or the terms of the liability are substantially modified, such an exchange or modification is treated as the derecognition of the original liability and the recognition of a new liability. The difference in the respective carrying amounts is recognized in the consolidated statement of operations.
Classification
Subsequent to initial recognition, financial instruments are measured according to the category to which they are classified. Financial instruments are measured at amortized cost unless they are classified as fair value through other comprehensive income, or FVOCI, classified as FVPL or designated as FVPL, in which case they are subsequently measured at fair value.
The classification of financial asset debt instruments is driven by our business model for managing the financial assets and their contractual cash flow characteristics. Assets that are held to collect contractual cash flows where those cash flows represent solely payments of principal and interest are measured at amortized cost. Equity instruments that are held for trading (including all equity derivative instruments) are classified as FVPL. Financial liabilities are measured at amortized cost, unless they are required to be measured at FVPL (such as instruments held for trading or derivatives) or we have opted to measure them at FVPL.
Currently, we classify cash, cash equivalents, trade receivables, other receivables, restricted cash, and long-term deposits as financial assets measured at amortized cost and trade payables, wages and benefits payable, royalty payment obligations, license acquisition payment obligations, other employee benefit liabilities and long-term debt as financial liabilities measured at amortized cost.
We classify the warrant liability as a financial liability at FVPL for which the variation in fair value is recorded in consolidated statement of operations. We previously held an investment in convertible debt that it classified as financial assets at FVPL in the year ended December 31, 2018.
Impairment of financial assets
The expected credit losses associated with its debt instruments carried at amortized cost is assessed on a forward-looking basis. The impairment methodology applied depends on whether there has been a significant increase in credit risk. For trade receivables, we apply the simplified approach permitted by IFRS 9, which requires lifetime expected losses to be recognized from initial recognition of the receivables.
Cash equivalents
Cash and cash equivalents comprise deposits in banks and highly liquid investments having an original maturity of 90 days or less when issued.
97
Impairment of long-lived assets
At the end of each reporting period, we review the carrying amounts of our capital, ROU and intangible assets to determine whether there is any indication that those assets have suffered an impairment loss. If impairment indicators exist, the recoverable amount of the asset is estimated in order to determine the extent of the impairment loss, if any. For intangible assets not yet available for use, we perform an impairment test annually at November 30, until amortization commences, whether or not there are impairment indicators. When it is not possible to estimate the recoverable amount of an individual asset, we estimate the recoverable amount of the CGU which represents the smallest identifiable group of assets that generates cash inflows that are largely independent of the cash inflows from other assets, groups of assets or CGUs to which the asset belongs. Where a reasonable and consistent basis of allocation can be identified, the corporate assets are also allocated to individual CGUs, or otherwise they are allocated to the smallest group of CGUs for which a reasonable and consistent allocation basis can be identified.
The recoverable amount is the higher of the fair value less costs to sell and value in use. In assessing value in use, the estimated future cash flows are discounted to their present value using a pre-tax discount rate that reflects current market assessments of the time value of money and the risks specific to the asset for which the estimates of future cash flows have not been adjusted.
An impairment loss is recognized when the carrying amount of an asset or a CGU exceeds its recoverable amount by the amount of this excess. An impairment loss is recognized immediately in profit or loss in the period during which the loss is incurred. Where an impairment loss subsequently reverses, the carrying amount of the asset or CGU is increased to the revised estimate of its recoverable amount; on reversal of an impairment loss, the increased carrying amount does not exceed the carrying amount that would have been determined had an impairment loss not been recognized for the asset or CGU in prior periods. A reversal of an impairment loss is recognized immediately in profit or loss.
Lease liabilities
At the commencement date of a lease, we recognize a lease liability measured at the present value of lease payments to be made over the lease term. The lease payments include fixed payments (including in-substance fixed payments) less any lease incentives, variable lease payments that depend on an index or a rate, and amounts expected to be paid under residual value guarantees. The lease payments also include the exercise price of a purchase option reasonably certain to be exercised by us and payments of penalties for terminating a lease, if the lease term reflects that we will be exercising the option to terminate. The variable lease payments that do not depend on an index or a rate are recognized as expense in the period on which the event or condition that triggers the payment occurs.
In calculating the present value of lease payments, we use the incremental borrowing rate at the lease commencement date if the interest rate implicit in the lease is not readily determinable. After the commencement date, the amount of a lease liability is increased to reflect the accretion of interest and reduced for the lease payments made. In addition, the carrying amount of a lease liability is remeasured if there is a modification, a change in the lease term, a change in the in-substance fixed lease payments or a change in the assessment whether the underlying asset will be purchased.
We apply the short-term lease recognition exemption to leases of 12 months or less, as well as the lease of low-value assets recognition exemption. Lease payments on short-term leases and leases of low-value assets are recognized as expense on a straight-line basis over the lease term.
98
Revenue recognition
To determine revenue recognition for contracts with customers, we perform the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. The five-step model is only applied to contracts when it is probable that we will collect the consideration we are entitled to in exchange for the goods or services we transfer. At contract inception, we assess the goods or services promised within each contract, determine those that are performance obligations, and assess whether each promised good or service is distinct. The transaction price that is allocated to the respective performance obligation is recognized as revenue when (or as) the performance obligation is satisfied.
Sale of goods
Revenue from sale of goods is recognized when the terms of a contract with a customer have been satisfied. This occurs when:
| • | The control over the product has been transferred to the customer; and |
| • | The product is received by the customer or transfer of title to the customer occurs upon shipment. |
Following delivery, the customer bears the risks of obsolescence and loss in relation to the goods. Revenue is recognized based on the price specified in the contract, net of estimated sales discounts and returns.
Rendering of services
Revenues from contracted services are generally recognized as the performance obligations are satisfied over time, and the related expenditures are incurred pursuant to the terms of the agreement. Contract revenue is recognized on a percentage of completion basis based on key milestones contained within the contract.
Licensing fees and milestone payments
Under IFRS 15, we determine whether our promise to grant a license provides the customer with either a right to access our intellectual property, or IP or a right to use our IP. A license will provide a right to access the intellectual property if there is significant development of the intellectual property expected in the future whereas for a right to use, the intellectual property is to be used in the condition it is at the time the license is signed. Revenue from a license that provides to a customer the right to use our IP is recognized at a point in time when the transfers to the licensee is completed and the license period begins. When a license provides access to our IP over a license term, the performance obligation is satisfied over time and, therefore, revenue is recognized over the term of the license arrangement. Milestone payments are immediately recognized as licensing revenue when the condition is met, if the milestone is not a condition to future deliverables and collectability is reasonably assured. Otherwise, they are recognized over the remaining term of the agreement or the performance period.
Royalty
Royalty revenues are recognized once the sale of products to which the royalties give rise occurs.
Rental revenue
We account for the lease or sub-lease with a tenant as an operating lease when we have not transferred substantially all of the risks and benefits of ownership of our property or leased property. Revenue recognition under an operating lease commences when the tenant has a right to use the leased asset, and the total amount of contractual rent to be received from the operating lease is recognized on a straight-line basis over the term of the lease. Rental revenue also includes recoveries of operating expenses and property taxes.
99
Share-based payments
We have a stock option plan and an RSU plan. The fair value of stock options granted is determined at the grant date using the Black-Scholes option pricing model and is expensed over the vesting period of the options. Awards with graded vesting are considered to be multiple awards for fair value measurement. The fair value of RSU is determined using the market value of our shares on the grant date. The expense associated with RSU awards that vest over time are recognized over the vesting period. When the vesting of RSU is dependent on meeting performance targets as well as a service requirement, we will estimate the outcome of the performance targets to determine the expense to recognize over the vesting period and revise those estimates until the final outcome is determined. An estimate of the number of awards that are expected to be forfeited is also made at the time of grant and revised periodically if actual forfeitures differ from those estimates.
Our policy is to issue new shares upon the exercise of stock options and the release of RSU for which conditions have been met.
Significant judgments and estimates
Going concern - In assessing whether the going concern assumption is appropriate and whether there are material uncertainties that may cast significant doubt about our ability to continue as a going concern, management must estimate future cash flows for a period of at least twelve months following the end of the reporting period by considering relevant available information about the future. We have considered a wide range of factors relating to expected cash inflows such as whether we will earn licensing and milestone revenues, obtain regulatory approval for commercialization of product candidates, if ever, and potential sources of debt and equity financing available to us. We have also estimated expected cash outflows such as operating and capital expenditures and debt repayment schedules, including the ability to delay uncommitted expenditures. These cash flow estimates are subject to uncertainty.
Accounting for loan modifications – When the terms of a loan are modified, we must evaluate whether the terms of the loan are substantially different in order to determine the accounting treatment. If they are considered to be substantially different, the modification will be accounted for as a derecognition of the carrying value of the pre-modified loan and the recognition of a new loan at its fair value. Otherwise, the changes will be treated as a modification which will result in adjusting the carrying amount to the present value of the modified cash flows using the original effective interest rate of the loan instrument. In assessing whether the terms of a loan are substantially different, we perform an analysis of the changes in the cash flows under the previous agreement and the new agreement and we also consider other modifications that have no cash flow impacts. In the context of the simultaneous modification to the terms of several loans with the same lender, we use judgment to determine if the cash flow analysis should be performed on the loans in aggregate or individually. Judgment is also used to evaluate the relative importance of additional rights given to the lender such as additional Board of Director seats and the extension of the term of the security compared to the quantitative analysis.
Functional currency – The functional currency of foreign subsidiaries is reviewed on an ongoing basis to assess if changes in the underlying transactions, events and conditions have resulted in a change. During the years ended December 31, 2020 the functional currency of the Pathogen Removal and Diagnostic Technologies Inc., or PRDT, subsidiary changed from £ to USD. In 2019 and 2018 no changes were deemed necessary. This assessment is also performed for new subsidiaries. When assessing the functional currency of a foreign subsidiary, management’s judgment is applied in order to determine, amongst other things, the primary economic environment in which an entity operates, the currency in which the activities are funded and the degree of autonomy of the foreign subsidiary from the reporting entity in its operations and financially. Judgment is also applied in determining whether the inter-company loans denominated in foreign currencies form part of our net investment in the foreign subsidiary. Considering such loans as part of the net investment in the foreign subsidiary results in foreign currency translation gains or losses from the translation of these loans being recorded in other comprehensive loss instead of the consolidated statement of operations.
100
Share-based compensation - On March 23, 2020, our board of directors approved a plan to seek shareholder approval to modify the exercise price of certain stock options as disclosed in note 18b in the consolidated financial statements. In order to determine when the expense related to this modification is recognized in our consolidated statement of operations, we evaluated the timing of notification to option holders, the timing and method of determining the exercise price and the service period. We further considered whether the holders of the stock options had sufficient understanding of the terms and conditions of the potentially revised awards, the degree of certainty of the approval for the repricing and whether the service period for earning the rights to the awards had commenced. We concluded that the definition of the grant date was not met but that the service period had commenced and therefore a preliminary calculation of the incremental fair value of the repricing of the awards was performed using assumptions as of March 31, 2020. On May 26, 2020, the conditions for a grant date were met and the options exercise price was revised to $15.21 and a final calculation to determine the incremental fair value of the repriced options was performed.
COVID-19 – The negative impact of the COVID-19 pandemic on our financial statements for year ended December 31, 2020 has been limited, however we were eligible for salary and rent subsidy programs from the Government of Canada under which it submitted claims. The subsidy programs may provide further financial support while the programs are available. Consistent within the global biopharmaceutical sector, some clinical programs may have been and may be impacted by the shift of resources within hospitals to COVID-19 and related matters, resulting in potential delays to recruitment or site initiation on our clinical and preclinical programs, and potentially causing an adjustment of certain development timelines and activities. The partial disruption caused by COVID-19 may continue to impact our operations, workforce and overall business by delaying the progress of our research and development programs, regulatory submissions and reviews, regulatory inspections, production and plasma collection activities and business and corporate development activities. There is uncertainty as to the duration of the COVID-19 pandemic and related government restrictions, including travel bans, the impact on our workforce, and the availability of donors, healthy subjects and patients for the conduct of clinical trials, and the effects of the COVID-19 pandemic, including on the global economy, continue to be fluid. Estimates and assumptions about future events and their effects cannot be determined with certainty and therefore require the exercise of judgment. As of the date of issuance of these interim financial statements, we are not aware of any specific event or circumstance that would require it to update our estimates, assumptions and judgments or revise the carrying value of our assets or liabilities, and we are unable to estimate the potential impact on our future business or our financial results as of the date of this filing. These estimates may change as new events occur and additional information is obtained and are recognized in the consolidated financial statements as soon as they become known.
Fair value of financial instruments – The individual fair values attributed to the different components of a financing transaction, are determined using valuation techniques. We use judgment to select the methods used to determine certain inputs/assumptions used in the models and the models used to perform the fair value calculations in order to determine, 1) the values attributed to each component of a transaction at the time of their issuance, 2) the fair value measurements for certain instruments that require subsequent measurement at fair value on a recurring basis and 3) the fair value of financial instruments subsequently carried at amortized cost. When the determination of the fair value of a new loan is required, discounted cash flow techniques which includes inputs that are not based on observable market data and inputs that are derived from observable market data are used. When determining the appropriate discount rates to use, we seek comparable interest rates where available. If unavailable, we use those considered appropriate for the risk profile of a Company in the industry.
In determining the fair value of the warrants issued in November 2020 presented as a warrant liability in the consolidated statement of financial position at December 31, 2020, and considered to be a level 3 measurement, we made assumptions on unobservable inputs used in the valuation model that have an important impact on the resulting fair value computed.
101
Notably, we estimated the timing and the amounts of equity financings it expects to complete before the expiry of those warrants. The fair value computed could be higher if our actual equity financing needs are higher than those expected. We also estimated the future volatility of the common shares of Liminal for the contractual life of the warrants. To do so, we used the historical volatility its own shares and of comparable company in the same industry as a starting basis for this estimate and also considered whether there are factors that would indicate that the historical volatility is not indicative of the future. In addition, we applied an illiquidity discount rate on the resulting Black-Shole pricing model to reflect that the November 2020 warrants are not publicly traded instruments and therefore the ability to sell them is limited. In establishing the illiquidity discount rate, we considered the remaining life of the warrants and the volatility assumption for the underlying share. The fair value of the warrants could be higher if we had selected a higher volatility assumption and a lower illiquidity discount rate.
The fair value estimates could be significantly different because of the use of judgment and the inherent uncertainty in estimating the fair value of these instruments that are not quoted in an active market.
Leases - We determine the lease term as the non-cancellable term of the lease, together with any periods covered by an option to extend the lease if it is reasonably certain to be exercised, or any periods covered by an option to terminate the lease, if it is reasonably certain that this option will not be exercised.
We have the option, under some of our leases, to lease the assets for additional terms of up to fifteen years. Judgement is applied in evaluating whether it is reasonably certain to exercise the option to renew. That is, all relevant factors that create an economic incentive for us to exercise the renewal are considered. After the commencement date, the lease term is reassessed if there is a significant event or change in circumstances that is within our control and affects our ability to exercise (or not to exercise) the option to renew.
The renewal period is included as part of the lease term for a manufacturing plant lease since we estimated it is reasonably certain to exercise due to the importance of this asset to our operations, the limited availability on the market of a similar asset with similar rental terms and the related cost of moving the production equipment to another facility.
Uncertainty over income tax treatments - R&D tax credits for the current period and prior periods are measured at the amount we expect to recover, based on our best estimate and judgment, of the amounts it expects to receive from the tax authorities as at the reporting date, either in the form of income tax refunds or refundable grants. However, there are uncertainties as to the interpretation of the tax legislation and regulations, in particular regarding what constitutes eligible R&D activities and expenditures, as well the amount and timing of recovery of these tax credits. In order to determine whether the expenses we incur are eligible for R&D tax credits, we must use judgment and may resort to complex techniques, which makes the recovery of tax credits uncertain. As a result, there may be a significant difference between the estimated timing and amount recognized in the consolidated financial statements in respect of tax credits receivable and the actual amount of tax credits received as a result of the tax administrations' review of matters that were subject to interpretation. The amounts recognized in the consolidated financial statements are based on our best estimates and in our best possible judgment, as noted above.
Assessing the recoverable amount of long-lived assets - We evaluate the recoverable value of long-lived assets when indicators of impairment arise or as part of the annual impairment test, if they are intangible assets not yet available for use. The recoverable value is the higher of the value in use and the FVLCD.
Long-lived assets include capital assets, ROU assets and intangible assets such as patents and licenses and other rights. Some of these rights are considered not available for use until regulatory approval to commercialize the product candidate is obtained.
When calculating the net recoverable amounts, we make estimates and assumptions regarding the outcome of certain future events, future cash flows and their timing.
102
When determining the FVLCD, significant estimates we made include amongst others, the outcome of the exercise we have undertaken in evaluating the potential alternatives for the Ryplazim® CGU, including the probability of completing a sale or closing those activities; the operating cash outflows to support those operations until one of the alternative strategies is executed; the outcome of the FDA review of the BLA for our Ryplazim® product candidate and the timing of completion of this review; if we will be able to benefit from the monetization of a Priority Review Voucher, if received, and what would be the amount received upon its monetization; and whether some assets, liabilities and commitments could potentially be excluded from the activities sold and for those commitments that could be retained, the possibility of reducing those commitments and what would be their settlement amount. In addition, when calculating the FVLCD of an asset or a group of assets for which selling price information for comparable assets are not readily available, we also must make assumptions regarding the value it may recuperate from its sale.
A plus or minus 10% change in the probability weighted terminal value would impact the impairment we recorded on the Ryplazim® CGU by $ 3,638.
In determining the value in use for the IVIG CGU at December 31, 2018, we made a series of estimates at the time regarding the time period in which the we could possibly resume the activities of this CGU and for earning revenues from the IVIG product candidate, if approved.
Share-based compensation – The RSU expense recognized for RSU in which the performance conditions have not yet been met, is based on an estimation of the probability of successful achievement of a number of performance conditions, many of which depend on research, regulatory process and business development outcomes which are difficult to predict, as well as the timing of their achievement. The final expense is only determinable when the outcome is known.
To determine the fair value of stock options on a given date, we must determine the assumptions that will be used as inputs to the Black-Scholes option pricing model, including the assumption regarding the future volatility of our common shares for the expected life of the stock options. We use the historical volatility as a starting basis for the estimate and also considers whether there are factors that would indicate that the past volatility is not indicative of the future volatility. In making this assessment, we consider changes in our activities and other factors such as a significant share consolidation. As the volatility is an assumption that has a significant impact on the calculated value of a stock option, the impact of this estimate can significantly impact the share-based payment expense over the vesting period of an award.
Valuation of deferred income tax assets – To determine the extent to which deferred income tax assets can be recognized, we estimate the amount of probable future taxable profits that will be available against which deductible temporary differences and unused tax losses can be utilized. We exercise judgment to determine the extent to which realization of future taxable benefits is probable, considering the history of taxable profits, budgets and forecasts and availability of tax strategies.
103
Financial instruments
Use of financial instruments
The financial instruments that we used result from our operating and investing activities, namely in the form of accounts receivables and payables, and from our financing activities resulting usually in the issuance of long‑term debt. We do not use financial instruments for speculative purposes and have not issued or acquired derivative financial instruments for hedging purposes. The following table presents the carrying amounts of our financial instruments at December 31, 2020 and 2019.
| | | | | | 2020 | | | 2019 | |
Financial assets | | | | | | | | | | | | |
Cash and cash equivalents | | | | | | $ | 45,075 | | | $ | 61,285 | |
Trade receivables | | | | | | | 1,664 | | | | 1,677 | |
Restricted cash | | | | | | | 178 | | | | 169 | |
Long-term deposits | | | | | | | 137 | | | | 143 | |
Financial liabilities | | | | | | | | | | | | |
Trade payables | | | | | | $ | 9,153 | | | $ | 10,496 | |
Wages and benefits payable | | | | | | | 3,083 | | | | 5,593 | |
Royalty payment obligations | | | | | | | 3,355 | | | | 3,148 | |
License acquisition payment obligation | | | | | | | - | | | | 1,302 | |
Warrant liability | | | | | | | 11,640 | | | | - | |
Long-term debt | | | | | | | 40,532 | | | | 8,834 | |
Impact of financial instruments in the consolidated statements of operations
The following line items in the consolidated statement of operations for the quarter and the year ended December 31, 2020 include income, expense, gains and losses relating to financial instruments:
| • | loss on extinguishments of liabilities |
| • | change in fair value of financial instruments measured at fair value through profit or loss |
| • | foreign exchange gains and losses. |
JOBS Act Exemptions and Foreign Private Issuer Status
We qualify as an “emerging growth company” as defined in the JOBS Act. An emerging growth company may take advantage of specified reduced reporting and other burdens that are otherwise applicable generally to public companies. This includes an exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act. We may take advantage of this exemption for up to five years or such earlier time that we are no longer an emerging growth company. We will cease to be an emerging growth company if we have more than $1.07 billion in total annual gross revenue, have more than $700.0 million in market value of our common shares held by non-affiliates or issue more than $1.0 billion of non-convertible debt over a three-year period. We may choose to take advantage of some but not all of these reduced burdens.
We will not take advantage of the extended transition period provided under Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Since IFRS makes no distinction between public and private companies for purposes of compliance with new or revised accounting standards, the requirements for our compliance as a private company and as a public company are the same.
Additionally, we report under the Exchange Act as a non-U.S. company with foreign private issuer status. Even after we no longer qualify as an emerging growth company, as long as we qualify as a foreign private issuer under the Exchange Act, we will be exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including:
| • | the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; |
| • | the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; |
104
| • | the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K, upon the occurrence of specified significant events; and |
| • | Regulation FD, which regulates selective disclosures of material information by issuers. |
B.Liquidity and Capital Resources
Overview
Presently our funding requirements comprise those of our small molecules and plasma-derived therapeutics segments as well as our Corporate activities. We are currently evaluating potential alternatives aimed at minimizing the plasma-derived therapeutics segment cash burn which may result in divestment in whole or in part of this business, and other course of action including the closure of the Ryplazim® related operations in order to focus our resources on the small molecules segment. As the timeline to evaluate the different alternatives available to us and the range in cash expenditures or proceeds can be widely different depending on strategy chosen, including the offers we may receive from third parties, it is difficult to predict our cash needs over the next year.
Generally speaking, our primary uses of cash for our small molecules segment are to fund our ongoing research and development activities. We expect our expenses to increase in connection with these activities, particularly as we continue the research and development of our portfolio of compounds and continue or initiate clinical trials. As 2021 progresses and following the successful completion of our fezagepras phase 1 MAD study, our goal is to secure funding to launch a phase 2 IPF clinical trial.
As previously disclosed, we are assessing strategic alternatives for our plasma-derived therapeutics business, which could result in the divestiture in whole or in part of that business, or in the closure of that business. If we choose to continue with the plasma-derived operations until we are able to divest, funding needs may include the costs to manufacture Ryplazim® and to seek marketing approval. In addition, if we obtain marketing approval, and have not divested the business, we could incur significant commercialization expenses related to program sales, marketing, manufacturing and distribution to the extent that such sales, marketing and distribution are not the responsibility of potential collaborators as a result of licensing or partnering deals, if any. If our BLA for Ryplazim® is approved by the FDA, we may be also eligible to receive a PRV from the FDA, if we have not divested the business at the point of a potential approval. If received, we anticipate seeking to monetize any PRV received in 2021 to help offset such expenditures. Cash inflows from this business could include the proceeds from entering into a transaction with a third party, if we do so.
Furthermore, we expect to continue to incur additional costs associated with operating as a public company. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs, clinical trials or future commercialization efforts.
Identifying potential product candidates and conducting nonclinical studies and clinical trials is a time-consuming, expensive and uncertain process that takes many years to complete. Success in the generation of the necessary data or results required to obtain marketing approval and achieve product sales cannot be certain. In addition, successful commercialisation of our product candidates cannot be certain and any resulting revenue derived from product sales would not arise for many years, if at all.
Until such time that we can generate substantial product revenue, if ever, we will need to finance our cash needs through a combination of equity offerings, debt financings, collaborations, strategic alliances and licensing arrangements.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, shareholder ownership interest may be diluted, and the terms of any additional securities may include liquidation or other preferences that adversely affect the rights of shareholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
If we raise funds through additional collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates, or to grant licenses on terms that may not be favourable to us.
105
If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts, or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Liquidity position at December 31, 2020 and analysis of going concern
At December 31, 2020, we had a positive working capital position of $49.3 million which comprised $45.1 million of cash and cash equivalents. The increase in our liquidities since September 30, 2020 reflects the private placement we closed on November 3, 2020, in which a U.S. public investment fund and SALP participated equally, generating approximate net proceeds of $36.9 million (US$27.7 million) for the issuance of 5,757,894 common shares, 557,894 pre-funded warrants, and 6,315,788 warrants. The pre-funded warrants and the warrants each give the holder the right to purchase one common share at an exercise price of US$0.001 and US$5.50 per share respectively. The pre-funded warrants and the warrants were exercisable immediately upon issuance and have a term of five years.
Despite this recent funding, this financial position will not provide us sufficient cash runway to fund our operating activities and meet our contractual and financial obligations for a period of at least 12 months from December 31, 2020.
In our cash management efforts, we have been operating at a low spending level, pacing our investments on new research programs, and reducing infrastructure costs where possible, while we continue taking steps to further transition our company to focus on the development of our small molecule product candidates. Our liquidity resources are allocated in priority towards the fezagepras phase 1 MAD study and maintaining our plasma-derived therapeutics segment operations, while minimizing its expenditures as we evaluate various options and decide on which one to enact.
We are pursuing a number of financing initiatives that could potentially extend our cash runway, if completed. Potential sources of funding include the key ones identified below:
| • | We are continuing to evaluate potential strategic collaborations with upfront or continuous funding support; |
| • | We are continuing to evaluate avenues to monetize non-core assets; and |
| • | We will consider raising funds through the issuance of equity instruments. |
Until we are successful in completing one or more significant financing transactions that may change our financial condition (which may not be available on acceptable terms, if at all), our current circumstances indicate the existence of a material uncertainty that may cast significant doubt about our ability to continue as a going concern. The perception that we may not be able to continue as a going concern may also make it more difficult to operate our business due to concerns about our ability to meet our contractual obligations and may make it more difficult to obtain a reasonable value on assets we may decide to sell. Further, if we are unable to secure additional capital, we may be required to curtail our research and development initiatives and take additional measures to reduce costs in order to conserve our cash in amounts sufficient to sustain operations and meet our obligations. These measures could cause significant delays in our preclinical, clinical and regulatory efforts, which are critical to the realization of our business plan. See “Item 3.D—Risk Factors”.
The audited consolidated financial statements as of December 31, 2020 do not include any adjustments to the amounts and classification of assets and liabilities that might be necessary should we be unable to continue as a going concern. Such adjustments could be material.
Debt Facility
Prior Indebtedness
On November 14, 2018, we entered into an omnibus amendment agreement with SALP, extending the maturity dates of all four outstanding loans with SALP. As part of the consideration for the extension of the maturity dates of the indebtedness under the loan agreements, we cancelled 100,117 existing warrants and granted 128,056 warrants to SALP, bearing a term of eight years and exercisable at a per share price equal to $1,000.00.
We also entered into an amendment agreement to the Fourth Loan Agreement on February 22, 2019 securing an additional amount of up to US$15.0 million from SALP under the Fourth Loan Agreement.
106
Restructuring Transactions
On April 23, 2019, we completed transactions pursuant to a debt restructuring agreement we entered into on April 15, 2019 with SALP and certain of our subsidiaries, or the Restructuring Agreement. In accordance with the Restructuring Agreement, (i) SALP acquired 15,050,312 of our common shares, or the New Common Shares, at a price per common share of $15.21, or the Transaction Price, for a total purchase price of $228.9 million, which was satisfied by the cancellation of outstanding indebtedness owed by us, and (ii) certain Warrants to purchase our common shares held by SALP were amended, with new warrants being issued, or the New Warrants, exercisable for 168,735 common shares at a per-share exercise price equal to the Transaction Price. Under the Restructuring Agreement, all but $10.0 million of the outstanding debt we owed to SALP in the aggregate amount of $238.9 million was converted into common shares. We also entered into a consolidated loan agreement with SALP on April 23, 2019, relating to future indebtedness.
Line of Credit
On November 11, 2019, we entered into an amendment to our April 23, 2019 consolidated loan agreement with SALP to include a non-revolving $75.0 million secured line of credit, or the LOC. The LOC limit available to draw upon was automatically reduced by the amounts of net proceeds generated, upon the occurrence of the sale of the bioseparations business to KKR. On September 14, 2020, we drew down $29.1 million, which represented the entire balance available on the LOC.
Secured convertible debentures
Concurrently with the Fairhaven acquisition that closed on July 17, 2020, we issued secured convertible debentures, or SCD, to certain former Fairhaven shareholders, for an aggregate principal amount of $2.4 million and bearing an interest rate of 8% per annum, compounded quarterly. The SCD are due on the earlier of i) March 31, 2022, the maturity date, unless converted into our common shares prior to the maturity date or ii) upon a change of control event. The SCD are secured by all the assets of Fairhaven and rank in priority to the term loans issued under consolidated loan agreement with SALP. At any time prior to the maturity date, the SCD holders have the right to convert the SCD into our common shares. Liminal has the right to convert the SCD into our common shares under certain pre-determined events. The five-trading day VWAP of Liminal’s common shares immediately preceding the date of any conversion will be used to determine the number of common shares of the Company that will be issued. At any time prior to the maturity date, the holders have a collective right to purchase additional SCD issued by us for an aggregate principal amount of up to $5.7 million with substantially the same terms and conditions as set out in the original SCD. If the pre-determined events allowing us to trigger the conversion of the SCD occur prior to the maturity date, we have the right to require the holders of the SCD to purchase additional SCD for an aggregate principal amount of up to $5.7 million, which would then be converted into our common shares.
107
Cash flow analysis
The following major cash flow components are presented on a total company basis, inclusive of continuing and discontinued operations.
The summarized consolidated statements of cash flows for the year ended December 31, 2020 and the corresponding periods in 2019 and 2018 are presented below.
| | Year ended December 31 | | Change | |
| | 2020 | | | 2019 | | | 2018 | | | 2020 vs 2019 | | | 2019 vs 2018 | |
Cash flows used in operating activities | | $ | (75,917 | ) | | $ | (99,390 | ) | | $ | (82,454 | ) | | $ | 23,473 | | | $ | (16,936 | ) |
Cash flows from financing activities | | | 57,405 | | | | 117,919 | | | | 72,158 | | | | (60,514 | ) | | | 45,761 | |
Cash flows from (used in) investing activities | | | 2,305 | | | | 36,096 | | | | (5,859 | ) | | | (33,791 | ) | | | 41,955 | |
Net change in cash and cash equivalents during the year | | | (16,207 | ) | | | 54,625 | | | | (16,155 | ) | | | (70,832 | ) | | | 70,780 | |
Net effect of currency exchange rate on cash and cash equivalents | | | (3 | ) | | | (729 | ) | | | 378 | | | | 726 | | | | (1,107 | ) |
Cash and cash equivalents, beginning of the year | | | 61,285 | | | | 7,389 | | | | 23,166 | | | | 53,896 | | | | (15,777 | ) |
Cash and cash equivalents, end of the year | | $ | 45,075 | | | $ | 61,285 | | | $ | 7,389 | | | $ | (16,210 | ) | | $ | 53,896 | |
Cash flows used in operating activities decreased by $23.5 million during the year ended December 31, 2020 compared to the same period in 2019. The decrease can be explained by a reduction in R&D expenses and by the receipt of grants from the Canadian government through programs to support businesses during the COVID‑19 pandemic and a reduction in payments to suppliers compared to in the prior year when we settled payments in arrears following the receipt of funding during the quarter ended June 30, 2019. These decreases were partially offset by an increase in directors’ and officers’ insurance costs.
Cash flows from financing activities decreased by $60.5 million during the year ended December 31, 2020 compared to the same period in 2019 as gross proceeds raised from equity financing declined by $78.8 million, since in 2020 we raised gross proceeds of $39.9 million in a private placement in November 2020 compared to the gross proceeds raised in the April 2019 private placements and in the rights offering in June 2019 of $75.0 million and $39.4 million respectively. The decline was partially offset by an increase of $11.7 million from financings where long-term debt was issued, mainly due to our draw down of $29.1 million in September 2020 on the non-revolving line of credit we had with SALP, representing the entire balance available, which resulted in the issuance of the second term loan. The second term loan bears an annual interest rate of 10% compounded monthly and payable quarterly and matures on April 23, 2024.
In 2019 and 2018, some of our proceeds from the issuance of shares came from shares issued under an ATM equity distribution agreement or EDA, entered into in November 2018, under which we were able, at our discretion and from time to time, subject to conditions in the EDA, to offer common shares through ATM issuances on the TSX for aggregate proceeds not exceeding $31 million. This agreement provided that common shares were to be sold at market prices prevailing at the time of sale. For the year ended December 31, 2018, a total of 1,945 common shares were issued under the ATM, at an average price of $386.12 per share, for aggregate gross proceeds of $0.8 million and total net proceeds of $0.7 million. For the year ended December 31, 2019, a total of 12,865 common shares were issued under the ATM at an average price of $327.55 per share, for aggregate gross proceeds of $4.2 million and total net proceeds of $4.0 million. The ATM facility was suspended concurrently with the debt restructuring in April 2019 and subsequently expired in April 2020.
Cash flows from investing activities decreased by $33.8 million during the year ended December 31, 2020 compared to the same period in 2019 as the proceeds we received from the sale of our bioseparations business, net of transaction costs paid, in 2020, as adjustments to the initial purchase price and following the resolution of a tax matter was significantly lower to the initial payment received upon the closing of the sale, net of the cash divested and transaction costs by $36.0 million.
108
C.Research and Development, Patents and Licences
For a discussion of our research and development activities, see “Item 4.B—Business Overview” and “Item 5.A—Operating Results.”
D.Trend Information
Other than as disclosed elsewhere in this annual report, we are not aware of any trends, uncertainties, demands, commitments or events for the period from January 1, 2020 to December 31, 2020 that are reasonably likely to have a material adverse effect on our net revenues, income, profitability, liquidity or capital resources, or that caused the disclosed financial information to be not necessarily indicative of future operating results or financial conditions. For a discussion of trends, see “Item 4.B.—Business overview,” “Item 5.A.—Operating results,” and “Item 5.B.—Liquidity and capital resources.”
E.Off-balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet financing arrangements or any relationships with unconsolidated entities or financial partnerships, including entities sometimes referred to as structured finance or special purpose entities, that were established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
F.Tabular Disclosure of Contractual Obligations
The timing and expected contractual outflows required to settle our financial obligations recognized in the consolidated statement of financial position at December 31, 2020 and unrecognized purchase obligations and commitments are presented in the table below:
| | | | | | Contractual Cash flows | |
| | Carrying amount | | | Less than 1 year | | | 1-3 years | | | 3 - 5 years | | | More than 5 years | | | Total | |
Accounts payable and accrued liabilities 1) | | $ | 16,835 | | | $ | 16,835 | | | $ | - | | | $ | - | | | $ | - | | | $ | 16,835 | |
Long-term portion of royalty payment obligations | | | 107 | | | | - | | | | 51 | | | | 51 | | | | 229 | | | | 331 | |
Lease liabilities | | | 33,452 | | | | 7,434 | | | | 13,957 | | | | 13,328 | | | | 30,725 | | | | 65,444 | |
Long-term portion of other employee benefit liabilities | | | 206 | | | | - | | | | 206 | | | | - | | | | - | | | | 206 | |
Long-term debt 2) | | | 40,532 | | | | 3,945 | | | | 10,649 | | | | 40,353 | | | | - | | | | 54,947 | |
Purchase obligations and commitments | | n/a | | | | 11,762 | | | | 8,990 | | | | 8,441 | | | | 16,114 | | | | 45,307 | |
| | $ | 91,132 | | | $ | 39,976 | | | $ | 33,853 | | | $ | 53,732 | | | $ | 30,954 | | | $ | 183,070 | |
1) Short term portions of the royalty payment obligations and of other employee benefit liabilities are included in the account payable and accrued liabilities.
2) Under the terms of the consolidated loan agreement (see “restructuring transactions”), SALP may decide to cancel a portion of the principal value of the loans as payment upon the exercise of their 168,735 warrants #10 and 3,947,367 November 2020 warrants. The maximum repayment due on the loan has been included in the above table.
Royalties
SALP has a right to receive a 2% royalty on future revenues relating to patents of a specified small molecule product candidate and analogues, existing as of the date of the agreement was signed. The obligation under this royalty agreement is secured by all of our assets until the expiry of the last patent anticipated in 2033.
109
In the normal course of business, we enter into license agreements for the market launching or commercialization of product candidates, if approved. Under these licenses, including the ones mentioned above, we have committed to pay royalties ranging generally between 0.5% and 12.0% of net sales from products we may commercialize, if approved, and 3% of license revenues in regard to certain small molecule product candidates.
Commitments
We signed a long-term manufacturing contract with the CDMO which provides us with additional manufacturing capacity. In connection with this contract, we have committed to a minimum annual spending of $9.0 million for 2021 to 2030 (the end of the initial term) which includes all expenditures under the contract. As of December 31, 2020, the remaining payments under the CDMO contract was $83.7 million or $38.2 million after deduction of the minimum lease payments under the contract which are recognized in the consolidated financial statements as a lease liability following the adoption of IFRS 16.
At December 31, 2020, total commitment remaining under the agreement with the CDMO that is not recognized in the lease liability is included in the tabular disclosure of contractual obligations presented above under “purchase obligations and commitments”.
G.Safe Harbor
This Annual Report contains forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Exchange Act and as defined in the Private Securities Litigation Reform Act of 1995. See “Special Note Regarding Forward-Looking Statements.”
Item 6.Directors, Senior Management and Employees.
A.Directors and Senior Management.
The following table sets forth certain information with respect to our executive officers and directors, including their ages as of December 31, 2020:
Name of Executive Officers | | Age | | Position(s) |
Bruce Pritchard | | 48 | | Chief Executive Officer |
Patrick Sartore | | 46 | | President |
Moira Daniels | | 59 | | Head of Regulatory Affairs and Quality Assurance |
Marie Iskra | | 45 | | General Counsel |
Martin Leclerc | | 58 | | Chief Talent Officer |
Murielle Lortie | | 51 | | Chief Financial Officer |
Name of Non-Employee Directors | | Age | | Position(s) |
Simon Best(1)(2) | | 64 | | Lead Independent Director |
Gary J. Bridger(3) | | 57 | | Director |
Neil A. Klompas(1) | | 49 | | Director |
Alek Krstajic(2) | | 57 | | Chair |
Eugene Siklos | | 57 | | Director |
Timothy Steven Wach(1) | | 60 | | Director |
(1) Member of the audit, risk and finance committee.
(2) Member of the HR and corporate governance committee.
(3) Member of the science and technology committee.
Unless otherwise indicated, the current business addresses for our executive officers and directors is 440 Armand-Frappier Blvd., Suite 300, Laval, Québec, Canada H7V 4B4. The current business address for Bruce Pritchard and Moira Daniels is Park Ground Floor, Unit 1, Iconix Park, London Road, Sawston, Cambridge CB22 3EG.
Each executive officer serves at the discretion of our board of directors and holds office until his or her successor is duly elected or qualified or until his or her earlier resignation or removal.
110
Executive Officers
Bruce Pritchard, Chief Executive Officer. Mr. Bruce Pritchard was appointed as Chief Executive Officer, effective November 13, 2020. Prior to that he was one of two Chief Operating Officers. In this role, he was responsible for the company’s discovery, clinical development, program management, CMC and business development functions related to Small Molecule Therapeutics. Mr. Bruce Pritchard has served as our Chief Operating Officer from August 2014 to November 2020. Mr. Pritchard joined us as Chief Financial Officer of PBL in 2006 and was promoted to our Chief Financial Officer in July 2008, relinquishing that post in November 2015. Mr. Pritchard also served as Interim Chief Financial Officer from August 2017 to September 2019. A Heriot-Watt University graduate, Mr. Pritchard holds a Bachelor of Arts in Accountancy and Computer Science and is qualified as a Member of the Institute of Chartered Accountants of Scotland. Mr. Pritchard was appointed a Fellow of the Institute of Directors in 2014.
Patrick Sartore, President. Mr. Patrick Sartore was appointed as President, effective November 13, 2020. Prior to that, he was one of two Chief Operating Officers. In this role, he was responsible for the company’s commercial, marketing, clinical development, manufacturing, and corporate & business development functions, related to Plasma-Derived Therapeutics. Mr. Patrick Sartore has served as our Chief Operating Officer, North America from May 2019 to November 2020. Mr. Sartore joined us in November 2006 as Senior Legal Counsel – Intellectual Property and was nominated as our Corporate Secretary in December 2007. Mr. Sartore held the position of General Counsel and Corporate Secretary from May 2013 to May 2015, at which date he was appointed Chief Legal Officer and Corporate Secretary. Mr. Sartore was previously employed by Univalor Inc. as Legal Counsel and Leger Robic Richard, L.L.P., a firm specializing in Intellectual Property, Corporate and Commercial Law, as an associate attorney. Mr. Sartore also holds a Bachelor of Science, with Distinction, from Concordia University.
Moira Daniels, Head of Regulatory Affairs and Quality Assurance. Ms. Moira Daniels has served as our Head of Regulatory Affairs and Quality Assurance since November 2019. Ms. Daniels has held multiple senior roles within the pharmaceutical industry over the past 35 years and brings significant experience in product development and regulatory affairs, as well as change management. Prior to joining us, Ms. Daniels served as a Vice President at UCB Pharma from August 2015 to October 2019. Ms. Daniels held senior positions in Regulatory Affairs at PAREXEL from November 2013 to September 2015 and at AstraZeneca from August 2001 to October 2013. Ms. Daniels holds a B.Sc. in Biochemistry and Physiology and an M.B.A. from Henley Management College.
Marie Iskra, General Counsel. Ms. Marie Iskra has served as our General Counsel since September 2019. As General Counsel, Ms. Iskra oversees the legal, compliance, intellectual property and corporate governance functions. Ms. Iskra is also our Corporate Secretary. Ms. Iskra has served in various roles since September 2013, including as Associate General Counsel and Senior Legal Counsel. Ms. Iskra was previously employed as Director of Legal Affairs for Jubilant Draximage Inc. as and Jubilant HollisterStier General Partnership from 2009 to 2013 as Legal Counsel for Draxis Health Inc. from 2005 to 2009. Ms. Iskra holds an L.L.B. from the Université de Montreal and is a member of the Québec Bar. Ms. Iskra also holds a Certificate Arts and Sciences from the Université de Montreal.
