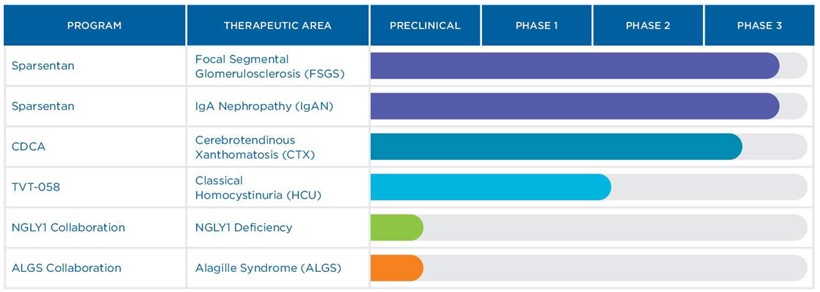
pharmacodynamics and clinical effects in patients with classical HCU. At this time, topline data from the ongoing Phase 1/2 study are expected to become available in the third quarter of 2021. TVT-058 has been granted Rare Pediatric Disease designation for treatment of cystathionine B-synthase deficiency homocystinuria and Fast Track designation for treatment of cystathionine B-synthase deficiency homocystinuria by the FDA, as well as orphan drug designation in the United States for the treatment of homocystinuria and Europe for the treatment of homocystinuria. It is estimated that there are at least 3,500 people living with HCU in the US with similar numbers in Europe. We acquired TVT-058 as part of the November 2020 acquisition of Orphan Technologies Limited.
Cooperative Research and Development Agreements, or CRADAs
We are a participant in two CRADAs, which form a multi-stakeholder approach to pool resources with leading experts, and incorporate the patient perspective early in the identification and development process. We have partnered with the National Institutes of Health’s National Center for Advancing Translational Sciences and leading patient advocacy organizations, NGLY1.org and Alagille Syndrome Alliance, aimed at the identification of potential small molecule therapeutics for NGLY1 deficiency and Alagille syndrome, respectively. There are no treatment options currently approved for these diseases.
We currently sell the following products:
| | • | | Chenodal (chenodeoxycholic acid) is approved in the United States for the treatment of patients suffering from gallstones in whom surgery poses an unacceptable health risk due to disease or advanced age. Chenodal has also been the standard of care for CTX patients for more than three decades and we are currently pursuing adding this indication to the label. |
| | • | | Cholbam (cholic acid) is approved in the United States for the treatment of bile acid synthesis disorders due to single enzyme defects and is further indicated for adjunctive treatment of patients with certain peroxisomal disorders. |
| | • | | Thiola and Thiola EC (tiopronin) are approved in the United States for the prevention of cystine (kidney) stone formation in patients with severe homozygous cystinuria. On June 28, 2019, we announced that the U.S. Food and Drug Administration approved 100 mg and 300 mg tablets of Thiola EC, a new enteric-coated formulation of Thiola, to be used for the treatment of cystinuria. Thiola EC offers the potential for administration with or without food, and the ability to reduce the number of tablets necessary to manage cystinuria. Thiola EC became available to patients in July 2019. |
Recent Developments
Certain Unaudited Preliminary 2020 Financial Results
Although our financial results as of and for the fourth quarter and full year ended December 31, 2020 are not yet finalized, based on currently available information, we expect net product sales for the fourth quarter of 2020 to be approximately $51.0 million, net product sales for the full year ended December 31, 2020 to be approximately $198.3 million, operating expenses for the full year ended December 31, 2020 to be approximately $374.5 million (of which approximately $97.1 million is related to in process research and development expense associated with our acquisition of Orphan Technologies Limited in November 2020), and our cash, cash equivalents and available-for-sale debt securities as of December 31, 2020 to be approximately $361.6 million.
The preliminary results set forth above are based on management’s initial review of our operations for the quarter and full year ended December 31, 2020 and are subject to revision based upon our year-end closing procedures and the completion and external audit of the our year-end financial statements. Our independent registered public accounting firm has not audited these preliminary financial results. Actual results may differ materially from these preliminary results as a result of the completion of year-end closing procedures, final adjustments, and other developments arising between now and the time that our financial results are finalized. In addition, these preliminary results are not a comprehensive statement of our financial results for the fourth quarter or full year ended December 31, 2020, should not be viewed as a substitute for full, audited financial statements prepared in accordance with generally accepted accounting principles, and are not necessarily indicative of our results for any future period.
Interim Results from Phase 3 DUPLEX Study
In February 2021, we announced that our ongoing pivotal Phase 3 DUPLEX Study of sparsentan in focal segmental glomerulosclerosis (“FSGS”) achieved its pre-specified interim FSGS partial remission of proteinuria endpoint (“FPRE”) after 36 weeks of treatment. Consistent with prior guidance, we are providing limited data from the interim analyses to maintain trial integrity in the ongoing study.