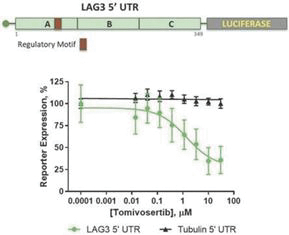
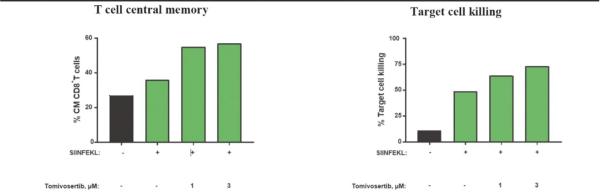
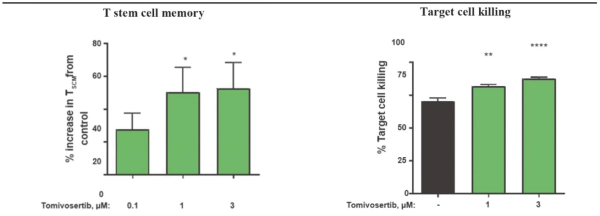
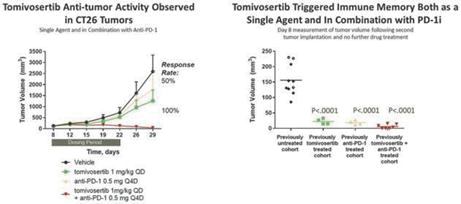
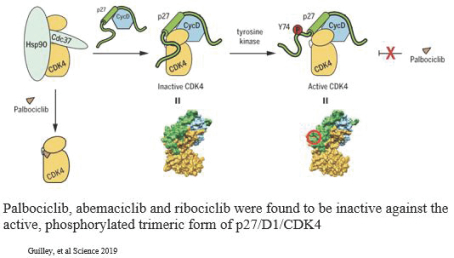
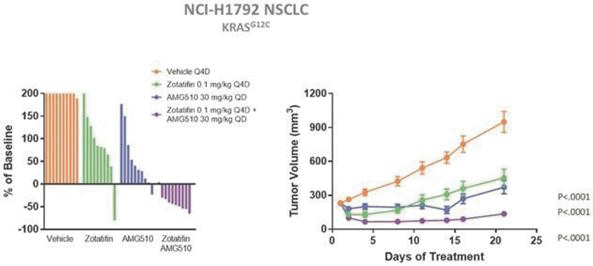
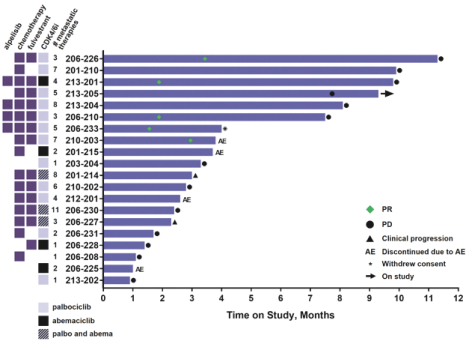
experience and scientific resources provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public and private research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. Many of our competitors, either alone or with their collaborators have significantly greater financial, technical, manufacturing, marketing, sales and supply resources or experience than we do. Accordingly, our competitors may be more successful than us in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining approval for treatments and achieving widespread market acceptance, rendering our treatments obsolete or non-competitive. Merger and acquisition activity in the biotechnology and biopharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. These companies also compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials and acquiring technologies complementary to, or necessary for, our programs. We also face competition from such companies in seeking any future potential collaborations to partner our product candidates. Our commercial opportunity could be substantially limited if our competitors develop and commercialize products that are more effective, safer, less toxic, more convenient or less expensive than our comparable products. The key competitive factors affecting the success of all of our product candidates, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic and other competition and the availability of reimbursement from government and other third-party payors.