Martin Leclerc, Chief Talent Officer. Mr. Martin Leclerc has served as our Chief Talent Officer since May 2019 and previously served as our Vice President, Human Resources from May 2017 to May 2019. Prior to joining us, Mr. Leclerc was Global Leader, Total Rewards for CAE Inc. since October 2010. After working as a consulting actuary for more than 10 years at the beginning of his career, he has since held a number of senior positions in the human resources field within large global corporations. Mr. Leclerc has a Bachelor of Science degree in Mathematics from Université de Montreal. Mr. Leclerc completed all requirements and became a Fellow of the Society of Actuaries and of the Canadian Institute of Actuaries and has worked as an actuary for several years before working in the broader human resources field.
111
Murielle Lortie, Chief Financial Officer. Ms. Murielle Lortie has served as our Chief Financial Officer since September 2019. Ms. Lortie previously served as Vice President of Finance from September 2018 to August 2019. Ms. Lortie is also Director of Profound Medical Corp. since November 2020. Prior to joining us, Ms. Lortie was employed as Vice President & Chief Financial Officer as well as Advisor to the CEO, Global Strategy, Mergers & Acquisitions at Pharmascience Inc. from March 2014 to October 2017. Ms. Lortie also consulted for Cirque du Soleil from March to September 2018. Previous to this, Ms. Lortie held senior positions in finance at Bristol Myers Squibb from 2004 to 2014, including Vice-President of Finance for Bristol Myers Squibb Canada Co. and Global Director of Finance supporting BMS Headquarters. Ms. Lortie also served as a director on the board of directors of Bellus Health from February 2015 to May 2017 and Pharmascience Barbados Ltd and Pharmascience International Ltd from 2015 to 2017. Mrs. Lortie is a Chartered Professional Accountant and member of the Ordre des comptables professionnels agréés du Québec. Mrs. Lortie holds a Graduate Diploma in Accountancy from Concordia University and a Bachelor of Business Administration from Bishop’s University.
Non-Employee Directors
Simon Best. Prof. Best has served as a member of our board of directors since May 2014. Prof. Best also served as our interim President and Chief Executive Office from December 2018 to April 2019. Prof. Best has experience as the founder and/or Chief Executive Officer of four biotechnology companies between 1992 and 2012, and as a Chairman or board member of major industry bodies including the UK BioIndustry Association (BIA) and the US Biotechnology Industry Organization (BIO). Since 2012, he has played a pivotal role in building pioneering Digital Healthcare companies – in Pathology with PathXL which was acquired by Philips Healthcare in May 2016 – subsequently with BrainWaveBank, which develops EEG tools and analytics for at-home use to validate digital biomarkers, stratify patients for clinical trials and monitor them to facilitate the development of novel drugs for neuro-degenerative disorders. He also served as a director of EvoFem Inc. from November 2015 to December 2017. He was previously an Entrepreneur-In-Residence and Venture Partner with TVM Capital in Munich and Dubai from 2007-2010 and co-founded Par Equity LLP in Edinburgh in 2007 of which he remains a Non-Executive Partner. Prof. Best holds an M.B.A. from the London Business School and an Honorary Doctorate and B.Mus from York University. In 2007, he was elected a Fellow of the Royal Society of Edinburgh. In 2008, he was awarded an OBE by Queen Elizabeth II and appointed a Visiting Professor of Medicine by the University of Edinburgh. We believe that Prof. Best’s extensive experience in the life science and investment industries qualifies him to serve as a member of our board of directors.
Gary J. Bridger. Dr. Bridger has served as a member of our board of directors since May 2019. Dr. Bridger has also served as a member of the board of directors of X4 Pharmaceuticals, Inc. since October 2018. From February 2015 to December 2017, Dr. Bridger served as a consultant to Xenon Pharmaceuticals Inc., a biopharmaceutical company, where he previously served as the Executive Vice President of Research and Development from January 2013 to February 2015. From October 2013 to October 2015, Dr. Bridger served as a Managing Director at Five Corners Capital Inc. From June 2010 to June 2012, Dr. Bridger served as a venture partner at Venture West Capital Management, a venture capital firm. From November 2006 to December 2007, Dr. Bridger served as Senior Vice President of Research and Development of Genzyme Corporation, a biotechnology company, which was acquired by Sanofi, S.A. In June 1996, Dr. Bridger co-founded AnorMED Inc., a biopharmaceutical company, and was its Vice President of Research and Development and Chief Scientific Officer from 2000 until its acquisition by Genzyme in November 2006. Dr. Bridger has served on the board of directors of Aquinox Pharmaceuticals, Inc. from 2013 until August 2019. Dr. Bridger holds a Ph.D. in Organic Chemistry from the University of Manchester Institute of Science and Technology. We believe that Dr. Bridger’s experience as a director and executive officer at numerous biopharmaceutical companies qualifies him to serve as a member of our board of directors.
Alek Krstajic. Mr. Krstajic is the former Chief Executive Officer of Wind Mobile, which was sold to Shaw Communications in 2016 for $1.6 Billion. Previously he was the founder and Chief Executive Officer of Public Mobile, which was purchased by Telus in 2013. Mr. Krstajic has also served as the President of Bell Mobility and a Senior Vice President of Rogers Communications. In the process, Mr. Krstajic and his team fundamentally changed the wireless market in Canada from a three player market with high prices to a sustainable four player market, providing Canadians with an alternative and competitive pricing and a potential path to better value. As well as being an operator, Mr. Krstajic has shown a level of expertise managing regulatory processes to positive outcomes. Mr. Krstajic sits on a number of boards, and has received numerous awards and recognitions, including Canada’s Top 40 under 40 and a Queen Elizabeth II Diamond Jubilee Medal for his service to Canada.
112
Eugene Siklos. Mr. Siklos is the President of Thomvest Asset Management. Formerly he was the Vice President, Head of Investments for Export Development Canada (“EDC”), which he joined in 2009. In 2012, Mr. Siklos was appointed head of its direct investing activities and in 2017, was appointed head of EDC’s overall investing activities. During his time at EDC, the capital committed to fund direct, private equity and venture capital investments doubled in size to over C$2 billion, while achieving both market returns and mandate objectives for its shareholder, the Government of Canada. Mr. Siklos holds an MBA degree from Harvard Business School and has over 30 years of financial and business experience, including as a corporate finance professional with Morgan Stanley and Merrill Lynch. Mr. Siklos has served on numerous Boards of Directors, including Paragon Pharmacies (TSX), Copperleaf Technologies, BTI Systems, and ecobee.
Timothy Steven Wach. Mr. Wach has served as a member of our board of directors since May 2019. Since January 2015 until the end of February 2020, Mr. Wach served as Managing Director and Board Member of Taxand. In 1989, Mr. Wach joined Gowling Lafleur Henderson LLP as an associate and was a partner from 1992 to 2014. Mr. Wach also has experience in government relations and tax policy, having twice served in the Tax Policy Branch of the Canadian Department of Finance in Ottawa, most recently as Director of Legislative Development and Chief Legislative Counsel from 2009 to 2011. In that role, he chaired the Interdepartmental Legislation Review Committee, the committee responsible for the preparation of Canada's federal income tax legislation for submission to Parliament. We believe that Mr. Wach’s experience with respect to tax and finance qualifies him to serve as a member of our board of directors.
Family Relationships
There are no family relationships among any of our executive officers or directors.
B.Compensation.
Executive Compensation
Summary Compensation Table
The following table provides a summary of compensation earned during each of the financial years ended on December 31, 2020 and 2019 by the Named Executive Officers, or NEOs, in accordance with applicable rules and regulations. Unless otherwise specified, the executive and directors’ compensation is presented in U.S. Dollars. For the convenience of the reader, in this Annual Report, unless otherwise indicated, translations from Canadian Dollars into U.S. Dollars were made at the rate of $1.00 to US$0.7442, which is the average rate for the 2020 fiscal year, (2019 average rate: $1.00=US$0.7525). Translations from Great Britain Pounds or GBP into U.S. Dollars were made at the rate of 1 GPB to US$1.2861 which is the average rate for the 2020 fiscal year, (2019 average rate: 1 GBP = US$1.2747). Such U.S. Dollar amounts are not necessarily indicative of the amounts of U.S. Dollars that could actually have been purchased upon exchange of Canadian Dollars or GBP at the dates indicated.
Summary Compensation Table | |
Name and Principal Position | | Year | | Salary (US$) | | | | Share Awards(1) (US$) | | | | Option Awards(2) (US$) | | | | Non-equity Incentive Plan Compensation(3) (US$) | | | | All Other Compensation(4) (US$) | | | | Total Compensation (US$) | |
Bruce Pritchard | | 2020 | | | 437,274 | | (6) | | — | | | | | 909,941 | | | | | 54,659 | | (6) | | | 43,727 | | (6) | | | 1,445,601 | |
CEO(5) | | 2019 | | | 433,398 | | (6) | | | 893,744 | | (7) | | | 2,465,614 | | | | | 390,058 | | (6) | | | 51,838 | | (6) | | | 4,234,652 | |
Patrick Sartore | | 2020 | | | 409,310 | | (9) | | — | | | | | 606,853 | | | | | 51,164 | | '(9) | | | 22,418 | | (10) | | | 1,089,745 | |
President(8) | | 2019 | | | 371,292 | | (9) | | | 729,398 | | (7) | | | 2,465,614 | | | | | 262,434 | | '(9) | | | 25,936 | | (10) | | | 3,854,674 | |
Murielle Lortie | | 2020 | | | 307,913 | | (9) | | — | | | | | 496,029 | | | | | 39,536 | | '(9) | | | 16,941 | | (10) | | | 860,419 | |
CFO(10) | | 2019 | | | 245,817 | | (9) | | | 66,822 | | (7) | | | 260,540 | | | | | 101,588 | | '(9) | | | 18,031 | | (10) | | | 692,798 | |
Kenneth Galbraith | | 2020 | | | 496,443 | | (9) | | — | | | | | 1,056,812 | | | | — | | | | | 158,405 | | (10) | | | 1,711,660 | |
Former CEO(11) | | 2019 | | | 368,815 | | (12) | | — | | | | | 8,826,695 | | | | | 262,499 | | | | | 33,326 | | | | | 9,491,335 | |
(1) | In accordance with SEC rules, this column reflects the aggregate grant date fair value of the share awards granted during the fiscal year. The fair value of the RSU awards was determined using the price of common shares underlying the RSUs on the grant date (2019 – US$0.23 ($0.30); this price is pre share consolidation). The amounts in this column do not represent the actual amounts paid to or realized by our NEOs during the years ended December 31, 2020 and 2019. |
113
(2) | In accordance with SEC rules, this column reflects the aggregate grant date fair value of the option awards granted during the fiscal year as well as the impact of the Re-Pricing 2020, as defined below, of stock options granted in 2019, where applicable. The fair value of the option awards, and of the Re-pricing 2020, was estimated using the Black Scholes option pricing model, a common valuation methodology which uses the same assumptions for determining the equity-based compensation expense in the Company's financial statements for the year-ended December 31, 2020 in accordance with IFRS. See Note 18b of the Notes to our Consolidated Financial Statements for a discussion of assumptions made by us in determining the aggregate grant date fair value of our option awards. The amounts in this column do not represent the actual amounts paid to or realized by our NEOs during the years ended December 31, 2020 and 2019. |
(3) | Represents the cash bonus earned for each financial year, pursuant to our Short-Term Incentive Plan, or STIP. For additional information, see the “Narrative Disclosure to Summary Compensation Table – Short-Term Incentive Plan” section below. |
(4) | The following table provides the details for the amounts reported for 2020 for each NEO: |
Name | | Car Allowance (US$) | | | Pension Contributions (US$) | | | Professional & Membership Dues (US$) | | | Relocation Expenses (US$) | | | Total (US$) | |
Bruce Pritchard | | — | | | | 43,727 | | | — | | | — | | | | 43,727 | |
Patrick Sartore | | — | | | | 20,466 | | | | 1,952 | | | — | | | | 22,418 | |
Murielle Lortie | | — | | | | 15,396 | | | | 1,545 | | | — | | | | 16,941 | |
Ken Galbraith | | | 19,137 | | | | 24,822 | | | — | | | | 114,446 | | | | 158,405 | |
(5) | Mr. Pritchard was appointed as CEO on November 13, 2020. Prior to this appointment, he was COO, Small Molecule Therapeutics and, prior to that, COO, International. |
(6) | Paid in pounds sterling (GBP) and converted at the following exchange rate: 1 GBP=US$1.2861 (2020 average); 1 GBP=US$1.2747 (2019 average) |
(7) | These amounts represent the value of the time-vested RSUs granted under the 2019 Special Retention Program. |
(8) | Mr. Sartore was appointed as President on November 13, 2020. Prior to this appointment, he was COO, Plasma-Derived Therapeutics and, prior to that, COO, North America. |
(9) | Paid in Canadian dollars (CAD) and converted at the following exchange rate: $1.00=US$0.7442 (2020 average); $1=US$0.7525 (2019 average) |
(10) | Mrs. Lortie was appointed as CFO on September 1, 2019. Prior to this appointment, she was Vice President, Finance. |
(11) | Mr. Galbraith was hired as CEO on April 23, 2019. He resigned as CEO on November 13, 2020 but remained employed to support the transition. |
(12) | Amount includes $29,555 of board fees that Mr. Galbraith was entitled to receive as compensation as a member of the board of directors and committees of the board until his appointment as CEO on April 23, 2019. Mr. Galbraith received no compensation for his role of executive director starting on April 23, 2019. |
Narrative Disclosure to Summary Compensation Table
Base Salary
We aim to provide a salary established based on the level of responsibility relative to other positions in our company and relative to base salaries paid by the organizations in our peer group as well as the performance of us and of the NEOs. It is an important component of our ability to attract and retain executives who have the leadership and management skills to drive the further growth and success of our business.
114
Short-Term Incentive Plan
We provide our NEOs with the STIP, which aims to engage, recognize and reward their contributions to reaching our annual objectives. Having a significant portion of all executives’ annual incentive determined on the basis of the same corporate performance objectives reinforces the teamwork of the members of our executive team and motivates them to achieve widespread success for the whole organization. The corporate short-term performance objectives against which our annual performance is evaluated are established each year by the HR and corporate governance committee, in conjunction with the CEO.
The STIP targets for the NEOs are reviewed regularly to ensure that they remain competitive with those of our peer group. Target incentive levels (as a percentage of base salary) for the NEOs were the following in 2020:
Name | | Short-Term Incentive Target | |
Bruce Pritchard | | 50% | |
Patrick Sartore | | 50% | |
Murielle Lortie | | 50% | |
Kenneth Galbraith | | 75% | |
At the beginning of 2020, corporate objectives were established for the year, each objective being assigned a relative weight. The board of directors, with the support of the HR and corporate governance committee was responsible for determining and approving a payout factor that would recognize our performance against the corporate objectives for the year. As a result of this assessment, the payout factor for 2020 was 25%.
The resulting payout amounts are included in the Summary Compensation Table above.
Long-Term Incentives
Long-term incentive awards are designed to reward the creation of value for our shareholders while providing a vehicle to attract and retain talented and skilled executives. Under the Omnibus Incentive Plan, adopted in 2019 and replacing the previous Long Term Incentive Plan, or LTIP, we grant stock options to our executives to ensure alignment of their interests with those of our shareholders, by creating focus on activities and development projects which will increase share price performance over time. It is also our practice to award an initial grant of stock options to new hires to attract qualified and skilled executives. Outstanding awards granted under the previous LTIP will continue to be governed by the terms of the LTIP until such awards are exercised, expire, or are otherwise terminated or cancelled. The previous LTIP provided stock options and RSUs.
Stock Options
We granted stock options in 2020 to the NEOs at a level reflecting the executive’s performance and the desired positioning of total compensation versus the comparator group in accordance with our compensation policy. Additionally, in June and August 2019, we cancelled certain outstanding options that were issued to our NEOs and employees prior to June 2019, as the exercise price of these options was significantly above the market price, such that it was highly unlikely that the options would ever be exercised. In compensation for the option holders’ agreement to the cancellation, they received the new options in June 2019.
Re-pricing of stock options granted in 2019
In May 2020, we reduced in the exercise price of an aggregate amount of 1,975,289 Options originally issued on June 4, 2019 and June 19, 2019 at $36.00 per Common Share and $27.00 per Common Share respectively to $15.21 (the “Re-Pricing 2020”). This repricing was completed to maintain the incentive nature of the stock options granted previously to the key active personnel of the Company to focus on the Company’s future performance.
Restricted Share Units
No RSUs were granted in 2020. All outstanding RSUs vested at the end of January 2021 as they met the time-vesting criteria.
The value of options granted or repriced in 2020 are included in the Summary Compensation Table above.
115
Perquisites and Other Benefits
We provide our NEOs with a competitive executive benefits package which is designed to attract and retain qualified executives. This package includes a capital accumulation retirement plan, including company contributions equal to 5% of each NEO’s base salary (10% for Bruce Pritchard), group insurance, reimbursement of annual medical examinations and annual professional memberships. Variations to these programs exist between NEOs due to local market conditions. The value of all such benefits conferred to the NEOs in 2020 is described in the Summary Compensation Table.
Employment Agreements
Below are descriptions of the employment agreements with our NEOs. See the section titled “—Termination and Change of Control Benefits” for information about the benefits each NEO is entitled to in the event that such executive’s employment is terminated by us without cause or in the event that the executive’s employment is terminated by us between six months before and 12 months after a change of control.
Bruce Pritchard. We entered into an employment agreement with Bruce Pritchard that became effective on July 31, 2019. Mr. Pritchard’s employment agreement provided for his employment as Chief Operating Officer, International for an unspecified duration. This employment agreement was not modified upon his appointment as CEO on November 13, 2020. Mr. Pritchard’s employment agreement established his base salary at GBP 340,000. Mr. Pritchard’s employment agreement further provides that he is eligible to earn an annual target performance bonus upon attainment of objectives to be determined by our board of directors, with a bonus target of 50%, with the potential of reaching a maximum of 100% of base salary. Mr. Pritchard also received a one-time grant of 253,542 stock options in connection with the entering into of the employment agreement. Mr. Pritchard is entitled to reimbursement for all necessary and reasonable business expenses incurred in connection with his duties in accordance with our generally applicable policies.
Patrick Sartore. We entered into an employment agreement with Patrick Sartore that became effective on July 22, 2019. Mr. Sartore’s employment agreement provides for his employment as Chief Operating Officer, North America, for an unspecified duration. This employment agreement has not been modified upon his appointment as President on November 13, 2020. Mr. Sartore’s employment agreement established his base salary at $465,000, which was increased to $550,000 as of September 1, 2019. Mr. Sartore’s employment agreement further provides that he is eligible to earn an annual target performance bonus upon attainment of objectives to be determined by our board of directors, with a bonus target of 50%, with the potential of reaching a maximum of 100% of base salary. Mr. Sartore also received a one-time grant of 253,542 stock options in connection with the entering into of the employment agreement. Mr. Sartore is entitled to reimbursement for all necessary and reasonable business expenses incurred in connection with his duties in accordance with our generally applicable policies.
Murielle Lortie. We entered into an employment agreement with Murielle Lortie that became effective on August 12, 2019. Mrs. Lortie’s employment agreement provides for her employment as Chief Financial Officer for an unspecified duration. Mrs. Lortie’s employment agreement established her base salary at $380,000, which was since increased to $425,000 as of April 1, 2020. Mrs. Lortie’s employment agreement further provides that she is eligible to earn an annual target performance bonus upon attainment of objectives to be determined by our board of directors, with a bonus target of 50%, with the potential of reaching a maximum of 100% of base salary. Mrs. Lortie’s bonus was subject to a minimum of $135,000 for 2019. Mrs. Lortie also received a one-time grant of 41,250 stock options in connection with the entering into of the employment agreement. Mrs. Lortie is entitled to reimbursement for all necessary and reasonable business expenses incurred in connection with her duties in accordance with our generally applicable policies.
116
Kenneth H. Galbraith. We entered into an employment agreement with our former Chief Executive Officer, Kenneth H. Galbraith that became effective on April 23, 2019. Mr. Galbraith’s employment agreement provided for his employment as Chief Executive Officer. Mr. Galbraith’s employment agreement established his base salary at US$500,000, which was prorated for the period after which Mr. Galbraith became our Chief Executive Officer. Mr. Galbraith’s employment agreement further provided that he was eligible to earn an annual target performance bonus upon attainment of objectives to be determined by our board of directors, with a bonus target of 75%, with the potential of reaching a maximum of 150% of base salary. Mr. Galbraith also received a one-time grant of 887,400 stock options in connection with the entering into of the employment agreement. Mr. Galbraith was entitled to reimbursement for all necessary and reasonable business expenses incurred in connection with his duties in accordance with our generally applicable policies.
Mr. Kenneth Galbraith resigned as CEO on November 13, 2020. The Company and Mr. Galbraith reached an agreement on the conditions of his end of employment. Mr. Galbraith remained as an advisor to the Company until February 12, 2021 in order to assist on the transition; for that period of time, Mr. Galbraith’s base salary was reduced by 25%. All other terms of his employment contract remained unchanged.
Termination and Change of Control Benefits
Our employment agreements with our NEOs include termination and change of control benefits in the event that the NEO’s employment is terminated by us without cause or in the event that the NEO’s employment is terminated by us between six months before and 12 months after a change of control. A change of control is deemed to occur upon a change of ownership resulting from the acquisition of a majority of our common shares.
The employment agreements provide for the payment of a lump sum equivalent to the value of 12 months of salary and benefit coverage (excluding disability) and 12 months of accelerated vesting of unvested stock options for termination without cause.
If the NEO’s employment is terminated by us between six months before and 12 months after a change of control, the NEOs’ employment agreements provide for the payment of a lump sum equivalent to the value of 18 months of salary and benefit coverage (excluding disability), 150% of the NEO’s then current annual target bonus under the short-term incentive plan and accelerated vesting of all unvested stock options.
Clawback Policy
In the event that we are required to prepare an accounting restatement due to a material non-compliance of the Company, as a result of misconduct, with any financial reporting requirement under the securities laws and the NEO knowingly engaged in the misconduct, was grossly negligent in engaging in the misconduct, knowingly failed to prevent the misconduct or was grossly negligent in failing to prevent the misconduct, the NEO shall reimburse us the amount of any payment in settlement of the award earned or accrued by him during the 12-month period following the first public issuance or filing with the Autorité des Marchés Financiers (whichever first occurred) of the financial document that contained such material noncompliance.
Indemnification
By-law No. 1 provides for indemnification of each of our directors and executive officers to the fullest extent permitted by the Canada Business Corporations Act, or CBCA.
We have entered into indemnity agreements with each director and officer providing that if such director or officer is or was involved in any threatened, pending or completed proceeding by reason of the fact that such director or officer is or was a director or officer of the Company or is or was serving at our request as a director or officer of another entity, including service with respect to employee benefit plans, whether the basis of such proceeding is an alleged action in an official capacity while serving as a director or officer, such director or officer will be indemnified and held harmless by us to the fullest extent authorized by and in the manner set forth in the CBCA against all expense, liability and loss reasonably incurred or suffered by such director or officer in connection therewith. Under such indemnity agreements, we may indemnify any of our directors or officers in connection with a proceeding (or part thereof) initiated by such director or officer only if such proceeding (or part thereof) is authorized by the board of directors or if such proceeding is a successful proceeding, in whole or in part, by a director or officer for claims under an indemnity agreement.
117
Outstanding Equity Awards at Fiscal Year End
The following table indicates, for each NEO, stock option grants outstanding as of December 31, 2020.
Outstanding Equity Awards at Fiscal Year End(9) |
| | Option Awards |
Name | | Grant Date | | Number of securities underlying unexercised options (#) (Exercisable) | | | Number of securities underlying unexercised options (#) (Unexercisable) | | | | Option exercise price (US$/sh) | | | | Option expiration date |
Bruce Pritchard | | June 4, 2019 | | | 73,959 | | | | 179,583 | | (1) | | 15.21 | | (2) | | June 4, 2029 |
| | December 7, 2020 | | | 50,000 | | | | 100,000 | | (3) | | 4.27 | | | | December 7, 2030 |
Patrick Sartore | | June 4, 2019 | | | 73,959 | | | | 179,583 | | (1) | | 15.21 | | (2) | | June 4, 2029 |
| | December 7, 2020 | | | 25,000 | | | | 50,000 | | (3) | | 4.27 | | | | December 7, 2030 |
Murielle Lortie | | June 19, 2019 | | | 3,238 | | | | 4,259 | | (4) | | 15.21 | | (2) | | June 19, 2029 |
| | September 3, 2019 | | | 12,891 | | | | 28,359 | | (5) | | 11.99 | | (2) | | September 3,2029 |
| | June 8, 2020 | | — | | | | 58,000 | | (6) | | 14.06 | | (2) | | June 8, 2030 |
| | December 7, 2020 | | | 5,000 | | | | 10,000 | | (7) | | 4.27 | | | | December 7, 2030 |
Kenneth Galbraith | | September 2, 2016 | | 33 | | | — | | | | | 2,800.00 | | (2) | | September 2,2021 |
| | May 18, 2017 | | 69 | | | — | | | | | 2,070.00 | | (2) | | May 18, 2027 |
| | December 4, 2018 | | 182 | | | — | | | | | 770.00 | | (2) | | December 4, 2028 |
| | June 4, 2019 | | | 149,874 | | | | 737,526 | | (10) | | 15.21 | | (2) | | June 4, 2029 |
(1) | These stock options vest monthly over the 6-year period following the grant date in accordance with the following vesting schedule, based on the number of months of service completed after the grant date. |
Months | | Vesting % | | | Months | | Vesting % | | | Months | | Vesting % | |
1 | | 0.93% | | | 25 | | 32.18% | | | 49 | | 76.39% | |
2 | | 1.85% | | | 26 | | 33.80% | | | 50 | | 77.78% | |
3 | | 2.78% | | | 27 | | 35.42% | | | 51 | | 79.17% | |
4 | | 3.70% | | | 28 | | 37.04% | | | 52 | | 80.56% | |
5 | | 4.63% | | | 29 | | 38.66% | | | 53 | | 81.94% | |
6 | | 5.56% | | | 30 | | 40.28% | | | 54 | | 83.33% | |
7 | | 6.48% | | | 31 | | 41.90% | | | 55 | | 84.72% | |
8 | | 7.41% | | | 32 | | 43.52% | | | 56 | | 86.11% | |
9 | | 8.33% | | | 33 | | 45.14% | | | 57 | | 87.50% | |
10 | | 9.26% | | | 34 | | 46.76% | | | 58 | | 88.89% | |
11 | | 10.19% | | | 35 | | 48.38% | | | 59 | | 90.28% | |
12 | | 11.11% | | | 36 | | 58.33% | | | 60 | | 91.67% | |
13 | | 12.04% | | | 37 | | 59.72% | | | 61 | | 92.36% | |
14 | | 12.96% | | | 38 | | 61.11% | | | 62 | | 93.06% | |
15 | | 13.89% | | | 39 | | 62.50% | | | 63 | | 93.75% | |
16 | | 14.81% | | | 40 | | 63.89% | | | 64 | | 94.44% | |
17 | | 15.74% | | | 41 | | 65.28% | | | 65 | | 95.14% | |
18 | | 16.67% | | | 42 | | 66.67% | | | 66 | | 95.83% | |
19 | | 17.59% | | | 43 | | 68.06% | | | 67 | | 96.53% | |
20 | | 18.52% | | | 44 | | 69.44% | | | 68 | | 97.22% | |
21 | | 19.44% | | | 45 | | 70.83% | | | 69 | | 97.92% | |
22 | | 20.37% | | | 46 | | 72.22% | | | 70 | | 98.61% | |
23 | | 21.30% | | | 47 | | 73.61% | | | 71 | | 99.31% | |
24 | | 30.56% | | | 48 | | 75.00% | | | 72 | | 100.00% | |
118
(2) | In Canadian dollars. Prior to October 8, 2020, the exercise prices of stock options were determined in Canadian dollar. |
(3) | These stock options vest monthly over the 3-year period spanning from December 7, 2020 to December 7, 2023. |
(4) | These stock options vest monthly over 3-year period spanning from June 19, 2020 to June 19, 2023. |
(5) | These stock options vest monthly over 3-year period spanning from September 3, 2020 to September 3, 2023. |
(6) | These stock options vest 25% upon the grant’s first anniversary, and then monthly over the following 3-year period. |
(7) | These stock options vest 50% on July 31, 2021 and 50% on December 31, 2022. |
(8) | These stock options vest annually over the 5-year period following the grant date in accordance with the following vesting schedule. |
Months | | Vesting % | |
12 | | 0.00% | |
24 | | 4.44% | |
36 | | 13.33% | |
48 | | 56.67% | |
60 | | 95.56% | |
72 | | 100.00% | |
(9) | For information on vesting acceleration of the equity awards described above upon a NEO’s termination of employment, see the “Executive Compensation – Executive Compensation Elements – Termination and Change of Control Benefits” section above. |
Director Compensation
Director Compensation Program
The following table describes the director compensation program that is currently in effect for the 12-month period starting June 8, 2020, or the 2020-2021 Mandate, and the director compensation program that was in effect for the period spanning from June 19, 2019 through June 8, 2020, or the 2019-2020 Mandate. Cash retainers are paid on a quarterly basis and no attendance fee is paid to the members for attending the meetings of our board of directors and meetings of our standing committees. Under the director compensation program for the 2020-2021 Mandate, 20,000 stock options are granted to newly appointed directors and vest over a 3-year period. 10,000 stock options are granted for subsequent grants and vest over one-year period.
119
Director Compensation Program | |
| | 2020-2021 Mandate (US$) | | | 2019-2020 Mandate (US$) | |
Chair of Board | | | | | | | | |
Annual Retainer (inclusive of committee retainers/fees) | | $ | 115,351 | | | $ | 131,688 | |
| | 10,000 stock options | | | 15,846 stock options(2) | |
Lead Independent Director | | | | | | | | |
Annual Retainer (inclusive of committee retainers/fees) | | $ | 100,467 | | | $ | 112,875 | |
| | 10,000 stock options | | | 15,846 stock options(2) | |
Non-Executive Directors | | | | | | | | |
Annual Retainer | | $ | 55,815 | | | $ | 56,438 | |
| | 10,000 stock options | | | 15,846 stock options(2) | |
Committee Chairs | | | | | | | | |
Additional Annual Retainer | | | | | | | | |
Audit, Risk and Finance | | $ | 26,047 | | | $ | 26,338 | |
HR and Corporate Governance(3) | | — | | | — | |
Science and Technology(4) | | $ | 26,047 | | | — | |
Attendance Fees | | No attendance fees | | | No attendance fees | |
Non-Chair Committee Members | | | | | | | | |
Additional Annual Retainer | | | | | | | | |
Audit, Risk and Finance | | $ | 13,024 | | | $ | 13,169 | |
HR and Corporate Governance(3) | | $ | 11,163 | | | $ | 11,288 | |
Science and Technology(4) | | — | | | — | |
Attendance Fees | | No attendance fees | | | No attendance fees | |
(1) | Options vest quarterly over 1-year period spanning from August 1st, 2020 to May 1st, 2021. | |
(2) | Options vest quarterly over 3-year period spanning from September 1, 2019 to June 1, 2022. | |
(3) | Included in Lead Independent Director annual retainer. | |
(4) | The new science and technology committee was established on July 1st, 2020. | |
Director Compensation Table
The following table sets forth information concerning compensation accrued or paid to our non-employee directors during the year ended December 31, 2020 for their service on our Board. Mr. Kenneth Galbraith, who was an executive director from April 23, 2019 to November 13, 2020 received no additional compensation for such roles. His compensation is set forth in the Summary Compensation Table above.
Director Compensation Table | |
| | Fees earned or paid in cash | | | | | | | | | | | | | |
Name | | Board Annual Cash Retainer (US$) | | | Committee Annual Cash Retainer (US$) | | | Attendance Fee (US$) | | Option awards(1)(2) (US$) | | | All other compensation (US$) | | | | Total Compensation (US$) | |
Current Members | | | | | | | | | | | | | | | | | | | | | | | |
Simon Best(3) | | | 95,816 | | | | 11,899 | | | — | | | 92,805 | | | | 15,956 | | (4) | | | 216,476 | |
Gary Bridger(5) | | | 51,164 | | | | 16,791 | | | — | | | 92,805 | | | | 157,678 | | (6) | | | 318,438 | |
Neil A. Klompas(7) | | | 51,164 | | | | 23,876 | | | — | | | 92,805 | | | — | | | | | 167,845 | |
Alek Krstajic(8) | | | 38,451 | | | — | | | — | | | 214,330 | | | — | | | | | 252,781 | |
Eugene Siklos(9) | | — | | | — | | | — | | — | | | — | | | | — | |
Timothy Steven Wach | | | 51,164 | | | | 18,450 | | | — | | | 92,805 | | | — | | | | | 162,419 | |
Previous Members | | | | | | | | | | | | | | | | | | | | | | | |
Stefan Clulow(10) | | | 72,249 | | | — | | | — | | | 92,805 | | | — | | | | | 165,054 | |
Zachary Newton(11) | | | 32,559 | | | | 6,512 | | | — | | | 92,805 | | | — | | | | | 131,875 | |
120
(1) | In accordance with SEC rules, this column reflects the aggregate grant date fair value of the option awards granted during the fiscal year. The fair value of the option awards was estimated using the Black Scholes option pricing model, a common valuation methodology which uses the same assumptions for determining the equity-based compensation expense in the Company's financial statements for the year-ended December 31, 2020 in accordance with IFRS. See Note 18b of the Notes to Consolidated Financial Statements in this Annual Report for a discussion of assumptions made by us in determining the aggregate grant date fair value of our option awards. The amounts in this column do not represent the actual amounts paid to or realized by our directors during the year ended December 31, 2020. | |
(2) | Include the incremental value of stock options granted in 2019, as a result of the Re-Pricing 2020. | |
(3) | Prof. Best is our Lead Independent Director and Chair of the HR and corporate governance committee. | |
(4) | Of this amount, (i) US$2,040 represents the incremental value of stock options granted in 2019 to Prof. Best for his role and Interim President and CEO, as a result of the Re-Pricing 2020. Prof. Best held such position from December 2018 to April 2019, and (ii) US$13,916 represents the aggregate of a reimbursement of 2017 and 2019 tax penalties with Canada Revenue Agency and 2017 tax penalties with Quebec Revenue Agency. | |
(5) | Dr. Bridger was member of the HR and corporate governance committee until June 8, 2020. Dr. Bridger was appointed chair of the science and technology committee on July 1st, 2020. | |
(6) | This amount, paid in U.S. dollar, represents the compensation Dr. Bridger received pursuant to his consultancy agreement entered into in October 2019 with us, as amended from time to time. | |
(7) | Mr. Klompas is Chair of the audit, risk and finance committee. | |
(8) | Mr. Krstajic was appointed Chair of our board of directors on September 1, 2020 and member of the HR and corporate governance committee on September 17, 2020. | |
(9) | Mr. Siklos was appointed on board of directors on September 1, 2020. He was member of the HR and corporate governance committee from September 1st to September 17, 2020. Mr. Siklos waived the compensation he is entitled to receive as director on our board of directors. | |
(10) | Mr. Clulow resigned from our board of directors on September 1, 2020. | |
(11) | Mr. Newton resigned from our board of directors on September 1, 2020. | |
(12) | The following table indicates the number of option awards outstanding for each current and former director as of December 31, 2020. No share awards were provided to directors during the year ended December 31, 2020. | |
Name | | Options Outstanding (#) | |
Simon Best | | | 27,781 | |
Gary Bridger | | | 25,846 | |
Neil Klompas | | | 25,846 | |
Alek Krstajic | | | 20,000 | |
Eugene Siklos | | — | |
Timothy Steven Wach | | | 25,846 | |
Stefan Clulow | | 313 | |
Zachary Newton | | 129 | |
C.Board Practices.
Our board of directors is responsible for our stewardship and strategic direction. It does not actively manage but rather supervises the management of our business and affairs, to ensure a consistent focus on increasing shareholder value. In exercising their powers and discharging their duties, our directors shall (a) act honestly and in good faith with a view to the best interests of the company; and (b) exercise the care, diligence and skill that a reasonably prudent person would exercise in comparable circumstances.
121
Our board of directors currently consists of six members and one vacant seat. The following table sets forth the names of our directors, the years of their initial appointment as directors and the expiration dates of their current term.
Name | | Current Position | | Year of Initial Appointment | | Term Expiration Year |
Simon Best | | Lead Independent Director | | 2014 | | n/a |
Gary J. Bridger | | Director | | 2019 | | n/a |
Neil A. Klompas | | Director | | 2019 | | n/a |
Alek Krstajic | | Chair | | 2020 | | n/a |
Eugene Siklos | | Director | | 2020 | | n/a |
Timothy Steven Wach | | Director | | 2019 | | n/a |
Director Independence
As a foreign private issuer, under the listing requirements and rules of Nasdaq, we are not required to have independent directors on our board of directors, except to the extent that our audit committee is required to consist of independent requirements, subject to certain phase-in schedules. However, our board of directors has determined that, under current listing requirements and rules of Nasdaq (which we are not subject to) and taking account any applicable committee independence standards four of our six directors, Prof. Best, Mr. Klompas, Mr. Krstajic and Mr. Wach, are independent directors. In accordance with the Nasdaq Listing Rules, a director shall be considered independent if she/he does not have any relationship which, in the opinion of the board, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. In addition, in accordance with Nasdaq Listing Rules, to be considered independent, a director shall not be subject to any of the mandatory bars to independence set forth in Rule 5605(a) of the Nasdaq Listing Rules. In making such determination, our board of directors considered the relationships that each non-employee director has with us and all other facts and circumstances our board of directors deemed relevant in determining the director’s independence, including the number of ordinary shares beneficially owned by the director and his or her affiliated entities (if any).
On an annual basis, each director has to declare whether he is independent, and all such declarations are reviewed by the HR and corporate governance committee, which then makes recommendations to our board of directors in respect of our board of director’s determination on the independence of the directors. We have not instigated a board tenure policy given the length of tenure of the directors that currently serve on our board of directors, which is well below the average tenure of directors of Canadian reporting issuers.
Board Committees
Our board of directors has two standing committees: the audit, risk and finance committee and the HR and corporate governance committee.
Audit, risk and finance committee
The audit, risk and finance committee currently consists of Messrs. Best, Klompas and Wach.
Mr. Klompas serves as chair of the audit, risk and finance committee. All of the members of the audit, risk and finance committee are independent directors and financially literate within the meaning of the Canadian Securities Administrators rules. The members also qualify as “audit committee financial experts” as defined by the SEC.
The audit, risk and finance committee is mainly responsible for the four following fundamental matters:
| • | our financial reporting process and internal control systems; |
| • | our process to identify and manage risks: |
| • | the internal and external audit process; and |
| • | our communication system to provide an open avenue of communication among the external auditors, our financial and senior management, our internal auditing department (if any), and our board of directors. |
122
The audit, risk and finance committee reviews the interim and annual consolidated financial statements, management's discussion and analysis and other legally required public disclosure documents containing financial information. In its oversight of the services rendered by the external auditors, the audit, risk and finance committee (i) discusses with the external auditor its responsibilities in performing the audit, its determination of areas of significant audit risk and related risk mitigation procedures, and reviews and approves its annual audit plan and associated fees; (ii) discusses with the external auditor the key accounting risks and significant judgments made by management; (iii) receives written confirmation from the external auditor of its independence; (iv) pre-approves all additional engagements with the external auditor (including any non-audit services); and (v) assesses, annually, the external auditor’s performance. The audit, risk and finance committee is also involved in the assessment and selection of our external auditor.
The audit, risk and finance committee also oversees the financial reporting processes and internal controls. This comprises keeping abreast of how our management's addresses changes in the operations and transactions to ensure that they are appropriately reflected in the financial disclosure documents and the internal controls put in place to address these changes. As part of this, the audit, risk and finance committee reviews the activities of the internal audit department, including its organizational structure, resources and budget, the internal audit plan for the year and their report on their review and testing of ICFR and disclosure controls. It also considers any recommendations made by the external auditors regarding internal controls. In addition, the audit, risk and finance committee reviews the quarterly internal reporting package prepared by management, understanding the key variances from budget and the impact on the financial condition.
Finally, in the accomplishment of its financial oversight, the audit, risk and finance committee reviews and discusses our short and long-term financial plans, including financial results and the management of tax matters.
The audit, risk and finance committee meets as often as one or more members of the audit, risk and finance committee deems necessary, but in any event meets at least four scheduled times per year.
The HR and corporate governance committee
The HR and corporate governance committee consists of Messrs. Best, Krstajic and Wach. Prof. Best serves as the chair of the HR and corporate governance committee. The primary responsibility of the HR and corporate governance committee with respect to human resources and compensation are mainly to:
| • | review short and long-term corporate objectives relevant to the compensation of the chief executive officer and evaluate chief executive officer performance in meeting these objectives; |
| • | review the compensation of the chief executive officer, other senior executive officers and director compensation; |
| • | review the design and ensure implementation of incentive-compensation and equity-based plans; |
| • | ensure that appropriate mechanisms are in place regarding succession planning for the chief executive officer; and |
| • | perform any other activities delegated by our board of directors consistent with the responsibilities and duties relating to compensation. |
The primary responsibility of the HR and corporate governance committee with respect to corporate governance will be mainly to:
| • | assess and establish the size and composition of our board of directors and its committees, and identify candidates with the right skills for the nomination of directors; |
| • | develop and review an appropriate procedure to periodically evaluate the performance of our board of directors, its committees and their members; |
| • | develop and monitor the continuing education programs for directors; |
| • | review the mandate of our board of directors and its committees, of their chairs and of the lead independent director; |
123
| • | review annually our Code of Ethics and Business Conduct as well as any other of our policies as deemed appropriate; and |
| • | perform any other activities delegated by our board of directors consistent with the responsibilities and duties relating to corporate governance. |
The HR and corporate governance committee meets regularly throughout the year. At the end of each meeting or whenever deemed necessary, the HR and corporate governance committee may hold an informal in-camera session without the presence of management.
Other Corporate Governance Matters
The Sarbanes-Oxley Act, as well as related rules subsequently implemented by the SEC, requires foreign private issuers, including our company, to comply with various corporate governance practices. In addition, Nasdaq rules provide that foreign private issuers may follow home country practice in lieu of the Nasdaq corporate governance standards, subject to certain exceptions and except to the extent that such exemptions would be contrary to U.S. federal securities laws.
Because we are a foreign private issuer, our members of our board of directors, executive board members and senior management are not subject to short-swing profit and insider trading reporting obligations under Section 16 of the Exchange Act. They will, however, be subject to the obligations to report changes in share ownership under Section 13 of the Exchange Act and related SEC rules.
D.Employees.
As of December 31, 2020, we had 251 full-time employees. None of our employees are represented by collective bargaining agreements. We believe that we maintain good relations with our employees. At each date shown, we had the following number of full time employees, broken out by function.
| | At December 31, |
| | 2020 | | 2019 | | 2018 |
Function: | | | | | | |
Research and development | | | | | | |
Small molecule therapeutics segment | | 29 | | 39 | | 47 |
Plasma-derived therapeutics segment | | 134 | | 166 | | 216 |
Plasma collection centers | | 34 | | 33 | | 31 |
Bioseparations segment | | - | | - | | 83 |
General and administrative | | 54 | | 62 | | 77 |
Total | | 251 | | 300 | | 454 |
E.Share Ownership.
