UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-Q
(Mark One)
☒ QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the quarterly period ended March 31, 2022
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission File Number: 001-37785
Reata Pharmaceuticals, Inc.
(Exact Name of Registrant as Specified in its Charter)
| | |
Delaware | | 11-3651945 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer
Identification No.) |
| | |
5320 Legacy Drive
Plano, Texas | | 75024 |
(Address of principal executive offices) | | (Zip Code) |
(972) 865-2219
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Securities Exchange Act of 1934:
| | | | |
Title of each class | | Trading Symbol(s) | | Name of each exchange on which registered |
Class A Common Stock, Par Value $0.001 Per Share | | RETA | | NASDAQ Global Market |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, an emerging growth company, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | |
Large accelerated filer | | ☒ | | Accelerated filer | | ☐ |
Non-accelerated filer | | ☐ | | Smaller reporting company | | ☐ |
Emerging growth company | | ☐ | | | | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
As of May 4, 2022, the registrant had 31,543,735 shares of Class A common stock, $0.001 par value per share, and 4,919,249 shares of Class B common stock, $0.001 par value per share, outstanding.
TABLE OF CONTENTS
i
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Quarterly Report on Form 10-Q contains forward-looking statements that involve substantial risks and uncertainties. We make such forward-looking statements pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 and other federal securities laws. In this Quarterly Report on Form 10-Q, all statements, other than statements of historical or present facts, including statements regarding our future financial condition, future revenues, projected costs, prospects, business strategy, and plans and objectives of management for future operations, are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “believe,” “will,” “may,” “might,” “estimate,” “continue,” “anticipate,” “intend,” “target,” “project,” “model,” “should,” “would,” “plan,” “expect,” “predict,” “could,” “seek,” “goals,” “potential,” and similar terms or expressions that concern our expectations, strategy, plans, or intentions. These forward-looking statements include, but are not limited to, statements about:
•our expectations regarding the timing, costs, conduct, and outcome of our clinical trials, including statements regarding the timing of the initiation and availability of data from such trials;
•the timing and likelihood of regulatory filings and approvals for our product candidates;
•whether regulatory authorities determine that additional trials or data are necessary in order to accept a new drug application for review and/or approval;
•our ability to obtain funding for our operations, including funding necessary to complete further development and commercialization of our product candidates;
•our plans to research, develop, and commercialize our product candidates;
•the manufacturing, supply, and commercialization of our product candidates, if approved;
•the rate and degree of market acceptance of our product candidates;
•our expectations regarding the potential market size and the size of the patient populations for our product candidates, if approved for commercial use, and the potential market opportunities for commercializing our product candidates;
•the success of competing therapies that are or may become available;
•our expectations regarding our ability to obtain and maintain intellectual property protection for our product candidates;
•the ability to license additional intellectual property relating to our product candidates and to comply with our existing license agreements;
•our ability to maintain and establish relationships with third parties, such as contract research organizations (CROs), contract manufacturing organizations, suppliers, and distributors;
•our ability to maintain and establish collaborators with development, regulatory, and commercialization expertise;
•our ability to attract and retain key scientific or management personnel;
•our ability to grow our organization and increase the size of our facilities to meet our anticipated growth;
•the accuracy of our estimates regarding expenses, future revenue, capital requirements, and needs for additional financing;
•our expectations related to the use of our available cash;
•our ability to develop, acquire, and advance product candidates into, and successfully complete, clinical trials;
1
•the initiation, timing, progress, and results of future preclinical studies and clinical trials, and our research and development programs;
•the impact of governmental laws and regulations and regulatory developments in the United States and foreign countries;
•developments and projections relating to our competitors and our industry; and
•the impact of the coronavirus disease (COVID-19) on our clinical trials, our supply chain, and our operations; and
•other risks and uncertainties, including those described under the heading “Risk Factors” included in our most recent Annual Report on Form 10-K for the year ended December 31, 2021, filed with the U.S. Securities and Exchange Commission (SEC) on February 28, 2022.
Any forward-looking statements in this Quarterly Report on Form 10-Q reflect our current views with respect to future events or to our future financial performance and involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance, or achievements to be materially different from any future results, performance, or achievements expressed or implied by these forward-looking statements. Given these uncertainties, you should not place undue reliance on these forward-looking statements.
You should read this Quarterly Report on Form 10-Q and the documents that we have filed as exhibits to this Quarterly Report on Form 10-Q completely and with the understanding that our actual future results may be materially different from what we expect. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
2
DEFINED TERMS
Unless the context requires otherwise, references to “Reata,” “the Company,” “we,” “us,” or “our” in this Quarterly Report on Form 10-Q refer to Reata Pharmaceuticals, Inc. and its subsidiaries. We also have used several other terms in this Quarterly Report on Form 10-Q, most of which are explained or defined below.
| | |
Abbreviated Term | | Defined Term |
AbbVie | | AbbVie Inc. |
ADPKD | | Autosomal dominant polycystic kidney disease |
ADL | | Activities of Daily Living |
AE | | Adverse event |
ALS | | Amyotrophic lateral sclerosis |
ATP | | Adenosine triphosphate |
bardoxolone | | Bardoxolone methyl |
BXLS | | Blackstone Life Sciences, LLC |
CKD | | Chronic kidney disease |
CMC | | Chemistry manufacturing controls |
COVID-19 | | Coronavirus disease |
CRL | | Complete Response Letter |
CRO | | Contract research organization |
DPNP | | Diabetic peripheral neuropathic pain |
eGFR | | Estimated glomerular filtration rate |
EMA | | European Medicines Agency |
ESKD | | End stage kidney disease |
Exchange Act | | Securities Exchange Act of 1934 |
FA | | Friedreich’s ataxia |
FDA | | United States Food and Drug Administration |
GFR | | Glomerular filtration rate |
Kyowa Kirin | | Kyowa Kirin Co., Ltd. |
LTIP Plan | | Second Amended and Restated Long Term Incentive Plan |
MAA | | Marketing Authorization Application |
mFARS | | Modified Friedreich’s Ataxia Rating Scale |
NDA | | New Drug Application |
PGIC | | Patient global impression of change |
PK | | Pharmacokinetic |
Registrational trial | | An adequate and well-controlled trial designed to be sufficient to apply for regulatory approval of a drug candidate, although notwithstanding the Company’s design a regulatory agency may determine that further clinical studies or data are required |
RSU | | Restricted Stock Unit |
SAE | | Serious adverse event |
SEC | | U.S. Securities and Exchange Commission |
U.S. GAAP | | Accounting principles generally accepted in the United States |
3
PART I - FINANCIAL INFORMATION
Item 1. Financial Statements.
Reata Pharmaceuticals, Inc.
Consolidated Balance Sheets
(in thousands, except share data)
| | | | | | | | |
| | March 31, 2022 | | | December 31, 2021 | |
| | (unaudited) | | | | |
Assets | | | | | | |
Cash and cash equivalents | | $ | 531,979 | | | $ | 590,258 | |
Prepaid expenses and other current assets | | | 5,357 | | | | 6,217 | |
Total current assets | | | 537,336 | | | | 596,475 | |
Property and equipment, net | | | 11,202 | | | | 11,604 | |
Operating lease right-of-use-assets | | | 131,178 | | | | 126,777 | |
Other assets | | | 152 | | | | 160 | |
Total assets | | $ | 679,868 | | | $ | 735,016 | |
Liabilities and stockholders’ equity | | | | | | |
Accounts payable | | | 9,015 | | | | 13,505 | |
Accrued direct research liabilities | | | 14,930 | | | | 14,249 | |
Other current liabilities | | | 13,778 | | | | 21,450 | |
Operating lease liabilities, current | | | 5,142 | | | | 3,142 | |
Deferred revenue | | | 755 | | | | 1,648 | |
Total current liabilities | | | 43,620 | | | | 53,994 | |
Other long-term liabilities | | | 5 | | | | 0 | |
Operating lease liabilities, noncurrent | | | 136,445 | | | | 132,891 | |
Liability related to sale of future royalties, net | | | 372,013 | | | | 362,142 | |
Total noncurrent liabilities | | | 508,463 | | | | 495,033 | |
Commitments and contingencies | | | | | | |
Stockholders’ equity: | | | | | | |
Common stock A, $0.001 par value:
500,000,000 shares authorized; issued and outstanding – 31,525,514 and
31,478,197 at March 31, 2022 and December 31, 2021, respectively | | | 31 | | | | 31 | |
Common stock B, $0.001 par value:
150,000,000 shares authorized; issued and outstanding – 4,919,249 and
4,919,249 at March 31, 2022 and December 31, 2021, respectively | | | 5 | | | | 5 | |
Additional paid-in capital | | | 1,457,222 | | | | 1,441,584 | |
Accumulated deficit | | | (1,329,473 | ) | | | (1,255,631 | ) |
Total stockholders’ equity | | | 127,785 | | | | 185,989 | |
Total liabilities and stockholders’ equity | | $ | 679,868 | | | $ | 735,016 | |
See accompanying notes.
4
Reata Pharmaceuticals, Inc.
Unaudited Consolidated Statements of Operations
(in thousands, except share and per share data)
| | | | | | | | |
| | Three Months Ended | |
| | March 31 | |
| | 2022 | | | 2021 | |
Collaboration revenue | | | | | | |
License and milestone | | $ | 893 | | | $ | 795 | |
Other revenue | | | 21 | | | | 149 | |
Total collaboration revenue | | | 914 | | | | 944 | |
Expenses | | | | | | |
Research and development | | | 39,804 | | | | 34,880 | |
General and administrative | | | 24,841 | | | | 20,704 | |
Depreciation | | | 308 | | | | 274 | |
Total expenses | | | 64,953 | | | | 55,858 | |
Other income (expense), net | | | (9,772 | ) | | | (12,556 | ) |
Loss before taxes on income | | | (73,811 | ) | | | (67,470 | ) |
Benefit from (provision for) taxes on income | | | (31 | ) | | | 15 | |
Net loss | | $ | (73,842 | ) | | $ | (67,455 | ) |
Net loss per share—basic and diluted | | $ | (2.03 | ) | | $ | (1.86 | ) |
Weighted-average number of common shares used in
net loss per share basic and diluted | | | 36,412,621 | | | | 36,203,631 | |
See accompanying notes.
5
Reata Pharmaceuticals, Inc.
Unaudited Consolidated Statements of Stockholders’ Equity
(in thousands, except share and per share data)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Three Months Ended March 31, 2022 | |
| | Common Stock A | | | Common Stock B | | | Additional
Paid-In | | | Total
Accumulated | | | Total
Stockholders’ | |
| | Shares | | | Amount | | | Shares | | | Amount | | | Capital | | | Deficit | | | Equity | |
Balance at December 31, 2021 | | | 31,478,197 | | | $ | 31 | | | | 4,919,249 | | | $ | 5 | | | $ | 1,441,584 | | | $ | (1,255,631 | ) | | $ | 185,989 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (73,842 | ) | | | (73,842 | ) |
Compensation expense
related to stock options | | | — | | | | — | | | | — | | | | — | | | | 15,444 | | | | — | | | | 15,444 | |
Exercise of options | | | — | | | | — | | | | 9,375 | | | | — | | | | 194 | | | | — | | | | 194 | |
Issuance of common stock upon
vesting of restricted stock units | | | 35,107 | | | | — | | | | 2,835 | | | | — | | | | — | | | | — | | | | — | |
Conversion of common
stock Class B to Class A | | | 12,210 | | | | — | | | | (12,210 | ) | | | — | | | | — | | | | — | | | | — | |
Balance at March 31, 2022 | | | 31,525,514 | | | $ | 31 | | | | 4,919,249 | | | $ | 5 | | | $ | 1,457,222 | | | $ | (1,329,473 | ) | | $ | 127,785 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Three Months Ended March 31, 2021 | |
| | Common Stock A | | | Common Stock B | | | Additional
Paid-In | | | Total
Accumulated | | | Total
Stockholders’ | |
| | Shares | | | Amount | | | Shares | | | Amount | | | Capital | | | Deficit | | | Equity | |
Balance at December 31, 2020 | | | 31,109,154 | | | $ | 31 | | | | 5,044,931 | | | $ | 5 | | | $ | 1,375,640 | | | $ | (958,245 | ) | | $ | 417,431 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (67,455 | ) | | | (67,455 | ) |
Compensation expense
related to stock options | | | — | | | | — | | | | — | | �� | | — | | | | 14,679 | | | | — | | | | 14,679 | |
Exercise of options | | | — | | | | — | | | | 112,423 | | | | — | | | | 4,678 | | | | — | | | | 4,678 | |
Issuance of common stock upon
vesting of restricted stock units | | | | | | | | | 3,302 | | | | | | | | | | | | | |
Conversion of common
stock Class B to Class A | | | 251,102 | | | | — | | | | (251,102 | ) | | | — | | | | — | | | | — | | | | — | |
Balance at March 31, 2021 | | | 31,360,256 | | | $ | 31 | | | | 4,909,554 | | | $ | 5 | | | $ | 1,394,997 | | | $ | (1,025,700 | ) | | $ | 369,333 | |
See accompanying notes.
