UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| | x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | | For the fiscal year ended December 31, 2007 |
or
| | ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | | For the transition period from ______________ to ______________ |
Commission file number: 0-19986
CELL GENESYS, INC.
(Exact name of registrant as specified in its charter)
| | |
| Delaware | | 94-3061375 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. employer identification number) |
| |
500 Forbes Blvd., South San Francisco, CA | | 94080 |
| (Address of principal executive offices) | | (Zip Code) |
(650) 266-3000
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12 (b) of the Act:
| | |
Title of each class | | Name of exchange on which registered |
| Common Stock, $.001 Par Value | | The NASDAQ Stock Market LLC |
| Preferred Shares Purchase Rights | | |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check One):
| | | | | | | | | | |
| Large accelerated filer | | ¨ | | | | | Accelerated filer | | x | |
| Non-accelerated filer | | ¨ | | | (Do not check if a smaller reporting company) | | Smaller reporting company | | ¨ | |
Indicate by check mark whether the registrant is a shell company, as defined in Rule 12b-2 of the Exchange Act. Yes ¨ No x
As of June 29, 2007, the last business day of the registrant’s most recently completed second fiscal quarter the approximate aggregate market value of shares held by non-affiliates of the registrant (based on the closing sale price of shares on the NASDAQ Global Market on June 29, 2007) was $242.3 million, which excludes 613,111 shares of common stock held by directors and officers, and any stockholders whose ownership exceeded ten percent of the shares outstanding at June 29, 2007. Exclusion of these shares does not constitute a determination that each such person is an affiliate of the registrant.
As of February 27, 2008, the number of outstanding shares of the registrant’s Common Stock was 78,755,234.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Proxy Statement for the 2008 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K. Such Proxy Statement will be filed within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K.
TABLE OF CONTENTS
2
PART I
Statements made in this document other than statements of historical fact, including statements about us and our subsidiaries and our respective clinical trials, research programs, product pipelines, current and potential corporate partnerships, licenses and intellectual property, the adequacy of capital reserves and anticipated operating results and cash expenditures, are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. As forward-looking statements, they are subject to a number of uncertainties that could cause actual results to differ materially from the statements made, including risks associated with the success of research and product development programs, the issuance and validity of patents, the development and protection of proprietary technologies, the ability to raise capital, operating expense levels and the ability to establish and retain corporate partnerships and other risks. Reference is made to discussions about risks associated with product development programs, intellectual property and other risks that may affect us under Item 1A, “Risk Factors” below. We do not undertake any obligation to update forward-looking statements. The following should be read in conjunction with our consolidated financial statements located elsewhere in this Annual Report on Form 10-K for the year ended December 31, 2007 and other documents filed by us from time to time with the Securities and Exchange Commission.
In this Annual Report on Form 10-K, “Cell Genesys,” “we,” “us,” “our” and “the Registrant” refer to Cell Genesys, Inc.
Overview
We are a biotechnology company focused on the development and commercialization of novel biological therapies for patients with cancer. We are currently developing cell-based cancer immunotherapies and oncolytic virus therapies to treat different types of cancer. Our clinical stage cancer programs involve cell- or viral-based products that have been modified to impart disease-fighting characteristics that are not found in conventional chemotherapeutic agents. As part of our GVAX™ cancer immunotherapy programs, we are conducting two Phase 3 clinical trials in prostate cancer. We initiated our two Phase 3 clinical trials for GVAX immunotherapy for prostate cancer in July 2004 and June 2005, respectively, each under a Special Protocol Assessment, or SPA, with the United States Food and Drug Administration, or FDA. In May 2006, we were granted Fast Track designation for GVAX immunotherapy for prostate cancer by the FDA. Fast Track designation can potentially facilitate development and expedite the review of Biologics License Applications, or BLAs. The first of these two Phase 3 clinical trials, referred to as VITAL-1, is now fully enrolled. In January 2008 the Independent Data Monitoring Committee, or IDMC, completed a pre-planned interim analysis for VITAL-1 in the timeframe originally estimated and recommended that the study continue. In collaboration with investigators at Johns Hopkins University we are conducting Phase 2 trials in pancreatic cancer and Phase 1 and Phase 2 trials in leukemia and myelodysplastic syndrome. In our oncolytic virus therapies program, which we are developing in part through a global alliance with Novartis AG, we are conducting a multiple dose Phase 1 clinical trial of CG0070 in recurrent bladder cancer.
Cell Genesys, Inc. was incorporated in the State of Delaware in 1988. Our common stock trades on the NASDAQ Global Market under the symbol “CEGE.” Our principal executive offices are located at 500 Forbes Boulevard, South San Francisco, California 94080, and our phone number is (650) 266-3000. Our Internet home page is located athttp://www.cellgenesys.com;however, the information in, or that can be accessed through, our home page is not part of this report. Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to these reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act are available, free of charge, on or through our Internet home page as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission, or SEC.
3
In 2001, we spun out our central nervous system gene therapy technology into Ceregene, Inc., in which we now have a minority ownership position. Ceregene is continuing to develop gene therapies for the treatment of neurological disorders including Parkinson’s disease, Alzheimer’s disease, and Amyotrophic Lateral Sclerosis, or ALS, commonly known as “Lou Gehrig’s disease.”
In February 2003, our shelf registration statement on Form S-3 was declared effective by the SEC under the Securities Act of 1933, as amended, or the Securities Act, which allowed us to offer up to $150.0 million of securities on short notice in one or more public offerings registered under the Securities Act. Between March 2004 and September 2006 we used this shelf registration to raise net proceeds of $82.2 million through the sale of 10.7 million shares of our common stock. In April 2007, we used the remaining registered amount under this shelf registration to raise net proceeds of $55.4 million in a registered direct offering of 10.8 million shares of our common stock at $5.55 per share and warrants to purchase 2.2 million shares of our common stock at a price of $7.18 per share from selected institutional investors.
In March 2006, we entered into a Committed Equity Financing Facility, the 2006 CEFF, with Kingsbridge Capital Limited, pursuant to which Kingsbridge committed to purchase, subject to certain conditions, up to the lesser of 8.7 million shares of our common stock or an aggregate of $75.0 million during the three-year period following entry into the 2006 CEFF. In connection with the 2006 CEFF, we issued a warrant to Kingsbridge to purchase 0.4 million shares of our common stock at a price of $9.12 per share exercisable beginning on September 14, 2006 for a period of five years thereafter. In 2006, we received net proceeds of $27.9 million from the sale of 6.3 million shares of our common stock under the 2006 CEFF. In 2007, we received net proceeds of $7.1 million from the sale of 2.4 million shares of common stock under the 2006 CEFF, which concluded the 2006 CEFF.
In February 2007, we entered into a new CEFF, the 2007 CEFF, with Kingsbridge, pursuant to which Kingsbridge committed to purchase, subject to certain conditions, up to the lesser of 11.6 million shares of our common stock or an aggregate of $75.0 million during the three-year period following entry into the 2007 CEFF. In connection with the 2007 CEFF, we issued a warrant to Kingsbridge to purchase 0.4 million shares of our common stock at a price of $4.68 per share exercisable beginning on September 5, 2007 for a period of five years thereafter. In 2007, we received net proceeds of $23.0 million from the sale of 7.1 million shares of our common stock under the 2007 CEFF.
On May 16, 2007, our new shelf registration statement on Form S-3 was declared effective by the SEC under the Securities Act, which allows us to offer up to $150.0 million of securities on short notice in one or more public offerings under the Securities Act.
In December 2007, we sold for $12.0 million all of our assets, intellectual property and previously established licensing agreements relating to our lentiviral gene delivery technology, commonly referred to as lentiviral vectors, to GBP IP, LLC, an affiliate of GBP Capital, the majority shareholder in privately held Lentigen Corporation. As part of the asset sale agreement we retained our rights to use the technology for research and development purposes including potential future use with our cancer immunotherapy products.
We ended 2007 with $147.3 million in cash, cash equivalents and short-term investments, including $2.9 million of restricted cash and investments. We have maintained our financial position through strategic management of our resources including access to debt and equity financing and funding from various corporate collaborations and licensing agreements.
A major portion of our operating expenses to date is related to the research and development of our GVAX cancer immunotherapy and oncolytic virus therapy programs. During 2007, 2006, and 2005, our research and development expenses were $106.1 million, $96.3 million and $92.4 million, respectively. We expect that our research and development expenditures and headcount will likely remain approximately at the current levels in
4
the near term. We intend to maintain our strong commitment to research and development as an essential component of our oncology product development effort involving biological therapies for cancer and may also license additional potential products from outside parties.
Our Clinical Pipeline
| | | | | | |
Product Candidates | | Targeted Indication | | Status | | Commercialization Rights |
| GVAX Cancer Immunotherapies: | | | | | | |
| Prostate Cancer | | Prostate cancer | | Phase 3 | | Cell Genesys |
| Pancreatic Cancer | | Pancreatic cancer | | Phase 2 | | Cell Genesys |
| Leukemia | | Acute myelogenous leukemia | | Phase 2 | | Cell Genesys |
| | Chronic myelogenous leukemia | | Phase 2 | | Cell Genesys |
| | Myelodysplastic syndrome | | Phase 1 | | Cell Genesys |
| Oncolytic Virus Therapy: | | | | | | |
| CG0070 | | Bladder cancer | | Phase 1 | | Cell Genesys/Novartis |
Our GVAX™ Cancer Immunotherapy Program
Our GVAX immunotherapies are cancer treatments designed to stimulate the patient’s immune system to effectively fight cancer. GVAX cancer immunotherapies are comprised of tumor cells that are genetically modified to secrete an immune-stimulating cytokine known as granulocyte-macrophage colony-stimulating factor, or GM-CSF, and are then irradiated for safety. Since GVAX cancer immunotherapies consist of whole tumor cells, the cancer patient’s immune system can be activated against multiple tumor cell components, or antigens, potentially resulting in greater clinical benefit than if the immunotherapy consisted of only a single tumor cell component. Additionally, the secretion of GM-CSF by the modified tumor cells can greatly enhance the immune response by recruiting and activating dendritic cells at the injection site, a critical step in the optimal response by the immune system to any immunotherapy product. The antitumor immune response which occurs throughout the body following administration of a GVAX product can potentially result in the destruction of tumor cells that persist or recur following surgery, radiation therapy or chemotherapy treatment.
More than 600 patients have received our GVAX cancer immunotherapies in multiple Phase 1 and 2 clinical trials to date, and the immunotherapies have been shown to have a favorable side effect profile that avoids many of the toxicities associated with conventional cancer therapies. GVAX cancer immunotherapies can be conveniently administered in an outpatient setting as an injection into the skin, a site where immune cells, including in particular dendritic cells, can be optimally accessed and activated. Our GVAX cancer immunotherapies are being tested as non patient-specific, or allogeneic, products. We intend to develop these immunotherapies as “off-the-shelf” pharmaceutical products.
GVAX Immunotherapy for Prostate Cancer
Our GVAX immunotherapy for prostate cancer is a non patient-specific product comprised of two genetically-modified prostate cancer cell lines. We intend to develop and manufacture this immunotherapy as an “off-the-shelf” pharmaceutical for use after hormonal therapy for advanced-stage prostate cancer. Prostate cancer is the second leading cause of cancer death in men in the United States, with approximately 30,000 men dying each year from the disease. When a man is diagnosed with early-stage prostate cancer, he is treated with either a prostatectomy, which is the surgical removal of the prostate, or radiation therapy. If the patient relapses, he is treated with hormone therapy to suppress testosterone in order to reduce the growth of the tumor. When the hormone therapy fails, the patient may or may not be treated with chemotherapy depending upon whether the disease has spread, or metastasized, to other parts of the body. We have designed our Phase 3 clinical trials to evaluate whether GVAX immunotherapy for prostate cancer can benefit patients who have ceased responding to, or have become refractory to, hormone therapy and have metastatic disease.
5
We have completed five Phase 1 and Phase 2 clinical trials of our GVAX immunotherapy for prostate cancer in approximately 200 patients with various stages of recurrent prostate cancer, and the immunotherapy has had a favorable safety profile in each trial. These clinical trials include two Phase 2 clinical trials in hormone-refractory prostate cancer, or HRPC, patients with radiologic evidence of metastatic, or spreading disease, which is the target population for our current Phase 3 trials. These trials were designed to evaluate the safety and efficacy of the immunotherapy, as well as treatment regimens for Phase 3 clinical trials.
In April 2007, we reported final, updated results from our second multi-center Phase 2 trial of GVAX immunotherapy for prostate cancer which evaluated escalating doses of the immunotherapy in 80 patients with metastatic HRPC. Additional follow-up of the 22 patients who received the dose that is comparable to that being employed in our ongoing Phase 3 program indicates that the median survival is 35 months. We previously reported final median survival results from our first multi-center Phase 2 trial of GVAX immunotherapy for prostate cancer in 34 patients with metastatic HRPC that showed a median survival of 35 months for the 10 patients receiving the dose comparable to the Phase 3 dose. These consistent median survival results from the two independent multi-center Phase 2 clinical trials compare favorably to the previously published overall median survival of approximately 19 months for metastatic HRPC patients treated with Taxotere® (docetaxel) chemotherapy plus prednisone, the current standard of care for these patients. Our ongoing Phase 3 program is designed to confirm this potential survival benefit of GVAX immunotherapy for prostate cancer.
At the June 2007 meeting of the American Society of Clinical Oncology, or ASCO, we reported follow-up data from the ongoing Phase 1 clinical trial in patients with advanced prostate cancer receiving GVAX immunotherapy for prostate cancer administered in combination with ipilimumab (MDX-010), a fully human anti-CTLA-4 antibody that is being jointly developed by Medarex, Inc. and Bristol-Myers Squibb Company. The reported data included median follow-up of 18 months on the first 12 patients enrolled in the trial. Of the six patients who received the two highest doses, antitumor activity was observed in five patients, including prostate-specific antigen, or PSA, declines of greater than 50% that were maintained in four of these patients for at least six months, with the longest response to date at approximately 16 months. Clinical evidence of antitumor activity was observed in four of these five PSA responders, including complete resolution of multiple lesions on bone scan in two patients, resolution of abdominal lymph node disease by CT scan in one patient and improvement in bone pain in another patient. The five patients with PSA declines experienced either Grade 2 or 3 immune-mediated endocrine deficiencies similar in type to those previously reported with ipilimumab therapy, and such deficiencies were successfully treated with standard hormone replacement therapy. Importantly, the PSA declines could not be consistently correlated with declines in adrenal androgens and there was no induction of the alpha-21-hydroxylase auto-antibody that is seen in 90% of cases of auto-immune adrenal insufficiency. Two patients requiring thyroid replacement therapy were successfully tapered off after recovery of thyroid function, with one patient subsequently maintaining a PSA response. One patient who received the highest dose of ipilimumab tested in the trial developed a Grade 3 dose-limiting pulmonary alveolitis that responded to steroid treatment. Immunomonitoring studies showed that the combination therapy enhanced T cell and dendritic cell activity, which was more pronounced at the higher dose levels. Evaluation of antibody responses shows that the combination therapy can induce antibody responses to a broad array of previously identified cancer-associated antigens including PSMA, NY-ESO-1 and filamin-B, and that these responses were patient-specific with respect to the pattern of antibodies detected in different patients.
At the ASCO Genitourinary Cancer Symposium in February 2008, we reported the results of an analysis examining the potential association between immune responses to GVAX immunotherapy for prostate cancer and increased patient survival in our second multi-center Phase 2 trial of GVAX immunotherapy for prostate cancer. More than 400 patient-specific GVAX-induced antibody responses were identified in the sera of the treated patients by three different biochemical techniques confirming, as previously reported, that GVAX treatment results in a broad, multi-antigen immune response. An ongoing analysis of these GVAX-induced antibody responses has shown that at least two of the antibody responses are associated with increased patient survival. Of the 80 patients enrolled in the trial, the sera of 65 patients (the total number for whom adequate sera were available) were examined to determine each patient’s immune response to two specific antigens, HLA-A24 and
6
FLJ14668, following GVAX treatment. Thirty-four of 65 patients demonstrated an FLJ14668-specific antibody immune response. These 34 patients had a median survival of 43 months, compared to a median survival of 21 months achieved by the patients who did not generate anti-FLJ14668 antibodies (p=0.002). Twenty-two of these 65 patients received a dose of GVAX immunotherapy for prostate cancer comparable to that being evaluated in ongoing Phase 3 clinical trials. Of these 22 patients, 16 patients (73 percent) mounted an immune response to FLJ14668. These 16 patients achieved a median survival of 44.9 months. As previously reported, the median survival for all 22 patients in this treatment group was 35 months. Finally, of the 58 patients who were HLA-A24 genotype negative and therefore potentially able to mount anti-HLA-A24 specific antibody responses, 30 patients were found to be anti-HLA-A24 antibody positive. These 30 patients had a median survival of 43 months, compared to a median survival of 18 months in the patients who did not generate anti-HLA-A24 antibodies (p=0.05). Importantly, the apparent associations between the presence of these two specific antibody responses and survival were shown by multivariate analysis to be independent of both dose and duration of treatment.
We are conducting two Phase 3 clinical trials of GVAX immunotherapy for prostate cancer in metastatic HRPC. The first Phase 3 clinical trial, referred to as VITAL-1, commenced in July 2004 and compares GVAX immunotherapy for prostate cancer to Taxotere chemotherapy administered with prednisone in metastatic HRPC patients who are asymptomatic with respect to cancer-related pain. The VITAL-1 trial is designed to demonstrate superior survival in the patients receiving GVAX cancer immunotherapy compared to patients receiving Taxotere plus prednisone therapy. VITAL-1 is now fully enrolled with a total of 626 patients. In January 2008 we announced that the Independent Data Monitoring Committee, or IDMC, for VITAL-1 completed a pre-planned interim analysis in the timeframe originally estimated and recommended that the study continue. As is customary to preserve study blinding, the IDMC provided us no additional information other than the recommendation to continue the trial. We expect that there will be a sufficient number of events to conduct the final analysis in the second half of 2009. The second Phase 3 clinical trial, referred to as VITAL-2, commenced in June 2005 and compares GVAX immunotherapy for prostate cancer plus Taxotere chemotherapy to Taxotere chemotherapy plus prednisone with respect to a survival benefit in metastatic HRPC patients with cancer-related pain. We expect to enroll approximately 600 patients in the VITAL-2 trial and have set a goal for completing enrollment in the first half of 2009 at which time we expect that there will be enough events to conduct a pre-planned interim analysis. VITAL-1 and VITAL-2 are both being conducted in the United States, Canada and Europe.
We received a Special Protocol Assessment, or SPA, from the FDA for VITAL-1 in May 2004 and for VITAL-2 in May 2005. Under this procedure, a sponsor may seek the FDA’s agreement on the design and analysis of a clinical trial intended to form the primary basis of an effectiveness claim. If the FDA agrees in writing, its agreement may not be changed after the trial begins except in limited circumstances, such as the FDA determining that a substantial scientific issue essential to determining the safety or effectiveness of the product was identified after the trial had begun. If the outcome of the trial is successful, the sponsor will ordinarily be able to rely on it as the basis for approval with respect to effectiveness. While we have received the FDA’s agreement on a SPA for each of our two Phase 3 trials assessing GVAX immunotherapy for prostate cancer, there can be no assurance that these trials will have successful outcomes or that we will ultimately receive approval for this product. We currently manufacture GVAX immunotherapy for prostate cancer for Phase 3 clinical trials in our Hayward, California manufacturing facility, which operates in accordance with cGMP regulations, and plan to manufacture product for the potential market launch of this immunotherapy in the same facility. We have recently begun to develop a strategy to achieve optimal reimbursement for GVAX immunotherapy for prostate cancer and have conducted preliminary market research for this product.
We were granted Fast Track designation for GVAX immunotherapy for prostate cancer by the FDA in May 2006. Fast Track designation, which was mandated by the FDA Modernization Act of 1997, can potentially facilitate development and expedite the review of Biologics License Applications, or BLAs. Fast Track designation is reserved for products that demonstrate the potential to treat a serious or life-threatening condition and the potential to address unmet medical needs for that condition.
7
GVAX Immunotherapy for Pancreatic Cancer
Our GVAX immunotherapy for pancreatic cancer is a non patient-specific product. A Phase 2 clinical trial of GVAX immunotherapy for pancreatic cancer is currently being conducted by the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, or Johns Hopkins, in 60 patients with resectable pancreatic cancer who received the immunotherapy after surgical resection of their tumor and standard adjuvant radiation and chemotherapy. We reported updated results from this trial at the January 2007 ASCO Gastrointestinal Cancers Symposium that showed a median overall survival of 26.8 months. This compares favorably with published historical data from multiple single-arm and randomized studies in patients undergoing pancreatic cancer surgery and adjuvant therapy for whom the median survival has been reported to be in the range of 17 to 22 months, including the most recently reported results for gemcitabine chemotherapy. Of note, 53 of the 60 patients were considered high risk, based on the unfavorable finding that their cancer had spread to regional lymph nodes. New data reported in June 2007 included a median disease-free survival of approximately 16 months which compares favorably to the 13 months disease-free survival recently reported for gemcitabine adjuvant therapy. Moreover, a comparison of the median overall survival of 60 patients from this trial with patients at Johns Hopkins with operable pancreatic cancer who underwent surgery and adjuvant therapy without receiving GVAX indicated that the median overall survival of the latter group was approximately 21 months, or nearly six months shorter than the patients in the Phase 2 trial. The analysis of survival data further showed that the potential additive benefit of GVAX following surgery and adjuvant therapy was present during the first three years of the study although GVAX dosing did not continue beyond 18 months, suggesting that ongoing booster administrations of GVAX should be evaluated. Our collaborators at Johns Hopkins have initiated a follow-up Phase 2 clinical trial of GVAX in resectable pancreatic cancer to evaluate booster administration as well as a Phase 2 trial in metastatic pancreatic cancer evaluating GVAX in combination with Erbitux® and cyclophosphamide.
The Phase 2 trial described above was prompted by results from an initial Phase 1 clinical trial also conducted by Johns Hopkins. Data from the Phase 1 trial, which evaluated GVAX immunotherapy for pancreatic cancer in combination with surgery and standard adjuvant radiation and chemotherapy, demonstrated prolongation of disease-free survival in three of eight patients who received the two highest immunotherapy doses after surgical resection of their tumors followed by standard adjuvant radiation and chemotherapy. The most recently updated data revealed that these three patients remained alive and disease-free at least eight years after their respective diagnoses. In July 2004, studies were published inThe Journal of Experimental Medicinedescribing the immune response to the cancer immunotherapy in these three patients which indicated that patient-specific T cell immunity had been induced in these patients, but not in patients whose disease had progressed and who died.
GVAX Cancer Immunotherapy for Leukemia
Our GVAX cancer immunotherapy for leukemia is a non patient-specific GVAX cancer immunotherapy product. Clinical trials are being conducted to evaluate this GVAX cancer immunotherapy administered after initial chemotherapy pre- and post-hematopoietic stem cell transplantation in patients with newly-diagnosed acute myelogenous leukemia, or AML, after treatment with Gleevec® (imatinib mesylate) for more than one year in patients with chronic myelogenous leukemia, or CML, and as a single agent in myelodysplastic syndrome, or MDS. The goal of GVAX immunotherapy in these settings is to reduce or eliminate residual disease after standard chemotherapy or Gleevec therapy.
Updated data from a Phase 2 clinical trial in AML of GVAX immunotherapy for leukemia combined with autologous (derived from a person’s own body) leukemia cells, which has enrolled 54 patients, were presented at the May 2005 ASCO meeting. The preliminary findings of this trial indicated that this cancer immunotherapy was well tolerated and may reduce residual leukemic cells that persist after chemotherapy, as indicated by decreased levels of WT-1, a leukemia-associated genetic marker that is detectable in over 95 percent of patients with active AML. Eleven of 16 patients tested to date were reported to have decreased WT-1 levels in their
8
peripheral blood following the initiation of immunotherapy. Furthermore, two-year relapse-free survival after a single pre-transplant immunotherapy was greater in these 11 patients compared to those that did not have decreases in WT-1 (73% for the 11 patients with a decreased level of WT-1 versus 0% for those patients that did not have a decrease in WT-1, p=0.03).
Findings from a Phase 2 clinical trial in CML of GVAX immunotherapy for leukemia were presented at the June 2006 ASCO meeting. The trial was conducted by Johns Hopkins. In this trial, a total of 19 CML patients with molecular evidence of persistent leukemia following at least one year of Gleevec® (imatinib mesylate) therapy were treated with GVAX immunotherapy while continuing to receive Gleevec. Updated results showed that the addition of GVAX immunotherapy to Gleevec therapy reduced persistent leukemic disease in 10 of 19 patients as demonstrated by a complete disappearance (five patients) or a greater than one log (90%) reduction (five patients) in bcr-abl, which is a validated genetic marker found on the leukemic cells. Reductions of bcr-abl have been previously shown to be strongly associated with improved progression-free survival in CML patients treated with Gleevec. The responses were ongoing in all but one of the ten responders, with a median follow-up from treatment initiation of 14 months. Of the remaining nine patients, only one patient has developed cytogenetic progression on therapy. Treatment with GVAX immunotherapy for leukemia was well tolerated.
In May 2007, we announced three new clinical trials for GVAX immunotherapy for leukemia that are now under way in collaboration with Johns Hopkins. The new trials are based on results in an initial Phase 2 study of the product in patients with CML and include: (i) a randomized Phase 2 trial in 56 patients with CML who have persistent molecular evidence of disease following Gleevec (imatinib) therapy that will compare the combination of GVAX plus continued Gleevec to the combination of interferon-alpha, GM-CSF plus continued Gleevec with respect to the levels of bcr-abl, a well-established marker of residual leukemia; (ii) an extension study of the initial Phase 2 trial in patients with CML that will evaluate the efficacy of a second course of GVAX in the 11 of 19 patients who failed to achieve a sustained complete response; and (iii) a Phase 1 trial in 18 patients with poor risk myelodysplastic syndrome.
Our Oncolytic Virus Therapies Program
Our oncolytic virus therapies program utilizes adenovirus, one of the viruses responsible for the common cold, to create viruses that can kill cancer cells. The virus is engineered to selectively replicate in targeted cancer cells, thereby killing these cells and leaving healthy normal cells largely unharmed. The virus replicates in cancer cells until the cancer cells can no longer contain the virus and burst. The tumor cell is destroyed and the newly created viruses are believed to spread to neighboring cancer cells to continue the cycle of viral replication and tumor cell destruction.
In July 2003, we announced a global alliance with Novartis AG for the development and commercialization of oncolytic virus therapies. Under the agreement, we also acquired exclusive worldwide rights to certain oncolytic virus therapy products and certain related intellectual property of Genetic Therapy, Inc., or GTI, an affiliate of Novartis, as well as related intellectual property of Novartis. Our alliance with Novartis thereby provides us with additional oncolytic virus therapy product opportunities at the preclinical stage of development.
CG0070 Oncolytic Virus Therapy for Recurrent Bladder Cancer
CG0070, an oncolytic virus therapy, has been evaluated in numerous preclinical studies. CG0070 is the first “armed” oncolytic virus therapy developed by us, so-named because it has been engineered to secrete GM-CSF, an immune-stimulating hormone, which also serves as the adjuvant in our GVAX cancer immunotherapy platform. As a result, CG0070 can potentially destroy cancer cells by two different mechanisms: direct cell-killing by the virus and immune-mediated cell-killing stimulated by GM-CSF. In early 2005, we announced that an Investigational New Drug, or IND, application filed with the FDA for CG0070 had become effective. We initiated a Phase 1 clinical trial in patients with recurrent bladder cancer in April 2005. In July 2006, we announced that enrollment has been opened for an expanded multi-center Phase 1 clinical trial of CG0070 to
9
evaluate escalating multiple-dose regimens of CG0070 and to include up to 45 additional patients who have failed previous therapy with Bacillus Calmette-Guerin, or BCG, the current standard therapy for recurrent bladder cancer.
Government Regulations
FDA and Other Foreign Regulation
Prescription pharmaceutical products and biologics are subject to extensive pre- and post-marketing regulation by the FDA including regulations that govern the testing, manufacturing, safety, efficacy, labeling, storage, record-keeping, advertising and promotion of the products under the Federal Food, Drug and Cosmetic Act and the Public Health Services Act, and by comparable agencies in most foreign countries. The process required by the FDA before a new drug or biologic may be marketed in the U.S. generally involves the following:
| | • | | completion of preclinical laboratory and animal testing; |
| | • | | submission of an IND application, which must become effective before clinical trials may begin; |
| | • | | performance of adequate and well controlled human clinical trials to establish the safety and efficacy of the proposed drug’s or biologic’s intended use; and |
| | • | | approval by the FDA of a New Drug Application, or NDA, in the case of a drug, or of a Biologics License Application, or BLA, for a biologic. |
Foreign countries have similar requirements.