For information regarding the share ownership of our directors and executive officers, see “Item 6.B.—Compensation” and “Item 7.A.—Major Shareholders.”
Item 7.Major Shareholders and Related Party Transactions.
A.Major shareholders.
The following table sets forth information with respect to the beneficial ownership of our common shares as of March 15, 2021 for:
| • | each beneficial owner of more than five percent (5%) of our outstanding common shares; |
| • | each of our executive officers; |
| • | each of our directors; and |
| • | all of our directors and executive officers as a group. |
124
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include common shares that can be acquired within 60 days of March 15, 2021. The percentage ownership information shown in the table is based upon 29,943,839 common shares outstanding as of March 15, 2021.
Except as otherwise indicated, all of the shares reflected in the table are common shares and all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
In computing the number of common shares beneficially owned by a person and the percentage ownership of that person, we deemed outstanding common shares subject to options and warrants held by that person that are immediately exercisable or exercisable within 60 days of March 15, 2021. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. The information in the table below is based on information known to us or ascertained by us from public filings made by the shareholders. Except as otherwise indicated, addresses of the directors, executive officers and named beneficial owners are in care of Liminal BioSciences Inc., 440 Armand-Frappier Blvd., Suite 300, Laval, Québec, Canada H7V 4B4. The current address for Moira Daniels and Bruce Pritchard is Park Ground Floor, Unit 1, Iconix Park, London Road, Sawston, Cambridge CB22 3EG.
Name and Address | | Number of Shares Beneficially Owned | | | Percentage of Shares Beneficially Owned | |
5% or Greater Shareholders | | | | | | | | |
Structured Alpha LP(1) | | | 23,992,231 | | | 70.4 | |
Executive Officers and Directors | | | | | | | | |
Bruce Pritchard | | * | | | * | |
Patrick Sartore | | * | | | * | |
Murielle Lortie | | * | | | * | |
Kenneth Galbraith | | * | | | * | |
Simon Best | | * | | | * | |
Gary J. Bridger | | * | | | * | |
Neil A. Klompas | | * | | | * | |
Alek Krstajic | | * | | | * | |
Eugene Siklos | | * | | | * | |
Timothy Wach | | * | | | * | |
All current executive officers and directors as a group (12 persons)(4) | | | 550,504 | | | | 1.8 | |
___________________
* Represents beneficial ownership of less than 1%.
(1) | Consists of (a) 19,876,129 common shares and (b) 4,116,102 common shares that SALP has the right to acquire within 60 days following December 31, 2020 pursuant to the exercise of warrants. Thomvest Asset Management Ltd. is the general partner of SALP and may be deemed to beneficially own, have sole power to vote and sole power to dispose of these common shares. The address for SALP and Thomvest Asset Management Ltd. is 65 Queen Street West, Suite 2400, Toronto Ontario M5H 2M8. |
(2) | Consists of (a) 26,537 common shares and (b) 523,967 common shares that all officers and directors as a group have the right to acquire within 60 days following March 15, 2021 pursuant to the exercise of stock options (521,988) and restricted share units (1,979). |
As of March 15, 2021, we estimate that approximately 10.82% of our outstanding common shares were held in the United States by 11 holders of record. The actual number of holders is greater than these numbers of record holders and includes beneficial owners whose common shares are held in street name by brokers and other nominees. This number of holders of record also does not include holders whose shares may be held in trust by other entities.
125
B.Related Party Transactions.
Since January 1, 2019, we have engaged in the following transactions with our directors, executive officers and holders of more than five percent (5%) of our outstanding voting securities and their affiliates, which we refer to as our related-parties. All of the transactions have been reviewed and approved by our board of directors or another independent committee of the board.
Transactions with Structured Alpha LP (by its partner, Thomvest Asset Management Ltd.)
Prior Indebtedness
We had previously entered into a first loan agreement with SALP, or the First Loan Agreement, on September 10, 2013, which provided for an original issued discount loan having a face value of $15.7 million. This loan was amended and restated on July 31, 2014, March 31, 2015 and February 29, 2016 with an additional increase in the face value of $50.4 million, and further amended from time to time. We and SALP subsequently entered into a second loan agreement, or the Second Loan Agreement, on July 31, 2014, which provided for an original issued discount loan with a face value of $31.3 million. This loan was amended and restated on March 31, 2015 and February 29, 2016, and further amended from time to time.
On April 27, 2017, we entered into a third loan agreement with SALP, or the Third Loan Agreement, which provided for an original issued discount loan with a face value of $39.2 million. This loan was amended from time to time. We subsequently entered into a fourth loan agreement with SALP, or the Fourth Loan Agreement, on November 31, 2017, which provided for a delayed-drawn term loan of up to US$80 million, as amended from time to time.
We and SALP then entered into an omnibus amendment agreement dated November 14, 2018, extending the maturity dates of the First Loan Agreement, the Second Loan Agreement, the Third Loan Agreement and the Fourth Loan agreement.
On February 22, 2019, we entered into an amendment agreement to the Fourth Loan Agreement securing an additional amount of up to US$15 million from SALP under the Fourth Loan Agreement.
The issuance or modification of loans were generally done concurrently with the issuance or modification of warrants.
Restructuring Transactions
In April and June 2019, we entered into a series of related arrangements to restructure our outstanding indebtedness, reduce our interest and certain other payment obligations, and raise sufficient cash to build a robust balance-sheet for the next phase of our development, or the Restructuring Transactions.
On April 23, 2019, we completed transactions pursuant to a debt restructuring agreement we entered into on April 15, 2019 with SALP and certain of our subsidiaries, or the Restructuring Agreement. Upon execution of the Restructuring Agreement, (i) SALP acquired 15,050,312 of our common shares, or the New Common Shares, at a price per common share of $15.21, or the Transaction Price, for a total purchase price of $228.9 million, which was satisfied by the cancellation of outstanding indebtedness in such amount owed by us to SALP, and (ii) certain Warrants to purchase our common shares held by SALP were amended, with new warrants being issued, or the New Warrants, exercisable for 168,735 common shares at a per-share exercise price equal to the Transaction Price. In connection with the Restructuring Transactions, in the event that SALP owns less than a majority of our issued and outstanding common shares, SALP was granted the right to (i) nominate the number of directors to our board of directors that represents the same percentage of the total number of directors to be elected as the percentage of our issued and outstanding common shares owned by SALP at the record date for the applicable meeting of our shareholders, and (ii) nominate two directors to our board of directors for so long as it owns at least 10% of our issued and outstanding common shares. Our directors, Alek Krstajic and Eugene Siklos were nominated pursuant to this right.
126
Debt Conversion
The New Common Shares have been issued to SALP in connection with the Restructuring Agreement. Under the Restructuring Agreement, all but $10 million of the outstanding debt we owed to SALP in the aggregate amount of $238,887,948 was converted into common shares. We also entered into a consolidated loan agreement with SALP on April 23, 2019, relating to future indebtedness secured against our assets, or the Consolidated Loan Agreement.
Warrant Amendments
The New Warrants have been issued to SALP in connection with the Restructuring Agreement. Prior to the closing of the transactions described above, SALP owned the following Warrants:
| • | 100,000 warrants, each of which entitled SALP to acquire one common share upon the payment of an exercise price equal to $520 per Warrant, or Warrants 1; |
| • | 20,277 Warrants, each of which entitled SALP to acquire one common share upon the payment of an exercise price equal to an aggregate of $15,653,138 or the satisfaction of the loan made by SALP to us on September 10, 2013 in the principal amount of $10 million (plus interest accrued thereon), or Warrants 2; |
| • | 128,057 Warrants, each of which entitled SALP to acquire one common share upon the payment of an exercise price equal to $1,000 per Warrant, or Warrants 8; and |
| • | 19,402 Warrants, each of which entitled SALP to acquire one series A preferred share of ours, or a Series A Preferred Share, upon the payment of an exercise price equal to $156.36 per Warrant, or Warrants 9. |
Pursuant to the Restructuring Agreement, (i) the exercise price of Warrants 1, 2, 8 and 9 was amended to be the Transaction Price, and (ii) the terms of Warrant 9 were amended so that each Warrant 9 becomes exercisable for a common share instead of a Series A Preferred Share. The New Warrants may be exercised in whole or in part at SALP’s option until April 23, 2027, at the Transaction Price per New Warrant.
See “Item 4.A.—History and Development of the Company” for more information relating to the Restructuring Transactions.
Loan Agreement
Under the Consolidated Loan Agreement, all indebtedness we owed to SALP was reduced to a principal amount of $10 million of secured indebtedness to be used by us for the purpose of providing short-term working capital, paying transaction expenses, general corporate purposes and certain specified permitted debt payments.
Line of Credit
On November 11, 2019, we entered into an amendment to our Consolidated Loan Agreement with SALP to provide us with a non-revolving $75.0 million secured line of credit, or the LOC. The Consolidated Loan Agreement between SALP and us dated as of April 23, 2019 has been amended to incorporate the terms of the LOC. As a result of the sale of Prometic Bioseparations Ltd, in November 2019 the LOC was reduced to $29.1 million. In September 2020, we drew down the available balance of $29.1 million under the LOC. The principal amount of $29.1 million borrowed under the LOC bears a stated interest of 10%, payable quarterly, and matures on April 23, 2024.
Royalties
We have also granted SALP a right to receive low single digit royalties on future revenues relating to patents existing as of the date of the agreement of certain compounds and analogues, including fezagepras. The obligation under this royalty agreement is secured by all of our assets until the expiry of the last patent anticipated in 2033. For additional information on this arrangement, please see “Item 4.—Information on the Company.”
127
Private Placement
As part of the Restructuring Transactions, we issued 4,931,161 of our common shares in the Private Placement at a subscription price of $15.21 per common share for gross proceeds of $75 million. SALP’s participation in the Private Placement was for gross proceeds of $25 million and the participation in the Private Placement by Consonance was for gross proceeds of $50 million. In connection with the Private Placement, we also entered into a registration rights agreement for the benefit of Consonance.
Registration Rights Agreement
Consonance Capital Management
On April 23, 2019, we entered into a registration rights agreement with Consonance in connection with the Private Placement. Pursuant to the registration rights agreement, we agreed to use our commercially reasonable efforts to have a registration statement registering the common shares purchased by Consonance in the Private Placement declared effective. We also agreed, among other things, to cause such registration statement to remain effective and to assist Consonance with offerings of such securities. On March 17, 2020, we amended the registration rights agreement to provide that we will use reasonable best efforts to have a registration statement declared effective by the SEC. We filed with the SEC a registration statement on Form F-1 to register the common shares held by Consonance Capital Master Account LP and P Consonance Opportunities Ltd., which was declared effective by the SEC on September 30, 2020. We also filed with the SEC a post-effective amendment no. 1 on Form F-3 to registration statement on Form F-1, which was effective on December 7, 2020.
In addition, if we propose to register (including, for this purpose, a registration effected by us for shareholders other than Consonance) any of our securities under the Securities Act in connection with the public offering of such securities solely for cash (other than a rights offering or pursuant to a registration statement on Form S-4, F-4 or S-8), we are obligated, at such time, to promptly give each of Consonance notice of such registration. Upon the request of each of Consonance given within 10 days after such notice is given by us, we are obligated, subject to underwriter requirements, to use commercially reasonable efforts to cause to be registered all of the registrable securities that each such holder has requested to be included in such registration. We have the right to terminate or withdraw any registration initiated by us before the effectiveness of such registration, whether or not any such holder has elected to include registrable securities in such registration. The expenses of such withdrawn registration will be borne by us.
We have also agreed, among other things, to indemnify each of Consonance from certain liabilities and to bear all fees and expenses incident to the registration of our common shares purchased by each of Consonance in the Private Placement, including fees, charges and disbursements of counsel to Consonance.
Structured Alpha LP and Armistice Capital Master Fund Ltd.
On October 29, 2020, we, SALP, and Armistice entered into a registration right agreement in connection to the Securities Purchase Agreement, as amended in November 2020. The Company has agreed to file a resale registration statement with the SEC within 30 days of the closing of the offering for purposes of registering the resale of the common shares issuable in connection with the offering and the common shares underlying the warrants and pre-funded warrants issuable in the offering. We filed with the SEC a registration statement on Form F-3 to register the common shares held by SALP and Armistice, which was declared effective by the SEC on December 9, 2020.
Share Purchase Agreement with Fairhaven Pharmaceuticals Inc.
In July 2020, we acquired all of the issued and outstanding shares in the capital of Fairhaven, a company with a preclinical research program of small molecule antagonists, and completed a concurrent private placement with each of Genesys, AmorChem LP and AmorChem II of $2.4 million aggregate principal amount of secured convertible debentures.
128
Under the terms of the acquisition, we will satisfy the aggregate purchase price of up to $8 million by the issuance of our common shares al to the shareholders of Fairhaven, being Genesys, AmorChem LP, MSBi Valorisation Inc. and The Royal Institution for the Advancement of Learning/McGill University subject to the achievement of certain pre-determined milestones. Investigational therapies developed in this program target a key chemoattractant and activator of eosinophils, which play a key role in Type 2 inflammation-driven diseases through tissue repair and resolution of inflammation, or the Fairhaven R&D Program.
In July, 2020, we issued an aggregate of 202,308 of our common shares, representing $3.6 million of the aggregate purchase price, to the Sellers, at a price per share equal to the five-trading day VWAP of our common shares on the Toronto Stock Exchange (the "TSX"). The remainder of the aggregate purchase price payable by us to the Sellers is issuable upon, and subject to, the achievement of certain pre-determined research and development milestones prior to the fifth anniversary of the closing date of the acquisition, including $1.3 million upon the selection by us of a compound for development and $3.1 million upon the first dosing of a human in a Phase 1 clinical trial with a compound subject to the acquisition agreement. Any future shares to be issued as part of the milestone payments will be issued at a price per share equal to the five-trading day VWAP of our common shares prior to the achievement of such milestone events.
The acquisition constitutes a "related party transaction" within the meaning of Regulation 61-101 – Protection of Minority Security Holders in Special Transactions ("Regulation 61-101"). We relied on the exemptions from the valuation and minority shareholder approval requirements contained in sections 5.5(a) and 5.7(1)(a) of Regulation 61-101 in respect of related party participation in the acquisition, as neither the fair market value of the shares distributed to, nor the consideration paid by, related parties, exceeded 25% of our market capitalization.
Each of Mr. Kenneth Galbraith and Mr. Gary Bridger, or the Directors, served on the board of directors of both us and Fairhaven prior to the closing of the acquisition. In addition, the Directors are officers of, and owned all of the shares in the capital of, Five Corners Capital Inc., a former shareholder of Fairhaven immediately prior to the closing of the acquisition. Pursuant to a corporate reorganization of Fairhaven which occurred prior to the closing of the acquisition, Fairhaven repurchased all of the shares held by Five Corners Capital Inc. in the capital of Fairhaven, such that Five Corners Capital Inc. was no longer a shareholder of Fairhaven on the date of closing of the acquisition. Our independent directors reviewed and unanimously approved the acquisition.
Share Purchase Agreement with SALP and Armistice Capital Master Fund Ltd.
On October 29, 2020, we, Structured Alpha LP, or SALP, and Armistice Capital Master Fund Ltd., or Armistice, entered into a Securities Purchase Agreement, as amended in November 2020, or the Purchase Agreement, and a Registration Rights Agreement, or the RRA, as part of a private placement, which we refer to as the Private Placement. Pursuant to the Purchase Agreement, the selling shareholders purchased in a private placement an aggregate of (i) 5,757,894 common shares, (ii) prefunded warrants to purchase up to 557,894 common shares at an exercise price of US$0.001 per share, which we refer to as the Prefunded Warrants, and (iii) warrants to purchase up to 7,894,734 common shares at an exercise price of US$5.50 per share, which we refer to as the Warrants. The warrants are subject to a beneficial ownership limitation of 4.99% (subject to the right of the selling shareholder to increase or decrease such beneficial ownership limitation upon notice to us, provided that such limitation cannot exceed 9.99% and provided that such increase shall not be effective until 61 days after such notice is delivered), which does not permit Armistice to exercise that portion of the warrants that would result in Armistice and its affiliates owning, after exercise, a number of common shares in excess of the beneficial ownership limitation.
Board Observation Right and Director Nomination Agreement
In connection with the Restructuring Transactions, in the event that SALP owns less than a majority of our issued and outstanding common shares, SALP was granted the right to (i) nominate the number of directors to our board of directors that represents the same percentage of the total number of directors to be elected as the percentage of our issued and outstanding common shares owned by SALP at the record date for the applicable meeting of our shareholders, and (ii) nominate two directors to our board of directors for so long as it owns at least 10% of our issued and outstanding common shares. Our directors, Alek Krstajic and Eugene Siklos were nominated pursuant to this right.
129
In connection with the Private Placement, we entered into an agreement with Consonance pursuant to which Consonance were granted the right to collectively designate one observer to our board of directors until our Nasdaq Listing, and from and after such time, have the right to nominate one director for election to our board of directors.
Employment Agreements
We have entered into compensation, including grants of stock options and other equity awards, termination and change of control arrangements with our named executive officers, which are described under the sections entitled “Item 6.B.—Compensation—Executive Compensation—Employment Agreements” and “Item 6.B.—Compensation—Executive Compensation—Termination and Change of Control Benefits.”
Bargaining Process
The Company was informed on January 20, 2021 that the Quebec’s Tribunal Administratif du Travail had approved the unionization request made by the production technicians and team leaders of Prometic Bioproduction inc. This group of approximately 25 employees will be represented by the local section 1991-P of the United Food and Commerce Workers (UFCW) union. The process to negotiate a first bargaining agreement between this group and the Company is expected to start during the first few months of 2021.
Directors and Officers Liability Insurance
We maintain directors’ and officers’ liability insurance policies for the liability of our directors and officers arising out of the performance of their duties and for our liability arising out of securities claims. The policies provide coverage in respect of a maximum total liability of US$15,000,000 (primary and excess liability insurance), subject to a general retention of US$5,000,000 per loss, and includes specific exclusions, which are usually contained in insurance policies of this nature. We also maintain a concurrent excess liability insurance for the liability of our directors and officers arising out of the performance of their duties, with limits of liability of US$5,000,000 and subject to specific exclusions which are usually contained in insurance policies of this nature.
D&O Indemnification Agreements
By-law No. 1 provides for indemnification of each of our directors and executive officers to the fullest extent permitted by the Canada Business Corporations Act, or CBCA.
We have entered into indemnity agreements with each director and officer providing that if such director or officer is or was involved in any threatened, pending or completed proceeding by reason of the fact that such director or officer is or was a director or officer of the Company or is or was serving at our request as a director or officer of another entity, including service with respect to employee benefit plans, whether the basis of such proceeding is an alleged action in an official capacity while serving as a director or officer, such director or officer will be indemnified and held harmless by us to the fullest extent authorized by and in the manner set forth in the CBCA against all expense, liability and loss reasonably incurred or suffered by such director or officer in connection therewith. Under such indemnity agreements, we may indemnify any of our directors or officers in connection with a proceeding (or part thereof) initiated by such director or officer only if such proceeding (or part thereof) is authorized by the board of directors or if such proceeding is a successful proceeding, in whole or in part, by a director or officer for claims under an indemnity agreement.
C.Interests of Experts and Counsel.
Not applicable.
Item 8.Financial Information.
A.Consolidated Statements and Other Financial Information.
Consolidated Financial Statements
Our consolidated financial statements are appended at the end of this Annual Report, starting at page F-1, and incorporated herein by reference.
130
Dividend Distribution Policy
We have not paid any dividends on our outstanding common shares, and we have no current intention to declare dividends on our common shares in the foreseeable future. Any decision to pay dividends on our common shares in the future will be at the discretion of our board of directors and will depend on, among other things, our results of operations, current and anticipated cash requirements and surplus, financial condition, any future contractual restrictions and financing agreement covenants, solvency tests imposed by corporate law and other factors that our board of directors may deem relevant. We are subject to covenants under our non-revolving LOC with SALP that places restrictions on our ability to pay dividends.
Legal Proceedings
We are, in the course of our business, subject to lawsuits and other claims. On April 15, 2019, we announced our intention to enter into a series of related arrangements to restructure its outstanding indebtedness, reduce our interest and certain other payment obligations, and raise sufficient cash to build a robust balance-sheet for the next phase of our development (collectively, the “Refinancing Transactions”), which included (i) an offering of Common Shares, (ii) the conversion of approximately $229 million of the outstanding debt into Common Shares, (iii) the adjustment of the terms of certain outstanding warrants and (iv) a rights offering to all shareholders whereby each shareholder received one right for each Common Share held, with each right entitling the holder thereof to subscribe for 20 Common Shares for aggregate proceeds of up to $75 million.
On March 2, 2021, we were served with an action instituted by multiple individual shareholder plaintiffs (the “Plaintiffs”) against the Company, Structured Alpha LP (“SALP”), Thomvest Asset Management (“Thomvest”), Consonance Capital Management LP (“Consonance”), as well as the directors (the “Directors”) that were on the Company’s Board on March 31, 2019 or on April 15, 2019 and certain officers of the Company (the “D&Os”, together with Liminal, SALP, Thomvest and Consonance, the “Defendants”).
The Plaintiffs allege, among other things, that, as part of the Refinancing Transactions, the Defendants (i) undervalued certain products, (ii) reduced certain of their operational activities, (iii) artificially devalued certain assets in order for them to be written off the balance sheet, (iv) conducted their business in a manner that prevented them from obtaining financing from certain parties and (v) never properly disclosed their financial difficulties, the alleged collective result of which was, among other things, that SALP and Thomvest were able to take control of the Company to the detriment of the minority shareholders.
The Plaintiffs seek almost $700 million in damages, approximately $664 million of which is based on the loss of future value of our shares.
The Company believes that the Plaintiffs’ claims are completely without merit and intends to vigorously defend itself. Defence and settlement costs associated with such lawsuits and claims can be substantial, even when these lawsuits and claims have no merit. Due to the inherent uncertainty of the litigation process, the resolution of any particular legal proceeding could have an adverse effect on the Company’s operating results or financial performance.
Liminal Former Employee Dispute
On June 6, 2019, a former employee of ours filed a suit in the United States District Court, District of Connecticut alleging claims for declaratory judgment, breach of contract, breach of the covenant of good faith and fair dealing, unjust enrichment, and violation of Conn. Gen. Stat. § 31-72 for unpaid wages and benefits, based upon the former employee’s employment contract and the terms of employee benefit programs. The matter has been resolved and the District of Connecticut action was dismissed with prejudice on August 13, 2020.
From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. Although the results of litigation and claims cannot be predicted with certainty, we currently believe that the final outcome of these ordinary course matters will not have a material adverse effect on our business. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
B.Significant Changes
Not applicable.
131
Item 9.The Offer and Listing.
A.Offer and Listing Details
On July 28, 1998, our common shares began trading on the TSX under the trading symbol “PLI” and, on October 7, 2019, began trading under the trading symbol “LMNL.” Prior to July 28, 1998, there was no public trading market for our common shares. On November 18, 2019, our common shares began trading on Nasdaq under the trading symbol “LMNL.” On August 5, 2020, we voluntarily delisted our common shares from the TSX.
B.Plan of Distribution.
Not applicable.
C.Markets.
On July 28, 1998, our common shares began trading on the TSX under the trading symbol “PLI” and, on October 7, 2019, began trading under the trading symbol “LMNL.” On November 18, 2019, our common shares began trading on Nasdaq under the trading symbol “LMNL.” On August 5, 2020, we voluntarily delisted our common shares from the TSX.
D.Selling Shareholders.
Not applicable.
E.Dilution.
Not applicable.
F.Expenses of the Issue.
Not applicable.
Item 10.Additional Information.
A.Share Capital.
Not applicable.
B.Memorandum and Articles of Association.
See Exhibit 2.1 to this Annual Report on Form 20-F for a summary of certain material provisions of our articles of incorporation, as amended, bylaws, as amended, and certain related sections of the Canada Business Corporations Act. See Exhibit 1.1 to this Annual Report on Form 20-F for our articles of incorporation, as amended, and Exhibits 1.2, 1.3 and 1.4 for our bylaws, as amended.
C.Material Contracts.
For information on our material contracts, please see “Item 4.—Information on the Company,” and “Item 7.B.—Related Party Transactions” of this Annual Report.
132
D.Exchange Controls.
Competition Act
Limitations on the ability to acquire and hold our common shares may be imposed by the Competition Act (Canada). This legislation establishes a pre-merger notification regime for certain types of merger transactions that exceed certain statutory shareholding and financial thresholds. Transactions that are subject to notification cannot be closed until the required materials are filed and the applicable statutory waiting period has expired or been waived by the Commissioner of Competition, or the Commissioner. Further, the Competition Act (Canada) permits the Commissioner to review any acquisition of control over or of a significant interest in us, whether or not it is subject to mandatory notification. This legislation grants the Commissioner jurisdiction, for up to one year, to challenge this type of acquisition before the Canadian Competition Tribunal if it would, or would be likely to, substantially prevent or lessen competition in any market in Canada.
Investment Canada Act
The Investment Canada Act requires notification and, in certain cases, advance review and approval by the Government of Canada of an investment to establish a new Canadian business by a non-Canadian or of the acquisition by a non-Canadian of “control” of a “Canadian business”, all as defined in the Investment Canada Act. Generally, the threshold for advance review and approval will be higher in monetary terms for a member of the World Trade Organization. The Investment Canada Act generally prohibits the implementation of such a reviewable transaction unless, after review, the relevant minister is satisfied that the investment is likely to be of net benefit to Canada.
The Investment Canada Act contains various rules to determine if there has been an acquisition of control. For example, for purposes of determining whether an investor has acquired control of a corporation by acquiring shares, the following general rules apply, subject to certain exceptions. The acquisition of a majority of the voting shares of a corporation is deemed to be acquisition of control of that corporation. The acquisition of less than a majority but one-third or more of the voting shares of a corporation is presumed to be an acquisition of control of that corporation unless it can be established that, on the acquisition, the corporation is not controlled in fact by the acquiror through the ownership of voting shares. The acquisition of less than one-third of the voting shares of a corporation is deemed not to be acquisition of control of that corporation.
In addition, under the Investment Canada Act, national security review on a discretionary basis may also be undertaken by the federal government in respect of a much broader range of investments by a non-Canadian to “acquire, in whole or in part, or to establish an entity carrying on all or any part of its operations in Canada, with the relevant test being whether such an investment by a non-Canadian could be “injurious to national security.” The Minister of Industry has broad discretion to determine whether an investor is a non-Canadian and therefore may be subject to national security review. Review on national security grounds is at the discretion of the federal government and may occur on a pre- or post-closing basis.
See “Item 10.E.—Taxation” for additional information regarding the material U.S. federal income tax consequences relating to the ownership and disposition of our common shares by U.S. Holders (as defined thereto).
Any of these provisions may discourage a potential acquirer from proposing or completing a transaction that may have otherwise presented a premium to our shareholders. We cannot predict whether investors will find our company and our common shares less attractive because we are governed by foreign laws.
133
E.Taxation.
Certain Material U.S. Federal Income Tax Considerations
The following discussion describes the material U.S. federal income tax consequences relating to the ownership and disposition of common shares by U.S. Holders (as defined below). This discussion applies to U.S. Holders that hold our common shares as capital assets. This discussion is based on the U.S. Internal Revenue Code of 1986, as amended, or the Code, U.S. Treasury regulations promulgated thereunder and administrative and judicial interpretations thereof, all as in effect on the date hereof and all of which are subject to change, possibly with retroactive effect. This discussion does not address all of the U.S. federal income tax consequences that may be relevant to specific U.S. Holders in light of their particular circumstances or to U.S. Holders subject to special treatment under U.S. federal income tax law (such as certain financial institutions, insurance companies, broker-dealers and traders in securities or other persons that generally mark their securities to market for U.S. federal income tax purposes, tax-exempt entities, retirement plans, regulated investment companies, real estate investment trusts, certain former citizens or residents of the United States, persons who hold our common shares as part of a “straddle”, “hedge”, “conversion transaction”, “synthetic security” or integrated investment, persons that have a “functional currency” other than the U.S. dollar, persons that own directly, indirectly or through attribution 10% or more of the voting power of our shares, corporations that accumulate earnings to avoid U.S. federal income tax, persons subject to special tax accounting rules under Section 451(b) of the Code, persons subject to special tax accounting rules under Section 451(b) of the Code, partnerships and other pass-through entities, and investors in such pass-through entities). This discussion does not address any U.S. state or local or non-U.S. tax consequences or any U.S. federal estate, gift or alternative minimum tax consequences.
As used in this discussion, the term “U.S. Holder” means a beneficial owner of our common shares that is, for U.S. federal income tax purposes, (1) an individual who is a citizen or resident of the United States, (2) a corporation (or entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof, or the District of Columbia, (3) an estate the income of which is subject to U.S. federal income tax regardless of its source or (4) a trust (x) with respect to which a court within the United States is able to exercise primary supervision over its administration and one or more United States persons have the authority to control all of its substantial decisions or (y) that has elected under applicable U.S. Treasury regulations to be treated as a domestic trust for U.S. federal income tax purposes.
If an entity treated as a partnership for U.S. federal income tax purposes holds our common shares, the U.S. federal income tax consequences relating to an investment in our common shares will depend in part upon the status and activities of such entity and the particular partner. Any such entity should consult its own tax advisor regarding the U.S. federal income tax consequences applicable to it and its partners of the purchase, ownership and disposition of our common shares. Persons considering an investment in our common shares should consult their own tax advisors as to the particular tax consequences applicable to them relating to the purchase, ownership and disposition of our common shares, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Passive Foreign Investment Company Consequences
In general, a corporation organized outside the United States will be treated as a passive foreign investment company, or PFIC, for any taxable year in which either (1) at least 75% of its gross income is “passive income”, or the “PFIC income test”, or (2) on average at least 50% of its assets, determined on a quarterly basis, are assets that produce passive income or are held for the production of passive income, the “PFIC asset test”. Passive income for this purpose generally includes, among other things, dividends, interest, royalties, rents, and gains from the sale or exchange of property that gives rise to passive income. Assets that produce or are held for the production of passive income generally include cash, even if held as working capital or raised in a public offering, marketable securities, and other assets that may produce passive income. Generally, in determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account.
134
Although we do not believe that we were a PFIC for the year ending December 31, 2020, our determination is based on an interpretation of complex provisions of the law, which are not addressed in a significant number of administrative pronouncements or rulings by the Internal Revenue Service. Accordingly, there can be no assurance that our conclusions regarding our status as a PFIC for the 2020 taxable year will not be challenged by the Internal Revenue Service and, if challenged, upheld in appropriate proceedings. In addition, because PFIC status is determined on an annual basis and generally cannot be determined until the end of the taxable year, there can be no assurance that we will not be a PFIC for the current taxable year. Because we may continue to hold a substantial amount of cash and cash equivalents, and because the calculation of the value of our assets may be based in part on the value of our common shares, which may fluctuate considerably, we may be a PFIC in future taxable years. Even if we determine that we are not a PFIC for a taxable year, there can be no assurance that the IRS will agree with our conclusion and that the IRS would not successfully challenge our position. Our status as a PFIC is a fact-intensive determination made on an annual basis. Accordingly, our U.S. counsel expresses no opinion with respect to our PFIC status and also expresses no opinion with regard to our expectations regarding our PFIC status.
If we are a PFIC in any taxable year during which a U.S. Holder owns our common shares, the U.S. Holder could be liable for additional taxes and interest charges under the “PFIC excess distribution regime” upon (1) a distribution paid during a taxable year that is greater than 125% of the average annual distributions paid in the three preceding taxable years, or, if shorter, the U.S. Holder’s holding period for our common shares, and (2) any gain recognized on a sale, exchange or other disposition, including a pledge, of our common shares, whether or not we continue to be a PFIC. Under the PFIC excess distribution regime, the tax on such distribution or gain would be determined by allocating the distribution or gain ratably over the U.S. Holder’s holding period for our common shares. The amount allocated to the current taxable year (i.e., the year in which the distribution occurs or the gain is recognized) and any year prior to the first taxable year in which we are a PFIC will be taxed as ordinary income earned in the current taxable year. The amount allocated to other taxable years will be taxed at the highest marginal rates in effect for individuals or corporations, as applicable, to ordinary income for each such taxable year, and an interest charge, generally applicable to underpayments of tax, will be added to the tax.
If we are a PFIC for any year during which a U.S. Holder holds our common shares, we must generally continue to be treated as a PFIC by that holder for all succeeding years during which the U.S. Holder holds our common shares, unless we cease to meet the requirements for PFIC status and the U.S. Holder makes a “deemed sale” election with respect to our common shares. If the election is made, the U.S. Holder will be deemed to sell our common shares it holds at their fair market value on the last day of the last taxable year in which we qualified as a PFIC, and any gain recognized from such deemed sale would be taxed under the PFIC excess distribution regime. After the deemed sale election, the U.S. Holder’s common shares would not be treated as shares of a PFIC unless we subsequently become a PFIC.
If we are a PFIC for any taxable year during which a U.S. Holder holds our common shares and one of our non-U.S. corporate subsidiaries is also a PFIC (i.e., a lower-tier PFIC), such U.S. Holder would be treated as owning a proportionate amount (by value) of the shares of the lower-tier PFIC and would be taxed under the PFIC excess distribution regime on distributions by the lower-tier PFIC and on gain from the disposition of shares of the lower-tier PFIC even though such U.S. Holder would not receive the proceeds of those distributions or dispositions. Each U.S. Holder is advised to consult its tax advisors regarding the application of the PFIC rules to our non-U.S. subsidiaries.
If we are a PFIC, a U.S. Holder will not be subject to tax under the PFIC excess distribution regime on distributions or gain recognized on our common shares if such U.S. Holder makes a valid “mark-to-market” election for our common shares. A mark-to-market election is available to a U.S. Holder only for “marketable stock”.
135
Our common shares will be marketable stock as long as they remain listed on the Nasdaq and are regularly traded, other than in de minimis quantities, on at least 15 days during each calendar quarter. If a mark-to-market election is in effect, a U.S. Holder generally would take into account, as ordinary income each year, the excess of the fair market value of our common shares held at the end of such taxable year over the adjusted tax basis of such common shares. The U.S. Holder would also take into account, as an ordinary loss each year, the excess of the adjusted tax basis of such our common shares over their fair market value at the end of the taxable year, but only to the extent of the excess of amounts previously included in income over ordinary losses deducted as a result of the mark-to-market election. The U.S. Holder’s tax basis in our common shares would be adjusted to reflect any income or loss recognized as a result of the mark-to-market election. Any gain from a sale, exchange or other disposition of our common shares in any taxable year in which we are a PFIC would be treated as ordinary income and any loss from such sale, exchange or other disposition would be treated first as ordinary loss (to the extent of any net mark-to-market gains previously included in income) and thereafter as capital loss.
A mark-to-market election will not apply to our common shares for any taxable year during which we are not a PFIC, but will remain in effect with respect to any subsequent taxable year in which we become a PFIC. Such election will not apply to any non-U.S. subsidiaries that we may organize or acquire in the future. Accordingly, a U.S. Holder may continue to be subject to tax under the PFIC excess distribution regime with respect to any lower-tier PFICs that we may organize or acquire in the future notwithstanding the U.S. Holder’s mark-to-market election for our common shares.
The tax consequences that would apply if we are a PFIC would also be different from those described above if a U.S. Holder were able to make a valid qualified electing fund, or QEF, election. At this time, we do not expect to provide U.S. Holders with the information necessary for a U.S. Holder to make a QEF election, prospective investors should assume that a QEF election will not be available.
Each U.S. person that is an investor of a PFIC is generally required to file an annual information return on IRS Form 8621 containing such information as the U.S. Treasury Department may require. The failure to file IRS Form 8621 could result in the imposition of penalties and the extension of the statute of limitations with respect to U.S. federal income tax.
The U.S. federal income tax rules relating to PFICs are very complex. Prospective U.S. investors are strongly urged to consult their own tax advisors with respect to the impact of PFIC status on the purchase, ownership and disposition of our common shares, the consequences to them of an investment in a PFIC, any elections available with respect to our common shares and the IRS information reporting obligations with respect to the purchase, ownership and disposition of the common shares of a PFIC.
Distributions
Subject to the discussion above under “Passive Foreign Investment Company Consequences”, a U.S. Holder that receives a distribution with respect to our common shares generally will be required to include the gross amount of such distribution in gross income as a dividend when actually or constructively received to the extent of the U.S. Holder’s pro rata share of our current and/or accumulated earnings and profits (as determined under U.S. federal income tax principles). To the extent a distribution received by a U.S. Holder is not a dividend because it exceeds the U.S. Holder’s pro rata share of our current and accumulated earnings and profits, it will be treated first as a tax-free return of capital and reduce (but not below zero) the adjusted tax basis of the U.S. Holder’s common shares. To the extent the distribution exceeds the adjusted tax basis of the U.S. Holder’s common shares, the remainder will be taxed as capital gain. Because we may not account for our earnings and profits in accordance with U.S. federal income tax principles, U.S. Holders should expect all distributions to be reported to them as dividends. Distributions on our common shares that are treated as dividends generally will constitute income from sources outside the United States for foreign tax credit purposes and generally will constitute passive category income. Such dividends will not be eligible for the “dividends received” deduction generally allowed to corporate shareholders with respect to dividends received from U.S. corporations.
136
Dividends paid by a “qualified foreign corporation” are eligible for taxation for certain non-corporate U.S. Holders at a reduced capital gains rate rather than the marginal tax rates generally applicable to ordinary income provided that certain requirements are met. However, if we are a PFIC for the taxable year in which the dividend is paid or the preceding taxable year (see discussion above under “Passive Foreign Investment Company Consequences”), we will not be treated as a qualified foreign corporation, and therefore the reduced capital gains tax rate described above will not apply. Each U.S. Holder is advised to consult its tax advisors regarding the availability of the reduced tax rate on dividends with regard to its particular circumstances.
A non-United States corporation (other than a corporation that is classified as a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation (a) if it is eligible for the benefits of a comprehensive tax treaty with the United States which the Secretary of Treasury of the United States determines is satisfactory for purposes of this provision and which includes an exchange of information provision, or (b) with respect to any dividend it pays on our common shares that are readily tradable on an established securities market in the United States. We believe that we qualify as a resident of Canada for purposes of, and are eligible for the benefits of, the U.S.-Canada Treaty, although there can be no assurance in this regard. Further, the IRS has determined that the U.S.-Canada Treaty is satisfactory for purposes of the qualified dividend rules and that it includes an exchange of information provision. Therefore, subject to the discussion above under “Passive Foreign Investment Company Consequences”, if the U.S.-Canada Treaty is applicable, such dividends will generally be “qualified dividend income” in the hands of individual U.S. Holders, provided that certain conditions are met, including holding period and the absence of certain risk reduction transactions.
Sale, Exchange or Other Disposition of our common shares
Subject to the discussion above under “Passive Foreign Investment Company Consequences”, a U.S. Holder generally will recognize capital gain or loss for U.S. federal income tax purposes upon the sale, exchange or other disposition of our common shares in an amount equal to the difference, if any, between the amount realized (i.e., the amount of cash plus the fair market value of any property received) on the sale, exchange or other disposition and such U.S. Holder’s adjusted tax basis in our common shares. Such capital gain or loss generally will be long-term capital gain taxable at a reduced rate for noncorporate U.S. Holders or long-term capital loss if, on the date of sale, exchange or other disposition, our common shares were held by the U.S. Holder for more than one year. Any capital gain of a non-corporate U.S. Holder that is not long-term capital gain is taxed at ordinary income rates. The deductibility of capital losses is subject to limitations. Any gain or loss recognized from the sale or other disposition of our common shares will generally be gain or loss from sources within the United States for U.S. foreign tax credit purposes.
Medicare Tax
Certain U.S. Holders that are individuals, estates or trusts and whose income exceeds certain thresholds generally are subject to a 3.8% tax on all or a portion of their net investment income, which may include their gross dividend income and net gains from the disposition of our common shares. If you are a United States person that is an individual, estate or trust, you are encouraged to consult your tax advisors regarding the applicability of this Medicare tax to your income and gains in respect of your investment in our common shares.
Information Reporting and Backup Withholding
U.S. Holders may be required to file certain U.S. information reporting returns with the IRS with respect to an investment in our common shares, including, among others, IRS Form 8938 (Statement of Specified Foreign Financial Assets). As described above under “Passive Foreign Investment Company Consequences”, each U.S. Holder who is a shareholder of a PFIC must file an annual report containing certain information. U.S. Holders paying more than US$100,000 for our common shares may be required to file IRS Form 926 (Return by a U.S. Transferor of Property to a Foreign Corporation) reporting this payment. Substantial penalties may be imposed upon a U.S. Holder that fails to comply with the required information reporting.
137
Dividends on and proceeds from the sale or other disposition of our common shares may be reported to the IRS unless the U.S. Holder establishes a basis for exemption. Backup withholding may apply to amounts subject to reporting if the holder (1) fails to provide an accurate United States taxpayer identification number or otherwise establish a basis for exemption, or (2) is described in certain other categories of persons. However, U.S. Holders that are corporations generally are excluded from these information reporting and backup withholding tax rules. Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules generally will be allowed as a refund or a credit against a U.S. Holder’s U.S. federal income tax liability if the required information is furnished by the U.S. Holder on a timely basis to the IRS.
U.S. Holders should consult their own tax advisors regarding the backup withholding tax and information reporting rules.
Certain Canadian Federal Income Tax Considerations
The following summary describes, as of the date hereof, the material Canadian federal income tax considerations generally applicable to shareholders who are a beneficial owner of our common shares and who, at all relevant times, for the purposes of the application of the Income Tax Act (Canada) and the Income Tax Regulations (collectively, the “Canadian Tax Act”), (1) is not, and is not deemed to be, resident in Canada for purposes of the Canadian Tax Act and any applicable income tax treaty or convention; (2) deals at arm’s length with us; (3) is not affiliated with us; (4) does not use or hold, and is not deemed to use or hold, common shares in a business or part of a business carried on in Canada; (5) has not entered into, with respect to the common shares, a “derivative forward agreement”, as that term is defined in the Canadian Tax Act and (6) holds the common shares as capital property (a “Non-Canadian Holder”). This summary does not apply to a Non-Canadian Holder that is an insurer carrying on an insurance business in Canada and elsewhere or an “authorized foreign bank”, as that term is defined in the Canadian Tax Act. Such Non‑Canadian Holders should consult their tax advisors for advice having regards to their particular circumstances.
