UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| x | Annual Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the fiscal year ended December 31, 2012
OR
| o | | Transition Report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the transition period from to
Commission File Number 000-24537
DYAX CORP.
(Exact name of registrant as specified in its charter)
Delaware (State of Incorporation) | | 04-3053198 (IRS Employer Identification No.) |
| | | |
55 Network Drive, Burlington, Massachusetts 01803 (Address of principal executive offices and zip code) |
| |
Registrant's telephone number, including area code: (617) 225-2500 |
| Securities registered pursuant to Section 12(b) of the Act: |
| |
Title of each class: Common Stock, $.01 Par Value | Name of each exchange on which registered: The NASDAQ Stock Market LLC (NASDAQ Global Market) |
| | |
| Securities registered pursuant to Section 12(g) of the Act: None |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes o No x
Indicate by checkmark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x or No ¨
Indicate by checkmark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of "large accelerated filer," "accelerated filer," and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer o | Accelerated filer x | Non-accelerated filer o (do not check if a smaller reporting company) | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
The aggregate market value of the registrant's common stock held by nonaffiliates of the registrant as of the last business day of the registrant's most recently completed fiscal second quarter, June 30, 2012, based on the last reported sale price of the registrant's common stock of $2.13 per share was $179,193,701. The number of shares outstanding of the registrant's Common Stock, $.01 par value, as of February 15, 2013, was 99,527,059.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant's Definitive Proxy Statement for its 2012 Annual Meeting of Shareholders scheduled to be held on May 9, 2013, which Definitive Proxy Statement will be filed with the Securities and Exchange Commission not later than 120 days after the registrant's fiscal year-end of December 31, 2012, are incorporated by reference into Part III of this Form 10-K.
As used in this Form 10-K, "Dyax," "the Company," "we," "our," and "us" refer to Dyax Corp., except where the context otherwise requires or as otherwise indicated.
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements, including statements regarding:
| | ● | the potential benefits and commercial potential of KALBITOR® (ecallantide) for its approved indication and any additional indications; |
| | ● | our commercialization of KALBITOR, including revenues, costs and the potential benefits of new sales initiatives; |
| | ● | the potential for regulatory approval for KALBITOR in markets outside the United States; |
| | ● | plans and anticipated timing for pursuing additional indications and uses for ecallantide and other product candidates to address plasma kallikrein (bradykinin) mediated angioedemas; |
| | ● | plans to enter into additional collaborative and licensing arrangements for ecallantide and for other compounds in development; |
| | ● | estimates of potential markets for our products and product candidates; |
| | ● | the sufficiency of our cash, cash equivalents and short-term investments; and |
| | ● | expected future revenues, operating results and cash flows. |
Statements that are not historical facts are based on our current expectations, beliefs, assumptions, estimates, forecasts and projections for our business and the industry and markets in which we compete. We often use the words or phrases of expectation or uncertainty like “guidance,” "believe," "anticipate," "plan," "expect," "intend," "project," "future," "may," "will," "could," "would" and similar words to help identify forward-looking statements. These statements are not guarantees of future performance and are subject to certain risks, uncertainties, and assumptions that are difficult to predict; therefore, actual results may differ materially from those expressed or forecasted in any such forward-looking statements. Such risks and uncertainties include, but are not limited to, those discussed later in this report under the section entitled "Risk Factors". Unless required by law, we undertake no obligation to update publicly any forward-looking statements, whether because of new information, future events or otherwise. Readers should carefully review the risk factors set forth in this and other reports or documents we file from time to time with the Securities and Exchange Commission.
DYAX CORP.
ANNUAL REPORT ON FORM 10-K
For the year ended December 31, 2012
INDEX
| Item No. | | | Page |
| |
| PART I |
| 1. | | | 3 |
| 1A. | | | 20 |
| 1B. | | | 39 |
| 2. | | | 39 |
| 3. | | | 39 |
| 4. | | | 39 |
| |
| PART II |
| 5. | | | 40 |
| 6. | | | 41 |
| 7. | | | 42 |
| 7A. | | | 54 |
| 8. | | | 55 |
| 9. | | | 89 |
| 9A. | | | 89 |
| 9B. | | | 89 |
| |
| PART III |
| 10. | | | 90 |
| 11. | | | 90 |
| 12. | | | 90 |
| 13. | | | 91 |
| 14. | | | 91 |
| |
| PART IV |
| 15. | | | 91 |
| | | | 96 |
PART I
We are a biopharmaceutical company with two business elements:
| | ● | Plasma Kallikrein-Mediated Angioedema Portfolio |
The principal focus of our strategy is to identify, develop and commercialize treatments for angioedemas that are identified as plasma kallikrein-mediated, which we refer to as PKM angioedemas, including hereditary angioedema (HAE) and idiopathic angioedema.
We discovered and developed KALBITOR (ecallantide) using our phage display technology, and since February 2010, we have been selling it in the United States for the treatment of acute attacks of HAE. Outside of the United States, we have established partnerships to obtain regulatory approval for and to commercialize KALBITOR in certain markets and we are evaluating opportunities in others.
We are expanding our franchise for the treatment of PKM angioedemas in the following ways:
| | ● | Development of diagnostic strategies to assist in the differentiation between histamine-mediated and PKM angioedema. |
| | ● | Continuing our development of DX-2930, a fully human monoclonal antibody inhibitor of plasma kallikrein, which could be a candidate to prophylactically treat PKM angioedemas. |
| | ● | Phage Display Licensing and Funded Research Program |
We leverage our proprietary phage display technology through our Licensing and Funded Research Program, referred to as the LFRP. This program has provided us a portfolio of product candidates being developed by our licensees, which currently includes 13 product candidates in various stages of clinical development, including three in Phase 3 trials, for which we are eligible to receive future royalties and/or milestone payments. The LFRP generated approximately $12.5 million of revenue for us in 2012. To the extent that our licensees commercialize some of the Phase 3 product candidates, our revenues under the LFRP are expected to experience growth beginning in 2014.
PKM ANGIOEDEMA PORTFOLIO
We are focused on identifying and developing treatments for patients who experience PKM angioedema. Using our phage display technology, we developed ecallantide a compound shown in vitro to be a high affinity, high specificity inhibitor of human plasma kallikrein. Plasma kallikrein, an enzyme found in blood, produces bradykinin, a protein that causes blood vessels to enlarge or dilate, which can cause swelling known as angioedema. Plasma kallikrein is believed to be a key component in the regulation of inflammation and contact activation pathways. Excess plasma kallikrein activity is thought to play a role in a number of inflammatory diseases, including PKM angioedemas such as HAE and idiopathic angioedema.
We have three key areas of activity in our PKM angioedema portfolio:
| | ● | HAE and KALBITOR. In February 2010, we began selling KALBITOR in the United States for treatment of acute attacks of HAE in patients 16 years of age and older. We are selling KALBITOR on our own in the United States. Working with international partners, we intend to seek approval for and commercialize KALBITOR for HAE and other angioedema indications in markets outside of the United States. We have entered into agreements for others to develop and commercialize subcutaneous ecallantide for the treatment of HAE and other angioedema indications throughout Europe, Japan, China and countries in Latin America and the Middle East. |
| | ● | Identification of PKM angioedemas. In order to expand our PKM angioedema portfolio, we have launched a program to identify one or more diagnostic strategies that will assist in the differentiation of PKM angioedema from histamine-mediated angioedema, and facilitate appropriate treatment. We have developed laboratory tests for which the process of clinical validation has commenced. These tools are expected to be relevant to both normal C1esterase inhibitor (C1-INH) and C1-INH -deficient patients and will enable the identification of PKM angioedema, including Type III HAE and angioedema of unknown origin, or idiopathic angioedema. |
| | ● | DX-2930 - Antibody for PKM angioedemas. Based on our knowledge of angioedema and the kallikrein-kinin pathway, we are investigating the use of a fully human monoclonal antibody that is an inhibitor of plasma kallikrein and which could be a candidate to treat prophylactically PKM angioedemas. After completing a series of pharmacokinetic, tolerability and preclinical studies, we believe DX-2930 may be effective for prophylactically treating these indications. We expect to file an Investigational New Drug application (IND) for this antibody in mid-2013. |
HAE AND KALBITOR
HAE is a rare, genetic disorder characterized by severe, debilitating and often painful swelling, which can occur in the abdomen, face, hands, feet and airway. HAE is caused by a deficiency of C1-INH activity, a naturally occurring molecule that inhibits plasma kallikrein, a key mediator of inflammation, and other serine proteases in the blood. It is estimated that HAE affects between 1 in 10,000 to 1 in 50,000 people around the world. Based upon HAE patient association registries, we estimate there is an addressable target population of approximately 6,500 patients in the United States.
Our product, ecallantide, was approved by the FDA under the brand name KALBITOR for treatment of HAE in patients 16 years of age and older regardless of anatomic location. KALBITOR, a potent, selective and reversible plasma kallikrein inhibitor, was the first subcutaneous HAE treatment approved in the United States.
United States Sales and Marketing
We have a commercial organization to support sales of KALBITOR in the United States, including a field-based team of approximately 30 professionals, consisting of sales representatives, market access and field advocates and corporate account directors. At this time, our commercial organization is sized to market KALBITOR in the United States, where patients are treated primarily by a limited number of specialty physicians, consisting mainly of allergists and immunologists.
KALBITOR Access®
To facilitate access to KALBITOR in the United States, we have established the KALBITOR Access program, designed as a one-stop point of contact for information about KALBITOR. This program offers treatment support services for patients with HAE and their healthcare providers. KALBITOR case managers provide comprehensive product and disease information, treatment site coordination, financial assistance for qualified patients and reimbursement facilitation services.
Distribution
During 2012, KALBITOR was distributed through a limited network of wholesale and specialty pharmacy arrangements with the following entities:
| | ● | US Bioservices Corporation (US Bio), serves as a specialty pharmacy for KALBITOR and also administers KALBITOR Access, which provides comprehensive call center services for patients and healthcare providers seeking information and access to KALBITOR; and |
| | ● | ASD Specialty Healthcare Inc. (ASD) serves as a wholesale distributor for KALBITOR to treating hospitals in the United States. |
| | ● | Walgreens Infusion Services, Inc. (Walgreens) provides eligible HAE patients with on-demand nursing services for the home administration of KALBITOR by healthcare professionals, as well as treatment at Walgreens’ infusion centers. This agreement has a term through December 2013 and will renew annually unless amended or terminated by the parties. |
In January 2013, we elected to open our network to include a limited number of additional specialty pharmacies. In addition to the agreements mentioned above, we now have an arrangement with Accredo Health Group, Inc., and may enter into additional arrangements as appropriate to provide HAE patients with broader opportunities to access KALBITOR.
Post-Marketing Commitments
As part of the product approval of KALBITOR by the United States Food and Drug Administration (FDA), we have implemented a Risk Evaluation and Mitigation Strategy (REMS) program to communicate the risk of anaphylaxis and the importance of distinguishing between hypersensitivity reaction and HAE attack symptoms. To communicate these risks, through February 2012, a "Dear Healthcare Professional" letter was provided to doctors identified as likely to prescribe KALBITOR and treat HAE patients.
In February 2010, we initiated a 4-year, Phase 4 observational study which is being conducted with up to 200 HAE patients to evaluate immunogenicity and hypersensitivity with exposure to KALBITOR for treatment of acute attacks of HAE.
Single-Injection KALBITOR Formulation
We are currently in the process of developing a more convenient formulation of ecallantide, which is intended to allow for a single subcutaneous injection of KALBITOR, instead of the current three subcutaneous injection formulation. We completed a bioequivalence clinical study which successfully demonstrated bioequivalence between the current formulation and the new single-injection formulation. We expect to file a supplemental Biologics License Application (BLA) with the FDA by mid-2013.
Manufacturing
We have established a commercial supply chain, consisting of third parties to manufacture, test and transport KALBITOR. All third party manufacturers involved in the KALBITOR manufacturing process are required to comply with current good manufacturing practices, or cGMPs.
To date, ecallantide drug substance used in the production of KALBITOR has been manufactured in the United Kingdom by Fujifilm Diosynth Biotechnologies (UK) Ltd. (Fujifilm). Under our agreement with Fujifilm, they have committed to be available to manufacture bulk drug substance through 2020.
The shelf-life of our frozen ecallantide drug substance is four years. Ecallantide drug substance is filled, labeled and packaged into the final form of KALBITOR drug product by Jubilant Hollister-Stier Contract Manufacturing Services at its facilities in Spokane, Washington under a commercial supply agreement. This process is known in the industry as the "fill and finish" process. KALBITOR in its "filled and finished" form has additional refrigerated shelf-life of four years.
Our current inventory of filled drug product, together with drug substance inventory, when filled, is sufficient to supply all ongoing studies relating to ecallantide and to meet anticipated KALBITOR market demand into 2016.
Ecallantide Outside of the United States
In markets outside of the United States, we intend to work with international partners to seek approval and commercialize ecallantide for HAE and other angioedema indications. We have entered into license or collaboration agreements with several such companies, which have regulatory capabilities, distribution systems and sales capabilities in their designated territories.
CMIC – In Japan, we have an agreement with CMIC Co., Ltd to develop and commercialize subcutaneous ecallantide for the treatment of HAE and other angioedema indications. Under the terms of the agreement, we received a $4.0 million upfront payment. We will also be eligible to receive up to $102 million in development and sales milestones for ecallantide in HAE and other angioedema indications and royalties of 20%-24% of net product sales. CMIC is solely responsible for all costs associated with development, regulatory activities, and commercialization of ecallantide for all angioedema indications in Japan. CMIC will purchase drug product from us on a cost-plus basis for clinical and commercial supply.
CMIC has a clinical development plan that was established in consultation with the Japanese regulatory authorities. CMIC has completed a twelve patient pharmacokinetic study and, to fulfill submission requirements, the company is required to complete an open-label study of ten patients which commenced in the second half of 2012. Assuming successful completion of the open-label study, CMIC plans to commercialize subcutaneous ecallantide for the treatment of HAE in Japan in 2014.
CVie – In February 2013, we entered into a strategic partnership with CVie Therapeutics (CVie), a subsidiary of Lee’s Pharmaceutical Holdings Ltd. in China, for the development and commercialization of KALBITOR in the treatment of HAE and other angioedema indications in China.
Under the terms of the exclusive license agreement, we will receive an upfront payment and we are eligible to receive future development, regulatory and sales milestones. We are also eligible to receive royalties on net product sales. CVie is solely responsible for all costs associated with development, regulatory activities, and the commercialization of KALBITOR in their licensed territories. Additionally, CVie will purchase drug product from us on a cost-plus basis for commercial supply.
Novellus – In January 2013, we entered into a strategic partnership with Novellus Biopharma AG for the development and commercialization of KALBITOR for the treatment of HAE and other angioedema indications in select countries in Latin America, including Argentina, Brazil, Chile, Colombia, Mexico and Venezuela.
Under the terms of the exclusive license agreement, we will receive an upfront payment and we are eligible to receive future regulatory and sales milestones. We are also eligible to receive royalties on net product sales. Novellus is solely responsible for all costs associated with development, regulatory activities, and the commercialization of KALBITOR in their licensed territories. Additionally, Novellus will purchase drug product from us on a cost-plus basis for commercial supply.
Taiba – In May 2012, we granted exclusive distribution rights to taiba ME (referred to as Taiba), under which they will obtain registration and reimbursement approval and commercialize ecallantide for HAE in certain countries in the Middle East. Under the terms of the agreement, we will provide Taiba with drug supply at a price equal to 60% of net sales within the licensed territory. The initial supply of drug to Taiba was made in January 2013.
Sigma-Tau – We have a collaboration agreement with Sigma-Tau Rare Diseases S.A. (as successor-in-interest to Defiante Farmaceutica S.A.), a subsidiary of Sigma-Tau SpA, to develop and commercialize subcutaneous ecallantide for the treatment of HAE and other therapeutic indications throughout Europe, Russia, Australia and New Zealand. Under the terms of the agreement, as amended, Sigma-Tau made an aggregate of $7.0 million in upfront payments to us and also purchased 787,647 shares of our common stock for an aggregate purchase price of $3.0 million. We are eligible to receive development and sales milestones related to ecallantide and royalties equal to 41% of net sales of product, as adjusted for product costs. Under the terms of amendments to our agreement that eliminated Sigma-Tau’s rights to the Middle East, Latin America and the Caribbean territories, we agreed to make payments to Sigma-Tau ranging from 5%-12.5% of the amounts received by us as a result of any product sales in certain countries in these territories.
Sigma-Tau is responsible for the costs associated with regulatory approval and commercialization in the licensed territories. In addition, we and Sigma-Tau will share equally the costs for all development activities for future indications developed in partnership with Sigma-Tau.
IDENTIFICATION OF PKM ANGIOEDEMAS
We have a program that has identified several diagnostic strategies that will assist in the differentiation of PKM angioedema from histamine-mediated angioedema, and facilitate appropriate treatment. We have developed laboratory tests for which clinical validation has commenced. These tools are expected to be relevant to both normal C1-INH and C1-INH deficient patients and will enable the identification of PKM angioedema, including Type III HAE and angioedema of unknown origin, or idiopathic angioedema.
PLASMA KALLIKREIN ANTIBODY – DX-2930
We are currently in preclinical development of DX-2930, a potent and specific fully human monoclonal antibody that is an inhibitor of plasma kallikrein and which would be a candidate to prophylactically treat PKM angioedema. DX-2930 has the potential for a subcutaneous formulation, with a half-life which could enable less frequent dosing than currently available prophylactic therapies and an advantageous immunogenicity profile. We have completed a series of preliminary preclinical pharmacokinetic and tolerability studies and found DX-2930 to have relevant activity in animal models. We expect to file an IND for this antibody in mid-2013.
LICENSING AND FUNDED RESEARCH PROGRAM
We believe that our phage display libraries, which we have developed using our core technology and know-how, represent a leading technology in antibody discovery. We leverage our proprietary phage display technology and libraries through our LFRP licenses and collaborations. To date, we have recognized more than $185 million of revenue under the LFRP, primarily related to license fees and milestones, including approximately $12.5 million in 2012. Our LFRP has the potential for substantially greater revenues if and when product candidates that are discovered by our licensees receive marketing approval and are commercialized.
LFRP Product Development
Currently, 18 product candidates generated by our licensees or collaborators under our LFRP portfolio are in clinical development and one product has received marketing approval from the FDA. We will receive future milestones and/or royalties from our licensees and collaborators for 13 of the 18 product candidates that are currently in clinical development, including three in Phase 3 and four in Phase 2 trials, to the extent these product candidates advance in development and are ultimately commercialized. Furthermore, we believe our licensees and collaborators have 13 additional product candidates in various stages of preclinical development. Our licensees and collaborators are responsible for all costs associated with development of these product candidates. To the extent that our licensees commercialize some of the Phase 3 product candidates, our revenues under the LFRP are expected to experience growth beginning in 2014.
The chart below, which is based on information publicly disclosed by our licensees, provides a summary of the clinical stage product candidates under the LFRP for which we are eligible to receive future milestones and/or royalties to the extent these candidates are developed and commercialized. Certain of these product candidates are in multiple clinical trials for various indications.
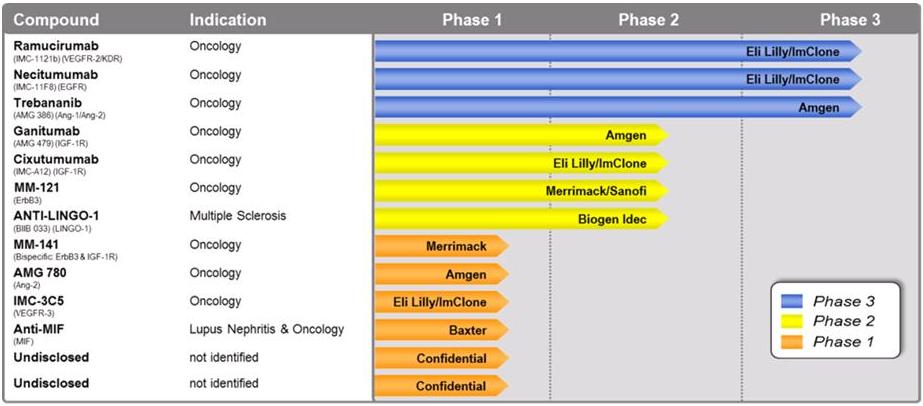
The types of licenses and collaborations that we have entered into under the LFRP have one of three distinct structures:
| | ● | Library Licenses. Under our library license program, we grant our licensees rights to use our phage display libraries in connection with their internal discovery and therapeutic development programs. These libraries are protected by a patent portfolio in which the last patent is scheduled to expire in 2024. We also provide these licensees with related materials and training so that they may rapidly identify compounds that bind with high affinity to therapeutic targets. The period during which our licensees may use our libraries is typically limited to a 4 to 5 year term. Library license agreements contain up-front license fees, annual maintenance fees, milestone payments based on successful product development, and royalties based on any future product sales. We have approximately 20 library licensees, including Amgen, Aveo, Bayer Schering, Biogen Idec, Boehringer Ingelheim, CSL Behring, ImClone Systems (a wholly-owned subsidiary of Eli Lilly), Kadmon, Merck Serono, Novo Nordisk, sanofi and Emergent BioSolutions (formerly known as Emergent Trubion). |
| | ● | Funded Research. Under our funded research program, we have performed funded research for various collaborators using our phage display libraries to identify, characterize and optimize antibodies that bind to disease targets provided by the collaborators. Funded research agreements provide for fees, technical and development milestones, and royalties based on any future product sales. Our funded research collaborators with products currently in development include Baxter Healthcare, Biogen Idec, Merck Serono, Merrimack Pharmaceuticals, and Emergent BioSolutions. |
| | ● | Patent Licenses. Under our patent license program, we previously granted other biopharmaceutical and pharmaceutical companies non-exclusive licenses to use certain of our phage display patents to discover and develop biologic compounds for use in specified fields. The last of these patents expired in November 2012 and we do not anticipate entering into future agreements for this patent portfolio. In addition, certain existing patent licenses will no longer have a royalty obligation. We do not expect the expiration of these patents to have a material impact on our LFRP business. |
We expect to continue to enter into library licenses and funded research agreements to maximize the strategic value of our LFRP.
Cross-Licensed Technology
The use of our antibody library involves technology that we have cross-licensed from other biotechnology companies, including Affimed Therapeutics AG, Affitech A/S, Biosite, Inc. (now owned by Alere Inc.), Cambridge Antibody Technology Limited or CAT (now known as MedImmune Limited and owned by AstraZeneca), Domantis Limited (a wholly-owned subsidiary of GlaxoSmithKline), Genentech, Inc. and XOMA Ltd. Under the terms of our cross-license agreement with CAT, we are required to pay milestone and low single-digit royalty payments to CAT in connection with antibody products developed and commercialized by our licensees. These payments are passed through to CAT from our licensees. None of our other cross-license agreements contain financial obligations applicable to our LFRP licensees or collaborators.
HealthCare Royalty Partners
In August 2012, we completed an agreement with an affiliate of HealthCare Royalty Partners, formerly Cowen Healthcare Partners (HC Royalty) that we entered into in December 2011 to refinance our existing loans with HC Royalty. At December 31, 2012, the aggregate principal amount of the new loan was $81.2 million, consisting of a $21.9 million Tranche A Loan and a $59.3 million Tranche B Loan. The loans bear interest at a rate of 12% per annum, payable quarterly. The loans will mature in August 2018, and can be repaid without penalty beginning in August 2015.
In connection with the loans, we entered into a security agreement granting HC Royalty a security interest in the intellectual property related to the LFRP, and the revenues generated through our licenses of the intellectual property related to the LFRP. The security agreement does not apply to our internal drug development or to any of our co-development programs for HAE.
Under the terms of the loan agreement, we are required to repay the loans based on the annual net LFRP receipts. Until September 30, 2016, required payments are equal to the sum of 75% of the first $15.0 million in specified annual LFRP receipts and 25% of specified annual LFRP receipts over $15.0 million. After September 30, 2016, and until the maturity date or the complete repayment of the loans, HC Royalty will receive 90% of all specified LFRP receipts. If the HC Royalty portion of LFRP receipts for any quarter exceeds the interest for that quarter, then the principal balance will be reduced. Any unpaid principal will be due upon the maturity of the loans. If the HC Royalty portion of LFRP revenues for any quarterly period is insufficient to cover the cash interest due for that period, the deficiency may be added to the outstanding principal or paid in cash. After five years from the dates of the Tranche A Loan and the Tranche B Loan, respectively, we must repay to HC Royalty all additional accumulated principal above the original loan amounts of $21.7 million and $58.8 million, respectively.
OUR PHAGE DISPLAY TECHNOLOGY
What Is Phage Display?
Living organisms, such as viruses, have the ability to display a foreign gene product, or protein, on their surfaces. Based on this ability of organisms to display proteins, our scientists in the late 1980s invented protein phage display, a novel method to individually display up to tens of billions of human antibodies, peptides or small proteins on the surface of a small bacterial virus called a bacteriophage, or phage. Using phage display, we have built large collections, or libraries, of antibodies, small proteins or peptides that we use to rapidly identify those compounds that bind with high affinity and high specificity to targets of interest.
Through the use of our proprietary phage display technology, we have been able to establish a broad discovery platform to identify compounds that interact with a wide array of targets, including membrane proteins and circulating proteins which have been shown to be involved in pathologic processes. Our discovery capabilities have been further enhanced through automation, which has enabled us to evaluate a large number of molecules binding to each target. In this way we can rapidly identify and select a specific antibody, peptide or small protein with the desired biochemical and biological characteristics. While our discovery research efforts are focused primarily on monoclonal antibodies, we are also testing the in vitro and in vivo activity of several of our peptide and small protein compounds.
Scientists can use phage display to improve the speed and cost effectiveness of drug discovery and optimization. Phage display offers important advantages over, and can be used to improve, other drug discovery technologies that are currently employed to identify biopharmaceutical leads.
Over the past several years, we have brought on-line high-throughput automated capacity, developed antibody phage display libraries that are a leading technology in antibody discovery, and successfully implemented a strategy under which, as of today, we believe we have obtained freedom to operate in the antibody phage display area through cross-licenses with Affimed Therapeutics, Affitech, Biosite, CAT, Domantis, Genentech and XOMA. As a result of these activities, we now have an industry-leading technology that allows us to identify fully human antibodies with high specificity and high affinity and to move product candidates rapidly into both in vitro testing and optimization.
Although we use this technology to advance our own internal development activities, we also leverage it broadly through licenses and collaborations so that other biopharmaceutical and pharmaceutical companies can use it to discover and develop biopharmaceutical leads.
Our phage display process generally consists of the following steps:
| | ● | Generating a phage display library |
| | ● | Screening the phage display library against a target of interest |
| | ● | Evaluating the selected compounds that bind to the target of interest |
Generating a Phage Display Library
The generation of a phage display library is based upon a single protein framework and contains tens of billions of variants of this protein. The first step in generating a library is the selection of the protein framework upon which the library will be created. This selection is based on the desired product properties, such as structure, size, stability, or lack of immunogenicity. We then determine which amino acids in the framework will be varied, but do not vary amino acids that contribute to the framework structure. We also control the exact numbers and types of different amino acids that are varied, so that the resulting phage display library consists of a diverse set of chemical entities, each of which retains the desired physical and chemical properties of the original framework.
The next step is the creation of a collection of genes that encode the designed variations of the framework protein. We can easily generate diverse collections of up to hundreds of millions of different synthetic DNA sequences. Each new DNA sequence, or gene, encodes a single protein sequence that may be displayed on the surface of the individual phage that contains this gene. The scientists combine the new DNA sequences with phage genome DNA and certain enzymes so that the new DNA is inserted into a specific location of the phage genome. The result is that the new protein is displayed on the phage surface fused to one of the naturally occurring phage proteins. The phage acts as a physical link between the displayed protein and its gene.
In addition to fused synthetic DNA sequences, we may also use cDNA, or genomic DNA, which are sequences that represent all of the expressed genes in a cell or organism, to create a library. We have also inserted genes from antibody expressing human cells into the phage genome. Using these genes, we have constructed phage display libraries that express tens of billions of different human antibodies on the phage surface. From one of these libraries, individual antibody fragments can be selected and used to express highly specific human monoclonal antibodies.
The new phage genome is then transferred into laboratory bacteria, where the phage genome directs the bacterial cells to produce thousands of copies of each new phage. The collection of phage displaying multiple antibodies, peptides or small proteins is referred to as a phage display library. Because we can reproduce the phage display library by infecting a new culture of laboratory bacteria to produce millions of additional copies of each phage, we can use each library for a potentially unlimited number of selections.
Screening the Phage Display Library Against a Target of Interest
We can then select binding compounds with high affinity and high specificity by exposing the library to a specified target of interest and isolating the various phage that display compounds that bind to the target. Each individual phage contains the gene encoding one potential binding compound, and once its displayed protein is selected in the screening procedure, it can be retrieved and amplified by growth in laboratory bacteria.
To identify specific binders from a phage display library, we expose the library to the target under desired binding conditions. The target may be attached to a fixed surface, such as the bottom of a tube, or a bead, allowing removal of phage that do not express binding compounds that recognize the target. Once these unbound phage are washed away, the phage containing the selected binding compounds can be released from the target. Since the phage are still viable, they can be amplified rapidly by again infecting bacteria. The capacity of the phage to replicate itself is an important feature that makes it particularly well suited for rapid discovery of specific binding compounds. We can amplify a single phage by infecting bacteria and producing millions of identical phage in one day.
If the binding affinities of the compounds identified in an initial screening for a target are not considered sufficiently high, information derived from the binding compounds identified in the initial screening can be used to design a new focused library. The design, construction and screening of a second generation library, known as affinity maturation, can lead to increases of 10- to 100-fold or more in the affinity of the binding compounds for the target.
Evaluating the Selected Compounds That Bind to the Target of Interest
Screening phage display libraries generally results in the identification of one or more groups of related binding compounds such as antibodies, peptides or small proteins. These groups of compounds are valuable in providing information about which chemical features are necessary for binding to the target with affinity and specificity, as well as which features can be altered without affecting binding. Using DNA sequencing, we can determine the amino acid sequences of the binding compounds and identify the essential components of desired binding properties by comparing similarities and differences in such sequences. If desired, scientists can further optimize the binding compounds by building additional phage display libraries based on these key components and repeating this process. We can complete the entire selection process in several weeks. We can produce small amounts of the binding compound by growing and purifying the phage. For production of larger amounts, we can remove the gene from the phage DNA and place it into a standard recombinant protein expression system. Alternatively, if the identified binding compound is sufficiently small, it can be chemically synthesized. These binding compounds can be evaluated for desired properties including affinity, specificity and stability under conditions that will be encountered during its intended use. From each group of compounds, scientists can identify, develop and test a compound with the desired properties for utility as a biopharmaceutical, diagnostic, research reagent or affinity separations product.
The entire phage display process for identifying compounds that bind to targets of interest is nearly identical whether the ultimate product is to be used for biopharmaceuticals, diagnostics, research reagents or separations, which allows for an efficient use of scientific resources across a broad array of commercial applications.