Preclinical Testing. The activities required before a pharmaceutical agent may be marketed begin with preclinical testing. Preclinical tests include laboratory evaluation of potential products and animal studies to assess the potential safety and efficacy of the product and its formulations. The results of these studies and other information, including chemistry, manufacturing and controls, must be submitted to the FDA or comparable foreign agencies and regulatory bodies as part of an application which must be reviewed and approved before proposed clinical testing can begin. Clinical trials involve the administration of the investigational new drug to healthy volunteers or to patients under the supervision of a qualified principal investigator. Clinical trials are conducted in accordance with Good Clinical Practices under protocols that detail the objectives of the study, the parameters to be used to monitor safety, and the efficacy criteria to be evaluated. Each protocol must be submitted for regulatory approval. Further, each clinical study must be conducted under the auspices of an independent institutional review board at the institution at which the study is conducted. The institutional review board considers, among other things, ethical factors and the safety of human subjects. In addition, certain protocols involving the use of genetically modified products must also be reviewed by the Recombinant DNA Advisory Committee of the National Institutes of Health as well as similar bodies in many European countries.
Clinical Trial Phases. Typically, human clinical trials are conducted in three phases that may overlap. In Phase 1, clinical trials are conducted with a small number of patients to determine the early safety profile and pharmacology of the new therapy. In Phase 2, clinical trials are conducted with groups of patients afflicted with a specific disease in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. In Phase 3, large scale, multicenter, comparative clinical trials are conducted with patients afflicted with a target disease in order to provide enough data for the statistical proof of efficacy and safety required by the FDA and other regulatory agencies. In the case of products for life-threatening diseases, the initial human testing is generally done in the target patients rather than in healthy volunteers. Since these patients are already afflicted with the target disease, it is possible that such studies may provide some results traditionally obtained in Phase 2 clinical trials. These trials are frequently referred to as Phase 1/2 clinical trials. Although the preliminary Phase 1/2 and Phase 2 clinical trials of our GVAX cancer immunotherapies and oncolytic virus therapies have shown a generally favorable safety profile to date, there can be no assurance that such therapies or products will be
10
tolerated at higher doses or that the clinical efficacy or safety of such therapy or product will be demonstrated in later stage testing.
Marketing Approvals. The results of the preclinical and clinical testing, together with chemistry, manufacturing and controls information, are submitted to regulatory agencies in order to obtain approval to commence commercial sales. In responding to such an application, regulatory agencies may grant marketing approval, request additional information or further research, or deny the application if they determine that the application does not satisfy their regulatory approval criteria. Approval for a pharmaceutical or biologic product may not be granted on a timely basis, if at all, or if granted may not cover all the clinical indications for which approval is sought, or may contain significant limitations in the form of warnings, precautions or contraindications with respect to conditions of use.
In the United States we have utilized the procedure called a Special Protocol Assessment, or SPA, for GVAX immunotherapy for prostate cancer. Under this procedure, a sponsor may seek the FDA’s agreement on the design and analysis of a clinical trial intended to form the primary basis of an effectiveness claim. If the FDA agrees in writing, its agreement may not be changed after the trial begins except in limited circumstances, such as the FDA determining that a substantial scientific issue essential in determining the safety or effectiveness of the product was identified after the trial had begun. If the outcome of the trial is successful, the sponsor will ordinarily be able to rely on it as the basis for approval with respect to effectiveness. While we have received FDA’s agreement on a SPA for each of our Phase 3 VITAL-1 and VITAL-2 trials, there can be no assurance that these trials will have a successful outcome or that we will ultimately receive approval for this product.
Satisfaction of pre-market approval requirements for new drugs and biologics typically takes several years, and the actual time required may vary substantially based upon the type, complexity and novelty of the product or targeted disease. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures upon our activities. Success in early stage clinical trials or with prior versions of products does not assure success in later stage clinical trials. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval.
Post-Marketing Regulations. Once approved, regulatory agencies may withdraw the product approval if compliance with pre- and/or post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. In addition, they may require post-marketing studies, referred to as Phase 4 studies, to monitor the effect of an approved product, and may limit further marketing of the product based on the results of these post-market studies. The FDA and other foreign regulatory agencies have broad post-market regulatory and enforcement powers, including the ability to levy fines and penalties, suspend or delay issuance of approvals, seize or recall products, or withdraw approvals.
Manufacturing. Our manufacturing facilities are subject to periodic inspection by the FDA, the United States Drug Enforcement Administration, or DEA, and other domestic and foreign authorities where applicable, and must comply with cGMP regulations. Manufacturers of biologics also must comply with general biological product standards. Failure to comply with the statutory and regulatory requirements subjects the manufacturer to possible legal or regulatory action, such as suspension of manufacturing, seizure of product, or mandatory or voluntary recall of a product. Adverse experiences with the product must be reported to the FDA and foreign agencies and could result in the imposition of market restrictions through labeling changes or in product removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval.
Advertising and Promotion. With respect to both pre- and post-market product advertising and promotion, the FDA and similar foreign agencies impose a number of complex regulations on entities that advertise and promote pharmaceuticals and biologics, which include, among other things, standards and regulations relating to direct-to-consumer advertising, off-label promotion, industry sponsored scientific and educational activities, and
11
promotional activities involving the Internet. These agencies have very broad enforcement authority and failure to abide by these regulations can result in penalties including the issuance of a warning letter directing the entity to correct deviations from requisite standards, a requirement that future advertising and promotional materials be pre-cleared by the FDA or relevant foreign agencies, and foreign, state and federal civil and criminal investigations and prosecutions.
Other Government Regulations
We are subject to various laws and regulations regarding laboratory practices, the experimental use of animals, and the use and disposal of hazardous or potentially hazardous substances in connection with our research. In each of these areas, as above, the government has broad regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products, and withdraw approvals, any one or more of which could have a material adverse effect upon us.
In addition to laws and regulations enforced by the FDA, we are also subject to comparable foreign regulations, regulation under National Institutes of Health guidelines, as well as under the Controlled Substances Act, the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other present and potential foreign, federal, state or local laws and regulations as our research and development involves the controlled use of hazardous materials, chemicals, viruses and various radioactive compounds.
Manufacturing
Manufacture of our products for clinical trials does not require an FDA license, although the FDA and other regulatory authorities may at any time inspect our manufacturing facility. Our Hayward, California manufacturing facility, which we operate according to cGMP regulations, consists of 51,000 square feet of manufacturing space and 50,000 square feet of laboratory and office space. Our Hayward manufacturing facility currently has the capacity to manufacture products for Phase 3 trials of our prostate cancer immunotherapy and we believe that it will also have the capacity to support market launch.
Corporate Collaborations
Novartis AG
In July 2003, we announced a global alliance between Novartis AG and ourselves for the development and commercialization of oncolytic virus therapies. Under the agreement, we also acquired exclusive worldwide rights to certain oncolytic virus therapy products and related intellectual property of Genetic Therapy, Inc., or GTI, an affiliate of Novartis, as well as certain related intellectual property of Novartis. We also received a payment of $28.5 million from Novartis to be dedicated to the further development of several oncolytic virus therapy products developed by both ourselves and GTI, for which Novartis has certain marketing options. In exchange, we issued to Novartis and GTI 1,999,840 shares of our common stock, with the result that Novartis became the holder of approximately five percent (as of the time of the issuance) of our outstanding common stock. In addition, the agreement provides the basis for the sharing of future additional development costs and potential profits for certain oncolytic virus products on a worldwide basis. Upon the exercise of certain options by Novartis, development costs and profits are to be shared on an approximately equal basis in the United States. Novartis will be responsible for the development costs for markets outside the United States and pay us a royalty on potential future sales outside the United States. Novartis is also required to reimburse us on a cost-plus basis for products that we manufacture for them to sell outside of the United States.
In September 2004, the terms of our agreement with Novartis were amended to include the grant to us of a non-exclusive worldwide perpetual license to all patent rights of Novartis relating to GM-CSF, a component of our GVAX cancer immunotherapies, in the field of gene therapy. This license bears a low single digit royalty.
12
Also included in the agreement was acknowledgment that certain GVAX cancer immunotherapy products, such as our GVAX immunotherapy for prostate cancer, would not require this license and hence would not be subject to future royalty payments to Novartis.
Medarex, Inc.
In May 2003, we entered into a research and development collaboration with Medarex, Inc. to evaluate combination therapy with our GVAX immunotherapy for prostate cancer and Medarex’s anti-CTLA-4 antibody called ipilimumab. Preclinical studies indicate that ipilimumab may enhance the activity of GVAX cancer immunotherapies. We initiated a Phase 1 trial of this combination therapy in September 2004 which was expanded in April 2007 to treat up to a total of approximately 25 to 30 patients. The costs of this ongoing trial are shared equally by us and Medarex which is now jointly developing ipilimumab with Bristol-Meyers Squibb Company.
GBP IP, LLC
In December 2007, we sold for $12.0 million all of our assets, intellectual property and previously established licensing agreements relating to our lentiviral gene delivery technology, commonly referred to as lentiviral vectors, to GBP IP, LLC, an affiliate of GBP Capital, the majority shareholder in privately held Lentigen Corporation. We received full payment of $12.0 million in December 2007. As part of the asset sale agreement we retained our rights to use the technology for research and development purposes including potential future use with our cancer immunotherapy products.
Patents and Trade Secrets
The patent positions and proprietary rights of pharmaceutical and biotechnology firms, including us, are generally uncertain and involve complex legal and factual questions. We believe there will continue to be significant litigation in the industry regarding patent and other intellectual property rights.
As of December 31, 2007, we had 392 U.S. and non-U.S. patents issued or granted to us or available for use by us based on licensing arrangements and 268 U.S. and non-U.S. applications pending in our name or available for use by us based on licensing arrangements. We are currently prosecuting our patent applications in various patent offices around the world, but we cannot be certain whether any given patent application filed by us or our licensors will result in the issuance of a patent or if any given patent issued to us or our licensors will later be challenged and invalidated. Nor can we be certain whether any given patent that may be issued to us or our licensors will provide any significant proprietary protection to our products and business.
Litigation or other proceedings may also be necessary to enforce or defend our proprietary rights and patents. To determine who was first to make an invention claimed in a United States patent application or patent and thus be entitled to a patent, the United States Patent and Trademark Office, or USPTO, can declare an interference proceeding. In Europe, patents can be revoked through opposition or nullity proceedings. In the United States patents may be revoked or invalidated in court actions or in reexamination proceedings in the USPTO. Such litigation or proceedings could result in substantial cost or distraction to us, or result in an adverse decision as to our or our licensors’ patent applications and patents. We are not currently involved in any interference proceedings concerning our or our licensors’ patent applications and patents. We may be involved in such proceedings in the future.
Our commercial success depends in part on not infringing the patents or proprietary rights of others and not breaching licenses granted to us. Competitors may have filed patent applications and obtained patents and may in the future file patent applications and obtain patents relevant to our products and technologies. We are aware of competing intellectual property relating to both our programs in cancer immunotherapies and oncolytic viruses. While we currently believe that we have the necessary freedom to operate in these areas, there can be no
13
assurance that others will not challenge our position in the future. Litigation to defend our position could be costly and time consuming. We also cannot be certain that we will be successful. We may be required to obtain a license from the prevailing party in order to continue the portion of our business that relates to the proceeding. We may also be required to obtain licenses to other third-party technologies, genes or other product components necessary in order to market our products. Such licenses may not be available to us on acceptable terms or on any terms and we may have to discontinue that portion of our business. Any failure to license any technologies or genes required to commercialize our technologies or products at reasonable cost may have a material adverse effect on our business, results of operations, financial condition, cash flow and future prospects. We are not currently involved in any litigation concerning our competitors’ patent applications and patents. We may be involved in such litigation in the future.
We also rely on unpatented trade secrets and improvements, unpatented know-how and continuing technological innovation to develop and maintain our competitive position. No assurance can be given that others will not independently develop substantially equivalent proprietary information and techniques, or otherwise gain access to our trade secrets or disclose such technology, or that we can meaningfully protect our rights to our unpatented trade secrets.
We require our employees and consultants to execute confidentiality agreements upon the commencement of employment and consulting relationships with us. These agreements provide that all confidential information developed by or made known to an individual during the course of the employment or consulting relationship generally must be kept confidential. In the case of employees, the agreements provide that all inventions conceived by the individual, while employed by us, relating to our business are our exclusive property. While we have implemented reasonable business measurements to protect confidential information, these agreements may not provide meaningful protection for our trade secrets in the event of unauthorized use or disclosure of such information.
Competition
We face substantial competition in the development of products for cancer and other diseases. This competition from other manufacturers of the same types of products and from manufacturers of different types of products designed for the same uses is expected to continue in both U.S. and international markets. Cancer immunotherapies and oncolytic virus therapies, our two primary focus areas, are rapidly evolving areas in the biotechnology industry and are expected to undergo many changes in the coming years as a result of technological advances. We are currently aware of a number of groups that are developing cancer immunotherapies and oncolytic virus therapies including early-stage and established biotechnology companies, pharmaceutical companies, academic institutions, government agencies and research institutions. Examples in the cancer immunotherapy area include Dendreon Corporation, which is undertaking a Phase 3 trial in prostate cancer and has filed a BLA with the FDA, and Onyvax Ltd., which has commenced Phase 2 trials in prostate cancer. Antigenics, Inc., Genitope Corporation, Oncothyreon Inc. and Favrille, Inc. are also developing immunotherapy products for other types of cancers. We face competition from these groups in areas such as recruiting employees, acquiring technologies that might enhance our ability to commercialize products, establishing relationships with certain research and academic institutions, enrolling patients in clinical trials and seeking program partnerships and collaborations with larger pharmaceutical companies. It is possible that our competitors could achieve earlier market commercialization, could have superior patent protection, or could have safer, more effective or more cost-effective products. These factors could render our potential products less competitive, which could adversely affect our business.
Human Resources
As of December 31, 2007, we employed 302 people, of whom 21 hold Ph.D. degrees and six hold M.D. degrees. As of December 31, 2007, 251 employees were engaged in research, development and manufacturing operations, and 51 employees support business development, intellectual property, finance and other
14
administrative functions. Many of our management have had prior product development experience in the biotechnology and pharmaceutical industries.
Our success will depend in large part upon our ability to attract and retain employees. We compete for qualified employees with other companies, research and academic institutions, government entities and other organizations. We believe that our employee relations are good.
Executive Officers
Our executive officers and their ages as of February 28, 2008, were as follows:
| | | | |
Name | | Age | | Position |
Stephen A. Sherwin, M.D. | | 59 | | Chairman of the Board and Chief Executive Officer |
Sharon E. Tetlow | | 48 | | Senior Vice President and Chief Financial Officer |
Robert J. Dow, MBChB | | 57 | | Senior Vice President, Medical Affairs and Chief Medical Officer |
Carol C. Grundfest | | 53 | | Senior Vice President, Regulatory Affairs and Portfolio Management |
Christine B. McKinley | | 54 | | Senior Vice President, Human Resources |
Michael W. Ramsay | | 51 | | Senior Vice President, Operations |
Robert H. Tidwell | | 64 | | Senior Vice President, Corporate Development |
Peter K. Working, Ph.D. | | 59 | | Senior Vice President, Research and Development |
Marc L. Belsky | | 52 | | Vice President, Finance and Chief Accounting Officer |
Kristen M. Hege, M.D. | | 44 | | Vice President, Clinical Research |
Dr. Sherwin, chairman of the board and chief executive officer, joined Cell Genesys in March 1990. Dr. Sherwin has served as chief executive officer since the Company’s inception, and in March 1994 he was elected to the additional position of chairman of the board of directors. From 1983 to 1990, Dr. Sherwin held various positions at Genentech, Inc., a biotechnology company, most recently as vice president of clinical research. Prior to 1983, Dr. Sherwin was on the staff of the National Cancer Institute. Dr. Sherwin currently serves as the chairman of the board of Ceregene, Inc., a former subsidiary of Cell Genesys, which he co-founded in 2001. Dr. Sherwin was also a co-founder of Abgenix, Inc, another former subsidiary of Cell Genesys, which was acquired by Amgen in 2006. He is also a director of Neurocrine Biosciences, Inc. and Rigel Pharmaceuticals, Inc. Dr. Sherwin, who also serves as a board member and chair of the emerging companies section of the Biotechnology Industry Organization, holds a B.A. in biology from Yale University, an M.D. from Harvard Medical School and is board-certified in internal medicine and medical oncology.
Ms. Tetlow, senior vice president and chief financial officer, joined Cell Genesys in June 2005. Between 2004 and 2005, Ms. Tetlow was a venture partner at Apax Partners, a private equity firm. From 1999 to 2004, Ms. Tetlow was chief financial officer for diaDexus, a pharmacogenomics company. From 1998 to 1999, she was chief financial officer at Reprogen, and prior to that, between 1988 and 1998, she held senior financial management positions in other biotechnology companies including Terrapin Technologies, Inc. (now Telik, Inc.), Synergen (now part of Amgen, Inc.) and Genentech, Inc. Ms. Tetlow received a Master of Business Administration from the Graduate School of Business, Stanford University, and a Bachelor of Arts and Science from the University of Delaware.
Dr. Dow, senior vice president, medical affairs and chief medical officer, joined Cell Genesys in March 2005. Prior to joining Cell Genesys, from 2002 to 2005, Dr. Dow served as chief executive officer at Biolitec Pharma ltd, a UK biotechnology company wholly-owned by Biolitec AG of Germany. From 1997 to 2002, Dr. Dow held senior executive positions with Quantanova and Scotia Holdings, plc. From 1995 to 1997, Dr. Dow was Head of Global Drug Development with Hoffmann-La Roche, and from 1982 to 1995 he held senior executive positions in drug development with Syntex Corporation. Dr. Dow holds a B.Sc. in Medical Science from the University of St. Andrews and his medical qualification, an MBChB degree, from the University of Dundee in Scotland. He also is a Fellow of the Royal College of Physicians of Edinburgh.
15
Ms. Grundfest, senior vice president, regulatory affairs and portfolio management, joined Cell Genesys in July 2003. Prior to joining Cell Genesys, Ms. Grundfest served as an independent consultant providing advice, analysis and recommendations regarding the regulation and approval of pharmaceutical products in the United States from 2000 to 2003. From 1998 to 2000, Ms. Grundfest served as executive director of project management and strategic planning at Systemix, Inc. and Genetic Therapy, Inc. (affiliates of Novartis AG). Ms. Grundfest also held senior regulatory positions with Roche Global Development and Syntex from 1990 to 1996, as well as served as assistant vice president, research and development at the Pharmaceutical Research and Manufacturers of America from 1982 to 1990. Ms. Grundfest received an M.H.S. in environmental health sciences from The Johns Hopkins University, School of Public Health and a B.S. in biology from Stanford University.
Ms. McKinley, senior vice president, human resources, joined Cell Genesys in August 1994. From 1985 to 1994, she was with Nellcor Puritan Bennett, Inc., where the last position she held was corporate human resources director. Previously, Ms. McKinley also worked at Genentech, Inc. from 1978 to 1984 in various human resource positions. She received a B.A. in psychology from the University of California, Santa Barbara.
Mr. Ramsay, senior vice president, operations, joined Cell Genesys in January 2002. Prior to joining Cell Genesys, Mr. Ramsay served as a vice president of manufacturing at ALZA Corporation from 1999 to 2001. Mr. Ramsay also held various positions from 1992 to 1999 with SEQUUS Pharmaceuticals, including vice president of manufacturing operations, as well as various positions at Syntex Corporation focusing on manufacturing, product development and regulatory affairs from 1978 to 1991. Mr. Ramsay holds a Bachelor of Pharmacy from the University of Nottingham in the United Kingdom.
Mr. Tidwell, senior vice president, corporate development, joined Cell Genesys in August 2000. Prior to joining Cell Genesys, Mr. Tidwell was vice president of business development at Calydon, Inc. from 1998 to 2000. Mr. Tidwell has also held various management positions with such companies as Boston Life Sciences, where he served as chief operating officer from 1993 to 1994, Genetics Institute, where he was vice president of marketing and business development from 1988 to 1993, and Eli Lilly and Company, where he held various positions including director of worldwide pharmaceutical licensing, between 1969 and 1985. Mr. Tidwell holds an M.B.A. from The Ohio State Graduate School of Business and a Bachelor of Pharmacy from The Ohio State School of Pharmacy.
Dr. Working, senior vice president, research and development, joined Cell Genesys in September 2001. Prior to joining Cell Genesys, from 1999 to 2001, Dr. Working served as vice president of analytical and non-clinical sciences and principal scientist at ALZA Corporation. From 1992 to 1999, Dr. Working was with SEQUUS Pharmaceuticals, where the last position he held was vice president of research and development. From 1988 to 1992 he was with Genentech, Inc., where he served as a senior toxicologist and head of the Experimental Toxicology Group in the Department of Safety Evaluation. Dr. Working holds Ph.D., M.S. and B.S. degrees from the University of California, Davis and an M.A. degree from the University of California, San Francisco.
Mr. Belsky, vice president, finance and chief accounting officer, joined Cell Genesys in December 2006. Prior to joining Cell Genesys, Mr. Belsky served as vice president, Global Visa Commerce from 2005 to 2006, for Visa International. Mr. Belsky held senior management positions in other companies including chief financial officer at Active Aero Group from 2003 to 2005 and DataWave Systems, Inc. from 2001 to 2003, as well as senior finance and banking positions at Michigan National Corporation from 1986 to 2001. Mr. Belsky started his career as an auditor with Coopers & Lybrand. Mr. Belsky is a certified public accountant, a certified treasury professional, and received an M.B.A. from the University of Michigan and a B.S. in accounting from Wayne State University.
Dr. Hege, vice president, clinical research joined Cell Genesys in January 1994 as a medical post-doctoral research fellow. She transferred to the clinical research department in 1996 where she has worked on all of our clinical programs. For the past 12 years, she has worked in the clinical research department, and was promoted to vice president in 2004. In addition to her work at Cell Genesys, Dr. Hege has held a clinical faculty appointment
16
at University of California, San Francisco, or UCSF, since 1997 in the adult leukemia and bone marrow transplant program. Dr. Hege received a B.A. in biochemistry from Dartmouth College, an M.D. from UCSF, and subspecialty training at UCSF and Harvard. She is board-certified in medical oncology and board eligible in internal medicine and hematology.
Medical Advisory Board
We have established a medical advisory board that includes several prominent leaders in the field of oncology. As of December 31, 2007, the board consisted of the following individuals:
| | |
Name | | Scientific Position |
Bruce Chabner, M.D. | | Clinical Director Massachusetts General Hospital Cancer Center Professor of Medicine, Harvard Medical School |
| |
Jordan U. Gutterman, M.D. | | Chief of the Section of Cellular and Molecular Growth Regulation Department of Molecular Therapeutics Professor of Medicine University of Texas M.D. Anderson Cancer Center |
| |
I. Craig Henderson, M.D. | | Adjunct Professor of Hematology/Oncology University of California, San Francisco |
| |
Ronald Levy, M.D. | | Robert K. Summy and Helen K. Summy Professor of Medicine Chief of the Division of Oncology Stanford University School of Medicine |
| |
William Nelson, M.D., Ph.D. | | Associate Professor of Oncology, Pathology, Pharmacology and Medicine, and Urology Sidney Kimmel Comprehensive Cancer Center The Johns Hopkins University |
| |
John T. Potts, Jr., M.D. | | Director of Research, Massachusetts General Hospital Physician-in-Chief Emeritus Jackson Distinguished Professor of Clinical Medicine Harvard Medical School |
Dr. Potts, who is also a member of our board of directors, serves as a liaison between the medical advisory board and the board of directors, making periodic reports on the findings of the medical advisory board to the board of directors.
17
Investors in Cell Genesys, Inc. should carefully consider the risks described below before making an investment decision. The risks described below may not be the only ones relating to our company. This description includes any material changes to and supersedes the description of the risk factors associated with our business previously disclosed in Item 1A of our Quarterly Report on Form 10-Q for the three months ended September 30, 2007. Additional risks that we currently believe are immaterial may also impair our business operations. Our business, results of operation, financial condition, cash flow and future prospects and the trading price of our common stock and our abilities to repay our convertible notes could be harmed as a result of any of these risks, and investors may lose all or part of their investment. In assessing these risks, investors should also refer to the other information contained or incorporated by reference in this Annual Report on Form 10-K, including our consolidated financial statements and related notes, and our other filings from time to time with the SEC.
Risks Related to Our Company
Our products are in developmental stage, are not approved for commercial sale and may never receive regulatory approval or become commercially viable.
All of our potential cancer immunotherapies and oncolytic virus therapies are in research and development. We have not generated any revenues from the sale of products, and we do not expect to generate any revenues from product sales for at least the next several years. Our products currently under development will require significant additional research and development efforts, including extensive preclinical and clinical testing and regulatory approval, prior to commercial use. There are many reasons that potential products that appear promising at an early stage of research or development do not result in commercially successful products. Clinical trials may be suspended or terminated if safety or efficacy issues are identified, if our investigators or we fail to comply with regulations governing clinical trials or for other reasons. Our research and development efforts may not be successful. Even if developed, our products may not receive regulatory approval or be successfully manufactured in commercial quantities at reasonable cost, or successfully introduced and marketed at prices that would permit us to operate profitably.
Clinical testing is a lengthy, costly and uncertain process that may not demonstrate that our cancer immunotherapies and oncolytic virus therapies are safe or effective, or result in regulatory approval.
Our GVAX cancer immunotherapies and oncolytic virus therapies are currently being tested in human clinical trials to determine their safety and efficacy. Clinical trials are very costly and time-consuming, especially the larger Phase 3 clinical trials such as the VITAL-1 and VITAL-2 trials of our GVAX immunotherapy for prostate cancer. The VITAL-1 and VITAL-2 trials of our GVAX immunotherapy for prostate cancer are our first Phase 3 clinical trials. Although we have recently completed patient recruitment for the VITAL-1 trial, we cannot be certain if and when any of our current clinical trials will be successfully completed. Because our programs use new technologies, existing preclinical and clinical data on the safety and efficacy of our programs is limited. The results of preclinical or earlier stage clinical trials do not necessarily predict safety or efficacy in humans, and our products in later stage clinical trials may fail to show desired safety and efficacy, despite having progressed through preclinical or early clinical trials. Serious and potentially life-threatening side effects may be discovered during preclinical and clinical testing of our potential products or thereafter. We, the United States Food and Drug Administration, or FDA, foreign regulatory authorities or the Institutional Review Boards at our research institutions may suspend or terminate any clinical trials of our products at any time, and we cannot guarantee that we will be permitted to undertake additional human clinical trials for any of our products.
Even if clinical trials are successful, we might not obtain regulatory approval for any indication. Preclinical and clinical data can be interpreted in many different ways, and the FDA or foreign regulatory officials could interpret data that we consider promising differently, which could delay or prevent regulatory approval. Moreover, the degree of regulatory risk as well as the commercialization risk is generally greater for products based on new technologies, such as our GVAX cancer immunotherapies and oncolytic virus therapies.
18
We also cannot guarantee that adequate numbers of patients can be recruited for our clinical trials. Many factors affect patient enrollment in clinical trials, including, but not limited to:
| | • | | the size of the patient population; |
| | • | | the proximity of patients to clinical sites; |
| | • | | the willingness of local patients to participate; |
| | • | | the eligibility criteria for the trial; |
| | • | | competing clinical trials; and |
| | • | | new therapies approved for the conditions that we are investigating. |
In addition to delays in patient enrollment, other unforeseen developments could prevent or delay completion of a clinical trial and increase our costs, which could also prevent or delay filing or approval of marketing applications and/or prevent or delay any eventual commercial sale of the therapy that is the subject of the trial. The delay, suspension or termination of a clinical trial could harm our business and future prospects. Such developments include, but are not limited to:
| | • | | delays in obtaining regulatory approvals to commence or continue a study; |
| | • | | delays in identifying and reaching agreement on acceptable terms with prospective clinical trial sites; |
| | • | | competition with ongoing clinical trials; |
| | • | | manufacturing delays or limitations on manufacturing capacity; |
| | • | | lack of effectiveness during clinical trials; |
| | • | | unforeseen safety issues; |
| | • | | uncertain dosing issues; |
| | • | | inability to monitor patients adequately during or after treatment; |
| | • | | failure by us or our investigators to comply with the FDA or other health authority regulations governing clinical trials; |
| | • | | an inability or unwillingness of medical investigators to follow our clinical protocols; and |
| | • | | lack of prior experience of regulatory agencies with our new technologies. |
Each of our two Phase 3 clinical trials of GVAX immunotherapy for prostate cancer involves a comparison to a Taxotere chemotherapy regimen, which is the currently approved standard of care for this patient group. However, there can be no assurance that this chemotherapy regimen will continue to be commonly used to treat these patients in the future. Should another therapy be shown to be more effective than the Taxotere chemotherapy regimen, we may need to conduct additional comparative clinical trials in the future. The delay, suspension or termination of a clinical trial for any reason could harm our business and future prospects.
Failure to comply with foreign regulatory requirements governing human clinical trials and marketing approval for drugs and devices could prevent us from conducting our clinical trials or selling our products in foreign markets, which may impair our operating results and financial condition.
For development and marketing of drugs and biologics outside the United States, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country and may require us to perform additional testing and expend additional resources. The time required to obtain approvals in some countries outside the United States may be longer than that required to obtain FDA approval, and we may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA
19
does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other countries or by the FDA. Failure to comply with these regulatory requirements or obtain required approvals could impair our ability to conduct clinical trials in foreign markets or commercially develop foreign markets for our products and may harm our business and future prospects.