This summary is based on the current provisions of the Canadian Tax Act, and an understanding of the current administrative policies of the Canada Revenue Agency published in writing prior to the date hereof. It takes into account all specific proposals to amend the Canadian Tax Act and the Canada-United States Tax Convention (1980), as amended (the “Canada-U.S. Tax Treaty”), publicly announced by or on behalf of the Minister of Finance (Canada) prior to the date hereof (the “Proposed Amendments”) and assumes that all Proposed Amendments will be enacted in the form proposed. However, no assurances can be given that the Proposed Amendments will be enacted as proposed, or at all. This summary does not otherwise take into account or anticipate any changes in law or administrative policy or assessing practice whether by legislative, regulatory, administrative or judicial action nor does it take into account tax legislation or considerations of any province, territory or foreign jurisdiction, which may differ from those discussed herein.
This summary is of a general nature only and is not, and is not intended to be, legal or tax advice to any particular shareholder, and no representations with respect to the income tax consequences to any particular shareholder are made. This summary is not exhaustive of all Canadian federal income tax considerations. Accordingly, you should consult your own tax advisor with respect to your particular circumstances.
Generally, for purposes of the Canadian Tax Act, all amounts relating to the acquisition, holding or disposition of the common shares must be converted into Canadian dollars based on the exchange rates as determined in accordance with the Canadian Tax Act. The amount of any dividends, capital gains or capital losses realized by a Non-Canadian Holder may be affected by fluctuations in the Canadian exchange rate.
138
Dividends
Dividends paid or credited on the common shares or deemed to be paid or credited on the common shares to a Non-Canadian Holder will be subject to Canadian withholding tax at the rate of 25%, subject to any reduction in the rate of withholding to which the Non-Canadian Holder is entitled under any applicable income tax treaty or convention between Canada and the country in which the Non-Canadian Holder is resident. For example, under the Canada-U.S. Tax Treaty, where dividends on the common shares are considered to be paid to or derived by a Non-Canadian Holder that is a beneficial owner of the dividends and is a U.S. resident for the purposes of, and is entitled to benefits of, the Canada-U.S. Tax Treaty, the applicable rate of Canadian withholding tax is generally reduced to 15%. We will be required to withhold the applicable withholding tax from any dividend and remit it to the Canadian government for the Non-Canadian Holder’s account.
Dispositions
A Non-Canadian Holder will not be subject to tax under the Canadian Tax Act on any capital gain realized on a disposition or deemed disposition of a common share, unless the common shares are “taxable Canadian property” to the Non-Canadian Holder for purposes of the Canadian Tax Act at the time of disposition and the Non-Canadian Holder is not entitled to relief under an applicable income tax treaty or convention between Canada and the country in which the Non-Canadian Holder is resident.
Generally, the common shares will not constitute “taxable Canadian property” to a Non-Canadian Holder at a particular time provided that the common shares are listed at that time on a “designated stock exchange” (as defined in the Canadian Tax Act), which includes the TSX and the Nasdaq, unless at any particular time during the 60-month period that ends at that time.
| • | at least 25% of the issued shares of any class or series of our capital stock was owned by or belonged to any combination of (a) the Non-Canadian Holder, (b) persons with whom the Non-Canadian Holder does not deal at arm’s length, and (c) partnerships in which the Non-Canadian Holder or a person described in (b) holds a membership interest directly or indirectly through one or more partnerships, and |
| • | more than 50% of the fair market value of the common shares was derived, directly or indirectly, from one or any combination of : (i) real or immoveable property situated in Canada, (ii) “Canadian resource properties” (as that term is defined in the Canadian Tax Act), (iii) “timber resource properties” (as that term is defined in the Canadian Tax Act) and (iv) options in respect of, or interests in, or for civil law rights in, property in any of the foregoing whether or not the property exists. |
Notwithstanding the foregoing, in certain circumstances, common shares could be deemed to be “taxable Canadian property.” Non-Canadian Holders whose common shares may constitute “taxable Canadian property” should consult their own tax advisors.
F.Dividends and Paying Agents.
Not applicable.
G.Statement by Experts.
Not applicable.
139
H.Documents on Display.
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and under those requirements will file reports with the SEC. Those reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an Annual Report on Form 20-F containing financial statements that have been examined and reported on, with and opinion expressed by an independent registered public accounting firm.
We maintain a corporate website at www.liminalbiosciences.com. We intend to post our Annual Report on Form 20-F on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report. We have included our website address in this Annual Report solely as an inactive textual reference.
The SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as us, that file electronically with the SEC.
With respect to references made in this Annual Report to any contract or other document of our company, such references are not necessarily complete and you should refer to the exhibits attached or incorporated by reference to this Annual Report for copies of the actual contract or document.
I.Subsidiary Information
Not required.
Item 11.Quantitative and Qualitative Disclosures About Market Risk.
Financial risk management
We have exposure to credit risk, liquidity risk and market risk. Our Board of Directors has the overall responsibility for the oversight of these risks and reviews our policies on an ongoing basis to ensure that these risks are appropriately managed.
i) Credit risk:
Credit risk is the risk of financial loss to our company if a customer, partner or counterparty to a financial instrument fails to meet its contractual obligations, and arises principally from the Company’s cash, investments, receivables and share purchase loan to a former officer. The carrying amount of the financial assets represents the maximum credit exposure.
We mitigate credit risk through its reviews of new customer’s credit history before extending credit and conducts regular reviews of its existing customers’ credit performance. We evaluate at each reporting period, the lifetime expected credit losses on our accounts receivable balances based on the age of the receivable, credit history of the customers and past collection experience.
ii) Liquidity risk:
Liquidity risk is the risk that we will not be able to meet financial obligations as they come due. We manage our liquidity risk by continuously monitoring forecasts and actual cash flows. Our current liquidity situation is discussed in the liquidity and contractual obligation section of this MD&A.
iii) Market risk:
Market risk is the risk that changes in market prices, such as interest rates and foreign exchange rates, will affect our income or the value of its financial instruments.
140
a) Interest risk:
Our interest-bearing financial liabilities have fixed rates and as such there is limited exposure to changes in interest payments as a result of interest rate risk.
b) Foreign exchange risk:
We are exposed to the financial risk related to the fluctuation of foreign exchange rates. We operate in the U.S. and the U.K., and previously had operations in the Isle of Man (discontinued operations) and a portion of our expenses incurred are in U.S. dollars and in pounds sterling (£). Historically, the majority of our revenues have been in U.S. dollars and in £, continuing operations revenues are in U.S. dollars, which serve to mitigate a portion of the foreign exchange risk relating to the expenditures. Financial instruments that have exposed us to foreign exchange risk have been cash and cash equivalents, short-term investments, receivables, trade and other payables, lease liabilities, licence payment obligations and the amounts drawn on the credit facility. We manage foreign exchange risk by holding foreign currencies we received to support forecasted cash outflows in foreign currencies.
Item 12.Description of Securities Other than Equity Securities.
A.Debt Securities.
Not applicable.
B.Warrants and Rights.
Not applicable.
C.Other Securities.
Not applicable.
D.American Depositary Shares.
Not applicable.
PART II
Item 13.Defaults, Dividend Arrearages and Delinquencies.
Not applicable.
Item 14.Material Modifications to the Rights of Security Holders and Use of Proceeds.
Not applicable.
Item 15.Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our chief executive officer and chief financial officer, after evaluating the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) of the Exchange Act) as of December 31, 2020, have concluded that, as of such date, our disclosure controls and procedures were effective and ensured that information required to be disclosed by us in reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our chief executive officer and chief financial officer, to allow timely decisions regarding required disclosure and is recorded, processed, summarized and reported within the time periods specified by the SEC’s rules and forms.
141
Our disclosure controls and procedures are designed to provide reasonable assurance of achieving the desired control objectives. Our management recognizes that any control system, no matter how well designed and operated, is based upon certain judgments and assumptions and cannot provide absolute assurance that its objectives will be met. Similarly, an evaluation of controls cannot provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, have been detected.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal controls over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) and for the assessment of the effectiveness of our internal control over financial reporting. Under the supervision and with the participation of our chief executive officer (principal executive officer) and deputy chief executive officer (principal financial officer), management assessed our internal control over financial reporting based upon the framework in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this assessment, our management has concluded that our internal control over financial reporting was effective as of December 31, 2020.
Attestation Report of the Registered Public Accounting Firm
Not applicable to emerging growth companies.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the period covered by this annual report that have materially affected, or that are reasonably likely to materially affect, our internal control over financial reporting.
Item 16.Reserved.
Not applicable.
Item 16A.Audit Committee Financial Expert.
Our board of directors has determined that all the members of our audit, risk and finance committee qualify as “audit committee financial experts” as defined by SEC rules and have the requisite financial sophistication under the applicable rules and regulations of the Nasdaq. All of the members of the audit, risk and finance committee are independent as such term is defined in Rule 10A-3 under the Exchange Act and under the listing standards of the Nasdaq Stock Market.
Item 16B.Code of Ethics.
We have adopted a Code of Ethics and Business Conduct, or the Code of Conduct, that is applicable to all of our employees, executive officers and directors. A copy of the Code of Conduct is available on our website at www.liminalbiosciences.com.
142
Item 16C.Principal Accountant Fees and Services.
PricewaterhouseCoopers LLP served as our independent registered public accounting firm for the 2019 audit and 2020 under PCAOB standards. Our accountant billed the following fees to us for professional services in each of those fiscal years:
| | Year Ended December 31, |
| | (thousands of Canadian dollars) |
| | PricewaterhouseCoopers LLP |
| | 2020 | 2019 |
Audit Fees | | $ | | 660,000 | | | $ | | 770,000 | | (1) |
Audit-Related Fees | | | | 94,908 | | | | | 153,730 | | |
Tax Fees | | | - | | | | - | | |
Other Fees | | | | 62,000 | | | | | 20,800 | | |
Total | | $ | | 816,908 | | | $ | | 944,530 | | |
___________________
(1) | These fees include $325,000 related to the audit of our financial statements for the year ended December 31, 2018 under PCAOB standards. |
“Audit Fees” are the aggregate fees billed for the audit of our annual financial statements. This category also includes services that only our independent external auditor can reasonably provide, such as consents and assistance with and review of documents filed with the SEC.
“Audit-Related Fees” are the aggregate fees billed for assurance and related services that are reasonably related to the performance of the audit and are not reported under Audit Fees.
“All Tax Fees” consist of tax consultations, such as advice in connection with employees’ taxation arising from share-based compensation.
“Other Fees” consist of advisory services relating to the adoption or application of IFRS and translation services.
Audit and Non-Audit Services Pre-Approval Policy
To ensure the independence and objectivity of our external auditors, the provision of all non-audit services by the external auditors are pre-approved by our audit, risk and finance committee, other than non-audit services: (i) that were not recognized as non-audit services at the time of the engagement, (ii) that are promptly brought to the attention of our audit, risk and finance committee and approved, prior to the completion of the audit, by our audit, risk and finance committee or by one or more of its members to whom authority to grant such approvals has been delegated by the audit, risk and finance committee, and (iii) the aggregate amount of which is reasonably expected to constitute no more than 5% of the total amount of fees paid by us to out external auditor during the fiscal year in which the services are provided.
Item 16D.Exemptions from the Listing Standards for Audit Committees.
Not applicable.
Item 16E.Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
Not applicable.
Item 16F.Change in Registrant’s Certifying Accountant.
Not applicable.
Item 16G.Corporate Governance.
As a “foreign private issuer,” as defined by the SEC, we are permitted to follow home country corporate governance practices, instead of certain corporate governance practices required by the Nasdaq for U.S. domestic issuers.
143
While we intend to follow most Nasdaq corporate governance listing standards, we intend to follow Canadian corporate governance practices of in lieu of Nasdaq corporate governance listing standards as follows:
| • | Exemption from quorum requirements applicable to meetings of shareholders. Such quorum requirements are not required under Canadian law. In accordance with generally accepted business practice, our by-laws provide alternative quorum requirements that are generally applicable to meetings of shareholders. |
| • | Exemption from the requirement to obtain shareholder approval for certain issuances of securities pursuant to Nasdaq Rule 5635, including in connection with an acquisition of the stock or assets of another company. |
| • | Exemption from requirement to adopt a formal written audit committee charter specifying the items enumerated in Nasdaq Rule 5605(c)(1). Under Nasdaq Rule 5605(c)(1), an audit committee must be directly responsible for the appointment, compensation, retention and oversight of the work of any registered public accounting firm engaged. Our audit committee charter provides that the auditors be appointed by, and their compensation be approved by, our board of directors and be ratified by our shareholders, as required under Canadian law. |
Although we currently intend to comply with the Nasdaq corporate governance rules applicable other than as noted above, we may in the future decide to use the foreign private issuer exemption with respect to some or all the other Nasdaq corporate governance rules.
We intend to take all actions necessary for us to maintain compliance as a foreign private issuer under the applicable corporate governance requirements of the Sarbanes-Oxley Act, the rules adopted by the SEC and the Nasdaq corporate governance rules and listing standards.
As a foreign private issuer, our directors and senior management are not subject to short-swing profit and insider trading reporting obligations under Section 16 of the Exchange Act. They will, however, be subject to the obligations to report changes in share ownership under Section 13 of the Exchange Act and related SEC rules.
Item 16H.Mine Safety Disclosure.
Not applicable.
PART III
Item 17.Financial Statements.
See pages F-1 through F-71 of this Annual Report on Form 20-F.
Item 18.Financial Statements.
Not applicable.
144
Item 19.Exhibits.
| | | | | | | | | | |
| | | | Incorporated by Reference |
Exhibit | | Description | | Schedule/ Form | | File Number | | Exhibit | | File Date |
1.1 | | Articles of Incorporation of Liminal BioSciences Inc., as amended, as currently in effect. | | Form S-8 | | 333-235692 | | 4.1 | | 12/23/2019 |
1.2 | | By-Law No. 1 of Liminal BioSciences Inc., as amended and currently in effect. | | Form S-8 | | 333-235692 | | 4.2 | | 12/23/2019 |
1.3 | | By-Law No. 2 of Liminal BioSciences Inc., as currently in effect. | | Form S-8 | | 333-235692 | | 4.3 | | 12/23/2019 |
1.4 | | By-Law No. 3 of Liminal BioSciences Inc., as currently in effect. | | Form S-8 | | 333-235692 | | 4.4 | | 12/23/2019 |
2.1* | | Description of Securities | | | | | | | | |
4.1 | | Registration Rights Agreement, dated April 23, 2019 | | Form 40-F | | 001-39131 | | 99.69 | | 11/12/2019 |
4.2 | | Amendment and Waiver to Registration Right Agreement, dated March 17, 2020 | | Form 20-F | | 001-39131 | | 4.2 | | 03/18/2020 |
4.3† | | Amended and Restated Stock Option Plan, dated May 10, 2017 | | Form 40-F | | 001-39131 | | 99.59 | | 11/12/2019 |
4.4† | | Amended and Restated Stock Option Plan, dated June 7, 2018 | | Form 40-F | | 001-39131 | | 99.61 | | 11/12/2019 |
4.5† | | Restricted Share Unit Plan, dated May 6, 2009 | | Form 40-F | | 001-39131 | | 99.60 | | 11/12/2019 |
4.6† | | Restricted Share Unit Plan, dated May 9, 2018 | | Form 40-F | | 001-39131 | | 99.62 | | 11/12/2019 |
4.7† | | Omnibus Incentive Plan, dated May 7, 2019 | | Form 40-F | | 001-39131 | | 99.125 | | 11/12/2019 |
4.8† | | Spin-Off Shareholder Rights Plan Agreement between Prometic Life Sciences Inc., Computershare Trust Company of Canada, Prometic Biosciences Inc., Prometic Bioproduction Inc. and Prometic Biotherapeutics Inc., dated March 22, 2018 | | Form 40-F | | 001-39131 | | 99.64 | | 11/12/2019 |
4.9† | | Shareholder Rights Plan Agreement between Prometic Life Sciences Inc. and Computershare Trust Company of Canada, dated March 22, 2018 | | Form 40-F | | 001-39131 | | 99.65 | | 11/12/2019 |
4.10 | | Third Omnibus Amendment Agreement between Structured Alpha LP, Prometic Life Sciences Inc., Prometic Biotherapeutics Inc., Prometic Bioseparations Ltd, Prometic Biosciences Inc., Prometic Bioproduction Inc., NantPro Biosciences, LLC, Prometic Plasma Resources Inc., Prometic Pharma SMT Holdings Limited, Prometic Pharma SMT Limited, Prometic Biotherapeutics Ltd, Telesta Therapeutics Inc. and Prometic Plasma Resources (USA) Inc., dated November 14, 2018 | | Form 40-F | | 001-39131 | | 99.66 | | 11/12/2019 |
4.11 | | Board Observation Rights and Director Nomination Agreement, dated April 23, 2019 | | Form 40-F | | 001-39131 | | 99.68 | | 11/12/2019 |
4.12 | | Restructuring Agreement between Structured Alpha LP, Prometic Life Sciences Inc., Prometic Biotherapeutics Inc., Prometic Bioseparations Ltd, Prometic Biosciences Inc., Prometic Bioproduction Inc., NantPro Biosciences, LLC, Prometic Plasma Resources Inc., Prometic Pharma SMT Holdings Limited, Prometic Pharma SMT Limited, Prometic Biotherapeutics Ltd, Telesta Therapeutics Inc. and Prometic Plasma Resources (USA) Inc., dated April 15, 2019 | | Form 40-F | | 001-39131 | | 99.70 | | 11/12/2019 |
4.13 | | Private Placement Subscription Agreement, dated April 15, 2019 | | Form 40-F | | 001-39131 | | 99.71 | | 11/12/2019 |
| | | | | | | | | | |
4.14 | | Consolidated Loan Agreement between Structured Alpha LP, Prometic Life Sciences Inc., Prometic Biotherapeutics Inc., Prometic Bioseparations Ltd, Prometic Biosciences Inc., Prometic Bioproduction Inc., NantPro Biosciences, LLC, Prometic Plasma Resources Inc., Prometic Pharma SMT Holdings Limited, Prometic Pharma SMT Limited, Prometic Biotherapeutics Ltd, Telesta Therapeutics Inc. and Prometic Plasma Resources (USA) Inc., dated April 23, 2019 | | Form 40-F | | 001-39131 | | 99.72 | | 11/12/2019 |
4.15 | | Share Purchase Agreement, among Liminal BioSciences Inc. and Gamma Biosciences GP LLC, dated November 3, 2019 | | Form 6-K | | 001-39131 | | 99.1 | | 11/18/2019 |
4.16 | | First Amendment to the Consolidated Loan Agreement between Structured Alpha LP, Liminal BioSciences Inc., Prometic BioTherapeutics Inc., Liminal R&D BioSciences Inc., Prometic Bioproduction Inc., NantPro Biosciences, LLC, Prometic Plasma Resources Inc., Liminal BioSciences Holdings Limited, Liminal BioSciences Limited, Prometic Biotherapeutics Ltd, Telesta Therapeutics Inc. and Prometic Plasma Resources (USA) Inc., dated November 11, 2019 | | Form 6-K | | 001-39131 | | 99.5 | | 11/18/2019 |
4.17# | | Assignment of rights entered into in the City of Montreal, province of Quebec, among Prometic Biosciences Inc., Innovon Pharmaceuticals Inc., Pierre Laurin and Prometic Life Sciences Inc., dated October 17, 2001 | | Form 20-F | | 001-39131 | | 4.18 | | 03/20/2020 |
4.18# | | Assignment Agreement, among Prometic Biosciences Inc., Innovon Pharmaceuticals Inc., Pierre Laurin and Prometic Life Sciences Inc., dated May 23, 2012 | | Form 20-F/A | | 001-39131 | | 4.19 | | 08/04/2020 |
4.19# | | Amendment to Entente PBI-1101, PBI–1402, among Prometic Biosciences Inc., Innovon Pharmaceuticals Inc., Pierre Laurin and Prometic Life Sciences Inc., dated December 14, 2015 | | Form 20-F/A | | 001-39131 | | 4.20 | | 08/04/2020 |
4.20 | | Consent and Acknowledgment, among Prometic Biosciences Inc., Innovon Pharmaceuticals Inc., Pierre Laurin and Prometic Pharma SMT Limited, dated December 14, 2015 | | Form 20-F/A | | 001-39131 | | 4.21 | | 08/04/2020 |
4.21# | | Plasma Purchase Agreement between Prometic Plasma Resources Inc. and Interstate Blood Bank, Inc., dated November 6, 2015 | | Form 20-F/A | | 001-39131 | | 4.22 | | 08/04/2020 |
4.22# | | Amendment No. 1 to Plasma Purchase Agreement between Prometic Plasma Resources Inc. and Interstate Blood Bank, Inc., dated February 8, 2019 | | Form 20-F/A | | 001-39131 | | 4.23 | | 08/04/2020 |
4.23# | | Amended and Restated License Agreement PPPS Process between Prometic Life Sciences Inc. and Hematech Biotherapeutics Inc., dated May 7, 2012 | | Form 20-F | | 001-39131 | | 4.23 | | 03/20/2020 |
4.24# | | Amendment No. 1 to Amended and Restated License Agreement PPPS Process between Prometic Life Sciences Inc. and Hematech Biotherapeutics Inc., dated December 16, 2016 | | Form 20-F/A | | 001-39131 | | 4.25 | | 08/04/2020 |
4.25# | | Amended and Restated Contract Manufacturing Agreement between Prometic Life Sciences Inc. and Hematech Biotherapeutics Inc., dated May 7, 2012 | | Form 20-F/A | | 001-39131 | | 4.33 | | 08/04/2020 |
4.26#* | | Letter Agreement between Liminal BioSciences Inc. and Hematech Biotherapeutics Inc., dated February 17, 2021 | | | | | | | | |
4.27# | | Master Services Agreement between Prometic Life Sciences Inc. and Cangene Corporation doing business as Emergent BioSolutions, dated May 15, 2015 | | Form 20-F | | 001-39131 | | 4.25 | | 03/20/2020 |
4.28# | | Royalty Stream Purchase Agreement, among Prometic Life Sciences Inc., Prometic Pharma SMT Limited, Prometic Biosciences Inc. and each of their affiliates and Structured Alpha LP, dated April 30, 2018 | | Form 20-F | | 001-39131 | | 4.26 | | 03/20/2020 |
| | | | | | | | | | |
4.29# | | Absorbent Supply Agreement, among Prometic Bioseparations Ltd and Prometic Bioproduction Inc., dated June 18, 2019 | | Form 20-F/A | | 001-39131 | | 4.28 | | 08/04/2020 |
4.30# | | Plasma Purchase Agreement between Prometic Plasma Resources Inc. and Biotest Pharmaceuticals Corporation, dated June 26, 2017 | | Form 20-F/A | | 001-39131 | | 4.29 | | 08/04/2020 |
4.31 | | Notice of Assignment of Plasma Purchase Agreement, between Biotest Pharmaceuticals Corporation and Grifols Worldwide Operations Limited, dated December 10, 2018 | | Form 20-F/A | | 001-39131 | | 4.30 | | 08/04/2020 |
4.32# | | Amendment No. 1 to Plasma Purchase Agreement between Prometic Plasma Resources Inc. and Grifols Worldwide Operations Limited, dated December 31, 2018 | | Form 20-F/A | | 001-39131 | | 4.31 | | 08/04/2020 |
4.33# | | Amendment No. 2 to Plasma Purchase Agreement between Prometic Plasma Resources Inc. and Grifols Worldwide Operations Limited, dated November 11, 2019 | | Form 20-F/A | | 001-39131 | | 4.32 | | 08/04/2020 |
4.34# | | Share Purchase Agreement, dated as of July 17, 2020, among the Registrant and the investors listed therein. | | Form F-1/A | | 001-39131 | | 4.6 | | 09/14/2020 |
4.35# | | Form of Secured Convertible Debenture, dated as of July 17, 2020, issued by the Registrant | | Form F-1/A | | 001-39131 | | 4.7 | | 09/14/2020 |
4.36#* | | Amended and Restated License Agreement between Fairhaven Pharmaceuticals Inc., The Royal Institution of the Advancement of Learning/McGill University and Florida Institute of Technology, Inc., dated May 16, 2018 | | | | | | | | |
4.37 | | Securities Purchase Agreement dated October 29, 2020, by and among the Company and the Purchasers | | Form 6-K | | 001-39131 | | 99.1 | | 11/02/2020 |
4.38 | | Amendment No.1 to Securities Purchase Agreement dated November 25, 2020, by and among the company and the Purchasers | | Form 6-K | | 001-39131 | | 99.1 | | 11/27/2020 |
4.39 | | Registration Rights Agreement, dated October 29, 2020, by and among the Company and the Purchasers | | Form 6-K | | 001-39131 | | 99.2 | | 11/02/2020 |
4.40 | | Form of Warrant to Purchase Common Shares | | Form 6-K | | 001-39131 | | 99.3 | | 11/02/2020 |
8.1* | | List of subsidiaries of the Registrant | | | | | | | | |
12.1* | | Certification by the Principal Executive Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | | | | | | | | |
12.2* | | Certification by the Principal Financial Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | | | | | | | | |
13.1** | | Certification by the Principal Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | | | | | | | | |
13.2** | | Certification by the Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | | | �� | | | | | |
15.1* | | Consent of PricewaterhouseCoopers LLP | | | | | | | | |
101.INS* | | XBRL Instance Document | | | | | | | | |
101.SCH* | | XBRL Taxonomy Extension Schema Document | | | | | | | | |
101.CAL* | | XBRL Taxonomy Extension Calculation Linkbase Document | | | | | | | | |
101.LAB* | | XBRL Taxonomy Extension Label Linkbase Document | | | | | | | | |
| | | | | | | | | | |
101.PRE* | | XBRL Taxonomy Extension Presentation Linkbase Document | | | | | | | | |
101.DEF* | | XBRL Taxonomy Extension Definition Linkbase Document | | | | | | | | |
| | |
* | Filed herewith. |
** | Furnished herewith. |
† | Indicates a management contract or any compensatory plan, contract or arrangement. |
# | Certain portions of this exhibit (indicated by asterisks) have been omitted because they are not material and would likely cause competitive harm to Liminal BioSciences Inc. if publicly disclosed. |

Report of Independent Registered Public Accounting Firm
To the Board of Directors and the Shareholders of Liminal BioSciences Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated statements of financial position of Liminal BioSciences Inc. and its subsidiaries (together, the Company) as of December 31, 2020 and 2019, and the related consolidated statements of operations, comprehensive loss, changes in equity and cash flows for the each of the three years in the period ended December 31, 2020, including the related notes (collectively referred to as the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2020 and 2019, and its financial performance and its cash flows for each of the three years in the period ended December 31, 2020 in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Substantial Doubt About the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in note 1 to the consolidated financial statements, the Company has suffered recurring losses from operations and has a net deficit, which together raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Change in Accounting Principle
As discussed in Note 4 to the consolidated financial statements, the Company changed the manner in which it accounts for leases in 2019.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
F-1
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ PricewaterhouseCoopers LLP
Montréal, Quebec, Canada
March 24, 2021
We have served as the Company’s auditor since 2019.
F-2
LIMINAL BIOSCIENCES INC.
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION
(In thousands of Canadian dollars)
At December 31 | | 2020 | | | 2019 | |
| | | | | | | | |
ASSETS (note 16) | | | | | | | | |
Current assets | | | | | | | | |
Cash and cash equivalents | | $ | 45,075 | | | $ | 61,285 | |
Accounts receivable and others (note 7) | | | 4,081 | | | | 4,086 | |
Income tax receivable | | | — | | | | 9,214 | |
Inventories (note 8) | | | 9,377 | | | | 7,532 | |
Prepaids | | | 14,486 | | | | 12,733 | |
Total current assets | | | 73,019 | | | | 94,850 | |
| | | | | | | | |
Other long-term assets (note 9) | | | 1,353 | | | | 1,170 | |
Capital assets (note 10) | | | 18,791 | | | | 21,471 | |
Right-of-use assets (note 11) | | | 8,557 | | | | 33,254 | |
Intangible assets (note 12) | | | 15,492 | | | | 13,846 | |
Deferred tax assets (note 26) | | | 572 | | | | 507 | |
Total assets | | $ | 117,784 | | | $ | 165,098 | |
| | | | | | | | |
LIABILITIES | | | | | | | | |
Current liabilities | | | | | | | | |
Accounts payable and accrued liabilities (note 13) | | $ | 16,835 | | | $ | 22,808 | |
Current portion of lease liabilities (note 14) | | | 6,946 | | | | 8,290 | |
Current portion of long-term debt (note 16) | | | — | | | | 165 | |
Total current liabilities | | | 23,781 | | | | 31,263 | |
| | | | | | | | |
Long-term portion of lease liabilities (note 14) | | | 26,506 | | | | 29,947 | |
Long-term warrant liability (note 15) | | | 11,640 | | | | — | |
Long-term debt (note 16) | | | 40,532 | | | | 8,669 | |
Other long-term liabilities (note 17) | | | 313 | | | | 285 | |
Total liabilities | | $ | 102,772 | | | $ | 70,164 | |
| | | | | | | | |
EQUITY | | | | | | | | |
Share capital (note 18a) | | $ | 977,261 | | | $ | 932,951 | |
Contributed surplus (note 18b) | | | 39,877 | | | | 43,532 | |
Warrants (note 18c) | | | 95,856 | | | | 95,856 | |
Accumulated other comprehensive loss | | | (2,846 | ) | | | (3,099 | ) |
Deficit | | | (1,087,049 | ) | | | (967,051 | ) |
Equity attributable to owners of the parent | | | 23,099 | | | | 102,189 | |
Non-controlling interests (note 19) | | | (8,087 | ) | | | (7,255 | ) |
Total equity | | | 15,012 | | | | 94,934 | |
Total liabilities and equity | | $ | 117,784 | | | $ | 165,098 | |
Going concern (note 1), Commitments (note 31), Subsequent event (note 33)
The accompanying notes are an integral part of the consolidated financial statements.
F-3
LIMINAL BIOSCIENCES INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands of Canadian dollars except for per share amounts)
Years ended December 31 | | 2020 | | | 2019 | | | 2018 | |
Revenues (note 21) | | $ | 3,317 | | | $ | 4,904 | | | $ | 24,633 | |
| | | | | | | | | | | | |
Expenses | | | | | | | | | | | | |
Cost of sales and other production expenses (note 8) | | | 2,033 | | | | 2,763 | | | | 25,707 | |
Research and development expenses (note 22a) | | | 56,826 | | | | 75,114 | | | | 84,858 | |
Administration, selling and marketing expenses (note 22a) | | | 38,552 | | | | 45,283 | | | | 29,448 | |
Loss (gain) on foreign exchange | | | (668 | ) | | | (1,451 | ) | | | 4,696 | |
Finance costs (note 22b) | | | 8,982 | | | | 14,056 | | | | 22,041 | |
Loss (gain) on extinguishments of liabilities (notes 16, 18a) | | | (79 | ) | | | 92,374 | | | | (33,626 | ) |
Change in fair value of financial instruments measured at fair value through profit or loss (note 15) | | | (850 | ) | | | (1,140 | ) | | | 1,000 | |
Impairment losses (note 24) | | | 20,859 | | | | 12,366 | | | | 149,952 | |
Share of losses of an associate (note 25) | | | — | | | | — | | | | 22 | |
Net loss from continuing operations before taxes | | $ | (122,338 | ) | | $ | (234,461 | ) | | $ | (259,465 | ) |
| | | | | | | | | | | | |
Current | | $ | (136 | ) | | $ | (348 | ) | | $ | (5,822 | ) |
Deferred | | | (65 | ) | | | 111 | | | | (13,815 | ) |
Income tax recovery on continuing operations (note 26) | | | (201 | ) | | | (237 | ) | | | (19,637 | ) |
Net loss from continuing operations | | $ | (122,137 | ) | | $ | (234,224 | ) | | $ | (239,828 | ) |
| | | | | | | | | | | | |
Discontinued operations, net of taxes | | | | | | | | | | | | |
Gain on sale of subsidiaries (note 6) | | | 3,380 | | | | 26,346 | | | | — | |
Net income from discontinued operations (note 6) | | | — | | | | 1,125 | | | | 1,932 | |
Net loss | | $ | (118,757 | ) | | $ | (206,753 | ) | | $ | (237,896 | ) |
| | | | | | | | | | | | |
Net income (loss) attributable to: | | | | | | | | | | | | |
Non-controlling interests - continuing operations (note 19) | | $ | (832 | ) | | $ | (1,044 | ) | | $ | (42,530 | ) |
Owners of the parent | | | | | | | | | | | | |
- Continuing operations | | | (121,305 | ) | | | (233,180 | ) | | | (197,298 | ) |
- Discontinued operations | | | 3,380 | | | | 27,471 | | | | 1,932 | |
| | $ | (117,925 | ) | | $ | (205,709 | ) | | $ | (195,366 | ) |
| | | | | | | | | | | | |
Net loss | | $ | (118,757 | ) | | $ | (206,753 | ) | | $ | (237,896 | ) |
| | | | | | | | | | | | |
Income (loss) per share | | | | | | | | | | | | |
Attributable to the owners of the parent basic and diluted (note 27): | | | | | | | | | | | | |
From continuing operations | | $ | (4.96 | ) | | $ | (14.52 | ) | | $ | (238.28 | ) |
From discontinued operations | | | 0.14 | | | | 1.71 | | | | 2.33 | |
Total loss per share | | $ | (4.83 | ) | | $ | (12.81 | ) | | $ | (235.95 | ) |
Weighted average number of outstanding shares (in thousands) (note 27) | | | 24,438 | | | | 16,062 | | | | 828 | |
The accompanying notes are an integral part of the consolidated financial statements.
F-4
LIMINAL BIOSCIENCES INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In thousands of Canadian dollars)
Years ended December 31 | | 2020 | | | 2019 | | | 2018 | |
Net Loss | | $ | (118,757 | ) | | $ | (206,753 | ) | | $ | (237,896 | ) |
| | | | | | | | | | | | |
Other comprehensive income (loss) | | | | | | | | | | | | |
Items that may be subsequently reclassified to profit and loss: | | | | | | | | | | | | |
Exchange differences on translation of foreign operations from continuing operations | | | 253 | | | | 294 | | | | (462 | ) |
Exchange differences on translation of foreign operations from discontinued operations (note 6) | | | — | | | | (692 | ) | | | 832 | |
Reclassification of exchange differences on translation of foreign operations sold to consolidated statement of operations (note 6) | | | — | | | | (1,449 | ) | | | — | |
Total other comprehensive income (loss) | | $ | 253 | | | $ | (1,847 | ) | | $ | 370 | |
| | | | | | | | | | | | |
Total comprehensive loss | | $ | (118,504 | ) | | $ | (208,600 | ) | | $ | (237,526 | ) |
| | | | | | | | | | | | |
Total comprehensive income (loss) attributable to: | | | | | | | | | | | | |
Non-controlling interests | | $ | (832 | ) | | $ | (1,044 | ) | | $ | (42,530 | ) |
Owners of the parent | | | | | | | | | | | | |
- Continuing operations | | | (121,052 | ) | | | (232,886 | ) | | | (197,760 | ) |
- Discontinued operations | | | 3,380 | | | | 25,330 | | | | 2,764 | |
Total comprehensive loss | | $ | (118,504 | ) | | $ | (208,600 | ) | | $ | (237,526 | ) |
The accompanying notes are an integral part of the consolidated financial statements.
F-5
LIMINAL BIOSCIENCES INC.
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
(In thousands of Canadian dollars)
| | Equity (deficiency) attributable to owners of the parent | | | | | | | | | |
| | Share capital | | | Contributed surplus | | | Warrants | | | Foreign currency translation reserve | | | Deficit | | | Total | | | Non- controlling interests | | | Total equity (deficiency) | |
| | $ | | | $ | | | $ | | | $ | | | $ | | | $ | | | $ | | | $ | |
Balance at January 1, 2018 | | | 575,150 | | | | 16,193 | | | | 73,944 | | | | (1,622 | ) | | | (541,571 | ) | | | 122,094 | | | | 21,447 | | | | 143,541 | |
Net loss | | | - | | | | - | | | | - | | | | - | | | | (195,366 | ) | | | (195,366 | ) | | | (42,530 | ) | | | (237,896 | ) |
Foreign currency translation reserve | | | - | | | | - | | | | - | | | | 370 | | | | - | | | | 370 | | | | - | | | | 370 | |
Issuance of shares (note 18a) | | | 6,340 | | | | - | | | | - | | | | - | | | | - | | | | 6,340 | | | | - | | | | 6,340 | |
Share-based payments expense (note 18b) | | | - | | | | 6,722 | | | | - | | | | - | | | | - | | | | 6,722 | | | | - | | | | 6,722 | |
Exercise of stock options (note 18b) | | | 1,073 | | | | (438 | ) | | | - | | | | - | | | | - | | | | 635 | | | | - | | | | 635 | |
Shares issued pursuant to restricted share units plan (note 18b) | | | 554 | | | | (554 | ) | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | |
Issuance of warrants (note 18c) | | | - | | | | - | | | | 21,352 | | | | - | | | | - | | | | 21,352 | | | | - | | | | 21,352 | |
Share and warrant issuance cost | | | - | | | | - | | | | - | | | | - | | | | (581 | ) | | | (581 | ) | | | - | | | | (581 | ) |
Effect of changes in the ownership of a subsidiary and funding arrangements on non-controlling interests (note 19) | | | - | | | | - | | | | - | | | | - | | | | (18,170 | ) | | | (18,170 | ) | | | 14,541 | | | | (3,629 | ) |
Balance at December 31, 2018 | | | 583,117 | | | | 21,923 | | | | 95,296 | | | | (1,252 | ) | | | (755,688 | ) | | | (56,604 | ) | | | (6,542 | ) | | | (63,146 | ) |
Net loss | | | - | | | | - | | | | - | | | | - | | | | (205,709 | ) | | | (205,709 | ) | | | (1,044 | ) | | | (206,753 | ) |
Foreign currency translation reserve | | | - | | | | - | | | | - | | | | (398 | ) | | | - | | | | (398 | ) | | | - | | | | (398 | ) |
Reclassification of exchange differences on translation of foreign operations to consolidated statement of operations (note 6) | | | - | | | | - | | | | - | | | | (1,449 | ) | | | - | | | | (1,449 | ) | | | - | | | | (1,449 | ) |
Issuance of shares (note 18a) | | | 349,834 | | | | - | | | | - | | | | - | | | | - | | | | 349,834 | | | | - | | | | 349,834 | |
Share-based payments expense (note 18b) | | | - | | | | 22,030 | | | | - | | | | - | | | | - | | | | 22,030 | | | | - | | | | 22,030 | |
Share-based compensation paid in cash (note 18b) | | | - | | | | (421 | ) | | | - | | | | - | | | | - | | | | (421 | ) | | | - | | | | (421 | ) |
Issuance of warrants (note 18c) | | | - | | | | - | | | | 560 | | | | - | | | | - | | | | 560 | | | | - | | | | 560 | |
Share issuance cost (note 18a) | | | - | | | | - | | | | - | | | | - | | | | (5,323 | ) | | | (5,323 | ) | | | - | | | | (5,323 | ) |
Effect of funding arrangements on non-controlling interests (note 19) | | | - | | | | - | | | | - | | | | - | | | | (331 | ) | | | (331 | ) | | | 331 | | | | - | |
Balance at December 31, 2019 | | | 932,951 | | | | 43,532 | | | | 95,856 | | | | (3,099 | ) | | | (967,051 | ) | | | 102,189 | | | | (7,255 | ) | | | 94,934 | |
Net loss | | | - | | | | - | | | | - | | | | - | | | | (117,925 | ) | | | (117,925 | ) | | | (832 | ) | | | (118,757 | ) |
Foreign currency translation reserve | | | - | | | | - | | | | - | | | | 253 | | | | - | | | | 253 | | | | - | | | | 253 | |
Issuance of shares (note 18a) | | | 31,755 | | | | - | | | | - | | | | - | | | | - | | | | 31,755 | | | | - | | | | 31,755 | |
Share-based payments expense (note 18b) | | | - | | | | 6,234 | | | | - | | | | - | | | | - | | | | 6,234 | | | | - | | | | 6,234 | |
Exercise of stock options (note 18b) | | | 167 | | | | (85 | ) | | | - | | | | - | | | | - | | | | 82 | | | | - | | | | 82 | |
Shares issued pursuant to restricted share units plan (note 18b) | | | 9,764 | | | | (9,764 | ) | | | - | | | | - | | | | - | | | | - | | | | | | | | - | |
Share-based compensation paid in cash (note 18b) | | | - | | | | (40 | ) | | | - | | | | - | | | | - | | | | (40 | ) | | | - | | | | (40 | ) |
Issuance of warrants (note 18c) | | | | | | | | | | | 2,623 | | | | | | | | | | | | 2,623 | | | | | | | | 2,623 | |
Exercise of warrants (note 18c) | | | 2,624 | | | | - | | | | (2,623 | ) | | | - | | | | - | | | | 1 | | | | - | | | | 1 | |
Share issuance cost (note 18a) | | | - | | | | - | | | | - | | | | - | | | | (2,073 | ) | | | (2,073 | ) | | | - | | | | (2,073 | ) |
Balance at December 31, 2020 | | | 977,261 | | | | 39,877 | | | | 95,856 | | | | (2,846 | ) | | | (1,087,049 | ) | | | 23,099 | | | | (8,087 | ) | | | 15,012 | |
The accompanying notes are an integral part of the consolidated financial statements
F-6
LIMINAL BIOSCIENCES INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands of Canadian dollars)
Years ended December 31 | | 2020 | | | 2019 | | | 2018 | |
Cash flows used in operating activities | | | | | | | | | | | | |
Net loss from continuing operations for the year | | $ | (122,137 | ) | | $ | (234,224 | ) | | $ | (239,828 | ) |
Net income from discontinued operations for the year | | | 3,380 | | | | 27,471 | | | | 1,932 | |
Adjustments to reconcile net loss to cash flows used in operating activities: | | | | | | | | | | | | |
Finance costs and foreign exchange | | | 8,307 | | | | 12,809 | | | | 25,282 | |
Change in operating and finance lease inducements and obligations | | | — | | | | — | | | | 2,565 | |
Loss (gain) from disposition of capital and intangible assets | | | (15 | ) | | | 196 | | | | 513 | |
Share of losses of an associate | | | — | | | | — | | | | 22 | |
Non-cash issuance of warrants (note 15) | | | 2,228 | | | | — | | | | — | |
Gain on sale of subsidiaries (note 6) | | | (3,380 | ) | | | (26,346 | ) | | | — | |
Change in fair value of financial instruments measured at fair value through profit or loss (note 15) | | | (850 | ) | | | (1,140 | ) | | | 1,000 | |
Impairment losses (note 24) | | | 20,859 | | | | 12,366 | | | | 149,952 | |
Loss (gain) on extinguishments of liabilities (notes 16, 18a) | | | (79 | ) | | | 92,374 | | | | (33,626 | ) |
Deferred income taxes (note 26) | | | — | | | | 87 | | | | (13,815 | ) |
Share-based payments expense (note 18b) | | | 6,194 | | | | 21,609 | | | | 6,722 | |
Depreciation of capital assets (note 10) | | | 2,779 | | | | 3,734 | | | | 4,086 | |
Depreciation of right-of-use assets (note 11) | | | 4,578 | | | | 4,913 | | | | — | |
Amortization of intangible assets (note 12) | | | 1,090 | | | | 1,259 | | | | 1,372 | |
| | | (77,046 | ) | | | (84,892 | ) | | | (93,823 | ) |
Change in non-cash working capital items | | | 1,129 | | | | (14,498 | ) | | | 11,369 | |
| | $ | (75,917 | ) | | $ | (99,390 | ) | | $ | (82,454 | ) |
Cash flows from financing activities | | | | | | | | | | | | |
Proceeds from share issuances (with or without warrants) (note 18a) | | | 39,960 | | | | 118,785 | | | | 751 | |
Proceeds from long-term debt (with or without warrants) (notes 16, 18c) | | | 31,533 | | | | 19,859 | | | | 79,105 | |
Repayment of principal on long-term debt (note 16) | | | (165 | ) | | | (988 | ) | | | (3,184 | ) |
Repayment of interest on long-term debt (note 16) | | | (1,879 | ) | | | (3,540 | ) | | | (3,934 | ) |
Exercise of options (note 18b) | | | 82 | | | | — | | | | 635 | |
Proceeds from exercise of pre-funded warrants (note 18c) | | | 1 | | | | — | | | | — | |
Payments of principal on lease liabilities (note 14) | | | (7,069 | ) | | | (7,563 | ) | | | — | |
Payment of interest on lease liabilities (note 14) | | | (2,098 | ) | | | (1,767 | ) | | | — | |
Debt, share and warrants issuance costs | | | (2,960 | ) | | | (6,867 | ) | | | (970 | ) |
Payments of principal under finance leases | | | — | | | | — | | | | (245 | ) |
| | $ | 57,405 | | | $ | 117,919 | | | $ | 72,158 | |
Cash flows used in investing activities | | | | | | | | | | | | |
Additions to capital assets | | | (966 | ) | | | (2,741 | ) | | | (3,786 | ) |
Additions to intangible assets | | | (1,080 | ) | | | (1,703 | ) | | | (1,342 | ) |
Proceeds from sale of discontinued operations business, net of cash divested | | | 4,555 | | | | 43,958 | | | | — | |
Transaction costs paid relating to the sale of discontinued operations business | | | (787 | ) | | | (4,228 | ) | | | — | |
Proceeds from disposal of capital assets | | | 133 | | | | — | | | | — | |
Acquisition of convertible debt | | | — | | | | — | | | | (955 | ) |
Release of restricted cash | | | — | | | | 65 | | | | — | |
Interest received | | | 450 | | | | 745 | | | | 224 | |
| | $ | 2,305 | | | $ | 36,096 | | | $ | (5,859 | ) |
| | | | | | | | | | | | |
Net change in cash and cash equivalents during the year | | | (16,207 | ) | | | 54,625 | | | | (16,155 | ) |
Net effect of currency exchange rate on cash and cash equivalents | | | (3 | ) | | | (729 | ) | | | 378 | |
Cash and cash equivalents, beginning of year | | | 61,285 | | | | 7,389 | | | | 23,166 | |
Cash and cash equivalents, end of year | | $ | 45,075 | | | $ | 61,285 | | | $ | 7,389 | |
Comprising of: | | | | | | | | | | | | |
Cash | | | 45,075 | | | | 41,761 | | | | 7,389 | |
Cash equivalents | | | — | | | | 19,524 | | | | — | |
| | $ | 45,075 | | | $ | 61,285 | | | $ | 7,389 | |
Cash flows from discontinued operations presented in note 6
The accompanying notes are an integral part of the consolidated financial statements.