Advantages of Phage Display Technology in Therapeutic Drug Discovery
We believe our phage display technology has the following advantages over other drug discovery technologies:
| | ● | Diversity and abundance. Many of our phage display libraries contain billions of potential binding compounds that are rationally-designed variations of a particular antibody, peptide or small protein framework. The size and diversity of our libraries significantly increases the likelihood of identifying binding compounds with high affinity and high specificity for the target. Once we generate libraries, we can reproduce them rapidly in phage and use them for an unlimited number of screenings. |
| | ● | Speed and cost effectiveness. We can construct phage display libraries in a few months and rapidly select binding compounds for characterization in screening assays. Conventional or combinatorial chemistry approaches require between several months and several years to complete this process. Similarly, mouse and human-mouse technologies generally require four to six months to identify an antibody. As a result, our phage display technology can significantly reduce the time and expense required to identify an antibody, peptide or small protein with desired binding characteristics. |
| | ● | Automated parallel screening. In an automated format, we can apply our phage display technology to many targets simultaneously to discover specific, high-affinity proteins, including human monoclonal antibodies, for each target. In contrast, human-mouse antibody technologies identify antibodies that bind to a single target per test group of mice and are difficult to automate. Among antibody technologies, phage display is particularly well suited for functional genomic applications, due to the large number of genetic targets that need to be screened for specific antibodies. |
| | ● | Rapid optimization. We screen phage display libraries to identify binding compounds with high affinity and high specificity for the desired target and can design and produce successive generations of phage display libraries to further optimize the leads. We have demonstrated between 10- and 1000-fold improvement in binding affinity with second-generation phage display libraries. |
COMPETITION
The biopharmaceutical industry is characterized by continuous intense competition and rapid technological change. New developments occur constantly and are expected to continue to occur. Discoveries or commercial developments by our competitors or others may render some or all of our technologies, products or potential products obsolete or non-competitive.
In our PKM angioedema portfolio, our principal focus is on the development and commercialization of therapeutic and diagnostic products in angioedema indications. Therefore our principal competition is companies that either are already marketing products in those indications or are developing new products for those indications, as described below.
For KALBITOR as a treatment for HAE, our principal competitors include:
| | ● | Manufacturers of corticosteroids, including danazol, which have been used historically and are still used to treat prophylactically a significant number of identified HAE patients. |
| | ● | Shire plc— Shire markets its bradykinin receptor antagonist, known as Firazyr® (icatibant), which is administered subcutaneously. Firazyr is approved in the US, Europe, and certain other countries for the treatment of acute HAE attacks in adult patients. The US and EU labels allow for patients to self-administer Firazyr following training by their healthcare provider. Firazyr has orphan drug designations from the FDA and in Europe. |
| | ● | CSL Behring— CSL Behring markets a plasma-derived C1-esterase inhibitor, known as Berinert®, which is administered intravenously. Berinert is approved in the US for the treatment of acute abdominal, facial or laryngeal attacks of HAE in adults and adolescents, and has orphan drug designation from the FDA. The FDA has also approved labeling for Berinert to include self-administration after proper training by a healthcare professional. Berinert is also approved in the EU, Japan and several rest-of-world markets for the treatment of acute attacks of HAE. CSL Behring announced in May 2012 that they had commenced an international Phase I/II study of a volume-reduced subcutaneous formulation of C1-INH which will evaluate the pharmacokinetics, pharmacodynamics and safety of various doses of C1-INH. |
| | ● | ViroPharma Inc. — ViroPharma markets a plasma-derived C1-esterase inhibitor, known as Cinryze®, which is administered intravenously. Cinryze is approved in the US for routine prophylaxis against angioedema attacks in adolescent and adult patients with HAE, and has orphan drug designation from the FDA. The FDA has also approved patient labeling for Cinryze to include self-administration for routine prophylaxis once a patient is properly trained by his or her healthcare provider. ViroPharma has also received approval in the EU where the product is approved for the treatment and pre-procedure prevention of angioedema attacks in adults and adolescents with HAE, and routine prevention of angioedema attacks in adults and adolescents with severe and recurrent attacks of HAE, who are intolerant to or insufficiently protected by oral prevention treatments or patients who are inadequately managed with repeated acute treatment. The EU approval includes a self-administration option for appropriately trained patients. ViroPharma is conducting two Phase 2 trials evaluating subcutaneous administration of Cinryze. |
| | ● | Pharming Group NV— Pharming markets a recombinant C1-esterase inhibitor, known as Ruconest™, which is administered intravenously. Ruconset is approved in the EU for the treatment of acute HAE attacks in adult patients. In November 2012, Pharming and its US partner Santarus announced positive topline data from their Phase 3 trial for Ruconest. Pharming's recombinant C1-esterase inhibitor has Fast Track status from the FDA and orphan drug designations from the FDA and in Europe. |
Other competitors for the treatment of HAE are companies that are developing small molecule plasma kallikrein inhibitors, including BioCryst.
Additionally, a significant number of companies compete with our LFRP in the antibody technology space by offering licenses and/or research services to pharmaceutical and biotechnology companies. Specifically, our phage display technology is one of several in vitro display technologies available to generate libraries of compounds that can be leveraged to discover new antibody products. Companies that compete with us in the display technology space include BioInvent, XOMA, Adimab and several others. Additional platforms that pharmaceutical and biotechnology companies use to identify antibodies that bind to a desired target are in vivo technology platforms which use direct immunization of mice or other species to generate fully human antibodies. Competitors in this space include GenMab, arGEN-X and several others. There are also a number of new technologies directed to the generation of candidate compounds with novel scaffolds that may possess similar properties to monoclonal antibodies.
In addition to the technologies described above, many pharmaceutical companies have either acquired antibody discovery technologies or developed humanized murine antibodies derived from hybridomas. Pharmaceutical companies also develop orally available small molecule compounds directed to the targets for which we and others are seeking to develop antibody, peptide and/or protein products.
Furthermore, we may also experience competition from companies that have acquired or may acquire other technologies from universities and other research institutions and that may impact our competitive position.
PATENTS AND PROPRIETARY RIGHTS
Our success is significantly dependent upon our ability to obtain patent protection for our products and technologies, to defend and enforce our issued patents, including patents related to phage display, and to avoid the infringement of patents issued to others. Our policy generally is to file for patent protection on methods and technology useful for the display of binding molecules and on biopharmaceutical, diagnostic and separation product candidates.
Our proprietary position in the field of phage display is based upon patent rights, technology, proprietary information, trade secrets and know-how. Our patents and patent applications related to specific aspects of our phage display libraries, including claiming our currently licensed antibody phage display libraries and methods of making and using such libraries include issued patents in the United States, Europe, Australia, Canada, and Japan, and pending patent applications in the United States, Europe and other countries. These patent rights are expected to expire in 2021 or later (not including any term extension from the addition of patent term adjustment by the US Patent and Trademark Office). Patent rights claiming our currently licensed antibody libraries include United States Patent No. 8,288,322, which expires February 10, 2023 and issued patents in Australia, Canada and Europe. We have filed a petition in the United States Patent Office for reconsideration and recalculation of the patent term adjustment for United States Patent No. 8,288,322 based on an erroneous calculation of the patent's term by the United States Patent Office. This petition, which is expected to be successful based on recent district court rulings, could extend the patent's expiration date by 2,473 days, from the current expiry of February 10, 2023 to January 24, 2028. Additionally, patent rights claiming our currently licensed peptide libraries include United States Patent No. 7,413,537, which expires March 1, 2014 and issued patents in Canada, Japan and Europe. We have filed suit in the United States District Court for the District of Columbia to obtain a patent term adjustment for United States Patent No. 7,413,537 based on an erroneous calculation of the patent's term by the United States Patent Office. This action, which is expected to be successful based on a recent ruling by the United States Court of Appeals for the Federal Circuit, could extend the patent's expiration date by 1,614 days, from the current expiry of March 1, 2014 to May 1, 2017.
With respect to KALBITOR (ecallantide), our patent rights include United States Patent Nos. 5,795,865, which expires August 18, 2015; 5,994,125, which expires January 11, 2014: 6,057,287, which expires August 18, 2015: 6,333,402, which expires January 11, 2014: 7,064,107, which expires June 6, 2023: 7,153,829, which expires July 2, 2023: 7,166,576, which expires September 27, 2024: 7,235,530, which expires September 27, 2024: 7,276,480, which expires June 6, 2023; 7,628,983, which expires February 11, 2015: 7,718,617, which expires August 26, 2024: 7,811,991, which expires February 26, 2024; 7,704,949, which expires June 6, 2023; 7,851,442 which expires September 9, 2023; 8,034,775 which expires June 6, 2023; and European Patent Nos. 0739355 which expires January 11, 2015; 1,531,791 which expires June 6, 2023; and 1941867 which expires June 6, 2023, as well as issued patents in Australia, Canada and Japan, claiming sequences of peptides that have human kallikrein inhibitory activity, including the sequence for KALBITOR, and polynucleotide sequences encoding these peptides, as well as methods of using such peptides.
For our other therapeutic product candidates, we file for patent protection on groups of antibodies, peptides and small proteins that we identify using phage display.
There are no legal challenges to our phage display patent rights or our other issued or pending patent now pending in the United States. However, European Patent No. 1,578,903, a patent directed to certain phage display embodiments, is presently under opposition at the European Patent Office. We cannot assure that additional challenges will not be brought in the future. We plan to protect our patent rights in a manner consistent with our product development and business strategies. If we bring legal action against an alleged infringer of any of our patents, we expect the alleged infringer to claim that our patent is invalid, not infringed, or not enforceable for one or more reasons, thus subjecting that patent to a judicial determination of infringement, validity and enforceability. In addition, in certain situations, an alleged infringer could seek a declaratory judgment of non-infringement, invalidity or unenforceability of one or more of our patents. We cannot be sure that we will have sufficient resources to enforce or defend our patents against any such challenges or that a challenge will not result in an adverse judgment against us or the loss of one or more of our patents. Uncertainties resulting from the initiation and continuation of any patent or related litigation, including those involving our patent rights, could have a material adverse effect on our ability to maintain and expand our licensing program and collaborations, and to compete in the marketplace.
Our phage display patent rights are central to our non-exclusive patent licensing program and our performance under our related agreement with Healthcare Royalty Partners. We offer non-exclusive licenses under our phage display patent rights to companies and non-profit institutions in the fields of therapeutics, diagnostics and other select fields. In jurisdictions where we have not applied for, obtained, or maintained patent rights, we will be unable to prevent others from developing or selling products or technologies derived using phage display. In addition, in jurisdictions where we have phage display patent rights, we cannot assure that we will be able to prevent others from selling or importing products or technologies derived using phage display.
We are aware that other parties have patents and pending applications to various products and processes relating to phage display technology. Through licensing our phage display patent rights, we have secured a limited ability to practice under some of the third party patent rights relating to phage display technology. These rights are a result of our standard license agreement, which contains a covenant by the licensee that it will not sue us under the licensee's phage display improvement patents. In addition, we have sought and obtained affirmative rights of license or ownership under certain patent rights relating to phage display technology owned by other parties. For example, in addition to our amended license agreement with CAT, we have entered into licensing agreements with Affimed Therapeutics, Affitech, Biosite, Domantis and Genentech, Inc. under which we granted each of those companies rights to practice our phage display patents and in return received rights to practice under their phage display related patents. These types of agreements in which each party licenses technology to the other are referred to as cross-licensing agreements. We have also entered into a cross-licensing agreement with XOMA Ireland Limited under which we received a license to use XOMA's antibody expression technology to develop antibody products for ourselves and our collaborators. We also received a license from XOMA to produce antibodies. In exchange we agreed to pay XOMA a license fee and a royalty in connection with the sale of any of our antibody products. We also granted XOMA a license to our phage display patents and agreed to provide them with limited quantities of our antibody phage display libraries.
Under the terms of our amended and restated license agreement with CAT, we were granted a worldwide license under their antibody phage display patents to discover and develop antibody products. In consideration for this license, CAT is eligible to receive milestone payments and low single-digit royalty payments in connection with antibody products developed and commercialized by us or our licensees under the agreement.
Under the agreement, we also granted CAT a worldwide license to use our antibody libraries to discover and develop antibody products. In consideration for this license, we will receive no milestone payments but are eligible to receive a low single-digit royalty payments on antibody products developed by CAT or its licensees under the agreement.
The preclinical study and clinical testing, manufacture, labeling, storage, record keeping, advertising, promotion, export, and marketing, among other things, of our products and product candidates, including KALBITOR, are subject to extensive regulation by governmental authorities in the United States and other countries. In the United States, pharmaceutical products are regulated by the FDA under the Federal Food, Drug, and Cosmetic Act and other laws, including, in the case of biologics, the Public Health Service Act. KALBITOR is regulated by the FDA as a biologic. Biologics require the submission of a BLA, and approval by FDA prior to being marketed in the United States. Manufacturers of biologics may also be subject to state regulation. Failure to comply with FDA requirements, both before and after product approval, may subject us and/or our partners, contract manufacturers, and suppliers to administrative or judicial sanctions, including FDA refusal to approve applications, issuance of warning letters, product recalls, product seizures, total or partial suspension of production activities or distribution, fines and/or criminal prosecution.
The steps required before a biologic may be approved for marketing in the United States generally include:
| | ● | preclinical laboratory tests and animal tests; |
| | ● | submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may commence; |
| | ● | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product; |
| | ● | submission to the FDA of a BLA; |
| | ● | FDA pre-approval inspection of product manufacturers; and |
| | ● | FDA review and approval of BLA. |
Preclinical studies include laboratory evaluation, as well as animal studies to assess the potential safety and efficacy of the product candidate. Preclinical safety tests must be conducted in compliance with FDA regulations relating to good laboratory practices. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND, which must become effective before human clinical trials may be commenced. An IND application will automatically become effective 30 days after receipt by the FDA, unless the FDA before that time raises concerns about the drug candidate or the conduct of the trials as outlined in the IND. The IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed. We cannot assure you that submission of an IND will result in FDA authorization to commence clinical trials or that once commenced, other concerns will not arise.
Clinical trials involve the administration of the investigational product to healthy volunteers or to patients, under the supervision of qualified principal investigators. Each clinical study at each participating clinical site must be reviewed and approved by an independent institutional review board, prior to the recruitment of subjects.
Clinical trials are typically conducted in three sequential phases, but the phases may overlap and different trials may be initiated with the same drug candidate within the same phase of development in similar or differing patient populations.
Phase 1 studies may be conducted in a limited number of patients, but are usually conducted in healthy volunteer subjects. The drug candidate is usually tested for safety and, as appropriate, for absorption, metabolism, distribution, excretion, pharmaco-dynamics and pharmaco-kinetics.
Phase 2 usually involves studies in a larger, but still limited patient population to evaluate preliminarily the efficacy of the drug candidate for specific, targeted indications; to determine dosage tolerance and optimal dosage; and to identify possible short-term adverse effects and safety risks.
Phase 3 studies are undertaken to further evaluate clinical efficacy of a drug candidate against specific endpoints and to test further for safety within an expanded patient population at geographically dispersed clinical study sites. Phase 1, Phase 2 or Phase 3 testing might not be completed successfully within any specific time period, if at all, with respect to any of our product candidates. Results from one trial are not necessarily predictive of results from later trials. Furthermore, the FDA may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
The results of the preclinical studies and clinical trials, together with other detailed information, including information on the manufacture and composition of the product candidate, are submitted to the FDA as part of a BLA requesting approval to market the product candidate. Under the Prescription Drug User Fee Act, as amended, the required fees payable to the FDA for reviewing a BLA, as well as annual fees for commercial manufacturing establishments and for approved products, can be substantial. Each BLA submitted to the FDA for approval is typically reviewed for administrative completeness and reviewability within 45 to 60 days following submission of the application. If found complete, the FDA will "file" the BLA, thus triggering a full review of the application. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable. The FDA's established goals for the review of a BLA are six months for Priority applications and 12 months for Standard applications, whereupon a review decision is to be made. The FDA, however, may not approve a drug within these established guidelines and its review goals are subject to change from time to time. Further, the outcome of the review, even if generally favorable, may not result in an actual approval but an "action letter" may be issued that describes additional work that must be done before the application can be approved. Before approving a BLA, the FDA may inspect the facilities at which the product is manufactured and will not approve the product for marketing unless current Good Manufacturing Practices, or cGMP, compliance is satisfactory. The FDA may deny approval of a BLA if applicable statutory or regulatory criteria are not satisfied, or may require additional testing or information, which can delay the approval process. FDA approval of any application may include many delays or never be granted. If a product is approved, the approval will impose limitations on the indicated uses for which the product may be marketed, may require that warning statements be included in the product labeling, and may require that additional studies be conducted following approval as a condition of the approval, and may impose restrictions and conditions on product distribution, prescribing or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. To market a product for other indicated uses, or to make certain manufacturing or other changes requires additional FDA review and approval of a BLA Supplement or new BLA. Further post--marketing testing and surveillance to monitor the safety or efficacy of a product is required. Also, product approvals may be withdrawn if compliance with regulatory standards is not maintained or if safety or manufacturing problems occur following initial marketing. In addition new government requirements may be established that could delay or prevent regulatory approval of our product candidates under development.
Under the Patient Protection and Affordable Care Act of 2010 an abbreviated approval process is currently available for generic or “follow-on” biologic products that are demonstrated to be biosimilar to or interchangeable with an FDA-licensed biological product. The FDA has issued guidance documents on biosimilar product development. The FDA will consider multiple factors as part of its biosimilarity assessment, including, but not limited to, the product’s complexity, formulation, and stability; as well as usefulness of biochemical and functional characterizations. Although it is unclear how the abbreviated approval process will impact our business, it could have a material adverse impact as follow-on products may be significantly less costly to bring to market and may be priced significantly lower than our products would be.
Both before and after the FDA approves a product, the manufacturer and the holder or holders of the BLA for the product are subject to comprehensive regulatory oversight. For example, quality control and manufacturing procedures must conform to cGMP requirements, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to spend time, money and effort to maintain cGMP compliance.
Orphan Drug Designation
We have received orphan drug designation from the FDA for KALBITOR. Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a "rare disease or condition," which generally is a disease or condition that affects fewer than 200,000 individuals in the United States. Orphan drug designation must be requested before submitting a BLA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are publicly disclosed by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning the FDA may not approve any other applications to market the same drug for the same indication for a period of seven years, except in limited circumstances.
Regulation Pertaining to Sales and Marketing
We are subject to various federal and state laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws generally prohibit a prescription drug manufacturer from soliciting, offering, receiving, or paying any remuneration to generate business, including the purchase or prescription of a particular drug. Although the specific provisions of these laws vary, their scope is generally broad and there may be no regulations, guidance or court decisions that clarify how the laws apply to particular industry practices. There is therefore a possibility that our practices might be challenged under the anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third party payors (including Medicare and Medicaid) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws. Violations of fraud and abuse laws may be punishable by criminal or civil sanctions, including fines and civil monetary penalties, and exclusion from federal health care programs (including Medicare and Medicaid). Federal and state authorities are paying increased attention to enforcement of these laws within the pharmaceutical industry and private individuals have been active in alleging violations of the laws and bringing suits on behalf of the government under the federal civil False Claims Act. If we were subject to allegations concerning, or were convicted of violating, these laws, our business could be harmed.
Laws and regulations have been enacted by the federal government and various states to regulate the sales and marketing practices of pharmaceutical manufacturers. These laws and regulations generally limit financial interactions between manufacturers and health care providers or require disclosure to the government and public of such interactions. The laws include federal “sunshine” provisions enacted in 2010 as part of the comprehensive federal health care reform legislation. The sunshine provisions apply to pharmaceutical manufacturers with products reimbursed under certain government programs and require those manufacturers to disclose annually to the federal government (for re-disclosure to the public) certain payments made to physicians and certain other healthcare practitioners or to teaching hospitals. State laws may also require disclosure of pharmaceutical pricing information and marketing expenditures. Many of these laws and regulations contain ambiguous requirements. Given the lack of clarity in laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent federal and state laws and regulations. Outside the U.S., other countries have implemented requirements for disclosure of financial interactions with healthcare providers and additional countries may also consider or implement such laws.
Federal and state governments also have pursued direct methods to reduce the cost of drugs for which they pay. We participate in state government-managed Medicaid programs as well as certain other qualifying federal and state government programs whereby discounts and mandatory rebates are provided to participating state and local government entities. We also participate in other programs with government entities, the most significant of which are the U.S. Department of Defense and the U.S. Department of Veterans Affairs. These entities receive minimum discounts based on a defined “non-federal average manufacturer price” for purchases. Additional programs in which we participate provide mandatory discounts for outpatient medicines purchased by certain Public Health Service entities and “disproportionate share” hospitals (hospitals meeting certain criteria regarding the percentage of needy population served).
In connection with several of these government programs, we are required to report prices to various government agencies. Pricing calculations vary among programs. The calculations are complex and are often subject to interpretation by the reporting entities, government agencies and the courts. Our methodologies for calculating these prices could be challenged under false claims laws or other laws. We could make a mistake in calculating reported prices and required discounts, which could result in retroactive liability to government agencies. Government agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. If we make these mistakes or if governmental agencies make these changes, we could face, in addition to prosecution under federal and state false claims laws, substantial liability and civil monetary penalties, exclusion of our products from reimbursement under government programs, criminal fines or imprisonment, or prosecutors may impose a Corporate Integrity Agreement, Deferred Prosecution Agreement, or similar arrangement.
Foreign Regulation
In addition to regulations in the United States, we are subject to a variety of foreign regulatory requirements governing human clinical trials and marketing approval for drugs. The foreign regulatory approval process includes all of the risks associated with FDA approval set forth above, as well as additional country-specific regulations. Whether or not we obtain FDA approval for a product, we must obtain further approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
For example, under European Union regulatory systems, we may submit marketing authorizations either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions, and the holder of a national marketing authorization may submit an application to the remaining member states.
Reimbursement
Sales of pharmaceutical products depend in significant part on the coverage and reimbursement policies of government programs, including Medicare and Medicaid in the United States, and other third-party payers. These health insurance programs may restrict coverage of some products by using payer formularies under which only selected drugs are covered, variable co-payments that make drugs that are not preferred by the payer more expensive for patients, and by using utilization management controls, such as requirements for prior authorization or prior failure on another type of treatment. Payers may especially impose these obstacles to coverage for higher-priced drugs, and consequently KALBITOR may be subject to payer-driven restrictions.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices and/or reimbursement of medicinal products for human use. A member state may approve a specific price or level of reimbursement for the medicinal product, or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market.
In furtherance of our efforts to facilitate patient access to KALBITOR in the United States, we have created the KALBITOR Access program, a treatment support service for patients with HAE and their healthcare providers. KALBITOR case managers provide education about HAE and KALBITOR and help facilitate solutions for reimbursement, coverage and treatment site coordination.
OUR CORPORATE INFORMATION
We are a Delaware corporation, incorporated in 1989, and merged with Protein Engineering Corporation in 1995. Our principal executive offices are located at 55 Network Drive, Burlington, Massachusetts 01803, and our telephone number is (617) 225-2500. Our web site address is www.dyax.com.
Segment Information
We provide financial information by geographical area in Note 13 to our Consolidated Financial Statements included in Item 8 of this report. We are incorporating that information into this section by this reference.
As of February 1, 2013, we had 125 employees, including 15 Ph.D.’s and/or M.D.’s. Approximately 43 of our employees are in research and development, and 82 in marketing, business development and administration. Our workforce is non-unionized, and we believe that our relations with employees are good.
We make our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to these reports filed pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 available without charge through our website, www.dyax.com, as soon as reasonably practicable after filing them with the Securities and Exchange Commission. Information contained on the website is not part of this report.
You should carefully consider the following risk factors before you decide to invest in our Company and our business because these risk factors may have a significant impact on our business, operating results, financial condition, and cash flows. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business operations. If any of the following risks actually occurs, our business, financial condition and results of operations could be materially and adversely affected.
Risks Related To Our Business
We have a history of net losses, expect to incur additional net losses and may never achieve or sustain profitability.
We have incurred net losses on an annual basis since our inception. As of December 31, 2012 we had an accumulated deficit of approximately $506.2 million. We expect to incur additional net losses in 2013 as our research, development, preclinical testing, clinical trial and commercial activities continue.
Although we generate revenue from product sales and through our LFRP, it is possible that we will not generate sufficient revenue to become profitable. To become profitable, we, alone or with our collaborators, must either generate higher product sales from the commercialization of KALBITOR, increase licensing receipts under the LFRP, or reduce costs. It is possible that we will never have sufficient product sales revenue or receive sufficient royalties on our licensed product candidates or licensed technology in order to achieve or sustain future profitability.
Our revenues and operating results have fluctuated significantly in the past, and we expect this to continue in the future.
Our revenues and operating results have fluctuated significantly on a quarterly and year to year basis. We expect these fluctuations to continue in the future. Fluctuations in revenues and operating results will depend on:
| | ● | the amount of future sales of KALBITOR and related costs to manufacture and sell the product; |
| | ● | the cost and timing of our research and development, manufacturing and commercialization activities; |
| | ● | the establishment of new collaboration and licensing arrangements; |
| | ● | the timing and results of clinical trials, including a failure to receive the required regulatory approvals to commercialize ecallantide in additional indications and other product candidates; |
| | ● | the timing, receipt and amount of payments, if any, from current and prospective collaborators and licensees, including the completion of certain milestones; and |
| | ● | revenue recognition and other generally accepted accounting policies. |
Our revenues and costs in any period are not reliable indicators of our future operating results. If the revenues we recognize are less than the revenues we expect for a given fiscal period, then we may be unable to reduce our expenses quickly enough to compensate for the shortfall. In addition, our fluctuating operating results may fail to meet the expectations of securities analysts or investors which may cause the price of our common stock to decline.
We may need additional capital in the future and may be unable to generate the capital that we will need to sustain our operations.
We require significant capital to fund our operations to commercialize KALBITOR and to develop and commercialize other product candidates and ecallantide in other indications. Our future capital requirements will depend on many factors, including:
| | ● | future sales levels of KALBITOR and any other commercial products and the profitability of such sales, if any; |
| | ● | the timing and cost to develop, obtain regulatory approvals for and commercialize other product candidates and additional indications for ecallantide; |
| | ● | maintaining or expanding our existing collaborative and license arrangements and entering into additional arrangements on terms that are favorable to us; |
| | ● | the amount and timing of milestone and royalty payments from our collaborators and licensees related to their progress in developing and commercializing products; |
| | ● | the decision to manufacture, or have third parties manufacture, the materials used in KALBITOR and any other product candidates; |
| | ● | competing technological and market developments; |
| | ● | the progress of our development programs; |
| | ● | the costs of prosecuting, maintaining, defending and enforcing our patents and other intellectual property rights; |
| | ● | the amount and timing of additional capital equipment purchases; and |
| | ● | the overall condition of the financial markets. |
We expect that existing cash, cash equivalents and investments together with anticipated cash flow from product sales and existing product development, collaborations and license fees will be sufficient to support our current operations into 2014. We will need additional funds if our cash requirements exceed our current expectations or if we generate less revenue than we expect.
We may seek additional funding through collaborative arrangements, public or private financings, or other means. We may not be able to obtain financing on acceptable terms or at all, and we may not be able to enter into additional collaborative arrangements. Arrangements with collaborators or others may require us to relinquish rights to certain of our technologies, product candidates or products. The terms of any financing may adversely affect the holdings or the rights of our stockholders and if we are unable to obtain funding on a timely basis, we may be required to curtail significantly our research, development or commercialization programs which could adversely affect our business prospects.
We depend heavily on the success of our lead product, KALBITOR, which was approved in the United States for treatment of acute attacks of HAE in patients 16 years and older.
Our ability to generate product sales will depend on commercial success of KALBITOR in the United States and whether physicians, patients and healthcare payers view KALBITOR as therapeutically effective relative to cost. We initiated the commercial launch of KALBITOR in the United States in February 2010.
The commercial success of KALBITOR and our ability to generate and increase product sales and positive cash flow will depend on multiple factors, including the following:
| | ● | the number of patients with HAE who are diagnosed with the disease and identified to us; |
| | ● | the number of patients with HAE who may be treated with KALBITOR; |
| | ● | acceptance of KALBITOR in the medical community; |
| | ● | the frequency of HAE patients' use of KALBITOR to treat their acute attacks of HAE; |
| | ● | HAE patients' ability to obtain and maintain sufficient coverage or reimbursement by third-party payers for the use of KALBITOR; |
| | ● | our ability to effectively market and distribute KALBITOR in the United States; |
| | ● | competition from other products that treat HAE; |
| | ● | the maintenance of marketing approval in the United States and the receipt and maintenance of marketing approval from foreign regulatory authorities; |
| | ● | our maintenance of commercial manufacturing capabilities through third-party manufacturers; and |
| | ● | our ability to maintain sufficient inventories to supply KALBITOR for patient use. |
If we are unable to develop substantial sales of KALBITOR in the United States and commercialize ecallantide in additional countries or if we are significantly delayed or limited in doing so, our business prospects would be adversely affected.
Because the target patient population of KALBITOR for treatment of HAE is small and has not been definitively determined, we must be able to successfully identify HAE patients and achieve a significant market share in order to achieve or maintain profitability.
The prevalence of HAE patients, which has been estimated at approximately 1 in 10,000 to 1 in 50,000 people around the world, has not been definitively determined. There can be no guarantee that any of our programs will be effective at identifying HAE patients, and the number of HAE patients in the United States may turn out to be lower than expected or patients may not utilize treatment with KALBITOR for all or any of their acute HAE attacks, all or any of which would adversely affect our results of operations and business prospects.
If HAE patients are unable to obtain and maintain reimbursement for KALBITOR from government health administration authorities, private health insurers and other organizations, KALBITOR may be too costly for regular use and our ability to generate product sales would be harmed.
We may not be able to sell KALBITOR on a profitable basis or our profitability may be reduced if we are required to sell our product at lower than anticipated prices or if reimbursement is unavailable or limited in scope or amount. KALBITOR is more expensive than traditional drug treatments and most patients require some form of third party insurance coverage and/or patient assistance provided by us in order to afford its cost. Our future revenues and profitability will be adversely affected if HAE patients cannot depend on governmental, private and other third-party payers, such as Medicare and Medicaid in the United States or country specific governmental organizations, to defray the cost of KALBITOR. If these entities refuse to provide coverage and reimbursement with respect to KALBITOR or determine to provide a lower level of coverage and reimbursement than anticipated, KALBITOR may be too costly for general use, and physicians may not prescribe it.
In addition to potential restrictions on insurance coverage, the amount of reimbursement for KALBITOR may also reduce our ability to profitably commercialize KALBITOR. In the United States and elsewhere, there have been, and we expect there will continue to be, actions and proposals to control and reduce healthcare costs. Government and other third-party payers are challenging the prices charged for healthcare products and increasingly limiting and attempting to limit both coverage and level of reimbursement for prescription drugs.
It is possible that we will never have significant KALBITOR sales revenue in order to achieve or sustain future profitability.
We may not be able to maintain or expand market acceptance among the medical community or patients for KALBITOR, which would prevent us from achieving or maintaining profitability in the future.
We cannot be certain that KALBITOR will continue to maintain or gain additional market acceptance among physicians, patients, healthcare payers, and others. Although we have received regulatory approval for KALBITOR in the United States, such approval does not assure future revenue. We cannot predict whether physicians, other healthcare providers, government agencies or private insurers will continue to determine that KALBITOR is safe and therapeutically effective relative to cost. Medical doctors' willingness to prescribe, and patients' willingness to accept, KALBITOR depends on many factors, including prevalence and severity of adverse side effects in both clinical trials and commercial use, effectiveness of our marketing strategy and the pricing of KALBITOR, publicity concerning our products or competing products, HAE patient's ability to obtain and maintain third-party coverage or reimbursement, and availability of alternative treatments. In addition, the number of acute attacks that are treated with KALBITOR will vary from patient to patient depending upon a variety of factors.
