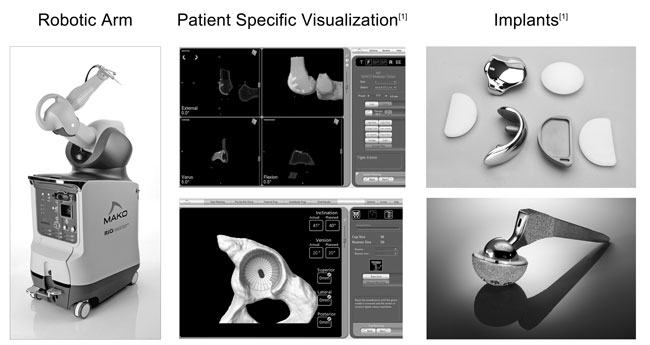
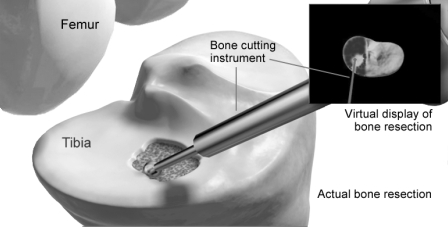
increase in procedure revenue was attributable to an increase in MAKOplasty procedures performed during the year ended December 31, 2010 as compared with the year ended December 31, 2009. There were 3,485 MAKOplasty procedures performed during the year ended December 31, 2010 compared to 1,602 MAKOplasty procedures performed during the year ended December 31, 2009. The increase in MAKOplasty procedures performed was driven by the continued adoption of knee MAKOplasty, both in terms of utilization per commercial site and total commercial installed base. Revenue for the year ended December 31, 2010 was reduced by $2.2 million, of which $1.2 million related to system sales prior to the fourth quarter, for the deferral of system revenue related to the first year warranty and maintenance services provided by MAKO. This deferred revenue will be recognized in service revenue over a twelve-month period. In accordance with our revenue recognition policy, recognition of revenue on unit sales of our TGS was deferred until delivery of the RIO system, which we commercially released in the first quarter of 2009. We expect our revenue to continue to increase as unit sales of our RIO system increase in future periods and the number of MAKOplasty procedures performed increases in future periods.
We loan instrumentation to our customers, which are used to perform MAKOplasty procedures in conjunction with using the RIO system. These loaned instrument sets, or implant instruments, are comprised of tools and equipment that facilitate the implantation of our implants. Implant instruments loaned to customers are not part of the tangible product sold and title of loaned instrument remains with the Company. To better reflect the economics of the implant instruments and enhance comparability with other companies in our industry, depreciation expense on implant instruments has been reclassified from cost of revenue – procedures to selling, general and administrative expense beginning in the first quarter of 2010. Depreciation expense for implant instruments was $464,000, $250,000 and $134,000 for the years ended December 31, 2010, 2009 and 2008, respectively.
Table of Contents
Research and Development. Research and development expense was $15.0 million for the year ended December 31, 2010, compared to $13.1 million for the year ended December 31, 2009. The increase of $1.9 million, or 14%, was primarily due to an increase in research and development activities associated with on-going development of our RIO system, our MAKO implant systems and potential future products, including our hip MAKOplasty application and associated implant systems. We expect our research and development expense to increase as we continue to expand our research and development activities, including the support of existing products and the research of potential future products, including our hip MAKOplasty application and associated implant systems.
Depreciation and Amortization. Depreciation and amortization expense was $3.0 million for the year ended December 31, 2010, compared to $2.0 million for the year ended December 31, 2009. The increase of $1.0 million, or 56%, was primarily due to an increase in depreciation of property and equipment as a result of purchases made during 2010 and 2009 due to the growth in our business, the expansion of our facilities in 2010 to accommodate an increase in employees necessary to support such growth.
Interest and Other Income. Interest and other income was $317,000 for the year ended December 31, 2010, compared to $432,000 for the year ended December 31, 2009. The decrease of $115,000, or 27%, was primarily due to lower yields realized on our cash, cash equivalents and investments for the year ended December 31, 2010 compared with the same period of 2009 which is attributable to a cash investment strategy which emphasizes the security of the principal invested and fulfillment of liquidity needs.
Income Taxes. No federal income taxes were recognized for the year ended December 31, 2010 and 2009, due to net operating losses in each period. State and local income taxes were $68,000 for the year ended December 31, 2010, compared to $56,000 for the year ended December 31, 2009. Income taxes recognized to date have not been significant due to net operating losses we have incurred in each period since our inception in November 2004. In addition, no current or deferred income taxes were recorded for the years ended December 31, 2010 and 2009, as all income tax benefits were fully offset by a valuation allowance against our net deferred income tax assets.
Year Ended December 31, 2009 Compared to the Year Ended December 31, 2008
Revenue. Revenue was $34.2 million for the year ended December 31, 2009, compared to $2.9 million for the year ended December 31, 2008. The increase in revenue of $31.3 million was primarily due to $14.7 million of revenue from 19 unit sales of our RIO system and the recognition of approximately $11.3 million of revenue from 17 previously deferred unit sales of our TGS. In accordance with our revenue recognition policy, recognition of revenue on unit sales of our TGS was deferred until delivery of the RIO system, which we commercially released in the first quarter of 2009. Prior to 2009, recognized revenue was primarily generated from the sale of implants and disposable products utilized in knee MAKOplasty procedures. Total revenue was also positively impacted by a $5.1 million increase in procedure revenue attributable to an increase in knee MAKOplasty procedures performed during the year ended December 31, 2009 as compared with the year ended December 31, 2008. There were 1,602 knee MAKOplasty procedures performed during the year ended December 31, 2009 compared to 601 knee MAKOplasty procedures performed during year ended December 31, 2008.
Cost of Revenue. Cost of revenue was $21.5 million for the year ended December 31, 2009, compared to $3.3 million for the year ended December 31, 2008. The increase in cost of revenue of $18.2 million was primarily due to the cost of revenue from 19 unit sales of our RIO system, the recognition of the direct cost of revenue from 17 previously deferred unit sales of our TGS, including the cost of providing the RIO system upgrades, as described in the “Critical Accounting Policies and Significant Judgments and Estimates” section above, and an increase in knee MAKOplasty procedures performed.
Selling, General and Administrative. Selling, general and administrative expense was $32.1 million for the year ended December 31, 2009, compared to $23.3 million for the year ended December 31, 2008. The increase of $8.8 million, or 38%, was primarily due to an increase in sales, marketing and operations costs
75
Table of Contents
associated with the production and commercialization of our products and an increase in general and administrative costs to support growth and costs associated with operating as a public company. Selling, general and administrative expense for the year ended December 31, 2009 also included $3.3 million of stock-based compensation expense compared to $1.9 million for the year ended December 31, 2008. The increase in stock-based compensation expense was primarily due to additional option and restricted stock grants made in 2009.
Research and Development. Research and development expense was $13.1 million for the year ended December 31, 2009, compared to $12.5 million for the year ended December 31, 2008. The increase of $655,000, or 5%, was primarily due to an increase in research and development activities associated with on-going development of our RIO system, our MAKO implant systems and potential future products. This was partially offset by a nonrecurring charge of $949,000 incurred in the first quarter of 2008 associated with the vesting in full, upon completion of our IPO in February 2008, of restricted common stock issued pursuant to business consultation agreements entered into in December 2004.
Depreciation and Amortization. Depreciation and amortization expense was $2.0 million for the year ended December 31, 2009, compared to $1.8 million for the year ended December 31, 2008. The increase of $123,000, or 7%, was primarily due to an increase in depreciation of property and equipment as a result of purchases made during 2009 and 2008.
Interest and Other Income. Interest and other income was $432,000 for the year ended December 31, 2009, compared to $988,000 for the year ended December 31, 2008. The decrease of $556,000, or 56%, was primarily due to lower yields realized on our cash, cash equivalents and investments for the year ended December 31, 2009 compared with the same period of 2008.
Interest and Other Expense. Interest and other expense was $3,000 for the year ended December 31, 2009, compared to $110,000 for the year ended December 31, 2008. Through February 2008, interest and other expense consisted primarily of the amortization of a $590,000 discount associated with a deferred license fee payment of $4.0 million which had been fully amortized and paid upon the completion of our IPO in February 2008.
Income Taxes. No federal income taxes were recognized for the year ended December 31, 2009 and 2008, due to net operating losses in each period. State and local income taxes were $56,000 for the year ended December 31, 2009. Income taxes recognized to date have not been significant due to net operating losses we have incurred in each period since our inception in November 2004. In addition, no current or deferred income taxes were recorded for the year ended December 31, 2009 and 2008, as all income tax benefits were fully offset by a valuation allowance against our net deferred income tax assets.
Liquidity and Capital Resources
| | | | | | | | | | | | | | | | |
(in thousands) | | | | | | | | | | | | | | | | |
| | 2010 | | Change | | 2009 | | Change | | 2008 | |
Cash and cash equivalents | | $ | 27,108 | | $ | 9,949 | | | 17,159 | | $ | (45,388 | ) | $ | 62,547 | |
Short-term investments | | | 46,401 | | | 1,715 | | | 44,686 | | | 43,609 | | | 1,077 | |
Long-term investments | | | 23,283 | | | 13,915 | | | 9,368 | | | 9,368 | | | ― | |
Total cash, cash equivalents, and investments | | $ | 96,792 | | $ | 25,579 | | $ | 71,213 | | $ | 7,589 | | $ | 63,624 | |
| | | | | | | | | | | | | | | | |
Cash used in operating activities | | $ | (30,292 | ) | $ | 15,280 | | $ | (45,572 | ) | $ | (15,752 | ) | $ | (29,820 | ) |
Cash used in investing activities | | | (20,076 | ) | | 34,104 | | | (54,180 | ) | | (50,631 | ) | | (3,549 | ) |
Cash provided by financing activities | | | 60,317 | | | 5,953 | | | 54,364 | | | (31,937 | ) | | 86,301 | |
Net increase (decrease) in cash and cash equivalents | | $ | 9,949 | | $ | 55,337 | | $ | (45,388 | ) | $ | (98,320 | ) | $ | 52,932 | |
We have incurred net losses and negative cash flow from operating activities for each period since our inception in November 2004. As of December 31, 2010, we had an accumulated deficit of $152.9 million and have financed our operations principally through the sale of our equity securities.
76
Table of Contents
In November 2010, we completed a public offering of our common stock, issuing 6,325,000 shares at a price per share of $9.44, resulting in net proceeds of approximately $59.3 million, after expenses.
As of December 31, 2010, we had approximately $96.8 million in cash, cash equivalents and investments. Our cash and investment balances are held in a variety of interest bearing instruments, including notes and bonds from U.S. government agencies, certificates of deposit and investment grade rated U.S. corporate debt.
Net Cash Used in Operating Activities
Net cash used in operating activities primarily reflects the net loss for those periods, which was reduced in part by depreciation and amortization, stock-based compensation, inventory write-downs and property and equipment write-downs. For the year ended December 31, 2010, inventory write-downs of $1.7 million and property and equipment write-downs of $1.2 million were incurred primarily due to the write-off of excess RESTORIS Classic implants and instrumentation as discussed in “Factors Which May Influence Future Results of Operations” above. Net cash used in operating activities was also affected by changes in operating assets and liabilities. Included in changes in operating assets and liabilities for the year ended December 31, 2010 are approximately $6.1 million of increases to inventory necessitated by increased sales of implants and disposable products to support the growth of knee MAKOplasty and preparation for the anticipated launch of our hip MAKOplasty application, $5.0 million of increases to accounts receivable due to increased sales in 2010, which was partially offset by $4.0 million of increases to other accrued liabilities and accrued compensation and employee benefits. Included in changes in operating assets and liabilities for the year ended December 31, 2009 are approximately $11.0 million and $3.6 million of decreases to the deferred revenue balance and deferred cost of revenue balance, respectively, due to the recognition of 17 previously deferred unit sales of our TGS, $7.4 million of increases in inventory necessitated by the commercial release of the RIO system, the commercial release of the RESTORIS MCK implant system and increased sales of implants and disposable products and $3.8 million of increases in accounts receivable due to increased sales in the fourth quarter of 2009 as compared to the fourth quarter of 2008. In accordance with our revenue recognition policy, recognition of revenue and direct cost of revenue associated with the unit sales of our TGS was deferred until delivery of the RIO system, which we commercially released in the first quarter of 2009.
Net Cash Used in Investing Activities
Net cash used in investing activities for the year ended December 31, 2010 was primarily attributable to the purchase of investments of $65.8 million and purchases of property and equipment of $2.6 million, which was partially offset by proceeds of $49.7 million from sales and maturities of investments. Purchases of investments in 2010 were primarily driven by net proceeds of $59.3 million received in a public offering of our common stock in November 2010. Net cash used in investing activities for the year ended December 31, 2009 was primarily attributable to the purchase of investments of $60.0 million and purchases of property and equipment of $790,000, which was partially offset by proceeds of $6.7 million from sales and maturities of investments.
Net Cash Provided by Financing Activities
Net cash provided by our financing activities for the year ended December 31, 2010 was primarily attributable to net proceeds of $59.3 million received in public offering of our common stock in November 2010. Net cash provided by our financing activities for the year ended December 31, 2009 was primarily attributable to net proceeds of $54.3 million received in connection with our equity financing in August 2009.
Operating Capital and Capital Expenditure Requirements
To date, we have not achieved profitability. We anticipate that we will continue to incur substantial net losses for at least the next two or three years as we expand our sales and marketing capabilities in the orthopedic products market, continue to commercialize our RIO system and RESTORIS MCK multicompartmental knee implant system, continue research and development of existing and future products, including our hip
77
Table of Contents
MAKOplasty application and associated implant systems, and continue development of the corporate infrastructure required to sell and market our products and support operations. We also expect to experience increased cash requirements for inventory and property and equipment in conjunction with the continued commercialization of our RESTORIS MCK multicompartmental knee implant system and our RIO system and introducing other potential future applications, including our hip MAKOplasty application and associated implant systems.
