UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of the Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): March 6, 2015
Kura Oncology, Inc.
(Exact Name of Registrant as Specified in its Charter)
| Delaware | 000-53058 | 61-1547851 | ||
(State of Incorporation) | (Commission File Number) | (IRS Employer Identification No.) |
11119 N. Torrey Pines Road, Suite 125
La Jolla, CA 92037
(Address of principal executive offices)
Registrant’s telephone number, including area code: (858) 500-8800
Zeta Acquisition Corp. III
c/o Equity Dynamics Inc.
666 Walnut Street, Suite 2116
Des Moines, Iowa 50309
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ¨ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ¨ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ¨ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ¨ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
| Item 1.01 | Entry into a Material Definitive Agreement. |
The disclosures set forth in Item 2.01 hereof are hereby incorporated by reference into this Item 1.01.
| Item 2.01 | Completion of Acquisition or Disposition of Assets. |
Pursuant to an Agreement and Plan of Merger dated March 6, 2015, or the Merger Agreement, by and among Zeta Acquisition Corp. III, which, unless otherwise indicated, we refer to as the Company, we, our and us; Kura Operations, Inc., a Delaware corporation and wholly-owned subsidiary of the Company, or Merger Sub; and Kura Oncology, Inc., a Delaware corporation, which, unless otherwise indicated, we refer to as Kura; Merger Sub merged with and into Kura, with Kura remaining as the surviving entity and a wholly-owned operating subsidiary of the Company. This transaction is referred to throughout this report as the “Merger.” The Merger was effective on March 6, 2015, upon the filing of a Certificate of Merger with the Secretary of State of the State of Delaware. As part of the Merger, Kura changed its name to Kura Operations, Inc. A copy of the Merger Agreement is filed herewith as Exhibit 2.1, and is incorporated herein by reference.
Immediately following the Merger, a newly organized wholly-owned subsidiary of the Company named “Kura Oncology, Inc.”, or Name Change Merger Sub, merged with and into the Company, leaving the Company as the surviving corporation. We refer to this transaction as the “Name Change Merger.” In connection with the Name Change Merger, we relinquished our corporate name “Zeta Acquisition Corp. III” and assumed in its place the name “Kura Oncology, Inc.” The Name Change Merger and name change became effective on March 6, 2015, upon the filing of a Certificate of Ownership and Merger with the Secretary of State of the State of Delaware. A copy of the Certificate of Ownership and Merger is filed herewith as Exhibit 3.4, and is incorporated herein by reference.
At the effective time of the Merger, or the Effective Time, the legal existence of Merger Sub ceased and each share of Kura common stock that was issued and outstanding immediately prior to the Effective Time was automatically exchanged for 0.5 shares of our common stock, which we refer to as the Exchange. We issued an aggregate of 14,508,177 shares of our common stock upon the Exchange. In addition, at the Effective Time, we assumed Kura’s 2014 Equity Incentive Plan and concurrently approved the amendment and restatement of the Kura 2014 Equity Incentive Plan pursuant to our Amended and Restated 2014 Equity Incentive Plan, or 2014 plan, effective upon the date that is 20 days after the mailing of a definitive Schedule 14C information statement to our pre-Merger stockholders. As of the Effective Time, there were no outstanding options to purchase shares of Kura common stock under the Kura 2014 Equity Incentive Plan.
Immediately following the Effective Time, pursuant to the terms of a Redemption Agreement dated March 6, 2015, or the Redemption Agreement, by and among the Company and its pre-Merger stockholders, we completed the closing of a redemption of 5,000,000 shares of our common stock, or the Redemption, from our pre-Merger stockholders for consideration of $70,000, plus professional costs related to the transaction, not to exceed $30,000. The 5,000,000 shares constituted all of the issued and outstanding shares of our capital stock, on a fully-diluted basis, immediately prior to the Merger. A copy of the Redemption Agreement is filed herewith as Exhibit 10.10, and is incorporated herein by reference.
Upon completion of the Merger and the Redemption, the former stockholders of Kura held 100% of the outstanding shares of our capital stock. Unless otherwise indicated in this Current Report on Form 8-K, or this report, all share and per share figures reflect the exchange of each share of Kura common stock then outstanding for 0.5 shares of our common stock at the Effective Time of the Merger; however, the share and per share numbers in the financial statements of Kura filed herewith as Exhibit 99.1 are not adjusted to give effect to the Merger.
As a condition to the Merger, we entered into an Indemnity Agreement with our former officers and directors, or the Indemnity Agreement, pursuant to which we agreed to indemnify such former officers and directors for actions taken by them in their official capacities relating to the consideration, approval and consummation of the Merger and certain related transactions. A copy of the Indemnity Agreement is filed herewith as Exhibit 10.11, and is incorporated herein by reference.
The Merger is being accounted for as a capital transaction. Upon the effectiveness of the Merger, the Company’s business became the operation of Kura and its business. Immediately following the Effective Time, our board of directors, which immediately prior to the Effective Time consisted of John Pappajohn and Matthew P. Kinley, appointed Troy E. Wilson, Ph.D., J.D., who was President and Chief Executive Officer of Kura, as our Chairman, President and Chief Executive Officer and as a director to serve on our board of directors. At the Effective Time, Mr. Pappajohn and Mr. Kinley resigned from all of their positions as officers of the Company and Mr. Pappajohn resigned from his position as a director of the Company. In addition, immediately following the Effective Time, our board of directors appointed Heidi Henson, who was the Chief Financial Officer and Secretary of Kura, as our Chief Financial Officer and Secretary; Yi Liu, Ph.D., who was the Chief Scientific Officer of Kura, as our Chief Scientific Officer; Antonio Gualberto, M.D., Ph.D., who was the Chief Medical Officer of Kura, as our Chief Medical Officer; Annette North, who was the Senior Vice President, General Counsel of Kura, as our
2
Senior Vice President, General Counsel; and Pingda Ren, Ph.D., who was the Senior Vice President, Chemistry and Pharmaceutical Sciences of Kura, as our Senior Vice President, Chemistry and Pharmaceutical Sciences. On March 17, 2015, which is the eleventh day following the date that we filed with the Securities and Exchange Commission, or SEC, and transmitted to our stockholders prior to the Merger, a Schedule 14f-1 reporting a change in the majority of our directors, or the New Board Effective Date, Robert E. Hoffman will be appointed to our board of directors to serve on our board of directors with Dr. Wilson, and Mr. Kinley will resign from our board of directors as of such date.
Prior to the Merger, Kura sold to accredited investors approximately $60.0 million of its shares of common stock, or 18,971,136 shares at a price of $3.16 per share, which included $7.5 million in principal and $0.1 million in accrued interest from the conversion of Kura’s then outstanding convertible promissory notes. We refer to this transaction as the Private Placement and the number of shares stated in the preceding sentence does not reflect the Exchange in the Merger. The price per share in the Private Placement, as adjusted for the Exchange in the Merger, would be $6.32 per share of our post-Merger common stock. Also, Kura granted the investors in the Private Placement registration rights requiring Kura or any successor to register those shares of Kura common stock (which were exchanged for shares of our common stock, along with the rest of the outstanding shares of Kura capital stock, except for dissenting shares, at the Effective Time) for public resale, as described in more detail below. The then existing stockholders of Kura who agreed to become parties to the registration rights agreement also became entitled to such registration rights, subject to specified differences in the agreement between the rights of new investors and existing stockholders. The Private Placement closed immediately prior to the filing of a Certificate of Merger with the Secretary of State of the State of Delaware, on March 6, 2015.
The Merger Agreement has been filed as Exhibit 2.1 to this Current Report on Form 8-K to provide investors and security holders with information regarding its terms. It is not intended to provide any other factual information about the Company or Kura. The representations, warranties and covenants contained in the Merger Agreement were made only for the purposes of the Merger Agreement and as of specified dates, were solely for the benefit of the parties to the Merger Agreement, and may be subject to limitations agreed upon by the contracting parties. The representations and warranties may have been made for the purposes of allocating contractual risk between the parties to the Merger Agreement instead of establishing these matters as facts, and may be subject to standards of materiality applicable to the contracting parties that differ from those applicable to investors. Investors are not third-party beneficiaries under the Merger Agreement and should not rely on the representations, warranties and covenants or any descriptions thereof as characterizations of the actual state of facts or condition of the Company, Kura or any of their respective subsidiaries or affiliates. In addition, the assertions embodied in the representations and warranties contained in the Merger Agreement are qualified by information in confidential disclosure schedules provided by the Company and Merger Sub and Kura, which are not being filed with this Current Report on Form 8-K as permitted by the SEC’s rules and regulations. Accordingly, investors should not rely on the representations and warranties as characterizations of the actual state of facts, since (i) they were made only as of the date of the Merger Agreement or a prior, specified date, (ii) in some cases they are subject to qualifications with respect to materiality, knowledge and/or other matters, and (iii) they may be modified in important part by the underlying disclosure schedule. Moreover, information concerning the subject matter of the representations and warranties may change after the date of the Merger Agreement, which subsequent information may or may not be fully reflected in the Company’s public disclosures.
Kura announced the Private Placement and the Merger in a press release dated March 12, 2015, which has been attached as Exhibit 99.3 to this Current Report on Form 8-K.
DESCRIPTION OF THE BUSINESS OF KURA ONCOLOGY, INC.
Overview
We were originally incorporated in the State of Delaware in November 2007 under the name “Zeta Acquisition Corp. III.” Prior to the Merger, Zeta Acquisition Corp. III was a “shell” company registered under the Securities Exchange Act of 1934, as amended, or the Exchange Act, with no specific business plan or purpose until it began operating the business of Kura through the Merger transaction on March 6, 2015. Kura was incorporated in the State of Delaware in August 2014 to focus primarily on discovering and developing personalized therapeutics for the treatment of solid tumors and blood cancers. Effective upon the Merger, a wholly-owned subsidiary of Zeta Acquisition Corp. III merged with and into Kura, and Kura continues as the operating subsidiary of Zeta Acquisition Corp. III. As used herein, unless otherwise indicated, the words the “Company,” “we,” “us,” and “our” refer to the current Delaware corporation operating the business of Kura as a wholly-owned subsidiary, which business will continue as the business of Zeta Acquisition Corp. III.
We are a clinical stage biopharmaceutical company discovering and developing personalized therapeutics for the treatment of solid tumors and blood cancers. We focus on the development of small molecule drug candidates that target cell signaling pathways that are important to driving the progression of certain cancers. We aim to employ molecular diagnostics to identify patients with cancers who are likely to benefit from our targeted drug candidates.
3
Advancements in cancer genetics and new molecular diagnostic tools are helping define why some patients respond to a particular therapy while other patients receive little to no clinical benefit. This new era in cancer drug discovery and development offers the potential for innovative treatments that are safer and more effective for patients with particular cancers. We aim to improve patient outcomes and contribute to the reduction in healthcare cost by matching targeted therapeutics to the patients who will benefit the most. We are developing drugs designed to inhibit the mutated or abnormally functioning cellular pathways that drive cancer growth and intend to pair them with molecular diagnostics to identify those patients with tumors most likely to respond to treatment.
Our lead drug candidate, tipifarnib, is an inhibitor of protein farnesylation that we intend to evaluate in Phase 2 clinical studies as a treatment for patients with solid tumors with HRAS mutations as well as patients with peripheral T-cell lymphoma. Our pipeline includes two preclinical programs (1) orally-available small molecule inhibitors of extracellular-signal-regulated kinases 1 and 2 (ERK1/2), including KO-947 and other backup compounds in development for the treatment of patients with activating mutations in or other dysregulation of the mitogen-activated protein kinase (MAPK) signaling pathway, including mutations in KRAS, BRAF and NRAS and (2) orally available, small molecule inhibitors of the menin-MLL interaction, which are currently in lead optimization as a treatment for patients with acute leukemias involving translocations or partial tandem duplications of the mixed lineage leukemia (MLL) gene.
Strategy
Our strategy is to acquire, develop, and commercialize innovative anti-cancer agents in oncology indications with significant unmet medical need. The critical components of our strategy include the following:
Focus on Oncology.
The oncology market is characterized by a number of disorders with high rates of disease recurrence and a limited response from current therapies or treatments. New oncology product candidates that address unmet medical needs or provide efficacy and safety profiles superior to those of standard of care have the potential for expedited regulatory review and, if approved, could be positioned to experience rapid adoption rates. We believe that the combination of molecularly-targeted cancer therapies and companion diagnostics to identify patients whose cancers are dependent on these targeted cell signaling pathways presents the potential for improved patient outcomes.
Focus on Compounds Where Improved Outcomes are Associated with Specific Biomarkers.
Our strategy is to prioritize those programs for which strong scientific and clinical hypotheses exist to link improved patient outcomes with specific biomarkers. Significant progress has been made in the identification of molecular targets and pathways that more narrowly specify the causes of cancer and explain the variability in responses to different therapies by subsets of patients with a particular cancer or tumor type. We believe that the identification of such patient subsets and the correlation of their specific characteristics to the drug candidate under development should increase the clinical benefit and the probability of success in our clinical trials. We believe such patient identification should also enable us to design clinical trials that may be completed more rapidly and, if successful, to achieve clinical outcomes for the targeted group that are more beneficial to the patients as well as more attractive to physicians and healthcare payors.
Leverage Companion Diagnostics to Realize Positive Clinical Outcomes.
Our development strategy is based on our belief that we can utilize effective companion diagnostics to identify patient subsets that will derive greater benefit from our product candidates. We intend to partner development of these companion diagnostics for use in clinical trials and, if successful, for commercialization of our product candidates. We have the ability to select from a number of diagnostic technology platforms and providers when choosing a partner for our programs under development.
Advance our Product Candidates in Clinical Proof-of-Concept Studies.
We plan to initiate two Phase 2 clinical trials of our lead product candidate, tipifarnib in 2015. The first trial, which we plan to initiate in the second quarter of 2015, will be in patients with solid tumors characterized by HRAS mutations and the second trial, which we plan to initiate in the third quarter of 2015, will be in patients with peripheral T-cell lymphoma. We intend to maximize the likelihood of success in those trials by: (1) using genetic analysis to identify one or more target patient populations that are more likely to respond to and benefit from tipifarnib and (2) evaluating biomarkers as indications of efficacy. We intend to advance our ERK1/2 program and our menin-MLL program through to clinical development pending successful completion of research activities and preclinical studies.
4
Seek and Maintain Significant Development and Commercial Rights.
We believe it is important to maintain significant development and commercial rights to our product candidates. For many cancer indications, there are a relatively small number of oncologists practicing in each of the major pharmaceutical markets and an even smaller number of oncology key opinion leaders who significantly influence the types of drugs prescribed in cancer therapy. We believe that we can reach these oncology markets effectively with a relatively small sales and marketing organization focused on these physicians and oncology key opinion leaders. As a result, we plan to seek to retain significant development and commercial rights to our products, which will enable us to retain the vast majority of the revenues from and commercial and economic value of our product candidates.
Cancer Background
Cancer is the second leading cause of death in the United States. The American Cancer Society (ACS) estimated that, in 2014, there would be approximately 1.7 million new cases of cancer and approximately 585,000 deaths from cancer in the U.S. The World Health Organization estimated that 8.2 million people worldwide died of cancer in 2012. Despite advances in cancer diagnostics and treatment the unmet medical need remains high. According to ACS, cancer is a general name for a group of over 100 diseases.
Despite significant disease variability, cancer in general originates from defects in the cell’s genetic code, or DNA, which disrupt the mechanisms that normally prevent uncontrolled cell growth, proliferation, invasion and programmed cell death. Cancer cells that arise in other tissues or organs are referred to as solid tumors. Cancerous cells that arise in the lymphatic system and bone marrow are referred to as hematological tumors. Increasingly, doctors are using diagnostic tests that identify genetic defects that may make a tumor more or less sensitive to a particular therapy in order to select better treatment options for patients with that disease. As genetic testing in cancer becomes a more routine practice, we are learning that many cancers arising in diverse sites in the body may share the same type of genetic alterations. For example, a mutation in a gene called BRAF is found in the majority of patients with metastatic melanoma, but it is also found in subsets of patients with colorectal cancer, lung cancer and other malignancies.
The most common methods of treating patients with cancer are surgery, radiation and drug therapy. A cancer patient often receives treatment with a combination of these methods. Surgery and radiation therapy are particularly effective when the disease is localized. Physicians generally use systemic drug therapies when the cancer has spread beyond the primary site or cannot otherwise be treated through surgery. The goal of drug therapy is to damage and kill cancer cells or to interfere with the molecular and cellular processes that control the development, growth and survival of cancer cells. In many cases, drug therapy entails the administration of several different drugs in combination. Over the past several decades, drug therapy has been evolving from non-specific drugs that kill both healthy and cancerous cells, such as cytotoxic therapies, to drugs that target specific molecular pathways or cellular processes involved in cancer and, more recently, to therapeutics that target specific activating alterations that are the “drivers” of cancer.
Cytotoxic Therapies. The earliest approach to pharmacological cancer treatment was to develop drugs referred to as cytotoxic drugs that kill rapidly proliferating cancer cells through non-specific mechanisms, such as deterring cellular metabolism or causing damage to cellular components required for survival and rapid growth. While these drugs have been effective in the treatment of some cancers, many unmet medical needs for the treatment of cancer remain. Also, cytotoxic drug therapies act in an indiscriminate manner, killing healthy cells as well as those that are cancerous, thereby causing significant side effects and tolerability issues for patients. Due to their mechanism of action, many cytotoxic drugs have a narrow dose range above which the toxicity causes unacceptable or even fatal levels of damage to healthy cells and below which the drugs are not effective in eradicating cancer cells.
Targeted Therapies. Advances in biology and understanding of cancer have led to the development of drugs, referred to as targeted therapeutics, which are designed to attack either a target that causes uncontrolled growth of cancer cells due to a specific genetic alteration primarily found in tumors but not in normal cells, or a target that cancer cells are more dependent on for their growth than normal cells. Targeted therapeutics are designed to preferentially kill cancer cells and spare normal cells and thus, in principle, they should exhibit enhanced efficacy and patients should experience fewer treatment-related side effects. Researchers and clinical oncologists now often incorporate genetic assessments into clinical trials and routine care with the hope of directing patients to medicines, which may have a greater chance of treating their cancers effectively. Furthermore, through the use of genetic testing, it is possible to develop drugs for defined subsets of patients, and to look for patients whose tumor types harbor genetically similar alterations. As such, doctors may begin to identify tumors and select therapies based on the type of mutations they share, rather than the part of the body from which they arise. Such a system should afford more efficient drug development, the opportunity for robust clinical responses and a better understanding of the underlying mechanisms of cancer.
5
Disease and Market Overview
We are focused on developing targeted therapeutics for the treatment of solid tumors and blood cancers. We are evaluating our lead product candidate, tipifarnib, a farnesyl transferase inhibitor, as a potential treatment for certain solid tumors, including thyroid cancer, head and neck cancers, urothelial carcinomas and salivary cancers, with HRAS mutations. Collectively, cancers that have an HRAS mutation are estimated to have an annual incidence of approximately 8,000 patients in the U.S. and, in general, patients with these cancers have poor prognosis and limited options for treatment. We are also evaluating tipifarnib as a potential treatment for patients with peripheral T-cell lymphoma, which has an annual incidence of approximately 7,000-10,000 patients in the U.S. Although several drugs have been approved by the U.S. Food and Drug Administration, or FDA, for treatment of relapsed or refractory PCTL, these drugs are associated with relatively low objective response rates and relatively short durations of response. Accordingly, we believe the treatment of relapsed/refractory PTCL remains a significant unmet medical need.
We are advancing a set of compounds that inhibit the activity of extracellular-signal-regulated kinases 1 and 2 (ERK1/2), including our lead candidate KO-947 as well as backup compounds, as a potential treatment for patients with tumors that have mutations in the Ras-MAP kinase pathway, including lung cancers, colorectal cancers, pancreatic cancers and melanoma. According to the National Cancer Institute, there are estimated to be over 43,000 cases of pancreatic cancer, 125,000 cases of colorectal cancer and over 188,000 cases of NSCLC diagnosed each year in the United States. We believe this corresponds to approximately 42,000 cases of KRAS mutant pancreatic cancer, 55,000 cases of KRAS mutant CRC, and 58,000 cases of KRAS mutant NSCLC each year in the United States. According to the American Cancer Society, the annual incidence of melanoma patients is estimated at 75,000 patients in the United States, with approximately 40%-60% of those patients having BRAF mutations and an additional 15-20% of those patients having NRAS mutations. As ERK inhibitors target the RAS/RAF/MEK/ERK pathway, which is activated with a BRAF mutation, they may also have the potential for activity not only in patients with BRAF-mutant melanoma but also in patients with tumors that harbor mutations in the NRAS gene, who currently have no adequate treatment option and poor prognosis.
We are also advancing a set of compounds that inhibit the interaction between the proteins menin and MLL for the treatment of mixed lineage leukemias-rearranged (MLL-r) and mixed lineage leukemias-partial tandem duplications (MLL-PTD), two genetically-defined subsets of acute leukemias that affect both adults and children. The annual incidence of MLL-r and MLL-PTD patients is estimated to be 3,200 patients in the United States, and those patients currently have limited options other than chemotherapy.
Clinical Programs and Pipeline
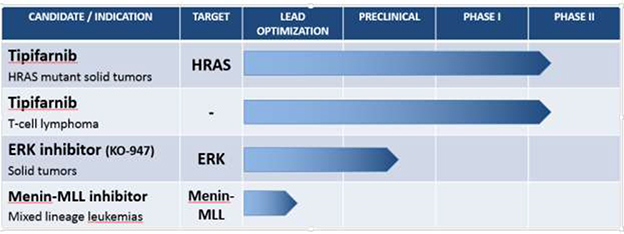
Tipifarnib – An Oral Farnesyl Transferase Inhibitor
Overview
Tipifarnib is a new chemical entity we in-licensed in December 2014 from Janssen Pharmaceutica NV, an affiliate of Johnson and Johnson. Tipifarnib is a novel, patented small molecule inhibitor of protein farnesylation, a key cell signaling process implicated in cancer initiation and development. Tipifarnib has been studied in more than 5,000 patients, including more than 600 patients at the dose and schedule we intend to use in our Phase 2 trials. At that dose and schedule, tipifarnib exhibited a manageable side effect profile and was generally well tolerated.
6
Although tipifarnib has demonstrated compelling and durable anti-cancer activity in certain patients and a well-established safety profile, its activity has not been sufficient in any patient subset to support marketing approval by the FDA. However, clinical and preclinical data suggest that, in the right context, tipifarnib has the potential to provide significant benefit to cancer patients with limited treatment options. Leveraging advances in next-generation sequencing as well as emerging information about cancer genetics, we will seek to identify patients most likely to benefit from tipifarnib. We plan to initiate a Phase 2 trial in patients who have tumors characterized by HRAS mutations in the second quarter of 2015 and a second Phase 2 trial in patients with peripheral T-cell lymphomas in the third quarter of 2015.
HRAS Mutant Tumors – Market Opportunity
RAS proteins are GTPase enzymes that are involved in regulating cell division in response to growth factor stimulation. HRAS is a member of the RAS family, which includes two other proto-oncogenes: KRAS and NRAS. Collectively, the three RAS genes constitute one of the most frequently mutated families of oncogenes in human cancers. Although HRAS mutations are less common overall relative to KRAS and NRAS mutations, they have a relatively high prevalence in cancers of the upper aerodigestive tract, skin, thyroid and urinary bladder.
We believe the sum of these patient subsets, defined by the presence of an HRAS mutation, represents a significant potential patient population.
Farnesyl transferase inhibitors (FTIs) such as tipifarnib prevent protein farnesylation, a key cell signaling process implicated in cancer initiation and development. Tipifarnib has been shown to inhibit HRAS function. Specifically, by blocking HRAS farnesylation and subsequent membrane localization, tipifarnib inhibits oncogenic, HRAS-driven cellular transformationin vitro andin vivo. Earlier studies of FTIs were based on the hypothesis that FTIs would be generally active in RAS driven tumors. However, FTIs showed no significant antitumor activity in patients with advanced solid tumors such as lung, pancreatic and colon cancers, which mainly harbor KRAS mutations, and although the FTIs have demonstrated responses in certain patients with acute myeloid leukemia, the activity of the compound has not been shown to correlate with NRAS mutations. We believe the refractory nature of RAS-driven tumors to treatment with FTIs has been attributed to mechanisms of resistance that are available to tumors with KRAS and NRAS mutations but not to those tumors with HRAS mutations.
HRAS as a Human Oncogene
The HRAS protein is a GTPase that is involved in regulating cell division in response to growth factor stimulation. Growth factors act by binding cell surface receptors that span the cell’s plasma membrane. Once activated, receptors stimulate signal transduction events in the cytoplasm, a process by which proteins and second messengers relay signals from outside the cell to the cell nucleus and instruct the cell to grow or divide. HRAS is an early player in many signal transduction pathways. HRAS acts as a molecular on/off switch – once it is turned on it recruits and activates proteins necessary for the propagation of the receptor’s signal. In certain tumors, mutations in HRAS or its upstream effectors cause it to be permanently on, resulting in persistent activation of downstream growth and proliferation signals that drive tumor cell growth. FTIs work to prevent the aberrant growth and proliferation of cells that are dependent on these signaling pathways by switching HRAS off.
Clinical Significance of HRAS
The role of HRAS in patients with Costello syndrome illustrates its potential as a human oncogene. At least five inherited mutations in the HRAS gene have been identified in people with Costello syndrome. Each of these mutations changes an amino acid in a critical region of the HRAS protein. The mutations associated with Costello syndrome lead to the production of an HRAS protein that is permanently active. Instead of triggering cell growth in response to particular signals from outside the cell, the overactive protein directs cells to grow and divide constantly. This uncontrolled cell division can result in the formation of noncancerous and cancerous tumors beginning in early childhood.
Transitional cell carcinoma of the bladder frequently occurs in adolescents with Costello syndrome, a presentation that is rare in the general population. Sporadic bladder tumors occurring in young patients without Costello syndrome also have a high frequency of HRAS mutation, but otherwise, lack extensive genetic alterations. Furthermore, HRAS mutations are present at all disease stages of bladder cancer and are detected in low-grade non-muscle invasive transitional tumors. These pieces of clinical evidence point to HRAS as a key protein involved in tumorigenesis in both Costello syndrome and, by extension, in the broader population.
7
Preclinical Data Supporting Tipifarnib as an Inhibitor of HRAS Function
Tipifarnib inhibits cell proliferation of mutant HRAS transformed NIH3T3 cells with an IC50 value of 1.7 nM, but it does not inhibit parental NIH3T3 cells that have normal HRAS up to a concentration of 500 nM. In a panel of human tumor cell lines, tipifarnib inhibited two HRAS mutant cell lines with IC50 values of 1.7 and 5.2 nM, respectively. Tumor cell lines with KRAS or NRAS mutations displayed a range of sensitivities, ranging from ~ 10 nM to > 500 nM. In murine xenograft models, tipifarnib inhibited HRAS mutated model much more potently (86% tumor growth inhibition at 25 mg/kg bid) than KRAS mutated tumor models (10% tumor growth inhibition at 25 mg/kg bid).
Further support for using a farnesyl transferase inhibitor to treat tumors driven by mutant HRAS is provided by studies evaluating the two-stage DMBA/TPA model of mouse skin carcinogenesis. This model of mouse skin carcinogenesis has been used to study mechanisms of epithelial tumor development by oncogenic HRAS. Treatment of the model with the farnesyl transferase inhibitor, SCH66336, induced near-complete regression of papillomas of TPA-treated HRASG12V knock-in mice. Such data support the notion that farnesyl transferase inhibitors such as tipifarnib should be re-evaluated as targeted agents for HRAS-driven cancers.
Clinical Development in HRAS Mutant Tumors
We have designed a clinical trial to test the hypothesis that tipifarnib can be used as a treatment for advanced tumors with a known HRAS mutation. We expect to initiate this Phase 2 trial in the second quarter of 2015. The trial will enroll 2 cohorts of 18 patients each. Cohort 1 will enroll subjects with malignant thyroid tumors with HRAS mutations, independently of thyroid histology. Any subject with a non-hematological HRAS mutant tumor who meets eligibility criteria may be enrolled in Cohort 2. The study has null (H0) and of-interest (H1) hypotheses of 10% and 30% response rate. This trial has a two-stage study design to minimize the number of study subjects treated if tipifarnib were not sufficiently efficacious. If one or no objective response is observed in a cohort after the first 11 evaluable patients, the cohort will be closed to further enrollment. If more than one response is observed in the cohort, 7 additional subjects will be enrolled (stage 2). Treatment will be considered of further interest if at least 4 responses are observed in a cohort (out of 18 subjects). Tumor response assessments will be conducted according to RECIST v1.1 criteria (confirmation of response is required), but in order to expedite the response assessment of the initial 11 evaluable patients, tipifarnib will be considered not sufficiently efficacious if no confirmed objective tumor responses are observed in the study cohort prior to 6 months from the time of enrollment of the last of the 11 evaluable subjects.
Peripheral T-cell Lymphoma Opportunity
We intend to initiate a Phase 2 human clinical trial to evaluate tipifarnib as a treatment for patients with peripheral T-cell lymphoma (PTCL) in the third quarter of 2015.
Lymphoma is the most common blood cancer. The two main forms of lymphoma are Hodgkin lymphoma and non-Hodgkin lymphoma (NHL). Lymphoma occurs when cells of the immune system called lymphocytes grow and multiply uncontrollably. Cancerous lymphocytes can travel to many parts of the body, including the lymph nodes, spleen, bone marrow, blood, or other organs, and form tumors. The body has two main types of lymphocytes that can develop into lymphomas: B-lymphocytes (B-cells) and T-lymphocytes (T-cells).
Peripheral T-cell lymphoma (PTCL) consists of a group of rare and usually aggressive (fast-growing) NHLs that develop from mature T-cells. Most T-cell lymphomas are PTCLs, which collectively account for about 10 percent to 15 percent of all NHL cases, corresponding to an annual incidence of 7,000-10,000 patients per year in the United States. By some estimates, the incidence of PTCL is growing significantly, and the increasing incidence may be driven by an aging population.
PTCLs are sub-classified into various subtypes, each of which are typically considered to be separate diseases based on their distinct clinical differences. Most of these subtypes are rare; the three most common subtypes of PTCL, peripheral T-cell lymphoma not otherwise specified (PTCL-NOS), anaplastic large-cell lymphoma (ALCL), and angioimmunoblastic T-cell lymphoma (AITL), that collectively account for approximately 70 percent of all PTCLs in the United States.
Treatment Options for PTCL
For most PTCL subtypes, the frontline treatment regimen is typically combination chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone), EPOCH (etoposide, vincristine, doxorubicin, cyclophosphamide, prednisone), or other multi-drug regimens.
8
Patients who relapse or are refractory to frontline treatments are typically treated with gemcitabine in combination with other chemotherapies, including vinorelbine (Navelbine®) and doxorubicin (Doxil®) in a regimen called GND, or other chemotherapy regimens such as DHAP (dexamethasone, cytarabine, cisplatin) or ESHAP (etoposide, methylprednisolone, cytarabine, and cisplatin).
Because most patients with PTCL will relapse, some oncologists recommend giving high-dose chemotherapy followed by an autologous stem cell transplant to some patients who had a good response to their initial chemotherapy. Recent, non-cytotoxic therapies that have been approved for relapsed or refractory PCTL, such as pralatrexate, romidepsin and belinostat, are associated with relatively low objective response rates (25-27% ORR) and relatively short durations of response (8.2-9.4 months). Accordingly, we believe the treatment of relapsed/refractory PTCL remains a significant unmet medical need.
Previous Phase II Experience with Tipifarnib in the Treatment of PTCL
A prior Phase 2 trial of tipifarnib was conducted at the Mayo Clinic in adult patients with relapsed or refractory lymphoma. Ninety-three patients (42 aggressive, 15 indolent, and 36 HL/T) were enrolled in the study, and patients received tipifarnib 300 mg twice daily on days 1-21 of each 28-day cycle. The median age of patients was 62 years (range, 18-91 years). A total of 71% of patients had stage IV disease. The median number of prior regimens was five (range, 1-17). The majority of patients were diagnosed with diffuse large B-cell lymphoma (DLBCL) (40%; 37 of 93) or Hodgkin lymphoma (HL) (20%; 19 of 93). See Table A below.
The overall response rate (ORR) for all patients was 20.4% (19 of 93), with 7% (6 of 93) complete responses (CR) and 14% (13 of 93) partial responses (PR). In the groups of aggressive, indolent, and HL/T-cell types of lymphoma, the ORRs were 17%, 7%, and 31%, respectively.
In the 19 responders, the median response duration was 7.5 months with a mean of 15.8 months. The median response duration was 11.3 months, 2 months, and 7.5 months for the groups of aggressive, indolent, and HL/T-cell lymphomas, respectively.
The highest ORR (31%) was demonstrated in the HL/T cell lymphoma group. Within that group, the ORR was 21% (4 of 19) in patients with HL and 50% (6 of 12) in T-cell Non Hodgkin Lymphoma (NHL).
The median time to progression (TTP) was 3.6 months for all patients and 3.2 months for the HL/T-cell lymphoma groups, respectively. Five patients in the HL/T-cell lymphoma group received treatment for more than 30 months with several patients receiving treatment for 60+ months.
The median overall survival (OS) was 14.8 months for all patients and 6.4 months, 20.6 months, and 19.7 months for the aggressive, indolent, and HL/T-cell lymphoma groups, respectively.
Table A: Phase 2 Clinical Trial of tipifarnib in Adult Patients with Relapsed or Refractory Lymphoma.
| Disease Type | n(%) | CR, n (%) | PR, n (%) | ORR, (%) (95% CI) | Median DR (95% CI) | Median TTP (95% CI) | Median OS (95% CI) | |||||||
All patients | 93 | 6 (7) | 13 (14) | 20 (13-30) | 7.5 (4.9-18.5) | 3.6 (2.1-4.5) | 14.8 (7.6-17.8) | |||||||
Aggressive B-cell lymphoma group | 42 | 0 | 7(17) | 17 (7-31) | 11.3 (4.9-17.1) | 2.8 (1.7-4.2) | 6.4 (4.1-10.7) | |||||||
DLBCL | 37 (88) | 0 | 7(19) | 19 | — | — | — | |||||||
MCL | 4 (10) | 0 | 0 | 0 | — | — | — | |||||||
FL III | 1 (2) | 0 | 0 | 0 | — | — | — |
9
Indolent B-cell lymphoma group | 15 | 0 | 1 (7) | 7 (0.2-32) | 2 (NR) | 5.2 (4-9.2) | 20.6 (NR) | |||||||
Chronic lymphocytic Leukemia/small lymphocytic lymphoma | 5 (33) | 0 | 0 | 0 | — | — | — | |||||||
Extranodal marginal zone | 1 (7) | 0 | 0 | 0 | — | — | — | |||||||
FL grade I | 3 (20) | 0 | 0 | 0 | — | — | — | |||||||
FL grade II | 6 (40) | 0 | 1 | 17 | — | — | — | |||||||
HL/T group | 36 | 6 (17) | 5 (14) | 31 (16-48) | 7.5 (3.2-29.8) | 3.2 (1.9-5.8) | 19.7 (9-60) | |||||||
HL | 19 (53) | 2 (11) | 2 (11) | 21 | — | — | — | |||||||
Mycosis fungoides | 4 (11) | 0 | 2 (50) | 50 | — | — | — | |||||||
Peripheral T-cell, unspecified | 8 (22) | 3 (38) | 1 (13) | 50 | — | — | — | |||||||
Anaplastic large cell, cutaneous | 3 (8) | 1 (33) | 0 | 33 | — | — | — | |||||||
Anaplastic large cell, systemic | 2 (6) | 0 | 0 | 0 | — | — | — |
| — | indicates not applicable ; and NR, not reported |
Tipifarnib was generally well tolerated on this dose and schedule. Three patients with aggressive lymphoma died on study of progressive disease, but there were no deaths related to tipifarnib treatment. The grade 3 or 4 toxicities were primarily reversible myelosuppression, with 11% anemia, 37% neutropenia, and 32% thrombocytopenia.