We will need substantial additional funds to continue operations, and our ability to generate funds depends on many factors beyond our control.
We will need substantial additional funds for existing and future preclinical and clinical trials, to continue research and development activities, for lease obligations related to our manufacturing and headquarter facilities, for principal and interest payments related to our debt financing obligations, for potential settlements with tax authorities and to establish marketing capabilities for any products we may develop. We will need to raise additional capital to further fund our operations.
Our future capital requirements will depend on, and could increase as a result of, many factors, including, but not limited to:
| | • | | the progress and scope of our internally funded research, development, clinical, manufacturing and commercialization activities; |
| | • | | our ability to establish new collaborations and the terms of those collaborations; |
| | • | | competing technological and market developments; |
| | • | | the time and cost of regulatory approvals; |
| | • | | the extent to which we choose to commercialize our future products through our own sales and marketing capabilities; |
| | • | | our ability to reach favorable resolutions with respect to potential tax assessments; |
| | • | | the costs we incur in obtaining, defending and enforcing patent and other proprietary rights or gaining the freedom to operate under the patents of others; |
| | • | | our success in acquiring and integrating complementary products, technologies or businesses; and |
| | • | | the extent to which we choose to expand and develop our manufacturing capacities, including manufacturing capacities necessary to meet potential commercial requirements. |
If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research, development, manufacturing or clinical activities or be required to sell or merge the company or otherwise substantially modify or cease some or all of our current operations.
We plan to raise additional funds through additional equity or debt financings, collaborative business relationships, or otherwise, but we may not be able to do any of the foregoing on favorable terms, or at all.
Because of our capital requirements, we will need to raise additional funds. From time to time, we may seek to access the public or private debt and equity markets. We also may seek to raise additional capital through outlicensing technologies or third-party collaborations. If adequate funds are not available on favorable terms, or at all, we may be required to delay, reduce the scope of, or eliminate one or more of our research, development, manufacturing or clinical activities.
Alternatively, we may need to seek funds through arrangements with collaborative partners or others that require us to relinquish rights to technologies or product candidates that we would otherwise seek to develop or commercialize ourselves. These arrangements could harm our business, results of operations, financial condition, cash flow or future prospects. Currently, we do not have collaborative partners for the further development of our
20
GVAX cancer immunotherapies. Although we are in active discussions with potential partners for our GVAX immunotherapy for prostate cancer, we may not be successful in entering into collaborative partnerships on favorable terms, if at all. Certain of our oncolytic virus therapy products are subject to future commercialization rights belonging to Novartis, which at its option may provide further development funding for these products. However, we can give no assurance that our alliance with Novartis will continue, as Novartis periodically has the option of terminating the alliance at its discretion. Also, there is no assurance that Novartis will provide further development funding. Our efforts to raise capital through such outlicensing activities may fail. Failure to enter into new corporate relationships may harm our business.
The successful growth of our business may depend, in part, on our ability to find third-party collaborators to assist or share in the costs of product development.
Our strategy for the development and commercialization of our proprietary product candidates may include the formation of collaborative arrangements with third parties. Potential third parties include pharmaceutical and biotechnology companies, academic institutions and other entities. Third party collaborators may assist us in:
| | • | | funding or performing research, preclinical development, clinical trials and manufacturing; |
| | • | | seeking and obtaining regulatory approvals; and |
| | • | | successfully commercializing existing and future product candidates. |
If we are not able to establish further collaboration agreements, we may be required to undertake product development and commercialization at our own expense. Such an undertaking may limit the number of product candidates that we will be able to develop, significantly increase our capital requirements and place additional strain on our internal resources. Our failure to enter into additional collaborations could materially harm our business, financial condition, results of operations, cash flow or future prospects.
In addition, our dependence on licensing, collaboration and other agreements with third parties may subject us to a number of risks. These agreements may not be on terms that ultimately prove favorable to us and may require us to relinquish certain rights in our technologies and product candidates. To the extent we agree to work exclusively with one collaborator in a given area, our opportunities to collaborate with other entities could be curtailed. Lengthy negotiations with potential new collaborators may lead to delays in the research, development or commercialization of product candidates. The decision by our collaborators to pursue alternative technologies or the failure of our collaborators to develop or commercialize successfully any product candidate to which they have obtained rights from us could materially harm our business, financial condition, results of operations, cash flow or future prospects.
We expect to continue to incur substantial losses and negative cash flow from operations and may not become profitable in the future.
We have incurred an accumulated deficit since our inception. As of December 31, 2007, our accumulated deficit was $491.1 million. Our accumulated deficit would be substantially higher absent the gains we have realized on sales of our Abgenix common stock. We expect to incur substantial operating losses for at least the next several years and potentially longer. This is due primarily to the expansion of development programs, clinical trials and manufacturing activities and, to a lesser extent, general and administrative expenses, at a time when we have yet to realize any product revenues. We also have substantial lease obligations related to our manufacturing and headquarter facilities. We expect that losses will fluctuate from quarter to quarter and that these fluctuations may be substantial. We cannot guarantee that we will successfully develop, manufacture, commercialize or market any products, or that we will ever achieve positive cash flow or profitability.
21
Our substantial indebtedness could harm our financial condition.
We have a significant amount of debt. As of December 31, 2007, we had approximately $145.0 million aggregate principal amount of outstanding convertible notes due in 2011. Our annual interest payment on this note is approximately $4.5 million. Our substantial indebtedness could harm our business, results of operations, financial condition, cash flow and future prospects. For example, it could:
| | • | | make it more difficult for us to satisfy our obligations with respect to the convertible notes; |
| | • | | increase our vulnerability to general adverse economic and industry conditions; |
| | • | | limit our ability to raise or borrow additional funds for future working capital, capital expenditures, research and development and other general corporate requirements; and |
| | • | | limit our flexibility to react to changes in our business and the industry in which we operate. |
We plan to use potential future operating losses and our federal and state net operating loss carryforwards to offset taxable income from revenue generated from operations or corporate collaborations. However, our ability to use net operating loss carryforwards could be limited as a result of issuances of equity securities.
We plan to use our current year operating losses to offset taxable income from any revenue generated from operations or corporate collaborations. To the extent that our taxable income exceeds any current year operating losses, we plan to use our net operating loss carryforwards to offset income that would otherwise be taxable. However, under the Tax Reform Act of 1986, the amount of benefits from our net operating loss carryforwards may be impaired or limited if we incur a cumulative ownership change of more than 50%, as interpreted by the IRS, over a three year period. As a result, our use of federal net operating loss carryforwards could be limited by the provisions of Section 382 of the Internal Revenue Code, depending upon the timing and amount of additional equity securities that we issue. State net operating loss carryforwards may be similarly limited. Any such disallowances may result in greater tax liabilities than we would incur in the absence of such a limitation and any increased liabilities could adversely affect our business, results of operations, financial condition and cash flow.
Our ability to manufacture our products is uncertain, which may delay or impair our ability to develop, test and commercialize our products.
We have built our own manufacturing facility for the manufacture of products for clinical trials and to support the potential commercial launch of our GVAX cancer immunotherapy product candidates, but the successful manufacture of our potential products is subject to a number of risks and uncertainties. The manufacturing techniques and process controls, as well as the product release specifications, required for our GVAX cancer immunotherapies and oncolytic virus therapies are more complex and less well-established than those required for other biopharmaceutical products, such as small molecules, therapeutic proteins and monoclonal antibodies. We may not be able to develop the necessary techniques and process controls in a timely manner, if at all, to manufacture and evaluate our products effectively to meet the demands of regulatory agencies, clinical testing and commercial production. Advances in manufacturing techniques may render our facility and equipment inadequate or obsolete.
Our ability to manufacture products is also subject to various licensing and regulatory requirements. Our manufacturing facility is subject to the licensing requirements of, and inspection by, the FDA, United States Drug Enforcement Administration, the California Department of Health Services and foreign regulatory authorities. Failure to obtain or maintain these licenses or to meet the inspection criteria of these agencies would disrupt our manufacturing processes and adversely affect our ongoing clinical trials and our business and future prospects, results of operations, financial condition and cash flow. We operate our manufacturing facility according to current Good Manufacturing Practices, or cGMP, regulations for the manufacture of products for clinical trials. Our manufacturing facility is subject to inspection by the FDA and other regulatory bodies to ensure compliance with cGMP. Any failure to follow and document our adherence to such cGMP regulations or
22
satisfy other manufacturing and product release regulatory requirements may lead to significant delays in the availability of products for commercial use or clinical study, may result in the termination or hold on a clinical study, or may delay or prevent filing or approval of marketing applications for our products. Failure to comply with applicable regulations may also result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our products, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could harm our business.
We have at times encountered and/or may in the future encounter problems with the following:
| | • | | our ability to “scale-up” our manufacturing facility by a significant factor over the current level of production within our planned timelines and budget; |
| | • | | achieving consistent and acceptable production yield and costs; |
| | • | | meeting product release specifications; |
| | • | | shortages of qualified personnel; |
| | • | | shortages of raw materials; |
| | • | �� | shortages of key contractors or contract manufacturers; |
| | • | | ongoing compliance with cGMP regulations and other expectations from the FDA and other regulatory bodies; |
| | • | | equipment shortage or malfunction; and |
| | • | | inability to validate new equipment or processes in a timely manner. |
Any of these factors could harm our product development, delay commercialization, or prevent us from producing our products in a sufficient quantity to meet the requirements for product launch or future demand. If we are unable to manufacture our products for any reason, our options for outsourcing manufacturing are currently limited. We are unaware of available contract manufacturing facilities on a worldwide basis in which our GVAX immunotherapy product candidates can be manufactured under cGMP regulations, a requirement for all pharmaceutical products. It would take a substantial period of time for a contract manufacturing facility that has not been producing our particular products to begin successfully producing them under cGMP regulations. Logistical arrangements for wide-spread distribution of our products for clinical and commercial purposes may prove to be impractical or prohibitively expensive, which could hinder our ability to commercialize our products.
Due to our reliance on clinical trial arrangements with public and private medical institutions, clinical research organizations, other vendors, and individual investigators, we may not be able to directly control the timing, conduct, expense and quality of some of our clinical trials.
We have arrangements with a number of public and private medical institutions, clinical research organizations, other vendors, and individual investigators, for the conduct of human clinical trials for our GVAX cancer immunotherapy programs and oncolytic virus therapies. In some cases, trials are conducted by institutions without our direct control or monitoring. The early termination of any of these clinical trial arrangements, the failure of these institutions to comply with the regulations and requirements governing clinical trials, or the reliance upon results of trials that we have not directly conducted or monitored could hinder the progress of our clinical trial programs and our development programs. If any of these relationships are terminated, the clinical trials might not be completed, and the result might not be valuable.
23
If our proposed products are not effectively protected by issued patents we will be more vulnerable to competitors and our business could be harmed.
We rely heavily on the development and protection of our intellectual property portfolio. As of December 31, 2007, we had 392 U.S. and non-U.S. patents issued or granted to us or available for use by us based on licensing arrangements and 268 U.S. and non-U.S. applications pending in our name or available for use by us based on licensing arrangements. The patent positions of pharmaceutical and biotechnology firms, including ours, are generally uncertain and involve complex legal and factual questions. We believe that there will continue to be significant litigation in the industry regarding patent and other intellectual property rights. We cannot be certain whether any given patent application filed by us or our licensors will result in the issuance of a patent or if any given patent issued to us or our licensors will later be challenged and invalidated. Nor can we be certain whether any given patent that may be issued to us or our licensors will provide any significant proprietary protection to our products and business. Competitors may have filed patent applications or received patents and may file additional patent applications and obtain additional patents that may prevent patents being issued to us or our licensors or limit the scope of those patents. Depending upon their filing date, patent applications in the United States are confidential until the patent applications are published, typically eighteen months after their filing date, or patents are issued. Outside the United States, patent applications are confidential until they are published, typically eighteen months after their first filing date. In addition, publication of discoveries in the scientific or patent literature tends to lag behind actual discoveries by several to many months. Accordingly, we cannot be sure that we or our licensors were the first creator of inventions covered by our or our licensors’ pending patent applications or issued patents or that we or our licensors were the first to file patent applications for these inventions. In addition, because patents have a limited life, subject to potential extensions, patents issued to us or our licensors may have expired prior to or have limited term remaining after the first commercial sale of a related product. As a result, the commercial value of these patents to our products and business might be limited.
Our freedom to operate may be challenged by others and we may have to engage in litigation to determine the scope and validity of competitors’ patents and proprietary rights, which, if we do not prevail, could harm our business, results of operations, financial condition, cash flow and future prospects.
Our commercial success depends in part on not infringing the patents or proprietary rights of others, not breaching licenses granted to us and ensuring that we have the necessary freedom to operate, to develop and commercialize our products. Competitors may have filed patent applications and obtained patents and may in the future file patent applications and obtain patents relating to our products and technologies. We are aware of competing intellectual property relating to both our GVAX cancer immunotherapy and oncolytic virus therapy technologies and products. While we believe we have the necessary freedom to operate for both of these programs and are aware of no valid issued patents that we believe would prevent us from developing and commercializing the products we are currently developing in these programs, others may challenge our position. From time to time we have received communications from third parties claiming to have conflicting rights relating to components of our products and technologies. Regardless of their ultimate merit, any infringement or other intellectual property claims against our products and technologies may be expensive and time-consuming to litigate and may divert management attention. If any such claim were successful, we could be required to obtain licenses to a third party’s technologies, patents or other proprietary rights or to their biological or chemical reagents in order to develop and market our products. Moreover, we may choose to voluntarily seek such a license in order to avoid the expense and uncertainty of fully defending our position. In either event, such a license may not be available to us on acceptable terms or on any terms, and we may have to discontinue that portion of our business, or such third party may seek an injunction to prevent us from practicing their proprietary technology. In addition, to the extent we license our intellectual property to other parties, we may incur expenses as a result of contractual agreements in which we indemnify those licensing our technologies against losses incurred if practicing our intellectual property infringes upon the proprietary rights of others. The failure to license any technologies or biological or chemical reagents required to develop or commercialize our technologies or products at reasonable cost may harm our business, results of operations, financial condition, cash flow and future prospects.
24
We may have to engage in litigation, which could result in substantial cost or distraction, to enforce or defend our patents and which, if we do not prevail, could harm our business and make us more vulnerable to competition.
In the future, we may have to engage in litigation to enforce or defend our proprietary rights and patents. To determine who was first to make an invention claimed in a U.S. patent application or patent and thus be entitled to a patent, the USPTO can declare an interference proceeding. In Europe, patents can be revoked through opposition or nullity proceedings. In the United States, patents may be revoked or invalidated in court actions or in reexamination proceedings in front of the USPTO. Such litigation or proceedings could result in substantial cost or distraction to us. These proceedings could potentially result in an adverse decision as to our or our licensors’ patent applications and patents.
We cannot predict the outcome of interference, reexamination, opposition or nullity proceedings or patent litigation that we may become involved with in the future. An adverse result in any of these proceedings could have an adverse effect on our intellectual property position in the technologies to which the patent applications or patents involved in the proceedings are directed and on our business in related areas. If we lose in any such proceeding, our patents or patent applications that are the subject matter of the proceeding may be invalidated or may not be issued as patents. We also may be required to obtain a license from the prevailing party in order to continue the portion of our business that relates to the proceeding. Such license may not be available to us on acceptable terms or on any terms, and we may have to discontinue that portion of our business.
Our competitive position may be impaired by our limited ability to protect and control unpatented trade secrets, know-how and other technological innovation.
We have a limited ability to protect and control unpatented trade secrets, know-how and other technological innovation. Our competitors may independently develop similar or better proprietary information and techniques and disclose them publicly. Also, others may gain access to our trade secrets, and we may not be able to meaningfully protect our rights to our unpatented trade secrets. In addition, confidentiality agreements and other measures may not provide meaningful protection for our trade secrets in the event of unauthorized use or disclosure of such information. Failure to protect and control such trade secrets, know-how and innovation could harm our competitive position.
Inventions or processes discovered by our outside scientific collaborators or consultants may not become our property, which may affect our competitive position.
We rely on the continued availability of outside scientific collaborators to perform research for us. These relationships generally may be terminated at any time by the collaborator, typically by giving 30 days notice. These scientific collaborators are not our employees. As a result, we have limited control over their activities and can expect that only limited amounts of their time will be dedicated to our activities. Our arrangements with these collaborators, as well as those with our scientific consultants, provide that any rights we obtain as a result of their research efforts will be subject to the rights of the research institutions for which they work. In addition, some of these collaborators have consulting or other advisory arrangements with other entities that may conflict with their obligations to us. For these reasons, inventions or processes discovered by our scientific collaborators or consultants may not become our property.
Our competitors may develop therapies for the diseases that we are targeting that are safer, more effective, less expensive, or otherwise more advanced than ours or they may commercialize competing products more rapidly than we do, which may adversely affect our competitive position.
Many companies pursue programs for the treatment of cancer. Our competitors include large biotechnology or pharmaceutical companies, such as Amgen Inc., Bristol-Myers Squibb Company, Genentech, Inc., Novartis AG, Hoffmann-La Roche Inc. and sanofi-aventis Group, which have greater experience and resources than we do in developing products, undertaking preclinical testing and human clinical trials of new pharmaceutical products,
25
obtaining FDA and other regulatory approvals of products, and manufacturing and marketing new therapies. We also compete with other biotechnology companies, which have prostate cancer immunotherapy products in various stages of clinical development, such as Dendreon Corporation and Onyvax, Ltd.
Some competitors are pursuing product development strategies that are similar to ours, particularly with respect to our cancer immunotherapy and oncolytic virus therapy programs. Certain products of our competitors are in more advanced stages of product development, clinical trials and regulatory filings. We compete with other companies and institutions for clinical trial participants, which could reduce our ability to recruit participants for our clinical trials, delay clinical trials and adversely affect our ability to bring a product to market prior to our competitors or in our projected timeline. Our competitors may develop technologies and products that are more effective than ours, or that would render our technology and products less competitive or obsolete.
Our competitive position and those of our competitors can vary based on the performance of products in clinical trials. In addition, our competitors may obtain patent protection or FDA or other regulatory approvals and commercialize products more rapidly than we do, which may impact future sales of any products that we are able to commercialize. We also may not have the access that some of our competitors have to materials necessary to support the research, development or manufacture of planned therapies. If we are permitted by the FDA or other regulatory agencies to commence commercial sales of products, we will also be competing with respect to marketing capabilities and manufacturing efficiency, areas in which we have limited or no experience. We expect that competition among products approved for sale will be based, among other things, on:
| | • | | severity of any side effects compared to competing products; |
| | • | | availability of third-party coverage and reimbursement; |
| | • | | sales, marketing and distribution capabilities. |
Our competitive position also depends upon our ability to attract and retain qualified personnel, develop proprietary products or processes, and secure sufficient funding for the often-lengthy period between product conception and commercial sales.
The commercial success of any products that we may develop will depend upon the degree of market acceptance of our products among physicians, patients, health care payers, private health insurers and the medical community.
Our ability to commercialize any products that we may develop will be highly dependent upon the extent to which these products gain market acceptance among physicians, patients, health care payers, such as Medicare and Medicaid, private health insurers, including managed care organizations and group purchasing organizations, and the medical community. If these products do not achieve an adequate level of acceptance, we may not generate adequate product revenues, if at all, and we may not become profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend upon a number of factors, including, but not limited to:
| | • | | the effectiveness, or perceived effectiveness, of our products in comparison to competing products; |
| | • | | the existence of any significant side effects, as well as their severity in comparison to any competing products; |
26
| | • | | potential advantages over alternative treatments; |
| | • | | the ability to offer our products for sale at competitive prices; |
| | • | | relative convenience and ease of administration; |
| | • | | the strength of marketing and distribution support; and |
| | • | | sufficient third-party coverage or reimbursement. |
To the extent we depend on strategic partners to sell, market or distribute our products, we will have reduced control over the sales, marketing and distribution of our future products.
We have no experience in sales, marketing or distribution of biopharmaceutical products. We may in the future rely on the sales, marketing and distribution expertise of potential corporate partners for our initial products.
If we choose to rely on strategic partners for the sale, marketing or distribution of our future products, we will have less control over the sales, marketing and distribution of our products and will depend heavily upon their abilities and dedication to our products. These future strategic partnerships may not be available on favorable terms, if at all, or they could harm our business.
We may in the future be exposed to product liability claims, which could harm our business, results of operations, financial condition and cash flow.
Clinical trials or marketing of any of our potential products may expose us to liability claims resulting from the use of our products. These claims might be made by clinical trial participants and associated parties, consumers, health care providers, sellers of our products or others. A claim, particularly resulting from a clinical trial, or a product recall could harm our business, results of operations, financial condition, cash flow and future prospects.
Insurance coverage is increasingly more difficult and costly to obtain or maintain.
We currently maintain a certain amount of insurance with respect to each of our clinical trials as well as insurance to reduce our direct exposure to certain other business risks, but may not be able to maintain insurance or obtain sufficient coverage at reasonable rates, as premiums are generally increasing and coverage is narrowing in scope. As a result, we may be required to assume more risk in the future or make significant expenditures to maintain our current levels of insurance. Any inability to maintain insurance at an acceptable cost, or at all, could also result in a breach of terms of our product license agreements or could prevent or inhibit the clinical testing or commercialization of our products or otherwise affect our business, results of operation, financial condition, cash flow and future prospects. If we are subject to third-party claims or suffer a loss or damage in excess of our insurance coverage, we may be required to pay for claims, losses and damages in excess of our insurance limits. Furthermore, any claims made on our insurance policies may affect our ability to obtain or maintain future insurance coverage at reasonable costs, if at all.
Our business, financial condition, results of operations, cash flow and future prospects could suffer as a result of future strategic acquisitions and investments.
We may engage in future acquisitions or investments that could dilute our existing stockholders or cause us to incur contingent liabilities, commitments, debt or significant expense. From time to time, in the ordinary course of business, we evaluate potential acquisitions or investments in related businesses, products or technologies.
27
Future acquisitions could subject us to a number of risks, including, but not limited to:
| | • | | the loss of key personnel and business relationships; |
| | • | | difficulties associated with assimilating and integrating the new personnel, intellectual property and operations of the acquired companies; |
| | • | | the potential disruption of our ongoing business; |
| | • | | the expense associated with maintenance of diverse standards, controls, procedures, employees and clients; |
| | • | | the diversion of managerial, financial and other resources from the development of our own proprietary technology; and |
| | • | | our inability to generate revenue from acquired technology sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs. |
As a result of such risks, an acquisition or investment could harm our business, results of operation, financial condition, cash flow and future prospects.
Our operations are vulnerable to interruption by fire, earthquake, power loss, telecommunications failure, terrorist activity and other events beyond our control, which could harm our business.
Our facilities have, in the past, been subject to electrical blackouts as a result of a shortage of available electrical power. Future blackouts could disrupt the operations of our facilities. Most of our facilities are located in seismically active regions. We have not undertaken a systematic analysis of the potential consequences to our business and financial results from a major earthquake, fire, power loss, terrorist activity or other disasters and do not have a recovery plan for such disasters. We are unable to predict the effects of any such event, but the effects could harm our business, results of operations, financial condition, cash flow and future prospects. In addition, we do not carry sufficient insurance to compensate us for actual losses from interruption of our business that may occur, and any losses or damages incurred by us could harm our business.
We depend on our key technical and management personnel to advance our technology, and the loss of these personnel could impair the development of our products.
We rely and will continue to rely on our key management and scientific staff. Because all employees are employed at-will, they can leave at any time. The loss of key personnel or the failure in our industry to recruit necessary additional qualified personnel could harm our business and results of operations. There is intense competition from other companies, research and academic institutions and other organizations for qualified personnel. We may not be able to continue to attract and retain the qualified personnel necessary for the development of our business. We will need to continue to recruit experts in the areas of clinical testing, manufacturing, regulatory, finance, marketing and distribution and to develop additional expertise in our existing personnel. If we do not succeed in hiring or retaining necessary personnel or developing this expertise, our business could suffer significantly.
Various materials that we use are purchased from single qualified suppliers, which could result in our inability to secure sufficient materials to conduct our business.
Critical materials used in our manufacturing operations are subject to a supplier qualification program. In the event that a material or supplier is no longer appropriate to support our cGMP operations, we may incur significant additional expenses to find and qualify alternate materials and/or suppliers. Because the suppliers of key components and materials for a product must be carefully tested, qualified and named in our filings with the FDA and other health authorities, significant delays can occur if a new supplier is required. There is no guarantee that the FDA or other health authorities will deem the alternative materials and/or suppliers to be comparable, which may require us to perform additional and/or extended clinical studies and could delay product approval.
28
In addition, some of the materials which we purchase for use in our manufacturing operations are sole-sourced, meaning only one known supplier exists or is qualified for our use. In the event of a significant interruption of sole-sourced supplies, the quantity of our inventory may not be adequate to complete our clinical trials or to launch our potential products.
The prices of our common stock and convertible senior notes are likely to continue to be volatile in the future.
The stock prices of biopharmaceutical and biotechnology companies, including ours, have historically been highly volatile. Since January 1, 2005, our stock price has fluctuated between a high closing price of $7.98 on March 31, 2006 and a low closing price of $1.81 on January 31, 2008. The market has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. In addition, as our convertible senior notes are convertible into shares of our common stock, volatility or depressed prices of our common stock could have a similar effect on the trading price of the notes. Also, interest rate fluctuations can affect the price of our convertible senior notes. The following factors, among others, may affect the prices of our common stock and notes:
| | • | | announcements of data from, or material developments in, our clinical trials or those of our competitors, including delays in the commencement, progress or completion of a clinical trial; |
| | • | | fluctuations in our financial results; |
| | • | | announcements of technological innovations or new therapeutic products by us or our competitors, including innovations or products by our competitors that may require us to redesign, and therefore delay, our clinical trials to account for those innovations or products; |
| | • | | announcements of changes in governmental regulation affecting us or our competitors; |
| | • | | announcements of regulatory approval, disapproval, delays or suspensions of our or our competitors’ products; |
| | • | | announcements of new collaborative relationships by us or our competitors; |
| | • | | developments in patent or other proprietary rights affecting us or our competitors; |
| | • | | public concern as to the safety of products developed by us or other biotechnology and pharmaceutical companies; |
| | • | | material developments related to our minority interest in Ceregene, Inc.; |
| | • | | fluctuations in price and volume in the stock market in general, or in the trading of the stock of biopharmaceutical and biotechnology companies in particular, that are unrelated to our operating performance; |
| | • | | issuances of securities in equity, debt or other financings or issuances of common stock upon conversion of our convertible senior notes or exercise of warrants; |
| | • | | the favorable or unfavorable resolution of potential tax assessments; |
| | • | | sales of common stock by existing stockholders; and |
| | • | | the perception that such issuances by us or sales by existing stockholders could occur. |
Our stockholders may be diluted by the conversion of outstanding convertible senior notes.
In October and November 2004 we issued and sold $145.0 million aggregate principal amount of notes, which are convertible into our common stock, initially at the conversion price of $9.10 per share, equal to a conversion rate of 109.8901 shares per $1,000 principal amount of notes, subject to adjustments for stock
29
dividends, stock splits, and other similar events. The holders of the notes may choose at any time to convert their notes into common stock. The number of shares of common stock issuable upon conversion of the notes, and therefore the dilution of existing common stockholders, could increase as a result of an event triggering the antidilution rights of the notes, including certain acquisitions in which 10% or more of the consideration paid for our common stock in the transaction is in the form of cash or securities that are not freely tradable. Conversion of our convertible senior notes would result in issuance of additional shares of common stock, diluting existing common stockholders.
Our stockholders may be diluted, and the prices of our securities may decrease, by the exercise of outstanding stock options and warrants or by future issuances of securities.
We may issue additional common stock, preferred stock, restricted stock units, or securities convertible into or exchangeable for our common stock. Furthermore, substantially all shares of common stock for which our outstanding stock options or warrants are exercisable are, once they have been purchased, eligible for immediate sale in the public market. The issuance of additional common stock, preferred stock, restricted stock units, or securities convertible into or exchangeable for our common stock or the exercise of stock options or warrants would dilute existing investors and could adversely affect the price of our securities.
We have adopted anti-takeover defenses that could make it difficult for another company to acquire control of us or could limit the price investors might be willing to pay for our stock.
Certain provisions of our certificate of incorporation, bylaws, debt instruments and Delaware law could make it more difficult for a third party to acquire us, even if doing so would benefit our stockholders. These provisions include the adoption of a Stockholder Rights Plan, commonly known as a “poison pill.” Under the Stockholder Rights Plan, we made a dividend distribution of one preferred share purchase right for each share of our common stock outstanding as of August 21, 1995 and each share of our common stock issued after that date. In July 2000, we made certain technical changes to amend the plan and extended the term of such plan until 2010. The rights are exercisable only if an acquirer purchases 15% or more of our common stock or announces a tender offer for 15% or more of our common stock. Upon exercise, holders other than the acquirer may purchase our stock at a discount. Our Board of Directors may terminate the rights plan at any time or under certain circumstances redeem the rights. Because the rights may substantially dilute the stock ownership of a person or group attempting to take us over without the approval of our Board of Directors, the plan could make it more difficult for a third party to acquire us (or a significant percentage of our outstanding capital stock) without first negotiating with our Board of Directors regarding such acquisition. These provisions and certain provisions of the Delaware General Corporation Law may have the effect of deterring hostile takeovers or otherwise delaying or preventing changes in our management or in the control of our company, including transactions in which our stockholders might otherwise receive a premium over the fair market value of our common stock.
