UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| | | |
þ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES
EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2007
Commission file number: 001-33966
MAKO Surgical Corp.
(Exact Name of Registrant as Specified in Its Charter)
| | | |
| Delaware | | 20-1901148 |
| (State or Other Jurisdiction of Incorporation or Organization) | | (I.R.S. Employer Identification No.) |
| | | |
| 2555 Davie Road, Ft. Lauderdale, FL | | 33317 |
| (Address of Principal Executive Offices) | | (Zip Code) |
(954) 927-2044
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | | |
| Title of Class | | Name of Exchange on Which Registered |
| | | |
| Common stock, $0.001 par value per share | | The NASDAQ Global Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yeso Noþ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yeso Noþ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 (the “Exchange Act”) during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yeso Noþ
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filero | Accelerated filero | Non-accelerated filerþ | Smaller reporting companyo |
(Do not check if a smaller reporting company)
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act)
Yeso Noþ
The aggregate market value of the common stock held by non-affiliates of the registrant as of March 14, 2008 was approximately $99,321,369 (based on a closing price of $10.09 per share on The NASDAQ Global Market as of such date).
As of March 14, 2008, the registrant had outstanding 18,459,633 shares of common stock.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Company’s definitive proxy statement for the 2008 annual meeting of stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K.
MAKO Surgical Corp.
INDEX TO FORM 10-K
We have received or applied for trademark registration of and/or claim trademark rights, including in the following marks that appear in this report: “MAKO Surgical Corp.,” “MAKOplasty®,” “Tactile Guidance System” and “TGS,” as well as in the MAKO Surgical Corp. “MAKO” logo, whether standing alone or in connection with the words “MAKO Surgical Corp.” All other trademarks, trade names and service marks appearing in this report are the property of their respective owners. Unless the context requires otherwise, the terms “registrant,” “company,” “we,” “us” and “our” refer to MAKO Surgical Corp.
2
FORWARD-LOOKING STATEMENTS
This report contains forward-looking statements within the meaning of the U.S. federal securities laws. Statements that are not historical facts, including statements about our beliefs and expectations, are forward-looking statements. Forward-looking statements include statements generally preceded by, followed by or that include the words “believe,” “could,” “expect,” “intend,” “may,” “anticipate,” “plan,” “predict,” “potential,” “estimate” or similar expressions. These statements include, but are not limited to, statements related to:
| | • | | the timing and number of planned new product introductions; |
| |
| | • | | market acceptance of the MAKOplasty solution; |
| |
| | • | | the effect of anticipated changes in the size, health and activities of population on demand for our products; |
| |
| | • | | assumptions and estimates regarding the size and growth of certain market segments; |
| |
| | • | | our ability and intent to expand into international markets; |
| |
| | • | | the timing and anticipated outcome of clinical studies; |
| |
| | • | | assumptions concerning anticipated product developments and emerging technologies; |
| |
| | • | | the future availability of implants and components of our Tactile Guidance System from third-party suppliers, including single-source suppliers; |
| |
| | • | | the viability of maintaining our licensed intellectual property or our ability to obtain additional licenses necessary to our growth; |
| |
| | • | | the anticipated adequacy of our capital resources to meet the needs of our business; |
| |
| | • | | our continued investment in new products and technologies; |
| |
| | • | | the ultimate marketability of products currently being developed; |
| |
| | • | | the ability to implement new technologies successfully; |
| |
| | • | | future declarations of cash dividends; |
| |
| | • | | our ability to sustain sales and earnings growth; |
| |
| | • | | our goals for sales and earnings growth; |
| |
| | • | | our success in achieving timely approval or clearance of products with domestic and foreign regulatory entities; |
| |
| | • | | the stability of certain foreign economic markets; |
| |
| | • | | the impact of anticipated changes in the medical device industry and our ability to react to and capitalize on those changes; |
| |
| | • | | our ability to take advantage of technological advancements; and |
| |
| | • | | the impact of any managerial changes. |
Forward-looking statements reflect our current expectations and are not guarantees of performance. These statements are based on management’s beliefs and assumptions, which in turn are based on currently available information. Important assumptions relating to these forward-looking statements include, among others, assumptions regarding demand for our products, expected pricing levels, raw material costs, the timing and cost of planned capital expenditures, competitive conditions and general economic conditions. You are cautioned that reliance on any forward-looking statement involves risks and uncertainties.
3
Although we believe that the assumptions on which the forward-looking statements contained herein are based are reasonable, any of those assumptions could prove to be inaccurate given the inherent uncertainties as to the occurrence or nonoccurrence of future events. There can be no assurance that the forward-looking statements contained in this report will prove to be accurate. The inclusion of a forward-looking statement in this report should not be regarded as a representation by us that our objectives will be achieved.
Forward-looking statements also involve risks and uncertainties, which could cause actual results to differ materially from those contained in any forward-looking statement. Many of these factors are beyond our ability to control or predict and could, among other things, cause actual results to differ from those contained in forward-looking statements made in this report and presented elsewhere by management from time to time. Such factors, among others, may have a material adverse effect on our business, financial condition and results of operations and may include, but are not limited to, factors discussed under Item 1A, Risk Factors, and the following:
| | • | | changes in general economic conditions and interest rates; |
| |
| | • | | changes in the availability of capital and financing sources; |
| |
| | • | | changes in competitive conditions and prices in our markets; |
| |
| | • | | changes in the relationship between supply of and demand for our products; |
| |
| | • | | fluctuations in costs of raw materials and labor; |
| |
| | • | | changes in other significant operating expenses; |
| |
| | • | | decreases in sales of our principal product lines; |
| |
| | • | | slow downs or inefficiencies in our product research and development efforts; |
| |
| | • | | increases in expenditures related to increased government regulation of our business; |
| |
| | • | | developments adversely affecting our potential sales activities outside the United States; |
| |
| | • | | increases in cost-containment efforts by group purchasing organizations; |
| |
| | • | | loss of key management and other personnel or inability to attract such management and other personnel; |
| |
| | • | | increases in costs of retaining a direct sales force and building a network of independent orthopedic product agents and distributors of our products; |
| |
| | • | | unanticipated expenditures related to any future litigation; and |
| |
| | • | | unanticipated intellectual property expenditures required to develop and market our products. |
We caution you not to place undue reliance on these forward-looking statements that speak only as of the date they were made. We do not undertake any obligation to release any revisions to these forward-looking statements publicly to reflect events or circumstances after the date of this report or to reflect the occurrence of unanticipated events.
4
PART I
ITEM 1. BUSINESS
Overview
We are a medical device company that markets our advanced robotic-arm solution and orthopedic implants for minimally invasive orthopedic knee procedures. We offer MAKOplasty, an innovative, restorative surgical solution that enables orthopedic surgeons to consistently, reproducibly and precisely treat patient-specific, early to mid-stage osteoarthritic knee disease.
Z-KAT, Inc. was formed in 1997 to develop and commercialize computer-assisted surgery, or CAS, applications. Z-KAT acquired, developed and commercialized certain CAS intellectual property and technology assets and also acquired and developed, but did not commercialize, certain haptic robotic intellectual property and technology assets. At the direction of its board of directors and shareholders, Z-KAT formed MAKO Surgical Corp. and incorporated it in Delaware in November 2004, initially as a wholly owned subsidiary, to develop and commercialize unique applications combining CAS with haptic robotics in the medical field of orthopedics. In February 2008, our common stock began trading on The NASDAQ Global Market under the ticker symbol “MAKO” and we closed our initial public offering, or IPO.
MAKOplasty is performed using our proprietary, U.S. Food and Drug Administration, or FDA, cleared Tactile Guidance System, or TGS. Our TGS includes an interactive tactile robotic-arm platform that utilizes tactile-guided robotic-arm technology and patient-specific visualization to prepare the knee joint for the insertion and alignment of our resurfacing implants through a keyhole incision in a minimally invasive, bone-preserving and tissue-sparing procedure. We believe MAKOplasty will empower physicians to address the needs of the large and growing, yet underserved population of patients with early to mid-stage osteoarthritic knee disease who desire a restoration of quality of life and reduction of pain, but for whom current surgical treatments are not appropriate or desirable due to the highly invasive nature of such procedures, the slow recovery and the substantial costs of rehabilitation, medication and hospitalization.
Unlike conventional knee replacement surgery, which requires extraction and replacement of the entire joint, MAKOplasty enables resurfacing of the specific diseased compartment of the joint, preserving significantly more soft tissue and healthy bone of the knee. We believe localized resurfacing can be optimized using the robotic-arm technology of our TGS, which offers consistently reproducible precision to surgeons to achieve optimal implant placement and alignment. We believe that the tissue-sparing and bone-conserving techniques enabled with MAKOplasty can offer substantial advantages to patients, surgeons and healthcare providers. Because of the minimally invasive nature of the procedure, smaller incisions are possible, which lead to less tissue loss and faster recoveries, thereby reducing the overall costs of rehabilitation, medication and hospitalization. In addition, because more of the patient’s natural anatomy is preserved and less trauma is inflicted on the knee, we believe that patients who undergo MAKOplasty have the potential to experience better functionality and more natural knee movements, thereby achieving an improved post-operative quality of life. Finally, because our TGS is easy to use, we believe that our MAKOplasty solution makes resurfacing procedures accessible to orthopedic surgeons with a broad range of training and skills and has the potential to lead to greater adoption of knee resurfacing solutions for early to mid-stage osteoarthritis of the knee.
5
In May 2005, we obtained 510(k) marketing clearance from the FDA for a patient-specific visualization system with a robotic arm that was an earlier version of our TGS. In November 2005, we obtained 510(k) marketing clearance from the FDA for version 1.0 of our TGS. In January 2008, we received 510(k) marketing clearance from the FDA for version 1.2 of our TGS, which incorporates several upgrades developed and introduced since the commercial introduction of version 1.0. We commercially launched version 1.2 in the first quarter of 2008 and plan to launch version 2.0 of our TGS in the first half of 2009, subject to regulatory clearances or approvals, which we may not receive. As part of the sales contract, existing TGS customers are entitled to receive a replacement version 2.0 unit at no additional charge, with the exception of one customer who has the right to receive it at a discounted price. As of December 31, 2007, we commercially installed six TGS units, five of which achieved customer acceptance, and installed two non-commercial additional units for research and evaluation purposes. As of December 31, 2007, 181 MAKOplasty procedures had been performed since commercial introduction in June 2006. We are currently conducting a post-market study of MAKOplasty, which is aimed at demonstrating the accuracy of the placement and alignment of our implants and the clinical value of the MAKOplasty procedure. We released preliminary results of this study in the first quarter of 2008. We have an intellectual property portfolio of more than 200 licensed or owned patents and patent applications relating to the areas of computer-assisted surgery, robotics, haptics and implants.
To date, we have generated revenue primarily from the sale of implants and disposable products to several significant customers. Although we have generated revenue from sales of our current version of the TGS, we are unable to recognize such revenue until we have fulfilled our contractual obligation to deliver version 2.0 of our TGS to customers.
Industry Background
The Growing Osteoarthritis Problem
Osteoarthritis is a common medical condition that leads to the degeneration of joints from aging and repetitive stresses, resulting in a loss of the flexibility, elasticity and shock-absorbing properties of the joints. As osteoarthritis disease progresses, the cartilage and other soft tissues protecting the surfaces of key joints in the body, including knees, hips and shoulders, deteriorate, resulting in substantial and chronic joint pain, numbness and loss of motor function. This pain can be overwhelming for patients and can have significant physical, psychological, quality of life and financial implications. According to estimates by the National Institutes of Health, or NIH, 21 million people in the U.S., or 12.1% of the U.S. population age 25 and older, suffer from osteoarthritis.
6
Compelling demographic trends, such as the growing, aging and more active population and rising obesity rates are expected to be key drivers in the continued growth of osteoarthritis. The NIH projects that by 2030, 20% of Americans, or approximately 72 million people, will be 65 years or older and will be at high risk of developing osteoarthritis. According to Frost & Sullivan, it is estimated that in 2007 there were 73.7 million obese people in the U.S. and by 2012, as many as 88 million Americans will suffer from obesity. According to the American Journal of Epidemiology, obese women had nearly four times the risk of suffering from osteoarthritis of the knee as non-obese women, and obese men had nearly five times the risk of suffering from osteoarthritis of the knee as non-obese men.
For the most severe cases of osteoarthritis, in which patients suffer from extreme pain, reconstructive joint surgery may be required. Reconstructive joint surgery involves the removal of the bone area surrounding the affected joint and the insertion of one or more manufactured implants as a replacement for the affected bone. According to Knowledge Enterprises, Inc., the joint replacement product market as a whole, including knees, hips, elbows, wrists, digits and shoulders, is estimated to have approached $9 billion worldwide in 2004. According to Frost & Sullivan, the U.S. joint implant market was nearly $6 billion in 2006, and is expected to grow to nearly $10 billion by 2013, with knee and hip implant systems representing the two largest sectors.
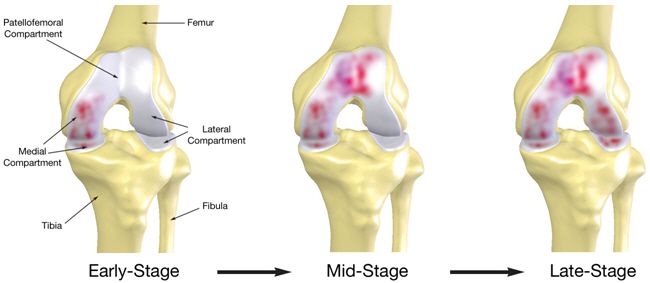
Market for Osteoarthritis of the Knee
The knee joint consists of the medial, patellofemoral and lateral compartments. As depicted below by the shaded diseased areas of the knee joint, osteoarthritis of the knee usually begins with the deterioration of the soft tissue and cartilage in the medial compartment and progresses to either or both the patellofemoral and lateral compartments. The progression of osteoarthritis of the knee can take many years, and even in the early-stages, it can result in substantial pain for the patient and a reduction in the quality of life.
7
According to Datamonitor, in 2006 there were approximately 15 million people in the U.S. with osteoarthritis of the knee. The growth of osteoarthritis of the knee among the U.S. population is expected to accelerate as the increasingly active population ages and obesity rates increase. As a result of this substantial clinical need, the market for orthopedic knee procedures in the U.S. has experienced tremendous growth over the past decade. According to Frost & Sullivan, the U.S. market for total knee replacement and knee resurfacing procedures was greater than $2.7 billion in 2006, and is expected to grow at approximately 8% per year to more than $4.6 billion by 2013. In addition to the substantial costs of the procedure itself, total knee replacement and resurfacing procedures represent significant incremental costs to the healthcare system. These include costs associated with rehabilitation, medication, hospitalization and, over the long-term, costs incurred as a result of replacements or revisions that may be required due to wear and tear or improper placement.
Current Orthopedic Knee Arthroplasty Approaches
To date, arthroplasty options for treating osteoarthritis of the knee have been limited to either total knee replacement surgery or knee resurfacing procedures.
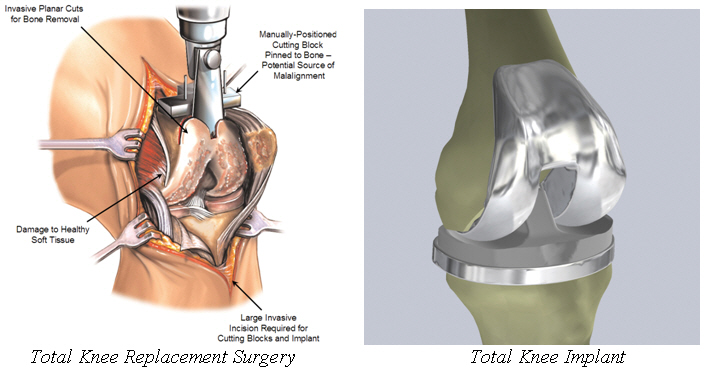
Total Knee Replacement.Currently, most people who choose to surgically address osteoarthritis of the knee elect to undergo total knee replacement surgery. Total knee replacement is a highly invasive surgical procedure in which a patient’s diseased knee joint is removed and replaced with a manufactured replacement knee joint comprised of several components that attempt to mimic the normal function of the knee joint. The procedure requires a large incision ranging from 4 to 12 inches to accommodate the complex scaffold of cutting blocks and jigs required to execute the blunt, planar cuts involved in total knee replacement surgery and to prepare the knee for insertion of the large implants. Both internal and external soft-tissue damage is significant in this procedure as the entire knee joint is fully exposed and much of the bone and tissue surrounding it are removed. The bone cuts are also extensive, presenting a large surface area for bone bleeding. The implants are typically manufactured out of metal, ceramic or polymers and have an approximate useful life of between 15 and 20 years before they usually are revised or replaced.
The figures below illustrate a conventional total knee replacement surgery and implant:
8
Despite its long history as an established and effective orthopedic procedure, total knee replacement surgery is not an ideal option for many patients suffering from early to mid-stage, unicompartmental or multicompartmental degeneration of the knee. Some of the principal limitations of total knee replacement surgeries include:
| | • | | highly invasive nature of the surgical procedure, which requires a large incision ranging from 4 to 12 inches to prepare and implant the large implants; |
| |
| | • | | significant damage to the bone and tissue surrounding the joint; |
| |
| | • | | substantial bone bleeding; |
| |
| | • | | required removal of all three compartments of the knee, regardless of which compartments are actually diseased; |
| |
| | • | | extended and often painful recovery time and rehabilitation; |
| |
| | • | | reduced mobility and range of motion; and |
| |
| | • | | likely implant replacement or revision in approximately 15 to 20 years when the implant reaches the end of its useful life. |
For these and other reasons, many people who are eligible for total knee replacement surgery elect not to undergo or postpone the procedure, choosing instead to suffer significant pain and limited mobility.
Unicompartmental Knee Resurfacing.Unicompartmental knee resurfacing is a less invasive arthroplasty procedure in which only the arthritic region of the knee is removed and a small implant is inserted to resurface the diseased compartment of the knee. Unicompartmental knee resurfacing procedures are ideal for patients with early to mid-stage osteoarthritis and are aimed at sparing the healthy bone, cartilage and other soft tissues typically removed in a conventional total knee replacement procedure. Today, these procedures are generally performed manually and require a level of training, expertise and precision that significantly exceeds what is required for the typical total knee replacement surgery. Orthopedic Network News has estimated that approximately 49,000 unicompartmental knee resurfacing procedures were performed in 2006 in the U.S.
Unicompartmental knee resurfacing is a potentially more desirable procedure than total knee replacement surgery for patients suffering from early to mid-stage degeneration of the knee because it preserves more of the patient’s natural anatomy and results in less trauma to the patient. As a result, patients experience less tissue loss and faster recoveries. However, despite the potential clinical, quality of life and cost benefits of the procedure, it has achieved only limited adoption to date, in part, as a result of the following limitations that make performing the procedure very difficult:
| | • | | the restricted room to maneuver and impeded line of sight due to the smaller incision and minimally invasive nature of the procedure which make it difficult to insert, place and align the implant properly; and |
| |
| | • | | the complex process of removing portions of the bone and resurfacing the knee joint in preparation for the implant. |
9
The difficulties in manually executing a unicompartmental knee resurfacing procedures can result in inaccurate implant alignment, which can lead to reduced range of motion and premature implant failure. In light of the difficulties, many physicians choose not to recommend the procedure and many patients choose either to live with the osteoarthritic pain or to undergo total knee replacement surgery. According to Medtech Insight, LLC, some experts estimate that between 5% to 20% of patients who underwent total knee replacement surgeries had osteoarthritis in only one compartment of the knee, which we believe may qualify them as appropriate candidates for a unicompartmental implant.
Introduction of Minimally Invasive Surgery
Over the past thirty years, one of the most significant medical trends has been the development of minimally invasive methods of performing surgical procedures. Compared to traditional, open surgical techniques, minimally invasive techniques offer potentially superior benefits for patients, surgeons and hospitals. For patients, these techniques result in reduced procedure-related pain and less scarring at the incision site leading to faster recovery times and shorter post-operative hospital stays, as well as better aesthetic outcomes. For the surgeon, these techniques reduce procedure-related complications and have the potential to reduce risks associated with more invasive procedures. For the hospital, these procedures can result in reduced hospital stays for faster recovery times and lower rates of complications.
Despite the many benefits of minimally invasive techniques, however, they also present several notable limitations due to the restricted surgical space, including:
| | • | | restricted vision at the anatomical site; |
| |
| | • | | cumbersome handling of surgical instruments; |
| |
| | • | | difficult hand-eye coordination; and |
| |
| | • | | limited tactile feedback. |
Minimally invasive approaches have seen substantial adoption in various surgical fields where procedures can be performed within existing anatomical cavities of the human body. However, because of the limitations of minimally invasive techniques, they have been less successful for complex surgical procedures requiring cutting and replacement of large anatomical parts that nevertheless require precision and control.
10
Introduction of Robotics into Other Surgical Fields
We believe that the application of robotics technologies in minimally invasive surgical procedures represents the next generation in the evolution of the surgical technique. These technologies are being developed to provide surgeons with a more precise, repeatable and controlled ability to perform complex procedures by offering increased visual acuity and greatly improved tactile feedback. These characteristics empower surgeons to better control their surgical technique and limit the margin of error.
With the assistance of robotics technology, an increasing number of surgeons have been able to perform procedures previously limited to a small subset of highly-skilled surgeons. In addition, robotics technology has allowed these procedures to be performed in a more minimally invasive manner, requiring only small incisions, which result in reduced procedure related trauma, fewer infections and post-procedure complications, and reduced recovery and hospitalization periods.
To date, robotics technology has been successfully applied in a variety of diverse fields including urology, gynecology, cardiothoracic surgery and catheter-based interventional cardiology and radiology. The success of robotics technologies in these applications has led to the growing adoption and commercialization of these technologies in the medical world.
The Use of Robotics in Orthopedic Surgical Procedures
Despite the success of robotics technology in other medical fields, only limited applications have been commercialized in the field of orthopedics to date, although we are aware of current orthopedic robotic development by other companies. Some orthopedic companies have introduced instruments that are smaller than their predecessors, which are marketed as “minimally invasive,” but these instruments still require large incisions to perform the surgical procedure. Orthopedic companies have also introduced computer assisted surgery, or CAS, systems that are designed for use in open procedures. However, while these systems do provide a minimally invasive means of viewing the anatomical site, their benefits are marginal because they do not improve a surgeon’s ability to make consistently reproducible and precise surgical movements through a small keyhole incision.
We believe that the limitations of currently available surgical options for knee disease have created a sizeable market for treatment of a large, growing and underserved population of patients with early to mid-stage osteoarthritis of the knee. We believe that robotics technology is the key to enabling surgeons to perform the kind of minimally invasive knee surgery that results in restoration of function and improved post-operative outcomes for such patients.
11
The MAKO Solution
We have designed our MAKOplasty solution to provide the consistently reproducible precision, accuracy and dexterity necessary for a surgeon to successfully perform minimally invasive orthopedic arthroplasty procedures on the knee despite a limited field of vision in a confined anatomical space. Our MAKOplasty solution is composed of two critical components: the TGS, which consists of the proprietary tactile robotic-arm and our patient-specific visualization system that provides both pre-operative and intra-operative guidance to the surgeon, and the MAKO implant portfolio that is designed for minimally invasive restoration of the diseased compartment of the joint. By integrating robotic-arm and patient-specific visualization technology with the touch and feel of the surgeon’s skilled hand, MAKOplasty is designed to enable a level of surgical precision and accuracy that is beyond the scope of the typical surgeon’s freehand capabilities, which we believe will result in broad adoption of our technologies by orthopedic knee surgeons and better outcomes for patients. We believe MAKOplasty offers the following key benefits to patients, surgeons and hospitals:
| | • | | Minimally Invasive Targeted Knee Arthroplasty. MAKOplasty enables surgeons to isolate and resurface just the diseased compartment of the knee joint through a minimally invasive keyhole incision, rather than replacing the entire joint. The precision of our robotic-arm technology makes such minimally invasive targeted treatment possible by eliminating the complex scaffold of cutting blocks and jigs that would otherwise be required to execute the blunt, planar bone cuts and insert the large implants involved in conventional total knee replacement surgery or a manually executed resurfacing procedure. We believe that our solution will make minimally invasive orthopedic procedures, like unicompartmental resurfacing, a viable option for a greatly expanded pool of patients and physicians. |
| |
| | • | | Consistently Reproducible Precision. We believe that MAKOplasty will reduce the variability of procedure outcomes and increase efficacy through the consistently reproducible precision provided by our computer assisted and tactile robotic arm technology. We believe that the precision of our cutting process and placement and alignment of implants leads to significantly improved and reliable results, compared to conventional, manually executed unicompartmental resurfacing procedures. The surgeon retains control of the actual movements of the robotic arm within a pre-established volume of space, the tactile “safety zone,” which is tracked and bounded by our TGS. We believe that the tactile safety zone enables improved placement and alignment of the implant, while the 3-D visualization enables the procedure to be performed through a small incision without direct visualization. We believe that this consistently reproducible precision will enable physicians to be trained in the use of MAKOplasty in a relatively short period of time and also increase the number of physicians who are willing and able to perform unicompartmental resurfacing procedures. |
| |
| | • | | Ease of Use. We believe that our TGS leverages and complements the surgical skills and techniques already familiar to the surgeon, while providing substantial incremental control and precision that has not previously been possible. The customized, patient-specific visualization system guides the surgeon through each step of the surgical procedure, while the tactile “safety zone” ensures that the surgeon does not apply the bone cutting instrument beyond the intended area of the knee joint. |
12
| | • | | Improved Restorative Post-Operative Outcomes. Due to the minimally invasive nature of the procedure, we believe that patients who undergo MAKOplasty are likely to experience less tissue loss, less visible scarring and a faster recovery, thereby reducing the cost of rehabilitation, physical therapy, medication and hospitalization. In addition, because more of the patient’s natural anatomy is preserved and less trauma is inflicted on the knee, patients who undergo MAKOplasty have the potential to experience better mobility, comfort, range of motion and more natural knee movements to achieve an improved post-operative quality of life. |
| |
| | • | | Reduced Costs for Patients and Hospitals. The minimally invasive nature of the MAKOplasty solution aids hospitals and patients in reducing costs by shortening hospital stays and recovery periods and reducing the amount of rehabilitation and medication. |
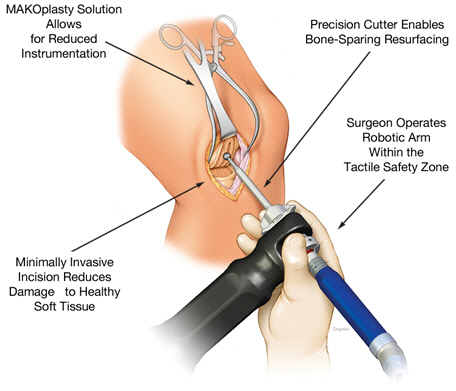
The comprehensive nature of the MAKOplasty solution also provides hospitals with all the implants and disposable products necessary to perform the procedures. We believe that our complete knee arthroplasty solution represents a substantial improvement over currently available approaches that we hope will lead to rapid adoption in the marketplace.
The figure below illustrates a MAKOplasty unicompartmental resurfacing procedure.
13
Our Strategy
Our goal is to drive sales of our TGS and generate recurring revenue through sales of implants, disposable products and service contracts by establishing MAKOplasty as the preferred surgical procedure for patients with early to mid-stage, unicompartmental and multicompartmental degeneration of the knee. We believe that we can achieve this objective by working with hospitals to demonstrate key benefits of MAKOplasty, such as consistently reproducible surgical precision, improved post-operative outcomes and reduced healthcare costs. Our strategy includes the following key elements:
| | • | | Focus on key physicians and thought leaders to encourage early adoption of our MAKOplasty solution. We plan to initially focus our marketing efforts on key orthopedic surgeons who currently perform the majority of unicompartmental knee procedures or who are actively involved in the development of minimally invasive orthopedic approaches. We also plan to focus our marketing efforts on the hospitals with which these key surgeons are affiliated and engage them to promote the benefits of MAKOplasty. Our strategy is to convince hospitals that through early adoption of MAKOplasty and purchase of our TGS, they can reinforce their reputations as leading institutions for the treatment of early to mid-stage osteoarthritis of the knee. |
| |
| | • | | Expand the market for unicompartmental knee resurfacing. We plan to expand the market for unicompartmental knee resurfacing procedures by encouraging use of the procedure for patients who, given only conventional surgical alternatives, would have opted for total knee replacement surgery or no surgery at all. Our current application of MAKOplasty is for unicompartmental knee resurfacing procedures using either an inlay knee implant system or onlay knee implant system, allowing us to accommodate varied patient profiles and surgeon preferences. The addition of onlay knee implants to our offerings helps accommodate additional patient profiles and surgeon preferences. We believe that the potential benefits of our MAKOplasty solution and the combination of these product offerings will facilitate our efforts to expand and capture the market for unicompartmental knee resurfacing. |
| |
| | • | | Drive volume sales of implants and disposable products for installed TGS units. Following the initial installation of our TGS at a given hospital, we intend to expand the number of orthopedic surgeons who use our TGS and work with the hospitals and their surgeons to promote patient education about the benefits of MAKOplasty. Our goal is to increase usage per system to drive higher volume sales of our implants and disposable products. |
| |
| | • | | Expand our product offerings to multicompartmental implants. We believe that a key to growing our business is expanding the application of MAKOplasty to resurfacing procedures that address mid-stage multicompartmental degeneration of the knee. This modular application of MAKOplasty to multicompartmental resurfacing procedures will allow orthopedic surgeons to treat degenerative osteoarthritis of the knee from early-stage, unicompartmental degeneration through mid-stage, multicompartmental degeneration with a single knee implant system. To achieve this goal, we are developing the next version of our TGS, which will include improved surgical planning and execution software and customized bone cutting instruments. We are also developing new modular implants, as well as strengthening our intellectual property rights as necessary to support these new offerings. We believe that this expanded product offering should position us as a leading company in the field of early to mid-stage orthopedic knee procedures, offering a complete range of minimally invasive solutions for the treatment of osteoarthritis of the knee. |
14
| | • | | Demonstrate the clinical and financial value proposition of MAKOplasty. We intend to collaborate with leading surgeons and early-adopting hospitals through such programs as the MAKOplasty Knee Center of Excellence to build clinical and financial data that support the benefits of MAKOplasty. The MAKOplasty Knee Center of Excellence is a program developed in conjunction with participating hospitals to educate surgeons and patients regarding the benefits of MAKOplasty. As part of the collaborative program, participating hospitals maintain and provide us with certain clinical and financial data that we use to support the business case for the MAKOplasty solution. Our goal is to obtain clinical data further supporting the value of MAKOplasty unicompartmental resurfacing procedures, as well as the accuracy and longevity of such implant placements, while demonstrating to hospitals the top and bottom line financial benefits of our MAKOplasty solution. |
Our Products
Our proprietary technology consists of two components: our TGS and our knee implants for use in the resurfacing procedures.
Tactile Guidance System
The centerpiece of MAKOplasty is the TGS, a proprietary tactile robotic-arm and patient-specific visualization system that provides both pre-operative and intra-operative guidance to the surgeon, enabling minimally invasive, tissue-sparing bone removal and implant insertion. Our TGS consists of two elements: a tactile robotic arm utilizing an integrated bone-cutting instrument and a patient-specific visualization component.
The figures below identify the key components of the tactile robotic arm and stereo tracking system and instruments:
15
Tactile Robotic-Arm System.The tactile robotic-arm system consists of the key components identified in the figures above and incorporates the following specifications, features and benefits:
| | • | | Tactile Robotic Arm— The tactile robotic arm is designed to respond fluidly to movements initiated by the surgeon operating the bone cutting instrument. We have designed the robotic arm with five degrees of freedom which enables the robotic arm to achieve substantial dexterity and range of movement. The robotic arm helps enforce a tactile safety zone that is established by the patient-specific visualization system by providing tactile resistance when the boundaries of the tactile safety zone are reached. This tactile resistance helps ensure that the surgeon does not apply the bone cutting instrument beyond the intended area of the knee joint. |
| |
| | • | | Controller— The controller is the electronic hardware and firmware component of our computing system which interfaces with our proprietary surgical planning and execution software to allow the surgeon to safely guide the tactile robotic arm. The controller governs the basic, low-level functions of the tactile robotic arm, such as the tactile constraints and the safety circuit. |
| |
| | • | | Stereo Tracking System Camera and Instruments— During a MAKOplasty procedure, the location of the tactile safety zone is updated continuously based on bone tracking data supplied to the computer system by an infrared stereo tracking system, which consists of a special camera that is directed toward a series of spheres and arrays placed in the patient’s anatomy by bone pins. The tracking system assists the TGS in locating and physically tracking the patient’s anatomy and coordinating its real-time position with the cutting instrument of the robotic arm. It has a refresh rate of approximately 30 — 60 hz (cycles/second), providing the TGS with a sufficient flow of information regarding movements by both the patient and the robotic arm to ensure optimal cutting and placement. Our TGS updates the tactile safety zone output forces at a rate of 2,000 Hz (cycles/second), enabling it to adjust for movements of the tracked anatomy by dynamically adjusting the position of the tactile safety zone. As a result, the surgeon can freely move the robotic arm within the defined space, but encounters tactile resistance as the boundaries of such space are reached. |
| |
| | • | | End Effector— The end effector is the mechanical component by which the bone cutting instrument is attached to the tactile robotic arm. It is designed to ensure the secure placement of the bone cutting instrument, while providing the flexibility necessary for the surgeon to manipulate the instrument. |
| |
| | • | | Bone Cutting Instrument with Disposable Cutting Tip— The bone cutting instrument is integrated into the tactile robotic arm at the end effector. This instrument is composed of a high-speed motor and a component that houses a variety of single-use bone cutting tips. The design of the bone cutting instrument allows the surgeon to grip it in a manner similar to holding a pen-like cutting tool, making it easy to manipulate the instrument in the patient’s anatomy. The cutting tip is the disposable end tip of the bone cutting instrument that makes contact with the knee joint and actually removes the bone for placement of the implant in accordance with the pre-operative plan. In combination with our tactile robotic arm, the bone cutting instrument enables the smooth precision and accuracy necessary for resurfacing procedures. |
| |
| | • | | Portable Base Console— The base component of our tactile robotic arm is a mobile unit that enables the portability of the tactile robotic arm from one operating room to another. The base controller houses the controller and various electrical and mechanical components that help power the tactile robotic arm. Its design enables the console to be situated next to the patient during surgery and the tactile robotic arm to be conveniently positioned over the patient’s anatomy. |
16
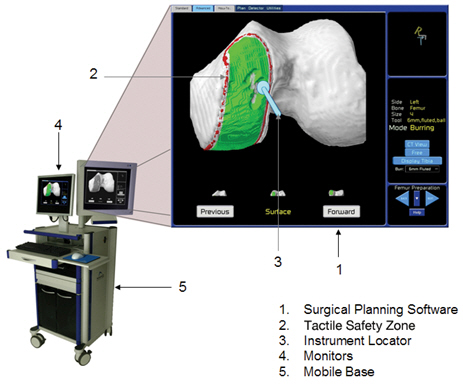
The figure below identifies the key components of the patient-specific visualization system:
Patient-Specific Visualization System.Our patient-specific visualization system is a vital part of our ability to deliver minimally invasive surgical procedures for the knee. The surgical team uses our system pre-operatively to plan and intra-operatively to guide the surgical procedure. It consists of the key components identified in the figure above and incorporates the following specifications, features and benefits:
| | • | | Surgical Planning and Execution Software— Our surgical planning and execution software, which is integrated into our patient-specific visualization system, is used during the pre-operative surgical planning process to visualize and map the exact portion of bone to be removed and resurfaced, define the anatomical boundaries of the tactile safety zone and plan the optimal placement and alignment of our implants. During the procedure, the visualization system guides the surgeon through each specific, well-defined surgical technique and displays in real time each current and planned surgical activity. |
| |
| | • | | Tactile Safety Zone— While the robotic arm enforces a tactile safety zone by providing tactile resistance when the boundaries of the tactile safety zone are reached, our patient-specific visualization system provides a visual representation of the tactile safety zone and provides additional visual and auditory cues when the boundaries of such tactile safety zone are reached. The combination of this tactile resistance and patient-specific visualization helps ensure that the surgeon does not apply the bone cutting instrument beyond the intended area of the knee joint. |
17
| | • | | Instrument Locator— The instrument locator provides visual guidance on the position of the bone cutting instrument and other surgical instruments in relation to the patient’s anatomy. |
| |
| | • | | Monitors— Prior to surgery, patients undergo a conventional CT-scan that captures an image of the diseased knee joint. This CT-image is uploaded to the patient-specific visualization system, where a MAKO clinical technical specialist processes the image for display as a 3-D volume in space corresponding to the implant shape and placement overlaid onto the CT-image of the patient’s knee joint. This patient-specific visualization of our implant overlaid onto an image of the patient’s actual knee joint helps the surgeon to pre-operatively plan the procedure, by providing information which enables the surgeon to determine the optimal placement, alignment and sizing of the implant and establishing the boundaries of the tactile safety zone. During surgery, each monitor projects an active 3-D computer graphics visualization of the patient’s knee joint, showing the areas of the bone that are actually removed as the procedure progresses. The user can also change the viewpoint and zoom level of the visualization as the procedure progresses to focus on different portions of the anatomy. |
| |
| | • | | Mobile Base— The base component of our patient-specific visualization system is a mobile unit that enables the portability of the patient-specific visualization system from one operating room to another. It houses our computer hardware and our surgical planning and execution software and various electrical and mechanical components that help power the visualization system. |
Version 1.0 of the Tactile Guidance System
In November 2005, we obtained 510(k) marketing clearance from the FDA for version 1.0 of our TGS for use with an inlay knee implant system, as described below. We subsequently developed and introduced several upgrades to our TGS, including improvements to our surgical planning software as well as changes to certain instrumentation to make the device easier to use. We determined that these modifications, embodied in version 1.1 of our TGS, did not require the submission of a new 510(k) application.
Version 1.2 of the Tactile Guidance System
In January 2008, we obtained 510(k) marketing clearance from the FDA for version 1.2 of our TGS, which became commercially available in the first quarter of 2008. Version 1.2 reflects further refinement of the basic instrumentation set and features a customized bone cutting instrument and new surgical planning software applications necessary to support unicompartmental resurfacing procedures using a tibial onlay knee implant system.
As part of our ongoing efforts to improve our TGS, we research, develop and launch product iterations from time to time. We have commenced development of a software application, expected to launch by the end of 2008, which, as version 1.3, will enable integration of components of both our inlay and onlay knee implant systems into a single MAKO-branded unicompartmental implant system, for use with our TGS. We do not anticipate that these modifications will require the submission of a new 510(k) application.
18
Future Versions of the Tactile Guidance System
We are developing version 2.0 of our TGS. We expect that version 2.0 will represent an important expansion from the first generation of our TGS, enabling application of MAKOplasty to multicompartmental resurfacing procedures, allowing orthopedic surgeons to treat degenerative osteoarthritis from early-stage, unicompartmental degeneration through mid-stage, multicompartmental degeneration with a modular knee implant system. In addition, we currently plan to incorporate in version 2.0 the following improvements, which we believe will allow us to offer the benefits of MAKOplasty to more patients:
| | • | | improved dexterity and range of motion in the robotic arm to allow additional degrees of freedom in the movement of the robotic arm; |
| |
| | • | | more efficient physical configuration of the patient-specific visualization system, robotic arm, customized bone cutting instruments and electronic components; |
| |
| | • | | improvement of the tracking system for monitoring movements by the patient and the robotic arm; |
| |
| | • | | intelligent implant planning features that will aid the surgeon in achieving optimal patient-specific alignments; |
| |
| | • | | redesign of certain components to make them more accessible for service repairs and easier to replace; and |
| |
| | • | | sophisticated industrial design and state-of-the art user interface. |
We plan to commercially release version 2.0 of our TGS in the first half of 2009, subject to regulatory clearances or approvals, which we may not receive. In addition, we intend to apply for a European Union CE marking.
Knee Implants
The second component of MAKOplasty is the implant that is designed for insertion and cementation in a minimally invasive manner. We currently offer both an inlay knee implant system and an onlay knee implant system for unicompartmental resurfacing procedures.
Inlay Implant for Use in Unicompartmental Procedures
The inlay knee implant system is composed of a rounded, anatomically-shaped femoral component that attaches to the sculpted surface of the femur and a flat polymer component that fits into a “pocket” that has been sculpted in the tibial bone using the TGS. Both the femoral and tibial components are offered in multiple sizes to best accommodate the size and shape of the patient’s knee. Patients with relatively good tibial bone quality, including a sufficiently thick and appropriately located bed of hardened sclerotic tibial bone, are generally candidates for our inlay implants.