Of particular relevance to our planned Phase 2 clinical trial in PTCL are the results observed in the patients with T-cell non-Hodgkin lymphoma. Although the trial enrolled only small numbers of patients, a 41% response rate (7 responses out of 17 patients) was observed in patients with T-cell non-Hodgkin lymphoma, including 4 objective responses out of 8 patients with PTCL (3 CR and 1 PR). We believe the results observed from this Phase 2 trial suggests that tipifarnib can be administered for prolonged periods and may produce durable responses as a single agent in relapsed lymphoma in a group of patients who were heavily pretreated with a median of 5 prior therapies.
The five year survival for patients with PTCL is low – roughly 35% by most published records – and few treatment options are able to provide a durable treatment effect. Treatments in the relapsed or refractory setting are not very effective. Therefore, National Comprehensive Cancer Network guidelines currently recommend that patients seek participation in a clinical trial for the initial treatment.
Clinical Development in Peripheral T-cell Lymphoma
Based on the promising results observed in the Phase 2 lymphoma study, we have designed a clinical trial to test the hypothesis that tipifarnib can be used as a treatment for patients with relapsed or refractory PTCL. We expect to initiate this Phase 2 trial in the third quarter of 2015. The trial is a two-stage design for a total number of 18 patients. If one or no objective response is observed after the first 11 evaluable patients (stage 1), the study will be closed to further enrollment. If more than one response is observed, 7 additional patients will be enrolled (stage 2). Treatment will be considered of further interest if at least 4 responses are observed (out of 18 patients). Tumor response assessments will be conducted according to the International Workshop Criteria for the assessment of responses in lymphoma.
10
ERK Inhibitor Program
Overview
We are advancing a set of novel, orally bioavailable small molecule inhibitors of extracellular-signal-regulated kinases 1 and 2 (ERK1/2), including our lead candidate KO-947 as well as backup compounds, as a potential treatment for patients with tumors that have mutations in the RAS-MAP kinase pathway, including lung cancers, colorectal cancers, pancreatic cancers and melanoma. The compounds, including KO-947 and backup compounds, represent new chemical entities we acquired pursuant to an agreement effective December 23, 2014 from Araxes Pharma LLC.
The high frequency of activating mutations in components of the mitogen-activated protein kinase (MAPK) pathway found in cancer provides strong rationale for targeting the MAPK pathway and, specifically, ERK. The MAPK pathway is responsible for receiving growth-promoting signals from outside the cell and translating these signals within the cell into programs that affect cell growth and proliferation. When external growth factors activate cell surface receptor tyrosine kinases (RTKs), the MAPK pathway acts inside the cell to relay these growth signals through a series of signaling molecules, including the RAS, RAF, MEK, and ERK family of kinases. ERK kinase is the final signaling kinase of the MAPK pathway. See Figure 1.
Figure 1: MAPK pathway
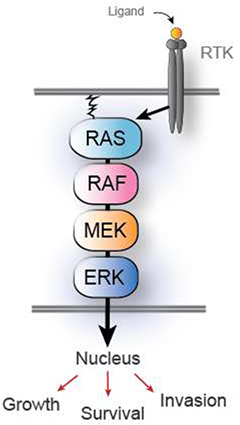
Many cancers harbor genetic mutations in components of the MAPK pathway, especially in protein kinases, that lock transformed cells in a pro-growth state, even in the absence of external growth signals. Studies have shown that such aberrations in the MAPK pathway, including mutations in KRAS, BRAF, and other components of the pathway, are frequent contributors to the development of cancer in humans. Targeted cancer drugs, such as inhibitors of the proteins BRAF and MEK, that have been designed to turn off MAPK signaling by inhibiting specific protein kinases are effective, particularly in melanomas where the MAPK circuit is aberrantly active. We believe that a therapeutic drug candidate that can block signaling of the MAPK pathway through inhibition of ERK should reduce or prevent cancer growth and may have a beneficial effect for patients.
11
As part of our ERK inhibitor program, we are advancing KO-947, which is an orally-available inhibitor of ERK that has nanomolar cellular potency in tumor cells with mutations in BRAF, NRAS or KRAS and induces tumor regressions in xenograft models at doses that are well tolerated. Because KO-947 targets ERK, a protein kinase essential to signaling through the MAPK pathway, it has the potential to selectively kill tumor cells bearing activating mutations in this critical pathway. KO-947 is currently in IND enabling studies, and we anticipate filing an IND in the first quarter of 2016. In addition, we are also advancing other ERK inhibitors as backup compounds to KO-947 and, if we elect to advance one of those compounds to IND-enabling studies, we would anticipate filing an IND on such compound in the first half of 2016.
Opportunity for Kura Oncology
We have focused on the discovery and development of ERK inhibitors and selected KO-947 as a potential product candidate because we believe that ERK inhibitors have two important potential advantages as therapeutics:
| • | Potential to effectively treat patients with mutations in the KRAS gene — a large and growing group of patients with lung, colorectal, pancreatic and other cancers who today have no effective therapy, and who have been identified with greater frequency due to recently approved diagnostic guidelines, and |
| • | Potential to effectively treat patients with metastatic melanoma who receive “first-generation” BRAF or MEK inhibitors, but who develop resistance due to reactivation of ERK pathway signaling. KO-947 could prevent resistance through this mechanism and may thus cause responses of greater duration than the ones seen with first generation inhibitors and extend progression-free survival. |
We acquired our ERK inhibitor program from Araxes Pharma based in La Jolla, California. Scientists at Araxes Pharma designed our ERK inhibitors using structure-guided drug discovery approaches to model chemical structures that would inhibit the ERK protein kinase but spare inhibition of closely related kinases. These molecules were then synthesized and tested in assays to verify their ability to inhibit ERK as well as to inhibit MAPK pathway signaling.
Market Overview: Solid Tumors with KRAS Mutations Represent a Significant Unmet Medical Need
Activating mutations in the KRAS gene are commonly found in a wide variety of tumor types. Among cancer indications with large patient populations, KRAS mutations are found in approximately 90 percent of pancreatic cancers, approximately 40 percent of colorectal cancers and approximately 25 percent of non-small cell lung cancers (NSCLC). According to the American Cancer Society, there are estimated to be over 43,000 cases of pancreatic cancer, 125,000 cases of colorectal cancer and over 188,000 cases of NSCLC diagnosed each year in the United States. We believe this corresponds to approximately 42,000 cases of KRAS mutant pancreatic cancer, 55,000 cases of KRAS mutant CRC, and 58,000 cases of KRAS mutant NSCLC each year in the United States. These cancers typically present relatively late in their clinical course, when locally directed therapy (surgery and radiation) is not curative. The treatment of locally advanced and metastatic cancers represents a significant unmet medical need.
Therapeutic Rationale for KRAS Mutant Tumors
In its normal, non-mutant form, the KRAS protein plays a key role in the promotion and regulation of cell growth and division. The KRAS protein acts in a keystone position in the MAPK pathway. This pathway is responsible for receiving growth-promoting signals from outside the cell and communicating those signals within the cell so that the cell can respond appropriately to the cell growth signals.
Studies have shown that disruptions to the MAPK pathway, either by mutations in KRAS or other components of the pathway, are frequent contributors to the development of cancer in humans. Certain mutations in KRAS promote cancer by putting the KRAS protein into a constitutively active state, which promotes the uncontrolled cell growth and division that are the hallmarks of cancer. We believe that a therapeutic drug candidate that can inhibit signaling through the MAPK pathway should reduce or prevent cancer growth and may have a beneficial effect for patients.
Therapeutics have been successfully developed against other components of the MAPK pathway, including the BRAF inhibitors vemurafenib (ZELBORAF®) and dabrafenib (TAFINLAR®) and the MEK inhibitor trametinib (MEKINIST®), each of which has received approval from the FDA for treatment of BRAFV600E mutant melanoma. However, patients with melanoma frequently develop resistance to these drugs, and the drugs do not have potent activity in patients with KRAS mutations. Accordingly, oncologists and patients are still in need of a therapeutic agent that can inhibit signaling through the MAPK signaling pathway and provide benefit to patients.
12
Preclinical Data for KO-947 for KRAS Mutant Solid Tumors
Our lead candidate in our ERK inhibitor program, KO-947, demonstrates potent inhibition of the ERK kinase and high selectivity relative to a panel of approximately 400 kinases. KO-947 has also shown promising activity in both cell culture and xenograft animal models of KRAS mutant tumors.
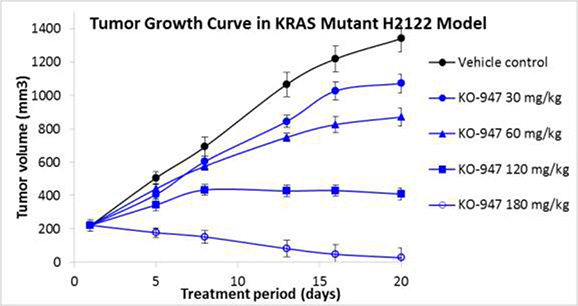
Xenograft tumors were grown subcutaneously in mice, followed by oral treatment with the ERK inhibitor or control. Treated animals showed full tumor regression, while vehicle control treated animals showed rapid tumor growth. In addition, KO-947 was well tolerated at all dose levels with no apparent body weight loss in the mice, which is a surrogate measure for toxicity.
Market Overview: Melanoma Tumors with Acquired Resistance to BRAF and MEK Inhibitors Represent a Significant Unmet Medical Need
Specific inhibitors of RAF and MEK kinases have been developed to target BRAF- and RAS-mutant tumors. In particular, the FDA has approved the BRAF inhibitors vemurafenib (ZELBORAF®) and dabrafenib (TAFINLAR®) as well as the MEK inhibitor trametinib (MEKINIST®) for the treatment of BRAFV600E-mutant metastatic melanoma. Although these approvals are encouraging, durable responses in patients are limited, as median time to disease progression is approximately 6-7 months and resistance is often associated with pathway reactivation of the ERK signaling pathway.
According to the American Cancer Society in 2014, the annual incidence of diagnosed melanoma is 76,000 cases in the United States, and nearly 9,500 melanoma deaths occur in the each year in the United States. Mutations that activate the RAS/RAF/MEK/ERK pathway are common in melanoma, with BRAF mutations in 40% to 60%, and NRAS mutations in 15-20% of melanoma patients, suggesting the therapeutic potential for agents that target this pathway in melanoma. As ERK inhibitors target the RAS/RAF/MEK/ERK pathway, which is activated with BRAF mutation, they may also have the potential for activity not only in patients with BRAF-mutant melanoma, but also in patients with tumors that harbor mutations in the NRAS gene, who currently have no adequate treatment option and poor prognosis.
Preclinical Data for ERK Product Candidate for Melanoma with Acquired Resistance to BRAF and MEK Inhibitors
There is a strong rationale to develop ERK inhibitors for tumors that are resistant to other inhibitors of the MAPK pathway. Selective BRAF and MEK inhibitors have shown clinical efficacy in patients with melanoma. However, the majority of responses are transient, and resistance is often associated with pathway reactivation of the extracellular signal-regulated kinase (ERK) signaling pathway. In preclinical studies, ERK inhibitors have demonstrated promising activity in both cell culture and xenograft animal models of tumors resistant to BRAF and MEK inhibitors.
13
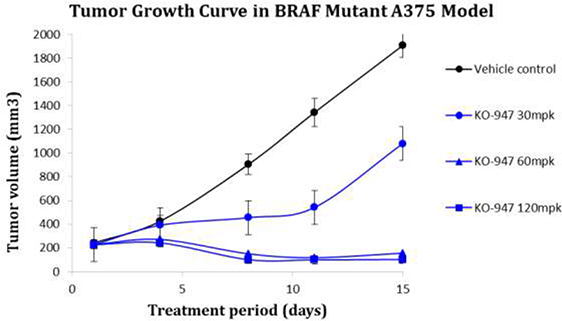
In particular, xenograft tumors were grown subcutaneously in mice, followed by oral treatment with ERK inhibitor or control. Treated animals showed full tumor regression at tolerated doses, while vehicle control treated animals showed rapid tumor growth. Based on these preclinical efficacy data in both KRAS and BRAF mutant tumor models, we have advanced KO-947 into IND-enabling studies, and we continue to evaluate additional ERK inhibitors as potential backup compounds to KO-947.
Menin-MLL Program
Overview
We are developing orally bioavailable small molecule inhibitors of the menin-MLL interaction for the treatment ofMLL-rearranged (MLL-r) acute leukemias, a genetically defined subtype of the two most common forms of acute leukemia, acute myeloid leukemia, or AML, and acute lymphoblastic leukemia, or ALL.
Background on Mixed Lineage Leukemias
MLL-r leukemias are an aggressive subtype of two of the most common forms of acute leukemia, ALL and AML. The estimated five-year overall survival rate for adult patients with the MLL-r subtype of AML ranges from approximately 5% to 24%, and the total annual incidence of MLL-r leukemias in all patients in the U.S. and Europe has been estimated at approximately 5,000 patients. Patients with MLL-r leukemias are routinely diagnosed using existing technologies that are commonly used in clinical settings. As a result, there is high awareness of MLL-r leukemias among oncologists. The disease predominantly occurs in two different demographics – an adult population and an infant/pediatric population. While they share a common genetic alteration, the adult disease is frequently a secondary leukemia resulting from prior chemotherapy for a different, unrelated cancer, and the childhood disease arises de novo. MLL-r leukemias are caused by a chromosomal translocation involving the MLL gene.
Mixed lineage leukemia gene-partial tandem duplication (MLL-PTD) is a subset of acute myeloid leukemia (AML). MLL-PTD typically confers a worse prognosis with shortened overall and event free survival in childhood and adult AML.
The annual incidence of MLL-r and MLL-PTD patients is estimated to be 3,200 patients in the United States, and those patients currently have limited options other than chemotherapy. There are no approved therapies specifically indicated for either the MLL-r or MLL-PTD leukemias. Physicians treat these hematological cancers with therapies approved for other acute leukemias and malignancies. Patients with AML and ALL typically are treated with intensive multi-agent chemotherapy and high risk patients are treated with an allogeneic stem cell transplant. However, some patients, especially those who are older, are too fragile for any of these treatments and, as a result, have very few treatment options. Accordingly, we believe the treatment of MLL-r and MLL-PTD leukemias remains a significant unmet medical need.
14
Targeting the MLL-Menin Interaction
The mixed lineage leukemia (MLL) gene is a common target of chromosomal translocations found in patients with AML and ALL, which affects both children and adults. Fusion of MLL with one of over 50 different partner genes forms oncogenes encoding MLL fusion proteins, which play a causative role in the onset, development and progression of MLL.
The effect of MLL fusion proteins on the development and progression of leukemia is critically dependent on their direct interaction with menin, a protein encoded by the Multiple Endocrine Neoplasia 1 (MEN1) gene. Menin is a tumor suppressor protein, which directly controls cell growth in endocrine organs. Binding of menin to MLL fusion proteins upregulates expression of target genes involved in the malignant transformation of blood cells. In contrast, mutations to MLL fusion proteins that block association with menin abrogate the development of acute leukemia in mice. These findings demonstrate that menin functions as an essential oncogenic co-factor of MLL fusion proteins, and it implies that the menin-MLL interaction represents a valuable target for molecular therapy.
We have licensed from the University of Michigan a class of small molecule inhibitors of the menin-MLL fusion protein interaction that specifically bind to menin with nanomolar potency. By blocking menin – MLL fusion protein interactions, these compounds effectively reverse MLL fusion protein-mediated leukemic transformation by down regulating the expression of target genes required for MLL-fusion protein oncogenic activity. These compounds also selectively block proliferation and induce both apoptosis and differentiation of leukemia cells harboring MLL translocations.
Opportunity for Kura Oncology
Our menin-MLL development program is aimed at identifying product candidates with the potential to effectively treat patients with MLL-r leukemias – a subset of adult and pediatric patients who today have no effective therapy – as well as MLL-PTD leukemias, a subset of acute myeloid leukemias that have no effective therapy.
License and Asset Purchase Agreements
Janssen Pharmaceutica NV
We entered into a license agreement with Janssen Pharmaceutica NV, or Janssen, on December 18, 2014, which grants us exclusive global rights to develop and commercialize tipifarnib in the field of oncology and includes the right to grant sublicenses. We are obligated under the license agreement to use commercially reasonable efforts to develop and commercialize tipifarnib in oncology and, with the exception of the transfer to us without cost of Janssen’s existing inventory of tipifarnib material, we are responsible for all future development and commercialization costs for tipifarnib in oncology. Under the license agreement, Janssen has a first right to negotiate for an exclusive license back from Kura to develop and commercialize tipifarnib on terms to be negotiated in good faith. Janssen may exercise this right of first negotiation during the 60-day period following completion of a Phase 2 clinical trial of tipifarnib in HRAS mutant patients in oncology and delivery by Kura to Janssen of a complete data package from such clinical trial.
Under the terms of the license agreement, on January 20, 2015 we issued a convertible promissory note in the principal amount of $1,000,000 to Johnson & Johnson Innovation—JJDC, Inc., which automatically converted into shares of Kura common stock in the Private Placement. When and if commercial sales of tipifarnib begin, we are obligated to pay Janssen tiered royalties of low double digit percentages of our net sales, depending on the amount of our net sales, with standard provisions for royalty offsets in the event of generic competition or compulsory licenses. We are also required to make regulatory milestone payments to Janssen of up to $25 million in the aggregate, if specified regulatory approvals are achieved for the first indication and additional payments for each subsequent indication if specified regulatory approvals are achieved. In addition, we are required to make sales milestone payments of up to $50 million in the aggregate if specified sales thresholds are surpassed. If we grant sublicenses under the license from Janssen, we are required to pay to Janssen a percentage of any upfront, lump-sum or milestone payments received from our sublicensee, subject to certain exclusions for regulatory milestone payments due under the license agreement.
The license agreement with Janssen will remain in effect until the expiration of all of our royalty and sublicense revenue obligations to Janssen, determined on a product-by-product and country-by-country basis, unless we elect to terminate the license agreement earlier. If we fail to meet our obligations under the license agreement and are unable to cure such failure within specified time periods, Janssen can terminate the license agreement, resulting in a loss of our rights to tipifarnib.
15
Araxes Pharma LLC
We entered into an asset purchase agreement with Araxes Pharma LLC, or Araxes, on December 23, 2014, under which we purchased all of Araxes’ patent rights in the ERK program, including KO-947 and additional backup compounds, and related intellectual property. When and if commercial sales of a product candidate covered by the purchased patent rights begin, we are obligated to pay Araxes tiered royalties of low single digit percentages of our net sales, depending on the amount of our net sales with standard provisions for royalty offsets. We are also required to make development and regulatory milestone payments to Araxes of up to $9.7 million in the aggregate if specified development events and regulatory approvals are achieved. Under the terms of the asset purchase agreement, on December 23, 2014 we issued a convertible promissory note in the principal amount of $500,000 to Araxes, which automatically converted into shares of Kura common stock in the Private Placement.
Competition
The development and commercialization of new products to treat cancer is intensely competitive and subject to rapid and significant technological change. While we believe that our knowledge, experience and scientific resources provide us with competitive advantages, we face substantial competition from major pharmaceutical companies, specialty pharmaceutical companies, and biotechnology companies worldwide. Many of our competitors have significantly greater financial, technical, and human resources. Smaller and early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. As a result, our competitors may discover, develop, license or commercialize products before or more successfully than we do.
We face competition with respect to our current product candidates, and will face competition with respect to future product candidates, from segments of the pharmaceutical, biotechnology and other related markets that pursue approaches to targeting molecular alterations and signaling pathways associated with cancer. Our competitors may obtain regulatory approval of their products more rapidly than we do or may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our product candidates. Our competitors may also develop drugs that are more effective, more convenient, less costly, or possessing better safety profiles than our products, and these competitors may be more successful than us in manufacturing and marketing their products.
In addition, we will need to develop our product candidates in collaboration with diagnostic companies, and we will face competition from other companies in establishing these collaborations. Our competitors will also compete with us in recruiting and retaining qualified scientific, management and commercial personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Furthermore, we also face competition more broadly across the market for cost-effective and reimbursable cancer treatments. The most common methods of treating patients with cancer are surgery, radiation and drug therapy, including chemotherapy, hormone therapy and targeted drug therapy or a combination of such methods. There are a variety of available drug therapies marketed for cancer. In many cases, these drugs are administered in combination to enhance efficacy. While our product candidates, if any are approved, may compete with these existing drug and other therapies, to the extent they are ultimately used in combination with or as an adjunct to these therapies, our product candidates may not be competitive with them. Some of these drugs are branded and subject to patent protection, and others are available on a generic basis. Insurers and other third-party payors may also encourage the use of generic products or specific branded products. We expect that if our product candidates are approved, they will be priced at a premium over competitive generic, including branded generic, products. As a result, obtaining market acceptance of, and gaining significant share of the market for, any of our product candidates that we successfully introduce to the market will pose challenges. In addition, many companies are developing new therapeutics, and we cannot predict what the standard of care will be as our product candidates progress through clinical development.
Tipifarnib Competition
While there are currently no approved drugs targeting farnesyltransferase, we are aware of a number of compounds that are now or have previously been in clinical development, including Merck’s lonafarnib, Bristol-Myers Squibb’s BMS-214662, Astellas Pharma’s (formerly OSI) CP-609,754, and AstraZeneca’s AZD3409. To our knowledge, there are no ongoing clinical trials evaluating any of these agents for the treatment of cancer. However, the initiation of clinical development of these agents in an oncology setting could become competitively significant, and if tipifarnib or our other product candidates do not offer sustainable advantages over competing products, we may not be able to successfully compete against current and future competitors.
16
Even if we are successful in developing our product candidates, the resulting products would compete with a variety of established drugs in targeted therapeutic indication of peripheral T-cell lymphoma, including belinostat (Beleodaq®) and pralatrexate (Folotyn®), marketed by Spectrum Pharmaceuticals, romidepsin (Istodax®), marketed by Celgene, and brentuximab vedotin (Adcetris®) (for anaplastic large-cell lymphoma), marketed by Seattle Genetics. Although there are currently no drugs approved specifically for the treatment of HRAS-mutant solid tumors, there are a number of targeted therapies approved for the treatment of thyroid cancer, including AstraZeneca’s vandetanib (Caprelsa®), Bayer’s sorafenib (Nexavar®), Exelixis’ cabozantinib (Cometriq®) and Eisai’s lenvatinib (Lenvima®).
ERK Inhibitor Competition
While there are currently no approved drugs targeting extracellular-signal regulated kinase (ERK), we are aware of a number of compounds that are in clinical development, including Merck’s SCH772984, Roche/Genentech’s GDC-0994, Celgene’s CC-90003, and BioMed Valley Discoveries’ BVD-523. Furthermore, it is possible that other companies are also engaged in discovery or preclinical development of compounds targeting ERK. These competitors, if successful in clinical development, may achieve clinical activity, regulatory approval and market adoption in advance of our compounds, constraining the ability of our compounds to gain significant market share. Although we believe that our ERK inhibitors, including KO-947, present several potential advantages relative to these aforementioned candidates, including potency against and selectivity for ERK as demonstrated in preclinical studies, these results may not translate to superior therapeutic benefit in clinical trials.
Menin-MLL Inhibitor Competition
There are no drugs approved or in clinical trials targeting the Menin-MLL protein-protein interaction. Although there are no targeted therapies approved specifically for the treatment of MLL-rearranged leukemias, there are a number of products in clinical development, including Epizyme’s EPZ-5676 and Novartis’s midostaurin, as well as Pfizer’s palbociclib (IBRANCE®), which has received accelerated approval in combination with letrozole, for the treatment of postmenopausal women with estrogen receptor-positive, human epidermal growth factor receptor 2-negative (ER+/HER2-) advanced breast cancer as initial endocrine-based therapy for their metastatic disease.
Commercialization
We have not yet established a sales, marketing or product distribution infrastructure because our lead candidates are still in discovery, preclinical or early clinical development. We anticipate that we will aim to retain commercial rights in North America for any of our product candidates for which we may in the future receive marketing approvals. We may also seek to retain commercial rights in Europe for any of our product candidates for which we may in the future receive marketing approvals. We currently anticipate that, if and when appropriate, we will seek to access the North American or European oncology markets through a focused, specialized, internal sales force.
Subject to receiving marketing approvals, we expect to commence commercialization activities by building a focused internal sales and marketing team in North America to sell our products. We may also build a focused internal sales and marketing team in Europe to sell our products. We believe that such an approach will enable us to address the community of oncologists who are the key specialists in treating the patient populations for which our current product candidates are being developed. Outside of regions where we maintain commercial rights, we may enter into distribution and other marketing arrangements with third parties for any of our product candidates that obtain marketing approval in foreign jurisdictions.
We also aim to build a marketing and sales management force to create and implement marketing strategies for any products that we may in the future market through our own sales teams and to oversee and support our sales force. We anticipate that our goals for any such marketing force include developing educational initiatives with respect to any approved products and establishing relationships with thought leaders in relevant fields of medicine.
We currently expect that any third parties with which we may collaborate in the future on the development of any commercial companion diagnostics for use with our therapeutic products will most likely hold the commercial rights to those diagnostic products. We expect that we would coordinate closely with any future diagnostic collaborators in connection with the marketing and sale of such diagnostic products and our related therapeutic products.
Manufacturing
We do not own or operate, and currently have no plans to establish, any manufacturing facilities. We currently rely, and expect to continue to rely, on third parties for the manufacture of our product candidates for preclinical and clinical testing as well as for commercial manufacture of any products that we may commercialize. Under our license agreement with Janssen, Janssen has provided us with its existing inventory of clinical supply of tipifarnib, which we believe will support our planned
17
Phase 2 clinical trials of tipifarnib. Janssen also provided us with its existing inventory of the crude drug substance and bulk key intermediate for manufacture of drug substance for tipifarnib. If needed, we aim to engage, by entering into a supply agreement or through another arrangement, third party manufacturers to provide us with additional tipifarnib clinical supply. For all of our product candidates, we aim to identify and qualify manufacturers to provide the active pharmaceutical ingredient and fill-and-finish services prior to submission of a new drug application (NDA) to the FDA.
We generally expect to rely on third parties for the manufacture of any companion diagnostics we or our collaborators may develop.
Intellectual Property
Our commercial success depends in part on our ability to obtain and maintain proprietary or intellectual property protection for our product candidates and our core technologies, including novel biomarker and diagnostic discoveries and other know-how, to operate without infringing on the proprietary rights of others and to prevent others from infringing our proprietary or intellectual property rights. We expect that we will seek to protect our proprietary and intellectual property position by, among other methods, licensing or filing our own U.S., international and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development and implementation of our business. We also rely on trade secrets, know-how and continuing technological innovation to develop and maintain our proprietary and intellectual property position, which we generally seek to protect through contractual obligations with third parties.
We currently, and expect that we will continue to, file or license patent applications directed to our key product candidates in an effort to establish intellectual property positions regarding new chemical entities relating to these product candidates, as well as uses of new chemical entities in the treatment of various cancers. We also intend to seek patent protection, if available, with respect to biomarkers that may be useful in selecting the right patient population for use of any of our product candidates. We own or in-license a patent portfolio consisting of over 20 patent families, including issued U.S. patents and their respective counterparts in a number of foreign jurisdictions, pending U.S. patent applications, pending applications under the Patent Cooperation Treaty and corresponding pending patent applications in a number of foreign jurisdictions. The issued U.S. patents cover tipifarnib and the issued patents covering composition of matter of tipifarnib are expected to expire in 2016 without patent term extension. The pending patent applications pertain to our ERK program and our Menin-MLL program. We would expect that any patents that may issue from the pending U.S. patent applications directed to our ERK product candidate would likely start to expire in 2034; however, any and all of these patent applications may not result in issued patents.
In addition to the patent applications that we have filed to date, we plan to continue to expand our intellectual property portfolio by filing patent applications directed to dosage forms, methods of treatment and additional inhibitor compounds of oncology molecular targets and their derivatives. Specifically, we anticipate that we will seek patent protection in the United States and internationally for novel compositions of matter covering the compounds, the chemistries and processes for manufacturing these compounds, their intermediates and/or metabolites, the use of these compounds in a variety of therapies and the use of biomarkers for patient selection for these compounds. However, these or other patent applications that we may file or license from third parties may not result in the issuance of patents, and any issued patents may cover limited claims that reduce their value and/or may be challenged, invalidated or circumvented. See “Risk Factors—Rights Related to Our Intellectual Property.”
In addition to patents, we also rely upon unpatented trade secrets and know-how and continuing technological innovation to develop and maintain our competitive position. We seek to protect our proprietary information, in part, using confidentiality agreements with our collaborators, scientific advisors, employees and consultants, and invention assignment agreements with our employees and selected consultants, scientific advisors and collaborators. The confidentiality agreements are designed to protect our proprietary information and, in the case of agreements or clauses requiring invention assignment, to grant us ownership of technologies that are developed through a relationship with a third-party.
Orange Book Listing
In seeking approval for a drug through an NDA, applicants are required to list with the FDA certain patents whose claims cover the applicant’s product. Upon approval, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Any applicant who files an ANDA seeking approval of a generic equivalent version of a drug listed in the Orange Book or a Section 505(b)(2) NDA referencing a drug listed in the Orange Book must certify to the FDA that (1) no patent information on the drug product that is the subject of the application has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the
18
drug product for which the application is submitted. This last certification is known as a paragraph IV certification. A notice of the paragraph IV certification must be provided to each owner of the patent that is the subject of the certification and to the holder of the approved NDA to which the ANDA or Section 505(b)(2) application refers. The applicant may also elect to submit a “section viii” statement certifying that its proposed label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent.
If the NDA holder for the reference drug and/or patent owners assert a patent challenge directed to one of the Orange Book listed patents within 45 days of the receipt of the paragraph IV certification notice, the FDA is prohibited from approving the application until the earlier of 30 months from the receipt of the paragraph IV certification, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the applicant. The ANDA or Section 505(b)(2) application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the reference drug has expired as described in further detail below.
Non-Patent Exclusivity
In addition to patent exclusivity, the holder of an NDA for a listed drug may be entitled to a period of non-patent exclusivity, during which the FDA cannot approve an ANDA or Section 505(b)(2) application that relies on the listed drug. For example, a pharmaceutical manufacturer may obtain five years of non-patent exclusivity upon FDA approval of a new chemical entity, or NCE, which is a drug that contains an active moiety that has not been approved by the FDA in any other NDA. An “active moiety” is defined as the molecule or ion responsible for the drug substance’s physiological or pharmacologic action. During the five year exclusivity period, the FDA cannot accept for filing any ANDA seeking approval of a generic version of that drug or any Section 505(b)(2) NDA for the same active moiety and that relies on the FDA’s findings regarding that drug, except that the FDA may accept an application for filing after four years if the follow-on applicant makes a paragraph IV certification. Five-year NCE exclusivity does not block the submission, review or approval of a 505(b)(1) NDA.
Patent Term Extension
After NDA approval, owners of relevant drug patents may apply for up to a five-year patent extension. The allowable PTE is calculated as half of the drug’s testing phase—the time between IND application and NDA submission—plus all of the review phase—the time between NDA submission and approval up to a maximum of five years. The time can be shortened if the FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years.
For patents that might expire during the application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the U.S. PTO must determine that approval of the drug covered by the patent for which a patent extension is being sought is likely. Interim patent extensions are not available for a drug for which an NDA has not been submitted.
Government Regulation
FDA Approval Process
In the United States, pharmaceutical products are subject to extensive regulation by FDA. The Federal Food, Drug and Cosmetic Act and other federal and state statutes and regulations govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending new drug applications, or NDAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
Pharmaceutical product development for a new product or certain changes to an approved product in the U.S. typically involves preclinical laboratory and animal tests, the submission to FDA of an investigational new drug application, or IND, which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with
19
federal regulations and requirements, including good laboratory practices. The results of preclinical testing are submitted to FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls, and a proposed clinical trial protocol. Long-term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with federal regulations; (ii) in compliance with good clinical practice, or GCP, an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors; as well as (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to FDA as part of the IND.
FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, the initial introduction of the drug into healthy human patients, the drug is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence of effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug for a particular indication, dosage tolerance and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In most cases FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible.
After completion of the required clinical testing, an NDA is prepared and submitted to FDA. FDA approval of the NDA is required before marketing of the product may begin in the U.S. The NDA must include the results of all preclinical, clinical and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture and controls. The cost of preparing and submitting an NDA is substantial, and the fees are typically increased annually.
FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, FDA begins an in-depth review. FDA has agreed to certain performance goals in the review of new drug applications to encourage timeliness. Most applications for standard review drug products are reviewed within twelve months from submission; most applications for priority review drugs are reviewed within eight months from submission. Priority review can be applied to drugs that FDA determines offer major advances in treatment, or provide a treatment where no adequate therapy exists. The review process for both standard and priority review may be extended by FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission.
FDA may also refer applications for novel drug products, or drug products that present difficult questions of safety or efficacy, to an outside advisory committee—typically a panel that includes clinicians and other experts—for review, evaluation and a recommendation as to whether the application should be approved. FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations.
Before approving an NDA, FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, FDA will inspect the facility or the facilities at which the drug is manufactured. FDA will not approve the product unless compliance with current good manufacturing practice, or GMP—a quality system regulating manufacturing—is
20
satisfactory and the NDA contains data that provide substantial evidence that the drug is safe and effective in the indication studied.
After FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for FDA to reconsider the application. If, or when, those deficiencies have been addressed to FDA’s satisfaction in a resubmission of the NDA, FDA will issue an approval letter. FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Fast Track Designation and Accelerated Approval
FDA is required to facilitate the development, and expedite the review, of drugs that are intended for the treatment of a serious or life-threatening disease or condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the Fast Track program, the sponsor of a new drug candidate may request that FDA designate the drug candidate for a specific indication as a Fast Track drug concurrent with, or after, the filing of the IND for the drug candidate. FDA must determine if the drug candidate qualifies for Fast Track Designation within 60 days of receipt of the sponsor’s request.
Under the Fast Track program and FDA’s accelerated approval regulations, FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions, or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow FDA to withdraw the drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to priority review by FDA.