The Committed Equity Financing Facility, or CEFF, that we entered into with Kingsbridge in February 2007 may not be available to us if we elect to make a drawdown, may require us to make additional “blackout” or other payments to Kingsbridge, and may result in dilution to our stockholders.
The 2007 CEFF entitles us to sell and obligates Kingsbridge to purchase, from time to time over a period of three years, shares of our common stock for cash consideration, subject to certain conditions and restrictions. As of December 31, 2007, there were approximately 4.5 million shares remaining that may be sold under this CEFF. Kingsbridge will not be obligated to purchase shares under the CEFF unless certain conditions are met, which include a minimum price for our common stock; the accuracy of representations and warranties made to Kingsbridge; compliance with laws; effectiveness of the registration statement filed by us with the SEC; and the continued listing of our stock on the NASDAQ Global Market. In addition, Kingsbridge is permitted to terminate the CEFF if it obtains actual knowledge that a material and adverse event has occurred affecting our business, operations, properties or financial condition. If we are unable to access funds through the CEFF, or if the CEFF is terminated by Kingsbridge, we may be unable to access capital on favorable terms, or at all.
30
We are entitled, in certain circumstances, to deliver a blackout notice to Kingsbridge to suspend the registration statement filed by us with the SEC and prohibit Kingsbridge from selling shares under the registration statement. We may deliver a blackout notice, for example, at a time when we believe there may be a potential material development, premature disclosure of which would not be in the interests of our company or stockholders. If we deliver a blackout notice in the 30 calendar days following the settlement of a drawdown, or if the registration statement is not effective in circumstances not permitted by the agreement, then we must make a payment to Kingsbridge, or issue Kingsbridge additional shares in lieu of this payment, calculated on the basis of the number of shares held by Kingsbridge (exclusive of shares that Kingsbridge may hold pursuant to exercise of the Kingsbridge warrant) and the change in the market price of our common stock during the period in which the use of the registration statement is suspended. If the trading price of our common stock declines during a suspension of the registration statement, the blackout or other payment could be significant.
If we continue to sell shares to Kingsbridge under the CEFF, or issue shares in lieu of a blackout payment, it will dilute the holdings of our current stockholders, and may result in downward pressure on the price of our common stock. If we draw down under the CEFF, we will issue shares to Kingsbridge at a discount of up to 10% from the volume weighted average price of our common stock. If we draw down amounts under the CEFF when our share price is decreasing, we will need to issue more shares to raise the same amount than if our stock price was higher. Issuances in the face of a declining share price will cause even more dilution than if our share price were stable or increasing, and may further decrease our share price.
If we fail to maintain an effective system of internal controls, we may not be able to accurately report our financial results, maintain investor confidence or prevent fraud.
Effective internal controls are necessary for us to provide reliable financial reports, maintain investor confidence and prevent fraud. As our operations have grown, as well as part of our examination of our internal systems in response to Sarbanes-Oxley requirements, we have discovered in the past, and may in the future discover, areas of our internal controls that could be improved. None of these issues have risen to the level that we were unable to attest to the effectiveness of our internal controls when we were required to do so. We cannot be certain that any measures that we take to improve our internal controls will ensure that we implement and maintain adequate controls over our financial processes and reporting in the future. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm our operating results or cause us to fail to meet our reporting obligations. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
Accounting pronouncements may impact our results of operations and financial position.
U.S. generally accepted accounting principles and related implementation guidelines and interpretations can be highly complex and involve subjective judgments. Changes in these rules or their interpretation, the adoption of new pronouncements or the application of existing pronouncements to changes in our business could significantly alter our financial statements and results of operations.
For example, at its July 25, 2007 meeting, the Financial Accounting Standards Board, or FASB, agreed to issue for comment a proposed FASB Staff Position, FSP, addressing convertible instruments that may be settled in cash upon conversion, including instruments commonly referred to as Instrument C from EITF Issue No. 90-19, Convertible Bonds with Issuer Option to Settle for Cash upon Conversion, requiring the issuer to settle the principal amount of an Instrument C Security in cash and the conversion spread in cash or net shares at the issuer’s option. We understand that the proposed FSP would require bifurcation of the conversion option from the debt instrument, classification of the conversion option in equity, and then accretion of the resulting discount on the debt to result in additional interest expense being reported in the income statement. Since we cannot predict the outcome of the final FSP, we believe that if the FASB determines that we should account for Instrument C securities in the manner described above, the accounting for convertible senior notes would be
31
affected and the impact to our financial statements and results of operations would need to be evaluated to determine the impact, if any.
Risks Related to Our Industry
In order for our products to be offered to the public, they must undergo extensive clinical testing and receive approval from the FDA and other regulatory agencies, which could delay or prevent the commercialization of our products.
Human therapeutic products must undergo rigorous preclinical and clinical testing and other premarket approval procedures by the FDA and similar authorities in foreign countries. Preclinical tests include laboratory evaluation of potential products and animal studies to assess the potential safety and efficacy of the product and its formulations. Initiation of clinical trials requires approval by health authorities. Clinical trials involve the administration of the investigational new drug to healthy volunteers or to patients under the supervision of a qualified principal investigator. Depending on the country in which the study is conducted, clinical trials are subject to compliance with the FDA and the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practices and the European Clinical Trials Directive under protocols that detail the objectives of the study, the parameters to be used to monitor safety and the efficacy criteria to be evaluated. Other national, foreign and local regulations may also apply. The developer of the drug must provide information relating to the characterization and controls of the product before administration to the patients participating in the clinical trials. This requires developing approved assays of the product to test before administration to the patient and during the conduct of the trial. In addition, developers of pharmaceutical products must provide periodic data regarding clinical trials to the FDA and other health authorities, and these health authorities or our Independent Data Monitoring Committees may issue a clinical hold upon, or terminate, a trial if they do not believe, or cannot confirm, that the trial can be conducted without unreasonable risk to the trial participants. The FDA or foreign health authorities could issue a clinical hold with respect to any of our clinical trials in the future. The results of the preclinical testing and clinical testing, together with chemistry, manufacturing and controls information, are submitted to the FDA and other health authorities in the form of a new drug application for a pharmaceutical product and in the form of a biologics license application for a biological product, requesting approval to commence commercial sales.
In responding to a new drug application or a biologics license application, the FDA or foreign health authorities may grant marketing approvals, request additional information or further research, or deny the application if it determines that the application does not satisfy its regulatory approval criteria. Regulatory approval of a new drug application, biologics license application, or supplement is never guaranteed, and the approval process can take several years and is extremely expensive. The FDA and foreign health authorities have substantial discretion in the drug and biologics approval processes. Despite the time and expense incurred, failure can occur at any stage, and we could encounter problems that cause us to abandon clinical trials or to repeat or perform additional preclinical, clinical or manufacturing-related studies. Approvals may not be granted on a timely basis, if at all, and if granted may not cover all the clinical indications for which we may seek approval. Also, an approval might contain significant limitations in the form of warnings, precautions or contraindications with respect to conditions of use.
In addition, during the course of the development and testing of human therapeutic products, improvements may be made and may have been made to processes, formulations or manufacturing methods or employ different manufacturing facilities. Such changes may be made to improve the product’s potential efficacy, make it easier to manufacture at scale, reduce variability, improve ability to meet specification, or for other reasons. As a result, certain of the products that are currently being tested in clinical trials are not identical to those used in previous clinical trials from which clinical data have been previously reported or may vary throughout the course of a clinical trial. Certain studies may be required in order to demonstrate the comparability of the products if additional manufacturing changes are introduced. There is no guarantee that the results of studies using the current versions of the products will be as successful as the results of earlier studies conducted using different versions of the products.
32
Even if our products are approved by regulatory authorities, if we fail to comply with ongoing regulatory requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain marketing approval, along with the manufacturing processes, post-approval preclinical, manufacturing, clinical and safety data and promotional activities for such product, will be subject to continual review and periodic inspections by the FDA and other regulatory bodies. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Later discovery of previously unknown problems with our products including unanticipated adverse events of unanticipated severity or frequency, manufacturer or manufacturing problems, or failure to comply with regulatory requirements, may result in restrictions on such products or manufacturing processes, withdrawal of the products from the market, additional clinical trials, additional marketing application requirements, voluntary or mandatory recall, fines, suspension of regulatory approvals, product seizures or detention, injunctions or the imposition of civil or criminal penalties.
We are subject to federal, state, local and foreign laws and regulations, and complying with these may cause us to incur significant costs.
Our research, product development and manufacturing activities involve the controlled use of hazardous materials, and we may incur significant costs as a result of the need to comply with numerous laws and regulations. We are subject to laws and regulations enforced by the FDA, the DEA, the CDHS, foreign health authorities and other regulatory statutes including the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Food, Drug and Cosmetic Act, the Resource Conservation and Recovery Act, and other current and potential federal, state, local and foreign laws and regulations governing the use, manufacture, storage, handling and disposal of our products, materials used to develop and manufacture our products, and resulting waste products.
We cannot completely eliminate the risk of contamination or injury, by accident or as the result of intentional acts from these materials. In the event of an accident, we could be held liable for any damages that result, and any resulting liability could exceed our resources. We do not carry insurance for potential exposures which could result from these risks. We may also be required to incur significant costs to comply with environmental laws and regulations in the future.
The continuing efforts of governmental and third-party payers to contain or reduce the costs of healthcare may restrict pricing of our products and reimbursement from third party payers, which may reduce demand for and the profitability of our products.
Because of the considerable pressure to reduce the cost of biotechnology and pharmaceutical products, there is uncertainty related to the extent to which third-party payers will cover and pay for newly approved therapies. Our potential products represent a new mode of therapy, and while the cost-benefit ratio of the products may be favorable, we expect that the costs associated with our products will be substantial. Acceptance and sales of our future products will depend in part upon coverage and third party payment amounts from third-party payers, including government agencies, private health care insurers and other health care payers, such as health maintenance organizations, and self-insured employee plans. Reimbursement from these organizations may become more restrictive in the future. Our proposed products may not be considered cost-effective by third-party payers. Insurance coverage might not be provided by third-party payers at all or may be provided only after substantial delay. Even if such coverage is provided, the approved third-party payment amounts might not be sufficient to permit widespread acceptance of our products. Any such cost control initiatives could lessen our ability to commercialize our products, decrease the price that we would receive for products that are commercialized, if any, and may impede the ability of patients using our products to obtain reimbursement under their insurance programs.
33
The pricing of our future products in the United States and elsewhere may also be influenced in part by government restrictions on the pricing and profitability of prescription pharmaceuticals. In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement more rigorous provisions relating to government payment levels. While we cannot predict whether the government will adopt any such legislative or regulatory proposals, the announcement or adoption of these proposals could impair our business, results of operations, financial condition, cash flow and future prospects.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
We maintain our corporate headquarters in South San Francisco, California, in a facility consisting of approximately 154,000 square feet of research and development and administrative space. Our cGMP manufacturing facility in Hayward, California, consists of 51,000 square feet of manufacturing space and 50,000 square feet of laboratory and office space. It is designed to produce one or more types of products at a scale suitable for Phase 3 trials and potential commercial market launch, and is currently producing our GVAX immunotherapy for prostate cancer product. Our approximately 12,000 square-foot facility in Memphis, Tennessee is a centrally located facility which we intend to use in the future as a centralized product distribution center. We lease all of our facilities.
None.
| ITEM 4. | SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS |
None.
34
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY AND RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Our common stock trades on the NASDAQ Global Market under the symbol “CEGE.” The following table shows, for the periods indicated, the high and low closing prices per share of our common stock as reported by the NASDAQ Global Market. We did not declare or pay any cash dividends with respect to our common stock during any of the periods indicated below and do not expect to pay cash dividends on our common stock in the foreseeable future.
| | | | | | |
Year Ended December 31, 2007: | | High | | Low |
First Quarter | | $ | 4.20 | | $ | 2.81 |
Second Quarter | | | 6.86 | | | 3.35 |
Third Quarter | | | 4.07 | | | 3.22 |
Fourth Quarter | | | 3.74 | | | 2.17 |
| | |
Year Ended December 31, 2006: | | High | | Low |
First Quarter | | $ | 7.98 | | $ | 5.36 |
Second Quarter | | | 7.79 | | | 4.92 |
Third Quarter | | | 5.24 | | | 4.32 |
Fourth Quarter | | | 4.69 | | | 3.39 |
As of January 31, 2008, there were approximately 588 holders of record and approximately 30,000 beneficial holders of our common stock. On February 27, 2008, the last reported sales price on the NASDAQ Global Market for our common stock was $2.47. The market for our common stock is highly volatile.
We did not repurchase any shares of our equity securities or declare any dividends during the year ended December 31, 2007. There are no restrictions under our outstanding convertible senior note agreement or other agreements that prohibit payment of dividends.
35
STOCKHOLDER RETURN COMPARISON
The following graph shows the total stockholder return of an investment of $100 cash on December 31, 2002 for (i) our common stock, (ii) the NASDAQ Global Market—US Index and (iii) the NASDAQ Biotechnology Index. The stock price performance shown on the graph is not necessarily indicative of future price performance.
Performance Measurement Comparison
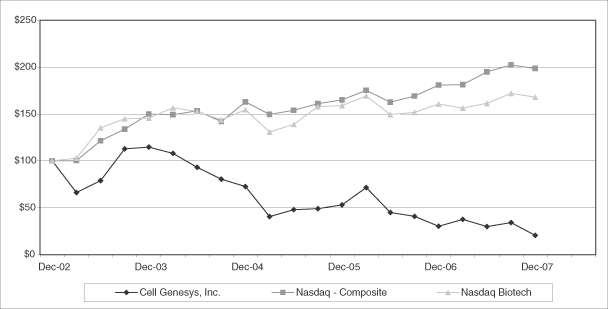
The information required by this item regarding equity compensation plans is incorporated by reference to the information in Item 12 of this Annual Report on Form 10-K.
36
| ITEM 6. | SELECTED FINANCIAL DATA |
The following selected financial information has been derived from our audited consolidated financial statements. The information below is not necessarily indicative of results of future operations, and should be read together withItem 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations”and the consolidated financial statements and related notes included in Item 8 of this Annual Report on Form 10-K in order to fully understand factors that may affect the comparability of the information presented below.
| | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | | | 2004 | | | 2003 | |
| | | (In thousands, except per share amounts) | |
Consolidated Statement of Operations Data: | | | | | | | | | | | | | | | | | | | | |
Revenue | | $ | 1,380 | | | $ | 1,364 | | | $ | 4,584 | | | $ | 11,458 | | | $ | 18,128 | |
Total operating expenses | | | 126,532 | * | | | 114,387 | * | | | 111,097 | | | | 110,061 | | | | 111,276 | |
Gain on sale of Abgenix, Inc. common stock | | | — | | | | 62,677 | | | | 55,123 | | | | 12,160 | | | | 12,638 | |
Net loss | | | (99,274 | )* | | | (82,929 | )* | | | (64,939 | ) | | | (97,411 | ) | | | (56,406 | ) |
Net loss attributed to common stockholders | | | (99,274 | )* | | | (82,929 | )* | | | (64,943 | ) | | | (97,511 | ) | | | (56,636 | ) |
Basic and diluted net loss per common share | | | (1.39 | )* | | | (1.67 | )* | | | (1.43 | ) | | | (2.23 | ) | | | (1.48 | ) |
| |
| | | December 31, | |
| | | 2007 | | | 2006 | | | 2005 | | | 2004 | | | 2003 | |
| | | (In thousands) | |
Consolidated Balance Sheet Data: | | | | | | | | | | | | | | | | | | | | |
Cash, cash equivalents and short-term investments, including restricted cash and investments | | $ | 147,306 | | | $ | 154,074 | | | $ | 129,139 | | | $ | 174,227 | | | $ | 158,917 | |
Total assets | | | 273,392 | | | | 291,167 | | | | 366,975 | | | | 435,139 | | | | 460,502 | |
Total current liabilities | | | 34,565 | | | | 51,314 | | | | 69,385 | | | | 77,923 | | | | 94,296 | |
Long-term obligations, excluding current portion | | | 56,278 | | | | 51,326 | | | | 52,093 | | | | 51,013 | | | | 146,634 | |
Convertible senior notes | | | 145,000 | | | | 145,000 | | | | 145,000 | | | | 145,000 | | | | — | |
Redeemable convertible preferred stock | | | — | | | | — | | | | — | | | | 1,897 | | | | 2,706 | |
Accumulated deficit | | | (491,115 | ) | | | (391,841 | ) | | | (308,912 | ) | | | (243,973 | ) | | | (146,562 | ) |
Stockholders’ equity | | | 37,549 | | | | 43,527 | | | | 100,497 | | | | 159,306 | | | | 216,866 | |
| * | As discussed inNote 1, “Organization and Summary of Significant Accounting Policies”to the consolidated financial statements included under Item 8 of this Annual Report on Form 10-K, effective January 1, 2006, we changed our method of accounting for stock-based compensation pursuant to Statement of Financial Accounting Standards No. 123(R), “Share-Based Payment”. |
37
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION |
Statements made in this Item other than statements of historical fact, including statements about us and our subsidiaries and our respective clinical trials, research programs, product pipelines, current and potential corporate partnerships, licenses and intellectual property, the adequacy of capital reserves and anticipated operating results and cash expenditures, are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. As forward-looking statements, they are subject to a number of uncertainties that could cause actual results to differ materially from the statements made, including risks associated with the success of research and product development programs, the issuance and validity of patents, the development and protection of proprietary technologies, the ability to raise capital, operating expense levels and the ability to establish and retain corporate partnerships and other risks. Reference is made to discussions about risks associated with product development programs, intellectual property and other risks that may affect us underItem 1A, “Risk Factors”above. We do not undertake any obligation to update any forward-looking statement. The following should be read together with our consolidated financial statements and related notes located elsewhere in this Annual Report on Form 10-K for the year ended December 31, 2007 and other documents filed by us from time to time with the SEC.
Overview
We are a biotechnology company focused on the development and commercialization of novel biological therapies for patients with cancer. We are currently developing cell-based cancer immunotherapies and oncolytic virus therapies to treat different types of cancer. Our clinical stage cancer programs involve cell- or viral-based products that have been modified to impart disease-fighting characteristics that are not found in conventional chemotherapeutic agents. As part of our GVAX™ cancer immunotherapy programs, we are conducting two Phase 3 clinical trials in prostate cancer. We initiated our two Phase 3 clinical trials for GVAX immunotherapy for prostate cancer in July 2004 and June 2005, respectively, each under a Special Protocol Assessment, or SPA, with the United States Food and Drug Administration, or FDA. In May 2006, we were granted Fast Track designation for GVAX immunotherapy for prostate cancer by the FDA. Fast Track designation can potentially facilitate development and expedite the review of Biologics License Applications, or BLAs. The first of these two Phase 3 clinical trials, referred to as VITAL-1, is now fully enrolled. In January 2008 the Independent Data Monitoring Committee, or IDMC, for VITAL-1 completed a pre-planned interim analysis in the timeframe originally estimated and recommended that the study continue. In collaboration with investigators at Johns Hopkins University we are conducting Phase 2 trials in pancreatic cancer and Phase 1 and Phase 2 trials in leukemia and myelodysplastic syndrome. In our oncolytic virus therapies program, which we are developing in part through a global alliance with Novartis AG, we are conducting a multiple dose Phase 1 clinical trial of CG0070 in recurrent bladder cancer.
Critical Accounting Policies and the Use of Estimates
The accompanying discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements and related disclosures, which we have prepared in accordance with U.S. generally accepted accounting principles. The preparation of these consolidated financial statements requires management to make estimates, assumptions and judgments that affect the reported amounts in our consolidated financial statements and accompanying notes. These estimates form the basis for making judgments about the carrying values of assets and liabilities. Actual results may differ from these estimates. We consider certain accounting policies related to revenue recognition, income taxes and stock-based compensation to be critical accounting policies.
Revenue recognition
Our revenues are derived principally from research and licensing agreements with collaborators. Revenue under such collaboration agreements typically includes up-front payments, cost reimbursements, milestone payments and license fees. We evaluate whether the delivered element under these arrangements has value to our
38
customer on a stand-alone basis and whether objective and reliable evidence of fair value of the undelivered item exists. Deliverables that do not meet these criteria are treated as one unit of accounting for the purposes of revenue recognition.
Up-front payments: Up-front payments from our research collaborations include payments for licenses, technology transfer and access rights. Non-refundable up-front license fees and other payments under collaboration agreements where we cannot establish standalone value for the delivered license and where we have continuing involvement following the execution of the collaboration agreement, are deferred and recognized on a straight-line or ratable method over the period of our continuing involvement unless we determine that another methodology is more appropriate. During 2005, we recognized revenue from a non-refundable up-front payment under our global alliance with Novartis AG for the development of certain oncolytic virus therapies based upon when the underlying development expenses were incurred, rather than a ratable method, as we determined that the expense method was more appropriate for this agreement. The revenues recorded under the Novartis AG alliance approximated the related development expenses that were incurred in the respective periods. We recognize cost reimbursement revenue under collaborative agreements as the related research and development costs are incurred, as provided for under the terms of these agreements.
Milestones: Payments for milestones that are based on the achievement of substantive and at-risk performance criteria are recognized in full upon achievement of the incentive milestone events in accordance with the terms of the agreement. Incentive milestone payments are triggered either by the results of our research efforts or by events external to us, such as regulatory approval to market a product or the achievement of specified sales levels by a marketing partner. As such, the incentive milestones are substantially at risk at the inception of the agreement, and the amounts of the payments assigned thereto are commensurate with the milestone achieved. Upon the achievement of an incentive milestone event, we have no future performance obligations related to that milestone payment.
License fees: Non-refundable license fees where we have completed all obligations at the execution of the arrangement are recognized as revenue upon execution of the technology licensing agreement when delivery has occurred, collectibility is reasonably assured and the price is fixed and determinable.
Income taxes
In June 2006, the Financial Accounting Standards Board issued Interpretation No. 48, Accounting for Uncertainty in Income Taxes, or FIN 48. FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an entity’s financial statements in accordance with Statement of Financial Accounting Standards No. 109, Accounting for Income Taxes, or FAS 109, and prescribes a recognition threshold and measurement attribute for financial statement disclosure of tax positions taken or expected to be taken on a tax return. Additionally, FIN 48 provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition.
On January 1, 2007, we adopted the provisions of FIN 48. As a result of the implementation of FIN 48, we did not recognize any adjustment in the liability for unrecognized income tax benefits and, therefore, implementation of FIN 48 did not result in a cumulative adjustment to accumulated deficit. At the adoption date of January 1, 2007, we had $25.0 million of unrecognized tax benefits, all of which would affect our effective tax rate if recognized. At December 31, 2007, we had $4.0 million of unrecognized tax benefits, all of which would affect our effective tax rate if recognized. As a result of a final settlement with the Internal Revenue Service, or IRS, during May 2007, unrecognized tax benefits decreased by $21.0 million in the second quarter of 2007. Our policy is to account for interest and penalties related to income tax matters in the income tax provision in the consolidated statement of operations. Accrued interest and penalties related to income tax matters are included within the related tax liability line in the consolidated balance sheet. At the adoption date of January 1, 2007, we had $10.4 million of accrued interest and zero penalties related to tax contingencies recorded in the consolidated balance sheet. As of December 31, 2007, we had accrued $2.2 million of interest and zero penalties related to tax contingencies.
39
Income tax benefits previously recorded have been based on a determination of deferred tax assets and liabilities and any valuation allowances that might be required against these deferred tax assets. We record a valuation allowance to reduce deferred tax assets to the amounts that are more likely than not to be realized. We considered anticipated future taxable income, and potential tax planning strategies in assessing the need for valuation allowances. Certain of these determinations require judgment on the part of management. If we determine that we will be able to realize deferred tax assets in the future in excess of the carrying value of our net deferred tax assets, adjustments to the deferred tax assets will increase income by reducing tax expense in the period that such determination is reached. Likewise, if we determine that we will not be able to realize all or part of the carrying value of our net deferred tax assets in the future, adjustments to the deferred tax assets will decrease income by increasing tax expense in the period that such determination is reached. Significant estimates are required in determining our income tax benefits. Various internal and external factors may have favorable or unfavorable effects on our future effective tax rate. These factors include, but are not limited to, changes in tax laws and regulations, our future levels of spending for research and development, and changes in our overall level of pre-tax earnings or losses.
Stock-based compensation
Our operating expenses included the cost of stock-based compensation awarded to our employees. Prior to 2006, we accounted for stock-based compensation plans under the recognition and measurement provisions of Accounting Principles Board Opinion No. 25. Beginning January 1, 2006, we accounted for employee stock-based compensation in accordance with Statement of Financial Accounting Standards No. 123 (revised 2004), “Share-Based Payment,” or FAS 123R. Under the provisions of FAS 123R, we estimate the fair value of our employee stock-based awards at the date of grant using the Black-Scholes option valuation model, which requires the use of certain subjective assumptions. When establishing an estimate of the expected term of an award, we consider our historical stock option exercise experience including forfeitures, our post vesting termination pattern and the term of the options outstanding. As required under the accounting rules, we review our valuation assumptions at each grant date and, as a result, we are likely to change our valuation assumptions used to value employee stock-based awards granted in future periods.
FAS 123R requires that employee stock-based compensation costs be recognized over the requisite service period, or the vesting period, in a manner similar to all other forms of compensation paid to employees. Accordingly, for the year ended December 31, 2007, we recognized $6.5 million of stock-based compensation expense in operating expenses with an allocation of $5.0 million to research and development and $1.5 million to general and administrative expenses. For the year ended December 31, 2006, we recognized $5.9 million of stock-based compensation expense in operating expenses with an allocation of $4.6 million to research and development and $1.3 million to general and administrative expenses. There was no stock-based compensation expense related to employee stock options and employee stock purchases recognized during fiscal year 2005. We adopted FAS 123R on a modified prospective basis. The allocation of employee stock-based compensation costs to each operating expense line is estimated based on specific employee headcount information at each grant date and revised, if necessary, in future periods if actual employee headcount information differs materially from those estimates. As a result, the amount of employee stock-based compensation costs we record in future periods in each operating expense line may differ significantly from what we have recorded in the current period. As of December 31, 2007, total compensation cost related to nonvested stock options not yet recognized was $4.4 million, which is expected to be allocated to stock-based compensation expense over a weighted-average period of 25 months, and total compensation cost related to nonvested restricted stock units not yet recognized was $1.7 million, which is expected to be allocated to stock-based compensation expense over a weighted-average period of seven months. No compensation expense will be recognized for restricted stock units that do not vest.
SeeNote 1, “Organization and Summary of Significant Accounting Policies” and Note 9, “Stockholder’s Equity and Stock-Based Compensation” of Notes to Consolidated Financial Statementsincluded under Item 8 of this Annual Report on Form 10-K for further information.
40
Recently issued accounting standards
In September 2006, the Financial Accounting Standards Board issued Statement of Financial Accounting Standards No. 157, “Fair Value Measurements,” or FAS 157. FAS 157 defines fair value, establishes a market-based framework or hierarchy for measuring fair value under GAAP, and expands disclosures about fair value measurements. FAS 157 is effective for financial statements issued for fiscal years beginning after November 15, 2007, and interim periods within those fiscal years. We have evaluated the impact of adopting FAS 157 on our consolidated financial statements and do not expect any impact on our results of operations or financial position.
In February 2007, the FASB issued SFAS No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities—Including an Amendment of FASB Statement 115,” or FAS 159. FAS 159 permits entities to choose to measure many financial instruments and certain other items at fair value. Entities that elect the fair value option will report unrealized gains and losses in earnings at each subsequent reporting date. The fair value option may be elected on an instrument-by-instrument basis, with few exceptions. FAS 159 also establishes presentation and disclosure requirements to facilitate comparisons between companies that choose different measurement attributes for similar assets and liabilities. FAS 159 is effective for financial statements issued for fiscal years beginning after November 15, 2007. We have evaluated the impact of adopting FAS 159 on our consolidated financial statements and do not expect any impact on our results of operations or financial position.
In June 2007, the FASB ratified Emerging Issues Task Force, or EITF, Issue No. 07-3, “Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities,” or Issue 07-3, which addresses the accounting for nonrefundable advance payments. The EITF concluded that nonrefundable advance payments for goods or services to be received in the future for use in research and development activities should be deferred and capitalized. The capitalized amounts should be expensed as the related goods are delivered or the services are performed. If an entity’s expectations change such that it does not expect it will need the goods to be delivered or the services to be rendered, capitalized nonrefundable advance payments should be charged to expense. Issue 07-3 is effective for new contracts entered into during fiscal years beginning after December 15, 2007, including interim periods within those fiscal years. We have evaluated the impact of adopting Issue 07-3 on our consolidated financial statements and do not expect any impact on our results of operations or financial position.
In December 2007, the FASB ratified the final consensuses in Emerging Issues Task Force Issue No. 07-1,“Accounting for Collaborative Arrangements,” or Issue 07-1, which requires certain income statement presentation of transactions with third parties and of payments between parties to the collaborative arrangement, along with disclosure about the nature and purpose of the arrangement. Issue 07-1 is effective for us beginning January 1, 2009. We have evaluated the impact of adopting Issue 07-1 on our consolidated financial statements and do not expect any impact on our results of operations or financial position.