F-7
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
1. | Nature of operations and going concern |
Liminal BioSciences Inc. or Liminal, or the Company, is incorporated under the Canada Business Corporations Act and is a publicly traded clinical stage biotechnology company (Nasdaq symbol: LMNL) focused on discovering and developing novel small molecule drug candidates for the treatment of patients suffering from respiratory fibrotic diseases and other fibrotic or inflammatory diseases that have high unmet medical need. Liminal has a deep understanding of certain biological targets and pathways that have been implicated in the fibrotic process, including fatty acid receptors 1, or FFAR1 also known as G-protein-coupled receptor 40, or GPR40, a related receptor G-protein-coupled receptor 84, or GPR84, and peroxisome proliferator-activated receptors, or PPARs.
Liminal’s lead small molecule segment product candidate, fezagepras or PBI 4050, is being developed for the treatment of idiopathic pulmonary fibrosis, or IPF. The plasma‑derived therapeutics segment leverages Liminal’s experience in bioseparation technologies used to isolate and purify biopharmaceuticals from human plasma. With respect to this second platform, the Company is focused on the development of its plasma-derived product candidate Ryplazim® (plasminogen) or Ryplazim®, a highly purified glu-plasminogen derived from human plasma that acts as a plasminogen replacement therapy for patients deficient in plasminogen protein.
The Company’s head office is located at 440, Boul. Armand-Frappier, suite 300, Laval, Québec, Canada, H7V 4B4. Liminal has Research and Development facilities in Canada, the U.K. and the U.S. and manufacturing facilities in Canada.
On July 5, 2019, the Company performed a one thousand-to-one share consolidation of its common shares, stock options, restricted share units and warrants. The quantities and per unit prices presented in these audited annual consolidated financial statements have been retroactively adjusted to give effect to the share consolidation.
On October 7, 2019, the Company formerly named Prometic Life Sciences Inc. changed its name to Liminal BioSciences Inc. and the Company’s TSX stock symbol changed from PLI to LMNL. On November 12, 2019 the Company listed on the Nasdaq exchange with the Nasdaq symbol LMNL and effective August 5, 2020, the Company voluntarily delisted from the TSX.
On November 25, 2019 the Company sold the majority of its bioseparations business to a third party. These activities are presented as discontinued operations in the annual consolidated financial statements. Details on this transaction and the results from discontinued operations are disclosed in note 6. The prior period results from discontinued operations have been reclassified and presented in the consolidated statements of operations.
Structured Alpha LP (“SALP”) has been Liminal’s majority and controlling shareholder since the debt restructuring on April 23, 2019 (note 16) and is considered Liminal’s parent entity for accounting purposes. Thomvest Asset Management Ltd. is the general partner of SALP and the ultimate controlling parent, for accounting purposes, of Liminal is The 2003 TIL Settlement. Prior to this date, Liminal did not have a controlling parent.
The consolidated financial statements for the year ended December 31, 2020 have been prepared in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board, or IFRS, on a going concern basis, which presumes the Company will continue its operations for the foreseeable future and will be able to realize its assets and discharge its liabilities and commitments in the ordinary course of business.
F-8
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
During the year ended December 31, 2020, the Company incurred a net loss of $118.8 million ($206.8 million for the year ended December 31, 2019 which included a loss on extinguishment of liabilities of $92.4 million) and had negative operating cash flows of $75.9 million ($99.4 million for the year ended December 31, 2019). In addition, at December 31, 2020, the Company had a working capital of $49.2 million ($63.6 million at December 31, 2019) and an accumulated deficit of $1,087.0 million ($967.1 million at December 31, 2019). Given Liminal's main activities continue to be in the R&D stage, management has concluded it will need additional sources of financing to ensure it has sufficient funds to continue its operations for at least the next twelve months.
Until the Company completes a significant financing, it continues operating at a low spending level, pacing investments on new research programs, and reducing infrastructure cost, where possible. The need to complete multiple financing transactions is likely to continue until the Company can generate sufficient product revenues to finance its cash requirements. Meanwhile, management may revert to a variety of sources for financing future cash needs including public or private equity offerings, debt financings, strategic collaborations, business and asset divestitures and grant funding amongst others. Despite the Company’s efforts to obtain the necessary funding and improve profitability of its operations, there can be no assurance of its success in doing so, especially with respect to its access to further funding on acceptable terms, if at all.
These circumstances indicate the existence of a material uncertainty that may cast substantial doubt about the Company’s ability to continue as a going concern. If the Company is unable to secure additional capital, it may be required to curtail its research and development initiatives and take additional measures to reduce costs in order to conserve its cash in amounts sufficient to sustain operations and meet its obligations. These measures could cause significant delays in the Company’s preclinical, clinical and regulatory efforts, which are critical to the realization of its business plan. These financial statements do not include any adjustments to the amounts and classification of assets and liabilities that might be necessary should the Company be unable to continue as a going concern. Such adjustments could be material.
2. | Significant accounting policies |
Statement of compliance
These consolidated financial statements have been prepared in accordance with IFRS and were approved by the Board of Directors on March 23, 2021.
Basis of measurement
The consolidated financial statements have been prepared on a historical cost basis, except for the warrant liability which has been measured at fair value. Certain assets may be carried at their net realizable value or at their recoverable amount if they have been subject to impairment.
Functional and presentation currency
The consolidated financial statements are presented in Canadian dollars, which is also the Company’s functional currency.
F-9
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Basis of consolidation
The consolidated financial statements include the accounts of Liminal BioSciences Inc., and those of its subsidiaries. The Company’s subsidiaries at December 31, 2020, 2019 and 2018 are as follows:
Name of subsidiary | Segment activity | Place of incorporation and operation | Proportion of ownership interest held by group | |
| | | 2020 | | 2019 | | 2018 | |
Fairhaven Pharmaceuticals Inc. | Small molecule therapeutics | Quebec, Canada | 100% | | nil | | nil | |
Liminal R&D BioSciences Inc. | Small molecule therapeutics | Quebec, Canada | 100% | | 100% | | 100% | |
Liminal BioSciences Holdings Limited | Small molecule therapeutics | Cambridge, United Kingdom | 100% | | 100% | | 100% | |
Liminal BioSciences Limited | Small molecule therapeutics | Cambridge, United Kingdom | 100% | | 100% | | 100% | |
Prometic Pharma SMT B.V | Small molecule therapeutics | Amsterdam, Netherlands | 100% | | 100% | | N/A | |
Prometic Bioproduction Inc. | Plasma-derived therapeutics | Quebec, Canada | 100% | | 100% | | 100% | |
Prometic Plasma Resources Inc. | Plasma-derived therapeutics | Winnipeg, Canada | 100% | | 100% | | 100% | |
Telesta Therapeutics Inc. | Plasma-derived therapeutics | Quebec, Canada | 100% | | 100% | | 100% | |
NantPro Biosciences, LLC | Plasma-derived therapeutics | Delaware, U.S. | 73% | | 73% | | 73% | |
Prometic Biotherapeutics Inc. | Plasma-derived therapeutics | Delaware, U.S. | 100% | | 100% | | 100% | |
Prometic Plasma Resources USA Inc. | Plasma-derived therapeutics | Delaware, U.S. | 100% | | 100% | | 100% | |
Prometic Biotherapeutics Ltd | Plasma-derived therapeutics | Cambridge, United Kingdom | 100% | | 100% | | 100% | |
Prometic Biotherapeutics B.V. | Plasma-derived therapeutics | Amsterdam, Netherlands | 100% | | 100% | | N/A | |
Pathogen Removal and Diagnostic Technologies Inc. | Corporate | Delaware, U.S. | 77% | | 77% | | 77% | |
Prometic Bioseparations Ltd | Discontinued operations | Isle of Man, British Isles | nil | | nil | | 100% | |
Prometic Manufacturing Inc. | Discontinued operations | Quebec, Canada | nil | | nil | | 100% | |
The Company consolidates investees when, based on the evaluation of the substance of the relationship with the Company, it concludes that it controls the investees. The Company controls an investee when it is exposed, or has rights, to variable returns from its involvement with the investee and has the ability to affect those returns through its power over the investee. The financial statements of the subsidiaries are prepared for the same reporting period as the parent Company, using consistent accounting policies. All intra-group transactions, balances, income and expenses are eliminated in full upon consolidation.
When a subsidiary is not wholly-owned the Company recognizes the non-controlling interests’ share of the net assets and results of operations in the subsidiary. When the proportion of the equity held by non-controlling interests’ changes without resulting in a change of control, the carrying amount of the controlling and non-controlling interest are adjusted to reflect the changes in their relative interests in the subsidiary. In these situations, the Company recognizes directly in equity the effect of the change in ownership of a subsidiary on the non-controlling interests. Similarly, after recognizing its share of the operating losses, the non-controlling interest is adjusted for its share of the equity contribution made by Liminal that does not modify the interest held by either party. The offset to this adjustment is recorded in the deficit. The effect of these transactions is presented in the consolidated statement of changes in equity.
Financial instruments
Recognition and derecognition
Financial instruments are recognized in the consolidated statement of financial position when the Company becomes a party to the contractual obligations of the instrument. On initial recognition, financial instruments are recognized at their fair value plus, in the case of financial instruments not at fair value through profit or loss (“FVPL”), transaction costs that are directly attributable to the acquisition or issue of financial instruments. Financial assets are subsequently derecognized when payment is received in cash or other financial assets or if the debtor is discharged of its liability.
F-10
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
A financial liability is derecognized when the obligation under the liability is discharged or cancelled or expires. When an existing liability is replaced by another from the same creditor on substantially different terms, or the terms of the liability are substantially modified, such an exchange or modification is treated as the derecognition of the original liability and the recognition of a new liability. The difference in the respective carrying amounts is recognized in the consolidated statement of operations.
Classification
Subsequent to initial recognition, financial instruments are measured according to the category to which they are classified. Financial instruments are measured at amortized cost unless they are classified as fair value through other comprehensive income, or FVOCI, classified as FVPL or designated as FVPL, in which case they are subsequently measured at fair value.
The classification of financial asset debt instruments is driven by the Company’s business model for managing the financial assets and their contractual cash flow characteristics. Assets that are held to collect contractual cash flows where those cash flows represent solely payments of principal and interest are measured at amortized cost. Equity instruments that are held for trading (including all equity derivative instruments) are classified as FVPL. Financial liabilities are measured at amortized cost, unless they are required to be measured at FVPL (such as instruments held for trading or derivatives) or the Company has opted to measure them at FVPL.
Currently, the Company classifies cash, cash equivalents, trade receivables, other receivables, restricted cash, and long-term deposits as financial assets measured at amortized cost and trade payables, wages and benefits payable, royalty payment obligations, license acquisition payment obligations, other employee benefit liabilities and long-term debt as financial liabilities measured at amortized cost.
The Company classifies the warrant liability as a financial liability at FVPL for which the variation in fair value is recorded in consolidated statement of operations. The Company previously held an investment in convertible debt that it classified as financial assets at FVPL in the year ended December 31, 2018.
Impairment of financial assets
The expected credit losses associated with its debt instruments carried at amortized cost is assessed on a forward-looking basis. The impairment methodology applied depends on whether there has been a significant increase in credit risk. For trade receivables, the Company applies the simplified approach permitted by IFRS 9, which requires lifetime expected losses to be recognized from initial recognition of the receivables.
Cash equivalents
Cash and cash equivalents comprise deposits in banks and highly liquid investments having an original maturity of 90 days or less when issued.
Inventories
Inventories of raw materials and finished goods are valued at the lower of cost and net realizable value. Cost is determined on a first in, first out basis. The cost of manufactured inventories comprises all costs that are directly attributable to the manufacturing process, such as raw materials, direct labour and manufacturing overhead based on normal operating capacity. Net realizable value is the estimated selling price in the ordinary course of business and the estimated selling costs except for raw materials for which it is determined using replacement cost.
F-11
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Capital assets
Capital assets are recorded at cost less any government assistance, accumulated depreciation and accumulated impairment losses, if any. Depreciation is calculated on a straight-line basis over the estimated useful lives of the assets, as described below.
Capital asset | | | | | | | | | Period |
Buildings and improvements | | | | | | | 20 years |
Leasehold improvements | | | The lower of the lease term and the useful life |
Production and laboratory equipment | | | | | | | 5 - 20 years |
Furniture | | | | | | | 5 - 10 years |
Computer equipment | | | | | | | 3 - 5 years |
The estimated useful lives, residual values and depreciation methods are reviewed annually with the effect of any changes in estimates accounted for on a prospective basis. The gain or loss arising on the disposal or retirement of a capital asset is determined as the difference between the sales proceeds and its carrying amount and is recognized in profit or loss.
Government assistance
Government assistance programs, including investment tax credits on research and development expenses and salary and rent subsidies are reflected as reductions to the cost of the assets or to the expenses to which they relate and are recognized when there is reasonable assurance that the assistance will be received and all attached conditions are complied with.
Right-of-use assets
The Company recognizes a right-of-use, or ROU, asset at the commencement date of a lease which is when the date at which the underlying asset is available for use. Right-of-use assets are measured at cost, less any accumulated depreciation and impairment losses, and adjusted for any remeasurement of lease liabilities. The cost of right-of-use assets includes the amount of lease liabilities recognized, initial direct costs incurred, and lease payments made at or before the commencement date less any lease incentives received. Unless the Company is reasonably certain to obtain ownership of the leased asset at the end of the lease term, the recognized right-of-use asset is depreciated on a straight-line basis over the shorter of its estimated useful life and the lease term.
Intangible assets
Intangible assets include acquired rights such as licenses for product manufacturing and commercialization, donor lists, external patent costs and software costs. They are carried at cost less accumulated amortization. Amortization commences when the intangible asset is available for use and is calculated over the estimated useful lives of the intangible assets acquired using the straight-line method. The maximum period used for each category of intangible asset are presented in the table below. The estimated useful lives and amortization method are reviewed annually, with the effect of any changes in estimates being accounted for on a prospective basis. The amortization expense is recognized in the consolidated statement of operations in the expense category consistent with the function of the intangible assets.
Intangible asset | | | | | | | | | Period |
Licenses and other rights | | | | | | | | | 30 years |
Donor lists | | | | | | | | | 10 years |
Patents | | | | | | | | | 20 years |
Software | | | | | | | | | 5 years |
F-12
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Impairment of long-lived assets
At the end of each reporting period, the Company reviews the carrying amounts of its capital, ROU and intangible assets to determine whether there is any indication that those assets have suffered an impairment loss. If impairment indicators exist, the recoverable amount of the asset is estimated in order to determine the extent of the impairment loss, if any. For intangible assets not yet available for use, an impairment test is performed annually at November 30, until amortization commences, whether or not there are impairment indicators. When it is not possible to estimate the recoverable amount of an individual asset, the Company estimates the recoverable amount of the cash-generating unit, or CGU, which represents the smallest identifiable group of assets that generates cash inflows that are largely independent of the cash inflows from other assets, groups of assets or CGUs to which the asset belongs. Where a reasonable and consistent basis of allocation can be identified, the corporate assets are also allocated to individual CGUs, or otherwise they are allocated to the smallest group of CGUs for which a reasonable and consistent allocation basis can be identified.
The recoverable amount is the higher of the fair value less costs to sell and value in use. In assessing value in use, the estimated future cash flows are discounted to their present value using a pre-tax discount rate that reflects current market assessments of the time value of money and the risks specific to the asset for which the estimates of future cash flows have not been adjusted.
An impairment loss is recognized when the carrying amount of an asset or a CGU exceeds its recoverable amount by the amount of this excess. An impairment loss is recognized immediately in profit or loss in the period during which the loss is incurred. Where an impairment loss subsequently reverses, the carrying amount of the asset or CGU is increased to the revised estimate of its recoverable amount; on reversal of an impairment loss, the increased carrying amount does not exceed the carrying amount that would have been determined had an impairment loss not been recognized for the asset or CGU in prior periods. A reversal of an impairment loss is recognized immediately in profit or loss.
Investment in an associate
Investments in associates are accounted for using the equity method. An associate is an entity over which the Company has significant influence. Under the equity method, the investment in the associate is carried on the consolidated statement of financial position at cost plus post acquisition changes in the Company’s share of net assets of the associate.
The consolidated statement of operations reflects the Company’s share of the results of operations of the associate.
At each balance sheet date, management considers whether there is objective evidence of impairment in associates. If there is such evidence, management determines the amount of impairment to record, if any, in relation to the associate.
When the level of influence over an associate changes either following a loss of significant influence over the associate, or the obtaining of control over the associate or when an investment in a financial asset accounted for under IFRS 9 becomes subject to significant influence, the Company measures and recognizes its investment at its fair value. Any difference between the carrying amount of the associate at the time of the change in influence and the fair value of the investment, and proceeds from disposal if any, is recognized in profit or loss.
F-13
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Lease liabilities
At the commencement date of a lease, the Company recognizes a lease liability measured at the present value of lease payments to be made over the lease term. The lease payments include fixed payments (including in-substance fixed payments) less any lease incentives, variable lease payments that depend on an index or a rate, and amounts expected to be paid under residual value guarantees. The lease payments also include the exercise price of a purchase option reasonably certain to be exercised by the Company and payments of penalties for terminating a lease, if the lease term reflects the Company exercising the option to terminate. The variable lease payments that do not depend on an index or a rate are recognized as expense in the period on which the event or condition that triggers the payment occurs.
In calculating the present value of lease payments, the Company uses the incremental borrowing rate at the lease commencement date if the interest rate implicit in the lease is not readily determinable. After the commencement date, the amount of a lease liability is increased to reflect the accretion of interest and reduced for the lease payments made. In addition, the carrying amount of a lease liability is remeasured if there is a modification, a change in the lease term, a change in the in-substance fixed lease payments or a change in the assessment whether the underlying asset will be purchased.
The Company applies the short-term lease recognition exemption to leases of 12 months or less, as well as the lease of low-value assets recognition exemption. Lease payments on short-term leases and leases of low-value assets are recognized as expense on a straight-line basis over the lease term.
Revenue recognition
To determine revenue recognition for contracts with customers, Liminal performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. The five-step model is only applied to contracts when it is probable that we will collect the consideration we are entitled to in exchange for the goods or services we transfer. At contract inception, the Company assesses the goods or services promised within each contract, determines those that are performance obligations, and assesses whether each promised good or service is distinct. The transaction price that is allocated to the respective performance obligation is recognized as revenue when (or as) the performance obligation is satisfied.
Sale of goods
Revenue from sale of goods is recognized when the terms of a contract with a customer have been satisfied. This occurs when:
| • | The control over the product has been transferred to the customer; and |
| • | The product is received by the customer or transfer of title to the customer occurs upon shipment. |
Following delivery, the customer bears the risks of obsolescence and loss in relation to the goods. Revenue is recognized based on the price specified in the contract, net of estimated sales discounts and returns.
Rendering of services
Revenues from contracted services are generally recognized as the performance obligations are satisfied over time, and the related expenditures are incurred pursuant to the terms of the agreement. Contract revenue is recognized on a percentage of completion basis based on key milestones contained within the contract.
F-14
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Licensing fees and milestone payments
The Company determines whether the Company's promise to grant a license provides its customer with either a right to access the Company’s intellectual property ("IP") or a right to use the Company’s IP. A license will provide a right to access the intellectual property if there is significant development of the intellectual property expected in the future whereas for a right to use, the intellectual property is to be used in the condition it is at the time the license is signed. Revenue from a license that provides a customer the right to use the Company’s IP is recognized at a point in time when the transfers to the licensee is completed and the license period begins. When a license provides access to the Company's IP over a license term, the performance obligation is satisfied over time and, therefore, revenue is recognized over the term of the license arrangement. Milestone payments are immediately recognized as licensing revenue when the condition is met, if the milestone is not a condition to future deliverables and collectability is reasonably assured. Otherwise, they are recognized over the remaining term of the agreement or the performance period.
Royalty revenue
Royalty revenues are recognized once the sale of products to which the royalties gives rise occurs.
Rental revenue
The Company accounts for the lease or sub-lease with its tenant as an operating lease when the Company has not transferred substantially all of the risks and benefits of ownership of its property or leased property. Revenue recognition under an operating lease commences when the tenant has a right to use the leased asset, and the total amount of contractual rent to be received from the operating lease is recognized on a straight-line basis over the term of the lease. Rental revenue also includes recoveries of operating expenses and property taxes.
Research and development expenses
Expenditure on research activities is recognized as an expense in the period during which it is incurred. An internally generated intangible asset arising from development (or from the development phase of an internal project) is recognized if, and only if, all of the following have been demonstrated:
| • | the technical feasibility of completing the intangible asset so that it will be available for use or sale; |
| • | the intention to complete the intangible asset and use or sell it; |
| • | the ability to use or sell the intangible asset; |
| • | how the intangible asset will generate probable future economic benefits; |
| • | the availability of adequate technical, financial and other resources to complete the development and to use or sell the intangible asset; and |
| • | the ability to measure reliably the expenditures attributable to the intangible asset during its development. |
To date, the Company has not capitalized any development costs.
Research and development expenses presented in the consolidated statement of operations comprise the costs to manufacture the plasma-derived product candidates used in pre-clinical tests and clinical trials. It also includes the cost of product candidates used in our small molecule clinical trials such as PBI-4050, external consultants supporting the clinical trials and pre-clinical tests, employee compensation and other operating expenses involved in research and development activities.
F-15
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Foreign currency translation
Transactions and balances
Transactions in foreign currencies are initially recorded by the Company and its entities at their respective functional currency rates prevailing at the date of the transaction. Monetary assets and liabilities denominated in foreign currencies are retranslated at the functional currency spot rate of exchange at the reporting date. All differences are taken to the consolidated statement of operations. Non-monetary items that are measured in terms of historical cost in a foreign currency are translated using the exchange rates at the dates when the initial transactions took place.
Group companies
The assets and liabilities of foreign operations are translated into Canadian dollars at the rate of exchange prevailing at the reporting date and their statements of operations are translated at exchange rates prevailing at the dates of the transactions. The exchange differences arising on the translation are recognized in other comprehensive loss. On disposal of a foreign operation, the component of other comprehensive loss relating to that particular foreign operation is reclassified from the consolidated statement of comprehensive loss to the consolidated statement of operations as part of the gain or loss on the disposal of the foreign operation.
Income taxes
The Company uses the liability method of accounting for income taxes. Deferred income tax assets and liabilities are recognized in the consolidated statement of financial position for the future tax consequences attributable to differences between the consolidated financial statements carrying values of existing assets and liabilities and their respective income tax bases. Deferred income tax assets and liabilities are measured using income tax rates expected to apply when the assets are realized or the liabilities are settled. The effect of a change in income tax rates is recognized in the year during which these rates change. Deferred income tax assets are recognized to the extent that it is probable that future tax profits will allow the deferred tax assets to be recovered. When uncertainties exist over income tax treatments, the Company applies the guidance in IFRIC 23, Uncertainty over income tax treatments when evaluating its income tax provisions.
Share-based payments
The Company has a stock option plan and a restricted share unit plan. The fair value of stock options granted is determined at the grant date using the Black-Scholes option pricing model and is expensed over the vesting period of the options. Awards with graded vesting are considered to be multiple awards for fair value measurement. The fair value of Restricted Share Units, or RSU, is determined using the market value of the Company’s shares on the grant date. The expense associated with RSU awards that vest over time are recognized over the vesting period. When the vesting of RSU is dependent on meeting performance targets as well as a service requirement, the Company will estimate the outcome of the performance targets to determine the expense to recognize over the vesting period, and revise those estimates until the final outcome is determined. An estimate of the number of awards that are expected to be forfeited is also made at the time of grant and revised periodically if actual forfeitures differ from those estimates.
The Company’s policy is to issue new shares upon the exercise of stock options and the release of RSU for which conditions have been met.
F-16
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Assets held for sale and discontinued operations
The Company classifies non-current assets and disposal groups as held for sale if their carrying amounts will be recovered principally through a sale rather than through continuing use. Such non-current assets and disposal groups classified as held for sale are measured at the lower of their carrying amount and their fair value less cost to sell. Costs to sell are the incremental costs directly attributable to the sale, excluding finance costs and income tax expense. Such assets are only presented as held for sale when the sale is highly probable and the assets or disposal group are available for immediate sale in their present condition. Actions required to complete the sale should indicate that it is unlikely that significant changes to the sale will be made or that the sale will be withdrawn. Management must be committed to the sale, which should be expected to qualify for recognition as a completed sale within one year from the date of classification.
Capital assets included as part of the assets held for sale are not depreciated once classified as held for sale. Assets and liabilities classified as held for sale are presented separately as current items in the consolidated statement of financial position.
The results of discontinued operations are presented net of tax in the consolidated statement of operations. Incremental cost related to the disposition and income taxes are allocated to discontinued operations. The discontinued operations also include the gain or loss on the disposal, which will also include the reclassification of historical exchange differences on translation of foreign operations sold. The results of discontinued operations exclude the allocation of the corporate finance costs and general corporate overheads in the forms of management fees if those costs will continue to be incurred by Liminal following the disposition. The prior period results from discontinued operations have been reclassified and presented in the consolidated statements of operations.
Earnings per share (EPS)
The Company presents basic and diluted earnings per share, or EPS, data for its common shares. Basic EPS is calculated by dividing the profit or loss attributable to common shareholders of the Company by the weighted average number of common shares outstanding during the year adjusted for any bonus element. Diluted EPS is determined by adjusting the weighted average number of common shares outstanding for the effects of all dilutive potential common shares, which comprise warrants, stock options, RSU and common shares that would be issued upon the conversion of the secured convertible debentures into shares.
Share and warrant issue expenses
The Company records share and warrant issue expenses as an increase to the deficit.
3. | Significant accounting judgements and estimation uncertainty |
The preparation of these consolidated financial statements requires the use of judgments, estimates and assumptions that affect the reported amounts of revenues, expenses, assets and liabilities and the accompanying disclosures. The uncertainty that is often inherent in estimates and assumptions could result in material adjustments to assets or liabilities affected in future periods.
F-17
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Significant judgments
Going concern - In assessing whether the going concern assumption is appropriate and whether there are material uncertainties that may cast significant doubt about the Company’s ability to continue as a going concern, management must estimate future cash flows for a period of at least twelve months following the end of the reporting period by considering relevant available information about the future. Management has considered a wide range of factors relating to expected cash inflows such as the forecasted product sales, whether the Company will earn other significant revenues, obtain regulatory approval for commercialization of its product candidate Ryplazim® and the potential sources of debt and equity financing available to it. Management has also estimated expected cash outflows such as operating and capital expenditures and debt repayment schedules, including the ability to delay uncommitted expenditures. These cash flow estimates are subject to uncertainty.
Accounting for loan modifications – When the terms of a loan are modified, management must evaluate whether the terms of the loan are substantially different in order to determine the accounting treatment. If they are considered to be substantially different, the modification will be accounted for as a derecognition of the carrying value of the pre-modified loan and the recognition of a new loan at its fair value. Otherwise, the changes will be treated as a modification which will result in adjusting the carrying amount to the present value of the modified cash flows using the original effective interest rate of the loan instrument. In assessing whether the terms of a loan are substantially different, management performs a quantitative analysis of the changes in the cash flows under the previous agreement and the new agreement and also considers other modifications that have no cash flow impact. In the context of the simultaneous modification to the terms of several loans with the same lender, management uses judgment to determine if the cash flow analysis should be performed on the loans in aggregate or individually. Judgment is also used to evaluate the relative importance of additional rights given to the lender such as additional Board of Director seats and the extension of the term of the security compared to the quantitative analysis.
Functional currency – The functional currency of foreign subsidiaries is reviewed on an ongoing basis to assess if changes in the underlying transactions, events and conditions have resulted in a change. During the year ended December 31, 2020, the functional currency of the Pathogen Removal and Diagnostic Technologies Inc., or PRDT, subsidiary changed from £ to USD. In 2019 and 2018 no changes in functional currency of the subsidiaries were deemed necessary. This assessment is also performed for new subsidiaries. When assessing the functional currency of a foreign subsidiary, management’s judgment is applied in order to determine, amongst other things, the primary economic environment in which an entity operates, the currency in which the activities are funded and the degree of autonomy of the foreign subsidiary from the reporting entity in its operations and financially. Judgment is also applied in determining whether the inter-company loans denominated in foreign currencies form part of the parent Company’s net investment in the foreign subsidiary. Considering such loans as part of the net investment in the foreign subsidiary results in foreign currency translation gains or losses from the translation of these loans being recorded in other comprehensive loss instead of the consolidated statement of operations.
F-18
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Share-based compensation - On March 23, 2020, the board of directors of the Company approved a plan to seek shareholder approval to modify the exercise price of certain stock options as disclosed in note 18b. In order to determine when the expense related to this modification is recognized in the consolidated statement of operations, management evaluated the timing of notification to option holders, the timing and method of determining the exercise price and the service period. Management further considered whether the holders of the stock options had sufficient understanding of the terms and conditions of the potentially revised awards, the degree of certainty of the approval for the repricing and whether the service period for earning the rights to the awards had commenced. Management concluded that the definition of the grant date was not met but that the service period had commenced and therefore a preliminary calculation of the incremental fair value of the repricing of the awards was performed using assumptions as of March 31, 2020. On May 26, 2020, the conditions for a grant date were met and the options exercise price was revised to $15.21 and a final calculation to determine the incremental fair value of the repriced options was performed.
Estimates and assumptions
COVID-19 – The negative impact of the COVID-19 pandemic on the financial statements for year ended December 31, 2020 has been limited, however the Company is eligible for salary and rent subsidy programs from the Government of Canada under which it submitted claims (note 22). The subsidy programs may provide further financial support while the programs are available. Consistent within the global biopharmaceutical sector, some clinical programs may have been and may be impacted by the shift of resources within hospitals to COVID-19 and related matters, resulting in potential delays to recruitment or site initiation on our clinical and preclinical programs, and potentially causing an adjustment of certain development timelines and activities. The partial disruption caused by COVID-19 may continue to impact the Company’s operations, workforce and overall business by delaying the progress of our research and development programs, regulatory submissions and reviews, regulatory inspections, production and plasma collection activities and business and corporate development activities. There is uncertainty as to the duration of the COVID-19 pandemic and related government restrictions, including travel bans, the impact on our workforce, and the availability of donors, healthy subjects and patients for the conduct of clinical trials, and the effects of the COVID-19 pandemic, including on the global economy, continue to be fluid.
Estimates and assumptions about future events and their effects cannot be determined with certainty and therefore require the exercise of judgment. As of the date of issuance of these financial statements, the Company is not aware of any specific event or circumstance that would require it to update its estimates, assumptions and judgments or revise the carrying value of its assets or liabilities, and the Company is unable to estimate the potential impact on its future business or our financial results as of the date of this filing. These estimates may change as new events occur and additional information is obtained and are recognized in the consolidated financial statements as soon as they become known.
F-19
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Fair value of financial instruments – The individual fair values attributed to the different components of a financing transaction, are determined using valuation techniques. Management uses judgment to select the methods used to determine certain inputs/assumptions used in the models and the models used to perform the fair value calculations in order to determine, 1) the values attributed to each component of a transaction at the time of their issuance, 2) the fair value measurements for certain instruments that require subsequent measurement at fair value on a recurring basis and 3) the fair value of financial instruments subsequently carried at amortized cost. When the determination of the fair value of a new loan is required, discounted cash flow techniques which includes inputs that are not based on observable market data and inputs that are derived from observable market data are used. When determining the appropriate discount rates to use, Management seeks comparable interest rates where available. If unavailable, it uses those considered appropriate for the risk profile of a Company in the industry.
In determining the fair value of the warrants issued in November 2020 (note 16, 18c), presented as a warrant liability in the consolidated statement of financial position, and considered to be a level 3 measurement, the company made assumptions on unobservable inputs used in the valuation model that have an important impact on the resulting fair value computed.
Notably, the Company estimated the timing and the amounts of equity financings it expects to complete before the expiry of those warrants. The fair value computed could be higher if the actual equity financing needs of the company are higher than those expected. The company also estimated the future volatility of the common shares of Liminal for the contractual life of the warrants. To do so, the Company used the historical volatility its own shares and of comparable companies in the same industry as a starting basis for this estimate and also considered whether there are factors that would indicate that the historical volatility is not indicative of the future. In addition, the company applied an illiquidity discount rate on the resulting Black-Shole pricing model to reflect that the November 2020 warrants are not publicly traded instruments and therefore the ability to sell them is limited. In establishing the illiquidity discount rate, the Company considered the remaining life of the warrants and the volatility assumption for the underlying share. The fair value of the warrants could be higher if we had selected a higher volatility assumption and a lower illiquidity discount rate.
The fair value estimates could be significantly different because of the use of judgment and the inherent uncertainty in estimating the fair value of these instruments that are not quoted in an active market.
Leases - The Company determines the lease term as the non-cancellable term of the lease, together with any periods covered by an option to extend the lease if it is reasonably certain to be exercised, or any periods covered by an option to terminate the lease, if it is reasonably certain that this option will not be exercised.
The Company has the option, under some of its leases to lease the assets for additional terms of up to fifteen years. Judgement is applied in evaluating whether it is reasonably certain that the Company will exercise the option to renew. That is, all relevant factors that create an economic incentive for it to exercise the renewal are considered. After the commencement date, the lease term is reassessed if there is a significant event or change in circumstances that is within the Company’s control and affects its ability to exercise (or not to exercise) the option to renew.
The renewal period is included as part of the lease term for a manufacturing plant lease since the Company estimated it is reasonably certain to exercise due to the importance of this asset to its operations, the limited availability on the market of a similar asset with similar rental terms and the related cost of moving the production equipment to another facility.
F-20
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Uncertainty over income tax treatments - R&D tax credits for the current period and prior periods are measured at the amount the Company expects to recover, based on its best estimate and judgment, of the amounts it expects to receive from the tax authorities as at the reporting date, either in the form of income tax refunds or refundable grants. However, there are uncertainties as to the interpretation of the tax legislation and regulations, in particular regarding what constitutes eligible R&D activities and expenditures, as well the amount and timing of recovery of these tax credits. In order to determine whether the expenses it incurs are eligible for R&D tax credits, the Company must use judgment and apply to complex techniques, which makes the recovery of tax credits uncertain. As a result, there may be a significant difference between the estimated timing and amount recognized in the consolidated financial statements in respect of tax credits receivable and the actual amount of tax credits received as a result of the tax administrations' review of matters that were subject to interpretation. The amounts recognized in the consolidated financial statements are based on the best estimates of the Company and in its best possible judgment, as noted above.
Assessing the recoverable amount of long-lived assets - The Company evaluates the recoverable value of long-lived assets when indicators of impairment arise or as part of the annual impairment test, if they are intangible assets not yet available for use. The recoverable value is the higher of the value in use and the fair value less costs of disposal or FVLCD.
Long-lived assets include capital assets, ROU assets and intangible assets such as patents and licenses and other rights. Some of these rights are considered not available for use until regulatory approval to commercialize the product candidate is obtained.
When calculating the net recoverable amounts for the impairments discussed in note 24, management proceeded to make estimates and assumptions regarding the outcome of certain future events, future cash flows and their timing.
When determining the FVLCD, significant estimates made include amongst others, the outcome of the exercise it has undertaken in evaluating the potential alternatives for the Ryplazim® CGU, including the probability of completing a sale or closing those activities; the operating cash outflows to support those operations until one of the alternative strategies is executed; the outcome of the FDA review of the Company’s Biological License Application, or BLA for its Ryplazim® product candidate and the timing of completion of this review; if the Company will be able to benefit from the monetization of a Priority Review Voucher, if received, and what would be the amount received upon its monetization; and whether some assets, liabilities and commitments could potentially be excluded from the activities sold and for those commitments that could be retained, the possibility of reducing those commitments and what would be their settlement amount. In addition, when calculating the FVLCD of an asset or a group of assets for which selling price information for comparable assets are not readily available, management also must make assumptions regarding the value it may recuperate from its sale.
A plus or minus 10% change in the probability weighted terminal value would impact the impairment recorded on the Ryplazim® CGU by $ 3,638.
In determining the value in use for the IVIG CGU at December 31, 2018, management made a series of estimates at the time regarding the time period in which the Company could possibly resume the activities of this CGU and for earning revenues from the IVIG product candidate, if approved.
When the recoverable amounts are calculated using a discounted cash flow model, the estimated cash flows are discounted to their net present value using a pre-tax discount rate that includes a risk premium specific to the line of business.
F-21
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Share-based compensation - The expense recognized for those RSU which the performance conditions have not been met, is based on an estimation of the probability of successful achievement of a number of performance conditions, many of which depend on research, regulatory process and business development outcomes which are difficult to predict, as well as the timing of their achievement. The final expense is only determinable when the outcome is known.
To determine the fair value of stock options on a given date, the Company must determine the assumptions that will be used as inputs to the Black-Scholes option pricing model, including the assumption regarding the future volatility of the common shares of Liminal for the expected life of the stock options. The Company uses the historical volatility as a starting basis for the estimate and also considers whether there are factors that would indicate that the past volatility is not indicative of the future volatility. In making this assessment, management considers changes in Liminal’s activities and other factors such as a significant share consolidation. As the volatility is an assumption that has a significant impact on the calculated value of a stock option, the impact of this estimate can significantly impact the share-based payment expense over the vesting period of an award.
Valuation of deferred income tax assets – To determine the extent to which deferred income tax assets can be recognized, management estimates the amount of probable future taxable profits that will be available against which deductible temporary differences and unused tax losses can be utilized. Management exercises judgment to determine the extent to which realization of future taxable benefits is probable, considering the history of taxable profits, budgets and forecasts and availability of tax strategies.
4. | Change in standards, interpretations and accounting policies |
a) | Adoption of new accounting standards |
The accounting policies used in these annual consolidated financial statements are consistent with those applied by the Company in its December 31, 2019 and 2018 audited annual consolidated financial statements except for the amendments to certain accounting standards which are relevant to the Company and were adopted by the Company as of January 1, 2019 and January 1, 2020 as described below.
IFRS 16, Leases or IFRS 16
IFRS 16 replaces IAS 17, Leases, or IAS 17. IFRS 16 provides a single lessee accounting model, requiring the recognition of assets and liabilities for all leases, unless the lease term is less than 12 months, or the underlying asset has a low value. IFRS 16 substantially carries forward the lessor accounting in IAS 17 with the distinction between operating leases and finance leases being retained.
Effective January 1, 2019, the Company adopted IFRS 16 using the modified retrospective approach and accordingly the information presented for 2018 has not been restated. The cumulative effect of initially applying the standard is recognized at the date of initial application. The current and long-term portions of operating and finance lease inducements and obligations presented in the statement of financial position at December 31, 2018, reflect the accounting treatment under IAS 17 and related interpretations.
The Company elected to use the transitional practical expedient allowing the standard to be applied only to contracts that were previously identified as leases under IAS 17 and IFRIC 4, Determining whether an arrangement contains a lease at the date of initial application. The Company applied the definition of a lease under IFRS 16 to contracts entered into or changed on or after January 1, 2019.
F-22
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The Company also elected to record right-of-use assets for leases previously classified as operating leases under IAS 17 based on the corresponding lease liability, adjusted for prepaids or liabilities existing at the date of the transition that relate to the lease. When measuring lease liabilities, the Company discounted lease payments using its incremental borrowing rate at January 1, 2019. The weighted average discount rate applied to the total lease liabilities recognized on transition was 18.54%. For leases that were previously classified as finance leases under IAS 17, the carrying amount of the right-of-use asset and the lease liability at the date of adoption was established as the carrying amount of the lease asset classified in capital assets and the finance lease obligation at December 31, 2018. These assets and liabilities are grouped under right-of-use assets and lease liabilities as of January 1, 2019 and IFRS 16 applies to these leases as of that date.