If KALBITOR fails to maintain or gain additional market acceptance, we may not be able to market and sell it successfully, which would limit our ability to generate revenue and adversely affect our results of operations and business prospects.
Competition and technological change may make our potential products and technologies less attractive or obsolete.
We compete in industries characterized by continuous intense competition and rapid technological change. New developments occur and are expected to continue to occur constantly at a rapid pace. Discoveries or commercial developments by our competitors or others may render some or all of our technologies, products or potential products obsolete or non-competitive.
In our PKM angioedema portfolio, our principal focus is on the development and commercialization of human therapeutic products. We plan to conduct research and development programs to develop and test product candidates and demonstrate to appropriate regulatory agencies that these products are safe and effective for therapeutic use in particular indications. Therefore our principal competition going forward, as further described below, is companies that either are already marketing products in those indications or are developing new products for those indications. Many of our competitors have greater financial resources and experience than we do.
For KALBITOR as a treatment for HAE, our principal competitors include:
| | ● | Manufacturers of corticosteroids, including danazol, which have been used historically and are still used to treat prophylactically a significant number of identified HAE patients. |
| | ● | Shire plc— Shire markets its bradykinin receptor antagonist, known as Firazyr® (icatibant), which is administered subcutaneously. Firazyr is approved in the US, Europe, and certain other countries. Firazyr is approved in these markets for the treatment of acute HAE attacks in adult patients. The US and EU labels allow for patients to self-administer Firazyr following training by their healthcare provider. Firazyr has orphan drug designations from the FDA and in Europe. |
| | ● | CSL Behring— CSL Behring markets a plasma-derived C1-esterase inhibitor, known as Berinert®, which is administered intravenously. Berinert is approved in the US for the treatment of acute abdominal, facial or laryngeal attacks of HAE in adults and adolescents, and has orphan drug designation from the FDA. The FDA has also approved labeling for Berinert to include self-administration after proper training by a healthcare professional. Berinert is also approved in the EU, Japan and several rest-of-world markets for the treatment of acute attacks of HAE. CSL Behring announced in May 2012 that they had commenced an international Phase I/II study of a volume-reduced subcutaneous formulation of C1-INH which will evaluate the pharmacokinetics, pharmacodynamics and safety of various doses of C1-INH. |
| | ● | ViroPharma Inc. — ViroPharma markets a plasma-derived C1-esterase inhibitor, known as Cinryze®, which is administered intravenously. Cinryze is approved in the US for routine prophylaxis against angioedema attacks in adolescent and adult patients with HAE, and has orphan drug designation from the FDA. The FDA has also approved patient labeling for Cinryze to include self-administration for routine prophylaxis once a patient is properly trained by his or her healthcare provider. ViroPharma has also received approval in the EU where the product is approved for the treatment and pre-procedure prevention of angioedema attacks in adults and adolescents with HAE, and routine prevention of angioedema attacks in adults and adolescents with severe and recurrent attacks of HAE, who are intolerant to or insufficiently protected by oral prevention treatments or patients who are inadequately managed with repeated acute treatment. The EU approval includes a self-administration option for appropriately trained patients. ViroPharma is conducting two Phase 2 trials evaluating subcutaneous administration of Cinryze. |
| | ● | Pharming Group NV— Pharming markets a recombinant C1-esterase inhibitor, known as Ruconest™, which is administered intravenously. Ruconset is approved in the EU for the treatment of acute HAE attacks in adult patients. In November 2012, Pharming and its US partner Santarus announced positive topline data from their Phase 3 trial for Ruconest. Pharming's recombinant C1-esterase inhibitor has Fast Track status from the FDA and orphan drug designations from the FDA and in Europe. |
Other competitors for the treatment of HAE are companies that are developing small molecule plasma kallikrein inhibitors, including BioCryst.
Additionally, a significant number of companies compete with us in the antibody technology space by offering licenses and/or research services to pharmaceutical and biotechnology companies. Specifically, our phage display technology is one of several in vitro display technologies available to generate libraries of compounds that can be leveraged to discover new antibody products. Companies that compete with us in the display technology space include BioInvent, XOMA, Adimab and several others. Additional platforms that pharmaceutical and biotechnology companies use to identify antibodies that bind to a desired target are in vivo technology platforms which use direct immunization of mice or other species to generate fully human antibodies. Competitors in this space include GenMab, arGEN-X and several others. There are also a number of new technologies directed to the generation of candidate compounds with novel scaffolds that may possess similar properties to monoclonal antibodies.
In addition to the technologies described above, many pharmaceutical companies have either acquired antibody discovery technologies or developed humanized murine antibodies derived from hybridomas. Pharmaceutical companies also develop orally available small molecule compounds directed to the targets for which we and others are seeking to develop antibody, peptide and/or protein products.
Furthermore, we may also experience competition from companies that have acquired or may acquire other technologies from universities and other research institutions and that may impact our competitive position.
If we fail to comply with continuing regulations, we could lose our approvals to market KALBITOR, and our business would be adversely affected.
We cannot guarantee that we will be able to maintain our regulatory approval for KALBITOR in the United States. We and our current and future partners, contract manufacturers and suppliers are subject to rigorous and extensive regulation by the FDA, other federal and state agencies, and governmental authorities in other countries. These regulations continue to apply after product approval, and cover, among other things, testing, manufacturing, quality control, labeling, advertising, promotion, adverse event reporting requirements, and export of biologics.
As a condition of approval for marketing KALBITOR in the United States and other jurisdictions, the FDA or governmental authorities in those jurisdictions may require us to conduct additional clinical trials. For example, in connection with the approval of KALBITOR in the United States, we have agreed to conduct a Phase 4 clinical study to evaluate immunogenicity and hypersensitivity with exposure to KALBITOR for treatment of acute attacks of HAE. The FDA can propose to withdraw approval if new clinical data or information shows that KALBITOR is not safe for use or determines that such study is inadequate. We are required to report any serious and unexpected adverse experiences and certain quality problems with KALBITOR to the FDA and other health agencies. We, the FDA or another health agency may have to notify healthcare providers of any such developments. The discovery of any previously unknown problems with KALBITOR or its manufacturer may result in restrictions on KALBITOR and the manufacturer or manufacturing facility, including withdrawal of KALBITOR from the market. Certain changes to an approved product, including the way it is manufactured or promoted, often require prior regulatory approval before the product as modified may be marketed.
Our third-party manufacturing facilities were subjected to inspection prior to grant of marketing approval and are subject to continued review and periodic inspections by the regulatory authorities. Any third party we would use to manufacture KALBITOR for sale must also be licensed by applicable regulatory authorities. Although we have established a corporate compliance program, we cannot guarantee that we or our third party vendors are and will continue to be in compliance with all applicable laws and regulations. Failure to comply with the laws, including statutes and regulations, administered by the FDA or other agencies could result in:
| | ● | administrative and judicial sanctions, including warning letters; |
| | ● | fines and other civil penalties; |
| | ● | withdrawal of a previously granted approval; |
| | ● | interruption of production; |
| | ● | product recall or seizure; injunctions; and |
The discovery of previously unknown problems with a product, including KALBITOR, or the facility used to produce the product could result in a regulatory authority imposing restrictions on us, or could cause us to voluntarily adopt such restrictions, including withdrawal of KALBITOR from the market.
If we do not maintain our regulatory approval for KALBITOR in the United States, our results of operations and business prospects will be materially harmed.
If the use of KALBITOR harms people, or is perceived to harm patients even when such harm is unrelated to KALBITOR, our regulatory approvals could be revoked or otherwise negatively affected and we could be subject to costly and damaging product liability claims.
The testing, manufacturing, marketing and sale of drugs for use in humans exposes us to product liability risks. Side effects and other problems from using KALBITOR could:
| | ● | lessen the frequency with which physicians decide to prescribe KALBITOR; |
| | ● | encourage physicians to stop prescribing KALBITOR to their patients who previously had been prescribed KALBITOR; |
| | ● | cause serious adverse events and give rise to product liability claims against us; and |
| | ● | result in our need to withdraw or recall KALBITOR from the marketplace. |
Some of these risks are unknown at this time.
We have tested KALBITOR in only a limited number of patients. As more patients begin to use KALBITOR, new risks and side effects may be discovered, and risks previously viewed as inconsequential could be determined to be significant. Previously unknown risks and adverse effects of KALBITOR may also be discovered in connection with unapproved and unsolicited, or off-label, uses of KALBITOR. We do not promote, or in any way support or encourage the promotion of KALBITOR for off-label uses in violation of relevant law, but current regulations allow physicians to use products for off-label uses. In addition, we expect to study ecallantide in diseases other than HAE in controlled clinical settings, and expect independent investigators to do so as well. In the event of any new risks or adverse effects discovered as new patients are treated for HAE, regulatory authorities may modify or revoke their approvals and we may be required to conduct additional clinical trials, make changes in labeling of KALBITOR, reformulate KALBITOR or make changes and obtain new approvals for our and our suppliers' manufacturing facilities. We may also experience a significant drop in the potential sales of KALBITOR, an increase in costs, experience harm to our reputation and the reputation of KALBITOR in the marketplace or become subject to government investigations or lawsuits, including class actions. Any of these results could decrease or prevent any sales of KALBITOR or substantially increase the costs and expenses of commercializing and marketing KALBITOR.
We may be sued by people who use KALBITOR, whether as a prescribed therapy, during a clinical trial, during an investigator initiated study, or otherwise. Any informed consents or waivers obtained from people who enroll in our trials or use KALBITOR may not protect us from liability or litigation. Our product liability insurance may not cover all potential types of liabilities or may not cover certain liabilities completely. Moreover, we may not be able to maintain our insurance on acceptable terms. In addition, negative publicity relating to the use of KALBITOR or a product candidate, or to a product liability claim, may make it more difficult, or impossible, for us to market and sell KALBITOR. As a result of these factors, a product liability claim, even if successfully defended, could have a material adverse effect on our business, financial condition or results of operations.
During the course of treatment, patients may suffer adverse events, including death, for reasons that may or may not be related to KALBITOR. Such events could subject us to costly litigation, require us to pay substantial amounts of money to injured patients, delay, negatively impact or end our opportunity to receive or maintain regulatory approval to market KALBITOR, or require us to suspend or abandon our commercialization efforts. Even in a circumstance in which we do not believe that an adverse event is related to KALBITOR, the investigation into the circumstance may be time consuming, costly or may be inconclusive. These investigations may interrupt our sales efforts, delay our regulatory approval process in other countries, or impact and limit the type of regulatory approvals KALBITOR receives or maintains.
Although we obtained regulatory approval of KALBITOR for the treatment of acute attacks of HAE in patients 16 years and older in the United States, we may be unable to obtain regulatory approval for ecallantide in any other territory.
Governments in countries outside the United States also regulate drugs distributed in such countries and facilities in such countries where such drugs are manufactured, and obtaining their approvals can also be lengthy, expensive and highly uncertain. The approval process varies from country to country and the requirements governing the conduct of clinical trials, product manufacturing, product licensing, pricing and reimbursement vary greatly from country to country. In certain jurisdictions, we are required to finalize operational, reimbursement, price approval and funding processes prior to marketing our products. We may not receive regulatory approval for ecallantide in countries other than the United States on a timely basis, if ever. Even if approval is granted in any such country, the approval may require limitations on the indicated uses for which the drug may be marketed. Failure to obtain regulatory approval for ecallantide in territories outside the United States could have a material adverse effect on our business prospects.
If we are unable to maintain effective sales, marketing and distribution capabilities, or to enter into agreements with third parties to do so, we will be unable to successfully commercialize KALBITOR.
We are marketing and selling KALBITOR ourselves in the United States and have only limited experience with marketing, sales or distribution of drug products. If we are unable to adequately establish the capabilities to sell, market and distribute KALBITOR, either ourselves or by entering into agreements with others, or to maintain such capabilities, we will not be able to successfully sell KALBITOR. In that event, we will not be able to generate significant product sales. We cannot guarantee that we will be able to establish and maintain our own capabilities or enter into and maintain any marketing or distribution agreements with third-party providers on acceptable terms, if at all.
In the United States, we sell KALBITOR through a limited distribution network. This distribution network includes wholesale, specialty pharmacy and service arrangements, including a call center to support KALBITOR’s commercialization, and a service agreement with Walgreens, which provides nursing services for home administration of KALBITOR. Additionally, in 2013, we plan to expand our distribution network through a limited number of additional specialty pharmacy arrangements. Our distributors do not set or determine demand for KALBITOR. We expect our distribution arrangements to continue for the foreseeable future through an extension or replacement of our current agreements. Our ability to successfully commercialize KALBITOR will depend, in part, on the extent to which we are able to provide adequate distribution of KALBITOR to patients through our distributors. It is possible that our distributors could change their policies or fees, or both, at some time in the future. This could result in their refusal to distribute smaller volume products such as KALBITOR, or cause higher product distribution costs, lower margins or the need to find alternative methods of distributing KALBITOR. Although we have contractual remedies to mitigate these risks for the term of the contract with ABSG and we also believe we can find alternative distributors on relatively short notice, our product sales during that period of time may suffer and we may incur additional costs to replace a distributor. A significant reduction in product sales to our distributors, any cancellation of orders they have made with us or any failure to pay for the products we have shipped to them could materially and adversely affect our results of operations and financial condition.
We have hired sales and marketing professionals for the commercialization of KALBITOR throughout the United States. Even with these sales and marketing personnel, we may not have the necessary size and experience of the sales and marketing force and the appropriate distribution capabilities necessary to successfully market and sell KALBITOR. Establishing and maintaining sales, marketing and distribution capabilities are expensive and time-consuming. Our expenses associated with building up and maintaining the sales force and distribution capabilities may be disproportional compared to the revenues we may be able to generate on sales of KALBITOR. We cannot guarantee that we will be successful in commercializing KALBITOR and a failure to do so would adversely affect our business prospects.
If we market KALBITOR in a manner that violates healthcare fraud and abuse laws, we may be subject to civil or criminal penalties.
In addition to FDA and related regulatory requirements, we are subject to health care "fraud and abuse" laws, such as the federal False Claims Act, the anti-kickback provisions of the federal Social Security Act, and other state and federal laws and regulations. Federal and state anti-kickback laws prohibit, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce, or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid, or other federally or state financed health care programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, patients, purchasers and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchasing, or recommending may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. Pharmaceutical companies have been prosecuted under these laws for a variety of alleged promotional and marketing activities, such as allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product; reporting to pricing services inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in promotion for uses that the FDA has not approved, or "off-label" uses, that caused claims to be submitted to Medicaid for non-covered off-label uses; and submitting inflated best price information to the Medicaid Rebate Program.
Although based on their medical judgment, physicians are permitted to prescribe products for indications other than those cleared or approved by the FDA, manufacturers are prohibited from promoting their products for such off-label uses. We market KALBITOR in the U.S. according to its FDA approved label for acute attacks of HAE in patients 16 years and older and provide promotional materials to physicians regarding the use of KALBITOR for this indication. Although we believe our marketing, promotional materials do not constitute off-label promotion of KALBITOR, the FDA may disagree. If the FDA determines that our promotional materials, training or other activities constitute off-label promotion of KALBITOR, it could request that we modify our training or promotional materials or other activities or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they believe that the alleged improper promotion led to the submission and payment of claims for an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. Even if it is later determined we are not in violation of these laws, we may be faced with negative publicity, incur significant expenses defending our position and have to divert significant management resources from other matters.
The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer's products from reimbursement under government programs, criminal fines, and imprisonment. Even if we are not determined to have violated these laws, government investigations into these issues typically require the expenditure of significant resources and generate negative publicity, which would also harm our financial condition. Because of the breadth of these laws and the narrowness of the safe harbors and because government scrutiny in this area is high, it is possible that some of our business activities could come under that scrutiny.
In recent years, several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, and file periodic reports with the state or make periodic public disclosures on sales, marketing, pricing, clinical trials, and other activities. Many of these requirements are new and uncertain, and the penalties for failure to comply with these requirements are unclear. Nonetheless, although we have established compliance policies that comport with the Code of Interactions with Healthcare Providers adopted by Pharmaceutical Research Manufacturers of America (PhRMA Code) and the Office of Inspector General's (OIG) Compliance Program Guidance for Pharmaceutical Manufacturers, if we are found not to be in full compliance with these laws, we could face enforcement action and fines and other penalties, and could receive adverse publicity.
The FDA or similar agencies in other jurisdictions may require us to restrict the distribution or use of KALBITOR or other future products or take other potentially limiting or costly actions if we or others identify side effects after the product is on the market.
The FDA has required that we implement a REMS for KALBITOR and conduct post-marketing studies to assess a risk of hypersensitivity reactions, including anaphylaxis. The REMS consisted of a communication plan to healthcare providers which was completed in February 2012. The FDA and other regulatory agencies could impose new requirements or change existing regulations or promulgate new ones at any time that may affect our ability to obtain or maintain approval of KALBITOR or future products or require significant additional costs to obtain or maintain such approvals. For example, the FDA or similar agencies in other jurisdictions may require us to restrict the distribution or use of KALBITOR if we or others identify side effects after KALBITOR and/or future products are on the market. Changes in KALBITOR's approval or restrictions on its use could make it difficult to achieve market acceptance, and we may not be able to market and sell KALBITOR or continue to sell it, successfully, or at all, which would limit our ability to generate product sales and adversely affect our results of operations and business prospects.
We rely on third-party manufacturers to produce our preclinical and clinical drug supplies and commercial supplies of KALBITOR and we intend to rely on third parties to produce any future approved product candidates. Any failure by a third-party manufacturer to produce supplies for us may delay or impair our ability to develop, obtain regulatory approval for or commercialize our product candidates.
We have relied upon a small number of third-party manufacturers for the manufacture of our product candidates for preclinical, clinical testing and commercial purposes and intend to continue to do so in the future. As a result, we depend on collaborators, partners, licensees and other third parties to manufacture clinical and commercial scale quantities of our biopharmaceutical candidates in a timely and effective manner and in accordance with government regulations. If these third party arrangements are not successful, it will adversely affect our ability to develop, obtain regulatory approval for or commercialize our product candidates.
We have identified only a few vendors with facilities that are capable of producing material for preclinical, clinical studies and for commercial purposes and we cannot assure you that they will be able to supply sufficient clinical materials on a timely basis during the clinical development or commercialization of our biopharmaceutical candidates. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured product candidates ourselves, including reliance on the third party for regulatory compliance and quality assurance, the possibility of breach of the manufacturing agreement by the third party because of factors beyond our control (including a failure to synthesize and manufacture our product candidates in accordance with our product specifications) and the possibility of termination or nonrenewal of the agreement by the third party, based on its own business priorities, at a time that is costly or damaging to us. In addition, the FDA and other regulatory authorities require that our product candidates be manufactured according to cGMP and similar foreign standards. Any failure by our third-party manufacturers to comply with cGMP or failure to scale up manufacturing processes, including any failure to deliver sufficient quantities of product candidates in a timely manner, could lead to a delay in, or failure to obtain, regulatory approval of any of our product candidates.
In addition, as our drug development pipeline increases and matures, we will have a greater need for clinical trial and commercial manufacturing capacity. We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of our product candidates and we currently have no plans to build our own clinical or commercial scale manufacturing capabilities. To meet our projected needs for commercial manufacturing, third parties with whom we currently work will need to increase their scale of production or we will need to secure alternate suppliers.
We are dependent on a single contract manufacturer to produce ecallantide drug substance and another to fill, label and package ecallantide drug product into the final form, which may adversely affect our ability to commercialize KALBITOR and other potential ecallantide products.
We currently rely on Fujifilm to produce the bulk drug substance used in the manufacture of KALBITOR and other potential ecallantide products. In addition, ecallantide drug substance is filled, labeled and packaged into the final form of KALBITOR drug product by Hollister-Stier under a commercial supply agreement. Our business, therefore, faces risks of difficulties with, and interruptions in, performance by Fujifilm and Hollister-Stier, the occurrence of which could adversely impact the availability and/or sales of KALBITOR and other potential ecallantide products in the future. The failure of Fujifilm or Hollister-Stier to supply manufactured product on a timely basis or at all, or to manufacture our drug substance in compliance with our specifications or applicable quality requirements or in volumes sufficient to meet demand could adversely affect our ability to sell KALBITOR and other potential ecallantide products, could harm our relationships with our collaborators or customers and could negatively affect our revenues and operating results. If the operations of Fujifilm or Hollister-Stier are disrupted, we may be forced to secure alternative sources of supply, which may be unavailable on commercially acceptable terms, cause delays in our ability to deliver products to our customers, increase our costs and negatively affect our operating results.
In addition, failure to comply with applicable good manufacturing practices and other governmental regulations and standards could be the basis for action by the FDA or corresponding foreign agency to withdraw approval for KALBITOR or any other product previously granted to us and for other regulatory action, including recall or seizure, fines, imposition of operating restrictions, total or partial suspension of production or injunctions.
We currently have a long-term commercial supply agreement with Fujifilm, under which FujiFilm has committed to be available to manufacture ecallantide drug substance through 2020. In addition, we believe that our existing supply of ecallantide drug substance will be sufficient to supply all ongoing studies relating to ecallantide and KALBITOR and to meet anticipated market demand into 2016. These estimates are subject to changes in market conditions and other factors beyond our control. If Fujifilm or Hollister-Stier is unable to dependably meet our demands for ecallantide drug substance or product, it could adversely affect our ability to further develop and commercialize KALBITOR and other potential ecallantide products, generate revenue from product sales, increase our costs and negatively affect our operating results.
Any new biopharmaceutical product candidates we develop must undergo rigorous clinical trials which could substantially delay or prevent their development or marketing.
In addition to KALBITOR, we are evaluating ecallantide in further indications and are developing DX-2930, another potential biopharmaceutical product. Before we can commercialize any biopharmaceutical product candidate, we must engage in a rigorous clinical trial and regulatory approval process mandated by the FDA and analogous foreign regulatory agencies. This process is lengthy and expensive, and approval is never certain. Positive results from preclinical studies and early clinical trials do not ensure positive results in late stage clinical trials designed to permit application for regulatory approval. We cannot accurately predict when planned clinical trials will begin or be completed. Many factors affect patient enrollment, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, alternative therapies, competing clinical trials and new drugs approved for the conditions that we are investigating. As a result of all of these factors, our future trials may take longer to enroll patients than we anticipate. Such delays may increase our costs and slow down our product development and the regulatory approval process. Our product development costs will also increase if we need to perform more or larger clinical trials than planned. The occurrence of any of these events will delay our ability to commercialize products, generate revenue from product sales and impair our ability to become profitable, which may cause us to have insufficient capital resources to support our operations.
Products that we or our collaborators develop could take a significantly longer time to gain regulatory approval than we expect or may never gain approval. If we or our collaborators do not receive these necessary approvals, we will not be able to generate substantial product or royalty revenues and may not become profitable. We and our collaborators may encounter significant delays or excessive costs in our efforts to secure regulatory approvals. Factors that raise uncertainty in obtaining these regulatory approvals include the following:
| | ● | we or our collaborators must demonstrate through clinical trials that the proposed product is safe and effective for its intended use; |
| | ● | we have limited experience in conducting the clinical trials necessary to obtain regulatory approval; and |
| | ● | data obtained from preclinical and clinical activities are subject to varying interpretations, which could delay, limit or prevent regulatory approvals. |
Regulatory authorities may delay, suspend or terminate clinical trials at any time if they believe that the patients participating in trials are being exposed to unacceptable health risks or if they find deficiencies in the clinical trial procedures. There is no guarantee that we will be able to resolve such issues, either quickly, or at all. In addition, our or our collaborators' failure to comply with applicable regulatory requirements may result in criminal prosecution, civil penalties and other actions that could impair our ability to conduct our business.
We rely on third parties to conduct clinical trials and to perform certain regulatory processes, which may adversely affect our ability to commercialize any biopharmaceuticals that we may develop.
We have hired experienced clinical development and regulatory staff to develop and supervise our clinical trials and regulatory processes. However, we will remain dependent upon third party contract research organizations to carry out some of our clinical and preclinical research studies for the foreseeable future. As a result, we have had and will continue to have less control over the conduct of the clinical trials, the timing and completion of the trials, the required reporting of adverse events and the management of data developed through the trials than would be the case if we were relying entirely upon our own staff. Communicating with outside parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. Outside parties may have staffing difficulties, may undergo changes in priorities or may become financially distressed, adversely affecting their willingness or ability to conduct our trials. We may also experience unexpected cost increases that are beyond our control.
Problems with the timeliness or quality of the work of a contract research organization may lead us to seek to terminate the relationship and use an alternative service provider. However, changing our service provider may be costly and may delay our trials, and contractual restrictions may make such a change difficult or impossible. Additionally, it may be impossible to find a replacement organization that can conduct our trials in an acceptable manner and at an acceptable cost.
Government regulation of drug development is costly, time consuming and fraught with uncertainty, and our products in development cannot be sold if we do not gain regulatory approval.
We and our licensees and partners conduct research, preclinical testing and clinical trials for our product candidates. These activities are subject to extensive regulation by numerous state and federal governmental authorities in the United States, such as the FDA, as well as foreign countries, such as the EMEA in European countries, Canada and Australia. Currently, we are required in the United States and in foreign countries to obtain approval from those countries' regulatory authorities before we can manufacture (or have our third-party manufacturers produce), market and sell our products in those countries. The FDA and other United States and foreign regulatory agencies have substantial authority to fail to approve commencement of, suspend or terminate clinical trials, require additional testing and delay or withhold registration and marketing approval of our product candidates.
Obtaining regulatory approval has been and continues to be increasingly difficult and costly and takes many years; and, if obtained, is costly to maintain. With the occurrence of a number of high profile safety events with certain pharmaceutical products, regulatory authorities, and in particular the FDA, members of Congress, the United States Government Accountability Office (GAO), Congressional committees, private health/science foundations and organizations, medical professionals, including physicians and investigators, and the general public are increasingly concerned about potential or perceived safety issues associated with pharmaceutical and biological products, whether under study for initial approval or already marketed.
This increasing concern has produced greater scrutiny, which may lead to fewer treatments being approved by the FDA or other regulatory bodies, as well as restrictive labeling of a product or a class of products for safety reasons, potentially including a boxed warning or additional limitations on the use of products, pharmacovigilance programs for approved products or requirement of risk management activities related to the promotion and sale of a product.
If regulatory authorities determine that we or our licensees or partners conducting research and development activities on our behalf have not complied with regulations in the research and development of a product candidate, new indication for an existing product or information to support a current indication, then they may not approve the product candidate or new indication or maintain approval of the current indication in its current form or at all, and we will not be able to market and sell it. If we were unable to market and sell our product candidates, our business and results of operations would be materially and adversely affected.
Product liability and other claims arising in connection with the testing of our product candidates in human clinical trials may reduce demand for our products or result in substantial damages.
We face an inherent risk of product liability exposure related to KALBITOR and the testing of our product candidates in human clinical trials.
An individual may bring a product liability claim against us if KALBITOR or one of our product candidates causes, or merely appears to have caused, an injury. Moreover, in some of our clinical trials, we test our product candidates in indications where the onset of certain symptoms or "attacks" could be fatal. Although the protocols for these trials include emergency treatments in the event a patient appears to be suffering a potentially fatal incident, patient deaths may nonetheless occur. As a result, we may face additional liability if we are found or alleged to be responsible for any such deaths.
These types of product liability claims may include but are not limited to:
| | ● | decreased demand for KALBITOR or any other product candidates; |
| | ● | injury to our reputation; |
| | ● | withdrawal of clinical trial volunteers; |
| | ● | related litigation costs; and |
| | ● | substantial monetary awards to plaintiffs. |
Although we currently maintain product liability insurance, we may not have sufficient insurance coverage, and we may not be able to obtain sufficient coverage at a reasonable cost. Our inability to obtain product liability insurance at an acceptable cost or to otherwise protect against potential product liability claims could prevent or inhibit the commercialization of any products that we or our collaborators develop, including KALBITOR. If we are successfully sued for any injury caused by our products or processes, then our liability could exceed our product liability insurance coverage and our total assets.
If we fail to establish and maintain strategic license, research and collaborative relationships, or if our collaborators are not able to successfully develop and commercialize product candidates, our ability to generate revenues could be adversely affected.
Our business strategy includes leveraging certain product candidates, as well as our proprietary phage display technology, through collaborations and licenses that are structured to generate revenues through license fees, technical and clinical milestone payments, and royalties. We have entered into, and anticipate continuing to enter into, collaborative and other similar types of arrangements with third parties to develop, manufacture and market drug candidates and drug products.
In addition, for us to continue to receive any significant payments from our LFRP related licenses and collaborations and generate sufficient revenues to meet the required payments under our agreement with HC Royalty, the relevant product candidates must advance through clinical trials, establish safety and efficacy, and achieve regulatory approvals, obtain market acceptance and generate revenues.
Reliance on license and collaboration agreements involves a number of risks as our licensees and collaborators:
| | ● | are not obligated to develop or market product candidates discovered using our phage display technology; |
| | ● | may not perform their obligations as expected, or may pursue alternative technologies or develop competing products; |
| | ● | control many of the decisions with respect to research, clinical trials and commercialization of product candidates we discover or develop with them or have licensed to them; |
| | ● | may terminate their collaborative arrangements with us under specified circumstances, including, for example, a change of control, with short notice; and |
| | ● | may disagree with us as to whether a milestone or royalty payment is due or as to the amount that is due under the terms of our collaborative arrangements. |
We cannot be assured we will be able to maintain our current licensing and collaborative efforts, nor can we assure the success of any current or future licensing and collaborative relationships. An inability to establish new relationships on terms favorable to us, work successfully with current licensees and collaborators, or failure of any significant portion of our LFRP related licensing and collaborative efforts would result in a material adverse impact on our business, operating results and financial condition.
Our success depends significantly upon our ability to obtain and maintain intellectual property protection for our products and technologies and upon third parties not having or obtaining patents that would limit or prevent us from commercializing any of our products.
We face risks and uncertainties related to our intellectual property rights. For example:
| | ● | we may be unable to obtain or maintain patent or other intellectual property protection for any products or processes that we may develop or have developed; |
| | ● | third parties may obtain patents covering the manufacture, use or sale of these products or processes, which may prevent us from commercializing any of our products under development globally or in certain regions; or |
| | ● | our patents or any future patents that we may obtain may not prevent other companies from competing with us by designing their products or conducting their activities so as to avoid the coverage of our patents. |
Patent rights relating to our phage display technology are central to our LFRP. In countries where we do not have and/or have not applied for phage display patent rights, we will be unable to prevent others from using phage display or developing or selling products or technologies derived using phage display. In addition, in jurisdictions where we have phage display patent rights, we may be unable to prevent others from selling or importing products or technologies derived elsewhere using phage display. Any inability to protect and enforce such phage display patent rights, whether by any inability to license or any invalidity of our patents or otherwise, could negatively affect future licensing opportunities and revenues from existing agreements under the LFRP.
In all of our activities, we also rely substantially upon proprietary materials, information, trade secrets and know-how to conduct our research and development activities and to attract and retain collaborators, licensees and customers. Although we take steps to protect our proprietary rights and information, including the use of confidentiality and other agreements with our employees and consultants and in our academic and commercial relationships, these steps may be inadequate, these agreements may be violated, or there may be no adequate remedy available for a violation. Also, our trade secrets or similar technology may otherwise become known to, or be independently developed or duplicated by, our competitors.