If a submission is granted Fast Track Designation, the sponsor may engage in more frequent interactions with FDA, and FDA may review sections of the NDA before the application is complete. This rolling review is available if the applicant provides, and FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, FDA’s time period goal for reviewing an application does not begin until the last section of the NDA is submitted. Additionally, Fast Track Designation may be withdrawn by FDA if FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
Breakthrough Therapy Designation
FDA is also required to expedite the development and review of the application for approval of drugs that are intended to treat a serious or life-threatening disease or condition where preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. Under the
21
Breakthrough Therapy program, the sponsor of a new drug candidate may request that FDA designate the drug candidate for a specific indication as a breakthrough therapy concurrent with, or after, the filing of the IND for the drug candidate. FDA must determine if the drug candidate qualifies for Breakthrough Therapy designation within 60 days of receipt of the sponsor’s request.
Orphan Drug Designation and Exclusivity
The Orphan Drug Act provides incentives for the development of products intended to treat rare diseases or conditions. Under the Orphan Drug Act, the FDA may grant orphan designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug available in the United States for this type of disease or condition will be recovered from sales of the product. If a sponsor demonstrates that a drug is intended to treat a rare disease or condition, the FDA will grant orphan designation for that product for the orphan disease indication, assuming that the same drug has not already been approved for the indication for which the sponsor is seeking orphan designation. If the same drug has already been approved for the indication for which the sponsor is seeking orphan designation, the sponsor must present a plausible hypothesis of clinical superiority in order to obtain orphan designation. Orphan designation must be requested before submitting an NDA. After the FDA grants orphan designation, the FDA discloses the identity of the therapeutic agent and its potential orphan use.
Orphan designation may provide manufacturers with benefits such as research grants, tax credits, PDUFA application fee waivers, and eligibility for orphan drug exclusivity. If a product that has orphan designation subsequently receives the first FDA approval of the active moiety for that disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which for seven years prohibits the FDA from approving another product with the same active ingredient for the same indication, except in limited circumstances. Orphan drug exclusivity will not bar approval of another product under certain circumstances, including if a subsequent product with the same active ingredient for the same indication is shown to be clinically superior to the approved product on the basis of greater efficacy or safety, or providing a major contribution to patient care, or if the company with orphan drug exclusivity is not able to meet market demand. Further, the FDA may approve more than one product for the same orphan indication or disease as long as the products contain different active ingredients. Moreover, competitors may receive approval of different products for the indication for which the orphan drug has exclusivity or obtain approval for the same product but for a different indication for which the orphan drug has exclusivity.
In the European Union, orphan drug designation also entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity is granted following drug or biological product approval. This period may be reduced to 6 years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Post-Approval Requirements
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
Adverse event reporting and submission of periodic reports are required following FDA approval of an NDA. FDA also may require post-marketing testing, known as Phase 4 testing, risk evaluation and mitigation strategies, or REMS, and surveillance to monitor the effects of an approved product, or FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, drug manufacture, packaging and labeling procedures must continue to conform to current good manufacturing practices, or cGMPs, after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with FDA and certain state agencies. Registration with FDA subjects entities to periodic unannounced inspections by FDA, during which the Agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality-control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
22
Pediatric Information
Under the Pediatric Research Equity Act, or PREA, NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. FDA may grant full or partial waivers, or deferrals, for submission of data. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted.
The Best Pharmaceuticals for Children Act, or BPCA, provides NDA holders a six-month extension of any exclusivity—patent or non-patent—for a drug if certain conditions are met. Conditions for exclusivity include FDA’s determination that information relating to the use of a new drug in the pediatric population may produce health benefits in that population, FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the statutory timeframe. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
FDA Regulation of Companion Diagnostics
Our drug products may rely upon in vitro companion diagnostics for use in selecting the patients that we believe will respond to our cancer therapeutics. If safe and effective use of a therapeutic product depends on an in vitro diagnostic, FDA generally will require approval or clearance of the diagnostic at the same time that FDA approves the therapeutic product. This policy is described in an August 2014 FDA guidance document.
FDA has required in vitro companion diagnostics intended to select the patients who will respond to cancer treatment to obtain a pre-market approval, or PMA, for that diagnostic simultaneously with approval of the drug. We believe that FDA will require PMA approval of one or more in vitro companion diagnostics to identify patient populations suitable for our cancer therapies. The review of these in vitro companion diagnostics in conjunction with the review of our cancer treatments involves coordination of review by FDA’s Center for Drug Evaluation and Research and by FDA’s Center for Devices and Radiological Health.
The PMA process, including the gathering of clinical and nonclinical data and the submission to and review by FDA, can take several years or longer. It involves a rigorous premarket review during which the applicant must prepare and provide FDA with reasonable assurance of the device’s safety and effectiveness and information about the device and its components regarding, among other things, device design, manufacturing and labeling. PMA applications are subject to an application fee. In addition, PMAs for certain devices must generally include the results from extensive preclinical and adequate and well-controlled clinical trials to establish the safety and effectiveness of the device for each indication for which FDA approval is sought. In particular, for a diagnostic, the applicant must demonstrate that the diagnostic produces reproducible results when the same sample is tested multiple times by multiple users at multiple laboratories. As part of the PMA review, FDA will typically inspect the manufacturer’s facilities for compliance with the Quality System Regulation, or QSR, which imposes elaborate testing, control, documentation and other quality assurance requirements.
PMA approval is not guaranteed, and FDA may ultimately respond to a PMA submission with a not approvable determination based on deficiencies in the application and require additional clinical trial or other data that may be expensive and time-consuming to generate and that can substantially delay approval. If FDA’s evaluation of the PMA application is favorable, FDA typically issues an approvable letter requiring the applicant’s agreement to specific conditions, such as changes in labeling, or specific additional information, such as submission of final labeling, in order to secure final approval of the PMA. If FDA concludes that the applicable criteria have been met, FDA will issue a PMA for the approved indications, which can be more limited than those originally sought by the applicant. The PMA can include post-approval conditions that FDA believes necessary to ensure the safety and effectiveness of the device, including, among other things, restrictions on labeling, promotion, sale and distribution.
After a device is placed on the market, it remains subject to significant regulatory requirements. Medical devices may be marketed only for the uses and indications for which they are cleared or approved. Device manufacturers must also establish registration and device listings with FDA. A medical device manufacturer’s manufacturing processes and those of its suppliers are required to comply with the applicable portions of the QSR, which cover the methods and documentation of the design, testing, production, processes, controls, quality assurance, labeling, packaging and shipping of medical devices. Domestic facility records and manufacturing processes are subject to periodic unscheduled inspections by FDA. FDA also may inspect foreign facilities that export products to the United States.
Failure to comply with applicable regulatory requirements can result in enforcement action by FDA, which may include any of the following sanctions: warning letters, fines, injunctions, civil or criminal penalties, recall or seizure of current or
23
future products, operating restrictions, partial suspension or total shutdown of production, denial of submissions for new products, or withdrawal of PMA approvals.
Clinical Trials and IDEs
A clinical trial is almost always required to support a PMA application. In some cases, one or more smaller Investigational Device Exemption (IDE) studies may precede a pivotal clinical trial intended to demonstrate the safety and efficacy of the investigational device.
All clinical studies of investigational devices must be conducted in compliance with the FDA’s requirements. If an investigational device could pose a significant risk to patients pursuant to FDA regulations, the FDA must approve an IDE application prior to initiation of investigational use. IVD trials usually do not require an IDE, as the FDA does not judge them to be a significant risk because the results do not affect the patients in the study. However, for a trial where the IVD result directs the therapeutic care of patients with cancer, we believe that the FDA may consider the investigation to present significant risk and require an IDE application.
An IDE application must be supported by appropriate data, such as laboratory test results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The FDA typically grants IDE approval for a specified number of patients. A non-significant risk device does not require FDA approval of an IDE. Both significant risk and non-significant risk investigational devices require approval from IRBs at the study centers where the device will be used.
During the critical trial, the sponsor must comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting and record keeping. The investigators must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of investigational devices and comply with all reporting and record keeping requirements. Prior to granting PMA approval, the FDA typically inspects the records relating to the conduct of the study and the clinical data supporting the PMA application for compliance with applicable requirements.
Although the QSR does not fully apply to investigational devices, the requirement for controls on design and development does apply. The sponsor also must manufacture the investigational device in conformity with the quality controls described in the IDE application and any conditions of IDE approval that the FDA may impose with respect to manufacturing.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our product candidates to the extent we choose to sell any products outside of the United States. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. As in the United States, post-approval regulatory requirements, such as those regarding product manufacture, marketing, or distribution would apply to any product that is approved outside the United States.
Government authorities in the United States, at the federal, state and local level, and in other countries, extensively regulate, among other things, the research, development, testing, manufacture, including any manufacturing changes, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, import and export of pharmaceutical products, such as those we are developing.
Additional Regulations and Environmental Matters
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws restrict certain marketing practices in the pharmaceutical industry in recent years. These laws, which generally will not be applicable to us or our product candidates unless and until we obtain FDA marketing approval for any of our product candidates, include transparency laws, anti-kickback statutes, false claims statutes and regulation regarding providing drug samples.
The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. Violations of the federal anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs.
24
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws.
The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Further, sales of any of our product candidates that may be approved will depend, in part, on the extent to which the cost of the product will be covered by third party payors. Third party payors may limit coverage to an approved list of products, or formulary, which might not include all drug products approved by the FDA for an indication. Any product candidates for which we obtain marketing approval may not be considered medically necessary or cost-effective by third party payors, and we may need to conduct expensive pharmacoeconomic studies in the future to demonstrate the medical necessity and/or cost effectiveness of any such product. The U.S. government, state legislatures and foreign governments have shown increased interest in implementing cost containment programs to limit government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Continued interest in and adoption of such controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals such as the drug candidates we are developing.
In addition to regulatory schemes that apply, or may in the future apply, to our business, we are or may become subject to various environmental, health and safety laws and regulations governing, among other things, laboratory procedures and any use and disposal by us of hazardous or potentially hazardous substances in connection with our research and development activities. We do not presently expect such environmental, health and safety laws or regulations to materially impact our present or planned future activities.
Employees
As of the date of this report, we have 12 full-time employees and three part-time employees, including six employees with M.D. or Ph.D. degrees. None of our employees is represented by a labor union or covered by a collective bargaining agreement. We consider our relationship with our employees to be good.
Facilities
We occupy approximately 1,560 rentable square feet of office and laboratory space in La Jolla, California under a sublease that expires in August 2016. We also occupy two offices in Cambridge, Massachusetts under sublease that expires in October 2016. We believe that our facilities are sufficient to meet our current needs and that suitable additional space will be available as and when needed.
Legal Proceedings
Neither we nor our subsidiaries are currently a party to, nor is our property the subject of, any material legal proceedings.
FORWARD-LOOKING STATEMENTS
Statements in this Current Report on Form 8-K that are not descriptions of historical facts are forward-looking statements that are based on management’s current expectations and are subject to risks and uncertainties that could negatively affect our business, operating results, financial condition and stock price. We have attempted to identify forward-looking statements by terminology including “anticipates,” “believes,” “can,” “continue,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “should,” “will,” “would” or the negative of these terms or other comparable terminology. Factors that could cause actual results to differ materially from those currently anticipated include those set forth in the section titled “Risk Factors” including, in particular, risks relating to:
| • | the initiation, cost, timing, progress and results of our research and development activities, clinical trials and preclinical studies; |
| • | the early stage of products under development; |
25
| • | the timing of and our ability to obtain and maintain regulatory approval of our existing product candidates, any product candidates that we may develop, and any related restrictions, limitations, and/or warnings in the label of any approved product candidates; |
| • | our plans to research, develop and commercialize our future product candidates; |
| • | our ability to attract collaborators with development, regulatory and commercialization expertise; |
| • | our ability to obtain and maintain intellectual property protection for our product candidates; |
| • | our ability to successfully commercialize our product candidates; |
| • | the size and growth of the markets for our product candidates and our ability to serve those markets; |
| • | the rate and degree of market acceptance of any future products; |
| • | the success of competing drugs that are or become available; |
| • | government regulation; |
| • | regulatory developments in the United States and other countries; |
| • | the performance of our third-party suppliers and manufacturers and our ability to obtain alternative sources of raw materials; |
| • | our ability to obtain additional financing; |
| • | our use of the proceeds from our recently completed private placement; |
| • | our expectations regarding the period during which we qualify as an emerging growth company under the Jumpstart Our Business Startups Act of 2012, or the JOBS Act; |
| • | the accuracy of our estimates regarding expenses, future revenues, capital requirements and the need for additional financing; and |
| • | our ability to attract and retain key management, scientific or clinical personnel. |
These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described in the section titled “Risk Factors.” Moreover, we operate in a very competitive and rapidly-changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this Current Report on Form 8-K may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements. You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. Moreover, except as required by law, neither we nor any other person assumes responsibility for the accuracy and completeness of the forward-looking statements. We undertake no obligation to update publicly any forward-looking statements for any reason after the date of this Current Report on Form 8-K to conform these statements to actual results or to changes in our expectations.
RISK FACTORS
Investing in our common stock involves a high degree of risk. In addition to the other information set forth in this Current Report on Form 8-K, you should carefully consider the factors discussed below when considering an investment in our common stock. If any of the events contemplated by the following discussion of risks should occur, our business, results of operations and financial condition could suffer significantly. As a result, you could lose some or all of your investment in our common stock. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business.
Risks Related to Our Financial Position and Need For Additional Capital
We expect to incur losses over the next several years and may never achieve or maintain profitability.
We expect that it will be many years, if ever, before we have a product candidate ready for commercialization. To date, we have financed our operations primarily through equity and debt financings. We expect to continue to incur significant
26
expenses and increasing operating losses for the foreseeable future. The net losses we incur may fluctuate significantly from quarter to quarter. We anticipate that our expenses will increase substantially if and as we:
| • | continue development of our product candidates; |
| • | initiate clinical trials for our product candidates; |
| • | seek marketing approvals for our product candidates; |
| • | establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval; |
| • | maintain, expand and protect our intellectual property portfolio; |
| • | hire additional personnel; |
| • | add operational, financial and management information systems and personnel, including personnel to support our product development and planned future commercialization efforts; and |
| • | incur increased costs as a result of operating as a public company. |
To become and remain profitable, we must develop and eventually commercialize a product or products with significant market potential. This will require us to be successful in a range of challenging activities, including completing clinical trials of our product candidates, obtaining marketing approval from the FDA for these product candidates and manufacturing, marketing and selling those products for which we may obtain marketing approval. We may never succeed in these activities and, even if we do, may never generate revenues that are significant or large enough to achieve profitability. If we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would decrease the value of the company and could impair our ability to raise capital, maintain our discovery and preclinical development efforts, expand our business or continue our operations and may require us to raise additional capital that may dilute your ownership interest. A decline in the value of our company could also cause you to lose all or part of your investment.
Our limited operating history may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
We are an early-stage clinical development company. Kura was incorporated in August 2014 and commenced operations in the fourth quarter of 2014. Our operations to date have been limited to organizing and staffing our company, business planning, raising capital, identifying and acquiring potential product candidates, undertaking preclinical studies and preparing to undertake clinical studies of our most advanced product candidate, tipifarnib. We have not yet demonstrated our ability to commence or successfully complete any clinical trials, including those clinical trials in support of FDA approval, obtain marketing approvals, manufacture a commercial scale product, or arrange for a third-party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. Medicines, on average, take 10 to 15 years to be developed from the time they are discovered to the time they are available for treating patients. Consequently, any predictions you make about our future success or viability based on our short operating history to date may not be as accurate as they could be if we had a longer operating history.
In addition, as a new business, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors. We will need to transition from a company with a research focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
We are a clinical-stage company with no approved products and no historical product revenue. Consequently, we expect that our financial and operating results will vary significantly from period to period.
We are a clinical-stage company that has incurred losses since its inception and expect to continue to incur substantial losses in the foreseeable future. Biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of uncertainty. We expect our actual financial condition and operating results to fluctuate significantly from quarter-to-quarter or year-to-year due to a variety of factors, many of which are beyond our control. Factors relating to our business that may contribute to these fluctuations include:
| • | the success of our clinical trials through all phases of clinical development; |
| • | delays in the commencement, enrollment and timing of clinical trials; |
27
| • | our ability to secure and maintain collaborations, licensing or other arrangements for the future development and/or commercialization of our product candidates, as well as the terms of those arrangements; |
| • | our ability to obtain, as well as the timeliness of obtaining, additional funding to develop our product candidates; |
| • | the results of clinical trials or marketing applications for product candidates that may compete with our product candidates; |
| • | competition from existing products or new products that may receive marketing approval; |
| • | potential side effects of our product candidates that could delay or prevent approval or cause an approved drug to be taken off the market; |
| • | any delays in regulatory review and approval of our product candidates; |
| • | our ability to identify and develop additional product candidates; |
| • | the ability of patients or healthcare providers to obtain coverage or sufficient reimbursement for our products; |
| • | our ability, and the ability of third parties such as Clinical Research Organizations, or CROs, to adhere to clinical study and other regulatory requirements; |
| • | the ability of third-party manufacturers to manufacture our product candidates and key ingredients needed to conduct clinical trials and, if approved, successfully commercialize our products; |
| • | the costs to us, and our ability as well as the ability of any third-party collaborators, to obtain, maintain and protect our intellectual property rights; |
| • | costs related to and outcomes of any future intellectual property litigation; |
| • | our ability to adequately support future growth; |
| • | our ability to attract and retain key personnel to manage our business effectively; and |
| • | our ability to build our finance infrastructure and, to the extent required, improve our accounting systems and controls. |
Accordingly, the likelihood of our success must be evaluated in light of many potential challenges and variables associated with a clinical-stage company, many of which are outside of our control, and past operating or financial results should not be relied on as an indication of future results. Fluctuations in our operating and financial results could cause our share price to decline. It is possible that in some future periods, our operating results will be above or below the expectations of securities analysts or investors, which could also cause our share price to decline.
We may be unable to raise additional funds when needed. Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of equity offerings and debt financings. We do not have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our
28
stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect rights of our stockholders as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional funds when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts.
Our ability to use our net operating tax loss carryforwards and certain other tax attributes may be limited.
Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change” (generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income may be limited. As a result of the Private Placement and other transactions that have occurred over the past three years, we may trigger an “ownership change” limitation. In addition, we may experience ownership changes in the future as a result of subsequent shifts in our stock ownership. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards and other pre-change tax attributes to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us.
Risks Related to the Discovery and Development of Our Product Candidates
Our discovery and preclinical development is focused on the development of targeted therapeutics for patients with genetically defined cancers, which is a rapidly evolving area of science, and the approach we are taking to discover and develop drugs is novel and may never lead to marketable products.
The discovery and development of targeted drug therapeutics for patients with genetically defined cancers is an emerging field, and the scientific discoveries that form the basis for our efforts to discover and develop product candidates are relatively new. The scientific evidence to support the feasibility of developing product candidates based on these discoveries is both preliminary and limited. The patient populations for our product candidates are not completely defined but are substantially smaller than the general treated cancer population, and we will need to screen and identify these patients. Successful identification of patients is dependent on several factors, including achieving certainty as to how specific genetic alterations respond to our product candidates and developing companion diagnostics to identify such genetic alterations. Furthermore, even if we are successful in identifying patients, we cannot be certain that the resulting patient populations will be large enough to allow us to successfully commercialize our products and achieve profitability. Therefore, we do not know if our approach of treating patients with genetically defined cancers will be successful, and if our approach is unsuccessful, our business will suffer.
Our research and development programs and product candidates are at an early stage of development. As a result we are unable to predict if or when we will successfully develop or commercialize our product candidates.
Our clinical-stage product candidate, tipifarnib, as well as our other pipeline assets are at an early stage of development and will require significant investment and regulatory approvals prior to commercialization. We currently have no product candidates beyond Phase 2 clinical trials. We anticipate commencing a Phase 2 clinical trial of tipifarnib in advanced solid tumors with the HRAS mutation in the second quarter of 2015 and a Phase 2 clinical trial in peripheral T-cell lymphoma in the third quarter of 2015. Our lead candidate in our ERK program, KO-947, is in IND-enabling pre-clinical development, and our backup compounds in the ERK program as well as our other programs, including our Menin-MLL program, are in earlier stages of development. Each of our product candidates will require additional clinical and preclinical development, management of clinical, preclinical and manufacturing activities, obtaining regulatory approval, obtaining manufacturing supply, building of a commercial organization, substantial investment and significant marketing efforts before we generate any revenues from product sales. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates. In addition, our product development programs contemplate the development of companion diagnostics. Companion diagnostics are subject to regulation as medical devices and we may be required to obtain marketing approval for accompanying companion diagnostics before we may commercialize our product candidates.
We cannot be certain that such clinical development of tipifarnib or any of our other product candidates will be successful or that we will obtain regulatory approval or be able to successfully commercialize any of our product candidates and generate revenue. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and the clinical trial process may fail to demonstrate that our product candidates are safe and effective for their
29
proposed uses. Any such failure could cause us to abandon further development of any one or more of our product candidates and may delay development of other product candidates. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and initial clinical trials. Tipifarnib has been studied in more than 5,000 patients, including more than 600 patients at the dose and schedule we intend to use in our Phase 2 trials. At that dose and schedule, tipifarnib exhibited a manageable side effect profile and was generally well tolerated. In prior studies tipifarnib demonstrated anti-cancer activity in certain patient subsets. However the anti-cancer activity observed was not sufficient to support marketing approval by the FDA in the indication in which it was sought. Although we are designing our clinical trials to target the patient subsets who we believe are most likely to benefit from treatment with tipifarnib, there is no guarantee that our clinical trials will be successful. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. Any delay in, or termination of, our clinical trials will delay and possibly preclude the filing of any new drug applications, or NDAs, with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenue.
We have not previously submitted an NDA to the FDA, or similar drug approval filings to comparable foreign authorities, for any product candidate, and we cannot be certain that any of our product candidates will receive regulatory approval. Further, our product candidates may not receive regulatory approval even if they are successful in clinical trials. If we do not receive regulatory approvals for our product candidates, we may not be able to continue our operations. Even if we successfully obtain regulatory approvals to market one or more of our product candidates, our revenues will be dependent, in part, upon our or our future collaborators’ ability to obtain regulatory approval of the companion diagnostics to be used with our product candidates, if required, as well as the size of the markets in the territories for which we gain regulatory approval and have commercial rights. If the markets for patient subsets that we are targeting are not as significant as we estimate, we may not generate significant revenues from sales of such products, if approved.
Difficulty in enrolling patients could delay or prevent clinical trials of our product candidates. We may find it difficult to enroll patients in our Phase 2 clinical trial for tipifarnib given that we do not know how many patients share the HRAS mutations tipifarnib is expected to inhibit.
Identifying and qualifying patients to participate in clinical studies of our product candidates is critical to our success. The timing of our clinical studies depends in part on the speed at which we can recruit patients to participate in testing our product candidates, and we may experience delays in our clinical trials if we encounter difficulties in enrollment. The patient population for our product candidates is not completely defined, but it is substantially smaller than other cancer indications, because we are looking for the same type of genetic alterations across different tumor types and the number of patients with these alterations may be small. For example, with respect to tipifarnib, we do not know how many patients will have the target HRAS mutations that tipifarnib is expected to inhibit.
In addition to the potentially small populations, the eligibility criteria of our clinical trials will further limit the pool of available study participants as we will require that patients have specific characteristics that we can measure or to assure their disease is either severe enough or not too advanced to include them in a study. Additionally, the process of finding and diagnosing patients may prove costly. We also may not be able to identify, recruit, and enroll a sufficient number of patients to complete our clinical studies because of the perceived risks and benefits of the product candidate under study, the availability and efficacy of competing therapies and clinical trials, the proximity and availability of clinical study sites for prospective patients, and the patient referral practices of physicians. If patients are unwilling to participate in our studies for any reason, the timeline for recruiting patients, conducting studies, and obtaining regulatory approval of potential products may be delayed.
If we experience delays in the completion of, or termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenue from any of these product candidates could be delayed or prevented. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process, and jeopardize our ability to commence product sales and generate revenue. Any of these occurrences may harm our business, financial condition, and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates, including:
| • | unforeseen safety issues or adverse side effects; |
| • | failure of our companion diagnostics in identifying patients; |
| • | modifications to protocols of our clinical trials resulting from FDA or institutional review board, or IRB, decisions; and |
| • | ambiguous or negative interim results of our clinical trials, or results that are inconsistent with earlier results. |
30
We may not be successful in our efforts to build a pipeline of product candidates.
A key element of our strategy is to build a pipeline of small molecule product candidates that inhibit cancer signaling targets where we believe outcomes can be improved by using molecular diagnostics to identify those patients whose tumors have the genetic mutations most likely to respond to treatment, and to progress those product candidates through clinical development for the treatment of a variety of different types of cancer. We may not be able to develop product candidates that are safe and effective inhibitors of all or any of these targets. Even if we are successful in building a product pipeline, the potential product candidates that we identify may not be suitable for clinical development for a number of reasons, including causing harmful side effects or demonstrating other characteristics that indicate a low likelihood of receiving marketing approval or achieving market acceptance. If our methods of identifying potential product candidates fail to produce a pipeline of potentially viable drug candidates, then our success as a business will be dependent on the success of fewer potential product candidates, which introduces risks to our business model and potential limitations to any success we may achieve.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome. We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
The risk of failure for all of our product candidates is high. Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must complete preclinical development and then conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Clinical testing is expensive, difficult to design and implement and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. Further, the results of preclinical studies and early clinical trials of our product candidates may not be predictive of the results of later-stage clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products. For instance, the FDA issued a non-approval letter for tipifarnib in acute myelogenous leukemia, in June 2005. It is impossible to predict when or if any of our product candidates will prove effective or safe in humans or will receive regulatory approval.
We may experience delays in our clinical trials and we do not know whether planned clinical trials will begin or enroll patients on time, need to be redesigned or be completed on schedule, if at all. We have submitted the first tipifarnib Phase 2 study plan to the FDA and we expect to submit the second study plan to the FDA in the second quarter of 2015. If the FDA has comments that we are required to address, the initiation of one or both Phase 2 studies may be delayed. There can be no assurance that FDA will not put any of our product candidates on clinical hold in the future. We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to receive marketing approval or commercialize our product candidates. Clinical trials may be delayed, suspended or prematurely terminated because costs are greater than we anticipate or for a variety of reasons, such as:
| • | delay or failure in reaching agreement with FDA or a comparable foreign regulatory authority on a trial design that we are able to execute; |
| • | delay or failure in obtaining authorization to commence a trial or inability to comply with conditions imposed by a regulatory authority regarding the scope or design of a clinical trial; |
| • | delays in reaching, or failure to reach, agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites; |
| • | inability, delay, or failure in identifying and maintaining a sufficient number of trial sites, many of which may already be engaged in other clinical programs; |
| • | delay or failure in recruiting and enrolling suitable subjects to participate in a trial; |
| • | delay or failure in having subjects complete a trial or return for post-treatment follow-up; |
| • | clinical sites and investigators deviating from trial protocol, failing to conduct the trial in accordance with regulatory requirements, or dropping out of a trial; |
| • | lack of adequate funding to continue the clinical trial, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional clinical studies and increased expenses associated with the services of our CROs and other third parties; |
| • | clinical trials of our product candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs; |
| • | the number of patients required for clinical trials of our product candidates may be larger than we anticipate, |
31
enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials at a higher rate than we anticipate; |
| • | we may experience delays or difficulties in the enrollment of patients whose tumors harbor the specific genetic alterations that our product candidates are designed to target; |
| • | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; |
| • | we may have difficulty partnering with experienced contract research organizations (CROs) that can screen for patients whose tumors harbor the applicable genetic alterations and run our clinical trials effectively; |
| • | regulators or IRBs may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks; |
| • | the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; or |
| • | there may be changes in governmental regulations or administrative actions. |
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our product candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
| • | be delayed in obtaining marketing approval for our product candidates; |
| • | not obtain marketing approval at all; |
| • | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| • | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings that would reduce the potential market for our products or inhibit our ability to successfully commercialize our products; |
| • | be subject to additional post-marketing restrictions and/or testing requirements; or |
| • | have the product removed from the market after obtaining marketing approval. |
Our product development costs will also increase if we experience delays in testing or marketing approvals. We do not know whether any of our preclinical studies or clinical trials will need to be restructured or will be completed on schedule, or at all. Significant preclinical or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
We may not be successful in advancing the clinical development of our product candidates, including tipifarnib.
In order to execute on our strategy of advancing the clinical development of our product candidates, we have designed our Phase 2 clinical trials of tipifarnib, and expect to design future trials, to include patients whose tumors harbor the applicable genetic alterations that we believe contribute to particular cancer subsets. Our goal in doing this is to enroll patients who have the highest probability of responding to the drug, in order to show early evidence of clinical efficacy. If we are unable to include patients whose tumors harbor the applicable genetic alterations, or if our product fails to work as we expect, our ability to assess the therapeutic effect, seek participation in FDA expedited review and approval programs, including Breakthrough Therapy, Fast Track Designation, Priority Review and Accelerated Approval, or otherwise to seek to accelerate clinical development and regulatory timelines, could be compromised, resulting in longer development times, larger trials and a greater likelihood of not obtaining regulatory approval. In addition, because the natural history of different tumor types is variable, we will need to study our product candidates, including tipifarnib, in clinical trials specific for a given tumor type and this may result in increased time and cost. Even if our product candidate demonstrates efficacy in a particular tumor type, we cannot guarantee that any product candidate, including tipifarnib, will behave similarly in all tumor types, and we will be required to obtain separate regulatory approvals for each tumor type we intend a product candidate to treat. If any of our clinical trials are unsuccessful, our business will suffer.
Preclinical and clinical testing of tipifarnib that has been conducted to date may not have been performed in compliance with applicable regulatory standards, which could lead to increased costs or material delays for their further development.
32
We have only recently licensed the rights to develop our lead product candidate, tipifarnib, from Janssen, and the development of tipifarnib prior to our license was conducted wholly by Janssen or any third parties with which it had contracted. As a result, we were not involved with nor did we have any control over any of those development activities. Because we had no input on Janssen’s development activities relating to tipifarnib, we may discover that all or certain elements of the trials and studies it performed have not been in compliance with applicable regulatory standards or have otherwise been deficient, particularly relative to current requirements as development of tipifarnib began in the 1990’s. Any such deficiency in the prior development of tipifarnib may adversely affect our ability to obtain regulatory approval for tipifarnib. We and Janssen are in the process of transitioning the development program documentation and databases from studies previously conducted by Janssen to us. We cannot assure you that our efforts to transition all of the necessary documentation from Janssen will be completed on a timely basis, or at all. If we are unable to successfully complete the transition of Janssen’s tipifarnib development documentation to us on a timely basis, our development plans may be delayed, which could harm our business, prospects, financial condition and results of operations.
If serious adverse events or unacceptable side effects are identified during the development of our product candidates, we may need to abandon or limit our development of some of our product candidates.
If our product candidates are associated with undesirable side effects in preclinical or clinical trials or have characteristics that are unexpected, we may need to interrupt, delay or abandon their development or limit development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Tipifarnib has been studied in more than 5,000 patients, including more than 600 patients at the dose and schedule we intend to use in our Phase 2 trials. At that dose and schedule, tipifarnib exhibited a manageable side effect profile and was generally well tolerated. The most common hematologic adverse events of any grade were neutropenia (low white blood cell count), anemia and thrombocytopenia (low platelet count). The most common non-hematologic adverse events of any grade were gastrointestinal system disorders (nausea, anorexia, diarrhea and vomiting, and abdominal pain) fatigue, and fever.
Treatment discontinuation with this regimen was approximately 20%. There is no guarantee that additional or more severe side effects will not be identified through further clinical studies. Rights to develop tipifarnib in certain non-oncology indications have been granted by Janssen to EB Pharma, a subsidiary of Eiger BioPharmaceuticals. Janssen may grant rights to other non-oncology indications to other third parties. Undesirable side effects may be identified in clinical trials that EB Pharma or any other third party may conduct in non-oncology indications, which may negatively impact the development, commercialization or potential value of tipifarnib. These or other drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Many compounds developed in the biopharmaceutical industry that initially showed promise in early-stage testing for treating cancer have later been found to cause side effects that prevented further development of the compound. Any of these occurrences may harm our business, financial condition and prospects significantly.
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we must focus on a limited number of research programs and product candidates and on specific indications. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future discovery and preclinical development programs and product candidates for specific indications may not yield any commercially viable products.
Failure to successfully validate, develop and obtain regulatory approval for companion diagnostics for our product candidates could harm our drug development strategy and operational results.
As one of the central elements of our business strategy and clinical development approach, we seek to screen and identify subsets of patients with a genetic alteration who may derive meaningful benefit from our development product candidates. To achieve this, our product development program is dependent on the development and commercialization of a companion diagnostic by us or by third party collaborators. Companion diagnostics are developed in conjunction with clinical programs for the associated product and are subject to regulation as medical devices. Each agency that approves a product will independently need to approve the companion diagnostic before or concurrently with its approval of the product candidate, and before a product can be commercialized. The approval of a companion diagnostic as part of the product label will limit the use of the product candidate to only those patients who express the specific genetic alteration it was developed to detect. We may also experience delays in developing a sustainable, reproducible and scalable manufacturing process or transferring that process to
33
commercial partners or negotiating insurance reimbursement plans, all of which may prevent us from completing our clinical trials or commercializing our products on a timely or profitable basis, if at all.
Companion diagnostics are subject to regulation by FDA and comparable foreign regulatory authorities as medical devices and require separate clearance or approval prior to their commercialization. To date, FDA has required premarket approval of all companion diagnostics for cancer therapies. We and our third-party collaborators may encounter difficulties in developing and obtaining approval for these companion diagnostics. Any delay or failure by us or third- party collaborators to develop or obtain regulatory approval of a companion diagnostic could delay or prevent approval of our related product candidates.
Failure by us or our third-party collaborators to successfully commercialize companion diagnostics developed for use with our product candidates could harm our ability to commercialize these product candidates.
Even if we or our companion diagnostic collaborators successfully obtain regulatory approval for the companion diagnostics for our product candidates, our collaborators:
| • | may not perform their obligations as expected; |
| • | may not pursue commercialization of companion diagnostics for our therapeutic product candidates that achieve regulatory approval; |
| • | may elect not to continue or renew commercialization programs based on changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition, that divert resources or create competing priorities; |
| • | may not commit sufficient resources to the marketing and distribution of such product or products; and |
| • | may terminate their relationship with us. |
Additionally, we or our collaborators may encounter production difficulties that could constrain the supply of the companion diagnostics, affect the ease of use, affect the price or have difficulties gaining acceptance of the use of the companion diagnostics in the clinical community.
If companion diagnostics for use with our product candidates fail to gain market acceptance, our ability to derive revenues from sales of our product candidates could be harmed. If we or our collaborators fail to commercialize these companion diagnostics, we may not be able to enter into arrangements with another diagnostic company to obtain supplies of an alternative diagnostic test for use in connection with our product candidates or do so on commercially reasonable terms, which could adversely affect and delay the development or commercialization of our product candidates.
