UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
x | | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2006 |
or |
o | | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to |
Commission file number 000-19319
Vertex Pharmaceuticals Incorporated
(Exact name of registrant as specified in its charter)
Massachusetts | | 04-3039129 |
(State or other jurisdiction of
incorporation or organization) | | (I.R.S. Employer
Identification No.) |
130 Waverly Street
Cambridge, Massachusetts | | 02139-4242 |
(Address of principal executive offices) | | (Zip Code) |
Registrant’s telephone number, including area code (617) 444-6100
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Title of Each Class | | | | Name of Each Exchange on Which Registered | |
Common Stock, $0.01 Par Value Per Share
Rights to Purchase Series A Junior Participating Preferred Stock | | The Nasdaq Global Select Market |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes x No o
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes o No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer x | | Accelerated filer o | | Non-accelerated filer o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate) based on the last reported sale price of the common stock on June 30, 2006 was $2.3 billion.
As of February 26, 2007, the registrant had 126,650,456 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive Proxy Statement for the 2007 Annual Meeting of Stockholders to be held on May 31, 2007 are incorporated by reference into Part III of this Annual Report on Form 10-K.
FORM 10-K
TABLE OF CONTENTS
The “Company,” “Vertex,” “we” and “us” as used in this Annual Report on Form 10-K, refer to Vertex Pharmaceuticals Incorporated, a Massachusetts corporation, and its subsidiaries.
“Vertex” is a registered trademark of Vertex. “Agenerase,” “Lexiva” and “Telzir” are registered trademarks of GlaxoSmithKline plc. Other brands, names and trademarks contained in this Annual Report are the property of their respective owners.
PART I
ITEM 1. BUSINESS
OVERVIEW
We are in the business of discovering, developing and commercializing small molecule drugs for the treatment of serious diseases. We have built a drug discovery capability that integrates biology, chemistry, biophysics, automation and information technologies, with a goal of making the drug discovery process more efficient and productive. A Vertex-discovered compound for the treatment of HIV infection, fosamprenavir calcium, is being marketed by our collaborator GlaxoSmithKline plc as Lexiva in the United States and Telzir in Europe. We currently are concentrating most of our drug development resources on four drug candidates: telaprevir (VX-950) for the treatment of hepatitis C virus, or HCV, infection, VX-702 for the treatment of rheumatoid arthritis and other inflammatory diseases, VX-770 for the treatment of cystic fibrosis, or CF, and VX-883 for the treatment of bacterial infection.
Our lead drug candidate is telaprevir, an oral hepatitis C protease inhibitor, and one of the most advanced of a new class of antiviral treatments in development targeting HCV infection, a serious and life-threatening disease. We are conducting three major Phase 2b clinical trials of telaprevir. We expect that these three Phase 2b clinical trials together will evaluate sustained viral response rates in approximately 1,000 patients infected with genotype 1 HCV, the most prevalent form of HCV, including treatment-naïve patients and patients who did not achieve sustained viral response with current therapies. We expect the clinical results from these Phase 2b clinical trials to provide important information supporting the design and initiation in the second half of 2007 of a Phase 3 clinical program for telaprevir. The United States Food and Drug Administration, or FDA, has granted “Fast-Track” designation to telaprevir. In June 2006, we entered into a collaboration agreement with Janssen Pharmaceutica, N.V., or Janssen, a Johnson & Johnson company, relating to telaprevir. Under our agreement with Janssen, we have retained exclusive commercial rights to telaprevir in North America and will lead the clinical development program. Janssen will be responsible for the commercialization of telaprevir, including the manufacture of its own commercial supply of telaprevir for the Janssen territories, which include the territories outside of North America and the Far East. Janssen has agreed to be responsible for 50% of drug development costs under the development program for North America and the Janssen territories and to make additional contingent milestone payments for the successful development, approval and launch of telaprevir.
In addition to telaprevir, we currently are focusing on:
· VX-702, a p38 MAP kinase inhibitor, which we currently are investigating for the treatment of rheumatoid arthritis, or RA. We are conducting a 12-week, 120 patient Phase 2a clinical trial in patients with RA to evaluate the safety, tolerability and anti-inflammatory effects of VX-702 on a background of methotrexate. In January 2007, we commenced a Thorough QTc study, which is a type of clinical trial required for all small molecule drug candidates prior to the initiation of Phase 3 clinical trials. Depending on the results of the 12-week Phase 2a clinical trial and the Thorough QTc study, we plan to conduct a larger six-month Phase 2 clinical trial of VX-702 on a background of methotrexate.
· VX-770, a cystic fibrosis transmembrane regulator, or CFTR, potentiator, which we are investigating for the treatment of CF. We recently completed a Phase 1 clinical trial of VX-770 in healthy volunteers and in patients with CF and plan to initiate a Phase 2 clinical trial of VX-770 in patients with CF in the second quarter of 2007.
· VX-883, a Vertex-discovered dual-mechanism investigational antibiotic, which is in preclinical development for the treatment of patients with bacterial infection. Depending on the outcomes of our preclinical activities, we plan to initiate a Phase 1 clinical trial of this drug candidate in 2007.
Our pipeline also includes several drug candidates that are being developed by our collaborators. The most advanced of these drug candidates is MK-0457 (VX-680), an Aurora kinase inhibitor that is being
1
developed by Merck & Co., Inc. for the treatment of cancer. In December 2006, Merck announced results from the Phase 1 clinical trials of MK-0457 (VX-680), which showed that MK-0457 (VX-680) demonstrated clinical activity in select patients with leukemias and myeloproliferative disorders, and initiated a pivotal Phase 2 clinical trial of MK-0457 (VX-680) in patients with treatment-resistant chronic myelogenous leukemia, or CML, and Philadelphia chromosome-positive acute lymphocytic leukemia, or Ph+ ALL, containing the T315I BCR-ABL mutation. We also are collaborating with Merck on the development of other Aurora kinase inhibitors.
We plan to continue adding promising drug candidates to our development pipeline through our ongoing commitment to discovery research. As we have matured as a company by advancing our drug candidates to later stage development, we have chosen to retain greater development control and commercial rights to some of our drug candidates in order retain a greater proportion of the potential value of those drug candidates if they are successfully commercialized. In the future, we expect that we will retain control of clinical development and the right to market some of our drug candidates in therapeutic and geographic areas where we have or believe we can build sufficient expertise to independently market the drug candidate, if it is approved, on a cost-effective basis.
In order to complete the clinical development program for telaprevir and prepare to market telaprevir in North America if it is approved, we are investing significant resources to expand our capabilities with respect to clinical development, regulatory affairs and quality control, and to build and manage a commercial supply chain. We also expect to incur significant costs in 2007 to manufacture registration batches of telaprevir and build telaprevir commercial inventory.
OUR STRATEGY
Our goal is to become a fully integrated pharmaceutical company with industry-leading capabilities in research, development and commercialization of pharmaceutical products. The key elements of our strategy are:
Focus on the development and commercialization of telaprevir. We plan to invest significant resources in the clinical development and preparation for launch of telaprevir. We have designed a comprehensive clinical development program for telaprevir consisting of multiple concurrent clinical trials that will study the profile of telaprevir. In addition, we are investing in building and managing a commercial supply chain for telaprevir, including purchasing significant quantities of long lead-time raw materials and identifying and entering into relationships with third-party manufacturers, in order to support a timely launch of telaprevir if we are successful in completing development and obtaining marketing approval.
Capitalize on the advances in our telaprevir clinical program to build our general drug development and commercialization capabilities. We believe that the progress we made in 2006 and in particular the advances we made in the development of telaprevir have positioned us to expand our general drug development and commercialization capabilities. In 2007, we plan to invest in key areas—including clinical development, regulatory affairs, quality control, commercial operations and commercial supply chain management—that will be necessary in order to complete development of telaprevir, to seek approval for telaprevir and to commercialize telaprevir if we are successful in obtaining marketing approval. We expect that these capabilities also will support realization of additional drug candidates that may progress through our pipeline.
Invest in research and development and retain a greater proportion of rights to proprietary drug candidates. We intend to continue to invest significant amounts in our research and development programs. In the future, we expect that we will fund a greater proportion of our research programs than in past years, using internal funds rather than collaborator funds. We believe that this strategy will ultimately allow us to retain greater development control of, and commercial rights to, those proprietary drug candidates that may meet our strategic internal investment criteria as in effect from time to time.
Support a broad-based portfolio of drug candidates, including the introduction of multiple drug candidates into development. We have elected to diversify our research and development activities across a relatively
2
broad array of investment opportunities, due in part to the high risks associated with the biotechnology and pharmaceutical business. We plan to continue to add promising potential drug candidates to our development pipeline through our continuing commitment to discovery research.
Continue existing and establish new collaborations to develop and commercialize selected drug candidates. Collaborations with pharmaceutical companies have played an important role in helping us advance our drug discovery capabilities as well as to grow and advance our drug candidate pipeline. Collaborations provide us with financial support and other valuable resources for our research programs, development resources for our drug candidates and marketing and sales support for our products. We plan to continue to rely on collaborators to develop and commercialize certain of our drug candidates either worldwide, in collaboration with us, or in markets in which we are not currently concentrating our resources.
License and acquire technologies, resources and products. In addition to collaborations, we also seek opportunistically to license and acquire technologies, resources and drugs that have the potential to strengthen our drug discovery platform, pipeline and commercial capabilities.
Appropriately manage our capital resources in order to execute our strategy. We are focused on maintaining a financial profile that will enable us to execute our business strategy. In 2006, we increased our cash, cash equivalents and marketable securities balance and reduced our outstanding debt as a result of payments received from collaborators and a series of capital transactions that offset significant operating expenses. We expect to continue to pursue a general financing strategy that may lead us to undertake one or more additional capital transactions.
COMMERCIAL PRODUCT AND CLINICAL DEVELOPMENT PROGRAMS
The drug candidates on which we are currently concentrating most of our drug development resources, along with the drugs and drug candidates we have developed or are developing with collaborators, are set forth in the following table. In addition, we are currently conducting clinical trials or engaging in preclinical activities with respect to a number of additional drug candidates.
Drug or Drug Candidate | | Clinical Indication(s) | | Phase | | Marketing Rights (Region) |
Principal Areas of Focus | | | | | | |
telaprevir (VX-950) | | Chronic HCV infection | | Phase 2b | | Vertex (North America);
Mitsubishi (Far East); and
Janssen (Rest of World) |
VX-702 | | Rheumatoid arthritis and other inflammatory diseases | | Phase 2 | | Vertex (Worldwide, except for Far East); and
Kissei (Far East, co-exclusive with Vertex in certain Far East countries) |
VX-770 | | Cystic fibrosis | | Phase 1 | | Vertex (Worldwide) |
VX-883 | | Bacterial infection | | Preclinical | | Vertex (Worldwide) |
Collaborator-Led Programs | | | | | | |
Lexiva/Telzir* | | HIV infection and AIDS | | Marketed | | GlaxoSmithKline (Worldwide) |
MK-0457 (VX-680) | | Oncology | | Phase 2 | | Merck (Worldwide) |
MK-6592 (VX-667) | | Oncology | | Phase 1 | | Merck (Worldwide) |
AVN-944 (VX-944) | | Oncology | | Phase 1 | | Avalon Pharmaceuticals (Worldwide) |
VX-409 and backup compounds | | Pain | | Preclinical | | GlaxoSmithKline (Worldwide) |
* Fosamprenavir calcium is marketed under the trade names “Lexiva” in North America and “Telzir” in the European Union. Lexiva/Telzir is a prodrug of amprenavir (marketed as “Agenerase”), our first drug for the treatment of HIV infection and AIDS. Lexiva/Telzir has replaced Agenerase in worldwide markets.
3
Principal Areas of Focus
Telaprevir (VX-950) (investigational oral hepatitis C protease inhibitor for the treatment of chronic HCV infection)
Telaprevir, our most advanced drug candidate, is an oral hepatitis C protease inhibitor. Telaprevir is designed to inhibit NS3-4A serine protease, an enzyme thought to be necessary for HCV replication. The FDA has granted “Fast-Track” designation to telaprevir. We are conducting three major Phase 2b clinical trials of telaprevir, which we refer to as the PROVE clinical trials, as part of a global development program to determine the safety and antiviral activity of telaprevir. We expect the clinical results from these Phase 2b clinical trials to provide important information supporting the design and initiation in the second half of 2007 of a Phase 3 clinical program for telaprevir. We believe that the current PROVE clinical trials have the potential to generate sufficient safety and efficacy data in a broad range of genotype 1 HCV patients, together with safety data from the proposed Phase 3 program, to support a New Drug Application, or NDA, filing in late 2008. If efficacy data from the Phase 3 program is required for the NDA, the NDA filing may be later than 2008.
Under our agreement with Janssen, we have retained exclusive commercial rights to telaprevir in North America and will lead the clinical development program in North America and the Janssen territories. Janssen has the right to market telaprevir in the rest of the world, except for Japan and certain Far East countries, where we are collaborating with Mitsubishi Pharma Corporation. Janssen has agreed to be responsible for 50% of drug development costs under the development program for North America and the Janssen territories and to make additional contingent milestone payments for the successful development, approval and launch of telaprevir. Janssen will be responsible for the commercialization of telaprevir, including the manufacture of its own commercial supply of telaprevir, outside of North America and the Far East.
In 2007, we anticipate a clinical trial exploring twice-daily dosing of telaprevir will be initiated and that Janssen will conduct this clinical trial through Tibotec, a separate Johnson & Johnson company. In addition in 2007, we expect to expand the clinical development of telaprevir into patients with genotype 2 and genotype 3 HCV infection. In 2006, Mitsubishi conducted a Phase 1 clinical trial of telaprevir in the Far East. Mitsubishi is designing a Phase 2 clinical program for telaprevir in the Far East.
We have begun to identify and enter into commercial relationships with third-party manufacturers that will be necessary in order to manufacture commercial quantities of telaprevir. In 2007, we expect to manufacture registration batches of telaprevir, and invest in telaprevir commercial supply in order to support a timely launch if we are successful in obtaining regulatory marketing approval for telaprevir.
Telaprevir was discovered in our collaboration, now ended, with Eli Lilly and Company. We hold worldwide rights to all other second-generation HCV protease inhibitors discovered by us during our collaboration with Eli Lilly. We will owe Eli Lilly royalties on any future sales of telaprevir, if approved, and certain other HCV protease inhibitors.
Background: Treatment of Chronic Hepatitis C Virus Infection
HCV infection causes chronic inflammation in the liver. The World Health Organization estimates that there are as many as 170 million people chronically infected with HCV worldwide and that an additional 3 million to 4 million people are infected each year. Reports published by the American Association for the Study of Liver Disease have estimated that approximately 3.4 million people in the United States are chronically infected with HCV, and the American Liver Foundation estimates that 8,000 to 10,000 people in the United States die as a result of HCV infection each year.
Currently, there is no vaccine available to prevent HCV infection. The current standard treatment for genotype 1 HCV infection is a combination of pegylated interferon, or peg-IFN, and ribavirin, or RBV, administered for up to 48 weeks. This treatment regimen is associated with significant side effects, including fatigue, flu-like symptoms, depression and anemia. Among patients who begin treatment, approximately 50% of patients infected with genotype 1 HCV, the most common HCV genotype in the
4
United States, fail to either complete treatment or show a long-term sustained response to therapy. As a result, we believe new safe and effective treatment options for HCV infection are needed.
Telaprevir Development Program
We are conducting three major Phase 2b clinical trials of telaprevir. PROVE 1 is ongoing in the United States and PROVE 2 is ongoing in European Union, both in treatment-naïve patients. PROVE 3 has commenced and is being conducted with patients in North America and the European Union who did not achieve sustained viral response with previous interferon-based treatments. PROVE 1 and PROVE 2 are fully enrolled, and we commenced patient enrollment in PROVE 3 in January 2007.
PROVE 1 and PROVE 2
We expect that together, the PROVE 1 and PROVE 2 clinical trials will evaluate rates of sustained viral response, or SVR, in approximately 580 treatment-naïve patients infected with genotype 1 HCV, including patients who will receive telaprevir and patients in the control arms. SVR is defined as undetectable viral levels 24 weeks after all treatment has ceased.
A description of each of the clinical trial arms for the PROVE 1 and PROVE 2 clinical trials, including the intended number of patients in each trial, is set forth in the following table:
Treatment Regimen | | | | Number of
Patients
(treatment naïve)
PROVE 1 | | Number of
Patients
(treatment naïve)
PROVE 2 | | Total | |
12-week regimens of telaprevir in combination with peg-IFN and RBV | | | 20 | | | | 80 | | | 100 | |
12-week regimens of telaprevir in combination with only peg-IFN | | | 0 | | | | 80 | | | 80 | |
12-week regimens of telaprevir in combination with peg-IFN and RBV, followed by 12 weeks of therapy with peg-IFN and RBV | | | 80 | | | | 80 | | | 160 | |
12-week regimens of telaprevir in combination with peg-IFN and RBV, followed by 36 weeks of therapy with peg-IFN and RBV | | | 80 | | | | 0 | | | 80 | |
48-weeks of therapy with peg-IFN and RBV | | | 80 | | | | 80 | | | 160 | |
Total | | | 260 | | | | 320 | | | 580 | |
The PROVE 1 and PROVE 2 clinical trials together have the following four key objectives:
· to evaluate the optimal SVR rate that can be achieved with telaprevir therapy in combination with peg-IFN and RBV;
· to evaluate the optimal treatment duration for telaprevir combination therapy;
· to evaluate the role of RBV in telaprevir-based therapy; and
· to evaluate the safety of telaprevir in combination with peg-IFN and RBV.
In the PROVE 1 and PROVE 2 clinical trials, patients receive telaprevir in a tablet formulation at a dose of 750 mg every eight hours for 12 weeks. The PROVE 1 clinical trial is double-blinded and placebo-controlled, and the PROVE 2 clinical trial is partially-blinded and placebo-controlled.
In December 2006, we announced results from a planned interim safety and antiviral activity analysis that was conducted and reviewed by the independent data monitoring committee overseeing the PROVE 1 clinical trial. As of the cut-off date of the interim analysis, a total of 250 patients had been enrolled in the PROVE 1 clinical trial and received at least one dose of telaprevir or placebo. In the data reported, the patients in all three telaprevir-containing arms (approximately 175 patients) were pooled together and the results were compared to the results in the control arm of peg-IFN and RBV and placebo (approximately 75 patients).
At the time of the data cut-off for the safety analysis, approximately 100 patients had completed 12 weeks on-study and more than 200 patients had completed eight weeks. The most common adverse
5
events were similar in type between the two groups. Of these, the adverse events that were more commonly reported in the telaprevir arms included gastrointestinal disorders and rash. In the telaprevir groups, 9% of patients had discontinued treatment due to adverse events, including 3% as a result of rash, compared to 3% of patients who discontinued treatment due to adverse events in the control arm. The difference between the two groups is due to the greater number of discontinuations due to rash, gastrointestinal disorders, and anemia in the telaprevir arms compared to the control arm. Serious adverse events were noted in 3% of patients in the telaprevir groups and 1% of patients in the control group.
The table below summarizes available HCV preliminary results at week 12:
| Treatment Assignment | | | Patients who had undetectable HCV levels
(less than 10 IU/mL) at week 12 |
telaprevir + peg-IFN + RBV | | 65 of 74 (88%) |
placebo + peg-IFN + RBV | | 17 of 33 (52%) |
PROVE 3
In the PROVE 3 clinical trial, we expect to evaluate SVR rates in approximately 440 patients infected with genotype 1 HCV located in North America and the European Union who did not achieve SVR with previous interferon-based treatments. We expect to complete enrollment in this double-blinded and placebo-controlled clinical trial by the end of the second quarter of 2007. Patients in the telaprevir arms of the PROVE 3 clinical trial will receive telaprevir in a tablet formulation at a dose of 750 mg every eight hours for 12 weeks or 24 weeks.
A description of each of the clinical trial arms of the PROVE 3 clinical trial, including the intended number of patients in the trial, is set forth in the following table:
Treatment Regimen | | | | Number of
Patients
(treatment failure)
PROVE 3 | |
12-week regimens of telaprevir in combination with peg-IFN and RBV, followed by 12 weeks of therapy with peg-IFN and RBV | | | 110 | | |
24-week regimens of telaprevir in combination with only peg-IFN | | | 110 | | |
24-week regimens of telaprevir in combination with peg-IFN and RBV, followed by 24 weeks of therapy with peg-IFN and RBV | | | 110 | | |
48-weeks of therapy with peg-IFN and RBV | | | 110 | | |
Total | | | 440 | | |
The PROVE 3 clinical trial has the following two key objectives:
· to evaluate the SVR rate that can be achieved with telaprevir therapy in combination with peg-IFN and RBV in patients who have not achieved SVR with previous interferon-based treatments; and
· to evaluate the safety profile of telaprevir administered in a 24-week course of therapy of telaprevir in combination with peg-IFN and RBV.
Previous Trials
From late 2004 through early 2006, we conducted a series of small-scale clinical trials of telaprevir in healthy volunteers and HCV-infected patients designed to evaluate safety, pharmacokinetics and antiviral activity of telaprevir. The first of these trials, a Phase 1 clinical trial of telaprevir in healthy volunteers, was completed in 2004. The next two trials were Phase 1b trials conducted in 2005—a trial of telaprevir as monotherapy completed early in 2005, and a trial of telaprevir dosed together with peg-IFN completed later in 2005. We also conducted a small Phase 2a combination therapy clinical trial of telaprevir dosed with peg-IFN and RBV, which was completed in 2006. The results of these earlier trials supported the design and initiation of the ongoing PROVE clinical trials. The results of these trials represent clinical treatment of small numbers of patients who were initially dosed in clinical trials of telaprevir for short durations and may not be predictive of patient outcomes in large clinical trials evaluating telaprevir.
6
VX-702 (oral p38 MAP kinase inhibitor for the treatment of RA and other inflammatory diseases)
VX-702 is our oral p38 mitogen-activated protein, or MAP, kinase inhibitor, which we are currently investigating for the treatment of RA. In March 2006, we reported data from our three-month Phase 2 clinical trial of VX-702 in patients with RA. In this trial, VX-702 demonstrated statistically significant clinical effects on signs and symptoms of RA as measured by ACR20 criteria, as well as several other widely used clinical measures. However, we believe that the levels of improvement shown in this trial did not warrant further development of VX-702 as a single agent for the treatment of RA. We are conducting a 12-week, 120-patient Phase 2a clinical trial in patients with RA to evaluate the safety, tolerability and anti-inflammatory effects of VX-702 on a background of methotrexate. In January 2007, we started a Thorough QTc study of VX-702. The FDA requires that companies conduct a Thorough QTc study of any small molecule drug candidate prior to the commencement of Phase 3 clinical trials. Depending on the results of the 12-week Phase 2a clinical trial and the Thorough QTc study, we plan to conduct a larger six-month Phase 2 clinical trial in patients with RA of VX-702 on a background of methotrexate.
We hold worldwide development and commercialization rights to VX-702, except for Japan and certain Far East countries, where we are collaborating with Kissei Pharmaceutical Co., Ltd. In 2006, Kissei completed a Phase 1 clinical trial of VX-702 in Japan in RA.
Background
p38 MAP Kinase Inhibitors for Inflammatory Diseases
The MAP kinases are a family of structurally-related human enzymes involved in intracellular signaling pathways that enable cells to respond to their environment. The p38 MAP kinase is involved in a variety of cellular processes, including the onset and progression of inflammation. When activated, the p38 MAP kinase triggers production of multiple cytokines, including interleukin-1, or IL-1, TNF-alpha and interleukin-6, or IL-6. Excess levels of IL-1 and TNF-alpha are associated with a number of acute and chronic inflammatory diseases.
We have extensive preclinical and clinical experience with p38 MAP kinase inhibitors, which we believe may be a useful new class of oral anti-inflammatory drugs.
Rheumatoid Arthritis
Rheumatoid arthritis, a systemic disease, is the most common form of inflammatory arthritis. RA has a prevalence of about 1% of the worldwide population and an annual incidence of 3 cases per 10,000 adults. RA causes pain, swelling and loss of function in affected joints. The disease is often accompanied by significant morbidity and mortality. Patients with RA also have a significant impairment in their quality of life.
The current first line standard treatment for RA is administration of a disease-modifying anti-rheumatic drug, or DMARD, most commonly methotrexate. Injectable anti-tumor necrosis factor, or TNF, agents such as Enbrel® (etanercept), Humira® (adalimumab) and Remicade® (infliximab), generally on a background of methotrexate, are used when disease activity is not controlled by DMARDs and non-steroidal anti-inflammatory drugs. We believe that an oral agent that successfully targets TNF production may provide an attractive treatment option for patients with RA.
VX-702 Development Program
In March 2006, we obtained results from our 315-patient three-month Phase 2 clinical trial of VX-702, in RA which we refer to as the VeRA trial. A total of 278 patients completed 12 weeks of treatment in this double-blind, randomized and placebo-controlled trial. Patients received either 5 mg or 10 mg of VX-702 once daily, or placebo. In addition to VX-702, patients could receive certain DMARDs, but could not receive methotrexate or anti-TNF therapies. At the end of 12 weeks, patients completed dosing with VX-702 and were evaluated for improvement in clinical signs and symptoms according to American College of Rheumatology criteria, or ACR20. ACR20 is a standardized measure based on a patient’s
7
attainment of at least a 20% improvement in ACR-specified indicators of RA activity. In comparing differences in outcomes between treatments in a clinical trial, statistical significance testing is used to establish the probability that the observed differences did not occur by chance. The result of statistical testing is often defined in terms of a “p-value,” with a level of 0.05 or less considered to be a statistically significant difference.
In this clinical trial, treatment with VX-702 led to a dose-dependent, statistically significant increase in week 12 ACR20 response rates, the primary endpoint of the clinical trial. Thirty percent of patients receiving placebo, 38% of patients receiving 5 mg daily of VX-702 and 40% of patients receiving 10 mg daily of VX-702 achieved an ACR20 response at week 12 (p-value=0.04; Jonckheere-Terpstra test for increasing dose-response). In addition, 32% of placebo patients, 41% of 5 mg VX-702-treated patients and 44% of 10 mg VX-702-treated patients achieved a EULAR (moderate or good) response (p=0.01). EULAR is another standardized measure of indicators of RA under criteria specified by the European League Against Rheumatism. Dose-dependent statistically significant effects also were seen on tender joint counts (p=0.007), swollen joint counts (p=0.003), disease activity score (DAS28; p=0.02) and morning stiffness (p=0.03).
In 2004, we completed a Phase 2a double-blind, randomized, placebo-controlled, dose-escalation clinical trial of VX-702 for the treatment of patients with acute coronary syndrome, or ACS, undergoing percutaneous coronary intervention, or PCI, such as stent placement. p38 MAP kinase regulates the production of key inflammatory cytokines implicated in the pathogenesis of ACS. This Phase 2a trial of VX-702 was designed to evaluate the safety, tolerability and pharmacokinetics of VX-702 in 45 patients with unstable angina and elevated levels of c-reactive protein, a marker of inflammation measured in the blood, undergoing PCI. In the Phase 2a ACS trial, there were no clinically significant differences between treatment and placebo groups with respect to adverse events, and VX-702 met pre-established safety and pharmacokinetic objectives.
Clinical trials of a number of other p38 MAP kinase inhibitors have demonstrated dose-dependent elevations in liver enzymes, which generally are thought to be markers for liver injury. In 2004, we completed a 28-day trial of VX-702 in healthy volunteers designed specifically to evaluate the effect of VX-702 on liver enzymes. This trial showed some transient elevations in liver enzymes in a small number of subjects. However, the magnitude of those enzyme elevations did not reach clinical significance and did not require discontinuation of dosing. The enzyme levels returned to normal during continued dosing.
In the VeRA trial, premature discontinuations for adverse events across the trial arms were: placebo (2%), 5 mg (3%) and 10 mg (5%). No clinically significant adverse effects were seen on laboratory parameters, including liver function tests. The most common adverse events that led to treatment discontinuation in patients receiving VX-702 were seen in two patients each and were: gastroenteritis, nausea/vomiting, rash, and renal impairment (increased serum creatinine levels to 1.2 to 1.5 times upper limit of normal). The most common adverse events were generally mild or moderate and were: infection (5% of placebo patients and 10% of VX-702 patients), gastrointestinal disorders (6% placebo and 8% VX-702), and skin disorders (0% placebo and 9% VX-702). In electrocardiograms conducted during the VeRA trial, increases in QT interval (a measure of electrical conduction in the heart) were seen in the VX-702 treatment groups, but no patient in the VeRA trial experienced a clinically significant (60 msec or approximately 15%) increase in the Fridericia rate-corrected QT interval. We expect that Thorough QTc trial currently underway will provide additional information on the effects of VX-702 on electrical conduction in the heart.
VX-770 (investigational oral CFTR potentiator for the treatment of cystic fibrosis)
VX-770 is a small molecule drug candidate designed to potentiate the gating activity of the CFTR protein, a chloride ion transporter on the cell surface that is functionally defective in patients with cystic fibrosis. We submitted an Investigational New Drug, or IND, application with the FDA and completed a Phase 1 clinical trial of VX-770 in 2006. VX-770 received both “Fast Track” and “Orphan Drug” designations from the FDA in 2006.
8
VX-770 was discovered by us in our ongoing research collaboration with Cystic Fibrosis Foundation Therapeutics Incorporated, or CFFT, and with the support and participation of the Cystic Fibrosis Foundation. We hold worldwide development and commercialization rights to VX-770. We would be required to pay CFFT royalties on any future sales of VX-770.
Cystic fibrosis is a genetic disease afflicting approximately 30,000 people in the United States. The symptoms of cystic fibrosis, particularly the development of thick mucous that causes lung tissue inflammation and, ultimately, irreversible lung damage, are caused by defects in the CFTR protein. A leading hypothesis is that mucous accumulates in the lung due to improper water and salt (including chloride ion) transport across the cell surface membrane. Using our expertise in ion channels, including high-content cell assays and medicinal chemistry, we have identified selective ion channel modulators for potential application to the treatment of CF. VX-770 may work by increasing the frequency with which the CFTR channel is open, which could result in an increase in chloride transport across the cell membrane. In laboratory studies involving bronchial epithelial cells isolated from cystic fibrosis patients, our researchers have demonstrated that potentiator compounds may improve CFTR function.
In 2006, we completed a Phase 1 clinical trial of VX-770 in 63 individuals, including healthy volunteers and patients with CF. Healthy volunteers in the Phase 1 trial received escalating doses of VX-770 for treatment durations of up to 14 days, and patients with CF received single doses of VX-770. A rash was observed in some subjects during the multi-dose arm of the trial. However, we believe that the trial results support a Phase 2 clinical trial of VX-770. We plan to initiate this Phase 2 clinical trial of VX-770 in patients with CF in the second quarter of 2007.
VX-883 (gyrase inhibition for the treatment of bacterial infection)
VX-883 is a novel, Vertex-discovered dual-mechanism investigational antibiotic currently in preclinical development that targets both DNA gyrase and topoisomerase IV. DNA gyrase and topoisomerase IV are enzymes that are essential to bacteria during the replication process. DNA gyrase and topoisomerase IV inhibitors already on the market have proven to be potent, broad-spectrum antibiotics and are used to treat a variety of common Gram-positive and Gram-negative bacterial infections in various treatment settings. While existing gyrase and topoisomerase IV inhibitors work by interacting with the GyrA and ParC subunits of DNA gyrase and topoisomerase IV, VX-883 targets the GyrB and ParE subunits. VX-883 is active, in vitro, against Gram-positive and Gram-negative bacterial pathogens prevalent in both community and hospital settings, including certain pathogens that are less susceptible to other classes of antibiotics, such as agents targeting the other subunits of gyrase and topoisomerase IV. Accordingly, we believe that VX-883 may be useful in treating infections caused by drug resistant bacteria, including methicillin-resistant Staphylococcus aureus, commonly referred to as MRSA, a major and growing problem with currently marketed antibiotics. Depending on the outcomes of specific preclinical activities we are conducting in early 2007, we plan to initiate a Phase 1 clinical trial of VX-883 later in 2007.
We hold worldwide development and commercial rights to VX-883.
Collaborator-Led Programs
Lexiva/Telzir: HIV protease inhibition for the treatment of HIV/AIDS (GlaxoSmithKline plc)
Infection with HIV can lead to AIDS, a severe, life-threatening impairment of the immune system. According to the Joint United Nations Programme on HIV/AIDS, an estimated 39.5 million people worldwide were living with HIV in 2006. The United States National Institutes of Health has estimated that there may be as many as 950,000 individuals in the United States infected with HIV. There are four classes of antiviral drugs approved for the treatment of HIV infection and AIDS: nucleoside reverse transcriptase inhibitors, such as AZT and 3TC; non-nucleoside reverse transcriptase inhibitors, such as efavirenz; the fusion inhibitor enfuvirtide; and HIV protease inhibitors, or HIV PIs. HIV PIs are used as part of combination regimens for the treatment of HIV. HIV PIs block the cleavage of HIV polyproteins into active proteins, and result in the production of non-infectious viral particles. The HIV PI ritonavir has
9
been shown to significantly boost the levels of certain other PIs in the bloodstream and therefore co-administration of HIV PIs with ritonavir has become progressively more frequent in clinical practice as a strategy for achieving maximum antiviral activity, reducing the likelihood of treatment failure (viral breakthrough), and lowering the overall pill count for patients. Sales of HIV PIs in the United States exceeded $2.0 billion (excluding ritonavir) in 2006, an increase of approximately 14% from 2005. The United States market for HIV PIs is highly competitive, with a number of HIV PIs currently on the market.
Our HIV PI, fosamprenavir calcium, is marketed by our collaborator GlaxoSmithKline under the name Lexiva in the United States and under the name Telzir in the European Union. Lexiva/Telzir was co-discovered by us and GlaxoSmithKline and was developed by GlaxoSmithKline under our collaboration with them. GlaxoSmithKline has worldwide marketing rights for Lexiva/Telzir, and we have the right to conduct certain promotional and educational activities for Lexiva/Telzir in the United States and the European Union. We also have the right, which we have not exercised, to supply bulk drug substance to GlaxoSmithKline. We receive royalties on GlaxoSmithKline’s sales of Lexiva/Telzir.
Lexiva/Telzir is a prodrug of amprenavir, which also was discovered and developed under our collaboration with GlaxoSmithKline and marketed under the name Agenerase. Lexiva/Telzir has replaced Agenerase in worldwide markets. A prodrug is an inactive compound that is metabolized by the body to become the active drug. Due to the physical properties of prodrugs such as Lexiva/Telzir, it is possible to achieve a higher effective dose of the active drug for each prodrug pill administered, resulting in a smaller pill burden for patients.
In September 2006, GlaxoSmithKline announced with us at the 46th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy, or ICAAC, data from a randomized, open-label, multicenter international Phase 3b clinical trial comparing the safety and efficacy of Lexiva in combination with ritonavir to lopinavir in combination with ritonavir. The analysis of this clinical trial presented at ICAAC describes the virologic response and resistance patterns associated with virologic failure. At 48 weeks, four percent of the 434 patients in the Lexiva/ritonavir arm and five percent of the 444 patients in the lopinavir/ritonavir arm experienced virologic failure, which was defined as either a failure to achieve plasma HIV-1 RNA of less than 400c/mL by week 24, or confirmed rebound in plasma HIV-1 RNA to greater than 400 c/mL.
Lexiva was launched in the United States in late 2003, and in certain European Union countries in the third quarter of 2004. In 2006, Lexiva generated the third largest sales revenues among HIV PI inhibitors in the United States, excluding ritonavir, and it currently holds an approximate 11% share of the United States HIV PI market based on total prescriptions (also excluding ritonavir). Lexiva/Telzir is currently approved for sale in approximately 45 countries worldwide, including the United States, France, Germany, Spain, Italy, the United Kingdom and Canada.
MK-0457 (VX-680) and MK-6592 (VX-667): Aurora kinase inhibition for the treatment of cancer (Merck & Co., Inc.)
We are collaborating with Merck in the area of Aurora kinase inhibitors, including MK-0457 (VX-680), MK-6592 (VX-667) and additional potential follow-on compounds. Aurora kinases are enzymes thought to play multiple roles in the development and progression of cancer, acting as regulators of cell proliferation, transforming normal cells into cancer cells and downregulating p53, one of the body’s natural tumor suppressors. MK-0457 (VX-680) is a potent inhibitor of Aurora kinases and of flt-3 kinase, a receptor tyrosine kinase that is known to be inappropriately activated in several different types of leukemia. We believe that inhibitors of Aurora kinases may be useful as highly targeted treatments for a range of oncology indications.
As part of the collaboration, we conducted a joint research program with Merck to characterize MK-0457 (VX-680) activity across a broad range of cancer types and to identify additional drug candidates targeting the Aurora kinases. Merck holds worldwide development and commercialization rights to
10
MK-0457 (VX-680), MK-6592 (VX-667) and certain additional compounds identified during the research program.
MK-0457 (VX-680)
In December 2006, Merck initiated a pivotal Phase 2 clinical trial of MK-0457 (VX-680) in patients with treatment-resistant chronic myelogenous leukemia, or CML, and Philadelphia chromosome-positive acute lymphocytic leukemia, or Ph+ ALL, containing the T315I BCR-ABL mutation based on encouraging results from a Phase 1 clinical trial of MK-0457 (VX-680). This clinical trial is expected to enroll approximately 270 patients. In the trial, MK-0457 (VX-680) will be given as a five-day intravenous infusion every two to three weeks to evaluate both safety and efficacy.
In December 2006, Merck also reported the results of its Phase 1 clinical trial for MK-0457 (VX-680) in patients with CML, in patients with Ph+ ALL with the T315I BCR-ABL mutation and in patients with refractory JAK-2 positive myeloproliferative diseases. Merck reported that the Phase 1 dose escalation clinical trial evaluated 44 adult patients with advanced leukemias and myeloproliferative disorders who were treated with MK-0457 (VX-680) given as a five-day intravenous infusion every two-to-three weeks. Out of the 15 patients with refractory CML, nine patients had a T315I BCR-ABL mutation. Eight of these nine T315I patients had either a hematologic and/or cytogenetic response to MK-0457 (VX-680) following multiple cycles of treatment. The six patients without the T315I BCR-ABL mutation (of the 15 patients with refractory CML) did not exhibit any clinical responses to administration of MK-0457 (VX-680).
Merck also reported that two patients in the clinical trial with Ph+ ALL carrying the T315I mutation had either hematologic and/or cytogenetic responses, including one patient who had a clinical response with a full molecular remission. Six of nine patients with myeloproliferative disorders having the V617F activating mutation in JAK-2 also had clinical responses. These clinical responses were consistent with drug effects observed in leukemic cells.
The Phase 1 clinical trial of MK-0457 (VX-680) was designed primarily to evaluate the safety of VX-680 given as a five-day continuous infusion, to determine maximum tolerated dose and dose-limiting toxicities, and to assess pharmacokinetics and pharmacodynamics. Merck reported that no drug-related non-hematological toxicities were observed with MK-0457 (VX-680) in the clinical trial and therefore a maximum-tolerated dose has not yet been established. Side effects were observed in the clinical trial and included a lower white blood cell count, nausea, hair loss, and inflammation in the mouth.
Merck also is conducting a Phase 2a clinical trial of MK-0457 (VX-680) in patients with lung cancer, a Phase 1 clinical trial of MK-0457 (VX-680) in patients with recurrent or non-responsive solid tumors and a Phase 1 clinical trial of MK-0457 (VX-680) in patients with solid tumor and/or colorectal cancer.
Vertex researchers published the three-dimensional atomic structure of Aurora-A kinase in 2002, and published the structure of flt-3 kinase in January 2004. We also presented preclinical data in a number of research and medical venues in 2003 suggesting that MK-0457 (VX-680) should be further investigated to determine its potential to treat several different cancer types for which there are currently few or no available treatments. In a paper published in February 2004, our researchers reported demonstrating for the first time that a selective small molecule inhibitor of Aurora kinase MK-0457 (VX-680) inhibited tumor growth and induced tumor regression in xenograft models of human pancreatic and colon cancer. In addition, our researchers have presented data showing that MK-0457 (VX-680) prolonged survival and induced sustained remission in an oncogene-driven model of acute myelocytic leukemia.
MK-6592 (VX-667)
In 2005, Merck selected MK-6592 (VX-667), a second Aurora kinase inhibitor, for preclinical development. In December 2006, Merck initiated a Phase 1 clinical trial of MK-6592 (VX-667) in oncology patients with advanced solid tumors.
11
AVN-944 (VX-944): IMPDH inhibition for the treatment of cancer (Avalon Pharmaceuticals, Inc.)
Our collaborator Avalon Pharmaceuticals is developing AVN-944 (VX-944), an IMPDH inhibitor, for the treatment of advanced hematological malignancies, such as leukemia, lymphoma or myeloma. Inosine 5-monophosphate dehydrogenase, or IMPDH, is an enzyme thought to be critical for the synthesis of quanosine triphosphate, a molecule required for DNA synthesis and cellular signalling. IMPDH is over-expressed in many cancer cells, especially for hemotological malignancies. Reports in medical literature and presentations at scientific conferences provide a clinical rationale for the development of IMPDH inhibitors for the treatment of hematologic malignancies. Results from certain preclinical studies of AVN-944 (VX-944) indicated that AVN-944 (VX-944) inhibited the in vitro proliferation of lymphoid and myeloid cells, the principal cells involved in the most common types of human leukemias. AVN-944 (VX-944) also significantly prolonged survival in a model of aggressive mouse leukemia. In a single-dose, dose-escalation Phase 1 clinical trial of AVN-944 (VX-944) in healthy volunteers, data indicated that AVN-944 (VX-944) was orally bioavailable.
In January 2006, Avalon initiated a Phase 1 clinical trial in the United States of AVN-944 (VX-944) in patients with advanced hematological cancer. In December 2006, Avalon announced interim results from an ongoing open-label, repeat dose-escalation clinical trial designed to evaluate the safety and tolerability of AVN-944 (VX-944) in patients with advanced hematologic malignancies and to determine the optimal dose for Phase 2 clinical trials. Avalon reported that AVN-944 (VX-944) demonstrated a statistically meaningful impact on IMPDH and other certain genes that are known to be involved with cancer, including nucleotide biosynthesis, energy and metabolism, DNA replication, apoptosis and cell cycle control.
Avalon holds worldwide development and commercialization rights to AVN-944 (VX-944).
VX-409: Selective sodium channel modulation for the treatment of pain (GlaxoSmithKline plc)
GlaxoSmithKline is leading the development of VX-409, an oral, subtype-selective sodium channel modulator, and certain additional back-up compounds as potential drug candidates for the treatment of pain. VX-409 and the backup compounds were discovered through our San Diego-based ion channel research program using the capabilities and proprietary technologies that are unique to that site.
GlaxoSmithKline holds worldwide development and commercialization rights to VX-409 and certain backup compounds.
Other Programs
We have a number of additional drug candidates. These drug candidates include the following drug candidates for which we may consider entering into collaborative arrangements in order to advance their development.
VX-692 (gyrase inhibition for the treatment of bacterial infection)
VX-692 is a novel, Vertex-discovered investigational antibiotic, which targets both DNA gyrase and topoisomerase IV and is active, in vitro, against Gram-positive and Gram-negative bacterial pathogens. We are currently evaluating VX-692 in preclinical development. We hold worldwide development and commercial rights to VX-692.
VX-166 (caspase inhibition for the treatment of liver disease and sepsis)
VX-166 is a novel, Vertex-discovered inhibitor of multiple caspases. Inhibition of caspases has been shown to inhibit apoptosis, a critical component in the pathology of several liver diseases and the onset and progression of sepsis. We have demonstrated in animal models that administration of VX-166 may be useful in the treatment of such indications as acute liver disease and sepsis. We hold worldwide rights to VX-166.
12
VX-765 (ICE inhibition for the treatment of inflammatory diseases)
We discovered and have completed certain development activities with respect to two interleukin-1 converting enzyme, or ICE, inhibitors for the treatment of inflammatory diseases, VX-765 and pralnacasan. ICE is an enzyme that controls the release of active IL-1 (one of two forms of IL-1) and IL-18, which have been correlated with disease states in a number of acute and chronic inflammatory diseases. Phase 1 clinical trials of VX-765 in healthy volunteers demonstrated a dose-dependent decrease in levels of IL-18, the first time this has been demonstrated for any therapeutic agent. During 2005, we completed the clinical portion of our four week, Phase 2a clinical trial of VX-765, in 68 patients with psoriasis. We believe these results of preclinical studies and clinical trials to date warrant further investigation of VX-765.