Before we and our collaborators can market some of our processes or products, we and our collaborators may need to obtain licenses from other parties who have patent or other intellectual property rights covering those processes or products. Third parties have patent rights related to phage display, particularly in the area of antibodies. While we have gained access to key patents in the antibody area through the cross licenses with Affimed Therapeutics AG, Affitech AS, Biosite Incorporated (now owned by Alere Inc.), Cambridge Antibody Technology Limited or CAT (now known as MedImmune Limited and owned by AstraZeneca), Domantis Limited (a wholly-owned subsidiary of GlaxoSmithKline), Genentech, Inc. and XOMA Ireland Limited, other third party patent owners may contend that we need a license or other rights under their patents in order for us to commercialize a process or product. In addition, we may choose to license patent rights from other third parties. In order for us to commercialize a process or product, we may need to license the patent or other rights of other parties. If a third party does not offer us a needed license or offers us a license only on terms that are unacceptable, we may be unable to commercialize one or more of our products. If a third party does not offer a needed license to our collaborators and as a result our collaborators stop work under their agreement with us, we might lose future milestone payments and royalties, which would adversely affect us. If we decide not to seek a license, or if licenses are not available on reasonable terms, we may become subject to infringement claims or other legal proceedings, which could result in substantial legal expenses. If we are unsuccessful in these actions, adverse decisions may prevent us from commercializing the affected process or products and could require us to pay substantial monetary damages.
We seek affirmative rights of license or ownership under existing patent rights relating to phage display technology of others. For example, through our patent licensing program, we have secured a limited freedom to practice some of these patent rights pursuant to our standard license agreement, which contains a covenant by the licensee that it will not sue us under certain of the licensee's phage display improvement patents. We cannot guarantee, however, that we will be successful in enforcing any agreements from our licensees, including agreements not to sue under their phage display improvement patents, or in acquiring similar agreements in the future, or that we will be able to obtain commercially satisfactory licenses to the technology and patents of others. If we cannot obtain and maintain these licenses and enforce these agreements, this could have a material adverse impact on our business.
Proceedings to obtain, enforce or defend patents and to defend against charges of infringement are time consuming and expensive activities. Unfavorable outcomes in these proceedings could limit our patent rights and our activities, which could materially affect our business.
Obtaining, protecting and defending against patent and proprietary rights can be expensive. For example, if a competitor files a patent application claiming technology also invented by us, we may have to participate in an expensive and time-consuming interference proceeding before the United States Patent and Trademark Office to address who was first to invent the subject matter of the claim and whether that subject matter was patentable. Moreover, an unfavorable outcome in an interference proceeding could require us to cease using the technology or to attempt to license rights to it from the prevailing party. Our business would be harmed if a prevailing third party does not offer us a license on terms that are acceptable to us. In patent offices outside the United States, we may be forced to respond to third party challenges to our patents.
The issues relating to the validity, enforceability and possible infringement of our patents present complex factual and legal issues that we periodically reevaluate. Third parties have patent rights related to phage display, particularly in the area of antibodies. While we have gained access to key patents in the antibody area through our cross-licensing agreements with Affimed, Affitech, Biosite, Domantis, Genentech, XOMA and CAT, other third party patent owners may contend that we need a license or other rights under their patents in order for us to commercialize a process or product. In addition, we may choose to license patent rights from third parties. While we believe that we will be able to obtain any needed licenses, we cannot assure you that these licenses, or licenses to other patent rights that we identify as necessary in the future, will be available on reasonable terms, if at all. If we decide not to seek a license, or if licenses are not available on reasonable terms, we may become subject to infringement claims or other legal proceedings, which could result in substantial legal expenses. If we are unsuccessful in these actions, adverse decisions may prevent us from commercializing the affected process or products. Moreover, if we are unable to maintain the covenants with regard to phage display improvements that we obtain from our licensees through our patent licensing program and the licenses that we have obtained to third party phage display patent rights, it could have a material adverse effect on our business.
We would expect to incur substantial costs in connection with any litigation or patent proceeding. In addition, our management's efforts would be diverted, regardless of the results of the litigation or proceeding. An unfavorable result could subject us to significant liabilities to third parties, require us to cease manufacturing or selling the affected products or using the affected processes, require us to license the disputed rights from third parties or result in awards of substantial damages against us. Our business will be harmed if we cannot obtain a license, can obtain a license only on terms we consider to be unacceptable or if we are unable to redesign our products or processes to avoid infringement.
In all of our activities, we substantially rely on proprietary materials and information, trade secrets and know-how to conduct research and development activities and to attract and retain collaborative partners, licensees and customers. Although we take steps to protect these materials and information, including the use of confidentiality and other agreements with our employees and consultants in both academic commercial relationships, we cannot assure you that these steps will be adequate, that these agreements will not be violated, or that there will be an available or sufficient remedy for any such violation, or that others will not also develop the same or similar proprietary information.
Failure to meet our HC Royalty debt service obligations could adversely affect our financial condition and our loan agreement obligations could impair our operating flexibility.
Our loans from an affiliate of HC Royalty have an aggregate principal balance of $81.2 million at December 31, 2012. The loans bear interest at a rate of 12% per annum, payable quarterly, will mature in August 2018, and can be repaid without penalty beginning in August 2015.
In connection with this loan, we have entered into a security agreement granting HC Royalty a security interest in the intellectual property related to the LFRP and the revenues generated by us through licenses of our intellectual property related to the LFRP. We are required to repay the loans based on a percentage of LFRP related revenues, including royalties, milestones, and license fees received by us under the LFRP. If the LFRP revenues for any quarterly period are insufficient to cover the cash interest due for that period, the deficiency may be added to the outstanding loan principal or paid in cash by us. In the event of certain changes of control or mergers or sales of all or substantially all of our assets, any or all of the loans may become due and payable at HC Royalty’s option, including a prepayment premium obligation which will expire in August 2015. We must comply with certain loan covenants which if not observed could make all loan principal, interest and all other amounts payable under the loans immediately due and payable.
Our obligations under the HC Royalty agreement require that we dedicate a substantial portion of cash flow from our LFRP receipts to service the loans, which will reduce the amount of cash flow available for other purposes while the loan is outstanding. If the LFRP fails to generate sufficient receipts to fund quarterly principal and interest payments to HC Royalty, we will be required to fund such obligations from cash on hand or from other sources, further decreasing the funds available to operate our business. In the event that amounts due under the loans is accelerated, payment would significantly reduce our cash, cash equivalents and short-term investments and we may not have sufficient funds to pay the debt if any of it is accelerated.
As a result of the security interest granted to HC Royalty, we are restricted in our ability to sell our rights to part or all of those assets, or take certain other actions, without first obtaining permission from HC Royalty. This requirement could delay, hinder or condition our ability to enter into corporate partnerships or strategic alliances with respect to these assets.
The obligations and restrictions under the HC Royalty agreement may limit our operating flexibility, make it difficult to pursue our business strategy and make us more vulnerable to economic downturns and adverse developments in our business.
If we lose or are unable to hire and retain qualified personnel, then we may not be able to develop our products or processes.
We are highly dependent on qualified scientific and management personnel, and we face intense competition from other companies and research and academic institutions for qualified personnel. If we lose an executive officer, a manager of one of our principal business units or research programs, or a significant number of any of our staff or are unable to hire and retain qualified personnel, then our ability to develop and commercialize our products and processes may be delayed which would have an adverse effect on our business, financial condition, and results of operations.
We use and generate hazardous materials in our business, and any claims relating to the improper handling, storage, release or disposal of these materials could be time-consuming and expensive.
Our phage display research and development involves the controlled storage, use and disposal of chemicals and solvents, as well as biological and radioactive materials. We are subject to foreign, federal, state and local laws and regulations governing the use, manufacture and storage and the handling and disposal of materials and waste products. Although we believe that our safety procedures for handling and disposing of these hazardous materials comply with the standards prescribed by laws and regulations, we cannot completely eliminate the risk of contamination or injury from hazardous materials. If an accident occurs, an injured party could seek to hold us liable for any damages that result and any liability could exceed the limits or fall outside the coverage of our insurance. We may not be able to maintain insurance on acceptable terms, or at all. We may incur significant costs to comply with current or future environmental laws and regulations.
Our business is subject to risks associated with international contractors and exchange rate risk.
None of our business is conducted in currencies other than the United States dollar. We do, however, rely on an international contract manufacturer for the production of our drug substance for ecallantide. We recognize foreign currency gains or losses arising from our transactions in the period in which we incur those gains or losses. As a result, currency fluctuations among the United States dollar and the currencies in which we do business have caused foreign currency transaction gains and losses in the past and will likely do so in the future. Because of the variability of currency exposures and the potential volatility of currency exchange rates, we may suffer significant foreign currency transaction losses in the future due to the effect of exchange rate fluctuations.
Compliance with changing regulations relating to corporate governance and public disclosure may result in additional expenses.
Keeping abreast of, and in compliance with, changing laws, regulations, and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, new SEC regulations, and NASDAQ Global Market rules, have required an increased amount of management attention and external resources. We intend to invest all reasonably necessary resources to comply with evolving corporate governance and public disclosure standards, and this investment may result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities.
We may not succeed in acquiring technology and integrating complementary businesses.
We may acquire additional technology and complementary businesses in the future. Acquisitions involve many risks, any one of which could materially harm our business, including:
| | ● | the diversion of management's attention from core business concerns; |
| | ● | the failure to exploit acquired technologies effectively or integrate successfully the acquired businesses; |
| | ● | the loss of key employees from either our current business or any acquired businesses; and |
| | ● | the assumption of significant liabilities of acquired businesses. |
We may be unable to make any future acquisitions in an effective manner. In addition, the ownership represented by the shares of our common stock held by our existing stockholders will be diluted if we issue equity securities in connection with any acquisition. If we make any significant acquisitions using cash consideration, we may be required to use a substantial portion of our available cash. If we issue debt securities to finance acquisitions, then the debt holders would have rights senior to the holders of shares of our common stock to make claims on our assets and the terms of any debt could restrict our operations, including our ability to pay dividends on our shares of common stock. Acquisition financing may not be available on acceptable terms, or at all. In addition, we may be required to amortize significant amounts of intangible assets in connection with future acquisitions. We might also have to recognize significant amounts of goodwill that will have to be tested periodically for impairment. These amounts could be significant, which could harm our operating results.
Risks Related To Our Common Stock
Our common stock may continue to have a volatile public trading price and low trading volume.
The market price of our common stock has been highly volatile. Since our initial public offering in August 2000 through February 25, 2013, the price of our common stock on the NASDAQ Global Market has ranged between $54.12 and $1.05. The market has experienced significant price and volume fluctuations for many reasons, some of which may be unrelated to our operating performance.
Many factors may have an effect on the market price of our common stock, including:
| | ● | public announcements by us, our competitors or others; |
| | ● | developments concerning proprietary rights, including patents and litigation matters; |
| | ● | publicity regarding actual or potential clinical results or developments with respect to products or compounds we or our collaborators are developing; |
| | ● | regulatory decisions in both the United States and abroad; |
| | ● | public concern about the safety or efficacy of new technologies; |
| | ● | issuance of new debt or equity securities; |
| | ● | general market conditions and comments by securities analysts; and |
| | ● | quarterly fluctuations in our revenues and financial results. |
While we cannot predict the effect that these factors may have on the price of our common stock, these factors, either individually or in the aggregate, could result in significant variations in price during any given period of time.
Anti-takeover provisions in our governing documents and under Delaware law may make an acquisition of us more difficult.
We are incorporated in Delaware. We are subject to various legal and contractual provisions that may make a change in control of us more difficult. Our board of directors has the flexibility to adopt additional anti-takeover measures.
Our charter authorizes our board of directors to issue up to 1,000,000 shares of preferred stock and to determine the terms of those shares of stock without any further action by our stockholders. If the board of directors exercises this power to issue preferred stock, it could be more difficult for a third party to acquire a majority of our outstanding voting stock. Our charter also provides staggered terms for the members of our board of directors. This may prevent stockholders from replacing the entire board in a single proxy contest, making it more difficult for a third party to acquire control of us without the consent of our board of directors. Our equity incentive plans generally permit our board of directors to provide for acceleration of vesting of options granted under these plans in the event of certain transactions that result in a change of control. If our board of directors used its authority to accelerate vesting of options, then this action could make an acquisition more costly, and it could prevent an acquisition from going forward.
Section 203 of the Delaware General Corporation Law prohibits a person from engaging in a business combination with any holder of 15% or more of its capital stock until the holder has held the stock for three years unless, among other possibilities, the board of directors approves the transaction. This provision could have the effect of delaying or preventing a change of control of Dyax, whether or not it is desired by or beneficial to our stockholders.
The provisions described above, as well as other provisions in our charter and bylaws and under the Delaware General Corporation Law, may make it more difficult for a third party to acquire our company, even if the acquisition attempt was at a premium over the market value of our common stock at that time.
| UNRESOLVED STAFF COMMENTS |
None.
As of January 2012, we are leasing approximately 45,000 square feet of space in Burlington, Massachusetts in a facility which serves as our corporate headquarters and laboratory facility. Our lease will expire in 2022, although we have an option to extend our lease for an additional five year term. We have provided the lessor with a Letter of Credit of approximately $1.1 million.
Through January 2012, we leased approximately 43,000 square feet of space in Cambridge, Massachusetts. This facility served as our corporate headquarters and laboratory facility. The lease was terminated on January 28, 2012. We had provided the lessor with a Letter of Credit of approximately $1.3 million which was fully released in the first quarter of 2012.
We are not a party to any material legal proceedings.
Not applicable.
PART II
| MARKET FOR THE COMPANY'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Our common stock is traded on The NASDAQ Global Market under the symbol DYAX. As of February 20, 2013, there were approximately 148 common stockholders of record.
The following table sets forth, for the periods indicated, the high and low selling prices for our common stock as reported on NASDAQ Global Market:
| | | High | | | Low | |
| Fiscal year ended December 31, 2012 | | | | | | |
| First Quarter | | $ | 1.67 | | | $ | 1.27 | |
| Second Quarter | | $ | 2.20 | | | $ | 1.45 | |
| Third Quarter | | $ | 2.88 | | | $ | 2.08 | |
| Fourth Quarter | | $ | 3.74 | | | $ | 2.24 | |
| | | High | | | Low | |
| Fiscal year ended December 31, 2011 | | | | | | |
| First Quarter | | $ | 2.23 | | | $ | 1.44 | |
| Second Quarter | | $ | 2.33 | | | $ | 1.51 | |
| Third Quarter | | $ | 2.12 | | | $ | 1.10 | |
| Fourth Quarter | | $ | 1.53 | | | $ | 1.17 | |
We have never declared or paid cash dividends on our capital stock. We currently intend to retain our future earnings, if any, for use in our business and therefore do not anticipate paying cash dividends in the foreseeable future. Payment of future dividends, if any, will be at the discretion of our Board of Directors after taking into account various factors, including our financial condition, operating results, current and anticipated cash needs and plans for expansion.
We provide equity compensation plan information in Item 12, "Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters". We are incorporating that information into this section by this reference.
The following table summarizes certain selected consolidated financial data, which should be read in conjunction with "Management's Discussion and Analysis of Financial Condition and Results of Operations" and our consolidated financial statements and related notes included elsewhere in this Form 10-K. The selected consolidated financial data at December 31, 2012 and 2011, and for the years ended December 31, 2012, 2011 and 2010 have been prepared from our audited financial statements included in this Form 10-K. The selected consolidated financial data at December 31, 2010, 2009 and 2008, and for the years ended December 31, 2009 and 2008, have been prepared from our audited financial statements not included in this Annual Report on Form 10-K.
| | | December 31, | |
| | | 2012 | | | 2011 | | | 2010 | | | 2009 | | | 2008 | |
| | | (In thousands, except share and per share data) | |
| Consolidated Statement of Operations Data: | | | | | | | | | | | | | | | |
| Revenues: | | | | | | | | | | | | | | | |
| Product sales, net | | $ | 39,783 | | | $ | 22,884 | | | $ | 8,835 | | | $ | — | | | $ | — | |
| Development and license fee revenues | | | 14,867 | | | | 25,853 | | | | 42,564 | | | | 21,643 | | | | 43,429 | |
| Total revenues | | | 54,650 | | | | 48,737 | | | | 51,399 | | | | 21,643 | | | | 43,429 | |
| | | | | | | | | | | | | | | | | | | | | |
| Cost of product sales | | | 2,152 | | | | 1,223 | | | | 505 | | | | — | | | | — | |
| Research and development expenses | | | 30,028 | | | | 34,676 | | | | 31,522 | | | | 46,587 | | | | 68,077 | |
| Selling, general and administrative expenses | | | 39,915 | | | | 37,740 | | | | 33,583 | | | | 25,843 | | | | 22,663 | |
| Restructuring costs | | | 1,440 | | | | — | | | | — | | | | 2,331 | | | | 4,631 | |
| Impairment of fixed assets | | | — | | | | — | | | | — | | | | 955 | | | | 352 | |
| Total operating expenses | | | 73,535 | | | | 73,639 | | | | 65,610 | | | | 75,716 | | | | 95,723 | |
| Loss from operations | | | (18,885 | ) | | | (24,902 | ) | | | (14,211 | ) | | | (54,073 | ) | | | (52,294 | ) |
| Other (expense) income, net | | | (10,380 | ) | | | (9,697 | ) | | | (10,292 | ) | | | (8,346 | ) | | | (5,910 | ) |
| Loss on extinguishment of debt | | | — | | | | — | | | | — | | | | — | | | | (8,264 | ) |
| Net loss | | $ | (29,265 | ) | | $ | (34,599 | ) | | $ | (24,503 | ) | | $ | (62,419 | ) | | $ | (66,468 | ) |
| Basic and diluted net loss per share | | $ | (0.30 | ) | | $ | (0.35 | ) | | $ | (0.26 | ) | | $ | (0.90 | ) | | $ | (1.08 | ) |
| Shares used in computing basic and diluted net loss per share | | | 98,991,056 | | | | 98,731,289 | | | | 93,267,850 | | | | 69,151,841 | | | | 61,626,095 | |
| | | December 31, | |
| | | 2012 | | | 2011 | | | 2010 | | | 2009 | | | 2008 | |
| | | (In thousands) | |
| Consolidated Balance Sheet Data: | | | | | | | | | | | | | | | |
| Cash and cash equivalents | | $ | 20,018 | | | $ | 31,468 | | | $ | 18,601 | | | $ | 29,386 | | | $ | 27,668 | |
| Short-term investments | | | 9,028 | | | | 26,036 | | | | 58,783 | | | | 23,009 | | | | 30,792 | |
| Working capital | | | 24,431 | | | | 48,186 | | | | 67,869 | | | | 34,126 | | | | 40,736 | |
| Total assets | | | 55,486 | | | | 83,375 | | | | 92,431 | | | | 64,801 | | | | 75,075 | |
| Long-term obligations, less current portion | | | 78,992 | | | | 75,372 | | | | 56,474 | | | | 58,749 | | | | 48,499 | |
| Accumulated deficit | | | (506,186 | ) | | | (476,921 | ) | | | (442,322 | ) | | | (417,819 | ) | | | (355,400 | ) |
| Total stockholders' equity (deficit) | | | (51,560 | ) | | | (27,399 | ) | | | 2,633 | | | | (38,602 | ) | | | (20,044 | ) |
The Company has revised the classification for $4.9 million of inventory from current assets to non-current other assets for the year ended December 31, 2011, to correct the classification of inventory based on the projected sale of inventory beyond the Company's normal operating cycle. This reclassification is reflected in working capital at December 31, 2011 in the chart above.
| MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
We are a biopharmaceutical company with two business elements:
| | ● | Plasma Kallikrein-Mediated Angioedema Portfolio |
The principal focus of our efforts is to identify, develop and commercialize treatments for angioedemas that are identified as plasma kallikrein-mediated, which we refer to as PKM angioedemas, including hereditary angioedema (HAE) and idiopathic angioedema.
We developed KALBITOR (ecallantide) on our own, and since February 2010, we have been selling it in the United States for the treatment of acute attacks of HAE. Outside of the United States, we have established partnerships to obtain regulatory approval for and to commercialize KALBITOR in certain markets and we are evaluating opportunities in others.
We are expanding our franchise for the treatment of PKM angioedemas in the following ways:
| | ● | Development of diagnostic strategies to assist in the differentiation between histamine-mediated and PKM angioedema. |
| | ● | Continuing our development of DX-2930, a fully human monoclonal antibody inhibitor of plasma kallikrein, which could be a candidate to prophylactically treat PKM angioedemas. |
| | ● | Phage Display Licensing and Funded Research Program |
We leverage our proprietary phage display technology through our Licensing and Funded Research Program, referred to as the LFRP. This program has provided us a portfolio of product candidates being developed by our licensees, which currently includes 13 product candidates in various stages of clinical development, including three in Phase 3 trials, for which we are eligible to receive future royalties and/or milestone payments. The LFRP generated approximately $12.5 million of revenue for us in 2012. To the extent that our licensees commercialize some of the Phase 3 product candidates, our revenues under the LFRP are expected to experience growth beginning in 2014.
PKM ANGIOEDEMA PORTFOLIO
We are focused on identifying and developing treatments for patients who experience PKM angioedema. Using our phage display technology, we developed ecallantide, a compound shown in vitro to be a high affinity, high specificity inhibitor of human plasma kallikrein. Plasma kallikrein, an enzyme found in blood, produces bradykinin, a protein that causes blood vessels to enlarge or dilate, which can cause swelling known as angioedema. Plasma kallikrein is believed to be a key component in the regulation of inflammation and contact activation pathways. Excess plasma kallikrein activity is thought to play a role in a number of inflammatory diseases, including PKM angioedemas such as HAE and idiopathic angioedema.
We have three key areas of activity in our PKM angioedema portfolio:
| | ● | HAE and KALBITOR. In February 2010, we began selling KALBITOR in the United States for treatment of acute attacks of HAE in patients 16 years of age and older. We are selling KALBITOR on our own in the United States. Working with international partners, we intend to seek approval for and commercialize KALBITOR for HAE and other angioedema indications in markets outside of the United States. We have entered into agreements for others to develop and commercialize subcutaneous ecallantide for the treatment of HAE and other angioedema indications throughout Europe, Japan, China and countries in Latin America and the Middle East. |
| | ● | Identification of PKM angioedemas. In order to expand our PKM angioedema portfolio, we have launched a program to identify one or more diagnostic strategies that will assist in the differentiation of PKM angioedema from histamine-mediated angioedema, in order to direct appropriate treatment. We have developed laboratory tests for which the process of clinical validation has commenced. These tools are expected to be relevant to both normal C1esterase inhibitor (C1-INH) and C1-INH deficient patients and will enable the identification of PKM angioedema, including Type III HAE and angioedema of unknown origin, or idiopathic angioedema. |
| | ● | DX-2930 - Antibody for PKM angioedemas. Based on our knowledge of angioedema and the kallikrein-kinin pathway, we are investigating the use of a fully human monoclonal antibody that is an inhibitor of plasma kallikrein and which could be a candidate to treat prophylactically PKM angioedemas. After completing a series of pharmacokinetic, tolerability and preclinical studies, we believe DX-2930 may be effective for prophylactically treating these indications. We expect to file an Investigational New Drug application (IND) for this antibody in mid-2013. |
LICENSING AND FUNDED RESEARCH PROGRAM
We believe that our phage display libraries, which we have developed using our core technology and know-how, represent a leading technology in antibody discovery. We leverage our proprietary phage display technology and libraries through our LFRP licenses and collaborations. To date, we have recognized more than $185 million of revenue under the LFRP, primarily related to license fees and milestones, including approximately $12.5 million of revenue in 2012. The LFRP has the potential for substantially greater revenues, if and when product candidates that are discovered by our licensees receive marketing approval and are commercialized.
LFRP Product Development
Currently, 18 product candidates generated by our licensees or collaborators under the LFRP portfolio are in clinical development and one product has received market approval from the FDA. We will receive future milestones and/or royalties from our licensees and collaborators for 13 of the 18 product candidates that are currently in clinical development, including three in Phase 3 and four in Phase 2 trials, to the extent these product candidates advance in development and are ultimately commercialized. Furthermore, our licensees and collaborators have 13 additional product candidates in various stages of preclinical development. Our licensees and collaborators are responsible for all costs associated with development of these product candidates. To the extent that our licensees commercialize some of the Phase 3 product candidates, our revenues under the LFRP are expected to experience growth beginning in 2014.
Under loan arrangements with affiliates of HC Royalty, we have obtained debt funding which has a principal balance of $81.2 million as of December 31, 2012, secured exclusively by the LFRP, which is described more fully in Note 8 to Notes to Consolidated Financial Statements filed under Item 8 of this Form 10-K.
RESULTS OF OPERATIONS
Revenues. Total revenues for 2012 were $54.7 million, compared with $48.7 million in 2011 and $51.4 million in 2010.
Product Sales. We began commercializing KALBITOR in the United States in 2010 for treatment of acute attacks of HAE in patients 16 years of age and older. We sell KALBITOR to our distributors, and we recognize revenue when title and risk of loss have passed to the distributor, typically upon delivery. Due to the specialty nature of KALBITOR, the limited number of patients, limited return rights and contractual limits on inventory levels, we anticipate that distributors will carry inventory that is in line with ordinary business needs. Although fluctuations can occur due to the acute nature of HAE attacks, generally distributors do not hold inventory of more than 60 days of anticipated demand.
We record product sales net of allowances and accruals related to trade prompt pay discounts, government rebates, a patient financial assistance program, product returns and other applicable allowances. In 2012, product sales of KALBITOR increased to $39.8 million, net of product discounts and allowances of $3.5 million, compared to product sales of $22.9 million, net of product discounts and allowances of $1.1 million during 2011 and product sales of $8.8 million, net of product discounts and allowances of $458,000 during 2010. The 2012 and 2011 increases in product sales were primarily due to a significant increase in the volume of KALBITOR units sold, as well as a price increase in each of those years.
Provisions for product sales allowances reduced gross product sales as follows (in thousands):
| | | 2012 | | | 2011 | | | 2010 | |
| | | | | | | | | | |
| Total gross product sales | | $ | 43,251 | | | $ | 23,999 | | | $ | 9,293 | |
| | | | | | | | | | | | | |
| Prompt pay and other discounts | | $ | (1,722 | ) | | $ | (831 | ) | | $ | (205 | ) |
| Government rebates and chargebacks | | | (1,243 | ) | | | (249 | ) | | | (239 | ) |
| Returns | | | (503 | ) | | | (35 | ) | | | (14 | ) |
Product sales allowances | | $ | (3,468 | ) | | $ | (1,115 | ) | | $ | (458 | ) |
Total product sales, net | | $ | 39,783 | | | $ | 22,884 | | | $ | 8,835 | |
| | | | | | | | | | | | | |
Total product sales allowances as a percent of gross product sales | | | 8.0 | % | | | 4.6 | % | | | 4.9 | % |
Development and License Fees. We also derive revenues from licensing, funded research and development fees, including milestone payments from our licensees and collaborators, in amounts that fluctuate from year-to-year due to the timing of the licensing and clinical activities of our collaborators and licensees. This revenue was $14.9 million in 2012, $25.9 million in 2011 and $42.6 million in 2010.
Development and license fee revenue in 2011 included $10.5 million recognized under our agreement, as amended, with Sigma-Tau, compared with $204,000 and $2.2 million recognized in 2012 and 2010, respectively (see Note 3, Significant Transactions – Sigma-Tau).
Development and license fee revenue in 2010 included the recognition of $11.3 million from the sale of rights to royalties and other payments related to Xyntha, a product developed by one of our licensees under the LFRP, and $13.8 million of previously deferred revenue associated with the Cubist license that was fully recognized during 2010 based upon Cubist's termination of its ecallantide development program.
Cost of Product Sales. We incurred $2.2 million of costs associated with product sales during 2012, $1.2 million during 2011 and $505,000 during 2010. This primarily includes the cost of testing, filling, packaging and distributing the KALBITOR product, as well as a royalty due on net sales of KALBITOR. Costs associated with the manufacture of KALBITOR prior to FDA approval were previously expensed when incurred and, accordingly, were not included in the cost of product sales during 2010, 2011 and 2012. The supply of KALBITOR produced prior to FDA approval met commercial needs through the third quarter of 2012. When this supply was depleted during the fourth quarter of 2012, our cost of product sales increased, reflecting more of the cost of manufacturing KALBITOR product. During 2013, we expect the cost of product sales to reflect the full cost of KALBITOR manufacturing.
Research and Development. Our research and development expenses are summarized as follows:
| | | Years Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
| | | (In thousands) | |
| KALBITOR development costs | | $ | 16,371 | | | $ | 21,474 | | | $ | 17,157 | |
| Other research and development expenses | | | 12,070 | | | | 10,753 | | | | 11,975 | |
| LFRP pass-through fees | | | 1,587 | | | | 2,449 | | | | 2,390 | |
Total | | $ | 30,028 | | | $ | 34,676 | | | $ | 31,522 | |
Our research and development expenses arise primarily from compensation and other related costs for our personnel dedicated to research, development, medical and pharmacovigilence activities, costs of post-approval studies and commitments and KALBITOR life cycle management, as well as fees paid and costs reimbursed to outside parties to conduct research and clinical trials.
KALBITOR development costs decreased in 2012 compared to 2011 primarily due to the discontinuation during June 2012 of the clinical study of the use of ecallantide for the treatment of ACE inhibitor-induced angioedema. This Phase 2 clinical study was initiated during 2011 and the increase in 2011 costs over 2010 was primarily due to this study. In addition, costs associated with obtaining regulatory approval for the treatment of HAE in territories outside the United States were $32,000, $2.0 million and $1.2 million during 2012, 2011 and 2010, respectively. These amounts were reimbursed by Sigma Tau and such payments are recorded as development and license fee revenue in our results of operations.
The 2012 increase in other research and development costs from 2011 was due primarily to the pre-clinical, scale-up activities for DX-2930, a fully human monoclonal antibody inhibitor of plasma kallikrein, which could be a candidate to treat prophylactically PKM angioedemas. The 2011 decrease in other research and development costs from 2010 was due to a shift in internal efforts from early stage development programs to KALBITOR development.
Research and development expenses may increase in future years, due to costs associated with our clinical study developing DX-2930 as a therapeutic candidate. Until the clinical studies are further advanced, we are not able to predict the future clinical costs that may be incurred for the development of this candidate.
Selling, General and Administrative. Our selling, general and administrative expenses consist primarily of the sales and marketing costs of commercializing KALBITOR, costs of our management and administrative staff, as well as expenses related to business development, protecting our intellectual property, administrative occupancy, professional fees and the reporting requirements of a public company. Selling, general and administrative expenses were $39.9 million in 2012 compared to $37.7 million in 2011 and $33.6 million in 2010.
The 2012 and 2011 increases in costs are primarily due to the expansion of infrastructure to support KALBITOR commercial efforts, primarily sales and marketing programs, and additional legal expenses to protect our intellectual property.
We do not anticipate a significant change in the trend for selling, general and administrative expenses in 2013.
Restructuring. In February 2012, we implemented a realignment of our business which included a workforce reduction. As a result, we recorded restructuring charges of approximately $1.4 million.
