UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For The Fiscal Year Ended December 31, 2014
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
COMMISSION FILE NUMBER: 001-34256
HEARTWARE INTERNATIONAL, INC.
(Exact name of registrant as specified in its charter)
| | |
| Delaware | | 26-3636023 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) |
500 Old Connecticut Path
Framingham, Massachusetts 01701
+1 508 739 0950
(Address of principal executive offices) (Zip Code)
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | |
Title of Each Class | | Name of Each Exchange on which Registered |
| Common Stock, $0.001 Par Value Per Share | | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes x No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to thisForm 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” inRule 12b-2 of the Exchange Act.
| | | | | | |
| Large accelerated filer | | x | | Accelerated filer | | ¨ |
| | | |
| Non-accelerated filer | | ¨ (Do not check if a smaller reporting company) | | Smaller reporting company | | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the registrant’s outstanding common stock held by non-affiliates computed by reference to the closing sale price of the common stock reported on the NASDAQ Stock Market as of June 30, 2014, the last business day of the registrant’s second fiscal quarter, was approximately $1.07 billion.
As of February 25, 2015, the registrant had 17,238,508 shares of common stock, par value $0.001, issued and outstanding.
Documents Incorporated By Reference
Portions of the registrant’s definitive proxy statement to be delivered to stockholders in connection with the registrant’s 2015 Annual Meeting of Stockholders are incorporated herein by reference in Part III of this Annual Report on Form 10-K to the extent stated herein. Such proxy statement will be filed with the Securities and Exchange Commission within 120 days of the registrant’s fiscal year ended December 31, 2014.
Table of Contents
References
Unless the context requires otherwise, references in this Annual Report on Form 10-K to “HeartWare,” “the Company,” “HeartWare Group,” “we,” “us” and “our” refer to HeartWare International, Inc. and its consolidated direct and indirect subsidiaries.
Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These forward-looking statements are based on our management’s beliefs, assumptions and expectations and on information currently available to our management. Generally, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,” “potential” and similar expressions intended to identify forward-looking statements, which generally are not historical in nature. All statements that address operating or financial performance, events or developments that we expect or anticipate will occur in the future are forward-looking statements, including without limitation:
| | • | | our ability to implement systemic improvements necessary to satisfactorily address the observations cited in the June 2, 2014 warning letter we received from the United States Food and Drug Administration (“FDA”); |
| | • | | our expectations with respect to submissions to and approvals from regulatory bodies, such as the FDA; |
| | • | | our ability to operate our business in compliance with regulatory requirements and to implement appropriate corrective and preventive actions; |
| | • | | our expectations with respect to our clinical trials, including enrollment in, completion of, or outcomes of our clinical trials as well as approval of new clinical trials and continued access or supplemental protocols with respect to our existing clinical trials; |
| | • | | our expectations with respect to the integrity or capabilities of our intellectual property position; |
| | • | | our ability and plans to commercialize our existing products; |
| | • | | our ability and plans to develop and commercialize new products and the expected features, functionalities and benefits of these products; |
| | • | | our estimates regarding our capital requirements and financial performance, including earnings fluctuation and cash availability; and |
| | • | | our ability to manage the costs and achieve the benefits of our strategic initiatives including acquired companies and technologies. |
Our management believes that these forward-looking statements are reasonable as and when made. However, you should not place undue reliance on our forward-looking statements because they speak only as of the date when made. We do not assume any obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required by federal securities laws and the rules and regulations of the Securities and Exchange Commission. We may not actually achieve the plans, projections or expectations disclosed in our forward-looking statements, and actual results, developments or events could differ materially from those disclosed in the forward-looking statements. Forward-looking statements are subject to a number of risks and uncertainties, including without limitation those described in Part I, Item 1A. “Risk Factors” and elsewhere in this report and those described from time to time in our filings with the Securities and Exchange Commission. Investors should read this entire Annual Report on Form 10-K and consult their respective financial, legal or other professional advisers in relation to the subject matter herein, especially as it pertains to our risks and uncertainties outlined in Part I, Item 1A. “Risk Factors” of this Annual Report on Form 10-K, together with the information provided in our other public filings with the Securities and Exchange Commission.
1
Corporate Information
HeartWare International, Inc. was incorporated in Delaware on July 29, 2008 and became the successor to HeartWare Limited, an Australian corporation, on November 13, 2008, as a result of a redomiciliation of HeartWare Limited from Australia to Delaware. Following the redomiciliation, HeartWare International, Inc. became the ultimate parent company of the HeartWare Group. The ordinary shares of HeartWare Limited traded on the Australian Securities Exchange (the “ASX”) from January 31, 2005 until November 13, 2008 when interests in HeartWare International, Inc. started trading on the ASX in the form of CHESS Depositary Interests, or CDIs, each representing one thirty-fifth of a share of our common stock. On February 24, 2009, common shares of HeartWare International, Inc. were listed for trading on the NASDAQ Global Market and commenced trading on the following day. On September 17, 2013, HeartWare was officially delisted from the ASX meaning that interests in HeartWare are no longer publicly traded on the ASX.
We further discuss our corporate history under “Business—Corporate History.”
Our corporate headquarters is located at 500 Old Connecticut Path, Framingham, Massachusetts. Our telephone number is 1-508-739-0950. Our website address iswww.heartware.com. We make available on this website, free of charge, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports as soon as reasonably practicable after we electronically file or furnish these materials to the Securities and Exchange Commission. We have included our website address in this Annual Report on Form 10-K as an inactive textual reference only. The information on, or that can be accessed through, our website is not part of this Annual Report on Form 10-K.
In addition to our corporate headquarters, our principal facilities include our manufacturing and operations facility in Miami Lakes, Florida and our distribution and customer service facility in Hannover, Germany. As of December 31, 2014, we had approximately 585 employees worldwide.
Currency
Unless indicated otherwise in this Annual Report on Form 10-K, all references to “$”, “U.S.$” or “dollars” refer to United States dollars, the lawful currency of the United States of America. References to “AU$” refer to Australian dollars, the lawful currency of the Commonwealth of Australia. References to “€” or “Euros” means Euros, the single currency of Participating Member States of the European Union. References to “£” or “British Pounds” refer to British pound sterling, the lawful currency of the United Kingdom.
Trademarks
HEARTWARE®, HVAD®, MVAD®, Pal™, CIRCULITE®, SYNERGY® and various company logos are the trademarks of the Company. All other trademarks and trade names mentioned in this Annual Report on Form 10-K are the property of their respective owners.
Part I
Item 1. Business
Overview
HeartWare is a medical device company that develops and manufactures miniaturized implantable heart pumps, or ventricular assist devices, to treat patients suffering from advanced heart failure. We have one operating segment, which designs, manufactures and markets our medical devices. We are headquartered in Framingham, Massachusetts and have primary facilities in Miami Lakes, Florida and Hannover, Germany.
The HeartWare Ventricular Assist System (the “HVAD System”), which includes a ventricular assist device (“VAD”) or blood pump, patient accessories and surgical tools, is designed to provide circulatory support for
2
patients in the advanced stage of heart failure. The core of the HVAD System is a proprietary continuous flow blood pump, the HVAD pump, which is a full-output device capable of pumping up to 10 liters of blood per minute. The HVAD System is designed to be implanted adjacent to the heart, avoiding abdominal surgery, which is generally required to implant similar devices.
Heart failure is a chronic disease that results in the heart’s pumping power being weaker than normal. In a healthy person, the left ventricle of the heart pumps oxygenated blood into the aorta and the blood is then circulated throughout the body until it returns through the venous system to the right side of the heart, which then pumps the blood into the lungs where it is re-oxygenated. If the left ventricle is not working properly, the oxygenated blood is not fully cleared from the lungs and the blood is not circulated effectively. If the muscle of the left ventricle is damaged or is not working efficiently, the ventricle will tend to compensate by working harder in an effort to supply adequate blood flow into the aorta. The increased effort generally results in dilation or enlargement of the ventricle, rather than increased blood flow. This dilation then makes it harder for the heart to contract effectively, which results in even lower blood flow and increased effort and further dilation of the ventricle. This progressive, degenerative process generally continues until the patient becomes debilitated and eventually dies from inadequate clearing of the lungs and inadequate flow of oxygenated blood throughout the body. The inadequate lung clearance or lung congestion is why the advanced stages of heart failure are called congestive heart failure, or CHF.
In November 2012, we received approval from the United States Food and Drug Administration (“FDA”) for the HVAD System as a bridge to heart transplantation in patients with end-stage heart failure. The HVAD System has been available in the European Union since receiving CE marking in 2009. In May 2012, we received an expanded European label for long-term use of the HVAD System in all patients at risk of death from refractory, end-stage heart failure. As of December 31, 2014, there have been over 7,000 implants of the HVAD System in patients at over 270 health care sites in 41 countries.
Bridge-to-transplant
FDA approval for a bridge-to-transplant (“BTT”) indication was based on the results of our pivotal ADVANCE clinical trial, an FDA approved Investigational Device Exemption (“IDE”) study designed to evaluate the HVAD System as a bridge to heart transplantation for patients with end-stage heart failure, as well as a Continued Access Protocol (“CAP”). Under ADVANCE, 140 patients at 30 hospitals in the U.S. received the HeartWare investigational device between August 2008 and February 2010. The ADVANCE study achieved 94% survival at 6 months and successfully met its primary endpoint of establishing non-inferiority between the investigational device and comparator arm of the study, which was derived from contemporaneous patients from the Interagency Registry for Mechanically Assisted Circulatory Support (“INTERMACS”) [p<0.0001]. Four supplemental allotments of patients have been granted by the FDA under a CAP, encompassing more than 250 additional patients.
To help assure the continued safety and effectiveness of an approved device, FDA requires a post-approval study (“PAS”) as a condition of approval under 21 CFR 814.82(a)(2) to assess device performance in a real-world setting. HeartWare’s PAS is a registry consisting of 600 post-approval patients who receive an HVAD and an additional 600 post-approval control patients derived from a contemporaneous group of continuous flow, intracorporeal left VAD (“LVAD”) patients entered into the INTERMACS database. The data for both arms of the study will be entered into the INTERMACS registry by the implanting centers. Other post approval commitments include the transfer of patients from the ADVANCE IDE study into a post approval database as well as an obligation to continue training sites in accordance with an approved training program.
Destination Therapy
In May 2012, we completed enrollment in our clinical trial named “ENDURANCE” for a destination therapy indication. Designed to enroll up to 450 patients at 50 U.S. hospitals, the non-inferiority study is a
3
randomized, controlled, unblinded, multi-center clinical trial to evaluate the use of the HVAD System as a destination therapy in advanced heart failure patients. The study population was selected from patients with end-stage heart failure who have not responded to standard medical management and who are ineligible for heart transplantation. Patients in the study were randomly selected to receive either the HVAD System or, as part of a control group, an alternative ventricular assist device approved by the FDA for destination therapy, in a 2:1 ratio. Each patient receiving the HVAD System or control VAD was followed to the primary endpoint at two years, and will undergo subsequent follow-up for a five-year period post implant.
On August 27, 2013, the FDA approved an Investigational Device Exemption (“IDE”) Supplement allowing HeartWare to commence enrollment in an additional patient cohort for the ENDURANCE clinical trial. In this supplemental cohort, HeartWare intends to enroll up to 310 patients receiving the HVAD System, as well as up to an additional 155 control patients using a randomization scheme consistent with the ENDURANCE protocol. Patients will be followed for 12 months after implant. In 2016, HeartWare intends to incorporate the data from both this supplemental cohort and ENDURANCE into an anticipated PMA Application seeking approval of the HVAD System for the destination therapy indication.
Other Clinical Activities
In the fourth quarter of 2013, HeartWare received approval from the Japanese Pharmaceuticals and Medical Devices Agency to commence a clinical study in Japan for market authorization for a BTT indication. In the third quarter of 2014, we completed enrollment in the study (6 patients at 5 clinical sites).
In addition, in the fourth quarter of 2013, HeartWare received conditional approval from the FDA for a prospective, controlled, unblinded, multi-center clinical trial to evaluate the thoracotomy implant technique for the HVAD System. We began enrollment in this study in January 2015.
MVAD System
Beyond the HVAD System, we are developing our next generation miniaturized device, known as the MVAD System. The MVAD System is based on the same technology platform as the HVAD System, but adopts an axial flow, rather than a centrifugal flow, configuration and is being developed in multiple designs. The MVAD pump is less than one-half the size of the HVAD pump and can provide partial or full support. The MVAD platform is designed to allow for a variety of configurations and surgical placements with the goal of further reducing surgical invasiveness while producing superior clinical results. In December 2014, we initiated submissions to commence a CE Mark study at 9 international sites and are preparing regulatory submissions to commence an IDE study in the United States.
CircuLite
On December 1, 2013, we acquired CircuLite, Inc. CircuLite is the developer of the SYNERGY Circulatory Support System, a partial support system designed to treat less sick, ambulatory, chronic heart failure patients who are not yet inotrope-dependent. The SYNERGY Surgical System is designed for long-term support and is intended to reduce the heart’s workload while improving blood flow to vital organs. SYNERGY experienced issues that arose after its commercial release and caused the loss of its CE marking in the European Union in March 2014. In January 2015, we discontinued development of the SYNERGY micropump and have focused our efforts on a version of our MVAD pump for our partial-assist program. Following the necessary clinical trials and regulatory approvals, we plan to re-launch the system in Europe and will focus on building experience at a small number of centers of excellence, refining training techniques and implementing additional system upgrades. The next generation endovascular system, which will be implanted collaboratively by cardiologists and surgeons in a hybrid catheterization (“cath”) lab setting, offers an interventional approach to circulatory support. While our HVAD and MVAD Systems offer minimally invasive treatment to end-stage heart failure patients, the SYNERGY Circulatory Support System offers even less invasive and ultimately interventional options to earlier-stage heart failure patients.
4
Operations
We began generating commercial revenue from sales of the HVAD System in January 2009 and have incurred net losses in each year since our inception. We expect our losses to continue as we expand our pipeline through continued research and development into next generation products, continue our clinical trials, enhance our infrastructure and expand commercial markets both inside and outside of the United States.
We have funded our operations primarily through product revenue, the issuance of convertible notes and the issuance of shares of our common stock. On December 15, 2010, we issued convertible senior notes with an aggregate principal amount of $143.75 million pursuant to the terms of an indenture dated as of December 15, 2010. The convertible senior notes are senior unsecured obligations of the Company. The convertible senior notes bear interest at a rate of 3.5% per annum, payable semi-annually in arrears on June 15 and December 15 of each year. The convertible senior notes will mature on December 15, 2017, unless earlier repurchased or converted. The notes offering was completed pursuant to a prospectus supplement, dated December 9, 2010, to a shelf registration statement on Form S-3 that was previously filed with the SEC and which was declared effective on December 9, 2010.
Most recently, in March 2013, we completed a public offering of 1,725,000 shares of our common stock, including the underwriters’ exercise of their over-allotment option to purchase 225,000 shares, at an offering price of $86.45 per share for aggregate gross proceeds of approximately $149.1 million. After fees and related expenses, net proceeds from the offering were approximately $141 million. The offering was completed pursuant to a prospectus supplement, dated March 12, 2013, to a shelf registration statement on Form S-3 that was previously filed with the SEC and which was declared effective on December 9, 2010.
On January 30, 2014, we filed a new shelf registration statement with the SEC on Form S-3. This shelf registration statement allows us to offer and sell from time to time, in one or more series or issuances and on terms that we will determine at the time of the offering, any combination and amount of the securities described in the prospectus contained in the registration statement or in the prospectus supplement filed with respect to a particular offering.
Market Opportunity
Heart Failure
Heart failure is one of the leading causes of death in the developed world. The American Heart Association estimates that heart failure affects over 5.5 million people in the United States, while the European Society of Cardiology reports a prevalence of at least 10 million in European countries. Heart failure is a cardiovascular disease with both an increasing incidence and death rate worldwide. In the United States, approximately 670,000 new cases are diagnosed annually and approximately 57,000 patient deaths are attributed to advanced heart failure.
Our Target Markets—Class III and Class IV Patients
Our technologies target certain classes of advanced heart failure patients, specifically Class III and IV patients as defined by the New York Heart Association (“NYHA”). We believe that there is a significant market opportunity for ventricular assist devices, or VADs, that are smaller, easier to implant, easier to use and/or more reliable than the other devices that are currently available. We also believe there is a significant market opportunity for any device that, relative to existing therapies, demonstrates superior patient outcomes at a lower cost.
It is estimated that there are approximately 5 million Class III heart failure patients worldwide. Of these 5 million patients, we estimate that approximately 1 million patients are severely impacted by congestive heart failure, or CHF, but are not yet nearing the end stages of the disease. While these patients suffer on a daily basis,
5
they do not need the same full support as the sicker, later-stage Class IV patients and they may be less willing to undergo the more invasive procedure required for the placement of the typical VAD. We believe that up to one-third of these 1 million patients could be candidates for a partial support system, such as the SYNERGY Circulatory Support System, placed in a less invasive surgical approach because of the potential for reduced surgical risk and shorter post-operative recovery periods.
CHF Treatment Options
Although many pharmacological therapies and pacing devices that are designed to stimulate the heart have proven to be effective at prolonging the quality and duration of a patient’s life, these treatments and devices do not halt the progression of CHF. Pharmacologic management of CHF focuses primarily on improving the overall pump function of the heart while slowing the rate of CHF progression. For later stage Class III and Class IV patients, some investigations have suggested that the increase in patient survival rates using medical therapy is limited and that optimal medical therapy has not been demonstrated to stop or reverse the effects of CHF. Other approaches, such as devices that allow physicians to restrict or reduce the size of the heart and cell based therapy, are either in the early development stages or have not yet achieved outcomes that we believe would lead most physicians to consider these technologies as viable solutions.
Heart transplantation is the current primary therapy for refractory advanced heart failure and ultimately provides the best recovery of cardiac function. Heart transplantation is an effective and accepted surgical procedure that can result in end-stage heart failure patients resuming relatively normal lives for a period usually expected to be ten years or longer. However, the therapy is significantly constrained by the limited number of available donor hearts. Also, many patients with heart failure are ineligible for heart transplantation because of factors such as age or the presence of other diseases.
VAD Treatment for Advanced Heart Failure
Circulatory assist devices are designed to take over some or all of the pumping function of the heart by mechanically pumping blood into the aorta. Implantation of circulatory assist devices is the only therapy other than transplantation that has been shown to rehabilitate a patient from NYHA Class IV to Class I or II. A November 2001 article inThe New England Journal of Medicineon a study entitled “Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure,” or the REMATCH study, concluded that “the use of a left ventricular assist device in patients with advanced heart failure resulted in a clinically meaningful survival benefit and an improved quality of life. A left ventricular assist device is an acceptable alternative therapy in selected patients who are not candidates for cardiac transplantation.” The conclusions in this study have since been reconfirmed in a number of subsequent similar studies with VADs, including a bridge-to-transplant study and a destination therapy study reported in August 2007 and November 2009 articles, respectively, inThe New England Journal of Medicine. These conclusions were further echoed in the bridge-to-transplant study for the HVAD System, as described by Aaronsonet al. in the June 2012 article inCirculation. In summary, the HVAD pump was associated with “high rates of 180 day success and survival” with “a favorable adverse event profile” and significantly improved functional capacity and quality of life as not previously reported with any other “drug or device therapy for advanced heart failure.”
A large population of end-stage heart failure patients can benefit from VAD therapy, such as our HVAD System. Within this population there are generally four applications for VADs: “bridge-to-transplant” therapy, “bridge-to-decision” therapy, “destination therapy” and “bridge-to-recovery” therapy. Currently, the HVAD System is approved for the “bridge-to-transplant” population. Each year, the number of heart failure patients in need of a heart transplant exceeds the number of donor hearts that become available. According to the Organ Procurement and Transplantation Network, or OPTN, and Scientific Registry of Transplant Recipients, or SRTR, 2,431 heart transplants were conducted in the United States in 2014, and as of February 2015, 3,986 people are currently listed for heart transplant. The OPTN/SRTR 2013 Annual Data Report reported 44% of the patients transplanted were on a VAD as a bridge-to-transplant. In light of the survival benefit of VADs over inotropes, the
6
volume of patients on inotropic therapy at transplant has diminished while the use of VADs has increased. Bridging the patient to transplant with a VAD provides the clinicians time to stabilize the patient until a suitable donor heart becomes available. We expect this percentage of patients on the waiting list who receive VAD support as a bridge-to-transplant to continue to increase as surgeons and cardiologists become more familiar with the technology, and confidence in the procedure grows in line with improving clinical data and device reliability.
Our Solution and Products
Proprietary Pump Technology
The HVAD System features the smallest, full-output centrifugal pump designed to be implanted in the chest, directly adjacent to the heart. At the core of our technology platform is our proprietary “hybrid” system for suspending the impeller, which is the only moving part within the pump. The impeller is suspended within the pump housing by the opposing forces of passive magnets and hydrodynamic thrust generated by the pump impeller, which circulates a cushion of blood. Once power is applied to the device and the impeller begins to rotate, there are no points of mechanical contact within the pump, thus providing a wearless pumping system.
We believe the hybrid suspension system has several important advantages over traditional technologies. The elimination of the internal mechanical bearings which are characteristic of second generation devices removes all points of mechanical friction or contact within the pump. We believe that this removal of contact should lead both to longer term reliability of the device and to a potential reduced risk of physical damage to blood cells as they pass through the pump. Our hybrid suspension technology also establishes a miniaturization path, which we believe will allow us to significantly downsize our pump technology without compromising clinical performance. We believe competing pump designs which rely on either active magnetic or hydrodynamic forces alone face various physical constraints that may limit their ability to downsize without sacrificing performance.
The HeartWare HVAD System
The first product in our portfolio, the HVAD System, is comprised of the HVAD pump, a small, permanently implantable VAD, patient accessories and surgical tools. The HVAD pump is capable of generating up to 10 liters of blood flow per minute. With a displaced volume of only 50 cubic centimeters and a mass of 140 grams, the HVAD pump is the only approved full-output pump implantable in the pericardial space, directly adjacent to the heart. It is also the only pump designed to be implanted above the diaphragm in all eligible patients. We believe the implanting in the pericardial space generally leads to shorter surgery time and a less invasive procedure relative to alternative devices, which are normally implanted in the abdomen. In January, 2015, we began enrollment in our clinical trial related to the thoracotomy implant technique for the HVAD System, which, if successful, will provide an even less invasive procedure.
Device reliability of the HVAD System is designed to be enhanced through the use of dual motor stators with independent drive circuitry, allowing a seamless transition between dual and single stator mode if required. The pump’s inflow cannula is integrated with the device itself, providing proximity between the heart and the pumping mechanism, facilitating ease of implant and helping to ensure optimal blood flow characteristics. The use of a wide-bladed impeller and the clear flow paths through the pump are designed to help reduce the risk of pump-induced damage to blood cells.
The HVAD System has been approved for sale in Europe since early 2009. In November 2012, we received approval from the FDA for the HVAD System as a bridge to heart transplantation in patients with end-stage heart failure.
7
The HeartWare MVAD System
The MVAD System is comprised of similar components, surgical tools and peripherals as the HVAD System, but is differentiated significantly by the MVAD pump. The MVAD pump is a miniaturized blood pump intended for chronic heart failure patients. The device is a full-output axial flow pump with a fully suspended rotor and a displacement volume of less than one half of that of the HVAD pump. The pre-clinical Good Laboratory Practices (“GLP”) in-vivo studies completed in September 2011 have shown the MVAD pump to have similar comparable blood flow characteristics to the HVAD pump. The MVAD pump is designed for pericardial implantation and initial human clinical trials are expected to commence in 2015. We plan to introduce our next generation controller, Pal, with the MVAD pump in clinical trials and separately for use with the current HVAD pump. Pal is a one-piece wearable controller and battery designed for an active patient lifestyle.
We believe it is likely that more patients will be willing to undergo a shorter, less invasive surgical procedure that may result in quicker recoveries and hospital discharge. We have taken advantage of the versatility of the MVAD pump design with multiple configurations specific to less invasive implantation procedures. This development has been supported by over 100 in-vivo studies. These devices may expand the potential pool of chronic heart failure patients.
Before the MVAD System will be available for commercial sale, we will need to achieve the following milestones:
| | • | | completion of the system development including next generation peripherals (e.g., controller, batteries, power adapters); |
| | • | | approval of and successful completion of a clinical trial; and |
| | • | | receipt of regulatory approvals for commercialization. |
The CircuLite SYNERGY System
HeartWare acquired CircuLite on December 1, 2013 and with it, CircuLite’s SYNERGY Circulatory Support System. The SYNERGY Surgical System is designed to be implanted through a right, mini-thoracotomy procedure and does not require a sternotomy or cardiopulmonary bypass. With this approach, the inflow cannula is placed in the left atrium, and the outflow graft is attached to the subclavian artery. A small pump is then placed in a pacemaker-like pocket and attached to the inflow cannula and outflow graft, which connects to a wearable, external controller and battery pack. SYNERGY experienced issues that arose after its commercial release and caused the loss of its CE marking in the European Union in March 2014. In January 2015, we discontinued development of CircuLite’s proprietary micropump and focused our efforts on a version of our MVAD pump for our partial-assist program. CircuLite pioneered the partial-assist approach and despite challenges, demonstrated that this technique can enhance the quality of life for a less sick group of heart failure patients, which is believed to be a larger population than the end-stage heart failure patients that we currently treat with our full-support VADs. The next generation endovascular system, which will be implanted collaboratively by cardiologists and surgeons in a hybrid cath lab setting, offers an interventional approach to circulatory support. While our HVAD and MVAD Systems offer minimally invasive treatment to end-stage heart failure patients, the SYNERGY Circulatory Support System offers even less invasive and ultimately interventional options to earlier-stage heart failure patients.
Enhanced Quality of Life with Implantable Devices
Currently, the HVAD System and all commercially available VADs are powered by a controller, batteries and other power sources carried external to the body. Power is transferred to the implanted pump via a thin electrical cable, called a driveline, which exits the patient’s skin in the abdominal area. We are working to develop an implantable system utilizing transcutaneous energy transfer (“TET”) or wireless power that will eliminate the need for a percutaneous driveline. A TET system contains a wearable power management system
8
that is inductively coupled to an implanted pump controller and electronics that includes a rechargeable battery. The patient can remove the wearable power management system and enjoy a high quality lifestyle while the system is powered by the implanted battery.
Prior to the third quarter of 2014, we worked collaboratively with Dualis MedTech GmbH, a subsidiary of AVRA Surgical, Inc., on the development of a wireless energy transfer system. Currently, we are working internally to develop a commercial, implantable system.
Our Business Strategy
Our primary goal, above all else, is to focus on optimizing outcomes of patients being treated for advanced heart failure. To this end, we are leading innovation in the VAD sector and are also striving to develop and maintain a proprietary technology platform that enables the development of a pipeline of ever-smaller heart pumps that will reduce procedural invasiveness, and adverse events while simultaneously increasing the number of patients who can benefit from our products. In addition, we intend to explore technologies and therapies for the general treatment of heart failure.
We believe that our technology portfolio provides us with a significant competitive advantage in the market. To capitalize on that advantage, we are pursuing the following plan:
Expand Market Penetration outside of the U.S.—We sell to VAD centers and distributors throughout Europe and in other countries outside the U.S. With the receipt of the HVAD System’s CE Marking in January 2009, we began to develop the necessary infrastructure to support commercial sales in Europe. Throughout 2014, we continued to expand our infrastructure to support commercial activity and have generated sales through 2014 from customers in over 40 countries. In the future, we intend to build wider distribution channels and ordering systems to deliver our products to the European market on a wider commercial scale as well as increase the number of countries in which we have approval to sell our device commercially.
Expand U.S. Market Penetration—Our goal is to continue to expand U.S. market penetration for the HVAD System for a bridge-to-transplant system. Our focus is to continue to establish and maintain commercial sites at all of the major U.S. health centers that support VAD implantation. In the U.S., we currently have approximately 110 commercial sites.
We also intend to seek an expanded indication for the HVAD System to include destination therapy. In May 2012, we completed enrollment in our ENDURANCE destination therapy clinical trial. Each test patient has now been followed to the primary endpoint of two years, and will be followed for a subsequent five-year period post-implant. On August 27, 2013, the FDA approved an Investigational Device Exemption (“IDE”) Supplement allowing HeartWare to commence enrollment in an additional patient cohort for the ENDURANCE clinical trial. Patients have been, or will be, followed for 12 months after implant. HeartWare intends to incorporate the data from both this supplemental cohort and ENDURANCE into an anticipated pre-market approval application seeking approval of the HVAD System for the destination therapy indication.
Focus on continuous product development—In parallel with the clinical development of the HVAD System, we plan to advance the development of our next generation products, such as the MVAD System, the SYNERGY System and a TET system, and to enhance our peripheral equipment. We expect assessment and development and/or enhancement work for the MVAD System, the SYNERGY System and the TET system and peripheral equipment to continue throughout 2015. We plan to introduce our next generation controller, Pal, with the MVAD pump in clinical trials and separately for use with the current HVAD pump. Pal is a one-piece wearable controller and battery designed for an active patient lifestyle.
Partner with leading professionals in the fields of cardiovascular surgery around the world—We have established relationships with leading professionals in the field of cardiovascular surgery and heart centers around the world and will continue to expand this network. We believe these relationships are key to our growth as they help to drive clinical awareness of our products.
9
Explore complementary or alternative therapies and technologies—We intend to explore business development opportunities including strategic alliances, joint ventures, and acquisitions that might complement or expand our market opportunities. In mid-2012, we acquired World Heart Corporation primarily to expand our intellectual property portfolio and in late 2013, we acquired CircuLite to develop a partial support system to treat less sick, chronic heart failure patients.
Sales and Marketing
Our sales and marketing strategy is to educate and promote the benefits of HeartWare’s family of circulatory support systems for the treatment of clinical heart failure among a variety of health care professionals. We market directly to clinicians and medical facilities as well as through medical device distributors in some markets outside of the U.S.
We work with a broad spectrum of health care industry participants to promote the clinical benefits of our device, including hospital administrators, cardiologists, surgeons, nurses, perfusionists, insurers and government payer policy representatives. A key to the development of our business is enhancing patient outcomes via effective training and clinical end-user support programs and resources.
To support our sales and marketing strategy, we have recruited and trained experienced territory managers and clinical specialists. This field team supports customers by supporting implant procedures, providing technical information and training, resolving clinical issues, and educating health care professionals on the benefits of HeartWare systems. In addition, we partner with leading physicians in the field to proctor and preceptor new physicians on the use of our devices in their centers and to present clinical and technical data about our system at scientific symposia, congresses, and trade shows, as well as publish in peer reviewed cardiovascular journals.
Our product management team conducts market research on end-user preferences and unmet needs, identifies areas of improvement in our product offering and services, and works with research and development on new technologies that meet newly identified needs that are not addressed with our current platform of products.
We sell our products primarily to large hospitals and distributors. No customer accounted for more than 10% of total revenue in fiscal years 2014, 2013, and 2012. Approximately 46%, 49% and 75% of our 2014, 2013 and 2012 revenues, respectively, were derived from international sales. Globally, over 7,000 implants have been performed using the HVAD System. The HVAD System has been implanted in patients at over 270 health care sites in 41 countries.
Intellectual Property
We rely on a combination of patents, trade secrets, trademarks and copyrights, together with non-disclosure and confidentiality agreements, to protect our proprietary rights in our technologies.
As of December 31, 2014, we had 86 U.S. patents, 24 Australian patents, 23 Japanese patents, 11 patents issued in each of Great Britain, Germany, and France, as well as patents issued in the Italy, Netherlands, Spain, Austria, Belgium, China, Korea, Canada, Israel, Hong Kong and Turkey. We also have 97 pending U.S. non-provisional patent applications and a number of international patent applications filed with various national and regional patent offices including the Patent Cooperation Treaty, Japan, Europe, Australia, China, Canada, Hong Kong, India, Korea and Israel.
10
Our U.S. and foreign issued patents and patent applications cover fundamental technologies underlying our hemodynamically and physiologically compatible full-output, long-term circulatory assist devices and partial support technologies. The main technologies claimed in patents and patent applications include:
| | • | | use of dual stators in a blood pump; |
| | • | | the combination of passive magnetic bearings and hydrodynamic thrust bearings; |
| | • | | channels or wide-bladed impellers in a blood pump; |
| | • | | the use of ceramic between an impeller and motor stator; |
| | • | | flow estimation based on impeller speed and viscosity; |
| | • | | use of platinum alloy for blood pump impellers; |
| | • | | alternative axial flow pump technologies; |
| | • | | physiological response and pump control algorithms; |
| | • | | wireless energy transfer systems; and |
| | • | | blood pump system cannulae. |
Major patents and pending patent applications covering technologies for our HVAD System are scheduled to expire at various times between 2016 and 2027. Patents and patent applications covering technologies for our MVAD System are scheduled to expire at various times between 2024 and 2030.
We actively monitor our intellectual property position and periodically review new developments to identify prudent extensions to our patent portfolio. We plan to file additional patent applications on inventions that we believe are patentable and important to our business. We may also license or acquire patents from third parties that may enhance or expand our development activities. Accordingly, we intend to pursue and defend aggressively patent protection on our proprietary technologies. We have also entered into and may in the future enter into settlement agreements pursuant to which third parties or their successors or assigns may commercialize competing technologies or products that would have otherwise been precluded by our patents subject to the agreement.
Despite our efforts, we may be subject to challenges, with or without merit, regarding our patents or other intellectual property. The medical device industry is characterized by a large number of patents and by frequent and substantial intellectual property litigation. Our products and technologies could infringe, or other persons could allege that our products and technologies infringe, upon the proprietary rights of third parties. If third parties successfully assert infringement or other claims against us, we may not be able to sell our products. In addition, patent or intellectual property disputes or litigation may be costly, result in product development delays or divert the efforts and attention of our management and technical personnel. If a dispute or litigation arises, we may seek to enter into a royalty or licensing arrangement. However, such an arrangement may not be available on commercially acceptable terms, if at all. We may decide, in the alternative, to litigate the claims or to design around the patented or otherwise proprietary technology. At this time we are not party to any material legal proceedings that relate to patents or proprietary rights.
Our intellectual property also includes non-patented technology, processes and procedures, and technical knowledge and know-how accumulated or acquired since inception, all of which are significant to our competitive position. It is our policy to enter into confidentiality, non-disclosure and intellectual property assignment agreements with employees and consultants to help ensure that we can protect our rights in developed proprietary technology and prohibit the disclosure of any confidential information or trade secrets.
11
Government Regulation
United States
Our products are regulated by the FDA as a Class III medical device under the U.S. Food, Drug, and Cosmetic Act. FDA regulations govern:
| | • | | product design and development; |
| | • | | product safety and effectiveness; |
| | • | | premarket approval (“PMA”); |
| | • | | advertising and promotion; |
| | • | | product sales and post-market activities; |
| | • | | medical device (adverse event) reporting; and |
| | • | | field corrective actions (e.g., recalls). |
Premarket Approval
Each of our devices are regulated as a Class III medical device. PMA approval from the FDA is required before marketing of a Class III medical device in the United States can commence and the process of obtaining PMA can be costly, lengthy and uncertain. In November 2012, we received approval for the HVAD System as a bridge to heart transplantation in patients with end-stage heart failure.
The Company recently completed its ENDURANCE trial relating to destination therapy and is working to complete the supplemental cohort to the ENDURANCE trial approved in August 2013. A separate PMA submission for the HVAD System for a destination therapy indication will be required. A PMA application must be supported by extensive data including, but not limited to, technical, preclinical and clinical trials to demonstrate the safety and effectiveness of the device to the FDA’s satisfaction. Among other information, the PMA application must also contain a full description of the device and its components, a full description of the methods, facilities and controls used for manufacturing, and proposed device and patient labeling.
If the FDA determines that a PMA application is complete, the FDA accepts the application and then begins an in-depth review of the submitted information. The FDA, by statute and regulation, has 180 days to review an accepted PMA application, although the review and response process generally occurs over a significantly longer period of time, often more than a year, and can take up to several years. HeartWare’s receipt of an FDA warning letter in June 2014 may further delay an FDA review and response. During the review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a pre-approval inspection of our and our key suppliers’ facilities to evaluate compliance with quality system
12
regulations. They will also conduct a Bioresearch Monitoring (“BIMO”) inspection of the clinical trial including some of the clinical data sites. To help assure the continued safety and effectiveness of an approved device, the FDA may also require a post-approval study as a condition of approval. Post-approval studies were required as a condition of approval for the HVAD System as a bridge to heart transplantation.
Under the FDA Safety and Innovation Act (Public Law 112-144) which included the Medical Device User Fee Amendments of 2012, the fee to submit a PMA application for FY 2014 was $258,520. User fees are expected to rise over time until the law sunsets in 2017. We qualified for a small business exemption that allowed us to file our first PMA application at no charge, however we presently do not qualify for the exemption. PMA supplements are required for modifications to the manufacturing process, labeling, use and design of a device that is approved through the premarket approval process. PMA supplements often require submission of the same type of information as a PMA application except that the supplement is limited to information needed to support any changes from the device covered by the original PMA. The changes to design may require significant testing, validation and documentation and may be associated with significant review times from 30 days to 180 days and fees ranging from approximately $4,100 up to approximately $39,000 with the exception of the Panel Track Supplement (which includes clinical data) is $193,890.
A post-approval study (“PAS”) may also be required and could be a clinical or non-clinical study required in the PMA approval order and is intended to gather specific information to address questions about the post-market performance and physician experience with an approved medical device. Concurrent with PMA approval of the HVAS for bridge-to-transplant indication, the FDA has required us to complete a PAS as a condition of approval under 21 CFR 814.82(a)(2) to assess device performance in a real-world setting. HeartWare’s PAS is a registry consisting of 600 patients who receive an HVAD and an additional 600 control patients derived from a contemporaneous group of continuous flow, intracorporeal LVAD patients entered into the INTERMACS database. The data for both arms of the study will be entered into the INTERMACS registry by the implanting centers. Other post approval commitments include the transfer of patients from the ADVANCE IDE study into a post approval database as well as an obligation to continue training sites in accordance with an approved training program.
Pervasive and Continuing FDA Regulation
Clinical trials require extensive recordkeeping and reporting requirements. Our clinical trials must be conducted under the oversight of an institutional review board at the relevant clinical trial site and in accordance with applicable regulations and policies including, but not limited to, the FDA’s good clinical practice, or GCP, requirements. We, the trial data safety monitoring board, the FDA or the institutional review board at each clinical trial site may suspend a clinical trial at any time for various reasons, including a belief that the risks to study patients outweigh the anticipated benefits.
Both before and after FDA approval, numerous regulatory requirements apply. These include:
| | • | | quality system regulation, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures during the design, manufacturing and commercialization phases; |
| | • | | regulations which govern product labels and labeling, prohibit the promotion of products for unapproved or “off-label” uses and impose other restrictions on labeling and promotional activities; |
| | • | | medical device reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; |
| | • | | regulations which govern medical device tracking; and |
| | • | | notices of correction or removal and recall regulations. |
13
Advertising and promotion of medical devices are also regulated by the Federal Trade Commission and by state regulatory and enforcement authorities. Recently, some promotional activities for FDA-regulated products have resulted in enforcement actions brought under healthcare reimbursement laws and consumer protection statutes. In addition, under the federal Lanham Act, competitors and others can initiate litigation relating to advertising claims.