19
Onlay Implant for Use in Unicompartmental Procedures
We received 510(k) marketing clearance for version 1.2 of our TGS from the FDA in January 2008. Version 1.2 of our TGS can be used with either our inlay or onlay knee implant systems. The addition of onlay knee implants to our offerings helps accommodate different patient profiles and surgeon preferences. The MAKO onlay knee implant system consists of a femoral component and a flat polymer component that is backed by a metal support. The metal support is placed horizontally on a planar surface prepared on the tibia using the TGS, supported by the tibial cortical rim, rather than fitted into a pocket of the tibia. The onlay knee implant system is designed to accommodate patients who lack sufficient quality tibia sclerotic bone bed. Some surgeons also prefer to utilize the tibial cortical rim support in all cases. We plan to offer both the femoral and tibial components in multiple sizes to best accommodate the size and shape of the patient’s knee.
We have commenced development of and received 510(k) clearance for a single MAKO-branded unicompartmental implant system for use with our TGS that integrates components of both our inlay and onlay knee implant systems. Because of our TGS’s technical design and programming, only our knee implant systems may be used effectively with our TGS. In addition, purchasers of our TGS are contractually required to purchase all implants and disposable products used in MAKOplasty procedures from us.
20
Modular Implants for Use in Multicompartmental Procedures
We are currently in the process of developing a proprietary modular knee implant system for use with version 2.0 of our TGS, which we expect to offer on a commercial basis in the first half of 2009, subject to the receipt of regulatory clearance or approval. This line of implants would allow an orthopedic surgeon to treat degenerative osteoarthritis of the knee from early-stage, unicompartmental degeneration through mid-stage, multicompartmental degeneration with a modular implant system. We believe that modular components are key to the successful execution of minimally invasive knee surgeries because they can be more easily inserted into the knee joint through smaller incisions than a single, complete device. They can also be positioned independently to better accommodate the specific contours of the patient’s anatomy. We are planning development of the modular knee implant system for targeted release in the first half of 2009, subject to regulatory clearance or approval. We expect to seek 510(k) marketing clearance from the FDA, but it is possible that pre-market approval, or PMA, may be required if the new implant is not eligible for 510(k) marketing clearance, in which case our commercial release would likely be delayed. See “Regulatory Requirements of the U.S. Food and Drug Administration” below.
Disposable Products
Our TGS utilizes disposable products such as the arrays, bone pins and spheres used in our tracking system, irrigation clips and tubes that cool the cutting instruments, a boot used to position the patient’s leg, drapes to cover the robotic arm and other items that require disposal after each use. Disposables are not only a potential source of recurring revenue, but also an opportunity to differentiate our product platform from those of less comprehensive solutions offered by competitors.
Future Potential Applications
We believe that with further research and development, our robotic-arm technology has the potential to serve as a platform technology with applications in other areas of the body, such as the hip, shoulder and spine. However, we are not currently pursuing applications of MAKOplasty outside of the knee, and to date, we have not conducted significant research or development for these other potential applications. Moreover, our products do not have marketing clearance from the FDA or any other regulatory approvals for applications outside of the knee.
Sales and Marketing
We are currently building a sales and marketing organization comprised of a direct sales force and a network of independent orthopedic product agents and distributors, who primarily generate leads for us, to commercialize and market MAKOplasty in the U.S. As of March 21, 2008, our sales and marketing group had a total of 30 employees, including six direct sales representatives, who are responsible for sales and marketing activity throughout the U.S. We expect to increase the number of sales and marketing personnel as we continue to expand our business.
21
Our sales and marketing goals are to drive capital equipment sales of our TGS and generate recurring revenue through sales of implants, disposable products and service contracts. To achieve these goals, we must promote early adoption of MAKOplasty by leading surgeons and hospitals and build demand for the procedure among patients through the following sales and marketing strategy:
| | • | | Target High Volume Orthopedic Facilities.Our sales representatives actively target hospitals with strong orthopedic reputations and significant knee replacement and resurfacing practices. We believe that early adoption by such leading hospitals will help us to seed the market for MAKOplasty and provides the validation and visibility necessary for more widespread adoption. |
| |
| | • | | Establish and Promote MAKOplasty Knee Centers of Excellence.The MAKOplasty Knee Center of Excellence is a joint marketing program that we promote in collaboration with participating hospitals to educate surgeons and patients regarding the benefits of MAKOplasty and to coordinate our public relations strategy. As part of the program, hospitals agree to maintain and provide us with certain clinical and financial data that we use in support of our business case for the MAKOplasty solution. As of December 31, 2007, we entered into four co-marketing agreements with hospitals to establish MAKOplasty Knee Centers of Excellence. |
| |
| | • | | Drive Patient Demand for MAKOplasty.We plan to expand our marketing efforts to include direct-to-patient marketing. We believe that patients are becoming increasingly more involved in the healthcare decision-making process and have the potential to influence the adoption of new procedures such as MAKOplasty. Currently, our representatives support hospitals participating in the MAKOplasty Knee Center of Excellence program in their efforts to publicize the benefits of MAKOplasty and educate patients. |
The generation of recurring revenue through sales of our implants, disposable products and service contracts is an important part of the MAKOplasty business model. We anticipate that recurring revenue will constitute an increasing percentage of our total revenue as we leverage each new installation of our TGS to generate recurring sales of implants and disposable products. To enhance our generation of recurring revenue, purchasers of our TGS are contractually required to purchase all implants and disposable products used in MAKOplasty procedures from us. In addition, because of our TGS’s technical design and programming, only our knee implant systems may be used effectively with our TGS. We also offer a four-year supplemental service contract that provides enhanced levels of maintenance and support services related to our TGS beyond the basic warranty period. We also offer protection against technological obsolescence, which requires us to upgrade the installed version of our TGS to version 2.0 and provide all interim software and hardware version enhancements.
22
We provide training to surgeons and hospital staff on the use of the TGS. Our customers also receive pre-operative and intra-operative support from our on-site clinical and technical representatives who provide clinical and technical support in connection with each MAKOplasty procedure. The representative helps set up the equipment, participates in the pre-operative planning process and is present in the operating room with the surgeon, facilitating the surgeon’s use of the TGS. By increasing familiarity with the system and helping ensure safe and proper usage of our equipment and products by surgeons and hospitals, we hope to promote seamless adoption of MAKOplasty. The presence of our representatives in the surgical theater also provides us with immediate feedback and understanding of our customers’ preferences and requirements in clinical conditions.
Research and Development
Continued innovation through research and development is critical to our future success. Substantially all of our research and development activity is performed internally. As of March 21, 2008, our research and development team, which is based at our headquarters in Ft. Lauderdale, Florida, consisted of 53 employees. We have assembled an experienced team with recognized expertise in advanced robotics, software, instrumentation and orthopedic knee implants. Although we do not currently have plans to increase the size of our research and development team significantly, we may do so in the future, depending on the progress of our ongoing research and development efforts.
Our principal research and development goal is to enable use of MAKOplasty for both unicompartmental and multicompartmental knee resurfacing procedures. To that end, we are working to improve the dexterity and range of motion in the robotic arm of our TGS and developing upgraded surgical planning software to facilitate multicompartmental resurfacing procedures. We expect to incorporate these improvements in version 2.0 of the TGS. We are also researching customized bone cutting instruments and alternative tracking systems that may be more robust, easier to use, fit better into the busy operating room environment and have improved tracking performance. Similarly, we are researching and developing a modular knee implant system that would allow a single knee implant system to treat multiple stages of osteoarthritis of the knee from early-stage unicompartmental degeneration through mid-stage, multicompartmental degeneration.
We have historically spent a significant portion of our capital resources on research and development. Our research and development expenses were $8.3 million in fiscal year 2007, $5.2 million in fiscal year 2006 and $2.6 million in fiscal year 2005.
Manufacturing and Assembly
The MAKOplasty solution includes both off-the-shelf and custom-made components produced to our specifications by various third parties. We purchase major components of our TGS, including the computer hardware, the camera used in connection with our tracking system, robotic controller components, the high-speed bone cutting instrumentation, the molded plastic and machined metal parts, and the various electro-mechanical components that support the robotic-arm system from a number of third-party suppliers. We internally develop the software components of our TGS. We then assemble and integrate these various hardware components with our proprietary software to complete each TGS. By assembling the final product at our facility, we are able to perform stringent quality assurance inspection and testing on each TGS to best control the quality of the final product prior to shipment. We also purchase fully manufactured and pre-packaged implants from third-party suppliers. A portion of our Ft. Lauderdale facility is presently dedicated to these warehousing, assembly, testing and inspection activities.
23
Other than our proprietary software, single source suppliers currently provide us with all major components of the TGS, including the bone cutting instrument, and our current offering of implants.
We generally purchase our components through purchase orders and do not have long-term contracts with most of our suppliers. We have, however, entered into a long-term contractual arrangement, including both supply and license agreements, with Encore Medical, L.P., the supplier of our onlay knee implant system. Under the supply agreement, Encore provides us with the desired quantity of implants in accordance with a fixed pricing schedule. Our supply contract with Stelkast (a business division of Trigon Incorporated), the supplier of our inlay knee implants, expired in September 2007, and we currently purchase our inlay knee implants from Stelkast pursuant to purchase orders. We do, however, have a long-term license agreement with Stelkast. Under the license agreements, Stelkast granted us a non-exclusive license and Encore granted us an exclusive license to the design of the respective implants for use with the TGS and the right to sublicense for the manufacture of components. We have also entered into a long-term agreement with Symmetry Medical, Inc. to manufacture, label and package knee implant and instrument systems, pursuant to which we plan to have Symmetry supply us with one or more of inlay knee implants, and onlay knee implants and related instrumentation, subject to final agreement on pricing. Our agreement with Symmetry also contemplates the development and manufacture of new implant designs in the future.
Our supply agreement with Encore expires on the date we are able and ready to make and sell onlay implants independently, under our own label and own 510(k) clearance, but no later than February 28, 2010, which we may extend by one year periods. The supply agreement with Encore terminates automatically upon the termination of the corresponding license agreement that we have entered into with Encore. In addition, Encore may terminate its supply agreement at any time Encore ceases to manufacture the onlay implants based on a bona fide product safety, efficacy or regulatory concern or upon Encore’s six months written notice to us that Encore elects to cease manufacturing onlay implants for any other reason. Our agreement with Symmetry continues until terminated. We may terminate the agreement with Symmetry for any reason upon 180 days notice, and Symmetry may terminate the agreement for any reason upon one year’s notice.
We intend to achieve improvements in our manufacturing operations and in our cost of sales by improving our procurement and third-party manufacturing processes. We also intend to upgrade our management information systems and implement new quality assurance, inventory and cost controls to improve the efficiency of our manufacturing operations, maintain product quality, reduce our cost of sales and increase our profitability.
24
Our operations and those of the third-party suppliers and manufacturers we use are subject to extensive regulation by the FDA under its Quality System Regulations, or QSRs, as well as numerous post-market requirements. Our operations and those of third-party suppliers and manufacturers may also be subject to international regulatory requirements in the event we expand our operations or business overseas. Our facility is FDA registered and we believe is compliant with FDA’s QSR. We have instituted a quality management system to evaluate and monitor compliance internally and by our third-party suppliers and manufacturers. Our facility and the facilities of the third-party suppliers and manufacturers we use are subject to periodic, announced and unannounced inspections by regulatory authorities, including the FDA and other governmental agencies. To date, our facilities have not been inspected by any regulatory authorities. We did pass a BSi certification audit of our Quality System to ISO 13485:2003 in preparation for CE marking. BSi will be doing surveillance audits once a year to make sure we continue to be in compliance.
Intellectual Property
We must develop, maintain and protect the proprietary aspects of our products and technologies to remain competitive in the marketplace. Our intellectual property portfolio includes rights to patents, patent applications and other intellectual property that we wholly-own or license from others. We seek patent and other intellectual property protection in the U.S. and internationally for our products and technologies where available and when appropriate.
We also rely on other forms of intellectual property rights, including copyright, trademark, trade secrets and know-how, to develop, maintain and protect the proprietary aspects of our products and technologies. We require our employees and consultants to execute confidentiality agreements in connection with their employment or consulting relationships with us. We also require our employees and consultants to disclose and assign to us all inventions conceived during the term of their employment or engagement while using our property or which relate to our business.
Despite measures taken to protect our intellectual property, unauthorized parties may attempt to copy aspects of our products or to obtain and use information that we regard as proprietary. In addition, our competitors may independently develop similar technologies. Although patents may provide some degree of protection for our intellectual property, patent protection involves complex legal and factual determinations and is therefore uncertain.
Wholly-Owned Patent Applications
As of January 1, 2008, we held 19 wholly-owned pending U.S. patent applications. All of these patent applications are either used in our current products or relate to core technologies used in our products, such as CAS, robotics, haptics and implants. The first of our currently pending patent applications was filed in October 2003 and should expire in October 2023, exclusive of any statutory extensions or reductions. None of our patent applications has yet issued. As of January 1, 2008, we also held 21 foreign patent applications. We are also pursuing additional U.S. and foreign patent applications on key inventions to enhance our intellectual property portfolio.
25
Patents and Patent Applications Licensed from Third Parties
As of January 1, 2008, we had licensed rights to 118 U.S. and 47 foreign third-party granted patents, and we had licensed rights to 22 U.S. and 40 foreign third-party pending patent applications. The majority of these patents and applications are either used in our current products or relate to core technologies used in our products, such as CAS, robotics, haptics and implants. We also have rights to additional third-party patents and intellectual property that relate to our core technologies, but are not currently used in our products. Nine of the licensed U.S. patents and three related foreign patents will expire by the end of 2009. Of these, four licensed U.S. patents and three related foreign patents will expire during 2008 and five licensed U.S. patents will expire by the end of 2009. Two of these U.S. patents and all three related foreign patents are method patents related to CAS, and three of these U.S. patents relate to robotic technology. These five U.S. patents and the related foreign patents are considered material to our intellectual property portfolio because they potentially enable us to exclude others from practicing the claimed technology. The last licensed patent will expire in 2024.
License Arrangements with Z-KAT
Our principal licensing arrangement is with Z-KAT, from whom we license or sublicense core technologies in CAS, haptics and robotics. In connection with our formation in November 2004, we were granted an exclusive, irrevocable, non-terminable license or sublicense to all intellectual property owned or licensed by Z-KAT in the field of medical orthopedic surgery to the extent Z-KAT’s licenses from third parties were exclusive. Our license from Z-KAT includes a limited license to Z-KAT’s CAS and haptic robotic intellectual property portfolio for exclusive use in the field of orthopedics, subject to a prior license to Biomet Manufacturing Corp. to use Z-KAT’s CAS intellectual property, but not its haptic robotic intellectual property, in the field of orthopedics. Because of the prior license to Biomet and pursuant to our license with Z-KAT, we cannot use the CAS intellectual property on a stand-alone basis; we can only use the CAS intellectual property in combination with robotics technology. Z-KAT’s license also granted to us the sole right to prosecute and maintain all Z-KAT patents and patent applications that are licensed to us. In 2006, we obtained the right to take enforcement action against all third parties with respect to any intellectual property rights held by Z-KAT in the field of orthopedics. We have granted back to Z-KAT a fully paid, royalty-free, nonexclusive sublicense to our intellectual property portfolio in all fields other than orthopedic surgery. Through these and other arrangements, we have rights to Z-KAT’s wholly-owned and third-party licensed intellectual property portfolio, which includes a wide suite of intellectual property in the areas of haptic robotics and patient-specific visualization.
License Arrangements with Other Third Parties
In September 2005, we entered into a license agreement with Integrated Surgical Systems, Inc. pursuant to which we obtained an exclusive, worldwide license to patented technology relating to bone registration and tracking for use in the field of human interactive robotics in orthopedics and a nonexclusive license in the field of orthopedics generally. We paid a one-time licensing fee that provides a fully paid, worldwide license for the life of the licensed patents.
26
In March 2006, we entered into a license agreement with IBM that covers a number of technologies related to the application of computers and robotics to surgery. Under the terms of this agreement, we have a nonexclusive, worldwide license to any IBM patents and patent applications with effective filing dates prior to March 31, 2011 in the field of robotic devices primarily designed for surgery in the medical field of orthopedics and/or primarily designed for spinal surgery in the medical field of neurology. We are obligated to make royalty payments based on the sale of each robotic product covered by the IBM patents. The IBM license agreement will terminate upon the expiration of the last licensed patent.
In May 2006, we entered into a sublicense agreement with SensAble Technologies, Inc. The sublicense grants nonexclusive rights in the field of CAS to a patent directed to core haptic technology that SensAble licensed from MIT. The sublicense also included an option to license or sublicense five additional patents, which we exercised in May 2007. We paid a one-time sublicensing fee (and a one-time option fee) that provides a fully paid, worldwide license for the life of the licensed patents. A subsequent dispute concerning this sublicense is discussed in Item 3, Legal Proceedings, and in Item 8, Financial Statements and Supplementary Data, Note 6 to the Financial Statements, of this report.
Competition
Our success depends on convincing hospitals, surgeons and patients to utilize the robotic-arm technology embodied in both our current version of the TGS to perform unicompartmental resurfacing and our planned version 2.0 of the TGS to perform multicompartmental resurfacing of the knee. We face competition from large, well-known companies, principally Zimmer Holdings, Inc., DePuy Orthopedics, Inc., a Johnson & Johnson company, Stryker Corporation, and Biomet, Inc., that dominate the market for orthopedic products. Each of these companies, as well as other companies like Smith & Nephew, Inc., which introduced the Journey Deuce Bi-Compartmental Knee System in July 2007, offers conventional instruments and implants for use in conventional total and partial knee replacement surgeries as well as unicompartmental resurfacings procedures, which may compete with our MAKOplasty solution and negatively impact sales of our TGS. A number of these and other companies also offer CAS systems for use in arthroplasty procedures that provide a minimally invasive means of viewing the anatomical site.
Currently, we are not aware of any well-known orthopedic companies that broadly offer robotics technology in combination with CAS. All of these companies, however, have the ability to acquire and develop robotics technology that may compete with our TGS. We are aware of certain early stage companies developing CAS and robotic applications in orthopedics and others commercializing customized implants and instruments for early and mid-stage arthroplasty solutions. In addition, Biomet has a license from Z-KAT to intellectual property rights in computer assisted surgery, or CAS intellectual property, for use in the field of orthopedics. The license is non-exclusive with respect to use of CAS intellectual property in combination with robotics technology and exclusive with respect to all other uses within the field of orthopedics, which could enable them to compete with us.
27
We also face competition from other medical device companies that may seek to extend robotics technology and minimally invasive approaches and products that they have developed for use in other parts of the human anatomy to minimally invasive arthroplasty of the knee. Even if these companies currently do not have an established presence in the field of minimally invasive surgery for the knee, they may attempt to apply their robotics technology to the field of knee replacement and resurfacing procedures to compete directly with us.
Even if our TGS becomes commercially successful, our implant products may face substantial competition from implants offered by the well-known companies currently in the market for orthopedic products. We have designed our products so that our TGS only works effectively with our implant products. We also contractually require purchasers of our TGS to use only our implants in connection with the TGS. We cannot guarantee, however, that these measures will be effective or that our customers will agree to such contracts in the future. Accordingly, if use of our TGS becomes more prevalent, competitors may attempt to market their implant products for use with the TGS and compete directly with our implant products.
We believe that the principal competitive factors in our market include:
| | • | | the safety and efficacy of the procedure and product offerings, as documented through published studies and other clinical reports; |
| |
| | • | | product benefits, including the ability to offer orthopedic surgeons a complete solution for minimally invasive orthopedic knee procedures; |
| |
| | • | | the strength of acceptance and adoption by orthopedic surgeons and hospitals; |
| |
| | • | | the ability to deliver new product offerings and enhanced technology to expand or improve upon existing applications through continued research and development; |
| |
| | • | | the quality of training, services and clinical support provided to surgeons and hospitals; |
| |
| | • | | the cost of product offerings and the availability of product coverage and reimbursement from third-party payors, insurance companies and others parties; |
| |
| | • | | the ability to provide proprietary products protected by strong intellectual property rights; and |
| |
| | • | | the ability to offer products that are intuitive and easy to learn and use. |
28
Many of our competitors have significantly greater financial, human and other resources than we do, and have established relationships with healthcare professionals, customers and third-party payors. In addition, many of our competitors have established sales networks, greater resources for product development, additional lines of products and the ability to offer financial incentives such as rebates, bundled products or discounts on other product lines that we cannot provide. Our products could also be rendered obsolete or uneconomical by technological advances developed by one or more of our competitors. These competitive factors may negatively affect our ability to convince individuals to utilize our TGS and implant products and result in our inability to acquire technology, products and businesses from third parties to develop our current and planned versions of the TGS and related products.
Regulatory Requirements of the U.S. Food and Drug Administration
Our research, development and clinical programs, as well as our manufacturing and marketing operations, are subject to extensive regulation in the U.S. and other countries. Most notably, all of our products sold in the U.S. are subject to regulation as medical devices under the Federal Food, Drug, and Cosmetic Act, or the FDCA, as implemented and enforced by the FDA. The FDA governs the following activities that we perform or that are performed on our behalf, to ensure that medical products we manufacture, promote and distribute domestically or exported internationally are safe and effective for their intended uses:
| | • | | product design, preclinical and clinical development and manufacture; |
| |
| | • | | product premarket clearance and approval; |
| |
| | • | | product safety, testing, labeling and storage; |
| |
| | • | | record keeping procedures; |
| |
| | • | | product marketing, sales and distribution; and |
| |
| | • | | post-marketing surveillance, complaint handling, medical device reporting, reporting of deaths, serious injuries or device malfunctions and repair or recall of products. |
FDA Premarket Clearance and Approval Requirements
Unless an exemption applies, each medical device we wish to commercially distribute in the U.S. will require either premarket notification, or 510(k), clearance or approval of a premarket approval application, or PMA, from the FDA. The FDA classifies medical devices into one of three classes. Class I devices, considered to have the lowest risk, are those for which safety and effectiveness can be assured by adherence to the FDA’s general regulatory controls for medical devices, which include compliance with the applicable portions of the FDA’s QSR, facility registration and product listing, reporting of adverse medical events, and appropriate, truthful and non-misleading labeling, advertising, and promotional materials (General Controls). Class II devices are subject to the FDA’s General Controls, and any other special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device (Special Controls). Manufacturers of most class II and some class I devices are required to submit to the FDA a premarket notification under Section 510(k) of the FDCA requesting permission to commercially distribute the device. This process is generally known as 510(k) clearance. Devices deemed by the FDA to pose the greatest risks, such as life-sustaining, life-supporting or implantable devices, or devices that have a new intended use, or use advanced technology that is not substantially equivalent to that of a legally marketed device, are placed in class III, requiring approval of a PMA.
29
Certain of our currently marketed products, such as our TGS, are class II devices marketed pursuant to 510(k) clearances. In January 2008, we obtained 510(k) marketing clearance from the FDA for version 1.2 of our TGS. We originally submitted a Special 510(k) application in September 2007, which the FDA subsequently indicated was converted to a Traditional 510(k) application. On November 1, 2007, the FDA provided us with a letter requesting additional information in which the FDA, among other things, asked us to justify our proposed use of the terms “haptic” and “robot” in the labeling of version 1.2 of our TGS. Through subsequent correspondence and communications, the FDA indicated that we needed to use the term “tactile” in lieu of “haptic” and the term “robotic-arm” in lieu of “robotic,” as appropriate, when these terms are used to market our products and in order to obtain timely clearance of our 510(k) submission. The FDA granted 510(k) clearance for version 1.2 of our TGS with those terms. See Item 1A, Risk Factors, “Risks Related to Our Business — We are currently required by the FDA to refrain from using certain terms to label and market our products, which could harm our ability to market and commercialize our current and future products.”
Our current regulatory strategy anticipates that version 1.3 of our TGS will not require submission of a 510(k) application. For version 2.0 of our TGS, we anticipate submitting a 510(k) application to obtain FDA clearance once development is substantially complete. We hope to substantially complete development of version 2.0 sometime in late 2008 or early 2009 and to submit a 510(k) application soon thereafter. However, the FDA may require us to submit extensive additional data to support clearance for use in multi-compartmental knee resurfacing procedures. Due to this indication, FDA also may require us to submit a PMA for version 2.0 of the TGS.
510(k) Clearance Pathway
To obtain 510(k) clearance, we must submit a premarket notification demonstrating that the proposed device is “substantially equivalent” to a legally marketed “predicate device” that is either in class I or class II, or to a class III device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of a PMA. A Special 510(k) is an abbreviated 510(k) application which can be used to obtain clearance for certain types of device modification such as modifications that do not affect the intended use of the device or alter the device’s fundamental scientific technology. A Special 510(k) generally requires less information and data than a complete, or Traditional 510(k). In addition, a Special 510(k) application often takes a shorter period of time, which could be as short as 30 days, than a Traditional 510(k) clearance application, which can be used for any type of 510(k) device. FDA’s 510(k) clearance pathway usually takes from three to twelve months, but may take significantly longer. The FDA may require additional information, including clinical data, to make a determination regarding substantial equivalence. There is no guarantee that the FDA will grant 510(k) clearance for our future products and failure to obtain necessary clearances for our future products would adversely affect our ability to grow our business.
30
Medical devices can be marketed only for the indications for which they are cleared or approved. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a new or major change in its intended use, will require a new 510(k) clearance or, depending on the modification, PMA approval. The FDA requires each manufacturer to determine whether the proposed change requires submission of a 510(k) or a PMA, but the FDA can review any such decision and can disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination, the FDA can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or PMA approval is obtained. Also, in these circumstances, we may be subject to significant regulatory fines or penalties. We have made and plan to continue to make additional product enhancements to our TGS and other products that we believe do not require new 510(k) clearances. We cannot assure you that the FDA would agree with any of our decisions not to seek 510(k) clearance or PMA approval.
PMA Approval Pathway
A PMA must be submitted to the FDA if the device cannot be cleared through the 510(k) process, or is not otherwise exempt from the FDA’s premarket clearance and approval requirements. A PMA must generally be supported by extensive data, including, but not limited to, technical, preclinical, clinical trials, manufacturing and labeling, to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device for its intended use. No device that we are marketing to date has required premarket approval. During the review period, the FDA will typically request additional information or clarification of the information already provided. Also, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. The FDA may or may not accept the panel’s recommendation. In addition, the FDA will generally conduct a pre-approval inspection of our or our third-party manufacturers’ or suppliers’ manufacturing facility or facilities to ensure compliance with the QSR.
New PMAs or PMA supplements are required for modifications that affect the safety or effectiveness of the device, including, for example, certain types of modifications to the device’s indication for use, manufacturing process, labeling and design. PMA supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA and may not require as extensive clinical data or the convening of an advisory panel. None of our products is currently approved under a PMA approval. However, we may in the future develop devices which will require the approval of a PMA. There is no guarantee that the FDA will grant PMA approval of our future products and failure to obtain necessary approvals for our future products would adversely affect our ability to grow our business.
31
Clinical Trials
Clinical trials are generally required to support a PMA application and are sometimes required for 510(k) clearance. Such trials generally require an investigational device exemption application, or IDE, approved in advance by the FDA for a specified number of patients and study sites, unless the product is deemed a non-significant risk device eligible for more abbreviated IDE requirements. A significant risk device is one that presents a potential for serious risk to the health, safety or welfare of a patient and either is implanted, used in supporting or sustaining human life, substantially important in diagnosing, curing, mitigating or treating disease or otherwise preventing impairment of human health, or otherwise presents a potential for serious risk to a subject. Clinical trials are subject to extensive monitoring, recordkeeping and reporting requirements. Clinical trials must be conducted under the oversight of an institutional review board, or IRB, for the relevant clinical trial sites and must comply with FDA regulations, including but not limited to those relating to good clinical practices. To conduct a clinical trial, we also are required to obtain the patient’s informed consent in form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations. We, the FDA or the IRB could suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and efficacy of the device or may otherwise not be sufficient to obtain FDA clearance or approval to market the product in the U.S. Similarly, in Europe the clinical study must be approved by a local ethics committee and in some cases, including studies with high-risk devices, by the ministry of health in the applicable country.
Post-Market Study
To date, none of our submissions to the FDA have required the submission of clinical data. However, we are conducting a post-market study of MAKOplasty aimed at demonstrating the accuracy of the placement and alignment of our implants to further support the clinical value of the MAKOplasty procedure. We released the preliminary results of this study in the first quarter of 2008. Currently, we are conducting this study, known as a post-market study, at only one site, Holy Cross Hospital, but we may expand this to additional sites in the future. We are conducting this study as a “non-significant risk” study. As a result, we do not believe that we are required to obtain FDA approval of an IDE. However, we did receive the approval of the Holy Cross Hospital IRB and obtained informed consents from all study subjects. Holy Cross Hospital IRB policy required us to obtain IRB approval and informed consent for patient data confidentiality reasons only. If the FDA disagrees with our determination that the study is a “non-significant risk” study, the FDA could require us to stop the study and could take enforcement action against us.
Pervasive and Continuing Regulation
After a device is placed on the market, numerous regulatory requirements continue to apply. In addition to the requirements below, the Medical Device Reporting, or MDR, regulations require that we report to the FDA any incident in which our products may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. MAKO has submitted four MDRs to the FDA to date. See Item 1A, Risk Factors, “Risks Related to Regulatory Compliance,” for further information regarding our reporting obligations under MDR regulations. Additional regulatory requirements include:
| | • | | product listing and establishment registration, which helps facilitate FDA inspections and other regulatory action; |
| |
| | • | | QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the design and manufacturing process; |
32
| | • | | labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label use or indication; |
| |
| | • | | clearance of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use of one of our cleared devices; |
| |
| | • | | approval of product modifications that affect the safety or effectiveness of one of our approved devices; |
| |
| | • | | post-approval restrictions or conditions, including post-approval study commitments; |
| |
| | • | | post-market surveillance regulations, which apply, when necessary, to protect the public health or to provide additional safety and effectiveness data for the device; |
| |
| | • | | the FDA’s recall authority, whereby it can ask, or under certain conditions order, device manufacturers to recall from the market a product that is in violation of governing laws and regulations; |
| |
| | • | | regulations pertaining to voluntary recalls; and |
| |
| | • | | notices of corrections or removals. |
We must also register with the FDA as a medical device manufacturer and must obtain all necessary state permits or licenses to operate our business. As a manufacturer, we are subject to announced and unannounced inspections by the FDA to determine our compliance with FDA’s QSR and other regulations. We have not yet been inspected by the FDA. We believe that we are in substantial compliance with QSR and other regulations. Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include any of the following sanctions:
| | • | | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| |
| | • | | customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; |
| |
| | • | | operating restrictions or partial suspension or total shutdown of production; |
33
| | • | | refusing or delaying requests for 510(k) clearance or PMA approvals of new products or modified products; |
| |
| | • | | withdrawing 510(k) clearances or PMA approvals that have already been granted; |
| |
| | • | | refusal to grant export approval for our products; or |
| |
| | • | | criminal prosecution. |
International Marketing Approvals
International sales of medical devices are subject to foreign government regulations, which vary substantially from country to country. The time required to obtain approval by a foreign country may be longer or shorter than that required for FDA clearance or approval, and the requirements may differ.
The European Union has adopted numerous directives and standards regulating the design, manufacture, clinical trials, labeling and adverse event reporting for medical devices. Each European Union member state has implemented legislation applying these directives and standards at the national level. Other countries, such as Switzerland, have voluntarily adopted laws and regulations that mirror those of the European Union with respect to medical devices. Devices that comply with the requirements of the laws of the relevant member state applying the applicable European Union directive are entitled to bear CE conformity marking and, accordingly, can be commercially distributed throughout the member states of the European Union and other countries that comply with or mirror these directives. The method of assessing conformity varies depending on the type and class of the product, but normally involves a combination of self-assessment by the manufacturer and a third-party assessment by a “Notified Body,” an independent and neutral institution appointed to conduct conformity assessment. This third-party assessment consists of an audit of the manufacturer’s quality system and clinical information, as well as technical review of the manufacturer’s product. An assessment by a Notified Body in one country within the European Union is required in order for a manufacturer to commercially distribute the product throughout the European Union. In addition, compliance with ISO 13845 on quality systems issued by the International Organization for Standards, among other standards, establishes the presumption of conformity with the essential requirements for a CE marking. In addition, many countries apply requirements in their reimbursement, pricing or health care systems that affect companies’ ability to market products.
Health Care Laws and Regulations
Third-Party Reimbursement
In the U.S. and elsewhere, health care providers that perform surgical procedures using medical devices such as ours generally rely on third-party payors, including governmental payors such as Medicare and Medicaid and private payors, to cover and reimburse all or part of the cost of the products. Consequently, sales of medical devices are dependent in part on the availability of reimbursement to the customer from third- party payors.
34
The manner in which reimbursement is sought and obtained varies based upon the type of payor involved and the setting in which the product is furnished and utilized. In general, third-party payors will provide coverage and reimbursement for medically reasonable and necessary procedures and tests that utilize medical devices and may provide separate payments for the implanted or disposable devices themselves. Most payors, however, will not pay separately for capital equipment, such as our TGS. Instead, payment for the cost of using the capital equipment is considered to be covered as part of payments received for performing the procedure. In determining payment rates, third-party payors are increasingly scrutinizing the prices charged for medical products and services in comparison to other therapies. Our products, and the procedures in which our products are used, may not be reimbursed by these third-party payors at rates sufficient to allow us to sell our products on a competitive and profitable basis.
In addition, in many foreign markets, including the countries in the European Union, pricing of medical devices is subject to governmental control. In the U.S., there have been, and we expect that there will continue to be, a number of federal and state proposals to limit payments by governmental payors for medical devices, and the procedures in which medical devices are used. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
Medicare and Medicaid
The Medicare program is a federal health benefit program administered by the Centers for Medicare and Medicaid Services, or CMS, that covers and pays for certain medical care items and services for eligible elderly, blind and disabled individuals, and individuals with end stage renal disease. The Medicaid program is a federal-state partnership under which states receive matching federal payments to fund healthcare services for the poor. Because we expect that a significant percentage of MAKOplasty patients will be Medicare beneficiaries, and because some private commercial health insurers and some state Medicaid programs may follow the coverage and payment policies for Medicare, Medicare’s coverage and payment policies are significant to our business.
Medicare coverage for procedures using our technology currently exists in the hospital inpatient setting, which falls under Part A of the Medicare program. Under Medicare Part A, Medicare reimburses acute care hospitals a flat prospectively determined payment amount for beneficiaries receiving covered inpatient services in an acute care hospital. This method of payment is known as the prospective payment system, or PPS. Under PPS, the prospective payment for a patient’s stay in an acute care hospital is determined by the patient’s condition and other patient data and procedures performed during the inpatient stay using a classification system known as diagnosis-related groups, or DRGs. As of October 1, 2007, CMS implemented a revised version of the DRG system that uses 745 Medicare Severity DRGs, or MS-DRGs, instead of the approximately 540 DRGs Medicare previously used. The MS-DRGs are intended to account more accurately for the patient’s severity of illness when assigning each patient’s stay to a payment classification. Medicare pays a fixed amount to the hospital based on the MS-DRG into which the patient’s stay is classified, regardless of the actual cost to the hospital of furnishing the procedures, items and services that the patient’s condition requires.
35
Accordingly, acute care hospitals generally do not receive direct Medicare reimbursement under PPS for the specific costs incurred in purchasing medical devices. Rather, reimbursement for these costs is deemed to be included within the MS-DRG-based payments made to hospitals for the services furnished to Medicare-eligible inpatients in which the devices are utilized. For cases involving unusually high costs, a hospital may receive additional “outlier” payments above the pre-determined amount. In addition, there is a mechanism by which new technology services can apply to Medicare for additional payments above the pre-determined amount, although such requests have not been granted frequently.
Because PPS payments are based on predetermined rates and may be less than a hospital’s actual costs in furnishing care, acute care hospitals have incentives to lower their inpatient operating costs by utilizing products, devices and supplies that will reduce the length of inpatient stays, decrease labor or otherwise lower their costs. For each MS-DRG, a relative weight is calculated representing the average resources required to care for cases grouped in that particular MS-DRG relative to the average resources used to treat cases in all MS-DRGs. MS-DRG relative weights are recalculated every year to reflect changes in technology and medical practice in a budget neutral manner. Under the MS-DRG payment system, there can be significant delays in obtaining adequate reimbursement amounts for hospitals for new technologies such that reimbursement may be insufficient to permit broad acceptance by hospitals.
We believe that there are existing reimbursement codes that can be used for MAKOplasty procedures performed in the hospital inpatient setting. Procedures for hospital inpatient billing are referenced by international classifications of diseases, clinical modification, or ICD-9-CM, volume 3 procedure codes. Knee arthroplasty is billed under ICD-9-CM code 81.54 (“Total Knee Replacement”), which is assigned to MS-DRG 469 (“Major Joint Replacement or Reattachment of Lower Extremity with Complication or Comorbidity”) and MS-DRG 470 (“Major Joint Replacement or Reattachment of Lower Extremity without Major Complication or Comorbidity”). We anticipate that Medicare will continue to reimburse hospitals under MS-DRGs 469 and 470 for MAKOplasty procedures, but CMS can revise MS-DRG assignments from year to year.
In addition to payments to hospitals for procedures using our technology, Medicare makes separate payments to physicians for their professional services. The American Medical Association, or AMA, has developed a coding system known as the Current Procedural Terminology, or CPT, codes, which have been adopted by the Medicare program to describe and develop payment amounts for certain physician services. The Medicare physician fee schedule uses CPT codes (and other codes) as part of the determination of allowable payment amounts to physicians. In determining appropriate payment amounts for surgeons, CMS receives guidance from the AMA regarding the relative technical skill level, level of resources used, and complexity of a new surgical procedure. Generally, the FDA approval of a new product is necessary, but not necessarily sufficient, for the designation of a new procedure code for a new surgical procedure using that product. Codes are assigned by either the AMA (for CPT codes) or CMS (for Medicare-specific codes) and new codes usually become effective on January 1st of each year. Physicians performing procedures using our technology submit bills under CPT code 27446 (“Arthroplasty, knee, condyle and plateau; medial OR lateral compartment”). We anticipate that third-party payors will continue to reimburse physicians under this code for services performed in connection with MAKOplasty procedures.
36
Commercial Insurers
In addition to the Medicare program, many private payors look to CMS policies as a guideline in setting their coverage policies and payment amounts. The current coverage policies of these private payors may differ from the Medicare program, and the payment rates they make may be higher, lower, or the same as the Medicare program. A decrease of, or limitation on, reimbursement payments for doctors and hospitals by CMS or other agencies may affect coverage and reimbursement determinations by many private payors. Additionally, some private payors do not follow the Medicare guidelines, and those payors may reimburse only a portion of the costs associated with the use of our products, or not at all.
Fraud and Abuse Laws
Because of the significant federal funding involved in Medicare and Medicaid, Congress and the states have enacted, and actively enforce, a number of laws whose purpose is to eliminate fraud and abuse in federal health care programs. Our business is subject to compliance with these laws.
Anti-Kickback Statutes and Federal False Claims Act
The federal healthcare programs’ Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program such as Medicare or Medicaid. The definition of “remuneration” has been broadly interpreted to include anything of value, including for example gifts, certain discounts, the furnishing of free supplies, equipment or services, credit arrangements, payments of cash and waivers of payments. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. Penalties for violations include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal healthcare programs. In addition, some kickback allegations have been claimed to violate the Federal False Claims Act, discussed in more detail below.
The Anti-Kickback Statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements, Congress authorized the Office of Inspector General of the U.S. Department of Health and Human Services, or OIG, to issue a series of regulations, known as “safe harbors.” These safe harbors, issued by the OIG beginning in July 1991, set forth provisions that, if all their applicable requirements are met, will assure healthcare providers and other parties that they will not be prosecuted under the Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the OIG.
37
Many states have adopted laws similar to the Anti-Kickback Statute. Some of these state prohibitions apply to referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
Government officials have focused their enforcement efforts on marketing of healthcare services and products, among other activities, and recently have brought cases against companies, and certain sales, marketing and executive personnel, for allegedly offering unlawful inducements to potential or existing customers in an attempt to procure their business.
Another development affecting the healthcare industry is the increased use of the federal Civil False Claims Act and, in particular, actions brought pursuant to the False Claims Act’s “whistleblower” or “qui tam” provisions. The False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government, and to share in any monetary recovery. In recent years, the number of suits brought against healthcare providers by private individuals has increased dramatically. In addition, various states have enacted false claim laws analogous to the Civil False Claims Act, although many of these state laws apply where a claim is submitted to any third-party payor and not merely a federal healthcare program.
When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties of between $5,500 to $11,000 for each separate false claim. There are many potential bases for liability under the False Claims Act. Liability arises, primarily, when an entity knowingly submits, or causes another to submit, a false claim for reimbursement to the federal government. The False Claims Act has been used to assert liability on the basis of inadequate care, kickbacks and other improper referrals, and improper use of Medicare numbers when detailing the provider of services, in addition to the more predictable allegations as to misrepresentations with respect to the services rendered. In addition, companies have been prosecuted under the False Claims Act in connection with alleged off-label promotion of products. Our future activities relating to the reporting of wholesale or estimated retail prices for our products, the reporting of discount and rebate information and other information affecting federal, state and third-party reimbursement of our products, and the sale and marketing of our products, may be subject to scrutiny under these laws. We believe our current consulting agreements with physicians represent legitimate compensation for needed documented services actually furnished to us.