Results of Operations
Revenue
Revenues were $1.4 million in 2007, compared to $1.4 million in 2006 and $4.6 million in 2005. The following table shows our revenues by source for the periods indicated:
| | | | | | | | | |
| | | 2007 | | 2006 | | 2005 |
| | | (in thousands) |
Novartis AG | | $ | — | | $ | — | | $ | 2,031 |
sanofi-aventis Group | | | 1,000 | | | 1,000 | | | 2,000 |
Ceregene, Inc. | | | 13 | | | 83 | | | 69 |
Other | | | 367 | | | 281 | | | 484 |
| | | | | | | | | |
| | $ | 1,380 | | $ | 1,364 | | $ | 4,584 |
| | | | | | | | | |
41
Revenues for both 2007 and 2006 included $1.0 million in connection with our gene activation technology license agreement with sanofi-aventis Group for gene activated erythropoietin, compared to revenues of $2.0 million in 2005 which included $1.0 million in milestone revenue under the same agreement. We recorded contract revenue of $13,000 in 2007 compared to $0.1 million in both 2006 and 2005 for services provided to Ceregene of which we own a minority position. Revenues for 2005 included $2.0 million from Novartis AG in connection with our global alliance for the development and commercialization of oncolytic virus therapies. There was no revenue in 2007 and 2006 from Novartis AG as the $28.5 million payment was fully recognized by December 31, 2005.
Research and development expenses
Research and development expenses were $106.1 million in 2007 compared to $96.3 million in 2006 and $92.4 million in 2005. These increases can be attributed to the increase in Phase 3 clinical activities for our GVAX immunotherapy program for prostate cancer in Europe, our expansion in clinical trials and other product development activities in both our GVAX cancer immunotherapy and oncolytic virus therapy programs. Research and development expenses included $5.0 million in 2007, and $4.6 million in 2006, in non-cash stock-based compensation expense related to FAS 123R. In July 2004, we announced the commencement of our VITAL-1 trial, which compares GVAX prostate cancer immunotherapy to Taxotere chemotherapy in patients with advanced prostate cancer without cancer-related pain. In June 2005, we announced the commencement of our VITAL-2 trial, which compares GVAX prostate cancer immunotherapy plus Taxotere chemotherapy to Taxotere chemotherapy alone in advanced prostate cancer patients with cancer-related pain.
We continue to deploy the majority of our research and development resources to advance GVAX immunotherapy for prostate cancer. Expenses related to GVAX immunotherapy for leukemia, GVAX immunotherapy for pancreatic cancer, the oncolytic virus therapy CG0070 and other potential product candidates in preclinical studies are a minor proportion of our spending. We expect that our research and development expenditures and headcount will remain approximately at current levels in the near term. However, this depends on a number of factors, including progress in research and development and clinical trials.
Biopharmaceutical products, such as those being developed by us, may take 10 to 15 years to research, develop and bring to market in the United States. Drug development in the United States is a process that includes several steps regulated by the FDA. The process begins with the filing of an IND application, which, if successful, allows for a clinical study of the potential new medicine. Clinical development typically involves three phases of study: Phase 1, 2 and 3. Costs for each phase are generally larger than the preceding phase, as the size of the clinical trial (number of patients) grows. The most significant costs associated with clinical development are the Phase 3 trials, as they tend to be the longest and largest studies conducted during the drug development process. We currently have one product in development for which we have initiated Phase 3 studies. However, the successful development of our products is highly uncertain. Estimates of product completion dates and completion costs can vary significantly for each product and are difficult to predict. Completion of clinical trials, including the VITAL-1 and VITAL-2 trials that we initiated in July 2004 and June 2005, respectively, may take several years or more. The length of time generally varies substantially according to the type, complexity, novelty and intended use of the product candidate. However, we estimate that clinical trials of the type we generally conduct are usually completed over the following timelines:
| | |
Clinical Phase | | Estimated
Completion Period |
Phase 1 | | 1-3 years |
Phase 2 | | 1-3 years |
Phase 3 | | 2-5 years |
Many factors may delay our commencement and speed of completion of clinical trials, including the size and number of patients participating in the trial, the duration of patient follow-up required, the number of clinical
42
sites at which the trial is conducted, competing trials and the length of time required to locate and enroll suitable patient subjects. Various statutes and regulations also govern or influence the manufacturing, safety, labeling, storage, record keeping and marketing of each product. The lengthy process of seeking these approvals, and the subsequent compliance with applicable statutes and regulations, require the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, regulatory approvals could materially adversely affect our business. In responding to an NDA or a BLA, the FDA may grant marketing approval, request additional information or deny the application if it determines that the application does not provide an adequate basis for approval. There can be no assurance that any approval required by the FDA or other regulatory body will be obtained on a timely basis, if at all. For additional discussion of the risks and uncertainties associated with completing development of potential products, see “Item 1A. Risk Factors” above.
Included below is a summary of products and the related stage of development for each product in clinical development. The information in the column labeled “Estimated Completion of Ongoing Phase” constitutes forward-looking statements regarding timing of completion of product development phases. Our estimates of timing of completion of these trials are based on typical times of completion for trials of that type at such phases of development. The actual timing of completion of these phases of our clinical trials could differ materially from the estimates provided in the table due to the number of patients enrolled in the trial, the number of clinical trial sites involved, the time needed to fully enroll the trial, the time required for patient follow-up and other factors. Longer time frames for the completion of certain trials may be the result of requirements to measure patient survival. In addition, it is possible that any of these ongoing clinical trials may never be completed due to the occurrence of unacceptable treatment-related side effects, lack of clinical efficacy, insufficient supply of product for these clinical trials and other factors. For a discussion of the risks and uncertainties associated with the timing of completing a product development phase, see “Item 1A. Risk Factors” above.
| | | | |
Treatment | | Phase of
Development | | Estimated
Completion
of Ongoing Phase |
GVAX Cancer Immunotherapies: | | | | |
Prostate Cancer | | Phase 3 | | 2009-2010 |
Pancreatic Cancer | | Phase 2 | | 2008-2009 |
Leukemia | | Phase 2 | | 2008-2009 |
| | |
Oncolytic Virus Therapy: | | | | |
CG0070 (Recurrent Bladder Cancer) | | Phase 1 | | 2008-2009 |
General and administrative expenses
General and administrative expenses consist primarily of compensation, including non-cash, stock-based compensation, for employees in executive and operational functions, including finance, legal, information technology, human resources and business development. Other significant costs include facilities costs and professional fees for accounting and legal services, including legal services associated with obtaining and maintaining patents. General and administrative expenses were $20.4 million in 2007 compared to $18.1 million in 2006 and $16.3 million in 2005. The increase in 2007 compared to 2006 is primarily due to the continuing increase in infrastructure costs associated with product development and other business activities. The increase in 2006 compared to 2005 is primarily due to recording $1.3 million in 2006 of non-cash stock-based compensation expense related to adoption of FAS 123R. Future spending for general and administrative costs is expected to remain approximately at current levels in the near term.
Restructuring charges
In June 2005, we announced the implementation of a strategic restructuring of our business operations to focus resources on our most advanced and most promising product development programs. In November 2005, we sold our San Diego, California, manufacturing facility for viral products to Genzyme Corporation for
43
$3.2 million. We recorded a charge of $2.4 million in 2005 related to our restructuring decisions, including $1.5 million for workforce reduction initiatives, $0.3 million to reduce the carrying value of the San Diego manufacturing facility and $0.6 million for lease termination and other expenses. During 2006, we decreased the restructuring accrual by $0.1 million due to the extension of a sublease, and paid all remaining obligations.
Gain on sale of Abgenix common stock
During 2006, we sold all of our remaining 3.0 million shares of Abgenix common stock for a gain of $62.7 million. During 2005, we recorded a gain of $55.1 million associated with our sale of 3.7 million shares of Abgenix common stock.
Interest and other income
Interest and other income was $9.0 million in 2007 compared to $7.5 million in 2006 and $3.0 million in 2005. The increases are primarily attributed to higher average cash and short-term investment balances. In addition, we received $1.0 million from Laboratoire Serono S.A., or Serono, for our agreement to withdraw from a patent dispute between us and Serono relating to gene activation technology in December 2007. The amount of this settlement is included in “Interest and other income” in 2007.
Interest expense
Interest expense was $10.3 million in 2007 compared to $10.5 million in 2006 and $10.7 million in 2005. In October and November 2004, we issued $145.0 million aggregate principal amount of our 3.125% Convertible Senior Notes due in 2011 and used a portion of those proceeds to repay bank debt totaling $95.0 million. We recorded interest expense related to our Convertible Senior Notes, including amortization of related debt issuance costs, of $5.3 million in each of 2007, 2006 and 2005. In addition, we recorded interest expense associated with our South San Francisco, California capital lease obligation of $5.1 million in 2007, $5.2 million in 2006 and $5.3 million in 2005.
Income taxes
In July 2005, the IRS issued a Notice of Proposed Adjustment, or NOPA, seeking to disallow $48.7 million of net operating losses that we deducted for the 2000 fiscal year and seeking a $3.4 million penalty for substantial underpayment of tax in the year ended December 31, 2000. We responded to the IRS in September 2005, disagreeing with the conclusions reached by the IRS in the NOPA and seeking to resolve this matter with the Appeals Office of the IRS. In May 2007, we reached final settlement regarding this matter with the IRS in the amount of $3.3 million with respect to the fiscal years ended December 31, 2000, 2001 and 2002. This amount was comprised of $2.3 million in federal tax and $1.0 million in related interest. No penalty was assessed.
We recorded a tax benefit of $25.9 million in 2007 compared to a tax provision of $29.6 million in 2006 and $5.9 million in 2005. The tax benefit recorded for the year ended December 31, 2007 is related to the reversal of $26.4 million of previously accrued income taxes as a result of the final settlement with the IRS, in May 2007, offset by additional accrued interest for tax contingencies. The tax provision in 2006 relates to a realized gain on the sale of 3.0 million shares of Abgenix common stock and $2.8 million related to additional interest recorded for tax contingencies. The tax provision recorded in 2005 relates to the realized gain on the sale of 3.7 million shares of Abgenix common stock and $2.7 million of additional interest for tax contingencies, partially offset by tax benefits related to unrealized gains on Abgenix common stock.
It is reasonably possible that we will close certain years to examination under the relevant statute of limitations which may further decrease our liability for unrecognized tax benefits by approximately $4.0 million
44
in the next 12 months. We file tax returns in the U.S., U.K. and California. In general, the years 2004 through 2007, remain open to examination for U.S. and U.K. purposes, and 2000 through 2007 for California purposes. At December 31, 2007, we had federal net operating loss carryforwards of $447.6 million which will expire in the years beginning 2008 through 2027 if not utilized.
Deferred Revenue
In December 2007, we sold for $12.0 million all of our assets, intellectual property and previously established licensing agreements relating to our lentiviral gene delivery technology, commonly referred to as lentiviral vectors to GBP IP, LLC, an affiliate of GBP Capital, the majority shareholder in privately held Lentigen Corporation. We received full payment of $12.0 million in December 2007. As part of the sales agreement we retained our rights to use the technology for research and development purposes including potential future use with our cancer immunotherapy products. We applied EITF Issue No. 00-21, “Revenue Arrangements with Multiple Deliverables”, in evaluating the appropriate accounting for this agreement. We identified the delivery of biological materials (including certain GMP-compliant materials), intellectual property and previously established licensing agreements related to our lentiviral gene delivery technology as the primary deliverables under this sales agreement and concluded that these deliverables should be accounted for as a single unit of accounting based upon the determination that these deliverables did not have stand-alone value and can not be separated. Therefore, we have deferred recognition of revenue until we have performed all of our obligations under the agreement which we expect to complete during the first quarter of 2008.
Liquidity and Capital Resources
At December 31, 2007, we had $147.3 million in cash, cash equivalents and short-term investments, of which $2.9 million was classified as restricted and was related to letters of credit for our corporate headquarters facility in South San Francisco, California and our cGMP manufacturing facility in Hayward, California. Information regarding the classification of these assets is included inNote 4, “Investments” of Notes to Consolidated Financial Statementsincluded under Item 8 of this Annual Report on Form 10-K. We have maintained our financial position through strategic management of our resources including access to debt and equity financing, funding from various corporate collaborations and licensing agreements, and the sale of Abgenix common stock.
Equity Financing
In February 2003, our shelf registration statement on Form S-3 was declared effective by the SEC under the Securities Act, which allowed us to offer up to $150.0 million of securities on short notice in one or more public offerings registered under the Securities Act. We used this shelf registration in March 2004 to complete a public offering of 4.9 million shares of our common stock, resulting in net proceeds of $57.2 million. We next used this shelf registration in September 2006 to complete an underwritten public offering of 5.8 million shares of our common stock, resulting in net proceeds of $25.0 million. In April 2007, we used the remaining registered amount under this shelf registration to raise net proceeds of $55.4 million in a registered direct offering of 10.8 million shares of our common stock at $5.55 per share and warrants to purchase 2.2 million shares of our common stock at a price of $7.18 per share from selected institutional investors. These warrants are exercisable beginning October 12, 2007 for a period of four and a half years thereafter. The fair value of the warrants, which was recorded in additional paid-in capital, was determined to be $5.3 million on the date of issuance using the Black-Scholes option valuation model applying the following assumptions: (i) a risk-free rate of 4.66%, (ii) an expected term of 5.0 years, (iii) no dividend yield and (iv) volatility of 57%.
In March 2006, we entered into a Committed Equity Financing Facility, the 2006 CEFF, with Kingsbridge, pursuant to which Kingsbridge committed to purchase, subject to certain conditions, up to the lesser of 8.7 million shares of our common stock or an aggregate of $75.0 million during the three year period following entry into the 2006 CEFF.
45
In connection with the 2006 CEFF, we issued a warrant to Kingsbridge to purchase 0.4 million shares of our common stock at a price of $9.12 per share exercisable until September 14, 2011. The fair value of the warrant was determined on the date of issuance using the Black-Scholes option valuation model applying the following assumptions: (i) a risk-free rate of 4.68%, (ii) an expected term of 5.5 years, (iii) no dividend yield and (iv) volatility of 57%. The estimated fair value of this warrant was $1.3 million which was recorded as a contra-equity amount in additional paid-in capital in March 2006. In 2006, we received net proceeds of $27.9 million from the sale of 6.3 million shares of our common stock under the 2006 CEFF. In 2007, we received net proceeds of $7.1 million from the sale of 2.4 million shares of our common stock under the 2006 CEFF, which concluded the 2006 CEFF. Since inception of the 2006 CEFF, we received cumulative net proceeds of $35.0 million from the sale of 8.7 million shares of our common stock.
In February 2007, we entered into a new CEFF, the 2007 CEFF, with Kingsbridge, pursuant to which Kingsbridge committed to purchase, subject to certain conditions, up to the lesser of 11.6 million shares of our common stock or an aggregate of $75.0 million during the three year period following entry into the 2007 CEFF. In connection with the 2007 CEFF, we issued a warrant to Kingsbridge to purchase 0.4 million shares of our common stock at a price of $4.68 per share exercisable beginning on September 5, 2007 for a period of five years thereafter. The fair value of the warrant was determined on the date of issuance using the Black-Scholes option valuation model applying the following assumptions: (i) a risk-free rate of 4.80%, (ii) an expected term of 5.5 years, (iii) no dividend yield and (iv) volatility of 55%. The estimated fair value of this warrant was $0.6 million which was recorded as a contra-equity amount in additional paid-in capital in February 2007. During the year ended December 31, 2007, we received net proceeds of $23.0 million from the sale of 7.1 million shares of our common stock under the 2007 CEFF.
On May 16, 2007, our new shelf registration statement on Form S-3 was declared effective by the SEC under the Securities Act, which allows us to offer up to $150.0 million of securities on short notice in one or more public offerings under the Securities Act. As of December 31, 2007, the entire amount under this shelf is available for issuance.
Debt Financing
In October and November 2004, we issued and sold a total of $145.0 million aggregate principal amount of 3.125% Convertible Senior Notes due in 2011 in a private placement. We received approximately $139.9 million in proceeds after deducting the initial purchasers’ discount and estimated offering expenses, which we used, in part, to repay bank debt totaling $95.0 million. Under certain circumstances, we may redeem some or all of the convertible senior notes on or after November 1, 2009 at a redemption price equal to 100% of the principal amount of the notes. Holders of the notes may require us to repurchase some or all of their notes if a fundamental change (as defined in the indenture) occurs, at a repurchase price equal to 100% of the principal amount of the notes, plus accrued and unpaid interest (and additional amounts, if any) to, but not including, the repurchase date. The notes are convertible into our common stock, initially at the conversion price of $9.10 per share, equal to a conversion rate of approximately 109.8901 shares per $1,000 principal amount of notes, subject to adjustments for stock dividends, stock splits and other similar events.
Net Cash Used in Operating Activities
Net cash used in operating activities was $89.7 million in 2007 compared to $91.7 million in 2006 and $105.9 million in 2005. The decrease in cash used in 2007 compared to 2006 is primarily due to the one time receipt of $12 million from the sale of our intellectual property in December 2007, partially offset by the increased costs associated with Phase 3 clinical activities in Europe. The decrease in 2006 compared to 2005 was primarily due to lower costs resulting from the strategic restructuring of our business operations and exit payments made in 2005. We expect cash requirements for operating activities to remain approximately at current levels in the near term. The timing of cash requirements may vary from period to period depending on our research and development activities, including our planned preclinical and clinical trials, obligations related to our existing manufacturing and headquarter facilities, and future requirements to establish commercial capabilities for any products that we may develop.
46
Net Cash Provided by Investing Activities
Net cash provided by investing activities was $4.6 million in 2007 compared to net cash provided by investing activities of $8.4 million in 2006 and $100.9 million in 2005. Net cash provided by investing activities in 2007 decreased by $3.8 million compared to 2006 primarily due to purchases of short-term investments exceeding maturities of short-term investments in 2007 as compared to 2006. Capital expenditures of $4.5 million in 2007 were partially offset by $2.2 million of proceeds from the sale of property and equipment. Cash provided by net short-term investment activities was $96.1 million lower in 2006 compared to 2005 due to purchases of short-term investments exceeding sales and maturities in 2006. Cash inflows related to the sale of Abgenix common stock were $65.4 million for 3.0 million shares in 2006 and $58.5 million for 3.7 million shares in 2005. Cash inflows for 2005 also include $3.2 million in connection with the sale of our San Diego manufacturing facility for viral products to Genzyme Corporation. Capital expenditures were $2.0 million in 2006 compared to $2.2 million in 2005.
As of December 31, 2007, we did not have any direct exposure to financial assets backed by subprime mortgage loans. We do not invest in asset backed securities backed by subprime mortgages, secured liquidity notes, structured investments backed by subprime mortgage loans or collateralized debt obligations.
Net Cash Provided by Financing Activities
Net cash provided by financing activities was $85.0 million in 2007 compared to $52.9 million in 2006 and $0.8 million in 2005. Net cash provided by financing activities increased by $32.1 million in 2007 primarily related to net proceeds of $55.4 million in a registered direct offering from the sale of 10.8 million shares of our common stock at $5.55 per share and warrants to purchase 2.2 million shares of our common stock at a price of $7.18 per share, and $30.1 million from the sale of 9.5 million shares of our common stock to Kingsbridge under our 2006 and 2007 CEFF. Cash flows in 2006 included net proceeds of $27.9 million from the sale of 6.3 million shares of our common stock to Kingsbridge under our 2006 CEFF and net proceeds of $25.0 million from the sale of 5.8 million shares of our common stock under an underwritten public offering. Net cash provided in financing activities was $0.8 million for 2005, primarily related to our employee stock plans, partially offset by principal payments under our capital lease obligation.
Operating Leases
We lease certain of our facilities and equipment under non-cancelable operating leases. These leases, including the Hayward and Memphis facility leases, expire at various dates through 2017, and some contain options for renewal. Our South San Francisco headquarters facility lease is recorded as a capital lease as a result of certain amendments that required us to fund the costs of certain structural components of the facility.
In May 2007, we sold a portion of our leasehold improvements and equipment located in our Memphis facility for $2.2 million in cash. These leasehold improvements and equipment related to manufacturing activities that are no longer being carried out at this facility. Additionally, we amended our existing lease for the Memphis facility with the landlord and the buyer entered into a separate lease with the landlord for a majority portion of the facility. We continue to lease the remaining portion of the facility for our product distribution center. The net book value of the assets sold was $0.8 million, resulting in a gain on sale of $1.4 million. In November 2005, in connection with the sale of our San Diego, California, manufacturing facility for viral products to Genzyme Corporation and the termination of the related facility leases, we retired $13.6 million of leasehold improvements and manufacturing assets and $10.2 million of related accumulated depreciation. Also, in November 2005, we terminated the lease for our former corporate headquarters facility in Foster City, California, and retired $5.0 million of associated leasehold improvements, equipment and related accumulated depreciation.
47
Our long-term contractual obligations at December 31, 2007 were as follows:
| | | | | | | | | | | | | | | |
| | | | | Payment Due |
| | | Total | | 2008 | | 2009 and 2010 | | 2011 and 2012 | | 2013 and
thereafter |
| | | (in thousands) |
Convertible senior notes and related interest | | $ | 163,124 | | $ | 4,531 | | $ | 9,062 | | $ | 149,531 | | $ | — |
South San Francisco capital lease obligation | | | 80,219 | | | 6,204 | | | 14,250 | | | 15,226 | | | 44,539 |
Operating leases | | | 25,562 | | | 1,668 | | | 3,868 | | | 5,551 | | | 14,475 |
| | | | | | | | | | | | | | | |
| | $ | 268,905 | | $ | 12,403 | | $ | 27,180 | | $ | 170,308 | | $ | 59,014 |
| | | | | | | | | | | | | | | |
Under certain circumstances, we may redeem some or all of the convertible senior notes on or after November 1, 2009 at a redemption price equal to 100% of the principal amount of the notes. Holders of the notes may require us to repurchase some or all of their notes if a fundamental change (as defined in the indenture) occurs, at a repurchase price equal to 100% of the principal amount of the notes, plus accrued and unpaid interest (and additional amounts, if any) to, but not including, the repurchase date. The notes are convertible into our common stock, initially at the conversion price of $9.10 per share, equal to a conversion rate of 109.8901 shares per $1,000 principal amount of notes, subject to adjustments for stock dividends, stock splits and other similar events.
We estimate that our cash to be used in operating activities during 2008 will be approximately $100 million to $105 million. This estimated use of cash excludes capital expenditures and the cost of any potential acquisitions, and does not reflect the potential offset by equity or debt financings or major new collaborative ventures or potential proceeds from the 2007 CEFF. Our capital requirements depend on numerous factors, including: the progress and scope of our internally funded research, development, clinical, manufacturing and commercialization activities; our ability to establish new collaborations and the terms of those collaborations; competing technological and market developments; the time and cost of regulatory approvals; and various other factors that we discuss underItem 1A. “Risk Factors”above. Our ongoing development programs and any increase in the number and size of programs and trials and proportion of patients enrolled outside North America will reduce our current cash resources and potentially create further need to raise additional capital. Therefore, we will continue to consider financing alternatives including collaborative ventures and potential equity and debt financings.
While we believe that our current liquidity position will be sufficient to meet our cash needs for at least the next year, we will need to raise substantial additional funds in order to complete our pending and planned trials over their multi-year course before we will obtain product revenue, if any, from such products. Accordingly, we will need to raise additional capital to preserve our liquidity, and our ability to generate funds depends on many factors beyond our control including conditions in the capital markets. The sources of liquidity available to us include payments from potential partners and/or licensees of our potential products and technologies, and private or public placement of our equity securities, warrants, debt securities or depositary shares. We regularly consider the conditions of capital markets, dilution, stockholder value and tax consequences of each type of financing on stockholders. Certain of the financing options available to us may have negative consequences to stockholders such as dilution. Given the volatile nature of the capital markets, decisions to raise capital may require actions that would impose a negative consequence in order to reduce or minimize a more significant negative consequence to stockholders.
Off-Balance Sheet Arrangements
As of December 31, 2007, we did not have any off-balance sheet arrangements, as defined in Item 303(a)(4)(ii) of Regulation S-K promulgated by the SEC.
48
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Interest Rate Risk
We are exposed to interest rate sensitivity on our investments in debt securities and our outstanding fixed rate debt. The objective of our investment activities is to preserve principal, while at the same time maximizing yields without significantly increasing risk. To achieve this objective, we invest in highly liquid, investment grade and government debt securities. Our investments in debt securities are subject to interest rate risk. To minimize the exposure due to an adverse shift in interest rates, we invest in short-term securities and our goal is to maintain an average maturity of less than one year. We have the means and intent to hold these securities to maturity, mitigating any interest rate risk which we have estimated, as of December 31, 2007, to be less than $0.3 million from an immediate one percent increase in interest rates. The following table provides information about our financial instruments that are sensitive to changes in interest rates.
Interest Rate Sensitivity
Principal Amount by Expected Maturity and Average Interest Rate
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
As of December 31, 2007 | | 2008 | | | 2009 | | | 2010 | | | 2011 | | | 2012
Thereafter | | | Total | | | Fair Value
December 31,
2007 |
| | | (Dollars in thousands) |
Total Investment Securities Excluding Asset Backed | | $ | 114,098 | | | $ | — | | | $ | — | | | $ | — | | | $ | — | | | $ | 114,098 | | | $ | 114,394 |
Average Interest Rate | | | 5.04 | % | | | — | | | | — | | | | — | | | | — | | | | 5.04 | % | | | — |
Asset Backed Securities(i) | | | | | | | | | | | | | | | | | | | | | | $ | 26,426 | | | $ | 26,452 |
Average Interest Rate | | | | | | | | | | | | | | | | | | | | | | | 3.85 | % | | | — |
Fixed Interest Rate Convertible Senior Notes | | $ | 4,531 | | | $ | 4,531 | | | $ | 4,531 | | | $ | 149,531 | | | $ | — | | | $ | 163,124 | | | $ | 105,828 |
Average Interest Rate | | | 3.125 | % | | | 3.125 | % | | | 3.125 | % | | | 3.125 | % | | | — | | | | 3.125 | % | | | — |
| | | | | | | |
As of December 31, 2006 | | 2007 | | | 2008 | | | 2009 | | | 2010 | | | 2011
Thereafter | | | Total | | | Fair Value
December 31,
2006 |
| | | (Dollars in thousands) |
Total Investment Securities Excluding Asset Backed | | $ | 123,968 | | | $ | 603 | | | $ | — | | | $ | — | | | $ | — | | | $ | 124,571 | | | $ | 124,608 |
Average Interest Rate | | | 5.35 | % | | | 4.63 | % | | | — | | | | — | | | | — | | | | 5.35 | % | | | — |
Asset Backed Securities(ii) | | | | | | | | | | | | | | | | | | | | | | $ | 23,836 | | | $ | 23,837 |
Average Interest Rate | | | | | | | | | | | | | | | | | | | | | | | 3.25 | % | | | — |
Fixed Interest Rate Convertible Senior Notes | | $ | 4,531 | | | $ | 4,531 | | | $ | 4,531 | | | $ | 4,531 | | | $ | 149,531 | | | $ | 167,655 | | | $ | 110,449 |
Average Interest Rate | | | 3.125 | % | | | 3.125 | % | | | 3.125 | % | | | 3.125 | % | | | 3.125 | % | | | 3.125 | % | | | — |
| (i) | Asset backed securities have various contractual maturity dates ranging from 2008 to 2011. The expected maturity dates for these securities are throughout 2008 and differ from the contractual maturity dates because the issuers of these securities have, in some circumstances, the right to prepay the obligations. |
| (ii) | Asset backed securities have various contractual maturity dates ranging from 2007 to 2010. The expected maturity dates for these securities range from 2007 to 2008 and differ from the contractual maturity dates because the issuers of these securities have, in some circumstances, the right to prepay the obligations. |
49
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Cell Genesys, Inc.
We have audited Cell Genesys, Inc.’s internal control over financial reporting as of December 31, 2007, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Cell Genesys, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Cell Genesys, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2007, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Cell Genesys, Inc. as of December 31, 2007 and 2006, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2007, and our report dated February 27, 2008 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Palo Alto, California
February 27, 2008
50
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Cell Genesys, Inc.
We have audited the accompanying consolidated balance sheets of Cell Genesys, Inc. as of December 31, 2007 and 2006, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2007. These financial statements are the responsibility of Cell Genesys, Inc.’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Cell Genesys at December 31, 2007 and 2006, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2007, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 1 to the consolidated financial statements, Cell Genesys, Inc. changed its method of accounting for stock-based compensation as of January 1, 2006, in accordance with guidance provided in Statement of Accounting Standards No. 123(R), “Share-Based Payment”, and its method of accounting for uncertain tax positions as of January 1, 2007, in accordance with guidance provided in Financial Accounting Standards Board Interpretation No. 48, “Accounting for Uncertainty in Income Taxes”.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the effectiveness of Cell Genesys’s internal control over financial reporting as of December 31, 2007, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 27, 2008 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Palo Alto, California
February 27, 2008
51
CELL GENESYS, INC.