Interest Expense. Interest expense, which is primarily derived from our loan arrangement with HC Royalty, was $10.5 million, $10.3 million and $11.9 million in 2012, 2011 and 2010, respectively. Based on the modification of the HC Royalty loan in December 2011, under which we received additional proceeds of $20 million, we recorded interest expense in 2012 using the effective interest rate method for the different tranches of the loans. The effective interest rate is now approximately 13%. In 2011 and 2010, we recorded interest expense at the stated interest rate for each tranche, which in total was approximately 17.4%. The higher 2010 expense was also due to additional interest expense under the HC Royalty loan of approximately $1.4 million for payments in connection with the sale of our rights to royalties and other payments related to the Xyntha product.
Interest and Other Income. Interest income was $28,000, $184,000 and $209,000 in 2012, 2011 and 2010, respectively. The 2012 decrease in interest income was primarily due to lower cash and investment balances during the year.
In 2010, income of $1.5 million was recognized from several grants received under the Qualifying Therapeutic Discovery Project program. Under this program, the Internal Revenue Service, in conjunction with the Department of Health and Human Services, approved our applications for projects that showed significant potential to produce new and cost-saving therapies, support jobs and increase U.S. competitiveness. All proceeds from this grant were classified as Other Income in the Statement of Operations.
Net Loss. For the year ended December 31, 2012, the net loss was $29.3 million or $0.30 per share, as compared to $34.6 million or $0.35 per share in 2011, and $24.5 million or $0.26 per share in 2010.
LIQUIDITY AND CAPITAL RESOURCES
| | | December 31, | |
| | | 2012 | | | 2011 | |
| | | (In thousands) | |
Cash and cash equivalents | | $ | 20,018 | | | $ | 31,468 | |
Short-term investments | | | 9,028 | | | | 26,036 | |
Total cash, cash equivalents and investments | | $ | 29,046 | | | $ | 57,504 | |
The following table summarizes our cash flow activity:
| | | Years Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
| | | (In thousands) | |
Net cash used in operating activities | | $ | (28,482 | ) | | $ | (36,452 | ) | | $ | (34,131 | ) |
Net cash provided by (used in) investing activities | | | 14,343 | | | | 30,609 | | | | (35,409 | ) |
Net cash provided by financing activities | | | 2,689 | | | | 18,710 | | | | 58,755 | |
Net (decrease) increase in cash and cash equivalents | | $ | (11,450 | ) | | $ | 12,867 | | | $ | (10,785 | ) |
We require cash to fund our operating activities, make capital expenditures, acquisitions and investments, and service debt. Through December 31, 2012, we have funded our operations through the sale of equity securities, which have provided aggregate net cash proceeds since inception of approximately $399 million, and from borrowed funds under our loan agreement with HC Royalty, which are secured by certain assets associated with our LFRP. In addition, we generate funds from product sales and development and license fees. Our excess funds are currently invested in short-term investments primarily consisting of United States Treasury notes and bills and money market funds backed by the United States Treasury.
Operating Activities.
In 2012, the principal use of cash in our operations was to fund our $29.3 million net loss. Of this net loss, certain costs were non-cash charges, such as depreciation and amortization costs of $1.1 million and stock-based compensation expense of $3.6 million. In addition to non-cash charges, we also had cash outflows due to changes in other operating assets and liabilities, including an increase in inventory of $2.9 million, an increase in prepaid and other assets of $2.8 million and a decrease in deferred revenue of $4.1 million, primarily associated with revenue recognized for LFRP licenses that was previously deferred.
In 2011, the principal use of cash in our operations was to fund our $34.6 million net loss. Of this net loss, certain costs were non-cash charges, such as depreciation and amortization costs of $1.6 million and stock-based compensation expense of $4.0 million. In addition to non-cash charges, we also had cash outflows due to changes in other operating assets and liabilities, including an increase in inventory of $5.2 million, an increase in accounts payable and accrued expenses of $1.6 million and a decrease in deferred revenue of $5.4 million, due primarily to the recognition of previously deferred revenue related to our collaborations to commercialize ecallantide outside of the United States.
In 2010, the principal use of cash in our operations was to fund our $24.5 million net loss. Of this net loss, certain costs were non-cash charges, such as depreciation and amortization costs of $1.6 million and stock-based compensation expense of $4.1 million. In addition to non-cash charges, we also had a net change in other operating assets and liabilities of $16.3 million, including a decrease in accounts payable and accrued expenses of $2.5 million, an increase in accounts receivable of $2.6 million, and a decrease in deferred revenue of $8.8 million. The change in deferred revenue is primarily due to the recognition of $13.8 million of revenue associated with the 2010 termination of the license and collaboration agreement with Cubist, offset by additional deferred revenue of $7.0 million from new collaborations to commercialize ecallantide outside the United States.
Investing Activities.
Our investing activities for 2012 primarily consisted of $23.0 million of investments which matured, offset by the purchase of $6.1 million of investments, as well as a decrease of $1.3 million in restricted cash resulting from the release of the letter of credit that had been issued as a security deposit under the lease of our previous facility in Cambridge, Massachusetts. These are offset by the purchase of $4.1 million of fixed assets primarily made up of leasehold improvements for the new Burlington facility, of which $2.6 million was covered by a tenant improvement allowance and $1.4 million was financed through an equipment loan arrangement.
Our investing activities for 2011 consisted of approximately $35.5 million of investment maturities, offset by the purchase of $3.0 million of investments. In addition, during 2011, we expended approximately $1.7 million in leasehold improvements related to the build-out of the new Burlington facility, of which $925,000 was reimbursed by the Landlord in 2011. The residual balance was reimbursed by the Landlord during 2012, as referenced above. In conjunction with the Burlington lease agreement, we have provided the landlord a letter of credit of $1.1 million to secure our obligations under the lease.
Our investing activities for 2010 consisted of the purchase of approximately $82.8 million of investments, offset by $47.0 million of investment maturities, as well as a decrease of $700,000 in restricted cash from the contractual reduction of the letter of credit that serves as our security deposit for the lease of our facility in Cambridge, Massachusetts.
Financing Activities.
Our financing activities for 2012 consisted $1.4 million from the issuance of common stock under the Employee Stock Purchase Plan and the Employee Stock Option Plan, a drawdown of $1.4 million under an equipment loan arrangement, as well as the repayment of long-term debt totaling $135,000, primarily to HC Royalty. See below for more details on our loan with HC Royalty.
Our financing activities for 2011 consisted of net proceeds of $19.9 million of new debt from an affiliate of HC Royalty, as well as principal repayments of long-term debt totaling $1.7 million, consisting of capital lease payments and $1.1 million to HC Royalty.
Our financing activities for 2010 consisted of net proceeds of $61.1 million from the sale of 20,186,132 shares of our common stock, as well as principal repayments of long-term debt totaling $2.8 million, consisting of capital lease payments and $1.9 million to HC Royalty.
We expect to continue to manage our cash requirements by completing additional partnerships, collaborations, and financial and strategic transactions. We expect that existing cash, cash equivalents, and short-term investments together with anticipated cash flow from existing development, collaborations and license agreements and product sales of KALBITOR will be sufficient to support our current operations into 2014. We will need additional funds if our cash requirements exceed our current expectations or if we generate less revenue than we expect during this period. We may seek additional funding through our collaborative arrangements and public or private financings. We may not be able to obtain financing on acceptable terms or at all, and we may not be able to enter into additional collaborative arrangements. Arrangements with collaborators or others may require us to relinquish rights to certain of our technologies, product candidates or products. The terms of any financing may adversely affect the holdings or the rights of our stockholders. If we need additional funds and are unable to obtain funding on a timely basis, we would curtail significantly our research, development or commercialization programs in an effort to provide sufficient funds to continue our operations, which could adversely affect our business prospects.
HealthCare Royalty Partners
In August 2012, we completed an agreement that we entered into with an affiliate of HC Royalty in December 2011 to refinance our existing loans with HC Royalty. At December 31, 2012, the aggregate principal amount of the new loan was $81.2 million, consisting of a $21.9 million Tranche A Loan and a $59.3 million Tranche B Loan. The loans bear interest at a rate of 12% per annum, payable quarterly. The loans will mature in August 2018, and can be repaid without penalty beginning in August 2015.
In connection with the loans, we entered into a security agreement granting HC Royalty a security interest in the intellectual property related to the LFRP, and the revenues generated through our licenses of the intellectual property related to the LFRP. The security agreement does not apply to our internal drug development or to any of our co-development programs for HAE.
Under the terms of the loan agreement, we are required to repay the loans based on the annual net LFRP receipts. Until September 30, 2016, required payments are equal to the sum of 75% of the first $15.0 million in specified annual LFRP receipts and 25% of specified annual LFRP receipts over $15.0 million. After September 30, 2016, and until the maturity date or the complete repayment of the loans, HC Royalty will receive 90% of all specified LFRP receipts. If the HC Royalty portion of LFRP receipts for any quarter exceeds the interest for that quarter, then the principal balance will be reduced. Any unpaid principal will be due upon the maturity of the loans. If the HC Royalty portion of LFRP revenues for any quarterly period is insufficient to cover the cash interest due for that period, the deficiency may be added to the outstanding principal or paid in cash. In December 2016 and August 2017, five years from the issuance dates of the Tranche A and Tranche B Loans, we must repay all additional accumulated principal above the original loan amounts of $21.7 million and $58.8 million, respectively.
Tranche A Loan
In December 2011, we entered into an agreement with an affiliate of HC Royalty and received a loan of $20 million (Tranche A Loan) and a commitment to refinance the amounts outstanding under our March 2009 amended and restated loan agreement (the March 2009 loan agreement) at a reduced interest rate in August 2012. The Tranche A Loan was unsecured and accrued interest at an annual rate of 13% through August 2012, at which time the Tranche A Loan and its accrued interest was combined with 102% of the unpaid principal and accrued interest outstanding under the March 2009 loan agreement upon the closing of the Tranche B Loan.
Upon execution of the Tranche A Loan, the terms of the Original Loans (defined below) were determined to be modified under ASC 470. Accordingly, during the year ended December 31, 2012, interest expense on the Loan is being recorded in the Company’s financial statements at an effective interest rate of 13%.
Upon modification of the debt arrangement, the note payable balance related to the Tranche A Loan was reduced by $193,000 to reflect payment of legal fees in conjunction with the loan; these fees are being accreted over the life of the Loan, through August 2018.
Tranche B Loan
In 2008 and 2009, we entered into loan agreements with an affiliate of HC Royalty that provided aggregate loan proceeds of $65.0 million (the Original Loans). These loans had an outstanding principal and accrued interest balance of $57.6 million at the time of their refinancing in August 2012 (Tranche B Loan). The Original Loans bore interest at an annual rate of 17.4%, payable quarterly, and were secured by the Company’s LFRP.
In connection with the Original Loans, the Company issued affiliates of HC Royalty warrants to purchase shares of our common stock. In August 2008, the Company issued warrants to purchase 250,000 shares of our common stock at an exercise price of $5.50 per share. This warrant expires in August 2016 and became exercisable in August 2009. We estimated the relative fair value of the warrant to be $853,000 on the date of issuance, using the Black-Scholes valuation model, assuming a volatility factor of 83.64%, risk-free interest rate of 4.07%, an eight-year expected term and an expected dividend yield of zero. In March 2009, we issued HC Royalty a second warrant to purchase 250,000 shares of the Company’s common stock at an exercise price of $2.87 per share. This warrant expires in August 2016 and became exercisable in March 2010. We estimated the relative fair value of the warrant to be $477,000 on the date of issuance, using the Black-Scholes valuation model, assuming a volatility factor of 85.98%, risk-free interest rate of 2.77%, a seven-year, four-month expected term and an expected dividend yield of zero. The relative fair values of the warrants as of the date of issuance are recorded in additional paid-in capital on our consolidated balance sheets.
The cash proceeds from the Original Loans were recorded as a note payable on our consolidated balance sheet. The note payable balance was reduced by $1.3 million for the fair value of the warrants issued, and by $580,000 for payment of HC Royalty’s legal fees in conjunction with the Tranche B Loan. Prior to the December 2011 issuance of the Tranche A Loan, each of these amounts was being accreted over the life of the note through August 2016. Subsequent to the modification of the debt arrangement in December 2011, the unamortized portion of these amounts is being accreted over the life of the Loan through August 2018.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements with the exception of operating leases.
Contractual Obligations
Contractual obligations represent future cash commitments and liabilities under agreements with third parties, and exclude contingent liabilities for which we cannot reasonably predict future payment. The following chart represents our total contractual obligations, including principal and interest, at December 31, 2012, aggregated by type:
| | | Payments due by period | |
| Contractual obligations | | Total | | | Less than 1 year | | | 1-2 years | | | 2-3 years | | | 3-4 years | | | 4-5 years | | | More than 5 years | |
| | | | | | (In thousands) | | | | | | | |
Note Payable (1) | | $ | 125,022 | | | $ | 6,088 | | | $ | 11,364 | | | $ | 14,907 | | | $ | 26,961 | | | $ | 64,561 | | | $ | 1,141 | |
Leasehold improvement arrangements | | | 888 | | | | 98 | | | | 98 | | | | 98 | | | | 98 | | | | 98 | | | | 398 | |
| Equipment lease line | | | 1,503 | | | | 515 | | | | 515 | | | | 473 | | | | — | | | | — | | | | — | |
Operating lease obligations | | | 14,914 | | | | 1,503 | | | | 1,605 | | | | 1,615 | | | | 1,581 | | | | 1,588 | | | | 7,022 | |
| Patient services (2) | | | 4,535 | | | | 1,635 | | | | 1,450 | | | | 1,450 | | | | — | | | | — | | | | — | |
Obligations for research, development and manufacturing (3) | | | 1,483 | | | | 1,277 | | | | 87 | | | | 77 | | | | 21 | | | | 21 | | | | — | |
Total contractual obligations | | $ | 148,345 | | | $ | 11,116 | | | $ | 15,119 | | | $ | 18,620 | | | $ | 28,661 | | | $ | 66,268 | | | $ | 8,561 | |
| (1) | These amounts represent projected future principal and interest payments to HC Royalty based on our current LFRP projections, which are subject to uncertainties based on the timing and amounts of cash receipts under the LFRP. See Notes to the Financial Statements, Note 8 of Item 8 "Financial Statements and Supplementary Data" |
| (2) | These amounts represent the cash commitment for the KALBITOR Access program, which offers treatment support services for patients with HAE and their healthcare providers. |
| (3) | These amounts represent the cash commitment due on research, development and manufacturing contracts. We will not owe any royalties or milestones in connection with these contracts. |
Critical Accounting Estimates
Our discussion and analysis of our results of operations and liquidity and capital resources are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses. On an ongoing basis, we evaluate our estimates and judgments, including those related to revenue recognition for licensing and collaboration arrangements, gross to net reserves, receivable collectibles, royalty interest obligations, clinical trial accruals, useful lives with respect to long-lived and intangible assets and valuation of common stock, related stock options, and deferred tax assets. We base our estimates on historical and anticipated results and trends and on various other assumptions that we believe are reasonable under the circumstances, including assumptions as to future events. These estimates form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. By their nature, estimates are subject to an inherent degree of uncertainty. Actual results may differ from our estimates. We believe that our judgment and assumptions with respect to the following significant accounting policies are most critical to the accounting estimates used in the preparation of our consolidated financial statements.
Revenue Recognition. Our principal sources of revenue are product sales of KALBITOR, license fees, funding for research and development, and milestones and royalties derived from collaboration and license agreements. In all instances, revenue is recognized only when the price is fixed or determinable, persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, collectability of the resulting receivable is reasonably assured and we have fulfilled the respective performance obligation.
Product Sales and Allowances
Revenues from product sales are recognized when title and risk of loss have passed to the customer. As such, product sales are recorded upon delivery of KALBITOR to the customer, our specialty and wholesale distributors. We establish reserves for trade distributor and prompt pay discounts, government rebates, a patient assistance programs, product returns and other applicable allowances. Reserves are based on estimates of the amount earned or to be claimed on the related sales. Our estimates are based on KALBITOR patient data, actual sales data, rebate claims and product returns. If actual results vary, we may need to adjust these estimates, which could have an effect on earnings in the period of the adjustment.
In addition to the discounts and rebates described above and classified as a reduction of revenue, we also maintain service contracts for patient service initiatives. We have established that the services are at fair value and represent a separate and identifiable benefit related to these services and, accordingly, have classified them as selling, general and administrative expense. If we had concluded that we did not receive a separate identifiable benefit or have sufficient evidence that the fair value did not exist for these services, we would have been required to classify these costs as a reduction of revenue.
Development and License Fee Revenues
We make significant assumptions and estimates relating to revenue recognition, which include the expected term of the agreement and around the best estimate of selling price of individual deliverables for multiple element arrangements. Our assumptions and estimates may prove to be inaccurate. Therefore, although we make every effort to ensure the accuracy of our estimates, any significant unanticipated changes in our estimates could have a material impact on revenues and our results of operations.
Collaboration Agreements. The Company enters into collaboration agreements with other companies for the research and development of therapeutic, diagnostic and separations products. The terms of the agreements may include non-refundable signing and licensing fees, funding for research and development, payments related to manufacturing services, milestone payments and royalties on any product sales derived from collaborations. These multiple element arrangements are analyzed to determine how the deliverables, which often include license and performance obligations such as research, steering committee and manufacturing services, are separated into units of accounting.
Before January 1, 2011, we evaluated license arrangements with multiple elements in accordance with ASC, 605-25 Revenue Recognition – Multiple-Element Arrangements. In October 2009, the FASB, issued ASU 2009-13 Revenue Arrangements with Multiple Deliverables, or ASU 2009-13, which amended the accounting standards for certain multiple element arrangements to:
| | ● | Provide updated guidance on whether multiple elements exist, how the elements in an arrangement should be separated and how the arrangement considerations should be allocated to the separate elements; |
| | ● | Require an entity to allocate arrangement consideration to each element based on a selling price hierarchy, also called the relative selling price method, where the selling price for an element is based on vendor-specific objective evidence (VSOE), if available; vendor objective evidence (VOE), if available and VSOE is not available; or the best estimate of selling price (BESP), if neither VSOE or VOE is available; |
| | ● | Eliminate the use of the residual method and require an entity to allocate arrangement consideration using the selling price hierarchy. |
The Company evaluates all deliverables within an arrangement to determine whether or not they provide value to the licensee on a stand-alone basis. Based on this evaluation, the deliverables are separated into units of accounting. If VSOE or VOE is not available to determine the fair value of a deliverable, the Company determines the best estimate of selling price associated with the deliverable. The arrangement consideration, including upfront license fees and funding for research and development, is allocated to the separate units based on relative fair value.
VSOE is based on the price charged when an element is sold separately and represents the actual price charged for that deliverable. When VSOE cannot be established, we attempt to establish the selling price of the elements of a license arrangement based on VOE. VOE is determined based on third party evidence for similar deliverables when sold separately. In circumstances when the Company charges a licensee for pass-through costs paid to external vendors for development services, these costs represent VOE.
When we are unable to establish the selling price of an element using VSOE or VOE, management determines BESP for that element. The objective of BESP is to determine the price at which we would transact a sale if the element within the license agreement was sold on a stand-alone basis. Our process for establishing BESP involves management’s judgment and considers multiple factors including discounted cash flows, estimated direct expenses and other costs and available data.
Based on the value allocated to each unit of accounting within an arrangement, upfront fees and other guaranteed payments are allocated to each unit based on relative value. The appropriate revenue recognition method is applied to each unit and revenue is accordingly recognized as each unit is delivered.
For agreements entered into prior to 2011, revenue related to upfront license fees was spread over the full period of performance under the agreement, unless the license was determined to provide value to the licensee on a stand-alone basis and the fair value of the undelivered performance obligations, typically including research or steering committee services was determinable.
Steering committee services that were not inconsequential or perfunctory and were determined to be performance obligations were combined with other research services or performance obligations required under an arrangement, if any, to determine the level of effort required in an arrangement and the period over which the Company expected to complete its aggregate performance obligations.
Whenever the Company determined that an arrangement should be accounted for as a single unit of accounting, it determined the period over which the performance obligations would be completed. Revenue is recognized using either an efforts-based or time-based (i.e. straight line) proportional performance method. The Company recognizes revenue using an efforts-based proportional performance method when the level of effort required to complete its performance obligations under an arrangement can be reasonably estimated and such performance obligations are provided on a best-efforts basis. Direct labor hours or full-time equivalents are typically used as the measurement of performance.
If the Company cannot reasonably estimate the level of effort to complete its performance obligations under an arrangement, then revenue under the arrangement is recognized on a straight-line basis over the period the Company is expected to complete its performance obligations.
Many of the Company's collaboration agreements entitle it to additional payments upon the achievement of performance-based milestones. For all milestones achieved prior to 2011, substantive milestones were included in the Company's revenue model when achievement of the milestone was met. Milestones that were tied to regulatory approval were not considered probable of being achieved until such approval was received. All milestones achieved after January 1, 2011 which are determined to be substantive milestones are recognized as revenue in the period in which they are met in accordance with Accounting Standards Update (ASU) No. 2010-17, Revenue Recognition – Milestone Method. Milestones tied to counter-party performance are not included in the Company’s revenue model until the performance conditions are met. Milestones determined to be non-substantive are allocated to each unit of accounting within an arrangement when met. The allocation of the milestone to each unit is based on relative value and revenue related to each unit is recognized accordingly.
Royalty revenue is recognized upon the sale of the related products provided the Company has no remaining performance obligations under the arrangement.
Costs of revenues related to product development and license fees are classified as research and development in the consolidated statements of operations and comprehensive loss.
Library Licenses. Standard terms of the proprietary phage display library agreements generally include non-refundable signing fees, license maintenance fees, development milestone payments, product license payments and royalties on product sales. Signing fees and maintenance fees are generally recognized on a straight-line basis over the term of the agreement. As milestones are achieved under a phage display library license, a portion of the milestone payment, equal to the percentage of the performance period completed when the milestone is achieved, multiplied by the amount of the milestone payment, will be recognized. The remaining portion of the milestone will be recognized over the remaining performance period on a straight-line basis. If the Company has no future obligations under the license, milestone payments under these license arrangements are recognized when the milestone is achieved. Product license payments are recognized as revenue when the license is issued if the Company has no future obligations under the agreement. If there are future obligations under the agreement, product license payments are recognized as revenue only to the extent of the fair value of the license. Amounts paid in excess of fair value are recognized in a manner similar to milestone payments. Royalty revenue is recognized upon the sale of the related products provided the Company has no remaining performance obligations under the arrangement.
Patent Licenses. The Company previously licensed its phage display patents on a non-exclusive basis to third parties for use in connection with the research and development of therapeutic, diagnostic, and other products. The last of these patents expired in November 2012. Even after patent expiration, the Company generally remains eligible under these patent licenses to receive milestones and/or royalties for products discovered prior to patent expiration, although certain existing patent licenses will no longer have a royalty obligation. The Company does not expect the expiration of these patents to have a material impact on its LFRP business.
Standard terms of the patent rights agreements include non-refundable signing fees, non-refundable license maintenance fees, development milestone payments and/or royalties on product sales. Signing fees and maintenance fees are generally recognized on a straight line basis over the term of the agreement or through the date of patent expiry, if shorter, except that in the case of perpetual patent licenses for which fees were recognized immediately if it was determined that the Company had no future obligations under the agreement and the payments were made upfront. As the Company has no remaining performance obligations under their patent license agreements, milestones are recognized as revenue in the period in which the milestone is achieved, and royalty revenue is recognized upon the sale of the related products.
Share-Based Compensation. We apply the provisions of Accounting Standards Codification (ASC) 718, "Accounting for Stock-Based Compensation" which required us to recognize the expense related to the fair value of stock-based compensation awards in our consolidated statement of operations. ASC 718 requires companies to estimate the fair value of stock-based awards on the date of grant using an option-pricing model. We use the Black-Scholes option pricing model. A number of assumptions are used by the Black-Scholes option-pricing model to compute the grant date fair value, including expected price volatility, option term, risk-free interest rate, and dividend yield. Expected volatilities are based on historical volatilities of our stock. The expected option term is derived from historical data on exercise behavior. The dividend yield is based on historical dividend payments.
The risk-free rate for periods within the contractual life of the option is based on the United States treasury yield curve in effect at the time of grant. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods in our consolidated statement of operations. We recognize expense on a straight-line basis over the requisite service period for each separately vesting portion of the award as if the award was, in-substance, multiple awards. The equity-based compensation expense recorded in future income statements could fluctuate based on the terms of the awards, the assumptions used in the valuation model, or the status of those employees receiving awards.
Note Payable Interest Expense. We record interest expense associated with our note payable with HC Royalty based on the effective interest rate. This calculation is based on management’s best estimate of future cash receipts expected from our LFRP. See HealthCare Royalty Partners in the Liquidity and Capital Resources section of Management’s Discussion and Analysis for more information on this debt arrangement.
Recent Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (FASB) or other standard setting bodies, which we adopt as of the specified effective date. Unless otherwise discussed, we believe that the impact of recently issued standards that are not yet effective will not have a material impact on our financial position or results of operations upon adoption.
| QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK |
None.
| FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
Index to Consolidated Financial Statements
To the Board of Directors and Stockholders of Dyax Corp.:
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations and comprehensive loss, changes in stockholders' equity (deficit) and cash flows present fairly, in all material respects, the financial position of Dyax Corp. and its subsidiaries at December 31, 2012 and 2011, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2012 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2012 based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Report on Internal Control over Financial Reporting appearing in Item 9A. Our responsibility is to express opinions on these financial statements and on the Company’s internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Boston, Massachusetts
March 4, 2013
Consolidated Balance Sheets
| | | December 31, 2012 | | | December 31, 2011 | |
| | | (In thousands, except share data) | |
| ASSETS | |
| Current assets: | | | | | | |
| Cash and cash equivalents | | $ | 20,018 | | | $ | 31,468 | |
| Short-term investments | | | 9,028 | | | | 26,036 | |
Accounts receivable, net | | | 7,507 | | | | 6,092 | |
| Inventory | | | 4,085 | | | | 2,121 | |
| Current portion of restricted cash | | | — | | | | 1,266 | |
| Other current assets | | | 2,159 | | | | 4,968 | |
| Total current assets | | | 42,797 | | | | 71,951 | |
| Fixed assets, net | | | 5,329 | | | | 4,881 | |
| Restricted cash | | | 1,100 | | | | 1,100 | |
| Other assets | | | 6,260 | | | | 5,443 | |
| Total assets | | $ | 55,486 | | | $ | 83,375 | |
| LIABILITIES AND STOCKHOLDERS' EQUITY (DEFICIT) | |
| Current liabilities: | | | | | | | | |
| Accounts payable and accrued expenses | | $ | 12,472 | | | $ | 15,318 | |
| Current portion of deferred revenue | | | 5,449 | | | | 6,637 | |
| Current portion of long-term obligations | | | 445 | | | | 101 | |
| Other current liabilities | | | — | | | | 1,709 | |
| Total current liabilities | | | 18,366 | | | | 23,765 | |
| Deferred revenue | | | 6,402 | | | | 9,265 | |
| Note payable | | | 78,061 | | | | 75,372 | |
| Long-term obligations | | | 931 | | | | — | |
| Deferred rent and other long-term liabilities | | | 3,286 | | | | 2,372 | |
| Total liabilities | | | 107,046 | | | | 110,774 | |
| Commitments and Contingencies (Notes 8, 11, 14) | | | | | | | | |
| Stockholders' equity (deficit): | | | | | | | | |
Preferred stock, $0.01 par value; 1,000,000 shares authorized; 0 shares issued and outstanding | | | | | | | — | |
Common stock, $0.01 par value; 200,000,000 shares authorized; 99,482,629 and 98,798,065 shares issued and outstanding at December 31, 2012 and 2011, respectively | | | 995 | | | | 988 | |
| Additional paid-in capital | | | 453,625 | | | | 448,527 | |
| Accumulated deficit | | | (506,186 | ) | | | (476,921 | ) |
| Accumulated other comprehensive income | | | 6 | | | | 7 | |
| Total stockholders' equity (deficit) | | | (51,560 | ) | | | (27,399 | ) |
| Total liabilities and stockholders' equity (deficit) | | $ | 55,486 | | | $ | 83,375 | |
The accompanying notes are an integral part of the consolidated financial statements.
Consolidated Statements of Operations and Comprehensive Loss
| | | Years Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
| | | (In thousands, except share and per share data) | |
| Revenues: | | | | | | | | | |
| Product sales, net | | $ | 39,783 | | | $ | 22,884 | | | $ | 8,835 | |
| Development and license fee revenues | | | 14,867 | | | | 25,853 | | | | 42,564 | |
| Total revenues | | | 54,650 | | | | 48,737 | | | | 51,399 | |
| | | | | | | | | | | | | |
| Costs and expenses: | | | | | | | | | | | | |
| Cost of product sales | | | 2,152 | | | | 1,223 | | | | 505 | |
| Research and development expenses | | | 30,028 | | | | 34,676 | | | | 31,522 | |
| Selling, general and administrative expenses | | | 39,915 | | | | 37,740 | | | | 33,583 | |
| Restructuring costs | | | 1,440 | | | | — | | | | — | |
| Total costs and expenses | | | 73,535 | | | | 73,639 | | | | 65,610 | |
| Loss from operations | | | (18,885 | ) | | | (24,902 | ) | | | (14,211 | ) |
| Other income (expense): | | | | | | | | | | | | |
| Interest and other income | | | 111 | | | | 554 | | | | 1,645 | |
| Interest expense | | | (10,491 | ) | | | (10,251 | ) | | | (11,937 | ) |
| | | | | | | | | | | | | |
| Total other expense, net | | | (10,380 | ) | | | (9,697 | ) | | | (10,292 | ) |
| Net loss | | | (29,265 | ) | | | (34,599 | ) | | | (24,503 | ) |
| Other comprehensive (loss) income: | | | | | | | | | | | | |
| Unrealized gain (loss) on investments | | | (1 | ) | | | (37 | ) | | | 29 | |
| Comprehensive loss | | | (29,266 | ) | | | (34,636 | ) | | | (24,474 | ) |
| Basic and diluted net loss per share: | | | | | | | | | | | | |
| Net loss | | $ | (0.30 | ) | | $ | (0.35 | ) | | $ | (0.26 | ) |
| Shares used in computing basic and diluted net loss per share | | | 98,991,056 | | | | 98,731,289 | | | | 93,267,850 | |
The accompanying notes are an integral part of the consolidated financial statements.