In executing our current business plan, we believe our existing cash, cash equivalents and investment balances, and interest income we earn on these balances will be sufficient to meet our anticipated cash requirements for at least the next twelve months. To the extent our available cash, cash equivalents and investment balances are insufficient to satisfy our operating requirements, we will need to seek additional sources of funds, including selling additional equity, debt or other securities or entering into a credit facility, or modify our current business plan. The sale of additional equity and convertible debt securities may result in dilution to our current stockholders. If we raise additional funds through the issuance of debt securities, these securities may have rights senior to those of our common stock and could contain covenants that could restrict our operations and issuance of dividends. We may also require additional capital beyond our currently forecasted amounts. Any required additional capital, whether forecasted or not, may not be available on reasonable terms, or at all. If we are unable to obtain additional financing, we may be required to reduce the scope of, delay or eliminate some or all of our planned research, development and commercialization activities, which could materially harm our business and results of operations.
Because of the numerous risks and uncertainties associated with the development of medical devices and the current economic situation, we are unable to estimate the exact amounts of capital outlays and operating expenditures necessary to complete the development of our products and successfully deliver commercial products to the market. Our future capital requirements will depend on many factors, including but not limited to the following:
| | |
| • | the revenue generated by sales of our current and future products; |
| | |
| • | the expenses we incur in selling and marketing our products; |
| | |
| • | the costs and timing of regulatory clearance or approvals for upgrades or changes to our existing products as well as future products; |
| | |
| • | the rate of progress, cost and success of on-going product development activities; |
| | |
| • | the emergence of competing or complementary technological developments; |
| | |
| • | the costs of filing, prosecuting, defending and enforcing any patent or license claims and other intellectual property rights, or participating in litigation related activities; |
| | |
| • | the future unknown impact of recently enacted healthcare legislation; |
| | |
| • | the acquisition of businesses, products and technologies, although we currently have no understandings, commitments or agreements relating to any material transaction of this type; and |
| | |
| • | general economic conditions and interest rates. |
Contractual Obligations
The following table summarizes our outstanding contractual obligations as of December 31, 2010 and the effect those obligations are expected to have on our liquidity and cash flows in future periods:
78
Table of Contents
| | | | | | | | | | | | | | | | |
(in thousands) | | Payment Due by Period | |
| | | | December 31, | | After |
| | Total | | 2011 | | 2012-2013 | | 2014-2015 | | 2015 | |
Contractual Obligations | | | | | | | | | | | | | | | | |
Minimum royalty payments – licenses | | $ | 14,150 | | $ | 1,069 | | $ | 3,538 | | $ | 4,068 | | $ | 5,475 | |
Operating lease – real estate | | | 5,823 | | | 290 | | | 689 | | | 1,161 | | | 3,683 | |
Purchase commitments and obligations | | | 8,005 | | | 8,005 | | | ― | | | ― | | | ― | |
Development agreement obligations | | | 1,000 | | | 750 | | | 250 | | | ― | | | ― | |
Total | | $ | 28,978 | | $ | 10,114 | | $ | 4,477 | | $ | 5,229 | | $ | 9,158 | |
Our commitments for minimum royalty payments relate to payments under various licenses and sublicenses as discussed in Item 8, Financial Statements and Supplementary Data, Note 7 to the Financial Statements. Our commitments for operating leases relate to the lease for our headquarters in Fort Lauderdale, Florida. Our commitments for purchase commitments and obligations include an estimate of open purchase orders and contractual obligations in the ordinary course of business, including commitments with contract manufacturers and suppliers, for which we have not received the goods or services. Our commitments for development agreement obligations relate to payments under development agreements as discussed in Item 8, Financial Statements and Supplementary Data, Notes 5 and 7 to the Financial Statements.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
| |
ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Our exposure to market risk is confined to our cash, cash equivalents, investments and exchange rate risk on international sales. The goals of our cash investment policy are the security of the principal invested and fulfillment of liquidity needs, with the need to maximize value being an important consideration. To achieve our goals, we maintain a portfolio of cash equivalents and investments in a variety of securities including notes and bonds from U.S. government agencies, certificates of deposit and investment grade rated U.S. corporate debt. The securities in our investment portfolio are not leveraged and are classified as available-for-sale. We currently do not hedge interest rate exposure or exchange rate risk. We do not believe that a variation in market rates of interest would significantly impact the value of our investment portfolio. We do not believe that a variation in the value of the U.S. dollar relative to foreign currencies would significantly impact our results of operations.
79
Table of Contents
| |
ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
MAKO SURGICAL CORP.
Index to the Financial Statements
80
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
MAKO Surgical Corp.
We have audited the accompanying balance sheets of MAKO Surgical Corp. as of December 31, 2010 and 2009, and the related statements of operations, redeemable convertible preferred stock and stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2010. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of MAKO Surgical Corp. at December 31, 2010 and 2009, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2010, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), MAKO Surgical Corp.’s internal control over financial reporting as of December 31, 2010, based on criteria established inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 10, 2011 expressed an unqualified opinion thereon.
| |
| /s/ ERNST & YOUNG LLP |
| Certified Public Accountants |
Boca Raton, Florida | |
March 10, 2011 | |
81
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
MAKO Surgical Corp.
We have audited MAKO Surgical Corp.’s internal control over financial reporting as of December 31, 2010, based on criteria established inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). MAKO Surgical Corp.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, MAKO Surgical Corp. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of MAKO Surgical Corp. as of December 31, 2010 and 2009, and the related statements of operations, redeemable convertible preferred stock and stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2010 of MAKO Surgical Corp. and our report dated March 10, 2011 expressed an unqualified opinion thereon.
| |
| /s/ ERNST & YOUNG LLP |
| Certified Public Accountants |
Boca Raton, Florida | |
March 10, 2011 | |
82
Table of Contents
MAKO SURGICAL CORP.
Balance Sheets
(in thousands, except share and per share data)
| | | | | | | |
| | December 31, | |
| | 2010 | | 2009 | |
ASSETS | | | | | | | |
Current Assets: | | | | | | | |
Cash and cash equivalents | | $ | 27,108 | | $ | 17,159 | |
Short-term investments | | | 46,401 | | | 44,686 | |
Accounts receivable | | | 11,560 | | | 6,536 | |
Inventory | | | 10,504 | | | 10,190 | |
Prepaids and other assets | | | 1,283 | | | 532 | |
Total current assets | | | 96,856 | | | 79,103 | |
Long-term investments | | | 23,283 | | | 9,368 | |
Property and equipment, net | | | 9,212 | | | 6,205 | |
Intangible assets, net | | | 7,530 | | | 4,234 | |
Other assets | | | 198 | | | 193 | |
Total assets | | $ | 137,079 | | $ | 99,103 | |
| | | | | | | |
LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | |
Current Liabilities: | | | | | | | |
Accounts payable | | $ | 1,518 | | $ | 1,159 | |
Accrued compensation and employee benefits | | | 5,546 | | | 3,709 | |
Other accrued liabilities | | | 5,064 | | | 2,872 | |
Deferred revenue | | | 3,071 | | | 548 | |
Total current liabilities | | | 15,199 | | | 8,288 | |
| | | | | | | |
Deferred revenue, non-current | | | 109 | | | 21 | |
Total liabilities | | | 15,308 | | | 8,309 | |
| | | | | | | |
Commitments and contingencies (Note 7) | | | ― | | | ― | |
| | | | | | | |
Stockholders’ equity: | | | | | | | |
Preferred stock, $0.001 par value; 27,000,000 authorized; 0 shares issued and outstanding as of December 31, 2010 and 2009 | | | ― | | | ― | |
Common stock, $0.001 par value; 135,000,000 authorized; 39,945,467 and 33,036,378 shares issued and outstanding as of December 31, 2010 and 2009, respectively | | | 40 | | | 33 | |
Additional paid-in capital | | | 274,712 | | | 204,977 | |
Accumulated deficit | | | (152,882 | ) | | (114,195 | ) |
Accumulated other comprehensive loss | | | (99 | ) | | (21 | ) |
Total stockholders’ equity | | | 121,771 | | | 90,794 | |
Total liabilities and stockholders’ equity | | $ | 137,079 | | $ | 99,103 | |
See accompanying notes.
83
Table of Contents
MAKO SURGICAL CORP.
Statements of Operations
(in thousands, except per share data)
| | | | | | | | | | |
| | Years Ended December 31, | |
| | 2010 | | 2009 | | 2008 | |
Revenue: | | | | | | | | | | |
Procedures | | $ | 17,620 | | $ | 7,550 | | $ | 2,457 | |
Systems – RIO | | | 24,928 | | | 14,715 | | | ― | |
Systems – TGS, previously deferred | | | ― | | | 11,297 | | | ― | |
Service and other | | | 1,748 | | | 646 | | | 487 | |
Total revenue | | | 44,296 | | | 34,208 | | | 2,944 | |
Cost of revenue: | | | | | | | | | | |
Procedures | | | 5,960 | | | 3,087 | | | 1,387 | |
Systems – RIO | | | 11,171 | | | 9,032 | | | 1,692 | |
Systems – RIO upgrades | | | ― | | | 5,183 | | | ― | |
Systems – TGS, previously deferred | | | ― | | | 3,606 | | | ― | |
Service and other | | | 1,042 | | | 546 | | | 233 | |
Total cost of revenue | | | 18,173 | | | 21,454 | | | 3,312 | |
Gross profit (loss) | | | 26,123 | | | 12,754 | | | (368 | ) |
Operating costs and expenses: | | | | | | | | | | |
Selling, general and administrative | | | 47,041 | | | 32,072 | | | 23,292 | |
Research and development | | | 14,975 | | | 13,127 | | | 12,472 | |
Depreciation and amortization | | | 3,043 | | | 1,951 | | | 1,828 | |
Total operating costs and expenses | | | 65,059 | | | 47,150 | | | 37,592 | |
Loss from operations | | | (38,936 | ) | | (34,396 | ) | | (37,960 | ) |
Interest and other income | | | 317 | | | 432 | | | 988 | |
Interest and other expenses | | | ― | | | (3 | ) | | (110 | ) |
Loss before income taxes | | | (38,619 | ) | | (33,967 | ) | | (37,082 | ) |
Income tax expense | | | 68 | | | 56 | | | ― | |
Net loss | | | (38,687 | ) | | (34,023 | ) | | (37,082 | ) |
Accretion of preferred stock | | | ― | | | ― | | | (44 | ) |
Dividends on preferred stock | | | ― | | | ― | | | (521 | ) |
Net loss attributable to common stockholders | | $ | (38,687 | ) | $ | (34,023 | ) | $ | (37,647 | ) |
Net loss per share – Basic and diluted attributable to common stockholders | | $ | (1.13 | ) | $ | (1.22 | ) | $ | (2.20 | ) |
Weighted average common shares outstanding – Basic and diluted | | | 34,349 | | | 27,806 | | | 17,096 | |
See accompanying notes.
84
Table of Contents
MAKO SURGICAL CORP.
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit)
(in thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | Redeemable
Convertible Preferred | | Common
Shares | | Stock
Amount | | Additional
Paid-in
Capital | | Accumulated
Deficit | | Other
Comprehensive
Income (Loss) | | Total
Stockholders’
Equity (Deficit) | |
| | Shares | | Amount | | | | | | | |
Balance at December 31, 2007 | | | 33,164 | | $ | 59,487 | | | 1,871 | | $ | 2 | | $ | — | | $ | (42,843 | ) | $ | 4 | | $ | (42,837 | ) |
Issuance of common stock in initial public offering | | | — | | | — | | | 5,100 | | | 5 | | | 43,789 | | | — | | | — | | | 43,794 | |
Issuance of common stock in equity financing | | | — | | | — | | | 6,451 | | | 7 | | | 39,726 | | | — | | | — | | | 39,733 | |
Issuance of common stock upon exercise of options | | | — | | | — | | | 62 | | | — | | | 46 | | | — | | | — | | | 46 | |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | 1,467 | | | — | | | — | | | 1,467 | |
Restricted common stock compensation expense | | | — | | | — | | | 256 | | | — | | | 1,856 | | | — | | | — | | | 1,856 | |
Accretion to redemption value of Series A, B and C redeemable convertible preferred stock | | | — | | | 44 | | | — | | | — | | | (44 | ) | | — | | | — | | | (44 | ) |
Accrued dividends on Series A, B and C redeemable convertible preferred stock | | | — | | | 521 | | | — | | | — | | | (274 | ) | | (247 | ) | | — | | | (521 | ) |
Conversion of Series A, B and C redeemable convertible preferred shares into common shares | | | (33,164 | ) | | (53,667 | ) | | 10,945 | | | 11 | | | 53,656 | | | — | | | — | | | 53,667 | |
Reclassification of accrued dividends on redeemable convertible preferred stock to additional paid-in capital | | | — | | | (6,385 | ) | | — | | | — | | | 6,385 | | | — | | | — | | | 6,385 | |
Change in unrealized gain on available-for-sale securities | | | — | | | — | | | — | | | — | | | — | | | — | | | 50 | | | 50 | |
Net loss | | | — | | | — | | | — | | | — | | | — | | | (37,082 | ) | | — | | | (37,082 | ) |
Total comprehensive loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (37,032 | ) |
Balance at December 31, 2008 | | | — | | $ | — | | | 24,685 | | $ | 25 | | $ | 146,607 | | $ | (80,172 | ) | $ | 54 | | $ | 66,514 | |
(continued)
85
Table of Contents
MAKO SURGICAL CORP.