In addition, the Company elected to apply the practical expedient to account for leases for which the lease term ends within 12 months of the date of initial application as short-term leases for which it is not required to recognize a right-of-use asset and a corresponding lease liability. The Company also elected to not apply IFRS 16 when the underlying asset in a lease is of low value.
The Company has elected, for the class of assets related to the lease of building space, not to separate non-lease components from lease components, and instead account for each lease component and any associated non-lease components as a single lease component.
The table below shows which line items of the consolidated financial statements were affected by the adoption of IFRS 16 and the impact. There was no net impact on the deficit.
| | | | | | | Adjustments | | | | | |
| | | As reported as at | | | for the transition | | | Balance as at | |
| | | December 31, 2018 | | | to IFRS 16 | | | January 1, 2019 | |
Assets | | | | | | | | | | | | | |
Prepaids | | | $ | 1,452 | | | $ | (84 | ) | | $ | 1,368 | |
Capital assets (note 10) | | | | 41,113 | | | | (1,043 | ) | | | 40,070 | |
Right-of-use assets (note 11) | | | | — | | | | 39,149 | | | | 39,149 | |
Liabilities | | | | | | | | | | | | | |
Accounts payable and accrued liabilities (note 13) | | | $ | 31,855 | | | $ | (2,499 | ) | | $ | 29,356 | |
Current portion of lease liabilities (note 14) | | | | — | | | | 8,575 | | | | 8,575 | |
Long-term portion of lease liabilities (note 14) | | | | — | | | | 34,126 | | | | 34,126 | |
Long-term portion of operating and finance lease inducements and obligations | | | | 1,850 | | | | (1,850 | ) | | | — | |
Other long-term liabilities (note 17) | | | | 5,695 | | | | (330 | ) | | | 5,365 | |
F-23
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Prior to adopting IFRS 16, the total minimum operating lease commitments as at December 31, 2018 were $74,977. The decrease between the total of the minimum lease payments set out in Note 31 of the audited annual consolidated financial statements for the year ended December 31, 2018 and the total lease liabilities recognized on adoption of $42,701 was principally due to the effect of discounting on the minimum lease payments. The amount also decreased slightly due to the fact that certain costs that are contractually committed under lease contracts, but which do not qualify to be accounted for as a lease liability, such as variable lease payments not tied to an index or rate, were previously included in the lease commitment table whereas they are not included in the calculation of the lease liabilities. These impacts were partially offset by the inclusion of lease payments beyond minimum commitments relating to reasonably certain renewal periods that had not yet been exercised as at December 31, 2018 which effect is to increase the liability. Right-of-use assets at transition have been measured at an amount equal to the corresponding lease liabilities, adjusted for any prepaid or accrued rent relating to that lease.
The consolidated statement of operations for the year ended December 31, 2019 and onwards were impacted by the adoption of IFRS 16 as the recording of depreciation of the right-of-use assets continues to be recorded in the same financial statement line items as it was previously while the implicit financing component of leasing agreements is now recorded under finance costs. The impact is not simply in the form of a reclassification but also in terms of measurement, which are very much affected by the discount rates used and whether the Company has included renewal periods when calculating the lease liability.
The consolidated cash flow statement for the year ended December 31, 2019 and onwards were also impacted since the cash flows attributable to the lease component of the lease agreements are now shown as payments of principal and interest on lease liabilities which are now part of cash flows from financing activities.
IFRIC 23, Uncertainty over income tax treatments or IFRIC 23
IFRIC 23 clarifies how the recognition and measurement requirements of IAS 12 – Income Taxes are applied where there is uncertainty over income tax treatments. The Interpretation is effective for annual periods beginning on or after January 1, 2019 and was adopted by the Company on that date. The Company assessed the impact of this Interpretation and concluded that it had no impact on the amounts recorded in its consolidated statements of financial position on the date of adoption.
Amendments to IFRS 3, Business Combinations or IFRS 3
The amendments to IFRS 3 clarifies the definition of a business and includes an optional concentration test to determine whether an acquired set of activities and assets is a business. These amendments were adopted on January 1, 2020 and are applied prospectively to acquisitions made on or after this date.
b) | New Standards and interpretations not yet adopted |
The IFRS accounting standards, amendments, and interpretations that the Company reasonably expects may have a material impact on the disclosures, the financial position or results of operations of the Company when applied at a future date are as follows:
Amendment to IFRS 16, Leases or IFRS 16 for COVID-19-Related Rent Concessions - IFRS 16 has been revised to incorporate an amendment issued by the IASB in May 2020. The amendment permits lessees not to assess whether particular COVID-19-related rent concessions are lease modifications and, instead, account for those rent concessions as if they were not lease modifications. In addition, the amendment to IFRS 16 provides specific disclosure requirements regarding COVID-19-related rent concessions. The amendment is effective for annual reporting periods beginning on or after June 1, 2020 and earlier application is permitted. Presently, the Company has not benefited from COVID-19 related rent concessions.
F-24
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Amendments to IAS 37, Provisions, Contingent Liabilities and Contingent Assets or IAS 37 - IAS 37 has been revised to specify which costs an entity includes in determining the cost of fulfilling a contract for the purpose of assessing whether the contract is onerous. The amendments are effective for annual reporting periods beginning on or after January 1, 2022. The cumulative effect of initially applying the amendment, if any, will be recorded as an adjustment to the opening retained earnings and comparative periods will not be restated. Earlier application is permitted.
Amendment to IFRS 9 Financial Instruments or IFRS 9 - IFRS 9 has been revised to clarify the fees an entity includes when assessing whether the terms of a new or modified financial liability are substantially different from the terms of the original financial liability. The amendment is effective for annual reporting periods beginning on or after January 1, 2022 and is to be applied to financial liabilities that are modified after the date of adoption. Earlier application is permitted.
Amendments to IAS 1, Presentation of Financial Statements or IAS 1 - IAS 1 has been revised to clarify how to classify debt and other liabilities as current or non-current. The amendments help to determine whether, in the statement of financial position, debt and other liabilities with an uncertain settlement date should be classified as current (due or potentially due to be settled within one year) or non-current. The amendments also include clarifying the classification requirements for debt an entity might settle by converting it into equity. The amendments are applicable retrospectively and is effective for annual reporting periods beginning on or after January 1, 2023 with earlier application permitted.
At the present time, the Company does not expect the amendments to IFRS 16, IAS 37, IFRS 9 and IAS 1 will have a significant effect on its financial statements when these amendments are adopted by the Company. This assessment may change as we approach the various dates of adoption as additional amendments may be issued and new transactions occur.
5. | Acquisition of Fairhaven Pharmaceuticals Inc. |
Pursuant to a share purchase agreement, or SPA, dated July 17, 2020, the Company acquired 100% of the issued and outstanding common shares of Fairhaven Pharmaceuticals Inc., or Fairhaven, a company with a preclinical research program of small molecule antagonists. As consideration for the acquisition, the Company issued 202,308 common shares. Upon achievement of certain pre-determined research and development milestones prior to July 17, 2025, the Company may be obligated to make additional payments in the form of common shares totalling up to $4,374. The number of shares to be issued, if any, upon completion of a milestone, will be calculated using the five-trading day volume weighted average trading price, or VWAP of the Company’s common shares on Nasdaq prior to the achievement of such milestone events.
As Fairhaven did not meet the definition of a business under IFRS 3, the acquisition has been accounted for as an asset acquisition, the total cost of the net assets acquired being the fair value of the consideration paid. The shares issued were recorded at a fair value of $3,441, based on the closing price of Liminal’s common shares at the date of the transaction. The transaction costs of $308 incurred by the Company were capitalized and allocated to the net assets acquired. Any future milestone payments would be recognized if and when the triggering event occurs.
F-25
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The consideration paid and the allocation thereof to the net assets acquired was as follows:
Cost of acquisition | | | | |
Fair value of common shares issued | | $ | 3,441 | |
Cash payment | | | 50 | |
Total consideration paid | | $ | 3,491 | |
Transaction fees | | | 308 | |
Total cost of acquisition | | $ | 3,799 | |
| | | | |
Net assets acquired | | | | |
Current assets | | $ | 217 | |
Licenses and other rights (note 12) | | | 3,796 | |
Current liabilities | | | (214 | ) |
Total net assets acquired | | $ | 3,799 | |
6. | Discontinued operations |
On November 25, 2019, the Company sold two subsidiaries in its bioseparations segment, representing the majority of its bioseparations operations and all of the bioseparations revenues. This transaction was part of the Company’s goal to monetize non-core assets to focus resources on the small molecules segment. This disposal has been presented as discontinued operations with the revenues and costs relating to ceased activities being reclassified and presented retrospectively in the consolidated statements of operations, statements of comprehensive loss, statements of cash flows and notes to the financial statements as discontinued operations. The 2019 and 2018 years in the segmented information note were also restated to present the existing segments as of the reporting date.
Gain on the sale of subsidiaries
The details of the gain on sale of subsidiaries during the years ended December 31, 2020 and 2019 is provided in the table bellow:
Year ended December 31 | | | | 2020 | | | 2019 | |
Fair value of the consideration received and receivable: | | $ | 3,380 | | | $ | 51,927 | |
Less: | | | | | | | |
Carrying amount of net assets sold | | — | | | | (22,015 | ) |
Transaction costs | | — | | | | (5,015 | ) |
Add: Reclassification of foreign currency translation reserve from other comprehensive income into the statement of operations | | — | | | | 1,449 | |
Gain on sale of subsidiaries (income tax $nil) | | $ | 3,380 | | | $ | 26,346 | |
F-26
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
As of December 31, 2019, the Company had received $50,752 in cash and recorded an amount receivable of $1,175. This amount was received in the beginning of 2020 and then later during the year, an additional amount of 3,380 in proceeds was recorded and received upon resolution of a taxation matter. In the event the operations sold achieve certain yearly performance criteria during the three years following the transaction, the additional cash payments may be received. As of the date of these consolidated financial statements, the aggregate cash consideration that could still be earned until the end of the performance period is $19,129 (£11,000,000). At the time of the sale and as at December 31, 2020 and 2019, the fair value of the contingent consideration was determined to be $nil as its receipt is dependent on future target achievement that is out of the Company’s influence and is primarily dependent on the growth of the operations sold.
Results and cash flows from discontinued operations
The net income from discontinued operations for the years ended December 31, 2019 and 2018 are presented below:
Year ended December 31 | | | | 2019 | | | 2018 | |
Revenues | | | | | $ | 22,499 | | | $ | 22,741 | |
| | | | | | | | | | | |
Expenses | | | | | | | | | | | |
Cost of sales and other production expenses | | | | | | 11,347 | | | | 12,295 | |
Research and development expenses | | | | | | 5,926 | | | | 6,808 | |
Administration, selling and marketing expenses | | | | | | 3,387 | | | | 2,084 | |
Loss (gain) on foreign exchange | | | | | | (64 | ) | | | (15 | ) |
Finance costs | | | | | | 737 | | | | 19 | |
Net income before income taxes | | | | | $ | 1,166 | | | $ | 1,550 | |
Income tax expense (recovery): | | | | | | | | | | | |
Current | | | | | | 65 | | | | (382 | ) |
Deferred | | | | | | (24 | ) | | | — | |
Total income tax expense (recovery) | | | | | | 41 | | | | (382 | ) |
Net income from discontinued operations | | | | | $ | 1,125 | | | $ | 1,932 | |
The cash flows from the discontinued operations and the gain on sale of subsidiaries for the years ended December 31, 2020, 2019 and 2018 are presented in the following table:
Year ended December 31 | | 2020 | | | 2019 | | | 2018 | |
Cash flows from operating activities | | | $ | — | | | $ | 6,327 | | | $ | 1,379 | |
Cash flows used in financing activities | | | | — | | | | (866 | ) | | | — | |
Cash flows from (used in) investing activities* | | | | 3,768 | | | | 39,690 | | | | (1,752 | ) |
Cash generated (used) during the year | | | $ | 3,768 | | | $ | 45,151 | | | $ | (373 | ) |
Net effect of currency exchange rate on cash | | | | — | | | | 54 | | | | 41 | |
Total cash generated (used) by discontinued operations | | | $ | 3,768 | | | $ | 45,205 | | | $ | (332 | ) |
F-27
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
*Cash flows from investing activities for the year ended December 31, 2020 include proceeds received from the sales of the discontinued operations business of $4,555, net of the transaction cost paid of $787. Cash flows from investing activities for the period ended December 31, 2019 include the proceeds from the sale of the discontinued operations business (net of the cash disposed) of $43,958 deduction made of the transaction costs paid of $4,228.
The carrying amounts of assets and liabilities sold on the date of the transaction are as follows:
| | | | | | | | | | | |
Cash | | | | | | | | | $ | 6,794 | |
Accounts receivable | | | | | | | | | | 1,148 | |
Inventories | | | | | | | | | | 8,313 | |
Prepaids | | | | | | | | | | 236 | |
Other long-term assets | | | | | | | | | | 48 | |
Capital assets | | | | | | | | | | 8,483 | |
Right-of-use assets | | | | | | | | | | 3,300 | |
Intangible assets | | | | | | | | | | 370 | |
Deferred tax assets | | | | | | | | | | 12 | |
Total assets | | | | | | | | | $ | 28,704 | |
Accounts payable and accrued liabilities | | | | | | | | | | 2,163 | |
Deferred revenue | | | | | | | | | | 370 | |
Current portion of lease liabilities | | | | | | | | | | 809 | |
Long-term portion of deferred revenues | | | | | | | | | | 87 | |
Long-term portion of lease liabilities | | | | | | | | | | 3,260 | |
Total liabilities | | | | | | | | | $ | 6,689 | |
Net assets sold | | | | | | | | | $ | 22,015 | |
7. | Accounts receivable and others |
| | | | | | | December 31, | | | December 31, | |
| | | | | | | 2020 | | | 2019 | |
Trade receivables | | | | $ | 943 | | | $ | 44 | |
Tax credits and government grants receivable | | | | | 1,808 | | | | 1,546 | |
Sales taxes receivable | | | | | 431 | | | | 863 | |
Restricted cash | | | | | 178 | | | | — | |
Other receivables | | | | | 721 | | | | 1,633 | |
| | | | | | | $ | 4,081 | | | $ | 4,086 | |
Restricted cash is composed of a guaranteed investment certificate, bearing interest at 0.35% per annum (at December 31, 2019, bearing interest at 0.35%), pledged as collateral for a letter of credit to a landlord.
F-28
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
| | | | | | | December 31, | | | December 31, | |
| | | | | | | 2020 | | | 2019 | |
Raw materials | | | | | | | $ | 9,138 | | | $ | 7,175 | |
Finished goods | | | | | | | | 239 | | | | 357 | |
| | | | | | | $ | 9,377 | | | $ | 7,532 | |
Inventories sold in the amount of $1,102, $2,315 and $23,136 were recognized in cost of sales and other production expenses from continuing operations, and inventories sold in the amount of $nil, $10,126 and $10,295 were included in the results from discontinued operations during the years ended December 31, 2020, 2019 and 2018 respectively. Inventory write-downs included in cost of sales and other production expenses from continuing operations for the year ended December 31, 2018 were $2,028 while inventory write-downs for 2019 and 2020 were insignificant. Inventory write-downs affecting the results from discontinued operations were $nil, $642 and $981 during the years ended December 31, 2020, 2019 and 2018 respectively.
| | | | | | | December 31, | | | December 31, | |
| | | | | | | 2020 | | | 2019 | |
Restricted cash (note 7) | | | | | | | $ | — | | | $ | 169 | |
Long-term deposits | | | | | | | | 137 | | | | 143 | |
Tax credits receivable | | | | | | | | 1,216 | | | | 858 | |
| | | | | | | $ | 1,353 | | | $ | 1,170 | |
F-29
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
| | | | | | | | | | Production | | | Furniture and | | | | | |
| | Land and | | | Leasehold | | | and laboratory | | | computer | | | | | |
| | Buildings | | | improvements | | | equipment | | | equipment | | | Total | |
Cost | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2018 | | $ | 4,567 | | | $ | 16,034 | | | $ | 38,885 | | | $ | 3,786 | | | $ | 63,272 | |
Impact of adopting IFRS 16 1) | | | — | | | | — | | | | (1,170 | ) | | | — | | | | (1,170 | ) |
Balance at January 1, 2019 | | $ | 4,567 | | | $ | 16,034 | | | $ | 37,715 | | | $ | 3,786 | | | $ | 62,102 | |
Additions | | | — | | | | 61 | | | | 712 | | | | 202 | | | | 975 | |
Disposals | | | — | | | | (5 | ) | | | (109 | ) | | | (14 | ) | | | (128 | ) |
Sold - discontinued operations (note 6) | | | — | | | | (7,307 | ) | | | (5,774 | ) | | | (744 | ) | | | (13,825 | ) |
Effect of foreign exchange differences | | | — | | | | (225 | ) | | | (127 | ) | | | (7 | ) | | | (359 | ) |
Balance at December 31, 2019 | | | 4,567 | | | | 8,558 | | | | 32,417 | | | | 3,223 | | | | 48,765 | |
Additions | | | — | | | | 214 | | | | 295 | | | | 550 | | | | 1,059 | |
Disposals | | | — | | | | (1,380 | ) | | | (2,791 | ) | | | (404 | ) | | | (4,575 | ) |
Effect of foreign exchange differences | | | — | | | | (43 | ) | | | (17 | ) | | | (4 | ) | | | (64 | ) |
Balance at December 31, 2020 | | $ | 4,567 | | | $ | 7,349 | | | $ | 29,904 | | | $ | 3,365 | | | $ | 45,185 | |
| | | | | | | | | | | | | | | | | | | | |
Accumulated depreciation | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2018 | | $ | 414 | | | $ | 4,421 | | | $ | 15,071 | | | $ | 2,253 | | | $ | 22,159 | |
Impact of adopting IFRS 16 1) | | | — | | | | — | | | | (127 | ) | | | — | | | | (127 | ) |
Balance at January 1, 2019 | | $ | 414 | | | $ | 4,421 | | | $ | 14,944 | | | $ | 2,253 | | | $ | 22,032 | |
Depreciation expense | | | 195 | | | | 786 | | | | 2,136 | | | | 617 | | | | 3,734 | |
Disposals | | | — | | | | (2 | ) | | | (106 | ) | | | (14 | ) | | | (122 | ) |
Impairments (note 24) | | | — | | | | 559 | | | | 6,408 | | | | 103 | | | | 7,070 | |
Sold - discontinued operations (note 6) | | | — | | | | (2,297 | ) | | | (2,550 | ) | | | (495 | ) | | | (5,342 | ) |
Effect of foreign exchange differences | | | — | | | | (38 | ) | | | (36 | ) | | | (4 | ) | | | (78 | ) |
Balance at December 31, 2019 | | $ | 609 | | | $ | 3,429 | | | $ | 20,796 | | | $ | 2,460 | | | $ | 27,294 | |
Depreciation expense | | | 195 | | | | 710 | | | | 1,450 | | | | 424 | | | | 2,779 | |
Disposals | | | — | | | | (1,380 | ) | | | (2,527 | ) | | | (404 | ) | | | (4,311 | ) |
Impairments (note 24) | | | — | | | | 167 | | | | 498 | | | | — | | | | 665 | |
Effect of foreign exchange differences | | | — | | | | (25 | ) | | | (9 | ) | | | 1 | | | | (33 | ) |
Balance at December 31, 2020 | | $ | 804 | | | $ | 2,901 | | | $ | 20,208 | | | $ | 2,481 | | | $ | 26,394 | |
| | | | | | | | | | | | | | | | | | | | |
Carrying amounts | | | | | | | | | | | | | | | | | | | | |
At December 31, 2020 | | $ | 3,763 | | | $ | 4,448 | | | $ | 9,696 | | | $ | 884 | | | $ | 18,791 | |
At December 31, 2019 | | | 3,958 | | | | 5,129 | | | | 11,621 | | | | 763 | | | | 21,471 | |
1) The balance of fixed assets capitalized as finance lease assets under IAS 17 was transferred to right-of-use assets upon adoption of IFRS 16 (note 4).
The depreciation expense for the year ended December 31, 2018 was $4,086.
Certain investments in equipment are eligible for government grants. The government grants receivable are recorded in the same period as the eligible additions and are credited against the capital asset addition. During the year ended December 31, 2020, the Company recognized $nil ($694 during the year ended December 31, 2019) in government grants against the equipment cost.
Impairment losses of $665 were recorded on capital assets during the year ended December 31, 2020 ($7,070 during the year ended December 31, 2019, $5,689 during the year ended December 31, 2018). Details of these impairments are provided in note 24.
F-30
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
| | | | | | Production | | | | | | | | | |
| | | | | | and laboratory | | | | | | | | | |
| | Buildings | | | equipment | | | Other | | | Total | |
Transfer from capital assets on adoption of IFRS 16 (note 10) | | $ | — | | | $ | 1,043 | | | $ | — | | | $ | 1,043 | |
Initial recognition of assets under operating leases on adoption of IFRS 16 | | | 37,552 | | | | 460 | | | | 94 | | | | 38,106 | |
Balance at January 1, 2019 | | $ | 37,552 | | | $ | 1,503 | | | $ | 94 | | | $ | 39,149 | |
Additions | | | 2,331 | | | | — | | | | 49 | | | | 2,380 | |
Lease modifications and other remeasurements | | 36 | | | | — | | | | — | | | 36 | |
Depreciation expense | | | (4,274 | ) | | | (592 | ) | | | (47 | ) | | | (4,913 | ) |
Sold - discontinued operations (note 6) | | | (3,300 | ) | | | — | | | | — | | | | (3,300 | ) |
Effect of foreign exchange differences | | | (99 | ) | | | 1 | | | | — | | | | (98 | ) |
Net book value as at January 1, 2020 | | $ | 32,246 | | | $ | 912 | | | $ | 96 | | | $ | 33,254 | |
Additions | | | 378 | | | | 151 | | | | 15 | | | | 544 | |
Lease modifications and other remeasurements | | | (1,998 | ) | | | — | | | | — | | | | (1,998 | ) |
Depreciation expense | | | (3,956 | ) | | | (561 | ) | | | (61 | ) | | | (4,578 | ) |
Impairment (note 24) | | | (18,553 | ) | | | (70 | ) | | | (4 | ) | | | (18,627 | ) |
Effect of foreign exchange differences | | | (31 | ) | | | (6 | ) | | | (1 | ) | | | (38 | ) |
Net book value at December 31, 2020 | | $ | 8,086 | | | $ | 426 | | | $ | 45 | | | $ | 8,557 | |
F-31
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
| | | | Licenses and other rights | | | Patents | | | Software | | | Total | |
Cost | | | | | | | | | | | | | | | | | | |
Balance at January 1, 2019 | | | | $ | 160,782 | | | $ | 6,997 | | | $ | 3,286 | | | $ | 171,065 | |
Additions | | | | | — | | | | 728 | | | | 467 | | | | 1,195 | |
Sold - discontinued operations (note 6) | | | | | (2,505 | ) | | | (842 | ) | | | (47 | ) | | | (3,394 | ) |
Disposals | | | | | — | | | | (524 | ) | | | (39 | ) | | | (563 | ) |
Effect of foreign exchange differences | | | | | (9 | ) | | | (50 | ) | | | (19 | ) | | | (78 | ) |
Balance at December 31, 2019 | | | | $ | 158,268 | | | $ | 6,309 | | | $ | 3,648 | | | $ | 168,225 | |
Additions | | | | | 3,796 | | | | 668 | | | | 29 | | | | 4,493 | |
Disposals | | | | | — | | | | (179 | ) | | | (362 | ) | | | (541 | ) |
Effect of foreign exchange differences | | | | | — | | | | (15 | ) | | | (9 | ) | | | (24 | ) |
Balance at December 31, 2020 | | | | $ | 162,064 | | | $ | 6,783 | | | $ | 3,306 | | | $ | 172,153 | |
| | | | | | | | | | | | | | | | | | |
Accumulated amortization | | | | | | | | | | | | | | | | | | |
Balance at January 1, 2019 | | | | $ | 147,356 | | | $ | 2,838 | | | $ | 1,068 | | | $ | 151,262 | |
Amortization expense | | | | | 410 | | | | 403 | | | | 446 | | | | 1,259 | |
Disposals | | | | | — | | | | (364 | ) | | | (9 | ) | | | (373 | ) |
Impairments (note 24) | | | | | 4,528 | | | | 761 | | | | 7 | | | | 5,296 | |
Sold - discontinued operations (note 6) | | | | | (2,418 | ) | | | (570 | ) | | | (36 | ) | | | (3,024 | ) |
Effect of foreign exchange differences | | | | | (6 | ) | | | (29 | ) | | | (6 | ) | | | (41 | ) |
Balance at December 31, 2019 | | | | $ | 149,870 | | | $ | 3,039 | | | $ | 1,470 | | | $ | 154,379 | |
Amortization expense | | | | | 178 | | | | 353 | | | | 559 | | | | 1,090 | |
Disposals | | | | | — | | | | (23 | ) | | | (333 | ) | | | (356 | ) |
Impairments (note 24) | | | | | 480 | | | | 1,072 | | | | 15 | | | | 1,567 | |
Effect of foreign exchange differences | | | | | — | | | | (12 | ) | | | (7 | ) | | | (19 | ) |
Balance at December 31, 2020 | | | | $ | 150,528 | | | $ | 4,429 | | | $ | 1,704 | | | $ | 156,661 | |
| | | | | | | | | | | | | | | | | | |
Carrying amounts | | | | | | | | | | | | | | | | | | |
At December 31, 2020 | | | | $ | 11,536 | | | $ | 2,354 | | | $ | 1,602 | | | $ | 15,492 | |
At December 31, 2019 | | | | | 8,398 | | | | 3,270 | | | | 2,178 | | | | 13,846 | |
At December 31, 2020, intangible assets include $7,106 pertaining to a reacquired right from a licensee; these rights are not yet available for use and consequently their amortization has not commenced (note 17a,ii).
Impairment losses of $1,567, $5,296 and $142,609 were recorded on certain licenses and patents during the years ended December 31, 2020, 2019 and 2018 respectively (note 24).
The amortization expense for the year ended December 31, 2018 was $1,372.
F-32
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
13. | Accounts payable and accrued liabilities |
| | | | | | | | |
| | | | | | | December 31, 2020 | | | December 31, 2019 | |
Trade payables | | | | | | | $ | 9,153 | | | $ | 10,496 | |
Wages and benefits payable | | | | | | | | 3,083 | | | | 5,593 | |
Current portion of royalty payment obligations (note 17) | | | | | | | | 3,248 | | | | 3,043 | |
Current portion of license acquisition payment obligation (note 17) | | | | | | | | — | | | | 1,302 | |
Current portion of other employee benefit liabilities (note 17) | | | | | | | | 1,351 | | | | 2,374 | |
| | | | | | | $ | 16,835 | | | $ | 22,808 | |
The transactions affecting the lease liabilities during the years ended December 31, 2020 and 2019 were as follows:
| | | | | | | 2020 | | | 2019 | |
Transfer of finance lease from operating and finance lease inducements and obligations | | $ | — | | | $ | 846 | |
initial recognition of lease liabilities under operating leases on adoption of IFRS 16 | | | — | | | | 41,855 | |
Balance at January 1 | | $ | 38,237 | | | $ | 42,701 | |
Additions | | | 544 | | | | 2,823 | |
Interest expense | | | 6,030 | | | | 7,068 | |
Payments | | | (9,167 | ) | | | (9,330 | ) |
Derecognized - discontinued operations (note 6) | | | — | | | | (4,069 | ) |
Lease modification and other remeasurements | | | (1,934 | ) | | | — | |
Effect of foreign exchange differences | | | (258 | ) | | | (956 | ) |
Balance at December 31 | | $ | 33,452 | | | $ | 38,237 | |
Less current portion of lease liabilities | | | 6,946 | | | | 8,290 | |
Long-term portion of lease liabilities | | $ | 26,506 | | | $ | 29,947 | |
The interest expense on lease liabilities is included as part of finance costs in the consolidated statement of operations.
F-33
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
2020
As part of the consideration for the private placement completed on November 3, 2020 (note 18a, 18c) where SALP and another investor participated equally, the Company issued 6,315,788 warrants that expire on November 3, 2025. On November 25, 2020, the Company issued 1,578,946 additional warrants with the same terms and conditions as described above for no additional consideration, following an amendment to the private placement agreement. Both of these issuances combined will be referred to as the November 2020 warrants. Each warrant can be exercised to acquire one common share at an exercise price initially set at US$5.50 and that can be reduced if equity financings are completed at a lower price before its expiry. The November 2020 warrants do not meet the definition of an equity instrument since the exercise price is denominated in US$ which is different than the functional currency of Liminal which is the CA$. Consequently, they are accounted for as a financial instrument, presented as a warrant liability in the consolidated statement of financial position and carried at fair value through profit or loss.
The fair value of the warrants issued on November 3, 2020 and then on November 25, 2020 were $10,263 and $2,227 respectively. The portion of the total issuance cost pertaining to the private placement allocated to the issuance of the November 3, 2020 warrants of $709 and the fair value of the additional warrants issued on November 25, 2020 were recorded in the consolidated statement of operation transactions in financing costs and administration, selling and marketing expenses respectively. The fair value of the warrant liability of the November 2020 warrants was $11,640 at December 31, 2020 ($5,820 for the November 2020 warrants held by SALP). The gain of $850 resulting from the change in fair value of the warrants since their issuance was recognized in the statement of operations for the year ended December 31, 2020.
The fair value of the November 2020 warrants on the various dates discussed above was calculated using a Black-Scholes option pricing model in a Monte Carlo simulation in order to evaluate the downward adjustment mechanism to the exercise price. The assumptions used at the different valuation dates are provided in the table below:
| | | December 31, | | | November 25, | | | November 3, | |
| | | | | 2020 | | | 2020 | | | 2020 | |
Underlying common share fair value (in US$) | | $ | 4.20 | | | $ | 3.97 | | | $ | 4.30 | |
Remaining life until expiry | | | 4.8 | | | | 4.9 | | | | 5.0 | |
Volatility | | | 49.0 | % | | | 49.0 | % | | | 49.0 | % |
Risk-free interest rate | | | 0.34 | % | | | 0.38 | % | | | 0.39 | % |
Expected dividend rate | | | — | | | | — | | | | — | |
Fair value of a warrant calculated using a Black-Sholes pricing model (in US$) | | $ | 1.41 | | | $ | 1.29 | | | $ | 1.53 | |
Fair value of exercise price adjustment mechanism (in US$) | | $ | 0.22 | | | $ | 0.24 | | | $ | 0.22 | |
Illiquidity discount | | | 29.0 | % | | | 29.0 | % | | | 30.0 | % |
Fair value of a warrant (in US$) | | $ | 1.16 | | | $ | 1.08 | | | $ | 1.22 | |
Fair value of a warrant (in CA$) | | $ | 1.47 | | | $ | 1.41 | | | $ | 1.62 | |
F-34
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
2019
As consideration for the modification of the terms of the loan agreements between Liminal and SALP on November 14, 2018, the Company had a commitment to issue warrants, or Warrants #9, to SALP on or before March 20, 2019. The exact number of warrants to be issued was based on the number of warrants necessary to increase the ownership of SALP to 19.99% on a fully diluted basis at the date of issuance.
On February 22, 2019, the Company further amended the fourth loan agreement with SALP with the addition of two tranches, one of US$10 million and another one of US$5 million, that were drawn on February 22, 2019 and March 22, 2019 respectively. As consideration for the modification to the fourth loan agreement, the Company amended the terms applicable at the time of issuance of Warrants #9 to reduce the originally agreed exercise price from $1,000.00 to $156.36 per preferred share and to issue the Warrants #9 concurrently with this modification. Accordingly, the Company issued 19,402 warrants on February 22, 2019. Each warrant entitles the holder to acquire one preferred share (note 18c) at a price of $156.36 per preferred share and expires on February 22, 2027. The Warrants #9 did not meet the definition of an equity instrument since the underlying preferred shares qualify as a liability instrument, and therefore they were accounted for as a financial instrument carried at fair value through profit or loss and were presented in the consolidated statement of financial position as a warrant liability.
The change in fair value of the warrant liability between December 31, 2018, when it was valued at $157 and prior to its modification on February 22, 2019, in the amount of $218 was recorded in the consolidated statement of operations. The Company recorded the increase in fair value of the warrants of $1,137 resulting from the reduction of the exercise price of Warrants #9 on February 22, 2019 against the two additional tranches of the credit facility, treating the increase as financing fees. The change in fair value of the warrant liability between February 22, 2019, after the modification, and March 31, 2019 was an increase of $11 and a decrease in fair value of $1,369 (a gain) between March 31, 2019 to April 23, 2019. Both variations were recorded in the consolidated statements of operations. The estimated fair value of these warrants at April 23, 2019 was $153.
As part of the debt restructuring agreement entered into on April 23, 2019 (note 16), all the outstanding warrants belonging to SALP, including the Warrants #9, were cancelled and replaced by new warrants (note 18c). The cancellation and the issuance of new warrants was treated as a modification. Following this modification, the Warrants #9 no longer meet the definition of a liability instrument and the Company reclassified the fair value of the Warrants #9 as of April 23, 2019 of $153 from warrant liability to warrants classified as equity.
The fair value of Warrants #9 on the various dates was calculated using a Black-Scholes option pricing model with the assumptions provided in the table below. In order to estimate the fair value of the underlying preferred share, the Company used the market price of Liminal’s common shares at the measurement date, discounted for the fact that the preferred shares are illiquid. The value of the discount was calculated using a European put option model to sell a common share of Liminal at the price of $1,000.00 or $156.36 per share in 20 years.
| | | | | April 23, | | | February 22, | | | December 31, | |
| | | | | 2019 | | | 2019 | | | 2018 | |
Underlying preferred share fair value | | | | | | 32.43 | | | | 152.15 | | | | 130.00 | |
Number of warrants issued | | | | | | 19,402 | | | | 19,402 | | | | 14,088 | |
Volatility | | | | | | 55.6 | % | | | 48.1 | % | | | 44.5 | % |
Risk-free interest rate | | | | | | 1.66 | % | | | 1.84 | % | | | 2.82 | % |
Remaining life until expiry | | | | | | 7.8 | | | | 8.0 | | | | 7.9 | |
Expected dividend rate | | | | | | — | | | | — | | | | — | |
F-35
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The transactions during the year ended December 31, 2020 and 2019 and the carrying value of the long-term debt at December 31, 2020 and 2019 were as follows:
| | | | | | | 2020 | | | 2019 | |
Balance at January 1 | | | | $ | 8,834 | | | $ | 125,804 | |
Stated and accreted interest | | | | | 2,209 | | | | 7,874 | |
Drawdown on non-revolving line of credit (second term loan) | | | | | 29,123 | | | | — | |
Drawdown on credit facility | | | | | — | | | | 18,677 | |
Issuance of secured convertible debentures | | | | | 2,410 | | | | — | |
Repayment of principal through share issuance | | | | | — | | | | (141,536 | ) |
Repayment of principal | | | | | (165 | ) | | | (988 | ) |
Repayment of stated interest | | | | | (1,879 | ) | | | (3,540 | ) |
Foreign exchange revaluation on Credit Facility balance | | | | | — | | | | (1,311 | ) |
Extinguishment of loans following a debt modification | | | | | — | | | | (4,667 | ) |
Recognition of loans following a debt modification | | | | | — | | | | 8,521 | |
Balance at December 31 | | | | | | | $ | 40,532 | | | $ | 8,834 | |
At December 31, 2020 and 2019, the carrying amount of the debt comprised the following loans:
| | | | | | | December 31, | | | December 31, | |
| | | | | | | 2020 | | | 2019 | |
First term loan having a principal of $10,000 maturing on April 23, 2024 bearing stated interest of 8% per annum (effective interest rate of 15.05%) 2) | | | | $ | 8,910 | | | $ | 8,669 | |
Second term loan having a principal of $29,123 maturing on April 23, 2024 bearing stated interest of 10% per annum (effective interest rate of 10.47%) 2) | | | | | 29,123 | | | | — | |
Secured convertible debentures having an aggregate principal amount of $2,410 maturing on March 31, 2022 bearing stated interest of 8% per annum (effective interest rate of 8.24%) 1) | | | | | 2,499 | | | | — | |
Non-interest bearing government term loan repayable in equal monthly installments of $82 until January 31, 2020 with an effective interest rate of 8.8% | | | | | — | | | | 165 | |
| | | | | | | $ | 40,532 | | | $ | 8,834 | |
Less current portion of long-term debt | | | | | — | | | | (165 | ) |
Long-term portion of long-term debt | | | | $ | 40,532 | | | $ | 8,669 | |
1) The secured convertible debentures are secured by all the assets of Fairhaven. The Company’s security interest created pursuant to its consolidated loan agreement with SALP, its parent, is subordinated to the security interest on the Fairhaven assets.
2) The first and second term loans issued under the consolidated loan agreement with SALP are secured by all the assets of the Company and require that certain covenants be respected including maintaining an adjusted working capital ratio.
F-36
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
2020
Concurrently with the Fairhaven acquisition that closed on July 17, 2020, the Company issued secured convertible debentures, or SCD, to certain former Fairhaven shareholders, for an aggregate principal amount of $2,410 and bearing an interest rate of 8% per annum, compounded quarterly. The SCD are due on the earlier of i) March 31, 2022, the maturity date, unless converted into common shares of the Company prior to the maturity date or ii) upon a change of control event. The SCD are secured by all the assets of Fairhaven and rank in priority to the term loans issued under consolidated loan agreement with SALP. At any time prior to the maturity date, the SCD holders have the right to convert the SCD into common shares of the Company. Liminal has the right to convert the SCD into common shares under certain pre-determined events. The five-trading day VWAP of Liminal’s common shares immediately preceding the date of any conversion will be used to determine the number of common shares of the Company that will be issued. The SCD were recorded as financial liabilities. The conversion features were determined to have no value.
At any time prior to the maturity date, the holders, shall have a collective right to purchase additional SCD issued by the Company for an aggregate principal amount of up to $5,740 with substantially the same terms and conditions as set out in the original SCD. If the pre-determined events allowing the Company to trigger the conversion of the SCD occur prior to the maturity date, the Company has the right to require the holders of the SCD to purchase additional SCD for an aggregate principal amount of up to $5,740, which would then be converted into common shares.
On November 11, 2019, the consolidated loan agreement with SALP was amended to provide for a non-revolving line of credit bearing the same terms and conditions as the first term loan. On September 14, 2020, the Company drew down $29,123 on the non-revolving line of credit representing the entire balance available, which resulted in the issuance of the second term loan. The second term loan bears an annual interest rate of 10% compounded monthly and payable quarterly and matures on April 23, 2024.
At December 31, 2020, the Company was in compliance with all of its covenants under its long-term debt agreement.
2019
On February 22, 2019, the Company amended the fourth loan agreement, or credit facility, with the addition of two tranches of US$10 million and US$5 million which the Company drew on February 22 and March 22, 2019 respectively. Those two tranches bear interest at an annual rate of 8.5% payable quarterly. Concurrently with the amendment, the Company agreed to reduce the exercise price of Warrants #9 from $1,000.00 to $156.36 per preferred share and to immediately issue those warrants (note 15). The incremental fair value of the warrant liability of $1,137 due to this change was recognized as deferred financing fees related to the additional two tranches received. The Company recorded the credit facility draws on February 22, 2019 and March 22, 2019 at their fair value at the transaction date less the associated transaction costs and financing fees of $45 and $1,137, respectively, for a net amount of $18,677.
F-37
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
On April 23, 2019, the Company entered into a debt restructuring agreement with the long-term debt holder whereby the entirety of the principal on the credit facility plus a portion of the interest due, the entirety of the First and Second Original Issue Discount, or OID loans and the majority of the Third OID loan would be repaid by Liminal by the issuance of common shares, at a conversion price, rounded to the nearest two decimals, of $15.21 per common share. Consequently, the US$95 million of principal plus interest due on the credit facility was reduced to $663 and the aggregate face value of the three OID loans was reduced by $99,552 to $10,000 with the remaining balance of the Third OID loan modified into an interest-bearing loan, which is referred to as the first term loan, at a stated interest of 10% payable quarterly. This resulted in the reduction of the long-term debt recorded on the consolidated statement of financial position by $141,536. The Company issued 15,050,312 common shares on that date which were recorded in share capital at a value of $228,915. The difference between the carrying amount of the debt converted into common shares and the increase in the value of the share capital is recognized as a loss on extinguishment of a loan of $87,379. The balance of interest due on the credit facility of $663 was paid in cash.
Following the debt restructuring agreement, SALP became Liminal’s majority and controlling shareholder and was thereafter considered Liminal’s parent entity for accounting purposes.
Pursuant to the debt restructuring, the Company cancelled the warrants previously held by SALP and replaced them with new warrants having an exercise price rounded to the nearest two decimals of $15.21 per common share, expiring on April 23, 2027 (note 18c). The incremental fair value of the replacement warrants was recognized in warrants equity and as part of the loss on the debt extinguishment together with the legal fees incurred to finalize all the related legal agreements.
The modification in terms of the remaining balance of the Third OID loan of $10,000, resulting in the first term loan, was accounted for as an extinguishment of the long-term debt and the re-issuance of a new interest-bearing loan. The difference between the carrying amount of the loan extinguished of $4,667 and the fair value of the first term loan of $8,521 recognized was recorded as a loss on debt extinguishment of $3,854. The fair value of the modified loan was determined using a discounted cash flow model with a market interest rate of 15.1%.
As a result of this transaction and the extinguishments of liabilities that occurred earlier in the beginning of 2019 following payments made to suppliers by the issuance of equity (note 18a), the consolidated statement of operations for the year ended December 31, 2019, includes a loss on extinguishment of liabilities of $92,374 detailed as follows:
Loss on extinguishment of liabilities due to April 23, 2019 loan modification | | | | |
Comprising the following elements: | | | | |
Debt to equity conversion | | $ | 87,379 | |
Expensing of financing fees on loan extinguishment | | | 653 | |
Extinguishment of previous loan | | | (4,667 | ) |
Recognition of modified loan | | | 8,521 | |
Expensing of increase in the fair value of the warrants (note 18c) | | | 408 | |
Loss on extinguishment of liabilities due to April 23, 2019 loan modification | | $ | 92,294 | |
Loss on extinguishment of liabilities to suppliers (note 18a) | | | 80 | |
Loss on extinguishments of liabilities | | $ | 92,374 | |
F-38
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
2018
In November 2017, the Company entered into a credit facility agreement bearing interest of 8.5% per annum expiring on November 30, 2019. The credit facility comprised two US$40 million tranches which became available to draw down once certain conditions were met. The drawdowns on the available tranches were limited to US$10 million per month.
As part of the agreement, the Company issued 54,000 warrants on November 30, 2017, or Warrants #7, to the holder of the long-term debt in consideration for the credit facility. Further details concerning the warrants are provided in note 18c. At each drawdown, the value of the proceeds drawn are allocated to the debt and the warrants classified as equity based on their fair value.