Risks Related to Regulatory Approval of Our Product Candidates and Other Legal Compliance Matters
If we are not able to obtain, or if there are delays in obtaining, required regulatory approvals, we will not be able to commercialize our product candidates, and our ability to generate revenue will be materially impaired.
Our product candidates must be approved by FDA pursuant to a new drug application, or NDA, in the United States and by the European Medicines Agency, or EMA, and similar regulatory authorities outside the United States prior to commercialization. The process of obtaining marketing approvals, both in the United States and abroad, is expensive and takes many years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Failure to obtain marketing approval for a product candidate will prevent us from commercializing the product candidate. We have not received approval to market any of our product candidates from regulatory authorities in any jurisdiction. We have no experience in filing and supporting the applications necessary to gain marketing approvals and expect to rely on third-party CROs to assist us in this process. Securing marketing approval requires the submission of extensive preclinical and clinical data and supporting information to regulatory authorities for each therapeutic indication to establish the product candidate’s safety and efficacy. Securing marketing approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the regulatory authorities. Our product candidates may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit commercial use. Regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies. In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent marketing approval of a product candidate. Changes in marketing approval policies during the
34
development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may also cause delays in or prevent the approval of an application.
If we experience delays in obtaining approval or if we fail to obtain approval of our product candidates, the commercial prospects for our product candidates may be harmed and our ability to generate revenues will be materially impaired.
We may not be able to benefit from available regulatory exclusivity periods if another company obtains regulatory approval for tipifarnib before we do.
As the composition of matter patents covering tipifarnib expire in 2016 in the United States and in countries in Europe, our commercial strategy for tipifarnib relies on obtaining patents covering methods of use of tipifarnib and on non-patent regulatory exclusivity. In the United States, a pharmaceutical manufacturer may obtain five years of non-patent exclusivity upon FDA approval of an NDA for a new chemical entity, or NCE, which is a drug that contains an active moiety that has not been approved by the FDA in any other NDA. An “active moiety” is defined as the molecule or ion responsible for the drug substance’s physiological or pharmacologic action. During the five year exclusivity period, the FDA cannot accept for filing any ANDA seeking approval of a generic version of that drug or any Section 505(b)(2) NDA for the same active moiety and that relies on the FDA’s findings regarding that drug, except that the FDA may accept an application for filing after four years if the follow-on applicant makes a paragraph IV certification. EB Pharma has licensed rights from Janssen to develop tipifarnib in certain indications outside of our exclusive field of oncology and Janssen may license rights to other non-oncology indications to other third parties. If EB Pharma or another third party obtains regulatory approval for tipifarnib in a non-oncology indication before we obtain regulatory approval in one of our oncology indications, the five year exclusivity period would commence on the date upon which EB Pharma or another third party obtains regulatory approval, and as a result, the period of regulatory exclusivity to which we may be entitled may be reduced or eliminated and the commercial prospects for tipifarnib would be harmed as a result.
Additionally, if EB Pharma or another third party obtains approval of tipifarnib for another indication outside of oncology, EB Pharma or the other third party may sell tipifarnib at a lower price, which could adversely affect the price at which we could sell tipifarnib for oncology indications.
We may not be able to obtain orphan drug exclusivity for the product candidates for which we seek it, which could limit the potential profitability of such product candidates.
Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as orphan drugs. Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals in the United States. Generally, if a product with an orphan drug designation subsequently receives the first marketing approval for the indication for which it receives the designation, then the product is entitled to a period of marketing exclusivity that precludes the applicable regulatory authority from approving another marketing application for the same drug for the same indication during the exclusivity period. The applicable period is seven years in the United States and ten years in Europe. The European exclusivity period can be reduced to six years if a drug no longer meets the criteria for Orphan Drug Designation or if the drug is sufficiently profitable so that market exclusivity is no longer justified. Orphan Drug Exclusivity may be lost if FDA or EMA determines that the request for designation was materially defective, if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition.
We expect that we may in the future pursue an orphan drug designation for at least some of our product candidates, including tipifarnib. However, obtaining an orphan drug designation can be difficult, and we may not be successful in doing so for any of our product candidates. Even if we were to obtain orphan drug exclusivity for a product candidate, that exclusivity may not effectively protect the product from the competition of different drugs for the same condition, which could be approved during the exclusivity period. Additionally, after an orphan drug is approved, the FDA could subsequently approve another application for the same drug for the same condition if the FDA concludes that the later drug is shown to be safer, more effective or makes a major contribution to patient care. The failure to obtain an orphan drug designation for any drug candidates we may develop for the treatment of rare cancers, and/or the inability to maintain that designation for the duration of the applicable exclusivity period, could reduce our ability to make sufficient sales of the applicable drug candidate to balance our expenses incurred to develop it, which would have a negative impact on our operational results and financial condition.
If we obtain an orphan drug designation and FDA approval of tipifarnib for an oncology indication, we would be entitled to seven years of marketing exclusivity for that orphan drug indication. However, if a competitor obtained approval of a generic form of tipifarnib for another indication, physicians would not be prevented from prescribing the generic drug for the orphan indication during the period of marketing exclusivity. Such prescribing practices could adversely affect the sales of tipifarnib for the orphan indication.
35
A Fast Track Designation by FDA, even if granted for any of our product candidates, may not lead to a faster development or regulatory review or approval process and does not increase the likelihood that our product candidates will receive marketing approval.
We do not currently have Fast Track Designation for any of our product candidates but intend to seek such designation. If a drug is intended for the treatment of a serious or life-threatening condition and the drug demonstrates the potential to address unmet medical needs for this condition, the drug sponsor may apply for FDA Fast Track Designation. FDA has broad discretion whether or not to grant this designation. Even if we believe a particular product candidate is eligible for this designation, we cannot assure you that FDA would decide to grant it. Even if we do receive Fast Track Designation, we may not experience a faster development process, review or approval compared to conventional FDA procedures. FDA may withdraw Fast Track Designation if it believes that the designation is no longer supported by data from our clinical development program. Many drugs that have received Fast Track Designation have failed to obtain drug approval.
A Breakthrough Therapy Designation by FDA, even if granted for any of our product candidates, may not lead to a faster development or regulatory review or approval process, and does not increase the likelihood that our product candidates will receive marketing approval.
We do not currently have Breakthrough Therapy Designation for any of our product candidates, but we may seek such designation. A Breakthrough Therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For drugs that have been designated as Breakthrough Therapies, interaction and communication between FDA and the sponsor can help to identify the most efficient path for development.
Designation as a Breakthrough Therapy is within the discretion of FDA. Accordingly, even if we believe, after completing early clinical trials, that one of our product candidates meets the criteria for designation as a Breakthrough Therapy, FDA may disagree and instead determine not to make such designation. In any event, the receipt of a Breakthrough Therapy designation for a product candidate may not result in a faster development process, review or approval compared to drugs considered for approval under conventional FDA procedures and does not assure ultimate approval by FDA. In addition, even if one or more of our product candidates qualify as Breakthrough Therapies, FDA may later decide that such product candidates no longer meet the conditions for qualification and rescind such designations.
Failure to obtain marketing approval in international jurisdictions would prevent our product candidates from being marketed abroad.
In order to market and sell our products in the European Union and many other jurisdictions, we or our third-party collaborators must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. The regulatory approval process outside the United States generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the United States, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. We or our third-party collaborators may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all. Approval by FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by FDA. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our products in any market.
Any product candidate for which we obtain marketing approval will be subject to extensive post-marketing regulatory requirements and could be subject to post-marketing restrictions or withdrawal from the market, and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our products, when and if any of them are approved.
Our product candidates and the activities associated with their development and commercialization, including their testing, manufacture, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, current good manufacturing practices, or cGMP, requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, including periodic inspections by FDA and other regulatory authority, requirements regarding the distribution of samples to physicians and recordkeeping.
36
FDA may also impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of the product. FDA closely regulates the post-approval marketing and promotion of drugs to ensure drugs are marketed only for the approved indications and in accordance with the provisions of the approved labeling. FDA imposes stringent restrictions on manufacturers’ communications regarding use of their products and if we promote our products beyond their approved indications, we may be subject to enforcement action for off-label promotion. Violations of the Federal Food, Drug, and Cosmetic Act relating to the promotion of prescription drugs may lead to investigations alleging violations of federal and state healthcare fraud and abuse laws, as well as state consumer protection laws.
In addition, later discovery of previously unknown adverse events or other problems with our products, manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may yield various results, including:
| • | restrictions on such products, manufacturers or manufacturing processes; |
| • | restrictions on the labeling or marketing of a product; |
| • | restrictions on product distribution or use; |
| • | requirements to conduct post-marketing studies or clinical trials; |
| • | warning or untitled letters; |
| • | withdrawal of the products from the market; |
| • | refusal to approve pending applications or supplements to approved applications that we submit; |
| • | recall of products; |
| • | fines, restitution or disgorgement of profits or revenues; |
| • | suspension or withdrawal of marketing approvals; |
| • | refusal to permit the import or export of our products; |
| • | product seizure; or |
| • | injunctions or the imposition of civil or criminal penalties. |
Non-compliance with European Union requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties. Similarly, failure to comply with the European Union’s requirements regarding the protection of personal information can also lead to significant penalties and sanctions.
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute any products for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:
| • | the federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid; |
| • | the federal False Claims Act imposes criminal and civil penalties, including civil whistleblower orqui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its |
37
implementing regulations, also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| • | federal law requires applicable manufacturers of covered drugs to report payments and other transfers of value to physicians and teaching hospitals, which includes data collection and reporting obligations; and |
| • | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers. |
Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion of products from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Recently enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidates for which we obtain marketing approval.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for physician-administered drugs. In addition, this legislation provided authority for limiting the number of drugs that will be covered in any therapeutic class. Cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for any approved products. While the MMA only applies to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the ACA, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms.
Among the provisions of the ACA of importance to our potential product candidates are the following:
| • | an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents; |
| • | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; |
| • | expansion of healthcare fraud and abuse laws, including the False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance; |
| • | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices; |
| • | extension of manufacturers’ Medicaid rebate liability; |
38
| • | expansion of eligibility criteria for Medicaid programs; |
| • | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
| • | new requirements to report financial arrangements with physicians and teaching hospitals; |
| • | a new requirement to annually report drug samples that manufacturers and distributors provide to physicians; and |
| • | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. These changes included aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding.
We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our products.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
Governments outside the United States tend to impose strict price controls, which may adversely affect our revenues, if any.
In some countries, particularly the countries of the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. If reimbursement for our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be harmed, possibly materially.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could harm our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our discovery, preclinical development or production efforts. Our failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
39
Risks Related to Our Dependence on Third Parties
We expect to rely on third-party contractors and organizations to conduct our clinical trials, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials.
We will rely on third party contractors, clinical data management organizations, independent contractors, medical institutions and clinical investigators to support our pre-clinical development activities and conduct our clinical trials, including our Phase 2 clinical trials of tipifarnib. These agreements may terminate for a variety of reasons, including a failure to perform by the third parties. If we are required to enter into alternative arrangements, our product development activities would be delayed.
We compete with many other companies, some of which may be our competitors, for the resources of these third parties. Large pharmaceutical companies often have significantly more extensive agreements and relationships with such third-party providers, and such third-party providers may prioritize the requirements of such large pharmaceutical companies over ours. The third parties on whom we rely may terminate their engagements with us at any time, which may cause delay in the development and commercialization of our product candidates. If any such third party terminates its engagement with us or fails to perform as agreed, we may be required to enter into alternative arrangements, which would result in significant cost and delay to our product development program. Moreover, our agreements with such third parties generally do not provide assurances regarding employee turnover and availability, which may cause interruptions in the research on our product candidates by such third parties.
Our reliance on these third parties to conduct our clinical trials will reduce our control over these activities but will not relieve us of our responsibilities. For example, we will remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, FDA and other regulatory authorities require us to comply with standards, commonly referred to as good clinical practices, or GCPs, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. We are also required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within specified timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Additionally, we expect to rely substantially on third-party data managers for our clinical trial data. There is no assurance that these third parties will not make errors in the design, management or retention of our data or data systems. There is no assurance that these third parties will pass FDA or other regulatory audits, which could delay or prevent regulatory approval.
If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
We will depend on third parties for the manufacture of our product candidates for preclinical and clinical testing and expect to continue to do so for commercialization. This reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or products at an acceptable cost and quality, which could delay, prevent or impair our development or commercialization efforts.
We do not own or operate facilities for the manufacture of our product candidates, and we do not have any manufacturing personnel. We currently have no plans to build our own clinical or commercial scale manufacturing capabilities. Janssen has provided us with its existing inventory of clinical supply of tipifarnib. Janssen also provided us with its existing inventory of crude drug substance and bulk key intermediate for manufacture of drug substance for tipifarnib. A portion of the clinical supply of tablets of tipifarnib provided by Janssen have a non-uniform surface where the film coating on the tablets has worn away to a varying degree. We believe this surface erosion is a cosmetic defect only and has no impact on patient safety or the effectiveness of the tablets, and an insignificant impact on taste masking, and that this clinical supply will support our planned Phase 2 clinical trials for tipifarnib. However there is no guarantee that clinical trial participants will accept all the tablets and that our existing clinical supply will be sufficient for our planned Phase 2 clinical trials or for any unanticipated extension of our planned Phase 2 clinical trials. If we are required to manufacture additional clinical supplies our planned Phase 2 clinical trials may be delayed. We rely, and expect to continue to rely, on third parties, for the manufacture of our other product candidates for preclinical and clinical testing. We will rely on third parties as well for commercial manufacture if any of our product candidates receive marketing approval. This reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or products or such quantities at an acceptable cost or quality, which could delay, prevent or impair our development or commercialization efforts. We also expect to rely on other third parties to store and distribute drug supplies for our clinical trials.
40
We also expect to rely on third-party manufacturers or third-party collaborators for the manufacture of commercial supply of any other product candidates for which our collaborators or we obtain marketing approval.
Any performance failure on the part of our existing or future manufacturers or distributors could delay clinical development or marketing approval of our product candidates or commercialization of our products, producing additional losses and depriving us of potential product revenue.
We may be unable to establish any agreements with third-party manufacturers or to do so on acceptable terms. Even if we are able to establish agreements with third-party manufacturers, reliance on third-party manufacturers entails additional risks, including:
| • | reliance on the third party for regulatory compliance and quality assurance; |
| • | the possible breach of the manufacturing agreement by the third party; |
| • | the possible misappropriation of our proprietary information, including our trade secrets and know-how; and |
| • | the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us. |
Third-party manufacturers may not be able to comply with cGMP regulations or similar regulatory requirements outside the United States. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our products.
Our product candidates and any products that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us.
Our current and anticipated future dependence upon others for the manufacture of our product candidates or products may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis.
Risks Related to the Commercialization of Our Product Candidates
Even if any of our product candidates receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
If any of our product candidates receives marketing approval, it may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. For example, current cancer treatments like chemotherapy and radiation therapy are well established in the medical community, and doctors may continue to rely on these treatments to the exclusion of our product candidates. In addition, physicians, patients and third-party payors may prefer other novel products to ours. If our product candidates do not achieve an adequate level of acceptance, we may not generate significant product revenues and we may not become profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, including:
| • | the efficacy and safety and potential advantages and disadvantages compared to alternative treatments; |
| • | our ability to offer our products for sale at competitive prices; |
| • | the convenience and ease of administration compared to alternative treatments; |
| • | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
| • | the strength of our marketing and distribution support; |
| • | the availability of third-party coverage and adequate reimbursement, including patient cost-sharing programs such as copays and deductibles; |
| • | our ability to develop or partner with third-party collaborators to develop companion diagnostics; |
| • | the prevalence and severity of any side effects; and |
| • | any restrictions on the use of our products together with other medications. |
41
We currently have no marketing and sales force. If we are unable to establish effective sales or marketing capabilities or enter into agreements with third parties to sell or market our product candidates, we may not be able to effectively sell or market our product candidates, if approved, or generate product revenues.
We currently do not have a marketing or sales team for the marketing, sales and distribution of any of our product candidates that are able to obtain regulatory approval. In order to commercialize any product candidates, we must build on a territory-by-territory basis marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. If our product candidates receive regulatory approval, we intend to establish an internal sales or marketing team with technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be expensive and time consuming and will require significant attention of our executive officers to manage. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of any of our products that we obtain approval to market. With respect to the commercialization of all or certain of our product candidates, we may choose to collaborate, either globally or on a territory-by-territory basis, with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. If we are unable to enter into such arrangements when needed on acceptable terms or at all, we may not be able to successfully commercialize any of our product candidates that receive regulatory approval or any such commercialization may experience delays or limitations. If we are not successful in commercializing our product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
We face substantial competition, which may result in others discovering, developing or commercializing competing products before or more successfully than we do.
The development and commercialization of new drug products is highly competitive. We face competition with respect to our current product candidates, and will face competition with respect to any product candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. There are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of products for the treatment of the disease indications for which we are developing our product candidates. Some of these competitive products and therapies are based on scientific approaches that are the same as or similar to our approach, and others are based on entirely different approaches. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Specifically, there are a large number of companies developing or marketing treatments for cancer, including many major pharmaceutical and biotechnology companies, which may directly compete with tipifarnib, KO-947 and any future product candidates. See “Description of the Business of Kura Oncology, Inc.—Competition.”
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market and or slow our regulatory approval. In addition, our ability to compete may be affected in many cases by insurers or other third-party payors seeking to encourage the use of generic products. Generic products are currently on the market for the indications that we are pursuing, and additional products are expected to become available on a generic basis over the coming years. If our product candidates achieve marketing approval, we expect that they will be priced at a significant premium over competitive generic products.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
The insurance coverage and reimbursement status of newly-approved products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for new or current products could limit our ability to market those products and decrease our ability to generate revenue.
42
The availability and extent of reimbursement by governmental and private payors is essential for most patients to be able to afford expensive treatments. Sales of our product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, the principal decisions about reimbursement for new medicines are typically made by the Centers for Medicare & Medicaid Services, or CMS, an agency within the U.S. Department of Health and Human Services, as CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare. Private payors tend to follow CMS to a substantial degree. It is difficult to predict what CMS will decide with respect to reimbursement for fundamentally novel products such as ours, as there is no body of established practices and precedents for these new products. Reimbursement agencies in Europe may be more conservative than CMS. For example, a number of cancer drugs have been approved for reimbursement in the United States and have not been approved for reimbursement in certain European countries. Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe, Canada, and other countries has and will continue to put pressure on the pricing and usage of our product candidates. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. In general, the prices of medicines under such systems are substantially lower than in the United States. Other countries allow companies to fix their own prices for medicines, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our products may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenues and profits.
Moreover, increasing efforts by governmental and third-party payors, in the United States and abroad, to cap or reduce healthcare costs may cause such organizations to limit both coverage and level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of any of our product candidates, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products into the healthcare market.
In addition to CMS and private payors, professional organizations such as the National Comprehensive Cancer Network and the American Society of Clinical Oncology can influence decisions about reimbursement for new medicines by determining standards for care. In addition, many private payors contract with commercial vendors who sell software that provide guidelines that attempt to limit utilization of, and therefore reimbursement for, certain products deemed to provide limited benefit to existing alternatives. Such organizations may set guidelines that limit reimbursement or utilization of our products.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for any product candidates or products that we may develop; |
| • | injury to our reputation and significant negative media attention; |
| • | withdrawal of clinical trial participants; |
| • | significant costs to defend the related litigation; |
| • | substantial monetary awards to trial participants or patients; |
| • | loss of revenue; |
| • | reduced resources of our management to pursue our business strategy; and |
43
| • | the inability to commercialize any products that we may develop. |
Our current product liability insurance coverage may not be adequate to cover all liabilities that we may incur. We may need to increase our insurance coverage as we expand our clinical trials or if we commence commercialization of our product candidates. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
Risks Related to Our Intellectual Property
If we are unable to obtain and maintain intellectual property protection for our technology and products, or if the scope of the intellectual property protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired.
We intend to rely upon a combination of regulatory exclusivity periods, patents, trade secret protection, confidentiality agreements, and license agreements to protect the intellectual property related to our current product candidates and development programs. If the breadth or strength of protection provided by any patents, patent applications or future patents we may own, license, or pursue with respect to any of our current or future product candidates or products is threatened, it could threaten our ability to commercialize any of our current or future product candidates or products. Further, if we encounter delays in our development efforts, the period of time during which we could market any of our current or future product candidates or products under any patent protection we obtain would be reduced. Given the amount of time required for the development, testing and regulatory review of new product candidates or products, patents protecting such candidates might expire before or shortly after such product candidates or products are commercialized.
Our patent rights may not protect our patent protected products and product candidates if competitors devise ways of making products that compete with us without legally infringing our patent rights. For example, our patent rights in tipifarnib are limited in ways that affect our ability to exclude third parties from competing against us. In particular, the patent term for the composition of matter patents covering the active pharmaceutical ingredient, or API, of tipifarnib expire in 2016 in the United States, countries in Europe and other jurisdictions. Composition of matter patents on APIs are generally considered to be the strongest form of intellectual property protection because such patents provide protection without regard to any particular method of use or manufacture or formulation of the API used. Patent term extension may be available in the US to account for regulatory delays in obtaining human marketing approval for tipifarnib however, only one patent may be extended per marketed compound. Under our license agreement with Janssen, we and Janssen agree to cooperate in obtaining available patent term extensions. We and Janssen may not reach agreement and no patent term extension may be obtained. Additionally, the applicable authorities, including the USPTO and the FDA, and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to patents, or may grant more limited extensions than requested. If this occurs, our competitors who obtain the requisite regulatory approval can offer products with the same API as tipifarnib so long as the competitors do not infringe any method of use or formulations patents that we may hold. Competitors may take advantage of our investment in development and trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
We expect that following expiration of composition of matter patents and any regulatory exclusivity we are able to obtain, competitors may manufacture and sell generic versions of tipifarnib, at a lower price, which would reduce tipifarnib’s revenues. In certain jurisdictions, legislation mandates generic substitution for brand name drugs.
We depend on our licensors to prosecute and maintain patents and patent applications that are material to our business. Any failure by our licensors to effectively protect these intellectual property rights could adversely impact our business and operations.
We have licensed patent rights from third parties for some of our development programs, including tipifarnib from Janssen and compounds in our Menin-MLL program from the University of Michigan. As a licensee of third parties, we rely on these third parties to file and prosecute patent applications and maintain patents and otherwise protect the licensed intellectual property under some of our license agreements. We have not had and do not have primary control over these activities for certain of our patents or patent applications and other intellectual property rights. We cannot be certain that such activities by third parties have been or will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents or other intellectual property rights. Pursuant to the terms of the license agreements with some of our licensors, the licensors may have the right to control enforcement of our licensed patents or defense of any claims asserting the invalidity of these patents and even if we are permitted to pursue such enforcement or defense, we will require the cooperation of our licensors. We cannot be certain that our licensors will allocate sufficient resources or prioritize their or our enforcement of such patents or defense of such claims to protect our interests in the licensed patents. Even if we are not a party
44
to these legal actions, an adverse outcome could harm our business because it might prevent us from continuing to license intellectual property that we may need to operate our business.
With respect to the patent portfolio for tipifarnib, which is in-licensed from Janssen, Janssen maintains rights to prosecute and maintain patents and patent applications within the portfolio as well as to assert such patents against infringers within and outside the scope of our license, and to defend such patents against claims of invalidity and unenforceability. Although we have rights to consult with Janssen on actions taken as well as back-up rights of prosecution and enforcement, rights to tipifarnib granted to another licensee, such as EB Pharma, could potentially influence Janssen’s interests in the exercise of its prosecution, maintenance and enforcement rights in a manner that may favor the interests of such other licensee as compared with us.
If we breach any of the agreements under which we license from third parties the commercialization rights to our product candidates, we could lose license rights that are important to our business and our operations could be materially harmed.
We have in-licensed from Janssen the use, development and commercialization rights in oncology indications for our lead product candidate, tipifarnib. We have also in-licensed rights to potential product candidates in other programs in our pipeline. As a result, our current business plans are dependent upon our satisfaction of certain conditions to the maintenance of the Janssen agreement and the rights we license under it and our other in-license agreements. The Janssen license agreement provides that we are subject to diligence obligations relating to the commercialization and development of tipifarnib, milestone payments, royalty payments and other obligations. If we fail to comply with any of the conditions or obligations or otherwise breach the terms of our license agreement with Janssen, or any of our other license agreements or license agreements we may enter into on which our business or product candidates are dependent, Janssen or other licensors may have the right to terminate the applicable agreement in whole or in part and thereby extinguish our rights to the licensed technology and intellectual property and/or any rights we have acquired to develop and commercialize certain product candidates, including, with respect to our license agreement with Janssen, tipifarnib. The loss of the rights licensed to us under our license agreement with Janssen, or our other license agreements or any future license agreement that we may enter granting us rights on which our business or product candidates are dependent, would eliminate our ability to further develop the applicable product candidates and would materially harm our business, prospects, financial condition and results of operations.
The patent applications of pharmaceutical and biotechnology companies involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our patent position.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. For example, India and China do not allow patents for methods of treating the human body. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued which protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted and may also affect patent litigation. The U.S. Patent and Trademark Office, or U.S. PTO, recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition.
Moreover, we may be subject to a third-party preissuance submission of prior art to the U.S. PTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the
45
scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.
Even if our owned and licensed patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our owned or licensed patents by developing similar or alternative technologies or products in a non-infringing manner.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and licensed patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the U.S. PTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and/or applications. We have systems in place to remind us to pay these fees, and we employ an outside firm and rely on our outside counsel to pay these fees due to non-U.S. patent agencies. The U.S. PTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
Because competition in our industry is intense, competitors may infringe or otherwise violate our issued patents, patents of our licensors or other intellectual property. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their patents. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, in whole or in part, construe the patent’s claims narrowly or refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly. We may also elect to enter into license agreements in order to settle patent infringement claims or to resolve disputes prior to litigation, and any such license agreements may require us to pay royalties and other fees that could be significant. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability, and the ability of our collaborators, to develop, manufacture, market and sell our product candidates and use our proprietary technologies without infringing the proprietary rights of third parties. There is considerable intellectual property litigation in the biotechnology and pharmaceutical industries. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our products and technology, including interference or derivation proceedings before the U.S. PTO. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future.
46
If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our products and technology. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
We may not be successful in obtaining or maintaining necessary rights for our development pipeline through acquisitions and in-licenses.
Presently we have rights to intellectual property to develop tipifarnib in the field of oncology, including patents and patent applications we exclusively licensed from Janssen, as well as exclusive worldwide licenses for all therapeutic indications for other potential product candidates currently in our pipeline, including in our Menin-MLL program. Because our programs may involve additional product candidates that may require the use of proprietary rights held by third parties, the growth of our business may depend in part on our ability to acquire, in-license or use these proprietary rights. Additionally, a companion diagnostic may require that we or a third-party collaborator developing the diagnostic acquire use or proprietary rights held by third parties. We may be unable to acquire or in-license any compositions, methods of use, or other third-party intellectual property rights from third parties that we identify. The licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies are also pursuing strategies to license or acquire third-party intellectual property rights that we may consider attractive. These established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities.
For example, we may collaborate with U.S. and foreign academic institutions to accelerate our discovery and preclinical development work under written agreements with these institutions. Typically, these institutions provide us with an option to negotiate a license to any of the institution’s rights in technology resulting from the collaboration. Regardless of such right of first negotiation for intellectual property, we may be unable to negotiate a license within the specified time frame or under terms that are acceptable to us. If we are unable to do so, the institution may offer the intellectual property rights to other parties, potentially blocking our ability to pursue our program.
In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment. If we are unable to successfully obtain rights to required third-party intellectual property rights, our business, financial condition and prospects for growth could suffer.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and product candidates, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We seek to protect our confidential proprietary information, in part, by entering into confidentiality and invention or patent assignment agreements with our employees and consultants, however, we cannot be certain that such agreements have been entered into with all relevant parties. Moreover, to the extent we enter into such agreements, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
Risks Related to Employee Matters, Managing Growth and Macroeconomic Conditions
We currently have a limited number of employees, are highly dependent on our Chief Executive Officer and our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel.
47
We are an early-stage clinical development company with a limited operating history, and, as of the date of this report, we have only 15 employees. We are highly dependent on the expertise of Troy E. Wilson, our President and Chief Executive Officer, Antonio Gualberto, our Chief Medical Officer, Yi Liu, our Chief Scientific Officer, and Pindga Ren, our Senior Vice President, Chemistry and Pharmaceutical Sciences, as well as the other principal members of our management, scientific and clinical team. Although we have entered into employment letter agreements with our executive officers, each of them may terminate their employment with us at any time. We do not maintain “key person” insurance for any of our executives or other employees. Additionally, Dr. Wilson currently also serves as President and Chief Executive Officer of Avidity Nanomedicines, LLC. As a result, Dr. Wilson is not able to devote all of his business time and attention to our business. Conflicts may arise in the future if there are competing demands on Dr. Wilson’s time and attention and our business may be harmed as a result.
Recruiting and retaining qualified scientific, clinical, manufacturing and sales and marketing personnel will also be critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our discovery and preclinical development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
We expect to expand our development and regulatory capabilities and potentially implement sales, marketing and distribution capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of regulatory affairs and commercial, if any of our product candidates receives marketing approval, sales, marketing and distribution. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources and the limited experience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. The recent global financial crisis caused extreme volatility and disruptions in the capital and credit markets. A severe or prolonged economic downturn, such as the recent global financial crisis, could result in a variety of risks to our business, including our ability to raise additional capital when needed on acceptable terms, if at all. This is particularly true in Europe, which is undergoing a continued severe economic crisis. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our CROs, collaborators and third-parties on whom we rely are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and we may incur substantial costs to attempt to recover or reproduce the data. If any disruption or security breach resulted in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and/or the further development of our product candidates could be delayed.
48
Our operations are vulnerable to interruption by natural disasters, power loss, terrorist activity and other events beyond our control, the occurrence of which could materially harm our business.
Businesses located in California have, in the past, been subject to electrical blackouts as a result of a shortage of available electrical power, and any future blackouts could disrupt our operations. We are vulnerable to a major earthquake, wildfire and other natural disasters, and we have not undertaken a systematic analysis of the potential consequences to our business as a result of any such natural disaster and do not have an applicable recovery plan in place. We do not carry any business interruption insurance that would compensate us for actual losses from interruption of our business that may occur, and any losses or damages incurred by us could cause our business to materially suffer.
Risks Related to Ownership of our Common Stock
There is currently no market for our common stock and there can be no assurance that any market will ever develop. You may therefore be unable to re-sell shares of our common stock at times and prices that you believe are appropriate.
Our common stock is not listed on a national securities exchange, an over-the-counter market or any other exchange. Therefore, there is no trading market, active or otherwise, for our common stock and our common stock may never be included for trading on any stock exchange, automated quotation system or any over-the-counter market. Accordingly, our common stock is highly illiquid and you will likely experience difficulty in re-selling such shares at times and prices that you may desire.
Our common stock may not be eligible for listing or quotation on any securities exchange.
We do not currently meet the initial listing standards of any national securities exchange and our common stock is not quoted for sale on any over-the-counter trading system. We cannot assure you that we will be able to meet the initial listing standards of any national securities exchange, or, if we do meet such initial listing standards, that we will be able to maintain any such listing. Further, the national securities exchanges have adopted so-called “seasoning” rules that require that we meet certain requirements, including prescribed periods of time trading over-the-counter and minimum filings of periodic reports with the SEC, before we are eligible to apply for listing on such national securities exchanges. We intend to contact an authorized market maker for an over-the-counter quotation system for sponsorship of our common stock, but we cannot guarantee that such sponsorship will be approved and our common stock listed and quoted for sale. Even if our common stock is quoted for sale on an over-the-counter quotation system, buyers may be insufficient in numbers to allow for a robust market and it may prove impossible to sell your shares. In addition, an investor may find it difficult to obtain accurate quotations as to the market value of our common stock. In addition, if we fail to meet the criteria set forth in SEC regulations, various requirements would be imposed by law on broker-dealers who sell our securities to persons other than established customers and accredited investors. Consequently, such regulations may deter broker-dealers from recommending or selling our common stock, which may further affect its liquidity. This would also make it more difficult for us to raise additional capital. Further, an unestablished trading market for our common stock may also impair our ability to raise capital by selling additional equity in the future, and may impair our ability to enter into strategic partnerships or acquire companies or products by using shares of our common stock as consideration.
The price of our common stock may be volatile and may be influenced by numerous factors, some of which are beyond our control.
If a market for our common stock develops, its market price could fluctuate substantially due to a variety of factors, some of which may be beyond our control. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this Current Report on Form 8-K, these factors include:
| • | the product candidates we seek to pursue, and our ability to obtain rights to develop, commercialize and market those product candidates; |
| • | our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial; |
| • | actual or anticipated adverse results or delays in our clinical trials; |
| • | our failure to commercialize our product candidates, if approved; |
| • | unanticipated serious safety concerns related to the use of any of our product candidates; |
| • | adverse regulatory decisions; |
49
| • | additions or departures of key scientific or management personnel; |
| • | changes in laws or regulations applicable to our product candidates, including without limitation clinical trial requirements for approvals; |
| • | disputes or other developments relating to patents and other proprietary rights and our ability to obtain patent protection for our product candidates; |
| • | our dependence on third parties, including CROs as well as our potential partners that produce companion diagnostic products; |
| • | failure to meet or exceed any financial guidance or expectations regarding development milestones that we may provide to the public; |
| • | actual or anticipated variations in quarterly operating results, liquidity or other indicators of our financial condition; |
| • | failure to meet or exceed the estimates and projections of the investment community; |
| • | overall performance of the equity markets and other factors that may be unrelated to our operating performance or the operating performance of our competitors, including changes in market valuations of similar companies; |
| • | conditions or trends in the biotechnology and biopharmaceutical industries; |
| • | introduction of new products offered by us or our competitors; |
| • | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
| • | our ability to maintain an adequate rate of growth and manage such growth; |
| • | issuances of debt or equity securities; |
| • | sales of our common stock by us or our stockholders in the future, or the perception that such sales could occur; |
| • | trading volume of our common stock; |
| • | ineffectiveness of our internal control over financial reporting or disclosure controls and procedures; |
| • | general political and economic conditions; |
| • | effects of natural or man-made catastrophic events; and |
| • | other events or factors, many of which are beyond our control. |
In addition, the stock market in general, and the stocks of small-cap biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. These events may also lead to securities litigation, which can be expensive and time-consuming to defend, regardless of the merit or outcome. The realization of any of the above risks or any of a broad range of other risks, including those described in these “Risk Factors,” could have a dramatic and material adverse impact on the market price of our common stock.
The designation of our common stock as a “penny stock” would limit the liquidity of our common stock.