38
However, engagement of physician consultants by orthopedic medical device manufacturers has recently been subject to heightened scrutiny, and has resulted in four of the major orthopedic medical device implant manufacturers entering deferred prosecution agreements with the federal government and agreeing to pay substantial amounts to the federal government in settlement of Anti-Kickback Statute allegations, and all such companies submitting to supervision by a court-appointed monitor throughout the term of the 18-month agreements. In this environment, our engagement of physician consultants in product development and product training and education could subject us to similar scrutiny. We are unable to predict whether we would be subject to actions under the False Claims Act or a similar state law, or the impact of such actions. However, the costs of defending such claims, as well as any sanctions imposed, could have a material adverse effect on our financial performance.
As part of our internal compliance program, we review our sales and marketing materials, contracts and programs with counsel, and require employees and marketing representatives to participate in regular training. We also have adopted and train our personnel on the code of conduct for Interactions with Health Care Professionals promulgated by the Advanced Medical Technology Association, or AdvaMed, a leading trade association representing medical device manufacturers. However, we cannot rule out the possibility that the government or other third parties could interpret these laws differently and challenge one or more of our activities under these laws.
HIPAA and Other Fraud and Privacy Regulations
Among other things, the Health Insurance Portability and Accountability Act of 1996, or HIPAA, created two new federal crimes: healthcare fraud and false statements relating to healthcare matters. The HIPAA health care fraud statute prohibits, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment and/or exclusion from government-sponsored programs. The HIPAA false statements statute prohibits, among other things, knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines and/or imprisonment.
In addition to creating the two new federal healthcare crimes, regulations implementing HIPAA also establish uniform standards governing the conduct of certain electronic healthcare transactions and protecting the security and privacy of individually identifiable health information maintained or transmitted by healthcare providers, health plans and healthcare clearinghouses, which are referred to as “covered entities.” Three standards have been promulgated under HIPAA’s regulations: the Standards for Privacy of Individually Identifiable Health Information, which restrict the use and disclosure of certain individually identifiable health information, the Standards for Electronic Transactions, which establish standards for common healthcare transactions, such as claims information, plan eligibility, payment information and the use of electronic signatures, and the Security Standards, which require covered entities to implement and maintain certain security measures to safeguard certain electronic health information. Although we are not a covered entity and therefore not directly subject to these standards, we expect that our customers generally will be covered entities and may ask us to contractually comply with certain aspects of these standards. While the government intended this legislation to reduce administrative expenses and burdens for the healthcare industry, our compliance with certain provisions of these standards entails significant costs for us.
39
In addition to federal regulations issued under HIPAA, some states have enacted privacy and security statutes or regulations that, in some cases, are more stringent than those issued under HIPAA. In those cases, it may be necessary to modify our planned operations and procedures to comply with the more stringent state laws. If we fail to comply with applicable state laws and regulations, we could be subject to additional sanctions.
Employees
As of March 21, 2008, we had 133 employees, 30 of whom were engaged directly in sales and marketing, 53 in research and development, 17 in assembly, manufacturing and service, 15 in regulatory, clinical affairs and quality activities and 18 in general administrative and accounting activities. None of our employees is covered by a collective bargaining agreement, and we consider our relationship with our employees to be good.
Available Information
From our Internet website,http://www.makosurgical.com, you may obtain additional information about us including:
| | • | | Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, including amendments to these reports, and other documents as soon as reasonably practicable after we file them with the Securities and Exchange Commission, or the SEC; |
| |
| | • | | Beneficial ownership reports filed by officers, directors and principal security holders under Section 16(a) of the Securities Exchange Act of 1934, as amended, or the Exchange Act; and |
| |
| | • | | Corporate governance information that includes our |
| | • | | Corporate Governance Guidelines |
| |
| | • | | Audit Committee Charter |
| |
| | • | | Compensation Committee Charter |
| |
| | • | | Corporate Governance and Nominating Committee Charter |
| |
| | • | | Code of Business Conduct and Ethics |
| |
| | • | | Information on how to communicate directly with our board of directors |
We will also provide printed copies of any of these documents to any stockholder upon request.
40
ITEM 1A. RISK FACTORS
The following risk factors and other information included in this report should be carefully considered. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently treat as immaterial also may impair our business operations. If any of the following risks occur, our business, financial condition, operating results and cash flows could be materially adversely affected.
Risks Related to Our Business
We are an early-stage medical device company with a limited operating history and our business may not become profitable.
We are an early-stage medical device company with a limited operating history. Our only current or planned products with 510(k) marketing clearance from the FDA are versions 1.0 and 1.2 of our Tactile Guidance System, or TGS, and inlay and onlay implant systems for use in unicompartmental knee resurfacing procedures. We also may not be successful in our research and development efforts for version 2.0 of the TGS and a modular knee implant system, which would allow multicompartmental knee resurfacing procedures. The future success of our business depends on our ability to develop and obtain regulatory clearances or approvals for these products, especially version 2.0 of the TGS and the modular knee implant system, which we may be unable to do in a timely manner, or at all. Our success and ability to generate revenue or be profitable also depends on our ability to establish our sales and marketing force, generate product sales and control costs, all of which we may be unable to do. We have a limited history of operations upon which you can evaluate our business and our operating expenses are increasing. Our lack of any significant operating history also limits your ability to make a comparative evaluation of us, our products and our prospects.
We have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future.
We have sustained net losses in every fiscal year since our inception in 2004, including a net loss of $20.7 million for the year ended December 31, 2007. As of December 31, 2007, we had an accumulated deficit of $42.8 million. We expect to continue to incur significant operating losses as we increase our sales and marketing activities and otherwise continue to invest capital in the development of our products and our business generally. We also expect that our general and administrative expenses will increase due to additional operational and regulatory burdens associated with operating as a public company. We are required to defer revenue associated with sales of our TGS units until we have fulfilled our contractual obligation to deliver version 2.0 of our TGS to the customer. Therefore, our deferred revenue will be higher in the short term and we may not be able to recognize some or any of our deferred revenue until we have satisfied all obligations for delivery of product upgrades, which we may be unable to do. Our losses have had and will continue to have an adverse effect on our stockholders’ deficit and working capital. Any failure to achieve and maintain profitability would continue to have an adverse effect on our stockholders’ deficit and working capital and could result in a decline in our stock price or cause us to cease operations.
41
We rely heavily on intellectual property that we license from others, and if we are unable to maintain these licenses or obtain additional licenses that we may need, our ability to compete will be harmed.
We rely heavily on intellectual property that we license or sublicense from others, including patented technology that is integral to our TGS and implants. As of January 1, 2008, we had licensed rights to 118 U.S. and 47 foreign third-party granted patents, and we had licensed rights to 22 U.S. and 40 foreign third-party pending patent applications. The majority of these patents and applications are either used in our current products or relate to core technologies used in our products, such as computer assisted surgery, or CAS, robotics, haptics and implants. Nine of the licensed U.S. patents (and three related foreign patents) will expire by the end of 2009. Two of these U.S. patents (and all three related foreign patents) are CAS patents, and three of these U.S. patents relate to robotic technology. These five U.S. patents (and the foreign patents) are considered material to our intellectual property portfolio because they potentially enable us to exclude others from practicing the claimed technology. Our portfolio also includes 19 wholly-owned pending U.S. patent applications, 21 pending foreign applications and other intellectual property that is wholly-owned by us. We are particularly dependent on our licensing arrangements with Z-KAT, Inc., or Z-KAT, from whom we license or sublicense, among other things, core technologies in CAS, and haptics and robotics. We also rely on our licensing arrangement with Stelkast (a business division of Trigon Incorporated), pursuant to which we have specified rights to the design of our inlay knee implant system, and our licensing arrangement with Encore Medical, L.P., pursuant to which we have certain rights to the design of our onlay knee implant system. Any of these or other third parties may terminate a license in the event that we fail to make required payments or for other causes. In the event a third party terminates a license agreement, we cannot assure you that we could acquire another license to adequately replace the product, technology or method covered by the terminated license. If we fail to maintain our current licenses, our ability to compete in the knee implant market will be harmed.
In addition, as we enhance our current product offerings and develop new ones, including version 2.0 of our TGS and a modular knee implant system, we may find it advisable or necessary to seek additional licenses from third parties who hold patents covering technology or methods used in these products. If we cannot obtain these additional licenses, we could be forced to design around those patents at additional cost or abandon the product altogether. As a result, our ability to grow our business and compete in the knee implant market may be harmed.
42
We are currently required by the FDA to refrain from using certain terms to label and market our products, which could harm our ability to market and commercialize our current or future products.
On September 28, 2007, we submitted a Special 510(k) application to the FDA for version 1.2 of our TGS which the FDA indicated was converted to a Traditional 510(k) application. On November 1, 2007, the FDA provided us with a letter requesting additional information in which the FDA, among other things, asked us to justify our proposed use of the terms “haptic” and “robot” in the labeling of version 1.2 of our TGS. Through subsequent correspondence and communications, the FDA indicated that we needed to use the term “tactile” in lieu of “haptic” and the term “robotic-arm” in lieu of “robotic,” as appropriate, when these terms are used to market our products and in order to obtain timely clearance of our 510(k) submission for version 1.2. The FDA granted 510(k) clearance in January 2008 for version 1.2 of our TGS with those terms. Because the FDA currently requires us to use the terms “tactile” or “robotic-arm,” we have revised the promotional and labeling materials for our existing TGS products, and may need to consider the use of modified language for our future products. As a result, our ability to market and commercialize our products and our growth may be harmed.
Modifications to our currently FDA-cleared products or the introduction of new products may require new regulatory clearances or approvals or require us to recall or cease marketing our current products until clearances or approvals are obtained.
In November 2005, we obtained 510(k) marketing clearance from the FDA for version 1.0 of our TGS for use with our FDA-cleared inlay implant system. We were not required to obtain premarket approval, or PMA. We were also not required to conduct any clinical trials in support of our application for 510(k) marketing clearance. Modifications to our products, however, may require new regulatory approvals or clearances or require us to recall or cease marketing the modified products until these clearances or approvals are obtained. Any modification to one of our 510(k)-cleared products that would constitute a major change in its intended use, or any change that could significantly affect the safety or effectiveness of the device would require us to obtain a new 510(k) clearance and may even, in some circumstances, require the submission of a PMA, if the change raises complex or novel scientific issues or the product has a new intended use. The FDA requires every manufacturer to make the determination regarding the need for a new 510(k) submission in the first instance, but the FDA may review any manufacturer’s decision. Since obtaining 510(k) marketing clearance for version 1.0 of our TGS, we developed and commercially introduced several upgrades to our TGS that we believe did not require additional clearances or approvals. Our Special 510(k) application for version 1.2, which the FDA converted to a Traditional 510(k) application and cleared in January 2008, incorporated these upgrades. We may make additional modifications in the future to our TGS without seeking additional clearances or approvals if we believe such clearances or approvals are not necessary. If the FDA disagrees and requires new clearances or approvals for the modifications, we may be required to recall and stop marketing our products as modified, which could cause us to redesign our products, conduct clinical trials to support any modifications, and pay significant regulatory fines or penalties. Any of these actions would harm our operating results.
We may not be successful in obtaining 510(k) marketing clearances or other required approvals for version 2.0 of our TGS and the modular knee implant system, which version 2.0 will be designed to support. Obtaining clearances and approvals can be a difficult and time consuming process, and we may not be able to obtain any of these or other clearances or approvals in a timely manner, or at all. In addition, the FDA may not approve or clear these products for the indications that are necessary or desirable for successful commercialization or could require clinical trials to support any modifications. Any delay or failure in obtaining required clearances or approvals would adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our future growth.
43
Moreover, clearances and approvals are subject to continual review, and the later discovery of previously unknown problems can result in product labeling restrictions or withdrawal of the product from the market. The loss of previously received approvals or clearances, or the failure to comply with existing or future regulatory requirements could reduce our sales, profitability and future growth prospects.
We depend on the success of a single line of products for our revenue, which could impair our ability to achieve profitability.
We expect to derive most of our revenue from capital sales of our TGS units, recurring sales of implants and disposable products required for each MAKOplasty procedure, and service plans that are sold with our TGS units. Currently, the only line of products that has been commercially introduced or received 510(k) marketing clearance is versions 1.0 and 1.2 of our TGS and the inlay and onlay knee implant systems for use in unicompartmental knee resurfacing procedures. Our future growth and success is dependent on the commercial introduction of version 2.0 of our TGS and a modular knee implant system, which would allow application of MAKOplasty to multicompartmental knee resurfacing procedures. We are in the early stages of development of these products and will need to obtain regulatory clearances or approvals which we may be unable to do on a timely basis, or at all. If we are unable to complete development of these products, obtain regulatory clearances or approvals or achieve commercial acceptance of MAKOplasty for multicompartmental knee resurfacing procedures, our revenue would be adversely affected and we would not become profitable.
If our MAKOplasty solution does not gain market acceptance, we will not be able to generate the revenue necessary to develop a sustainable, profitable business.
Achieving patient, surgeon and hospital acceptance of MAKOplasty as the preferred method of treating early to mid-stage osteoarthritis of the knee is crucial to our success. We believe MAKOplasty represents a fundamentally new way of performing arthroplasty of the knee, employing computer-assisted robotic-arm technology and a patient-specific visualization system to resurface only the diseased areas of the knee joint. The orthopedic market has been traditionally slow to adopt new products and treatment practices. We believe that if surgeons and hospitals do not adopt the concept of computer-assisted robotics-enabled technology and do not perceive such technology as having significant advantages over conventional arthroplasty procedures, patients will be less likely to accept or be offered MAKOplasty and we will fail to meet our business objectives. Surgeons’ and hospitals’ perceptions of such technology having significant advantages are likely to be based on a determination that, among other factors, our products are safe, cost-effective and represent acceptable methods of treatment. Even if we can prove the clinical value of MAKOplasty through clinical trials, surgeons may elect not to use our products for any number of other reasons. For example, surgeons may continue to recommend total knee replacement surgery simply because such surgery is already widely accepted.
44
In addition, surgeons may be slow to adopt our products because of the perceived liability risks arising from the use of new products. Hospitals may not accept MAKOplasty because the TGS is a piece of capital equipment, representing a significant portion of a hospital’s budget. Our TGS may not be cost-efficient if hospitals are not able to perform a significant volume of MAKOplasty procedures. If MAKOplasty fails to achieve market acceptance for any of these or other reasons, we will not be able to generate the revenue necessary to develop a sustainable, profitable business.
We have only limited clinical data to support the value of MAKOplasty, which may make patients, surgeons and hospitals reluctant to purchase our products.
We believe that patients, surgeons and hospitals will only accept MAKOplasty or purchase our products if they believe that MAKOplasty is a safe and effective procedure with advantages over competing products and conventional procedures. To date, we have collected only limited, short-term clinical data with which to assess MAKOplasty’s clinical value. As of December 31, 2007, 181 MAKOplasty procedures had been performed since commercial introduction in 2006. Two additional procedures were intended as MAKOplasty procedures, but in each case, the procedure concluded as total knee replacements when our TGS failed to perform as intended or was improperly operated during surgery. We reported both of these incidents to the FDA pursuant to medical device reporting, or MDR, regulations. In January 2008, we reported a third incident in which a patient suffered a post-operative bone fracture at the insertion site of the bone pins. In February 2008, we reported a fourth incident in which a bone pin broke off below the bone surface of the tibia during a procedure. See “Risks Related to Regulatory Compliance.” We have not collected, and are not aware that others have collected, any long-term clinical data regarding the clinical value of MAKOplasty. The results of short-term studies, such as our post-market study, do not necessarily predict long-term clinical results. If longer-term or more extensive clinical studies that may be performed by us or others indicate that MAKOplasty is a less safe or less effective procedure than our current data suggest, patients may choose not to undergo, and surgeons may choose not to, perform MAKOplasty. Furthermore, unsatisfactory patient outcomes or patient injury could cause negative publicity for our products, particularly in the early phases of product introduction. The FDA could also rescind our marketing clearances if future results and experience indicate that our products cause unexpected or serious complications or other unforeseen negative effects. See “Risks Related to Regulatory Compliance.” Surgeons may be slow to adopt our products if they perceive liability risks arising from the use of these new products. As a result, patients, surgeons and hospitals may not accept MAKOplasty or our products and we may fail to become profitable and may be subject to significant legal liability.
We have limited sales and marketing experience and capabilities, which could impair our ability to achieve profitability.
We have limited experience as a company in the sales and marketing of our products. We may not be successful in marketing and selling our products in the U.S. through our direct sales force with assistance from independent orthopedic product agents and distributors. Our sales and marketing organization is supported by clinical and technical representatives who provide training, clinical and technical support and other services to our customers before and during the surgery. To reach our revenue targets, we need to expand and strengthen our U.S. direct sales force.
45
Developing a sales and marketing organization is expensive and time consuming and an inability to develop such an organization in a timely manner could delay the successful adoption of our products. Additionally, any sales and marketing organization that we develop may be competing against the experienced and well-funded sales and marketing organizations of some of our competitors. We will face significant challenges and risks in developing our sales and marketing organization, including, among others:
| | • | | our ability to recruit, train and retain adequate numbers of qualified sales and marketing personnel; |
| |
| | • | | the ability of sales personnel to obtain access to leading surgeons and persuade adequate numbers of hospitals to purchase our products; |
| |
| | • | | costs associated with hiring, maintaining and expanding a sales and marketing organization; and |
| |
| | • | | government scrutiny with respect to promotional activities in the healthcare industry. |
If we are unable to develop and maintain these sales and marketing capabilities, we may be unable to generate revenue and may not become profitable.
Surgeons, hospitals and orthopedic product agents and distributors may have existing relationships with other medical device companies that make it difficult for us to establish new relationships with them, and as a result, we may not be able to sell and market our products effectively.
We believe that to sell and market our products effectively, we must establish relationships with key surgeons and hospitals in the field of orthopedic knee surgery. Many of these key surgeons and hospitals already have long-standing relationships with large, better-known companies that dominate the medical devices industry through collaborative research programs and other relationships. Because of these existing relationships, some of which may be contractually enforced, surgeons and hospitals may be reluctant to adopt MAKOplasty, particularly if MAKOplasty competes with or has the potential to compete with products supported through their own collaborative research program or by these existing relationships. Even if these surgeons and hospitals purchase our TGS, they may be unwilling to enter into collaborative relationships with us to promote joint marketing programs such as the MAKOplasty Knee Center of Excellence or to provide us with clinical and financial data.
In addition to our direct sales force, we work with a network of independent orthopedic product agents and distributors that primarily generate sales leads for us. If these product agents and distributors believe that their relationship with us is less beneficial than other relationships they may have with more established or well-known medical device companies, they may be unwilling to continue their relationships with us, making it more difficult for us to sell and market our products effectively.
46
Because the markets for our products are highly competitive, customers may choose to purchase our competitors’ products, resulting in reduced revenue and harm to our financial results.
MAKOplasty requires the use of new robotics technology, and we face competition from large, well-known companies, principally Zimmer Holdings, Inc., DePuy Orthopedics, Inc., a Johnson & Johnson company, Stryker Corporation, and Biomet, Inc., that dominate the market for orthopedic products. Each of these companies, as well as other companies like Smith & Nephew, Inc., which introduced the Journey Deuce Bi-Compartmental Knee System in July 2007, offers conventional instruments and implants for use in conventional total and partial knee replacement surgeries as well as unicompartmental resurfacing procedures, which may compete with our MAKOplasty solution and negatively impact sales of our TGS. A number of these and other companies also offer CAS systems for use in arthroplasty procedures that provide a minimally invasive means of viewing the anatomical site.
Currently, we are not aware of any well-known orthopedic company that broadly offers robotics technology in combination with computer assisted surgery. All of these companies, however, have the ability to acquire and develop robotics technology that may compete with our TGS. We are aware of certain early stage companies developing CAS and robotic applications in orthopedics and others commercializing customized implants and instruments for early- and mid-stage arthroplasty solutions. In addition, Biomet has a license from Z-KAT to intellectual property rights in computer assisted surgery, or CAS intellectual property, for use in the field of orthopedics. The license is non-exclusive with respect to use of CAS intellectual property in combination with robotics technology and exclusive with respect to all other uses within the field of orthopedics, which could enable them to compete with us.
We also may face competition from other medical device companies that may seek to extend robotics technology and minimally invasive approaches and products that they have developed for use in other parts of the human anatomy to minimally invasive arthroplasty of the knee. Even if these other companies currently do not have an established presence in the field of minimally invasive surgery for the knee, they may attempt to apply their robotics technology to the field of knee replacement and resurfacing procedures to compete directly with us.
Many of these medical device competitors enjoy competitive advantages over us, including:
| | • | | significantly greater name recognition; |
| |
| | • | | longer operating histories; |
| |
| | • | | established exclusive relations with healthcare professionals, customers and third-party payors; |
| |
| | • | | established distribution networks; |
47
| | • | | additional lines of products and the ability to offer rebates or bundle products to offer higher discounts or incentives to gain a competitive advantage; |
| |
| | • | | greater experience in conducting research and development, manufacturing, clinical trials, obtaining regulatory clearance for products and marketing approved products; and |
| |
| | • | | greater financial and human resources for product development, sales and marketing and patent litigation. |
Moreover, our competitors in the medical device industry make significant investments in research and development, and innovation is rapid and continuous. If new products or technologies emerge that provide the same or superior benefits as our products at equal or lesser cost, they could render our products obsolete or unmarketable. Because our products can have long development and regulatory clearance or approval cycles, we must anticipate changes in the marketplace and the direction of technological innovation and customer demands. In addition, we face increasing competition from well-financed orthopedic companies in our attempts to acquire such new technologies, products and businesses. As a result, we cannot be certain that surgeons will use our products to replace or supplement established surgical procedures or that our products will be competitive with current or future products and technologies resulting in reduced revenue and harm to our financial results.
If we do not timely achieve our development goals for new versions of our TGS or our implants, the commercialization of these products will be delayed and our business and financial results may be adversely affected.
The success of our business is dependent on our ability to develop new products, to introduce enhancements to our existing products and to develop these new products and enhancements within targeted time frames and budgets. We may not be successful in our research and development efforts for version 2.0 of our TGS and a modular knee implant system, for which we are targeting commercial introduction in the first half of 2009, subject to regulatory clearances. The actual timing of these product releases can vary dramatically compared to our estimates for reasons that may or may not be within our control, including clearance or approval to market these products by the FDA. Customers may forego purchases of our existing products and purchase our competitors’ products as a result of delays in the introduction of our new products and enhancements or failure by us to offer innovative products or enhancements at competitive prices and in a timely manner. Announcements of new products by us or by competitors may also result in a delay in or cancellation of purchasing decisions in anticipation of such new products. Any such losses of new customers would harm our business and financial results. In addition, most customers who purchase our TGS are entitled by contract to receive version 2.0 of our TGS and all interim software and hardware version enhancements at no additional cost. Until we deliver version 2.0 of our TGS to the customer, we are required to defer all revenue associated with the sale of the TGS. Any delay in or failure to deliver version 2.0 of our TGS to our existing customers could result in the loss of such accounts and a delay or inability to recognize revenue associated with the initial sale of the TGS to the customer.
48
If we fail to develop, acquire or secure a customized bone cutting instrument, we may not be able to develop future iterations of version 2.0 of our TGS, and as a result, our business and financial results may be adversely affected.
A key element of our TGS is the bone cutting instrument that attaches to the end of the robotic arm. The current version of the bone cutting instrument is supplied by a third-party manufacturer that supplies substantially the same instrument to other customers for use in other parts of the anatomy. We believe that to successfully develop and market version 2.0 of our TGS for use in multicompartmental resurfacing procedures, we must develop, acquire or secure a supplier for a customized bone cutting instrument that provides greater durability and is custom-fit for use with version 2.0 of our TGS in multicompartmental resurfacing procedures. Alternatively, we may need to collaborate or enter into partnerships with strategic partners to provide us with such technology. We cannot assure you that we would be able to develop, acquire or secure a supplier for a customized bone cutting instrument or enter into collaborations or partnerships on terms that are favorable to us, or at all. If we are not able to do so, we may not be able to develop and commercialize version 2.0 of our TGS and our business and financial results will be adversely affected.
We may not have sufficient funding to complete the development and commercialization of our existing products.
Our operations have consumed substantial amounts of cash since our inception. We expect to continue to spend substantial amounts of cash on expansion of our sales and marketing organization, research and development, and commercialization of our products. We believe that the net proceeds from our IPO that closed in February 2008, together with our future sales, existing cash and cash equivalent balances and interest we earn on these balances will be sufficient to meet our anticipated cash requirements for approximately the next 16-18 months. However, actual capital requirements may change and will depend on many factors, including:
| | • | | the success of our research and product development efforts; |
| |
| | • | | the expenses we incur in selling and marketing our products; |
| |
| | • | | the costs and timing of regulatory clearances for upgrades or changes to our products; |
| |
| | • | | the cash generated by sales of our products; |
| |
| | • | | the emergence of competing or complementary technological developments; |
| |
| | • | | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights, or participating in litigation-related activities; |
| |
| | • | | the terms and timing of any collaborative, licensing or other arrangements that we may establish; and |
| |
| | • | | the acquisition of businesses, products and technologies. |
49
Any additional financing may be dilutive to stockholders or may require us to grant a lender a security interest in our assets. If adequate funds are not available to us, we may have to delay or abandon development or commercialization of some of our products. We also may have to reduce marketing, customer support or other resources devoted to our products. Any of these factors could harm our business and financial results.
Our reliance on third-party suppliers, including single source suppliers, for our implants and nearly all components of our TGS could harm our ability to meet demand for our products in a timely and cost effective manner.
We rely on third-party suppliers to manufacture and supply our implants and nearly all components used in our TGS, other than software. We currently rely on a number of sole source suppliers, such as Stelkast (a business division of Trigon Incorporated), for our inlay knee implant system, Encore Medical, L.P., for our onlay knee implant system and The Anspach Effort, Inc., for our bone cutting instrument. We generally do not have long-term contracts with our suppliers, other than with Encore. We have long-term design and licensing agreements, however, with Stelkast and Encore that provide us with certain rights to the design and manufacture of the implants, and a long-term agreement with Symmetry Medical, Inc., which contemplates the manufacture, label and packaging of knee implant systems and related instrumentation and the potential development of new implant and instrument designs in the future. Because we do not have long-term contracts, our suppliers generally are not required to provide us with any guaranteed minimum production levels. As a result, we cannot assure you that we will be able to obtain sufficient quantities of key components in the future. In addition, our reliance on third-party suppliers involves a number of risks, including, among other things:
| | • | | suppliers may fail to comply with regulatory requirements or make errors in manufacturing components that could negatively affect the efficacy or safety of our products or cause delays in shipments of our products; |
| |
| | • | | we or our suppliers may not be able to respond to unanticipated changes in customer orders, and if orders do not match forecasts, we or our suppliers may have excess or inadequate inventory of materials and components; |
| |
| | • | | we may be subject to price fluctuations due to a lack of long-term supply arrangements for key components; |
| |
| | • | | we may experience delays in delivery by our suppliers due to changes in demand from us or their other customers; |
| |
| | • | | we or our suppliers may lose access to critical services and components, resulting in an interruption in the manufacture, assembly and shipment of our systems; |
| |
| | • | | fluctuations in demand for products that our suppliers manufacture for others may affect their ability or willingness to deliver components to us in a timely manner; |
50
| | • | | our suppliers may wish to discontinue supplying components or services to us for risk management reasons; |
| |
| | • | | we may not be able to find new or alternative components or reconfigure our system and manufacturing processes in a timely manner if the necessary components become unavailable; and |
| |
| | • | | our suppliers may encounter financial hardships unrelated to our demand for components, which could inhibit their ability to fulfill our orders and meet our requirements. |
If any of these risks materialize, it could significantly increase our costs and impact our ability to meet demand for our products. If we are unable to satisfy commercial demand for our TGS or implants in a timely manner, our ability to generate revenue would be impaired, market acceptance of our products could be adversely affected, and customers may instead purchase or use our competitors’ products. In addition, we could be forced to secure new or alternative components through a replacement supplier. Securing a replacement supplier could be difficult, especially for complex components such as motors, encoders, brakes and certain TGS components that are manufactured in accordance with our custom specifications. The introduction of new or alternative components may require design changes to our system that are subject to FDA and other regulatory clearances or approvals. We may also be required to assess the new manufacturer’s compliance with all applicable regulations and guidelines, which could further impede our ability to manufacture our products in a timely manner. As a result, we could incur increased production costs, experience delays in deliveries of our products, suffer damage to our reputation and experience an adverse effect on our business and financial results.
We have limited experience in assembling and testing our products and may encounter problems or delays in the assembly of our products or fail to meet certain regulatory requirements that could result in a material adverse effect on our business and financial results.
We have limited experience in assembling and testing our products, including the current version of our TGS, and no experience in doing so on a commercial scale. The current version of our TGS is complex and requires the integration of a number of separate components and processes. Version 2.0 of our TGS, if developed in the future as currently planned, is likely to be even more complex. To become profitable, we must assemble and test the TGS in commercial quantities in compliance with regulatory requirements and at an acceptable cost. Increasing our capacity to assemble and test our products on a commercial scale will require us to improve internal efficiencies. We may encounter a number of difficulties in increasing our assembly and testing capacity, including:
| | • | | managing production yields; |
| |
| | • | | maintaining quality control and assurance; |
| |
| | • | | providing component and service availability; |
51
| | • | | maintaining adequate control policies and procedures; |
| |
| | • | | hiring and retaining qualified personnel; and |
| |
| | • | | complying with state, federal and foreign regulations. |
If we are unable to satisfy commercial demand for our TGS due to our inability to assemble and test our TGS, our business and financial results, including our ability to generate revenue, would be impaired, market acceptance of our products could be materially adversely affected and customers may instead purchase or use, our competitors’ products.
Any failure in our efforts to train surgeons or hospital staff could result in lower than expected product sales and potential liabilities.
A critical component of our sales and marketing efforts is the training of a sufficient number of surgeons and hospital staff to properly use our TGS. We rely on surgeons and hospital staff to devote adequate time to learn to use our products. Convincing surgeons and hospital staff to dedicate the time and energy necessary for adequate training in the use of our system is challenging, and we cannot assure you we will be successful in these efforts. If surgeons or hospital staff are not properly trained, they may misuse or ineffectively use our products. If nurses or other members of the hospital staff are not adequately trained to assist in using our TGS, surgeons may be unable to use our products. Insufficient training may result in unsatisfactory patient outcomes, patient injury and related liability or negative publicity, which could have an adverse effect on our product sales or create substantial potential liabilities.
We will likely experience extended and variable sales cycles, which together with the unit price of the TGS and our revenue recognition policies, could cause significant variability in our results of operations for any given quarter.
Our TGS will likely have a lengthy sales cycle because it involves a major piece of capital equipment, the purchase of which will generally require the approval of senior management at hospitals, inclusion in the hospitals’ budget process for capital expenditures and, in some instances, a certificate of need from the state or other regulatory clearance. As a result, we expect that a relatively small number of units will be installed each quarter. Based on our limited experience, we estimate that this sales cycle may take between seven and twelve months from the point of initial identification and contact with a qualified surgeon until closing of the purchase with the hospital. Sales of TGS units may also be subject to a customer acceptance period, during which the customer may return the TGS unit to us subject to a penalty. Although we believe that training can be accomplished in a relatively short period of time, there may be situations where training of physicians and staff may last an additional month or more after installation. In addition, the introduction of new products could adversely impact our sales cycle as customers take additional time to assess the capital products. Because of the lengthy sales cycle, the unit price of the TGS and the relatively small number of units installed each quarter, each installation of a TGS can represent a significant component of our revenue for a particular quarter, particularly in the near term and during any other periods in which our sales volume is relatively low.
52
Moreover, we are required to defer revenue associated with our TGS until we have fulfilled our contractual obligation to deliver version 2.0 of our TGS to our customers. The deferral of revenue will result in even greater fluctuations in our reporting of quarterly revenue. As a result, in future quarters our operating results could fall below the expectations of securities analysts or investors, in which event our stock price would likely decrease. These fluctuations also mean that you will not be able to rely upon our operating results in any particular period as an indication of future performance.
Other factors that may contribute to fluctuations in our operating results may include:
| | • | | timing and level of expenditures associated with new product development activities; |
| |
| | • | | delays in shipment due, for example, to cancellations by customers, natural disasters or labor disturbances; |
| |
| | • | | delays or unexpected difficulties in the manufacturing processes of our suppliers or in our assembly process; |
| |
| | • | | timing of the announcement, introduction and delivery of new products or product upgrades by us and by our competitors; |
| |
| | • | | timing and level of expenditures associated with expansion of sales and marketing activities and our overall operations; |
| |
| | • | | disruptions in the supply or changes in the costs of raw materials, labor, product components or transportation services; and |
| |
| | • | | changes in third-party coverage and reimbursement, changes in government regulation, or a change in a customer’s financial condition or ability to obtain financing. |
These factors are difficult to forecast and may contribute to substantial fluctuations in our quarterly revenue and substantial variation from our projections, particularly during the periods in which our sales volume is low. Moreover, many of our expenses, such as office leases and certain personnel costs, are relatively fixed. We may be unable to adjust spending quickly enough to offset any unexpected revenue shortfall. Accordingly, any shortfall in revenue may cause significant variation in operating results in any quarter. Based on the above factors, we believe that quarter-to-quarter comparisons of our operating results are not a good indication of our future performance. These and other potential fluctuations also mean that you will not be able to rely upon our operating results in any particular period as an indication of future performance.
53
If we receive a significant number of warranty claims or our TGS units require significant amounts of service after sale, our costs will increase and our business and financial results will be adversely affected.
We currently warrant each TGS against defects in materials and workmanship for a period of approximately 12 months from the installation of our product by a customer. We also expect to provide technical and other services to customers beyond the warranty period pursuant to a supplemental service plan that we sell for our TGS. We have a limited history of commercial placements from which to judge our rate of warranty claims. If product returns or warranty claims are significant or exceed our expectations, we could incur unanticipated reductions in sales or additional expenditures for parts and service. In addition, our reputation could be damaged and our products may not achieve market acceptance. While we have established accruals for liability associated with product warranties, unforeseen warranty exposure in excess of those accruals could negatively impact our business and financial results.
We could become subject to product liability claims, product recalls and other field or regulatory actions that could be expensive, divert management’s attention and harm our business.
Our business exposes us to potential liability risks, product recalls and other field or regulatory actions that are inherent in the manufacturing, marketing and sale of medical device products. We may be held liable if our TGS or implants cause injury or death or is found otherwise unsuitable or defective during usage. Our TGS incorporates mechanical, electrical and optical parts, complex computer software and other sophisticated components, any of which can contain errors or failures. Complex computer software is particularly vulnerable to errors and failures, especially when first introduced. In addition, new products or enhancements to our existing products may contain undetected errors or performance problems that, despite testing, are discovered only after installation.
If any of our products are defective, whether due to design or manufacturing defects, improper use of the product or other reasons, we may be required to notify regulatory authorities and, in some circumstances, to recall the product at our expense. In particular, we are required to submit an MDR report to the FDA for any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. In 2007, we submitted MDRs to the FDA reporting two procedures that were intended as MAKOplasty procedures, but concluded as total knee replacements when a component of our TGS failed to perform as intended or was improperly operated during surgery. In January 2008, we reported a third incident in which a patient suffered a post-operative bone fracture at the insertion site of the bone pins. In February 2008, we reported a fourth incident in which a bone pin broke off below the bone surface of the tibia during a procedure.
54
In the future, we may experience additional events that may require reporting to the FDA pursuant to the MDR regulations. See “Risks Related to Regulatory Compliance.” A required notification to a regulatory authority could result in an investigation by regulatory authorities of our products, which could in turn result in product recalls, restrictions on the sale of the products, civil or criminal penalties and other field corrective action. In addition, because our products are designed to be used to perform complex surgical procedures, defects could result in a number of complications, some of which could be serious and could harm or kill patients. The adverse publicity resulting from any of these events could cause surgeons or hospitals to review and potentially terminate their relationships with us. Regulatory investigations or product recalls could also result in our incurring substantial costs, losing revenue, and implementing a change in the design, manufacturing process or the indications for which our products may be used, each of which would harm our business.
It is also possible that defects in the design, manufacture or labeling of our products could result in a product liability claim. The medical device industry has historically been subject to extensive litigation over product liability claims. A product liability claim, regardless of its merit or eventual outcome, could result in significant legal defense costs. Although we maintain product liability insurance, the coverage is subject to deductibles and limitations, and may not be adequate to cover future claims. Additionally, we may be unable to maintain our existing product liability insurance in the future at satisfactory rates or adequate amounts. A product liability claim, regardless of its merit or eventual outcome could result in:
| | • | | decreased demand for our products; |
| |
| | • | | injury to our reputation; |
| |
| | • | | diversion of management’s attention; |
| |
| | • | | significant costs of related litigation; |
| |
| | • | | payment of substantial monetary awards by us; |
| |
| | • | | product recalls or market withdrawals; |
| |
| | • | | a change in the design, manufacturing process or the indications for which our products may be used; |
| |
| | • | | loss of revenue; and |
| |
| | • | | an inability to commercialize our products under development. |
If hospitals, surgeons and other healthcare providers are unable to obtain coverage or reimbursement from third-party payors for MAKOplasty procedures, hospitals may not purchase our TGS and surgeons may not perform MAKOplasty, which would harm our business and financial results.
Our ability to successfully commercialize MAKOplasty depends significantly on the availability of coverage and reimbursement from third-party payors, including governmental programs such as Medicare and Medicaid as well as private insurance and private health plans. Reimbursement is a significant factor considered by hospitals in determining whether to acquire new capital equipment such as our technology. Although our customers have been successful in obtaining coverage and reimbursement, we cannot assure you that procedures using our technology will be covered or reimbursed by third-party payors in the future.
55
We anticipate that in the U.S. our products will be purchased primarily by hospitals, which bill various third-party payors, including governmental healthcare programs, such as Medicare, and private insurance plans for procedures using our technology. Ensuring adequate Medicare reimbursement can be a lengthy and expensive endeavor and we cannot provide assurance that we will be successful. In addition, the U.S. Congress may pass laws that impact coverage and reimbursement for healthcare services, including Medicare reimbursement to physicians and hospitals. Many private payors look to Medicare’s coverage and reimbursement policies in setting their coverage policies and reimbursement amounts. If the Centers for Medicare and Medicaid Services, or CMS, the federal agency that administers the Medicare program, or Medicare contractors limit payments to hospitals or surgeons for MAKOplasty procedures, private payors may similarly limit payments. In addition, state legislatures may enact laws limiting or otherwise affecting the level of Medicaid reimbursements. As a result, hospitals may not purchase our TGS and surgeons may choose not to perform MAKOplasty, and, as a result, our business and financial results would be adversely affected.
Medicare pays acute care hospitals a prospectively determined amount for inpatient operating costs under the Medicare hospital inpatient prospective payment system, or PPS. Under the Medicare hospital inpatient PPS, the prospective payment for a patient’s stay in an acute care hospital is determined by the patient’s condition and other patient data and procedures performed during the inpatient stay using a classification system known as diagnosis-related groups, or DRGs. As of October 1, 2007, CMS, implemented a revised version of the DRG system that uses 745 Medicare Severity DRGs, or MS-DRGs, instead of the approximately 540 DRGs Medicare previously used. The MS-DRGs are intended to account more accurately for the patient’s severity of illness when assigning each patient’s stay to a payment classification. Medicare pays a fixed amount to the hospital based on the MS-DRG into which the patient’s stay is assigned, regardless of the actual cost to the hospital of furnishing the procedures, items and services provided. Accordingly, acute care hospitals generally do not receive direct Medicare reimbursement under PPS for the specific costs incurred in purchasing medical devices. Rather, reimbursement for these costs is deemed to be included within the MS-DRG-based payments made to hospitals for the services furnished to Medicare-eligible inpatients in which the devices are utilized. Accordingly, a hospital must absorb the cost of our products as part of the payment it receives for the procedure in which the device is used. In addition, physicians that perform procedures in hospitals are paid a set amount by Medicare for performing such services under the Medicare physician fee schedule. Medicare payment rates for both systems are established annually.
56
At this time, we do not know the extent to which hospitals and physicians would consider third-party reimbursement levels adequate to cover the cost of our products. Failure by hospitals and surgeons to receive an amount that they consider to be adequate reimbursement for procedures in which our products are used could deter them from purchasing or using our products and limit our sales growth. In addition, pre-determined MS-DRG payments or Medicare physician fee schedule payments may decline over time, which could deter hospitals from purchasing our products or physicians from using them. If hospitals are unable to justify the costs of our products or physicians are not adequately compensated for procedures in which our products are utilized, they may refuse to purchase or use them, which would significantly harm our business.