CONSOLIDATED BALANCE SHEETS
| | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| | | (In thousands, except par
value and share data) | |
| ASSETS | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 23,588 | | | $ | 23,692 | |
Short-term investments | | | 120,828 | | | | 127,492 | |
Prepaid expenses and other current assets | | | 3,932 | | | | 3,481 | |
| | | | | | | | |
Total current assets | | | 148,348 | | | | 154,665 | |
Restricted cash and investments | | | 2,890 | | | | 2,890 | |
Property and equipment, net | | | 119,011 | | | | 129,643 | |
Unamortized debt issuance costs and other assets | | | 3,143 | | | | 3,969 | |
| | | | | | | | |
Total assets | | $ | 273,392 | | | $ | 291,167 | |
| | | | | | | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 4,268 | | | $ | 2,360 | |
Accrued compensation and benefits | | | 7,730 | | | | 5,820 | |
Deferred revenue | | | 12,000 | | | | — | |
Other accrued liabilities | | | 8,852 | | | | 6,378 | |
Current portion of accrued income taxes | | | 4 | | | | 35,410 | |
Current portion of capital lease obligation | | | 1,711 | | | | 1,346 | |
| | | | | | | | |
Total current liabilities | | | 34,565 | | | | 51,314 | |
Other liabilities | | | 3,451 | | | | 2,851 | |
Non-current portion of accrued income taxes | | | 6,192 | | | | — | |
Non-current portion of capital lease obligation | | | 46,635 | | | | 48,475 | |
Convertible senior notes | | | 145,000 | | | | 145,000 | |
| | |
Commitments and contingencies | | | | | | | | |
Stockholders’ equity: | | | | | | | | |
Preferred stock, $.001 par value: 5,000,000 shares authorized; none issued and outstanding in 2007 and 2006, respectively | | | — | | | | — | |
Common stock, $.001 par value: 150,000,000 shares authorized; 78,473,876 and 57,853,953 shares issued and outstanding in 2007 and 2006, respectively; 396,107 subscribed in 2006 | | | 78 | | | | 58 | |
Additional paid-in capital | | | 528,674 | | | | 436,888 | |
Stock subscription receivable | | | — | | | | (1,200 | ) |
Accumulated other comprehensive loss | | | (88 | ) | | | (378 | ) |
Accumulated deficit | | | (491,115 | ) | | | (391,841 | ) |
| | | | | | | | |
Total stockholders’ equity | | | 37,549 | | | | 43,527 | |
| | | | | | | | |
Total liabilities and stockholders’ equity | | $ | 273,392 | | | $ | 291,167 | |
| | | | | | | | |
See accompanying notes
52
CELL GENESYS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| | | (In thousands, except per share data) | |
Revenue | | $ | 1,380 | | | $ | 1,364 | | | $ | 4,584 | |
Operating expenses: | | | | | | | | | | | | |
Research and development | | | 106,131 | | | | 96,346 | | | | 92,405 | |
General and administrative | | | 20,401 | | | | 18,123 | | | | 16,342 | |
Restructuring charges | | | — | | | | (82 | ) | | | 2,350 | |
| | | | | | | | | | | | |
Total operating expenses | | | 126,532 | | | | 114,387 | | | | 111,097 | |
| | | | | | | | | | | | |
Loss from operations | | | (125,152 | ) | | | (113,023 | ) | | | (106,513 | ) |
Other income (expense): | | | | | | | | | | | | |
Gain on sale of Abgenix, Inc. common stock | | | — | | | | 62,677 | | | | 55,123 | |
Gain (loss) on sale of property and equipment | | | 1,306 | | | | (2 | ) | | | 27 | |
Interest and other income | | | 9,021 | | | | 7,497 | | | | 3,031 | |
Interest expense | | | (10,331 | ) | | | (10,465 | ) | | | (10,679 | ) |
| | | | | | | | | | | | |
Loss before income taxes | | | (125,156 | ) | | | (53,316 | ) | | | (59,011 | ) |
Income tax benefit (provision) | | | 25,882 | | | | (29,613 | ) | | | (5,928 | ) |
| | | | | | | | | | | | |
Net loss | | | (99,274 | ) | | | (82,929 | ) | | | (64,939 | ) |
Dividend in kind to preferred stockholders | | | — | | | | — | | | | (4 | ) |
| | | | | | | | | | | | |
Net loss attributed to common stockholders | | $ | (99,274 | ) | | $ | (82,929 | ) | | $ | (64,943 | ) |
| | | | | | | | | | | | |
Basic and diluted net loss per share | | $ | (1.39 | ) | | $ | (1.67 | ) | | $ | (1.43 | ) |
| | | | | | | | | | | | |
Weighted average shares of common stock outstanding-basic and diluted | | | 71,255 | | | | 49,728 | | | | 45,434 | |
| | | | | | | | | | | | |
See accompanying notes
53
CELL GENESYS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Common Stock | | Additional
Paid-In
Capital | | | Accumulated
Other
Comprehensive
Income (Loss) | | | Accumulated
Deficit | | | Common
Stock
Subscription
Receivable | | | Total
Stockholders’
Equity | |
| | | Shares | | Amounts | | | | | |
| | | (In thousands, except price per share amounts) | |
Balances at December 31, 2004 | | 44,978 | | $ | 45 | | $ | 372,014 | | | $ | 31,220 | | | $ | (243,973 | ) | | $ | — | | | $ | 159,306 | |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | — | | | — | | | — | | | | — | | | | (64,939 | ) | | | — | | | | (64,939 | ) |
Change in net unrealized loss on available-for-sale securities, net of taxes | | — | | | — | | | — | | | | 2,443 | | | | — | | | | — | | | | 2,443 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | | (62,496 | ) |
Issuance of common stock upon exercise of stock options and pursuant to the Employee Stock Purchase Plan | | 305 | | | 1 | | | 1,595 | | | | — | | | | — | | | | — | | | | 1,596 | |
Non-employee stock-based compensation | | — | | | — | | | 195 | | | | — | | | | — | | | | — | | | | 195 | |
Conversion of 152 preferred shares into common shares | | 276 | | | — | | | 1,900 | | | | — | | | | — | | | | — | | | | 1,900 | |
Dividend to preferred stockholders | | — | | | — | | | (4 | ) | | | — | | | | — | | | | — | | | | (4 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balances at December 31, 2005 | | 45,559 | | | 46 | | | 375,700 | | | | 33,663 | | | | (308,912 | ) | | | — | | | | 100,497 | |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | — | | | — | | | — | | | | — | | | | (82,929 | ) | | | — | | | | (82,929 | ) |
Change in net unrealized gain on available-for-sale securities, net of taxes | | — | | | — | | | — | | | | (34,041 | ) | | | — | | | | — | | | | (34,041 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | | (116,970 | ) |
Issuance of common stock upon exercise of stock options and pursuant to the Employee Stock Purchase Plan | | 256 | | | — | | | 1,196 | | | | — | | | | — | | | | — | | | | 1,196 | |
Issuance of common stock upon drawdown of committed equity financing facility at $3.07-$6.00 per share, net of issuance costs of $0.1 million | | 6,289 | | | 6 | | | 27,845 | | | | — | | | | — | | | | — | | | | 27,851 | |
Issuance of warrant in connection with committed equity financing facility | | — | | | — | | | 1,324 | | | | — | | | | — | | | | — | | | | 1,324 | |
Financing costs related to warrant issued in connection with committed equity financing facility | | — | | | — | | | (1,324 | ) | | | — | | | | — | | | | — | | | | (1,324 | ) |
Issuance of common stock related to public offering, net of issuance costs of $0.3 million | | 5,750 | | | 6 | | | 25,017 | | | | — | | | | — | | | | — | | | | 25,023 | |
Stock-based compensation | | — | | | — | | | 5,930 | | | | — | | | | — | | | | — | | | | 5,930 | |
Common stock subscription receivable | | — | | | — | | | 1,200 | | | | — | | | | — | | | | (1,200 | ) | | | — | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balances at December 31, 2006 | | 57,854 | | | 58 | | | 436,888 | | | | (378 | ) | | | (391,841 | ) | | | (1,200 | ) | | | 43,527 | |
See accompanying notes
54
CELL GENESYS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY—(Continued)
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Common Stock | | Additional
Paid-In
Capital | | | Accumulated
Other
Comprehensive
Income (Loss) | | | Accumulated
Deficit | | | Common
Stock
Subscription
Receivable | | | Total
Stockholders’
Equity | |
| | | Shares | | Amounts | | | | | |
| | | (In thousands, except price per share amounts) | |
Balances at December 31, 2006 | | 57,854 | | $ | 58 | | $ | 436,888 | | | $ | (378 | ) | | $ | (391,841 | ) | | $ | (1,200 | ) | | $ | 43,527 | |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | — | | | — | | | — | | | | — | | | | (99,274 | ) | | | — | | | | (99,274 | ) |
Change in net unrealized loss on available-for-sale securities, net of taxes | | — | | | — | | | — | | | | 284 | | | | — | | | | — | | | | 284 | |
Foreign currency translation adjustment | | — | | | — | | | — | | | | 6 | | | | — | | | | — | | | | 6 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | | (98,984 | ) |
Issuance of common stock upon exercise of stock options and pursuant to the Employee Stock Purchase Plan | | 322 | | | — | | | 913 | | | | — | | | | — | | | | — | | | | 913 | |
Issuance of common stock upon drawdown of committed equity financing facility at $2.83-$4.08 per share, net of issuance costs of $0.1 million | | 9,487 | | | 9 | | | 30,133 | | | | — | | | | — | | | | — | | | | 30,142 | |
Issuance of warrant in connection with committed equity financing facility | | — | | | — | | | 623 | | | | — | | | | — | | | | — | | | | 623 | |
Financing costs related to warrant issued in connection with committed equity financing facility | | — | | | — | | | (623 | ) | | | — | | | | — | | | | — | | | | (623 | ) |
Issuance of common stock related to registered direct offering, net of issuance costs of $4.6 million | | 10,811 | | | 11 | | | 50,130 | | | | — | | | | — | | | | — | | | | 50,141 | |
Issuance of warrant in connection with registered direct offering | | — | | | — | | | 5,282 | | | | — | | | | — | | | | — | | | | 5,282 | |
Stock-based compensation | | — | | | — | | | 6,528 | | | | — | | | | — | | | | — | | | | 6,528 | |
Common stock subscription receivable | | — | | | — | | | (1,200 | ) | | | — | | | | — | | | | 1,200 | | | | — | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balances at December 31, 2007 | | 78,474 | | $ | 78 | | $ | 528,674 | | | $ | (88 | ) | | $ | (491,115 | ) | | $ | — | | | $ | 37,549 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
See accompanying notes
55
CELL GENESYS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| | | (In thousands) | |
Cash flows from operating activities: | | | | | | | | | | | | |
Net loss | | $ | (99,274 | ) | | $ | (82,929 | ) | | $ | (64,939 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
Depreciation and amortization | | | 14,356 | | | | 14,521 | | | | 16,204 | |
(Gain) loss on disposal of property and equipment | | | (1,306 | ) | | | 2 | | | | (27 | ) |
Gain on sale of Abgenix, Inc. common stock | | | — | | | | (62,677 | ) | | | (55,123 | ) |
Stock-based compensation expense | | | 6,528 | | | | 5,930 | | | | 137 | |
Deferred income tax provision | | | — | | | | 26,815 | | | | 3,270 | |
Impairment of long-lived assets | | | — | | | | 193 | | | | 280 | |
Changes in operating assets and liabilities: | | | | | | | | | | | | |
Prepaid expenses and other assets | | | 290 | | | | (178 | ) | | | 185 | |
Accounts payable | | | 1,908 | | | | 458 | | | | (986 | ) |
Accrued compensation and benefits | | | 1,910 | | | | 1,421 | | | | (672 | ) |
Deferred revenue | | | 12,000 | | | | — | | | | (2,031 | ) |
Accrued facility exit costs | | | — | | | | — | | | | (6,092 | ) |
Other accrued liabilities | | | 3,076 | | | | 1,967 | | | | 1,197 | |
Accrued income taxes | | | (29,214 | ) | | | 2,798 | | | | 2,658 | |
| | | | | | | | | | | | |
Net cash used in operating activities | | | (89,726 | ) | | | (91,679 | ) | | | (105,939 | ) |
| | | | | | | | | | | | |
Cash flows from investing activities: | | | | | | | | | | | | |
Purchases of short-term investments | | | (262,904 | ) | | | (183,511 | ) | | | (109,730 | ) |
Maturities of short-term investments | | | 269,852 | | | | 103,825 | | | | 51,066 | |
Sales of short-term investments | | | — | | | | 24,521 | | | | 99,629 | |
Conversion of restricted cash and investments | | | — | | | | 4 | | | | 406 | |
Capital expenditures | | | (4,542 | ) | | | (2,046 | ) | | | (2,189 | ) |
Proceeds from sale of property and equipment | | | 2,207 | | | | 138 | | | | 3,255 | |
Proceeds from sale of Abgenix, Inc. common stock | | | — | | | | 65,425 | | | | 58,506 | |
| | | | | | | | | | | | |
Net cash provided by investing activities | | | 4,613 | | | | 8,356 | | | | 100,943 | |
| | | | | | | | | | | | |
Cash flows from financing activities: | | | | | | | | | | | | |
Net proceeds from registered direct offering | | | 55,423 | | | | — | | | | — | |
Net proceeds from committed equity financing facility | | | 30,142 | | | | 27,851 | | | | — | |
Net proceeds from issuance of public offering | | | — | | | | 25,023 | | | | — | |
Proceeds from exercise of stock options | | | 913 | | | | 1,196 | | | | 1,596 | |
Payments under capital lease obligation | | | (1,475 | ) | | | (1,193 | ) | | | (786 | ) |
| | | | | | | | | | | | |
Net cash provided by financing activities | | | 85,003 | | | | 52,877 | | | | 810 | |
| | | | | | | | | | | | |
Net decrease in cash and cash equivalents | | | (110 | ) | | | (30,446 | ) | | | (4,186 | ) |
Effect of exchange rates on cash and cash equivalents | | | 6 | | | | — | | | | — | |
Cash and cash equivalents, beginning of the year | | | 23,692 | | | | 54,138 | | | | 58,324 | |
| | | | | | | | | | | | |
Cash and cash equivalents, end of the year | | $ | 23,588 | | | $ | 23,692 | | | $ | 54,138 | |
| | | | | | | | | | | | |
Supplemental cash flow information: | | | | | | | | | | | | |
Interest paid | | $ | 10,582 | | | $ | 10,158 | | | $ | 9,997 | |
| | | | | | | | | | | | |
Income tax paid | | $ | 2,338 | | | $ | — | | | $ | — | |
| | | | | | | | | | | | |
See accompanying notes
56
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
In this Annual Report, “Cell Genesys,” “we,” “us,” “our” and “the Registrant” refer to Cell Genesys, Inc.
1. Organization and Summary of Significant Accounting Policies
Business activity
We have focused our research and product development efforts on biological therapies for patients with cancer. Our objective is to develop and commercialize cell-based immunotherapies and oncolytic virus therapies to treat different types of cancer. Our current clinical-stage programs include GVAX cancer immunotherapies and oncolytic virus therapies.
Principles of consolidation
Our consolidated financial statements include the accounts of Cell Genesys and all wholly-owned subsidiaries. Intercompany balances and transactions have been eliminated.
Use of estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make judgments, assumptions and estimates that affect the amounts reported in our consolidated financial statements and accompanying notes. Actual results could differ from those estimates. Management makes estimates when preparing the financial statements including those related to revenue recognition, accrued but unbilled expenses for clinical trials, income taxes, long-term service contracts, stock-based compensation and contingencies.
Concentrations of risk
We are subject to concentration of risk from our investments. Risk for investments is managed by the purchase of investment grade securities and the diversification of the investment portfolio among issuers and maturities.
Revenue recognition
Our revenues are derived principally from research and licensing agreements with collaborators. Revenue under such collaboration agreements typically includes up-front payments, cost reimbursements, milestone payments and license fees. We evaluate whether the delivered element under these arrangements has value to our customer on a stand-alone basis and whether objective and reliable evidence of fair value of the undelivered item exists. Deliverables that do not meet these criteria are treated as one unit of accounting for the purposes of revenue recognition.
Up-front payments: Up-front payments from our research collaborations include payments for licenses, technology transfer and access rights. Non-refundable up-front license fees and other payments under collaboration agreements where we cannot establish standalone value for the delivered license and where we have continuing involvement following the execution of the collaboration agreement, are deferred and recognized on a straight-line or ratable method over the period of our continuing involvement unless we determine that another methodology is more appropriate. During 2005, we recognized revenue from a non-refundable up-front payment under our global alliance with Novartis AG for the development of certain oncolytic virus therapies based upon when the underlying development expenses were incurred, rather than a ratable method, as we determined that the expense method was more appropriate for this agreement. The
57
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
revenues recorded under the Novartis AG alliance approximated the related development expenses that were incurred in the respective periods. We recognize cost reimbursement revenue under collaborative agreements as the related research and development costs are incurred, as provided for under the terms of these agreements.
Milestones: Payments for milestones that are based on the achievement of substantive and at-risk performance criteria are recognized in full upon achievement of the incentive milestone events in accordance with the terms of the agreement. Incentive milestone payments are triggered either by the results of our research efforts or by events external to us, such as regulatory approval to market a product or the achievement of specified sales levels by a marketing partner. As such, the incentive milestones are substantially at risk at the inception of the agreement, and the amounts of the payments assigned thereto are commensurate with the milestone achieved. Upon the achievement of an incentive milestone event, we have no future performance obligations related to that milestone payment.
License fees: Non-refundable license fees where we have completed all obligations at the execution of the arrangement are recognized as revenue upon execution of the technology licensing agreement when delivery has occurred, collectibility is reasonably assured and the price is fixed and determinable.
Property and equipment
Property and equipment are stated at cost and depreciated using the straight-line method over the estimated useful lives of the assets, generally five to 15 years. Repair and maintenance costs are expensed as incurred. Computer equipment is depreciated over a life of three years. Property and equipment leased under capital leases are amortized over the shorter of the useful lives or the lease term. Amortization of capitalized leased equipment is included in depreciation expense. Leasehold improvements are stated at cost and amortized over the shorter of the useful lives or the lease term. Costs for assets under construction are classified as “construction in process” and such costs are reclassified to an appropriate fixed asset classification and depreciated when the asset is placed into service.
Long-lived assets
Our policy regarding long-lived assets is to evaluate the recoverability of our assets when the facts and circumstances suggest that the assets may be impaired. This assessment of fair value is performed based on the estimated undiscounted cash flows compared to the carrying value of the assets. If the future cash flows (undiscounted and without interest charges) are less than the carrying value, a write-down would be recorded to reduce the related asset to its estimated fair value.
Unamortized debt issuance costs
Unamortized debt issuance costs relate to our convertible senior notes and are amortized over the life of the related debt. Amortization expense totaled $0.7 million in each of the years ended December 31, 2007, 2006 and 2005, respectively, and is reported as interest expense.
Cash, cash equivalents and short-term investments
We invest our excess cash and short-term investments, including restricted cash and investments, with high credit quality United States and foreign financial institutions, and government and corporate issuers. We limit the amount of credit exposure to any one issuer. We consider all highly liquid investments with insignificant interest rate risk with original maturities of less than three months when purchased to be cash equivalents. All
58
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
investments are denominated in U.S. dollars. We record our investments at fair market value, based on quoted market prices.
Our debt securities are classified as available-for-sale and carried at fair value. Management considers our investments in debt securities to be available for use in current operations. As a result, all investments in debt securities are classified as current assets, even if the remaining maturity of the investment is more than one year beyond the balance sheet date. The cost of securities sold is based on the specific identification method. Realized gains and losses and declines in value on securities classified as available-for-sale that are judged to be other than temporary are included in interest and other income (loss). Unrealized gains and losses on securities classified as available-for-sale are recorded in accumulated other comprehensive income, net of tax. We determine the appropriate classification of debt securities at the time of purchase and re-evaluate such designation as of each balance sheet date.
We regularly review all of our investments for potential “other-than-temporary” declines in fair value. We review the cause of the impairment, the creditworthiness of the security issuers, the number of securities in an unrealized loss position, as well as the severity and duration of the unrealized losses. When we determine that the decline in fair value of an investment below its net carrying value is other-than-temporary, we reduce the carrying value of the securities by recognizing a loss in the amount of such decline. No such reductions have been required during the past three years.
Restricted cash and investments as of December 31, 2007 relate to outstanding letters of credit which secure our leased corporate headquarters facility in South San Francisco, California, and our leased cGMP manufacturing facility in Hayward, California.
Fair value of financial instruments
The carrying amounts of financial instruments such as cash equivalents, prepaid expenses and other current assets, accounts payable, accrued expenses and other current liabilities approximate fair value because of the short maturities of these instruments. The estimated fair value of our convertible senior notes is determined by using available market information and valuation methodologies that correlate fair value with the market price of our common stock which fair value is provided by a third party financial institution. The fair value of our convertible senior notes as of December 31, 2007 and 2006 was approximately $105.8 million and $110.4 million, respectively.
Foreign Currency Translation
Our subsidiary located in the United Kingdom operates using the local currency as the functional currency. Accordingly, all assets and liabilities of this subsidiary are translated using exchange rates in effect at the end of the period, and revenues and expenses are translated using average exchange rates for the period. The resulting translation adjustments are presented as a separate component of accumulated other comprehensive loss.
Research and development costs
Costs incurred in research and development activities are expensed as incurred. Research and development expenses include, but are not limited to, payroll and personnel expenses, lab expenses, clinical trial and related clinical manufacturing costs, facilities and overhead costs.
59
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Net loss per share
Basic net loss per share is calculated using the weighted average number of shares of common stock outstanding during the period. Diluted net loss per share includes the impact of potentially dilutive securities. As our potentially dilutive securities were anti-dilutive for all years presented, such securities have been excluded from the computation of shares used in computing diluted net loss per share. These outstanding securities consisted of the following (in thousands):
| | | | |
| | | December 31, |
| | | 2007 | | 2006 |
Convertible senior notes | | 15,934 | | 15,934 |
| | | | |
Outstanding stock options | | 9,429 | | 8,368 |
| | | | |
Warrants to purchase common stock | | 2,959 | | 375 |
| | | | |
Comprehensive loss
Comprehensive loss is comprised of net loss and other comprehensive income (loss). Other comprehensive income (loss) includes certain changes to our stockholders’ equity that are excluded from net loss, such as unrealized gains or losses on our available-for-sale securities, including our holdings of Abgenix, Inc. common stock in 2006, net of related tax and foreign currency translation adjustments. The following table presents the calculation of comprehensive loss (in thousands):
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
Net loss | | $ | (99,274 | ) | | $ | (82,929 | ) | | $ | (64,939 | ) |
Other comprehensive income (loss): | | | | | | | | | | | | |
Increase in unrealized gain on investments, net of taxes of zero, $0.6 million, $21.7 million in 2007, 2006 and 2005, respectively | | | 284 | | | | 1,063 | | | | 32,297 | |
Increase in foreign currency translation gain, net | | | 6 | | | | — | | | | — | |
Less: reclassification adjustment for gains recognized in net loss, net of related tax of, $27.5 million and $25.0 million, in 2006 and 2005, respectively | | | — | | | | (35,104 | ) | | | (29,854 | ) |
| | | | | | | | | | | | |
Comprehensive loss | | $ | (98,984 | ) | | $ | (116,970 | ) | | $ | (62,496 | ) |
| | | | | | | | | | | | |
Income taxes
On January 1, 2007, we adopted FASB Interpretation No. 48, Accounting for Uncertainty in Income Taxes, or FIN 48, an interpretation of SFAS No. 109, Accounting for Income Taxes, or FAS 109, which clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements in accordance with FAS 109 by prescribing a minimum recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. FIN 48 also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition.
As a result of the implementation of FIN 48, we did not recognize any adjustment in the liability for unrecognized tax benefits and, therefore, implementation of FIN 48 did not result in a cumulative adjustment to accumulated deficit. At the adoption date of January 1, 2007, we had $25.0 million of unrecognized tax benefits,
60
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
all of which would affect our effective tax rate if recognized. At December 31, 2007, we had $4.0 million of unrecognized tax benefits, all of which would affect our effective tax rate if recognized. As a result of a final settlement in May 2007 with the Internal Revenue Service, or IRS, unrecognized tax benefits decreased by $21.0 million in the second quarter of 2007. See Note 10, “Income Taxes” for further information. Our policy is to account for interest and penalties related to income tax matters in the income tax provision in the consolidated statement of operations. Accrued interest and penalties related to income tax matters are included within the related tax liability line in the consolidated balance sheet. At the adoption date of January 1, 2007, we had $10.4 million of accrued interest and zero penalties related to tax contingencies recorded in the consolidated balance sheet. As of December 31, 2007, we had accrued $2.2 million of interest and zero penalties related to tax contingencies.
Segment reporting
Our operations are treated as one operating segment, as we report profit and loss information only on an aggregate basis to the chief operating decision-makers.
Stock-based compensation
On January 1, 2006, we adopted Statement of Financial Accounting Standards No. 123 (revised 2004), “Share-Based Payment”, or FAS 123R, which supersedes our previous accounting under APB Opinion No. 25, “Accounting for Stock Issued to Employees”, or APB 25. FAS 123R requires the recognition of compensation expense, using a fair-value based method, for costs related to all share-based payments including stock options and stock issued under our employee stock plans. FAS 123R requires companies to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense on a straight-line basis over the requisite service periods in our Consolidated Statements of Operations. We adopted FAS 123R using the modified prospective transition method, which requires that compensation expense be recognized in the financial statements for all awards granted after the date of adoption as well as for existing awards for which the requisite service has not been rendered as of the date of adoption. The modified prospective transition method does not require restatement of prior periods to reflect the impact of FAS 123R.
We adopted the alternative transition method provided in FASB Staff Position No. 123R-3, “Transition Election Related to Accounting for the Tax Effects of Share-Based Payment Awards.” We have adopted the simplified method to calculate the beginning balance of the additional paid-in-capital pool, or APIC pool, of the excess tax benefit, and to determine the subsequent impact on the APIC pool and Consolidated Statements of Cash Flows of the tax effects of employee stock-based compensation awards that were outstanding upon our adoption of FAS 123R.
Prior to the adoption of FAS 123R, we accounted for stock-based awards to employees and directors using the intrinsic value method in accordance with APB 25 as allowed under Statement of Accounting Standards No. 123, “Accounting for Stock-Based Compensation”, or FAS 123. Under the intrinsic value method, no employee stock-based compensation expense had been recognized in our Consolidated Statements of Operations for any period prior to our adoption of FAS 123R on January 1, 2006, as the exercise price of the stock options granted to employees and directors equaled the fair market value of the underlying stock on the date of grant. See Note 9, “Stockholders’ Equity and Stock-Based Compensation” for further information.
61
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Reclassifications
Certain prior year amounts have been reclassified to conform to the current year presentation. We reclassified gain (loss) on sale of property and equipment from interest and other income and presented it as a separate line item on our consolidated statements of operations. The reclassification had no impact on our total operating expenses or our net loss.
Recent accounting pronouncements
In September 2006, the Financial Accounting Standards Board, or FASB, issued Statement of Financial Accounting Standards No. 157, “Fair Value Measurements,” or FAS 157. FAS 157 defines fair value, establishes a market-based framework or hierarchy for measuring fair value under GAAP, and expands disclosures about fair value measurements. FAS 157 is effective for financial statements issued for fiscal years beginning after November 15, 2007, and interim periods within those fiscal years. We have evaluated the impact of adopting FAS 157 on our consolidated financial statements and do not expect any impact on our results of operations or financial position.
In February 2007, the FASB issued SFAS No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities—Including an Amendment of FASB Statement 115,” or FAS 159. FAS 159 permits entities to choose to measure many financial instruments and certain other items at fair value. Entities that elect the fair value option will report unrealized gains and losses in earnings at each subsequent reporting date. The fair value option may be elected on an instrument-by-instrument basis, with few exceptions. FAS 159 also establishes presentation and disclosure requirements to facilitate comparisons between companies that choose different measurement attributes for similar assets and liabilities. FAS 159 is effective for financial statements issued for fiscal years beginning after November 15, 2007. We have evaluated the impact of adopting FAS 159 on our consolidated financial statements and do not expect any impact on our results of operations or financial position.
In June 2007, the FASB ratified Emerging Issues Task Force, or EITF, Issue No. 07-3, “Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities,” or Issue No. 07-3, which addresses the accounting for nonrefundable advance payments. The EITF concluded that nonrefundable advance payments for goods or services to be received in the future for use in research and development activities should be deferred and capitalized. The capitalized amounts should be expensed as the related goods are delivered or the services are performed. If an entity’s expectations change such that it does not expect it will need the goods to be delivered or the services to be rendered, capitalized nonrefundable advance payments should be charged to expense. Issue 07-3 is effective for new contracts entered into during fiscal years beginning after December 15, 2007, including interim periods within those fiscal years. We have evaluated the impact of adopting Issue 07-3 on our consolidated financial statements and do not expect any impact on our results of operations or financial position.