Compliance with regulatory requirements is enforced through periodic, unannounced facility inspections by the FDA. At the conclusion of an FDA inspection, the inspector may provide observations identifying areas of potential regulatory concern. Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA. Enforcement actions may include any of the following sanctions against us:
| | • | | warning letters or untitled letters; |
| | • | | fines, injunction and civil penalties; |
| | • | | recall or seizure of our products; |
| | • | | customer notification, or orders for repair, replacement or refund; |
| | • | | operating restrictions, partial suspension or total shutdown of production or clinical trials; |
| | • | | refusing our request for premarket approval of new products; |
| | • | | withdrawing premarket approvals that are already granted; and |
FDA Warning Letter
We received a warning letter from the FDA, dated June 2, 2014, following an inspection of our Miami Lakes, Florida facility conducted in January 2014. The FDA letter cited four categories for us to address: (1) procedures for validating device design, including device labeling; (2) procedures for implementing corrective and preventive action (CAPA); (3) maintaining records related to investigations; and (4) validation of computer software used as part of production or quality systems. The warning letter did not require any action by physicians or patients and did not restrict the use of our devices.
We sent the FDA our initial response to the warning letter within the required fifteen business days of receipt, and committed to undertaking certain quality system improvements and providing the FDA with periodic updates. During 2014, we commenced implementing systemic changes and organizational enhancements to address the four warning letter items and related quality systems. We have established teams to review and address the items cited by the FDA and have engaged external subject matter experts to assist in assessment and remediation efforts. As we complete this comprehensive review of our quality systems, it is possible that we may need to take additional actions including the possibility of voluntary product recalls when necessary to ensure patient safety and effective performance of the HVAD System.
European Union
The primary regulatory environment in Europe is that of the European Union, or EU which consists of 28 member states in Europe. In 2014, two EU directives that covered medical devices—Directive 93/42/EEC covering medical devices and Directive 90/385/EEC for active implantable medical devices—became EU regulations, which augmented and clarified earlier directives. The EU also has numerous standards that govern and harmonize the national laws and standards regulating the design, manufacture, clinical trials, labeling, adverse event reporting and post market surveillance activities for medical devices that are marketed in member states. Medical devices that comply with the requirements of the national law of the member state in which they are first marketed will be entitled to bear CE Marking, indicating that the device conforms to applicable regulatory requirements, and, accordingly, can be commercially marketed within EEC states and other countries
14
that recognize this mark for regulatory purposes. We received CE Marking for the HVAD System in January 2009. In May 2012, we received an expanded label for long-term use of the HVAD System in patients at risk of death from refractory, end-stage heart failure.
Australia
In Australia, the Therapeutic Goods Administration, or TGA, is responsible for administering the Australian Therapeutics Goods Act. The Office of Devices, Blood and Tissues is the department within the TGA responsible for devices. The TGA recognizes five classes of medical devices and HeartWare’s circulatory assist device falls under the category of “active implantable medical devices.”
The Australian Register of Therapeutic Goods, or ARTG, controls the legal supply of therapeutic goods in Australia. The ARTG is the register of information about therapeutic goods for human use that may be imported, supplied in, or exported from Australia. Any use of an unapproved medical device in humans, even in pilot trials, requires an exemption from the requirement for inclusion on the ARTG.
In March 2011, we received approval from the TGA to sell the HVAD System commercially in Australia.
Other International Regulations
We are also subject to international regulations in other countries where our products are sold. We currently have sales to customers in a variety of countries outside of the EU, U.S. and Australia. These regulations relate to product standards, packaging and labeling requirements, import restrictions, tariff regulations, duties and tax requirements. Many of the regulations applicable to our products in these countries are similar to those of the FDA. The national health or social security organizations of certain countries require our products to be qualified before they can be marketed in those countries. In certain countries, doctors may request use of our product under compassionate or emergency use or special access programs prior to approval. These programs tend to be limited and patient-specific and do not replace premarket approval.
In order to be positioned for access to European and other international markets, we sought and obtained certification under the International Standards Organization (“ISO”) 13485 standards. ISO 13485 is a set of integrated requirements, which when implemented, form the foundation and framework for an effective quality management system. These standards were developed and published by the ISO, a worldwide federation of national bodies, founded in Geneva, Switzerland in 1947. ISO has more than 90 member countries and ISO certification is widely regarded as essential to enter Western European markets.
Healthcare Regulation
Recent healthcare policy changes
In response to perceived increases in health care costs in recent years, there have been and continue to be proposals by the federal government, state governments, regulators and third-party payors to control these costs and, more generally, to reform the U.S. healthcare system. For example, on March 23, 2010, the Patient Protection and Affordable Care Act (the “Affordable Care Act”) was signed into law. On March 30, 2010, a companion bill, the Health Care and Education Reconciliation Act of 2010 (the “Reconciliation Act”) was also signed into law. Among other things, the Affordable Care Act and the Reconciliation Act (collectively, the “Acts”), when taken together, impose a 2.3% excise tax on the sale of certain medical devices, including our devices, which took effect January 1, 2013. In addition, it is possible that standard setters or regulators may address certain unique aspects of the accounting for the Acts in the future.
Regulations related to prohibiting “kickbacks” and false claims and protecting patient confidentiality
A federal law commonly known as the “Anti-Kickback Law,” and several similar state and foreign laws, prohibit payments that are intended to induce physicians or others either to refer patients or to acquire or arrange
15
for or recommend the acquisition of healthcare products or services. These laws constrain our sales, marketing and other promotional activities by limiting the kinds of financial arrangements, including sales programs, we may have with hospitals, physicians or other potential purchasers of medical devices. Other federal and state laws generally prohibit individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent, or for items or services that were not provided as claimed. Because we may provide some coding and billing information to purchasers of the HVAD System and our other products, and because we cannot assure that the government will regard any billing errors that may be made as inadvertent, these laws are potentially applicable to us. Anti-kickback and false claims laws prescribe civil and criminal penalties for noncompliance, which can be substantial.
Data Privacy
There are a number of federal and state and foreign laws protecting the confidentiality of certain patient health information, including patient records, and restricting the use and disclosure of that protected information. In particular, the U.S. Department of Health and Human Services promulgated patient privacy rules under the Health Insurance Portability and Accountability Act of 1996, or HIPAA. These privacy rules protect medical records and other personal health information by limiting their use and disclosure, giving individuals the right to access, amend and seek accounting of their own health information and limiting most use and disclosures of health information to the minimum amount reasonably necessary to accomplish the intended purpose. If we are found to be in violation of the privacy rules under HIPAA or similar laws, we could be subject to civil or criminal penalties.
Transparency of payments
A section of the Affordable Care Act known as the Sunshine Act requires applicable manufacturers of drugs and devices to report annually for publication certain payments and other transfers of value to physicians and teaching hospitals as well as certain ownership interests held by physicians. Pursuant to recently issued regulations, applicable manufacturers, including the Company, began to submit reports in 2014 with respect to payments and transfers occurring in 2013. Reports are now due annually. Certain states and foreign countries have similar statutes and regulations requiring reporting of payments to healthcare providers. We are establishing processes and procedures to capture and report payments to physicians and teaching hospitals.
Other Regulation
The Dodd-Frank Wall Street Reform and Consumer Protection Act includes certain disclosure requirements regarding the use of “conflict minerals” originating from the Democratic Republic of Congo and adjoining countries and procedures regarding a manufacturer’s efforts to prevent the sourcing of “conflict minerals” whether or not these products are manufactured by third parties. The conflict minerals include tin, tantalum, tungsten and gold, and their derivatives. These new requirements could affect the pricing, sourcing and availability of minerals used in the manufacture of our products. There will be additional costs associated with complying with the disclosure requirements, such as costs related to determining the source of any conflict minerals used in our products. Our supply chain is complex and we may be unable to verify the origins for all metals used in our products. We may also encounter challenges with our customers and stockholders if we are unable to certify that our products are conflict free.
Third Party Reimbursement
In the United States, hospitals and doctors generally rely on third-party payers, such as Medicare, Medicaid, private health insurance plans and self-funded employers, to pay or reimburse for all or part of the cost of medical devices and the related surgical procedures. In the United States, heart failure represents Medicare’s greatest area of spending.
16
In 2011, the Center for Medicare and Medicaid Services, or CMS, established reimbursement rates for the treatment of patients with LVADs, with major complications and comorbidities (“MS-DRG 1”) and without major complications and comorbidities (“MS-DRG 2”). Most patients that receive VADs and all patients that receive heart transplants are eligible for MD-DRG 1 reimbursement. Under current payment rates, the national average Medicare payment to CMS-certified centers for MS-DRG 1 procedures is approximately $150,000. Actual payments are subject to other variables such as an application of a wage index, indirect medical education costs, cost outliers, and disproportionate share payments for each institution. In addition, when VAD patients are discharged from the hospital and then readmitted for transplantation, hospitals may qualify for 2 separate MS-DRG 1 or 2 payments.
We believe that our products are Medicare-eligible and therefore that they should be entitled to reimbursement. On October 30, 2013, CMS issued a Decision Memo for Ventricular Assist Devices for Bridge-to-Transplant and Destination Therapy (CAG-00432R), which updated the national coverage determination (“NCD”) for bridge-to-transplant and destination therapy VADs. The updated NCD, among other things, attempts to clarify BTT and DT patient selection criteria. The updated NCD clarified that BTT patients, without an exemption, must be active on the Organ Procurement and Transplantation Network’s waitlist for a heart in order to be eligible for Medicare or Medicaid reimbursement. Since the HVAD System is currently only approved in the U.S. for BTT patients, this update to the NCD creates a subset of potential HeartWare BTT patients who may no longer be eligible for Medicare and Medicaid reimbursement.
Most private insurance providers have implemented U.S. policies for circulatory assist devices, including Blue Cross and Blue Shield Plans, Aetna, Cigna, United Healthcare and others, and these private insurance providers have their own reimbursement criteria for their members, which can be found in their VAD medical policies. Generally, these third-party payors do not impose the active listing requirements contained in the NCD. However, they may not cover medical devices during an ongoing clinical trial; even though a trial may be categorized as a Medicare approved IDE Category B2 clinical trial. All of our sites in the U.S. bridge-to-transplant and destination therapy clinical trials received Medicare and third party reimbursement to some extent. Our staff includes reimbursement and government policy professionals whose objectives include improving insurance reimbursement outcomes for the HVAD System.
International reimbursement varies from country to country and often hospital to hospital. The European system is more effective at focusing resource intensive procedures in a small number of centers within each country and LVADs fall into that category of resource intensive procedures. In those hospitals that perform VAD implantation, we believe that there are adequate budgets to purchase circulatory assist devices although governmental austerity programs could impact available funding. As in the United States, we believe that in Europe physicians and patients drive the decision as to which VAD to purchase. In many jurisdictions, a favorable health technology assessment is required prior to obtaining reimbursement for a medical device. These assessments are often difficult to conduct and can be time consuming and expensive.
Competition
Competition in the VAD industry is expected to increase as better devices become available. We believe that our products compete primarily on their safety and efficacy as a treatment for congestive heart failure as compared to other devices and other treatments. Other factors that affect our ability to effectively compete in the VAD market is our ability to obtain necessary regulatory approvals to market the device in the U.S., the price of our device and the ability of healthcare providers to secure reasonable reimbursement rates. We believe that over time, smaller, less invasive, reliable and durable devices will emerge as the preferred alternatives for the treatment of congestive heart failure. In the long run, we believe our continued competitive success will depend on our ability to enhance patient outcomes and develop innovate products.
Our principal competitors in the implantable cardiac assist space include Thoratec Corporation, Jarvik Heart, Inc., ReliantHeart Inc., Berlin Heart GmbH, and Sunshine Heart, Inc., and a range of other smaller, specialized medical device companies with devices at varying stages of development.
17
Research and Development
Research and development costs include activities related to the research, development, design, testing, and manufacturing of prototypes of our products as well as costs associated with certain clinical and regulatory activities. We expect our research and development expenses to continue to increase as we continue to carry out research and develop improvements to the HVAD System, research the application of, and develop our MVAD heart pump technology as well as the SYNERGY System in a variety of designs and variations, enhance our peripheral product offerings, conduct additional pre-approval and post-approval clinical trials and hire additional employees. For the years ended December 31, 2012, 2013 and 2014 we incurred research and development expenses of $83.5 million, $102.5 million, and $122.4 million respectively.
Manufacturing and Assembly
Our manufacturing activities to date,and for the foreseeable future, will continue to consist primarily of process development, component assembly, quality control testing and sustaining engineering. Most of the components of the HVAD System are manufactured by third parties, including the center post, pump housing and impeller. Some critical components, including the controller, are manufactured solely by an outside supplier and are essentially provided to us as a finished good ready-for-sale as part of our HVAD System.
In order to sell our product commercially in the European Union, we are required to meet certain regulatory standards. In October 2008, we received a Certificate of Registration from British Standard Institution (BSI) certifying that the Company’s Quality Management System complies with the requirements of ISO 13485:2003. It signifies that HeartWare has established a comprehensive quality system that conforms to the International Organization for Standardization (“ISO”) 13485:2003 requirements. The ISO 13485:2003 standard is fully recognized in many countries as a measure of quality. In January 2009, we received a Full Quality Assurance Certificate, CE 540273 from BSI. It signifies that the HVAD System designed and manufactured by HeartWare conforms with the provisions of Council Directive for Active Implantable Medical Devices, 90/385/EEC, Annex 2, Section 3.2 at every stage, from design to final controls. In order to maintain these certifications we must show through annual surveillance audits conducted by the British Standard Institute (BSI) that HeartWare’s Quality System remains compliant with the requirements of ISO 13485 and applicable standards. Our Miami Lakes facility was inspected and approved by the FDA prior to the FDA’s approval to manufacture the HVAD System in the U.S. Our new Miami Lakes facility was inspected and approved by the FDA in June 2013.
We do not presently have supply agreements with many of our key suppliers and we have not secured second source suppliers for all of our supplies.
Employees
As of December 31, 2014, we had 585 employees, of whom 399 employees are engaged in operations activities including research and development, quality assurance and manufacturing, 119 are engaged in marketing, sales, clinical and regulatory activities and 67 are engaged in finance, legal and other administrative functions. None of our employees are represented by a labor union or covered by a collective bargaining agreement other than employees in France that are subject to national, collective bargaining agreements. We consider our relations with our employees to be good.
Corporate History
HeartWare International, Inc. was incorporated in Delaware on July 29, 2008 as a wholly-owned subsidiary of HeartWare Limited, a corporation incorporated in Australia on November 26, 2004. On November 13, 2008, HeartWare Limited completed its redomiciliation from Australia to Delaware pursuant to certain schemes of arrangement approved by an Australian court. In connection with this redomiciliation, each holder of HeartWare Limited ordinary shares was issued one share of HeartWare International, Inc. common stock in exchange for
18
every 35 ordinary shares of HeartWare Limited. As a result, HeartWare Limited became a wholly-owned subsidiary of HeartWare International, Inc., and HeartWare International, Inc. became the parent company of the HeartWare Group. The ordinary shares of HeartWare Limited traded on the on the ASX from January 31, 2005 until November 13, 2008 when the interests in HeartWare International, Inc. started trading on the ASX in the form of CHESS Depositary Interests, or CDIs. On February 24, 2009, common shares of HeartWare International, Inc. were listed for trading on the NASDAQ Global Market and commenced trading on the following day. On September 17, 2013, HeartWare was officially delisted from the ASX meaning that interests in HeartWare are no longer publicly traded on the ASX.
HeartWare Limited became the parent of our operating subsidiary, HeartWare, Inc., on January 24, 2005. HeartWare, Inc. is a Delaware corporation which was incorporated on April 8, 2003 under the name Perpetual Medical, Inc., and which changed its name to HeartWare, Inc. on July 10, 2003. Since July 10, 2003, HeartWare, Inc. has operated the business formerly owned and operated by Kriton Medical, Inc., which had been developing the HeartWare System since approximately 1995. In May 2003, Kriton filed for protection from creditors under Chapter 11 of the United States Bankruptcy Code. On July 10, 2003, HeartWare, Inc. purchased substantially all of the assets of Kriton free and clear of any and all liens, security interests, encumbrances and claims.
19
Item 1A. Risk Factors
Our business faces many risks. We believe the risks described below are material risks facing the Company. However, these risks may not be the only risks we face. Additional unknown risks, or risks that we currently consider immaterial, may also impair our business operations. If any of the events or circumstances described below actually occurs, our business, financial condition or results of operations could suffer, and the trading price of our shares could decline significantly. Investors should consider the specific risk factors discussed below, together with the cautionary statements under the caption “Forward-Looking Statements” and the other information and documents that we file from time to time with the Securities and Exchange Commission.
Risks Related to Our Financial Standing
We have incurred operating losses since our inception and anticipate that we will continue to incur operating losses for the foreseeable future. Our ability to achieve profitability from a current net loss level will depend on our ability to increase gross revenue, manage our expenditures and reduce the per unit cost of producing the HVAD System by increasing our customer orders and manufacturing volume.
We have incurred net losses since our inception, including net losses of $19.4 million, $59.3 million and $87.7 million for the fiscal years ended December 31, 2014, 2013 and 2012, respectively. As of December 31, 2014, our accumulated deficit was $348.7 million. Currently, we only have one product, the HVAD System, approved for sale. We continue to incur substantial clinical trial expenditures, significant research and development costs and costs related to our operations. We expect to continue to incur significant operating losses for the foreseeable future as we incur costs associated with:
| | • | | conducting multiple clinical trials, including preapproval trials for new products, such as MVAD, and new indications, such as destination therapy, and post approval trials for existing products; |
| | • | | researching and developing next generation products and peripherals, such as the CircuLite SYNERGY System, as well as incremental improvements to and sustaining engineering for existing products and peripherals; |
| | • | | integrating and developing acquired and licensed technology; |
| | • | | building our service capabilities to meet growing customer demand; |
| | • | | growing, maintaining and protecting our intellectual property; |
| | • | | seeking and maintaining regulatory approvals and operating our quality systems; |
| | • | | expanding our sales and marketing capabilities both internationally and in the U.S.; |
| | • | | manufacturing product and increasing our manufacturing capabilities to meet rising demand; |
| | • | | broadening our infrastructure in order to meet the needs of our growing operations; and |
| | • | | complying with the requirements related to being a public company in the United States. |
To become and remain profitable, we must succeed in a range of challenging activities, including all of the activities listed above. We also must, among other things, continue to reduce the per unit cost of our products. If we are unable to increase sales and simultaneously reduce assembly, raw material, component and manufacturing overhead costs, our ability to achieve profitability will continue to be constrained. If we do achieve profitability, we may not be able to sustain it.
20
We have a significant amount of indebtedness consisting primarily of our convertible senior notes. We may not be able to generate enough cash flow from our operations to service or pay principal and interest on our indebtedness, which could adversely affect our business, financial condition or results of operations. Furthermore, we may incur additional indebtedness or refinance our current indebtedness in the future, which could also adversely affect our business, financial condition or results of operations. The conversion of our convertible senior notes at the election of the holders, to the extent we settle the conversion in cash, could impact our liquidity and to the extent we settle in stock, could dilute our existing stockholders.
As of December 31, 2014, our indebtedness under our 3.5% Convertible Senior Notes due December 15, 2017 in the principal amount of $143.75 million totaled $114.8 million, net of discounts. Generally, holders may convert their Convertible Notes at their option only upon satisfaction of one or more of the conditions relating to the sale price of our common stock, the trading price per $1,000 principal amount of Convertible Notes or specified corporate events. Upon conversion, we will pay or deliver, as the case may be, cash, shares of our common stock or a combination thereof, at our election. Our ability to make payments on, or to refinance, our Convertible Notes, to incur future indebtedness, and to fund planned capital expenditures, research and development efforts, working capital, acquisitions and other general corporate purposes, depends on our ability to generate or access cash in the future. This, to a certain extent, is subject to general economic, financial, competitive, legislative, regulatory, clinical and other factors, some of which are beyond our control. If we do not generate sufficient cash flow from operations, or if future borrowings are not available to us in an amount sufficient to pay our indebtedness, including payments of principal upon conversion of the Convertible Notes or upon their maturity, or to fund our liquidity needs, we may be forced to refinance all or a portion of our indebtedness, including the Convertible Notes, on or before their maturity, sell assets, reduce or delay capital expenditures, seek to raise additional capital or take other similar actions. We may not be able to affect any of these actions on commercially reasonable terms or at all. Our ability to refinance our indebtedness will depend on our financial condition at the time, the restrictions in the instruments governing our indebtedness and other factors, including market conditions. In addition, in the event of a default with respect to the Convertible Notes, the holders of the Convertible Notes and/or the trustee under the indenture governing the notes may accelerate the payment of our obligations under the notes, which could have a material adverse effect on our business, financial condition or results of operations. Our inability to generate sufficient cash flow to satisfy our debt service obligations, or to refinance or restructure our obligations on commercially reasonable terms or at all, would likely have an adverse effect, which could be material, on our business, financial condition and results of operations.
In addition, our significant indebtedness combined with our other financial obligations and contractual commitments could have other important consequences. For example, it could:
| | • | | make us more vulnerable to adverse changes in general U.S. and worldwide economic, industry and competitive conditions; |
| | • | | make us more vulnerable to adverse changes in government regulation and reimbursement; |
| | • | | limit our flexibility in planning for, or reacting to, changes in our business and our industry; |
| | • | | place us at a competitive disadvantage compared to our competitors who have less debt; and |
| | • | | limit our ability to borrow additional amounts for working capital, capital expenditures, acquisitions, debt service requirements, execution of our business strategy or other purposes. |
Any of these factors could materially and adversely affect our business, financial condition or results of operations. In addition, if we incur additional indebtedness, which we are not prohibited from doing under the terms of the indenture governing the convertible senior notes, the risks related to our business and our ability to service our indebtedness would increase.
In the event the conditional conversion feature of the notes is triggered, holders of the Convertible Notes will be entitled to convert the Convertible Notes at any time during specified periods at their option. If one or more holders elect to convert their Convertible Notes, unless we elect to satisfy our conversion obligation by delivering
21
solely shares of our common stock (other than cash in lieu of any fractional share), we would be required to settle a portion or all of our conversion obligation through the payment of cash, which could adversely affect our liquidity. In addition, even if holders do not elect to convert their notes, we could be required under applicable accounting rules to reclassify all or a portion of the outstanding principal of the notes as a current rather than long-term liability, which would result in a material reduction of our net working capital. The terms of our convertible senior notes permit us to settle them, upon conversion by the holders thereof, in cash, stock, or a combination thereof. To the extent we use stock for settlement, our existing stockholders may be diluted.
We may need substantial additional funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts.
We currently spend more cash than we generate from operating revenue. Depending on a range of outcomes, especially our achievement and continuation of regulatory approval of our products and the growth of revenue, we may need to seek additional funding in the future. Additional funding may not be available on terms favorable to us, or at all. If we raise additional funding through the issuance of equity or convertible debt securities, our shares may suffer dilution. If we are unable to secure additional funding, our product development programs and our commercialization efforts would be delayed or reduced or may cease entirely.
We currently rely entirely on sales of our sole product, the HVAD System, to generate revenue. Our existing and future products may not achieve or sustain market acceptance. In addition, any factors that negatively impact sales of this product will adversely affect our business, financial condition and results of operations.
Our sole marketed product is the HVAD System, which we introduced to the European market in January 2009 and which received regulatory approval in the U.S. in late 2012. We expect to continue to derive substantially all of our revenue for the next few years from the sale of this product and its related devices. Accordingly, our ability to generate revenue is entirely reliant on our ability to market and sell this product. We expect to begin to derive limited revenue as a result of MVAD systems purchased by participating clinical trial sites pursuant to a qualified U.S. IDE study; however, clinical trials of the next generation MVAD System may adversely impact revenue derived from the current generation HVAD System.
Even if we are able to obtain and maintain the necessary regulatory approvals in all jurisdictions to commercialize the HVAD System or other products that we may develop, our products may not gain or sustain market acceptance among physicians, patients, health care payers or the medical community.
The degree of market acceptance of any of the devices that we may develop and commercialize will depend on a number of factors, including: the prevalence and severity of any adverse events or side effects especially as it relates to survival, quality of life, and patient management; potential advantages over alternative treatments or competitive products; the strength and perceived advantages of our peripherals such as the monitor, controller and batteries; and sufficient third party coverage or reimbursement.
If we fail to obtain and maintain adequate level of reimbursement for our products by third party payers, there may be no commercially viable markets for our products or the markets may be much smaller than expected.
Although our customers have generally achieved reimbursement for the purchase of our products to date, the availability and levels of reimbursement by governmental and other third party payers affect the market for our products. Reimbursement and health care payment systems vary significantly by country, and include both government sponsored health care and private insurance. Payers may attempt to limit coverage and the level of reimbursement of new therapeutic products or experimental devices. Government and other third party payers also continually attempt to contain or reduce the costs of health care by challenging prices charged for health care products and services. Often, reimbursement is determined independently of and only following product approval, and may need to be renewed on a regular basis.
22
To obtain reimbursement or pricing approval in some countries, we may be required to produce clinical and economic data, which may involve one or more clinical trials, that compares the cost-effectiveness of our products to other available therapies. In addition, the efficacy, safety, performance and cost-effectiveness of our products in comparison to any competing products may determine the availability and level of reimbursement for our products.
We believe that future reimbursement may be subject to increased restrictions both in the United States and in international markets. Future legislation, regulation or reimbursement policies of third party payers may adversely affect the demand for our existing products as well as products currently under development and limit our ability to sell our products on a profitable basis. We cannot predict how pending or future legislative and regulatory proposals would influence the manner in which medical devices, including ours, are purchased or covered and reimbursed.
On October 30, 2013, CMS issued a Decision Memo for Ventricular Assist Devices for bridge-to-transplant (“BTT”) and destination therapy (“DT”) (CAG-00432R), which updated the national coverage determination (“NCD”) for bridge-to-transplant and destination therapy VADs. The updated NCD, among other things, attempts to clarify BTT and DT patient selection criteria. The updated NCD clarified that BTT patients, without an exemption, must be active on the Organ Procurement and Transplantation Network’s waitlist for a heart in order to be eligible for Medicare or Medicaid reimbursement. Since the HVAD System is currently only approved in the U.S. for BTT patients, the update to the NCD creates a subset of potential HeartWare BTT patients who may no longer be eligible for Medicare and Medicaid reimbursement. The update also mandates affiliations with heart transplant centers, which limits VAD implantation at alternate care facilities. Therefore, the update may adversely affect our revenue, earnings, business or results of operation.
If reimbursement for our products is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels, market acceptance of our products would be impaired and our future revenue would be materially adversely affected. Often reimbursement is not available for products used in clinical trials as the relevant insurance providers may refuse to provide reimbursement for trial products on the basis that the products are “experimental” or “investigational” and do not have the requisite regulatory approvals. As we develop next generation products, this requirement may materially adversely affect our revenue, earnings, business and stock price.
Investors could lose confidence in our financial reports, and the value of our shares may be adversely affected, if our internal controls over financial reporting are found not to be effective by management or by our independent registered public accounting firm or if we make disclosure of existing or potential significant deficiencies or material weaknesses in those controls.
Management’s assessment of our internal controls over financial reporting is discussed in Item 9A of this Annual Report on Form 10-K for the year ended December 31, 2014. The Company’s management, with the participation of the Company’s Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of the Company’s disclosure controls and procedures, and internal control over financial reporting as of December 31, 2014. Based on that evaluation, the Company’s Chief Executive Officer and Chief Financial Officer concluded that the Company’s disclosure controls and procedures, and internal control over financial reporting are effective as of December 31, 2014. Our independent registered public accounting firm has issued their attestation report on our internal control over financial reporting, which is included in Item 8 of this Annual Report on Form 10-K for the year ended December 31, 2014.
We continue to evaluate our existing internal controls over financial reporting against the standards adopted by the Public Company Accounting Oversight Board, or PCAOB. During the course of our ongoing evaluation of the internal controls, we may identify areas requiring improvement and will design enhanced processes and controls to address any issues identified through this review. As we continue to commercialize our products, we will need to enhance our accounting and financial controls functions, particularly as they relate to accounting for
23
revenue and inventory, and we will need to add more personnel to our financial reporting group. Remediating any deficiencies, significant deficiencies or material weaknesses that have been or could be identified by us or our independent registered public accounting firm may require us to incur significant costs and expend significant time and management resources. We cannot assure you that any of the measures we implement to remedy any deficiencies will effectively mitigate or remedy deficiencies. The existence of one or more deficiencies or weaknesses could affect the accuracy and timing of our financial reporting. Investors could lose confidence in our financial reports, and the value of our shares may be adversely affected if our internal controls over financial reporting are found not to be effective by management or by our independent registered public accounting firm or if we make disclosure of existing or potential significant deficiencies or material weaknesses in those controls.
Risks Related to Regulatory Approval
In November 2012, we received approval to market the HVAD System in the U.S. as a bridge to heart transplantation. Our future success depends heavily on our ability to maintain FDA approval to market our existing product for our initial indication, obtain FDA approval for additional indications, and to market our pipeline products in the U.S. and around the world.
The process of obtaining and maintaining marketing approval or clearance from the FDA and other regulatory authorities for our existing and future products, or enhancements or modifications to these products, could:
| | • | | take a significant period of time; |
| | • | | require the expenditure of substantial resources; |
| | • | | involve rigorous pre-clinical and clinical testing; |
| | • | | require changes to our products; |
| | • | | require corrective action, including recalls, with respect to products already distributed; and |
| | • | | result in limitations on the indicated uses of the products. |
Assuming we are able to file the required regulatory premarket approval applications for additional indications for our HVAD System as well as for the MVAD System and other pipeline products, there can be no assurance that we will receive the required approvals , or, if we do receive the required approvals, that we will receive them on a timely basis or that we will otherwise be able to satisfy the conditions of approval, if any. In particular, because of the FDA warning letter received by HeartWare in June 2014, we are under additional regulatory scrutiny, which could increase the risk of delay. The failure to receive product approval by the FDA, or any significant delay in receipt, will have a material adverse effect on our business, financial condition or results of operations.
If we are unable to commence or complete successfully our clinical trials, or if we experience significant delays in the successful commencement or completion of our clinical trials, our ability to obtain regulatory approval to commercialize our products, and our ability to generate revenue, will be materially adversely affected.
Regulatory approvals to sell our existing and future products both in and outside of the U.S. typically require clinical trials, which can be time consuming, unpredictable and expensive. Significant technical, bench and preclinical testing may be required prior to submitting for regulatory authorization to commence a clinical trial. The cost, timing and outcome of any of these trials or testing may be unfavorable or may be insufficient to obtain the required approvals.
Completion of any of our clinical trials, including our ongoing destination therapy and thoracotomy trials, and initiation of new clinical trials, including our trials for our next generation MVAD System and trials for the
24
SYNERGY System, could be delayed or adverse events during a trial could cause us to amend, repeat or terminate the trial. If this were to happen, our costs associated with the trial will increase, and it will take us longer to obtain regulatory approvals and to commercialize the product, or we may never obtain regulatory approvals. Our clinical trials may also be suspended or terminated at any time by regulatory authorities, the data safety and monitoring board, site investigational review boards, or by us including during the closing stages of enrollment of the trial and the subsequent patient data follow-up period in the event that, for example, there should be an unacceptable level of adverse clinical events such as stroke, bleeding or pump exchanges. Any failure or significant delay in completing clinical trials for our products may materially harm our financial results and the commercial prospects for our products.
The completion of any of our clinical trials could be substantially delayed or prevented by several factors, including: slower than expected rates of patient recruitment and enrollment, including as a result of study inclusion and exclusion criteria; our competitors undertaking similar clinical trials at the same time as us, or having functionally comparable products that have received approval for sale; physicians or patients preferring to use approved devices or other experimental treatments or devices rather than our devices; prevalence and severity of adverse events, such as thrombus or stroke rates, and other unforeseen safety issues; and governmental and regulatory delays or changes in regulatory requirements, policies or guidelines.
Our manufacturing facilities and the manufacturing facilities of our suppliers must comply with applicable regulatory requirements. If we or our suppliers fail to achieve and maintain regulatory approval for these or additional manufacturing facilities, our business and our results of operations would be harmed.
Completion of our clinical trials and commercialization of our products require access to, or the development of, manufacturing facilities that meet and maintain applicable U.S. and international regulatory standards to manufacture a sufficient supply of our products. In addition, the FDA must approve facilities that manufacture our products for U.S. commercial purposes, as well as the manufacturing processes and specifications for the product, with similar, additional approvals required in order to achieve and maintain CE marking in Europe or regulatory approvals in other jurisdictions. Suppliers of components and products used to manufacture our products must also comply with FDA and foreign regulatory requirements, which often require significant time, money, resources, record-keeping and quality assurance efforts and subject us and our suppliers to potential regulatory inspections and stoppages. If we or our suppliers fail to comply with the regulatory requirements for our manufacturing operations, our commercialization efforts could be delayed or suspended, which would harm our business and our results of operations. Our receipt of an FDA warning letter increases this risk as we are under a higher level of regulatory scrutiny as we work to update and improve our quality system.
We may not meet regulatory quality standards applicable to our manufacturing and quality processes, which could have an adverse effect on our business, financial condition or results of operations.
Even after products have received marketing approval or clearance from the FDA or other regulatory bodies, those product approvals and clearances can be withdrawn due to failure to comply with regulatory standards or the occurrence of problems following initial approval whether identified through a required post-approval study or through medical device reporting. As a device manufacturer, we are required to demonstrate and maintain compliance with a variety of regulatory requirements, including the FDA’s Quality System Regulation, or “QSR.” The QSR is a complex regulatory scheme that covers the methods and documentation of the design, testing, control, manufacturing, labeling, quality assurance, packaging, storage and shipping of our products, including trend analysis and corrective and preventative actions. In addition, the U.S. federal medical device reporting regulations require us to provide information to the FDA whenever there is evidence that reasonably suggests that a device may have caused or contributed to a death or serious injury or, if a malfunction were to occur, could cause or contribute to a death or serious injury. Compliance with applicable regulatory requirements is subject to continual review and is rigorously monitored through periodic inspections by the FDA. Our failure to comply with the QSR or to take satisfactory corrective action in response to an adverse QSR
25
inspection or complaint information could result in enforcement actions, including a public warning letter, a shutdown of or restrictions on our manufacturing operations, delays in approving or clearing a product, refusal to permit the import or export of our products, a recall or seizure of our products, fines, injunctions, civil or criminal penalties, or other sanctions, any of which could cause our business and operating results to materially suffer. The FDA has issued and could issue in the future warning letters or other communications to the Company. If we fail to satisfy or remediate the matters discussed in warning letters or communications, the FDA could take further enforcement actions as described above. Further, related remediation could require a substantial amount of money and time and could precipitate field actions, all of which could harm our business and our results of operations.
In the European Union, we are required to maintain certain ISO certifications in order to sell our products and must undergo periodic inspections by notified bodies to obtain and maintain these certifications. If we fail to continue to comply with ISO regulations, European Union organizations may withdraw clearance to market, require a product recall or take other enforcement action.
Product deficiencies could result in field actions, recalls, substantial costs and write-downs; this could also lead to delay or termination of ongoing trials.
Our products are subject to various regulatory guidelines, involve complex technologies and are approved for a specified life. Identified quality problems, such as failure of critical components including batteries, controllers or driveline connectors, or the failure of third parties to supply us with sufficient conforming quantities of these products or components, could lead to adverse clinical events that could cause us to amend, repeat or terminate clinical trials, or impact the availability of our product in the marketplace. In addition, product improvements, product redundancies or failure to sell product before it expires could result in scrapping or expensive rework of product and our business, financial or results of operations could suffer. Product complaints, quality issues and necessary corrective and preventative actions could result in communications to customers or patients, field actions, the scrapping, rework, recall or replacement of product, substantial costs and write-offs, and harm to our business reputation and financial results. Further these activities could adversely affect our relationships with our customers or affect our reputation which could materially adversely affect our earnings, results and financial viability.
We operate in multiple regulatory environments that require costly and time consuming approvals.
Even if we obtain regulatory approvals in specific jurisdictions to commercialize the HVAD System or other products that we may develop, sales of our products in other jurisdictions will be subject to regulatory requirements that vary from country to country. The time and cost required to obtain approvals from these countries may be longer or shorter than that required for FDA approval, and requirements for licensing may differ from those of the FDA. Some jurisdictions, such as Japan, may even require that we conduct additional trials. Laws and regulations regarding the manufacture and sale of our products are subject to future changes, as are administrative interpretations and policies of regulatory agencies. If we fail to comply with applicable foreign, federal, state or local market laws or regulations, we could be subject to enforcement actions. Enforcement actions could include product seizures, recalls, withdrawal of clearances or approvals, and civil and criminal penalties, which in each case would harm our business.
Risks Related to Operations
We have limited manufacturing capabilities and personnel, and if our manufacturing facilities are unable to provide an adequate supply of products, our growth could be limited and our business could be harmed.
We currently manufacture our HVAD System at our facilities in Miami Lakes, Florida. If there were a disruption to our manufacturing facilities or the surrounding area, for example, due to a hurricane, we would have no other means of manufacturing our HVAD System until we were able to restore the manufacturing capability at our facility or develop alternative manufacturing facilities.
26
If we are unable to produce sufficient quantities of our HVAD System for sale or for use in our current and planned clinical trials, or if our manufacturing process yields substandard product, our development and commercialization efforts would be delayed. In addition, if a regulatory authority approves or clears a new product or a new indication for the HVAD System, we may not be able to manufacture such product in the quantities needed to meet the increased commercial demand on a timely basis. Even if we are able to produce sufficient quantities of our products, we may not be able to attain sufficient profitability on that production or any resultant sales.
We currently have limited resources, facilities and experience to commercially manufacture our products. In order to produce our products in the quantities that we anticipate will be required to meet anticipated market demand, we will need to increase the production process and efficiency over the current level of production. There are significant technical and regulatory challenges to increasing manufacturing capacity and efficiency, and continuing to develop commercial-scale manufacturing facilities will require the investment of additional funds and hiring and retaining additional management and technical personnel who have the necessary manufacturing and operational experience. We may not successfully complete any required increase in a timely or economically viable manner, or at all. If we are unable to do so, we may not be able to produce the HVAD System or other products that we may develop, in sufficient quantities to meet future demand.
If we are unable to manufacture a sufficient or consistent supply of the HVAD System or other products we are developing, or if we cannot do so efficiently or pursuant to regulatory standards, our revenue, business and financial prospects would be adversely affected.
We rely on specialized suppliers for certain components and materials, and we do not have second-source suppliers for all of our components.
We depend on a number of suppliers to successfully manufacture sufficient quantities of the components we use in our products, both our existing commercial products and our products in development. We rely on suppliers for various critical components including the center post, housing and impeller that are assembled into our primary product, the HVAD System, as well as finished products that comprise our peripheral and external equipment included in the HVAD System. Similar relationships exist in relation to our MVAD System. Lead times for our components are significant and can be up to as long as sixteen weeks and many of our components are manufactured to very tight tolerances and specifications. We do not presently have supply agreements with the vast majority of our key suppliers but have extensive purchase orders in place with these vendors.