Our common stock may be deemed a “penny stock” (as that term is defined under Rule 3a51-1 of the Exchange Act) in any market that may develop in the future. Generally, a “penny stock” is a common stock that is not listed on a securities exchange and trades for less than $5.00 per share. Prices often are not available to buyers and sellers and the market may be very limited. Penny stocks in start-up companies are among the riskiest equity investments. Broker-dealers who sell penny stocks must provide purchasers of these stocks with a standardized risk-disclosure document prepared by the SEC. The document provides information about penny stocks and the nature and level of risks involved in investing in the penny stock
50
market. A broker must also provide purchasers with bid and offer quotations and information regarding broker and salesperson compensation and make a written determination that the penny stock is a suitable investment for the purchaser and obtain the purchaser’s written agreement to the purchase. Many brokers choose not to participate in penny stock transactions. Because of the penny stock rules, there may be less trading activity in penny stocks in any market that develops for our common stock in the future and stockholders are likely to have difficulty selling their shares.
FINRA sales practice requirements may limit a stockholder’s ability to buy and sell our stock.
The Financial Industry Regulatory Authority, or FINRA, has adopted rules requiring that, in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending speculative or low-priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA has indicated its belief that there is a high probability that speculative or low-priced securities will not be suitable for at least some customers. If these FINRA requirements are applicable to us or our securities, they may make it more difficult for broker-dealers to recommend that at least some of their customers buy our common stock, which may limit the ability of our stockholders to buy and sell our common stock and could have an adverse effect on the market for and price of our common stock.
The shares of common stock issued in the Merger are “restricted securities” and, as such, may not be sold except in limited circumstances.
None of the shares of common stock that we issued in the Merger, which constitute all of our outstanding shares of common stock as of the date of this Current Report on Form 8-K, have been registered under the Securities Act or registered or qualified under any state securities laws. The shares of common stock that issued in the Merger were issued pursuant to exemptions contained in and under those laws. Accordingly, such shares of common stock are “restricted securities” as defined in Rule 144 under the Securities Act and must, therefore, be held indefinitely unless registered under applicable federal and state securities laws, or an exemption is available from the registration requirements of those laws. The shares in electronic book-entry form representing the shares of common stock issued in the Merger reflect their restricted status.
We have agreed to register the shares of common stock issued in the Merger. There can be no assurance, however, that the SEC will declare the registration statement effective, thereby enabling the shares of common stock issued in the Merger to be freely tradable. In addition, Rule 144 under the Securities Act, which permits the resale, subject to various terms and conditions, of limited amounts of restricted securities after they have been held for six months, will not immediately apply to our common stock because we were at one time designated as a “shell company” under SEC regulations. Pursuant to Rule 144(i), securities issued by a current or former shell company that otherwise meet the holding period and other requirements of Rule 144 nevertheless cannot be sold in reliance on Rule 144 until one year after the date on which the issuer filed current “Form 10 information” (as defined in Rule 144(i)) with the SEC reflecting that it ceased being a shell company and, provided that at the time of a proposed sale pursuant to Rule 144, the issuer has satisfied certain reporting requirements under the Exchange Act. We believe this requirement to file Form 10 information has been satisfied by the filing of this Current Report on Form 8-K. Because, as a former shell company, the reporting requirements of Rule 144(i) will apply regardless of holding period, the restrictive legends on the shares in electronic book-entry form representing the shares of common stock issued in the Merger cannot be removed except in connection with an actual sale that is subject to an effective registration statement under, or an applicable exemption from the registration requirements of the Securities Act.
If we are unable to register in a timely manner the shares of common stock that we issued to stockholders in the Merger, then the ability to re-sell shares of our common stock will be delayed.
We expect to prepare and file a registration statement with the SEC registering the resale of the shares of our common stock issued in connection with the Merger. There are many reasons, including some over which we have little or no control, which could keep the registration statement from being declared effective by the SEC, including delays resulting from the SEC review process and comments raised by the SEC during that process. Accordingly, in the event that the registration statement is not declared effective within these timeframes, the shares of common stock proposed to be covered by such registration statement will not be eligible for resale until the registration statement is effective or an exemption from registration, such as Rule 144, becomes available. In addition, we have agreed to pay damages to the investors in the Private Placement if we do not satisfy certain deadlines and requirements in connection with the registration statement, as specified in the Registration Rights Agreement.
Because we became a reporting company under the Exchange Act by means other than a traditional underwritten initial public offering, we may not be able to attract the attention of research analysts at major brokerage firms.
51
Because we did not become a reporting company by conducting an underwritten initial public offering of our common stock, and because we will not be listed on a national securities exchange, security analysts of brokerage firms may not provide coverage of our company. In addition, investment banks may be less likely to agree to underwrite secondary offerings on our behalf than they might if we became a public reporting company by means of an underwritten initial public offering, because they may be less familiar with our company as a result of more limited coverage by analysts and the media, and because we became public at an early stage in our development. The failure to receive research coverage or support in the market for our shares will have an adverse effect on our ability to develop a liquid market for our common stock.
Because the Merger was a reverse merger, the registration statement we expect to file with respect to the shares of common stock received by stockholders in the Merger might be subject to heightened scrutiny by the SEC, and we may not be able to attract the attention of major brokerage firms.
Additional risks may exist as a result of our becoming a public reporting company through a “reverse merger.” Certain SEC rules are more restrictive when applied to reverse merger companies, such as the ability of stockholders to re-sell their shares of common stock pursuant to Rule 144, and the SEC may subject the registration statement we expect to file with respect to the shares of our common stock received by stockholders in the Merger to heightened scrutiny. In addition, securities analysts of major brokerage firms may not provide coverage of our capital stock or business. Because we became a public reporting operating company through a reverse merger, there is no incentive for brokerage firms to recommend the purchase of our common stock. We cannot assure you that brokerage firms will want to provide analyst coverage of our capital stock or business in the future.
The resale of shares covered by a registration statement could adversely affect the market price of our common stock in the public market, should one develop, which result would in turn negatively affect our ability to raise additional equity capital.
The sale, or availability for sale, of our common stock in the public market may adversely affect the prevailing market price of our common stock and may impair our ability to raise additional capital by selling equity or equity-linked securities. We expect to prepare and file a registration statement with the SEC registering the resale of the shares of our common stock issued in connection with the Merger. Once effective, the registration statement will permit the resale of these shares at any time. The resale of a substantial number of shares of our common stock in the public market could adversely affect the market price for our common stock and make it more difficult for you to sell shares of our common stock at times and prices that you feel are appropriate. Furthermore, we expect that, because there will be a large number of shares registered pursuant to a registration statement, selling stockholders will continue to offer shares covered by such registration statement for a significant period of time, the precise duration of which cannot be predicted. Accordingly, the adverse market and price pressures resulting from an offering pursuant to a registration statement may continue for an extended period of time and continued negative pressure on the market price of our common stock could have a material adverse effect on our ability to raise additional equity capital.
We will incur increased costs associated with, and our management will need to devote substantial time and effort to, compliance with public company reporting and other requirements.
As a public company, and particularly if and after we cease to be an “emerging growth company” or a “smaller reporting company,” we will incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, the rules and regulations of the SEC and any national securities exchange to which we may be subject in the future impose numerous requirements on public companies, including requirements relating to our corporate governance practices, with which we will need to comply. Further, we are required to, among other things, file annual, quarterly and current reports with respect to our business and operating results. Based on currently available information and assumptions, we estimate that we will incur approximately $450,000 in expenses on an annual basis as a direct result of these requirements, and we expect that the amount of such expenses may be increased by an additional $200,000 during our first year operating as a public reporting company. Our management and other personnel will need to devote substantial time to gaining expertise regarding operations as a public company and compliance with applicable laws and regulations, and our efforts and initiatives to comply with those requirements could be expensive.
Kura was not subject to requirements to establish, and did not establish, internal control over financial reporting and disclosure controls and procedures prior to the Merger. Our management team and board of directors will need to devote significant efforts to maintaining adequate and effective disclosure controls and procedures and internal control over financial reporting in order to comply with applicable regulations, which may include hiring additional legal, financial reporting and other finance and accounting staff and engaging consultants to assist in designing and implementing such procedures. Additionally, any of our efforts to improve our internal controls and design, implement and maintain an adequate system of disclosure controls may not be successful and will require that we expend significant cash and other resources.
52
If we fail to maintain proper and effective internal controls, our ability to produce accurate and timely financial statements could be impaired, which could harm our operating results, our ability to operate our business and investors’ views of us.
We will be required to comply with Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act. Section 404 of the Sarbanes-Oxley Act requires public companies to conduct an annual review and evaluation of their internal controls and attestations of the effectiveness of internal controls by independent auditors. Ensuring that we have adequate internal financial and accounting controls and procedures in place so that we can produce accurate financial statements on a timely basis is a costly and time-consuming effort that will need to be evaluated frequently. Our failure to maintain the effectiveness of our internal controls in accordance with the requirements of the Sarbanes-Oxley Act could have a material adverse effect on our business. We could lose investor confidence in the accuracy and completeness of our financial reports, which could have an adverse effect on the price of our common stock. In addition, if our efforts to comply with new or changed laws, regulations, and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
We are an emerging growth company and a smaller reporting company, which will allow us to take advantage of certain reduced disclosure obligations as a public reporting company that may make our common stock less attractive to investors.
We are an “emerging growth company” under the JOBS Act and a “smaller reporting company” as defined in applicable rules under the Exchange Act. As an emerging growth company and a smaller reporting company, we are eligible to take advantage of certain extended accounting standards and exemptions from various reporting requirements that are not available to public reporting companies that do not qualify for those classifications. For instance, are exempt from any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and financial statements, commonly known as an “auditor discussion and analysis;” we are not required to hold a nonbinding advisory stockholder vote on executive compensation or any golden parachute payments not previously approved by stockholders; we are not required to comply with the requirement of auditor attestation of management’s assessment of internal control over financial reporting, which is required for some other public reporting companies by Section 404 of the Sarbanes-Oxley Act of 2002; we are eligible for reduced disclosure obligations regarding executive compensation in our periodic and annual reports; and we are eligible for reduced financial statement disclosure in any registration statements under the Securities Act or reports under the Exchange Act that we may file. For as long as we continue to be an emerging growth company and/or a smaller reporting company, which we anticipate will be for the foreseeable future, we expect that we will take advantage of the reduced disclosure obligations available to us as a result of those respective classifications. As a result, our publicly available disclosure may not be as robust or comprehensive as that of other public reporting companies that do not qualify for those classifications.
Further, as an emerging growth company, we can elect to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to take advantage of this extended transition period and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. As a result, changes in rules of U.S. generally accepted accounting principles or their interpretation, the adoption of new guidance or the application of existing guidance to changes in our business could significantly affect our financial position and results of operations.
We are not subject to compliance with rules requiring the adoption of certain corporate governance measures and as a result our stockholders have limited protections against interested director transactions, conflicts of interest and similar matters.
The Sarbanes-Oxley Act of 2002, as well as rule changes enacted by the SEC, the New York and American Stock Exchanges and the NASDAQ Stock Market, as a result of Sarbanes-Oxley, require the implementation of various measures relating to corporate governance. These measures are designed to enhance the integrity of corporate management and the securities markets and apply to securities that are listed on those exchanges or the NASDAQ Stock Market. Because we are not presently required to comply with many of the corporate governance provisions we have not yet adopted these measures.
We do not currently have independent audit or compensation committees. As a result, our directors have the ability, among other things, to determine their own level of compensation. Until we comply with such corporate governance measures, regardless of whether such compliance is required, the absence of such standards of corporate governance may leave our stockholders without protections against interested director transactions, conflicts of interest and similar matters.
53
We have broad discretion in the use of our cash and may not use our cash effectively, which could adversely affect our results of operations.
Our management has broad discretion in the application of our cash resources. Because of the number and variability of factors that will determine our use of our cash resources, our management might not apply our cash in ways that ultimately increase the value of our common stock. The failure by our management to apply our cash effectively could harm our business. Pending their use, we may invest our cash in short-term, investment-grade, interest-bearing securities. These investments may not yield a favorable return to our stockholders. If we do not invest or apply our cash in ways that enhance stockholder value, we may fail to achieve expected financial results, which could cause our stock price to decline.
Management and our board of directors beneficially own a substantial amount of our outstanding equity securities and will be able to exert substantial control over us.
Our executive officers and directors beneficially own a substantial percentage of our outstanding equity securities. Accordingly, if they act as a group, our executive officers and directors will be able to significantly influence all business decisions, including with respect to such matters as amendments to our charter, other fundamental corporate transactions such as mergers, asset sales and the sale of us, and otherwise will be able to significantly influence our business and affairs.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans or otherwise, could result in dilution to the percentage ownership of our stockholders and could cause our stock price to fall.
Even after giving effect to the funds raised by Kura in the Private Placement, we expect that significant additional capital will be needed in the future to continue our planned operations. To raise capital, we may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors in a prior transaction may be materially diluted by subsequent sales. Additionally, any such sales may result in material dilution to our existing stockholders, and new investors could gain rights, preferences and privileges senior to those of holders of our common stock. Further, any future sales of our common stock by us or resales of our common stock by our existing stockholders could cause the market price of our common stock to decline.
Pursuant to our 2014 plan, we are authorized to grant equity awards consisting of shares of our common stock to our employees, directors and consultants. Any future grants of options, warrants or other securities exercisable or convertible into our common stock, or the exercise or conversion of such shares, and any sales of such shares in the market, could have an adverse effect on the market price of our common stock.
We do not intend to pay cash dividends on our capital stock in the foreseeable future.
We have never declared or paid any dividends on our common stock and do not anticipate paying any dividends in the foreseeable future. Any future payment of cash dividends in the future would depend on our financial condition, contractual restrictions, solvency tests imposed by applicable corporate laws, results of operations, anticipated cash requirements and other factors and will be at the discretion of the our board of directors. Our stockholders should not expect that we will ever pay cash or other dividends on our outstanding capital stock.
MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion of the financial condition and results of operations of Kura Oncology, Inc. and its wholly-owned subsidiary should be read in conjunction with the financial statements and the notes to those statements filed as Exhibits 99.1 and 99.2 to this Current Report on Form 8-K. Some of the information contained in this discussion and analysis or set forth elsewhere in this report, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties. You should read the “Risk Factors” section of this report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
Effective as of March 6, 2015, we consummated the Merger and changed our name from “Zeta Acquisition Corp. III” to “Kura Oncology, Inc.”
54
We are a clinical stage biopharmaceutical company discovering and developing personalized therapeutics for the treatment of solid tumors and blood cancers. We focus on the development of small molecule drug candidates that target cell signaling pathways that are important to driving the progression of certain cancers. We aim to employ molecular diagnostics to identify patients with cancers who are likely to benefit from our targeted drug candidates.
Our lead drug candidate, tipifarnib, is a farnesyl transferase inhibitor that we intend to evaluate in Phase 2 clinical studies as a treatment for certain solid tumors, including thyroid, head and neck, urothelial, and salivary cancers, with mutations in the HRAS oncogene. Collectively, the annual incidence of these cancers containing HRAS mutations is approximately 8,000 patients per year in the United States, and, in general, patients with these cancers have poor prognoses and limited options for treatment. We are also evaluating tipifarnib as a potential treatment for patients with peripheral T-cell lymphoma, which has an annual incidence of approximately 7,000-10,000 patients in the United States.
We are advancing a set of compounds that inhibit the activity of extracellular-signal-regulated kinases 1 and 2 (ERK1/2), including our lead candidate KO-947 as well as backup compounds, as a potential treatment for patients with tumors that have mutations in or other dysregulation of the mitogen-activated protein kinase (MAPK) signaling pathway, including mutations in the proteins KRAS, BRAF and NRAS. The cancer indications that frequently harbor mutations in the MAPK pathway include lung cancer, colorectal cancer, pancreatic cancer, and melanoma. According to the American Cancer Society, there are estimated to be over 43,000 cases of pancreatic cancer, 125,000 cases of colorectal cancer and over 188,000 cases of non-small cell lung cancer, or NSCLC, diagnosed each year in the United States. We believe this corresponds to approximately 42,000 cases of KRAS mutant pancreatic cancer, 55,000 cases of KRAS mutant CRC, and 58,000 cases of KRAS mutant NSCLC each year in the United States. According to the American Cancer Society, the annual incidence of melanoma patients is estimated at 75,000 patients in the United States, with approximately 40%-60% of those patients having BRAF mutations and an additional 15-20% of those patients having NRAS mutations.
We are also advancing a set of orally available, small molecule compounds that inhibit the interaction between the proteins menin and MLL for the treatment of MLL-r and MLL-PTD, two genetically-defined subsets of acute leukemias that affect both adults and children. The annual incidence of MLL-r and MLL-PTD patients is estimated to be 3,200 patients in the United States, and those patients currently have limited options other than chemotherapy.
Kura has incurred net losses since its inception on August 22, 2014. Kura’s net loss and accumulated deficit was $3.7 million for the year ended December 31, 2014. Substantially all of Kura’s net losses resulted from costs incurred in connection with its research and development programs and from general and administrative costs associated with its operations. Kura was founded in August 2014 by the former co-founders and executive team of Intellikine, Inc., a company that discovered and advanced into human clinical testing three drug candidates against molecular targets on the phosphoinositide-3 kinase (PI3K) pathway and that was acquired in January 2012 by Takeda America Holdings, Inc.
We expect to continue to incur significant expenses and increasing operating losses for at least the next several years as we continue the clinical development of, and seek regulatory approval for our product candidates. Our net losses may fluctuate significantly from quarter to quarter and year to year. We will need to raise capital for the further development of our existing product candidates and we may also need to raise additional funds sooner than expected to pursue other development activities related to our other pipeline programs. As of December 31, 2014, we had a cash balance of $1.1 million. We may seek to obtain additional financing in the future through the issuance of our common stock, through other equity or debt financings or through collaborations or partnerships with other companies. We may not be able to raise additional capital on terms acceptable to us, or at all, and any failure to raise capital as and when needed could compromise our ability to execute on our business plan.
Recent Developments
Private Placement
Prior to the Merger, Kura sold to accredited investors approximately $60.0 million of its shares of common stock, or 18,971,136 shares, at a price of $3.16 per share, which included approximately $7.5 million in principal and $0.1 million in accrued interest from the conversion of Kura’s then outstanding convertible promissory notes. Also, Kura granted the investors in the Private Placement registration rights requiring Kura or any successor to register those shares of Kura common stock (which were exchanged for shares of our common stock, along with the rest of the outstanding shares of Kura capital stock, except for dissenting shares, at the Effective Time) for public resale, as described in more detail below. The then existing stockholders of Kura who agreed to become parties to the registration rights agreement also became entitled to such registration rights, subject to specified differences in the agreement between the rights of new investors and existing stockholders. The
55
Private Placement closed immediately prior to the filing of a Certificate of Merger with the Secretary of State of the State of Delaware, on March 6, 2015.
Reverse Merger
On March 6, 2015, pursuant to the Merger Agreement, Merger Sub merged with and into Kura, with Kura remaining as the surviving entity and a wholly-owned operating subsidiary of the Company. The Merger was effective as of March 6, 2015, upon the filing of a Certificate of Merger with the Secretary of State of the State of Delaware. As part of the Merger, Kura changed its name to Merger Sub.
At the Effective Time, the legal existence of Merger Sub ceased and each share of Kura common stock that was issued and outstanding immediately prior to the Effective Time was automatically exchanged for 0.5 shares of our common stock. We issued an aggregate of 14,508,177 shares of our common stock upon such exchange of the outstanding shares of Kura common stock. In addition, at the Effective Time, we assumed Kura’s 2014 Equity Incentive Plan and concurrently approved the amendment and restatement of the Kura 2014 Equity Incentive Plan pursuant to our 2014 plan, effective upon the date that is 20 days after the mailing of a definitive Schedule 14C information statement to our pre-Merger stockholders. As of the Effective Time, there were no outstanding options to purchase shares of Kura common stock under the Kura 2014 Equity Incentive Plan.
Immediately following the Effective Time, pursuant to the terms of the Redemption Agreement, we completed the closing of a redemption of 5,000,000 shares of our common stock from our then-current stockholders in consideration of $70,000, plus professional costs related to the transaction, not to exceed $30,000. The 5,000,000 shares constituted all of the issued and outstanding shares of our capital stock, on a fully-diluted basis, immediately prior to the Merger.
Kura is considered the accounting acquirer in the Merger and will account for the transaction as a capital transaction because Kura’s former stockholders received 100% of the voting rights in the combined entity and Kura’s senior management represents all of the senior management of the combined entity.
Financial Overview of Kura
Research and Development Expenses
Since its inception, Kura has focused on the research and development of its product programs. Kura’s research and development expenses consist primarily of:
| • | salaries and related expenses for personnel; |
| • | license costs associated with our compounds; |
| • | expenses related to preclinical studies and chemistry; |
| • | other consulting fees paid to third parties; and |
| • | travel, facilities, depreciation, insurance and other expenses. |
Research and development expenses are expensed as they are incurred. As of December 31, 2014, Kura had incurred an aggregate of approximately $2.7 million in research and development expenses related to the in-licensing and development of its product candidates and pipeline programs. To date, its tipifarnib program represents the largest portion of its research and development expense.
We cannot determine with certainty the timing of initiation, the duration or the completion costs of current or future preclinical studies and clinical trials of our product candidates. At this time, due to the inherently unpredictable nature of preclinical and clinical development, we are unable to estimate with any certainty the costs we will incur and the timelines we will require in the continued development of our product candidates and our other pipeline programs. Clinical and preclinical development timelines, the probability of success and development costs can differ materially from expectations. Our future research and development expenses will depend on the preclinical and clinical success of each product candidate that we develop, as well as ongoing assessments of the commercial potential of such product candidates. In addition, we cannot forecast which product candidates may be subject to future collaborations, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
56
Completion of clinical trials may take several years or more, and the length of time generally varies according to the type, complexity, novelty and intended use of a product candidate. The cost of clinical trials may vary significantly over the life of a project as a result of differences arising during clinical development, including, among others:
| • | per patient trial costs; |
| • | the number of trials required for approval; |
| • | the number of sites included in the trials; |
| • | the length of time required to enroll suitable patients; |
| • | the number of doses that patients receive; |
| • | the number of patients that participate in the trials; |
| • | the drop-out or discontinuation rates of patients; |
| • | the duration of patient follow-up; |
| • | potential additional safety monitoring or other studies requested by regulatory agencies; |
| • | the number and complexity of analyses and tests performed during the trial; |
| • | the phase of development of the product candidate; and |
| • | the efficacy and safety profile of the product candidate. |
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and benefits for employees in executive, finance, business development and support functions. Other significant general and administrative expenses include the costs associated with obtaining and maintaining Kura’s patent portfolio, professional fees for accounting, auditing, consulting and legal services, travel and allocated facilities.
We expect that our general and administrative expenses will increase in the future as we expand our operating activities, maintain and expand our patent portfolio.
Other Income (Expense)
Other income (expense) consists primarily of management fee income and non-cash interest expense. Management fee income is earned in accordance with the management services agreement with Kura’s affiliated company Araxes. Interest expense consists of interest accrued on its convertible notes.
Income Taxes
Kura has incurred net losses and has not recorded any U.S. federal or state income tax benefits for the losses as they have been offset by valuation allowances.
Critical Accounting Policies and Significant Judgments and Estimates of Kura
Management’s discussion and analysis of Kura’s financial condition and results of operations are based on its financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires it to make estimates and judgments that affect the reported amounts of assets, liabilities, and expenses and the disclosure of contingent assets and liabilities in its financial statements. On an ongoing basis, Kura evaluates its estimates and judgments, including those related to accrued expenses and stock-based compensation. Kura bases its estimates on historical experience, known trends and events, and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While Kura’s significant accounting policies are described in more detail in the notes to its financial statements appearing elsewhere in this Current Report on Form 8-K, Kura believes the following accounting policies are the most critical to the judgments and estimates used in the preparation of its financial statements.
Convertible Notes and Derivative Accounting
57
At inception, Kura performs an assessment of all embedded features of a debt instrument to determine if 1) such features should be bifurcated and separately accounted for, and, 2) if bifurcation requirements are met, whether such features should be classified and accounted for as equity or liability. The fair value of the embedded feature is measured initially, included as a liability on the balance sheet, and remeasured each reporting period. Any changes in fair value are recorded in the statement of operations. Kura monitors, on an ongoing basis, whether events or circumstances could give rise to a change in its classification of embedded features.
Kura accounts for its convertible notes, that may be settled in cash upon conversion (including partial cash settlement), by separating the liability and equity components of the instruments in a manner that reflects its nonconvertible debt borrowing rate. Kura determines the carrying amount of the liability component by measuring the fair value of similar debt instruments that do not have the conversion feature. If a similar debt instrument does not exist, Kura estimates the fair value by using assumptions that market participants would use in pricing a debt instrument, including market interest rates, credit standing, yield curves and volatilities. Determining the fair value of the debt component requires the use of accounting estimates and assumptions. These estimates and assumptions are judgmental in nature and could have a significant impact on the determination of the debt component and the associated non-cash interest expense.
Kura assigns a value to the debt component of its convertible notes equal to the estimated fair value of similar debt instruments without the conversion feature, which resulted in us recording the debt at a discount. Kura amortizes the debt discount over the life of the convertible notes as additional non-cash interest expense utilizing the effective interest method.
Research and Development Expenses
Kura makes estimates of its accrued expenses as of each balance sheet date in its financial statements based on facts and circumstances known to it at that time. If the actual timing of the performance of services or the level of effort varies from the estimate, Kura will adjust the accrual accordingly. Nonrefundable advance payments for goods and services, including fees for process development or manufacturing and distribution of clinical supplies that will be used in future research and development activities, are deferred and recognized as expense in the period that the related goods are consumed or services are performed. Payments that Kura makes in connection with in-licensed technology for a particular research and development project that have no alternative future uses (in other research and development projects or otherwise) and therefore no separate economic values are expensed as research and development costs at the time such costs are incurred.
Results of Operations of Kura
The Year Ended December 31, 2014
The following table sets forth Kura’s results of operations for the year ended December 31, 2014:
| Year Ended December 31, 2014 | ||||
Research and development | $ | 2,653,000 | ||
General and administrative | 1,281,000 | |||
Other income, net | 263,000 | |||
Research and Development Expenses. Kura’s research and development expenses were $2.7 million for the year ended December 31, 2014. Research and development expenses for the year ended December 31, 2014 were primarily comprised of $1.8 million in license fees related to the acquisition of in-process research and development. In addition, other research and development expenses included $0.4 million of payroll related expenses and $0.2 million of share-based compensation, as well as other expenses as Kura expanded its operations.
General and Administrative Expenses. General and administrative expenses were $1.3 million for the year ended December 31, 2014. General and administrative expenses are comprised of $0.3 million of payroll related expenses, $0.6 million of professional and consulting fees and a $0.3 million gift to the Leukemia and Lymphoma Society in connection with Kura’s license agreement with University of Michigan.
Management Fee Income, Related Party. Management fee income, related party was $0.3 million for the year ended December 31, 2014. In accordance with the management services agreement with Araxes, Kura receives a fixed monthly fee of $0.1 million for management services. The agreement has an initial term expiring on December 31, 2015 and renews automatically for additional consecutive one-year periods. The agreement may be terminated by either party with a notice of at least 30 days prior to December 31, 2015 or the expiration of the then-renewal term.
58
Interest Expense. Interest expense was $37,000 for the year ended December 31, 2014. The interest expense incurred during the year ended December 31, 2014 is primarily related to the convertible notes, which were converted into shares of Kura’s common stock in connection with the Private Placement.
Liquidity and Capital Resources
Kura has incurred losses since inception on August 22, 2014 and negative cash flows from operating activities for the year ended December 31, 2014. As of December 31, 2014, Kura had an accumulated deficit of $3.7 million. We anticipate that we will continue to incur net losses for the foreseeable future as we continue the development and potential commercialization of our product candidates.
Kura has funded its operations primarily through the sale of convertible notes. Since inception through December 31, 2014, Kura has raised cash proceeds of $2.0 million from the sale of convertible notes. As of December 31, 2014, Kura had a cash balance of $1.1 million. Since December 31, 2014, Kura received proceeds of $4.0 million related to the issuance of convertible notes. Prior to the Merger, on March 6, 2015, Kura closed the Private Placement in which it sold to accredited investors approximately $60.0 million of its shares of common stock, which included approximately $7.5 million in principal and $0.1 million in accrued interest from the conversion of its then outstanding convertible promissory notes. Additionally, all of the shares issued in the Private Placement, along with the other shares of Kura that were outstanding immediately prior to the Merger, were exchanged for shares of our common stock at the Effective Time. For a more detailed discussion of the Private Placement and the Merger, see “Recent Developments – Private Placement” and “Recent Developments – Reverse Merger” above.
While we believe that our existing cash resources will be sufficient to fund our cash requirements for the next 12 months, we will require significant additional financing in the future to continue to fund our operations. We may seek to obtain additional financing in the future through equity or debt financings or through collaborations or partnerships with other companies. If we are unable to obtain additional financing on commercially reasonable terms, our business, financial condition and results of operations will be materially adversely affected.
The following table provides a summary of Kura’s net cash flow activity for the period set forth below:
| Year Ended December 31, 2014 | ||||
Net cash used in operating activities | $ | (849,000 | ) | |
Net cash used in investing activities | (28,000 | ) | ||
Net cash provided by financing activities | 2,001,000 | |||
Net increase in cash | 1,124,000 | |||
Cash used in operating activities
Cash used in operating activities was $0.8 million for the year ended December 31, 2014. Cash used in operating activities during the year ended December 31, 2014 primarily consisted of $3.7 million of net losses incurred. Cash used in operating activities was further adjusted for non-cash items such as an asset acquisition of $0.5 million, share-based compensation expenses of $0.2 million and net cash inflows from a change in our operating assets and liabilities of $2.1 million.
Cash used in investing activities
Net cash used in investing activities was $28,000 for the year ended December 31, 2014, which consisted of the purchase of fixed assets.
Cash provided by financing activities
Net cash provided from financing activities was $2.0 million for the year ended December 31, 2014. Net cash provided from financing activities for the year ended December 31, 2014 resulted from proceeds of $2.0 million from the issuance of a convertible note.
Operating Capital Requirements
To date, we have not generated any revenues from product sales, and we do not have any approved products. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate significant revenue from product
59
sales unless and until we obtain regulatory approval of and commercialize one of our current or future product candidates. We anticipate that we will continue to incur losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our product candidates, and begin to commercialize any approved products. We are subject to all of the risks incident in the development of new therapeutic products, and we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. We anticipate that we will need substantial additional funding in connection with our continuing operations.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of stock offerings, debt financings, collaborations, strategic partnerships and licensing arrangements. We do not have any committed external source of funds. Additional capital may not be available on reasonable terms, if at all. To the extent that we raise additional capital through the sale of stock or convertible debt securities, the ownership interest of our stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders. Debt financing, if available, may involve agreements that include increased fixed payment obligations and covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, declaring dividends, selling or licensing intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. If we raise additional funds through collaborations, strategic partnerships or licensing arrangements with third parties, we may have to relinquish valuable rights to our product candidates, including our other technologies, future revenue streams or research programs, or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through stock offerings or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and commercialize our product candidates even if we would otherwise prefer to develop and commercialize such product candidates ourself.
Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. The amount and timing of future funding requirements, both near- and long-term, will depend on many factors, including, but not limited to:
| • | the outcome, timing and cost of regulatory approvals by the FDA and comparable foreign regulatory authorities, including the potential for the FDA or comparable foreign regulatory authorities to require that we perform more studies than, or evaluate clinical endpoints other than, those that we currently expect; |
| • | the timing and costs associated with manufacturing our product candidates for clinical trials, preclinical studies and, if approved, for commercial sale; |
| • | the number and characteristics of product candidates that we pursue; |
| • | the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| • | Our need to expand our research and development activities, including our need and ability to hire additional employees; |
| • | the effect of competing technological and market developments; and |
| • | the cost of establishing sales, marketing and distribution capabilities for any products candidates for which we may receive regulatory approval. |
If we cannot expand our operations or otherwise capitalize on our business opportunities because we lack sufficient capital, our business, financial condition and results of operations could be materially adversely affected.
60
Contractual Obligations and Commitments of Kura
The following table summarizes Kura’s contractual obligations and commitments as of December 31, 2014 that will affect its future liquidity:
| Total | Less than 1 Year | 1-3 Years | 3-5 Years | More than 5 Years | ||||||||||||||||
Operating lease obligations(1) | $ | 199,000 | $ | 111,000 | $ | 88,000 | $ | — | $ | — | ||||||||||
Guaranteed charitable gift | $ | 285,000 | $ | 95,000 | $ | 190,000 | $ | — | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total | $ | 484,000 | $ | 206,000 | $ | 278,000 | $ | — | — | |||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
| (1) | In August 2014, Kura entered into a multi-year non-cancelable building sublease for its facility in San Diego, California. The sublease expires in August 2016. In September 2014, Kura entered into a multi-year non-cancelable building lease for office space in Cambridge, Massachusetts. The lease expires in October 2016. |
Kura’s commitment for operating leases relates to its leases of office space in San Diego, California and Cambridge, Massachusetts.
Kura enters into contracts in the normal course of business with clinical sites for the conduct of clinical trials, CROs for preclinical research studies, professional consultants for expert advice and other vendors for clinical supply manufacturing or other services. These contracts generally provide for termination on notice, and therefore are cancelable contracts and not included in the table of contractual obligations and commitments.
Kura has in-license and asset purchase agreements under which it is obligated to make payments if and when specified development, regulatory approval and sales threshold milestones are achieved. The milestone payment obligations are not included in the table of contractual obligations and commitments if the amount and timing of such obligations are unknown or uncertain.
On February 15, 2015, Kura entered into a Sponsored Research Agreement with the University of Michigan under which it will sponsor up to $2.7 million of research at the University of Michigan over a three-year period. Kura will receive a non-exclusive right to any technology developed under the agreement and has an option right for an exclusive right to any such licenses developed under the agreement. The agreement allows for termination with notice at any time by Kura. In the event of termination by Kura prior to the second anniversary of the agreement, other than due to breach by the University of Michigan, Kura will be required to pay costs budgeted through the second anniversary up to $2.0 million of the sponsored research amount.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements (as defined by applicable regulations of the SEC) that are reasonably likely to have a current or future material effect on our financial condition, results of operations, liquidity, capital expenditures or capital resources.
Other Information
JOBS Act
On April 5, 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies.
We are in the process of evaluating the benefits of relying on other exemptions and reduced reporting requirements provided by the JOBS Act. Subject to certain conditions set forth in the JOBS Act, as an “emerging growth company,” we intend to rely on certain of these exemptions, including without limitation with respect to, (1) providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act and (2) complying with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding a supplement to the auditor’s report providing additional information about the audit and the financial statements, known as the auditor discussion and analysis. We will remain an “emerging growth company” until the earliest of (a) the last day of the fiscal year in which we have total annual gross revenues of $1 billion or more, (b) the last day of our fiscal year following the fifth anniversary of the date of the completion of our initial public offering, (c) the date on which we have issued more than $1 billion in non-convertible debt during the previous three years or (d) the date on which we are deemed to be a large accelerated filer (i.e., a seasoned issuer with public float of $700 million or more) under the rules of the SEC.
Recently Adopted Accounting Pronouncements
See “Notes to Financial Statements—Note 3—Recent Accounting Pronouncements” of Kura’s financial statements.