In December 2007, the FASB ratified the final consensuses in Emerging Issues Task Force Issue No. 07-1, “Accounting for Collaborative Arrangements,” or Issue 07-1, which requires certain income statement presentation of transactions with third parties and of payments between parties to the collaborative arrangement, along with disclosure about the nature and purpose of the arrangement. Issue 07-1 is effective for us beginning January 1, 2009. We have evaluated the impact of adopting Issue 07-1 on our consolidated financial statements and do not expect any impact on our results of operations or financial position.
2. Restructuring Charges
In June 2005, we commenced the implementation of a strategic restructuring of our business operations to focus resources on our most advanced and most promising product development programs. We have deployed
62
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
the majority of our resources to advance GVAX immunotherapy for prostate cancer, which is currently in Phase 3 development, as well as GVAX immunotherapy for pancreatic cancer and GVAX immunotherapy for leukemia, both of which are in Phase 2 development. In our oncolytic virus therapies program, we are conducting a multiple dose Phase 1 clinical trial of CG0070 in recurrent bladder cancer, which could be evaluated in multiple types of cancer in the future. We discontinued certain clinical programs including patient-specific GVAX immunotherapy programs for lung cancer and myeloma. In the oncolytic virus therapy program, we discontinued further development of CG7870 for prostate cancer and early-stage research programs for the development of new oncolytic virus therapies, as well as research efforts in anti-angiogenesis gene therapy for cancer.
In connection with this restructuring, in November 2005, we sold our San Diego, California, manufacturing facility to Genzyme Corporation for $3.2 million. We recorded a charge of $2.4 million in 2005 related to our restructuring decisions, including $1.5 million for workforce reduction initiatives, $0.3 million to reduce the carrying value of the San Diego manufacturing facility and $0.6 million for lease termination and other expenses. During 2006, we decreased the restructuring accrual by $0.1 million, due to the extension of a sublease, and paid all remaining obligations.
3. Collaborative and License Agreements
We have derived substantially all of our revenues from collaborative and license agreements, as shown in the following table (in thousands):
| | | | | | | | | |
| | | Year Ended December 31, |
| | | 2007 | | 2006 | | 2005 |
Novartis AG | | $ | — | | $ | — | | $ | 2,031 |
sanofi-aventis Group | | | 1,000 | | | 1,000 | | | 2,000 |
Ceregene, Inc. | | | 13 | | | 83 | | | 69 |
Other | | | 367 | | | 281 | | | 484 |
| | | | | | | | | |
| | $ | 1,380 | | $ | 1,364 | | $ | 4,584 |
| | | | | | | | | |
Global alliance with Novartis AG
On July 23, 2003, we announced a global alliance with Novartis AG for the development and commercialization of oncolytic virus therapies. Under the agreement, we acquired exclusive worldwide rights to certain oncolytic virus therapy products and related intellectual property of Genetic Therapy, Inc., or GTI, an affiliate of Novartis, as well as certain related intellectual property of Novartis, and received an up-front payment of $28.5 million from Novartis. This payment was dedicated to the further development of certain of our existing oncolytic virus therapy products and those products acquired from GTI, for which products both we and Novartis have future commercialization rights. In exchange, we issued to Novartis and GTI 1,999,840 shares of our common stock. Of the $28.5 million up-front payment received from Novartis, we recorded $18.5 million for the 1,999,840 common shares issued, based upon the fair market value of such shares, and $10.0 million to deferred revenue, which we amortized to revenue over the related development period based on when the underlying development expenses were incurred. The agreement also provides the basis for the sharing of additional development costs and profits in the future related to potential products on a worldwide basis upon the exercise of certain options by Novartis. We recognized $2.0 million in revenue under this agreement in 2005. As of December 31, 2005, we had recognized all revenues associated with the $28.5 million up-front payment received from Novartis.
63
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Gene activation technology licenses
In February 1997, we executed a license agreement with Aventis, now sanofi-aventis Group, for gene-activated erythropoietin, or EPO, and a second undisclosed protein. In late 2000, sanofi-aventis Group informed us of its intention to terminate the portion of this license agreement that relates to the second undisclosed protein. The agreement for gene-activated EPO provides for up to $26.0 million in milestone payments, as well as annual maintenance fees and any royalties on future sales of gene-activated EPO anywhere in the world. As of December 31, 2007, we had received approximately $27.2 million under this license agreement, which included certain milestone payments relating to the development of gene-activated EPO which sanofi-aventis Group is developing in collaboration with Transkaryotic Therapies, Inc., now owned by Shire Pharmaceuticals Group plc. We recognized revenues of $1.0 million, $1.0 million, and $2.0 million in 2007, 2006 and 2005, respectively, pursuant to the agreement.
GBP IP, LLC Technology and Intellectual Property Agreement
In December 2007, we sold for $12.0 million all of our assets, intellectual property and previously established licensing agreements relating to our lentiviral gene delivery technology, commonly referred to as lentiviral vectors, to GBP IP, LLC, an affiliate of GBP Capital, the majority shareholder in privately held Lentigen Corporation. We received full payment of $12.0 million in December 2007. As part of the sales agreement we retained our rights to use the technology for research and development purposes including potential future use with our cancer immunotherapy products. We applied EITF Issue No. 00-21, “Revenue Arrangements with Multiple Deliverables”, in evaluating the appropriate accounting for this agreement. We identified the delivery of biological materials (including certain GMP-compliant materials), intellectual property and previously established licensing agreements related to our lentiviral gene delivery technology as the primary deliverables under this sales agreement and concluded that these deliverables should be accounted for as a single unit of accounting based upon the determination that these deliverables did not have stand-alone value and can not be separated. Therefore, we have deferred recognition of revenue until we have performed all of our obligations under the agreement which we expect to complete during the first quarter of 2008.
Other collaborations
As a result of the aforementioned asset sale to GBP IP, LLC, as of December 2007, we no longer have licensing agreements relating to our proprietary lentiviral vector technologies. These collaborations had enabled us to receive monetary reimbursement for providing viral vector technologies to companies such as the Clontech division of Becton, Dickinson and Company and Invitrogen Corporation that commercialize these technologies for the research market.
64
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
4. Investments
The following is a summary of our available-for-sale securities at December 31, 2007 and 2006 (in thousands):
| | | | | | | | | | | | | |
December 31, 2007 | | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | | Estimated
Fair Value |
Money market funds | | $ | 20,018 | | $ | — | | $ | — | | | $ | 20,018 |
Corporate notes | | | 93,475 | | | 314 | | | (19 | ) | | | 93,770 |
Asset backed securities | | | 26,426 | | | 26 | | | — | | | | 26,452 |
U.S. government agencies | | | 605 | | | 1 | | | — | | | | 606 |
| | | | | | | | | | | | | |
| | $ | 140,524 | | $ | 341 | | $ | (19 | ) | | $ | 140,846 |
| | | | | | | | | | | | | |
Classified as: | | | | | | | | | | | | | |
Cash equivalents | | | | | | | | | | | | $ | 20,018 |
Short-term investments | | | | | | | | | | | | | 120,828 |
| | | | | | | | | | | | | |
| | | | | | | | | | | | $ | 140,846 |
| | | | | | | | | | | | | |
| | | | |
December 31, 2006 | | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | | Estimated
Fair Value |
Money market funds | | $ | 20,953 | | $ | — | | $ | — | | | $ | 20,953 |
Corporate notes | | | 100,416 | | | 44 | | | (3 | ) | | | 100,457 |
Asset backed securities | | | 23,836 | | | 4 | | | (3 | ) | | | 23,837 |
U.S. government agencies | | | 3,202 | | | — | | | (4 | ) | | | 3,198 |
| | | | | | | | | | | | | |
| | $ | 148,407 | | $ | 48 | | $ | (10 | ) | | $ | 148,445 |
| | | | | | | | | | | | | |
Classified as: | | | | | | | | | | | | | |
Cash equivalents | | | | | | | | | | | | $ | 20,953 |
Short-term investments | | | | | | | | | | | | | 127,492 |
| | | | | | | | | | | | | |
| | | | | | | | | | | | $ | 148,445 |
| | | | | | | | | | | | | |
As of December 31, 2007, unrealized losses set forth above were primarily due to increases in interest rates. The gross unrealized losses in our portfolio of investments represent approximately 0.01% of the total fair value of the portfolio. Gross realized gains on the sale of investment securities were $62.7 million and $55.1 million for the years ended December 31, 2006 and 2005, respectively. We sold 3.0 million and 3.7 million shares of Abgenix stock resulting in net proceeds of $65.4 million and $58.5 million in 2006 and 2005, respectively, and we no longer own any Abgenix shares. There were no gross realized gains on the sale of investment securities for the year ended December 31, 2007.
65
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The amortized cost and estimated fair value of our available-for-sale securities by contractual maturity are shown below (in thousands):
| | | | | | |
December 31, 2007 | | Amortized
Cost | | Estimated
Fair Value |
Debt securities maturing: | | | | | | |
In one year or less | | $ | 114,098 | | $ | 114,394 |
Asset backed securities* | | | 26,426 | | | 26,452 |
| | | | | | |
| | $ | 140,524 | | $ | 140,846 |
| | | | | | |
| * | Asset backed securities have various contractual maturity dates ranging from 2008 to 2011. The expected maturity dates for these securities are throughout 2008 and differ from the contractual maturity dates because the issuers of these securities have, in some circumstances, the right to prepay the obligations. |
5. Property and Equipment
Property and equipment consists of the following (in thousands):
| | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
Machinery, furniture and equipment | | $ | 30,803 | | | $ | 29,609 | |
Leasehold improvements | | | 101,553 | | | | 103,461 | |
Property and equipment under capital lease obligation | | | 52,361 | | | | 52,361 | |
Construction in process | | | 3,261 | | | | 1,343 | |
| | | | | | | | |
| | | 187,978 | | | | 186,774 | |
Accumulated depreciation and amortization | | | (68,967 | ) | | | (57,131 | ) |
| | | | | | | | |
| | $ | 119,011 | | | $ | 129,643 | |
| | | | | | | | |
In May 2007, we sold a portion of our leasehold improvements and equipment located in our Memphis facility for $2.2 million in cash. Such property was related to manufacturing activities which were no longer being carried out at this facility. The net book value of the assets sold was $0.8 million, resulting in a gain on sale of $1.4 million.
Accumulated amortization related to capital lease obligations was $17.0 million and $13.5 million as of December 31, 2007 and 2006, respectively.
6. Convertible Senior Notes and Other Debt Financings
In October 2004, we entered into a purchase agreement with initial purchasers relating to the private placement of $110.0 million aggregate principal amount of our 3.125% Convertible Senior Notes due in 2011. We granted the initial purchasers a 30-day option to purchase up to an additional $35.0 million principal amount of the notes, which the purchasers elected to exercise in full in November 2004. We received approximately $139.9 million in net proceeds, after deducting the initial purchasers’ discount and estimated offering expenses. We used a portion of the net proceeds to repay bank debt of $60.0 million related to an asset-backed debt financing obligation acquired from Fleet Bank in December 2001 in connection with the construction of our manufacturing facility in Hayward, California, and to repay $35.0 million in term loans acquired in September 2003 from Silicon Valley Bank. We recorded interest expense including the amortization of debt issuance costs
66
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
related to our convertible senior notes of $5.3 million for the each of the three years ended December 31, 2007, 2006 and 2005. Interest on the notes is payable on May 1 and November 1 each year until the notes are converted or redeemed. The notes are due in 2011.
Under certain circumstances, we may redeem some or all of the notes on or after November 1, 2009 at a redemption price equal to 100% of the principal amount of the notes. Holders of the notes may require us to repurchase some or all of their notes if a fundamental change (as defined in the indenture governing the notes) occurs, at a repurchase price equal to 100% of the principal amount of the notes, plus accrued and unpaid interest (and additional amounts, if any) to the repurchase date. The notes are convertible into our common stock, initially at the conversion price of $9.10 per share, equal to a conversion rate of 109.8901 shares per $1,000 principal amount of notes, subject to adjustments for stock dividends, stock splits, and other similar events.
7. Leases
Operating leases
We lease certain of our facilities and equipment under non-cancelable operating leases which generally require us to make minimum lease payments as well as to reimburse the lessor for real estate taxes, insurance and maintenance expenses. These leases, including the Hayward and Memphis facility leases, expire on various dates through 2017 and some contain options for renewal. Rent expense under operating leases was $4.4 million, $3.8 million, and $5.0 million in 2007, 2006, and 2005, respectively. In November 2005, we terminated two facility leases in connection with the sale of our San Diego manufacturing facility to Genzyme Corporation and the closure of our former headquarters in Foster City. In May 2007, we sold a portion of our leasehold improvements and equipment located in our Memphis facility. We amended our existing lease for the Memphis facility with the landlord, and the buyer entered into a separate lease with the landlord for a majority portion of the facility. We continue to lease the remaining portion of the facility as our product distribution center for our Phase 3 clinical trials for GVAX immunotherapy for prostate cancer.
Capital lease obligation
During 2002, we amended the lease for our headquarters facility in South San Francisco, California to fund the costs of certain structural components of the facility. As a result of this lease amendment, we were required to account for this lease as a capital lease. As of December 31, 2007, we had $48.3 million of capital lease obligations and $35.4 million of related capital lease assets, net of accumulated amortization.
67
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Future minimum payments under non-cancelable operating leases and the capital lease obligation as of December 31, 2007 are as follows (in thousands):
| | | | | | | |
| | | Operating
Leases | | Capital
Lease
Obligation | |
Year ending December 31: | | | | | | | |
2008 | | $ | 1,668 | | $ | 6,204 | |
2009 | | | 1,846 | | | 7,018 | |
2010 | | | 2,022 | | | 7,232 | |
2011 | | | 2,721 | | | 7,482 | |
2012 | | | 2,830 | | | 7,744 | |
2013 and beyond | | | 14,475 | | | 44,539 | |
| | | | | | | |
Total minimum payments | | $ | 25,562 | | | 80,219 | |
| | | | | | | |
Less: Amount representing interest | | | | | | (31,873 | ) |
| | | | | | | |
Present value of future debt payments | | | | | | 48,346 | |
Less: Current portion of future payments | | | | | | (1,711 | ) |
| | | | | | | |
Noncurrent portion of future payments | | | | | $ | 46,635 | |
| | | | | | | |
8. Redeemable Convertible Preferred Stock
In January 2000, we issued shares of Series B redeemable convertible preferred stock pursuant to a call option granted in connection with a previous offering. The number of shares of common stock issuable upon conversion of the shares of Series B preferred stock issued in January 2000 was determined by dividing the market value of the shares to be converted by the lower of a fixed conversion price of $14.53 per share (subject to antidilution provisions), or the average of certain trading prices during the 10 trading days preceding such date of conversion.
In January 2005, all of the 152 then-outstanding shares of our Series B redeemable convertible preferred stock automatically converted into an aggregate of 0.3 million shares of our common stock at a conversion price of $6.895 per share. This conversion occurred in accordance with their terms on the five-year anniversary of their issuance, according to a predetermined formula. Following the conversion, no shares of our Series B preferred stock remained outstanding.
9. Stockholders’ Equity and Stock-Based Compensation
Common stock
In September 2006, we completed an underwritten public offering and sold 5.8 million shares of our common stock, resulting in net proceeds of $25.0 million. These offerings were pursuant to our shelf registration statement filed in February 2003, which allowed us to offer up to $150.0 million of securities in one or more public offerings. In April 2007, we used the remaining registered amount under this shelf registration statement to raise net proceeds of $55.4 million in a registered direct offering of 10.8 million shares of our common stock at $5.55 per share and warrants to purchase 2.2 million shares of our common stock at a price of $7.18 per share from selected institutional investors.
On May 16, 2007, our new shelf registration statement was declared effective by the Securities and Exchange Commission under the Securities Act of 1933, as amended, which allows us to offer up to $150.0
68
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
million of securities on short notice in one or more public offerings under the Securities Act of 1933, as amended. As of December 31, 2007, no securities have been sold under this shelf registration statement.
Committed Equity Financing Facility
In March 2006, we entered into a Committed Equity Financing Facility, or 2006 CEFF, with Kingsbridge Capital Limited, or Kingsbridge, pursuant to which Kingsbridge committed to purchase, subject to certain conditions, up to the lesser of 8.7 million shares of our common stock or an aggregate of $75.0 million during the following three years. Under the 2006 CEFF, we were able to draw down in tranches of up to a maximum of 2.5 percent of the closing market value of our common stock on the last trading day prior to the commencement of the drawdown, or $15.0 million, whichever is less, subject to certain conditions. The purchase price of these shares was discounted between 6 to 10 percent from the volume weighted average price of our common stock for each of the eight trading days following the election to sell shares. Kingsbridge was not obligated to purchase shares at prices below $3.00 per share or at a price below 85% of the closing market value of our common stock on the trading day immediately preceding the commencement of the drawdown.
In connection with the 2006 CEFF, we issued to Kingsbridge a warrant to purchase 0.4 million shares of our common stock at a price of $9.12 per share exercisable beginning on September 14, 2006 and exercisable for a period of five years thereafter. The fair value of the warrant was determined on the date of issuance using the Black-Scholes option valuation model applying the following assumptions: (i) a risk-free rate of 4.68%, (ii) an expected term of 5.5 years, (iii) no dividend yield and (iv) volatility of 57%. The estimated fair value of this warrant was $1.3 million which was recorded as a contra-equity amount in additional paid-in capital in March 2006. In 2006, we received net proceeds of $27.9 million from the sale of 6.3 million shares of our common stock under the 2006 CEFF. In 2007, we received net proceeds of $7.1 million from the sale of 2.4 million shares of our common stock under the 2006 CEFF, which concluded the 2006 CEFF. Since inception of the 2006 CEFF, we received cumulative net proceeds of $35.0 million from the sale of 8.7 million shares of our common stock.
In February 2007, we entered into a new CEFF, or 2007 CEFF, with Kingsbridge, pursuant to which Kingsbridge committed to purchase, subject to certain conditions, up to the lesser of 11.6 million shares of our common stock or an aggregate of $75.0 million during the following three years. Under the 2007 CEFF, we are able to draw down in tranches of up to a maximum of 2.5 percent of our closing market value of our common stock on the last trading day prior to the commencement of the drawdown, or $15.0 million, whichever is less, subject to certain conditions. In addition, subject to the $15.0 million limit, we can issue up to 3.5% of our market capitalization once per fiscal quarter. The purchase price of these shares is discounted between 6 to 10 percent from the volume weighted average price of our common stock for each of the eight trading days following the election to sell shares. Kingsbridge is not obligated to purchase shares at prices below $1.75 per share or at a price below 85% of the closing share price of our stock on the trading day immediately preceding the commencement of the drawdown.
In connection with the 2007 CEFF, we issued to Kingsbridge a warrant to purchase 0.4 million shares of our common stock at a price of $4.68 per share exercisable beginning on September 5, 2007 for a period of five years thereafter. The fair value of the warrant was determined on the date of issuance using the Black-Scholes option valuation model applying the following assumptions: (i) a risk-free rate of 4.80%, (ii) an expected term of 5.5 years, (iii) no dividend yield and (iv) volatility of 55%. The estimated fair value of this warrant was $0.6 million which was recorded as a contra-equity amount in additional paid-in capital in February 2007. During the year ended December 31, 2007, we received net proceeds of $23.0 million from the sale of 7.1 million shares of our common stock under the 2007 CEFF.
69
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Stock option plans
Prior to May 2005, we had five approved stock option plans: the 1989, 1992, and 1998 Incentive Stock Option Plans, the 2001 Nonstatutory Option Plan and the 2001 Non-Employee Directors Stock Option Plan, or collectively, the Prior Plans. Under the Prior Plans, incentive stock options and non-qualified stock options were granted to eligible employees, directors and consultants to purchase shares of our common stock at no less than the fair market value of the underlying common stock as of the date of grant. Options granted under these plans have a maximum term of ten years and generally vest over four years at the rate of 25 percent one year from the date of grant and 1/48 monthly thereafter. The 1998 Incentive Stock Option Plan replaced the 1989 and 1992 Incentive Stock Option Plans which expired and were retired in 1999 and 2002, respectively.
Amended 2005 Equity Incentive Plan: In May 2005, our stockholders approved the 2005 Equity Incentive Plan, or the 2005 Plan, at which time 1.0 million new shares of common stock were authorized for issuance. The 2005 Plan replaced our 1998 Incentive Stock Option Plan, the 2001 Nonstatutory Option Plan and the 2001 Non-Employee Directors Stock Option Plan. Upon approval of the 2005 Plan, shares in the Prior Plans that had been reserved but not issued were reserved for issuance under the 2005 Plan. Since such approval, shares that would otherwise return to the Prior Plans as a result of option cancellations are rolled into and are reserved for issuance under the 2005 Plan. No additional grants are made under the Prior Plans. On June 19, 2007, our stockholders approved an amendment to the 2005 Plan to increase the number of shares reserved for issuance by 3.5 million.
The 2005 Plan permits the granting of incentive and non-statutory stock options, restricted stock, stock appreciation rights, performance units and performance shares and other stock awards to eligible employees, directors and consultants. We generally grant options to purchase shares of common stock under the 2005 Plan at no less than the fair market value of the underlying common stock as of the date of grant. Options granted under the 2005 Plan have a maximum term of ten years and generally vest over four years at the rate of 25 percent of total shares underlying the option upon the one year anniversary of the date of grant and 1/48 of the total shares monthly thereafter. As of December 31, 2007, there were 4.5 million shares available for grant under the 2005 Plan.
Adoption of FAS 123R
Stock-based compensation expense recognized during 2007 and 2006 was calculated based on awards ultimately expected to vest and has been reduced for estimated forfeitures. FAS 123R requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
Total stock-based compensation expense recognized under FAS 123R was as follows (in thousands, except per share data):
| | | | | | | | |
| | | Year Ended
December 31,
2007 | | | Year Ended
December 31,
2006 | |
Research and development | | $ | 5,058 | | | $ | 4,640 | |
General and administrative | | | 1,470 | | | | 1,290 | |
| | | | | | | | |
Total stock-based compensation expense | | $ | 6,528 | | | $ | 5,930 | |
| | | | | | | | |
Effect on earnings per share-basic and diluted | | $ | (0.09 | ) | | $ | (0.12 | ) |
| | | | | | | | |
As of December 31, 2007, total compensation cost related to nonvested stock options not yet recognized was $4.4 million, which is expected to be allocated to expense over a weighted-average period of 25 months, and
70
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
total compensation cost related to restricted stock units not yet recognized was $1.7 million, which is expected to be allocated to expense over a weighted-average period of seven months.
Pro Forma Information for Period Prior to Adoption of FAS 123R
The following pro forma net loss and net loss per share were determined as if we had accounted for employee stock-based compensation for our employee stock plans under the fair value method prescribed by FAS 123 for the periods indicated (in thousands except per share data):
| | | | |
| | | Year Ended
December 31,
2005 | |
Net loss attributed to common stockholders, as reported | | $ | (64,943 | ) |
Deduct: | | | | |
Stock-based employee compensation expense determined under fair value method for all awards, net of related tax effects | | | (9,369 | ) |
| | | | |
Pro forma net loss | | $ | (74,312 | ) |
| | | | |
Loss per share: | | | | |
Basic and diluted loss per common share, as reported | | $ | (1.43 | ) |
Basic and diluted pro forma loss per common share | | $ | (1.64 | ) |
Employee Stock-Based Compensation Valuation Assumptions
The compensation expense related to stock options recognized under FAS 123R was determined using the Black-Scholes option valuation model. Option valuation models require the input of subjective assumptions and these assumptions can vary over time. The weighted-average assumptions used were as follows:
| | | | | | | | | | | | | | | | | | |
| | | Options | | | Employee Stock
Purchase Plan | |
| | | Year Ended December 31, | | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | | | 2007 | | | 2006 | | | 2005 | |
Dividend yield | | 0.00 | % | | 0.00 | % | | 0.00 | % | | 0.00 | % | | 0.00 | % | | 0.00 | % |
Annual risk free rate of return | | 4.52 | % | | 4.82 | % | | 3.82 | % | | 4.86 | % | | 4.37 | % | | 2.51 | % |
Expected volatility | | 0.55 | | | 0.57 | | | 0.60 | | | 0.48 | | | 0.48 | | | 0.60 | |
Expected term (years) | | 5.2 | | | 5.1 | | | 4.0 | | | 1.4 | | | 1.0 | | | 0.5 | |
In estimating the expected term, we considered our historical stock option exercise experience including forfeitures, our post vesting termination pattern and the term of the options outstanding. The expected term of employee stock purchase plan rights is the average of the remaining purchase periods under each offering period. The annual risk free rate of return was based on the U.S. Treasury constant maturity rates with similar terms to the expected term of the stock option awards or of the employee stock purchase plan rights. We based our determination of expected volatility on our historical stock price volatility over the expected term. The fair value of the restricted stock units was estimated based upon the closing sales price of our common stock on the grant date. These restricted stock units will be fully vested and become unforfeitable on the first anniversary of the grant date. No compensation expense will be recognized for restricted stock units that do not vest.
71
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Stock option activity
A summary of the status of our stock option plan as of December 31, 2007, 2006 and 2005 and changes during the periods then ended is presented in the table below:
| | | | | | | | | | | | | | | | | | |
| | | Year ended December 31, |
| | | 2007 | | 2006 | | 2005 |
| | | Shares | | | Weighted
Average
Exercise
Price | | Shares | | | Weighted
Average
Exercise
Price | | Shares | | | Weighted
Average
Exercise
Price |
| | | (In thousands) | | | | | (In thousands) | | | | | (In thousands) | | | |
Outstanding, beginning of year | | 8,368 | | | $ | 10.08 | | 8,087 | | | $ | 11.18 | | 7,822 | | | $ | 12.23 |
Granted | | 1,542 | | | $ | 3.20 | | 1,640 | | | $ | 5.83 | | 1,758 | | | $ | 6.27 |
Exercised | | (7 | ) | | $ | 5.38 | | (74 | ) | | $ | 4.83 | | (106 | ) | | $ | 4.44 |
Forfeited or expired | | (1,340 | ) | | $ | 9.95 | | (1,285 | ) | | $ | 11.87 | | (1,387 | ) | | $ | 11.39 |
| | | | | | | | | | | | | | | | | | |
Outstanding, end of year | | 8,563 | | | $ | 8.86 | | 8,368 | | | $ | 10.08 | | 8,087 | | | $ | 11.18 |
| | | | | | | | | | | | | | | | | | |
Exercisable, end of year | | 6,237 | | | $ | 10.41 | | 6,040 | | | $ | 11.33 | | 5,772 | | | $ | 12.26 |
| | | | | | | | | | | | | | | | | | |
Weighted average grant date fair value | | | | | $ | 1.70 | | | | | $ | 3.15 | | | | | $ | 3.07 |
The total fair value of options that vested during the year ended December 31, 2007 was $4.6 million. The total intrinsic value of options exercised during the year ended December 31, 2007 was $5,000. Cash proceeds from the exercise of stock options were $40,000 for the year ended December 31, 2007. As of December 31, 2007, the aggregate intrinsic value of the stock options outstanding was zero.
The following table summarizes information about stock options outstanding as of December 31, 2007:
| | | | | | | | | | | | |
| | | Options Outstanding | | Options Exercisable |
Range of
Exercise Price | | Number
Outstanding | | Weighted-
Average
Remaining
Contractual
Life
(in years) | | Weighted-
Average
Exercise
Price | | Number
Exercisable | | Weighted-
Average
Exercise
Price |
| | | (In thousands) | | | | | | (In thousands) | | |
| $ 2.30-$ 3.40 | | 1,249 | | 9.1 | | $ | 3.08 | | 288 | | $ | 3.10 |
| $ 3.44-$ 5.80 | | 1,357 | | 6.4 | | $ | 4.55 | | 902 | | $ | 4.59 |
| $ 5.81-$ 6.19 | | 1,253 | | 7.1 | | $ | 6.04 | | 692 | | $ | 6.01 |
| $ 6.26-$ 7.50 | | 1,304 | | 5.7 | | $ | 6.87 | | 1,018 | | $ | 6.92 |
| $ 7.94-$11.95 | | 1,281 | | 4.9 | | $ | 9.94 | | 1,238 | | $ | 9.99 |
| $12.04-$16.06 | | 1,226 | | 5.1 | | $ | 14.33 | | 1,206 | | $ | 14.34 |
| $16.85-$42.63 | | 893 | | 3.2 | | $ | 21.33 | | 893 | | $ | 21.33 |
| | | | | | | | | | | | |
| | 8,563 | | 6.0 | | $ | 8.86 | | 6,237 | | $ | 10.41 |
| | | | | | | | | | | | |
Restricted Stock Units
On June 29, 2007, we issued 32,000 restricted stock units to our outside Directors under our 2005 Equity Incentive Plan, as amended, at a grant date fair value of $3.35. These restricted stock units will be fully vested on the first anniversary of the grant date. In accordance with FAS 123R, the fair value of the restricted stock units
72
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
was estimated based upon the closing sales price of our common stock on the grant date. Such value is being amortized over the one-year vesting term of the award, net of estimated forfeitures.