Our first generation ICE inhibitor, pralnacasan, was developed in collaboration with Sanofi-Aventis (then Aventis). Phase 2 clinical trials of pralnacasan conducted by Aventis suggested that treatment with pralnacasan produced positive anti-inflammatory effects in patients with RA and led to dose-dependent suppression of the production of IL-1. In 2003, Aventis and Vertex voluntarily suspended the clinical development of pralnacasan pending full analysis of findings that emerged from a nine-month nonclinical toxicology study. In that nonclinical study, high doses of pralnacasan were associated with the development of fibrosis in circumscribed areas of the liver of one species of animal.
Merimepodib (VX-497) (IMPDH inhibition for the treatment of autoimmune diseases)
We currently are completing the data analysis phase of our Phase 2b clinical trial of merimepodib, an oral, small molecule inhibitor of inosine 5-monophosphate dehydrogenase, or IMPDH, for the treatment of HCV infection. The goal of this clinical trial, referred to as the METRO trial, was to evaluate the safety, pharmacokinetics and efficacy of merimepodib in combination with peg-IFN and RBV. In the HCV field, we currently are focusing our efforts on the development of direct antivirals such as telaprevir. We currently do not plan to conduct additional merimepodib clinical trials in HCV infection after the METRO trial is completed. Significant research on the IMPDH pathway has demonstrated its potential utility in transplant and other autoimmune indications.
RESEARCH PROGRAMS
We believe that our integrated drug design approach has significantly enhanced our ability to discover and develop small molecule drug candidates directed at biologically complex targets, including novel targets identified by genomic research. We believe that our approach has been validated through our ability to interest prospective collaborators in our research output and by our success in moving drug candidates into clinical trials. We have recently decided to focus on several core disease areas, in order to expand and develop our expertise and leadership in specific disease areas and to permit a framework for portfolio planning and execution. Currently, the four disease areas of highest priority to us are: infectious diseases; immunological/inflammatory diseases; cancer; and neurological diseases and disorders. These disease areas were selected by mapping our research strengths onto disease areas with high unmet need, with an emphasis on indications where we believe we, independently or in collaboration with other pharmaceutical companies, will be able to discover, develop, and commercialize transforming medicines consistent with our core purpose. Within each disease area, we intend to specialize in specific indications.
Integrated Drug Innovation Approach. Our drug design platform integrates biology, pharmacology, drug metabolism and pharmacokinetics, toxicology, material sciences, biophysics, medicinal chemistry and process chemistry, automation and information technologies in a coordinated and simultaneous fashion throughout the discovery process. The goal of our integrated, interdisciplinary approach is to make the drug discovery and development process more efficient and productive.
Focused Drug Discovery in Target-Rich Gene Families. We have pioneered a novel approach to drug discovery in target-rich gene families, which are groups of genes with similar sequences that code for structurally similar proteins. We organize and cluster targets within a gene family according to how they
13
interact with chemical inhibitors, which allows us to use high-throughput screening technologies, informatics and medicinal chemistry to rapidly identify drug-like classes of compounds in parallel for multiple targets. Along with this approach, we use a variety of biological and chemical methodologies that interrogate the function of newly discovered proteins in order to focus our drug discovery and development efforts on the most promising targets within the most promising gene families. We believe that our systematic application of this drug discovery approach is increasing the speed and efficiency of drug design efforts directed at novel biological targets, and is securing valuable intellectual property for us in gene families of interest.
Technology Platform
We employ a variety of technologies and use information from a number of different scientific disciplines as part of our integrated technology platform. The most significant of them are as follows.
· Functional Genomics. We use functional genomics techniques, such as gene knock-out mice, to help guide target selection and test the potential of chemical compounds in disease models. We also use antisense, siRNA, dominant negative cell lines, transcriptional profiling, proteomics and other biological approaches to better characterize the role played by specific targets in cellular processes.
· Biophysics. We generate atomic structural information on molecular targets using X-ray crystallography and nuclear magnetic resonance, or NMR, spectroscopy to guide design and optimization of lead classes of drugs.
· Computer-based Modeling. We apply advanced proprietary computational modeling tools to guide the evaluation and selection of compounds for synthesis. During our virtual screening process, which we refer to as in silico screening, candidate compounds are selected for synthesis and further investigation. We use proprietary algorithms to sort and filter compounds for specific properties in order to seek compounds that are more likely to become development candidates.
· Pharmacokinetics and Pharmacology. We employ a number of approaches to obtain predictive information on the bioavailability, pharmacokinetic profile and efficacy of potential drug candidates. These approaches include in vitro metabolism and toxicological studies and in vivo assessment of leads in hypothetically predictive animal models.
· Assay Development. We use modern cell biology, enzymology, and screening techniques to develop high-throughput assays which provide high-quality information to support drug discovery. We also are utilizing our assay capabilities to develop novel proprietary in vitro assays to rapidly establish ADME/toxicology profiles for compounds in our screening library.
· High-Throughput Screening. We conduct assays for most enzyme and receptor targets using very high-throughput screening approaches, many of which are proprietary. These assays enable us to rapidly generate large numbers of lead compounds and drug candidates across targets from many different gene families. These approaches integrate compound management, plate replication with miniaturized screening, hit (potential lead) identification and follow-up.
· Material Sciences. By applying advanced principles of physical chemistry and materials science at the interface between preclinical drug discovery and pharmaceutical product development, we are attempting to ensure that the drugs in our pipeline have excellent Chemistry, Manufacturing and Controls properties, or CMC properties. CMC refers to the FDA requirement that all drugs be manufactured, stored and administered in a controlled fashion. Our materials scientists study the atomic level structures of drug candidates, as well as their physical and chemical properties. Utilizing this knowledge together with molecular engineering design principles, the properties of a drug candidate are adjusted to improve the resultant active pharmaceutical ingredient’s profile. Advances in materials science have been applied with great success to the consumer products industries, the electronics industry and the medical device industry in products such as plasma televisions, computer chips and artificial bone replacements. Leveraging advanced materials science technologies in the pharmaceutical industry to the selection and development of drug
14
candidates offers the potential for a higher rate of success in clinical development, decreased time to market, and enhanced product performance.
· Instrumentation. We have a dedicated research and application development group, which is responsible for designing and building automated solutions to address many of the more repetitive and labor-intensive aspects and processes of drug discovery. For example, most of our ion channel research is conducted using E-VIPR, our proprietary screening technology that uses fluorescent probes and waves of electrical stimulation to study ion channels. E-VIPR provides an automated, high-throughput platform that enables us to collect high quality data at speeds up to a thousand times faster than patch clamping.
Current Research Programs
Our past drug discovery efforts have produced a variety of drug candidates that are currently in preclinical or clinical development. We believe our ongoing research programs continue to create potential value for us by generating new drug candidates in areas of significant unmet medical need. These programs include research targeting certain kinases, ion channels, g-protein coupled receptors and HCV protease inhibitors.
Kinase Program
We have a broad-based drug discovery effort targeting the human protein and lipid kinase family, of which there are more than 500 members. These kinases are enzymes that play a key role in transmitting signals between and within cells. Kinases exert their effect by phosphorylating other proteins, which then become activated and perform a specific function. Kinase activity has been implicated in many major diseases, including cancer and autoimmune, inflammatory, cardiovascular, metabolic, and neurological diseases. As a result, we believe that kinases are ideal targets for therapeutic intervention. The clinical success of the oncology drugs Gleevec® (imatinib mesylate) and Tarceva® (erlotinib) offer examples of how small molecule kinase inhibitors can be tailored to address specific diseases. Our extensive drug discovery efforts involving numerous targets in the kinase gene family continue to refine our understanding of kinase biology and the design of kinase inhibitors. Our researchers have determined the atomic structure of more than 25 kinase drug targets and hundreds of kinase/inhibitor co-complexes. This information is of critical importance in the design of selective inhibitors for ongoing research projects. We also have designed a diverse library of proprietary kinase inhibitors and we continue to expand that library. We apply all of these tools with the objective of determining and optimizing new chemical scaffolds against targets of interest in the area of kinase inhibition.
From 2000 through 2006, we received funding and support for our research efforts within the kinase target family under our collaboration with Novartis. In accordance with the terms of the collaboration agreement, we retain the rights to all the drug candidates and intellectual property developed under that collaboration.
In 2004, we entered into a collaboration with Merck for the development of MK-0457 (VX-680), an Aurora kinase inhibitor, in cancer, and for continuing collaborative research through mid-2006 in the area of Aurora kinase inhibition. Under this collaboration agreement, Merck is currently conducting Phase 2 clinical trials of MK-0457 (VX-680) and a Phase 1 clinical trial of MK-6592 (VX-667).
We are continuing investigation of several other kinase inhibitors that play a role in the development and progression of cancer, inflammation and autoimmune disease, infectious diseases and neurodegenerative diseases. The most advanced of these inhibitors are selective JAK3 inhibitors, which we believe have the potential to be used in treatments for transplantation, psoriasis and RA.
Membrane Target Programs (Ion Channels and GPCRs)
We are conducting a broad-based drug discovery program targeting the ion channel family. Ion channels are a gene family of more than 650 proteins that act as cellular gatekeepers, controlling the flow of ions across cell membranes. The ion channel family contains numerous drugable targets representing
15
potential therapeutic intervention points for a variety of indications, including cystic fibrosis, pain and inflammatory, cardiovascular and metabolic diseases. Existing therapies such as amlodipine and nifedipine, which are calcium channel blockers for the treatment of hypertension, and lamotrigine and carbamazepine, which are sodium channel inhibitors for the treatment of epilepsy, provide a strong rationale for developing drugs targeting ion channels.
Our ion channel research extends across several ion channel subfamilies, including sodium channels, potassium channels, calcium channels, and chloride channels and is principally focused at present on the design and development of small-molecule drugs for the treatment of pain and cystic fibrosis. For example, specific sodium channels have been shown to increase in expression and function in peripheral nerve cells at the site of injury, making them novel and attractive targets for the treatment of neuropathic pain. We are utilizing our expertise in assay development and screening to advance discovery efforts within the ion channel family.
We have an ongoing research collaboration with CFFT targeting the CFTR protein. Improper functioning of CFTR may cause the accumulation of water and salt, including chloride ions, that is believed to be the cause of mucous accumulation in the lungs of cystic fibrosis patients. This collaboration with CFFT has produced VX-770, a potentiator of CFTR function that is intended to increase the flux of chloride ions through CFTR. VX-770 is currently in clinical testing. Our continuing research efforts in this collaboration are focused on the identification of possible CFTR corrector compounds that may work to increase the number of ion channels in certain lung cell membranes of patients with CF as well as potentiator back up compounds that will increase the flux of chloride ions through CFTR.
G-protein coupled receptors, or GPCRs, represent an additional gene family comprising over 1000 distinct proteins that transduce extracellular signals to the inside of the cell and regulate a diversity of functions in both normal and pathological conditions. A study has estimated that approximately 25% of FDA-approved drugs target GPCRs. Our program is aimed at several type 1 and type 2 GPCR targets in the areas of pain and nervous system disorders.
Additional Discovery Efforts
We plan to utilize our proprietary gene family-based platform and experience in structure-based drug design to pursue targets in other medically important gene families, such as proteases. Among other things, we have significant efforts underway targeting follow up HCV protease inhibitors to strengthen our HCV research and development franchise.
CORPORATE COLLABORATIONS
We have entered into corporate collaborations with pharmaceutical and other companies and organizations that provide financial and other resources, including capabilities in research, development, manufacturing, and sales and marketing, to support our research and development programs. At present, we have the following major corporate collaborations.
Janssen Pharmaceutica, N.V.
On June 30, 2006, we entered into a license, development, manufacturing and commercialization agreement with Janssen. Under the collaboration agreement, we will collaborate with Janssen to develop and commercialize telaprevir.
Under the terms of the collaboration agreement, we will retain exclusive commercial rights to telaprevir in North America and will continue to lead the development plan for telaprevir in North America and the Janssen territories. Janssen received exclusive rights to commercialize telaprevir outside of North America and the Far East. In connection with the execution of the collaboration agreement, we received an up-front payment of $165 million in July 2006. In addition, we could receive additional contingent milestone payments, which could total up to $380 million if telaprevir is successfully developed, approved and launched. Janssen has agreed to be responsible for 50% of drug development costs under
16
the development program for North America and the Janssen territories. Each of the parties to the collaboration agreement will be responsible for drug supply in their respective territories. The collaboration agreement also includes a tiered royalty averaging a mid-20% range, as a percentage of net sales in the Janssen territories, depending upon successful commercialization. In addition, Janssen will be responsible for certain third party royalties in its territories. Janssen may terminate the collaboration agreement upon six months’ notice to us. In such an event, all manufacturing, commercialization and intellectual property rights to telaprevir under the collaboration agreement will revert to us.
As part of the collaboration agreement, following regulatory approval and commercialization of telaprevir in both North America and Janssen’s territory, we will establish with Tibotec, also a Johnson & Johnson company, a global health initiative to increase the prevention, diagnosis, treatment and cure of HCV infection, to be principally directed toward developing countries.
GlaxoSmithKline plc
In 1993, we entered into a collaboration with GlaxoSmithKline covering the research, development and commercialization of HIV protease inhibitors, including Agenerase (amprenavir), Lexiva/Telzir (fosamprenavir calcium) and brecanavir (VX-385). Under the original agreement, GlaxoSmithKline had exclusive rights to develop and commercialize our HIV PIs in all parts of the world except the Far East. In 2003, we amended the agreement to add the Far East to GlaxoSmithKline’s territory for development and commercialization of Lexiva/Telzir. GlaxoSmithKline pays us a royalty on all sales of the HIV PIs covered by the agreement. We have retained certain bulk drug manufacturing rights and certain co-promotion rights in the territories licensed to GlaxoSmithKline. We began earning a royalty from GlaxoSmithKline in 1999 on sales of Agenerase, in the fourth quarter of 2003 on sales of Lexiva, and in the third quarter of 2004 on sales of Telzir. Lexiva and Tezir have replaced Agenerase in worldwide markets.
GlaxoSmithKline initiated development of a third HIV PI discovered under the collaboration known as brecanavir (VX-385). GlaxoSmithKline bore all development costs for brecanavir, and paid us $1.0 million in development stage milestones. On December 15, 2006, GlaxoSmithKline formally notified us that it would discontinue clinical development of brecanavir, which had advanced to Phase 2 clinical trials. Currently, there are no drug candidates being developed under this collaboration, and we do not anticipate any additional milestone payments under this collaboration.
GlaxoSmithKline has the right to terminate its agreement with us without cause upon 12 months’ notice. Termination of the agreement by GlaxoSmithKline will relieve it of its obligation to make further commercialization and development milestone and royalty payments, and will end any license granted by us to GlaxoSmithKline under the agreement. In June 1996, we and GlaxoSmithKline obtained a worldwide, non-exclusive license under certain G.D. Searle & Co. (now owned by Pharmacia/Pfizer) patents in the area of HIV protease inhibition. We pay Searle a royalty based on sales of Lexiva/Telzir.
In December 2005, we entered into a separate collaboration agreement with GlaxoSmithKline for the development and commercialization of VX-409 and certain back-up compounds. Under the terms of the agreement, GlaxoSmithKline has the exclusive right and license to develop and commercialize VX-409 and the back-up compounds worldwide. The agreement provides for a $20 million up-front license payment, which was paid in December 2005, and potentially additional development and commercial milestone payments assuming the development of VX-409 and back-up compounds in major pharmaceutical markets across a range of indications. GlaxoSmithKline will also pay us royalties on annual net sales of any pharmaceutical products commercialized under the agreement. Prior to commercial launch of any drug that is covered by the agreement, GlaxoSmithKline can terminate the agreement without cause upon six months’ notice to us. Following commercial launch, GlaxoSmithKline can terminate the agreement on one year’s notice, unless the termination is the result of a safety issue associated with a drug arising from the collaboration, in which case GlaxoSmithKline may terminate immediately upon notice.
17
Merck & Co., Inc.
In June 2004, we entered into a global collaboration with Merck to develop and commercialize MK-0457 (VX-680), our lead Aurora kinase inhibitor, for the treatment of cancer, and to conduct research targeting the discovery of an additional Aurora kinase inhibitory compound or compounds to follow MK-0457 (VX-680). Merck made an up-front license payment of $20 million in June 2004, and provided research funding of $15.8 million between June 2004 and September 2006. In addition, the agreement provides for as much as $350 million in milestone payments, including up to $130 million for the successful development of MK-0457 (VX-680) in the first oncology indication and additional milestone payments for development of MK-0457 (VX-680) and follow-on compounds in subsequent major oncology indications. In 2005, Merck selected MK-6592 (VX-667), a second drug candidate covered by the collaboration, for development. Under the agreement, Merck made two milestone payments totaling $19.5 million in 2005 and three milestone payments totaling $36.3 million in 2006. Under the agreement, Merck is responsible for worldwide clinical development and commercialization of MK-0457 (VX-680) and follow-on candidates (including MK-6592 (VX-667)) and will pay us royalties on any product sales. Merck may terminate the agreement at any time without cause upon 90 days’ advance written notice, except that six months’ advance written notice is required for termination at any time when a product has marketing approval in a major market and the termination is not the result of a safety issue.
Cystic Fibrosis Foundation Therapeutics Incorporated
In May 2004, we entered into a collaboration agreement with CFFT providing funding for our late-stage cystic fibrosis drug discovery effort. The agreement subsequently was amended to extend the term of the drug discovery effort to March 31, 2008 and to include additional development stage funding for specified VX-770 development activities through the end of 2007. The agreement, as amended, provides that CFFT will pay up to $32.4 million to us for research and development activities. Under the amended agreement, we retain the right to develop and commercialize any compounds discovered in the course of the research collaboration, and we will pay a royalty to CFFT on the net sales of any drugs discovered in the collaboration. CFFT provided $21.3 million in research support payments related to the agreement in aggregate in 2004 and 2005, and the amended agreement provides for additional funding from CFFT for further research during the period from January 1, 2006 through March 31, 2008 directed toward CFTR corrector compounds. CFFT also made a $1.5 million milestone payment to us upon advancement of the first compound from the research program into clinical development. CFFT has the right to terminate the agreement without cause, effective on June 30, 2007, upon 60 days’ prior written notice.
Mitsubishi Pharma Corporation
In June 2004, we entered into a license, development and commercialization agreement with Mitsubishi for the development and commercialization of telaprevir, in Japan and certain other Far East countries. Under the terms of the agreement, Mitsubishi has the right to develop and commercialize telaprevir in its territory. Under the agreement, we are entitled to receive up to $33 million in payments from Mitsubishi through Phase 2 clinical development, including an up-front license fee, development milestone payments and contributions to certain drug development costs incurred by us for telaprevir. Further cost sharing beyond Phase 2 clinical development will be determined by Mitsubishi and us based on the design of registration trials for telaprevir. We will also be entitled to royalties on sales of telaprevir, if approved, in Mitsubishi’s territory. Mitsubishi may terminate the agreement at any time without cause upon 60 days’ prior written notice. In 2006, Mitsubishi conducted a Phase 1 clinical trial of telaprevir in the Far East. Mitsubishi is designing a Phase 2 clinical program of telaprevir in the Far East.
18
Kissei Pharmaceutical Co., Ltd.
In September 1997, we entered into a collaboration agreement with Kissei to identify and develop compounds that target p38 MAP kinase. The research phase of the collaboration ended on June 30, 2000, and we have received the full amount of research funding specified under the agreement. We are working with Kissei to develop and commercialize VX-702, which was discovered during our p38 MAP kinase research collaboration. Kissei has exclusive rights to develop and commercialize VX-702 in Japan and certain Far East countries, and co-exclusive rights (with us) in China, Taiwan and South Korea. We retain exclusive marketing rights outside the Far East. Under our agreement, Kissei will pay us development milestone payments for the successful development of VX-702 in the Far East, including $2.5 million paid in 2005 upon Kissei’s submission of regulatory filings in preparation for Phase 1 clinical trials of VX-702 in Japan. Kissei is providing a portion of the funding for our clinical trials of VX-702 in patients with RA. If VX-702 is approved for sale in Kissei’s territory, we will have the option to supply Kissei with bulk drug substance for manufacture by Kissei into drug product. We will receive drug supply payments or royalties on any product sales.
Avalon Pharmaceuticals, Inc.
In February 2005, we entered into a license agreement with Avalon for the development and commercialization of the IMPDH inhibitor AVN-944 (VX-944) for the treatment of cancer. Under the agreement, Avalon has the exclusive worldwide right and responsibility to develop and commercialize AVN-944 (VX-944) for the treatment of cancer. Avalon made a $5.0 million up-front license payment to us and has agreed to make additional milestone payments to us for the successful development of AVN-944 (VX-944) in multiple oncology indications. Avalon will pay us royalties on any product sales. The agreement provides us with certain rights to co-promote AVN-944 (VX-944). Neither party has the right to terminate the agreement other than for cause.
INTELLECTUAL PROPERTY
We actively seek protection for our products and proprietary information by means of United States and foreign patents, trademarks, and copyrights, as appropriate. In addition, we rely upon trade secret protection and contractual arrangements to protect certain of our proprietary information and products. In addition to patents and pending patent applications that relate to potential drug targets, compounds we are developing to modulate those targets, and methods of making or using those compounds, we have several patents and pending patent applications directed to proprietary elements of our drug discovery platform. For example, some of these “platform” patents and applications claim our proprietary E-VIPR ion channel screening technology.
Much of our technology and many of our processes depend upon the knowledge, experience and skills of key scientific and technical personnel. To protect our rights to our proprietary know-how and technology, we require all employees, as well as our consultants and advisors when feasible, to enter into confidentiality agreements that require disclosure and assignment to us of ideas, developments, discoveries and inventions made by these employees, consultants and advisors in the course of their service to us.
Patents and Pending Patent Applications
We hold issued patents and pending patent applications in the United States, and in foreign countries we deem appropriate, covering intellectual property developed as part of each of our advanced research, development and commercial programs. Our intellectual property holdings include but are not limited to:
· United States and foreign patents and pending foreign patent applications covering telaprevir, and many other HCV protease inhibitors.
· United States patents and pending applications covering assays useful to evaluate potential inhibitors of HCV protease, including patents and applications covering the X-ray crystal structures of HCV protease and the use of those structures to develop HCV protease inhibitors.
19
· United States and foreign patents and foreign patent applications covering a class of chemical compounds that includes VX-702 as well as compositions including VX-702 and similar compounds and the use of those compounds to treat p38 MAP kinase related disorders.
· United States and foreign patent applications covering potentiators of the CFTR protein, including VX-770 and many other related compounds, and the uses of those potentiators to treat CF.
· United States and foreign patent applications covering bacterial gyrase inhibitors including VX-883 and VX-692 and the use of these compounds for the treatment of bacterial infections.
· United States and foreign patents that cover classes of chemical compounds, pharmaceutical formulations and uses of the same for treating HIV infection and AIDS. These patents include specific coverage for fosamprenavir and its pharmaceutical formulations, methods of manufacture and methods to treat HIV infection. In addition we have a non-exclusive, worldwide license under certain patent applications claiming HIV PIs. We have an issued patent in the United States and foreign patents and foreign applications covering amprenavir and related compounds.
· United States and foreign patents and pending United States and foreign patent applications covering inhibitors of multiple kinase proteins.
· United States and foreign patent applications covering modulators of sodium ion channels and uses thereof, including VX-409 and many other related compounds.
· United States and foreign patents and pending foreign patent applications covering classes of chemical compounds, pharmaceutical compositions containing such compounds, and methods of using those compounds to treat or prevent IMPDH-mediated diseases, including HCV infection. These patents cover merimepodib (VX-497) and AVN-944 (VX-944), their combination with certain other therapeutic agents and their uses for IMPDH-mediated diseases.
· United States and foreign patents and pending foreign patent applications covering several different classes of compounds useful as inhibitors of ICE, as well as pharmaceutical compositions containing those compounds and methods of using those compounds to treat ICE-related diseases. These patents and applications include a series of patents and applications purchased from Sanofi S.A. in July 1997, including a United States patent that covers DNA sequences encoding ICE. We also have applications pending in the United States and other countries claiming VX-765, pralnacasan (VX-740) and related compounds.
· Foreign patents and pending United States and foreign patent applications covering many caspase inhibitors including VX-166.
From time to time we enter into non-exclusive license agreements for proprietary third-party technology used in connection with our research activities. These license agreements typically provide for the payment by us of a license fee, but may also include terms providing for milestone payments or royalties for the development and/or commercialization of our drug products arising from the related research. For example, we have entered into a non-exclusive license arrangement with Chiron Corporation for rights to technology in the HCV area that may provide Chiron with certain developmental milestone payments and royalty payments based on future sales of telaprevir if approved.
MANUFACTURING
As we advance our proprietary drug candidates through clinical development toward commercialization, we will be required to continue to build our manufacturing, logistics, supply chain and quality assurance resources. We currently rely on a worldwide network of third-party manufacturers to manufacture and distribute our drug candidates for clinical trials, and we expect that we will continue to do so to meet our commercial supply needs for those drugs, if they are approved for sale. Commercial manufacturing of Lexiva/Telzir is being done by GlaxoSmithKline.
20
We have retained manufacturing and commercialization responsibilities for telaprevir in North America. In 2007, we expect to undertake significant efforts to prepare for the commercial supply and marketing of telaprevir, in support of a timely and effective commercial product launch in subsequent years if we are successful in obtaining regulatory marketing approval. Establishing the commercial supply chain for telaprevir is a multi-step international endeavor involving the purchase of several raw materials, the application of certain manufacturing processes requiring significant lead times, the conversion of active pharmaceutical ingredient to tablet form and the packaging of tablets for distribution. We expect to source raw materials, drug substance and drug product, including finished packaging, from third parties located in the Far East, the European Union and the United States, and we are currently establishing and expanding third-party relationships in this regard. Establishing and providing quality assurance for this global supply chain requires a significant financial commitment, experienced personnel and the creation or expansion of numerous third-party contractual relationships. Because of the significant lead times involved in our supply chain for telaprevir, we may have less flexibility to adjust our supply in response to changes in demand than if we had shorter lead times. We have successfully completed the technical development work for the Phase 3 and commercial formulation of telaprevir. While we believe that there are multiple third parties that are capable of providing the materials and services we need in order to manufacture and distribute telaprevir, if it is approved for sale, some of these services are in high demand and capacity is constrained. As a result there can be no assurance that we will be able to establish or maintain these relationships on commercially reasonable terms.
We believe that entering into arrangements with multiple third-party manufacturers will reduce our risk of supply chain disruption by limiting our reliance on any one manufacturer. In addition, we are in the process of transferring technical information regarding the manufacture of telaprevir to Janssen so that Janssen will be able to manufacture telaprevir, if approved, for sale in Janssen’s territories and as a secondary supply source for us. There is no assurance, however, that we will be able to establish second sources for each stage of manufacturing of telaprevir or that any second source will be able to produce sufficient quantities of a particular material in the required timeframe to avoid a supply chain disruption if there is a problem with one of our suppliers.
We are focusing resources on the development of systems and processes to track, monitor and oversee our third-party manufacturers’ activities. We evaluate the performance of our third-party manufacturers and confirm their continuing capabilities to meet our needs efficiently and economically. Third-party manufacturing facilities, both foreign and domestic, are subject to inspections by or under the authority of the FDA and by or under the authority of other federal, state, local or foreign authorities. Any failure by any of our third-party manufacturers to pass any inspection could adversely affect our ability to launch telaprevir in a timely manner, if it is approved for sale, or adversely affect our ability to continue to distribute telaprevir after launch.
We have established a quality assurance program intended to ensure that our third-party manufacturers and service providers produce materials and provide services, when applicable, in accordance with the FDA’s current Good Manufacturing Practices, or cGMP, and other applicable regulations. We will need to increase our quality assurance resources in connection with the commercial launch of any drug product.
The production of our drug candidates is based in part on technology that we believe to be proprietary. Where applicable, we license this technology to our third-party manufacturers to enable them to manufacture the various forms of our drug candidates for us. However, in the course of their services, a third-party manufacturer may develop process technology related to the manufacture of our drug candidates that the manufacturer owns, either independently or jointly with us. This might increase our reliance on that manufacturer or require us to obtain a license from that manufacturer if we wish to have our drug candidates manufactured by other suppliers utilizing the same process.
21
COMPETITION
We are engaged in fields characterized by extensive research efforts, rapid technological progress and intense competition. There are many public and private companies, including pharmaceutical companies, chemical companies and biotechnology companies, engaged in developing products for the same human therapeutic applications as those we are targeting. Many of our competitors have substantially greater financial, technical and human resources than we do and are more experienced in the development of new drugs than we are. In order for us to compete successfully, we may need to demonstrate improved safety, efficacy, ease of manufacturing and market acceptance of our products over the products of our competitors that have received or will receive regulatory approval for marketing.
We face competition based on the safety and efficacy of our drug candidates, the timing and scope of regulatory approvals, the availability and cost of supply, marketing and sales capabilities, reimbursement coverage, price, patent position and other factors. Our competitors may develop or commercialize more effective, safer or more affordable products than we are able to develop or commercialize or obtain more effective patent protection. As a result, our competitors may commercialize products more rapidly or effectively than we do, which would adversely affect our competitive position, the likelihood that our drug candidates, if approved, will achieve initial market acceptance and our ability to generate meaningful revenues from those drugs. Even if our drug candidates are approved and achieve initial market acceptance, competitive products may render our drugs obsolete or noncompetitive. If any such drug is rendered obsolete, we may not be able to recover the expenses of developing and commercializing that drug. With respect to all of our drugs and drug candidates, we are aware of existing treatments and numerous drug candidates in development by our competitors.
HCV Infection
We are aware of numerous companies that are attempting to develop new treatments for HCV infection, including Schering-Plough Corporation, InterMune, Inc. in collaboration with Roche, and Gilead Sciences in collaboration with Achillion Pharmaceuticals, which are developing HCV protease inhibitors:
· Schering-Plough is developing SCH 503034, an orally available HCV protease inhibitor. SCH 503034 received “Fast Track” designation from the FDA in January 2006. Schering-Plough has reported that it is conducting a Phase 2 clinical trial with approximately 350 patients. In 2005, Schering-Plough commenced this clinical trial of SCH 503034 in combination with peg-IFN, with and without RBV, for 24 or 48 weeks in patients with chronic HCV genotype 1 who were nonresponders to previous peg-IFN and RBV combination therapy. In addition, in January 2007 Schering-Plough disclosed the initiation of a Phase 2 clinical trial of SCH 503034 in combination with peg-IFN and RBV in which it expects to enroll approximately 400 treatment-naïve patients.
· InterMune in collaboration with Roche is developing ITMN-191, a HCV protease inhibitor. In December 2006, InterMune announced the initiation of a Phase 1a clinical trial of ITMN-191 in healthy volunteers.
· Gilead Sciences and Achillion Pharmaceuticals are collaborating on the development of HCV protease inhibitors. In February 2007, Gilead and Achillion announced the discontinuation of the development of GS 9132 (ACH-806), which had been their lead HCV protease inhibitor drug candidate, based on preliminary data from a Phase 1b/2 clinical trial. In February 2007 they also announced they were evaluating additional compounds that they were investigating for the treatment of HCV infection.
22
In addition to the protease inhibitor drug candidates, companies are developing polymerase inhibitors, a drug discovery target distinct from protease inhibitors, for the treatment of HCV. The HCV polymerase is responsible for synthesizing viral RNA during HCV replication. Companies that are in Phase 2 clinical development with polymerase inhibitors include:
· Idenix Pharmaceuticals, Inc. in collaboration with Novartis is developing valopicitabine, an orally available inhibitor of HCV RNA polymerase. Idenix has two ongoing Phase 2b clinical trials, which were initiated in 2005, evaluating the combination of valopicitabine and peg-IFN in patients infected with HCV who previously failed to respond to antiviral treatment and treatment-naïve patients. In addition, in October 2006, Idenix announced that it had recently initiated an additional 12-week 90-patient Phase 2 clinical trial of valopicitabine, referred to as a drug/drug interaction trial, to assess valopicitabine when administered in combination with peg-IFN and RBV.
· ViroPharma Incorporated in collaboration with Wyeth is developing HCV-796, a non-nucleoside polymerase inhibitor, which is in Phase 2 clinical trials.
· Roche is developing R1626, a polymerase inhibitor, which is in Phase 2 clinical trials.
We are aware of numerous other compounds targeting HCV that are in clinical trials, and we believe that there are many additional potential HCV treatments in research or early development.
We believe that there is a potential for new oral drug candidates, if approved, to be administered together with or without peg-IFN and RBV. We expect that in the future we may explore the potential for other combination therapies and in particular a combination where all the necessary drugs could be administered orally.
RA
There are currently several biological tumor necrosis factor-alpha (TNF-a) inhibitors approved by the FDA for the treatment of RA including Enbrel® (etanercept), Humira® (adalimumab), and Remicade® (infliximab). Additionally, Orencia® (abatacept), an inhibitor of T-cell activation, and Rituxan® (rituximab), a monoclonal antibody, are now marketed for the treatment of RA.
There is a wide range of drug candidates in numerous classes being investigated for the treatment of RA. Examples include the IL-6 monoclonal antibody Actemra® (tocilizumab) (Roche/Chugai), and the TNF-a inhibitor certolizumab (UCB). There are also a number of p38 MAP kinase inhibitors under investigation for the potential treatment of RA. In addition to Vertex, other companies with open or planned Phase 2 p38 MAP programs in RA include GlaxoSmithKline, Pfizer, and Roche.
CF
Several companies are engaged in the process of developing treatments for CF. For example, PTC Therapeutics, Inc. recently has an ongoing Phase 2 clinical trials for PTC124, a drug candidate that targets nonsense genetic mutations that can cause cystic fibrosis in some populations. Altus Pharmaceuticals, Inc. is developing ALTU-135, an orally-delivered enzyme replacement therapy for the treatment of pancreatic insufficiency, a condition that affects many CF patients. Inspire Pharmaceuticals Inc. currently is conducting Phase 3 clinical trials of denufosol tetrasodium, an inhaled molecule that activates an alternate chloride channel in the airway of patients with CF.
HIV
The United States market for HIV PIs is highly competitive, with a number of competitive PIs currently on the market. The two leading HIV PIs in the United States are Bristol-Myers Squibb Company’s Reyataz® and Abbott Laboratories’ Kaletra®. In 2006, Lexiva was the third largest (measured in terms of sales revenue) HIV PI inhibitor in the United States, excluding ritonavir, and it currently holds
23
an approximate 11% share of the United States HIV PI market based on total prescriptions (also excluding ritonavir).
In the field of HIV protease inhibition, Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Inc., Johnson & Johnson and Pfizer Inc., among others, have other HIV PIs drug candidates in various stages of development. In addition to the currently marketed protease inhibitors, each of these compounds and others that may be in research or development may eventually compete with Lexiva/Telzir.
GOVERNMENT REGULATION
Our development, manufacture and potential sale of therapeutics are subject to extensive regulation by United States and foreign governmental authorities. In particular, pharmaceutical products are subject to rigorous preclinical, nonclinical and clinical testing and to other approval requirements by the FDA in the United States under the Food, Drug and Cosmetic Act, and by comparable agencies in most foreign countries. In addition to prohibiting the sale and distribution of pharmaceutical products prior to regulatory approval, the FDA and comparable agencies in most foreign countries prohibit the pre-approval promotion of investigational drugs as being safe and effective for the indication for which it is under investigation. Although we have summarized the FDA process below, other countries may have different approval processes with which we will need to comply if we seek to conduct clinical trials or obtain marketing approval in those countries. In addition, even if we ultimately intend to seek initial marketing approval in the United States, we may conduct early clinical trials in other countries, for a variety of reasons, and therefore the submission of our initial IND in the United States may not occur until after one or more foreign-sited clinical trials have been initiated.
Approval Process
As an initial step in the FDA regulatory approval process, toxicity studies in animals and other studies typically are conducted to help identify potential safety problems that might be associated with administration of the drug candidate being tested over the period of time planned for the initial human trials. For certain diseases, animal models exist that are believed to be predictive of efficacy in humans. For such diseases, a drug candidate typically is tested for efficacy in that animal model. The results of these nonclinical safety and disease model studies are submitted to the FDA as a part of the IND submission, which is submitted prior to commencement of human clinical testing in the United States. For several of our drug candidates, no appropriately predictive animal model exists. As a result, no in vivo evidence of efficacy will be available until those drug candidates progress to human clinical trials. A variety of nonclinical studies in a number of animal species, and other nonclinical studies, ordinarily are conducted while human clinical trials are underway, to provide supplemental toxicology and other information. This information as well as the results from the early clinical trials will provide a foundation for the design of broader and more lengthy human clinical trials.
Clinical trials typically are conducted in three sequential phases, although the phases may overlap. Phase 1 frequently begins with the initial introduction of the drug candidate into healthy human subjects prior to introduction into patients. The drug candidate may then be tested in a relatively small number of patients for safety on a preliminary basis, dosage tolerance, absorption, bioavailability, biodistribution, metabolism, excretion, clinical pharmacology and, if possible, for early information on effectiveness. Phase 2 typically involves trials in a small sample of the intended patient population to assess the efficacy of the drug for a specific indication, to determine dose tolerance and the optimal dose range and to gather additional information relating to safety and potential adverse effects. Phase 3 trials are undertaken to further evaluate clinical safety and efficacy in an expanded patient population at geographically dispersed trial sites, to determine the overall risk-benefit ratio of the drug candidate and to provide an adequate basis for proposed physician labeling. Each trial is conducted in accordance with standards set forth in protocols that detail the objectives of the trial, the parameters to be used to monitor safety and the efficacy criteria to be evaluated. For clinical trials in the United States, each protocol must be submitted to the FDA as part of the IND submission. Further, each clinical trial must be evaluated by an independent
24
Institutional Review Board, or IRB, at each institution at which the trial will be conducted. The IRBs will consider, among other things, ethical factors, the safety of human subjects and the possible liability of the institution.
Data from nonclinical testing and clinical trials are submitted to the FDA in a New Drug Application, or NDA, for United States marketing approval. The process of completing nonclinical and clinical testing and obtaining FDA approval for a new drug is likely to take a number of years and require the expenditure of substantial resources. Preparing an NDA involves considerable data collection, verification, analysis and expense, and there can be no assurance that approval will be granted on a timely basis, if at all. The approval process is affected by a number of factors, including the severity of the targeted disease, the availability of alternative treatments and the risks and benefits demonstrated in clinical trials. The FDA may deny an NDA if applicable regulatory criteria are not satisfied or may require additional testing or information. Among the conditions for marketing approval is the requirement that the prospective manufacturer’s quality control and manufacturing procedures conform to the FDA’s cGMP regulations, which must be followed at all times. In complying with standards set forth in these regulations, manufacturers must continue to expend time, money and effort in the area of production and quality control to ensure full technical compliance. Manufacturing establishments, both foreign and domestic, also are subject to inspections by or under the authority of the FDA and by or under the authority of other federal, state, local agencies or foreign authorities.
Under the FDA Modernization Act of 1997, the FDA may grant “Fast Track” designation to facilitate the development of a drug intended for the treatment of a serious or life-threatening condition if the drug demonstrates, among other things, the potential to address an unmet medical need. The benefits of Fast Track designation include scheduled meetings with the FDA to receive input on development plans, the option of submitting an NDA in sections (rather than submitting all components simultaneously), and the option of requesting evaluation of trials using surrogate endpoints. Fast Track designation does not necessarily lead to a priority review or accelerated approval of a drug candidate by the FDA. Telaprevir and VX-770 have received Fast Track designation by the FDA.
Timing to Approval
We estimate that it generally takes 10 to 15 years, or possibly longer, to discover, develop and bring to market a new pharmaceutical product in the United States as outlined below:
Phase: | | | | Objective: | | Estimated Duration: | |
Discovery | | Lead identification and target validation | | 2 to 4 years | |
Preclinical | | Initial toxicology for preliminary identification of risks for humans; gather early pharmacokinetic data | | 1 to 2 years | |
Phase 1 | | Evaluate safety in humans; study how the drug candidate works, metabolizes and interacts with other drugs | | 1 to 2 years | |
Phase 2 | | Establish effectiveness of the drug candidate and its optimal dosage; continue safety evaluation | | 2 to 4 years | |
Phase 3 | | Confirm efficacy, dosage regime and safety profile of the drug candidate; submit NDA | | 2 to 4 years | |
FDA approval | | Approval by the FDA to sell and market the drug for the approved indication | | 6 months to 2 years | |
A drug candidate may fail at any point during this process. Animal and other nonclinical studies typically are conducted during each phase of human clinical trials.
Post-approval Studies
Even after FDA approval has been obtained, further studies, including post-approval trials, may be required to provide additional data on safety and will be required to gain approval for the sale of a product as a treatment for clinical indications other than those for which the product initially was approved. Also,
25
the FDA will require post-approval reporting to monitor the side effects of the drug. Results of post-approval programs may limit or expand the indications for which the drug product may be marketed. Further, if there are any requests for modifications to the initial FDA approval for the drug, including changes in indication, manufacturing process, labeling or manufacturing facilities, a supplemental NDA may be required to be submitted to the FDA or we may elect to seek changes and submit a supplemental NDA to obtain approval.
Other Regulations
Pursuant to the Drug Price Competition and Patent Term Restoration Act of 1984, under certain conditions a sponsor may be granted marketing exclusivity for a period of five years following FDA approval. During this period, third parties would not be permitted to obtain FDA approval for a similar or identical drug through an Abbreviated NDA, which is the application form typically used by manufacturers seeking approval of a generic drug. The statute also allows a patent owner to extend the term of the patent for a period equal to one-half the period of time elapsed between the submission of an IND and the filing of the corresponding NDA plus the period of time between the filing of the NDA and FDA approval. We intend to seek the benefits of this statute, but there can be no assurance that we will be able to obtain any such benefits.
Whether or not FDA approval has been obtained, approval of a drug product by regulatory authorities in foreign countries must be obtained prior to the commencement of commercial sales of the product in such countries. Historically, the requirements governing the conduct of clinical trials and product approvals, and the time required for approval, have varied widely from country to country.
The FDA may grant orphan drug designation to drugs intended to treat a “rare disease or condition” that affects fewer than 200,000 individuals in the United States. Orphan drug designation must be requested before submitting an application for marketing authorization. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. If a product that has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Also, competitors may receive approval of different drugs or biologics for the indications for which the orphan product has exclusivity.
Pharmaceutical companies also are subject to various federal and state laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws make it illegal for any entity or person to solicit, offer, receive, or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented, for payment to third party payors, including Medicare and Medicaid, claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services.
In addition to the statutes and regulations described above, we are also subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other federal, state, local and foreign regulations, now or hereafter in effect.
26
OTHER MATTERS
Employees
As of December 31, 2006, we had 962 employees (945 full time, 17 part time), including 707 in research and development and 255 in general and administrative functions. The number of our full time employees increased by 17% during 2006, from 806 on December 31, 2005. We expect to increase further our headcount in 2007 as we invest in expanding our drug development and commercialization capabilities. Of our employees, 84 were located at our U.K. research and development facilities, 163 were located at our facility in San Diego, California and 715 were based at our Cambridge, Massachusetts headquarters. Our scientific staff members have diversified experience and expertise in molecular and cell biology, biochemistry, animal pharmacology, synthetic organic chemistry, protein X-ray crystallography, protein nuclear magnetic resonance spectroscopy, microbiology, computational chemistry, biophysical chemistry, medicinal chemistry, clinical pharmacology and clinical medicine. Our employees are not covered by a collective bargaining agreement, and we consider our relations with our employees to be good.
Information Available on the Internet
Our internet address is www.vrtx.com. Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and all amendments to those reports, are available to you free of charge through the “Investors” section of our website as soon as reasonably practicable after those materials have been electronically filed with, or furnished to, the Securities and Exchange Commission.
Corporate Information
Vertex was incorporated in Massachusetts in 1989, and our principal executive offices are located at 130 Waverly Street, Cambridge, Massachusetts 02139. We have research sites located in San Diego, California, Iowa City, Iowa and Milton Park, U.K.