A royalty agreement between the Company and holder of long-term debt became effective upon drawing on the second tranche of the credit facility and then was subsequently modified as part of the loan modification discussed below. The proceeds to be received upon the first three draws on the second US$40 million tranche was increased from US$10.0 million to US$11.5 million to include the consideration paid by the holder for the royalty commitment (note 31).
In 2018, the Company drew on the remaining US$60 million available on the credit facility throughout the year, bringing the cumulative draws from US$20 million at December 31, 2017 to US$80 million at December 31, 2018.
The table below summarizes by quarter, the impact of the various drawdowns and the royalty proceeds on the consolidated financial statements:
| | | | | | | | | | Allocation of Proceeds | |
Quarter | | US$ proceeds | | | CA$ equivalent* | | | Debt * | | | Warrants * | | | Royalty liability* | |
Q1 2018 | | | 20,000,000 | | | | 25,155,000 | | | | 19,585,372 | | | | 5,569,628 | | | | — | |
Q2 2018 | | | 11,500,000 | | | | 14,768,300 | | | | 12,881,631 | | | | 1,886,669 | | | | — | |
Q3 2018 | | | 23,000,000 | | | | 29,808,690 | | | | 27,144,445 | | | | 2,531,438 | | | | 132,807 | |
Q4 2018 | | | 10,000,000 | | | | 13,280,100 | | | | 12,109,314 | | | | 1,170,786 | | | | — | |
*Exceptionally for this table Canadian dollars are not rounded to thousands of dollars.
For the August and September 2018 draws, the holder of the long-term debt used the set-off of principal right under the OID loan agreements to settle $3,917 (US$3 million) of the amounts due to the Company under the royalty agreement by reducing the face value of the second OID loan from $21,172 to $17,255. As a result, the cash proceeds received for those two draws were $25,892.
These transactions were accounted for as an extinguishment of a portion of the OID loan and the difference between the adjustment to the carrying value of the loan of $2,639 and the reduction in the face value of the OID loan of $3,917, was recorded as a loss on extinguishment of liabilities of $1,278.
F-39
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
On November 14, 2018, the Company and SALP modified the terms of the four loan agreements to extend the maturity date of the credit facility from November 30, 2019 to September 30, 2024 and all three OID loans from July 31, 2022 to September 30, 2024. Interest on amounts outstanding on the credit facility to be payable quarterly at an annual rate of 8.5% during the period of the extension. As of July 31, 2022, the OID loans would be restructured into cash paying loans bearing interest at an annual rate of 10%, payable quarterly. The outstanding face values of the OID loans at that date would become the principal amounts of the restructured loans. As additional consideration for the extension of the maturity dates, Liminal agreed to cancel 100,117 existing warrants (Warrants #3 to 7) and issue replacement warrants to SALP, bearing a term of 8 years and exercisable at a per share price equal to $1,000.00 (note 18c). The exact number of warrants to be granted to be set at a number that will result in the holder of the long-term debt having a 19.99% fully-diluted ownership level in Liminal upon issuance of the warrants, which are to be issued no later than March 20, 2019. On November 30, 2018, Warrants #3 to 7 were cancelled and 128,057 warrants to purchase common shares, or Warrants #8, representing a portion of the replacement warrants, were issued. At the end of the agreed upon measurement period for calculating the number of new warrants to be issued, Liminal would issue the remaining replacement warrant under a new series of warrants, or Warrants #9, which would give the holder the right to acquire preferred shares (notes 15 and 18a). The SALP also obtained the Company’s best efforts to support the election of a second representative of the lender to the Board of directors of the Company, and the extension of the security to the royalty agreement.
Management assessed the changes made to the previous agreements and determined that the modification should be accounted for as an extinguishment of the previous loans and the recording of new loans at their fair value determined as of the date of the modification. The fair value of the modified loans, determined using a discounted cash flow model with a market interest rate of 20.1%, was $107,704. Any cost or fees incurred with this transaction were recognized as part of the gain on extinguishment, including legal fees incurred in the amount of $434 and the improvements to the terms of the warrants. To determine this value, the Company estimated the fair value of the vested warrants (Warrants #3 to 7) and the fair value of the new warrants, excluding the 6,000 warrants that were associated with the last draw on the credit facility that occurred on November 22, 2018. The incremental fair value was $8,778 of which $338 pertains to Warrants #9 (note 15).
In addition, the fees incurred in regards of the credit facility, that were previously recorded in the consolidated statement of financial position as other long-term assets and were being amortized and recognized in the consolidated statement of operations over the original term of the credit facility, were recognized as part of the gain on extinguishment for an amount of $3,245.
As a result of this transaction and the extinguishments of debt that occurred earlier in the year following the use of the set-off of principal right by SALP, the consolidated statement of operations for the year ended December 31, 2018, includes a gain on extinguishment of liabilities of $33,626 detailed as follows:
Gain on extinguishment of liabilities due to November 14, 2018 debt modification | | | | |
Comprising the following elements: | | | | |
Extinguishment of previous loans | | $ | (155,055 | ) |
Expensing of deferred financing fees on credit facility | | | 3,245 | |
Recognition of modified loans | | | 107,704 | |
Expensing of increase in the fair value of the warrants | | | 8,778 | |
Warrants proceeds | | | (10 | ) |
Expensing of legal fees incurred with the debt modification | | | 434 | |
Gain on extinguishment of liabilities due to November 14, 2018 debt modification | | $ | (34,904 | ) |
Loss on extinguishment of liabilities due to set-off of principal | | | 1,278 | |
Gain on extinguishments of liabilities | | $ | (33,626 | ) |
F-40
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
17. | Other long-term liabilities |
| | December 31, | | | December 31, | |
| | 2020 | | | 2019 | |
Royalty payment obligations (a) | | $ | 3,355 | | | $ | 3,148 | |
License acquisition payment obligation (b) | | | — | | | | 1,302 | |
Other employee benefit liabilities | | | 1,557 | | | | 2,554 | |
| | $ | 4,912 | | | $ | 7,004 | |
Less: | | | | | | | | |
Current portion of royalty payment obligations (note 13) | | | (3,248 | ) | | | (3,043 | ) |
Current portion of license acquisition payment obligation (note 13) | | | — | | | | (1,302 | ) |
Current portion of other employee benefit liabilities (note 13) | | | (1,351 | ) | | | (2,374 | ) |
| | $ | 313 | | | $ | 285 | |
a) | Royalty payment obligations |
i) Royalty payment obligations to SALP
During the second quarter of 2018, the Company signed a royalty agreement with SALP at the same time as certain conditions pertaining to the second advance of the credit facility were modified. As a result of the agreement, the Company obtained the right to receive US$1.5 million milestone payments upon each draw of the second tranche of the credit facility in exchange for increasing royalty entitlements on future revenues relating to patents existing as of the date of the agreement of PBI-1402 and analogues, including PBI-4050. The agreement includes a minimum royalty payment of US$5,000 per quarter until approximately 2033 and a liability of $132 was recognized in the consolidated statement of financial position at December 31, 2020 representing the discounted value of the minimum royalty payments to be made until the expiry of the patents covered by the agreement, using a discount rate of 18.57% ($131 at December 31, 2019). In the case where royalties based on revenues became payable, the minimum royalty previously paid would be deducted from future remittances.
On November 14, 2018, as part of the debt modification agreement, the royalty rate was increased from 1.5% to 2% on future revenues relating to the specified patents and the right to receive the final US$1.5 million milestone payment was foregone.
ii) Royalty payment obligation for reacquired rights
As part of the consideration given by the Company in 2016 for the reacquisition of the rights to 50% of the worldwide profits pertaining to the sale of plasminogen for the treatment of plasminogen congenital deficiency which were previously granted to a licensee under a license agreement, the Company agreed to make royalty payments on the sales of plasminogen for congenital deficiency, using a rate of 5% up to a total of US$2.5 million. If by December 2020 the full royalty obligation has not been paid, the unpaid balance will become due. The Company has recognized a royalty payment obligation of $3,185 (US$2.5 million) in the consolidated statement of financial position at December 31, 2020 ($2,978; US$2.3 million at December 31, 2019), representing the discounted value of the expected royalty payments to be made until December 2020, using a discount rate of 9.2%. Subsequent to December 31, 2020, the payment terms of the licence agreement were modified resulting in the amount due being paid in instalments until August 15, 2021.
F-41
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
b) | Licence acquisition payment obligation |
In consideration for acquiring a license in January 2018, the Company agreed to pay an equivalent of US$3 million; US$1 million on the date of the transaction, and US$1 million on both the first and second anniversary of the transaction, to be settled in common shares of the Company. At December 31, 2020, the license is fully paid (licence payment obligation of $1,302 as at December 31, 2019).
18. | Share capital and other equity instruments |
a)Share capital
Authorized and without par value
Common shares: unlimited number authorized, participating, carrying one vote per share, entitled to dividends.
Preferred shares: unlimited number authorized, issuable in one or more series.
| - | Series A preferred shares: unlimited number authorized, no par value, non-voting, ranking in priority to the common shares, entitled to the same dividends as the common shares, non-transferable, redeemable at the redemption amount offered for the common shares upon a change in control event. |
Changes in the issued and outstanding common shares during the year ended December 31, 2020 and 2019 were as follows:
| | 2020 | | | 2019 | |
| | Number | | | Amount | | | Number | | | Amount | |
Balance - beginning of year | | | 23,313,164 | | | $ | 932,951 | | | | 720,306 | | | $ | 583,117 | |
Issued to acquire assets | | | 299,141 | | | | 4,681 | | | | 4,420 | | | | 1,326 | |
Exercise of stock options (note 18b) | | | 5,391 | | | | 167 | | | | — | | | | — | |
Exercise of pre-funded warrants (note 18c) | | | 557,894 | | | | 2,624 | | | | — | | | | — | |
Shares issued pursuant to a restricted share units plan (note 18b) | | | 10,355 | | | | 9,764 | | | | — | | | | — | |
Shares issued pursuant to debt restructuring | | | — | | | | — | | | | 15,050,312 | | | | 228,915 | |
Shares issued for cash | | | 5,757,894 | | | | 27,074 | | | | 7,536,654 | | | | 118,648 | |
Shares released from escrow | | | — | | | | — | | | | — | | | | 400 | |
Shares issued in payment to suppliers | | | — | | | | — | | | | 1,472 | | | | 545 | |
Balance - end of year | | | 29,943,839 | | | $ | 977,261 | | | | 23,313,164 | | | $ | 932,951 | |
2020
On January 29, 2020, the Company issued 96,833 common shares as a consideration for the final payment for the licence acquired in January 2018. This transaction was accounted for as an extinguishment of the license acquisition payment obligation (note 17b) and the difference between the carrying value of the liability of $1,319 and the amount recorded for the shares issued of $1,240, which were valued at the market price of the shares on their date of issuance, was recorded as a gain on extinguishment of liabilities of $79 during the year ended December 31, 2020.
On July 17, 2020 the Company issued 202,308 common shares in payment for the acquisition of Fairhaven, which has been accounted for as an asset acquisition (note 5). The common shares issued were valued at the market price of the shares, on their date of issuance for an aggregate value of $3,441.
On November 3, 2020, the Company completed a private placement for a total gross proceed of $39,960 in exchange for the issuance of 5,757,894 common shares, 557,894 prefunded warrants (note 18c) and 6,315,788 warrants (note 15, 18c). SALP’s participation in the private placement was for gross proceeds of $19,980.
F-42
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The total gross proceeds were allocated to the warrant liability based on its fair value of $10,263 on that date with the residual value being allocated between the common shares and the pre‑funded warrants. The value attributed to the common share was $27,074. The total transaction costs of $2,755 were allocated to the three instruments issued based on their relative fair values. The amount allocated to the common shares and the pre-funded warrants, of $2,048, was recognized in the deficit.
On December 30, 2020, the 557,894 pre-funded warrants were exercised resulting in the issuance of 557,894 common shares and the receipt of $1 in cash. An amount of $2,623 was reclassified from warrants to common shares.
2019
In November 2018, the Company entered into an At-the-Market, or ATM, Equity Distribution Agreement, or EDA, under which the Company was able, at its discretion and from time to time, subject to conditions in the EDA, to offer common shares through ATM issuances on the TSX for aggregate proceeds not exceeding $31 million. The agreement provided that common shares were to be sold at market prices prevailing at the time of sale. The Company issued a total of 12,865 common shares at an average price of $327.55 per share under the ATM in January and February 2019, for aggregate gross proceeds of $4,214, less transaction costs of $248 recorded in deficit, for total net proceeds of $3,966. This ATM facility expired in April 2020.
On January 29, 2019, the Company issued 4,420 common shares in settlement of second payment due for the license acquisition payment obligation (note 17) and recorded $1,326 in share capital based on the market value of the shares on that date.
On February 25 and 27, 2019, the Company issued a total of 1,472 common shares in payment for amounts due to certain suppliers. This transaction was accounted for as an extinguishment of liabilities and the difference between the carrying value of the accounts payable of $465 and the amount recorded for the shares issued of $545, which were valued at the market price of the shares on their date of issuance, was recorded as a loss on extinguishment of liabilities of $80.
As part of the settlement agreement concluded in April 2019 with a former CEO of the Company, common shares held in escrow as security for a share purchase loan of $400 to a former CEO were released and the loan extinguished in exchange for the receipt of a payment of $137, representing the fair value of the shares at the time of the settlement.
On April 23, 2019, the Company issued 15,050,312 common shares as part of the debt restructuring (note 16). The shares issued in relation with the debt restructuring contained trading restrictions and accordingly, the Company determined that their quoted price did not fairly represent the value of the shares issued. As such, the issued shares were recorded at fair value using a market approach under a level 2 fair value measurement of $15.21 per share, resulting in a value of the shares issued of $228,915. The fair value was based on a share issuance for cash on the same date with a non-related party. The difference between the adjustment to the carrying value of the loan of $141,536 and the amount recorded for the shares issued of $228,915 was recorded as a loss on extinguishment of a loan of $87,379.
Concurrently with the debt restructuring, the Company closed two private placements for 4,931,161 common shares at a subscription price rounded to the nearest two decimals of $15.21 for gross proceeds of $75,000, less transaction costs of $4,802 recorded in deficit, for total net proceeds of $70,198. SALP’s participation in the private placement was for gross proceeds to the Company of $25,000.
F-43
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
In May 2019, the Company announced a rights offering to the holders of its common shares at the close of business on May 21, 2019 to subscribe for up to 20 additional common shares, for each share they held, for a subscription price rounded to the nearest two decimals of $15.21 per common share. In June 2019, the Company issued 2,592,628 common shares for gross proceeds of $39,434 as part of the right offerings less transactions costs of $271 recorded in deficit, for total net proceeds of $39,163.
b) | Contributed surplus (Share-based payments) |
Stock options
The Company has established a stock option plan for its directors, officers, employees and service providers. The plan provides that the aggregate number of shares reserved for issuance at any time under the plan may not exceed 3,749,714 common shares and the maximum number of common shares, which may be reserved for issuance to any individual, may not exceed 5% of the outstanding common shares. The stock options issued under the plan may be exercised over a period not exceeding ten years from the date they were granted. All stock options granted since May 2017 have a contractual life of 10 years. Stock options issued prior to May 2017 had a life of five years.
The vesting period of the stock options varies from immediate vesting to vesting over a period not exceeding 6 years. Participants meeting certain service and age requirements may see the vesting of certain awards accelerate upon retirement. The vesting conditions are established by the Board of Directors on the grant date. The exercise price is based on the weighted average share price for the five business days prior to the grant.
For stock options having a CA$ exercise price, the changes in the number of stock options outstanding during the years ended December 31, 2020, 2019 and 2018 were as follows:
| | | 2020 | | | 2019 | | 2018 | |
| | | | | | | Weighted | | | | | | | Weighted | | | | | | Weighted | |
| | | | | | | average | | | | | | | average | | | | | | average | |
| | | | | | | exercise price | | | | | | | exercise price | | | | | | exercise price | |
| | | Number | | | (in CA$) | | | Number | | | (in CA$) | | Number | | | (in CA$) | |
Balance - beginning of year | | | | 2,209,864 | | | $ | 38.72 | | | | 21,625 | | | $ | 1,464.49 | | | 14,256 | | | $ | 1,782.70 | |
Granted | | | | 436,570 | | | | 14.06 | | | | 2,218,810 | | | | 33.13 | | | 10,837 | | | | 755.97 | |
Forfeited | | | | (153,982 | ) | | | 19.33 | | | | (16,774 | ) | | | 159.61 | | | (377 | ) | | | 1,933.34 | |
Exercised | | | | (5,391 | ) | | | 15.21 | | | | — | | | | — | | | (1,681 | ) | | | 376.10 | |
Cancelled | | | | — | | | | — | | | | (11,713 | ) | | | 1,237.94 | | | — | | | | — | |
Expired | | | | (1,506 | ) | | | 2,462.46 | | | | (2,084 | ) | | | 1,176.20 | | | (1,410 | ) | | | 408.43 | |
Repriced - options before repricing | | | | (1,929,685 | ) | | | 35.14 | | | | — | | | | — | | | — | | | | — | |
Repriced - options after repricing | | | | 1,929,685 | | | | 15.21 | | | | — | | | | — | | | — | | | | — | |
Balance - end of year | | | | 2,485,555 | | | $ | 18.70 | | | | 2,209,864 | | | $ | 38.72 | | | 21,625 | | | $ | 1,464.49 | |
F-44
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
For options having a US$ exercise price, the changes in the number of stock options outstanding during the year ended December 31, 2020 were as follows:
| | | | | | | | | | | Weighted | |
| | | | | | | | | | | average | |
| | | | | | | | | | | exercise price | |
| | | | | | | Number | | | (in US$) | |
Balance - beginning of year | | | | | | | | — | | | $ | — | |
Granted | | | | | | | | 305,000 | | | | 4.70 | |
Balance - end of year | | | | | | | | 305,000 | | | $ | 4.70 | |
2020
In March 2020, Liminal’s board of directors approved a plan to reduce the exercise price of the stock options issued in June 2019, held by active employees and directors at the time of the repricing. On May 26, 2020, a revised exercise price, pending approval, of $15.21 was determined, changing the exercise price to the higher of (i) $15.21 and (ii) the five trading-day VWAP of Liminal common shares on the repricing date. On June 8, 2020, the repricing of 1,929,685 of the outstanding stock options having exercise prices of $27.00 and $36.00 to the revised exercise price was approved at the Company’s annual shareholder meeting.
Although the stock options were not repriced until May 26 2020, management concluded that the service period for employees and directors to earn the modified awards had commenced from the date the Company informed the holders of these stock options of the repricing proposal and the expense resulting from the repricing plan should be recognized starting from that date. Using the revised exercise price of $15.21, the Company calculated the final incremental fair value of the repricing on the grant date of May 26, 2020 to be $3,000. This incremental fair-value will be amortized from the services commencement date of March 25 over the remaining vesting period of the repriced options. The incremental grant date fair value of the repriced options was estimated based on the Black-Scholes option-pricing model calculated before and after the effect of the repricing. The following Black-Scholes assumption were used:
Expected dividend rate | | — | |
Expected volatility of share price | | 93.2 | % |
Risk-free interest rate | | 0.4 | % |
Expected life in years | | 6.3 | |
Weighted average grant date incremental fair value | $ | 1.55 | |
In June 2020, 436,570 stock options, having an exercise price of $14.06 and vesting over a period of up to four years, were issued to employees and directors. In October 2020, 20,000 stock options, having an exercise price of US$10.80 and vesting over a period of three years were issued to a new director. In December 2020, 285,000 stock options having an exercise price of US$4.27, of which 95,000 stock options vested immediately and the remaining stock options vest over a period up to three years, were issued to key management.
During the year ended December 31, 2020, 5,391 stock options were exercised resulting in cash proceeds of $82 and a transfer from contributed surplus to share capital of $85. The weighted average share price on the date of exercise of the stock options during the year ended December 31, 2020 was $18.47.
F-45
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
2019
In January 2019, 1,622 stock options were granted at an exercise price of $300.00 and vesting on December 31, 2019. On June 4, 2019, 1,794,224 stock options were granted to management at a strike price of $36.00 of which 248,825 stock options vested immediately and the remaining vest over a period up to six years. On June 19, 2019, 251,714 stock options were issued at a strike price of $27.00 of which 60,717 stock options vested immediately and the remaining vest over a period up to four years. On September 3, 2019, 71,250 stock options were issued at a strike price of $11.99 and on December 3, 2019, 100,000 stock options were issued at a strike price of $7.86, both of these grants having a vesting period of up to four years. The weighted average grant date fair value of the stock options issued in 2019 was $12.74.
In June and August 2019, the Company cancelled the options that were issued prior to June 2019, as the exercise price of these options were so above the market price at the time, that it was highly unlikely that they would ever be exercised. In compensation for their agreement to the cancellation, key management and employees, received the new options granted to them in June 2019 discussed above. Consequently, 11,084 stock options with a weighted average exercise price of $1,256.73 were cancelled. There was no exercise of stock options in 2019.
2018
During the year ended December 31, 2018, 10,837 stock options having a contractual term of 10 years and a vesting period of up to four years were granted.
During the year ended December 31, 2018, 1,681 stock options were exercised resulting in cash proceeds of $635 and a transfer from contributed surplus to share capital of $438. The weighted average share price on the date of exercise of the options during the year ended December 31, 2018 was $1,044.16.
The Company uses the Black-Scholes option pricing model to calculate the fair value of stock options at the date of grant. The weighted average inputs into the model and the resulting grant date fair values during the years ended December 31, 2020, 2019 and 2018 were as follows:
| | | | | 2020 | | | 2019 | | | 2018 | |
Expected dividend rate | | | | | | — | | | | — | | | | — | |
Expected volatility of share price | | | | | | 100.5 | % | | | 45.0 | % | | | 66.1 | % |
Risk-free interest rate | | | | | | 0.5 | % | | | 1.4 | % | | | 2.1 | % |
Expected life in years | | | | | | 6.7 | | | | 7.2 | | | | 7.9 | |
Weighted average grant date fair value | | | | | $ | 8.66 | | | $ | 12.74 | | | $ | 221.64 | |
F-46
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
At December 31, 2020, stock options issued and outstanding denominated in CA$ and US$ by range of exercise price are as follows:
| | | | | | | Weighted average | | | Weighted | | | | | | | Weighted | |
Range of exercise | | | | | | remaining | | | average | | | | | | | average | |
price for stock option | | Number | | | contractual life | | | exercise price | | | Number | | | exercise price | |
issued in CA$ | | outstanding | | | (in years) | | | (CA$) | | | exercisable | | | (CA$) | |
$7.86 - $11.99 | | | 171,250 | | | | 8.8 | | | $ | 9.58 | | | | 47,266 | | | $ | 9.81 | |
$ | 14.06 | | | 402,980 | | | | 9.4 | | | | 14.06 | | | | 20,000 | | | | 14.06 | |
$ | 15.21 | | | 1,854,541 | | | | 8.4 | | | | 15.21 | | | | 480,577 | | | | 15.21 | |
$27.00 - $3,170.00 | | | 56,784 | | | | 8.1 | | | | 193.03 | | | | 55,734 | | | | 183.86 | |
| | | | 2,485,555 | | | | 8.6 | | | $ | 18.70 | | | | 603,577 | | | $ | 30.32 | |
| | | | | | | Weighted average | | | Weighted | | | | | | | Weighted | |
Range of exercise | | | | | | remaining | | | average | | | | | | | average | |
price for stock option | | Number | | | contractual life | | | exercise price | | | Number | | | exercise price | |
issued in US$ | | outstanding | | | (in years) | | | (US$) | | | exercisable | | | (US$) | |
$ | 4.27 | | | 285,000 | | | | 9.9 | | | $ | 4.27 | | | | 95,000 | | | $ | 4.27 | |
$ | 10.80 | | | 20,000 | | | | 9.8 | | | | 10.80 | | | | — | | | | — | |
| | | | 305,000 | | | | 9.9 | | | $ | 4.70 | | | | 95,000 | | | $ | 4.27 | |
A share-based payment compensation expense of $6,169 was recorded for the stock options for the year ended December 31, 2020 ($12,212 and $3,372 for the year ended December 31, 2019 and 2018 respectively).
Restricted share units, or RSU
The Company has established an equity-settled restricted share units plan for executive officers of the Company, as part of its incentive program designed to align the interests of its executives with those of its shareholders, and in accordance with its long-term incentive plan. The vesting conditions are established by the Board of Directors on the grant date. Participants meeting certain service and age requirements may see the vesting of certain awards accelerate upon retirement. Each vested RSU gives the right to receive a common share.
Changes in the number of RSU outstanding during the years ended December 31, 2020, 2019 and 2018 were as follows:
| | | | | | | | | | | | | | | |
| | | | | 2020 | | | 2019 | | | 2018 | |
Balance - beginning of year | | | | | | 17,565 | | | | 18,299 | | | | 9,799 | |
Granted | | | | | | — | | | | 12,564 | | | | 10,329 | |
Expired | | | | | | — | | | | — | | | | (1,578 | ) |
Forfeited | | | | | | (46 | ) | | | (409 | ) | | | (19 | ) |
Released | | | | | | (10,355 | ) | | | — | | | | (232 | ) |
Paid in cash | | | | | | (2,948 | ) | | | (8,396 | ) | | | — | |
Cancelled | | | | | | — | | | | (4,493 | ) | | | — | |
Balance - end of year | | | | | | 4,216 | | | | 17,565 | | | | 18,299 | |
F-47
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
2020
During the first quarter of 2020, 2,948 RSU were paid in cash resulting in a reduction to contributed surplus of $40. As at December 31, 2020, all 4,216 outstanding RSU were vested. Share-based payment compensation expense of $65 was recorded during the year ended December 31, 2020.
2019
On January 31, 2019, the Company granted 12,564 RSU at a grant price of $300.00 and a one-year vesting period. On May 30, 2019, the Company decided to vest the 12,564 RSU and the employees were given the choice to receive the then current value of the shares in cash or to receive the shares at a later date. As a result, 8,396 RSU were released and paid in cash resulting in a reduction to contributed surplus of $421.
On May 7, 2019 the 12,886 performance‑based RSU pertaining to the “2017-2019” cycle and the “2018-2020” cycle were modified by removing the performance conditions and converting them into time-vesting RSU. The quantity modified into time-vesting units was equivalent to the 100% achievement range whereby in the past, the outcome of the performance conditions could go from zero to 150%. Historically, the Company has always reported the quantity of RSU outstanding as the maximum number of shares that could be issued under the plan. This change resulted in the cancellation of 4,305 units.
At December 31, 2019, 13,262 vested RSU and 4,303 unvested RSU were outstanding. Share-based payments compensation expense of $9,818 was recorded during the year ended December 31, 2019.
2018
On December 4, 2018, the Company granted 10,329 RSU to management (the “2018-2020 RSU”) with a time period to meet the vesting conditions extending to December 31, 2020. The grant included 2,374 units that vest at a rate of 33.3% at the end of each year and become available for release at the time of vesting, and 7,955 units that have performance-based conditions with a scaling payout depending on performance (ranging from 0% to 150%). These 2018-2020 performance-based RSU have since been converted into time-vesting RSU at 100% in 2019 as mentioned above.
Share-based payments compensation expense of $3,350 was recorded during the year ended December 31, 2018.
Share-based payments expense
The total share-based payments compensation expense, comprising the above-mentioned expenses for stock options and RSU, has been included in the consolidated statements of operations for the years ended December 31, 2020, 2019 and 2018 as indicated in the following table:
| | | | | | | | | | | | | | | |
| | | | | 2020 | | | 2019 | | | 2018 | |
Cost of sales and other production expenses | | | | | $ | 40 | | | $ | 107 | | | $ | 299 | |
Research and development expenses | | | | | | 2,946 | | | | 7,137 | | | | 2,295 | |
Administration, selling and marketing expenses | | | | | | 3,248 | | | | 14,786 | | | | 4,128 | |
| | | | | $ | 6,234 | | | $ | 22,030 | | | $ | 6,722 | |
F-48
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The following table summarizes the changes in the number of warrants outstanding with an exercise price in CA$ during the years ended December 31, 2020 and 2019:
| | | 2020 | | | | | 2019 | |
| | | | | | | | | Weighted | | | | | | | | | | | Weighted | |
| | | | | | | | | average | | | | | | | | | | | average | |
| | | | | | | | | exercise price | | | | | | | | | | | exercise price | |
| | | Number | | | | | (CA$) | | | | | Number | | | | | (CA$) | |
Balance of warrants - beginning of year | | | 172,735 | | | | | $ | 84.33 | | | | | | 153,611 | | | | | $ | 1,028.35 | |
Issued for cash | | | — | | | | | | — | | | | | | 19,402 | | | | | | 156.36 | |
Cancelled - loan modification | | | — | | | | | | — | | | | | | (168,735 | ) | | | | | 872.51 | |
Issued - loan modification | | | — | | | | | | — | | | | | | 168,735 | | | | | | 15.21 | |
Expired | | | — | | | | | | — | | | | | | (278 | ) | | | | | 6,390.00 | |
Balance of warrants - end of year | | | 172,735 | | | | | $ | 84.33 | | | | | | 172,735 | | | | | $ | 84.33 | |
Balance of warrants exercisable - end of year | | | 172,735 | | | | | $ | 84.33 | | | | | | 170,735 | | | | | $ | 50.17 | |
The following table summarizes the changes in the number of warrants outstanding with an exercise price in US$ during the year ended December 31, 2020:
| | | | | | | 2020 | |
| | | | | | | | | | | | | | | Weighted | |
| | | | | | | | | | | | | | | average | |
| | | | | | | | | | | | | | | exercise price | |
| | | | | | | | | Number | | | | | (US$) | |
Balance of warrants - beginning of year | | | | | | | | | — | | | | | $ | — | |
Issued for cash | | | | | | | | | 6,873,682 | | | | | | 5.05 | |
Issued for no consideration | | | | | | | | | | 1,578,946 | | | | | | 5.50 | |
Exercised | | | | | | | | | (557,894 | ) | | | | | — | |
Balance of warrants - end of year | | | | | | | | | 7,894,734 | | | | | $ | 5.50 | |
Balance of warrants exercisable - end of year | | | | | | | | | 7,894,734 | | | | | $ | 5.50 | |
2020
As a consideration to the private placement on November 3, 2020 (note 18a), the Company issued 6,315,788 warrants (note 15) and 557,894 pre-funded warrants. The gross proceeds allocated to the pre-funded warrants was $2,623. The pre-funded warrants exercise price was US$0.001 and a term of five years.
On November 25, 2020, the Company issued 1,578,946 additional warrants with the same terms and conditions as described above, following an amendment to the private placement agreement. On December 30, 2020, the pre-funded warrants were fully exercised and 557,894 common shares were issued (note 18a).
F-49
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
2019
On February 22, 2019, pursuant to modifying the fourth loan agreement, the Company issued 19,402 warrants, Warrants #9, having an exercise price of $156.36 (note 15). Warrants #9 do not meet the definition of an equity instrument since the underlying preferred shares qualify as a liability instrument, and therefore they must be accounted for as a financial instrument carried at fair value through profit or loss (note 15).
On April 23, 2019, as part of the debt restructuring (note 16), 168,735 warrants (Warrants #1, 2, 8 and 9) were cancelled and replaced with an equivalent number of new warrants, Warrants #10, that will be exercisable at an exercise price of $15.21 per common share and expire on April 23, 2027. The increase in the fair value of the replacement warrants compared to those cancelled was $408 at the date of the modification and was recorded in shareholders’ equity – warrants with the corresponding expense recorded as part of the loss on extinguishment of liabilities due to the debt restructuring.
2018On November 14, 2018, an agreement was signed between the Company and the holder of the long-term debt to extend the maturity of the three OID loans and the Credit Facility (note 16). As part of the cost for the debt modification, the Company proceeded on November 30, 2018 to cancel 100,117 existing warrants (Warrants #3 to 7) and replace them with 128,057 new warrants (Warrants #8), each giving the holder the right to acquire one common share at an exercise price of $1000.00 per share, paid either in cash or in consideration of the lender’s cancellation of an equivalent amount of the face value of an OID loan. The warrants expire on November 30, 2026. A payment of $10 was received from the holder of the long-term debt as part of this transaction. The increase in the fair value of the replacement warrants compared to those cancelled was $8,440 at the date of the modification. This value in addition to the payment received was recorded in shareholders’ equity – warrants and the corresponding debit was recorded against the gain on extinguishment of liabilities relating to the debt modification.
The warrants outstanding as at December 31, 2020, their exercise price in CA$ or in US$, expiry rate and the overall weighted average exercise price in both currency are as follows:
| | | | | Number | | | Expiry date | | Exercise price (CA$) | |
| | | | | | 4,000 | | | January 2023 | | | 3,000.00 | |
| | | | | | 168,735 | | | April 2027 | | | 15.21 | |
Warrants outstanding with an exercise price in CA$ | | | 172,735 | | | | | $ | 84.33 | |
| | | | | Number | | | Expiry date | | Exercise price (US$) | |
Warrants outstanding with an exercise price in US$ | | | 7,894,734 | | | November 2025 | | $ | 5.50 | |
F-50
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
19. | Non-controlling interests |
The Company held less than 100% interest in the following three entities during the last three fiscal years. The interest in these subsidiaries at December 31, 2020, 2019 and 2018 was as follows:
Name of subsidiary | Segment activity | Place of incorporation and operation | Proportion of ownership interest held by group |
Prometic Bioproduction Inc. 1) | Plasma-derived therapeutics | Quebec, Canada | 100% |
Pathogen Removal and Diagnostic Technologies Inc. | Corporate | Delaware, U.S. | 77% |
NantPro Biosciences, LLC 2) | Plasma-derived therapeutics | Delaware, U.S. | 73% |
1) The non-controlling interest, or NCI, in Prometic Bioproduction Inc. or PBP owned 13% of the common shares until April 2018, when the Company acquired these shares in the subsidiary in exchange for 4,712,422 common shares of the Company. Consequently, $15,278 was recognized in the deficit to reflect Liminal’s increase in the ownership of the subsidiary, representing the difference in value between the $3,629 of equity issued in payment of the 13% ownership acquired and $11,649 of total net liabilities attributed to the NCI at the date of the transaction that was derecognized from the statement of financial position. Until that time, the NCI in PBP was attributed its share of the operating results and the financial position of the entity. The loss allocated to the NCI of PBP during the four first months of 2018 was $927.
2) Following a change in the Company’s strategic plans that resulted in the recording of an impairment of the assets of NantPro Biosciences, LLC or Nantpro, during the year ended December 31, 2018 (note 24), NantPro wound up its activities in 2019. This resulted in reduced operating costs in 2019 compared to 2018 and $nil operating costs in 2020. The carrying value of NantPro’s assets or liabilities was $nil at December 31, 2020 and 2019 and consequently, the share of the NCI in the NantPro statement of financial position is $nil at December 31, 2020 and 2019.
The summarized statements of financial position for Pathogen Removal and Diagnostic Technologies Inc, or PRDT, and the summarized statements of operations for PRDT and NantPro are provided below. This information is based on amounts before inter-company eliminations.
Summarized statements of financial position for PRDT
| | | | | | | December 31, | | | December 31, | |
| | | | | | | 2020 | | | 2019 | |
Receivables (current) | $ | 233 | | | $ | 9 | |
Capital and intangible assets (long-term) | | 113 | | | | 156 | |
Trade and other payables (current) | | (877 | ) | | | (748 | ) |
Intercompany loan | | (16,846 | ) | | | (15,956 | ) |
Total equity (negative equity) | $ | (17,377 | ) | | $ | (16,539 | ) |
Attributable to non-controlling interests | $ | (8,087 | ) | | $ | (7,255 | ) |
The share of the NCI in PRDT’s statement of financial position represents an asset on the Company’s consolidated statement of financial position.
F-51
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Summarized statement of operations of PRDT
Year ended December 31 | | 2020 | | | 2019 | | | 2018 | |
Royalty revenues | | | | $ | 572 | | | $ | 585 | | | $ | 839 | |
Royalty expenses | | | | | (128 | ) | | | (132 | ) | | | (190 | ) |
Research and development expenses | | | | | (196 | ) | | | (215 | ) | | | (179 | ) |
Administration and other expenses | | | | | (1,506 | ) | | | (896 | ) | | | (1,001 | ) |
Impairment loss | | | | | — | | | | (129 | ) | | | — | |
Net loss and comprehensive loss | | | | $ | (1,258 | ) | | $ | (787 | ) | | $ | (531 | ) |
Attributable to non-controlling interests | | | | $ | (832 | ) | | $ | (713 | ) | | $ | (641 | ) |
Summarized statement of operations of NantPro:
Year ended December 31 | | | | 2019 | | | 2018 | |
Research and development expenses | | | | | | $ | (1,213 | ) | | $ | (10,556 | ) |
Administration and other expenses | | | | | | | (13 | ) | | | (131 | ) |
Impairment loss | | | | | | | — | | | | (141,025 | ) |
Net loss and comprehensive loss | | | | | | $ | (1,226 | ) | | $ | (151,712 | ) |
Attributable to non-controlling interests | | | | | | $ | (331 | ) | | $ | (40,962 | ) |
For all years presented, the losses from continuing operations allocated to the NCI in the consolidated statements of operations, per subsidiary are as follows:
| | | | | 2020 | | | 2019 | | | 2018 | |
Consolidated statements of operations: | | | | | | | | | | | | | | |
Prometic Bioproduction Inc. | | | | $ | — | | | $ | — | | | $ | (927 | ) |
Pathogen Removal and Diagnostic Technologies Inc. | | | | | (832 | ) | | | (713 | ) | | | (641 | ) |
NantPro Biosciences, LLC | | | | | — | | | | (331 | ) | | | (40,962 | ) |
Total non-controlling interests | | | | $ | (832 | ) | | $ | (1,044 | ) | | $ | (42,530 | ) |
The NantPro NCI’s share in the funding of the subsidiary by Liminal was $nil for the year ended December 31, 2020 ($331 for the year ended December 31, 2019 and $2,892 for the year ended December 31, 2018) and has been presented in the consolidated statements of changes in equity.
F-52
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The Company defines its capital as shareholders’ equity including warrants presented as a liability and long-term debt (including the current portion) less cash and cash equivalents.
| | | | | | | December 31, | | | December 31, | |
| | | | | | | 2020 | | | 2019 | |
Warrant liability | | | | | | | $ | 11,640 | | | $ | — | |
Lease liabilities | | | | | | | | 33,452 | | | | 38,237 | |
Long-term debt | | | | | | | | 40,532 | | | | 8,834 | |
Total equity | | | | | | | | 15,012 | | | | 94,934 | |
Cash and cash equivalents | | | | | | | | (45,075 | ) | | | (61,285 | ) |
Total capital | | | | | | | $ | 55,561 | | | $ | 80,720 | |
The Company manages its capital resources to fund the growth and development of its business and to ensure it has sufficient liquidities to support the working capital required to maintain its ability to continue as a going concern and to pay long-term obligations upon maturity. The Company monitors its ability to meet its financial obligations and evaluates funding requirements by forecasting cash requirements. Financial covenants of existing debt agreements, including capital requirements (note 16) are reviewed by management on an ongoing basis to monitor compliance.
At the present time, the Company favors financing by issuing equity instruments in order to minimize future financial obligations, however it considers all sources of financing reasonably available, including but not limited to the issuance of equity instruments, new debt and the sale of assets. The Company considers the cost of capital, the terms and conditions and the dilutive effect on shareholders when considering the different forms financings that it may prevail upon.
21. | Revenues from continuing operations |
| | | | | 2020 | | | 2019 | | | 2018 | |
Revenues from the sale of goods | | | | | $ | 2,593 | | | $ | 4,734 | | | $ | 23,874 | |
Royalty revenues | | | | | | 572 | | | | — | | | | — | |
Revenues from the rendering of services | | | | | | 6 | | | | 34 | | | | 260 | |
Rental revenue | | | | | | 146 | | | | 136 | | | | 499 | |
| | | | | $ | 3,317 | | | $ | 4,904 | | | $ | 24,633 | |
All the rental revenues are generated from subleasing right-of-use assets.
F-53
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
22. | Supplemental information regarding the consolidated statements of operations |
a) Government assistance
For the year ended December 31, 2020, the Company recognized $7,199 and $597 of government grants in connection with the Canada Emergency Wage Subsidy program and the Canada Emergency Rent Subsidy program respectively, two new subsidies program created by the Government of Canada in 2020 in response to the COVID-19 pandemic that the Company benefits from. The Company also recognized research and development tax credits during the years ended December 31, 2020, 2019 and 2018. These grants were recorded as a reduction of salary expenses and other related charges and are recognized as follows in the consolidated statement of operations:
Year ended December 31 | | 2020 | | | 2019 | | | 2018 | |
Government grants recognized in cost of sales and other production expenses: | | | | | | | | | | | | |
Salary subsidy | | | | | $ | 682 | | | $ | — | | | $ | — | |
Rent subsidy | | | | | | 49 | | | | — | | | | — | |
| | | | | $ | 731 | | | $ | — | | | $ | — | |
Government grants recognized in research and development expenses: | | | | | | | | | | | | |
Salary subsidy | | | | | $ | 5,093 | | | $ | — | | | $ | — | |
Rent subsidy | | | | | | 485 | | | | — | | | | — | |
Research and development tax credits | | | 1,758 | | | | 572 | | | | 3,175 | |
| | $ | 7,336 | | | $ | 572 | | | $ | 3,175 | |
Government grants recognized in administration, selling and marketing expenses: | | | | | | | | | | | | |
Salary subsidy | | | | | $ | 1,457 | | | $ | — | | | $ | — | |
Rent subsidy | | | | | | 63 | | | | — | | | | — | |
| | $ | 1,520 | | | $ | — | | | $ | — | |
b) Finance costs
Year ended December 31 | | 2020 | | | 2019 | | | 2018 | |
Interest accretion on long-term debt | | $ | 2,209 | | | $ | 7,874 | | | $ | 18,856 | |
Amortization of fees for credit facility | | | — | | | | 10 | | | | 2,625 | |
Financing fees on warrant liability | | | | | | 709 | | | | — | | | | — | |
Other interest expense, transaction and bank fees | | | 485 | | | | 594 | | | | 886 | |
Interest expense on lease liabilities | | | 6,030 | | | | 7,068 | | | | — | |
Interest income | | | (451 | ) | | | (753 | ) | | | (307 | ) |
| | $ | 8,982 | | | $ | 14,793 | | | $ | 22,060 | |
The table above includes financing costs from continuing and discontinued operations. Financing costs from discontinued operations for the year ended December 31, 2019 were $737 and mainly represented interest expense on lease liabilities (19$ for the year ended December 31, 2018) (note 6).