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit)
(in thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | Redeemable
Convertible Preferred | | Common
Shares | | Stock
Amount | | Additional
Paid-in
Capital | | Accumulated
Deficit | | Other
Comprehensive
Income (Loss) | | Total
Stockholders’
Equity (Deficit) | |
| | Shares | | Amount | | | | | | | |
Balance at December 31, 2008 | | | — | | $ | — | | | 24,685 | | $ | 25 | | $ | 146,607 | | $ | (80,172 | ) | $ | 54 | | $ | 66,514 | |
Issuance of common stock in equity financing | | | — | | | — | | | 8,050 | | | 8 | | | 54,300 | | | — | | | — | | | 54,308 | |
Issuance of common stock under employee stock purchase plan | | | — | | | — | | | 72 | | | — | | | 455 | | | — | | | — | | | 455 | |
Issuance of common stock upon exercise of options and warrants | | | — | | | — | | | 140 | | | — | | | 149 | | | — | | | — | | | 149 | |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | 3,032 | | | — | | | — | | | 3,032 | |
Restricted common stock compensation expense | | | — | | | — | | | 145 | | | — | | | 982 | | | — | | | — | | | 982 | |
Receipt of 56,045 shares delivered in payment of payroll taxes | | | — | | | — | | | (56 | ) | | — | | | (492 | ) | | — | | | — | | | (492 | ) |
Equity financing costs | | | — | | | — | | | — | | | — | | | (56 | ) | | — | | | — | | | (56 | ) |
Change in unrealized gain (loss) on available-for-sale securities | | | — | | | — | | | — | | | — | | | — | | | — | | | (75 | ) | | (75 | ) |
Net loss | | | — | | | — | | | — | | | — | | | — | | | (34,023 | ) | | — | | | (34,023 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (34,098 | ) |
Balance at December 31, 2009 | | | — | | | — | | | 33,036 | | | 33 | | | 204,977 | | | (114,195 | ) | | (21 | ) | | 90,794 | |
Issuance of common stock in equity financing | | | — | | | — | | | 6,325 | | | 6 | | | 59,277 | | | — | | | — | | | 59,283 | |
Issuance of common stock under employee stock purchase plan | | | — | | | — | | | 86 | | | — | | | 765 | | | — | | | — | | | 765 | |
Issuance of common stock upon exercise of options and warrants | | | — | | | — | | | 199 | | | — | | | 611 | | | — | | | — | | | 611 | |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | 5,027 | | | — | | | — | | | 5,027 | |
Restricted common stock compensation expense | | | — | | | — | | | 97 | | | — | | | 1,344 | | | — | | | — | | | 1,344 | |
Receipt of 28,307 shares delivered in payment of payroll taxes | | | — | | | — | | | (28 | ) | | — | | | (342 | ) | | — | | | — | | | (342 | ) |
Issuance of stock to a related party for intangible assets | | | — | | | — | | | 230 | | | 1 | | | 3,053 | | | — | | | — | | | 3,054 | |
Change in unrealized loss on available-for-sale securities | | | — | | | — | | | — | | | — | | | — | | | — | | | (78 | ) | | (78 | ) |
Net loss | | | — | | | — | | | — | | | — | | | — | | | (38,687 | ) | | — | | | (38,687 | ) |
Total comprehensive loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (38,765 | ) |
Balance at December 31, 2010 | | | — | | $ | — | | | 39,945 | | $ | 40 | | $ | 274,712 | | $ | (152,882 | ) | $ | (99 | ) | $ | 121,771 | |
See accompanying notes.
86
Table of Contents
MAKO SURGICAL CORP.
Statements of Cash Flows
(in thousands)
| | | | | | | | | | |
| | Years Ended December 31, | |
| | 2010 | | 2009 | | 2008 | |
Operating activities: | | | | | | | | | | |
Net loss | | $ | (38,687 | ) | $ | (34,023 | ) | $ | (37,082 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | |
Depreciation | | | 2,445 | | | 1,769 | | | 1,502 | |
Amortization of intangible assets | | | 1,070 | | | 682 | | | 660 | |
Stock-based compensation | | | 6,371 | | | 4,014 | | | 3,323 | |
Inventory write-down | | | 1,701 | | | 1,081 | | | 730 | |
Amortization of premium on investment securities | | | 480 | | | 188 | | | ― | |
Loss on asset impairment | | | 1,248 | | | 51 | | | ― | |
Accrued interest expense on deferred license fee | | | ― | | | ― | | | 45 | |
Changes in operating assets and liabilities: | | | | | | | | | | |
Accounts receivable | | | (5,024 | ) | | (3,809 | ) | | (514 | ) |
Inventory | | | (6,087 | ) | | (7,358 | ) | | (7,056 | ) |
Prepaid and other assets | | | (751 | ) | | (49 | ) | | (173 | ) |
Other assets | | | (57 | ) | | (16 | ) | | (7 | ) |
Accounts payable | | | 359 | | | (650 | ) | | 298 | |
Accrued compensation and employee benefits | | | 1,837 | | | 1,371 | | | 1,305 | |
Other accrued liabilities | | | 2,192 | | | (1,411 | ) | | 1,603 | |
Deferred cost of revenue | | | ― | | | 3,608 | | | (2,682 | ) |
Deferred revenue | | | 2,611 | | | (11,020 | ) | | 8,228 | |
Net cash used in operating activities | | | (30,292 | ) | | (45,572 | ) | | (29,820 | ) |
Investing activities: | | | | | | | | | | |
Purchase of investments | | | (65,828 | ) | | (59,961 | ) | | (1,990 | ) |
Proceeds from sales and maturities of investments | | | 49,692 | | | 6,721 | | | 4,047 | |
Acquisition of property and equipment | | | (2,628 | ) | | (790 | ) | | (1,606 | ) |
Acquisition of intangible assets | | | (1,312 | ) | | (150 | ) | | ― | |
Payment of deferred license fee | | | ― | | | ― | | | (4,000 | ) |
Net cash used in investing activities | | | (20,076 | ) | | (54,180 | ) | | (3,549 | ) |
Financing activities: | | | | | | | | | | |
Proceeds from issuance of common stock in equity financing, net of underwriting fees | | | 59,708 | | | 54,861 | | | 40,202 | |
Equity financing costs | | | (425 | ) | | (609 | ) | | (469 | ) |
Proceeds from initial public offering of common stock, net of underwriting fees | | | ― | | | ― | | | 47,430 | |
Initial public offering costs | | | ― | | | ― | | | (908 | ) |
Proceeds from employee stock purchase plan | | | 765 | | | 455 | | | ― | |
Exercise of common stock options and warrants for cash | | | 611 | | | 149 | | | 46 | |
Payment of payroll taxes relating to vesting of restricted stock | | | (342 | ) | | (492 | ) | | ― | |
Net cash provided by financing activities | | | 60,317 | | | 54,364 | | | 86,301 | |
Net increase (decrease) in cash and cash equivalents | | | 9,949 | | | (45,388 | ) | | 52,932 | |
Cash and cash equivalents at beginning of year | | | 17,159 | | | 62,547 | | | 9,615 | |
| | | | | | | | | | |
Cash and cash equivalents at end of year | | $ | 27,108 | | $ | 17,159 | | $ | 62,547 | |
Non-cash investing and financing activities: | | | | | | | | | | |
Receipt of 28,307 and 56,045 shares of common stock delivered in payment of payroll taxes for the years ended December 31, 2010 and 2009, respectively | | $ | 342 | | $ | 492 | | $ | ― | |
Transfers of inventory to property and equipment | | | 2,259 | | | 3,760 | | | 999 | |
Issuance of stock to a related party for intangible assets | | | 3,054 | | | ― | | | ― | |
Accretion of redeemable convertible preferred stock | | | ― | | | ― | | | 44 | |
Accrued dividends on redeemable convertible preferred stock | | | ― | | | ― | | | 521 | |
Conversion of redeemable convertible preferred stock into 10,945,080 common shares | | | ― | | | ― | | | 53,667 | |
Reclassification of accrued dividends on redeemable convertible preferred stock to additional paid-in capital | | | ― | | | ― | | | 6,385 | |
Reclassification of deferred initial public offering costs to additional paid-in capital | | | ― | | | ― | | | 3,636 | |
See accompanying notes.
87
Table of Contents
MAKO SURGICAL CORP.
Notes to Financial Statements
1. Description of the Business
MAKO Surgical Corp. (the “Company” or “MAKO”) is an emerging medical device company that markets its advanced robotic arm solution and orthopedic implants for orthopedic procedures called MAKOplasty®. The Company was incorporated in the State of Delaware on November 12, 2004 and is headquartered in Fort Lauderdale, Florida. The Company’s common stock trades on The NASDAQ Global Select Market under the ticker symbol “MAKO.”
In November 2010, the Company completed a public offering of its common stock, issuing 6,325,000 shares at a price per share of $9.44, resulting in net proceeds of approximately $59.3 million, after expenses.
2. Summary of Significant Accounting Policies
Basis of Presentation and Use of Estimates
The financial statements have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. The accounting estimates that require management’s most significant, difficult and subjective judgments include revenue recognition, allowance for doubtful accounts, inventory valuation, valuation allowance for deferred income tax assets, impairment of long-lived assets and the determination of stock-based compensation. Actual results could differ significantly from these estimates.
Liquidity and Operations
In executing its current business plan, the Company believes its existing cash, cash equivalents and investment balances and interest income earned on these balances will be sufficient to meet its anticipated cash requirements for at least the next twelve months. To the extent the Company’s available cash, cash equivalents and investment balances are insufficient to satisfy its operating requirements, the Company will need to seek additional sources of funds, including selling additional equity, debt or other securities or entering into a credit facility, or modifying its current business plan. The sale of additional equity and convertible debt securities may result in dilution to the Company’s current stockholders. If the Company raises additional funds through the issuance of debt securities, these securities may have rights senior to those of its common stock and could contain covenants that could restrict the Company’s operations and issuance of dividends. The Company may also require additional capital beyond its currently forecasted amounts. Any required additional capital, whether forecasted or not, may not be available on reasonable terms, or at all. If the Company is unable to obtain additional financing, the Company may be required to reduce the scope of, delay or eliminate some or all of its planned research, development and commercialization activities, which could materially harm its business and results of operations.
Concentrations of Credit Risk and Other Risks and Uncertainties
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash and cash equivalents, investments, and accounts receivable. The Company’s cash and cash equivalents are held in demand and money market accounts at four large financial institutions. The Company’s investments are held in a variety of interest bearing instruments, including notes and bonds from U.S. government agencies, certificates of deposit and investment grade rated U.S. corporate debt at three large financial institutions. Such deposits are generally in excess of insured limits. The Company has not experienced any historical losses on its deposits of cash and cash equivalents.
88
Table of Contents
The Company may perform credit evaluations of its customers’ financial condition and, generally, requires no collateral from its customers. The Company will provide an allowance for doubtful accounts when collections become doubtful but has not experienced any credit losses to date.
The Company is subject to risks common to emerging companies in the medical device industry including, but not limited to: new technological innovations, dependence on key personnel, dependence on key suppliers, changes in general economic conditions and interest rates, protection of proprietary technology, compliance with changing government regulations and taxes, uncertainty of widespread market acceptance of products, access to credit for capital purchases by our customers, product liability and the need to obtain additional financing. The Company’s products include components subject to rapid technological change. Certain components used in manufacturing have relatively few alternative sources of supply and establishing additional or replacement suppliers for such components cannot be accomplished quickly. The inability of any of these suppliers to fulfill the Company’s supply requirements may negatively impact future operating results. While the Company has ongoing programs to minimize the adverse effect of such uncertainty and considers technological change in estimating the net realizable value of its inventory, uncertainty continues to exist.
The Company expects to derive most of its revenue from capital sales of its RIO system, future applications to the RIO system, including the RIO-enabled hip application, recurring sales of implants and disposable products required for each MAKOplasty procedure, and service plans that are sold with the RIO system. If the Company is unable to achieve commercial acceptance of MAKOplasty or obtain regulatory clearances or approvals for future products, including products to treat other joints of the human body, its revenue would be adversely affected and the Company would not become profitable.
The Company’s current versions of its RIO® Robotic Arm Interactive Orthopedic (“RIO”) system, which is the second generation of its Tactile Guidance System™ (“TGS™”), its RESTORIS ® unicompartmental and RESTORIS MCK multicompartmental knee implant systems and its TGS have been cleared by the U.S. Food and Drug Administration (“FDA”). Certain products currently under development by the Company will require clearance or approval by the FDA or other international regulatory agencies prior to commercial sale. There can be no assurance that the Company’s products will receive the necessary clearances or approvals. If the Company were to be denied such clearance or approval or such clearance or approval were delayed, it could have a material adverse impact on the Company.
Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 280,Segment Reporting, establishes standards for reporting information about operating segments. Operating segments are defined as components of an enterprise about which separate financial information is available that is evaluated regularly by the chief operating decision maker, or decision making group, in deciding how to allocate resources and in assessing performance. The Company’s chief operating decision maker is its CEO. The Company’s CEO reviews financial information presented on an aggregate basis for purposes of allocating resources and evaluating financial performance. The Company has one business activity and there are no segment managers who are held accountable for operations, operating results and plans for products or components below the aggregate Company level. Accordingly, the Company reports as a single operating segment. To date, substantially all of the Company’s revenue is from companies located in the United States. No one customer accounted for more than 10% of the Company’s total revenue for the years ended December 31, 2010 and 2009. The following table presents information about the Company’s revenue by significant customer for the year ended December 31, 2008:
89
Table of Contents
| | | | |
(in thousands) | | Year Ended
December 31, | |
| | 2008 | |
Company A | | $ | 417 | |
Company B | | | 277 | |
Company C | | | 493 | |
Company D | | | 274 | |
Others | | | 996 | |
Net Revenue | | $ | 2,457 | |
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity at date of purchase of 90 days or less to be cash equivalents.
Fair Value of Financial Instruments
Carrying amounts of certain of the Company’s financial instruments, including cash and cash equivalents, investments, accounts receivable and other accrued liabilities approximate fair value due to their short maturities or market rates of interest.
Allowance for Doubtful Accounts
The Company regularly reviews customer balances by considering factors such as historical experience, credit quality, the age of the accounts receivable balances and current economic conditions that may affect a customer’s ability to pay. The Company has not experienced any collectability issues to date and has no allowance for doubtful accounts or write-offs to date in the accompanying financial statements.
Inventory
Inventory is stated at the lower of cost or market value on a first-in, first-out basis. Inventory costs include direct materials, direct labor and manufacturing overhead. The Company reviews its inventory periodically to determine net realizable value and considers product upgrades in its periodic review of realizability. The Company writes down inventory, if required, based on forecasted demand and technological obsolescence. These factors are impacted by market and economic conditions, technology changes and new product introductions and require estimates that may include uncertain elements.
Beginning with the fourth quarter of 2008, manufacturing overhead costs have been capitalized and included in inventory. As of December 31, 2010 and 2009, capitalized manufacturing overhead included in inventory was approximately $1.7 million and $1.1 million, respectively. Prior to 2009, such overhead costs were fully expensed as selling, general and administrative expense as capitalizable amounts were not significant.
Property and Equipment
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation of property and equipment is computed using the straight-line method over their estimated useful lives of two to seven years. Leasehold improvements are amortized on a straight-line basis over the lesser of their useful life or the term of the lease and are included in depreciation expense in the accompanying statements of operations. Upon retirement or sale, the cost and related accumulated depreciation are removed from the balance sheet and the resulting gain or loss is reflected in operations. Maintenance and repairs are charged to operations as incurred.