Consolidated Statements of Changes in Stockholders' Equity (Deficit)
For the years ended December 31, 2012, 2011 and 2010
(In thousands, except share data)
| | | | | | | | | | | | | | | Accumulated | | | | |
| | | Common Stock | | | Additional | | | | | | Other | | | | |
| | | | | | Par | | | Paid-in | | | Accumulated | | | Comprehensive | | | | |
| | | Shares | | | Value | | | Capital | | | Deficit | | | Income | | | Total | |
Balance at January 1, 2010 | | | 78,074,052 | | | $ | 781 | | | $ | 378,421 | | | $ | (417,819 | ) | | $ | 15 | | | $ | (38,602 | ) |
Exercise of stock options | | | 148,369 | | | | 1 | | | | 258 | | | | — | | | | — | | | | 259 | |
| Issuance of common stock for employee stock purchase plan | | | 99,934 | | | | 1 | | | | 186 | | | | — | | | | — | | | | 187 | |
Sale of common stock | | | 20,186,132 | | | | 202 | | | | 60,931 | | | | — | | | | — | | | | 61,133 | |
| Compensation expense associated with stock options | | | — | | | | — | | | | 4,130 | | | | — | | | | — | | | | 4,130 | |
| Unrealized gain on investments | | | — | | | | — | | | | — | | | | — | | | | 29 | | | | 29 | |
Net loss | | | — | | | | — | | | | — | | | | (24,503 | ) | | | — | | | | (24,503 | ) |
Balance at December 31, 2010 | | | 98,508,487 | | | | 985 | | | | 443,926 | | | | (442,322 | ) | | | 44 | | | | 2,633 | |
Exercise of stock options | | | 896 | | | | — | | | | 2 | | | | — | | | | — | | | | 2 | |
| Issuance of common stock for employee stock purchase plan | | | 137,167 | | | | 1 | | | | 216 | | | | — | | | | — | | | | 217 | |
Sale of common stock | | | 151,515 | | | | 2 | | | | 321 | | | | — | | | | — | | | | 323 | |
| Compensation expense associated with stock options | | | — | | | | — | | | | 4,062 | | | | — | | | | — | | | | 4,062 | |
| Unrealized loss on investments | | | — | | | | — | | | | — | | | | — | | | | (37 | ) | | | (37 | ) |
Net loss | | | — | | | | — | | | | — | | | | (34,599 | ) | | | — | | | | (34,599 | ) |
Balance at December 31, 2011 | | | 98,798,065 | | | | 988 | | | | 448,527 | | | | (476,921 | ) | | | 7 | | | | (27,399 | ) |
Exercise of stock options | | | 590,590 | | | | 6 | | | | 1,313 | | | | — | | | | — | | | | 1,319 | |
| Issuance of common stock for employee stock purchase plan | | | 93,974 | | | | 1 | | | | 122 | | | | — | | | | — | | | | 123 | |
| Compensation expense associated with stock options | | | — | | | | — | | | | 3,663 | | | | — | | | | — | | | | 3,663 | |
| Unrealized loss on investments | | | — | | | | — | | | | — | | | | — | | | | (1 | ) | | | (1 | ) |
Net loss | | | — | | | | — | | | | — | | | | (29,265 | ) | | | — | | | | (29,265 | ) |
Balance at December 31, 2012 | | | 99,482,629 | | | $ | 995 | | | $ | 453,625 | | | $ | (506,186 | ) | | $ | 6 | | | $ | (51,560 | ) |
The accompanying notes are an integral part of the consolidated financial statements.
Consolidated Statements of Cash Flows
| | | Years Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
| | | (In thousands) | |
| Cash flows from operating activities: | | | | | | | | | |
Net loss | | $ | (29,265 | ) | | $ | (34,599 | ) | | $ | (24,503 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
Amortization of investment premium/discount | | | 64 | | | | 229 | | | | 76 | |
| Depreciation and amortization of fixed assets and intangible assets | | | 1,060 | | | | 1,417 | | | | 1,484 | |
Non-cash interest expense | | | 2,784 | | | | 1,955 | | | | 945 | |
(Gain) loss on disposal of fixed assets | | | (110 | ) | | | — | | | | 43 | |
| Compensation expenses associated with stock-based compensation plans | | | 3,640 | | | | 4,030 | | | | 4,098 | |
Provision for doubtful accounts | | | (80 | ) | | | 35 | | | | 15 | |
Non-cash other income | | | (68 | ) | | | — | | | | — | |
| Changes in operating assets and liabilities | | | | | | | | | | | | |
Accounts receivable | | | (1,335 | ) | | | (810 | ) | | | (2,608 | ) |
Prepaid research and development and other assets | | | 2,809 | | | | (1,720 | ) | | | (1,143 | ) |
Inventory | | | (2,916 | ) | | | (5,201 | ) | | | (992 | ) |
Accounts payable and accrued expenses | | | (2,113 | ) | | | 1,426 | | | | (2,478 | ) |
Deferred revenue | | | (4,051 | ) | | | (5,434 | ) | | | (8,794 | ) |
Long-term deferred rent | | | 685 | | | | 1,417 | | | | (258 | ) |
Other | | | 414 | | | | 803 | | | | (16 | ) |
Net cash used in operating activities | | | (28,482 | ) | | | (36,452 | ) | | | (34,131 | ) |
| Cash flows from investing activities: | | | | | | | | | | | | |
Purchase of investments | | | (6,057 | ) | | | (3,021 | ) | | | (82,824 | ) |
Proceeds from maturity of investments | | | 23,000 | | | | 35,502 | | | | 47,003 | |
Purchase of fixed assets | | | (4,066 | ) | | | (1,694 | ) | | | (326 | ) |
Proceeds from sale of fixed assets | | | 200 | | | | — | | | | 38 | |
Restricted cash | | | 1,266 | | | | (178 | ) | | | 700 | |
Net cash provided by (used in) investing activities | | | 14,343 | | | | 30,609 | | | | (35,409 | ) |
| Cash flows from financing activities: | | | | | | | | | | | | |
Net proceeds from common stock offerings | | | — | | | | 323 | | | | 61,133 | |
Proceeds from note payable | | | 1,382 | | | | 19,850 | | | | — | |
Repayment of long-term obligations | | | (135 | ) | | | (1,682 | ) | | | (2,824 | ) |
| Proceeds from the issuance of common stock under employee stock purchase plan and exercise of stock options | | | 1,442 | | | | 219 | | | | 446 | |
Net cash provided by financing activities | | | 2,689 | | | | 18,710 | | | | 58,755 | |
| Effect of foreign currency translation on cash balances | | | — | | | | — | | | | — | |
Net (decrease) increase in cash and cash equivalents | | | (11,450 | ) | | | 12,867 | | | | (10,785 | ) |
Cash and cash equivalents at beginning of the period | | | 31,468 | | | | 18,601 | | | | 29,386 | |
Cash and cash equivalents at end of the period | | $ | 20,018 | | | $ | 31,468 | | | $ | 18,601 | |
| Supplemental disclosure of cash flow information: | | | | | | | | | | | | |
Interest paid | | $ | 9,364 | | | $ | 9,108 | | | $ | 12,211 | |
The accompanying notes are an integral part of the consolidated financial statements.
Notes to Consolidated Financial Statements
Dyax Corp. (Dyax or the Company) is a biopharmaceutical company with two business elements:
| | ● | Plasma Kallikrein-Mediated Angioedema Portfolio |
The principal focus of the Company's efforts is to identify, develop and commercialize treatments for angioedemas that are identified as plasma kallikrein-mediated, which the Company refers to as PKM angioedemas, including hereditary angioedema (HAE) and idiopathic angioedema.
The Company developed KALBITOR® (ecallantide) on its own and since February 2010, the Company has been selling it in the United States for the treatment of acute attacks of HAE. Outside of the United States, the Company has established partnerships to obtain regulatory approval for and to commercialize KALBITOR in certain markets and is evaluating opportunities in others.
The Company is expanding its franchise for the treatment of PKM angioedemas in the following ways:
| | ● | Development of diagnostic strategies to assist in the differentiation between histamine-mediated and PKM angioedema. |
| | ● | Continuing the development of DX-2930, a fully human monoclonal antibody inhibitor of plasma kallikrein, which could be a candidate to treat prophylactically PKM angioedemas. |
| | ● | Phage Display Licensing and Funded Research Program |
The Company leverages its proprietary phage display technology through its Licensing and Funded Research Program, referred to as the LFRP. This program has provided the Company a portfolio of product candidates being developed by its licensees, which currently includes 13 product candidates in various stages of clinical development, including three in Phase 3 trials, for which we are eligible to receive future royalties and / or milestone payments.
The Company expects that existing cash, cash equivalents, and short-term investments, together with anticipated cash flow from existing development, collaborations and license agreements and product sales of KALBITOR, will be sufficient to support the Company's current operations into 2014. If over the longer term, the Company's cash requirements exceed its current expectations or if the Company generates less revenue than it expects, the Company will need additional funds. The Company may seek additional funding through collaborative arrangements, public or private financings, or other means. However, the Company may not be able to obtain financing on acceptable terms or at all, and the Company may not be able to enter into additional collaborative arrangements. Arrangements with collaborators or others may require the Company to relinquish rights to certain of its technologies, product candidates or products. The terms of any financing may adversely affect the holdings or the rights of the Company's stockholders. If the Company needs additional funds and it is unable to obtain funding on a timely basis, the Company may be required to significantly curtail its research, development or commercialization programs in an effort to provide sufficient funds to continue its operations, which could adversely affect its business prospects.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
2. Accounting Policies
Basis of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and the Company's European research subsidiaries Dyax S.A. and Dyax BV. All inter-company accounts and transactions have been eliminated.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make certain estimates and assumptions that affect the amounts of assets and liabilities reported and disclosure of contingent assets and liabilities at the dates of the financial statements and the reported amounts of revenue and expenses during the reporting periods. The significant estimates and assumptions in these financial statements include revenue recognition, product sales allowances, useful lives with respect to long lived assets, valuation of stock options, accrued expenses and tax valuation reserves. Actual results could differ from those estimates.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash, cash equivalents, short-term investments and trade accounts receivable. At December 31, 2012 and 2011, approximately 86% and 61% , respectively, of the Company's cash, cash equivalents and short-term investments were invested in money market funds backed by U.S. Treasury obligations, U.S. Treasury notes and bills, and obligations of United States government agencies held by one financial institution. The Company maintains balances in various operating accounts in excess of federally insured limits.
The Company provides most of its services and licenses its technology to pharmaceutical and biomedical companies worldwide, and makes all product sales to its distributors. Concentrations of credit risk with respect to trade receivable balances associated the Company’s development and license fee revenue are usually limited on an ongoing basis, due to the diverse number of licensees and collaborators comprising the Company's customer base. Trade receivable balances associated with the Company’s product sales are comprised of a few customers due to the Company’s limited distribution network. The Company completes ongoing credit evaluations of their customers. As of December 31, 2012, two customers accounted for 50% (Walgreens) and 34% (US Bio) of the accounts receivable balance. Two customers accounted for approximately 43% (US Bio) and 34% (Walgreens), of the Company's accounts receivable balance as of December 31, 2011.
The Company’s accounts receivable balance was net of $0 and $115,000 of allowance for doubtful accounts as of December 31, 2012 and 2011, respectively.
Cash and Cash Equivalents
All highly liquid investments purchased with an original maturity of ninety days or less are considered to be cash equivalents. Cash and cash equivalents consist principally of cash, money market and U.S. Treasury funds.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Investments
Short-term investments primarily consist of investments with original maturities greater than ninety days and remaining maturities less than one year at period end. The Company has also classified its investments with maturities beyond one year as short-term, based on their highly liquid nature and because such marketable securities represent the investment of cash that is available for current operations. The Company considers its investment portfolio of investments available-for-sale. Accordingly, these investments are recorded at fair value, which is based on quoted market prices. As of December 31, 2012, the Company's investments consisted of U.S. Treasury notes and bills with an amortized cost and estimated fair value of $9.0 million, and had an unrealized gain of $6,000. As of December 31, 2011, the Company's investments consisted of United States Treasury notes and bills with an estimated fair value and amortized cost of $26.0 million, and had an unrealized gain of $7,000, which is recorded in other comprehensive income on the accompanying consolidated balance sheet.
Inventories
Inventories are stated at the lower of cost or market with cost determined under the first-in, first-out, or FIFO, basis. The Company evaluates inventory levels and would write-down inventory that is expected to expire prior to being sold, inventory that has a cost basis in excess of its expected net realizable value, inventory in excess of expected sales requirements, or inventory that fails to meet commercial sale specifications, through a charge to cost of product sales. Included in the cost of inventory are employee stock-based compensation costs capitalized under ASC 718.
Fixed Assets
Property and equipment are recorded at cost and depreciated over the estimated useful lives of the related assets using the straight-line method. Laboratory and production equipment, furniture and office equipment are depreciated over a three to seven year period. Leasehold improvements are stated at cost and are amortized over the lesser of the non-cancelable term of the related lease or their estimated useful lives. Leased equipment is amortized over the lesser of the life of the lease or their estimated useful lives. Maintenance and repairs are charged to expense as incurred. When assets are retired or otherwise disposed of, the cost of these assets and related accumulated depreciation and amortization are eliminated from the balance sheet and any resulting gains or losses are included in operations in the period of disposal.
The Company records all proceeds received from the lessor for tenant improvements under the terms of its operating lease as deferred rent. The amounts are amortized on a straight-line basis over the term of the lease as an offset to rent expense.
Impairment of Long-Lived Assets
The Company reviews its long-lived assets for impairment whenever events or changes in business circumstances indicate that the carrying amount of assets may not be fully recoverable or that the useful lives of these assets are no longer appropriate. Each impairment test is based on a comparison of the undiscounted cash flow to the recorded value of the asset. If impairment is indicated, the asset is written down to its estimated fair value on a discounted cash flow basis.
Revenue Recognition
The Company’s principal sources of revenue are product sales of KALBITOR, license fees, funding for research and development, and milestones and royalties derived from collaboration and license agreements. In all instances, revenue is recognized only when the price is fixed or determinable, persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, collectability of the resulting receivable is reasonably assured and the Company has no further performance obligations.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Product Sales and Allowances
Product Sales. Product sales are generated from the sale of KALBITOR to the Company’s wholesale and specialty distributors, and are recorded upon delivery when title and risk of loss have passed to the customer. Product sales are recorded net of applicable reserves for trade prompt pay and other discounts, government rebates, patient assistance programs, product returns and other applicable allowances.
Product Sales Allowances. The Company establishes reserves for trade distributor and prompt pay discounts, government rebates, patient assistance programs, product returns and other applicable allowances. Reserves established for these discounts and allowances are classified as a reduction of accounts receivable (if the amount is payable to the customer) or a liability (if the amount is payable to a party other than the customer).
Allowances against receivable balances primarily relate to prompt payment discounts and are recorded at the time of sale, resulting in a reduction in product sales revenue. Accruals related to government rebates, patient financial assistance programs, product returns and other applicable allowances are recognized at the time of sale, resulting in a reduction in product sales revenue and the recording of an increase in accrued expenses.
The Company maintains service contracts with its distributors. Accounting standards related to consideration given by a vendor to a customer, including a reseller of a vendor’s product, specify that each consideration given by a vendor to a customer is presumed to be a reduction of the selling price. Consideration should be characterized as a cost if the company receives, or will receive, an identifiable benefit in exchange for the consideration, and fair value of the benefit can be reasonably estimated. The Company has established that patient support services are at fair value and represent a separate and identifiable benefit related to these services and, accordingly, has classified them as selling, general and administrative expense.
Prompt Payment and Other Discounts. The Company offers a prompt payment discount to its United States distributors. Since the Company expects that these distributors will take advantage of this discount, the Company accrues 100% of the prompt payment discount that is based on the gross amount of each invoice, at the time of sale. The accrual is adjusted quarterly to reflect actual earned discounts.
Government Rebates and Chargebacks. The Company estimates reductions to product sales for Medicaid and Veterans' Administration (VA) programs and the Medicare Part D Coverage Gap Program, as well as with respect to certain other qualifying federal and state government programs. The Company estimates the amount of these reductions based on KALBITOR patient data, actual sales data and rebate claims. These allowances are adjusted each period based on actual experience.
Medicaid rebate reserves relate to the Company’s estimated obligations to states under the established reimbursement arrangements of each applicable state. Rebate accruals are recorded during the same period in which the related product sales are recognized. Actual rebate amounts are determined at the time of claim by the state, and the Company will generally make cash payments for such amounts after receiving billings from the state.
VA rebates or chargeback reserves represent the Company’s estimated obligations resulting from contractual commitments to sell products to qualified healthcare providers at a price lower than the list price charged to the Company’s distributor. The distributor will charge the Company for the difference between what the distributor pays for the product and the ultimate selling price to the qualified healthcare provider. Rebate accruals are established during the same period in which the related product sales are recognized. Actual chargeback amounts for Public Health Service are determined at the time of resale to the qualified healthcare provider from the distributor, and the Company will generally issue credits for such amounts after receiving notification from the distributor.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
The Company offers a financial assistance program, which involves the use of a patient voucher, for qualified KALBITOR patients in order to aid a patient’s access to KALBITOR. The Company estimates its liability from this voucher program based on actual redemption rates.
Although allowances and accruals are recorded at the time of product sale, certain rebates are typically paid out, on average, up to six months or longer after the sale. Reserve estimates are evaluated quarterly and if necessary, adjusted to reflect actual results. Any such adjustments will be reflected in the Company’s operating results in the period of the adjustment.
Product Returns. Allowances for product returns are recorded during the period in which the related product sales are recognized, resulting in a reduction to product revenue. The Company does not provide its distributors with a general right of product return. The Company permits returns if the product is damaged or defective when received by customers or if the product has expired. The Company estimates product returns based upon actual returns history and data provided by a distributor.
Development and License Fee Revenues
Collaboration Agreements. The Company enters into collaboration agreements with other companies for the research and development of therapeutic, diagnostic and separations products. The terms of the agreements may include non-refundable signing and licensing fees, funding for research and development, payments related to manufacturing services, milestone payments and royalties on any product sales derived from collaborations. These multiple element arrangements are analyzed to determine how the deliverables, which often include license and performance obligations such as research, steering committee and manufacturing services, are separated into units of accounting.
Before January 1, 2011, the Company evaluated license arrangements with multiple elements in accordance with ASC, 605-25 Revenue Recognition – Multiple-Element Arrangements. In October 2009, the FASB issued ASU 2009-13 Revenue Arrangements with Multiple Deliverables, or ASU 2009-13, which amended the accounting standards for certain multiple element arrangements to:
| | ● | Provide updated guidance on whether multiple elements exist, how the elements in an arrangement should be separated and how the arrangement considerations should be allocated to the separate elements; |
| | ● | Require an entity to allocate arrangement consideration to each element based on a selling price hierarchy, also called the relative selling price method, where the selling price for an element is based on vendor-specific objective evidence (VSOE), if available; vendor objective evidence (VOE), if available and VSOE is not available; or the best estimate of selling price (BESP), if neither VSOE or VOE is available; |
| | ● | Eliminate the use of the residual method and require an entity to allocate arrangement consideration using the selling price hierarchy. |
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
The Company evaluates all deliverables within an arrangement to determine whether or not they provide value to the licensee on a stand-alone basis. Based on this evaluation, the deliverables are separated into units of accounting. If VSOE or VOE is not available to determine the fair value of a deliverable, the Company determines the best estimate of selling price associated with the deliverable. The arrangement consideration, including upfront license fees and funding for research and development, is allocated to the separate units based on relative fair value.
VSOE is based on the price charged when an element is sold separately and represents the actual price charged for that deliverable. When VSOE cannot be established, the Company attempts to establish the selling price of the elements of a license arrangement based on VOE. VOE is determined based on third party evidence for similar deliverables when sold separately. In circumstances when the Company charges a licensee for pass-through costs paid to external vendors for development services, these costs represent VOE.
When the Company is unable to establish the selling price of an element using VSOE or VOE, management determines BESP for that element. The objective of BESP is to determine the price at which the Company would transact a sale if the element within the license agreement was sold on a stand-alone basis. The Company’s process for establishing BESP involves management’s judgment and considers multiple factors including discounted cash flows, estimated direct expenses and other costs and available data.
Based on the value allocated to each unit of accounting within an arrangement, upfront fees and other guaranteed payments are allocated to each unit based on relative value. The appropriate revenue recognition method is applied to each unit and revenue is accordingly recognized as each unit is delivered.
For agreements entered into prior to 2011, revenue related to upfront license fees was spread over the full period of performance under the agreement, unless the license was determined to provide value to the licensee on a stand-alone basis and the fair value of the undelivered performance obligations, typically including research or steering committee services was determinable.
Steering committee services that were not inconsequential or perfunctory and were determined to be performance obligations were combined with other research services or performance obligations required under an arrangement, if any, to determine the level of effort required in an arrangement and the period over which the Company expected to complete its aggregate performance obligations.
Whenever the Company determined that an arrangement should be accounted for as a single unit of accounting, it determined the period over which the performance obligations would be completed. Revenue is recognized using either an efforts-based or time-based (i.e. straight-line) proportional performance method. The Company recognizes revenue using an efforts-based proportional performance method when the level of effort required to complete its performance obligations under an arrangement can be reasonably estimated and such performance obligations are provided on a best-efforts basis. Direct labor hours or full-time equivalents are typically used as the measurement of performance.
If the Company cannot reasonably estimate the level of effort to complete its performance obligations under an arrangement, then revenue under the arrangement is recognized on a straight-line basis over the period the Company is expected to complete its performance obligations.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Many of the Company's collaboration agreements entitle it to additional payments upon the achievement of performance-based milestones. For all milestones achieved prior to 2011, substantive milestones were included in the Company's revenue model when the milestone was achieved. Milestones that were tied to regulatory approval were not considered probable of being achieved until such approval was received. All milestones achieved after January 1, 2011 which are determined to be substantive milestones are recognized as revenue in the period in which they are met in accordance with Accounting Standards Update (ASU) No. 2010-17, Revenue Recognition – Milestone Method. Milestones tied to counter-party performance are not included in the Company’s revenue model until performance conditions are met. Milestones determined to be non-substantive are allocated to each unit of accounting within an arrangement when met. The allocation of the milestone to each unit is based on relative value and revenue related to each unit is recognized accordingly.
Royalty revenue is recognized upon the sale of the related products provided the Company has no remaining performance obligations under the arrangement.
Costs of revenues related to product development and license fees are classified as research and development in the consolidated statements of operations and comprehensive loss.
Library Licenses. Standard terms of the proprietary phage display library agreements generally include non-refundable signing fees, license maintenance fees, development milestone payments, product license payments and royalties on product sales. Signing fees and maintenance fees are generally recognized on a straight line basis over the term of the agreement as deliverables within these arrangements are determined to not provide the licensee with value on a stand-alone basis and therefore are accounted for as a single unit of accounting. As milestones are achieved under a phage display library license, a portion of the milestone payment, equal to the percentage of the performance period completed when the milestone is achieved, multiplied by the amount of the milestone payment, will be recognized. The remaining portion of the milestone will be recognized over the remaining performance period on a straight-line basis. If the Company has no future obligations under the license, milestone payments under these license arrangements are recognized when the milestone is. Product license payments, which are optional to the licensee, are substantive and therefore are excluded from the initial allocation of the arrangement consideration. These payments are recognized as revenue when the license is issued upon exercise of the licensee’s option, if the Company has no future obligations under the agreement. If there are future obligations under the agreement, product license payments are recognized as revenue only to the extent of the fair value of the license. Amounts paid in excess of fair value are recognized in a manner similar to milestone payments. Royalty revenue is recognized upon the sale of the related products provided the Company has no remaining performance obligations under the arrangement.
Payments received that have not met the appropriate criteria for revenue recognition are recorded as deferred revenue.
Patent Licenses. The Company previously licensed its phage display patents on a non-exclusive basis to third parties for use in connection with the research and development of therapeutic, diagnostic, and other products. The last of these patents expired in November 2012. Even after patent expiration, the Company generally remains eligible under these patent licenses to receive milestones and/or royalties for products discovered prior to patent expiration, although certain existing patent licenses will no longer have a royalty obligation. The Company does not expect the expiration of these patents to have a material impact on its LFRP business.
Standard terms of the patent rights agreements include non-refundable signing fees, non-refundable license maintenance fees, development milestone payments and/or royalties on product sales. Signing fees and maintenance fees are generally recognized on a straight line basis over the term of the agreement or through the date of patent expiry, if shorter, except that in the case of perpetual patent licenses for which fees were recognized immediately if it was determined that the Company had no future obligations under the agreement and the payments were made upfront. As the Company has no remaining performance obligations under their patent license agreements, milestones are recognized as revenue in the period in which the milestone is achieved, and royalty revenue is recognized upon the sale of the related products.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Cost of Product Sales
Cost of product sales includes costs to procure, manufacture and distribute KALBITOR and manufacturing royalties. Costs associated with the manufacture of KALBITOR prior to regulatory approval were expensed when incurred as a research and development cost and accordingly, KALBITOR units sold during the years ended December 31, 2012, 2011 and 2010 do not reflect the full cost of drug manufacturing.
Research and Development
Research and development costs include all direct costs, including salaries and benefits for research and development personnel, outside consultants, costs of clinical trials, sponsored research, clinical trials insurance, other outside costs, depreciation and facility costs related to the development of drug candidates. Costs of revenues related to product development and license fees are classified as research and development in the consolidated statements of operations and comprehensive loss.
Income Taxes
The Company utilizes the asset and liability method of accounting for income taxes in accordance with ASC 740. Under this method, deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the carrying amounts and the tax basis of assets and liabilities using the enacted statutory tax rates. At December 31, 2012 and 2011, there were no unrecognized tax benefits.
The Company accounts for uncertain tax positions using a "more-likely-than-not" threshold for recognizing and resolving uncertain tax positions. The evaluation of uncertain tax positions is based on factors that include, but are not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. The Company evaluates uncertain tax positions on a quarterly basis and adjusts the level of the liability to reflect any subsequent changes in the relevant facts surrounding the uncertain positions.
Translation of Foreign Currencies
Assets and liabilities of the Company's foreign subsidiaries are translated at period end exchange rates. Amounts included in the statements of operations are translated at the average exchange rate for the period. All currency translation adjustments are recorded to other income (expense) in the consolidated statement of operations. For the year ended December 31, 2012 the Company recorded other income of $15,000 and for the years ended December 31, 2011 and 2010 the Company recorded other expense of $3,000 and $32,000, respectively, for the translation of foreign currency.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Share-Based Compensation
The Company’s share-based compensation program consists of share-based awards granted to employees in the form of stock options and restricted stock units, as well as its 1998 Employee Stock Purchase Plan, as amended (the Purchase Plan). The Company’s share-based compensation expense is recorded in accordance with ASC 718.
Income or Loss Per Share
The Company presents two earnings or loss per share (EPS) amounts, basic and diluted in accordance with ASC 260. Basic earnings or loss per share is computed using the weighted average number of shares of common stock outstanding. Diluted net loss per share does not differ from basic net loss per share since potential common shares from the exercise of stock options, warrants or rights under the Purchase Plan are anti-dilutive for the years ended December 31, 2012, 2011 and 2010, and therefore, are excluded from the calculation of diluted net loss per share.
The weighted average of stock options and warrants outstanding totaled 12,278,657, 11,456,758 and 10,067,486 at December 31, 2012, 2011, and 2010, respectively.
Comprehensive Income (Loss)
The Company accounts for comprehensive income (loss) under ASC 220, Comprehensive Income, which established standards for reporting and displaying comprehensive income (loss) and its components in a full set of general purpose financial statements. The statement requires that all components of comprehensive income (loss) be reported in a financial statement that is displayed with the same prominence as other financial statements.
Accumulated other comprehensive income (loss) consists entirely of changes in unrealized gains (losses) on investments.
Business Segments
The Company discloses business segments under ASC 280, Segment Reporting. The statement established standards for reporting information about operating segments and disclosures about products and services, geographic areas and major customers. The Company operates as one business segment within predominantly one geographic area.
Recent Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the FASB or other standard setting bodies, which are adopted by the Company as of the specified effective date. Unless otherwise discussed, the Company believes that recently issued standards that are not yet effective will not have a material impact on its financial position or results of operations upon adoption.
3. SIGNIFICANT TRANSACTIONS
Sigma-Tau
In June 2010, the Company entered into a strategic collaboration agreement with Sigma-Tau Rare Diseases S.A. (as successor-in-interest to Defiante Farmaceutica S.A.) (Sigma-Tau) to develop and commercialize subcutaneous ecallantide for the treatment of HAE and other therapeutic indications throughout Europe, North Africa, the Middle East and Russia. In December 2010, the original agreement was amended to expand the partnership to commercialize KALBITOR for the treatment of HAE in Australia and New Zealand (the first amendment). In May 2011, the Company further amended its agreement with Sigma-Tau to include development and commercialization rights in Latin America (excluding Mexico), the Caribbean and certain Asian territories (the second amendment). Three subsequent amendments to this agreement eliminated rights that Dyax had previously granted to Sigma-Tau for the Asian territories, the Middle East, North Africa, Latin America and the Caribbean. This collaboration arrangement now includes rights throughout Europe, Russia, Australia and New Zealand.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Under the terms of the original agreement, Sigma-Tau made a $2.5 million upfront payment. In addition, Sigma-Tau purchased 636,132 shares of the Company's common stock at a price of $3.93 per share, which represented a 50% premium over the 20-day average closing price through June 17, 2010, for an aggregate purchase price of $2.5 million.
Under the terms of the first amendment, Sigma-Tau made an additional $500,000 upfront payment to the Company and also purchased 151,515 shares of the Company's common stock at a price of $3.30 per share, which represented a 50% premium over the 20-day average closing price through December 20, 2010, for an aggregate purchase price of $500,000. Both payments were received in January 2011.
Under the terms of a second amendment, Sigma-Tau made an additional upfront payment of $4.0 million in 2011 and was required to make an additional $3.0 million non-refundable payment to the Company by December 31, 2011. Under a third amendment, upon elimination of Sigma-Tau’s rights to certain Asian territories, the $3.0 million payment obligation was eliminated, as were the future milestones and royalties related to these territories.
Under the terms of the fourth and fifth amendments, Sigma-Tau’s rights to the Middle East, Latin America and the Caribbean were eliminated. The Company agreed to make payments to Sigma-Tau ranging from 5%-12.5% of the amounts received by the Company as a result of any future product sales for certain countries in these territories.
The Company is eligible to receive up to $100 million in development and sales milestones related to ecallantide and royalties equal to 41% of net sales of product, as adjusted for product costs, in all licensed territories. Sigma-Tau will pay costs associated with regulatory approval and commercialization in the licensed territories. In addition, the Company and Sigma-Tau will share equally the costs for all development activities for optional future indications developed in partnership with Sigma-Tau in the territories covered under the initial Sigma-Tau agreement. The partnership agreement may be terminated by Sigma-Tau, at will, upon 6 months’ prior written notice. Either party may terminate the partnership agreement in the event of an uncured material breach or declaration or filing of bankruptcy by the other party.
Prior to the second amendment in May 2011, revenue related to this multiple element arrangement was being recognized in accordance with ASC 605. The Company evaluated the terms of the second amendment relative to the entire arrangement and determined the amendment to be a material modification to the existing agreement for financial reporting purposes. As a result, the Company evaluated the entire arrangement under the guidance of ASU No. 2009-13 which was adopted in 2011.
Under the terms of the original agreement and first amendment, the Company analyzed this multiple element arrangement in accordance with ASC 605 and evaluated whether the performance obligations under this agreement, including the product license and development, steering committee, and manufacturing services should be accounted for as a single unit or multiple units of accounting. The Company determined that there were two units of accounting. The first unit of accounting included the product license, the committed future development services and the steering committee involvement. These deliverables were grouped into one unit of accounting due to the lack of objective and reliable evidence of fair value. The second unit of accounting related to the manufacturing services, and was determined to meet all of the criteria to be a separate unit of accounting. The Company had the ability to estimate the scope and timing of its involvement in the future development of the program, as the Company's obligations under the development period are clearly defined. Therefore, the Company recognized revenue related to the first unit of accounting utilizing a proportional performance model based on the actual effort performed in proportion to the total estimated level of effort. Under this model, the Company estimated the level of effort to be expended over the term of the agreement and recognized revenue based on the lesser of the amount calculated based on proportional performance of total expected revenue or the amount of non-refundable payments earned. As of the date of the second amendment, $4.8 million of revenue had been recognized for the first unit of accounting and $2.4 million of deferred revenue remained. To date, no revenue has been recognized related to manufacturing services, as no such services have been provided.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
As the second amendment represented a material modification to the existing agreement under applicable accounting rules, the Company re-evaluated the entire arrangement under ASU No. 2009-13, and determined all undelivered items under the agreement and divided them into separate units of accounting based on whether the deliverable provided stand-alone value to the licensee. These units of accounting consist of (i) the license to develop and commercialize ecallantide for the treatment of HAE and other therapeutic indications in the territories granted under the original agreement and first amendment, (ii) the license to develop and commercialize ecallantide for the treatment of HAE and other therapeutic indications in the territories granted under the second amendment, (iii) steering committee services and (iv) committed future development services. The Company then determined the best estimate selling price (BESP) for the license and steering committee services and the fair value of committed future development services was determined using vendor objective evidence. The Company’s process for determining BESP involves management’s judgment and includes factors such as discounted cash flows, estimated direct expenses and other costs and available data.