We have second-source suppliers for some, but not all, of our components. In particular, we do not have second-source suppliers for our controllers, battery chargers and monitors. Our reliance on third-party suppliers also subjects us to other risks that could harm our business, including:
| | • | | we do not believe that we are a major customer of some of our suppliers, in terms of the volume of components and materials that we purchase, and these suppliers may therefore give other customers’ needs higher priority than ours or discontinue or modify components based on demand from other customers; |
| | • | | we may not be able to obtain adequate supply in a timely manner or on commercially reasonable terms; |
| | • | | some of our components are extraordinarily complex and must be manufactured to extremely tight tolerances and specifications with the result that our suppliers, especially new suppliers, may make errors in manufacturing that could negatively affect the efficacy or safety of our products or cause our components not to be delivered on time or at all or to be delivered outside of our specifications; |
| | • | | the availability of second-source suppliers may be extremely limited or their implementation as a supplier may be lengthy due to the tight tolerances and specifications in which we typically operate; |
| | • | | switching components or changes to our components, specifications or designs may require product redesign and regulatory submissions, which can lead to production interruptions; and |
27
| | • | | our suppliers may encounter financial hardships unrelated to our demand, which could inhibit their ability to fulfill our orders and meet our requirements. |
Any interruption or delay in obtaining products from our third-party suppliers, or our inability to obtain products from alternate sources at acceptable prices in a timely manner, could impair our ability to meet the demand of our customers and cause them to cancel orders or switch to competing products.
While we have identified second-source suppliers for other key components, we have not entered into written agreements with these suppliers and we cannot be certain that we will be able to maintain our manufacturing schedule without undue delay or substantial cost if any of these arrangements is terminated.
Additionally, we may experience problems or delays in our own manufacturing and assembly processes, which may be harmful to our financial status or reputation and, therefore, make it more difficult or expensive for us to continue with or enter into relationships with specialized suppliers. Our business plan is predicated on maintaining strong relationships and favorable supply arrangements with a series of external parties to manufacture components of our HVAD System and other products we may develop, such as the MVAD System. If we are unsuccessful in this regard or are unable to secure or maintain agreements with these manufacturers on favorable terms or at all, then our ability to commercialize our technology and expand our operations will be dramatically impaired.
We use external consultants and engineers to help us develop new products and peripherals as well as components for our current products and peripherals. Many of these external projects anticipate innovation or technology development which does not currently exist and are funded on a time and materials basis. As a result, the ability of third party contractors to develop the necessary technology in accordance with established budgets and timelines is uncertain. Failure to timely execute a development program could result in the delay or abandonment of a new product or component or increase the cost of the activity. These outcomes could adversely impact our business, prospects and financial condition.
Risks Related to Future Growth
If we are unable to manage our expected growth, we may not be able to meet market demand, generate expected benefits from the opportunities available to us, satisfy quality regulations or commercialize our products.
We expect to continue to expand our operations and grow our research and development, product development, quality, regulatory, manufacturing, sales, marketing and administrative operations. This expansion has placed, and is expected to continue to place, a significant strain on our management, infrastructure, information technology, operational and financial resources. To manage continued growth and to commercialize our products, we will be required to improve existing operational, quality and financial systems, procedures and controls and expand, train and manage our employee base. In addition, we will need to manage relationships with various third parties and external entities participating in our research and development efforts, clinical trials, quality systems, manufacturers, suppliers and other organizations, including various regulatory bodies in the U.S. and other jurisdictions. We have begun to implement systemic changes and organizational improvements to address the items raised by the FDA in our warning letter. We may not be able to implement needed improvements in an efficient and timely manner and may discover deficiencies in existing systems and controls, which may require remediation such as field actions. Our failure to accomplish any of these tasks could materially harm our business and prospects.
Further, we have aggressively expanded, and expect to continue to expand, into foreign markets. Expansion into additional foreign markets imposes additional burdens on our small executive and administrative personnel, research, regulatory and sales departments and generally limited managerial resources. Our efforts to introduce our current or future products into additional foreign markets may not be successful, in which case we may have
28
expended significant resources without realizing the expected benefit. Ultimately, the investments required for expansion into additional foreign markets could exceed the returns, if any, generated from this expansion. The taxation and customs requirements, together with other applicable laws and regulations of foreign jurisdictions, can be inherently complex and subject to differing interpretation by local authorities. We are subject to the risk that either we have misinterpreted applicable laws and regulations or that foreign authorities may take inconsistent, unclear or changing positions on local law, customs practices or rules. In the event that we have misinterpreted local requirements, or that foreign authorities take positions contrary to ours, we may incur liabilities that may differ materially from the amounts accrued in our financial statements.
Destination therapy procedures represent an increasing share of ventricular assist device implants. Although we are currently conducting destination therapy trials, we may be unable to submit for approval of a destination therapy indication for several years.
Hospitals must meet specific regulatory or reimbursement requirements in order to perform destination therapy procedures. These requirements and national coverage determinations may change from time to time. We are currently conducting clinical trials to study the use of the HVAD System for destination therapy. If reimbursement is reduced or unavailable for clinical trial cases or physicians decline to use our products for destination therapy in the future, our market opportunities will be diminished and our business and stock price may be adversely impacted. The number of destination therapy procedures actually performed depends on many factors, most of which are out of our direct control, including:
| | • | | the number of sites approved for destination therapy by relevant regulatory agencies; |
| | • | | the clinical outcomes, and physician presentations of clinical outcomes, of our destination therapy trials and destination therapy procedures, in general; |
| | • | | implanting surgeons’ and referring cardiologists’ commitment to destination therapy; |
| | • | | the economics of ventricular assist devices for destination therapy at individual hospitals, which includes the costs of the VAD and related pre- and post-operative procedures and treatment and their reimbursement; and |
| | • | | the economics of hospitals not conducting a destination therapy procedure, including the costs and related reimbursements of long-term hospitalization of advance heart failure patients and alternative therapies. |
The different outcomes of these and other factors, and their timing, may have a material and adverse effect on our future results.
In addition, our primary competitor has received a destination therapy indication for its product. If physicians grow accustomed to that device for destination therapy and become unwilling to use our device for this indication, our ability to participate in and benefit from this opportunity may suffer.
If we fail to successfully introduce next generation products and improvements to our existing product, our future growth may suffer.
As part of our strategy, we intend to develop and introduce a number of next generation products and make enhancements to our existing products. We also intend to develop new indications for our existing products. If we are slow in bringing new products to market or otherwise fail to successfully develop, manufacture, design clinical trials for, introduce or commercialize any of these new products, product improvements or new indications on a timely basis, or if they are not well accepted by the market, our future growth may suffer. For example, we are developing a next generation pump based on our MVAD System technology, designing a new and improved controller and conducting a clinical trial for a thoracotomy placement, among others. If we are not successful in these efforts, among others, our future business opportunities and growth potential will suffer.
29
Business development activities are inherently risky, and integrating our operations with businesses we may acquire may be difficult and, if unsuccessfully executed, may have a material adverse effect on our business.
We may, selectively, from time to time engage in business development activities, such as strategic acquisitions, like our acquisitions of World Heart Corporation and CircuLite, Inc., investments and alliances in order to complement or expand our current business or enter into a new product area. These transactions can involve significant challenges and risks, including that the transaction does not advance our business strategy or fails to produce a satisfactory return on our investment. While our evaluation of any potential acquisition or investment includes business, clinical, legal and financial due diligence with the goal of identifying and evaluating the material risks involved, we may be unsuccessful in ascertaining or evaluating all risks. These plans are also subject to the availability of appropriate opportunities and competition from other companies seeking similar opportunities. Moreover, the success of any strategic effort may be affected by a number of factors, including our ability to properly assess and value the potential business opportunity to finalize the development of the acquired technology and to commercialize successfully the final product.
Each acquisition involves the integration of a separate company that was previously operated independently and has different systems, processes, policies and cultures. The process of combining companies may be disruptive to our businesses and may cause an interruption of, or a loss of momentum in, both of the businesses. Further, these challenges could materially harm our stock price, business, financial condition or results of operations. If we are unable to successfully integrate strategic acquisitions in a timely manner, our business and our growth strategies could be negatively affected. Even if we are able to successfully complete the integration of the operations of other companies or businesses we may acquire in the future, we may not be able to realize all or any of the benefits that we expect to result from the integration, either in monetary terms or in a timely manner.
We may choose to license or acquire products or technologies, in addition to or instead of developing them ourselves. We cannot be certain that these efforts will be successful or that we will realize any revenue from them.
We license or acquire products and technologies under licensing, purchasing and other agreements. In addition to active internal and external research and development efforts, we may seek, from time to time, to license or acquire new products or technologies to supplement or replace products or technologies. License and acquisition of technology involves numerous risks, including: the inability to successfully license or acquire the desired product or technology; the incurrence of significant financial commitments to third parties; and the risks associated with third parties terminating licenses arrangements if we do not perform as required under the agreements.
In addition, third parties may breach or terminate their license agreements with us or fail to conduct their activities in connection with our relationships in a timely manner. If we or our counterparties terminate or breach any of our licenses we may:
| | • | | lose our right to develop and market certain intellectual property; |
| | • | | experience delays in the development or commercialization of our product; |
| | • | | litigate or arbitrate disputes, both of which are time-consuming and expensive and have uncertain outcomes; |
| | • | | incur liability for damages; and |
| | • | | be unable to obtain any other similar licenses on acceptable terms, if at all. |
Any of these factors could materially harm our stock price, business, financial condition or results of operations.
30
Expansion into medical centers that have not historically used our products may incur long sales and training cycles that may cause our product sales and operating results to vary significantly from quarter-to-quarter.
Our VAD systems have lengthy sales cycles and we may incur substantial sales and marketing expenses and expend significant effort without making a sale. Even after making the decision to purchase our VAD systems, our customers often deploy our products slowly, and this time period may be extended if our products are acquired on a consignment basis, as is the case for some of our customers. In addition, cardiac centers that buy the majority of our products are usually led by cardiac surgeons who are heavily recruited by competing centers or by centers looking to increase their profiles. When one of these surgeons moves to a new center we sometimes experience a temporary but significant reduction in purchases by the departed center while it replaces its lead surgeon. As a result, it is difficult for us to predict the quarter in which customers may purchase our VAD systems and our product sales and operating results may vary significantly from quarter to quarter. In addition, product purchases often lag initial expressions of interest in our product by new centers as training of the VAD team and internal hospital administrative procedures are typically required prior to the initial implant procedures.
Risks Related to Competition
We compete against companies that have longer operating histories, more established or approved products and greater resources than we do, which may prevent us from achieving further market penetration or improving operating results.
Competition in the medical device industry is intense. Our products will compete against products offered by public companies, such as Thoratec Corporation and Sunshine Heart, Inc., as well as several private companies, such as Jarvik Heart, Inc. and ReliantHeart Inc. Some of these competitors have significantly greater financial and human resources than we do, have established reputations or approved products or significantly greater name recognition than we do, and have significantly larger and more established distribution channels and sales and marketing capabilities than we do. For example, Thoratec Corporation has received marketing approval in the U.S. for HeartMate II for both destination and bridge-to-transplant indications, whereas the HVAD System is currently only approved for the bridge-to-transplant indication. Thoratec Corporation has also commenced clinical trials for its next generation device prior to our initiation of clinical trials for the MVAD System, our next generation device. We are likely to compete with new companies in the future as additional competitors enter the market. We also face competition from other medical therapies, which may focus on our target markets as well as competition from manufacturers of pharmaceutical treatments and other devices that are currently in development.
Our ability to compete effectively depends upon our ability to distinguish our company and our products from our competitors and their products. Factors affecting our competitive position include:
| | • | | the availability of other products and procedures, such as heart transplants and pharmaceuticals; |
| | • | | product performance and design; |
| | • | | sales, marketing and distribution capabilities; |
| | • | | comparable clinical outcomes; |
| | • | | success and timing of new product development and introductions; |
| | • | | penetration into existing and new geographic markets; and |
| | • | | intellectual property protection. |
31
The competition for qualified personnel is particularly intense in our industry. In addition, we have added or made changes to executive personnel during 2014 and may continue to do so as our needs evolve. If we are unable to retain or hire executive and other key personnel, we may not be able to sustain or grow our business.
Our ability to operate successfully and manage our potential future growth depends significantly upon our ability to attract, retain and motivate highly skilled and qualified research, technical, clinical, regulatory, sales, marketing, managerial, legal and financial personnel. We have hired and expect to continue to hire a substantial number of employees in these areas and others in order to support U.S. commercialization and the expected growth in our global business. During 2014, we filled key open positions, including Chief Medical Officer and Vice President, Quality and Design Assurance. However, we face intense competition for qualified personnel, and we may not be able to attract, retain and motivate these individuals. We compete for talent with numerous companies, as well as universities and non-profit research organizations. Our future success also depends on the personal efforts and abilities of the principal members of our senior management and scientific staff to provide strategic direction, management of our operations and maintenance of a cohesive and stable environment. Although we have employment and incentive compensation agreements with all of our executive officers and incentive and compensation plans for our other personnel providing them with various economic incentives to remain employed with us, these incentives may not be sufficient to retain them. We do not maintain key man life insurance on the lives of any of the members of our senior management. The loss of key personnel for any reason or our inability to hire, retain and motivate additional qualified personnel in the future could prevent us from sustaining or growing our business.
Risks Related to Product Liability
We manufacture a Class III device implanted in the heart that subjects us to numerous risks. Product liability claims could damage our reputation or adversely affect our business.
There are risks associated with implanting our device in end stage heart failure patients, including, but not limited to, death, bleeding, stroke, device malfunction and other adverse events. Should our customers experience an increase in adverse events, they may reduce their usage or purchase of our device. In addition, should our patients experience injury due to these events, they or regulatory authorities may pursue legal or administrative action against us. Any of these occurrences could have a materially adverse impact on our operations and financial results and conditions as well as customer confidence in us or our products. Specifically, product liability and similar claims may be expensive to defend and may result in large judgments against us. A product liability or other damages claim, product recall or product misuse, regardless of the ultimate outcome, could require us to spend significant time and money in litigation or to pay significant damages and could seriously harm our business. We maintain limited clinical trial insurance and product liability insurance. We cannot be certain that insurance will be sufficient to cover all claims that may be made against us. Our insurance policies generally must be renewed on an annual basis. We may not be able to maintain or increase insurance on acceptable terms or at reasonable costs. Claims against us, regardless of their merit, could result in significant awards against us that could materially adversely harm our business, financial condition, results of operations or prospects. A product liability or other damages claim, product recall or product misuse involving any type of VAD, but especially involving one of ours, could also materially and adversely damage our reputation and the perception of VADs generally and affect our ability to attract and retain customers, irrespective of whether or not the claim or action was meritorious.
Risks Related to the Economy and Public Policy Relevant to our Business
Adverse changes in general economic conditions in the United States and overseas could adversely affect us.
We are subject to the risks arising from adverse changes in general economic market conditions. Many global economies remain sluggish as they recover from a severe recession and unprecedented turmoil. The U.S. and other developed economies continue to suffer from market volatility, difficulties in the financial
32
services sector, tight credit markets, concerns of inflation, reduced corporate profits and capital spending, significant job losses or slower than expected job creation, reduced consumer spending, and continuing economic uncertainties. The turmoil and the uncertainty about future economic conditions could negatively impact our current and prospective customers, adversely affect the financial ability of governments and health insurers to pay claims, adversely impact our expenses and ability to obtain financing of our operations, cause delays or other problems with key suppliers and increase the risk of counterparty failures. Since the use of the HVAD System in a patient is generally dependent on the availability of third-party reimbursement and normally requires the patient to make a significant co-payment, the pace of the global recovery will continue to influence our potential customers and may negatively affect customer orders. Similarly, the impacts of the challenging economy on our existing customers may cause some of them to cease purchasing HVAD Systems and this will reduce our revenue, which in turn will make it more difficult to achieve the per unit cost-savings which are expected to be attained through increases in our manufacturing volume.
The severe recession impacted the financial stability of many public and private health insurers. As a result, insurers continue to scrutinize claims more rigorously and delay or deny reimbursement more often. Although VAD procedures occur in relatively limited numbers, the per procedure reimbursement levels may draw the attention of third-party payers. Since the sale of the HVAD System is generally dependent on the availability of third-party reimbursement, any delay or decline in reimbursement will adversely affect our revenue.
Global market and economic conditions may exacerbate certain risks affecting our business.
International markets, especially Europe, represent a major part of our present business. Approximately 46% of our 2014 revenues were derived from international sales and a significant amount of our marketing efforts are focused on European countries. Although not materially impacted to date, our accounts receivable in certain European countries may be subject to significant payment delays due to government funding and reimbursement practices or limited financial flexibility of our distributors. European governments have announced or implemented austerity measures to constrain the overall level of government expenditures, which may include reforming health care coverage and reducing health care costs. These measures will continue to exert pressure on our customers and may impact their ability to pay for product on a timely basis or to maintain their current purchasing patterns. These adverse market and economic conditions could reduce our product sales and revenue, result in additional allowances, or reduce credit sales to our distribution network. In addition, some European and other payers require health technology assessments or economic cost-benefit analyses to be conducted by a manufacturer or third party analysis group in order to obtain or maintain reimbursement of medical devices. These analyses can be expensive and time consuming, and may not produce outcomes favorable to us. Adverse or delayed outcomes will adversely affect our revenue. Similar obstacles may delay our expansion into new markets or reduce the expected benefits of entry into new markets.
Fluctuations in foreign currency exchange rates could adversely affect our financial results.
Changes in foreign currency exchange rates can affect the value of our assets, liabilities, costs and revenue. In 2014, approximately 43% of our revenue was sourced from international sales denominated in foreign currencies, mainly in Europe and principally in Euros, while most of our expenditures are incurred in U.S. dollars.
With limited exceptions, our international sales will be denominated in Euros or in local currencies, not U.S. dollars, and fluctuations in foreign currency exchange rates, especially an appreciation of the U.S. dollar against major international currencies, will materially impact our revenue and earnings. Due to the size and stage of development of our operations and revenue, we do not presently mitigate our exposure to exchange risk to a significant extent other than by holding the majority of our funds in U.S. dollars or U.S. dollar denominated investments.
33
Healthcare policy changes, including the Patient Protection and Affordable Care Act, may have a material adverse effect on us.
In response to perceived increases in health care costs in recent years, there have been and continue to be proposals by the federal government, state governments, regulators and third-party payors to control these costs and, more generally, to reform the U.S. healthcare system. Certain of these proposals could limit the prices we are able to charge for our products or the amounts of reimbursement available for our products and could limit the acceptance and availability of our products. Moreover, as discussed in the paragraph below, the Affordable Care Act imposes significant taxes on medical device makers such as us. The Affordable Care Act and other proposals could have a material adverse effect on our financial position and results of operations.
On March 23, 2010, the Affordable Care Act was signed into law by President Obama. On March 30, 2010, a companion bill, the Health Care and Education Reconciliation Act of 2010 (the “Reconciliation Act”) was also signed into law by President Obama. Among other things, the Affordable Care Act and the Reconciliation Act (collectively, the “Acts”), when taken together, impose a 2.3% excise tax on the sale of certain medical devices. In addition, it is possible that standard setters or regulators may address certain unique aspects of the accounting for the Acts in the future. Other elements of this legislation such as comparative effectiveness research, an independent payment advisory board, transparency requirements, payment system reforms including shared savings pilots and other provisions could meaningfully change the way healthcare is developed and delivered, and may materially impact numerous aspects of our business.
We are subject to federal and state laws prohibiting “kickbacks” and false or fraudulent claims, which, if violated, could subject us to substantial penalties. Foreign jurisdictions in which we operate may have similar laws. Other laws such as the Foreign Corrupt Practices Act (the “FCPA”) and the U.K. Bribery Act prohibit improper payments to government officials to induce the purchase of products or similar actions. Any challenge to or investigation into our practices under these laws are costly to defend, might result in fines and penalties and could cause adverse publicity, thus causing harm to our business and operations. In addition, we can be held liable for our distributors’ failure to comply with these laws.
A federal law commonly known as the Anti-Kickback Law, and several similar state and foreign laws, prohibit payments that are intended to induce physicians or others either to refer patients or to acquire or arrange for or recommend the acquisition of healthcare products or services. These laws constrain our sales, marketing and other promotional activities by limiting the kinds of financial arrangements, including sales programs, we may have with hospitals, physicians or other potential purchasers of medical devices. Other federal and state laws generally prohibit individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent, or for items or services that were not provided as claimed. Because we may provide some coding and billing information to purchasers of the HVAD System and our other products, and because we cannot assure that the government will regard any billing errors that may be made as inadvertent, these laws are potentially applicable to us. In addition, these laws are potentially applicable to us because we may provide training reimbursement and other payments to healthcare professionals and institutions. Anti-kickback and false claims laws prescribe civil and criminal penalties for noncompliance, which can be substantial.
The FCPA and the United Kingdom’s Bribery Act prohibit improper payments to government officials to induce inappropriate behavior. In many jurisdictions, hospitals are owned or operated by governmental authorities, and physicians and administrators who are employed by the hospital may be considered to be a government official. As a result, certain relationships with our customers could expose us to liability under these statutes. Corrupt practices and anti-bribery laws prescribe civil and criminal penalties for noncompliance, which can be substantial. Even an unsuccessful challenge or investigation into our practices is costly to defend, and could cause adverse publicity, and thus could have a material adverse effect on our business, financial condition or results of operations.
In addition, under certain circumstances, we may be liable for the actions of our distributors to the extent they do not comply with these laws.
34
Risks Related to Intellectual Property and Confidential Information
We may not be able to effectively protect our intellectual property rights which could have an adverse effect on our business, financial condition or results of operations.
Our success depends in part on our ability to obtain and maintain protection in the United States and other countries of the intellectual property relating to or incorporated into our technology and products. Our patent portfolio consists of internally developed technology as well as patents and patent applications which we licensed or acquired from others. In addition, from time to time, we also acquire or license technology from third parties. As a result, we may have less complete knowledge and records with respect to the development and ownership of acquired and third party technology, patents and intellectual property than we would otherwise have for technology, patents and intellectual property developed internally by us.
Our pending and future patent applications may not issue as patents or, if issued, may not issue in a form that will provide us with any meaningful protection or any competitive advantage. Even if issued, existing or future patents may be challenged, narrowed, invalidated or circumvented, which could limit our ability to stop competitors from developing and marketing similar products or limit the length of terms of patent protection we may have for our products. Further, other companies may design around technologies we have patented, licensed or developed. Moreover, changes in patent laws or their interpretation in the United States and other countries could also diminish the value of our intellectual property or narrow the scope of our patent protection. In addition, the legal systems of certain countries do not favor the aggressive enforcement of patents, and the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar to ours.
We may need to initiate lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive and, if we lose, could cause us to lose some of our intellectual property rights, which would harm our ability to compete in the market.
We rely on patents to protect a portion of our intellectual property and our competitive position. Patent law relating to the scope of claims in the technology fields in which we operate is still evolving and, consequently, patent positions in the medical device industry are generally uncertain. In order to protect or enforce our patent rights, we may initiate patent litigation against third parties, such as infringement suits or interference proceedings. Litigation may be necessary to:
| | • | | assert claims of infringement; |
| | • | | protect our trade secrets or know-how; or |
| | • | | determine the enforceability, scope and validity of the proprietary rights of others. |
Any lawsuits that we initiate could be expensive, take significant time and divert management’s attention from other business concerns. Litigation also puts our patents at risk of being invalidated or interpreted narrowly. Additionally, we may provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially valuable. The occurrence of any of these events may have a material adverse effect on our business, financial condition and results of operations.
Claims that our current or future products infringe or misappropriate the proprietary rights of others could adversely affect our ability to sell those products and cause us to incur additional costs.
Substantial litigation over intellectual property rights exists in the medical device industry. We expect that we could be increasingly subject to third-party infringement claims as our revenue increases, the number of
35
technology holders grows and the functionality of products and technology in different industry segments overlaps. Third parties may currently have, or may eventually be issued, patents on which our current or future products or technologies may allegedly infringe.
There can be no certainty that litigation will not arise in relation to third party intellectual property or, if it does arise, whether or not it will be determined in a manner which is favorable to us. Any litigation, regardless of its outcome, would likely result in the expenditure of significant financial resources and the diversion of management’s time and resources. In addition, litigation in which we are accused of infringement may cause negative publicity, adversely impact prospective customers, cause product shipment delays, prohibit us from manufacturing, marketing or selling our current or future products, require us to develop non-infringing technology, make substantial payments to third parties or enter into royalty or license agreements, which may not be available on acceptable terms or at all. If a successful claim of infringement were made against us and we could not develop non-infringing technology or license the infringed or similar technology on a timely and cost-effective basis, our revenue may decrease substantially and we could be exposed to significant liability. A court could enter orders that temporarily, preliminarily or permanently enjoin us or our customers from making, using, selling, offering to sell or importing our current or future products, or could enter an order mandating that we undertake certain remedial activities. Claims that we have misappropriated the confidential information or trade secrets of third parties can have a similar negative impact on our reputation, business, financial condition or results of operations.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patented technology, we rely on a combination of non-patented proprietary technology, trade secrets, processes and procedures, technical knowledge and know-how accumulated or acquired since inception. Despite these measures, any of our intellectual property rights could be challenged, invalidated, circumvented or misappropriated. We generally seek to protect this information by confidentiality, non-disclosure and assignment of invention agreements with our employees, consultants, scientific advisors and third parties. These agreements may be breached, and we may not have adequate remedies for any such breach. In addition, our trade secrets may be disclosed to or otherwise become known or be independently developed by competitors. To the extent that our employees, consultants or contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. If our intellectual property is disclosed or misappropriated, it would harm our ability to protect our rights and have a material adverse effect on our business, financial condition and results of operations.
We may be subject to claims that we or our employees have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of former employers of our employees.
We employ individuals who were previously employed at other medical device companies, including our competitors or potential competitors. To the extent that our employees are involved in research areas that are similar to those in which they were involved with their former employers, we may be subject to claims that such employees have inadvertently or otherwise used or disclosed the alleged trade secrets or other proprietary information of the former employers. Litigation may be necessary to defend against such claims.
Security breaches and other disruptions could compromise our information and expose us to liability, which could cause our business and reputation to suffer.
In the ordinary course of our business and as required by regulatory authorities, we collect and store sensitive data, including certain patient tracking information and information related to our clinical sites. We also maintain in confidence intellectual property, proprietary business information and personally identifiable information of our employees, on our networks. The secure processing, maintenance and transmission of this information is critical to our operations and business strategy. Despite our security measures, such as limiting
36
access to information and utilizing password protection, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Any breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any access, disclosure or other loss of information could result in legal claims or proceedings, regulatory penalties, liability under laws that protect the privacy of personal information, disruption of our operations and the services we provide to customers, damage to our reputation, and a loss of confidence in our products and services, which could adversely affect our business/operating margins, revenues and competitive position.
If we are found to have violated laws protecting the confidentiality of patient health information, we could be subject to civil or criminal penalties, which could increase our liabilities and harm our reputation or our business.
There are a number of federal and state and foreign laws protecting the confidentiality of certain patient health information, including patient records, and restricting the use and disclosure of that protected information. In particular, the U.S. Department of Health and Human Services promulgated patient privacy rules under the Health Insurance Portability and Accountability Act of 1996, or HIPAA. These privacy rules protect medical records and other personal health information by limiting their use and disclosure, giving individuals the right to access, amend and seek accounting of their own health information and limiting most use and disclosures of health information to the minimum amount reasonably necessary to accomplish the intended purpose. If we are found to be in violation of the privacy rules under HIPAA or similar laws (to the extent applicable to us), we could be subject to civil or criminal penalties, which could increase our liabilities, harm our reputation and have a material adverse effect on our business, financial condition or results of operations. European privacy laws are generally more stringent than similar laws in the U.S. Since a significant amount of our revenue arises in Europe, we may be at risk should we fail to comply with local requirements even if we have complied with U.S. regulations.
Risks Related to Our Common Stock
The price of our common stock may fluctuate significantly.
Shares of our common stock were listed for trading on The NASDAQ Stock Market LLC on February 24, 2009. Trading commenced the following day. Prior to that time, there had been no public market for our common stock in the United States. The closing price of our shares of common stock traded on The NASDAQ Stock Market LLC has ranged from U.S. $69.43 to U.S. $104.66 in the period from January 1, 2014 to December 31, 2014. The price of our common stock could fluctuate significantly for many reasons, including future announcements or new information concerning us or our competitors, reimbursement, or the potential market for our products.
In addition, stock markets in general and the market for shares of healthcare stocks in particular, have experienced extreme price and volume fluctuations in recent years, fluctuations that frequently have been unrelated to the operating performance of the affected companies. These broad market fluctuations may adversely affect the market price of our shares. The market price of our shares could decline below its current price and the market price of our shares may fluctuate significantly in the future. These fluctuations may be unrelated to our performance.
We do not intend to pay cash dividends on our common stock in the foreseeable future.
We have never declared or paid any cash dividends on our shares, and we currently do not anticipate paying any cash dividends in the foreseeable future. We intend to retain any earnings to finance the development and expansion of our products and business. Accordingly, our stockholders will not realize a return on their investment unless the trading price of our shares appreciates.
37
Anti-takeover provisions in our charter documents and Delaware law may discourage a third party from acquiring us, which could limit our stockholders’ opportunities to sell their shares at a premium.
Certain provisions of our Certificate of Incorporation and By-laws may be considered as having an anti-takeover effect, such as those provisions establishing a classified board of directors, consisting of three classes of directors, and requiring that directors be removed only for cause, authorizing the board of directors to issue from time to time any series of preferred stock and fix the designation, powers, preferences and rights of the shares of such series of preferred stock, prohibiting stockholders from acting by written consent in lieu of a meeting, requiring advance notice of stockholder intention to put forth director nominees or bring up other business at a stockholders’ meeting, and prohibiting stockholders from calling a special meeting of stockholders. We are also subject to Section 203 of the Delaware General Corporation Law, which in general prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years following the date that the stockholder became an interested stockholder, unless certain conditions specified therein are satisfied. These provisions could have the effect of depriving our stockholders of an opportunity to sell their shares at a premium over prevailing market prices by discouraging third parties from seeking to obtain control of us in a tender offer or similar transaction.
We may undergo an “ownership change” for U.S. federal income tax purposes, which would limit our ability to utilize net operating losses from prior tax years.
For U.S. federal income tax purposes, we have incurred net losses since our inception. If we undergo an “ownership change” for U.S. federal income tax purposes, our ability to utilize net operating loss carry-forwards from prior years to reduce taxable income in future tax years might be limited by operation of the Internal Revenue Code, either by limiting the amount of net operating losses that can be utilized to offset taxable income in a given year, or in total over the entire carry-forward period. Certain changes in the ownership of our common stock may result in an ownership change sufficient to limit the availability of our net operating losses.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
We relocated our corporate headquarters to 500 Old Connecticut Path, Framingham, Massachusetts in the first quarter of 2014. In addition to our corporate headquarters, our principal facilities include our manufacturing and operations facility in Miami Lakes, Florida and our distribution and customer service facility in Hannover, Germany. We also occupy a small space in Arden Hills, Minnesota for research and development purposes and, as a result of our 2013 acquisition of CircuLite, continue to rent space in Teaneck, New Jersey and Aachen, Germany.
Our corporate headquarters in Framingham, Massachusetts consists of approximately 58,000 square feet of company space and is primarily used for administrative and ancillary laboratory purposes, including development testing. The initial lease term is seven years with an option to renew for a period of fifty-seven months, but in no event beyond September 30, 2025. We no longer occupy our previous corporate headquarters in Framingham, Massachusetts, which consisted of approximately 21,300 square feet, in the aggregate, but continue to be party to various operating leases through June 30, 2015 for that space.
On December 9, 2010, we entered into a lease for a facility in Miami Lakes, Florida as part of our planned expansion to support our efforts to prepare for U.S. commercialization. The facility is used primarily for manufacturing, research and development and administrative functions. Under the lease, which was amended in November 2012 to add a small amount of additional space, we rent approximately 132,000 square feet for a period ending February 28, 2022, with an option to renew for two five-year terms.
38
Our facility in Hannover, Germany is approximately 3,900 square feet. The lease commenced on October 11, 2010 with an initial term of two years and has been extended for one additional three-year term.
On August 19, 2014, we entered into a lease for a facility in Arden Hills, Minnesota consisting of about 2,400 square feet to support certain research and development activities. The Minnesota lease has a two-year term with an option to extend for an additional two years.
Through our acquisition of CircuLite in December 2013, we assumed a lease for a facility in Teaneck, New Jersey used primarily for research and development and administrative functions. The lease, which covers approximately 22,000 square feet, was entered into in December 2012 and expires in October 2020. We also assumed a collection of leases that CircuLite entered into between September 2007 and February 2013 for office space, research and development functions and manufacturing purposes in two adjacent properties in Aachen, Germany. We have initiated a plan to close our facilities located in Aachen, Germany on or by March 31, 2015. A separate facility in Aachen, Germany, which was originally intended to be used as office and research and development space and which is approximately 8,300 square feet, is covered under an operating lease that expires on October 31, 2017.
We believe that the facilities noted above are suitable and adequate for our current needs.
Item 3. Legal Proceedings
The Company is not a party to any material pending legal proceedings at the date of filing of this Annual Report on Form 10-K.
Item 4. Mine Safety Disclosures
Not applicable.
39
Part II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our shares of common stock trade on the NASDAQ Stock Market under the symbol “HTWR”. Our shares of common stock also traded in the form of CHESS Depositary Interests (“CDIs”), each CDI representing one thirty-fifth of a share of our common stock, on the Australian Securities Exchange (“ASX”) under the symbol “HIN” since November 13, 2008. At our request, our CDIs were suspended from quotation on September 10, 2013 and on September 17, 2013 our CDIs were removed from the official list of ASX. There are no CDIs currently outstanding.
The following table sets forth, for the periods indicated, the high and low closing prices for our common stock on NASDAQ.
| | | | | | | | |
Period | | High | | | Low | |
Fiscal Year 2014: | | | | | | | | |
First Quarter | | $ | 104.66 | | | $ | 90.83 | |
Second Quarter | | | 95.42 | | | | 81.26 | |
Third Quarter | | | 93.44 | | | | 76.89 | |
Fourth Quarter | | | 86.03 | | | | 69.43 | |
Fiscal Year 2013: | | | | | | | | |
First Quarter | | $ | 94.22 | | | $ | 85.42 | |
Second Quarter | | | 99.19 | | | | 82.89 | |
Third Quarter | | | 97.14 | | | | 73.19 | |
Fourth Quarter | | | 96.67 | | | | 69.99 | |
As of February 25, 2015, we had 17,238,458 shares of common stock issued and outstanding and there were 76 holders of record of our common stock.
We have not declared or paid any cash dividends on our shares, and we currently do not anticipate paying any cash dividends in the foreseeable future. Our convertible notes were issued pursuant to the terms of an Indenture dated December 15, 2010. The Indenture does not contain any covenants or restrictions on the payments of dividends. We intend to retain any earnings to finance the development and expansion of our products and business.
40
Stock Price Performance Graph
The graph presents the cumulative total stockholder return on an investment in our common stock, the NASDAQ Composite Index (U.S. companies only) and the Morningstar Medical Devices Index for the period from December 31, 2009 to December 31, 2014. The graph assumes the value of an investment in our common stock was $100 on December 31, 2009, and the reinvestment of all dividends, if any.
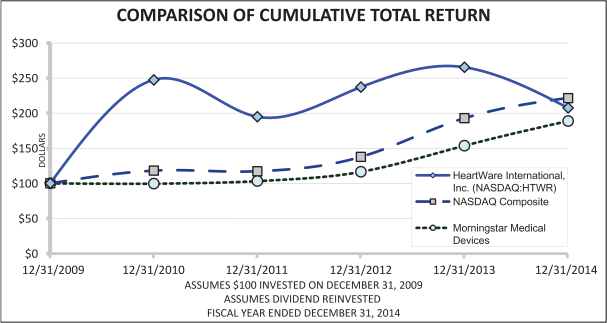
| | | | | | | | | | | | | | | | | | | | | | | | |
Company/Market/Peer Group | | 12/31/2009 | | | 12/31/2010 | | | 12/31/2011 | | | 12/31/2012 | | | 12/31/2013 | | | 12/31/2014 | |
HeartWare International, Inc.