F-54
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
c) Employee compensation expense
Year ended December 31 | | 2020 | | | 2019 | | | 2018 | |
Wages and salaries | | $ | 32,410 | | | $ | 48,846 | | | $ | 46,775 | |
Employer's benefits | | | 5,443 | | | | 8,263 | | | | 8,377 | |
Share-based payments expense | | | 6,234 | | | | 22,030 | | | | 6,722 | |
| | $ | 44,087 | | | $ | 79,139 | | | $ | 61,874 | |
The Company maintains a defined contribution pension plan for its permanent employees. The Company matches the contributions made by employees who elect to participate in the plan up to a maximum percentage of their annual salary. The Company’s contributions recognized as an expense for the year ended December 31, 2020 amounted to $1,055 ($1,495 and $1,635 for the years ended December 31, 2019 and 2018 respectively).
| | | | | 2020 | | | 2019 | | | 2018 | |
Intangible assets (note 12) | | | | | $ | 1,567 | | | $ | 5,296 | | | $ | 142,609 | |
Capital assets (note 10) | | | | | | 665 | | | | 7,070 | | | | 5,689 | |
Right-of-use assets (note 11) | | | | | | 18,627 | | | | — | | | | — | |
Option to purchase equipment | | | | | | — | | | | — | | | | 653 | |
Investment in an associate (note 25) | | | | | | — | | | | — | | | | 1,182 | |
Deferred revenue | | | | | | — | | | | — | | | | (181 | ) |
| | | | | $ | 20,859 | | | $ | 12,366 | | | $ | 149,952 | |
2020
At the end of 2020, in reviewing its portfolio of compounds in the small molecule therapeutics segment, the Company identified impairment indicators for certain patents. One of the patent families impaired concerned a molecule that had entered into a phase 1 clinical trial in 2019 that was subsequently discontinued after the review of the pharmacokinetic data for the first three cohorts obtained. Following additional pre-clinical studies conducted in 2020 to further the Company’s understanding of the mechanism of action, or MOA, lead to findings that the MOA included engaging a receptor which has been known in other products which engage the same receptor to occasionally cause undesirable side effects. Subsequently, management decided that the preclinical and clinical development activities associated with demonstrating that such molecule did not induce such side effects would be both time-consuming and costly and therefore the future development has been suspended. Another patent family impaired concerned another molecule that is licensed for development with a third party, whose research and development work we believe to be delayed from the agreed upon timelines and is unlikely to perform significant development in the near future. Further, the development of another compound was deprioritized, as the Company wishes to prioritize development of its lead compound fezagepras, as well as GPR84 and OXER1 drug candidates, which led to the impairment of the related patents. These small molecules patents were written down to their net recoverable amount of $nil, as both the FVLCD and the value in use were determined to be insignificant, resulting in an impairment of $1,072 for the year ended December 31, 2020 (note 12).
F-55
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Subsequent to December 31, 2020, the Company announced it has undertaken to evaluate potential alternatives aimed at minimizing the plasma-derived therapeutics segment cash burn which may result in divestment in whole or part of this business, or other courses of action including but not limited to the closure of the Ryplazim® related operations, in order to focus our resources on the small molecules segment.
As the capital, intangible and ROU assets in the Ryplazim® CGU were no longer to be used as originally planned, management proceeded to review them for impairment and writing them down to their net recoverable value determined as the FVLCD using a market approach. The Ryplazim® CGU includes the assets involved in production, R&D and commercialization activities relating to the Ryplazim® product candidate that has yet to receive regulatory approval for commercialization. The Ryplazim® CGU evaluated excluded the assets pertaining the plasma collection activities since these can generate distinct cash inflows and could potentially be divested separately from the Ryplazim® assets. The plasma collection assets were not considered impaired.
The FVLCD was calculated using a discounted cash flow model for one year and a terminal value of $58.1 million using a post-tax discount rate of 7.75%. The fair value computed by management is considered as a level 3 computation in the fair value hierarchy under IFRS 13, Fair value measurement. As part of this valuation exercise, management needed to make several key assumptions which affected the cash inflows and outflows considered in the model. The significant estimates used in determining the FVLCD are disclosed in note 3.
As a result of this exercise, the Company recorded an impairment of $665 on capital assets (note 10), $18,553 on ROU assets (note 11) and $480 on intangible assets (note 12), respectively, representing an aggregate impairment of $19,698 on these plasma-derived therapeutic assets for the year ended December 31, 2020.
During the year, the Company recorded other impairments amounting to $89.
2019
During the year 2019, the Company, evaluated its intellectual property and the related market opportunities in the context of the Company’s financial situation and has made further decisions about the areas the Company will or will not pursue.
One of these decisions affecting our plasma-derived therapeutic segment was to no longer pursue further indications relating to the human-plasma protein plasminogen. As such, the Company decided it would retain sufficient staff to complete and resubmit a BLA, for congenital plasminogen deficiency and to build ongoing manufacturing supply, but then it would cease all R&D activities in the plasma-derived therapeutics segment not relating to Ryplazim®. Because of this, the Company’s long-term production forecasts for plasminogen were reduced and it was decided that one of its planned manufacturing facilities and a technical transfer facility would no longer be required. The Company also decided to close its R&D facility in Rockville, MD by the end of 2020. Consequently, the capital and intangible assets in the Plasma-derived therapeutics segment that were no longer to be used as originally planned were reviewed for impairment and written-down to their net recoverable value determined as the FVLCD using a market approach. The Company assessed the resale value of the property, plant and equipment, the licenses and patents, in their present condition, less cost of disposal and consequently, recorded an impairment of $7,070 and $4,535 on capital assets and intangible assets, respectively for the year ended December 31, 2019.
In reviewing its portfolio of compounds in the small molecule therapeutics segment, the Company identified compounds that where not within the areas of fibrosis on which it intends to focus and evaluated the net recoverable value of those related patents as $nil, determined as the fair value less cost of disposal using a market approach. An impairment on intangible assets of $634 was recognized for the year ended December 31, 2019.
F-56
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
As a result of the bioseparations business sale, some intellectual property including patents retained by the company are no longer expected to be developed. The company evaluated the net recoverable value of those patents is $nil, using a FVLCD using a market approach. An impairment on intangible assets of $127 was recognized for the year ended December 31, 2019.
2018
As a result of various events affecting the Company during 2018, including; 1) the delay of the commercial launch of Ryplazim® following the identification by the FDA of a number of changes required in the Chemistry, Manufacturing and Controls, or CMC, section of the BLA submission for congenital plasminogen deficiency, 2) the Company’s limited financial resources since the fourth quarter of 2018, which significantly delayed manufacturing expansion plans and resulted in the Company focusing its resources on the resubmission of the Ryplazim® BLA; 3) the recognition of the larger than anticipated commercial opportunities for Ryplazim®, and 4) the change in executive leadership in December 2018, the Company modified its strategic plans during the fourth quarter to focus all available plasma-derived therapeutic segment resources on the manufacturing and development of Ryplazim®, for the treatment of congenital plasminogen deficiency and other indications.
These changes and their various impacts prompted Management to perform an impairment test of the IVIG cash generating unit, which includes assets such as the licenses held by NantPro and Prometic Biotherapeutics inc. amongst others, manufacturing equipment located at its Canadian manufacturing facilities and the CDMO facility at December 31, 2018, and to review whether other assets pertaining to follow-on proteins might be impaired.
In regards to the IVIG CGU, in light of the substantial work, time and investment required to complete a robust CMC package for IVIG prior to the BLA filing, the limited resources available to complete the CMC section and the reduction of the forecasted IVIG production capacity at all plants would significantly delay the commercialisation of IVIG compared to previous timelines and as a result, cash inflows beginning beyond 2023 were not considered in the determination of the value in use due to the inherent uncertainty in forecasting cash flows beyond a five year period, as required by IAS 36, Impairment of Assets. As a result, the value in use for the IVIG CGU was $nil. Management also evaluated the FVLCD and determined that this value would also approximate $nil.
Consequently, impairment losses for the carrying amounts of the NantPro license and a second license acquired in January 2018, giving the rights to use IVIG clinical data and the design plans for a plant with a production capacity in excess of current needs, of $141,025 and $1,584, respectively, were recorded.
The Company acquired an option to purchase equipment located in Europe in January 2018 whose purchase was settled by the issuance of common shares as described in note 19a. An impairment was subsequently recorded on the option to purchase equipment in the amount of $653 since the likelihood of exercising this option is low in view of the current manufacturing and production plans.
Finally, an impairment of $5,689 was recorded on IVIG production equipment, to reduce its value to the FVLCD.
Management also reviewed the carrying amount of its investment in ProThera, as this represents an investment in follow-on proteins the Company had acquired, since the resources for further advancement of these assets are currently limited due to the focus on Ryplazim®.
The uncertainty of future cash flows for product candidates that have not yet commenced phase 1 trials was an important consideration is making these estimates. As a result, the Company recorded an impairment on its investment in an associate of $1,182. The value in use and the fair value less cost to sell of the investment in an associate were estimated to approximate $nil.
F-57
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Of the impairment losses recognized for the year ended December 31, 2018, $148,770 pertain to the plasma-derived therapeutics segment. The remainder of the impairment losses recognized did not belong to any segment.
25. Investment in an associate
During the quarter ended September 30, 2018, the Company concluded it exerted significant influence over ProThera Biologics, Inc., or ProThera, a company headquartered in Rhode Island, U.S.A., since August 15, 2018. As such, ProThera became an associate as well as a related party from that date and consequently, the equity investment in ProThera was accounted for using the equity method (note 2). ProThera is a biotherapeutics company developing methods for using Inter-alpha Inhibitor Proteins, or IaIP, to treat severe inflammation associated with infection, trauma and disease.
As of December 31, 2018, Liminal held 15.2% of the outstanding common shares of Prothera having a historical cost of $1,204. It also held an investment in convertible debt of ProThera. At December 31, 2018, the Company had invested $1,181 (US$ 866,000) in convertible debt of Prothera Biologics Inc. The convertible debt was convertible at the option of the issuer or the holder into preferred shares of ProThera, denominated in U.S. dollars and earning interest at 8.0% per annum, to be received at the date of maturity which was January 3, 2020.
As required when significant influence over an investment is obtained, the investment must be measured at fair value as of the date it became an associate. A fair value approach was applied by management in determining the fair value of the identifiable assets and liabilities of ProThera.
From August 15 until December 31, 2018, the associate reported losses of $144 of which $22 were recorded in the statement of operation as the Company’s share of the loss and comprehensive loss of an associate. During the fourth quarter of 2018, following changes to the Company’s strategic plans, an impairment of the investment in the associate, in the amount of $1,182 was recognized (note 24), reducing the carrying amount of the investment in the associate to $nil at December 31, 2018. The fair value of the investment in the convertible debt was estimated at $nil at December 31, 2018 and the loss of $1,181 is included as part of the change in fair value of financial instruments measure at fair value through profit or loss in the statement of operations.
On January 3, 2019, the convertible debt was converted into preferred shares of ProThera by the issuer.
In February 2019, the Company decided that it was no longer part of its strategy to pursue the development of Inter-alpha Inhibitor proteins and undertook discussions with ProThera to terminate the various corporate and commercial agreements it had in place with ProThera. The Company determined that, from that point on, it no longer had significant influence over ProThera and therefore changed its accounting for its investment in ProThera’s common shares as an investment in an associate to that of a financial asset at fair value through profit and loss. The fair value of such financial asset was evaluated at $nil at that time. Any transactions between the Company and ProThera as of that date are no longer considered as a related party transaction. During December 2019, Liminal transferred the preferred shares it held back to ProThera in consideration for the termination of the agreement.
F-58
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Changes in the carrying amount of the investment in an associate from the date it was initially recognized as an associate on August 15, 2018 to December 31, 2018 are as follows:
Loss and comprehensive loss of an associate from August 15 to December 31, 2018 | | | | | | | | $ | 144 | |
Share of losses of an associate | | | | | | | | | — | |
| | | | | | | | | | |
Historical cost of the investment in an associate | | | | | | | | | 1,204 | |
Less: | | | | | | | | | | |
Share of losses of an associate | | | | | | | | | — | |
Impairment on investment in an associate (note 24) | | | | | | | | | 1,182 | |
Carrying amount of the investment in an associate as at December 31, 2018 | | | | | | | | $ | 22 | |
The income tax recovery reported in the consolidated statement of operations for the years ended December 31, 2020, 2019 and 2018 are as follows:
| | 2020 | | | 2019 | | | 2018 | |
Current income taxes recovery | | $ | (136 | ) | | $ | (348 | ) | | $ | (5,822 | ) |
Deferred income taxes (recovery) | | | (65 | ) | | | 111 | | | | (13,815 | ) |
Income tax recovery from continuing operations | | | (201 | ) | | | (237 | ) | | | (19,637 | ) |
Income taxes from discontinued operations (note 6) | | | — | | | | 41 | | | | (382 | ) |
Total income tax recovery | | $ | (201 | ) | | $ | (196 | ) | | $ | (20,019 | ) |
F-59
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The following table provides a reconciliation of the income tax recovery calculated at the combined statutory income tax rate to the income tax recovery for both continuing and discontinued operations, recognized in the consolidated statements of operations.
| | 2020 | | | 2019 | | | 2018 | |
Net loss before tax from continuing operations | | $ | (122,338 | ) | | $ | (234,461 | ) | | $ | (259,465 | ) |
Net income before tax from discontinued operations | | | 3,380 | | | | 27,512 | | | | 1,550 | |
Combined Canadian statutory income tax rate | | | 26.5 | % | | | 26.6 | % | | | 26.7 | % |
Income tax recovery at combined income tax rate | | | (31,524 | ) | | | (55,048 | ) | | | (68,863 | ) |
| | | | | | | | | | | | |
Increase (decrease) in income taxes resulting from: | | | | | | | | | | | | |
Unrecorded potential tax benefit arising from current-period losses and other deductible temporary differences | | | 33,238 | | | | 31,962 | | | | 29,693 | |
Effect of tax rate differences in foreign subsidiaries | | | 1,101 | | | | 4,989 | | | | 4,481 | |
Non-deductible or taxable items | | | (157 | ) | | | (696 | ) | | | 6,074 | |
Change in tax rate | | | (1,455 | ) | | | 1,609 | | | | 242 | |
Write-off of previously recognized tax losses | | | - | | | | — | | | | 22,415 | |
Non-deductible loss (taxable gain) on debt renegociation | | | - | | | | 24,572 | | | | (8,784 | ) |
Research and development tax credit | | | (494 | ) | | | (740 | ) | | | (5,072 | ) |
Non-taxable gain on disposition of subsidiary (note 6) | | | (896 | ) | | | (6,903 | ) | | | — | |
Other | | | (14 | ) | | | 59 | | | | (205 | ) |
Income tax recovery | | $ | (201 | ) | | $ | (196 | ) | | $ | (20,019 | ) |
The following table presents the nature of the deferred tax assets and liabilities that make up the balance at December 31, 2020 and 2019.
| | Intangible assets | | | R&D expenses | | | Losses | | | Other | | | Total | |
As at January 1, 2019 Deferred tax assets | | $ | — | | | $ | (618 | ) | | $ | — | | | $ | 12 | | | $ | (606 | ) |
Charged (credited) to profit and loss | | | — | | | | 111 | | | | — | | | | (24 | ) | | | 87 | |
Derecognized - discontinued operations (note 6) | | | — | | | | — | | | | — | | | | 12 | | | | 12 | |
As at December 31, 2019 Deferred tax assets | | $ | — | | | $ | (507 | ) | | $ | — | | | $ | — | | | $ | (507 | ) |
Charged (credited) to profit and loss | | | — | | | | (65 | ) | | | — | | | | — | | | | (65 | ) |
As at December 31, 2020 Deferred tax assets | | $ | — | | | $ | (572 | ) | | $ | — | | | $ | — | | | $ | (572 | ) |
F-60
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Available temporary differences not recognized at December 31, 2020 and 2019 are as follows:
| | | | | | | 2020 | | | 2019 | |
Tax losses (non-capital) | | | | | | | $ | 569,542 | | | $ | 416,816 | |
Tax losses (capital) | | | | | | | | 305 | | | | — | |
Unused research and development expenses | | | | | | | | 84,556 | | | | 115,491 | |
Undeducted financing expenses | | | | | | | | 27,053 | | | | 21,258 | |
Interest expenses carried forward | | | | | | | | 32,475 | | | | 5,358 | |
Trade and other payable | | | | | | | | 1,640 | | | | 4,022 | |
Capital assets | | | | | | | | 3,392 | | | | 4,673 | |
Intangible assets | | | | | | | | 68,329 | | | | 81,899 | |
Start-up expense | | | | | | | | 5,358 | | | | 4,569 | |
Unrealized loss on exchange rate | | | | | | | | 5,430 | | | | 6,612 | |
Lease obligations | | | | | | | | 15,494 | | | | 44 | |
Other | | | | | | | | 1,071 | | | | 894 | |
| | | | | | | $ | 814,646 | | | $ | 661,636 | |
At December 31, 2020, the Company has non-capital losses of $574,465 of which $569,542 are available to reduce future taxable income for which the benefits have not been recognized. These losses expire at various dates from 2027 to 2040 except for the non-capital losses in the U.K. and U.S. losses that arose after 2017 which do not expire. Capital losses arising in Canada can only be utilized to shelter future capital gains. At December 31, 2020, the Company also has unused research and development expenses of $86,715 of which $84,556 are available to reduce future taxable income for which the benefits have not been recognized. These deductible expenses can be carried forward indefinitely.
At December 31, 2020, the Company also had unused federal tax credits available to reduce future income tax in the amount of $19,300 expiring between 2023 and 2040. Those credits have not been recorded and no deferred income tax assets have been recognized in respect to those tax credits.
F-61
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The unused non-capital losses expire as indicated in the table below:
| | | | | Canada | | | Foreign | |
At December 31, 2020 | | | | | Federal | | | Provincial | | | Countries | |
Losses carried forward expiring in: | | | | | | | | | | | | | | | |
2027 | | | | | $ | 3,510 | | | $ | 3,495 | | | $ | 4,774 | |
2028 | | | | | | — | | | | — | | | | 5,526 | |
2029 | | | | | | 76 | | | | 76 | | | | 2,658 | |
2030 | | | | | | 977 | | | | 977 | | | | 5,371 | |
2031 | | | | | | 855 | | | | 855 | | | | 6,380 | |
2032 | | | | | | 4,215 | | | | 4,208 | | | | — | |
2033 | | | | | | 13,763 | | | | 13,758 | | | | — | |
2034 | | | | | | 20,172 | | | | 22,219 | | | | 2,537 | |
2035 | | | | | | 45,396 | | | | 45,308 | | | | 13,086 | |
2036 | | | | | | 25,800 | | | | 25,695 | | | | 23,296 | |
2037 | | | | | | 35,788 | | | | 35,779 | | | | 32,071 | |
2038 | | | | | | 18,720 | | | | 18,746 | | | | — | |
2039 | | | | | | 47,365 | | | | 47,388 | | | | — | |
2040 | | | | | | 71,964 | | | | 72,036 | | | | — | |
| | | | | $ | 288,601 | | | $ | 290,540 | | | $ | 95,699 | |
Not expiring - UK | | | | | | — | | | | — | | | | 126,075 | |
Not expiring - US (post 2017) | | | | | | — | | | | — | | | | 64,091 | |
| | | | | $ | 288,601 | | | $ | 290,540 | | | $ | 285,865 | |
As a result of the conversion of the parent's debt into Liminal shares on April 23, 2019, more than 50% of the issued shares of Liminal were owned by a single shareholder at December 31, 2020. US tax rules impose restrictions that will impact how $236,701 of losses are available to shelter income in future taxation years. As a result of the US restrictions, approximately $111,834 of losses will no longer be available to the company and are not presented in the available tax loss table presented above. The utilization of the remainder of the company's available US tax losses included under Foreign tax loss carryforwards above are subject to restrictions and management is evaluating strategies to be able to benefit from them. The Company has $34,923 of U.S. tax loss carryforwards which arose after April 23, 2019 not subject to these limitations. A deferred tax asset has not been recognized for any loss carryforwards at December 31, 2020.
27. | Basic and diluted earnings per share |
The Company presents basic and diluted earnings per share, or EPS, data for its common shares. Basic EPS is calculated by dividing the profit or loss attributable to common shareholders of the Company by the weighted average number of common shares outstanding during the period, adjusted for any bonus element.
The numbers for the average basic and diluted shares outstanding for all the periods presented in the consolidated statements of operations have been adjusted in order to reflect the effect of the bonus element of the Rights Offering that occurred in June 2019 and the share consolidation that took place on July 5, 2019 (note 18).
For the years ended December 31, 2020, 2019 and 2018, all warrants, stock options and RSU were anti-dilutive since the Company reported net losses from continuing operations. The secured convertible debentures issued in 2020 are also anti-dilutive.
F-62
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The Company has two operating segments at December 31, 2020 which are the small molecule therapeutics segment, and the plasma-derived therapeutics segment. In previous financial statements, the Company also presented results for the bioseparations segment but since the sale of the bioseparation business on November 25, 2019 (note 5), those operations are being presented as discontinued operations in the consolidated statements of operations. The segmented information for prior periods has been restated to present the existing segments at the reporting date.
Small molecule therapeutics: The segment is focused on the discovery and development of novel small molecule drug candidates for the treatment of patients suffering from respiratory fibrotic diseases and other fibrotic or inflammatory diseases that have a high unmet medical need. Our lead small product candidate, fezagepras, is currently being developed for idiopathic pulmonary fibrosis.
Plasma-derived therapeutics: The segment leverages Liminal’s experience in bioseparation technologies used to isolate and purify biopharmaceuticals from human plasma. With respect to this second platform, the Company is focused on the development of its plasma-derived investigative drug Ryplazim® (plasminogen), or Ryplazim®, a highly purified glu-plasminogen derived from human plasma that acts as a plasminogen replacement therapy for patients deficient in plasminogen protein.
The reconciliation to the consolidated statement of operations column includes the elimination of intercompany transactions between the segments and the remaining activities not included in the above segments. These expenses generally pertain to public entity reporting obligations, investor relations, financing and other corporate office activities.
The accounting policies of the segments are the same as the accounting policies of the Company. The operating segments results include intercompany transactions between the segments which are done in a manner similar to transactions with third parties.
F-63
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
a) Revenues and expenses by operating segments:
For the year ended December 31, 2020 | | Small molecule therapeutics | | | Plasma- derived therapeutics | | | Reconciliation to statement of operations | | | Total | |
Revenues | | $ | 5 | | | $ | 2,599 | | | $ | 713 | | | $ | 3,317 | |
| | | | | | | | | | | | | | | | |
Expenses | | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | — | | | | 1,905 | | | | 128 | | | | 2,033 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | | 106 | | | | 27,427 | | | | — | | | | 27,533 | |
R&D - Other expenses | | | 13,107 | | | | 16,239 | | | | (53 | ) | | | 29,293 | |
Administration, selling and marketing expenses | | | 3,269 | | | | 6,532 | | | | 28,751 | | | | 38,552 | |
Segment loss | | $ | (16,477 | ) | | $ | (49,504 | ) | | $ | (28,113 | ) | | $ | (94,094 | ) |
Gain on foreign exchange | | | | | | | | | | | | | | | (668 | ) |
Finance costs | | | | | | | | | | | | | | | 8,982 | |
Gain on extinguishments of liabilities | | | | | | | | | | | | | | | (79 | ) |
Change in fair value of financial instruments measured at fair value through profit or loss | | | | | | | | | | | | | | | (850 | ) |
Impairment losses | | | | | | | | | | | | | | | 20,859 | |
Net loss before income taxes from continuing operations | | | | | | | | | | | | | | $ | (122,338 | ) |
Other information | | | | | | | | | | | | | | | | |
Depreciation and amortization | | $ | 1,007 | | | $ | 6,735 | | | $ | 705 | | | $ | 8,447 | |
Share-based payment expense | | | 2,153 | | | | 558 | | | | 3,523 | | | | 6,234 | |
For the year ended December 31, 2019 | | Small molecule therapeutics | | | Plasma- derived therapeutics | | | Reconciliation to statement of operations | | | Total | |
Revenues | | $ | 34 | | | $ | 4,736 | | | $ | 134 | | | $ | 4,904 | |
| | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | — | | | | 2,633 | | | | 130 | | | | 2,763 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | | 132 | | | | 37,107 | | | | (195 | ) | | | 37,044 | |
R&D - Other expenses | | | 15,419 | | | | 22,366 | | | | 285 | | | | 38,070 | |
Administration, selling and marketing expenses | | | 4,709 | | | | 8,368 | | | | 32,206 | | | | 45,283 | |
Segment loss | | $ | (20,226 | ) | | $ | (65,738 | ) | | $ | (32,292 | ) | | $ | (118,256 | ) |
Gain on foreign exchange | | | | | | | | | | | | | | | (1,451 | ) |
Finance costs | | | | | | | | | | | | | | | 14,056 | |
Loss on extinguishments of liabilities | | | | | | | | | | | | | | | 92,374 | |
Change in fair value of financial instruments measured at fair value through profit or loss | | | | | | | | | | | | | | | (1,140 | ) |
Impairment losses | | | | | | | | | | | | | | | 12,366 | |
Net loss before income taxes from continuing operations | | | | | | | | | | | | | | $ | (234,461 | ) |
Other information | | | | | | | | | | | | | | | | |
Depreciation and amortization | | $ | 779 | | | $ | 7,400 | | | $ | 679 | | | $ | 8,858 | |
Share-based payment expense | | | 4,782 | | | | 4,390 | | | | 12,369 | | | | 21,541 | |
F-64
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
For the year ended December 31, 2018 | | Small molecule therapeutics | | | Plasma- derived therapeutics | | | Reconciliation to statement of operations | | | Total | |
Revenues | | $ | — | | | $ | 24,521 | | | $ | 112 | | | $ | 24,633 | |
| | | | | | | | | | | | | | | | |
Cost of sales and other production expenses | | | — | | | | 25,297 | | | | 410 | | | | 25,707 | |
Manufacturing and purchase cost of product candidates used for R&D activities | | | 1,692 | | | | 37,107 | | | | (132 | ) | | | 38,667 | |
R&D - Other expenses | | | 14,234 | | | | 31,727 | | | | 230 | | | | 46,191 | |
Administration, selling and marketing expenses | | | 3,522 | | | | 10,393 | | | | 15,533 | | | | 29,448 | |
Segment loss | | $ | (19,448 | ) | | $ | (80,003 | ) | | $ | (15,929 | ) | | $ | (115,380 | ) |
Loss on foreign exchange | | | | | | | | | | | | | | | 4,696 | |
Finance costs | | | | | | | | | | | | | | | 22,041 | |
Gain on extinguishments of liabilities | | | | | | | | | | | | | | | (33,626 | ) |
Share of losses of an associate | | | | | | | | | | | | | | | 22 | |
Impairment losses | | | | | | | | | | | | | | | 149,952 | |
Change in fair value of financial instruments measured at fair value through profit or loss | | | | | | | | | | | | | | | 1,000 | |
Net loss before income taxes from continuing operations | | | | | | | | | | | | | | $ | (259,465 | ) |
Other information | | | | | | | | | | | | | | | | |
Depreciation and amortization | | $ | 480 | | | $ | 3,644 | | | $ | 415 | | | $ | 4,539 | |
Share-based payment expense | | | 1,270 | | | | 1,524 | | | | 3,606 | | | | 6,400 | |
Information by geographic area
b) Capital, intangible and right-of-use assets by geographic area
| | | 2020 | | | 2019 | |
Canada | | | | | | | $ | 28,231 | | | $ | 48,309 | |
United Kingdom | | | | | | | | 2,092 | | | | 3,141 | |
United States | | | | | | | | 12,517 | | | | 17,121 | |
| | | $ | 42,840 | | | $ | 68,571 | |
c) Revenues by location from continuing operations
| | | | 2020 | | | 2019 | | | 2018 | |
United States | | | | | $ | 2,073 | | | $ | 3,023 | | | $ | 22,854 | |
Canada | | | | | | 672 | | | | 1,881 | | | | 1,519 | |
United Kingdom | | | | | | 572 | | | | — | | | | — | |
Norway | | | | | | — | | | | — | | | | 260 | |
| | | | | $ | 3,317 | | | $ | 4,904 | | | $ | 24,633 | |
F-65
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Revenues are attributed to countries based on the location of customers.
The Company derives significant revenues from certain customers. During the year ended December 31, 2020, there is one customer in the plasma-derived therapeutics segment who accounted for 63% of total revenue from continuing operations. During the year ended December 31, 2019, there were two customers in the Plasma-derived therapeutics segment who accounted for 97% (62% and 35% respectively) of total revenue from continuing operations. For the year ended December 31, 2018, there were two customers in the Plasma-derived therapeutics segment who accounted for 93% (57% and 36% respectively) of total revenues for continuing operations.
29. | Related party transactions |
Balances and transactions between the Company and its subsidiaries, which are related parties of the Company, have been eliminated on consolidation and are not disclosed in this note. Details of transactions between the Company and other related parties are disclosed below and in other notes accordingly to the nature of the transactions. These transactions have been recorded at the exchange amount, meaning the amount agreed to between the parties.
Following the debt modification on November 14, 2018, the Company assessed whether SALP, the holder of the debt, had gained significant influence for accounting purposes, despite holding less than 20% of voting rights. The Company deemed that qualitative factors were significant enough to conclude that the holder of the debt had gained significant influence over the Company and had become a related party. SALP subsequently became Liminal’s parent Company following the debt restructuring completed on April 23, 2019.
All material transactions with SALP are disclosed in notes 15, 16, 17a, 19a and 19c where the transactions are disclosed and otherwise in this note.
During the year ended December 31, 2020, the Company recorded an interest expense and paid interest on the loan with its parent, SALP, of $2,121 and of $1,879 respectively ($7,831 and $7,831, respectively for the year ended December 31, 2019). The Company also recorded professional fee expenses, incurred by the parent and recharged to the Company, during the year ended December 31, 2019 of $469, all of which were paid as of December 31, 2019, $nil for the year ended December 31, 2020.
A former CEO had a share purchase loan outstanding in the amount of $400 at December 31, 2018. The loan bore interest at prime plus 1% and had a maturity date of the earlier of (i) March 31, 2019 or (ii) 30 days preceding a targeted Nasdaq or New York Stock Exchange listing date of Liminal’s shares. As part of the settlement agreement concluded in April 2019 with a former CEO of the Company, common shares held in escrow as security for a share purchase loan of $400 were released and the loan extinguished in exchange for the receipt of a payment of $137, representing the fair value of the shares at the time of the settlement.
F-66
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
30. | Compensation of key management personnel |
The Company’s key management personnel comprise the external directors, officers and executives which included 20 individuals in 2020, 28 individuals in 2019 and 25 individuals in 2018. The remuneration of the key management personnel during the years ended December 31, 2020, 2019 and 2018 was as follows:
| | | | | | | | | | | | | | | |
| | | | | 2020 | | | 2019 | | | 2018 | |
Current employee benefits1) | | | | | $ | 6,153 | | | $ | 10,083 | | | $ | 5,953 | |
Pension costs | | | | | | 115 | | | | 267 | | | | 268 | |
Share-based payments | | | | | | 4,917 | | | | 16,842 | | | | 3,685 | |
Termination benefits | | | | | | 319 | | | | 2,919 | | | | 3,651 | |
| | | | | $ | 11,504 | | | $ | 30,111 | | | $ | 13,557 | |
1) Current employee benefits include salaries, bonuses, other employee benefits other than those listed in the table and director fees paid in cash.
Royalties
SALP has a right to receive a 2% royalty on future revenues relating to patents of a specified small molecule product candidate and analogues, existing as of the date of the agreement was signed. The obligation under this royalty agreement is secured by all the assets of the Company until the expiry of the last patent covered by this agreement, anticipated in 2033.
In the normal course of business, the Company enters into license agreements for the market launching or commercialization of products. Under these licenses, including the ones mentioned above, the Company has committed to pay royalties ranging generally between 0.5% and 12.0% of net sales from products it commercializes and 3% of license revenues in regard to certain small molecule product candidates.
Other commitments
The Company signed a long-term manufacturing contract with a contract development and manufacturing organization, or CDMO, which provides the Company with additional manufacturing capacity. In connection with this contract, the Company has committed to a minimum annual spending of $9,000 for 2021 to 2030 (the end of the initial term) which includes all expenditures under the contract. As of December 31, 2020, the remaining payment under the CDMO contract was $83,700 or $38,236 after deduction of the minimum lease payments under the contract recognized in the consolidated statement of financial position as a lease liability (note 14). At December 31, 2020, total commitment remaining under the agreement with the CDMO that are not recognized in the lease liability are as follows:
| | | | Within 1 year | | | | | 2 - 5 years | | | | | Later than 5 years | | | | | Total | |
CDMO operating expense commitment | | | | | 4,691 | | | | | | 17,431 | | | | | | 16,114 | | | | | | 38,236 | |
The Company has one plasma purchase agreement whereby it has committed to purchase $7,071 of plasma in the year ended December 31, 2021.
F-67
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
32. | Financial instruments and financial risk management |
a)Fair value
As at December 31, 2020 and 2019, the fair value of financial liabilities for which fair value disclosure is required was the royalty payment obligation, the licence acquisition payment obligations and the long-term debt. The fair value of those liabilities approximated the carrying amount of such instruments.
The fair value of financial liabilities at December 31, 2020 was calculated using a discounted cash flow model and the market interest rates specific to the term of the debt instruments ranging from 8.24% to 15.05% (8.83% to 15.05% at December 31, 2019).
Fair value hierarchy
Financial instruments recorded at fair value on the consolidated statements of financial position are classified using a fair value hierarchy that reflects the significance of the inputs used in making the measurements. The fair value hierarchy has the following levels:
Level 1 – valuation based on quoted prices observed in active markets for identical assets or liabilities.
Level 2 – valuation techniques based on inputs that are quoted prices of similar instruments in active markets; quoted prices for identical or similar instruments in markets that are not active; inputs other than quoted prices used in a valuation model that are observable for that instrument; and inputs that are derived principally from or corroborated by observable market data by correlation or other means.
Level 3 – valuation techniques with significant unobservable market inputs.
A financial instrument is classified to the lowest level of the hierarchy for which a significant input has been considered in measuring fair value.
Cash, cash equivalents, and restricted cash are considered to be level 1 fair value measurements.
The long-term receivables, royalty payment obligation, license acquisition payment obligations, and long-term debt are level 2 measurements.
The investment in convertible debt of Prothera and the warrant liability are considered to be a level 3 measurements. Further discussion regarding assumptions used in determining their fair values are discussed in notes 3, 15 and 25 respectively.
b)Financial risk management
The Company has exposure to credit risk, liquidity risk and market risk. The Company’s Board of Directors has the overall responsibility for the oversight of these risks and reviews the Company’s policies on an ongoing basis to ensure that these risks are appropriately managed.
Credit risk:
Credit risk is the risk of financial loss to the Company if a customer, partner or counterparty to a financial instrument fails to meet its contractual obligations, and arises principally from the Company’s cash and receivables. The carrying amount of the financial assets represents the maximum credit exposure.
The Company mitigates credit risk through its reviews of new customer’s credit history before extending credit and conducts regular reviews of its existing customers’ credit performance. We evaluate at each reporting period, the lifetime expected credit losses on our accounts receivable balances based on the age of the receivable, credit history of the customers and past collection experience.
F-68
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
Following the sale of its bioseparations business, the Company has limited product sales from its plasma-derived therapeutics segment and as such the Company’s exposure to customer credit risk is limited.
Liquidity risk:
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they come due. The Company manages its liquidity risk by continuously monitoring forecasts and actual cash flows. The Company’s current liquidity situation is discussed in note 1.
The following table presents the contractual maturities of the financial liabilities as of December 31, 2020:
| | | | | | | | Contractual Cash flows | |
| | | | Carrying amount | | | Less than 1 year | | | 1-3 years | | | 3 - 5 years | | | More than 5 years | | | Total | |
Accounts payable and accrued liabilities 1) | | | | $ | 16,835 | | | $ | 16,835 | | | $ | — | | | $ | — | | | $ | — | | | $ | 16,835 | |
Long-term portion of royalty payment obligations | | | | | 107 | | | | — | | | | 51 | | | | 51 | | | | 229 | | | | 331 | |
Lease liabilities | | | | | 33,452 | | | | 7,434 | | | | 13,957 | | | | 13,328 | | | | 30,725 | | | | 65,444 | |
Long-term portion of other employee benefit liabilities | | | | | 206 | | | | — | | | | 206 | | | | — | | | | — | | | | 206 | |
Long-term debt 2) | | | | | 40,532 | | | | 3,945 | | | | 10,649 | | | | 40,353 | | | | — | | | | 54,947 | |
| | | | $ | 91,132 | | | $ | 28,214 | | | $ | 24,863 | | | $ | 53,732 | | | $ | 30,954 | | | $ | 137,763 | |
1) Short term portions of the royalty payment obligations and of other employee benefit liabilities are included in the account payable and accrued liabilities.
2) Under the terms of the consolidated loan agreement (note 16), SALP may decide to cancel a portion of the principal value of the loans as payment upon the exercise of their 168,735 warrants #10 and 3,947,367 November 2020 warrants. The maximum repayment due on the loans have been included in the above table.
Market risk:
Market risk is the risk that changes in market prices, such as interest rates and foreign exchange rates, will affect the Company’s income or the value of its financial instruments.
i)Interest risk
The Company’s interest-bearing financial liabilities have fixed rates and as such, there is limited exposure to changes in interest payments as a result of interest rate risk.
ii)Foreign exchange risk:
The Company is exposed to the financial risk related to the fluctuation of foreign exchange rates. The Company has had operations in the U.S. and the U.K. during the past years and therefore a portion of its expenses incurred are in U.S. dollars and in pounds sterling (£). The majority of the Company’s revenues that are part of its continuing operations have been in U.S. dollars which serve to mitigate a portion of the U.S. foreign exchange risk relating to the expenditures. Financial instruments that have exposed the Company to foreign exchange risk have been cash and cash equivalents receivables, trade and other payables, lease liabilities, licence payment obligations and the amounts drawn on the US$ credit facility. The Company manages foreign exchange risk by holding foreign currencies it received to support forecasted cash outflows in foreign currencies.
F-69
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
As at December 31, 2020 and 2019, the Company’s net exposure to currency risk through assets and liabilities denominated respectively in US$ and £ was as follows:
| | | 2020 | | | 2019 | |
| | | Amount | | | Equivalent in | | | Amount | | | Equivalent in | |
Exposure in US$ | | | in U.S. dollar | | | full CDN dollar | | | in U.S. dollar | | | full CDN dollar | |
Cash and cash equivalents | | | | 17,281,338 | | | | 22,018,153 | | | | 26,032,017 | | | | 33,883,273 | |
Accounts receivable | | | | 886,211 | | | | 1,129,121 | | | | 159,604 | | | | 207,741 | |
Other long-term assets | | | | 45,428 | | | | 57,880 | | | | 45,428 | | | | 59,129 | |
Accounts payable and accrued liabilities | | | | (6,023,877 | ) | | | (7,675,022 | ) | | | (7,209,564 | ) | | | (9,383,969 | ) |
Lease liabilities | | | | (10,918,525 | ) | | | (13,911,293 | ) | | | (22,426,384 | ) | | | (29,140,152 | ) |
Other long-term liabilities | | | | (162,000 | ) | | | (206,404 | ) | | | — | | | | — | |
Net exposure | | | | 1,108,575 | | | | 1,412,435 | | | | (3,398,899 | ) | | | (4,373,978 | ) |
| | | 2020 | | | 2019 | |
| | | Amount | | | Equivalent in | | | Amount | | | Equivalent in | |
Exposure in £ | | | in £ | | | full CDN dollar | | | in £ | | | full CDN dollar | |
Cash and cash equivalents | | | | 4,619,225 | | | | 8,032,832 | | | | 279,840 | | | | 480,233 | |
Accounts receivable | | | | 230,588 | | | | 400,993 | | | | 713,078 | | | | 1,223,713 | |
Income tax receivable | | | | — | | | | — | | | | 5,369,467 | | | | 9,214,542 | |
Accounts payable and accrued liabilities | | | | (983,697 | ) | | | (1,710,649 | ) | | | (971,763 | ) | | | (1,667,642 | ) |
Lease liabilities | | | | (271,623 | ) | | | (472,352 | ) | | | (350,783 | ) | | | (601,979 | ) |
Net exposure | | | | 3,594,493 | | | | 6,250,824 | | | | 5,039,839 | | | | 8,648,867 | |
Based on the above net exposures as at December 31, 2020, and assuming that all other variables remain constant, a 10% depreciation or appreciation of the CA$ against the US$ would result in a decrease or an increase of the consolidated net loss of approximately $141 while a 10% depreciation or appreciation of the CA$ against the £ would result in a decrease or an increase of the total comprehensive loss of approximately $625. The Company has not hedged its exposure to currency fluctuations.
The Company is, in the course of its business, subject to lawsuits and other claims. On April 15, 2019, the Company announced its intention to enter into a series of related arrangements to restructure its outstanding indebtedness, reduce its interest and certain other payment obligations, and raise sufficient cash to build a robust balance-sheet for the next phase of its development, collectively the refinancing transactions, which included (i) an offering of common shares through private placements for gross proceeds of $75,000 (note 18a), (ii) the conversion of approximately $229.0 million of the outstanding SALP debt into common shares (note 16), (iii) the adjustment of the terms of certain outstanding warrants (note 16) and (iv) a rights offering to all shareholders whereby each shareholder received one right for each common share held (note 18a). The restructuring transaction occurred on April 23, 2019.
On March 2, 2021, the Company was served with an action instituted by multiple individual shareholder plaintiffs, or the plaintiffs, against the Company, SALP, the directors that were on the Company’s Board on March 31, 2019 or on April 15, 2019, certain officers of the Company and other parties involved with the above refinancing transactions, together referred to as the defendants.
F-70
LIMINAL BIOSCIENCES INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2020
(In thousands of Canadian dollars, except for per share amounts)
The plaintiffs allege, among other things, that, as part of the refinancing transactions, the defendants (i) undervalued certain products, (ii) reduced certain of their operational activities, (iii) artificially devalued certain assets in order for them to be written-off in the consolidated statement of financial position, (iv) conducted their business in a manner that prevented them from obtaining financing from certain parties and (v) never properly disclosed their financial difficulties, the alleged collective result of which was, among other things, that SALP and Thomvest Asset Management were able to take control of the Company to the detriment of the minority shareholders.
The plaintiffs seek almost $700 million in damages, approximately $664 million of which is based on the loss of future value of the Company’s shares.
The Company believes that the plaintiffs’ claims are completely without merit and intends to vigorously defend itself. Defence and settlement costs associated with such lawsuits and claims can be substantial, even when these lawsuits and claims have no merit. Due to the inherent uncertainty of the litigation process, the resolution of any particular legal proceeding could have an adverse effect on the Company’s operating results or financial performance. No provisions have been recorded in the consolidated financial statements in regards to these claims.
F-71
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
Liminal BioSciences Inc.
/s/ Bruce Pritchard
Name: Bruce Pritchard
Title: Chief Executive Officer
(Principal Executive Officer)
Date: March 24, 2021