6
Reata Pharmaceuticals, Inc.
Unaudited Consolidated Statements of Cash Flows
(in thousands)
| | | | | | | | |
| | Three Months Ended | |
| | March 31 | |
| | 2022 | | | 2021 | |
Operating activities | | | | | | |
Net loss | | $ | (73,842 | ) | | $ | (67,455 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | |
Depreciation | | | 308 | | | | 274 | |
Amortization of debt issuance costs and imputed interest | | | 0 | | | | 1,714 | |
Non-cash interest expense on liability related to sale of future royalty | | | 9,871 | | | | 10,925 | |
Stock-based compensation expense | | | 15,444 | | | | 14,679 | |
Changes in operating assets and liabilities: | | | | | | |
Income tax receivable and payable | | | 0 | | | | (22 | ) |
Prepaid expenses, other current assets and other assets | | | 877 | | | | 1,330 | |
Accounts payable | | | (4,525 | ) | | | 3,629 | |
Accrued direct research, other current and long-term liabilities | | | (8,914 | ) | | | (9,301 | ) |
Operating lease obligations | | | 3,489 | | | | 11 | |
Deferred revenue | | | (893 | ) | | | (795 | ) |
Net cash used in operating activities | | | (58,185 | ) | | | (45,011 | ) |
Investing activities | | | | | | |
Purchases of property and equipment | | | (288 | ) | | | (193 | ) |
Net cash used in investing activities | | | (288 | ) | | | (193 | ) |
Financing activities | | | | | | |
Exercise of options | | | 194 | | | | 4,678 | |
Net cash provided by financing activities | | | 194 | | | | 4,678 | |
Net decrease in cash and cash equivalents | | | (58,279 | ) | | | (40,526 | ) |
Cash and cash equivalents at beginning of year | | | 590,258 | | | | 818,150 | |
Cash and cash equivalents at end of period | | $ | 531,979 | | | $ | 777,624 | |
Non-cash activity: | | | | | | |
Right-of-use assets obtained in exchange for lease obligations | | $ | 4,885 | | | $ | 0 | |
Purchases of equipment in accounts payable, accrued direct research, other current, and long-term liabilities | | $ | 2,258 | | | $ | 28 | |
Acquisition of property and equipment through tenant improvement allowance | | $ | 0 | | | $ | 2,495 | |
See accompanying notes.
7
Reata Pharmaceuticals, Inc.
Notes to Unaudited Consolidated Financial Statements
1. Description of Business
Reata Pharmaceuticals, Inc.’s (Reata, the Company, we, us, or our) mission is to identify, develop, and commercialize innovative therapies that change patients’ lives for the better. The Company focuses on small-molecule therapeutics with novel mechanisms of action for the treatment of severe, life-threatening diseases with few or no approved therapies. The Company’s lead programs are omaveloxolone in a rare neurological disease called Friedreich’s ataxia (FA) and bardoxolone methyl (bardoxolone) in rare forms of chronic kidney disease (CKD). Both of the Company’s lead product candidates activate the transcription factor Nrf2 to normalize mitochondrial function, restore redox balance, and resolve inflammation. Because mitochondrial dysfunction, oxidative stress, and inflammation are features of many diseases, the Company believes omaveloxolone, bardoxolone, and our next-generation Nrf2 activators have many potential clinical applications. Reata possesses exclusive, worldwide rights to develop, manufacture, and commercialize omaveloxolone, bardoxolone, and our next-generation Nrf2 activators, excluding certain Asian markets for bardoxolone in certain indications, which are licensed to Kyowa Kirin Co., Ltd. (Kyowa Kirin). In addition, we are developing RTA 901, the lead product candidate from our Hsp90 modulator program, in neurological indications. We are the exclusive licensee of RTA 901 and have worldwide commercial rights.
The Company’s consolidated financial statements include the accounts of all majority-owned subsidiaries. Accordingly, the Company’s share of net earnings and losses from these subsidiaries is included in the consolidated statements of operations. Intercompany profits, transactions, and balances have been eliminated in consolidation.
Prior period reclassifications
Certain prior period amounts in the consolidated financial statements have been reclassified to conform to the current period presentation. Specifically, Operating lease obligations have been reclassed out of Accrued direct research, other current and long-term liabilities in prior periods to conform with the current period presentation on the consolidated statements of cash flows.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying unaudited consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (U.S. GAAP) for interim financial information and with the instructions to Form 10-Q and Article 10 of Regulation S-X. Accordingly, they do not include all of the information and notes required by U.S. GAAP for complete financial statements. In the opinion of management, all adjustments (consisting of normal recurring adjustments) considered necessary for a fair presentation have been included. Operating results for the three months ended March 31, 2022 are not necessarily indicative of the results that may be expected for the year ending December 31, 2022. The consolidated balance sheet at December 31, 2021, has been derived from the audited consolidated financial statements at that date but does not include all of the information and footnotes required by U.S. GAAP for complete financial statements. For further information, refer to the annual consolidated financial statements and footnotes thereto of the Company.
Summary of Significant Accounting Policies
The significant accounting policies used in the preparation of these condensed consolidated financial statements for the three months ended March 31, 2022 are consistent with those discussed in Note 2 to the consolidated financial statements in the Company’s Annual Report on Form 10-K for the year ended December 31, 2021.
8
3. Collaboration Agreements
Subsequent to the 2019 reacquisition of certain rights originally licensed to AbbVie Inc. (AbbVie) (see AbbVie below), the Company’s collaboration revenue and deferred revenue have been generated primarily from licensing fees and reimbursements for expenses received under our exclusive license with Kyowa Kirin (the Kyowa Kirin Agreement).
Kyowa Kirin
In December 2009, the Company entered into an exclusive license with Kyowa Kirin to develop and commercialize bardoxolone in the licensed territory. The terms of the agreement include payment to the Company of a nonrefundable, up-front license fee of $35.0 million and additional development and commercial milestone payments. As of March 31, 2022, the Company has received $50.0 million related to regulatory development milestone payments from Kyowa Kirin and has the potential in the future to achieve another $47.0 million from regulatory milestones and $140.0 million from commercial milestones. The Company also has the potential to achieve tiered royalties ranging from the low teens to the low 20 percent range, depending on the country of sale and the amount of annual net sales, on net sales by Kyowa Kirin in the licensed territory. The Company is participating on a joint steering committee with Kyowa Kirin to oversee the development and commercialization activities related to bardoxolone. Any future milestones and royalties received are subject to mid to lower single digit percent declining tiered commissions to certain consultants as compensation for negotiations of the Kyowa Kirin Agreement.
The up-front payment and regulatory milestones are accounted for as a single unit of accounting. The Company regularly evaluates its remaining performance obligation under the Kyowa Kirin Agreement. Accordingly, revenue may fluctuate from period to period due to changes to its estimated performance obligation period and variable considerations. The Company began recognizing revenue related to the up-front payment upon execution of the Kyowa Kirin Agreement.
In March 2021, the Company’s performance obligation period under the Kyowa Kirin Agreement was extended to June 2022 , which decreased quarterly revenue recognition by approximately $0.4 million prospectively.
On July 27, 2021, Kyowa Kirin submitted a New Drug Application (NDA) in Japan to the Ministry of Health, Labour and Welfare for bardoxolone for improvement of renal function in patients with Alport syndrome. Based on this submission, the Company earned a $5.0 million milestone payment, variable consideration previously considered constrained, under the Kyowa Kirin Agreement. As a result, the Company recorded $4.7 million in collaboration revenue, a cumulative catch-up for the portion of this milestone that was satisfied in prior periods, and $0.3 million in deferred revenue that will be recognized over the remaining performance obligation period, ending in June 2022.
AbbVie
In September 2010, the Company entered into a license agreement with AbbVie (the AbbVie License Agreement) for an exclusive license to develop and commercialize bardoxolone in the Licensee Territory (as defined in the AbbVie License Agreement).
In December 2011, the Company entered into a collaboration agreement with AbbVie (the Collaboration Agreement) to jointly research, develop, and commercialize the Company’s portfolio of second and later generation oral Nrf2 activators.
In October 2019, the Company and AbbVie entered into an Amended and Restated License Agreement (the Reacquisition Agreement) pursuant to which the Company reacquired the development, manufacturing, and commercialization rights concerning its proprietary Nrf2 activator product platform originally licensed to AbbVie in the AbbVie License Agreement and the Collaboration Agreement. In exchange for such rights, the Company agreed to pay AbbVie $330.0 million, all of which has subsequently been paid. Additionally, the Company will pay AbbVie an escalating, low single-digit royalty on worldwide net sales, on a product-by-product basis, of omaveloxolone and certain next-generation Nrf2 activators. The execution of the Reacquisition Agreement ended our performance obligations under the Collaboration Agreement.
9
The Company recognized interest expense related to the Reacquisition Agreement of approximately $1.7 million, during the three months ended March 31, 2021. As of March 31, 2022, the Company has fully satisfied its payable to AbbVie, therefore 0 interest expense was recognized for the three months ended March 31, 2022.
4. Liability Related to Sale of Future Royalties
On June 24, 2020, the Company closed on the Development and Commercialization Funding Agreement with an affiliate of Blackstone Life Sciences, LLC (BXLS), which provides funding for the development and commercialization of bardoxolone for the treatment of CKD caused by Alport syndrome, autosomal dominant polycystic kidney disease (ADPKD), and certain other rare CKD indications in return for future royalties (the Development Agreement). The Development Agreement includes a $300.0 million payment by an affiliate of BXLS in return for various percentage royalty payments on worldwide net sales of bardoxolone, once approved in the United States or certain specified European countries, by Reata and its licensees, other than Kyowa Kirin. The royalty percentage will initially be in the mid-single digits and, in future years, can vary between higher-mid single digit percentages to low-single digit percentages depending on various milestones, including indication approval dates, cumulative royalty payments, and cumulative net sales. Pursuant to the Development Agreement, we have granted BXLS a security interest in substantially all of our assets. After a bardoxolone product approval has been obtained by the Company, the Company is obligated to make certain minimum cumulative payment amounts in 2025 through 2033, but only until BXLS has achieved certain internal rate of return target.
In addition, concurrent with the Development Agreement, the Company entered into a common stock purchase agreement (the Purchase Agreement) with affiliates of BXLS to sell an aggregate of 340,793 shares of the Company’s Class A common stock at $146.72 per share for a total of $50.0 million.
The Company concluded that there were 2 units of accounting for the consideration received, comprised of the liability related to the sale of future royalties and the common shares. The Company allocated the $300.0 million from the Development Agreement and $50.0 million from the Purchase Agreement between the two units of accounting on a relative fair value basis at the time of the transaction. The Company allocated $294.5 million, which includes $0.8 million in transaction costs incurred, in transaction consideration to the liability, and $55.5 million to the common shares. The Company determined the fair value of the common shares based on the closing stock price on the June 24, 2020, the closing date of the Development Agreement. The effective interest rate under the Development Agreement, including transaction costs, is approximately 13.8%. The Company reassessed the expected royalty payments and lowered our previous estimate of future sales for which royalties will be paid. Accordingly, we have prospectively adjusted and recognized lower non-cash interest expense using a 10.9% effective interest rate, as of March 31, 2022.
The following table shows the activity within the liability related to sale of future royalties for the three months ended March 31, 2022:
| | | |
| Liability Related to Sale of Future Royalties | |
| (in thousands) | |
Balance at December 31, 2021 | $ | 362,928 | |
Non-cash interest expense recognized | | 9,855 | |
Balance at March 31, 2022 | | 372,783 | |
Less: Unamortized transaction cost | | (770 | ) |
Carrying value at March 31, 2022 | $ | 372,013 | |
10
5. Other Income (Expense), Net
| | | | | | | | |
| | Three Months Ended | |
| | March 31 | |
| | 2022 | | | 2021 | |
| | (in thousands) | |
Other income (expense), net | | | | | | |
Investment income | | $ | 132 | | | $ | 80 | |
Interest expense | | | 0 | | | | (1,714 | ) |
Non-cash interest expense on liability
related to sale of future royalty | | | (9,871 | ) | | | (10,925 | ) |
Other income (expense) | | | (33 | ) | | | 3 | |
Total other income (expense), net | | $ | (9,772 | ) | | $ | (12,556 | ) |
Investment Income
Interest income consists primarily of interest generated from our cash and cash equivalents.
Interest Expense
Interest expense consists primarily of the imputed interest from amount due to AbbVie under the Reacquisition Agreement.
Non-Cash Interest Expense on Liability Related to Sale of Future Royalties
Non-cash interest expense consists of recognition of interest expense based on the Company’s current estimate of future royalties expensed to be paid over the estimated term of the Development Agreement.
Other Income (Expense)
Other income (expense) consists primarily of gains and losses on foreign currency exchange.