(NASDAQ: HTWR) | | $ | 100.00 | | | $ | 246.88 | | | $ | 194.53 | | | $ | 236.68 | | | $ | 264.73 | | | $ | 207.02 | |
NASDAQ Composite-Total Returns | | $ | 100.00 | | | $ | 118.02 | | | $ | 117.04 | | | $ | 137.47 | | | $ | 192.62 | | | $ | 221.02 | |
Morningstar Medical Devices | | $ | 100.00 | | | $ | 99.58 | | | $ | 103.14 | | | $ | 116.29 | | | $ | 153.38 | | | $ | 188.41 | |
41
Equity Compensation Plans
The following table sets forth information regarding the Company’s Equity Compensation Plans as of December 31, 2014:
| | | | | | | | | | | | |
Plan Category | | Number of
securities
to be
issued
upon
exercise of
outstanding
options,
warrants
and rights
(a) | | | Weighted
average
exercise
price of
outstanding
options,
warrants
and rights
(b) (1) | | | Number of securities
remaining available for
future issuance under
equity compensation plans
(excluding securities
reflected in column (a))
(c) | |
Equity compensation plans approved by security holders: | | | | | | | | | | | | |
HeartWare International, Inc. Employee Stock Option Plan | | | 53,771 | | | $ | 27.55 | (2) | | | — | (3) |
HeartWare International, Inc. 2008 Stock Incentive Plan (4) | | | 111,108 | | | $ | 21.20 | | | | — | (3) |
HeartWare International, Inc. 2012 Incentive Award Plan (5) | | | 528,332 | | | $ | 2.44 | | | | 721,519 | (6) |
| | | |
Equity compensation plans not approved by security holders: | | | | | | | | | | | | |
Non-Plan options | | | 2,857 | | | $ | 21.41 | (2) | | | N/A | |
| (1) | The weighted average exercise price of all outstanding options is $48.32 and the weighted average remaining term of all outstanding options is 4.39 years. |
| (2) | The exercise price has been converted to U.S. dollars using the spot rate at December 31, 2014. |
| (3) | Upon stockholder approval of the 2012 Incentive Award Plan in May 2012, no additional grants may be made under these Plans. |
| (4) | Outstanding awards under the 2008 Stock Incentive Plan include 74,421 restricted stock units with exercise prices of $0 and 36,687 options with exercise prices equal to the fair value of our common stock on the date of grant. The weighted average exercise price of the outstanding options was $64.21 at December 31, 2014. |
| (5) | Outstanding awards under the 2012 Incentive Award Plan consist of 514,332 restricted stock units with exercise prices of $0 and 14,000 options with exercise prices equal to the fair value of our common stock on the date of grant. The weighted average exercise price of the outstanding options was $91.95 at December 31, 2014. |
| (6) | Under the terms of the 2012 Incentive Award Plan, the total number of shares of our common stock initially reserved for issuance is 1,375,000, provided that the total number of shares of our common stock that may be issued pursuant to awards other than options, SARs or other awards for which the holder pays the intrinsic value existing as of the date of grant whether directly or by forgoing a right to receive a payment from the Company, is 1,275,000. |
42
Item 6. Selected Financial Data
The following selected consolidated statement of operations data for the years ended December 31, 2014, 2013 and 2012 and the selected consolidated balance sheet data as of December 31, 2014 and 2013 have been derived from our audited consolidated financial statements included elsewhere in this Annual Report on Form 10-K. The following selected consolidated statement of operations data for the years ended December 31, 2011 and 2010 and balance sheet data as of December 31, 2012, 2011 and 2010 have been derived from our audited consolidated financial statements which are not included in this Annual Report on Form 10-K. The selected consolidated financial data set forth below should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” below and our audited consolidated financial statements and notes thereto appearing elsewhere in this Annual Report on Form 10-K.
| | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | |
| (In thousands, except per share data) | | 2014 | | | 2013 | | | 2012 | | | 2011 | | | 2010 | |
Consolidated Statement of Operations Data: | | | | | | | | | | | | | |
Revenue, net | | $ | 278,420 | | | $ | 207,929 | | | $ | 110,922 | | | $ | 82,764 | | | $ | 55,164 | |
Cost of revenue | | | 92,195 | | | | 76,468 | | | | 51,023 | | | | 32,932 | | | | 24,441 | |
Selling, general and administrative expenses | | | 87,177 | | | | 76,524 | | | | 53,945 | | | | 42,314 | | | | 26,642 | |
Research and development expenses | | | 122,432 | | | | 102,483 | | | | 83,548 | | | | 50,149 | | | | 33,108 | |
Change in fair value of contingent consideration | | | (23,260 | ) | | | — | | | | — | | | | — | | | | — | |
Other expense, net (a) | | | 18,682 | | | | 11,298 | | | | 10,124 | | | | 12,424 | | | | 370 | |
Net loss | | | (19,366 | ) | | | (59,311 | ) | | | (87,718 | ) | | | (55,055 | ) | | | (29,397 | ) |
Basic and diluted net loss per share | | | (1.14 | ) | | | (3.69 | ) | | | (6.15 | ) | | | (3.94 | ) | | | (2.17 | ) |
| | | | | | | | | | | | | | | | | | | | |
| | | As of December 31, | |
| (In thousands) | | 2014 | | | 2013 | | | 2012 | | | 2011 | | | 2010 | |
Consolidated Balance Sheet Data: | | | | | | | | | | | | | | | | | | | | |
Cash, cash equivalents and short-term investments | | $ | 178,481 | | | $ | 200,476 | | | $ | 102,808 | | | $ | 163,182 | | | $ | 213,478 | |
Total assets | | | 423,813 | | | | 429,827 | | | | 206,499 | | | | 240,732 | | | | 267,577 | |
Convertible senior notes, net of discounts (b) | | | 114,803 | | | | 107,125 | | | | 100,315 | | | | 94,277 | | | | 88,922 | |
Contingent liabilities (c) | | | 43,740 | | | | 67,000 | | | | — | | | | — | | | | — | |
Total stockholders’ equity | | | 208,534 | | | | 198,607 | | | | 68,211 | | | | 126,784 | | | | 167,764 | |
| (a) | In the years ended December 31, 2014, 2013, 2012 and 2011, other expense includes approximately $13.1 million, $12.2 million, $11.4 million and $10.7 million, respectively, of interest expense associated with our 3.5% convertible senior notes due December 15, 2017. |
| (b) | At December 31, 2014, 2013, 2012, 2011 and 2010, the aggregate principal amount of our 3.5% convertible senior notes due December 15, 2017 was $143.75 million. |
| (c) | On December 1, 2013, we acquired CircuLite, Inc. using a combination of cash and stock. In addition to the initial consideration paid at closing, the former CircuLite securityholders may be entitled to receive up to an additional $320 million in shares of HeartWare common stock (or cash, in certain cases, at our discretion) upon the achievement of five specified performance milestones and royalty payments. This liability is carried at its estimated fair value at the reporting dates. |
43
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations should be read together with our consolidated financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K. This discussion and analysis contains forward-looking statements that involve risks, uncertainties, judgments and assumptions. You should review the “Risk Factors” section of this Annual Report on Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis. Certain abbreviated key terms have the meanings defined elsewhere in this Annual Report on Form 10-K.
Overview
HeartWare is a medical device company that develops and manufactures miniaturized implantable heart pumps, or ventricular assist devices, to treat patients suffering from advanced heart failure.
The HeartWare Ventricular Assist System (the “HVAD System”), which includes a ventricular assist device (“VAD”) or blood pump, patient accessories and surgical tools, is designed to provide circulatory support for patients in the advanced stage of heart failure. The core of the HVAD System is a proprietary continuous flow blood pump, the HVAD pump, which is a full-output device capable of pumping up to 10 liters of blood per minute. The HVAD System is designed to be implanted adjacent to the heart, avoiding abdominal surgery, which is generally required to implant similar devices.
In November 2012, we received approval from the United States Food and Drug Administration (“FDA”) for the HVAD System as a bridge to heart transplantation in patients with end-stage heart failure. The HVAD System has been available in the European Union since receiving CE marking in 2009. In May 2012, we received an expanded European label for long-term use of the HVAD System in all patients at risk of death from refractory, end-stage heart failure. As of December 31, 2014, there have been over 7,000 implants of the HVAD System in patients at over 270 health care sites in 41 countries.
On August 27, 2013, the FDA approved an Investigational Device Exemption (“IDE”) Supplement allowing HeartWare to commence enrollment in an additional patient cohort for the ENDURANCE clinical trial. In this supplemental cohort, HeartWare intends to enroll up to 310 patients receiving the HVAD System, as well as up to an additional 155 control patients using a randomization scheme consistent with the ENDURANCE protocol. Patients will be followed for 12 months after implant. In 2016, HeartWare intends to incorporate the data from both this supplemental cohort and ENDURANCE into an anticipated PMA Application seeking approval of the HVAD System for the destination therapy indication.
MVAD System
We continue to expand our pipeline through research and development into next generation products and peripherals and through ongoing and new clinical trials and to expand our presence in commercial markets outside of the United States. Among these activities, we are developing our next generation miniaturized device, the MVAD System. The MVAD System is based on the same technology platform as the HVAD System but adopts an axial flow, rather than a centrifugal flow, configuration and is being developed in multiple designs. The MVAD pump is less than one-half the size of the HVAD pump and can provide partial or full support. The MVAD platform is designed to allow for a variety of configurations and surgical placements with the goal towards further reduction of surgical invasiveness while producing superior clinical results. In December 2014, we initiated submissions to commence a CE Mark study at 9 international sites and are preparing regulatory submissions to commence an IDE study in the United States.
CircuLite
On December 1, 2013, we acquired CircuLite, Inc. CircuLite is the developer of the SYNERGY Circulatory Support System, a partial support system designed to treat less sick, ambulatory, chronic heart failure patients
44
who are not yet inotrope-dependent. The SYNERGY Surgical System is designed for long-term support and is intended to reduce the heart’s workload while improving blood flow to vital organs. The system is currently undergoing an upgrade to resolve issues that arose after its commercial release and caused the loss of its CE marking in the European Union in March 2014. In January 2015, we discontinued development of the SYNERGY micropump and have focused our efforts on a version of our MVAD pump for our partial-assist program. Following the necessary clinical trials and regulatory approvals, we plan to re-launch the system in Europe and will focus on building experience at a small number of centers of excellence, refining training techniques and implementing additional system upgrades. The next generation endovascular system, which will be implanted collaboratively by cardiologists and surgeons in a hybrid catheterization (“cath”) lab setting, offers an interventional approach to circulatory support. While our HVAD and MVAD Systems offer minimally invasive treatment to end-stage heart failure patients, the SYNERGY Circulatory Support System offers even less invasive and ultimately interventional options to earlier-stage heart failure patients.
FDA Warning Letter
We received a warning letter from the FDA, dated June 2, 2014, following an inspection of our Miami Lakes, Florida facility conducted in January 2014. The FDA letter cited four categories for us to address: (1) procedures for validating device design, including device labeling; (2) procedures for implementing corrective and preventive action (CAPA); (3) maintaining records related to investigations; and (4) validation of computer software used as part of production or quality systems. The warning letter did not require any action by physicians or patients and did not restrict the use of our devices.
We sent the FDA our initial response to the warning letter within the required fifteen business days of receipt, and committed to undertaking certain quality system improvements and providing the FDA with periodic updates. During 2014, we commenced implementing systemic changes and organizational enhancements to address the four warning letter items and related quality systems. We have established teams to review and address the items cited by the FDA and have engaged external subject matter experts to assist in assessment and remediation efforts. As we complete this comprehensive review of our quality systems, it is possible that we may need to take additional actions including the possibility of voluntary product recalls when necessary to ensure patient safety and effective performance of the HVAD System.
Recent Urgent Medical Device Corrective Actions
On April 30, 2014, we implemented a corrective action to update patients and clinicians with respect to a driveline connector correction communicated to all of our clinical sites back in December 2013. The update provided instruction to both patients and clinicians advising them that should the locking mechanism fail to engage, or the driveline becomes disconnected from the controller, clinicians should promptly call their HeartWare representative to arrange a permanent repair. This corrective action was ongoing as of December 31, 2014.
Also on April 30, 2014, we implemented a corrective action to notify clinicians and patients of an observed increase in complaints related to earlier-than-expected battery depletion and routine battery handling. This notification provided information to assist patients and clinicians with monitoring battery performance, recognizing abnormal behaviors and reinforcing proper power management of the HVAD System. Subsequently, on July 30, 2014, we extended this field action to include a voluntary recall of certain older batteries. The recall instructed sites to replace certain older batteries in the field upon patients’ routine visits in order to further mitigate the potential risks associated with premature battery depletion. This corrective action was ongoing as of December 31, 2014.
In February 2015, we expanded a 2013 voluntary Field Safety Corrective Action, by initiating a voluntary medical device recall of certain older controllers distributed in the U.S. during the ADVANCE and ENDURANCE clinical trial periods. The affected controllers exhibit a higher susceptibility to electrostatic discharge than newer, commercial controllers. This recall was ongoing as of December 31, 2014.
45
During 2014, we recorded charges aggregating $3.6 million for estimated costs associated with the battery and controller recalls discussed above.
Summary of Recent Financial Performance
Revenue was $278.4 million in 2014 compared to $207.9 million in 2013. This increase reflects 44% revenue growth in the United States, where our HVAD System is labeled for a bridge-to-transplantation indication, and 24% internationally where the HVAD System is more broadly indicated for general long-term heart failure patients. In each case, revenue growth reflected continued market penetration within existing customer accounts and to a lesser extent revenue contributed from newly added customers. As of December 31, 2014, the Company had approximately 110 customers in the United States and approximately 160 customers internationally.
We realized an increase in gross margin percentage to 67% in 2014 compared to 63% in 2013, which was primarily a result of production efficiencies driven by increased revenue and manufacturing throughput.
Combined selling, general, administrative, research and development expenses in 2014 increased to $209.6 million, compared to $179.0 million in 2013. The net increase includes expansion of sales and marketing activities, external warning-letter remediation costs, research and development pipeline activities and increased clinical activity as we make preparations for human clinical testing for the MVAD System and associated peripherals as well as expanded indications for the HVAD System.
Our financial results are more fully described inResults of Operations below.
Critical Accounting Policies and Estimates
We prepare our financial statements in accordance with accounting principles generally accepted in the United States. We are required to adopt various accounting policies and to make estimates and assumptions in preparing our financial statements that affect the reported amounts of our assets, liabilities, revenue and expenses. On an ongoing basis, we evaluate our estimates and assumptions. We base our estimates on our historical experience to the extent practicable and on various other assumptions that we believe are reasonable under the circumstances and at the time they are made. If our assumptions prove inaccurate or if our future results are not consistent with our historical experience, we may be required to make adjustments in our policies that affect our reported results. Our significant accounting policies are disclosed in Note 3 to the financial statements included in this report.
Our most critical accounting policies and estimates include: revenue recognition, inventory capitalization and valuation, accounting for share-based compensation, measurement of fair value, valuation of tax assets and liabilities, reserves, long-lived assets, intangible assets and goodwill, and contingent consideration. We also have other key accounting policies that are less subjective and, therefore, their application is less subject to variations that would have a material impact on our reported results of operations. The following is a discussion of our most critical policies, as well as the estimates and judgments involved.
Revenue recognition
We recognize revenue from product sales in accordance with FASB ASC 605—Revenue Recognition. Revenue from product sales is recognized when persuasive evidence of an arrangement exists, substantially all the risks and rewards of ownership have transferred to our customers, the selling price is fixed and collection is reasonably assured and there are no further obligations to customers. Sales from products are not subject to rights of return and, historically, actual sales returns have not been significant. We sell products through our direct sales force and through distributors. Sales through distributors are recognized as revenue upon sale to the distributor as these sales are considered to be final and no right of return or price protection exists. Sales to customers, when not made on consignment, are recognized upon shipment. A significant portion of our revenue is generated on a
46
consignment basis. Revenue from products sold on a consignment basis is recognized on the date the consigned product is implanted or otherwise consumed. In limited circumstances, we rent peripheral equipment to patients. We recognize revenue from this arrangement when a contract is entered into with the patient’s insurer over the term the equipment is rented.
Inventory
We expense costs relating to the production of inventories as research and development (“R&D”) expense in the period incurred until such time as we believe future commercialization is considered probable and future economic benefit is expected to be recognized, which generally is reliant upon receipt of regulatory approval. We then begin to capitalize subsequent inventory costs relating to that product. Inventories are stated at the lower of cost or market. Cost is determined on a first-in, first out, or FIFO, method. Work-in-process and finished goods include direct and indirect labor and manufacturing overhead. Finished goods include product which is ready-for-use and which is held by us or by our customers on a consignment basis.
We review our inventory for excess or obsolete items and write-down obsolete or otherwise unmarketable inventory to its estimated net realizable value. Obsolescence may occur due to product expiring or product improvements rendering previous versions obsolete. The extent to which product improvements will cause obsolescence of existing inventory is difficult to determine as the rate of customer acceptance is dependent on many factors. We make judgments and estimates on matters, including forecasted sales volume. Our estimates and judgments in this area are subject to uncertainty and may differ from our actual experience in the future, which could have a material effect on recorded inventory values.
We include in inventory materials and finished goods that are held for sale. Certain materials and finished goods held in inventory may be used in research and development activities and are expensed as part of research and development costs when consumed.
Share-Based Compensation
We recognize share-based compensation expense in connection with our share-based awards based on the estimated fair value of the awards on the date of grant, net of estimated forfeitures, using an accelerated accrual method over the vesting period. Therefore, we only recognize compensation cost for those awards expected to vest over the service period of the award. We estimate the forfeiture rate based on our historical experience of forfeitures. If our actual forfeiture rate is materially different from our estimate, share-based compensation expense could be significantly different from what we have recorded in the current period.
Calculating share-based compensation expense requires the input of highly subjective judgment and assumptions, including forfeiture rates, estimates of expected life of the share-based award, stock price volatility and risk-free interest rates. The assumptions used in estimating the fair value of our share-based awards represent our best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As a result, if factors change and we use different assumptions, our share-based compensation expense could be materially different in the future.
We value restricted stock units, or RSUs, at their intrinsic value on the date of grant. We estimate the fair value of our stock options using a Black-Scholes option pricing model. When appropriate, we estimate the expected life of a stock option by averaging the contractual term of the stock option (up to 10 years) with the associated vesting term (typically 4 years). We estimate the volatility of our shares on the date of grant considering several factors, including the historical volatility of our publicly-traded shares. We estimate the risk-free interest rate based on rates in effect for United States government bonds with terms similar to the expected lives of the stock options, at the time of grant.
We have issued share-based awards with performance-based vesting criteria. Achievement of the milestones must be probable before we begin recording share-based compensation expense. At each reporting date, we
47
review the likelihood that these awards will vest and if the vesting is deemed probable, we begin to recognize compensation expense at that time. In the period that achievement of the performance based criteria is deemed probable, U.S. GAAP requires the immediate recognition of all previously unrecognized compensation since the original grant date. As a result, compensation expense recorded in the period that achievement is deemed probable could include a substantial amount of previously unrecorded compensation expense related to the prior periods. If ultimately performance goals are not met, for any share-based awards where vesting was previously deemed probable, previously recognized compensation cost will be reversed.
Fair Value Measurements
FASB ASC 820—Fair Value Measurements and Disclosures defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. FASB ASC 820 requires disclosures about the fair value of all financial instruments, whether or not recognized, for financial statement purposes. Disclosures about the fair value of financial instruments are based on pertinent information available to us as of the reporting dates. Accordingly, the estimates presented in our financial statements are not necessarily indicative of the amounts that could be realized on disposition of the financial instruments.
FASB ASC 820 specifies a hierarchy of valuation techniques based on whether the inputs to those valuation techniques are observable or unobservable. Observable inputs reflect market data obtained from independent sources, while unobservable inputs reflect market assumptions. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurement) and the lowest priority to unobservable inputs (Level 3 measurement).
The three levels of the fair value hierarchy are as follows:
Level 1—Quoted prices for identical instruments in active markets.
Level 2—Quoted prices for similar instruments in active markets; quoted prices for identical or similar instruments in markets that are not active; and model-derived valuations whose inputs are observable or whose significant value drivers are observable.
Level 3—Instruments with primarily unobservable value drivers.
The assumptions used in calculating the fair value of financial instruments represent our best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As a result, the use of different estimates or assumptions would result in a higher or lower fair value and different amounts being recorded in our financial statements. Calculating fair value utilizing Level 3 inputs requires the input of highly subjective judgment and assumptions.See Note 6 to the accompanying financial statements.
Income Taxes
We account for income taxes in accordance with the liability method presented by FASB ASC 740—Income Taxes. Under this method, deferred income taxes are determined based on the estimated future tax effects of differences between the financial statement and tax basis of assets and liabilities given the provisions of enacted tax laws. Deferred income tax provisions and benefits are based on changes to the assets or liabilities from year to year. In providing for deferred taxes, we consider tax regulations of the jurisdictions in which we operate, estimates of future taxable income, and available tax planning strategies. If tax regulations, operating results or the ability to implement tax-planning strategies vary, adjustments to the carrying value of deferred tax assets and liabilities may be required. Valuation allowances are recorded related to deferred tax assets based on the “more likely than not” criteria of FASB ASC 740. Through December 31, 2014, we have historically concluded that a full valuation allowance is required to offset our net deferred tax assets. We operate within multiple taxing jurisdictions and are subject to audit in those jurisdictions. Because of the complex issues involved, any claims could require an extended period to resolve.
48
FASB ASC 740 requires that we recognize the financial statement benefit of a tax position only after determining that the relevant tax authority would more likely than not sustain the position following an audit. For tax positions meeting the “more likely than not” threshold, the amount recognized in the financial statements is the largest benefit that has a greater than 50 percent likelihood of being realized upon ultimate settlement with the relevant tax authority.
Reserves
Management must make estimates and assumptions to determine the amount of reserves to record in the financial statements. If any of these decisions proves incorrect, our consolidated financial statements could be materially and adversely affected.
We maintain allowances for doubtful accounts for estimated losses that may result from an inability to collect payments owed to us for product sales. We regularly review the allowance by considering factors such as historical experience, the age of the accounts receivable balances and current economic conditions that may affect a customer’s ability to pay.
Certain patient accessories sold with the HVAD System are covered by a limited warranty ranging from one to two years. Estimated contractual warranty obligations are recorded as an expense when the related revenue is recognized and are included in cost of revenue on our consolidated statements of operations. Factors that affect estimated warranty liability include the number of units sold, historical and anticipated rates of warranty claims, cost per claim, and vendor supported warranty programs. We periodically assess the adequacy of our recorded warranty liabilities and adjust the amounts as necessary. The costs to repair or replace products associated with product recalls and voluntary service campaigns are recorded when they are determined to be probable and reasonably estimable as a cost of revenue and are not included in our warranty liability.
Long-Lived Assets, Purchased Intangible Assets and Goodwill
We evaluate the carrying value of our long-lived assets, including purchased intangible assets, whenever events, changes in business circumstances or our planned use of long-lived assets indicate that their carrying amounts may not be fully recoverable or that their useful lives are no longer appropriate. If these facts and circumstances exist, we assess for recovery by comparing the carrying values of long-lived assets with their future undiscounted net cash flows. If the comparison indicates that impairment exists, impairment losses are recorded for the excess of the carrying value over the fair value of the long-lived assets based on discounted cash flows. Significant management judgment is required in the forecast of future operating results that are used in the preparation of expected undiscounted cash flows.
We also evaluate the carrying value of intangible assets (not subject to amortization) related to in-process research and development (“IPR&D”) assets which are considered to be indefinite-lived until the completion or abandonment of the associated research and development projects. Accordingly, amortization of the IPR&D assets does not occur until the product reaches commercialization. During the period the assets are considered indefinite-lived, they are tested for impairment on an annual basis, as well as between annual tests if we become aware of any events occurring or changes in circumstances that indicate that the fair values of the IPR&D assets are less than their carrying amounts. If and when development is complete, which generally occurs when regulatory approval to market the product is obtained, the associated IPR&D assets are deemed definite-lived and are then amortized based on their estimated useful lives at that point in time. If the related project is terminated or abandoned, we may have a full or partial impairment related to the IPR&D assets, calculated as the excess of their carrying value over fair value.
We test goodwill for impairment on an annual basis in the fourth quarter of each fiscal year or more frequently if we believe indicators of impairment exist. The performance of the test involves a two-step process. The first step requires comparing the fair value of the reporting unit to its net book value, including goodwill. A potential impairment exists if the fair value of the reporting unit is lower than its net book value. The second step
49
of the process is only performed if a potential impairment exists, and it involves comparing the aggregate fair value of the reporting unit’s net assets other than goodwill to the fair value of the reporting unit as a whole. Goodwill is considered impaired, and an impairment charge is recorded, if the excess of the fair value of the reporting unit over the fair value of the net assets is less than the carrying value of goodwill.
Contingent Consideration
On December 1, 2013, we acquired CircuLite, Inc. In addition to initial consideration paid at closing, the former CircuLite securityholders may be entitled to receive additional shares of HeartWare common stock (or cash, in certain cases, at our discretion) upon the achievement of specified regulatory and commercial milestones, not to exceed $320 million in the aggregate over a ten-year period. The estimated fair value of the contingent payments is recorded as a liability and is remeasured at each reporting period. The estimated fair value is calculated using a discounted cash flow model utilizing significant unobservable inputs including future revenue projections, the probability of achieving each of the potential milestones and an estimated discount rate commensurate with the risks of the expected cash flows attributable to the various milestones. Material changes in any of these inputs could result in a significantly higher or lower fair value measurement and commensurate changes to this liability. Changes in the fair value of the contingent payments are recorded on a separate line item on our consolidated statements of operations.See Note 6 to the accompanying financial statements.
Results of Operations
The following is a description of significant components of our operations, including significant trends and uncertainties that we believe are important to an understanding of our business and results of operations.
The results of operations for CircuLite are included in our consolidated statements of operations subsequent to the December 1, 2013 date of acquisition.
Fiscal Years 2014 and 2013
Revenue, net
In 2014 and 2013, we generated revenue through commercial sales and clinical trials. Net revenue for the years ended December 31, 2014 and 2013 was as follows:
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | Change | |
| | | (in thousands) | | | | |
Revenue, net | | $ | 278,420 | | | $ | 207,929 | | | | 34 | % |
| | | | | | | | | | | | |
Revenue in 2014, reflected 44% growth in the United States, where our HVAD System is labeled for a bridge-to-transplantation indication, and 24% internationally where the HVAD System is more broadly indicated for general long-term heart failure patients. In each case, revenue growth reflected continued market penetration within existing customer accounts and to a lesser extent revenue contributed from newly added customers.
Our U.S. revenue was $151.3 million in 2014 compared to $105.3 million in 2013. We recognized revenue on a total of 1,394 HVAD in the U.S. in 2014 compared to 978 pumps in 2013. The U.S. revenue in 2014 included 191 HVAD Systems as part of our supplemental patient cohort for the ENDURANCE clinical trial.
Our international revenue was $127.1 million in 2014 compared to $102.6 million in 2013. A total of 1,357 HVAD pumps were sold internationally in 2014 compared to 1,101 pumps sold in 2013.
Changes in foreign currency exchange rates favorably impacted net revenue by approximately $0.9 million, or 0.4%, in 2014 compared to 2013. In 2014, approximately 43% of our net revenue was denominated in foreign currencies including principally the Euro and British Pound compared to 45% in 2013. Movements in foreign currency exchange rates have had an effect on our reported revenue amounts in the past and could have a significant favorable or unfavorable impact on our reported revenue amounts in the future.
50
We expect to continue to generate and grow commercial revenue from product sales as we further expand our sales and marketing efforts on a global basis. Future product sales are dependent on many factors, including perception of product performance and market acceptance among physicians, patients, health care payers and the medical community as well as our capacity to meet customer demand by manufacturing sufficient quantities of our products.
Cost of Revenue
Cost of revenue includes costs associated with manufacturing and distributing our products and consists of direct materials, labor and overhead expenses allocated to the manufacturing process, provisions for excess or obsolete inventory, and shipping costs. Cost of revenue totaled approximately $92.2 million and $76.5 million in 2014 and 2013, respectively.
Gross profit and gross margin percentage for the years ended December 31, 2014 and 2013 were as follows:
| | | | | | | | |
| | | 2014 | | | 2013 | |
| | | (in thousands) | |
Gross profit | | $ | 186,225 | | | $ | 131,461 | |
| | | | | | | | |
Gross margin % | | | 67 | % | | | 63 | % |
| | | | | | | | |
The increase in gross margin percentage for 2014 compared to 2013 was primarily a result of production efficiencies driven by increased revenue and manufacturing throughput resulting in 5.0 percentage points of improvement, partially offset by 1.3 percentage points resulting from increases in reserve allowances including the battery and controller recalls noted above.
Selling, General and Administrative
Selling, general and administrative expenses include costs associated with selling and marketing our products and the general corporate administration of the Company. These costs are primarily related to salaries and wages and related employee costs, travel, marketing, external consultants and contractors, legal and accounting fees and general infrastructure costs, and include all operating costs not associated with or otherwise classified as research and development costs or cost of revenue.
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | Change | |
| | | (in thousands) | | | | |
Selling, general and administrative expenses | | $ | 87,177 | | | $ | 76,524 | | | | 14 | % |
| | | | | | | | | | | | |
% of operating expenses, excluding changes in fair value of contingent consideration | | | 42 | % | | | 43 | % | | | | |
| | | | | | | | | | | | |
The net increase of $10.7 million included approximately a net increase of $2.3 million of restructuring charges, primarily related to our acquisition of CircuLite. These expenses included lease exit costs associated with facilities we vacated in Massachusetts and New Jersey, severance costs and asset impairment charges. The remainder of the increase resulted primarily from commercial expansion and included $8.5 million of salaries and related costs associated with headcount growth, and $2.7 million of increased travel, conferences, tradeshows and other marketing expenditures. We also experienced an increase in non-cash share-based compensation expense of $1.4 million and an increase in medical device excise taxes of $1.4 million as a result of the Affordable Care Act (discussed below). These increases were partially offset by reductions across all other areas of spending, including allocated facility and overhead costs, of $5.6 million. In 2010, the Affordable Care Act and the Health Care and Education Reconciliation Act were signed into law. Among other things, these Acts, when taken together, impose a 2.3% excise tax on the U.S. sales of certain medical devices, including our devices, which became effective January 1, 2013. We have included this tax expense in selling, general and
51
administrative expenses on our consolidated statements of operations. We have not invoiced our customers for this tax as a separate charge, and the tax is not included as an element of revenue. The statutory rate of the medical device excise tax is 2.3% of revenue on initial sales of finished medical products sold in the United States.
We expect our selling, general and administrative expenses to continue to increase in 2015 compared to 2014 as we continue to expand our sales and distribution capabilities in an effort to increase market penetration on a global basis as well as enhance our administrative capabilities to support our overall corporate growth.
Research and Development
Research and development expenses are the direct and indirect costs associated with developing our products prior to commercialization, including the costs of operating clinical trials, and are expensed as incurred. These expenses fluctuate based on project level activity and consist primarily of salaries and wages and related employee costs of our research and development, clinical and regulatory staffs, external research and development costs, and materials and expenses associated with clinical trials. Research and development expenses also include costs associated with our compliance with FDA regulations. Additional costs include travel, facilities and overhead allocations.
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | Change | |
| | | (in thousands) | | | | |
Research and development expenses | | $ | 122,432 | | | $ | 102,483 | | | | 19 | % |
| | | | | | | | | | | | |
% of operating expenses, excluding changes in fair value of contingent consideration | | | 58 | % | | | 57 | % | | | | |
| | | | | | | | | | | | |
The $19.9 million increase in research and development expenses included approximately $7.8 million related to clinical and regulatory operations spend, including ongoing clinical trial studies, regulatory management and warning letter-related spending, $7.1 million in increased salaries and related costs associated with headcount growth, an increase in non-cash share-based compensation of $0.8 million and $0.5 million in fees related to the cancellation of a development agreement. In connection with our acquisition of CircuLite, we recorded restructuring charges aggregating $1.1 million, including contract termination fees and severance costs. All other research and development spending aggregated to an increase of approximately $2.6 million.
During the fourth quarter of 2014 we recorded an impairment charge of $2.6 million related to in-process research and development that was recorded in connection with our acquisition of CircuLite in December 2013. The charge resulted from a decision to discontinue development of the acquired SYNERGY micropump which we intend to replace with a version of our MVAD pump. During the fourth quarter of 2013, we recorded an impairment charge totaling $3.7 million to write-off goodwill and in-process research and development that was recorded in 2012 in connection with our acquisition of World Heart.See Note 6 in the accompanying consolidated financial statements for additional information.
Included in the amounts above for 2014 are incremental expenses of $5.4 million incurred in connection with the warning letter we received from the FDA in June 2014. We expect our warning letter-related expenses to remain high into the first quarter of 2015, but should decrease thereafter as we complete some of the resource intensive activities for which we have contracted external consultants and advisors.
We expect that research and development expenses will continue to represent a significant portion of our operating expenses for the foreseeable future as we continue to incur substantial development costs related to our next generation products, including the Pal controller, the MVAD System, the SYNERGY System and certain early research initiatives. We also expect to incur substantial costs for clinical trials for the HVAD System in new markets and expanded indications and for the MVAD System both in Europe and the United States, as well as ongoing clinical trial expenses associated with bridge-to-transplant post-approval study requirements and ongoing patient follow-up related to the ENDURANCE clinical trial.
52
Change in Fair Value of Contingent Consideration
On December 1, 2013, we acquired CircuLite, Inc. using a combination of cash and stock. In addition to initial consideration paid at closing, the former CircuLite securityholders may be entitled to receive additional shares of HeartWare common stock (or cash, in certain cases, at our discretion) upon the achievement of five specified performance milestones and royalty payments. We calculate the estimated fair value of the contingent consideration on a quarterly basis. In 2014, adjustments aggregating $23.3 million were recorded for the net decrease in the estimated fair value of the contingent consideration since December 31, 2013. The change in fair value of the contingent consideration in 2014 was due to several factors, including a $16.6 million reduction due to the likelihood of not achieving the performance milestone conditions related to the re-launch of the acquired form of the SYNERGY Surgical System following its loss of CE marking in the European Union in March 2014. In addition, we have discontinued development of the SYNERGY micropump and have focused our efforts on a version of our MVAD pump for our partial-assist program. As a result of this shift, we updated our future projections for the SYNERGY System and reassessed the probabilities of attaining certain contingent milestone payments under the terms of the merger agreement. Our updated analysis resulted in an aggregate $17.5 million decrease in the fair value of the contingent consideration related to certain milestones and royalty payments. This overall decrease in fair value was partially offset by an increase of $10.8 million due to accretion of the liability due to the passage of time.
Determining the estimated fair value of the contingent consideration requires significant management judgment or estimation. The estimated fair value is calculated using the income approach, with significant inputs that include various revenue assumptions, discount rates and applying a probability to each outcome. Material changes in any of these inputs could result in a significantly higher or lower fair value measurement. Potential valuation adjustments will be made in future accounting periods as additional information becomes available, including, among other items, progress toward developing the SYNERGY System, as well as revenue and milestone targets as compared to our current projections. Adjustments associated with changes in the estimated fair value of the contingent consideration are presented on a separate line item on our consolidated statements of operations and will be similarly presented in future accounting periods.
Foreign Exchange
We generate a substantial portion of our revenue and collect receivables in foreign currencies. Fluctuations in the exchange rate of the U.S. dollar against the Euro, British Pound and Australian dollar can result in foreign currency exchange gains and losses that may significantly affect our financial results. Continued fluctuation of these exchange rates could result in financial results that are not comparable from quarter to quarter.
In 2014, our net foreign exchange losses totaled approximately $5.0 million compared to net losses of approximately $0.1 million in 2013. During the second half of 2014, the Euro weakened significantly relative to the U.S. dollar. This was the primary contributor to the net foreign exchange losses experienced in 2014.
In 2014 and 2013, the majority of our realized and unrealized foreign exchange gains and losses resulted from the settlement of certain balance sheet accounts, primarily accounts receivable that were denominated in foreign currencies, and the remeasurement to U.S. dollars at period end of certain balance sheet accounts, denominated in foreign currencies, primarily the Euro. We expect to continue to realize foreign exchange gains and losses for the foreseeable future as a significant portion of our sales is denominated in foreign currencies. We do not currently utilize foreign currency contracts to manage foreign exchange risks.
Interest Expense
Interest expense in 2014 and 2013 primarily consisted of interest incurred on the principal amount of our convertible senior notes issued in December 2010, amortization of the related discount and amortization of the portion of the deferred financing costs allocated to the debt component. The convertible senior notes bear interest
53
at a rate of 3.5% per annum. The discount on the convertible senior notes and the deferred financing costs are being amortized to interest expense through the December 15, 2017 maturity date of the convertible senior notes using the effective interest method.
Interest expense was approximately $13.1 million and $12.2 million in 2014 and 2013, respectively. Interest incurred on the principal amount of the convertible senior notes at the 3.5% coupon rate was approximately $5.0 million in 2014 and 2013. Non-cash amortization of the discount and deferred financing costs totaled approximately $8.1 million and $7.2 million in 2014 and 2013, respectively.
Investment Income, net
Investment income is primarily derived from investments and cash and short-term deposit accounts held in the U.S. as well as note receivable interest on a strategic investment in an early stage privately-held company. The amortization of premium on our investments is also included in investment income, net. Investment income, net was approximately $0.7 million and $0.4 million in 2014 and 2013, respectively. The increase was primarily due to interest earned on the aforementioned note receivable. We continue to experience low interest rates on our deposits and available-for-sale investments.
Income Taxes
We are subject to taxation in the United States and jurisdictions outside of the United States. These jurisdictions have different marginal tax rates. Foreign earnings are considered to be permanently reinvested in operations outside the U.S. and therefore are not subject to U.S. income taxes until repatriated. As of December 31, 2014, we had no unrepatriated foreign earnings. We have incurred significant U.S. losses since inception, however, changes in issued capital and share ownership, as well as other factors, may limit our ability to utilize any net operating loss carry-forwards, and therefore a 100% valuation allowance has been recorded against our net deferred tax assets. In 2014 and 2013, our tax provision includes estimated foreign taxes in jurisdictions where wholly-owned subsidiaries may be subject to current taxes.
In accordance with ASC 740 we continue to record and evaluate tax positions for recognition using a more-likely-than-not threshold, and those tax positions eligible for recognition are measured as the largest amount of tax benefit that is greater than 50% likely of being realized upon the effective settlement with a taxing authority that has full knowledge of all relevant information.
Fiscal Years 2013 and 2012
Revenue, net
In November 2012, we received approval from the FDA for the HVAD System as a bridge to heart transplantation in patients with end-stage heart failure. This approval resulted in substantially increased sales in the United States compared to 2012 sales. 2013 sales reflected commercial activities both in the United States and internationally, while 2012 sales were derived from a mix of clinical trial activities in the United States and ongoing commercial sales of our HVAD system internationally. In 2013, domestic revenue comprised approximately 51% of our net revenue compared to approximately 25% in 2012.
Net revenue for the years ended December 31, 2013 and 2012 was as follows:
| | | | | | | | | | | | |
| | | 2013 | | | 2012 | | | Change | |
| | | (in thousands) | | | | |
Revenue, net | | $ | 207,929 | | | $ | 110,922 | | | | 87 | % |
| | | | | | | | | | | | |
In 2013, our U.S. revenue increased approximately $77.7 million, or 281%, to $105.3 million compared to U.S. revenue of approximately $27.6 million in 2012. A total of 978 HVAD pumps were sold in the U.S. in 2013
54
compared to 292 pumps sold in 2012. International revenue increased in 2013 by approximately $19.3 million, or 23%, to $102.6 million compared to international revenue of approximately $83.3 million in 2012. A total of 1,101 HVAD pumps were sold internationally in 2013 compared to 925 pumps sold internationally in 2012.
Changes in foreign currency exchange rates favorably impacted net revenue by approximately $2.0 million, or 1.8%, in 2013 compared to 2012. In 2013, approximately 45% of our net revenue was denominated in foreign currencies including principally the Euro and British pound.
Cost of Revenue
Cost of revenue totaled approximately $76.5 million and $51.0 million in 2013 and 2012, respectively.
Gross profit and gross margin percentage for the years ended December 31, 2013 and 2012 were as follows:
| | | | | | | | |
| | | 2013 | | | 2012 | |
| | | (in thousands) | |
Gross profit | | $ | 131,461 | | | $ | 59,899 | |
| | | | | | | | |
Gross margin % | | | 63 | % | | | 54 | % |
| | | | | | | | |
The increase in gross margin percentage for 2013 compared to 2012 was primarily a result of production efficiencies resulting from fixed overhead costs spread over a greater number of units produced and sold, and to a lesser extent an increase in the average per unit selling price in 2013 attributable to the introduction of commercial product in the United States during the fourth quarter of 2012. Compared to 2012 higher unit sales prices contributed 2.2% of the gross margin increase, with the balance of the increase related to the described production efficiencies.
Selling, General and Administrative
Selling, general and administrative expenses for the years ended December 31, 2013 and 2012 were as follows:
| | | | | | | | | | | | |
| | | 2013 | | | 2012 | | | Change | |
| | | (in thousands) | | | | |
Total selling, general and administrative expenses | | $ | 76,524 | | | $ | 53,945 | | | | 42 | % |
| | | | | | | | | | | | |
% of operating expenses | | | 43 | % | | | 39 | % | | | | |
| | | | | | | | | | | | |
During 2013, we continued to experience significant growth as we expanded our sales and distribution capabilities, especially in the U.S. in connection with the commercial launch of the HVAD System subsequent to receiving FDA approval in November 2012. We also experienced increased administrative costs as we expanded our administrative capabilities to support overall corporate growth.
The increase of $22.6 million resulted primarily from commercial expansion and included $8.0 million of salaries and related costs associated with headcount growth, $5.0 million of increased travel, conferences, tradeshows and other marketing expenditures, $3.5 million of professional fees and $2.0 million of non-cash share-based compensation expense. In addition, we incurred excise taxes of $2.4 million as a result of the Reconciliation Act (discussed above) and $0.6 million in severance costs in connection with the acquisition of CircuLite.
55
Research and Development
Research and development expenses for the years ended December 31, 2013 and 2012 were as follows:
| | | | | | | | | | | | |
| | | 2013 | | | 2012 | | | Change | |
| | | (in thousands) | | | | |
Total research and development expenses | | $ | 102,483 | | | $ | 83,548 | | | | 23 | % |
| | | | | | | | | | | | |
% of operating expenses | | | 57 | % | | | 61 | % | | | | |
| | | | | | | | | | | | |
The $18.9 million increase was primarily due to a $9.8 million increase in development project costs, including consumables, outside engineering, consultants and contractors. We also experienced a $7.0 million increase in salaries and related costs associated with headcount growth, and an increase in non-cash share-based compensation of $1.7 million. These increases were partially offset by a decrease in costs related to clinical trials of $5.4 million.