Notwithstanding current or future FDA clearances, if granted, third-party payors may deny reimbursement if the payor determines that a therapeutic medical device is unnecessary, inappropriate, not cost-effective or experimental, or is used for a non-approved indication. Although we are not aware of any potential customer that has declined to purchase our TGS based upon third-party payors’ reimbursement policies, cost control measures adopted by third-party payors may have a significant effect on surgeries performed using MAKOplasty or as to the levels of reimbursement. All third-party payors, whether governmental or private, whether inside the U.S. or outside, are developing increasingly sophisticated methods of controlling healthcare costs. These cost control methods include prospective payment systems, capitated rates, benefit redesigns, pre-authorization or second opinion requirements prior to major surgery, an emphasis on wellness and healthier lifestyle interventions and an exploration of other cost-effective methods of delivering healthcare. These cost control methods also potentially limit the amount which healthcare providers may be willing to pay for medical technology which could, as a result, adversely affect our business and financial results. In addition, in the U.S., no uniform policy of coverage and reimbursement for medical technology exists among all these payors. Therefore, coverage and reimbursement for medical technology can differ significantly from payor to payor.
There also can be no assurance that current levels of reimbursement will not be decreased or eliminated in the future, or that future legislation, regulation, or reimbursement policies of third-party payors will not otherwise adversely affect the demand for our products or our ability to sell products on a profitable basis. Our customers are currently using existing reimbursement codes for knee arthroplasty. Knee arthroplasty performed in the hospital inpatient setting is currently assigned to MS-DRG 469 (“Major Joint Replacement or Reattachment of Lower Extremity with Major Complication or Comorbidity”) and MS-DRG 470 (“Major Joint Replacement or Reattachment of Lower Extremity without Major Complication of Comorbidity”), and surgeons currently bill Current Procedural Terminology, or CPT, code 27446 (“Arthroplasty, knee, condyle and plateau; medial OR lateral compartment”) for services performed in connection with procedures using our technology. If unicompartmental and multicompartmental knee resurfacing procedures gain market acceptance and the number of such procedures increases, CMS and other payors may establish billing codes for unicompartmental and multicompartmental knee resurfacing procedures that provide for a smaller reimbursement amount than knee arthroplasty, which could adversely affect our financial results and business.
57
In international markets, market acceptance of our products will likely depend in large part on the availability of reimbursement within prevailing healthcare payment systems. Reimbursement and healthcare payment systems in international markets vary significantly by country, and by region in some countries, and include both government- sponsored healthcare and private insurance. We may not obtain international reimbursement approvals in a timely manner, if at all. In addition, even if we do obtain international reimbursement approvals, the level of reimbursement may not be enough to commercially justify expansion of our business into the approving jurisdiction. To the extent we or our customers are unable to obtain coverage or reimbursement for procedures using our technology in major international markets in which we seek to market and sell our technology, our international revenue growth would be harmed, and our business and results of operations would be adversely affected.
We may attempt to acquire new products or technologies, and if we are unable to successfully complete these acquisitions or to integrate acquired businesses, products, technologies or employees, we may fail to realize expected benefits or harm our existing business.
Our success will depend, in part, on our ability to expand our product offerings and grow our business in response to changing technologies, customer demands and competitive pressures. In some circumstances, we may determine to do so through the acquisition of complementary businesses, products or technologies rather than through internal development. The identification of suitable acquisition candidates can be difficult, time consuming and costly, and we may not be able to successfully complete identified acquisitions. Furthermore, even if we successfully complete an acquisition, we may not be able to successfully integrate newly acquired organizations, products or technologies into our operations, and the process of integration could be expensive, time consuming and may strain our resources. Consequently, we may not achieve anticipated benefits of the acquisitions, which could harm our existing business. In addition, future acquisitions could result in potentially dilutive issuances of equity securities or the incurrence of debt, contingent liabilities or expenses, or other charges such as in-process research and development, any of which could harm our business and materially adversely affect our financial results or cause a reduction in the price of our common stock.
We depend on key employees, and if we fail to attract and retain employees with the expertise required for our business, we cannot grow or achieve profitability.
We are highly dependent on members of our senior management and research and development staff, in particular Maurice R. Ferré, M.D., our President and Chief Executive Officer, and Rony A. Abovitz, our Senior Vice President and Chief Technology Officer. Our future success will depend in part on our ability to retain these key employees and to identify, hire and retain additional qualified personnel with expertise in research and development and sales and marketing. Competition for qualified personnel in the medical device industry is intense, and finding and retaining qualified personnel with experience in our industry is very difficult. We believe that there are only a limited number of individuals with the requisite skills to serve in many of our key positions, and we compete for key personnel with other medical equipment and software manufacturers and technology companies, as well as universities and research institutions. It is increasingly difficult to hire and retain these persons, and we may be unable to replace key persons if they leave or fill new positions requiring key persons with appropriate experience. A significant portion of our compensation to our key employees is in the form of stock option grants. A prolonged depression in our stock price could make it difficult for us to retain our employees and recruit additional qualified personnel.
58
We do not maintain, and do not currently intend to obtain, key employee life insurance on any of our personnel other than Dr. Ferré. Although we have obtained key-man insurance covering Dr. Ferré in the amount of $2,000,000, this would not fully compensate us for the loss of Dr. Ferré’s services. Dr Ferré may terminate his employment at will at any time with 30 days notice or immediately upon the occurrence of certain events. Each of our other officers and key employees may terminate his or her employment at will at any time with 60 days notice or immediately upon the occurrence of certain events. The loss of key employees, the failure of any key employee to perform or our inability to attract and retain skilled employees, as needed, could harm our business.
If we do not effectively manage our growth, we may be unable to successfully develop, market and sell our products.
Our future revenue and operating results will depend on our ability to manage the anticipated growth of our business. We have experienced significant growth in the scope of our operations and the number of our employees since our inception. This growth has placed significant demands on our management, as well as our financial and operations resources. In order to achieve our business objectives, we must continue to grow. However, continued growth presents numerous challenges, including:
| | • | | implementing appropriate operational and financial systems and controls; |
| |
| | • | | expanding manufacturing and assembly capacity and increasing production; |
| |
| | • | | developing our sales and marketing infrastructure and capabilities; |
| |
| | • | | improving our information systems; |
| |
| | • | | identifying, attracting and retaining qualified personnel in our areas of activity; and |
| |
| | • | | hiring, training, managing and supervising our personnel. |
We cannot be certain that our systems, controls, infrastructure and personnel will be adequate to support our future operations. Any failure to effectively manage our growth could impede our ability to successfully develop, market and sell our products and our business will be harmed.
59
If we decide to market and sell MAKOplasty internationally, we would be subject to various risks relating to our international activities, which could adversely affect our business and financial results.
Although currently we do not actively market or sell our products abroad, we may actively pursue such markets in the future. If we were to conduct business outside the U.S., we would be exposed to risks separate and distinct from those we face in our U.S. operations. Our international business may be adversely affected by changing economic conditions in foreign countries. In addition, because international sales would most likely be denominated in the functional currency of the country where the product is being shipped, increases or decreases in the value of the U.S. dollar relative to foreign currencies could affect our results of operations. Engaging in international business inherently involves a number of other difficulties and risks, including:
| | • | | export restrictions and controls and other government regulation relating to technology; |
| |
| | • | | the availability and level of reimbursement within prevailing foreign healthcare payment systems; |
| |
| | • | | pricing pressures that we may experience internationally; |
| |
| | • | | compliance with existing and changing foreign regulatory laws and requirements; |
| |
| | • | | foreign laws and business practices favoring local companies; |
| |
| | • | | longer payment cycles; |
| |
| | • | | shipping delays; |
| |
| | • | | difficulties in enforcing agreements and collecting receivables through certain foreign legal systems; |
| |
| | • | | political and economic instability; |
| |
| | • | | potentially adverse tax consequences, tariffs and other trade barriers; |
| |
| | • | | international terrorism and anti-American sentiment; |
| |
| | • | | difficulties and costs of staffing and managing foreign operations; and |
| |
| | • | | difficulties in enforcing intellectual property rights. |
Our exposure to each of these risks may increase our costs, impair our ability to market and sell our products and require significant management attention, resulting in harm to our business and financial results.
60
Our operations are currently conducted primarily at a single location in Florida, which may be at risk from hurricanes, storm, fire, terror attacks or other disasters.
We currently conduct all of our management activities, most of our research and development activities and assemble all of our products at a single location in Ft. Lauderdale, Florida. We have taken various precautions to safeguard our facilities, such as obtaining insurance, establishing health and safety protocols and securing off-site storage of computer data. However, a casualty due to a hurricane, storm or other natural disasters, a fire, terrorist attack, or other unanticipated problems at this location could cause substantial delays in our operations, delay or prevent assembly of our TGS units and shipment of our implants, damage or destroy our equipment and inventory, and cause us to incur substantial expenses. Our insurance does not cover losses caused by certain events such as floods or other activities and may not be adequate to cover our losses in any particular case. Any damage, loss or delay could seriously harm our business and have an adverse affect on our financial results.
Certain of our directors, executive officers and key employees have an interest in Z-KAT that could pose potential conflicts of interest, which could harm our business.
Certain of our directors, executive officers and key employees hold, in the aggregate, approximately 17% of the equity interests in Z-KAT. We are heavily dependent on intellectual property that we license or sublicense from Z-KAT and have entered into various licensing and related arrangements with Z-KAT. Each of these individuals may face potential conflicts of interest regarding these licensing transactions as a result of their interests in Z-KAT. Dr. Ferré may face additional conflicts of interest regarding these licensing and related arrangements if he serves on the board of directors of Z-KAT. We do not have existing arrangements to address these potential conflicts of interest, cannot assure you that any conflicts will be resolved in our favor, and as a result, our business could be harmed.
Risks Related to Our Intellectual Property
If we, or the third parties from whom we license intellectual property, are unable to secure and maintain patent or other intellectual property protection for the intellectual property contained in our products, our ability to compete will be harmed.
Our commercial success depends, in part, on obtaining patent and other intellectual property protection for the technologies contained in our products. The patent positions of medical device companies, including ours, can be highly uncertain and involve complex and evolving legal and factual questions. Our patent position is uncertain and complex, in part, because of our dependence on intellectual property that we license from others. If we, or the third parties from whom we license intellectual property, fail to obtain adequate patent or other intellectual property protection for intellectual property contained in our products, or if any protection is reduced or eliminated, others could use the intellectual property contained in our products, resulting in harm to our competitive business position. In addition, patent and other intellectual property protection may not provide us with a competitive advantage against competitors that devise ways of making competitive products without infringing any patents that we own or have rights to.
As of January 1, 2008, our portfolio includes 19 wholly-owned pending U.S. patent applications, 21 pending foreign applications and other intellectual property that is wholly-owned by us. As of January 1, 2008, we had licensed rights to 118 U.S. and 47 foreign third-party granted patents, and we had licensed rights to 22 U.S. and 40 foreign third-party pending patent applications. U.S. patents and patent applications may be subject to interference proceedings and U.S. patents may be subject to reexamination proceedings in the U.S. Patent and Trademark Office. Foreign patents may be subject to opposition or comparable proceedings in the corresponding foreign patent offices.
61
Any of these proceedings could result in either loss of the patent or denial of the patent application, or loss or reduction in the scope of one or more of the claims of the patent or patent application. Changes in either patent laws or in interpretations of patent laws may also diminish the value of our intellectual property or narrow the scope of our protection. Interference, reexamination and opposition proceedings may be costly and time-consuming, and we, or the third parties from whom we license intellectual property, may be unsuccessful in defending against such proceedings. Thus, any patents that we own or license may provide limited or no protection against competitors. In addition, our pending patent applications and those we may file in the future may have claims narrowed during prosecution or may not result in patents being issued. Even if any of our pending or future applications are issued, they may not provide us with adequate protection or any competitive advantages. Our ability to develop additional patentable technology is also uncertain.
Non-payment or delay in payment of patent fees or annuities, whether intentional or unintentional, may also result in the loss of patents or patent rights important to our business. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as do the laws of the U.S., particularly in the field of medical products and procedures.
If we are unable to prevent unauthorized use or disclosure of our proprietary trade secrets and unpatented know-how, our ability to compete will be harmed.
Proprietary trade secrets, copyrights, trademarks and unpatented know-how are also very important to our business. We rely on a combination of trade secrets, copyrights, trademarks, confidentiality agreements and other contractual provisions and technical security measures to protect certain aspects of our technology, especially where we do not believe that patent protection is appropriate or obtainable. We require our employees and consultants to execute confidentiality agreements in connection with their employment or consulting relationships with us. We also require our employees and consultants to disclose and assign to us all inventions conceived during the term of their employment or engagement while using our property or which relate to our business. However, these measures may not be adequate to safeguard our proprietary intellectual property. Our employees, consultants, contractors, outside clinical collaborators and other advisors may unintentionally or willfully disclose our confidential information to competitors. In addition, confidentiality agreements may be unenforceable or may not provide an adequate remedy in the event of unauthorized disclosure. Enforcing a claim that a third party illegally obtained and is using our trade secrets is expensive and time consuming, and the outcome is unpredictable. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Unauthorized parties may also attempt to copy or reverse engineer certain aspects of our products that we consider proprietary. As a result, third parties may be able to use our proprietary technology or information, and our ability to compete in the market would be harmed.
62
We could become subject to patent and other intellectual property litigation that could be costly, result in the diversion of management’s attention, require us to pay damages and force us to discontinue selling our products.
The medical device industry is characterized by competing intellectual property and a substantial amount of litigation over patent and other intellectual property rights. In particular, the fields of orthopedic implants, CAS, haptics and robotics are well established and crowded with the intellectual property of competitors and others. A number of companies in our market, as well as universities and research institutions, have issued patents and have filed patent applications which relate to the use of CAS.
Determining whether a product infringes a patent involves complex legal and factual issues, and the outcome of a patent litigation action is often uncertain. We have not conducted an extensive search of patents issued to third parties, and no assurance can be given that third-party patents containing claims covering our products, parts of our products, technology or methods do not exist, have not been filed or could not be filed or issued. Because of the number of patents issued and patent applications filed in our technical areas, our competitors or other third parties, including third parties from whom we license intellectual property, may assert that our products and the methods we employ in the use of our products are covered by U.S. or foreign patents held by them. In addition, because patent applications can take many years to issue and because publication schedules for pending applications vary by jurisdiction, there may be applications now pending of which we are unaware and which may result in issued patents which our current or future products infringe. Also, because the claims of published patent applications can change between publication and patent grant, there may be published patent applications that may ultimately issue with claims that we infringe. There could also be existing patents that one or more of our products or parts may infringe and of which we are unaware. As the number of competitors in the market for CAS and robotics assisted knee implant systems grows, and as the number of patents issued in this area grows, the possibility of patent infringement claims against us increases. In certain situations, we or third parties, such as Z-KAT from whom we license intellectual property may determine that it is in our best interests or their best interests to voluntarily challenge a third party’s products or patents in litigation or other proceedings, including patent interferences or reexaminations. Pursuant to our licensing arrangement with Z-KAT, we have the right to prosecute, control and maintain all Z-KAT patents and intellectual property rights that are licensed to us within the field of orthopedic surgery. Z-KAT retains the right to prosecute, control and maintain its patent and intellectual property rights outside the field of orthopedic surgery, subject to certain conditions. For example, Z-KAT must notify us prior to taking any action to enforce their patent or intellectual property rights. To help ensure that Z-KAT has the resources necessary for proper prosecution and defense of any litigation arising from such enforcement action, our agreement with Z-KAT also requires that it enter into an engagement letter with competent counsel and deposit funds into an escrow account, for use by us to take over the litigation or action in the event Z-KAT is unable or unwilling to conduct proper prosecution and defense of such litigation or action. Despite these arrangements, we may have no control over Z-KAT’s decisions regarding enforcement actions outside the field of orthopedic surgery. As a result, we may become involved in unwanted litigation that could be costly, result in diversion of management’s attention, require us to pay damages and force us to discontinue selling our products.
63
On November 26, 2007, we received a letter from counsel to SensAble Technologies, Inc. alleging that we infringed certain of its patents and breached a confidentiality provision in the Sublicense Agreement, dated May 24, 2006, pursuant to which we license certain patents from SensAble. In the letter, SensAble alleged, among other things, that we exceeded the scope of our licensed field of computer-assisted surgery by using the technology for, among other things, pre-operative planning and post-operative follow-up. SensAble also alleged that we infringed one or more claims in five U.S. patents that are not among the patents licensed to us pursuant to the Sublicense Agreement. Although SensAble has not commenced any legal action against us as of the date of this report, it may do so in the future and at this time we cannot determine the ultimate outcome of any such legal action. Any such legal action could result in our inability to manufacture and sell our TGS, as currently sold, which could have a material adverse effect on our company. See also Item 3, Legal Proceedings, and Item 8, Financial Statements and Supplementary Data, Note 6 to the Financial Statements.
Infringement actions and other intellectual property claims and proceedings, whether with or without merit, may cause us to incur substantial costs and could place a significant strain on our financial resources, divert the attention of management from our business and harm our reputation. Some of our competitors may be able to sustain the costs of complex patent or intellectual property litigation more effectively than we can because they have substantially greater resources.
We cannot be certain that we will successfully defend against allegations of infringement of third-party patents and intellectual property rights. In the event that we become subject to a patent infringement or other intellectual property lawsuit and if the other party’s patents or other intellectual property were upheld as valid and enforceable and we were found to infringe the other party’s patents or violate the terms of a license to which we are a party, we could be required to pay damages. We could also be prevented from selling our products unless we could obtain a license to use technology or processes covered by such patents or were able to redesign the product to avoid infringement. A license may not be available at all or on commercially reasonable terms or we may not be able to redesign our products to avoid infringement. Modification of our products or development of new products could require us to conduct clinical trials and to revise our filings with the FDA and other regulatory bodies, which would be time-consuming and expensive. In these circumstances, we may be unable to sell our products at competitive prices or at all, our business and operating results could be harmed and our stock price may decline. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
64
We may be subject to damages resulting from claims that our employees or we have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees were previously employed at universities or other medical device companies, including our competitors or potential competitors. We could in the future be subject to claims that these employees, or we, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending against such claims, a court could order us to pay substantial damages and prohibit us from using technologies or features that are essential to our products and processes, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers. In addition, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize certain potential products, which could severely harm our business. Even if we are successful in defending against these claims, such litigation could result in substantial costs and be a distraction to management.
Risks Related to Regulatory Compliance
If we fail to comply with the extensive government regulations relating to our business, we may be subject to fines, injunctions and other penalties that could harm our business.
Our medical device products and operations are subject to extensive regulation by the FDA, pursuant to the Federal Food, Drug, and Cosmetic Act, or FDCA, and various other federal, state and foreign governmental authorities. Government regulations and foreign requirements specific to medical devices are wide ranging and govern, among other things:
| | • | | design, development and manufacturing; |
| |
| | • | | testing, labeling and storage; |
| |
| | • | | clinical trials; |
| |
| | • | | product safety; |
| |
| | • | | marketing, sales and distribution; |
| |
| | • | | premarket clearance or approval; |
| |
| | • | | record keeping procedures; |
| |
| | • | | advertising and promotions; |
| |
| | • | | recalls and field corrective actions; |
65
| | • | | post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or serious injury; and |
| |
| | • | | product export. |
In the U.S., before we can market a new medical device, or a new use of, or claim for, or significant modification to, an existing product, we must first receive either premarket clearance under Section 510(k) of the FDCA, or approval of a PMA from the FDA, unless an exemption applies. In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology and safety and effectiveness, in order to clear the proposed device for marketing. Clinical data is sometimes required to support substantial equivalence. The PMA approval pathway requires an applicant to demonstrate the safety and effectiveness of the device based, in part, on data obtained in clinical trials. Both of these processes can be expensive and lengthy and entail significant user fees, unless exempt. The FDA’s 510(k) clearance process usually takes from three to 12 months, but it can last longer. The process of obtaining PMA approval is much more costly and uncertain than the 510(k) clearance process. It generally takes from one to three years, or even longer, from the time the PMA application is submitted to the FDA until an approval is obtained. There is no assurance that we will be able to obtain FDA clearance or approval for any of our new products on a timely basis, or at all.
The FDA, state, foreign and other governmental authorities have broad enforcement powers. Our failure to comply with applicable regulatory requirements could result in governmental agencies or a court taking action, including any of the following sanctions:
| | • | | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| |
| | • | | customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; |
| |
| | • | | operating restrictions or partial suspension or total shutdown of production; |
| |
| | • | | refusing or delaying requests for 510(k) clearance or PMA approvals of new products or modified products; |
| |
| | • | | withdrawing 510(k) clearances or PMA approvals that have already been granted; |
| |
| | • | | refusal to grant export approval for our products; or |
| |
| | • | | criminal prosecution. |
66
Failure to obtain regulatory approval in additional foreign jurisdictions will prevent us from expanding the commercialization of our products abroad.
Currently, our primary market is the U.S. market for knee resurfacing procedures. We are, however, exploring international markets on a limited basis and may expand our overseas sales and marketing efforts in the future. If we were to expand our sales and marketing efforts to foreign jurisdictions, we would have to obtain separate regulatory approvals from those foreign jurisdictions. The approval procedure varies among jurisdictions and can involve substantial additional testing. Approval or clearance by the FDA does not ensure approval by regulatory authorities in other jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign jurisdictions or by the FDA. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval in addition to other risks. In addition, the time required to obtain foreign approval may differ from that required to obtain FDA approval, and we may not obtain foreign regulatory approvals on a timely basis, if at all. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any foreign market, which would harm our potential for future growth.
If we or our third-party manufacturers or suppliers fail to comply with the FDA’s Quality System Regulation, our manufacturing operations could be interrupted and our product sales and operating results could suffer.
We and some of our third-party manufacturers and suppliers are required to comply with the FDA’s Quality System Regulation, or QSR, which covers the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our products. We and our manufacturers and suppliers are also subject to the regulations of foreign jurisdictions regarding the manufacturing process if we market our products overseas. The FDA enforces the QSR through periodic and unannounced inspections of manufacturing facilities. To date, our facilities have not been subject to any inspections by regulatory authorities. We did pass a BSi certification audit of our Quality System to ISO 13485:2003 in preparation for CE marking. BSi will be doing surveillance audits once a year to make sure we continue to be in compliance. We anticipate that we and certain of our third-party manufacturers and suppliers will be subject to inspections by regulatory authorities in the future. If our facilities or those of our manufacturers or suppliers fail to take satisfactory corrective action in response to an adverse QSR inspection, the FDA could take enforcement action, including any of the following sanctions:
| | • | | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| |
| | • | | customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; |
| |
| | • | | operating restrictions or partial suspension or total shutdown of production; |
| |
| | • | | refusing or delaying requests for 510(k) clearance or PMA approvals of new products or modified products; |
67
| | • | | withdrawing 510(k) clearances or PMA approvals that have already been granted; |
| |
| | • | | refusal to grant export approval for our products; or |
| |
| | • | | criminal prosecution. |
Any of these sanctions could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be required to bear other costs or take other actions that may have a negative impact on our future sales and our ability to generate profits.
Our products may in the future be subject to product recalls that could harm our reputation, business operations and financial results.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design, or manufacture or labeling. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is a reasonable probability that the device would cause serious injury or death. In addition, foreign governmental bodies have the authority to require the recall of our products in the event of material deficiencies or defects in design or manufacture. Manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us or one of our distributors could occur as a result of component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our financial condition and results of operations. We may initiate certain voluntary recalls involving our products in the future. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA. If we determine that certain of those recalls do not require notification of the FDA, the FDA may disagree with our determinations and require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA could take enforcement action, including any of the following sanctions for failing to report the recalls when they were conducted:
| | • | | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| |
| | • | | customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; |
| |
| | • | | operating restrictions or partial suspension or total shutdown of production; |
| |
| | • | | refusing or delaying our requests for 510(k) clearance or PMA approvals of new products or modified products; |
| |
| | • | | withdrawing 510(k) clearances or PMA approvals that have already been granted; |
| |
| | • | | refusal to grant export approval for our products; or |
| |
| | • | | criminal prosecution. |
68
Any of these sanctions could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be required to bear other costs or take other actions that may have a negative impact on our future sales and our ability to generate profits.
If our products, or malfunction of our products, cause or contribute to a death or a serious injury, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA medical device reporting, or MDR, regulations, we are required to report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. In addition, all manufacturers placing medical devices in European Union markets are legally bound to report any serious or potentially serious incidents involving devices they produce or sell to the relevant authority in whose jurisdiction the incident occurred. In 2007, we submitted MDRs to report two procedures that were intended as MAKOplasty procedures, but concluded as total knee replacements when a component of our TGS failed to perform as intended or was improperly operated during surgery. In January 2008, we reported a third incident in which a patient suffered a post-operative bone fracture at the insertion site of the bone pins. In February 2008, we reported a fourth incident in which a bone pin broke off below the bone surface of the tibia during a procedure. In the future, we may experience additional events that may require reporting to the FDA pursuant to the MDR regulations. Any adverse event involving our products could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection, mandatory recall or other enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results. In addition, failure to report such adverse events to appropriate government authorities on a timely basis, or at all, could result in an enforcement action against us.
We may be subject to fines, penalties or injunctions if we are determined to be promoting the use of our products for unapproved or “off-label” uses, resulting in damage to our reputation and business.
Our promotional materials and training methods must comply with FDA and other applicable laws and regulations, including the prohibition of the promotion of a medical device for a use that has not been cleared or approved by FDA. Use of a device outside its cleared or approved indications is known as “off-label” use. We believe that the specific surgical procedures for which our products are marketed fall within the scope of the surgical applications that have been cleared by the FDA. However, physicians may use our products off-label, as the FDA does not restrict or regulate a physician’s choice of treatment within the practice of medicine. However, if the FDA determines that our promotional materials or training constitutes promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine and criminal penalties.
69
It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and adoption of the products would be impaired. Although our policy is to refrain from statements that could be considered off-label promotion of our products, the FDA or another regulatory agency could disagree and conclude that we have engaged in off-label promotion. In addition, the off-label use of our products may increase the risk of injury to patients, and, in turn, the risk of product liability claims. Product liability claims are expensive to defend and could divert our management’s attention and result in substantial damage awards against us.
Federal regulatory reforms may adversely affect our ability to sell our products profitably.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the clearance or approval, manufacture and marketing of a medical device. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be.
Without limiting the generality of the foregoing, Congress has recently enacted, and the President has signed into law, the Food and Drug Administration Amendments Act of 2007, or the Amendments. This law requires, among other things, that the FDA propose, and ultimately implement, regulations that will require manufacturers to label medical devices with unique identifiers unless a waiver is received from the FDA. Once implemented, compliance with those regulations may require us to take additional steps in the manufacture of our products and labeling. These steps may require additional resources and could be costly. In addition, the Amendments will require us to, among other things, pay annual establishment registration fees to the FDA for each of our FDA registered facilities.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws and regulations and could face substantial penalties if we are unable to fully comply with such laws.
While we do not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, many healthcare laws and regulations apply to our business. For example, we could be subject to healthcare fraud and abuse and patient privacy regulation and enforcement by both the federal government and the states in which we conduct our business. The healthcare laws and regulations that may affect our ability to operate include:
| | • | | the federal healthcare programs’ Anti-Kickback Statute, which prohibits, among other things, persons or entities from soliciting, receiving, offering or providing remuneration, directly or indirectly, in return for or to induce either the referral of an individual for, or the purchase order or recommendation of, any item or service for which payment may be made under a federal healthcare program such as the Medicare and Medicaid programs; |
70
| | • | | federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent, or are for items or services not provided as claimed, and which may apply to entities like us to the extent that our interactions with customers may affect their billing or coding practices; |
| |
| | • | | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which established new federal crimes for knowingly and willfully executing a scheme to defraud any healthcare benefit program or making false statements in connection with the delivery of or payment for healthcare benefits, items or services, as well as leading to regulations imposing certain requirements relating to the privacy, security and transmission of individually identifiable health information; and |
| |
| | • | | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, and state laws governing the privacy of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
The orthopedic medical device industry is, and in recent years has been, under heightened scrutiny as the subject of government investigations and enforcement actions involving manufacturers who allegedly offered unlawful inducements to potential or existing customers in an attempt to procure their business, including specifically arrangements with physician consultants. We have arrangements with surgeons, hospitals and other entities which may be subject to scrutiny. For example, we have consulting agreements with orthopedic surgeons using or considering the use of our TGS, knee implants and disposable products, for assistance in product development, and professional training and education, among other things. Payment for these consulting services sometimes is in the form of stock options or royalties rather than per hour or per diem amounts that would require verification of time worked. In addition, we sometimes allow hospitals a period of evaluation of our products at no charge. If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from the Medicare and Medicaid programs and the curtailment or restructuring of our operations. Any penalties, damages, fines, exclusions, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results. The risk of our being found in violation of these laws is increased by the fact that many of these laws are broad and their provisions are open to a variety of interpretations. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. If the surgeons or other providers or entities with whom we do business are found to be non-compliant with applicable laws, they may be subject to sanctions, which could also have a negative impact on our business.
71
Risks Related to Ownership of our Common Stock
We expect that the price of our common stock will fluctuate substantially, which could lead to losses for stockholders, possibly resulting in class action securities litigation.
Prior to our IPO in February 2008, there was no public market for shares of our common stock. Since the IPO, our common stock has experienced low trading volumes. An active public trading market may not develop or, if developed, may not be sustained. The market price for our common stock will be affected by a number of factors, including:
| | • | | the receipt, denial or timing of regulatory clearances or approvals of our products or competing products; |
| |
| | • | | changes in policies affecting third-party coverage and reimbursement in the U.S. and other countries; |
| |
| | • | | ability of our products, if they receive regulatory clearance, to achieve market success; |
| |
| | • | | the performance of third-party contract manufacturers and component suppliers; |
| |
| | • | | our ability to develop sales and marketing capabilities; |
| |
| | • | | our ability to manufacture our products to commercial standards; |
| |
| | • | | the success of any collaborations we may undertake with other companies; |
| |
| | • | | our ability to develop, introduce and market new or enhanced versions of our products on a timely basis; |
| |
| | • | | actual or anticipated variations in our results of operations or those of our competitors; |
| |
| | • | | announcements of new products, technological innovations or product advancements by us or our competitors; |
| |
| | • | | developments with respect to patents and other intellectual property rights; |
| |
| | • | | sales of common stock or other securities by us or our stockholders in the future; |
72
| | • | | additions or departures of key scientific or management personnel; |
| |
| | • | | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| |
| | • | | trading volume of our common stock; |
| |
| | • | | changes in earnings estimates or recommendations by securities analysts, failure to obtain analyst coverage of our common stock or our failure to achieve analyst earnings estimates; |
| |
| | • | | developments in our industry; and |
| |
| | • | | general market conditions and other factors unrelated to our operating performance or the operating performance of our competitors. |
In addition, the stock prices of many companies in the medical device industry have experienced wide fluctuations that have often been unrelated to the operating performance of these companies. We expect our stock price to be similarly volatile. These broad market fluctuations may continue and could harm our stock price. Following periods of volatility in the market price of a company’s securities, stockholders have often instituted class action securities litigation against those companies. Class action securities litigation, if instituted against us, could result in substantial costs and a diversion of our management resources, which could significantly harm our business.
Securities analysts may issue negative reports, which may have a negative impact on the market price of our common stock.
The trading market for our common stock may be affected in part by the research and reports that industry or financial analysts publish about us or our business. It may be difficult for companies such as ours, with smaller market capitalizations, to continue to attract securities analysts that will cover our common stock. If one or more of the analysts who elects to cover us downgrades our stock, our stock price would likely decline rapidly. If one or more of these analysts ceases coverage of our company, we could lose visibility in the market, which in turn could cause our stock price to decline. This could have a negative effect on the market price of our stock.
Our principal stockholders, directors and executive officers own a large percentage of our voting stock, which allows them to exercise significant influence over matters subject to stockholder approval.
Our executive officers, directors and principal stockholders holding 5% or more of our outstanding common stock beneficially own or control approximately 47% of the outstanding shares of our common stock. Accordingly, these executive officers, directors and principal stockholders, acting as a group, have substantial influence over the outcome of corporate actions requiring stockholder approval, including the election of directors, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transaction. These stockholders may also delay or prevent a change of control or otherwise discourage a potential acquirer from attempting to obtain control of us, even if such a change of control would benefit our other stockholders. This significant concentration of stock ownership may adversely affect the trading price of our common stock due to investors’ perception that conflicts of interest may exist or arise.
73
We have not paid dividends in the past and do not expect to pay dividends in the future.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all future earnings for the operation and expansion of our business and, therefore, do not anticipate declaring or paying cash dividends in the foreseeable future. The payment of dividends will be at the discretion of our board of directors and will depend on our results of operations, capital requirements, financial condition, prospects, contractual arrangements, any limitations on payments of dividends present in any of our future debt agreements, and other factors our board of directors may deem relevant. If we do not pay dividends, a return on your investment will only occur if our stock price appreciates.
Sales of a substantial number of shares of our common stock in the public market, or the perception that they may occur, may depress the market price of our common stock.
Of the approximately 18.5 million shares of our common stock currently outstanding, up to approximately 13.3 million shares held by existing holders prior to our IPO will generally become available for sale in the public market following expiration or termination of 180-day lock-up agreements or upon exercise by certain holders of available registration rights. For more information regarding registration rights of our stockholders, see Item 5, Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities, “Registration Rights,” in this report.
The lock-up agreements delivered by our executive officers, directors and substantially all of our stockholders and option holders in connection with our IPO provide that Morgan Stanley & Co. Incorporated and J.P. Morgan Securities Inc., on behalf of the underwriters, in their sole discretion, may release those parties, at any time or from time to time and without notice, from their obligation not to dispose of shares of common stock on or before August 12, 2008. Sales of substantial amounts of our common stock in the public market, or the perception that substantial sales may be made, could cause the market price of our common stock to decline. These sales might also make it more difficult for us to sell equity securities at a time and price that we deem appropriate.
We are obligated to develop and maintain proper and effective internal control over financial reporting, and we may not complete our analysis of our internal control over financial reporting in a timely manner or this internal control may not be determined to be effective, which may adversely affect investor confidence in our company and, as a result, the value of our common stock.
We are required, pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting beginning in our second annual report on Form 10-K for the fiscal year ending December 31, 2008. This assessment will need to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting, as well as a statement that our auditors have issued a report on our internal control over financial reporting.
74
We are just beginning the costly and challenging process of compiling this documentation before we perform the testing and evaluation needed to comply with Section 404. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control is effective unless remediation occurs and is satisfactorily tested prior to December 31, 2008. If we are unable to assert that our internal control over financial reporting is effective, or if our auditors are unable to express an opinion on the effectiveness of our internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, which would have a material adverse effect on the price of our common stock. Failure to comply with the new rules might make it more difficult for us to obtain certain types of insurance, including director and officer liability insurance, and we might be forced to accept reduced policy limits and coverage and/or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on committees of our board of directors, or as executive officers.
In addition, as a public company, we will incur significant additional legal, accounting and other expenses that we did not incur as a private company, and our administrative staff will be required to perform additional tasks. For example, we have increased the size of our accounting staff, updated our accounting systems and procedures, revised the roles and duties of our board committees, retained a transfer agent, adopted an insider trading policy and are bearing all of the internal and external costs of preparing and distributing periodic public reports in compliance with our obligations under the securities laws. In addition, we are in the process of adopting disclosure controls and procedures. Changing laws, regulations and standards relating to corporate governance and public disclosure, and related regulations implemented by the SEC, and The NASDAQ Global Market, are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. We are investing resources to comply with evolving laws, regulations and standards, and this investment will result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
75
ITEM 2. PROPERTIES
We lease approximately 20,000 square feet of office and warehouse space in Ft. Lauderdale, Florida, which is used as our headquarters and for the assembly of our products. Our lease expires on July 31, 2011. Thereafter, we have the right to renew our lease for two three-year terms upon prior written notice and the fulfillment of certain conditions. We believe that this facility will be adequate to meet our needs through July 2008, but additional space will be required in the future to accommodate our anticipated growth. We anticipate securing, before July 2008, additional office and warehouse space of approximately 16,000 square feet adjacent to our facility under a lease having term and renewal provisions comparable to the provisions of our current lease.
ITEM 3. LEGAL PROCEEDINGS
On November 26, 2007, we received a letter from counsel to SensAble Technologies, Inc. alleging that we infringed certain of its patents and breached a confidentiality provision in the Sublicense Agreement, dated May 24, 2006, pursuant to which we license certain patents from SensAble. In the letter, SensAble alleged, among other things, that we exceeded the scope of our licensed field of computer-assisted surgery by using the technology for, among other things, pre-operative planning and post-operative follow-up. SensAble also alleged that we infringed one or more claims in five U.S. patents that are not among the patents licensed to us pursuant to the Sublicense Agreement.
We have investigated SensAble’s allegations, and, based on the opinion of counsel, we believe that if SensAble initiates a lawsuit against us, a court should find that our TGS does not infringe any of the SensAble patents identified in the November 26, 2007 letter. We have communicated our belief to SensAble. SensAble has not commenced any legal action against us, but may do so in the future. The letter from counsel to SensAble stated that unless we, among other things, cease and desist from alleged infringement of its patents or pay additional licensing fees, including a proposed licensing fee of $30 million for additional patents not included in the Sublicense Agreement, SensAble intends to bring a lawsuit against us. We intend to vigorously defend ourselves against these allegations in the event of a lawsuit. We cannot predict the outcome of this matter at this time.
ITEM 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
None
76
PART II
| | |
| ITEM 5. | | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market for Our Common Stock
Our common stock began trading on The NASDAQ Global Market under the symbol “MAKO” on February 14, 2008. Prior to that date, there was no identifiable public market for our common stock.
Our stock transfer records indicated that as of March 14, 2008, there were approximately 80 holders of record of our common stock.
Dividend Policy
We have never declared dividends or paid any cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings to support our operations and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future. Any future determination related to dividend policy will be made at the discretion of our board of directors.
Reverse Stock Split and Conversion of Preferred Stock
In connection with our IPO, we effected a one-for-3.03 reverse split of our common stock on February 8, 2008. On the effective date of the reverse split, each 3.03 shares of issued and outstanding common stock were combined into one share of common stock. In accordance with the terms of each series of preferred stock, all outstanding shares of our preferred stock were converted to common stock immediately prior to the closing of our IPO on February 20, 2008 at a conversion price that was adjusted to reflect the reverse split.
All amounts related to our issued and outstanding common stock and options to purchase common stock in this report and in the accompanying financial statements and the related notes have been retroactively adjusted to give effect to the one-for-3.03 reverse split.
Equity Compensation Plan Information
We had one equity compensation plan as of December 31, 2007, the 2004 Stock Incentive Plan, or 2004 Plan, which was approved by our stockholders in December 2004.
77
Information at fiscal year-end 2007 about issuances of common stock under the 2004 Plan follows.
| | | | | | | | | | | | | |
| | | | | | | | | | | (c) |
| | | (a) | | (b) | | Number of Shares of |
| | | Number of Shares of | | Weighted- | | Our Common Stock Remaining |
| | | Our Common Stock | | Average | | Available for Future Issuance |
| | | to be Issued Upon | | Exercise Price of | | Under Equity Compensation |
| | | Exercise of | | Outstanding | | Plans (Exceeding Securities |
| Plan Category | | Outstanding Options | | Options | | Reflected in Col (a)) |
| 2004 Stock Incentive Plan | | | 1,916,525 | | | $ | 4.81 | | | | 84,728 | |
| Equity compensation plans not approved by our security holders | | None | | | None | | | None | |
| TOTAL | | | 1,916,525 | | | $ | 4.81 | | | | 84,728 | |
In January 2008, our board of directors and stockholders approved the MAKO Surgical Corp. 2008 Omnibus Incentive Plan, or the 2008 Omnibus Incentive Plan. The 2008 Omnibus Incentive Plan was effective upon the consummation of our IPO and will expire January 9, 2018 unless earlier terminated by the board of directors. The aggregate number of shares of our common stock that may be issued initially pursuant to stock awards under the 2008 Omnibus Incentive Plan is 1,084,703 shares. Awards under the plan may be made in the form of: stock options, which may be either incentive stock options or non-qualified stock options; stock appreciation rights; restricted stock; restricted stock units; dividend equivalent rights; performance shares; performance units; cash-based awards; other stock-based awards, including unrestricted shares; and any combination of the foregoing.
In January 2008, our board of directors and stockholders approved the MAKO Surgical Corp. 2008 Employee Stock Purchase Plan, or the 2008 Employee Stock Purchase Plan. The plan was effective upon the consummation of our IPO. The 2008 Employee Stock Purchase Plan authorizes the issuance of 625,000 shares of common stock for purchase by our eligible employees or any of our participating affiliates. The shares of common stock issuable under the 2008 Employee Stock Purchase Plan may be authorized but unissued shares, treasury shares or shares purchased on the open market. The purchase price for an offering period may not be less than 85% of the fair market value of our common stock on the first trading day of the offering period or the day on which the shares are purchased, whichever is lower.