6. Leases
The Company headquarters is located in Plano, Texas, where it leases approximately 122,000 square feet of office space. The Company leases additional space located in Irving, Texas, where it leases approximately 34,890 square feet of office and laboratory space.
On February 4, 2022, the Company extended the lease for the office and laboratory space in Irving, Texas, to October 31, 2024, with an option to extend for a fixed twelve-month period.
On March 8, 2022, the Company extended the lease for the Plano office to December 31, 2023.
The Company has an additional lease of a single-tenant, build-to-suit building of approximately 327,400 square feet of office and laboratory space located in Plano, Texas with an initial lease term of 16 years. The Company entered into the lease agreement on October 15, 2019 (the 2019 Lease Agreement), and at the Company’s option, it may renew the lease for two consecutive five-year renewal periods or one ten-year renewal period. On December 15, 2021, the Company obtained control of the space, and, accordingly, the Company recorded related right-of-use assets and the lease liabilities during the fourth quarter of 2021. The Company recorded the liability associated with the 2019 Lease Agreement at the present value of the lease payments not yet paid, using the discount rate as of the commencement date. As the discount rate implicit in the 2019 Lease Agreement was not readily determinable, the Company utilized its incremental borrowing rate. The renewals are not assumed in the determination of the lease term, since they are not deemed to be reasonably assured at the inception of the lease. At inception, the Company recorded $124.5 million as a right-of-use asset, which represented a lease liability of $133.2 million, net of $8.7 million of lease incentives recognized.
11
For the three months ended March 31, 2022, the Company paid $0.8 million for amounts included in the measurement of lease liabilities. During the three months ended March 31, 2022, and 2021, the Company recorded total rent expense of $4.3 million and $0.8 million, respectively.
Supplemental balance sheet and other information related to the Company’s operating leases is as follows:
| | | | | | | | | | |
| | | | As of March 31, | |
| | | | 2022 | | | 2021 | |
Weighted-average remaining lease term (in years) | | | 15.6 | | | | 1.5 | |
Weighted-average discount rate | | | | | 6.5 | % | | | 8.1 | % |
Maturities of lease liabilities by fiscal year for the Company’s operating leases:
| | | | |
| | As of March 31, 2022 | |
| | (in thousands) | |
2022 (remaining nine months) | | $ | 9,748 | |
2023 (1) | | | 10,638 | |
2024 | | | 7,427 | |
2025 | | | 13,737 | |
Thereafter | | | 196,049 | |
Total lease payments (1) | | | 237,599 | |
Less: Imputed interest | | | (96,012 | ) |
Present value of lease liabilities | | $ | 141,587 | |
(1) Above table assumes one year rent abatement is applied beginning in June 2023 following United States Food and Drug Administration (FDA) approval of omaveloxolone.
7. Income Taxes
The following table summarizes income tax (benefit) expense and effective income tax rate:
| | | | | | | | |
| | Three Months Ended | |
| | March 31 | |
| | 2022 | | | 2021 | |
| | (in thousands, except for percentage data) | |
Benefit from (provision for) taxes on income | | $ | (31 | ) | | $ | 15 | |
Effective income tax rate | | | 0.0 | % | | | 0.0 | % |
The Company’s effective tax rate for the three months ended March 31, 2022, varies with the statutory rate primarily due to changes in the valuation allowance related to certain deferred tax assets generated or utilized in the applicable period.
Deferred tax assets are regularly reviewed for recoverability by jurisdiction and valuation allowances are established based on historical and projected future taxable losses and the expected timing of the reversals of existing temporary differences. The Company has recorded valuation allowances against the majority of its deferred tax assets as of March 31, 2022, and the Company expects to maintain these valuation allowances until there is sufficient evidence that future earnings can be achieved, which is uncertain at this time.
12
8. Stock-Based Compensation
The following table summarizes time-based and performance-based stock compensation expense reflected in the consolidated statements of operations:
| | | | | | | | |
| | Three Months Ended | |
| | March 31 | |
| | 2022 | | | 2021 | |
| | (in thousands) | |
Research and development | | $ | 7,606 | | | $ | 6,808 | |
General and administrative | | | 7,838 | | | | 7,871 | |
Total stock compensation expense | | $ | 15,444 | | | $ | 14,679 | |
Restricted Stock Units (RSUs)
The following table summarizes RSU activity as of March 31, 2022, under the Second Amended and Restated Long Term Incentive Plan (LTIP Plan) agreement:
| | | | | | | | |
| | Number of
RSUs | | | Weighted-Average
Grant Date Fair
Value | |
Outstanding at January 1, 2022 | | | 809,145 | | | $ | 66.91 | |
Granted | | | 513,559 | | | | 27.36 | |
Vested | | | (37,942 | ) | | | 110.88 | |
Forfeited | | | (67,917 | ) | | | 71.76 | |
Outstanding at March 31, 2022 | | | 1,216,845 | | | $ | 48.58 | |
As of March 31, 2022, total unrecognized compensation expense related to RSU and performance-based RSUs awards that were deemed probable of vesting was approximately $40.8 million, which excludes 148,000 shares of unvested performance-based RSUs that were deemed not probable of vesting totaling unrecognized stock-based compensation expense of $14.2 million.
Stock Options
The following table summarizes stock option activity as of March 31, 2022, under the LTIP Plan and standalone option agreements:
| | | | | | | | |
| | Number of
Options | | | Weighted-
Average
Price | |
Outstanding at January 1, 2022 | | | 4,743,180 | | | $ | 86.06 | |
Granted | | | 1,110,981 | | | | 27.78 | |
Exercised | | | (9,375 | ) | | | 21.25 | |
Forfeited | | | (175,492 | ) | | | 114.33 | |
Expired | | | (7,468 | ) | | | 145.19 | |
Outstanding at March 31, 2022 | | | 5,661,826 | | | $ | 73.78 | |
Exercisable at March 31, 2022 | | | 3,013,168 | | | $ | 59.05 | |
As of March 31, 2022, total unrecognized compensation expense related to stock options was approximately $88.4 million, which excludes 568,450 shares of unvested performance-based stock options that were deemed not probable of vesting totaling unrecognized stock-based compensation expense of $49.4 million.
The total intrinsic value of all outstanding options and exercisable options as of March 31, 2022 was $24.5 million and $18.9 million, respectively.
13
The number of weighted average options that were not included in the diluted earnings per share calculation because the effect would have been anti-dilutive represented 6,878,671 and 4,766,386 shares as of March 31, 2022 and 2021, respectively.
9. Employee Benefit Plans
In 2010, we adopted an Employee Investment Plan, qualified under Section 401(k) of the Internal Revenue Code, which is a retirement savings plan covering substantially all of our U.S. employees (the Plan). The Plan is administered under the “safe harbor” provision of ERISA. Under the Plan, an eligible employee may elect to contribute a percentage of their salary on a pre-tax basis, subject to federal statutory limitations. Beginning in January 2019, the Company implemented a discretionary employer matching contribution of $1.00 for every $1.00 contributed by a participating employee up to $7,000 and $6,000 annually in 2022 and 2021, respectively, which such matching contributions become fully vested after four years of service. The Company recorded expense of $1.3 million and $0.8 million for the three months ended March 31, 2022 and 2021, respectively, which includes the Company’s contributions and administrative costs.
10. Commitments and Contingencies
Litigation
From time to time, the Company is a party to legal proceedings in the course of its business, including the matters described below. The outcome of any such legal proceedings, regardless of the merits, is inherently uncertain. In addition, litigation and related matters are costly and may divert the attention of our management and other resources that would otherwise be engaged in other activities. If the Company were unable to prevail in any such legal proceedings, its business, results of operations, liquidity and financial condition could be adversely affected. The Company recognizes accruals for litigations to the extent that it can conclude that a loss is both probable and reasonably estimable and recognizes legal expenses as incurred.
Bardoxolone Securities Litigation
Four putative stockholders of the Company filed complaints for alleged violations of the federal securities laws against the Company and certain of its executives, including its Chief Executive Officer, its Chief Operating Officer and Chief Financial Officer, and its Chief Innovation Officer (in one of the suits), three of the complaints were filed in the United States District Court for the Eastern District of Texas, and one was filed in the District of New Jersey. The complaints allege, among other things, that the Company (and certain of its executives) made false and misleading statements regarding the sufficiency of the Phase 3 CARDINAL study to support an NDA for bardoxolone in the treatment of CKD caused by Alport syndrome, and the Company’s interactions with the FDA concerning the study. The complaints filed in the United States District Court for the Eastern District of Texas, were consolidated on April 22, 2022, and the complaint filed in the District of New Jersey on February 18, 2022, which subsequently was voluntarily dismissed. The plaintiffs seek, among other things, a class action designation, an award of damages, and costs and expenses, including attorney fees and expert fees. The Company currently expects a single, consolidated, amended complaint to be filed in the future.
The Company believes that the allegations contained in the complaints are without merit and intends to defend the cases. The Company cannot predict at this point the length of time that these actions will be ongoing or the liability, if any, which may arise therefrom.
Derivative Lawsuit
An alleged stockholder of the Company filed a derivative action in the Court of Chancery of the State of Delaware against all of the directors of the Company and naming the Company as a nominal defendant. The plaintiff asserts claims in the complaint of breach of fiduciary duty and unjust enrichment concerning the alleged payment of excessive compensation to the non-employee directors of the Company between fiscal years 2019 and 2021. The plaintiff seeks, among other things, an order awarding damages and costs and expenses, including attorneys and expert fees, and directing the Board of Directors to reform and improve its corporate governance and internal procedures relating to the award of non-employee director compensation.
14
The defendants believe that the allegations contained in the complaint are without merit and intend to defend the case. The Company cannot predict at this point the length of time that this action will be ongoing or the liability, if any, which may arise therefrom.
Indemnifications
Accounting Standards Codification 460, Guarantees, requires that, upon issuance of a guarantee, the guarantor must recognize a liability for the fair value of the obligations it assumes under that guarantee.
As permitted under Delaware law and in accordance with the Company’s bylaws, officers and directors are indemnified for certain events or occurrences, subject to certain limits, while the officer or director is or was serving in such capacity. The maximum amount of potential future indemnification is unlimited; however, the Company has obtained director and officer insurance that limits its exposure and may enable recoverability of a portion of any future amounts paid. The Company believes the fair value for these indemnification obligations is minimal. Accordingly, the Company has not recognized any liabilities relating to these obligations as of March 31, 2022.
The Company has certain agreements with licensors, licensees, collaborators, and vendors that contain indemnification provisions. In such provisions, the Company typically agrees to indemnify the licensor, licensee, collaborator, or vendor against certain types of third-party claims. The Company accrues for known indemnification issues when a loss is probable and can be reasonably estimated. There were 0 accruals for expenses related to indemnification issues for any period presented.
11. Subsequent Events
On May 6, 2022, an alleged stockholder of the Company filed a derivative action in the Court of Chancery of the State of Delaware against all of the directors of the Company and naming the Company as a nominal defendant. See Note 10, Commitments and Contingencies of Notes to Unaudited Consolidated Financial Statements for a description of the litigation.
15
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and related notes and other financial information appearing in this Quarterly Report on Form 10-Q. Some of the information contained in this discussion and analysis or set forth elsewhere in this Quarterly Report on Form 10-Q, including information with respect to our plans and strategy for our business, operations, and product candidates, includes forward-looking statements that involve risks and uncertainties. Factors that may cause actual results to differ materially from current expectations include, among other things, those described under the headings “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements” and discussed elsewhere in this Quarterly Report on Form 10-Q.
Overview
We are a clinical-stage biopharmaceutical company focused on identifying, developing, and commercializing innovative therapies that change patients’ lives for the better. We concentrate on small-molecule therapeutics with novel mechanisms of action for the treatment of severe, life-threatening diseases with few or no approved therapies. Our lead programs are omaveloxolone in FA and bardoxolone in rare forms of CKD. Both of our lead product candidates activate the transcription factor Nrf2 to normalize mitochondrial function, restore redox balance, and resolve inflammation. Because mitochondrial dysfunction, oxidative stress, and inflammation are features of many diseases, we believe omaveloxolone, bardoxolone, and our next-generation Nrf2 activators have many potential clinical applications. We possess exclusive, worldwide rights to develop, manufacture, and commercialize omaveloxolone, bardoxolone, and our next-generation Nrf2 activators, excluding certain Asian markets for bardoxolone in certain indications, which are licensed to Kyowa Kirin. In addition, we are developing RTA 901, the lead product candidate from our Hsp90 modulator program, in neurological indications. We are the exclusive licensee of RTA 901 and have worldwide commercial rights.
Recent Key Developments
Omaveloxolone for Friedreich’s Ataxia
The FDA has granted Fast Track Designation, Orphan Drug Designation, and Rare Pediatric Disease Designation to omaveloxolone for the treatment of FA. In March 2022, we completed rolling submission of an NDA to the FDA for omaveloxolone for the treatment of patients with FA. This NDA is supported by the efficacy and safety data from the MOXIe Part 2 trial and additional supporting data from the MOXIe Part 1 and MOXIe Extension trials.