During the fourth quarter of 2013, we recorded an impairment charge totaling $3.7 million to write-off goodwill and in-process research and development that was recorded in 2012 in connection with our acquisition of World Heart. Subsequent to an evaluation of the ongoing research and development efforts surrounding the technology, we determined we would discontinue further development efforts needed to commercialize the MiFlow technology, other than with respect to certain know-how which has been incorporated into our development of the MVAD System.
Foreign Exchange
In 2013, our net foreign exchange losses totaled approximately $0.1 million compared to net gains of approximately $1.2 million in 2012. In 2013 and 2012, the majority of our realized and unrealized foreign exchange gains and losses resulted from the settlement of certain balance sheet accounts, primarily accounts receivable that were denominated in foreign currencies, and the remeasurement to U.S. dollars at period end of certain balance sheet accounts, denominated in foreign currencies, primarily the Euro. We expect to continue to realize foreign exchange gains and losses for the foreseeable future as a significant portion of our sales is denominated in foreign currencies. We do not currently utilize foreign currency contracts to manage foreign exchange risks.
Interest Expense
Interest expense in 2013 and 2012 primarily consisted of interest incurred on the principal amount of our convertible senior notes issued in December 2010, amortization of the related discount and amortization of the portion of the deferred financing costs allocated to the debt component. The convertible senior notes bear interest at a rate of 3.5% per annum. The discount on the convertible senior notes and the deferred financing costs are being amortized to interest expense through the December 15, 2017 maturity date of the convertible senior notes using the effective interest method.
Interest expense was approximately $12.2 million and $11.4 million in 2013 and 2012, respectively. Interest incurred on the principal amount of the convertible senior notes at the 3.5% coupon rate was approximately $5.0 million in 2013 and 2012. Non-cash amortization of the discount and deferred financing costs totaled approximately $7.2 million and $6.4 million in 2013 and 2012, respectively.
Investment Income, net
Investment income was primarily derived from investments and cash and short-term deposit accounts held in the U.S. The amortization of premium on our investments is also included in investment income, net.
56
Investment income, net was approximately $0.4 million and $0.2 million in 2013 and 2012, respectively. Due to our public offering of our common stock completed in March 2013, which resulted in net proceeds of approximately $141.0 million, we maintained higher average balances during 2013 compared to 2012.
Income Taxes
In 2013 and 2012, income tax expense related primarily to income earned by a foreign subsidiary. Foreign earnings were considered to be permanently reinvested in operations outside the U.S. and therefore we did not provide for U.S. income taxes on these unrepatriated foreign earnings.
Liquidity and Capital Resources
As of December 31, 2014, our cash and cash equivalents combined with short term investments were approximately $178.5 million as compared to $200.5 million at December 31, 2013.
Following is a summary of our cash flow activities for the years ended December 31, 2014, 2013 and 2012:
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | 2012 | |
| | | (in thousands) | |
Net cash used in operating activities | | $ | (17,174 | ) | | $ | (22,223 | ) | | $ | (59,862 | ) |
Net cash (used in) provided by investing activities | | | (46,556 | ) | | | (46,321 | ) | | | 72,825 | |
Net cash provided by financing activities | | | 721 | | | | 145,649 | | | | 2,509 | |
Effect of exchange rate changes on cash and cash equivalents | | | 3,075 | | | | (146 | ) | | | (808 | ) |
| | | | | | | | | | | | |
Net increase (decrease) in cash and cash equivalents | | $ | (59,934 | ) | | $ | 76,959 | | | $ | 14,664 | |
| | | | | | | | | | | | |
Cash Used in Operating Activities
Cash used in operating activities for the year ended December 31, 2014 included a net loss of approximately $19.4 million, adjustments for non-cash items of $22.3 million and cash used in working capital of $20.1 million. The net loss was driven by normal operating activities including the sale of the HVAD System in the United States and abroad, increased expenditures on research and development as well as increased administrative costs. Adjustments for non-cash items primarily consisted of $23.5 million of share-based compensation, $8.4 million of depreciation and amortization on long-lived assets, $7.7 million for the amortization of the discount on our convertible senior notes, $3.3 million related to impairment of intangible assets and fixed assets and $1.0 million loss on an equity investment. The above non-cash adjustments were partially offset by a $23.3 million decrease in fair value of contingent consideration related to the CircuLite acquisition. The decrease in cash from changes in working capital included $16.3 million for the purchase and manufacture of inventories, $11.8 million in increased trade accounts receivable, and $4.3 million for the payment of trade accounts payable. These amounts were partially offset by a decrease in prepaid expenses and other assets of $4.0 million and an increase in accrued liabilities of $7.3 million.
Cash used in operating activities for the year ended December 31, 2013 included a net loss of approximately $59.3 million and non-cash adjustments to net loss totaling approximately $40.9 million, which primarily consisted of $21.9 million of share-based compensation, $6.8 million for the amortization of the discount on our convertible senior notes, $7.0 million of depreciation and amortization on long-lived assets, and $3.7 million related to impairment of goodwill and intangible assets. Also included in cash used in operating activities in 2013 are approximately $4.9 million in increased prepaid expenses, $4.3 million for the purchase and manufacture of inventories and $2.4 million in increased trade accounts receivables. These amounts were partially offset by an increase in other accrued liabilities of $4.0 million and an increase in trade accounts payable of $3.6 million.
Cash used in operating activities for the year ended December 31, 2012 included a net loss of approximately $87.7 million and non-cash adjustments to net loss totaling approximately $31.8 million, which primarily
57
consisted of $18.8 million of share-based compensation, $6.0 million for the amortization of the discount on our convertible senior notes and $5.0 million of depreciation and amortization on long-lived assets. Also included in cash used in operating activities in 2012 are approximately $10.4 million in increased trade accounts receivable, $7.9 million for the purchase and manufacture of inventories and $0.9 million for prepaid expenses. These amounts were partially offset by an increase in trade accounts payable of $7.0 million, an increase in other accrued liabilities of $7.6 million and an increase in deferred rent of $0.7 million.
Cash (Used in) Provided by Investing Activities
In 2014, net cash used by investing activities included $39.0 million for the purchase (net of maturities) of available-for sale securities, $6.7 million used to acquire property, plant and equipment and $1.6 million for intellectual property. In 2014, we received approximately $0.7 million upon the release of a portion of the security deposit on one of our facility leases.
In 2013, net cash used by investing activities included $22.6 million for the purchase (net of maturities) of available-for sale securities, $20.0 million used for our acquisition of CircuLite and other strategic investments, $3.4 million used to acquire property, plant and equipment and $0.7 million received upon the sale of certain property, plant and equipment in connection with the closure of our Australian facility. Other investing activities in 2013 used cash of approximately $1.1 million.
In 2012, net cash provided by investing activities included $75.3 million received upon maturity (net of purchases) of available-for sale securities, $3.7 million received upon the acquisition of World Heart, $4.6 million used to acquire property, plant and equipment, $1.0 million used to acquire patents and $0.8 million paid for a security deposit on a facility lease.
Cash Provided by Financing Activities
On March 12, 2013, we entered into an Underwriting Agreement (the “Underwriting Agreement”) with J.P. Morgan Securities LLC, as representative of the several underwriters named in the Underwriting Agreement (the “Underwriters”), pursuant to which we agreed to sell and the Underwriters agreed to purchase, subject to and upon terms and conditions set forth therein, an aggregate of 1,500,000 shares of our common stock at a net sales price of $81.9114 per share (the public offering price of $86.45 per share minus the underwriting discount). We also granted the Underwriters an option to purchase 225,000 additional shares of our common stock at the public offering price less the underwriting discount, which the Underwriters exercised in full on March 13, 2013. The closing of the offering occurred on March 18, 2013. After fees and related expenses, net proceeds from the offering were approximately $141.0 million. The offering was completed pursuant to a prospectus supplement, dated March 12, 2013, to a shelf registration statement on Form S-3 that was previously filed with the SEC and which was declared effective on December 9, 2010.
In 2014, 2013 and 2012, we received approximately $0.9 million, $4.9 million and $2.7 million, respectively, from the exercise of stock options.
Operating Capital and Capital Expenditure Requirements
We have incurred substantial operating losses to date and anticipate that we will continue to consume cash and incur substantial net losses as we expand our sales and marketing capabilities, develop new products and seek regulatory approvals for expanded indications of the HVAD System in the U.S. In 2015, cash on hand is expected to primarily be used to fund our ongoing operations, including:
| | • | | expanding our sales and marketing capabilities on a global basis; |
| | • | | growing market penetration particularly in U.S.; |
| | • | | continued product development, including development of the MVAD pump and Pal controller; |
58
| | • | | pre-clinical and clinical costs relating to prospective first human implants of the MVAD pump, and clinical trials related to expanded indications of the HVAD System; |
| | • | | development of the SYNERGY System, including the next generation endovascular system; |
| | • | | regulatory and other compliance functions, including activities to enhance our quality systems in response to the warning letter we received from the FDA in June 2014; |
| | • | | expand work in process and finished goods inventory to support ongoing operations; |
| | • | | planned investments in infrastructure to support our growth; and |
| | • | | general working capital. |
Our convertible notes bear interest at a rate of 3.5% per annum, payable semi-annually in arrears on June 15 and December 15 of each year. To date, all interest payments have been paid on a timely basis. Based on the outstanding principal amount of our convertible senior notes at December 31, 2014, the semi-annual interest payments due on June 15 and December 15, 2015 will be approximately $2.5 million each. These amounts are expected to be paid from cash on hand.
We believe cash on hand and investment balances as of December 31, 2014 are sufficient to support our planned operations for at least the next twelve months. At December 31, 2014, approximately $4.0 million of our cash on hand was held in foreign locations, including Australia, Germany and the United Kingdom. To date, the Company has not had unremitted foreign earnings and has not incurred U.S. federal and state income taxes related to repatriated earnings. As our operations in our foreign subsidiaries grow, we may generate foreign earnings. Any repatriation of those earnings to the United States would likely result in us incurring federal and state income taxes. We currently plan to permanently reinvest any earnings of our foreign subsidiaries.
Because of the numerous risks and uncertainties associated with the development of medical devices, we are unable to estimate the exact amounts of capital outlays and operating expenditures necessary to maintain regulatory approvals, fund commercial expansion, and develop and obtain regulatory approvals for new products. Our future capital requirements will depend on many factors, including but not limited to the following:
| | • | | implementation of systemic improvements necessary to satisfactorily address the observations cited in the June 2, 2014 warning letter we received from the FDA; |
| | • | | commercial acceptance of our products; |
| | • | | reimbursement of our products by governmental agencies and third party payers; |
| | • | | costs to manufacture and ensure regulatory compliance of our products; |
| | • | | expenses required to operate multiple clinical trials; |
| | • | | further product research and development for next generation products and expanding indications for our products as well as efforts to sustain and implement incremental improvements to existing products; |
| | • | | expanding our sales and marketing capabilities on a global basis; |
| | • | | broadening our infrastructure in order to meet the needs of our growing operations, including regulatory compliance; |
| | • | | expenses related to funding and integrating strategic investments, acquisitions and collaborative arrangements; |
| | • | | payment of the 2.3% excise tax on gross revenue from the sale of our medical devices in the United States imposed by the Patient Protection and Affordable Care Act; |
| | • | | payment of our convertible notes on maturity if not converted or refinanced; and |
| | • | | complying with the requirements related to being a public company in the U.S. |
59
Contractual Obligations
At December 31, 2014, our contractual financial obligations and commitments by due dates were as follows:
| | | | | | | | | | | | | | | | | | | | |
| | | Total | | | Less
than 1
year | | | 1-3 years | | | 3-5 years | | | Thereafter | |
| | | (in thousands) | |
Convertible senior notes | | $ | 158,844 | | | $ | 5,031 | | | $ | 153,813 | | | $ | — | | | $ | — | |
Operating lease obligations | | | 24,124 | | | | 3,618 | | | | 7,221 | | | | 7,415 | | | | 5,870 | |
Purchase obligations | | | 37,327 | | | | 36,398 | | | | 610 | | | | 319 | | | | — | |
Other | | | 1,552 | | | | 160 | | | | 203 | | | | 200 | | | | 989 | |
| | | | | | | | | | | | | | | | | | | | |
Total | | $ | 221,847 | | | $ | 45,207 | | | $ | 161,847 | | | $ | 7,934 | | | $ | 6,859 | |
| | | | | | | | | | | | | | | | | | | | |
From time to time we invest in certain development stage entities in connection with research activities. The above table does not reflect certain contingent milestone payments in connection with these arrangements as the amounts and timing of payment are indeterminate at this time.
On December 1, 2013, we acquired CircuLite, Inc. using a combination of cash and stock. In addition to initial consideration paid at closing, the former CircuLite securityholders may be entitled to receive additional shares of HeartWare common stock (or cash, in certain cases, at our discretion) upon the achievement of five specified performance milestones and royalty payments. The above table does not reflect these milestone payments, which may be payable over the next 10 years as the timing and amount of the milestone payments are indeterminate at this time. The maximum amount of the aggregate milestone payments could be $320 million. As of December 31, 2014, the fair value of the contingent payments was approximately $43.7 million and is reflected on our consolidated balance sheet.
As of December 31, 2014, our potential liability for uncertain tax positions was approximately $3.2 million, including interest. Due to the degree of uncertainty regarding these liabilities, we are unable to make a reasonably reliable estimate of the amount and period in which these liabilities might be realized.
Off-Balance Sheet Arrangements
The Company has no off-balance sheet arrangements.
60
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Market risk represents the risk of changes in the value of market risk sensitive instruments caused by fluctuations in interest rates, foreign exchange rates and commodity prices. Changes in these factors could cause fluctuations in our results of operations and cash flows.
Interest Rate Risk
Our exposure to interest rate risk is currently confined to interest earnings on our cash and cash equivalents that are invested in highly liquid money market funds, short-term time deposits, short-term bank notes and short-term commercial paper. The primary objective of our investment activities is to preserve our capital to fund operations. We also seek to generate reasonable income from our investments without assuming significant risk. We do not presently use derivative financial instruments in our investment portfolio. Our cash and investments policy emphasizes liquidity and preservation of principal over other portfolio considerations.
If interest rates rise, the market value of our investment portfolio may decline, which could result in a loss if we choose or are forced to sell an investment before its scheduled maturity. We do not utilize derivative financial instruments to manage interest rate risks.
Our convertible senior notes do not bear interest rate risk as the notes were issued with a fixed interest rate of 3.5% per annum.
Foreign Currency Rate Fluctuations
We conduct business in foreign countries. For U.S. reporting purposes, we translate all assets and liabilities of our non-U.S. entities at the period-end exchange rate and revenue and expenses at the average exchange rates in effect during the periods. The net effect of these translation adjustments is shown in the accompanying consolidated financial statements as a component of stockholders’ equity.
We generate a significant portion of our revenue and collect receivables in foreign currencies. Fluctuations in the exchange rate of the U.S. dollar against major foreign currencies, including the Euro, British Pound and Australian dollar, can result in foreign currency exchange gains and losses that may significantly impact our financial results. These foreign currency transaction and translation gains and losses are presented as a separate line item on our consolidated statements of operations. Continued fluctuation of these exchange rates could result in financial results that are not comparable from quarter to quarter. We do not currently utilize foreign currency contracts to mitigate the gains and losses generated by the re-measurement of non-functional currency assets and liabilities but do hold cash reserves in currencies in which those reserves are anticipated to be expended.
61
Item 8. Financial Statements and Supplementary Data
HEARTWARE INTERNATIONAL, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
62
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
HeartWare International, Inc.
We have audited the accompanying consolidated balance sheets of HeartWare International, Inc. (a Delaware corporation) and subsidiaries (the “Company”) as of December 31, 2014 and 2013, and the related consolidated statements of operations, comprehensive loss, changes in stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014. We also have audited the Company’s internal control over financial reporting as of December 31, 2014, based on criteria established in the 2013Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Annual Report. Our responsibility is to express an opinion on these financial statements and an opinion on the Company’s internal control over financial reporting based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of HeartWare International, Inc. and subsidiaries as of December 31, 2014 and 2013, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on criteria established in the 2013Internal Control—Integrated Framework issued by COSO.
/s/ Grant Thornton LLP
Fort Lauderdale, Florida
March 2, 2015
63
HEARTWARE INTERNATIONAL, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except per share data)
| | | | | | | | |
| | | December 31, | |
| | | 2014 | | | 2013 | |
ASSETS | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 102,946 | | | $ | 162,880 | |
Short-term investments | | | 75,535 | | | | 37,596 | |
Accounts receivable, net | | | 38,041 | | | | 28,052 | |
Inventories | | | 54,046 | | | | 40,876 | |
Prepaid expenses and other current assets | | | 5,975 | | | | 11,205 | |
| | | | | | | | |
Total current assets | | | 276,543 | | | | 280,609 | |
Property, plant and equipment, net | | | 19,036 | | | | 18,562 | |
Goodwill | | | 61,390 | | | | 61,596 | |
In-process research and development | | | 32,850 | | | | 35,500 | |
Other intangible assets, net | | | 17,807 | | | | 15,975 | |
Deferred financing costs, net | | | 1,552 | | | | 1,964 | |
Long-term investments and other assets | | | 14,635 | | | | 15,621 | |
| | | | | | | | |
Total assets | | $ | 423,813 | | | $ | 429,827 | |
| | | | | | | | |
LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 13,322 | | | $ | 17,914 | |
Other accrued liabilities | | | 36,589 | | | | 35,276 | |
| | | | | | | | |
Total current liabilities | | | 49,911 | | | | 53,190 | |
Convertible senior notes, net | | | 114,803 | | | | 107,125 | |
Contingent liabilities (See Notes 4 and 6) | | | 43,740 | | | | 67,000 | |
Other long-term liabilities | | | 6,825 | | | | 3,905 | |
| | |
Commitments and contingencies—See Note 17 | | | | | | | | |
Stockholders’ equity: | | | | | | | | |
Preferred stock—$.001 par value; 5,000 shares authorized; no shares issued and outstanding at December 31, 2014 and 2013, respectively | | | — | | | | — | |
Common stock—$.001 par value; 25,000 shares authorized; 17,156 and 16,878 shares issued and outstanding at December 31, 2014 and 2013, respectively | | | 17 | | | | 17 | |
Additional paid-in capital | | | 565,609 | | | | 535,817 | |
Accumulated deficit | | | (348,719 | ) | | | (329,353 | ) |
Accumulated other comprehensive loss: | | | | | | | | |
Cumulative translation adjustments | | | (8,112 | ) | | | (7,859 | ) |
Unrealized loss on investments | | | (261 | ) | | | (15 | ) |
| | | | | | | | |
Total accumulated other comprehensive loss | | | (8,373 | ) | | | (7,874 | ) |
| | | | | | | | |
Total stockholders’ equity | | | 208,534 | | | | 198,607 | |
| | | | | | | | |
Total liabilities and stockholders’ equity | | $ | 423,813 | | | $ | 429,827 | |
| | | | | | | | |
The accompanying notes are an integral part of these financial statements.
64
HEARTWARE INTERNATIONAL, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share data)
| | | | | | | | | | | | |
| | | Years Ended December 31, | |
| | | 2014 | | | 2013 | | | 2012 | |
Revenue, net | | $ | 278,420 | | | $ | 207,929 | | | $ | 110,922 | |
Cost of revenue | | | 92,195 | | | | 76,468 | | | | 51,023 | |
| | | | | | | | | | | | |
Gross profit | | | 186,225 | | | | 131,461 | | | | 59,899 | |
| | | |
Operating expenses: | | | | | | | | | | | | |
Selling, general and administrative | | | 87,177 | | | | 76,524 | | | | 53,945 | |
Research and development | | | 122,432 | | | | 102,483 | | | | 83,548 | |
Change in fair value of contingent consideration | | | (23,260 | ) | | | — | | | | — | |
| | | | | | | | | | | | |
Total operating expenses | | | 186,349 | | | | 179,007 | | | | 137,493 | |
Loss from operations | | | (124 | ) | | | (47,546 | ) | | | (77,594 | ) |
| | | |
Other income (expense): | | | | | | | | | | | | |
Foreign exchange (loss) gain | | | (4,952 | ) | | | (70 | ) | | | 1,224 | |
Interest expense | | | (13,133 | ) | | | (12,224 | ) | | | (11,404 | ) |
Investment income, net | | | 666 | | | | 370 | | | | 243 | |
Other, net | | | (1,263 | ) | | | 626 | | | | (187 | ) |
| | | | | | | | | | | | |
Loss before taxes | | | (18,806 | ) | | | (58,844 | ) | | | (87,718 | ) |
Income tax expense | | | 560 | | | | 467 | | | | — | |
| | | | | | | | | | | | |
Net loss | | $ | (19,366 | ) | | $ | (59,311 | ) | | $ | (87,718 | ) |
| | | | | | | | | | | | |
Net loss per common share—basic and diluted | | $ | (1.14 | ) | | $ | (3.69 | ) | | $ | (6.15 | ) |
| | | | | | | | | | | | |
Weighted average shares outstanding—basic and diluted | | | 16,992 | | | | 16,066 | | | | 14,252 | |
| | | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
65
HEARTWARE INTERNATIONAL, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In thousands)
| | | | | | | | | | | | |
| | | Years Ended December 31, | |
| | | 2014 | | | 2013 | | | 2012 | |
Net loss | | $ | (19,366 | ) | | $ | (59,311 | ) | | $ | (87,718 | ) |
Other comprehensive income (loss) | | | | | | | | | | | | |
Foreign currency translation adjustments | | | (253 | ) | | | 180 | | | | (408 | ) |
Unrealized (loss) gain on investments | | | (246 | ) | | | 9 | | | | (1 | ) |
| | | | | | | | | | | | |
Comprehensive loss | | $ | (19,865 | ) | | $ | (59,122 | ) | | $ | (88,127 | ) |
| | | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
66
HEARTWARE INTERNATIONAL, INC.
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY
(In thousands, except per share data)
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Common Shares,
$.001 Par Value Per Share | | | Additional
Paid-In
Capital | | | Accumulated
Deficit | | | Accumulated
Other
Comprehensive
Loss | | | Total | |
| | | Shares
Issued | | | Amount | | | | | |
Balance, December 31, 2011 | | | 14,114 | | | $ | 14 | | | $ | 316,748 | | | $ | (182,324 | ) | | $ | (7,654 | ) | | $ | 126,784 | |
Issuance of common stock in connection with acquisition of World Heart | | | 83 | | | | — | | | | 6,942 | | | | — | | | | — | | | | 6,942 | |
Issuance of common stock in connection with license agreement and acquisition of intellectual property | | | 13 | | | | — | | | | 1,098 | | | | — | | | | — | | | | 1,098 | |
Issuance of common stock pursuant to share-based awards | | | 372 | | | | 1 | | | | 2,708 | | | | — | | | | — | | | | 2,709 | |
Share-based compensation | | | — | | | | — | | | | 18,805 | | | | — | | | | — | | | | 18,805 | |
Net loss | | | — | | | | — | | | | — | | | | (87,718 | ) | | | — | | | | (87,718 | ) |
Other comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | (409 | ) | | | (409 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2012 | | | 14,582 | | | | 15 | | | | 346,301 | | | | (270,042 | ) | | | (8,063 | ) | | | 68,211 | |
Issuance of common stock pursuant to public offering, net of offering costs | | | 1,725 | | | | 2 | | | | 140,977 | | | | — | | | | — | | | | 140,979 | |
Issuance of common stock in connection with acquisition of CircuLite | | | 226 | | | | — | | | | 21,794 | | | | — | | | | — | | | | 21,794 | |
Issuance of common stock pursuant to share-based awards | | | 345 | | | | — | | | | 4,871 | | | | — | | | | — | | | | 4,871 | |
Share-based compensation | | | — | | | | — | | | | 21,874 | | | | — | | | | — | | | | 21,874 | |
Net loss | | | — | | | | — | | | | — | | | | (59,311 | ) | | | — | | | | (59,311 | ) |
Other comprehensive income | | | — | | | | — | | | | — | | | | — | | | | 189 | | | | 189 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2013 | | | 16,878 | | | | 17 | | | | 535,817 | | | | (329,353 | ) | | | (7,874 | ) | | | 198,607 | |
Issuance of common stock in connection with an intellectual property agreement | | | 50 | | | | — | | | | 5,000 | | | | — | | | | — | | | | 5,000 | |
Issuance of common stock in connection with acquisition of CircuLite | | | 3 | | | | — | | | | 329 | | | | — | | | | — | | | | 329 | |
Issuance of common stock pursuant to share-based awards | | | 225 | | | | — | | | | 921 | | | | — | | | | — | | | | 921 | |
Share-based compensation | | | — | | | | — | | | | 23,542 | | | | — | | | | — | | | | 23,542 | |
Net loss | | | — | | | | — | | | | — | | | | (19,366 | ) | | | — | | | | (19,366 | ) |
Other comprehensive income | | | — | | | | — | | | | — | | | | — | | | | (499 | ) | | | (499 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2014 | | | 17,156 | | | $ | 17 | | | $ | 565,609 | | | $ | (348,719 | ) | | $ | (8,373 | ) | | $ | 208,534 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
67
HEARTWARE INTERNATIONAL, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| | | | | | | | | | | | |
| | | Years Ended December 31, | |
| | | 2014 | | | 2013 | | | 2012 | |
CASH FLOWS FROM OPERATING ACTIVITIES | | | | | | | | | | | | |
Net loss | | $ | (19,366 | ) | | $ | (59,311 | ) | | $ | (87,718 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
Depreciation of property, plant and equipment | | | 6,674 | | | | 6,451 | | | | 4,826 | |
Amortization of intangible assets | | | 1,724 | | | | 598 | | | | 204 | |
Impairment of fixed assets | | | 607 | | | | — | | | | — | |
Impairment of goodwill and intangible assets | | | 2,650 | | | | 3,726 | | | | — | |
Share-based compensation expense | | | 23,542 | | | | 21,874 | | | | 18,805 | |
Amortization of premium on investments | | | 810 | | | | 635 | | | | 555 | |
Amortization of discount on convertible senior notes | | | 7,678 | | | | 6,809 | | | | 6,039 | |
Amortization of deferred financing costs | | | 412 | | | | 365 | | | | 324 | |
Change in fair value of contingent consideration | | | (23,260 | ) | | | — | | | | — | |
Other | | | 1,498 | | | | 467 | | | | 1,028 | |
Change in operating assets and liabilities: | | | | | | | | | | | | |
Accounts receivable | | | (11,786 | ) | | | (2,372 | ) | | | (10,390 | ) |
Inventories | | | (16,284 | ) | | | (4,331 | ) | | | (7,909 | ) |
Prepaid expenses and other current assets | | | 3,985 | | | | (4,850 | ) | | | (867 | ) |
Accounts payable | | | (4,300 | ) | | | 3,564 | | | | 6,991 | |
Other accrued liabilities | | | 7,324 | | | | 3,975 | | | | 7,589 | |
Other long-term liabilities | | | 918 | | | | 177 | | | | 661 | |
| | | | | | | | | | | | |
Net cash used in operating activities | | | (17,174 | ) | | | (22,223 | ) | | | (59,862 | ) |
CASH FLOWS FROM INVESTING ACTIVITIES | | | | | | | | | | | | |
Purchases of investments | | | (58,864 | ) | | | (28,905 | ) | | | (28,230 | ) |
Maturities of investments | | | 19,870 | | | | 6,346 | | | | 103,548 | |
Acquisition of World Heart, net of cash acquired | | | — | | | | — | | | | 3,687 | |
Acquisition of CircuLite, net of cash acquired | | | — | | | | (9,985 | ) | | | — | |
Investment in unconsolidated affiliate | | | — | | | | (10,000 | ) | | | — | |
Additions to property, plant and equipment, net | | | (6,721 | ) | | | (2,692 | ) | | | (4,406 | ) |
Additions to patents | | | (1,555 | ) | | | (814 | ) | | | (1,024 | ) |
Cash received from (paid for) security deposits | | | 714 | | | | (271 | ) | | | (750 | ) |
| | | | | | | | | | | | |
Net cash (used in) provided by investing activities | | | (46,556 | ) | | | (46,321 | ) | | | 72,825 | |
CASH FLOWS FROM FINANCING ACTIVITIES | | | | | | | | | | | | |
Proceeds from issuance of common stock | | | — | | | | 149,126 | | | | — | |
Payment of common stock issuance costs | | | — | | | | (8,148 | ) | | | — | |
Repayment of promissory note | | | (200 | ) | | | (200 | ) | | | (200 | ) |
Proceeds from exercise of stock options | | | 921 | | | | 4,871 | | | | 2,709 | |
| | | | | | | | | | | | |
Net cash provided by financing activities | | | 721 | | | | 145,649 | | | | 2,509 | |
Effect of exchange rate changes on cash and cash equivalents | | | 3,075 | | | | (146 | ) | | | (808 | ) |
| | | | | | | | | | | | |
CHANGE IN CASH AND CASH EQUIVALENTS | | | (59,934 | ) | | | 76,959 | | | | 14,664 | |
CASH AND CASH EQUIVALENTS—BEGINNING OF PERIOD | | | 162,880 | | | | 85,921 | | | | 71,257 | |
| | | | | | | | | | | | |
CASH AND CASH EQUIVALENTS—END OF PERIOD | | $ | 102,946 | | | $ | 162,880 | | | $ | 85,921 | |
| | | | | | | | | | | | |
Supplemental disclosure of cash flow information: | | | | | | | | | | | | |
Cash paid for interest | | $ | 5,040 | | | $ | 5,049 | | | $ | 5,040 | |
| | | | | | | | | | | | |
Cash paid for income taxes | | $ | 96 | | | $ | — | | | $ | — | |
| | | | | | | | | | | | |
Supplemental disclosure of non-cash investing and financing activities: | | | | | | | | | | | | |
Shares issued to acquire World Heart | | $ | — | | | $ | — | | | $ | 6,942 | |
| | | | | | | | | | | | |
Shares issued to acquire CircuLite | | $ | 329 | | | $ | 21,794 | | | $ | — | |
| | | | | | | | | | | | |
Contingent consideration related to acquisition of CircuLite | | $ | — | | | $ | 67,000 | | | $ | — | |
| | | | | | | | | | | | |
Shares issued to acquire intellectual property | | $ | 5,000 | | | $ | — | | | $ | 1,098 | |
| | | | | | | | | | | | |
Non-cash increase to patents and intellectual property | | $ | 2,000 | | | $ | 5,000 | | | $ | — | |
| | | | | | | | | | | | |
Transfers from inventory to property, plant and equipment | | $ | 1,153 | | | $ | 2,072 | | | $ | 1,766 | |
| | | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
68
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1. Description of Business
HeartWare International, Inc., referred to in these notes collectively with its subsidiaries as “we,” “our,” “us,” “HeartWare,” “the HeartWare Group” or the “Company”, is a medical device company that develops, manufactures and markets miniaturized implantable heart pumps, or ventricular assist devices, to treat patients suffering from advanced heart failure.
The HeartWare Ventricular Assist System (the “HVAD System”), which includes a ventricular assist device (“VAD”) or blood pump, patient accessories and surgical tools, is designed to provide circulatory support for patients in the advanced stage of heart failure. The core of the HVAD System is a proprietary continuous flow blood pump, the HVAD pump, which is a full-output device capable of pumping up to 10 liters of blood per minute. The HVAD System is designed to be implanted adjacent to the heart, avoiding abdominal surgery, which is generally required to implant similar devices.
In November 2012, we received approval from the United States Food and Drug Administration (“FDA”) for the HVAD System as a bridge to heart transplantation in patients with end-stage heart failure. The HVAD System has been available in the European Union since receiving CE marking in 2009. In May 2012, we received an expanded European label for long-term use of the HVAD System in all patients at risk of death from refractory, end-stage heart failure. As of December 31, 2014, there have been over 7,000 implants of the HVAD System in patients at over 270 health care sites in 41 countries
On August 27, 2013, the FDA approved an Investigational Device Exemption (“IDE”) Supplement allowing HeartWare to commence enrollment in an additional patient cohort for the ENDURANCE clinical trial. In this supplemental cohort, HeartWare intends to enroll up to 310 patients receiving the HVAD System, as well as up to an additional 155 control patients using a randomization scheme consistent with the ENDURANCE protocol. Patients will be followed for 12 months after implant. In 2016, HeartWare intends to incorporate the data from both this supplemental cohort and ENDURANCE into an anticipated PMA Application seeking approval of the HVAD System for the destination therapy indication.
We continue to expand our pipeline through research and development into next generation products and peripherals and through ongoing and new clinical trials and to expand our presence in commercial markets outside of the United States. Among these activities, we are developing our next generation miniaturized device, the MVAD System. The MVAD System is based on the same technology platform as the HVAD System but adopts an axial flow, rather than a centrifugal flow, configuration and is being developed in multiple designs. The MVAD pump is less than one-half the size of the HVAD pump and can provide partial or full support. The MVAD platform is designed to allow for a variety of configurations and surgical placements with the goal towards further reduction of surgical invasiveness while producing superior clinical results. In December 2014, we initiated submissions to commence a CE Mark study at 9 international sites and are preparing regulatory submissions to commence an IDE study in the United States.
We are headquartered in Framingham, Massachusetts. We have operating facilities in Miami Lakes, Florida, Hannover, Germany, and Arden Hills, Minnesota.
HeartWare International, Inc. shares trade on the NASDAQ Stock Market under the symbol of “HTWR”.
Note 2. Liquidity
We have funded our operations primarily through product revenue, the issuance of shares of our common stock and the issuance of convertible notes. At December 31, 2014, we had approximately $179.7 million of cash, cash equivalents and investments. Our cash, cash equivalents and investments are expected to be used
69
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
primarily to fund our ongoing operations including expanding our sales and marketing capabilities on a global basis, research and development (including clinical trials) of new and existing products, components and accessories, regulatory and other compliance functions as well as for general working capital. We believe our cash, cash equivalents and investment balances are sufficient to support our planned operations for at least the next twelve months.
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States, which contemplate continuation of the Company as a going concern. We have incurred substantial losses from operations since our inception, and such losses have continued through December 31, 2014. At December 31, 2014, we had an accumulated deficit of approximately $348.7 million.
Note 3. Significant Accounting Policies
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the HeartWare Group. All inter-company balances and transactions have been eliminated in consolidation. We hold certain investments in small privately-held development-stage entities which are included in other assets on our consolidated balance sheets. In accordance with FASB ASC 810, we analyzed the investments to determine whether the investments are variable interests or interests that give us a controlling financial interest in a variable interest entity (“VIE”). As of December 31, 2014, we determined there were no VIEs required to be consolidated, because we are not the primary beneficiary, as we do not have the power to direct the most meaningful activities of the VIE. Investments where we do not exercise operating and financial control are accounted for under the equity method or cost method depending on our ownership interest.
Accounting Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States (“U.S. GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting periods. Actual results could differ from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents, which primarily consist of money market funds, are recorded in the consolidated balance sheets at cost, which approximates fair value. All highly liquid investments with an original maturity of three months or less at the date of purchase are considered to be cash equivalents.
Investments
Our investments classified as available-for-sale are stated at fair value with unrealized gains and losses reported in accumulated other comprehensive loss within stockholders’ equity. We classify our available-for-sale investments as short-term if their remaining time to maturity at purchase is beyond three months, but less than twenty-four months. Investments with maturities at purchase beyond one year, but less than twenty-four months, may be classified as short-term based on their highly liquid nature and because such marketable securities represent the investment of cash that is available for current operations. Interest on investments classified as available-for-sale is included in investment income, net. Premiums paid on our short-term investments are amortized over the remaining term of the investment and such amortization is included in investment income, net.
70
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Receivables
Accounts receivable consists of amounts due from the sale of our HVAD System to our customers, which include hospitals, health research institutions and medical device distributors. We grant credit to customers in the normal course of business, but do not require collateral or any other security to support credit sales. Our receivables are geographically dispersed, with a significant portion from customers located in Europe and other foreign countries. At December 31, 2014, no customer had an accounts receivable balance greater than 10% of our total accounts receivable. At December 31, 2013, one customer had an accounts receivable balance greater than 10% of total accounts receivable representing approximately 15% of our total accounts receivable.
We maintain allowances for doubtful accounts for estimated losses that may result from an inability to collect payments owed to us for product sales. We regularly review the allowance by considering factors such as historical experience, the age of the accounts receivable balances and local economic conditions that may affect a customer’s ability to pay. Account balances are charged off against the allowance after appropriate collection efforts are exhausted and we feel it is probable that the receivable will not be recovered.
The following table summarizes the change in our allowance for doubtful accounts for the years ended December 31, 2014, 2013 and 2012:
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | 2012 | |
| | | (in thousands) | |
Beginning balance | | $ | 495 | | | $ | 750 | | | $ | 500 | |
Charges (reversals) to expense | | | 181 | | | | (255 | ) | | | 250 | |
Charge-offs | | | (5 | ) | | | — | | | | — | |
| | | | | | | | | | | | |
Ending balance | | $ | 671 | | | $ | 495 | | | $ | 750 | |
| | | | | | | | | | | | |
As of December 31, 2014 and 2013, we did not have an allowance for returns.
Inventories
Inventories are stated at the lower of cost or market. Cost is determined using a first-in, first-out, or FIFO, method. Work-in-process and finished goods manufactured or assembled by us include direct and indirect labor and manufacturing overhead. Finished goods include product which is ready-for-use and which is held by us or by our customers on a consignment basis.
We review our inventory for excess or obsolete inventory and write-down obsolete or otherwise unmarketable inventory to its estimated net realizable value. Obsolescence may occur due to product expiring or product improvements rendering previous versions obsolete.
Property, Plant and Equipment
We record property, plant and equipment and leasehold improvements at historical cost. Expenditures for maintenance and repairs are recorded to expense; additions and improvements are capitalized. We generally provide for depreciation using the straight-line method at rates that approximate the estimated useful lives of the assets. Leasehold improvements are amortized on a straight-line basis over the shorter of the useful life of the improvement or the remaining term of the lease.
71
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Property, plant and equipment, net consists of the following:
| | | | | | | | | | |
| | | Estimated
Useful Lives | | December 31, | |
| | | | 2014 | | | 2013 | |
| | | | | (in thousands) | |
Property, plant and equipment | | | | | | | | | | |
Machinery and equipment | | 1.5 to 7 years | | $ | 21,279 | | | $ | 19,790 | |
Leasehold improvements | | 3 to 10 years | | | 9,070 | | | | 7,131 | |
Office equipment, furniture and fixtures | | 5 to 7 years | | | 2,206 | | | | 1,294 | |
Purchased software | | 1 to 7 years | | | 6,474 | | | | 5,057 | |
| | | | | | | | | | |
| | | | | 39,029 | | | | 33,272 | |
Less: accumulated depreciation | | | | | (19,993 | ) | | | (14,710 | ) |
| | | | | | | | | | |
| | | | $ | 19,036 | | | $ | 18,562 | |
| | | | | | | | | | |
Depreciation expense was $6.7 million, $6.5 million, and $4.8 million for the years ended December 31, 2014, 2013 and 2012, respectively.