Registration Rights
Under our registration rights agreement with purchasers of our convertible preferred stock, the holders of 10,945,080 shares of common stock, which were issued upon conversion of 4,498,745 shares of Series A redeemable convertible preferred stock, 15,151,516 shares of Series B redeemable convertible preferred stock and 13,513,514 shares of Series C redeemable convertible preferred stock in connection with our IPO, have the following registration rights with respect to their registrable shares of common stock:
Demand Registration Rights.At any time beginning six months after the consummation of the offering, the holders of at least 10% of the registrable shares of common stock can request that we file up to two registration statements registering all or a portion of their registrable shares, provided that the net offering price for such registration is at least $5,000,000. These registration rights are subject to additional conditions and limitations, including the right of the underwriters to limit the number of shares included in any such registration under certain circumstances.
78
Form S-3 Registration Rights.If we are eligible to file a registration statement on Form S-3, each holder of registrable shares has the right to demand that we file a registration statement, including a shelf registration statement, for such holder on Form S-3 so long as the aggregate offering price of securities to be sold under the registration statement on Form S-3 is at least $500,000. There is no limit to the number of registrations on Form S-3 that holders may request of us, provided that we are not required to effect more than two registrations during any six consecutive month period.
“Piggyback” Registration Rights.Whenever we propose to file a registration statement under the Securities Act of 1933, other than with respect to a registration related to employee benefit plans, debt securities or corporate reorganizations, the holders of registrable shares are entitled to notice of the registration and have the right to include their registrable shares in such registration.
Expenses of Registration.We are required to pay all fees and expenses, other than underwriting discounts and commissions, relating to all demand registrations, Form S-3 registrations and piggyback registrations.
Lock-Up Agreements
We, along with our directors, executive officers and substantially all of our other stockholders, option holders and warrant holders, have agreed with the underwriters of our IPO that for a period of 180 days following the date of the final prospectus, we or they will not offer, sell, assign, transfer, pledge, contract to sell or otherwise dispose of or hedge any shares of our common stock or any securities convertible into or exchangeable for shares of common stock, subject to specified exceptions. J.P. Morgan Securities Inc. and Morgan Stanley & Co. Incorporated may, in their sole discretion, at any time without prior notice, release all or any portion of the shares from the restrictions in any such agreement.
Unregistered Sales of Equity Securities
During fiscal year 2007, we sold and issued the following unregistered securities:
| | • | | We granted options to purchase an aggregate of 1,001,695 shares of our common stock, at a weighted-average exercise price of $8.30 per share, to our employees pursuant to our 2004 Stock Incentive Plan, or 2004 Plan. During this period, options to purchase an aggregate of 31,814 shares of our common stock related to our 2004 Plan were cancelled without being exercised. Also during this period, 1,306 options were exercised under our 2004 Plan. |
79
| | • | | We granted 330,033 restricted shares of our common stock pursuant to the 2004 Plan. |
| |
| | • | | We sold 13,513,514 shares of Series C redeemable convertible preferred stock. |
The grants of options to purchase shares of our common stock and restricted shares of our common stock pursuant to the 2004 Plan were deemed to be exempt from registration under the Securities Act of 1933, as amended, by virtue of (a) Rule 701 promulgated thereunder in that they were offered and sold pursuant to a written compensatory benefit plan, as provided by Rule 701 or (2) Section 4(2) of the Securities Act of 1933, as amended, as transactions by an issuer not involving any public offering. The offer and sale of shares of convertible preferred stock to purchase shares of our common stock were deemed to be exempt from registration under the Securities Act of 1933, as amended, by virtue of Rule 506 of Regulation D promulgated thereunder for limited offers and sales of securities.
Uses of Proceeds from Sale of Registered Securities
Our IPO was effected through a registration statement on Form S-1 (File No. 333-146162), that was declared effective by the SEC on February 14, 2008. We registered 5,100,000 shares of our common stock with an aggregate offering price of $51 million, all of which shares we sold. The offering was completed after the sale of all 5,100,000 shares. Morgan Stanley & Co. Incorporated and J.P. Morgan Securities Inc. were the joint book-running managing underwriters of our IPO and Cowen and Company and Wachovia Securities acted as co-managers. The underwriters elected not to exercise their over-allotment option. We paid $3.6 million of the proceeds in underwriting discounts and commissions, and we incurred an additional $3.7 million of expenses, of which approximately $2.7 million was incurred during the fiscal year ended December 31, 2007 and $1.0 million was incurred subsequent to the fiscal year end. None of the expenses were paid, directly or indirectly, to directors, officers or persons owning 10% or more of our common stock, or to our affiliates.
We currently intend to use the aggregate proceeds of $47.4 million, net of underwriting discounts and commissions, from our IPO as follows:
| | • | | Approximately $14.0 — $20.0 million for the expansion of our sales and marketing activities; |
| |
| | • | | Approximately $12.0 — $18.0 million for continuation of our research and development activities; |
| |
| | • | | Approximately $4.0 million payment to IBM, as required upon the IPO of our common stock under the terms of our licensing agreement with IBM; and |
| |
| | • | | The remainder to fund working capital and other general corporate purposes, including the expenses associated with our IPO. |
Management has broad discretion over the uses of the proceeds of the IPO. As of December 31, 2007, the proceeds were not yet available to us. As of March 14, 2008, no significant amount of the proceeds had been used. Pending the uses described above, we plan to invest the net proceeds in U.S. government securities and other short-term, investment-grade, interest-bearing instruments or high-grade corporate notes.
80
Issuer Purchases of Equity Securities
We did not repurchase any shares of our capital stock during the fourth quarter of fiscal year 2007.
ITEM 6. SELECTED FINANCIAL DATA
The following table sets forth certain financial data with respect to our business. The information set forth below is not necessarily indicative of results of future operations and should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Item 7 and the financial statements and related notes thereto in Item 8.
We were formed in November 2004 to be the successor of the CAS and haptic robotics business of Z-KAT, Inc., a company founded in 1997 to develop CAS technologies. Z-KAT is considered to be our “Predecessor.” The balance sheet and statements of operations data for the periods prior to and including November 11, 2004 refer to the Predecessor. The statement of operations data for the period from January 1, 2004 through November 11, 2004 have been derived from the audited statement of operations of the Predecessor. The statement of operations and balance sheet data for the fiscal year ended December 31, 2003, have been derived from the unaudited financial statements of the Predecessor.
The balance sheet and statements of operations data subsequent to November 11, 2004 refer to operations subsequent to our formation, and these periods are referred to as the Company.
Because the Predecessor financial statements were significantly different from the Company, the Predecessor and the Company financial statements are not comparable and, accordingly, the accompanying selected financial data below are presented separated by a vertical black line.
81
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | The Company | | | | Predecessor | |
| | | | | | | | | | | | | | Nov. 12, | | | | Jan. 1, | | | | |
| | | | | 2004 | | | | 2004 | | | | |
| | | Year Ended | | Through | | | | Through | | | Year Ended | |
| | | December 31, | | Dec. 31, | | | | Nov. 11, | | | December 31, | |
| | | 2007 | | | 2006 | | | 2005 | | 2004 | | | | 2004 | | | 2003 | |
Statements of Operations Data | | | | | | | | | | | | | | | | | | | | | | | | |
| Revenue | | $ | 771,362 | | | $ | 62,571 | | | $ | — | | $ | — | | | | $ | 1,648,342 | | | $ | 1,380,128 | |
| Cost of revenue | | | 582,914 | | | | 76,547 | | | | — | | | — | | | | | 1,085,523 | | | | 1,010,803 | |
| | | | | | | | | | | | | | | | | | | |
| Gross profit (loss) | | | 188,448 | | | | (13,976 | ) | | | — | | | — | | | | | 562,819 | | | | 369,325 | |
| | | | | | | | | | | | | | | | | | | |
| Operating costs and expenses: | | | | | | | | | | | | | | | | | | | | | | | | |
| Selling, general and administrative | | | 12,042,690 | | | | 5,022,685 | | | | 2,735,901 | | | 630,048 | | | | | 2,642,028 | | | | 4,244,414 | |
| Research and development | | | 8,268,803 | | | | 5,192,453 | | | | 2,581,828 | | | 402,899 | | | | | 1,453,685 | | | | 3,530,298 | |
| Depreciation and amortization | | | 1,296,881 | | | | 644,082 | | | | 98,519 | | | 5,727 | | | | | 429,694 | | | | 564,720 | |
| | | | | | | | | | | | | | | | | | | |
| Total operating costs and expenses | | | 21,608,374 | | | | 10,859,220 | | | | 5,416,248 | | | 1,038,674 | | | | | 4,525,407 | | | | 8,339,432 | |
| | | | | | | | | | | | | | | | | | | |
| Loss from operations | | | (21,419,926 | ) | | | (10,873,196 | ) | | | (5,416,248 | ) | | (1,038,674 | ) | | | | (3,962,588 | ) | | | (7,970,107 | ) |
| Interest and other income | | | 1,073,280 | | | | 476,578 | | | | 269,231 | | | — | | | | | 868 | | | | 8,682 | |
| Interest and other expenses | | | (311,608 | ) | | | (220,219 | ) | | | — | | | — | | | | | (479,959 | ) | | | (17,240 | ) |
| | | | | | | | | | | | | | | | | | | |
| Net loss | | $ | (20,658,254 | ) | | $ | (10,616,837 | ) | | $ | (5,147,017 | ) | $ | (1,038,674 | ) | | | $ | (4,441,679 | ) | | $ | (7,978,665 | ) |
| | | | | | | | | | | | | | | | | | | |
| Net loss attributable to common stockholders | | $ | (24,318,028 | ) | | $ | (12,493,183 | ) | | $ | (6,288,297 | ) | $ | (1,060,713 | ) | | | $ | (5,221,774 | ) | | $ | (8,828,815 | ) |
| | | | | | | | | | | | | | | | | | | |
| Net loss per share: Basic and diluted attributable to common stockholders (1) | | $ | (14.74 | ) | | $ | (8.03 | ) | | $ | (4.18 | ) | $ | (2.39 | ) | | | $ | (0.63 | ) | | $ | (1.23 | ) |
| | | | | | | | | | | | | | | | | | | |
Weighted average common shares
outstanding: Basic and diluted (2) | | | 1,649,365 | | | | 1,555,287 | | | | 1,502,761 | | | 443,868 | | | | | 8,234,560 | | | | 7,202,176 | |
| | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| | | The Company | | | Predecessor | |
| | | | | | As of | |
| | | As of December 31 | | | December 31 | |
| | | 2007 | | | 2006 | | | 2005 | | | 2004 | | | 2003 | |
Balance Sheet Data: | | | | | | | | | | | | | | | | | | | | |
| Cash and cash equivalents | | $ | 9,615,027 | | | $ | 2,108,015 | | | $ | 6,145,266 | | | $ | 1,959,079 | | | $ | 257,939 | |
| Short-term investments | | | 3,083,980 | | | | 1,399,763 | | | | 10,097,020 | | | | — | | | | — | |
| Total assets | | | 29,190,306 | | | | 12,753,581 | | | | 17,435,073 | | | | 2,403,518 | | | | 5,717,405 | |
| Long-term debt, net of current portion | | | — | | | | — | | | | — | | | | — | | | | 1,021,681 | |
| Redeemable convertible preferred stock | | | 59,486,700 | | | | 25,910,622 | | | | 24,034,276 | | | | 2,866,073 | | | | 13,597,736 | |
| Accumulated deficit | | | (42,843,131 | ) | | | (19,366,087 | ) | | | (6,819,675 | ) | | | (1,038,674 | ) | | | (23,409,631 | ) |
| Total stockholders’ deficit | | | (42,837,231 | ) | | | (19,436,916 | ) | | | (6,887,600 | ) | | | (700,080 | ) | | | (12,528,142 | ) |
| | | |
| (1) | | The basic and diluted net loss per share computation excludes potential common shares upon exercise of options to purchase common stock as their effect would be anti-dilutive. See Item 8, Financial Statements and Supplementary Data, Note 2 to the Financial Statements, for a detailed explanation of the determination of shares used in computing basic and diluted loss per share. |
| |
| (2) | | Weighted average common shares outstanding and per share amounts have been retroactively adjusted to give effect to a one-for-3.03 reverse stock split of our common stock effected on February 8, 2008. |
82
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read together with our financial statements and related notes appearing elsewhere in this Form 10-K. This discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. You should review Item 1A, Risk Factors, of this report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements described in the following discussion and analysis.
Overview
We are a medical device company that markets our advanced robotic arm solution and orthopedic implants for minimally invasive orthopedic knee procedures. We offer MAKOplasty, an innovative, restorative surgical solution that enables orthopedic surgeons to consistently, reproducibly and precisely treat patient-specific, early to mid-stage osteoarthritic knee disease. In February 2008, our common stock began trading on The NASDAQ Global Market under the ticker symbol “MAKO” and we closed our IPO.
Through December 31, 2007, our revenue was primarily generated from the sale of our implants and disposable products utilized in MAKOplasty procedures. In accordance with our revenue recognition policy, as more fully described in the “Critical Accounting Policies and Significant Judgments and Estimates” section below, upon customer acceptance of the sale of our TGS we defer recognition of the related revenue and cost of revenue until delivery of version 2.0 of the TGS, which is anticipated in the first half of 2009, subject to regulatory clearances or approvals. We have incurred net losses in each year since our inception, and as of December 31, 2007, we had an accumulated deficit of $42.8 million. We expect to continue to incur significant operating losses as we increase our sales and marketing activities and otherwise continue to invest capital in the development and expansion of our products and our business generally. We also expect that our general and administrative expenses will increase due to additional operational and regulatory costs and burdens associated with operating as a public company.
Key milestones and goals in the development of our business include the following:
| | • | | In 2004 and 2005, we were a development stage company primarily engaged in research and development for key aspects of our core technology and establishment of our intellectual property portfolio. |
| |
| | • | | In May 2005, we obtained 510(k) marketing clearance from the FDA for a patient-specific visualization system with a robotic arm that was an earlier version of our TGS. In November 2005, we obtained 510(k) marketing clearance from the FDA for version 1.0 of our TGS. |
83
| | • | | In 2006, we completed development of the first version of our TGS and commenced the commercialization of our MAKOplasty solution. We entered into contracts with third-party manufacturers and suppliers for the manufacturing of key components of our TGS and implants, and began assembly of our products at our headquarters in Fort Lauderdale, Florida. Our first TGS was installed in June 2006. As of December 31, 2007, 181 MAKOplasty procedures had been performed since its commercial introduction in June 2006. |
| |
| | • | | In January 2008, we obtained 510(k) marketing clearance from the FDA for version 1.2 of our TGS. |
| |
| | • | | In February 2008, we completed our IPO of common stock, issuing a total of 5.1 million shares at an issue price of $10.00 per share, for proceeds, before expenses, of $51.0 million. |
| |
| | • | | We commenced development of a TGS software application to enable a single MAKO-branded unicompartmental implant system, combining our inlay and onlay system. We intend to commercially introduce this software enhanced version 1.3 and the MAKO-branded implant by the end of 2008. We have received a 510(k) clearance for the MAKO-branded implant, and we do not anticipate that TGS version 1.3 will require a 510(k) clearance. |
| |
| | • | | We are currently developing version 2.0 of our TGS and modular implants, which would allow multicompartmental knee resurfacing procedures. We intend to commercially introduce version 2.0 of the TGS and the modular implants in the first half of 2009, subject to regulatory clearances or approvals. If we were to be denied such clearances or approvals or if such clearances or approvals were delayed, it could have a material adverse impact on our results of operations. |
We believe that the key to growing our business is expanding the application of MAKOplasty to multicompartmental resurfacing procedures employing implants that address mid-stage, multicompartmental degeneration. To successfully commercialize our products and grow our business, we must gain market acceptance for MAKOplasty.
Factors Which May Influence Future Results of Operations
The following is a description of factors which may influence our future results of operations, including significant trends and challenges that we believe are important to an understanding of our business and results of operations.
Revenue
Revenue is generated from unit sales of our TGS, including installation services, training and upgrades and enhancements, from sales of implants and disposable products, and by providing extended warranty services. To date, we have generated revenue primarily from the sale of implants and disposable products utilized in MAKOplasty procedures, the majority of which is from several significant customers. The recognition of revenue associated with the sale of TGS units is dependent upon the delivery of version 2.0 of our TGS, as more fully described in the “Critical Accounting Policies and Significant Judgments and Estimates” section below.
84
Future revenue from sales of our products are difficult to predict and will only modestly reduce our continued and increasing losses resulting from selling, general and administrative expenses, research and development, and other activities for the next several years.
The generation of recurring revenue through sales of our implants, disposable products and service contracts are an important part of the MAKOplasty business model. We anticipate that recurring revenue will constitute an increasing percentage of our total revenue as we leverage each new installation of our TGS to generate recurring sales of implants and disposable products and as we expand our implant product offering.
Cost of Revenue
Cost of revenue primarily consists of the direct costs associated with the manufacture of TGS units, implants and disposable products for which revenue has been recognized in accordance with our revenue recognition policy discussed below. Costs associated with providing services are expensed as incurred. Cost of revenue also includes the cost associated with establishing at the time of installation an accrual for the TGS standard one-year warranty liability and royalties related to the sale of products covered by licensing arrangements.
The cost of revenue associated with the sale of TGS units is deferred until the recognition of the related revenue. In addition, we expect that deferred costs of revenue associated with the sale of TGS will be higher during the deferral period due to the additional costs associated with providing hardware enhancements and upgrades through and including the delivery of version 2.0 of our TGS as described in the “Critical Accounting Policies and Significant Judgments and Estimates” section below.
Selling, General and Administrative Expenses
Our selling, general and administrative expenses consist primarily of compensation, including stock-based compensation, for sales, operations, regulatory, quality, executive, finance, legal and administrative personnel. Other significant expenses include costs associated with sales and marketing activities, marketing and advertising materials, professional fees for legal and accounting services, consulting fees, travel expenses, facility and related operating costs, and recruiting expenses. Our selling, general and administrative expenses are expected to increase due to the cost associated with the expected commercial launch of version 2.0 of our TGS, our modular implant system and disposable products, increased number of employees necessary to support our continued growth in operations, and the additional operational and regulatory burdens and costs associated with operating as a publicly traded company. In addition, we expect to incur additional costs associated with protecting our intellectual property rights as necessary to support our future product offerings.
85
Research and Development Expenses
Costs related to research, design and development of products are charged to research and development expense as incurred. These costs include direct salary costs for research and development personnel including stock-based compensation, cost for materials used in research and development activities and costs for outside services. Research and development expenses are expected to increase as we develop version 2.0 of our TGS and our modular implant system.
Subject to regulatory clearances or approvals, version 2.0 of our TGS will enable the use of our modular implant system and will include:
| | • | | improved dexterity and range of motion in the robotic arm to allow additional degrees of freedom in the movement of the robotic arm; |
| |
| | • | | more efficient physical configuration of the patient-specific visualization system, robotic arm, customized bone cutting instruments and electronic components; |
| |
| | • | | improvement of the tracking system for monitoring movements by the patient and robotic arm; |
| |
| | • | | intelligent implant planning features that will aid the surgeon in achieving optimal patient-specific alignments; |
| |
| | • | | redesign of certain components to make them more accessible for service repairs and easier to replace; and |
| |
| | • | | sophisticated industrial design and state-of-the-art user interface. |
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities as of the date of the financial statements as well as the reported expenses during the reporting periods. The accounting estimates that require our most significant, difficult and subjective judgments include revenue recognition, allowance for doubtful accounts, inventory impairment charges, accrual for warranty costs, valuation allowance for deferred tax assets and liabilities, impairment of long-lived assets and the determination of stock-based compensation. We evaluate our estimates and judgments on an ongoing basis. Actual results may differ materially from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Item 8, Financial Statements and Supplementary Data, Note 2 to the Financial Statements, we believe that the following accounting policies and estimates are most critical to a full understanding and evaluation of our reported financial results.
86
Revenue Recognition
We generate revenue from unit sales of our TGS, including installation services, training and upgrades and enhancements, from sales of implants and disposable products, and by providing extended warranty services. Because our TGS includes software that is essential to the functionality of the system, we account for the sale of the TGS pursuant to the American Institute of Certified Public Accountants’ Statement of Position No. 97-2,Software Revenue Recognition(“SOP 97-2”), as amended.
We recognize product revenue for unit sales of the TGS when there is persuasive evidence of a sales arrangement, the fee is fixed or determinable, collection of the fee is probable and delivery has occurred as prescribed by SOP 97-2. For all sales, we use either a signed agreement or a binding purchase order as evidence of an arrangement. Such arrangements typically require customer acceptance of the system, which is evidenced by the receipt of a form executed by the customer indicating acceptance of the TGS unit. The customer acceptance period is typically defined as a certain number of surgical procedures over a certain period which typically does not exceed three months. Sales arrangements contain several elements, including elements requiring us to provide upgrades and enhancements to the TGS unit, including related software on a when and if available basis. Payments received upon customer acceptance of TGS units are recorded as deferred revenue due to the significance of the undelivered elements. The direct cost of revenue associated with the sale of TGS units is recorded as deferred cost of revenue. The deferred revenue and deferred cost of revenue associated with the sale of TGS units will be recognized in our statement of operations if and when we have satisfied all related revenue recognition criteria, which include the delivery of version 2.0 of the TGS, which is anticipated to be in the first half of 2009, subject to regulatory clearances or approvals.
For sales arrangements with multiple elements, we allocate arrangement consideration to TGS units, upgrades, enhancements and services based upon vendor specific objective evidence, or VSOE, of fair value of the respective elements. As we are in the early stages of commercialization, VSOE of fair value does not exist for all of the undelivered elements. Accordingly, all revenue and cost of revenue associated with the sale of the TGS are deferred until the earlier of (1) delivery of all elements or (2) establishment of VSOE of fair value for all undelivered elements.
Product revenue from the sale of implants and disposable products is recognized as revenue in accordance with Staff Accounting Bulletin No. 104,Revenue Recognition, when persuasive evidence of an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable and collectibility is reasonably assured. The implants and disposable products are a separate unit of accounting from the TGS as (1) they have value to the customer on a standalone basis, (2) objective and reliable evidence of the fair value of the item exists and (3) no right of return exists once the implants and disposable products are implanted or consumed. Accordingly, as our implants and disposable products are sold for each procedure, the revenue and costs associated with the sale of our implants and disposable products are recognized at the time of sale.
87
Service revenue, which consists primarily of extended warranty services for the TGS hardware, is deferred and recognized ratably over the service period, until no further obligation exists. Costs associated with providing services are expensed when incurred.
Our agreements with customers do not contain product return rights beyond the customer acceptance period which is typically defined as a certain number of surgical procedures over a certain period of time and which typically does not exceed three months.
For purposes of obtaining clinical and technical feedback on the current version of our TGS, we also enter into consignment programs with certain customers. We anticipate that our participation in these programs will remain limited and is not part of our long-term business strategy. Under the terms of such programs, we retain title to the TGS unit, while the customer has use of the TGS and purchases our implants and disposable products. We may provide unspecified upgrades to the TGS product during the term of each program when and if available. The TGS units associated with our consignment programs are recorded as property and equipment and are depreciated over their estimated useful life of two years. Depreciation and warranty expense attributable to TGS consignment units are recorded as cost of revenue. The revenue associated with the sale of implants and disposable products to customers under consignment programs is recognized as revenue when persuasive evidence of an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable and collectibility is reasonably assured.
Allowance for Doubtful Accounts
The allowance for doubtful accounts is based on our assessment of the collectibility of customer accounts. We regularly review the allowance by considering factors such as historical experience, credit quality, the age of the accounts receivable balances, and current economic conditions that may affect a customer’s ability to pay. We have not experienced any collectibility issues to date and have no allowance for doubtful accounts, provision for doubtful accounts receivable or write offs to date in the accompanying financial statements included in Item 8, Financial Statements and Supplementary Data, of this report.
Inventory Impairment Charges
Inventory is stated at the lower of cost or market value on a first-in, first-out basis. Inventory costs include direct materials and direct labor. We review our inventory periodically to determine net realizable value. We write down inventory, if required, based on forecasted demand and technological obsolescence. These factors are impacted by market and economic conditions, technology changes and new product introductions and require estimates that may include uncertain elements.
Accrual for Warranty Costs
Upon installation of a TGS unit, we establish an accrual for the estimated costs associated with providing a standard one-year warranty for defects in materials and workmanship. Due to our limited history of commercial placements of TGS units, the estimation of warranty costs is subjective; however, costs incurred to date have not been significantly different from the estimate.
88
Valuation Allowance for Deferred Income Tax Assets and Liabilities
Deferred income tax assets and liabilities are determined based on the differences between financial reporting and income tax bases of assets and liabilities, using income tax rates expected to be in effect when the differences will reverse. Valuation allowances are established when necessary to reduce the net deferred tax assets to the amounts expected to be realized. A full valuation allowance has been recorded in the accompanying financial statements relating to all our net deferred income tax assets.
Impairment of Long-Lived Assets
We evaluate our long-lived assets for indicators of impairment by comparison of the carrying amounts to future net undiscounted cash flows expected to be generated by such assets when events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. Should an impairment exist, the impairment loss would be measured based on the excess carrying value of the asset over the asset’s fair value or discounted estimate of future cash flows. We have not recorded any such impairment losses to date.
Determination of Stock-Based Compensation
Effective January 1, 2006, we adopted the fair value provisions of Statement of Financial Accounting Standards No. 123 Revised,Share-Based Payment(“SFAS 123(R)”). SFAS 123(R) requires the recognition of compensation expense, using a fair-value based method, for costs related to all share-based payments including stock options. SFAS 123(R) requires companies to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model.
We adopted SFAS 123(R) using the modified retrospective transition method, which requires the restatement of financial statements for prior periods. Prior to the adoption of SFAS 123(R), we accounted for stock-based compensation arrangements by recording compensation expense based on the estimated fair-value of stock-based awards in accordance with Statement of Financial Accounting Standards No. 123,Accounting for Stock Based Compensation. The impact of SFAS 123(R) on prior periods was not significant.
We account for stock-based compensation arrangements with non-employees in accordance with the Emerging Issues Task Force (“EITF”) Abstract No. 96-18,Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling Goods or Services.We record the expense of such services based on the estimated fair value of the equity instrument using the Black-Scholes-Merton pricing model. The value of the equity instrument is charged to expense over the term of the service agreement.
89
We selected the Black-Scholes-Merton pricing model to determine the fair value of stock options. The determination of the fair value of stock-based payment awards on the date of grant using an option-pricing model will be affected by our stock price as well as assumptions regarding a number of complex and subjective variables. These variables include our expected stock price volatility over the term of the awards, actual and projected employee stock option exercise behaviors, risk-free interest rates, forfeitures and expected dividends.
Included in our 2007 option grants are approximately 462,000 options which are subject to performance conditions based on the achievement of certain future performance metrics. Upon satisfaction of the performance condition, the options will vest ratably quarterly over a period of 4 years. Through December 31, 2007, we began recognizing compensation expense on approximately 296,000 performance options as both the terms of the performance conditions had been established and it was probable that the performance condition will be satisfied. Once the performance conditions are established and it is probable the performance conditions will be met, we will begin recognizing compensation expense on the remaining performance options.
During the year ended December 31, 2007, we recognized $453,000 of compensation expense relating to stock option grants, leaving $3.9 million to be recognized in future periods. Total unrecognized compensation cost will be adjusted for future changes in estimated forfeitures, and is expected to be recognized over a remaining weighted average period of 2.45 years as of December 31, 2007.
In July 2005 and May 2006, we issued a total of 446,287 shares of restricted common stock to our CEO and 49,504 shares of unrestricted common stock to an entity affiliated with the CEO in exchange for promissory notes from the CEO totaling approximately $631,000 representing the fair value of shares on the date of issuance, approximately 50% of which were nonrecourse. The promissory notes accrued interest at a rate of 8% per annum, with 25% of the restricted stock vesting immediately and the remainder vesting monthly over 48 months as service is provided. The restricted stock was pledged as collateral against the promissory notes. In March 2007, we issued 82,508 shares of restricted common stock to our CEO at a purchase price of $2.48 per share, the estimated fair value at the date of issuance, in exchange for a promissory note of $205,000, 50% of which was nonrecourse, and a pledge agreement. The March 2007 restricted stock, pledge agreement and promissory note were issued under terms substantially similar to the July 2005 and May 2006 restricted stock issuances. Because it was unclear as to whether the recourse portion had substance as of the dates of issuance of the restricted stock and the promissory notes, we determined to treat the entire amount of the promissory notes related to the restricted stock as nonrecourse for accounting purposes. A nonrecourse note issued for restricted stock is in substance an option to acquire the stock. Accordingly, we recorded compensation expense of approximately $73,000 and $90,000 for the years ended December 31, 2006 and 2005 under stock option accounting guidance, and the promissory notes and the restricted stock were not recorded in the financial statements for those periods.
90
In September 2007, we forgave outstanding loans to our CEO of approximately $1.1 million, including accrued interest of $113,000, which represents all loans outstanding to our CEO. Of this amount, $949,000 was associated with the issuances of the restricted stock noted above and $200,000 was associated with employee loans. In connection with the forgiveness of the loans, 35,244 shares of common stock were surrendered by our CEO to us to pay for the payroll taxes associated with the taxable income from the forgiveness of the loans. The forgiveness resulted in a modification to the original terms of the restricted stock-based award with a charge of approximately $395,000 recorded in the financial statements in the third quarter of 2007. The remaining unrecognized compensation expense of approximately $533,000 relating to the unvested restricted stock will be recorded in the financial statements over the remaining vesting period, along with the related vested common stock.
Compensation expense related to the CEO restricted stock was approximately $774,000 for the year ended December 31, 2007, of which $77,000 was incurred in the eight months prior to the modification, $395,000 was incurred due to the modification, and $302,000 was incurred subsequent to the modification.
All common share and per share amounts have been retroactively adjusted to give effect to a one-for-3.03 reverse stock split of our common stock effected on February 8, 2008.
Acquisitions of Assets from Predecessor
Z-KAT, Inc. was formed in 1997 to develop and commercialize computer assisted surgery, or CAS, applications. Z-KAT formed MAKO Surgical Corp. in November 2004, to develop and commercialize unique applications combining CAS with haptic robotics in the medical field of orthopedics. Z-KAT is considered to be our Predecessor. In December 2004, pursuant to a contribution agreement, we acquired substantially all of Z-KAT’s tangible assets and a majority of Z-KAT’s CAS technology assets not required for Z-KAT’s retained CAS business, and all of its haptic robotic research and development technology inventory. We were granted a limited license to Z-KAT’s CAS and haptic robotic intellectual property portfolio for exclusive use in the field of orthopedics, subject to a prior license to a strategic partner of Z-KAT to use Z-KAT’s CAS intellectual property, but not its haptic robotic intellectual property, in the field of orthopedics. The contribution agreement, including the Z-KAT license, was made in exchange for approximately 1,410,000 shares of common stock, 1,999,000 shares of Series A redeemable convertible preferred stock, and warrants to purchase 190,000 shares of common stock at an exercise price of $3.00 per share.
Pursuant to the December 2004 contribution of the Z-KAT license, we obtained the right to manage and maintain the Z-KAT patent portfolio and assumed the obligation to pay a ratable portion (among all licensees) of all maintenance fees, patent costs and applicable net annual minimum royalties to Z-KAT’s licensors. For the majority of applicable licenses, our ratable portion for the intellectual property fees, costs and net annual minimum royalties has been 50% since consummation of the Z-KAT license.
91
In December 2006, we entered into an addendum to the contribution agreement. Under the addendum, Z-KAT assigned to us its right to receive required royalty payments from two prior third-party CAS intellectual property licensees; and we assumed the obligation to pay the annual minimum royalty to a third-party CAS licensor due to the importance of maintaining the licensed rights. There was no change in licensed intellectual property rights as a result of the addendum.
Results of Operations
Year Ended December 31, 2007 Compared to the Year Ended December 31, 2006
Revenue.Revenue was $771,000 for the year ended December 31, 2007, compared to $63,000 for the year ended December 31, 2006 and was primarily generated from the sale of implants and disposable products utilized in MAKOplasty procedures. The increase in revenue of $708,000 was primarily due to an increase in MAKOplasty procedures performed during the year ended December 31, 2007. The first MAKOplasty procedure was performed in June 2006, and 168 procedures were performed during the year ended December 31, 2007 compared to 13 procedures performed during the year ended December 31, 2006. The increase was also attributable to a $54,000 increase in other revenue, which consists primarily of service revenue on extended warranty services. We expect our revenue to increase as the number of MAKOplasty procedures performed increases in future periods. The deferred revenue balance was $3.4 million and $700,000 as of December 31, 2007 and 2006, respectively. The increase in the deferred revenue balance is primarily related to four unit sales of our TGS. Deferred revenue related to unit sales of our TGS will be recognized in our statement of operations if and when we have satisfied all related revenue recognition criteria, which includes the delivery of version 2.0 of the TGS, which is anticipated to be in the first half of 2009, subject to regulatory clearances or approvals.
Cost of Revenue.Cost of revenue was $583,000 for the year ended December 31, 2007, compared to $77,000 for the year ended December 31, 2006. The increase in cost of revenue of $506,000 was primarily due to an increase in MAKOplasty procedures performed, the establishment of warranty accruals on sales of TGS units and royalties incurred on sales of TGS units during the year ended December 31, 2007. The increase was also attributable to a $25,000 increase in other cost of revenue, which consists primarily of cost of service revenue on extended warranty services. We expect our cost of revenue to increase as the number of MAKOplasty procedures performed increases in future periods. In addition, anticipated increases in sales of TGS units will result in an increase in cost of sales as a result of the corresponding increase in royalty and warranty expense. The deferred cost of revenue balance was $926,000 and $210,000 as of December 31, 2007 and 2006, respectively. The increase in the deferred cost of revenue balance is primarily related to four unit sales of our TGS. Deferred cost of revenue related to unit sales of our TGS will be recognized in our statement of operations if and when we have satisfied all related revenue recognition criteria, which includes the delivery of version 2.0 of the TGS, which is anticipated to be in the first half of 2009, subject to regulatory clearances or approvals.
92
Selling, General and Administrative.Selling, general and administrative expense was $12.0 million for the year ended December 31, 2007, compared to $5.0 million for the year ended December 31, 2006. The increase of $7 million, or 140%, was primarily due to a $4.2 million increase in compensation expense associated with increased selling, marketing and administrative personnel, which includes a $929,000 increase in stock-based compensation due primarily to the modification of the CEO’s restricted stock discussed in “Determination of Stock-Based Compensation” above and additional option and restricted stock grants made in 2007, a $1.6 million increase in general overhead costs due to an increase in marketing activities and increases in facility and travel related costs, a $427,000 increase in recruiting and relocation costs, and a $770,000 increase in professional fees. We expect our selling, general and administrative expense to increase substantially due to our planned increase in the number of employees necessary to support the commercial launch of version 1.2 of our TGS in the first quarter of 2008, sales and marketing costs associated with the anticipated commercial launch of version 2.0 of our TGS in the first half of 2009, continued growth in operations and the costs associated with operating as a public company.
Research and Development.Research and development expense was $8.3 million for the year ended December 31, 2007, compared to $5.2 million for the year ended December 31, 2006. The increase of $3.1 million, or 60%, was primarily due to a $2.1 million increase in compensation expense associated with the increased number of research and development employees and a $1.0 million increase in material, supply and other expenses used in research and development activities. The increases in compensation expense and material and supplies were related to the development of versions 1.2 and 2.0 of our TGS and our unicondylar and modular implant systems. We expect our research and development expense to increase as we continue to expand our research and development activities, including the development of version 2.0 of our TGS and our modular implant system.
Depreciation and Amortization.Depreciation and amortization expense was $1.3 million for the year ended December 31, 2007, compared to $644,000 for the year ended December 31, 2006. The increase of $653,000, or 101%, was primarily due to a $478,000 increase in depreciation of property and equipment due to purchases made in 2007, and a $175,000 increase in amortization associated with the license of $5.4 million of intangible assets from a license agreement entered into in March 2006 with IBM. The license agreement with IBM provides a license in our field of business to IBM’s patent portfolio and is stated net of a discount estimated at $590,000 less accumulated amortization of the discount to date associated with a deferred payment of $4.0 million paid upon completion of our IPO in February 2008.
Interest and Other Income.Interest income was $1.1 million for the year ended December 31, 2007, compared to $477,000 for the year ended December 31, 2006. The increase of $597,000, or 125%, was primarily due to an increase in short-term investments from the net proceeds of the issuance of our Series C redeemable convertible preferred stock in February 2007. We expect our interest income to increase as a result of the investment of the net proceeds of our IPO.
Interest and Other Expense.Interest and other expense was $312,000 for the year ended December 31, 2007, compared to $220,000 for the year ended December 31, 2006. The increase of $92,000, or 42%, was primarily due to the amortization of the $590,000 discount on the intangible assets licensed under the IBM license agreement entered into in March 2006 as discussed above.
93
Income Taxes.No income taxes were recognized for the year ended December 31, 2007 and 2006, due to net operating losses in each period. In addition, no current or deferred income taxes were recorded for the year ended December 31, 2007 and 2006, as all income tax benefits were fully offset by a valuation allowance against our net deferred income tax assets.
Year Ended December 31, 2006 Compared to the Year Ended December 31, 2005
Revenue.Revenue was $63,000 for the year ended December 31, 2006, compared to $0 for the year ended December 31, 2005, and was primarily generated from the sale of implants and disposal products utilized in MAKOplasty procedures. Revenue was recognized in 2006 as performance of MAKOplasty procedures commenced in 2006. In 2006, 13 MAKOplasty procedures were performed.
Cost of Revenue.Cost of revenue was $77,000 for the year ended December 31, 2006, compared to $0 for the year ended December 31, 2005. Cost of revenue was recognized in 2006 as performance of MAKOplasty procedures commenced in 2006. Cost of revenue for the year ended December 31, 2006 also included the expense associated with the estimated warranty costs and royalties associated with the sales of the TGS.
Selling, General and Administrative.Selling, general and administrative expense was $5.0 million for the year ended December 31, 2006, compared to $2.7 million for the year ended December 31, 2005. The increase of $2.3 million, or 85%, was primarily due to a $1.4 million increase in compensation expense associated with increased headcount for selling, marketing, operations, and administrative personnel, a $736,000 increase in general overhead costs due to higher marketing, facility, and travel related costs, a $145,000 increase in employee recruiting costs for sales and marketing and administrative personnel, and a $63,000 increase in legal expenses related to patent filings and general corporate matters.
Research and Development.Research and development expense was $5.2 million for the year ended December 31, 2006, compared to $2.6 million for the year ended December 31, 2005. The increase of $2.6 million, or 100%, was primarily due to a $1.3 million increase in compensation expense associated with the increased number of research and development employees, a $540,000 increase in material and supplies and a $728,000 increase in outside services. The increases in compensation expense, material and supplies and outside services were related to the development and support of the current version of our TGS and development efforts for version 1.2 of our TGS.
Depreciation and Amortization.Depreciation and amortization expense was $644,000 for the year ended December 31, 2006, compared to $99,000 for the year ended December 31, 2005. The increase of $545,000, or 551%, was primarily due to a $451,000 increase in amortization associated with the license of $5.4 million of intangible assets from a license agreement entered into in March 2006 with IBM, and a $94,000 increase in depreciation of property and equipment due to purchases in 2006. The license agreement with IBM provides a license in our field of business to IBM’s patent portfolio and is stated net of a discount estimated at $590,000 less accumulated amortization of the discount to date associated with a deferred payment of $4.0 million paid upon completion of our IPO in February 2008.
94
Interest and Other Income.Interest income was $477,000 for the year ended December 31, 2006, compared to $269,000 for the year ended December 31, 2005. The increase of $208,000, or 77%, was primarily due to higher average short-term investment balances during 2006 due to the net proceeds of our $20.0 million Series B redeemable convertible preferred stock offering in July 2005.
Interest and Other Expense.Interest and other expense was $220,000 for the year ended December 31, 2006, compared to $0 for the year ended December 31, 2005. The increase was primarily due to the amortization of the $590,000 discount on the intangible assets licensed in the IBM license agreement entered into in March 2006.
Income Taxes.No income taxes were recognized for the years ended December 31, 2006 and 2005, due to net operating losses in each period. In addition, no current or deferred income taxes were recorded for the years ended December 31, 2006 and 2005, as all income tax benefits were fully offset by a valuation allowance against our net deferred income tax assets.