61
Quantitative and Qualitative Disclosures about Market Risk
Interest Rate Risk
Kura’s cash balance as of December 31, 2014 consisted of cash held in an operating account that earns nominal interest income. All of Kura’s long-term debt was converted into shares of its common stock in connection with the Private Placement; therefore, there was no or minimal interest rate risk
Internal Control Over Financial Reporting
Pursuant to Section 404(a) of the Sarbanes-Oxley Act, commencing the year following our first annual report required to be filed with the SEC, our management will be required to report on the effectiveness of our internal control over financial reporting. To comply with the requirements of being a reporting company under the Exchange Act, we may need to upgrade our systems, including information technology, implement additional financial and management controls, reporting systems and procedures and hire additional accounting and finance staff.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth information regarding beneficial ownership of our common stock as of March 6, 2015, after giving effect to the Merger (including the Redemption) and the Private Placement, by:
| • | each person or group who is known by us to beneficially own more than 5% of our common stock; |
| • | each director and director-elect; |
| • | our named executive officers; and |
| • | all executive officers, directors and directors-elect as a group. |
We have determined beneficial ownership in accordance with the rules of the SEC. Except as indicated by the footnotes below, we believe, based on the information furnished to us, that the persons and entities named in the table below have sole voting and investment power with respect to all shares of common stock that they beneficially own, subject to applicable community property laws.
Applicable percentage ownership is based on 14,508,177 shares of common stock outstanding at March 6, 2015, after giving effect to the Merger (including the Redemption) and the Private Placement. In computing the number of shares of common stock beneficially owned by a person and the percentage ownership of that person, we deemed to be outstanding all shares of common stock subject to options, warrants or other convertible securities held by that person or entity that are currently exercisable or will be exercisable on or before May 5, 2015, which is 60 days after March 6, 2015. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. Except as otherwise noted below, the address for each person or entity is c/o Kura Oncology, Inc., 11119 N. Torrey Pines Road, Suite 125, La Jolla, California 92037.
Beneficial Owner | Title | Shares of Common Stock Beneficially Owned (#)(1) | Percentage of Common Stock Beneficially Owned (%)(1) | |||||||
Directors, Directors-Elect and Named Executive Officers | ||||||||||
Troy E. Wilson, Ph.D.(2) | Chairman, President and Chief Executive Officer | 2,168,727 | 14.95 | % | ||||||
Matthew P. Kinley | Director | — | * | |||||||
Robert E. Hoffman | Director-Elect | 9,494 | * | |||||||
All current executive officers, directors and directors-elect as a group (8 persons)(3) | 4,391,521 | 30.27 | % | |||||||
| Other 5% or More Stockholders | ||||||||||
62
Entities affiliated with FMR LLC(4) | 1,846,519 | 12.73 | % | |||||
EcoR1 Capital, LLC(5) | 1,450,000 | 9.99 | % | |||||
ARCH Venture Fund VIII, L.P.(6) | 1,344,937 | 9.27 | % | |||||
Pingda Ren, Ph.D.(7) | 773,982 | 5.33 | % | |||||
Yi Liu, Ph.D.(8) | 749,999 | 5.17 | % | |||||
Kevan Shokat(9) | 750,000 | 5.17 | % |
| * | Represents beneficial ownership of less than 1% of the shares of common stock. |
| (1) | Beneficial ownership is determined in accordance with SEC rules, and includes any shares as to which the stockholder has sole or shared voting power or investment power, and also any shares which the stockholder has the right to acquire within 60 days of March 6, 2015, whether through the exercise or conversion of any stock option, convertible security, warrant or other right. The indication herein that shares are beneficially owned is not an admission on the part of the stockholder that he, she or it is a direct or indirect beneficial owner of those shares. |
| (2) | Consists of (a) 12,500 shares of common stock owned by the 2013If I Ran the Circus Irrevocable Trust for the benefit of Aidan Eliasson, a trust for the benefit of Dr. Wilson’s minor child, (b) 12,500 shares of common stock owned by the 2013If I Ran the Circus Irrevocable Trust for the benefit of Ethan Eliasson, a trust for the benefit of Dr. Wilson’s minor child, (c) 1,736,991 shares of restricted common stock and common stock owned byRed Fish Blue Fish Revocable Trust, dated December 31, 2012, 1,458,334 shares of which are subject to a right of repurchase by us as of May 5, 2015, and (d) 406,736 shares of common stock owned by Araxes. Dr. Wilson is the trustee ofRed Fish Blue Fish Revocable Trust, dated December 31, 2012 and as such has the dispositive power and control over the securities held by such trust. |
| (3) | Consists of the shares identified in footnotes (2), (7), and (8) and includes 698,813 shares of restricted common stock and common stock owned by four other executive officers, directors-elect and/or entities affiliated with such executive officers or directors-elect, 537,762 shares of which are subject to a right of repurchase by us as of May 5, 2015. |
| (4) | Consists of (a) 1,520,587 shares of common stock owned by Fidelity Select Portfolios: Biotechnology Portfolio, or Fidelity Select, and (b) 325,932 shares of common stock owned by Fidelity Advisor Series VII: Fidelity Advisor Biotechnology Fund, or Fidelity Advisor. Fidelity Select has an address at c/o Brown Brothers Harriman & Co, 525 Washington Blvd., Jersey City, NJ 07310 and Fidelity Advisor has an address at c/o State Street Bank & Trust, P.O. Box 5756, Boston, MA 02206. |
| (5) | Consists of (a) 918,000 shares of common stock owned by EcoR1 Capital Fund Qualified, L.P. and (b) 532,000 shares of common stock owned by EcoR1 Capital Fund, L.P. EcoR1 Capital, LLC has an address at 409 Illinois Street, San Francisco, CA 94158. |
| (6) | Consists of shares held of record by ARCH Venture Fund VIII, L.P., or ARCH VIII. ARCH Venture Partners VIII, L.P., or the GPLP, as the sole general partner of ARCH VIII, may be deemed to beneficially own certain of the shares held of record by ARCH VIII. The GPLP disclaims beneficial ownership of all shares held of record by ARCH VIII in which the GPLP does not have an actual pecuniary interest. ARCH Venture Partners VIII, LLC, or the GPLLC, as the sole general partner of the GPLP, may be deemed to beneficially own certain of the shares held of record by ARCH VIII. The GPLLC disclaims beneficial ownership of all shares held of record by ARCH VIII in which it does not have an actual pecuniary interest. Keith Crandell, Clinton Bybee and Robert Nelsen are the managing directors of the GPLLC, and may be deemed to beneficially own certain of the shares held of record by ARCH VIII. The managing directors disclaim beneficial ownership of all shares held of record by ARCH VIII in which they do not have an actual pecuniary interest. ARCH Venture Fund VIII, L.P. has an address at 8725 West Higgins Road, Suite 290, Chicago, IL 60631. |
| (7) | Consists of (a) 734,375 shares of restricted common stock owned by Pingda Ren, Ph.D., 625,000 shares of which are subject to a right of repurchase by us as of May 5, 2015, (b) 23,983 shares of common stock owned by Pingda Ren, Ph.D., (c) 7,812 shares of common stock owned by Pingda Ren, Custodian for Evan T. Ren, of which Dr. Ren has dispositive power and control, and (d) 7,812 shares of common stock owned by Pingda Ren, Custodian for Oliver T. Ren, of which Dr. Ren has dispositive power and control. |
| (8) | Consists of (a) 734,375 shares of restricted common stock owned by Yi Liu, Ph.D., 625,000 shares of which are subject to a right of repurchase by us as of May 5, 2015, (b) 7,812 shares of common stock owned by Yi Liu, Custodian for Max Liu, of which Dr. Liu has dispositive power and control, and (c) 7,812 shares of common stock owned by Yi Liu, Custodian for Nicholas Liu, of which Dr. Liu has dispositive power and control. |
63
| (9) | Consists of 750,000 shares of restricted common stock owned by Kevan Shokat, 625,000 shares of which are subject to a right of repurchase by us as of May 5, 2015. |
MANAGEMENT AND DIRECTORS
At the Effective Time, John Pappajohn resigned from our board of directors and effective immediately following the Effective Time, Troy E. Wilson, Ph.D., J.D. was appointed to our board of directors, and together with Matthew P. Kinley, constitute our board of directors as of March 6, 2015. Effective upon the eleventh day after we file with the SEC and mail to our stockholders prior to the Merger a Schedule 14f-1 reporting a change in the majority of our directors, our board of directors will be reconstituted by the appointment of Robert E. Hoffman to serve with Dr. Wilson as directors, and the resignation of Matthew P. Kinley as a director. Our executive management team was also reconstituted immediately following the Effective Time by the appointment of Dr. Wilson as our President and Chief Executive Officer, Heidi Henson as our Chief Financial Officer and Secretary, Yi Liu, Ph.D. as our Chief Scientific Officer, Antonio Gualberto, M.D., Ph.D. as our Chief Medical Officer, Annette North as our Senior Vice President, General Counsel, and Pingda Ren, Ph.D. as our Senior Vice President, Chemistry and Pharmaceutical Sciences, and the resignation of Mr. Pappajohn and Mr. Kinley from all of their positions as officers. The following table sets forth the name and positions of each of our directors, directors-elect and executive officers after the Merger.
Executive Officers and Directors
The following table sets forth certain information concerning our executive officers, directors and directors-elect as of March 6, 2015:
Name | Age | Position | ||
| Executive Officers | ||||
Troy Wilson, Ph.D., J.D. | 46 | Chairman, President and Chief Executive Officer | ||
Heidi Henson | 49 | Chief Financial Officer and Secretary | ||
Yi Liu, Ph.D. | 47 | Chief Scientific Officer | ||
Antonio Gualberto, M.D., Ph.D. | 50 | Chief Medical Officer | ||
Annette North | 49 | Senior Vice President, General Counsel | ||
Pingda Ren, Ph.D. | 46 | Senior Vice President, Chemistry and Pharmaceutical Sciences | ||
Non-Employee Directors and Directors-Elect | ||||
Matthew P. Kinley | 47 | Director | ||
Robert E. Hoffman | �� | 49 | Director-Elect |
Executive Officers
Troy Wilson, Ph.D., J.D. has served as our President and Chief Executive Officer and as the chairman of our board of directors since the Merger in March 2015 and has served as the President and Chief Executive Officer of Kura, as well as a member of Kura’s board of directors, since August 2014. Dr. Wilson co-founded Kura in August 2014. Dr. Wilson has served as President and Chief Executive Officer of Wellspring Biosciences LLC, a private biopharmaceutical company, and its parent company Araxes Pharma LLC since July 2012 and as President and Chief Executive Officer of Avidity NanoMedicines LLC, a private biopharmaceutical company, since November 2012. Dr. Wilson served as the President and Chief Executive Officer and a member of the board of directors of Intellikine, Inc., a private biopharmaceutical company, from April 2007 to January 2012 and from August 2007 to January 2012, respectively, until its acquisition by Takeda Pharmaceuticals. He has also been a member of the board of directors of Puma Biotechnology, Inc., a public biopharmaceutical company, since October 2013, a member of the board of directors of Zosano Pharma, Inc., a public biopharmaceutical company, since June 2014, and a member of the board of managers of Araxes Pharma LLC, a private biopharmaceutical company, since May 2012, a member of the board of managers of Avidity NanoMedicines LLC since November 2012 and a member of the board of managers of Wellspring Biosciences LLC since May 2012. He holds a J.D. from New York University and graduated with a Ph.D. in bioorganic chemistry and a B.A. in biophysics from the University of California, Berkeley. Our board of directors believes that Dr. Wilson’s experience in the pharmaceutical industry and his experience serving in executive roles and on other boards of directors qualify him to serve on our board of directors, including as the chairman.
64
Heidi Henson has served as our Chief Financial Officer and Secretary since the Merger in March 2015 and has served as the Chief Financial Officer and Secretary of Kura since August 2014. Ms. Henson has also served as Chief Financial Officer and Secretary of Wellspring Biosciences LLC, a private biopharmaceutical company, and its parent company Araxes Pharma LLC, since July 2012. From 2007 to March 2012, Ms. Henson served as the Vice President, Finance at Intellikine, Inc., a private biopharmaceutical company, until its acquisition by Takeda Pharmaceuticals. Ms. Henson has served as an independent financial consultant for several years assisting with various start-up activities for early stage companies, SEC reporting and Sarbanes-Oxley implementation and compliance. Ms. Henson previously served as Director of Finance at Anadys Pharmaceuticals, Inc., a public biopharmaceutical company, and held a number of management positions with Fair Isaac & Co., Inc. (formally HNC Software, Inc.), a public software company. Ms. Henson began her career in auditing at PricewaterhouseCoopers LLP, a public accounting firm, where she served both public and private companies. She received a Bachelor’s of Accountancy from the University of San Diego and is a Certified Public Accountant.
Yi Liu, Ph.D. has served as our Chief Scientific Officer since the Merger in March 2015 and has served as the Chief Scientific Officer of Kura since October 2014. Dr. Liu co-founded Kura in August 2014. Prior to that, Dr. Liuco-founded and served as Chief Scientific Officer of Wellspring Biosciences LLC, a private biopharmaceutical company, from July 2012 to September 2014. Dr. Liu also co-founded Intellikine, Inc., a private biopharmaceutical company, where he served as Vice President of Drug Discovery from 2007 to May 2012, until its acquisition by Takeda Pharmaceuticals. Prior to Intellikine, Dr. Liu was the head of the drug design group at the Genomics Institute of the Novartis Research Foundation. Earlier in his career, he held senior scientist positions at both SGX Pharmaceuticals, Inc., a public biopharmaceutical company which was acquired by Eli Lilly and Company in 2008, and Curagen Corporation, a public biopharmaceutical development company. Dr. Liu received his Ph.D. in Biochemistry from Princeton University, his MSc in computational chemistry from Beijing University and his BE in Chemical Engineering from Tsinghua University.
Antonio Gualberto, M.D., Ph.D. has served as our Chief Medical Officer since the Merger in March 2015 and has served as the Chief Medical Officer of Kura since October 2014. Dr. Gualberto co-founded Kura in August 2014. From June 2012 to September 2014, Dr. Gualberto served as the head of the global clinical development center for oncology at EMD Serono, Inc., the biopharmaceutical subsidiary in the United States of Merck KGaA, Darmstadt, Germany, a global pharmaceutical and chemical group. Prior to this, from September 2010 to April 2012, Dr. Gualberto served as a group head of clinical research for the Takeda Oncology Company, a private biopharmaceutical company. From October 1999 to August 2010 Dr. Gualberto served in varying roles at Pfizer, Inc., a public pharmaceutical company, including Senior Director, Clinical Development and Medical Affairs, and Global Clinical Leader. He has also held several academic positions including, from October 2008 to June 2012, an adjunct appointment of associate professor of pathology and laboratory medicine at The Warren Alpert Medical School of Brown University. Dr. Gualberto received his B.S. from Trinidad College and M.D. and Ph.D. degrees from the University of Seville in Spain. He received postgraduate fellowship training at Case Western Reserve University and the University of North Carolina at Chapel Hill Lineberger Comprehensive Cancer Center.
Annette North has served as our Senior Vice President, General Counsel since the Merger in March 2015 and has served as the Senior Vice President, General Counsel of Kura since January 2015. Ms. North also serves as General Counsel and Secretary of Wellspring Biosciences LLC and its parent company Araxes Pharma LLC. Prior to joining us, Ms. North served as Senior Vice President and General Counsel of Ambit Biosciences Corporation, a public biopharmaceutical company, from June 2013 to January 2015, during which time Ambit completed its initial public offering and was acquired by Daiichi Sankyo Company Limited. From January 2009 to December 2014, Ms. North was an independent legal consultant to a number of life sciences companies. From 2000 to 2008, Ms. North served as General Counsel and held a number of other positions at SGX Pharmaceuticals, Inc., a public biopharmaceutical company which was acquired by Eli Lilly and Company in 2008. Earlier in her career, Ms. North served as Senior Director of Operations and Legal at Axys Pharmaceuticals, Inc., a biopharmaceutical company, and Director of Legal Affairs at Sequana Therapeutics, Inc., a biopharmaceutical company. Ms. North received both her Bachelor of Commerce and her Bachelor of Laws from the University of Melbourne, Australia.
Pingda Ren, Ph.D. has served as our Senior Vice President of Chemistry and Pharmaceutical Sciences since the Merger in March 2015 and has served as the Senior Vice President of Chemistry and Pharmaceutical Sciences of Kura since October 2014. Dr. Ren co-founded Kura in August 2014. Prior to that, Dr. Renco-founded and served as Senior Vice President of Chemistry of Wellspring Biosciences LLC, a private biopharmaceutical company, from July 2012 to September 2014. Dr. Ren also co-founded Intellikine, Inc., a private biopharmaceutical company, where he served as Vice President of Chemistry from 2007 to May 2012, until its acquisition by Takeda Pharmaceuticals. Prior to Intellikine, Dr. Ren was a Senior Research Investigator in Genomics Institute of the Novartis Research Foundation. Earlier in his career, Dr. Ren was a Senior Research Chemist at Albany Molecular Research Inc., a public global contract research and manufacturing organization. Dr. Ren earned his B.A and Ph.D. of Chemistry from Fudan University in China. He completed his postdoctoral research with Professor Huw M. L. Davies at State University of New York at Buffalo.
Non-Employee Directors and Directors-Elect
Matthew P. Kinley served as our Chief Financial Officer and Secretary since our inception in 2007 until the closing of the Merger in March 2015. In addition, he has served a member of our board of directors since our inception in 2007, but has
65
agreed to resign as a director effective upon the eleventh day following the date we file with the SEC and mail to our stockholders prior to the Merger a Schedule 14f-1 reporting a change in the majority of our directors. Mr. Kinley currently serves as the Senior Vice President of Equity Dynamics, Inc., a financial consulting firm, and Pappajohn Capital Resources, a venture capital firm. He has served as such since 1995. Mr. Kinley served as President and as a director of Healthcare Acquisition Corp. from April 2005 through August 2007. Healthcare Acquisition Corp. is now the public company known as PharmAthene, Inc., which trades under the symbol “NYSE MKT:PIP”. Mr. Kinley has been involved in the financing and development of more than 25 companies in the past ten years. Mr. Kinley currently serves as a director of American CareSource Holdings, Inc. which trades under the symbol “NASDAQ:ANCI”. From 1990 through 1995, Mr. Kinley was manager and held various positions at KPMG Peat Marwick, working on tax, audit and merger and acquisition issues. He currently serves on the board of directors of several private companies. Mr. Kinley also serves on the College of Business Executive Advisory Board and the Entrepreneurial Center Advisory Board at the University of Northern Iowa. Mr. Kinley received his B.A. in Business, with highest honors, from the University of Northern Iowa in May 1990. Mr. Kinley’s experience serving in executive roles and on other boards of directors qualify him to serve on our board of directors until the director-elect takes office.
Robert E. Hoffman. Mr. Hoffman has served as Senior Vice President, Finance and Chief Financial Officer of Arena Pharmaceuticals, Inc., or Arena, a public biopharmaceutical company, since June 2012. Mr. Hoffman served as the Vice President, Finance and Chief Financial Officer of Arena from August 2011 to June 2012 and previously from December 2005 to March 2011. Mr. Hoffman served as Vice President, Finance and Chief Accounting Officer of Arena from June 2004 to December 2005, as Vice President, Finance of Arena from April 2000 to June 2004, and as Controller of Arena from August 1997 to April 2000. From March 2011 to August 2011, Mr. Hoffman served as Chief Financial Officer for Polaris Group, a biopharmaceutical drug company. Mr. Hoffman is a member of the board of directors of CombiMatrix Corporation, a molecular diagnostics company, and MabVax Therapeutics Holdings, Inc., a biopharmaceutical company. Mr. Hoffman serves as a member of the Financial Accounting Standards Board’s Small Business Advisory Committee and the steering committee of the Association of Bioscience Financial Officers. Mr. Hoffman is also a member and a former director and President of the San Diego Chapter of Financial Executives International. Mr. Hoffman holds a B.B.A. from St. Bonaventure University, and is licensed as a C.P.A. (inactive) in the State of California. Our board of directors believes that Mr. Hoffman’s experience in the biopharmaceutical industry and his experience serving in executive roles qualify him to serve on our board of directors.
Scientific Advisors
Kevan Shokat, Ph.D.Professor Shokat is currently an Investigator of the Howard Hughes Medical Institute, Chair of the Department of Cellular and Molecular Pharmacology at the University of California at San Francisco. He is also a Professor in the Department of Chemistry at the University of California at Berkeley. After receiving his Ph.D. in Organic Chemistry at UC Berkeley with Professor Peter Schultz, and post-doctoral work in immunology at Stanford University with Professor Chris Goodnow, Dr. Shokat began his independent research career at Princeton University where he was promoted from Assistant to Associate Professor in four years. He has received numerous awards including being named a Fellow of several prestigious research foundations including the Pew Foundation, Searle Foundation, Sloan Foundation, Glaxo-Wellcome Foundation, and the Cotrell Foundation. He has also received the Eli Lilly Award, given to the most promising biological chemist in the country under the age of 37. He was inducted into the National Academy of Sciences (2010), the Institute of Medicine (2011), and the American Academy of Arts and Sciences (2011).
Frank McCormick, PhD, FRS. Dr. McCormick is currently Professor Emeritus of the UCSF Helen Diller Family Comprehensive Cancer Center. Prior to joining the UCSF faculty, Dr. McCormick pursued cancer-related work with several Bay Area biotechnology firms and held positions with Cetus Corporation (Director of Molecular Biology, 1981-1990; Vice President of Research, 1990-1991) and Chiron Corporation, where he was Vice President of Research from 1991 to 1992. In 1992 he founded Onyx Pharmaceuticals, a company dedicated to developing new cancer therapies, and served as its Chief Scientific Officer until 1996. At Onyx Pharmaceuticals, Dr. McCormick initiated and led drug discovery efforts that led to the approval of Sorafenib in 2005 for treatment of renal cell cancer, and for liver cancer in 2007, and the approval of ONYX-015 in 2006 in China for treatment of nasopharyngeal cancer. Dr. McCormick holds the David A. Wood Chair of Tumor Biology and Cancer Research at UCSF. Dr. McCormick is the author of over 285 scientific publications and holds 20 issued patents. He also served as President, 2012-2013 for the American Association for Cancer Research (AACR). More recently, he has taken a leadership role at the Frederick National Lab for Cancer Research, overseeing an NCI supported national effort to develop therapies against Ras-driven cancers.
Compensation Committee Interlocks and Insider Participation
We currently do not have a compensation committee. None of our executive officers serves as a member of the board of directors or compensation committee of any other entity that has one or more executive officers serving as a member of our board of directors.
66
Board Composition and Election of Directors
Terms of Office
Our amended and restated certificate of incorporation, which we expect to file on or about the date that is 20 calendar days from the date we expect to file with the SEC and mail to our stockholders prior to the Merger a definitive Schedule 14C reporting the adoption of the amended and restated certificate of incorporation by our stockholders prior to the Merger, and our amended and restated bylaws, which became effective upon the Merger, provide that the authorized number of directors may be changed only by resolution of our board of directors. We currently have authorized two directors. We do not currently have a classified board.
Director Independence
Our securities are not listed on a national securities exchange or on any inter-dealer quotation system which has a requirement that a majority of directors be independent. We evaluate independence, however, by the standards for director independence set forth in the NASDAQ Marketplace Rules. Under Rules 5605 and 5615 of the NASDAQ Marketplace Rules, a majority of a listed company’s board of directors must be comprised of independent directors, subject to certain phase-in exceptions. In addition, NASDAQ Marketplace Rules require that, subject to specified exceptions, each member of a listed company’s audit, compensation and governance and nominating committees be independent and that audit committee members also satisfy independence criteria set forth in Rule 10A-3 under the Exchange Act. Under Rule 5605(a)(2) of the NASDAQ Marketplace Rules, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
Based upon information requested from and provided by each director concerning their background, employment and affiliations, including family relationships, our board of directors has determined that Mr. Hoffman, who will serve on our board of directors as the New Board Effective Date, does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that Mr. Hoffman is “independent” as that term is defined under Rule 5605(a)(2) of the NASDAQ Marketplace Rules. Dr. Wilson and Mr. Kinley, our current directors, are or have been employed by us during 2015 and are therefore not independent under NASDAQ Marketplace Rules.
Committees of the Board of Directors
Our board of directors does not currently have an audit committee, a compensation committee or a nominating and governance committee but intends to establish an audit committee, a compensation committee and a nominating and corporate governance committee. Each committee will operate under a charter to be approved by our board of directors.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors and executive officers, and persons who own more than ten percent of a registered class of our equity securities, to file with the SEC initial reports of ownership and reports of changes in ownership of our common stock and other equity securities. Officers, directors and greater than ten percent stockholders are required by SEC regulations to furnish us with copies of all Section 16(a) forms they file.
To our knowledge, based solely on a review of the copies of such reports furnished to us and written representations regarding the filing of required reports, we believe that all Section 16(a) filing requirements applicable to our directors, executive officers and greater-than-ten-percent beneficial owners with respect to fiscal 2014 were met.
Code of Ethics
We have not yet adopted a code of ethics, which would apply to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. Our board of directors plans to adopt a code of ethics.
Board Structure
We have chosen to combine the chief executive officer and chairman of the board of directors positions. We believe that this board of directors leadership structure is the most appropriate for us. Because we are a small company, it is more efficient to have the leadership of the board of directors in the same hands as the chief executive officer. The challenges faced by us at this stage – obtaining financing and implementing our business and marketing plan – are most efficiently dealt with by one person who is familiar with both the operational aspects as well as the strategic aspects of our business.
Board Assessment of Risk
Our board of directors oversees our risk management function. Our management keeps the board of directors apprised of material risks and provides directors access to all information necessary for them to understand and evaluate how these risks
67
interrelate and how management addresses those risks. If the identified risk poses an actual or potential conflict with management, our non-employee directors may conduct the assessment. Currently, the primary risks affecting us are access to financing and the conduct of our clinical trials.
Board Diversity
While we do not have a formal policy on diversity, our board of directors considers diversity to include the skill set, background, reputation, type and length of business experience of our board of directors members, as well as, a particular nominee’s contributions to that mix. Our board of directors believes that diversity brings a variety of ideas, judgments and considerations that can benefit our stockholders and us. Although there are many other factors, the board of directors primarily seeks individuals with experience in the design and conduct of clinical trials and other aspects of life science companies.
Indemnification of Directors and Officers
Our pending amended and restated certificate of incorporation will limit our directors’ liability to the fullest extent permitted under Delaware corporate law. Delaware corporate law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except for liability:
| • | for any breach of the director’s duty of loyalty to us or our stockholders; |
| • | for acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of the law; |
| • | under Section 174 of the Delaware General Corporation Law for the unlawful payment of dividends; or |
| • | for any transaction from which the director derives an improper personal benefit. |
These provisions eliminate our rights and those of our stockholders to recover monetary damages from a director for breach of his fiduciary duty of care as a director except in the situations described above. The limitations summarized above, however, do not affect our ability or that of our stockholders to seek non-monetary remedies, such as an injunction or rescission, against a director for breach of his fiduciary duty.
Section 145 of the Delaware General Corporation Law provides a corporation with the power to indemnify any officer or director acting in his capacity as our representative who is, or is threatened to be, made a party to any lawsuit or other proceeding for expenses, judgment and amounts paid in settlement in connection with such lawsuit or proceeding. The indemnity provisions apply whether the action was instituted by a third party or was filed by one of our stockholders. The Delaware General Corporation Law provides that Section 145 is not exclusive of other rights to which those seeking indemnification may be entitled under any bylaw, agreement, vote of stockholders or disinterested directors or otherwise. We have provided for this indemnification in our pending amended and restated certificate of incorporation because we believe that it is important to attract qualified directors and officers.
We have entered into indemnification agreements with each of our executive officers and directors that require us to indemnify such persons against any and all expenses, including judgments, fines or penalties, attorney’s fees, witness fees or other professional fees and related disbursements and other out-of-pocket costs incurred, in connection with any action, suit, arbitration, alternative dispute resolution mechanism, investigation, inquiry or administrative hearing, whether threatened, pending or completed, to which any such person may be made a party by reason of the fact that such person is or was a director, officer, employee or agent of our company, provided that such director or officer acted in good faith and in a manner that the director or officer reasonably believed to be in, or not opposed to, our best interests. The indemnification agreements also set forth procedures that will apply in the event of a claim for indemnification thereunder. We believe that these provisions and agreements are necessary to attract and retain qualified persons as directors and officers.
Insofar as indemnification by us for liabilities arising under the Securities Act may be permitted to our directors, officers or persons controlling us pursuant to provisions of our pending amended and restated certificate of incorporation and amended and restated bylaws, or otherwise, we have been advised that in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. In the event that a claim for indemnification by such director, officer or controlling person of us in the successful defense of any action, suit or proceeding is asserted by such director, officer or controlling person in connection with the securities being offered, we will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by us is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
68
At the present time, there is no pending litigation or proceeding involving a director, officer, employee or other agent of ours in which indemnification would be required or permitted. We are not aware of any threatened litigation or proceeding, which may result in a claim for such indemnification.
Stockholder Communications
Although we do not have a formal policy regarding communications with our board of directors, stockholders may communicate with the board of directors by writing to us at 11119 N. Torrey Pines Road, Suite 125, La Jolla, California 92037, Attention: Chief Executive Officer. Stockholders who would like their submission directed to a member of the board of directors may so specify, and the communication will be forwarded, as appropriate.
Other Information
We are required to file periodic reports, proxy statements and other information with the SEC. You may read and copy this information at the Public Reference Room of the SEC, 100 F. Street, N.E., Washington, D.C. 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. You may also obtain a copy of these reports by accessing the SEC’s website at http://www.sec.gov. You may also send communications to our board of directors at: Kura Oncology, Inc., 11119 N. Torrey Pines Road, Suite 125, La Jolla, California 92037, Attention: Board of Directors.
EXECUTIVE OFFICER AND DIRECTOR COMPENSATION
Our named executive officers for the year ended December 31, 2014 consist solely of our principal executive officer, Troy Wilson, Ph.D., J.D., our President and Chief Executive Officer. None of our other executive officers received total compensation in excess of $100,000 for the year ended December 31, 2014. Unless we specifically indicate otherwise, all share and per share numbers included in this “Executive Officer and Director Compensation” section have been adjusted as necessary to reflect the exchange of shares in the Merger.
Summary Compensation Table
Name and principal position | Year | Salary ($) | Stock awards ($)(2) | All other compensation ($)(3) | Total ($) | |||||||||||||||
Troy Wilson, Ph.D., J.D.(1) | 2014 | 82,500 | 3,500 | 409 | 86,409 | |||||||||||||||
| (1) | Dr. Wilson served as Kura’s President and Chief Executive Officer commencing on August 29, 2014. |
| (2) | In accordance with SEC rules, this column reflects the aggregate fair value of the stock awards granted during 2014 computed as of their respective grant dates in accordance with Financial Accounting Standard Board Accounting Standards Codification Topic 718 for stock-based compensation transactions (ASC 718). Assumptions used in the calculation of these amounts are included in Note 2 to our financial statements appearing elsewhere in this Current Report on Form 8-K. These amounts do not reflect the actual economic value that will be realized by the named executive officer upon the vesting of the stock awards or the sale of the common stock underlying such stock awards. |
| (3) | This column reflects amounts paid by us on behalf of the named executive officer pursuant to an executive disability policy. For more information regarding these benefits, see below under “—Perquisites, Health, Welfare and Retirement Benefits.” |
69
Annual Base Salary
The base salary of our named executive officers is generally determined and approved at the beginning of each year or, if later, in connection with the commencement of employment of the executive, by our board of directors. The following represents the 2014 annual base salary, which became effective in October 2014, for our named executive officer.
Name | 2014 Base Salary ($) | |||
Troy Wilson, Ph.D., J.D. | 330,000 | |||
Bonus Compensation
From time to time our board of directors may approve bonuses for our named executive officers based on individual performance, company performance or as otherwise determined appropriate.
Pursuant to Dr. Wilson’s executive employment agreement, he is eligible for an annual discretionary bonus of up to 40% of his annual base salary based upon our and Dr. Wilson’s achievement of objectives and milestones as determined by the board of directors. In 2014, Dr. Wilson did not receive or earn any bonus.
Equity-Based Incentive Awards
Our equity-based incentive awards are designed to align our interests and the interests of our stockholders with those of our employees and consultants, including our named executive officers. The board of directors is responsible for approving equity grants.
We have historically used restricted stock awards as the primary incentive for long-term compensation to our named executive officer. We may grant equity awards at such times as our board of directors determines appropriate. Our executives generally are awarded an initial equity grant in connection with their commencement of employment. Additional grants may occur periodically in order to specifically incentivize executives with respect to achieving certain corporate goals or to reward executives for exceptional performance.
Prior to the Merger, Kura granted all restricted stock awards pursuant to its 2014 Equity Incentive Plan. Such restricted stock awards generally vest over a four-year period and may be subject to acceleration of vesting under certain termination and change of control events. In connection with the Merger, we assumed Kura’s 2014 Equity Incentive Plan and concurrently approved the amendment and restatement of the Kura 2014 Equity Incentive Plan pursuant to our 2014 plan, effective upon the date that is 20 days after the mailing of a definitive Schedule 14C information statement to our pre-Merger stockholders. We will continue to grant equity incentive awards under the terms of the 2014 plan. The terms of our equity plan is described below under “—Equity Benefit Plans.”
On August 29, 2014, the Kura board of directors granted a restricted stock award to Dr. Wilson for 3,500,000 shares of Kura common stock, with the shares vesting in equal monthly installments over the following four years, subject to Dr. Wilson’s continued service with us. Prior to the Merger, such shares were transferred to three trusts affiliated with Dr. Wilson. In connection with the Exchange, such shares became shares of our common stock.
Agreements with our Named Executive Officers
Below is a written description of our executive employment agreement with our named executive officer, Dr. Wilson. Dr. Wilson’s employment is “at will” and may be terminated at any time.
Dr. Wilson. We entered into an executive employment agreement with Dr. Wilson, which was effective as of October 1, 2014, setting forth the terms of his employment as our President and Chief Executive Officer. Pursuant to the agreement, Dr. Wilson is entitled to an initial annual base salary of $330,000 and is eligible for an annual discretionary bonus of up to 40% of his annual base salary based upon our and Dr. Wilson’s achievement of objectives and milestones as determined by the board of directors.
Potential Payments upon Termination and Change of Control
Regardless of the manner in which our named executive officer’s service terminates, the named executive officer is entitled to receive amounts earned during his term of service, including salary and unused vacation pay.
Dr. Wilson. Pursuant to his executive employment agreement, if we terminate Dr. Wilson’s employment without cause or he resigns for good reason (i) more than 59 days before or 12 months after the closing of a corporate transaction, subject to his execution of an effective release and waiver of claims in favor of us, Dr. Wilson will receive a cash lump-sum payment in an amount equal to 12 months of Dr. Wilson’s then annual base salary or (ii) within 59 days prior to or within 12 months following the closing of a corporate transaction, subject to his execution of an effective release and waiver of claims in favor of us, Dr. Wilson will receive (1) a cash lump-sum payment in an amount equal to 12 months of Dr. Wilson’s then annual base salary; (2) a cash lump-sum payment in an amount equal to Dr. Wilson’s full target bonus amount for services to be performed during the year in which the corporate transaction occurs; (3) payment for continued health benefits under COBRA for up to 12 months; and (4) accelerated vesting of all of his outstanding stock awards in full.