During the year ended December 31, 2014, we ceased activities at the former headquarters of CircuLite in Teaneck, New Jersey and vacated the facility. We recorded an impairment charge of $0.6 million related to certain office equipment and software at the facility upon their discontinued use. This amount is included in selling, general and administrative expenses on our condensed consolidated statements of operations.
During the year ended December 31, 2013, we ceased certain development activities performed in our Australian facility, and relocated those activities to the United States. We sold a portion of the fixed assets at our Australian facility while others were written-off upon their discontinued use. The aggregate loss on disposal of fixed assets sold or written-off in 2013 in connection with this action was $0.5 million. This amount is included in research and development expenses on our consolidated statements of operations.
We enter into agreements with medical centers participating in our U.S. clinical trials under which we loan certain equipment, including patient monitors, to the center to be used throughout the trials. The equipment loaned to the centers is classified as a long-lived asset and included as a component of property, plant and equipment (machinery and equipment) on our consolidated balance sheets. Depreciation expense on equipment subject to these agreements is classified in cost of revenue and is computed using the straight-line method based on the estimated useful life of three years.
We also enter into short-term cancellable rental agreements with certain commercial customers for components of the HVAD System, including patient monitors and controllers. Under the terms of such agreements, we provide the equipment to the customers, but we retain title to the equipment. Equipment subject to rental agreements is classified as a long-lived asset and included as a component of property, plant and equipment (machinery and equipment). Depreciation expense on equipment subject to these agreements is classified in cost of revenue and is computed using the straight-line method based on the estimated useful life of fifteen months.
The net carrying value of equipment subject to the agreements discussed above was approximately $0.8 million and $1.3 million as of December 31, 2014 and 2013, respectively.
Deferred Financing Costs
Costs incurred in connection with the issuance of our convertible senior notes were allocated between the liability component and the equity component as further discussed in Note 10. The issuance costs allocated to
72
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
convertible senior notes was capitalized within deferred financing costs, net on our consolidated balance sheets. These costs are being amortized using the effective interest method through December 15, 2017, the maturity date of the notes, and such amortization expense is reflected in interest expense on our consolidated statements of operations. The amount of amortization was approximately $0.4 million for each of the years ended December 31, 2014 and 2013. The amount of accumulated amortization at December 31, 2014 and 2013 was approximately $1.4 million and $1.0 million, respectively.
Revenue Recognition
We recognize revenue from product sales in accordance with FASB ASC 605—Revenue Recognition. Revenue from product sales is recognized when persuasive evidence of an arrangement exists, substantially all the risks and rewards of ownership have transferred to our customers, the selling price is fixed and collection is reasonably assured and there are no further obligations to customers. Sales from products are not subject to rights of return and, historically, actual sales returns have not been significant. We sell products through our direct sales force and through distributors. Sales through distributors are recognized as revenue upon sale to the distributor as these sales are considered to be final and no right of return or price protection exists. Sales to customers, when not made on consignment, are recognized upon shipment. A significant portion of our sales are made on a consignment basis. Revenue from products sold on a consignment basis is recognized on the date the consigned product is implanted or otherwise consumed. In limited circumstances, we rent peripheral equipment to patients. We recognize revenue from this arrangement when a contract is entered into with the patient’s insurer over the term the equipment is rented.
Shipping fees billed to customers are included in revenue and the related shipping costs are included in cost of revenue. Value added taxes and other similar types of taxes collected from customers in connection with the sale of our products are recorded on a net basis and are not included in revenue.
Product Warranty
Certain patient accessories sold with the HVAD System are covered by a limited warranty ranging from one to two years. Estimated contractual warranty obligations are recorded as an expense when the related revenue is recognized and are included in cost of revenue on our consolidated statements of operations. Factors that affect the estimated warranty liability include the number of units sold, historical and anticipated rates of warranty claims, cost per claim, and vendor supported warranty programs. We periodically assess the adequacy of our recorded warranty liabilities and adjust the amounts as necessary. The amount of the liability recorded is equal to the estimated costs to repair or otherwise satisfy claims made by customers. Accrued warranty is included as a component of other accrued liabilities on our consolidated balance sheets.
Share-Based Compensation
We recognize share-based compensation expense in connection with our share-based awards based on the estimated fair value of the awards on the date of grant, net of estimated forfeitures, using an accelerated accrual method over the vesting period. Therefore, we only recognize compensation cost for those awards expected to vest over the service period of the award. We estimate the forfeiture rate based on our historical experience of forfeitures. If our actual forfeiture rate is materially different from our estimate, share-based compensation expense could be significantly different from what we have recorded in the current period.
Calculating share-based compensation expense requires the input of highly subjective judgment and assumptions, including forfeiture rates, estimates of expected life of the share-based award, stock price volatility and risk-free interest rates. The assumptions used in calculating the fair value of share-based awards represent our best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As a result, if factors change and we use different assumptions, our share-based compensation expense could be materially different in the future.
73
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
We value restricted stock units (“RSUs”) at their intrinsic value on the date of grant. We estimate the fair value of our stock options using a Black-Scholes option pricing model. When appropriate, we estimate the expected life of a stock option by averaging the contractual term of the stock option (typically 10 years) with the associated vesting term (typically 4 years). We estimate the volatility of our shares on the date of grant considering several factors, including the historical volatility of our publicly-traded shares. We estimate the risk-free interest rate based on rates in effect for United States government bonds with terms similar to the expected lives of the stock options, at the time of grant.
We have issued share-based awards with performance-based vesting criteria. Achievement of the milestones must be “probable” before we begin recording share-based compensation expense. At each reporting date, we review the likelihood that these awards will vest and if the vesting is deemed probable, we begin to recognize compensation expense at that time. In the period that achievement of the performance based criteria is deemed probable, U.S. GAAP requires the immediate recognition of all previously unrecognized compensation since the original grant date. As a result, compensation expense recorded in the period that achievement is deemed probable could include a substantial amount of previously unrecorded compensation expense related to the prior periods. If ultimately performance goals are not met, for any share-based awards where vesting was previously deemed probable, previously recognized compensation cost will be reversed.
Valuation of Long-Lived Assets and Purchased Intangible Assets
We evaluate the carrying value of our long-lived assets, including purchased intangible assets, whenever events, changes in business circumstances or our planned use of long-lived assets indicate that their carrying amounts may not be fully recoverable or that their useful lives are no longer appropriate. If these facts and circumstances exist, we assess for recovery by comparing the carrying values of long-lived assets with their future undiscounted net cash flows. If the comparison indicates that impairment exists, impairment losses are recorded for the excess of the carrying value over the fair value of the long-lived assets based on discounted cash flows. Significant management judgment is required in the forecast of future operating results that are used in the preparation of expected undiscounted cash flows. In 2014, we ceased activities at CircuLite’s former headquarters in Teaneck, New Jersey and vacated the facility. We recorded an impairment charge of $0.6 million related to certain office equipment and software at the facility upon their discontinued use. This amount is included in selling, general and administrative expenses on our consolidated statements of operations. No impairments of similar long-lived assets were identified during the years ended December 31, 2013 or 2012.
We also evaluate the carrying value of intangible assets (not subject to amortization) related to in-process research and development (“IPR&D”) assets which are considered to be indefinite-lived until the completion or abandonment of the associated research and development projects. Accordingly, amortization of the IPR&D assets does not occur until the product reaches commercialization. During the period the assets are considered indefinite-lived, they are tested for impairment on an annual basis, as well as between annual tests if we become aware of any events occurring or changes in circumstances that indicate that the fair values of the IPR&D assets are less than their carrying amounts. If and when development is complete, which generally occurs when regulatory approval to market the product is obtained, the associated IPR&D assets are deemed definite-lived and are then amortized based on their estimated useful lives at that point in time. If the related project is terminated or abandoned, we may have a full or partial impairment related to the IPR&D assets, calculated as the excess of their carrying value over fair value.
During the fourth quarter of 2014 we performed an impairment review of our in-process research and development and recorded an impairment charge of $2.6 million related to in-process research and development acquired in 2013. In the fourth quarter of 2013, we recorded an impairment charge of $2.5 million related to in-process research and development acquired in 2012.
74
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Goodwill
We test goodwill for impairment on an annual basis in the fourth quarter of each fiscal year or more frequently if we believe indicators of impairment exist. The performance of the test involves a two-step process. The first step requires comparing the fair value of the reporting unit to its net book value, including goodwill. A potential impairment exists if the fair value of the reporting unit is lower than its net book value. The second step of the process is only performed if a potential impairment exists, and it involves comparing the aggregate fair value of the reporting unit’s net assets other than goodwill to the fair value of the reporting unit as a whole. Goodwill is considered impaired, and an impairment charge is recorded, if the excess of the fair value of the reporting unit over the fair value of the net assets is less than the carrying value of goodwill.
Based on the results of our annual impairment review in the fourth quarter of 2014, we concluded that goodwill was not impaired in 2014. In 2013, we recorded an impairment charge of $1.2 million to write-off goodwill that was recorded in 2012 in connection with our acquisition of World Heart.
Contingent Consideration
On December 1, 2013, we acquired CircuLite, Inc. In addition to initial consideration paid at closing, the former CircuLite securityholders may be entitled to receive additional shares of HeartWare common stock (or cash, in certain cases, at our discretion) upon the achievement of specified regulatory and commercial milestones, not to exceed $320 million in the aggregate over a ten-year period. The estimated fair value of the contingent payments is recorded as a liability and is remeasured at each reporting period. The estimated fair value is calculated using a discounted cash flow model utilizing significant unobservable inputs including future revenue projections, the probability of achieving each of the potential milestones and an estimated discount rate commensurate with the risks of the expected cash flows attributable to the various milestones. Material changes in any of these inputs could result in a significantly higher or lower fair value measurement and commensurate changes to this liability. Changes in the fair value of the contingent payments are recorded on a separate line item on our consolidated statements of operations.
Income Taxes
We account for income taxes in accordance with FASB ASC 740—Income Taxes. Under this method, deferred tax assets and liabilities are provided for differences between the carrying amounts of assets and liabilities in the consolidated financial statements and the tax bases of assets and liabilities that will result in future taxable or deductible amounts. The deferred tax assets and liabilities are measured using the enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Income tax expense is computed as the tax payable or refundable for the period, plus or minus the change during the period in deferred tax assets and liabilities. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred taxes will not be realized.
FASB ASC 740 requires that we recognize the financial statement benefit of a tax position only after determining that the relevant tax authority would more likely than not sustain the position following an audit. For tax positions meeting the “more likely than not” threshold, the amount recognized in the financial statements is the largest benefit that has a greater than 50 percent likelihood of being realized upon ultimate settlement with the relevant tax authority.
We recognize interest and penalties related to unrecognized tax benefits within the provision for income taxes line on our consolidated statements of operations.
75
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Translation of Foreign Currency
Assets and liabilities of our non-U.S. entities are translated at the period-end exchange rate and revenue and expenses are translated at the average exchange rates in effect during the respective periods. Equity transactions are translated at the spot rates on the dates of the original transactions. The net effect of these translation adjustments is shown in the accompanying consolidated financial statements as a component of stockholders’ equity, titled accumulated other comprehensive loss. Items in accumulated other comprehensive loss are not tax affected as we have incurred a net loss in each period since inception.
While most of the transactions of our domestic and international operations are denominated in the respective local currency, some transactions are denominated in other currencies. Transactions denominated in other currencies are accounted for in the respective local currency at the time of the transaction. Upon settlement of this type of transaction, any foreign currency gains or losses are included in our consolidated statements of operations.
Research and Development
Research and development costs, including new product development programs, regulatory compliance and clinical research, are expensed as incurred.
Marketing and Advertising Costs
Marketing, advertising and promotional costs are expensed when incurred. Advertising expenses were immaterial to our results of operations for the years ended December 31, 2014, 2013 and 2012.
Leases
We lease all of our administrative and manufacturing facilities. We recognize rent expense on a straight-line basis over the terms of our leases. Any scheduled rent increases, rent holidays and other related incentives are recognized on a straight-line basis over the terms of the leases. The difference between the cash rental payments and the straight-line recognition of rent expense over the terms of the leases results in a deferred rent asset or liability. As of December 31, 2014, the long-term portion of our deferred rent liability of approximately $3.5 million is included in other long-term liabilities on our consolidated balance sheets.
Fair Value Measurements
The carrying amounts reported in the consolidated balance sheets for cash and cash equivalents, accounts receivable, accounts payable and other accrued liabilities approximate their fair value based on the short-term maturity of these instruments.See Note 6 (Fair Value Measurements) and Note 10 (Debt) for more information.
Vendor Concentration
For the years ended December 31, 2014, 2013 and 2012, we purchased approximately 72%, 70%, and 67%, respectively, of our inventory components and supplies from three vendors. In addition, one of the three vendors supplies consulting services and material used in research and development activities. As of December 31, 2014, 2013 and 2012, the amounts due to these vendors totaled approximately $5.4 million, $5.8 million and $5.4 million, respectively.
76
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Concentration of Credit Risk and other Risks and Uncertainties
Financial instruments that potentially expose us to concentrations of credit risk consist primarily of cash and cash equivalents, investments and trade accounts receivable. Cash and cash equivalents are primarily on deposit with financial institutions in the United States and these deposits generally exceed the amount of insurance provided by the Federal Deposit Insurance Corporation (the “FDIC”). The Company has not experienced any historical losses on its deposits of cash and cash equivalents. Our investments consist of investment grade rated corporate and government agency debt and time deposits.
Concentration of credit risk with respect to our trade accounts receivable from our customers is primarily limited to hospitals, health research institutions and medical device distributors. Credit is extended to our customers based on an evaluation of a customer’s financial condition, and collateral is not required.
We are subject to certain risks and uncertainties including, but not limited to, our ability to achieve profitability, to generate cash flow sufficient to satisfy our indebtedness, to run clinical trials in order to receive and maintain FDA and foreign regulatory approvals for our products, our ability to adequately and timely address issues raised by FDA inspections, our ability to identify and correct quality issues in a timely manner and at a reasonable cost, the ability to achieve widespread acceptance of our products, our ability to manufacture our products in a sufficient volume and at a reasonable cost, the ability to protect our proprietary technologies and develop new products, the risks associated with operating in foreign countries, and general competitive and economic conditions. Changes in any of the preceding areas could have a material adverse effect on our business, results of operations or financial position.
New Accounting Standards
In September 2012, the FASB issued ASU No. 2012-02,Intangibles—Goodwill and Other (Topic 350), Testing Indefinite-Lived Intangible Assets for Impairment, which provides an entity the option first to assess qualitative factors to determine whether the existence of events and circumstances indicates that it is more likely than not that an indefinite-lived intangible asset is impaired. If, after assessing the totality of events and circumstances, an entity concludes that it is not more likely than not that the indefinite-lived intangible asset is impaired, then the entity is not required to take further action. The qualitative assessment is optional, allowing companies to go directly to the quantitative assessment. ASU No. 2012-02 is effective for our annual and interim impairment tests performed subsequent to January 1, 2013. The adoption of ASU No. 2012-02 did not affect our consolidated financial position, results of operations or cash flows.
In July 2013, the FASB issued ASU No. 2013-11,Income Taxes (Topic 740): Presentation of an Unrecognized Tax Benefit When a Net Operating Loss Carryforward, a Similar Tax Loss, or a Tax Credit Carryforward Exists (a consensus of the FASB Emerging Issues Task Force). U.S. GAAP does not include explicit guidance on the financial statement presentation of an unrecognized tax benefit when a net operating loss carryforward, a similar tax loss, or a tax credit carryforward exists. The amendments in this ASU state that an unrecognized tax benefit, or a portion of an unrecognized tax benefit, should be presented in the financial statements as a reduction to a deferred tax asset for a net operating loss carryforward, a similar tax loss, or a tax credit carryforward, except as follows: To the extent a net operating loss carryforward, a similar tax loss, or a tax credit carryforward is not available at the reporting date under the tax law of the applicable jurisdiction to settle any additional income taxes that would result from the disallowance of a tax position or the tax law of the applicable jurisdiction does not require the entity to use, and the entity does not intend to use, the deferred tax asset for such purpose, the unrecognized tax benefit should be presented in the financial statements as a liability and should not be combined with deferred tax assets. This ASU applies to all entities that have unrecognized tax benefits when a net operating loss carryforward, a similar tax loss, or a tax credit carryforward exists at the reporting date. The amendments in this ASU are effective for fiscal years, and interim periods within those years, beginning after December 15, 2013, with early adoption permitted. The amendments should be applied
77
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
prospectively to all unrecognized tax benefits that exist at the effective date, although retrospective application is permitted. The adoption of ASU No. 2013-11 effective January 1, 2014 did not have a material effect on our consolidated financial position, results of operations or cash flows.
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-09,Revenue from Contracts with Customers (Topic 606) (“ASU 2014-09”). The updated standard is a new comprehensive revenue recognition model that requires revenue to be recognized in a manner that depicts the transfer of goods or services to a customer at an amount that reflects the consideration expected to be received in exchange for those goods or services. ASU 2014-09 is effective for fiscal years, and interim periods within those years, beginning after December 15, 2016, and early adoption is not permitted. The updated standard becomes effective for us in the first quarter of our fiscal year ending December 31, 2017. We have not yet selected a transition method, and we are currently evaluating the effect that the ASU 2014-09 will have on our consolidated financial statements and related disclosures.
In January 2015, the FASB issued ASU No. 2015-01,Income Statement—Extraordinary and Unusual Items (Subtopic 225-20): Simplifying Income Statement Presentation by Eliminating the Concept of Extraordinary Items. The FASB issued this ASU as part of its initiative to reduce complexity in accounting standards. This ASU eliminates from U.S. GAAP the concept of extraordinary items. Subtopic 225-20 required that an entity separately classify, present, and disclose extraordinary events and transactions. Presently, an event or transaction is presumed to be an ordinary and usual activity of the reporting entity unless evidence clearly supports its classification as an extraordinary item. If an event or transaction meets the criteria for extraordinary classification, an entity is required to segregate the extraordinary item from the results of ordinary operations and show the item separately in the income statement, net of tax, after income from continuing operations. The entity also is required to disclose applicable income taxes and either present or disclose earnings-per-share data applicable to the extraordinary item. The amendments in this ASU are effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2015. A reporting entity may apply the amendments prospectively. A reporting entity also may apply the amendments retrospectively to all prior periods presented in the financial statements. Early adoption is permitted provided that the guidance is applied from the beginning of the fiscal year of adoption. The adoption of ASU No. 2015-01 is not expected to have a material effect on our consolidated financial position, results of operations or cash flows.
Note 4. Acquisitions
CircuLite, Inc.
On December 1, 2013, we entered into an Agreement and Plan of Merger (the “Merger Agreement”) pursuant to which we acquired CircuLite. At the effective time of the merger, all of the issued and outstanding shares of CircuLite capital stock (other than any shares of capital stock held by CircuLite or its subsidiary immediately before the effective time of the Merger and any dissenting shares of CircuLite capital stock) automatically converted into the right to receive an upfront payment and certain contingent merger consideration, in accordance with the terms of the Merger Agreement, as described in our Current Report on Form 8-K filed with the Securities and Exchange Commission on December 2, 2013.
In connection with the acquisition of CircuLite, we agreed to pay $30 million consisting of approximately $18 million in shares of HeartWare common stock, par value $0.001 per share (the “Common Stock”), equal to approximately 230,000 shares of Common Stock (the “Closing Payment”), and approximately $12 million in cash to repay outstanding CircuLite indebtedness and pay certain transaction liabilities and expenses. We funded the cash payment at closing with our existing cash balances. In accordance with the terms of the Merger Agreement, a volume weighted average of the per share prices of Common Stock during the 60 consecutive trading days ending on (and including) November 27, 2013 was used to determine the number of shares of
78
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Common Stock issued in connection with the closing. For accounting purposes, these shares were valued as of closing at approximately $22 million based upon the closing price of our common stock on the trading day prior to closing. In addition to the Closing Payment, the former CircuLite securityholders may be entitled to receive additional shares of Common Stock (or cash, in certain cases, at our discretion) upon the achievement of specified performance milestones (the “Contingent Payments”). The Contingent Payments were recorded as a liability at the estimated acquisition-date fair value of approximately $67.0 million. Fair value was estimated using a discounted cash flow model utilizing significant unobservable inputs including the probability of achieving each of the potential milestones and an estimated discount rate commensurate with the risks of the expected cash flows attributable to the various milestones.
The acquisition-date fair value of the consideration transferred is as follows:
| | | | |
| | | Total Acquisition
Date Fair Value
(in thousands) | |
Cash transferred, including acquisition costs and repayment of debt | | $ | 11,780 | |
Shares of common stock issued | | | 22,328 | |
Contingent consideration | | | 67,000 | |
| | | | |
Total consideration transferred | | $ | 101,108 | |
| | | | |
We paid $5.7 million in transaction related liabilities and expenses and repaid $6.1 million in debt on behalf of CircuLite, which are included as cash transferred in the table above.
The transaction was accounted for as a business combination under the acquisition method of accounting in accordance with the Financial Accounting Standards Board’s Accounting Standards Codification Topic 805,Business Combinations. Accordingly, the tangible assets and identifiable intangible assets acquired and liabilities assumed were recorded at fair value as of the date of acquisition, with the remaining purchase price recorded as goodwill.
The following table summarizes the estimated fair values of the assets acquired and liabilities assumed at the date of acquisition on December 1, 2013 (in thousands):
| | | | |
Cash and cash equivalents | | $ | 1,795 | |
Identified intangible assets | | | 5,500 | |
In-process research and development | | | 35,500 | |
Goodwill | | | 61,576 | |
Other assets acquired | | | 2,724 | |
| | | | |
Total assets acquired | | | 107,095 | |
Other liabilities assumed | | | (5,987 | ) |
| | | | |
Total net assets acquired | | $ | 101,108 | |
| | | | |
The identified intangible assets consist of customer relationships and trade names. These assets are being amortized on a straight-line basis over their estimated economic useful lives ranging from 15-20 years.
In-process research and development (“IPR&D”) is principally the estimated fair value of the SYNERGY System, with assigned values to be allocated among the various IPR&D assets acquired. IPR&D is recorded as an indefinite-lived asset until put into commercial use, upon which each applicable IPR&D asset becomes classified as developed technology and is amortized over the estimated period of economic benefit. We recognized a partial IPR&D impairment charge of $2.6 million in the fourth quarter of 2014 as a result of our decision in January 2015 to discontinue development of the pump component of the Synergy, and to replace the pump with a version of our MVAD pump.
79
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Goodwill, which largely represents the potential economic benefits of a technology that could expand our product portfolio and the patient population we can address, is calculated as the difference between the acquisition-date fair value of the consideration transferred and the fair values of the assets acquired and liabilities assumed. The goodwill is not expected to be deductible for income tax purposes. Goodwill is recorded as an indefinite-lived asset and is not amortized. Goodwill will be reviewed for impairment on an annual basis in the fourth quarter of our subsequent fiscal years or sooner if indications of impairment arise.
We incurred legal, consulting and other costs related to the acquisition aggregating approximately $2.8 million, which were expensed as incurred and are included in selling, general and administrative expenses in our consolidated statements of operations. The results of operations for CircuLite are included in our consolidated statements of operations subsequent to the December 1, 2013 date of acquisition. CircuLite’s results of operations for the period from December 1, 2013 to December 31, 2013 represented approximately $3.2 million of our consolidated net loss for the year ended December 31, 2013 and included approximately $0.6 million for restructuring costs.
The following unaudited pro forma information presents the combined results of operations for the years ended December 31, 2013 and 2012 as if we had completed the CircuLite acquisition at the beginning of 2012. The pro forma financial information is provided for comparative purposes only and is not necessarily indicative of what actual results would have been had the acquisition occurred on the date indicated, nor do they give effect to synergies, cost savings, fair market value adjustments, immaterial amortization expense and other changes expected to result from the acquisition. Accordingly, the pro forma financial results do not purport to be indicative of consolidated results of operations as of the date hereof, for any period ended on the date hereof, or for any other future date or period.
| | | | | | | | |
| | | 2013 | | | 2012 | |
| | | (in thousands) | |
Revenue | | $ | 209,792 | | | $ | 112,214 | |
Income before taxes | | | (77,973 | ) | | | (106,636 | ) |
Net loss | | | (78,599 | ) | | | (105,307 | ) |
World Heart Corporation
On August 2, 2012, we completed the acquisition of 100% of the outstanding shares of World Heart Corporation (“World Heart”) for consideration of approximately 83,000 shares of HeartWare International common stock, valued at approximately $6.9 million. The fair value of the shares issued was determined on the basis of the closing market price of HeartWare International common stock on the acquisition date. The acquisition expands our intellectual property and technology portfolio. In accordance with accounting standards for business combinations, we accounted for the acquisition of World Heart under the acquisition method. Under the acquisition method, the assets and liabilities assumed at the date of acquisition were recorded in the consolidated financial statements at their respective fair values at the date of acquisition. The excess of the purchase price over the fair value of the acquired net assets has been recorded as goodwill. World Heart’s results of operations are included in our consolidated financial statements from the date of acquisition.
80
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
The determination of the estimated fair value of the acquired assets and liabilities required management to make significant estimates and assumptions. We determined the fair value by applying established valuation techniques, based on information that management believed to be relevant to this determination. We also hired an independent third party to assist in the valuation of purchased intangible assets and goodwill. The following table summarizes the purchase price allocation of the fair value of the assets acquired and liabilities assumed at the date of acquisition (in thousands):
| | | | |
Assets | | | | |
Short-term: | | | | |
Cash and cash equivalents | | $ | 3,689 | |
Other current assets | | | 1,116 | |
Long-term: | | | | |
Property, plant and equipment | | | 307 | |
In-process research and development | | | 2,536 | |
Patents | | | 1,327 | |
Goodwill | | | 1,190 | |
| | | | |
Total assets acquired | | | 10,165 | |
Liabilities | | | | |
Current | | | 1,964 | |
Non-current | | | 1,258 | |
| | | | |
Net assets acquired | | $ | 6,943 | |
| | | | |
Patents are being amortized on a straight-line basis over the estimated useful life of 16 years.
As discussed in Note 8, during the fourth quarter of 2013 we recorded an impairment charge totaling $3.7 million to write-off goodwill of $1.2 million and in-process research and development of $2.5 million recorded in connection with our acquisition of World Heart.
All legal, consulting and other costs related to the acquisition aggregating approximately $1.1 million have been expensed as incurred and are included in selling, general and administrative expenses in our statements of operations. Pro forma results of operations have not been presented because the effect of this acquisition was not material our consolidated financial position, results of operations or cash flows.
Note 5. Investments
We have cash investment policies that limit investments to investment grade rated securities. At December 31, 2014 and 2013, all of our investments were classified as available-for-sale and carried at fair value. At December 31, 2014 and 2013, our short-term investments had maturity dates of less than twenty-four months and our long-term investments had maturity dates within thirty-six months.
81
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
The amortized cost and fair value of our investments, with gross unrealized gains and losses, were as follows:
At December 31, 2014
| | | | | | | | | | | | | | | | |
| | | Amortized
Cost Basis | | | Gross
Unrealized
Gains | | | Gross
Unrealized
Losses | | | Aggregate
Fair Value | |
| | | (in thousands) | |
Short-term investments: | | | | | | | | | | | | | | | | |
Corporate debt | | $ | 51,241 | | | $ | 8 | | | $ | (244 | ) | | $ | 51,005 | |
U.S. government agency debt | | | 15,000 | | | | — | | | | (25 | ) | | | 14,975 | |
Certificates of deposit | | | 9,555 | | | | — | | | | — | | | | 9,555 | |
| | | | | | | | | | | | | | | | |
Total short-term investments | | $ | 75,796 | | | $ | 8 | | | $ | (269 | ) | | $ | 75,535 | |
| | | | | | | | | | | | | | | | |
Long-term investments: | | | | | | | | | | | | | | | | |
Certificates of deposit | | $ | 1,225 | | | $ | — | | | $ | — | | | $ | 1,225 | |
| | | | | | | | | | | | | | | | |
Total long-term investments | | $ | 1,225 | | | $ | — | | | $ | — | | | $ | 1,225 | |
| | | | | | | | | | | | | | | | |
At December 31, 2013
| | | | | | | | | | | | | | | | |
| | | Amortized
Cost Basis | | | Gross
Unrealized
Gains | | | Gross
Unrealized
Losses | | | Aggregate
Fair Value | |
| | | (in thousands) | |
Short-term investments: | | | | | | | | | | | | | | | | |
Corporate debt | | $ | 32,221 | | | $ | 3 | | | $ | (18 | ) | | $ | 32,206 | |
Certificates of deposit | | | 5,390 | | | | — | | | | — | | | | 5,390 | |
| | | | | | | | | | | | | | | | |
Total short-term investments | | $ | 37,611 | | | $ | 3 | | | $ | (18 | ) | | $ | 37,596 | |
Long-term investments: | | | | | | | | | | | | | | | | |
Certificates of deposit | | $ | 1,225 | | | $ | — | | | $ | — | | | $ | 1,225 | |
| | | | | | | | | | | | | | | | |
Total long-term investments | | $ | 1,225 | | | $ | — | | | $ | — | | | $ | 1,225 | |
| | | | | | | | | | | | | | | | |
In the years ended December 31, 2014 and 2013, we did not have any realized gains or losses on our investments. At December 31, 2014 and 2013, none of our available-for-sale investments had been in a continuous loss position for more than twelve months. As of December 31, 2014, a total of 22 individual securities had been in an unrealized loss position for twelve months or less and the losses were determined to be temporary.
Note 6. Fair Value Measurements
FASB ASC 820—Fair Value Measurements and Disclosures, defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. FASB ASC 820 requires disclosures about the fair value of all financial instruments, whether or not recognized, for financial statement purposes. Disclosures about the fair value of financial instruments are based on pertinent information available to us as of the reporting dates. Accordingly, the estimates presented in these consolidated financial statements are not necessarily indicative of the amounts that could be realized on disposition of the financial instruments.
82
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
FASB ASC 820 specifies a hierarchy of valuation techniques based on whether the inputs to those valuation techniques are observable or unobservable. Observable inputs reflect market data obtained from independent sources, while unobservable inputs reflect market assumptions. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurement) and the lowest priority to unobservable inputs (Level 3 measurement).
The three levels of the fair value hierarchy are as follows:
Level 1—Quoted prices for identical instruments in active markets.
Level 2—Quoted prices for similar instruments in active markets; quoted prices for identical or similar instruments in markets that are not active; and model-derived valuations whose inputs are observable or whose significant value drivers are observable.
Level 3—Instruments with primarily unobservable value drivers.
We review the fair value hierarchy classification on a quarterly basis. Changes in the ability to observe valuation inputs may result in a reclassification of levels of certain securities within the fair value hierarchy. There were no transfers between Level 1, Level 2 and Level 3 during the years ended December 31, 2014, 2013 or 2012.
The following tables represent the fair value of our financial assets and financial liabilities measured at fair value on a recurring basis and which level was used in the fair value hierarchy.
| | | | | | | | | | | | | | | | | | | | |
At December 31, 2014 | |
| | | Carrying
Value | | | Fair
Value | | | Fair Value Measurements at the Reporting Date Using | |
| | | | | Level 1 | | | Level 2 | | | Level 3 | |
| | | (in thousands) | |
Assets | | | | |
Short-term investments | | $ | 75,535 | | | $ | 75,535 | | | $ | — | | | $ | 75,535 | | | $ | — | |
Long-term investments | | | 1,225 | | | | 1,225 | | | | — | | | | 1,225 | | | | — | |
| | | | | |
Liabilities | | | | | | | | | | | | | | | | | | | | |
Convertible senior notes | | | 114,803 | (1) | | | 153,978 | | | | — | | | | 153,978 | | | | — | |
Contingent consideration | | | 43,740 | | | | 43,740 | | | | — | | | | — | | | | 43,740 | |
Royalties | | | 962 | | | | 962 | | | | — | | | | — | | | | 962 | |
Lease exit costs | | | 1,207 | | | | 1,207 | | | | — | | | | — | | | | 1,207 | |
| | | | | | | | | | | | | | | | | | | | |
At December 31, 2013 | |
| | | Carrying
Value | | | Fair
Value | | | Fair Value Measurements at the Reporting Date Using | |
| | | | | Level 1 | | | Level 2 | | | Level 3 | |
| | | (in thousands) | |
Assets | | | | | | | | | | | | | | | | | | | | |
Short-term investments | | $ | 37,596 | | | $ | 37,596 | | | $ | — | | | $ | 37,596 | | | $ | — | |
Long-term investments | | | 1,225 | | | | 1,225 | | | | — | | | | 1,225 | | | | — | |
| | | | | |
Liabilities | | | | | | | | | | | | | | | | | | | | |
Convertible senior notes | | | 107,125 | (1) | | | 174,117 | | | | — | | | | 174,117 | | | | — | |
Contingent consideration | | | 67,000 | | | | 67,000 | | | | — | | | | — | | | | 67,000 | |
Royalties | | | 999 | | | | 999 | | | | — | | | | — | | | | 999 | |
| (1) | The carrying amount of our convertible senior notes is net of unamortized discount.See Note 10 (Debt) for more information. |
83
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Our Level 2 financial assets and liabilities include available-for-sale investments and our convertible senior notes. The fair value of our available-for-sale investments and our convertible senior notes was determined using quoted prices (including trade data) for the instruments in markets that are not active. The fair value of our convertible senior notes is presented for disclosure purposes only.
Financial assets and liabilities are considered Level 3 when their fair values are determined using pricing models, discounted cash flow methodologies, or similar techniques, and at least one significant model assumption or input is unobservable. Our Level 3 financial liabilities include the following:
| | • | | Contingent consideration—Determining the fair value of the contingent consideration related to our acquisition of CircuLite in December 2013 requires significant management judgment or estimation. The estimated fair value is calculated using the income approach, with significant inputs that include various revenue assumptions, discount rates and applying a probability to each outcome. Material changes in any of these inputs could result in a significantly higher or lower fair value measurement. The fair value of the contingent consideration is remeasured at the estimated fair value at each reporting period. Actual amounts paid may differ from the obligations recorded. |
| | • | | Royalties—Royalties represent future royalty payments to be made over the next 15 years pursuant to agreements related to intellectual property licensed or acquired by World Heart Corporation, which we acquired in August 2012. Determination of fair value requires significant management judgment or estimation. The royalty payment obligations were valued using a discounted cash flow model, the future minimum royalty payment amounts and discount rates commensurate with our market risk and the terms of the obligations. |
| | • | | Lease exit costs—In the first quarter of 2014 we ceased the use of CircuLite’s former headquarters in Teaneck, New Jersey, which was subject to an operating lease that runs through the end of 2020, and we recorded a liability equal to the estimated fair value of the remaining lease payments as of the cease-use date. The fair value was estimated based upon the discounted present value of the remaining lease payments, considering future estimated sublease income, estimated broker fees and required tenant improvements. This estimated fair value requires management judgment. The fair value of this liability will be remeasured at estimated fair value at each reporting period. Actual amounts paid may differ from the obligation recorded. |
The following table summarizes the change in fair value, as determined by Level 3 inputs, of the contingent consideration for the year ended December 31, 2014:
| | | | |
| | | Contingent
Consideration | |
| | | (in thousands) | |
Beginning balance | | $ | 67,000 | |
Payments | | | — | |
Change in fair value | | | (23,260 | ) |
| | | | |
Ending balance | | $ | 43,740 | |
| | | | |
The change in fair value of the contingent consideration for the year ended December 31, 2014 was due to several factors, including a $16.6 million reduction due to the likelihood of not achieving the performance milestone conditions related to the re-launch of the acquired form of the SYNERGY Surgical System following its loss of CE marking in the European Union in March 2014. In addition, we have discontinued development of the SYNERGY mircopump and have focused our efforts on a version of our MVAD pump for our partial-assist program. As a result of this shift in our development efforts, we updated our future projections for the
84
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
SYNERGY System and reassessed the probabilities of attaining certain contingent milestone payments under the terms of the merger agreement. Our updated analysis resulted in an aggregate $17.5 million decrease in the fair value of the contingent consideration related to certain milestones and royalty payments. This overall decrease in fair value was partially offset by an increase of $10.8 million due to accretion of the liability due to the passage of time. Adjustments associated with the change in fair value of contingent consideration are presented on a separate line item on our consolidated statements of operations. Potential valuation adjustments will be made in future accounting periods as additional information becomes available, including, among other items, progress toward developing the SYNERGY System, as well as revenue and milestone targets as compared to our current projections, with the impact of these adjustments being recorded in our consolidated statements of operations.
The following table summarizes the change in fair value, as determined by Level 3 inputs, of the royalties in the year ended December 31, 2014:
| | | | |
| | | Royalties | |
| | | (in thousands) | |
Beginning balance | | $ | 999 | |
Payments | | | (116 | ) |
Change in fair value | | | 79 | |
| | | | |
Ending balance | | $ | 962 | |
| | | | |
The expense associated with the change in fair value of the royalty payment obligations is included in research and development expenses on our consolidated statements of operations.
The following table summarizes the change in fair value, as determined by Level 3 inputs, of the lease exit costs in the year ended December 31, 2014:
| | | | |
| | | Lease Exit
Costs | |
| | | (in thousands) | |
Beginning balance | | $ | — | |
Accruals | | | 1,676 | |
Payments | | | (518 | ) |
Change in fair value | | | 49 | |
| | | | |
Ending balance | | $ | 1,207 | |
| | | | |
The expense associated with the change in fair value of the lease exit costs is included in selling, general and administrative expenses on our consolidated statements of operations.
85
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
The following table presents quantitative information about the inputs and valuation methodologies used for our fair value measurements classified in Level 3 of the fair value hierarchy as of December 31, 2014:
| | | | | | | | |
| | | Valuation Methodology | | Significant Unobservable Input | | Weighted Average
(range, if applicable) | |
Contingent consideration | | Probability weighted income approach | | Milestone dates Discount rate Probability of occurrence | |
| 2019 to 2022
17.0% to 24.0% 0% to 100% |
|
| | | |
Royalties | | Discounted cash flow | | Discount rate | | | 4.8% to 7.8% | |
| | | |
Lease exit costs | | Discounted cash flow | | Sublease start date Sublease rate Discount rate | |
| November 2015
$26.50/square foot 3.5% |
|
Assets That Are Measured at Fair Value on a Nonrecurring Basis
Non-financial assets such as intangible assets, goodwill and property, plant, and equipment are evaluated for impairment annually or when indicators of impairment exist. In the first quarter of 2014, we recorded an impairment charge of $0.6 million related to certain office equipment and software and in the fourth quarter of 2014, we recorded an impairment charge of $2.6 million related to in-process research and development (see Note 8). Impairment charges of $3.7 million were recorded in the fourth quarter of 2013 (see Note 8). No impairment charges were recorded in 2012. Non-financial assets such as identified intangibles acquired in connection with our acquisition of World Heart in August 2012 and CircuLite in December 2013 are measured at fair value using Level 3 inputs, which include discounted cash flow methodologies, or similar techniques, when there is limited market activity and the determination of fair value requires significant judgment or estimation.
Note 7. Inventories
Components of inventories are as follows:
| | | | | | | | |
| | | December 31, | |
| | | 2014 | | | 2013 | |
| | | (in thousands) | |
Raw material | | $ | 28,688 | | | $ | 21,761 | |
Work-in-process | | | 10,240 | | | | 8,206 | |
Finished goods | | | 15,118 | | | | 10,909 | |
| | | | | | | | |
| | $ | 54,046 | | | $ | 40,876 | |
| | | | | | | | |
Finished goods inventories includes inventory held on consignment at customer sites of $5.8 million and $4.6 million, at December 31, 2014 and 2013, respectively.