27
EXECUTIVE OFFICERS AND DIRECTORS
The names, ages and positions held by our executive officers and directors are as follows:
Name | | | | Age | | Position |
Joshua S. Boger, Ph.D. | | 55 | | President and Chief Executive Officer |
John J. Alam, M.D. | | 45 | | Executive Vice President, Medicines Development, and Chief Medical Officer |
Victor A. Hartmann, M.D. | | 57 | | Executive Vice President, Strategic and Corporate Development |
Peter Mueller, Ph.D. | | 50 | | Executive Vice President, Drug Innovation and Realization, and Chief Scientific Officer |
Ian F. Smith, C.P.A., A.C.A. | | 41 | | Executive Vice President and Chief Financial Officer |
Kenneth S. Boger, M.B.A., J.D. | | 60 | | Senior Vice President and General Counsel |
Richard C. Garrison | | 58 | | Senior Vice President and Catalyst |
Johanna Messina Power, C.P.A | | 34 | | Vice President and Corporate Controller |
Eric K. Brandt | | 44 | | Director |
Roger W. Brimblecombe, Ph.D., D.Sc. | | 77 | | Director |
Stuart J.M. Collinson, Ph.D. | | 47 | | Director |
Eugene H. Cordes, Ph.D. | | 70 | | Director |
Matthew W. Emmens | | 55 | | Director |
Bruce I. Sachs | | 47 | | Director |
Charles A. Sanders, M.D. | | 75 | | Director |
Eve E. Slater, M.D., F.A.C.C. | | 61 | | Director |
Elaine S. Ullian | | 59 | | Director |
Dr. Joshua Boger is a founder of Vertex. He has been Chief Executive Officer since 1992. He was our Chairman of the Board from 1997 until May 2005. He was our President from our inception in 1989 until December 2000, and was again appointed our President in 2005. He was our Chief Scientific Officer from 1989 until May 1992. Dr. Boger has been a director since Vertex’s inception. Prior to founding Vertex in 1989, Dr. Boger held the position of Senior Director of Basic Chemistry at Merck Sharp & Dohme Research Laboratories in Rahway, New Jersey, where he headed both the Department of Medicinal Chemistry of Immunology & Inflammation and the Department of Biophysical Chemistry. Dr. Boger holds a B.A. in chemistry and philosophy from Wesleyan University and M.S. and Ph.D. degrees in chemistry from Harvard University. Dr. Boger is the brother of Mr. Kenneth Boger, our Senior Vice President and General Counsel.
Dr. Alam currently serves as our Executive Vice President, Medicines Development, and Chief Medical Officer, a position he has held since February 2006. From January 2001 to February 2006, he served as our Senior Vice President of Drug Evaluation and Approval. From October 1997 to January 2001, he was our Vice President of Clinical Development. From 1991 to 1997, Dr. Alam held a variety of positions with Biogen, Inc., including Director of Medical Research and Program Executive (beta interferon) for Avonex. Prior to joining Biogen, Dr. Alam was a Research Fellow at the Dana Farber Cancer Institute and completed an internal medicine residency at The Brigham and Women’s Hospital in
28
Boston. Dr. Alam holds an M.D. from Northwestern University Medical School and an S.B. in chemical engineering from the Massachusetts Institute of Technology.
Dr. Hartmann, our Executive Vice President, Strategic and Corporate Development, joined Vertex in February 2005. From 2000 to 2005, Dr. Hartmann served as the Senior Vice President, Global Business Development and Licensing, for Novartis Pharma AG. He served as Vice President, Head of Scientific and Business Evaluation at Novartis from 1999 to 2000. Dr. Hartmann served as Vice President and Head of Global Project Management of Sandoz Pharma Ltd. (later Novartis AG) from 1995 to 1999. Dr. Hartmann joined Sandoz as Vice President and Head of Clinical Research and Development in 1994. Dr. Hartmann received his medical degree from the University of Bonn, Germany, and a bachelor’s degree from Macalester College.
Dr. Mueller currently serves as our Executive Vice President, Drug Innovation and Realization, and Chief Scientific Officer, a position he has held since February 2006. From July 2003 to February 2006, Dr. Mueller was our Chief Scientific Officer and Senior Vice President, Drug Discovery and Innovation. Prior to joining Vertex, Dr. Mueller was the Senior Vice President, Research and Development, of Boehringer Ingleheim Pharmaceuticals, Inc., with responsibility for the development of all drug candidates in the company’s worldwide portfolio in North America. He led research programs in the areas of immunology, inflammatory cardiovascular disease and gene therapy on a global basis. During his time with Boehringer Ingelheim, Dr. Mueller oversaw the discovery of numerous development candidates and held several positions in basic research, medicinal chemistry and management. Dr. Mueller received both an undergraduate degree and a Ph.D. in chemistry at the Albert Einstein University of Ulm, Germany, where he also holds a Professorship in Theoretic Organic Chemistry. He completed fellowships in quantum pharmacology at Oxford University and in biophysics at Rochester University.
Mr. Smith currently serves as our Executive Vice President and Chief Financial Officer, a position he has held since February 2006. From November 2003 to February 2006, he was our Senior Vice President and Chief Financial Officer, and from October 2001 to November 2003, he served as our Vice President and Chief Financial Officer. Prior to joining Vertex, Mr. Smith served as a partner in the Life Science and Technology Practice Group of Ernst & Young LLP, an accounting firm, from 1999 to 2001. Mr. Smith initially joined Ernst & Young’s U.K. firm in 1987, and then joined its Boston office in 1995. Mr. Smith currently is a member of the Board of Directors of Acorda Therapeutics, Inc, Epix Pharmaceuticals, Inc. and TolerRx Inc. Mr. Smith holds a B.A. in accounting and finance from Manchester Metropolitan University, U.K., is a member of the American Institute of Certified Public Accountants and is a Chartered Accountant of England and Wales.
Mr. Kenneth Boger currently serves as our Senior Vice President and General Counsel, a position he has held since 2001. He came to Vertex from the law firm of Kirkpatrick & Lockhart LLP, now known as Kirkpatrick & Lockhart Preston Gates Ellis LLP, where he was a partner specializing in business and corporate law and was a member of the firm’s Management Committee. Prior to the merger of Kirkpatrick & Lockhart with the Boston law firm of Warner & Stackpole LLP in 1999, Mr. Boger was a partner at Warner & Stackpole, where he served on its Executive Committee from 1988 to 1997. Mr. Boger holds an A.B. in history from Duke University, an M.B.A. from the Graduate School of Business at the University of Chicago, and a J.D. from Boston College Law School. Mr. Boger is the brother of Dr. Joshua Boger, our President and Chief Executive Officer.
Mr. Garrison, our Senior Vice President and Catalyst, joined Vertex in that role in December 2005. From June 2001 to December 2005, Mr. Garrison was the founder and President of Bink Inc., a strategic consulting firm. Prior to that, Mr. Garrison was the Chairman and CEO of Ingalls, Quinn & Johnson, one of New England’s largest advertising agencies, for 18 years. Mr. Garrison holds a B.A. in English from Princeton University.
Ms. Messina Power currently serves as our Vice President and Corporate Controller. Since joining the Company in 1999, Ms. Messina Power also has served us as Assistant Corporate Controller (from 1999 to 2000) and Corporate Controller (from 2000 to 2006). Prior to joining us, Ms. Messina Power was employed
29
as an accountant by PricewaterhouseCoopers LLP, an accounting firm, from 1995 to 1999. She holds a B.S. in accounting from Boston College, and is a Certified Public Accountant.
Mr. Brandt has been a member of our Board of Directors since 2003. He is the President, Chief Executive Officer and a member of the Board of Directors of Avanir Pharmaceuticals, which he joined in 2005. Prior to joining Avanir, Mr. Brandt held various positions at Allergan Inc. from 1999 to 2005, including Executive Vice President, Finance and Technical Operations and Chief Financial Officer from February 2005 to September 2005, Executive Vice President, Finance, Strategy and Business Development, and Chief Financial Officer from 2003 until February 2005, and Corporate Vice President and Chief Financial Officer from May 1999 to 2003. From January 2001 to January 2002, he also assumed the duties of President, Global Consumer Eye Care Business, at Allergan. Prior to that, he held various positions with the Boston Consulting Group, most recently serving as Vice President and Partner, and a senior member of the BCG Health Care practice. Mr. Brandt also currently serves as a director of Dentsply International Inc. Mr. Brandt holds a B.S. in chemical engineering from the Massachusetts Institute of Technology and an M.B.A. from Harvard University.
Dr. Brimblecombe has been a member of our Board of Directors since 1993 and a member of the Board of Vertex Pharmaceuticals (Europe) Ltd. since 2005. He served as Chairman of Vanguard Medica plc from 1991 to 2000, of Core Group plc from 1997 to 1999, of Oxford Asymmetry International plc from 1997 to 2000 and pSivida Ltd. From 2002 to 2007. From 1979 to 1990, he held various Vice Presidential posts in SmithKline & French Laboratories’ research and development organization, including Vice President R&D for Europe and Japan. He is currently a Partner in MVM Life Science Partners LLP and a director of Tissue Science Laboratories plc (listed on the AIM market in the United Kingdom). He holds Ph.D. and D.Sc. degrees in pharmacology from the University of Bristol, England.
Dr. Collinson has been a member of our Board of Directors since July 2001. He currently serves as a Partner at Forward Ventures. Prior to our merger with Aurora Biosciences Corporation in 2001, Dr. Collinson served as the President, Chief Executive Officer and Chairman of the Board of Aurora. Dr. Collinson held senior management positions with Glaxo Wellcome from December 1994 to June 1998, most recently serving as Co-Chairman, Hospital and Critical Care Therapy Management Team and Director of Hospital and Critical Care. Dr. Collinson received his Ph.D. in physical chemistry from the University of Oxford, England and his M.B.A. from Harvard University.
Dr. Cordes has been a member of our Board of Directors since 2005, and a member of the Company’s Scientific Advisory Board since 1996. Dr. Cordes was the Chairman of Vitae Pharmaceuticals, Inc., a position he held from January 2002 to March 2006. Prior to joining Vitae Pharmaceuticals, Dr. Cordes was a professor of pharmacy at the University of Michigan. Dr. Cordes received a B.S. degree in chemistry from the California Institute of Technology and a Ph.D. in biochemistry from Brandeis University.
Mr. Emmens has been a member of our Board of Directors since 2004. Mr. Emmens is the Chief Executive Officer, Chairman of the Executive Committee and a member of the Board of Directors of Shire Pharmaceuticals Group plc. Before joining Shire in 2003, Mr. Emmens served as president of Merck KGaA’s global prescription pharmaceuticals business in Darmstadt, Germany. In 1999, he joined Merck KGaA and established EMD Pharmaceuticals, its United States prescription pharmaceutical business. Mr. Emmens held the position of President and Chief Executive Officer at EMD Pharmaceuticals from 1999 to 2001. Prior to this, Mr. Emmens held various positions, including Chief Executive Officer, at Astra Merck, Inc. as well as several positions at Merck & Co., Inc. Mr. Emmens also currently serves as a director of Incyte Corporation. Mr. Emmens received a B.S. degree in business management from Farleigh Dickinson University.
Mr. Sachs has been a member of our Board of Directors since 1998. He is a General Partner at Charles River Ventures. From 1998 to 1999, he served as Executive Vice President and General Manager of Ascend Communications, Inc. From 1997 until 1998, Mr. Sachs served as President and CEO of Stratus Computer, Inc. From 1995 to 1997, he served as Executive Vice President and General Manager of the Internet Telecom Business Group at Bay Networks, Inc. From 1993 to 1995, he served as President and
30
Chief Executive Officer at Xylogics, Inc. Mr. Sachs also currently serves as a director of BigBand Networks, Inc. Mr. Sachs holds a B.S.E.E. in electrical engineering from Bucknell University, an M.E.E. in electrical engineering from Cornell University, and an M.B.A. from Northeastern University.
Dr. Sanders has been a member of our Board of Directors director since 1996, has served as our lead outside director since 2003 and has served as our Chairman since May 2006. He retired in 1994 as Chief Executive Officer and in 1995 as Chairman of Glaxo Inc. From 1990 to 1995, he served as a member of the board of Glaxo plc. From 1981 to 1989, Dr. Sanders held a number of positions at Squibb Corporation, including that of Vice Chairman. Dr. Sanders has previously served on the boards of Merrill Lynch, Reynolds Metals Co., Morton International Inc., Fisher Scientific International and Biopure Corporation. He is also a director of Cephalon Corporation, Genentech, Inc. and Icagen, Inc. Dr. Sanders had his undergraduate education at University of Texas, and earned an M.D. from the University of Texas Southwestern Medical School.
Dr. Slater has been a member of our Board of Directors since 2004. Dr. Slater is board-certified in internal medicine and cardiology and has extensive experience in the pharmaceutical industry, including 19 years in senior management positions at Merck Research Laboratories. Most recently, she was Assistant Secretary for Health at the United States Department of Health and Human Services, or HHS, where she served as Health and Human Services Secretary Tommy Thompson’s chief health policy advisor. Prior to joining HHS, Dr. Slater held senior management positions at Merck Research Laboratories from 1983 to 2001, including Senior Vice President of External Policy, Vice President of Corporate Public Affairs, Senior Vice President of Clinical and Regulatory Development, Executive Director of Biochemistry and Molecular Biology and Senior Director of Biochemical Endocrinology. Dr. Slater also serves as a director of VaxGen, Inc., Phase Forward Incorporated and Theravance, Inc. Dr. Slater is a graduate of Vassar College and received her M.D. from Columbia University’s College of Physicians and Surgeons.
Ms. Ullian has been a member of our Board of Directors since 1997. Since 1996, she has served as President and Chief Executive Officer of Boston Medical Center. From 1994 to 1996, she served as President and Chief Executive Officer of Boston University Medical Center Hospital. From 1987 to 1994, Ms. Ullian served as President and Chief Executive Officer of Faulkner Hospital. She also serves as a director of Thermo Fisher Scientific Inc. and Valeant Pharmaceuticals, Inc. Ms. Ullian holds a B.A. in political science from Tufts University and a M.P.H. from the University of Michigan.
31
SCIENTIFIC ADVISORY BOARD
Our Scientific Advisory Board consists of individuals with demonstrated expertise in various fields who advise us concerning long-term scientific planning, research and development. The Scientific Advisory Board also evaluates our research programs, recommends personnel to us and advises us on technological matters. The members of the Scientific Advisory Board, which is chaired by Dr. Mark Murcko, our Chief Technology Officer, are:
Mark Murcko, Ph.D. | | Vice President and Chief Technology Officer, Vertex Pharmaceuticals Incorporated |
Peter Mueller, Ph.D. | | Chief Scientific Officer and Executive Vice President, Drug Innovation and Realization, Vertex Pharmaceuticals Incorporated |
Paul S. Anderson, Ph.D. | | Retired Vice President, Drug Discovery, Bristol-Myers Squibb Company |
Steven J. Burakoff, M.D. | | Laura and Isaac Perlmutter Professor, New York University School of Medicine; Director, New York University Cancer Institute; Director, Skirball Institute of Biomolecular Medicine, New York University School of Medicine |
Lewis C. Cantley, Ph.D. | | Chief, Division of Signal Transduction at Beth Israel Deaconess Medical Center and the Harvard Institutes of Medicine |
Eugene H. Cordes, Ph.D. | | Chairman, Vitae Pharmaceuticals, Inc.; former Professor of Pharmacy, College of Pharmacy and Adjunct Professor of Chemistry, College of Literature, Science and the Arts, University of Michigan, Ann Arbor |
Stephen C. Harrison, Ph.D. | | Higgins Professor of Biochemistry, Harvard University; Investigator, Howard Hughes Medical Institute; Professor of Biological Chemistry and Molecular Pharmacology and Professor of Pediatrics, Harvard Medical School |
Jeremy R. Knowles, D. Phil. | | Amory Houghton Professor of Chemistry and Biochemistry, Harvard University |
Robert T. Schooley, M.D. | | Professor and Head of the Division of Infectious Diseases, University of California, San Diego |
Roger Tsien, Ph.D. | | Investigator, Howard Hughes Medical Institute; Professor of Pharmacology and Professor of Chemistry and Biochemistry, University of California, San Diego |
Other than Dr. Murcko and Dr. Mueller, who are employed by Vertex, and Dr. Cordes, who is a member of our Board of Directors, none of the members of the Scientific Advisory Board is employed by Vertex, and members may have other commitments to or consulting or advisory contracts with their employers or other entities that may restrict their availability. Accordingly, such persons are expected to devote only a small portion of their time to us. In addition to our Scientific Advisory Board, we have established consulting relationships with a number of scientific and medical experts who advise us on a project-specific basis.
32
ITEM 1A. RISK FACTORS
Risk Factors
Investing in our common stock involves a high degree of risk, and you should carefully consider the risks and uncertainties described below in addition to the other information included or incorporated by reference in this Annual Report on Form 10-K. If any of the following risks actually occurs, our business, financial condition or results of operations would likely suffer, possibly materially. In that case, the trading price of our common stock could fall.
WE EXPECT TO INCUR FUTURE LOSSES, AND WE MAY NEVER BECOME PROFITABLE.
We have incurred significant operating losses each year since our inception, including net losses of $206.9 million, $203.4 million and $166.2 million during 2006, 2005 and 2004, respectively, and expect to incur a significant operating loss in 2007. We believe that operating losses will continue beyond 2007, even if we receive significant future payments under our existing and future collaborative agreements, because we are planning to make significant investments in research and development, and because we will incur significant selling, general and administrative expenses in the course of researching, developing and commercializing our drug candidates. These net losses have had and will continue to have an adverse effect on, among other things, our stockholders’ equity, total assets and working capital. We expect that losses will fluctuate from quarter to quarter and year to year, and that such fluctuations may be substantial. We cannot predict when we will become profitable, if at all.
WE DEPEND HEAVILY ON THE SUCCESS OF OUR LEAD DRUG CANDIDATE, TELAPREVIR, WHICH IS STILL UNDER DEVELOPMENT. IF WE ARE UNABLE TO COMMERCIALIZE TELAPREVIR, OR EXPERIENCE SIGNIFICANT DELAYS IN DOING SO, OUR BUSINESS WILL BE MATERIALLY HARMED.
We are investing a significant portion of our time, personnel and financial resources in the development of telaprevir, which is currently in Phase 2b clinical trials. The clinical development and commercial success of telaprevir will depend on several factors, including the following:
· successful completion and favorable outcome of clinical trials;
· ongoing discussions with the FDA and comparable foreign authorities regarding the scope and design of our clinical trials;
· receipt and timing of marketing approvals for telaprevir from the FDA and similar foreign regulatory authorities;
· establishing and maintaining commercial manufacturing arrangements for telaprevir with third-party manufacturers;
· launching commercial sales of telaprevir by us and our collaborators; and
· acceptance of telaprevir, if approved, in the medical community and with third-party payors.
If the data from our ongoing clinical trials or non-clinical studies regarding the safety or efficacy of telaprevir are not favorable, we may be forced to delay or terminate the clinical development of telaprevir, which would materially harm our business. Further, even if we gain marketing approvals from the FDA and similar foreign regulatory authorities in a timely manner, we cannot be sure that telaprevir will be accepted by purchasers in the pharmaceutical market. If the results of clinical trials of telaprevir, the anticipated or actual timing of marketing approvals for telaprevir, or the market acceptance of telaprevir, if approved, do not meet the expectations of investors or public market analysts, the market price of our common stock would likely decline.
33
MANY OF OUR DRUG CANDIDATES ARE STILL IN THE EARLY STAGES OF DEVELOPMENT, AND ALL OF OUR DRUG CANDIDATES REMAIN SUBJECT TO CLINICAL TESTING AND REGULATORY APPROVAL. IF WE ARE UNABLE TO SUCCESSFULLY DEVELOP AND TEST OUR DRUG CANDIDATES, WE WILL NOT BE SUCCESSFUL.
The success of our business depends primarily upon our ability, and our collaborators’ ability, to develop and commercialize our drug candidates, including telaprevir, successfully. Our drug candidates are in various stages of development and must satisfy rigorous standards of safety and efficacy before they can be approved by the FDA or other regulatory authorities for sale. To satisfy these standards, we and/or our collaborators must engage in expensive and lengthy testing of our drug candidates. Despite our efforts, our drug candidates may not:
· offer therapeutic or other improvement over existing competitive drugs;
· be proven safe and effective in clinical trials;
· meet applicable regulatory standards;
· be capable of being produced in commercial quantities at acceptable costs; or
· if approved for commercial sale, be successfully commercialized.
Positive results in preclinical studies of a drug candidate may not be predictive of similar results in humans during clinical trials, and promising results from earlier clinical trials of a drug candidate may not be replicated in later clinical trials. Findings in nonclinical studies conducted concurrently with clinical trials could result in abrupt changes in our development activities, including the possible cessation of development activities associated with a drug candidate. Furthermore, results from our clinical trials may not meet the level of statistical significance required by the FDA or other regulatory authorities for approval of a drug candidate.
We and many other companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in early-stage development. Accordingly, the results from the completed preclinical studies and clinical trials and ongoing clinical trials for our drug candidates may not be predictive of the results we may obtain in later stage trials, and may not be predictive of the likelihood of approval of a drug candidate for commercial sale.
IF WE ARE UNABLE TO OBTAIN UNITED STATES AND/OR FOREIGN REGULATORY APPROVAL, WE WILL BE UNABLE TO COMMERCIALIZE OUR DRUG CANDIDATES.
Our drug candidates are subject to extensive governmental regulations relating to their development, clinical trials, manufacturing and commercialization. Rigorous preclinical testing and clinical trials and an extensive regulatory approval process are required in the United States and in most other countries prior to the commercial sale of our drug candidates. Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays. It is possible that none of the drug candidates we are developing independently, or in collaboration with others, will be approved for marketing.
We have limited experience in conducting and managing the late-stage clinical trials necessary to obtain regulatory approvals, including approval by the FDA. The time required to complete clinical trials and for the FDA and other countries’ regulatory review processes is uncertain and typically takes many years. Our analysis of data obtained from preclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. We may also encounter unanticipated delays or increased costs due to government regulation from future legislation or administrative action or changes in FDA policy during the period of drug development, clinical trials and FDA regulatory review.
Any delay in obtaining or failure to obtain required approvals could materially adversely affect our ability to successfully commercialize any drug candidate. Furthermore, any regulatory approval to market a drug may be subject to unexpected limitations on the indicated uses for which we may market the drug. These limitations may limit the size of the market for the drug.
34
We are also subject to numerous foreign regulatory requirements governing the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process includes all of the risks associated with the FDA approval process described above, as well as risks attributable to the satisfaction of foreign requirements. Approval by the FDA does not ensure approval by regulatory authorities outside the United States. Foreign jurisdictions may have different approval procedures than those required by the FDA and may impose additional testing requirements for our drug candidates.
IF CLINICAL TRIALS FOR OUR DRUG CANDIDATES ARE PROLONGED OR DELAYED, WE MAY BE UNABLE TO COMMERCIALIZE OUR DRUG CANDIDATES ON A TIMELY BASIS, WHICH WOULD REQUIRE US TO INCUR ADDITIONAL COSTS AND WOULD DELAY OUR RECEIPT OF ANY PRODUCT REVENUE.
We cannot predict whether or not we will encounter problems with any of our completed, ongoing or planned clinical trials that will cause us or regulatory authorities to delay or suspend clinical trials, or delay the analysis of data from our completed or ongoing clinical trials. Any of the following could delay the clinical development of our drug candidates:
· ongoing discussions with the FDA or comparable foreign authorities regarding the scope or design of our clinical trials;
· delays in receiving or the inability to obtain required approvals from the independent institutional review board at one or more of the institutions at which a clinical trial is conducted or other reviewing entities at clinical sites selected for participation in our clinical trials;
· delays in enrolling volunteers or patients into clinical trials;
· a lower than anticipated retention rate of volunteers or patients in clinical trials;
· the need to repeat clinical trials as a result of inconclusive results or unforeseen complications in testing;
· inadequate supply or deficient quality of drug candidate materials or other materials necessary for the conduct of our clinical trials;
· unfavorable FDA inspection and review of a clinical trial site or records of any clinical or preclinical investigation;
· serious and unexpected drug-related side effects experienced by participants in our clinical trials; or
· the placement by the FDA of a clinical hold on a trial.
Our ability to enroll patients in our clinical trials in sufficient numbers and on a timely basis will be subject to a number of factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites, the availability of effective treatments for the relevant disease, the number of other clinical trials competing for patients in the same indication and the eligibility criteria for the clinical trial. In addition, subjects may drop out of our clinical trials, and thereby possibly impair the validity or statistical significance of the trials. Delays in patient enrollment or unforeseen drop-out rates may result in increased costs and longer development times. While all or a portion of these additional costs may be covered by payments under our collaborative agreements, we bear all of the costs for our development candidates for which we have no financial support from a collaborator.
We, the FDA or other applicable regulatory authorities may suspend clinical trials of a drug candidate at any time if we or they believe the subjects or patients participating in such clinical trials are being exposed to unacceptable health risks or for other reasons.
In addition, it is impossible to predict whether legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes, if any, may be. If we experience any such problems, we may not have the financial resources to continue development of the drug candidate that is affected or the development of any of our other drug candidates.
35
IF OUR COMPETITORS BRING SUPERIOR DRUGS TO MARKET OR BRING THEIR DRUGS TO MARKET BEFORE WE DO, WE MAY BE UNABLE TO FIND A MARKET FOR OUR DRUG CANDIDATES.
Our drug candidates in development may not be able to compete effectively with drugs that are currently on the market or new drugs that may be developed by others. There are many other companies developing drugs for the same indications that we are pursuing in development. In order to compete successfully in these areas, we must demonstrate improved safety, efficacy and ease of manufacturing and gain market acceptance over competing drugs that may receive regulatory approval before or after our drug candidates, and over those that currently are marketed. Many of our competitors, including major pharmaceutical companies such as GlaxoSmithKline, Wyeth, Pfizer, Roche, Amgen, Novartis, Johnson & Johnson and Schering-Plough possess substantially greater financial, technical and human resources than we possess. In addition, many of our competitors have significantly greater experience than we have in conducting preclinical and nonclinical testing and human clinical trials of drug candidates, scaling up manufacturing operations and obtaining regulatory approvals of drugs and manufacturing facilities. Accordingly, our competitors may succeed in obtaining regulatory approval for drugs more rapidly than we do. If we obtain regulatory approval and launch commercial sales of our drug candidates, we also will compete with respect to manufacturing efficiency and sales and marketing capabilities, areas in which we currently have limited experience.
IF OUR PROCESSES AND SYSTEMS ARE NOT COMPLIANT WITH REGULATORY REQUIREMENTS, WE COULD BE SUBJECT TO DELAYS IN FILING NEW DRUG APPLICATIONS OR RESTRICTIONS ON MARKETING OF DRUGS AFTER THEY HAVE BEEN APPROVED.
We currently are developing drug candidates for regulatory approval for the first time since our inception, and are in the process of implementing regulated processes and systems required to obtain and maintain regulatory approval for our drug candidates. Certain of these processes and systems for conducting clinical trials and manufacturing material must be compliant with regulatory requirements before we can apply for regulatory approval for our drug candidates. These processes and systems will be subject to continual review and periodic inspection by the FDA and other regulatory bodies. If we are unable to achieve compliance in a timely fashion, we may experience delays in filing for regulatory approval for our drug candidates. In addition, any later discovery of previously unknown problems or safety issues with approved drugs or manufacturing processes, or failure to comply with regulatory requirements, may result in restrictions on such drugs or manufacturing processes, withdrawal of drugs from the market, the imposition of civil or criminal penalties or a refusal by the FDA and/or other regulatory bodies to approve pending applications for marketing approval of new drugs or supplements to approved applications, any of which could have a material adverse effect on our business. In addition, we are a party to collaboration agreements that transfer responsibility for complying with specified regulatory requirements, such as filing and maintenance of marketing authorizations and safety reporting, to our collaborator. If our collaborators do not fulfill these regulatory obligations, any drugs for which we or they obtain approval may be withdrawn from the market, which would have a material adverse effect on our business.
EVEN IF WE OBTAIN REGULATORY APPROVALS, OUR DRUG CANDIDATES WILL BE SUBJECT TO ONGOING REGULATORY REVIEW. IF WE FAIL TO COMPLY WITH CONTINUING UNITED STATES AND APPLICABLE FOREIGN REGULATIONS, WE COULD LOSE THOSE APPROVALS, AND OUR BUSINESS WOULD BE SERIOUSLY HARMED.
Even if we receive regulatory approval of any drug candidates that we are developing, we will be subject to continuing regulatory review, including the review of clinical results that are reported after our drug candidates become commercially available, approved drugs. Since drugs are more widely used by patients once approval has been obtained, side effects and other problems may be observed after approval that were not seen or anticipated during pre-approval clinical trials. In addition, the manufacturers and the manufacturing facilities we engage to make any of our drug candidates will also be subject to periodic
36
review and inspection by the FDA. The subsequent discovery of previously unknown problems with the drug, manufacturers or manufacturing facilities may result in restrictions on the drug, manufacturers or facilities, including withdrawal of the drug from the market or our inability to use the facilities to make our drug. If we fail to comply with applicable continuing regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approval, product recalls and seizures, operating restrictions and criminal prosecutions.
OUR DRUG DEVELOPMENT EFFORTS ARE DATA-DRIVEN AND THEREFORE POTENTIALLY SUBJECT TO ABRUPT CHANGES IN EXPECTED OUTCOMES.
Small molecule drug discovery and development involve, initially, the identification of chemical compounds that may have promise as treatments for specific diseases. Once identified as drug candidates, compounds are subjected to years of testing in a laboratory setting, in animals and in humans. Our ultimate objective is to determine whether the drug candidates have physical characteristics, both intrinsically and in animal and human systems and include a toxicological profile, that are compatible with clinical and commercial success in treatment of the disease being targeted. Throughout this process, experiments are conducted and data are gathered that could reinforce a decision to move to the next step in the investigation process for a particular drug candidate, could result in uncertainty over the proper course to pursue, or could result in the termination of further drug development efforts with respect to the compound being evaluated. We monitor the results of our discovery research and our nonclinical studies and clinical trials and regularly evaluate and re-evaluate our portfolio investments with the objective of balancing risk and potential return in view of new data and scientific, business and commercial insights. This process can result in relatively abrupt changes in focus and priority as new information comes to light and we gain additional insights into ongoing programs and potential new programs.
WE DEPEND ON OUR COLLABORATORS TO WORK WITH US TO DEVELOP, MANUFACTURE AND COMMERCIALIZE MANY OF OUR DRUG CANDIDATES.
We have granted development and commercialization rights to telaprevir to Janssen (worldwide other than North America and Far East) and to Mitsubishi (Far East). We also have granted Far East rights to VX-702 to our collaborator Kissei. We expect to receive significant financial support under our Janssen collaboration agreement, as well as meaningful technical and manufacturing contributions to the telaprevir program. The success of some of our key in-house programs, such as for telaprevir and VX-702, is dependent upon the continued financial and other support that our collaborators have agreed to provide.
For some drug candidates on which we are not currently focusing our development efforts, we have granted worldwide rights to a collaborator, such as our MK-0457 (VX-680) and MK-6592 (VX-667) collaboration with Merck, our Lexiva/Telzir and VX-409 collaborations with GlaxoSmithKline and our AVN-944 (VX-944) collaboration with Avalon.
The success of our collaborations depends on the efforts and activities of our collaborators. Each of our collaborators has significant discretion in determining the efforts and resources that it will apply to the collaboration. Our existing collaborations may not be scientifically or commercially successful.
The risks that we face in connection with these existing and any future collaborations include the following:
· Our collaboration agreements are subject to termination under various circumstances, including, as in the case of our agreement with Janssen, termination without cause. Any such termination could have an adverse material effect on our financial condition and/or delay the development and commercial sale of our drug candidates, including telaprevir.
· Our collaborators may change the focus of their development and commercialization efforts. Pharmaceutical and biotechnology companies historically have re-evaluated their development and commercialization priorities following mergers and consolidations, which have been common in recent years in these industries. The ability of some of our drug candidates to reach their potential
37
could be limited if our collaborators decrease or fail to increase development or commercialization efforts related to those drug candidates.
· Our collaboration agreements may have the effect of limiting the areas of research and development that we may pursue, either alone or in collaboration with third parties.
· Our collaborators may develop and commercialize, either alone or with others, drugs that are similar to or competitive with the drugs or drug candidates that are the subject of the collaboration with us.
IF WE ARE UNABLE TO ATTRACT AND RETAIN COLLABORATORS FOR THE DEVELOPMENT AND COMMERCIALIZATION OF OUR DRUGS AND DRUG CANDIDATES, WE MAY NOT BE ABLE TO FUND OUR DEVELOPMENT AND COMMERCIALIZATION ACTIVITIES.
Our collaborators have agreed to fund portions of our research and development programs and/or to conduct the development and commercialization of specified drug candidates and, if they are approved, drugs. In exchange, we have given them technology, sales and marketing rights relating to those drugs and drug candidates. Some of our corporate collaborators have rights to control the planning and execution of drug development and clinical programs including for our drug candidates MK-0457 (vx-680), MK-6592 (vx-667), VX-409 and AVN-944 (VX-944). Our collaborators may exercise their control rights in ways that may negatively affect the timing and success of those programs. Our collaborations are subject to termination rights by the collaborators. If any of our collaborators were to terminate its relationship with us, or fail to meet its contractual obligations, that action could have a material adverse effect on our ability to undertake research, to fund related and other programs and to develop, manufacture and market any drugs that may have resulted from the collaboration. We expect to seek additional collaborative arrangements, which may not be available to us, to develop and commercialize our drug candidates in the future. Even if we are able to establish acceptable collaborative arrangements in the future, they may not be successful.
OUR INVESTMENT IN THE CLINICAL DEVELOPMENT AND MANUFACTURE OF COMMERCIAL SUPPLY OF TELAPREVIR MAY NOT RESULT IN ANY BENEFIT TO US IF TELAPREVIR IS NOT APPROVED FOR COMMERCIAL SALE.
We are investing significant resources in the clinical development of telaprevir. In 2006, we increased our investment in telaprevir to support our Phase 2b clinical development program. Telaprevir is the first drug candidate for which we expect to perform all activities related to late stage development, drug supply, registration and commercialization in a major market. Even though telaprevir is a Phase 2b drug candidate, we are planning for and investing significant resources now in preparation for Phase 3 clinical trials, application for marketing approval, commercial supply and sales and marketing. We also expect to incur significant costs in 2007 to manufacture registration batches and invest in telaprevir commercial supply. Our engagement in these resource-intensive activities could make it more difficult for us to maintain our portfolio focus, and puts significant investment at risk if we do not obtain regulatory approval and successfully commercialize telaprevir in North America. There is no assurance that our development of telaprevir will lead successfully to regulatory approval, or that obtaining regulatory approval will lead to commercial success. If telaprevir is not approved for commercial sale or if its development is delayed for any reason, our full investment in telaprevir may be at risk and our business and financial condition could be materially adversely affected.
WE DEPEND ON THIRD-PARTY MANUFACTURERS TO DISTRIBUTE CLINICAL TRIAL MATERIALS FOR CLINICAL TRIALS AND EXPECT TO CONTINUE TO RELY ON THEM TO MEET OUR COMMERCIAL SUPPLY NEEDS FOR ANY DRUG CANDIDATE THAT IS APPROVED FOR SALE. WE MAY NOT BE ABLE TO ESTABLISH OR MAINTAIN THESE RELATIONSHIPS AND ARE SUBJECT TO SUPPLY DISRUPTIONS OUTSIDE OF OUR CONTROL.
If we are successful in advancing our proprietary drug candidates through clinical development, we plan to establish and maintain a commercial supply chain and build our logistics and quality control
38
capabilities. We currently are relying on a worldwide network of third-party manufacturers to manufacture and distribute our drug candidates for clinical trials, and we expect that we will continue to do so to meet our commercial supply needs for these drugs, if they are approved for sale. As a result of our reliance on these third-party manufacturers and suppliers, including sole source suppliers of certain components of our drug candidates and drugs, we may be subject to significant supply disruptions outside of our control.
We have retained manufacturing and commercialization responsibilities for telaprevir in North America. Establishing the commercial supply chain for telaprevir is a multi-step international endeavor involving the purchase of several raw materials, the application of certain manufacturing processes requiring significant lead times, the conversion of active pharmaceutical ingredient to tablet form and the packaging of tablets for distribution. We expect to source raw materials, drug substance and drug product, including finished packaging, from third parties located in the Far East, the European Union and the United States, and we are currently establishing and expanding third-party relationships in this regard. Establishing and providing quality assurance for this global supply chain requires a significant financial commitment, experienced personnel and the creation or expansion of numerous third-party contractual relationships. While we believe that there are multiple third parties that are capable of providing the materials and services that we need in order to manufacture and distribute telaprevir, if it is approved for sale, some of these services are in high demand and capacity is constrained. There can be no assurance that we will be able to establish and maintain this commercial supply chain on commercially reasonable terms in order to support a timely launch of telaprevir or at all.
We plan to identify and enter into commercial relationships with multiple third-party manufacturers in order to reduce the risk of supply chain disruption by limiting our reliance on any one manufacturer. In addition, we are in the process of transferring technical information regarding the manufacture of telaprevir to Janssen so that Janssen will be able to manufacture telaprevir, if approved, for sale in Janssen’s territories and as a secondary source for us. There is no assurance, however, that we will be able to establish second sources for each stage of manufacturing of telaprevir, or any other drug or drug candidate, or that any second source will be able to produce sufficient quantities in the required timeframe to avoid a supply chain disruption if there is a problem with one of our suppliers.
Even if we successfully establish arrangements with third-party manufacturers, supply disruptions may result from a number of factors including shortages in product raw materials, labor or technical difficulties, regulatory inspections or restrictions, shipping or customs delays or any other performance failure by any third-party manufacturer on which we rely.
Any supply disruptions could impact the timing of our clinical trials and the commercial launch of any approved pharmaceutical drugs. Furthermore, we may be required to modify our production methods to permit us to economically manufacture our drugs for commercial launch and sale. Upon approval of a pharmaceutical drug for sale, if any, we similarly may be at risk of supply chain disruption for our commercial drug supply. These modifications may require us to reevaluate our resources and the resources of our third-party manufacturers, which could result in abrupt changes in our production methods and supplies. The production of our drug candidates is based in part on technology that we believe to be proprietary. We have licensed this technology to enable our third-party manufacturers to manufacture drug candidates for us. However, in the course of their services, a contract manufacturer may develop process technology related to the manufacture of our drug candidates that the manufacturer owns, either independently or jointly with us. This would increase our reliance on that manufacturer or require us to obtain a license from that manufacturer in order to have our products manufactured by other suppliers utilizing the same process.
WE RELY ON THIRD PARTIES TO CONDUCT OUR CLINICAL TRIALS, AND THOSE THIRD PARTIES MAY NOT PERFORM SATISFACTORILY, INCLUDING FAILING TO MEET ESTABLISHED DEADLINES FOR THE COMPLETION OF SUCH TRIALS.
We do not have the ability to independently conduct clinical trials for our drug candidates, and we rely on third parties such as contract research organizations, medical institutions and clinical investigators to
39
enroll qualified patients and conduct our clinical trials. Our reliance on these third parties for clinical development activities reduces our control over these activities. Accordingly, these third-party contractors may not complete activities on schedule, or may not conduct our clinical trials in accordance with regulatory requirements or our trial design. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be required to replace them. Although we believe that there are a number of other third-party contractors we could engage to continue these activities, it may result in a delay of the affected trial. Accordingly, our efforts to obtain regulatory approvals for and commercialize our drug candidates may be delayed.
IF WE ARE UNABLE TO DEVELOP INDEPENDENT SALES AND MARKETING CAPABILITIES OR ESTABLISH THIRD-PARTY RELATIONSHIPS FOR THE COMMERCIALIZATION OF OUR DRUG CANDIDATES, WE WILL NOT BE ABLE TO SUCCESSFULLY COMMERCIALIZE OUR DRUG CANDIDATES EVEN IF WE ARE ABLE TO OBTAIN REGULATORY APPROVAL.
We currently have no experience as a company in sales and marketing or with respect to pricing and obtaining adequate third-party reimbursement for drugs. GlaxoSmithKline currently has exclusive sales and marketing rights to Lexiva/Telzir. We will need to either develop marketing capabilities and an independent sales force or enter into arrangements with third parties to sell and market any of our drug candidates if they are approved for sale by regulatory authorities.
In order to market telaprevir in North America if it is approved, we intend to build a marketing organization and a direct sales force, which will require substantial efforts and significant management and financial resources. During 2007, we intend to commit significant personnel and financial resources to this effort, staging our commitments to the extent possible in consideration of the ongoing telaprevir development timeline. We will need to devote significant effort, in particular, to recruiting individuals with experience in the sales and marketing of pharmaceutical products. Competition for personnel with these skills is intense and may be particularly difficult for us since telaprevir is still an investigational drug candidate. In addition, if we develop our own marketing and sales capability, we may be competing with other companies that currently have experienced and well-funded marketing and sales operations. As a result, we may not be able to successfully develop our own marketing capabilities or independent sales force for telaprevir in North America in order to support an effective launch of telaprevir if it is approved for sale.
We have granted commercialization rights to other pharmaceutical companies with respect to certain of our drug candidates in certain geographic locations, including telaprevir (Janssen worldwide except for North America and the Far East, and Mitsubishi in the Far East), MK-0457 (VX-680) and MK-6592 (VX-667) (Merck worldwide) and AVN-944 (VX-944) (Avalon worldwide). To the extent that our collaborators have commercial rights to our drugs, any revenues we receive from any approved drugs, will depend primarily on the sales and marketing efforts of others. We do not know whether we will be able to enter into additional third-party sales and marketing arrangements with respect to any of our other drug candidates on acceptable terms, if at all, or whether we will be able to leverage the sales and marketing capabilities we intend to build for telaprevir in order to market and sell any other drug candidate if it is approved for sale.
WE MAY NEED TO RAISE ADDITIONAL CAPITAL THAT MAY NOT BE AVAILABLE.
We expect to incur substantial research and development and related supporting expenses as we design and develop existing and future compounds, undertake clinical trials of drug candidates resulting from such compounds, and build our manufacturing, regulatory, development and commercial capabilities. We also expect to incur substantial administrative and commercialization expenses in the future. We may need to make significant capital investment in building our manufacturing capacity and creating pre-launch inventory for one or more of our drug candidates. We anticipate that we will finance these substantial cash needs with:
· cash received from our existing collaborative agreements;
40
· cash received from new collaborative agreements;
· Lexiva/Telzir royalty revenue;
· existing cash reserves, together with interest earned on those reserves; and
· future product sales to the extent that we market drugs directly.
We expect that funds from these sources will be sufficient to fund our planned activities for at least the next eighteen months from the date of this filing. If we need additional capital, it will be necessary to raise additional funds through public offerings or private placements of equity or debt securities or other methods of financing. Even if our financial resources are sufficient to meet our short or intermediate term needs, we may still decide, as we have in the past, to raise additional funds when we believe financial market conditions are favorable. Any equity financings could result in dilution to our then-existing security holders. Any debt financing, if available at all, may be on terms that, among other things, restrict our ability to pay dividends and interest (although we do not intend to pay dividends for the foreseeable future). The required interest payments associated with any significant additional debt financing could materially adversely affect our ability to service our convertible subordinated notes and convertible senior subordinated notes. The terms of any additional debt financing may also, under certain circumstances, restrict or prohibit us from making interest payments on our convertible subordinated notes. If adequate funds are not available, we may be required to curtail significantly or discontinue one or more of our research, drug discovery or development programs (including clinical trials), or attempt to obtain funds through arrangements with collaborators or others that may require us to relinquish rights to certain of our technologies, drugs or drug candidates. Additional financing may not be available on acceptable terms, if at all.
IF WE FAIL TO EXPAND OUR HUMAN RESOURCES AND MANAGE OUR GROWTH EFFECTIVELY, OUR BUSINESS MAY SUFFER.
We expect that if our clinical drug candidates continue to progress in development, we continue to build our commercial organization and our drug discovery efforts continue to generate drug candidates, we will require significant additional investment in personnel, management systems and resources. For example, the number of our full time employees increased by 17% in 2006, and we expect to experience significant growth in 2007. Our ability to commercialize our drug candidates, achieve our research and development objectives, and satisfy our commitments under our collaboration agreements depends on our ability to respond effectively to these demands and expand our internal organization to accommodate additional anticipated growth. If we are unable to manage our growth effectively, there could be a material adverse effect on our business.
RISKS ASSOCIATED WITH OUR INTERNATIONAL BUSINESS RELATIONSHIPS COULD MATERIALLY ADVERSELY AFFECT OUR BUSINESS.
