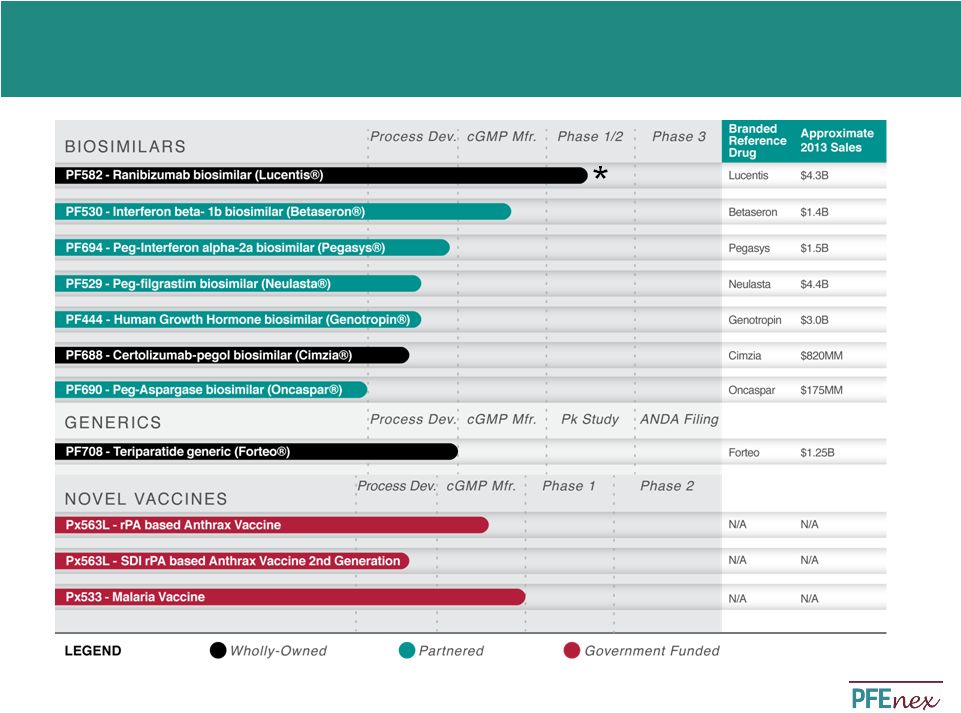
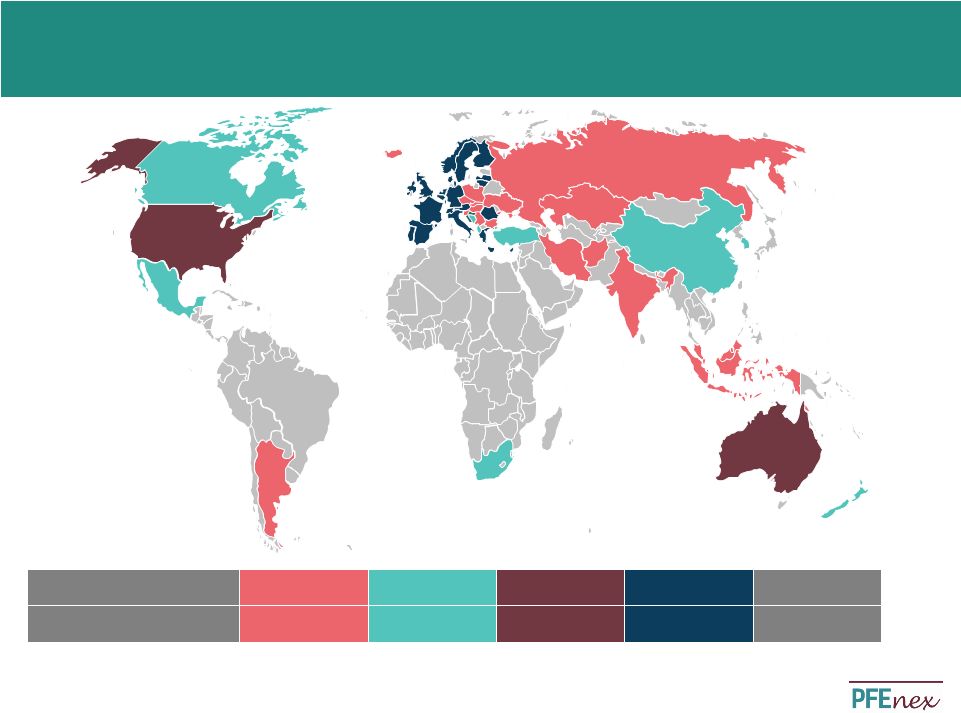
19 Overview of US/EU Biosimilar Regulation US Biosimilar Regulatory Path EU Biosimilar Regulatory Path • 351(k) U.S. biosimilar approval pathway established in 2010 – Five FDA guidance documents provide additional clarity on path • Benefits of 351(k) = abbreviated development • Bioanalytical assessment is a key element of biosimilar market authorization – Defines degree of similarity and whether 351(k) pathway is available for the proposed product • FDA Guidance adds clarity regarding pathway timing/requirements – Similar to EU process: totality of evidence – Specific delineation and outcomes of meetings with the FDA in fourth guidance document – Unlike generics, patent disputes do not trigger 30 month delay; FDA review of 351(k) submission may continue during any litigation • Additional interchangeability guidance expected in 2014 • CHMP/437/04 biosimilar approval pathway established in 2005 – Biosimilar guidance on quality as well as non-clinical and clinical issues, including immunogenicity – Additional product-class specific annex guidelines issued including: • EMA approval based on totality of evidence similar to FDA 351(k) pathway • First biosimilar approved in 2006 (Omnitrope) • As of 2014 20 biosimilar products have been approved by EMA – Price discount between 8% and 23% of innovator in for biosimilars 2010 – Avg. 71% filgrastim biosimilar penetrance in EU – >50% EPO biosimilar penetrance in Germany – >90% EPO and filgrastim biosimilar penetrance in Hungary • Australia, Canada, Japan, South Korea and South Africa have adopted a biosimilar path very similar or idem quot to the EU – Abbreviated pathway with limited preclinical and clinical studies, with reference to innovator product Wang, J. and Chow, S. On the Regulatory Approval Pathway of Biosimilar Products. Pharmaceuticals, 5 (4). April 2012, pp. 353-368. Alliance for Safe Biologic Medicines. Alliance for Safe Biologic Medicines: Information Center. Alliance for Safe Biologic Medicines. http://www.safebiologics.org/info-center.php. |