The Company loans instrumentation to its customers, who use the instrumentation to perform MAKOplasty procedures in conjunction with using the RIO system. These loaned instrument sets are comprised
90
Table of Contents
of tools and equipment that facilitate the implantation of the Company’s implants (“Implant Instruments”). Implant Instruments loaned to customers are not part of the tangible product sold and title of Implant Instruments remains with the Company. Accordingly, Implant Instruments are classified as a long-lived asset and included as a component of property and equipment. Undeployed Implant Instruments are carried at cost, net of allowances for excess and obsolete instruments. Implant Instruments in the field are carried at cost less accumulated depreciation. Depreciation is computed using the straight-line method based on an estimated useful life of five years. The Company reviews instruments for impairment whenever events or changes in circumstances indicate that the carrying value of an instrument may not be recoverable. To better reflect the economics of the Implant Instruments and enhance comparability with other companies in our industry, depreciation expense on Implant Instruments has been reclassified from cost of revenue – procedures to selling, general and administrative expense beginning in the first quarter of 2010. Depreciation expense for implant instruments was $464,000, $250,000 and $134,000 for the years ended December 31, 2010, 2009 and 2008, respectively.
Prior to 2010, Implant Instruments were included as components of a RIO system sale and undeployed Implant Instruments were classified as inventory. Beginning in the first quarter of 2010, Implant Instruments are no longer included as components of a RIO system sale. To better reflect the economics of the Implant Instruments no longer being sold, undeployed Implant Instruments have been reclassified from inventory to property and equipment. As of December 31, 2010, approximately $1.2 million of undeployed Implant Instruments have been included as property and equipment.
The Company also enters into RIO system consignment arrangements for clinical evaluation and clinical research purposes with terms ranging from one to three years. Under the terms of such arrangements, the Company installs a RIO system at the customer site and retains title to the RIO system, while the customer has use of the RIO system and purchases the Company’s implants and disposables products. Depreciation expense on consigned RIO systems and instruments is classified in selling, general and administrative expense and is computed using the straight-line method based on the estimated useful life of three years. As of December 31, 2010, the Company had three consigned RIO systems.
Service and demonstration RIO systems and instruments consist of RIO systems, associated instrumentation, service tools and equipment, and MAKOplasty procedure models used for sales demonstrations, surgeon training, and temporary RIO system placements at customer sites under warranty and extended warranty agreements. Service and demonstration RIO systems and instruments are classified as a long-lived asset and included as a component of property and equipment. Depreciation expense on service and demonstration RIO systems and instruments is classified in selling, general and administrative expense and is computed using the straight-line method based on an estimated useful life of three years.
Intangible Assets
The Company’s intangible assets are comprised of patents, patent applications and licenses to intellectual property rights. These intangible assets are carried at cost, net of accumulated amortization. Amortization is recorded using the straight-line method over their respective useful lives, which range from 5 to 13 years based on the respective anticipated lives of the underlying patents and patent applications.
Impairment of Long-Lived Assets
The Company evaluates its long-lived assets for indicators of impairment by comparison of the carrying amounts to future net undiscounted cash flows expected to be generated by such assets when events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. Should an impairment exist, the impairment loss would be measured based on the excess carrying value of the asset over the asset’s fair value or estimated discounted future cash flows.
91
Table of Contents
Revenue Recognition
Revenue is generated: from (1) unit sales of the Company’s RIO system, including associated instrumentation, installation services and training; (2) sales of implants and disposable products; and (3) sales of warranty and maintenance services. The Company recognizes revenue in accordance with Accounting Standards Codification (“ASC”) 605-10-S99,Revenue Recognition, when persuasive evidence of an arrangement exists, the fee is fixed or determinable, collection of the fee is probable and delivery has occurred. For all sales, the Company uses either a signed agreement or a binding purchase order as evidence of an arrangement.
The Company’s multiple-element arrangements are generally comprised of the following elements that qualify as separate units of accounting: (1) RIO system sales; (2) sales of implants and disposable products; and (3) warranty and maintenance services on the RIO system hardware. The Company’s revenue recognition policies generally result in revenue recognition at the following points:
| | |
| 1. | RIO system sales: Revenues related to RIO system sales are recognized upon installation of the system and training of at least one surgeon, which typically occurs prior to or concurrent with the RIO system installation. |
| | |
| 2. | Procedure revenue: Revenues from the sale of implants and disposable products utilized in MAKOplasty procedures are recognized at the time of sale (i.e., at the time of the related surgical procedure). |
| | |
| 3. | Service revenue: Revenues from warranty and maintenance services on the RIO system hardware are deferred and recognized ratably over the service period until no further obligation exists. Sales of the Company’s RIO system generally include a one-year warranty and maintenance obligation for services. Upon installation of the RIO system, the Company defers the revenue attributable to the warranty and maintenance obligation and recognizes it ratably over the warranty and maintenance period. Costs associated with providing warranty and maintenance services are expensed to cost of revenue as incurred. |
A portion of the Company’s end-user customers acquire the RIO system through a leasing arrangement with one of a number of qualified third-party leasing companies. In these instances, the Company typically sells the RIO system to the third-party leasing company, and the end-user customer enters into an independent leasing arrangement with the third-party leasing company. The Company recognizes RIO system revenue for a RIO system sale to a third-party leasing company on the same basis as a RIO system sale directly to an end-user customer. The Company sells implants and disposable products utilized in MAKOplasty procedures directly to end-user customers under a separate agreement.
The Company’s sales contracts generally do not provide the customer with a right of return. If such a right is provided, all related revenues would be deferred until such right expires or is waived. In a limited number of RIO system sales, the Company’s agreement with a customer provides for a customer acceptance period, which typically does not exceed three months, following which the customer may either accept or return the RIO system. No system revenue is recognized for these RIO system sales until the customer has unconditionally accepted the RIO system.
Effective January 1, 2010, the Company early adopted the Financial Accounting Standards Board (“FASB”) Accounting Standard Update No. 2009-13,Multiple-Deliverable Revenue Arrangements—a consensus of the FASB Emerging Issues Task Force (“ASU 2009-13”) and Update No. 2009-14,Certain Revenue Arrangements That Include Software Elements, a consensus of the FASB Emerging Issues Task Force (“ASU 2009-14”) on a prospective basis for applicable transactions originating or materially modified after December 31, 2009. In accordance with ASU 2009-13 (as codified under ASC 605-25,Multiple-Element Arrangements) and ASU 2009-14, the Company allocates arrangement consideration to the RIO systems and associated instrumentation, its implants and disposables and its warranty and maintenance services based upon the relative selling-price method. Under this method, revenue is allocated at the time of sale to all deliverables based on their relative selling price using a specific hierarchy. The hierarchy is as follows: vendor-specific objective evidence (“VSOE”) of fair value of the respective elements, third-party evidence of selling price (“TPS”), or best estimate of selling price (“ESP”).
92
Table of Contents
The Company uses ESP for its RIO system, TPS for its implants and disposables and VSOE of fair value for its warranty and maintenance services to allocate arrangement consideration. VSOE of fair value is based on the price charged when the element is sold separately. TPS is established by evaluating largely interchangeable competitor products or services in stand-alone sales. ESP is established by determine the price at which the Company would transact a sale if the product was sold on a stand-alone basis. The Company determines ESP for its systems by considering multiple factors including, but not limited to, geographies, type of customer, and market conditions. The Company regularly reviews ESP and maintains internal controls over the establishment and updates of these estimates.
The Company’s RIO system includes software that is essential to the functionality of the product. Since the RIO system’s software and non-software components function together to deliver the RIO system’s essential functionality, they are considered one deliverable that is excluded from the software revenue recognition guidance under ASU 2009-13 and ASU 2009-14.
Prior to the adoption of ASU 2009-13 and ASU 2009-14, the Company accounted for the sale of the RIO systems and associated instrumentation pursuant to ASC 985-605,Software – Revenue Recognition, which required the Company to allocate arrangement consideration to the RIO systems and associated instrumentation based upon VSOE of fair value of the respective elements. Had the new accounting guidance been applied to revenue at the beginning of 2009, the resultant revenue and net loss for the year ended December 31, 2009 would have been substantially the same.
Subsequent to December 31, 2008, the Company no longer manufactures TGS units, to which associated TGS sales arrangements required it to provide upgrades and enhancements, through and including the delivery of the RIO system. The Company commercially released the RIO system in the first quarter of 2009. Sales arrangements for RIO systems do not require the Company to provide upgrades and enhancements. As a result, revenues related to RIO system sales are generally recognized upon installation of the system and training of at least one surgeon.
For sales of TGS units through December 31, 2008, VSOE of fair value was not established for upgrades and enhancements (through and including delivery of the RIO system), which the TGS sales arrangements required the Company to provide. Accordingly, prior to delivery of the RIO system, sales of TGS units were recorded as deferred revenue and the direct cost of revenue associated with the sale of TGS units was recorded as deferred cost of revenue. Revenue for all previously deferred TGS sales was recognized in the Company’s statement of operations during the year ended December 31, 2009, upon delivery of the RIO system. As of December 31, 2010, the deferred revenue balance consists primarily of deferred service revenue as discussed below.
Costs associated with establishing an accrual for royalties covered by licensing arrangements related to the sale of RIO systems are expensed upon installation and are included in cost of revenue - systems, in the statements of operations.
93
Table of Contents
Deferred Revenue and Deferred Cost of Revenue
Deferred revenue consists of deferred service revenue and deferred system revenue. Deferred service revenue results from the advance payment for services to be delivered over a period of time, usually in one-year increments. Service revenue is recognized ratably over the service period. Deferred system revenue arises from timing differences between the installation of RIO systems and satisfaction of all revenue recognition criteria consistent with the Company’s revenue recognition policy. Deferred revenue expected to be realized within one year is classified as a current liability. The deferred revenue balance as of December 31, 2010 consists primarily of deferred service revenue for warranty and maintenance services on the RIO system hardware.
Foreign Currency Transactions
Gains or losses from foreign currency transactions are included in interest and other income. To date, realized gains and losses recognized from foreign currency transactions were not significant.
Research and Development Costs
Costs related to research, design and development of products are charged to research and development expense as incurred. These costs include direct salary costs for research and development personnel, costs for materials used in research and development activities and costs for outside services.
Shipping and Handling Costs
Costs incurred for shipping and handling are included in cost of revenue at the time the expense is incurred.
Software Development Costs
Software development costs are included in research and development and are expensed as incurred. After technological feasibility is established, material software development costs are capitalized. The capitalized cost is then amortized on a straight-line basis over the estimated product life, or on the ratio of current revenue to total projected product revenue, whichever is greater. To date, the period between achieving technological feasibility, which the Company has defined as the establishment of a working model which typically occurs when the verification and validation testing is complete, and the general availability of such software has been short and software development costs qualifying for capitalization have been insignificant. Accordingly, the Company has not capitalized any software development costs to date.
Stock-Based Compensation
The Company recognizes compensation expense for its stock-based awards in accordance with ASC 718, Compensation-Stock Compensation. ASC 718 requires the recognition of compensation expense, using a fair value based method, for costs related to all stock-based payments including stock options. ASC 718 requires companies to estimate the fair value of stock-based payment awards on the date of grant using an option-pricing model.
The Company accounts for stock-based compensation arrangements with non-employees in accordance with the ASC 505-50, Equity-Based Payments to Non-Employees. The Company records the expense of such services based on the estimated fair value of the equity instrument using the Black-Scholes-Merton pricing model. The value of the equity instrument is charged to expense over the term of the service agreement.
See Note 8 for a detailed discussion of the various stock option plans and related stock-based compensation.
94
Table of Contents
Advertising Costs
Advertising costs are expensed as incurred. Advertising costs were approximately $1.6 million, $1.3 million and $1.4 million for the years ended December 31, 2010, 2009 and 2008, respectively.
Income Taxes
The Company accounts for income taxes under ASC 740, Income Taxes. Deferred income taxes are determined based upon differences between financial reporting and income tax bases of assets and liabilities and are measured using the enacted income tax rates and laws that will be in effect when the differences are expected to reverse. The Company recognizes any interest and penalties related to unrecognized tax benefits as a component of income tax expense.
Due to uncertainty surrounding realization of the deferred income tax assets in future periods, the Company has recorded a 100% valuation allowance against its net deferred tax assets. If it is determined in the future that it is more likely than not that the deferred income tax assets are realizable, the valuation allowance will be reduced.
Operating Leases
Rental payments and incentives are recognized on a straight-line basis over the life of a lease. See Note 7 for further discussion of operating leases.
Net Loss Per Share
The Company calculated net loss per share in accordance with ASC 260, Earnings per Share. Basic earnings per share (“EPS”) is calculated by dividing the net income or loss attributable to common stockholders by the weighted average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted EPS is computed by dividing the net income or loss attributable to common stockholders by the weighted average number of common shares outstanding for the period and the weighted average number of dilutive common stock equivalents outstanding for the period determined using the treasury stock method. The following table sets forth potential shares of common stock that are not included in the calculation of diluted net loss per share because to do so would be anti-dilutive as of the end of each period presented:
| | | | | | | | | | |
(in thousands) | | December 31, | |
| | 2010 | | 2009 | | 2008 | |
Stock options outstanding | | | 4,405 | | | 3,478 | | | 2,193 | |
Warrants to purchase common stock | | | 2,039 | | | 2,065 | | | 2,076 | |
Unvested restricted stock | | | 503 | | | 222 | | | 267 | |
Reclassifications
Certain reclassifications have been made to the prior periods’ statement of operations to conform to the current period’s presentation. The Company reclassified depreciation expense on its Implant Instruments from cost of revenue – procedures to selling, general and administrative expense as described in greater detail in “Property and Equipment” above. In addition, the Company reclassified its income tax expense from selling, general and administrative expense to income tax expense.
3. Investments
The Company’s investments are classified as available-for-sale. Available-for-sale securities are carried at fair value, with the unrealized gains and losses included in other comprehensive income within stockholders’
95
Table of Contents
equity. Realized gains and losses, interest and dividends, amortization of premium and discount on investment securities and declines in value determined to be other-than-temporary on available-for-sale securities are included in interest and other income. During the years ended December 31, 2010, 2009 and 2008, realized gains and losses recognized on the sale of investments were not significant. The cost of securities sold is based on the specific identification method.