We have manufacturing, collaborative and clinical trial relationships, and we and our collaborators are seeking approval for our drug candidates, outside the United States. In addition, we expect that if telaprevir is approved for commercial sale, a significant portion of our commercial supply chain, including sourcing of raw materials and manufacturing, will be located in the Far East and European Union. Consequently, we are, and will continue to be, subject to risks related to operating in foreign countries. Risks associated with conducting operations in foreign countries include:
· differing regulatory requirements for drug approvals in foreign countries;
· unexpected changes in tariffs, trade barriers and regulatory requirements;
· economic weakness, including inflation, or political instability in particular foreign economies and markets;
· compliance with tax, employment, immigration and labor laws for employees living or travelling abroad;
41
· foreign taxes, including withholding of payroll taxes;
· foreign currency fluctuations, which could result in increased operating expenses or reduced revenues, and other obligations incident to doing business or operating a subsidiary in another country;
· workforce uncertainty in countries where labor unrest is more common than in the United States;
· production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
· business interruptions resulting from geo-political actions, including war and terrorism.
These and other risks associated with our international operations may materially adversely affect our ability to attain or maintain profitable operations.
IF WE LOSE OUR TECHNOLOGICAL ADVANTAGES, WE MAY NOT BE ABLE TO COMPETE IN THE MARKETPLACE.
We believe that our integrated drug discovery capability gives us a technological advantage over our competitors. However, the pharmaceutical research field is characterized by rapid technological progress and intense competition. As a result, we may not realize the expected benefits from these technologies. For example, a large pharmaceutical company, with significantly more resources than we have, could pursue a systematic approach to the discovery of drugs based on gene families, using proprietary drug targets, compound libraries, novel chemical approaches, structural protein analysis and information technologies. Such a company might identify broadly applicable compound classes faster and more effectively than we do. Further, we believe that interest in the application of structure-based drug design, parallel drug design and related approaches has accelerated as the strategies have become more widely understood. Businesses, academic institutions, governmental agencies and other public and private research organizations are conducting research to develop technologies that may compete with those we use. It is possible that our competitors could acquire or develop technologies that would render our technology obsolete or noncompetitive. For example, a competitor could develop information technologies that accelerate the atomic-level analysis of potential compounds that bind to the active site of a drug target, and predict the absorption, toxicity, and relative ease-of-synthesis of candidate compounds. If we were unable to access the same technologies at an acceptable price, our business could be adversely affected.
THE LOSS OF THE SERVICES OF KEY EMPLOYEES OR THE FAILURE TO HIRE QUALIFIED EMPLOYEES WOULD NEGATIVELY IMPACT OUR BUSINESS AND FUTURE GROWTH.
Because our drug discovery and development activities are highly technical in nature, we require the services of highly qualified and trained scientists who have the skills necessary to conduct these activities. In addition, as we attempt to grow our capabilities with respect to clinical development, regulatory affairs, quality control and sales and marketing, we will need to attract and retain employees with experience in these fields. Our future success will depend in large part on the continued services of our key scientific and management personnel. We have entered into employment agreements with some individuals and provide compensation-related benefits to all of our key employees that vest over time and therefore induce them to remain with us. However, the employment agreements can be terminated by the employee on relatively short notice. The value to employees of stock-related benefits that vest over time—such as options and restricted stock—will be significantly affected by movements in our stock price that we cannot control, and may at any point in time be insufficient to counteract more lucrative offers from other companies.
42
We face intense competition for our personnel from our competitors, our collaborators and other companies throughout our industry. Moreover, the growth of local biotechnology companies and the expansion of major pharmaceutical companies into the Boston area have increased competition for the available pool of skilled employees, especially in technical fields, and the high cost of living in the Boston and San Diego areas makes it difficult to attract employees from other parts of the country. A failure to retain, as well as hire, train and effectively integrate into our organization a sufficient number of qualified scientists and professionals would negatively affect our business and our ability to grow our business. In addition, the level of funding under certain of our collaborative agreements depends on the number of our employees performing research and/or development under those agreements. If we cannot hire and retain the required personnel, funding received under the agreements may be reduced.
IF OUR PATENTS DO NOT PROTECT OUR DRUGS, OR OUR DRUGS INFRINGE THIRD-PARTY PATENTS, WE COULD BE SUBJECT TO LITIGATION AND SUBSTANTIAL LIABILITIES.
We have numerous patent applications pending in the United States, as well as foreign counterparts in other countries. Our success will depend, in significant part, on our ability to obtain and maintain United States and foreign patent protection for our drugs, their uses and our processes, to preserve our trade secrets and to operate without infringing the proprietary rights of third parties. We do not know whether any patents will issue from any of our patent applications or, even if patents issue or have issued, that the issued claims will provide us with any significant protection against competitive products or otherwise be valuable commercially. Legal standards relating to the validity of patents and the proper scope of their claims in the pharmaceutical field are still evolving, and there is no consistent law or policy regarding the valid breadth of claims in biopharmaceutical patents or the effect of prior art on them. If we are not able to obtain adequate patent protection, our ability to prevent competitors from making, using and selling similar drugs will be limited. Furthermore, our activities may infringe the claims of patents held by third parties. Defense and prosecution of infringement or other intellectual property claims, as well as participation in other inter-party proceedings, can be expensive and time-consuming, regardless of whether or not the outcome is favorable to us. If the outcome of any such litigation or proceeding were adverse, we could be subject to significant liabilities to third parties, could be required to obtain licenses from third parties or could be required to cease sales of affected drugs, any of which outcomes could have a material adverse effect on our business.
WE DO NOT KNOW WHETHER LEXIVA/TELZIR WILL CONTINUE TO BE COMPETITIVE IN THE MARKET FOR HIV PROTEASE INHIBITORS.
We currently receive royalties from sales of Lexiva/Telzir under our collaboration with GlaxoSmithKline. Lexiva/Telzir’s share of the worldwide protease inhibitor market may decrease due to competitive forces and market dynamics. Other HIV PIs including Bristol-Myers Squibbs’ Reyataz® and Abbott Laboratories’ Kaletra®, and a number of other products are on the market for the treatment of HIV infection and AIDS. Other drugs are still in development by our competitors, including Bristol-Myers Squibb, Boehringer Ingelheim and Johnson & Johnson, which may have better efficacy, fewer side effects, easier administration and/or lower costs than Lexiva/Telzir. Moreover, the growth in the worldwide market for HIV PIs has, to a certain extent, occurred as a result of early and aggressive treatment of HIV infection with a protease inhibitor-based regimen. Changes in treatment strategy, in which treatment is initiated later in the course of infection, or in which treatment is more often initiated with a regimen that does not include a protease inhibitor, may result in reduced use of HIV PIs. As a result, the total market for HIV PIs may decline, decreasing the sales potential of Lexiva/Telzir. Further, although we provide education efforts related to the promotion of Lexiva/Telzir in the United States and key markets in Europe, GlaxoSmithKline directs the majority of the marketing and sales efforts and the positioning of Lexiva/Telzir in the overall market, and we have little control over the direction or success of those efforts. GlaxoSmithKline has the right to terminate its agreement with us without cause upon twelve months’ notice, and would have no obligation to pay further royalties to us upon any such termination.
43
IF PHYSICIANS, PATIENTS AND THIRD-PARTY PAYORS DO NOT ACCEPT OUR FUTURE DRUGS, WE MAY BE UNABLE TO GENERATE SIGNIFICANT REVENUE, IF ANY.
Even if our drug candidates obtain regulatory approval, they may not gain market acceptance among physicians, patients and health care payors. Physicians may elect not to recommend our drugs for a variety of reasons including:
· the timing of the market introduction of competitive drugs;
· lower demonstrated clinical safety and efficacy compared to other drugs;
· lack of cost-effectiveness;
· lack of availability of reimbursement from third-party payors;
· convenience and ease of administration;
· prevalence and severity of adverse side effects;
· other potential advantages of alternative treatment methods; and
· ineffective marketing and distribution support.
If our approved drugs fail to achieve market acceptance, we will not be able to generate significant revenue.
IF THE GOVERNMENT AND OTHER THIRD-PARTY PAYORS FAIL TO PROVIDE COVERAGE AND ADEQUATE PAYMENT RATES FOR OUR FUTURE DRUGS, OUR REVENUE AND PROSPECTS FOR PROFITABILITY WILL BE HARMED.
In both domestic and foreign markets, our sales of any future drugs will depend in part upon the availability of reimbursement from third-party payors. Such third-party payors include government health programs such as Medicare, managed care providers, private health insurers and other organizations. These third-party payors are increasingly attempting to contain healthcare costs by demanding price discounts or rebates and limiting both the types and variety of drugs that they will cover and the amounts that they will pay for these drugs. As a result, they may not cover or provide adequate payment for our future drugs. We might need to conduct post-marketing studies in order to demonstrate the cost-effectiveness of any future drugs to such payors’ satisfaction. Such studies might require us to commit a significant amount of management time and financial and other resources. Our future drugs might not ultimately be considered cost-effective. Adequate third-party reimbursement might not be available to enable us to maintain price levels sufficient to realize an appropriate return on investment in product development.
Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on payments allowed for lower-cost products that are already reimbursed, may be incorporated into existing payments for other products or services, and may reflect budgetary constraints and/or imperfections in Medicare or Medicaid data used to calculate these rates. Net prices for drugs may be reduced by mandatory discounts or rebates required by government health care programs. In addition, legislation has been introduced in Congress that, if enacted, would permit more widespread importation of drugs from foreign countries into the United States, which may include importation from countries where the drugs are sold at lower prices than in the United States. Such legislation, or similar regulatory changes or relaxation of laws that restrict imports of drugs from other countries, could reduce the net price we receive for our marketed drugs.
44
OUR BUSINESS HAS A SUBSTANTIAL RISK OF PRODUCT LIABILITY CLAIMS. IF WE ARE UNABLE TO OBTAIN APPROPRIATE LEVELS OF INSURANCE, A PRODUCT LIABILITY CLAIM COULD ADVERSELY AFFECT OUR BUSINESS.
Our business exposes us to significant potential product liability risks that are inherent in the development, manufacturing and sales and marketing of human therapeutic products. We currently have clinical trial insurance and will seek to obtain product liability insurance prior to the sales and marketing of any of our drug candidates. However, our insurance may not provide adequate coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to maintain current amounts of insurance coverage or obtain additional or sufficient insurance at a reasonable cost to protect against losses that could have a material adverse effect on us. If a claim is brought against us, we might be required to pay legal and other expenses to defend the claim, as well as uncovered damages awards resulting from a claim brought successfully against us. Furthermore, whether or not we are ultimately successful in defending any such claims, we might be required to direct significant financial and managerial resources to such defense, and adverse publicity is likely to result.
IF WE DO NOT COMPLY WITH LAWS REGULATING THE PROTECTION OF THE ENVIRONMENT AND HEALTH AND HUMAN SAFETY, OUR BUSINESS COULD BE ADVERSELY AFFECTED.
Our research and development efforts involve the controlled use of hazardous materials, chemicals and various radioactive compounds. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials. Although we maintain workers’ compensation insurance to cover us for costs we may incur due to injuries to our employees resulting from the use of these materials, this insurance may not provide adequate coverage against potential liabilities. Due to the small amount of hazardous materials that we generate, we have determined that the cost to secure insurance coverage for environmental liability and toxic tort claims far exceeds the benefits. Accordingly, we do not maintain any insurance to cover pollution conditions or other extraordinary or unanticipated events relating to our use and disposal of hazardous materials. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of these laws or regulations.
WE HAVE ADOPTED ANTI-TAKEOVER PROVISIONS THAT MAY FRUSTRATE ANY ATTEMPT TO REMOVE OR REPLACE OUR CURRENT MANAGEMENT.
Our corporate charter and by-law provisions and stockholder rights plan may discourage certain types of transactions involving an actual or potential change of control of Vertex that might be beneficial to us or our security holders. Our charter provides for staggered terms for the members of the Board of Directors. Our by-laws grant the directors a right to adjourn annual meetings of stockholders, and certain provisions of the by-laws may be amended only with an 80% stockholder vote. Pursuant to our stockholder rights plan, each share of common stock has an associated preferred share purchase right. The rights will not trade separately from the common stock until, and are exercisable only upon, the acquisition or the potential acquisition through tender offer by a person or group of 15% or more of the outstanding common stock. We may issue shares of any class or series of preferred stock in the future without stockholder approval and upon such terms as our Board of Directors may determine. The rights of the holders of common stock will be subject to, and may be adversely affected by, the rights of the holders of any class or series of preferred stock that may be issued in the future. As a result, stockholders or other parties may find it more difficult to remove or replace our current management.
45
OUR STOCK PRICE MAY FLUCTUATE BASED ON FACTORS BEYOND OUR CONTROL.
Market prices for securities of companies such as Vertex are highly volatile. Within the twelve months ended December 31, 2006, our common stock traded between $26.50 and $45.38 per share. The market for our stock, like that of other companies in the biotechnology field, has from time to time experienced significant price and volume fluctuations that are unrelated to our operating performance. The future market price of our securities could be significantly and adversely affected by factors such as:
· announcements of results of clinical trials or nonclinical studies;
· announcements of financial results and other operating performance measures, or capital structuring activities;
· technological innovations or the introduction of new drugs by our competitors;
· government regulatory action;
· public concern as to the safety of drugs developed by others;
· developments in patent or other intellectual property rights or announcements relating to these matters;
· developments in domestic and international governmental policy or regulation, for example relating to intellectual property rights; and
· developments relating specifically to other companies and market conditions for pharmaceutical and biotechnology stocks in general.
OUR OUTSTANDING INDEBTEDNESS MAY MAKE IT MORE DIFFICULT TO OBTAIN ADDITIONAL FINANCING OR REDUCE OUR FLEXIBILITY TO ACT IN OUR BEST INTERESTS.
As of December 31, 2006, we had approximately $42.1 million in aggregate principal amount of 5% Convertible Subordinated Notes due in September 2007 and approximately $59.6 million in aggregate principal amount of 5.75% Convertible Senior Subordinated Notes due in February 2011 outstanding. In February 2007, we announced that we will redeem the 5.75% Convertible Senior Subordinated Notes due in February 2011 on March 5, 2007 after which our outstanding indebtedness will be significantly reduced. Notwithstanding the redemption of our convertible senior subordinated notes in March 2007, the level of our indebtedness will affect us by:
· exposing us to fixed rates of interest, which may be in excess of prevailing market rates;
· making it more difficult to obtain additional financing for working capital, capital expenditures, debt service requirements or other purposes;
· constraining our ability to react quickly in an unfavorable economic climate or to changes in our business or the pharmaceutical industry; and
· requiring the dedication of a substantial portion of our expected cash flow to service of our indebtedness, thereby reducing the amount of expected cash flow available for other purposes.
OUR ESTIMATES OF OUR LIABILITY UNDER OUR KENDALL SQUARE LEASE MAY BE INACCURATE.
We leased a 290,000 square foot facility in Kendall Square, Cambridge, Massachusetts in January 2003 for a 15-year term. We currently are occupying approximately 120,000 square feet of the facility. We have sublease arrangements in place for the remaining rentable square footage of the facility. In determining our obligations under the lease for the portion of the facility that we are not occupying, we have made certain assumptions relating to the time necessary to sublease the space after the expiration of the initial subleases, projected future sublease rental rates and the anticipated durations of future subleases. Our
46
estimates have changed in the past, and may change in the future, resulting in additional adjustments to the estimate of liability, and the effect of any such adjustments could be material.
GOVERNMENT INVESTIGATIONS OR LITIGATION AGAINST OUR COLLABORATORS COULD IMPACT OUR BUSINESS.
The federal government, certain state governments and private payors are investigating and have begun to file actions against numerous pharmaceutical and biotechnology companies alleging that the reporting of prices for pharmaceutical products has resulted in a false and overstated Average Wholesale Price, or AWP, which in turn is alleged to have improperly inflated the reimbursement paid by Medicare beneficiaries, insurers, state Medicaid programs, medical plans and others to health care providers who prescribed and administered those products. Some payors are also alleging that pharmaceutical and biotechnology companies are not reporting their “best price” to the states under the Medicaid program. In addition, recent government litigation against pharmaceutical companies has focused on allegations of off-label promotion in connection with the filing of false claims for government reimbursement. In any AWP cases or other cases brought by the government where our collaborators or licensees are named as defendants with respect to any products licensed from us, the outcome of the case could have a material adverse effect on our financial results.
SALES OF ADDITIONAL SHARES OF OUR COMMON STOCK COULD CAUSE THE PRICE OF OUR COMMON STOCK TO DECLINE.
Sales of substantial amounts of our common stock in the open market, or the availability of such shares for sale, could adversely affect the price of our common stock. In addition, the issuance of common stock upon exercise of any outstanding option or the conversion of any of our outstanding convertible debt would be dilutive, and may cause the market price for a share of our common stock to decline. As of December 31, 2006, we had approximately 126.1 million shares of common stock issued and outstanding. We also had outstanding options to purchase approximately 14.3 million shares of common stock with a weighted average exercise price of $26.44 per share. Our outstanding notes were convertible into approximately 4.4 million shares of common stock with a weighted average conversion price of $22.87 per share. Outstanding options and convertible notes may be exercised or converted, as the case may be, if the market price of our common stock exceeds the applicable exercise or conversion price.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K and, in particular, the description of our Business set forth in Item 1, the Risk Factors set forth in this Item 1A and our Management’s Discussion and Analysis of Financial Condition and Results of Operations set forth in Item 7 contain or incorporate a number of forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, including statements regarding:
· our expectations regarding clinical trials, development timelines and regulatory authority filings for telaprevir and other drug candidates under development by us and our collaborators;
· our expectations regarding the number of patients that will be evaluated, the anticipated date by which enrollment will be completed and the data that will be generated by ongoing and planned clinical trials, and the ability to use that data for the design and initiation of further clinical trials and to support regulatory filings, including potentially an NDA for telaprevir;
· our expectations regarding the scope and timing of ongoing and potential future clinical trials, including the ongoing Phase 2b clinical trials and expected Phase 3 clinical program for telaprevir, the ongoing and planned clinical trials of VX-702, the planned Phase 2 clinical trial of VX-770, the planned Phase 1 clinical trial of VX-883 and the ongoing clinical trials of MK-0457 (VX-680), MK-6592 (VX-667) and AVN-944 (VX-944);
47
· our expectations regarding the future market demand and medical need for telaprevir and our other drug candidates;
· our plans to fund a greater proportion of our research programs than in past years with internal funds, and our beliefs regarding the benefits of this strategy;
· our beliefs regarding the support provided by clinical trials and preclinical and nonclinical studies of our drug candidates for further investigation, clinical trials or potential use as a treatment of those drug candidates;
· our business strategy;
· our planned investments in our drug development and commercialization capabilities and telaprevir, including our expected 2007 research and development expenses for commercial supply investment in telaprevir;
· the focus of our drug development efforts;
· the establishment, development and maintenance of collaborative relationships;
· our ability to use our research programs to identify and develop new potential drug candidates;
· our ability to increase our headcount and scale up our drug development and commercialization capabilities;
· our estimates regarding obligations associated with a lease of a facility in Kendall Square, Cambridge, Massachusetts;
· the potential for the acquisition of new and complementary technologies, resources and drugs or drug candidates; and
· our liquidity.
Any or all of our forward-looking statements in this Annual Report may turn out to be wrong. They can be affected by inaccurate assumptions we might make or by known or unknown risks and uncertainties. Many factors mentioned in our discussion in this Annual Report will be important in determining future results. Consequently, no forward-looking statement can be guaranteed. Actual future results may vary materially.
We also provide a cautionary discussion of risks and uncertainties under “Risk Factors” in Item 1A of this Annual Report. These are factors that we think could cause our actual results to differ materially from expected results. Other factors besides those listed there could also adversely affect us.
Without limiting the foregoing, the words “believes,” “anticipates,” “plans,” “expects” and similar expressions are intended to identify forward-looking statements. There are a number of factors that could cause actual events or results to differ materially from those indicated by such forward-looking statements, many of which are beyond our control, including the factors set forth under “Item 1A. Risk Factors.” In addition, the forward-looking statements contained herein represent our estimate only as of the date of this filing and should not be relied upon as representing our estimate as of any subsequent date. While we may elect to update these forward-looking statements at some point in the future, we specifically disclaim any obligation to do so to reflect actual results, changes in assumptions or changes in other factors affecting such forward-looking statements.
ITEM 1B. UNRESOLVED STAFF COMMENTS
We have not received any written comments that have not been resolved from the Securities and Exchange Commission regarding our filings under the Securities Exchange Act of 1934, as amended.
48
ITEM 2. PROPERTIES
We lease an aggregate of approximately 650,000 square feet of laboratory and office space in nine facilities in Cambridge, Massachusetts. The leases have expiration dates ranging from 2009 to 2018. We have the option to extend the lease for our headquarters facility at 130 Waverly Street, Cambridge, where we lease approximately 100,000 square feet of space, for one additional term of five years, ending in 2015, with respect to one portion of the building, and for up to two additional terms of five years, ending in 2019, for the other portion of the building. The lease for approximately 192,000 square feet of laboratory and office space at 200 Sidney Street, located adjacent to our headquarters, will expire in 2010. We have the option to extend that lease for up to two additional consecutive ten-year terms. The lease for our 21 Erie Street facility for 21,000 square feet of office space, which we entered into in November 2006, expires in May 2012 with the option to extent for two additional consecutive five-year terms.
The lease for our Kendall Square, Cambridge, Massachusetts facility will expire in 2018. We have the option to extend that lease for two consecutive terms of ten years each. We have subleased approximately 145,000 square feet of the facility, and are using the remaining approximately 120,000 square feet of space leased in the facility for our research operations. The subleases are for terms ending in 2011 and 2012 with extension options to 2015 and 2018. One of the subleases has certain termination provisions beginning in 2010.
We also lease approximately 81,200 square feet of laboratory and office space in San Diego, California. The lease for this space will expire on August 31, 2008. We have the option to extend this lease for up to two additional terms of five years each.
We lease approximately 22,000 square feet of laboratory and office space in Milton Park, Abingdon, England, for our United Kingdom business and research and development activities under a lease expiring in 2013, with a right of early termination exercisable by us in 2008.
We believe our facilities are adequate for our current needs.
ITEM 3. LEGAL PROCEEDINGS
We are not a party to any material legal proceedings. We are not a party to any litigation in any court with any governmental authority, and management is not aware of any contemplated proceeding by any governmental authority against the Company.
ITEM 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
There were no matters submitted to a vote of security holders during the quarter ended December 31, 2006.
49
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is traded on The Nasdaq Global Select Market under the symbol “VRTX.” The following table sets forth for the periods indicated the high and low sale prices per share of our common stock as reported by Nasdaq:
Year Ended December 31, 2005: | | | | High | | Low | |
First quarter | | $ | 11.99 | | $ | 9.20 | |
Second quarter | | 17.06 | | 8.61 | |
Third quarter | | 22.68 | | 15.33 | |
Fourth quarter | | 29.24 | | 20.31 | |
| | | | | | | | | |
Year Ended December 31, 2006: | | | | | | | |
First quarter | | $ | 44.71 | | $ | 26.50 | |
Second quarter | | 40.00 | | 29.00 | |
Third quarter | | 37.10 | | 29.75 | |
Fourth quarter | | 45.38 | | 32.50 | |
| | | | | | | | | |
As of February 26, 2007, there were 1,515 holders of record of our common stock.
Performance Graph
CUMULATIVE TOTAL RETURN
Based on initial investment of $100 on December 31, 2001
with dividends reinvested (fiscal years ending December 31)
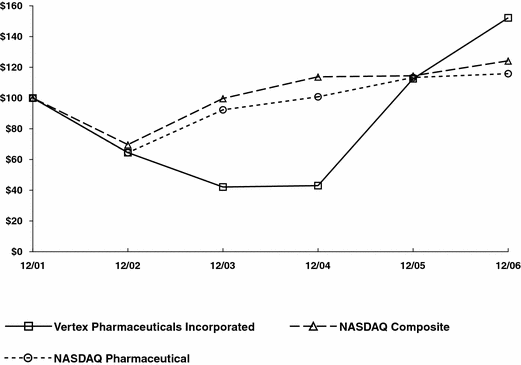
50
Dividends
We have never declared or paid any cash dividends on our common stock, and we currently expect that future earnings, if any, will be retained for use in our business.
Issuer Repurchases of Equity Securities
The table set forth below shows all repurchases of securities by us during the three months ended December 31, 2006:
Period | | | | Total Number
of Shares
Purchased | | Average Price
Paid per Share | | Total Number of Shares
Purchased as part of
publicly announced
Plans or Programs | | Maximum Number of
Shares that may yet
be purchased under
publicly announced
Plans or Programs | |
Oct. 1, 2006 to Oct. 31, 2006 | | | 13,486 | | | | $ | 0.01 | | | | — | | | | — | | |
Nov. 1, 2006 to Nov. 30, 2006 | | | 5,427 | | | | $ | 0.01 | | | | — | | | | — | | |
Dec. 1, 2006 to Dec. 31, 2006 | | | 4,071 | | | | $ | 0.01 | | | | — | | | | — | | |
Under the terms of our 1996 Stock and Option Plan and 2006 Stock and Option Plan, we may award shares of restricted stock to our employees and consultants. These shares of restricted stock typically are subject to a lapsing right of repurchase by us. We may exercise this right of repurchase in the event that a restricted stock recipient’s service to us is terminated. If we exercise this right, we are required to repay the purchase price paid by or on behalf of the recipient for the repurchased restricted shares, which typically is the par value per share of $0.01. Repurchased shares are returned to the applicable Stock and Option Plan under which they were issued. Shares returned to the 2006 Stock and Option Plan are available for future awards under the terms of that plan.
Unregistered Sales of Equity Securities
On February 2, 2007, we called our 2011 Notes for redemption. The redemption date is March 5, 2007. As of February 28, 2007, we had converted $29.4 million of aggregate principal amount of 2011 Notes into 2,173,745 shares of our common stock. The conversions were exempt from registration under the Securities Act of 1933, as amended, pursuant to Section 3(a)(9) thereof as they were exchanges with our existing security holders where no commission or other remuneration was paid.
51
ITEM 6. SELECTED FINANCIAL DATA
The following unaudited selected consolidated financial data for each of the five years in the period ended December 31, 2006 are derived from our audited consolidated financial statements. These data should be read in conjunction with our audited consolidated financial statements and related notes that are included elsewhere in this Annual Report on Form 10-K and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in Item 7 below.
| | Year Ended December 31, | |
| | 2006 | | 2005 | | 2004 | | 2003 | | 2002 | |
| | (In thousands, except per share amounts) | |
Consolidated Statements of Operations Data: | | | | | | | | | | | |
Revenues: | | | | | | | | | | | |
Royalties | | $ | 41,208 | | $ | 32,829 | | $ | 17,322 | | $ | 9,002 | | $ | 10,054 | |
Collaborative and other research and development revenues | | 175,148 | | 128,061 | | 85,395 | | 60,139 | | 84,716 | |
Total revenues | | 216,356 | | 160,890 | | 102,717 | | 69,141 | | 94,770 | |
Costs and expenses: | | | | | | | | | | | |
Royalty payments | | 12,170 | | 10,098 | | 5,649 | | 3,126 | | 3,334 | |
Research and development expenses | | 371,713 | | 248,540 | | 192,162 | | 199,636 | | 198,338 | |
Sales, general and administrative expenses | | 57,860 | | 43,990 | | 42,139 | | 39,082 | | 41,056 | |
Restructuring and other expense | | 3,651 | | 8,134 | | 17,574 | | 91,824 | | — | |
Total costs and expenses | | 445,394 | | 310,762 | | 257,524 | | 333,668 | | 242,728 | |
Loss from operations | | (229,038 | ) | (149,872 | ) | (154,807 | ) | (264,527 | ) | (147,958 | ) |
Other income/(expense), net | | 15,069 | | (5,332 | ) | (7,994 | ) | (1,886 | ) | 11,000 | |
Realized gain on sale of investment(1) | | 11,183 | | — | | — | | — | | — | |
Loss on exchange of convertible subordinated notes(2)(3) | | (5,151 | ) | (48,213 | ) | — | | — | | — | |
Loss on retirement of convertible subordinated notes(4) | | — | | — | | (3,446 | ) | — | | — | |
Loss from continuing operations before cumulative effect of changes in accounting principles | | (207,937 | ) | (203,417 | ) | (166,247 | ) | (266,413 | ) | (136,958 | ) |
Income from discontinued operations(5): | | | | | | | | | | | |
Gain on sales of assets | | — | | — | | — | | 70,339 | | — | |
Income (loss) from discontinued operations | | — | | — | | — | | (693 | ) | 28,337 | |
Total income from discontinued operations | | — | | — | | — | | 69,646 | | 28,337 | |
Loss before cumulative effect of change in accounting principle | | $ | (207,937 | ) | $ | (203,417 | ) | $ | (166,247 | ) | $ | (196,767 | ) | $ | (108,621 | ) |
Cumulative effect of a change in accounting principle—FAS 123(R)(6) | | 1,046 | | — | | — | | — | | — | |
Net loss | | $ | (206,891 | ) | $ | (203,417 | ) | $ | (166,247 | ) | $ | (196,767 | ) | $ | (108,621 | ) |
Basic and diluted loss per common share before cumulative effect of a change in accounting principle | | $ | (1.84 | ) | $ | (2.28 | ) | $ | (2.12 | ) | $ | (2.56 | ) | $ | (1.43 | ) |
Basic and diluted cumulative effect of a change in accounting principle per common share. | | 0.01 | | — | | — | | — | | — | |
Basic and diluted net loss per common share | | $ | (1.83 | ) | $ | (2.28 | ) | $ | (2.12 | ) | $ | (2.56 | ) | $ | (1.43 | ) |
Basic and diluted weighted average number of common shares outstanding | | 113,221 | | 89,241 | | 78,571 | | 77,004 | | 75,749 | |
52
| | December 31, | |
| | 2006 | | 2005 | | 2004 | | 2003 | | 2002 | |
| | (In thousands) | |
Consolidated Balance Sheets Data: | | | | | | | | | | | |
Cash, cash equivalents and marketable securities | | $ | 761,752 | | $ | 407,510 | | $ | 392,320 | | $ | 583,164 | | $ | 634,984 | |
Other current assets | | 66,780 | | 23,898 | | 14,392 | | 10,642 | | 21,588 | |
Restricted cash | | 30,258 | | 41,482 | | 49,847 | | 26,061 | | 26,091 | |
Property and equipment, net | | 61,535 | | 54,533 | | 64,225 | | 80,083 | | 95,991 | |
Other non-current assets | | 1,254 | | 21,575 | | 24,669 | | 24,461 | | 37,066 | |
Total assets | | $ | 921,579 | | $ | 548,998 | | $ | 545,453 | | $ | 724,411 | | $ | 815,720 | |
Deferred revenue | | $ | 150,184 | | $ | 32,300 | | $ | 66,086 | | $ | 59,517 | | $ | 58,486 | |
Accrued restructuring and other expense | | 33,073 | | 42,982 | | 55,843 | | 69,526 | | — | |
Other current liabilities | | 110,640 | | 54,443 | | 50,161 | | 47,795 | | 52,709 | |
Collaborator development loan (due 2008) | | 19,997 | | 19,997 | | 19,997 | | 32,460 | | 5,000 | |
Other long-term obligations | | — | | — | | 2,925 | | 7,268 | | 5,944 | |
Convertible subordinated notes(2)(4) | | 42,102 | | 42,102 | | 82,552 | | 315,000 | | 315,000 | |
Convertible senior subordinated notes(2)(3)(4)(7) | | 59,648 | | 117,998 | | 232,448 | | — | | — | |
Stockholders’ equity | | 505,935 | | 239,176 | | 35,441 | | 192,845 | | 378,581 | |
Total liabilities and stockholders’ equity | | $ | 921,579 | | $ | 548,998 | | $ | 545,453 | | $ | 724,411 | | $ | 815,720 | |
(1) During 2006 we sold 817,749 shares of Altus Pharmaceuticals common stock for approximately $11.7 million, and the Altus warrants for approximately $18.3 million. As a result of the sales of Altus common stock and warrants, we recorded a realized gain on a sale of investment of $11.2 million
(2) In the third quarter of 2005, holders of 5% Convertible Subordinated Notes due in September 2007 (“2007 Notes”) exchanged $40.5 million in aggregate principal amount of 2007 Notes, plus interest, for approximately 2.5 million shares of newly issued common stock. As a result of this exchange, we incurred a non-cash charge of approximately $36.3 million. In separate transactions, in the fourth quarter of 2005, holders of 5.75% Convertible Senior Subordinated Notes due in February 2011 (“2011 Notes”) exchanged $114.5 million in aggregate principal amount of 2011 Notes, plus interest, for approximately 8.1 million shares of newly issued common stock. As a result of this exchange, a non-cash charge of approximately $11.9 million was incurred. These charges relate to the incremental shares issued in the transactions over the number of shares that would have been issued upon conversion of the notes at the conversion prices set forth therein.
(3) In the third quarter of 2006, holders of 2011 Notes exchanged $58.3 million in aggregate principal amount of 2011 Notes, plus accrued interest, for approximately 4.1 million shares of newly issued common stock. As a result of this exchange, we incurred a non-cash charge of approximately $5.2 million. This charge relates to the incremental shares issued in the transaction over the number of shares that would have been issued upon conversion of the notes at the conversion prices set forth therein.
(4) During 2004, we issued approximately $232.4 million in aggregate principal amount of 2011 Notes in exchange for an equal principal amount of our outstanding 2007 Notes. The Company recorded a charge related to the write-off of the unamortized deferred issuance costs applicable to the 2007 Notes retired.
(5) We sold certain assets and liabilities of our Discovery Tools and Services business in two independent transactions in March and December 2003. In October 2001, the FASB issued Statement No. 144, “Accounting for the Impairment of Long-Lived Assets” (“FAS 144”). Pursuant to FAS 144, the Statement of Operations data shown above give effect to the disposition of the assets sold, accounting for such assets as discontinued operations.
(6) Financial Accounting Standards Board Statement No. 123(R), “Share-Based Payment” (“FAS 123(R)”) requires us to recognize expense only for restricted shares expected to vest, and this results in us being required to estimate forfeitures on grant date. In connection with the adoption of FAS 123(R) we recorded a $1.0 million benefit due to the cumulative effect of estimating forfeitures on the grant date rather than recording them as they occur.
(7) In February 2007, we announced that we will redeem our 2011 Notes on March 5, 2007.
53
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
We are in the business of discovering, developing and commercializing small molecule drugs for the treatment of serious diseases. We have built a drug discovery capability that integrates biology, chemistry, biophysics, automation and information technologies, with a goal of making the drug discovery process more efficient and productive. Our most advanced drug candidate, telaprevir, is being investigated for the treatment of hepatitis C virus infection in three major Phase 2b clinical trials. We are investing significant resources to expand our capabilities in clinical development, regulatory affairs, quality control and commercial operations and to build and manage a commercial supply chain in preparation for the Phase 3 development and the potential commercial launch of telaprevir. We also expect to incur significant costs in 2007 to manufacture registration batches of telaprevir and build a telaprevir commercial inventory. We have a number of other drug candidates, including candidates targeting rheumatoid arthritis, cystic fibrosis, bacterial infection, cancer and pain, that are being evaluated in preclinical studies or clinical trials either by us or in collaboration with other pharmaceutical companies. Our HIV protease inhibitor, fosamprenavir calcium, is being marketed by our collaborator GlaxoSmithKline plc as Lexiva in the United States and Telzir in Europe.
We currently are concentrating most of our drug development resources on four drug candidates: telaprevir, for the treatment of HCV infection; VX-702, for the treatment of rheumatoid arthritis and other inflammatory diseases; VX-770, for the treatment of cystic fibrosis; and VX-883, for the treatment of bacterial infection. In 2007, we expect to continue Phase 2b and commence Phase 3 clinical trials of telaprevir, commence Phase 2 clinical trials of VX-702 and VX-770 and commence a Phase 1 clinical trial of VX-883. We have retained the right to commercialize telaprevir in North America and to commercialize VX-702 (except for the Far East), VX-770 and VX-883 worldwide. Our pipeline also includes several drug candidates that are being developed by our collaborators. The most advanced of these drug candidates is MK-0457 (VX-680), an Aurora kinase inhibitor that is being developed by Merck & Co., Inc. for the treatment of cancer. We have a number of other drug candidates in development and a broad-based discovery effort.
Our net loss for 2006 was $206.9 million, or $1.83 per basic and diluted common share, and we expect to incur substantial operating losses in the future. In 2007, we expect that our research and development expenses will be higher than those in 2006, as we continue to incur research and development costs related to telaprevir and our other drug candidates, establish a commercial supply chain and build telaprevir commercial inventory to support markets where we expect to launch telaprevir, if approved, and build our general drug development and commercialization capabilities.
Business Focus
We have elected to diversify our research and development activities across a relatively broad array of investment opportunities, due in part to the high risks associated with the biotechnology and pharmaceutical business. This diversification strategy requires more significant financial resources than would be required if we pursued a more limited approach. We are expending significant resources on development and commercialization of the drug candidates for which we currently have principal clinical development responsibility, in those markets where we have commercial rights. We rely on collaborators to develop and commercialize certain of our other drug candidates either worldwide or in the markets upon which we are not currently focused.
To date, we have relied on pharmaceutical company collaborators to develop and market our drug candidates that have advanced to late stage clinical trials or commercialization. Telaprevir is the first drug candidate for which we expect to perform all activities related to late stage development, drug supply, registration and commercialization in a major market. We have limited experience in Phase 3 clinical development, supply chain management, or pharmaceutical sales and marketing, and we are building those
54
capabilities as we advance telaprevir through clinical development. Even though telaprevir is a Phase 2b drug candidate, we are planning for and investing significant resources now in preparation for Phase 3 clinical trials, application for marketing approval, commercial supply and sales and marketing. Our engagement in these resource-intensive activities could make it more difficult for us to maintain our portfolio focus, and puts significant investment at risk if we do not obtain regulatory approval and successfully commercialize telaprevir in North America. While we attempt to stage our investments in each drug candidate to coincide to some degree with the occurence of risk-reducing events associated with the development of that drug candidate, we may not be able through this approach to reduce significantly the overall financial risk associated with our drug development activities. There is no assurance that our development of telaprevir will lead successfully to regulatory approval, or that obtaining regulatory approval will lead to commercial success.
In the past, we have sought collaborator funding for a significant portion of our research activities, which required that we grant to those collaborators significant rights to develop and commercialize drug candidates generated by that research. In the future, we expect that we will fund a greater proportion of our research programs than in past years, using internal funds rather than collaborator funds. We believe that this strategy will ultimately allow us to retain greater development control of, and commercial rights with respect to, those proprietary drug candidates that may meet our strategic internal investment criteria as in effect from time to time.
Discovery and Development Process
Discovery and development of a new pharmaceutical product is a lengthy and resource-intensive process, which may take 10 to 15 years or more. Throughout this entire process, potential drug candidates are subjected to rigorous evaluation, driven in part by stringent regulatory considerations, designed to generate information concerning efficacy, proper dosage levels and a variety of other physical and chemical characteristics that are important in determining whether a drug candidate should be approved for marketing. The toxicity characteristics and profile of drug candidates at varying dose levels administered for varying periods of time also are monitored and evaluated during the nonclinical and clinical development process. Most chemical compounds that are investigated as potential drug candidates never progress into formal development, and most drug candidates that do advance into formal development never become commercial products. A drug candidate’s failure to progress or advance may be the result of any one or more of a wide range of adverse experimental outcomes including, for example, the lack of sufficient efficacy against the disease target, the lack of acceptable absorption characteristics or other physical properties, difficulties in developing a cost-effective manufacturing or formulation methods or the discovery of toxicities or side effects that are unacceptable for the disease indication being treated.
Given the uncertainties of the research and development process, it is not possible to predict with confidence which, if any, of our current research and development efforts will result in a marketable pharmaceutical product. We monitor the results of our discovery research and our nonclinical and clinical trials and frequently evaluate our portfolio investments in light of new data and scientific, business and commercial insights with the objective of balancing risk and potential. This process can result in relatively abrupt changes in focus and priority as new information becomes available and we gain additional insights into ongoing programs and potential new programs.
Clinical Development Programs
Our development of telaprevir illustrates our focus on maintaining greater development control of our drug candidates. We currently are conducting later stage clinical trials of telaprevir and expect to initiate a Phase 3 program for of telaprevir in the second half of 2007. Designing and coordinating large-scale clinical trials to determine the efficacy and safety of drug candidates and support the submission of an NDA requires significant financial resources, along with extensive technical and regulatory expertise and infrastructure.
55
We are conducting three major Phase 2b clinical trials of telaprevir. PROVE 1 is ongoing in the United States and PROVE 2 is ongoing in European Union, both in treatment-naïve patients. PROVE 3 has commenced and is being conducted with patients in North America and the European Union who did not achieve SVR with previous interferon-based treatments. We expect the results from these clinical trials to provide important information supporting the design and initiation of the Phase 3 clinical program for telaprevir.
In 2007, we plan to conduct Phase 2 clinical trials of VX-702 in patients with RA, a Phase 2 clinical trial of VX-770 in patients with CF, and a Phase 1 clinical trial of VX-883.
Each of these programs requires a significant investment of financial and personnel resources, time and expertise by us and/or any program collaborators to realize its full clinical and commercial value. Development investment at this stage is subject to the considerable risk that any one or more of these drug candidates will not advance to product registration. Each drug candidate could fail to progress or advance due to a wide range of adverse experimental outcomes, placing our investment in the drug candidate at risk. While we attempt to stage our investments to mitigate these financial risks, drug discovery and development by its nature is a very risky undertaking and staging of investment is not always possible or desirable. We expect to continue to evaluate and prioritize investment in our clinical development programs based on the emergence of new clinical and nonclinical data in each program throughout 2007 and in subsequent years.
Manufacturing and Commercialization Strategy
We have retained manufacturing and commercialization responsibilities for telaprevir in North America. In 2007, we expect to undertake significant efforts to prepare for the commercial supply and marketing of telaprevir, in support of a timely and effective commercial product launch in subsequent years if we are successful in obtaining regulatory marketing approval. Establishing the commercial supply chain for telaprevir is a multi-step international endeavor involving the purchase of several raw materials, the application of certain manufacturing processes requiring significant lead times, the conversion of active pharmaceutical ingredient to tablet form and the packaging of tablets for distribution. We expect to source raw materials, drug substance and drug product, including finished packaging, from third parties located in the Far East, the European Union and the United States, and we are currently establishing and expanding third-party relationships in this regard. Establishing and providing quality assurance for this global supply chain requires a significant financial commitment, experienced personnel and the creation or expansion of numerous third-party contractual relationships. Because of the significant lead times involved in our supply chain for telaprevir, we may have less flexibility to adjust our supply in response to changes in demand than if we had shorter lead times. We have successfully completed the technical development work for the Phase 3 and commercial formulation of telaprevir. We expect that the level of our investment in commercial supply of telaprevir, including costs related to building our internal infrastructure and costs related to third-party manufacturing relationships, will increase significantly in 2007. While most of this investment will relate specifically to telaprevir, and is at risk if telaprevir does not advance successfully to registration, we expect that the organization and expertise that we build as part of this process will contribute to our development as a pharmaceutical company, and that it could serve the advancement of our other, earlier-stage drug candidates if they progress to a commercial manufacturing stage.
Similarly, we currently have limited experience as a company in sales and marketing or with respect to pricing and obtaining adequate third-party reimbursement for drugs. In order to market telaprevir in North America if it is approved, we intend to build a marketing organization and a direct sales force, which will require substantial effort and significant management and financial resources. During 2007, we intend to commit significant personnel and financial resources to this effort, staging our commitments to the extent possible in consideration of the ongoing telaprevir development timeline. We will need to devote significant effort, in particular, to recruiting individuals with experience in the sales and marketing of pharmaceutical products. Competition for personnel with these skills is intense and may be particularly difficult for us since telaprevir is still an investigational drug candidate. Although our investment in this
56
infrastructure might be lost if telaprevir is not approved or if approval is significantly delayed, we would expect our sales and marketing infrastructure for telaprevir, if telaprevir is successfully developed and commercialized, to be useful to us if and when we commercialize any additional drugs.
Financing Strategy
At December 31, 2006, we had $761.8 million of cash, cash equivalents and marketable securities, $42.1 million in principal amount of 5% Convertible Subordinated Notes due September 2007, which we refer to as the 2007 Notes, and $59.6 million in principal amount of 5.75% Convertible Senior Subordinated Notes due in February 2011, which we refer to as the 2011 Notes. Because we have incurred losses from our inception and expect to incur losses for the foreseeable future, we are dependent in large part on our continued ability to raise significant funding to finance our research and development operations, our creation of a commercial infrastructure and our overhead, and to meet our long-term contractual commitments and obligations. In the past, we have secured funds principally through capital market transactions, strategic collaborative agreements, proceeds from the disposition of assets, investment income and the issuance of stock under our employee benefit programs. For example, we received $165.0 million in July 2006 as an up-front payment under our collaboration agreement with Janssen and we completed an offering of 10 million shares of our common stock in September 2006, resulting in net proceeds to us of approximately $313.7 million. In addition, we decreased the aggregate outstanding principal amount of our convertible debt by $58.4 million in 2006 through the issuance of approximately 4.1 million shares of our common stock.