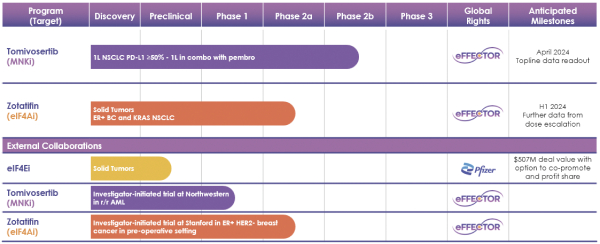
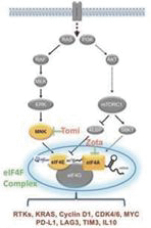
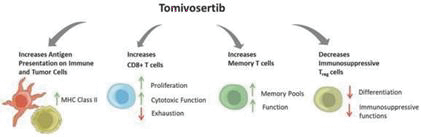
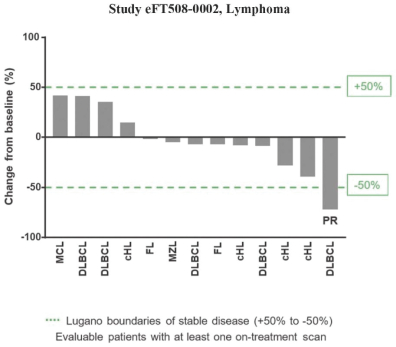
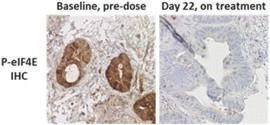
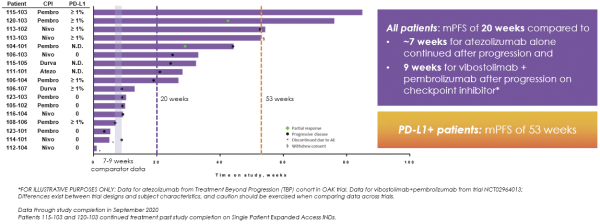
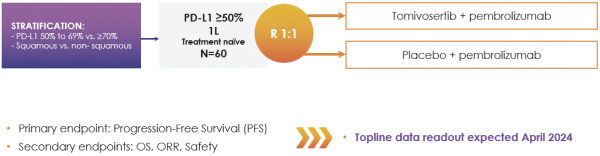
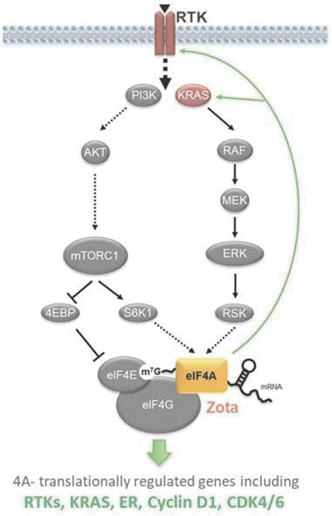
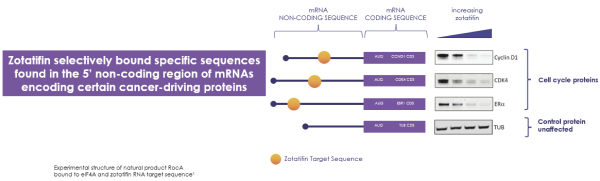
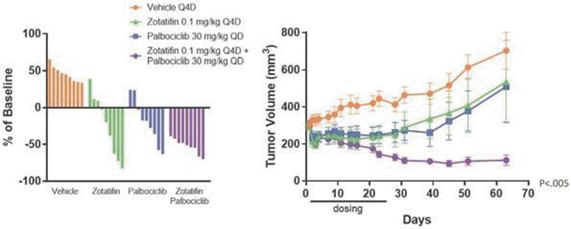
If any of our product candidates are approved in oncology indications such as NSCLC or breast cancer, they will compete with small molecule therapies, biologics, cell-based therapies and traditional chemotherapy, or a combination of any such methods. With respect to tomivosertib, we are aware of AUM Biosciences which is developing a MNK inhibitor, AUM001 in solid tumors. In addition, several novel combinations with FDA-approved PD-1 or PD-L1 inhibitors are being developed in NSCLC. These include, among others, anti-TIGIT and anti-LAG3 therapies from Bristol-Myers Squibb Co., Gilead Sciences, GSK plc, Merck & Co, Novartis AG and Roche. With respect to zotatifin, we are aware of PIC Therapeutics which is developing pre-clinical small molecules targeting the eukaryotic translation initiation factors (eIFs). Moreover, there are currently several marketed drugs and product candidates in development for the treatment of ER+ breast cancer that may compete with zotatifin, including fulvestrant marketed by AstraZeneca plc, elacestrant marketed by Menarini, capivasertib, an AKT inhibitor being marketed by AstraZeneca plc, oral SERDS being developed by Arvinas, Inc., AstraZeneca plc, Eli Lilly and Co, Eisai Co., Ltd, Roche and Zentalis Pharmaceuticals, Inc., among others.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, marketing and export and import of drug products. Generally, before a new drug can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific for each regulatory authority, submitted for review and approved by the regulatory authority. A new drug must be approved by the FDA through the NDA process before it may be legally marketed in the United States. We, along with any third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval of our products and product candidates. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. Drug Development Process
In the United States, the FDA regulates drugs under the federal Food, Drug, and Cosmetic Act (“FDCA”) and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with
117