86
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 8. Goodwill, In-Process Research and Development and Other Intangible Assets, Net
Goodwill
The carrying amount of goodwill and the change in the balance for the years ended December 31, 2014 and 2013 is as follows:
| | | | | | | | |
| | | 2014 | | | 2013 | |
| | | (in thousands) | |
Beginning balance | | $ | 61,596 | | | $ | 1,190 | |
Additions | | | — | | | | 61,576 | |
Impairment | | | — | | | | (1,190 | ) |
Foreign currency translation impact | | | (206 | ) | | | 20 | |
| | | | | | | | |
Ending balance | | $ | 61,390 | | | $ | 61,596 | |
| | | | | | | | |
In 2012, we acquired World Heart and recorded $1.2 million of goodwill. In 2013, we acquired CircuLite and recorded $61.6 million of goodwill.See Note 4 for additional information. Goodwill has been assigned to the Company’s single reporting unit, which is the single operating segment by which the chief decision maker manages the Company.See Note 16 for additional information. Goodwill is not deductible for U.S. tax purposes.
Based on our annual impairment review in the fourth quarter of 2014, we concluded that goodwill was not impaired in 2014. During the fourth quarter of 2013, we recorded an impairment charge totaling $3.7 million to write-off goodwill of $1.2 million and in-process research and development of $2.5 million that was recorded in 2012 in connection with our acquisition of World Heart. Subsequent to an evaluation of the ongoing research and development efforts surrounding the MiFlow technology, we determined we would discontinue further development efforts needed to commercialize the technology. As a result of this decision, an impairment charge was recorded. These amounts are included in research and development expenses on our consolidated statements of operations.
In-Process Research and Development
The carrying value of our in-process research and development assets, which relate to the development and potential commercialization of certain acquired technologies, consisted of the following at December 31, 2014 and 2013:
| | | | | | | | |
| | | 2014 | | | 2013 | |
| | | (in thousands) | |
SYNERGY System technology | | $ | 32,850 | | | $ | 35,500 | |
| | | | | | | | |
As discussed above, during the fourth quarter of 2013, we recorded an impairment charge of $2.5 million to write-off the value of the in-process research and development, which was recorded in connection with our acquisition of World Heart. This amount is included in research and development expenses on our consolidated statements of operations.
In December 2013, we acquired CircuLite and recorded $35.5 million of in-process research and development.See Note 4 for additional information. The in-process research and development has an indefinite life. At the time the economic life becomes determinable (upon project completion or abandonment) the amount will be amortized over its expected remaining life. During the fourth quarter of 2014 we performed an impairment review of this in-process research and development and recorded an impairment charge of
87
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
$2.6 million. The fair value of the IPR&D asset was determined using the multi-period excess earnings method which is equal to the present value of the incremental after-tax cash flows attributable to that intangible asset. The impairment charge is included in research and development expenses on our consolidated statements of operations.
Other Intangible Assets
Other intangible assets, net consisted of the following:
| | | | | | | | |
| | | December 31, | |
| | | 2014 | | | 2013 | |
| | | (in thousands) | |
Patents | | $ | 5,310 | | | $ | 3,754 | |
Purchased intangible assets | | | | | | | | |
Trade names | | | 3,700 | | | | 3,700 | |
Customer relationships | | | 1,800 | | | | 1,800 | |
Acquired technology rights | | | 9,925 | | | | 7,925 | |
| | | | | | | | |
| | | 20,735 | | | | 17,179 | |
Less: Accumulated amortization—Patents | | | (1,118 | ) | | | (800 | ) |
Less: Accumulated amortization—Purchased intangible assets | | | (1,810 | ) | | | (404 | ) |
| | | | | | | | |
| | $ | 17,807 | | | $ | 15,975 | |
| | | | | | | | |
Our other intangible assets are amortized using the straight-line method over their estimated useful lives as follows:
| | | | |
Patents | | | 15 years | |
Purchased intangible assets | | | | |
Trade names | | | 15 years | |
Customer relationships | | | 20 years | |
Acquired technology rights | | | 6 to 16 years | |
Following satisfaction of a pre-specified milestone in the fourth quarter of 2014, we were obligated to pay an additional $2.0 million under a certain patent assignment and license agreement. The $2.0 million, which is payable in cash or shares of our common stock, was recorded as additional acquired technology rights and accrued at December 31, 2014 in other long term liabilities on our consolidated balance sheets. We currently intend to settle this liability by issuing shares of our common stock in 2015.
Following satisfaction of a pre-specified milestone in December 2013, we were obligated to pay an additional $5.0 million under a certain patent assignment and license agreement. The $5.0 million, which was payable in cash or share of our common stock, was recorded as additional acquired technology rights and accrued at December 31, 2013 in other accrued liabilities on our consolidated balance sheets. We settled this liability through the issuance of 50,330 shares of our common stock in the first quarter of 2014.
Amortization expense for the years ended December 31, 2014, 2013 and 2012 was $1.7 million, $0.6 million and $0.2 million, respectively.
Estimated amortization expense for each of the five succeeding fiscal years based upon our intangible asset portfolio at December 31, 2014 is approximately $2.0 million.
88
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Note 9. Other Balance Sheet Information
Long-Term Investment
In October 2013, we invested $10 million in an early stage privately-held company focused on the development of novel, minimally invasive heart therapies in the form of a convertible promissory note with an interest rate of 6% per annum (the “Note”). Pursuant to the terms of the Note, on October 7, 2014 (the maturity date), the privately held company elected to convert all unpaid principal and interest on the Note (less applicable taxes) into shares of its preferred stock. This investment is carried at cost and is included in long-term investments and other assets on our consolidated balance sheets. The carrying value of this investment was $10.5 million and $10.0 million at December 31, 2014 and 2013, respectively.
The fair value of this investment has not been estimated as of December 31, 2014 and 2013. As of December 31, 2014, the investee company had limited liquidity and is in the process of raising additional capital to continue its operations. However, there can be no assurance this effort will be successful. We considered determination of the fair value of this investment to be impracticable as it represents an equity interest in an early stage privately-held company, and we have limited access to information that would assist with determining fair value. We believe there have been no significant events or changes in circumstances that may have a significant adverse effect on the fair value of the investment. Events may occur or information may become available, including the inability of the investee company to raise sufficient additional capital, that could require us to write-off all or a portion of this asset, which could have a material adverse effect on our consolidated results of operations.
Other Accrued Liabilities
Other accrued liabilities consist of the following:
| | | | | | | | |
| | | December 31, | |
| | | 2014 | | | 2013 | |
| | | (in thousands) | |
Accrued payroll and other employee costs | | $ | 13,404 | | | $ | 10,840 | |
Accrued milestone payment | | | — | | | | 5,000 | |
Accrued material purchases | | | 4,284 | | | | 4,325 | |
Accrued warranty | | | 4,685 | | | | 2,498 | |
Accrued research and development costs | | | 2,663 | | | | 2,307 | |
Accrued product recall costs | | | 1,888 | | | | — | |
Accrued professional fees | | | 1,624 | | | | 2,428 | |
Accrued VAT | | | 1,637 | | | | 1,329 | |
Accrued restructuring costs | | | 1,266 | | | | 245 | |
Other accrued expenses | | | 5,138 | | | | 6,304 | |
| | | | | | | | |
| | $ | 36,589 | | | $ | 35,276 | |
| | | | | | | | |
Accrued Payroll and Other Employee Costs
Accrued payroll and other employee costs included year-end employee bonuses of approximately $7.9 million and $6.6 million at December 31, 2014 and 2013, respectively.
89
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Accrued Warranty
The following table summarizes changes in our warranty liability for the years ended December 31, 2014, 2013 and 2012:
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | 2012 | |
| | | (in thousands) | |
Beginning balance | | $ | 2,498 | | | $ | 543 | | | $ | 203 | |
Accrual for warranty expense | | | 4,141 | | | | 2,721 | | | | 921 | |
Warranty costs incurred during the period | | | (1,954 | ) | | | (766 | ) | | | (581 | ) |
| | | | | | | | | | | | |
Ending balance | | $ | 4,685 | | | $ | 2,498 | | | $ | 543 | |
| | | | | | | | | | | | |
During 2014 and 2013, increases in warranty reserves resulted from the substantial increase in commercial sales activity, taking into consideration our historical return and replacement experience.
Accrued Product Recall Costs
The costs to repair or replace products associated with product recalls and voluntary service campaigns are recorded when they are determined to be probable and reasonably estimable as a cost of revenue and are not included in our warranty liability. No such costs were incurred in 2013 or 2012. In April 2014, we implemented Field Corrective Action Notification following an observed increase in complaints related to earlier-than-expected battery depletion and routine battery handling. This notification provided information to assist patients and clinicians to monitor battery performance, recognize abnormal behaviors and reinforce proper power management of the HVAD System. We increased our warranty liability in the first quarter of 2014 to account for an anticipated higher level of battery returns likely to be associated with increased battery performance awareness following implementation of the field safety corrective action. On July 30, 2014, we extended the corrective action to include a voluntary recall of certain older batteries. The recall instructs sites to replace certain older batteries in the field upon patients’ routine visits in order to further mitigate the potential risks associated with premature battery depletion.
In February 2015, we expanded a 2013 voluntary Field Safety Corrective Action, by initiating a voluntary medical device recall of certain older controllers distributed in the U.S. during the ADVANCE and ENDURANCE clinical trial periods. The affected controllers exhibit a higher susceptibility to electrostatic discharge than newer, commercial controllers. This recall was ongoing as of December 31, 2014.
During 2014, we recorded charges aggregating $3.6 million for estimated costs associated with the battery and controller recalls discussed above.
Accrued Restructuring Costs
The following table summarizes changes in our accrued restructuring costs for the year ended December 31, 2014:
| | | | | | | | | | | | | | | | |
| | | Facility Leases | | | Severance and
Related | | | Contract
Termination | | | Total | |
| | | (in thousands) | |
Beginning balance | | $ | — | | | $ | 245 | | | $ | — | | | $ | 245 | |
Restructuring charges | | | 2,204 | | | | 715 | | | | 688 | | | | 3,607 | |
Payments | | | (818 | ) | | | (833 | ) | | | (688 | ) | | | (2,339 | ) |
Adjustments to estimated obligations | | | (168 | ) | | | (127 | ) | | | — | | | | (295 | ) |
Change in fair value | | | 48 | | | | — | | | | — | | | | 48 | |
| | | | | | | | | | | | | | | | |
Ending balance | | $ | 1,266 | | | $ | — | | | $ | — | | | $ | 1,266 | |
| | | | | | | | | | | | | | | | |
90
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
The restructuring obligations reflected above resulted from the following actions:
Facility Closures
In the first quarter of 2014 we ceased the use of CircuLite’s former headquarters in Teaneck, New Jersey, which was subject to an operating lease that runs through the end of 2020 (see Note 11). In connection with this action, we recorded a $1.7 million liability equal to the estimated fair value of the remaining lease obligation as of the cease-use date (see Note 6). In the first quarter of 2014, we also relocated our corporate headquarters and ceased activities at our former headquarters in Framingham, Massachusetts. In connection with this action, we recorded a $0.5 million liability equal to the aggregate of the remaining payments on the lease for our former headquarters as of the cease-use date. Both of these items are included in selling, general and administrative expenses on our consolidated statements of operations.
Severance Agreements
In 2014, we incurred various costs related to the integration of CircuLite’s operations, including severance costs aggregating $0.6 million, the majority of which were recorded in the first quarter of 2014. We recorded $0.4 million in research and development expenses and the remaining $0.2 million in selling, general and administrative expenses on our consolidated statements of operations.
Contract Termination
As a result of anticipated design modifications to the SYNERGY System and our decision to move manufacturing of the SYNERGY System to our Miami Lakes facility, we terminated a supply agreement with a vendor in Germany for the purchase of components necessary to produce the prior-to-modification version of the SYNERGY System. In connection with the termination of this supply agreement, we recorded a charge of $0.7 million in the first quarter of 2014, which is included in research and development expenses on our consolidated statements of operations.
Note 10. Debt
Convertible Senior Notes
On December 15, 2010, we completed the sale of 3.5% convertible senior notes due 2017 (the “Convertible Notes”) for an aggregate principal amount of $143.75 million pursuant to the terms of an Indenture dated December 15, 2010 (the “Indenture”). The Convertible Notes are the senior unsecured obligations of the Company. The Convertible Notes bear interest at a rate of 3.5% per annum, payable semi-annually in arrears on June 15 and December 15 of each year. The Convertible Notes will mature on December 15, 2017, unless earlier repurchased by us or converted.
The Convertible Notes offering was completed pursuant to a prospectus supplement, dated December 9, 2010, to a shelf registration statement on Form S-3 that was previously filed with the SEC and which was declared effective on December 9, 2010.
The Convertible Notes will be convertible at an initial conversion rate of 10 shares of our common stock per $1,000 principal amount of Convertible Notes, which corresponds to an initial conversion price of $100.00 per share of our common stock. The conversion rate is subject to adjustment from time to time upon the occurrence of certain events.
Prior to June 15, 2017, holders may convert their Convertible Notes at their option only upon satisfaction of one or more of the conditions specified in the Indenture relating to the (i) sale price of our common stock, (ii) the
91
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
trading price per $1,000 principal amount of Convertible Notes or (iii) specified corporate events. As of the date of this report on Form 10-K, none of the events that would allow holders to convert their Convertible Notes have occurred. On or after June 15, 2017 until the close of business of the business day immediately preceding the date the Convertible Notes mature, holders may convert their Convertible Notes at any time, regardless of whether any of the foregoing conditions have been met. Upon conversion, we will pay or deliver, as the case may be, cash, shares of our common stock or a combination thereof, at our election.
We may not redeem the Convertible Notes prior to maturity. Holders of the Convertible Notes may require us to purchase for cash all or a part of their Convertible Notes at a repurchase price equal to 100% of the principal amount of the Convertible Notes to be repurchased, plus accrued and unpaid interest, upon the occurrence of certain fundamental changes (as defined in the Indenture) involving the Company. The Indenture does not contain any financial or operating covenants or restrictions on the payments of dividends, the incurrence of indebtedness or the issuance or repurchase of securities by us or any of our subsidiaries.
The Indenture contains customary terms and nonfinancial covenants and defines events of default. If an event of default (other than certain events of bankruptcy, insolvency or reorganization) involving the Company occurs and is continuing, the Trustee (by notice to the Company) or the holders of at least 25% in principal amount of the outstanding Convertible Notes (by notice to the Company and the Trustee) may declare 100% of the principal of and accrued and unpaid interest, if any, on all the Convertible Notes to be due and payable. In case of certain events of bankruptcy, insolvency or reorganization, involving the Company, 100% of the principal of and accrued and unpaid interest on the Convertible Notes will automatically become due and payable. Notwithstanding the foregoing, the Indenture provides that, to the extent we elect, the sole remedy for an event of default relating to certain failures by us to comply with certain reporting covenants in the Indenture consists exclusively of the right to receive additional interest on the Convertible Notes.
In accordance with ASC 470-20,Debt with Conversion and Other Options, which applies to certain convertible debt instruments that may be settled in cash or other assets, or partially in cash, upon conversion, we recorded the long-term debt and equity components on our Convertible Notes separately on the issuance date. The amount recorded for long-term debt was determined by measuring the fair value of a similar liability that does not have an associated equity component. The measurement of fair value required the Company to make estimates and assumptions to determine the present value of the cash flows of the Convertible Notes, absent the conversion feature. This treatment increased interest expense associated with our Convertible Notes by adding a non-cash component to interest expense in the form of amortization of a debt discount calculated based on the difference between the 3.5% cash coupon rate and the effective interest rate on debt borrowing of approximately 12.5%. The discount is being amortized to interest expense through the December 15, 2017 maturity date of the Convertible Notes using the effective interest method and is included in interest expense on our consolidated statements of operations. Additionally, we allocated the costs related to issuance of the Convertible Notes on the same percentage as the long-term debt and equity components, such that a portion of the costs is allocated to the long-term debt component and the equity component included in additional paid-in capital. The portion of the costs allocated to the long-term debt component is presented as deferred financing costs, net on our consolidated balance sheets. These deferred financing costs are also being amortized to interest expense through the December 15, 2017 maturity date of the Convertible Notes using the effective interest method and the amortization is included in interest expense on our consolidated statements of operations
92
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
The Convertible Notes and the equity component, which is recorded in additional paid-in-capital, consisted of the following:
| | | | | | | | |
| | | December 31, | |
| | | 2014 | | | 2013 | |
| | | (in thousands) | |
Principal amount | | $ | 143,750 | | | $ | 143,750 | |
Unamortized discount | | | (28,947 | ) | | | (36,625 | ) |
| | | | | | | | |
Net carrying amount | | $ | 114,803 | | | $ | 107,125 | |
| | | | | | | | |
Equity component | | $ | 55,038 | | | $ | 55,038 | |
| | | | | | | | |
Based on the initial conversion rate of 10 shares of our common stock per $1,000 principal amount of Convertible Notes, which corresponds to an initial conversion price of $100.00 per share of our common stock, the number of shares issuable upon conversion of the Convertible Notes is 1,437,500. The value of these shares, based on the closing price of our common stock on December 31, 2014 of $73.43 per share, was approximately $105.6 million. The fair value of our Convertible Notes as presented in Note 6 was approximately $154.0 million at December 31, 2014.
Interest expense related to the Convertible Notes consisted of interest due on the principal amount, amortization of the discount and amortization of the portion of the deferred financing costs allocated to the long-term debt component. For the years ended December 31, 2014 and 2013, interest expense related to the Convertible Notes was as follows:
| | | | | | | | |
| | | 2014 | | | 2013 | |
| | | (in thousands) | |
Stated amount at 3.5% coupon rate | | $ | 5,031 | | | $ | 5,031 | |
Amortization of discount | | | 7,678 | | | | 6,809 | |
Amortization of deferred financing costs | | | 412 | | | | 365 | |
| | | | | | | | |
| | $ | 13,121 | | | $ | 12,205 | |
| | | | | | | | |
Promissory Note
In connection with our acquisition of World Heart, we assumed a promissory note with an outstanding balance of $600,000, which matured on December 2, 2014. Interest on the promissory note was payable annually in arrears on December 2 of each year at a rate of 4.5% per annum. Principal payments of $200,000 were also due on December 2 of each year through maturity. At December 31, 2014, no further amounts are payable in connection with the promissory note. The amount of interest expense incurred in the years ended December 31, 2014, 2013 and 2012 was approximately $8,000, $17,000 and $11,000, respectively.
Note 11. Leases
Corporate Headquarters
On October 17, 2013, we entered into a lease for our corporate headquarters in Framingham, Massachusetts that commenced in January 2014. The facility is used primarily for office and ancillary laboratory purposes including development testing. Under the lease we rent approximately 58,000 square feet of company space and approximately 4,000 square feet of common space for an initial seven year period, with an option to renew for a period of fifty-seven months, but in no event beyond September 30, 2025. Annual base rent of approximately
93
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
$1.3 million is payable monthly starting January 1, 2015. Annual base rent is subject to periodic increases beginning March 1, 2017 and will increase to approximately $1.5 million per year for the final four years of the lease. A security deposit of $0.3 million was paid in connection with the lease, which is included in other assets on our consolidated balance sheets.
Under the lease for our former headquarters in Framingham, Massachusetts, which was last amended on July 30, 2012, we rented approximately 21,300 square feet. Effective January 1, 2013, base rent obligations were approximately $0.4 million per year. The lease term for approximately 17,800 square feet ended on December 31, 2014, while the lease term for the remaining 3,500 square feet ends on June 30, 2015. In connection with the move to our corporate headquarters, we recorded a $0.5 million liability in the first quarter of 2014, which equaled the aggregate of the remaining payments on the lease for our former headquarters as of the cease-use date (see Note 9).
Florida Facility
On December 9, 2010, we entered into a lease for our facility in Miami Lakes, Florida. The facility is used primarily for manufacturing, research and development and administrative functions. Under the lease, which was amended in November 2012 and July 2013, we rent approximately 132,000 square feet for a period ending February 28, 2022, with an option to renew for two five-year terms. Effective with the July 2013 amendment, base rent payments are $10.00 per square foot and are subject to a 3% annual escalation on March 1 of each subsequent year. The lease is secured by a security deposit of $1.25 million in the form of an unconditional stand-by letter of credit. The letter of credit is supported by a certificate of deposit for the same amount, which is included in other assets on our consolidated balance sheets.
New Jersey Facility
In connection with the acquisition of CircuLite in December 2013, we assumed a noncancelable operating lease that CircuLite entered into for its headquarters in Teaneck, New Jersey in December 2012. Under the lease, we rent approximately 22,200 square feet mixed use office space for a period ending October 2020. The lease provides for a fixed monthly rent, plus utilities, with a six-month rent abatement during the first year. Base rent obligations are approximately $0.6 million per year and subject to a 2% annual escalation starting on September 1, 2014. Pursuant to the lease agreement, we are required to maintain cash on deposit of $0.8 million, which is included in other assets on our consolidated balance sheets.
In the first quarter of 2014, we initiated a plan to close this facility. The facility closure was accounted for in accordance with ASC 420Exit or Disposal Cost Obligations, pursuant to which we recorded a liability equal to the fair value of the remaining lease payments as of the cease-use date. The fair value of this liability was estimated to be $1.7 million at the initial valuation date and was based upon the discounted present value of remaining lease rentals for the space no longer occupied, considering future estimated sublease income, estimated broker fees and required tenant improvements (see Note 6 and Note 9).
Other Facilities
In addition to the leases discussed above, we have entered into various operating lease agreements for miscellaneous office and research space and equipment. The duration of these agreements is typically twelve to thirty-six months from origination. The aggregate base annual rental payment on these leases is less than $0.3 million.
94
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Rent expense was approximately $3.6 million, $2.9 million, and $2.7 million in 2014, 2013 and 2012, respectively. Future minimum rental commitments under non-cancelable operating lease agreements with remaining terms of at least one year as of December 31, 2014 are as follows:
| | | | |
| | | Operating
Leases | |
| | | (in thousands) | |
Year Ending December 31, | | | | |
2015 | | $ | 3,618 | |
2016 | | | 3,559 | |
2017 | | | 3,662 | |
2018 | | | 3,664 | |
2019 | | | 3,751 | |
Thereafter | | | 5,870 | |
| | | | |
Total minimum lease payments | | $ | 24,124 | |
| | | | |
Aachen Germany Facility Closure
In January 2015, we initiated a plan to close our facilities located in Aachen, Germany. One facility is covered under an operating lease that ends on October 31, 2017. Base rent obligations are approximately $12,000 per month. The facility closure, which is targeted for completion on or by March 31, 2015, will be accounted for in accordance with ASC 420—Exit or Disposal Cost Obligations, pursuant to which we will record a liability equal to the fair value of the remaining lease payments as of the cease-use date. Fair value will be determined based upon the discounted present value of remaining lease rentals for the space no longer occupied, considering future estimated sublease income, estimated broker fees and required tenant improvements. We currently estimate that the total lease charge will be approximately $0.4 million.
Closure of this facility will result in a write-off of leasehold improvements, furnishings and other fixed assets that will not be transferred to our other facilities of approximately $0.6 million and a charge for non-cancellable purchase obligations of approximately $0.5 million. In January 2015, employees at this facility were notified of the closure and elimination of their positions. Certain of the employees have been asked to relocate. The estimated employee-related costs associated with severance obligations will be approximately $0.6 million. Each of these estimates is subject to further assessment and analysis. We anticipate recording these charges in the quarter ending March 31, 2015.
Note 12. Stockholders’ Equity
Preferred Stock
We are authorized to issue up to 5,000,000 shares of preferred stock, $.001 par value per share. Our board of directors is authorized, subject to any limitations prescribed by law, to provide for the issuance of the shares of preferred stock in series, and by filing a certificate pursuant to the applicable law of the state of Delaware, to establish from time to time the number of shares to be included in each such series, and to fix the designation, powers, preferences and rights of the shares of each such series and any qualifications, limitations or restrictions thereof. No shares of preferred stock have been issued or are outstanding.
95
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Common Stock
We are authorized to issue up to 25,000,000 shares of common stock, $.001 par value per share. As of December 31, 2014, we had 17,155,833 shares outstanding. Holders are entitled to one vote for each share of common stock (or its equivalent).
Shares of our common stock reserved at December 31, 2014, for possible future issuance are as follows:
| | | | |
| | | (in thousands) | |
Convertible senior notes | | | 1,768 | |
Equity award plans | | | 1,418 | |
| | | | |
| | | 3,186 | |
| | | | |
See the Consolidated Statement of Stockholders’ Equity for details related to our equity transactions.
2013 Public Offering
On March 12, 2013, we entered into an Underwriting Agreement (the “Underwriting Agreement”) with J.P. Morgan Securities LLC, as representative of the several underwriters named in the Underwriting Agreement (the “Underwriters”), pursuant to which we agreed to sell and the Underwriters agreed to purchase, subject to and upon terms and conditions set forth therein, an aggregate of 1,500,000 shares of our common stock at a net sales price of $81.9114 per share (the public offering price of $86.45 per share minus the underwriting discount). We also granted the Underwriters an option to purchase 225,000 additional shares of our common stock at the public offering price less the underwriting discount, which the Underwriters exercised in full on March 13, 2013. The closing of the offering occurred on March 18, 2013. After fees and related expenses, net proceeds from the offering were approximately $141.0 million.
The offering was completed pursuant to a prospectus supplement, dated March 12, 2013, to a shelf registration statement on Form S-3 that was previously filed with the SEC and which was declared effective on December 9, 2010. This shelf registration statement expired on December 9, 2013.
On January 30, 2014, we filed a shelf registration statement with the SEC on Form S-3. This shelf registration statement allows us to offer and sell from time to time, in one or more series or issuances and on terms that we will determine at the time of the offering any combination and amount of the securities described in the prospectus contained in the registration statement or in the prospectus supplement filed with respect to a particular offering. An aggregate of 530,816 shares of our common stock were registered for issuance pursuant to various prospectus filings on January 30, 2014 in connection with the CircuLite acquisition. As of December 31, 2014, there remained 248,872 shares reserved for potential issuance in connection with future contingent milestone payments under the terms of the merger agreement (seeNote 4).
Note 13. Share-Based Compensation
We allocate share-based compensation expense to cost of revenue, selling, general and administrative expense and research and development expense based on the award holder’s employment function. For the years ended December 31, 2014, 2013 and 2012, we recorded share-based compensation expenses as follows:
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | 2012 | |
| | | (in thousands) | |
Cost of revenue | | $ | 2,032 | | | $ | 2,539 | | | $ | 3,152 | |
Selling, general and administrative | | | 13,573 | | | | 12,184 | | | | 10,195 | |
Research and development | | | 7,937 | | | | 7,151 | | | | 5,458 | |
| | | | | | | | | | | | |
| | $ | 23,542 | | | $ | 21,874 | | | $ | 18,805 | |
| | | | | | | | | | | | |
96
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Deferred tax benefits attributed to our share-based compensation expense are not recognized in the accompanying consolidated financial statements because we are in a net operating loss position and a full valuation allowance is maintained for all net deferred tax assets. We receive a tax deduction for certain stock option exercises during the period the options are exercised, and for the vesting of restricted stock units during the period the restricted stock units vest. For stock options, the amount of the tax deduction is generally for the excess of the fair market value of our shares of common stock over the exercise price of the stock options at the date of exercise. For restricted stock units, the amount of the tax deduction is generally for the fair market value of our shares of common stock at the vesting date. Excess tax benefits are not included in the accompanying consolidated financial statements because we are in a net operating loss position and a full valuation allowance is maintained for all net deferred tax assets.
Equity Plans
We have issued share-based awards to employees, non-executive directors and outside consultants through various approved plans and outside of any formal plan. New shares are issued upon the exercise of share-based awards.
Upon receipt of stockholder approval on May 31, 2012, we adopted the HeartWare International, Inc. 2012 Incentive Award Plan (“2012 Plan”). The 2012 Plan provides for the grant of incentive stock options, non-qualified stock options, restricted stock, restricted stock units, performance awards, dividend equivalent rights, deferred stock, deferred stock units, stock payments and stock appreciation rights (collectively referred to as “Awards”), to our directors, employees and consultants. Under the terms of the 2012 Plan, the total number of shares of our common stock initially reserved for issuance under Awards is 1,375,000, provided that the total number of shares of our common stock that may be issued pursuant to “Full Value Awards” (Awards other than options, SARs or other awards for which the holder pays the intrinsic value existing as of the date of grant whether directly or by forgoing a right to receive a payment from the Company) is 1,275,000. As of December 31, 2014, 125,149 shares have been issued upon vesting of Awards issued under the 2012 Plan and Awards with respect to 528,332 shares were issued and outstanding under the 2012 Plan. Subsequent to adoption of the 2012 Plan, no new awards will be granted under our prior plans. Any outstanding awards under the prior will continue to be subject to the terms and conditions of the plan under which they were granted.
Stock Options
Each option allows the holder to subscribe for and be issued one share of our common stock at a specified price, which is generally the quoted market price of our common stock on the date the option is issued. Options generally vest on a pro-rata basis on each anniversary of the issuance date within four years of the date the option is issued. Options may be exercised after they have vested and prior to the specified expiry date provided applicable exercise conditions are met, if any. The expiry date can be for periods of up to ten years from the date the option is issued.
The fair value of each option is estimated on the date of grant using the Black-Scholes option pricing model using the assumptions established at that time. The following table includes the weighted average assumptions used for options issued in the years ended December 31, 2014, 2013 and 2012.
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | 2012 | |
Dividend yield | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % |
Expected volatility | | | 39.00 | % | | | 40.00 | % | | | 57.00 | % |
Risk-free interest rate | | | 1.65 | % | | | 1.15 | % | | | 1.00 | % |
Estimated holding period (years) | | | 5.00 | | | | 6.25 | | | | 6.25 | |
97
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Information related to options granted under all of our plans at December 31, 2014 and activity during the year then ended is as follows (certain amounts in U.S.$ were converted from AU$ at the then period-end spot rate):
| | | | | | | | | | | | | | | | |
| | | Number of
Options
(in thousands) | | | Weighted
Average
Exercise
Price | | | Weighted
Average
Remaining
Contractual Life
(Years) | | | Aggregate
Intrinsic
Value
(in thousands) | |
Outstanding at December 31, 2013 | | | 133 | | | $ | 42.82 | | | | | | | | | |
Granted | | | 7 | | | | 88.84 | | | | | | | | | |
Exercised | | | (32 | ) | | | 29.25 | | | | | | | | | |
Forfeited | | | — | | | | — | | | | | | | | | |
Expired | | | (1 | ) | | | 86.58 | | | | | | | | | |
| | | | | | | | | | | | | | | | |
Outstanding at December 31, 2014 | | | 107 | | | $ | 48.32 | | | | 4.39 | | | $ | 3,063 | |
| | | | | | | | | | | | | | | | |
Exercisable at December 31, 2014 | | | 87 | | | $ | 39.75 | | | | 3.52 | | | $ | 3,051 | |
| | | | | | | | | | | | | | | | |
The aggregate intrinsic values at December 31, 2014 noted in the table above represent the number of in-the-money options outstanding or exercisable multiplied by the closing price of our common stock traded on NASDAQ less the respective weighted average exercise price at period end.
The weighted average grant date fair value per share of options granted in the years ended December 31, 2014, 2013 and 2012 was $32.41, $38.51, and $43.83, respectively.
The total intrinsic value of options exercised during the years ended December 31, 2014, 2013 and 2012 was approximately $1.9 million, $10.3 million, and $3.7 million, respectively. Cash received from options exercised in the years ended December 31, 2014, 2013 and 2012 was approximately $0.9 million, $4.9 million and $2.7 million.
At December 31, 2014, there was approximately $0.2 million of unrecognized compensation expense, net of estimated forfeitures, related to non-vested option awards. The expense is expected to be recognized over a weighted average period of 0.7 years.
Restricted Stock Units
Each restricted stock unit (“RSU”) represents a contingent right to receive one share of our common stock. RSUs generally vest on a pro-rata basis on each anniversary of the issuance date over three or four years or vest in accordance with performance-based criteria. The RSUs with performance-based vesting criteria vest in one or more tranches contingent upon the achievement of pre-determined milestones related to the development of our products, the achievement of certain prescribed clinical and regulatory objectives, the achievement of specific financial performance measures or similar metrics. There is no consideration payable on the vesting or exercise of RSUs issued under the plans. Upon vesting, the RSUs are exercised automatically and settled in shares of our common stock.
98
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Information related to RSUs at December 31, 2014 and activity during the year then ended is as follows:
| | | | | | | | | | | | |
| | | Number of
Units
(in thousands) | | | Weighted
Average
Remaining
Contractual
Life
(Years) | | | Aggregate
Intrinsic Value
(in thousands) | |
Outstanding at December 31, 2013 | | | 476 | | | | | | | | | |
Granted | | | 363 | | | | | | | | | |
Vested/Exercised | | | (193 | ) | | | | | | | | |
Forfeited | | | (57 | ) | | | | | | | | |
| | | | | | | | | | | | |
Outstanding at December 31, 2014 | | | 589 | | | | 1.51 | | | $ | 43,232 | |
| | | | | | | | | | | | |
Exercisable at December 31, 2014 | | | — | | | | — | | | $ | — | |
| | | | | | | | | | | | |
The aggregate intrinsic value at December 31, 2014 noted in the table above represents the closing price of our common stock traded on NASDAQ multiplied by the number of RSUs outstanding.
At December 31, 2014, 26,705 of the RSUs outstanding are subject to performance-based vesting criteria as described above.
The total intrinsic value of RSUs vested during the years ended December 31, 2014, 2013 and 2012 was approximately $15.4 million, $16.4 million, and $25.6 million, respectively.
The fair value of each RSU award equals the closing price of our common stock on the date of grant. The weighted average grant date fair value per share of RSUs granted during the years ended December 31, 2014, 2013 and 2012 was $97.50, $91.21, and $83.13, respectively.
At December 31, 2014, we had approximately $24.9 million of unrecognized compensation expense, net of estimated forfeitures, related to non-vested RSU awards. The expense is expected to be recognized over a weighted average period of 1.5 years.
On February 23, 2015, our board of directors approved the grant of an aggregate of 280,000 RSUs to a group of employees, including officers of the Company. Approximately 230,000 of the RSUs granted in February 2015 will vest on a pro-rata basis on each anniversary of the issuance date over four years, while the remainder is subject to performance-based vesting criteria.
Note 14. Income Taxes
During 2014, we were subject to income taxes on foreign taxable income in certain jurisdictions. The 2014 income tax provision of $0.5 million related primarily to foreign income taxes.
Income (loss) before taxes on a geographic basis during 2014 was as follows:
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | 2012 | |
| | | (in thousands) | |
United States | | $ | (20,684 | ) | | $ | (58,063 | ) | | $ | (82,463 | ) |
Non-U.S. | | | 1,878 | | | | (781 | ) | | | (5,255 | ) |
| | | | | | | | | | | | |
| | $ | (18,806 | ) | | $ | (58,844 | ) | | $ | (87,718 | ) |
| | | | | | | | | | | | |
99
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Our effective tax rate of less than 1% differs from the statutory United States federal income tax rate of 34% for all periods presented due primarily to the valuation allowance on deferred tax assets, and differences in foreign tax rates.
The primary components of net deferred tax assets and liabilities at December 31, 2014 and 2013 were as follows:
| | | | | | | | |
| | | 2014 | | | 2013 | |
| | | (in thousands) | |
Deferred tax assets: | | | | | | | | |
U.S. losses carried forward | | $ | 128,216 | | | $ | 179,503 | |
Non-U.S. losses carried forward | | | 4,227 | | | | 5,039 | |
| | | | | | | | |
Total net operating losses carried forward | | | 132,443 | | | | 184,542 | |
Research and development credit | | | — | | | | 2,114 | |
Equity awards | | | 9,902 | | | | 7,152 | |
Other deferred tax assets | | | 11,935 | | | | 4,812 | |
| | | | | | | | |
Gross deferred tax assets | | | 154,280 | | | | 198,620 | |
Deferred tax liabilities: | | | | | | | | |
Convertible debt | | | (10,632 | ) | | | (13,171 | ) |
Purchased intangible assets | | | (13,549 | ) | | | (14,143 | ) |
| | | | | | | | |
Net deferred tax assets | | | 130,099 | | | | 171,306 | |
Less: valuation allowance | | | (130,099 | ) | | | (171,306 | ) |
| | | | | | | | |
Net deferred tax asset/(liability) | | $ | — | | | $ | — | |
| | | | | | | | |
FASB ASC 740—Income Taxes requires that a valuation allowance be established to reduce a deferred tax asset to its realizable value when it is more likely than not that all or a portion of a deferred tax asset will not be realized. A review of all available positive and negative evidence needs to be considered, including the utilization of past tax credits and length of carry-back and carry-forward periods, reversal of temporary differences, tax planning strategies, our current and past performance, the market environment in which we operate, and the evaluation of tax planning strategies to generate future taxable income.
At December 31, 2014 and 2013, we had gross deferred tax assets in excess of deferred tax liabilities of $130.1 million and $171.3 million, respectively. We determined that it is not “more likely than not” that substantially all of our deferred tax assets will not be realized and therefore we should apply a valuation allowance to reduce our net deferred tax assets to their estimated realizable value. The valuation allowance primarily relates to the deferred tax assets arising from operating loss carry-forwards. The valuation allowance on our net deferred tax assets decreased by approximately $41.2 million for the year ended December 31, 2014 and increased by approximately $97.3 million and $31.4 million for the years ended December 31, 2013 and 2012, respectively.
We completed an evaluation of our net operating loss and credit carry-forwards as outlined under section 382, which resulted in a deferred tax asset reduction of approximately $56.4 million due to limitations on our U.S. net operating loss carry-forwards. Such deferred tax asset had been fully reserved. We have adjusted our net operating loss and credit carry-forwards according to the results of this evaluation.
Net operating losses representing excess tax benefits attributable to share based compensation are not included in the table of deferred tax assets and liabilities shown above because they have not been realized for financial statement purposes. Pursuant to ASC 718, excess tax benefits attributable to share based compensation
100
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
will only be recorded to additional paid-in capital when they are realized through a reduction of taxes payable. As of December 31, 2014, the portion of the federal and state net operating loss related to share based compensation is approximately $34.4 million and $24.1 million, respectively.
At December 31, 2014, we had unexpired net operating loss carry-forwards of approximately $386.1 million and $206.3 million for U.S. federal and state income tax purposes, respectively, which are available to offset future taxable income and begin to expire starting in 2024 through 2034. We also have foreign tax loss carry-forwards of approximately $20.0 million that do not expire.
We operate within multiple taxing jurisdictions and are subject to audit in those jurisdictions. Because of the complex issues involved, any claims can require an extended period to resolve.
Uncertain tax positions
The amount of gross unrecognized tax benefits as of December 31, 2014 and December 31, 2013 was $3.2 million and $3.6 million, respectively. The fiscal years 2011 through 2013 are considered open tax years (however, any year with net operating loss carryforwards remain open to adjustment) in U.S. federal and state and Australian tax jurisdictions. The fiscal years 2010 through 2013 are considered open tax years for German and United Kingdom tax jurisdictions. The fiscal years 2011 through 2013 are considered open tax years for tax jurisdictions in France.