In February 2007, we announced that we will redeem our 2011 Notes on March 5, 2007. The 2011 Notes are convertible into shares of our common stock at the option of the holder at a price equal to $14.94 per share. We expect the holders of the 2011 Notes will elect to convert their notes into stock, in which case we will issue approximately 4.0 million shares. We will be required to repay any 2011 Notes that are not converted at the rate of $1,003.19 per $1,000 principal amount, which includes principal and interest that will accrue to the redemption date.
In order to fund our research, development and manufacturing activities, particularly for later stage drug candidates, we expect to continue to pursue a general financing strategy that may lead us to undertake one or more additional capital transactions, which may or may not be similar to transactions in which we have engaged in the past. We cannot be sure that any such financing opportunities will be available on acceptable terms if at all.
Collaborations and Collaborative Revenues
Collaborations have been and will continue to be an important component of our business strategy. We receive royalty revenues from GlaxoSmithKline on their sales of our HIV protease inhibitor fosamprenavir calcium, which is marketed as Lexiva in the United States and Telzir in Europe. In June 2006, we entered into a collaboration agreement with Janssen Pharmaceutica, N.V., a Johnson & Johnson company, relating to telaprevir. In July 2006, Janssen paid us a non-refundable license payment of $165 million. Under our agreement with Janssen, we have retained exclusive commercial rights to telaprevir in North America and will lead the global clinical development program. Janssen has agreed to be responsible for 50% of the drug development costs under the development program for telaprevir in North America and the Janssen territories, to pay us additional contingent milestone payments based on successful development, approval and launch of telaprevir, and to be responsible for the commercialization of telaprevir outside of North America and the Far East.
57
Our pipeline also includes several drug candidates that are being developed by our collaborators, including:
· MK-0457 (VX-680) and MK-6592 (VX-677), which are being investigated by Merck for oncology indications;
· AVN-944 (VX-944), which is being investigated by Avalon Pharmaceuticals, Inc. for oncology indications; and
· VX-409 and back-up compounds, which are being investigated by GlaxoSmithKline for pain indications.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations is based upon our consolidated financial statements prepared in accordance with generally accepted accounting principles in the United States, or GAAP. The preparation of these financial statements requires us to make certain estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reported periods. These items are monitored and analyzed by management for changes in facts and circumstances, and material changes in these estimates could occur in the future. Changes in estimates are reflected in reported results for the period in which they become known. We base our estimates on historical experience and various other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from our estimates if past experience or other assumptions do not turn out to be substantially accurate.
We believe that our application of the accounting policies for restructuring, revenue recognition, research and development expenses, investments and stock-based compensation, all of which are important to our financial condition and results of operations, require significant judgments and estimates on the part of management. Our accounting polices, including the ones discussed below, are more fully described in Note B, “Accounting Policies,” to our consolidated financial statements included in this Annual Report on Form 10-K.
Revenue Recognition
We recognize revenue in accordance with the SEC’s Staff Accounting Bulletin No. 101, “Revenue Recognition in Financial Statements” (“SAB 101”), as amended by SEC Staff Accounting Bulletin No. 104, “Revenue Recognition” (“SAB 104”), and for revenue arrangements entered into after June 30, 2003, Emerging Issues Task Force Issue No. 00-21, “Revenue Arrangements with Multiple Deliverables” (“EITF 00-21”).
Our revenues are generated primarily through collaborative research, development, manufacture and commercialization agreements. The terms of these agreements typically include payment to us of one or more of the following: non-refundable up-front license fees, research and development funding, milestone payments and royalties on product sales.
Agreements containing multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the collaborator and whether there is objective and reliable evidence of fair value of the undelivered obligation(s). The consideration received is allocated among the separate units based on each unit’s fair value or using the residual method, and the applicable revenue recognition criteria are applied to each of the separate units.
We recognize revenues from non-refundable, up-front license fees on a straight-line basis over the contracted or estimated period of performance, which is typically the research or development term. Research and development funding is recognized as earned, ratably over the period of effort.
58
Substantive milestones realized in collaboration arrangements are recognized as earned when the corresponding payment is reasonably assured, subject to the following policies in those circumstances where we have obligations remaining after achievement of the milestone:
· In those circumstances where collection of a substantive milestone is reasonably assured, we have remaining obligations to perform under the collaboration arrangement and we have sufficient evidence of fair value for our remaining obligations, we consider the milestone payment and the remaining obligations to be separate units of accounting. In these circumstances, we use the residual method under EITF 00-21 to allocate revenue among the milestones and the remaining obligations.
· In those circumstances where collection of a substantive milestone is reasonably assured, we have remaining obligations to perform under the collaboration arrangement, and we do not have sufficient evidence of fair value for our remaining obligations, we consider the milestone payment and the remaining obligations under the contract as a single unit of accounting. In those circumstances where the collaboration does not require specific deliverables at specific times or at the end of the contract term, but rather our obligations are satisfied over a period of time, substantive milestones are recognized over the period of performance. This typically results in a portion of the milestone payment being recognized as revenue on the date the milestone is achieved equal to the applicable percentage of the performance period that has elapsed as of the date the milestone is achieved, with the balance being deferred and recognized over the remaining period of performance.
We evaluate whether milestones are substantive at the inception of the agreement based on the contingent nature of the milestone, specifically reviewing factors such as the scientific and other risks that must be overcome to achieve the milestone as well as the level of effort and investment required. Milestones that are not considered substantive and do not meet the separation criteria are accounted for as license payments and recognized on a straight-line basis over the remaining period of performance.
Payments received after performance obligations are met completely are recognized when earned.
Royalty revenues are recognized based upon actual and estimated net sales of licensed products in licensed territories as provided by the licensee and are recognized in the period the sales occur. Differences between actual royalty revenues and estimated royalty revenues, which have not historically been significant, are reconciled and adjusted for in the quarter they become known.
Research and Development Expenses
All research and development expenses, including amounts funded by research and development collaborations, are expensed as incurred. Research and development expenses are comprised of costs incurred in performing research and development activities, including salaries and benefits; stock-based compensation expense; laboratory supplies; contract services, including clinical trial and pharmaceutical development costs; expenses associated with the commercial supply investment in telaprevir (due to telaprevir’s stage of development); and infrastructure costs, including facilities costs and depreciation. When third-party service providers’ billing terms do not coincide with our period-end, we are required to make estimates of the costs, including clinical trial costs, contract services and investment in commercial supply, incurred in a given accounting period and record accruals at period-end. We base our estimates on our knowledge of the research and development programs, services performed for the period, past history for related activities and the expected duration of the third-party service contract, where applicable.
Restructuring Expense
We record liabilities associated with restructuring activities based on estimates of fair value in the period the liabilities are incurred, in accordance with Financial Accounting Standards Board Statement No. 146, “Accounting for Costs Associated with Exit or Disposal Activities” (“FAS 146”). The liability for accrued restructuring expense of $33.1 million at December 31, 2006 is related to that portion of our
59
facility in Kendall Square, Cambridge, Massachusetts that we are not occupying and do not intend to occupy. This liability is calculated by applying our best estimate of our net ongoing obligation. As prescribed by FAS 146, we use a probability-weighted discounted cash-flow analysis to calculate the amount of this liability. The probability-weighted discounted cash-flow analysis is based on management’s assumptions and estimates of our ongoing lease obligations, including contractual rental commitments, build-out commitments and building operating costs, and estimates of income from subleases, based on the term and timing of such subleases. We discount the estimated cash flows using a discount rate of approximately 10%. These cash flow estimates are reviewed and may be adjusted in subsequent periods. Adjustments are based, among other things, on management’s assessment of changes in factors underlying the estimates. Because our estimate of the liability includes the application of a discount rate to reflect the time-value of money, the estimate will increase simply as a result of the passage of time, even if all other factors remain unchanged.
Our estimates of our restructuring liability have changed in the past, and it is possible that our assumptions and estimates will change in the future, resulting in additional adjustments to the amount of the estimated liability. The effect of any such adjustments could be material. For example, we currently have two subleases for portions of the Kendall Square facility with remaining terms of five and six years, respectively, and we have made certain estimates and assumptions relating to future sublease terms following the expiration of the current subleases. Market variability may require adjustments to those assumptions in the future. We will review our assumptions and judgments related to the lease restructuring on at least a quarterly basis until the Kendall Square lease is terminated or expires, and make whatever modifications we believe are necessary, based on our best judgment, to reflect any changed circumstances.
Stock-based Compensation Expense
We adopted the provisions of Statement of Financial Accounting Standards Board No. 123(R), “Share-Based Payment” (“FAS 123(R)”), on January 1, 2006. FAS 123(R) requires us to measure compensation cost of stock-based compensation at the grant date, based on the fair value of the award, including estimated forfeitures, and to recognize that cost as an expense ratably over the employee’s requisite service period (generally the vesting period of the equity award). Prior to January 1, 2006, we accounted for stock-based compensation to employees in accordance with Accounting Principles Board Opinion No. 25, “Accounting for Stock Issued to Employees” (“APB 25”), and related interpretations. We also followed the disclosure requirements of Statement of Financial Accounting Standards No. 123, “Accounting for Stock-Based Compensation” (“FAS 123”). We elected to adopt the modified prospective transition method as provided by FAS 123(R) and accordingly, financial statement amounts for the periods prior to January 1, 2006 that are presented in this Form 10-K have not been restated to reflect the fair value method.
Under FAS 123(R), we determine the fair value of awarded stock options and shares issued under the employee stock purchase plan using the Black-Scholes valuation model. The Black-Scholes valuation model requires us to make certain assumptions and estimates concerning our stock price volatility, the rate of return of risk-free investments, the expected term of the awards, and our anticipated dividends. In determining the amount of expense to be recorded, we also are required to exercise judgment to estimate forfeiture rates for awards, based on the probability that employees will complete the required service period. If actual forfeitures differ significantly from our estimates, our results could be materially affected.
Altus Investment
In 2004 and 2005, we assessed our investment in Altus Pharmaceuticals Inc., which we accounted for using the cost method, on a quarterly basis to determine if there had been any decrease in the estimated fair value of that investment below its $18.9 million carrying value that might have required us to write down the cost basis of the asset. In 2004 and 2005, we did not identify facts or circumstances that would cause us to determine that the cost basis of our interest in Altus should have been changed. If any adjustment to the estimated fair value of the investment had reflected a decline in the value of the
60
investment below its cost, we would have considered the evidence available to us, including the duration and extent to which the decline was other-than-temporary. If the decline had been considered other-than-temporary, the cost basis of the investment would have been written down to fair value as a new cost basis and the amount of the write-down would have been included in the consolidated statements of operations. Altus completed an initial public offering of its common stock in January 2006. In 2006, we sold 817,749 shares of Altus common stock and warrants to purchase 1,962,494 shares of Altus common stock for $30.0 million, resulting in realized net gain of $11.2 million, based on the difference between the proceeds of the sales and the carrying value of the asset.
RESULTS OF OPERATIONS
Year Ended December 31, 2006 Compared with Year Ended December 31, 2005
Our net loss for 2006 was $206.9 million, or $1.83 per basic and diluted common share, compared to a net loss for 2005 of $203.4 million, or $2.28 per basic and diluted common share. Included in our net loss for 2006 was stock-based compensation expense of $39.1 million, restructuring expense of $3.7 million, loss on exchange of convertible subordinated notes of $5.2 million, gains related to an investment of $11.2 million and the effect of a cumulative benefit of accounting change, related to the adoption of FAS 123(R) of $1.0 million. Included in our net loss for 2005 was stock-based compensation expense of $4.6 million, net restructuring expense of $8.1 million and loss on exchange of convertible subordinated notes of $48.2 million.
While our net loss for 2006 increased by only $3.5 million as compared to 2005, our revenues and expenses changed significantly period to period. In particular, our research and development expenses increased by $123.2 million from 2005 to 2006, including a $28.4 million increase in stock-based compensation expense. Overall, our total costs and expenses increased by $134.6 million from 2005 to 2006. These increased costs and expenses were partially offset by the $55.5 million increase in revenues from 2005 to 2006. Our net loss per basic and diluted common share decreased in 2006 from 2005 as a result of an increase in the basic and diluted weighted average number of common shares outstanding from 89.2 million shares to 113.2 million shares, which offset the increase in the net loss.
Revenues
Total revenues increased to $216.4 million in 2006 compared to $160.9 million in 2005. In 2006, revenues were comprised of $41.2 million in royalties and $175.1 million in collaborative and other research and development revenues, as compared with $32.8 million in royalties and $128.1 million in collaborative and other research and development revenues in 2005.
Royalty revenues increased by $8.4 million, or 26%, from 2005 to 2006. Royalties consist of Lexiva/Telzir (fosamprenavir calcium) royalty revenues and a small amount of Agenerase (amprenavir) royalty revenues. Fosamprenavir calcium is marketed under the trade name Lexiva in the United States and Telzir in the European Union. Royalty revenues are based on actual and estimated worldwide net sales of Lexiva/Telzir and Agenerase. The increase in royalty revenues was due to the increase in Lexiva/Telzir sales. By the end of 2005, Lexiva/Telzir had largely replaced Agenerase in worldwide markets.
61
Collaborative and other research and development revenues increased $47.1 million, or 37%, from 2005 to 2006. The table presented below is a summary of revenues from collaborative arrangements for 2006 as compared with 2005.
| | 2006 | | 2005 | |
| | (In thousands) | |
Collaborative and other research and development revenues: | | | | | |
Janssen | | $ | 68,004 | | $ | — | |
Merck | | 58,705 | | 24,428 | |
Novartis | | 17,585 | | 53,082 | |
CFFT | | 12,636 | | 14,490 | |
GlaxoSmithKline | | 2,434 | | 20,000 | |
Other | | 15,784 | | 16,061 | |
Total collaborative and other research and development revenues | | $ | 175,148 | | $ | 128,061 | |
In 2006, we entered into one new major collaboration agreement, with Janssen, which resulted in $68.0 million of revenues in 2006, including:
· an amortized portion of the $165.0 million up-front payment;
· payments from Janssen to fund a portion of telaprevir development costs; and
· a milestone of $15.0 million.
Our revenues from Merck increased by $34.3 million in 2006 over 2005 levels as the result of increased revenues recognized from milestone payments offset by decreased research funding. We recognized lower revenues from Novartis due to the completion of our research collaboration with Novartis. Our revenues from our GlaxoSmithKline collaboration were higher in 2005 due to our full recognition in 2005 of the $20.0 million up-front license payment paid under that agreement.
In 2007, we expect that our revenues from Janssen will increase significantly as a result of the recognition over a full year of an amortized portion of the up-front payment made to us by Janssen in 2006, a full year of telaprevir development cost sharing under our collaboration agreement with Janssen and potentially additional milestone payments. We expect that the increased Janssen revenues will be partially offset by decreased revenues from Novartis and Merck as a result of the completion of the research programs with these collaborators. Merck is continuing to develop drug candidates identified under the collaboration, and we may receive additional revenues in 2007 from Merck related to milestone payments. We do not expect to receive any further revenues from Novartis under our collaboration agreement. We expect that for the foreseeable future the revenues and funding from collaborations that support our development-stage compounds, such as the Janssen and Merck collaborations, will provide a proportionately higher level of financial support for our research and development activities than revenues and funding from research collaboration agreements.
Costs and Expenses
Royalty Payments
Royalty payments increased $2.1 million, or 21%, to $12.2 million in 2006 from $10.1 million in 2005. Royalty payments relate to a royalty we pay to a third party on sales of Lexiva/Telzir and Agenerase. The increased royalty payments relate to the increased royalty revenues we received in 2006 as compared to 2005.
Research and Development Expenses
Research and development expenses increased $123.2 million, or 50%, to $371.7 million in 2006, including stock-based compensation expense of $32.0 million, from $248.5 million in 2005, including stock-based compensation of $3.6 million. The increase in research and development expenses was primarily the result of increased development investment to support the global Phase 2b clinical
62
development program for telaprevir, as well as $27.3 million of investment in commercial supply of telaprevir, together with a $28.4 million increase in stock-based compensation expense. The cost of developing the commercial supply for telaprevir is considered a research and development expense due to telaprevir’s stage of development. Development expenses increased by $101.3 million, accounting for 82% of the aggregate increase in research and development expenses. Research expenses increased by $21.9 million, of which $13.9 million was increased stock-based compensation expense.
Research and development expenses consist primarily of salary and benefits, stock-based compensation expense, laboratory supplies, contractual services (including pharmaceutical development and clinical trial materials costs), commercial supply investment in telaprevir, and infrastructure costs, including facilities costs and depreciation. Set forth below is a summary that reconciles our total research and development expenses for 2006 and 2005:
| | 2006 | | 2005 | | $ Change | | % Change | |
| | (in thousands) | |
Research Expenses: | | | | | | | | | | | |
Salary and benefits | | $ | 45,546 | | $ | 40,019 | | $ | 5,527 | | | 14 | % | |
Stock-based compensation expense | | 15,879 | | 1,979 | | 13,900 | | | 702 | % | |
Laboratory supplies and other direct expenses | | 23,103 | | 20,877 | | 2,226 | | | 11 | % | |
Contractual services | | 6,640 | | 7,619 | | (979 | ) | | (13 | )% | |
Infrastructure costs | | 51,479 | | 50,285 | | 1,194 | | | 2 | % | |
Total research expenses | | $ | 142,647 | | $ | 120,779 | | $ | 21,868 | | | | | |
Development Expenses: | | | | | | | | | | | |
Salary and benefits | | $ | 40,424 | | $ | 27,202 | | $ | 13,222 | | | 49 | % | |
Stock-based compensation expense | | 16,123 | | 1,588 | | 14,535 | | | 915 | % | |
Laboratory supplies and other direct expenses | | 19,041 | | 11,674 | | 7,367 | | | 63 | % | |
Contractual services | | 86,146 | | 61,188 | | 24,958 | | | 41 | % | |
Commercial supply investment in telaprevir | | 27,332 | | — | | 27,332 | | | 100 | % | |
Infrastructure costs | | 40,000 | | 26,109 | | 13,891 | | | 53 | % | |
Total development expenses | | $ | 229,066 | | $ | 127,761 | | $ | 101,305 | | | | | |
Total Research and Development Expenses: | | | | | | | | | | | |
Salary and benefits | | $ | 85,970 | | $ | 67,221 | | $ | 18,749 | | | 28 | % | |
Stock-based compensation expense | | 32,002 | | 3,567 | | 28,435 | | | 797 | % | |
Laboratory supplies and other direct expenses | | 42,144 | | 32,551 | | 9,593 | | | 29 | % | |
Contractual services | | 92,786 | | 68,807 | | 23,979 | | | 35 | % | |
Commercial supply investment in telaprevir | | 27,332 | | — | | 27,332 | | | 100 | % | |
Infrastructure costs | | 91,479 | | 76,394 | | 15,085 | | | 20 | % | |
Total research and development expenses | | $ | 371,713 | | $ | 248,540 | | $ | 123,173 | | | | | |
To date we have incurred in excess of $1.5 billion in research and development costs associated with drug discovery and development. In 2007, we expect to focus our development investment on telaprevir, while continuing to advance the development of VX-702, VX-770, and VX-883. We expect research and development expenses in 2007 to be greater than in 2006 due to increased investment in clinical development, as we advance our core programs, as well as increased costs for the investment in commercial supply of telaprevir drug product in advance of obtaining regulatory marketing approval. In 2007, we expect to incur research and development expenses of approximately $110 million to $130 million for commercial supply investment in telaprevir. We are making this investment to support a timely commercial product launch if we are successful in completing development of telaprevir and obtaining marketing approval. In addition, we expect that our combined research and development expenses will increase in future periods as we add personnel and capabilities to support the planned development of our lead drug candidates. However, our anticipated 2007 research and development expenses could vary materially from our projections, depending on the occurrence and timing of clinical trials and clinical trial results.
63
The successful development of our drug candidates is highly uncertain and subject to a number of risk factors. The duration of clinical trials may vary substantially according to the type, complexity and novelty of the drug candidate. The FDA and comparable agencies in foreign countries impose substantial requirements on the introduction of therapeutic pharmaceutical products, typically requiring lengthy and detailed laboratory and clinical testing procedures, sampling activities and other costly and time-consuming procedures. Data obtained from preclinical, nonclinical and clinical activities at any step in the testing process may be adverse and lead to discontinuation or redirection of development activity. Data obtained from these activities also are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The duration and cost of discovery, preclinical studies, nonclinical studies and clinical trials may vary significantly over the life of a project and are difficult to predict. Therefore, accurate and meaningful estimates of the ultimate costs to bring our drug candidates to market are not available. The most significant costs associated with drug discovery and development are those costs associated with Phase 2 and Phase 3 clinical trials. Given the uncertainties related to development, we are currently unable to reliably estimate when, if ever, our drug candidates will generate revenue and net cash inflows.
Sales, General and Administrative Expenses
Sales, general and administrative expenses increased $13.9 million, or 32%, to $57.9 million in 2006 from $44.0 million in 2005. This increase is the result of increased headcount to support our growth as we advance our drug candidates, particularly telaprevir, into late-stage development. We expect that our sales, general and administration expenses in 2007 will be significantly higher than in 2006, because we are planning to build our capabilities in late-stage development, drug supply, registration and commercialization of pharmaceutical products, as we advance telaprevir through clinical development.
Restructuring Expense
We recorded net restructuring expense of $3.7 million in 2006 compared to a restructuring expense of $8.1 million in 2005. The expense in 2006 resulted primarily from imputed interest and build-out costs related to the restructuring liability. The expense for 2005 included a credit against the portion of restructuring liability relating to the portion of the Kendall Square facility that we decided in 2005 to occupy, offset by (i) the estimated incremental net ongoing lease obligation associated with the portion of the Kendall Square facility that we are not occupying and do not intend to occupy and (ii) imputed interest costs relating to the restructuring liability.
The activity related to the restructuring liability for 2006 is as follows (in thousands):
| | Liability as of
December 31, 2005 | | Cash Payments
in 2006 | | Cash Received
from Subleases
in 2006 | | Additional
Charge
in 2006 | | Liability as of
December 31,
2006 | |
Lease restructuring liability | | | $ | 42,982 | | | | $ | (21,607 | ) | | | $ | 8,047 | | | | $ | 3,651 | | | | $ | 33,073 | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
The activity related to the restructuring liability for 2005 is as follows (in thousands):
| | Liability as of
December 31, 2004 | | Cash Payments
in 2005 | | Cash Received
from Subleases
in 2005 | | Credit for
portion of
facility
Vertex
decided to
occupy | | Additional
Charge
in 2005 | | Liability as of
December 31,
2005 | |
Lease restructuring liability | | | $ | 55,843 | | | | $ | (24,229 | ) | | | $ | 3,234 | | | | $ | (10,018 | ) | | | $ | 18,152 | | | | $ | 42,982 | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
In accordance with FAS 146, we review our estimates and assumptions with respect to the Kendall Square lease on at least a quarterly basis, and will make whatever modifications we believe necessary to reflect any changed circumstances, based on our best judgment, until the termination of the lease. Our estimates have changed in the past, and may change in the future, resulting in additional adjustments to the estimate of liability, and the effect of any such adjustments could be material. Because our estimate of the liability includes the application of a discount rate to reflect the time-value of money, the estimate of the liability will increase each quarter simply as a result of the passage of time.
64
Non-Operating Items
Interest income increased $11.0 million, or 92%, to $23.0 million in 2006 from $12.0 million in 2005. The increase was the result of higher invested funds and portfolio yields.
Interest expense decreased $9.4 million, or 54%, to $8.0 million in 2006 from $17.3 million in 2005, because we reduced the level of our outstanding debt in 2006 from 2005.
In 2006, we sold 817,749 shares of the common stock of Altus Pharmaceuticals, Inc. for $11.7 million and warrants to purchase 1,962,494 shares of Altus common stock for $18.3 million, resulting in a realized gain of $11.2 million.
In addition, as a result of the issuance during 2005 and 2006 of common stock in exchange for a portion of our 2007 Notes and 2011 Notes, we recorded non-cash charges of $5.2 million in 2006 and $48.2 million in 2005. These charges related to the incremental shares issued in the transactions over the number of shares that would have been issued upon the conversion of the notes under their original terms.
In connection with the adoption of FAS 123(R), during 2006 we recorded a $1.0 million benefit from the cumulative effect of changing from recording forfeitures related to restricted stock awards as they occurred, to estimating forfeitures during the service period.
Year Ended December 31, 2005 Compared with Year Ended December 31, 2004
Our net loss for 2005 was $203.4 million, or $2.28 per basic and diluted common share, compared to a net loss for 2004 of $166.2 million, or $2.12 per basic and diluted common share. Our loss in 2005 included restructuring expense of $8.1 million and a charge of $48.2 million for the exchange of newly issued common stock for a portion of our outstanding convertible notes. Our loss in 2004 included restructuring expense of $17.6 million and a charge for the retirement of convertible notes of $3.4 million.
Our net loss for 2005 increased by $37.2 million compared to 2004, and our revenues and expenses changed significantly from period to period. Our total revenues increased by $58.2 million from 2004 to 2005. This increase was largely offset by a $56.4 million increase in research and development expenses. In addition, our net loss in 2005 included a $48.2 million charge on exchange of convertible notes for which there was no comparable charge in 2004. Our net loss per basic and diluted share increased due to an increase in our net loss partially offset by an increase in the basic and diluted weighted average number of common shares outstanding from 78.6 million shares to 89.2 million shares.
Revenues
Total revenues increased to $160.9 million in 2005 compared to $102.7 million in 2004. In 2005, revenues were comprised of $32.8 million in royalties and $128.1 million in collaborative and other research and development revenues, as compared with $17.3 million in royalties and $85.4 million in collaborative and other research and development revenues in 2004.
Royalty revenues increased by $15.5 million, or 90%, from 2004 to 2005. Royalties consisted of Lexiva/Telzir (fosamprenavir calcium) and Agenerase (amprenavir) royalty revenue. The increase in royalty revenues was due to an increase in Lexiva/Telzir sales.
Collaborative and other research and development revenues increased $42.7 million, or 50%, in 2005 as compared with 2004. The increase in collaborative and other research and development revenues was due to revenues related to collaboration agreements we entered into in 2004 and in 2005. In 2005, we entered into collaboration agreements with Avalon Pharmaceuticals and GlaxoSmithKline. Under these collaboration agreements, in 2005 we earned $5.0 million in up-front license fees from Avalon and $20.0 million in up-front license fees from GlaxoSmithKline. In 2004, we entered into collaboration agreements with CFFT, Mitsubishi Pharma Corporation, and Merck. We received $19.5 million in 2005 from two milestone payments made by Merck in our Aurora kinase inhibitor collaboration. We recognized revenues of approximately $7.3 million in 2005 related to these milestones. We earned $2.5 million in 2005 in milestone revenues from Kissei related to the completion of regulatory filings in preparation for Phase 1 clinical development of VX-702 in Japan. The table presented below is a summary of revenues from collaborative arrangements for 2005 as compared with 2004.
65
| | 2005 | | 2004 | |
| | (In thousands) | |
Collaborative and other research and development revenues: | | | | | |
Novartis | | $ | 53,082 | | $ | 50,497 | |
Merck | | 24,428 | | 8,367 | |
GlaxoSmithKline | | 20,000 | | — | |
CFFT | | 14,490 | | 6,792 | |
Other | | 16,061 | | 19,739 | |
Total collaborative and other research and development revenues | | $ | 128,061 | | $ | 85,395 | |
Costs and Expenses
Royalty Payments
Royalty payments increased $4.4 million, or 79%, to $10.1 million in 2005 from $5.6 million in 2004. Royalty payments relate to a royalty we pay to a third party on sales of Lexiva/Telzir and Agenerase. The increased royalty payments related to the increased royalty revenues we received in 2005 as compared to 2004.
Research and Development Expenses
Research and development expenses increased $56.4 million, or 29%, to $248.5 million in 2005, from $192.2 million in 2004. The increase in research and development expenses in 2005 as compared with 2004 was primarily a result of investment in our clinical development programs for telaprevir and VX-702. Development expenses accounted for 87%, or $48.9 million, of the aggregate increase in research and development expenses. In 2005, we incurred costs for Phase 2-enabling activities for telaprevir, we completed enrollment in a 315-patient Phase 2 clinical trial of VX-702 for the treatment of RA, and we continued a Phase 2b clinical trial, referred to as the METRO trial, of merimepodib (VX-497). We initiated the METRO trial during 2004. During 2004, we also completed a Phase 1a trial of telaprevir, and began a Phase 1b evaluation of telaprevir in patients with chronic HCV infection.
66
Research and development expenses consisted primarily of salary and benefits (including stock-based compensation expense), laboratory supplies, contractual services and infrastructure costs, including facilities costs and depreciation. Set forth below is a summary that reconciles our total research and development expenses for 2005 and 2004:
| | 2005 | | 2004 | | $ Change | | % Change | |
| | (In thousands) | |
Research Expenses: | | | | | | | | | | | |
Salary and benefits | | $ | 40,877 | | $ | 36,772 | | $ | 4,105 | | | 11 | % | |
Laboratory supplies and other direct expenses | | 20,877 | | 19,052 | | 1,825 | | | 10 | % | |
Contractual services | | 7,619 | | 8,857 | | (1,238 | ) | | (14 | )% | |
Infrastructure costs | | 51,406 | | 48,595 | | 2,811 | | | 6 | % | |
Total research expenses | | $ | 120,779 | | $ | 113,276 | | $ | 7,503 | | | | | |
Development Expenses: | | | | | | | | | | | |
Salary and benefits | | $ | 28,119 | | $ | 20,493 | | $ | 7,626 | | | 37 | % | |
Laboratory supplies and other direct expenses | | 11,674 | | 7,600 | | 4,074 | | | 54 | % | |
Contractual services | | 61,188 | | 28,837 | | 32,351 | | | 112 | % | |
Infrastructure costs | | 26,780 | | 21,956 | | 4,824 | | | 22 | % | |
Total development expenses | | $ | 127,761 | | $ | 78,886 | | $ | 48,875 | | | | | |
Total Research and Development Expenses: | | | | | | | | | | | |
Salary and benefits | | $ | 68,996 | | $ | 57,265 | | $ | 11,731 | | | 20 | % | |
Laboratory supplies and other direct expenses | | 32,551 | | 26,652 | | 5,899 | | | 22 | % | |
Contractual services | | 68,807 | | 37,694 | | 31,113 | | | 83 | % | |
Infrastructure costs | | 78,186 | | 70,551 | | 7,635 | | | 11 | % | |
Total research and development expenses | | $ | 248,540 | | $ | 192,162 | | $ | 56,378 | | | | | |
Sales, General and Administrative Expenses
Sales, general and administrative expenses of $44.0 million in 2005 remained consistent with 2004 expenses of $42.1 million.
Restructuring Expense
We recorded net restructuring expense of $8.1 million in 2005 compared to $17.6 million in 2004. The net restructuring expense in 2005 includes a $10.0 million credit to the restructuring liability made when we decided in mid-2005 to occupy and use a portion of the Kendall Square facility for our operations, which was offset by (i) the estimated incremental net ongoing lease obligations associated with the portion of the Kendall Square facility that we do not intend to occupy and (ii) imputed interest costs relating to the restructuring liability. The additional restructuring expense in 2004 primarily resulted from revising our estimates and assumptions about when we would identity and receive subtenants and imputing an interest charge for the related restructuring liability.
The activity related to the restructuring liability for 2005 is as follows (in thousands):
| | Liability as of
December 31,
2004 | | Cash Payments
in
2005 | | Cash Received
from Subleases
in
2005 | | Credit for
portion of
facility
Vertex
decided to
occupy | | Additional
Charge
in
2005 | | Liability as of
December 31,
2005 | |
Lease restructuring liability | | | $ | 55,843 | | | | $ | (24,229 | ) | | | $ | 3,234 | | | | $ | (10,018 | ) | | | $ | 18,152 | | | | $ | 42,982 | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
67
The activity related to the restructuring liability for 2004 is as follows (in thousands):
| | Liability as of
December 31,
2003 | | Cash
Payments
in 2004 | | Cash received from
sublease, net of
operating costs
in 2004 | | Additional
Charge in
2004 | | Liability as of
December 31,
2004 | |
Lease restructuring liability and other
operating lease liability | | | $ | 69,526 | | | | $ | (31,550 | ) | | | $ | 293 | | | | $ | 17,574 | | | | $ | 55,843 | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Non-Operating Items
Interest income increased $1.7 million to $12.0 million in 2005 from $10.3 million in 2004. The increase was mainly the result of higher returns on invested funds.
Interest expense decreased $1.0 million, or 5%, to $17.3 million in 2005 from $18.3 million in 2004 as a result of the exchange of newly issued stock for a portion of our outstanding convertible debt in the second half of 2005.
In addition, as a result of the issuance during 2005 of common stock in exchange for convertible subordinated notes, we recorded a non-cash charge of $48.2 million. This charge related to the incremental shares issued in the transactions over the number of shares that would have been issued upon the conversion of the notes under their original terms.
LIQUIDITY AND CAPITAL RESOURCES
We have incurred operating losses since our inception and historically have financed our operations principally through public and private offerings of our equity and debt securities, strategic collaborative agreements that include research and/or development funding, development milestones and royalties on the sales of products, investment income and proceeds from the issuance of stock under our employee benefit programs.
At December 31, 2006, we had cash, cash equivalents and marketable securities of $761.8 million, which was an increase of $354.2 million from $407.5 million at December 31, 2005. The increase was primarily a result of:
· $313.7 million in net proceeds from our September 2006 public offering of common stock;
· $165.0 million from an up-front payment we received in connection with signing the Janssen agreement;
· $52.4 million from the issuance of common stock under our employee benefit plans; and
· $30.0 million from the sale of shares of Altus Pharmaceuticals Inc. common stock and warrants to purchase Altus common stock.
These cash inflows were partially offset by the significant cash expenditures we made in 2006 related to research and development expenses and sales, general and administrative expenses. Capital expenditures for property and equipment during 2006 were $32.4 million.
At December 31, 2006, we had $42.1 million in aggregate principal amount of the 2007 Notes and $59.6 million in aggregate principal amount of the 2011 Notes outstanding. The 2007 Notes are due in September 2007 and are convertible into common stock at the option of the holder at a price equal to $92.26 per share, subject to adjustment under certain circumstances. In February 2007, we announced that we will redeem our 2011 Notes on March 5, 2007. The 2011 Notes are convertible into shares of our common stock at the option of the holder at a price equal to $14.94 per share. We expect the holders of the 2011 Notes will elect to convert their notes into stock, in which case we will issue approximately 4.0 million. We will be required to repay any 2011 Notes that are not converted at the rate of $1,003.19 per $1,000 principal amount, which includes principal and interest that will accrue to the redemption date.
68
In August 2006, we exchanged approximately 4.1 million shares of newly issued common stock for approximately $58.3 million in aggregate principal amount of then outstanding 2011 Notes, plus accrued interest. As a result of this exchange we incurred a non-cash charge of $5.2 million, which related to the incremental shares issued in the transaction over the number that would have been issued upon the conversion of the notes under the original conversion terms.
Our accrued restructuring expense of $33.1 million at December 31, 2006 relates to the portion of the Kendall Square facility that we do not intend to occupy and includes other lease obligations, recorded at net present value. In 2006, we made cash payments of $21.6 million against the accrued expense and received $8.0 million in sublease rental payments. We expect to make cash payments of approximately $21.7 million against the accrued expense in 2007 and receive $7.5 million in sublease rental payments. We review our estimates underlying our accrued restructuring expense on at least a quarterly basis, and the amount of the accrued expense, and consequently any expected future payment, could change with any change in our estimates.
The net increase of $117.9 million in deferred revenue for the year ended December 31, 2006 was a result of deferring a portion of the $165.0 million up-front license payment from Janssen, offset by recognition of revenues in 2006 related to cash payments received from collaborators, primarily Novartis and Merck, in previous periods, which were deferred and recognized over our period of performance in accordance with our revenue recognition policy.
We also achieved $57.8 million in milestones related to our collaboration agreements during 2006. Consistent with our revenue recognition policy, we recognized $56.9 million of this amount and have deferred recognition of the remainder, which will be recognized over the remaining period of our performance under the collaboration agreement.
At December 31, 2006, we had $20.0 million in loans outstanding under the loan facility established under our collaboration with Novartis, which is repayable, without interest, in May 2008.
We expect to continue to make significant investments in our pipeline, particularly in clinical trials of telaprevir, VX-702, VX-770 and VX-883, in our effort to prepare for potential registration, regulatory approval and commercial launch of our existing and future drug candidates. We also expect to continue to make a significant investment in the commercial supply of telaprevir in order to manufacture sufficient quantities of drug product in advance of obtaining regulatory marketing approval, to support a timely commercial product launch if we are successful in completing the development of telaprevir and obtaining marketing approval. We expect to incur losses on a quarterly and annual basis for the foreseeable future.
The adequacy of our available funds to meet our future operating and capital requirements will depend on many factors, including the number, breadth and prospects of our discovery and development programs, the costs and timing of obtaining regulatory approvals for any of our drug candidates and our decisions regarding manufacturing and commercial investments. Collaborations have been and will continue to be an important component of our business strategy.
As part of our strategy for managing our capital structure, we have from time to time adjusted the amount and maturity of our debt obligations through new issues, privately negotiated transactions and market purchases, depending on market conditions and our perceived needs at the time. We expect to continue pursuing a general financial strategy that may lead us to undertake one or more additional capital transactions. Any such capital transactions may or may not be similar to transactions in which we have engaged in the past.
We believe that our current cash, cash equivalents and marketable securities will be sufficient to fund our projected operating requirements for at least the next eighteen months. To the extent that our current cash, cash equivalents and marketable securities, in addition to the above-mentioned sources, are not sufficient to fund our activities, it will be necessary to raise additional funds through public offerings or private placements of our securities or other methods of financing. We also will continue to manage our capital structure and consider all financing opportunities, whenever they may occur, that could strengthen
69
our long-term liquidity profile. There can be no assurance that any such financing opportunities will be available on acceptable terms, if at all.
CONTRACTUAL COMMITMENTS AND OBLIGATIONS
The first part of the following table sets forth commitments and obligations that have been recorded on our consolidated balance sheets at December 31, 2006. Certain other obligations and commitments, while not required under GAAP to be included in the consolidated balance sheets, may have a material impact on liquidity. We have presented these items, all of which we have entered into in the ordinary course of business, in the table below in order to present a more complete picture of our financial position and liquidity.
| | 2007 | | 2008-
2009 | | 2010-
2011 | | 2012
and later | | Total | |
| | (in thousands) | |
Commitments and Obligations Recorded on the Consolidated Balance Sheets at December 31, 2006: | | | | | | | | | | | |
Collaborator development loans | | $ | — | | $ | 19,997 | | $ | — | | $ | — | | $ | 19,997 | |
Convertible subordinated notes | | 101,750 | | — | | — | | — | | 101,750 | |
Additional Commitments and Obligations at December 31, 2006: | | | | | | | | | | | |
Facilities operating leases | | 40,008 | | 76,287 | | 59,555 | | 171,483 | | 347,333 | |
Research and development and other commitments | | 1,094 | | 624 | | — | | — | | 1,718 | |
Total contractual commitments and obligations | | $ | 142,852 | | $ | 96,908 | | $ | 59,555 | | $ | 171,483 | | $ | 470,798 | |
Commitments and Obligations Recorded on the Consolidated Balance Sheets at December 31, 2006
The collaborator development loans in the table above represent indebtedness to Novartis in the amount of approximately $20.0 million, which will be repayable without interest in May 2008.
At December 31, 2006, we had $42.1 million in aggregate principal amount of 2007 Notes and $59.6 million in aggregate principal amount of 2011 Notes outstanding. In February 2007, we announced that we will redeem our 2011 Notes on March 5, 2007. The 2011 Notes are convertible at the option of the holder at a price equal to $14.94 per share. The principal amount of the 2011 Notes are shown in the preceding table as payable in 2007. However, we expect that the entire principal amount of these notes will be converted into common stock prior to March 5, 2007.
Additional Commitments and Obligations Not Required to be Recorded on Consolidated Balance Sheets at December 31, 2006
At December 31, 2006, our future minimum commitments and contractual obligations included facilities operating leases and contractual commitments related to our research and development programs. These items are not required under GAAP to be recorded on our consolidated balance sheets. They are disclosed in the table presented above and described more fully in the following paragraphs in order to provide a more complete picture of our financial position and liquidity at December 31, 2006.
Our future minimum commitments under our Kendall Square lease for the period commencing January 1, 2007, including lease payments, are $22.7 million for 2007, $47.3 for 2008 and 2009, $47.6 million for 2010 and 2011 and $171.2 million through the expiration of the lease in 2018. These amounts are included in the table above. We are using for our operations approximately 40% of the Kendall Square facility. We have entered into two subleases for the remaining rentable square footage at the Kendall Square facility to offset our on-going contractual lease obligations. The subleases will expire in 2011 and 2012 and contain options to extend through 2015 and 2018, respectively. One of the subleases has certain termination provisions beginning in 2010. The future minimum committed income from the
70
subleases is $8.2 million for 2007, $16.3 million for 2008 and 2009, $12.6 million for 2010 and 2011 and $1.7 million for years thereafter. These amounts are not offset against our obligations set forth in the table above. See Note E, “Restructuring” to our consolidated financial statements included in this Annual Report on Form 10-K.
Commitments under research and development programs represent contractual commitments entered into for materials and services in the normal course of business.
Our table detailing contractual commitments and obligations does not include severance pay obligations to certain of our executive officers in the event of a not-for-cause termination under existing employment contracts. The cash amount for which the we might be liable upon any such termination, based on current executive pay and bonus levels, could range from $0 to $1.5 million.
Recent Accounting Pronouncements
In February 2007, the Financial Accounting Standards Board (“FASB”) issued Statement No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities” (“FAS 159”). FAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The objective of FAS 159 is to reduce both complexity in accounting for financial instruments and the volatility in earnings caused by measuring related assets and liabilities differently.
GAAP have required different measurement attributes for different assets and liabilities that can create artificial volatility in earnings. The FASB has stated it believes that FAS 159 helps to mitigate this type of accounting-induced volatility by enabling companies to report related assets and liabilities at fair value, which would likely reduce the need for companies to comply with detailed rules regarding hedge accounting.
FAS 159 also establishes presentation and disclosure requirements designed to facilitate comparisons between companies that choose different measurement attributes for similar types of assets and liabilities.
For example, FAS 159 requires companies to provide additional information that will help investors and other users of financial statements to more easily understand the effect of the company’s choice to use fair value on its earnings. It also requires companies to display the fair value of those assets and liabilities for which the company has chosen to use fair value on the face of the balance sheet. FAS 159 does not eliminate disclosure requirements included in other accounting standards, including requirements for disclosures about fair value measurements included in FASB Statement No. 157, “Fair Value Measurements” (“FAS 157”), and FASB No. 107, “Disclosures about Fair Value of Financial Instruments.”
FAS 159 will be effective as of the beginning of our first fiscal year beginning after November 15, 2007. Early adoption is permitted as of the beginning of the previous fiscal year provided that a company makes that choice in the first 120 days of the relevant fiscal year and also elects to apply the provisions of FAS 157.
In September 2006, FASB issued FAS 157. FAS 157 provides guidance for using fair value to measure assets and liabilities and requires additional disclosure about the use of fair value measures, the information used to measure fair value, and the effect fair-value measurements have on earnings. FAS 157 does not require any new fair value measurements. FAS 157 will be effective for us beginning January 1, 2008. We currently are evaluating the effect of FAS 157 on our consolidated financial statements.
In June 2006, FASB issued FASB Interpretation No. 48, “Accounting for Uncertainty in Income Taxes—an interpretation of FASB Statement No. 109” (“FIN 48”). FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements in accordance with Statement of Financial Accounting Standards No. 109, “Accounting for Income Taxes.” FIN 48 prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. FIN 48 also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure, and
71
transition. FIN 48 is effective for fiscal years beginning after December 15, 2006. We currently are evaluating FIN 48 and believe the adoption of FIN 48 will not have a material effect on our consolidated financial statements.