Liquidity and Capital Resources
| | | | | | | | | | | | | | | | | | | | | |
| | | 2007 | | | Change | | | 2006 | | | Change | | | 2005 | |
| Cash and cash equivalents | | $ | 9,615,027 | | | $ | 7,507,012 | | | | 2,108,015 | | | $ | (4,037,251 | ) | | $ | 6,145,266 | |
| Short-term investments | | | 3,083,980 | | | | 1,684,217 | | | | 1,399,763 | | | | (8,697,257 | ) | | | 10,097,020 | |
| | | | | | | | | | | | | | | | |
| Total cash, cash equivalents, and short-term investments | | $ | 12,699,007 | | | $ | 9,191,229 | | | $ | 3,507,778 | | | $ | (12,734,508 | ) | | $ | 16,242,286 | |
| | | | | | | | | | | | | | | | |
| Cash used in operating activities | | $ | (15,380,647 | ) | | $ | (5,964,719 | ) | | $ | (9,415,928 | ) | | $ | (4,405,886 | ) | | $ | (5,010,042 | ) |
| Cash provided by (used in) investing activities | | | (3,910,548 | ) | | | (9,296,663 | ) | | | 5,386,115 | | | | 16,217,446 | | | | (10,831,331 | ) |
| Cash provided by (used in) financing activities | | | 26,798,207 | | | | 26,805,645 | | | | (7,438 | ) | | | (20,034,998 | ) | | | 20,027,560 | |
| | | | | | | | | | | | | | | | |
| Net increase (decrease) in cash and cash equivalents | | $ | 7,507,012 | | | $ | 11,544,263 | | | $ | (4,037,251 | ) | | $ | (8,223,438 | ) | | $ | 4,186,187 | |
| | | | | | | | | | | | | | | | |
We have incurred net losses and negative cash flow from operating activities for each period since our inception in November 2004. As of December 31, 2007, we had an accumulated deficit of $42.8 million and financed our operations principally through the sale of Series A, Series B and Series C redeemable convertible preferred stock. Through December 31, 2007, we received net proceeds of $52.2 million from the issuance of Series A, Series B and Series C redeemable convertible preferred stock.
As of December 31, 2007, we had $12.7 million in cash, cash equivalents and short-term investments. Our cash and investment balances are held in a variety of interest bearing instruments, including U.S. treasury notes, certificates of deposit and auction rate securities.
In February 2008, we completed our IPO of common stock, issuing a total of 5.1 million shares at an issue price of $10.00 per share, for proceeds, before expenses, of $51.0 million. In conjunction with the completion of the IPO, in February 2008, all of our outstanding Series A redeemable convertible preferred stock, Series B redeemable convertible preferred stock and Series C redeemable convertible preferred stock was converted into 10,945,080 shares of common stock, as adjusted for a one-for-3.03 reverse stock split.
95
As of December 31, 2007, we held $1.6 million of AAA rated auction rate securities issued by two separate funds. These securities are classified as short-term investments in our balance sheet. In February 2008, the auction rate securities experienced failed auctions that limited the liquidity of these securities. Based on current market conditions, it is likely that auction failures will continue. As a result, we are unable to determine when a successful auction will occur and the auction rate securities will be liquidated. We believe the carrying value is fully recoverable as auction rate securities are fully collateralized by assets held by the fund; however, we will continue to monitor the investments for any potential impairment.
Net Cash Used in Operating Activities
Net cash used in operating activities primarily reflects the net loss for those periods, which was reduced in part by depreciation and amortization, stock-based compensation, accrued interest and changes in operating assets and liabilities. Included in the changes in operating assets for the years ended December 31, 2007 and 2006 are $2.7 million and $700,000, respectively, of increases to the deferred revenue balance partially offset by increases in the deferred cost of revenue balance. The increases to the deferred revenue balance are primarily related to unit sales of our TGS. Deferred revenue related to unit sales of our TGS will be recognized in the statement of operations upon satisfaction of all related revenue recognition criteria. Additionally, net cash used in operating activities in 2007 was reduced by the increase in inventory and accounts receivable balances necessitated by increased sales of our TGS units and sales of implants and disposable products, and was partially offset by the increase in our accounts payable and accrued liability accounts as our increased operations require higher levels of purchasing and also to IPO costs incurred during 2007.
Net Cash Provided by (Used in) Investing Activities
Net cash provided by (used in) investing activities primarily relates to the proceeds and purchases of short-term investments as we manage our investment portfolio to provide interest income and liquidity. Investing activities were reduced by the purchase of property and equipment as we invest in the infrastructure of our growing company and acquisition of intangible assets, including the license of intellectual property rights from IBM in 2006.
Net Cash Provided by Financing Activities
Net cash provided by our financing activities was primarily attributable to the issuance of Series B redeemable convertible preferred stock during the year ended December 31, 2005, and the issuance of Series C redeemable convertible preferred stock during the year ended December 31, 2007. The net cash provided by financing activities for the year ended December 31, 2007 was partially offset by deferred IPO costs incurred and capitalized during the period.
96
Operating Capital and Capital Expenditure Requirements
To date, we have not achieved profitability. We anticipate that we will continue to incur substantial net losses for at least the next several years as we develop version 2.0 of our TGS and our modular implant system, expand our sales and marketing capabilities in the orthopedics product market and continue to develop the corporate infrastructure required to sell and market our products and operate as a public reporting company. We also expect to experience increased cash requirements for inventory and property and equipment in conjunction with the expected commercial launch of version 2.0 of our TGS and modular implant system in the first half of 2009.
We believe our existing cash, cash equivalents and investment balances, including the net proceeds received upon completion of our IPO, and interest income we earn on these balances will be sufficient to meet our anticipated cash requirements through at least the next 12 months from December 31, 2007. If our available cash, cash equivalents and short-term investment balances and net proceeds from the IPO are insufficient to satisfy our operating requirements, we may seek to sell additional equity or debt securities or enter into a credit facility. The sale of additional equity and debt securities may result in dilution to our stockholders. If we raise additional funds through the issuance of debt securities, these securities will have rights senior to those of our common stock and could contain covenants that would restrict our operations. We may require additional capital beyond our currently forecasted amounts. Any such required additional capital may not be available on reasonable terms, or at all. If we are unable to obtain additional financing, we may be required to reduce the scope of, delay or eliminate some or all of our planned research, development and commercialization activities, which could materially harm our business and results of operations.
As noted in the “Revenue Recognition” section above, sales arrangements for our TGS contain several elements, including elements requiring us to provide upgrades and enhancements to the TGS unit, including hardware and related software on a when and if available basis. As of December 31, 2007, four TGS customers are entitled to receive an upgrade to version 2.0 of the TGS at no additional charge and one customer has the right to receive the upgrade at a discounted price. All of these customer rights to receive the upgrades through version 2.0 of the TGS are on a when and if available basis. We are not obligated to provide upgrades for the two TGS units under consignment programs for clinical and technical feedback. For sales of TGS units to date, the costs to provide upgrades up to and including the delivery of version 2.0 of the TGS are estimated to not exceed $250,000 in total per customer TGS unit. Payments received upon customer acceptance of TGS units are recognized as deferred revenue until all related revenue recognition criteria is satisfied. We anticipate ultimately recognizing a positive margin on the sales of TGS units to date, including the satisfaction of the remaining upgrades through the final deliverable of version 2.0 of the TGS, which is anticipated to be in the first half of 2009, subject to regulatory clearances or approvals. If we are not able to deliver version 2.0 of the TGS, customers would retain the original TGS unit sold and we would not be obligated to refund the purchase price of the TGS unit.
97
We are in the process of developing version 2.0 of our TGS. If completion of version 2.0 of our TGS unit is unsuccessful or delayed, or regulatory clearances or approvals are denied or delayed, it could have a material adverse impact on our results of operations and financial position and we may be unable to recognize any revenue associated with sales of our TGS. No right of return exists on sales of prior versions of our TGS if we are unable to complete and deliver version 2.0 of our TGS.
We have various license and royalty agreements which are more fully discussed in Item 8, Financial Statements and Supplementary Data, Note 6 to the Financial Statements. Royalty payments related to each of these existing agreements are anticipated to range between 1% and 7% of future sales of our TGS units, TGS components thereof and/or products and are recognized as cost of revenue as incurred. Some of these royalty payments are subject to certain minimum annual royalty payments as shown in the “Contractual Obligations” section below. The license and royalty agreements are not expected to impact our future product development efforts.
Because of the numerous risks and uncertainties associated with the development of medical devices, such as version 2.0 of our TGS and modular implant system, we are unable to estimate the exact amounts of capital outlays and operating expenditures necessary to complete the development of the products and successfully deliver a commercial product to the market. Our future capital requirements will depend on many factors, including but not limited to the following:
| | • | | the expenses we incur in selling and marketing our products; |
| |
| | • | | the costs and timing of regulatory clearance; |
| |
| | • | | the revenue generated by sales of our future products; |
| |
| | • | | the rate of progress and cost of development activities; |
| |
| | • | | the success of our research and development efforts; |
| |
| | • | | the emergence of competing or complementary technological developments; |
| |
| | • | | the costs of filing, prosecuting, defending and enforcing any patent or license claims and other intellectual property rights, or participating in litigation-related activities; and |
| |
| | • | | the acquisition of businesses, products and technologies, although we currently have no understandings, commitments or agreements relating to any of these types of transactions. |
98
Contractual Obligations
The following table summarizes our outstanding contractual obligations as of December 31, 2007 and the effect those obligations are expected to have on our liquidity and cash flows in future periods:
| | | | | | | | | | | | | | | | | | | | | |
| | | Payment Due by Period | |
| | | | | | | December 31, | | | After | |
| | | Total | | | 2008 | | | 2009-2010 | | | 2011-2012 | | | 2012 | |
Contractual Obligations | | | | | | | | | | | | | | | | | | | | |
| Operating lease – real estate | | $ | 775,000 | | | $ | 208,000 | | | $ | 436,000 | | | $ | 131,000 | | | $ | — | |
| Capital leases – furniture | | | 24,000 | | | | 13,000 | | | | 11,000 | | | | — | | | | — | |
| IBM deferred license fee(1) | | | 4,000,000 | | | | 4,000,000 | | | | — | | | | — | | | | — | |
| Minimum royalty payments – licenses | | | 4,058,000 | | | | 631,000 | | | | 1,350,000 | | | | 1,260,000 | | | | 817,000 | |
| | | | | | | | | | | | | | | | |
| Total | | $ | 8,857,000 | | | $ | 4,852,000 | | | $ | 1,797,000 | | | $ | 1,391,000 | | | $ | 817,000 | |
| | | | | | | | | | | | | | | | |
Our commitments for operating leases relate to the lease for our headquarters in Fort Lauderdale, Florida. Our commitments for minimum royalty payments relate to payments under various licenses and sublicenses as discussed in Item 8, Financial Statements and Supplementary Data, Note 6 to the Financial Statements.
| | | |
| (1) | | In March 2006, we entered into a license agreement with IBM in exchange for a payment of $2.0 million upon execution of the agreement and pursuant to which we were required to make a $4.0 million payment in the form of a deferred license fee to IBM upon the closing of the initial public offering of our common stock or other change in control events (e.g., acquisition or change in voting ownership greater than 50.01%). In February 2008, upon completion of our initial public offering, the Company paid the $4.0 million deferred license fee due to IBM. |
Recent Accounting Pronouncements
In July 2006, the Financial Accounting Standards Board, or FASB, issued Interpretation No. 48,Accounting for Uncertainty in Income Taxes, an interpretation of FASB Statement No. 109, or FIN 48. FIN 48 clarifies the accounting for uncertainty in income taxes by prescribing the recognition threshold a tax position is required to meet before being recognized in the financial statements. It also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. FIN 48 is effective for fiscal years beginning after December 15, 2006. We adopted the provisions of FIN 48 effective January 1, 2007. No cumulative adjustment to our accumulated deficit was required upon adoption, and there was no effect of adoption.
In September 2006, the SEC issued Staff Accounting Bulletin No. 108,Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements, or SAB 108. SAB 108 provides guidance on the consideration of the effects of prior year misstatements in quantifying current year misstatements for the purpose of a materiality assessment. SAB 108 establishes an approach that requires quantification of financial statement errors based on the effects on each of our balance sheets and statement of operations and the related financial statement disclosures. We adopted SAB 108 effective January 1, 2007. We have determined that the adoption of SAB 108 had no effect on our results of operations and financial position.
99
In September 2006, the FASB issued Statement of Financial Accounting Standards No. 157,Fair Value Measurements, or SFAS 157. SFAS 157 provides guidance for using fair value to measure assets and liabilities. It also responds to investors’ requests for expanded information about the extent to which companies measure assets and liabilities at fair value, the information used to measure fair value and the effect of fair value measurements on earnings. SFAS 157 applies whenever other standards require (or permit) assets or liabilities to be measured at fair value, and does not expand the use of fair value in any new circumstances. SFAS 157 is effective for financial statements issued for fiscal years beginning after November 15, 2007 and is required to be adopted effective January 1, 2008. We are currently evaluating the effect that the adoption of SFAS 157 will have on our results of operations and financial position. We are currently evaluating the impact of SFAS 157, but do not expect the adoption of SFAS 157 to have a material impact on our financial statements.
In February 2007, the FASB issued Statement of Financial Accounting Standards No. 159,The Fair Value Option for Financial Assets and Financial Liabilities — Including an amendment of FASB Statement No. 115, or SFAS 159. SFAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The objective of SFAS 159 is to reduce both complexity in accounting for financial instruments and the volatility in earnings caused by measuring related assets and liabilities differently. Most of the provisions in Statement 159 are elective; however, the amendment to FASB Statement No. 115,Accounting for Certain Investments in Debt and Equity Securities, applies to all entities with available-for-sale and trading securities. SFAS 159 is effective as of the beginning of an entity’s first fiscal year beginning after November 15, 2007. We are currently evaluating the impact of SFAS 159, but do not expect the adoption of SFAS 159 to have a material impact on our financial statements.
In June 2007, the FASB issued EITF Issue No. 07-03,Accounting for Nonrefundable Advance Payments for Goods or Services Received for Use in Future Research and Development Activities, or EITF 07-03. This EITF requires that nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities should be deferred and capitalized. Such amounts should be recognized as an expense as the related goods are delivered or the related services are performed. Entities should continue to evaluate whether they expect the goods to be delivered or services to be rendered. If an entity does not expect the goods to be delivered or services to be rendered, the capitalized advance payment should be charged to expense. EITF 07-03 is effective for financial statements issued for fiscal years beginning after December 15, 2007, and interim periods within those fiscal years. Earlier adoption is permitted. We are currently evaluating the impact of EITF 07-03, but do not expect the adoption of EITF 07-03 to have a material impact on our financial statements.
100
In December 2007, the FASB issued SFAS No. 141 (revised 2007),Business Combinations, or SFAS 141(R). SFAS 141(R) retains the fundamental requirements of the original pronouncement requiring that the purchase method be used for all business combinations. SFAS 141(R) defines the acquirer as the entity that obtains control of one or more businesses in the business combination, establishes the acquisition date as the date that the acquirer achieves control and requires the acquirer to recognize the assets acquired, liabilities assumed and any noncontrolling interest at their fair values as of the acquisition date. SFAS 141(R) also requires that acquisition-related costs be recognized separately from the acquisition. SFAS 141(R) will become effective for us on January 1, 2009. We are currently evaluating the impact that SFAS 141(R) will have on our financial statements.
In December 2007, the FASB issued SFAS No. 160,Noncontrolling Interests in Consolidated Financial Statements, or SFAS 160. SFAS 160 establishes accounting and reporting standards for ownership interests in subsidiaries held by parties other than the parent, the amount of consolidated net income attributable to the parent and to the noncontrolling interest, changes in a parent’s ownership interest and the valuation of retained noncontrolling equity investments when a subsidiary is deconsolidated. SFAS 160 also establishes reporting requirements that provide sufficient disclosures that clearly identify and distinguish between the interests of the parent and the interests of the noncontrolling owners. SFAS 160 will become effective for us beginning in the first quarter of 2009. We are currently evaluating the impact that SFAS 160 will have on our financial statements.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenue or expenses, results of operations, liquidity, capital expenditures or capital resources.
101
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our exposure to market risk is confined to our cash, cash equivalents and short-term investments that have maturities or interest reset dates of less than one year. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and fiduciary control of cash and investments. We also seek to maximize income from our investments without assuming significant risk. To achieve our goals, we maintain a portfolio of cash equivalents and investments in a variety of securities. The securities in our investment portfolio are not leveraged, are classified as available for sale and are, due to their very short-term nature, subject to minimal interest rate risk. We currently do not hedge interest rate exposure. Because of the short-term maturities of our investments, we do not believe that an increase in market rates would have any material negative impact on the value of our investment portfolio.
As of December 31, 2007, we held $1.6 million of AAA rated auction rate securities issued by two separate funds. These securities are classified as short-term investments in our balance sheet. In February 2008, the auction rate securities experienced failed auctions that limited the liquidity of these securities. Based on current market conditions, it is likely that auction failures will continue. As a result, we are unable to determine when a successful auction will occur and the auction rate securities will be liquidated. We believe the carrying value is fully recoverable as auction rate securities are fully collateralized by assets held by the fund; however, we will continue to monitor the investments for any potential impairment.
102
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
MAKO SURGICAL CORP.
Index to the Financial Statements
103
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
MAKO Surgical Corp.
We have audited the accompanying balance sheets of MAKO Surgical Corp. as of December 31, 2007 and 2006, and the related statements of operations, redeemable convertible preferred stock and stockholders’ deficit, and cash flows for each of the three years in the period ended December 31, 2007. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing auditing procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of MAKO Surgical Corp. at December 31, 2007 and 2006, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2007, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 2 to the financial statements, MAKO Surgical Corp. adopted SFAS No. 123(R), “Share-Based Payment,” applying the modified retrospective method on January 1, 2006.
/s/ Ernst & Young LLP
Certified Public Accountants
Fort Lauderdale, Florida
March 28, 2008
104
MAKO SURGICAL CORP.
Balance Sheets
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| ASSETS | | | | | | | | |
| Current Assets: | | | | | | | | |
| Cash and cash equivalents | | $ | 9,615,027 | | | $ | 2,108,015 | |
| Short-term investments | | | 3,083,980 | | | | 1,399,763 | |
| Accounts receivable | | | 2,212,697 | | | | 578,340 | |
| Inventory | | | 2,346,351 | | | | 926,031 | |
| Due from related party | | | 29,485 | | | | 97,736 | |
| Employee loans | | | — | | | | 225,155 | |
| Prepaids and other assets | | | 281,010 | | | | 84,835 | |
| | | | | | | |
| Total current assets | | | 17,568,550 | | | | 5,419,875 | |
| Deferred cost of revenue | | | 926,342 | | | | 210,349 | |
| Restricted cash | | | 100,000 | | | | 125,000 | |
| Property and equipment, net | | | 2,321,097 | | | | 1,216,464 | |
| Intangible assets, net | | | 5,476,836 | | | | 5,656,186 | |
| Note receivable – related party | | | 69,649 | | | | 125,707 | |
| Deferred initial public offering costs | | | 2,727,832 | | | | — | |
| | | | | | | |
| Total assets | | $ | 29,190,306 | | | $ | 12,753,581 | |
| | | | | | | |
| | | | | | | | | |
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | | | | | | | | |
| Current Liabilities: | | | | | | | | |
| Accounts payable | | $ | 1,511,347 | | | $ | 433,319 | |
| Accrued compensation and employee benefits | | | 1,033,414 | | | | 510,547 | |
| Other accrued liabilities | | | 2,679,938 | | | | 1,015,458 | |
| Deferred revenue | | | 50,000 | | | | 29,167 | |
| Accrued license fee | | | 3,955,305 | | | | 3,620,551 | |
| | | | | | | |
| Total current liabilities | | | 9,230,004 | | | | 5,609,042 | |
| Long-term liabilities: | | | | | | | | |
| Deferred revenue | | | 3,310,833 | | | | 670,833 | |
| | | | | | | |
| Total liabilities | | | 12,540,837 | | | | 6,279,875 | |
| Commitments and contingencies | | | | | | | | |
| Redeemable convertible preferred stock: | | | | | | | | |
| Series A redeemable convertible preferred stock, $0.001 par value; 5,000,000 shares authorized; 4,498,745 shares issued and outstanding; liquidation value of $4,498,745; including accrued cumulative dividends of $1,054,185 and $681,909 as of December 31, 2007 and 2006, respectively | | | 4,806,125 | | | | 4,171,890 | |
| | | | | | | | | |
| Series B redeemable convertible preferred stock, $0.001 par value; 16,500,000 shares authorized; 15,151,516 shares issued and outstanding; liquidation value of $20,000,000; including accrued cumulative dividends of $3,162,032 and $1,822,901 as of December 31, 2007 and 2006, respectively | | | 23,101,855 | | | | 21,738,732 | |
| | | | | | | | | |
| Series C. redeemable convertible preferred stock, $0.001 par value; none authorized, issued and outstanding as of December 31, 2006; 13,600,000 shares authorized; 13,513,514 shares issued and outstanding; liquidation value of $30,000,000; including accrued cumulative dividends of $1,647,456 as of December 31, 2007 | | | 31,578,720 | | | | — | |
| | | | | | | |
| | | | | | | | | |
| Total redeemable convertible preferred stock | | | 59,486,700 | | | | 25,910,622 | |
| | | | | | | |
| Stockholders’ deficit: | | | | | | | | |
| Common stock, $0.001 par value; 60,000,000 shares authorized as of December 31, 2007, and 50,000,000 shares authorized as of December 31, 2006; 1,870,603 and 1,555,938 shares issued and outstanding as of December 31, 2007 and 2006, respectively | | | 1,871 | | | | 1,556 | |
| Note receivable from stockholder and related interest | | | — | | | | (70,574 | ) |
| Accumulated deficit | | | (42,843,131 | ) | | | (19,366,087 | ) |
| Accumulated other comprehensive income (loss) | | | 4,029 | | | | (1,811 | ) |
| | | | | | | |
| Total stockholders’ deficit | | | (42,837,231 | ) | | | (19,436,916 | ) |
| | | | | | | |
| Total liabilities and stockholders’ deficit | | $ | 29,190,306 | | | $ | 12,753,581 | |
| | | | | | | |
See accompanying notes.
105
MAKO SURGICAL CORP.
Statements of Operations
| | | | | | | | | | | | | |
| | | Years Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| Revenue: | | | | | | | | | | | | |
| Products | | $ | 717,798 | | | $ | 62,571 | | | $ | — | |
| Tactile Guidance System | | | — | | | | — | | | | — | |
| Other | | | 53,564 | | | | — | | | | — | |
| | | | | | | | | | |
| Total revenue | | | 771,362 | | | | 62,571 | | | | — | |
| Cost of revenue: | | | | | | | | | | | — | |
| Products | | | 197,086 | | | | 18,780 | | | | — | |
| Tactile Guidance System | | | 360,525 | | | | 57,767 | | | | — | |
| Other | | | 25,303 | | | | — | | | | — | |
| | | | | | | | | | |
| Total cost of revenue | | | 582,914 | | | | 76,547 | | | | — | |
| | | | | | | | | | |
| Gross profit (loss) | | | 188,448 | | | | (13,976 | ) | | | — | |
| | | | | | | | | | |
| Operating costs and expenses: | | | | | | | | | | | | |
| Selling, general and administrative | | | 12,042,690 | | | | 5,022,685 | | | | 2,735,901 | |
| Research and development | | | 8,268,803 | | | | 5,192,453 | | | | 2,581,828 | |
| Depreciation and amortization | | | 1,296,881 | | | | 644,082 | | | | 98,519 | |
| | | | | | | | | | |
| Total operating costs and expenses | | | 21,608,374 | | | | 10,859,220 | | | | 5,416,248 | |
| | | | | | | | | | |
| Loss from operations | | | (21,419,926 | ) | | | (10,873,196 | ) | | | (5,416,248 | ) |
| Interest and other income | | | 1,073,280 | | | | 476,578 | | | | 269,231 | |
| Interest and other expenses | | | (311,608 | ) | | | (220,219 | ) | | | — | |
| | | | | | | | | | |
| Net loss | | | (20,658,254 | ) | | | (10,616,837 | ) | | | (5,147,017 | ) |
| Accretion of preferred stock | | | (300,911 | ) | | | (267,319 | ) | | | (257,474 | ) |
| Dividends on preferred stock | | | (3,358,863 | ) | | | (1,609,027 | ) | | | (883,806 | ) |
| | | | | | | | | | |
| Net loss attributable to common stockholders | | $ | (24,318,028 | ) | | $ | (12,493,183 | ) | | $ | (6,288,297 | ) |
| | | | | | | | | | |
| Net loss per share – Basic and diluted attributable to common stockholders | | $ | (14.74 | ) | | $ | (8.03 | ) | | $ | (4.18 | ) |
| | | | | | | | | | |
| Weighted average common shares outstanding – Basic and diluted | | | 1,649,365 | | | | 1,555,287 | | | | 1,502,761 | |
| | | | | | | | | | |
See accompanying notes.
106
MAKO SURGICAL CORP.
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Deficit
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | Note | | | | | | | | | | | |
| | | Redeemable | | | | | | | | | | | Additional | | | Receivable | | | | | | | Other | | | Total | |
| | | Convertible Preferred | | | Common | | | Stock | | | Paid-in | | | from | | | Accumulated | | | Comprehensive | | | Stockholders’ | |
| | | Shares | | | Amount | | | Shares | | | Amount | | | Capital | | | Stockholder | | | Deficit | | | Income (Loss) | | | Deficit | |
| |
| Balance at December 31, 2004 | | | 4,346,235 | | | $ | 2,866,250 | | | | 1,457,342 | | | $ | 1,457 | | | $ | 267,137 | | | $ | — | | | $ | (1,038,674 | ) | | $ | — | | | $ | (770,080 | ) |
| Issuance of common stock for consulting services | | | — | | | | — | | | | 47,442 | | | | 47 | | | | 60,329 | | | | — | | | | — | | | | — | | | | 60,376 | |
| Issuance of common stock for stockholder note and accrued interest | | | — | | | | — | | | | 49,504 | | | | 50 | | | | 65,298 | | | | (65,348 | ) | | | — | | | | — | | | | — | |
| Receipt of subscription receivable for Series A redeemable convertible preferred stock and common stock warrants | | | 53,000 | | | | 53,000 | | | | — | | | | — | | | | 174 | | | | — | | | | — | | | | — | | | | 174 | |
| Issuance of Series A redeemable convertible preferred stock and warrants for common stock for cash, net of issuance costs of $7,000 | | | 99,510 | | | | 92,446 | | | | — | | | | — | | | | 640 | | | | — | | | | — | | | | — | | | | 640 | |
| Issuance of Series B redeemable convertible preferred stock for cash, net of issuance costs of $119,000 | | | 15,151,516 | | | | 19,881,300 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Employee share-based compensation expense | | | — | | | | — | | | | — | | | | — | | | | 113,718 | | | | — | | | | — | | | | — | | | | 113,718 | |
| Accretion to redemption value of Series A and B redeemable convertible preferred stock | | | — | | | | 257,474 | | | | — | | | | — | | | | (257,474 | ) | | | — | | | | — | | | | — | | | | (257,474 | ) |
| Accrued dividends on Series A and B redeemable convertible preferred stock | | | — | | | | 883,806 | | | | — | | | | — | | | | (249,822 | ) | | | — | | | | (633,984 | ) | | | — | | | | (883,806 | ) |
| Change in unrealized loss on available-for-sale securities | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (4,131 | ) | | | (4,131 | ) |
| Net loss and comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (5,147,017 | ) | | | — | | | | (5,147,017 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (5,151,148 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2005 | | | 19,650,261 | | | | 24,034,276 | | | | 1,554,288 | | | | 1,554 | | | | — | | | | (65,348 | ) | | | (6,819,675 | ) | | | (4,131 | ) | | | (6,887,600 | ) |
| Issuance of common stock upon exercise of options | | | — | | | | — | | | | 1,650 | | | | 2 | | | | 1,098 | | | | — | | | | — | | | | — | | | | 1,100 | |
| Employee share-based compensation expense | | | — | | | | — | | | | — | | | | — | | | | 170,033 | | | | — | | | | — | | | | — | | | | 170,033 | |
| Interest on note receivable from stockholder | | | — | | | | — | | | | — | | | | — | | | | 5,226 | | | | (5,226 | ) | | | — | | | | — | | | | — | |
| Addendum to asset contribution with Z-KAT | | | — | | | | — | | | | — | | | | — | | | | (176,357 | ) | | | — | | | | (44,691 | ) | | | — | | | | (221,048 | ) |
| Stock issuance costs for Series C redeemable convertible preferred stock sale | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (8,538 | ) | | | — | | | | (8,538 | ) |
| Accretion to redemption value of Series A and B redeemable convertible preferred stock | | | — | | | | 267,319 | | | | — | | | | — | | | | — | | | | — | | | | (267,319 | ) | | | — | | | | (267,319 | ) |
| Accrued dividends on Series A and B redeemable convertible preferred stock | | | — | | | | 1,609,027 | | | | — | | | | — | | | | — | | | | — | | | | (1,609,027 | ) | | | — | | | | (1,609,027 | ) |
| Change in unrealized loss on available-for-sale securities | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 2,320 | | | | 2,320 | |
| Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (10,616,837 | ) | | | — | | | | (10,616,837 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (10,614,517 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2006 | | | 19,650,261 | | | $ | 25,910,622 | | | | 1,555,938 | | | $ | 1,556 | | | $ | — | | | $ | (70,574 | ) | | $ | (19,366,087 | ) | | $ | (1,811 | ) | | $ | (19,436,916 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
(continued)
107
MAKO SURGICAL CORP.
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Deficit
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | Note | | | | | | | | | | | |
| | | Redeemable | | | | | | | | | | | Additional | | | Receivable | | | | | | | Other | | | Total | |
| | | Convertible Preferred | | | Common | | | Stock | | | Paid-in | | | from | | | Accumulated | | | Comprehensive | | | Stockholders’ | |
| | | Shares | | | Amount | | | Shares | | | Amount | | | Capital | | | Stockholder | | | Deficit | | | Income (Loss) | | | Deficit | |
| |
| Balance at December 31, 2006 | | | 19,650,261 | | | $ | 25,910,622 | | | | 1,555,938 | | | $ | 1,556 | | | $ | — | | | $ | (70,574 | ) | | $ | (19,366,087 | ) | | $ | (1,811 | ) | | $ | (19,436,916 | ) |
| Issuance of Series C redeemable convertible preferred stock, net of issuance costs of $84,000 | | | 13,513,514 | | | | 29,916,304 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Issuance of common stock upon exercise of options | | | — | | | | — | | | | 1,306 | | | | 1 | | | | 1,661 | | | | — | | | | — | | | | — | | | | 1,662 | |
| Employee share-based compensation expense | | | — | | | | — | | | | — | | | | — | | | | 530,592 | | | | — | | | | — | | | | — | | | | 530,592 | |
| Interest on note receivable from stockholder | | | — | | | | — | | | | — | | | | — | | | | 3,760 | | | | (3,760 | ) | | | — | | | | — | | | | — | |
| Modification of restricted stock | | | — | | | | — | | | | 300,084 | | | | 300 | | | | 394,480 | | | | 74,334 | | | | — | | | | — | | | | 469,114 | |
| Return of 35,244 shares due to modification of CEO restricted stock | | | — | | | | — | | | | (35,244 | ) | | | (35 | ) | | | (391,892 | ) | | | — | | | | — | | | | — | | | | (391,927 | ) |
| Restricted common stock compensation expense | | | — | | | | — | | | | 48,519 | | | | 49 | | | | 302,383 | | | | — | | | | — | | | | — | | | | 302,432 | |
| Accretion to redemption value of Series A, B and C redeemable convertible preferred stock | | | — | | | | 300,911 | | | | — | | | | — | | | | (300,911 | ) | | | — | | | | — | | | | — | | | | (300,911 | ) |
| Accrued dividends on Series A, B and C redeemable convertible preferred stock | | | — | | | | 3,358,863 | | | | — | | | | — | | | | (540,073 | ) | | | — | | | | (2,818,790 | ) | | | — | | | | (3,358,863 | ) |
| Change in unrealized gain on available-for-sale securities | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 5,840 | | | | 5,840 | |
| Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (20,658,254 | ) | | | — | | | | (20,658,254 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (20,652,414 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2007 | | | 33,163,775 | | | $ | 59,486,700 | | | | 1,870,603 | | | $ | 1,871 | | | $ | — | | | $ | — | | | $ | (42,843,131 | ) | | $ | 4,029 | | | $ | (42,837,231 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
See accompanying notes.
108
MAKO SURGICAL CORP.
Statements of Cash Flows
| | | | | | | | | | | | | |
| | | Years Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
Operating activities: | | | | | | | | | | | | |
| Net loss | | $ | (20,658,254 | ) | | $ | (10,616,837 | ) | | $ | (5,147,017 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
| Depreciation | | | 677,538 | | | | 175,311 | | | | 80,149 | |
| Amortization of intangible assets | | | 645,374 | | | | 470,082 | | | | 18,370 | |
| Stock-based compensation | | | 1,227,804 | | | | 170,033 | | | | 174,094 | |
| Inventory writedown | | | 7,918 | | | | 35,821 | | | | — | |
| Loss on disposal of assets | | | — | | | | 2,533 | | | | — | |
| Loss on asset impairment | | | 13,759 | | | | — | | | | — | |
| Accrued interest expense on deferred license fee | | | 304,971 | | | | 210,913 | | | | — | |
| Changes in operating assets and liabilities: | | | | | | | | | | | | |
| Accounts receivable | | | (1,634,357 | ) | | | (578,340 | ) | | | — | |
| Inventory | | | (1,428,238 | ) | | | (605,402 | ) | | | (87,950 | ) |
| Due from related party | | | 68,251 | | | | (97,736 | ) | | | — | |
| Employee loans | | | 225,155 | | | | (225,155 | ) | | | — | |
| Prepaid and other assets | | | (196,175 | ) | | | 214,498 | | | | (28,729 | ) |
| Other assets | | | (560,601 | ) | | | (405,349 | ) | | | — | |
| Accounts payable | | | 1,078,028 | | | | 353,423 | | | | 14,717 | |
| Accrued compensation and employee benefits | | | 522,867 | | | | 385,452 | | | | (25,405 | ) |
| Other accrued liabilities | | | 1,664,480 | | | | 509,851 | | | | (8,271 | ) |
| Due to related party | | | — | | | | (115,026 | ) | | | — | |
| Deferred revenue | | | 2,660,833 | | | | 700,000 | | | | — | |
| | | | | | | | | | |
| Net cash used in operating activities | | | (15,380,647 | ) | | | (9,415,928 | ) | | | (5,010,042 | ) |
Investing activities: | | | | | | | | | | | | |
| Purchase of short-term investments | | | (15,158,741 | ) | | | (600,000 | ) | | | (11,099,419 | ) |
| Proceeds from sales and maturities of short-term investments | | | 13,480,364 | | | | 9,299,577 | | | | 998,268 | |
| Acquisition of property and equipment | | | (1,782,171 | ) | | | (1,193,462 | ) | | | (175,180 | ) |
| Acquisition of intangible assets | | | (450,000 | ) | | | (2,120,000 | ) | | | (555,000 | ) |
| | | | | | | | | | |
| Net cash provided by (used in) investing activities | | | (3,910,548 | ) | | | 5,386,115 | | | | (10,831,331 | ) |
Financing activities: | | | | | | | | | | | | |
| Proceeds from issuance of common stock and warrants | | | — | | | | — | | | | 640 | |
| Deferred initial public offering costs | | | (2,727,832 | ) | | | — | | | | — | |
| Proceeds from issuance of Series A. redeemable convertible preferred stock, net of stock issuance costs | | | — | | | | — | | | | 145,620 | |
| Proceeds from issuance of Series B redeemable convertible preferred stock, net of stock issuance costs | | | — | | | | �� | | | | 19,881,300 | |
| Proceeds from issuance of Series C redeemable convertible preferred stock, net of stock issuance costs | | | 29,916,304 | | | | (8,538 | ) | | | — | |
| Exercise of common stock options for cash | | | 1,662 | | | | 1,100 | | | | — | |
| Payment of CEO payroll taxes relating to restricted common stock modification | | | (391,927 | ) | | | — | | | | — | |
| | | | | | | | | | |
| Net cash provided by (used in) financing activities | | | 26,798,207 | | | | (7,438 | ) | | | 20,027,560 | |
| | | | | | | | | | |
| |
| Net increase (decrease) in cash and cash equivalents | | | 7,507,012 | | | | (4,037,251 | ) | | | 4,186,187 | |
| Cash and cash equivalents at beginning of year | | | 2,108,015 | | | | 6,145,266 | | | | 1,959,079 | |
| | | | | | | | | | |
| |
| Cash and cash equivalents at end of year | | $ | 9,615,027 | | | $ | 2,108,015 | | | $ | 6,145,266 | |
| | | | | | | | | | |
Supplemental disclosures of cash flow information | | | | | | | | | | | | |
| Interest paid | | $ | — | | | $ | 6,775 | | | $ | — | |
| | | | | | | | | | |
Noncash investing and financing activities: | | | | | | | | | | | | |
| Accretion of preferred stock | | $ | 300,911 | | | $ | 267,319 | | | $ | 257,474 | |
| Accrued dividends on preferred stock | | | 3,358,863 | | | | 1,609,027 | | | | 883,806 | |
| Licensing of intellectual property | | | 29,783 | | | | 3,409,638 | | | | — | |
| Deferred license fee payable | | | 29,783 | | | | 3,409,638 | | | | — | |
| Common stock issued for note receivable | | | — | | | | — | | | | 63,000 | |
| Interest on note receivable for common stock | | | 3,760 | | | | 5,226 | | | | 2,348 | |
| Receipt of 35,244 shares of restricted common stock as reimbursement for payment of CEO payroll taxes | | | 391,927 | | | | — | | | | — | |
| Acquisition of assets from affiliate: | | | | | | | | | | | | |
| Note receivable | | | — | | | | 55,707 | | | | — | |
See accompanying notes.
109
MAKO SURGICAL CORP.
Notes to Financial Statements
1. Description of the Business
MAKO Surgical Corp. (the “Company” or “MAKO”) is an emerging medical device company providing innovative surgical solutions to the orthopedic knee arthroplasty market. The Company was incorporated in the State of Delaware on November 12, 2004 and is headquartered in Fort Lauderdale, Florida.
As discussed in Note 11, in February 2008, the Company effected a one for 3.03 reverse split of its issued and outstanding common stock, which has been retroactively reflected in these financial statements.
Predecessor
Z-KAT, Inc. (the “Predecessor” or “Z-KAT”) was formed in 1997 to develop and commercialize computer assisted surgery (“CAS”) applications. Z-KAT formed MAKO in November 2004, to develop and commercialize unique applications combining CAS with haptic robotics in the medical field of orthopedics. As more fully described in Note 4, in December 2004, pursuant to a contribution agreement (the “Contribution Agreement”), the Company acquired substantially all of Z-KAT’s tangible assets and a majority of Z-KAT’s CAS technology assets not required for Z-KAT’s retained CAS business, and all of its haptic robotic research and development technology inventory.
2. Summary of Significant Accounting Policies
Basis of Presentation and Use of Estimates
The financial statements have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. The accounting estimates that require management’s most significant, difficult and subjective judgments include revenue recognition, allowance for doubtful accounts, accrual for warranty costs, inventory impairment charges, valuation allowance for deferred tax assets, impairment of long-lived assets and the determination of stock-based compensation. Actual results could differ significantly from these estimates.
110
Liquidity and Operations
The Company has incurred net losses and negative cash flow from operating activities each year since inception. In order to continue its operations and achieve its business objectives, the Company must achieve profitability or obtain additional debt or equity financing. Any such required additional capital may not be available on reasonable terms, or at all. If we are unable to obtain additional financing, we may be required to reduce the scope of, delay, or eliminate some or all of, our planned research, development and commercialization activities, which could materially harm our business and results of operations. See Note 11 for discussion regarding the Company’s initial public offering (“IPO”) in 2008. The failure of the Company to win widespread acceptance of its products by hospitals, physicians and patients could have a material adverse effect on the Company’s business, results of operations, future cash flows and financial condition.
Concentrations of Credit Risk and Other Risks and Uncertainties
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash and cash equivalents and short-term investments. Cash and cash equivalents are deposited in demand and money market accounts at two large financial institutions. Such deposits are generally in excess of insured limits. The Company has not experienced any losses on its deposits of cash and cash equivalents.
As of December 31, 2007, the Company held $1.6 million of AAA rated auction rate securities issued by two separate funds. These securities are classified as short-term investments in the Company’s balance sheet. In February 2008, the auction rate securities experienced failed auctions that limited the liquidity of these securities. Based on current market conditions, it is likely that auction failures will continue. As a result, the Company is unable to determine when a successful auction will occur and the auction rate securities will be liquidated. The Company believes the carrying value is fully recoverable as auction rate securities are fully collateralized by assets held by the fund; however, the Company will continue to monitor the investments for any potential impairment.
The Company is subject to risks common to companies in the medical device industry including, but not limited to: new technological innovations, dependence on key personnel, dependence on key suppliers, protection of proprietary technology, compliance with government regulations, uncertainty of widespread market acceptance of products, product liability and the need to obtain additional financing. The Company’s products include components subject to rapid technological change. Certain components used in manufacturing have relatively few alternative sources of supply, and establishing additional or replacement suppliers for such components cannot be accomplished quickly. The inability of any of these suppliers to fulfill the Company’s supply requirements may negatively impact future operating results. While the Company has ongoing programs to minimize the adverse effect of such uncertainty and considers technological change in estimating its inventory net realizable value, uncertainty continues to exist.