The amortized cost and fair value of short and long-term investments, with gross unrealized gains and losses, were as follows:
As of December 31, 2010
| | | | | | | | | | | | | |
(in thousands) | | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair
Value | |
Short-term investments: | | | | | | | | | | | | | |
U.S. government agencies | | $ | 34,483 | | $ | 4 | | $ | (16 | ) | $ | 34,471 | |
Certificates of deposit | | | 10,453 | | | 1 | | | (28 | ) | | 10,426 | |
U.S. corporate debt | | | 1,501 | | | 3 | | | ― | | | 1,504 | |
Long-term investments: | | | | | | | | | | | | | |
U.S. government agencies | | | 17,251 | | | 2 | | | (8 | ) | | 17,245 | |
Certificates of deposit | | | 6,095 | | | ― | | | (57 | ) | | 6,038 | |
Total investments | | $ | 69,783 | | $ | 10 | | $ | (109 | ) | $ | 69,684 | |
As of December 31, 2009
| | | | | | | | | | | | | |
(in thousands) | | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair
Value | |
Short-term investments: | | | | | | | | | | | | | |
U.S. government agencies | | $ | 32,860 | | $ | 31 | | $ | (24 | ) | $ | 32,867 | |
Certificates of deposit | | | 10,297 | | | 1 | | | (25 | ) | | 10,273 | |
U.S. corporate debt | | | 1,532 | | | 14 | | | ― | | | 1,546 | |
Long-term investments: | | | | | | | | | | | | | |
U.S. government agencies | | | 5,418 | | | ― | | | (18 | ) | | 5,400 | |
Certificates of deposit | | | 2,462 | | | ― | | | (10 | ) | | 2,452 | |
U.S. corporate debt | | | 1,506 | | | 10 | | | ― | | | 1,516 | |
Total investments | | $ | 54,075 | | $ | 56 | | $ | (77 | ) | $ | 54,054 | |
As of December 31, 2010 and December 31, 2009, all short-term investments had maturity dates of less than one year. As of December 31, 2010 and December 31, 2009, all long-term investments had maturity dates between one and three years.
96
Table of Contents
The fair values of the Company’s investments based on the level of inputs are summarized below:
| | | | | | | | | | | | | |
(in thousands) | | Fair Value Measurements at the Reporting Date Using | |
| | December
31, 2010 | | Quoted
Prices in
Active
Markets for
Identical
Assets
(Level 1) | | Significant
Other
Observable
Inputs
(Level 2) | | Significant
Unobservable
Inputs
(Level 3) | |
Short-term investments: | | | | | | | | | | | | | |
U.S. government agencies | | $ | 34,471 | | $ | 34,471 | | $ | ― | | $ | ― | |
Certificates of deposit | | | 10,426 | | | 10,426 | | | ― | | | ― | |
U.S. corporate debt | | | 1,504 | | | 1,504 | | | ― | | | ― | |
Long-term investments: | | | | | | | | | | | | | |
U.S. government agencies | | | 17,245 | | | 17,245 | | | ― | | | ― | |
Certificates of deposit | | | 6,038 | | | 6,038 | | | ― | | | ― | |
Total investments | | $ | 69,684 | | $ | 69,684 | | $ | ― | | $ | ― | |
No investments measured at fair value on a recurring basis used Level 3 or significant unobservable inputs for the year ended December 31, 2010. There have been no transfers between Level 1 and Level 2 measurements during the year ended December 31, 2010.
4. Selected Balance Sheet Components
| | | | | | | |
(in thousands) | | December 31, | |
| | 2010 | | 2009 | |
Inventory: | | | | | | | |
Raw materials | | $ | 2,522 | | $ | 2,770 | |
Work-in-process | | | 972 | | | 932 | |
Finished goods | | | 7,010 | | | 6,488 | |
Total inventory | | $ | 10,504 | | $ | 10,190 | |
The Company incurred inventory write-offs totaling approximately $1.7 million and $1.1 million during the years ended December 31, 2010 and 2009, respectively.
For the year ended December 31, 2010, the Company wrote-off $2.6 million, or $(0.08) per basic and diluted share, of excess RESTORIS unicompartmental knee implant system (“RESTORIS Classic”) implants and related instrumentation. Excess implants of $1.4 million was charged to cost of revenue – procedures, cancellation charges of $208,000 was charged to cost of revenue – service and other and excess instrumentation of $1.0 million was charged to selling, general and administrative expenses. These charges were necessitated by the rapid adoption of the RESTORIS MCK multicompartmental knee implant system (“RESTORIS MCK”) and the corresponding decline in the usage of RESTORIS Classic. RESTORIS Classic was introduced in the third quarter of 2008 and was modeled after existing well-known unicompartmental designs. In connection with the launch of the RIO system, in the second quarter of 2009, the Company launched its next generation RESTORIS MCK which was designed as a premium addition to the RESTORIS product family with the goal of delivering a more natural feeling knee by preserving bone and providing anatomical features such as high flexion.
Write-offs of $1.1 million in 2009 primarily relate to technology changes associated with the launch of the RIO system in 2009 and disposal of spare TGS inventory associated with the launch of the RIO system.
97
Table of Contents
The Company reviews its inventory periodically to determine net realizable value and considers product upgrades in its periodic review of realizability. Depending on demand for the Company’s products and technical obsolescence, additional future write-offs of the Company’s inventory may occur.
| | | | | | | | | | |
(in thousands) | | December 31, | | Estimated
Useful Life | |
| | 2010 | | 2009 | | |
Property and equipment: | | | | | | | | | | |
Consigned RIO systems and instruments | | $ | 3,287 | | $ | 2,551 | | | 3-5 years | |
Service and demo RIO systems and instruments | | | 4,029 | | | 2,354 | | | 3 years | |
Computer equipment and software | | | 2,960 | | | 2,160 | | | 3-5 years | |
Manufacturing and laboratory equipment | | | 1,945 | | | 1,654 | | | 3-5 years | |
Undeployed implant instruments | | | 1,226 | | | ― | | | See note 2 | |
Office furniture and equipment | | | 1,038 | | | 882 | | | 7 years | |
Leasehold improvements | | | 738 | | | 607 | | | Lesser of 5-10 years or lease term | |
| | | 15,223 | | | 10,208 | | | | |
Less accumulated depreciation and amortization | | | (6,011 | ) | | (4,003 | ) | | | |
Total property and equipment, net | | $ | 9,212 | | $ | 6,205 | | | | |
| | | | | | | |
(in thousands) | | December 31, | |
| | 2010 | | 2009 | |
Other accrued liabilities: | | | | | | | |
Accrued royalties | | $ | 833 | | $ | 413 | |
Accrued consulting fees | | | 792 | | | 231 | |
Other | | | 3,439 | | | 2,228 | |
| | $ | 5,064 | | $ | 2,872 | |
98
Table of Contents
5. Related Party Transactions
Securities Purchase Agreement
In October 2008, the Company entered into a Securities Purchase Agreement for an equity financing of up to approximately $60 million, with initial gross proceeds of approximately $40.2 million, which the Company closed on October 31, 2008, and conditional access to an additional $20 million. The financing resulted in net proceeds to the Company of approximately $39.7 million, after expenses of approximately $525,000. In connection with the financing, the Company issued and sold to the participating investors 6,451,613 shares of its common stock at a purchase price of $6.20 per share and issued to participating investors, at the purchase price of $0.125 per warrant, warrants to purchase 1,290,323 shares of common stock at an exercise price of $7.44 per share. The warrants became exercisable on April 29, 2009 and have a seven-year term.
Subject to the Company’s satisfaction of certain business related milestones before December 31, 2009, the Company had the right (the “Call Right”) to require certain participants in the financing to purchase an additional $20 million of common stock and warrants to purchase common stock. The Company did not exercise its Call Right, which expired on December 31, 2009, to require these participants to purchase an additional $20 million of common stock. At the initial closing, the investors that agreed to provide the additional $20 million investment received warrants to purchase an additional 322,581 shares of common stock at a purchase price of $0.125 per warrant and an exercise price of $6.20 per share. These warrants became exercisable on December 31, 2009 and have a seven-year term.
The participating investors consisted of eleven accredited investors, six of which were existing stockholders of the Company who were deemed to be affiliates of the Company by virtue of their being represented on the Company’s Board of Directors or by virtue of their Board membership.
Asset Purchase Agreement
In February 2010, the Company completed the acquisition of substantially all of the intellectual property portfolio of Z-Kat, Inc. (“Z-Kat”). The terms of the Asset Purchase Agreement between the Company and Z-Kat (the “Asset Purchase Agreement”) terminated the Company’s prior licenses with Z-Kat, including Z-Kat’s nonexclusive sublicense to the Company’s intellectual property portfolio, and transferred to the Company ownership rights to certain intellectual property assets for core technologies in computer assisted surgery (“CAS”), haptics and robotics, including U.S. and foreign patents and patent applications, proprietary software and documentation, trade secrets and trademarks owned by Z-Kat, and certain contractual and other rights to patents, patent applications and other intellectual property licensed to Z-Kat under licenses. In connection with the acquisition, the Company also entered into a new sublicense agreement with Z-Kat (the “License Agreement”) pertaining to certain intellectual property for technologies in CAS licensed by Z-Kat. The License Agreement was terminated by MAKO in December 2010. Certain of the Company’s rights under the Asset Purchase Agreement remain subject to any prior license granted by Z-Kat, including a license to Biomet Manufacturing Corp. In consideration for consummation of the transactions contemplated by the Asset Purchase Agreement and License Agreement, the Company issued 230,458 shares of its unregistered common stock to Z-Kat in a private placement, which was treated as a related party transaction because certain directors and executive officers of the Company had a material interest in Z-Kat by virtue of their ownership of Z-Kat stock. The Company and Z-Kat are not entities under common control. The Asset Purchase Agreement and License Agreement were approved by the independent members of the board of directors and audit committee of the Company. The value of the intellectual property acquired under the Asset Purchase Agreement of $3.1 million was based on the closing price per share of $13.25 of the Company’s common stock on February 25, 2010, the date the transaction was closed, and was recorded as an intangible asset and is being amortized over its estimated useful life of eight years.
License Agreement
In August 2009, the Company entered into a License Agreement (the “Sensor Agreement”) with a third-party sensor company associated with the potential future development of intellectual property and technology
99
Table of Contents
related to sensing devices in orthopedics. The Sensor Agreement required an initial payment of $50,000 and required future payments in the event that the Company decided to enter into a licensing and supply agreement with the third-party sensor company following the end of the research and development period. In August 2010, the Company exercised its option to enter into a non-exclusive license and supply agreement for an upfront payment of $250,000 (the “Non-Exclusive License Payment”). The Non-Exclusive License Payment was recorded as an intangible asset and will be amortized over its estimated useful life of ten years. In October 2010, the Company exercised its option to enter into an exclusive license and supply agreement which required an upfront payment of $500,000 and a future payment of $250,000 which is anticipated to be paid in the first quarter of 2011 (the “Exclusive License Payments”). The Exclusive License Payments will be recorded as an intangible asset and amortized over its estimated useful life of approximately five years. The Sensor Agreement was treated as a related party transaction because certain directors of the Company had a material interest in the third-party sensor company by virtue of their ownership of the third-party sensor company’s outstanding stock. The Company and the third-party sensor company are not entities under common control.
6. Intangible Assets
The Company’s intangible assets are comprised of purchased patents, patent applications and licenses to intellectual property rights (the “Licenses”). The Licenses are amortized on a straight line basis over their estimated useful lives which range from approximately 5 to 13 years. See Note 7 for additional discussion of Licenses.
The following tables present details of MAKO’s intangible assets:
| | | | | | | | | | | | | |
| | December 31, | |
(in thousands) | | 2010 | | 2009 | |
| | Amount | | Weighted
Average
Amortization
Period | | Amount | | Weighted
Average
Amortization
Period | |
Licenses | | $ | 7,880 | | | 9.4 | | $ | 6,679 | | | 9.9 | |
Z-Kat intellectual property (see Note 5) | | | 3,166 | | | 8.0 | | | ― | | | | |
| | | 11,046 | | | 9.0 | | | 6,679 | | | 9.9 | |
Less: accumulated amortization | | | (3,516 | ) | | | | | (2,445 | ) | | | |
Intangible assets, net | | $ | 7,530 | | | | | $ | 4,234 | | | | |
Amortization expense related to intangible assets was approximately $1.1 million, $682,000 and $660,000 for the years ended December 31, 2010, 2009 and 2008, respectively.
The estimated future amortization expense of intangible assets for the next five years as of December 31, 2010 is as follows:
| | | | |
(in thousands) | | | | |
2011 | | $ | 1,279 | |
2012 | | | 1,279 | |
2013 | | | 1,273 | |
2014 | | | 1,245 | |
2015 | | | 1,198 | |
Total | | $ | 6,274 | |
100
Table of Contents
7. Commitments and Contingencies
Operating Leases
In September 2010, the Company entered into an expanded ten year operating lease for the Company’s existing headquarters in Fort Lauderdale, Florida, to allow for expansion to support anticipated growth, and terminated the previous lease. Under the new lease, the Company has the option to renew its facility lease for two consecutive five year periods. The lease provides for periodic rent increases and requires the Company to pay the operating costs including taxes, insurance and maintenance. Rent expense on a straight-line basis was $624,000, $613,000 and $498,000 for the years ended December 31, 2010, 2009 and 2008, respectively. The rent expense for the years ended December 31, 2010, 2009 and 2008 included the Company’s monthly variable operating costs of the facility.
Future minimum lease commitments, excluding monthly variable operating costs, under the Company’s operating lease as of December 31, 2010 are as follows:
| | | | |
(in thousands) | | | | |
2011 | | $ | 290 | |
2012 | | | 337 | |
2013 | | | 352 | |
2014 | | | 504 | |
2015 | | | 657 | |
Thereafter | | | 3,683 | |
Total | | $ | 5,823 | |
Purchase Commitments
At December 31, 2010, the Company was committed to make future purchases for inventory related items and instrumentation under various purchase arrangements with fixed purchase provisions aggregating approximately $8.0 million.
License and Royalty Agreements
The Asset Purchase Agreement with Z-Kat includes licenses with third-party intellectual property rights for which the Company is obligated to make ongoing royalty payments of 2% on the sale of certain products or components thereof and minimum annual payments totaling $75,000.
See Note 5 for further discussion of the Asset Purchase Agreement.