We are continuing to complete the regulatory procedures and submissions required prior to filing a Marketing Authorization Application (MAA) in Europe for approval of omaveloxolone for the treatment of patients with FA. We have secured agreement on our Pediatric Investigation Plan with the Pediatric Committee, and we also received European Medicines Agency (EMA) Follow-Up Protocol Assistance feedback regarding our nonclinical and chemistry manufacturing controls (CMC) programs. The EMA feedback indicated that there were no identified impediments to our planned MAA submission and included agreement that certain nonclinical studies, including 2- year carcinogenicity study data, may be submitted after approval. We plan to submit an MAA to the European Medicines Agency for omaveloxolone in the fourth quarter of 2022.
RTA 901 for Neurological Indications, Including Diabetic Peripheral Neuropathic Pain
In the first quarter of 2022 we initiated additional Phase 1 clinical pharmacology studies of RTA 901, including a drug-drug interaction study. Following completion of these Phase 1 studies, we plan to initiate a randomized, double-blind placebo-controlled Phase 2 trial of RTA 901 in diabetic patients with peripheral neuropathic pain in the fourth quarter of 2022.
Bardoxolone in Patients with CKD Caused by Alport Syndrome
We received a Complete Response Letter (CRL) from the FDA in February 2022 with respect to its review of our NDA for bardoxolone in the treatment of patients with CKD caused by Alport syndrome. The CRL indicated the FDA cannot approve the NDA in its present form. We will continue to work with the FDA to confirm our next steps on our Alport syndrome program.
16
In October 2021, we submitted an MAA to the EMA for bardoxolone in the treatment of patients with CKD caused by Alport syndrome. In the first quarter of 2022, we received the 120-day list of questions from the EMA. We are in the process of preparing our responses. We requested a 90-day extension for our responses which was granted by the EMA. The timeline for the EMA’s review cycle was therefore extended.
Bardoxolone in Patients with Autosomal Dominant Polycystic Kidney Disease (ADPKD)
We are currently enrolling patients in FALCON, a Phase 3, international, multi-center, randomized, double-blind, placebo-controlled trial studying the safety and efficacy of bardoxolone in patients with ADPKD, randomized one-to-one to active drug or placebo. FALCON is enrolling patients in a broad range of ages with an estimated glomerular filtration rate (eGFR) between 30 and 90 mL/min/1.73 m2. More than 550 patients are currently enrolled in the trial.
In the first quarter of 2022, we submitted a protocol amendment to the FALCON Phase 3 trial of bardoxolone in patients with ADPKD with the FDA and other relevant health authorities. As agreed with the FDA prior to submission of the amendment, we recently had a Type A meeting to discuss key changes made to the FALCON protocol. Based on the discussion during the meeting and the meeting minutes, the Division stated that the proposed primary endpoint of eGFR change from baseline at Week 108 (8 weeks after planned drug discontinuation at Week 100) was reasonable since the available data suggest that bardoxolone’s acute pharmacodynamic effect on eGFR should be largely resolved. The Division provided guidance on handling of data from patients who completed Year 2 of the study before the protocol amendment and so did not have an eGFR assessment at Week 108. This included direction on imputing missing data for these patients in the primary analysis. The Division stated that, in addition to the primary endpoint, it will be important to demonstrate that the treatment effect accrues over time to support a claim that bardoxolone slows the loss of kidney function in patients with ADPKD and provided guidance on the statistical methodologies for the exploratory eGFR slope analyses. The FDA also confirmed that if FALCON is positive, it could support registration of bardoxolone in ADPKD.
Update on Adjustments to Operations Due to COVID-19
When COVID-19 emerged as a global pandemic in the first quarter of 2020, Reata was quick to respond and was an early adopter of a work-from-home policy, with the exception of the laboratory that continued to operate throughout under strict safety protocols. For all remote employees, we provided appropriate workstation equipment as well as training and resources to support employees’ mental and emotional wellbeing. In the second quarter of 2021, Reata relaxed its policy and permitted employees to return to the office as required. Although the Omicron variant temporarily caused us to again restrict office attendance, in February 2022 we permitted modified work schedules to meet business and personal needs.
Background: Our Programs
The following chart outlines each of our programs by indication and phase of development:

1Rolling NDA submission completed in March 2022.
2DPNP: Diabetic peripheral neuropathic pain.
17
3On February 25, 2022, we received a CRL from the FDA. We will continue to work with the FDA to confirm our next steps on our Alport syndrome program. MAA in EU is under review.
4AYAME trial conducted in Japan by our strategic collaborator in CKD, Kyowa Kirin. Kyowa Kirin expects the last patient visit in the second half of 2022.
5Based on the outcome of AYAME and FALCON trials, and our discussions with the FDA regarding the bardoxolone program, we will decide future development plans for bardoxolone in additional forms of CKD.
Programs in Neurological Diseases
We are developing omaveloxolone for the treatment of patients with FA, an inherited, debilitating, and degenerative neuromuscular disorder that is usually diagnosed during adolescence and can ultimately lead to premature death. In March 2022, we completed rolling submission of our NDA for omaveloxolone for the treatment of patients with FA. Because mitochondrial dysfunction is a key feature of many neuromuscular diseases, we believe omaveloxolone may be broadly applicable to treat neurological diseases by activating Nrf2 to normalize and improve mitochondrial function and adenosine triphosphate (ATP) production. We plan to pursue the development of omaveloxolone and our other Nrf2 activators for one or more additional neurological diseases.
We are also developing RTA 901 for the treatment of neurological diseases. RTA 901 is a highly potent and selective C-terminal modulator of Hsp90, which has a critical role in mitochondrial function, protein folding, and inflammation. RTA 901 has demonstrated profound efficacy in a wide range of animal models of neurological disease, including diabetic neuropathy, neuroinflammation, and neuropathic pain. We plan to initiate a randomized, placebo-controlled Phase 2 trial in DPNP in the fourth quarter of 2022.
Omaveloxolone in Patients with Friedreich’s Ataxia
Patients with FA experience progressive loss of coordination, muscle weakness, and fatigue, which commonly progress to motor incapacitation and wheelchair reliance. Based on literature and proprietary research, we believe FA affects approximately 5,000 children and adults in the United States and 22,000 individuals globally. According to data provided by IQVIA in 2020, there are approximately 4,000 projected patients diagnosed with FA in the United States. The FDA has granted Orphan Drug Designation, Fast Track Designation, and Rare Pediatric Disease Designation to omaveloxolone for the treatment of FA. The European Commission has granted Orphan Drug Designation in Europe to omaveloxolone for the treatment of FA.
Diagnosis of FA typically occurs by genetic testing, and most people in the United States with FA are diagnosed in their teens and early twenties. Patients with FA experience progressive loss of coordination, muscle weakness, and fatigue that commonly results in motor incapacitation, with patients requiring a wheelchair in their twenties. The mean age of death for patients with FA is in the mid-thirties. Childhood-onset FA can occur as early as age five, is more common than later-onset FA, and normally involves more rapid disease progression. Currently, there are no approved therapies for the treatment of FA.
MOXIe Part 2 Trial Results
Part 2 of our Phase 2 trial, called MOXIe (MOXIe Part 2), was an international, multi-center, double-blind, placebo-controlled, randomized, Registrational trial that enrolled 103 patients with FA at 11 trial sites in the United States, Europe, and Australia. MOXIe Part 2 was one of the largest global, interventional trials ever completed in FA. Patients were randomized one-to-one to omaveloxolone or placebo. MOXIe Part 2 was completed in October 2019. The primary analysis population excluded patients with pes cavus (n=82), a musculoskeletal foot deformity that may interfere with the patient’s ability to perform some components of the neurological exam used to score the primary endpoint of the trial. Safety analyses were evaluated in the all-randomized population (n=103).
The primary endpoint for the trial was the change in the Modified Friedreich’s Ataxia Rating Scale (mFARS) score for omaveloxolone relative to placebo after 48 weeks of treatment. Omaveloxolone treatment demonstrated statistically significant evidence of efficacy for the primary endpoint of the trial, producing a placebo-corrected -2.40 point mean improvement in mFARS (n=82; p=0.014). Patients treated with omaveloxolone experienced a mean improvement in mFARS of -1.55 points from baseline, while patients treated with placebo experienced a mean worsening in mFARS of +0.85 points from baseline. Further, the observed placebo-corrected improvements in mFARS were time-dependent, increasing over the course of treatment with the largest improvement observed after 48 weeks of treatment.
18
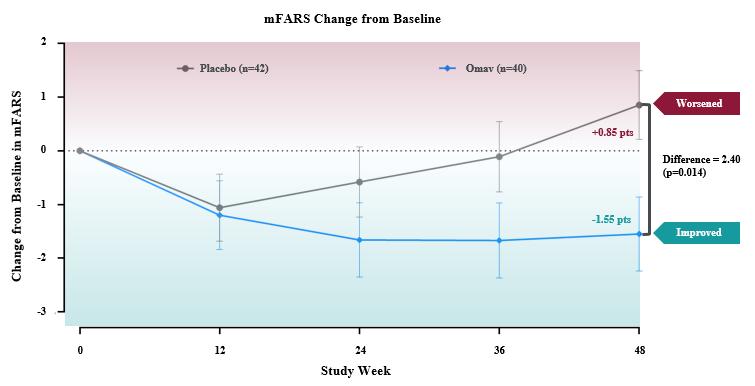
Additionally, all secondary endpoints either favored the omaveloxolone arm or were neutral. Patients on omaveloxolone experienced a nominal improvement in the Activities of Daily Living (ADL) questionnaire, with all nine questions favoring the omaveloxolone arm. On average, ADL scores for patients on omaveloxolone did not change from baseline, while placebo-treated patients worsened. Both patient global impression of change (PGIC) and clinical global impression of change numerically favored omaveloxolone, and improvement in PGIC correlated with the observed improvement in mFARS.
Omaveloxolone was reported to be generally well-tolerated. Four (8%) omaveloxolone patients and two (4%) placebo patients discontinued trial drug due to an adverse event (AE). The reported AEs were generally mild to moderate in intensity, and the most common AEs (i.e., reported in > 10% of omaveloxolone-treated patients) observed more frequently (>5% difference) in omaveloxolone compared to placebo were headache, nausea, increased aminotransferases, fatigue, abdominal pain, diarrhea, oropharyngeal pain, muscle spasms, back pain, and decreased appetite. Increases in aminotransferases are a pharmacological effect of omaveloxolone. In preclinical studies, omaveloxolone has been shown to increase production of aminotransferases in vitro, which we believe are related to restoration of mitochondrial function. In MOXIe Part 2, the aminotransferase increases were associated with improvements (reductions) in total bilirubin and were not associated with any evidence of liver injury.
In MOXIe Part 2, the overall rate of serious adverse events (SAEs) was low, with five patients in the omaveloxolone group and three patients in the placebo group reporting SAEs. No new safety signals were identified, and the reported SAEs were sporadic and generally expected in FA patients. In the patients who reported SAEs while receiving omaveloxolone, none led to discontinuation.
MOXIe Extension Trial
The open-label MOXIe Extension trial is ongoing, with a total of 149 patients enrolled (57 patients from MOXIe Part 1 and 92 patients from MOXIe Part 2). A total of 73 out of 75 (97%) patients without pes cavus who completed MOXIe Part 2 were enrolled in the MOXIe Extension, including 39 patients previously randomized to placebo (the placebo-to-omaveloxolone group) and 34 patients previously randomized to omaveloxolone (the omaveloxolone-to-omaveloxolone group). Due to the COVID-19 pandemic, not all patients had mFARS assessments performed at each time point.
19
Delayed-Start Analysis Results from August 2021 Data Cut-Off
The intent of the post-hoc Delayed-Start Analysis is to evaluate whether omaveloxolone has a persistent effect on FA disease course. Conceptually, this analysis evaluates whether the treatment effect that was observed in the placebo-controlled MOXIe Part 2 trial is maintained in the MOXIe Extension trial when all patients are receiving omaveloxolone. If the treatment effect is maintained between those originally randomized to placebo (the placebo-to-omaveloxolone group) versus those originally randomized to omaveloxolone (the omaveloxolone-to-omaveloxolone group), then it demonstrates evidence of a persistent effect on the course of the disease. If the treatment effect is not maintained, and the patients originally randomized to placebo are able to achieve the same absolute response and “catch up” to the patients initially randomized to omaveloxolone, the results are consistent with a symptomatic treatment that does not affect the underlying course of the disease.
Two timepoints were used in the analysis. The first timepoint was at Week 48, the final week of treatment in the placebo-controlled MOXIe Part 2 trial. The second timepoint was at Week 72 of the open-label MOXIe Extension in which all patients received omaveloxolone. A non-inferiority test was used to evaluate if the difference in mFARS between groups observed at the first timepoint was maintained or non-inferior at the second timepoint. The analysis methods, including the specified non-inferiority margin, were based on literature (Liu-Seifert, 2015a, 2015b). When comparing treatment groups using this methodology, maintaining a negative difference between treatment groups in mFARS is evidence of a persistent treatment effect.