We evaluate tax positions for recognition using a more-likely-than-not recognition threshold, and those tax positions eligible for recognition are measured as the largest amount of tax benefit that is greater than 50% likely of being realized upon the effective settlement with a taxing authority that has full knowledge of all relevant information. A reconciliation of the beginning and ending amount of gross unrecognized tax benefits is as follows:
| | | | |
Uncertain tax position | | Year ended
December 31,
2014 | |
| | | (in thousands) | |
Unrecognized tax benefits—beginning of the year | | $ | 3,567 | |
Gross increases/(decrease)—prior year | | | 17 | |
Gross increases/(decrease)—current year | | | (356 | ) |
| | | | |
Unrecognized tax benefits—end of the year | | $ | 3,228 | |
| | | | |
Included in the balance of unrecognized tax benefits at December 31, 2014, are $1.8 million of tax benefits that, if recognized, would impact the effective tax rate. The remainder of the unrecognized tax benefits would increase our net operating loss carry-forwards and would not impact the effective tax rate, so long as we continue to maintain a full valuation allowance. We anticipate that no material amounts of unrecognized tax benefits will either expire or be settled in the next 12 months of the reporting date. Additionally, no uncertain tax positions had been identified prior to 2014.
Note 15. Net Loss Per Share
Basic net loss per common share was computed by dividing net loss for the period by the weighted-average number of common shares outstanding for each respective period. Diluted net loss per common share adjusts basic net loss per common share for the dilutive effects of share-based awards as determined under the treasury stock method, our convertible senior notes as determined under the if-converted method, and other potentially
101
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
dilutive instruments only in the periods in which the effect is dilutive. Due to our net loss for all periods presented, all potentially dilutive instruments were excluded because their inclusion would have been anti-dilutive. The following instruments were excluded from the calculation of diluted weighted average shares outstanding, as their effect would be anti-dilutive.
| | | | | | | | | | | | |
Common shares issuable upon: | | 2014 | | | 2013 | | | 2012 | |
| | | (in thousands) | |
Conversion of convertible senior notes | | | 1,438 | | | | 1,438 | | | | 1,438 | |
Exercise or vesting of share-based awards | | | 696 | | | | 608 | | | | 838 | |
Note 16. Business Segment, Geographic Areas and Major Customers
For financial reporting purposes, we have one reportable segment which designs, manufactures and markets medical devices for the treatment of advanced heart failure. Products are distributed to customers located in the United States through our clinical trials and as commercial products, as commercial products to customers in Europe and under special access in other countries. Product sales attributed to a country or region are based on the location of the customer to whom the products are sold. Long-lived assets are primarily held in the United States.
Product sales by geographic location for the years ended December 31, 2014, 2013 and 2012 are as follows:
| | | | | | | | | | | | |
| | | 2014 | | | 2013 | | | 2012 | |
| | | (in thousands) | |
United States | | $ | 151,335 | | | $ | 105,345 | | | $ | 27,650 | |
Germany | | | 63,629 | | | | 54,793 | | | | 40,001 | |
International, excluding Germany | | | 63,456 | | | | 47,791 | | | | 43,271 | |
| | | | | | | | | | | | |
| | $ | 278,420 | | | $ | 207,929 | | | $ | 110,922 | |
| | | | | | | | | | | | |
The percentage of our revenue generated in the U.S. increased in 2014 and 2013 as compared to 2012 due to receipt in November 2012 of FDA approval to sell the HVAD System commercially in the U.S.
As a significant portion of our revenue is generated outside of the U.S., we are dependent on favorable economic and regulatory environments for our products in Europe and other countries outside of the U.S. For the years ended December 31, 2014, 2013 and 2012, no customers individually accounted for more than 10% of product sales.
Note 17. Commitments and Contingencies
We received a warning letter from the FDA, dated June 2, 2014, following an inspection of our Miami Lakes, Florida facility conducted in January 2014. The FDA letter cited four categories for us to address: (1) procedures for validating device design, including device labeling; (2) procedures for implementing corrective and preventive action (“CAPA”); (3) maintaining records related to investigations; and (4) validation of computer software used as part of production or quality systems. The warning letter did not require any action by physicians or patients and did not restrict use of our devices.
We sent the FDA our initial response to the warning letter within the required fifteen business days of receipt, and committed to undertaking certain quality system improvements and providing the FDA with periodic updates. During 2014, we commenced implementing systemic changes and organizational enhancements to
102
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
address the four warning letter items and related quality systems. We have established teams to review and address the items cited by the FDA and have engaged external subject matter experts to assist in assessment and remediation efforts.
At December 31, 2014, we had purchase order commitments of approximately $36.1 million related to product costs, supplies, services and property, plant and equipment purchases. Many of our materials and supplies require long lead times. Our purchase order commitments reflect materials that may be received up to one year from the date of order.
In addition to the above, we have entered into employment agreements with all of our executive officers. These contracts do not have a fixed term and are constructed on an at-will basis. Some of these contracts provide executives with the right to receive certain additional payments and benefits if their employment is terminated after a change of control, as defined in such agreements.
From time to time we invest in certain development stage entities in connection with research activities. Certain contingent milestone payments in connection with these arrangements have not been accrued in the accompanying consolidated financial statements as the amounts are indeterminate at this time.
The taxation and customs requirements, together with other applicable laws and regulations of certain foreign jurisdictions, can be inherently complex and subject to differing interpretation by local authorities. We are subject to the risk that either we have misinterpreted applicable laws and regulations, or that foreign authorities may take inconsistent, unclear or changing positions on local law, customs practices or rules. In the event that we have misinterpreted any of the above, or that foreign authorities take positions contrary to ours, we may incur liabilities that may differ materially from the amounts accrued in the accompanying consolidated financial statements.
Litigation
From time to time we may be involved in litigation or other contingencies arising in the ordinary course of business. Except as set forth below, and based on the information presently available, management believes there are no contingencies, claims or actions, pending or threatened, the ultimate resolution of which will have a material adverse effect on our financial position, liquidity or result of operations.
In accordance with FASB ASC 450, Contingencies, we accrue loss contingencies including costs of settlement, damages and defense related to litigation to the extent they are probable and reasonably estimable. Otherwise, we expense these costs as incurred. If the estimate of a probable loss is a range and no amount within the range is more likely, we accrue the minimum amount of the range.
On February 24, 2010, we received a letter from two holders of Series A Preferred Stock in HeartWare, Inc., an indirect subsidiary of HeartWare International, Inc. These holders requested various financial and other information regarding HeartWare, Inc. for the purpose of determining the Company’s compliance with their rights as holders of Series A Preferred Stock, including whether a liquidation event has occurred since inception in 2003. HeartWare, Inc. issued Series A-1 and Series A-2 Preferred Stock to certain equity holders of Kriton Medical, Inc. when HeartWare, Inc. purchased out of bankruptcy substantially all of the assets of Kriton in July 2003. The Series A-1 and Series A-2 Preferred Stock do not have voting or dividend rights but, prior to the settlement described below, entitled the holders thereof to receive, upon certain liquidation events of HeartWare, Inc. (but not the liquidation of or change of control of HeartWare International, Inc.), an amount equal to $10 per share of Series A-1 and $21 per share of Series A-2. The aggregate liquidation preference payment obligation totaled approximately $15 million.
103
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
On June 27, 2011, HeartWare International, Inc. and HeartWare, Inc., along with HeartWare’s directors, certain officers and a significant stockholder, were named as defendants in a putative class action lawsuit filed in Massachusetts state court by two other Series A Preferred Stockholders on behalf of all holders of Series A Preferred Stock. The complaint alleged that the defendants breached their fiduciary and contractual obligations to Series A Preferred Stockholders by preventing them from receiving a payment of the liquidation preference in connection with certain corporate transactions, including a transaction in 2005 in which HeartWare, Inc. was acquired by HeartWare Limited, a subsidiary of HeartWare International, Inc. The plaintiffs sought monetary damages, interest, costs and limited equitable relief. We do not believe HeartWare International, Inc., HeartWare, Inc. or any of our directors, officers or stockholders have abrogated the rights, or in any way failed to satisfy obligations owed to, any of our stockholders, including holders of Series A Preferred Stock. On February 3, 2012, counsel for plaintiffs and defendants entered into a Memorandum of Understanding to settle the matter. Defendants agreed to pay up to $1.1 million to participating putative class members in exchange for a full and unconditional release of all claims asserted in the litigation, including any and all claims arising from any right to receive a payment upon any liquidation or deemed liquidation event that has arisen or may arise in the future. On March 22, 2012, the parties filed with the court a stipulation of settlement formalizing the settlement agreement. Shortly thereafter, plaintiffs caused notice of the settlement to be made to putative class members. Following a hearing on July 25, 2012, the court entered judgment granting plaintiffs’ motion to finally approve the settlement, including the full and unconditional release of all present and future claims to receive the liquidation preference, and dismissed the case with prejudice.
At December 31, 2011, we determined that settlement of the litigation discussed above was probable and that the reasonably estimable settlement amount was $1.1 million. Accordingly, we recorded a liability for the $1.1 million and a $0.2 million receivable from one of the co-defendants, who was a related party. On September 4, 2012, the settlement was funded after receiving $0.8 million from our insurance carrier in connection with the settlement of this litigation. We recorded the insurance recovery as a reduction to selling, general and administrative expenses in our statement of operations in 2012.
Contingent Consideration and Milestone Payments
In December 2013, we acquired CircuLite using a combination of cash, stock and post-acquisition milestone and royalty payments. The post-acquisition payments are payable based upon the achievement of five specified performance milestones and revenue over periods ranging from 8-10 years subsequent to the acquisition date. The maximum amount of the aggregate post-acquisition payments could be $320 million. As of December 31, 2014, the fair value of this contingent consideration was estimated to be $43.7 million (see Note 6).
License and Development Agreements
From time to time, we license rights to technology or intellectual property from third parties. These licenses may require us to pay upfront payments as well as development or other payments upon successful completion of preclinical, clinical, regulatory or revenue milestones. In addition, these agreements may require us to pay royalties on sales of products arising from the licensed technology or intellectual property. Because the achievement of these milestones is not reasonably estimable, we have not recorded a liability in the accompanying consolidated financial statements for any of these contingencies.
Note 18. Guarantees
On December 16, 2008, we entered into a Deed of Cross Guarantee (the “Deed”) by and among the Group’s then-existing entities; HeartWare International, Inc., HeartWare Pty. Limited (formerly HeartWare Limited) and HeartWare Inc., whereby the companies have agreed to cross-guarantee each other’s liabilities. The Deed was
104
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
established as a condition to obtaining financial reporting relief under ASIC Class Order 98/1418 which provided relief for us from the requirement to prepare and lodge audited accounts for HeartWare Pty. Limited in Australia. HeartWare International, Inc. is the holding entity, HeartWare, Inc. is the alternative Trustee and HeartWare Pty. Limited is a member of the Closed Group for purposes of the Class Order.
Note 19. Retirement Savings Plan
We have established a 401(k) plan in which substantially all of our U.S. employees are eligible to participate. Contributions made by employees are limited to the maximum allowable for U.S. federal income tax purposes. Beginning in April 2010, we commenced a matching program whereby we match employee contributions at a rate of 100% of applicable contributions up to 3% of included compensation plus 50% of applicable contributions up to the next 2% of included compensation. Our contributions to the 401(k) plan were approximately $1.4 million, $1.1 million and $0.8 million for the years ended December 31, 2014, 2013 and 2012.
Note 20. Quarterly Financial Information (Unaudited)
The following table presents selected quarterly financial information for the periods indicated. This information has been derived from our unaudited quarterly consolidated financial statements, which in the opinion of management include all adjustments (consisting only of normal recurring adjustments) necessary for a fair presentation of such information. The quarterly per share data presented below was calculated separately and may not sum to the annual figures presented in the consolidated financial statements. These operating results are also not necessarily indicative of results for any future period.
| | | | | | | | | | | | | | | | |
| | | Three Months Ended | |
| | | March 31 | | | June 30 | | | September 30 | | | December 31 | |
| | | (in thousands, except per share data) | |
2014 | | | | | | | | | | | | | | | | |
Revenue, net | | $ | 66,472 | | | $ | 70,131 | | | $ | 68,608 | | | $ | 73,209 | |
Gross profit | | | 43,557 | | | | 47,176 | | | | 45,631 | | | | 49,860 | |
Net income (loss) | | | (19,444 | ) | | | 8,364 | | | | (7,370 | ) | | | (916 | ) |
Net income (loss) per common share—basic (1) | | $ | (1.15 | ) | | $ | 0.49 | | | $ | (0.43 | ) | | $ | (0.05 | ) |
Net income (loss) per common share—diluted (1) | | $ | (1.15 | ) | | $ | 0.48 | | | $ | (0.43 | ) | | $ | (0.05 | ) |
Weighted average shares outstanding—basic | | | 16,934 | | | | 16,989 | | | | 17,007 | | | | 17,037 | |
Weighted average shares outstanding—diluted | | | 16,934 | | | | 17,305 | | | | 17,007 | | | | 17,037 | |
| | | | |
2013 | | | | | | | | | | | | | | | | |
Revenue, net | | $ | 49,239 | | | $ | 50,836 | | | $ | 54,800 | | | $ | 53,054 | |
Gross profit | | | 30,459 | | | | 31,970 | | | | 35,271 | | | | 33,761 | |
Net loss | | | (12,959 | ) | | | (12,934 | ) | | | (11,371 | ) | | | (22,048 | ) |
Net loss per common share—basic and diluted (1) | | $ | (0.87 | ) | | $ | (0.79 | ) | | $ | (0.69 | ) | | $ | (1.33 | ) |
Weighted average shares outstanding—basic and diluted | | | 14,860 | | | | 16,370 | | | | 16,439 | | | | 16,574 | |
| (1) | Net income (loss) per common share for each quarter is computed using the weighted-average number of shares outstanding during that quarter while net loss per common share for the full year is computed using the weighted-average number of shares outstanding during the year. Thus, the sum of the four quarters’ net income (loss) per common share may not equal the full-year loss per share. |
105
HEARTWARE INTERNATIONAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
Significant amounts in per quarter information listed above include:
| | • | | Net loss for the quarter ended December 31, 2014 included $2.6 million for the impairment of certain purchased intangible assets and a $9.1 million decrease in the fair value of contingent consideration associated with the acquisition of CircuLite. |
| | • | | Net loss for the quarter ended September 30, 2014 included a $3.6 million decrease in the fair value of contingent consideration associated with the acquisition of CircuLite. |
| | • | | Net income for the quarter ended June 30, 2014 included a $13.7 million decrease in the fair value of contingent consideration associated with the acquisition of CircuLite. |
| | • | | Net loss for the quarter ended March 31, 2014 included $4.1 million in restructuring costs and a $3.1 million increase in the fair value of contingent consideration associated with the acquisition of CircuLite. |
| | • | | Net loss for the quarter ended December 31, 2013 included the impairment of certain purchased intangible assets aggregating $3.7 million, $3.1 million of CircuLite operating expenses, which includes $0.6 million of severance costs, and $2.3 million of transaction costs associated with the acquisition of CircuLite. |
| | • | | Net loss for the quarter ended March 31, 2014 included foreign exchange gains of $0.2 million. |
| | • | | Net loss for the quarters ended September 30 and December 31, 2014 included foreign exchange losses of $3.3 million and $1.8 million, respectively. |
| | • | | Net loss for the quarters ended March 31 and June 30, 2013 included foreign exchange losses of $1.9 million and $0.6 million, respectively. |
| | • | | Net loss for the quarters ended September 30 and December 31, 2013 included foreign exchange gains of $2.1 million and $0.3 million, respectively. |
| | • | | Net income (loss) for the quarters ended March 31, June 30, September 30 and December 31, 2014 included share-based compensation expense of approximately $4.4 million, $6.5 million, $6.4 million and $6.2 million, respectively. |
| | • | | Net loss for the quarters ended March 31, June 30, September 30 and December 31, 2013 included share-based compensation expense of approximately $4.4 million, $4.9 million, $7.1 million and $5.3 million, respectively. |
Note 21. Subsequent Events
We have evaluated events and transactions that occurred subsequent to December 31, 2014 through the date the financial statements were issued, for potential recognition or disclosure in the accompanying consolidated financial statements. As disclosed in Note 11, in January 2015, we initiated a plan to close our facilities located in Aachen, Germany. We currently estimate that the aggregate amount of charges related to this action will be approximately $2.1 million, which we anticipate recording in the quarter ending March 31, 2015. Except for this event, we did not identify any events or transactions that should be recognized or disclosed in the accompanying consolidated financial statements.
106
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of the Chief Executive Officer and the Chief Financial Officer, carried out an evaluation required by the Securities Exchange Act of 1934, as amended (the “Exchange Act”), of the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rule13a-15(e) of the Exchange Act, as of December 31, 2014. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2014, our disclosure controls and procedures were effective to provide reasonable assurance that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and to provide reasonable assurance that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosures.
Management’s Annual Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Our internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on our financial statements.
Under the supervision and with the participation of our Chief Executive Officer and our Chief Financial Officer, management conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2014 based on the framework inInternal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on that evaluation, management concluded that our internal control over financial reporting was effective as of December 31, 2013.
Our independent registered public accounting firm, Grant Thornton LLP, has issued a report on our internal control over financial reporting, which is included in Item 8 of this Annual Report on Form 10-K.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the fiscal quarter ended December 31, 2014, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations on Controls and Procedures
Our management, including the Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all error and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute
107
assurance that all control issues and instances of fraud, if any, have been detected. Thus, misstatements due to error or fraud may occur and not be detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of controls.
Item 9B. Other Information
None.
108
Part III
Item 10. Directors, Executive Officers and Corporate Governance
Our executive officers and their respective ages as of December 31, 2014 are as follows:
| | | | | | |
Name | | Age | | | Position |
Douglas Godshall | | | 50 | | | Director, President and Chief Executive Officer |
Jeffrey LaRose | | | 52 | | | Executive Vice President and Chief Scientific Officer |
Katrin Leadley | | | 52 | | | Chief Medical Officer |
Peter McAree | | | 50 | | | Senior Vice President and Chief Financial Officer |
Larry Knopf | | | 53 | | | Senior Vice President, General Counsel and Secretary |
James Schuermann | | | 46 | | | Senior Vice President, Sales and Marketing |
Mark Strong | | | 43 | | | Senior Vice President, Research & Development and Quality Assurance |
Robert Yocher | | | 63 | | | Senior Vice President, Regulatory Affairs |
Biographical Summaries
Douglas Godshall. Mr. Godshall has been our President and Chief Executive Officer since September 2006 and a director since October 2006. Prior to joining HeartWare, Mr. Godshall served in various executive and managerial positions at Boston Scientific Corporation, where he had been employed since 1990, including as a member of Boston Scientific’s Operating Committee and since January 2005, as President, Vascular Surgery. Mr. Godshall also spent five years as Vice President, Business Development, at Boston Scientific, where he was focused on acquisition strategies for the cardiology, electrophysiology, neuroradiology and vascular surgery divisions. Mr. Godshall, since March 2012, serves on the board of directors of pSivida Corp., a public company traded on both the NASDAQ Stock Market and the ASX that specializes in the development of miniaturized, injectable drug delivery systems and, since May of 2013, Vital Therapies, Inc. a public company traded on the NASDAQ Stock Market that develops cell based therapies for the treatment of liver disease. Additionally, since May 2014, Mr. Godshall serves on the Board of Directors of the Medical Device Manufacturers Association, a national trade association. Mr. Godshall has a Bachelor of Arts in Business from Lafayette College and Masters of Business Administration from Northeastern University in Boston, Massachusetts.
Jeffrey LaRose. Mr. LaRose is our Chief Scientific Officer and has been with the Company since its inception. Prior to joining HeartWare, since April 1999, he was involved in the development of HeartWare’s technology through his employment with Kriton Medical, which the Company acquired in 2003. He is responsible for all aspects of the design and physiological controls for HeartWare’s left ventricular assist device, the HVAD System. Mr. LaRose also leads the development of our miniaturization technology and has twenty years of experience in hydraulic technology development including roles with AEA Technology Engineering Software and Babcock and Wilcox. He holds a Master of Science in Mechanical Engineering from the University of Akron, Ohio.
Dr. Katrin Leadley. Dr. Leadley joined HeartWare in September 2014 as our Chief Medical Officer responsible for all medical and clinical affairs, including the design and execution of HeartWare’s clinical trial program. Prior to joining HeartWare, Dr. Leadley served as the Chief Medical Officer of JenaValve Technology from 2011 until September 2014. From 2003 until 2011, Dr. Leadley held a variety of leadership positions at Boston Scientific Corporation, a global medical device company. From 2009 until 2011, Dr. Leadley served as Boston Scientific’s Global Senior Medical Director, Clinical Services where she provided medical and scientific leadership for the development of clinical research programs. She also served as its Medical Director, Clinical services from 2005 until 2009 and its Associate Medical Director from 2003 until 2005. Prior to joining Boston Scientific, Dr. Leadley held managerial positions at several other life sciences companies based in California, including Advanced Stent Technologies, Pulmonx and Quintiles/The Lewin Group in San Francisco. She has authored numerous scientific and medical publications and presented at industry conferences and events around the world. Dr. Leadley earned her medical degree at Ludwig-Maximillian University Medical School in Munich. She was also awarded a National Institute of Health Postdoctoral Fellowship by the School of Public Health at the University of California at Berkeley.
109
Peter McAree. Mr. McAree joined HeartWare in July 2012 as our Senior Vice President, Chief Financial Officer and Treasurer. Previously, he served as Senior Vice President and Chief Financial Officer of Caliper Life Sciences, Inc. from April 2008 through November 2011, after having held the position of Vice President of Finance since 2003. Mr. McAree was Chief Financial Officer of Zymark Corporation from May 2000, until the acquisition of Zymark by Caliper in 2003. Having also served in financial leadership positions in other industries, Mr. McAree began his career with Arthur Andersen, Boston, where he held various positions over nearly a decade. He received his B.S. in Accountancy from Bentley University, and is a licensed Certified Public Accountant in Massachusetts.
Lawrence Knopf. Mr. Knopf joined HeartWare in March 2011 as our Senior Vice President, General Counsel and Secretary. Mr. Knopf has overall responsibility for the Company’s legal, reimbursement and compliance functions. Between 1993 and 2010, Mr. Knopf served in a variety of legal positions at Boston Scientific Corporation, a global medical device company. From 2007, Mr. Knopf was Senior Vice President and Deputy General Counsel, from 1994, Vice President and Assistant General Counsel and from 1993, Assistant General Counsel. Previously, Mr. Knopf was a corporate associate at the Boston law firms of Bingham McCutchen, LLP and Gaston & Snow. Mr. Knopf received a Juris Doctor from the University of Michigan School of Law and holds a Bachelor of Science, Accounting and Political Science, from The Wharton School of the University of Pennsylvania. He is admitted to the Bar in Massachusetts, New York and Connecticut and passed the Certified Public Accountant examination in Connecticut.
James Schuermann.Mr. Schuermann joined HeartWare in September 2007 as our Senior Vice President, Sales and Marketing. Mr. Schuermann has overall responsibility for HeartWare’s global sales and marketing activities. Mr. Schuermann has over 20 years of sales and marketing experience in the medical device arena. Prior to joining HeartWare, Mr. Schuermann spent nine years in sales and marketing at Boston Scientific Corporation and held various management positions including Director of Marketing. Before joining Boston Scientific, he spent 5 years in medical sales and sales management at Sherwood Davis & Geck and 3 years in marketing at Armstrong World Industries. Mr. Schuermann received his undergraduate degree in marketing from Kelley School of Business, Indiana University, Bloomington, and his MBA from Ageno School of Business, Golden Gate University, San Francisco.
Mark Strong. Mr. Strong joined HeartWare on September 30, 2013 and currently serves HeartWare as Senior Vice President, Research and Development and Quality. Prior to joining HeartWare, he held a variety of research and development leadership positions at Boston Scientific Corporation, a global medical device company. From 2011 to 2013, Mr. Strong served as Boston Scientific’s Vice President of Research and Development for CRV, its largest business unit. He also served as Director, Systems Engineering and Product Development Process from 2009 to 2011 and Director of Design Engineering from 2006 to 2009. From 1993 until 2006, Mr. Strong held a variety of research and development and engineering positions at Boston Scientific. Mr. Strong holds a B.S. in electrical engineering and biomedical engineering from North Dakota State University. In addition, he earned a Master’s degree in electrical engineering at the University of Minnesota and an MBA at the University of St. Thomas in St. Paul, Minnesota.
Robert Yocher.Mr. Yocher, our Senior Vice President of Regulatory Affairs, joined HeartWare in June 2011 and has overall responsibility for HeartWare’s regulatory activities. Between 1999 and 2011, Mr. Yocher was Vice President, Regulatory Affairs and Corporate Quality Compliance at Genzyme Corporation, a global biopharmaceuticals and medical device company. Previously, Mr. Yocher worked in a variety of senior regulatory, quality and clinical positions at EDAP Technomed, Inc., a urological medical device company, BioField Corporation, a development stage medical device company, and Dornier Medical Systems, a therapeutic device company, among others. Mr. Yocher holds a Master of Health Science, Public Health Microbiology and Epidemiology, from Quinnipiac University and a Bachelor of Arts, Microbiology/Chemistry, from the University of Connecticut.
110
Other Information
We have a code of business conduct and ethics that applies to each director, officer and employee of the Company, including the executive, financial and accounting officers. Our code of conduct is available in the “Corporate Governance” section on our website atwww.heartware.com.
We expect to make all required disclosures regarding any amendments to, or waivers from, this Code of Conduct on our website.
The other information required by this Item 10 is incorporated herein by reference to the applicable information in our definitive proxy statement for our 2015 annual meeting of stockholders to be filed with the Securities and Exchange Commission or is to be included in Item 10 of an amendment to this Annual Report on Form 10-K to be filed with the Securities and Exchange Commission.
Item 11. Executive Compensation
The information required by this Item 11 is incorporated herein by reference to the applicable information in our definitive proxy statement for our 2015 annual meeting of stockholders to be filed with the Securities and Exchange Commission, including the information set forth under the caption “Executive Compensation” or is to be included in Item 11 of an amendment to this Annual Report on Form 10-K to be filed with the Securities and Exchange Commission.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item 12 is incorporated herein by reference to the applicable information in our definitive proxy statement for our 2015 annual meeting of stockholders to be filed with the Securities and Exchange Commission, including the information set forth under the caption “Security Ownership of Certain Beneficial Owners and Management” or is to be included in Item 12 of an amendment to this Annual Report on Form 10-K to be filed with the Securities and Exchange Commission.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item 13 is incorporated herein by reference to the applicable information in our definitive proxy statement for our 2015 annual meeting of stockholders to be filed with the Securities and Exchange Commission, including the information set forth under the captions “Certain Relationships and Related Transactions, and Director Independence”, “Policies and Procedures for Review and Approval of Related Party Transactions”, “Corporate Governance” and “Compensation Committee Interlocks and Insider Participation,” or is to be included in Item 13 of an amendment to this Annual Report on Form 10-K to be filed with the Securities and Exchange Commission.
Item 14. Principal Accounting Fees and Services
The information required by this Item 14 is incorporated herein by reference to the applicable information in our definitive proxy statement for our 2015 annual meeting of stockholders to be filed with the Securities and Exchange Commission, including the information set forth under the captions “Principal Accounting Fees and Services” and “Audit Committee’s Pre-Approval Policy,” or is to be included in Item 14 of an amendment to this Annual Report on Form 10-K to be filed with the Securities and Exchange Commission.
111
Part IV.
Item 15. Exhibits, Financial Statement Schedules
The following documents are filed as part of this Annual Report on Form 10-K:
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets
Consolidated Statements of Operations
Consolidated Statements of Comprehensive Loss
Consolidated Statement of Stockholders’ Equity
Consolidated Statements of Cash Flows
Notes to Consolidated Financial Statements
| | 2. | Financial Statement Schedules: |
Required schedule information is included in the Notes to Consolidated Financial Statements or is omitted because it is either not required or not applicable.
See Exhibit Index
112
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | |
| | | | HeartWare International, Inc. |
| | | |
| Date: March 2, 2015 | | | | By | | /s/ Douglas Godshall |
| | | | | | Name: Douglas Godshall |
| | | | | | Title: President and Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | |
| Name | | Title | | Date |
| | |
/s/ Douglas Godshall Douglas Godshall | | President, Chief Executive Officer and Director (Principal Executive Officer) | | March 2, 2015 |
| | |
/s/ Peter McAree Peter McAree | | Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | | March 2, 2015 |
| | |
/s/ C. Raymond Larkin, Jr. C. Raymond Larkin, Jr. | | Chairman and Director | | March 2, 2015 |
| | |
/s/ Timothy Barberich Timothy Barberich | | Director | | March 2, 2015 |
| | |
/s/ Cynthia Feldmann Cynthia Feldmann | | Director | | March 2, 2015 |
| | |
/s/ Seth Harrison Seth Harrison | | Director | | March 2, 2015 |
| | |
/s/ Robert Stockman Robert Stockman | | Director | | March 2, 2015 |
| | |
/s/ Robert Thomas Robert Thomas | | Director | | March 2, 2015 |
| | |
/s/ Denis Wade Denis Wade | | Director | | March 2, 2015 |
113
Exhibit Index
| | |
Exhibit No. | | Description |
| |
| 2.1 | | Agreement and Plan of Merger, dated as of December 1, 2013, by and among HeartWare International, Inc., Chronos Merger Sub Inc., CircuLite, Inc., and Shareholder Representative Services LLC, as securityholder representative. (10) |
| |
| 3.1 | | Certificate of Incorporation of HeartWare International, Inc.(3) |
| |
| 3.2 | | Bylaws of HeartWare International, Inc. (3) |
| |
| 10.01 | | Amended and Restated Employment Agreement, dated as of December 16, 2009, by and between HeartWare International, Inc. and Douglas Godshall (9) + |
| |
| 10.02 | | Amended and Restated Employment Agreement, dated as of December 16, 2009, by and between HeartWare, Inc. and Jeffrey LaRose (16) + |
| |
| 10.03 | | Employment Agreement, dated as of December 5, 2008, between HeartWare, Inc. and James Schuermann (4) + |
| |
| 10.05 | | Form of Amendment to Employment Agreement (for Section 16 officers), dated December 2010 (23) + |
| |
| 10.06 | | Form of Deed of Indemnity, Access and Insurance Agreement for directors and executive officers (1) + |
| |
| 10.07 | | Letter of Appointment as a Director of the Company dated December 1, 2006 between HeartWare Limited and Robert Stockman (1) + |
| |
| 10.08 | | Letter of Appointment as a Director of the Company dated December 15, 2004 between HeartWare Limited and Robert Thomas (1) + |
| |
| 10.09 | | Letter of Appointment as a Director of the Company dated December 15, 2004 between HeartWare Limited and Denis Wade (1) + |
| |
| 10.10 | | Letter of Appointment as a Director of the Company dated September 3, 2008 between HeartWare International, Inc. and Ray Larkin (11) + |
| |
| 10.11 | | Letter of Appointment as a Director of the Company dated April 16, 2008 between HeartWare International, Inc. and Timothy J. Barberich (12) + |
| |
| 10.12 | | Letter of Appointment as a Director of the Company dated December 21, 2011 between HeartWare International, Inc. and Cynthia Feldmann (2) + |
| |
| 10.13 | | HeartWare International, Inc. 2008 Stock Incentive Plan (5) + |
| |
| 10.14 | | HeartWare International, Inc. Employee Stock Option Plan (6) + |
| |
| 10.15 | | HeartWare International, Inc. Restricted Stock Unit Plan (7) + |
| |
| 10.16 | | Form of HeartWare International, Inc. Incentive Option Terms (8) + |
| |
| 10.17 | | Nonstatutory Stock Option Notice and Agreement to 2008 Stock Incentive Plan (20) + |
| |
| 10.18 | | Restricted Stock Units Notice and Agreement to 2008 Stock Incentive Plan (21) + |
| |
| 10.19 | | Sublease agreement, dated as of October 17, 2013, by and between The TJX Companies, Inc. and HeartWare International, Inc. (32) |
| |
| 10.20 | | Business Lease, dated December 27, 2006, between HeartWare, Inc. and Atlantic-Philadelphia Realty LLC (13) |
| |
| 10.21 | | First Amendment to Business Lease, dated December 27, 2006, between HeartWare, Inc. and Atlantic-Philadelphia Realty LLC, dated August 19, 2008 (14) |
114
| | |
Exhibit No. | | Description |
| |
| 10.22 | | Second Amendment to Business Lease, dated December 27, 2006, between HeartWare, Inc. and Atlantic-Philadelphia Realty LLC, dated August 9, 2010 (22) |
| |
| 10.23 | | Third amendment to Business Lease, dated December 27, 2006, between HeartWare, Inc. and Atlantic-Philadelphia Realty LLC, dated June 30, 2011 (26) |
| |
| 10.24 | | Fourth Amendment to business lease, dated December 27, 2006, between HeartWare, Inc. and Atlantic-Philadelphia Realty LLC, dated July 30, 2012 (15) |
| |
| 10.25 | | Lease Agreement dated December 8, 2010 by and between MCP EWE LLC, as Landlord, HeartWare, Inc., as Tenant, and guaranteed by HeartWare International, Inc., as Guarantor (24) |
| |
| 10.26 | | First Amendment to Lease Agreement dated December 8, 2010 by and between MCP EWE LLC, as Landlord, HeartWare, Inc., as Tenant, and guaranteed by HeartWare International, Inc., as Guarantor, dated November 30, 2012 between The Graham Companies, as successor in interest to MCP EWE LLC, and HeartWare, Inc. (34) |
| |
| 10.27 | | Indenture dated as of December 15, 2010 between the Company and Wilmington Trust FSB, as trustee (17) |
| |
| 10.28 | | First Supplemental Indenture dated as of December 15, 2010 between the Company and Wilmington Trust FSB, as trustee (18) |
| |
| 10.29 | | Form of 3.50% Convertible Senior Notes due 2017 (19) |
| |
| 10.30 | | Separation Agreement and General Release between HeartWare, Inc. and Lauren Farrell dated as of February 26, 2013 (33) + |
| |
| 10.31 | | Offer letter, dated as of March 21, 2011, between HeartWare, Inc. and Lawrence J. Knopf (27) + |
| |
| 10.32 | | Offer letter, dated as of June 6, 2011, between HeartWare, Inc. and Robert E. Yocher (28) + |
| |
| 10.33 | | Offer Letter, dated as of June 18, 2012, between HeartWare, Inc. and Peter McAree (25) + |
| |
| 10.34 | | Offer letter, dated as of September 23, 2013, between HeartWare, Inc. and Mark Strong (35) + |
| |
| 10.35 | | Promotion letter, effective as of July 21, 2014, between HeartWare, Inc. and Mark Strong (36)+ |
| |
| 10.36 | | Employment Contract, dated as of September 1, 2014, between HeartWare GmbH and Katrin Leadley, M.D. (37)+ |
| |
| 10.37 | | HeartWare International, Inc. 2012 Incentive Award Plan (29) |
| |
| 10.38 | | Form of HeartWare International Inc. 2012 Incentive Award Plan Stock Option Notice and Award Agreement (30) |
| |
| 10.39 | | Form of HeartWare International Inc. 2012 Incentive Award Plan Restricted Stock Unit Notice and Award Agreement (31) |
| |
| 10.40 | | Registration Rights Agreement, dated as of December 1, 2013, by and among HeartWare International, Inc., Shareholder Representative Services LLC, as securityholder representative, and the other parties thereto. (10) |
| |
| 21.1 | | List of Subsidiaries * |
| |
| 23.1 | | Consent of Independent Registered Public Accounting Firm * |
| |
| 31.1 | | Certification pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of
1934 * |
| |
| 31.2 | | Certification pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of
1934 * |
115
| | |
Exhibit No. | | Description |
| |
| 32.1 | | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 ** |
| |
| 32.2 | | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 ** |
| |
| 101 | | The following materials from the Company’s Annual Report on Form 10-K for the year ended December 31, 2014, formatted in XBRL (Extensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Comprehensive Loss, (iv) Consolidated Statement of Stockholders’ Equity, (v) Consolidated Statements of Cash Flows, and (vi) Notes to the Consolidated Financial Statements. |
| (1) | Incorporated by reference to the respective exhibits filed with the Company’s Registration Statement on Form 10 (File No. 000-52595) filed with the Securities and Exchange Commission on April 30, 2007. |
| (2) | Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on February 28, 2012. |
| (3) | Incorporated by reference to the respective exhibits filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on November 13, 2008. |
| (4) | Incorporated by reference to Exhibit 99 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 5, 2008. |
| (5) | Incorporated by reference to Appendix 12 to the Information Memorandum contained in the Company’s Proxy Statement on Form DEF 14A filed with the Securities and Exchange Commission on September 22, 2008. |
| (6) | Incorporated by reference to Appendix 9 to the Information Memorandum contained in the Company’s Proxy Statement on Form DEF 14A filed with the Securities and Exchange Commission on September 22, 2008. |
| (7) | Incorporated by reference to Appendix 10 to the Information Memorandum contained in the Company’s Proxy Statement on Form DEF 14A filed with the Securities and Exchange Commission on September 22, 2008. |
| (8) | Incorporated by reference to Exhibit 99.4 to the Company’s Registration Statement on Form S-8 (File No. 333-155359) filed with the Securities and Exchange Commission on November 13, 2008. |
| (9) | Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 18, 2009. |
| (10) | Incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 2, 2013. |
| (11) | Incorporated by reference to Exhibit 10.27 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 26, 2009. |
| (12) | Incorporated by reference to Exhibit 10.28 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 26, 2009. |
| (13) | Incorporated by reference to Exhibit 10.40 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 28, 2008. |
| (14) | Incorporated by reference to Exhibit 10.27 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 23, 2010. |
| (15) | Incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 9, 2012. |
| (16) | Incorporated by reference to Exhibit 10.6 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 23, 2010. |
| (17) | Incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 15, 2010. |
| (18) | Incorporated by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 15, 2010. |
116
| (19) | Incorporated by reference to Exhibit 4.3 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 15, 2010. |
| (20) | Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 6, 2010. |
| (21) | Incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 6, 2010. |
| (22) | Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on August 19, 2010. |
| (23) | Incorporated by reference to Exhibit 10.08 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 24, 2011. |
| (24) | Incorporated by reference to Exhibit 10.29 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 24, 2011. |
| (25) | Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 8, 2012. |
| (26) | Incorporated by reference to Exhibit 10.35 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 5, 2011. |
| (27) | Incorporated by reference to Exhibit 10.38 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 27, 2012. |
| (28) | Incorporated by reference to Exhibit 10.39 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 27, 2012. |
| (29) | Incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 8, 2012. |
| (30) | Incorporated by reference to Exhibit 99.2 to the Registrant’s Registration Statement on Form S-8 (File No. 333-184358) filed with the Securities and Exchange Commission on October 10, 2012. |
| (31) | Incorporated by reference to Exhibit 99.3 to the Registrant’s Registration Statement on Form S-8 (File No. 333-184358) filed with the Securities and Exchange Commission on October 10, 2012. |
| (32) | Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 7, 2013. |
| (33) | Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 3, 2013. |
| (34) | Incorporated by reference to Exhibit 10.29 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 27, 2013. |
| (35) | Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on October 31, 2014. |
| (36) | Incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on October 31, 2014. |
| (37) | Incorporated by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on October 31, 2014. |
| + | Management contract or compensatory plan or arrangement. |
117