On July 30, 2007, we issued 844,550 restricted stock units to all employees under our 2005 Equity Incentive Plan, as amended, at a grant date fair value of $3.51. These restricted stock units will be fully vested on the first anniversary of the grant date. In accordance with FAS 123R, the fair value of the restricted stock units was estimated based upon the closing sales price of our common stock on the grant date. Such value is being amortized over the one-year vesting term of the award, net of estimated forfeitures.
Information with respect to restricted stock units as of December 31, 2007 was as follows (in thousands, except per share amount):
| | | | | | |
| | | Number of
Shares | | | Weighted Average
Grant-Date Fair
Value |
Outstanding at December 31, 2006 | | — | | | $ | — |
Granted | | 877 | | | $ | 3.50 |
Forfeited | | (11 | ) | | $ | 3.51 |
| | | | | | |
Outstanding at December 31, 2007 | | 866 | | | $ | 3.50 |
| | | | | | |
Employee stock purchase plan
The 2002 Employee Stock Purchase Plan, or the Purchase Plan, was approved by the stockholders in June 2002. The Purchase Plan allows eligible employees to purchase our common stock at 85 percent of the fair value at certain specified dates. Employee contributions are limited to 10 percent of compensation or $25,000, whichever is less. On June 20, 2006, our stockholders approved an amendment to the Purchase Plan to increase the maximum number of shares of common stock authorized for issuance under the plan automatically on the first day of each year by an amount equal to the lesser of (a) 0.3 million shares, (b) 1/2 percent of the outstanding shares on such date, or (c) an amount determined by the Board of Directors. As of December 31, 2007, a total of 1.1 million shares of common stock have been authorized for issuance under the Purchase Plan. As of December 31, 2007, 1.0 million shares have been issued pursuant to the Purchase Plan. During 2007, 0.3 million shares were sold under the plan for a total price of $0.9 million. The compensation expense related to the plan during 2007 was $0.7 million.
Non-employee stock-based compensation
We recorded $0.1 million in each of 2007, 2006 and 2005 for non-employee stock-based compensation for grants of stock options to consultants. These amounts were based upon the fair value of the vested portion of the grants. Additional compensation will be recorded in future periods for the remaining unvested portion of these option grants.
Stockholder rights plan
In July 1995, the Board of Directors approved a stockholder rights plan under which stockholders of record on August 21, 1995 received one preferred share purchase right for each outstanding share of our common stock. In July 2000, we made certain technical changes to amend the plan and extend the life of the plan until 2010. The rights are exercisable only if an acquirer purchases 15 percent or more of our common stock or announces a tender offer for 15 percent or more of our common stock. Upon exercise, holders other than the acquirer may
73
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
purchase our stock at a discount. The Board of Directors may terminate the rights plan at any time or, under certain circumstances, redeem the rights.
Common shares reserved for future issuance
As of December 31, 2007, we had reserved shares of common stock for potential future issuance as follows: 15.9 million shares upon conversion of convertible senior notes, 14.1 million shares for exercises under our stock option plans and stock purchase plan, 4.5 million shares for future CEFF purchases by Kingsbridge, 0.8 million shares for the warrants issued under the 2006 and 2007 CEFF, and 2.2 million shares for the warrants issued under the registered direct offering.
10. Income Taxes
In July 2005, the IRS issued a Notice of Proposed Adjustment, or NOPA, seeking to disallow $48.7 million of net operating losses that we deducted for the 2000 fiscal year and seeking a $3.4 million penalty for substantial underpayment of tax in the year ended December 31, 2000. We responded to the IRS in September 2005, disagreeing with the conclusions reached by the IRS in the NOPA and seeking to resolve this matter at the Appeals Office level of the IRS. In May 2007, we reached final settlement regarding this matter with the IRS in the amount of $3.3 million with respect to the fiscal years ended December 31, 2000, 2001 and 2002. This amount was comprised of $2.3 million in federal tax and $1.0 million in related interest. No penalty was assessed.
Our provision for income taxes consists of the following (in thousands):
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
Current: | | | | | | | | | | | | |
Federal | | $ | — | | | $ | (2,076 | ) | | $ | (2,009 | ) |
State | | | (523 | ) | | | (722 | ) | | | (649 | ) |
Foreign | | | (4 | ) | | | — | | | | — | |
| | | | | | | | | | | | |
| | | (527 | ) | | | (2,798 | ) | | | (2,658 | ) |
| | | | | | | | | | | | |
Deferred: | | | | | | | | | | | | |
Federal | | | — | | | | (26,815 | ) | | | (3,270 | ) |
State | | | — | | | | — | | | | — | |
| | | | | | | | | | | | |
| | | — | | | | (26,815 | ) | | | (3,270 | ) |
| | | | | | | | | | | | |
Other: | | | | | | | | | | | | |
Federal | | | 20,604 | | | | — | | | | — | |
State | | | 5,805 | | | | — | | | | — | |
| | | | | | | | | | | | |
| | | 26,409 | | | | — | | | | — | |
| | | | | | | | | | | | |
Income tax benefit (provision) | | $ | 25,882 | | | $ | (29,613 | ) | | $ | (5,928 | ) |
| | | | | | | | | | | | |
The tax benefit recorded for the year ended December 31, 2007 is related to the reversal of $26.4 million in May 2007 of previously accrued income taxes as a result of the final settlement with the IRS, offset by additional accrued interest for tax contingencies. The tax provision recorded in 2006 primarily relates to the realized gain on the sale of 3.0 million shares of Abgenix common stock and $2.8 million of additional interest for tax contingencies. The tax provision recorded in 2005 relates to the realized gain on the sale of 3.7 million shares of
74
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Abgenix common stock and $2.7 million of additional interest for tax contingencies partially offset by tax benefits related to unrealized gains on Abgenix common stock.
A reconciliation of our recorded income tax benefit (provision) to the U.S. statutory rate follows (in thousands):
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
Tax benefit at U.S. statutory rate | | $ | 43,804 | | | $ | 18,661 | | | $ | 20,654 | |
Change in valuation allowance | | | (46,200 | ) | | | (20,969 | ) | | | (23,357 | ) |
IRS settlement | | | 26,409 | | | | — | | | | — | |
Research and development tax credits | | | 3,188 | | | | 2,865 | | | | 2,739 | |
Stock-based compensation | | | (912 | ) | | | (531 | ) | | | — | |
Tax effect of realized and unrealized gains on available-for-sale-securities recorded in other comprehensive income | | | — | | | | (26,815 | ) | | | (3,270 | ) |
Interest on tax contingencies | | | (523 | ) | | | (2,798 | ) | | | (2,658 | ) |
Other | | | 116 | | | | (26 | ) | | | (36 | ) |
| | | | | | | | | | | | |
Benefit (provision) for income taxes | | $ | 25,882 | | | $ | (29,613 | ) | | $ | (5,928 | ) |
| | | | | | | | | | | | |
As of December 31, 2007, we had net operating loss carryforwards for federal income tax purposes of $447.6 million, which will expire on various dates between 2008 and 2027, if not utilized. As of December 31, 2007, we had federal research and development tax credits of $19.7 million, which will expire on various dates between 2008 and 2027. As of December 31, 2007, we had net operating loss carryforwards for California state income tax purposes of $124.8 million, which will expire on various dates between 2012 and 2017. As of December 31, 2007, we had California state research and development tax credits of $20.7 million, which do not expire. We also had Manufacturer Investment Credits of $0.1 million which expire in 2010 and 2011. Utilization of the net operating loss and credit carryforwards may be subject to substantial annual limitation due to ownership change provisions of the Internal Revenue Code of 1986 and similar state provisions. The annual limitation may result in the expiration of net operating losses and credits before utilization. To the extent net operating loss carryforwards, when realized, relate to non-qualified stock option deductions, the resulting benefits will be credited to stockholders’ equity.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets and liabilities are as follows (in thousands):
| | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
Deferred tax assets: | | | | | | | | |
Net operating loss carryforwards | | $ | 163,767 | | | $ | 125,366 | |
Research credit carryforwards | | | 33,221 | | | | 29,262 | |
Capitalized research and development, net of amortization | | | 17,343 | | | | 13,366 | |
Deferred revenue | | | 4,884 | | | | — | |
Other accruals and reserves | | | 10,951 | | | | 3,771 | |
| | | | | | | | |
Net deferred tax assets | | | 230,166 | | | | 171,765 | |
Valuation allowance | | | (230,166 | ) | | | (171,765 | ) |
| | | | | | | | |
Net deferred tax assets | | $ | — | | | $ | — | |
| | | | | | | | |
75
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. The valuation allowance increased by $58.4 million, $52.8 million and $25.4 million, in 2007, 2006 and 2005, respectively.
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows:
| | | | |
| | | Amount
(in thousands) | |
Balance at January 1, 2007 | | $ | 24,980 | |
Additions for tax positions of current year | | | — | |
Additions for tax positions of prior years | | | — | |
Reductions for tax positions of prior years | | | (18,601 | ) |
Settlement with tax authorities | | | (2,338 | ) |
| | | | |
Balance at December 31, 2007 | | $ | 4,041 | |
| | | | |
We file tax returns in the U.S., U.K. and California. In general, the years 2004 through 2007, remain open to examination for U.S. and U.K. purposes, and 2000 through 2007 for California purposes. It is reasonably possible that we will close certain years to examination under the relevant statute of limitations which may further decrease our liability for unrecognized tax benefits by approximately $4.0 million in the next 12 months.
The nature of these matters is uncertain and subject to change. As a result, the amount of our liability for certain of these matters could exceed or be less than the amount of our current estimates, depending on the outcome of these matters. An outcome of such matters different than previously estimated could materially impact our financial position or results of operations in the year of resolution.
11. 401(k) Plan
We sponsor a defined-contribution savings plan under Section 401(k) of the Internal Revenue Code covering all full time employees and part-time employees who work at least 20 hours per week, or 401K Plan. Participating employees may contribute up to the annual Internal Revenue Service contribution limit. Our 401K Plan also provides for employer matching contributions up to an annual limit of $3,000 per employee. Our 401K Plan is intended to qualify under Section 401 of the Internal Revenue Code so that contributions by the employees and by us, and income earned on the contributions are not taxable to employees until withdrawn from the plan. Contributions by us are tax deductible when made. At the discretion of each participant, the assets of our 401K Plan are invested in any of seventeen different investment options.
The employer matching contribution is invested in the same investment options selected by the employee for their individual contributions. The employer matching contributions vest ratably over three years. We contributed $0.6 million, $0.8 million and $0.9 million in employer matching contributions in 2007, 2006 and 2005, respectively.
12. Related Party
The accounting policies we apply to our transactions with our related parties are consistent with those applied in transactions with independent third parties.
Ceregene: Ceregene, Inc., or Ceregene, was previously our majority-owned subsidiary. In August 2004, July 2005, and April 2006, respectively, Ceregene announced the initial, second and third closings of itsSeries B preferred stock financing. We participated through the pro rata conversion of an outstanding bridge loan to Ceregene together with related accrued interest into shares of Ceregene’s Series B preferred stock. In January
76
CELL GENESYS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
2007, we participated in Ceregene’s Series C preferred financing by acquiring 1.8 million shares of Ceregene’s Series C in exchange for an exclusive license to Ceregene of certain of our intellectual property. As a result, we own approximately 16% of Ceregene on a fully diluted basis as of December 31, 2007. We did not record a gain on the exchange because the assets exchanged with Ceregene were nonmonetary and the carrying value of the intangible assets we used as consideration had no carrying value and no determinable fair value.
We account for our investment in Ceregene under the equity method of accounting for investments since we have the ability to exercise significant influence as our Chairman of the Board of Directors and Chief Executive Officer, or CEO, is also the Chairman of Ceregene’s Board of Directors. In 2007, we recorded revenue from Ceregene of $13,000. In both 2006 and 2005, we recorded revenue from Ceregene of $0.1 million. We did not recognize losses from Ceregene, nor do we expect to recognize future losses from Ceregene, as the net book value of our investment in Ceregene is zero.
We had guaranteed certain secured indebtedness of Ceregene totaling less than $0.1 million until May 2007. We had accrued less than $0.1 million related to this guarantee as of December 31, 2006. As of December 31, 2007, we have recorded no liability for this guarantee since our obligation expired in May 2007.
Caliper Life Sciences: The former Chairman and CEO of Xenogen Corporation, or Xenogen, a related party, which was acquired by Caliper Life Sciences, Inc., or Caliper, in August 2006, is now on the Board of Directors of Caliper and is also a member of our Board of Directors. We paid approximately $0.2 million in each of the years ended December 31, 2007, 2006 and 2005 for license fees to Xenogen Corporation.
13. Selected Quarterly Financial Information (Unaudited)
| | | | | | | | | | | | | | | | |
| | | Quarter Ended | |
Quarterly Results of Operations | | March 31,
2007 | | | June 30,
2007 | | | September 30,
2007 | | | December 31,
2007 | |
| | | | | | (Unaudited) | | | | |
| | | (In thousands, except per share amounts) | |
Revenue | | $ | 1,273 | | | $ | 3 | | | $ | 2 | | | $ | 102 | |
Research and development expenses | | | 24,015 | | | | 25,030 | | | | 28,629 | | | | 28,457 | |
General and administrative expenses | | | 5,245 | | | | 4,782 | | | | 5,114 | | | | 5,260 | |
Net loss | | | (29,449 | ) | | | (1,891 | ) | | | (34,538 | ) | | | (33,396 | ) |
Basic and diluted net loss per share | | | (0.49 | ) | | | (0.03 | ) | | | (0.46 | ) | | | (0.43 | ) |
| |
| | | Quarter Ended | |
Quarterly Results of Operations | | March 31,
2006 | | | June 30,
2006 | | | September 30,
2006 | | | December 31,
2006 | |
| | | | | | (Unaudited) | | | | |
| | | (In thousands, except per share amounts) | |
Revenue | | $ | 176 | | | $ | 1,046 | | | $ | 52 | | | $ | 90 | |
Research and development expenses | | | 25,314 | | | | 23,203 | | | | 23,196 | | | | 24,633 | |
General and administrative expenses | | | 5,045 | | | | 4,308 | | | | 4,335 | | | | 4,435 | |
Restructuring charges | | | (17 | ) | | | (40 | ) | | | — | | | | (25 | ) |
Gain on sale of Abgenix, Inc. common stock | | | 62,677 | | | | — | | | | — | | | | — | |
Net income (loss) | | | 4,014 | | | | (27,892 | ) | | | (28,801 | ) | | | (30,250 | ) |
Basic net income (loss) per common share | | | 0.09 | | | | (0.60 | ) | | | (0.57 | ) | | | (0.54 | ) |
Diluted net income (loss) per common share | | | 0.08 | | | | (0.60 | ) | | | (0.57 | ) | | | (0.54 | ) |
Basic and diluted net income (loss) per share is computed independently for each of the quarters presented. Therefore, the sum of quarterly basic and diluted per share amounts may not equal annual basic and diluted net income (loss) per share amounts.
77
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
Not applicable.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
We have performed an evaluation under the supervision and with the participation of our management, including our Chief Executive Officer, or CEO, and Chief Financial Officer, or CFO, of the effectiveness of our disclosure controls and procedures, as defined in Rule 13a-15(e) under the Securities Exchange Act of 1934 (the Exchange Act). Based on that evaluation, our management, including our CEO and CFO, concluded that our disclosure controls and procedures were effective as of December 31, 2007 to provide reasonable assurance that information required to be disclosed by us in the reports filed or submitted by us under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. There was no change in our internal controls during the fiscal quarter ended December 31, 2007 that materially affected, or is reasonably likely to materially affect, our internal controls over financial reporting.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control system was designed to provide reasonable assurance to our management and Board of Directors regarding the preparation and fair presentation of published financial statements.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2007. In making this assessment, it used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control — Integrated Framework. Based on our assessment we believe that, as of December 31, 2007, our internal control over financial reporting is effective based on those criteria.
| ITEM 9B. | OTHER INFORMATION |
Not applicable.
78
PART III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
(a) The information required by this Item concerning our directors is incorporated by reference to our Definitive Proxy Statement for the 2008 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days after the end of our 2007 fiscal year (the “2008 Proxy Statement”).
(b) The information required by this Item concerning our executive officers is set forth in the section entitled“Executive Officers”in Part I of this Form 10-K and is incorporated by reference into this section.
We have adopted a code of ethics that applies to all of our employees, including our principal executive officer, our principal financial officer and our principal accounting officer. This code of ethics, which is part of our Code of Business Conduct and Ethics that applies to all of our employees, is posted on our website and can be accessed from the following link:http://www.cellgenesys.com/investing-business-conduct.shtml.
We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding any amendment to, or waiver from, a provision of this code of ethics by posting such information on our website, at the address and location specified above.
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this Item is incorporated by reference to our 2008 Proxy Statement.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this item regarding security ownership of certain beneficial owners and management, as well as equity compensation plans, is incorporated by reference to the information set forth in the sections “Beneficial Owners and Management’s Ownership of Cell Genesys Stock” and “Equity Compensation Plan Information” in our 2008 Proxy Statement.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this Item is incorporated by reference to our 2008 Proxy Statement.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this Item is incorporated by reference to our 2008 Proxy Statement.
79
PART IV
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
(a) The following documents are included as part of this Annual Report on Form 10-K.
1. Index to Financial Statements
2. Financial Statement Schedules
All financial statement schedules are omitted because they are not applicable or not required or because the required information is included in the financial statements or notes thereto.
3. Exhibits
| | | | |
Number | | Note | | Description |
| 3.1 | | (1) | | Restated Certificate of Incorporation. |
| | |
| 3.2 | | (2) | | Certificate of Amendment to Restated Certificate of Incorporation. |
| | |
| 3.3 | | (1) | | Certificate of Designation of Rights, Preferences and Privileges of Series A Participating Preferred Stock. |
| | |
| 3.4 | | (1) | | Certificate of Amendment to Certificate of Designation of Rights, Preferences and Privileges of Series A Participating Preferred Stock. |
| | |
| 3.5 | | (1) | | Certificate of Designations, Preferences and Rights of Series B Convertible Preferred Stock. |
| | |
| 3.6 | | (3) | | Amended and Restated Bylaws dated October 17, 2007. |
| | |
| 4.1 | | (4) | | Amended and Restated Preferred Shares Rights Agreement, dated as of July 26, 2000 between the Registrant and Fleet National Bank. |
| | |
| 4.2 | | (5) | | Indenture dated as of October 20, 2004 by and between the Registrant and U.S. Bank National Association. |
| | |
| 10.1† | | (6) | | Form of Indemnification Agreement for Directors and Officers. |
| | |
| 10.2 | | (7) | | License Agreement dated August 13, 1990 between the Registrant and the University of North Carolina at Chapel Hill. |
| | |
| 10.3 | | (8) | | License Agreement dated June 28, 1991 between the Registrant and the University of Utah Research Foundation. |
| | |
| 10.4† | | (9) | | Amended Employment Agreement between the Registrant and Stephen A. Sherwin, M.D. |
| | |
| 10.5 | | (10) | | Research and Development Leases between the Registrant and Drawbridge/Forbes LLC, dated March 3, 2001. |
| | |
| 10.6† | | (11) | | 2001 Nonstatutory Stock Option Plan. |
| | |
| 10.7† | | (12) | | Form of Change of Control Severance Agreement. |
80
| | | | |
Number | | Note | | Description |
| 10.8† | | (13) | | Amended and Restated 1998 Incentive Stock Plan. |
| | |
| 10.9* | | (14) | | License Agreement dated June 7, 2002 between Transkaryotic Therapies, Inc. and the Registrant |
| | |
| 10.10* | | (15) | | Patent Assignment and License Agreement dated July 23, 2003 between the Registrant, Novartis AG and Genetic Therapy Inc. |
| | |
| 10.11* | | (16) | | Product Development and Option Agreement dated July 23, 2003 between the Registrant and Novartis Pharma AG. |
| | |
| 10.12 | | (2) | | Lease Agreement dated June 29, 2000 between Lincoln-RECP Industrial OPCO, LLC and the Registrant, First Amendment to Lease Agreement dated January 2, 2001 between F & S Hayward, LLC and the Registrant, and Lease Agreement dated January 7, 2002 between F & S Hayward II, LLC and the Registrant for premises located at 24570 Clawiter Road, Hayward, California. |
| | |
| 10.13 | | (17) | | Standstill and Registration Rights Agreement dated July 23, 2003 between the Registrant, Novartis AG and Genetic Therapy, Inc. |
| | |
| 10.14 | | (18) | | Resale Registration Rights Agreement dated as of October 20, 2004 among the Registrant and J.P. Morgan Securities Inc. and Lehman Brothers Inc., as representatives of the initial purchaser. |
| | |
| 10.15† | | (19) | | Contract of Employment dated February 25, 2005 between the Registrant and Robert J. Dow. |
| | |
| 10.16† | | (20) | | Change of Control Severance Agreement dated May 2, 2005 between the Registrant and Robert J. Dow. |
| | |
| 10.17† | | (21) | | 2005 Equity Incentive Plan, as amended. |
| | |
| 10.18† | | (22) | | 2002 Employee Stock Purchase Plan, as amended. |
| | |
| 10.19 | | (23) | | Common Stock Purchase Agreement dated March 14, 2006 between the Registrant and Kingsbridge Capital Limited. |
| | |
| 10.20 | | (24) | | Registration Rights Agreement dated March 14, 2006 between the Registrant and Kingsbridge Capital Limited. |
| | |
| 10.21 | | (25) | | Warrant Issued to Kingsbridge Capital Limited. |
| | |
| 10.22 | | (26) | | Common Stock Purchase Agreement dated February 5, 2007 between the Registrant and Kingsbridge Capital Limited. |
| | |
| 10.23 | | (27) | | Registration Rights Agreement dated February 5, 2007 between the Registrant and Kingsbridge Capital Limited. |
| | |
| 10.24 | | (28) | | Warrant Issued to Kingsbridge Capital Limited. |
| | |
| 10.25 | | (29) | | Form of Subscription Agreement, dated as of April 11, 2007, between the Registrant and the investor signatories thereto. |
| | |
| 10.26 | | (30) | | Form of Warrant. |
| | |
| 10.27 | | (31) | | Form of Restricted Stock Unit Award Agreement. |
| | |
| 10.28 | | (32) | | Form of Stock Option Award Agreement. |
| | |
| 10.29 | | (33) | | Technology and Intellectual Property Agreement between the Registrant and GBP IP, LLC. |
| | |
| 12.1 | | (34) | | Computation of Ratio of Earnings to Fixed Charges and Ratio of Earnings to Combined Fixed Charges and Preferred Stock Dividend Requirements. |
| | |
| 21.1 | | (34) | | Subsidiaries of the Registrant. |
81
| | | | |
Number | | Note | | Description |
| 23.1 | | (34) | | Consent of Independent Registered Public Accounting Firm. |
| | |
| 31.1 | | (34) | | Certification of Chief Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under Sarbanes-Oxley Act of 1934, as amended. |
| | |
| 31.2 | | (34) | | Certification of Chief Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under Sarbanes-Oxley Act of 1934, as amended. |
| | |
| 32.1 | | (35) | | Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| | |
| 32.2 | | (35) | | Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| * | Confidential treatment has been granted with respect to specific portions of this exhibit. |
| † | Management compensatory plan or arrangement required to be filed as an exhibit pursuant to Item 15(c) of Form 10-K. |
| (1) | Incorporated by reference to the same numbered exhibit filed with the Registrant’s Registration Statement on Form S-3/A (Reg. No. 333-102122) filed with the SEC on January 30, 2003. |
| (2) | Incorporated by reference to the same numbered exhibit filed with the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2005. |
| (3) | Incorporated by reference to Exhibit 3.1 filed with the Registrant’s Current Report on Form 8-K dated October 23, 2007. |
| (4) | Incorporated by reference to the Registrant’s Form 8-A12G/A dated July 28, 2000. |
| (5) | Incorporated by reference to Exhibit 4.1 filed with the Registrant’s Registration Statement on Form S-3 (Reg. No. 333-121732) filed with the SEC on December 29, 2004. |
| (6) | Incorporated by reference to Exhibit 10.2 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2007. |
| (7) | Incorporated by reference to Exhibit 10.6 filed with the Registrant’s Registration Statement on Form S-1 (Reg. No. 33-46452) as amended. |
| (8) | Incorporated by reference to Exhibit 10.9 filed with the Registrant’s Registration Statement on Form S-1 (Reg. No. 33-46452) as amended. |
| (9) | Incorporated by reference to Exhibit 10.20 filed with the Registrant’s Annual Report on Form 10-K for the year ended December 31, 1992. |
| (10) | Incorporated by reference to Exhibit 10.2 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2001. |
| (11) | Incorporated by reference to Exhibit 4.2 filed with the Registrant’s Registration Statement on Form S-8 (Reg. No. 333-91796) filed with the SEC on July 2, 2002. |
| (12) | Incorporated by reference to Exhibit 10.3 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2007. |
| (13) | Incorporated by reference to Exhibit 10.2 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2003. |
| (14) | Incorporated by reference to Exhibit 10.21 filed with the Registrant’s Quarterly Report on Form 10-Q/A for the quarter ended June 30, 2002 (filed July 30, 2003). |
| (15) | Incorporated by reference to Exhibit 10.3 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2003. |
| (16) | Incorporated by reference to Exhibit 10.4 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2003. |
| (17) | Incorporated by reference to Exhibit 10.5 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2003. |
| (18) | Incorporated by reference to Exhibit 10.1 filed with the Registrant’s Registration Statement on Form S-3 (Reg. No. 333-121732) filed with the SEC on December 29, 2004. |
82
| (19) | Incorporated by reference to Exhibit 10.1 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2005. |
| (20) | Incorporated by reference to Exhibit 10.2 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2005. |
| (21) | Incorporated by reference to Exhibit 10.3 filed with the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2007. |
| (22) | Incorporated by reference to Exhibit 10.1 filed with the Registrant’s Current Report on Form 8-K dated June 21, 2006. |
| (23) | Incorporated by reference to Exhibit 10.1 filed with the Registrant’s Current Report on Form 8-K dated March 15, 2006. |
| (24) | Incorporated by reference to Exhibit 10.2 filed with the Registrant’s Current Report on Form 8-K dated March 15, 2006. |
| (25) | Incorporated by reference to Exhibit 10.3 filed with the Registrant’s Current Report on Form 8-K dated March 15, 2006. |
| (26) | Incorporated by reference to Exhibit 10.1 filed with the Registrant’s Current Report on Form 8-K dated February 5, 2007. |
| (27) | Incorporated by reference to Exhibit 10.2 filed with the Registrant’s Current Report on Form 8-K dated February 5, 2007. |
| (28) | Incorporated by reference to Exhibit 10.3 filed with the Registrant’s Current Report on Form 8-K dated February 5, 2007. |
| (29) | Incorporated by reference to Exhibit 10.1 filed with the Registrant’s Current Report on Form 8-K dated April 11, 2007. |
| (30) | Incorporated by reference to Exhibit 4.1 filed with the Registrant’s Current Report on Form 8-K dated April 11, 2007. |
| (31) | Incorporated by reference to Exhibit 10.1 filed with the Registrant’s Current Report on Form 8-K dated August 1, 2007. |
| (32) | Incorporated by reference to Exhibit 4.2 filed with the Registrant’s Registration Statement on Form S-8 (Reg. No. 333-127158) filed with the SEC on August 3, 2005. |
| (33) | Incorporated by reference to Exhibit 10.1 filed with the Registrant’s Current Report on Form 8-K dated December 20, 2007. |
83
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, this 28th day of February 2008.
| | |
| CELL GENESYS, INC. |
| |
| By: | | /S/ SHARON E. TETLOW |
| | Sharon E. Tetlow Senior Vice President and Chief Financial Officer (Principal Financial Officer) |
| |
| By: | | /S/ MARC L. BELSKY |
| | Marc L. Belsky Vice President, Finance and Chief Accounting Officer (Principal Accounting Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | |
Signature | | Title | | Date |
| | |
/S/ STEPHEN A. SHERWIN, M.D. Stephen A. Sherwin, M.D. | | Chairman of the Board and Chief Executive Officer(Principal Executive Officer) | | February 28, 2008 |
| | |
/S/ SHARON E. TETLOW Sharon E. Tetlow | | Senior Vice President and Chief Financial Officer(Principal Financial Officer) | | February 28, 2008 |
| | |
/S/ MARC L. BELSKY Marc L. Belsky | | Vice President, Finance and Chief Accounting Officer(Principal Accounting Officer) | | February 28, 2008 |
| | |
/S/ DAVID W. CARTER David W. Carter | | Director | | February 28, 2008 |
| | |
/S/ NANCY M. CROWELL Nancy M. Crowell | | Director | | February 28, 2008 |
| | |
/S/ JAMES M. GOWER James M. Gower | | Director | | February 28, 2008 |
| | |
/S/ JOHN T. POTTS, JR., M.D. John T. Potts, Jr., M.D. | | Director | | February 28, 2008 |
| | |
/S/ THOMAS E. SHENK, PH.D. Thomas E. Shenk, Ph.D. | | Director | | February 28, 2008 |
| | |
/S/ EUGENE L. STEP Eugene L. Step | | Director | | February 28, 2008 |
| | |
/S/ INDER M. VERMA, PH.D. Inder M. Verma, Ph.D. | | Director | | February 28, 2008 |
| | |
/S/ DENNIS L. WINGER Dennis L. Winger | | Director | | February 28, 2008 |
84