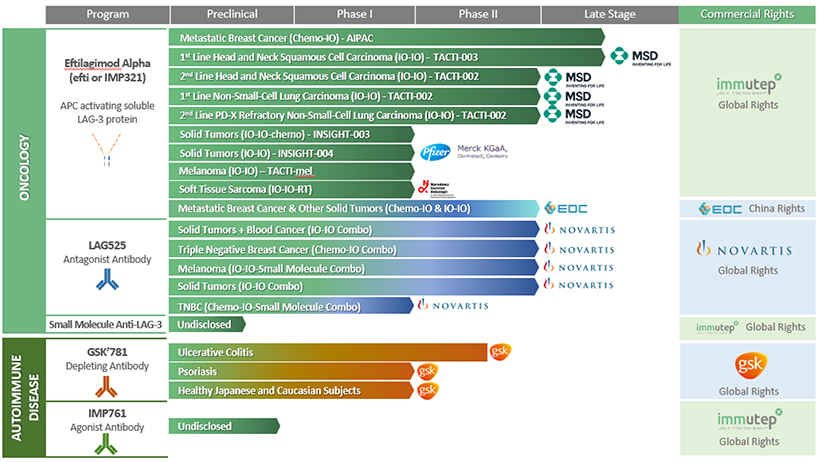
Our lead product candidate, eftilagimod alpha (IMP321 or efti), is being developed as a cancer therapeutic. Efti’s MOA stimulates and augments the human body’s natural immune response to fight cancer tumors, and it is a member of a class of drugs known as “antigen presenting cell (APC) activators.” Other types of APC activators include toll like receptor (TLR) agonists, stimulator of interferon genes (STING) agonists, CD40 agonists, or oncolytic viral therapies. Efti’s unique MOA leads to the activation of APCs, such as monocytes and dendritic cells, which results in increased T cell levels.
We are aware of other companies that are developing cancer therapeutics in the same specific indications we are currently targeting and may target in the future. Some of these competitors are developing APC product candidates, other immune-modulating therapeutics that promote an immunological response against cancer and therapies targeting patients who have received prior anti-PD-1/PD-L1 therapies. These companies include, but are not limited to Birdie Biotech, Inc., AstraZeneca PLC, Arcus Biosciences Inc., Curevac NV, Gilead Sciences Inc., GlaxoSmithKline plc, Hoffmann-La Roche Ltd., Idera Pharmaceuticals, Inc., AbbVie, Inc., Incyte Corporation, ImmunityBio, Inc, Merck & Co, Idera Pharmaceuticals, Inc., Nektar Therapeutics, Eisai Co Ltd. and Regeneron Pharmaceuticals, Inc.
Current treatments for metastatic breast cancer, include chemotherapies/cytotoxics, parp inhibitors, CDK4/6 inhibitors, angiogenesis inhibitors, nonsteroidal aromatase inhibitors and immunotherapies. Efti is developed for a specific subset of metastatic breast cancer, specifically human epidermal growth factor receptor 2-negative (HER2-), metastatic breast cancer patients that have previously undergone endocrine based therapy (possibly with CDK4/6 therapy and/or PI3Kinase inhibitors) and are eligible to receive chemotherapy. Efti is also being investigated in patients with recurrent/metastatic head and neck squamous cell carcinomas (R/M HNSCC). Current treatments for recurrent HNSCC include combination therapy with cetuximab, cisplatin, and 5-fluoruracil (5-FU). More recently, immunotherapy through PD-1 blockade has become an option, either alone or in combination with platinum plus 5-FU. Similarly in non-EGFR/ALK targetable metastatic non-small cell lung cancer, another indication where efti is tested, platinum based chemotherapies were mostly replaced as 1st line therapy by anti-PD-(L)1 monotherapies or anti-PD-(L)1 + chemotherapy combination treatments in recent years. Despite all the progress made in these three indications, there is still a high unmet medical need as the median overall survival in these indications is still between 12-24 months dependent on the indication.
Many competitors, or potential competitors, either alone, or with their strategic partners, have substantially greater financial, technical and human resources than we do. Therefore, our competitors may be more successful than us in obtaining approval for treatments and achieving widespread market adoption which may render our treatments obsolete or non-competitive. In addition, many of our competitors have significantly greater experience than we have in undertaking preclinical studies and human clinical trials of new pharmaceutical products, obtaining FDA and other regulatory approvals of products for use in health care, manufacturing and marketing and selling approved products. Our competitors may also obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours.
We expect our product candidates, if approved and commercialized, to compete with other products on a number of factors including, but not limited to, product safety and efficacy, time to market, price, insurance coverage and reimbursement by third-party payors, extent of adverse side effects, and convenience of treatment. We may not be able to effectively compete in any of these areas.
Regulatory Authorities
Our ongoing research and development, clinical, regulatory, commercial and manufacturing activities of our pharmaceutical products are subject to extensive regulation by numerous governmental authorities, including (i) in Australia, principally the Therapeutics Goods Administration, or TGA; (ii) in the United States, principally the Food and Drug Administration, or FDA; and (iii) in Europe, principally the European Medicines Agency, or EMA and local competent authorities, human research ethic committee (HREC), ethics committees (ECs), institutional research boards (IRBs) and other regulatory authorities at federal, state or local levels. We, along with our third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval and post-approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our product candidates.
United States – FDA process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. Regulations that govern the testing, manufacturing, safety, efficacy, labeling, storage, record keeping, advertising and promotion of such products under the Federal Food, Drug and Cosmetic Act, the Public Health Service Act, and their implementing regulations. The process of obtaining FDA approval and achieving and maintaining compliance with applicable laws and regulations requires the expenditure of substantial time and financial resources. Failure to comply with applicable FDA or other requirements may result in refusal to approve pending applications, a clinical hold, warning letters, civil or criminal penalties, recall or seizure of products, partial or total suspension of production or withdrawal of the product from the market. FDA approval is required before any new drug or biologic, including a new use of a previously approved drug, can be marketed in the United States. All applications for FDA approval must contain, among other things, information relating to safety and efficacy, stability, manufacturing, processing, packaging, labeling and quality control.
32