In May 2005, the FASB issued FAS No. 154, “Accounting Changes and Error Corrections” (“FAS 154”). FAS 154 replaces APB Opinion No. 20, “Accounting Changes” and Statement No. 3, “Reporting Accounting Changes in Interim Financial Statements.” FAS No. 154 requires retrospective application to prior periods’ financial statements of changes in accounting principle, unless it is impractible to determine either the period-specific effects or the cumulative effect of the change. We adopted SFAS 154 beginning on January 1, 2006. Its adoption did not have a material effect on our consolidated financial statements.
In November 2005, FASB issued FSP FAS 115-1 and FAS 124-1, “The Meaning of Other-Than-Temporary Impairment and Its Application to Certain Investments” (“FSP FAS 115-1”), which provides guidance on determining when investments in certain debt and equity securities are considered impaired, whether an impairment is other-than-temporary, and on measuring such impairment loss. FSP FAS 115-1 also includes accounting considerations subsequent to the recognition of an other-than-temporary impairment and requires certain disclosures about unrealized losses that have not been recognized as other-than-temporary impairments. FSP FAS 115-1 is required to be applied to reporting periods beginning after December 15, 2005. We adopted FSP FAS 115-1 in the first quarter of 2006. Adoption of FSP FAS 115-1 did not have a material effect on the our consolidated results of operations or financial condition.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
As part of our investment portfolio, we own financial instruments that are sensitive to market risks. The investment portfolio is used to preserve our capital until it is required to fund operations, including our research and development activities. None of these market risk-sensitive instruments are held for trading purposes. We do not have derivative financial instruments in our investment portfolio.
Interest Rate Risk
We invest our cash in a variety of financial instruments, principally securities issued by the United States government and its agencies, investment grade corporate bonds and notes and money market instruments. These investments are denominated in United States dollars. All of our interest-bearing securities are subject to interest rate risk, and could decline in value if interest rates fluctuate. Substantially all of our investment portfolio consists of marketable securities with active secondary or resale markets to help ensure portfolio liquidity, and we have implemented guidelines limiting the term-to-maturity of our investment instruments. Due to the conservative nature of these instruments, we do not believe that we have a material exposure to interest rate risk.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information required by this Item 8 is contained on pages F-1 through F-42 of this Annual Report on Form 10-K.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES
(1) Evaluation of Disclosure Controls and Procedures. The Company’s chief executive officer and chief financial officer, after evaluating the effectiveness of the Company’s disclosure controls and procedures (as defined in Exchange Act Rule 13a-15(c)) as of the end of the period covered by this Annual
72
Report on Form 10-K, have concluded that, based on such evaluation, the Company’s disclosure controls and procedures were effective. In designing and evaluating the disclosure controls and procedures, the Company’s management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and the Company’s management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
(2) Management’s Annual Report on Internal Control over Financial Reporting. The management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) and Rule 15d-15(f) promulgated under the Securities Exchange Act of 1934 as a process designed by, or under the supervision of, the Company’s principal executive and principal financial officers and effected by the Company’s Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. The Company’s internal control over financial reporting includes those policies and procedures that:
· pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company;
· provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and
· provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2006. In making this assessment, it used the criteria set forth in the Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on its assessment, the Company’s management has concluded that, as of December 31, 2006, the Company’s internal control over financial reporting is effective based on those criteria.
The assessment of the Company’s management of the effectiveness of the Company’s internal control over financial reporting as of December 31, 2006, has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated below.
(3) Changes in Internal Controls. During the quarter ended December 31, 2006, there were no changes in our internal control over financial reporting, that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
(4) Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Vertex Pharmaceuticals Incorporated
We have audited management’s assessment, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting, that Vertex Pharmaceuticals Incorporated maintained effective internal control over financial reporting as of December 31, 2006, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Vertex Pharmaceuticals Incorporated’s
73
management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting. Our responsibility is to express an opinion on management’s assessment and an opinion on the effectiveness of the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, evaluating management’s assessment, testing and evaluating the design and operating effectiveness of internal control, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, management’s assessment that Vertex Pharmaceuticals Incorporated maintained effective internal control over financial reporting as of December 31, 2006, is fairly stated, in all material respects, based on the COSO criteria. Also, in our opinion, Vertex Pharmaceuticals Incorporated maintained, in all material respects, effective internal control over financial reporting as of December 31, 2006, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Vertex Pharmaceuticals Incorporated as of December 31, 2006 and 2005, and the related consolidated statements of operations, stockholders’ equity and comprehensive loss, and cash flows for each of the two years in the period ended December 31, 2006 of Vertex Pharmaceuticals Incorporated and our report dated February 27, 2007 expressed an unqualified opinion thereon.
| | /s/ Ernst & Young LLP |
Boston, Massachusetts | | |
February 27, 2007 | | |
ITEM 9B. OTHER INFORMATION
Not applicable.
74
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information regarding directors required by this Item 10 will be included in the definitive Proxy Statement for our 2007 Annual Meeting of Stockholders, or 2007 Proxy Statement, under “Election of Directors” and “Information Regarding the Board of Directors and its Committees” and is incorporated herein by reference. Other information required by this Item 10 will be included in the 2007 Proxy Statement under “Section 16(a) Beneficial Ownership Reporting Compliance” and “Code of Conduct and Ethics” and is incorporated herein by reference. The information regarding executive officers required by this Item is included in Part I of this Annual Report on Form 10-K.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item 11 will be included in the 2007 Proxy Statement under “Executive Compensation,” and “Compensation Committee Interlocks and Insider Information,” and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item 12 will be included in the 2007 Proxy Statement under “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” and is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item 13 will be included in the 2007 Proxy Statement under “Election of Directors” and “Transactions with Related Persons” and is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this Item 14 will be included in the 2007 Proxy Statement under “Independent Registered Public Accounting Firm” and is incorporated herein by reference.
75
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
(a)(1) The Financial Statements required to be filed by Items 8 and 15(c) of Form 10-K, and filed herewith, are as follows:
| | Page Number in
this Form 10-K | |
Reports of Independent Registered Public Accounting Firms | | | F-1, F-2 | | |
Consolidated Balance Sheets as of December 31, 2006 and 2005 | | | F-3 | | |
Consolidated Statements of Operations for the years ended December 31, 2006, 2005 and 2004 | | | F-4 | | |
Consolidated Statements of Stockholders’ Equity and Comprehensive Loss for the years ended December 31, 2006, 2005 and 2004 | | | F-5 | | |
Consolidated Statements of Cash Flows for the years ended December 31, 2006, 2005 and 2004 | | | F-6 | | |
Notes to Consolidated Financial Statements | | | F-7 | | |
(a)(2) Financial Statement Schedules have been omitted because they are either not applicable or the required information is included in the consolidated financial statements or notes thereto.
(a)(3) Exhibits.
Exhibit
Number | | Exhibit Description | | Filed with
this report | | Incorporated by
Reference herein
from–Form
or Schedule | | Filing Date/
Period Covered | | SEC File/
Reg. Number | |
| 3.1 | | | Restated Articles of Organization of Vertex, filed with the Secretary of State of The Commonwealth of Massachusetts on July 31, 1991. | | | | | | | 10-K
(Exhibit 3.1) | | | March 26, 1998 | | | 000-19319 | | |
| 3.2 | | | Certificate of Vote of Directors Establishing a Series of a Class of Stock, filed with the Secretary of State of The Commonwealth of Massachusetts on July 31, 1991. | | | | | | | 10-K
(Exhibit 3.3) | | | March 26, 1998 | | | 000-19319 | | |
| 3.3 | | | Articles of Amendment of the Articles of Organization of Vertex, filed with the Secretary of State of The Commonwealth of Massachusetts on May 17, 1995. | | | | | | | S-3
(Exhibit 3.3) | | | April 1, 2005 | | | 333-123731 | | |
| 3.4 | | | Articles of Amendment of the Articles of Organization of Vertex, filed with the Secretary of State of The Commonwealth of Massachusetts on June 4, 1997. | | | | | | | 10-K
(Exhibit 3.2) | | | March 26, 1998 | | | 000-19319 | | |
| 3.5 | | | Articles of Amendment of the Articles of Organization of Vertex, filed with the Secretary of State of The Commonwealth of Massachusetts on May 21, 2001 | | | | | | | S-4
(Exhibit 3.4) | | | May 23, 2001 | | | 333- 61480 | | |
| 3.6 | | | By-laws of Vertex, as amended and restated as of May 11, 2005. | | | | | | | 10-Q
(Exhibit 3.1) | | | August 9, 2005 | | | 000-19319 | | |
| 4.1 | | | Specimen stock certificate. | | | | | | | S-1
(Exhibit 4.1) | | | July 18, 1991 | | | 33- 40966 | | |
| 4.2 | | | Rights Agreement, dated as of July 1, 1991. | | | | | | | S-1
(Exhibit 4.2) | | | July 5, 1991 | | | 33-40966 | | |
| 4.3 | | | First Amendment to Rights Agreement, dated as of February 21, 1997. | | | | | | | 10-K
(Exhibit 4.3) | | | March 28, 1997 | | | 000-19319 | | |
| 4.4 | | | Second Amendment to Rights Agreement, dated as of June 30, 2001. | | | | | | | 10-Q
(Exhibit 4.4) | | | August 14, 2001 | | | 000-19319 | | |
| 4.5 | | | Indenture, dated as of September 19, 2000, between Vertex and State Street Bank and Trust Company. | | | | | | | 10-Q
(Exhibit 4.1) | | | November 13, 2000 | | | 000-19319 | | |
76
| 4.6 | | | Supplemental Indenture, dated as of December 12, 2000, between Vertex and State Street Bank and Trust Company. | | | | | | | S-3/A
(Exhibit 4.2) | | | January 17, 2001 | | | 333-49844 | | |
| 4.7 | | | Indenture, dated February 13, 2004, between Vertex and U.S. Bank National Association. | | | | | | | 8-K
(Exhibit 4.1) | | | February 23, 2004 | | | 000-19319 | | |
| 4.8 | | | Indenture, dated as of September 17, 2004, between Vertex and U.S. Bank National Association. | | | | | | | 8-K
(Exhibit 10.2) | | | September 17, 2004 | | | 000-19319 | | |
| 10.1 | | | Research and Development Agreement, dated as of September 10, 1997, between Vertex and Kissei Pharmaceutical Co. Ltd.† | | | | | | | 10-Q
(Exhibit 10.1) | | | November 12, 1997 | | | 000-19319 | | |
| 10.2 | | | Research Agreement and License Agreement, both dated December 16, 1993, between Vertex and Burroughs Wellcome Co.† | | | | | | | 10-K
(Exhibit 10.16) | | | Year Ended
December 31, 1993 | | | 000-19319 | | |
| 10.3 | | | First Revised and Restated Research and Early Development Agreement, dated as of February 3, 2004, between Vertex and Novartis Pharma AG.† | | | | | | | 10-K/A
(Exhibit 10.35) | | | September 8, 2004 | | | 000-19319 | | |
| 10.4 | | | License, Development and Commercialization Agreement, dated as of June 11, 2004, between Vertex and Mitsubishi Pharma Corporation.† | | | | | | | 8-K/A
(Exhibit 99.2) | | | September 10, 2004 | | | 000-19319 | | |
| 10.5 | | | Research, Development and Commercialization Agreement, dated as of May 24, 2004, between Vertex and Cystic Fibrosis Foundation Therapeutics Incorporated.† | | | | | | | 8-K/A
(Exhibit 99.2) | | | September 10, 2004 | | | 000-19319 | | |
| 10.6 | | | Amendment to Research, Development and Commercialization Agreement, dated as of January 6, 2006, between Vertex and Cystic Fibrosis Foundation Therapeutics Incorporated.† | | | | | | | 10-K
(Exhibit 10.9) | | | March 16, 2006 | | | 000-19319 | | |
| 10.7 | | | Second Amendment to Research, Development and Commercialization Agreement, dated as of March 17, 2006, between Vertex and Cystic Fibrosis Foundation Therapeutics Incorporated.† | | | | | | | 10-Q
(Exhibit 10.1) | | | May 10, 2006 | | | 000-19319 | | |
| 10.8 | | | Exclusive Research Collaboration, License and Commercialization Agreement, dated as of June 21, 2004, between Vertex Pharmaceuticals Incorporated and Merck & Co., Inc.† | | | | | | | 8-K/A
(Exhibit 99.4) | | | September 10, 2004 | | | 000-19319 | | |
| 10.9 | | | Letter Agreement, dated June 26, 2006, by and between Merck & Co., Inc. and Vertex Pharmaceuticals Incorporated. | | | | | | | 10-Q
(Exhibit 10.2) | | | August 9, 2006 | | | 000-19319 | | |
| 10.10 | | | Research, License and Commercialization Agreement, dated as of December 12, 2005, between Vertex and Glaxo Group Limited.† | | | | | | | 10-K
(Exhibit 10.11) | | | March 16, 2006 | | | 000-19319 | | |
| 10.11 | | | License, Development, Manufacturing and Commercialization Agreement, dated June 30, 2006, by and between Vertex Pharmaceuticals Incorporated and Janssen Pharmaceutica, N.V. † | | | | | | | 10-Q
(Exhibit 10.1) | | | August 9, 2006 | | | 000-19319 | | |
| 10.12 | | | Lease, dated as of March 3, 1995, between Fort Washington Realty Trust and Vertex. | | | | | | | 10-K
(Exhibit 10.15) | | | Year Ended
December 31, 1994 | | | 000-19319 | | |
| 10.13 | | | First Amendment to Lease, dated as of December 29, 1995, between Fort Washington Realty Trust and Vertex. | | | | | | | 10-K
(Exhibit 10.15) | | | Year Ended
December 31, 1995 | | | 000-19319 | | |
| 10.14 | | | Second Amendment to Lease, dated as of June 13, 1997, between Fort Washington Realty Trust and Vertex. | | | | | | | (10-K
(Exhibit 10.20) | | | March 26, 1998 | | | 000-19319 | | |
| 10.15 | | | Third, Fourth and Fifth Amendments to Lease between Fort Washington Realty Trust and Vertex.† | | | | | | | (10-K
(Exhibit 10.14) | | | March 26, 2001 | | | 000-19319 | | |
77
| 10.16 | | | Lease, dated as of September 17, 1999, between Trustees of Fort Washington Realty Trust and Vertex.† | | | | | | | 10-Q
(Exhibit 10.27) | | | November 15, 1999 | | | 000-19319 | | |
| 10.17 | | | Lease, dated as of January 18, 2001, between Kendall Square, LLC and Vertex.† | | | | | | | (10-K
(Exhibit 10.16) | | | March 26, 2001 | | | 000-19319 | | |
| 10.18 | | | Agreement for Lease, dated as of November 4, 1998, between Milton Park Limited, Vertex and Vertex Pharmaceuticals (Europe) Limited. | | | | | | | 10-K
(Exhibit 10.21) | | | March 30, 1999 | | | 000-19319 | | |
| 10.19 | | | 1991 Stock Option Plan, as amended and restated as of September 14, 1999.* | | | | | | | 10-K
(Exhibit 10.1) | | | March 3, 2000 | | | 000-19319 | | |
| 10.20 | | | 1994 Stock and Option Plan, as amended and restated as of September 14, 1999.* | | | | | | | 10-K
(Exhibit 10.2) | | | March 3, 2000 | | | 000-19319 | | |
| 10.21 | | | 1996 Stock and Option Plan, as amended and restated as of March 14, 2005.* | | | | | | | 10-K
(Exhibit 10.3) | | | March 16, 2005 | | | 000-19319 | | |
| 10.22 | | | Form of Stock Option Agreement under 1996 Stock and Option Plan.* | | | | | | | 8-K
(Exhibit 10.1) | | | February 9, 2005 | | | 000-19319 | | |
| 10.23 | | | Form of Restricted Stock Agreement under 1996 Stock and Option Plan—Annual Vesting.* | | | | | | | 8-K
(Exhibit 10.2) | | | February 9, 2005 | | | 000-19319 | | |
| 10.24 | | | Form of Restricted Stock Agreement under 1996 Stock and Option Plan—Performance Accelerated Restricted Stock.* | | | | | | | 8-K
(Exhibit 10.3) | | | February 9, 2005 | | | 000-19319 | | |
| 10.25 | | | Vertex Pharmaceuticals Incorporated 2006 Stock and Option Plan.* | | | | | | | 8-K
(Exhibit 10.1) | | | May 15, 2006 | | | 000-19319 | | |
| 10.26 | | | Form of Stock Option Grant under 2006 Stock and Option Plan.* | | | | | | | 8-K
(Exhibit 10.2) | | | May 15, 2006 | | | 000-19319 | | |
| 10.27 | | | Form of Restricted Stock Award under 2006 Stock and Option Plan.* | | | | | | | 8-K
(Exhibit 10.3) | | | May 15, 2006 | | | 000-19319 | | |
| 10.28 | | | Form of Restricted Stock Award (Performance Accelerated Restricted Stock) under 2006 Stock and Option Plan.* | | | | | | | 8-K
(Exhibit 10.4) | | | May 15, 2006 | | | 000-19319 | | |
| 10.29 | | | Executive Employment Agreement, dated as of November 1, 1994, between Vertex and Joshua S. Boger.* | | | | | | | 10-K
(Exhibit 10.6) | | | Year Ended
December 31, 1994 | | | 000-19319 | | |
| 10.30 | | | Amendment to Employment Agreement, dated as of May 12, 1995, between Vertex and Joshua S. Boger.* | | | | | | | 10-Q
(Exhibit 10.1) | | | Quarter Ended
June 30, 1995 | | | 000-19319 | | |
| 10.31 | | | Second Amendment to Employment Agreement, dated as of November 8, 2004, between Vertex and Joshua S. Boger.* | | | | | | | 10-Q
(Exhibit 10.9) | | | November 9, 2004 | | | 000-19319 | | |
| 10.32 | | | Amended and Restated Employment Agreement, dated as of November 8, 2004, between Vertex and Ian F. Smith.* | | | | | | | 10-Q
(Exhibit 10.13) | | | November 9, 2004 | | | 000-19319 | | |
| 10.33 | | | Amended and Restated Employment Agreement, dated as of November 8, 2004, between Vertex and Kenneth S. Boger.* | | | | | | | 10-Q
(Exhibit 10.11) | | | November 9, 2004 | | | 000-19319 | | |
| 10.34 | | | Employment Agreement, dated as of February 15, 2005, between Vertex and Victor Hartmann.* | | | | | | | 10-Q
(Exhibit 10.2) | | | May 9, 2005 | | | 000-19319 | | |
| 10.35 | | | Form of Letter Agreement, dated as of March 7, 2003, between Vertex and each of John J. Alam and Peter Mueller.* | | | | | | | 10-K
(Exhibit 10.32) | | | March 31, 2003 | | | 000-19319 | | |
| 10.36 | | | Form of Amendment to Letter Agreement, dated as of November 8, 2004, between Vertex and each of John J. Alam and Peter Mueller.* | | | | | | | 10-Q
(Exhibit 10.7) | | | November 9, 2004 | | | 000-19319 | | |
78
| 10.37 | | | Form of Restricted Stock Agreement between Vertex and each of the individuals listed on Schedule 1 thereto.* | | | | | | | 10-Q
(Exhibit 10.8) | | | November 9, 2004 | | | 000-19319 | | |
| 10.38 | | | Letter Agreement, dated as of December 5, 2005, between Vertex and Richard C. Garrison.* | | | | | | | 10-K
(Exhibit 10.35) | | | March 16, 2006 | | | 000-19319 | | |
| 10.39 | | | Change of Control Letter Agreement, dated as of December 12, 2005, between Vertex and Richard C. Garrison.* | | | | | | | 10-K
(Exhibit 10.36) | | | March 16, 2006 | | | 000-19319 | | |
| 10.40 | | | Amendment to Change of Control Letter Agreement, dated as of December 12, 2005, between Vertex and Richard C. Garrison.* | | | | | | | 10-K
(Exhibit 10.37) | | | March 16, 2006 | | | 000-19319 | | |
| 10.41 | | | Form of Employee Non-Disclosure and Inventions Agreement.* | | | | | | | S-1
(Exhibit 10.4) | | | May 30, 1991 | | | 33-40966 | | |
| 10.42 | | | Vertex Employee Compensation Plan* | | | X | | | | | | | | | | | | |
| 10.43 | | | Vertex Pharmaceuticals Non-Employee Board Compensation* | | | X | | | | | | | | | | | | |
| 21.1 | | | Subsidiaries of Vertex. | | | X | | | | | | | | | | | | |
| 23.1 | | | Consent of Independent Registered Public Accounting Firm Ernst & Young LLP. | | | X | | | | | | | | | | | | |
| 23.2 | | | Consent of Independent Registered Public Accounting Firm PricewaterhouseCoopers LLP. | | | X | | | | | | | | | | | | |
| 31.1 | | | Certification of the Chief Executive Officer under Section 302 of the Sarbanes-Oxley Act of 2002. | | | X | | | | | | | | | | | | |
| 31.2 | | | Certification of the Chief Financial Officer under Section 302 of the Sarbanes-Oxley Act of 2002. | | | X | | | | | | | | | | | | |
| 32.1 | | | Certification of the Chief Executive Officer and the Chief Financial Officer under Section 906 of the Sarbanes-Oxley Act of 2002. | | | X | | | | | | | | | | | | |
* Management contract, compensatory plan or agreement.
† Confidential portions of these documents have been filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment.
79
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | VERTEX PHARMACEUTICALS INCORPORATED |
March 1, 2007 | | By: | /s/ JOSHUA S. BOGER |
| | | Joshua S. Boger |
| | | Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Name | | | | Title | | Date |
/s/ JOSHUA S. BOGER | | Director, President and Chief Executive Officer (Principal Executive Officer) | | March 1, 2007 |
Joshua S. Boger |
/s/ IAN F. SMITH | | Executive Vice President and Chief Financial Officer (Principal Financial Officer) | | March 1, 2007 |
Ian F. Smith |
/s/ JOHANNA MESSINA POWER | | Vice President and Corporate Controller (Principal Accounting Officer) | | March 1, 2007 |
Johanna Messina Power |
/s/ ERIC K. BRANDT | | Director | | March 1, 2007 |
Eric K. Brandt |
/s/ ROGER W. BRIMBLECOMBE | | Director | | March 1, 2007 |
Roger W. Brimblecombe |
/s/ STUART J. COLLINSON | | Director | | March 1, 2007 |
Stuart J. Collinson |
/s/ EUGENE H. CORDES | | Director | | March 1, 2007 |
Eugene H. Cordes |
/s/ MATTHEW W. EMMENS | | Director | | March 1, 2007 |
Matthew W. Emmens |
/s/ BRUCE I. SACHS | | Director | | March 1, 2007 |
Bruce I. Sachs |
/s/ CHARLES A. SANDERS | | Director | | March 1, 2007 |
Charles A. Sanders |
/s/ EVE E. SLATER | | Director | | March 1, 2007 |
Eve E. Slater |
/s/ ELAINE S. ULLIAN | | Director | | March 1, 2007 |
Elaine S. Ullian |
80
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of Vertex Pharmaceuticals Incorporated
We have audited the accompanying consolidated balance sheets of Vertex Pharmaceuticals Incorporated as of December 31, 2006 and 2005, and the related consolidated statements of operations, stockholders’ equity and comprehensive loss, and cash flows for the each of the two years in the period ended December 31, 2006. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Vertex Pharmaceuticals Incorporated at December 31, 2006 and 2005, and the consolidated results of its operations and its cash flows for each of the two years in the period ended December 31, 2006, in conformity with U.S. generally accepted accounting principles.
As discussed in Notes B and D to the consolidated financial statements, on January 1, 2006, the Company adopted the provisions of Statement of Financial Accounting Standards No. 123(R), Share-Based Payment.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the effectiveness of Vertex Pharmaceuticals Incorporated’s internal control over financial reporting as of December 31, 2006, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 27, 2007 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
February 27, 2007
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
Vertex Pharmaceuticals Incorporated:
In our opinion, the accompanying consolidated statements of operations, of stockholders’ equity and comprehensive loss and of cash flows for the year ended December 31, 2004 present fairly, in all material respects, the results of operations and cash flows of Vertex Pharmaceuticals Incorporated and its subsidiaries for the year ended December 31, 2004 in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit. We conducted our audit of these statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
/s/ PricewaterhouseCoopers LLP
Boston, Massachusetts
March 15, 2005
F-2
VERTEX PHARMACEUTICALS INCORPORATED
Consolidated Balance Sheets
(In thousands, except share and per share amounts)
| | December 31, | |
Assets | | 2006 | | 2005 | |
Current assets: | | | | | |
Cash and cash equivalents | | $ | 213,171 | | $ | 78,045 | |
Marketable securities, available for sale, current portion | | 491,455 | | 283,112 | |
Accounts receivable | | 62,923 | | 20,595 | |
Prepaid expenses | | 3,857 | | 3,303 | |
Total current assets | | 771,406 | | 385,055 | |
Marketable securities, available for sale, excluding current portion | | 57,126 | | 46,353 | |
Restricted cash | | 30,258 | | 41,482 | |
Property and equipment, net | | 61,535 | | 54,533 | |
Investments | | — | | 18,863 | |
Other assets | | 1,254 | | 2,712 | |
Total assets | | $ | 921,579 | | $ | 548,998 | |
Liabilities and Stockholders’ Equity | | | | | |
Current liabilities: | | | | | |
Accounts payable | | $ | 15,368 | | $ | 6,210 | |
Accrued expenses and other current liabilities | | 91,359 | | 42,061 | |
Accrued interest | | 1,905 | | 3,184 | |
Deferred revenues, current portion | | 33,889 | | 31,449 | |
Accrued restructuring expense, current portion | | 4,735 | | 14,351 | |
Convertible subordinated notes, current portion | | 42,102 | | — | |
Convertible senior subordinated notes, current portion | | 59,648 | | — | |
Other obligations | | 2,008 | | 2,988 | |
Total current liabilities | | 251,014 | | 100,243 | |
Accrued restructuring expense, excluding current portion | | 28,338 | | 28,631 | |
Collaborator development loan | | 19,997 | | 19,997 | |
Deferred revenues, excluding current portion | | 116,295 | | 851 | |
Convertible subordinated notes, excluding current portion | | — | | 42,102 | |
Convertible senior subordinated notes, excluding current portion | | — | | 117,998 | |
Total liabilities | | 415,644 | | 309,822 | |
Commitments and contingencies (Note K and Note R) | | | | | |
Stockholders’ equity: | | | | | |
Preferred stock, $0.01 par value; 1,000,000 shares authorized; none issued and outstanding at December 31, 2006 and 2005, respectively | | — | | — | |
Common stock, $0.01 par value; 200,000,000 shares authorized; 126,121,473 and 108,153,149 shares issued and outstanding at December 31, 2006 and 2005, respectively | | 1,244 | | 1,081 | |
Additional paid-in capital | | 1,702,128 | | 1,243,960 | |
Deferred compensation, net | | — | | (13,408 | ) |
Accumulated other comprehensive loss | | (962 | ) | (2,873 | ) |
Accumulated deficit | | (1,196,475 | ) | (989,584 | ) |
Total stockholders’ equity | | 505,935 | | 239,176 | |
Total liabilities and stockholders’ equity | | $ | 921,579 | | $ | 548,998 | |
The accompanying notes are an integral part of the consolidated financial statements.
F-3
VERTEX PHARMACEUTICALS INCORPORATED
Consolidated Statements of Operations
(In thousands, except per share amounts)
| | Years Ended December 31, | |
| | 2006 | | 2005 | | 2004 | |
Revenues: | | | | | | | |
Royalties | | $ | 41,208 | | $ | 32,829 | | $ | 17,322 | |
Collaborative and other research and development revenues | | 175,148 | | 128,061 | | 85,395 | |
Total revenues | | 216,356 | | 160,890 | | 102,717 | |
Costs and expenses: | | | | | | | |
Royalty payments | | 12,170 | | 10,098 | | 5,649 | |
Research and development expenses (including stock-based compensation expense: 2006—$32,002, 2005—$3,567, 2004—$1,366)(1) | | 371,713 | | 248,540 | | 192,162 | |
Sales, general and administrative expenses (including stock-based compensation expense: 2006—$7,135, 2005—$1,065, 2004—$295)(1) | | 57,860 | | 43,990 | | 42,139 | |
Restructuring expense | | 3,651 | | 8,134 | | 17,574 | |
Total costs and expenses | | 445,394 | | 310,762 | | 257,524 | |
Loss from operations | | (229,038 | ) | (149,872 | ) | (154,807 | ) |
Interest income | | 23,024 | | 11,994 | | 10,323 | |
Interest expense | | (7,955 | ) | (17,326 | ) | (18,317 | ) |
Realized gain on sale of investment | | 11,183 | | — | | — | |
Loss on exchange of convertible subordinated notes | | (5,151 | ) | (48,213 | ) | — | |
Loss on retirement of convertible subordinated notes | | — | | — | | (3,446 | ) |
Loss before cumulative effect of a change in accounting principle | | $ | (207,937 | ) | $ | (203,417 | ) | $ | (166,247 | ) |
Cumulative effect of a change in accounting principle—FAS 123(R)(1) | | 1,046 | | — | | — | |
Net loss | | $ | (206,891 | ) | $ | (203,417 | ) | $ | (166,247 | ) |
Basic and diluted loss per common share before cumulative effect of a change in accounting principle | | $ | (1.84 | ) | $ | (2.28 | ) | $ | (2.12 | ) |
Basic and diluted cumulative effect of a change in accounting principle per common share | | 0.01 | | — | | — | |
Basic and diluted net loss per common share | | $ | (1.83 | ) | $ | (2.28 | ) | $ | (2.12 | ) |
Basic and diluted weighted average number of common shares outstanding | | 113,221 | | 89,241 | | 78,571 | |
(1) The Company adopted Financial Accounting Standards Board Statement No. 123(R), “Share-Based Payment,” using a modified prospective method. See Note D to the Consolidated Financial Statements, “Stock-based Compensation,” for further details.
The accompanying notes are an integral part of the consolidated financial statements.
F-4
VERTEX PHARMACEUTICALS INCORPORATED
Consolidated Statements of Stockholders’ Equity and Comprehensive Loss
(In thousands)
| | | | | | | | | | Accumulated | | | | | | | |
| | | | | | Additional | | | | Other | | | | Total | | | |
| | Common Stock | | Paid-In | | Deferred | | Comprehensive | | Accumulated | | Stockholders’ | | Comprehensive | |
| | Shares | | Amount | | Capital | | Compensation | | Income (Loss) | | Deficit | | Equity | | Income (Loss) | |
Balance, December 31, 2003 | | 78,025 | | | 780 | | | | 810,407 | | | | (1,112 | ) | | | 2,690 | | | | (619,920 | ) | | | 192,845 | | | | | | |
Net change in unrealized holding losses on marketable securities | | | | | | | | | | | | | | | | | (4,269 | ) | | | | | | | (4,269 | ) | | | $ | (4,269 | ) | |
Translation adjustments | | | | | | | | | | | | | | | | | 205 | | | | | | | | 205 | | | | 205 | | |
Net loss | | | | | | | | | | | | | | | | | | | | | (166,247 | ) | | | (166,247 | ) | | | (166,247 | ) | |
Comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | | | | | | | $ | (170,311 | ) | |
Issuances of common stock: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Benefit plans | | 2,740 | | | 27 | | | | 23,425 | | | | (12,206 | ) | | | | | | | | | | | 11,246 | | | | | | |
Amortization of deferred compensation | | | | | | | | | | | | | 1,661 | | | | | | | | | | | | 1,661 | | | | | | |
Balance, December 31, 2004 | | 80,765 | | | 807 | | | | 833,832 | | | | (11,657 | ) | | | (1,374 | ) | | | (786,167 | ) | | | 35,441 | | | | | | |
Net change in unrealized holding losses on marketable securities | | | | | | | | | | | | | | | | | (868 | ) | | | | | | | (868 | ) | | | $ | (868 | ) | |
Translation adjustments | | | | | | | | | | | | | | | | | (631 | ) | | | | | | | (631 | ) | | | (631 | ) | |
Net loss | | | | | | | | | | | | | | | | | | | | | (203,417 | ) | | | (203,417 | ) | | | (203,417 | ) | |
Comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | | | | | | | $ | (204,916 | ) | |
Issuances of common stock: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Equity Offering | | 13,513 | | | 135 | | | | 165,251 | | | | | | | | | | | | | | | | 165,386 | | | | | | |
Convertible Subordinated Notes exchanged | | 10,578 | | | 106 | | | | 203,424 | | | | | | | | | | | | | | | | 203,530 | | | | | | |
Benefit plans | | 3,297 | | | 33 | | | | 41,453 | | | | (6,172 | ) | | | | | | | | | | | 35,314 | | | | | | |
Amortization of deferred compensation | | | | | | | | | | | | | 4,421 | | | | | | | | | | | | 4,421 | | | | | | |
Balance, December 31, 2005 | | 108,153 | | | 1,081 | | | | 1,243,960 | | | | (13,408 | ) | | | (2,873 | ) | | | (989,584 | ) | | | 239,176 | | | | | | |
Net change in unrealized holding gains on marketable securities | | | | | | | | | | | | | | | | | 1,704 | | | | | | | | 1,704 | | | | $ | 1,704 | | |
Translation adjustments | | | | | | | | | | | | | | | | | 207 | | | | | | | | 207 | | | | 207 | | |
Net loss | | | | | | | | | | | | | | | | | | | | | (206,891 | ) | | | (206,891 | ) | | | (206,891 | ) | |
Comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | | | | | | | $ | (204,980 | ) | |
Issuances of common stock: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Equity Offering | | 10,000 | | | 100 | | | | 313,618 | | | | | | | | | | | | | | | | 313,718 | | | | | | |
Convertible Subordinated Notes exchanged | | 4,065 | | | 41 | | | | 64,197 | | | | | | | | | | | | | | | | 64,238 | | | | | | |
Benefit plans | | 3,903 | | | 22 | | | | 55,670 | | | | | | | | | | | | | | | | 55,692 | | | | | | |
Reversal of deferred compensation | | | | | | | | | (13,408 | ) | | | 13,408 | | | | | | | | | | | | — | | | | | | |
Stock-based compensation expense | | | | | | | | | 39,137 | | | | | | | | | | | | | | | | 39,137 | | | | | | |
Cumulative effect of a change in accounting principle—FAS 123(R) | | | | | | | | | (1,046 | ) | | | | | | | | | | | | | | | (1,046 | ) | | | | | |
Balance, December 31, 2006 | | 126,121 | | | 1,244 | | | | 1,702,128 | | | | — | | | | (962 | ) | | | (1,196,475 | ) | | | 505,935 | | | | | | |
The accompanying notes are an integral part of the consolidated financial statements.
F-5
VERTEX PHARMACEUTICALS INCORPORATED
Consolidated Statements of Cash Flows
(In thousands)
| | Years Ended December 31, | |
| | 2006 | | 2005 | | 2004 | |
Cash flows from operating activities: | | | | | | | |
Net loss | | $ | (206,891 | ) | $ | (203,417 | ) | $ | (166,247 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | |
Depreciation and amortization | | 25,868 | | 27,289 | | 29,640 | |
Stock-based compensation expense | | 39,137 | | 4,632 | | 1,661 | |
Other non-cash based compensation expense | | 3,341 | | 2,898 | | 2,504 | |
Cumulative effect of a change in accounting principle | | (1,046 | ) | — | | — | |
Loss on disposal of property and equipment | | 10 | | 344 | | 43 | |
Realized loss (gain) on marketable securities | | (7,579 | ) | 60 | | (423 | ) |
Realized gain on warrants | | (3,520 | ) | — | | — | |
Charge for exchange of convertible subordinated notes | | 5,151 | | 48,213 | | — | |
Charge for retirement of a portion of 2007 convertible subordinated notes | | — | | — | | 3,446 | |
Changes in operating assets and liabilities: | | | | | | | |
Accounts receivable | | (42,328 | ) | (8,704 | ) | (4,567 | ) |
Prepaid expenses and other current assets | | (554 | ) | (802 | ) | 817 | |
Accounts payable | | 9,158 | | (450 | ) | (5,646 | ) |
Accrued expenses and other current liabilities | | 48,523 | | 4,262 | | 2,362 | |
Accrued restructuring | | (9,909 | ) | (12,861 | ) | (13,683 | ) |
Accrued interest | | 280 | | 268 | | 1,407 | |
Deferred revenue | | 117,884 | | (33,786 | ) | 6,569 | |
Net cash used in operating activities | | (22,475 | ) | (172,054 | ) | (142,117 | ) |
Cash flows from investing activities: | | | | | | | |
Purchases of marketable securities | | (508,085 | ) | (236,489 | ) | (148,506 | ) |
Sales and maturities of marketable securities | | 302,265 | | 243,410 | | 292,351 | |
Sale of warrants | | 18,369 | | — | | — | |
Expenditures for property and equipment | | (32,417 | ) | (16,959 | ) | (12,538 | ) |
Restricted cash | | 11,224 | | 8,365 | | (23,786 | ) |
Investments and other assets | | 173 | | (59 | ) | (136 | ) |
Net cash (used in) provided by investing activities | | (208,471 | ) | (1,732 | ) | 107,385 | |
Cash flows from financing activities: | | | | | | | |
Issuances of common stock from employee benefit plans, net | | 52,363 | | 32,205 | | 8,742 | |
Issuances of common stock from stock offering, net | | 313,672 | | 165,386 | | — | |
Principal payments on capital leases and other obligations | | — | | — | | (12,563 | ) |
Issuance costs related to 2011 convertible senior subordinated notes | | — | | (16 | ) | (4,805 | ) |
Exchange costs | | (170 | ) | (119 | ) | — | |
Net cash provided by (used in) financing activities | | 365,865 | | 197,456 | | (8,626 | ) |
Effect of changes in exchange rates on cash | | 207 | | (631 | ) | 205 | |
Net increase (decrease) in cash and cash equivalents | | 135,126 | | 23,039 | | (43,153 | ) |
Cash and cash equivalents—beginning of period | | 78,045 | | 55,006 | | 98,159 | |
Cash and cash equivalents—end of period | | $ | 213,171 | | $ | 78,045 | | $ | 55,006 | |
Supplemental disclosure of cash flow information: | | | | | | | |
Cash paid for interest | | $ | 7,212 | | $ | 16,077 | | $ | 15,597 | |
Cash paid for taxes | | $ | — | | $ | — | | $ | — | |
The accompanying notes are an integral part of the consolidated financial statements.
F-6
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements
A. The Company
Vertex Pharmaceuticals Incorporated (“Vertex” or the “Company”) is in the business of discovering, developing and commercializing small molecule drugs for the treatment of serious diseases. The Company intends to continue investing in and building capabilities in research, development and commercialization of pharmaceutical products while it advances its drug candidates to market. Vertex earns royalty revenues from the sale of Lexiva/Telzir (fosamprenavir calcium), an HIV protease inhibitor for the treatment of HIV.
The Company currently is concentrating most of its drug development resources on four drug candidates: telaprevir (VX-950) for the treatment of hepatitis C virus infection; VX-702 for the treatment of rheumatoid arthritis and other inflammatory diseases; VX-770 for the treatment of cystic fibrosis; and VX-883 for the treatment of bacterial infection. In June 2006, the Company entered into a collaboration agreement with Janssen Pharmaceutica, N.V., a Johnson & Johnson company, relating to telaprevir. Under the collaboration agreement, the Company has retained exclusive commercial rights to telaprevir in North America and will lead the development program. Janssen has agreed to be responsible for 50% of the drug development costs under the development program for North America and the Janssen territories. The Company’s pipeline also includes several drug candidates that are being developed by its collaborators including Merck & Co., Inc., GlaxoSmithKline plc and Avalon Pharmaceuticals, Inc.
Vertex is subject to risks common to companies in its industry including, but not limited to, rapid technological change and competition, uncertain protection of proprietary technology, the dependence on the success of the Company’s lead drug candidate telaprevir, uncertainty about clinical trial outcomes, the need to comply with government regulations, share price volatility, the need to obtain additional funding, uncertainties relating to pharmaceutical pricing and reimbursement, dependence on collaborative relationships, potential product liability and limited experience in drug development, manufacturing, and sales and marketing. The Company expects to incur operating losses for the foreseeable future, as a result of expenditures for its research, development and commercialization programs.
B. Accounting Policies
Basis of Presentation
The consolidated financial statements reflect the operations of the Company and its wholly-owned subsidiaries. All significant intercompany balances and transactions have been eliminated. The Company operates in one segment, Pharmaceuticals, and all revenues are from United States operations.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires management to make certain estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements, and the reported amounts of revenues and expenses during the reported periods. Significant estimates in these consolidated financial statements have been made in connection with the calculation of revenues, research and development expenses, stock-based compensation expense, investments and restructuring expense. The Company bases its estimates on historical experience and various other assumptions that management believes to be reasonable under the circumstances. Actual results could differ from those estimates. Changes in estimates are reflected in reported results in the period in which they become known.
F-7
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
B. Accounting Policies (Continued)
Reclassification in the Preparation of Financial Statements
Certain amounts in prior years’ financial statements have been reclassified to conform to the current presentation. The reclassifications had no effect on the reported net loss.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentration of credit risk consist principally of money market funds and marketable securities. The Company places these investments with highly rated financial institutions, and, by policy, limits the amounts of credit exposure to any one financial institution. These amounts at times may exceed federally insured limits. The Company has not experienced any credit losses in these accounts and does not believe it is exposed to any significant credit risk on these funds. The Company has no foreign exchange contracts, option contracts or other foreign exchange hedging arrangements.
To date, the Company’s revenues have been generated from a limited number of customers in the biotechnology and pharmaceuticals industries in the United States, Europe and Japan. In 2006, the Company had significant revenue transactions with Janssen, Merck and GlaxoSmithKline that accounted for 31%, 27% and 20% respectively, of the Company’s total revenues. In 2005, the Company had significant revenue transactions with Novartis Pharma AG, GlaxoSmithKline and Merck that accounted for 33%, 33% and 15% respectively, of the Company’s total revenues. In 2004, revenue transactions with Novartis and GlaxoSmithKline accounted for 49% and 19%, respectively, of the Company’s total revenues.
Receivables from Janssen and GlaxoSmithKline represented approximately 63% and 22%, respectively, of the Company’s accounts receivable balance at December 31, 2006. Receivables from GlaxoSmithKline and Novartis represented approximately 46% and 22%, respectively, of the Company’s accounts receivable balance at December 31, 2005. Management believes that credit risks associated with these collaborators are not significant.
Cash and Cash Equivalents
The Company considers all highly liquid investments with original maturities of three months or less at the date of purchase to be cash equivalents. Cash equivalents consist principally of money market funds and debt securities. Changes in cash and cash equivalents may be effected by shifts in investment portfolio maturities as well as by actual cash receipts and disbursements.
Marketable Securities
Marketable securities consist of investments in municipal bond securities, U.S. government agency securities, U.S. government-sponsored enterprises securities, high-grade corporate bonds and asset-backed securities that are classified as available-for-sale. The Company classifies marketable securities available to fund current operations as current assets on the consolidated balance sheets. Marketable securities are classified as long-term assets on the consolidated balance sheets if (i) they have been in an unrealized loss position for longer than one year and (ii) the Company has the ability and intent to hold them (a) until the carrying value is recovered and (b) such holding period may be longer than one year. Marketable securities are stated at fair value with their unrealized gains and losses included as a component of accumulated other comprehensive loss, which is a separate component of stockholders’ equity, until such gains and losses are realized. The fair value of these securities is based on quoted market prices. If a decline in the fair value is considered other-than-temporary, based on available evidence, the unrealized loss is transferred from other comprehensive loss to the consolidated statements of operations. There were no write-downs of marketable securities in 2006, 2005 or 2004. Realized gains and losses are determined on
F-8
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
B. Accounting Policies (Continued)
the specific identification method and are included in interest income in the consolidated statements of operations.
Stock-Based Compensation
The Company adopted Financial Accounting Standards Board Statement No. 123(R), “Share-Based Payment” (“FAS 123(R)”), as of January 1, 2006. FAS 123(R) revises Financial Accounting Standards Board Statement No. 123, “Accounting for Stock-Based Compensation” (“FAS 123”), supersedes Accounting Principles Board (“APB”) Opinion No. 25, “Accounting for Stock Issued to Employees” (“APB 25”), and amends Financial Accounting Standards Board Statement Statement No. 95, “Statement of Cash Flows” (“FAS 95”). FAS 123(R) requires companies to expense the fair value of employee stock options and other forms of stock-based employee compensation over the employees’ service periods. Compensation cost is measured at the fair value of the award at the grant date, including estimated forfeitures, and is adjusted to reflect actual forfeitures and the outcomes of certain conditions.