The upfront fee of $4.0 million, the non-refundable payment of $3.0 million due in December 2011 and $2.4 million of previously deferred revenue under the Sigma-Tau contracts were allocated to the units of accounting based upon relative fair value. The $9.2 million allocated to the licenses was recognized during the second quarter of 2011, as the licenses had been delivered.
In the fourth quarter of 2011, based on the terms of the third amendment, Sigma-Tau’s $3.0 million payment obligation was eliminated and the full $3.0 million in revenue was recorded as a reduction of revenue. In addition, during the fourth quarter of 2011, the Company recognized $1.0 million related to a regulatory filing milestone for the Australian territory which was determined to be substantive based on the level of effort and involvement required by the Company for the achievement of this milestone.
Revenue related to steering committee services of $190,000 was deferred and is being recognized under the proportional performance model, as meetings are held through the estimated development period for ecallantide in the Sigma-Tau territories. Revenues associated with future committed development services will be recognized as incurred and billed to Sigma-Tau for reimbursement. As future milestones are achieved and to the extent they involve substantial effort on the Company’s part, revenue will be recognized in the period in which the milestone is achieved. The manufacturing services were determined to represent a contingent deliverable and, as such, have been excluded from the current revenue model.
The Company recognized revenue of approximately $204,000, $10.5 million and $2.2 million related to the Sigma-Tau agreement, as amended, for the years ended December 31, 2012, 2011 and 2010, respectively. As of December 31, 2012 and 2011, the Company has deferred $95,000 and $158,000, respectively, of revenue related to this arrangement, which is recorded in deferred revenue on the accompanying consolidated balance sheets at such dates. The deferred revenue balance at December 31, 2012, relates to the joint steering committee obligation which is estimated to continue until 2014. As of December 31, 2012 and 2011, the Company had receivable balances due from Sigma-Tau of $8,000 and $65,000, respectively.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
CMIC
In 2010, the Company entered into an agreement with CMIC Co., Ltd, (CMIC) to develop and commercialize subcutaneous ecallantide for the treatment of HAE and other angioedema indications in Japan.
Under the terms of the agreement, the Company received a $4.0 million upfront payment. The Company is also eligible to receive up to $102 million in development and sales milestones for ecallantide in HAE and other angioedema indications and royalties of 20%-24% of net product sales. CMIC is solely responsible for all costs associated with development, regulatory activities, and commercialization of ecallantide for all angioedema indications in Japan. CMIC will purchase drug product from the Company on a cost-plus basis for clinical and commercial supply.
The Company analyzed this multiple element arrangement in accordance with ASC 605 and evaluated whether the performance obligations under this agreement, including the product license, development of ecallantide for the treatment of HAE and other angioedema indications in Japan, steering committee, and manufacturing services should be accounted for as a single unit or multiple units of accounting. The Company determined that there were two units of accounting. The first unit of accounting includes the product license, the committed future development services and the steering committee involvement. The second unit of accounting relates to the manufacturing services. At this time the scope and timing of the future development of ecallantide for the treatment of HAE and other indications in the CMIC territory are the joint responsibility of the Company and CMIC and therefore, the Company cannot reasonably estimate the level of effort required to fulfill its obligations under the first unit of accounting. As a result, the Company is recognizing revenue under the first unit of accounting on a straight-line basis over the estimated development period of ecallantide for the treatment of HAE in the CMIC territory through 2016.
The Company recognized revenue of approximately $755,000, $595,000 and $148,000 related to this agreement for the years ended December 31, 2012, 2011 and 2010, respectively. As of December 31, 2012 and 2011, the Company has deferred approximately $2.5 million and $3.3 million, respectively, of revenue related to this arrangement, which is recorded in deferred revenue on the accompanying consolidated balance sheets.
Sale of Xyntha Royalty Rights
In 2010, the Company sold its rights to royalties and other payments related to the commercialization of the product Xyntha®, which was developed by one of the Company's licensees under the Company's LFRP. Under the terms of this sale, the Company received an upfront cash payment of $9.8 million and earned additional milestones payments of $1.5 million in 2010 and $500,000 in 2011, based on product sales for each respective year. A portion of the cash payments received were required to be applied to the Company's loan with Healthcare Royalty Partners (see Note 8 – Note Payable), totaling a $2.2 million principal reduction and interest expense of $1.4 million. A similar proportion of the $500,000 sales milestone payment was also applied to the loan. The Company determined that it had no substantive future obligations under the arrangement. During the years ended December 31, 2010 and 2011,the Company recognized $11.3 million and $500,000 of revenue under this arrangement, respectively.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Cubist Pharmaceuticals Inc.
In 2008, the Company entered into an exclusive license and collaboration agreement with Cubist Pharmaceuticals, Inc. (Cubist), for the development and commercialization in North America and Europe of the intravenous formulation of ecallantide for the reduction of blood loss during surgery. Under this agreement, Cubist assumed responsibility for all further development and costs associated with ecallantide in the licensed indications in the Cubist territory and the Company received $17.5 million in upfront license and milestone fees.
In 2010, Cubist announced its plan to stop investing in the clinical development of ecallantide and terminated the 2008 agreement. Based upon Cubist's decision, $13.8 million of deferred revenue was recognized as revenue during the year ended December 31, 2010, as the development period had ended.
4. Fair Value Measurements
The following tables present information about the Company's financial assets that have been measured at fair value as of December 31, 2012 and 2011, in thousands, and indicate the fair value hierarchy of the valuation inputs utilized to determine such fair value. In general, fair values determined by Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities. Fair values determined by Level 2 inputs utilize observable inputs other than Level 1 prices, such as quoted prices, for similar assets or liabilities, quoted prices in markets that are not active or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the related assets or liabilities. Fair values determined by Level 3 inputs are unobservable data points for the asset or liability, and includes situations where there is little, if any, market activity for the asset or liability.
| Description | | December 31, 2012 | | | Quoted Prices in Active Markets (Level 1) | | | Significant Other Observable Inputs (Level 2) | | | Significant Unobservable Inputs (Level 3) | |
| Assets: | | | | | | | | | | | | |
| Cash equivalents | | $ | 15,910 | | | $ | 15,910 | | | $ | — | | | $ | — | |
| Marketable debt securities | | | 9,028 | | | | — | | | | 9,028 | | | | — | |
| Total | | $ | 24,938 | | | $ | 15,910 | | | $ | 9,028 | | | $ | — | |
| Description | | December 31, 2011 | | | Quoted Prices in Active Markets (Level 1) | | | Significant Other Observable Inputs (Level 2) | | | Significant Unobservable Inputs (Level 3) | |
| Assets: | | | | | | | | | | | | |
| Cash equivalents | | $ | 8,825 | | | $ | 8,825 | | | $ | — | | | $ | — | |
| Marketable debt securities | | | 26,036 | | | | — | | | | 26,036 | | | | — | |
| Total | | $ | 34,861 | | | $ | 8,825 | | | $ | 26,036 | | | $ | — | |
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
The following tables summarize the Company’s marketable securities at December 31, 2012 and 2011, in thousands:
| | | December 31, 2012 | |
| Description | | Amortized Cost | | | Gross Unrealized Gains | | | Gross Unrealized Losses | | | Fair Value | |
| US Treasury Bills and Notes (due within 1 year) | | $ | 9,022 | | | $ | 6 | | | $ | — | | | $ | 9,028 | |
US Treasury Bills and Notes (due after 1 year through 2 years) | | | — | | | | — | | | | — | | | | — | |
| Total | | $ | 9,022 | | | $ | 6 | | | $ | — | | | $ | 9,028 | |
| | | December 31, 2011 | |
| Description | | Amortized Cost | | | Gross Unrealized Gains | | | Gross Unrealized Losses | | | Fair Value | |
| US Treasury Bills and Notes (due within 1 year) | | $ | 23,013 | | | $ | 7 | | | $ | — | | | $ | 23,020 | |
US Treasury Bills and Notes (due after 1 year through 2 years) | | | 3,016 | | | | — | | | | — | | | | 3,016 | |
| Total | | $ | 26,029 | | | $ | 7 | | | $ | -- | | | $ | 26,036 | |
As of December 31, 2012 and 2011, the Company's cash equivalents which are invested in money market funds are valued based on Level 1 inputs. As of December 31, 2012 and 2011, the Company’s short-term investments consisted of United States Treasury notes and bills which are valued based on Level 2 inputs. The Company has also classified its investments with maturities beyond one year as short-term, based on their highly liquid nature and because such marketable securities represent the investment of cash that is available for current operations.
The carrying amounts reflected in the consolidated balance sheets for cash, cash equivalents, accounts receivable, other current assets, accounts payable and accrued expenses and other current liabilities approximate fair value due to their short-term maturities.
5. Inventory
In 2009, the Company received marketing approval of KALBITOR from the FDA. Costs associated with the manufacture of KALBITOR prior to regulatory approval were expensed when incurred, and therefore were not capitalized as inventory. The supply of KALBITOR produced prior to FDA approval met commercial needs through 2012.
Subsequent to FDA approval, all costs associated with the manufacture of KALBITOR have been recorded as inventory. Inventory on-hand that will be sold beyond the Company's normal operating cycle is classified as non-current and grouped with other assets on the Company's balance sheet. As of December 31, 2012 and 2011, approximately $5.9 million and $4.9 million of inventory, respectively, is classified as non-current.
Inventory consists of the following (in thousands):
| | | December 31, 2012 | | | December 31, 2011 | |
| Raw Materials | | $ | 1,116 | | | $ | 1,429 | |
| Work in Progress | | | 8,274 | | | | 5,474 | |
| Finished Goods | | | 599 | | | | 119 | |
| Total | | $ | 9,989 | | | $ | 7,022 | |
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
The Company has revised the classification for $4.9 million of inventory from current assets to non-current other assets for the year ended December 31, 2011, to correct the classification of inventory based on the projected sale of inventory beyond the Company's normal operating cycle. The Company concluded this error was not material to the prior period financial statements.
Fixed assets consist of the following:
| | | December 31, | |
| | | 2012 | | | 2011 | |
| | | (In thousands) | |
| Laboratory equipment | | $ | 7,628 | | | $ | 9,103 | |
| Furniture and office equipment | | | 1,619 | | | | 1,095 | |
| Software and computers | | | 4,763 | | | | 4,445 | |
| Leasehold improvements | | | 4,502 | | | | 6,845 | |
| Construction in process | | | 197 | | | | 3,960 | |
| Total | | | 18,709 | | | | 25,448 | |
| Less: accumulated depreciation and amortization | | | (13,380 | ) | | | (20,567 | ) |
| | | $ | 5,329 | | | $ | 4,881 | |
Depreciation expense for the years ended December 31, 2012, 2011, and 2010 was approximately $1.1 million, $1.4 million and $1.5 million, respectively.
During 2012, the company retired fixed assets of $8.4 million with accumulated depreciation of $8.3 million, which resulted in a net gain on retirement of $ 110,000 due to proceeds received of $200,000. There was no significant fixed asset disposal activity in 2011.
7. Accounts Payable and Accrued Expenses
Accounts payable and accrued expenses consist of the following:
| | | December 31, | |
| | | 2012 | | | 2011 | |
| | | (In thousands) | |
| Accounts payable | | $ | 3,557 | | | $ | 2,927 | |
| Accrued employee compensation and related taxes | | | 4,018 | | | | 4,529 | |
| Accrued external research and development and contract manufacturing | | | 1,652 | | | | 1,591 | |
| Accrued legal | | | 518 | | | | 214 | |
| Accrued leasehold improvements | | | — | | | | 2,472 | |
| Accrued sales allowances | | | 1,052 | | | | 525 | |
| Other accrued liabilities | | | 1,675 | | | | 3,060 | |
| | | $ | 12,472 | | | $ | 15,318 | |
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Long-term obligations and note payable consists of the following:
| | | December 31, | |
| | | 2012 | | | 2011 | |
| | | (In thousands) | |
| Note payable | | $ | 78,061 | | | $ | 75,372 | |
| Obligations under equipment loan | | | 1,376 | | | | — | |
| Obligation under leasehold improvement arrangements | | | — | | | | 101 | |
| Total | | $ | 79,437 | | | $ | 75,473 | |
| Less: current portion | | | (445 | ) | | | (101 | ) |
| Long-term obligations | | $ | 78,992 | | | $ | 75,372 | |
Estimated future payments under the Company's long-term obligations and note payable as of December 31, 2012 are as follows:
| | | (In thousands) |
| 2013 | | $ | 6,603 | |
| 2014 | | | 11,880 | |
| 2015 | | | 15,379 | |
| 2016 | | | 26,961 | |
| 2017 | | | 64,561 | |
| Thereafter | | | 1,141 | |
| Total estimated future payments | | | 126,525 | |
| Less: amount representing interest | | | (43,493) | |
| Present value of estimated future payments | | | 83,032 | |
| Less: current portion | | | (445) | |
| Less: unamortized portion of discount and deferred interest expense | | | (3,595) | |
| Long-term obligations and note payable | | $ | 78,992 | |
HealthCare Royalty Partners
In August 2012, the Company completed the second closing under an agreement with an affiliate of HealthCare Royalty Partners, formerly Cowen Healthcare Partners (HC Royalty), that the Company entered into in December 2011 to refinance its existing loans from HC Royalty. At December 31, 2012, the aggregate principal amount of the new loan was $81.2 million, consisting of a $21.9 million Tranche A Loan and a $59.3 million Tranche B Loan (collectively, the “Loan”). The Loan bears interest at a rate of 12% per annum, payable quarterly. The Loan will mature in August 2018, and can be repaid without penalty beginning in August 2015.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
In connection with the Loan, the Company entered into a security agreement granting HC Royalty a security interest in the intellectual property related to the LFRP, and the revenues generated by the Company through the licenses of the intellectual property related to the LFRP. The security agreement does not apply to the Company's internal drug development or to any of the Company's co-development programs for ecallantide.
Under the terms of the agreement, the Company is required to repay the Loan based on the annual net LFRP receipts. Until September 30, 2016, required payments are equal to the sum of 75% of the first $15.0 million in specified annual LFRP receipts and 25% of specified annual LFRP receipts over $15.0 million. After September 30, 2016, and until the maturity date or the complete repayment of the Loan, HC Royalty will receive 90% of all included LFRP receipts. If the HC Royalty portion of LFRP receipts for any quarter exceeds the interest for that quarter, then the principal balance will be reduced. Any unpaid principal will be due upon the maturity of the Loan. If the HC Royalty portion of LFRP revenues for any quarterly period is insufficient to cover the cash interest due for that period, the deficiency may be added to the outstanding principal or paid in cash by the Company. After five years from the dates of the Tranche A Loan and the Tranche B Loan, respectively, the Company must repay to HC Royalty all additional accumulated principal above the original loan amounts of $21.7 million and $58.8 million, respectively.
Tranche A Loan
Under the terms of the agreement, the Company received a loan of $20 million (Tranche A Loan) in December 2011 and a commitment to refinance the amounts their outstanding under the Company’s March 2009 amended and restated loan agreement (the March 2009 Loan) at a reduced interest rate in August 2012. The Tranche A Loan was unsecured and accrued interest at an annual rate of 13% through August 2012.
Upon execution of the Tranche A Loan, the terms of the Original Loans (defined below) were determined to be modified under ASC 470. During the year ended December 31, 2012, interest expense on the Loan is being recorded in the Company’s financial statements at an effective interest rate of 13%.
Upon modification of the Original Loans, the note payable balance related to the Tranche A Loan was reduced by $193,000 to reflect payment of the lender’s legal fees in conjunction with the Tranche A Loan; these fees are being accreted over the life of the Loan, through August 2018.
Tranche B Loan
In August 2012, the Company completed a second closing with an affiliate of HC Royalty to refinance approximately $57.6 million outstanding under the March 2009 Loan under the same terms as Tranche A Loan (Tranche B Loan).
Original Loans
In 2008 and 2009, the Company entered into loan agreements with an affiliate of HC Royalty that provided aggregate loan proceeds of $65.0 million (the Original Loans), which had an outstanding principal and accrued interest balance of $57.6 million at the time of their refinancing in August 2012. The Original Loans bore interest at an annual rate of 17.4%, payable quarterly, and were secured by the Company’s LFRP.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
In connection with the Original Loans, the Company issued affiliates of HC Royalty warrants to purchase shares of the Company’s common stock. In August 2008, the Company issued warrants to purchase 250,000 shares of the Company’s common stock at an exercise price of $5.50 per share. This warrant expires in August 2016 and became exercisable in August 2009. The Company estimated the relative fair value of the warrant to be $853,000 on the date of issuance, using the Black-Scholes valuation model, assuming a volatility factor of 83.64%, risk-free interest rate of 4.07%, an eight-year expected term and an expected dividend yield of zero. In March 2009, the Company issued HC Royalty a second warrant to purchase 250,000 shares of the Company’s common stock at an exercise price of $2.87 per share. This warrant expires in August 2016 and became exercisable in March 2010. The Company estimated the relative fair value of the warrant to be $477,000 on the date of issuance, using the Black-Scholes valuation model, assuming a volatility factor of 85.98%, risk-free interest rate of 2.77%, a seven-year, four-month expected term and an expected dividend yield of zero. The relative fair values of the warrants as of the date of issuance are recorded in additional paid-in capital on the Company's consolidated balance sheets.
The cash proceeds from the Original Loans were recorded as a note payable on the Company's consolidated balance sheet. The note payable balance was reduced by $1.3 million for the fair value of the warrants issued, and by $580,000 for payment of HC Royalty’s legal fees in conjunction with the Original Loan. Prior to the December 2011 issuance of the Tranche A Loan, each of these amounts was being accreted over the life of the note through August 2016. Subsequent to the modification of the debt arrangement in December 2011, the unamortized portion of these amounts is being accreted over the life of the Loan through August 2018.
The Loan principal balance at December 31, 2012 and 2011 was $81.2 million and $76.7 million, respectively. For financial reporting purposes, the Loan is adjusted for discounts associated with the debt issuance, including warrants and fees.
Activity under the Loan is presented for financial reporting purposes, as follows (in thousands):
| | | | |
| | | 2012 | | | 2011 | |
| Balance, January 1 | | $ | 75,372 | | | $ | 56,406 | |
| Accretion of discount | | | 198 | | | | 246 | |
| Loan activity: | | | | | | | | |
| Net proceeds from Tranche A Loan | | | — | | | | 20,000 | |
| Discount on Tranche A Loan | | | (43 | ) | | | (150 | ) |
| Interest Expense | | | 10,199 | | | | 9,932 | |
| Payments applied to principal | | | (96 | ) | | | (1,129 | ) |
| Payments applied to interest | | | (7,569 | ) | | | (8,224 | ) |
| Accrued interest payable | | | — | | | | (1,709 | ) |
| Balance, December 31 | | $ | 78,061 | | | $ | 75,372 | |
The estimated fair value of the note payable was $81.2 million at December 31, 2012 which was calculated based on level 3 inputs due to the limited availability of comparable data points. The note payable was valued using expected cash flows discounted at our estimate of the currently available market interest rate.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Obligations Under Equipment Loan Arrangements
In June 2012, the Company entered into an equipment lease line of credit for up to $3 million with Silicon Valley Bank. When drawn, the note bears interest at a 6% annual rate. The Company drew down $1.4 million from this line, which will be financed over a 3-year term. The outstanding balance of this loan was $1.4 million as of December 31, 2012.
Obligation Under Leasehold Improvement Arrangement
In 2001, the Company entered into an agreement to lease laboratory and office space in Cambridge, Massachusetts. Under the terms of the agreement, the landlord loaned the Company approximately $2.4 million to be used towards the cost of leasehold improvements. The loan’s interest rate was 12% per annum and was payable in 98 equal monthly installments through February 2012, at which time it was paid in full. As of December 31, 2011, there was $101,000 outstanding under the loan.
In 2011, the Company entered into a lease agreement for new premises located in Burlington, Massachusetts and in January 2012, the Company relocated its operations to the new facility. The new premises, consisting of approximately 45,000 rentable square feet of office and laboratory facilities, serves as the Company’s principal offices and corporate headquarters. The term of the lease is ten years, and the Company has rights to extend the term for an additional five years at fair market value subject to specified terms and conditions. The aggregate minimum lease commitment over the ten year term of the new lease is approximately $15.0 million. The Company has provided the landlord a Letter of Credit of $1.1 million to secure its obligations under the lease, which is classified as restricted cash on the Company’s balance sheet at December 31, 2011 and 2012.
Under the terms of the lease agreement, the landlord has provided the Company with a tenant improvement allowance of $2.6 million which was used towards the cost of leasehold improvements. Through December 31, 2012, the Company capitalized approximately $4.5 million in leasehold improvements associated with the Burlington facility. Costs reimbursed under the tenant improvement allowance have been recorded as deferred rent and are being amortized as a reduction to rent expense over the lease term. All rent payments due over the term of the lease are being expensed on a straight-line basis in the Company’s statement of operations.
In January 2012, the Company’s lease agreement associated with its former facility terminated and the $1.3 million Letter of Credit which secured the Company’s obligations under the lease was released. This Letter of Credit was classified on the Company’s balance sheet as restricted cash at December 31, 2011.
Gross minimum future lease payments under the Company's non-cancelable operating leases as of December 31, 2012 are as follows:
| | | (In thousands) |
| 2013 | | $ | 1,503 | |
| 2014 | | | 1,605 | |
| 2015 | | | 1,615 | |
| 2016 | | | 1,581 | |
| 2017 | | | 1,588 | |
| Thereafter | | | 7,022 | |
| Total | | $ | 14,914 | |
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Rent expense for the years ended December 31, 2012, 2011, and 2010 was approximately $1.6 million, $3.4 million and $3.6 million, respectively. Rent expense for the years ended December 31, 2011 and 2010 is reflected as net of sublease payments of $194,000 and $1.5 million, respectively.
In February 2012, the Company realigned its business structure and implemented a number of strategic and operational initiatives. As part of these initiatives, the Company terminated certain early stage, preclinical research and development programs and a workforce reduction was implemented. As a result of the restructuring, during the year ended December 31, 2012, the Company recorded one-time charges of approximately $1.4 million, which included severance and benefits related charges of approximately $1.2 million, outplacement costs of approximately $120,000, stock compensation expense of $55,000 for amendments to the exercise schedules to certain options and other exit costs of $90,000. All restructuring costs were paid as of December 31, 2012 and no additional charges are expected in future periods related to this restructuring.
10. Stockholders' Deficit
Preferred Stock: As of December 31, 2012 and 2011, there were a total of 1,000,000 shares of $0.01 par value preferred stock authorized with 950,000 shares undesignated and 50,000 shares designated as Series A Junior Participating Preferred Stock.
Common Stock: In January 2011, the Company issued 151,515 shares of its common stock for an aggregate purchase price of $500,000 in connection with an amendment to a strategic partnership (see Note 3, Significant Transactions - Sigma Tau).
Stock-Based Compensation Expense
The Company measures compensation cost for all stock awards at fair value on date of grant and recognition of compensation over the service period for awards expected to vest. The fair value of stock options was determined using the Black-Scholes valuation model. Such value is recognized as expense over the service period, net of estimated forfeitures and adjusted for actual forfeitures. The estimation of stock options that will ultimately vest requires significant judgment. The Company considers many factors when estimating expected forfeitures, including historical experience. Actual results and future changes in estimates may differ substantially from the Company's current estimates.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
The following table reflects stock compensation expense recorded, net of amounts capitalized into inventory (in thousands):
| | | Year Ended December 31, | |
| Compensation expense related to: | | 2012 | | | 2011 | | | 2010 | |
Equity incentive plan | | $ | 3,549 | | | $ | 3,984 | | | $ | 4,073 | |
Employee stock purchase plan | | | 91 | | | | 46 | | | | 57 | |
| | | $ | 3,640 | | | $ | 4,030 | | | $ | 4,130 | |
| Stock-based compensation expense charged to: | | | | | | | | | | | | |
| Research and development | | $ | 839 | | | $ | 1,193 | | | $ | 1,466 | |
| | | | | | | | | | | | | |
| General and administrative | | $ | 2,746 | | | $ | 2,837 | | | $ | 2,664 | |
| | | | | | | | | | | | | |
| Restructuring charges | | $ | 55 | | | $ | — | | | $ | — | |
Stock-based compensation of $23,000, $31,000 and $31,000 was capitalized into inventory for the each of the years ended December 31, 2012, 2011 and 2010, respectively. Capitalized stock-based compensation is recognized into cost of product sales when the related product is sold.
Stock-based compensation expense for the year ended December 31, 2012, 2011 and 2010 included $467,000, $273,000 and $261,000, respectively, related to the modification of certain stock options.
Valuation Assumptions for Stock Options
For the years ended December 31, 2012, 2011 and 2010, 5,897,221, 2,624,160 and 2,042,180 stock options were granted, respectively. The fair value of each option was estimated on the date of grant using the Black-Scholes option-pricing model with the following assumptions:
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
| Expected Option Term (in years) | | | 6 – 8 | | | | 5.5 – 6 | | | | 5.5 | |
| Risk-free interest rate | | | 0.86 % - 1.37% | | | | 1.28% - 2.39% | | | | 1.76 % - 2.68% | |
| Expected dividend yield | | | 0 | | | | 0 | | | | 0 | |
| Volatility factor | | | 70% - 73% | | | | 74% - 75% | | | | 74% - 76% | |
Valuation Assumptions for Employee Stock Purchase Plans
The fair value of shares issued under the employee stock purchase plan was estimated on the commencement date of each offering period using the Black-Scholes option-pricing model with the following assumptions:
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
| Expected Option Term (in years) | | | 0.5 | | | | 0.5 | | | | 0.5 | |
| Risk-free interest rate | | | 0.9% - 0.12% | | | | 0.07% - 0.11% | | | | 0.15% - 0.22% | |
| Expected dividend yield | | | 0 | | | | 0 | | | | 0 | |
| Volatility factor | | | 45% - 54% | | | | 37% - 72% | | | | 40% - 50% | |
Expected volatilities are based on historical volatilities of our common stock; the expected life represents the weighted average period of time that options granted are expected to be outstanding giving consideration to vesting schedules and our historical exercise and cancellation patterns; and the risk-free rate is based on the United States Treasury yield curve in effect at the time of grant for periods corresponding with the expected life of the option.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Equity Incentive Plan
The Company's 1995 Equity Incentive Plan (the Equity Plan), as amended, is an equity plan under which equity awards, including awards of restricted stock and incentive and nonqualified stock options to purchase shares of common stock may be granted to employees, consultants and directors of the Company by action of the Compensation Committee of the Board of Directors. Options are generally granted at the current fair market value on the date of grant, generally vest ratably over a 48-month period, and expire within ten years from date of grant. Restricted stock units are generally granted at the current fair market value on the date of grant, generally vest over a four year period in equal installments on each anniversary of the grant date. The Equity Plan is intended to attract and retain employees and to provide an incentive for employees, consultants and directors to assist the Company to achieve long-range performance goals and to enable them to participate in the long-term growth of the Company.
At the Annual Meeting of Stockholders held in May 2012 (the Annual Meeting), the Company’s shareholders approved a stock option exchange program for employees, executive officers and non-employee directors. Under this option exchange program, outstanding options to purchase an aggregate of 4,192,310 shares of the Company’s common stock were exchanged for new options to purchase an aggregate of 2,473,596 shares of the Company’s common stock at an exercise price equal to $2.06 per share, which was the closing price of the Company’s common stock on the grant date of June 20, 2012. All new options issued in the option exchange program are subject to a new, extended vesting schedule, the terms of which differ depending on the holder’s status as an executive, director or non-executive employee. The new options have a term equal to the greater of (i) the term of the original options for which they were exchanged, or (ii) five years from date of grant. The new options had a fair value approximately equal to the fair value of the surrendered options, based on a Black-Scholes option pricing model applied immediately prior to commencement of the exchange program. Accordingly, approximately $46,000 of expense was recorded during the year ended December 31, 2012 related to the modification of the exchanged options.
Also at the Annual Meeting, the Company’s shareholders approved an amendment to the Equity Plan to increase the number of shares of common stock available for issuance under the plan by 5,000,000 shares less the net number of shares, if any, returned to the Equity Plan for issuance following the option exchange program. Accordingly, 3,281,286 additional shares of common stock, constituting the approved 5,000,000 share increase net of the 1,718,714 shares that were returned to the Equity Plan as a result of the option exchange program, became available for issuance under the plan.
At December 31, 2012, a total of 6,734,731 shares were available for future grants under the Equity Plan.
Stock Option Activity
The following table summarizes stock option activity for the year ended December 31, 2012:
| | | Number of Options | | | Weighted-Avg. Exercise Price | | | Weighted-Avg. Remaining Contractual Life | | | Aggregate Intrinsic Value (in thousands) | |
Outstanding as of December 31, 2011 | | | 11,054,126 | | | $ | 3.41 | | | | 6.69 | | | $ | 22 | |
Granted at fair market value * | | | 5,897,221 | | | $ | 1.83 | | | | | | | | | |
Exercised | | | (590,590 | ) | | $ | 2.23 | | | | | | | | | |
Forfeited * | | | (5,640,435 | ) | | $ | 2.49 | | | | | | | | | |
Expired | | | (191,848 | ) | | $ | 4.44 | | | | | | | | | |
Outstanding as of December 31, 2012 | | | 10,528,474 | | | $ | 2.28 | | | | 6.84 | | | $ | 13,864 | |
| | | | | | | | | | | | | | | | | |
Exercisable as of December 31, 2012 | | | 4,275,812 | | | $ | 2.85 | | | | 5.69 | | | $ | 3,965 | |
| | | | | | | | | | | | | | | | | |
| Vested and unvested expected to vest as of December 31, 2012 | | | 10,425,737 | | | $ | 2.29 | | | | 6.81 | | | $ | 13,659 | |
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
*Included in this option activity are 4,192,310 stock options to purchase shares of the Company’s common stock that were forfeited and exchanged for 2,473,596 new option grants in conjunction with the Company’s stock option exchange program in June 2012.
The aggregate intrinsic value in the table above represents the total intrinsic value of the options outstanding, options exercisable and options vested and unvested which are expected to vest, based on the Company's common stock closing price of $3.48 as of December 31, 2012, which would have been received by the option holders had all option holders exercised their options and sold the underlying common stock as of that date. The total number of in-the-money options exercisable as of December 31, 2012 was 3,244,365.
The weighted average grant date fair values of options, as determined under ASC 718, granted during the years ended December 31, 2012, 2011 and 2010 were $1.00, $1.17 and $2.07 per share, respectively. The total intrinsic value of options exercised during years ended December 31, 2012, 2011 and 2010 was approximately $375,000, $0, and $120,000, respectively. The total cash received from employees as a result of employee stock option exercises during the years ended December 31, 2012, 2011 and 2010 was approximately $1.3 million, $2,000, and $260,000, respectively.