In March 2006, the Company entered into a license agreement that covers a number of technologies related to the application of computers and robotics to surgery in exchange for a payment of $2 million upon execution of the agreement (the “Upfront License Fee”) and a deferred payment of $4 million payable upon a change of control, as defined (e.g., IPO, acquisition or change in voting ownership greater than 50.01%) (the “Deferred License Fee”). The license also requires royalty payments of 2% of the selling price of each RIO system. The Upfront License Fee and net present value of the Deferred License Fee were included in intangible assets in the accompanying balance sheets. The net present value of the Deferred License Fee obligation was approximately $3.4 million, net of a discount of $590,000 and was recorded as a long-term debt obligation as the Company believed it was probable at the inception of the agreement that the contingent obligation would become payable. The net present value of the Deferred License Fee was determined using an incremental borrowing rate of 8% and an expected payment date of approximately two years from the effective date of the license agreement. The discount on the debt obligation was being amortized over the estimated term of the Deferred License Fee obligation as interest expense which was approximately $45,000 for the year ended December 31, 2008, in the accompanying statements of operations. In February 2008, the Company paid the $4 million Deferred License Fee due upon completion of the Company’s IPO.
In May 2009, the Company entered into a license agreement for patents relating to its RIO system (the “Robotic Arm License”). The Robotic Arm License requires running royalty payments of 1.8% on sales of Company’s RIO systems and requires annual minimum royalty payments on sales of the Company’s RIO systems
101
Table of Contents
inclusive of the running royalty payments. The minimum running royalties are estimated to be approximately $1.0 million for the year ended December 31, 2010, and increase annually thereafter through 2013. The minimum running royalties for the year ended December 31, 2013 and for each subsequent year through the term of the agreement are estimated to be approximately $1.7 million annually.
Effective as of January 2010, the Company entered into a research and license agreement (the “Research and License Agreement”) associated with potential future products for use with the Company’s RIO system. The Research and License Agreement required an upfront payment of $300,000 for research services upon the execution of the agreement and requires an additional $500,000 to be paid quarterly over two years beginning in January 2011. The payments for research services are being expensed as research and development expense on a straight-line basis from January 2010 through December 2012, the period in which research services are being performed under the Research and License Agreement. The Research and License Agreement required a $200,000 upfront license payment upon execution of the agreement, running royalty payments of 3% on sales of products and processes covered under the Agreement and requires minimum royalty payments (inclusive of running royalty payments) not to exceed $250,000 over the term of agreement.
The Company has other license agreements related to current product offerings and research and development projects. Upfront license fees paid for these agreements total approximately $2.1 million. Royalty payments related to these agreements are anticipated to range between 1% and 6% of future sales of the Company’s RIO system and components thereof and/or products. These royalty payments are subject to certain minimum annual royalty payments as shown in the schedule below. The terms of these license agreements continue until the related licensed patents and intellectual property rights expire, which is expected to range between 6 and 16 years. The net expense related to the Company’s license and royalty agreements was approximately $2.0 million, $1.5 million and $525,000 for the years ended December 31, 2010, 2009 and 2008, respectively.
As of December 31, 2010, future annual minimum royalty payments under the licenses and sublicenses are anticipated to be as follows:
| | | | |
(in thousands) | | | | |
2011 | | $ | 1,069 | |
2012 | | | 1,554 | |
2013 | | | 1,984 | |
2014 | | | 2,034 | |
2015 | | | 2,034 | |
Thereafter | | | 5,475 | |
| | $ | 14,150 | |
Development Agreements
In June 2009, the Company entered into a Research and Development License and Supply Agreement (the “R&D Agreement”) associated with a potential future implant system for RIO-enabled hip MAKOplasty procedures. The R&D Agreement required an up-front payment of $450,000, and requires future milestone payments based on development progress. The aggregate milestone payments the Company is obligated to pay under the R&D Agreement are $1.6 million assuming the achievement of all development milestones. Through December 31, 2010, the Company paid the $450,000 up-front payment and $1.1 million of milestone payments which became due upon the achievement of the related milestones. The aggregate up-front payment and milestone payments of $2.0 million the Company is required to pay under the R&D Agreement were recognized as research and development expense on a straight-line basis from June 2009 through August 2010, the period in which development services were performed under the R&D Agreement. As of December 31, 2010, the Company had an accrued liability of $500,000 which will become payable upon the achievement of the final milestone under the R&D Agreement, which the Company believes is probable.
102
Table of Contents
In October 2010, the Company entered into a Strategic Alliance Agreement (the “Pipeline Agreement”) with Pipeline Biomedical Holdings, LLC (“Pipeline”) to develop and supply potential future advanced implant technologies for use with the Company’s RIO system, including the development of a MAKO-branded RESTORIS family of hip implant systems for use with the hip MAKOplasty application. Upon execution of the Pipeline Agreement on October 1, 2010, the Company issued and delivered to Pipeline 203,417 unregistered restricted shares of its common stock (the “Pipeline Shares”) as consideration for the rights granted to MAKO under the Pipeline Agreement. The restricted shares vest upon achievement of certain performance conditions within six months of the effective date of the Pipeline Agreement. If the performance conditions are not achieved, the Company may terminate the Pipeline Agreement at its option subject to a breakup fee not to exceed $800,000 (the “Breakup Fee”). The value of the Pipeline Shares will be recognized as a component of research and development expense on a straight line basis over 33 months from the effective date of the Pipeline Agreement through June 30, 2013 – the period over which Pipeline is expected to perform development services under the Pipeline Agreement. In accordance with ASC 505-50-30-30; however, no research and development expense associated with the services under the Pipeline Agreement will be recognized for the Pipeline Shares until achievement of the performance conditions. Although the Company believes the performance conditions will be met by Pipeline, as of December 31, 2010, the Company accrued $400,000 in expense for its obligation under the Breakup Fee. Upon achievement of the performance conditions, the Company will record an adjustment for the difference between the amount accrued to date for the Breakup Fee obligation and the ratable portion of the expense to be recognized for the Pipeline Shares from the effective date of the Pipeline Agreement to the achievement of the performance conditions. The total amount of expense to be recognized for the Pipeline Shares will be determined on the date the performance conditions are achieved. The Company has no further obligation beyond the previously issued Pipeline Shares to fund Pipeline’s research of implant technologies under the Pipeline Agreement. The Pipeline Agreement contains provisions under which Pipeline will supply the Company implants developed under the Pipeline Agreement. Subsequent to December 31, 2010, the performance conditions were achieved and the Pipeline Shares vested.
Contingencies
The Company is a party to legal contingencies or claims arising in the normal course of business, none of which the Company believes is material to its financial position, results of operations or cash flows.
8. Stockholders’ Equity
Preferred Stock
As of December 31, 2010 and 2009, the Company was authorized to issue 27,000,000 shares of $0.001 par value preferred stock. As of December 31, 2010 and 2009, there were no shares of preferred stock issued or outstanding. All shares of Series A, B and C redeemable convertible preferred stock that were issued and outstanding as of December 31, 2007 converted into 10,945,080 shares of common stock upon closing of the Company’s IPO in February 2008.
Common Stock
As of December 31, 2010 and 2009, the Company was authorized to issue 135,000,000 shares of $0.001 par value common stock. Common stockholders are entitled to dividends as and if declared by the Board of Directors, subject to the rights of holders of all classes of stock outstanding having priority rights as to dividends. There have been no dividends declared to date on the common stock. The holder of each share of common stock is entitled to one vote.
In December 2004, the Company issued 189,768 shares of restricted common stock to certain consultants (the “Consultant Restricted Stock”). The Consultant Restricted Stock vested in tranches upon the Company’s achievement of certain business milestones and any unvested restricted stock vested immediately upon completion of an initial public offering of common stock. Upon vesting, the Company recorded a consulting expense equal to the estimated fair value of the Company’s common stock on the date of vesting. As of January 1,
103
Table of Contents
2008, 94,884 shares of the Consultant Restricted Stock were unvested. Upon closing of the IPO in February 2008, the vesting of the remaining 94,884 shares of Consultant Restricted Stock was accelerated and the Company recognized $949,000 of compensation expense associated with the accelerated vesting of the Consultant Restricted Stock during the year ended December 31, 2008 based on the IPO price of $10.00 per share.
Comprehensive Loss
Comprehensive loss is defined as the change in equity from transactions and other events and circumstances other than those resulting from investments by owners and distributions to owners. For the years ended December 31, 2010, 2009 and 2008, the Company recorded comprehensive losses of approximately $38.8 million, $34.1 million and $37.0 million, respectively. The difference between comprehensive loss and net loss for the years ended December 31, 2010, 2009 and 2008 is due to changes in unrealized gains and losses on the Company’s available-for-sale securities.
401K Plan
The Company maintains a qualified deferred compensation plan under Section 401K of the Internal Revenue Code, covering substantially all full-time employees, which permits employees to contribute up to 84% of pre-tax annual compensation up to annual statutory limitations. The discretionary company match for employee contributions to the plan is 25% of up to the first 6% of the participant’s earnings contributed to the plan. The discretionary company match commenced in 2008 and to date has not been significant.
Employee Stock Purchase Plan
In January 2008, the Company’s Board of Directors and stockholders approved the MAKO Surgical Corp. 2008 Employee Stock Purchase Plan (the “2008 Employee Stock Purchase Plan”). The 2008 Employee Stock Purchase Plan became effective upon closing of the IPO. The 2008 Employee Stock Purchase Plan authorizes the issuance of 625,000 shares of the Company’s common stock for purchase by eligible employees of the Company or any of its participating affiliates. The shares of common stock issuable under the 2008 Employee Stock Purchase Plan may be authorized but unissued shares, treasury shares or shares purchased on the open market. The purchase price for a purchase period may not be less than 85% of the fair market value of the Company’s common stock on the first trading day of the applicable purchase period or the last trading day of such purchase period, whichever is lower. During the year ended December 31, 2010, the Company issued approximately 86,000 shares under the 2008 Employee Stock Purchase Plan. As of December 31, 2010, there were approximately 467,000 shares reserved for future grant under the 2008 Employee Stock Purchase Plan.
Stock Option Plans and Stock-Based Compensation
The Company recognizes compensation expense for its stock-based awards in accordance with ASC 718,Compensation-Stock Compensation. ASC 718 requires the recognition of compensation expense, using a fair value based method, for costs related to all stock-based payments including stock options. ASC 718 requires companies to estimate the fair value of stock-based payment awards on the date of grant using an option-pricing model.
During the years ended December 31, 2010, 2009 and 2008, stock-based compensation expense was $6.4 million, $4.0 million and $3.3 million respectively. Included within stock-based compensation expense for the year ended December 31, 2010 were $4.8 million related to stock option grants, $1.3 million related to the partial vesting of shares of restricted stock granted to the Company’s CEO at various dates from 2006 through 2010, and $256,000 related to employee stock purchases under the 2008 Employee Stock Purchase Plan.
In December 2004, the Company’s stockholders approved the Company’s 2004 Stock Incentive Plan (the “2004 Plan”). Under the 2004 Plan, the Board of Directors was authorized to grant restricted common stock and options to purchase shares of common stock to employees, directors and consultants. No further awards will be made under the 2004 Plan. In January 2008, the Company’s Board of Directors and stockholders approved the
104
Table of Contents
MAKO Surgical Corp. 2008 Omnibus Incentive Plan (the “2008 Plan,” and together with the 2004 Plan, the “Plans”). The 2008 Plan became effective upon the closing of the IPO and will expire January 9, 2018 unless earlier terminated by the Board of Directors. Awards under the 2008 Plan may be made in the form of: stock options, which may be either incentive stock options or non-qualified stock options; stock appreciation rights; restricted stock; restricted stock units; dividend equivalent rights; performance shares; performance units; cash-based awards; other stock-based awards, including unrestricted shares; and any combination of the foregoing.
Generally, the Company’s outstanding stock options vest over four years. Stock options granted to certain non-employee directors prior to 2010 generally vest over three years; however, stock options granted to non-employee directors after January 1, 2010 generally vest over one year. Continued vesting typically terminates when the employment or consulting relationship ends. Vesting generally begins on the date of grant.
The 2008 Plan contains an evergreen provision whereby the authorized shares increase on January 1st of each year in an amount equal to the least of (1) four percent (4%) of the total number of shares of the Company’s common stock outstanding on December 31st of the preceding year, (2) 2.5 million shares and (3) a number of shares determined by the Company’s Board of Directors that is lesser than (1) and (2). The number of additional shares authorized under the 2008 Plan on January 1, 2010 and 2011 was approximately 1,330,000 and 1,618,000, respectively.
Under the terms of the Plans, the maximum term of options intended to be incentive stock options granted to persons who own at least 10% of the voting power of all outstanding stock on the date of grant is 5 years. The maximum term of all other options is 10 years. Options issued under the 2008 Plan that are forfeited or expire will again be made available for issuing grants under the 2008 Plan. Options issued under the 2004 Plan that are forfeited or expire will not be made available for issuing grants under the 2008 Plan. All future equity awards will be made under the Company’s 2008 Plan.
As of December 31, 2010, the Company had reserved shares of common stock for the issuance of common stock under the 2008 Employee Stock Purchase Plan, the exercise of warrants and the issuance of options granted under the 2008 Plan as follows:
| | | | |
(in thousands) | | | | |
2008 Employee Stock Purchase Plan | | | 625 | |
Warrants to purchase common stock | | | 2,076 | |
2008 Plan | | | 3,413 | |
| | | 6,114 | |
Only employees are eligible to receive incentive stock options. Non-employees may be granted non-qualified options. The Board of Directors has the authority to set the exercise price of all options granted, subject to the exercise price of incentive stock options being no less than 100% of the estimated fair value, as determined by the Board of Directors, of a share of common stock on the date of grant; and no less than 85% of the estimated fair value for non-qualified stock options, except for an employee or non-employee with options who owns more than 10% of the voting power of all classes of stock of the Company, in which case the exercise price shall be no less than 110% of the fair market value per share on the grant date. Options become exercisable as determined by the Board of Directors.