In accordance with Financial Accounting Standards Board Statement No. 148, “Accounting for Stock-Based Compensation, Transition and Disclosure” (“FAS 148”), for periods prior to January 1, 2006, the Company adopted the disclosure-only provisions of FAS 123 and also applied APB 25 and related interpretations in accounting for all stock awards granted to employees. Under APB 25, for periods prior to January 1, 2006, provided that other criteria are met, when the exercise price of options equaled the market price of the common stock on the date of grant, no compensation expense was recognized. Also in accordance with APB 25 the Company was not required to record compensation expense for shares issued under the ESPP. Prior to January 1, 2006 and in accordance with APB 25, the Company recorded stock-based compensation expense related to restricted stock awards over the related vesting period for an amount equal to the difference between the price per share of restricted stock issued and the fair value of the Company’s common stock at the date of grant or issuance. The Company recorded forfeitures of restricted stock as they occurred.
Please refer to Note D, “Stock-based Compensation,” for further information.
Restructuring
The Company records costs and liabilities associated with exit and disposal activities, as defined in Financial Accounting Standards Board Statement No. 146, “Accounting for Costs Associated with Exit or Disposal Activities” (“FAS 146”), at fair value in the period the liability is incurred. In periods subsequent to initial measurement, changes to a liability are measured using the credit-adjusted risk-free discount rate applied in the initial period. In 2006, 2005 and 2004, the Company recorded costs and liabilities for exit and disposal activities related to a restructuring plan in accordance with FAS 146. The liability is evaluated and adjusted as appropriate on at least a quarterly basis for changes in circumstances.
Please refer to Note E, “Restructuring,” for further information.
Revenue Recognition
The Company recognizes revenue in accordance with the Securities and Exchange Commission’s (“SEC”) Staff Accounting Bulletin No. 101, “Revenue Recognition in Financial Statements” (“SAB 101”), as amended by SEC Staff Accounting Bulletin No. 104, “Revenue Recognition” (“SAB 104”), and for revenue arrangements entered into after June 30, 2003, Emerging Issues Task Force Issue No. 00-21, “Revenue Arrangements with Multiple Deliverables” (“EITF 00-21”).
The Company’s revenues are generated primarily through collaborative research, development and commercialization agreements. The terms of these agreements typically include payment to Vertex of one
F-9
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
B. Accounting Policies (Continued)
or more of the following: non-refundable, up-front license fees, funding of research and/or development efforts, milestone payments and royalties on product sales.
Agreements containing multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of fair value of the undelivered obligation(s). The consideration received is allocated among the separate units based on their respective fair values or the residual method, and the applicable revenue recognition criteria are applied to each of the separate units.
The Company recognizes revenues from non-refundable, up-front license fees on a straight-line basis over the contracted or estimated period of performance, which is typically the research or development term. Research and development funding is recognized as earned, ratably over the period of effort.
Substantive milestones realized in collaboration arrangements are recognized as earned when the corresponding payment is reasonably assured, subject to the following policies in those circumstances where the Company has obligations remaining after achievement of the milestone:
· In those circumstances where collection of a substantive milestone is reasonably assured, the Company has remaining obligations to perform under the collaboration arrangement and the Company has sufficient evidence of fair value for its remaining obligations, management considers the milestone payment and the remaining obligations to be separate units of accounting. In these circumstances, the Company uses the residual method under EITF 00-21 to allocate revenue among the milestones and the remaining obligations.
· In those circumstances where collection of a substantive milestone is reasonably assured, the Company has remaining obligations to perform under the collaboration arrangement and the Company does not have sufficient evidence of fair value for its remaining obligations, management considers the milestone payment and the remaining obligations under the contract as a single unit of accounting. In those circumstances where the collaboration does not require specific deliverables at specific times or at the end of the contract term, but rather the Company’s obligations are satisfied over a period of time, substantive milestones are recognized over the period of performance. This typically results in a portion of the milestone payment being recognized as revenue on the date the milestone is achieved equal to the applicable percentage of the performance period that has elapsed as of the date the milestone is achieved, with the balance being deferred and recognized over the remaining period of performance.
The Company evaluates whether milestones are substantive at the inception of the agreement based on the contingent nature of the milestone, specifically reviewing factors such as the scientific and other risks that must be overcome to achieve the milestone as well as the level of effort and investment required. Milestones that are not considered substantive and do not meet the separation criteria are accounted for as license payments and recognized on a straight-line basis over the remaining period of performance.
Payments received after performance obligations are met completely are recognized when earned.
Royalty revenues are recognized based upon actual and estimated net sales of licensed products in licensed territories, as provided by the licensee, and are recognized in the period the sales occur. Differences between actual royalty revenues and estimated royalty revenues, which have not historically been significant, are reconciled and adjusted for in the quarter during which they become known.
Research and Development Expenses
All research and development expenses, including amounts funded by research collaborations, are expensed as incurred. Research and development expenses are comprised of costs incurred in performing
F-10
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
B. Accounting Policies (Continued)
research and development activities, including salaries and benefits; laboratory supplies; contract services, including clinical trial costs and pharmaceutical development costs; stock-based compensation expense; and infrastructure costs, including facilities costs and depreciation. Due to telaprevir’s stage of development, costs related to the investment in its commercial supply are included in research and development expenses. The Company’s collaborators have funded portions of the Company’s research and development programs related to specific drug candidates and research targets, including, in 2006, telaprevir, VX-702, VX-770, kinases and certain cystic fibrosis research targets; and in 2005, telaprevir, VX-702, kinases and certain cystic fibrosis research targets; and in 2004, telaprevir, kinases, caspase inhibitors, and certain cystic fibrosis research targets.
The following table details the research and development expenses incurred by the Company for collaborator-sponsored and Company-sponsored programs (collaborator-sponsored programs are those in which a collaborator has funded at least a portion of the related program expenses, such as the telaprevir program) for 2006, 2005 and 2004 (in thousands):
| | 2006 | | 2005 | | 2004 | |
| | Research | | Development | | Total | | Research | | Development | | Total | | Research | | Development | | Total | |
Collaborator-sponsored | | | $ | 39,021 | | | | $ | 178,253 | | | $ | 217,274 | | | $ | 68,194 | | | | $ | 72,101 | | | $ | 140,295 | | | $ | 62,181 | | | | $ | 28,294 | | | $ | 90,475 | |
Company-sponsored | | | 103,626 | | | | 50,813 | | | 154,439 | | | 52,585 | | | | 55,660 | | | 108,245 | | | 51,095 | | | | 50,592 | | | 101,687 | |
Total | | | $ | 142,647 | | | | $ | 229,066 | | | $ | 371,713 | | | $ | 120,779 | | | | $ | 127,761 | | | $ | 248,540 | | | $ | 113,276 | | | | $ | 78,886 | | | $ | 192,162 | |
The total research and development expenses for 2006, 2005 and 2004 includes $32.0 million, $3.6 million and $1.4 million, respectively, of stock-based compensation expense.
Property and Equipment
Property and equipment are recorded at cost. Depreciation and amortization is provided using the straight-line method over the estimated useful life of the related asset, generally four to seven years for furniture and equipment and three to five years for computers and software. Leasehold improvements are amortized using the straight-line method over the lesser of the useful life of the improvements or the remaining life of the associated lease. Major additions and betterments are capitalized; maintenance and repairs to an asset that do not improve or extend its life are charged to operations. When assets are retired or otherwise disposed of, the assets and related allowances for depreciation and amortization are eliminated from the accounts and any resulting gain or loss is reflected in the Company’s consolidated statements of operations.
Investments
Investments include long-term investments recorded using the cost method of accounting. When the Company holds an ownership interest in an entity of less than 20%, and does not have the ability to exercise significant influence over the entity’s operating activities, the Company accounts for its investment using the cost method. If any adjustment to the fair value of an investment reflects a decline in the value of that investment below its cost, the Company considers the evidence available to it, including the duration and extent to which the market value of the investment has been less than cost, to evaluate the extent to which the decline is other-than-temporary. If the decline is considered other-than-temporary, the cost basis of the investment is written down to fair value as a new cost basis and the amount of the write-down is included in the Company’s consolidated statements of operations. There were no write-downs of investments in 2006, 2005 or 2004. Please refer to Note I, “Altus Investment,” for further information about the Company’s investment in Altus Pharmaceuticals, Inc.
F-11
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
B. Accounting Policies (Continued)
Income Taxes
Deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the financial statement carrying amounts and the income tax bases of assets and liabilities. A valuation allowance is applied against any net deferred tax asset if, based on the weighted available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
Debt Issuance Costs
Debt issuance costs incurred to complete Vertex’s convertible subordinated note offerings are deferred and included in other assets on the consolidated balance sheets. The costs are amortized based on the effective interest method over the term of the related debt issuance. The amortization expense is included in interest expense on the consolidated statements of operations. Unamortized costs related to exchanged debt is transferred from other assets to additional paid-in capital on the consolidated balance sheets.
Stock Offering Costs
Expenses incurred in connection with common stock issuances are recorded as an offset to additional paid-in capital on the consolidated balance sheets.
Comprehensive Loss
Comprehensive loss consists of net loss and other comprehensive loss, which includes foreign currency translation adjustments and unrealized gains and losses on certain marketable securities. For purposes of comprehensive loss disclosures, the Company does not record tax provisions or benefits for the net changes in foreign currency translation adjustment, as the Company intends to permanently reinvest undistributed earnings in its foreign subsidiary.
Foreign Currency Translation
The functional currency of the Company’s foreign subsidiary is the local currency. Assets and liabilities of the foreign subsidiary are re-measured into U.S. dollars at rates of exchange in effect at the end of the year. Revenue and expense amounts are re-measured using the average exchange rates for the period. Net unrealized gains and losses resulting from foreign currency translation are included in other comprehensive loss, which is a separate component of stockholders’ equity. Included in other comprehensive loss is a net unrealized gain related to foreign currency translation of $189,000 at December 31, 2006, a net unrealized loss related to foreign currency translation of $18,000 at December 31, 2005 and a net unrealized gain related to foreign currency translation of $613,000 at December 31, 2004.
Basic and Diluted Net Loss per Common Share
Basic net loss per share is based upon the weighted average number of common shares outstanding during the period, excluding restricted stock that has been issued but is not yet vested. Diluted net loss per share is based upon the weighted average number of common shares outstanding during the period plus additional weighted average common equivalent shares outstanding during the period when the effect is dilutive. Common equivalent shares result from the exercise of outstanding stock options (the proceeds of
F-12
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
B. Accounting Policies (Continued)
which are then assumed to have been used to repurchase outstanding stock using the treasury stock method), the assumed conversion of convertible notes and the vesting of unvested restricted shares of common stock. Common equivalent shares have not been included in the net loss per share calculations because their effect would have been anti-dilutive. Total potential gross common equivalent shares consisted of the following (in thousands, except per share amounts):
| | At December 31, | |
| | 2006 | | 2005 | | 2004 | |
Stock Options | | 14,279 | | 14,669 | | 15,820 | |
Weighted-average exercise price (per share) | | $ | 26.44 | | $ | 22.84 | | $ | 22.67 | |
Convertible Notes | | 4,449 | | 8,354 | | 16,454 | |
Weighted-average conversion price (per share) | | $ | 22.87 | | $ | 19.16 | | $ | 19.15 | |
Unvested restricted shares | | 1,764 | | 1,521 | | 1,399 | |
New Accounting Pronouncements
In February 2007, the Financial Accounting Standards Board (“FASB”) issued Statement No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities” (“FAS 159”). FAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The objective of FAS 159 is to reduce both complexity in accounting for financial instruments and the volatility in earnings caused by measuring related assets and liabilities differently.
Generally accepted accounting principles have required different measurement attributes for different assets and liabilities that can create artificial volatility in earnings. FASB has indicated it believes that FAS 159 helps to mitigate this type of accounting-induced volatility by enabling companies to report related assets and liabilities at fair value, which would likely reduce the need for companies to comply with detailed rules for hedge accounting.
FAS 159 also establishes presentation and disclosure requirements designed to facilitate comparisons between companies that choose different measurement attributes for similar types of assets and liabilities.
For example, FAS 159 requires companies to provide additional information that will help investors and other users of financial statements to more easily understand the effect of the company’s choice to use fair value on its earnings. It also requires entities to display the fair value of those assets and liabilities for which the company has chosen to use fair value on the face of the balance sheet. FAS 159 does not eliminate disclosure requirements included in other accounting standards, including requirements for disclosures about fair value measurements included in FASB Statement No. 157, “Fair Value Measurements” (“FAS 157”), and FASB Statement No. 107, “Disclosures about Fair Value of Financial Instruments.”
FAS 159 is effective as of the beginning of a company’s first fiscal year beginning after November 15, 2007. Early adoption is permitted as of the beginning of the previous fiscal year provided that the company makes that choice in the first 120 days of that fiscal year and also elects to apply the provisions of FAS 157.
In September 2006, FASB issued FAS 157. FAS 157 provides guidance for using fair value to measure assets and liabilities and requires additional disclosure about the use of fair value measures, the information used to measure fair value, and the effect fair-value measurements have on earnings. FAS 157 does not require any new fair value measurements. FAS 157 will be effective for the Company beginning
F-13
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
B. Accounting Policies (Continued)
January 1, 2008. The Company is currently evaluating the effect of FAS 157 on its consolidated financial statements.
In June 2006, FASB issued FASB Interpretation No. 48, “Accounting for Uncertainty in Income Taxes—an interpretation of FASB Statement No. 109” (“FIN 48”). FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements in accordance with Statement of Financial Accounting Standards No. 109, “Accounting for Income Taxes.” FIN 48 prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. FIN 48 also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure, and transition. FIN 48 is effective for fiscal years beginning after December 15, 2006. The Company is currently evaluating FIN 48 and believes the adoption of FIN 48 will not have a material effect on the Company’s consolidated financial statements.
In May 2005, FASB issued Statement No. 154, “Accounting Changes and Error Corrections (“FAS 154”).” FAS 154 replaces APB Opinion No. 20, “Accounting Changes” and Statement No.3, “Reporting Accounting Changes in Interim Financial Statements.” FAS 154 requires retrospective application to prior periods’ financial statements of changes in accounting principle, unless it is impractible to determine either the period-specific effects or the cumulative effect of the change. The Company adopted FAS 154 beginning on January 1, 2006. Its adoption did not have a material effect on the Company’s consolidated financial statements.
In November 2005, FASB issued a FASB Staff Position (“FSP”) on FAS 115-1 and FAS 124-1, “The Meaning of Other-Than-Temporary Impairment and Its Application to Certain Investments” (“FSP FAS 115-1”), which provides guidance on determining when investments in certain debt and equity securities are considered impaired, whether an impairment is other-than-temporary, and on measuring such impairment loss. FSP FAS 115-1 also includes accounting considerations subsequent to the recognition of an other-than-temporary impairment and requires certain disclosures about unrealized losses that have not been recognized as other-than-temporary impairments. FSP FAS 115-1 is required to be applied to reporting periods beginning after December 15, 2005. The Company adopted FSP FAS 115-1 in the first quarter of 2006. Adoption of FSP FAS 115-1 did not have a material effect on the Company’s consolidated financial statements.
C. Common and Preferred Stock
Stock and Option Plans
At December 31, 2006, the Company had four stock-based employee compensation plans: the 1991 Stock Option Plan (the “1991 Plan”), the 1994 Stock and Option Plan (the “1994 Plan”), the 1996 Stock and Option Plan (the “1996 Plan”) and the 2006 Stock and Option Plan (the “2006 Plan,” and together with the 1991 Plan, the 1994 Plan, and the 1996 Plan, collectively, the “Stock and Option Plans”), and one Employee Stock Purchase Plan (the “ESPP”).
Under the 2006 Plan, the Company may issue restricted stock and options to its employees, directors and consultants for services. Stock options may be granted under the 2006 Plan either as options intended to qualify as “incentive stock options” (“ISOs”) under the Internal Revenue Code or as non-qualified stock options (“NQSOs”). Each option granted under the 2006 Plan has an exercise price equal to the fair market value of the underlying common stock on the date of grant. For options issued to current employees, the date of grant is the date the option grant is approved by the Company’s Board of Directors. For grants to new employees, the date of grant is the employee’s first day of employment. The price per
F-14
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
C. Common and Preferred Stock (Continued)
share of restricted stock granted to employees is equal to $0.01, the par value of the Company’s common stock. Vesting of options and restricted stock generally is ratable over specified periods, usually four years, and is determined by the Company’s Board of Directors. All options awarded under the 2006 Plan expire not more than ten years from the grant date.
Stock options granted under the 1991 Plan, the 1994 Plan and the 1996 Plan were granted either as ISOs or NQSOs. Under the 1991 Plan, stock options could only be granted to employees (including officers and directors who were employees) and to consultants of the Company (NQSOs only). Under the 1994 Plan and the 1996 Plan, stock rights, which may be (i) ISOs when Internal Revenue Code requirements are met, (ii) NQSOs, or (iii) shares of common stock or the opportunity to make a direct purchase of shares of common stock, could be granted to employees (including officers and directors who are employees) and consultants, advisors and non-employee directors (NQSOs and stock awards only). Under the 1991 Plan and the 1994 Plan, ISOs could only be granted at a price not less than the fair market value of the common stock on the date of the grant, and NQSOs could be granted at an exercise price established by the Board of Directors, which could have been less than, equal to or greater than the fair value of the common stock on the date of the grant. Stock options granted under the 1996 Plan could not have been granted at a price less than the fair market value of the common stock on the date of grant. Vesting is ratable over specified periods for all plans, is generally four or five years, and was determined by the Board of Directors. ISOs granted under the 1991 Plan, the 1994 Plan and the 1996 Plan must expire not more than ten years from the date of grant.
The ESPP permits eligible employees to enroll in a twelve-month offering period comprising two six-month purchase periods. Participants may purchase shares of the Company’s common stock, through payroll deductions, at a price equal to 85% of the fair market value of the common stock on the first day of the applicable twelve-month offering period, or the last day of the applicable six-month purchase period, whichever is lower. Purchase dates under the ESPP occur on or about May 14 and November 14 of each year.
The Company reserved an aggregate of 8,000,000 shares under the 1991 Plan and 1994 Plan. The Company reserved 22,000,000 shares under the 1996 Plan and 7,302,380 shares under the 2006 Plan. At December 31, 2006, the Company had approximately 14,279,000 stock options outstanding and approximately 1,764,000 outstanding and unvested restricted shares. At December 31, 2006, the Company had approximately 5,807,000 shares of common stock available for grants under the 2006 Plan, and no shares were available for grants under the 1991 Plan, the 1994 Plan or the 1996 Plan. As of December 31, 2006, approximately 507,000 shares remained available for future purchases under the ESPP and approximately 179,000 shares remained available for grant under the 401(k) Plan.
Rights
Each Vertex shareholder also holds one share purchase right (a “Right”) for each share of common stock owned. Each Right entitles the holder to purchase from the Company one half of one-hundredth of a share of Series A junior participating preferred stock, $0.01 par value (the “Junior Preferred Shares”), of the Company at a price of $135 per one half of one-hundredth of a Junior Preferred Share, subject to adjustment (the “Purchase Price”). The Rights are not exercisable until after the acquisition by a person or group of 15% or more of the outstanding common stock (an “Acquiring Person”), or after the announcement of an intention to make or the commencement of a tender offer or exchange offer, the consummation of which would result in the beneficial ownership by a person or group of 15% or more of the outstanding common stock (the earlier of such dates being called the “Distribution Date”). Until the
F-15
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
C. Common and Preferred Stock (Continued)
Distribution Date (or earlier redemption or expiration of the Rights), the Rights will be traded with, and only with, the common stock. Until a Right is exercised, the Right will not entitle the holder thereof to any rights as a stockholder.
If any person or group becomes an Acquiring Person, each holder of a Right, other than Rights beneficially owned by the Acquiring Person, will thereafter have the right to receive upon exercise and payment of the Purchase Price that number of shares of common stock having a market value of two times the Purchase Price and, if the Company is acquired in a business combination transaction or 50% or more of its assets are sold, each holder of a Right will thereafter have the right to receive upon exercise and payment of the Purchase Price that number of shares of common stock of the acquiring company that at the time of the transaction will have a market value of two times the Purchase Price.
At any time after any person becomes an acquiring person and prior to the acquisition by such person or group of 50% or more of the outstanding common stock, the Board of Directors of the Company may cause the Rights (other than Rights owned by such person or group) to be exchanged, in whole or in part, for common stock or Junior Preferred Shares, at an exchange rate of one share of common stock per Right or one half of one-hundredth of a Junior Preferred Share per Right.
At any time prior to the acquisition by a person or group of beneficial ownership of 15% or more of the outstanding common stock, the Board of Directors of the Company may redeem the Rights at a price of $0.01 per Right.
The Rights have certain anti-takeover effects, in that they will cause substantial dilution to a person or group that attempts to acquire a significant interest in Vertex on terms not approved by the Board of Directors.
D. Stock-based Compensation
For the twelve months ended December 31, 2006
On January 1, 2006, Vertex adopted FAS 123(R), using the modified prospective method, pursuant to which the Company applies the provisions of FAS 123(R) to its consolidated financial statements on a going-forward basis. The modified prospective transition method requires the application of the accounting standard as of January 1, 2006, the first day of Vertex’s 2006 fiscal year. Prior periods have not been restated. FAS 123(R) requires companies to recognize share-based payments to employees as compensation expense using the “fair value” method. Under the fair value recognition provisions of FAS 123(R), stock-based compensation cost, measured at the grant date based on the fair value of the award, is recognized as expense ratably over the service period, which generally is the vesting period of the award. The fair value of stock options and shares purchased pursuant to the ESPP is calculated using the Black- Scholes valuation model. The fair value of restricted stock is based on intrinsic value on the date of grant. The expense recognized over the service period includes an estimate of awards that will be forfeited. Prior to adoption of FAS 123(R), Vertex recorded the impact of forfeitures of restricted stock as they occurred. In connection with the adoption of FAS 123(R) during 2006, Vertex recorded a $1.0 million benefit from the cumulative effect of changing from recording forfeitures related to restricted stock awards as they occurred to estimating forfeitures during the service period.
Stock-based compensation expense recognized during 2006 includes: (a) stock option and restricted stock awards granted after December 31, 2005, based on the grant-date fair value, and ESPP awards with the offering periods commencing May 15, 2006 and November 15, 2006, in accordance with the provisions of FAS 123(R); (b) stock option and restricted stock awards granted prior to but not yet vested as of
F-16
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
D. Stock-based Compensation (Continued)
December 31, 2005, based on the grant-date fair value estimated in accordance with the provisions of FAS 123; and (c) ESPP awards with offering periods commencing May 15, 2005 and November 15, 2005, based on the grant-date fair value estimated in accordance with the provisions of FAS 123. Stock-based compensation expense recognized during the year ended December 31, 2006 reflects estimated forfeitures of awards.
The estimated fair value of Vertex’s stock-based awards, less estimated forfeitures, is amortized on a ratable basis over the awards’ service periods. No equity compensation cost was capitalized during the year ended December 31, 2006.
The effect of recording stock-based compensation in 2006 was as follows (in thousands):
| | Year Ended
December 31, 2006 | |
Stock-based compensation expense by type of award: | | | | | |
Stock options | | | $ | 29,804 | | |
Restricted shares | | | 7,065 | | |
ESPP | | | 2,268 | | |
Total stock-based compensation expense | | | $ | 39,137 | | |
Effect of stock-based compensation expense on income by line item: | | | | | |
Research and development expenses | | | $ | 32,002 | | |
Sales, general and administrative expenses | | | 7,135 | | |
Total stock-based compensation expense | | | $ | 39,137 | | |
Cumulative effect of a change in accounting principle—FAS 123(R) | | | (1,046 | ) | |
Net stock-based compensation expense included in net loss | | | $ | 38,091 | | |
As a result of the adoption of FAS 123(R) the Company’s:
· net loss before cumulative effect of a change in accounting principle for 2006 is greater by $31.3 million;
· net loss for 2006 is greater by $30.3 million;
· basic and diluted net loss per share for 2006 is greater by $0.28 before cumulative effect of a change in accounting principle; and
· basic diluted net loss per share for 2006 is greater by $0.27.
Stock Options
All options granted during 2006, 2005 and 2004 were granted with exercise prices equal to the fair market value of the Company’s common stock on the date of grant and the options had weighted average grant date fair values, measured on the grant date, of $20.08, $7.11 and $5.00, respectively.
In accordance with FAS 123(R), the Company recorded stock-based compensation expense of $29.8 million in 2006 related to stock options. As of December 31, 2006, there was $51.9 million of total unrecognized compensation cost, net of estimated forfeitures, related to unvested options granted under the Stock and Option Plans. That cost is expected to be recognized over a weighted-average period of 2.59 years.
F-17
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
D. Stock-based Compensation (Continued)
The Company uses the Black-Scholes valuation model to estimate the fair value of stock options at the grant date. The Black-Scholes valuation model uses the option exercise price as well as estimates and assumptions related to the expected price volatility of the Company’s stock, the period during which the options will be outstanding, the rate of return on risk-free investments, and the expected dividend yield for the Company’s stock to estimate the fair value of a stock option on the grant date. The Company validates its estimates and assumptions through consultations with independent third parties having relevant expertise.
The fair value of each option granted under the Stock and Option Plans during 2006 was estimated on the date of grant using the Black-Scholes option pricing model with the following weighted average assumptions:
| | 2006 | |
Expected stock price volatility | | 57.10 | % |
Risk-free interest rate | | 4.74 | % |
Expected term | | 5.64 years | |
Expected annual dividends | | — | |
The weighted-average valuation assumptions were determined as follows:
· Expected stock price volatility: In 2006, the Company changed its method of estimating expected volatility from relying exclusively on historical volatility to relying exclusively on implied volatility. Options to purchase the Company’s stock with remaining terms of greater than one year are regularly traded in the market. Expected stock price volatility is calculated using the trailing one month average of daily implied volatilities prior to grant date.
· Risk-free interest rate: The Company bases the risk-free interest rate on the interest rate payable on U.S. Treasury securities in effect at the time of grant for a period that is commensurate with the assumed expected option term.
· Expected term of options: The expected term of options represents the period of time options are expected to be outstanding. The Company uses historical data to estimate employee exercise and post-vest termination behavior. The Company believes that all groups of employees exhibit similar exercise and post-vest termination behavior and therefore does not stratify employees into multiple groups in determining the expected term of options.
· Expected annual dividends: The estimate for annual dividends is $0.00, because the Company has not historically paid, and does not intend for the foreseeable future to pay, a dividend.
F-18
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
D. Stock-based Compensation (Continued)
The following table summarizes information related to the outstanding and vested options during 2006:
| | Stock
Options | | Weighted-Average
Exercise Price | | Weighted-Average
Remaining
Contractual Life | | Aggregate
Intrinsic Value | |
| | (In thousands) | | | | (In years) | | (In thousands) | |
Outstanding at December 31, 2005 | | | 14,669 | | | | $ | 22.84 | | | | | | | | | | |
Granted | | | 3,205 | | | | 35.45 | | | | | | | | | | |
Exercised | | | (2,968 | ) | | | 15.65 | | | | | | | | | | |
Forfeited | | | (345 | ) | | | 17.79 | | | | | | | | | | |
Expired | | | (282 | ) | | | 65.93 | | | | | | | | | | |
Outstanding at December 31, 2006 | | | 14,279 | | | | $26.44 | | | | 6.09 | | | | $ | 209,721 | | |
Exercisable at December 31, 2006 | | | 9,486 | | | | $27.02 | | | | 4.90 | | | | $ | 151,007 | | |
Total exercisable or expected
to vest | | | 13,671 | | | | $26.37 | | | | 5.97 | | | | $ | 204,005 | | |
The aggregate intrinsic value in the table above represents the total pre-tax amount, net of exercise price, which would have been received by option holders if all option holders had exercised all options with an exercise price lower than the market price on December 29, 2006 (the last trading day of 2006), based on the average of the high and low price of the Company’s common stock of $37.51 on that date.
The total intrinsic value (the amount by which the fair market value exceeded the exercise price) of stock options exercised during 2006, 2005 and 2004 was $63.4 million, $18.4 million and $2.3 million, respectively. The total cash received from employees as a result of employee stock option exercises during 2006, 2005 and 2004 was $46.5 million, $28.3 million and $5.2 million, respectively.
The Company settles employee stock option exercises with newly issued common shares.
Restricted Stock
The Company recorded stock-based compensation expense of approximately $7.1 million, $4.4 million and $1.7 million for 2006, 2005 and 2004, respectively, related to restricted shares outstanding during those periods.
As of December 31, 2006, there was $18.6 million of total unrecognized stock-based compensation expense, net of estimated forfeitures, related to unvested restricted stock granted under the Stock and Option Plans. That cost is expected to be recognized over a weighted-average period of 2.53 years.
F-19
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
D. Stock-based Compensation (Continued)
The following table summarizes the restricted stock activity of the Company during 2006:
| | Restricted
Stock | | Weighted-Average
Grant Date Fair Value | |
| | (Shares in thousands) | | (per Share) | |
Outstanding and unvested at December 31, 2005 | | | 1,521 | | | | $ | 11.02 | | |
Granted | | | 592 | | | | 35.46 | | |
Vested | | | (266 | ) | | | 11.83 | | |
Cancelled | | | (83 | ) | | | 18.98 | | |
Outstanding and unvested at December 31, 2006 | | | 1,764 | | | | $18.72 | | |
The total fair value of the shares vesting during 2006, 2005 and 2004 (measured on the date of vesting) was $9.9 million, $4.7 million and $0.3 million, respectively.
Employee Stock Purchase Plan
The stock-based compensation expense related to the ESPP for 2006 is $2.3 million. As of December 31, 2006, there was $1.3 million of total unrecognized compensation expense, net of estimated forfeitures, related to ESPP shares. That cost is expected to be recognized during 2007.
During 2006, the following shares were issued to employees under the ESPP (shares in thousands):
| | Year Ended
December 31, 2006 | |
Number of shares | | | 335 | | |
Average price paid | | | $ | 17.58 | | |
| | | | | | |
The weighted average fair value of each purchase right granted during 2006, 2005 and 2004 was $13.07, $6.42 and $5.34, respectively. The following table reflects the weighted average assumptions used in the Black-Scholes valuation model for 2006:
| | 2006 | |
Expected stock price volatility | | 55.84 | % |
Risk-free interest rate | | 4.99 | % |
Expected term | | 0.75 years | |
Expected annual dividends | | — | |
The expected stock price volatility for ESPP offerings beginning before the fourth quarter of 2005 is based on historical volatility, while the volatility for offerings beginning in the fourth quarter of 2005 and the second quarter of 2006 is based on implied volatility. The expected term represents purchases and purchase periods that take place within the offering period. The Company bases the risk-free interest rate on the interest rate payable on U.S. Treasury securities in effect at the time of grant for a period that is commensurate with the assumed expected term. The expected annual dividends estimate is $0.00, because the Company has not historically paid, and does not for the foreseeable future intend to pay, a dividend.
For Periods Prior to the Adoption of FAS 123(R)
In accordance with FAS 148, for periods prior to January 1, 2006, the Company adopted the disclosure-only provisions of FAS 123 and also applied APB 25 and related interpretations in accounting
F-20
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
D. Stock-based Compensation (Continued)
for all stock awards granted to employees. Under APB 25, provided that other criteria were met, when the exercise price of stock options granted to employees equaled the market price of the common stock on the date of the grant, no compensation expense was recognized. Additionally, under APB 25, the Company was not required to record compensation expense for the cost of options or shares issued under the ESPP. Accordingly, no expense related to options or ESPP shares was recorded prior to January 1, 2006, except for $0.2 million in stock-based compensation expense in 2005 related to the acceleration of options in accordance with one executive’s severance agreement.
Prior to January 1, 2006, the Company recorded stock-based compensation expense related to restricted stock awards over the related vesting period for an amount equal to the difference between the price per share of restricted stock issued and the fair value of the Company’s common stock at the date of grant or issuance. Prior to January 1, 2006, the Company recorded forfeitures of restricted stock as they occurred.
The following table illustrates the effect on net loss and net loss per share for the years ended December 31, 2005 and 2004 if the fair value recognition provisions of FAS 123 had been applied to the Company’s stock-based employee compensation in such periods. Employee stock-based compensation expense was amortized on a straight-line basis, because the Company’s valuation of options subject to FAS 123 assumed a single weighted-average expected term for each award. Included in employee stock-based compensation expense for the year ended December 31, 2005 is expense related to the modification of certain stock awards in accordance with an officer’s severance agreement.
| | Year Ended December 31, | |
| | 2005 | | 2004 | |
| | (In thousands, except per
share data) | |
Net loss attributable to common stockholders, as reported | | $ | (203,417 | ) | $ | (166,247 | ) |
Add: Employee stock-based compensation expense included in net loss, net of tax | | 4,632 | | 1,661 | |
Deduct: Total stock-based employee compensation expense determined under the fair value based method for all awards, net of tax | | (38,217 | ) | (39,504 | ) |
Pro forma net loss | | $ | (237,002 | ) | $ | (204,090 | ) |
Basic and diluted net loss per common share, as reported | | $ | (2.28 | ) | $ | (2.12 | ) |
Basic and diluted net loss per common share, pro forma | | $ | (2.66 | ) | $ | (2.60 | ) |
The weighted average fair value of options granted during 2005 and 2004 was $7.11 and $5.00, respectively. The fair value of each stock option granted during the years ended December 31, 2005 and 2004 was estimated on the date of grant using the Black-Scholes option pricing model with the following weighted-average assumptions:
| | Year Ended December 31, | |
| | 2005 | | 2004 | |
Expected stock price volatility | | 60.00 | % | 60.00 | % |
Risk-free interest rate | | 3.78 | % | 2.95 | % |
Expected term | | 4.20 years | | 4.00 years | |
Expected annual dividends | | — | | — | |
F-21
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
D. Stock-based Compensation (Continued)
The weighted average fair value of purchase rights granted during 2005 and 2004 was $6.42 and $5.34, respectively. The fair value of each ESPP purchase right outstanding during the years ended December 31, 2005 and 2004 was estimated on the date of subscription using the Black-Scholes option pricing model with the following weighted-average assumptions
| | Year Ended December 31, | |
| | 2005 | | 2004 | |
Expected stock price volatility | | 59.00 | % | 65.00 | % |
Risk-free interest rate | | 3.59 | % | 1.35 | % |
Expected term | | 0.43 years | | 0.80 years | |
Expected annual dividends | | — | | — | |
E. Restructuring
On June 10, 2003, Vertex adopted a plan to restructure its operations to coincide with its increasing internal emphasis on advancing drug candidates through clinical development to commercialization. The restructuring was designed to re-balance the Company’s relative investments in research and development to better support the Company’s long-term strategy. The restructuring plan included a workforce reduction, write-offs of certain assets and a decision not to occupy approximately 290,000 square feet of specialized laboratory and office space in Cambridge, Massachusetts under lease to Vertex (the “Kendall Square Lease”). The Kendall Square Lease commenced in January 2003 and has a 15-year term. In the second quarter of 2005, the Company revised its assessment of its real estate requirements and decided to use approximately 120,000 square feet of the facility subject to the Kendall Square Lease (the “Kendall Square Facility”) for its operations, beginning in 2006. The Company is now occupying this portion of the Kendall Square Facility. The remaining rentable square footage of the Kendall Square Facility currently is subleased to third parties.
In accordance with FAS 146, the Company’s initial estimate of its liability for net ongoing costs associated with the Kendall Square Lease obligation was recorded in the second quarter of 2003 at fair value. The restructuring expense incurred from the second quarter of 2003 through the end of the first quarter of 2005 (i.e., immediately prior to the Company’s decision to use a portion of the Kendall Square Facility for its operations) relates to the estimated incremental net ongoing lease obligations associated with the entire Kendall Square Facility, together with imputed interest costs relating to the restructuring liability. The restructuring expense incurred in the period beginning in the second quarter of 2005 continues to be estimated in accordance with FAS 146, but relates only to the portion of the building that the Company currently does not intend to occupy for its operations. The remaining lease obligations, which are associated with the portion of the Kendall Square Facility that the Company occupies and uses for its operations, are recorded as rental expense in the period incurred. The Company reviews its assumptions and estimates quarterly and updates its estimates of this liability as changes in circumstances require. As required by FAS 146, the expense and liability recorded is calculated using probability- weighted discounted cash-flows of the Company’s estimated ongoing lease obligations, including contractual rental and build-out commitments, net of estimated sublease rentals, offset by related sublease costs.
In estimating the expense and liability under its Kendall Square Lease obligation, the Company estimated (i) the costs to be incurred to satisfy rental and build-out commitments under the lease (including operating costs), (ii) the lead-time necessary to sublease the space, (iii) the projected sublease rental rates, and (iv) the anticipated durations of subleases. The Company validates its estimates and assumptions through consultations with independent third parties having relevant expertise. The Company
F-22
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
E. Restructuring (Continued)
uses a credit-adjusted risk-free rate of approximately 10% to discount the estimated cash flows. The Company will review its estimates and assumptions on at least a quarterly basis, until the termination of the Kendall Square Lease, and will make whatever modifications management believes necessary, based on the Company’s best judgment, to reflect any changed circumstances. The Company’s estimates have changed in the past, and may change in the future, resulting in additional adjustments to the estimate of liability, and the effect of any such adjustments could be material. Because the Company’s estimate of the liability includes the application of a discount rate to reflect the time-value of money, the estimate of the liability will increase each quarter simply as a result of the passage of time. Changes to the Company’s estimate of the liability are recorded as additional restructuring expense/(credit).
The restructuring accrual of $33.1 million at December 31, 2006 relates solely to the portion of the Kendall Square Facility that the Company does not intend to use for its operations and includes other lease obligations, recorded at net present value. The Company classified $4.7 million of the total amount at December 31, 2006 as short-term, and $28.3 million as long-term. The short-term portion of the restructuring liability represents the net amount the Company expects to pay in 2007.
In 2006, the Company recorded net restructuring expense of $3.7 million, which was primarily attributable to imputed interest and to build-out costs relating to the restructuring liability.
Activity with respect to the restructuring liability for 2006 was as follows (in thousands):
| | Liability as of
December 31, 2005 | | Cash
Payments
in 2006 | | Cash received
from subleases
in 2006 | | Additional Charge
in 2006 | | Liability
as of
December 31,
2006 | |
Lease restructuring liability | | | $ | 42,982 | | | $ | (21,607 | ) | | $ | 8,047 | | | | $ | 3,651 | | | | $ | 33,073 | | |
| | | | | | | | | | | | | | | | | | | | | | | | |
In 2005, the Company recorded net restructuring expense of $8.1 million. This net expense includes a $10.0 million credit to the restructuring liability made when the Company decided to occupy and use a portion of the Kendall Square Facility, which was offset by (i) the estimated incremental net ongoing lease obligations associated with the portion of the Kendall Square Facility that the Company does not intend to occupy and (ii) imputed interest costs relating to the restructuring liability. The portion of the $18.2 million additional charge in 2005 that was for incremental lease obligations was related to the revision of certain key estimates and assumptions about operating costs, including real estate taxes associated with the portion of the Kendall Square Facility that the Company does not intend to occupy. Activity with respect to the restructuring liability for 2005 was as follows (in thousands):
| | Liability as of
December 31, 2004 | | Cash Payments
in 2005 | | Cash received
from subleases
in 2005 | | Credit for
portion of
facility Vertex
decided to
occupy in
2005 | | Additional Charge
in 2005 | | Liability
as of
December 31,
2005 | |
Lease restructuring liability | | | $ | 55,843 | | | | $ | (24,229 | ) | | | $ | 3,234 | | | | $ | (10,018 | ) | | | $ | 18,152 | | | | $ | 42,982 | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
F-23
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
E. Restructuring (Continued)
In 2004, the Company recorded $17.6 million of additional restructuring expense, which primarily resulted from the revision of estimates and assumptions about when subtenants would be identified and secured and imputing an interest charge for the related restructuring liability. Activity with respect to the restructuring liability for 2004 was as follows (in thousands):
| | Liability as of
December 31, 2003 | | Cash Payments
in 2004 | | Cash received
from sublease, net
of operating costs
in 2004 | | Additional
Charge in
2004 | | Liability as of
December 31, 2004 | |
Lease restructuring and other operating lease liability | | | $ | 69,526 | | | | $ | (31,550 | ) | | | $ | 293 | | | | $ | 17,574 | | | | $ | 55,843 | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
In 2003, the Company recorded restructuring and other related expenses of $91.8 million. The $91.8 million includes $78.7 million of lease restructuring expense, $6.0 million of lease operating expense incurred prior to the decision not to occupy the Kendall Square Facility, $2.6 million for severance and related employee transition benefits and $4.5 million for a write-off of leasehold improvements and other assets.
The activity related to restructuring and other liability for the twelve months ended December 31, 2003 is presented below (in thousands):
| | Charge for the Twelve
Months Ended
December 31, 2003 | | Cash Payments
in 2003 | | Non-cash
Write-off
in 2003 | | Liability
as of
December 31, 2003 | |
Lease restructuring and other operating lease expense | | | $ | 84,726 | | | | $ | (15,200 | ) | | | $ | — | | | | $ | 69,526 | | |
Employee severance, benefits and related costs | | | 2,616 | | | | (2,616 | ) | | | — | | | | — | | |
Leasehold improvements and asset impairments | | | 4,482 | | | | — | | | | (4,482 | ) | | | — | | |
Total | | | $ | 91,824 | | | | $ | (17,816 | ) | | | $ | (4,482 4,482 | ) | | | $ | 69,526 | | |
F-24
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
F. Marketable Securities
A summary of cash, cash equivalents and marketable securities is shown below (in thousands):
December 31, 2006 | | | | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value | |
(Maturities stated are effective maturities) | | | | | | | | | | | | | |
Cash and cash equivalents | | | | | | | | | | | | | |
Cash and money market funds | | $ | 200,059 | | | $ | — | | | | $ | — | | | $ | 200,059 | |
Corporate debt securities | | 13,115 | | | 1 | | | | (4 | ) | | 13,112 | |
Total cash and cash equivalents | | $ | 213,174 | | | $ | 1 | | | | $ | (4 | ) | | $ | 213,171 | |
Marketable securities | | | | | | | | | | | | | |
Municipal bonds, due after 10 years | | $ | 1,802 | | | $ | — | | | | $ | — | | | $ | 1,802 | |
U.S. government securities | | | | | | | | | | | | | |
Due within 1 year | | 18,026 | | | 4 | | | | (136 | ) | | 17,894 | |
Due after 1 year through 5 years | | 53,440 | | | 51 | | | | (389 | ) | | 53,102 | |
Total US government securities | | 71,466 | | | 55 | | | | (525 | ) | | 70,996 | |
Corporate debt securities | | | | | | | | | | | | | |
Due within 1 year | | 409,044 | | | 169 | | | | (240 | ) | | 408,973 | |
Due after 1 year through 5 years | | 67,417 | | | 49 | | | | (656 | ) | | 66,810 | |
Total corporate debt securities | | 476,461 | | | 218 | | | | (896 | ) | | 475,783 | |
Total marketable securities | | $ | 549,729 | | | $ | 273 | | | | $ | (1,421 | ) | | $ | 548,581 | |
Total cash, cash equivalents and marketable securities | | $ | 762,903 | | | $ | 274 | | | | $ | (1,425 | ) | | $ | 761,752 | |
F-25
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
F. Marketable Securities (Continued)
December 31, 2005 | | | | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value | |
(Maturities stated are effective maturities) | | | | | | | | | | | | | |
Cash and cash equivalents | | | | | | | | | | | | | |
Cash and money market funds | | $ | 73,288 | | | $ | 1 | | | | $ | — | | | $ | 73,289 | |
Corporate debt securities | | 4,757 | | | — | | | | (1 | ) | | 4,756 | |
Total cash and cash equivalents | | $ | 78,045 | | | $ | 1 | | | | $ | (1 | ) | | $ | 78,045 | |
Marketable securities | | | | | | | | | | | | | |
Municipal bonds, due after 10 years | | $ | 8,370 | | | $ | — | | | | $ | — | | | $ | 8,370 | |
US government securities | | | | | | | | | | | | | |
Due within 1 year | | 23,275 | | | — | | | | (205 | ) | | 23,070 | |
Due after 1 year through 5 years | | 41,658 | | | 5 | | | | (702 | ) | | 40,961 | |
Total US government securities | | 64,933 | | | 5 | | | | (907 | ) | | 64,031 | |
Corporate debt securities | | | | | | | | | | | | | |
Due within 1 year | | 184,841 | | | 10 | | | | (753 | ) | | 184,098 | |
Due after 1 year through 5 years | | 72,622 | | | 5 | | | | (1,212 | ) | | 71,415 | |
Due after 5 years through 10 years | | 1,553 | | | — | | | | (2 | ) | | 1,551 | |
Total corporate debt securities | | 259,016 | | | 15 | | | | (1,967 | ) | | 257,064 | |
Total marketable securities | | $ | 332,319 | | | $ | 20 | | | | $ | (2,874 | ) | | $ | 329,465 | |
Total cash, cash equivalents and marketable securities | | $ | 410,364 | | | $ | 21 | | | | $ | (2,875 | ) | | $ | 407,510 | |
The Company has marketable securities of $491.5 million and $283.1 million classified as current assets on the consolidated balance sheets as of December 31, 2006 and 2005, respectively, and $57.1 million and $46.4 million classified as long term-assets on the consolidated balance sheets as of December 31, 2006 and 2005, respectively.