As of December 31, 2012 future compensation cost related to non-vested stock options is approximately $6.9 million and will be recognized over an estimated weighted average period of approximately 2.16 years.
The following table summarizes unvested stock option activity for the year ended December 31, 2012:
| | | Non-vested Number of Options | |
Unvested balance at December 31, 2011 | | | 3,566,032 | |
Granted at fair market value | | | 5,897,221 | |
Vested | | | (1,762,466 | ) |
Forfeited | | | (1,448,125 | ) |
Unvested balance at December 31, 2012 | | | 6,252,662 | |
The total fair value of options vested during the years ended December 31, 2012, 2011 and 2010 were $3.0 million, $3.7 million and $4.1 million, respectively.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Restricted Stock Unit Activity
The fair market value of restricted stock units is expensed over the period of vesting. Restricted stock units generally vest over a four year period in equal installments on each anniversary of the grant date. The fair value of restricted stock units was determined based on the closing market price of the underlying stock on the grant date.
The following table summarizes our RSU activity:
| | | Shares | | | Weighted-Avg Fair Value | |
Unvested balance at December 31, 2011 | | | — | | | $ | — | |
Granted at fair market value | | | 380,832 | | | $ | 1.48 | |
Vested | | | — | | | $ | — | |
Forfeited | | | (7,083 | ) | | $ | 1.48 | |
| | | | | | | | | |
Unvested balance at December 31, 2012 | | | 373,749 | | | $ | 1.48 | |
As of December 31, 2012, there was $328,000 of total unrecognized compensation cost related to non-vested restricted stock units. This expense, net of forfeitures is expected to be recognized over a weighted average period of approximately 3.15 years. Of the 373,749 unvested shares, the Company estimates that 313,465 will vest.
Employee Stock Purchase Plan
The Company's Purchase Plan allows employees to purchase shares of the Company's common stock at a discount from fair market value. Under this Plan, eligible employees may purchase shares during six-month offering periods commencing on June 1 and December 1 of each year at a price per share of 85% of the lower of the fair market value price per share on the first or last day of each six-month offering period. Participating employees may elect to have up to 10% of their base pay withheld and applied toward the purchase of such shares, subject to the limitation of 875 shares per participant per quarter. The rights of participating employees under the Purchase Plan terminate upon voluntary withdrawal from the Purchase Plan at any time or upon termination of employment. The compensation expense in connection with the Plan for the years ended December 31, 2012, 2011 and 2010 was approximately $91,000, $46,000 and $57,000, respectively. There were 93,974 and 137,167 shares purchased under the Plan during the years ended December 31, 2012 and 2011, respectively. At December 31, 2012, a total of 362,939 shares were reserved and available for issuance under this Plan.
11. Employee Savings and Retirement Plans
The Company has an employee savings and retirement plan (the "Retirement Plan"), qualified under Section 401(k) of the Internal Revenue Code, covering substantially all of the Company's employees. Employees may elect to contribute a portion of their pretax compensation to the Retirement Plan up to the annual maximum allowed under the Retirement Plan. Employees are 100% vested in company matching contributions which have been 50% of employee contributions up to 6% of eligible pay. For the years ended December 31, 2012, 2011 and 2010, the Company's matching contributions amounted to $440,000, $431,000 and $410,000, respectively.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
Deferred tax assets and deferred tax liabilities are recognized based on temporary differences between the financial reporting and tax basis of assets and liabilities using future enacted rates. A valuation allowance is recorded against deferred tax assets for which the Company determines that it does not meet the criteria under ASC 740.
The provision for income taxes for continuing operations was calculated at rates different from the United States federal statutory income tax rate for the following reasons:
| | | 2012 | | | 2011 | | | 2010 | |
| Statutory federal income taxes | | | 34.0 | % | | | 34.0 | % | | | 34.0 | % |
| State income taxes, net of federal benefit | | | (3.3 | %) | | | (2.0 | %) | | | 1.7 | % |
| General business credit | | | 3.8 | % | | | 5.9 | % | | | 4.7 | % |
| Stock compensation cancellations | | | (6.8 | %) | | | — | | | | (1.8 | )% |
| Other | | | (1.7 | %) | | | (1.2 | %) | | | (1.1 | )% |
| Federal true up and expiring NOLs and research credits | | | (8.6 | %) | | | (3.7 | %) | | | (8.0 | )% |
| Valuation allowance | | | (17.4 | %) | | | (33.0 | %) | | | (29.5 | )% |
| Effective income tax rate | | | — | % | | | — | % | | | — | % |
The principal components of the Company's deferred tax assets and liabilities at December 31, 2012, 2011 and 2010, respectively are as follows:
| | | 2012 | | | 2011 | | | 2010 | |
| | | (In thousands) | |
| Net Deferred Tax Asset: | | | | | | | | | |
| Allowance for doubtful accounts | | $ | — | | | $ | 44 | | | $ | 18 | |
| Depreciation and amortization | | | 255 | | | | 2,023 | | | | 1,848 | |
| Accrued expenses | | | 303 | | | | 173 | | | | 151 | |
| Capitalized research and development costs | | | 8,192 | | | | — | | | | — | |
| Other | | | 1,394 | | | | 861 | | | | (205 | ) |
| Stock based compensation | | | 2,127 | | | | 3,615 | | | | 2,561 | |
| Deferred revenue | | | 4,223 | | | | 5,898 | | | | 8,249 | |
| Research credit carryforwards | | | 63,259 | | | | 61,999 | | | | 58,772 | |
| Net operating loss carryforwards | | | 124,507 | | | | 124,558 | | | | 116,351 | |
| Total gross deferred tax asset | | | 204,260 | | | | 199,171 | | | | 187,745 | |
| Valuation allowance | | | (204,260 | ) | | | (199,171 | ) | | | (187,745 | ) |
| Net deferred tax asset | | $ | — | | | $ | — | | | $ | — | |
As of December 31, 2012 and 2011, the Company had federal tax net operating loss carryforwards (NOLs) of $344.9 million and $339.9 million, respectively, available to reduce future taxable income, which expire at various times beginning in 2018 through 2032. The Company also had federal research and experimentation and orphan drug credit carryforwards of approximately $58.1 million and $57.2 million as of December 31 2012 and 2011, respectively, available to reduce future tax liabilities which will expire at various dates beginning in 2018 through 2032. The Company had state tax net operating loss carryforwards of approximately $138.1 million and $170.7 million as of December 31, 2012 and 2011, respectively, available to reduce future state taxable income, which will expire at various dates beginning in 2013 through 2032. The Company also had state research and development and investment tax credit carryforwards of approximately $7.8 million and $7.2 million as of December 31, 2012 and 2011, respectively, available to reduce future tax liabilities which expire at various dates beginning in 2015 through 2027.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
The Company has recorded a deferred tax asset of approximately $1.8 million at December 31, 2012 and 2011, reflecting the benefit of deductions from the exercise of stock options which has been fully reserved until it is more likely than not that the benefit will be realized. The benefit from this deferred tax asset will be recorded as a credit to additional paid-in capital if and when realized through a reduction of cash taxes.
As required by ASC 740, management of the Company has evaluated the positive and negative evidence bearing upon the reliability of its deferred tax assets, which are comprised principally of NOL carry forwards, research and experimentation credit carry forwards, and capitalized start up expenditures and research and development expenditures amortizable over ten years straight-line. Management has determined at this time that it is more likely than not that the Company will not recognize the benefits of its federal and state deferred tax assets and, as a result, a valuation allowance of approximately $204.3 million and $199.2 million has been established at December 31, 2012 and 2011, respectively.
The Company accounts for uncertain tax positions using a "more-likely-than-not" threshold for recognizing and resolving uncertain tax positions. The evaluation of uncertain tax positions is based on factors that include, but are not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. The Company evaluates uncertain tax positions on a quarterly basis and adjusts the level of the liability to reflect any subsequent changes in the relevant facts surrounding the uncertain positions. As of December 31, 2012 , the Company had no unrecognized tax benefits or liabilities and had no accrued interest or penalties related to uncertain tax positions.
Utilization of the NOLs and tax credit carryforwards may be subject to a substantial annual limitation due to ownership change limitations that have occurred previously or that could occur in the future, as provided by Section 382 of the Internal Revenue Code of 1986 (Section 382), as well as similar state and foreign provisions. Ownership changes may limit the amount of NOLs and tax credit carryforwards that can be utilized annually to offset future taxable income and tax, respectively. In general, an ownership change, as defined by Section 382, results from transactions that increase the ownership of 5% shareholders or public groups in the stock of a corporation by more than 50 percent in the aggregate over a three-year period. During 2012, the Company completed a study through December 31, 2012, to determine whether any ownership change has occurred since the Company's formation and has determined that transactions have resulted in two ownership changes, as defined by Section 382. There could be additional ownership changes in the future that could further limit the amount of NOLs and tax credit carryforwards that the Company can utilize.
As of December 31, 2012, the Company’s federal tax NOLs available to reduce future taxable income without limitation are $277.7 million, which expire at various times beginning in 2024 through 2032. The Company’s federal research and experimentation and orphan drug credit carryforward as of December 31, 2012 available to reduce future tax liabilities without limitation are $53.3 million, which will expire at various dates beginning in 2024 through 2032. In addition the Company has NOLs and federal tax credits that are restricted and expire at various times beginning in 2018 through 2024. These restricted NOLs and federal tax credits of $67.2 million and $4.8 million, respectively, may be utilized in part, subject to an annual limitation.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
A full valuation allowance has been provided against the Company's NOL carryforwards and research and development credits and, if an adjustment is required, this adjustment would be offset by an adjustment to the valuation allowance. Thus there would be no impact to the consolidated balance sheet or statement of operations if an adjustment were required.
The tax years 1998 through 2012 remain open to examination by major taxing jurisdictions to which the Company is subject, which are primarily in the United States, as carryforward attributes generated in years past may still be adjusted upon examination by the Internal Revenue Service or state tax authorities if they have or will be used in a future period. The Company is currently not under examination in any jurisdictions for any tax years.
The Company discloses business segments under ASC 280, Segment Reporting. The statement established standards for reporting information about operating segments in annual financial statements of public enterprises and in interim financial reports issued to shareholders. It also established standards for related disclosures about products and services, geographic areas and major customers. The Company operates as one business segment in one geographic area.
Novellus – In January 2013, the Company entered into a strategic partnership with Novellus Biopharma AG for the development and commercialization of KALBITOR for the treatment of HAE and other angioedema indications in select countries in Latin America, including Argentina, Brazil, Chile, Colombia, Mexico and Venezuela.
Under the terms of the exclusive license agreement, Dyax will receive an upfront payment and is eligible to receive future regulatory and sales milestones. Dyax is also eligible to receive royalties on net product sales. Novellus is solely responsible for all costs associated with development, regulatory activities, and the commercialization of KALBITOR in their licensed territories. Additionally, Novellus will purchase drug product from Dyax on a cost-plus basis for commercial supply.
CVie – In February 2013, the Company entered into a strategic partnership with CVie Therapeutics (CVie), a subsidiary of Lee’s Pharmaceutical Holdings Ltd. in China, for the development and commercialization of KALBITOR in the treatment of HAE and other angioedema indications in China.
Under the terms of the exclusive license agreement, Dyax will receive an upfront payment and is eligible to receive future development, regulatory and sales milestones. Dyax is also eligible to receive royalties on net product sales. CVie is solely responsible for all costs associated with development, regulatory activities, and the commercialization of KALBITOR in their licensed territories. Additionally, CVie will purchase drug product from Dyax on a cost-plus basis for commercial supply.
As of December 31, 2012, there were no active legal proceedings that were expected to be material to the Company. The Company makes provisions for claims specifically identified for which it believes the likelihood of an unfavorable outcome is probable and reasonably estimable. The Company records at least the minimum estimated liability related to claims where there is a range of loss and the loss is considered probable and no estimate within the range is better than any other. As additional information becomes available, the Company assesses the potential liability related to its pending claims and revises its estimates. Future revisions in the estimates of the potential liability could materially impact the results of operations and financial position. The Company maintains insurance coverage that limits the exposure for any single claim as well as total amounts incurred per policy year, and it believes that its insurance coverage is adequate. The Company makes every effort to use the best information available in determining the level of liability reserves.
Dyax Corp. and Subsidiaries
Notes to Consolidated Financial Statements (Continued)
16.Unaudited Quarterly Operating Results
The following is a summary of unaudited quarterly results of operations for the years ended December 31, 2012 and 2011:
| Year ended December 31, 2012 | | First Quarter | | | Second Quarter | | | Third Quarter | | | Fourth Quarter | |
| | | (in thousands, except share and per share) | |
Revenue | | $ | 11,489 | | | $ | 14,030 | | | $ | 13,101 | | | $ | 16,030 | |
(Loss) Income from operations | | $ | (8,747 | ) | | $ | (5,389 | ) | | $ | (2,667 | ) | | $ | (2,082 | ) |
Net loss | | $ | (11,282 | ) | | $ | (7,939 | ) | | $ | (5,210 | ) | | $ | (4,834 | ) |
| Shares used in computing basic and diluted net loss per share | | | 98,798,426 | | | | 98,820,699 | | | | 99,069,928 | | | | 99,271,225 | |
| Basic and diluted net loss per share: | | $ | (0.11 | ) | | $ | (0.08 | ) | | $ | (0.05 | ) | | $ | (0.05 | ) |
| Year ended December 31, 2011 | | First Quarter | | | Second Quarter | | | Third Quarter | | | Fourth Quarter | |
| | | (in thousands, except share and per share) | |
Revenue | | $ | 8,214 | | | $ | 21,875 | | | $ | 10,132 | | | $ | 8,516 | |
Income (loss) from operations | | $ | (8,769 | ) | | $ | 2,428 | | | $ | (7,577 | ) | | $ | (10,984 | ) |
Net (loss) income | | $ | (11,265 | ) | | $ | (76 | ) | | $ | (9,723 | ) | | $ | (13,535 | ) |
| Shares used in computing basic and diluted net loss per share | | | 98,689,795 | | | | 98,721,889 | | | | 98,748,086 | | | | 98,764,384 | |
| Basic and diluted net loss per share: | | $ | (0.11 | ) | | $ | (0.00 | ) | | $ | (0.10 | ) | | $ | (0.14 | ) |
| CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in the Company's reports under the Securities Exchange Act of 1934, as amended (the Exchange Act), is recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms, and that such information is accumulated and communicated to management, including our Chief Executive Officer (CEO) and Chief Financial Officer (CFO), as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, as the Company's are designed to do, and management necessarily was required to apply its judgment in evaluating the risk related to controls and procedures.
In connection with the preparation of this Form 10-K, as of December 31, 2012, an evaluation was performed under the supervision and with the participation of our management, including the CEO and CFO, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rule 13a-15(e) under the Exchange Act). Based on that evaluation, our management concluded that our disclosure controls and procedures were effective at a reasonable assurance level as of December 31, 2012. These conclusions were communicated to the Audit Committee.
Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) under the Exchange Act. Our internal control system is designed to provide reasonable assurance to the Company's management and Board of Directors regarding the preparation and fair presentation of published financial statements. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Our management has assessed the effectiveness of our internal control over financial reporting as of December 31, 2012. In making its assessment of internal control over financial reporting, management used the criteria set forth by the Committee of Sponsoring Organizations (COSO) of the Treadway Commission in Internal Control — Integrated Framework. Based on this assessment, our management concluded that our internal control over financial reporting was effective as of December 31, 2012 based on the criteria set forth by COSO in Internal Control — Integrated Framework.
The effectiveness of the Company’s internal control over financial reporting as of December 31, 2012, has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which appears in Item 8 above.
Change in Internal Control Over Financial Reporting — There were no changes in our internal control over financial reporting that occurred during our last fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
PART III
| DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Portions of the response to this item are incorporated herein by reference from the discussion responsive thereto under the captions "Election of Directors—Nominees for Director", "Section 16(a) Beneficial Ownership Reporting Compliance", "Executive Officers" and "Corporate Governance—Board and Committee Matters" in the Company's Definitive Proxy Statement relating to the 2013 Annual Meeting of Stockholders (the 2013 Proxy Statement).
We have adopted a Code of Business Conduct and Ethics (the code of ethics) that applies to all of our directors, officers and employees. The code of ethics is available on our website at www.dyax.com. In addition, if we make any substantive amendments to the code of ethics or grant any waiver, including any implicit waiver, from a provision of the code to any of our executive officers or directors, we will disclose the nature of such amendment or waiver as required by applicable law.
The response to this item is incorporated herein by reference from the discussion responsive thereto under the following captions in the 2013 Proxy Statement: "Executive Compensation" and "Corporate Governance—Compensation Committee Interlocks and Insider Participation."
| SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The response to this item is incorporated herein by reference in part from the discussion responsive thereto under the caption "Share Ownership" in the 2013 Proxy Statement.
The following table provides information about the securities authorized for issuance under the Company's equity compensation plans as of December 31, 2012:
Equity Compensation Plan Information
| Plan Category | | Number of securities to be issued upon exercise of outstanding options, warrants and rights | | | Weighted average exercise price of outstanding options, warrants and rights | | | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | |
| | | (a) | | | (b) | | | (c) | |
Equity compensation plans approved by security holders(1) | | | 10,528,474 | | | $ | 2.28 | | | | 7,097,670 | |
Equity compensation plans not approved by security holders: | | | — | | | | — | | | | — | |
Totals: | | | 10,528,474 | (2) | | $ | 2.28 | | | | 7,097,670 | (3) |
| (1) | Consists of the Amended and Restated 1995 Equity Incentive Plan, as amended, and the 1998 Employee Stock Purchase Plan, as amended. |
| (2) | Does not include purchase rights currently accruing under the 1998 Employee Stock Purchase Plan, because the purchase price (and therefore the number of shares to be purchased) will not be determined until the end of the purchase period, which is May 31, 2013. |
| (3) | Includes 362,939 shares issuable under the 1998 Employee Stock Purchase Plan, of which up to 50,000 of purchase rights are issuable in connection with the current offering period which ends on May 31, 2013. The remaining shares consist of 362,939 under the 1995 Amended and Restated Equity Incentive Plan. The plan may be amended, suspended, or terminated by the Compensation Committee of the Board of Directors at any time, subject to any required stockholder approval. |
| CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The response to this item is incorporated herein by reference from the discussion responsive thereto under the caption "Election of Directors—Certain Relationships and Related Transactions" and "Corporate Governance – Board and Committee Matters" in the 2013 Proxy Statement.
| PRINCIPAL ACCOUNTING FEES AND SERVICES |
The response to this item is incorporated herein by reference from the discussion responsive thereto under the captions "Corporate Governance—Board and Committee Matters" and "Audit Committee Report – Audit Fees" in the 2013 Proxy Statement.
PART IV
| EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
(a) 1. FINANCIAL STATEMENTS
The financial statements are included under Part II, Item 8 of this Report.
2. FINANCIAL STATEMENTS SCHEDULE
Schedules are omitted because they are not applicable, or are not required, or because the information is included in the consolidated financial statements and notes thereto.
The exhibits are listed below under Part IV, Item 15(b) of this Report.
Exhibit No. | | Description |
| 3.1(a) | | Amended and Restated Certificate of Incorporation of the Company. Filed as Exhibit 3.1 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2008 and incorporated herein by reference. |
| 3.1(b) | | Certificate of Amendment of the Company’s Amended and Restated Certificate of Incorporation. Filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K (File No. 000-24537) filed May 13, 2011 and incorporated herein by reference. |
| 3.2 | | Amended and Restated By-laws of the Company. Filed as Exhibit 3.2 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2008 and incorporated herein by reference. |
| 3.3 | | Certificate of Designations Designating the Series A Junior Participating Preferred Stock of the Company. Filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K (File No. 000-24537) filed on June 27, 2001 and incorporated herein by reference. |
| 4.1 | | Specimen Common Stock Certificate. Filed as Exhibit 4.1 to the Company’s Registration Statement on Form S-1 (File No. 333-37394) and incorporated herein by reference. |
| 4.3 | | Form of Warrant issued to Cowen Healthcare Royalty Partners, L.P. on August 5, 2008 and March 18, 2009. Filed as an exhibit to Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2008 and incorporated herein by reference. |
| 10.1(a) | | Amended and Restated 1995 Equity Incentive Plan. Filed as Exhibit 99.1 to the Company’s Registration Statement on Form S8 (File No. 000-024537) for the quarter ended June 28, 2012 and incorporated herein by reference. |
| 10.1(b) | | Form of the Company’s Incentive Stock Option Certificate under the Company’s Amended and Restated 1995 Equity Incentive Plan for all U.S. employees, including its executive officers. Filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2004 and incorporated herein by reference. |
| 10.1(c) | | Form of the Company’s Nonstatutory Stock Option Certificate under the Company’s Amended and Restated 1995 Equity Incentive Plan for its U.S. employees, including its executive officers. Filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2004 and incorporated herein by reference. |
| 10.1(d) | | Form of the Company’s Nonstatutory Stock Option Certificate under the Company’s Amended and Restated 1995 Equity Incentive Plan for its non-employee directors. Filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2004 and incorporated herein by reference. |
| 10.1(e) | | Form of the Company’s Restricted Stock Unit Certificate under the Company’s Amended and Restated 1995 Equity Incentive Plan for all U.S. employees, including its executive officers. Filed as exhibit 10.1(e) to the Company’s Annual Report on Form 10-K (File No. 000-24537) for the year ended December 31, 2011 and incorporated herein by reference. |
| 10.2 | | 1998 Employee Stock Purchase Plan, as amended on March 25, 2009. Filed as Exhibit 10.2 to the Company’s Annual Report on Form 10-K (File No. 000-24537) for the year ended December 31, 2009 and incorporated herein by reference. |
| 10.3* | | Form of Indemnification Agreement by and between certain directors and executive officers of the Company and the Company. Filed as Exhibit 10.32 to the Company’s Registration Statement on Form S-1 (File No. 333-37394) and incorporated herein by reference. |
| 10.4 | | Amended and Restated Registration Rights Agreement, dated as of February 12, 2001, between holders of the Company’s capital stock named therein and the Company. Filed as Exhibit 10.33 to the Company’s Annual Report on Form 10-K (File No. 000-24537) for the year ended December 31, 2000 and incorporated herein by reference. |
| 10.5 † | | Amended and Restated License Agreement between XOMA Ireland Limited and the Company dated as of October 27, 2006. Filed as Exhibit 10.20(b) to the Company’s Annual Report on Form 10-K (File No. 000-24537) for the year ended December 31, 2007 and incorporated herein by reference. |
| 10.6 † | | Amended and Restated License Agreement by and between the Company and MedImmune Limited, formerly Cambridge Antibody Technology Limited (“MedImmune”), dated as of July 26, 2012. Filed as Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2012 and incorporated herein by reference. |
| 10.7(a) † | | Distribution Agreement by and between the Company and ASD Specialty Healthcare Inc. dated as of November 19, 2009. Filed as Exhibit 10.21 to the Company’s Annual Report on Form 10-K (File No. 000-24537) for the year ended December 31, 2009 and incorporated herein by reference. |
| 10.7(b) † | | First Amendment to Distribution Agreement by and between the Company and ASD Specialty Healthcare Inc. dated as of August 25, 2011. Filed as exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2011 and incorporated herein by reference. |
| 10.7(c) | | Second Amendment to Distribution Agreement by and between the Company and ASD Specialty Healthcare Inc. dated as of July 31, 2012. Filed herewith. |
| 10.7(d) | | Third Amendment to Distribution Agreement by and between the Company and ASD Specialty Healthcare Inc. dated as of December 3, 2012. Filed herewith. |
| 10.7(e) † | | Fourth Amendment to Distribution Agreement by and between the Company and ASD Specialty Healthcare Inc. dated as of December 19, 2012. Filed herewith. |
| 10.8(a) † | | Distribution Agreement by and between the Company and Walgreens Infusion Services, Inc. dated as of August 31, 2011; as amended by a First Amendment to Distribution Agreement dated August 31, 2012. Filed herewith. |
| 10.8(b) † | | Agreement for Services Related to KALBITOR® by and between the Company and Walgreens Infusion Services, Inc. effective as of September 1, 2011; as amended by a First Amendment to Agreement for Services Related to KALBITOR® dated August 31, 2012. Filed herewith. |
| 10.9 † | | Royalty Interest Purchase Agreement by and between the Company and KGH Domestic III, LP dated as of April 16, 2010. Filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended June 30, 2010, as amended, and incorporated herein by reference. |
| 10.10 † | | Joint Development and License Agreement by and between the Company and Defiante Farmaceutica S.A., a subsidiary of the pharmaceutical company Sigma-Tau SpA dated as of June 18, 2010. Filed as Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended June 30, 2010, as amended, and incorporated herein by reference. |
| 10.11(a) † | | Amendment No. 1 to Joint Development and License Agreement by and between the Company and Defiante Farmaceutica S.A., a subsidiary of the pharmaceutical company Sigma-Tau SpA dated as of December 21, 2010. Filed as Exhibit 10.27 to the Company’s Annual Report on Form 10-K (File No. 000-24537) for the year ended December 31, 2010 and incorporated herein by reference. |
| 10.11(b) † | | Amendment No. 2 to Joint Development and License Agreement by and between the Company and Defiante Farmaceutica S.A., a subsidiary of the pharmaceutical company Sigma-Tau SpA dated as of May 27, 2011. Filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended June 30, 2011 and incorporated herein by reference. |
| 10.11(c) | | Third Amendment, dated December 26, 2011, to Joint Development and License Agreement by and between the Company and Defiante Farmaceutica S.A., a subsidiary of the pharmaceutical company Sigma-Tau SpA dated as of June 18, 2010. Filed as exhibit 10.14(c) to the Company’s Annual Report on Form 10-K (File No. 000-24537) for the year ended December 31, 2011 and incorporated herein by reference. |
| 10.11(d) | | Fourth Amendment to Joint Development and License Agreement by and between the Company and Sigma-Tau Rare Diseases S.A., as successor-in-interest to Defiante Farmaceutica S.A., dated as of May 29, 2012. Filed as Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended June 30, 2012 and incorporated herein by reference. |
| 10.11(e) | | Fifth Amendment to Joint Development and License Agreement by and between the Company and Sigma-Tau Rare Diseases S.A., as successor-in-interest to Defiante Farmaceutica S.A., dated as of June 21, 2012. Filed as Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended June 30, 2012 and incorporated herein by reference. |
| 10.12† | | Product Development and License Agreement by and between the Company and CMIC Co. Ltd. dated as of September 28, 2010. Filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2010 and incorporated herein by reference. |
| 10.13* | | Executive Retention Agreement by and between the Company and Gustav Christensen dated as of December 22, 2010. Filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 000-24537) filed on December 23, 2010 and incorporated herein by reference. |
10.14* | | Form of Executive Retention Agreement for executive officers other than the CEO. Filed as Exhibit 10.2 to the Company’s Current Report on Form 8-K (File No. 000-24537) filed on December 23, 2010 and incorporated herein by reference. |
10.15(a) | | Lease, dated as of July 14, 2011, by and between the Company and Netview 5 and 6 LLC. Filed as Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended September 30, 2011 and incorporated herein by reference. |
10.15(b) | | First Amendment to Lease, dated as of November 4, 2011, by and between the Company and Netview 5 and 6 LLC. Filed as exhibit 10.18(b) to the Company’s Annual Report on Form 10-K (File No. 000-24537) for the year ended December 31, 2011 and incorporated herein by reference. |
10.15(c) | | Second Amendment to Lease, dated as of January 5, 2012, by and between the Company and Netview 5 and 6 LLC. Filed as Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended March 31, 2012 and incorporated herein by reference. |
10.16† | | Loan Agreement by and between the Company and LFRP Investors, L.P. dated as of December 29, 2011. Filed as exhibit 10.19 to the Company’s Annual Report on Form 10-K (File No. 000-24537) for the year ended December 31, 2011 and incorporated herein by reference. |
10.17 | | Agreement by and between the Company and Fujifilm Diosynth Biotechnologies UK Limited, dated as of June 29, 2012. Filed as Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q (File No. 000-24537) for the quarter ended June 30, 2012 and incorporated herein by reference. |
| 21.1 | | Subsidiaries of the Company. Filed herewith. |
| 23.1 | | Consent of PricewaterhouseCoopers LLP, an independent registered public accounting firm. Filed herewith. |
| 31.1 | | Certification of Chief Executive Officer Pursuant to §240.13a-14 or §240.15d-14 of the Securities Exchange Act of 1934, as amended. Filed herewith. |
| 31.2 | | Certification of Chief Financial Officer Pursuant to §240.13a-14 or §240.15d-14 of the Securities Exchange Act of 1934, as amended. Filed herewith. |
| 32.1 | | Certification pursuant to 18 U.S.C. Section 1350. Filed herewith. |
| 101** | | The following materials from the Company’s Annual Report on Form 10-K for the year ended December 31, 2011 formatted in Extensible Business Reporting Language (XBRL): (i) Consolidated Balance Sheets as of December 31, 2011 and December 31, 2010, (ii) Consolidated Statements of Operations and Comprehensive Loss for the for the years ended December 31, 2011, 2010 and 2009, (iii) Consolidated Statements of Changes in Stockholders’ Equity (Deficit) for the years ended December 31, 2011, 2010 and 2009 (iv) Consolidated Statements of Cash Flows for the for the years ended December 31, 2011, 2010 and 2009, and (v) Notes to Consolidated Financial Statements, tagged as blocks of text. |
| * | Indicates a contract with management. |
| ** | Pursuant to Rule 406T of Regulation S-T, these interactive data files are deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities and Exchange Act of 1934, as amended, and otherwise are not subject to liability under those sections. |
| † | This Exhibit has been filed separately with the Commission pursuant to an application for confidential treatment. The confidential portions of this Exhibit have been omitted and are marked by an asterisk. |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Company has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, this 4th day of March, 2013.
| | DYAX CORP. |
| | |
| | |
| | By: | /s/ Gustav A. Christensen |
| | | Gustav A. Christensen |
| | | Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Company in the capacities and on the dates indicated.
| Name | | Title | | Date |
| /s/ Gustav A. Christensen | | President and Chief Executive Officer, and | | |
| Gustav A. Christensen | | (Principal Executive Officer) and Director | | March 4, 2013 |
| | | | | |
| /s/ George Migausky | | Executive Vice President and Chief Financial | | |
| George Migausky | | Officer (Principal Financial Officer and | | March 4, 2013 |
| | | Principal Accounting Officer) | | |
| | | | | |
| /s/ Henry E. Blair | | Chairman of the Board of Directors | | |
| Henry E. Blair | | | | March 1, 2013 |
| | | | | |
| /s/ Ron Cohen | | Director | | |
| Ron Cohen | | | | March 1, 2013 |
| | | | | |
| /s/ James W. Fordyce | | Director | | |
| James W. Fordyce | | | | March 1, 2013 |
| | | | | |
| /s/ Mary Ann Gray | | Director | | |
| Mary Ann Gray | | | | March 1, 2013 |
| | | | | |
| /s/ Thomas L. Kempner | | Director | | |
| Thomas L. Kempner | | | | March 1, 2013 |
| | | | | |
| /s/ Marc Kozin | | Director | | |
| Marc Kozin | | | | March 1, 2013 |
| | | | | |
| /s/ David J. McLachlan | | Director | | |
| David J. McLachlan | | | | March 1, 2013 |
| | | | | |
| /s/ Paolo Pucci | | Director | | |
| Paolo Pucci | | | | March 1, 2013 |
96