The Delayed-Start Analysis used in clinical modules in our initial NDA rolling submission for omaveloxolone was updated as of August 2021. In this updated analysis 58 of 73 patients from MOXIe Part 2 without pes cavus who enrolled into MOXIe Extension had at least 72 weeks of exposure in MOXIe Extension, and 28 of these patients had at least 120 weeks of exposure in the Moxie Extension.
Results of this analysis demonstrated that the between-group difference in mFARS observed at the end of the placebo-controlled MOXIe Part 2 period (least squares mean difference = -2.25 ± 1.07) was preserved at MOXIe Extension Week 72 in the delayed-start period (LS mean difference = -3.51 ± 1.45). Consistent with a persistent treatment effect on disease, the upper limit of the 90% CI for the difference estimate was less than zero (-0.615), meeting the threshold for demonstrating significant evidence of non-inferiority.
Delayed-Start Analysis Primary Endpoint (Non-Inferiority Test)1
| | | | | |
| Placebo-Controlled Week 48 (Δ1) | Delayed-Start Week Ex. 72 (Δ2) | |
Difference (LS Mean ± SE) | -2.25 ± 1.07 p=0.037 | -3.51 ± 1.45 p=0.016 | |
Estimate = Δ2 – 0.5 × Δ1 | -2.39 ± 1.38 | | |
Upper Limit of 1-sided 90% CI for Estimate | -0.615 | | |
1Non-Inferiority test performed using a MMRM analysis with a Toeplitz covariance structure. | |
The graphical representation of changes from baseline in mFARS for omaveloxolone and placebo groups shows the separation at the end of the placebo-controlled period is maintained in the open-label period at Extension Week 72 and beyond.
20
Change from Baseline in mFARS (Patients without Pes Cavus)
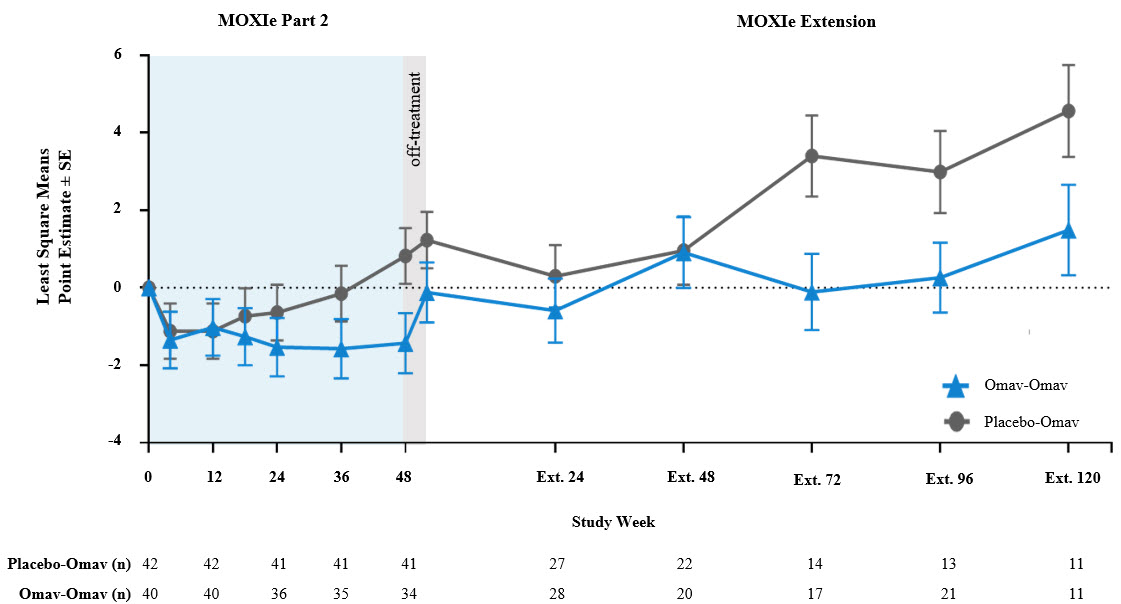
Many of the visits at Week 48 and Week 72 of the MOXIe Extension were scheduled during the initial peak of COVID-19 cases during Spring to Fall 2020. The mFARS assessment must be conducted in the clinic, and many in-clinic visits did not occur due to COVID-19 related travel restrictions and site closures during this period. Apart from the data at MOXIe Extension Week 48, parallel trajectories were seen in LS Mean mFARS change from baseline between the placebo-to-omaveloxolone group and the omaveloxolone-to-omaveloxolone group in MOXIe Extension.
A longitudinal analysis was also performed to calculate annualized slopes incorporating all available data from the MOXIe Extension, which showed similar mean slopes in mFARS for the placebo-to-omaveloxolone group (0.45 ± 0.38 points per year) when compared to the omaveloxolone-to-omaveloxolone group (0.27 ± 0.59 points per year) with no significant difference between slopes (difference = -0.18 ± 0.67; p=0.79). In MOXIe Extension, omaveloxolone-treated patients have been progressing at a rate that is >75% less than the approximately two points per year that patients progressed in a recent large natural history study (Patel, 2016).
Results from the Delayed-Start Analysis indicate a persistent omaveloxolone treatment effect on the disease course of FA. Patients who received omaveloxolone during the double-blind MOXIe Part 2 had a benefit that could not be achieved by patients initially randomized to placebo who began omaveloxolone one year later in MOXIe Extension. Notably, patients previously randomized to omaveloxolone in MOXIe Part 2 continued to show mean mFARS values that were similar to their original baseline after over three years of treatment.
No new safety signals have been identified in the MOXIe Extension trial.
21
Regulatory Interactions
In the third quarter of 2021, we completed our pre-NDA meeting with the FDA. The purpose of the pre-NDA meeting was to discuss the content of Reata’s planned NDA submission including the nonclinical data and CMC packages, data standard plan, and the overall content plan. In the meeting, we stated that we believed that the MOXIe data, along with the Delayed-Start Analysis, would provide sufficient clinical data to support a full approval. The FDA stated that the proposed primary and supportive efficacy data appear reasonable, though the Delayed-Start Analysis was viewed as exploratory. The FDA noted that the ability of the data to support full approval, and the adequacy of the data and the determination of which data may be supportive of efficacy, would be a matter of review. In response to our other questions about the contents of the NDA, the FDA exercised its discretion based on the seriousness of the indication and unmet medical need, subject to review, to permit us to submit the results of certain clinical pharmacology and nonclinical studies after approval. The additional studies include a thorough QT study with omaveloxolone, nonclinical metabolite toxicity studies, and six-month and two-year nonclinical carcinogenicity studies. The need for a drug-drug interaction study with a moderate CYP3A inducer will be established upon review of the adequacy of our submitted physiological based pharmacokinetic (PK) model.
On November 18, 2021, the FDA granted omaveloxolone Fast Track Designation for the treatment of FA, providing eligibility for FDA programs such as Priority Review and rolling submission of the NDA, if relevant criteria are met. The FDA granted our request for a rolling submission, and in March 2022, we completed submission of the NDA. This NDA is supported by the efficacy and safety data from the MOXIe Part 2 trial and additional supporting data from the MOXIe Part 1 and MOXIe Extension trials.
We are continuing to complete the regulatory procedures and submissions required prior to filing a MAA in Europe for approval of omaveloxolone for the treatment of patients with FA. We have secured agreement on our Pediatric Investigation Plan with the Pediatric Committee, and we also received EMA Follow-Up Protocol Assistance feedback regarding our nonclinical and CMC programs. The EMA feedback indicated that there were no identified impediments to our planned MAA submission and included agreement that certain nonclinical studies, including 2- year carcinogenicity study data, may be submitted after approval. We plan to submit an MAA to the European Medicines Agency for omaveloxolone in the fourth quarter of 2022.
Omaveloxolone in Other Neurological Indications
Omaveloxolone is a promising platform molecule. Because mitochondrial dysfunction is a key feature of many neurological and neuromuscular diseases, we believe omaveloxolone may be broadly applicable to treat such diseases by activating Nrf2 to normalize and improve mitochondrial function and ATP production.
Based on our understanding of the pathophysiology of neurological diseases characterized by mitochondrial dysfunction, inflammation, and oxidative stress, we believe omaveloxolone may be applicable to diseases such as progressive supranuclear palsy, Parkinson’s disease, frontotemporal dementia, Huntington’s disease, amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and epilepsy. Consistent with this, we have observed promising activity of omaveloxolone and our other Nrf2 activators in preclinical models of many of these diseases.
Our Nrf2 activators reduced seizure frequency in refractory, progressive epilepsy models and restored mitochondrial function in patient biopsy samples and preclinical models of FA, ALS, familial and sporadic Parkinson’s disease, and frontotemporal dementia. In clinical trials, improvements in neuromuscular function have been observed in FA patients treated with omaveloxolone as assessed by mFARS, and improvements in mitochondrial function, as measured by reductions in blood lactate and heart rate, have been observed in patients with primary mitochondrial disease. Accordingly, we believe that omaveloxolone has the potential to treat a number of neurological and neuromuscular diseases that currently have few or no effective therapies, and we plan to pursue the development of omaveloxolone and our other Nrf2 activators for one or more of these diseases.
RTA 901 in Neurological Diseases
RTA 901 is the lead product candidate from our Hsp90 modulator program, which includes highly potent and selective C-terminal modulators of Hsp90. We have observed favorable activity of RTA 901 in a range of preclinical models of neurological disease, including models of diabetic neuropathy, neuroinflammation, and neuropathic pain.
22
Historically, other companies have explored N-terminal Hsp90 inhibitors for cancer therapeutics; however, this approach has been associated with multiple AEs including peripheral neuropathy and ocular toxicity. Binding at the C-terminus of Hsp90 leads to increased transcription of Hsp70, a cytoprotective and molecular chaperone gene, which facilitates cell survival in response to stress without the deleterious activities of N-terminal inhibition.
In preclinical rodent disease models, we observed that RTA 901 administered orally once-daily rescued existing nerve function, restored thermal and mechanical sensitivity, and improved nerve conductance velocity and mitochondrial function. These effects are dose-dependent, reversible, and Hsp70-dependent.
We completed a Phase 1 SAD/MAD trial of oral, once-daily RTA 901 in healthy adult volunteers to evaluate the safety, tolerability, and PK profile. The PK was approximately dose-proportional up to the highest doses evaluated with a half-life ranging from two to nine hours. Human exposures easily exceeded the exposures necessary for efficacy in multiple animal models. No safety or tolerability concerns were reported. In the first quarter of 2022, we initiated additional Phase 1 clinical pharmacology studies of RTA 901, including a drug-drug interaction study. Following completion of these Phase 1 studies, we plan to initiate a randomized, double-blind placebo-controlled Phase 2 trial of RTA 901 in diabetic patients with peripheral neuropathic pain in the fourth quarter of 2022. We are the exclusive licensee of RTA 901 and have worldwide commercial rights.
Programs in Chronic Kidney Disease
We and Kyowa Kirin, our strategic collaborator in CKD in Japan, are developing bardoxolone for the treatment of CKD in multiple indications, including CKD caused by Alport syndrome, ADPKD, and type 1 and 2 diabetic CKD. CKD is characterized by a progressive worsening in the rate at which the kidney filters waste products from the blood, called the glomerular filtration rate (GFR). eGFR is an estimate of GFR that nephrologists use to track the decline in kidney function and progression of CKD. When GFR gets too low, patients develop end-stage kidney disease (ESKD) and require dialysis or a kidney transplant to survive.
We received a CRL from the FDA in February 2022 with respect to its review of our NDA for bardoxolone in the treatment of patients with CKD caused by Alport syndrome. We will continue to work with the FDA to confirm our next steps on our Alport syndrome program. Kyowa Kirin is currently conducting its registrational AYAME trial of bardoxolone in diabetic (types 1 and 2) CKD in Japan.
Bardoxolone in Patients with CKD Caused by Alport Syndrome
Alport syndrome is a rare, genetic form of CKD caused by mutations in the genes encoding type IV collagen, which is a major structural component of the glomerular basement membrane in the kidney. The kidneys of patients with Alport syndrome progressively lose the capacity to filter waste products out of the blood, which can lead to ESKD and the need for chronic dialysis treatment or a kidney transplant. Alport syndrome affects both children and adults and can manifest as early as the first decade of life and causes average annual declines in eGFR of approximately four to five mL/min/1.73 m2. In patients with the most severe forms of the disease, approximately 50% progress to dialysis by age 25, 90% by age 40, and nearly 100% by age 60. There are currently no approved therapies to treat CKD caused by Alport syndrome.
The Alport Syndrome Foundation has estimated that Alport syndrome affects approximately 30,000 to 60,000 people in the United States. According to data provided by IQVIA in 2020, there are approximately 14,000 projected patients diagnosed with Alport syndrome in all stages of CKD in the United States. However, recent literature suggests that a large number of patients with Alport syndrome are either undiagnosed or misdiagnosed with other forms of CKD.