105
Table of Contents
Activity under the Plans is summarized as follows:
| | | | | | | | | | |
(in thousands, except per share data) | | | | | Outstanding Options | |
| | Shares/Options
Available
For Grant | | Number of
Options | | Weighted
Average
Exercise
Price | |
Balance at December 31, 2009 | | | 174 | | | 3,478 | | | 6.71 | |
Shares reserved | | | 1,330 | | | ― | | | ― | |
Restricted stock issued | | | (175 | ) | | ― | | | ― | |
Shares swapped under the 2008 Plan | | | 2 | | | ― | | | ― | |
Options granted | | | (1,183 | ) | | 1,183 | | | 12.41 | |
Options exercised | | | ― | | | (180 | ) | | 3.48 | |
Options forfeited under the 2004 Plan | | | ― | | | (20 | ) | | 6.96 | |
Options forfeited under the 2008 Plan | | | 56 | | | (56 | ) | | 8.75 | |
Balance at December 31, 2010 | | | 204 | | | 4,405 | | $ | 8.35 | |
The options outstanding and exercisable, by exercise price, at December 31, 2010 were as follows:
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | Options Outstanding | | Options Exercisable | |
(in thousands,
except per
share data) | | Number
Of
Options | | Weighted
Average
Remaining
Contractual
Life (Years) | | Weighted
Average
Exercise
Price | | Aggregate Intrinsic
Value (1) | | Number
Of
Options | | Weighted
Average
Remaining
Contractual
Life (Years) | | Weighted
Average
Exercise
Price | | Aggregate Intrinsic
Value (1) | |
Range of Exercise Prices: | | | | | | | | | | | | | | | | | | | | | | | | | |
$0.67 | | | 233 | | | | | $ | 0.67 | | | | | | 233 | | | | | $ | 0.67 | | | | |
$1.27 – $2.48 | | | 664 | | | | | $ | 1.75 | | | | | | 647 | | | | | $ | 1.73 | | | | |
$6.90 – $8.06 | | | 1,410 | | | | | $ | 7.94 | | | | | | 616 | | | | | $ | 7.96 | | | | |
$8.27 – 11.41 | | | 949 | | | | | $ | 10.50 | | | | | | 604 | | | | | $ | 10.52 | | | | |
$11.95 – 15.12 | | | 1,149 | | | | | $ | 12.44 | | | | | | 211 | | | | | $ | 12.39 | | | | |
| | | 4,405 | | | 7.54 | | $ | 8.35 | | $ | 30,272 | | | 2,311 | | | 6.72 | | $ | 6.56 | | $ | 20,023 | |
| | |
| (1) | The aggregate intrinsic value represents the total pre-tax intrinsic value, based on the Company’s closing stock price of $15.22 on December 31, 2010, which would have been received by the option holders had all option holders exercised their options as of that date. |
As of December 31, 2010, approximately 4,308,000 options were vested and expected to vest at a weighted average exercise price of $8.31 per share, a weighted average contractual life of 7.5 years and aggregate intrinsic value of $29.8 million.
The weighted average fair values of options granted were $6.48, $4.43 and $5.08 for the years ended December 31, 2010, 2009 and 2008, respectively. The total fair value of shares vested was approximately $4.6 million, $2.8 million and $1.3 million during the years ended December 31, 2010, 2009 and 2008, respectively. The total intrinsic value of options exercised was $1.6 million, $1.0 million and $491,000 for the years ended December 31, 2010, 2009 and 2008.
The Company records stock-based compensation expense on a straight-line basis over the vesting period. As of December 31, 2010, there was total unrecognized compensation cost of approximately $10.5 million, net of estimated forfeitures, related to non-vested stock option grants to the Company’s employees and non-employee directors. The unrecognized compensation cost will be adjusted for future changes in estimated forfeitures, and is expected to be recognized over a remaining weighted average period of 2.5 years as of December 31, 2010.
106
Table of Contents
On February 4, 2010, the Company issued 100,000 shares of restricted stock to its CEO at a fair value of $1.2 million, or $11.95 per share, on the date of issuance. The restricted stock will vest over a four-year period. On April 13, 2010, the Company issued 75,000 shares of restricted stock to its CEO at a fair value of approximately $476,000 on the date of issuance. The April 13, 2010 grant is subject to performance conditions based on the achievement of certain performance metrics. Upon satisfaction of the performance conditions, 50% of the shares will vest on March 13, 2013 and 50% of the shares will vest on March 13, 2014. For the year ended December 31, 2010, 28,307 shares of common stock were surrendered by the CEO to the Company to cover payroll taxes associated with the taxable income from the vesting of restricted stock previously granted to the Company’s CEO. As of December 31, 2010, 879,723 shares of restricted stock granted to the Company’s CEO were issued and outstanding.
Restricted stock activity for the year ended December 31, 2010 is as follows:
| | | | | | | |
(in thousands, except per share data) | | Shares | | Weighted Average
Fair Value | |
Unvested shares at December 31, 2009 | | 222 | | | $ | 8.86 | |
Unvested shares at December 31, 2010 | | 300 | | | $ | 7.64 | |
Shares granted in 2010 | | 175 | | | | 9.55 | |
Shares vested in 2010 | | 97 | | | | 10.30 | |
As of December 31, 2010, the remaining stock-based compensation expense for the restricted stock awards was approximately $2.3 million, which will be recognized on a straight line basis over a remaining weighted average period of 2.38 years.
The Company uses the Black-Scholes-Merton pricing model to determine the fair value of stock options. The determination of the fair value of stock-based payment awards on the date of grant using a pricing model is affected by our stock price as well as assumptions regarding a number of complex and subjective variables. These variables include our expected stock price volatility over the term of the awards, actual and projected employee stock option exercise behaviors, risk-free interest rates and expected dividends.
The estimated grant date fair values of the employee stock options were calculated using the Black-Scholes-Merton valuation model, based on the following assumptions:
| | | | | | | | | | |
Stock Option Plans | | Years Ended December 31, | |
| | 2010 | | 2009 | | 2008 | |
Risk-free interest rate | | | 2.04% - 3.36% | | | 1.99% - 3.53% | | | 1.59% - 3.62% | |
Expected life | | | 6.25 years | | | 6.25 years | | | 6.25 years | |
Expected dividends | | | ― | | | ― | | | ― | |
Expected volatility | | | 50.15% - 50.74% | | | 54.43% - 57.71% | | | 56.36% - 58.31% | |
The Company estimates the fair value of each share of stock which will be issued under the 2008 Employee Stock Purchase Plan based upon its stock prices at the beginning of each offering period using a Black-Scholes-Merton pricing model and amortizes that value to expense over the plan purchase period. The fair values determined for the years ended December 31, 2010, 2009 and 2008, as well as the assumptions used in calculating those values are as follows:
107
Table of Contents
| | | | | | | | | | |
2008 Employee Stock Purchase Plan | | Year Ended
December 31, | | Year Ended
December 31, | | Year Ended
December 31, | |
| | 2010 | | 2009 | | 2008 | |
Fair Value | | $ | 2.21 - $3.68 | | $ | 1.82 - $2.54 | | $ | 1.82 - $1.89 | |
Assumptions | | | | | | | | | | |
Risk-free interest rate | | | 0.12% - 0.60% | | | 0.60% - 3.20% | | | 1.87% - 3.29% | |
Expected life | | | 0.25 years | | | 0.25 years | | | 0.25 years | |
Expected dividends | | | ― | | | ― | | | ― | |
Expected volatility | | | 34.50% - 52.08% | | | 34.50% - 60.68% | | | 57.05% - 60.68% | |
Risk-Free Interest Rate. The risk-free rate is based on U.S. Treasury zero-coupon issues with remaining terms similar to the expected term on the options.
Weighted-Average Expected Term. The expected term of options granted is determined using the average period the stock options are expected to remain outstanding and is based on the options vesting term, contractual terms and historical exercise and vesting information used to develop reasonable expectations about future exercise patterns and post-vesting employment termination behavior. The expected term of the 2008 Employee Stock Purchase Plan is equal to the duration of the purchase period.
Dividend Yield. The Company has never declared or paid any cash dividends and does not plan to pay cash dividends in the foreseeable future, and, therefore, used an expected dividend yield of zero in the valuation model.
Volatility. Since the Company was a private entity until February 2008 with no historical data regarding the volatility of its common stock, the expected volatility used for the years ended December 31, 2010, 2009 and 2008, is based on volatility of similar entities, referred to as “guideline” companies. In evaluating similarity, the Company considered factors such as industry, stage of life cycle and size.
Forfeitures. ASC 718 requires the Company to estimate forfeitures at the time of grant, and revise those estimates in subsequent periods if actual forfeitures differ from those estimates. The Company uses historical data to estimate pre-vesting option forfeitures and records stock-based compensation expense only for those awards that are expected to vest. All stock-based payment awards are amortized on a straight-line basis over the requisite service periods of the awards, which are generally the vesting periods. If the Company’s actual forfeiture rate is materially different from its estimate, the stock-based compensation expense could be significantly different from what the Company has recorded in the accompanying periods.
Warrants
In December 2004, the Company issued at the purchase price of $0.03 per share warrants to purchase 462,716 shares of common stock. The warrants are immediately exercisable at an exercise price of $3.00 per share, with the exercise period expiring in December 2014. As of December 31, 2010 and 2009, 425,915 and 451,916 warrants were outstanding and exercisable, respectively.
In October 2008, the Company issued warrants to purchase 1,290,323 shares of common stock at a purchase price of $0.125 per warrant and an exercise price of $7.44 per share. The warrants became exercisable on April 29, 2009 and have a seven-year term. As of December 31, 2010, all the warrants were outstanding and exercisable.
In October 2008, the Company issued warrants to purchase 322,581 shares of common stock at a purchase price of $0.125 per warrant and an exercise price of $6.20 per share. These warrants became exercisable on December 31, 2009 and have a seven-year term. As of December 31, 2010, all the warrants were outstanding and exercisable.
108
Table of Contents
9. Income Taxes
The provision for income taxes is as follows:
| | | | | | | | | | |
(in thousands) | | Years Ended | |
| | December 31,
2010 | | December 31,
2009 | | December 31,
2008 | |
Current income taxes: | | | | | | | | | | |
Federal | | $ | ― | | $ | ― | | $ | ― | |
State | | | 68 | | | 56 | | | ― | |
Total current income taxes | | | 68 | | | 56 | | | ― | |
Deferred income taxes | | | (14,184 | ) | | (12,593 | ) | | (13,031 | ) |
Change in valuation allowance | | | 14,184 | | | 12,593 | | | 13,031 | |
Provision for income taxes | | $ | 68 | | $ | 56 | | $ | ― | |
The Company accounts for income taxes under ASC 740,Income Taxes. Deferred income taxes are determined based upon differences between financial reporting and income tax bases of assets and liabilities and are measured using the enacted income tax rates and laws that will be in effect when the differences are expected to reverse. The Company recognizes any interest and penalties related to unrecognized tax benefits as a component of income tax expense.
No current or deferred income taxes were recorded for the years ended December 31, 2010, 2009 and 2008, as the Company’s income tax benefits were fully offset by a corresponding increase to the valuation allowance against its net deferred income tax assets.
At December 31, 2010, 2009 and 2008, the Company had federal and state net operating loss carryforwards of approximately $129.6 million, $100.2 million and $60 million, respectively, available to offset future taxable income. These net operating loss carryforwards will expire in varying amounts from 2024 through 2030.
The Tax Reform Act of 1986 limits the annual utilization of net operating loss and tax credit carryforwards, following an ownership change of the Company. Note that as a result of the Company’s equity financings in recent years, the Company underwent changes in ownership for purposes of the Tax Reform Act.
109
Table of Contents
Deferred income taxes reflect the net tax effect of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s net deferred income taxes are as follows:
| | | | | | | |
(in thousands) | | December 31, | |
| | 2010 | | 2009 | |
Current deferred income tax assets: | | | | | | | |
Deferred revenue | | $ | 1,256 | | $ | 212 | |
Reserves | | | 619 | | | ― | |
Accrued expenses | | | 502 | | | ― | |
Total current deferred income tax assets | | | 2,377 | | | 212 | |
| | | | | | | |
Noncurrent deferred income tax assets: | | | | | | |
Net operating loss carryforwards | | | 51,163 | | | 38,650 | |
Amortization | | | 591 | | | 404 | |
Other | | | 10 | | | 481 | |
Total noncurrent deferred income tax assets | | | 51,764 | | | 39,535 | |
| | | | | | | |
Current deferred income tax liabilities: | | | | | | | |
Other deferred income tax liabilities | | | ― | | | (2 | ) |
Total current deferred income tax liabilities | | | ― | | | (2 | ) |
| | | | | | | |
Noncurrent deferred income tax liabilities: | | | | | | | |
Other deferred income tax liabilities | | | (213 | ) | | ― | |
Total noncurrent deferred income tax liabilities | | | (213 | ) | | ― | |
| | | | | | | |
Less valuation allowance | | | (53,928 | ) | | (39,745 | ) |
Total deferred income tax assets, net | | $ | ― | | $ | ― | |
Due to uncertainty surrounding realization of the deferred income tax assets in future periods, the Company has recorded a 100% valuation allowance against its net deferred tax assets. If it is determined in the future that it is more likely than not that the deferred income tax assets are realizable, the valuation allowance will be reduced.
110
Table of Contents
The reconciliation of the income tax provision computed at the U.S. federal statutory rate to income tax provision is as follows:
| | | | | | | | | | |
| | Years Ended | |
| | December 31,
2010 | | December 31,
2009 | | December 31,
2008 | |
Tax at U.S. statutory rate | | | (35.00 | )% | | (35.00 | )% | | (35.00 | )% |
State taxes, net of federal impact | | | (4.49 | )% | | (3.28 | )% | | (3.26 | )% |
Non-deductible items | | | 5.46 | % | | 2.92 | % | | 3.13 | % |
Return to provision differences | | | (2.44 | )% | | ― | | | ― | |
Change in valuation allowance | | | 36.66 | % | | 35.13 | % | | 35.14 | % |
Other, net | | | (0.19 | )% | | 0.23 | % | | (0.01 | )% |
Effective income tax rate | | | 0.00 | % | | 0.00 | % | | 0.00 | % |
In accordance with ASC 740, the Company has decided to classify any interest and penalties as a component of income tax expense. To date, there have been no interest or penalties charged to the Company in relation to the underpayment of income taxes. The Company’s primary tax jurisdictions are in the United States and in multiple state jurisdictions. The tax years from 2005 through 2010 remain open and are subject to examination by the appropriate governmental agencies.