The Company’s current version of its Tactile Guidance System (“TGS”) and its unicompartmental implants have been cleared by the U.S. Food and Drug Administration (“FDA”). Certain products currently under development by the Company, such as future versions of its TGS and implants, will require clearance by the FDA or other international regulatory agencies prior to commercial sales. There can be no assurance that the Company’s products will receive the necessary clearance. If the Company were to be denied such clearance or such clearance was delayed, it could have a material adverse impact on the Company.
111
The Company may perform credit evaluations of its customers’ financial condition and, generally, requires no collateral from its customers. The Company provides an allowance for doubtful accounts when required but has not experienced any losses to date. To date, the majority of revenue recognized by the Company was from several significant customers.
FASB Statement No. 131,Disclosures about Segments of an Enterprise and Related Information, establishes standards for reporting information about operating segments. Operating segments are defined as components of an enterprise about which separate financial information is available that is evaluated regularly by the chief operating decision maker, or decision making group, in deciding how to allocate resources and in assessing performance. The Company’s chief operating decision maker is its CEO. The Company’s CEO reviews financial information presented on an aggregate basis for purposes of allocating resources and evaluating financial performance. The Company has one business activity and there are no segment managers who are held accountable for operations, operating results and plans for products or components below the aggregate Company level. Accordingly, the Company reports as a single operating segment. To date, all of the Company’s revenue is from companies located in the United States. The following table presents information about the Company’s revenue by customer:
| | | | | | | | | | | | | |
| | | Years Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| Company A | | $ | 330,650 | | | $ | 59,543 | | | $ | — | |
| Company B | | | 161,176 | | | | — | | | | — | |
| Company C | | | 126,411 | | | | — | | | | — | |
| Others | | | 99,561 | | | | 3,028 | | | | — | |
| | | | | | | | | | |
| Net Revenue | | $ | 717,798 | | | $ | 62,571 | | | $ | — | |
| | | | | | | | | | |
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity at date of purchase of 90 days or less to be cash equivalents.
Restricted Cash
Restricted cash amounts include amounts deposited as collateral in connection with the Company’s facility operating lease which requires that specific cash amounts be set aside. Restricted cash amounts were $100,000 and $125,000 at December 31, 2007 and 2006, respectively.
Fair Value of Financial Instruments
Carrying amounts of certain of the Company’s financial instruments, including cash and cash equivalents, short-term investments, accounts and notes receivable, accrued license fees and others approximate fair value due to their short maturities or market rates of interest.
112
Short-Term Investments
The Company’s short-term investments are classified as available-for-sale. Available-for-sale securities are carried at fair value based on quoted market prices, with the unrealized gains and losses included in other comprehensive income (loss) within stockholders’ deficit. Realized gains and losses and declines in value judged to be other-than-temporary on available-for-sale securities are included in interest and other expense. Interest and dividends on securities classified as available-for-sale are included in interest and other income. The cost of securities sold is based on the specific identification method.
The amortized cost and fair value of short-term investments, with gross unrealized gains and losses, were as follows:
As of December 31, 2007
| | | | | | | | | | | | | | | | | |
| | | | | | | Gross | | | Gross | | | | |
| | | Amortized | | | Unrealized | | | Unrealized | | | Fair | |
| | | Cost | | | Gains | | | Losses | | | Value | |
| Variable auction rate securities | | $ | 1,550,000 | | | $ | — | | | $ | — | | | $ | 1,550,000 | |
| U.S. treasury notes | | | 1,504,951 | | | | 4,029 | | | | — | | | | 1,508,980 | |
| Certificates of deposit | | | 25,000 | | | | — | | | | — | | | | 25,000 | |
| | | | | | | | | | | | | |
| | | $ | 3,079,951 | | | $ | 4,029 | | | $ | — | | | $ | 3,083,980 | |
| | | | | | | | | | | | | |
As of December 31, 2006
| | | | | | | | | | | | | | | | | |
| | | | | | | Gross | | | Gross | | | | |
| | | Amortized | | | Unrealized | | | Unrealized | | | Fair | |
| | | Cost | | | Gains | | | Losses | | | Value | |
| Variable auction rate securities | | $ | 1,301,574 | | | $ | — | | | $ | (1,574 | ) | | $ | 1,300,000 | |
| Certificate of deposit | | | 100,000 | | | | — | | | | (237 | ) | | | 99,763 | |
| | | | | | | | | | | | | |
| | | $ | 1,401,574 | | | $ | — | | | $ | (1,811 | ) | | $ | 1,399,763 | |
| | | | | | | | | | | | | |
As of December 31, 2007 and 2006, all securities had maturities or interest reset dates of less than one year. Realized gains and losses to date have not been significant.
Allowance for Doubtful Accounts
The allowance for doubtful accounts is based on the Company’s assessment of the collectibility of customer accounts. The Company regularly reviews the allowance by considering factors such as historical experience, credit quality, the age of the accounts receivable balances and current economic conditions that may affect a customer’s ability to pay. The Company has not experienced any collectibility issues to date and has no allowance, provision for doubtful accounts receivable or write-offs to date in the accompanying financial statements.
113
Accrual for Warranty Costs
Upon installation of a TGS unit, the Company establishes an accrual for the estimated costs associated with providing a standard one-year warranty for defects in materials and workmanship. Due to the Company’s limited history of commercial placements of TGS units, the estimate of warranty costs is subjective; however, costs incurred to date have not been significantly different from the estimate.
Inventory
Inventory is stated at the lower of cost or market value on a first-in, first-out basis. Inventory costs include direct materials and direct labor. The Company reviews its inventory periodically to determine net realizable value and considers product upgrades in its periodic review of realizability. The Company writes down inventory, if required, based on forecasted demand and technological obsolescence. These factors are impacted by market and economic conditions, technology changes and new product introductions and require estimates that may include uncertain elements. Writedowns of inventory for the years ended December 31, 2007, 2006 and 2005 were approximately $8,000, $36,000 and $0, respectively.
Property and Equipment
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation of property and equipment is computed using the straight-line method over their estimated useful lives of two to seven years. Leasehold improvements and assets recorded under capital leases are amortized on a straight-line basis over the lesser of their useful life or the term of the lease and are included in depreciation expense in the accompanying statements of operations. Upon retirement or sale, the cost and related accumulated depreciation are removed from the balance sheet and the resulting gain or loss is reflected in operations. Maintenance and repairs are charged to operations as incurred.
Depreciation expense for the years ended December 31, 2007, 2006 and 2005 was approximately $678,000, $175,000 and $80,000, respectively.
Intangible Assets
The Company’s intangible assets are comprised of licenses to intellectual property rights. These intangible assets are carried at cost, net of accumulated amortization. Amortization is recorded using the straight-line method, over their respective useful lives (generally the life of underlying patents), which range from approximately 10 to 19 years.
Impairment of Long-Lived Assets
The Company evaluates its long-lived assets for indicators of impairment by comparison of the carrying amounts to future net undiscounted cash flows expected to be generated by such assets when events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. Should an impairment exist, the impairment loss would be measured based on the excess carrying value of the asset over the asset’s fair value or discounted estimate of future cash flows. The Company has not recorded any such impairment losses to date.
114
Revenue Recognition
Revenue is generated from unit sales of the TGS, including installation services, training, upgrades and enhancements, from sales of implants and disposable products, and by providing extended warranty services. The Company’s TGS product, upgrades and enhancements to those products include software that is essential to the functionality of the product and, accordingly, the Company accounts for the sale of the TGS pursuant to Statement of Position No. 97-2,Software Revenue Recognition(“SOP 97-2”), as amended.
The Company recognizes product revenue for sales of the TGS when there is persuasive evidence of an arrangement, the fee is fixed or determinable, collection of the fee is probable and delivery has occurred as prescribed by SOP 97-2. For all sales, the Company uses either a signed agreement or a binding purchase order as evidence of an arrangement. Such arrangements typically require customer acceptance of the system which is evidenced by the receipt by the Company of a form executed by the customer indicating their acceptance of the TGS unit. Such arrangements require the Company to provide upgrades and enhancements to the TGS unit on a when and if available basis. As of December 31, 2007, four TGS customers are entitled to receive an upgrade to version 2.0 of the TGS at no additional charge, and one customer has the right to receive it at a discounted price. All of these customer upgrade rights to receive the upgrades through version 2.0 of the TGS are on a when and if available basis.
Payments received upon customer acceptance of TGS units are recognized as deferred revenue. The direct cost of revenue associated with the sale of TGS units is recognized as deferred cost of revenue. Costs associated with establishing an accrual for the TGS standard one-year warranty liability and royalties related to the sale of TGS units covered by licensing arrangements are expensed as incurred. The Company anticipates ultimately recognizing a positive margin on the sales of TGS units to date, including the satisfaction of the remaining upgrades through the final deliverable of version 2.0 of the TGS, which is anticipated to be in the first half of 2009. If the Company is not able to deliver version 2.0 of the TGS, customers would retain the original TGS unit sold and the Company would not be obligated to refund the purchase price of the TGS unit.
For arrangements with multiple elements, the Company allocates arrangement consideration to TGS units, upgrades, enhancements and services based upon vendor specific objective evidence (“VSOE”) of fair value of the respective elements. As the Company is in the early stages of commercialization, VSOE of fair value does not yet exist for all the undelivered elements. Accordingly, all revenue and direct cost of revenue associated with the sale of the TGS are deferred until the earlier of (1) delivery of all elements or (2) establishment of VSOE of fair value for all undelivered elements.
115
Product revenue from the sale of implants and disposable products (the “Products”) is recognized as revenue in accordance with Staff Accounting Bulletin (“SAB”) No. 104,Revenue Recognition, when persuasive evidence of an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable and collectibility is reasonably assured. The Products are a separate unit of accounting from the TGS as (1) they have value to the customer on a standalone basis, (2) objective and reliable evidence of the fair value of the item exists and (3) no right of return exists once the Products are implanted or consumed. Accordingly, as the Company’s implants and disposable products are sold on a procedural basis, the revenue and costs associated with the sale of Products are recognized at the time of sale (i.e., at the time of the completion of the related surgical procedure).
Service revenue, which consists of extended warranty services on the TGS hardware, is deferred and recognized ratably over the service period until no further obligation exists. Costs associated with providing services are expensed when incurred.
The Company’s agreements with customers do not contain product return rights beyond the customer acceptance period, which is typically defined as a certain number of surgical procedures over certain period typically not exceeding three months.
For purposes of obtaining clinical and technical feedback on the current version of the TGS, the Company also enters into consignment programs with certain customers. Under the terms of such programs, the Company retains title to the TGS unit, while the customer has use of the TGS and purchases the Company’s implants and disposables products. The Company may provide unspecified upgrades to the product during the term of each program when and if available; however, the Company is not obligated to provide upgrades for the two TGS units under consignment programs as of December 31, 2007. The TGS units associated with the Company’s consignment programs are recorded within property and equipment and are depreciated over their estimated useful life of two years. Depreciation and warranty expense attributable to the TGS consignment units are recorded within cost of revenue. The revenue associated with the sale of Products to customers under consignment programs are recognized as revenue when persuasive evidence of an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable and collectibility is reasonably assured.
Deferred Revenue and Deferred Cost of Revenue
Deferred revenue consists of deferred product revenue and deferred service revenue. Deferred product revenue arises from timing differences between the shipment of product and satisfaction of all revenue recognition criteria consistent with the Company’s revenue recognition policy. Deferred service revenue results from the advance payment for services to be delivered over a period of time, usually in one year increments. Service revenue is recognized ratably over the service period. Deferred cost of revenue consists of the direct costs associated with the manufacture of units for which the revenue has been deferred in accordance with the Company’s revenue recognition policy. Deferred revenue and associated deferred cost of revenue expected to be realized within one year are classified as current liabilities and current assets, respectively.
116
Deferred Initial Public Offering Costs
Specific incremental costs directly associated with the Company’s IPO, primarily legal, accounting and printing costs, were deferred and charged directly to stockholders’ equity upon the closing of the IPO in February 2008. See Note 11 for discussion regarding the Company’s IPO.
Shipping and Handling Costs
Costs incurred for shipping and handling are included in cost of revenue at the time the related revenue is recognized. Amounts billed to customers for shipping and handling are reported as revenue.
Research and Development Costs
Costs related to research, design and development of products are charged to research and development expense as incurred. These costs include direct salary costs for research and development personnel, costs for materials used in research and development activities and costs for outside services.
Software Development Costs
Software development costs are included in research and development and are expensed as incurred. After technological feasibility is established, material software development costs are capitalized. The capitalized cost is then amortized on a straight-line basis over the estimated product life, or on the ratio of current revenue to total projected product revenue, whichever is greater. To date, the period between achieving technological feasibility, which the Company has defined as the establishment of a working model which typically occurs when the verification and validation testing is complete, and the general availability of such software has been short and software development costs qualifying for capitalization have been insignificant. Accordingly, MAKO has not capitalized any software development costs to date.
Stock-Based Compensation
Effective January 1, 2006, the Company adopted the fair value provisions of SFAS No. 123 Revised,Share-Based Payment(“SFAS 123(R)”). SFAS 123(R) requires the recognition of compensation expense, using a fair-value based method, for costs related to all share-based payments including stock options. SFAS 123(R) requires companies to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model.
The Company adopted SFAS 123(R) using the modified retrospective transition method, which requires the restatement of financial statements for prior periods. Prior to the adoption of SFAS 123(R), the Company accounted for stock-based compensation arrangements by recording compensation expense based on the estimated fair-value of stock-based awards in accordance with SFAS 123. The impact of SFAS 123(R) on prior periods was not significant.
117
The Company accounts for stock-based compensation arrangements with non-employees in accordance with the Emerging Issues Task Force (“EITF”) Abstract No. 96-18,Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling Goods or Services.The Company records the expense of such services based on the estimated fair value of the equity instrument using the Black-Scholes-Merton pricing model. The value of the equity instrument is charged to expense over the term of the service agreement.
See Note 8 for a detailed discussion of the various stock option plans and related stock-based compensation.
Advertising Costs
Advertising costs are expensed as incurred. Advertising costs have been insignificant to date.
Income Taxes
Deferred tax assets and liabilities are determined based on the differences between financial reporting and tax bases of assets and liabilities, using tax rates expected to be in effect when the differences will reverse. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized.
Operating Leases
Rental payments and incentives, if any, are recognized on a straight-line basis over the life of a lease. See Note 6 for further discussion on operating leases.
Net Loss Per Share
The Company calculated net loss per share in accordance with SFAS No. 128,Earnings per Share. Basic earnings per share (EPS) is calculated by dividing the net income or loss available to common stockholders adjusted for redeemable convertible preferred stock accretion and dividends by the weighted average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted EPS is computed by dividing the net income or loss available to common stockholders adjusted by the weighted average number of common shares outstanding for the period and the weighted average number of dilutive common stock equivalents outstanding for the period determined using the treasury-stock method. For purposes of this calculation, stock options (totaling 1,916,525, 947,950 and 629,567 as of December 31, 2007, 2006 and 2005, respectively), warrants (to purchase 462,716 shares of common stock as of December 31, 2007, 2006 and 2005), and redeemable convertible preferred stock (totaling 33,163,775, 19,650,261, and 19,650,261 as of December 31, 2007, 2006 and 2005, respectively) are considered to be common stock equivalents but are excluded in the calculation of diluted earnings per share as their effect is anti-dilutive. Accordingly, basic and diluted EPS are the same for all periods presented. The conversion of all of the Company’s redeemable convertible preferred stock into 10,945,080 shares of common stock in February 2008 will have a significant impact on subsequent EPS calculations. See Note 11 for further discussion of the conversion of the Company’s preferred stock in 2008.
118
Comprehensive Loss
Comprehensive loss is defined as the change in equity from transactions and other events and circumstances other than those resulting from investments by owners and distributions to owners. For the years ended December 31, 2007, 2006 and 2005, the Company recorded comprehensive losses of approximately $20,652,000, $10,615,000 and $5,151,000, respectively.
Recent Accounting Pronouncements
In July 2006, the Financial Accounting Standards Board (“FASB”) issued Interpretation No. 48,Accounting for Uncertainty in Income Taxes, an interpretation of FASB Statement No. 109 (“FIN 48”). FIN 48 clarifies the accounting for uncertainty in income taxes by prescribing the recognition threshold a tax position is required to meet before being recognized in the financial statements. It also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure, and transition. FIN 48 is effective for fiscal years beginning after December 15, 2006. The Company adopted the provisions of FIN 48 effective January 1, 2007. No cumulative adjustment to the Company’s accumulated deficit was required upon adoption and there was no effect of adoption.
In September 2006, the Securities and Exchange Commission issued Staff Accounting Bulletin No. 108,Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements(“SAB 108”). SAB 108 provides guidance on the consideration of the effects of prior year misstatements in quantifying current year misstatements for the purpose of a materiality assessment. SAB 108 establishes an approach that requires quantification of financial statement errors based on the effects on each of the Company’s balance sheets and statement of operations and the related financial statement disclosures. SAB 108 was adopted by the Company effective January 1, 2007. The Company has determined that the adoption of SAB 108 had no effect on its results of operations and financial position.
In September 2006, the FASB issued SFAS No. 157,Fair Value Measurements(“SFAS 157”). SFAS 157 provides guidance for using fair value to measure assets and liabilities. It also responds to investors’ requests for expanded information about the extent to which companies measure assets and liabilities at fair value, the information used to measure fair value, and the effect of fair value measurements on earnings. SFAS 157 applies whenever other standards require (or permit) assets or liabilities to be measured at fair value, and does not expand the use of fair value in any new circumstances. SFAS 157 is effective for financial statements issued for fiscal years beginning after November 15, 2007 and is required to be adopted by the Company effective January 1, 2008. The Company is currently evaluating the impact of SFAS 157, but does not expect the adoption of SFAS 157 to have a material impact on its financial statements.
119
In February 2007, the FASB issued SFAS No. 159,The Fair Value Option for Financial Assets and Financial Liabilities—Including an amendment of FASB Statement No. 115(“SFAS 159”). SFAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The objective of SFAS 159 is to reduce both complexity in accounting for financial instruments and the volatility in earnings caused by measuring related assets and liabilities differently. Most of the provisions in Statement 159 are elective; however, the amendment to FASB Statement No. 115,Accounting for Certain Investments in Debt and Equity Securities, applies to all entities with available-for-sale and trading securities. SFAS 159 is effective as of the beginning of an entity’s first fiscal year beginning after November 15, 2007. The Company is currently evaluating the impact of SFAS 159, but does not expect the adoption of SFAS 159 to have a material impact on its financial statements.
In June 2007, the FASB issued EITF Issue No. 07-03,Accounting for Nonrefundable Advance Payments for Goods or Services Received for Use in Future Research and Development Activities (“EITF 07-03”). This EITF requires that nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities should be deferred and capitalized. Such amounts should be recognized as an expense as the related goods are delivered or the related services are performed. Entities should continue to evaluate whether they expect the goods to be delivered or services to be rendered. If an entity does not expect the goods to be delivered or services to be rendered, the capitalized advance payment should be charged to expense. EITF 07-03 is effective for financial statements issued for fiscal years beginning after December 15, 2007, and interim periods within those fiscal years. Earlier adoption is permitted. The Company is currently evaluating the impact of EITF 07-03, but does not expect the adoption of EITF 07-03 to have a material impact on its financial statements.
In December 2007, the FASB issued SFAS No. 141 (revised 2007),Business Combinations (“SFAS 141(R)”). SFAS 141(R) retains the fundamental requirements of the original pronouncement requiring that the purchase method be used for all business combinations. SFAS 141(R) defines the acquirer as the entity that obtains control of one or more businesses in the business combination, establishes the acquisition date as the date that the acquirer achieves control and requires the acquirer to recognize the assets acquired, liabilities assumed and any noncontrolling interest at their fair values as of the acquisition date. SFAS 141(R) also requires that acquisition-related costs be recognized separately from the acquisition. SFAS 141(R) will become effective for the Company on January 1, 2009. The Company is currently evaluating the impact that SFAS 141(R) will have on its financial statements.
In December 2007, the FASB issued SFAS No. 160,Noncontrolling Interests in Consolidated Financial Statements(“SFAS 160”). SFAS 160 establishes accounting and reporting standards for ownership interests in subsidiaries held by parties other than the parent, the amount of consolidated net income attributable to the parent and to the noncontrolling interest, changes in a parent’s ownership interest and the valuation of retained noncontrolling equity investments when a subsidiary is deconsolidated. SFAS 160 also establishes reporting requirements that provide sufficient disclosures that clearly identify and distinguish between the interests of the parent and the interests of the noncontrolling owners. SFAS 160 will become effective for the Company beginning in the first quarter of 2009. The Company is currently evaluating the impact that SFAS 160 will have on its financial statements.
120
Reclassifications
Certain reclassifications have been made to the prior periods’ statements of operations and to the notes to the financial statements to conform to the current period’s presentation.
3. Selected Balance Sheet Components
The following table provides details of selected balance sheet items:
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
Inventory: | | | | | | | | |
| Raw materials | | $ | 974,450 | | | $ | 396,772 | |
| Work-in-process | | | 641,810 | | | | 371,764 | |
| Finished goods | | | 730,091 | | | | 157,495 | |
| | | | | | | |
| Total inventory | | $ | 2,346,351 | | | $ | 926,031 | |
| | | | | | | |
| | | | | | | | | | | | | |
| | | December 31, | | | Estimated | |
| | | 2007 | | | 2006 | | | Useful Life | |
Property and equipment: | | | | | | | | | | | | |
| Consigned TGS units and instruments | | $ | 605,833 | | | $ | 279,631 | | | 2 years | |
| TGS demo system | | | 402,600 | | | | — | | | 2 years | |
| Computer equipment and software | | | 1,057,209 | | | | 625,618 | | | 3-5 year | |
| Laboratory and manufacturing equipment | | | 570,531 | | | | 287,821 | | | 5 years | |
| Office furniture and equipment | | | 493,047 | | | | 249,763 | | | 7 years | |
| Leasehold improvements | | | 128,201 | | | | 32,420 | | | Lease Term | |
| | | | | | | | | | | |
| | | | 3,257,421 | | | | 1,475,253 | | | | | |
| Less accumulated depreciation and amortization | | | (936,324 | ) | | | (258,789 | ) | | | | |
| | | | | | | | | | | |
| Total property and equipment, net | | $ | 2,321,097 | | | $ | 1,216,464 | | | | | |
| | | | | | | | | | | |
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
Other accrued liabilities: | | | | | | | | |
| Accrued development services | | $ | 71,166 | | | $ | 362,866 | |
| Accrued legal fees | | | 800,852 | | | | 53,513 | |
| Other | | | 1,807,920 | | | | 599,079 | |
| | | | | | | |
| | | $ | 2,679,938 | | | $ | 1,015,458 | |
| | | | | | | |
121
4. Related Parties
Acquisitions of Assets From Predecessor
In December 2004, pursuant to the Contribution Agreement, the Company acquired substantially all of Z-KAT’s tangible assets and a majority of Z-KAT’s CAS technology assets not required for Z-KAT’s retained CAS business, and all of its haptic robotic research and development technology inventory. The Company was granted a limited license to Z-KAT’s CAS and haptic robotic intellectual property portfolio for exclusive use in the field of orthopedics, subject to a prior license to a strategic partner of Z-KAT to use Z-KAT’s CAS intellectual property, but not its haptic robotic intellectual property, in the field of orthopedics (the “Z-KAT License”). The Contribution Agreement, including the Z-KAT License, was made in exchange for approximately 1,410,000 shares of common stock, 1,999,000 shares of Series A redeemable convertible preferred stock, and warrants to purchase 190,000 shares of common stock at an exercise price of $3.00 per share. This acquisition was accounted for based on the Z-KAT carrying value of the assets acquired totaling approximately $652,000 as it was a transaction between entities under common control. The common stock was recorded at par and the remainder of the ascribed value was allocated to the Series A redeemable convertible preferred stock resulting in a discount to the potential redemption value of approximately $1,346,000 which is being accreted over the period from the date of issuance to the redemption date (see further discussion on redemption rights in Note 7).
Pursuant to the December 2004 contribution of the Z-KAT License, the Company obtained the right to manage and maintain the Z-KAT patent portfolio, and assumed the obligation to pay a ratable portion (among all licensees) of all maintenance fees, patent costs and applicable net annual minimum royalties to Z-KAT’s licensors. For the majority of applicable licenses, the Company’s ratable portion for the intellectual property fees, costs and net annual minimum royalties has been 50% since consummation of the Z-KAT License.
In December 2006, the Company entered into an Addendum to the Contribution Agreement (the “Addendum”). Under the Addendum, Z-KAT assigned to MAKO its right to receive required royalty payments from two prior third-party CAS intellectual property licensees; and we assumed the obligation to pay the annual minimum royalty to a third-party CAS licensor due to the importance of maintaining the licensed rights. There was no change in licensed intellectual property rights as a result of the Addendum.
Due From Related Party
At December 31, 2007 and 2006, due from related party represents amounts due from Z-KAT for various transactions.
Note Receivable — Related Party
At December 31, 2007 and 2006, the balance of the Z-KAT Note, with accrued interest, was approximately $70,000 and $126,000, respectively. The Z-KAT Note is being repaid through collections from Z-KAT sales of its products in its retained CAS business. As of December 31, 2007 and 2006, the Company believed that the Z-KAT Note was fully collectible. As of December 31, 2007 and 2006, Z-KAT’s holdings of MAKO common stock on a fully diluted and assumed converted basis were approximately 9.0% and 13.6%, respectively.
122
Employee Loans
During 2006, the Company issued $225,000 in employee loans to certain officers of the Company (the “Employee Loans”). The Employee Loans accrued interest at a rate of 4.0% per annum, compounded annually. The interest was paid biweekly. The Employee Loans and accrued interest were due upon the earlier of one year from the date of the Employee Loan or a liquidation event, as defined. The Employee Loans were fully repaid in April 2007. In May and June 2007, the Company issued $225,000 in employee loans to certain officers of the Company under terms that were substantially similar to the Employee Loans issued in 2006. In August and September 2007, the Company forgave the $225,000 of outstanding loans, including accrued interest, with a charge to the statement of operations.
Restricted Stock and Note Receivable from Related Party
In July 2005 and May 2006, the Company issued a total of 446,287 shares of restricted common stock to its CEO and 49,504 shares of unrestricted common stock to an entity affiliated with the CEO in exchange for promissory notes from the CEO totaling approximately $631,000 (representing the fair value of the shares on the date of issuance) approximately 50% of which was nonrecourse. The promissory notes accrue interest at a rate of 8% per annum, with 25% of the restricted stock vesting immediately and the remainder vesting monthly over 48 months as service is provided. The restricted stock was pledged as collateral against the promissory notes. In March 2007, the Company issued 82,508 shares of restricted common stock to its CEO at a purchase price of $2.48 per share (the estimated fair value at the date of issuance) in exchange for a promissory note of $205,000, 50% of which was nonrecourse and a pledge agreement. The March 2007 restricted stock, pledge agreement and promissory note were issued under terms substantially similar to the July 2005 and May 2006 restricted stock issuances. Because it was unclear as to whether the recourse portion had substance as of the dates of issuance of the restricted stock and the promissory notes, the Company has determined to treat the entire amount of the promissory notes related to the restricted stock as nonrecourse for accounting purposes. A nonrecourse note issued for restricted stock is in substance an option to acquire the stock. Accordingly, the Company recorded compensation expense of approximately $73,000 and $90,000 in the years ended December 31, 2006 and 2005, and the promissory notes and the restricted stock are not recorded in the accompanying financial statements. At December 31, 2006, the balance of the unrecognized promissory notes related to the restricted stock with accrued interest was approximately $699,000. The unrecognized compensation associated with the restricted stock is approximately $172,000 as of December 31, 2006. The compensation expense was determined under the Black-Scholes-Merton model assuming a risk free interest rate of 0.0% (as the interest rate on the promissory notes was greater than the risk free interest rate and the excess was not significant to the Black-Scholes-Merton valuation — risk free interest rate ranging from 4.08% to 4.96% less the stated interest rate of 8% implicit in the promissory notes), a volatility factor ranging from 57.1% to 66.5% and a 6.25 year estimated life.
123
The value of the common stock was initially determined by the Company’s board of directors and was validated as reasonable on a retrospective basis in a March 2007 valuation by an independent valuation firm. As the unrestricted stock does not contain vesting provisions, it is not subject to the pledge against the promissory notes and collectibility is reasonably assured, the Company recorded the portion of the promissory notes associated with the unrestricted stock as a component of stockholders’ deficit (principal of $63,000) in the accompanying financial statements and accrued interest at a rate of 8% per annum as an increase to additional paid-in-capital.
On September 5, 2007, the Company forgave approximately $1,149,000 of outstanding loans, including accrued interest of $113,000, to its CEO. Of this amount, which represents all loans outstanding to the Company’s CEO, $949,000 was associated with the issuances of the restricted and unrestricted stock and $200,000 was associated with the employee loans discussed above. In connection with the forgiveness of the loans, 35,244 shares of common stock were surrendered by the CEO to the Company to pay for the payroll taxes associated with the taxable income from the forgiveness of the loans. The forgiveness of the notes receivable resulted in a modification to the original award. Accordingly, the Company accounted for the modification by determining the amount of the incremental compensation charge to be recorded in accordance with paragraph 51 of SFAS 123(R). The original award, which was accounted for as a stock option, was revalued on the date of modification using the Black-Scholes-Merton model with current inputs for risk-free rate, volatility and market value. This calculated amount was compared to the fair value of the restricted stock award on the date of modification resulting in the incremental charge. Due to the forgiveness of the note, the Company ceased to record the award as a stock option and commenced the recording of the award as a restricted stock award. Accordingly, on the date of modification, the Company recognized the incremental charge for the portion of the vested shares and will record the additional portion related to the unvested shares over the remaining term. The forgiveness resulted in a modification to the original terms of the restricted stock-based award with a charge of approximately $395,000 recorded in the financial statements in September 2007. The remaining unrecognized compensation expense of approximately $533,000 relating to the unvested restricted stock will be recorded in the financial statements over the remaining vesting period, along with the related vested common stock. The compensation expense associated with the modification of the terms of the restricted stock was determined under the Black-Scholes-Merton model assuming a risk free interest rate of 0.0% (as the interest rate on the promissory notes was greater than the risk free interest rate and the excess was not significant to the Black-Scholes-Merton valuation — risk free interest rate of 4.29% less the stated interest rate of 8% implicit in the promissory notes), a volatility factor of 54.07% and an estimated life ranging from 4.10 to 5.80 years. The value of the common stock on the modification date was determined in an August 2007 valuation by an independent valuation firm.
In August 2007, the Company issued 247,524 shares of restricted stock to its CEO at an estimated fair value of $11.12 per share on the date of issuance. The restricted stock will vest over a four-year period.
124
Compensation expense related to the CEO restricted stock was approximately $774,000 for the year ended December 31, 2007, of which $77,000 was incurred in the eight months prior to the modification, $395,000 was incurred due to the modification, and $302,000 was incurred subsequent to the modification.
Outstanding restricted stock was comprised of the following vested and unvested shares as of December 31, 2007.
| | | | | | | | | |
| | | | | | | Weighted
Average | |
| | | Shares | | | Fair Value | |
| Vested shares | | | 313,360 | | | $ | 1.88 | |
| Unvested shares | | | 427,715 | | | | 6.76 | |
| | | | | | | | |
| Total | | | 741,075 | | | $ | 4.70 | |
| | | | | | | |
Outstanding restricted stock was comprised the following vested and unvested shares as of December 31, 2006.
| | | | | | | | | |
| | | | | | | Weighted
Average | |
| | | Shares | | | Fair Value | |
| Vested shares | | | 217,224 | | | $ | 1.27 | |
| Unvested shares | | | 229,063 | | | | 1.27 | |
| | | | | | | | |
| Total | | | 446,287 | | | $ | 1.27 | |
| | | | | | | |
5. Intangible Assets
The Company’s intangible assets are comprised of a purchased patent application and licenses to intellectual property rights, (the “Licenses”). The Licenses are amortized on a straight line basis over their estimated useful lives which range on average from 10 to 19 years. See Note 6 for additional discussion of Licenses.
The following tables present details of MAKO’s intangible assets:
| | | | | | | | | | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| | | | | | | Weighted | | | | | | | Weighted | |
| | | | | | | Average | | | | | | | Average | |
| | | | | | | Amortization | | | | | | | Amortization | |
| | | Amount | | | Period | | | Amount | | | Period | |
| Licenses | | $ | 6,549,421 | | | | 10.0 | | | $ | 6,084,638 | | | | 10.1 | |
| Patent | | | 60,000 | | | | 17.8 | | | | 60,000 | | | | 17.8 | |
| | | | | | | | | | | | | | | |
| | | | 6,609,421 | | | | 10.1 | | | | 6,144,638 | | | | 10.1 | |
| Less: accumulated amortization | | | (1,132,585 | ) | | | | | | | (488,452 | ) | | | | |
| | | | | | | | | | | | | | | |
| Intangible assets, net | | $ | 5,476,836 | | | | | | | $ | 5,656,186 | | | | | |
| | | | | | | | | | | | | | | |
Amortization expense related to intangible assets was approximately $645,000, $470,000 and $18,000 for the years ended December 31, 2007, 2006 and 2005, respectively.
125
The estimated future amortization expense of intangible assets for the next five years as of December 31, 2007 is as follows:
| | | | | |
| 2008 | | $ | 660,000 | |
| 2009 | | | 660,000 | |
| 2010 | | | 660,000 | |
| 2011 | | | 660,000 | |
| 2012 | | | 660,000 | |
| | | | |
| Total | | $ | 3,300,000 | |
| | | | |
6. Commitments and Contingencies
Operating Leases
The Company leases its facility under an operating lease that expires in July 2011. The Company has the option to renew its facility lease for two consecutive three year periods. Rent expense on a straight-line basis was $314,000, $250,000 and $185,000 for the years ended December 31, 2007, 2006 and 2005, respectively. The rent expense for the years ended December 31, 2007, 2006 and 2005 includes the Company’s monthly variable operating costs of the facility.
Future minimum lease commitments under the Company’s operating lease as of December 31, 2007 are approximately as follows:
| | | | | |
| 2008 | | $ | 208,000 | |
| 2009 | | | 215,000 | |
| 2010 | | | 221,000 | |
| 2011 | | | 131,000 | |
| | | | |
| | | $ | 775,000 | |
| | | | |
License and Royalty Agreements
As more fully described in Note 4, in December 2004 and December 2006, respectively, the Company entered into the Z-KAT License and the Addendum whereby MAKO was granted rights to certain intellectual property. The Z-KAT License included sublicenses to third-party intellectual property rights (the “Sublicenses”). The Z-KAT License is fully paid up as to licenses directly to Z-KAT. Under the Sublicenses, the Company is obligated to make ongoing royalty payments ranging from 2% to 5% on the sale of certain products and minimum annual payments totaling $575,000. By their terms, the Z-KAT License and the component Sublicenses generally continue until all of the licensed patents have expired, which, based on the licensed granted patents and presently pending patent applications is currently estimated to be December 2024.
In March 2006, the Company entered into a license agreement with International Business Machines Corporation (“IBM”) for a license, in the Company’s field of business, to IBM’s patent portfolio (the “IBM License”) in exchange for a payment of $2 million upon execution of the agreement (the “Upfront License Fee”) and a deferred payment of $4 million payable upon a change of control, as defined (e.g., IPO, acquisition or change in voting ownership greater than 50.01%) (the “Deferred License Fee”). The IBM License requires royalty payments of 2% of the selling price of each TGS unit.
126
The Upfront License Fee and net present value of the Deferred License Fee are included in intangible assets in the accompanying balance sheets. The net present value of the Deferred License Fee obligation was approximately $3,410,000, net of a discount of $590,000 and was recorded as a long-term debt obligation as the Company believed it was probable at the inception of the agreement that the contingent obligation would become payable. The net present value of the Deferred License Fee was determined using an incremental borrowing rate of 8% and an expected payment date of approximately two years from the effective date of the license agreement. The discount on the debt obligation is being amortized over the estimated term of the Deferred License Fee obligation as interest expense which was approximately $305,000 and $211,000 for the years ended December 31, 2007 and 2006, respectively, in the accompanying statements of operations. See Note 11 for further discussion regarding the Deferred License Fee.
The Company has other license agreements related to current product offerings and research and development projects. Upfront license fees paid for these agreements total approximately $1.1 million. Royalty payments related to these agreements are anticipated to range between 1% and 7% of future sales of the Company’s TGS units, TGS components thereof and/or products. These royalty payments are subject to certain minimum annual royalty payments as shown in the schedule below. The terms of these license agreements continue until the related licensed patents and intellectual property rights expire, which is expected to range between 9 and 17 years.
As of December 31, 2007, future annual minimum royalty payments under the licenses and sublicenses are as follows:
| | | | | |
| 2008 | | $ | 631,000 | |
| 2009 | | | 660,000 | |
| 2010 | | | 690,000 | |
| 2011 | | | 630,000 | |
| 2012 | | | 630,000 | |
| Thereafter | | | 817,000 | |
| | | | |
| | | $ | 4,058,000 | |
Contingencies
In November 2007, the Company received a letter from counsel to SensAble Technologies, Inc., a licensor to the Company, alleging that the Company infringed certain of its patents and breached a confidentiality provision in the SensAble License Agreement. In the letter, SensAble alleged, among other things, that the Company exceeded the scope of its licensed field of computer-assisted surgery by using the technology for, among other things, pre-operative planning and post-operative follow-up. SensAble also alleged that the Company infringed one or more claims in five U.S. patents that are not among the patents licensed to the Company pursuant to the SensAble Sublicense Agreement.
127
The Company investigated SensAble’s allegations, and, based on the opinion of counsel, it believes that if SensAble initiates a lawsuit against the Company, a court should find that its TGS does not infringe any of the SensAble patents identified in the November 2007 letter. The Company communicated its belief to SensAble. SensAble has not commenced any legal action against the Company, but may do so in the future. The letter from counsel to SensAble stated that unless the Company, among other things, ceases and desists from alleged infringement of SensAble’s patents or pay additional licensing fees, including a proposed licensing fee of $30 million for additional patents not included in the SensAble License Agreement, SensAble intends to bring a lawsuit against the Company. The Company intends to vigorously defend itself against these allegations in the event of a lawsuit. The Company cannot predict the outcome of this matter at this time.
The Company has been a party to other legal contingencies or claims arising in the normal course of business, none of which the Company believes is material to its financial position, results of operations or cash flows.
7. Redeemable Convertible Preferred Stock
As of December 31, 2007, 2006 and 2005, the Company had redeemable convertible preferred stock outstanding, as follows:
| | | | | | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| Authorized shares | | | 40,000,000 | | | | 40,000,000 | | | | 40,000,000 | |
| | | | | | | | | | |
| Outstanding shares: | | | | | | | | | | | | |
| Series A | | | 4,498,745 | | | | 4,498,745 | | | | 4,498,745 | |
| Series B | | | 15,151,516 | | | | 15,151,516 | | | | 15,151,516 | |
| Series C | | | 13,513,514 | | | | — | | | | — | |
| | | | | | | | | | |
| Total outstanding shares | | | 33,163,775 | | | | 19,650,261 | | | | 19,650,261 | |
| | | | | | | | | | |
| Liquidation amount: | | | | | | | | | | | | |
| Series A | | $ | 4,498,745 | | | $ | 4,498,745 | | | $ | 4,498,745 | |
| Series B | | | 20,000,000 | | | | 20,000,000 | | | | 20,000,000 | |
| Series C | | | 30,000,000 | | | | — | | | | — | |
| | | | | | | | | | |
| Total liquidation amount | | $ | 54,498,745 | | | $ | 24,498,745 | | | $ | 24,498,745 | |
| | | | | | | | | | |
| Cash proceeds, net of issuance costs | | | | | | | | | | | | |
| Series A | | $ | — | | | | — | | | | 145,620 | |
| Series B | | | — | | | | — | | | | 19,881,300 | |
| Series C | | | 29,907,767 | | | | — | | | | — | |
| | | | | | | | | | |
| Total proceeds, net of issuance costs | | $ | 29,907,767 | | | $ | — | | | $ | 20,026,920 | |
| | | | | | | | | | |
| Cumulative undeclared accrued dividends | | | | | | | | | | | | |
| Series A | | $ | 958,680 | | | $ | 681,909 | | | $ | 334,590 | |
| Series B | | | 2,819,736 | | | | 1,822,901 | | | | 561,193 | |
| Series C | | | 1,179,760 | | | | — | | | | — | |
| | | | | | | | | | |
| Total cumulative undeclared accrued |
| Dividends | | $ | 4,958,176 | | | $ | 2,504,810 | | | $ | 895,783 | |
| | | | | | | | | | |
128
Dividend Rights
The holders of the Company’s Series C, Series B and Series A redeemable convertible preferred stock were entitled to receive cash dividends in preference to the holders of the Company’s common stock, at the rate of $0.13 per share, $0.08 per share and $0.07 per share, respectively, per year of the outstanding original issue price amounts. Such dividends were cumulative whether or not declared by the Company’s Board of Directors. The Company recorded the cumulative undeclared accrued dividends in the accompanying statements of redeemable convertible preferred stock and stockholders’ deficit with a charge to additional paid-in capital until it was depleted to zero and the excess charged to accumulated deficit. As discussed under Conversion Rights, the cumulative undeclared accrued dividends were not included in the conversion price upon a qualified public offering unless declared and unpaid. No dividends were declared or paid. See Note 11 for additional discussion regarding dividends and conversion of the Company’s redeemable convertible preferred stock.