For purposes of the agreement described above:
| • | “cause” generally means with respect to Dr. Wilson, (1) being convicted of or pleading guilty ornolo contendere to a |
70
felony or any crime involving moral turpitude or dishonesty; (2) participating in a fraud or act of dishonesty against us; (3) materially breaching any agreement with us or any of our written policies, and not curing such breach within five days of our written notice of such breach; (4) engaging in conduct that demonstrates gross unfitness to serve; or (5) engaging in willful misconduct or refusing to comply with any lawful directive of us, and not curing such noncompliance within five days of our written notice of such noncompliance. |
| • | “good reason” generally means with respect to Dr. Wilson, if any of the following actions are taken by us without Dr. Wilson’s written consent: (1) a material reduction in Dr. Wilson’s base salary, unless pursuant to a generally applicable salary reduction program; (2) a material reduction in Dr. Wilson’s duties (including responsibilities and/or authorities); (3) if applicable, a material reduction in the authority, duties, or responsibilities of the supervisor to whom Dr. Wilson is required to report, including a requirement that the executive report to someone other than our chief executive officer; (4) relocation of Dr. Wilson’s principal place of employment to a place that increases his one-way commute by more than 50 miles; or (5) any other action or inaction that constitutes a material breach by us of Dr. Wilson’s employment agreement or other service agreement. |
| • | “corporate transaction” generally means the consummation, in a single transaction or is a series of related transactions, of (1) a sale, lease, or other disposition or all or substantially all of our consolidated assets; (2) a merger, consolidation, or similar transaction following which we are not the surviving entity, or (3) a merger, consolidation or similar transaction following which we are the surviving entity but the units outstanding immediately preceding the transaction are converted or exchanged into other property, whether in the form of securities, cash or otherwise. |
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth certain information regarding equity awards granted to our named executive officer that remain outstanding as of December 31, 2014.
| Stock Awards(1) | ||||||||
| Number of Shares or Units of Stock that Have Not Vested (#) | Market Value of Shares or Units of Stock that Have Not Vested ($) | |||||||
Troy Wilson, Ph.D., J.D. | 3,208,334 | (2) | 9,079,585 | (3) | ||||
| (1) | All of the outstanding stock awards were granted under and subject to the terms of the Kura 2014 Equity Incentive Plan which we amended and restated pursuant to our 2014 plan which is described below under “—Equity Benefit Plans.” All vesting of stock awards is subject to the executive’s continuous service with us through the vesting dates and the potential vesting acceleration described above under “—Potential Payments upon Termination and Change of Control.” |
| (2) | Represents the unvested portion of a restricted stock award originally granted to Dr. Wilson. The shares vest such that 1/48th of the 3,500,000 shares granted (or 72,916.67 shares) vest on the 29th day of the month, commencing on September 29, 2014 and ending on August 29, 2018. Such shares are currently held in the name of “Red Fish Blue Fish Revocable Trust, dated December 31, 2012,” an affiliated trust of Dr. Wilson. |
| (3) | Because our common stock was not traded on a public market on December 31, 2014, the market value has been determined based on a per-share common stock value of $2.83, which was the per share value of our common stock as determined by an independent valuation firm as of December 31, 2014. |
71
Perquisites, Health, Welfare and Retirement Benefits
Our named executive officer is eligible to participate in our employee benefit plans, including our medical, dental, vision, group life, disability and accidental death and dismemberment insurance plans, in each case on the same basis as all of our other employees. We provide a 401(k) plan to our employees, including our current named executive officer, as discussed in the section below entitled “—401(k) Plan.”
We generally do not provide perquisites or personal benefits to our named executive officer, except in limited circumstances. We do, however, pay the premiums for term life insurance and disability insurance for all of our employees, including our current named executive officer. In addition, we have an executive disability policy for our executive officers. Our board of directors may elect to adopt qualified or non-qualified benefit plans in the future if it determines that doing so is in our best interests.
401(k) Plan
We maintain a defined contribution employee retirement plan, or 401(k) plan, for our employees. Our named executive officer is eligible to participate in the 401(k) plan on the same basis as our other employees. The 401(k) plan is intended to qualify as a tax-qualified plan under Section 401(k) of the Code. The 401(k) plan provides that each participant may contribute up to the lesser of 100% of his or her compensation or the statutory limit, which is $17,500 for calendar year 2014. Participants that are 50 years or older can also make “catch-up” contributions, which in calendar year 2014 may be up to an additional $5,500 above the statutory limit. We currently do not make matching contributions into the 401(k) plan on behalf of participants. Participant contributions are held and invested, pursuant to the participant’s instructions, by the plan’s trustee.
Nonqualified Deferred Compensation
We do not maintain nonqualified defined contribution plans or other nonqualified deferred compensation plans. Our board of directors may elect to provide our officers and other employees with non-qualified defined contribution or other nonqualified deferred compensation benefits in the future if it determines that doing so is in our best interests.
Equity Benefit Plans
Amended and Restated 2014 Equity Incentive Plan
The board of directors and stockholders of Kura approved the Kura 2014 Equity Incentive Plan in August 2014 and we have approved the amendment and restatement of the Kura 2014 Equity Incentive Plan pursuant to our 2014 plan, effective upon the date that is 20 days after the mailing of a definitive Schedule 14C information statement to our pre-Merger stockholders. As of March 6, 2015, there were outstanding restricted stock awards covering 4,943,498 shares that were granted under the Kura 2014 Equity Incentive Plan. Upon the effectiveness of our 2014 plan, there will be 1,031,500 shares remaining available for the grant of stock awards under our 2014 plan.
Stock Awards.The 2014 plan provides for the grant of incentive stock options, or ISOs, nonstatutory stock options, or NSOs, stock appreciation rights, restricted stock awards, restricted stock unit awards, performance-based stock awards, and other forms of equity compensation, which we refer to collectively as stock awards, all of which may be granted to employees, including officers, non-employee directors and consultants of us and our affiliates. Additionally, the 2014 plan provides for the grant of performance cash awards. ISOs may be granted only to employees. All other awards may be granted to employees, including officers, and to non-employee directors and consultants.
Share Reserve.Initially, the aggregate number of shares of our common stock that may be issued pursuant to stock awards under the 2014 plan as restated is 5,975,000 shares. Additionally, the number of shares of our common stock reserved for issuance under our 2014 plan will automatically increase on January 1 of each year, beginning on January 1, 2016 and continuing through and including January 1, 2025, by 4% of the total number of shares of our capital stock outstanding on December 31 of the preceding calendar year, or a lesser number of shares determined by our board of directors. The maximum number of shares that may be issued upon the exercise of ISOs under our 2014 plan is 12,000,000 shares.
No person may be granted stock awards covering more than 1,000,000 shares of our common stock under our 2014 plan during any calendar year pursuant to stock options, stock appreciation rights and other stock awards whose value is determined by reference to an increase over an exercise or strike price of at least 100% of the fair market value on the date the stock award is granted. Additionally, no person may be granted in a calendar year a performance stock award covering more than 1,000,000 shares or a performance cash award having a maximum value in excess of $1,000,000. Such limitations are designed to help assure that any deductions to which we would otherwise be entitled with respect to such awards will not be subject to the $1,000,000 limitation on the income tax deductibility of compensation paid to any covered executive officer imposed by Section 162(m) of the Code.
If a stock award granted under the 2014 plan expires or otherwise terminates without being exercised in full, or is settled in cash, the shares of our common stock not acquired pursuant to the stock award again will become available for subsequent
72
issuance under the 2014 plan. In addition, the following types of shares under the 2014 plan may become available for the grant of new stock awards under the 2014 plan: (1) shares that are forfeited to or repurchased by us prior to becoming fully vested; (2) shares withheld to satisfy income or employment withholding taxes; or (3) shares used to pay the exercise or purchase price of a stock award. Shares issued under the 2014 plan may be previously unissued shares or reacquired shares bought by us on the open market.
Administration. Our board of directors, or a duly authorized committee thereof, has the authority to administer the 2014 plan. Our board of directors may also delegate to one or more of our officers the authority to (1) designate employees (other than other officers) to be recipients of certain stock awards, and (2) determine the number of shares of common stock to be subject to such stock awards. Subject to the terms of the 2014 plan, our board of directors or the authorized committee, referred to herein as the plan administrator, determines recipients, dates of grant, the numbers and types of stock awards to be granted and the terms and conditions of the stock awards, including the period of their exercisability and vesting schedule applicable to a stock award. Subject to the limitations set forth below, the plan administrator will also determine the exercise price, strike price or purchase price of awards granted and the types of consideration to be paid for the award.
The plan administrator has the authority to modify outstanding awards under our 2014 plan. Subject to the terms of our 2014 plan, the plan administrator has the authority to reduce the exercise, purchase or strike price of any outstanding stock award, cancel any outstanding stock award in exchange for new stock awards, cash or other consideration, or take any other action that is treated as a repricing under generally accepted accounting principles, with the consent of any adversely affected participant.
Stock Options. ISOs and NSOs are granted pursuant to stock option agreements adopted by the plan administrator. The plan administrator determines the exercise price for a stock option, within the terms and conditions of the 2014 plan, provided that the exercise price of a stock option generally cannot be less than 100% of the fair market value of our common stock on the date of grant. Options granted under the 2014 plan vest at the rate specified by the plan administrator.
The plan administrator determines the term of stock options granted under the 2014 plan, up to a maximum of 10 years. Unless the terms of an option holder’s stock option agreement provide otherwise, if an option holder’s service relationship with us, or any of our affiliates, ceases for any reason other than disability, death or cause, the option holder may generally exercise any vested options for a period of three months following the cessation of service. The option term may be extended in the event that exercise of the option following such a termination of service is prohibited by applicable securities laws or our insider trading policy. If an optionholder’s service relationship with us or any of our affiliates ceases due to disability or death, or an optionholder dies within a certain period following cessation of service, the optionholder or a beneficiary may generally exercise any vested options for a period of 12 months in the event of disability and 18 months in the event of death. In the event of a termination for cause, options generally terminate immediately upon the termination of the individual for cause. In no event may an option be exercised beyond the expiration of its term.
Acceptable consideration for the purchase of common stock issued upon the exercise of a stock option will be determined by the plan administrator and may include (1) cash, check, bank draft or money order, (2) a broker-assisted cashless exercise, (3) the tender of shares of our common stock previously owned by the optionholder, (4) a net exercise of the option if it is an NSO, and (5) other legal consideration approved by the plan administrator.
Unless the plan administrator provides otherwise, options generally are not transferable except by will, the laws of descent and distribution, or pursuant to a domestic relations order. An optionholder may designate a beneficiary, however, who may exercise the option following the optionholder’s death.
Tax Limitations On Incentive Stock Options. The aggregate fair market value, determined at the time of grant, of our common stock with respect to ISOs that are exercisable for the first time by an optionholder during any calendar year under all of our stock plans may not exceed $100,000. Options or portions thereof that exceed such limit will generally be treated as NSOs. No ISO may be granted to any person who, at the time of the grant, owns or is deemed to own stock possessing more than 10% of our total combined voting power or that of any of our affiliates unless (1) the option exercise price is at least 110% of the fair market value of the stock subject to the option on the date of grant, and (2) the term of the ISO does not exceed five years from the date of grant.
Restricted Stock Awards.Restricted stock awards are granted pursuant to restricted stock award agreements adopted by the plan administrator. Restricted stock awards may be granted in consideration for (1) cash, check, bank draft or money order, (2) services rendered to us or our affiliates, or (3) any other form of legal consideration. Common stock acquired under a restricted stock award may, but need not, be subject to a share repurchase option in our favor in accordance with a vesting schedule to be determined by the plan administrator. A restricted stock award may be transferred only upon such terms and
73
conditions as set by the plan administrator. Except as otherwise provided in the applicable award agreement, restricted stock that has not vested will be forfeited or repurchased by us upon the participant’s cessation of continuous service for any reason.
Restricted Stock Unit Awards.Restricted stock unit awards are granted pursuant to restricted stock unit award agreements adopted by the plan administrator. Restricted stock unit awards may be granted in consideration for any form of legal consideration. A restricted stock unit award may be settled by cash, delivery of stock, a combination of cash and stock as deemed appropriate by the plan administrator, or in any other form of consideration set forth in the restricted stock unit award agreement. Additionally, dividend equivalents may be credited in respect of shares covered by a restricted stock unit award. Except as otherwise provided in the applicable award agreement, restricted stock units that have not vested will be forfeited upon the participant’s cessation of continuous service for any reason.
Stock Appreciation Rights.Stock appreciation rights are granted pursuant to stock appreciation grant agreements adopted by the plan administrator. The plan administrator determines the strike price for a stock appreciation right, which generally cannot be less than 100% of the fair market value of our common stock on the date of grant. Upon the exercise of a stock appreciation right, we will pay the participant an amount equal to the product of (1) the excess of the per share fair market value of our common stock on the date of exercise over the strike price, multiplied by (2) the number of shares of common stock with respect to which the stock appreciation right is exercised. A stock appreciation right granted under the 2014 plan vests at the rate specified in the stock appreciation right agreement as determined by the plan administrator.
The plan administrator determines the term of stock appreciation rights granted under the 2014 plan, up to a maximum of ten years. Unless the terms of a participant’s stock appreciation right agreement provides otherwise, if a participant’s service relationship with us or any of our affiliates ceases for any reason other than cause, disability or death, the participant may generally exercise any vested stock appreciation right for a period of three months following the cessation of service. The stock appreciation right term may be further extended in the event that exercise of the stock appreciation right following such a termination of service is prohibited by applicable securities laws. If a participant’s service relationship with us, or any of our affiliates, ceases due to disability or death, or a participant dies within a certain period following cessation of service, the participant or a beneficiary may generally exercise any vested stock appreciation right for a period of 12 months in the event of disability and 18 months in the event of death. In the event of a termination for cause, stock appreciation rights generally terminate immediately upon the occurrence of the event giving rise to the termination of the individual for cause. In no event may a stock appreciation right be exercised beyond the expiration of its term.
Performance Awards.The 2014 plan permits the grant of performance-based stock and cash awards that may qualify as performance-based compensation that is not subject to the $1,000,000 limitation on the income tax deductibility of compensation paid to a covered executive officer imposed by Section 162(m) of the Code. To help assure that the compensation attributable to performance-based awards will so qualify, our board of directors can structure such awards so that stock or cash will be issued or paid pursuant to such award only after the achievement of certain pre-established performance goals during a designated performance period.
The performance goals that may be selected include one or more of the following: (1) earnings (including earnings per share and net earnings); (2) earnings before interest, taxes and depreciation; (3) earnings before interest, taxes, depreciation and amortization; (4) earnings before interest, taxes, depreciation, amortization and legal settlements; (5) earnings before interest, taxes, depreciation, amortization, legal settlements and other income (expense); (6) earnings before interest, taxes, depreciation, amortization, legal settlements, other income (expense) and stock-based compensation; (7) earnings before interest, taxes, depreciation, amortization, legal settlements, other income (expense), stock-based compensation and changes in deferred revenue; (8) total stockholder return; (9) return on equity or average stockholders’ equity; (10) return on assets, investment, or capital employed; (11) stock price; (12) margin (including gross margin); (13) income (before or after taxes); (14) operating income; (15) operating income after taxes; (16) pre-tax profit; (17) operating cash flow; (18) sales or revenue targets; (19) increases in revenue or product revenue; (20) expenses and cost reduction goals; (21) improvement in or attainment of working capital levels; (22) economic value added (or an equivalent metric); (23) market share; (24) cash flow; (25) cash flow per share; (26) share price performance; (27) debt reduction; (28) stockholders’ equity; (29) capital expenditures; (30) debt levels; (31) operating profit or net operating profit; (32) workforce diversity; (33) growth of net income or operating income; (34) billings; (35) bookings; (36) employee retention; (37) initiation of phases of clinical trials and/or studies by specific dates; (38) patient enrollment rates; (39) budget management; (40) submission to, or approval by, a regulatory body (including, but not limited to the FDA) of an applicable filing or a product candidate; (41) implementation or completion of projects or processes (including, without limitation, clinical trial initiation, clinical trial enrollment, clinical trial results, new and supplemental indications for existing products, regulatory filing submissions, regulatory filing acceptances, regulatory or advisory committee interactions, regulatory approvals, and product supply); (42) regulatory milestones; (43) progress of internal research or clinical programs; (44) progress of partnered programs; (45) partner satisfaction; (46) timely completion of clinical trials; (47) submission of INDs and NDAs and other regulatory achievements; (48) research progress, including the development of programs; (49) strategic partnerships or transactions (including in-licensing and out-licensing of intellectual property; and (50) to the extent
74
that an award is not intended to comply with Section 162(m) of the Code, other measures of performance selected by our board of directors.
The performance goals may be based on a company-wide basis, with respect to one or more business units, divisions, affiliates, or business segments, and in either absolute terms or relative to the performance of one or more comparable companies or the performance of one or more relevant indices. Unless specified otherwise (1) in the award agreement at the time the award is granted or (2) in such other document setting forth the performance goals at the time the goals are established, we will appropriately make adjustments in the method of calculating the attainment of performance goals as follows: (a) to exclude restructuring and/or other nonrecurring charges; (b) to exclude exchange rate effects; (c) to exclude the effects of changes to generally accepted accounting principles; (d) to exclude the effects of any statutory adjustments to corporate tax rates; (e) to exclude the effects of any “extraordinary items” as determined under generally accepted accounting principles; (f) to exclude the dilutive effects of acquisitions or joint ventures; (g) to assume that any business divested by us achieved performance objectives at targeted levels during the balance of a performance period following such divestiture; (h) to exclude the effect of any change in the outstanding shares of our common stock by reason of any stock dividend or split, stock repurchase, reorganization, recapitalization, merger, consolidation, spin-off, combination or exchange of shares or other similar corporate change, or any distributions to common stockholders other than regular cash dividends; (i) to exclude the effects of stock based compensation and the award of bonuses under our bonus plans; (j) to exclude costs incurred in connection with potential acquisitions or divestitures that are required to be expensed under generally accepted accounting principles; (k) to exclude the goodwill and intangible asset impairment charges that are required to be recorded under generally accepted accounting principles; (l) to exclude the effect of any other unusual, non-recurring gain or loss or other extraordinary item; and (m) to exclude the effects of the timing of acceptance for review and/or approval of submissions to the FDA or any other regulatory body. In addition, we retain the discretion to reduce or eliminate the compensation or economic benefit due upon attainment of the goals. The performance goals may differ from participant to participant and from award to award.
Other Stock Awards.The plan administrator may grant other awards based in whole or in part by reference to our common stock. The plan administrator will set the number of shares under the stock award and all other terms and conditions of such awards.
Changes to Capital Structure.In the event that there is a specified type of change in our capital structure, such as a stock split or recapitalization, appropriate adjustments will be made to (1) the class and maximum number of shares reserved for issuance under the 2014 plan, (2) the class and maximum number of shares by which the share reserve may increase automatically each year, (3) the class and maximum number of shares that may be issued upon the exercise of ISOs, (4) the class and maximum number of shares subject to stock awards that can be granted in a calendar year (as established under the 2014 plan pursuant to Section 162(m) of the Code), (5) the class and maximum number of shares that may be awarded to any non-employees director and (6) the class and number of shares and exercise price, strike price, or purchase price, if applicable, of all outstanding stock awards.
Corporate Transactions.In the event of certain specified significant corporate transactions, the plan administrator has the discretion to take any of the following actions with respect to stock awards:
| • | arrange for the assumption, continuation or substitution of a stock award by a surviving or acquiring entity or parent company; |
| • | arrange for the assignment of any reacquisition or repurchase rights held by us to the surviving or acquiring entity or parent company; |
| • | accelerate the vesting of the stock award and provide for its termination prior to the effective time of the corporate transaction; |
| • | arrange for the lapse of any reacquisition or repurchase right held by us; |
| • | cancel or arrange for the cancellation of the stock award in exchange for such cash consideration, if any, as our board of directors may deem appropriate; or |
| • | make a payment equal to the excess of (a) the value of the property the participant would have received upon exercise of the stock award over (b) the exercise price otherwise payable in connection with the stock award. |
Our plan administrator is not obligated to treat all stock awards, even those that are of the same type, in the same manner.
Under the 2014 plan, a corporate transaction is generally the consummation of (1) a sale or other disposition of all or substantially all of our assets, (2) a sale or other disposition of at least 90% of our outstanding securities, (3) a merger, consolidation or similar transaction following which we are not the surviving corporation, or (4) a merger, consolidation or
75
similar transaction following which we are the surviving corporation but the shares of our common stock outstanding immediately prior to such transaction are converted or exchanged into other property by virtue of the transaction.
Change of Control.The plan administrator may provide, in an individual award agreement or in any other written agreement between a participant and us that the stock award will be subject to additional acceleration of vesting and exercisability in the event of a change of control. Under the 2014 plan, a change of control is generally (1) the acquisition by a person or entity of more than 50% of our combined voting power other than by merger, consolidation or similar transaction; (2) a consummated merger, consolidation or similar transaction immediately after which our stockholders cease to own more than 50% of the combined voting power of the surviving entity; (3) a consummated sale, lease or exclusive license or other disposition of all or substantially of our assets; (4) a complete dissolution or liquidation of us, except for a liquidation into a parent corporation, or (5) when a majority of our board of directors becomes comprised of individuals who were not serving on our board of directors on the date of adoption of the 2014 plan, or the incumbent board, or whose nomination, appointment, or election was not approved by a majority of the incumbent board still in office.
Amendment and Termination.Our board of directors has the authority to amend, suspend, or terminate our 2014 plan, provided that such action does not materially impair the existing rights of any participant without such participant’s written consent. No ISOs may be granted after March 6, 2025, which is the tenth anniversary of the date our board of directors amended and restated our 2014 plan.
2015 Employee Stock Purchase Plan
Our board of directors and stockholders adopted the ESPP, which will become effective upon the date that is 20 days after the mailing of a definitive Schedule 14C information statement to our pre-Merger stockholders. The purpose of the ESPP is to retain the services of new employees and secure the services of new and existing employees while providing incentives for such individuals to exert maximum efforts toward our success and that of our affiliates.
Share Reserve. The ESPP authorizes the issuance of 25,000 shares of our common stock pursuant to purchase rights granted to our employees or to employees of any of our designated affiliates. The number of shares of our common stock reserved for issuance will automatically increase on January 1 of each calendar year, from January 1, 2016 through January 1, 2025 by the least of (1) 1% of the total number of shares of our common stock outstanding on December 31 of the preceding calendar year, (2) 2,000,000 shares, or (3) a number determined by our board of directors that is less than (1) and (2). The ESPP is intended to qualify as an “employee stock purchase plan” within the meaning of Section 423 of the Code. As of the date hereof, no shares of our common stock have been purchased under the ESPP.
Administration. Our board of directors, or a duly authorized committee thereof, has the authority to administer the ESPP. The ESPP is implemented through a series of offerings of purchase rights to eligible employees. Under the ESPP, we may specify offerings with durations of not more than 27 months, and may specify shorter purchase periods within each offering. Each offering will have one or more purchase dates on which shares of our common stock will be purchased for employees participating in the offering. An offering may be terminated under certain circumstances.
Payroll Deductions. Generally, all regular employees, including executive officers, employed by us or by any of our designated affiliates, may participate in the ESPP and may contribute, normally through payroll deductions, up to 15% of their earnings for the purchase of our common stock under the ESPP. Unless otherwise determined by our board of directors, common stock will be purchased for accounts of employees participating in the ESPP at a price per share equal to the lower of (1) 85% of the fair market value of a share of our common stock on the first date of an offering or (2) 85% of the fair market value of a share of our common stock on the date of purchase.
Limitations. Employees may have to satisfy one or more of the following service requirements before participating in the ESPP, as determined by our board of directors: (1) customarily employed for more than 20 hours per week, (2) customarily employed for more than five months per calendar year or (3) continuous employment with us or one of our affiliates for a period of time (not to exceed two years). No employee may purchase shares under the ESPP at a rate in excess of $25,000 worth of our common stock based on the fair market value per share of our common stock at the beginning of an offering for each year such a purchase right is outstanding. Finally, no employee will be eligible for the grant of any purchase rights under the ESPP if immediately after such rights are granted, such employee has voting power over 5% or more of our outstanding capital stock measured by vote or value pursuant to Section 424(d) of the Code.
Changes to Capital Structure. In the event that there occurs a change in our capital structure through such actions as a stock split, merger, consolidation, reorganization, recapitalization, reincorporation, stock dividend, dividend in property other than cash, large nonrecurring cash dividend, liquidating dividend, combination of shares, exchange of shares, change in corporate structure or similar transaction, the board of directors will make appropriate adjustments to (1) the number of shares
76
reserved under the ESPP, (2) the maximum number of shares by which the share reserve may increase automatically each year and (3) the number of shares and purchase price of all outstanding purchase rights.
Corporate Transactions. In the event of certain significant corporate transactions, including the consummation of: (1) a sale of all our assets, (2) the sale or disposition of 90% of our outstanding securities, (3) a merger or consolidation where we do not survive the transaction and (4) a merger or consolidation where we do survive the transaction but the shares of our common stock outstanding immediately prior to such transaction are converted or exchanged into other property by virtue of the transaction, any then-outstanding rights to purchase our stock under the ESPP may be assumed, continued or substituted for by any surviving or acquiring entity (or its parent company). If the surviving or acquiring entity (or its parent company) elects not to assume, continue or substitute for such purchase rights, then the participants’ accumulated payroll contributions will be used to purchase shares of our common stock within ten business days prior to such corporate transaction, and such purchase rights will terminate immediately.
Plan Amendments, Termination. Our board of directors has the authority to amend or terminate our ESPP, provided that except in certain circumstances any such amendment or termination may not materially impair any outstanding purchase rights without the holder’s consent. We will obtain stockholder approval of any amendment to our ESPP as required by applicable law or listing requirements.
Director Compensation
Historically, neither we nor Kura has paid cash or equity compensation to directors for their service on the board of directors. In 2014, we did not have any non-employee directors. We have reimbursed and will continue to reimburse all of our non-employee directors for their travel, lodging and other reasonable expenses incurred in attending meetings of our board of directors.
Our board of directors expects to adopt a compensation policy that is applicable to all of our non-employee directors.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table presents information regarding Kura’s equity compensation plans as of December 31, 2014. There are no equity compensation plans that have not been approved by Kura’s stockholders.
Plan Category | Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) | Weighted-average exercise price of outstanding options, warrants and rights (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a))(1) (c) | |||||||||
Equity compensation plans approved by stockholders: | ||||||||||||
2014 Equity Incentive Plan | — | (2) | $ | — | 1,113,000 | |||||||
Equity compensation plans not approved by stockholders: | �� | |||||||||||
None | ||||||||||||
| (1) | Does not reflect the adjustment in the number of shares as a result of the Merger. |
| (2) | Under the Kura 2014 Equity Incentive Plan, Kura has granted restricted stock awards covering 9,887,000 shares of its common stock. |
77
CERTAIN RELATIONSHIPS AND RELATED PERSON TRANSACTIONS
Since January 1, 2012, we and Kura have engaged in the following transactions with our respective directors, directors-elect, executive officers and holders of more than 5% of voting securities, which we refer to as principal stockholders, and affiliates or immediate family members of our respective directors, directors-elect, executive officers and principal stockholders, other than compensation arrangements, which are described in the section above titled “Executive Officer and Director Compensation.” We believe that all of these transactions were on terms as favorable as could have been obtained from unrelated third parties.
As described above, the following executive officers and directors held the following positions at Kura prior to the Merger:
| • | Troy Wilson, Ph.D., J.D., our President and Chief Executive Officer and chairman of our board of directors, was the President and Chief Executive Officer and a member of the board of directors of Kura prior to the Merger. |
| • | Heidi Henson, our Chief Financial Officer and Secretary, was the Chief Financial Officer and Secretary of Kura prior to the Merger. |
| • | Yi Liu, Ph.D., our Chief Scientific Officer, was the Chief Scientific Officer of Kura prior to the Merger. |
| • | Antonio Gualberto, M.D., Ph.D., our Chief Medical Officer, was the Chief Medical Officer of Kura prior to the Merger. |
| • | Annette North, our Senior Vice President, General Counsel, was the Senior Vice President, General Counsel of Kura prior to the Merger. |
| • | Pingda Ren, Ph.D., our Senior Vice President, Chemistry and Pharmaceutical Sciences, was the Senior Vice President, Chemistry and Pharmaceutical Sciences of Kura prior to the Merger. |
Employment Arrangements
We currently have written employment agreements with our executive officers. For information about our employment agreements with our named executive officers, refer to “Executive Officer and Director Compensation— Agreements with our Named Executive Officers.”
Convertible Note Financings
In October 2014, Kura entered into a note purchase agreement with Araxes pursuant to which Kura issued to Araxes a convertible promissory note in aggregate principal amount of $2.0 million, or the October 2014 note. Araxes is affiliated with the following director and executive officers of us and Kura: Troy Wilson, Ph.D., J.D., Heidi Henson, Yi Liu, Ph.D., Antonio Gualberto, M.D., Ph.D., Pingda Ren, Ph.D. and Annette North. The October 2014 note accrued interest at a rate of 8% per annum, compounded annually.
In January 2015, Kura entered into a note purchase agreement with certain investors, including certain executive officers and directors or entities affiliated with such individuals, pursuant to which Kura issued $3.0 million aggregate principal amount of convertible notes, or the January 2015 notes. The January 2015 notes accrued interest at a rate of 8% per annum, compounded annually.
The holders of the January 2015 notes included the following related parties:
Participants | Aggregate Principal Amount of Notes Converted | |||
Directors and Executive Officers | ||||
Troy Wilson, Ph.D., J.D.(1) | $ | 75,000 | ||
Heidi Henson | $ | 35,000 | ||
Pingda Ren, Ph.D. | $ | 150,000 | ||
Antonio Gualberto, M.D., Ph.D. | $ | 250,000 | ||
| (1) | Dr. Wilson participated through his affiliated family trust, Red Fish Blue Fish Revocable Trust, dated December 31, 2012. |
78
The October 2014 note and the January 2015 notes converted into shares of Kura common stock in connection with the Private Placement discussed in “Common Stock Issued in Private Placement in 2015” below.
Asset Purchase Agreement and Convertible Note
In December 2014, Kura entered into an asset purchase agreement with Araxes. For information about the asset purchase agreement with Araxes, refer to “Description of the Business of Kura Oncology, Inc. — License and Asset Purchase Agreements.”
In connection with the asset purchase agreement, Kura issued to Araxes a convertible promissory note in aggregate principal amount of $500,000, or the December 2014 note. The December 2014 note accrued interest at a rate of 8% per annum. The December 2014 note converted into shares of Kura common stock in connection with the Private Placement discussed in “Common Stock Issued in Private Placement in 2015” below.
Common Stock Issued in Private Placement in 2015
The following table summarizes Kura’s sales of its common stock on March 6, 2015 in the Private Placement to its executive officers, directors, directors-elect and beneficial owners of more than five percent of its voting securities. The purchase price of $3.16 per share (as adjusted to $6.32 after giving effect to the Merger) was the fair market value as determined by arms-length negotiations between sophisticated investors and Kura’s management and board of directors. In addition, the aggregate principal amount plus accrued interest of the October 2014 note, the December 2014 note and the January 2015 notes was converted into shares of Kura common stock at the purchase price of $3.16 per share (as adjusted to $6.32 after giving effect to the Merger). Kura received no additional consideration from the conversion of the October 2014 note, the December 2014 and the January 2015 notes.
Participants | Purchase Price of Common Stock | Principal Plus Accrued Interest of Convertible Notes Through Date of Conversion(1) | Shares of Common Stock Issued(2) | |||||||||||
Greater than 5% stockholders | ||||||||||||||
Entities affiliated with FMR LLC(3) | $ | 11,670,000 | $ | — | 3,693,038 | |||||||||
EcoR1 Capital, LLC(4) | $ | 9,164,000 | $ | — | 2,900,000 | |||||||||
ARCH Venture Fund VIII, L.P. | $ | 8,500,002 | $ | — | 2,689,874 | |||||||||
Directors, Directors-Elect and Executive Officers | ||||||||||||||
Troy Wilson, Ph.D., J.D. | $ | — | $ | 2,646,364 | (5) | 837,454 | (6) | |||||||
Heidi Henson | $ | — | $ | 35,368 | 11,192 | |||||||||
Pingda Ren, Ph.D. | $ | — | $ | 151,578 | 47,966 | |||||||||
Antonio Gualberto, M.D., Ph.D. | $ | — | $ | 252,630 | 79,946 | |||||||||
Robert E. Hoffman | $ | 60,002 | $ | — | 18,988 | |||||||||
| (1) | Per the terms of the convertible promissory notes, with respect to the conversion, interest was calculated through February 28, 2015. Interest accruing after February 28, 2015 was paid in cash. |
| (2) | Does not reflect the adjustment in the number of shares as a result of the Merger. |
| (3) | Includes (a) 3,041,174 shares purchased by Fidelity Select and (b) 651,864 shares purchased by Fidelity Advisor. |
| (4) | Includes (a) 1,836,000 shares purchased by EcoR1 Capital Fund Qualified, L.P. and (b) 1,064,000 shares purchased by EcoR1 Capital Fund, L.P. |
| (5) | Includes (a) $75,789 from a note owned by Dr. Wilson’s affiliated family trust, Red Fish Blue Fish Revocable Trust, dated December 31, 2012 and (b) $2,570,575 from notes owned by Araxes. |
| (6) | Includes (a) 23,982 shares purchased by Dr. Wilson’s affiliated family trust, Red Fish Blue Fish Revocable Trust, dated December 31, 2012 and (b) 813,472 shares purchased by Araxes. |
At the Effective Time of the Merger, on March 6, 2015, each share of Kura common stock outstanding immediately prior to the Effective Time was exchanged for 0.5 shares of our common stock. The following table summarizes the exchange of the
79
outstanding shares of Kura common stock at the Effective Time by our executive officers, directors, directors-elect and beneficial owners of more than five percent of our voting securities.