10. Selected Quarterly Data (Unaudited)
| | | | | | | | | | | | | |
(in thousands, except per share data) | | 2010 | |
| | Q1 | | Q2 | | Q3 | | Q4 (1) | |
Revenue | | $ | 7,249 | | $ | 10,251 | | $ | 12,014 | | $ | 14,782 | |
Gross profit | | | 3,253 | | | 6,580 | | | 7,455 | | | 8,835 | |
Loss from operations | | | (11,470 | ) | | (8,587 | ) | | (9,006 | ) | | (9,873 | ) |
Net loss attributable to common stockholders | | | (11,408 | ) | | (8,524 | ) | | (8,938 | ) | | (9,817 | ) |
Net loss per share – basic and diluted attributable to common stockholders | | | (0.34 | ) | | (0.26 | ) | | (0.27 | ) | | (0.26 | ) |
| | |
| (1) | During the fourth quarter of 2010, the Company determined that it had incorrectly recognized revenue and expenses associated with the initial warranty obligation and maintenance services included in all previous RIO system sales. Accordingly, in the fourth quarter of 2010, the Company recorded an adjustment to decrease revenue by $1.2 million, to reverse the accrual for its warranty and maintenance obligation by $552,000 and to increase net loss by $644,000, or $(0.02) per basic and diluted share. The adjustment arose over the quarters throughout 2009 and 2010 and did not materially affect the Company’s trend in earnings. As the adjustment was related to the correction of an error, the Company performed the analysis required by Staff Accounting Bulletin 99,Materiality, and Staff Accounting Bulletin 108,Considering the Effects of Prior Year Misstatements When Quantifying Misstatements in Current Year Financial Statements. Based on this analysis, the Company concluded that the effect of the error was not material to the quarters and years in the two year period ended December 31, 2010 from both a quantitative and qualitative perspective. |
111
Table of Contents
| | | | | | | | | | | | | |
(in thousands, except per share data) | | 2009 | |
| | Q1 (2) | | Q2 (2) | | Q3 | | Q4 | |
Revenue | | $ | 3,727 | | $ | 14,904 | | $ | 6,726 | | $ | 8,851 | |
Gross profit | | | 697 | | | 4,685 | | | 2,830 | | | 4,542 | |
Loss from operations | | | (9,102 | ) | | (6,491 | ) | | (9,447 | ) | | (9,356 | ) |
Net loss attributable to common stockholders | | | (8,885 | ) | | (6,424 | ) | | (9,439 | ) | | (9,275 | ) |
Net loss per share – basic and diluted attributable to common stockholders | | | (0.36 | ) | | (0.26 | ) | | (0.33 | ) | | (0.28 | ) |
| | |
| (2) | Revenue for the first and second quarters of 2009 includes approximately $2.5 million and $8.8 million, respectively, of revenue from previously deferred unit sales of our TGS. In accordance with our revenue recognition policy, recognition of revenue on unit sales of our TGS was deferred until delivery of the RIO system, which we commercially released in the first quarter of 2009. |
112
Table of Contents
| |
ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| |
ITEM 9A. | CONTROLS AND PROCEDURES |
In accordance with Rule 13a-15(b) of the Securities Exchange Act of 1934, or the Exchange Act, our management evaluated, with the participation of our chief executive officer and chief financial officer, or the Certifying Officers, the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rule 13a-15(e) under the Exchange Act) as of December 31, 2010. Based upon their evaluation of these disclosure controls and procedures, our Certifying Officers concluded that the disclosure controls and procedures were effective as of December 31, 2010 to provide reasonable assurance that information required to be disclosed by us in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time period specified in the SEC rules and forms, and to provide reasonable assurance that information required to be disclosed by us in the reports we file or submit under the Exchange Act is accumulated and communicated to our management, including our principal executive and principal financial officers, as appropriate, to allow timely decisions regarding required disclosure.
We believe that a controls system, no matter how well designed and operated, is based in part upon certain assumptions about the likelihood of future events, and therefore can only provide reasonable, not absolute, assurance that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) and 15d-15(f) promulgated under the Securities Exchange Act of 1934, as amended, as a process designed by, or under the supervision of, a company’s principal executive and financial officers, or the certifying officers, and effected by a company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Internal control over financial reporting includes policies and procedures that pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; provide reasonable assurance that transactions are recorded as necessary to permit preparation of our financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with the authorization of our board of directors and management; and provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements.
Under the supervision and with the participation of our management, including the certifying officers, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on this evaluation under the criteria established in Internal Control – Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2010. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The effectiveness of our internal control over financial reporting as of December 31, 2010 has been audited by our independent registered public account firm, as stated in their report, which is included herein.
113
Table of Contents
During the most recently completed fiscal quarter, there was no change in our internal control over financial reporting that has materially affected or is reasonably likely to materially affect, our internal control over financial reporting.
| |
ITEM 9B. | OTHER INFORMATION |
None
114
Table of Contents
PART III.
| |
ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by this item will be contained under the following headings in our definitive proxy statement to be filed in connection with our 2011 annual meeting of stockholders and, upon filing with the SEC, will be incorporated herein by reference:
| | |
| • | Section 16(a) Beneficial Ownership Reporting Compliance |
| | |
| • | Election of Directors |
| | |
| • | Board of Directors and Corporate Governance |
| | |
| • | Executive Officers |
| |
ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this item will be contained under the following headings in our definitive proxy statement to be filed in connection with our 2011 annual meeting of stockholders and, upon filing with the SEC, will be incorporated herein by reference:
| | |
| • | Director Compensation |
| | |
| • | Compensation Discussion and Analysis |
| | |
| • | Compensation Committee Report |
| | |
| • | Executive Compensation |
| |
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this item will be contained under the following heading in our definitive proxy statement to be filed in connection with our 2011 annual meeting of stockholders and, upon filing with the SEC, will be incorporated herein by reference:
The information under “Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities—Equity Compensation Plan Information” in this annual report on Form 10-K is also incorporated herein by reference.
| |
ITEM 13. | CERTAIN RELATIONSHIPS, RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE |
The information required by this item will be contained under the following heading in our definitive proxy statement to be filed in connection with our 2011 annual meeting of stockholders and, upon filing with the SEC, will be incorporated herein by reference:
| | |
| • | Board of Directors and Corporate Governance – Independent Directors |
| | |
| • | Certain Relationships and Related Person Transactions |
115
Table of Contents
| |
ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this item will be contained under the following heading in our definitive proxy statement to be filed in connection with our 2011 annual meeting of stockholders and, upon filing with the SEC, will be incorporated herein by reference:
| | |
| • | Ratification of the Appointment of Ernst & Young LLP as Independent Registered Public Accounting Firm |
116
Table of Contents
PART IV
| |
ITEM 15. | EXHIBITS, FINANCIAL STATEMENTS and FINANCIAL STATEMENT SCHEDULES |
| |
| (a) The following documents are filed as a part of this Annual Report on Form 10-K: |
| |
| 1.Financial Statements |
| |
| See Item 8, Financial Statements and Supplementary Data,Index to Financial Statements. |
| |
| 2.Financial Statement Schedules |
| |
No financial statement schedules are provided because the information called for is not required or is shown either in the financial statements or the notes thereto. |
|
| (b)Exhibits |
| | | |
Exhibit
No. | | Description | |
| | |
3.1 | | Third Amended and Restated Certificate of Incorporation of the Registrant, dated February 20, 2008 (2) |
| | |
3.2 | | Fourth Amended and Restated Bylaws of the Registrant effective October 31, 2008 (6) |
| | |
4.1 | | Securities Purchase Agreement by and among the Registrant and Investors named therein, dated as of October 28, 2008 (6) |
| | |
4.2 | | Form of Warrant (6) |
| | |
4.3 | | Form of Call Warrant (6) |
| | |
10.1 | | Form of Indemnity Agreement for Directors and Executive Officers (3) |
| | |
10.2+ | | 2004 Stock Incentive Plan and forms of agreements related thereto (3) |
| | |
10.3+ | | 2008 Omnibus Incentive Plan (3) |
| | |
10.4+ | | 2008 Employee Stock Purchase Plan (3) |
| | |
10.5+ | | Amended Employment Agreement, dated as of November 12, 2007, by and between Registrant and Maurice R. Ferré, M.D (3) |
| | |
10.6 | | Lease, by and between Registrant and Westport Business Park Associates LLP, dated September 8, 2010 (1) |
| | |
10.7+ | | Form of Incentive Stock Option Agreement related to the 2008 Omnibus Incentive Plan (4) |
| | |
10.8+ | | Employment Agreement between Registrant and Duncan Moffat, effective as of April 28, 2008 (5) |
| | |
10.9+ | | Form of Non-Qualified Stock Option Agreement related to the 2008 Omnibus Incentive Plan (5) |
| | |
10.10+ | | Form of Restricted Stock Unit Agreement related to the 2008 Omnibus Incentive Plan (5) |
| | |
10.11+ | | Form of Subscription Agreement related to the 2008 Employee Stock Purchase Plan (12) |
| | |
10.12+ | | Amendment to Amended Employment Agreement by and between Registrant and Maurice R. Ferré, M.D., effective February 13, 2009 (7) |
117
Table of Contents
| | |
10.13+ | | Amended and Restated Employment Agreement by and between Registrant and Fritz L. LaPorte, effective February 13, 2009 (7) |
| | |
10.14+ | | Amended and Restated Employment Agreement by and between Registrant and Menashe R. Frank, effective February 13, 2009 (7) |
| | |
10.15+ | | Amended and Restated Employment Agreement by and between Registrant and Steven J. Nunes, effective February 13, 2009 (7) |
| | |
10.16+ | | Employment Agreement by and between Registrant and Ivan Delevic, effective April 27, 2009 (8) |
| | |
10.17+ | | 2010 Leadership Cash Bonus Plan (9) |
| | |
10.18+ | | 2010 Performance Bonus Plan for S. Nunes – SVP of Sales & Marketing (9) |
| | |
10.19+ | | Second Amendment to Amended Employment Agreement by and between Registrant and Maurice R. Ferré, M.D., effective February 17, 2010 (9) |
| | |
10.20+ | | Employment Agreement between Registrant and James E. Keller, effective as of March 22, 2010 (10) |
| | |
10.21+ | | Employment Agreement between Registrant and Richard Leparmentier, effective as of March 29, 2010 (11) |
| | |
10.22+ | | First Amendment to Employment Agreement between Registrant and Ivan Delevic, effective as of April 13, 2010 (12) |
| | |
10.23+ | | Restricted Stock Agreement dated April 13, 2010 issued to Maurice R. Ferré (13) |
| | |
10.24+ | | 2011 Leadership Cash Bonus Plan (14) |
| | |
10.25+ | | Restricted Stock Agreement dated February 3, 2011 issued to Maurice R. Ferré (14) |
| | |
23 | | Consent of Independent Registered Public Accounting Firm (1) |
| | |
31.1 | | Certification of Chief Executive Officer pursuant to Rule 13a-14(a) of the Exchange Act (1) |
| | |
31.2 | | Certification of Chief Financial Officer pursuant to Rule 13a-14(a) of the Exchange Act (1) |
| | |
32.1 | | Certification of Chief Executive Officer pursuant to 18 U.S.C. §1350 (1) |
| | |
32.2 | | Certification of Chief Financial Officer pursuant to 18 U.S.C. §1350 (1) |
| | |
99.1 | | Registration Rights Agreement by and between Registrant and Z-Kat, Inc. dated February 25, 2010 (15) |
| |
(1) | Filed herewith |
| |
(2) | Incorporated by reference to Registrant’s Annual Report on Form 10-K for the period ended December 31, 2007 filed with the SEC on March 31, 2008 |
| |
(3) | Incorporated by reference to Registrant’s Registration Statement on Form S-1, as amended, filed with the SEC on September 19, 2007 (Registration No. 333-146162) |
| |
(4) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on February 26, 2008 |
| |
(5) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on April 29, 2008 |
| |
(6) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on October 30, 2008 |
118
Table of Contents
| |
(7) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on February 20, 2009 |
| |
(8) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on April 28, 2009 |
| |
(9) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on February 23, 2010 |
| |
(10) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on March 24, 2010 |
| |
(11) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on March 29, 2010 |
| |
(12) | Incorporated by reference to Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010 filed with the SEC on May 7, 2010 |
| |
(13) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on April 15, 2010 |
| |
(14) | Incorporated by reference to Registrant’s Current Report on Form 8-K filed with the SEC on February 4, 2011 |
| |
(15) | Incorporated by reference to Registrant’s Annual Report on Form 10-K for the period ended December 31, 2009 filed with the SEC on March 10, 2010 |
| |
+ | Indicates management contract or compensatory plan. |
119
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | |
| By: | /s/ Maurice R. Ferré, M.D. | |
| | President, Chief Executive Officer | |
| | and Chairman of the Board | |
| | (Principal Executive Officer) | |
Dated: March 10, 2011 | | | |
|
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated. |
| | | | |
Signature | | Title | | Date |
| | | | |
/s/ Maurice R. Ferré, M.D. | | President, Chief Executive Officer and Chairman of the Board (Principal Executive Officer) | | March 10, 2011 |
Maurice R. Ferré, M.D. | | | |
| | | | |
/s/ Fritz L. LaPorte | | Senior Vice President of Finance and Administration, Chief Financial Officer and Treasurer (Principal Accounting and Financial Officer) | | March 10, 2011 |
Fritz L. LaPorte | | | |
| | | |
| | | | |
/s/ S. Morry Blumenfeld, Ph.D. | | Director | | March 10, 2011 |
S. Morry Blumenfeld, Ph.D. | | | | |
| | | | |
/s/ Christopher C. Dewey | | Director | | March 10, 2011 |
Christopher C. Dewey | | | | |
| | | | |
/s/ Charles W. Federico | | Director | | March 10, 2011 |
Charles W. Federico | | | | |
| | | | |
/s/ John G. Freund, M.D. | | Director | | March 10, 2011 |
John G. Freund, M.D. | | | | |
| | | | |
/s/ Frederic H. Moll, M.D. | | Director | | March 10, 2011 |
Frederic H. Moll, M.D. | | | | |
| | | | |
/s/ Richard R. Pettingill | | Director | | March 10, 2011 |
Richard R. Pettingill | | | | |
| | | | |
/s/ William D. Pruitt | | Director | | March 10, 2011 |
William D. Pruitt | | | | |
| | | | |
/s/ John J. Savarese, M.D. | | Director | | March 10, 2011 |
John J. Savarese, M.D. | | | | |
120
Table of Contents
EXHIBIT INDEX
| | | |
Exhibit
No. | | Description | |
10.6 | | Lease, by and between Registrant and Westport Business Park Associates LLP, dated September 8, 2010 |
| | |
23 | | Consent of Independent Registered Public Accounting Firm |
| | | |
31.1 | | Certification of Chief Executive Officer pursuant to Rule 13a-14(a) of the Exchange Act |
| | | |
31.2 | | Certification of Chief Financial Officer pursuant to Rule 13a-14(a) of the Exchange Act |
| | | |
32.1 | | Certification of Chief Executive Officer pursuant to 18 U.S.C. §1350 |
| | | |
32.2 | | Certification of Chief Financial Officer pursuant to 18 U.S.C. §1350 |
121