On November 9, 2020, we reported interim results from the long-term extension EAGLE trial. EAGLE is an international, multi-center, open-label, extended access trial evaluating the longer-term safety and tolerability of bardoxolone in patients with CKD caused by Alport syndrome who participated in the CARDINAL trial or patients with ADPKD who participated in the FALCON trial. The change from baseline in eGFR was assessed for the 14 patients with Alport syndrome who were treated with bardoxolone for three years (two years in CARDINAL and one year in EAGLE), with four-week off-treatment periods occurring at Weeks 48 and 100. Bardoxolone treatment resulted in a mean on-treatment increase from baseline in eGFR of 11.5 mL/min/1.73 m2 at Year 1, 13.3 mL/min/1.73 m2 at Year 2, and 11.0 mL/min/1.73 m2 at Year 3.
23
In February 2022, we provided the FDA with an update on results from patients with CKD caused by Alport syndrome in the ongoing EAGLE trial. Mean increases in eGFR were observed at Week 12, Week 24, and Week 48 relative to Day 0 (before treatment) in EAGLE in patients who previously received placebo and initiated treatment with bardoxolone in EAGLE. Patients who previously received bardoxolone for two years in CARDINAL experienced similar mean increases in eGFR at all timepoints. For the 37 patients randomized to bardoxolone in CARDINAL who completed 48 weeks in EAGLE, bardoxolone treatment resulted in a mean on-treatment change from baseline in eGFR (relative to original CARDINAL baseline) of 9.2 mL/min/1.73 m2 at Year 1, 7.8 mL/min/1.73 m2 at Year 2, and 6.7 mL/min/1.73 m2 at Year 3.
A subset of patients with Alport syndrome (n=18) completed 96 weeks of treatment in EAGLE, which amounts to approximately four years of total treatment, and had a mean ± standard error change in eGFR from CARDINAL baseline of 5.5 ± 3.5 mL/min/1.73 m2. This sustained improvement of kidney function is notable when compared to the CARDINAL trial population’s expected yearly eGFR decline of 5.1 mL/min/1.73 m2, which was calculated based on five-year historical eGFR data collected before patients entered the trial. No new safety findings have been identified in the EAGLE trial.
On February 25, 2022, we received a CRL from the FDA with respect to its review of our NDA for bardoxolone in the treatment of patients with CKD caused by Alport syndrome. The CRL indicated that the FDA cannot approve the NDA in its present form. Based on its review, the FDA concluded that it does not believe the submitted data demonstrates that bardoxolone is effective in slowing the loss of kidney function in patients with Alport syndrome and reducing the risk of progression to kidney failure and has requested additional data to support the efficacy and safety of bardoxolone. Their conclusion was based on efficacy and safety concerns primarily set forth in the FDA’s briefing book and discussed at the Cardiovascular and Renal Drugs Advisory Committee meeting held on December 8, 2021.
The FDA stated that the issues could be resolved by providing evidence of effectiveness that includes evidence from an adequate and well-controlled study showing a clinically relevant effect on the rate of loss of kidney function in patients with Alport syndrome or, alternatively, an effect on a clinical outcome (i.e., an endpoint that captures how patients with Alport syndrome feel, function, or survive). In addition, the FDA stated that we would need to address whether bardoxolone has a clinically relevant effect on the QT interval and show that the demonstrated clinical benefits of bardoxolone outweigh its risks. The FDA welcomed continued discussion on the details of a path forward. We plan to work closely with the FDA to achieve our goal of bringing this important medicine to patients in the United States.
In October 2021, we submitted an MAA to the EMA for bardoxolone in the treatment of patients with CKD caused by Alport syndrome. In the first quarter of 2022, we received the 120-day list of questions from the EMA. We are in the process of preparing our responses. We requested a 90-day extension for our responses which was granted by the EMA. The timeline for the EMA’s review cycle was therefore extended.
Bardoxolone in Patients with Autosomal Dominant Polycystic Kidney Disease
ADPKD is a rare and serious hereditary form of CKD caused by a genetic defect in PKD1 or PKD2 genes leading to the formation of fluid-filled cysts in the kidneys and other organs. Cyst growth can cause the kidneys to expand up to five to seven times their normal volume, leading to pain and progressive loss of kidney function. Inflammation appears to play a role in cyst growth and is associated with disease progression in ADPKD.
ADPKD affects both men and women of all racial and ethnic groups and is the leading inheritable cause of kidney failure with an estimated diagnosed population of 140,000 patients and an estimated prevalent population of 400,000 patients in the United States. Despite current standard-of-care treatment, an estimated 50% of ADPKD patients progress to ESKD and require dialysis or a kidney transplant by 60 years of age. The only therapy currently approved for ADPKD is JYNARQUE® (tolvaptan), developed by Otsuka Pharmaceuticals Co., Ltd., which was approved in the United States in 2018 to slow kidney function decline in adults at risk of rapidly progressing ADPKD.
We are currently enrolling patients in FALCON, an international, multi-center, randomized, double-blind, placebo-controlled trial studying the safety and efficacy of bardoxolone in patients with ADPKD randomized one-to-one to active drug or placebo. FALCON is enrolling patients in a broad range of ages, with an eGFR between 30 and 90 mL/min/1.73 m2.
24
In the first quarter of 2022, we finalized a protocol amendment to FALCON, and we initiated submission of the amendment to the FDA and other relevant health authorities. The major protocol amendment changes include an increase in the sample size from 550 to 850 patients, addition of adolescent (12 to 17 years) patients with ADPKD for a total age range of 12 to 70 years, removal of the off-treatment period (Week 48 – Week 52) during Year 1, change of the primary endpoint of off-treatment eGFR change from baseline at Week 52 (or four weeks after drug discontinuation in Year 1) to eGFR change from baseline at Week 108 (eight weeks after planned drug discontinuation at Week 100). The protocol amendment also includes addition of an exploratory endpoint of eGFR change from baseline at Week 112 (12 weeks after planned drug discontinuation at Week 100), and addition of a sub study with ambulatory blood pressure monitoring.
As agreed with the FDA prior to submission of the amendment, we recently had a Type A meeting to discuss key changes made to the FALCON protocol. Based on the discussion during the meeting and the meeting minutes, the Division stated that the proposed primary endpoint of eGFR change from baseline at Week 108 (8 weeks after planned drug discontinuation at Week 100) was reasonable since the available data suggest that bardoxolone’s acute pharmacodynamic effect on eGFR should be largely resolved. The Division provided guidance on handling of data from patients who completed Year 2 of the study before the protocol amendment and so did not have an eGFR assessment at Week 108. This included direction on imputing missing data for these patients in the primary analysis. The Division stated that, in addition to the primary endpoint, it will be important to demonstrate that the treatment effect accrues over time to support a claim that bardoxolone slows the loss of kidney function in patients with ADPKD and provided guidance on the statistical methodologies for the exploratory eGFR slope analyses. The FDA also confirmed that if FALCON is positive, it could support registration of bardoxolone in ADPKD.
Pursuant to the protocol amendment, patients will be treated with bardoxolone or placebo for 100 weeks followed by a twelve-week withdrawal period. The trial will remain blinded until study completion. All patients will be asked to return at Week 108 independent of the time of study drug discontinuation. The secondary endpoint is the eGFR change from baseline at Week 100. More than 550 patients are currently enrolled in the trial.
AYAME Trial in Diabetic CKD Conducted by Kyowa Kirin
Upon completion of Kyowa Kirin's Phase 2 TSUBAKI trial of bardoxolone in patients with Stage 3 and 4 diabetic CKD in Japan, and after discussions with the Pharmaceuticals and Medical Devices Agency, Kyowa Kirin initiated a Phase 3 outcomes trial called AYAME in patients with Stage 3 or 4 diabetic CKD in Japan. The primary endpoint is time to onset of a ≥ 30% decrease in eGFR from baseline or ESKD. The secondary endpoints are time to onset of a ≥ 40% decrease in eGFR from baseline or ESKD, time to onset of a ≥ 53% decrease in eGFR from baseline or ESKD, time to onset of ESKD, and change in eGFR from baseline at each evaluation time point. Kyowa Kirin completed patient enrollment in AYAME in June 2019 and expects the last patient visit to occur in the second half of 2022.
U.S. Commercial Readiness
With the NDA submission of omaveloxolone complete, we are advancing commercial launch preparations in the United States. We are in the process of building the commercial infrastructure necessary to effectively support the commercialization of omaveloxolone for the treatment of FA, if and when we receive regulatory approval. Commercial launch preparation for omaveloxolone in FA will continue to advance in step with the regulatory progress.
Our ability to launch omaveloxolone is dependent on the successful filing and defense of an NDA and approval by the FDA. We have hired commercial leadership and intend to build the teams, infrastructure, systems, and processes necessary for the launch of omaveloxolone. This will include sales, marketing, market access, patient support, and distribution. Additionally, we are expanding quality and compliance functions to support commercialization. A trade name for omaveloxolone has been selected.
25
One challenge unique to rare-disease commercialization is patient identification due to the very small and sometimes heterogeneous disease populations. Our management team is experienced in maximizing patient identification for both clinical development and commercialization purposes in rare diseases. Commercial infrastructure for orphan products typically consists of a targeted, specialty sales force that calls on a limited and focused group of physicians and personnel involved in sales management, internal sales support, marketing, patient access, and distribution.
Ex-U.S. Commercial Readiness
Our ability to launch omaveloxolone is dependent on the successful filing and defense of an MAA and approval by EMA or other regulatory agencies. Outside of the United States, where appropriate and depending on the terms of our contractual arrangements, we plan, either alone, or with new collaboration partners, to commercialize our products. Our strategic collaborator Kyowa Kirin has all rights to commercialize bardoxolone in its territories. We are refining our strategy and market assessments with respect to potential launches in the EU, and we continue to evaluate market opportunities for our products in other global markets. Commercial launch preparation for omaveloxolone in FA outside of the United States will advance in step with the regulatory progress.
Corporate Overview
To date, we have focused most of our efforts and resources on developing our product candidates and conducting preclinical studies and clinical trials. We have historically financed our operations primarily through revenue generated from our collaborations with AbbVie and Kyowa Kirin, from sales of our securities, secured loans, and a strategic financing from BXLS. We have not received any payments or revenue from collaborations other than nonrefundable upfront, milestone, and cost sharing payments from our collaborations with AbbVie and Kyowa Kirin, from the Development Agreement with BXLS, and from reimbursements of expenses under the terms of our agreement with Kyowa Kirin. We have incurred losses in each year since our inception, other than in 2014. As of March 31, 2022, we had $532.0 million of cash and cash equivalents and an accumulated deficit of $1,329.5 million. We continue to incur significant research and development and other expenses related to our ongoing operations. Despite contractual product development commitments and the potential to receive future payments from Kyowa Kirin, we anticipate that we will continue to incur losses for the foreseeable future, and we anticipate that our losses will increase as we continue our development of, seek regulatory approval for, and potentially commercialize our product candidates. If we do not successfully develop and obtain regulatory approval of our existing product candidates or any future product candidates and effectively manufacture, market, and sell any products that are approved, we may never generate revenue from product sales. Furthermore, even if we do generate revenue from product sales, we may never again achieve or sustain profitability on a quarterly or annual basis. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ equity and working capital. Our failure to become and remain profitable could depress the market price of our Class A common stock and could impair our ability to raise capital, expand our business, diversify our product offerings, or continue our operations.
The probability of success for each of our product candidates and clinical programs and our ability to generate product revenue and become profitable depend upon a variety of factors, including the quality of the product candidate, clinical results, investment in the program, competition, manufacturing capability, commercial viability, and our collaborators’ ability to successfully execute our development and commercialization plans. We will also require additional capital through equity, debt, or royalty financings or collaboration arrangements in order to fund our operations and execute on our business plans, and there is no assurance that such financing or arrangements will be available to us on commercially reasonable terms or at all. For a description of the numerous risks and uncertainties associated with product development and raising additional capital, see “Risk Factors” included in our Annual Report on Form 10-K for the year ended December 31, 2021.
26
Financial Operations Overview
Revenue
Our revenue to date has been generated primarily from licensing fees received under our collaborative license agreements and reimbursements for expenses. We currently have no approved products and have not generated any revenue from the sale of products to date. In the future, we may generate revenue from product sales, royalties on product sales, reimbursements for collaboration services under our current collaboration agreements, or license fees, milestones, or upfront payments if we enter into any new collaborations or license agreements. We expect that our future revenue will fluctuate from quarter to quarter for many reasons, including the uncertain timing and amount of any such payments and sales.
Our license and milestone revenue has been generated primarily from the Kyowa Kirin Agreement, the AbbVie License Agreement, and the Collaboration Agreement and consists of upfront payments and milestone payments. License revenue recorded with respect to the Kyowa Kirin Agreement, the AbbVie License Agreement, and the Collaboration Agreement consists solely of the recognition of deferred revenue. Under our revenue recognition policy, collaboration revenue associated with upfront, non-refundable license payments received under our license and collaboration agreements are deferred and recognized ratably over the expected term of the performance obligations under each agreement. Under the Reacquisition Agreement, we no longer have performance obligations under the AbbVie License Agreement and the Collaboration Agreement. Under the Kyowa Kirin Agreement, we only expect to recognize the deferred revenue through June 2022.