Liquidation Rights
Upon any liquidation, dissolution or winding up of the Company, the holders of Series C redeemable convertible preferred stock were entitled to an amount equal to the liquidation amount of Series C redeemable convertible preferred stock (which was $2.22 per share) plus an amount equal to any dividends declared but unpaid thereon, if any, in preference to any distribution to holders of Series B redeemable convertible preferred stock, Series A redeemable convertible preferred stock or common stock. If the assets of the Company were insufficient to pay the Series C liquidation preference amounts, the available assets would have been distributed to the holders of Series C redeemable convertible preferred stock ratably in proportion to the preference amounts they would otherwise have been entitled to receive.
After the holders of Series C redeemable convertible preferred stock have been paid the amounts to which they shall be entitled, the holders of Series B redeemable convertible preferred stock shall be entitled to an amount equal to the liquidation amount of Series B redeemable convertible preferred stock (which was $1.32 per share) plus an amount equal to any dividends declared but unpaid thereon, if any, in preference to any distribution to Series A or common stock. If the assets of the Company were insufficient to pay the Series B redeemable convertible preferred stock liquidation preference amounts, the available assets would have been distributed to the holders of Series B redeemable convertible preferred stock ratably in proportion to the preference amounts they would otherwise have been entitled to receive.
After the holders of Series C redeemable convertible preferred stock and Series B redeemable convertible preferred stock would have been paid the amounts to which they were entitled, the holders of Series A redeemable convertible preferred stock were entitled to an amount equal to the liquidation amount of Series A redeemable convertible preferred stock (which was $1.00 per share) plus an amount equal to any dividends declared but unpaid thereon, if any, in preference to any distribution to common stock. If the assets of the Company were insufficient to pay the Series A redeemable convertible preferred stock liquidation preference amounts, the available assets would have been distributed to the holders of Series A redeemable convertible preferred stock ratably in proportion to the preference amounts they would otherwise have been entitled to receive.
129
After payment of the liquidation preference amounts, any remaining assets of the Company were to be distributed ratably to the holders of the Company’s common stock. The treatment of any particular transaction or series of related transactions as a liquidation event may have been waived upon the affirmative vote or written consent of the holders of at least seventy percent (70%), sixty-six and two-thirds percent (66 2/3%) and sixty percent (60%) of the outstanding Series C, Series B and Series A redeemable convertible preferred stockholders, respectively, each voting as a separate class (the “Respective Preferred Majority”).
Voting Rights
Each holder of preferred stock was entitled to the number of votes equal to the number of shares of common stock into which such holder’s shares of preferred stock could be converted as of the record date. The holders of shares of the preferred stock were entitled to vote on all matters on which the holders of common stock were entitled to vote. The holders of preferred stock were entitled to protective provisions that require the affirmative vote of the holders of at least a majority of the outstanding preferred stock voting as separate classes for certain actions of the Company.
Conversion Rights
At the option of the holder thereof, each share of preferred stock was convertible, at any time or from time to time, into fully paid and non-assessable shares of common stock determined by dividing the applicable original issue price for such series, plus declared and unpaid dividends thereon, by the applicable conversion price for such series.
Each share of preferred stock shall automatically be converted into shares of common stock immediately upon the earlier of (a) anytime upon the affirmative election of the holders of at least the Respective Preferred Majority of the then-outstanding shares of the preferred stock, or (b) immediately upon the closing of a firmly underwritten public offering pursuant to an effective registration statement filed under the Securities Act of 1933, as amended, the public offering price of which was not less than $13.45 per share (as adjusted for any stock splits, stock dividends, combinations, subdivisions or recapitalization) and which resulted in proceeds to the Company of at least $25.0 million. Upon conversion, accrued but undeclared dividends would be reversed and not paid. As of December 31, 2007 and 2006, each share of preferred stock would have converted into one share of common stock.
In September 2007, the Board of Directors authorized management to file a registration statement with the Securities and Exchange Commission for the Company to sell shares of its common stock to the public. See Note 11 for further discussion regarding the IPO.
Redemption Rights
The preferred stock was subject to mandatory redemption, at the option of the holders, on any date after July 1, 2010 upon the affirmative vote of the Respective Preferred Majority of the preferred stockholders. The redemption amount was equal to the original issue price per share plus accrued and unpaid dividends at the redemption date. The Company was accreting to additional paid-in-capital the carrying value (gross proceeds, net of stock issuance costs) of the preferred stock up to its redemption value over the period from the original issue price. During the years ended December 31, 2007, 2006 and 2005, accretion totaled approximately $301,000, $267,000 and $257,000, respectively. As of December 31, 2007, 2006 and 2005, the unaccreted discount was approximately $876,000, $1,086,000 and $1,354,000, respectively.
130
The Company accounted for the preferred stock in accordance with the SEC’sAccounting Series ReleaseNo. 268 and EITF D-98,Classification and Measurement of Redeemable Securities. The redeemable convertible preferred stock had a redemption feature that was outside the control of the Company and, accordingly, was classified outside stockholders’ deficit in the accompanying balance sheets and statements of redeemable convertible preferred stock and stockholders’ deficit.
8. Stockholders’ Deficit
Common Stock
As of December 31, 2006, the Company was authorized to issue 50,000,000 shares of $0.001 par value common stock. As of December 31, 2007, the Company was authorized to issue 60,000,000 shares of $0.001 par value common stock. Common stockholders are entitled to dividends as and when declared by the Board of Directors, subject to the rights of holders of all classes of stock outstanding having priority rights as to dividends. There have been no dividends declared to date. The holder of each share of common stock is entitled to one vote.
Under the Company’s 2004 Stock Incentive Plan (see further discussion under Stock Option Plan) (the “2004 Plan”), the Company has issued shares of common stock under restricted stock purchase agreements to its CEO (see further discussion in Note 4).
In December 2004, the Company issued 189,768 shares of restricted common stock to certain consultants (the “Consultant Restricted Stock”). The Consultant Restricted Stock vests 25% at the date of issuance, 25% on the Company’s closing of its Series B preferred stock financing, 25% upon a certain development milestone and 25% upon a clinical milestone. Upon vesting, the Company recorded consulting expense equal to the estimated fair value of the Company’s common stock on the date of vesting. Consulting expense associated with the vesting of the Consultant Restricted Stock was approximately, $0, $0, and $60,000 for the years ended December 31, 2007, 2006, and 2005, respectively. As of December 31, 2007 and 2006, 94,884 shares of the Consultant Restricted Stock were vested.
As of December 31, 2007, the Company had reserved shares of common stock for the conversion of redeemable convertible preferred stock, the exercise of warrants and the issuance of options granted under the 2004 Plan as follows:
| | | | | |
| Preferred stock | | | 10,945,080 | |
| Warrants to purchase common stock | | | 462,716 | |
| Stock option plans | | | 2,830,033 | |
| | | | |
| | | | 14,237,829 | |
| | | | |
131
As of December 31, 2006, the Company had reserved shares of common stock for the conversion of redeemable convertible preferred stock, the exercise of warrants and the issuance of options granted under the Company’s stock option plans as follows:
| | | | | |
| Preferred stock | | | 6,485,199 | |
| Warrants to purchase common stock | | | 462,716 | |
| Stock option plans | | | 1,856,435 | |
| | | | |
| | | | 8,804,350 | |
| | | | |
Stock Option Plans
In December 2004, the Company’s stockholders approved the 2004 Plan. Under the 2004 Plan, the Board of Directors is authorized to grant restricted common stock and options to purchase shares of common stock to employees, directors and consultants. See Note 11 for further discussion regarding the Company’s incentive plans.
Only employees are eligible to receive incentive stock options. Non-employees may be granted non-qualified options. The Board of Directors has the authority to set the exercise price of all options granted, subject to the exercise price of incentive stock options being no less than 100% of the estimated fair value, as determined by the Board of Directors, of a share of common stock on the date of grant; and no less than 85% of the estimated fair value for non-qualified stock options, except for an employee or non-employee with options who owns more than 10% of the voting power of all classes of stock of the Company, in which case the exercise price shall be no less than 110% of the fair market value per share on the grant date. Options become exercisable as determined by the Board of Directors.
Generally, the Company’s outstanding options vest over four years. However, certain options granted in 2004 vested on the date of grant. Continued vesting typically terminates when the employment or consulting relationship ends.
The maximum term of the options granted to persons who owned at least 10% of the voting power of all outstanding stock on the date of grant is 5 years. The maximum term of all other options is 10 years.
132
Activity under the 2004 Plan is summarized as follows:
| | | | | | | | | | | | | |
| | | Outstanding Options | |
| | | | | | | | | | | Weighted | |
| | | Shares/Options | | | | | | | Average | |
| | | Available | | | Number of | | | Exercise | |
| | | For Grant | | | Options | | | Price | |
Balance at December 31, 2004 | | | 241,406 | | | | 360,904 | | | $ | 0.67 | |
| Shares reserved | | | 1,254,125 | | | | — | | | $ | — | |
| Restricted stock issued | | | (413,283 | ) | | | — | | | $ | — | |
| Options granted | | | (284,900 | ) | | | 284,900 | | | $ | 1.24 | |
| Options forfeited | | | 16,237 | | | | (16,237 | ) | | $ | 0.76 | |
| | | | | | | | | | | |
Balance at December 31, 2005 | | | 813,585 | | | | 629,567 | | | $ | 0.91 | |
| Restricted stock issued | | | (82,508 | ) | | | — | | | $ | — | |
| Options granted | | | (344,884 | ) | | | 344,884 | | | $ | 1.27 | |
| Options exercised | | | — | | | | (1,650 | ) | | $ | 0.67 | |
| Options forfeited | | | 24,851 | | | | (24,851 | ) | | $ | 0.85 | |
| | | | | | | | | | | |
Balance at December 31, 2006 | | | 411,044 | | | | 947,950 | | | $ | 1.06 | |
| Shares reserved | | | 973,598 | | | | — | | | $ | — | |
| Restricted stock issued | | | (330,033 | ) | | | — | | | $ | — | |
| Options granted | | | (1,001,695 | ) | | | 1,001,695 | | | $ | 8.30 | |
| Options exercised | | | — | | | | (1,306 | ) | | $ | 1.27 | |
| Options forfeited | | | 31,814 | | | | (31,814 | ) | | $ | 3.51 | |
| | | | | | | | | | | |
Balance at December 31, 2007 | | | 84,728 | | | | 1,916,525 | | | $ | 4.81 | |
| | | | | | | | | | | |
Included in the 2007 option grants in the table above are approximately 462,000 options granted at an exercise price of $11.12 per share that are subject to performance conditions based on the achievement of certain future performance metrics. Upon satisfaction of the performance condition, the options will vest ratably quarterly over a period of four years. Through December 31, 2007, the Company began recognizing compensation expense on approximately 296,000 performance options as both the terms of the performance conditions were established and it was probable that the performance condition will be satisfied. Once the performance conditions are established and it is probable the performance conditions will be met, the Company will begin recognizing compensation expense on the remaining performance options. If the performance conditions are not met, no compensation cost is recognized and any previously recognized compensation cost is reversed.
In addition to the option grants in the table above, in August 2007, the Company committed to grant the CEO 198,019 incentive stock options upon the completion of an IPO by March 31, 2008 from a pool of newly reserved options to be created in 2008. See Note 11 for further discussion regarding the option grant to the CEO and the IPO.
133
The options outstanding and exercisable, by exercise price, at December 31, 2007 were as follows:
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Options Outstanding | | | Options Exercisable | |
| | | | | | | Weighted | | | | | | | | | | | Weighted | | | | |
| | | | | | | Average | | | Weighted | | | | | | | Average | | | Weighted | |
| | | Number | | | Remaining | | | Average | | | Number | | | Remaining | | | Average | |
| | | Of | | | Contractual | | | Exercise | | | Of | | | Contractual | | | Exercise | |
| | | Options | | | Life (Years) | | | Price | | | Options | | | Life (Years) | | | Price | |
Exercise Price: | | | | | | | | | | | | | | | | | | | | | | | | |
| $0.67 | | | 345,386 | | | | 6.97 | | | $ | 0.67 | | | | 342,526 | | | | 6.96 | | | $ | 0.67 | |
| $1.27 | | | 582,286 | | | | 7.99 | | | $ | 1.27 | | | | 298,305 | | | | 7.92 | | | $ | 1.27 | |
| $1.97 | | | 3,547 | | | | 9.00 | | | $ | 1.97 | | | | 371 | | | | 8.97 | | | $ | 1.97 | |
| $2.48 | | | 313,344 | | | | 9.23 | | | $ | 2.48 | | | | — | | | | — | | | $ | — | |
| $9.67 | | | 9,900 | | | | 9.43 | | | $ | 9.67 | | | | — | | | | — | | | $ | — | |
| $11.12 | | | 662,062 | | | | 9.65 | | | $ | 11.12 | | | | 13,820 | | | | 9.65 | | | $ | 11.12 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | 1,916,525 | | | | 8.59 | | | $ | 4.81 | | | | 655,022 | | | | 7.46 | | | $ | 1.16 | |
| | | | | | | | | | | | | | | | | | | | | | | |
The following is a summary of changes in unvested options granted under the 2004 Plan for the year ended December 31, 2007:
| | | | | | | | | |
| | | | | | | Weighted Average | |
| | | Number of | | | Grant Date Fair | |
| | | Options | | | Value | |
| Nonvested, December 31, 2006 | | | 515,615 | | | $ | 0.82 | |
| Granted | | | 1,001,695 | | | $ | 4.54 | |
| Vested | | | (222,687 | ) | | $ | 1.17 | |
| Exercised | | | (1,306 | ) | | $ | 0.82 | |
| Forfeited | | | (31,814 | ) | | $ | 2.12 | |
| | | | | | | |
| Nonvested, December 31, 2007 | | | 1,261,503 | | | $ | 3.68 | |
| | | | | | | |
The weighted average fair values of options granted were $4.54, $0.82, and $0.79 for the years ended December 31, 2007, 2006 and 2005, respectively. The total intrinsic value of exercisable options at December 31, 2007 is approximately $5,788,000. The total intrinsic value of outstanding options at December 31, 2007 is approximately $8,289,000.
The total fair value of shares vested was approximately $260,000, $79,000, and $0 during the years ended December 31, 2007, 2006 and 2005, respectively. Included in the 2007 option grants in the table above are approximately 166,000 options issued at an exercise price of $11.12 per share which are subject to performance conditions for which the performance condition had not been established and/or it was not probable that the performance condition would be satisfied as of December 31, 2007. The weighted average fair value of these options will be determined and compensation expense will begin to be recorded once these conditions are satisfied.
The Company utilized the option vesting period for recognizing compensation expense. For the options that vest upon grant, the Company records the entire related compensation expense immediately upon the date of grant. For all other options, the Company records compensation expense on a straight-line basis over the vesting period. As of December 31, 2007, there was total unrecognized compensation cost of approximately $3,926,000, adjusted for estimated forfeitures, related to non-vested stock-based payments granted to the Company’s employees and non-employee directors. Total unrecognized compensation cost will be adjusted for future changes in estimated forfeitures, and is expected to be recognized over a remaining weighted average period of 2.45 years as of December 31, 2007.
134
The Company uses the Black-Scholes-Merton pricing model to determine the fair value of stock options. The determination of the fair value of stock-based payment awards on the date of grant using a pricing model is affected by our stock price as well as assumptions regarding a number of complex and subjective variables. These variables include our expected stock price volatility over the term of the awards, actual and projected employee stock option exercise behaviors, risk-free interest rates and expected dividends.
The estimated grant date fair values of the employee stock options were calculated using the Black-Scholes-Merton valuation model, based on the following assumptions:
| | | | | | | | | | | | | |
| | | 2007 | | | 2006 | | | 2005 | |
| Risk-free interest rate | | | 4.50% - 5.14% | | | | 4.29% - 5.23% | | | | 3.83% - 4.59% | |
| Expected life | | 6.25 years | | 6.25 years | | 6.25 years |
| Expected dividends | | | — | | | | — | | | | — | |
| Expected volatility | | | 56.24% - 60.00% | | | | 59.23% - 64 .27% | | | | 65.46% - 68.31% | |
Risk-Free Interest Rate.The risk-free rate is based on U.S. Treasury zero-coupon issues with remaining terms similar to the expected term on the options.
Weighted-Average Expected Term.Under the Plan, the expected term of options granted is determined using the average period the stock options are expected to remain outstanding and is based on the options vesting term, contractual terms and historical exercise and vesting information used to develop reasonable expectations about future exercise patterns and post-vesting employment termination behavior.
Dividend Yield.The Company has never declared or paid any cash dividends and does not plan to pay cash dividends in the foreseeable future, and, therefore, used an expected dividend yield of zero in the valuation model.
Volatility.Since the Company was a private entity until February 2008 with no historical data regarding the volatility of its common stock, the expected volatility used for the years ended December 31, 2007, 2006 and 2005, is based on volatility of similar entities, referred to as “guideline” companies. In evaluating similarity, the Company considered factors such as industry, stage of life cycle and size.
Forfeitures.SFAS No. 123(R) also requires the Company to estimate forfeitures at the time of grant, and revise those estimates in subsequent periods if actual forfeitures differ from those estimates. The Company uses historical data to estimate pre-vesting option forfeitures and records stock-based compensation expense only for those awards that are expected to vest. All stock-based payment awards are amortized on a straight-line basis over the requisite service periods of the awards, which are generally the vesting periods. If the Company’s actual forfeiture rate is materially different from its estimate, the stock-based compensation expense could be significantly different from what the Company has recorded in the accompanying periods.
135
Warrants
In December 2004, in connection with the issuance of the Series A redeemable convertible preferred stock to third parties, the Company issued at the purchase price of $0.03 per share warrants to purchase 272,259 shares of common stock. In addition, the Company issued warrants to Z-KAT to purchase 190,457 shares of common stock as previously discussed in Note 4. The warrants are immediately exercisable at an exercise price of $3.00 per share, with the exercise period expiring in December 2014. The value of such warrants at issuance totaled approximately $99,000 and was recorded as a discount to the Series A preferred stock. The warrants were valued using the Black-Scholes-Merton model with a risk free interest rate of 3.9%, a term of ten years and a 69.55% volatility factor. The fair value of the underlying shares was estimated by management. As of December 31, 2007 and 2006, all the warrants were outstanding and exercisable, and none have been exercised.
9. Income taxes
The provision for income taxes is as follows:
| | | | | | | | | | | | | |
| | | Years Ended | |
| | | December 31, | | | December 31, | | | December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| Current income taxes: | | | | | | | | | | | | |
| Federal | | $ | — | | | $ | — | | | $ | — | |
| State | | | — | | | | — | | | | — | |
| | | | | | | | | | |
| Total current income taxes | | | — | | | | — | | | | — | |
| Deferred income taxes | | | (7,834,890 | ) | | | (4,011,314 | ) | | | (1,933,658 | ) |
| Less change in valuation allowance | | | 7,834,890 | | | | 4,011,314 | | | | 1,933,658 | |
| | | | | | | | | | |
| Provision for income taxes | | $ | — | | | $ | — | | | $ | — | |
| | | | | | | | | | |
The Company accounts for income taxes under SFAS 109. Deferred income taxes and liabilities are determined based upon differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse.
No current or deferred income taxes were recorded for the years ended December 31, 2007, 2006 and 2005, as the Company’s income tax benefits were fully offset by a corresponding increase to the valuation allowance against its net deferred income tax assets.
At December 31, 2007, 2006 and 2005, the Company had federal and state net operating loss carryforwards of approximately $32,900,000, $14,500,000 and $5,900,000, respectively, available to offset future taxable income. These net operating loss carryforwards will expire in varying amounts from 2024 through 2027.
136
The Tax Reform Act of 1986 limits the annual utilization of net operating loss and tax credit carryforwards, following an ownership change of the Company. Should the Company undergo such an ownership change, it is probable that there will be limitations placed on the amount of net operating loss carryforwards available for use in future years.
Deferred income taxes reflect the net tax effect of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s net deferred income taxes are as follows:
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| Current deferred income tax assets: | | | | | | | | |
| Deferred revenue | | $ | 19,290 | | | $ | 11,253 | |
| Accrued bonus | | | 4,946 | | | | 190,468 | |
| | | | | | | |
| Total current deferred income tax assets | | | 24,236 | | | | 201,721 | |
| | | | | | | | | |
| Noncurrent deferred income tax assets: | | | | | | | | |
| Net operating loss carryforwards | | | 12,678,400 | | | | 5,575,775 | |
| Deferred revenue | | | 1,277,151 | | | | 270,025 | |
| Amortization | | | 229,685 | | | | 116,371 | |
| Other | | | 326,443 | | | | 202,939 | |
| | | | | | | |
| Total noncurrent deferred income tax assets | | | 14,511,679 | | | | 6,165,110 | |
| | | | | | | | | |
| Noncurrent deferred income tax liabilities: | | | | | | | | |
| Deferred costs | | | (357,336 | ) | | | (81,142 | ) |
| Other deferred income tax liabilities | | | (58,000 | ) | | | — | |
| | | | | | | |
| Total noncurrent deferred income tax liabilities | | | (415,336 | ) | | | (81,142 | ) |
| | | | | | | | | |
| Less valuation allowance | | | (14,120,579 | ) | | | (6,285,689 | ) |
| | | | | | | |
| | | | | | | | | |
| Total deferred income tax assets, net | | $ | — | | | $ | — | |
| | | | | | | |
Due to uncertainty surrounding realization of the deferred income tax assets in future periods, the Company has recorded a 100% valuation allowance against its net deferred tax assets. If it is determined in the future that it is more likely than not that the deferred income tax assets are realizable, the valuation allowance will be reduced.
137
The reconciliation of the income tax provision computed at the U.S. federal statutory rate to income tax provision is as follows:
| | | | | | | | | | | | | |
| | | Years Ended | |
| | | December 31, | | | December 31, | | | December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| Tax at U.S. statutory rate | | | (35.00 | )% | | | (35.00 | )% | | | (35.00 | )% |
| State taxes, net of federal impact | | | (3.55 | )% | | | (3.50 | )% | | | (3.48 | )% |
| Non-deductible items | | | 0.28 | % | | | 0.72 | % | | | 0.91 | % |
| Change in valuation allowance | | | 37.93 | % | | | 37.78 | % | | | 37.57 | % |
| Other, net | | | 0.34 | % | | | 0.00 | % | | | 0.00 | % |
| | | | | | | | | | |
| Effective income tax rate | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % |
| | | | | | | | | | |
In July 2006, the FASB issued Interpretation No. 48 (FIN 48),Accounting for Uncertainty in Income Taxes. FIN 48 clarifies the accounting for uncertainty in income taxes by prescribing the recognition threshold a tax position is required to meet before being recognized in the financial statements. It also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure, and transition. The Company adopted the provisions of FIN 48 effective January 1, 2007. At the date of adoption of FIN 48, the Company had no unrecognized tax benefits and expected no significant changes in unrecognized tax benefits in the next 12 months. The adoption of this statement did not result in a cumulative accounting adjustment and did not impact the financial position, results of operations or cash flows. In accordance with FIN 48, paragraph 19, the Company has decided to classify any interest and penalties as a component of tax expense. To date, there have been no interest or penalties charged to the Company in relation to the underpayment of income taxes. The Company’s primary tax jurisdictions are the United States, Florida, California, Rhode Island, Ohio, New York, Georgia, Texas, Maryland, Oklahoma and Tennessee. The tax years from 2004 through 2007 remain open and are subject to examination by the appropriate governmental agencies.
138
10. Selected Quarterly Data (Unaudited)
| | | | | | | | | | | | | | | | | |
| | | 2007 | |
| | | Q1 | | | Q2 | | | Q3 | | | Q4 | |
| Revenue | | $ | 99,769 | | | $ | 106,174 | | | $ | 149,439 | | | $ | 415,980 | |
| Gross profit (loss) | | | 64,416 | | | | 58,851 | | | | (59,314 | ) | | | 124,495 | |
| Loss from operations | | | (3,655,543 | ) | | | (4,255,027 | ) | | | (5,981,487 | ) | | | (7,527,869 | ) |
| Net loss | | | (3,495,674 | ) | | | (4,014,966 | ) | | | (5,750,396 | ) | | | (7,397,218 | ) |
| Net loss attributable to common stockholders | | | (4,250,771 | ) | | | (4,968,500 | ) | | | (6,718,546 | ) | | | (8,380,211 | ) |
| Net loss per share - - basic and diluted attributable to common stockholders | | | (2.73 | ) | | | (3.19 | ) | | | (4.12 | ) | | | (4.53 | ) |
| | | | | | | | | | | | | | | | | |
| | | 2006 | |
| | | Q1 | | | Q2 | | | Q3 | | | Q4 | |
| Revenue | | $ | — | | | $ | 4,665 | | | $ | 30,166 | | | $ | 27,740 | |
| Gross profit (loss) | | | — | | | | 3,645 | | | | 2,386 | | | | (20,007 | ) |
| Loss from operations | | | (1,648,869 | ) | | | (2,381,660 | ) | | | (2,860,419 | ) | | | (3,982,248 | ) |
| Net loss | | | (1,486,139 | ) | | | (2,320,806 | ) | | | (2,816,136 | ) | | | (3,993,756 | ) |
| Net loss attributable to common stockholders | | | (1,944,296 | ) | | | (2,786,175 | ) | | | (3,288,826 | ) | | | (4,473,886 | ) |
| Net loss per share - - basic and diluted attributable to common stockholders | | | (1.25 | ) | | | (1.79 | ) | | | (2.11 | ) | | | (2.88 | ) |
11. Subsequent Events
In January 2008, the Company’s Board of Directors and its shareholders approved a one for 3.03 reverse split of its issued and outstanding common stock effected on February 8, 2008. Such reverse stock split has been retroactively reflected in the financial statements and notes thereto. As a result of the reverse split of the Company’s common stock, each share of the Company’s redeemable convertible preferred stock was convertible into 0.330033 of a share of common stock.
In January 2008, the Company’s Board of Directors and stockholders approved the MAKO Surgical Corp. 2008 Omnibus Incentive Plan (the “2008 Omnibus Incentive Plan”). The 2008 Omnibus Incentive Plan became effective upon the consummation of the Company’s IPO discussed below and will expire January 9, 2018 unless earlier terminated by the Board of Directors. The aggregate number of shares of the Company’s common stock that may be issued initially pursuant to stock awards under the 2008 Omnibus Incentive Plan is 1,084,703 shares. Awards under the plan may be made in the form of: stock options, which may be either incentive stock options or non-qualified stock options; stock appreciation rights; restricted stock; restricted stock units; dividend equivalent rights; performance shares; performance units; cash-based awards; other stock-based awards, including unrestricted shares; and any combination of the foregoing.
139
In January 2008, the Company’s Board of Directors and stockholders approved the MAKO Surgical Corp. 2008 Employee Stock Purchase Plan (the “2008 Employee Stock Purchase Plan”). The 2008 Employee Stock Purchase Plan became effective upon the consummation of the Company’s IPO discussed below. The 2008 Employee Stock Purchase Plan authorizes the issuance of 625,000 shares of the Company’s common stock for purchase by eligible employees of the Company or any of its participating affiliates. The shares of common stock issuable under the 2008 Employee Stock Purchase Plan may be authorized but unissued shares, treasury shares or shares purchased on the open market. The purchase price for an offering period may not be less than 85% of the fair market value of the Company’s common stock on the first trading day of the offering period or the day on which the shares are purchased, whichever is lower.
In February 2008, the Company completed its IPO of common stock, issuing a total of 5.1 million shares at an issue price of $10.00 per share, for proceeds to the Company, before expenses, of $51.0 million. As discussed in Note 8, upon completion of the IPO, the Company granted its CEO 198,019 incentive stock options. The options vest ratably quarterly over a four-year period starting on the grant date. The exercise price of the options equaled the fair value of the Company’s common stock on the grant date.
In conjunction with the completion of the Company’s IPO in February 2008, all of the Company’s outstanding Series A redeemable convertible preferred stock, Series B redeemable convertible preferred stock and Series C redeemable convertible preferred stock were converted into 10,945,080 shares of common stock, adjusted for the February 2008 reverse stock split, which was approved by the Company’s Board of Directors and the affirmative election of the holders of at least the Respective Preferred Majority of the then-outstanding shares of the preferred stock. In connection therewith, all remaining preferred stock discounts and accrued dividends were reclassified to additional paid-in capital.
In February 2008, the Company paid the $4 million Deferred License Fee due to IBM (as discussed in Note 6) upon completion of the Company’s IPO.
140
| |
| ITEM 9.CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None
ITEM 9A(T). CONTROLS AND PROCEDURES
In accordance with Rule 13a-15(b) of the Securities Exchange Act of 1934, or the Exchange Act, our management evaluated, with the participation of our chief executive officer and chief financial officer, or the Certifying Officers, the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rule 13a-15(e) under the Exchange Act) as of December 31, 2007. Based upon their evaluation of these disclosure controls and procedures, our Certifying Officers concluded that the disclosure controls and procedures were effective as of December 31, 2007 to provide reasonable assurance that information required to be disclosed by us in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time period specified in the SEC rules and forms, and to provide reasonable assurance that information required to be disclosed by us in the reports we file or submit under the Exchange Act is accumulated and communicated to our management, including our principal executive and principal financial officers, as appropriate, to allow timely decisions regarding required disclosure.
We believe that a controls system, no matter how well designed and operated, is based in part upon certain assumptions about the likelihood of future events, and therefore can only provide reasonable, not absolute, assurance that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected.
This annual report does not include a report of management’s assessment regarding internal control over financial reporting or a report of our registered public accounting firm due to a transition period established by rules of the SEC for newly public companies.
ITEM 9B. OTHER INFORMATION
None
141
PART III.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item will be contained in our definitive proxy statement to be filed in connection with our 2008 annual meeting of stockholders and is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item will be contained in our definitive proxy statement to be filed in connection with our 2008 annual meeting of stockholders and is incorporated herein by reference.
| | |
| ITEM 12. | | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this item will be contained in our definitive proxy statement to be filed in connection with our 2008 annual meeting of stockholders and is incorporated herein by reference.
| |
| ITEM 13. | CERTAIN RELATIONSHIPS, RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE |
The information required by this item will be contained in our definitive proxy statement to be filed in connection with our 2008 annual meeting of stockholders and is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item will be contained in our definitive proxy statement to be filed in connection with our 2008 annual meeting of stockholders and is incorporated herein by reference.
142
PART IV
| | |
| ITEM 15. | | EXHIBITS, FINANCIAL STATEMENTS and FINANCIAL STATEMENT SCHEDULES |
(a) The following documents are filed as a part of this Annual Report on Form 10-K:
1. Financial Statements
See Item 8, Financial Statements and Supplementary Data,Index to Financial Statements.
2. Financial Statement Schedules
No financial statement schedules are provided because the information called for is not required or is shown either in the financial statements or the notes thereto.
(b) Exhibits
| | | |
| Exhibit | | |
| No. | | Description |
| | | |
| 3.1 | | Third Amended and Restated Certificate of Incorporation of the Registrant, dated February 20, 2008 (1) |
| | | |
| 3.2 | | Third Amended and Restated Bylaws of the Registrant effective as of February 20, 2008 (1) |
| | | |
| 4.1 | | Second Amended and Restated Registration Rights Agreement, dated February 6, 2007, between the Registrant and certain of its stockholders (2) |
| | | |
| 10.1 | | Form of Indemnity Agreement for Directors and Executive Officers (2) |
| | | |
| 10.2+ | | 2004 Stock Incentive Plan and forms of agreements related thereto (2) |
| | | |
| 10.3+ | | 2008 Omnibus Incentive Plan (2) |
| | | |
| 10.4+ | | 2008 Employee Stock Purchase Plan and forms of agreements related thereto (2) |
| | | |
| 10.5+ | | Amended Employment Agreement, dated as of November 12, 2007, by and between Registrant and Maurice R. Ferré, M.D (2) |
| | | |
| 10.6+ | | Employment Agreement, dated as of January 1, 2005, by and between Registrant and Fritz L. LaPorte (2) |
| | | |
| 10.7+ | | Amendment to Employment Agreement, dated as of February 5, 2007, by and between Registrant and Fritz L. LaPorte (2) |
| | | |
| 10.8+ | | Employment Agreement, dated as of January 1, 2005, by and between Registrant and Rony Abovitz (2) |
| | | |
| 10.9+ | | Amendment to Employment Agreement, dated as of February 5, 2007, by and between Registrant and Rony Abovitz (2) |
143
| | | |
| Exhibit | | |
| No. | | Description |
| | | |
| 10.10+ | | Employment Agreement, dated as of January 1, 2005, by and between Registrant and Menashe R. Frank (2) |
| | | |
| 10.11+ | | Amendment to Employment Agreement, dated as of February 5, 2007, by and between Registrant and Menashe R. Frank (2) |
| | | |
| 10.12+ | | Employment Agreement, dated as of May 15, 2006, by and between Registrant and Steven J. Nunes (2) |
| | | |
| 10.13+ | | Amendment to Employment Agreement, dated as of February 5, 2007, by and between Registrant and Steven J. Nunes (2) |
| | | |
| 10.14# | | Consulting Agreement, by and between Registrant and Thomas M. Coon, M.D., dated as of April 6, 2007 (2) |
| | | |
| 10.15# | | Consulting Agreement, by and between Registrant and Martin W. Roche, M.D., dated August 12, 2005 (2) |
| | | |
| 10.16# | | Amendment to Consulting Agreement, by and between Registrant and Martin W. Roche, M.D., dated as of July 6, 2007 (2) |
| | | |
| 10.17# | | Development Agreement, by and between Registrant and Martin W. Roche, M.D., dated as of July 6, 2007 (2) |
| | | |
| 10.18# | | License Agreement, dated December 17, 2004, by and between Registrant and Z-KAT, Inc. (2) |
| | | |
| 10.19 | | Asset Contribution Agreement, dated December 17, 2004, by and between Registrant and Z-KAT, Inc. (2) |
| | | |
| 10.20# | | Addendum to Asset Contribution Agreement, dated December 28, 2006, by and between Registrant and Z-KAT, Inc. (2) |
| | | |
| 10.21 | | Amendment to Addendum to Asset Contribution Agreement, dated April 28, 2007, by and between Registrant and Z-KAT, Inc. (2) |
| | | |
| 10.22# | | License Agreement, by and between Registrant, International Business Machines Corporation and Z-KAT, Inc., dated as of March 29, 2006 (2) |
| | | |
| 10.23# | | License Agreement, by and between Registrant and Integrated Surgical Systems, Inc., dated September 1, 2005 (2) |
| | | |
| 10.24# | | Sublicense Agreement, by and between Registrant and SensAble Technologies, Inc., dated as of May 24, 2006, as amended and supplemented by that certain letter dated May 23, 2007 from SensAble Technologies, Inc. and by that certain letter dated May 23, 2007 from Registrant (2) |
| | | |
| 10.25# | | Research Agreement, by and between Registrant and University of Florida Board of Trustees, dated as of February 10, 2005 (2) |
| | | |
| 10.26# | | Amendment to Research Agreement, by and between Registrant and the University of Florida Board of Trustees, dated as of August 15, 2007 (2) |
| | | |
| 10.27# | | Exclusive License Agreement, by and between Registrant and University of Florida Research Foundation, dated August 15, 2007 (2) |
| | | |
| 10.28# | | Supply Agreement, by and between Registrant and Trigon Incorporated, dated as of September 13, 2005 (2) |
144
| | | |
| Exhibit | | |
| No. | | Description |
| | | |
| 10.29 | | License Agreement, by and between Registrant and Trigon Incorporated, dated September 14, 2005 (2) |
| | | |
| 10.30# | | Supply Agreement, by and between Registrant and Encore Medical, L.P., dated as of February 28, 2007 (2) |
| | | |
| 10.31# | | License Agreement, by and between Registrant and Encore Medical, L.P., dated as of December 14, 2006 (2) |
| | | |
| 10.32# | | Manufacturing Supply Agreement, by and between Registrant and Symmetry Medical, dated as of July 26, 2007 (2) |
| | | |
| 10.33 | | Letter of Agreement, by and between Registrant and The Anspach Effort, Inc., dated as of July 6, 2007 (2) |
| | | |
| 10.34 | | Multi-Tenant Lease, by and between Registrant and Westport Business Park Associates LLP, last dated January 31, 2006 (2) |
| | | |
| 10.35# | | Development Agreement by and between Registrant and Pro-Dex, Inc., dated December 12, 2007 (2) |
| | | |
| 10.36+ | | Form of Incentive Stock Option Agreement related to the 2008 Omnibus Incentive Plan (3) |
| | | |
| 23 | | Consent of Ernst & Young LLP, Independent Registered Public Accounting Firm (1) |
| | | |
| 31.1 | | Certification of Chief Executive Officer pursuant to Rule 13a-14(a) of the Exchange Act (1) |
| | | |
| 31.2 | | Certification of Chief Financial Officer pursuant to Rule 13a-14(a) of the Exchange Act (1) |
| | | |
| 32.1 | | Certification of Chief Executive Officer pursuant to18 U.S.C. §1350 (1) |
| | | |
| 32.2 | | Certification of Chief Financial Officer pursuant to 18 U.S.C. §1350 (1) |
| | | |
| (1) | | Filed herewith |
| |
| (2) | | Incorporated by reference to Registrant’s Registration Statement on Form S-1, as amended, filed with the SEC on September 19, 2007 |
| |
| (3) | | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on February 26, 2008 |
| |
| + | | Indicates management contract or compensatory plan. |
| |
| # | | Portions of the exhibit have been omitted pursuant to a request for confidential treatment. The omitted information has been filed separately with the SEC. |
145
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | |
| | By: | /s/ Maurice R. Ferré, M.D. | |
| | | President, Chief Executive Officer | |
| | | and Chairman of the Board
(Principal Executive Officer) | |
Dated: March 31, 2008
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| | | | | |
Signature | | Title | | Date |
| | | | | |
| | | | | |
| /s/ Maurice R. Ferré, M.D. | | President, Chief Executive Officer and | | March 31, 2008 |
Maurice R. Ferré, M.D. | | Chairman of the Board
(Principal Executive Officer) | | |
| | | | | |
| /s/ Fritz L. LaPorte | | Senior Vice President of Finance and Administration, | | March 31, 2008 |
Fritz L. LaPorte | | Chief Financial Officer and Treasurer
(Principal Accounting and Financial Officer) | | |
| | | | | |
| /s/ S. Morry Blumenfeld, Ph.D. | | Director | | March 31, 2008 |
S. Morry Blumenfeld, Ph.D. | | | | |
| | | | | |
| /s/ Gerald A. Brunk | | Director | | March 31, 2008 |
Gerald A. Brunk | | | | |
| | | | | |
| /s/ Marcelo G. Chao | | Director | | March 31, 2008 |
Marcelo G. Chao | | | | |
| | | | | |
| /s/ Christopher C. Dewey | | Director | | March 31, 2008 |
Christopher C. Dewey | | | | |
| | | | | |
| /s/ Charles W. Federico | | Director | | March 31, 2008 |
Charles W. Federico | | | | |
| | | | | |
| /s/ Frederic H. Moll, M.D. | | Director | | March 31, 2008 |
Frederic H. Moll, M.D. | | | | |
| | | | | |
| /s/ Michael P. Stansky | | Director | | March 31, 2008 |
Michael P. Stansky | | | | |
146
EXHIBIT INDEX
| | | |
| Exhibit | | |
| No. | | Description |
| | | |
| 3.1 | | Third Amended and Restated Certificate of Incorporation of the Registrant, dated February 20, 2008 |
| | | |
| 3.2 | | Third Amended and Restated Bylaws of the Registrant effective as of February 20, 2008 |
| | | |
| 23 | | Consent of Ernst & Young LLP, Independent Public Registered Accounting Firm |
| | | |
| 31.1 | | Certification of Chief Executive Officer pursuant to Rule 13a-14(a) of the Exchange Act |
| | | |
| 31.2 | | Certification of Chief Financial Officer pursuant to Rule 13a-14(a) of the Exchange Act |
| | | |
| 32.1 | | Certification of Chief Executive Officer pursuant to18 U.S.C. §1350 |
| | | |
| 32.2 | | Certification of Chief Financial Officer pursuant to18 U.S.C. §1350 |
147