F-26
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
F. Marketable Securities (Continued)
The Company reviews investments in marketable securities for other-than-temporary impairment whenever the fair value of an investment is less than amortized cost and evidence indicates that an investment’s carrying amount is not recoverable within a reasonable period of time. To determine whether an impairment is other-than-temporary, the Company considers whether it has the ability and intent to hold the investment until a market price recovery and considers whether the evidence indicating the cost of the investment is recoverable outweighs evidence to the contrary. Evidence considered in this assessment includes reasons for the impairment, compliance with the Company’s investment policy, the severity and the duration of the impairment and changes in value subsequent to year end. The following table summarizes the fair value and gross unrealized losses related to marketable securities, aggregated by investment category and length of time that individual securities have been in a continuous unrealized loss position as of December 31, (in thousands):
2006
| | Less than 12 months | | 12 months or more | | Total | |
| | Fair Value | | Gross
Unrealized
Loss | | Fair Value | | Gross
Unrealized
Loss | | Fair Value | | Gross
Unrealized
Loss | |
U.S. government securities | | | $ | 19,609 | | | | $ | (56 | ) | | $ | 34,163 | | | $ | (469 | ) | | $ | 53,772 | | | $ | (525 | ) | |
Corporate debt securities | | | 56,563 | | | | (95 | ) | | 67,517 | | | (805 | ) | | 124,080 | | | (900 | ) | |
Total | | | $ | 76,172 | | | | $ | (151 | ) | | $ | 101,680 | | | $ | (1,274 | ) | | $ | 177,852 | | | $ | (1,425 | ) | |
2005
| | Less than 12 months | | 12 months or more | | Total | |
| | Fair Value | | Gross
Unrealized
Loss | | Fair Value | | Gross
Unrealized
Loss | | Fair Value | | Gross
Unrealized
Loss | |
U.S. government securities | | $ | 20,680 | | | $ | (180 | ) | | $ | 40,916 | | | $ | (727 | ) | | $ | 61,596 | | | $ | (907 | ) | |
Corporate debt securities | | 151,521 | | | (710 | ) | | 94,573 | | | (1,258 | ) | | 246,094 | | | (1,968 | ) | |
Total | | $ | 172,201 | | | $ | (890 | ) | | $ | 135,489 | | | $ | (1,985 | ) | | $ | 307,690 | | | $ | (2,875 | ) | |
The Company owned 164 available-for-sale marketable securities at December 31, 2006. Of these 164 securities, there were 104 securities with losses.
F-27
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
F. Marketable Securities (Continued)
Unrealized losses in the portfolio relate to various debt securities including U.S. government securities, U.S. government-sponsored enterprise securities, corporate debt securities and asset-backed securities. For these securities, the unrealized losses are primarily due to increases in interest rates. The investments held by the Company are high investment grade and there were no adverse credit events. Because the Company has the ability and intent to hold these investments until a recovery of fair value, which may be maturity, the Company does not consider these investments to be other-than-temporarily impaired as of December 31, 2006 and 2005.
Gross realized gains and losses for 2006 were $4,000 and $88,000 respectively. Gross realized gains and losses for 2005 were $15,000 and $75,000, respectively. Gross realized gains and losses for 2004 were $628,000 and $205,000, respectively.
G. Restricted Cash
At December 31, 2006 and 2005, the Company held $30.3 million and $41.5 million respectively, in restricted cash. At December 31, 2006 and 2005 the balance was held in deposit with certain banks predominantly to collateralize conditional stand-by letters of credit in the names of the Company’s landlords pursuant to certain operating lease agreements.
H. Property and Equipment
Property and equipment consist of the following at December 31 (in thousands):
| | 2006 | | 2005 | |
Furniture and equipment | | $ | 97,638 | | $ | 98,387 | |
Leasehold improvements | | 74,875 | | 66,318 | |
Computers | | 19,733 | | 18,971 | |
Software | | 21,274 | | 18,683 | |
Total property and equipment, gross | | 213,520 | | 202,359 | |
Less accumulated depreciation and amortization | | 151,985 | | 147,826 | |
Total property and equipment, net | | $ | 61,535 | | $ | 54,533 | |
Depreciation and amortization expense for the years ended December 31, 2006, 2005 and 2004 was $25.4 million, $26.3 million and $28.4 million, respectively.
In 2006 and 2005, the Company wrote off certain assets that were fully depreciated and no longer utilized. There was no effect on the Company’s net property and equipment. Additionally, the Company wrote off or sold certain assets that were not fully depreciated. The net loss on disposal of those assets was $10,000 for 2006, $344,000 for 2005 and $43,000 for 2004.
I. Altus Investment
Altus Pharmaceuticals, Inc. (“Altus”) completed an initial public offering in January 2006. As of the completion of the offering, Vertex owned 817,749 shares of common stock and warrants to purchase 1,962,494 shares of common stock (the “Altus Warrants”). In addition, the Company, as of the completion
F-28
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
I. Altus Investment (Continued)
of the offering, held 450,000 shares of redeemable preferred stock, which are not convertible into common stock and which are redeemable for $10.00 per share plus annual dividends of $0.50 per share, which have been accruing since the redeemable preferred stock was issued in 1999, at Vertex’s option on or after December 31, 2010, or by Altus at any time. The Company was restricted from trading Altus securities for a period of six months following the initial public offering.
When the Altus securities trading restrictions expired, the Company sold the 817,749 shares of Altus common stock for approximately $11.7 million, resulting in a realized gain of approximately $7.7 million in August 2006. Additionally when the restrictions expired, the Company began accounting for the Altus Warrants as derivative instruments under the Financial Accounting Standards Board Statement No. FAS 133, “Accounting for Derivative Instruments and Hedging Activities” (“FAS 133”). In accordance with FAS 133, in the third quarter of 2006, the Company recorded the Altus Warrants on its consolidated balance sheet at a fair market value of $19.1 million and recorded an unrealized gain on the fair market value of the Altus Warrants of $4.3 million. In the fourth quarter of 2006 the Company sold the Altus Warrants for approximately $18.3 million, resulting in a realized loss of $0.7 million. As a result of the Company’s sales of Altus common stock and Altus Warrrants in 2006, the Company recorded a realized gain on a sale of investment of $11.2 million.
In accordance with the Company’s policy, as outlined in Note B, “Accounting Policies,” the Company assessed its investment in Altus, which it accounts for using the cost method, and determined that there had not been any adjustments to the fair values of that investment that would require the Company to write down the investment basis of the asset, in 2005 and 2006. The Company’s cost basis carrying value in its outstanding equity and warrants of Altus was $18.9 million at December 31, 2005.
J. Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities consist of the following at December 31 (in thousands):
| | 2006 | | 2005 | |
Research and development contract costs | | $ | 57,761 | | $ | 20,098 | |
Payroll and benefits | | 25,115 | | 15,832 | |
Professional fees | | 3,848 | | 4,816 | |
Other | | 4,635 | | 1,315 | |
Total | | $ | 91,359 | | $ | 42,061 | |
K. Commitments
The Company leases its facilities and certain equipment under non-cancelable operating leases. The Company’s leases have terms through April 2018. The term of the Kendall Square Lease began January 1, 2003 and lease payments commenced in May 2003. The Company had an obligation under the Kendall Square Lease, staged through 2006, to build-out the space into finished laboratory and office space. This lease will expire in 2018, and the Company has the option to extend the term for two consecutive terms of ten years each, ultimately expiring in 2038. The Company occupies and uses for its operations approximately 120,000 square feet of the Kendall Square Facility. The Company has sublease arrangements in place for the remaining rentable square footage of the Kendall Square Facility, with initial terms that expires in April 2011 and August 2012. See Note E, “Restructuring” for further information.
F-29
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
K. Commitments (Continued)
At December 31, 2006, future minimum commitments under facility operating leases with non-cancelable terms of more than one year (including commitments under the Kendall Square Lease) are as follows (in thousands):
Year | | | | Kendall Square
Lease | | Sublease income for
Kendall Square
Facility | | Other Operating
Leases | | Total Operating
Leases | |
2007 | | | $ | 22,718 | | | | $ | (8,156 | ) | | | $ | 17,290 | | | | $ | 31,852 | | |
2008 | | | 23,621 | | | | (8,156 | ) | | | 16,638 | | | | 32,103 | | |
2009 | | | 23,683 | | | | (8,156 | ) | | | 12,345 | | | | 27,872 | | |
2010 | | | 23,748 | | | | (8,156 | ) | | | 11,307 | | | | 26,899 | | |
2011 | | | 23,816 | | | | (4,466 | ) | | | 684 | | | | 20,034 | | |
Thereafter | | | 171,237 | | | | (1,747 | ) | | | 246 | | | | 169,736 | | |
Total minimum lease payments | | | $ | 288,823 | | | | $ | (38,837 | ) | | | $ | 58,510 | | | | $ | 308,496 | | |
Rental expense for 2006 was $26.7 million, which included $9.5 million related to the Kendall Square Facility. Rental expense for 2005 was $20.4 million, which included $4.7 million related to the space in the Kendall Square Facility that the Company occupied in 2006 in the Kendall Square Facility. For 2004, rental expense primarily related to facilities, excluding the Kendall Square Facility, was $16.3 million.
The Company has future contractual commitments in connection with its research and development programs. For 2007 and 2008 the amount committed under these contracts is $1.1 million and $0.6 million, respectively.
L. Convertible Subordinated Notes
On February 13, 2004, the Company issued approximately $153.1 million in aggregate principal amount of 5.75% Convertible Senior Subordinated Notes due in February 2011 (the “February 2011 Notes”) in exchange for an equal principal amount of its outstanding 5% Convertible Subordinated Notes due in September 2007 (the “2007 Notes”). On September 17, 2004, the Company issued approximately $79.3 million in aggregate principal amount of 5.75% Convertible Senior Subordinated Notes due in February 2011 (the “September 2011 Notes”) in exchange for an equal principal amount of its 2007 Notes. The terms of the September 2011 Notes are identical to those of the February 2011 Notes (the February 2011 Notes and the September 2011 Notes are referred to together as the “2011 Notes”).
The 2011 Notes are convertible, at the option of the holder, into common stock at a price equal to $14.94 per share, subject to adjustment under certain circumstances. The 2011 Notes bear interest at the rate of 5.75% per annum, and the Company is required to make semi-annual interest payments on the outstanding principal balance of the 2011 Notes on February 15 and August 15 of each year. On or after February 15, 2007, the Company may redeem the 2011 Notes at a redemption price equal to the principal amount plus accrued and unpaid interest, if any. The deferred issuance costs associated with the issuance of the 2011 Notes, which are classified as long-term other assets, were approximately $3.0 million for the February 2011 Notes and $1.9 million for the September 2011 Notes.
The 2007 Notes are convertible, at the option of the holder, into common stock at a price equal to $92.26 per share, subject to adjustment under certain circumstances. The 2007 Notes bear interest at the rate of 5% per annum, and the Company is required to make semi-annual interest payments on the outstanding principal balance of the 2007 Notes on March 19 and September 19 of each year. The 2007 Notes are redeemable by the Company at any time at specific redemption prices if the closing price of the
F-30
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
L. Convertible Subordinated Notes (Continued)
Company’s common stock exceeds 120% of the conversion price for at least 20 trading days within a period of 30 consecutive trading days. The deferred issuance costs associated with the original sale of the 2007 Notes were $9.3 million. Payment in full of the outstanding principal and accrued interest on these 2007 Notes is due in September 2007.
As a result of the 2004 exchanges of the 2007 Notes for 2011 Notes, the Company recorded a charge in the amount of $2.5 million on the retirement in February 2004 of $153.1 million in aggregate principal amount of the 2007 Notes, and a charge in the amount of $1.0 million on the retirement in September 2004 of $79.3 million in aggregate principal amount of the 2007 Notes. These charges represent that portion of the unamortized deferred issuance costs applicable to the amount of 2007 Notes retired.
In September 2005, the Company exchanged approximately 2.5 million shares of newly issued common stock for approximately $40.5 million in aggregate principal amount of then outstanding 2007 Notes, plus accrued interest. As a result of the exchange, the Company incurred a non-cash charge of $36.3 million in 2005. This charge is related to the incremental shares issued in the transaction over the number that would have been issued upon conversion of the 2007 Notes under their original terms, at the original conversion price of $92.26.
In the fourth quarter of 2005, the Company exchanged approximately 8.1 million shares of newly issued common stock for approximately $114.5 million in aggregate principal amount of then outstanding 2011 Notes, plus accrued interest. As a result of the exchange, the Company incurred a non-cash charge of $11.9 million in 2005. This charge is related to the incremental shares issued in the transaction over the number that would have been issued upon conversion of the 2011 Notes under their original terms, at the original conversion price of $14.94.
In the third quarter of 2006, the Company exchanged approximately 4.1 million shares of newly issued common stock for approximately $58.3 million in aggregate principal amount of then outstanding 2011 Notes plus accrued interest. As a result of this exchange, the Company incurred a non-cash charge of $5.2 million in 2006. This charge is related to the incremental shares issued in the transaction over the number that would have been issued upon the conversion of the 2011 Notes under their original terms, at the original conversion price of $14.94.
The following items related to the 2005 and 2006 exchanges were recorded as an offset to additional paid-in capital on the consolidated balance sheets: accrued interest, remaining unamortized issuance costs of the exchanged notes and issuance costs of the common stock.
For the year ended December 31, 2006, $0.5 million was amortized to interest expense for the issuance costs of the remaining 2007 Notes and the 2011 Notes. For the year ended December 31, 2005, $1.0 million was amortized to interest expense for the issuance costs of the remaining 2007 Notes and the 2011 Notes. For the year ended December 31, 2004, $1.3 million was amortized to interest expense for the issuance costs of the 2007 Notes and the 2011 Notes.
At December 31, 2006, there was approximately $42.1 million in aggregate principal amount of the 2007 Notes and approximately $59.6 million in aggregate principal amount of the 2011 Notes outstanding. At December 31, 2006, the 2007 Notes and the 2011 Notes had fair values of $41.3 million and $150.1 million, respectively, as obtained from a quoted market source.
In February 2007, the Company announced that it will redeem the 2011 notes on March 5, 2007. If the holders of the 2011 Notes all elect to convert their 2011 Notes into common stock at the conversion price of $14.94 per share, the Company will be required to issue approximately 4.0 million shares of the
F-31
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Company’s common stock in full satisfaction of the 2011 Notes. If the holders of the 2011 Notes do not elect to convert their 2011 Notes into common stock, the Company will be required to redeem the 2011 Notes at a redemption price of $1,003.19 per $1,000 principal amount, which includes principal and unpaid interest that will accrue through the redemption date.
M. Equity Offering
In September 2006, the Company completed a public offering of 10,000,000 shares of common stock, including the underwriters’ over-allotment of 900,000 shares, at a price of $33.00 per share. This transaction resulted in net proceeds of approximately $313.7 million to the Company.
In June 2005, the Company completed a public offering of 13,512,500 shares of common stock, including the underwriters’ over-allotment of 1,762,500 shares, at a price of $13.00 per share. This transaction resulted in net proceeds of approximately $165.4 million to the Company.
N. Income Taxes
For the years ended December 31, 2006, 2005 and 2004, there is no provision for income taxes included in the consolidated statements of operations.
The Company’s federal statutory income tax rate for 2006, 2005, and 2004 was 34%. The Company has incurred losses from operations but has not recorded an income tax benefit for 2006, 2005 and 2004, as the Company has recorded a valuation allowance against its net operating losses and other net deferred tax assets due to uncertainties related to the realizability of these tax assets.
The difference between the Company’s “expected” tax provision (benefit), as computed by applying the U.S. federal corporate tax rate of 34% to (loss) before provision for income taxes, and actual tax is reconciled as follows (in thousands):
| | 2006 | | 2005 | | 2004 | |
Loss before provision for income taxes | | $ | (206,891 | ) | $ | (203,417 | ) | $ | (166,247 | ) |
Expected tax benefit at 34% | | $ | (70,343 | ) | $ | (69,162 | ) | $ | (56,524 | ) |
State taxes, net of federal benefit | | (12,972 | ) | (12,754 | ) | (10,424 | ) |
Unbenefited operating losses | | 81,593 | | 64,262 | | 66,983 | |
Non-deductible expenses | | 1,817 | | 17,450 | | 40 | |
Other | | (95 | ) | 204 | | (75 | ) |
Income tax provision | | $ | — | | $ | — | | $ | — | |
For federal income tax purposes, as of December 31, 2006, the Company has net operating loss carryforwards of approximately $1,257.2 million, and $29.7 million of tax credits, which may be used to offset future federal income and tax liability, respectively. For state income tax purposes, the Company has net operating loss carryforwards of approximately $826.5 million, and $15.5 million of tax credits, which may be used to offset future state income and tax liability, respectively. These operating loss carryforwards began to expire in 2005, and the tax credit carryforwards began to expire in 2004. After consideration of all the evidence, both positive and negative, management has established a valuation allowance for the full amount of the 2006 net deferred tax asset since it is more likely than not that the net deferred tax asset will not be realized.
F-32
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
N. Income Taxes (Continued)
Deferred tax assets and liabilities are determined based on the difference between financial statement and tax bases using enacted tax rates in effect for the year in which the differences are expected to reverse. The components of the deferred taxes at December 31 were as follows (in thousands):
| | 2006 | | 2005 | |
Deferred Tax Assets: | | | | | |
Net operating loss | | $ | 352,014 | | $ | 278,765 | |
Tax credit carryforwards | | 39,981 | | 27,942 | |
Property, plant and equipment | | 16,130 | | 14,955 | |
Deferred revenue | | 343 | | 596 | |
Stock-based compensation | | 15,761 | | 243 | |
Capitalized research and development | | 15,591 | | 21,471 | |
Accrued expenses and other | | 12,580 | | 15,022 | |
Gross Deferred Tax Assets | | 452,400 | | 358,994 | |
Valuation Allowance | | (441,342 | ) | (347,936 | ) |
Total Deferred Tax Assets | | 11,058 | | 11,058 | |
Deferred Tax Liabilities: | | | | | |
Gain on Investment | | (11,058 | ) | (11,058 | ) |
Net Deferred Tax Assets/(Liabilities) | | $ | — | | $ | — | |
As discussed in Note D “Stock-based Compensation,” the Company adopted FAS 123(R) effective January 1, 2006 for stock-based compensation plans. Generally, tax return deductions are allowable on such arrangements, but, may arise in different amounts and periods from when compensation costs are recognized in the financial statements. Pursuant to FAS 123(R), if the tax return deduction for an award exceeds the cumulative compensation expense recognized in the financial statements, any excess tax benefit shall be recognized as additional paid-in capital when the deduction reduces income tax payable. Prior to adoption, the Company recognized deferred tax assets, along with an offsetting valuation allowance, for net operating loss carryforwards that included deductions for excess tax benefits from stock-based compensation. On adoption, the Company has chosen to derecognize the deferred tax asset for these excess tax deduction in the net operating loss carryforwards, along with the offsetting valuation allowance, adjusting the prior year footnote information accordingly. The net tax amount of the unrealized excess tax benefits as of December 31, 2006, no longer included and disclosed as a deferred tax asset, is approximately $111.7 million. The gross amount of this excess tax deduction in the net operating loss carryforward is approximately $284.0 million.
The valuation allowance increased by $93.4 million during 2006 due primarily to the increase in net operating losses from operations and tax credits.
Ownership changes, as defined by Internal Revenue Code, may limit the amount of net operating losses and research and experimentation credit carryforwards that can be utilized annually to offset future taxable income and taxes payable.
F-33
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
O. Significant Revenue Arrangements
The Company has formed strategic collaborations with pharmaceutical companies and other organizations in the areas of drug discovery, development, and commercialization. Research, development and commercialization agreements provide the Company with financial support and other valuable resources for its research programs, for the development of clinical drug candidates, and for the marketing and sales of products.
Collaborative Research, Development and Commercialization Agreements
In the Company’s collaborative research, development and commercialization programs the Company seeks to discover, develop and commercialize pharmaceutical products in conjunction with and supported by the Company’s collaborators. Collaborative research and development arrangements may provide research funding over an initial contract period with renewal and termination options that vary by agreement. The agreements may also include non-refundable, up-front license fees as well as milestone payments based on the achievement of a pre-agreed objective or the occurrence of a designated event. The agreements may also contain development expense reimbursement provisions, royalty rights or profit sharing rights, and manufacturing options. The Company has entered into significant research and development collaborations under terms that vary from agreement to agreement.
Janssen Pharmaceutica, N.V.
In June 2006, the Company entered into a collaboration agreement with Janssen for the development, manufacture and commercialization of telaprevir, the Company’s investigative HCV protease inhibitor. Under the agreement, Janssen has agreed to be responsible for 50% of the drug development costs incurred under the development program for the parties’ territories (North America for the Company, and the rest of the world, other than the Far East, for Janssen) and has exclusive rights to commercialize telaprevir in its territories including Europe, South America, the Middle East, Africa and Australia. Janssen made a $165 million up-front license payment to the Company in July 2006. Janssen has agreed to make additional contingent milestone payments, which could total up to $380 million if telaprevir is successfully developed, approved and launched. The agreement also provides the Company with royalties on any sales of telaprevir in the Janssen territories, with a tiered royalty averaging in the mid-20% range, as a percentage of net sales in the Janssen territories, depending upon successful commercialization of telaprevir. Each of the parties will be responsible for drug supply in their respective territories. However, the agreement provides for the purchase by Janssen from the Company of materials required for Janssen’s manufacture of the active pharmaceutical ingredient. In addition, Janssen will be responsible for certain third-party royalties on net sales in its territories. Janssen may terminate the agreement without cause at any time upon six months’ notice to the Company.
During 2006, the Company recognized $68.0 million in revenue under the Janssen agreement, which includes an amortized portion of the upfront payment, a milestone achievement of $15.0 million based on interim results of the Company’s first Phase 2b clinical trial of telaprevir, and funding of reimbursable drug development costs.
F-34
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
O. Significant Revenue Arrangements (Continued)
GlaxoSmithKline plc
In December 1993, the Company and GlaxoSmithKline entered into a collaboration agreement to research, develop and commercialize HIV protease inhibitors, including Agenerase (amprenavir), Lexiva/Telzir (fosamprenavir calcium) and brecanavir (VX-385). Under the collaboration agreement, GlaxoSmithKline agreed to pay the Company an up-front license payment, product research funding, development and commercialization milestone payments and royalty payments on sales of drugs developed under the collaboration.
The Company began earning a royalty from GlaxoSmithKline in 1999 on sales of Agenerase, in the fourth quarter of 2003 on sales of Lexiva, and in the third quarter of 2004 on sales of Telzir. In the fourth quarter of 2004, GlaxoSmithKline paid the Company a milestone payment of $1 million based on the initiation of Phase 2 clinical trials for brecanavir. In the third quarter of 2004, GlaxoSmithKline paid the Company a milestone payment of $1.5 million in connection with the regulatory approval of Telzir in the European Union.
On December 15, 2006, GlaxoSmithKline formally notified the Company that it would discontinue clinical development of brecanavir (VX-385), which had advanced to Phase 2 clinical trials. Currently, there are no drug candidates being developed under this collaboration and the Company does not anticipate any additional research funding or milestone payments under this collaboration. Under the original agreement, GlaxoSmithKline had exclusive rights to develop and commercialize Vertex’s HIV protease inhibitors in all parts of the world except the Far East. In 2003, the Company amended the agreement to add the Far East to GlaxoSmithKline’s territory for development and commercialization of Lexiva/Telzir. The Company has retained certain bulk drug manufacturing rights and certain product educational rights in territories licensed to GlaxoSmithKline. GlaxoSmithKline has the right to terminate its arrangement with the Company without cause upon twelve months’ notice. Termination of the agreement by GlaxoSmithKline will relieve it of its obligation to make further payments to the Company and will end any license granted to GlaxoSmithKline by Vertex under the agreement.
In June 1996, the Company and GlaxoSmithKline obtained a worldwide, non-exclusive license under certain G.D. Searle & Co. (“Searle,” now owned by Pharmacia/Pfizer) patents in the area of HIV protease inhibition. The Company pays Searle a royalty based on sales of Agenerase and Lexiva/Telzir.
In the fourth quarter of 2005, GlaxoSmithKline and the Company entered into a collaborative agreement to develop and commercialize VX-409 and certain back-up compounds, which are being investigated for the treatment of pain. Under the terms of the agreement, GlaxoSmithKline has the exclusive right and license to develop and commercialize VX-409 and certain back-up compounds worldwide. Vertex received a $20 million up-front license payment and could receive up to $385 million in additional development and commercial milestone payments assuming the development of VX-409 and back-up compounds in major pharmaceutical markets across a range of indications. GlaxoSmithKline will also pay Vertex royalties on annual net sales of any drugs commercialized under the agreement.
Revenues and royalties earned from GlaxoSmithKline under both agreements were $43.6 million, $52.8 million and $19.8 million in 2006, 2005 and 2004, respectively.
Merck & Co., Inc.
In June 2004, Vertex entered into a global collaboration with Merck & Co., Inc. to develop and commercialize MK-0457 (VX-680), Vertex’s lead Aurora kinase inhibitor, and additional follow-on compound(s), for the treatment of cancer. The Merck collaboration agreement provided for an up-front
F-35
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
O. Significant Revenue Arrangements (Continued)
license payment of $20 million, which was made in June 2004, and for research funding of $14 million over two years, ending in June 2006. In 2006, the Company agreed with Merck to extend the research program term and corresponding research funding for the parties’ ongoing research collaboration for an additional three months beyond the original research program termination date. Vertex could also receive as much as $350 million in milestone payments, including up to $130 million for the successful development of MK-0457 (VX-680) in the first oncology indication and additional milestone payments for development of MK-0457 (VX-680) and follow-on compounds in other major oncology indications. In 2005, Merck selected MK-6592 (VX-667), a follow-on compound, for development. Merck is responsible for worldwide clinical development and commercialization of MK-0457 (VX-680) and MK-6592 (VX-667), and any other Aurora kinase inhibitors discovered during the research program and has agreed to pay Vertex royalties on product sales. Merck may terminate the agreement without cause at any time upon 90 days’ advance written notice, except that six months’ advance written notice is required at any time when a product has marketing approval in a major market and the termination is not the result of a safety issue. In 2005, Vertex received two milestone payments from Merck totaling $19.5 million. In 2006, Vertex received three additional milestone payments from Merck totaling $36.3 million. Vertex recognized $58.7 million, $24.4 million and $8.4 million of revenue related to research support, milestone payments and the up-front license payment for this collaboration in 2006, 2005 and 2004, respectively.
Cystic Fibrosis Foundation Therapeutics Incorporated
In May 2004, Vertex entered into an agreement with Cystic Fibrosis Foundation Therapeutics Incorporated (“CFFT”) that provided funding through December 31, 2005 for Vertex’s late-stage cystic fibrosis drug discovery effort. In 2006, Vertex amended its agreement with CFFT to extend the term of the drug discovery effort to March 31, 2008 and to include additional development stage funding for specified VX-770 development activities through the end of 2007. The agreement, as amended, provides that CFFT will pay up to $32.4 million to Vertex for research and development activities. Under the amended agreement, Vertex retains the right to develop and commercialize VX-770 and any other compounds discovered in the research collaboration, and will pay royalties to CFFT on net sales of any drug candidate approved and commercialized under the collaboration.
In 2006, 2005 and 2004, Vertex recognized $12.6 million, $14.5 million and $6.8 million, respectively, in revenue related to its agreement with CFFT. During the first quarter of 2004, under an earlier agreement, Vertex earned revenue of $1.9 million. CFFT has the right to terminate the agreement without cause, effective June 30, 2007, upon 60 days’ prior written notice.
Novartis Pharma AG
In May 2000, the Company entered into an agreement with Novartis Pharma AG to collaborate on the discovery, development and commercialization of small molecule drugs directed at protein kinases. The agreement was amended in February 2004. Under the original agreement, the Company was responsible for drug discovery and clinical proof-of-concept testing of all drug candidates. Novartis agreed to pay the Company up to $200 million in research funding through April 2006, and to loan the Company up to $200 million on a non-interest-bearing basis to support clinical proof-of-concept studies. Development loans with respect to any drug candidates accepted by Novartis for development would be forgiven. Under the amended agreement, Vertex continued to receive research funding through April 2006 along with development milestone payments and royalties with respect to drug candidates selected by Novartis for development. Novartis held an option to develop drug candidates meeting certain pre-agreed criteria. Restrictions under the original agreement that limited Novartis’ right to pursue kinase research and
F-36
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
O. Significant Revenue Arrangements (Continued)
development outside the collaboration were removed, and the development loan facility was terminated. Following completion of the research term, Novartis’ development option with respect to all compounds discovered in the research program, none of which were then in development by Novartis, had expired. Vertex retains all rights to those candidates, as well as to all of the intellectual property it generated under the collaboration.
In November 2004, Novartis accepted VX-322 for preclinical development and made a $10 million milestone payment to Vertex that was recognized as revenue over the term of the contract. Novartis had exclusive worldwide development, manufacturing and commercialization rights to VX-322. That compound is no longer in development by Novartis, and Novartis’s worldwide development, manufacturing and commercialization rights to VX-322 have terminated.
In June 2004, the Company exercised its option under the amended agreement to develop MK-0457 (VX-680), and the Aurora kinases it targets, outside the Novartis collaboration and repaid approximately $12.5 million of unspent and uncommitted development loans previously advanced on account of MK-0457 (VX-680).
In 2006, 2005 and 2004, the Company recognized approximately $17.6 million, $53.1 million and $50.5 million, respectively, in revenue under this agreement. At December 31, 2006, there were approximately $20.0 million in remaining loans outstanding under the loan facility. These loans are repayable, without interest, in May 2008.
Mitsubishi Pharma Corporation
In June 2004, Vertex entered into a collaboration agreement with Mitsubishi Pharma Corporation, pursuant to which Mitsubishi agreed to provide financial and other support for the development and commercialization of telaprevir. Under the terms of the agreement, Mitsubishi has the right to develop and commercialize telaprevir in Japan and certain other Far East countries. The agreement provides for up to $33 million in payments by Mitsubishi to Vertex through Phase 2 clinical development, including an up-front license fee, development stage milestone payments and reimbursement of certain drug development costs for telaprevir. Further cost sharing beyond Phase 2 clinical development will be determined by Mitsubishi and Vertex based on the design of registration studies for telaprevir. The agreement also provides Vertex with royalties on any sales of telaprevir in the Mitsubishi territory. Mitsubishi may terminate the agreement at any time without cause upon 60 days’ prior written notice. In the fourth quarter of 2004, Mitsubishi paid the Company a milestone payment of $4.0 million for first dosing of telaprevir in a patient in the Phase 1b clinical trial in the United States. In the third quarter of 2006, Mitsubishi paid the Company a milestone payment of $3.0 million for the first dosing of telaprevir in a patient by Mitsubishi in the Mitsubishi territory. Vertex recognized $8.6 million, $3.4 million and $5.8 million in revenue under this agreement in 2006, 2005 and 2004, respectively. The revenue includes an amortized portion of the up-front payment, milestone achievements, and reimbursement of certain of Vertex’s expenses incurred in telaprevir development.
Kissei Pharmaceutical Co., Ltd.
The Company and Kissei Pharmaceutical Co., Ltd. (“Kissei”) are parties to an agreement to collaborate on the identification of inhibitors of p38 MAP kinase and the development of those compounds as novel, orally active drugs for the treatment of inflammatory and neurological diseases. Under the terms of the agreement, Kissei agreed to pay the Company up to $22 million comprised of a $4 million up-front license payment, $11 million of product research funding over three years and
F-37
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
O. Significant Revenue Arrangements (Continued)
$7 million of development and commercialization milestone payments. Additionally, Kissei agreed to reimburse the Company for certain development costs, including a portion of costs for Phase 2 trials of VX-702. Research funding ended under this program in June 2000, and the Company has received the full amount of research funding specified under the agreement. Kissei has exclusive rights to develop and commercialize VX-702 in Japan and certain Far East countries and co-exclusive rights in China, Taiwan and South Korea. The Company retains exclusive marketing rights outside the Far East and co-exclusive rights in China, Taiwan and South Korea. In addition, the Company will have the right to supply bulk drug material to Kissei for sale in its territory and will receive royalties or drug supply payments on future product sales, if any. In 2006, 2005 and 2004, approximately $6.4 million, $7.3 million and $3.5 million, respectively, was recognized as revenue under this agreement. The $7.3 million of revenue recognized in 2005 includes a $2.5 million milestone paid upon Kissei’s completion of regulatory filings in preparation for Phase 1 clinical development of VX-702 in Japan.
P. Employee Benefits
The Company has a 401(k) retirement plan (the “Vertex 401(k) Plan”) in which substantially all of its permanent employees are eligible to participate. Participants may contribute up to 60% of their annual compensation to the Vertex 401(k) Plan, subject to statutory limitations. The Company may declare discretionary matching contributions to the Vertex 401(k) Plan that are payable in the form of Vertex common stock. The match is paid in the form of fully vested interests in a Vertex common stock fund. Employees have the ability to transfer funds from the Company stock fund as they choose. The Company declared matching contributions to the Vertex 401(k) Plan as follows (in thousands):
| | 2006 | | 2005 | | 2004 | |
Discretionary matching contributions during the year ended December 31, | | $ | 3,341 | | $ | 2,894 | | $ | 2,492 | |
Shares issued during the year ended December 31, | | 91 | | 215 | | 239 | |
Shares issuable as of the year ended December 31, | | 28 | | 19 | | 57 | |
| | | | | | | | | | |
Q. Related Party Transactions
As of December 31, 2006, 2005 and 2004, the Company had a loan outstanding to a former officer of the Company in the amount of $36,000, $36,000, $97,000, respectively, which was initially advanced in April 2002. The loan balance is included in other assets on the consolidated balance sheets.
In 2001, the Company entered into a four year consulting agreement with a director of the Company for the provision of part-time consulting services over a period of four years, at the rate of $80,000 per year commencing in January 2002. The consulting agreement terminated in January 2006.
R. Contingencies
The Company has certain contingent liabilities that arise in the ordinary course of its business activities. The Company accrues a reserve for contingent liabilities when it is probable that future expenditures will be made and such expenditures can be reasonably estimated.
F-38
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
S. Guarantees
As permitted under Massachusetts law, Vertex’s Articles of Organization and Bylaws provide that the Company will indemnify certain of its officers and directors for certain claims asserted against them in connection with their service as an officer or director. The maximum potential amount of future payments that the Company could be required to make under these indemnification provisions is unlimited. However, the Company has purchased directors’ and officers’ liability insurance policies that could reduce its monetary exposure and enable it to recover a portion of any future amounts paid. No indemnification claims are currently outstanding and the Company believes the estimated fair value of these indemnification arrangements is minimal.
Vertex customarily agrees in the ordinary course of its business to indemnification provisions in agreements with clinical trials investigators and sites in its drug development programs, in sponsored research agreements with academic and not-for-profit institutions, in various comparable agreements involving parties performing services for the Company in the ordinary course of business, and in its real estate leases. The Company also customarily agrees to certain indemnification provisions in its drug discovery and development collaboration agreements. With respect to the Company’s clinical trials and sponsored research agreements, these indemnification provisions typically apply to any claim asserted against the investigator or the investigator’s institution relating to personal injury or property damage, violations of law or certain breaches of the Company’s contractual obligations arising out of the research or clinical testing of the Company’s compounds or drug candidates. With respect to lease agreements, the indemnification provisions typically apply to claims asserted against the landlord relating to personal injury or property damage caused by the Company, to violations of law by the Company or to certain breaches of the Company’s contractual obligations. The indemnification provisions appearing in the Company’s collaboration agreements are similar, but in addition provide some limited indemnification for its collaborator in the event of third party claims alleging infringement of intellectual property rights. In each of the cases above, the indemnification obligation generally survives the termination of the agreement for some extended period, although the obligation typically has the most relevance during the contract term and for a short period of time thereafter. The maximum potential amount of future payments that the Company could be required to make under these provisions is generally unlimited. The Company has purchased insurance policies covering personal injury, property damage and general liability that reduce its exposure for indemnification and would enable it in many cases to recover a portion of any future amounts paid. The Company has never paid any material amounts to defend lawsuits or settle claims related to these indemnification provisions. Accordingly, the Company believes the estimated fair value of these indemnification arrangements is minimal.
In March 2003, the Company sold certain assets of PanVera LLC to Invitrogen Corporation for approximately $97 million. In December 2003, the Company sold certain instrumentation assets to Aurora Discovery, Inc. for approximately $4.3 million. The agreements with the buyers each require the Company to indemnify the buyer against any loss it may suffer by reason of Vertex’s breach of certain representations and warranties, or failure to perform certain covenants, contained in such agreement. The representations, warranties and covenants contained in the agreements are of a type customary in agreements of this sort. The Company’s aggregate obligations under the indemnity contained in each agreement are, with a few exceptions which the Company believes are not material, capped at one-half of the applicable purchase price, and apply to claims under representations and warranties made within fifteen months after closing (which period has ended) although there is no corresponding time limit for claims made based on breaches of covenants. Neither Invitrogen or Aurora has made any claims to date under the applicable indemnities, and the Company believes that the estimated fair value of the remaining indemnification obligations is minimal.
F-39
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
S. Guarantees (Continued)
On February 10, 2004, Vertex entered into a Dealer Manager Agreement with UBS Securities LLC in connection with the exchange of approximately $153.1 million of the February 2011 Notes for approximately $153.1 million of 2007 Notes. On September 13, 2004, the Company entered into a second Dealer Manager Agreement with UBS Securities in connection with the exchange of approximately $79.3 million of the September 2011 Notes for approximately $79.3 million of 2007 Notes. Each of the Dealer Manager Agreements requires the Company to indemnify UBS Securities against any loss UBS Securities may suffer by reason of the Company’s breach of representations and warranties relating to the exchanges of the convertible notes, the Company’s failure to perform certain covenants in those agreements, the inclusion of any untrue statement of material fact in the disclosure materials provided to potential investors in the 2011 Notes, the omission of any material fact needed to make those materials not misleading, and any actions taken by the Company or its representatives in connection with the exchanges. The representations, warranties and covenants in the Dealer Manager Agreements are of a type customary in agreements of this sort. The Company believes the estimated fair value of these indemnification obligations is minimal.
On June 7, 2005 and September 14, 2006, the Company entered into Purchase Agreements with Merrill Lynch, Pierce, Fenner & Smith Incorporated, as the representative of the several underwriters named in such agreements, relating to the public offering and sale of shares of the Company’s common stock. The Purchase Agreement relating to each offering requires the Company to indemnify the underwriters against any loss they may suffer by reason of the Company’s breach of representations and warranties relating to that public offering, the Company’s failure to perform certain covenants in those agreements, the inclusion of any untrue statement of material fact in the prospectus used in connection with that offering, the omission of any material fact needed to make those materials not misleading, and any actions taken by the Company or its representatives in connection with the offering. The representations, warranties and covenants in the Purchase Agreement are of a type customary in agreements of this sort. The Company believes the estimated fair value of these indemnification obligations is minimal.
F-40
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
T. Quarterly Financial Data (unaudited)
(In thousands, except per share amounts)
| | Three Months Ended | |
| | March 31,
2006 | | June 30,
2006 | | Sept. 30,
2006 | | Dec. 31,
2006 | |
Revenues: | | | | | | | | | |
Royalties | | $ | 9,179 | | $ | 9,005 | | $ | 10,902 | | $ | 12,122 | |
Collaborative and other research and development revenues | | 29,908 | | 20,721 | | 42,387 | | 82,132 | |
Total revenues | | 39,087 | | 29,726 | | 53,289 | | 94,254 | |
Costs and expenses: | | | | | | | | | |
Royalty payments | | 2,995 | | 2,885 | | 3,113 | | 3,177 | |
Research and development expenses | | 75,202 | | 91,250 | | 96,115 | | 109,146 | |
Sales, general and administrative expenses | | 12,879 | | 14,370 | | 14,773 | | 15,838 | |
Restructuring expense | | 767 | | 443 | | 1,415 | | 1,026 | |
Total costs and expenses | | 91,843 | | 108,948 | | 115,416 | | 129,187 | |
Loss from operations | | (52,756 | ) | (79,222 | ) | (62,127 | ) | (34,933 | ) |
Interest income | | 3,980 | | 3,921 | | 5,330 | | 9,793 | |
Interest expense | | (2,357 | ) | (2,357 | ) | (1,767 | ) | (1,474 | ) |
Realized gain (loss) on sale of investment | | — | | — | | 7,663 | | (730 | ) |
Unrealized gain on warrants | | — | | — | | 4,250 | | — | |
Loss on exchange of convertible subordinated notes | | — | | — | | (5,151 | ) | — | |
Loss before cumulative effect of a change in accounting principle | | (51,133 | ) | (77,658 | ) | (51,802 | ) | (27,344 | ) |
Cumulative effect of a change in accounting principle—FAS 123(R) | | 1,046 | | — | | — | | — | |
Net loss | | $ | (50,087 | ) | $ | (77,658 | ) | $ | (51,802 | ) | $ | (27,344 | ) |
Basic and diluted loss before cumulative effect of a change in accounting principle per common share | | $ | (0.48 | ) | $ | (0.72 | ) | $ | (0.46 | ) | $ | (0.22 | ) |
Basic and diluted cumulative effect of a change in accounting principle per common share | | 0.01 | | — | | — | | — | |
Basic and diluted net loss per common share | | $ | (0.47 | ) | $ | (0.72 | ) | $ | (0.46 | ) | $ | (0.22 | ) |
Basic and diluted weighted average number of common shares outstanding | | 107,440 | | 108,523 | | 112,803 | | 123,942 | |
F-41
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
T. Quarterly Financial Data (unaudited) (Continued)
| | Three Months Ended | |
| | March 31,
2005 | | June 30,
2005 | | Sept. 30,
2005 | | Dec. 31,
2005 | |
Revenues: | | | | | | | | | |
Royalties | | $ | 6,153 | | $ | 7,467 | | $ | 9,466 | | $ | 9,743 | |
Collaborative and other research and development revenues | | 22,453 | | 24,854 | | 26,741 | | 54,013 | |
Total revenues | | 28,606 | | 32,321 | | 36,207 | | 63,756 | |
Costs and expenses: | | | | | | | | | |
Royalty payments | | 2,030 | | 2,489 | | 2,796 | | 2,783 | |
Research and development expenses | | 57,435 | | 59,357 | | 63,590 | | 68,158 | |
Sales, general and administrative expenses | | 9,627 | | 10,814 | | 10,738 | | 12,811 | |
Restructuring expense | | 1,914 | | (1,743 | ) | 1,565 | | 6,398 | |
Total costs and expenses | | 71,006 | | 70,917 | | 78,689 | | 90,150 | |
Loss from operations | | (42,400 | ) | (38,596 | ) | (42,482 | ) | (26,394 | ) |
Interest income | | 2,319 | | 2,247 | | 3,733 | | 3,695 | |
Interest expense | | (4,639 | ) | (4,639 | ) | (4,505 | ) | (3,543 | ) |
Loss on exchange of convertible subordinated notes | | — | | — | | (36,324 | ) | (11,889 | ) |
Net loss | | $ | (44,720 | ) | $ | (40,988 | ) | $ | (79,578 | ) | $ | (38,131 | ) |
Basic and diluted net loss per common share | | $ | (0.56 | ) | $ | (0.50 | ) | $ | (0.84 | ) | $ | (0.38 | ) |
Basic and diluted weighted average number of common shares outstanding | | 79,428 | | 82,274 | | 94,590 | | 100,535 | |
F-42