Research and Development Expenses
The largest component of our total operating expenses has historically been our investment in research and development activities, including the clinical development of our product candidates. From our inception through March 31, 2022, we have incurred a total of $1,129.7 million in research and development expense, a majority of which relates to the development of bardoxolone and omaveloxolone. We expect our research and development expense to continue to increase in the future as we advance our product candidates through clinical trials and expand our product candidate portfolio. The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming, and we consider the active management and development of our clinical pipeline to be crucial to our long-term success. The actual probability of success for each product candidate and preclinical program may be affected by a variety of factors, including the safety and efficacy data for product candidates, investment in the program, competition, manufacturing capability, and commercial viability.
Research and development expenses include:
•expenses incurred under agreements with clinical trial sites that conduct research and development activities on our behalf;
•expenses incurred under contract research agreements and other agreements with third parties;
•employee and consultant-related expenses, which include salaries, benefits, travel, and stock-based compensation;
•laboratory and vendor expenses related to the execution of preclinical and non-clinical studies and clinical trials;
•the cost of acquiring, developing, manufacturing, and distributing clinical trial materials;
•the cost of development, scale up, and process validation activities to support product registration; and
•facilities, depreciation, and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance, and other supply costs.
Research and development costs are expensed as incurred. Costs for certain development activities such as clinical trials are highly judgmental and are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors and our clinical sites.
27
We base our expense accruals related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and CROs that conduct and manage clinical trials on our behalf. The financial terms of these agreements vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing costs, we estimate the time period over which services will be performed and the level of effort to be expended in each period.
To date, we have not experienced material changes in our estimates of accrued research and development expenses after a reporting period. However, due to the nature of estimates, we cannot assure you that we will not make changes to our estimates in the future as we become aware of additional information about the status or conduct of our clinical trials and other research activities.
Currently, Kyowa Kirin has allowed us to conduct clinical studies of bardoxolone in certain rare forms of kidney diseases in Japan and has reimbursed us the majority of the costs for our CARDINAL study in Japan. Kyowa Kirin is the in-country caretaker in our FALCON study in Japan and we are reimbursing Kyowa Kirin for the costs of a certain number of patients in the study.
The following table summarizes our research and development expenses incurred during the three months ended March 31:
| | | | | | | | |
| | 2022 | | | 2021 | |
| | (in thousands) | |
Bardoxolone | | $ | 6,462 | | | $ | 11,386 | |
Omaveloxolone | | | 7,597 | | | | 2,473 | |
RTA 901 | | | 1,407 | | | | 706 | |
Other research and development expenses (1) | | | 24,338 | | | | 20,315 | |
Total research and development expenses | | $ | 39,804 | | | $ | 34,880 | |
(1) RTA 1701 expenses have been included in other research and development expenses due to development updates in the program.
The program-specific expenses summarized in the table above include costs that we directly allocate to our product candidates. Our other research and development expenses include salaries, benefits, stock-based compensation and preclinical, research, and discovery costs, which we do not allocate on a program-specific basis.
General and Administrative Expenses
General and administrative expenses consist primarily of employee-related expenses for executive, operational, finance, legal, compliance, and human resource functions. Other general and administrative expenses include personnel expense, facility-related costs, professional fees, accounting and legal services, depreciation expense, other external services, and expenses associated with obtaining and maintaining our intellectual property rights.
We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support our continued research and development and potential commercialization of our product candidates. We have also incurred, and anticipate incurring in the future, increased expenses associated with being a public company, including exchange listing and SEC requirements, director and officer insurance premiums, legal, audit and tax fees, compliance with the Sarbanes-Oxley Act, regulatory compliance programs, and investor relations costs. Additionally, if and when we believe the first regulatory approval of one of our product candidates appears likely, we anticipate an increase in payroll and related expenses as a result of our preparation for commercial operations, especially for the sales and marketing of our product candidates.
Other Income (Expense), Net
Other income (expense) includes interest and gains earned on our cash and cash equivalents, amortization of debt issuance costs, imputed interest on long term payables, foreign currency exchange gains and losses, and non-cash interest expense on liability related to the sale of future royalties.
28
Benefit from (Provision for) Taxes on Income
Provision for taxes on income consists of net loss, taxed at federal tax rates and adjusted for certain permanent differences. Realization of deferred tax assets is generally dependent upon future earnings by jurisdiction, of which the timing and amount are uncertain for the majority of our deferred tax assets, and valuation allowances are maintained against them. Changes in valuation allowances also affect the tax provision.
Results of Operations
Comparison of the Three Months Ended March 31, 2022 and 2021 (unaudited)
The following table sets forth our results of operations for the three months ended March 31:
| | | | | | | | | | | | | | | | |
| | 2022 | | | 2021 | | | Change $ | | | Change % | |
| | (in thousands, except for percentage data) | |
Collaboration revenue | | | | | | | | | | | | |
License and milestone | | $ | 893 | | | $ | 795 | | | $ | 98 | | | | 12 | |
Other revenue | | | 21 | | | | 149 | | | | (128 | ) | | | (86 | ) |
Total collaboration revenue | | | 914 | | | | 944 | | | | (30 | ) | | | (3 | ) |
Expenses | | | | | | | | | | | | |
Research and development | | | 39,804 | | | | 34,880 | | | | 4,924 | | | | 14 | |
General and administrative | | | 24,841 | | | | 20,704 | | | | 4,137 | | | | 20 | |
Depreciation | | | 308 | | | | 274 | | | | 34 | | | | 12 | |
Total expenses | | | 64,953 | | | | 55,858 | | | | 9,095 | | | | 16 | |
Other income (expense), net | | | (9,772 | ) | | | (12,556 | ) | | | 2,784 | | | | 22 | |
Loss before taxes on income | | | (73,811 | ) | | | (67,470 | ) | | | (6,341 | ) | | | (9 | ) |
Benefit from (provision for) taxes on income | | | (31 | ) | | | 15 | | | | (46 | ) | | ** | |
Net loss | | $ | (73,842 | ) | | $ | (67,455 | ) | | $ | (6,387 | ) | | | (9 | ) |
** Percentage not meaningful
Revenue
License and milestone revenue represented approximately 98% and 84% of total revenue for the three months ended March 31, 2022 and 2021, respectively, and consisted of the recognition of Kyowa Kirin deferred revenue. License and milestone revenue increased by 12% during the three months ended March 31, 2022, compared to the three months ended March 31, 2021, primarily due to the achievement of a regulatory milestone in July 2021, variable consideration previously considered constrained, under the Kyowa Kirin Agreement.
Other revenue was immaterial for the three months ended March 31, 2022, and 2021.
Expenses
The following table summarizes our expenses, including as a percentage of total expenses, for the three months ended March 31:
| | | | | | | | | | | | | | | | |
| | 2022 | | | % of Total
Expenses | | | 2021 | | | % of Total
Expenses | |
| | (in thousands, except for percentage data) | |
Research and development | | $ | 39,804 | | | | 61 | % | | $ | 34,880 | | | | 62 | % |
General and administrative | | | 24,841 | | | | 38 | % | | | 20,704 | | | | 37 | % |
Depreciation | | | 308 | | | | 1 | % | | | 274 | | | | 1 | % |
Total expenses | | $ | 64,953 | | | | | | $ | 55,858 | | | | |
29
Research and Development Expenses
Research and development expenses increased by $4.9 million, or 14%, for the three months ended March 31, 2022, compared to the three months ended March 31, 2021. The increase was primarily due to increased personnel and personnel-related costs to support the product development activities.
Research and development expenses, as a percentage of total expenses, was 61% and 62% for the three months ended March 31, 2022 and 2021, respectively. The decrease of 1% was due to the proportionately larger increase in general and administrative expenses, compared to research and development expenses.
General and Administrative Expenses
General and administrative expenses increased by $4.1 million, or 20%, for the three months ended March 31, 2022, compared to the three months ended March 31, 2021. The increase was primarily due to rent expense related to the new headquarters building lease that commenced in December 2021.
General and administrative expenses, as a percentage of total expenses, was 38% and 37%, for the three months ended March 31, 2022 and 2021, respectively. The increase of 1% was due to the proportionately larger increase in general and administrative expenses, compared to research and development expenses.
Other Income (Expense), Net
Other income (expense), net decreased by $2.8 million for the three months ended March 31, 2022, compared to the three months ended March 31, 2021. The decrease was primarily due to a decrease in effective interest rate in non-cash interest expense on liability related to the sale of future royalties.
We periodically reassess the expected royalty payments under the Development Agreement, and to the extent such payment is greater or less than the initial estimate, we adjust the amortization. Based on our review in the current first quarter of 2022, we lowered our previous estimate of future sales for which royalties will be paid. Accordingly, we have prospectively adjusted and recognized lower non-cash interest expense during the quarter ended March 31, 2022.
Benefit from (Provision for) Taxes on Income
Benefit from (Provision for) taxes on income was immaterial for the three months ended March 31, 2022 and 2021.
30
Liquidity and Capital Resources
Since our inception, we have funded our operations primarily through collaboration and license agreements, the sale of preferred and common stock, the sale of royalty interests, and secured loans. Through March 31, 2022, we have raised gross cash proceeds of $476.6 million through the sale of convertible preferred stock and $785.0 million from payments under license and collaboration agreements. We also obtained $1,222.1 million in net proceeds from our initial public offering, follow-on offerings, and the sale of our Class A common stock under the Purchase Agreement, and $299.0 million in net proceeds from the sale of future royalties under the Development Agreement. We have not generated any revenue from the sale of any products. As of March 31, 2022, we had available cash and cash equivalents of approximately $532.0 million. Our cash and cash equivalents are invested in accordance with our investment policy, primarily with a view to liquidity and capital preservation.
Cash Flows
The following table sets forth the primary sources and uses of cash for each of the three months ended March 31:
| | | | | | | | |
| | 2022 | | | 2021 | |
| | (in thousands) | |
| | | |
Net cash (used in) provided by: | | | | | | |
Operating activities | | $ | (58,185 | ) | | $ | (45,011 | ) |
Investing activities | | | (288 | ) | | | (193 | ) |
Financing activities | | | 194 | | | | 4,678 | |
Net change in cash and cash equivalents | | $ | (58,279 | ) | | $ | (40,526 | ) |
Operating Activities
Net cash used in operating activities was $58.2 million for the three months ended March 31, 2022, consisting primarily of a net loss of $73.8 million adjusted for non-cash items including stock-based compensation expense of $15.4 million, non-cash interest expense on liability related to sale of future royalty of $9.9 million, and a net decrease in operating assets and liabilities of $10.0 million. The significant items in the change in operating assets that impacted our use of cash in operations include a decrease of $4.5 million in accounts payable due to timing of payments, and a decrease of $8.9 million in direct research and other current and long-term liabilities, primarily due to annual bonus payments.
Net cash used in operating activities was $45.0 million for the three months ended March 31, 2021, consisting primarily of a net loss of $67.5 million adjusted for non-cash items including stock-based compensation expense of $14.7 million, non-cash interest expense on liability related to the sale of future royalties of $10.9 million, depreciation, amortization of issuance costs, imputed interest expense of $2.0 million, and a net increase in operating assets and liabilities of $5.1 million. The significant items in the change in operating assets that impacted our use of cash in operations include a decrease of $9.3 million in accrued direct research and other current and long-term liabilities primarily due to the change in timing of bonus payments from December to March of the following year, which began with the December 2020 payments being delayed to March 2021, and to the termination of PAH-related studies and the completion of CARDINAL and MOXIe clinical trials, offset by an increase in accounts payable of $3.6 million due to timing of payments.
Investing Activities
Net cash used in investing activities was $0.3 million and $0.2 million for the three months ended March 31, 2022 and 2021, respectively, consisting of purchases of property and equipment.
Financing Activities
Net cash provided by financing activities was $0.2 million and $4.7 million for the three months ended March 31, 2022 and 2021, respectively, consisting of stock options exercises.
31
Operating Capital Requirements
To date, we have not generated any revenue from product sales. We do not know when or whether we will generate any revenue from product sales. We do not expect to generate significant revenue from product sales unless and until we obtain regulatory approval of and commercialize one or more of our current or future product candidates. We anticipate that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our product candidates, and begin to commercialize any approved products. We are subject to all the risks related to the development and commercialization of novel therapeutics, including those described under the heading “Risk Factors” included in our most recent Annual Report on Form 10-K, and we may encounter unforeseen expenses, difficulties, complications, delays, and other unknown factors that may adversely affect our business. We continue to incur additional costs associated with operating as a public company. We anticipate that we will need substantial additional funding in connection with our continuing operations.