Participants | Number of Shares of Kura Common Stock Held Immediately Prior to Exchange | Number of Shares of Our Common Stock Held Immediately Following Exchange | ||||||
Greater than 5% stockholders | ||||||||
Entities affiliated with FMR LLC(1) | 3,693,038 | 1,846,519 | ||||||
EcoR1 Capital, LLC(2) | 2,900,000 | 1,450,000 | ||||||
ARCH Venture Fund VIII, L.P. | 2,689,874 | 1,344,937 | ||||||
Directors, Directors-Elect and Executive Officers | ||||||||
Troy Wilson, Ph.D., J.D.(3) | 4,337,454 | 2,168,727 | ||||||
Heidi Henson(4) | 511,192 | 255,596 | ||||||
Yi Liu, Ph.D.(5) | 1,500,000 | 749,999 | ||||||
Antonio Gualberto, M.D., Ph.D. | 679,946 | 339,973 | ||||||
Annette North | 187,500 | 93,750 | ||||||
Pingda Ren, Ph.D.(6) | 1,547,966 | 773,982 | ||||||
Robert E. Hoffman | 18,988 | 9,494 | ||||||
| (1) | Consists of (a) 3,041,174 shares of Kura common stock owned by Fidelity Select, which were exchanged for 1,520,587 shares of our common stock, and (b) 651,864 shares of Kura common stock owned by Fidelity Advisor, which were exchanged for 325,932 shares of our common stock. |
| (2) | Consists of (a) 1,836,000 shares of Kura common stock owned by EcoR1 Capital Fund Qualified, L.P., which were exchanged for 918,000 shares of our common stock, and (b) 1,064,000 shares of Kura common stock owned by EcoR1 Capital Fund, L.P., which were exchanged for 532,000 shares of our common stock. |
| (3) | Consists of (a) 25,000 shares of Kura common stock owned by the 2013If I Ran the Circus Irrevocable Trust for the benefit of Aidan Eliasson, a trust for the benefit of Dr. Wilson’s minor child, which were exchanged for 12,500 shares of our common stock, (b) 25,000 shares of Kura common stock owned by the 2013If I Ran the Circus Irrevocable Trust for the benefit of Ethan Eliasson, a trust for the benefit of Dr. Wilson’s minor child, which were exchanged for 12,500 shares of our common stock, (c) 3,473,982 shares of common stock owned byRed Fish Blue Fish Revocable Trust, dated December 31, 2012, which were exchanged for 1,736,991 shares of our common stock, and (d) 813,472 shares of common stock owned by Araxes, which were exchanged for 406,736 shares of our common stock. Dr. Wilson is the trustee ofRed Fish Blue Fish Revocable Trust, dated December 31, 2012 and as such has the dispositive power and control over the securities held by such trust. |
| (4) | Consists of (a) 501,192 shares of common stock owned by Heidi Henson, which were exchanged for 250,596 shares of our common stock, (b) 5,000 shares of common stock owned by Heidi Henson, Custodian for Emily Henson, of which Ms. Henson has dispositive power and control, which were exchanged for 2,500 shares of our common stock and (c) 5,000 shares of common stock owned by Heidi Henson, Custodian for Joshua Henson, of which Ms. Henson has dispositive power and control, which were exchanged for 2,500 shares of our common stock. |
| (5) | Consists of (a) 1,468,750 shares of common stock owned by Yi Liu, Ph.D., which were exchanged for 734,375 shares of our common stock, (b) 15,625 shares of common stock owned by Yi Liu, Custodian for Max Liu, of which Dr. Liu has dispositive power and control, which were exchanged for 7,812 shares of our common stock, and (c) 15,625 shares of common stock owned by Yi Liu, Custodian for Nicholas Liu, of which Dr. Liu has dispositive power and control, which were exchanged for 7,812 shares of our common stock. |
| (6) | Consists of (a) 1,516,716 shares of common stock owned by Pingda Ren, Ph.D., which were exchanged for 758,358 shares of our common stock, (b) 15,625 shares of common stock owned by Pingda Ren, Custodian for Evan T. Ren, of |
80
which Dr. Ren has dispositive power and control, which were exchanged for 7,812 shares of our common stock, and (c) 15,625 shares of common stock owned by Pingda Ren, Custodian for Oliver T. Ren, of which Dr. Ren has dispositive power and control, which were exchanged for 7,812 shares of our common stock.
The Redemption
Immediately following the Effective Time, pursuant to the terms of a Redemption Agreement dated March 6, 2015 by and among us and our then-current stockholders, we completed the closing of a redemption of 5,000,000 shares of our common stock from our then-current stockholders for consideration of $70,000, plus professional costs related to the transaction, not to exceed $30,000. The 5,000,000 shares constituted all of the issued and outstanding shares of our capital stock, on a fully-diluted basis, immediately prior to the Merger.
Registration Rights Agreement
At the closing of the Private Placement, Kura entered into a registration rights agreement with the investors in the Private Placement and also the existing stockholders of Kura who agreed to become parties to certain provisions of the agreement or who choose to become parties in the future, which covers substantially all of our outstanding shares of common stock as of the date of this filing. We assumed the registration rights agreement in connection with the Merger. Pursuant to the registration rights agreement and subject to the rules and regulations of the SEC, we have agreed to file a shelf registration statement covering the resale of the shares of our common stock held by the investors in the Private Placement and the shares of our common stock held by the former stockholders of Kura who are parties to the agreement. We are required to file the shelf registration statement within 60 days of the date of the registration rights agreement (May 5, 2015). In the event fewer than all of our outstanding shares of common stock can be registered pursuant to the so-called Rule 415 doctrine, priority will be given to the shares issued in the Private Placement.
We will be liable to each investor in the Private Placement (but not to the former stockholders of Kura who are parties to the agreement) for liquidated damages, on a 30-day basis, equal to 1.0% of the aggregate purchase price paid by the investor for the registrable shares of our common stock then held by the investor, subject to an overall cap of 5%, (i) if we fail to file the registration statement on time, (ii) if the registration statement is not declared effective within 120 days from the date of the registration rights agreement (or 150 days from Closing, if the registration statement is reviewed by the SEC), (iii) if we suspend (subject to limited suspension periods described below) or terminate the registration statement prior to the date which is the earlier of (x) the third anniversary of its effectiveness (or the third anniversary of the date on which all registrable shares (subject to certain limitations) are included therein, if later) and (y) the date on which all of the registrable shares cease to be registrable shares, or (iv) in the event one or more suspensions of the effectiveness of the registration statement exceeds 60 days in the aggregate during any 12-month period. We will be permitted to suspend the registration statement up to two times during any 12-month period provided such suspensions do not exceed 30 consecutive days or 60 days in the aggregate in any 12-month period, and a second suspension does not commence sooner than 30 days after the termination of the first suspension. Any suspension associated with our filing of an annual, periodic or current report, as required by the Exchange Act, will be permitted and will not be counted against the 60 day limitation. Any shares not registered due to the Rule 415 doctrine will not be subject to liquidated damages. Expenses with respect to the filing and effectiveness of such registration statement (but not selling expenses, or underwriter or agent compensation) will be paid by us, including expenses of one counsel for certain of the selling stockholders up to $25,000.
Lock-Up Provisions in Registration Rights Agreement
One of the provisions of the registration rights agreement that is applicable to the former stockholders of Kura who are parties to the agreement, other than the investors in the Private Placement, is a lock-up provision pursuant to which these stockholders agreed, subject to specified exceptions, not to sell, transfer, dispose of, contract to sell, sell any option or contract to purchase, or otherwise transfer or dispose of, directly or indirectly, any shares of our common stock or any securities convertible into or exercisable or exchangeable for shares of our common stock until the later of (a) 180 calendar days after the date on which our common stock is listed for trading on the New York Stock Exchange, the NYSE-Mkt, or the NASDAQ Stock Market or (b) the date that is twelve (12) months after the Closing (March 6, 2016). These lock-up provisions will not apply to, among other things, shares of common stock acquired in connection with any follow-on securities offerings by us or in open market transactions, or upon the exercise of stock options granted pursuant to our equity incentive plans, so long as the shares acquired upon exercise remain subject to the lock-up provisions in the agreement, or certain gifts and other transfers for estate-planning purposes or by stockholders who are entities to their limited partners, members or stockholders, as specified in the agreement. In the event that a former stockholder of Kura was also an investor in the Private Placement, then these lock-up provisions in the agreement will only apply with respect to the shares held by such stockholder that were not purchased in the Private Placement. Under the registration rights agreement, the investors in the Private Placement agreed, subject to specified exceptions, not to sell, transfer, dispose of, contract to sell, sell any option or contract to purchase, or otherwise transfer or
81
dispose of, directly or indirectly, any shares of our common stock or any securities convertible into or exercisable or exchangeable for shares of our common stock until the earlier of (a) the date on which our common stock is listed for trading on the OTC Bulletin Boards, OTCQB, OTCQX, the New York Stock Exchange, the NYSE-Mkt, or the NASDAQ Stock Market or (b) 180 calendar days following the date of the closing of the Private Placement. Certain investors in the Private Placement agreed under the Registration Rights Agreement to continue to hold at least 100 shares of our common stock until the date on which our shares are listed for trading on the New York Stock Exchange, the NYSE-Mkt, or the NASDAQ Stock Market.
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and certain of our officers. The indemnification agreements, our pending amended and restated certificate of incorporation and our amended and restated bylaws require us to indemnify our directors and officers to the fullest extent permitted by Delaware law. See “Indemnification of Directors and Officers.”
Indemnity Agreement
As a condition to the Merger, we entered into an Indemnity Agreement with our former officers and directors pursuant to which we agreed to indemnify such former officers and directors for actions taken by them in their official capacities relating to the consideration, approval and consummation of the Merger and certain related transactions.
Sublease Agreement
In August 2014, Kura entered into a sublease agreement with Wellspring Biosciences LLC, or Wellspring, a wholly-owned subsidiary of Araxes, which was amended in December 2014. For information about our sublease with Wellspring, refer to “Description of the Business of Kura Oncology, Inc. — Facilities.”
Services Agreements
In October 2014, Kura entered into a services agreement with Wellspring. Under the services agreement, Kura pays Wellspring for the provision of various services including research and development services.
In October 2014, Kura entered into a management services agreement with Araxes, under which Araxes pays Kura a fixed fee of $100,000 per month for the provision of management services including executive management services, general administrative services, financial and tax related services, development of intellectual property and collaboration services.
Policy for Approval of Related Person Transactions
We do not currently have a policy for the review and approval of related person transactions. We intend to adopt such a policy when we adopt an audit committee charter and establish an audit committee, which we expect will be responsible for reviewing and approving all transactions in which we are a participant and in which any parties related to us, including our executive officers, our directors, beneficial owners of more than 5% of our securities, immediate family members of the foregoing persons and any other persons whom our board of directors determines may be considered related parties under Item 404 of Regulation S-K, has or will have a direct or indirect material interest.
MARKET FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
There is not currently, and there has never been, any market for any of our securities. Our securities are not eligible for trading on any national securities exchange and are not quoted for sale on any over-the-counter markets, including the Over-the-Counter Bulletin Board.
As of March 6, 2015, after giving effect to the Merger (including the Redemption) and the Private Placement, we had 14,508,177 outstanding shares of common stock held by 357 holders of record and no outstanding shares of preferred stock.
We have never repurchased any of our equity securities.
DESCRIPTION OF CAPITAL STOCK
The following statements are qualified in their entirety by reference to the detailed provisions of our certificate of incorporation, pending amended and restated certificate of incorporation and amended and restated bylaws.
82
Capital Structure
We currently have authorized capital stock of 110,000,000 shares, of which 100,000,000 are designated as common stock, par value $0.0001 per share, and 10,000,000 shares are designated as preferred stock, par value $0.0001 per share. Under our amended and restated certificate of incorporation approved by our board of directors and stockholders prior to the Merger, which we expect to file 20 days after filing with the SEC and mailing to such stockholders a definitive Schedule 14C reporting the adoption of the amended and restated certificate of incorporation, we will have authorized capital stock consisting of 210,000,000 shares, of which 200,000,000 will be designated as common stock, par value $0.0001 per share, and 10,000,000 shares will be designated as preferred stock, par value $0.0001 per share.
As of March 6, 2015, after giving effect to the Merger (including the Redemption) and the Private Placement, 14,508,177 shares of our common stock and no shares of our preferred stock were issued and outstanding.
Common Stock
The holders of our common stock are entitled to one vote per share on matters on which our stockholders vote. Subject to any preferential dividend rights of any outstanding shares of preferred stock, holders of our common stock are entitled to receive dividends, if declared by our board of directors, out of funds that we may legally use to pay dividends. If we liquidate or dissolve, holders of our common stock are entitled to share ratably in our assets once our debts and any liquidation preference owed to any then-outstanding preferred stockholders are paid. Our restated certificate of incorporation does not provide our common stock with any redemption, conversion or preemptive rights.
Preferred Stock
As of March 6, 2015, no shares of our preferred stock are currently outstanding. If we issue preferred stock in the future, such preferred stock may have priority over common stock with respect to dividends and other distributions, including the distribution of assets upon liquidation. Our board of directors has the authority, without further stockholder authorization, to issue from time to time up to 10,000,000 shares of preferred stock in one or more series and to fix the terms, voting rights, designations, preferences, limitations or restrictions of each series. Although we have no present plans to issue any shares of preferred stock, the issuance of shares of preferred stock, or the issuance of rights to purchase such shares, could decrease the amount of earnings and assets available for distribution to the holders of common stock, could adversely affect the rights and powers, including voting rights, of the common stock, and could have the effect of delaying, deterring or preventing a change of control of us or an unsolicited acquisition proposal.
Dividend Policy
We have never paid cash dividends on any of our capital stock and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. We do not intend to pay cash dividends to holders of our common stock in the foreseeable future.
Registration Rights
On March 6, 2015, Kura entered into a registration rights agreement with the investors in the Private Placement and also the existing stockholders of Kura who agreed to become parties to certain provisions of the agreement or who choose to become parties in the future, which covers substantially all of our outstanding shares of common stock. We assumed the registration rights agreement in connection with the Merger.
The holders of an aggregate of 14,482,070 shares of our common stock, or their permitted transferees, are entitled to rights with respect to the registration of these shares under the Securities Act. Registration of these shares under the Securities Act would result in these shares becoming fully tradable without restriction under the Securities Act immediately upon the effectiveness of the initial registration statement, except for shares held by affiliates.
Resale Registration Rights
Pursuant to the registration rights agreement and subject to the rules and regulations of the SEC, we have agreed to file a shelf registration statement covering the resale of the shares of our common stock held by the investors in the Private Placement and the shares of our common stock held by the former stockholders of Kura who are parties to the agreement. We are required to file the shelf registration statement within 60 days of the date of the registration rights agreement (May 5, 2015). In the event fewer than all of our outstanding shares of common stock can be registered pursuant to the so-called Rule 415 doctrine, priority will be given to the shares issued in the Private Placement.
83
Registration of these shares under the Securities Act would result in the shares becoming saleable under the Securities Act immediately upon the effectiveness of such registration. Any sales of securities by holders of these shares could adversely affect the trading prices, if any, of our common stock.
We will be liable to each investor in the Private Placement (but not to the former stockholders of Kura who are parties to the agreement) for liquidated damages, on a 30-day basis, equal to 1.0% of the aggregate purchase price paid by the investor for the registrable shares of our common stock then held by the investor, subject to an overall cap of 5%, (i) if we fail to file the registration statement on time, (ii) if the registration statement is not declared effective within 120 days from the date of the registration rights agreement (July 4, 2015), or within 150 days from the registration rights agreement (August 3, 2015) if the registration statement is reviewed by the SEC, (iii) if we suspend (subject to limited blackout periods described below) or terminate the registration statement prior to the date which is the earlier of (x) the third anniversary of its effectiveness (or the third anniversary of the date on which all registrable shares (subject to certain limitations) are included therein, if later) and (y) the date on which all of the registrable shares cease to be registrable shares, or (iv) in the event one or more suspensions of the effectiveness of the registration statement exceeds 60 days in the aggregate during any 12-month period. We will be permitted to suspend the registration statement one or more times during any 12-month period provided such suspensions do not exceed 30 consecutive days or 60 days in the aggregate in any 12-month period.
Form S-3 Demand Registration Rights
Pursuant to the registration rights agreement, at any time after we become eligible to file a registration statement on Form S-3, subject to specified limitations set forth in the registration rights agreement, the holders of a specified percentage of the registrable shares of common stock then outstanding may request that we register on Form S-3 all or a portion of the registrable shares.
“Piggyback” Registration Rights
Pursuant to the registration rights agreement, if we propose to register any of our common stock in a firm commitment underwritten offering, the holders of registrable shares of our common stock will be entitled to notice of the registration and have the right to require us to register all or a portion of the registrable shares then held by them, subject to our right and the right of our underwriters to reduce the number of shares proposed to be registered in view of market conditions.
Expenses of Registration
We have agreed to pay all fees and expenses relating to the initial registration statement, as well as all Form S-3 demand registrations and piggyback registrations, including (i) up to $25,000 in fees of one special counsel for certain of the investors in connection with the filing of the initial registration statement and (ii) up to $25,000 in fees of one special counsel for certain of the investors in connection with the filing of one or more registration statements pursuant to the Form S-3 demand and piggyback registration rights.
Expiration of Registration Rights
The resale registration rights described above shall terminate upon the earlier of (1) the date on which all registrable shares have been effectively registered under the Securities Act and disposed of in accordance with such registration statement, and (2) the later of the third anniversary of the date (A) the initial registration statement is declared effective and (B) all registrable shares (subject to certain limitations) have been registered in the initial registration statement.
Anti-takeover Effects of Our Pending Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws
Our pending amended and restated certificate of incorporation and amended and restated bylaws contain certain provisions that may have anti-takeover effects, making it more difficult for or preventing a third party from acquiring control of us or changing our board of directors and management. According to our pending amended and restated certificate of incorporation and amended and restated bylaws, the holders of our common stock do not have cumulative voting rights in the election of our directors. The combination of the present ownership and control of 30.3% of our issued and outstanding common stock by our executive officers, directors and directors-elect as a group and the lack of cumulative voting, makes it more difficult for other stockholders to replace our board of directors or for a third party to obtain control of us by replacing our board of directors.
Delaware Anti-Takeover Law
84
We are subject to Section 203 of the Delaware General Corporation Law, or Section 203. Section 203 generally prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
| • | prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; |
| • | upon consummation of the transaction which resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced; or |
| • | at or subsequent to the time of the transaction, the business combination is approved by the board of directors of the corporation and authorized at an annual or special meeting of its stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of our outstanding voting stock that is not owned by the interested stockholder. |
In general, Section 203 defines a “business combination” to include mergers, asset sales and other transactions resulting in financial benefit to a stockholder and an “interested stockholder” as a person who, together with affiliates and associates, owns (or within three years, did own) 15% or more of a corporation’s voting stock.
Section 203 could prohibit or delay mergers or other takeover or change in control attempts not approved in advance by our board of directors and, accordingly, may discourage attempts to acquire us even though such a transaction may offer our stockholders the opportunity to sell their stock at a price above the prevailing market price.
Pending Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws
Provisions of our pending amended and restated certificate of incorporation and amended and restated bylaws may delay or discourage transactions involving an actual or potential change of control or change in our board of directors or our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our pending amended and restated certificate of incorporation and amended and restated bylaws:
| • | permit our board of directors to issue up to 10,000,000 shares of preferred stock, with any rights, preferences and privileges as they may designate (including the right to approve an acquisition or other change of control); |
| • | provide that the authorized number of directors may be changed only by resolution of our board of directors; |
| • | provide that directors may only be removed, subject to any limitation imposed by law, by the holders of at least 66 2/3% of the voting power of all of our then-outstanding shares of the capital stock entitled to vote generally at an election of directors; |
| • | provide that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; |
| • | require that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent or electronic transmission; |
| • | provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide advance notice in writing, and also specify requirements as to the form and content of a stockholder’s notice; |
| • | provide that special meetings of our stockholders may be called only by the chairman of our board of directors, our chief executive officer or by our board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors (whether or not there exists any vacancies); |
| • | do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose); and |
| • | provide that the Court of Chancery of the State of Delaware will be the sole and exclusive forum for (1) any derivative action or proceeding brought on our behalf, (2) any action asserting a claim of breach of a fiduciary duty owed by any of our directors or officers to us or our stockholders, (3) any action asserting a claim against the us arising pursuant to any provision of the Delaware General Corporation Law or our amended and restated certificate of incorporation or amended and restated bylaws, or (4) any action asserting a claim against us governed by the internal affairs doctrine. |
85
The amendment of any of these provisions, with the exception of the ability of our board of directors to issue shares of preferred stock and designate any rights, preferences and privileges thereto, would require the affirmative vote of the holders of at least 66 2/3% of the voting power of all of our then outstanding common stock.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is American Stock Transfer & Trust Company, LLC. The transfer agent and registrar’s address is 6201 15th Avenue, Brooklyn, NY 11219.
Recent Sales of Unregistered Securities
Set forth below is information regarding shares of common stock and convertible notes issued by us and by Kura within the past three years that were not registered under the Securities Act. Also included is the consideration, if any, received by us and by Kura for such shares and notes and information relating to the section of the Securities Act, or rule of the Securities and Exchange Commission, under which exemption from registration was claimed. The number of shares issued prior to the Merger described below in paragraphs A, C and D below do not reflect the exchange of shares in the Merger, which is further described in paragraph E below.
Original Issuances of Stock and Convertible Notes
A. Since August 29, 2014, Kura has issued an aggregate of 9,887,000 shares of Kura restricted common stock to certain founders and investors for aggregate consideration of $14,000.
B. Since October 8, 2014, Kura has issued convertible promissory notes having an aggregate principal amount of $7,500,000 to certain investors. In connection with the Private Placement described in paragraph D below, the principal amount of all outstanding convertible promissory notes, plus accrued interest, converted into Kura common stock on March 6, 2015.
C. On March 6, 2015, immediately prior to the Effective Time, Kura issued 18,971,136 shares of its common stock at a price of $3.16 per share, or an aggregate purchase price of approximately $60.0 million, which included approximately $7.5 million in principal and $0.1 million in accrued interest from the conversion of Kura’s then outstanding convertible promissory notes, to investors in the Private Placement. As part of the Private Placement, all outstanding convertible promissory notes, plus accrued interest thereon, described in paragraph B above, were converted into shares of Kura common stock at a price of $3.16 per share. Leerink Partners LLC acted as sole lead placement agent and National Securities Corporation and Livingston Securities LLC acted as co-agents for purposes of the sale of Kura common stock in the Private Placement. Entities affiliated with Leerink Partners LLC purchased an aggregate of 317,704 shares of Kura common stock in the Private Placement on the same terms as the other investors through the conversion of convertible promissory notes.
D. On March 6, 2015, concurrently with the Private Placement, Kura issued an aggregate of 158,226 shares of its common stock as partial consideration for its license agreement, as amended, with the University of Michigan.
E. On March 6, 2015, at the Effective Time, each share of Kura common stock that was issued and outstanding immediately prior to the Effective Time was automatically exchanged for 0.5 shares of our common stock. We issued an aggregate of 14,508,177 shares of our common stock upon such exchange of the outstanding shares of Kura common stock to Kura’s stockholders immediately prior to the Effective Time, which included no more than 35 non-accredited investors. In addition, at the Effective Time, we assumed the Kura 2014 Equity Incentive Plan.
Securities Act Exemptions
The offers, sales and issuances of the securities described in paragraph A above were deemed to be exempt from registration pursuant to either Rule 701 promulgated under the Securities Act as transactions pursuant to compensatory benefit plans or pursuant to Section 4(2) of the Securities Act as transactions by an issuer not involving a public offering. The shares of common stock issued upon the exercise of options are deemed to be restricted securities for purposes of the Securities Act.
The offers, sales and issuances of the securities described in paragraphs B, C, D and E above were deemed to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act (or Regulation D promulgated thereunder) as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions was an accredited investor within the meaning of Rule 501 of Regulation D under the Securities Act and had adequate access, through employment, business or other relationships, to information about the Registrant.
Shares Eligible for Future Sale
86
Future sales of substantial amounts of shares of our common stock, including shares issued upon the exercise of outstanding options, if any, in the public market or the possibility of these sales occurring could cause the prevailing market price for our common stock to fall or impair our ability to raise equity capital in the future. As of March 6, 2015, we had outstanding 14,508,177 shares of common stock. All of these shares are restricted securities under Rule 144, in that they were issued in private transactions not involving a public offering.
Restrictions on the Use of Rule 144 by Shell Companies or Former Shell Companies
Rule 144 is not available for the resale of securities initially issued by companies that are, or previously were, blank check companies like us, to their promoters or affiliates despite technical compliance with the requirements of Rule 144. Rule 144 also is not available for resale of securities issued by any shell companies (other than business combination-related shell companies) or any issuer that has been at any time previously a shell company. The SEC has provided an exception to this prohibition, however, if the following conditions are met:
| • | the issuer of the securities that was formerly a shell company has ceased to be a shell company; |
| • | the issuer of the securities is subject to the reporting requirements of Section 13 or 15(d) of the Exchange Act; |
| • | the issuer of the securities has filed all Exchange Act reports and materials required to be filed, as applicable, during the preceding 12 months (or such shorter period that the issuer was required to file such reports and materials), other than Form 8-K reports; and |
| • | at least one year has elapsed from the time that the issuer filed current Form 10 type information with the SEC reflecting its status as an entity that is not a shell company. |
As a result, none of our stockholders is currently able to sell shares of our common stock in reliance on Rule 144. Assuming we continue to meet the requirements set forth above, Rule 144 will become available to our stockholders one year after the date of this report. Our stockholders may currently resell their shares of our common stock only pursuant to a registration statement that has been declared effective under the Securities Act or pursuant to another exemption from registration.
Lock-Up Provisions in Registration Rights Agreement
The registration rights agreement contains lock-up provisions applicable to holders of our common stock. See “Certain Relationships and Related Person Transactions—Lock-Up Provisions in Registration Rights Agreement.”
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
The disclosures set forth in Item 4.01 below are hereby incorporated by reference into this Item 2.01.
| Item 3.02 | Unregistered Sales of Equity Securities |
The disclosures set forth in Item 2.01 above are hereby incorporated by reference into this Item 3.02.
| Item 4.01 | Changes in Registrant’s Certifying Accountant |
Effective at the Effective Time of the Merger, LWBJ, LLP, or LWBJ, was dismissed as the independent registered public accounting firm that audits the financial statements of the Company. Effective as of the Effective Time, our board of directors engaged Ernst & Young LLP, as the independent registered public accounting firm to audit the Company’s financial statements for the fiscal year ending December 31, 2015.
LWBJ’s audit report on the Company’s financial statements for the fiscal years ended December 31, 2014 and 2013 did not contain an adverse opinion or a disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principles.
During the fiscal years ended December 31, 2014 and 2013 and the subsequent interim period through the date of LWBJ’s dismissal, there were no disagreements with LWBJ on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedure, which disagreements, if not resolved to the satisfaction of LWBJ, would have caused it to make reference to the subject matter thereof in connection with its report.
87
During the fiscal years ended December 31, 2014 and 2013 and the subsequent interim period through the date of LWBJ’s dismissal, neither the Company nor anyone acting on its behalf consulted Ernst & Young LLP regarding the application of accounting principles to a specified transaction, either completed or proposed or the type of audit opinion that might be rendered on the Company’s financial statements.
The Company has provided LWBJ with a copy of this report prior to the filing hereof and has requested that LWBJ furnish to the Company a letter addressed to the Securities and Exchange Commission stating whether LWBJ agrees with the statements made by the Company in this report. LWBJ has furnished such letter, which letter is filed as Exhibit 16.1 hereto, as required by Item 304(a)(3) ofRegulation S-K.
| Item 5.01 | Changes in Control of Registrant |
The disclosures set forth in Item 2.01 above are hereby incorporated by reference into this Item 5.01.
| Item 5.02 | Departure of Directors or Certain Officers; Election of Directors; Appointment of Certain Officers; Compensatory Arrangements of Certain Officers |
At the Effective Time, John Pappajohn resigned from our board of directors and effective immediately following the Effective Time, Troy E. Wilson, Ph.D., J.D. was appointed to our board of directors, and together with Matthew P. Kinley, constitute our board of directors as of March 6, 2015. Effective upon the eleventh day after we file with the SEC and mail to our former stockholders a Schedule 14f-1 reporting a change in the majority of our directors, our board of directors will be reconstituted by the appointment of Robert E. Hoffman to serve with Dr. Wilson as our directors, and the resignation of Mr. Kinley as director.
Effective immediately following the Merger, our executive management team was also reconstituted by the appointment of Dr. Wilson as our President and Chief Executive Officer; Heidi Henson as our Chief Financial Officer and Secretary; Yi Liu, Ph.D. as our Chief Scientific Officer; Antonio Gualberto, M.D., Ph.D., as our Chief Medical Officer; Annette North as our Senior Vice President, General Counsel; and Pingda Ren, Ph.D., as our Senior Vice President, Chemistry and Pharmaceutical Sciences, effective upon the resignation of Mr. Pappajohn as our President and Mr. Kinley as our Chief Financial Officer and Secretary.
Biographical and other information regarding these individuals is provided under the caption “Management and Directors” in Item 2.01 above, which is incorporated by reference into this Item 5.02.
| Item 5.03 | Amendments to Articles of Incorporation or Bylaws; Change in Fiscal Year |
On March 6, 2015, we filed a Certificate of Ownership and Merger with the Secretary of State of the State of Delaware pursuant to which Kura Oncology, Inc., our newly created wholly-owned subsidiary, merged with and into us with us remaining as the surviving corporation, which we refer to as the Name Change Merger. In connection with the Name Change Merger, and as set forth in the Certificate of Ownership and Merger, we changed our corporate name to “Kura Oncology, Inc.” The Certificate of Ownership and Merger is filed herewith as Exhibit 3.4.
Effective March 6, 2015, prior to the Merger, our board of directors and stockholders approved, each by written consent, an amended and restated certificate of incorporation. We intend to prepare and file an information statement on Schedule 14C with the SEC to notify our former stockholders of this action. Twenty days after we mail the information statement, we expect to file the amended and restated certificate of incorporation with the Secretary of State of the State of Delaware. When the amended and restated certificate of incorporation becomes effective, we will have authorized capital stock of 210,000,000 shares, of which 200,000,000 shares will be designated as common stock, par value $0.0001 per share, and of which 10,000,000 shares will be designated as preferred stock, par value $0.0001 per share.
Effective March 6, 2015, prior to the Merger, our board of directors and stockholders approved, each by written consent, our amended and restated bylaws.
Please see the description of the amended and restated certificate of incorporation and amended and restated bylaws in Item 2.01 Completion of Acquisition or Disposition of Assets of this Current Report on Form 8-K in the section titled “Anti-takeover Effects of Our Pending Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws.” A copy of the amended and restated certificate of incorporation that we intend to file with the Secretary of State of the State of Delaware is filed herewith as Exhibit 3.2 and a copy of our amended and restated bylaws is filed herewith as Exhibit 3.5, each of which is incorporated hereby by reference.
| Item 5.06 | Change in Shell Company Status |
88
As described in Item 2.01 above, which is incorporated by reference into this Item 5.06, we ceased being a shell company (as defined in Rule 12b-2 under the Exchange Act) upon completion of the Merger.
| Item 9.01 | Financial Statements and Exhibits |
| (a) | Financial Statements of Businesses Acquired. In accordance with Item 9.01(a), audited financial statements for the year ended December 31, 2014, are filed with this Current Report on Form 8-K as Exhibit 99.1. |
| (b) | Pro Forma Financial Information. In accordance with Item 9.01(b), our unaudited pro forma financial statements are filed with this Current Report as Exhibit 99.2. |
| (c) | Shell Company Transactions. Reference is made to Items 9.01(a) and 9.01(b) and the exhibits referred to therein, which are incorporated herein by reference. |
| (d) | Exhibits. See Exhibit Index following the signature page of this Current Report on Form 8-K, which is incorporated by reference here. |
89
SIGNATURE
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| Dated: March 12, 2015 | KURA ONCOLOGY, INC. | |||||
| By: | /s/ Troy Wilson, Ph.D., J.D. | |||||
| Troy Wilson, Ph.D., J.D. | ||||||
| President and Chief Executive Officer | ||||||
90
EXHIBIT INDEX
Exhibit | Description | |
| 2.1 | Agreement and Plan of Merger, dated March 6, 2015, by and among the Registrant, Kura Operations, Inc. and Kura Oncology, Inc. | |
| 2.2 | Agreement and Plan of Merger, dated March 6, 2015, by and between the Registrant and Kura Oncology, Inc., relating to the name change of the Registrant. | |
| 3.1 | Certificate of Incorporation of the Registrant, as filed with the Secretary of State of the State of Delaware on November 16, 2007 (incorporated by reference to Exhibit 3.1 to the Registrant’s Registration Statement on Form 10 filed with the SEC on February 1, 2008 (File No. 000-54896)). | |
| 3.2 | Form of Amended and Restated Certificate of Incorporation of the Registrant, to be filed with the Secretary of State of the State of Delaware. | |
| 3.3 | Certificate of Merger relating to the Merger of Kura Operations, Inc. with and into Kura Oncology, Inc., filed with the Secretary of State of the State of Delaware on March 6, 2015. | |
| 3.4 | Certificate of Ownership and Merger relating to the merger of Kura Oncology, Inc. with and into the Registrant, filed with the Secretary of State of the State of Delaware on March 6, 2015, relating to the name change of the Registrant. | |
| 3.5 | Form of Amended and Restated Bylaws of the Registrant. | |
| 4.1 | Form of Common Stock certificate. | |
| 4.2 | Registration Rights Agreement, dated as of March 6, 2015, by and among the Kura Oncology, Inc. and the Investors listed on Schedule A thereto. | |
| 10.1+ | Kura Oncology, Inc. Amended and Restated 2014 Equity Incentive Plan and Forms of Stock Option Agreement, Notice of Exercise and Stock Option Grant Notice thereunder. | |
| 10.2+ | Form of Restricted Stock Purchase Agreement and Restricted Stock Purchase Award Notice under the Kura Oncology, Inc. Amended and Restated 2014 Equity Incentive Plan. | |
| 10.3+ | Kura Oncology, Inc. 2015 Employee Stock Purchase Plan. | |
91
| 10.4+ | Form of Indemnification Agreement by and between Kura Oncology, Inc. and each of its directors and officers. | |
| 10.5+ | Executive Employment Agreement, effective as of October 1, 2014, by and between Kura Oncology, Inc. and Troy Wilson, Ph.D., J.D. | |
| 10.6* | License Agreement, dated December 18, 2014, by and between Kura Oncology, Inc. and Janssen Pharmaceutica NV. | |
| 10.7* | Asset Purchase Agreement, dated December 23, 2014, by and between Kura Oncology, Inc. and Araxes Pharma LLC. | |
| 10.8 | Sublease, dated August 29, 2014, by and between Kura Oncology, Inc. and Wellspring Biosciences LLC. | |
| 10.9 | First Amendment to Sublease, dated December 18, 2014, by and between Kura Oncology, Inc. and Wellspring Biosciences LLC. | |
| 10.10 | Redemption Agreement dated as of March 6, 2015 by and between the Registrant and stockholders of the Registrant listed therein. | |
| 10.11 | Indemnity Agreement dated as of March 6, 2015 by and among the Registrant, Kura Oncology, Inc. and each of John Pappajohn and Matthew P. Kinley. | |
| 16.1 | Letter from LWBJ, LLP to the Securities and Exchange Commission, dated March 12, 2015. | |
| 21.1 | Subsidiaries. | |
| 99.1 | Audited financial statements of Kura Oncology, Inc. for the period from August 22, 2014 (inception) to December 31, 2014. | |
| 99.2 | Unaudited pro forma financial statements. | |
| 99.3 | Press Release, dated March 12, 2015. | |
| + | Indicates management contract or compensatory plan. |
| * | Confidential treatment has been requested with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission. |
92