In October 2019, we entered into the 2019 Lease Agreement, relating to a new headquarter building lease of approximately 327,400 square feet of office and laboratory space located in Plano, Texas.
•In December 2021, we obtained control of the building, and, accordingly, we recorded related right-of-use assets and the lease liabilities during the fourth quarter of 2021.
•We have paused the tenant improvement activities for the new headquarter building and are attempting to sublease the building. At this point, we will not spend the earlier-planned $50 million in capital expenditures. If at a future date we determine to move into the building, capital expenditures will need to be incurred based on our occupancy requirements at that time.
•The initial term of the lease is 16 years, with up to ten years of extension at our option. The annual base rent payment, which will begin in June 2022, will be determined based on the project cost, subject to an initial annual cap of approximately $13.3 million. Beginning in the third lease year, the base rent will increase 1.95% per annum each year. In addition to the annual base rent, we will pay for taxes, insurance, utilities, operating expenses, assessments under private covenants, maintenance and repairs, certain capital repairs and replacements, and building management fees.
In July 2021, Kyowa Kirin announced the submission of an NDA in Japan for bardoxolone for improvement of renal function in patients with Alport syndrome. We earned a $5.0 million milestone related to this event that was received and began to be recognized in the third quarter of 2021.
In December 2020, we closed a follow-on underwritten public offering of 2,000,000 shares of our Class A common stock for gross proceeds of $281.7 million. Net proceeds to us from the offering were approximately $277.5 million, after deducting underwriting discounts and commissions and offering expenses.
In June 2020, we closed on the Development Agreement and Purchase Agreement, each dated June 10, 2020, under which certain BXLS entities paid us an aggregate of $350.0 million in exchange for future royalties on bardoxolone and an aggregate of 340,793 shares of our Class A common stock at $146.72 per share.
Our longer term liquidity requirements will require us to raise additional capital, such as through additional equity, debt, or royalty financings or collaboration arrangements. Our future capital requirements will depend on many factors, including the receipt of milestones under our Kyowa Kirin Agreement and the timing of our expenditures related to clinical trials. We believe our existing cash and cash equivalents will be sufficient to enable us to fund our operations through the fourth quarter of 2024. However, we anticipate opportunistically raising additional capital before that time through equity offerings, collaboration or license agreements, additional debt financings, or royalty financings in order to maintain adequate capital reserves. In addition, we may choose to raise additional capital at any time for the further development of our existing product candidates and may also need to raise additional funds sooner to pursue other development activities related to additional product candidates. Decisions about the timing or nature of any financing will be based on, among other things, our perception of our liquidity and of the market opportunity to raise equity, debt, or royalty financing. Additional securities may include common stock, preferred stock, or debt securities. We may explore strategic collaborations or license arrangements for any of our product candidates. If we do explore any arrangements, there can be no assurance that any agreement will be reached, and we may determine to cease exploring a potential transaction for any or all of the assets at any time. If an agreement is reached, there can be no assurance that any such transaction would provide us with a material amount of additional capital resources.
32
Until we can generate a sufficient amount of revenue from our product candidates, if ever, we expect to finance future cash needs through public or private equity or debt offerings, loans, royalty financings, and collaboration or license transactions. Recent and continued volatility in global financial markets may reduce our ability to access capital, which could negatively affect our liquidity. Additional capital may not be available on reasonable terms, if at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back, or discontinue the development or commercialization of one or more of our product candidates. If we raise additional funds through the issuance of additional equity or debt securities, it could result in dilution to our existing stockholders or increased fixed payment obligations, and any such securities may have rights senior to those of our common stock. If we incur indebtedness or obtain royalty financing, we could become subject to covenants that would restrict our operations and potentially impair our competitiveness, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell, or license intellectual property rights, and other operating restrictions that could adversely affect our ability to conduct our business, and any such debt or royalty financing could be secured by some or all of our assets. Any of these events could significantly harm our business, financial condition, and prospects. For a description of the numerous risks and uncertainties associated with product development and raising additional capital, see “Risk Factors” included in our Annual Report on Form 10-K for the year ended December 31, 2021.
Our forecast of the period through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. Our future funding requirements, both near- and long-term, will depend on many factors, including, but not limited to:
•the scope, rate of progress, results, and cost of our clinical trials, preclinical testing, and other activities related to the development of our product candidates;
•the number and characteristics of product candidates that we pursue;
•the costs of development efforts for our product candidates that are not subject to reimbursement from our collaborators;
•the costs necessary to obtain regulatory approvals, if any, for our product candidates in the United States and other jurisdictions, and the costs of post-marketing studies that could be required by regulatory authorities in jurisdictions where approval is obtained;
•the continuation of our existing collaboration with Kyowa Kirin and entry into new collaborations and the receipt of any collaboration payments;
•the time and unreimbursed costs necessary to commercialize products in territories in which our product candidates are approved for sale;
•the revenue from any future sales of our products or for which we are entitled to a profit share, royalties, and milestones;
•the level of reimbursement or third-party payor pricing available to our products;
•the costs of obtaining third-party commercial supplies of our products, if any, manufactured in accordance with regulatory requirements;
•the costs associated with any potential loss or corruption of our information or data in a cyberattack on our computer systems or those of our suppliers, vendors, or collaborators who store or transmit our data;
•the costs associated with being a public company;
•any additional costs we incur, or delays in clinical trials we experience, associated with the COVID-19 pandemic; and
•the costs we incur in the filing, prosecution, maintenance, and defense of our patent portfolio and other intellectual property rights.
If we cannot expand our operations or otherwise capitalize on our business opportunities because we lack sufficient capital, our business, financial condition, and results of operations could be materially adversely affected.
33
Contractual Obligations and Commitments
We have various contractual obligations and other commitments that require payments at certain specified periods. The following table summarizes our contractual obligations and commitments as of March 31, 2022 (unaudited):
| | | | | | | | | | | | | | | | | | | | |
| | Payments due by period | |
| | Less than
1 year | | | 1 to 3
years | | | 4 to 5
years | | | 6 years and beyond | | | Total | |
| | (unaudited, in thousands) | |
Operating lease obligations(1) | | $ | 12,633 | | | $ | 17,448 | | | $ | 27,831 | | | $ | 179,687 | | | $ | 237,599 | |
Total contractual obligations | | $ | 12,633 | | | $ | 17,448 | | | $ | 27,831 | | | $ | 179,687 | | | $ | 237,599 | |
(1) Above table assumes one year rent abatement is applied beginning in June 2023 following FDA approval of omaveloxolone.
The terms of the Development Agreement require us to pay potential future royalty payments based on product development success. The above table excludes such obligations as the amount and timing of such obligations are unknown or uncertain, which are further described in Note 4, Liability Related to Sale of Future Royalties, to Consolidated Financial Statements contained in this Quarterly Report on Form 10-Q.
Clinical Trials
As of March 31, 2022, we have several on-going clinical trials in various stages. Under agreements with various CROs and clinical trial sites, we incur expenses related to clinical trials of our product candidates and potential other clinical candidates. The timing and amounts of these disbursements are contingent upon the achievement of certain milestones, patient enrollment, and services rendered or as expenses are incurred by the CROs or clinical trial sites. Therefore, we cannot estimate the potential timing and amount of these payments, and they have been excluded from the table above.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, and expenses and the disclosure of contingent assets and liabilities in our financial statements. On an ongoing basis, we evaluate our estimates and judgments, including those related to revenue recognition, accrued research and development expenses, income taxes, and stock-based compensation. We base our estimates on historical experience, known trends and events, and various other factors that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are described in Note 2 of Part I, Item 1 of this Quarterly Report on Form 10-Q and in Part I, Item 7, “Critical Accounting Policies and Significant Judgments and Estimates” in our Annual Report on Form 10-K. There have been no changes to our critical accounting policies and estimates since our Annual Report on Form 10-K for the year ended December 31, 2021.
Off-Balance Sheet Arrangements
Since our inception, we have not had any relationships with unconsolidated organizations or financial partnerships, such as structured finance or special purpose entities, that would have been established for the purpose of facilitating off-balance sheet arrangements, and we have not engaged in any other off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Recent Accounting Pronouncements
For a discussion of recent accounting pronouncements, please see Note 2, Summary of Significant Accounting Policies, of Notes to Consolidated Financial Statements contained in this Quarterly Report on Form 10-Q.
34
Item 3. Quantitative and Qualitative Disclosures About Market Risk.
We are exposed to market risks in the ordinary course of our business. These market risks are principally limited to interest rate fluctuations. We had cash and cash equivalents of $532.0 million at March 31, 2022, consisting primarily of funds in operating cash accounts. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes. Due to the short-term nature of our investment portfolio, we do not believe an immediate increase of 100 basis points in interest rates would have a material effect on the fair market value of our portfolio, and accordingly we do not expect a sudden change in market interest rates to affect materially our operating results or cash flows.
We contract with research, development, and manufacturing organizations and investigational sites globally. Generally, these contracts are denominated in United States dollars. However, we may be subject to fluctuations in foreign currency rates in connection with agreements not denominated in United States dollars. We do not hedge our foreign currency exchange rate risk.
Item 4. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of March 31, 2022. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934 (Exchange Act), means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized, and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of March 31, 2022, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in Internal Control Over Financial Reporting
There have been no changes in our internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) promulgated under the Exchange Act, during the three months ended March 31, 2022, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
35
PART II — OTHER INFORMATION
Item 1. Legal Proceedings.
For a discussion of material pending legal proceedings, please read Note 10, Commitments and Contingencies – Litigation, to our condensed consolidated financial statements included in Part I, Item I, “Financial Statements (Unaudited),” of this Quarterly Report on Form 10-Q, which is incorporated into this item by reference.
Item 1A. Risk Factors.
In addition to other information set forth in this Quarterly Report on Form 10-Q, you should carefully consider the risk factors and other cautionary statements described under the heading “Risk Factors” included in our Annual Report on Form 10-K for the year ended December 31, 2021, which could materially affect our businesses, financial condition, or future results. Additional risks and uncertainties currently unknown to us, or that we currently deem to be immaterial, also may materially adversely affect our business, financial condition, or future results. There have been no material changes in our risk factors from those described in the Annual Report on Form 10-K for the year ended December 31, 2021.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
None.
Item 3. Defaults Upon Senior Securities.
None.
Item 4. Mine Safety Disclosures.
None.
Item 5. Other Information.
None.
36
Item 6. Exhibits.
| | |
Exhibit Number | | Description |
| | |
3.1 | | Thirteenth Amended and Restated Certificate of Incorporation, dated May 11, 2016 (incorporated by reference to Exhibit 3.7 to the Company’s Form S-1 (File No. 333-208843), filed with the SEC on May 16, 2016). |
| | |
3.2 | | Second Amended and Restated Bylaws, dated as of December 7, 2016 (incorporated by reference to Exhibit 3.1 to the Company’s Form 8-K (File No. 001-37785), filed with the SEC on December 7, 2016). |
| | |
10.1*# | | Amendment No. 1 to Exclusive License Agreement, dated as of April 5, 2022, by and between the KU Center for Technology Commercialization, Inc. and the Registrant, dated as of, as amended. |
| | |
10.2*# | | Seventh Supplement to Exclusive License and Supply Agreement, dated as of February 28, 2022 between Reata Pharmaceuticals, Inc. and Kyowa Hakko Kirin Co., Ltd. |
| | |
31.1* | | Certification of Principal Executive Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| | |
31.2* | | Certification of Principal Financial Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| | |
32.1** | | Certification of Principal Executive Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| | |
32.2** | | Certification of Principal Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| | |
101.INS* | | Inline XBRL Instance Document - The cover page interactive data file does not appear in the interactive data file because its XBRL tags are embedded within the inline XBRL document |
| | |
101.SCH* | | Inline XBRL Taxonomy Extension Schema Document |
| | |
101.CAL* | | Inline XBRL Taxonomy Extension Calculation Linkbase Document |
| | |
101.DEF* | | Inline XBRL Taxonomy Extension Definition Linkbase Document |
| | |
101.LAB* | | Inline XBRL Taxonomy Extension Label Linkbase Document |
| | |
101.PRE* | | Inline XBRL Taxonomy Extension Presentation Linkbase Document |
| | |
104 | | Cover Page Interactive Data File (embedded within the Inline XBRL document) |
| | |
* Filed herewith.
** Furnished herewith.
# Information in this exhibit identified by three asterisks [***] is confidential and has been omitted pursuant to
Item 601(b)(10)(iv) of Regulation S-K because it is not material and is the type of information that the Company customarily treats as private or confidential. An unredacted copy of this exhibit will be furnished to the Securities and Exchange Commission on a supplemental basis upon request.
37
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | |
Date: May 10, 2022 | REATA PHARMACEUTICALS, INC. |
| | | |
| By: | | /s/ J. Warren Huff |
| Name: | | J. Warren Huff |
| Title: | | Chief Executive Officer |
| | | |
| By: | | /s/ Manmeet S. Soni |
| Name: | | Manmeet S. Soni |
| Title: | | Chief Operating Officer, Chief Financial Officer, and President |
38